UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
(Mark One)
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
OR
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report
Commission file number
(Exact name of registrant as specified in its charter and translation of Registrant’s name into English) |
The |
(Jurisdiction of incorporation or organization) |
(Address of principal executive offices) |
|
(Name, telephone, e-mail and/or facsimile number and address of company contact person) |
Securities registered or to be registered pursuant to Section 12(b) of the Act:
Title of each class: | Trading Symbol: | Name of each exchange on which registered: |
* Not for trading, but only in connection with the registration of the American Depositary Shares.
Securities registered or to be registered pursuant to Section 12(g) of the Act: None.
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act: None.
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report:
As of December 31, 2023
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
Yes ◻
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Accelerated filer ◻ | Non-accelerated filer ◻ | Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ◻
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
U.S. GAAP ◻ |
| Other ◻ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
Item 17 ◻ Item 18 ◻
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes
(APPLICABLE ONLY TO ISSUERS INVOLVED IN BANKRUPTCY PROCEEDINGS DURING THE PAST FIVE YEARS.)
Indicate by check mark whether the registrant has filed all documents and reports required to be filed by Section 12, 13 or 15(d) of the Securities Exchange Act of 1934 subsequent to the distribution of securities under a plan confirmed by a court.
Yes ☐ No ◻
TABLE OF CONTENTS
Page | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
39 | ||
39 | ||
40 | ||
104 | ||
105 | ||
105 | ||
105 | ||
106 | ||
118 | ||
121 | ||
121 | ||
122 | ||
122 | ||
122 | ||
130 | ||
152 | ||
162 | ||
163 | ||
DISCLOSURE OF A REGISTRANT’S ACTION TO RECOVER ERRONEOUSLY AWARDED COMPENSATION | 163 | |
163 | ||
163 | ||
166 | ||
168 | ||
168 | ||
168 | ||
169 | ||
169 | ||
169 | ||
169 | ||
169 | ||
169 | ||
169 | ||
170 | ||
170 | ||
170 | ||
170 | ||
173 | ||
173 |
ii
174 | ||
191 | ||
191 | ||
192 | ||
192 | ||
192 | ||
194 | ||
194 | ||
194 | ||
194 | ||
194 | ||
| 196 | |
|
| |
196 | ||
MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS | 196 | |
196 | ||
197 | ||
197 | ||
197 | ||
198 | ||
199 | ||
PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS | 199 | |
199 | ||
199 | ||
200 | ||
DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS | 200 | |
200 | ||
200 | ||
| 202 | |
|
| |
202 | ||
202 | ||
202 |
iii
Introduction
Unless otherwise indicated, “argenx,” “argenx SE,” “the Company,” “our company,” “we,” “us”, “our” our “Group” refer to argenx SE and its consolidated subsidiaries.
We own various trademark registrations and applications, and unregistered trademarks, including VYVGART®, VYVGART HYTRULO™, VYVDURA®, ARGENX™, ABDEG™, NHANCE™, SIMPLE ANTIBODY™, ARGENXMEDHUB™ and our corporate logo. Trade names, trademarks and service marks of other companies appearing in this annual report on Form 20-F (Annual Report) are the property of their respective holders. Solely for convenience, the trademarks and trade names in this Annual Report may be referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend to use or display other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship, any other companies.
VYVGART® (efgartigimod alfa) (VYVGART) has been approved in the United States (U.S.), Japan, Europe, the United Kingdom (UK), Israel, mainland China (Mainland China) and Canada for the intravenous (IV) treatment of generalized myasthenia gravis (gMG). We have now commercialized VYVGART in the U.S., several countries in the European Union (EU), Japan, Mainland China (through our partner Zai Lab Ltd (Zai Lab)), Israel (through Medison Pharma Ltd., Medison) and Canada (all such countries together including Iceland, Norway and Liechtenstein, the VYVGART Approved Countries).
VYVGART subcutaneous (SC) (efgartigimod alfa + hyaluronidase qvfc) (VYVGART SC) has been approved in the U.S. as VYVGART HYTRULO™ (VYVGART HYTRULO) and in Japan as VYVDURA® (VYVDURA) for the treatment of gMG. VYVGART SC has also been approved in the EU and the UK for the treatment of gMG. We have now commercialized VYVGART SC in the U.S. (as VYVGART HYTRULO) and in Germany. Pricing and reimbursement discussions for VYVGART SC remain ongoing in multiple other countries, including in the EU and Japan (as VYVDURA).
For both VYVGART and VYVGART SC, we are aiming for further approvals and we are working to expand commercialization in other jurisdictions.
Where not specified, references in this Annual Report to VYVGART should be read as references to VYVGART and/or VYVGART SC, including VYVGART HYTRULO in relation to the U.S. and VYVDURA in relation to Japan, depending on the context.
Our audited consolidated financial statements have been prepared in accordance with International Financial Reporting Standards (IFRS), as issued by the International Accounting Standards Board (IASB). Accordingly, our consolidated financial statements are presented in this Annual Report in U.S. dollars. All references in this Annual Report to “$,” “US$,” “U.S.$,” “U.S. dollars,” “dollars” and “USD” mean U.S. dollars and all references to “€,” “EUR,” and “euros” mean euros, unless otherwise noted. Throughout this Annual Report, references to ADSs mean American depositary shares (ADSs) or ordinary shares represented by ADSs, as the case may be.
Cautionary Statement with Respect to Forward-Looking Statements
This Annual Report contains certain forward-looking statements. A forward-looking statement is any statement that does not relate to historical facts or events or to facts or events as of the date of this Annual Report or that are derived from our management’s beliefs and assumptions based on information currently available to our management. Forward-looking statements are generally identified by the use of forward-looking words, such as “anticipate”, “aspire”, “believe”, “can”, “continue”, “could”, “estimate”, “expect”, “hope”, “intend”, “is designed to”, “look forward to”, “may”, “might”, “objective”, “plan”, “potential”, “project”, “predict”, “seek”, “should”, “target”, “will”, or other variations or the negative of such terms, or by discussion of strategy, although not all forward-looking statements contain these identifying words. These statements relate to our future results of operations and financial positions, prospects, developments, business strategies, plans and our objectives for future operations, results of clinical trials and regulatory approvals, and are based on analyses or forecasts of future developments and estimates of amounts not yet determinable.
iv
These forward-looking statements represent the view of management only as of the date of this Annual Report, and we disclaim any obligation to update forward-looking statements, except as may be otherwise required by law. The forward-looking statements in this Annual Report involve known and unknown risks, uncertainties and other factors that could cause our actual future results, performance and achievements to differ materially from those forecasted or suggested herein.
Forward-looking statements include, but are not limited to, statements about:
| ● | the initiation, timing, progress, development and results of clinical trials of our product candidates, including new indications, alternative dosing regimens and treatment modalities, including statements regarding when results or interim analysis of the clinical trials will be available or made public; |
| ● | the expansion of our business, including the further development of our sales and marketing abilities and our Immunology Innovation Program (IIP), and the value of our pipeline; |
| ● | the potential attributes and benefits of our products and product candidates, including new indications, alternative dosing regimens and treatment modalities, and their competitive position with respect to other alternative treatments; |
| ● | our ability to advance product candidates into, and successfully complete, clinical trials; |
| ● | our estimates of the number of patients who suffer from the diseases we are targeting and the number of patients that will enroll in our clinical trials; |
| ● | the commercialization of our products and product candidates, including new indications, alternative dosing regimens and treatment modalities, if approved; |
| ● | the anticipated timing of market authorizations of our products, including new indications, alternative dosing regimens and treatment modalities; |
| ● | the anticipated pricing and reimbursement of our products and product candidates, if approved; |
| ● | our plans to have various programs to help patients afford our products, including patient assistance and co-pay coupon programs for eligible patients; |
| ● | the timing or likelihood of regulatory filings and decisions for any products and product candidates, including new indications, alternative dosing regimens and treatment modalities; |
| ● | our ability to establish sales, marketing and distribution capabilities for any of our products and product candidates that achieve regulatory approval; |
| ● | our regulatory strategy and our ability to establish and maintain manufacturing arrangements for our products and product candidates; |
| ● | the scope and duration of protection, including any exclusivity period, we are able to establish and maintain for intellectual property rights covering our products and product candidates, platform and technology, including our intention to seek patent term extensions where available; |
| ● | our estimates regarding expenses, future revenues, cash burn, capital requirements and our needs for additional financing; |
| ● | our financial performance, including potential volatility in the price of our ordinary shares and ADSs; |
v
| ● | the rate and degree of market acceptance of our products and product candidates, if approved; |
| ● | the potential benefits of our current collaborations, including the possibility to access partner technology platforms or capabilities; |
| ● | our plans and ability to enter into collaborations for additional programs or product candidates; |
| ● | our plans and ability to enter into new distribution partnerships; |
| ● | the impact of government laws and regulations on our business; |
| ● | our expectations with respect to the timing and amount of any dividends; |
| ● | our plans regarding our supply chain, including our reliance on third parties, including contract manufacturing organizations (CMOs); and |
| ● | the implementation of our diversity, equity and inclusion policy, including our goal to further improve diversity on our board of directors (Board of Directors). |
These include changes in general economic and business conditions. You should refer to Item 3.D. “Risk Factors” of this Annual Report for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this Annual Report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this Annual Report and the documents that we reference in this Annual Report and have filed as exhibits to the Annual Report completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
Information regarding market and industry statistics contained in this Annual Report is included based on information available to us that we believe is accurate. Forecasts and other forward-looking information obtained from this available information is subject to the same qualifications and the additional uncertainties accompanying any estimates of future market size, revenue and market acceptance of products and services.
In addition, statements that include “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this Annual Report, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and you are cautioned not to unduly rely upon these statements.
vi
Summary of Risk Factors
Our ability to implement our business strategy is subject to numerous risks that you should be aware of before making an investment decision. These risks are described more fully below. These risks include, among others:
| ● | We have incurred significant losses since our inception and expect to incur losses for the foreseeable future. We may never achieve or sustain profitability. |
| ● | We may need to raise substantial additional funding which may not be available to us on acceptable terms or at all. |
| ● | Our assets, earnings and cash flows and the investment of our cash and cash equivalents may be subject to risks which may cause losses and affect the liquidity of these investments. |
| ● | We will face significant challenges in successfully commercializing our products and additional product candidates after they are launched. |
| ● | The commercial success of our products and product candidates, including in new indications or methods of administration, will depend on the degree of market acceptance. |
| ● | We face significant competition for our drug discovery and development efforts. |
| ● | Enacted and future legislation could impact demand for our products which could impact our business and future results of operations. |
| ● | We are subject to government pricing laws, regulation and enforcement. These laws affect the prices we may charge the government for our products and the reimbursement our customers may obtain from the government. Our failure to comply with these laws could harm our results, operations and/or financial conditions. |
| ● | We may not obtain or maintain adequate coverage or reimbursement status for our products and product candidates. |
| ● | If we fail to obtain orphan drug designation or we do not have valid and enforceable patents covering our products and product candidates and fail to obtain and/or maintain orphan drug exclusivity for our products or product candidates, our competitors may be able to sell products to treat the same conditions and our revenue may be reduced. |
| ● | We are subject to healthcare laws, regulation and enforcement. The failure to comply with these laws could harm our results, operations and/or financial conditions. |
| ● | All aspects of our business ranging from preclinical, clinical trials, marketing and commercialization are highly regulated and any delay by relevant regulatory authorities could jeopardize our development and approval process or result in other suspensions, refusals or withdrawal of approvals. |
| ● | We are subject to privacy laws, regulation and potential enforcement. Our failure to comply with these laws could harm our results, operations and/or financial conditions. |
| ● | Failure to successfully identify, select and develop VYVGART in other indications, or additional products or product candidates could impair our ability to grow. |
| ● | VYVGART has obtained regulatory approval in the VYVGART Approved Countries for the treatment of gMG. Our other products and product candidates – including additional indications or methods of use for efgartigimod, empasiprubart and ARGX-119 – are either in preclinical or clinical development or are pending marketing approval. |
vii
| ● | Our clinical trials have, and may in the future, fail, and even if they succeed, we may not obtain regulatory approval for our products and product candidates or regulatory approval may be delayed. |
| ● | Our products and product candidates may have serious adverse, undesirable or unacceptable side effects or even cause death, and we or others may identify undesirable or unacceptable side effects caused by VYVGART or any of our products or product candidates after they have received marketing approval. |
| ● | If our target patient population is smaller than expected, we are unable to successfully enroll and retain patients in our clinical trials, or experience significant delays in doing so, we may not realize the full commercial potential of any products or product candidates. |
| ● | We rely, and expect to continue to rely, on third parties to conduct some of our research activities and clinical trials and for parts of the development and commercialization of our existing and future research programs, products and product candidates. If our relationships with such third parties are not successful, our business may be adversely affected. |
| ● | Disruptions caused by our reliance on third parties for our manufacturing process may delay or disrupt our business, product development and commercialization efforts. |
| ● | Failure to adequately enforce or protect our intellectual property rights in products, product candidates and platform technologies could adversely affect our ability to maximize the value for patients in our marketed products and product candidates. |
| ● | If our trademarks and trade names are not adequately protected, we may not be able to build name recognition in our markets of interest. |
| ● | We may encounter difficulties efficiently managing our growth and our increasing development, regulatory and sales and marketing capabilities, which could disrupt our operations. |
| ● | The price of our ADSs and ordinary shares may be volatile and may fluctuate due to factors beyond our control. An active public trading market may not be sustained. |
| ● | Holders of our ADSs are not treated as holders of our ordinary shares and may be subject to limitations on the transfer of their ADSs and the withdrawal of the underlying ordinary shares. |
| ● | We are a Dutch European public company with limited liability (Societas Europaea or SE). The rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions. |
| ● | Claims of U.S. civil liabilities may not be enforceable against us or the members of our management and our Board of Directors. |
| ● | As a foreign private issuer, we are exempt from certain rules under U.S. securities laws and are permitted to file less information with the U.S. Securities and Exchange Commission (SEC) than a U.S. company. |
| ● | We may lose our foreign private issuer status which would then require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur significant legal, accounting and other expenses. |
| ● | If we were to be classified as a passive foreign investment company for U.S. federal income tax purposes, this could result in adverse U.S. tax consequences to certain U.S. holders. |
viii
PART I
ITEM 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS
Not applicable.
ITEM 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not applicable.
ITEM 3. KEY INFORMATION
A. [RESERVED]
B. CAPITALIZATION AND INDEBTEDNESS
Not applicable.
C. REASONS FOR THE OFFER AND USE OF PROCEEDS
Not applicable.
D. RISK FACTORS
Our business faces significant risks, including those described below. You should carefully consider all of the information set forth in this Annual Report and in our other filings with the SEC, including the following risk factors which we face and are faced by our industry. Our business, financial condition or results of operations could be materially and adversely affected if any of these risks occurs. These are not the only risks argenx faces. Additional risks and uncertainties not presently known to argenx or that it currently considers immaterial or not specific may also impair its business, results of operation and financial condition. This report also contains forward-looking statements that involve risks and uncertainties. Our actual results could differ materially and adversely from those anticipated in these forward-looking statements as a result of certain factors including the risks described below and elsewhere in this Annual Report and our other SEC filings. See “Cautionary Statement with Respect to Forward-Looking Statements.”
Risk Factors Related to argenx’s Financial Position and Need for Additional Capital
We have incurred significant losses since our inception and expect to incur losses for the foreseeable future. We may never achieve or sustain profitability.
Since our inception, we have incurred significant operating losses, totaling $2,405 million of cumulative losses. To date we have commercialized VYVGART for the treatment of gMG. We do not currently have any marketing approvals for any other product candidates or VYVGART in other indications. Our losses resulted principally from costs incurred in research and development, preclinical testing and clinical development of our research programs, and from general and administrative costs associated with commercial roll out and expansion. We intend to continue to conduct research and development, preclinical testing, clinical trials and regulatory compliance activities as well as the continued commercialization of VYVGART and other products candidates, for current and future indications, and we intend to continue our efforts to expand our sales, marketing and distribution infrastructure. These expenses, together with anticipated general and administrative expenses, may result in incurring further losses for the foreseeable future. We anticipate that our operating expenses will increase if and as we execute our strategic objectives and as we experience delays or encounter issues relating thereto, including failed clinical trials, ambiguous clinical trial results, safety issues or other regulatory challenges.
1
Although we have generated net product sales of $1.2 billion from global product net sales in fiscal year 2023, we can provide no assurances that we will be able to achieve or sustain profitability based on sales in that indication alone or that we will be able to receive regulatory approval of and commercialize VYVGART and VYVGART SC in other indications or in other countries. To become and remain profitable, we must succeed in developing and commercializing products that generate significant revenue. This will require us to be successful in a range of challenging activities, including completing preclinical testing and clinical trials of our products and our product candidates, discovering and developing additional products and product candidates, including new indications, obtaining regulatory approval, establishing manufacturing and marketing capabilities, obtaining funding or reimbursement for our products, and ultimately selling. Those activities are the drivers of our current path to profitability, however, we may not succeed in some or even all of these activities, and even if we do, we may not generate revenue that is significant enough to achieve profitability.
We may need to raise substantial additional funding which may not be available to us on acceptable terms or at all.
We have significant positions of cash and cash equivalents of $2,049 million and current financial assets of $1,131 million as of December 31, 2023. Developing products and product candidates, including new indications, and conducting clinical trials is time-intensive, expensive and risky. Our future capital requirements will depend on many factors, including: (i) the success, cost and timing of our development activities, preclinical testing and clinical trials for our product and product candidates, (ii) the time and costs involved in obtaining regulatory approvals and any delays we may encounter, including as we seek regulatory approval in additional jurisdictions or other indications, (iii) commercialization, manufacturing, sales and marketing of products and product candidates, (iv) securing adequate and uninterrupted supply chains, (v) the costs involved in growing our organization to the size needed to allow for the research, development and potential commercialization of our products or product candidates, (vi) the costs involved in filing patent applications and maintaining and enforcing patents or defending against claims or infringements raised by third parties, (vii) the maintenance of our existing collaboration agreements and entry into new collaboration agreements, and (viii) the amount of revenue, if any, we may derive either directly or in the form of royalty payments from future sales of our products or product candidates, if approved.
To finance our operations, we may need to raise additional capital through a combination of public or private equity or debt financings or other sources, which may include collaborations with third parties. For example, we completed a global offering in July 2023 whereby we raised $1.3 billion in gross proceeds from the sale of 1,917,715 ADSs at a price of $490.00 per ADS and the sale of 663,918 ordinary shares at a price of €436.37 per ordinary share. Our ability to raise additional funds on acceptable terms or at all will depend on financial, economic and market conditions and other factors, over which we may have no or limited control. If we are unable to raise additional capital if and when needed, or if the terms are not acceptable, our business strategy could be impacted, and we may be forced to delay, reduce or terminate the one or more of our research or development programs or the commercialization of any of our products or product candidates, including new indications, or be unable to expand our operations or otherwise capitalize on our business opportunities, all of which may have a material adverse impact on our business, financial condition and results of operations.
Our assets, earnings and cash flows and the investment of our cash and cash equivalents may be subject to risks which may cause losses and affect the liquidity of these investments.
As of December 31, 2023, we had cash and cash equivalents and current financial assets of $3.2 billion compared to $2.2 billion as at December 31, 2022. All of our available cash and cash equivalents and current financial assets are invested in either current accounts, savings accounts, term accounts or highly liquid money market funds. Any future investments may include term deposits, corporate bonds, commercial paper, certificates of deposit, government securities and money market funds in accordance with our cash investment policy. These investments may be subject to general credit, liquidity, market, inflation, foreign currency and interest rate risks and we may realize losses in the fair value of these investments or a complete loss of these investments, which would have a negative effect on our financial condition. The market risks associated with our cash flows and investment portfolio may adversely affect our results of operations, liquidity and financial condition.
2
Due to the international scope of our operations, our assets, earnings and cash flows are influenced by movements in exchange rates of several currencies, particularly between the U.S. dollar, euro and Japanese Yen. Our revenue from outside of the U.S. will increase as our products, whether commercialized by us or our business partners or our collaborators gain marketing approval in such jurisdictions. If the U.S. dollar weakens against a specific foreign currency, our revenues will increase, having a positive impact on net income, but our overall expenses will increase, having a negative impact. Conversely, if the U.S. dollar strengthens against a specific foreign currency, our revenues will decrease, having a negative impact on net income, but our overall expenses will decrease, having a positive impact. Continued volatility in foreign exchange rates is likely to impact our operating results and financial condition.
Risk Factors Related to Commercialization of argenx’s Products and Product Candidates, Including for New Indications
We will face significant challenges in successfully commercializing our products and additional product candidates after they are launched.
The commercialization of VYVGART in new indications or other approved product candidates, or entrance of any of our products or product candidates into new markets will require us to further expand our sales and marketing organization, enter into collaboration arrangements with third parties, outsource certain functions to third parties, or use some combination of each. We have built, and continue to expand, our sales forces in certain of the VYVGART Approved Countries and plan to further develop our sales and marketing capabilities to promote our products, and product candidates, including new indications, if and when marketing approval has been obtained in other relevant jurisdictions.
Even if we successfully expand our sales and marketing capabilities, either on our own or in collaboration with third parties, we may fail to launch or market our products effectively. Recruiting and training a specialized sales force is expensive and the costs of expanding an independent sales, marketing and/or promotion organization could be greater than we anticipate. We could further encounter difficulties in our sales or marketing, due to regulatory actions, shut-downs, work stoppages or strikes, approval delays, withdrawals, recalls, penalties, supply disruptions, shortages or stock-outs at our facilities or third-party facilities that we rely on, reputational harm, the impact to our facilities due to pandemics or natural or man-made disasters, including as a result of climate change, product liability, and/or unanticipated costs. In addition, recruiting and training a sales force is time-consuming and could delay any product launch. In the event that any such launch is delayed or does not occur for any reason, we would have prematurely or unnecessarily incurred these commercialization expenses, and our investment would be lost if we cannot retain or reposition our sales and marketing personnel.
We have entered into distribution agreements with Medison, Zai Lab, Genpharm Services FZ-LLC (Genpharm) and Handok Inc. (Handok) to perform sales and marketing services in Israel, Central and Eastern Europe, Mainland China, the Gulf Cooperation Council (GCC) and South Korea, respectively. Under these agreements, our product revenues or the profitability of these product revenues could be lower than if we were to market and sell the products that we develop ourselves. Such distribution agreements may place the commercialization of our products outside of our control, including over the amount or timing of resources that our distribution partners devote to our products. Furthermore, our distributors’ willingness or ability to comply with and complete their obligations under our arrangements may be adversely affected by business combinations or significant changes in our distributors’ business strategies. In addition, we may not succeed in entering into arrangements with third parties to sell and market our products or may be unable to do so on terms that are favorable to us.
3
The commercial success of our products and product candidates, including in new indications or methods of administration, will depend on the degree of market acceptance.
Our products and product candidates, including for new indications or methods of administration, if and when approved and available on the market, may never achieve an adequate level of acceptance by physicians, patients, the medical community, or healthcare payors for us to be profitable. This will depend on a number of factors, many of which are beyond our control, including, but not limited to:
| ● | the efficacy and safety as demonstrated by clinical trials and subsequent prevalence and severity of any side effects; |
| ● | approval may be for indications, dosage and methods of administration or patient populations that are not as broad as intended or desired; |
| ● | changes in the standard of care for the targeted indications for any product and product candidate; |
| ● | availability of alternative approved therapies; |
| ● | sales, marketing and distribution support; |
| ● | labeling may require significant use or distribution restrictions or safety warnings; |
| ● | potential product liability claims; |
| ● | acceptance by physicians, patients and healthcare payors of each product as safe, effective and cost-effective, and any subsequent changes thereof; |
| ● | relative convenience, ease of use, including administration, perceived dosing complexity and other perceived advantages over alternative and/or new products; |
| ● | patient continued commitment required to receive periodic in-center infusions; |
| ● | prevalence and severity of adverse events discovered before or after marketing approval has been received; |
| ● | consumer perceptions or publicity regarding our business or the safety and quality of our product and product candidates, clinical trials for new indications, or any similar products distributed by other companies; |
| ● | limitations, precautions or warnings listed in the summary of product characteristics, patient information leaflet, wording of package labeling or instructions for use, and any subsequent changes thereof; |
| ● | the cost of treatment with our products in relation to alternative and/or new treatments; |
| ● | the extent to which products are approved for inclusion and reimbursed on formularies of hospitals and managed care organizations, and any subsequent changes thereof; and |
| ● | whether our products are designated in the label, under physician treatment guidelines or under reimbursement guidelines as a first-line, second-line, third-line or last-line therapy, and any subsequent changes thereof. |
In addition, because we are developing our products and product candidates for the treatment of different indications, negative results in a clinical trial evaluating the efficacy and safety of a product or product candidate for one indication could negatively impact the perception of the efficacy and safety of such product or product candidate in a
4
different indication, which could have an adverse effect on our reputation, commercialization efforts and financial condition.
Moreover, efforts to educate the medical community and third-party payors on the benefits of our products and product candidates may require significant resources and may never be successful. If our product candidates or methods of use of existing products or new indications fail to gain market acceptance, this will have a material adverse impact on our ability to generate revenues. Even if some products achieve market acceptance, they may not be able to retain market acceptance and/or the market may prove not to be large enough to allow us to generate significant revenues.
We face significant competition for our drug discovery and development efforts.
The market for pharmaceutical products is highly competitive and characterized by rapidly growing understanding of disease biology, quickly changing technologies, strong intellectual property barriers to entry, and a multitude of companies involved in the creation, development, and commercialization of novel therapeutics. Many of these companies are highly sophisticated and often strategically collaborate with each other.
Competition in the autoimmune field is intense and involves multiple monoclonal antibodies (mAbs), other biologics and small molecules either already marketed or in development by many different companies including large pharmaceutical companies such as AbbVie, Inc. (AbbVie) (Humira/rheumatoid arthritis), Amgen, Inc. (Amgen) (Enbrel/rheumatoid arthritis), Biogen Inc, (Tysabr/multiple sclerosis), GlaxoSmithKline plc (GSK) (Benlysta/lupus), F. Hoffman-La Roche AG (Roche) (Rituxan/often used off label) and Janssen Pharmaceuticals, Inc. (Remicade/rheumatoid arthritis and Stelara/psoriasis). In addition, these and other pharmaceutical companies have mAbs or other biologics in clinical development for the treatment of autoimmune diseases.
Currently, our commercial revenue is generated by VYVGART, VYVGART HYTRULO and VYVGART SC in gMG. We face and expect to continue to face intense competition from other biopharmaceutical companies, who have launched or are developing products for the treatment of gMG and other autoimmune diseases, including products that are in the same class as VYVGART, as well as products that are similar to some of our product candidates. Competition for other (potential) future indications is also fierce, with significant development in almost all of the indications we are currently developing or planning to develop for our product or product candidates. For example, we are aware of several neonatal Fc receptor (FcRn) inhibitors that are in clinical development and one FcRn inhibitor, Rystiggo (rozanolixizumab-noli), which was approved in June 2023. We are also aware that AstraZeneca PLC is selling Soliris and Ultomiris for the treatment of adult patients with gMG who are AchR-AB+ and that UCB is selling Rystiggo for the treatment of adult patients with gMG who are AchR-AB+ or MuSK-AB+ and Zilbrysq for the treatment of adult patients with gMG who are AchR-AB+. Roche, Novartis AG, CSL Behring, Grifols, S.A., Curavac, Inc., Takeda Pharmaceutical Co Ltd, RemeGen Co, Immunovant, Inc., Cartesian Therapeutics, Inc., Horizon Therapeutics PLC, Regeneron Pharmaceuticals, Inc./Alnylam Pharmaceuticals, Inc. and Johnson & Johnson Innovation, Inc. (Johnson & Johnson), among others, are developing drugs that may have utility for the treatment of myasthenia gravis (MG).
Competitive product launches may erode future sales of our products, including our existing products and those currently under development, or result in unanticipated product obsolescence. Such launches continue to occur, and potentially competitive products are in various stages of development. We could also face competition for use of limited international infusion sites, particularly in new markets as competitors launch new products. We cannot predict with accuracy the timing or impact of the introduction of competitive products that treat diseases and conditions like those treated by our products or product candidates. In addition, our competitors and potential competitors compete with us in recruiting and retaining qualified scientific, clinical research and development and management personnel, establishing clinical trial sites, registering patients for clinical trials, as well as in acquiring technologies complementary to, or necessary for, the development of our products.
There can be no assurance that our competitors are not currently developing, or will not in the future develop, technologies and products that are equally or more effective, are more economically attractive, and can be administered more easily than any of our current or future technologies or products.
5
Competing products or technology platforms may gain faster or greater market acceptance than our products or technology platforms and medical advances or rapid technological development by competitors may result in our products and product candidates or technology platforms becoming non-competitive or obsolete before we are able to recover our research and development and commercialization expenses. If we, our products and product candidates or our technology platforms do not compete effectively, it is likely to have a material adverse effect on our business, financial condition and results of operation.
Our products and product candidates for which we have obtained or intend to seek approval as biological products, including for new indications, may face biosimilar competition.
In the U.S., the Biologics Price Competition and Innovation Act (BPCIA) created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with a U.S. Food and Drug Administration (FDA)-licensed reference biological product. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full biologics license application (BLA) for the competing product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of their product.
We believe that any of our product candidates approved as a biological product under a BLA should qualify for the 12-year period of exclusivity, as was the case with VYVGART and VYVGART HYTRULO. The regulatory exclusivity periods for VVVGART and VYVGART HYTRULO is expected to extend until December 2033 in the U.S. Regulatory protection in the EU (both orphan and data/marketing exclusivity) is expected to expire in August 2032 in the European Economic Area (EEA) and March 2033 in the UK. Following those periods of regulatory exclusivity, we must enforce our patent rights against biosimilar products that infringe the patent claims of these products. However, there is a risk that this exclusivity could be shortened due to congressional action or otherwise, or that the FDA will not consider our product candidates to be reference products for competing products, potentially creating the opportunity for competition by biosimilar products sooner than anticipated. Moreover, an interchangeable biosimilar product, once approved, may be substituted under existing state law for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products. Any non-interchangeable biosimilar products may also be substituted by a healthcare provider but, under existing law, will not be automatically substituted at the pharmacy. The extent of the impact of such substitution will depend on a number of marketplace and regulatory factors that are still developing.
In the EU, biosimilars are evaluated for marketing authorization pursuant to a set of general and product class-specific guidelines. In April 2023, the European Commission adopted a proposal to revise the EU’s pharmaceutical legislation. If adopted in the form proposed, a number of changes to the regulatory framework governing medicinal products in the EEA would occur, including a shortened period of data and market exclusivity. In addition, in an effort to spur biosimilar utilization and/or increase potential healthcare savings, some EU Member States have adopted, or are considering the adoption of, biosimilar uptake measures such as physician prescribing quotas or automatic pharmacy substitution of biosimilars for the corresponding reference products. Some EU Member States impose automatic price reductions upon market entry of one or more biosimilar competitors. In September 2022, the European Medicines Authority (EMA) and the EU Heads of Medicines’ Agencies issued a joint statement providing that biosimilar medicines approved in the EU are “interchangeable” with their reference products and other biosimilars of the same reference product. This statement could further contribute to the prescribing of biosimilars and to greater competition in Europe. While the degree of competitive effects of biosimilar competition differs among EU Member States and among products, the overall use of biosimilars and the rate at which product sales of innovative products are being affected by biosimilar competition is increasing.
6
Enacted and future legislation could impact demand for our products which could impact our business and future results of operations.
In the U.S., the UK, the EU and other jurisdictions, there have been a number of legislative and regulatory changes to the healthcare systems that could affect our future results of operations. Governmental regulations that mandate price controls or limitations on patient access to our products or establish prices paid by government entities or programs for our products could impact our business, and our future results of operations could be adversely affected by changes in such regulations or policies. For example, if the European Commission’s recent proposal to revise the EU’s pharmaceutical legislation is adopted in the form proposed, we may be affected by a decrease in data and market exclusivity for our products and product candidates in the EEA.
In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs in general and the cost of pharmaceuticals in particular. Healthcare reform initiatives in the U.S. culminated in the enactment of the Inflation Reduction Act (IRA) in August 2022, which allows, among other things, the U.S. Department of Health and Human Services (HHS) to directly negotiate the selling price of a statutorily specified number of drugs and biologics each year that the Centers for Medicare & Medicaid Services (CMS) reimburses under Medicare Part B and Part D. Only high-expenditure single-source biologics that have been approved for at least 11 years (7 years for single-source drugs) can qualify for negotiation, with the negotiated price taking effect two years after the selection year. In August 2023, the U.S. government announced the first 10 drugs to be subject to negotiation, although the Medicare drug price negotiation program is currently subject to legal challenges. Negotiations for Medicare Part D products begin in 2024 with the negotiated price taking effect in 2026, and negotiations for Medicare Part B products begin in 2024 with the negotiated price taking effect in 2028. HHS will announce the negotiated maximum fair price by September 1, 2024, and this price cap, which cannot exceed a statutory ceiling price, will come into effect on January 1, 2026.
The IRA also penalizes drug manufacturers that increase prices of Medicare Part D and Part B drugs at a rate greater than the rate of inflation. The IRA will also cap out-of-pocket spending for Medicare Part D enrollees and make other Part D benefit design changes beginning in 2024. Beginning in 2025, the IRA eliminates the coverage gap under Medicare Part D by significantly lowering the enrollee maximum out-of-pocket cost to $2,000 and by requiring manufacturers to subsidize, through a newly established manufacturer discount program, 10% of Part D enrollees’ prescription costs for brand drugs below the out-of-pocket maximum, and 20% once the out-of-pocket maximum has been reached (plans will also be required to cover 20% in this case). Although these discounts represent a lower percentage of enrollees’ costs than the current discounts required below the out-of-pocket maximum (that is, in the coverage gap phase of Part D coverage), the new manufacturer contribution required above the out-of-pocket maximum could be considerable for very high-cost patients and the total contributions by manufacturers to a Part D enrollee’s drug expenses may exceed those currently provided. These Part D design changes may also incentivize Part D plans to exclude certain drugs from their formularies, which could affect the supply, demand, and pricing of our product and product candidates.
The IRA permits the Secretary of HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years. HHS has and will continue to issue and update guidance as these programs are implemented. Manufacturers that fail to comply with the IRA may be subject to various penalties, some significant, including civil monetary penalties. The IRA also extends enhanced subsidies for individuals purchasing health insurance coverage in ACA (as defined below) marketplaces through plan year 2025. These provisions began taking effect progressively starting in 2023, although they may be subject to legal challenges. For example, the provisions related to the negotiation of selling prices of high-expenditure single-source drugs and biologics have been challenged in multiple lawsuits, Thus, while the full economic impact of IRA is unknown at this time, the law’s passage is likely to affect the pricing of our products and product candidates. In addition, in response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the Center for Medicare and Medicaid Innovation, which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether the models will be utilized in any health reform measures in the future. The adoption of restrictive price controls in new jurisdictions, more restrictive controls in existing jurisdictions, the adoption of these lower prices by commercial payors, or the failure to obtain or maintain timely or adequate pricing could also adversely impact revenue. We expect pricing pressures will continue globally.
7
Further, at the U.S. state level, legislatures are increasingly enacting laws and implementing regulations designed to control pharmaceutical and biological product pricing, including price or reimbursement constraints, discount requirements, marketing cost disclosure and price transparency reporting, and programs designed to encourage importation from other countries and bulk purchasing. States are also enacting laws modeled on federal policies, such as the IRA and the 340B drug discount program. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, including pharmaceuticals, which could result in reduced demand for our products and product candidates or additional pricing pressures.
The EU, on the other hand, has reopened the entire legislative framework for medicinal products. On April 26, 2023, the European Commission has published its proposal for a new directive (COM/2023/192 final) and a new regulation (COM/2023/193 final), which would revise and replace the existing general pharmaceutical legislation, including e.g., Directive 2001/83/EC, as well as Regulations (EC) No. 726/2004, No. 141/2000, or No. 1901/2006 (EU Pharmaceutical Legislation). This proposal is currently undergoing the ordinary legislative procedure in the European Parliament and Council of the European Union and is therefore still subject to changes. If at all, the EU Pharmaceutical Legislation is expected to be implemented at the earliest in the next few years. Prevention and mitigation of medicine shortages, simplification of the market entry of generics and biosimilars, the reduction of the regulatory burden (e.g., by increased digitalization) and the implementation of a new regime for data and / or market exclusivity (e.g., by reducing the minimum period while introducing factors that, if met, prolong protections for marketing authorization holders) are among the major objectives pursued by the European Commission. Pending the outcome of the legislative procedure, the impact could be positive with respect to certain regulatory processes. There could, however, also be a negative impact on innovative pharma and biotech companies such as argenx due to shorter baseline regulatory and orphan exclusivities if the proposal is not amended.
We are subject to government pricing laws, regulation and enforcement. These laws affect the prices we may charge the government for our products and the reimbursement our customers may obtain from the government. Our failure to comply with these laws could harm our results, operations and/or financial conditions.
In the U.S., we are required to participate in various government programs for our products to be reimbursed or purchased by the federal government. We participate in programs such as the Medicaid Drug Rebate Program, the 340B drug discount program, Medicare Part B, Medicare Part D and the U.S. Department of Veterans Affairs Federal Supply Schedule pricing program. The requirements vary by program, but among these and any other programs in which we participate, we are, among other things, required to enter into agreements with and calculate and report prices and other information to certain government agencies, charge no more than statutorily mandated ceiling prices and calculate and pay rebates and refunds for certain products.
The calculations are complex and are often subject to interpretation by us, governmental agencies and the courts. If we determine that the prices we reported were in error, we may be required to restate those prices and pay additional rebates or refunds to the extent we understated the rebate or overcharged the government due to the error. Additionally, there are penalties associated with submission of incorrect pricing or other data. We may incur significant civil monetary penalties if we are found to have knowingly submitted false prices or other information to the government, or to have charged 340B covered entities more than the statutorily mandated ceiling price. Certain failures to timely submit required data also could result in a civil monetary penalty for each day the information is late. We could also become subject to allegations under the False Claims Act and other laws and regulations. In addition, misreporting and failure to timely report data to CMS also can be grounds for CMS to terminate our Medicaid rebate agreement, pursuant to which we participate in the Medicaid Drug Rebate Program. In the event that CMS terminates our rebate agreement, no federal payments would be available under Medicaid or Medicare Part B for our covered outpatient drugs.
Recently enacted legislation in the U.S. has imposed additional rebates under government programs. For example, effective January 1, 2024, under the American Rescue Plan of 2021, rebates that manufacturers pay to state Medicaid programs was eliminated. Elimination of this cap may require pharmaceutical manufacturers to pay more in Medicaid rebates than they receive on the sale of products for products that have undergone substantial price increases. In addition, the Infrastructure Investment and Jobs Act, effective January 1, 2023, added a requirement for manufacturers of certain single-source drugs (including biologics and biosimilars) separately paid for under Medicare
8
Part B for at least 18 months and marketed in single-dose containers or packages (known as refundable single-dose containers or single-use package drugs) to provide annual refunds for any portions of the dispensed drug that are unused and discarded if those unused or discarded portions exceed an applicable percentage defined by statute or regulation. Manufacturers are subject to periodic audits and those that fail to pay refunds for their refundable single-dose containers or single-use package drugs shall be subject to civil monetary penalties. This requirement applies to VYVGART, and potentially other of our products in the future. As a result, we owe refunds to CMS starting this year. Although we will evaluate options to reduce the amount of refunds owed, pursuing any such actions will be time-consuming and costly. Even if we invest resources to reduce the amount of refunds owed to CMS, it is possible that we will be delayed or unsuccessful in achieving a reduction worthy of our investment.
Maintaining compliance with these government price reporting and discounting obligations is time-consuming and costly, and a failure to comply can result in substantial fines, penalties, all of which could adversely impact our financial results.
We may not obtain or maintain adequate coverage or reimbursement status for our products and product candidates.
Sales of VYVGART for gMG, VYVGART HYTRULO and our product candidates, if approved, will depend, in part, on the extent to which third-party payors, including government health programs in the U.S. (such as Medicare Parts B and D and Medicaid) and other countries, commercial health insurers, and managed care organizations, provide coverage and establish adequate reimbursement levels for such products and product candidates. In the U.S., no uniform policy of coverage and reimbursement for products exists among commercial third-party payors. Commercial third-party payors decide which products they will pay for and establish reimbursement levels. Commercial payors often rely upon Medicare coverage policy and payment limitations in setting their own coverage and reimbursement policies. However, decisions regarding the extent of coverage and amount of reimbursement to be provided for any product candidate that we develop through approval will be made on a plan-by-plan basis. One commercial payor’s determination to provide coverage for a product does not assure that other commercial payors will also provide coverage and adequate reimbursement for the product. Additionally, a commercial third-party payor’s decision to provide coverage for a drug does not imply that an adequate reimbursement rate will be approved. Each plan determines whether or not it will provide coverage for a product, what amount it will pay the manufacturer for the product, on what tier of its formulary the product will be placed and whether to require step therapy. The position of a product on a formulary generally determines the co-payment that a patient will need to make to obtain the product and can strongly influence the adoption of a product by patients and physicians.
Even under U.S. government healthcare programs such as Medicare and Medicaid, coverage and reimbursement policies can vary significantly. Medicare Part D is administered by commercial insurance companies under contract with the CMS. The many Part D plans operated by these companies vary considerably in their coverage and reimbursement policies, much like the commercial plans that these same companies offer, as described above. Medicare Part B and Medicaid coverage and reimbursement rates are more uniform, but even Medicaid programs vary from state to state in their coverage policies and reimbursement rates.
Patients who are prescribed treatments for their conditions and providers prescribing such services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of our products. Further, from time to time, typically on an annual basis, payment rates are updated and revised by third-party payors. Such updates could impact the demand for our products, to the extent that patients who are prescribed our products, if approved, are not separately reimbursed for the cost of the product.
The process for determining whether a third-party payor will provide coverage for a product may be separate from the process for setting the price of a product or for establishing the reimbursement rate that such a payor will pay for the product. Even if we do obtain adequate levels of reimbursement, third-party payors, such as government or private healthcare insurers, carefully review and increasingly question the coverage of, and challenge the prices charged for, products. Increasingly, third-party payors are requiring that biopharmaceutical companies provide them with predetermined discounts from list prices and are challenging the prices charged for products. We may also be required to conduct expensive pharmacoeconomic studies to justify the coverage and the amount of reimbursement for particular
9
medications. We cannot be sure that coverage and reimbursement will be available for any product that we commercialize and, if reimbursement is available, what the level of reimbursement will be.
Moreover, coverage policies and third-party payor reimbursement rates may change at any time. Therefore, even if favorable coverage and reimbursement status is attained for one or more products for which we receive marketing approval in one or more indications, less favorable coverage policies and reimbursement rates may be implemented in the future. For instance, even though favorable coverage and reimbursement status has been attained for VYVGART for the treatment of gMG in the U.S., access to VYVGART or for any other indication may be reduced or restricted by limited payer coverage due to treatment criteria, which may prevent us from realizing its full commercial potential. In addition, the coverage and reimbursement levels for our products for the treatment in one indication may have an adverse impact on the coverage and reimbursement levels of such products or product candidates in other indications for which marketing approval has previously been or may subsequently be obtained. Inadequate coverage or reimbursement may diminish or prevent altogether any significant demand for our products and/or may prevent us entirely from entering certain markets or indications, which would prevent us from generating significant revenues or becoming profitable, which would adversely affect our business, financials and results of operations.
In many foreign countries, pricing, coverage, and level of reimbursement of prescription drugs are subject to governmental control, and we and our collaborators may be unable to obtain coverage, pricing, and/or reimbursement on terms that are favorable to us or necessary for us or our collaborators to successfully commercialize our marketed products in those countries. In some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing and reimbursement vary widely from country to country, and may take into account the clinical effectiveness, cost, and service impact of existing, new, and emerging drugs and treatments. For example, the EU provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. Our results of operations may suffer if we or our collaborators are unable to market our products in foreign countries or if coverage and reimbursement for our marketed products in foreign countries is limited or delayed.
If we fail to obtain orphan drug designation or we do not have valid and enforceable patents covering our products and product candidates and fail to obtain and/or maintain orphan drug exclusivity for our products or product candidates, our competitors may be able to sell products to treat the same conditions and our revenue may be reduced.
Under the Orphan Drug Act, the FDA may designate a product as an orphan drug if it is intended to treat a rare disease or condition, defined as a patient population of fewer than 200,000 in the U.S., or a patient population greater than 200,000 in the U.S. where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the U.S. In the EU, after a recommendation from the EMA’s Committee for Orphan Medicinal Products, the EU Commission grants orphan drug designation to promote the development of products that are intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition either affecting not more than five in 10,000 persons in the EU or when, without incentives, it is unlikely that sales of the drug in the EU would be sufficient to justify the necessary investment in developing the drug or biological product. In each case there must be no satisfactory method of diagnosis, prevention or treatment of such condition, or, if such a method exists, the medicine must be of significant benefit to those affected by the condition.
In the U.S., orphan drug designation entitles a party to financial incentives such as tax advantages and user fee waivers. In addition, if a product receives the first FDA approval for the indication for which it has orphan designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application submitted by another applicant to market a same or similar biological product for the same indication for a period of seven years, except in limited circumstances. Whether a biological product is the same as another product is based on whether the two products have the same principal molecular structural features. Orphan designation does not, however, truncate the duration of the regulatory review and approval process.
In the EU, orphan drug designation entitles a party to financial incentives such as reduction of fees or fee waivers and 10 years of market exclusivity following drug or biological product approval. This period may be reduced to
10
six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. If we fail to obtain or if we lose orphan drug status for one or more of our products and product candidates, the aforementioned incentives and market exclusivity may not or no longer be available to us, which is likely to increase the overall cost of development and to decrease the competitive position of such product and product candidate including from biosimilars. Similar considerations apply in the UK.
We may from time to time seek orphan drug designation in the U.S. or the EU for certain indications addressed by our products and product candidates. For example, in September 2017, the FDA granted orphan drug designation for the use of efgartigimod for gMG, and upon approval of VYVGART, the FDA granted seven years of orphan drug exclusivity for VYVGART for the treatment of gMG in adult patients who are anti-acetylcholine receptor (AChR) antibody positive (AChR-AB+). In July 2022, the FDA granted orphan drug designation for VYVGART HYTRULO, and upon approval of VYVGART HYTRULO, the FDA granted seven years of orphan drug exclusivity for this product for the treatment of gMG in adult patients who are AChR-AB+. In January 2019, the FDA granted orphan drug designation for the use of efgartigimod for the treatment of primary immune thrombocytopenia (ITP) and for the use of cusatuzumab for the treatment of acute myeloid leukemia (AML), and in August 2021, the FDA granted orphan drug designation for the use of efgartigimod co-formulated with recombinant human hyaluronidase PH20 (rHuPH20) for the treatment of chronic inflammatory demyelinating polyneuropathy (CIDP). In June 2020, Japan’s Ministry of Health, Labour and Welfare (MHLW) granted orphan drug designation for the use of efgartigimod for the treatment of gMG and in January 2022, the MHLW granted approval of VYVGART for treatment of gMG. Furthermore, in December 2022, the MHLW granted orphan drug designation for the use of VYVGART for the treatment of ITP. The application for approval of VYVGART for treatment of ITP was filed for the first time, pioneering worldwide, but such approval is expected in the first quarter of 2024. With regard to these designations or future designations we may obtain, we may not be the first to obtain marketing approval of these drugs for such indication due to the uncertainties associated with developing therapeutic products, and we may not obtain orphan designation upon approval. In addition, exclusive marketing rights in the U.S. may be limited if we seek approval for an indication broader than the orphan-designated indication or may be lost if the FDA later determines that the request for designation was materially defective or if we are unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. Further, even if we obtain orphan drug exclusivity for a product, that exclusivity may not effectively protect the product from competition because different drugs with different active moieties or different principal molecular structural features can be approved for the same condition. Even after an orphan drug is approved, the FDA, EMA or other foreign regulator can subsequently approve the same drug with the same principal molecular structural features for the same condition if the regulator concludes that the later drug is safer, more effective, or makes a major contribution to patient care.
Risk Factors Related to Other Government Regulations
We are subject to healthcare laws, regulation and enforcement. The failure to comply with these laws could harm our results, operations and/or financial conditions.
Our current and future operations may be or may become directly, or indirectly through our customers and third-party payors, subject to various U.S. federal and state, EU, Japanese, Chinese, UK, Canadian, Israel and other jurisdictions’ healthcare laws including anti-kickback statutes, anti-bribery, anti-corruption provisions, anti-fraud statutes, false claims acts, including the U.S. federal Anti-Kickback Statute (AKS), the U.S. Federal Food, Drug, and Cosmetic Act (FDCA), False Claims Act and more. Healthcare providers, physicians and others play a primary role in the recommendation and prescription of any products for which we obtain marketing approval. These laws may impact, among other things, our proposed sales, marketing and education programs and constrain our business and financial arrangements with third-party payors, healthcare professionals who participate in our clinical research programs, healthcare professionals and others who recommend, purchase, or provide our approved products, and other parties through which we market, sell and distribute our products for which we obtain marketing approval.
In addition, our current and future operations are subject to other healthcare-related statutory and regulatory requirements and enforcement by regulatory authorities in jurisdictions in which we conduct our business. For example, the provision of benefits or advantages to physicians to induce or encourage the prescription, recommendation,
11
endorsement, purchase, supply, order or use of medical products is generally not permitted in countries that form part of the EU, or the UK. Some EU Member States have enacted laws explicitly prohibiting the provision of these types of benefits and advantages to induce or reward improper performance generally, and the UK has enacted similar restrictions through the Bribery Act 2010. Infringements of these laws can result in substantial fines and imprisonment, as well as associated reputational harm. We are also subject to EU Directive 2001/83/EC and the Human Medicines Regulations 2012. Any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business.
The shifting compliance environment and the need to maintain robust and expandable systems to comply with multiple jurisdictions with different compliance or reporting requirements increases the possibility that we or our collaborative partners may run afoul of one or more of the requirements. We continue to expand, enhance and refine our internal ethics and compliance function and program to ensure compliance with the different healthcare laws and regulations. The expansion and maintenance of an internal compliance program involves substantial costs and, notwithstanding our investment, mechanisms put in place to ensure compliance with applicable laws and regulations and our best efforts, the program may not be fully successful as there can be no assurance that our policies and procedures will be followed at all times or will effectively detect and/or prevent all compliance violations by our employees, consultants, subcontractors, agents and partners.
It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative investigations, penalties, damages, fines, disgorgement, imprisonment, exclusion of drugs from government funded healthcare programs, such as Medicare and Medicaid in the U.S., additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, reputational harm and the curtailment or restructuring of our operations. Managing such investigations and defending against or appealing any such actions or penalties can be costly and time-consuming and may require significant financial and personnel resources. Therefore, even if we are successful in managing any such governmental investigations and/or defending against or appealing any such actions or penalties that may be brought against or imposed upon us, our business may be impaired. Further, if any of the physicians or other healthcare providers or entities with whom we expect to do business is found to be not in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs. Efforts to ensure that our business arrangements with third parties comply with applicable healthcare laws and regulations also involves substantial costs.
The scope and enforcement of each of these laws is uncertain and subject to rapid change in the current environment of healthcare reform. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Ensuring business arrangements comply with applicable healthcare laws, as well as responding to possible investigations by government authorities, can be time and resource consuming and can divert a company’s attention from the business.
All aspects of our business ranging from preclinical, clinical trials, marketing and commercialization are highly regulated and any delay by relevant regulatory authorities could jeopardize our development and approval process or result in other suspensions, refusals or withdrawal of approvals.
Before we can commence clinical trials for a product candidate, we must complete extensive preclinical testing and clinical trials that support our investigational new drug (IND) or planned IND applications in the U.S. or Japan, or our clinical trial applications (CTAs) in the UK or in the EU, or a comparable application in other jurisdictions. We cannot be sure that we will be able to submit INDs or CTAs or comparable applications for our preclinical programs on the timelines we expect, if at all. We also cannot guarantee that submission of INDs or CTAs or comparable applications will result in the UK Medicines and Healthcare products Regulatory Agency (MHRA), the EMA, the FDA, the MHLW (collectively, the Relevant Regulatory Authorities) or other regulatory authorities allowing clinical trials to even begin.
12
Clinical trials must be conducted in accordance with Relevant Regulatory Authorities and other applicable regulatory authorities’ legal requirements and regulations and are subject to oversight by these governmental agencies and institutional review boards (IRBs) and ethics committees at the medical institutions where the clinical trials are conducted. In addition, clinical trials must be conducted in compliance with good clinical practices (GCPs) and with supplies of our products and product candidates produced under current good manufacturing practices (cGMPs) and other regulations. We could encounter delays if a clinical trial is suspended or terminated, by us, by the IRBs or ethics committees of the institutions in which such clinical trials are being conducted, by the data review committee or data safety monitoring board for such clinical trial by the Relevant Regulatory Authorities or other comparable regulatory authorities. Such authorities may impose a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or clinical trial site by the Relevant Regulatory Authorities or other applicable authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, including those relating to the class to which our products and product candidates belong, failure to demonstrate a benefit from using the product or product candidate, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
If we experience delays in the completion of, or termination of, any clinical trial of our products or product candidates, the costs of our clinical trials may increase, the commercial prospects of our products and product candidates may be harmed, and our ability to generate product revenues from any of these products and product candidates will be delayed. Significant clinical trial delays could also allow our competitors to bring products to market before we do or shorten any periods during which we have the exclusive right to commercialize our products and product candidates.
Moreover, we must obtain separate regulatory approvals in each jurisdiction where we want to market and approval by one regulatory authority does not ensure approval by any other regulatory authority. As approval procedures can vary among countries and may change over time, this can require additional clinical testing and the time required to obtain approval may differ. We can provide no assurances that such approval will be obtained on the timeline that we expect or at all. In addition, we anticipate to submit applications for approval of VYVGART in new indications, but can provide no assurances that such applications will be accepted or that we will receive approval on our anticipated timeline, or at all.
If VYVGART or any new formulations of VYVGART are not approved in one or more jurisdictions including beyond the VYVGART Approved Countries, or if such approvals are significantly delayed, it could have a material adverse effect on our business. It is possible that none of our other existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval in any other jurisdiction or indication.
Further, the Relevant Regulatory Authorities may impose extensive and ongoing unique regulatory requirements. For example, they:
| ● | may withdraw an approval or revoke a license; |
| ● | may refuse to grant approval, or may require additional data before granting approval, notwithstanding that approval may have been granted by another comparable foreign authority; |
| ● | may approve a product candidate for fewer or more limited indications or patient sub-segments than requested; or |
| ● | may grant approval contingent on the performance of costly post-marketing clinical trials, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product candidate; or |
| ● | may approve a product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. |
13
The costs of compliance with all Relevant Regulatory Authorities and applicable authorities regulations, requirements or guidelines could be substantial, and failure to comply could result in sanctions, including fines, injunctions, civil penalties, denial of applications for marketing authorization of our products, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could significantly increase our and/or our collaborative partners’ costs or delay the development and commercialization of our product candidates. At this time, we cannot guarantee or know the exact nature, precise timing and detailed costs of the efforts that will be necessary to complete the remainder of the development of our research programs and product candidates.
We are subject to privacy laws, regulation and potential enforcement. Our failure to comply with these laws could harm our results, operations and/or financial conditions.
Privacy laws, regulation and potential enforcement are particularly relevant to our business as we collect, store and process patient data, including sensitive health data as well as human biological samples such as blood or tissue, in the context of our clinical development activities, post-marketing approval monitoring obligations, and associated activities. We also collaborate on a regular basis with third parties where we may seek to use data collected by third parties on our or their behalf, or we may seek to share data collected by us with such third parties to further our research or commercial initiatives.
The EU General Data Protection Regulation (GDPR) imposes a broad range of strict requirements on companies, including with respect to cross-border transfers of personal data. The GDPR allows the imposition of substantial penalties in the event of non-compliance, including fines of up to €10 million or up to 2% of total worldwide annual turnover of the preceding fiscal year for certain comparatively minor offenses, or up to €20 million or up to 4% of total worldwide annual turnover if the preceding fiscal year for more serious offenses. We face uncertainty as to the exact interpretation of the requirements under the GDPR, and we may be unsuccessful in implementing all measures required by data protection authorities or courts in interpretation of the GDPR.
In addition, national laws of EU Member States may partially deviate from the GDPR and impose different obligations from country to country, so that we do not operate in a uniform legal landscape in the EU. Also, in the field of handling genetic data, the GDPR specifically allows EU Member States’ laws to impose additional and more specific requirements or restrictions, and European national laws have historically differed quite substantially in this field, leading to additional uncertainty.
Following its departure from the EU, the UK has maintained in force substantially equivalent provisions to the GDPR (UK GDPR). The UK Government has recently announced reforms to the regime. Similar concerns as those described above apply to our compliance with the UK GDPR and other UK data protection rules.
Privacy laws continue to evolve and expand in Europe. For example, Directive 2002/58/EC of the European Parliament and of the Council of July 12, 2002 (as amended, the e-Privacy Directive) required the EU Member States to implement laws to meet strict privacy requirements related to electronic communications, cookies and online monitoring, and other digital privacy. Violations of these requirements can result in administrative measures, including fines, or criminal sanctions. The EU is in the process of developing a new e-Privacy Regulation to replace the e-Privacy Directive, and the new e-Privacy Regulation may impose additional obligations and risk for our business.
Beyond the EU and UK, privacy and data protection laws and regulations continue to develop and expand around the world, including in other jurisdictions in which we operate, such as the U.S., Japan, and Canada. Such laws and regulations impose increasing restrictions and obligations on the processing of personal data, including sensitive personal data such as genetic data. For example, in the U.S., the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, imposes specific requirements relating to the privacy, security, and transmission of individually identifiable health information and new state privacy laws, such as the California Consumer Privacy Act of 2018 (CCPA) and the Washington My Health My Data Act of 2023, impose obligations on covered businesses, including, but not limited to, providing specific disclosures in privacy notices and affording residents certain rights related to their personal data. Additionally, effective as of
14
January 1, 2023, the California Privacy Rights Act of 2020 (CPRA) imposes additional obligations on companies covered by the legislation and will significantly modify the CCPA, including by expanding consumers’ rights with respect to certain sensitive personal information. The CPRA also creates a new state agency that will be vested with authority to implement and enforce the CCPA and the CPRA. Furthermore, in the U.S., similar laws were enacted in certain states and proposed in numerous others. The effects of the CCPA and the CPRA are potentially significant and may require us to modify our data collection or processing practices and policies and to incur substantial costs and expenses in an effort to comply and increase our potential exposure to regulatory enforcement and/or litigation. If we are investigated by a data protection authority, we may face fines and other penalties. Any such investigation or charges by such data protection authorities could have a negative effect on our existing business and on our ability to attract and retain new clients or pharmaceutical partners. We may also experience hesitancy, reluctance, or refusal by clients or pharmaceutical partners to continue to use our products and solutions due to the potential risk exposure as a result of the current (and, in particular, future) data protection obligations imposed on them by certain data protection authorities in interpretation of current law. Such clients or pharmaceutical partners may also view any alternative approaches to compliance as being too costly, too burdensome, too legally uncertain, or otherwise objectionable and therefore decide not to do business with us. Any of the foregoing could harm our business, prospects, financial condition and results of operations.
Failure to comply with anti-corruption laws and regulations, anti-money laundering laws and regulations, economic sanctions and/or export control regulations and other laws governing our operations such as in relation to sustainability could have an adverse impact on our business.
We are or may become subject to various laws and regulations regarding anti-corruption, anti-money laundering, economic sanctions, investment restrictions, anti-fraud and export control regulations issued by multiple jurisdictions. These include the UK Bribery Act 2010 and the U.S. Foreign Corrupt Practices Act of 1977, as amended, which prohibits, among other things, payments, offers, or promises made for the purpose of improperly influencing any act or decision of a foreign official. The nature of our business means that we engage in significant interactions with foreign officials. We are also subject to economic sanctions and export control rules and regulations imposed by, amongst others, the U.S. Department of the Treasury’s Office of Foreign Assets Control, other agencies of the U.S. government, HM Treasury and other agencies of the UK government, the EU, and the United Nations. Any change in export or import regulations, economic sanctions regulations or related legislation, shift in the enforcement or scope of existing regulations, or change in the countries, governments, persons or technologies targeted by such regulations, could decrease our ability to manufacture, import, export or sell our products internationally. Any limitation on our ability to manufacture, import, export or sell our products could adversely affect our business.
We have mechanisms in place to ensure compliance with such rules and regulations. However, there can be no assurance that our policies and procedures will be followed at all times or will effectively detect and/or prevent violations of applicable compliance regimes by our employees, consultants, sub-contractors, agents and partners. As a result, in the event of non-compliance, we could be subject to substantial civil or criminal penalties, including economic sanctions against us, incarceration for responsible employees and managers, the possible loss of export or import privileges, reputational harm, and resulting loss of revenue and profits, which could have a material adverse impact on our business, financial conditions and operations.
Moreover, a growing number of investors, regulators, self-regulatory organizations and other stakeholders have expressed an interest in setting Environmental, Social and Governance (ESG) goals and requiring the provision of new and more robust disclosure of steps taken to implement such goals. The related legislative landscape in the EU has been evolving accordingly. For example, on January 5, 2023, Directive (EU) 2022/2464 of the European Parliament and of the Council of December 14, 2022 amending Regulation (EU) No 537/2014, Directive 2004/109/EC, Directive 2006/43/EC and Directive 2013/34/EU, as regards corporate sustainability reporting (CSRD) came into force. This new directive strengthens the rules on the social and environmental information that companies must report. It expands the scope of companies required to report ESG information and increases the depth of required disclosures. The CSRD also introduces a ‘double materiality’ analysis, which requires companies to report on how sustainability issues might create financial risks for the company and on the company’s own impacts on people and the environment. The CSRD applies to large EU companies, EU parent companies of a “large group” and listed EU small or medium-sized companies. It also applies to non-EU companies that meet certain criteria, including an EU turnover threshold and an EU branch or
15
subsidiary. The specific information to be reported is set out in the European Sustainability Reporting Standards (ESRS). Companies subject to the CSRD will have to comply with the reporting requirements on a staggered basis depending on their category. For listed companies that qualify as large EU companies and which are already obliged to publish a non-financial statement, a mandatory sustainability report in accordance with the ESRS will be required for financial years starting on or after January 1, 2024. This means that in 2025, we will have to publish for the first time a sustainability report complying with requirements under the CSRD integrated in our annual report for the financial year 2024. The Dutch government is in the process of implementing the CSRD into Dutch legislation.
This means that in 2025, we will have to publish our first CSRD report covering the prescribed ESG information for the financial year 2024. The Dutch government is in the process of implementing the CSRD into Dutch legislation. In July 2023, a Dutch draft bill to implement certain elements of the CSRD (including the requirements for assurance of CSRD reports and applicability to listed companies) was published and submitted for public consultation, which consultation period ended in September 2023. Additionally, in November 2023, a Dutch draft decree implementing certain elements of the CSRD (including the CSRD disclosure obligations for in-scope companies, assurance rules and implementation timelines) was published and submitted for public consultation, which consultation period ended in December 2023. The Dutch government will need to progress the parliamentary debate and adoption of the aforementioned draft bill and decree in the near future given that the ultimate date for implementation of the CSRD within EU member states at a local level is July 6, 2024.
Similarly, the SEC adopted on March 6, 2024 final rules aimed at enhancing and standardizing climate-related disclosures relating to climate-related risks, Scope 1 and Scope 2 greenhouse gas emissions and climate-related financial metrics (SEC Climate Rules). As a foreign private issuer and large accelerated filer, we will need to begin complying with the disclosure requirements of the SEC Climate Rules in our Form 20-F for the fiscal year ending December 31, 2025, which will include quantitative and qualitative climate-related disclosures.
In response to new ESG initiatives and regulations we may voluntarily elect, or be required, to adopt strategies, policies, or procedures related to ESG matters. Reporting on ESG goals and objectives, including pursuant to the SEC Climate Rules, may cause us to expend significant capital and human resources, and could divert management’s attention from central operational matters. Reports could also lead to the disclosure of information that which may have a negative impact on our operations and reputation which may lead to additional exposure. Failure to accurately comply with any ESG reporting obligations may result in enforcement actions, sanctions, reputational harm or private litigation.
We may become exposed to liability and substantial expenses in connection with environmental compliance or remediation activities.
Our operations, including our research, development, testing and third-party manufacturing activities, are subject to numerous environmental, health and safety laws and regulations and for which we may become liable.
If we or one of our CMOs or third-party distributors, manufacturers, licensees or co-marketers fail to comply with such laws and regulations, such failure could result in substantial fines, penalties or other sanctions which could also bring significant reputational loss to our business.
We face a risk of environmental liability inherent in our current and historical activities, including liability relating to releases of our exposure to hazardous or biological materials. Furthermore, environmental, health and safety laws and regulations are becoming more stringent. Both us and our CMOs may be required to incur substantial expenses in connection with future environmental compliance or remediation activities, in which case, our production and development efforts may be interrupted or delayed, and our financial condition and results of operations may be materially adversely affected
16
Risk Factors Related to the Development and Clinical Testing of argenx’s Products and Product Candidates
Failure to successfully identify, select and develop VYVGART in other indications, or additional products or product candidates could impair our ability to grow.
Our long-term growth strategy entails developing and marketing additional products and product candidates, including VYVGART in new indications, which requires substantial resources, whether or not any product candidates or new indications are ultimately identified. The success of this strategy depends partly upon our ability to identify, select, develop, and ultimately, commercialize promising product candidates. We are heavily dependent on precise, accurate and reliable scientific data to identify, select and develop promising product candidates and products. Our business decisions may therefore be adversely influenced by inaccurate, improper or fraudulent scientific data, including data sourced from third parties. Any irregularities in the scientific data used by us to determine our focus in research and development of product and product candidates, could impair our ability to grow. Even with accurate scientific data, our technology platforms may fail to discover and to generate additional products and products candidates, that are suitable for further development.
Even if we identify additional product candidates, they may not be suitable for clinical development as a result of harmful side effects, limited efficacy or other characteristics that indicate that it is unlikely to be a product that will receive approval by the Relevant Regulatory Authorities and other comparable regulatory authorities or achieve market acceptance. For example, in November 2023, we announced that our ADVANCE-SC clinical trial evaluating VYVGART HYTRULO in adults with ITP did not meet the primary endpoint of a sustained platelet count response in chronic ITP patients. Secondary endpoints were also not met. In December 2023, we announced that topline data from the Phase 3 ADDRESS clinical trial evaluating SC efgartigimod in adults with pemphigus vulgaris (PV) and pemphigus foliaceus (PF) showed that the proportion of PV patients achieving the primary endpoint of complete remission on complete remission on minimal dose of steroids (CRmin) was not significantly different between SC efgartigimod and placebo. We consequently decided not to pursue additional development in pemphigus and plan to prioritize clinical development of efgartigimod in its ongoing severe autoimmune indications. If we do not successfully identify, develop and commercialize product candidates and VYVGART in new indications based upon our technological approach, we may not be able to obtain product or collaboration revenues in future periods.
VYVGART has obtained regulatory approval in the VYVGART Approved Countries for the treatment of gMG. Our other products and product candidates – including additional indications or methods of use for efgartigimod, empasiprubart and ARGX-119 – are either in preclinical or clinical development or are pending marketing approval.
To obtain the requisite regulatory approvals to market and sell any of our products and product candidates, we or our collaborators for such candidates must successfully demonstrate that our products are safe and effective in humans. Clinical trials are expensive and can take many years to complete, and their outcome is inherently uncertain. Further, success in early clinical trials or in one indication does not guarantee success in later clinical trials or in other indications.
The time required to obtain approval by the Relevant Regulatory Authorities is unpredictable but typically takes many years, if obtained at all, following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion or interpretation of the regulatory authorities. This lengthy approval process as well as the unpredictability of future clinical trial results may result in our failing to obtain regulatory approval to market any of our product candidates, including for new indications. We may experience delays in our ongoing or planned clinical trials, for a large variety of reasons outside our control in complying with regulatory approvals which can adversely affect the timing of trials, including as described in Item 3.D. “Risk Factors—Risk Factors Related to Other Government Regulations—All aspects of our business ranging from preclinical, clinical trials, marketing and commercialization are highly regulated and any delay by relevant regulatory authorities could jeopardize our development and approval process or result in other suspensions, refusals or withdrawal of approvals.”
If we are unable to obtain regulatory approval of our products and product candidates on a timely basis or at all, our business may be impacted.
17
Our clinical trials have, and may in the future, fail, and even if they succeed, we may not obtain regulatory approval for our products and product candidates or regulatory approval may be delayed.
Even if clinical trials are initiated, our development efforts may not be successful. Many of our clinical trials are blinded, which may cause us to incur significant expenses without any visibility as to the likelihood of successful results. Even if we obtain positive results from preclinical trials or initial clinical trials, we may not achieve the same success in future clinical trials.
Regulatory approval of our products or product candidates may be delayed or refused for many reasons, including:
| ● | the Relevant Regulatory Authorities may disagree with the design or implementation of our clinical trials; |
| ● | we may be unable to demonstrate, to the satisfaction of the FDA or comparable foreign regulatory authorities, that our product candidates are safe, pure, potent and effective for any of their proposed indications; |
| ● | we may be unable to demonstrate our product candidates’ clinical and other benefits outweigh their safety risks; |
| ● | the FDA may determine that clinical trial results are not generalizable to the U.S. population and/or U.S. medical practice based on the proportion and results of subjects outside of the U.S. where differences in patient management might affect the treatment response. Comparable foreign regulatory authorities may take a similar approach; |
| ● | the results of clinical trials may not meet the level of statistical significance required by the FDA or comparable foreign regulatory authorities for approval; |
| ● | the chemistry, manufacturing and controls information submitted in a marketing application is insufficient; and |
| ● | the facilities of third-party manufacturers with which we contract for the manufacture of our product candidates are not adequate to support approval of our product candidates. |
Any of these occurrences may harm our business, results of operations and financial condition significantly.
We could also experience operational challenges as we undertake an increasing number of clinical trials, including those conducted in countries outside the EU, UK and the U.S. that may subject us to further delays and expenses as a result of increased shipment costs, additional regulatory requirements and the engagement of non-EU, non-UK and non-U.S. contract research organizations (CROs), as well as expose us to risks associated with clinical investigators who are unknown to the Relevant Regulatory Authorities, and apply different standards of diagnosis, screening and medical care.
If we experience delays in the completion of, or termination of, any clinical trial of our products or product candidates, our commercial prospects may be harmed. Any delays in completing our clinical trials may increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates or result in the development of our product candidates being stopped early. Significant clinical trial delays could also allow our competitors to bring products to market before we do or shorten any periods during which we have the exclusive right to commercialize our products and product candidates.
18
If we decide to pursue accelerated approval for any of our product candidates, it may not lead to a faster development or regulatory review or approval process and does not increase the likelihood that our product candidates will receive marketing approval. If we are unable to obtain approval under an accelerated pathway, we may be required to conduct additional clinical trials beyond those that we contemplate, which could increase the expense of obtaining, reduce the likelihood of obtaining, and/or delay the timing of obtaining, necessary marketing approvals.
In the future, we may decide to pursue accelerated approval for one or more of our product candidates. For example, under the FDA’s accelerated approval program, the FDA may approve a drug or biological product for a serious or life-threatening disease or condition that provides a meaningful advantage over available therapies based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit. For products granted accelerated approval, post-marketing confirmatory clinical trials are required to verify and describe the anticipated effect on irreversible morbidity or mortality or other clinical benefit. These confirmatory clinical trials must be completed with due diligence, and the FDA may require that the clinical trial be designed, initiated, and/or fully enrolled prior to approval. If we were to pursue accelerated approval for a product candidate for a disease or condition, we would do so on the basis that there is no available therapy for that disease or condition. If standard of care were to evolve or if any of our competitors were to receive full approval on the basis of a confirmatory clinical trial for a drug or biological product for a disease or condition for which we are seeking accelerated approval before we receive accelerated approval, the disease or condition may no longer qualify as one for which there is no available therapy, and accelerated approval of our product candidate may not occur.
Moreover, the FDA may withdraw approval of any product candidate approved under the accelerated approval pathway if, for example:
| ● | the clinical trial or clinical trials required to verify the predicted clinical benefit of our product candidate fail to verify such benefit or do not demonstrate sufficient clinical benefit to justify the risks associated with such product; |
| ● | other evidence demonstrates that our product candidate is not shown to be safe or effective under the conditions of use; |
| ● | we fail to conduct any required post-approval trial of our product candidate with due diligence; or |
| ● | we disseminate false or misleading promotional materials relating to the relevant product candidate. |
Recently, the accelerated approval pathway has come under scrutiny within the FDA and by Congress. The FDA has put increased focus on ensuring that confirmatory studies are conducted with diligence and, ultimately, that such studies confirm the benefit. The recently enacted Food and Drug Omnibus Reform Act (FDORA) includes provisions related to the accelerated approval pathway. Pursuant to FDORA, the FDA is authorized to require a post-approval clinical trial to be underway prior to approval or within a specified time period following approval. FDORA also requires the FDA to specify conditions of any required post-approval clinical trial and requires sponsors to submit progress reports for required post-approval studies. In addition, FDORA enables the FDA to initiate enforcement action for the failure to conduct with due diligence a required post-approval clinical trial, including a failure to meet any required conditions specified by the FDA or to submit timely reports.
Failure to obtain accelerated approval for our product candidates could result in a longer time period to commercialization of such product candidate, if any, and could increase the cost of development of such product candidate and harm our competitive position in the marketplace.
19
Our products and product candidates may have serious adverse, undesirable or unacceptable side effects or even cause death, and we or others may identify undesirable or unacceptable side effects caused by VYVGART or any of our products or product candidates after they have received marketing approval.
Undesirable side effects that may be caused by our product candidates, or by the combination of our product candidates with other medical products, could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in more restrictive labeling or the delay or denial of regulatory approval by the Relevant Regulatory Authorities. While our preclinical trials and clinical trials for our product candidates to date show that our product candidates have generally been well tolerated from a risk-benefit perspective, we have observed adverse events and treatment emergent adverse events (TEAEs) in our clinical trials to date, and we may see additional adverse events and TEAEs in our ongoing and future clinical trials. Such side effects may be more serious than those observed to date, and as a result, our ongoing and future clinical trials may be negatively impacted. Moreover, as we seek to develop product candidates, including products in new indications, patients may experience new or more serious effects. Drug-related side effects could affect patient recruitment, the ability of enrolled patients to complete the clinical trial, result in potential product liability claims, damage sales of our existing products, result in significant reputational damage for us and our product development, and other issues including the delay of other programs.
Additionally, if we or others identify undesirable or unacceptable side effects caused by VYVGART or any of our other product candidates after they receive marketing approval, a number of potentially significant negative consequences could arise, including:
| ● | regulatory authorities may withdraw approvals or revoke licenses of such products and require us to take such products off the market; |
| ● | regulatory authorities may require the addition of labeling statements, specific warnings, or a contraindication or request the issuance of field alerts to physicians and pharmacies; |
| ● | regulatory authorities may require a medication guide outlining the risks of such side effects for distribution to patients, or a risk evaluation and mitigation strategy (REMS) plan to ensure that the benefits of the product outweigh its risks; |
| ● | we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| ● | we may be subject to limitations on how we may promote the product; |
| ● | sales of the product may decrease significantly; |
| ● | we may be subject to litigation, product liability claims, or criminal prosecution; and |
| ● | our reputation may suffer. |
Any of these events could negatively impact us, our collaborators or our potential future partners. Further, the Relevant Regulatory Authorities could require a change of label or even revoke the license, which could harm our reputation and have a material adverse effect on our ability to commercialize VYVGART.
If our target patient population is smaller than expected, we are unable to successfully enroll and retain patients in our clinical trials, or experience significant delays in doing so, we may not realize the full commercial potential of any products or product candidates.
Currently, we mainly develop products or product candidates for the treatment of rare diseases for which the target patient population can be small. If the actual number of patients with these disorders is smaller than we expected, we may encounter difficulties in enrolling sufficient patients in our clinical trials, thereby delaying or preventing
20
development and approval of our products or product candidates. Physicians, who are an important source of referral of patients for clinical trials, may also be less familiar with these rare diseases and may therefore fail to identify these conditions in their patients and therefore may not refer them to our clinical trials.
Patient enrollment, a significant factor in the timing of clinical trials, depends on many factors, including the size and nature of the patient population, eligibility criteria for the clinical trial, the proximity of patients to clinical sites, competition for patient recruitment from competing clinical trials, the design of the clinical protocol, the eligibility criteria for the clinical trials, the availability of alternate approved therapies for the indication the clinical trial is investigating, and clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies. We compete with other companies to enroll target patient populations, as set forth in Item 3.D. “Risk Factors—Risk Factors Related to Commercialization of argenx’s Products and Product Candidates, Including for New Indications—We face significant competition for our drug discovery and development efforts.” Even if product candidates obtain significant market share for their approved indications, because certain potential target populations are small, we may never recoup our investment in such product candidate without obtaining regulatory approval for additional indications for such product candidates.
Even once enrolled, we may be unable to retain a sufficient number of patients to complete any of our clinical trials. In addition, any negative results we may report in clinical trials of our drug candidates may make it difficult or impossible to recruit and retain patients in other clinical trials of that same drug candidate. Delays in the completion of any clinical trial of our product candidates will increase our costs, slow down our product candidate development and approval process and delay or potentially jeopardize our ability to commence product sales and generate revenue. In addition, some of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
In addition, certain of patients enrolled in our clinical trials are located in areas subject to conflict, hostilities or war, or other disruptive events outside of our control. See Item 3.D. “Risk Factors—Risk Factors Related to argenx’s Organization and Operations—‘Global geo- and socio-political threats and macro-economic uncertainty and other unforeseen political crises could materially and adversely affect our business and financial performance.’ and ‘We face risks related to natural disasters and public health issues, that could negatively affect our business and financial condition.”
Risk Factors Related to argenx’s Dependence on Third Parties
We rely, and expect to continue to rely, on third parties to conduct some of our research activities and clinical trials and for parts of the development and commercialization of our existing and future research programs, products and product candidates. If our relationships with such third parties are not successful, our business may be adversely affected.
We have relied upon and plan to continue to rely upon third parties, including independent clinical investigators, CROs, CMOs and other third-party service providers, to assist us in the conduct of certain of our research activities and clinical trials and to monitor and manage data for our ongoing preclinical trials and clinical trials. We also depend on our collaborators and on medical institutions and CROs to conduct our research activities and clinical trials in compliance with regulatory and legal requirements, including GCPs or GMPs, our standard operating procedures and our applicable protocols. Nevertheless, we are responsible for ensuring that each of our preclinical trials and clinical trials is conducted in accordance with the applicable protocol, legal and regulatory requirements and scientific standards, and our reliance on these third parties does not relieve us of our regulatory responsibilities. To the extent our collaborators or the CROs or investigators fail to enroll participants for our clinical trials, fail to conduct the clinical trial to GCP standards or in full compliance with legal and regulatory requirements or are delayed for a significant time in the execution of clinical trials, including achieving full enrollment, we may be affected by increased costs, program delays or both, which may harm our business.
In addition, we are, and expect to continue to be, dependent on partnerships with partners and licensees relating to the development and commercialization of our existing and future research programs, products and product candidates. We currently have collaborative research relationships with various pharmaceutical companies such as
21
AbbVie, Zai Lab, Genmab A/S (Genmab) and with various academic and research institutions worldwide for the development of product candidates resulting from such collaborations. We also have distribution agreements with Medison, Genpharm and Handok for the distribution of VYVGART. We had, have and will continue to have discussions on potential partnering opportunities with various pharmaceutical companies. If we fail to enter into or maintain collaborations on reasonable terms or at all, our ability to develop our existing or future research programs and product candidates and to commercialize our existing or future products could be delayed, the commercial potential of our products could change and our costs of development and commercialization could increase.
While we have agreements governing our relationships with these third parties, we have limited influence over their actual performance and control only certain aspects of their activities. If independent investigators, third-party service providers or CROs fail to devote sufficient resources to the development of our product candidates, or if their performance is substandard, it may delay or compromise the prospects for approval and commercialization of any product candidates that we develop. In addition, regulatory authorities enforce these GCPs through periodic inspections of trial sponsors, principal investigators and trial sites. If we, our investigators or any of our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the Relevant Regulatory Authorities or comparable regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. Upon inspection by a given regulatory authority, such regulatory authority may determine that our clinical trials do not fully comply with GCP regulations, which may require us to repeat clinical trials and delay the regulatory approval process. Our collaborative partners may not adhere or terminate collaboration agreements with all associated consequences or disagree on the interpretation of contractual terms. We may not be able to control our collaborative partners’ compliance with all applicable requirements for the commercialization of our products, which could adversely affect such commercialization and the profitability of such products. Failures by our collaborative partners to meet their contractual, regulatory, or other obligations to us or any disruption in the relationships between us and our collaborative partners, could have a material adverse effect on our product pipeline and business.
We face significant competition in establishing successful relationships with third-party service providers and appropriate collaborative partners. These third-party service providers may have contractual relationships with other entities, some of which may be our competitors, which may draw their time and resources away from our programs. In addition, some of our third-party service providers or CROs have the ability to terminate their respective agreements with us, and if such agreements terminate, we may not be able to enter into arrangements with alternative CROs or investigators or to do so on commercially reasonable terms. In addition, we may not be able to find appropriate collaboration partners. Our ability to reach a definitive agreement for a partnership will depend, among other things, upon an assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed partnership and the proposed collaborator’s evaluation of a number of factors. These factors may include the design or results of clinical trials, the likelihood of regulatory approval, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership regardless of the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar indications that may be available to collaborate on and whether such a partnership could be more attractive than the one with us.
Disruptions caused by our reliance on third parties for our manufacturing process may delay or disrupt our business, product development and commercialization efforts.
We do not have the ability to internally source the raw materials necessary to produce our products or product candidates, and do not currently have, nor do we plan to acquire, the infrastructure or capability internally to manufacture our products or product candidates and depend on a worldwide supply chain and third parties for both. Disruptions caused by our reliance on such third-party suppliers, service providers and manufacturers may delay or disrupt our business, product development and commercialization efforts.
22
Reliance on Third-Party Suppliers and Service Providers
For some of our raw materials, we rely on a single source of supply and there are limited supplies of the raw materials. If we were to experience an unexpected loss of supply of or if any supplier was unable to meet our demand for any of our products and product candidates, including for example if VYVGART is approved for additional indications, we could experience delays in our research or planned clinical trials or risk shortages in commercial supply which could materially impact our revenue potential. These issues could be made worse during a pandemic or due to geopolitical events, including trade disputes or economic sanctions enacted as a result of international conflict.
Additionally, certain of the raw materials required in the manufacture and the formulation of our products and product candidates may be derived from biological sources, including mammalian tissues, bovine serum and human serum albumin. There are certain European regulatory restrictions on using these biological source materials including rigorous testing requirements, which could limit or delay production. If there are changes in the regulation requirements that our suppliers are unable to meet, our clinical development or commercial activities may be delayed or interrupted.
We may not be able to engage a back-up or alternative supplier or service provider in a timely manner or at all if any of these third parties were to cease or interrupt production or otherwise fail to supply these materials, products, or services to us for any reasons, including due to regulatory requirements or actions (including recalls), adverse financial developments at or affecting the supplier, failure by the supplier to comply with cGMPs, contamination, business interruptions, or labor shortages or disputes. Interruptions in the supply of these materials, products or services may also result from international conflict, trade disputes or economic sanctions enacted by, or imposed on, the U.S., the UK, the EU or any other country or region.
Reliance on Third-Party Manufacturing
We rely on and expect to continue to rely on CMOs. We also rely on certain third parties to perform filling, finishing, distribution, laboratory testing and other services related to the manufacture of our products and product candidates.
Although we do not control the manufacturing process at our CMOs and are completely dependent on them for the production of our products and product candidates in accordance with relevant regulations (such as cGMPs), we are responsible for ensuring that our products comply with regulatory requirements. If our CMOs cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the Relevant Regulatory Authorities or other comparable regulatory authorities, our business could be adversely affected in a number of ways, including an inability to initiate or continue clinical trials of product candidates under development, delay in submitting regulatory applications, or receiving regulatory approvals for product candidates, including new indications, subjecting third-party manufacturing facilities to additional inspections by regulatory authorities, requirements to cease distribution or to recall batches of our products or product candidates and an inability to meet commercial demands for our marketed products.
We contract with Lonza Sales AG (Lonza) based in Slough, UK, Portsmouth, U.S, Singapore and Visp, Switzerland and FUJIFILM Diosynth Biotechnologies Denmark ApS (Fujifilm) based in Denmark for activities relating to the development of cell banks, development of our manufacturing processes and the manufacturing of drug substance, and use additional contract manufacturers to fill, test, label, package, store and distribute our (investigational) drug products. Our products and product candidates are biologics and require multiple processing steps that are more difficult than those required for most small molecule chemical pharmaceuticals. Problems with these manufacturing processes, such as capacity issues, or even minor deviations from the normal process or from the materials used in the manufacturing process, which may not be detectable by us in a timely manner, could lead to manufacturing failures or product defects, resulting in lot failures, product recalls, product liability claims and insufficient inventory.
We face risks inherent in relying on limited CMOs, as any failure in their ability to successfully manufacture our products or product candidates as described above or any disruption, such as a fire, pandemic, natural hazards or vandalism at the CMO could significantly interrupt our manufacturing capability. Alternative production plans in place or disaster-recovery facilities available to us may not be sufficient. In case of a disruption, we may have to establish
23
additional alternative manufacturing sources. This would require substantial investment on our part, which we may not be able to obtain on commercially acceptable terms or at all. Additionally, we may experience significant manufacturing delays as we build or locate replacement facilities and seek and obtain necessary regulatory approvals. If this occurs, we will be unable to satisfy manufacturing needs on a timely basis, if at all. Also, operating any new facilities may be more expensive than operating at our current facilities. Further, business interruption insurance may not adequately compensate us for any losses that may occur, and we would have to bear the additional cost of any disruption. For these reasons, a significant disruptive event of the manufacturing facility could have drastic consequences, including placing our financial stability at risk.
Accuracy and timing of our financial reporting is partially dependent on information received from third-party partners, which we do not control.
We have collaborated, and plan to continue to collaborate, with third parties, including distributor and licensing partners, on certain product candidates. As part of some of these collaborations, our collaboration partners are responsible for providing us with financial information regarding specific projects, including funds spent, liabilities incurred and expected future costs, on which we rely for our own financial reporting. If our collaboration partners fail to provide us with the necessary financial information within the agreed upon timeframes, or if such financial information proves inaccurate, it would adversely impact the timing and accuracy of our own financial reporting. Any inaccuracy in our financial reporting could cause investors to lose confidence in our financial reporting. This in turn may lead to reputational damage or affect our ability to obtain, and the terms of, any future financing, which may harm our business.
We and our third-party manufacturers and suppliers may become exposed to liability, fines, penalties or other sanctions and substantial expenses in connection with environmental compliance or remediation activities.
Our third-party manufacturers and suppliers operations, including research, development, testing and manufacturing activities, are subject to numerous environmental, health and safety laws and regulations. These laws and regulations govern, among other things, the controlled use, handling, release and disposal of and the maintenance of a registry for, hazardous materials and biological materials, laboratory procedures and exposure to pathogens. We do not have control over our manufacturers’ or suppliers’ compliance with environmental, health and safety laws and regulations. If we, or they fail to comply with such laws and regulations, we could be subject to liability, fines, penalties or other sanctions and incur substantial expenses to comply or remediate the activities.
We face a risk of environmental liability inherent in our current and historical activities, including liability relating to releases of or exposure to hazardous or biological materials. Environmental, health and safety laws and regulations are becoming more stringent. We may be required to incur substantial expenses in connection with future environmental compliance or remediation activities, in which case, our production and development efforts may be interrupted or delayed, and our financial condition and results of operations may be materially adversely affected.
Risk Factors Related to argenx’s Business and Industry
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, or insider trading violations, which could significantly harm our business.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with governmental regulations, comply with healthcare fraud and abuse and anti-kickback laws and regulations in the U.S. and other markets, or failure to report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of material information, including improper trading based upon, information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We maintain a global compliance program and remain focused on its evolution and enhancement. Our program includes efforts such as risk assessment and monitoring, fostering a culture encouraging employees and third parties to raise good
24
faith questions or concerns, and defined processes and systems for reviewing and remediating allegations and identified potential concerns. It is not always possible, however, to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
We may become exposed to costly and damaging liability claims.
We are exposed to potential product liability and professional indemnity risks that are inherent in the research, development, manufacturing, marketing and use of pharmaceutical products and marketing of human therapeutic products. The current and future use of products and product candidates by us and our collaborators in clinical trials and the sale of any approved products may further expose us to liability claims. If any of our products or product candidates were to cause adverse side effects during clinical trials or after approval of the product candidate, we may be exposed to substantial liabilities. These claims might be made by patients who use the product, healthcare providers, pharmaceutical companies, physicians, payors, caregivers, investors, employees, government agencies, or our collaborators or others selling such products. Physicians and patients may not comply with any warnings that identify known potential adverse effects and patients who should not use our product candidates. Any claims against us, regardless of their merit, could be difficult and costly to defend and could materially adversely affect the market for our products and product candidates or any prospects for commercialization of our products and product candidates. Any such claims, regardless of their merit, could also adversely affect our reputation and the trust that physician and patients place in our products. Product liability risk in the EU is likely to increase in the future if plaintiff-friendly reforms to the current EU legislation, which are currently at an advanced stage of the EU legislative process, are formally adopted.
Regardless of the merits or eventual outcome litigation or liability claims may result in:
| ● | decreased demand for our products due to negative public perception; |
| ● | damage to our reputation; |
| ● | withdrawal of clinical trial participants or difficulties in recruiting new clinical trial participants; |
| ● | initiation of investigations by regulators; |
| ● | costs to defend or settle the related litigation; |
| ● | a diversion of management’s time and our resources; |
| ● | substantial monetary awards to clinical trial participants or patients; |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| ● | loss of revenues from product sales; and |
| ● | the inability to successfully commercialize VYVGART and any of our other product candidates, if approved. |
Although we maintain product liability insurance, we may not be able to maintain insurance coverage at a reasonable cost or to obtain adequate insurance coverage to satisfy any liability that may arise. Product liability claims could delay or prevent completion of our clinical development programs. In addition, claims made by patients, healthcare professionals or others might not be fully covered by product liability insurance and could result in investigations of the safety of our products or product candidates or may result in recalls. If a successful product liability claim or series of claims is brought against us for uninsured liabilities or in excess of insured liabilities, our assets may
25
not be sufficient to cover such claims and our business, financial condition and results of operations would be adversely affected.
We may engage in strategic transactions, including acquisitions, collaborations, licenses or investments in other companies or technologies, and we may not realize the benefits of such transactions.
We may enter into strategic transactions, including acquisitions, collaborations, licenses or investments for or in other companies or technologies that complement or augment our existing business and facilitate our access to new products, research projects or geographical areas. However, we may not be able to identify appropriate targets or enter into such transactions under satisfactory conditions. In addition, we may need additional funding to finance these transactions including through issuances of public or private equity or convertible debt securities, which could be dilutive to our shareholders and ADS holders.
Integrating any newly acquired companies, business, technologies or products could be expensive, time-consuming, and may never be successful. Integration efforts often take a significant amount of time, place a significant strain on managerial, operational and financial resources, result in loss of key personnel and could prove to be more difficult or expensive than we predict. The diversion of our management’s attention and any delay or difficulties encountered in connection with any future transactions we may consummate could result in the disruption of our ongoing business or inconsistencies in standards and controls that could negatively affect our ability to maintain third-party relationships. We cannot assure that we will achieve the expected synergies to justify any such transaction, which could have a material adverse effect on our business, financial condition, results of operations and future growth prospects and our investors’ ability to realize on their investment.
Our business and operations could suffer in the event of system failures or unauthorized or inappropriate use of or access to our systems.
We are increasingly dependent on our information technology systems and infrastructure for our business. We collect, store and transmit sensitive information including intellectual property, proprietary business information, including highly sensitive clinical trial data, and personal data in connection with business operations. The secure maintenance of this information is critical to our operations and business strategy. Some of this information could be an attractive target of criminal attack or unauthorized access and use by third parties with a wide range of motives and expertise, including organized criminal groups, “hacktivists,” patient groups, disgruntled current or former employees and others. Cyber-attacks are of ever-increasing levels of sophistication, and despite our security measures, our information technology and infrastructure may be vulnerable to such attacks or may be breached, including due to employee error or malfeasance.
The pervasiveness of cybersecurity incidents in general and the risks of cyber-crime are complex and continue to evolve. Although we are making significant efforts to maintain the security and integrity of our information systems and are exploring various measures to manage the risk of a security breach or disruption, there can be no assurance that our security efforts and measures will be effective or that attempted security breaches or disruptions would not be successful or damaging. Despite the implementation of security measures, our internal computer systems and those of our contractors and consultants are vulnerable to damage or interruption from computer viruses, unauthorized or inappropriate access or use, natural disasters, pandemics, terrorism, war (including the ongoing conflict in Ukraine and the ongoing conflict in Israel and the Gaza Strip), and telecommunication and electrical failures. Such events could cause interruption of our operations. For example, the loss of pre-clinical trial data or data from completed or ongoing clinical trials for our product candidates could result in delays in our regulatory filings and development efforts, as well as delays in the commercialization of our products, and significantly increase our costs. To the extent that any disruption, security breach or unauthorized or inappropriate use or access to our systems were to result in a loss of or damage to our data, or inappropriate disclosure of confidential or proprietary information, including but not limited to patient, employee or vendor information, we could incur notification obligations to affected individuals and government agencies, liability, including potential lawsuits from patients, collaborators, employees, stockholders or other third parties and liability under foreign, federal and state laws that protect the privacy and security of personal data, and the development and potential commercialization of our product candidates could be delayed.
26
We are highly dependent on public perception of our products.
We are highly dependent upon consumer perceptions of the safety and quality of our products. We could be adversely affected if we, or any of our collaborators, are subject to negative publicity or if any of our products or any similar products distributed by other companies prove to be, or are asserted to be, harmful to patients, or for example, be deemed cruel to animals. Because of our dependence upon consumer perception, any adverse publicity associated with illness or other adverse effects resulting from patients’ use or misuse of our products or any similar products distributed by other companies could have a material adverse impact on our business, prospects, financial condition and results of operations.
Risk Factors Related to argenx’s Intellectual Property
Failure to adequately enforce or protect our intellectual property rights in products, product candidates and platform technologies could adversely affect our ability to maximize the value for patients in our marketed products and product candidates.
Our commercial success depends in part on obtaining and maintaining patents and other forms of intellectual property rights for our products, product candidates and platform technologies. Failure to protect or to obtain, maintain or extend adequate patent and other intellectual property rights, which may be challenging and costly, could adversely affect our ability to develop and market our products and product candidates and erode or negate any competitive advantage we may have.
We cannot be certain that patents will be issued or granted with respect to applications that are currently pending. The scope of patent protection that the European Patent Office and the U.S. Patent and the U.S. Patent and Trademark Office (USPTO) will grant with respect to the antibodies in our product pipeline is uncertain and may vary by jurisdiction. It is possible that the European Patent Office and the USPTO will not allow broad antibody claims that cover antibodies closely related to our products and product candidates as well as the specific antibody. As a result, upon receipt of EMA or FDA approval, competitors may be free to market molecules almost identical to ours if those competitors elected to engage and invest in a full clinical development program to establish the safety and efficacy of their molecule and secure that approval. If that would happen, then there is a risk of decreasing our market potential.
We and our current or future licensors, licensees or collaboration partners may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. Further, the issuance, scope, validity, enforceability and commercial value of our and our current or future licensors’, licensees’ or collaboration partners’ patent rights are highly uncertain. Moreover, in some circumstances, we may need to rely on patent procurement activities of our licensors, licensees or collaboration partners or obtain additional costly licenses. Such parties may not fully comply with applicable patent rules or disagree with us as to the prosecution, maintenance or enforcement of any patent rights. Even if patents do issue and such patents cover our products and product candidates, third parties may initiate proceedings challenging the validity, enforceability or scope of such patents, which may result in the patent claims being narrowed or invalidated. Our and our licensors’, licensees’ or collaboration partners’ patent applications cannot be enforced against third parties practicing the technology claimed in such applications unless and until a patent issues from such applications, and then only to the extent the issued claims cover the technology.
Furthermore, because patent applications are confidential for a period of time after filing, and some remain so until issued, we cannot be certain that we or our licensors were the first to file any patent application related to a product and product candidate. Even where we have a valid and enforceable patent, we may not be able to exclude others from practicing our invention where the other party can show that they used the invention in commerce before our filing date, or if the other party is able to obtain a compulsory license. In addition, after expiry of our regulatory exclusivities, we may not have a patent position to enforce if a biosimilar presents a ‘skinny label’ and introduces a product into commerce for unpatented uses, doses, and other important patient innovations described in our product labels. Any of the aforementioned situations could cause harm to our ability to protect our intellectual property, which in turn would allow competitors to market comparable products which could materially adversely affect our competitive position and as such our business, financial condition and results of operation.
27
We enjoy only limited geographical protection with respect to certain patents and may face difficulties in certain jurisdictions. We often file our first patent application (i.e., priority filing) at the UK Intellectual Property Office, the European Patent Office or the USPTO. International applications under the Patent Cooperation Treaty are usually filed within 12 months after the priority filing. We have so far not filed for patent protection in all national and regional jurisdictions where such protection may be available. If we fail to timely file a patent application in any such country or major market, we may be precluded from doing so at a later date. In addition, the grant proceeding of each national/regional patent may lead to situations in which applications might in some jurisdictions be refused by the relevant patent offices, while granted by others. Furthermore, competitors may use our and our licensors’ or collaboration partners’ technologies in jurisdictions where we have not obtained patent protection to develop their own products and, further, may export otherwise infringing products to territories where we and our licensors or collaboration partners have patent protection, but enforcement is not as strong as that in the U.S., UK and the EU. Finally, some countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties, and other countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent.
Issued patents could be found invalid or unenforceable if challenged in the applicable patent office or court.
Once granted, patents may remain open to invalidity challenges after allowance or grant, where third parties can raise objections against such granted patent. In the course of such proceedings, the patent owner may be compelled to limit the scope of the allowed or granted claims thus challenged or may lose the allowed or granted claims altogether.
To protect our competitive position, we may from time to time need to resort to adversarial proceedings in order to enforce or defend any intellectual property rights owned by or licensed to us, or to determine or challenge the scope or validity of intellectual property rights of third parties. Enforcement of intellectual property rights is difficult, unpredictable and expensive, and many of our or our licensors’ or collaboration partners’ adversaries in these proceedings may have the ability to dedicate substantially greater resources to prosecuting these legal actions than we or our licensors or collaboration partners can. In addition, adversarial proceedings involving our patents carries the risk that one or more of our patents will be held invalid or held unenforceable. Such an adverse ruling could allow third parties to commercialize our products immediately after the expiration of our regulatory protection or use our platform technologies, and then compete directly with us, without payment to us.
We may be subject to claims challenging the inventorship or ownership of our intellectual property or be required to make additional payments to secure intellectual property from collaborators.
Many of our consultants and employees, including our senior management, were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Some of these consultants and employees executed proprietary rights, non-disclosure and non-competition agreements in connection with such previous employment. Although we try to ensure that our consultants and employees do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these consultants and employees have used or disclosed confidential information or intellectual property of any such consultant’s or employee’s former employer or have breached their non-competition agreement. Additionally, many of our collaborators do not commit to assigning all intellectual property arising out of the collaboration to us and, instead, grant us options to acquire intellectual property or commit to making such intellectual property available to us at a fair price. As such, we or our licensors may have inventorship disputes arise from conflicting obligations of employees, consultants or others who are involved in developing our products and product candidates.
In addition, while it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with such party. Our and their assignment agreements may not be self-executing or may be breached and we may be forced to bring claims against third parties or defend claims they may bring against us to determine the ownership of what we regard as our intellectual property.
There is no guarantee we will be successful in defending such claims, which would result in us paying monetary damages, or lose valuable personnel or intellectual property rights.
28
Third-party intellectual property rights could adversely affect our ability to commercialize our products and product candidates.
Our competitive position may suffer if third-party intellectual property rights cover our products or product candidates or our manufacture or uses relevant to our development plans. In such cases, we may not be in a position to develop or commercialize products or product candidates unless we secure a license, design around, or successfully pursue costly and time-consuming proceedings to nullify or invalidate the third-party intellectual property right concerned or enter into a license agreement with the intellectual property right holder. If we were to challenge the validity of any issued U.S. patent in court, we would need to overcome a statutory presumption of validity that attaches to every U.S. patent. In the event that a patent is successfully asserted against us such that the patent is found to be valid and enforceable and infringed by our product, unless we obtain a license to such a patent, we could be prevented from continuing to develop or commercialize our product. Similarly, other companies have filed patent applications or have patents on the targets for certain of our products or their uses. There can be no assurance any such patents will not be asserted against us or that we will not need to seek licenses from such third parties.
It is also possible that we are unaware of relevant patents or applications or of relevant scientific discoveries. In general, patent applications in the U.S. and elsewhere are published approximately 18 months after the earliest filing from which priority is claimed, with such earliest filing date being commonly referred to as the priority date. Additionally, publications of discoveries in scientific literature often lag behind the actual discoveries. Therefore, patent applications covering our products, product candidates or platform technology could have been filed by others and relevant discoveries may have been made without our knowledge. Additionally, pending patent applications which have been published can, subject to certain limitations, be later amended in a manner that could cover our products or platform technologies.
Third-party intellectual property right holders, including our competitors, may actively bring infringement claims against us that we may not be able to successfully settle or otherwise resolve.
If we fail in any such dispute, we or our licensees may be prohibited from commercializing any of our products and product candidates that are held to be infringing for the remaining term of any valid and enforceable patents. We might, if possible, also be forced to redesign products and product candidates so that we no longer infringe the third-party intellectual property rights. We may be required to seek a license to any such technology that we are found to infringe, which license may not be available on commercially reasonable terms, or at all. Even if we or our licensors or collaboration partners obtain a license, it may be non-exclusive, thereby giving our competitors access to the same technologies licensed to us or our licensors or collaboration partners. In addition, if the breadth or strength of protection provided by our or our licensors’ or collaboration partners’ patents and patent applications is threatened, it could dissuade companies from collaborating with us to license, develop or commercialize current products and product candidates. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
We may not be successful in obtaining or maintaining necessary rights to our products and product candidates through acquisitions and in-licenses.
We may be unable to acquire or in-license third-party intellectual property rights that we identify as an appropriate strategic fit for our Company and necessary for our product candidates and technology. A number of more established companies with greater resources may pursue strategies to license or acquire third-party intellectual property rights that we may consider attractive.
We sometimes collaborate with U.S. and non-U.S. academic institutions to accelerate our preclinical research or development. Typically, these institutions provide us with an option to negotiate a license to any of the institution’s rights in technology resulting from the collaboration. Regardless of such option, we may be unable to negotiate a license within the specified timeframe or under terms that are acceptable to us, in which case the institution may offer the intellectual property rights to other parties, potentially blocking our ability to pursue our applicable product candidate or program.
29
In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment, in which case we may have to abandon development of that product candidate or program.
Existing license agreements impose various development, payment and other obligations. If we fail to comply with our obligations under these agreements, the licensor may have the right to terminate the license, which could result in us being unable to develop, manufacture, market and sell products that are covered by the licensed technology. Moreover, we could incur significant costs and/or disruption to our business and distraction of our management defending against any breach alleged by the licensor. Several of our existing license agreements are sub-licenses from third parties who are not the original licensors of the intellectual property at issue. If the licensors fail to comply with their obligations under these upstream license agreements, the original third-party licensor may have the right to terminate the original license, which may terminate the sublicense, causing us to lose our rights to the applicable intellectual property if we are unable to secure our own direct license with the owner of the relevant rights on reasonable terms.
Further, if disputes over intellectual property that we have licensed or our associated obligations prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected products and product candidates.
If our trademarks and trade names are not adequately protected, we may not be able to build name recognition in our markets of interest.
Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. Third parties may oppose or attempt to cancel our trademark applications or trademarks or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we may not be able to use these trademarks to market our products in those countries and could be forced to rebrand our products, which could result in loss of brand recognition and could require us to devote resources to advertising and marketing new brands. If we attempt to enforce our trademarks and assert trademark infringement claims, a court may determine that the marks we have asserted are invalid or unenforceable or that the party against whom we have asserted trademark infringement has superior rights to the marks in question. Over the long term, if we are unable to establish name recognition, we may not be able to compete effectively. If other entities use trademarks similar to ours in different jurisdictions, or have senior rights to ours, it could interfere with our use of our current trademarks throughout the world.
We may not be able to obtain protection under the U.S. Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Act) and similar non-U.S. legislation for extending the term of patents covering each of our products and product candidates.
Depending upon the timing, duration and conditions of FDA marketing approval of our product candidates, one or more of our U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Act and similar legislation in the EU and the Asia Pacific region. The Hatch-Waxman Act permits a patent term extension of up to five years for a patent covering an approved product as compensation for effective patent term lost during product development and the FDA regulatory review process. The patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, and only one patent applicable to an approved drug may be extended. However, we may not receive an extension if we fail to apply within applicable deadlines or prior to expiration of relevant patents or otherwise fail to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain patent term extension or the term of any such extension is less than we request, the period during which we can enforce our patent rights for that product will be shortened and our competitors may obtain approval to market competing products sooner than we expect.
30
Changes in patent laws or patent jurisprudence could diminish the value of patents in general, thereby impairing our ability to protect our products.
Changes in patent law and regulations in the various countries or jurisdictions or changes in the governmental bodies that enforce them or changes in how the relevant governmental authority enforces them may weaken our ability to obtain new patents or to enforce patents that we have licensed or that we may obtain in the future. We cannot predict future changes in the interpretation of patent laws or changes to patent laws that might be enacted into law by U.S. and foreign legislative bodies. For example, the U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. Relatedly, U.S. congressional representatives have introduced multiple draft bills this year that, if passed, may have a significant impact on U.S. patent laws. Such changes by the U.S. Congress, U.S. courts, USPTO, and the relevant law-making bodies in other countries may materially affect our patents or patent applications and our ability to obtain additional patent protection in the future.
We may be unable to protect the confidentiality of our trade secrets and know-how.
In addition to patent protection, we rely on trade secret protection for our proprietary information, including, for example, certain aspects of our llama immunization and antibody affinity maturation approaches. However, trade secrets are difficult to protect, and we have limited control over the protection of trade secrets used by our numerous licensors, collaborators and suppliers.
We require our employees, consultants, advisors and potential collaborators to enter into confidentiality agreements. Moreover, we put in place appropriate procedures to identify confidential material and restrict access to documentation. However, current or former employees, consultants, advisors and potential collaborators may unintentionally or willfully disclose our confidential information to competitors despite these procedures or in violation of our confidentiality agreements. In addition, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known to our competitors or inadvertently incorporated into the technology of others. Any disclosure, either intentional or unintentional, or misappropriation by third parties (such as through a cybersecurity breach) of our trade secrets or proprietary information could enable competitors to duplicate or surpass our technological achievements.
Enforcing a claim that a third party illegally obtained and is using trade secrets is expensive, time-consuming and the outcome is unpredictable, and the enforceability of confidentiality agreements may vary from jurisdiction to jurisdiction. Moreover, if any of our trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to prevent them from using that technology or information to compete with us.
Risk Factors Related to argenx’s Organization and Operations
Our future growth and ability to compete depends on retaining our key personnel and recruiting additional qualified personnel.
As a global organization in a highly competitive and specialized industry, our success depends upon the continued contributions of our key management, scientific, medical and technical personnel, many of whom have been instrumental for us and have substantial experience with our product and related technologies. These key management individuals include the members of our Board of Directors and senior management team. Difficulties in recruiting or the loss of key managers, scientific, medical or technical personnel could delay our research and development activities. In addition, it may be difficult to attract and retain highly qualified management, scientific and medical personnel, particularly if we expand into fields that will require additional skills.
As a Dutch company listed on Euronext Brussels in addition to the Nasdaq Global Select Market (Nasdaq), our remuneration practices and policies may be limited by local governance rules or shareholder guidance for EU companies. Such limitations may make it more difficult to successfully compete for key talent in a number of markets that have differing remuneration practices and policies as we are bound by more restrictive remuneration practices than
31
our competitors. For example, the Dutch Corporate Governance Code 2022 (DCGC) places certain limitations on the ability to grant equity incentives to non-executive directors, while Belgian law requires non-executive directors to receive part of their remuneration in the forms of shares, but not stock options. The DCGC also places limitations on amount of severance payment permitted in the event of dismissal. In addition, the U.S. has proposed legislation that imposes restrictions on our ability to prevent departing employees from competing with us following their departure. If finalized, such legislation could also adversely affect our ability to retain employees who may go to competitors with more resources than us and who are not bound by similar remuneration policies.
Many other biotechnology and pharmaceutical companies and academic institutions that we compete against for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than we do. Additionally, an inflationary environment, combined with the tight labor market for the recruitment and retention of skilled workers, could make it more costly for us to attract or retain employees. In order to meet the compensation expectations of our prospective and current employees due to inflationary factors, we may be required to increase our operating costs. Therefore, we might not be able to attract or retain these key persons on conditions that are economically acceptable.
Global geo- and socio-political threats and macro-economic uncertainty and other unforeseen political crises could materially and adversely affect our business and financial performance.
Many geo- and socio-political threats and macro-economic uncertainties are outside of our control, including general economic and market conditions, consumer and commercial credit availability, inflation, interest rates, unemployment, consumer debt levels, political crises, such as terrorist attacks, war and other political instability, economic sanctions, outbound investment restrictions and other challenges affecting the global economy, including the Russia-Ukraine and the Israel-Hamas conflicts, disruptions in supply chains, and changes in trade agreements which could adversely affect consumer confidence and disposable income levels, increase difficulty in forecasting our financial results and have other impacts on our business and financial performance. Such geo- and socio-political threats could also result in volatility in stock markets in general, causing our stock to have extreme price and volume fluctuations unrelated to our business and financial performance. For example, in 2023, we received approval for VYVGART in Israel through our partner Medison. It is not yet possible to predict or determine the ultimate consequence of the ongoing Israel-Hamas conflict on our business and financial performance.
Due to our international operations and the fact that we run clinical trials in a large number of jurisdictions, the eruption of global conflicts, such as the continuing conflict between Russia and Ukraine and the ongoing Israel-Hamas conflict, may negatively impact our ability to conduct or complete clinical trials in the affected regions, which could adversely affect our business and financial performance. On January 17, 2023, the U.S. Department of the Treasury’s Office of Foreign Assets Control issued General License 6C to replace General License 6B. General License 6C authorizes “clinical trials and other medical research activities” that would otherwise be prohibited by U.S. sanctions targeting Russia, and General License 6C does not have an expiration date. Following a risk assessment relating to the conflict between Russia and Ukraine, we increased target enrollment, which delayed expected topline data of SC efgartigimod for PV and PF to the second half of 2023. Additionally, the conflict between Russia and Ukraine and the sanctions imposed upon Russia by the U.S., the UK, and the EU, among others could disrupt:
| ● | the recruitment and enrollment of eligible patients who may not be able to travel safely to clinical trial sites or may be forced to withdraw for a number of reasons; |
| ● | the closure or destruction of clinical sites or treatment facilities; |
| ● | the ability to compensate patients or staff living in sanctioned countries; |
| ● | the manufacturing process for our products or supply chain, which could increase the costs of raw material and production costs; |
32
| ● | the ability to transport, deliver, supply and collect necessary materials, products or services to clinical trial sites or deliver them to third-party central laboratories’ for analysis; |
| ● | the ability to collect data from clinical trial sites and ensure the integrity of any data collected; |
| ● | the destruction or disruption of our data centers or our critical business or information technology systems; or |
| ● | the ability to submit data collected at Russian or Ukrainian sites due to the incompleteness or the fact that auditing by regulatory authorities was not fully possible. |
To date, other than as described above and elsewhere in this Annual Report, we have no indication that the conflict between Russia and Ukraine and the corresponding sanctions imposed on Russia will significantly hinder our clinical development activities performed in the affected regions or regulatory activities relevant for our pending or expected approval requests. Moreover, we do not generate revenues in Russia, and we gather more regular feedback from and to stakeholders and team members in Russia and Ukraine. However, we also perform development activities in a number of countries neighboring Russia and Ukraine and if the conflict were to escalate further and impact neighboring countries, it could impact our development activities in those countries.
Changes in U.S.-Mainland China relations, including tariffs, export controls, sanctions, and other regulations may adversely impact our collaboration with Zai Lab in Mainland China, Hong Kong, Taiwan and Macau (together, Greater China). The U.S. government has taken steps and continues to take steps with regard to U.S.- Mainland China relations that will impact companies with connections to the U.S. or Mainland China, including by imposing tariffs affecting certain products manufactured in mainland China, imposing certain sanctions on individuals and entities in the Mainland China, and issuing statements indicating enhanced review of companies with significant Mainland China-based operations. The scope of the impact of any such actions on the pharmaceutical industry or argenx remains unknown. Through argenx’s collaboration with Zai Lab, we have an interest in business operations in Greater China. Any unfavorable government policies on international trade, including tariffs, export controls, and/or increased scrutiny on companies with significant Mainland China-based operations, may impact the development and commercialization of products that contain argenx-licensed material.
Any new legislation, executive orders, tariffs, export controls, and/or other regulations that may be implemented, the renegotiation of existing trade agreements, and any retaliatory actions taken by the U.S. or Chinese governments due to the U.S.- Mainland China tensions could have an adverse effect on our business, including the development and commercialization of products containing argenx-licensed material.
We face risks related to natural disasters and public health issues, that could negatively affect our business and financial condition.
Our business could be adversely impacted by the effects of catastrophic global events including natural disasters such as an earthquake, fire, hurricane, tornado, flood or significant power outage and pandemics, such as the COVID-19 pandemic.
For example, the manufacturing of all of our products and product candidates requires using cells which are stored in a cell bank. We have one master cell bank for each product manufactured in accordance with cGMPs. However, it is possible that we could lose multiple cell banks and have our manufacturing significantly impacted by the need to replace these cell banks, which could materially adversely affect our business, prospects, financial condition and results of operations.
Public health issues could also negatively affect our business and financial condition. We operate and conduct our clinical trials globally. We cannot presently predict the scope and severity of any potential future business shutdowns or disruptions as a result of public health issues. If we or any of the third parties with whom we engage, including the suppliers, contract manufacturers, clinical trial sites, regulators and other third parties, were to experience shutdowns, quarantines, or other business disruptions due to natural disasters or global public health issues, it may impair our or our third-party partners’ ability to initiate clinical trials and recruit and retain patients, particularly if quarantine or travel
33
restrictions impede healthcare provider or patient movement, impact the usability of the data due to treatment interruptions and require protocol amendments. In addition, regulatory authorities may restrict their operations or be delayed in their operations during a pandemic, the outbreak of new variants or other public health issues, including further to travel restrictions which could adversely affect our ability to obtain regulatory approval for and to commercialize our products and product candidates and have a material adverse effect on our business and financial results.
We may encounter difficulties efficiently managing our growth and our increasing development, regulatory and sales and marketing capabilities, which could disrupt our operations.
We have grown significantly in the number of employees and scope of operations over recent years and expect to experience significant growth in the number of our employees and the scope of our operations also in the near future, particularly in the areas of drug research, drug development, regulatory affairs and sales and marketing. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. In particular, we must efficiently leverage our own sales and marketing capabilities in order to launch or market our products candidates effectively.
Due to our limited financial resources and the limited experience of our management team in managing a company with such anticipated growth, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. Our limited financial, manufacturing and management resources, could cause us to forgo or delay the pursuit of opportunities with potential product candidates that later prove to have greater market potential, fail to capitalize on viable commercial products or profitable market opportunities or relinquish rights to such product candidates through collaborations, licensing or royalty arrangements in circumstances where it would have been more advantageous for us to retain sole development and commercialization rights. Any inability to manage growth could delay the execution of our strategic objectives or disrupt our operations, which in turn could materially harm our business and prospects.
We have benefitted from certain research and development incentives. These could be impacted by changes in law (or interpretation), changes of fact (such as a change in ownership), or challenge by tax authorities.
As a company active in research and development, we have benefited from certain research and development tax incentives including tax credits and a payroll withholding tax exemption. We also expect to benefit from the Belgian innovation income deduction. The relevant tax authorities may challenge our eligibility for, or our calculation of, such tax incentives and, should such a challenge be successful, we may be liable for additional taxes, and penalties and interest related thereto, which could have an impact on our results of operations and future cash flows. In case of a change of control, we could be exposed to the risk of losing any unused tax credit and innovation income deduction. Furthermore, if any legislator decides to eliminate, or change the conditions for claiming such tax incentives, or reduce the scope or the rate of such incentives, any of which it could decide to do at any time, our results of operations could be adversely affected.
We are exposed globally to unanticipated changes in tax laws and regulations, adjustments to our tax provisions, exposure to additional tax liabilities, or forfeiture of our tax assets.
The determination of our provision for income taxes and other tax liabilities requires judgment, including the adoption of certain accounting policies and our determination of whether our deferred tax assets are, and will remain, fully available in future periods. We cannot guarantee that our interpretation of applicable tax laws or our structure will not be questioned by the relevant tax authorities, or that the relevant tax laws and regulations, or the interpretation thereof, including through tax rulings, by the relevant tax authorities, will not be subject to change. Any adverse outcome of such a review or change in law may lead to adjustments in the amounts recorded in our financial statements and could have a materially adverse effect on our operating results and financial condition.
Dealings between group companies are subject to transfer pricing regulations, which may be subject to change and could have a material impact. Compliance with these laws and regulations will be more challenging as we expand
34
our international operations, including in connection with potential approvals of our products and product candidates in Europe, Japan, the U.S. and elsewhere.
Our effective tax rates could be adversely affected, now or in the future, by changes in tax laws, treaties and regulations or the interpretation thereof by the relevant tax authorities in countries where we have material operations, including changes to the Belgian innovation income deduction, to the corporate income tax base, or to other tax incentives and the implementation of new tax incentives. A successful challenge to our qualifications for and application of these tax incentives by the tax authorities in Belgium or other country where we have material operations would have a significant impact on our effective tax rate and on our tax assets. An increase of the effective tax rates could have an adverse effect on our business, financial position, results of operations and cash flows.
In recent years, the Organisation of Economic Co-operation and Development (OECD) has worked on a project aimed at reforming the international tax system by, among other matters ensuring large multinational enterprises pay a minimum level of tax in each of the jurisdictions in which they operate (Pillar Two). In December 2021, the OECD released model rules in respect of Pillar Two (GloBE Rules). On December 14, 2022, the Council of the EU adopted Directive (EU) 2022/2523 implementing the GloBE Rules for multinational enterprise groups and large-scale domestic groups in the Union (Pillar Two Directive). The Pillar Two Directive was required to be implemented in the EU Member States’ national law by December 31, 2023. In addition, certain other jurisdictions in which the Group operates have either already enacted the GloBE Rules in their domestic law (such as Japan) or announced an intention to implement the GloBE Rules in their domestic law.
Based on current information, management expects that the Group could become subject to the Pillar Two Directive and domestic laws as early as 2025. Management does not expect the Pillar Two Directive and implementing domestic laws to have an impact on the Group in 2024. The Group is currently in the process of determining the impact, if any, for 2025 and onwards.
In addition, we may not be able to use, or changes in tax regulations may affect the use of, certain unrecognized tax assets or credits that we have built over the years. For instance, we have considerable material tax assets in Belgium and some of these tax assets may be forfeited in whole, or in part, as a result of various transactions, including corporate reorganizations or transactions relating to our shareholding structure, or their utilization may be restricted by statutory law or interpretation in the relevant jurisdiction.
Risks Related to the ADSs
The price of our ADSs and ordinary shares may be volatile and may fluctuate due to factors beyond our control. An active public trading market may not be sustained.
The stock markets in general, and biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. During 2023, the closing sales price of our ADSs representing our ordinary shares on Nasdaq fluctuated greatly, ranging from $334.23 to $548.43. The trading price of those securities depends on a number of factors, including those described in this “Risk Factors” section, many of which are beyond our control and may not be related to our operating performance, which may limit or prevent investors from readily selling their ADSs or ordinary shares and may otherwise negatively affect the liquidity of our ADSs and ordinary shares. Sales of a substantial number of ADSs or ordinary shares in the public market, or the perception that these sales might occur, could depress the market price of ADSs and ordinary shares and could impair the market price of our securities or our ability to raise capital through the sale of additional equity securities.
In addition, an active public trading market for our ADSs or our ordinary shares may not be sustained. Further, fluctuations in exchange rates may also impact the price of our ADSs and ordinary shares which may result in heavy trading by investors seeking to exploit such differences, or impact the proceeds holders receive.
35
If we fail to maintain an effective system of disclosure controls and internal control over financial reporting, our ability to comply with applicable regulations could be impaired, and the trading price of our ADSs may be negatively impacted.
We are required to comply with various corporate governance and financial requirements under the Sarbanes-Oxley Act of 2002, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the Nasdaq listing rules and requirements (Nasdaq Listing Rules), and other applicable securities rules and regulations. Pursuant to Section 404 of the Sarbanes-Oxley Act of 2002, we are required to furnish a report by management on, among other things, the effectiveness of our internal control over financial reporting and an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation. Moreover, any failure to maintain internal control over financial reporting or any material weaknesses or significant deficiency thereover, could result in a loss of investors’ in the accuracy, completeness and reliability of our financial statements, subject us to sanctions or investigations, or negatively impact the trading price of our ADSs.
Holders of our ADSs are not treated as holders of our ordinary shares and may be subject to limitations on the transfer of their ADSs and the withdrawal of the underlying ordinary shares.
ADSs are transferable on the books of the depositary. However, holders of ADSs are not treated as holders of our ordinary shares, unless they withdraw the ordinary shares underlying their ADSs in accordance with the deposit agreement and applicable laws and regulations. The depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary think it is advisable to do so because of any requirement of law, government or governmental body, or under any provision of the deposit agreement, or for any other reason. Temporary delays in the cancellation of ADSs and withdrawal of the underlying ordinary shares may arise because the depositary has closed its transfer books or we have closed our transfer books, the transfer of ordinary shares is blocked to permit voting at a shareholders’ meeting or we are paying a dividend on our ordinary shares. In addition, ADS holders may not be able to cancel their ADSs and withdraw the underlying ordinary shares when they owe money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
Holders of our ADSs do not have the same voting rights as the holders of our ordinary shares and may not receive voting materials in time to be able to exercise their right to vote.
Except as described in this Annual Report or any deposit agreements, holders of ADSs are not treated as our shareholders unless they withdraw the ordinary shares underlying their ADSs. The depositary, or its nominee, is the holder of the ordinary shares underlying the ADSs. Holders may vote them in person or by proxy in accordance with applicable laws and regulations and our Articles of Association. We cannot guarantee that holders of ADSs will receive the voting materials in time to ensure that they can instruct the depositary to vote the ordinary shares underlying their ADSs. Our shareholders are only entitled to participate in, and vote at, a general meeting of our shareholders (General Meeting), provided that their shares are recorded in their names at midnight (central European time) at the end of the 28th day preceding the date of such General Meeting. In addition, the depositary’s liability to holders of ADSs for failing to execute voting instructions or for the manner of executing voting instructions is limited by the deposit agreements. As a result, holders of our ADSs may not be able to exercise their right to give voting instructions or to vote in person or by proxy and they may not have any recourse against the depositary or us if their ordinary shares are not voted as they have requested or if their shares cannot be voted.
If securities or industry analysts cease coverage of us, or publish inaccurate or unfavorable research about our business, the price of our ADSs or ordinary shares and our trading volume could decline.
The trading market for the ADSs and ordinary shares depends in part on the research and reports that securities or industry analysts publish about us or our business. If no or too few securities or industry analysts cover us, the trading price of our ADSs and ordinary shares would likely be negatively affected. If one or more of the analysts who cover us
36
downgrade our ADSs or ordinary shares or publish inaccurate or unfavorable research about our business, the price of our ADSs or ordinary shares would likely decline.
Risks Related to being a Foreign Private Issuer or a Dutch Company
The risks in this subsection that relate to our status as a foreign private issuer will change if we lose our status as a foreign private issuer under U.S. law.
We are a Dutch European public company with limited liability (Societas Europaea or SE). The rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions.
We are a Dutch European public company with limited liability. The rights of shareholders and the responsibilities of members of our Board of Directors may be different from the rights and obligations of shareholders in companies governed by the laws of U.S. jurisdictions.
As a result of these differences between Dutch corporate law and our Articles of Association, on the one hand, and the U.S. federal and state laws, on the other hand, in certain instances, our shareholders and holders of our ADSs could receive less protection than they would as shareholders or ADS holders of a listed U.S. company.
Provisions of our Articles of Association might deter acquisition bids for us that might be considered favorable and prevent or frustrate any attempt to replace or remove the then Board of Directors.
Provisions of our Articles of Association may make it more difficult for a third party to acquire control of us or effect a change in our Board of Directors. We have adopted several provisions that may have the effect of making a takeover of our Company more difficult or less attractive. These provisions could discourage potential takeover attempts that other shareholders may consider to be in their best interest and could adversely affect the market price of our securities.
Holders of our ordinary shares outside the Netherlands, and holders of ADSs may not be able to exercise pre-emptive rights or preferential subscription rights, respectively.
In the event of an increase in our share capital, holders of our ordinary shares are generally entitled under Dutch law to full pre-emptive rights, unless these rights are excluded either by a resolution of the shareholders at a General Meeting, or by a resolution of the Board of Directors (if the Board of Directors has been designated by the shareholders at a General Meeting for this purpose).
However, making pre-emptive rights available to holders of ordinary shares or ADSs representing ordinary shares also requires compliance with applicable securities laws in the jurisdictions where holders of those securities are located, which we may be unable or unwilling to do. In particular, holders of ordinary shares or ADSs located in the U.S. would not be able to participate in a pre-emptive rights offering unless we registered the securities to which the rights relate under the U.S. Securities Act of 1933, as amended (Securities Act) or an exemption from the registration requirements. In addition, ADS holders would not be able to participate in a pre-emptive rights offering unless we made arrangements with the depositary to extend that offering to holders of ADSs, which we are not required to do.
We are not obligated to, and do not comply with, all the best practice provisions of the DCGC, which may affect shareholders’ rights.
As a Dutch public company with limited liability, we are subject to the DCGC. We do not comply with all the best practice provisions of the DCGC. As a Dutch company, we are required to disclose in our annual report, filed in the Netherlands, whether we comply with the provisions of the DCGC. If we do not comply with the provisions of the DCGC (for example, because of a conflicting Nasdaq Listing Rule or otherwise), we must list the reasons for any deviation from the DCGC in our annual report filed in the Netherlands.
37
Claims of U.S. civil liabilities may not be enforceable against us or the members of our management and our Board of Directors.
Substantially all of our assets are located outside the U.S. The majority of the members of our senior management team and our directors are not U.S. residents and we do not have significant assets in the U.S. As a result, it may not be possible, or may be very difficult, for investors to effect service of process within the U.S. upon such persons or to enforce against them or us in U.S. courts, including judgments predicated upon the civil liability provisions of the U.S. federal securities laws. There are no treaties between the U.S. with either the Netherlands or Belgium providing for the reciprocal recognition and enforcement of judgments, other than arbitration awards, in civil and commercial matters. Consequently, a final judgment for payment given by a court in the U.S. based on civil liability, whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in the Netherlands or in Belgium unless the underlying claim was re-litigated before a Dutch or Belgian court of competent jurisdiction. This will depend on the applicable Dutch or Belgian national rules. In light of the above, U.S. investors may not be able to enforce any judgments obtained in U.S. courts in civil and commercial matters, including judgments under the U.S. federal securities laws, against us, members of our management or our Board of Directors or certain experts named herein who are residents of the Netherlands, Belgium or countries other than the U.S. In addition, there is doubt as to whether a Dutch or Belgian court would impose civil liability on us or the members of our management or of our Board of Directors in an original action predicated solely upon the U.S. federal securities laws brought in a court of competent jurisdiction against us, our management or directors.
As a foreign private issuer, we are exempt from certain rules under U.S. securities laws and are permitted to file less information with the SEC than a U.S. company.
As a “foreign private issuer” defined in the SEC’s rules and regulations, we are exempt from certain provisions of the U.S. Securities Exchange Act of 1934, as amended (Exchange Act) that are applicable to U.S. domestic public companies.
We are subject to Dutch laws and regulations with regard to such matters. While we furnish quarterly unaudited financial information to the SEC on Form 6-K, the information we furnish to the SEC is less extensive and less timely compared to that required to be filed with the SEC by U.S. domestic issuers.
As a foreign private issuer, we are permitted to adopt certain home country governance practices rather than the corporate governance requirements of Nasdaq. These practices may afford less protection to shareholders than they would enjoy if we complied fully with corporate governance listing standards.
As a foreign private issuer listed on Nasdaq, we are subject to corporate governance listing standards. However, we are permitted to rely on home country governance requirements and certain exemptions thereunder. Certain of our corporate governance practices may differ significantly from other corporate governance listing standards.
We may lose our foreign private issuer status which would then require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur significant legal, accounting and other expenses.
In order to maintain our current status as a foreign private issuer, either (a) a majority of our ordinary shares must be either directly or indirectly owned of record by non-residents of the U.S. or (b)(i) a majority of our executive officers or directors may not be U.S. citizens or residents, (ii) more than 50% of our assets cannot be located in the U.S. and (iii) our business must be administered principally outside the U.S. As of February 1, 2024, we believe at least 50% of our outstanding ordinary shares were held by U.S. residents (assuming that all our ordinary shares represented by ADSs were held by residents of the U.S.).
The regulatory and compliance costs to us as a U.S. domestic issuer may be significantly higher than those we incur as a foreign private issuer. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain
38
coverage. These rules and regulations could also make it more difficult for us to attract and retain qualified members of our Board of Directors.
If we were to be classified as a passive foreign investment company for U.S. federal income tax purposes, this could result in adverse U.S. tax consequences to certain U.S. holders.
If our Company is classified as a passive foreign investment company (PFIC) for any taxable year, U.S. investors may be subject to adverse U.S. federal income tax consequences described below under “Taxation –U.S. Federal Income Tax Considerations – Passive Foreign Investment Company Rules.” Our Company will be classified as a PFIC for U.S. federal income tax purposes for any taxable year in which, taking into account a pro rata portion of the income and assets of 25% or more owned subsidiaries, either (i) at least 75% of its gross income consists of “passive income” or (ii) at least 50% of the average quarterly value of its assets is attributable to assets that produce, or are held for the production of, passive income.
Based on our historic and anticipated operations, the composition of our income and the projected composition and estimated fair market values of our assets, we do not believe that we were a PFIC for our most recent taxable year and do not expect to be classified as a PFIC for the current taxable year or for the foreseeable future. However, our status as a PFIC is a factual determination made on an annual basis, and we cannot provide any assurances regarding our PFIC status for the current or future taxable years.
ITEM 4. INFORMATION ON THE COMPANY
Our legal and commercial name is argenx SE. We were incorporated under the laws of the Netherlands on April 25, 2008, as a private company with limited liability (besloten vennootschap met beperkte aansprakelijkheid). From incorporation until August 28, 2009, our research and development activities were initially performed in the Netherlands, then Belgium, by argenx N.V. and its legal predecessors. Since August 28, 2009, all our research and development activities have been performed by our wholly-owned subsidiary, argenx BV, under a license provided by argenx N.V. Throughout this time, argenx BV assigned all resulting intellectual property to argenx N.V. On May 28, 2014, we converted to a Dutch public company with limited liability (naamloze vennootschap). On April 26, 2017, we converted to a Dutch European public company with limited liability (Societas Europaea or SE). On May 5, 2017, we transferred the legal ownership of all intellectual property rights of argenx SE to argenx BV, effective retroactively as of January 1, 2017. As a result, since January 1, 2017, (i) argenx BV holds all legal and economic ownership of our intellectual property rights, and (ii) the research and development agreement between argenx SE and argenx BV has been terminated.
Our official seat is in Rotterdam, the Netherlands, and our registered office is at Laarderhoogtweg 25, 1101 EB Amsterdam, the Netherlands. We are registered with the trade register of the Dutch Chamber of Commerce under number 24435214. Our European legal entity identifier number is 7245009C5FZE6G9ODQ71. Our telephone number is +31 (0) 10 70 38 441. Our website address is www.argenx.com. This website is not incorporated by reference in this Annual Report. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC at www.sec.gov. The registered agent for service of process in the U.S. is CT Corporation System, with an address at 111 8th Avenue, New York, NY 10011.
For information on our capital expenditure for the years ended December 31, 2023, 2022 and 2021, please see “Note 4—Property, plant and equipment” and “Note 5—Intangible assets” in our consolidated financial statements which are appended to our annual report on Form 20-F for the period ended December 31, 2023. We anticipate our capital expenditure in 2024 to be financed from the cash flows from operating activities and cash reserves. For more information on our capital expenditures and requirements, see Item 5.B. “Liquidity and Capital Resources—Cash Flows—Operating and Capital Expenditure Requirements.”
No takeover bid has been instigated by third parties in respect of our equity during the current or previous fiscal years.
39
B. | BUSINESS OVERVIEW |
We are a commercial-stage, global, fully-integrated biopharma company developing a deep pipeline of differentiated therapies for the treatment of severe autoimmune diseases. By combining our suite of antibody engineering technologies with the disease biology expertise of our research collaborators, we aim to translate immunology breakthroughs into a pipeline of novel antibody-based medicines through our discovery engine, the IIP. We developed and are commercializing the first approved FcRn blocker in the U.S., Japan, Israel, the EU, Mainland China and Canada. We are evaluating efgartigimod in multiple serious autoimmune diseases and advancing several earlier stage experimental medicines.
For additional information regarding our Company’s principal markets and revenue breakdown, see Item 5.A. “Operating Results” and “Note 15—Product net sales” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
2023 In Brief
Operational Highlights
At the start of 2023, we shared our key drivers for the year to continue our trajectory of value creation. First, we aimed to reach more patients globally with VYVGART, our first-in-class FcRn blocker. VYVGART and VYVGART SC are now approved in more than 30 countries or regions and by the end of 2023, we had treated over 6,000 gMG patients globally with our innovations.
gMG is just the beginning of our mission to transform autoimmunity. Our second key driver was to pioneer the FcRn class of medicines, including broadening the scope of indications we are evaluating with efgartigimod. As of the end of 2023, efgartigimod is approved or under regulatory review in three indications, including gMG, CIDP and ITP, and is being evaluated in more than 10 additional serious autoimmune indications. We are well on our way to achieve our ‘argenx 2025’ vision of efgartigimod being commercially available or in clinical development in 15 indications by 2025.
Third, we worked to advance our pipeline of differentiated immunology assets. Beyond efgartigimod, our wholly-owned clinical pipeline consists of empasiprubart (ARGX-117) targeting complement component 2 (C2) and ARGX-119 targeting muscle-specific kinase (MuSK). We believe both have potential as a novel treatment modality in multiple serious indications.
The fourth key driver was to build out our innovation ecosystem, serving our core mission to sustainably deliver immunology innovations to the patients who need them. We continue to invest in our IIP from which we drive pipeline expansion by collaborating with leading disease biologists who are researching first-in-class disease targets or pathways. Our IIP has a track record of success and nine programs have been tested in humans since our inception.
Reach More Patients Globally with VYVGART
| ● | VYVGART is now approved in the U.S., Japan, Europe, the UK, Israel, Mainland China and Canada for the treatment of gMG. VYVGART SC is now approved in the U.S., Europe, the UK and Japan for the treatment of gMG. This makes VYVGART the only gMG treatment available as both an IV and a simple SC injection, providing choice to patients in how and where they are treated. In 2023, we generated product net sales of $1.2 billion |
| ● | Pricing and reimbursement discussions for VYVGART and VYVGART SC remain ongoing in multiple jurisdictions, including in several countries in the EU |
| ● | We filed for approval of VYVGART for ITP in Japan and a decision is expected in the first quarter of 2024 |
40
| ● | A supplemental Biologics License Application (sBLA) for SC efgartigimod for CIDP has been accepted for priority review by the FDA, with a Prescription Drug User Fee Act (PDUFA) target date of June 21, 2024 |
| ● | We have filed for approval of VYVGART SC in Mainland China and we expect a decision on approval by the end of 2024 through our partnership with Zai Lab |
| ● | We entered into a VYVGART commercial and distribution agreement with Handok in South Korea (the Handok Agreement) |
| ● | We filed for approval of VYVGART for gMG in several jurisdictions and we are expecting multiple decisions by the end of 2024 |
Advance Extensive Pipeline
We continue to demonstrate breadth and depth within our immunology pipeline and have advanced multiple pipeline-in-a-product candidates. With efgartigimod, we are furthering our leadership in FcRn and we expect to be approved or in development in 15 autoimmune indications by 2025. Beyond efgartigimod, we are advancing earlier stage pipeline programs, including empasiprubart (C2 inhibitor) with Phase 2 proof-of-concept (POC) clinical trials ongoing in multifocal motor neuropathy (MMN), delayed graft function (DGF) and dermatomyositis (DM). In addition, we expect to initiate Phase 1b/2a clinical trials of ARGX-119, a MuSK agonist, in congenital myasthenic syndrome (CMS) and amyotrophic lateral sclerosis (ALS) in 2024.
Pioneer the FcRn Pathway with Efgartigimod
| ● | Neurology indications: |
| ● | ADHERE: following the positive topline results from the ADHERE clinical trial in CIDP, an sBLA for SC efgartigimod was submitted on December 21, 2023 and is under review by the FDA with a PDUFA date of June 21, 2024 |
| o | Clinical trial met primary endpoint (p=0.000039); SC efgartigimod demonstrated 61% reduction (HR: 0.39 95% CI: 0.25; 0.61) in risk of relapse versus placebo |
| ● | ALKIVIA: operationally seamless Phase 2/3 clinical trial is ongoing with SC efgartigimod for three subtypes of idiopathic inflammatory myopathies (Myositis), including immune-mediated necrotizing myopathy (IMNM), anti-synthetase syndrome (ASyS) and DM; analysis planned for first 30 patients of each subtype |
| ● | Registrational clinical trial in thyroid eye disease (TED) expected to start in first quarter of 2024 |
| ● | Hematology/rheumatology indications: |
| ● | ADVANCE-IV: positive clinical trial results formed basis of filing in Japan for ITP; topline results published in The Lancet in September 2023 |
| ● | ADVANCE-SC: topline data from SC efgartigimod in ITP announced on November 28, 2023 |
| o | Primary endpoint was not met (p=0.5081); 13.7% (17/124) of treated patients demonstrated sustained platelet count response compared to 16.2% (11/68) of placebo patients. Secondary endpoints were also not met |
| ● | RHO: Phase 2 POC clinical trial in primary Sjögren’s disease (SjD) is ongoing through our partnership with IQVIA Ltd (IQVIA) |
41
| ● | ALPHA: Phase 2 POC clinical trial in postural orthostatic tachycardia syndrome post-COVID-19 (PC-POTS) ongoing through our partnership with IQVIA |
| ● | Dermatology indications: |
| ● | ADDRESS: announced topline data of SC efgartigimod in PV and PF on December 20, 2023 |
| o | Primary endpoint was not met; proportion of PV patients achieving primary endpoint of CRmin was not significantly different between SC efgartigimod and placebo |
| o | Treatment with SC efgartigimod led to CRmin in 35.5% of patients compared to 30.3% with placebo (p=0.5956). Secondary endpoints were also not met |
| ● | BALLAD: in light of ADDRESS results and the comparable biology between PV and bullous pemphigoid (BP), we decided to stop enrollment of BALLAD. We will integrate key learnings from ADDRESS and data from already enrolled patients in BALLAD and we plan to communicate on a revised development plan before end of 2024 |
| ● | Nephrology indications: |
| ● | Membranous Nephrology (MN): Phase 2 POC clinical trial ongoing through our partnership with Zai Lab |
| ● | Lupus Nephritis (LN): Phase 2 POC clinical trial ongoing through our partnership with Zai Lab |
| ● | Antibody-mediated rejection (AMR): IND submission planned for the second quarter of 2024 |
Broaden Immunology Pipeline with Empasiprubart and ARGX-119
| ● | Empasiprubart (C2 inhibitor): |
| ● | ARDA: Phase 2 POC clinical trial ongoing of empasiprubart in MMN |
| o | In January 2024, we reported positive clinical data from the first cohort of the Phase 2 POC ARDA clinical trial establishing POC in MMN. Empasiprubart demonstrated a 91% reduction in the need for IV immunoglobulin (Ig, in IV formulation, IVIg) rescue compared to placebo [HR: 0.09 95% CI (0.02; 0.044)]. Safety profile was consistent with Phase 1 data |
| ● | Phase 2 POC clinical trials ongoing in DGF and DM |
| ● | ARGX-119 (MuSK agonist): |
| ● | Phase 1 dose-escalation clinical trial in healthy volunteers ongoing; Phase 1b/2a clinical trials expected to start in 2024 to assess early signal detection in patients with CMS and ALS, respectively |
Build out Innovation Ecosystem
| ● | In January 2024, we announced the nomination of four new pipeline candidates, including: ARGX-213 targeting FcRn and furthering argenx’s leadership in this new class of medicine; ARGX-121 and ARGX-220, which are first-in-class targets broadening argenx’s focus across the immune system; and ARGX-109, targeting IL-6, which plays an important role in inflammation. Preclinical work is ongoing in each candidate. |
42
| ● | We entered into a collaboration with Genmab to jointly discover, develop and commercialize novel therapeutic antibodies with applications in immunology, as well as in oncology therapeutic areas. |
Corporate Achievements
| ● | Mr. Steve Krognes joined our Board of Directors in February 2023 as a non-executive director and chairperson of the audit and compliance committee |
| ● | Mr. J. Donald deBethizy, who has served as a director since May 2015, was appointed to serve as vice-chairman of the Board of Directors as of February 2023 |
| ● | Karen Massey joined argenx as chief operating officer (COO) in March 2023 succeeding Keith Woods. Mr. Woods transitioned to serve as strategic advisor to the commercialization committee of the Board of Directors |
| ● | Expansion to 1,148 full-time employees (as of December 31, 2023) to support further growth of our business, including fully staffed commercial teams in the U.S., Europe, Japan and Canada |
2023 Financial Highlights
| ● | Product net sales of $1.2 billion |
| ● | Cash and cash equivalents and current financial assets of $3.2 billion enabling execution of our ambitious strategy objective |
| ● | Operating loss $425 million |
| ● | Loss $295 million |
| ● | $1.3 billion raised in gross proceeds in global offering of 2,581,633 ordinary shares (including ordinary shares represented by ADSs, which included the full exercise of the underwriters’ option to purchase 336,734 additional ADSs |
43
2024 Outlook
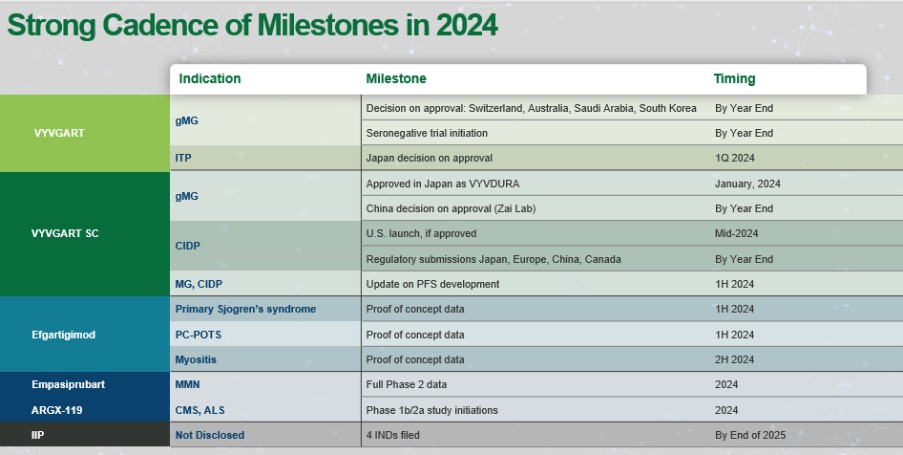
The table above is subject to risks and uncertainties that may materially impact the achievement of our 2024 outlook. For more information, please refer to Item 3.D. “Risk Factors” of this Annual Report for a discussion of such risks and uncertainties.
Our Medicines
VYVGART is a first-in-class antibody fragment targeting FcRn and is now approved for gMG in more than 30 countries globally for the treatment of gMG.
VYVGART SC is now approved in the U.S., the EU, the UK, and in Japan for the treatment of gMG. This makes VYVGART the only gMG treatment available as both an IV and simple SC injection.
Our Pipeline
| ● | Efgartigimod is a human IgG1 antibody region interacting with cell surface Fc receptors (Fc) fragment that is designed to target the FcRn and reduce immunoglobulin G (IgG). It is approved or under regulatory review in 3 indications, including gMG, CIDP and ITP, and is being evaluated in more than 10 additional serious autoimmune indications |
| ● | Empasiprubart (C2 inhibitor): empasiprubart is a novel complement inhibitor targeting C2, blocking the function of both the classical and lectin pathways while leaving the alternative pathway intact. We believe empasiprubart has the potential to be a pipeline-in-a-product candidate and is being evaluated in 3 serious autoimmune diseases |
| ● | ARGX-119 (MusK agonist): ARGX-119 is an agonist SIMPLE ANTIBODY™ to the MuSK receptor with potential in multiple neuromuscular indications. It is currently being evaluated in a Phase 1 dose escalation clinical trial in healthy volunteers |
| ● | Preclinical Candidates: Preclinical work is ongoing for each of the following candidates: |
| o | ARGX-213, targets FcRn, furthering argenx’s leadership in this new class of medicine |
44
| o | ARGX-121 and ARGX-220 are first-in-class targets broadening argenx’s focus across the immune system |
| o | ARGX-109, targets IL-6, which plays an important role in inflammation |
| ● | In addition to our wholly-owned pipeline, we have candidates that emerged from our IIP that we out-licensed to a partner for further development and for which we have milestone, royalty or profit-share agreements. These candidates include, amongst others: cusatuzumab (anti-CD70 antibody – Oncoverity), ARGX-112 (LP-0145 – anti-IL-22R antibody – LEO Pharma), ARGX-114 (AGMAB-101 – agonistic anti-MET antibody – Agomab) and ARGX-115 (ABBV-151 – anti-GARP antibody – AbbVie). |
IIP
Our IIP is central to our core business strategy of co-creation and innovation. The IIP also serves as our discovery engine to identify novel targets and together, in collaboration with our scientific and academic partners, to build potential new pipeline candidates. Every current pipeline candidate from both our wholly-owned and partnered pipeline emerged from an IIP collaboration. The IIP enables us to build our broad pipeline of products and product candidates and advance our long-term strategy to be a sustainable, integrated immunology company.
Examples of our IIP programs include:
| ● | Efgartigimod emerged from a collaboration with Professor Sally Ward at the University of Texas Southwestern Medical Center (UT Southwestern) and later became one of the blueprints for our IIP collaborations. Professor Ward’s research identified the crucial role that FcRn plays in maintaining and distributing IgGs throughout the body. Efgartigimod is a human IgG1 Fc fragment that is equipped with ABDEG™ mutations, which we in-licensed from UT Southwestern. These proprietary mutations modified efgartigimod to increase its affinity for FcRn while retaining the pH-dependent binding that is characteristic of FcRn interactions with its natural ligand, endogenous IgG. |
| ● | Empasiprubart was built in collaboration with Broteio Pharma B.V. (Broteio). Broteio was launched in 2017 with support from Professor Erik Hack and the University of Utrecht, to conduct research demonstrating preclinical POC of the mechanism of action of empasiprubart. Professor Hack is a renowned researcher in the role of inflammation in disease, specifically in the complement system, and has contributed research and expertise to the approval of 2 complement inhibitors. His understanding of the mild phenotype associated with a natural C2 deficiency and C2’s unique positioning at the junction of the classical and lectin pathways led to our interest in engineering empasiprubart, with our proprietary NHANCE™ mutations and LALA mutations. |
| ● | ARGX-119 was built in collaboration with the Leiden University Medical Center (LUMC) and New York University (NYU) with support from teams led by Professor Verschuuren and Professor Steve Burden, respectively. Both groups have world-class expertise in unraveling the biological mechanism of neuromuscular disease and translating these insights from the lab to the patient. |
Our Suite of Technologies
| ● | Through our IIP, we collaborate with scientific and academic partners to identify immunology breakthroughs and build potential pipeline candidates. This is done through co-creation. We bring to the collaboration our unique suite of antibody engineering technologies and experience in clinical development to complement our partners’ expertise in disease and target biology. |
| ● | SIMPLE ANTIBODY™ platform technology: Our proprietary SIMPLE ANTIBODY™ platform technology, based on the powerful llama immune system, allows us to exploit novel and complex disease biology targets. The platform sources antibody variable regions (V-regions) from the immune system of outbred llamas, each of which has a different genetic background. The llama produces highly diverse panels of antibodies with a high |
45
| human homology, or similarity, in their V-regions when immunized with targets of human disease. Our SIMPLE ANTIBODY™ platform technology allows us to access and explore a broad target universe while potentially minimizing the long timelines associated with generating antibody candidates using traditional methods. |
| ● | NHANCE™, ABDEG™, POTELLIGENT®, and dehydrated hereditary stomatocytosis (DHS) mutations focus on engineering the Fc region of antibodies in order to augment their intrinsic therapeutic properties. In addition, we obtained a non-exclusive research license and option from Chugai Pharmaceutical Co., Ltd. (Chugai) for the SMART-Ig® (‘Recycling Antibody’ and part of ‘Sweeping Antibody’) and ACT-Ig® (Antibody half-life extending) technologies. These technologies are designed to enable us to expand the therapeutic index of our product candidates, which is the ratio between toxic and therapeutic dose, by potentially modifying their half-life, tissue penetration, rate of disease target clearance and potency. In 2020, we also entered into a non-exclusive research agreement with the Clayton Foundation under which we may access the Clayton Foundation’s proprietary DHS mutations to extend the serum half-life of therapeutic antibodies. |
| ● | Halozyme’s ENHANZE® SC drug delivery technology: we have exclusive access to ENHANZE® for FcRn, C2 and four additional target nominations. The global collaboration and license agreement with Halozyme Therapeutics, Inc. (Halozyme) was announced in February 2019 and expanded in October 2020. The ENHANZE® technology has the potential to shorten drug administration time, reduce healthcare practitioner time and offer additional flexibility and convenience for patients. |
| ● | In April 2021, we entered into a collaboration and license agreement with Elektrofi, Inc. (Elektrofi) to explore Elektrofi’s high concentration technology for efgartigimod, and up to one additional target (Elektrofi Agreement). |
Our Products and Product Candidates
The following table summarizes key information on our portfolio of lead product and product candidates as of the date of this Annual Report.
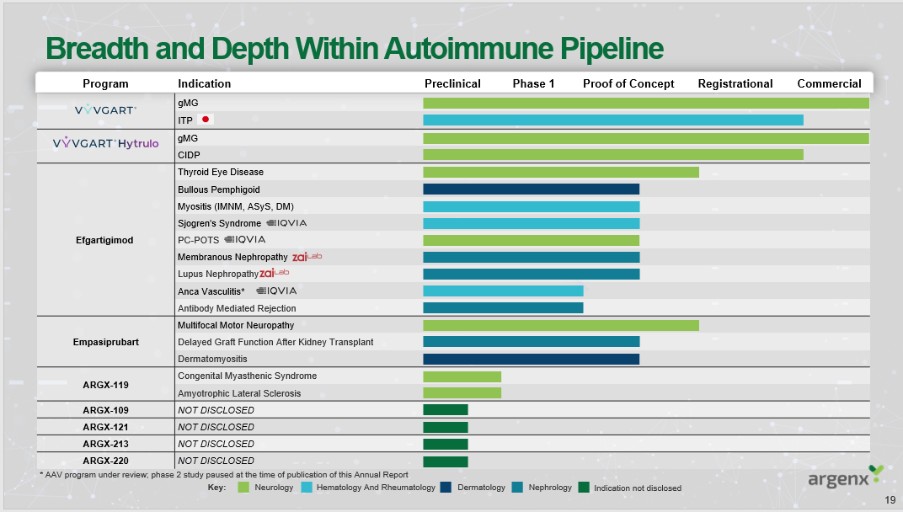
46
Our Programs
VYVGART
Approval in gMG
Our two approved medicines for gMG are VYVGART and VYVGART SC. VYVGART is a FcRn blocker approved for the treatment of adults with gMG who are AChR-AB+ in the U.S., the EU, Israel, the UK, Mainland China and Canada and for the treatment of adults with gMG who do not have sufficient response to steroids or non-steroidal immunosuppressive therapies (ISTs), including seronegative patients, in Japan. Our second product, VYVGART SC, is a subcutaneous combination of efgartigimod alfa and rHuPH20, Halozyme’s ENHANZE® SC drug delivery technology. It has been approved for the treatment of adults with gMG who are AChR-AB+ as VYVGART HYTRULO in the U.S. and VYVGART SC in the EU and the UK. It has also been approved as VYVDURA in Japan for the treatment of adults with gMG who do not have sufficient response to steroids or non-steroidal ISTs, including seronegative patients.
gMG is a rare and chronic autoimmune disease, often causing debilitating and potentially life-threatening muscle weakness. A key driver of gMG is the action of AChR autoantibodies at the neuromuscular junction. VYVGART, a human IgG1 antibody fragment that binds to FcRn, acts by reducing circulating IgG antibodies.
The approval of VYVGART is based on results from the global Phase 3 ADAPT clinical trial, which were published in the July 2021 issue of The Lancet Neurology (source: Howard JF Jr et al., Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, Phase 3 trial. Lancet Neurology. 2021; 20: 526-36).
The ADAPT clinical trial demonstrated significantly more AChR-AB+ gMG patients were responders on the Myasthenia Gravis Activities of Daily Living (MG-ADL) score following treatment with efgartigimod compared with placebo (67.7% vs. 29.7%; p<0.0001). Responders were defined as having at least a two-point improvement sustained for four or more consecutive weeks on the MG-ADL score. Additionally, 40% of patients treated with efgartigimod achieved minimal symptom expression defined as MG-ADL scores of zero (symptom free) or one, compared to 11.1% of patients who received placebo. Among AChR-AB+ responders, 84.1% showed clinically meaningful improvement on the MG-ADL score within the first two weeks of treatment. The safety profile of efgartigimod was comparable to placebo.
The approvals of VYVGART SC are based on positive results from the global Phase 3 ADAPT-SC bridging clinical trial.
The ADAPT-SC clinical trial established the efficacy of VYVGART SC by demonstrating a reduction in anti-AChR antibody levels comparable to VYVGART IV in adult gMG patients. The primary endpoint of noninferiority was met (p< 0.0001) and VYVGART SC demonstrated mean total IgG reduction of 66.4% from baseline at day 29, compared to 62.2% with VYVGART. Additional key secondary endpoints were met, which were consistent with efficacy measures from the ADAPT clinical trial identifying the correlation between total IgG reduction and clinical benefit in gMG. VYVGART SC has a demonstrated safety profile, consistent with the ADAPT IV clinical trial. As commonly observed with biologics administered subcutaneously, VYVGART SC showed injection site reactions. Such injection site reactions (ISRs) were mild to moderate and did not lead to treatment discontinuation.
Commercialization and Regulatory Plans
VYVGART has been approved in the U.S., Japan, Europe, Mainland China, Canada, the UK and Israel for the treatment of gMG. VYVGART launched in the U.S., Japan, Mainland China, Canada and some countries in Europe.
In Mainland China, VYVGART was added to the National Reimbursement Drug List (NRDL) in January 2024.
VYVGART SC has been approved in the U.S., in Europe, the UK and in Japan. VYVGART SC launched in the U.S. and in Germany.
47
Launches of both VYVGART and VYVGART SC in multiple jurisdictions and countries are planned following pricing and reimbursement negotiations.
We have established our own sales force in the U.S., Japan, Europe, Canada and the UK for VYVGART for the treatment of gMG. We plan to expand our own sales and marketing capabilities and promote our products and product candidates in other regions if we decide there is a business case to do so after regulatory approval has been obtained.
Development and commercialization may also be done through collaborations with third parties. In January 2021, we entered into an exclusive out-license agreement with Zai Lab (Zai Lab Agreement), a commercial-stage biopharmaceutical company, for the development and commercialization of efgartigimod in Greater China. Zai Lab announced approval of VYVGART in Mainland China in June 2023 for the treatment of adult gMG patients. Under the Zai Lab Agreement, we received and continue to be eligible for certain milestone payments and royalties based on annual net sales of efgartigimod in Greater China.
In October 2021, we announced an exclusive distribution agreement with Medison to commercialize efgartigimod for gMG in Israel (Medison Agreement). Medison filed for and obtained approval for VYVGART in Israel in April 2023. On June 6, 2022 we announced an exclusive multi-regional agreement with Medison to commercialize efgartigimod in 14 countries, including Poland, Hungary, Slovenia, Czech Republic, Romania, Bulgaria, Lithuania, Croatia, Slovakia, Estonia, Latvia, Greece, and Cyprus, for the treatment of adult patients with gMG (Medison Multi-Regional Agreement).
In January 2022, we entered into a partnership agreement with Genpharm, under which Genpharm shall purchase VYVGART from us for the resale in the GCC on an exclusive basis for Genpharm’s own account and own name (Genpharm Agreement).
In 2023, we entered into the Handok Agreement with Handok for the distribution of VYVGART in South Korea.
We intend to sign additional distribution partnerships for other territories.
For a discussion of total revenues by geographic market, please see “Note 18—Segment reporting” in our consolidated financial statements which are appended to this Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
Pre-Approval Access Program
We are committed to improving the lives of people suffering from rare diseases. We are driven to discover new treatment approaches in autoimmunity and fueled by the resilience of patients to urgently deliver them. We aim to do this in partnership; we listen to patients, supporters and advocacy communities, and we hear their stories. Their insights guide us as we develop our investigational therapies and motivate us to advance the understanding of rare diseases.
We implemented a pre-approval access program (PAA) on February 21, 2021 through which investigational therapies are made available in certain circumstances to treat gMG patients who are unable to participate in an ongoing clinical trial. In 2023, we approved access to the PAA for over 330 gMG patients in 14 countries. The PAA program remains open in countries where VYVGART is not yet launched or reimbursed.
Efgartigimod (formerly ARGX-113) Development
Mechanism of Action
As shown in Figure 1, efgartigimod is a human IgG1 Fc fragment equipped with our ABDEG™ mutations that is designed to target the FcRn and reduce IgG. FcRn is foundational to the immune system and functions to recycle IgG,
48
extending its serum half-life over other Igs that are not recycled by FcRn. IgGs that bind to FcRn are rescued from lysosomal degradation. By binding to FcRn, efgartigimod can reduce IgG recycling and increase IgG degradation.
Compared to alternative immunosuppressive approaches, such as B-lymphocyte (B-cell) depleting agents, efgartigimod acts in a highly selective manner. For efgartigimod, we now have an estimated 4,000 patients years of safety follow-up between clinical trials and real world experience. Efgartigimod has been observed to significantly reduce concentrations of all IgG subtypes without decreasing levels of other Igs or human serum albumin, which is also recycled by FcRn, discussed in more detail in Item 4.B. “Business Overview—Formulations” below.
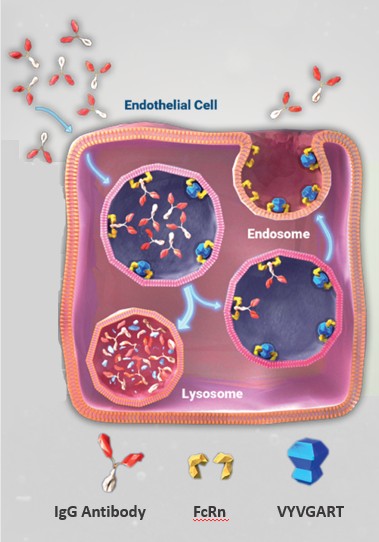
Figure 1: Efgartigimod’s mechanism of action blocks the recycling of IgG antibodies and removes them from circulation.
49
Based on its mechanism of action in targeting FcRn to selectively reducing IgGs, efgartigimod has the potential to address a multitude of severe autoimmune diseases where pathogenic IgGs are believed to be mediators of disease.
As of the end of 2023, we are evaluating efgartigimod in more than 10 serious autoimmune indications. We plan to expand efgartigimod into new indications and plan to be in 15 indications by 2025.
Indication Selection Strategy
We utilize the following strategy to select indications for efgartigimod:
| ● | We first start with a strong, unifying biological rationale. The indications in our pipeline are unified in that there exists a wide range of supportive evidence that demonstrates that each is IgG-mediated. This ranges from published literature, clinical trials with currently used therapies such as IVIg, PLEX, or Rituximab, and other experiments, such as passive transfer models. |
| ● | We also look at indications where a significant clinical or commercial opportunity exists. These are disease areas where there is a significant unmet need for innovation as patients are often not well-managed by current therapies and their respective side effects. |
| ● | Furthermore, for each indication, there is a defined path forward with established precedent for how to run POC and registrational clinical trials with generally accepted clinical and regulatory endpoints. |
| ● | Finally, as we work towards achieving our ‘argenx 2025’ vision, we select indications where there is a reasonable fit within our growing commercial activities. |
Formulations
Overview
We are developing two formulations of efgartigimod to address the needs of patients, physicians, and payors across indications and geographies, including IV efgartigimod (VYVGART) and SC efgartigimod (VYVGART SC).
IV (VYVGART)
We conducted a Phase 1 clinical trial in healthy volunteers to evaluate the safety, tolerability, pharmacokinetic (PK), pharmacodynamic (PD), and immunogenicity of single and multiple doses of efgartigimod. In the first part of the clinical trial, 30 subjects were randomized to receive a single dose of efgartigimod or placebo ranging from 0.2 mg/kg to 50 mg/kg. In the second part of the clinical trial, 32 subjects were randomized to receive multiple ascending doses (MADs) of efgartigimod or placebo up to a maximum of 25 mg/kg.
In the MAD part of the Phase 1 clinical trial, repeat administration of both 10 mg/kg and 25 mg/kg of efgartigimod every seven days, four doses in total, and 10 mg/kg every four days, six doses in total, was associated with a gradual reduction in levels of all four classes of IgG antibodies by 60% to 85%, with 10 mg/kg dose results shown in Figure 2. For all doses in the MAD part of the Phase 1 clinical trial, we observed the reduction in circulating IgG antibody levels to persist for more than four weeks after the last dose with levels below 50% at approximately three weeks and did not return to baseline levels for more than one month. PK analysis of serum baseline levels of efgartigimod indicates that it has a half-life of approximately three to four days with no drug accumulation following subsequent weekly dosing. The prolonged activity on the levels of IgG antibodies is consistent with the mechanism of action of efgartigimod and the effect of our proprietary ABDEG™ technology (detailed in Item 4.B. “Business Overview—Intellectual Property—Platform Technologies”) on increasing the intracellular recycling of efgartigimod. In
50
both the single and MAD portions, no significant reductions in immunoglobulin M (IgM), immunoglobulin A (IgA) or serum albumin were observed.
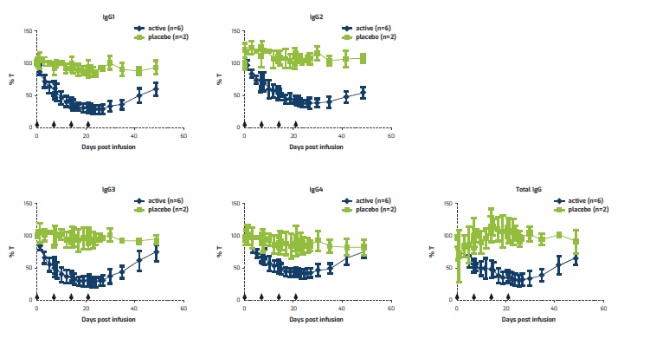
Figure 2: Reduction in the levels of four IgG antibody classes and total IgG levels in the MAD part of our Phase 1 clinical trial of efgartigimod in healthy volunteers at a dose of 10 mg/kg every seven days.
SC (VYVGART SC) - Partnership with Halozyme
In July 2019, we evaluated a first generation of SC efgartigimod that incorporates Halozyme’s ENHANZE® SC drug delivery technology in a Phase 1 clinical trial in healthy volunteers, which demonstrated retained PD profile of IV efgartigimod.
ENHANZE® has demonstrated across multiple FDA-approved products the ability to remove traditional limitations on the volume of biologics that can be delivered subcutaneously, potentially shortening drug administration time, reducing healthcare practitioner time, and offering additional flexibility and convenience for patients.
In 2020, we expanded the existing global collaboration and license agreement with Halozyme. Under the expansion, we gained the ability to access Halozyme’s ENHANZE® SC drug delivery technology for three additional exclusive targets upon nomination bringing the total to six potential targets under the collaboration. To date, two targets have been nominated including FcRn and C2.
In March 2022, we announced our Phase 3 ADAPT-SC clinical trial evaluating SC efgartigimod achieved the primary endpoint of total IgG reduction from baseline at day 29, demonstrating statistical non-inferiority to VYVGART IV formulation in gMG patients. Based on these results, we received approval of VYVGART SC for the treatment of adult patients with gMG in the U.S., the EU, the UK and Japan.
Currently, we are developing a pre-filled syringe presentation for the same SC formulation using the Halozyme technology, to allow for a convenient delivery and the potential for self-administration, reducing the healthcare practitioner time and further increasing flexibility and convenience for patients. As a next step in patient convenience, we have also started the development of a high-volume auto-injector.
SC – Partnership with Elektrofi
In April 2021, we entered into a collaboration and license agreement with Elektrofi to explore a high concentration technology for efgartigimod and up to one additional target. Please refer to Item 4.B. “Business Overview—License Agreements—Our Exclusive License with Elektrofi for efgartigimod” for more information.
51
Efgartigimod Indications
gMG
Overview
gMG is a rare and chronic autoimmune disease where IgG autoantibodies disrupt communication between nerves and muscles, causing debilitating and potentially life-threatening muscle weakness.
In MG, IgG autoantibodies either bind and occupy or cross-link and internalize the receptor on the muscle cells, thereby preventing the binding of acetylcholine, the signal sent by the nerve cell. In addition, these autoantibodies can cause destruction of the neuromuscular junction by recruiting complement, a potent cell-destroying mechanism of the human immune system. The muscle weakness associated with MG usually presents initially in ocular muscles and can then spread into a generalized form affecting multiple muscles, known as gMG. Approximately 85% of people with MG progress to gMG within 24 months (source: Behin et al. New Pathways and Therapeutics Targets in Autoimmune Myasthenia Gravis. J Neuromusc Dis 5. 2018. 265-277). MG in the ocular form initially causes droopy eyelids and blurred or double vision due to partial paralysis of eye movements. As MG becomes generalized it affects muscles in the neck and jaw, causing problems in speaking, chewing and swallowing. MG can also cause weakness in skeletal muscles leading to problems in limb function. In the most severe cases, respiratory function can be weakened to the point where it becomes life-threatening. These respiratory crises occur at least once in the lives of approximately 15% to 20% of MG patients. The U.S. prevalence of MG is estimated at approximately 20 cases per 100,000 (source: Philips et al, Ann NY Acad Sci. 2003).
Patients with confirmed AchR antibodies account for approximately 85% of the total gMG population (Behin et al. New Pathways and Therapeutics Targets in Autoimmune Myasthenia Gravis. J Neuromusc Dis 5. 2018. 265-277).
In May 2020, we announced positive topline results from the pivotal ADAPT clinical trial of efgartigimod for the treatment of gMG. The topline results from the ADAPT clinical trial showed that efgartigimod was well-tolerated, demonstrated clinically meaningful improvements in strength and quality of life measures, and provided the option of an individualized dosing schedule for gMG patients. The full Phase 3 ADAPT results were published in The Lancet Neurology in July 2021. The data from the ADAPT clinical trial and the subsequent open-label extension (OLE) (ADAPT+) formed the basis for the regulatory approvals of VYVGART in the U.S., Japan, the EU, Mainland China, Israel, the UK and Canada.
On March 22, 2022, we announced positive topline results from the Phase 3 ADAPT-SC s clinical trial, a registrational non-inferiority bridging clinical trial of SC efgartigimod for the treatment of gMG. SC efgartigimod achieved the primary endpoint of total IgG reduction from baseline at day 29, demonstrating statistical noninferiority to VYVGART IV formulation in gMG patients. Based on these results, we received regulatory approval in the U.S. in June 2023, in the EU in September 2023, in Japan in January 2024 and in the UK in February 2024.
Other clinical trials
We are currently evaluating alternative dosing regimens of IV efgartigimod in adult gMG patients in the ADAPT NXT clinical trial. In addition, a clinical trial of IV efgartigimod in pediatric gMG patients is ongoing. In 2022, a Phase 1 clinical trial evaluating the effect of efgartigimod or placebo on immune response to the polyvalent pneumococcal vaccine (PNEUMOVAX 23) was completed. In 2024, we plan to initiate registrational clinical trials to expand VYVGART label into broader MG populations, including in seronegative patients.
52
CIDP
Overview
CIDP is a chronic autoimmune disorder of peripheral nerves and nerve roots caused by an autoimmune-mediated destruction of the myelin sheath, or myelin producing cells, insulating the axon of the nerves and enabling speed of signal transduction. The cause of CIDP is unknown, but abnormalities in both cellular and humoral immunity have been shown. CIDP is a chronic and progressive disease: onset and progression occur over at least eight weeks in contrast with the more acute Guillain-Barré-syndrome. Demyelination and axonal damage in CIDP lead to loss of sensory and/or motor neuron function, which can lead to weakness, sensory loss, imbalance and/or pain. CIDP affects approximately 24,000 patients in the U.S.
Most CIDP patients require treatment, the majority currently with IVIg. Glucocorticoids and plasma exchange are used to a lesser extent as they are either limited by side effects upon chronic use, in the case of glucocorticoids, or invasiveness of the procedure and access, which is restricted to specialized centers in case of plasma exchange. Alternative immunosuppressant agents are typically reserved for patients ineligible for or refractory to IVIg, glucocorticoids or plasma exchange.
In July 2023, we announced positive topline results from the ADHERE clinical trial evaluating VYVGART SC (efgartigimod alfa and hyaluronidase-qvfc) in adults with CIDP. The clinical trial met its primary endpoint (p=0.000039), demonstrating a significantly lower risk of relapse with VYVGART SC compared to placebo (HR: 0.39 95% CI: 0.25; 0.61). 67% of patients in open-label Stage A demonstrated evidence of clinical improvement (ECI), indicating that IgG autoantibodies play a significant role in the underlying biology of CIDP.
VYVGART SC was well-tolerated with a safety profile that is consistent with prior clinical trials and the known profile of VYVGART. The most frequent treatment-related adverse event was ISRs, which occurred in a lower percentage of patients than previous VYVGART SC trials (20% in Stage A; 10% in Stage B). All ISRs were mild to moderate and resolved over time. 99% (226/249) of eligible patients continued to the ADHERE-Plus OLE clinical trial. Detailed data from ADHERE is expected to be presented at an upcoming medical meeting.
In December 2023, we submitted an sBLA to the FDA for SC efgartigimod for CIDP with a priority review voucher. The FDA accepted the sBLA for priority review, with a PDUFA target date of June 21, 2024.
Primary ITP
Overview
Primary ITP is an acquired autoimmune bleeding disorder, characterized by a low platelet count (<100×109/L) in the absence of other causes associated with thrombocytopenia. In most patients, IgG autoantibodies directed against platelet receptors can be detected. They accelerate platelet clearance and destruction, inhibit platelet production, and impair platelet function, resulting in increased risk of bleeding and impaired quality of life. Primary ITP is differentiated from secondary ITP, which is associated with other illnesses, such as infections or autoimmune diseases, or which occurs after transfusion or taking other drugs, such as cancer drugs. Platelet deficiency, or thrombocytopenia, can cause bleeding in tissues, bruising and slow blood clotting after injury. Patients may suffer from depression and fatigue as well as side effects of existing therapies, impairing their quality of life. Current therapeutic approaches include non-specific immunosuppression (e.g., steroids and rituximab), inhibition of platelet clearance (e.g., splenectomy, IVIg, anti-D globulin, and spleen tyrosine kinase inhibitor fostamatinib13) or stimulation of platelet production (e.g., thrombopoietin receptor agonist TPO-RA). Splenectomy remains the only treatment that provides sustained remission off therapy for one year or longer for a high proportion of patients. ITP affects approximately 72,000 patients in the U.S. (sources: Current Medical Research and Opinion, 25:12, 2961-2969; Am J Hematol. 2012 Sep; 87(9): 848–852; Pediatr Blood Cancer. 2012 Feb; 58(2): 216–220).
53
Phase 3 ADVANCE Clinical Trials
In 2019, the first of two registrational clinical trials, the ADVANCE clinical trial, was initiated to evaluate IV efgartigimod (VYVGART) for the treatment of primary ITP. The second registrational ADVANCE-SC clinical trial of SC efgartigimod for the treatment of primary ITP was initiated in 2020.
In May 2022, we announced positive Phase 3 data from the ADVANCE clinical trial. Primary endpoint was met, demonstrating that a significantly higher proportion of patients with chronic ITP receiving VYVGART (17/78; 21.8%) compared to placebo (2/40; 5%) achieved a sustained platelet response (p=0.0316), defined as having platelet counts greater than or equal to 50x109/L on at least four of the last six scheduled visits between weeks 19 and 24 of treatment. There was also a statistically significant separation from placebo in key platelet-derived secondary endpoints. Additional secondary endpoint data from the ADVANCE clinical trial are consistent with primary and secondary platelet-derived endpoints and provide additional context on metrics that often drive treatment decisions, including on International Working Group (IWG) responder status.
VYVGART was well-tolerated in this 24-week clinical trial and the observed safety and tolerability profile was consistent with previous clinical trials. Results from ADVANCE-IV clinical trial were published in The Lancet in September 2023. We filed for approval of VYVGART for ITP in Japan and an approval decision is expected in the first quarter of 2024.
In November 2023, results of the second registrational clinical trial as part of the ongoing ITP development program for VYVGART in adult patients with chronic and persistent ITP were announced. Patients were heavily pre-treated and 75% of patients had received three or more prior ITP therapies. The clinical trial did not meet the primary endpoint of a sustained platelet count response in chronic ITP patients. Secondary endpoints were also not met, including additional endpoints on IWG responder status and mean platelet count change from baseline.
VYVGART SC was well-tolerated in ADVANCE-SC; the observed safety and tolerability profile was consistent with ADVANCE-IV and the confirmed safety profile of VYVGART and VYVGART SC.
Pemphigus
Overview
PV is an autoimmune disorder associated with mucosal and skin blisters that lead to pain, difficulty swallowing and skin infection. This chronic, potentially life-threatening disease is triggered by IgG autoantibodies targeting desmoglein-1 and -3, which are present on the surface of keratinocytes and important for cell-to-cell adhesion in the epithelium. Autoantibodies targeting desmogleins result in loss of cell adhesion, the primary cause of blister formation in PV. Similar to MG and ITP, disease severity of pemphigus correlates to the amount of pathogenic IgGs targeting desmogleins. Currently, there are an estimated 19,000 pemphigus patients in the U.S., of which an estimated 13,100 patients are suffering from PV. Several disease activity measurements exist for the clinical evaluation of PV patients, including the pemphigus disease area index (PDAI), autoimmune bullous skin disorder intensity score, and the PV activity score (PVAS). The PDAI is reported to have the highest validity and is recommended for use in clinical trials of PV.
Phase 3 ADDRESS Clinical Trial
In 2020, the registrational ADDRESS clinical trial was initiated of SC efgartigimod for the treatment of PV and PF. This was a randomized, double-blinded, placebo-controlled clinical trial, where the objective was to assess efficacy, safety and tolerability in newly diagnosed or relapsing patients with moderate to severe pemphigus (total of 222 enrolled). Patients were randomized to receive either SC efgartigimod or placebo for 30 weeks. Patients started on concomitant steroids based on what we determined to be the optimized dosing regimen from the Phase 2 POC clinical trial. The primary endpoint assessed the proportion of patients who achieve sustained complete remission on a minimal steroid dose within 30 weeks. The ADDRESS clinical trial evaluated efficacy and safety, including the potential to drive fast onset of disease control and complete remission and the ability to taper corticosteroids.
54
Topline data from the Phase 3 ADDRESS clinical trial were announced in December 2023, in which the results show the proportion of PV patients achieving the primary endpoint of complete remission on CRmin was not significantly different between SC efgartigimod and placebo. We will not pursue additional development in pemphigus and we will prioritize clinical development of efgartigimod in its ongoing severe autoimmune indications.
BP
Overview
BP is the most common autoimmune blistering disease and is driven by autoantibodies affecting the skin. The disease typically affects elderly people and early key symptoms are itch and rash and patients develop fluid-filled blisters during disease progression. The prevalence of BP is 12 per 100,000 adults and the incidence increases with age. BP is associated with a high disease burden and can have a significant impact on the quality of life of patients. The mortality of BP in the U.S. is 2.4% or higher than the mortality in the general population of the same age. There are currently no approved therapies available for BP. First line treatment consists of topical or systemic corticosteroids, which result in substantial morbidity and increased mortality, conventional immunosuppressants as corticosteroid-sparing agents, rituximab and IVIg.
BP is a well characterized autoimmune disease in which the binding of autoantibodies to hemidesmosomal proteins, BP180 and BP230, initiates a cascade of inflammatory events resulting in blister formation. BP180 and BP230 are involved in the stable attachment of keratinocyte to the underlying matrix. The autoantibody actions include mechanical disruption of keratinocyte adhesion and cytokine release. Immune complex formation initiates complement activation leading to the recruitment mast cells, neutrophils, eosinophils and other immune cells and to the release of proteases and inflammatory mediators. All these effects, which start with the binding of the autoantibodies, induce the blistering observed in BP.
BALLAD Clinical Trial
We initiated the Phase 2/3 BALLAD registrational clinical trial evaluating SC efgartigimod in BP in 2022.
The clinical trial population are newly diagnosed and relapsing patients within one year from diagnosis. Patients are randomized 1-to-1 to receive efgartigimod or placebo for a total duration of 36 weeks. The primary endpoint is the proportion of participants in complete remission while off oral corticosteroids for at least eight weeks at week 36. Secondary endpoints relate to cumulative steroid doses, IGA BP score, time to achieving control of disease activity, change from baseline in average itch, and quality of life measures.
In light of ADDRESS results and the comparable biology between PV and BP, we decided to stop enrollment of BALLAD. We will integrate key learnings from ADDRESS and data from already enrolled patients in BALLAD and we plan to communicate on a revised development plan before end 2024.
Myositis
Overview
Myositis are a rare group of autoimmune diseases that can be muscle specific or affect multiple organs including the skin, joints, lung, gastrointestinal tract and heart. Myositis can be very severe and disabling and have a material impact on quality of life. Initially these Myositis were classified as either DM or polymyositis, but as the underlying pathophysiology of Myositis has become better understood, including through the identification of characteristic autoantibodies, new polymyositis subgroups have emerged. Two of these subtypes are IMNM and ASyS. Proximal muscle weakness is a unifying feature of each Myositis subset.
| ● | IMNM is characterized by skeletal muscle weakness due to muscle cell necrosis. The muscle weakness is typically symmetrical – on both sides of the body – and affects proximal muscles including hips, thighs, upper arms, shoulder and neck. The muscle weakness can be severe and lead to difficulty in completing daily tasks. |
55
| Characteristic autoantibodies of IMNM, include anti-signal recognition particle and anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase autoantibodies. |
| ● | ASyS is characterized by muscle inflammation, inflammatory arthritis, interstitial lung disease, thickening and cracking of the hands (“mechanic’s hands”) and Raynaud phenomenon. Autoantibodies associated with ASyS attack tRNA synthetase enzymes and include anti-Jo-1 and anti-PL1 and PL-12 most commonly. |
| ● | DM is characterized by muscle inflammation and degeneration and skin abnormalities, including heliotrope rash, Gottron papules, erythematous, calcinosis and edema. DM is associated with Myositis-specific autoantibodies, including anti-Mi-2, anti-MDA-5, anti-TIF-1γ and others. |
There are no current FDA-approved therapies for IMNM or ASyS. IVIg (Octagam 10%) was approved by the FDA for the treatment of DM in July 2021. Myositis patients are most often treated with high-dose steroids.
ALKIVIA Clinical Trial
We initiated the registrational ALKIVIA clinical trial of SC efgartigimod for the treatment of Myositis in 2022. The clinical trial plans to enroll approximately 240 patients in three Myositis subtypes, IMNM, ASyS and DM. The clinical trial will be conducted in two Phases, with an analysis of the Phase 2 portion of the clinical trial, including 30 patients of each subtype, followed by conduct of the Phase 3 portion of the clinical trial only if a signal is observed in the Phase 2 portion of the clinical trial.
The primary endpoint is the total improvement score (TIS) at the end of the treatment period. Key secondary endpoints include response rates at the end of treatment, time to response, and duration of response in TIS, as well as change from baseline in individual TIS components. Other secondary endpoints include quality of life and other functional scores.
An interim analysis of the first 30 patients in each subset is expected in the second half of 2024.
TED
TED is an autoimmune orbital disease associated with Graves’ disease and other autoimmune thyroid pathologies such as Hashimoto’s thyroiditis. TED is characterized by extraocular muscle enlargement, orbital adipose tissue expansion, and orbital inflammation, which can lead to proptosis, diplopia, or vision loss in severe cases. Persistent orbital symptoms often impair patient QoL long-term.
Substantial nonclinical and clinical evidence supports thyrotropin receptor (TSHR) autoantibodies as causative in the pathology of TED. Clinical evidence supports the removal of autoantibodies as a mechanism for the treatment of TED. By reducing immunoglobulin γ (IgGs), including TED-associated pathogenic IgG autoantibodies, efgartigimod is expected to ease disease manifestations. Additionally, IgG reduction could address the underlying hyperthyroidism. Side effects and tolerability issues with current therapies, including steroids and teprotumumab (only FDA-approved biologic), are treatment limiting for many patients based on comorbidities and a significant unmet need remains for safe and convenient therapies.
A registrational clinical trial evaluating efgartigimod in TED is expected to start in 2024.
56
SjD
Overview
SjD is a chronic, progressive autoimmune disease, characterized by lymphocytic infiltration and progressive destruction of exocrine glands. B-cells play a pivotal role in the development of the disease and this results amongst others in production of IgG autoantibodies, especially those which target SSA/Ro, SSB/La ribonuclear complexes. In addition to symptoms of dry eyes, dry mouth, chronic pain and fatigue, a substantial subset of patients suffer from extraglandular systemic disease. There are no FDA-approved treatments currently registered for the treatment of SjD.
Phase 2 RHO Clinical Trial (in partnership with IQVIA)
In 2023, we initiated a Phase 2 POC clinical trial evaluating IV efgartigimod for the treatment of SjD. The RHO clinical trial is a randomized, placebo-controlled, double-blind clinical trial evaluating IV efgartigimod. The clinical trial enrolled approximately 30 patients with at least moderate systemic disease (ESSDAI ≥5). Patients have to be on stable background treatment and positive for anti-SSA/Ro. At the end of the 24-week treatment period, participants who complete the clinical trial may roll over into an OLE. The primary endpoint is the proportion of responders to the Composite of Relevant Endpoints for SjD (CRESS; response on ≥3 out of 5 items) at week 24. Key secondary endpoints include change from baseline in the clinESSDAI (Clinical ESSDAI), ESSDAI (Eular Sjögrens Syndrome Disease Activity Index), and ESSPRI (Eular Sjögrens Patient Reported Index) scores.
RHO clinical trial results are expected in first half of 2024.
PC-POTS
Overview
PC-POTS has been emerging following SARS-Cov-2 infection in previously healthy patients. PC-POTS is a disorder of the autonomic nervous system that is characterized by a rise in heart rate when moving to a standing position and additional symptoms of shortness of breath, headache, fatigue, poor concentration, weakness and anxiety. The large majority of patients are women between 15 and 50 years of age. There is a strong association of PC-POTS to activating autoantibodies to autonomic G-protein coupled receptors, including the β1 and β2-adrenergic receptors and M2 and M3 muscarinic receptors. There are no current FDA-approved therapies and symptomatic treatments focus on blood volume, kidney sodium levels, heart rate reduction and vessel constriction.
Phase 2 POC ALPHA Clinical Trial (in partnership with IQVIA)
In 2022, we initiated the placebo-controlled Phase 2 POC ALPHA clinical trial of weekly IV efgartigimod for the treatment of de novo POTS triggered by COVID-19. The co-primary endpoints are COMPASS-31 and the Malmö POTS Symptom score at the end of the 24-week treatment period. Key secondary endpoints include change from baseline in PROMIS fatigue & cognitive function, as well as the Patient Global Impression of change and severity. Other secondary endpoints include quantitative autonomic testing and other functional scores.
Phase 2 POC ALPHA clinical trial results are expected in the first half of 2024.
LN
Overview
LN is an inflammatory autoimmune disease of the kidney and one of the most severe and common organ manifestations of the autoimmune disease systemic lupus erythematosus (SLE). In patients with SLE, approximately 25% to 50% have signs or symptoms of kidney disease at SLE onset. Approximately 40% to 60% of patients with SLE will develop renal involvement during the course of disease, with substantial morbidity or mortality. Pathogenic autoantibodies and complement deposits are critically involved in SLE pathogenesis and particularly LN, where renal
57
deposition of immune complexes is a hallmark of the disease. Autoantibodies associated with LN include anti-dsDNA, anti-C1q, anti-cardiolipin, anti-Smith and anti-nuclear antibodies. 10-30% of LN patients progress to end-stage renal disease. Oral corticosteroids and broad immunosuppressants are current standards of care but are not uniformly effective. Belimumab (Benlysta) and voclosporin (Lupkynis) are approved by the FDA for the treatment of LN.
Phase 2 POC Clinical Trial (in partnership with Zai Lab)
In 2023, we initiated a POC clinical trial to evaluate the efficacy and safety of IV efgartigimod in Chinese patients with active LN. The clinical trial plans to enroll approximately 60 patients with LN class III or IV (with or without class V).
The primary endpoint is the change in urine protein creatinine ratio (UPCR) from baseline to end of the treatment period. Key secondary endpoints include proportion of patients achieving complete and partial renal response (CRR and PRR, respectively) at the end of treatment period and time to CRR and PRR. Other secondary endpoints include additional efficacy measurements, PK, PD, immunogenicity, biomarkers, safety, and quality of life assessments.
MN
Overview
MN is an autoimmune, glomerular disease and one of the most common causes of nephrotic syndrome in adults. MN is characterized by thickening of the glomerular basement membrane caused by immune complex deposition. As many as 75% of MN patients have IgG autoantibodies against PLA2R. Data are highly suggestive of a causal relationship between anti-PLA2R Ab and MN pathogenesis. Other target antigens identified to date include thrombospondin type 1 domain-containing 7A (THSd7A), neural epidermal growth factor-like-1 (NELL-1), and semaphorin-3B (Sema3B). 20-30% of MN patients progress to end-stage renal disease. All MN patients receive optimal supportive care and patients at high risk for disease progression are additionally treated with broad immunosuppressants. There are no current approved therapies for MN.
Phase 2 POC Clinical Trial (in partnership with Zai Lab)
In 2023, we initiated a POC clinical trial to evaluate the efficacy and safety of IV efgartigimod in Chinese patients with primary MN (pMN). The clinical trial plans to enroll a maximum of 72 patients with pMN. The clinical trial will include two phases: a double-blinded period (DB) for the main clinical trial followed by an optional OLE period. The primary endpoint is the change in UPCR from baseline to end of the treatment period in the anti-PLA2R Ab seropositive population. Key secondary endpoints include change in UPCR from baseline to end of the treatment period in the overall population, proportion of participants achieving complete remission and partial remission at the end of the treatment period in the overall population and in the anti-PLA2R Ab seropositive population and time to complete remission and partial remission in the overall population and in the anti-PLA2R Ab seropositive population. Other secondary endpoints include additional efficacy measurements, PK, PD, immunogenicity, biomarkers, safety, and quality of life assessments.
Other Efgartigimod Indications
AMR
AMR is an autoimmune disease that affects transplanted organs and can contribute to allograft loss. AMR in kidney allografts is driven by donor specific antibodies (DSA), which often target human leukocyte antigens expressed by endothelial allograft cells. Through different mechanisms DSA can induce microvascular inflammation, a histopathological hallmark of AMR. Microvascular inflammation leads to loss in organ function which, if continued, can result in allograft loss. The unmet need for an efficacious treatment is very high, as evidenced by AMR being the leading cause of kidney transplant failure. There are currently no approved therapies for treating AMR.
58
AAV – in partnership with IQVIA
ANCA-associated vasculitis (AAV) is an autoimmune disease that is characterized by the inflammation and damaging of small blood vessels in the body. There a three different AAV subtypes; granulomatosis with polyangiitis, microscopic polyangiitis and eosinophilic granulomatosis with polyangiitis (EGPA). Polyangiitis or microscopic polyangiitis are often associated with the presence of PR3- or MPO-autoantibodies, respectively. These autoantibodies play a pivotal role in the disease, in which their binding to neutrophils initiates a series of inflammatory processes. Symptoms like fatigue, muscle pain, fever, abdominal pain, and blood in the urine are often observed, but many patients develop organ- or life-threatening disease where kidneys, lungs or the cardiovascular system are severely damaged. Multiple treatments are FDA-approved, with rituximab, on top of glucocorticoids, considered as main treatment for both induction and maintenance in AAV.
Partnerships for efgartigimod indications
Zai Lab Limited
Pursuant to the Zai Lab Agreement, Zai Lab obtained the exclusive right to develop and commercialize efgartigimod in Greater China. Zai Lab will also contribute patients to our global Phase 3 clinical trials of efgartigimod. Our Zai Lab strategic collaboration allows us to accelerate development of efgartigimod into new autoimmune indications with Zai Lab taking operational leadership of the Phase 2 POC clinical trials.
In 2022 Zai Lab initiated the Phase 2 POC clinical trials in MN and LN, which both fall within the emerging nephrology indications. This was done after having completed a Phase 1 PK/PD clinical trial to support the approval of efgartigimod for gMG in Mainland China, as well as obtaining regulatory approvals to enroll Chinese patient into our global Phase 3 clinical trials. In our collaboration with Zai Lab, we continue to evaluate additional POC clinical trials to initiate in the Greater China under the Zai Lab Agreement to accelerate the development of efgartigimod globally.
IQVIA
On December 2, 2021 we entered into a strategic asset development agreement (Asset Development Agreement) with IQVIA. Pursuant to the Asset Development Agreement, IQVIA shall perform asset and indication development services for efgartigimod through an advanced outsourcing model. Such services include, but are not limited to, overall product indication development strategy, design of clinical trial protocol, set-up, execution and oversight of clinical development plans for an indication for efgartigimod selected by us.
To enable and encourage fast and innovative delivery of the services by IQVIA, the Asset Development Agreement contains an innovative earn-back and bonus plan based upon the performance of IQVIA.
59
SjD, PC-POTS and AAV are the indications we identified to be further developed under the Asset Development Agreement.
Clinical Trial |
| Stage |
| Indication |
| Patients |
| Primary Endpoint |
| Status |
ADAPT | Registrational | gMG | The proportion of responders based on the Myasthenia Gravis Activities of Daily Living (MG-ADL) score | Marketed | ||||||
ADAPT-SC | Registrational | gMG | The proportion of responders based on the Myasthenia Gravis Activities of Daily Living (MG-ADL) score | Marketed | ||||||
ADHERE | Registrational | CIDP | 322 | The hazard ratio for the time to first adjusted INCAT deterioration | sBLA accepted by FDA | |||||
ADVANCE-IV | Registrational | ITP | The proportion of patients that achieved sustained platelet response | positive Topline Data | ||||||
ADVANCE-SC | Registrational | ITP | 131 | The proportion of patients that achieved sustained platelet response | Did not meet primary endpoint, analysis ongoing | |||||
ADDRESS | Registrational | PV and PF | 222 | The proportion of patients who achieve complete remission on a minimal steroid dose at 30 weeks | Did not meet primary enpoint, evaluation of efgart in PV and PF stopped. | |||||
BALLAD | Registrational | BP | The proportion of participants in complete remission while off oral corticosteroids for at least eight weks at week 36 | Analysis ongoing for path forward | ||||||
ALKIVIA | Registrational | Myositis | Appr.240 | The TIS at the end of the treatment period | Ongoing Interium analysis expected second half 2024 | |||||
RHO | PoC | Primary | Appr.30 | The proportion of responders to the Composite of Relevant Endpoints for Sjogren’s Syndrome (CRESS; response on 23 out of 5 items) at week 24. | Ongoing study results expected first half 2024 | |||||
ALPHA | PoC | PC-POST | 53 | The co-primary endpoints are 1) COMPASS-31 and 2) the Malmo POTS Symptom score at the end of the 24-week treatment period | Ongoing study results expected first half 2024 | |||||
In partnership with Zai Lab | PoC | LN | Appr.60 | The change in UPCR from baseline to end of the treatment period | Ongoing study results expected first half 2025 | |||||
In partnership with Zai Lab | PoC | MN | Appr.70 | The change in UPCR from baseline to end of the treatment period in the anti-PLAZR Ab seropositive population | Ongoing study results expected first half 2025 | |||||
Clinical trial to start in 2024 | Registrational | TED | ||||||||
Other Clinical Trails | PoC | AMR | IND submission planned for 2Q 2024 |
* | AAV program under review; phase 2 study paused at the time of publication of this Annual Report |
Empasiprubart (ARGX-117) Development
Mechanism of Action
Empasiprubart is a highly differentiated therapeutic mAb targeting C2 equipped with our proprietary NHANCE™ mutations. By addressing a novel target at the intersection of the complement and lectin pathways of the complement cascade, we believe empasiprubart represents a broad pipeline opportunity across several severe autoimmune indications. Activation of the classical and lectin pathway of complement may contribute to tissue damage and organ dysfunction in a number of autoimmune inflammatory diseases and ischemia-reperfusion conditions. Targeting C2 also leaves the alternative pathway of the complement system intact, which is an important component of the innate defense system.
Empasiprubart exhibits both pH- and calcium dependent binding. These unique characteristics enable empasiprubart to capture free C2 in circulation and release it in the endosome to be sorted for degradation in the lysosome. Empasiprubart is equipped with NHANCE™ mutations increasing its affinity for FcRn and allowing it to recycle back into circulation to capture more C2.
We obtained the rights to empasiprubart as part of our IIP. argenx and Broteio launched a collaboration in 2017 to conduct research, with support from the University of Utrecht, to demonstrate preclinical POC of the mechanism of
60
empasiprubart. Based on promising preclinical data generated under this collaboration agreement, we exercised the exclusive option to license the program and assumed responsibility for further development and commercialization.
In addition to an IV formulation, we have exclusive access to Halozyme’s ENHANZE® SC drug delivery technology for the C2 target.
|
|
|
Figure 3: LEFT: Empasiprubart exhibits both pH- and calcium dependent target binding. RIGHT: Empasiprubart is equipped with NHANCE™ mutations increasing its affinity for FcRn at acidic pH and allowing it to recycle back into circulation.
Empasiprubart Indications
MMN
Overview
MMN is a debilitating neuromuscular autoimmune disorder that is characterized by slowly progressive muscle weakness due to motor neuron degeneration. It mainly affects hands and forearms, mainly in males, and the median age of diagnosis is around 40 years. Diagnosis takes about a year and a half and is often misdiagnosed as ALS. There are estimated to be around 13,000 patients with MMN in the U.S. and this number is increasing.
Specific pathophysiologic characteristics of MMN include the presence of IgM autoantibodies against the ganglioside GM1 and conduction block, i.e., impaired propagation of action potentials along the axon. GM1 is widely expressed in the nervous system by neurons, particularly around the nodes of Ranvier, and Schwann cells.
IVIg is the only approved treatment for MMN and needs to be dosed frequently to address the disease’s progressive nature.
Phase 1 Data
We conducted a Phase 1 healthy volunteer clinical trial of IV and SC empasiprubart. This first-in-human clinical trial was a double-blind placebo-controlled clinical trial designed to assess the safety, tolerability, PK and PD of a broad dose range of empasiprubart in 102 healthy subjects. In the single ascending dose part, we evaluated 70 subjects and tested up to 80 mg/kg administered IV and up to 60mg/kg administered SC. In the MAD part of the clinical trial, we evaluated 32 subjects to understand the safety and tolerability of repeated administrations and in particular to generate a data-set to optimally inform a PK/PD model.
61
Both single and multiple administrations of empasiprubart or placebo had a favorable safety and tolerability profile supporting the investigation of clinical trial drug in patient clinical trials.
We observed a dose-dependent reduction of free C2 levels. After one dose of 30mg/kg empasiprubart, free C2 levels were reduced by 95% for more than 100 days. In the MAD part of the clinical trial, we could reach full complement blockade with more than 99% reduction of free C2 levels.
Following analysis of Phase 1 data, and the observed favorable safety and tolerability profile and consistent PK/PD profile, we launched a Phase 2 POC clinical trial in MMN in 2021.
Interim Results Phase 2 POC ARDA clinical trial
In June 2023, argenx announced its plan to advance to a second cohort with the Phase 2 ARDA clinical trial of empasiprubart in MMN. This decision followed a planned interim analysis of the first dose cohort by an Independent Data Monitoring Committee (IDMC) meeting.
The IDMC reviewed interim safety data from all patients (n=22) enrolled in the first cohort of the ARDA clinical trial, including nine patients who completed the full 16-week treatment period. The IDMC confirmed a favorable safety and tolerability profile of empasiprubart consistent with results from the Phase 1 clinical trial and recommended advancing to the second cohort. Combined with the early efficacy signals observed, supporting POC of empasiprubart in MMN, argenx started the second cohort of the ARDA clinical trial.
In January 2024, argenx announced positive data from the first cohort (n=22) of the Phase 2 POC ARDA clinical trial establishing POC in MMN. Empasiprubart demonstrated a 91% reduction in the need for IVIg rescue compared to placebo [HR: 0.09 95% CI (0.02; 0.044)].
In total, the ARDA clinical trial is expected to enroll 48 patients across two cohorts. The clinical trial’s objective, in addition to assessing safety and efficacy of empasiprubart, is to populate a PK/PD model to inform the Phase 3 clinical trial dose selection.
Phase 2 ARDA Clinical Trial Design
The Phase 2 POC ARDA clinical trial is a randomized, double-blinded, placebo-controlled multicenter clinical trial to evaluate the safety and tolerability, efficacy, PK, PD, and immunogenicity of two dose regimens of empasiprubart in adults with MMN. The clinical trial consists of an IVIg dependency and monitoring period and two 16-week treatment cohorts of 24 MMN patients receiving empasiprubart or placebo in a 2x1 randomization. The dosing for Cohort 2 was established after a planned interim analysis of the first nine patients to complete the 16-week treatment period from Cohort 1. The primary endpoint is safety and tolerability. Additional endpoints include time to IVIg retreatment, biomarker analyses of C2 levels, and changes in measurements on key functional scores (modified medical research council (mMRC)-10 sum score, grip strength, MMN-RODS) as well as several patient-reported quality of life outcome measures (fatigue severity score (FSS), chronic acquired polyneuropathy patient-reported index (CAP-PRI), and values of the patient global impression change (PGIC) scale).
DGF
Overview
DGF, a complication after kidney transplantation, is defined as the need for dialysis in the first week after transplant. DGF occurs in up to 40% of patients receiving a deceased donor graft, and is associated with worse long-term transplant outcomes. DGF is often the clinical representation of ischemia reperfusion injury, in which the classical and lectin complement pathways play an important role, as shown by compelling evidence from both (in-house) in vitro and in vivo preclinical, and clinical trials. There are currently no approved therapies to reduce DGF risk. Furthermore, there is a well-established process to measure kidney function and DGF, and to establish POC and achieve registration. On
62
this basis, combined with the significant unmet medical need, we have chosen DGF after kidney transplantation as second indication for empasiprubart.
Phase 2 POC VARVARA Clinical Trial
The Phase 2 POC VARVARA clinical trial was initiated in 2023 and is a randomized, placebo-controlled, double-blinded clinical trial to evaluate the efficacy, safety and tolerability of empasiprubart in improving allograft function in recipients at risk for DGF. The clinical trial will include approximately 102 recipients of an at-risk deceased donor kidney. After a short screening period of < 24 hours, patients are randomly assigned in a 1:1 ratio to receive two doses of empasiprubart IV or placebo, of which one dose is administered during transplantation and one a week later. Participants receive standardized background induction and maintenance immunosuppression. They are evaluated for 52 weeks, with one additional safety follow-up visit in week 64. The primary endpoint is the estimated glomerular filtration rate (eGFR) at six months. Key secondary endpoints include DGF risk, safety, and PK, PD and immunogenicity.
DM
Overview
DM is an idiopathic inflammatory myopathy characterized by muscle inflammation that causes progressive muscle weakness and is associated with various characteristic skin manifestations. Histopathological findings suggest that DM is a complement-mediated disease. The most common therapy for DM is the administration of steroids. IVIg is the only approved treatment for DM.
Phase 2 POC EMPACIFIC Clinical Trial
The EMPACIFIC clinical trial is a Phase 2 POC, randomized, double-blinded, placebo-controlled, multicenter clinical trial to evaluate the safety, tolerability, and efficacy of multiple dose regimens of IV empasiprubart in adults with dermatomyositis. A total of 56 adult participants with a clinical diagnosis of DM and active muscle disease will be randomized (1:1:1:1) to one of four treatment arms (three empasiprubart dose regimens and one placebo arm). Participants will receive loading doses on Days 1 and 8, followed by maintenance doses every four weeks until the end of the 52-week treatment phase. The primary objective is to evaluate safety and tolerability. The secondary objective is to evaluate clinical efficacy, using the mean TIS at weeks 13, 25, and 52 as endpoint.
ARGX-119 Development
ARGX-119 is a humanized agonist mAb that specifically targets and activates MuSK to promote maturation and stabilization of the neuromuscular junction (NMJ). We plan to develop ARGX-119 in a range of neuromuscular diseases including CMS, a rare hereditary subtype of MG, and ALS, both severe neuromuscular indications.
NMJs are specialized synapses formed between motor neurons and muscle cells, which are essential for the ability to move and breathe. At the NMJ, motor neurons release acetylcholine, which binds to AChRs on the muscle to initiate muscle contraction. Deficits in the NMJ can cause neuromuscular disorders, which can range in severity from mild to life-threatening skeletal muscle weakness. MuSK is an essential component for the formation and function of NMJs.
ARGX-119 is the first and highly specific agonist mAb targeting human MuSK being developed for patients with neuromuscular disease, such as CMS and ALS. This mAb is derived from llamas and discovered using the argenx SIMPLE ANTIBODY™ platform technology. We developed ARGX-119 through our IIP program in collaboration with the world leading key opinion leaders on MuSK and the neuromuscular junction, including Professor Steve Burden from NYU and Professor Verschuuren from LUMC. In collaboration with Professor Burden, it was shown that ARGX-119 holds promising preclinical POC data in Dok7 congenital myasthenic syndrome, observed in a mouse model bearing the most common patient mutation and in ALS using ALS patient derived NMJ on-a-chip models. Based on these data, clinical development for ARGX-119 was initiated as activation of MuSK by ARGX-119 may stabilize, mature, and
63
improve the function of the NMJ in patients with CMS or ALS, significantly reducing weakness and fatigability and improving quality of life.
A Phase 1 dose-escalation clinical trial in healthy volunteers started in 2023 and is ongoing. A Phase 1b and 2a clinical trial in CMS and ALS respectively are planned to start in 2024 to assess early signal detection in patients.
ARGX-109, ARGX-220, ARGX-121 and ARGX-213 Development
We continue to invest in our discovery engine, the IIP, to drive long-term sustainable pipeline growth. Through the IIP, four new pipeline candidates were nominated in 2023, including: ARGX-213 targeting FcRn and furthering argenx’ leadership in this new class of medicine; ARGX-121 and ARGX-220, which are first-in-class targets broadening argenx’ focus across the immune system; and ARGX-109, targeting IL-6, which plays an important role in inflammation. Preclinical work is ongoing for each candidate and we expect to file four IND applications by the end of 2025.
Antibody Engineering and Other Technology Capabilities
Our Proprietary SIMPLE ANTIBODY™ Platform
Our proprietary SIMPLE ANTIBODY™ platform technology sources V-regions from conventional antibodies existing in the immune system of outbred llamas. Outbred llamas are those that have been bred from genetically diverse parents, and each has a different genetic background. The llama produces highly diverse panels of antibodies with a high human homology in their V-regions when immunized with human disease targets. We then combine these llama V-regions with Fc regions of fully human antibodies, resulting in antibodies that we then produce in industry-validated production cell lines. The resulting antibodies are diverse and, due to their similarity to human antibodies, we believe they are well suited to human therapeutic use. With this breadth of antibodies, we are able to test many different epitopes. Being able to test many different epitopes with our antibodies enables us to search for an optimized combination of safety, potency and species cross-reactivity with the potential for maximum therapeutic effect on disease. These antibodies are often cross-reactive with the rodent version of chosen disease targets. This rodent cross-reactivity enables more efficient preclinical development of our product candidates because most animal efficacy models are rodent-based. By contrast, most other antibody discovery platforms start with antibodies generated in inbred mice or synthetic antibody libraries, approaches that we believe are limited by insufficient antibody repertoires and limited diversity, respectively. Our SIMPLE ANTIBODY™ platform technology allows us to access and explore a broad target universe, including novel and complex targets, while minimizing the long timelines associated with generating antibody candidates using traditional methods.
Our Antibody Engineering Technologies
Through licensing we have obtained access to a broad range of antibody engineering technologies. NHANCE™, ABDEG™, POTELLIGENT® and the DHS mutations focus on engineering the Fc region of antibodies, while SMART-Ig® and ACT-Ig® technologies allow to make sweeping antibodies.
Fc engineering can augment antibodies interactions with components of the immune system, thereby potentially expanding the therapeutic index of our product candidates by modifying their half-life, tissue penetration, rate of disease target clearance and potency. For example, our NHANCE™ and ABDEG™ engineering technologies enable us to modulate the interaction of the Fc region with FcRn, which is responsible for regulating half-life, tissue distribution and PD properties of IgG antibodies. Similarly, the POTELLIGENT engineering technology modulates the interaction of the antibody Fc region with receptors located on specialized immune cells known as natural killer (NK) cells. These NK cells can destroy the target cell, resulting in enhanced antibody-dependent cell-mediated cytotoxicity (ADCC).
64
NHANCE™ and ABDEG™: Modulation of Fc Interaction with FcRn.
An illustration of the FcRn-mediated antibody recycling mechanism is shown in Figure 4. [1] Serum proteins, including IgG antibodies, are routinely removed from the circulation by cell uptake. [2] Antibodies can bind to FcRn, which serves as a dedicated recycling receptor in the endosomes, which have an acidic environment, and then [3A] return to the circulation by binding with their Fc region to FcRn. [3B] Unbound antibodies end up in the lysosomes and are degraded by enzymes. Because this Fc/FcRn interaction is highly pH-dependent, antibodies tightly bind to FcRn at acidic pH (pH 6.0) in the endosomes but release again at neutral pH (pH 7. 4) in the circulation.
Figure 4: The FcRn-mediated recycling mechanism
65
NHANCE™
NHANCE™ refers to two mutations that we introduce into the Fc region of an IgG antibody. NHANCE™ is designed to extend antibody serum half-life and increase tissue penetration. In certain cases, it is advantageous to engineer antibodies that remain in the circulation longer, allowing them to potentially exert a greater therapeutic effect or be dosed less frequently. As shown in Figure 5, [1] NHANCE™ antibodies bind to FcRn with higher affinity, specifically under acidic pH conditions. [2] Due to these tighter bonds, NHANCE™ FcRn-mediated antibody recycling is strongly favored over lysosomal degradation, although some degradation does occur. [3] NHANCE™ allows a greater proportion of antibodies to return to the circulation potentially resulting in increased bioavailability and reduced dosing frequency. ARGX-109, empasiprubart and a number of our discovery-stage programs utilize NHANCE™.
Figure 5: NHANCE™ mutations favor the FcRn-mediated recycling of IgG antibodies.
ABDEG™
ABDEG™ refers to five mutations that we introduce in the Fc region that increase its affinity for FcRn at both neutral and acidic pH. In contrast to NHANCE™, ABDEG™-modified Fc regions remain bound to FcRn if the pH changes, occupying FcRn with such high affinity that they deprive endogenous IgG antibodies of their recycling mechanism, leading to enhanced clearance of such antibodies by the lysosomes. Some diseases mediated by IgG antibodies are directed against self-antigens. These self-directed antibodies are referred to as autoantibodies. We use our ABDEG™ technology to reduce the level of these pathogenic autoantibodies in the circulation by increasing the rate at which they are cleared by the lysosomes. ABDEG™ is a component in a number of our products and product candidates, including efgartigimod.
66
As shown in Figure 6, our ABDEG™ technology can also be used with our pH-dependent SIMPLE ANTIBODY™ generated antibodies in a mechanism referred to as sweeping. Certain antibodies generated through the SIMPLE ANTIBODY™ platform bind to their target in a pH-dependent manner. These antibodies [1] bind tightly to a target at neutral pH while in circulation, and [2] release the target at acidic pH in the endosome. [3] The unbound target is degraded in the lysosome. [4] However, when equipped with our ABDEG™ technology, the therapeutic antibodies remain tightly bound to FcRn at all pH levels and are not degraded themselves. Instead, they are returned to the circulation where they can bind new targets. We believe this is especially useful in situations where high levels of the target are circulating or where the target needs to be cleared very quickly from the system.
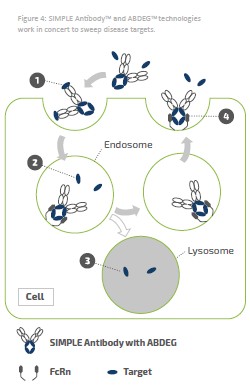
Figure 6: SIMPLE ANTIBODY™ and ABDEG™ platform technologies work in concert to sweep diseases targets.
POTELLIGENT®
POTELLIGENT® modulates the interaction of the Fc region with the Fc gamma receptor IIIa located on specialized immune cells, known as NK cells. These NK cells can destroy the target cell, resulting in enhanced ADCC. POTELLIGENT® changes the Fc structure by excluding a particular sugar unit such that it enables a tighter fit with the Fc gamma receptor IIIa. The strength of this interaction is a key factor in determining the killing potential of NK cells. An independent publication reported that the exclusion of this sugar unit of the Fc region increases the ADCC-mediated cell-killing potential of antibodies by 10- to 1000-fold. Cusatuzumab and ARGX-111 utilize POTELLIGENT® (source: Expert Opin Biol Ther 2006; 6:1161-1173; http://www.tandfonline.com/doi/full/10.1517/14712598.6.11.1161%20).
SMART-Ig®, ACT-Ig® and DHS
In 2020, we entered into a research license and option agreement with Chugai under which we may access Chugai’s SMART-Ig® and ACT-Ig® . In 2020, we also entered into a non-exclusive research agreement with the Clayton Foundation under which we may access the Clayton Foundation’s proprietary DHS mutations to extend the serum half-life of therapeutic antibodies.
Genmab collaboration
In 2023, we entered into a collaboration with Genmab to jointly discover, develop and commercialize novel therapeutic antibodies with applications in immunology, as well as in oncology therapeutic areas. The multiyear collaboration is expected to leverage the antibody engineering expertise and knowledge of disease biology of both companies to accelerate the identification and development of novel antibody therapeutic candidates with a goal to address unmet patient needs in immunology and cancer. Under the terms of the collaboration, argenx and Genmab each
67
have access to the suites of proprietary antibody technologies of both companies to advance the identification of lead antibody candidates against differentiated disease targets.
SC drug delivery technologies
We have exclusive access to Halozyme’s ENHANZE® SC drug delivery technology for the FcRn and C2 targets and four additional targets. ENHANZE® has the potential to shorten drug administration time, reduce healthcare practitioner time, and offer additional flexibility and convenience for patients.
In addition, in April 2021, we entered into the Elektrofi Agreement with Elektrofi to explore new SC formulations utilizing Elektrofi’s high concentration technology for efgartigimod, and up to one additional target.
For more information on our collaborations, please refer to Item 4.B. “Business Overview—Collaboration Agreements” and to Item 4.B. “Business Overview—License Agreements.”
Partnered Programs
For a description of collaboration and license agreements that we have entered into to further leverage our IIP, please refer to Item 4.B. “Business Overview—Collaboration Agreements” and to Item 4.B. “Business Overview—License Agreements.”
Strategy and objectives
| ● | Our goal is to deliver immunology innovations that are both first-in-class and best-in-class to transform the lives of people with serious autoimmune diseases. We do this by combining our leading antibody engineering capabilities with disease biology insights from our collaborators. Within this business model we plan to: |
| ● | Continue to execute our global launch in gMG. One of our goals of 2023 was to expand our global launch of VYVGART as the first approved neonatal FcRn blocker for the treatment of gMG beyond initial commercial regions of the U.S., Japan and EU. In 2023, we received approval for VYVGART in Israel (through our partner Medison), the UK, Mainland China (through our partner Zai Lab) and Canada and we aim for further approvals in additional jurisdictions. We have built our commercial infrastructure to support the commercialization of VYVGART in the U.S., Europe, Japan and Canada and will be prepared to expand this infrastructure to support the launch of VYVGART into new indications in some of these territories if and when we receive approval. |
| ● | Expand applications for our lead product efgartigimod beyond gMG. Our goal is to maximize the commercial potential of our existing products and product candidates by exploring additional indications, as well as formulations that may expand the target patient populations within existing indications. We are further developing our lead product, efgartigimod, for the treatment of more than 10 serious autoimmune indications. We expand the use of our products and product candidates in existing indications by developing new formulations and product generations, that may reach more patient groups by capturing different patient preferences and providing additional optionality with regards to dosing. |
| ● | Advance our pipeline of assets. In addition to new indications for efgartigimod, we plan to advance additional product candidates. In particular, we are advancing the clinical development of empasiprubart in MMN, DGF in the context of kidney transplants and DM. We are also advancing ARGX-119 into Phase 1b/2a clinical trials in CMS and ALS and beyond and plan to advance early-stage pipeline candidates towards IND filing by the end of 2025, as well as expand our pipeline of future product candidates through the IIP. |
| ● | Leverage our suite of technologies to seek strategic collaborations and maximize the value of our pipeline. Our suite of technologies and productive discovery capabilities have yielded several potential product candidates for which we seek to capture value, while maintaining our focus and discipline. We plan to collaborate on product candidates that we believe have promising utility in disease areas or patient populations but fall outside our |
68
| commercial franchises or are better served with the focus of a dedicated team in a spin-off company. In addition to collaborating on our products and product candidates, we may also elect to enter into collaborations for access to partner technology platforms or capabilities from which we can develop differentiated potential pipeline assets. |
| ● | Continue to build innovation into every step of our development, highlighted by our collaborative IIP translating immunology breakthroughs into medicines. Our IIP is our core business strategy connecting the specialized insight into disease- and target biology of our external scientific and academic collaborators with our unparalleled experience as antibody engineers. Co-creation has led to a deep pipeline of highly differentiated product candidates. Through our IIP, we hope to together transcend breakthrough research and publications to our ultimate and unifying mission of creating new potential treatment options for patients. |
Competitive position
We participate in a highly innovative industry characterized by a rapidly growing understanding of disease biology, quickly changing technologies, strong intellectual property barriers to entry, and a multitude of companies involved in the creation, development and commercialization of novel therapeutics. Many of these companies are highly sophisticated and often strategically collaborate with each other.
Competition in the autoimmune field is intense and involves multiple mAbs, other biologics and small molecules either already marketed or in development by many different companies, including large pharmaceutical companies. We compete with a wide range of biopharmaceutical companies, who are developing products for the treatment of gMG and other autoimmune diseases, including products that are in the same class as VYVGART, as well as products that are similar to some of our product candidates. We are aware of several FcRn inhibitors that are in clinical development or marketed. Competitive product launches may erode future sales of our products, including our existing products and those currently under development, or result in unanticipated product obsolescence. Such launches continue to occur, and potentially competitive products are in various stages of development. We could also face competition for use of limited international infusion sites, particularly in new markets as competitors launch new products. We cannot predict with accuracy the timing or impact of the introduction of competitive products that treat diseases and conditions like those treated by our products or product candidates. In addition, our competitors compete with us to recruit and retain qualified scientific and management personnel, establish clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, the development of our products. Please refer to Item 3.D. “Risk Factors—Risk Factors Related to Commercialization of argenx’s Products and Product Candidates, Including for New Indications—We face significant competition for our drug discovery and development efforts.” for further details on the competition we face.
Manufacturing and Supply
We utilize third-party contract manufacturers who act in accordance with the FDA’s cGMPs for the manufacture of drug substance and drug product. We continue to build our global network of contract manufacturers to support the development and commercialization of our products. We work with Lonza teams based in Slough, UK, Portsmouth, U.S., Singapore and Visp, Switzerland for activities relating to the development of cell banks, development of our manufacturing processes and the manufacturing of drug substance, thereby using validated and scalable systems broadly accepted in our industry. In 2022, we started our collaboration with Fujifilm based in Hillerød, Denmark, for activities relating to the large-scale manufacturing of efgartigimod drug substance. We use additional contract manufacturers to fill, label, package, store and distribute (investigational) drug products.
Intellectual Property
Introduction
We strive to protect and maintain exclusivity for the proprietary technologies that we believe are important to our business, patients and shareholders. We are focused on pursuing and maintaining patent protection intended to cover core platform technologies incorporated into, or used to produce, our product candidates and commercial products. We
69
will seek protection for our innovations in key global jurisdictions. We continue to focus our exclusivity strategies on all aspects of our assets, including our compositions of matter, methods of use for our approved products, and other inventions that are important to our business (e.g., the patient innovations described in our product labels/product inserts and our core manufacturing technologies).
Our intellectual property portfolio continues to grow and keep pace with the innovations arising from our discovery, development, and commercial efforts. We expect the total volume of patent positions under our management to increase with each year as our pipeline evolves. We currently oversee more than 500 pending applications and granted patents. More importantly, as we continue to innovate for patients, we will work to protect our patient innovations with new intellectual property filings to enable future reinvestment for patients.
In addition to patent protection, we rely on trademarks and trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection, including certain aspects of our llama immunization and antibody affinity maturation approaches.
Our commercial success depends in part upon our ability to obtain and maintain exclusivity, including regulatory exclusivities, patent, and other proprietary protection for commercially important technologies, inventions and know-how related to our business. We will defend and enforce our intellectual property rights, particularly our patent rights, and preserve the confidentiality of our trade secrets while operating without infringing valid and enforceable intellectual property rights of others. Specifically, we are materially dependent on regulatory, patent and other proprietary protection related to our core platform technologies, described in Item 4.B. “Business Overview—Intellectual Property—Platform Technologies”, our product candidates, as described in Item 4.B. “Business Overview—Intellectual Property—Our Internal Programs”, and Item 4.B. “Business Overview—Intellectual Property—Our Partnered Programs”.
The patent positions for biotechnology companies like us are generally uncertain and can involve complex legal, scientific and factual issues. In addition, the coverage claimed in a patent application can be significantly reduced before a patent is issued, and its scope can be reinterpreted and even challenged after issuance. As a result, we cannot guarantee that any of our platform technologies and product candidates will be protectable or remain protected by enforceable patents. We cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient proprietary protection from competitors. Any patents that we hold may be challenged, circumvented or invalidated by third parties.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the earliest date of filing a non-provisional patent application.
In the U.S., the term of a patent covering an FDA-approved drug may be eligible for a limited patent term extension under the Hatch-Waxman Act as compensation for the loss of patent term during the FDA regulatory review process as described in Item 4.B. “Business Overview—Regulation—Licensure and Regulation of Biologics in the U.S.” Similar provisions are available in the EU and in certain other jurisdictions to extend the term of a patent that covers an approved drug. It is possible that issued U.S. patents covering each of our product candidates may be entitled to patent term extensions. If our product candidates receive FDA approval, we intend to apply for patent term extensions, if available, to extend the term of patents that cover the approved product candidates. We also intend to seek patent term extensions in any jurisdictions where they are available, however, there is no guarantee that the applicable authorities, including the FDA, will agree with our assessment of whether such extensions should be granted, and if granted, the length of such extensions.
Platform Technologies
With regard to our platform technologies, we own or have intellectual property rights directed to our SIMPLE ANTIBODY™ discovery platform, the ABDEG™ and NHANCE™ technologies.
70
With regard to our SIMPLE ANTIBODY™ discovery platform, we have a broad patent portfolio providing exclusivity on the SIMPLE ANTIBODY™ platform. We expect to enjoy exclusivity under this patent portfolio until between 2029 and 2033.
With regard to the ABDEG™ platform, we co-own the technology with UT Southwestern and enjoy certain exclusive license rights. We have a broad patent portfolio covering the composition of matter and uses of certain FcRn antagonists to achieve certain biological effects. The composition of matter and other relevant patents in the U.S. expire in 2036 whereas in many other countries the base expiry date is 2034.
With regard to the NHANCE™ platform, we exclusively licensed two U.S. patents from UT Southwestern with composition of matter claims directed to an IgG molecule comprising a variant human Fc domain, and method of use claims directed to a method of blocking FcRn function in a subject by providing to the subject such an IgG molecule. The U.S. patents are expected to expire earliest in 2027 to 2028. The patent family also includes a granted European patent.
Our Internal Programs
Efgartigimod
Efgartigimod incorporates the ABDEG™ platform technology. We anticipate several more patient innovations to evolve during development for which we will seek additional patent protection.
Our ARGX-109 Product Candidate
With regard to our wholly-owned ARGX-109 product candidate, we have one patent family with composition of matter claims directed to ARGX-109. The patent family has a basic expiry date in 2033. We anticipate several more patient innovations to evolve during development for which we will seek additional patent protection. Furthermore, ARGX-109 incorporates or employs the SIMPLE ANTIBODY™ platform technology and the NHANCE™ platform technology.
Empasiprubart Product Candidate
With regard to the empasiprubart product candidate, we own or have rights to multiple patent families (including one in-licensed patent family from Broteio) with several granted patents and pending patent applications in multiple jurisdictions in North America, South America, the EU and Asia, directed to composition of matter claims and method of treatment claims. The in-licensed patent family from Broteio has granted patents in several countries/regions and has a basic expiry date in 2034. Additional patent families have granted patents with basic expiry dates in 2039 and 2040. We anticipate several more patient innovations to evolve during development for which we will seek additional patent protection. Empasiprubart product candidate incorporates or employs the NHANCE™ platform technology.
Our ARGX-119 Product Candidate
With regard to the ARGX-119 product candidate, we in-licensed patent families from/with NYU Langone Health, a U.S. medical center based in New York, and additional patent families from/with the LUMC, with a U.S. granted patent and several pending applications in multiple jurisdictions. We anticipate several more patient innovations to evolve during development for which we will seek additional patent protection.
Our ARGX-118 Product Candidate
With regard to the ARGX-118 product candidate, we co-own a patent portfolio with VIB vzw (VIB), an inflammation research center in Ghent, Brussels, and Ghent University, with one U.S. granted patent and pending patent applications in multiple jurisdictions in North America, South America, the EU and Asia. The patent family has a basic expiry date in 2039.
71
Our Partnered Programs
Our Cusatuzumab (ARGX-110) Product Candidate
With regard to the cusatuzumab product candidate, we have a broad patent portfolio that include claims to the composition of matter, uses of the molecule, and other important inventions. The issued U.S. patents expire in 2032 and 2033, without taking a potential patent term extension into account. Cusatuzumab incorporates or employs the SIMPLE ANTIBODY™ and POTELLIGENT® platform technologies.
Our ARGX-115 (ABBV-151) Product Candidate
With regard to the ARGX-115 (ABBV-151) product candidate that we co-own with, and exclusively license from, the Ludwig Institute for Cancer Research and UCL, we have a patent portfolio that includes a U.S. patent with a basic expiry date in 2034, without taking a potential patent term extension into account. There is a second family with meaningful patent coverage to the composition of matter and epitope claims that are expected to expire in 2036 and 2038. Furthermore, ARGX-115 (ABBV-151) incorporates or employs the SIMPLE ANTIBODY™ platform technology.
Our ARGX-112 (LP-0145) Product Candidate
With regard to the ARGX-112 (LP-0145) product candidate, we have one patent family with composition of matter claims directed to an antibody that binds human IL-22R. The patent family has a basic expiry date in 2037. Furthermore, ARGX-112 (LP-0145) incorporates the SIMPLE ANTIBODYTM platform technology.
Collaboration and Licenses
We follow a disciplined strategy to maximize the value of our pipeline. We plan to retain all development and commercialization rights to those products and product candidates that we believe we can commercialize successfully, if approved.
We have partnered, and plan to continue to partner, to develop products and product candidates that we believe have promising utility in disease areas or have patient populations that may benefit from resources of other biopharmaceutical companies. We expect to continue to collaborate selectively with pharmaceutical and biotechnology companies to leverage our platform technology and accelerate product candidate development.
We are also party to several license agreements under which we license patents, patent applications and other intellectual property to third parties. We have also entered into several license agreements under which we license patents, patent applications and other intellectual property from third parties. License agreements can relate to research and development and/or commercialization of the relevant product candidates (and technologies) or products. The licensed intellectual property covers some of our product candidates and some of the antibody engineering technologies that we use. Some of these licenses impose various diligence and financial payment obligations on us. We expect to continue to enter into these types of license agreements in the future.
We have entered into multiple collaboration agreements with pharmaceutical partners and license agreements, as described below.
Our Strategic Collaboration with Shire
In February 2012, we entered into a collaboration agreement with Shire AG (Shire, now known as Shire International GmbH) to discover, develop and commercialize novel human therapeutic antibodies against up to three targets to address diverse, rare and unmet diseases (Shire Collaboration Agreement). Pursuant to the Shire Collaboration Agreement, up through a specified period, we have granted Shire an exclusive option, against payment of a one-time option fee, to obtain all right, title and interest in any antibodies discovered under the collaboration. If Shire does not exercise its option with respect to any discovered antibody within a specified period, we are free to research,
72
develop and commercialize antibodies in relation to the applicable clinical trial target, subject to negotiation of a license from Shire.
OncoVerity for cusatuzumab
In 2022, we, the University of Colorado Anschutz Medical Campus and the University of Colorado Health (UCHealth) created an asset-centric spin-off, OncoVerity, Inc (OncoVerity), focused on optimizing and advancing the development of cusatuzumab, a novel anti-CD70 antibody, in AML. OncoVerity is an entity of co-creation, combining the extensive translational biology insights from Dr. Clayton Smith, M.D. from the University of Colorado with our experience on the CD70/CD27 pathway.
In 2023, we granted an exclusive license for cusatuzumab to OncoVerity and provided, together with a joint venture of UCHealth and University License Equity Holdings, Inc. on the University of Colorado Anschutz Medical Campus, $26.0 million in funding for ongoing clinical development of cusatuzumab.
Our Strategic Partnership with LEO Pharma for ARGX-112 (LP0145)
In May 2015, we entered into a collaboration agreement with LEO Pharma A/S (LEO Pharma) to develop and commercialize ARGX-112 (LP0145) for the treatment of dermatologic indications involving inflammation (LEO Pharma Collaboration Agreement). ARGX-112 (LP0145) employs our SIMPLE ANTIBODY™ technology and blocks the IL-22R in order to neutralize the signaling of cytokines implicated in autoimmune diseases of the skin. LEO Pharma funded more than half of all product development costs up to CTA approval of a first product in a Phase 1 clinical trial, with our share of such costs capped, which was achieved in April 2018. Since then, LEO Pharma has been solely responsible for funding the clinical development of the program. In May 2021, CTA approval of a Phase 2a clinical trial for LP0145 was received.
In September 2022, LEO Pharma, exercised its option to obtain, and was granted an exclusive, worldwide license to further develop and commercialize ARGX-112 against payment of a €5.0 million option fee to us. LEO Pharma assumed full responsibility for the continued development, manufacture and commercialization of such product and is subject to diligence obligations in respect of continuation of development and commercialization of such product. We are eligible to receive additional development, regulatory and commercial milestone payments in aggregate amount of up to €120.0 million, as well as tiered royalties on product sales at percentages ranging from the low single digits to the low teens, subject to customary reductions.
Unless earlier terminated, the term of the LEO Pharma Collaboration Agreement ends upon the later of (i) the expiration of the last license granted under the agreement, and (ii) the fulfilment of all payment obligations under the agreement. LEO Pharma may terminate the LEO Pharma Collaboration Agreement for any reason upon prior written notice to us. LEO Pharma’s royalty payment obligations expire, on a product-by-product and country-by-country basis, upon the later of (i) a time when no valid claims covering such product, and (ii) (a) in major market countries with no composition of matter patent covering such product, the expiration of the data exclusivity period or (b) in countries that are not major market countries, a double-digit number of years after the first commercial sale of such product sold in that country.
Our Strategic Partnership with Zai Lab for efgartigimod
Pursuant to the Zai Lab Agreement, Zai Lab obtained the exclusive right to develop and commercialize efgartigimod in Greater China. Zai Lab will also contribute patients to our global Phase 3 clinical trials of efgartigimod. Additionally, the collaboration with Zai Lab is expected to accelerate efgartigimod global development by initiating multiple Phase 2 POC clinical trials in new autoimmune indications under our supervision; first indications for such POC clinical trials are kidney conditions LN and MN.
Pursuant to the Zai Lab Agreement, we have received value worth $175.0 million from the Zai Lab Payments. We are also eligible to receive tiered royalties (mid-teen to low-twenties on a percentage basis) based on annual net sales of efgartigimod in Greater China.
73
Our Strategic Partnership with AbbVie for ARGX-115 (ABBV-151)
In April 2016, we entered into a collaboration agreement with AbbVie to develop and commercialize ARGX-115 (ABBV-151) as a cancer immunotherapy against the novel target glycoprotein A repetitions predominant (GARP) (the AbbVie Collaboration Agreement). ARGX-115 (ABBV-151) employs our SIMPLE ANTIBODY™ technology and works by stimulating a patient’s immune system after a tumor has suppressed the immune system by co-opting immunosuppressive cells such as regulatory T cells. Under the terms of the AbbVie Collaboration Agreement, we were responsible for conducting and funding all ARGX-115 (ABBV-151) research and development activities up to completion of IND enabling clinical trials.
AbbVie has exercised its option and obtained a worldwide, exclusive license to the ARGX-115 (ABBV-151) program to develop and commercialize products and has assumed development obligations, including the sole responsibility for all research, development and regulatory costs relating to ARGX-115 (ABBV-151)-based products. Subject to the continuing progress of ARGX-115 (ABBV-151) by AbbVie, we are eligible to receive development, regulatory and commercial milestone payments in aggregate amounts of up to $110 million, $190 million and $325 million, respectively, as well as tiered royalties on product sales at percentages ranging from the mid-single digits to the lower teens, subject to customary reductions.
Pursuant to the AbbVie Collaboration Agreement, we have the right, on a product-by-product basis, to co-promote ARGX-115 (ABBV-151) based products in the EEA and Switzerland and to combine the product with our own future oncology programs (if any). The co-promotion effort would be governed by a co-promotion agreement negotiated in good faith by the parties.
Unless earlier terminated upon mutual agreement, for material breach or as otherwise specified in the AbbVie Collaboration Agreement, the term of the license agreement ends, with respect to the ARGX-115 (ABBV-151) program, upon fulfilment of all payment obligations under the agreement.
AbbVie may terminate the AbbVie Collaboration Agreement for any reason upon prior written notice to us. AbbVie’s royalty payment obligations expire, on a product-by-product and country-by-country basis, on the date that is the later of (i) such time as there are no valid claims covering such product, (ii) expiration of regulatory or market exclusivity in respect of such product or (iii) 10 years after the first commercial sale of such product sold in that country under the AbbVie Collaboration Agreement.
Our Exclusive License with Elektrofi for efgartigimod
In April 2021, we entered into the Elektrofi Agreement with Elektrofi to explore new SC formulations utilizing Elektrofi’s high concentration technology for efgartigimod, and up to one additional target. The Elektrofi-enabled formulations are aimed to promote additional optionality for patients through at-home and self-administration capabilities.
Under the terms of the Elektrofi Agreement, we made an upfront payment and future milestones payments across both targets pending achievement of pre-defined development, regulatory, and commercial milestones. Elektrofi will also receive a mid-single digit royalty on sales of commercialized products.
Our collaboration with Genmab
In 2023, we entered into a collaboration with Genmab to jointly discover, develop and commercialize novel therapeutic antibodies with applications in immunology, as well as in oncology therapeutic areas. The multiyear collaboration is expected to leverage the antibody engineering expertise and knowledge of disease biology of both companies to accelerate the identification and development of novel antibody therapeutic candidates with a goal to address unmet patient needs in immunology and cancer. Under the terms of the collaboration, we and Genmab each have access to the suites of proprietary antibody technologies of both companies to advance the identification of lead antibody candidates against differentiated disease targets.
74
Our Non-Exclusive Research License and Option Agreement with Chugai for SMART-Ig® and ACT-Ig®
In September 2020, we entered into a non-exclusive research license and option agreement with Chugai, allowing us to access Chugai’s SMART-Ig® and ACT-Ig® engineering technologies for conducting feasibility clinical trials. These technologies are designed to enable us to make sweeping antibodies and expand the therapeutic index of our product candidates, which is the ratio between toxic and therapeutic dose, by potentially modifying their half-life, tissue penetration, rate of disease target clearance and potency.
Our Non-exclusive License with the Clayton Foundation for DHS mutations
In October 2020, we entered into a non-exclusive research agreement with the Clayton Foundation relating to the non-exclusive in-license for Clayton Foundation’s proprietary DHS mutations to extend the serum half-life of therapeutic candidates.
Our Exclusive License with Halozyme for ENHANZE®
In February 2019, we entered into an in-license agreement with Halozyme for the use of certain patents, materials and know-how owned by Halozyme and relating to its ENHANZE®, for application in the field of prevention and treatment of human diseases (the ENHANZE® License Agreement). Pursuant to the ENHANZE® License Agreement, we were granted exclusive rights to apply ENHANZE® to biologic products against pre-specified targets, in order to research, develop and commercialize SC formulations of our therapeutic antibody-based product candidates.
Our first therapeutic target for which we received an exclusive license from Halozyme was FcRn, which allows us to apply ENHANZE® to efgartigimod and any other product candidates selective and specific for FcRn. Moreover, the breadth of our exclusive license to FcRn precludes either Halozyme itself or any of its current or future partners from utilizing ENHANZE® in the context of an FcRn-targeted product. Our second therapeutic target for which we received an exclusive license from Halozyme was human C2 associated with the product candidate empasiprubart, which is being developed to treat severe autoimmune diseases. Pursuant to the ENHANZE® License Agreement, we also have the right to nominate future targets for an exclusive ENHANZE® license if the target in question has not already been licensed by Halozyme or is not already being pursued by Halozyme.
In October 2020, we expanded our collaboration with Halozyme for ENHANZE® drug delivery technology to include three additional exclusive targets upon nomination bringing the total to six potential targets. From the effective date of the ENHANZE® License Agreement, we have a seven-year period in which to conduct research and preclinical trials on other target-specific molecules in combination with ENHANZE® and may nominate up to four additional targets we have not yet nominated for an exclusive commercial license.
Pursuant to the ENHANZE® License Agreement, we have the right to grant sublicenses to our subsidiaries and to third parties both for research/preclinical work (for example, to subcontractors) and for development and commercialization. Halozyme provides dedicated specialist support to us which it has accrued over 10 years of licensing ENHANZE® to its collaborators.
Upon nomination of any future target for an exclusive commercialization license and confirmation by Halozyme that such a license is available, we will pay $12.5 million to Halozyme per target. We will be obligated to pay clinical development, regulatory and commercial milestones totaling $160.0 million for the first product that uses ENHANZE® and is specific for a given target. We are also obligated to pay Halozyme a percentage of net sales as a royalty of any licensed product that uses ENHANZE®. This royalty varies with net sales volume, ranging from the low to mid-single digits. The royalty obligations may be reduced by up to 50% under different circumstances including the requirement of a compulsory license from a government, the need to secure third-party licenses to enable sales of our products using the ENHANZE® technology, and/or the lack of patent coverage in a particular country. We have diligence obligations with respect to the continuation of development and commercialization of product candidates, but we are not obligated to utilize ENHANZE® for every product candidate directed to a given exclusive target(s).
75
We may terminate the ENHANZE® License Agreement at any time, either in its entirety or on a target-by-target basis, by sending Halozyme prior written notice. Absent early termination, the ENHANZE® License Agreement will automatically expire upon the expiry of our royalty payment obligations under the agreement. In the event the ENHANZE® License Agreement is terminated for any reason, the license granted to us would terminate but Halozyme would grant our sublicensees a direct license following such termination. In the event the ENHANZE® License Agreement is terminated other than for our breach, we would retain the right to sell licensed products then on hand for a certain period of time post-termination.
Our Exclusive License with Agomab Therapeutics NV (Agomab) for ARGX-114 (AGMB-101)
In March 2019, we entered into an exclusive out-license with Agomab for the use of certain patent rights relating to our proprietary suite of technologies for the development and commercialization of a series of agonistic anti-mesenchymal-epithelial transition factor (MET) SIMPLE ANTIBODYTM generated antibodies, including ARGX-114 (AGMB-101), a halofuginone-mimetic antibody directed against the MET receptor. Agomab is required to use commercially reasonable efforts to develop and commercialize at least one licensed product. In connection with our entry into this agreement, we received a profit-sharing certificate which entitles us to 20% of all distributions to Agomab’s shareholders (which shall be reduced to 10% following the filing of an IND and is subject to further adjustment upon the occurrence of certain financings). Upon the occurrence of a qualified initial public offering of Agomab, the profit-sharing certificate will automatically be converted into the equivalent number of ordinary shares in Agomab. This agreement is subject to mutual termination for material breach or insolvency and automatically expires upon the expiration of the last to expire of our licensed patent rights.
Our Exclusive License with Broteio for empasiprubart
In March 2017, we entered into a collaboration with Broteio in connection with our IIP, to develop an antibody against a novel target in the complement cascade, empasiprubart (Broteio Agreement). Under the Broteio Agreement, we are jointly developing the complement-targeted antibody to seek to establish preclinical POC using our proprietary suite of technologies. Upon successful completion of these clinical trials, we exercised an exclusive option to in-license the program in March 2018 and assumed responsibility for further development and commercialization. Pursuant to the Broteio Agreement, we are obligated to make milestone payments upon the occurrence of certain development milestones (up to an aggregate of €4.0 million), commercialization milestones (up to an aggregate of €10.0 million) and pay tiered royalties on net sales in the low single digits. We may terminate the Broteio Agreement for convenience upon 90 days prior written notice. The Broteio Agreement is also subject to mutual termination for material breach or insolvency and automatically expires upon the expiration of our financial obligations thereunder.
Our Exclusive License with VIB for ARGX-118
In November 2016, we entered into a collaboration under our IIP with VIB vzw (VIB) to develop antibodies against Galectin-10, the protein of Charcot-Ley-den Crystals, which play a major role in severe asthma and the persistence of mucus plugs, including ARGX-118 (VIB Agreement). Pursuant to the VIB Agreement, we are jointly developing antibodies against Galectin-10 using our proprietary suite of technologies. Upon successful completion of this initial research, we exercised an exclusive option to in-license the program and assumed responsibility for further development and commercialization. Under the VIB Agreement, including as amended in November 2018, we are obligated to make milestone payments upon the occurrence of certain development milestones (up to an aggregate of €4.0 million), commercialization milestones (up to an aggregate of €11.0 million) and pay tiered royalties on net sales in the low single digits. We may terminate the VIB Agreement for convenience upon 90 days prior written notice. The VIB Agreement is also subject to mutual termination for material breach, insolvency or certain patent challenges and automatically expires upon the expiration of VIB’s licensed patent rights.
Our Exclusive License with the University of Texas for NHANCE™ and ABDEG™
In February 2012, we entered into an exclusive in-license with the Board of Regents of the University of Texas System (UT BoR) for the use of certain patent rights relating to the NHANCE™ platform for any use worldwide (the
76
UT Agreement). The UT Agreement was amended on December 23, 2014 to also include certain additional patent rights relating to the ABDEG™ platform. Upon commercialization of any of our products that use the in-licensed patent rights, we will be obligated to pay UT BoR a percentage of net sales as a royalty until the expiration of any patents covering the product. This royalty varies with net sales volume and is subject to an adjustment for royalties we receive from a sublicensee of our rights under the UT Agreement, but in any event does not exceed 1%. In addition, we must make annual license maintenance payments to UT BoR until termination of the UT Agreement and we have assumed certain development and commercial milestone payment and reimbursement obligations. We also have diligence requirements with respect to development and commercialization of products which use the in-licensed patent rights.
Pursuant to the UT Agreement, we may grant sublicenses to third parties. If we receive any non-royalty income in connection with such sublicenses, we must pay UT BoR a percentage of such income varying from low-middle single digits to middle-upper single digits depending on the nature of the sublicense. Such fees are waived if a sublicensee agrees to pay the milestone payments as set forth in the UT Agreement.
We may unilaterally terminate the UT Agreement for convenience upon prior written notice. Absent early termination, the UT Agreement will automatically expire upon the expiration of all issued patents and filed patent applications within the patent rights covered by the UT Agreement. Our royalty payment obligations expire, on a product-by-product and country-by-country basis, at such time as there are no valid claims covering such product.
Our Non-Exclusive License with BioWa and Non-Exclusive Commercial Licenses with BioWa and Lonza for POTELLIGENT®
In October 2010, we entered into a non-exclusive license agreement with BioWa, Inc. (BioWa) for the use of certain patents and know-how owned by BioWa and relating to its POTELLIGENT® platform technology, for use in the field of prevention and treatment of human diseases (the BioWa Agreement). Pursuant to the BioWa Agreement, we are granted a non-exclusive right to use POTELLIGENT® to research and develop antibodies and products containing such antibodies using POTELLIGENT®.
In 2013 and 2014, we entered into non-exclusive license agreements for POTELLIGENT® CHOK1SV with BioWa and Lonza for the further development, manufacturing and commercialization of ARGX-110 and ARGX-111, respectively (the POTELLIGENT® License Agreements).
Upon commercialization of our products developed using POTELLIGENT®, we will be obligated to pay BioWa and Lonza a percentage of net sales of a licensed product as a royalty. This royalty varies with net sales volume, ranging in the low single digits, and it is reduced by half if during the following 10 years from the first commercial sale of the product in a country the last valid claim within the licensed patent(s) that covers the product expires or ends. In addition, we must make annual research license maintenance payments which cease with commencement of our royalty payments to BioWa. We have diligence requirements with respect to the continuation of development and commercialization of products. We have also assumed certain development, regulatory and commercial milestone payment obligations and must report on our progress toward achieving these milestones on an annual basis. Milestones to BioWa are to be paid on a commercial target-by-commercial target basis, and we are obligated to make milestone payments in aggregate amounts of up to $36.0 million per commercial target should we achieve annual global sales of over $1.0 billion.
Pursuant to the POTELLIGENT® License Agreements, we have the right to grant sublicenses to third parties. BioWa retains a right of first negotiation for the exclusive right to develop and commercialize, in certain countries only, any product we develop using POTELLIGENT®.
We may terminate the POTELLIGENT® License Agreements at any time by sending BioWa and Lonza prior written notice. Absent early termination, the POTELLIGENT® License Agreements will automatically expire upon the expiry of our royalty obligations under the POTELLIGENT® License Agreements. In the event a POTELLIGENT® License Agreement is terminated for any reason, the license granted to us would terminate but BioWa would grant our
77
sublicensees a direct license following such termination. In the event the POTELLIGENT® License Agreement is terminated other than for our breach or insolvency, we would retain the right to sell licensed products then on hand for a certain period of time post-termination.
Our non-exclusive license with Lonza for Multi-product GS Xceed-License
On February 4, 2015, we entered into a non-exclusive multi-product in-license agreement with Lonza (the Multi-Product License) for use of Lonza’s proprietary glutamine synthetase gene expression system known as GS Xceed™ consisting of Chinese hamster ovary cell line and the vectors for the manufacturing of drug substance (the System). The System is used for the manufacturing of, amongst others, efgartigimod, empasiprubart and ARGX-119.
Pursuant to the Multi-Product License, we have the right to grant sublicenses to certain pre-approved third parties without prior written consent of Lonza, but otherwise we must obtain Lonza’s prior written consent.
We have assumed certain development, regulatory and commercial milestone payment obligations to Lonza. We are required to pay such milestones using the System. We are obligated to make development, regulatory and commercial milestone payments to Lonza.
We may terminate the Multi-Product License on a product-by-product basis by giving Lonza prior written notice. Lonza may terminate the Multi-Product License solely in case of breach or insolvency events. Absent early termination, the Multi-Product License will automatically expire upon the expiry of the last valid claim for such product. We or our strategic partners would retain the right to sell the respective products then on hand post-termination.
Our Collaboration with Université Catholique de Louvain (UCL) and Sopartec S.A. (Sopartec) for GARP
In January 2013, we entered into a collaboration and exclusive product license agreement with UCL and its technology transfer company Sopartec to discover and develop novel human therapeutic antibodies against GARP (GARP Agreement). Pursuant to the GARP Agreement, each party is responsible for all of its own costs in connection with the activities assigned to it under a mutually agreed research plan.
In January 2015, we exercised the option we were granted under the GARP Agreement to enter into an exclusive, worldwide commercial in-license for use of certain GARP-related intellectual property rights owned by UCL and the Ludwig Institute for Cancer Research to further develop and commercialize licensed products, including the GARP-neutralizing antibody ARGX-115 which was discovered under the original collaboration (GARP License). Upon the expiration of the GARP Agreement, the GARP License will become a fully paid-up, perpetual worldwide exclusive license under the GARP intellectual property for any purpose, subject to UCL’s retention of non-commercial research rights.
Pursuant to the GARP License, we may grant sublicenses to third parties and affiliates of such third parties. In 2016, we entered into an exclusive collaboration and license agreement with AbbVie regarding ARGX-115. From any income we receive in connection with these sublicenses, such as in connection with AbbVie Collaboration Agreement, we must pay Sopartec a percentage of that income in the lower teen digit range. Royalty payment obligations expire on a product-by-product and country-by-country basis when there are no valid claims covering the ARGX-115 product. We also have diligence obligations with respect to the continued development and commercialization of ARGX-115 products.
Our Exclusive License with NYU Langone Health and LUMC for ARGX-119
In 2019 and 2020, we entered into collaboration and exclusive license agreements with NYU Langone Health and LUMC under our IIP to develop antibodies targeting the MuSK, for the treatment neuromuscular diseases, which play a major role at the neuromuscular junction (NYU and LUMC Agreements). Pursuant to the NYU and LUMC Agreements, we, NYU and LUMC jointly developed antibodies against MuSK using our proprietary suite of technologies. Under the NYU and LUMC Agreements, as amended, we are obligated to make milestone payments upon
78
the occurrence of certain development milestones, commercialization milestones and pay tiered royalties on net sales in the low single digits.
Distribution Agreements
We are parties to the Medison Agreement, the Medison Multi-Regional Agreement, Genpharm Agreement, and the Handok Agreement.
Trade Secret Protection
In addition to patent protection, we also rely on trade secret protection to ensure exclusivity for our proprietary information that is not amenable to, or that we do not consider appropriate for, patent protection, including, for example, certain aspects of our llama immunization and antibody affinity maturation approaches. However, trade secrets can be difficult to protect. Although we take steps to protect our proprietary information, including restricting access to our premises and our confidential information, as well as entering into agreements with our employees, consultants, advisors and potential collaborators, third parties may independently develop the same or similar proprietary information or may otherwise gain access to our proprietary information. As a result, we may be unable to meaningfully protect our trade secrets and proprietary information.
Regulation
Government authorities in the U.S., at the federal, state and local level, and in the EU and other countries and jurisdictions, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, and import and export of pharmaceutical products, including biological products. In addition, some jurisdictions regulate the pricing of pharmaceutical products. The processes for obtaining marketing approvals in the U.S. and in other countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations and other regulatory authorities, require the expenditure of substantial time and financial resources.
Licensure and Regulation of Biologics in the U.S.
In the U.S., biological products used for the prevention, treatment, or cure of a disease or condition in a human being are subject to regulation under the FDCA and its implementing regulations, with the exception that the section of the FDCA that governs the approval of drugs via new drug applications (NDAs) does not apply to the approval of biologics. Biologics are approved for marketing under provisions of the Public Health Service Act (PHSA) via biologics license applications (BLAs). However, the application process and requirements for approval of BLAs are very similar to those for NDAs. The failure to comply with the applicable U.S. requirements at any time during the product development process, including preclinical testing and clinical testing, the approval process or post-approval process may subject an applicant to delays in the conduct of a clinical trial, regulatory review and approval, and/or administrative or judicial sanctions. These sanctions may include, but are not limited to, the FDA’s refusal to allow an applicant to proceed with clinical testing, refusal to approve pending applications, license suspension or revocation, warning or untitled letters, adverse publicity, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines and civil or criminal investigations and penalties brought by the FDA or the Department of Justice or other governmental entities.
An applicant seeking approval to market and distribute a new biologic in the U.S. generally must satisfactorily complete each of the following steps:
| ● | preclinical laboratory tests, animal studies and formulation studies all performed in accordance with applicable regulations, including the GLPs; |
| ● | submission to the FDA of an IND application for human clinical testing, which must become effective before human clinical trials may begin; |
79
| ● | approval by an IRB representing each clinical site before each clinical trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials to establish the safety, potency and purity of the product candidate for each proposed indication, in accordance with GCPs; |
| ● | preparation and submission to the FDA of a BLA for a biological product requesting marketing for one or more proposed indications, including submission of detailed information on the manufacture and composition of the product in clinical development and proposed labeling; |
| ● | one or more FDA inspections of the manufacturing facility or facilities, including those of third parties, at which the product, or components thereof, are produced to assess compliance with cGMP requirements and to assure that the facilities, methods and controls are adequate to preserve the product’s identity, strength, quality and purity; |
| ● | FDA audits of the clinical trial sites to assure compliance with GCPs, and the integrity of clinical data in support of the BLA; |
| ● | payment of user fees and securing FDA approval of the BLA and licensure of the new biological product; and |
| ● | compliance with any post-approval requirements, including the potential requirement to implement a REMS and any post-approval studies required by the FDA; and preclinical trials and INDs. |
Before testing any biological product candidate in humans, the product candidate must undergo preclinical testing. Preclinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as animal studies to evaluate the potential for activity and toxicity. The conduct of the preclinical tests and formulation of the compounds for testing must comply with federal regulations and requirements. The results of the preclinical tests, together with manufacturing information and analytical data, are submitted to the FDA as part of an IND application. The IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about the product candidate or conduct of the proposed clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks, and places the proposed clinical trial on clinical hold. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns before the clinical trial can begin.
As a result, submission of the IND may result in the FDA not allowing the clinical trial to commence or on the terms originally specified by the sponsor in the IND. If the FDA imposes a partial or complete clinical hold, this action would delay either a proposed clinical trial or cause suspension of an ongoing clinical trial, or in the case of a partial clinical hold place limitations on the conduct of the clinical trial such as duration of treatment, until all outstanding concerns have been adequately addressed and the FDA has notified the company that investigation may proceed and then only under terms authorized by the FDA. This could cause significant delays or difficulties in completing planned clinical trials in a timely manner. The FDA may impose clinical holds on a biological product candidate at any time before or during clinical trials due to safety concerns or non-compliance.
Human Clinical Trials in Support of a BLA
Clinical trials involve the administration of the investigational product candidate to healthy volunteers or patients with the disease to be treated under the supervision of a qualified principal investigator in accordance with GCPs. Clinical trials are conducted under clinical trial protocols detailing, among other things, the objectives of the clinical trial, inclusion and exclusion criteria, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND.
A sponsor who wishes to conduct a clinical trial outside the U.S. may, but is not required to, obtain FDA clearance to conduct the clinical trial under an effective IND. If a foreign clinical trial is not conducted under an IND,
80
the sponsor may submit data from the clinical trial to the FDA in support of the BLA so long as the clinical trial is well-designed and well-conducted in accordance with GCPs, including review and approval by an independent ethics committee, and the FDA is able to validate the clinical trial data through an onsite inspection, if necessary.
Further, each clinical trial must be reviewed and approved by the IRB or, if applicable, the Ethics Committee, either centrally or individually at each institution at which the clinical trial will be conducted. The IRB or the Ethics Committee will consider, among other things, clinical trial design, patient informed consent, ethical factors and the safety of human subjects. An IRB must operate in compliance with FDA regulations. The FDA, IRB, the Ethics Committee or the clinical trial sponsor may suspend or discontinue a clinical trial at any time for various reasons, including a finding that the clinical trial is not being conducted in accordance with FDA requirements or the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive GCPs and the requirements for informed consent. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group may recommend continuation of the clinical trial as planned, changes in clinical trial conduct, or cessation of the clinical trial at designated check points based on access to certain data from the clinical trial.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined. Additional clinical trials may be required after approval.
| ● | Phase 1 clinical trials are initially conducted in a limited population to test the product candidate for safety, including adverse effects, dose tolerance, absorption, metabolism, distribution, excretion and PD in healthy humans or, on occasion, in patients, such as cancer patients. |
| ● | Phase 2 POC clinical trials are generally conducted in a limited patient population to identify possible adverse effects and safety risks, evaluate the efficacy of the product candidate for specific targeted indications and determine dose tolerance and optimal dosage. Multiple Phase 2 clinical trials POC may be conducted by the sponsor to obtain information prior to beginning larger Phase 3 clinical trials. |
| ● | Phase 3 clinical trials proceed if the Phase 2 POC clinical trials demonstrate that a dose range of the product candidate is potentially effective and has an acceptable safety profile. Phase 3 clinical trials are undertaken within an expanded patient population to gather additional information about safety and effectiveness necessary to evaluate the overall benefit-risk relationship of the drug and to provide an adequate basis for physician labeling. |
In some cases, the FDA may approve a BLA for a product candidate but require the sponsor to conduct additional clinical trials to further assess the product candidate’s safety and effectiveness after approval. Such post-approval clinical trials are typically referred to as Phase 4 clinical trials. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and to document a clinical benefit in the case of biologics approved under accelerated approval regulations. If the FDA approves a product while a company has ongoing clinical trials that were not necessary for approval, a company may be able to use the data from these clinical trials to meet all or part of any Phase 4 clinical trial requirement or to request a change in the product labeling. Failure to exhibit due diligence with regard to conducting required Phase 4 clinical trials could result in withdrawal of approval for products.
Progress reports detailing the results of the clinical trials, among other information, must be submitted at least annually to the FDA, and written IND safety reports must be submitted to the FDA and the investigators 15 days after the clinical trial sponsor determines the information qualifies for reporting for serious and unexpected suspected adverse events, findings from other clinical trials or animal or in vitro testing that suggest a significant risk for human subjects and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must also notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction as soon as possible but in no case later than seven calendar days after the sponsor’s initial receipt of the information.
81
A drug being studied in clinical trials may be made available to individual patients in certain circumstances. Pursuant to the 21st Century Cures Act, as amended, the manufacturer of an investigational drug for a serious disease or condition is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for individual patient access to such investigational drug. This requirement applies on the earlier of the first initiation of a Phase 2 POC or Phase 3 clinical trial of the investigational drug, or as applicable, 15 days after the drug receives a designation as a breakthrough therapy, fast track product, or regenerative advanced therapy.
Compliance with cGMP Requirements
Before approving a BLA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The PHSA emphasizes the importance of manufacturing control for products like biologics whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency, and purity of the final biological product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the biological product candidate does not undergo unacceptable deterioration over its shelf life.
Manufacturers and others involved in the manufacture and distribution of products must also register their establishments with the FDA and certain state agencies. Both domestic and foreign manufacturing establishments must register and provide additional information to the FDA upon their initial participation in the manufacturing process. Establishments may be subject to periodic unannounced inspections by government authorities to ensure compliance with the FDCA, cGMPs and other requirements. Manufacturers may have to provide, on request, electronic or physical records regarding their establishments.
Disclosure of Clinical Trial Information
Sponsors of clinical trials of FDA-regulated products, including biological products, are required to register and disclose certain clinical trial information on the website www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, clinical trial sites and investigators, and other aspects of a clinical trial are then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of clinical trials can be delayed in certain circumstances for up to two years after the date of completion of the clinical trial. Competitors may use this publicly available information to gain knowledge regarding the progress of clinical development programs as well as clinical trial design.
Review and Approval of a BLA
The results of product candidate development, preclinical testing and clinical trials, including negative or ambiguous results as well as positive findings, are submitted to the FDA as part of a BLA requesting a license to market the product. The BLA must contain extensive manufacturing information and detailed information on the composition of the product and proposed labeling as well as payment of a user fee.
The FDA has 60 days after submission of the application to conduct an initial review to determine whether the BLA is sufficient to file based on the agency’s threshold determination that it is sufficiently complete to permit substantive review. If the FDA determines the BLA is not sufficiently complete, it will refuse to file the BLA. Once the submission has been filed, the FDA begins an in-depth review of the application. Under the goals agreed to by the FDA under the PDUFA, the FDA has 10 months from the filing date in which to complete its initial review of a standard application and respond to the applicant, and six months from the filing date for a priority review of an application, if the BLA is not filed under the program. If the BLA is submitted under the program, additional two months are added to the review clock, whether standard or priority review for a total review time of 12 or 8 months, respectively. The FDA does not always meet its PDUFA goal dates for standard and priority reviews. The review process and the PDUFA goal date may also be extended by three months if the FDA so requests or if the applicant otherwise provides additional
82
information or clarification regarding information already provided in the submission which may be deemed as substantial information.
After the FDA’s evaluation of the application and accompanying information, including the results of any potential inspections of the manufacturing facilities and any FDA audits of clinical trial sites to assure compliance with GCPs, the FDA will issue an approval letter, or a complete response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. Under the PHSA, the FDA may approve a BLA if it determines that the product is safe, pure and potent and the facility where the product will be manufactured meets standards designed to ensure that it continues to be safe, pure and potent. If the application is not approved, the FDA will issue a complete response letter, which will identify the deficiencies in the application and the conditions that must be met in order to secure approval of the application, and when possible, will outline recommended actions the sponsor might take to obtain approval of the application. Sponsors that receive a complete response letter may submit to the FDA information that represents a complete response to the issues identified by the FDA, withdraw the application or request a hearing. The FDA will not approve an application until issues identified in the complete response letter have been addressed.
The FDA may also refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. In particular, the FDA may refer applications for novel biological products or biological products that present difficult questions of safety or efficacy to an advisory committee. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
If the FDA approves a new product, it may limit the approved indications for use of the product. It may also require that contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may call for post-approval studies, including Phase 4 clinical trials, to further assess the product’s safety after approval. The agency may also require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, including REMS, to help ensure that the benefits of the product outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals and elements to assure safe use (ETASU). ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patent registries. The FDA may prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs.
After approval, many types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Such regulatory reviews can result in denial or modification of the planned changes, or requirements to conduct additional tests or evaluations that can substantially delay or increase the cost of the planned changes.
Fast Track, Breakthrough Therapy and Priority Review Designations
The FDA is authorized to designate certain products for expedited review if they are intended to address an unmet medical need in the treatment of a serious or life-threatening disease or condition. These programs are referred to as fast track designation, breakthrough therapy designation and priority review designation.
The FDA may designate a product for fast track review if it is intended, whether alone or in combination with one or more other products, for the treatment of a serious or life-threatening disease or condition, and it demonstrates the potential to address unmet medical needs for such a disease or condition. For fast track products, sponsors may have greater interactions with the FDA and the FDA may initiate review of sections of a fast track product’s application before the application is complete. This rolling review may be available if the FDA determines, after preliminary evaluation of clinical data submitted by the sponsor, that a fast track product may be effective. The sponsor must also provide, and the FDA must approve, a schedule for the submission of the remaining information and the sponsor must
83
pay applicable user fees. However, the FDA’s goal for reviewing a rolling review does not begin until the last section of the application is submitted. Fast track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
A product may be designated as a breakthrough therapy if it is intended, either alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA may take certain actions with respect to breakthrough therapies, including holding meetings with the sponsor throughout the development process; providing timely advice to the product sponsor regarding development and approval; involving more senior staff in the review process; assigning a cross-disciplinary project lead for the review team; and taking other steps to design the clinical trials in an efficient manner.
The FDA may designate a product for priority review if it is a product that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. The FDA determines, on a case-by-case basis, whether the proposed product represents a significant improvement when compared with other available therapies. Significant improvement may be illustrated by evidence of increased effectiveness in the treatment of a condition, elimination or substantial reduction of a treatment-limiting product reaction, documented enhancement of patient compliance that may lead to improvement in serious outcomes and evidence of safety and effectiveness in a new subpopulation. A priority designation is intended to direct overall attention and resources to the evaluation of such applications, and to shorten the FDA’s goal for taking action on a marketing application from 10 months to 6 months.
Accelerated Approval Pathway
The FDA may grant accelerated approval to a product for a serious or life-threatening condition that provides meaningful therapeutic advantage to patients over existing treatments based upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on a clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality (IMM) and that is reasonably likely to predict an effect on IMM or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Products granted accelerated approval must meet the same statutory standards for safety and effectiveness as those granted traditional approval.
For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radio-graphic image, physical sign, or other measure that is thought to predict clinical benefit but is not itself a measure of clinical benefit. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. An intermediate clinical endpoint is a measurement of a therapeutic effect that is considered reasonably likely to predict the clinical benefit of a product, such as an effect on IMM. The FDA has limited experience with accelerated approvals based on intermediate clinical endpoints, but has indicated that such endpoints generally may support accelerated approval where the therapeutic effect measured by the endpoint is not itself a clinical benefit and basis for traditional approval, if there is a basis for concluding that the therapeutic effect is reasonably likely to predict the ultimate clinical benefit of a product.
The accelerated approval pathway is most often used in settings in which the course of a disease is long and an extended period of time is required to measure the intended clinical benefit of a product, even if the effect on the surrogate or intermediate clinical endpoint occurs rapidly. Thus, accelerated approval has been used extensively in the development and approval of products for treatment of a variety of cancers in which the goal of therapy is generally to improve survival or decrease morbidity and the duration of the typical disease course requires lengthy and sometimes large clinical trials to demonstrate a clinical or survival benefit.
The accelerated approval pathway is usually contingent on a sponsor’s agreement to conduct, in a diligent manner, a post-approval confirmatory clinical trial or studies to verify and describe the product’s clinical benefit. As a result, a product candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. These confirmatory clinical trials must be completed with due diligence, and the FDA may require that the confirmatory
84
clinical trial be designed, initiated, and/or fully enrolled prior to approval. Failure to conduct required post-approval studies, or confirm a clinical benefit during post-marketing studies, would allow the FDA to withdraw the product from the market on an expedited basis. Unless otherwise informed by the FDA, all promotional materials for product candidates approved under accelerated regulations are subject to prior review by the agency. FDORA, enacted in December 2022, included provisions related to the accelerated approval pathway. Pursuant to FDORA, the FDA is authorized to require a post-approval clinical trial to be underway prior to approval or within a specified time period following approval. FDORA also requires the FDA to specify the conditions of any required post-approval clinical trial not later than 180 days following approval and not less frequently than every 180 days thereafter until completion or termination of such clinical trial. Such conditions may include imposing milestones such as a target date of clinical trial completion or requiring sponsors to submit progress reports. FDORA also enables the FDA to initiate enforcement actions or criminal prosecutions for the failure to conduct with due diligence a required post-approval clinical trial, including a failure to meet any required conditions specified by the FDA or to submit timely reports.
Orphan Drug Designation
Orphan drug designation in the U.S. is designed to encourage sponsors to develop products intended for rare diseases or conditions. In the U.S., a rare disease or condition is statutorily defined as a condition that affects fewer than 200,000 individuals in the U.S. or that affects more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available the product for the disease or condition will be recovered from sales of the product in the U.S.
Orphan drug designation qualifies a company for tax credits and market exclusivity for seven years following the date of the product’s marketing approval if granted by the FDA and if it is the first FDA approval for that product for the disease for which it has such designation. An application for designation as an orphan product can be made any time prior to the filing of an application for approval to market the product. If the FDA grants orphan drug designation, the generic identity of the product and its potential orphan use are disclosed publicly by the FDA. The product must then go through the review and approval process like any other product in order to be marketed.
A sponsor may request orphan drug designation of a previously unapproved product or new orphan indication for an already marketed product. In addition, a sponsor of a product that is otherwise the same product as an already approved orphan drug may seek and obtain orphan drug designation for the subsequent product for the same rare disease or condition if it can present a plausible hypothesis that its product may be clinically superior to the first drug. Whether a large molecule product (i.e., a biological product) is the same as another product is based on whether the two products have the same principal molecular structural features. More than one sponsor may receive orphan drug designation for the same product for the same rare disease or condition, but each sponsor seeking orphan drug designation must file a complete request for designation.
If orphan drug exclusivity is granted by the FDA, the period of exclusivity begins on the date that the marketing application is approved by the FDA and applies only to the indication for which the product has been designated. The FDA may approve a second application for the same product for a different use or a second application for a clinically superior version of the product for the same use. The FDA cannot, however, approve the same product made by another sponsor for the same indication during the market exclusivity period unless it has the consent of the sponsor or the sponsor is unable to provide sufficient quantities of the product.
Post-Approval Regulation
If regulatory approval for marketing of a product or new indication for an existing product is obtained, the sponsor will be required to comply with all post-approval regulatory requirements as well as any post-approval requirements that the FDA has imposed as part of the approval process. The sponsor will be required to report certain adverse reactions and production problems to the FDA, provide updated safety and efficacy information and comply with requirements concerning advertising and promotional labeling. Manufacturers and other parties involved in the drug supply chain for prescription drug and biological products must also comply with product tracking and tracing requirements and for notifying the FDA of counterfeit, diverted, stolen and intentionally adulterated products or products that are otherwise unfit for distribution in the U.S. Manufacturers and certain of their subcontractors are required to
85
register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements upon manufacturers. Accordingly, the sponsor and its third-party manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with cGMPs and other regulatory requirements.
A biological product may also be subject to official lot release, meaning that the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official lot release, the manufacturer must submit samples of each lot, together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot, to the FDA. The FDA may in addition perform certain confirmatory tests on lots of some products before releasing the lots for distribution. Finally, the FDA will conduct laboratory research related to the safety, purity, potency and effectiveness of pharmaceutical products. Any distribution of prescription biological products and pharmaceutical samples must comply with the U.S. Prescription Drug Marketing Act and the PHSA.
Once an approval is granted, the FDA may revoke or suspend the approval of the BLA if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. FDA also has authority to require post-market studies, in certain circumstances, on reduced effectiveness of a product and may require labeling changes related to new reduced effectiveness information. Other potential consequences for a failure to maintain regulatory compliance include, among other things:
| ● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● | fines, untitled letters or warning letters or holds on post-approval clinical trials; |
| ● | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; or |
| ● | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Pharmaceutical products may be promoted only for the approved indications and in accordance with the provisions of the approved labeling. Although physicians may prescribe legally available products for unapproved uses or in patient populations that are not described in the product’s approved labeling (known as “off-label use”), companies with approved products may not market or promote such off-label uses. The FDA does not regulate the behavior of physicians in their choice of treatments, but the FDA regulations do impose stringent restrictions on manufacturers’ communications regarding off-label uses. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including investigation by federal and state authorities. Prescription biological product promotional materials must be submitted to the FDA in conjunction with their first use or first publication.
Pediatric Studies and Exclusivity
Under the Pediatric Research Equity Act of 2003 (as amended, PREA), a BLA or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the product for the claimed indications in all relevant pediatric sub-populations, and to support dosing and administration for each pediatric subpopulation for which
86
the product is safe and effective. Sponsors must also submit pediatric clinical trial plans prior to the assessment data. Those plans must contain an outline of the proposed pediatric clinical trial or studies the applicant plans to conduct, including clinical trial objectives and design, any deferral or waiver requests and other information required by regulation. The applicant, the FDA, and the FDA’s internal review committee must then review the information submitted, consult with each other and agree upon a final plan. The FDA or the applicant may request an amendment to the plan at any time.
The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements. Unless otherwise required by regulation, PREA does not apply to a biologic for an indication for which orphan designation has been granted, except that PREA will apply to an original BLA for a new active ingredient that is orphan-designated if the biologic is a molecularly targeted cancer product intended for the treatment of an adult cancer and is directed at a molecular target that FDA determines to be substantially relevant to the growth or progression of a pediatric cancer.
Pediatric exclusivity is another type of non-patent marketing exclusivity in the U.S. and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity. This six-month exclusivity may be granted if a BLA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity cover the product are extended by six months.
Biosimilars and Exclusivity
The Biologics Price Competition and Innovation Act (BPCIA) established a regulatory scheme authorizing the FDA to approve biosimilars and interchangeable biosimilars.
Under the BPCIA, an applicant may submit an application for licensure of a biologic product that is “biosimilar to” or “interchangeable with” a previously approved biological product or “reference product.” For the FDA to approve a biosimilar product, it must find that there are no clinically meaningful differences between the reference product and proposed biosimilar product in terms of safety, purity and potency. For the FDA to approve a biosimilar product as interchangeable with a reference product, the agency must find that the biosimilar product can be expected to produce the same clinical results as the reference product, and (for products administered multiple times) that the biologic and the reference biologic may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic.
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date of approval of the reference product. The FDA may not approve a biosimilar product until 12 years from the date on which the reference product was approved. Even if a product is considered to be a reference product eligible for exclusivity, another company could market a competing version of that product if the FDA approves a full BLA for such product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of their product. However, to rely on such exclusivities for establishing or protecting our market position is not without risk, as such laws are subject to changes by the legislature. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products. Products deemed interchangeable by the FDA may be substituted by pharmacies as dictated by individual state law.
U.S. Patent Term Restoration
Depending upon the timing, duration and specifics of FDA approval of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Act that permits restoration of the patent term of up to five years as compensation for patent term lost during the FDA regulatory review process. Patent-term restoration, however, cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date and only those claims covering such approved product, a method for using it or a method for
87
manufacturing it may be extended. The patent-term restoration period is generally one-half the time between the effective date of an IND and the submission date of a BLA plus the time between the submission date of a BLA and the approval of that application, except that the review period is reduced by any time during which the applicant failed to exercise due diligence. Only one patent applicable to an approved biologic is eligible for the extension and the application for the extension must be submitted within sixty (60) days of approval from FDA and prior to the expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we may apply for restoration of patent term for our currently owned or licensed patents to add patent life beyond its current expiration date, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant BLA.
Regulation and Procedures Governing Approval of Medicinal Products in the EU and the UK
In order to market any medicinal product outside of the U.S., a company also must comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of products. Whether or not it obtains FDA approval for a product, an applicant will need to obtain the necessary approvals by the comparable regulatory authorities before it can initiate clinical trials or marketing of the product in those countries or jurisdictions. Specifically, with respect to the EU, no medicinal product may be placed on the market of an EU Member State unless a marketing authorization has been issued by the competent authorities of that member state in accordance with Directive 2001/83/EC or a centralized marketing authorization has been granted in accordance with Regulation (EC) No 726/2004, read in conjunction with Regulation (EC) No 1901/2006 and Regulation (EC) No 1394/2007. Similar requirements apply in Great Britain. The process governing approval of medicinal products in the EU and Great Britain generally follows the same lines as in the U.S. It entails satisfactory completion of pharmaceutical development, pre-clinical trials and adequate and well-controlled clinical trials to establish the safety and efficacy of the medicinal product for each proposed indication. The EU also requires the submission to relevant competent authorities for clinical trials authorization and to the EMA or to competent authorities in EU Member States of a marketing authorization application (MAA) and granting of such MAA by these authorities before the product can be marketed and sold in the EU. Following the UK’s departure from the EU, a separate MAA is required in order to place medicinal products on the market in the Great Britain (England, Wales and Scotland) (under the Northern Ireland Protocol, the EU regulatory framework will continue to apply in Northern Ireland in this regard). Centralized EU marketing authorizations continue to be recognized, with new International Recognition Procedures anticipated.
Clinical Trial Approval
On January 31, 2022, the new Clinical Trials Regulation (EU) No 536/2014 (CTR) entered into application, and replaced the Clinical Trials Directive 2001/20/EC. The transitional provisions of the new CTR offered sponsors the possibility to choose between the requirements of the previous Directive and the new CTR if the request for authorization of a clinical trial was submitted by January 30, 2023. If the sponsor chose to submit under the previous Directive, the clinical trial continues to be governed by the Directive until three years after the new CTR became applicable (i.e., January 30, 2025). If a clinical trial continues for more than three years after the Regulation became applicable, the new CTR will at that time begin to apply to the clinical trial (i.e., from January 31, 2025). The new Regulation, which is directly applicable in all EU Member States, aims to simplify and streamline the approval of clinical trials in the EU. The main characteristics of the new CTR include: a streamlined application procedure via a single-entry point through the Clinical Trials Information System; a single set of documents to be prepared and submitted for the application as well as simplified reporting procedures for clinical trial sponsors; and a harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts (Part I contains scientific and medicinal product documentation and Part II contains the national and patient-level documentation). Part I is assessed by a coordinated review by the competent authorities of all EU Member States in which an application for authorization of a clinical trial has been submitted (Concerned Member States) of a draft report prepared by a reference member state. Part II is assessed separately by each Concerned Member State. Strict deadlines have also been established for the assessment of CTAs.
Prior to its exit from the EU, the UK implemented Directive 2001/20/EC into national law through the Medicines for Human Use (Clinical Trials) Regulations 2004 (as amended). However, implementation of the new EU
88
CTR took place after the UK’s departure from the EU, and so the new CTR described in the preceding paragraph does not apply to Great Britain. The MHRA, the UK medicines regulator, ran a consultation on reforms to the UK clinical trials legislation, which closed in March 2022. The outcome of that consultation was published in March 2023 and includes proposals to reform the clinical trials legislative framework, although content and timeline for reform are not yet determined. The future regulatory framework for clinical trials in the UK therefore remains uncertain.
Orphan Designation and Exclusivity
Regulation (EC) No. 141/2000 and Regulation (EC) No. 847/2000 provide that a product can be designated as an orphan drug by the EU Commission if its sponsor can establish: (1) that the product is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition, (2) either (i) the prevalence of the condition is not more than five in ten thousand persons in the EU when the application is made, or (ii) without incentives it is unlikely that the marketing of the product in the EU would generate sufficient return to justify the necessary investment in its development and (3) there exists no satisfactory method of diagnosis, prevention, or treatment of the condition in question that has been authorized in the EU or, if such method exists, the product has to be of a significant benefit compared to products available for the condition.
An orphan designation provides a number of benefits, including fee reductions and, regulatory assistance. If a marketing authorization is granted for an orphan medicinal product, this results in a ten-year period of market exclusivity. During this market exclusivity period, neither the EMA, the European Commission nor the EU Member States can accept an application or grant a marketing authorization for a “similar medicinal product.” A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. The market exclusivity period for the authorized therapeutic indication may, however, be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan designation because, for example, the product is sufficiently profitable not to justify market exclusivity. Market exclusivity may also be revoked in very select cases, such as if (i) it is established that a similar medicinal product is safer, more effective or otherwise clinically superior; (ii) the marketing authorization holder for the authorized orphan product consents to the second orphan application; or (iii) the marketing authorization holder for the authorized orphan product cannot supply enough orphan medicinal products. Orphan designation must be requested before submitting an application for marketing approval. Orphan designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
Since January 1, 2021, a separate process for orphan designation has applied in Great Britain. There is now no pre-marketing authorization orphan designation (as there is in the EU) and the application for orphan designation will be reviewed by the MHRA, at the time of an MAA for a UK or Great Britain marketing authorization. The criteria are the same as in the EU, save that they apply to Great Britain only (e.g., there must be no satisfactory method of diagnosis, prevention or treatment of the condition concerned in Great Britain, as opposed to the EU, and the prevalence of the condition must be no more than five in 10,000 persons in Great Britain).
Marketing Authorization
To obtain a marketing authorization for a product under the EU regulatory system, an applicant must submit an MAA, either to the EMA using the centralized procedure or to competent authorities in the EU using the other procedures (decentralized procedure, national procedure, or mutual recognition procedure). A marketing authorization may be granted only to an applicant established in the EU. Regulation (EC) No. 1901/2006 provides that prior to obtaining a marketing authorization in the EU, an applicant must demonstrate compliance with all measures included in an EMA-approved pediatric investigation plan (PIP), covering all subsets of the pediatric population, unless the EMA has granted a product-specific waiver, class waiver, or a deferral for one or more of the measures included in the PIP.
The centralized procedure provides for the grant of a single marketing authorization by the EU Commission that is valid for all EEA Member States. Pursuant to Regulation (EC) No. 726/2004, the centralized procedure is compulsory for specific products, including for medicines produced by certain biotechnological processes, products designated as orphan medicinal products, advanced therapy medicinal products (gene therapy, somatic cell therapy or
89
tissue engineered products) and products with a new active substance indicated for the treatment of certain diseases, including products for the treatment of cancer and auto-immune diseases and other immune dysfunctions and neurodegenerative disorders. The centralized procedure is optional for products that contain a new active substance for any other indications, which are a significant therapeutic, scientific or technical innovation and whose authorization would be in the interest of public health in the EU.
Under the centralized procedure, the EMA’s Committee for Medicinal Products for Human Use (CHMP) is responsible for conducting the assessment of a product to define its risk/benefit profile. The CHMP recommendation is then sent to the EU Commission, which adopts a decision binding in all EEA Member States. Under the centralized procedure, the maximum timeframe for the evaluation of an MAA is 210 days, excluding clock stops when additional information or written or oral explanation is to be provided by the applicant in response to questions asked by the CHMP. Clock stops may extend the timeframe of evaluation of an MAA considerably beyond 210 days. Accelerated evaluation may be granted by the CHMP in exceptional cases, when a medicinal product is of major interest from the point of view of public health and, in particular, from the viewpoint of therapeutic innovation. If the CHMP accepts such a request, the time limit of 210 days will be reduced to 150 days (excluding clock stops), but it is possible that the CHMP may revert to the standard time limit for the centralized procedure if it determines that it is no longer appropriate to conduct an accelerated assessment.
Following the departure of the UK from the EU, Great Britain is no longer covered by centralized marketing authorizations (under the Northern Ireland Protocol, centralized EU authorizations will continue to be recognized in Northern Ireland). All medicinal products with a current centralized authorization were automatically converted to UK marketing authorizations on January 1, 2021, and there is a further period to December 31, 2023, during which the MHRA may rely on a decision taken by the EU Commission on the approval of a new marketing authorization in the centralized procedure, in order to more quickly grant a new Great Britain marketing authorization European Commission Decision Reliance Procedure (ECDRP). A separate application is, however, still required. The December 31, 2023 date by which the ECDRP was due to draw to a close is currently subject to a public consultation. From January 1, 2024, a new International Recognition Procedure will become available, which is a new licensing route for medicines allowing the UK to recognize approvals from specified Reference Regulators, including the FDA and the EMA and EU member state competent authorities.
European Data and Market Exclusivity
In the EU, innovative medicinal products, approved on the basis of a complete independent data package, qualify for eight years of data exclusivity upon marketing authorization and an additional two years of market exclusivity. The data exclusivity, if granted, prevents generic or biosimilar applicants from referencing the innovator’s preclinical and clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization in the EU, for a period of eight years from the date on which the reference product was first authorized in the EU. During the additional two-year period of market exclusivity, a generic or biosimilar MAA can be submitted, and the innovator’s data may be referenced, but no generic or biosimilar product can be marketed in the EU until the expiration of the market exclusivity period. The overall ten-year period will be extended to a maximum of 11 years if, during the first 8 years of those 10 years, the marketing authorization holder obtains a marketing authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are determined to bring a significant clinical benefit in comparison with currently approved therapies. There is no guarantee that a product will be considered by the EMA to be an innovative medicinal product, and products may not qualify for data exclusivity. Even if a product is considered to be an innovative medicinal product so that the innovator gains the prescribed period of data exclusivity, another company nevertheless could also market another version of the product if such company obtained a marketing authorization based on an MAA with a complete independent data package of pharmaceutical tests, preclinical tests and clinical trials. Similar arrangements apply in the UK.
Periods of Authorization and Renewals
A marketing authorization is valid for five years, in principle, and it may be renewed after five years on the basis of a reevaluation of the risk benefit balance by the EMA for a centrally authorized product, or by the competent
90
authority of the authorizing member state for a nationally authorized product. Once renewed, the marketing authorization is valid for an unlimited period, unless the EU Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal period. Any marketing authorization that is not followed by the placement of the drug on the EU market (in the case of the centralized procedure) or on the market of the authorizing member state (for a nationally authorized product) within three years after authorization, or if the drug is removed from the market for three consecutive years, ceases to be valid. In Great Britain, centrally authorized products converted from EU to UK marketing authorizations will have the same renewal date.
Regulatory Requirements after Marketing Authorization
Following approval, the holder of the marketing authorization is required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of the medicinal product. These include compliance with the EU’s stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. In addition, the manufacturing of authorized products, for which a separate manufacturer’s license is mandatory, must also be conducted in strict compliance with the EMA’s GMP requirements and comparable requirements of other regulatory bodies in the EU, which mandate the methods, facilities and controls used in manufacturing, processing and packing of products to assure their safety and identity. Finally, the marketing and promotion of authorized products, including industry-sponsored continuing medical education and advertising directed toward the prescribers of products and/or the general public, are strictly regulated in the EU under Directive 2001/83/EC, as amended.
The aforementioned EU rules are generally applicable in the EEA, and similar arrangements apply in the UK.
Proposal for new EU Pharmaceutical Legislation
On April 26, 2023, the European Commission has published a proposal for a new directive (COM/2023/192 final) and a new regulation (COM/2023/193 final), which would revise and replace the existing general pharmaceutical legislation, including e.g., Directive 2001/83/EC, as well as Regulations (EC) No. 726/2004, No. 141/2000, or No. 1901/2006 (EU Pharmaceutical Legislation). This proposal is currently undergoing the ordinary legislative procedure in the European Parliament and Council of the European Union and is therefore still subject to changes. If at all, the EU Pharmaceutical Legislation is expected to be implemented at the earliest in the next few years. Prevention and mitigation of medicine shortages, simplification of the market entry of generics and biosimilars, the reduction of the regulatory burden (e.g. by increased digitalization) and the implementation of a new regime for data and / or market exclusivity (e.g. by reducing the minimum period while introducing factors that, if met, prolong protections for marketing authorization holders) are among the major objectives pursued by the European Commission.
Brexit and the Regulatory Framework in the UK
On January 31, 2020, the UK officially withdrew from the EU (Brexit). Provisionally since January 1, 2021 and formally since May 1, 2021, the EU and UK’s trade and cooperation agreement (TCA) includes specific provisions concerning pharmaceuticals, which include the mutual recognition of GMP, inspections of manufacturing facilities for medicinal products and GMP documents issued, but does not foresee wholesale mutual recognition of UK and EU pharmaceutical regulations. The UK has implemented EU legislation on the marketing, promotion and sale of medicinal products through the Human Medicines Regulations 2012 (as amended) and has not yet enacted significant legislative change in this area following its exit from the EU. The regulatory regime in Great Britain therefore largely aligns with current EU regulations. However, these regimes may diverge increasingly as time passes, now that Great Britain’s regulatory system is independent from the EU, and the TCA does not provide for mutual recognition of UK and EU pharmaceutical legislation. For example, as already explained, the new Clinical Trials Regulation which became effective in the EU on January 31, 2022 has not been implemented into UK law, and a separate application will need to be submitted for clinical trial authorization in the UK. Furthermore, the position in Northern Ireland differs in certain respects from that of the rest of the UK (England, Wales and Scotland) as some EU rules continue to be applicable to Northern Ireland following the UK’s departure from the EU.
91
Regulation and Procedures Governing Approval of Medicinal Products in Japan
In order to market any medical products in Japan, a company must comply with numerous and varying regulatory requirements in Japan regarding quality, safety and efficacy in the context, among other things, of clinical trials, marketing approval, commercial sales and distribution of products. A person who manufactures or markets medical products in Japan is subject to the supervision of the MHLW, primarily under the Act on Securing Quality, Efficacy and Safety of Pharmaceuticals and Medical Devices (Pharmaceutical and Medical Devices Act). This entails the satisfactory completion of pharmaceutical development, preclinical studies and adequate and well-controlled clinical trials to establish the safety and efficacy of the medical product for each proposed indication. It also requires the filing of a notification of clinical trials with the Pharmaceuticals and Medical Devices Agency (Japan) (PMDA) and the obtaining of marketing approval from the relevant authorities before the product can be marketed and sold in the Japanese market.
Business License
Under the Pharmaceutical and Medical Devices Act, a person is required to obtain from the MHLW a marketing license in order to conduct the business of marketing, leasing or providing medical products that are manufactured (or outsourced to a third party for manufacturing) or imported by such person.
Also, in order to conduct the business of manufacturing medical products which will be marketed in Japan, a person is required to obtain from the MHLW a manufacturing license for each manufacturing site.
Marketing Approval
Under the Pharmaceutical and Medical Devices Act, it is generally required to obtain marketing approval from the MHLW for the marketing of each medical product. An application for marketing approval must be made through the PMDA, which implements a marketing approval review.
Clinical Trial
Under the Pharmaceutical and Medical Devices Act, it is required to file notification of clinical trials with the PMDA. Also, the data of clinical trials and other pertinent data, which must be attached for an application for marketing approval, must be obtained in compliance with the standards established by the MHLW, such as GLPs and GCPs stipulated by the ministerial ordinances of the MHLW.
Regulatory Requirements after Marketing Approval
A marketing license-holder that has obtained marketing approval for a new pharmaceutical must have that pharmaceutical re-examined by the PMDA for a specified period after receiving marketing approval. Such re-examination period for VYVGART is stated to be 10 years after the marketing approval in January 2022. The purpose of this re-examination process is to ensure the safety and efficacy of a newly approved pharmaceutical by imposing on the marketing license-holder the obligation to gather clinical data for a certain period after the marketing approval was granted so that the PMDA has the opportunity to re-examine the product. Results of usage and other pertinent data must be attached for an application for a re-examination. A marketing license holder that has obtained a marketing approval is also required to investigate, among other things, the results of usage and to periodically report to the PMDA pursuant to the Pharmaceutical and Medical Devices Act.
Price Regulation
In Japan, public medical insurance systems cover virtually the entire Japanese population. The public medical insurance system, however, does not cover any medical product which is not listed on the National Health Insurance (NHI) price list published by the Minister of the MHLW. Accordingly, a marketing license-holder of medical products
92
must first have a new medical product listed on the NHI price list in order to obtain its coverage under the public medical insurance system. The NHI price list listed VYVGART in April 2022.
The NHI price of a medical product is determined either by price comparison of comparable medical products with necessary adjustments for innovativeness, usefulness or size of the market; or, in the absence of comparable medical products, by the cost calculation method, determined after considering of the opinion of the manufacturer. Prices on the NHI price list will be subject to revision, generally once every year, on the basis of the actual prices at which the medical products are purchased by medical institutions.
Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we may obtain regulatory approval. Even if our product candidates are approved for marketing, sales of such product candidates will depend, in part, on the extent to which third-party payors, including government health programs in the U.S. (such as Medicare and Medicaid), commercial health insurers, and managed care organizations, provide coverage and establish adequate reimbursement levels for such product candidates. Moreover, increasing efforts by governmental and third-party payors in the EU, the U.S. and other markets to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our product candidates. We expect to experience pricing pressures in connection with the sale of any of our product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription drugs and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products.
In the U.S. and markets in other countries, patients generally rely on third-party payors to reimburse all or part of the costs associated with their treatment. Adequate coverage and reimbursement from governmental healthcare programs, such as Medicare and Medicaid, and commercial payors is critical to new product acceptance. Patients are unlikely to use any product candidates we may develop unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of such product candidates.
Factors payors consider in determining reimbursement are based on whether the product is (i) a covered benefit under its health plan; (ii) safe, effective and medically necessary; (iii) appropriate for the specific patient; (iv) cost-effective; and (v) neither experimental nor investigational.
The Medicare and Medicaid programs increasingly are used as models for how private payors and other governmental payors develop their coverage and reimbursement policies for drugs and biologics. Some third-party payors may require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse healthcare providers who use such therapies. It is difficult to predict at this time what third-party payors will decide with respect to the coverage and reimbursement for our product candidates. No uniform policy for coverage and reimbursement for drug products exists among third-party payors in the U.S. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance. Each plan determines whether or not it will provide coverage for a product, what amount it will pay the manufacturer for the product, on what tier of its formulary the product will be placed and whether to require step therapy. The position of a product on a formulary generally determines the co-payment that a patient will need to make to obtain the product and can strongly influence the adoption of a product by patients and physicians. Third-party payors may limit coverage to specific products on a formulary, which might not include all of the approved products for a particular indication. Additionally, a payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. For example, the payor may require co-payments that patients find unacceptably high. Further, one payor’s determination to provide coverage for a product does not assure that other payors will also provide coverage and reimbursement for the product, and the level of coverage and reimbursement can differ significantly from payor to payor.
93
Third-party payors are increasingly challenging the price and examining the medical necessity and cost-effectiveness of medical products and services and imposing controls to manage costs, especially drugs when an equivalent generic drug or a less expensive therapy is available. It is possible that a third-party payor may consider our product candidate and other therapies as substitutable and only offer to reimburse patients for the less expensive product. Even if we show improved efficacy or improved convenience of administration with our product candidate, pricing of existing drugs may limit the amount we will be able to charge for our product candidate. These payors may deny or revoke the reimbursement status of a given drug product or establish prices for new or existing marketed products at levels that are too low to enable us to realize an appropriate return on our investment in product development. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize our product candidates and may not be able to obtain a satisfactory financial return on products that we may develop. Third-party payors may limit coverage to specific products on an approved list, also known as a formulary, which might not include all of the approved products for a particular indication.
In Mainland China, the newly created National Healthcare Security Administration (NHSA) an agency responsible for administering Mainland China’s social security system, organized a price negotiation with drug companies for certain new drugs that had not been included in the NRDL at the time of the negotiation in November 2019, which resulted in an average price reduction by over 60% for 70 of the 119 drugs that passed the negotiation. NHSA, together with other government authorities, review the inclusion or removal of drugs from Mainland China’s National Drug Catalog for Basic Medical Insurance, Work-related Injury Insurance and Maternity Insurance, or provincial or local medical insurance catalogues for the national medical insurance program regularly, and the tier under which a drug or device will be classified, both of which affect the amounts reimbursable to program participants for their purchases of those drugs. These determinations are made based on a number of factors, including price and efficacy. We may also be invited to attend the price negotiation with NHSA upon receiving regulatory approval in Mainland China, but we will likely need to significantly reduce our prices, and to negotiate with each of the provincial healthcare security administrations on reimbursement ratios. On the other hand, if the NHSA or any of its local counterpart includes our drugs and devices in the national RDL or provincial RDL, which may increase the demand for our drug candidates and devices, our potential revenue from the sales of our drug candidates and devices may still decrease as a result of lower prices. Moreover, eligibility for reimbursement in Mainland China does not imply that any drug or device will be paid for in all cases or at a rate that covers our costs, including licensing fees, research, development, manufacture, sale and distribution.
Furthermore, rules and regulations regarding reimbursement change frequently, in some cases at short notice, and we believe that changes in these rules and regulations are likely. Outside the U.S., we will face challenges in ensuring obtaining adequate coverage and payment for any product candidates we may develop. Pricing of prescription pharmaceuticals is subject to governmental control in many countries. In order to secure coverage and reimbursement for any product that might be approved for sale, we have needed and may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, and the cost of these studies would be in addition to the costs required to obtain FDA or other comparable marketing approvals. Conducting such studies could be expensive, involve additional risk and result in delays in our commercialization efforts. Even after pharmacogenomic studies are conducted, product candidates may not be considered medically necessary or cost-effective. A decision by a third-party payor not to cover any product candidates we may develop could reduce physician utilization of such product candidates once approved and have a material adverse effect on our sales, results of operations and financial condition. Third-party reimbursement and coverage may not be adequate to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. The insurance coverage and reimbursement status of newly approved products for orphan diseases is particularly uncertain, and failure to obtain or maintain adequate coverage and reimbursement for any such product candidates could limit our ability to generate revenue. Further, due to the COVID-19 pandemic, millions of individuals have lost/will lose employer-based insurance coverage, which may adversely affect our ability to commercialize our products. As noted above, in the U.S., we plan to have various programs to help patients afford our products, including patient assistance programs and co-pay coupon programs for eligible patients. More specifically, patients can enroll into MY VYVGART PATH™, a patient support program that provides personalized support from a nurse case manager and committed support team. In addition to providing support on questions on the treatment and on navigating the insurance process, the program provides a VYVGART Co-pay Program to eligible patients, aids in referring patients to charitable foundations that may be able to help with out-of-pocket costs and informs patients of financial assistance programs that may be available.
94
The containment of healthcare costs also has become a priority of U.S. federal, state and international governments and the prices of pharmaceuticals have been a focus in this effort. Governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the U.S. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that reimbursement will be available for any future product candidate that we commercialize and, if reimbursement is available, the level of reimbursement. In addition, many pharmaceutical manufacturers must calculate and report certain price reporting metrics to the government, such as average sales price and best price. Penalties may apply in some cases when such metrics are not submitted accurately and timely. Further, these prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our potential revenue from the sale of any products for which we may obtain approval. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more of our products for which we or our collaborators receive marketing approval, less favorable coverage policies and reimbursement rates may be implemented in the future. Obtaining and maintaining reimbursement status is time-consuming and costly.
In the EU, pricing and reimbursement schemes vary widely from country to country. Some countries provide that products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies (so called health technology assessments) in order to obtain reimbursement or pricing approval. For example, the EU provides options for its Member States to restrict the range of products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. EU Member States may approve a specific price for a product or may instead adopt a system of direct or indirect controls on the profitability of the company placing the product on the market. Other Member States allow companies to fix their own prices for products but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. Recently, many countries in the EU have increased the amount of discounts required on pharmaceuticals and these efforts could continue as countries attempt to manage healthcare expenditures, especially in light of the severe fiscal and debt crises experienced by many countries in the EU. The downward pressure on healthcare costs in general, particularly prescription products, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EU Member States and parallel trade (arbitrage between low-priced and high-priced Member States) can further reduce prices. Special pricing and reimbursement rules may apply to orphan drugs. Inclusion of orphan drugs in reimbursement systems tend to focus on the medical usefulness, need, quality and economic benefits to patients and the healthcare system as for any drug. Acceptance of any medicinal product for reimbursement may come with cost, use and often volume restrictions, which again can vary by country. In addition, results-based rules of reimbursement may apply. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products, if approved in those countries. Historically, products launched in the EU do not follow price structures of the U.S. and generally prices tend to be significantly lower.
Outside the U.S., international operations are generally subject to extensive governmental price controls and other market regulations, and we believe the increasing emphasis on cost-containment initiatives in Europe, Canada and other countries has and will continue to put pressure on the pricing and usage of our product candidates. In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for medical products but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside the U.S., the reimbursement for our products may be reduced compared with the U.S. and may be insufficient to generate commercially reasonable revenue and profits.
The delivery of healthcare in the EU, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than EU, law and policy.
95
National governments and health service providers have different priorities and approaches to the delivery of healthcare and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most EU Member States have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to commercialize any products for which we obtain marketing approval.
Government Pricing and Reimbursement Programs for Marketed Drugs in the U.S.
Medicaid, the 340B Drug Pricing Program, and Medicare
Federal law requires that a pharmaceutical manufacturer, as a condition of having its drug and biological products receive federal reimbursement under Medicaid and Medicare Part B, must pay rebates to state Medicaid programs for all units of its covered outpatient drugs dispensed to Medicaid beneficiaries and paid for by a state Medicaid program under either a fee-for-service arrangement or through a managed care organization. This federal requirement is effectuated through a Medicaid drug rebate agreement between the manufacturer and the Secretary of HHS. CMS administers the Medicaid drug rebate agreements, which provide, among other things, that the drug manufacturer will pay rebates to each state Medicaid agency on a quarterly basis and report certain price information on a monthly and quarterly basis. The rebates are based on prices reported to CMS by manufacturers for their covered outpatient drugs. For non-innovator products, generally generic drugs marketed under abbreviated NDAs, the rebate amount is 13% of the average manufacturer price (AMP) for the quarter. The AMP is the weighted average of prices paid to the manufacturer (1) directly by retail community pharmacies and (2) by wholesalers for drugs distributed to retail community pharmacies. For innovator products (i.e., drugs that are marketed under NDAs or BLAs), the rebate amount is the greater of 23.1% of the AMP for the quarter or the difference between such AMP and the best price for that same quarter. The best price is essentially the lowest price available to non-governmental entities after accounting for discounts and rebates. Innovator products may also be subject to an additional rebate that is based on the amount, if any, by which the product’s AMP for a given quarter exceeds the inflation-adjusted baseline AMP, which for most drugs is the AMP for the first full quarter after launch. Since 2017, non-innovator products are also subject to an additional rebate. To date, the rebate amount for a drug has been capped at 100% of the AMP; however, effective January 1, 2024, this cap will be eliminated, which means that a manufacturer could pay a total rebate amount on a unit of the drug that is greater than the average price the manufacturer receives for the drug.
The terms of participation in the Medicaid drug rebate program impose an obligation to correct the prices reported in previous quarters, as may be necessary. Any such corrections could result in additional or lesser rebate liability, depending on the direction of the correction. In addition to retroactive rebates, if a manufacturer were found to have knowingly submitted false information to the government, federal law provides for civil monetary penalties for failing to provide required information, late submission of required information, and false information.
A manufacturer must also participate in a federal program known as the 340B drug pricing program in order for federal funds to be available to pay for the manufacturer’s drug and biological products under Medicaid and Medicare Part B. Under this program, the participating manufacturer agrees to charge certain safety net healthcare providers no more than an established discounted price for its covered outpatient drugs. The formula for determining the discounted price is defined by statute and is based on the AMP and the unit rebate amount as calculated under the Medicaid drug rebate program, discussed above. Manufacturers are required to report pricing information to the Health Resources and Services Administration (HRSA) on a quarterly basis. HRSA has also issued regulations relating to the calculation of the ceiling price as well as imposition of civil monetary penalties for each instance of knowingly and intentionally overcharging a 340B covered entity.
Federal law also requires that manufacturers report data on a quarterly basis to CMS regarding the pricing of drugs that are separately reimbursable under Medicare Part B. These are generally drugs and biologics, such as injectable products, that are administered incident to a physician service and are not generally self-administered. The pricing information submitted by manufacturers is the basis for reimbursement to physicians and suppliers for drugs covered under Medicare Part B. Under the IRA, manufacturers are also required to provide quarterly rebates for certain single-source drugs and biologics (including biosimilars) covered under Medicare Part B with prices that increase faster than
96
the rate of inflation. This requirement started on January 1, 2023 for drugs approved on or before December 1, 2020 and begins six quarters after a drug is first marketed for all other drugs. As with the Medicaid drug rebate program, federal law provides for civil monetary penalties for failing to provide required information, late submission of required information, and false information.
Recently, the Infrastructure Investment and Jobs Act added a requirement, effective January 1, 2023, for manufacturers of certain single-source drugs (including biologics and biosimilars) separately paid for under Medicare Part B for at least 18 months and marketed in single-dose containers or packages (known as refundable single-dose containers or single-use package drugs) to provide annual refunds for any portions of the dispensed drug that are unused and discarded if those unused or discarded portions exceed an applicable percentage defined by statute or regulation. Manufacturers will be subject to periodic audits and those that fail to pay refunds for their refundable single-dose containers or single-use package drugs shall be subject to civil monetary penalties.
Medicare Part D provides prescription drug benefits for seniors and people with disabilities. Medicare Part D enrollees once had a gap in their coverage (between the initial coverage limit and the point at which catastrophic coverage begins) where Medicare did not cover their prescription drug costs, known as the coverage gap. However, beginning in 2019, Medicare Part D enrollees paid 25% of brand drug costs after they reached the initial coverage limit - the same percentage they were responsible for before they reached that limit - thereby closing the coverage gap from the enrollee’s point of view. Most of the cost of closing the coverage gap is being borne by innovator companies and the government through subsidies. Each manufacturer of drugs approved under NDAs or BLAs is required to enter into a Medicare Part D coverage gap discount agreement and provide a 70% discount on those drugs dispensed to Medicare Part D enrollees in the coverage gap, in order for its drugs to be reimbursed by Medicare Part D. Beginning in 2025, the IRA eliminates the coverage gap under Medicare Part D by significantly lowering the enrollee maximum out-of-pocket cost and requiring manufacturers to subsidize, through a newly established manufacturer discount program, 10% of Part D enrollees’ prescription costs for brand drugs above a deductible and below the out-of-pocket maximum, and 20% once the out-of-pocket maximum has been reached. Although these discounts represent a lower percentage of enrollees’ costs than the current discounts required below the out-of-pocket maximum (that is, in the coverage gap phase of Part D coverage), the new manufacturer contribution required above the out-of-pocket maximum could be considerable for very high-cost patients and the total contributions by manufacturers to a Part D enrollee’s drug expenses may exceed those currently provided. The IRA also requires manufacturers to provide annual Medicare Part D rebates for single-source drugs and biological products with prices that increase faster than the rate of inflation.
The IRA also allows HHS to directly negotiate the selling price of a statutorily specified number of drugs and biologics each year that CMS reimburses under Medicare Part B and Part D. Only high-expenditure single-source biologics that have been approved for at least 11 years (7 years for single-source drugs) can qualify for negotiation, with the negotiated price taking effect two years after the selection year. Negotiations for Medicare Part D products begin in 2024 with the negotiated price taking effect in 2026, and negotiations for Medicare Part B products begin in 2026 with the negotiated price taking effect in 2028. In August 2023, HHS announced the 10 Medicare Part D drugs and biologics that it selected for negotiations, and by October 1, 2023, each manufacturer of the selected drugs signed a manufacturer agreement to participate in the negotiations. HHS will announce the negotiated maximum fair price by September 1, 2024, and this price cap, which cannot exceed a statutory ceiling price will come into effect on January 1, 2026.
U.S. Federal Contracting and Pricing Requirements
Manufacturers are also required to make their covered drugs, which are generally drugs approved under NDAs or BLAs, available to authorized users of the Federal Supply Schedule (FSS) of the General Services Administration. The law also requires manufacturers to offer deeply discounted FSS contract pricing for purchases of their covered drugs by the Department of Veterans Affairs, the Department of Defense, the Coast Guard, and the Public Health Service (including the Indian Health Service) in order for federal funding to be available for reimbursement or purchase of the manufacturer’s drugs under certain federal programs. FSS pricing to those four federal agencies for covered drugs must be no more than the Federal Ceiling Price (FCP), which is at least 24% below the Non-Federal Average Manufacturer Price (Non-FAMP) for the prior year. The Non-FAMP is the average price for covered drugs sold to wholesalers or other middlemen, net of any price reductions.
97
The accuracy of a manufacturer’s reported Non-FAMPs, FCPs, or FSS contract prices may be audited by the government. Among the remedies available to the government for inaccuracies is recoupment of any overcharges to the four specified federal agencies based on those inaccuracies. If a manufacturer were found to have knowingly reported false prices, in addition to other penalties available to the government, the law provides for significant civil monetary penalties per incorrect item. Finally, manufacturers are required to disclose in FSS contract proposals all commercial pricing that is equal to or less than the proposed FSS pricing, and subsequent to award of an FSS contract, manufacturers are required to monitor certain commercial price reductions and extend commensurate price reductions to the government, under the terms of the FSS contract Price Reductions Clause. Among the remedies available to the government for any failure to properly disclose commercial pricing and/or to extend FSS contract price reductions is recoupment of any FSS overcharges that may result from such omissions.
Healthcare Law and Regulation
Healthcare providers and third-party payors play a primary role in the recommendation and prescription of pharmaceutical products that are granted marketing approval. Our current and future arrangements with providers, researchers, consultants, third-party payors and customers are subject to broadly applicable federal and state fraud and abuse, anti-kickback, false claims, transparency and patient privacy laws and regulations and other healthcare laws and regulations that may constrain our business and/or financial arrangements. Restrictions under applicable federal and state healthcare laws and regulations include, without limitation, the following:
| ● | the AKS prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering, or paying remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. A person or entity can be found guilty of violating the AKS without actual knowledge of the statute or specific intent to violate it. In addition, the government may assert that a claim including items or services resulting from a violation of the AKS constitutes a false or fraudulent claim for purposes of the federal False Claims Act or federal civil money penalties statute. Violations of the AKS carry potentially significant civil and criminal penalties, including imprisonment, fines, administrative civil monetary penalties, and exclusion from participation in federal healthcare programs. On December 2, 2020, the Office of Inspector General (OIG) published further modifications to the AKS. Under the final rules, OIG added safe harbor protections under the AKS for certain coordinated care and value-based arrangements among clinicians, providers, and others. This rule became effective January 19, 2021. We continue to evaluate what effect, if any, the rule will have on our business; |
| ● | the U.S. federal false claims and civil monetary penalties laws, including the civil False Claims Act and federal civil monetary penalty laws, which, among other things, impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the U.S. federal government, claims for payment or approval that are false or fraudulent, knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim or obligation to pay or transmit money to the federal government, or from knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the U.S. federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the AKS constitutes a false or fraudulent claim for purposes of the False Claims Act. Manufacturers can be held liable under the False Claims Act even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. The False Claims Act also permits a private individual acting as a “whistleblower” to bring qui tam actions on behalf of the federal government alleging violations of the False Claims Act and to share in any monetary recovery. When an entity is determined to have violated the federal civil False Claims Act, the government may impose civil fines and penalties for each false claim, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs; |
98
| ● | the U.S. federal Health Insurance Portability and Accountability Act of 1996 (HIPAA) which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or obtaining by means of false or fraudulent pretenses, representations, or promises, any of the money or property owned by, or under the custody or control of, any healthcare benefit program, regardless of the pay (e.g., public or private) or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for, healthcare benefits, items or services relating to healthcare matters; similar to the AKS, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 (HITECH) and its implementing regulations, and as amended again by the Omnibus Rule in 2013, which imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information without appropriate authorization by covered entities subject to the Final HIPAA Omnibus Rule, i.e., certain covered health plans, healthcare clearinghouses and healthcare providers, as well as their business associates, those independent contractors or agents of covered entities that perform certain services for or on their behalf involving the use or disclosure of individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions; |
| ● | the federal transparency requirements known as the federal Physician Payments Sunshine Act, under the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010 (collectively, the ACA), which requires certain manufacturers of drugs, devices, biologics and medical supplies to report annually to CMS information related to payments and other transfers of value made by that entity to physicians (currently defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain non-physician providers such as physician assistants and nurse practitioners and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in civil monetary penalties for all payments, transfers of value or ownership or investment interests that are not timely, accurately, and completely reported in an annual submission; |
| ● | federal government price reporting laws, which require us to calculate and report complex pricing metrics in an accurate and timely manner to government programs; |
| ● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
| ● | analogous state laws and regulations, including: state anti-kickback and false claims laws, which may apply to our business practices, including, but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state and local laws that require the licensure of sales representatives; state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures and pricing information; state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect; and state laws related to insurance fraud in the case of claims involving private insurers; and |
99
| ● | EU, UK and other foreign law equivalents, including reporting requirements detailing interactions with and payments to healthcare providers and data privacy and security laws and regulations that may be more stringent than those in the U.S. |
Some state laws require pharmaceutical companies to comply with the April 2003 Office of Inspector General Compliance Program Guidance for Pharmaceutical Manufacturers and/or the Pharmaceutical Research and Manufacturers of America’s Code on Interactions with Healthcare Professionals, in addition to requiring pharmaceutical manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures. Several states also impose other marketing restrictions or require pharmaceutical companies to make marketing or price disclosures to the state and require the registration of pharmaceutical sales representatives. State and foreign laws, including for example the GDPR, also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. There are ambiguities as to what is required to comply with these state requirements and if we fail to comply with an applicable state law requirement, we could be subject to penalties.
We have and will continue to spend substantial time and money to ensure that our business arrangements with third parties comply with applicable healthcare laws and regulations. Recent healthcare reform legislation has strengthened these federal and state healthcare laws. Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws.
Other laws that may affect our ability to operate include:
| ● | the anti-inducement law prohibits, among other things, the offering or giving of remuneration, which includes, without limitation, any transfer of items or services for free or for less than fair market value (with limited exceptions), to a Medicare or Medicaid beneficiary that the person know or should know is likely to influence the beneficiary’s selection of a particular supplier of items or services reimbursable by a federal or state governmental program; and |
| ● | European and other foreign law equivalents of each of the laws, including reporting requirements detailing interactions with and payments to healthcare providers. |
In the U.S., to help patients afford our approved product, we may utilize programs to assist them, including patient assistance programs and co-pay coupon programs for eligible patients. Government enforcement agencies have shown increased interest in pharmaceutical companies’ product and patient assistance programs, including reimbursement support services, and a number of investigations into these programs have resulted in significant civil and criminal settlements. In addition, at least one insurer has directed its network pharmacies to no longer accept co-pay coupons for certain specialty drugs the insurer identified. Our co-pay coupon programs could become the target of similar insurer actions. In addition, in November 2013, the CMS issued guidance to the issuers of qualified health plans sold through the ACA’s marketplaces encouraging such plans to reject patient cost-sharing support from third parties and indicating that the CMS intends to monitor the provision of such support and may take regulatory action to limit it in the future. The CMS subsequently issued a rule requiring individual market qualified health plans to accept third-party premium and cost-sharing payments from certain government-related entities. In September 2014, the OIG of the HHS issued a Special Advisory Bulletin warning manufacturers that they may be subject to sanctions under the AKS and/or civil monetary penalty laws if they do not take appropriate steps to exclude Part D beneficiaries from using co-pay coupons. Accordingly, companies exclude these Part D beneficiaries from using co-pay coupons. It is possible that changes in insurer policies regarding co-pay coupons and/or the introduction and enactment of new legislation or regulatory action could restrict or otherwise negatively affect these patient support programs, which could result in fewer patients using affected products, and therefore could have a material adverse effect on our sales, business, and financial condition.
Third-party patient assistance programs that receive financial support from companies have become the subject of enhanced government and regulatory scrutiny. The OIG has established guidelines that suggest that it is lawful for pharmaceutical manufacturers to make donations to charitable organizations who provide co-pay assistance to Medicare
100
patients, provided that such organizations, among other things, are bona fide charities, are entirely independent of and not controlled by the manufacturer, provide aid to applicants on a first-come basis according to consistent financial criteria and do not link aid to use of a donor’s product. However, donations to patient assistance programs have received some negative publicity and have been the subject of multiple government enforcement actions, related to allegations regarding their use to promote branded pharmaceutical products over other less costly alternatives. Specifically, in recent years, there have been multiple settlements resulting out of government claims challenging the legality of their patient assistance programs under a variety of federal and state laws. It is possible that we may make grants to independent charitable foundations that help financially needy patients with their premium, co-pay, and co-insurance obligations. If we choose to do so, and if we or our vendors or donation recipients are deemed to fail to comply with relevant laws, regulations or evolving government guidance in the operation of these programs, we could be subject to damages, fines, penalties, or other criminal, civil, or administrative sanctions or enforcement actions. We cannot ensure that our compliance controls, policies, and procedures will be sufficient to protect against acts of our employees, business partners, or vendors that may violate the laws or regulations of the jurisdictions in which we operate. Regardless of whether we have complied with the law, a government investigation could impact our business practices, harm our reputation, divert the attention of management, increase our expenses, and reduce the availability of foundation support for our patients who need assistance.
On November 30, 2020, the HHS published a regulation removing safe harbor protection for price reductions from pharmaceutical manufacturers to plan sponsors under Part D, either directly or through pharmacy benefit managers (PBMs), unless the price reduction is required by law. The rule also creates a new safe harbor for price reductions reflected at the point-of-sale, as well as a safe harbor for certain fixed fee arrangements between PBMs and manufacturers. The IRA delayed implementation of this change and new safe harbors for point-of-sale reductions in price for prescription pharmaceutical products and PBM service fees until January 1, 2032. Further, on December 31, 2020, CMS published a new rule, effective January 1, 2023, requiring manufacturers to ensure the full value of co-pay assistance is passed on to the patient or these dollars will count toward the AMP and Best Price calculation of the drug. On May 21, 2021, PhRMA sued the HHS in the U.S. District Court for the District of Columbia, to stop the implementation of the rule claiming that the rule contradicts federal law surrounding Medicaid rebates, and on May 17, 2022, the court vacated the rule.
Violations of these laws or any future enacted laws can subject us to criminal, civil and administrative sanctions including monetary penalties, damages, fines, disgorgement, individual imprisonment and exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, reputational harm, and we may be required to curtail or restructure our operations. Moreover, we expect that there will continue to be federal and state laws and regulations, proposed and implemented, that could impact our future operations and business.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. Ensuring that our internal operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to significant penalties, including administrative, civil and criminal penalties, damages, fines, disgorgement, the exclusion from participation in federal and state healthcare programs, individual imprisonment, reputational harm, and the curtailment or restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. Further, defending against any such actions can be costly and time-consuming, and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired. If any of the physicians or other providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs and imprisonment. If any of the above occur, our ability to operate our business and our results of operations could be adversely affected.
101
Healthcare Reform
In the U.S., the EU and other foreign jurisdictions, there have been a number of legislative and regulatory changes to the healthcare systems that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs and improve the quality of healthcare. For example, the ACA, effective since March 2010, is a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms.
Healthcare reforms that have been adopted, and that may be adopted in the future, could result in further reductions in coverage and levels of reimbursement for pharmaceutical products, increases in rebates payable under U.S. government rebate programs and additional downward pressure on pharmaceutical product prices. On September 9, 2021, the Biden administration published a wide-ranging list of policy proposals, most of which would need to be carried out by Congress, to reduce drug prices and drug payment. The HHS plan includes, among other reform measures, proposals to lower prescription drug prices, including by allowing Medicare to negotiate prices and disincentivize price increases, and to support market changes that strengthen supply chains, promote biosimilars and generic drugs, and increase price transparency. As discussed above, these initiatives culminated in the enactment of the IRA in August 2022, which allows, among other things, HHS to directly negotiate the selling price of statutorily specified number of drugs and biologics each year that CMS reimburses under Medicare Part B and Part D. Only high-expenditure single-source biologics that have been approved for at least 11 years (7 years for single-source drugs) can qualify for negotiation, with the negotiated price taking effect two years after the selection year. The negotiated prices, which will first become effective in 2026, will be capped at a statutory ceiling price. Negotiations for Medicare Part D products begin in 2024 with the negotiated price taking effect in 2026, and negotiations for Medicare Part B products begin in 2026 with the negotiated price taking effect in 2028. In August 2023, HHS announced the 10 Medicare Part D drugs and biologics that it selected for negotiations, and by October 1, 2023, each manufacturer of the selected drugs signed a manufacturer agreement to participate in the negotiations. HHS will announce the negotiated maximum fair price by September 1, 2024, and this price cap, which cannot exceed a statutory ceiling price will come into effect on January 1, 2026. The IRA also penalizes drug manufacturers that increase prices of Medicare Part B and Part D drugs at a rate greater than the rate of inflation. The IRA permits the Secretary of HHS to implement many of these provisions through guidance, as opposed to regulation, for the initial years. Manufacturers that fail to comply with the IRA may be subject to various penalties, including civil monetary penalties. The IRA also extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. These provisions will take began taking progressively starting in 2023, although they may be subject to legal challenges. For example, the provisions related to the negotiation of selling prices of high-expenditure single-source drugs and biologics have been challenged in multiple lawsuits. Thus, while it is unclear how the IRA will be implemented, it will likely have a significant impact on the pharmaceutical industry.
We expect that additional U.S. federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that the U.S. federal government will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
Further, legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical products. We cannot be sure whether additional legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will be changed, or what the impact of such changes on the marketing approvals, if any, of our product candidates, may be. In addition, increased scrutiny by the U.S. Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing conditions and other requirements.
Individual states in the U.S. have also become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations,
102
financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our products or put pressure on our product pricing, which could negatively affect our business, results of operations, financial condition and prospects.
In the EU, similar political, economic and regulatory developments may affect our ability to profitably commercialize our current or any future products. In addition to continuing pressure on prices and cost containment measures, legislative developments at the EU (such as the EU Pharmaceutical Legislation) or member state level may result in significant additional requirements or obstacles that may increase our operating costs. The delivery of healthcare in the EU, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than EU, law and policy. National governments and health service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most EU Member States have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to commercialize any products for which we obtain marketing approval. In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we or our collaborators are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or our collaborators are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability, which would adversely affect our business.
Environmental issues which may influence the use of our material fixed assets
Our primary research and development activities take place in our facilities in Zwijnaarde, Belgium. For these activities we require, and have obtained, the necessary environmental and biohazard permits from the responsible governments, required by us for the manner in which we use said facilities.
New shares created during 2023
As a result of the exercise of stock options and vesting of restricted stock units (RSUs) under our Equity Incentive Plan (as defined herein), 1,216,999 new shares were created in 2023. Equity Incentive Plan means the equity incentive plan as adopted by our Board of Directors on December 18, 2014, which was approved by a General Meeting on May 13, 2015, and subsequently amended at General Meetings on April 28, 2016 and November 25, 2019, respectively, and by the Board of Directors on December 18, 2019, November 5, 2020, December 15, 2021 and February 27, 2023.
On July 24, 2023, we closed an offering of 2,581,633 of our ordinary shares through a global offering. The global offering was comprised of an offering of ordinary shares represented by ADSs in the U.S. and certain other countries outside of the EEA and a simultaneous private placement of ordinary shares in the EEA and the UK. As a result, we received $1.3 billion of gross proceeds from this offering, decreased by $65.9 million of underwriter discounts and commissions, and offering expenses, of which $0.8 million has been deducted from equity. The total net cash proceeds from the offering amounted to $1.2 billion.
103
The following table shows the developments in our share capital for the fiscal year ending December 31, 2023 and on February 20, 2024:
Number of shares outstanding on December 31, 2021 |
| 51,668,315 |
Number of shares outstanding on December 31, 2022 | 55,395,856 | |
Exercise of stock options in 2023 | 1,137,439 | |
Vesting of RSUs | 79,560 | |
Global public offering in Euronext and Nasdaq on July 17, 2023 | 2,244,899 | |
Over-allotment option exercised by underwriters on July 19, 2023 | 336,734 | |
Number of shares outstanding on December 31, 2023 | 59,194,488 | |
Exercise of stock options in January 2024 | 106,617 | |
Exercise of stock options in February 2024 | 2,277 | |
Number of shares outstanding on February 20, 2024 | 59,302,232 |
C. | ORGANIZATIONAL STRUCTURE |
As of December 31, 2023, argenx SE, has two subsidiaries, argenx BV and argenx Benelux BV, based in Belgium and argenx BV has ten subsidiaries. The following table sets out for each of our principal subsidiaries, the country of incorporation, and percentage ownership and voting interest held by us (directly or indirectly through subsidiaries).
As per December 31, 2023 | |||||
Name |
| Country |
| Participation | |
argenx SE | The Netherlands | 100.00 | % | ||
argenx BV |
| Belgium |
| 100.00 | % |
argenx Benelux BV | Belgium | 100.00 | % | ||
argenx US, Inc. |
| USA |
| 100.00 | % |
argenx Switzerland, SA | Switzerland | 100.00 | % | ||
argenx Japan KK |
| Japan |
| 100.00 | % |
argenx France SAS | France | 100.00 | % | ||
argenx Germany GmbH | Germany | 100.00 | % | ||
argenx Canada Inc. | Canada | 100.00 | % | ||
argenx UK Ltd. | United Kingdom | 100.00 | % | ||
argenx Netherlands Services B.V. | The Netherlands | 100.00 | % | ||
argenx Italy S.r.l. | Italy | 100.00 | % | ||
argenx Spain S.L. | Spain | 100.00 | % | ||
The following chart provides an overview of the Group as of the date of this Annual Report. Percentages refer to both the share of capital and voting rights.
104
D.PROPERTY, PLANTS AND EQUIPMENT
In January 2021, we entered into a binding lease agreement related to the envisioned relocation of our Zwijnaarde facility to a newly built office in Zwijnaarde, with an annual base rent of $1.8 million, which will be operational in the third quarter of 2028, and with an initial term of 10.5 years. The total future cash flows related to this lease are represented below in “Note 22—Leases” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
We also lease office space in Amsterdam (the Netherlands), Boston (U.S.), Tokyo (Japan), Geneva (Switzerland), Munich (Germany), Issy Les Moulineaux (France), Vaughan Ontario (Canada), Gerrards Cross (UK) and Milan (Italy). In addition, our lease liabilities include a lease plan for company cars with maturity dates up to four years.
For a discussion of contractual obligations, please see “Note 29—Commitments” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
We have our principal executive, operational offices and laboratory space located in Zwijnaarde, Belgium. The following table sets forth our material leased facilities worldwide as of December 31, 2023:
Facility location |
| Use |
| Approx. size (m2) |
| Lease expiry |
Zwijnaarde, Belgium (leased) | Operations and Laboratory Space | 4,678 |
| September 30, 2028 | ||
Boston, Massachusetts (leased) | Office Space | 914 |
| August 31, 2030 | ||
Tokyo, Japan (leased) | Office Space | 546 |
| January 17, 2027 |
Environment, Health and Safety
Our primary research and development activities take place in our facilities in Zwijnaarde, Belgium. For these activities we require, and have obtained, the necessary environmental and biohazard permits from the responsible governments, required by us for the manner in which we use said facilities. See Item 3.D. “Risk Factors”.
ITEM 4A. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
The following “Operating and Financial Review and Prospects” should be read together with the information in our financial statements and related notes included elsewhere in this Annual Report. The following discussion is based on our financial information prepared in accordance with the IFRS, as issued by the IASB, which may differ in material respects from generally accepted accounting principles in other jurisdictions, including U.S. GAAP. The following discussion includes forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including but not limited to those described in Item 3.D. “Risk Factors” and elsewhere in this Annual Report. Please also see “Cautionary Statement Regarding Forward-Looking Statements” in this Annual Report.
105
A. OPERATING RESULTS
The review of the financial condition and results of operations of certain items from fiscal year ended December 31, 2021, and year-to-year comparisons between fiscal year ended December 31, 2022, and December 31, 2021, that are not included in this Annual Report can be found in Item 5 “Operating and Financial Review and Prospects” of our annual report on Form 20-F for the fiscal year ended December 31, 2022.
Overview
Since our inception in 2008, we have focused most of our financial resources and efforts towards developing our SIMPLE AntibodyTM Platform and antibody engineering technologies, identifying potential product candidates, establishing process, development and manufacturing capabilities for our product candidates and advancing multiple discovery programs into the clinic. In 2022, we executed on our global launch of VYVGART our first-in-class neonatal FcRn blocker for IV use, which is now approved in the U.S, Japan, Europe, Israel, Canada and China for gMG. In 2023, we executed on our global launch of VYVGART SC, the first-and-only neonatal FcRn blocker administered by subcutaneous injection, which is now approved in the U.S. and Europe. In 2023, the successful commercialization of VYVGART and VYVGART SC generated a global product net sales of $1.2 billion.
On our research and development, we continue towards advancing a deep pipeline of both clinical- and preclinical-stage product candidates for the treatment of severe autoimmune diseases. Leveraging our technology suite and clinical expertise, we have advanced several candidates into late-stage clinical development and we currently have multiple programs in the discovery stage.
As of December 31, 2023 and December 31, 2022, we had cash, cash equivalents and current financial assets of $3,180 million and $2,193 million, respectively.
Our statement of financial position shows our total assets of $4,542 million for the year ended December 31, 2023, compared to $3,134 million for the year ended December 31, 2022. The main reason for the material change in balance sheet total are the various equity financing rounds, completed over the periods covered by the financial statements.
Since our inception, we have incurred significant operating losses. For the years ended December 31, 2023 and 2022, we incurred total comprehensive losses of $295 million and $730 million, respectively. As of December 31, 2023, we had accumulated losses of $2,405 million.
Although we have generated revenue of $1.2 billion from global product net sales of VYVGART and VYVGART SC for gMG in the fiscal year ended December 31, 2023, we can provide no assurances that we will be able to achieve or sustain profitability based on product net sales in that indication alone or that we will be able to receive regulatory approval of and commercialize VYVGART or VYVGART SC in other indications or in other countries.
On December 17, 2021, the FDA approved efgartigimod, which is marketed as VYVGART, for the intravenous treatment of gMG in adult patients who are AChR-AB+, followed by Japanese PMDA approval (including seronegative patients) and approval the EU Commission in 2022 and China’s NMPA approval on July 30, 2023. On June 20, 2023, the FDA approved VYVGART SC for the subcutaneous treatment of gMG in adult patients who are AChR-AB+, followed by approval of the EU Commission on November 16, 2023. These are the only approved products we currently have.
We expect our expenses to continue to increase as we expand our global commercial infrastructure and drug product inventory for VYVGART and VYVGART SC for the treatment of gMG, the commercial launch of VYVGART SC for the treatment of CIDP if and when approval obtained, the advancement of our clinical-stage pipeline, including
106
ongoing registrational clinical trials across five indications of efgartigimod, and continued investment in our IIP. We anticipate that our expenses will increase if and as we:
Research and development activities:
| ● | Execute the phase 2 clinical trials of efgartigimod in SjD, PC-POTS and AMR |
| ● | Execute the phase 2 clinical trials with our partner Zai Lab in MN and LN |
| ● | Execute the seamless phase 2/3 clinical trials of efgartigimod in Myositis and BP |
| ● | Execute the phase 3 clinical trials of efgartigimod in MG seronegatives and Pediatric and TED |
| ● | Launch phase 2 and/or phase 3 in other indications with efgartigimod |
| ● | Execute the phase 2 clinical trials of empasiprubart in MMN, DGF and DM |
| ● | Execute the phase 1 clinical trial of ARGX-119 in healthy volunteers and the phase 1b / phase 2a clinical trials in CMS and ALS, respectively |
| ● | continue the research and development of our other clinical- and preclinical-stage product candidates and discovery stage programs; and |
| ● | seek regulatory approvals for any product candidates, including new indications, that successfully complete clinical trials. |
Pre-commercial and commercial activities:
| ● | further build our sales, marketing and distribution infrastructure and scale-up of manufacturing capabilities for the commercialization expansion of VYVGART and VYVGART SC and any product candidate, including new indications, for which we may obtain approval; and |
| ● | expand our global reach enabling us to commercialize any product candidates, including new indications, for which we may obtain regulatory approval. |
Other activities:
| ● | seek to enhance our technology platform and discover and develop additional product candidates; |
| ● | maintain, expand and protect our intellectual property portfolio, including litigation costs associated with defending against alleged patent infringement claims; |
| ● | add clinical, scientific, operational, financial and management information systems and personnel, including personnel to support our product development and potential future commercialization efforts; and |
| ● | experience any delays or encounter any issues, including failed studies, ambiguous clinical trial results, safety issues or other regulatory challenges. |
We expect that the costs of development and commercialization might also increase due to current and future collaborations with research and development partners as well as commercial partners.
107
Information pertaining to the years ended December 31, 2022 and December 31, 2021 was included in our annual reports on Form 20-F for the years ended December 31, 2022 and December 31, 2021, respecively, under Item 5 “Operating and Financial Review and Prospects,” which were filed with the SEC on March 16, 2023 and March 21, 2022, respectively.
Basis of Presentation
Foreign Currency Transactions
| 1. | Functional and presentation currency |
Items included in the consolidated financial statements of each of our entities are valued using the currency of their economic environment in which the entity operates. The consolidated financial statements are presented in USD ($), which is the Company’s presentation currency.
Revenue from sale of product
Revenue from the sale of goods is recognized at an amount that reflects the consideration that the Company expects to be entitled to receive in exchange for transferring goods to a customer, at the time when the customer obtains control of the goods rendered, i.e., when the customer has the ability to direct the use of the asset. The consideration that is committed in a contract with a customer can include fixed amounts, variable amounts, or both. The amount of the consideration may vary due to discounts, rebates, returns, chargebacks or other similar items. Contingent consideration is included in the transaction price when it is highly probable that the amount of revenue recognized is not subject to future significant reversals.
Our product net sales mainly consist of sales of VYVGART in the U.S., Japan, Europe and China and VYVGART SC in the U.S. and Europe. Product net sales are recognized once we satisfy the performance obligation at a point in time under the revenue recognition criteria in accordance with IFRS 15 “Revenue from contracts with customers”.
Revenue arising from the commercial sale of VYVGART and VYVGART SC is presented in the consolidated financial statements under “Note 15—Product net sales” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference. In accordance with IFRS 15 “Revenue from contracts with customers”, such revenue is recognized when the product is physically transferred, in accordance with the delivery and acceptance terms agreed with the customer. Payment of the transaction price is payable at the point the customer obtains the legal title to the goods.
Revenue from Collaborations and License Agreements
Revenues to date have consisted principally of milestones, license fees, non-refundable upfront fees and research and development service fees in connection with collaboration and license agreements.
We recognize revenue when the customer obtains control of promised goods or services, in an amount that reflects the consideration that we expect to receive in exchange for those goods and services. In order to determine revenue recognition for agreements that we determine to be in the scope of IFRS 15, we followed the IFRS 15 5-step model. The Company has currently two active collaboration and license agreements in scope of IFRS 15:
Zai Lab
For the collaboration agreement with Zai Lab the Company has assessed that there is more than one distinct performance obligation, being the transfer of a license and supply of clinical and commercial product. The Company concluded that these performance obligations are distinct in the context of the contract.
108
Therefore, the Company allocates the transaction price to all performance obligations identified. The transaction price of the agreement is composed of (i) a fixed part, that being an upfront payment in the form of newly issued Zai Lab shares, and a guaranteed, non-creditable, non-refundable payment and (ii) a milestone payment for approval of efgartigimod in the U.S. and the consideration received in return for the supply of clinical and commercial product.
The fixed part of the transaction price, as well as the milestone for approval of efgartigimod in the U.S. has been allocated to the transfer of a license performance obligation. The Company concluded that the license as of the effective date of the contract, being January 2021, has standalone value. As such, the Company concluded that the promise in granting the license to Zai Lab is to provide a right to use the entity’s intellectual property as it exists at the point in time at which the license is granted and therefore, revenue was recognized at a point in time.
Under the collaboration agreement, the Company provides clinical and commercial supply to Zai Lab. Company concludes to recognize such sales as revenue given that the Company acts as principal in the transaction as the risk related to inventory is born by the Company until the inventory is transferred to Zai Lab. The revenue related to clinical supply is recorded under line item “Collaboration revenue”. The revenue related to commercial supply is recorded under line item “product net sales” in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss). The income related to royalties is recorded under line item item “Collaboration revenue”.
AbbVie
For the collaboration agreement with AbbVie the Company has determined that the transfer of license combined with the performance of research and development activities represent one single performance obligation. The Company concluded that the license is not distinct in the context of the contract.
The transaction price is composed of a fixed part, that being an upfront license fee, and a variable part, being milestone payments and cost reimbursements of research and development activities delivered. Milestone payments are only included in the transaction price to the extent it is highly probable that a significant reversal in the amount of cumulative revenue recognition will not occur when the uncertainty associate with the variable consideration is subsequently resolved. Management estimates the amount to be included in the transaction price upon achievement of the milestone event. Sales-based milestones and sales-based royalties are a part of the Company’s arrangements but are not yet included in its revenues.
The transaction price has been allocated to the single performance obligation and revenues has been recognized over the estimated service period based on an input model, being the percentage of completion method. The upfront license fee has been fully recognized since 2021 as the performance obligation has been fulfilled at that time. Milestone payments that become highly probable after the performance obligation has been fulfilled are therefore recognized at that point in time.
Research and Development Expenses
Research and development expenses consist principally of:
| ● | External research and development expenses related to (i) chemistry, manufacturing and control costs for our product candidates, both for preclinical and clinical testing, all of which is conducted by specialized contract manufacturers, (ii) fees and other costs paid to CROs in connection with preclinical testing and the performance of clinical trials for our product candidates, (iii) costs associated with regulatory submissions and approvals, quality assurance (QA) and pharmacovigilance and (iv) costs associated with post-approval clincial trails. |
| ● | personnel expense related to compensation of research and development staff and related expenses, including salaries, benefits and share-based payment expenses; |
| ● | materials and consumables expenses; |
109
| ● | depreciation and amortization of tangible and intangible fixed assets used to develop our product candidates; and |
| ● | IT expenses; |
| ● | other expenses including, but not limited to costs associated with obtaining and maintaining patents and other intellectual property. |
We incur various external expenses under our collaboration and license agreements for material and services consumed in the discovery and development of our partnered product candidates.
Our Research and development expenses may vary substantially from period to period based on the timing of our research and development activities, including the timing of the initiation of clinical trials, production of product batches and enrolment of patients in clinical trials. Research and development expenses are expected to increase as we advance the clinical development of efgartigimod and empasiprubart and further advance the research and development of our other early-stage pipeline candidates. The successful development of our product candidates is highly uncertain. At this time, we cannot reasonably estimate the nature, timing and estimated costs of the efforts that will be necessary to complete the development of, or the period, if any, in which material net cash inflows may commence from any of our product candidates. This is due to numerous risks and uncertainties associated with developing drugs, as fully described in Item 3.D. “Risk Factors,” and including the uncertainty of:
| ● | the scope, rate of progress and expense of our research and development activities; |
| ● | the successful enrollment in, and completion of clinical trials; |
| ● | the ability to market, commercialize and achieve market acceptance for efgartigimod or any other product candidate that we may develop in the future, if approved; |
| ● | establishing and maintaining a continued acceptable safety profile for our product candidates; |
| ● | the terms, timing and receipt of regulatory approvals from applicable regulatory authorities; |
| ● | the successful completion of preclinical studies necessary to support IND applications in the U.S. or similar applications in other countries; |
| ● | the expense of filing, prosecuting, defending and enforcing patent claims and other intellectual property rights and our current and future collaborators continuing their collaborations with us. |
Selling, General and Administrative Expenses
Selling, general and administrative expenses consist primarily of:
| ● | personnel expenses relating to salaries and related costs for personnel, including share-based compensation, of our employees in executive, finance, business development, marketing, commercial and support functions; |
| ● | professional fees for business development, marketing, IT, audit, commercial, legal services and investor relations costs; |
| ● | Board of Directors expenses consisting of directors’ fees, travel expenses and share-based compensation for non-executive board members; |
| ● | costs associated with commercial launch of VYVGART and VYVGART SC for the treatment of gMG and marketing and promotional activities, pre-launch activities of VYVGART and VYVGART SC in other |
110
| indications and continued investment in supply chain and costs associated with pre-launch activities in other indications; |
| ● | allocated facilities costs; and |
| ● | other Selling, general and administrative expenses, including leasing costs, office expenses, travel costs. |
We expect our general and administrative expenses to increase as we continue to support our growth. Such costs include increases in our personnel, additional IT-related expenses, and expenses and costs associated with compliance with the regulations governing public companies. We expect our Selling and marketing expenses to increase due to marketing and promotional activities with respect to the ongoing commercial launch of VYVGART, VYVGART SC and preparation of commercial launch of our other product candidates.
Financial Income (Expense)
Financial income mainly reflects interest earned on our cash and cash equivalents and current financial assets and net gains on our cash and cash equivalents and current financial assets held at fair value through profit or loss. Financial expense corresponds mainly to net losses on cash and cash equivalents and current financial assets held at fair value through profit or loss.
Exchange Gains (Losses)
Our exchange gains (losses) relate to (i) our transactions denominated in foreign currencies, mainly in euro, and which generate exchange gains or losses and (ii) the translation at the reporting date of assets and liabilities denominated in foreign currencies into USD, which is our functional and presentation currency. For more information on currency exchange fluctuations on our business, please see “Note 26—Financial risk management—Foreign exchange risk” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference. We have no derivative financial instruments to hedge interest rate and foreign currency risk.
Income Tax Expense
We have a history of losses in certain jurisdictions, including Belgium and the Netherlands. We may continue to incur losses as we continue to invest in our clinical and pre-clinical development programs and our discovery platform, and we incur costs for various commercial launches and regulatory approvals. Consequently, we do not recognize any deferred tax asset regarding certain tax attributes on our consolidated statements of financial position.
We incur current income tax expense and recognize deferred tax assets in various subsidiaries in view of the transfer pricing policy set up between argenx BV and these subsidiaries.
For more information on income tax and deferred tax, please see “Note 24—Income taxes” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
111
Capitalization and Indebtedness
The table below sets forth our capitalization as of December 31, 2023 on an actual basis:
|
| As of December 31, | |
(in thousands of $) |
| 2023 (audited) | |
Total current debt (including current portion of non-current debt) |
| $ | — |
Guaranteed |
|
| — |
Secured |
|
| — |
Unguaranteed/unsecured |
|
| — |
Total non-current debt (excluding current portion of non-current debt) |
|
| — |
Guaranteed |
|
| — |
Secured |
|
| — |
Unguaranteed/unsecured |
|
| — |
Shareholder equity |
|
| 4,097,507 |
Share capital |
|
| 7,058 |
Share premium |
|
| 5,651,497 |
Legal reserve(s) (1) |
|
| 131,543 |
Retained earnings |
|
| (2,404,844) |
Other reserves |
|
| 712,253 |
Total |
| $ | 4,097,507 |
| (1) | Legal reserves are the amount of translation differences |
The table below sets forth our indebtedness as of December 31, 2023 on an actual basis:
(in thousands of $) |
| As of December 31, | |
A. Cash | $ | 20,744 | |
B. Cash equivalents (1) | 2,028,100 | ||
C. Other current financial assets (2) | 1,131,000 | ||
D. Liquidity (A + B + C) | 3,179,844 | ||
E. Current financial debt (including debt instruments, but excluding current portion of non-current financial debt) | — | ||
F. Current portion of non-current financial debt (3) | 4,646 | ||
G. Current financial indebtedness (E + F) | 4,646 | ||
H. Net current financial indebtedness (G - D) | (3,175,198) | ||
I. Non-current financial debt (excluding current portion and debt instruments) (3) | 15,354 | ||
J. Debt instruments | — | ||
K. Non-current trade and other payables | — | ||
L. Non-current financial indebtedness (I + J + K) | 15,354 | ||
M. Total financial indebtedness (H + L) | (3,159,844) | ||
| (1) | See “Note 11—Cash and cash equivalents” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference. |
| (2) | See “Note 10—Financial assets – current” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference. |
| (3) | Please note that financial debt balances as presented in the table above do not include any indirect or contingent indebtedness. For more information on the Company’s indirect and contingent indebtedness, please see “Note 29—Commitments” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference. |
As of December 31, 2023, current financial debt (as disclosed in item F in the table above) included current liabilities related to short-term leases in the amount of $4.6 million and non-current financial debt (as disclosed in item I in the table above) including non-current liabilities related to long-term leaes in the amount of $15.4 million.
112
More information is included in our consolidated financial statements and related notes included in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
Critical Accounting Estimates and Judgments
In the application of the Company’s accounting policies, which are described above, the Company is required to make judgments, estimates and assumptions about the carrying amounts of assets and liabilities that are not readily apparent from other sources. The estimates and associated assumptions are based on historical experience and other factors that are considered to be relevant. Actual results may differ from these estimates.
The estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimate is revised if the revision affects only that period or in the period of the revision and future periods if the revision affects both current and future periods.
Critical estimates in applying accounting policies
Gross to net adjustments
The product gross sales are subject to various deductions, which are primarily composed of rebates to government agencies, distributors, health insurance companies and managed healthcare organizations. These deductions represent estimates of the related obligations, requiring the use of judgment when estimating the effect of these sales deductions on product gross sales for a reporting period. These adjustments are deducted from product gross sales to arrive at product net sales. The significant components of variable consideration under revenue recognition policy summarizes the nature of these deductions and how the deduction is estimated, see “Note 2.17 —Product net sales” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference. After recording these, product net sales represent the Company’s best estimate of the cash that we expect to ultimately collect. If in future periods the actuals vary from prior period best estimates, this would affect revenue in the period of adjustment.
113
Results of Operation
Comparison of Years Ended December 31, 2023 and 2022
Year ended | |||||||||
December 31, | |||||||||
| 2023 |
| 2022 |
| % Change | ||||
(In thousands) | |||||||||
Product net sales |
| $ | 1,190,783 | $ | 400,720 | 197 | % | ||
Collaboration revenue |
|
| 35,533 |
| 10,026 | 254 | % | ||
Other operating income | 42,278 | 34,520 | 22 | % | |||||
Total operating income |
|
| 1,268,594 |
| 445,267 | 185 | % | ||
Cost of sales | (117,835) | (29,431) | 300 | % | |||||
Research and development expenses | (859,492) | (663,366) | 30 | % | |||||
Selling, general and administrative expenses |
|
| (711,905) |
| (472,132) | 51 | % | ||
Loss from investment in joint venture |
|
| (4,411) |
| (677) | 552 | % | ||
Total operating expenses | (1,693,643) | (1,165,607) | 45 | % | |||||
Operating loss |
|
| (425,049) |
| (720,340) | (41) | % | ||
Financial income |
|
| 107,386 |
| 27,665 | 288 | % | ||
Financial expense |
|
| (906) |
| (3,906) | (77) | % | ||
Exchange gains/(loss) | 14,073 |
| (32,732) | (143) | % | ||||
Loss for the year before taxes |
| $ | (304,496) | $ | (729,314) | (58) | % | ||
Income tax benefit | 9,443 | 19,720 | (52) | % | |||||
Loss for the year | $ | (295,053) | $ | (709,593) | (58) | % | |||
Weighted average number of shares outstanding | 57,169,253 | 54,381,371 | |||||||
Basic and diluted (loss) per share (in $) | (5.16) | (13.05) | |||||||
Product net sales
Year Ended | ||||||
December 31, | ||||||
(in thousands of $) |
| 2023 |
| 2022 | ||
United States | $ | 1,046,592 | $ | 377,659 | ||
Japan | 56,432 | 15,764 | ||||
EMEA | 72,852 | 7,297 | ||||
China | 14,907 | — | ||||
Total product net sales |
| $ | 1,190,783 |
| $ | 400,720 |
For the twelve months ended December 31, 2023, the product net sales were mainly related to sales of VYVGART in the U.S., Japan, EU and China and VYVGART SC in the U.S and Europe.
Year Ended | |||||||||
(in thousands of $) | December 31, | ||||||||
2023 | 2022 |
| 2021 | ||||||
Product gross sales | $ | 1,342,148 | 446,923 | - | |||||
Gross to net adjustment | (151,365) | (46,203) | - | ||||||
Product net sales | 1,190,783 | 400,720 | - | ||||||
114
Collaboration revenue
Year ended | |||||||||
December 31, | |||||||||
| 2023 |
| 2022 |
| % Change | ||||
(In thousands) | |||||||||
AbbVie | 30,000 | — | n.a. | % | |||||
Other | — | 5,365 | (100) | % | |||||
Milestone payments | 30,000 | 5,365 | 459 | % | |||||
Other | — | 424 | (100) | % | |||||
Research and development service fees | — | 424 | (100) | % | |||||
Zai Lab | 5,533 | 4,238 | 31 | % | |||||
Other collaboration revenues | 5,533 | 4,238 | 31 | % | |||||
Total collaboration revenue | $ | 35,533 | $ | 10,026 | 254 | % | |||
Our collaboration revenue increased by $26 million to $36 million for the year ended December 31, 2023, compared to $10 million for the year ended December 31, 2022. The collaboration revenue recognized in the year ended December 31, 2023 was mainly the result of the recognition of a $30 million development milestone related to the AbbVie collaboration agreement. The revenue recognized during the year ended December 31, 2022, from milestone payments primarily relates to €5 million triggered by the option exercised by LEO Pharma to enter into the LEO Pharma Collaboration Agreement for ARGX-112. The increase in revenue recognition from “Other collaboration revenues” of $1 million was primarily driven by the royalties on net sales of VYVGART in Greater China through Zai Lab.
Other operating income
Year ended | |||||||||
December 31, | |||||||||
| 2023 |
| 2022 |
| % Change | ||||
(In thousands) | |||||||||
Grants |
| $ | 2,538 | $ | 2,186 | 16 | % | ||
Research and development incentives |
|
| 27,815 |
| 19,502 | 43 | % | ||
Payroll tax rebates |
|
| 11,925 |
| 8,576 | 39 | % | ||
Change in fair value on non-current financial assets | — | 4,256 | (100) | % | |||||
Total |
| $ | 42,278 | $ | 34,520 | 22 | % | ||
Other operating income increased by $8 million to $42 million for the year ended December 31, 2023, compared to $35 million for the year ended December 31, 2022. The $8 million increase was primarily driven by:
| ● | the increase in research and development incentives due to a Belgian research and development tax incentive scheme, as a result of the overall increased research and development costs incurred. |
| ● | the increase in payroll tax rebates for the year ended December 31, 2023, as a result of higher research and development personnel expenses eligible for rebates for the year ended December 31, 2023; and |
| ● | a decrease of $4 million due to the fact that there was no change in fair value on our profit share in AgomAb for the year ended December 31, 2023; |
For more information regarding governmental policies that could affect our operations, see Item 4.B. “Business Overview—Healthcare Law and Regulation.”
115
Research and development expenses
Year ended | |||||||||
December 31, | |||||||||
| 2023 |
| 2022 |
| % Change | ||||
(In thousands) | |||||||||
Personnel expense |
| $ | 226,344 | $ | 162,010 | 40 | % | ||
External research and development expenses |
|
| 483,192 |
| 366,955 | 32 | % | ||
Materials and consumables |
|
| 4,057 |
| 2,396 | 69 | % | ||
Depreciation and amortization |
|
| 105,546 |
| 102,132 | 3 | % | ||
IT expenses | 19,935 | 12,678 | 57 | % | |||||
Other expenses |
|
| 20,418 |
| 17,194 | 19 | % | ||
Total |
| $ | 859,492 | $ | 663,366 | 30 | % | ||
Our Research and development expenses totaled $859 million and $663 million for the years ended December 31, 2023 and 2022, respectively. The increase of $196 million in fiscal year 2023 as compared to fiscal 2022 is primarily driven by Personnel expense and External research and development expenses.
Personnel expense primarily relates to internal and external personnel. The expense also includes share-based compensation expenses related to the grant of stock options and RSUs to our research and development employees. We employed on average 607 full-time equivalents in our research and development functions in the year ended December 31, 2023, compared to 475 in the year ended December 31, 2022.
Our External research and development expenses for the year ended December 31, 2023 totaled to $483 million, compared to $367 million for the year ended December 31, 2022. The expense reflects clinical trial costs and manufacturing expenses related to the development of our product candidate portfolio. The table below provides additional detail on our External research and development expenses by program:
Year ended | |||||||||
December 31, | |||||||||
| 2023 |
| 2022 |
| % Change | ||||
(In thousands) | |||||||||
efgartigimod |
| $ | 361,676 | $ | 280,572 | 29 | % | ||
cusatuzumab |
|
| 14,298 |
| 13,554 | 5 | % | ||
empasiprubart | 47,636 | 32,384 | 47 | % | |||||
Other programs (*) |
|
| 59,582 |
| 40,445 | 47 | % | ||
Total |
| $ | 483,192 | $ | 366,955 | 32 | % | ||
(*) Other programs include general expenses not allocated to specific program of $27 million in 2023 and $23 million in 2022.
External research and development expenses for our lead product candidate efgartigimod totaled $362 million for the year ended December 31, 2023, compared to $281 million for the year ended December 31, 2022. This increase corresponds primarily to manufacturing and clinical development activities in relation to:
| ● | the execution of two Phase 3 clinical trials in MG Ph3b and Pediatric |
| ● | the execution of two Phase 3 clinical trials in CIDP; |
| ● | the execution of two Phase 3 clinical trial in PV and PF; |
| ● | the execution of Phase 2 and 3 clinical trials in BP, Myositis, LN, MN, AMR, PC-POTS SjD, TED and AAV; |
| ● | the execution of multiple Phase 2 clinical trials in empasiprubart in MMN, DGF and DM |
| ● | the execution of one HV clinical trial in ARGX-119 |
116
| ● | the execution of pre-clinical activities |
External research and development expenses for empasiprubart totaled $48 million for the year ended December 31, 2023 compared to $32 million for the year ended December 31, 2022. This increase of $15 million was due to the ramp up of Ph2 clinical trials in MMN, DGF and DM and futher investments in Discovery activities.
External research and development expenses on other programs increased by $19 million to $60 million for the year ended December 31, 2023, compared to $40 million for the year ended December 31, 2022. Of the total External research and development expense, $27 million relates to general allocation of expenses.
Selling, general and administrative expenses \
Year ended | |||||||||
December 31, | |||||||||
| 2023 |
| 2022 |
| % Change | ||||
(In thousands) | |||||||||
Personnel expenses |
| $ | 303,033 | $ | 234,740 | 29 | % | ||
Marketing services |
|
| 202,146 |
| 115,950 | 74 | % | ||
Professional fees | 108,820 | 62,620 | 74 | % | |||||
Supervisory board |
|
| 8,362 |
| 6,912 | 21 | % | ||
Depreciation and amortization |
|
| 2,366 |
| 2,211 | 7 | % | ||
IT expenses | 20,408 | 17,431 | 17 | % | |||||
Other expenses | 66,770 | 32,268 | 107 | % | |||||
Total Selling, general and administrative expenses |
| $ | 711,905 | $ | 472,132 | 51 | % | ||
Our Selling, general and administrative expenses totaled $712 million and $472 million for the years ended December 31, 2023 and 2022, respectively. The increase in our Selling, general and administrative expenses for the year ended December 31, 2023 was principally resulting from:
| ● | increased professional and marketing fees, including promotional and marketing costs primarily due to the commercial launch of VYVGART and VYVGART SC; |
| ● | increased costs of the salary and wages and benefits to our selling, general and administrative employees due to planned increase in the headcount; |
| ● | increased costs associated with additional employees recruited to strengthen our selling, general and administrative activities, for the commercial launch of VYVGART and VYVGART SC; and |
| ● | continued investment in our IT infrastructure. |
We employed on average 681 full-time equivalents in our selling, general and administrative functions in the year ended December 31, 2023, compared to 442 in the year ended December 31, 2022.
Financial income and (expense)
For the year ended December 31, 2023, financial income amounted to $107 million compared to $28 million for the year ended December 31, 2022. The increase of $80 million in 2023 related primarily to higher interest.
For the year ended December 31, 2023, financial expense amounted to $1 million compared to $4 million for the year ended December 31, 2022.
117
Exchange gains (losses)
Exchange gains totaled $14 million for the year ended December 31, 2023, compared to exchange losses of $33 million for the year ended December 31, 2022. The decrease was mainly attributable to unrealized exchange rate gains on the cash, cash equivalents and current financial assets position in euro during the year ended December 31, 2023 as compared to unrealized exchange rate losses on the cash, cash equivalents and current financial assets position during the year ended December 31, 2022.
B. LIQUIDITY AND CAPITAL RESOURCES
Sources of Funds
Since our inception in 2008, we have invested most of our resources in developing our product candidates, building our intellectual property portfolio, developing our supply chain, conducting business planning, raising capital and providing general and administrative support for these operations. We currently have 2 products approved by the FDA and as of the year ended December 31, 2022, net product sales also started to contribute to the funding of our operations. To date, we have funded our operations through public and private placements of equity securities, upfront, milestone and expense reimbursement payments received from our collaborators, funding from governmental bodies and interest income from the investment of our cash, cash equivalents and financial assets. Through December 31, 2023, we have raised gross proceeds of $5.6 billion from private and public offerings of equity securities. We have made product net sales of $1.2 billion during the twelve months ended December 31, 2023.
Our cash flows may fluctuate, are difficult to forecast and will depend on many factors. On December 31, 2023, we had cash, cash equivalents and current financial assets of $3,180 million, compared to $2,193 million on December 31, 2022.
We have no ongoing material financing commitments, such as lines of credit or guarantees, that are expected to affect our liquidity over the next five years, other than a line of credit totalling to $7.2 million, leases and our commitments to Halozyme, Lonza and Fujifilm which are detailed in “Note 29—Commitments” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
For more information as to the risks associated with our future funding needs, see Item 3.D. “Risk Factors— Risk Factors Related to argenx’s Financial Position and Need for Additional Capital.”
For more information as to our financial instruments, please see “Note 26—Financial risk management” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
118
Cash Flows
Comparison for the Years Ended December 31, 2023 and 2022
The table below summarizes our cash flows for the years ended December 31, 2023 and 2022.
Year ended | |||||||||
December 31, | |||||||||
| 2023 |
| 2022 |
| Variance | ||||
(In thousands) | |||||||||
Cash and cash equivalents at beginning of the period | $ | 800,740 | $ | 1,334,676 | $ | (533,936) | |||
Net cash flows (used in) / from operating activities |
|
| (420,327) |
| (862,807) |
| 442,480 | ||
Net cash flows from / (used in) investing activities |
|
| 308,210 |
| (461,184) |
| 769,394 | ||
Net cash flows from / (used in) financing activities |
|
| 1,336,727 |
| 843,757 |
| 492,970 | ||
Exchange gains/(losses) on cash and cash equivalents |
|
| 23,494 |
| (53,702) |
| 77,196 | ||
Cash and cash equivalents at end of the period | $ | 2,048,844 | $ | 800,740 | $ | 1,248,104 | |||
Net cash used in operating activities
Net cash outflow used in our operating activities decreased by $442 million to a net outflow of $420 million for the year ended December 31, 2023, compared to a net outflow of $863 million for the year ended December 31, 2022.
The decrease in net cash outflow used in operating activities results primarily from an increase in net product sales related to VYVGART and VYVGART SC, partly offset by:
| (i) | the increase in Research and development expenses incurred in relation to the manufacturing and clinical development activities of efgartigimod and the advancement of other clinical, preclinical and discovery-stage product candidate; |
| (ii) | the increase in personnel expenses, marketing expenses and consulting expenses incurred for the commercial expansion of VYVGART and VYVGART SC; and |
| (iii) | the further increase in working capital as a result of our inventory levels, including prepaid inventory. |
Net cash used in / from investing activities
Investing activities for the year ended December 31, 2023, consist primarily of the net desinvestment of $272 million in current financial assets, and interests received, partly offset by payments related to regulatory and sales based milestones to Halozyme and investement in OncoVerity, resulting in a cash inflow of $308 million.
Investing activities for the year ended December 31, 2022, consists primarily of net investment of $369 million in current financial assets, and purchase of a priority review voucher for $102 million, partly offset by interests received, resulting in a cash outflow of $461 million.
Net cash provided by financing activities
Financing activities primarily consist of net proceeds from our private placements and public offerings of our securities and exercise of stock options. The net cash inflow from financing activities was $1,337 million for the year ended December 31, 2023, compared to a net cash inflow of $844 million for the year ended December 31, 2022. The net cash inflows were attributed to (i) $1.2 billion net cash proceeds from our global offering in July 2023, compared to $0.8 billion net cash proceeds from our global offering in February 2022 and (ii) $158 million proceeds received from the exercise of stock options in 2023, compared to $93 million for the year ended 2022.
119
Operating and Capital Expenditure Requirements
We have never achieved profitability and, as of December 31, 2023, we had accumulated losses of $2,405 million. Our losses resulted principally from costs incurred in research and development, preclinical testing and clinical development of our research programs, and from general and administrative costs associated with commercial roll out and expansion. We anticipate that our operating expenses will increase as we intend to continue to conduct research and development and continue our efforts to expand our sales, marketing and distribution infrastructure. Although we have generated net product sales of $1.2 billion from global product net sales of VYVGART and VYVGART SC for the treatment of gMG in fiscal year 2023, which supports our current path to profitability, we can provide no assurances that we will be able to achieve or sustain profitability based on this indication alone or that we will be able to receive regulatory approval of and commercialize VYVGART and VYVGART SC in other indications or in other countries.
On the basis of current assumptions, we expect that our existing cash and cash equivalents and current financial assets will enable us to fund our operating expenses and capital expenditure requirements through at least the next twelve months. Our future equity capital will depend on multiple factors. Because of the numerous risks and uncertainties associated with the development and commercialization of efgartigimod and our other product candidates and discovery stage programs and because the extent to which we may enter into collaborations with third parties for the development of these product candidates is unknown, we are unable to estimate the amounts of increased capital outlays and operating expenses associated with completing the research and development of our product candidates. Our future capital requirements for efgartigimod and our other product candidates and discovery stage programs will depend on many factors, including:
| ● | the progress, timing and completion of preclinical testing and clinical trials for our current or any future product candidates; |
| ● | the number of potential new product candidates we identify and decide to develop; |
| ● | the time and costs involved in obtaining regulatory approval for our product candidates and any delays we may encounter as a result of evolving regulatory requirements or adverse results with respect to any of our product candidates; |
| ● | selling and marketing activities undertaken in connection with the commercialization of VYVGART, VYVGART SC or potential commercialization of any of our current or any future product candidates, if approved, and costs involved in the creation of an effective sales and marketing organization; |
| ● | manufacturing activities undertaken for VYVGART, VYVGART SC and potential commercialization of any of our current or any future product candidates, if approved, and costs involved in the creation of an effective supply chain; |
| ● | the costs involved in growing our organization to the size needed to allow for the research, development and potential commercialization of our current or any future product candidates; |
| ● | the costs involved in filing patent applications and maintaining and enforcing patents or defending against claims or infringements raised by third parties; |
| ● | the maintenance of our existing collaboration agreements and entry into new collaboration agreements; |
| ● | developments related to the global economic uncertainties and political instability. |
For more information as to the risks associated with our future funding needs, see Item 3.D. “Risk Factors— Risk Factors Related to argenx’s Financial Position and Need for Additional Capital.”
120
Treasury and Liquidity Policy
The Company has adopted a policy whereby cash and cash equivalents and current financial assets are invested with several highly reputable banks and financial institutions. The Company holds its cash and cash equivalents mainly with different banks which are independently rated with a minimum rating of ‘A-’. The Company also holds short-term investment funds in the form of money market funds with a recommended investment horizon of six months or shorter but with a low historical volatility. These money market funds are highly liquid investments, can be readily convertible into a known amount of cash. The Company has adopted a policy whereby money market funds must have an average rating of “BBB” or higher.
For more information as to our treasury policy and liquidity, please see “Note 26—Financial risk management” in our consolidated financial statements which are appended to this Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
Working capital statement
In accordance with item 3.1 of Annex 11 of the Commission Delegated Regulation (EU) 2019/980 we make the following statement:
In our opinion, the working capital of the Company is sufficient for the Company’s present requirements, at least for a period of 12 months from the date of this Annual Report.
Off-Balance Sheet Arangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the applicable rules and regulations, such as relationships with unconsolidated entities or financial partnerships, which are often referred to as structured finance or special purpose entities, established for the purpose of facilitating financing transactions that are not required to be reflected on our balance sheets.
C. RESEARCH AND DEVELOPMENT, PATENTS AND LICENSES
For a discussion of our research and development policies, see Item 4 “Information on the Company” and Item 5 “Operating and Financial Review and Prospects”.
D. TREND INFORMATION
Other than as disclosed elsewhere in this Annual Report, we are not aware of any trends, uncertainties, demands, commitments or events for the current financial period that are reasonably likely to have a material effect on our net revenues, income, profitability, liquidity, capital resources or prospects, or that caused the disclosed financial information to be not necessarily indicative of future operating results or financial conditions.
Following the approval of VYVGART and VYVGART SC for the treatment of gMG in the U.S. by the FDA in 2021 and 2023 respectively, we transitioned from a clinical-stage to a commercial-stage biotechnology company. We have now commercialized VYVGART in U.S., the EU, Japan, China (through our partner Zai Lab), Israel (through our partner Medison) and Canada, and VYVGART SC in U.S and Germany. We are working to expand commercialization in other jurisdictions, and to launch new products and product candidates, including into new indications.
There has been no significant change in the financial performance or the financial position of the Group since the balance sheet date of December 31, 2023.
For more information, please refer to Item 4.B. “Business Overview”, Item 5.A. “Operating Results”, Item 5.B. “Liquidity and Capital Resources” and to “Note 29—Commitments” in our consolidated financial statements which are appended to this Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
121
E. CRITICAL ACCOUNTING ESTIMATES
See Item 5.A. “Operating Results—Critical Accounting Estimates”.
ITEM 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
A. DIRECTORS AND SENIOR MANAGEMENT
Our Board of Directors
As at December 31, 2023, we have a one-tier board structure consisting of one executive director and eight non-executive directors, and a senior management team (consisting of our CEO and senior personnel reporting directly to the CEO) responsible for the day-to-day operations. We have opted for this structure to allow for a division of responsibilities between our Board of Directors and our senior management team, keeping our Board of Directors at a manageable size whilst being able to involve some or all members of our senior management team in discussions with the Board of Directors if and when necessary.
In practice, all members of our senior management team are regularly involved in the discussions of our Board of Directors and its committees, in order to provide information and context to the various issues the Board of Directors needs to decide on. In addition to being present at meetings from time to time, our senior management and other senior leaders in the organization keep regular contact (face to face or via electronic means) with members of the Board of Directors and its committees.
Our Board of Directors had five formal meetings in the course of 2023. The meetings were held in the months February, May, July, October and December. The committees of the Board of Directors also convened regularly and at least once per quarter (please refer to Item 6.C. “Board Practices—Report Audit and Compliance Committee”, Item 6.C. “Board Practices—Report Audit and Compliance Committee”, Item 6.C. “Board Practices—Report Audit and Compliance Committee”, and Item 6.C. “Board Practices—Report Audit and Compliance Committee”).
All Board of Director meetings and all formal committee meetings were also attended by Mr. Van Hauwermeiren, as executive director. In addition, several members of the senior management team were invited to discuss specific items included on the Board of Directors and committee meetings’ agendas (please refer to Item 6.A “Directors and Senior Management—Attendance Record Board of Director Meetings”).
Set out below is a summary of certain provisions of Dutch corporate law as of the date of this Annual Report, as well as a summary of relevant information concerning our Board of Directors and certain provisions of our articles of association (Articles of Association) and the Board By-Laws.
This summary does not purport to give a complete overview and should be read in conjunction with and is qualified in its entirety by reference to the relevant provisions of Dutch law as in force on the date of this Annual Report, the Articles of Association and Board By-Laws. The Articles of Association are available in the governing Dutch language and an unofficial English translation thereof, and the Board By-laws are available in English, on our website.
122
The following table sets forth certain information with respect to the current members of our Board of Directors, including their ages as of December 31, 2023:
Name |
| Age |
| Position |
| Nationality |
| Date of Initial |
| Date of last |
| Term |
Tim Van Hauwermeiren | 51 | CEO and executive director | Belgium | July 15, 2008 | May 10, 2022 | 2026 | ||||||
Peter K. M. Verhaeghe | 65 | Non-executive director (chairperson) | Belgium | October 15, 2008 | May 10, 2022 | 2026 | ||||||
Werner Lanthaler(1) | 55 | Non-executive director (vice-chairperson) | Austria | July 9, 2014 | May 10, 2022 | 2024 | ||||||
Steve Krognes(1) | 55 | Non-executive director | U.S. and Norway | February 27, 2023 | February 27, 2023 | 2027 | ||||||
J. Donald deBethizy | 73 | Non-executive director | U.S. | May 13, 2015 | May 2, 2023 | 2025 | ||||||
Pamela Klein | 62 | Non-executive Director | U.S. | April 28, 2016 | May 12, 2020 | 2024 | ||||||
Anthony A. Rosenberg | 70 | Non-executive director | UK | April 26, 2017 | May 11, 2021 | 2025 | ||||||
James M. Daly | 62 | Non-executive director | U.S. | May 8, 2018 | May 10, 2022 | 2026 | ||||||
Camilla Sylvest | 51 | Non-executive director | Denmark | September 8, 2022 | September 8, 2022 | 2026 | ||||||
Ana Cespedes | 50 | Non-executive director | Spain | December 12, 2022 | December 12, 2022 | 2026 |
(1) | Werner Lanthaler resigned effective February 27, 2023 and was succeeded by Steve Krognes effective February 27, 2023. |
Peter K. M. Verhaeghe
Peter Verhaeghe has served as a member and chairperson of the board of arGEN-X B.V. since October 2008 and as non-executive director on our Board of Directors since July 2014.
Mr. Verhaeghe is the managing partner of VVGB Advocaten-Avocats, a corporate finance law and tax law firm, a position he has held since July 1999. He is currently lead counsel to a number of Belgian, Dutch, French, U.S. and Swiss life sciences companies. Mr. Verhaeghe has served on the boards of directors of Participatiemaatschappij Vlaanderen NV since May 2018 and miDiagnostics NV since April 2020. He has also served as chairman of the board of Haretis SA (Luxembourg) since March 2011 and as chairman of the LP & advisory committee of Bioqube Factory Fund I NV since September 2020. Mr. Verhaeghe previously served as a member of the board of directors of CzechPak Manufacturing s.r.o., Innogenetics NV (now Fujirebio Europe N.V.), Tibotec-Virco NV, and Biocartis SA. He was also the president of the board of directors of Merisant France SAS, a member of the management board of Merisant Company 2 sàrl., and chairman of the board of directors of PharmaNeuroBoost NV.
Mr. Verhaeghe holds a degree in law from the University of Leuven and an LL.M. degree from Harvard Law School.
Dr. Werner Lanthaler (until February 27, 2023)
Dr. Werner Lanthaler served as a member of our Board of Directors from July 2014 until February 27, 2023.
Dr. Lanthaler served as the CEO of Evotec SE until January 2024, a global drug discovery and development organization, a position he held since March 2009. He also serves on the supervisory board of AC Immune SA (Switzerland).
Dr. Lanthaler holds a degree in psychology, a Ph. D. in business administration from Vienna University of Economics and Business, and a Master’s degree in public administration from Harvard University.
123
Mr. Steve Krognes (effective February 27, 2023)
Mr. Krognes has served as a member of our Board of Directors and as a chairperson of our audit and compliance committee since February 27, 2023.
Mr. Krognes also serves on the boards of directors of Guardant Health, Inc., Denali Therapeutics, Inc., and Gritstone bio, Inc. In September 2023, he also was appointed to the board of directors of ClayvstBio. He previously served on the boards of directors of RLS Global AB and Corvus Pharmaceuticals, Inc. Mr. Krognes was the founding chief financial officer (CFO) of Denali Therapeutics, Inc. (Denali Therapeutics), from 2015 until retiring from that position in April 2022. In that role, he built and led the finance team as well as supervising the IT and facilities functions. He then joined the board of directors of Denali Therapeutics. Mr. Krognes led successful financings for Denali Therapeutics, including its initial public offering in 2017, and contributed significantly to the company’s strategy, growth and strong financial position. His extensive leadership experience in the biotech and pharmaceutical industries includes 12 years in total at Roche and Genentech, Inc., during which Mr. Krognes served as CFO of Genentech, Inc. for six years and global head of Roche’s mergers & acquisition team for six years. He also chaired the Genentech Access to Care Foundation and represented Genentech on the board and executive committee of the California Life Science Association. Before that, Mr. Krognes worked as an investment banker at Goldman Sachs, as a management consultant at McKinsey & Company, and as a venture capitalist in Scandinavia.
Mr. Krognes holds a Master’s of Business Administration (MBA) from Harvard Business School and a Bachelor’s of Science in economics from the Wharton School of the University of Pennsylvania.
Dr. J. Donald deBethizy
Dr. J. Donald deBethizy has served as a member of our Board of Directors since May 2015.
Dr. deBethizy has 30 years of experience in research and development, as well as financial, business and operating management, and board work in the biotechnology and consumer products industries.
He is the president of White City Consulting ApS, a consulting company that specializes in advising technology-focused companies. Dr. deBethizy currently serves on the boards of directors of Lophora ApS and Proterris, Inc. and as a board advisor for Cereno Scientific AB.
Previously, Dr. deBethizy served as president and CEO of Santaris Pharma A/S until October 2014, when the company was sold to Roche. From March 1997 to June 2012, Dr. deBethizy was co-founder and CEO of Targacept Inc. (Targacept), a U.S. biotechnology company listed on Nasdaq. From June 2012 to May 2013, he was special advisor to the chairman of Targacept’s board of directors. From May 2013 to November 2014, Dr. deBethizy served as executive chairman of Contera Pharma ApS until it was sold to Bukwang Pharma, and from July 2015 to November 2017, he served as chairman of Rigontec GmbH until it was sold to Merck Inc. He previously served as chairman of the boards of directors of Albumedix Ltd (sold to Sartorius AG in September 2022), Saniona AB, and TME Pharma NV and AG. Dr. deBethizy was also a member of the boards of directors of Asceneuron SA, Serendex Pharmaceuticals A/S, Enbiotix Inc., Ligocyte Pharmaceuticals until it was sold to Takeda Pharmaceutical Co Ltd, Biosource Inc., and NOXXON Pharma N.V. Dr. deBethizy has held adjunct appointments at Wake Forest University Babcock School of Management, Wake Forest University School of Medicine, and Duke University.
Dr. deBethizy holds a Bachelor’s of Science in biology from the University of Maryland, College Park and a Master’s of Science and a Ph.D. in toxicology from Utah State University.
Dr. Pamela Klein
Dr. Pamela Klein has served as a member of our Board of Directors since April 2016.
124
Since 2008, Dr. Klein has been a principal and founder of PMK BioResearch, a company offering strategic consulting in oncology drug development to corporate boards, management teams and the investment community. She has also been a venture partner in Ysios Capital Partners, SGIEC, S.A.U. since 2023. She currently serves as a member of the board of directors of several companies including I-Mab and Patrys Ltd; as well as various scientific advisor boards. In 2023, Dr. Klein also joined the boards of directors of Frontier Medicines Corp, Ona Therapeutics SL, and Sardona Therapeutics, Inc. Previously, Dr. Klein served on the board of directors of FStar Therapeutics, Inc. until March 2023, Jiya Acquisition Corp, and Spring Bank Pharmaceuticals, Inc. until its merger with F-Star Therapeutics in July 2020. Dr. Klein previously spent seven years at the National Cancer Institute as research director of the NCI-Navy Breast Center, after which she joined Genentech as vice president of development until 2001. She also served as chief medical officer for Intellikine, Inc., which was acquired by Takeda American Holdings.
Dr. Klein holds a Bachelor’s degree in biology from California State University and an M.D. from Stritch School of Medicine, Loyola University Chicago and is trained in internal medicine and medical oncology.
Anthony A. Rosenberg
Anthony A. Rosenberg has served as a member of our Board of Directors since April 2017.
He currently serves as CEO of TR Advisory Services GmbH, his own consultancy firm advising on business development, licensing, and mergers and acquisitions. Mr. Rosenberg also currently serves as chairman of the boards of directors of Oculis SA and Cullinan Oncology. Previously Mr. Rosenberg held the positions of Managing Director at MPM Capital, a venture capital firm (2015 until 2020); head of M&A and Licensing of Novartis International (2013 to 2015); and head of business development and licensing at Novartis Pharma (2005 to 2012). Mr. Rosenberg also previously served on the boards of directors of SiO2 Material Science (until March 2023), Radius Health Inc., TriNetX, Inc., iOmx Therapeutics AG, and Clinical Ink, Msc.
Mr. Rosenberg has a Bachelor of Science with honors from the University of Leicester and a Master’s of Science in physiology from the University of London.
James M. Daly
James M. Daly has served as a member of our Board of Directors since May 2018. Mr. Daly currently also serves as a director of Acadia Pharmaceuticals, Inc., Bellicum Pharmaceuticals, Inc., and Madrigal Pharmaceuticals, Inc. He was formerly a member of the board of Chimerix, Inc. and Halozyme.
In 1985, he joined GlaxoSmithKline where he held various positions, including senior vice president of the respiratory division with full responsibility for sales, marketing and medical affairs. Mr. Daly moved to Amgen in 2002 where he was senior vice president for the North America commercial operations until 2011. In 2012, he joined Incyte Corp, a publicly-traded company focused on oncology and inflammation, where he was chief commercial officer until June 2015.
Mr. Daly holds a Bachelor’s of Science and an MBA from the University at Buffalo, State University of New York.
Camilla Sylvest
Camilla Sylvest has served as a member of our Board of Directors since September 2022. Ms. Sylvest currently serves as the executive vice president of commercial strategy and corporate affairs of Novo Nordisk A/S.
Ms. Sylvest has more than 27 years of working experience within Novo Nordisk A/S and was based in Switzerland, Denmark, Germany, Malaysia, and Mainland China. Over the years, Ms. Sylvest has headed up Novo Nordisk A/S affiliates of growing size and complexity in Europe. She was also corporate vice president of the business area Oceania and Southeast Asia and senior vice president and general manager of the Novo Nordisk A/S region of Mainland China. Ms. Sylvest also serves as the vice chair of Danish Crown A/S.
125
Ms. Sylvest holds a Master’s of Science in economics from the University of Southern Denmark and an executive MBA from the Scandinavian Management Institute.
Ana Cespedes
Ana Cespedes has served as a member of our Board of Directors since December 2022. Ms. Cespedes is the COO of the International AIDS Vaccine Initiative (IAVI), a global organization dedicated to developing accessible vaccines and antibodies for infectious diseases.
Prior to joining IAVI, Ms. Cespedes held several roles at Merck KGaA, most recently serving as global head of strategy and engagement, government, and public affairs. She founded and led the global market access and pricing function for the company and worked with stakeholders to communicate the clinical, economic, and societal value of innovative medicines. Prior to that, Ms. Cespedes led the first integrated corporate affairs group at Serono Iberia and Merck Spain, was managing director of the Spanish branch of the company’s nonprofit organization, and worked as a senior consultant at Arthur Andersen. Ms. Cespedes is a founding member of the National Congress of Corporate Affairs in Spain, the London School of Economics Market Access Academy, and the Cooperation for Oncology Data. She is also the founder of Living Mindfulness S.L. Ms. Cespedes is also a member of the steering committee of ProPatiens Institute.
Ms. Cespedes holds a Bachelor’s of Pharmacy and a PhD from the Complutense University of Madrid, a Master in General Management (PDG) from IESE Business School and an Executive Certificate on Strategy and Innovation from the Massachusetts Institute of Technology.
Attendance Record Board of Director Meetings
In 2023, five Board of Directors meetings were held. The meeting attendance rate for our directors is set out in the table below.
Name |
| Number of meetings attended in to resignation, as applicable) |
| Attendance % |
|---|---|---|---|---|
Peter K. M. Verhaeghe (chairperson) | 5 | 100 % | ||
Tim Van Hauwermeiren | 5 | 100 % | ||
Werner Lanthaler(1) | 1 | 100 % | ||
Steve Krognes(1) | 4 | 100 % | ||
J. Donald deBethizy | 5 | 100 % | ||
Pamela Klein | 5 | 100 % | ||
Anthony A. Rosenberg | 5 | 100 % | ||
James M. Daly | 5 | 100 % | ||
Camilla Sylvest | 5 | 100 % | ||
Ana Cespedes | 5 | 100 % |
(1) | Werner Lanthaler resigned effective February 27, 2023 and was succeeded by Steve Krognes effective February 27, 2023. |
126
In 2023, all of the five Board of Directors meetings with solely the non-executive directors being present were held as closed sessions at the beginning or the end of other meetings. These meetings were attended by all non-executive directors appointed at such time.
Name |
| Number of meetings attended in |
| Attendance % |
|---|---|---|---|---|
Peter K. M. Verhaeghe (chairperson) | 5 | 100 % | ||
Werner Lanthaler(1) | 1 | 100 % | ||
Steve Krognes(10 | 4 | 100 % | ||
J. Donald deBethizy | 5 | 100 % | ||
Pamela Klein | 5 | 100 % | ||
Anthony A. Rosenberg | 5 | 100 % | ||
James M. Daly | 5 | 100 % | ||
Camilla Sylvest | 5 | 100 % | ||
Ana Cespedes | 5 | 100 % |
(1) | Werner Lanthaler resigned effective February 27, 2023 and was succeeded by Steve Krognes effective February 27, 2023. |
Activities
The agenda for the Board of Directors centers around the key business objectives for long-term value creation and the key risks involved, as well as the manner in which the senior management team implements our strategy including our research and development pipeline and the commercialization of our products, our culture to ensure proper monitoring by the non-executive directors, our financial position and finance readiness as well as the results of our subsidiaries, significant investment proposals, yearly budgets, the internal risk management and control system, diversity, equity and inclusion, succession planning and remuneration and appointment matters.
In 2023, specific attention was given to the statutory and governance topics including the long-term succession and contingency planning of the Board of Directors and senior management, leading to the appointment of Mr. Steve Krognes as non-executive director and chair of the audit and compliance committee and the renewal of the appointment of Mr. J. Donald deBethizy as non-executive director and his appointment as vice-chair of the Board of Directors. The Board of Directors furthermore discussed the long-term succession planning of the senior management team leading to the appointment of Ms. Karen Massey as our chief operating officer. The Board of Directors discussed the review and approval of forecasts, the Company’s product portfolios, business and corporate development, cybersecurity landscape, review and approval of consolidated financial statements, update research and developments, committee reports, financing of the Company and the approval of the proposed agendas and other meeting documents for our General Meeting, among other things.
Our Senior Management
The following table sets forth certain information with respect to the members of our senior management, including their ages, as of December 31, 2022:
Name |
| Age |
| Position |
| Nationality |
| Date of Initial |
Tim Van Hauwermeiren | 51 | CEO and Executive Director | Belgium | July 15, 2008 | ||||
Keith Woods(1) | 56 | COO | U.S. | April 5, 2018 | ||||
Karen Massey(1) | 45 | COO | Australia | March 13, 2023 | ||||
Karl Gubitz | 54 | CFO | South Africa | June 1, 2021 | ||||
Peter Ulrichts | 44 | Chief Scientific Officer | Belgium | January 1, 2023 | ||||
Malini Moorthy | 54 | General Counsel | Canada | February 14, 2022 | ||||
Arjen Lemmen | 39 | Vice-President Corporate Development & Strategy | The Netherlands | May 1, 2016 | ||||
Andria Wilk | 51 | Global Head of Quality | UK | January 13, 2020 | ||||
Luc Truyen | 59 | Chief Medical Officer | Belgium | April 1, 2022 |
| (1) | Keith Woods retired as COO effective March 13, 2023 and was succeeded by Karen Massey effective March 13, 2023. |
127
Tim Van Hauwermeiren
Tim Van Hauwermeiren co-founded our Company in 2008 and has served as our CEO since July 2008. He has served as a member of our Board of Directors since July 2014.
Mr. Van Hauwermeiren has more than 20 years of general management and business development experience across the life sciences and consumer goods sectors. He also serves on the boards of directors of iTeos Therapeutics, Inc. and RayzeBio, Inc.
Mr. Van Hauwermeiren holds a Bachelor’s of Science and Master’s of Science in bioengineering from Ghent University and an executive MBA from the Vlerick School of Management.
Keith Woods
Keith Woods served as our COO from April 2018 to March 2023, at which time he was succeeded by Karen Massey.
Mr. Woods transitioned to serve as an advisor to our Board of Directors. He has over 30 years of experience in the biopharmaceutical industry. Mr. Woods most recently served as senior vice president of North American operations for Alexion Pharmaceuticals, Inc. (Alexion). Within Alexion, he previously served as vice president and managing director of Alexion UK, overseeing all aspects of Alexion’s UK business, and as vice president of U.S. operations and executive director of sales. Prior to joining Alexion, Mr. Woods held various positions of increasing responsibility within Roche, Amgen, and Eisai Co., Ltd. over a span of 20 years. He holds a Bachelor’s of Science in marketing from Florida State University.
Karen Massey (effective March 13, 2023)
Karen Massey has served as our COO since March 2023.
Ms. Massey has over 20 years of experience in the pharmaceutical and biotechnology industry, including in commercial, product development, corporate strategy, and innovation roles. Prior to joining argenx, Ms. Massey was with Genentech (Roche Group) for over nine years, where she most recently served as senior vice president of product development and global clinical operations and previously held various commercial leadership roles across marketing and business operations, including as the vice president of the multiple sclerosis and neuromyelitis optica business. Ms. Massey started her biopharmaceutical career in marketing at Pfizer Inc., and returned there, after two years as a management consultant at Bain & Company, to take on leadership positions in corporate strategy and sales and as a commercial lead in Latin America.
Ms. Massey holds a Bachelor’s of Economics from the University of Sydney and an MBA from the NYU Stern School of Business.
Karl Gubitz
Karl Gubitz has served as our CFO since June 2021.
Mr. Gubitz previously worked at Pfizer Inc. for nearly 20 years, most recently as vice president of finance within the global oncology business. Within Pfizer Inc., Mr. Gubitz held country, regional, and global positions, and consistently delivered top-line growth. He managed teams of over 250 colleagues in financial leadership roles within the global internal medicine and global innovative products businesses. Prior to joining Pfizer Inc. in 2003, Mr. Gubitz held various management roles at PricewaterhouseCoopers LLP.
Mr. Gubitz holds an MBA from Henley Management College, a Bachelor’s degree in computing from the University of South Africa, and Bachelor’s of Commerce from the University of Pretoria.
128
Dr. Peter Ulrichts
Peter Ulrichts has served as our chief scientific officer since January 2023. In this role, he oversees the development of all clinical and pre-clinical compounds within our pipeline.
Dr. Ulrichts previously served in various roles at the Company since he joined us in 2010, including, most recently, as our head of clinical science. As a research scientist, Dr. Ulrichts was involved in the development of various therapeutic antibodies for the treatment of cancer and autoimmune diseases. In 2013, he headed the development of our FcRn antagonist efgartigimod until the first-in-human clinical trial. He subsequently transitioned to become the lead scientist of our efgartigimod program.
Dr. Ulrichts holds a Bachelor’s of Science in chemistry from Katholieke Universiteit Leuven, as well as a Master’s degree in Biotechnology and a Ph.D. in Biomedical Sciences, both from Ghent University.
Malini Moorthy
Malini Moorthy has served as our general counsel since February 2022.
She has over 25 years of extensive global legal and compliance experience in the biopharmaceutical and medical device industries. She was most recently senior vice president and chief deputy general counsel of legal, compliance, and government affairs at Medtronic plc, where she played a pivotal role in shaping and driving enterprise and functional strategies. Before joining Medtronic plc, Ms. Moorthy spent four years at Bayer Corporation as the head of global litigation and investigations and 10 years at Pfizer Inc., where she progressed to lead civil litigation globally. Ms. Moorthy began her career as a law firm associate, first with McCarthy Tétrault LLP and Genest Murray Desbrisay Lamek LLP in Toronto, Canada and then Salans LLP (now Dentons US LLP) in New York City.
She holds a Bachelor of Arts in political science and economics from the University of North Carolina at Chapel Hill and a Bachelor of Laws from the Faculty of Law at Queen’s University in Canada.
Luc Truyen
Luc Truyen has served as our chief medical officer since April 2022 and previously served as our head of research and development operations management from September 2021 to April 2022.
Prior to this, Dr. Truyen was with Johnson & Johnson (and its subsidiary companies) for over 20 years holding various leadership positions, primarily within neuroscience. In his most recent position prior to joining argenx, Dr. Truyen was global head of development and external affairs for neuroscience, managing the strategy and delivery of the early and late portfolio of assets for mood disorders, schizophrenia, and neurodegenerative and neuroinflammatory disorders. Besides Dr. Truyen’s strong track record in clinical development resulting in several globally innovative drug approvals, his broad-based experience also includes leading global clinical development operations for the whole Johnson & Johnson pharmaceutical group as well as serving as the head of research and development and chief medical officer of Janssen Alzheimer Immunotherapy Research & Development LLC, an internal spin-out from Johnson & Johnson.
Dr. Truyen holds an M.D. and Ph.D. in Neurology from the University of Antwerp.
Arjen Lemmen
Arjen Lemmen joined argenx in 2016 and has served as our vice president of corporate development & strategy since 2019. He has successfully executed several transactions including a number of programs within the IIP.
Prior to joining the Company, Mr. Lemmen served as a corporate finance specialist at Kempen & Co NV focusing on mergers and acquisitions, equity capital markets and strategic advisory transactions in the European life
129
sciences industry. He holds a Bachelor of Science in life science & technology from the University of Groningen and a Master of engineering management from Duke University.
Andria Wilk
Andria Wilk joined argenx as global head of quality in 2020. Ms. Wilk has more than 20 years of experience in QA within the pharmaceutical industry. Most recently, Ms. Wilk served as senior director, head of medical, regulatory & clinical QA (MRC QA) at H Lundbeck A/S (Lundbeck), where she managed the global MRC QA group based in the EU, US, and Asia. In this role, she was responsible for the global audit programs and QA support for all clinical trial and post-marketing activities and related computerized systems.
Prior to Lundbeck, she held various QA positions of increasing responsibility within AstraZeneca PLC, Takeda Global Research, Development Centre Europe, and Astellas Pharma Inc.
Ms. Wilk holds a joint Bachelor’s of Science in pharmacology and biochemistry, is a member of the Research Quality Association and observing board member of The European Forum for Good Clinical Practices.
General Information About Our Directors and Senior Management
As of the date of this Annual Report (or in any period before), none of the members of our Board of Directors and senior management has or has had a family relationship with any other member of our Board of Directors or senior management and there are no arrangement or understanding with major shareholders, customers, suppliers or others, pursuant to which any person referred to above was selected as a director or member of senior management. For further information regarding any arrangements by which our Board of Directors or senior management were appointed, see Item 7.B. “Related Party Transactions—Agreements with Our Senior Management”.
B. COMPENSATION
Remuneration Report and Compensation Statement
SAY-ON-PAY AND PROPOSED AMENDMENTS TO THE REMUNERATION POLICY
Introduction
In response to dissent expressed by shareholders on the 'say-on-pay’vote at the Company’s 2022 and 2023 annual General Meetings, we have engaged extensively with stakeholders, shareholders and proxy advisors. This group of stakeholders represented over 60% of the Company’s issued share capital. This has led to a proposal for a revised remuneration policy, which is expected to be published in draft form on or around March 21, 2024 (Draft 2024 Remuneration Policy), which requires approval at the Company’s annual General Meeting that will take place on May 7, 2024 (2024 General Meeting). Readers of this report are encouraged to read the Draft 2024 Remuneration Policy and corresponding explanatory notes, both of which will be made available on the Company’s website at https://www.argenx.com/investors/shareholder-meetings.
Although the following is not an exhaustive summary of the proposed changes to the Company’s current 2021 remuneration policy (the 2021 Remuneration Policy), which you are encouraged to read in full including the accompanying explanatory notes, the Company deems it relevant to bring to your attention the following key changes that will be proposed in the Draft 2024 Remuneration Policy.
3.4.1.1Non-executive pay
| ● | Stock options will no longer be granted to non-executive directors |
| ● | Non-executive pay will take the form of cash remuneration and equity remuneration in the form of RSUs |
130
| ● | Non-executive RSU grants will have a vesting period of one year and a holding period of three years after vest and as such underlying shares cannot be sold until after four years from the grant date |
| ● | Non-executive RSUs will be awarded based on a benchmarked target cash value, awarded in shares subject to the aforementioned holding requirements |
| ● | Minimum holding requirements extending at least two years beyond term of service will continue to apply |
Executive equity incentives
| ● | Performance share units (PSUs) will be introduced in the executive compensation plan, attaching financial and non-financial performance conditions to the vesting of the PSUs |
| ● | At least 50% of target executive equity pay-out will be performance based |
| ● | PSU performance conditions will link for at least 50% of their target value to financial targets |
| ● | Non-financial targets will relate to measurable sustainable long term value creating outcomes linked to the Company’s key value drivers: ‘innovation and pipeline development’ and ‘people and culture’ |
| ● | PSUs will not vest prior to the third anniversary of the grant date and only to the extent applicable performance conditions are met |
| ● | The target equity pay opportunities for the CEO, chief financial officer (CFO) and COO (together, NEOs) will continue to be set between the 50th percentile and 75th percentile of the reference group and will in any case not exceed 15x base cash compensation |
| ● | All equity grants will be subject to multi-year (at least three years) vesting periods and/or holding requirements |
| ● | Minimum holding requirements extending at least two years beyond term of service will continue to apply |
Executive short-term cash incentive
| ● | Short-term cash incentives will be linked to multiple strategically relevant targets, which, in turn, will be linked to clearly measurable outcomes |
| ● | At least 50% of short-term variable pay will be linked to financial targets |
| ● | The target cash pay opportunity (target and maximum), measurement and evaluation and pay-out will be disclosed |
| ● | Considering the rapid growth and development of the Company and the environment in which it operates, discretionary adjustment of the total variable pay within the set limits by the Board of Directors will be possible, but in the event this happens, a clear and detailed explanation of the use of such discretion will be included in the Company’s remuneration report |
The principles above will be applicable for remuneration granted and targets set after the approval of the Draft 2024 Remuneration Policy, which requires a majority vote of more than 75% at the 2024 General Meeting. If such majority is not achieved, the Company will, in accordance with Dutch law, be obliged to continue to apply the 2021 Remuneration Policy until a new policy gets approved at the 2024 General Meeting. You are encouraged to read this 2023 remuneration report in conjunction with the Draft 2024 Remuneration Policy and the accompanying explanatory notes.
131
It is noted that this 2023 remuneration report describes the application of the Company’s 2021 Remuneration Policy for the fiscal year 2023.
2023 Remuneration
The 2021 Remuneration Policy rewards contributions to achieving Company objectives and generating stakeholder value. The aim is to provide competitive remuneration packages that align with market practices in the key markets where the Company competes for talent. The Company conducts regular reviews (at least once every three years) of director and senior management members’ total remuneration (both in quantum and in program design) and makes comparisons against the Company’s reference companies. The 2021 Remuneration Policy and total compensation aligns or slightly exceeds the market median for fixed compensation, benefits, and short-term variable compensation. The long-term incentive component consists of equity grants, the size of which is positioned between the 50th and the 75th percentile of the global reference group. The 2021 Remuneration Policy was adopted at the 2021 General Meeting with a 76% majority vote and is available on the Company’s website at https://www.argenx.com/investors/governance/remuneration-policy.
Reference group - general
For the 2023 remuneration which was set following a benchmark exercise conducted in the August – September 2022 timeframe, the Company worked with an independent third party compensation advisor, AON Radford. The Company continued to benchmark against both US and European peer groups to account for being a global company competing for talent against European based and US based companies. The aim is to deliver globally competitive compensation supporting the execution of Company’s business strategy and aligning with long-term sustainable value creation for its stakeholders.
The following criteria were used to select the reference group for the 2023 remuneration as part of the Company’s benchmark performed in the third quarter of 2022, ahead of setting the long term incentive schemes for 2023 in December 2023 and the annual cash compensation for 2023 in first quarter of 2023:
| ● | Sector: Biotech and Pharmaceuticals |
| ● | Stage of development: Market |
| ● | Market Capitalization: primary ~1/3x – 3x argenx’s 30‐day average market value as of 5/20/22, secondary $5‐50 billion |
| ● | Headcount: primary ~1/3x – 3x the midpoint of argenx’s projected financial years ended 31 December, 2022 and 2023 headcounts, secondary 300‐2500 employees |
| ● | Revenue: less than $1 billion revenues |
| ● | Years public: less than 10 years since IPO |
With the goal of arriving at a sufficiently sized U.S. and EU peer group of companies disclosing detailed compensation information, a number of companies were added to the European peer group following a qualitative review by AON Radford to identify companies with relevant similarities in business model and therapeutic focus. This leads to the following selection of peer groups used by us in the 2022 benchmark for the 2023 compensation plans:
Note: for completeness’ sake, this is not the peer group the Company used in 2023 for its 2024 remuneration. The 2023 benchmark takes into account ongoing discussions and insight on the development of the 2021 Remuneration Policy and plans, as well as stakeholder feedback received. On or around the date of this report, the peer group for the
132
2024 remuneration will be reported on the Company’s website at https://www.argenx.com/investors/governance/remuneration-policy.
Company Name |
| Company Ticker |
| Country of Trade |
Abcam Plc | ABC | GBR | ||
Acadia Healthcare Company, Inc. | ACHC | USA | ||
ALK‐Abelló A/S | ALK.B | DNK | ||
Alnylam Pharmaceuticals, Inc. | ALNY | USA | ||
Amicus Therapeutics, Inc. | FOLD | USA | ||
Ascendis Pharma A/S | ASND | USA | ||
BeiGene, Ltd. | 6160 | USA | ||
Biohaven Pharmaceutical Holding Company Ltd. | BHVN | USA | ||
BioMarin Pharmaceutical Inc. | BMRN | USA | ||
BioNTech SE | BNTX | USA | ||
Blueprint Medicines Corp | BPMC | USA | ||
CRISPR Therapeutics AG | CRSP | USA | ||
Denali Therapeutics Inc | DNLI | USA | ||
Evotec SE | EVT | DEU | ||
Galapagos NV | GLPG | NLD | ||
Genmab A/S | GMAB | DNK | ||
Hikma Pharmaceuticals Plc | HIK | GBR | ||
Horizon Therapeutics Public Limited Company | HZNP | USA | ||
Idorsia Ltd | IDIA | CHE | ||
Incyte Corporation | INCY | USA | ||
Intellia Therapeutics, Inc. | NTLA | USA | ||
Intra‐Cellular Therapies, Inc. | ITCI | USA | ||
Ionis Pharmaceuticals, Inc. | IONS | USA | ||
Mirati Therapeutics, Inc. | MRTX | USA | ||
Neurocrine Biosciences, Inc. | NBIX | USA | ||
Recordati SpA | REC | ITA | ||
Sarepta Therapeutics, Inc. | SRPT | USA | ||
Seagen Inc. | SGEN | USA | ||
Swedish Orphan Biovitrum AB | SOBI | SWE | ||
UCB SA | UCB | BEL | ||
uniQure N.V. | QURE | USA | ||
Vifor Pharma AG | VIFN | CHE |
Award levels
Our Board of Directors sets award levels based on the outcome of our benchmarking exercise. Our remuneration policy, contains the following framework in this respect:
| Non-executive directors |
| Senior management team | |
Cash-based compensation | 50th percentile of the companies in the global reference group | 50th percentile of U.S. companies in the reference group for U.S.-based executives, and at or around the 75th percentile of EU companies in the reference group for EU-based executives | ||
Equity-based compensation | 50th percentile of the U.S. companies in the reference group | 50th to 75th percentile of the U.S. companies in the reference group |
133
NEO remuneration
This chapter contains a detailed overview of the remuneration paid for the year 2023 to the following NEOs: the CEO, the CFO and the COO. Of these NEOs, only the CEO is a statutory director of argenx. The remuneration of the NEOs in 2023 consisted of base salary, variable cash remuneration, company equity and benefits.
Executive Remuneration Policy
The majority of executive compensation is provided in the form of variable remuneration, which is a combination of performance dependent (short-term cash incentives, stock options) and service dependent (RSUs) compensation. Variable (short-term) compensation allows the Board of Directors to set challenging annual objectives aligning the priorities of the NEOs with the short-term strategic objectives of the Company. Company equity in the form of stock options provides an incentive to the NEOs to contribute to Company (stock price) value increase over the long-term (three years) vesting period of the stock options. Company equity in the form of RSUs also provides an incentive for value creation over the long-term (four years) vesting period of the RSUs. The combination of variable pay, stock options and RSUs ensures a balanced incentive for short term focus on and performance of near term strategic targets, while contributing to sustainable long-term value creation and ensuring long-term commitment (retention) of the executive. In addition, the Company provides market standard severance arrangements and pension and fringe benefits, including a corporate bonus of maximally €3,948 ($4,266) in accordance with Belgian practice. Moreover, in accordance with the DCGC, when determining the remuneration package of the executives, scenario analyses are performed annually and taken into account in setting the total remuneration levels and target and maximum awards under the short- and long-term incentive plans.
Total executive remuneration
The following table sets forth the total value of the remuneration paid to the NEOs for the last three years:
(in USD) | Base salary (1) | Base salary in % change vs the prior year (1) | Sign on bonus | Corporate bonus | Variable short-term incentive | Variable cash | Compensation in the form of stock options (2) | Compensation in the form of RSUs | Other benefits (3) | % fixed (of total) (4) | Total | |||||||||||
CEO - Tim Van Hauwermeiren | ||||||||||||||||||||||
2023 | 655,787 | 0% | — | — | 590,215 | 60% | 8,084,605 | (5) | 2,575,174 | 39,054 | 6% | 11,944,835 | ||||||||||
2022 | 638,901 | 10% | — | — | 766,682 | 60% | 4,174,684 | 2,159,689 | 38,342 | 9% | 7,778,298 | |||||||||||
2021 | 651,986 | 5% | — | 1,186 | 586,787 | 60% | 3,895,370 | 2,084,509 | 45,177 | 10% | 7,265,014 | |||||||||||
CFO - Karl Gubitz | ||||||||||||||||||||||
2023 | 516,043 | 6% | — | 3,556 | 260,866 | 40% | 2,626,062 | 1,287,587 | 62,798 | 12% | 4,756,913 | |||||||||||
2022 | 487,600 | 79% | — | 3,745 | 243,800 | 40% | 2,623,633 | 1,356,048 | 91,203 | 12% | 4,806,030 | |||||||||||
2021 | 271,646 | n.a. | (6) | — | 2,235 | 108,659 | 40% | 3,181,721 | 1,629,272 | 31,809 | 6% | 5,225,342 | ||||||||||
COO - Karen Massey (7) | ||||||||||||||||||||||
2023 | 481,471 | n.a. | 338,000 | (8) | 2,921 | 467,662 | 50% | 3,939,093 | 2,296,517 | 127,393 | 8% | 7,653,057 | ||||||||||
COO - Keith Woods (9) | ||||||||||||||||||||||
2023 | 305,022 | (48)% | — | — | — | — | — | — | 46,034 | 100% | 351,056 | |||||||||||
2022 | 583,774 | 5% | — | 3,745 | 583,774 | 50% | 2,601,982 | 1,364,014 | 205,032 | 15% | 5,342,321 | |||||||||||
2021 | 555,975 | 5% | — | 4,095 | 347,484 | 50% | 2,430,402 | 1,316,532 | 116,041 | 14% | 4,770,529 | |||||||||||
| (1) | The base salary of the CEO is paid in EUR (for 2023 base salary the exchange rate 1.0815 EUR/$ used in this table), the base salary of the COO is paid in CHF (for 2023 base salary exchange rate of 1.1135 EUR/CHF used in this table). The percentage presenting the change in salary is calculated using the currency of payment. |
| (2) | Amounts shown represent the expenses with respect to stock options measured using the Black Scholes formula. For a description of the assumptions used in valuing these awards, see “Note 13—Share-based payments” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference. |
| (3) | Other benefits consists of the lease of a company car, employer-paid medical insurance premiums, pension contributions, social security costs and other allowances. |
| (4) | Fixed compensation is considered as Base salary and Other benefits. |
(5) | Target pay level set in number of options and RSUs as part of benchmark performed in September of the prior year (target value $6,986,986, grant occurred on the first business day of July 2023. Share price increase between setting the grant using argenx’s 30-day average stock price of |
134
$366.58 as of July 22, 2022 and the share price of $389.73 at the date of grant) explains variation between target compensation level and the final calculation displayed in the table above. These amounts do not reflect the actual economic value realized by the beneficiary. Amounts shown represent the expenses with respect to the Stock Options awards granted in 2023 measured using the Black Scholes formula with unobservable assumptions. The assumptions used in the fair valuation differ between Belgian beneficiary versus non-Belgian beneficiary. The fair value of equity granted to Belgian beneficiaries was higher than that of non-Belgian beneficiaries resulting in Mr. Van Hauwermeiren’s stock based compensation expense to be higher than other beneficiaries. For a description of the assumptions used in valuing these awards, see “Note 13—Share-based payments” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
| (5) | Karl Gubitz joined as CFO in June 2021, and consequently no comparison for base salary 2021 to 2020 is possible, as well as comparison for base salary 2022 to 2021 being distorted. |
| (6) | Karen Massey joined as COO in March 2023, and consequently no comparisons to 2022 and before were possible, and Ms. Massey’s remuneration shows the remuneration paid for the period March 13, 2023 through December 31, 2023. Her variable pay pay-out has been pro-rated to reflect this as well. |
| (7) | In 2023, the Company paid a sign-on bonus to Karen Massey to allow the Company to make an overall competitive offer of employment and in recognition of lost corporate benefits as a result of early departure at Ms. Massey’s previous employer. Ensuring a competitive offer in this way and securing Ms. Massey as the Company’s new COO was deemed by the Board of Directors to be in the best interest of the Company and its stakeholders. |
| (8) | Keith Woods resigned as COO March 2023 and his employment relationship ended on June 30, 2023 and consequently the remuneration numbers show his remuneration for the period January 1, 2023 through June 30, 2023. No equity award or variable pay was paid to Mr. Woods in the year ended December 31, 2023. |
Base salary
In 2023, compared to 2022, the base salary of the NEOs was increased in line with the total argenx employee population merit increase guidelines (CEO +0%, CFO +6%, COO joined in 2023). These increases followed a review of the individual’s performance over the preceding year(s), in light of comprehensive analysis of benchmark data showing the relative positioning of base salaries compared to the relevant external and internal peers. This process ensures that the Company’s compensation packages are a fair reflection of individual performance while also remaining competitive and aligned with the market. The merit principles and base pay increase framework applied are identical to those applicable to all employees in the organization and are based on the individuals’ performance and contributions over the preceding period. From 2022 to 2023, our CEO declined to receive a base pay increase.
With respect to the CFO, the Board of Directors recognized outstanding performance in 2022, including the achievement and overachievement of short-term targets, and established that the CFO’s pay was below the midpoint of peer reviewed base pay for CFOs in the reference group. Consequently, and in line with pay practice applied consistently across all employees, the CFO’s base pay was increased with a merit increase and an additional increase to move the CFO closed to the benchmarked midpoint, totalling a 6% base pay increase in 2023 versus 2022.
Variable cash
The NEOs were eligible for a variable cash payment for the performance of pre-defined short term performance targets in 2023, with the target variable cash compensation set as a percentage of their base salary (60% for CEO, 50% for COO, 40% for CFO). The Board of Directors has set a cap of 200% pay-out per target, and a 200% overall pay-out cap. The Board of Directors evaluated the pay-out of each target, with ‘at target’, ‘maximum per target’ and ‘actual pay-out’ explained in detail in the table below. In addition, the Board of Directors has discretion to adjust the pay-out if the total outcome would not fairly represent pay-for-performance. If such discretion is used, it will be explained in detail in the remuneration report.
CEO
When considering the variable pay pay-out of the CEO, the Board of Directors primarily reviewed whether the key objectives of the Company’s business plan for 2023 were achieved. These key objectives were:
| (i) | delivering on the revenue targets for VYVGART by achieving the Company’s ambitious commercial business plan; |
| (ii) | growing and developing the pipeline for identified products, product candidates and indications as well as new innovations, building sustainable long term value creation potential for the Company: |
135
| a. | obtaining timely VYVGART subcutaneous approval |
| b. | subject to positive trial outcome, submitting high quality CIDP BLA in minimal time; and |
| c. | adding at least 3 new highly innovative programs to the pipeline, stretch target of 5 overachieved); and |
| (iii) | successful succession, hiring and onboarding of business-critical functions (including several new members of the Board of Directors and COO succession, plus a record number of new company hires across teams), delivering on the highly ambitious hiring plan and protecting and enhancing the Company’s culture through a period of explosive growth; and |
| (iv) | considering a number of expected clinical ‘moments of truth’ in relation to planned clinical trial readouts (CIDP, ITP, PV, MMN) and another critical year for commercial execution, the CEO needed to invest heavily in transparent and balanced communication, proactively and continuously ensuring data-based expectations and organizational resilience whilst retaining trust in the Company’s ability to execute. This needed to be achieved both externally (communications with investors) and internally as head of the Company’s senior management. |
Whereas the total target achievement of the CEO leads to a 125% of target pay-out (details provided below), the Board of Directors used its discretion to award an additional $98,368 (25% of target incentive) in recognition for the successful delivery of the Company’s business plan including the key objectives outlined above, and giving special recognition to the continued success of the commercial launches which well exceeded internal and external expectations. The Board of Directors deemed it in the interest of the Company and its stakeholders to reward the CEO for this high quality execution and its impact on the Company’s sustainable long term value creation trajectory.
The achievement of the targets as set out below, plus the discretionary upward adjustment have led to an overall payout of $590,215 of variable pay to the CEO, representing a pay-out of 150% of target pay-out and representing 75% of the maximum opportunity.
136
Personal targets set for the CEO, in addition to his overall responsibility for delivering the business plan, were the following:
Target | Measurement (how the Board of Directors evaluated the target) | % | Target pay-out | Max pay-out (USD) | Achievement | Actual pay-out (% of target) | Actual pay-out (USD) |
|---|---|---|---|---|---|---|---|
Line up the next wave of immunology breakthroughs: at least 5 new highly innovative programs entered the pipeline | Baseline: at least 3 new programs, Stretch: 5 or more | 25 | 98,368 | 196,736 | Overachieved Number of programs added significantly exceeded stretch target, warranting maximum pay-out of target. | 200% | 196,736 |
Proactively manage argenx’s clinical moments of truth ●Build organizational resilience ahead of key clinical trial readouts ●Ensure data based expectations internally and externally ahead of key clinical trial read-outs ●Protect and enhance investors’ trust in argenx’s ability to execute | External and internal trust in argenx’s ability to execute maintained, even in the context of some setbacks Support of key long term shareholders maintained Ability to attract and retain top talent preserved and/or enhanced | 25 | 98,368 | 196,736 | Achieved Continued support of key shareholders maintained, key talent retained and further key talent hired and onboarded, throughout significant wins (CIDP) and setbacks (ITP, PV) | 100% | 98,368 |
Future-proof company leadership, strengthen board effectiveness. Support successful board succession, maximally leverage the board as a resource | Successful selection, hiring and onboarding of new COO Continued access to talent, knowledge and expertise of departing COO, CMO and (founder) CSO, if feasible Ensured excellent onboarding of new board talent, positioned new board members for maximum impact
| 25 | 98,368 | 196,736 | Achieved Successful hiring and onboarding of top quality COO Retiring COO, CMO and founder CSO positioned for continued impact through long term Board of Directors’ committee advisory roles Successful onboarding of 3 new key Board of Directors positions | 100% | 98,368 |
Execute the 2023 hiring plan, delivering on successful onboarding of a record number of new hires (including 2022 hires, integrate what argenx scaled) in support of the company’s execution ambitions. Safeguard and enhance the corporate culture and align the entire employee base behind the strategic priorities | Company wide understanding of and support for the business plan and alignment around top priorities 2023 hiring plan delivered Record number of new hires throughout 2022 and 2023 successfully onboarded and embraced the argenx cultural values Corporate culture protected, no critical talent departures, voluntary turnover remained stable | 25 | 98,368 | 196,736 | Achieved Company business plan delivered through exceptional cross functional and cross regional collaboration and commitment of all employees Voluntary turnover rates remained relatively stable (less than 1% deviation from 2022 number) and on the low end of market averages (4,27% (2022) to 5% (2023)) Broad participation (295 argonauts across regions and functions) participated in newly launched dedicated forum designed to protect and enhance argenx’s company cultural pillars | 100% | 98,368 |
CFO
When considering the variable pay pay-out of the CFO, the Board of Directors primarily reviewed whether the following key objectives of the Company’s business plan for which the CFO had key responsibilities for 2023, were achieved. These key objectives were:
| (i) | delivering on the revenue targets for VYVGART (stretch target ‘revenue as per annual operating planning’ target significantly exceeded); |
137
| (ii) | considering VYVGART’s recent launch and continued challenges in a highly competitive environment, proactively and continuously ensure data-based external expectations around financial performance, while retaining trust in the Company’s ability to execute; and |
| (iii) | successful transitioning and onboarding of new Chairman of the Audit & Compliance Committee and continued strong audit performance (internal and external). |
The personal targets set for the CFO were the following:
Target | Measurement (how the Board of Directors evaluated the target) | % | Target pay-out | Max pay-out (USD) | Achievement | Actual pay-out (% of target) | Actual pay-out (USD) |
Raise at least $ 500 million of capital on favorable terms to finance the company’s increased ambition level and corresponding business plan | Achievement: at least $ 500 million raised on favourable terms Stretch: $ 750 million raised on favourable terms | 25 | 51,173 | 104,346 | Overachieved $1 billion+ raised on competitive terms, biggest biotech follow-on financing in the history of Nasdaq (at that time), revised business plan fully financed | 200% | 104,346 |
Ensure alignment of external and internal expectations around efgartigimod launch | Financial performance largely aligned or slightly above street expectations Continued support and retention of key shareholders and key talent by continuing to build on the company’s reputation for transparency and reliability | 25 | 51,173 | 104,346 | Achieved Four quarterly ‘beat and raise’ events without significant gaps between internal and external expectations | 100% | 52,173 |
Streamline and improve financial planning processes throughout the company, simplify where possible, significantly reduce time spent by non-financial staff on financial planning processes | Significant simplifications delivered across the company for financial planning and management processes Fewer distractions and increased focus on innovation | 25 | 52,173 | 104,346 | Achieved Broader company leadership recognized argenx financial planning process as best in class, delivering a simplified financial planning process with excellent outcomes, allowing the teams to focus on their core responsibilities while benefiting from high quality financial planning | 100% | 52,173 |
Protect and preserve company and critical assets, further build out working relationship with audit & compliance committee and ensure successful onboarding of new audit and compliance committee chairperson, support high quality internal and external audit processes ensuring excellence in transparency | Ensured excellent onboarding of new chairman of the audit and compliance committee, committee positioned for maximum impact | 25 | 52,173 | 104,346 | Achieved Successful onboarding of new audit and compliance committee chairperson, excellent working relationship with internal and external auditors facilitated, high quality processes and high levels of transparency led to clean audit outcomes | 100% | 52,173 |
COO
When considering the variable pay pay-out of the COO, the Board of Directors primarily reviewed whether the key commercial and operational objectives of the Company’s business plan for 2023 were achieved. These key objectives were:
| (i) | the new COO onboarding rapidly and successfully, positioning herself for high impact and designing her multi-year strategic plan; |
| (ii) | delivering on the revenue targets for VYVGART; and |
| (iii) | executing the succession, hiring and onboarding of business critical (commercial) functions including in new regions, delivering on a highly ambitious hiring plan while protecting and enhancing the Company’s culture through a period of explosive growth. |
138
The personal targets set for the COO, in addition to her overall responsibility for delivering commercial performance, were the following:
Target | Measurement (how the Board of Directors evaluated the target) | % | Target pay-out | Max pay-out (USD) | Achievement | Actual pay-out (% of target) | Actual pay-out (USD) |
Achieve annual operating plan targets for commercial revenue | At target: not disclosed Stretch: exceeding target by at least 10% | 35 | 109,121 | 218,242 | Overachieved $1.2 billion revenues achieved, significantly above internal and external expectations over $ 1 billion revenues in the US alone 4 quarterly beat & raise events | 200% | 218,242 |
Responsibly build out argenx’s global expansion plan across key non-US regions, fill key positions | Key aspects of business plan for non-U.S. regions delivered | 15 | 46,766 | 93,532 | Overachieved Stretch goals in business plan relating to non-us commercial expansion delivered, including successful Canada entry and first sales, successful execution of key distribution partnerships, robust business cases built for new regions, remarkable wins in Germany, Italy, Spain, successful launch in China | 200% | 93,532 |
Implement commercial operating model for consistent launch excellence, reflecting argenx’s culture and values | High impact operating model for commercial launches designed Put in place organizational design which sets us up for long term commercial success
| 25 | 77,944 | 155,887 | Achieved Successful internal restructuring of the commercial operating model which built cross-functional indication and field teams fully in line with the company’s cultural pillars and while significantly overachieving revenue targets Exceeded expectations right after joining, rapidly building real trust and support throughout the global organization and earning the full trust and support of the commercial (and field based) teams, setting the COO up for long term success and organizational impact | 100% | 77,944 |
Develop and gain Board of Directors approval of argenx 2030 commercial strategy, identifying key strategic options and investment scenario | Compelling 2030 vision designed with broad buy in from management team and endorsed by the Board of Directors | 25 | 77,944 | 155,887 | Achieved Built out high quality multi-year commercial strategy for future value creation, aligned the management team and the Board of Directors behind this strategic plan mission 2030, plan reviewed, vetted and approved by the Board of Directors | 100% | 77,944 |
Corporate bonus
All employees are eligible to annually earn a corporate bonus with a maximum of €3,948 ($4,266) per year, based on Company-wide goals. In 2023, the targets focused on (i) simplifying high-impact cross-functional processes, (ii) saving more on an undisclosed dollar amount in negotiated spend to advance financial responsibility and (iii) increased cybersecurity awareness. In 2023, the corporate bonus was achieved by 83.34% and a corresponding pay-out of €3,291/ $3,556 was made to all employees.
139
Equity
In 2023, the Company granted a mix of stock options and RSUs to the NEOs. The number of instruments to be granted in the course of 2023 were determined pursuant to the benchmark exercise performed with the help of AON Radford in September of 2022, where the equity compensation levels of CEO, CFO and COO roles within the Company’s reference group were reviewed. The target values for long term incentives were then converted into a number of stock options and a number of RSUs to be granted, using a Black Scholes valuation of $151,03 per stock option and a value of $366,58 per RSU, based on a 30-day average stock price used for the August 2022 benchmark. This number of equity instruments was then embedded into the equity allocation scheme for 2023. It is noted that as a result of the method of fixing the number of instruments based on the benchmark value and the time in between the benchmark and the grant, the value of the grant as ultimately reported will differ from the target value if the stock price has changed (positively or negatively) between the date of fixing the allocation scheme and the date of the grant. More specifically, if the stock price increases between date of setting the allocation scheme and the grant date, the Black Scholes value of the stock options will increase, assuming all other parameters stay stable. The Company is taking concrete steps to close the time gap between the benchmark and the grant date, as will be further explained in the Draft 2024 Remuneration Policy and accompanying explanatory notes.
Specifically for the COO, the Board of Directors decided to grant equity in excess of the base numbers for the COO role, as a means to attract the new COO in a highly competitive talent market. The Board of Directors deemed enabling Ms. Massey to join the Company of crucial importance for the Company’s long term succession planning.
The following table sets out the number, value and key terms of equity instruments granted to the NEOs in 2023:
RSUs granted in 2023 | Stock options granted in 2023 | ||||||||||||||||||||||||||||||||||
Name | # RSUs | Key terms | Value at grant | Benchmark value | # Stock options | Exercise price in € | Exercise price in $ | Key terms | Value at grant(5) | Benchmark value(5) | Total | ||||||||||||||||||||||||
Tim Van Hauwermeiren - CEO | |||||||||||||||||||||||||||||||||||
| 6,700 | (1) | $ | 2,575,174 | $ | 2,456,086 | 30,000 |
| € | 355.40 | $ | 387.35 | (3) | $ | 8,084,605 (6) | $ | 4,530,900 | $ | 10,659,779 | ||||||||||||||||
Karl Gubitz - CFO | |||||||||||||||||||||||||||||||||||
3,350 | (1) | 1,287,587 | 1,228,043 | 15,000 | 355.40 | 387.35 | (4) | 2,626,062 | 2,265,450 | 3,913,649 | |||||||||||||||||||||||||
Karen Massey - COO | |||||||||||||||||||||||||||||||||||
5,025 | (1) | 1,931,380 | 1,842,065 | 22,500 | 355.40 | 387.35 | (4) | 3,939,093 | 3,398,175 | 5,870,474 | |||||||||||||||||||||||||
Sign-on grant | 950 | (2) | 365,137 | 348,251 | — | — | — | — |
— | — | 365,137 | ||||||||||||||||||||||||
Keith Woods – COO | |||||||||||||||||||||||||||||||||||
— | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||
| (1) | RSUs vest and are settled in 4 equal instalments of 25% over a 4-year period. |
| (2) | RSUs vested on the date of the grant. |
| (3) | 1/3 stock options vests after year 1. 2/3 stock options vest in monthly instalments in year 2 and 3. Stock options are not exercisable until the 4th calendar year after the grant year. |
| (4) | 1/3 stock options vests after year 1. 2/3 stock options vest in monthly instalments in year 2 and 3. |
| (5) | Target pay level set in number of stock options and RSUs as part of benchmark performed in September of the prior year (target value $6,986,986, grant occurred on the first business day of July 2023), share price increase between setting the grant using argenx’s 30-day average stock price of $366.58 as of July 22, 2022 and the share price of $389.73 at the date of grant explains variation between target compensation level and the final calculation displayed in the table above. These amounts do not reflect the actual economic value realized by the beneficiary. Amounts shown represent the expenses with respect to the stock options awards granted in 2023 measured using the Black Scholes formula with unobservable assumptions. The assumptions used in the fair valuation differ between Belgian beneficiaries versus non-Belgian beneficiaries. The fair value of equity granted to Belgian beneficiaries was higher than that of non-Belgian beneficiaries resulting in Mr. Van Hauwermeiren’s stock based compensation expense to be higher than other beneficiaries. For a description of the assumptions used in valuing these awards, see Note 13 “Share-based payments” in our consolidated financial statements. |
| (6) | The reason that this amount is more than 2x the amount of the CFO, even though the number of equity instruments is exactly 2x that of the CFO, is due to different assumptions used in valuation applicable for Belgian based employees than for U.S. based employees. Please see footnote (1) for further details. |
140
The table below shows (i) the stock options held as of January 1, 2023, (ii) the stock options granted to the NEOs which vested during the year ended December 31, 2023, (iii) the number of stock options scheduled to vest in the years ending December 31, 2024, December 31, 2025 and December 31, 2026 and (iv) the respective exercise price of such stock options. Each stock option was granted pursuant to the Equity Incentive Plan:
Information regarding the reported financial year | ||||||||||||||||||||||||||||||
Opening | ||||||||||||||||||||||||||||||
|
|
|
|
|
|
| balance |
| During the Year |
| Closing balance | |||||||||||||||||||
Name of | Performance Period | Award | Vesting | End of | Exercise Period | Exercise | Stock options | Stock | Stock | Stock | Stock | Stock options | Stock options | Stock options | Stock options | |||||||||||||||
Tim Van Hauwermeiren, CEO | ||||||||||||||||||||||||||||||
14/12/2017 - 01/12/2020 | 12/14/2017 | (1) | 12/31/2020 | 01/01/2021 - 14/12/2027 | 21.17 | 72,500 | — | 72,500 | — | — | — | — | — | — | ||||||||||||||||
21/12/2018 - 01/12/2021 | 12/21/2018 | (1) | 12/31/2021 | 01/01/2022 - 21/12/2028 | 86.32 | 80,000 | — | — | — | — | — | — | 80,000 | — | ||||||||||||||||
20/12/2019 - 01/12/2022 | 12/20/2019 | (1) | 12/31/2022 | 01/01/2023 - 20/12/2029 | 135.75 | 80,000 | — | — | — | — | — | — | 80,000 | — | ||||||||||||||||
21/12/2020 - 01/12/2023 | 12/21/2020 | (1) | 12/31/2023 | 01/01/2024 - 21/12/2030 | 247.60 | 50,000 | — | — | — | 16,667 | — | — | 50,000 | — | ||||||||||||||||
24/12/2021 - 01/12/2024 | 12/24/2021 | (1) | 12/31/2024 | 01/01/2025 - 24/12/2031 | 309.20 | 25,000 | — | — | — | 8,334 | 8,333 | 8,333 | 25,000 | 25,000 | ||||||||||||||||
23/12/2022 - 01/12/2025 | 12/23/2022 | (1) | 12/31/2025 | 01/01/2026 - 23/12/2032 | 359.60 | 25,000 | — | — | — | 8,333 | 16,667 | 16,667 | 25,000 | 25,000 | ||||||||||||||||
03/07/2023 - 01/07/2026 | 7/3/2023 | (1) | 12/31/2026 | 01/01/2027 - 03/07/2033 | 355.40 | — | 30,000 | — | — | — | 30,000 | 30,000 | 30,000 | 30,000 | ||||||||||||||||
Total | 332,500 | 30,000 | 72,500 | — | 33,334 | 55,000 | 55,000 | 290,000 | 80,000 | |||||||||||||||||||||
Karl Gubitz, CFO | ||||||||||||||||||||||||||||||
01/07/2021 - 01/07/2024 | 7/1/2021 | (1) | n.a. | 01/07/2022 - 01/07/2031 | 255.10 | 24,000 | — | — | — | 8,000 | 4,667 | 4,667 | 24,000 | — | ||||||||||||||||
01/07/2022 - 01/07/2025 | 7/1/2022 | (1) | n.a. | 01/07/2023 - 01/07/2032 | 357.50 | 16,000 | — | — | — | 7,556 | 8,444 | 8,444 | 16,000 | — | ||||||||||||||||
03/07/2023 - 01/07/2026 | 7/3/2023 | (1) | n.a. | 03/07/2024 - 03/07/2033 | 355.40 | — | 15,000 | — | — | 15,000 | 15,000 | 15,000 | — | |||||||||||||||||
Total | 40,000 | 15,000 | — | — | 15,556 | 28,111 | 28,111 | 55,000 | — | |||||||||||||||||||||
Karen Massey, COO | ||||||||||||||||||||||||||||||
03/07/2023 - 01/07/2026 | 7/3/2023 | (1) | n.a. | 03/07/2024 - 03/07/2033 | 355.40 | — | 22,500 | — | — | — | — | 22,500 | 22,500 | — | ||||||||||||||||
Total | — | 22,500 | — | — | — | — | 22,500 | 22,500 | — | |||||||||||||||||||||
Keith Woods, former COO | ||||||||||||||||||||||||||||||
20/12/2019 - 01/12/2022 | 12/20/2019 | (1) | n.a. | 20/12/2020 - 20/12/2029 | 135.75 | 35,000 | — | 35,000 | — | — | — | — | — | — | ||||||||||||||||
21/12/2020 - 30/6/2023 | 12/21/2020 | (1) | n.a. | 21/12/2021 - 21/12/2030 | 247.60 | 50,000 | — | — | — | 16,667 | — | — | 50,000 | — | ||||||||||||||||
24/12/2021 - 30/6/2023 | 12/24/2021 | (1) | n.a. | 24/12/2022 - 31/03/2025 | 309.20 | 16,000 | — | — | — | 10,667 | — | — | 16,000 | — | ||||||||||||||||
23/12/2022 - 30/6/2023 | 12/23/2022 | (1) | n.a. | 23/12/2023 - 31/03/2026 | 359.60 | 16,000 | — | — | 10,667 | 5,333 | — | — | 5,333 | — | ||||||||||||||||
Total | 117,000 | — | 35,000 | 10,667 | 32,667 | — | — | 71,333 | — | |||||||||||||||||||||
(1) | 1/3rd of the stock options vests on the first anniversary of the date of grant and the remaining 2/3rd vest in equal instalments (24 in total) over the next two years, each time upon the 1st day of each next month. |
The table below shows (i) the RSUs held as of January 1, 2023, (ii) the RSUs granted to the NEOs which vested during the year ended December 31, 2023 and (iii) the number of RSUs scheduled to vest in the years ending December 31, 2024, December 31, 2025, December 31, 2026 and December 31, 2027. Each RSU was granted pursuant to the Equity Incentive Plan:
Information regarding the reported financial year | ||||||||||||||||||||||||
Opening | ||||||||||||||||||||||||
The main conditions of RSU plan | balance | During the Year | Closing balance | |||||||||||||||||||||
RSUs held | RSUs | RSUs | RSUs | RSUs | ||||||||||||||||||||
Name of | End of | at the | subject to | awarded | held at | subject to | ||||||||||||||||||
Directors, | Vesting | retention | beginning | RSUs | RSUs | RSUs | a service | and | the closing | a retention | ||||||||||||||
Position | Performance period | Award Date | date | period | of the year | awarded | Forfeited | vested | condition | unvested | of the year | period | ||||||||||||
Tim van Hauwermeiren, CEO | ||||||||||||||||||||||||
24/12/2021 - 24/12/2025 | 24/12/2021 | (1) | n.a. | 4,275 | — | — | 1,425 | — | 2,850 | 2,850 | — | |||||||||||||
23/12/2022 - 23/12/2026 | 23/12/2022 | (1) | n.a. | 5,700 | — | — | 1,425 | — | 4,275 | 4,275 | — | |||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 6,700 | — | — | 6,700 | 6,700 | — | ||||||||||||||
Total | 9,975 | 6,700 | — | 2,850 | — | 13,825 | 13,825 | — | ||||||||||||||||
Karl Gubitz, CFO | ||||||||||||||||||||||||
01/07/2021 - 01/07/2025 | 01/07/2021 | (1) | n.a. | 4,050 | — | — | 1,350 | — | 2,700 | 2,700 | — | |||||||||||||
01/07/2022 - 01/07/2026 | 01/07/2022 | (1) | n.a. | 3,600 | — | — | 900 | — | 2,700 | 2,700 | — | |||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 3,350 | — | — | — | 3,350 | 3,350 | — | |||||||||||||
Total | 7,650 | 3,350 | — | 2,250 | — | 8,750 | 8,750 | — | ||||||||||||||||
Karen Massey, COO | ||||||||||||||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 5,025 | — | — | — | 5,025 | 5,025 | — | |||||||||||||
N/A | 03/07/2023 | (2) | n.a. | — | 950 | — | — | — | 950 | 950 | — | |||||||||||||
Total | — | 5,975 | — | — | — | 5,975 | 5,975 | — | ||||||||||||||||
Keith Woods, former COO | ||||||||||||||||||||||||
24/12/2021 - 30/06/2023 | 24/12/2021 | (1) | n.a. | 2,700 | — | — | 2,700 | — | — | — | — | |||||||||||||
24/12/2021 - 30/06/2023 | 23/12/2022 | (1) | n.a. | 3,600 | — | 2,700 | 900 | — | — | — | — | |||||||||||||
Total | 6,300 | — | 2,700 | 3,600 | — | — | — | — | ||||||||||||||||
(1) RSUs vest over a period of four years with 1/4th of the total grant vesting at each anniversary of the date of grant. | ||||||||||||||||||||||||
(2) RSUs vest at date of grant. | ||||||||||||||||||||||||
Equity holding requirements for executives
In 2023, the Company implemented the following holding requirements for its executive team:
| ● | CEO: 3x base salary |
| ● | Other NEOs: 1x base salary |
141
The minimum equity stake has to be built up over a maximum of five years and continues to apply for the duration of employment and for two years thereafter.
Pension and fringe benefits
The benefits paid to the NEOs are jurisdiction dependent. For the CEO, these included benefits customary in the Belgian market, and which are standard components of Belgian based employees’ packages: pension contributions, a hospitalization insurance, a representation allowance and a company car. For the CFO, these included benefits customary in the U.S. market, and which are standard components of our U.S. based employees’ packages: a company administered health and 401k plan, with a 4% company match. For the COO, these included benefits customary in the Swiss market, and which are standard components of Switzerland based employees’ packages: car allowance, lunch allowance, health insurance allowance, representation allowance and pension contributions.
Severance arrangements
In accordance with our 2021 Remuneration Policy, the CEO has an 18 months’ notice period for termination (or alternatively, 12 months’ severance in lieu of notice). For our other NEOs, no contractual arrangements have been made for severance.
In fiscal year 2023, no severance payments were granted to the NEOs.
Treatment of leaver equity
With respect to Keith Woods, the Board of Directors determined his long-term equity incentives vested in full on June 30, 2023, consistent with the terms of his employment contract and a separate agreement made between him and the Company in which Mr. Woods’ agreed to stay on with the Company as long as necessary to identify, recruit and onboard a suitable replacement and to continue to contribute to long term value creation for the Company as a member of the commercialization committee (all as set out in a service agreement entered into between us and Mr. Woods, and for which no remuneration shall be paid):
| ● | all unvested stock options and RSUs granted prior to 2022 to and held by Mr. Woods vested on June 30, 2023, whereby Mr. Woods shall not be allowed to exercise stock options of which the vesting was accelerated pursuant to this resolution, or sell shares received pursuant to the settlement of RSUs of which the vesting was accelerated pursuant to this resolution, earlier than on the date on which such equity would normally have vested in accordance with the rules of the applicable argenx equity plan (assuming normal continuation of vesting in the situation where Mr. Woods would not have retired from the company). The sole exception to the aforementioned exercise/sell restriction shall be the sale of equity to the extent solely needed to cover tax liabilities directly following from the aforementioned accelerated vesting and/or settlement of equity; and |
| ● | equity granted to Mr. Woods in 2022 vested only through the first anniversary of the grant date and the remainder was forfeited as of December 31, 2023. |
Claw back policy
In the event that any variable remuneration (cash or equity) is paid to members of senior management, including the NEOs, based on financial information which later proves to be incorrect and leads to an accounting restatement (i) due to the material noncompliance of the Company with any financial reporting requirement under applicable securities laws, including any required accounting restatement to correct an error in previously issued financial statements of the Company that is material to the previously issued financial statements of the Company, or (ii) that corrects an error that is not material to previously issued financial statements of the company, but would result in a material misstatement if the error were corrected in the current period or left uncorrected in the current period, then the difference between the paid compensation and the compensation which would have been payable without such accounting restatement, shall be claimed back from the executive, all as further set out in the Executive Compensation Clawback Policy, as adopted by the Board of Directors on July 25, 2023.
142
In fiscal year 2023, no variable remuneration was clawed back and no variable remuneration was adjusted (retroactively).
Remuneration of other senior management members
For the purposes of the equity reporting by the Company under European legislation, all senior level employees reporting directly to the CEO qualify as the Company’s ‘senior management members’, and for the purposes of U.S. governance reporting requirements, as the Company’s ‘executives’. For that reason and in compliance with U.S. disclosure requirements, the remuneration disclosures in relation to this more extensive group of senior personnel (excluding the NEOs) in this remuneration report is presented on an aggregated basis, with the exception of equity remuneration, which is presented on an individual basis.
Aggregate compensation for other senior management members
The following table sets forth information regarding aggregate compensation paid to members of the senior management (other than the NEOs) during the fiscal year ended December 31, 2023.
| Compensation | |
($) | ||
Base salary | $ | 2,202,303 |
Corporate bonus | 17,790 | |
Variable short-term incentive | 1,134,786 | |
Compensation in the form of stock options |
| 13,333,334 |
Compensation in the form of RSUs | 5,534,702 | |
Other benefits |
| 882,154 |
TOTAL |
| 23,105,069 |
| (1) | Other benefits consists of the lease of a company car, employer-paid medical insurance premiums, pension contributions, social security costs and allowances. |
Equity for other senior management members granted in 2023
The following table sets forth information regarding stock option and RSU awards granted to members of the senior management during fiscal year ended December 31, 2023:
RSUs granted in 2023 | Stock options granted in 2023 | |||||||||||||||||||||
Name | # RSUs | Key terms | Value at grant | # Stock options | Exercise price | Exercise price in $ | Key terms | Value at grant (3) | Total | |||||||||||||
Peter Ulrichts |
| 3,350 | (1) | $ | 1,287,587 | 15,000 | € | 355.40 | $ | 387.35 | (2) | $ | 3,420,785 | $ | 4,708,372 | |||||||
Malini Moorthy | 3,350 | (1) | 1,287,587 | 15,000 | 355.40 | 387.35 | (2) | 2,626,062 | 3,913,649 | |||||||||||||
Luc Truyen | 3,350 | (1) | 1,287,587 | 15,000 | 355.40 | 387.35 | (2) | 3,420,785 | 4,708,372 | |||||||||||||
Arjen Lemmen | 3,350 | (1) | 1,287,587 | 15,000 | 355.40 | 387.35 | (2) | 2,626,062 | 3,913,649 | |||||||||||||
Andria Wilk |
| 1,000 | (1) | 384,354 | 4,600 | 355.40 | 387.35 | (2) | 1,239,639 | 1,623,994 | ||||||||||||
| (1) | RSUs vest and are settled in four equal instalments of 25% over a four-year period. |
| (2) | 1/3 of the RSUs vests after year 1. 2/3 vest in monthly instalments in year 2 and 3. |
| (3) | These amounts do not reflect the actual economic value realized by the beneficiary. Amounts shown represent the expenses with respect to the Stock options awards granted in 2023 measured using the Black Scholes formula with unobservable assumptions. The assumptions used in the fair valuation differ between Belgian beneficiaries versus non-Belgian beneficiaries. The fair value of equity granted to Belgian beneficiaries was higher than that of non-Belgian beneficiaries resulting in stock based compensation expense to be higher for Belgian beneficiaries than other beneficiaries. For a description of the assumptions used in valuing these awards, see “Note 13—Share-based payments” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference. |
143
The table below shows (i) the stock options held as of January 1, 2023, (ii) the stock options granted to members of senior management (other than the NEOs) which vested during the year ended December 31, 2023, (iii) the number of stock options scheduled to vest in the years ending December 31, 2024, December 31, 2025 and December 31, 2026 and (iv) the respective exercise price of such stock options. Each stock option was granted pursuant to the Equity Incentive Plan:
Information regarding the reported financial year | ||||||||||||||||||||||||||||
Opening balance | During the Year | Closing balance | ||||||||||||||||||||||||||
Name of | Performance | Award date | Vesting | End of | Exercise | Exercise | Stock options | Stock | Stock | Stock options | Stock options | Stock options | Stock options | Stock options | ||||||||||||||
Peter Ulrichts, CSO |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||
28/06/2018 - 01/06/2021 | 6/28/2018 | (1) | 12/31/2021 | 01/01/2022 - 28/06/2023 | 80.82 | 750 | — | 750 | — | — | — | — | — | |||||||||||||||
21/12/2018 - 01/12/2021 | 12/21/2018 | (1) | 12/31/2021 | 01/01/2022 - 21/12/2023 | 86.32 | 5,250 | — | 5,250 | — | — | — | — | — | |||||||||||||||
20/12/2019 - 01/12/2022 | 12/20/2019 | (1) | 12/31/2022 | 01/01/2023 - 20/12/2029 | 135.75 | 12,870 | — | 7,870 | — | — | — | 5,000 | — | |||||||||||||||
21/12/2020 - 01/12/2023 | 12/21/2020 | (1) | 12/31/2023 | 01/01/2024 - 21/12/2030 | 247.60 | 9,900 | — | — | 2,550 | — | — | 9,900 | — | |||||||||||||||
24/12/2021 - 01/12/2024 | 12/24/2021 | (1) | 12/31/2024 | 01/01/2025 - 24/12/2026 | 309.20 | 3,420 | — | — | 1,140 | 1,140 | 1,140 | 3,420 | 3,420 | |||||||||||||||
23/12/2022 - 01/12/2025 | 12/23/2022 | (1) | 12/31/2025 | 01/01/2026 - 23/12/2027 | 359.60 | 16,000 | — | — | 8,377 | 7,623 | 7,623 | 16,000 | 16,000 | |||||||||||||||
03/07/2023 - 01/07/2026 | 7/3/2023 | (1) | 12/31/2026 | 01/01/2027 - 03/07/2028 | 355.40 | — | 15,000 | — | — | 15,000 | 15,000 | 15,000 | 15,000 | |||||||||||||||
Total | 48,190 | 15,000 | 13,870 | 12,067 | 23,763 | 23,763 | 49,320 | 34,420 | ||||||||||||||||||||
Malini Morthy, Legal Counsil | ||||||||||||||||||||||||||||
01/04/2022 - 01/04/2025 | 4/1/2022 | (1) | n.a. | 01/04/2023 - 01/04/2032 | 282.50 | 24,000 | — | 7,500 | 13,333 | 10,667 | 10,667 | 16,500 | — | |||||||||||||||
03/07/2023 - 01/07/2026 | 7/3/2023 | (1) | n.a. | 03/07/2024 - 03/07/2033 | 355.40 | — | 15,000 | — | 0 | 15,000 | 15,000 | 15,000 | — | |||||||||||||||
Total | 24,000 | 15,000 | 7,500 | 13,333 | 25,667 | 25,667 | 31,500 | 0 | ||||||||||||||||||||
Luc Truyen, CMO | ||||||||||||||||||||||||||||
01/10/2021 - 01/10/2024 | 10/1/2021 | (1) | 12/31/2024 | 01/01/2025 - 01/10/2026 | 259.5 | 24,000 | — | — | 8,000 | 6,667 | 6,667 | 24,000 | 24,000 | |||||||||||||||
23/12/2022 - 01/12/2025 | 12/23/2022 | (1) | 12/31/2025 | 01/01/2026 - 23/12/2027 | 359.6 | 16,000 | — | — | 5,333 | 10,667 | 10,667 | 16,000 | 16,000 | |||||||||||||||
03/07/2023 - 01/07/2026 | 7/3/2023 | (1) | 12/31/2026 | 01/01/2027 - 03/07/2028 | 355.4 | — | 15,000 | — | — | 15,000 | 15,000 | 15,000 | 15,000 | |||||||||||||||
Total | 40,000 | 15,000 | — | 13,333 | 32,334 | 32,334 | 55,000 | 55,000 | ||||||||||||||||||||
Arjen Lemmen, Vice President of Corporate Development & Strategy | ||||||||||||||||||||||||||||
28/06/2018 - 01/06/2021 | 6/28/2018 | (1) | 12/31/2021 | 01/01/2022 - 28/06/2028 | 80.82 | 695 | — | — | — | — | — | 695 | — | |||||||||||||||
21/12/2018 - 01/12/2021 | 12/21/2018 | (1) | 12/31/2021 | 01/01/2022 - 21/12/2028 | 86.32 | 15,952 | — | — | — | — | — | 15,952 | — | |||||||||||||||
20/12/2019 - 01/12/2022 | 12/20/2019 | (1) | 12/31/2022 | 01/01/2023 - 20/12/2029 | 135.75 | 50,000 | — | 12,445 | — | — | — | 37,555 | — | |||||||||||||||
21/12/2020 - 01/12/2023 | 12/21/2020 | (1) | 12/31/2023 | 01/01/2024 - 21/12/2030 | 247.60 | 50,000 | — | — | 16,667 | — | — | 50,000 | — | |||||||||||||||
24/12/2021 - 01/12/2024 | 12/24/2021 | (1) | 12/31/2024 | 01/01/2025 - 24/12/2031 | 309.20 | 16,000 | — | — | 5,334 | 5,333 | 5,333 | 16,000 | 16,000 | |||||||||||||||
23/12/2022 - 01/12/2025 | 12/23/2022 | (1) | n.a. | 23/12/2023 - 23/12/2032 | 359.60 | 16,000 | — | — | 5,333 | 10,667 | 10,667 | 16,000 | 16,000 | |||||||||||||||
03/07/2023 - 01/07/2026 | 7/3/2023 | (1) | n.a. | 03/07/2024 - 03/07/2033 | 355.40 | — | 15,000 | — | 0 | 15,000 | 15,000 | 15,000 | 15,000 | |||||||||||||||
Total | 148,647 | 15,000 | 12,445 | 27,334 | 31,000 | 31,000 | 151,202 | 47,000 | ||||||||||||||||||||
Andria Wilk, Global Head of Quality | ||||||||||||||||||||||||||||
20/12/2019 - 01/12/2022 | 12/20/2019 | (1) | 12/31/2022 | 01/01/2023 - 20/12/2024 | 135.75 | 9,400 | — | 9,400 | — | — | — | — | — | |||||||||||||||
21/12/2020 - 01/12/2023 | 12/21/2020 | (1) | 12/31/2023 | 01/01/2024 - 21/12/2025 | 247.60 | 9,900 | — | — | 2,662 | — | — | 9,900 | — | |||||||||||||||
24/12/2021 - 01/12/2024 | 12/24/2021 | (1) | 12/31/2024 | 01/01/2025 - 24/12/2031 | 309.20 | 4,446 | — | — | 757 | 756 | 756 | 4,446 | 4,446 | |||||||||||||||
23/12/2022 - 01/12/2025 | 12/23/2022 | (1) | 12/31/2025 | 01/01/2026 - 23/12/2027 | 359.60 | 4,600 | — | — | 2,347 | 2,253 | 2,253 | 4,600 | 4,600 | |||||||||||||||
03/07/2023 - 01/07/2026 | 7/3/2023 | (1) | 12/31/2026 | 01/01/2027 - 03/07/2033 | 355.40 | — | 4,600 | — | 770 | 3,830 | 4,600 | 4,600 | 3,830 | |||||||||||||||
Total | 28,346 | 4,600 | 9,400 | 6,536 | 6,839 | 7,609 | 23,546 | 12,876 | ||||||||||||||||||||
| (1) | 1/3rd of the stock options vests on the first anniversary of the date of grant and the remaining 2/3rd vest in equal instalments (24 in total) over the next two years, each time upon the 1st day of each next month. |
144
The table below shows (i) the RSUs held as of January 1, 2023, (ii) the RSUs granted to members of senior management (other than the NEOs) which vested during the year ended December 31, 2023 and (iii) the number of RSUs scheduled to vest in the years ending December 31, 2024, December 31, 2025, December 31, 2026 and December 31, 2027. Each RSU was granted pursuant to the Equity Incentive Plan:
Information regarding the reported financial year | ||||||||||||||||||||||
Opening balance | During the Year | Closing balance | ||||||||||||||||||||
Name of | Performance period | Award | Vesting | End of | RSU’s held at | RSUs | RSUs | RSUs | RSUs | RSUs held | RSUs | |||||||||||
Peter Ulrichts, CSO |
|
|
|
|
|
|
|
|
|
|
| |||||||||||
24/12/2021 - 24/12/2025 | 12/24/2021 | (1) | n.a. | 570 | — | 190 | — | 380 | 380 | — | ||||||||||||
23/12/2022 - 23/12/2026 | 12/23/2022 | (1) | n.a. | 3,600 | — | 900 | — | 2,700 | 2,700 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 7/3/2023 | (1) | n.a. | — | 3,350 | — | 3,350 | 3,350 | — | |||||||||||||
Total | 4,170 | 3,350 | 1,090 | — | 6,430 | 6,430 | — | |||||||||||||||
Malini Morthy, Legal Counsil | ||||||||||||||||||||||
01/04/2022 - 01/04/2026 | 4/1/2022 | (1) | n.a. | 5,400 | — | 1,350 | — | 4,050 | 4,050 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 7/3/2023 | (1) | n.a. | — | 3,350 | — | 3,350 | 3,350 | — | |||||||||||||
Total | 5,400 | 3,350 | 1,350 | — | 7,400 | 7,400 | — | |||||||||||||||
Luc Truyen, CMO | ||||||||||||||||||||||
01/10/2021 - 01/10/2025 | 10/1/2021 | (1) | n.a. | 4,050 | — | 1,350 | — | 2,700 | 2,700 | — | ||||||||||||
23/12/2022 - 23/12/2026 | 12/23/2022 | (1) | n.a. | 3,600 | — | 900 | — | 2,700 | 2,700 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 7/3/2023 | (1) | n.a. | — | 3,350 | — | — | 3,350 | 3,350 | — | ||||||||||||
Total | 7,650 | 3,350 | 2,250 | — | 8,750 | 8,750 | — | |||||||||||||||
Arjen Lemmen, Vice President of Corporate Development & Strategy | ||||||||||||||||||||||
24/12/2021 - 24/12/2025 | 12/24/2021 | (1) | n.a. | 2,700 | — | 900 | — | 1,800 | 1,800 | — | ||||||||||||
23/12/2022 - 23/12/2026 | 12/23/2022 | (1) | n.a. | 3,600 | — | 900 | — | 2,700 | 2,700 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 7/3/2023 | (1) | n.a. | — | 3,350 | — | 3,350 | 3,350 | — | |||||||||||||
Total | 6,300 | 3,350 | 1,800 | — | 7,850 | 7,850 | — | |||||||||||||||
Andria Wilk, Global Head of Quality | ||||||||||||||||||||||
24/12/2021 - 24/12/2025 | 12/24/2021 | (1) | n.a. | 741 | — | 247 | — | 494 | 494 | — | ||||||||||||
23/12/2022 - 23/12/2026 | 12/23/2022 | (1) | n.a. | 1,000 | — | 250 | — | 750 | 750 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 7/3/2023 | (1) | n.a. | — | 1,000 | — | 1,000 | 1,000 | — | |||||||||||||
Total | 1,741 | 1,000 | 497 | — | 2,244 | 2,244 | — | |||||||||||||||
| (1) | RSUs vest over a period of four years with 1/4th of the total grant vesting at each anniversary of the date of grant. |
Non-Executive Remuneration
In accordance with the 2021 Remuneration Policy, the remuneration of the non-executive directors consists of (i) a fixed fee calculated on the basis of their membership or chairmanship of the Board of Directors and/or its committees and (ii) a long-term equity incentive in the form of stock options and RSUs. It is noted that as part of the changes proposed for the 2021 Remuneration Policy, subject to its approval at the 2024 General Meeting, the Company will no longer compensate non-executive directors with stock options, but only in the form of cash and RSUs, please refer to Item 6.B. “Compensation—Remuneration—Say-On-Pay and Proposed Amendments to the Remuneration Policy”.
145
Total non-executive remuneration
The following table sets forth the information regarding the compensation earned by the non-executive directors during fiscal year ended December 31, 2023:
| Fees earned |
|
| ||||||
or paid in | Stock option | RSU | |||||||
cash | awards | awards | Total | ||||||
Name |
| ($) |
| ($) |
| ($) |
| ($) | |
Peter K.M. Verhaeghe |
| 94,629 |
| 431,179 |
| 134,524 | 660,332 | ||
Werner Lanthaler |
| 11,716 |
| — |
| — | 11,716 | ||
Steve Krognes | 64,438 | 377,772 | 193,440 | 635,649 | |||||
Pamela Klein |
| 56,777 |
| 280,113 |
| 134,524 | 471,414 | ||
J. Donald deBethizy |
| 67,592 |
| 280,113 |
| 134,524 | 482,229 | ||
Anthony A. Rosenberg | 62,185 | 280,113 | 134,524 | 476,822 | |||||
James M. Daly |
| 67,592 |
| 280,113 |
| 134,524 | 482,229 | ||
Camilla Sylvest |
| 54,073 |
| 210,085 |
| 101,085 | 365,244 | ||
Ana Cespedes | 54,073 | 140,057 | 67,262 | 261,392 | |||||
Annual cash compensation
The Board of Directors has set the annual base remuneration, the annual remuneration for members of the audit and compliance committee, the research and development committee, the remuneration and nomination committee and the commercial committee and, in each case, the additional remuneration for the respective chairperson as follows:
in USD | |||||||||||||||||||||||||||||||||||
Relevant body |
| Position |
| Fees in EUR |
| Fees in USD | Peter | Werner | Steve | Pamela | J. Donald | Anthony A. | James M. | Camilla | Ana | ||||||||||||||||||||
Board of Directors |
| Chairperson |
| € | 75,000 |
| $ | 81,110 |
| $ | 81,110 |
| $ | — |
| $ | — |
| $ | — |
| $ | — |
| $ | — |
| $ | — |
| $ | — |
| $ | — |
Member | 45,000 | 48,666 | — | 8,111 | 44,611 | 48,666 | 48,666 | 48,666 | 48,666 | 48,666 | 48,666 | ||||||||||||||||||||||||
Audit & Compliance committee | Chairperson | 15,000 | 16,222 | — | 2,704 | 14,870 | — | — | — | — | — | — | |||||||||||||||||||||||
Member | 7,500 | 8,111 | 8,111 | — | — | — | — | 8,111 | 8,111 | — | — | ||||||||||||||||||||||||
Remuneration & Nomination committee | Chairperson | 10,000 | 10,815 | — | — | — | — | 10,815 | — | — | — | — | |||||||||||||||||||||||
Member | 5,000 | 5,407 | 5,407 | 901 | 4,957 | — | — | — | — | — | 5,407 | ||||||||||||||||||||||||
Commercial committee | Chairperson | 10,000 | 10,815 | — | — | — | — | — | — | 10,815 | — | — | |||||||||||||||||||||||
Member | 5,000 | 5,407 | — | — | — | — | — | 5,407 | — | 5,407 | — | ||||||||||||||||||||||||
Research & Development committee | Chairperson | 15,000 | 16,222 | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||
Member | 7,500 | 8,111 | — | — | — | 8,111 | 8,111 | — | — | — | — | ||||||||||||||||||||||||
Total | 94,628 | 11,716 | 64,438 | 56,777 | 67,592 | 62,184 | 67,592 | 54,073 | 54,073 | ||||||||||||||||||||||||||
| (1) | Mr. Lanthaler resigned from our Board of Directors following the board meeting of February 28, 2023 upon appointment and onboarding of Mr. Krognes. |
Compared to 2022, no changes were in 2023 made to the levels of cash compensation for the non-executive directors.
146
Equity compensation
In 2023, in accordance with the 2021 Remuneration Policy, the non-executive directors received grants of stock options and RSUs, as follows:
RSUs granted in 2023 | Stock options granted in 2023 | |||||||||||||||||||||
Name | Exercise price | Exercise price | ||||||||||||||||||||
Name | # RSUs | Key terms | Value at grant | # Stock options | in EUR | in USD | Key terms | Value at grant | Total | |||||||||||||
Peter Verhaeghe |
| 350 | (1) | $ | 134,524 | 1,600 | € | 355.40 | $ | 387.35 | (2) | $ | 431,179 | $ | 565,703 | |||||||
Werner Lanthaler | — | (1) | — | — | — | — | (2) | — | — | |||||||||||||
Steve Krognes | 525 | (1) | 193,440 | 2,400 | 355.40 | 387.35 | (2) | 377,772 | 571,212 | |||||||||||||
Pamela Klein | 350 | (1) | 134,524 | 1,600 | 355.40 | 387.35 | (2) | 280,113 | 414,637 | |||||||||||||
J. Donald deBethizy | 350 | (1) | 134,524 | 1,600 | 355.40 | 387.35 | (2) | 280,113 | 414,637 | |||||||||||||
Anthony A. Rosenberg | 350 | (1) | 134,524 | 1,600 | 355.40 | 387.35 | (2) | 280,113 | 414,637 | |||||||||||||
James M. Daly | 350 | (1) | 134,524 | 1,600 | 355.40 | 387.35 | (2) | 280,113 | 414,637 | |||||||||||||
Camilla Sylvest | 263 | (1) | 101,085 | 1,200 | 355.40 | 387.35 | (2) | 210,085 | 311,170 | |||||||||||||
Ana Cespedes |
| 175 | (1) | 67,262 | 800 | 355.40 | 387.35 | (2) | 140,057 | 207,319 | ||||||||||||
| (1) | RSUs vest and are settled in four equal instalments of 25% over a four-year period. |
| (2) | Stock options vest upon third anniversary of the grant. |
147
The table below shows (i) the stock options held at January 1, 2023, (ii) the stock options granted to the non-executive directors which have vested during the year ended December 31, 2023, (iii) the number of stock options scheduled to vest in the years ending December 31, 2024, December 31, 2025 and December 31, 2026 and (iv) the respective exercise price of such stock options:
Information regarding the reported financial year | ||||||||||||||||||||||||||||
Opening balance | During the Year | Closing balance | ||||||||||||||||||||||||||
Name of Directors | Performance period | Award date | Vesting date | End of retention period | Exercise period | Exercise price of stock option (€) | Stock options held at the beginning of the year | Stock options awarded | Stock options exercised | Stock options vested | Stock options subject to a service condition | Stock options awarded and unvested | Stock options held at the end of the year | Stock options subject to a retention period | ||||||||||||||
Peter K.M. Verhaeghe | ||||||||||||||||||||||||||||
30/09/2014 - 30/09/2017 | 30/09/2014 | (1) | 31/12/2017 | 01/01/2018 - 30/09/2024 | 3.95 | 1,969 | — | 1,969 | — | — | — | — | — | |||||||||||||||
30/09/2014 - 30/09/2017 | 30/09/2014 | (1) | 31/12/2017 | 01/01/2018 - 30/09/2024 | 2.44 | 2,885 | — | 2,885 | — | — | — | — | — | |||||||||||||||
18/12/2014 - 18/12/2017 | 18/12/2014 | (1) | 31/12/2017 | 01/01/2018 - 18/12/2024 | 7.17 | 5,000 | — | 3,000 | — | — | — | 2,000 | — | |||||||||||||||
18/06/2016 - 18/06/2019 | 18/06/2016 | (1) | 31/12/2019 | 01/01/2020 - 18/06/2026 | 11.38 | 10,000 | — | — | — | — | — | 10,000 | — | |||||||||||||||
21/12/2018 - 21/12/2021 | 21/12/2018 | (1) | 31/12/2021 | 01/01/2022 - 21/12/2028 | 86.32 | 10,000 | — | — | — | — | — | 10,000 | — | |||||||||||||||
20/12/2019 - 20/12/2022 | 20/12/2019 | (1) | 31/12/2022 | 01/01/2023 - 20/12/2029 | 135.75 | 10,000 | — | — | — | — | — | 10,000 | — | |||||||||||||||
21/12/2020 - 21/12/2023 | 21/12/2020 | (1) | 31/12/2023 | 01/01/2024 - 21/12/2030 | 247.60 | 10,000 | — | — | 3,333 | — | — | 10,000 | — | |||||||||||||||
24/12/2021 - 24/12/2024 | 24/12/2021 | (2) | 31/12/2024 | 01/01/2025 - 24/12/2031 | 309.20 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
23/12/2022 - 23/12/2025 | 23/12/2022 | (2) | 31/12/2025 | 01/01/2026 - 23/12/2032 | 359.60 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
03/07/2023 - 03/07/2026 | 03/07/2023 | (2) | 31/12/2026 | 01/01/2027 - 03/07/2033 | 355.40 | — | 1,600 | — | — | — | 1,600 | 1,600 | 1,600 | |||||||||||||||
Total | 55,254 | 1,600 | 7,854 | 3,333 | — | 7,000 | 49,000 | 7,000 | ||||||||||||||||||||
Werner Lanthaler | ||||||||||||||||||||||||||||
21/12/2018 - 21/12/2021 | 21/12/2018 | (1) | n.a. | 21/12/2019 - 21/12/2028 | 86.32 | 10,000 | — | 10,000 | — | — | — | — | — | |||||||||||||||
20/12/2019 - 20/12/2022 | 20/12/2019 | (1) | n.a. | 20/12/2020 - 20/12/2029 | 135.75 | 5,580 | — | 5,580 | — | — | — | — | — | |||||||||||||||
21/12/2020 - 21/12/2023 | 21/12/2020 | (1) | n.a. | 21/12/2021 - 21/12/2030 | 247.60 | 10,000 | — | 1,126 | 3,333 | — | — | 8,874 | — | |||||||||||||||
24/12/2021 - 24/12/2024 | 24/12/2021 | (2) | 01/12/2024 | 24/12/2022 - 24/12/2031 | 309.20 | 2,700 | — | — | — | — | — | 2,700 | — | |||||||||||||||
Total | 28,280 | — | 16,706 | 3,333 | — | — | 11,574 | — | ||||||||||||||||||||
Steve Krognes | ||||||||||||||||||||||||||||
03/04/2023 - 03/04/2026 | 03/04/2023 | (2) | 31/12/2026 | 03/04/2024 - 03/04/2033 | 340.70 | — | 2,400 | 2,400 | 2,400 | 2,400 | ||||||||||||||||||
Total | — | 2400 | — | — | — | 2,400 | 2,400 | 2400 | ||||||||||||||||||||
Pamela Klein | ||||||||||||||||||||||||||||
18/06/2015 - 18/06/2018 | 18/06/2015 | (1) | n.a. | 18/06/2016 - 18/06/2025 | 11.44 | — | — | — | — | — | — | — | — | |||||||||||||||
18/06/2016 - 18/06/2019 | 18/06/2016 | (1) | n.a. | 18/06/2017 - 18/06/2026 | 11.38 | — | — | — | — | — | — | — | — | |||||||||||||||
21/12/2018 - 21/12/2021 | 21/12/2018 | (1) | n.a. | 21/12/2019 - 21/12/2028 | 86.32 | 10,000 | — | 8,500 | — | — | — | 1,500 | — | |||||||||||||||
20/12/2019 - 20/12/2022 | 20/12/2019 | (1) | n.a. | 20/12/2020 - 20/12/2029 | 135.75 | 10,000 | — | — | — | — | — | 10,000 | — | |||||||||||||||
21/12/2020 - 21/12/2023 | 21/12/2020 | (1) | n.a. | 21/12/2021 - 21/12/2030 | 247.60 | 10,000 | — | — | 3,333 | — | — | 10,000 | — | |||||||||||||||
24/12/2021 - 24/12/2024 | 24/12/2021 | (2) | 31/12/2024 | 24/12/2022 - 24/12/2031 | 309.20 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
23/12/2022 - 23/12/2025 | 23/12/2022 | (2) | 31/12/2025 | 23/12/2023 - 23/12/2032 | 359.60 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
03/07/2023 - 03/07/2026 | 03/07/2023 | (2) | 31/12/2026 | 03/07/2024 - 03/07/2033 | 355.40 | — | 1,600 | — | — | — | 1,600 | 1,600 | 1,600 | |||||||||||||||
Total | 35,400 | 1,600 | 8,500 | 3,333 | — | 7,000 | 28,500 | 7,000 | ||||||||||||||||||||
J. Donald deBethizy | ||||||||||||||||||||||||||||
18/06/2016 - 18/06/2019 | 18/06/2016 | (1) | n.a. | 18/06/2017 - 18/06/2026 | 11.38 | 10,000 | — | — | — | — | — | 10,000 | — | |||||||||||||||
21/12/2018 - 21/12/2021 | 21/12/2018 | (1) | n.a. | 21/12/2019 - 21/12/2028 | 86.32 | 10,000 | — | — | — | — | — | 10,000 | — | |||||||||||||||
20/12/2019 - 20/12/2022 | 20/12/2019 | (1) | n.a. | 20/12/2020 - 20/12/2029 | 135.75 | 10,000 | — | — | — | — | — | 10,000 | — | |||||||||||||||
21/12/2020 - 21/12/2023 | 21/12/2020 | (1) | n.a. | 21/12/2021 - 21/12/2030 | 247.60 | 10,000 | — | — | 3,333 | — | — | 10,000 | — | |||||||||||||||
24/12/2021 - 24/12/2024 | 24/12/2021 | (2) | 31/12/2024 | 24/12/2022 - 24/12/2031 | 309.20 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
23/12/2022 - 23/12/2025 | 23/12/2022 | (2) | 31/12/2025 | 23/12/2023 - 23/12/2032 | 359.60 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
03/07/2023 - 03/07/2026 | 03/07/2023 | (2) | 31/12/2026 | 03/07/2024 - 03/07/2033 | 355.40 | — | 1,600 | — | — | — | 1,600 | 1,600 | 1,600 | |||||||||||||||
Total | 45,400 | 1,600 | — | 3,333 | — | 7,000 | 47,000 | 7,000 | ||||||||||||||||||||
A.A. Rosenberg | ||||||||||||||||||||||||||||
13/12/2016 - 13/12/2019 | 13/12/2016 | (1) | n.a. | 13/12/2017 - 13/12/2026 | 14.13 | 15,000 | — | — | — | — | — | 15,000 | — | |||||||||||||||
21/12/2018 - 21/12/2021 | 21/12/2018 | (1) | n.a. | 21/12/2019 - 21/12/2028 | 86.32 | 10,000 | — | — | — | — | — | 10,000 | — | |||||||||||||||
20/12/2019 - 20/12/2022 | 20/12/2019 | (1) | n.a. | 20/12/2020 - 20/12/2029 | 135.75 | 8,840 | — | — | — | — | — | 8,840 | — | |||||||||||||||
21/12/2020 - 21/12/2023 | 21/12/2020 | (1) | n.a. | 21/12/2021 - 21/12/2030 | 247.60 | 5,840 | — | 2,200 | 3,333 | — | — | 3,640 | — | |||||||||||||||
24/12/2021 - 24/12/2024 | 24/12/2021 | (2) | 31/12/2024 | 24/12/2022 - 24/12/2031 | 309.20 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
23/12/2022 - 23/12/2025 | 23/12/2022 | (2) | 31/12/2025 | 23/12/2023 - 23/12/2032 | 359.60 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
03/07/2023 - 03/07/2026 | 03/07/2023 | (2) | 31/12/2026 | 03/07/2024 - 03/07/2033 | 355.40 | — | 1,600 | — | — | — | 1,600 | 1,600 | 1,600 | |||||||||||||||
Total | 45,080 | 1600 | 2,200 | 3,333 | — | 7,000 | 44,480 | 7,000 | ||||||||||||||||||||
James M. Daly | ||||||||||||||||||||||||||||
28/06/2018 - 28/06/2021 | 28/06/2018 | (1) | n.a. | 28/06/2019 - 28/06/2028 | 80.82 | — | — | — | — | — | — | — | — | |||||||||||||||
21/12/2018 - 21/12/2021 | 21/12/2018 | (1) | n.a. | 21/12/2019 - 21/12/2028 | 86.32 | — | — | — | — | — | — | — | — | |||||||||||||||
20/12/2019 - 20/12/2022 | 20/12/2019 | (1) | n.a. | 20/12/2020 - 20/12/2029 | 135.75 | 10,000 | — | 10,000 | — | — | — | — | — | |||||||||||||||
21/12/2020 - 21/12/2023 | 21/12/2020 | (1) | n.a. | 21/12/2021 - 21/12/2030 | 247.60 | 10,000 | — | — | 3,333 | — | — | 10,000 | — | |||||||||||||||
24/12/2021 - 24/12/2024 | 24/12/2021 | (2) | 31/12/2024 | 24/12/2022 - 24/12/2031 | 309.20 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
23/12/2022 - 23/12/2025 | 23/12/2022 | (2) | 31/12/2025 | 23/12/2023 - 23/12/2032 | 359.60 | 2,700 | — | — | — | — | 2,700 | 2,700 | 2,700 | |||||||||||||||
03/07/2023 - 03/07/2026 | 03/07/2023 | (2) | 31/12/2026 | 03/07/2024 - 03/07/2033 | 355.40 | — | 1,600 | — | — | — | 1,600 | 1,600 | 1,600 | |||||||||||||||
Total | 25,400 | 1,600 | 10,000 | 3,333 | — | 7,000 | 17,000 | 7,000 | ||||||||||||||||||||
Camilla Sylvest | ||||||||||||||||||||||||||||
03/10/2022 - 03/10/2025 | 03/10/2022 | (2) | 31/12/2025 | 03/10/2023 - 03/10/2032 | 368.50 | 4,050 | — | — | — | — | 4,050 | 4,050 | 4,050 | |||||||||||||||
03/07/2023 - 03/07/2026 | 03/07/2023 | (2) | 31/12/2026 | 03/07/2024 - 03/07/2033 | 355.40 | — | 1,200 | — | — | — | 1,200 | 1,200 | 1,200 | |||||||||||||||
Total | 4,050 | 1,200 | — | — | — | 5,250 | 5,250 | 5,250 | ||||||||||||||||||||
Ana Cespedes | ||||||||||||||||||||||||||||
23/12/2022 - 23/12/2025 | 23/12/2022 | (2) | 31/12/2025 | 23/12/2023 - 23/12/2032 | 359.60 | 4,050 | — | — | — | — | 4,050 | 4,050 | 4,050 | |||||||||||||||
03/07/2023 - 03/07/2026 | 03/07/2023 | (2) | 31/12/2026 | 03/07/2024 - 03/07/2033 | 355.40 | — | 800 | — | — | — | 800 | 800 | 800 | |||||||||||||||
Total | 4,050 | 800 | — | — | — | 4,850 | 4,850 | 4,850 | ||||||||||||||||||||
(1) 1/3rd of the stock options vests on the first anniversary of the date of grant and the remaining 2/3rd vests in equal monthly instalments (24 in total) over the next two years, each time upon the 1st day of each next month | ||||||||||||||||||||||||||||
(2) stock options vests upon third anniversary of the grant | ||||||||||||||||||||||||||||
148
The table below shows (i) the RSUs held at January 1, 2023, (ii) the RSUs granted to the non-executive directors which have vested during the year ended December 31, 2023 and (iii) RSUs scheduled to vest in the years ending December 31, 2024, December 31, 2025, December 31, 2026 and December 31, 2027 (in number of RSUs):
Information regarding the reported financial year | ||||||||||||||||||||||
Opening balance | During the Year | Closing balance | ||||||||||||||||||||
Name of Directors | Performance period | Award date | Vesting date | End of retention period | RSU’s held at the beginning of the year | RSUs awarded | RSUs vested | RSUs subject to a service condition | RSU’s awarded and unvested | RSU’s held at the closing of the year | RSU’s subject to a retention period | |||||||||||
Peter K.M. Verhaeghe | ||||||||||||||||||||||
24/12/2021 - 24/12/2025 | 24/12/2021 | (1) | n.a. | 450 | — | 150 | — | 300 | 300 | — | ||||||||||||
23/12/2022 - 23/12/2026 | 23/12/2022 | (1) | n.a. | 600 | — | 150 | — | 450 | 450 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 350 | — | — | 350 | 350 | — | ||||||||||||
Total | 1,050 | 350 | 300 | — | 1,100 | 1,100 | — | |||||||||||||||
Werner Lanthaler | ||||||||||||||||||||||
24/12/2021 - 28/02/2023 | 24/12/2021 | (1) | n.a. | 450 | — | 450 | — | — | — | — | ||||||||||||
Total | 450 | — | 450 | — | — | — | — | |||||||||||||||
Steve Krognes | ||||||||||||||||||||||
03/04/2023 - 03/04/2027 | 03/04/2023 | (1) | n.a. | — | 525 | — | 525 | 525 | ||||||||||||||
Total | 0 | 525 | 0 | — | 525 | 525 | — | |||||||||||||||
Pamela Klein | ||||||||||||||||||||||
24/12/2021 - 24/12/2025 | 24/12/2021 | (1) | n.a. | 450 | — | 150 | — | 300 | 300 | — | ||||||||||||
23/12/2022 - 23/12/2026 | 23/12/2022 | (1) | n.a. | 600 | — | 150 | — | 450 | 450 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 350 | — | — | 350 | 350 | — | ||||||||||||
Total | 1,050 | 350 | 300 | — | 1,100 | 1,100 | — | |||||||||||||||
J. Donald deBethizy | ||||||||||||||||||||||
24/12/2021 - 24/12/2025 | 24/12/2021 | (1) | n.a. | 450 | — | 150 | — | 300 | 300 | — | ||||||||||||
23/12/2022 - 23/12/2026 | 23/12/2022 | (1) | n.a. | 600 | — | 150 | — | 450 | 450 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 350 | — | — | 350 | 350 | — | ||||||||||||
Total | 1,050 | 350 | 300 | — | 1,100 | 1,100 | — | |||||||||||||||
A.A. Rosenberg | ||||||||||||||||||||||
24/12/2021 - 24/12/2025 | 24/12/2021 | (1) | n.a. | 450 | — | 150 | — | 300 | 300 | — | ||||||||||||
23/12/2022 - 23/12/2026 | 23/12/2022 | (1) | n.a. | 600 | — | 150 | — | 450 | 450 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 350 | — | — | 350 | 350 | — | ||||||||||||
Total | 1,050 | 350 | 300 | — | 1,100 | 1,100 | — | |||||||||||||||
James M. Daly | ||||||||||||||||||||||
24/12/2021 - 24/12/2025 | 24/12/2021 | (1) | n.a. | 450 | — | 150 | — | 300 | 300 | — | ||||||||||||
23/12/2022 - 23/12/2026 | 23/12/2022 | (1) | n.a. | 600 | — | 150 | — | 450 | 450 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 350 | — | — | 350 | 350 | — | ||||||||||||
Total | 1,050 | 350 | 300 | — | 1,100 | 1,100 | — | |||||||||||||||
Camilla Sylvest | ||||||||||||||||||||||
03/10/2022 - 03/10/2026 | 03/10/2022 | (1) | n.a. | 900 | — | 225 | — | 675 | 675 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 263 | — | — | 263 | 263 | — | ||||||||||||
Total | 900 | 263 | 225 | — | 938 | 938 | — | |||||||||||||||
Ana Cespedes | ||||||||||||||||||||||
23/12/2022 - 23/12/2026 | 23/12/2022 | (1) | n.a. | 900 | — | 225 | — | 675 | 675 | — | ||||||||||||
03/07/2023 - 03/07/2027 | 03/07/2023 | (1) | n.a. | — | 175 | — | — | 175 | 175 | — | ||||||||||||
Total | 900 | 175 | 225 | — | 850 | 850 | — | |||||||||||||||
(1) RSUs vest over a period of four years with 1/4th of the total grant vesting at each anniversary of the date of grant. | ||||||||||||||||||||||
Holding requirements
In 2023, the Company implemented the following holding requirements for non-executive directors: 3x annual cash retainer.
The minimum equity stake is required to be built up over a maximum of five years and continues to apply for the duration of employment and for two years thereafter.
Severance arrangements
In fiscal year 2023, no severance payments were granted to the non-executive directors.
Non-executive equity treatment on departure
In 2023, the Company has updated the terms of the Equity Incentive Plan applicable to non-executive directors, with respect to leaver rules. In particular, and following shareholder feedback on the potential negative impact of having multi-year service based vesting requirements for non-executive director equity, the Equity Incentive Plan was updated to reflect that non-executive directors will lose their unvested equity if they are dismissed at a General Meeting, but not if they resign on their own initiative or if, at the end of their term, they do not apply for re-appointment. In the proposed
149
Draft 2024 Remuneration Policy, the Company is further developing this and proposed a one-year vest term combined with a three-year post-vest holding requirement for equity.
Applying the same principles as for prior board departures after full terms of service, for Mr. Lanthaler, the Company has agreed that equity granted during his eight-year term of service (there was no equity grant in 2022) is deemed vested, but that such equity is not exercisable other than after completion of the vesting terms set at grant. To address specifically the potential tax cost of vested but unexercisable equity, Mr. Lanthaler was granted the right to exercise or sell such portions of his vested equity to allow him to cover immediate tax liability resulting from the vesting of such equity.
PAY RATIOS
Overall pay ratios
The total expense for the non-equity remuneration paid to the CEO (being the only executive director on the Board of Directors) for the year ended December 31, 2023, equalled $1,285,056. The table below shows the evolution over the past five years of CEO compensation, the performance of the Company’s stock price and the median remuneration on a full-time equivalent basis (annualized for the employees who joined or left us during the year) of employees, other than the CEO:
Year ended December 31, | ||||||||||
| 2019 |
| 2020 |
| 2021 |
| 2022 |
| 2023 | |
Base salary of our CEO (EUR) | € | 525,000 | 525,000 | 551,250 | 606,368 | 606,368 | ||||
Base salary of our CEO (USD) | $ | 526,825 | 553,167 | 580,825 | 638,901 | 655,787 | ||||
Non-equity remuneration of our CEO (USD) (base salary, short-term cash incentive, pension contributions and other compensation elements) | $ | 1,001,891 | 1,144,301 | 1,285,136 | 1,443,925 | 1,285,056 | ||||
Non-equity median salary paid to our employees (USD) | $ | 121,603 | 163,062 | 157,349 | 153,193 | 159,500 | ||||
Ratio employee/CEO |
| 12% | 14% | 12% | 11% | 12% | ||||
Average compensation paid to non-executive director (USD) | $ | 60,372 | 57,925 | 54,484 | 48,587 | 59,230 | ||||
Number of employees on December 31 | 188 | 336 | 650 | 843 | 1,148 | |||||
Share price at end of year Euronext (EUR) on December 31 | € | 143.60 | 242.00 | 315.30 | 348.30 | 343.50 | ||||
Share price at end of year Euronext (USD) on December 31 | $ | 161.32 | 296.96 | 357.11 | 371.50 | 379.57 | ||||
The increase in the remuneration ratio between the CEO and other employees between 2022 and 2023 is caused by the increase in salary of employees when base salary of the CEO has been unchanged.
The comparison of non-equity compensation above is made between the compensation paid to the CEO, the Company’s sole executive director, and the median compensation paid to employees. The Company has opted to compare non-equity salaries, because whereas the number of stock options granted is linked to the overall size of remuneration packages granted, the value of equity components depends on the evolution of the Company’s share price, volatility and the risk-free rate, which is unknown at granting and as such the forward-looking valuation methods for stock options normally do not provide an accurate representation of actual economic value granted. In the assumptions used in the fair valuation differ between Belgian beneficiary versus non-Belgian beneficiary. For a description of the assumptions used in valuing these awards, please refer to “Note 13—Share-based payments” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
150
Regional pay ratios
Due to the global spread of employees over multiple continents, we deem it relevant to also include the above comparison separately to U.S. employees, EU employees and Japanese employees. Due to the overall higher compensation level in our business segment in the U.S. and Japan compared to the EU, there is a significant difference in the pay ratio when the CEO’s compensation is compared to the median compensation of all employees (the majority of which are EU citizens), as set out above, or compared to employees in the U.S. and Japan. The following information is provided for reference purposes:
Ratio of non-equity compensation of the median employee compared to the CEO for fiscal year ended December 31, 2023 | ||
All employees |
| 13% |
European employees | 9% | |
North-America Employees | 21% | |
Japan employees | 8% | |
Total employment costs (excluding any costs related to stock options and RSUs) paid in fiscal year 2023 was split between regions as follows:
Total employment costs in fiscal year ended December 31, 2023 | ||
(in millions of $) | ||
Europe | 159.2 | |
North-America | 130.6 | |
Japan | 12.8 | |
Share-based payment ratios
Year ended December 31, |
| ||||||||||
| 2019 |
| 2020 |
| 2021 |
| 2022 |
| 2023 |
| |
Stock options granted to the CEO |
| 80,000 |
| 50,000 |
| 25,000 | 25,000 | 30,000 | |||
Median stock options granted to our employees |
| 2,800 |
| 2,900 |
| 981 |
| 900 | 600 | ||
Ratio employee/CEO |
| 4 | % | 6 | % | 4 | % | 4 | % | 2% | % |
Average number of stock options granted to non-executive directors |
| 10,000 |
| 10,000 |
| 2,869 |
| 3,086 | 1,550 | ||
Median stock options granted to our employees |
| 2,800 |
| 2,900 |
| 981 |
| 900 | 600 | ||
Ratio employee/non-executive directors |
| 28 | % | 29 | % | 34 | % | 29 | % | 39% | % |
3.4.4OTHER DISCLOSURES
Remuneration by subsidiaries
In fiscal year 2023, no remuneration was granted and allocated by subsidiaries or other companies whose financials are consolidated, other than the regular remuneration payments made by the entities with whom members of senior management have their employment contracts.
No loans or guarantees
In fiscal year 2023, no loans were granted to members of senior management and non-executive directors and no guarantees or the like have been granted in favor of any member of senior management or Board of Directors.
Deviations
In fiscal year 2023, the Company did not deviate from the decision-making process for the implementation of the 2021 Remuneration Policy for members of senior management and non-executive directors and no temporary deviations took place from the 2021 Remuneration Policy.
151
Key terms of equity plan applicable to grants in 2023
Stock options granted pursuant to the Equity Incentive Plan shall vest with respect to one third of the shares upon the first anniversary of the date of grant, with the remaining two thirds vesting in 24 equal monthly instalments with the stock options fully vesting upon the third anniversary of the date of grant, subject, in each case, to the optionee’s continued status as a service provider. Stock options are exercisable when vested, and in any case not after the stock option expiration date included in each individual stock option grant, which is 10 years or in the case of Belgian tax resident employees, at their election either five years or 10 years from the date of grant.
Each stock option shall be granted with an exercise price equal to the fair market value upon the date of grant and shall have a term equal to five or 10 years from the date of grant. Optionees may prefer to elect the five-year period as this may limit their personal tax obligations in respect of the stock option in respect to the jurisdiction where stock options are taxed at grant, compared to a 10-year stock option. Stock options granted to Belgian tax resident beneficiaries (including the CEO) are not exercisable prior to the fourth year following the year of the grant. Stock options granted to non-executive directors vest at once on the third anniversary of the date of grant.
RSUs granted under the Equity Incentive Plan shall vest over a period of four years with respect to one fourth of the shares upon each anniversary of the date of grant. At the time of vesting, the holder of such RSUs receives shares in the share capital of the Company for free equal to the number equal of RSUs vested minus a certain number of shares required to cover employee taxes payable by us on behalf of the holder of RSUs, if applicable.
Unvested equity incentives shall vest in the event of a (i) sale, merger, consolidation, tender offer or similar acquisition of shares or other transaction or series of related transactions as a result of which a change in control occurs, (ii) sale or other disposition of all or substantially all of the Company’s assets or (iii) the Company’s dissolution and/or liquidation.
The Board of Directors, upon approval of a majority of the non-executive directors, may amend or terminate the Equity Incentive Plan or may amend the terms of the Equity Incentive Plan, or any outstanding stock options or RSUs, provided that the Company will compensate any affected individual for any direct negative impact of such amendment.
C. BOARD PRACTICES
Director Independence
As a foreign private issuer, under the Nasdaq Listing Rules, we are not required to have a majority independent directors on our Board of Directors, except that our audit and compliance committee is required to consist fully of independent directors. However, our Board of Directors has determined that, taking into account any applicable committee independence standards, all of our non-executive directors, including the members of our audit and compliance committee, are “independent directors” under Rule 10A-3 of the Exchange Act, the Nasdaq Listing Rules and the DCGC. In making such determination, our Board of Directors considered the relationships that each non-executive director has with us and all other facts and circumstances our Board of Directors deemed relevant in determining the director’s independence, including the number of ordinary shares beneficially owned by the director and his or her affiliated entities (if any).
The DCGC requires that the composition of non-executive directors is such that the members are able to operate independently and critically vis-à-vis one another, the executive directors, and any particular interests involved. As of the date of this Annual Report, all non-executive directors meet the independence criteria contained in the DCGC. Therefore, in the opinion of the non-executive directors, the composition of our non-executive directors complies with the independence requirements of best practice provisions 2.1.7 to 2.1.9 of the DCGC. Our Board of Directors has consequently also determined that all members of our committees are independent under the applicable rules of the DCGC.
152
As of the date of this Annual Report (or in any period before), none of the members of our Board of Directors and senior management has or has had a family relationship with any other member of our Board of Directors or senior management.
Directors may be suspended or removed by the shareholders at a General Meeting at any time, with or without cause, by means of a resolution passed by a simple majority of the votes cast. Pursuant to the Dutch Civil Code (Burgerlijk Wetboek) (DCC), executive directors may also be suspended by the board of directors. A suspension of an executive director by the board of directors may be discontinued by the shareholders at any time at a General Meeting.
Diversity
We value diversity among our colleagues as an integral component in building a sustainable growth platform and believe that a diverse workforce enhances our overall performance and success. We take pride in creating and sustaining a culture and environment where each of us can excel. We bring together people with diverse backgrounds experiences and functional expertise. By doing so, we broaden the scope of ideas and creativity essential to developing and delivering innovative therapies to patients. Acknowledging and benefiting from different perspectives promotes diversity of thought and empowers innovation. It also contributes to our commitment to improve the quality of lives of patients, wherefore we need teams with a healthy mix of contrasting perspectives and backgrounds that reflect the diverse communities we serve. We recognize that our people are our greatest strength. Fostering an inclusive work environment where everyone feels safe and encouraged to contribute leads to better work outcomes and supports high levels of employee commitment and retention. We aspire to be a consciously global company. Our success is built on, and dependent on true collaboration in cross-functional and often cross-regional teams in which open communication is encouraged and safeguarded. Everyone has a voice and is encouraged to contribute to the benefit of our common goals, irrespective of race, ethnicity, age, gender or cultural background. Good ideas as well as real concerns are taken seriously, regardless of who brings them forward.
We aim to foster an inclusive work environment in support of our strategic plan and priorities. We continue to raise the bar in this regard, and to commit to measures and goals designed to support our maturing company culture. We have set ourselves the goal of gender balance across all levels at argenx, including our Board of Directors.
In 2022, we adopted our current diversity, equity and inclusion policy, which sets out the basis for our inclusion, equity and diversity management throughout our organization in a way that we believe best supports our business objectives and our people. We monitor and annually report on relevant diversity, equity and inclusion metrics, initiatives and developments in this Annual Report and in our ESG reports, of which an updated version will be published on or around the date of this Annual Report.
Our policy is that we aim to balance our Board of Directors and senior management team in terms of gender, age, background, race, ethnicity, sexual orientation, experience and nationality as much as reasonably possible while still having our Board of Directors and senior management team composed of the best possible candidates overall. It has been and will remain our priority to have the best available specialists on our Board of Directors and in our senior management team, who make a balanced panel of directors and managers able to advise and guide argenx to further growth and success for all its stakeholders. This means we require a number of specialties and character traits to be present. We will seek to further improve diversity on our Board of Directors if and when proposing new appointments to our Board of Directors, whilst recognizing that, considering the specialist nature of our business, aspects other than diversity are relevant as well for the ultimate decision to select a board member.
Our plan of action to achieve our goal of gender balance includes a number of recruitment and development-related initiatives to promote balanced and diversified candidate pools as well as diversity amongst persons receiving promotion and development opportunities. We value our fair, inclusive recruitment process, which is standardized across the organization and focuses on pre-identified ‘what counts’ factors. The process involves a diverse group of colleagues from across the organization, who are provided with training to recognize existing biases. Recruitment decisions are based on a group evaluation of available candidates, to encourage different perspectives. Our onboarding program is designed to promote inclusion by building a strong social fabric across teams, functions and geographic locations. Once hired, employees are encouraged to participate in a personal development program aimed at building on their individual
153
strengths to benefit the broader team and taking into account their individual career aspirations. We offer opportunities for promotion, training and career development solely based on job-related, appropriate criteria such as skills, competencies, experience, aptitude and enthusiasm and giving account to each individual’s experience, ambitions and capabilities.
We will continue to implement our diversity, equity and inclusion policy by seeking new ways to improve and support diversity, equity and inclusion at the Company. We from time to time report on specific initiatives taken with respect to our diversity, equity and inclusion policy in our annual ESG report, of which an updated version will be published on or around the same date as this Annual Report.
In accordance with Dutch legislation, we report annually to the Social Economic Council (Sociaal-Economische Raad) whether or not we have complied with our diversity goals, and if we have not, the reasons for this.
| ● | As at December 31, 2023, our Board of Directors consisted of nine directors, including one executive director and eight non-executive directors. Of the directors who chose to disclose their gender, the Board of Directors contained five male directors and three female directors (non-executive directors), translating into a 55.55% male / 331/3% female balance for our full Board of Directors (compared to five males and three females (non-executive directors) (55.55%/331/3%) as of December 31, 2022) and a 62.5% male / 37.5% female balance for our non-executive directors (compared to 62.5% male/37.5% female as of December 31, 2022). As at December 31, 2023 and December 31, 2022, our Company leadership team consisted of 31 persons, comprised of a mix of 19 males and 12 females, (61% / 39% respectively). Our leadership consists of all full time employees reporting directly to our CEO, as well as all (other) leaders of our largest functions and projects. Each of these positions is characterized by a high impact across the organization, leading a global and cross functional team and having a global reach. We estimate that as of December 31, 2023, 58% of our workforce were female and 42% were male (compared to 63% female and 37% male as of December 31, 2022). |
Board Diversity Matrix (as of the date of this Annual Report)
Country of Principal Executive Offices | The Netherlands | ||||
Foreign Private Issuer in the U.S. | Yes | ||||
Disclosure of gender identity prohibited by Dutch Law | No | ||||
Total Number of Directors | 9 | ||||
Female | Male | Non-Binary | Did Not Disclose Gender identity | ||
Gender: Number of Directors | 3 | 5 | 0 | 1 | |
Demographic Background Categories | Number of Directors in Each Demographic Category | ||||
Underrepresented individual in home country jurisdiction | 1 | ||||
LGTBQ+ | 0 | ||||
Did not disclose demographic background | 8 | ||||
Role of the Board in Risk Oversight
Our Board of Directors is responsible for the oversight of our risk management activities and has specifically designated the audit and compliance committee to assist our Board of Directors in this task and prepare recommendations in this respect to the Board of Directors. While our Board of Directors oversees our risk management, our senior management is responsible for day-to-day risk management processes. Our Board of Directors expects our senior management to consider risk and risk management in each business decision, to proactively develop and monitor risk management strategies and processes for day-to-day activities and to effectively implement risk management strategies adopted by the Board of Directors. We believe this division of responsibilities is the most effective approach for addressing the risks we face.
154
Composition, Appointment and Dismissal
The Articles of Association provide that our Board of Directors will consist of our executive director(s) and non-executive directors. The number of executive directors must at all times be less than the number of non-executive directors. The number of directors, as well as the number of executive directors and non-executive directors, is determined by our Board of Directors, provided that the Board of Directors must consist of at least three members.
Our directors are appointed by the shareholders at a General Meeting for a period of four years as either executive directors or as non-executive directors. In accordance with best practice provision 2.2.1 of the DCGC, executive directors may be reappointed for periods not more than four years at a time. In accordance with best practice provision 2.2.2 of the DCGC, non-executive directors may be reappointed once for a period of four years, after which the non-executive director may be reappointed again for a period of two years, which reappointment may be extended by at most two years. In the event of a reappointment after an eight-year period, reasons will be given in the report of the Board of Directors. The Board of Directors is required to make one or more proposals for each seat on our Board of Directors to be filled. A resolution to nominate a director by our Board of Directors (with support from the remuneration and nomination committee) may be adopted by a simple majority of the votes cast. A nomination for appointment of an executive director must state the candidate’s age and the positions he or she holds, or has held, insofar as these are relevant for the performance of the duties of an executive director. The nomination must state the reasons for the nomination of the relevant person. A nomination for appointment of a non-executive director must state the candidate’s age, his or her profession, the number of shares he or she holds and the employment positions he or she holds, or has held, insofar as these are relevant for the performance of the duties of a non-executive director. Furthermore, the names of the legal entities of which he or she is already a supervisory board member or a non-executive member of the board shall be indicated; if those include legal entities which belong to the same group, a reference to that group will be sufficient. The nomination must state the reasons for the nomination of the relevant person.
Our Board of Directors designates one executive director as CEO. In addition, the Board of Directors may grant other titles to executive directors. Our Board of Directors also designates a non-executive director as chairperson of the Board of Directors and a non-executive director as vice chairperson of the Board of Directors. The legal relationship between an executive member of the Board of Directors and argenx will not be considered as an employment agreement. Employment agreements between an executive director and a Group company (other than argenx SE) are permitted. In the absence of an employment agreement, members of a board of directors generally do not enjoy the same protection as employees under Dutch labor law.
For a discussion of date of expiration of the current term of office and the period during which the person has served in that office, Item 6.A. “Directors, Senior Management and Employees—Directors and Senior Management”.
Except for the arrangements described in Item 7.B. “Related Party Transactions—Agreements with Our Senior Management”, there are no arrangements or understanding between us and any of the executive directors providing for benefits upon termination of their employment, other than as required by applicable law. In addition, the contracts between us and our non-executive directors do not provide for any benefits upon termination.
Committees
In accordance with the DCGC, our non-executive directors can set up specialized committees to analyze specific issues and advise the non-executive directors on those issues and prepare resolutions with respect thereto.
The committees are advisory bodies only, and the decision-making remains within the collegial responsibility of the Board of Directors. The non-executive directors determine the terms of reference of each committee with respect to the organization, procedures, policies and activities of the committee.
Our non-executive directors have established and appointed (i) an audit and compliance committee; and (ii) a remuneration and nomination committee.
155
The composition and function of these committees complies with all applicable requirements of Euronext Brussels, the DCGC, the Exchange Act, the exchange on which the ordinary shares and the ADSs are listed and U.S. SEC rules and regulations.
Only non-executive directors qualify for membership of these committees. The audit and compliance committee and the remuneration and nomination committee may not be chaired by the chairperson of the Board of Directors or by a former executive director of the Company.
In addition to the aforementioned legally required subcommittees, our Board of Directors may also opt to incorporate informal committees consisting of non-executive directors and other internal and external persons in argenx, in order to facilitate discussions and act as a sounding board on specific projects, as well as on a more permanent basis. Our Board of Directors has incorporated a research and development committee and a commercialization committee.
Audit and Compliance Committee
Our audit and compliance committee consists of four members: Steve Krognes (chairperson), effective February 27, 2023, Peter K. M. Verhaeghe, Anthony A. Rosenberg and James M. Daly. Mr. Lanthaler was a committee member and chairperson until February 27, 2023.
Our Board of Directors previously established that Mr. Verhaeghe, Mr. Rosenberg, Mr. Daly, Mr. Krognes satisfy the independence requirements set forth in Rule 10A-3 of the Exchange Act and that both Mr. Lanthaler (up until his resignation effective February 27, 2023) and Mr. Krognes qualify as “audit committee financial experts” as defined by SEC rules and Article 39 paragraph 1 of Directive 2014/56/EU of the European Parliament and of the Council of April 16, 2014 amending Directive 2006/43/EC on statutory audits of annual accounts and consolidated accounts and has the requisite financial sophistication under the Nasdaq Listing Rules. Further, our Board of Directors established that the composition of the audit and compliance committee meets the requirements under the Dutch Decree on Establishing Audit Committees.
Our audit and compliance committee assists our Board of Directors in overseeing the accuracy and integrity of our accounting and financial reporting processes and audits and reviews of our consolidated financial statements as well as non-financial statements (including ESG reporting), the implementation and effectiveness of an internal control system and our compliance with legal and regulatory requirements, the independent auditors’ qualifications and independence and the performance of the independent auditors. Our audit and compliance committee is also responsible for monitoring the status of, and compliance with, our global ethics and compliance program and meets with the head of our ethics and compliance function at least quarterly to discuss the status and overall effectiveness of the program as well as any issues or incidents that occurred and remedial actions needed (if applicable). The committee furthermore oversees climate-related risks and supervises the status of the Company’s cybersecurity program and regularly (at least quarterly) discusses the status thereof with our senior management team.
Our audit and compliance committee is governed by a charter that complies with the listing rules of the Nasdaq Global Market (the Nasdaq Listing Rules) and the DCGC and is publicly available on our website. It is responsible for, among other things, establishing methods and procedures for supervising, and where necessary requiring improvements of, our financial reporting, risk management, ethics and compliance and organization for the purpose of making appropriate recommendations to our Board of Directors in that regard.
Our audit and compliance committee meets as often as is required for its proper functioning, but at least four times a year and at least once a year meets separately with our independent auditor. See Item 6.C “Board Practices—Report Audit and Compliance Committee” for an overview of the number of meetings and attendance rates.
Our audit and compliance committee reports regularly to our Board of Directors on the exercise of its functions. It informs our Board of Directors about all areas in which action or improvement is necessary in its opinion and produces recommendations concerning the necessary steps or resolutions that need to be taken. The audit review and the reporting on that review cover us and our subsidiaries as a whole. The members of the audit and compliance committee are entitled to receive all information which they need for the performance of their function, from our Board of Directors
156
and employees. Every member of the audit and compliance committee shall exercise this right in consultation with the chairperson of the audit and compliance committee.
Report Audit and Compliance Committee
The audit and compliance committee reports regularly to our Board of Directors on the exercise of its functions. It informs our Board of Directors about all areas in which action or improvement is necessary in its opinion and produces recommendations concerning the necessary steps that need to be taken. The audit review and the reporting on that review cover the Company and its subsidiaries as a whole.
In 2023, the main points of discussion at the meetings were the 2022 consolidated financial statements and press release as well as interim consolidated financial statements and press releases, internal audit and external auditors’ reports, the, the review of quarterly forecasts, updates on tax priorities, compliance, cash management, CSRD readiness, the company’s ethics and compliance program, the company’s cyber security program and the company’s privacy program.
In 2023, five audit and compliance committee meetings were held. The meeting attendance rate for our directors is set out in the table below.
| Number of meetings attended in |
| ||
Name | 2023 since appointment | Attendance % | ||
Peter K. M. Verhaeghe | 5 | 100% | ||
Werner Lanthaler(1) | 1 | 100% | ||
Steve Krognes (chairperson)(1) | 4 | 100% | ||
Anthony A. Rosenberg | 5 | 100% | ||
James M. Daly | 5 | 100% |
| (1) | Werner Lanthaler resigned effective February 27, 2023 and was succeeded by Steve Krognes effective February 27, 2023. |
Remuneration and Nomination Committee
We have established a remuneration and nomination committee, which serves as both the remuneration committee and selection and appointment committee as prescribed by the DCGC. Our remuneration and nomination committee currently consists of three members: J. Donald deBethizy (chairperson), Peter K. M. Verhaeghe and Ana Cespedes.
Our remuneration and nomination committee is responsible for, among other things:
| ● | regularly reviewing the remuneration policy and practices in light of all relevant circumstances and benchmarks, and recommending to the non-executive directors the remuneration of the individual executive directors; |
| ● | advising our Board of Directors in respect of the remuneration for the non-executive directors; |
| ● | preparing the remuneration report to be included in our annual report; |
| ● | drawing up selection criteria and appointment procedures for directors and making proposals for appointment and re-appointment of the directors; |
| ● | periodically assessing the size and composition of our Board of Directors and making a proposal for a composition profile of the non-executive directors; |
157
| ● | periodically assessing the diversity (including gender diversity) on our Board of Directors and leadership teams, and taking into account any gaps between our then current diversity metrics and the goals specified in our diversity, equity and inclusion policy when making recommendations to the Board of Directors; |
| ● | periodically assessing the functioning of individual directors and reporting on this to the non-executive directors; and |
| ● | supervising the policy of the executive directors on the selection criteria and appointment procedures for senior management. |
In addition, our remuneration and nomination committee takes into account ESG when performing its duties, making sure that (i) ESG performance metrics are incorporated in the remuneration, (ii) ESG qualifications, experience, and expertise are taken into account in the director and executive nomination process, (iii) a culture of awareness and accountability for non-financial performance metrics is promoted, (iv) a diverse, equitable, and inclusive work environment is fostered, (v) our ESG reporting is in line with applicable regulatory requirements and industry best practices in these specific areas, and (vi) we maintain constructive dialogue with key stakeholders on ESG matters.
The remuneration and nomination committee consists of at least three members. The remuneration and nomination committee meets as often as is required for its proper functioning, but at least once per year to evaluate its functioning.
Report Remuneration and Nomination Committee
The remuneration and nomination committee assists the Board of Directors by, amongst other matters, regularly reviewing our remuneration policy, preparing remuneration proposals and periodically assessing the size and composition of the Board of Directors, as well as preparing the policy of the senior management team on the selection criteria and appointment procedures for senior management. During their deliberations in 2023, the main topics of discussion were the C-level long-term succession planning, the equity remuneration and holding guidelines, talent recruitment, the company’s clawback policy the outcome of our say-on-pay vote and the interactions with proxy advisors’ and investors’, prior to and following the negative say-on-pay vote at our annual General Meeting held on May 2, 2023 (2023 General Meeting).
In 2023, five formal remuneration and nomination committee meetings were held. The meeting attendance rate for our directors is set out in the table below.
| Number of meetings attended in |
| ||
Name | 2023 since appointment | Attendance % | ||
Peter K. M. Verhaeghe | 5 | 100% | ||
Ana Cespedes | 5 | 100% | ||
J. Donald deBethizy (chairperson) | 5 | 100% |
Informal subcommittees
Research and development committee
The research and development committee consists of members of our Board of Directors and other persons, which composition may vary from time to time. Currently, the research and development committee consists of two members who are also members of our Board of Directors: J. Donald deBethizy and Pamela Klein. Non-board member advisors of the research and development committee include David Lacey, Hans de Haard and Wim Parys. Ad-hoc participants to the committee meetings include a variety of employees and/or external advisors, depending on the needs of the committee and the topics under discussion.
158
The research and development committee is responsible for, among other things:
| ● | monitoring and overseeing our research and development goals, strategies and measures; |
| ● | serving as a sounding board to our research and development management, general management and Board of Directors; |
| ● | performing strategic reviews of our key research and development programs; |
| ● | reporting to our Board of Directors on the outcome of the strategic reviews; |
| ● | reviewing our scientific publication and communications plan; |
| ● | evaluating and challenging the effectiveness and competitiveness of our research and development endeavors; |
| ● | reviewing and discussing emerging scientific trends and activities critical to the success of our research and development; |
| ● | reviewing our clinical and preclinical product pipeline; and |
| ● | engaging in attracting, retaining and developing our senior research and development personnel. |
The research and development committee also pays specific attention to ESG duties when performing its duties. Amongst others, it help ensure that we meet our commitment to ensuring animal testing is carried out only when necessary and when no alternative methods are reasonably available and that we have policies and procedures in place to support high standards of animal welfare, minimizing pain and distress to research animals. Furthermore, it ensures a transparent reporting on animal testing practices and in R&D practices and helps ensure the prioritization of safety, dignity, and rights of clinical trial participants that informed consent is obtained in a transparent and ethically sound manner.
All members of the research and development committee shall have adequate industrial, academic and/or practical experience with the research and development of biopharmaceuticals.
One purpose of our research and development committee is to engage in discussion with our research and development personnel, and the committee’s responsibilities to carry out this purpose include, among others: monitoring the research and development activities, performing strategic reviews of the key research and development programs and reviewing the scientific publication plan, all with the intent to support our innovation mission.
Our research and development committee meets as often as is required for its proper functioning, but typically meets at least once prior to each meeting of our Board of Directors and reports regularly to our Board of Directors on the outcome of its deliberations, including any recommendations to the Board of Directors or the senior management team. The chairperson of our research and development committee reports to our Board of Directors on the research and development committee’s discussions and strategic advice after each meeting on all matters within its duties and responsibilities.
Report Research and Development Committee
The research and development committee functions as a sounding board to our research and development management, general management and the Board of Directors, and monitors our research and development goals, strategies and measures. In 2023, the committee held five formal meetings, in which it focused mainly on the vision and strategy on science, the Company’s research and development pipeline including its preclinical and clinical stage product-candidates, potential future indications for its commercial stage products and developments in relation to our IIP.
159
The meeting attendance rate for our directors is set out in the table below.
| Number of meetings attended in |
| ||
Name | 2023 since appointment | Attendance % | ||
J. Donald deBethizy | 5 | 100% | ||
Pamela Klein | 5 | 100% | ||
David Lacey (chairperson) | 5 | 100% |
Commercialization committee
Our commercialization committee consists of members of our Board of Directors and other persons, which composition may vary from time to time. As of the date of this Annual Report, the commercialization committee consists of three permanent members: James M. Daly (chairperson), Anthony A. Rosenberg and Camilla Sylvest. Keith Woods serves as a non-board member advisor of the committee.
The commercialization committee is responsible for, among other things:
| ● | reviewing and guiding the global sales and marketing strategy to ensure optimal product uptake and sustained growth and promoting innovation within commercialization efforts; |
| ● | overseeing the global product launch strategy and supervising all stages of product lifecycle; |
| ● | reviewing our partnerships and collaborations; |
| ● | reviewing and guiding the Company’s global medical affairs strategy; |
| ● | identifying and advising on potential risks associated with commercialization strategies and ensuring commercial strategies adhere to regulatory obligations; |
| ● | reviewing the ESG reporting on the topics which are relevant to the activities of the committee and providing comments thereto; and |
| ● | reporting to our Board of Directors on the outcome of the strategic reviews. |
The non-executive directors shall appoint and dismiss the members of the commercialization committee. All members of the commercialization committee shall have adequate industrial, academic and/or practical experience with the commercialization of (bio)pharmaceuticals.
Our commercialization committee meets as often as is required for its proper functioning and in practice meets at least once per quarter. The commercialization committee reports regularly to our Board of Directors on the outcome of its strategic reviews and any recommendations to the Board of Directors or senior management team.
Report Commercialization Committee
The commercialization committee functions as a sounding board on branded and unbranded strategic marketing plans for the Board of Directors. In 2023, the committee held five formal meetings, in which it focused mainly on the execution of our launch of VYVGART as well as the preparation for potential future launches, subject to obtaining further approvals.
160
The meeting attendance rate for our directors is set out in the table below.
| Number of meetings attended in |
| ||
Name | 2023 since appointment | Attendance % | ||
Anthony A. Rosenberg | 5 | 100% | ||
James M. Daly (chairperson) | 5 | 100% | ||
Camilla Sylvest | 5 | 100% |
Corporate Governance Practices
Our Board By-Laws, that describe, inter alia, the procedure for holding meetings of the Board of Directors, for the decision-making by the Board of Directors and the Board of Directors’ operating procedures.
In accordance with our Articles of Association, our Board of Directors meets at least once every three months to discuss the state of affairs within the Company and the expected developments.
Under our Board By-Laws, the members of our Board of Directors must endeavor, insofar as is possible, to ensure that resolutions are adopted unanimously. Where unanimity cannot be achieved and Dutch law, the Articles of Association or the Board By-Laws do not prescribe a larger majority, all resolutions of our Board of Directors must be adopted by a simple majority of the votes cast in a meeting at which at least a majority of the members of our Board of Directors then in office are present or represented. The Articles of Association provide that in case of a tie of votes, the chairperson does not have a casting vote and as such the proposal will be rejected in case of a tie.
Under the Board By-Laws, some specific matters require approval of the majority of the non-executive directors. These matters are set out in Schedule 1 of our Board By-Laws. Our Board By-Laws are available on our website. The non-executive directors may also determine that certain other matters shall require approval of a certain majority of the non-executive directors. Such matters shall be clearly specified and notified to the executive director(s) in writing.
Resolutions of the Board of Directors may also be adopted outside of a meeting in writing, provided that all directors in office (in respect of whom no conflict of interest exists as referred to in the Articles of Association) have consented in writing to this manner of decision-making. A director may issue a proxy for a specific Board of Directors meeting to another director in writing.
A director having a direct or indirect personal interest that conflicts with the interest of the Company and its affiliated enterprise has a conflict of interest. Each director shall inform all other directors of a conflict of interest without delay. A director shall not participate in the deliberations and decision-making process in relation to an item if he has a conflict of interest with respect thereto. In such case, the other directors shall resolve the item. In case because of this no resolution can be adopted by the executive directors, the non-executive directors will resolve on the matter. In case because of this no resolution can be adopted by the non-executive directors, the Board of Directors will resolve on the matter as if there were no conflict of interest.
The executive director(s) are required to be asked their vision on their own remuneration in accordance with best practice provision 3.2.2 but may not participate in the adoption of resolutions (including any deliberations in respect of such resolutions) relating to their remuneration.
161
Board Evaluation
The Board of Directors evaluates its functioning and the functioning of its committees and of each individual director annually. The evaluation process is performed with the help of an external professional board evaluation consultant. In 2023, the evaluation was performed by Nasdaq Governance Solutions. The evaluation includes preparing specific questionnaires focusing on the skills and competences most relevant to us, and the most material board topics and challenges we face. The written questionnaire is then followed up by one-to-one interviews with the representative of Nasdaq Governance Solutions with each member of the Board of Directors, followed by a debrief and discussion held with the external evaluator and the entire Board of Directors both in writing (in form of a report) and in the form of a live discussion of the evaluation report aimed at distilling specific learnings and conclusions.
Based on the self-evaluation performed, the non-executive directors concluded that the Board of Directors and its committees had properly discharged their responsibilities during 2023. The Board of Directors identified certain strengths and weaknesses and adopted a plan for further board development and succession in 2024. In general non-executive directors appreciate the high effectiveness of the Board and the functioning of its committees and consider that (i) the Board is high functioning, committed, open, transparent and very engaged and (ii) the Board committees are strong and work well.
D. EMPLOYEES
As of December 31, 2023, we had 1,148 employees and 309 consultants, which we refer to as “contingent workers.” At each date shown below, we had the following number of employees, broken out by department and geography:
At December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
Function: |
|
|
|
|
|
|
Research and development |
| 653 |
| 367 |
| 289 |
Selling, general and administrative |
| 495 |
| 476 |
| 361 |
Total |
| 1,148 |
| 843 |
| 650 |
Geography: |
|
|
| |||
Belgium |
| 355 |
| 363 |
| 296 |
U.S. | 454 | 340 | 276 | |||
Japan | 116 | 75 | 57 | |||
The Netherlands |
| 22 |
| — |
| — |
Switzerland | 28 |
| 15 |
| 9 | |
France | 40 | 11 | 3 | |||
Germany | 25 | 11 | 9 | |||
Canada | 16 | 5 | — | |||
UK | 37 | — | — | |||
Italy | 27 | — | — | |||
Spain | 20 | — | — | |||
Other - remote | 8 | 23 | — | |||
Total |
| 1,148 |
| 843 |
| 650 |
Collective bargaining agreements (CBAs) can be entered into in Belgium at the national, industry, or company levels. These CBAs are binding on both employers and employees. We have no trade union representation or CBAs at the company level, but we are subject to the national and chemical industry CBAs. The CBAs currently applicable to us relate to employment conditions such as wages, working time, job security, innovation and supplementary pensions. We have not had, and do not anticipate having, disputes on any of these subjects. CBAs may, however, change the employment conditions of our employees in the future and hence adversely affect our employment relationships.
162
E. SHARE OWNERSHIP
For information regarding the share ownership of our directors and members of our executive committee, see Item 6.B. “Compensation” and Item 7.A. “Major Shareholders”.
F. DISCLOSURE OF A REGISTRANT’S ACTION TO RECOVER ERRONEOUSLY AWARDED COMPENSATION
Not applicable.
ITEM 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A. MAJOR SHAREHOLDERS
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as of February 20, 2024 for:
| ● | each person who is known by us to own beneficially more than 3% of our total outstanding ordinary shares; |
| ● | each member of our Board of Directors and our senior management; and |
| ● | all members of our Board of Directors and our senior management as a group. |
Beneficial ownership is determined in accordance with the rules of the SEC, which may materially differ from other rules applicable to us. The SEC rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include ordinary shares that can be acquired within 60 days of February 20, 2024. The percentage ownership information shown in the table is based upon 59,302,232 ordinary shares outstanding as of February 20, 2024.
Except as otherwise indicated, all of the shares reflected in the table are ordinary shares and all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
163
In computing the number of ordinary shares beneficially owned by a person and the percentage ownership of that person, we deemed outstanding ordinary shares subject to options held by that person that are immediately exercisable or exercisable within 60 days of February 20, 2024. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. The information in the table below is based on information known to us or ascertained by us from public filings made by the shareholders.
Shares beneficially owned | |||||
Name of beneficial owner |
| Number |
| Percentage | |
3% or Greater Shareholders: |
|
|
|
|
|
FMR LLC (1) | 4,173,842 | 7.1 | % | ||
Blackrock, Inc. (2) | 4,036,853 | 6.8 | % | ||
Baillie Gifford & Co (3) | — | — | |||
6.2 (voting) | % | ||||
T. Rowe Price Group, Inc. (4) |
| 3,673,855 | 6.2 | % | |
Artisan Partners Limited Partnership (5) | 3,174,477 | 5.4 | % | ||
The Vanguard Group (6) | 1,978,464 | 4.2 | % | ||
Capital Research and Management Company (7) | — | — | % | ||
3.2 (voting) | % | ||||
Janus Henderson Group plc (8) | 1,784,723 | 3.0 | % | ||
Wellington Management Group LLP (9) | — | — | % | ||
3.0 (voting) | % | ||||
Directors and Senior Management: | |||||
Tim Van Hauwermeiren (10) |
| * | % | ||
Arjen Lemmen (11) | * | % | |||
Keith Woods (12) |
| * | % | ||
Karl Gubitz (13) |
| * | % | ||
Peter K.M. Verhaeghe (14) |
| * | % | ||
J. Donald deBethizy (15) |
| * | % | ||
Anthony A. Rosenberg (16) | * | % | |||
Luc Truyen (17) |
| * | % | ||
Peter Ulrichts (18) |
| * | % | ||
Malini Moorthy (19) |
| * | % | ||
Pamela Klein (20) |
| * | % | ||
Andria Wilk (21) |
| * | % | ||
Karen Massey |
| * | % | ||
Camilla Sylvest (22) |
| * | % | ||
Ana Cespedes (23) |
| * | % | ||
Werner Lanthaler (24) | * | % | |||
Steve Krognes (25) | * | % | |||
James M. Daly (26) | * | % | |||
All executive officers and directors as a group (18 persons) |
| 1.4 | % | ||
* | Indicates beneficial ownership of less than 1% of the total outstanding ordinary shares. |
(1) | Based on the most recently available Schedule 13G filed with the SEC on February 9, 2024. According to its Schedule 13G, FMR LLC reported having sole voting power over 4,017,222 ordinary shares and sole dispositive power over 4,173,842 ordinary shares. The Schedule 13G contained information as of December 31, 2023 and may not reflect current holdings of the Company’s stock. Members of the Johnson family, including Abigail P. Johnson, are the predominant owners, directly or through trusts, of Series B voting common shares of FMR LLC, representing 49% of the voting power of FMR LLC. The Johnson family group and all other Series B shareholders have entered into a shareholders’ voting agreement under which all Series B voting common shares will be voted in accordance with the majority vote of Series B voting common shares. Accordingly, through their ownership of voting common shares and the execution of the shareholders’ voting agreement, members of the Johnson family may be deemed, under the Investment Company Act of 1940, to form a controlling group with respect to FMR LLC. Neither FMR LLC nor Abigail P. Johnson has the sole power to vote or direct the voting of the shares owned directly by the various investment companies registered under the Investment Company Act advised by Fidelity Management & Research Company (FMR Co.), a wholly owned subsidiary of FMR LLC, which power resides with the Fidelity Funds’ Boards of Trustees. FMR Co. carries out the voting of the shares under written guidelines established by the Fidelity Funds’ Boards of Trustees. FMR LLC’s principal business office is located 245 Summer Street, Boston, MA 02210. |
164
(2) | Based on the most recently available Schedule 13G filed with the SEC on February 1, 2024. According to its Schedule 13G, BlackRock, Inc. reported having sole voting power over 3,767,146 ordinary shares and sole dispositive power over 4,036,853 ordinary shares. The Schedule 13G contained information as of December 31, 2023 and may not reflect current holdings of the Company’s stock. The address for BlackRock, Inc. is 50 Hudson Yards, New York, NY 10001. |
(3) | Based solely on the most recent transparency notification filed with the Dutch Authority for the Financial Markets (Stichting Autoriteit Financiële Markten) (AFM) as of February 20, 2024. Consists of 0 ordinary shares and voting rights on 2,966,216 ordinary shares. Other information regarding this shareholder’s beneficial ownership of our shares is not known to us or, to our knowledge, ascertainable from public filings. |
(4) | Based on the most recently available Schedule 13G filed with the SEC on February 14, 2024. According to its Schedule 13G, T. Rowe Price Associates, Inc. reported having sole voting power over 1,051,051 ADSs and sole dispositive power over 3,673,589 ADSs. The Schedule 13G contained information as of December 31, 2023 and may not reflect current holdings of the Company’s stock. The address for T. Rowe Price Associates, Inc. is 100 E. Pratt Street, Baltimore, MD 21202. |
(5) | Based on the most recently available Schedule 13G filed with the SEC on February 12, 2024. According to its Schedule 13G, Artisan Partners Limited Partnership (APLP), Artisan Investments GP LLC (Artisan Investments), Artisan Partners Holdings LP (Artisan Holdings), and Artisan Partners Asset Management Inc. (APAM) reported having shared voting power over 2,705,482 ordinary shares and shared dispositive power over 3,174,477 ordinary shares. APLP is an investment adviser registered under section 203 of the Investment Advisers Act of 1940. Artisan Holdings is the sole limited partner of APLP and the sole member of Artisan Investments. Artisan Investments is the general partner of APLP. APAM is the general partner of Artisan Holdings. The Schedule 13G contained information as of December 31, 2023 and may not reflect current holdings of the Company’s stock. The address for APLP, Artisan Investments, Artisan Holdings, and APAM is 875 East Wisconsin Avenue, Suite 800, Milwaukee, WI 53202. |
(6) | Based solely on the most recent transparency notification filed by The Vanguard Group, Inc. with the AFM as of February 20, 2024. Consists of 1,978,464 shares (on which, according to the AFM filing, no voting rights can be exercised by this shareholder). Other information regarding this shareholder’s beneficial ownership of our shares is not known to us or, to our knowledge, ascertainable from public filings. The address for The Vanguard Group, Inc. is the Vanguard Group, 100 Vanguard Blvd., Malvern, PA 19355. |
(7) | Based solely on the most recent transparency notification filed with the AFM as of February 20, 2024. Consists of voting rights on 119,041 ordinary shares and 1,765,665 ADSs. Other information regarding this shareholder’s beneficial ownership of our shares is not known to us or, to our knowledge, ascertainable from public filings. |
(8) | Based solely on the most recent transparency notification filed with the AFM as of February 20, 2024. Consists of 10,882 ordinary shares and 1,773,841 ADSs. Other information regarding this shareholder’s beneficial ownership of our shares is not known to us or, to our knowledge, ascertainable from public filings. |
(9) | Based solely on the most recent transparency notification filed with the AFM as of February 20, 2024. Consists of 0 ordinary shares and voting rights on 1,520,216 ordinary shares and 257,347 ADSs. Other information regarding this shareholder’s beneficial ownership of our shares is not known to us or, to our knowledge, ascertainable from public filings. |
(10) | Consists of 109,986 ordinary shares (of which 71,986 ordinary shares are held by Mr. Van Hauwermeiren directly and 38,000 ordinary shares are held indirectly by Mr. Van Hauwermeiren’s partner, Ms. Vissers, who holds the interest in these ordinary shares through an entity, Stichting Administratiekantoor Cinclus, which in turn holds the interest in these ordinary shares through the Belgian civil company (société civile/burgerlijke maatschap) “TVHNV”), and 240,555 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
(11) | Consists of 1,227 ordinary shares and 123,757 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024 (of which 12,445 stock options are subject to a mirror option contract pursuant to which the counterparty has the right to buy the shares underlying these stock options at the strike price as soon as these stock options become exercisable, subject to the specific terms of the contract). |
(12) | Consists of 3,307 ordinary shares and 71,333 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
(13) | Consists of 2,959 ordinary shares (including 500 ADSs) and 31,333 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
(14) | Consists of 450 ordinary shares and 42,000 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
(15) | Consists of 450 ordinary shares and 40,000 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
165
(16) | Consists of 450 ordinary shares and 37,480 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
(17) | Consists of 1,631 ordinary shares and 27,111 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
(18) | Consists of 515 ordinary shares and 27,208 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024 (of which 4,566 stock options are subject to a mirror option contract pursuant to which the counterparty has the right to buy the shares underlying these stock options at the strike price as soon as these stock options become exercisable, subject to the specific terms of the contract). |
(19) | Consists of 8,500 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024 and 1,350 shares issuable upon the settlement of restricted stock units vesting within 60 days of February 20, 2024. |
(20) | Consists of 450 ordinary shares and 21,500 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
(21) | Consists of 299 ordinary shares and 17,335 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024 (of which 6,083 stock options are subject to a mirror option contract pursuant to which the counterparty has the right to buy the shares underlying these stock options at the strike price as soon as these stock options become exercisable, subject to the specific terms of the contract). |
(22) | Consists of 225 ordinary shares. |
(23) | Consists of 225 ordinary shares. |
(24) | Consists of 600 ordinary shares and 4,000 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
(25) | Consists of 131 shares issuable upon the settlement of RSUs vesting within 60 days of February 20, 2024. |
(26) | Consists of 450 ordinary shares and 10,000 shares issuable upon the exercise of stock options that are immediately exercisable or exercisable within 60 days of February 20, 2024. |
Each of our shareholders is entitled to one vote per ordinary share. None of the holders of our shares have different voting rights from other holders of shares.
As of the date of this Annual Report, we are not directly or indirectly owned or controlled by any shareholder, whether individually or acting in concert. We are not aware of any arrangement that may, at a subsequent date, result in a change of control of the Company.
The number of record holders in the U.S. is not representative of the number of beneficial holders nor is it representative of where such beneficial holders are resident since many of these ordinary shares were held by brokers or other nominees. As of February 20, 2024, assuming that all of our ordinary shares represented by ADSs are held by residents of the U.S., we estimate that approximately 53.61% of our outstanding ordinary shares were held in the U.S. by approximately one institutional holder of record.
To our knowledge, and other than changes in percentage ownership as a result of the shares issued in connection with our initial and follow-on U.S. public offerings or publicly disclosed in AFM filings and any amendments thereof, there has been no significant change in the percentage ownership held by the major shareholders listed above.
B. RELATED PARTY TRANSACTIONS
As described under Item 4.B. “Business Overview—Our Exclusive License with Halozyme for ENHANZE®,” we are party to the ENHANZE License Agreement and may be required to make certain payments to Halozyme. In fiscal year 2022, we made $2.1 million in payments to Halozyme pursuant to the ENHANZE License Agreement. Our non-executive
166
director Mr. Daly is also a non-executive member of the board of directors of Halozyme. Mr. Daly did not participate in any discussions and decision making relating to the ENHANZE License Agreement.
Agreements with Our Senior Management.
There are no arrangements or understandings in place with major shareholders, customers, suppliers or others pursuant to which any member of our Board of Directors or senior management team has been appointed.
We have entered into a management agreement with Tim Van Hauwermeiren as our CEO, our sole executive director. The key terms of his agreement are as follows:
| Tim Van Hauwermeiren | ||
Base salary | $ | 655,787 | |
Cash bonus | maximum 60% of base salary based on previously determined bonus targets established by the non‑executive directors | ||
Pension contributions(1) | $ | 22,821 | |
Duration | Indefinite | ||
| (1) | Amounts shown represent pension contributions paid during the year-ended December 31, 2023. |
We may terminate Mr. Van Hauwermeiren’s services upon 18 months’ notice, or payment of 18 months’ pro-rated base compensation in lieu of notice. Mr. Van Hauwermeiren would be entitled to the same payment in lieu of notice in the event he terminates his services with us in circumstances in which it cannot reasonably be expected for him to continue providing services to us (and after our failure to remedy such conditions after being provided at least 14 days’ notice). Mr. Van Hauwermeiren would also be entitled to payment in lieu of notice in the event he terminated his services with us in certain cases of our failure to comply with obligations under applicable law or his agreement (and after our failure to remedy such non-compliance, if non-deliberate, after being provided at least 14 days’ notice). In these cases, there will be a full acceleration of the vesting of any outstanding stock options held by Mr. Van Hauwermeiren. There will be no notice period or payment in lieu of notice in certain cases of Mr. Van Hauwermeiren’s failure to comply with obligations under applicable law or his agreement. Mr. Van Hauwermeiren may be dismissed immediately as an executive director.
Karl Gubitz, our CFO, has an employment contract with our subsidiary, argenx US Inc., for an indefinite term.
Keith Woods, our COO, had an employment contract with our subsidiary, argenx US Inc., for an indefinite term. His employment contract ended in March 2023.
Karen Massey, our COO, joined argenx in March 2023 and has an employment contract with our subsidiary, argenx Switzerland SA, for an indefinite term.
Peter Ulrichts, our chief scientific officer, since January 2023, has an employment contract with our subsidiary, argenx BV, for an indefinite term.
Arjen Lemmen, our vice president corporate development and strategy, has an employment contract with our subsidiary, argenx BV, for an indefinite term. We may terminate his employment contract at any time, subject to a notice period and a severance payment of at least 12 months. Mr. Lemmen entered into a secondment agreement with argenx BV, under which Mr. Lemmen has been seconded from argenx BV to argenx US in the U.S. from August 1, 2022 until on or about July 31, 2024 (unless otherwise extended by the parties). In connection with his secondment, Mr. Lemmen receives a housing, schooling and cost of living allowance.
Andria Wilk, our global head of quality, has an employment contract with our subsidiary, argenx BV, for an indefinite term.
167
Malini Moorthy, our general counsel has an employment contract with our subsidiary, argenx US, for an indefinite term. Ms. Moorthy has also entered into a secondment agreement with argenx US, under which Ms. Moorthy has been seconded from argenx US to argenx BV and is based in Belgium for the period of April 1, 2023 through December 31, 2024 (unless otherwise extended by the parties).
Luc Truyen, our head of research and development management operations and our chief medical officer, has an employment contract with our subsidiary, argenx US, for an indefinite term. Mr. Truyen entered into a secondment agreement with argenx US, under which Mr. Truyen has been seconded from argenx US to argenx BV and is based in Belgium for the period of April 1, 2022 through November 30, 2026 (unless otherwise extended by the parties).
Indemnification Agreements
In connection with our initial U.S. public offering, we entered into indemnification agreements with each of our non-executive directors and each member of our senior management. We have entered into such agreements with each new non-executive director or member of our senior management when they have joined us since our initial U.S. public offering. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to non-executive directors, officers or persons controlling us pursuant to the foregoing provisions, we have been informed that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Transactions with Related Companies
From time to time, in the ordinary course of our business, we may contract for services from companies in which certain of the members of our senior management or directors may serve as director or advisor. The costs of these services are negotiated on an at arm’s length basis and none of these arrangements are material to us.
Related Party Transactions Policy
In connection with our initial U.S. public offering, we entered into a related party transaction policy. Our Code of Business Conduct and Ethics (Code of Conduct) and our Board Rules also include specific rules of transactions with related parties.
C. INTERESTS OF EXPERTS AND COUNSEL
Not applicable.
ITEM 8. FINANCIAL INFORMATION
A. CONSOLIDATED STATEMENTS AND OTHER FINANCIAL INFORMATION
Consolidated financial statements
Our consolidated financial statements, which are prepared in accordance with IFRS, as issued by the IASB, are appended at the end of this Annual Report, starting at page F-1, and incorporated herein by reference.
Legal proceedings
From time to time we may become involved in legal, governmental or arbitration proceedings or be subject to claims arising in the ordinary course of our business. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors. During the previous 12 months, there have not been any legal, governmental or arbitration proceedings (including any such proceedings which are pending or threatened of which we are aware) which may have, or have had in the recent past, significant effects on argenx and/or the Group’s financial position or profitability.
168
Dividend Distribution Policy
Our Board of Directors has declared a series of interim distributions on account of the Company’s freely distributable reserves for such amounts as was required to pay up the aggregate nominal value of all such shares that were issued to holders of vested RSUs, all in accordance with our Equity Incentive Plan. In accordance with Dutch law, our Board of Directors prepared and filed an interim simplified balance sheet demonstrating that there were sufficient freely distributable reserves for such interim distributions. Such interim simplified balance sheet was filed with the Dutch trade register. The aggregate amount of these interim distributions amounted to approximately €6,600 ($7,300) in 2023.
Other than these interim distributions, we have not paid or declared any cash dividends on our ordinary shares, and we do not anticipate paying any cash dividends in the foreseeable future. All of our outstanding shares have the same dividend rights. We intend to retain all available funds and any future earnings to fund the development and expansion of our business.
Even if future operations lead to significant levels of distributable profits, we currently intend that any earnings will be reinvested in our business and that cash dividends will not be paid until we have an established revenue stream to support continuing cash dividends. In addition, payment of any future dividends to shareholders would be subject to shareholder approval at a General Meeting, upon proposal of our Board of Directors, which proposal would be subject to the approval of the majority of the non-executive directors after taking into account various factors including our business prospects, cash requirements, financial performance and new product development.
Our Articles of Association, as available on our website, contain the provision on the distribution of profits in Article 20 (Profits, distributions and losses).
B. SIGNIFICANT CHANGES
For details regarding events subsequent to the reporting period, please see “Note 32—Events after the balance sheet date” in our consolidated financial statements which are appended to our Annual Report for the period ended December 31, 2023 and which are incorporated herein by reference.
ITEM 9. THE OFFER AND LISTING
A. OFFER AND LISTING DETAILS
See Item 4.A. “Information on the Company—History and Development of the Company”.
B. PLAN OF DISTRIBUTION
Not applicable.
C. MARKETS
The ADSs have been listed on Nasdaq under the symbol “ARGX” since May 18, 2017, and our ordinary shares have been listed on Euronext Brussels under the symbol “ARGX” since July 2014.
D. SELLING SHAREHOLDERS
Not applicable.
E. DILUTION
Not applicable.
169
F. EXPENSES OF THE ISSUE
Not applicable.
ITEM 10. ADDITIONAL INFORMATION
A. SHARE CAPITAL
Not applicable.
B. MEMORANDUM AND ARTICLES OF ASSOCIATION
Corporate Objectives
Please see “Exhibit 2.3—Corporate Objectives”, incorporated herein by reference.
Directors
Conflict of Interest
Please see “Exhibit 2.3—Board Members—Corporate Objectives”, incorporated herein by reference.
Remuneration
Please see “Exhibit 2.3—Board Members—Remuneration”, incorporated herein by reference.
Borrowing Powers
Please see “Exhibit 2.3—Board Members—Borrowing Powers”, incorporated herein by reference.
Rights, Preferences and Restrictions of Shares
Dividends and Other Distributions
Please see “Exhibit 2.3— Dividends and Other Distributions”, incorporated herein by reference.
Voting Rights
Please see “Exhibit 2.3—Shareholders’ Meetings and Consents—Quorum and Voting Requirements” and “Exhibit 2.3—Comparison of Dutch Corporate Law, our Articles of Association and Board of Directors By-Laws and DGCL—Voting Rights”, incorporated herein by reference.
Rights to Share in Company Profits
Please see “Exhibit 2.3—Dividends and Other Distributions—Rights to Share in Company Profits”, incorporated herein by reference.
Right to Surplus In the Event of Liquidation
Please see “Exhibit 2.3—Dividends and Other Distributions—Right to Surplus In the Event of Liquidation”, incorporated herein by reference.
170
Redemption Provisions
Please see “Exhibit 2.3—Dividends and Other Distributions—Redemption Provisions”, incorporated herein by reference.
Amendment of Articles of Association
Please see “Exhibit 2.3—Articles of Association and Dutch Law—Dividends and Other Distributions—Redemption Provisions”, incorporated herein by reference.
Shareholders’ Meetings and Consents
General Meetings, Voting Rights, and Admission
General Meetings are held at the place where the Company has its official seat, in Amsterdam or at Schiphol Airport (municipality of Haarlemmermeer), the Netherlands. The Articles of Association provide that at least one annual General Meeting shall be held within six months after the close of each fiscal year. Additional extraordinary General Meetings may be held whenever our Board of Directors deems such to be necessary. Shareholders representing alone or in aggregate at least one-tenth of our issued and outstanding share capital may, pursuant to the DCC, request that a General Meeting be convened. If our Board of Directors has not taken the steps necessary to ensure that a General Meeting will be held within the relevant statutory period after the request, the requesting persons may, at his/her/their request, be authorized by a court in preliminary relief proceedings to convene a General Meeting. The court shall disallow the application if it does not appear that the applicants have previously requested our Board of Directors to convene a General Meeting and our Board of Directors has not taken the necessary steps so that a General Meeting could be held within six weeks after the request.
Within three months of it becoming apparent to our Board of Directors that our equity has decreased to an amount equal to or lower than one-half of the paid-in and called-up capital, a General Meeting would be held to discuss any requisite measures.
We will give notice of any General Meeting by publication on our website and furthermore, to the extent required, in another manner in accordance with the applicable stock exchange regulations. The notice convening any General Meeting must include, among other items, an agenda indicating the place and date of the meeting, the items for discussion and voting, the proceedings for registration including the registration date, as well as any proposals for the agenda made by the Board of Directors or shareholders holding at least 3% of the issued share capital. For an annual General Meeting, the agenda shall include, among other things, the adoption of the annual accounts, appropriation of our profits and proposals relating to the composition of our Board of Directors, including the filling of any vacancies in our Board of Directors.
Pursuant to Dutch law, shareholders holding at least 3% of our issued and outstanding share capital have a right to request our Board of Directors to include items on the agenda of any General Meeting. Our Board of Directors must agree to these requests, provided that (i) the request was made in writing and motivated, and (ii) the request was received by the Chair of our Board of Directors at least 60 days prior to the date of a General Meeting.
No resolutions shall be adopted on items other than those which have been included in the agenda. In accordance with the DCGC, a shareholder may include an item on the agenda only after consulting our Board of Directors in that respect. If one or more shareholders intends to request that an item be put on the agenda that may result in a change in the company’s strategy, our Board of Directors may invoke a response time of a maximum of 180 days until the day of a General Meeting. In addition, pursuant to the DCC, our Board of Directors may invoke a statutory
171
cooling-off period up to a maximum of 250 days (wettelijke bedenktijd). For the Company, this means that the new rules will apply in case:
| ● | shareholders requesting our Board of Directors to have a General Meeting consider a proposal for the appointment, suspension or dismissal of one or more directors, or a proposal for the amendment of one or more provisions in the Articles of association relating thereto; or |
| ● | a public offering of shares in the capital of the Company is announced or made without the bidder and the Company having been reached agreement about the offering; and |
| ● | only if our Board of Directors also considers the relevant situation to be substantially contrary to the interests of the Company and its affiliated enterprises. |
If our Board of Directors invokes such a cooling-off period, this causes the powers of the General Meeting to appoint, suspend or dismiss directors (and to amend the Articles of Association in this respect) to be suspended.
General Meetings are presided over by the chairperson or, if he/she is absent, by the vice chairperson of the Board of Directors. If both the chairperson and the vice chairperson are absent, the non-executive directors present at the meeting shall appoint one of them to be chairperson. Board members may attend a General Meeting. In these meetings, they have an advisory vote. The chairperson of the meeting may decide at his/her discretion to admit other persons to the meeting.
The external auditor of the Company shall attend a General Meeting in which the annual accounts are discussed.
Our Board of Directors must give notice of a General Meeting, by at least such number of days prior to the day of the meeting as required by Dutch law, which is currently forty-two days.
Shareholders (as well as other persons with voting rights or meeting rights) may attend a General Meeting, to address the General Meeting and, in so far as they have such right, to exercise voting rights pro rata to its shareholding, either in person or by proxy. Shareholders may exercise these rights, if they are the holders of shares on the registration date which is currently the 28th day before the day of a General Meeting, and they or their proxy have notified our Board of Directors of their intention to attend a General Meeting in writing at the address and by the date specified in the notice of said meeting.
All shareholders, and each usufructuary and pledgee to whom the right to vote on our shares accrues, are entitled, in person or represented by a proxy authorized in writing, to attend and address a General Meeting and exercise voting rights pro rata to their shareholding. Shareholders may exercise their rights if they are the holders of our shares on the record date as required by Dutch law, which is currently the 28th day before the day of a General Meeting, and they or their proxy have notified us of their intention to attend such General Meeting in writing or by any other electronic means that can be reproduced on paper ultimately at a date set for that purpose by our Board of Directors which date may not be earlier than the seventh day prior to such General Meeting, specifying such person’s name and the number of shares for which such person may exercise the voting rights and/or meeting rights at such General Meeting. The convocation notice shall state the record date and the manner in which the persons entitled to attend a General Meeting may register and exercise their rights.
Each ordinary share confers the right on the holder to cast one vote at the General Meeting. Shareholders may vote by proxy. The voting rights attached to any shares held by us are suspended as long as they are held in treasury. Nonetheless, the holders of a right of usufruct (vruchtgebruik) in shares belonging to another and the holders of a right of pledge in respect of ordinary shares held by us are not excluded from any right they may have to vote on such ordinary shares, if the right of usufruct (vruchtgebruik) or the right of pledge was granted prior to the time such ordinary share was acquired by us. We may not cast votes in respect of a share in respect of which there is a right of usufruct (vruchtgebruik) or a right of pledge. Shares which are not entitled to voting rights pursuant to the preceding sentences
172
will not be taken into account for the purpose of determining the number of shareholders that vote and that are present or represented, or the amount of the share capital that is provided or that is represented at a General Meeting.
Decisions of the General Meeting are taken by an absolute majority of votes cast, except where Dutch law or the Articles of Association provide for a qualified majority or unanimity. In accordance with Dutch law and generally accepted business practices, our Articles of Association do not provide quorum requirements generally applicable to a General Meeting. To this extent, our practice varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock.
Members of our Board of Directors may attend a General Meeting in which they have an advisory role. The voting rights attached to shares are suspended as long as such shares are held by us.
Two General Meetings were held in 2023.
On February 27, 2023, an extraordinary General Meeting was held, to appoint Steve Krognes as a non-executive director to the Board of Directors for a term ending on the 2027 annual General Meeting.
At the 2023 General Meeting, our annual report and annual accounts for the fiscal year 2022 were approved, Mr. J. Donald deBethizy was reappointed as a non-executive director to the Board of Directors for a term of two years, and the Board of Directors was authorized to issue shares and grant rights to subscribe for shares in our share capital for up to 10% of the outstanding share capital at the date of the meeting and for a period of 18 months from the meeting and to limit or exclude statutory pre-emptive rights with regard to such (rights to subscribe for) shares, and the appointment of Deloitte Accountants B.V. as the Company’s auditor for the 2023 fiscal year was approved.
Limitations on the Right to Own Securities
Please see “Exhibit 2.3—Limitations on the Right to Own Securities”, incorporated herein by reference.
Comparison of Dutch Corporation Law, our Articles of Association and Board By-Laws and U.S. Corporate Law
Please see “Exhibit 2.3—Comparison of Dutch Corporate Law, our Articles of Association and Board of Directors By-Laws and DGCL”, incorporated herein by reference.
Change in the Capital
Please see “Exhibit 2.3—Change in the Capital”, incorporated herein by reference.
C. MATERIAL CONTRACTS
For additional information on our material contracts, please see Item 4 “Information on the Company”, Item 7.A. “Major Shareholders”, and Item 7.B. “Related Party Transactions”.
D. EXCHANGE CONTROLS
Under Dutch law, subject to the 1977 Sanction Act (Sanctiewet 1977) or otherwise by international sanctions, there are no exchange control restrictions on investments in, or payments on, shares (except as to cash amounts). There are no special restrictions in our Articles of Association or Dutch law that limit the right of shareholders who are not citizens or residents of the Netherlands to hold or vote shares.
173
E. TAXATION
This summary does not consider your particular circumstances. We urge you to consult your own independent tax advisors about the income, capital gains and/or transfer tax consequences to you in light of your particular circumstances of purchasing, holding and disposing of ordinary shares or ADSs.
Certain Material U.S. Federal Income Tax Considerations for U.S. Holders
The following discussion is a summary under present law of certain material U.S. federal income tax considerations relating to the ownership and disposition of ADSs by a U.S. holder (as defined below). This summary addresses only the U.S. federal income tax considerations for U.S. holders that hold ADSs as capital assets (generally, property held for investment) and use the U.S. dollar as their functional currency. This summary does not address all U.S. federal income tax matters that may be relevant to a particular U.S. holder and is not a substitute for tax advice. This summary does not address tax considerations applicable to a holder of ADSs that may be subject to special tax rules including, without limitation, banks, financial institutions or insurance companies, brokers, dealers or traders in securities, currencies, commodities, or notional principal contracts, traders in securities that elect to mark-to-market, tax-exempt entities or organizations, including “individual retirement accounts” or “Roth IRAs”, real estate investment trusts, regulated investment companies, persons that hold the ADSs as part of a “hedging,” “integrated” or “conversion” transaction or as a position in a “straddle”, partnerships (including entities or arrangements classified as partnerships for U.S. federal income tax purposes) or other pass-through entities (including S-corporations), or persons that will hold the ADSs through such an entity, certain former citizens or long-term residents of the U.S., persons that received the ADSs as compensation for the performance of services, persons subject to special tax accounting rules as a result of any item of gross income with respect to the shares being taken into account in an applicable financial statement, and holders that own directly, indirectly, or through attribution 10% or more of the voting power or value of our ordinary shares and ADSs. This summary does not address U.S. federal taxes other than the income tax (such as the Medicare surtax on net investment income, the estate, gift, or alternative minimum tax), any election to apply Section 1400Z-2 of the U.S. Internal Revenue Code of 1986, as amended (the Code) to gains recognized with respect to ADSs, or any U.S. state, local, or non-U.S. tax considerations of the ownership and disposition of ADSs.
For the purposes of this summary, a “U.S. holder” is a beneficial owner of ADSs that is (or is treated as), for U.S. federal income tax purposes, (i) an individual who is a U.S. citizen or resident, (ii) a corporation, or any other entity treated as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the U.S., any state thereof, or the District of Columbia, (iii) an estate, the income of which is subject to U.S. federal income taxation regardless of its source, or a trust, if a court within the U.S. is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of the substantial decisions of such trust.
If a partnership (or other entity or arrangement treated as a partnership for U.S. federal income tax purposes) holds ADSs, the U.S. federal income tax consequences relating to an investment in those ADSs will depend in part upon the status of the partner and the activities of the partnership. A partnership that holds ADSs should consult its tax advisor regarding the U.S. federal income tax considerations for it and for its partners of owning and disposing of ADSs in its and their particular circumstances.
In general, a U.S. holder that owns ADSs will be treated as the beneficial owner of the underlying shares represented by those ADSs for U.S. federal income tax purposes. Accordingly, no gain or loss will generally be recognized if a U.S. holder exchanges ADSs for the underlying shares represented by those ADSs. Persons considering an investment in the ADSs should consult their own taxad visors as to the particular tax consequences applicable to them relating to the ownership and disposition of ADSs, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Distributions
Although we do not currently plan to pay dividends, and subject to the discussion under Item 10.E. “Taxation —Certain Material U.S. Federal Income Tax Considerations for U.S. Holders—Passive Foreign Investment Company Considerations” below, the gross amount of distributions paid with respect to our ordinary shares including Dutch or
174
Belgian tax withheld therefrom, if any (other than pro rata distribution), generally will be included in a U.S. holder’s gross income as foreign source ordinary dividend income when actually or constructively received to the extent such distribution is paid out of our current and accumulated earnings and profits as determined under U.S. federal income tax principles. Distributions in excess of our current and accumulated earnings and profits will be treated as a non-taxable return of capital and will be applied against and reduce, the U.S. holder’s adjusted tax basis in ADSs (but not below zero) and distributions in excess of earnings and profits and a U.S. holder’s adjusted tax basis will generally be taxable to the U.S. holder as either long-term or short-term capital gain depending upon whether the U.S. holder has held the ADSs for more than one year as of the time such distribution is received. However, since we do not calculate our earnings and profits under U.S. federal income tax principles, it is expected that any distribution will be reported as a dividend, even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain.
Our dividends will not be eligible for the dividends-received deduction generally allowed to U.S. corporations. Dividends paid to non-corporate U.S. holders that satisfy a minimum holding period (during which they are not protected from the risk of loss) and certain other requirements may qualify for the preferential favorable tax rates applicable to qualified dividend income, provided that we are a “qualified foreign corporation” and we are not a PFIC as to the non-corporate U.S. holder in the taxable year of the dividend or the preceding taxable year. A qualified foreign corporation includes a non-U.S. corporation that is eligible for the benefits of a comprehensive income tax treaties with the U.S. A non-U.S. corporation also will be considered to be a qualified foreign corporation with respect to any dividend it pays on shares which are readily tradable on an established securities market in the U.S. Our ADSs are listed on the Nasdaq, which is an established securities market in the U.S., and we expect our ADSs to be readily tradable on the Nasdaq. However, there can be no assurance that the ADSs will be considered readily tradable on an established securities market in the U.S. in any taxable year. U.S. holders should consult their own tax advisors regarding the application of these rules given their particular circumstances.
If dividends are subject to Dutch or Belgian withholding tax, a U.S. holder may be entitled, subject to generally applicable limitations, to claim a U.S. foreign tax credit for Dutch or Belgian withholding tax imposed at the appropriate rate. U.S. holders who do not elect to claim a credit for any foreign income taxes paid or accrued during the taxable year may instead claim a deduction of such taxes. The rules relating to the foreign tax credit are complex and recent changes to the foreign tax credit rules that apply to foreign taxes paid or accrued in taxable years beginning after December 27, 2021 introduced additional requirements and limitations. Each U.S. holder should consult its own tax advisors regarding the foreign tax credit rules.
In general, the amount of a distribution paid to a U.S. holder in a foreign currency will be the dollar value of the foreign currency calculated by reference to the applicable exchange rate on the day the U.S. holder receives the distribution, regardless of whether the foreign currency is converted into USDs at that time. Any foreign currency gain or loss a U.S. holder realizes on a subsequent conversion of foreign currency into USDs will be U.S. source ordinary income or loss. If dividends received in a foreign currency are converted into USDs on the day they are received, a U.S. holder should not be required to recognize foreign currency gain or loss in respect of the dividend.
Sale, Exchange or Other Taxable Disposition of ADSs
Subject to the discussion under Item 10.E. “Taxation—Certain Material U.S. Federal Income Tax Considerations for U.S. Holders—Passive Foreign Investment Company Considerations” below, a U.S. holder will generally recognize capital gain or loss no the sale, exchange or other taxable disposition of ADSs in an amount equal to the difference between the amount realized from such sale or exchange and the U.S. holder’s adjusted basis in the ADSs, each amount determined in USD. The adjusted tax basis in ADSs generally will be equal to the USD cost of such ADSs. Any such capital gain or loss generally will be long-term capital gain or loss if the U.S. holder’s holding period for such ADSs exceeds one year as of the date of sale or other disposition. Long-term capital realized by a non-corporate U.S. holder is generally eligible for a preferential reduced rates. The deductibility of capital losses for U.S. federal income tax purposes is subject to certain limitations. Any such gain or loss that a U.S. holder recognizes generally will be treated as U.S. source income or loss for foreign tax credit limitation purposes.
175
Passive Foreign Investment Company Considerations
In general, a non-U.S. corporation will be classified as a passive foreign investment company, or PFIC, for any taxable year in which, after applying certain look-through rules with respect to certain dividends, rents, interest or royalties received from its affiliates and taking into account its proportionate share of the income and assets of its 25% or more owned subsidiaries, either: (i) at least 75% of its gross income is “passive income” or (ii) at least 50% of the average quarterly value of its total gross assets is attributable to cash in excess of working capital requirements or assets that produce “passive income” or are held for the production of “passive income”. Passive income for this purpose generally includes dividends, interest, royalties, rents, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income. While we are treated as a publicly traded company for these purposes, the value of our assets, including goodwill and other intangibles, will be based on their fair market value, which will depend on the market value of our ordinary shares and ADSs, which are subject to change.
Based on our historic and anticipated operations, the composition of our income and the projected composition and estimated fair market values of our assets, we do not believe that we were a PFIC for our most recent taxable year and do not expect to be classified as a PFIC for the current taxable year or for the foreseeable future. However, our possible status as a PFIC is a factual determination made annually after the close of each taxable year and, therefore, may be subject to change. Accordingly, there can be no assurance that we will not be a PFIC for any year in which a U.S. holder holds ADSs. The Company does not intend to provide any annual assessments of its PFIC status.
If we were to be classified as a PFIC for any taxable year during which a U.S. holder owns ADSs, gain recognized on a sale or other disposition (including certain pledges) of such U.S. holder’s ADSs would be allocated ratably over such U.S. holder’s holding period. Amounts allocated to the taxable year of the sale or disposition and to any year before we became a PFIC would be taxed as ordinary income and the amount allocated to each other taxable year would be subject to tax at the highest rate in effect for individuals or corporations, as appropriate, for that taxable year, and an interest charge will be imposed on the resulting tax liability for each such year. In addition, to the extent that distributions received by a U.S. holder on its ADSs in any taxable year exceed 125% of the average of the annual distributions on such holder’s ADSs received during the preceding three taxable years (or, if shorter, the U.S. holder’s holding period), such excess distributions will be subject to taxation in the same manner. Furthermore, dividends that are not excess distributions would not be eligible for the preferential tax rate applicable to qualified dividend income received by individuals and certain other non-corporate persons.
If the Company is a PFIC for any taxable year during which you own ADSs, the Company will generally continue to be treated as a PFIC with respect to you for all succeeding years during which you own the ADSs, even if the Company ceases to meet the threshold requirements for PFIC status. Certain elections may be available that will result in alternative treatments (such as mark-to-market treatment) of the Shares. U.S. holders should consult their own tax advisors concerning the Company’s possible PFIC status and the consequences to them if the Company were a PFIC for any taxable year, including whether any of these elections will be available, and, if so, what the consequences of the alternative treatments will be in your particular circumstances.
Backup Withholding and Information Reporting
U.S. holders generally will be subject to information reporting requirements with respect to dividends on ADSs and on the proceeds from the sale, exchange or disposition of the ADSs that are paid within the U.S. or through U.S.- related financial intermediaries, unless the U.S. holder is a corporation or other “exempt recipient.” In addition, U.S. holders may be subject to backup withholding on such payments, unless the U.S. holder provides a correct taxpayer identification number and a duly executed IRS Form W-9 or otherwise establishes an exemption. Backup withholding is not an additional tax, and the amount of any backup withholding will be allowed as a credit against a U.S. holder’s U.S. federal income tax liability and may entitle such holder to a refund, provided that the required information is timely furnished to the IRS.
176
Foreign Asset Reporting
Certain U.S. holders who are individuals and certain entities controlled by individuals may be required to report information relating to an interest in ADSs, subject to certain exceptions (including an exception for shares held in accounts maintained by U.S. financial institutions) by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their federal income tax return. Investors who fail to report required information could become subject to substantial penalties. U.S. holders are urged to consult their tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of the ADSs.
Material Dutch Tax Consequences
The following summary outlines certain material Dutch tax consequences in connection with the acquisition, ownership and disposal of the ADSs. All references in this summary to the Netherlands and Dutch law are to the European part of the Kingdom of the Netherlands and its law, respectively, only. The summary does not purport to present any comprehensive or complete picture of all Dutch tax aspects that could be of relevance to the acquisition, ownership and disposal of the ADSs by a (prospective) holder of the ADSs who may be subject to special tax treatment under applicable law. The summary is based on the tax laws and practice of the Netherlands as in effect on the date of this Annual Report, which are subject to changes that could prospectively or retrospectively affect the Dutch tax consequences.
This summary does not address the Dutch tax consequences for a holder of ADSs that is considered to be affiliated (gelieerd) to the company within the meaning of the Dutch Withholding Tax Act 2021 (Wet bronbelasting 2021). Generally, a holder of ADSs is considered to be affiliated to the company for these purposes if (i) it has a qualifying interest in the company, (ii) the company has a qualifying interest in such party, or (iii) a third party has a qualifying interest in both the company and such party. A party is equated with any collaborating group of parties of which it forms part. A qualifying interest is an interest that allows the holder to have a decisive influence over the other party’s decisions, in such a way that it is able to determine the activities of the other party. A party is in any case considered to have a qualifying interest in another party if it (directly or indirectly) owns more than 50 per cent. of the voting rights in such other party.
For purposes of Dutch income and corporate income tax, shares, or certain other assets, which may include depositary receipts in respect of shares, legally owned by a third party such as a trustee, foundation or similar entity or arrangement, a “Third Party”, may under certain circumstances have to be allocated to the (deemed) settlor, grantor or similar originator, the “Settlor”, or, upon the death of the Settlor, such Settlor’s beneficiaries, the “Beneficiaries”, in proportion to their entitlement to the estate of the Settlor of such trust or similar arrangement, the “Separated Private Assets”.
The summary does not address the Dutch tax consequences of a holder of the ADSs who is an individual and who has a substantial interest (aanmerkelijk belang) in the company. Generally, a holder of the ADSs will have a substantial interest in the company if such holder of the ADSs, whether alone or together with such holder’s spouse or partner and/or certain other close relatives, holds directly or indirectly, or as Settlor or Beneficiary of Separated Private Assets (i) (x) the ownership of, (y) certain other rights, such as usufruct, over, or (z) rights to acquire (whether or not already issued), shares (including the ADSs) representing 5% or more of the total issued and outstanding capital (or the issued and outstanding capital of any class of shares) of the company or (ii) (x) the ownership of, or (y) certain other rights, such as usufruct over, profit participating certificates (winstbewijzen) that relate to 5% or more of the annual profit of the company or to 5% or more of the liquidation proceeds of the company.
In addition, a holder of the ADSs has a substantial interest in the company if such holder, whether alone or together with such holder’s spouse or partner and/or certain other close relatives, has the ownership of, or other rights over, shares, or depositary receipts in respect of shares, in, or profit certificates issued by, the company that represent less than 5% of the relevant aggregate that either (a) qualified as part of a substantial interest as set forth above and where shares, or depositary receipts in respect of shares, profit certificates and/or rights there over have been, or are deemed to have been, partially disposed of, or (b) have been acquired as part of a transaction that qualified for non-recognition of gain treatment.
177
Furthermore, this summary does not address the Dutch tax consequences of a holder of the ADSs who:
| ● | is an individual and receives income or realizes capital gains in respect of the ADSs in connection with such holder’s employment activities or in such holder’s capacity as (former) board member or (former) supervisory board member; |
| ● | is a resident of any non-European part of the Netherlands; or |
| ● | falls within the scope of the Dutch Minimum Taxation Act 2024 (Wet minimumbelasting 2024). |
Dividend Withholding Tax
General
The Company is generally required to withhold dividend withholding tax imposed by the Netherlands at a rate of 15% on dividends distributed by the company in respect of our ordinary shares underlying the ADSs. The expression “dividends distributed by the company” as used herein includes, but is not limited to:
(a) | distributions in cash or in kind, deemed and constructive distributions and repayments of paid-in capital (gestort kapitaal) not recognized for Dutch dividend withholding tax purposes; |
(b) | liquidation proceeds, proceeds of redemption of our ordinary shares or, as a rule, consideration for the repurchase of our ordinary shares by the company in excess of the average paid-in capital recognized for Dutch dividend withholding tax purposes; |
(c) | the par value of our ordinary shares issued to a holder of our ordinary shares or an increase of the par value of our ordinary shares, to the extent that it does not appear that a contribution, recognized for Dutch dividend withholding tax purposes, has been made or will be made; and |
(d) | partial repayment of paid-in capital, recognized for Dutch dividend withholding tax purposes, if and to the extent that there are net profits (zuivere winst), unless (i) the shareholders at a General Meeting have resolved in advance to make such repayment and (ii) the par value of our ordinary shares concerned has been reduced by an equal amount by way of an amendment of our articles of association. |
Holders of the ADSs Resident in the Netherlands
A holder of the ADSs that is an individual that is resident or deemed to be resident in the Netherlands for Dutch tax purposes is generally entitled, subject to the anti-dividend stripping rules described below, to a full credit against its income tax liability, or a full refund, of the Dutch dividend withholding tax.
A holder of the ADSs that is a legal entity that is resident or deemed to be resident in the Netherlands for Dutch tax purposes is generally entitled, subject to the anti-dividend stripping rules described below, to a full credit against its corporate income tax liability of the Dutch dividend withholding tax. If and to the extent such legal entity cannot credit the full amount of Dutch dividend withholding tax in a given year, the Dutch dividend withholding tax may be carried forward and credited against its corporate income tax liability in subsequent years (without time limitation).
A holder of the ADSs that is a legal entity that is resident or deemed to be resident in the Netherlands for Dutch tax purposes that is exempt from Dutch corporate income tax but that is not qualifying exempt investment institution (vrijgestelde beleggingsinstelling), is generally entitled, subject to the anti-dividend stripping rules described below, to an exemption at source (subject to the completion of necessary procedural formalities) or a full refund of Dutch dividend withholding tax on dividends received.
178
The same generally applies to holders of the ADSs that are neither resident nor deemed to be resident in the Netherlands for Dutch tax purposes if the ADSs are attributable to a permanent establishment in the Netherlands of such non-resident holder.
Holders of the ADSs Resident Outside the Netherlands
A holder of the ADSs that is resident in a country for tax purposes with which the Netherlands has a tax treaty in effect, may, depending on the terms of such tax treaty and subject to the anti-dividend stripping rules described below, be eligible for a full or partial exemption from, or full or partial refund of, Dutch dividend withholding tax on dividends received.
A holder of the ADSs, that is a legal entity (a) tax resident in (i) an EU Member State, (ii) Iceland, Norway or Liechtenstein, or (iii) a country with which the Netherlands has concluded a tax treaty that includes an article on dividends and (b) that is in its state of residence under the terms of a tax treaty concluded with a third state, not considered to be resident for tax purposes in a country with which the Netherlands has not concluded a tax treaty that includes an article on dividends (i.e., not an EU Member State, Iceland, Norway or Liechtenstein), is generally entitled, subject to the anti-abuse rules and the anti-dividend stripping rules described below, to a full exemption from Dutch dividend withholding tax on dividends received if it holds an interest of at least 5% (in shares or, in certain cases, in voting rights) in the company or if it holds an interest of less than 5%, in either case where, had the holder of the ADSs been a Dutch resident, it would have had the benefit of the participation exemption (this may include a situation where another related party holds an interest of 5% or more in the company).
The full exemption from Dutch dividend withholding tax on dividends received by a holder of the ADSs, that is a legal entity (a) tax resident in (i) an EU Member State, (ii) Iceland, Norway or Liechtenstein, or (iii) a country with which the Netherlands has concluded a tax treaty that includes an article on dividends is not granted if (x) the interest held by such holder (i) is held with the avoidance of Dutch dividend withholding tax of another person as (one of) the main purpose(s) and (ii) forms part of an artificial structure or series of structures (such as structures which are not put into place for valid business reasons reflecting economic reality), or (y) the holder of ADSs has a similar function to a qualifying investment institution (fiscale beleggingsinstelling) or a qualifying exempt investment institution (vrijgestelde beleggingsinstelling).
A holder of the ADSs, that is an entity tax resident in (i) an EU Member State or (ii) Iceland, Norway or Liechtenstein, or (iii) in a jurisdiction which has an arrangement for the exchange of tax information with the Netherlands (and such holder as described under (iii) holds the ADSs as a portfolio investment (i.e., such holding is not acquired with a view to the establishment or maintenance of lasting and direct economic links between the holder of the ADSs and the company and does not allow the holder of the ADSs to participate effectively in the management or control of the company)), which is exempt from tax in its country of residence and does not have a similar function to a qualifying investment institution (fiscale beleggingsinstelling) or a qualifying exempt investment institution (vrijgestelde beleggingsinstelling), and that would have been exempt from Dutch corporate income tax if it had been a resident of the Netherlands, is generally entitled, subject to the anti-dividend stripping rules described below, to an exemption at source (subject to the completion of necessary procedural formalities) or a full refund of Dutch dividend withholding tax on dividends received. This exemption of full refund will in general benefit certain foreign pension funds, government agencies and certain government controlled commercial entities.
According to the anti-dividend stripping rules, no exemption, reduction, credit or refund of Dutch dividend withholding tax will be granted if the recipient of the dividend paid by the company is not considered the beneficial owner (uiteindelijk gerechtigde) of the dividend as defined in these rules. A recipient of a dividend is not considered the beneficial owner of the dividend if, as a consequence of a combination of transactions and tested at group level, (i) a person (other than the holder of the dividend coupon), directly or indirectly, partly or wholly benefits from the dividend, (ii) such person directly or indirectly retains or acquires a comparable interest in the ADSs, and (iii) such person is entitled to a less favorable exemption, refund or credit of dividend withholding tax than the recipient of the dividend distribution. The term “combination of transactions” includes transactions that have been entered into in the anonymity of a regulated stock market, the sole acquisition of one or more dividend coupons and the establishment of short-term rights or enjoyment on the ADSs (e.g., usufruct). The burden of proof to demonstrate that the recipient of a dividend
179
qualifies as the beneficial owner of such dividend lies with the recipient, unless the amount of the withheld dividend withholding tax in respect of such recipient in the relevant calendar is €1,000 or less.
Holders of the ADSs Resident in the U.S.
Dividends distributed by the company to U.S. resident holders of the ADSs that are eligible for benefits under the Convention between the Netherlands and the U.S. for the avoidance of Double Taxation and the Prevention of Fiscal Evasion with respect to Taxes and Income, dated December 18, 1992 as amended by the protocol of March 8, 2004 (U.S. Tax Treaty), generally will be entitled to a reduced dividend withholding tax rate of 5% in case of certain U.S. corporate shareholders owning at least 10% of the company’s total voting power. Certain U.S. pension funds and tax-exempt organizations may qualify for a complete exemption from Dutch dividend withholding tax.
Under the U.S. Tax Treaty such benefits are generally available to U.S. residents if such resident is the beneficial owner of the dividends, provided that such shareholder does not have an enterprise or an interest in an enterprise that is, in whole or in part, carried on through a permanent establishment or permanent representative in the Netherlands and to which enterprise or part of an enterprise the ADSs are attributable. A person may, however, not claim the benefits of the U.S. Tax Treaty if such person’s entitlement to such benefits is limited by the provisions of Article 26 (the limitation on benefits provision) of the U.S. Tax Treaty. The reduced dividend withholding tax rate can generally be applied at source upon the distribution of the dividends, provided that the proper forms have been filed in advance of the distribution. In the case of certain tax-exempt organizations, as a general rule, the so-called refund method applies; only when certain administrative conditions have been fulfilled may such tax-exempt organization use the exemption method.
Irrespective of meeting the conditions of the relevant provisions of the U.S. Tax Treaty, dividends distributed by the company to a U.S. resident holder (i) who is a legal entity resident in the U.S. and (ii) that is in the U.S. under the terms of a tax treaty with a third state not considered to be resident for tax purposes in a country with which the Netherlands has not concluded a tax treaty that includes an article on dividends (not being a Member State of the European Union, Iceland, Norway or Liechtenstein), are generally, subject to the anti-dividend stripping rules described above, fully exempt from Dutch dividend withholding tax if the U.S. resident holder of ADSs holds an interest of at least 5% in the company or if it holds an interest of less than 5%, in either case where, had the holder of ADSs been a Dutch resident, it would have had the benefit of the participation exemption (this may include a situation where another related party holds an interest of 5% or more in the company). The full exemption from Dutch dividend withholding tax on dividends received by a U.S. holder of ADSs that is a legal entity is however not granted if (x) the interest held by such U.S. holder (i) is held with the avoidance of Dutch dividend withholding tax of another person as (one of) the main purpose(s) and (ii) forms part of an artificial structure or series of structures (such as structures which are not put into place for valid business reasons reflecting economic reality) or (y) the U.S. holder of ADSs has a similar function to a qualifying investment institution (fiscale beleggingsinstelling) or a qualifying exempt investment institution (vrijgestelde beleggingsinstelling).
Taxes on Income and Capital Gains
Holders of the ADSs Resident in the Netherlands: Individuals
A holder of the ADSs, who is an individual resident or deemed to be resident in the Netherlands for Dutch tax purposes will be subject to regular Dutch income tax on the income derived from the ADSs and the gains realized upon the acquisition, redemption and/or disposal of the ADSs by the holder thereof, if:
(a) | such holder of the ADSs has an enterprise or an interest in an enterprise, to which enterprise the ADSs are attributable; and/or |
(b) | such income or capital gain forms “a benefit from miscellaneous activities” (resultaat uit overige werkzaamheden) which, for instance, would be the case if the activities with respect to the ADSs exceed “normal active asset management” (normaal, actief vermogensbeheer) or if income and gains are derived from the holding, whether directly or indirectly, of (a combination of) shares, debt claims or other rights (together, a “lucrative interest” (lucratief belang)) that the holder thereof has acquired under such circumstances that such |
180
income and gains are intended to be remuneration for work or services performed by such holder (or a related person), whether within or outside an employment relation, where such lucrative interest provides the holder thereof, economically speaking, with certain benefits that have a relation to the relevant work or services.
If either of the abovementioned conditions (a) or (b) applies, income derived from the ADSs and the gains realized upon the acquisition, redemption and/or disposal of the ADSs will in general be subject to Dutch income tax at the progressive rates up to 49.5%.
If the abovementioned conditions (a) and (b) do not apply, a holder of the ADSs who is an individual, resident or deemed to be resident in the Netherlands for Dutch tax purposes will not be subject to taxes on income and capital gains in the Netherlands. Instead, such individual is generally taxed at a flat rate of 36% on deemed income from “savings and investments” (sparen en beleggen), which deemed income is determined on the basis of the amount included in the individual’s “yield basis” (rendementsgrondslag) at the beginning of the calendar year (minus a tax-free threshold; the yield basis minus such threshold being the tax basis). For the 2024 tax year, the deemed income derived from savings and investments will be a percentage of the tax basis up to 6.04% that is determined based on the actual allocation of (i) savings, (ii) other investments, and (iii) debts/liabilities within the individual’s yield basis. The tax-free threshold for 2024 is €57,000. The percentages to determine the deemed income will be reassessed every year.
Holders of the ADSs Resident in the Netherlands: Corporate Entities
A holder of the ADSs that is resident or deemed to be resident in the Netherlands for corporate income tax purposes, and that is:
| ● | a corporation; |
| ● | another entity with a capital divided into shares; |
| ● | a cooperative (association); or |
| ● | another legal entity that has an enterprise or an interest in an enterprise to which the ADSs are attributable, |
but which is not:
| ● | a qualifying pension fund; |
| ● | a qualifying investment institution (fiscale beleggingsinstelling) or a qualifying exempt investment institution (vrijgestelde beleggingsinstelling); or |
| ● | another entity exempt from corporate income tax, will in general be subject to regular Dutch corporate income tax, generally levied at a rate of 25.8% (19% over profits up to and including €200,000) over income derived from the ADSs and the gains realized upon the acquisition, redemption and/or disposal of the ADSs, unless, and to the extent that, the participation exemption (deelnemingsvrijstelling) applies. |
Holders of the ADSs Resident Outside the Netherlands: Individuals
A holder of the ADSs who is an individual, not resident or deemed to be resident in the Netherlands will not be subject to any Dutch taxes on income derived from the ADSs and the gains realized upon the acquisition, redemption and/or disposal of the ADSs (other than the Dutch dividend withholding tax described above), unless:
(a) | such holder has an enterprise or an interest in an enterprise that is, in whole or in part, carried on through a permanent establishment (vaste inrichting) or a permanent representative (vaste vertegenwoordiger) in the Netherlands and to which enterprise or part of an enterprise, as the case may be, the ADSs are attributable; or |
181
(b) | such income or capital gain forms a “benefit from miscellaneous activities in the Netherlands” (resultaat uit overige werkzaamheden in Nederland) which would for instance be the case if the activities in the Netherlands with respect to the ADSs exceed “normal active asset management” (normaal, actief ver mogensbeheer) or if such income and gains are derived from the holding, whether directly or indirectly, of (a combination of) shares, debt claims or other rights (together, a “lucrative interest” (lucratief belang)) that the holder thereof has acquired under such circumstances that such income and gains are intended to be remuneration for work or services performed by such holder (or a related person), in whole or in part, in the Netherlands, whether within or outside an employment relation, where such lucrative interest provides the holder thereof, economically speaking, with certain benefits that have a relation to the relevant work or services. |
If either of the abovementioned conditions (a) or (b) applies, income or capital gains in respect of dividends distributed by the company or in respect of any gains realized upon the acquisition, redemption and/or disposal of the ADSs will in general be subject to Dutch income tax at the progressive rates up to 49.5%.
Holders of the ADSs Resident Outside the Netherlands: Legal and Other Entities
A holder of the ADSs, that is a legal entity, another entity with a capital divided into shares, an association, a foundation or a fund or trust, not resident or deemed to be resident in the Netherlands for corporate income tax purposes, will not be subject to any Dutch taxes on income derived from the ADSs and the gains realized upon the acquisition, redemption and/or disposal of the ADSs (other than the Dutch dividend withholding tax described above), unless:
| ● | such holder has an enterprise or an interest in an enterprise that is, in whole or in part, carried on through a permanent establishment (vaste inrichting) or a permanent representative (vaste vertegenwoordiger) in the Netherlands and to which enterprise or part of an enterprise, as the case may be, the ADSs are attributable; or |
| ● | such holder has a substantial interest (aanmerkelijk belang) in the company, that (i) is held with the avoidance of Dutch income tax of another person as (one of) the main purpose(s) and (ii) forms part of an artificial structure or series of structures (such as structures which are not put into place for valid business reasons reflecting economic reality).If one of the abovementioned conditions applies, income derived from the ADSs and the gains realized upon the acquisition, redemption and/or disposal of the ADSs will, in general, be subject to Dutch regular corporate income tax, levied at a rate of 25.8% (19% over profits up to and including €200,000), unless, and to the extent that, with respect to a holder as described under (a), the participation exemption (deelnemingsvrijstelling) applies. |
Gift, Estate and Inheritance Taxes
Holders of the ADSs Resident in the Netherlands
Gift tax may be due in the Netherlands with respect to an acquisition of the ADSs by way of a gift by a holder of the ADSs who is resident or deemed to be resident of the Netherlands at the time of the gift.
Inheritance tax may be due in the Netherlands with respect to an acquisition or deemed acquisition of the ADSs by way of an inheritance or bequest on the death of a holder of the ADSs who is resident or deemed to be resident of the Netherlands, or in case of a gift by an individual who at the date of the gift was neither resident nor deemed to be resident in the Netherlands, such individual dies within 180 days after the date of the gift, while that individual, at the time of the individual’s death, is resident or deemed to be resident in the Netherlands.
For purposes of Dutch gift and inheritance tax, an individual with the Dutch nationality will be deemed to be resident in the Netherlands if such individual has been resident in the Netherlands at any time during the 10 years preceding the date of the gift or such individual’s death. For purposes of Dutch gift tax, an individual not holding the Dutch nationality will be deemed to be resident of the Netherlands if such individual has been resident in the Netherlands at any time during the 12 months preceding the date of the gift.
182
Holders of the ADSs Resident Outside the Netherlands
No gift, estate or inheritance taxes will arise in the Netherlands with respect to an acquisition of the ADSs by way of a gift by, or on the death of, a holder of the ADSs who is neither resident nor deemed to be resident of the Netherlands, unless, in the case of a gift of the ADSs by an individual who at the date of the gift was neither resident nor deemed to be resident in the Netherlands, such individual dies within 180 days after the date of the gift, while being resident or deemed to be resident in the Netherlands.
Certain Special Situations
For purposes of Dutch gift, estate and inheritance tax, (i) a gift by a third party will be construed as a gift by the settlor, and (ii) upon the death of the settlor, as a rule such settlor’s beneficiaries will be deemed to have inherited directly from the settlor. Subsequently, such beneficiaries will be deemed the settlor, grantor or similar originator of the separated private assets for purposes of the Dutch gift, estate and inheritance tax in case of subsequent gifts or inheritances.
For the purposes of the Dutch gift and inheritance tax, a gift that is made under a condition precedent is deemed to have been made at the moment such condition precedent is satisfied. If the condition precedent is fulfilled after the death of the donor, the gift is deemed to be made upon the death of the donor.
Value Added Tax
No Dutch value added tax will arise in respect of or in connection with the subscription, issue, placement, allotment or delivery of the ADSs.
Other Taxes and Duties
No Dutch registration tax, capital tax, custom duty, transfer tax, stamp duty or any other similar documentary tax or duty, other than court fees, will be payable in the Netherlands in respect of or in connection with the subscription, issue, placement, allotment or delivery of the ADSs.
Residency
A holder of the ADSs will not be treated as a resident, or a deemed resident, of the Netherlands for tax purposes by reason only of the acquisition, or the holding, of the ADSs or the performance by the company under the ADSs.
Material Belgian Tax Consequences
The paragraphs below present a summary of certain Belgian federal income tax consequences of the ownership and disposal of ADSs by an investor that purchases such ADSs. The summary is based on laws, treaties and regulatory interpretations in effect in Belgium on the date of this Annual Report, all of which are subject to change, including changes that could have retroactive effect.
Investors should appreciate that, as a result of evolutions in law or practice, the eventual tax consequences may be different from what is stated below. The tax legislation of the investor’s country of residence may have an impact on the income received from the ADSs.
This summary does not purport to address all tax consequences of investments in, the ownership and disposal of ADSs, and does not take into account the specific circumstances of particular investors, some of which may be subject to special rules, or the tax laws of any country other than Belgium. This summary does not describe the tax treatment of investors that are subject to special rules, such as banks, insurance companies, collective investment undertakings, dealers in securities or currencies, persons that hold, or will hold, ADSs as a position in a straddle, share-repurchase transaction, conversion transactions, synthetic security or other integrated financial transactions. This summary does not address the tax regime applicable to ADSs held by Belgian tax residents through a fixed base or a permanent
183
establishment (PE) situated outside Belgium. This summary does not address the local taxes that may be due in connection with an investment in shares, other than the local surcharges which generally vary between 0% and 9% of the investor’s income tax liability in Belgium.
In addition to the assumptions mentioned above, it is also assumed in this discussion that for purposes of the domestic Belgian tax legislation, the owners of ADSs will be treated as the owners of the ordinary shares represented by such ADSs. However, this assumption has not been confirmed by or verified with the Belgian Tax Authorities.
Investors should consult their own advisors regarding the tax consequences of an investment in the ADSs in light of their particular situation, including the effect of any state, local or other national laws, treaties and regulatory interpretations thereof.
For the purposes of this summary, a resident investor is:
| ● | an individual subject to Belgian personal income tax (personenbelasting/impôt des personnes physiques), i.e. (i) an individual having its domicile in Belgium, (ii) when not having its domicile in Belgium, an individual having its seat of wealth in Belgium, or (iii) an individual assimilated to a resident for purposes of Belgian tax law; |
| ● | a company (as defined by Belgian tax law) subject to Belgian corporate income tax (vennootschapsbelasting/impôt des sociétés), i.e., a corporate entity having its principal establishment, administrative seat or effective place of management in Belgium (and that is not excluded from the scope of the Belgian corporate income tax). A company having its registered seat in Belgium shall be presumed, unless the contrary is proved, to have its principal establishment, administrative seat or effective place of management in Belgium; or |
| ● | a legal entity subject to the Belgian tax on legal entities (rechtspersonenbelasting/impôt des personnes morales), i.e., a legal entity other than a company subject to Belgian corporate income tax having its principal establishment, administrative seat or effective place of management in Belgium. |
A non-resident investor is any individual, company or legal entity that does not fall in any of the three previous classes.
Dividends
For Belgian income tax purposes, the gross amount of all benefits paid on or attributed to the ADSs is generally treated as a dividend distribution. By way of exception, the repayment of capital carried out in accordance with applicable Dutch company law provisions is not treated as a dividend distribution to the extent that such repayment is imputed on fiscal capital. This fiscal capital includes, in principle, the actual paid-up statutory share capital and, subject to certain conditions, the paid-up share premiums and the cash amounts subscribed to at the time of the issue of profit-sharing certificates. However, a repayment of capital is not fully imputed on fiscal capital if the company also has certain reserves. Indeed, in such case, a reimbursement of capital is proratedly imputed on, on the one hand, fiscal capital and, on the other hand, taxed reserves (whether or not incorporated in capital) and tax-exempt reserves incorporated in capital (according to a specific priority rule). The part imputed on the reserves is treated as a dividend distribution subject to applicable tax rules.
Belgian withholding tax of 30% is normally levied on dividends by any intermediary established in Belgium that is in any way involved in the processing of the payment of non-Belgian sourced dividends (e.g., a Belgian financial institution). This withholding tax rate is subject to such relief as may be available under applicable domestic or tax treaty provisions.
The Belgian withholding tax is calculated on the dividend amount after deduction of any non-Belgian dividend withholding tax.
184
In the case of a redemption of the ADSs, the redemption distribution (after deduction of the part of the fiscal capital represented by the redeemed ADSs) will be treated as a dividend subject to a Belgian withholding tax of 30%, subject to such relief as may be available under applicable domestic or tax treaty provisions. No withholding tax will be triggered if this redemption is carried out on a stock exchange and meets certain conditions.
In the event of our liquidation, any amounts distributed in excess of the fiscal capital will in principle be subject to the 30% withholding tax, subject to such relief as may be available under applicable domestic or tax treaty provisions.
Under Belgian law, non-Belgian dividend withholding tax is not creditable against Belgian income tax and is not reimbursable to the extent that it exceeds Belgian income tax. Please refer to Item 10.E. “Taxation —Certain Material U.S. Federal Income Tax Considerations for U.S. Holders—Passive Foreign Investment Company Considerations” for a description of withholding tax that may be imposed on dividends by the Netherlands.
Belgian Resident Individuals
For Belgian resident individuals who acquire and hold ADSs as a private investment, the Belgian dividend withholding tax fully discharges their personal income tax liability. They may nevertheless need to report the dividends in their personal income tax return if no intermediary established in Belgium was in any way involved in the processing of the payment of the non-Belgian sourced dividends. Moreover, even if an intermediary established in Belgium was involved, they can opt to report the income in their personal income tax return. If (and only if) the dividends are reported, they will normally be eligible for a tax exemption with respect to ordinary dividends in an amount of up to €833 (for income year 2024) per year and per taxpayer (Article 21, first subsection, 14°, of the Belgian Income Tax Code (ITC)). For the avoidance of doubt, all reported dividends (not only dividends distributed on our ADSs) are taken into account to assess whether the said maximum amount is reached. The abovementioned exempted amount is not applicable to redemption and liquidation dividends.
Where the beneficiary needs or, as applicable, opts to report them, dividends will normally be taxable at the lower of the generally applicable 30% Belgian withholding tax rate on dividends or, in case globalization is more advantageous, at the progressive personal income tax rates applicable to the taxpayer’s overall declared income. In addition, if the dividends are reported, the Belgian dividend withholding tax levied at source may be credited against the personal income tax due and is reimbursable to the extent that it exceeds the personal income tax due, provided that the dividend distribution does not result in a reduction in value of or a capital loss on our ADSs. The latter condition is not applicable if the individual can demonstrate that it has held ADSs in full legal ownership for an uninterrupted period of 12 months prior to the payment or attribution of the dividends.
For Belgian resident individual investors who acquire and hold the ADSs for professional purposes, the Belgian withholding tax does not fully discharge their Belgian income tax liability. Dividends received must be reported by the investor and will, in such a case, be taxable at the investor’s personal income tax rate increased with local surcharges. Belgian withholding tax levied may be credited against the personal income tax due and is reimbursable to the extent that it exceeds the income tax due, subject to two conditions: (i) the taxpayer must own the ADSs in full legal ownership on the dividend record date and (ii) the dividend distribution may not result in a reduction in value of or a capital loss on the ADSs. The latter condition is not applicable if the investor can demonstrate that it has held the full legal ownership of the ADSs for an uninterrupted period of 12 months prior to the payment or attribution of the dividends.
Belgian Resident Companies
Dividends received by Belgian resident companies are exempt from Belgian withholding tax provided that the investor satisfies the identification requirements in Article 117, §11 of the Royal Decree implementing the ITC.
For Belgian resident companies, the gross dividend income (after deduction of any non-Belgian withholding tax but including any Belgian withholding tax) must be declared in the corporate income tax return and will be subject to a corporate income tax rate of 25%, except that a reduced corporate income tax rate of 20% applies to small companies and medium sized enterprises (as defined by Article 1:24, §1 to §6 of the Belgian Code on Companies and Associations) on the first €100,000 of taxable profits (subject to certain conditions).
185
Belgian resident companies can generally (although subject to certain limitations) deduct 100% of the gross dividend received from their taxable income (Dividend Received Deduction) provided that at the time of a dividend payment or attribution: (i) the Belgian resident company holds ADSs representing at least 10% of our share capital or a participation with an acquisition value of at least €2,500,000 (it being understood that only one out of the two tests must be satisfied); (ii) the shares representing our share capital have been or will be held in full ownership for an uninterrupted period of at least one year; and (iii) the conditions described in Article 203 of the ITC (relating to the taxation of the underlying distributed income and the absence of abuse), or the Article 203 of the ITC Taxation Condition, are met (Conditions for Dividend Received Deduction).
Conditions (i) and (ii) above are, in principle, not applicable for dividends received by an investment company within the meaning of Article 2, §1, 5°, f) ITC. The Conditions for the application of the Dividend Received Deduction depend on a factual analysis and for this reason the availability of this regime should be verified upon each dividend distribution.
Any Belgian dividend withholding tax levied at source can be credited against the ordinary Belgian corporate income tax and is reimbursable to the extent it exceeds such corporate income tax, subject to two conditions: (i) the taxpayer must own the ADSs in full legal ownership on the dividend record date and (ii) the dividend distribution does not result in a reduction in value of or a capital loss on the ADSs. The latter condition is not applicable: (i) if the taxpayer can demonstrate that it has held the ADSs in full legal ownership for an uninterrupted period of 12 months immediately prior to the payment or attribution of the dividends or (ii) if, during that period, the ADSs never belonged to a taxpayer other than a Belgian resident company or a non-resident company that has, in an uninterrupted manner, invested the ADSs in a PE in Belgium.
Belgian Resident Organizations for Financing Pensions
For organizations for financing pensions (OFPs) i.e., Belgian pension funds incorporated under the form of an OFP (organisme voor de financiering van pensioenen/organisme de financement de pensions) within the meaning of Article 8 of the Belgian Law of October 27, 2006, dividend income generally does not constitute taxable income.
Dividends distributed through the intervention of a Belgian intermediary are generally subject to Belgian dividend withholding tax. If dividends are paid or attributed without the intervention of a Belgian intermediary, the applicable Belgian withholding tax will have to be reported and paid by the OFP to the Belgian tax administration.
The Belgian dividend withholding tax can in principle be credited against the OFPs’ corporate income tax and is reimbursable to the extent it exceeds the corporate income tax due. However, such Belgian withholding cannot be credited by an OFP if the shares on which the dividends are paid have not been held uninterruptedly in full ownership for at least 60 days, unless the OFP demonstrates that the dividends are not connected to an arrangement (or a series of arrangements) that is not genuine (kunstmatig/pas authentique) and has been put in place for the main purpose or one of the main purposes of obtaining this withholding tax credit.
Other Belgian Resident Taxable Legal Entities
For taxpayers subject to the Belgian income tax on legal entities, the Belgian dividend withholding tax in principle fully discharges their income tax liability. If the dividend is paid outside Belgium without the intervention of a Belgian paying agent and without the deduction of Belgian withholding tax, the legal entity is in principle required to declare and pay the 30% withholding tax to the Belgian tax authorities.
Belgian Non-Resident Individuals and Companies
Dividend payments on the ADSs through a professional intermediary in Belgium will, in principle, be subject to the 30% withholding tax, unless the shareholder is resident in a country with which Belgium has concluded a double taxation agreement and delivers the requested affidavit. Non-resident investors can also obtain an exemption of Belgian dividend withholding tax if they are the owners or usufructors of the ADSs and they deliver an affidavit confirming that they have not allocated the ADSs to business activities in Belgium and that they are non-residents, provided that the
186
dividend is paid through a Belgian credit institution, stock market company or recognized clearing or settlement institution.
If the ADSs are acquired by a non-resident investor in connection with a business in Belgium, the investor must report any dividends received, which are taxable at the applicable non-resident individual or corporate income tax rate, as appropriate. Any Belgian withholding tax levied at source can be credited against the non-resident individual or corporate income tax and is reimbursable to the extent it exceeds the income tax due, subject to two conditions: (i) the taxpayer must own the ADSs in full legal ownership on the dividend record date and (ii) the dividend distribution does not result in a reduction in value of or a capital loss on the ADSs. The latter condition is not applicable if (i) the non-resident individual or the non-resident company can demonstrate that the ADSs were held in full legal ownership for an uninterrupted period of 12 months immediately prior to the payment or attribution of the dividends or (ii) with regard to non-resident companies only, if, during the said period, the ADSs have not belonged to a taxpayer other than a resident company or a non-resident company which has, in an uninterrupted manner, invested the ADSs in a Belgian PE.
Non-resident companies that have invested the ADSs in a Belgian establishment can deduct up to 100% of the gross dividends included in their taxable profits if, at the date dividends are paid or attributed, the Conditions for Dividend Received Deduction are satisfied. Application of the Dividend Received Deduction depends, however, on a factual analysis to be made upon each distribution and its availability should be verified upon each distribution.
Capital Gains and Losses on ADSs
Belgian Resident Individuals
In principle, Belgian resident individuals acquiring the ADSs as a private investment should not be subject to Belgian capital gains tax on the disposal of the ADSs; capital losses are not tax deductible.
Capital gains realized in a private (i.e., non-professional) context on the transfer for consideration of shares by a private individual, are taxable at 33% (plus local surcharges) if the capital gain is deemed to be realized outside the scope of the normal management of the individual’s private estate. Capital losses are, however, not tax deductible in such event.
Capital gains realized in a private (i.e., non-professional) context on the transfer for consideration of shares of a Belgian company to a foreign company with its fiscal residency outside the EEA, by a private individual, who held alone or jointly with his/her family, directly or indirectly, more than 25% of the shares of that Belgian company, are taxable at a flat rate of 16.5% (plus local surcharges).
Gains realized by Belgian resident individuals upon the redemption of the ADSs or upon our liquidation are generally taxable as a dividend.
Belgian resident individuals who hold the ADSs for professional purposes are taxable at the ordinary progressive personal income tax rates (plus local surcharges) on any capital gains realized upon the disposal of the ADSs, except for ADSs held for more than five years, which are taxable at a flat rate of 16.5% (plus local surcharges). Capital losses on the ADSs incurred by Belgian resident individuals who hold the ADSs for professional purposes are in principle tax deductible.
Belgian Resident Companies
Belgian resident companies are normally not subject to Belgian capital gains taxation on gains realized upon the disposal of our ADSs provided that (i) the shares represent at least 10% of our share capital or a participation with an acquisition value of at least €2,500,000 (it being understood that only one out of the two tests must be satisfied), (ii) the Article 203 ITC Taxation Condition is satisfied and (iii) the ADSs have been held in full legal ownership for an uninterrupted period of at least one year immediately preceding the disposal.
187
If one of the above conditions is not met, the capital gains realized upon the disposal of our ADSs by a Belgian resident company are taxable at the ordinary corporate income tax rate of, currently, 25%, unless the reduced corporate income tax rate of 20% on the first €100,000 of taxable profits applies (see above).
Capital losses on our ADSs incurred by resident companies are as a general rule not tax deductible.
Our ADSs held in the trading portfolios (handelsportefeuille/portefeuille commercial) of qualifying credit institutions, investment enterprises and management companies of collective investment undertakings which are subject to the Royal Decree of 23 September 1992 on the annual accounts of credit institutions, investment firms and management companies of collective investment undertakings (Koninklijk besluit van 23 september 1992 op de jaarrekening van de kredietinstellingen, de beleggingsondernemingen en de beheervennootschappen van instellingen voor collectieve belegging/ arrêté royal du 23 septembre 1992 relatif aux comptes annuels des établissements de crédit, des entreprises d’investissement et des sociétés de gestion d’organismes de placement collectif) are subject to a different regime. The capital gains on such shares are taxable at the ordinary corporate income tax rate of 25%. Capital losses on such shares are tax deductible. Internal transfers to and from the trading portfolio are assimilated to a realization.
Capital gains realized by Belgian resident companies (both ordinary Belgian resident companies and qualifying credit institutions, investment enterprises and management companies of collective investment undertakings) upon the redemption of our ADSs or upon our liquidation are, in principle, subject to the same taxation regime as dividends. See Item 10.E. “Taxation —Material Belgian Tax Consequences”.
Belgian resident OFPs
OFPs are, in principle, not subject to Belgian capital gains taxation realized upon the disposal of the ADSs, and capital losses are not tax deductible.
Capital gains realized by Belgian OFPs upon the redemption of ADSs or upon our liquidation will in principle be taxed as dividends.
Other Belgian Taxable Legal Entities
Belgian resident legal entities subject to the legal entities income tax are, in principle, not subject to Belgian capital gains taxation on the disposal of ADSs.
Capital gains realized by Belgian resident legal entities upon the redemption of ADSs or upon our liquidation will in principle be taxed as dividends.
Capital losses on ADSs incurred by Belgian resident legal entities are not tax deductible.
Belgian Non-Resident Individuals and Companies
Non-resident individuals or companies are, in principle, not subject to Belgian income tax on capital gains realized upon disposal of the ADSs, unless such ADSs are held as part of a business conducted in Belgium through a Belgian establishment. In such a case, the same principles apply as described with regard to Belgian individuals (holding the shares for professional purposes) or Belgian companies.
Non-resident individuals who do not use the shares for professional purposes and who have their fiscal residence in a country with which Belgium has not concluded a tax treaty or with which Belgium has concluded a tax treaty that confers the authority to tax capital gains on the ADSs to Belgium, might be subject to tax in Belgium if the capital gains are obtained or received in Belgium and arise from transactions which are to be considered speculative or beyond the normal management of one’s private estate. See Item 10.E. “Taxation —Certain Material U.S. Federal Income Tax Considerations for U.S. Holders—Passive Foreign Investment Company Considerations”. Such non-resident individuals might therefore be obliged to file a tax return and should consult their own tax advisor.
188
Capital gains realized by non-resident individuals or non-resident companies upon the redemption of ADSs or upon our liquidation will, in principle, be subject to the same taxation regime as dividends.
Tax on Stock Exchange Transactions
Upon the issue of the ADSs (primary market), no Tax on Stock Exchange Transactions (taks op beursverrichtingen/taxe sur opérations de bourse) is due.
The purchase and the sale and any other acquisition or transfer for consideration of ADSs (secondary market transactions) is subject to the Tax on Stock Exchange Transactions if (i) it is executed in Belgium through a professional intermediary, or (ii) deemed to be executed in Belgium, which is the case if the order is directly or indirectly made to a professional intermediary established outside of Belgium, either by private individuals with habitual residence in Belgium, or legal entities for the account of their seat or establishment in Belgium (both, a Belgian Investor).
The Tax on Stock Exchange Transactions is levied at a rate of 0.35% of the purchase price, capped at €1,600 per transaction and per party.
A separate tax is due by each party to the transaction, and both taxes are collected by the professional intermediary. However, if the intermediary is established outside of Belgium, the tax will in principle be due by the Belgian Investor, unless that Belgian Investor can demonstrate that the tax has already been paid. Professional intermediaries established outside of Belgium can, subject to certain conditions and formalities, appoint a Belgian Stock Exchange Tax Representative, which will be liable for the Tax on Stock Exchange Transactions in respect of the transactions executed through the professional intermediary. If the Stock Exchange Tax Representative would have paid the Tax on Stock Exchange Transactions due, the Belgian Investor will, as per the above, no longer be the debtor of the Tax on Stock Exchange Transactions.
No Tax on Stock Exchange Transactions is due on transactions entered into by the following parties, provided they are acting for their own account: (i) professional intermediaries described in Article 2, 9° and 10° of the Belgian Law of August 2, 2002; (ii) insurance companies described in Article 2, §1 of the Belgian Law of July 9, 1975; (iii) professional retirement institutions referred to in Article 2,1° of the Belgian Law of October 27, 2006 concerning the supervision on institutions for occupational pension; (iv) collective investment institutions; (v) regulated real estate companies; and (vi) Belgian non-residents provided they deliver a certificate to their financial intermediary in Belgium confirming their non-resident status.
The EU Commission adopted on February 14, 2013 the Draft Directive on a Financial Transaction Tax (FTT). The Draft Directive currently stipulates that once the FTT enters into force, the Participating Member States shall not maintain or introduce taxes on financial transactions other than the FTT (or VAT as provided in the Council Directive 2006/112/EC of November 28, 2006 on the common system of value added tax). For Belgium, the Tax on Stock Exchange Transactions should thus be abolished once the FTT enters into force. Due to the lack of progress in the negotiations on the Draft Directive, the European Commission has announced that it would present a proposal for a new own resource based on the FTT by June 2024 (with a view to its introduction by January 1, 2026).
Annual Tax on Securities Accounts
A Law of 17 February 2021 introduced a new Belgian Annual Tax on Securities Accounts, which entered into effect on February 26, 2021. The Annual Tax on Securities Accounts is a subscription tax, levied on securities accounts and not on the holders thereof. A securities account is defined as an account on which financial instruments can be credited and debited.
The tax applies to securities accounts held both in Belgium and abroad when the account holder is a Belgian resident or when the account forms part of the assets of a Belgian establishment of a non-Belgian resident. The tax applies to natural persons residing in Belgium, as well as to companies and legal entities (subject to the tax for legal entities) that are established in Belgium.
189
The tax is also applicable to securities accounts held by non-Belgian residents (both natural persons and legal persons) if the securities account is held in Belgium. If the applicable double tax treaty however allocates the right to tax capital to the jurisdiction of residence, Belgium would be prevented from applying the Annual Tax on Securities Accounts to the Belgian securities accounts held by non-Belgian residents. As described above, the tax applies whether or not the account is held in Belgium if the account forms part of the assets of a Belgian establishment of a non-Belgian resident.
The Annual Tax on Securities Accounts is applicable to securities accounts of which the average value of the assets amounts to more than €1,000,000 during the reference period. In principle, this reference period starts on 1 October and ends on 30 September of the following year. The aforementioned threshold is assessed on the average value of the assets in the securities account at reference points within the reference period (in principle December 31st, March 31st, June 30th and September 30th). The threshold is assessed per securities account and not per account holder.
The applicable tax rate is 0.15%, which is levied on the average value of the assets held in the securities account that exceeds the €1,000,000 threshold. It is however limited to 10% of the difference between the average value and the threshold of €1,000,000, in order to avoid that the Annual Tax on Securities Accounts would result in reducing the value of the securities account below the €1,000,000 threshold.
The Annual Tax is in principle withheld, reported and paid by the Belgian intermediary. If the intermediary is established outside of Belgium, the tax must in principle be reported and paid by the account holder, unless the account holder can demonstrate that the tax has already been reported and paid by an intermediary. Intermediaries established outside of Belgium can, subject to certain conditions and formalities, appoint a Belgian Annual Tax on Securities Accounts Representative, which will be liable for reporting and paying the Annual Tax on Securities Accounts in respect of securities accounts in scope of the Annual Tax that are held through such intermediaries. If the Annual Tax on Securities Accounts Representative would have paid the Annual Tax on Securities Accounts due, the account holder will, as per the above, no longer be the debtor of the Annual Tax on Securities Accounts.
The Annual Tax on Securities Accounts is however not applicable to securities accounts held by certain categories of account holders active in the financial or fund sector, as listed in the relevant legislation (e.g., credit institutions, insurance companies, investment companies, and certain collective investment undertakings). These exemptions do however not apply if a non-qualifying third party has a direct or indirect claim on the value of the securities account.
Prospective investors are strongly advised to seek their own professional advice in relation to the possible impact of the Annual Tax on Securities Accounts on their own personal tax position.
Enforcement of Civil Liabilities
We are a European public company with limited liability (Societas Europaea or SE) incorporated under the laws of the Netherlands. Substantially all of our assets are located outside the U.S. As a result, it may not be possible for investors to effect service of process within the U.S. upon such persons or to enforce against them or us in U.S. courts, including judgments predicated upon the civil liability provisions of the federal securities laws of the U.S.
The U.S. and the Netherlands currently do not have a treaty providing for the reciprocal recognition and enforcement of judgments, other than arbitration awards, in civil and commercial matters. Consequently, a final judgment for payment given by a court in the U.S., whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in the Netherlands. In order to obtain a judgment which is enforceable in the Netherlands, the party in whose favor a final and conclusive judgment of the U.S. court has been rendered will be required to file its claim with a court of competent jurisdiction in the Netherlands. Such party may submit to the Dutch court the final judgment rendered by the U.S. court. This court will have a level of discretion in its assessment of the judgment rendered by the relevant U.S. court. On the basis of case law by the Dutch Supreme Court, Dutch courts will in principle have to give conclusive effect to a final and enforceable judgment of such court in respect of the contractual obligations thereunder without re-examination or re-litigation of the substantive matters adjudicated upon, provided that: (i) the U.S. court involved accepted jurisdiction on the basis of internationally recognized grounds to accept jurisdiction,
190
(ii) the proceedings before such court being in compliance with principles of proper procedure (behoorlijke rechtspleging), (iii) such judgment not being contrary to the public policy of the Netherlands and (iv) such judgment not being incompatible with a judgment given between the same parties by a Netherlands court or with a prior judgment given between the same parties by a foreign court in a dispute concerning the same subject matter and based on the same cause of action, provided such prior judgment fulfills the conditions necessary for it to be given binding effect in the Netherlands. Dutch courts may deny the recognition and enforcement of punitive damages or other awards that do not fit to the Dutch legal order. Moreover, a Dutch court may reduce the amount of damages granted by a U.S. court and recognize damages only to the extent that they are necessary to compensate actual losses or damages. Enforcement and recognition of judgments of U.S. courts in the Netherlands are solely governed by the provisions of the Dutch Civil Procedure Code.
Original actions or actions for the enforcement of judgments of U.S. courts relating to the civil liability provisions of the federal or state securities laws of the U.S. are not directly enforceable in Belgium. The U.S. and Belgium currently do not have a treaty providing for reciprocal recognition and enforcement of judgments, other than arbitral awards, in civil and commercial matters. Consequently, a final judgment for payment given by a court in the U.S., whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in Belgium. In order for a final judgment for the payment of money rendered by U.S. courts based on civil liability to produce any effect on Belgian soil, it is accordingly required that this judgment be recognized and be declared enforceable by a Belgian court pursuant to the relevant provisions of the PIL Code. Recognition or enforcement does not imply a review of the merits of the case and is irrespective of any reciprocity requirement. A U.S. judgment will, however, not be recognized or declared enforceable in Belgium if it infringes upon one or more of the grounds for refusal which are exhaustively listed in article 25 of the PIL Code. In addition to recognition or enforcement, a judgment by a federal or state court in the U.S. against us may also serve as evidence in a similar action in a Belgian court if it meets the conditions required for the authenticity of judgments according to the law of the state where it was rendered. In addition, with regard to enforcements by legal proceedings in Belgium (including the recognition of foreign court decisions in Belgium), a registration tax at the rate of 3% of the amount of the judgment is payable by the debtor, if the sum of money which the debtor is ordered to pay by a Belgian court, or by a foreign court judgment that is either (i) automatically enforceable and registered in Belgium, or (ii) rendered enforceable by a Belgian court, exceeds €12,500. The registration tax is payable by the debtor. The debtor is liable for the payment of the registration tax, in the proportion determined by the decision ordering payment or liquidation or determining priority for creditors made or established against it. The debtor(s) are jointly and severally liable in the event that they are ordered to pay jointly and severally. A stamp duty is payable as of the second certified copy of an enforcement judgment rendered by a Belgian court, with a maximum of €1,450.
Dutch and Belgian civil procedure differ substantially from U.S. civil procedure in a number of respects. Insofar as the production of evidence is concerned, U.S. law and the laws of several other jurisdictions based on common law provide for pre-trial discovery, a process by which parties to the proceedings may prior to trial compel the production of documents by adverse or third parties and the deposition of witnesses. Evidence obtained in this manner may be decisive in the outcome of any proceeding. No such pre-trial discovery process exists under Dutch or Belgian law.
Subject to the foregoing and service of process in accordance with applicable treaties, investors may be able to enforce in the Netherlands or Belgium judgments in civil and commercial matters obtained from U.S. federal or state courts. However, no assurance can be given that those judgments will be enforceable. In addition, it is doubtful whether a Dutch or Belgian court would accept jurisdiction and impose civil liability in an original action commenced in the Netherlands or Belgium and predicated solely upon U.S. federal securities laws.
F. DIVIDENDS AND PAYING AGENTS
Not applicable.
G. STATEMENT BY EXPERTS
Not applicable.
191
H. DOCUMENTS ON DISPLAY
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers. Accordingly, we are required to file reports and other information with the SEC. Those reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an Annual Report containing financial statements that have been examined and reported on, with an opinion expressed by an independent registered public accounting firm.
We maintain a corporate website at www.argenx.com. We make available on our website, free of charge, our Annual Report and the text of our reports on Form 6-K, including any amendments to these reports, as well as certain other SEC filings, as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report. We have included our website address in this Annual Report solely as an inactive textual reference.
The SEC maintains a website (www.sec.gov) that contains reports and other information regarding registrants, such as argenx SE, that file electronically with the SEC.
With respect to references made in this Annual Report to any contract or other document of argenx SE, such references are not necessarily complete and you should refer to the exhibits attached or included elsewhere to this Annual Report for copies of the actual contract or document.
I. SUBSIDIARY INFORMATION
Not applicable.
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We manage our exposure to market risks centrally. We coordinate our access to national and international financial markets and consider and manage continuously the financial risks concerning our activities. These risks relate to the adequacy of our equity and debt capitalization, the creditworthiness of our counterparties, our short-term liquidity, the impact of changes in interest rates on our investments and fluctuations in foreign currency exchange rates. We do not believe that other risks are material, including interest rate risk on borrowings, which are inapplicable as the Company has no financial debt. We do not buy or trade financial instruments for speculative purposes. For additional information on risk factors applicable to the Company, its business, financial condition and results of operations, please see Item 3.D. “Risk Factors.” See “Note 26—Financial risk management” to our consolidated financial statements included elsewhere in this Annual Report.
Capital risk
The Company manages its capital to ensure that it will be able to continue as a going concern. The capital structure of the Company consists of equity attributed to the holders of equity instruments of the Company, such as capital, reserves and accumulated losses as mentioned in the consolidated statements of changes in equity. The Company makes the necessary adjustments in the light of changes in the economic circumstances, risks associated to the different assets and the projected cash needs of the current and projected research activities. On December 31, 2023, cash and cash equivalents amounted to $2,048.8 million, current financial assets amounted to $1,131.0 million and total capital amounted to $5,658.6 million. The current cash situation and the anticipated cash generation and usage are the most important parameters in assessing the capital structure. The Company’s objective is to maintain the capital structure at a level to be able to finance its activities for at least 12 months. Cash income from existing and new partnerships is taken into account and, if needed and possible, the Company can issue new shares or enter into financing agreements.
192
Credit risk
Credit risk refers to the risk that a counterparty will default on its contractual obligations resulting in financial loss to the Company. The Company has adopted a policy of only dealing with creditworthy counterparties and obtaining sufficient collateral, where appropriate, as a means of mitigating the risk of financial loss from defaults. Concentrations in credit risk are determined based on an analysis of counterparties and their importance on the overall outstanding contractual obligations at year-end.
The Company has a limited number of collaboration and license partners and therefore has a significant concentration of credit risk. However, it has policies in place to ensure that credit exposure is kept to a minimum and significant concentrations of credit exposure are only granted for short periods of time to high credit quality collaboration partners.
The Company applied the IFRS 9 simplified approach to measuring expected credit losses which uses a lifetime expected loss allowance for all receivables. To measure the expected credit losses, receivables have been grouped based on credit risk characteristics and the days past due. The provision for expected credit losses was not significant given that there have been no credit losses over the last three years and the high-quality nature of the Company’s customers.
Cash and cash equivalents and current financial assets are invested with several highly reputable banks and financial institutions. The Company holds its cash and cash equivalents mainly with different banks which are independently rated with a minimum rating of ‘A-’. The Company also holds cash equivalents in the form of money market funds with a recommended investment horizon of 6 months or shorter but with a low historical volatility. These money market funds are highly liquid investments, can be readily convertible into a known amount of cash. The company has adopted a policy whereby money market funds must have an average rating of “BBB” or higher.
Liquidity risk
The Company manages liquidity risk by maintaining adequate reserves, by continuously monitoring forecast and actual cash flows, and by matching the maturity profiles of financial assets and liabilities.
The Company’s main sources of cash inflows are obtained through sale of commercial product, capital increases and collaboration agreements. This cash is invested in savings accounts, term accounts and short-term investment funds in the form of money market funds. These money market funds represent the majority of the Company’s available sources of liquidity. Since all of these are immediately tradable and convertible in cash they have no impact on the liquidity risk.
Interest rate risk
The only variable interest-bearing financial instruments are cash and cash equivalents and current financial assets. Changes in interest rates may cause variations in interest income and expense resulting from short-term interest-bearing assets. Interest rate cuts may have a negative impact on the interest income of the Company.
For the year ended December 31, 2023, if applicable interest rates would increase/decrease by 25 basis points, this would have a positive/negative impact of $7.9 million (compared to $6.2 million for the year ended December 31, 2022 and $1.7 million for the year ended December 31, 2021).
Foreign exchange risk
The Company undertakes transactions denominated in foreign currencies, causing exposures to exchange rate fluctuations. The Company is mainly exposed to the Euro, Japanese yen, British pound and Swiss franc. To limit this risk, the Company attempts to align incoming and outgoing cash flows in currencies other than USD.
193
The net exposure to exchange differences of the monetary assets (being cash, cash equivalents and current financial assets) of the Company at the end of the reporting period are as follows:
At December 31, | ||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 |
EUR |
| 923,773 |
| 613,866 |
| 591,887 |
JPY | 8,232 | 5,613 | 6,316 | |||
GBP |
| 7 |
| 59,026 |
| 1,237 |
CHF | 193 | 3,832 | 727 | |||
CAD | 266 | 657 | — | |||
SEK | 1 | 7 | — | |||
DKK |
| 9 |
| 6 | — | |
On December 31, 2023, if the EUR would have strengthened/weakened versus the USD by 10%, this would have had a negative/positive impact of $92.3 million, compared to $61.4 million and $53.8 million on December 31, 2022 and December 31, 2021, respectively. On December 31, 2023, if other currencies would have strengthen/weakened against the USD by 10%, this would have had no significant impact
ITEM 12. DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
A. DEBT SECURITIES
Not applicable.
B. WARRANTS AND RIGHTS
Not applicable.
C. OTHER SECURITIES
Not applicable.
D. AMERICAN DEPOSITARY SHARES
In connection with our initial public offering on Nasdaq, the Bank of New York Mellon, as depositary, registered and delivered ADSs. Each ADS represents one share (or a right to receive one share) deposited with ING Bank N.V., as custodian for the depositary in the Netherlands. Each ADS also represents any other securities, cash or other property which may be held by the depositary. The deposited shares together with our other securities, cash and other property held by the depositary, are referred to as the deposited securities. The depositary’s office at which the ADSs are administered is located at 101 Barclay Street, New York, New York 10286. The Bank of New York Mellon’s principal executive office is located at 225 Liberty Street, New York, New York 10286.
A deposit agreement among us, the depositary, ADS holders and all other persons indirectly or beneficially holding ADSs sets out ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADSs.
194
Fees and Charges
Persons depositing or withdrawing shares or ADS holders must pay: |
| For: |
$5.00 (or less) per 100 ADSs (or portion of 100 ADSs) |
| Issuance of ADSs, including issuances resulting from a distribution of shares or rights or other property |
| Cancellation of ADSs for the purpose of withdrawal, including if the deposit agreement terminates | |
$.05 (or less) per ADS |
| Any cash distribution to ADS holders |
A fee equivalent to the fee that would be payable if securities distributed to you had been shares and the shares had been deposited for issuance of ADSs |
| Distribution of securities distributed to holders of deposited securities (including rights) that are distributed by the depositary to ADS holders |
$.05 (or less) per ADS per calendar year |
| Depositary services |
Registration or transfer fees |
| Transfer and registration of shares on our share register to or from the name of the depositary or its agent when you deposit or withdraw shares |
Expenses of the depositary |
| Cable, telex and facsimile transmissions (when expressly provided in the deposit agreement) |
| Converting foreign currency to USDs | |
Taxes and other governmental charges the depositary or the custodian has to pay on any ADSs or shares underlying ADSs, such as stock transfer taxes, stamp duty or withholding taxes |
| As necessary |
Any charges incurred by the depositary or its agents for servicing the deposited securities |
| As necessary |
The depositary collects its fees for delivery and surrender of ADSs directly from investors depositing shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by deduction from cash distributions or by directly billing investors or by charging the book-entry system accounts of participants acting for them. The depositary may collect any of its fees by deduction from any cash distribution payable (or by selling a portion of securities or other property distributable) to ADS holders that are obligated to pay those fees. The depositary may generally refuse to provide fee-attracting services until its fees for those services are paid.
From time to time, the depositary may make payments to us to reimburse us for costs and expenses generally arising out of establishment and maintenance of the ADS program, waive fees and expenses for services provided to us by the depositary or share revenue from the fees collected from ADS holders. In performing its duties under the deposit agreement, the depositary may use brokers, dealers, foreign currency dealers or other service providers that are owned by or affiliated with the depositary and that may earn or share fees, spreads or commissions.
The depositary may convert currency itself or through any of its affiliates and, in those cases, acts as principal for its own account and not as agent, advisor, broker or fiduciary on behalf of any other person and earns revenue, including, without limitation, transaction spreads, that it will retain for its own account. The revenue is based on, among other things, the difference between the exchange rate assigned to the currency conversion made under the deposit agreement and the rate that the depositary or its affiliate receives when buying or selling foreign currency for its own account. The depositary makes no representation that the exchange rate used or obtained in any currency conversion under the deposit agreement will be the most favorable rate that could be obtained at the time or that the method by which that rate will be determined will be the most favorable to ADS holders, subject to the depositary’s obligations under the deposit agreement. The methodology used to determine exchange rates used in currency conversions is available upon request.
195
PART II
ITEM 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
Not applicable.
ITEM 14. MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS
On July 18, 2023, we entered into an Underwriting Agreement with J.P. Morgan Securities LLC, Morgan Stanley & Co. LLC, Goldman Sachs & Co. LLC, BofA Securities, Inc., Cowen and Company, LLC, as representatives of the several underwriters named therein, relating to a global offering of an aggregate of 2,244,899 ordinary shares of the Company, nominal value €0.10 per share, including ordinary shares represented by ADSs, comprised of (i) 1,580,981 ADSs at a public offering price of $490.00 per ADS in the U.S. and countries outside the EEA and (ii) 663,918 ordinary shares at an offering price of €436.37 per ordinary share in a concurrent private placement in the EEA to certain legal entities all of which are qualified investors within the meaning of Regulation 2017/1129 of the European Parliament and of the Council of June 14, 2017, as amended. The offering was made pursuant to our effective shelf registration statement on Form F-3ASR (File No. 333-258251) filed on July 29, 2021, as supplemented by a preliminary prospectus supplement dated July 17, 2023, filed with the SEC on July 17, 2023, and a final prospectus supplement dated July 18, 2023, filed with the SEC on July 20, 2023. In connection with this offering, we granted the underwriters a 30-day option to purchase up to 336,734 additional ordinary shares (which may be represented by ADSs), which was exercised in full. The net proceeds to us from the sale of the ADSs and ordinary shares in this offering, after deducting the underwriting discounts and commissions and estimated offering expenses payable by the Company, was $1.2 billion (€1.1 billion). The offering closed on July 24, 2023.
None of the underwriting discounts and commissions or offering expenses were paid to directors, officers or general partners of ours or their associates or to persons owning 10% or more of any class of our equity securities, or to any of our affiliates.
We have not used any of the net proceeds from the offering to make payments, directly or indirectly, to any director, officer or general partner of ours or to their associates, persons owning 10% or more of any class of our equity securities, or to any of our affiliates. We have invested the net proceeds from the offering in cash and cash equivalents and current financial assets. There has been no material change in our planned use of the net proceeds from the offering as described in our final prospectus supplement filed pursuant to Rule 424(b)(5) under the Securities Act with the SEC on July 20, 2023 (File No.333-258251). The registration statement was effective on July 29, 2021.
ITEM 15. CONTROLS AND PROCEDURES
A. | Disclosure Controls and Procedures |
Our management, with the participation of our CEO and CFO, has evaluated the effectiveness of the design and operation of our disclosure controls and procedures pursuant to Exchange Act Rule 13a-15(b) as of December 31, 2023. While there are inherent limitations to the effectiveness of any system of disclosure controls and procedures, including the possibility of human error and the circumvention or overriding of the controls and procedures, our disclosure controls and procedures are designed to provide reasonable assurance of achieving their objectives.
Based upon their evaluation, as of December 31, 2023, our CEO and CFO have concluded that the disclosure controls and procedures, in accordance with Exchange Act Rule 13a-15(e), are (i) effective at the level of reasonable assurance in ensuring that information required to be disclosed in the reports that are filed or submitted under the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the Commission’s rules and forms, and (ii) are effective at the level of reasonable assurance in ensuring that information to be disclosed in the reports that are filed or submitted under the Exchange Act is accumulated and communicated to the management of our company, including our CEO and CFO, to allow timely decisions regarding required disclosure.
196
B. | Management’s Annual Report on Internal Control Over Financial Reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as such term is defined in Rule 13a-15(f) and Rule 15d-15(f) under the Exchange Act). Our internal control over financial reporting is a process designed, under the supervision of our CEO and CFO, to provide reasonable assurance regarding the reliability of financial reporting and preparation of financial statements for external reporting purposes in accordance with IFRS, as issued by the IASB.
Our internal control over financial reporting includes policies and procedures that pertain to the maintenance of records that, in reasonable detail, accurately and fairly, reflect transactions and dispositions of assets, provide reasonable assurance that transactions are recorded in the manner necessary to permit the preparation of financial statements in accordance with IFRS, as issued by the IASB, and that receipts and expenditures are only carried out in accordance with the authorization of our management and directors, and provide reasonable assurance regarding the prevention or timely detection of any unauthorized acquisition, use or disposition of our assets that could have a material effect on our consolidated financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect all misstatements. Moreover, projections of any evaluation of the effectiveness of internal control to future periods are subject to a risk that controls may become inadequate because of changes in conditions and that the degree of compliance with the policies or procedures may deteriorate.
Our management has assessed the effectiveness of internal control over financial reporting based on the Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in 2013. Based on this assessment, our management has concluded that our internal control over financial reporting as of December 31, 2023 was effective.
| C. | Attestation Report of the Registered Public Accounting Firm |
The effectiveness of our internal control over financial reporting as of December 31, 2023 has been audited by Deloitte Accountants B.V., our independent registered public accounting firm. Their audit report, including their opinion on management’s assessment of internal control over financial reporting, is included in our audited consolidated financial statements included in this Annual Report.
| D. | Changes in Internal Control Over Financial Reporting |
During the period covered by this Annual Report, we have not made any change to our internal control over financial reporting that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
ITEM 16. [RESERVED]
ITEM 16A. AUDIT COMMITTEE FINANCIAL EXPERT
Our Board of Directors previously established that Mr. Verhaeghe, Mr. Rosenberg, Mr. Daly, Mr. Krognes and Mr. Lanthaler satisfy the independence requirements set forth in Rule 10A-3 of the Exchange Act and that both Mr. Lanthaler (up until his resignation effective February 27, 2023) and Mr. Krognes qualify as “audit committee financial experts” as defined by SEC rules and has the requisite financial sophistication under the Nasdaq Listing Rules.
ITEM 16B. CODE OF ETHICS
We adopted a Code of Conduct, that is applicable to all of our employees and directors. The Code of Conduct is available on our website at www.argenx.com/investors/governance/rules-codes-compliance. The audit and compliance committee of our Board of Directors is responsible for overseeing the Code of Conduct and is required to approve any
197
waivers of the Code of Conduct for employees and directors. We expect that any amendments to the Code of Conduct, and any waivers of its requirements, will be disclosed on our website.
ITEM 16C. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Deloitte Accountants B.V. has served as our independent registered public accounting firm for 2023 and 2022. Our accountants billed the following fees to us for professional services in each of those fiscal years:
Year Ended December 31, | ||||||
Fees |
| 2023 |
| 2022 | ||
in thousands of $ | ||||||
Audit fees (1) |
| $ | 1,979 |
| $ | 1,394 |
Audit-related fees |
| 330 |
| 380 | ||
Tax fees |
| — |
| — | ||
All other fees | — |
| — | |||
Total |
| $ | 2,309 |
| $ | 1,774 |
| (1) | Audit services performed by Deloitte Accountants B.V. as the external auditor referred to in Section 1 of the Dutch Accounting Firms Oversight Act (Wta) as well as by the Deloitte network |
“Audit fees” are the aggregate fees billed for the audit of our annual financial statements. This category also includes services that generally the independent accountants provide, such as consents and assistance with and review of documents filed with the SEC.
“Audit-related fees” are the aggregate fees billed for assurance and related services that are reasonably related to the performance of the audit and are not reported under Audit Fees. In 2023 and 2022, “Audit-Related Fees” also include fees billed for assurance and audit-related services regarding our public offerings on Nasdaq.
“Tax fees” are the aggregate fees billed for professional services rendered by the principal accountant for permissible tax related services.
“All other Fees” are any additional amounts billed for products and services provided by the principal accountant. No other fees were billed by Deloitte Accountants B.V. for the fiscal years ended December 31, 2023 and 2022.
Audit and Compliance Committee’s Pre-Approval Policies and Procedures
Our audit and compliance committee has responsibility for, among other things, appointing, setting compensation of and overseeing the work of our independent registered public accounting firm, or external auditor. In recognition of these responsibilities, the audit and compliance committee has adopted a policy governing the pre-approval of all audit and permitted non-audit services performed by our external auditor to ensure that the provision of such services does not impair the external auditor’s independence from us and our management. Unless a type of service to be provided by our external auditor has received general pre-approval from the audit and compliance committee, it requires specific pre-approval by the audit and compliance committee. The payment for any proposed services in excess of pre-approved cost levels requires specific pre-approval by the audit and compliance committee.
Pursuant to its pre-approval policy, the audit and compliance committee may delegate its authority to pre-approve services to the chairperson of the audit and compliance committee. The decisions of the chairperson to grant pre-approvals must be presented to the full audit committee at its next scheduled meeting. The audit and compliance committee may not delegate its responsibilities to pre-approve services to management.
198
The audit and compliance committee has considered the non-audit services provided by Deloitte Accountants B.V. as described above and believes that they are compatible with maintaining Deloitte Accountants B.V.’s independence as our external auditor. In accordance with Regulation S-X, Rule 2-01, paragraph (c)(7)(i), no fees for services were approved pursuant to any waivers of the pre-approval requirement.
ITEM 16D. EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES
Not applicable.
ITEM 16E. PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS
Not applicable.
ITEM 16F. CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT
Not applicable.
ITEM 16G. CORPORATE GOVERNANCE
As a foreign private issuer, the Nasdaq Listing Rules include certain accommodations in the corporate governance requirements that allow foreign private issuers to follow “home country” corporate governance practices in lieu of the otherwise applicable Nasdaq corporate governance standards. We intend to rely on certain exemptions for foreign private issuers and to follow Dutch corporate governance practices in lieu of the Nasdaq corporate governance rules.
The following is a summary of the significant ways in which our corporate governance practices differ from those required by the Nasdaq Listing Rules with which we are not required to comply:
| ● | Quorum at Shareholder Meetings. In accordance with Dutch law and generally accepted business practices in the Netherlands, our Articles of Association do not provide quorum requirements generally applicable to general meetings of shareholders. To that extent, our practice varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock. |
| ● | Solicitation of Proxies. Although we must provide shareholders with an agenda and other relevant documents ahead of any General Meeting, Dutch law does not have a regulatory regime for the solicitation of proxies, and the solicitation of proxies is not a generally accepted business practice in the Netherlands. Thus, our practice varies from the requirement of Nasdaq Listing Rule 5620(b). |
| ● | Shareholder Approval. We follow certain Dutch shareholder approval requirements for the issuance of securities in connection with certain events such as the acquisition of stock or assets of another company, the establishment of or amendments to equity-based compensation plans for employees, a change of control of us and certain private placements. To this extent, our practice varies from the requirements of Nasdaq Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events. |
199
| ● | Distribution of Annual Reports. We do not follow Nasdaq Listing Rule 5250(d), which requires companies to make available copies of their annual reports containing audited financial statements to their shareholders. The distribution of our annual reports to shareholders is not required under Dutch corporate law or Dutch securities laws. Furthermore, it is generally accepted business practice for Dutch companies not to distribute annual reports. In part, this is because the Dutch system of bearer shares has made it impractical to keep a current list of holders of the bearer shares in order to distribute the annual reports. Instead, we make our Annual Report available at our corporate head office in the Netherlands (and at the offices of our Dutch listing agent as stated in the convening notice for the meeting) no later than 42 days prior to convocation of any annual General Meeting. In addition, we post a copy of our annual reports on our website prior to our annual General Meeting. |
ITEM 16H. MINE SAFETY DISCLOSURE
Not applicable.
ITEM 16I. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
ITEM 16J. INSIDER TRADING POLICIES
Not required.
ITEM 16K. CYBERSECURITY
Information Security Risk Management and Strategy
Our approach to risk management is designed to identify, assess, prioritize and manage major risk exposures that could affect our ability to execute our corporate strategy and fulfill our business objectives. As part of our information security and privacy program, the Information Security and Management System (ISMS), we perform risk assessments in which we map and prioritize information security risks identified through the processes described below, including risks associated with our use of third-party service providers. These assessments inform our ISMS strategies and oversight processes and are included with other enterprise risks as part of our broader enterprise risk management. We view information security risks as one of the key risks categories we face. IT system vendors are subject to security review and audits. For more information regarding the cybersecurity-related risks we face, please refer to Item 3.D. “Risk Factors—Risk Factors Related to argenx’s Business and Industry—Our business and operations could suffer in the event of system failures or unauthorized or inappropriate use of or access to our systems.”
Our processes for assessing, identifying and managing information security risks and vulnerabilities are embedded across our business as part of our ISMS. Among other things, we conduct audits and tests of our information systems (including review and assessment by independent third-party advisors, who assess and report on the maturity of our security measures and help identify areas for continued focus and improvement) and review information security threat information published by government entities and other organizations in which we participate. We conduct training on data security matters for our employees to be aware and vigilant against potential data security risks and data privacy is incorporated into our overall compliance training, such as through privacy-specific training for employees and contractors. Phishing training is also implemented regularly, which includes mock phishing emails to test employee vigilance. In addition, employees are required to read and acknowledge information security policies that are relevant to their specific role. We also have implemented and maintain information security incident response plans, which include processes to triage, assess severity for, escalate, contain, investigate and remediate information security incidents, as well as to comply with potentially applicable legal obligations and mitigate brand and reputational damage.
200
Information Security Governance and Oversight
Our ISMS enables our Board of Directors to establish a mutual understanding with our senior management team of the effectiveness of our information security risk management practices and capabilities, including the division of responsibilities for reviewing our information security risk exposure and risk tolerance, tracking emerging information risks and ensuring proper escalation of certain key risks for periodic review by the Board of Directors and its committees. As part of its broader risk oversight activities, the Board of Directors oversees risks from information security threats, both directly and through the audit and compliance committee of the Board of Directors. The audit and compliance committee also oversees our internal control over financial reporting.
As an element of its cybersecurity oversight activities, the audit and compliance committee regularly reviews the results of our enterprise risk assessments, including information security risk assessments, as well as management's strategies to detect, monitor and manage such risks and related risk assessment and risk management policies. Our ISMS contains provisions regarding reporting to the Global Risk Management Committee. Additionally, the data protection officer (the DPO) provides regular updates to senior management, and the audit and compliance committee as a component of the audit and compliance committee’s compliance updates. The DPO also regularly reports to the Global Corporate Compliance Committee, the Global Risk Management Committee and the General Counsel on matters such as the status of the organizational privacy plan, data breaches and routine programs. In addition to these regularly scheduled updates from the DPO, the Global Head of Business Information Systems reports to the audit and compliance committee or the full Board of Directors, as appropriate, on how certain information security risks are being managed and progress towards agreed mitigation goals, as well as any potential material risks from cybersecurity threats that have been detected by the information security team.
Our information security team is responsible for day-to-day identification, assessment and management of the information security risks we face. Our Global Head of Business Information Systems has 32 years of experience in information management systems and the managers reporting to the Global Head of Business Information Systems have over 40 cumulative years of experience in information security. Our incident response and data breach procedures are designed for the timely detection, reporting, and investigation of all security incidents, as well as the timely notification of any reportable breaches (including any material cybersecurity incidents and personal data breaches) to the competent authorities and the timely communication to the affected individuals, where relevant. We maintain records of breaches on our quarterly corporate risk dashboard and our personal data breach register, and we monitor and regularly report our security and data breach metrics to senior management, including the audit and compliance committee of our Board of Directors, the Global Corporate Compliance Committee, and the Global Risk Management Committee. In addition to the ordinary-course Board of Directors and audit and compliance committee reporting and oversight described above, we also maintain disclosure controls and procedures designed for prompt reporting to the Board of Directors and timely public disclosure, as appropriate, of material events covered by our risk management framework, including information security risks.
201
PART III
ITEM 17. FINANCIAL STATEMENTS
Not applicable.
ITEM 18. FINANCIAL STATEMENTS
See pages F-1 through F-48 of this Annual Report.
ITEM 19. EXHIBITS
The exhibits listed in the Exhibit Index at the end of this Annual Report are filed as exhibits to this Annual Report.
|
| Incorporated by Reference | ||||||||
Exhibit |
| Description |
| Schedule/ |
| File Number |
| Exhibit |
| File Date |
1.1 |
| Form 20-F | 001-38097 |
| 1.1 | 03/16/2023 | ||||
1.2 | Form 20-F | 001-38097 | 1.2 | 03/16/2023 | ||||||
2.1 | Form F-1/A | 333-217417 | 4.1 | 05/16/2017 | ||||||
2.2 | Form of American Depositary Receipt (included in Exhibit 2.1) | |||||||||
2.3# | ||||||||||
4.1 | Leases dated April 1, 2016 between argenx BVBA and Bio-Incubator Gent 2 NV | Form F-1 | 333-217417 | 10.1 | 04/21/2017 | |||||
4.2** | Form F-1 | 333-217417 | 10.2 | 04/21/2017 | ||||||
4.3† | Form F-1 | 333-217417 | 10.3 | 04/21/2017 | ||||||
4.4#† | ||||||||||
4.5** | Form 20-F | 001-38097 | 4.5 | 03/26/2019 | ||||||
4.6 | Form 20-F | 001-38097 | 4.6 | 03/26/2019 | ||||||
202
4.7** | Form 20-F | 001-38097 | 4.7 | 03/30/2021 | ||||||
4.8 | Asset Purchase Agreement, dated November 29, 2022, by and between bluebird bio, Inc. and argenx BV | Form 6-K | 001-38097 | 1.1 | 11/30/2022 | |||||
4.9† | Form 20-F | 001-38097 | 4.9 | 03/16/2023 | ||||||
8.1 | Form 20-F | 001-38097 | 8.1 | 03/16/2023 | ||||||
12.1# | ||||||||||
12.2# | ||||||||||
13.1* | ||||||||||
13.2* | ||||||||||
15.1# | ||||||||||
97.1# | ||||||||||
101.INS# | Inline XBRL Instance Document | |||||||||
101.SCH# | Inline XBRL Taxonomy Extension Schema Document | |||||||||
101.CAL# | Inline XBRL Taxonomy Extension Calculation Linkbase Document | |||||||||
101.DEF# | Inline XBRL Taxonomy Extension Definition Linkbase Document | |||||||||
203
101.LAB# | Inline XBRL Taxonomy Extension Label Linkbase Document | |||||||||
101.PRE# | Inline XBRL Taxonomy Extension Presentation Linkbase Document | |||||||||
104 | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101) |
# | Filed herewith. |
* | Furnished herewith. |
† | Indicates a management contract or any compensatory plan, contract or arrangement. |
** | Confidential treatment status has been granted as to certain portions thereto, which portions are omitted and filed separately with the U.S. Securities and Exchange Commission. |
204
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this Annual Report on its behalf.
Date: March 21, 2024
ARGENX SE | |||
By: | /s/ Tim Van Hauwermeiren | ||
Name: | Tim Van Hauwermeiren | ||
Title: | Chief Executive Officer | ||
205
INDEX TO FINANCIAL STATEMENTS
Audited consolidated financial statements as of and for the years ended December 31, 2023, 2022 and 2021 | |
Reports of Independent Registered Public Accounting Firm (PCAOB ID No. | F-2 |
F-4 | |
F-6 | |
F-7 | |
F-8 | |
F-9 | |
F-10 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the Board of Directors of argenx SE
Opinion on the Financial Statements
We have audited the accompanying consolidated statements of financial position of argenx SE and subsidiaries (the "Company") as of December 31, 2023, 2022 and 2021, the related consolidated statements of profit or loss, comprehensive income (loss), cash flows, and changes in equity, for each of the three years in the period ended December 31, 2023, and the related notes (collectively referred to as the "financial statements"). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023, 2022 and 2021, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2023, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board (“IFRS”).
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 21, 2024, expressed an unqualified opinion on the Company's internal control over financial reporting.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current-period audit of the financial statements that was communicated or required to be communicated to the audit committee and that (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Gross-to-net adjustments in revenue – Refer to Note 14 & 15 to financial statements
Critical Audit Matter Description
The Company recognizes product net sales, relating to the sale of the products VYVGART and VYVGART HYTRULO. These product net sales are accounted for in accordance with IFRS 15 Revenue from Contracts with Customers (“IFRS 15”), whereby the sale of these products to customers is recognized for an amount that reflects the consideration to which the Company expects to be entitled in exchange for these goods. The majority of the product gross sales are in the United States of America, which are subject to reduction for significant components of variable
F-2
consideration primarily composed of rebates to government agencies, distributors, health insurance companies and managed healthcare organizations. Together, these deductions are referred to as gross-to-net (“GtN”) adjustments, as specified in Note 14 and 15 to the financial statements. The GtN adjustments that are recognized by the Company represent estimates of the related obligations that will be settled in a future period. The estimated amounts are based on contractual arrangements with healthcare authorities, government and state programs, and gross sales and third-party data.
We identified the GtN adjustments for product net sales in the United States of America as a critical audit matter, because of the significant effort spent on auditing the adjustments and the judgment required to obtain sufficient appropriate audit evidence that supports the Company’s estimate due to the reporting data being subject to a time lag.
How the Critical Audit Matter Was Addressed in the Audit
Our audit procedures related to the gross-to-net adjustments included the following, among others:
| ● | We evaluated the key revenue contracts and supply chain contracts, including evaluation of the accounting treatment of the GtN adjustments and the disclosures thereof in accordance with IFRS 15. |
| ● | We evaluated the independent service auditor reports for the service providers used by the Company to process gross-to-net adjustments on behalf of the Company. |
| ● | We evaluated the appropriateness and consistency of the Company’s methodology and assumptions in developing the GtN adjustments, including testing the completeness and accuracy of the underlying data used by management in their estimates. |
| ● | We performed detailed testing procedures on a selection of adjustments by reconciling them to underlying evidence. |
| ● | We performed recalculation procedures on the different components of management’s calculations. |
| ● | We evaluated the Company’s ability to estimate the GtN adjustments by evaluating the historical accuracy of estimates made in the prior year in relation to the actuals incurred in this year. |
/s/ Deloitte Accountants B.V.
Rotterdam, The Netherlands
March 21, 2024
We have served as the Company's auditor since 2015.
F-3
| Deloitte Accountants B.V. Wilhelminakade 1 3072 AP Rotterdam P.O.Box 2031 3000 CA Rotterdam Netherlands Tel: +31 (0)88 288 2888 Fax: +31 (0)88 288 9929 www.deloitte.nl |

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the Board of Directors of argenx SE
Opinion on Internal Control over Financial Reporting
We have audited the internal control over financial reporting of argenx SE and subsidiaries (the “Company”) as of December 31, 2023, based on criteria established in Internal Control — Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control — Integrated Framework (2013) issued by COSO.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated financial statements as of and for the year ended December 31, 2023, of the Company and our report dated March 21, 2024, expressed an unqualified opinion on those financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
4
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/
March 21, 2024
F-5
ARGENX SE
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION
As of | |||||||||||
December 31, | |||||||||||
(in thousands of $) |
| Note | 2023 |
| 2022 |
| 2021 | ||||
ASSETS |
|
|
|
|
|
|
|
|
|
| |
Non‑current assets |
|
|
|
|
|
| |||||
Property, plant and equipment |
| 4 | $ | |
| $ | |
| $ | | |
Intangible assets |
| 5 | |
| |
| | ||||
Deferred tax asset | 24 | | | | |||||||
Research and development incentive receivables | | | | ||||||||
Investment in joint venture |
| 27.1 | |
| |
| — | ||||
Prepaid expenses | 7 | | — | — | |||||||
Other non-current assets | 6 | | | | |||||||
Total non‑current assets |
|
| |
| |
| | ||||
Current assets |
|
|
|
|
|
|
| ||||
Inventories | 7 | $ | | $ | | $ | | ||||
Prepaid expenses |
| 8 | |
| |
| | ||||
Trade and other receivables | 9 | | | | |||||||
Research and development incentive receivables |
| |
| |
| — | |||||
Financial assets |
| 10 | |
| |
| | ||||
Cash and cash equivalents |
| 11 | |
| |
| | ||||
Total current assets |
| |
| |
| | |||||
TOTAL ASSETS |
|
| $ | |
| $ | |
| $ | | |
The accompanying notes form an integral part of these consolidated financial statements.
F-4
As of | ||||||||||||
December 31, | ||||||||||||
(in thousands of $) |
| Note |
| 2023 |
| 2022 |
| 2021 | ||||
EQUITY AND LIABILITIES |
|
|
|
|
|
|
|
|
|
|
| |
Equity |
| 12 |
|
|
|
|
|
| ||||
Equity attributable to owners of the parent |
|
|
|
|
|
|
| |||||
Share capital |
|
| $ | |
| $ | |
| $ | | ||
Share premium |
|
| |
| |
| | |||||
Translation differences | | | | |||||||||
Accumulated losses |
|
| ( |
| ( |
| ( | |||||
Other reserves |
|
| |
| |
| | |||||
Total equity |
|
| $ | |
| $ | |
| $ | | ||
Non-current liabilities |
|
|
|
| ||||||||
Provisions for employee benefits |
|
| |
| |
| | |||||
Lease liabilities | 22 | | | | ||||||||
Deferred tax liabilities | 24 | | | | ||||||||
Total non-current liabilities |
| | | | ||||||||
|
|
| ||||||||||
Current liabilities |
|
|
|
|
| |||||||
Lease liabilities | 22 | | | | ||||||||
Trade and other payables |
| 14 |
| |
| |
| | ||||
Tax liabilities | | | | |||||||||
Total current liabilities |
| | | | ||||||||
| ||||||||||||
Total liabilities |
|
|
| $ | |
| $ | |
| $ | | |
TOTAL EQUITY AND LIABILITIES |
|
|
| $ | |
| $ | |
| $ | | |
The accompanying notes form an integral part of these consolidated financial statements.
F-5
ARGENX SE
CONSOLIDATED STATEMENTS OF PROFIT OR LOSS
Year Ended | |||||||||||
December 31, | |||||||||||
(in thousands of $ except for shares and EPS) |
| Note |
| 2023 |
| 2022 |
| 2021 | |||
Product net sales | 15,18 | $ | | $ | | $ | — | ||||
Collaboration revenue | 16 | | | | |||||||
Other operating income |
| 17 |
| |
| |
| | |||
Total operating income |
|
|
| |
| |
| | |||
Cost of sales | ( | ( | — | ||||||||
Research and development expenses |
| 19 |
| ( |
| ( |
| ( | |||
Selling, general and administrative expenses |
| 20 |
| ( |
| ( |
| ( | |||
Loss from investment in joint venture | ( | ( | — | ||||||||
Total operating expenses | ( | ( | ( | ||||||||
Operating loss |
|
| $ | ( |
| $ | ( |
| $ | ( | |
Financial income |
| 23 |
| |
| |
| | |||
Financial expense | 23 | ( | ( | ( | |||||||
Exchange gains/(losses) |
| 23 |
| |
| ( |
| ( | |||
|
|
| |||||||||
Loss for the year before taxes |
|
|
| $ | ( |
| $ | ( |
| $ | ( |
Income tax benefit / (expense) |
| 24 |
| $ | |
| $ | |
| $ | ( |
Loss for the year |
|
|
| $ | ( |
| $ | ( |
| $ | ( |
Loss for the year attributable to: | |||||||||||
Owners of the parent | ( | $ | ( | $ | ( | ||||||
Weighted average number of shares outstanding |
|
|
| |
| |
| | |||
Basic and diluted (loss) per share (in $) |
| 25 |
| ( |
| ( |
| ( | |||
The accompanying notes form an integral part of these consolidated financial statements.
F-6
ARGENX SE
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
Year Ended | |||||||||||
December 31, | |||||||||||
(in thousands of $) |
| Note |
| 2023 |
| 2022 |
| 2021 | |||
Loss for the year |
|
| $ | ( |
| $ | ( |
| $ | ( | |
Items that may be reclassified subsequently to profit or loss, net of tax | |||||||||||
Currency translation differences, arisen from translating foreign activities | | ( | ( | ||||||||
Items that will not be reclassified subsequently to profit or loss, net of tax | |||||||||||
Fair value gain/(loss) on investments in equity instruments designated as at FVTOCI | 6 | ( | ( | ( | |||||||
Other comprehensive income/(loss), net of income | | ( | ( | ||||||||
Total comprehensive loss attributable to: | |||||||||||
Owners of the parent | $ | ( | $ | ( | $ | ( | |||||
The accompanying notes form an integral part of these consolidated financial statements.
F-7
ARGENX SE
CONSOLIDATED STATEMENTS OF CASH FLOWS
Year Ended | |||||||||||
December 31, | |||||||||||
(in thousands of $) |
| Note |
| 2023 |
| 2022 |
| 2021 | |||
Operating loss |
|
|
| $ | ( |
| $ | ( |
| $ | ( |
Adjustments for non-cash items |
|
|
|
|
|
|
|
| |||
Amortization of intangible assets |
| 5 |
| |
| |
| | |||
Depreciation of property, plant and equipment |
| 4 |
| |
| |
| | |||
Provisions for employee benefits |
|
| |
| |
| | ||||
Expense recognized in respect of share-based payments |
| 13 |
| |
| |
| | |||
Fair value gains on financial assets at fair value through profit or loss | 6 | — | ( | ( | |||||||
Non-cash revenue | — | — | ( | ||||||||
Loss from investment in joint venture | 27.1 | | | — | |||||||
Other non-cash expenses | | — | — | ||||||||
|
|
| $ | ( |
| $ | ( |
| $ | ( | |
Movements in current assets/liabilities |
|
|
|
|
|
|
|
| |||
(Increase)/decrease in trade and other receivables |
| 9 |
| ( |
| ( |
| ( | |||
(Increase)/decrease in inventories | 7 | ( | ( | ( | |||||||
(Increase)/decrease in other current assets |
|
|
| ( |
| ( |
| ( | |||
Increase/(decrease) in trade and other payables |
| 14 |
| |
| |
| | |||
Increase/(decrease) in deferred revenue — current |
|
| — |
| — |
| ( | ||||
Movements in non-current assets/liabilities | |||||||||||
(Increase)/decrease in other non‑current assets |
| 6 |
| ( |
| ( |
| ( | |||
(Increase)/decrease in non-current prepaid expense | 7 | ( | — | — | |||||||
Increase/(decrease) in deferred revenue — non-current | — | — | ( | ||||||||
Net cash flows used in operating activities, before interest and taxes | ( | ( | ( | ||||||||
Interest paid | ( | ( | ( | ||||||||
Income taxes paid | ( | ( | ( | ||||||||
Net cash flows used in operating activities |
|
|
| $ | ( |
| $ | ( |
| $ | ( |
Purchase of intangible assets |
| 5 |
| ( |
| ( |
| ( | |||
Purchase of property, plant and equipment |
| 4 |
| ( |
| ( |
| ( | |||
(Increase)/decrease in current financial assets |
| 10 |
| — |
| — |
| ( | |||
Purchase of current financial investments | 10 | ( | ( | — | |||||||
Sale of current financial investments | 10 | | | — | |||||||
Interest received | | | | ||||||||
Investment in joint venture |
|
| ( |
| ( |
| — | ||||
Net cash flows from / (used in) investing activities |
|
|
| $ | |
| $ | ( |
| $ | ( |
Principal elements of lease payments |
| 22 |
| ( |
| ( |
| ( | |||
Proceeds from issue of new shares, gross amount |
| 12 |
| |
| |
| | |||
Issue costs paid | 12 | ( | ( | ( | |||||||
Exchange (losses)/gains from currency conversion on proceeds from issue of new shares | ( | | | ||||||||
Payment of employee withholding taxes relating to restricted stock unit awards | ( | ( | — | ||||||||
Proceeds from exercise of stock options | 12 | | | | |||||||
Net cash flows from financing activities |
|
|
| $ | |
| $ | |
| $ | |
Increase/(decrease) in cash and cash equivalents |
|
|
| $ | |
| $ | ( |
| $ | |
Cash and cash equivalents at the beginning of the period |
|
|
| $ | |
| $ | |
| $ | |
Exchange gains/(losses) on cash and cash equivalents |
|
| $ | |
| $ | ( |
| $ | ( | |
Cash and cash equivalents at the end of the period |
|
|
| $ | |
| $ | |
| $ | |
The accompanying notes form an integral part of these consolidated financial statements.
F-8
ARGENX SE
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
Attributable to owners of the parent | |||||||||||||||||||||||||
|
|
|
|
| Share-based |
| Total |
|
| ||||||||||||||||
payment and | equity | ||||||||||||||||||||||||
|
|
| income tax | Fair value movement on | attributable |
| |||||||||||||||||||
deduction on | investment in equity | to owners | |||||||||||||||||||||||
Share | Share | Accumulated | Translation | share-based | instruments designated | of the | Total | ||||||||||||||||||
(in thousands of $) | capital | premium | losses |
| differences | payments | as at FVTOCI |
| parent | equity | |||||||||||||||
Balance at January 1, 2021 |
| $ | |
| $ | |
| $ | ( |
| $ | |
| $ | | $ | — |
| $ | |
|
| $ | | |
Loss for the year | ( | ( | ( | ||||||||||||||||||||||
Other comprehensive income / (loss) | ( | ( | ( | ( | |||||||||||||||||||||
Total comprehensive income / (loss) for the year |
| ( |
| ( | ( | ( |
|
| ( | ||||||||||||||||
Income tax benefit from excess tax deductions related to share-based payments | | |
|
| | ||||||||||||||||||||
Share-based payment |
| | |
|
| | |||||||||||||||||||
Issue of share capital |
| | | |
|
| | ||||||||||||||||||
Transaction costs for equity issue |
| ( | ( |
|
| ( | |||||||||||||||||||
Exercise of stock options |
| | | |
|
| | ||||||||||||||||||
Balance year ended December 31, 2021 |
| $ | | |
| $ | ( |
| $ | |
| $ | | $ | ( |
| $ | |
|
| $ | | |||
| $ | ||||||||||||||||||||||||
Loss for the year | ( | ( | ( | ||||||||||||||||||||||
Other comprehensive income / (loss) | ( | ( | ( | ( | |||||||||||||||||||||
Total comprehensive income / (loss) for the year |
| ( | ( | ( | ( | ( | |||||||||||||||||||
Income tax benefit from excess tax deductions related to share-based payments |
| | | | |||||||||||||||||||||
Share-based payment |
| | | | |||||||||||||||||||||
Issue of share capital |
| | | | | ||||||||||||||||||||
Transaction costs for equity issue | ( | ( | ( | ||||||||||||||||||||||
Exercise of stock options | | | | | |||||||||||||||||||||
Ordinary shares withheld for payment of employees’ withholding tax liability |
| ( | ( | ( | |||||||||||||||||||||
Balance year ended December 31, 2022 |
| $ | | | ( | | | ( | | | |||||||||||||||
Loss for the year | ( | ( | ( | ||||||||||||||||||||||
Other comprehensive income / (loss) | | ( | | | |||||||||||||||||||||
Total comprehensive income / (loss) for the year | ( |
| | ( | ( |
|
| ( | |||||||||||||||||
Income tax benefit from excess tax deductions related to share-based payments | | | | ||||||||||||||||||||||
Share-based payment |
| | |
|
| | |||||||||||||||||||
Issue of share capital |
| | | |
|
| | ||||||||||||||||||
Transaction costs for equity issue |
| ( | ( |
|
| ( | |||||||||||||||||||
Exercise of stock options |
| | | |
|
| | ||||||||||||||||||
Ordinary shares withheld for payment of employees’ withholding tax liability | ( | ( | ( | ||||||||||||||||||||||
Balance year ended December 31, 2023 |
| $ | |
| $ | |
| $ | ( |
| $ | | $ | | $ | ( |
| $ | |
|
| $ | | ||
Please refer to note 12 for more information on the share capital and movement in number of shares. See also note 13 for more information on the share-based payments.
The accompanying notes form an integral part of these consolidated financial statements.
F-9
ARGENX SE
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
| 1. | General information about the company |
argenx SE is a Dutch European public company with limited liability incorporated under the laws of the Netherlands. The company (COC 24435214) has its official seat in Rotterdam, the Netherlands, and its registered office is at Laarderhoogtweg 25, 1101 EB Amsterdam, the Netherlands. An overview of the company and its subsidiaries (the Company) are described in note 31.
argenx SE is a publicly traded company with ordinary shares listed on Euronext Brussels under the symbol “ARGX” since July 2014 and with American Depositary Shares listed on Nasdaq under the symbol “ARGX” since May 2017.
2. | Material accounting policy information |
The Company’s material accounting policies are summarized below.
2.1 | Statement of compliance and basis of preparation |
The consolidated financial statements are prepared in accordance with the International Financial Reporting Standards (IFRS), issued by the International Accounting Standards Board (IASB) and the interpretations issued by the IASB’s International Financial Reporting Interpretation Committee. The consolidated financial statements provide a general overview of the Company’s activities and the results achieved. They present fairly the entity’s financial position, its financial performance and cash flows, on a going concern basis.
The material accounting policy information applied in the preparation of the above consolidated financial statements are set out below. All amounts are presented in thousands of dollar, unless otherwise indicated, rounded to the nearest $ ‘000.
The consolidated financial statements have been approved for issue by the Company’s Board of Directors (the “Board”) on March 19, 2024.
2.2 | Adoption of new and revised standards |
New standards and interpretations applicable for the annual period beginning on January 1, 2023
| ● | Amendments to IAS 1 – Presentation of Financial Statements and IFRS Practice Statement 2 – Making Materiality Judgements. |
The amendments to IAS 1 require companies to disclose their material accounting policy information rather than their significant accounting policies. The amendments to IFRS Practice Statement 2 provide guidance on how to apply the concept of materiality to accounting policy disclosures. As result the Company revised its accounting policy disclosure in the consolidated financial statements and removed accounting policy information that the Company deemed to relate to immaterial transactions or other events or conditions.
No other standards and interpretations for the annual period beginning on January 1, 2023 have any material impact on the consolidated financial statements.
New standards and interpretations issued, but not yet applicable for the annual period beginning on January 1, 2023
| ● | Amendments to IAS 12 - issued International Tax Reform—Pillar Two Model Rules. |
F-10
On 23 May 2023, the International Accounting Standards Board (the IASB or Board) issued International Tax Reform – Pillar Two Model Rules – Amendments to IAS 12 which clarified the application of IAS 12 income taxes arising from tax law enacted or substantively enacted to implement the OECD/G20 Inclusive Framework on Base Erosion and Profit Shifting Pillar Two model rules. Based on current information, management expects that the Company could become subject to the Pillar Two Directive and implementing domestic laws as early as 2025. Thus, there is no impact for argenx in 2023. The company is currently in the process of determining the impact, if any, for 2025. Based on the preliminary analysis, we do not expect the Pillar Two Rules to have a material impact on our effective tax rate.
It is unclear if the Pillar Two model rules create additional temporary differences, whether to remeasure deferred taxes for the Pillar Two model rules, and which tax rate to use to measure deferred taxes. In response to this unclarity, the amendments mentioned above introduced a mandatory temporary exception to the requirements of IAS 12 under which a company does not recognise or disclose information about deferred tax assets and liabilities related to the Pillar Two model rules. We applied the temporary exception in financial year 2023.
We have not early adopted any other standard, interpretation, or amendment that has been issued but is not yet effective. Of the standards that are not yet effective, we expect no standard to have a material impact on the financial statements in the period of initial application.
2.3 | Basis of consolidation |
The consolidated financial statements include the financial statements of the Company and entities controlled by the Company (its subsidiaries). Control is achieved when the Company:
| ● | has power over the investee; |
| ● | is exposed, or has rights, to variable returns from its involvement with the investee; and |
| ● | has the ability to use its power to affect its returns. |
The Company reassesses whether or not it controls an investee if facts and circumstances indicate that there are changes to one or more of the three elements of control listed above.
The results of the subsidiaries are included in the consolidated statements of profit or loss and consolidated statements of other comprehensive income (loss) from the effective date of acquisition up to the date when control ceases to exist. When necessary, adjustments are made to the financial statements of subsidiaries to bring their accounting policies into line with those used by other members of the Group.
All intercompany transactions and unrealized gains on transactions between group companies are eliminated. Unrealised losses are also eliminated unless the transaction provides evidence of an impairment of the transferred asset.
2.4 | Foreign currency transactions |
2.4.1Functional and presentation currency
Items included in the consolidated financial statements of each of the entities are valued using the currency of their economic environment in which the entity operates. The consolidated financial statements are presented in USD ($), which is the Company’s presentation currency.
2.4.2Transactions and balances
Transactions in foreign currencies are translated at the exchange rate ruling at the date of the transaction. Monetary assets and liabilities denominated in foreign currencies are translated at the exchange rate ruling at
F-11
the reporting date. Foreign exchange differences arising on translation are recognized in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) as “Exchange gains /(losses)”. Non-monetary assets and liabilities denominated in foreign currencies are translated at the foreign exchange rate ruling at the date of the transaction.
2.4.3Financial statements of foreign entities
For foreign entities using a different functional currency than USD:
| ● | assets and liabilities for each balance sheet presented are translated at the closing rate at the date of the balance sheet. |
| ● | income and expenses for each statement presenting profit or loss and statements of other comprehensive income (loss) are translated at average exchange rates (unless this average is not a reasonable approximation of the cumulative effect of the rates prevailing on the transaction dates, in which case income and expenses are translated at the rate on the dates of the transactions). |
| ● | all resulting exchange differences are recognized in the statements of other comprehensive income (loss). |
2.5 | Intangible assets |
2.5.1Internally generated intangible assets
Expenditure on research activities is recognized as an expense in the period in which it is incurred.
Due to uncertainties inherent to the development and registration with the relevant healthcare authorities of its products, the Company estimates that the conditions for capitalization per IAS 38 can not be met before the regulatory procedures required by such healthcare authorities have been finalized. Also once regulatory approval has been obtained, an internally generated intangible asset arising from development is capitalized if, and only if, all of the criteria under IAS 38 have been demonstrated.
2.5.2Acquired In-Process R&D and Acquired R&D available for use
Upfront payments and development milestone payments for “Acquired In-Process R&D obtained through in-licensing arrangements are capitalized as intangible assets under “Acquired In-Process R&D” upon meeting the IAS 38 capitalization criteria. These intangibles are considered as intangible assets with definite useful lives and are carried at cost less accumulated impairment losses. “Acquired In-Process R&D” is not amortized, but is evaluated for potential impairment on an annual basis or when facts and circumstances warrant. Any impairment charge is recorded in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) under “Research and development expense”. Once an asset included in “Acquired In-Process R&D” has received marketing approval from a regulatory authority, it is recorded under “Acquired R&D available for use” category.
Regulatory milestone payments and sales-based milestone payments for R&D obtained through in-licensing arrangements acquired are capitalized intangible assets under “Acquired R&D available for use” upon meeting the IAS 38 capitalization criteria. All intangibles classified under “Acquired R&D available for use” are considered as intangible assets with finite useful lives and are carried at cost less accumulated amortization and accumulated impairment losses. “Acquired R&D available for use” is evaluated for potential impairment when the Company identifies indications based on facts and circumstances of the asset. Any impairment charge is recorded in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) under "Cost of sales". “Acquired R&D available for use” is amortized under “Cost of sales” on a straight-line basis over the estimated useful life, being the longer of the current patent protection life of the acquired R&D and patent protection life of the combined product.
F-12
2.5.3.Other intangible assets
Other intangible assets could include the Priority Review Voucher (“PRV”) which the Company can use to obtain the priority review by the FDA for one of its future regulatory submissions or may sell or transfer to a third party. The PRV is initially measured at cost and annually reviewed for impairment when events or circumstances indicate that the carrying value may not be recoverable. Any impairment charge is recorded in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) under “Research and development expenses.” Using the PRV results in amortization recorded in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) under “Research and development expenses” and subsequent derecognition of the intangible asset.
2.6 | Research and development incentives receivables |
The current and non-current research and development incentive receivables relate to refunds resulting from research and development incentives on Research and development expenses in Belgium and are credited to the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) under the line “Other operating income” when the relevant expenditure has been incurred and there is a reasonable assurance that the research and development incentives will be received.
2.7 | Inventories |
Inventories are carried at cost or net realisable value, whichever is lowest. Cost comprises of costs of purchase, costs of conversion and other costs incurred in bringing the inventories to their present location and condition. If the expected sales price less completion costs to execute sales (net realizable value) is lower than the carrying amount, a write-down is recognized for the amount by which the carrying amount exceeds its net realisable value
Included in inventory are products which could, besides commercial activities, be used in preclinical and clinical programs, and free-of-charge, compassionate use and pre-approval access program. These products are charged to “Research & development expenses” or “Selling, general and administrative expenses”, respectively, when dedicated to this channel.
We capitalize inventory costs associated with products prior to the regulatory approval of these products, or for inventory produced in production facilities not yet approved, when it is highly probable that the pre-approval inventories will be saleable. The determination to capitalize is based on the particular facts and circumstances relating to the expected regulatory approval of the product or production facility being considered. The assessment of whether or not the product is considered highly probable to be saleable is made and includes, but is not limited to, how far a particular product or facility has progressed along the approval process, any known safety or efficacy concern and other impediments.
Previously capitalized costs related to pre-launch inventories could be required to be written down upon a change in such judgement or due to a denial or delay of approval by regulatory bodies, a delay in commercialization or other potential factors, which will be recorded under “Research and development expenses” in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss).
2.8 | Trade and other receivables |
Trade and other receivables are designated as financial assets measured at amortized cost. They are initially measured either at their invoiced amounts or at transaction price, in the absence of a significant financing component less adjustments for estimated revenue deductions such as rebates, chargebacks and returns. All receivables are subsequently measured at amortized cost, which generally corresponds to nominal value less expected credit loss provision.
Loss allowance for expected credit losses are established using a simplified approach of forward-looking expected credit loss model (ECL), which includes possible default events on the trade receivables over the entire holding
F-13
period of the trade receivable. These provisions represent the difference between the trade receivable’s carrying amount in the consolidated statements of financial position and the estimated collectible amount. Charges for loss allowance for expected credit losses are recorded under “Selling, general and administrative expenses” in the consolidated statements of profit or loss and consolidated statements of other comprehensive income (loss).
2.9 | Current financial assets |
Current financial assets measured at amortized costs comprise of term accounts that have an initial maturity equal or less than 12 months, but exceeding 3 months.
Current financial assets measured at fair value through profit or loss comprise of money market funds.
Interests on Current financial assets is reported under Cash Flow from investment activities under “Interest received”.
2.10 | Cash and cash equivalents |
Cash are financial assets measured at amortized cost and comprise of cash at bank.
Cash equivalents measured at amortized cost comprise of term accounts that have an initial maturity of less than 3 months that are subject to an insignificant risk of changes in values. Those are used by the Company in the management of short-term commitments. Cash and cash equivalents exclude restricted cash, which is presented in the consolidated statements of financial position under the line “Other non-current assets”.
Cash equivalents measured at fair value through profit or loss comprise of money market funds that are readily convertible to cash and are subject to insignificant risk of changes in value. These financial assets are used by the Company in the management of the short-term commitments.
Interests on Cash equivalents is reported under Cash Flow from investment activities under “Interest received”.
2.11 | Trade and other payables |
Trade and other payables are comprised of liabilities that are due less than one year from the balance sheet date and are in general not interest bearing and settled on an ongoing basis during the financial year. They also include accrued expense related to the Company’s research and development activities, gross-to-net accruals and short-term employee benefits. Trade and other payables are initially measured at their transaction price, which are subsequent to initial recognition measured at amortized cost.
Short-term employee benefits include payables and accruals for salaries and bonuses to be paid to the employees of the Company. They are recognized as expenses for the period in which employees perform the corresponding services.
2.12 | Leases |
The Company assesses whether a contract is or contains a lease, at inception of the contract. The Company recognises a right-of-use asset and a corresponding lease liability with respect to all lease arrangements in which it is the lessee, except for short-term leases (defined as leases with a lease term of 12 months or less) and leases of low value assets. For these leases, the Company recognises the lease payments as an operating expense on a straight-line basis over the term of the lease unless another systematic basis is more representative of the time pattern in which economic benefits from the leased assets are consumed.
The lease liability is initially measured at the present value of the lease payments that are not paid at the commencement date, discounted by using the rate implicit in the lease. If this rate cannot be readily determined, the lessee uses its incremental borrowing rate. The lease liability is subsequently measured by increasing the carrying
F-14
amount to reflect interest on the lease liability (using the effective interest method) and by reducing the carrying amount to reflect the lease payments made. The lease liability is presented as a separate line in the consolidated statements of financial position.
The right-of-use assets comprise the initial measurement of the corresponding lease liability, lease payments made at or before the commencement day, less any lease incentives received and any initial direct costs. They are subsequently measured at cost less accumulated depreciation and impairment losses. Right-of-use assets are depreciated over the shorter period of lease term and useful life of the underlying asset. If a lease transfers ownership of the underlying asset or the cost of the right-of-use asset reflects that the Company expects to exercise a purchase option, the related right-of-use asset is depreciated over the useful life of the underlying asset. The right-of-use assets are presented in the consolidated statements of financial position under the caption “Property, plant and equipment”.
2.13 | Financial instruments |
Financial instruments are initially recognized either at fair value or at transaction price and subsequently measured at either amortized cost or fair value under IFRS 9 on the basis of both the Company’s model for managing the financial assets and the contractual cash flow characteristics of the financial asset. A financial asset is classified as current when the cash flows expected to flow from the instrument mature within one year.
Profit share in AgomAb Therapeutics NV: The Company holds investments in non-current financial assets, which based on IFRS 9, are designated as financial assets at fair value through profit or loss. The fair value of listed investments is based upon the closing price of such securities at each reporting date. As there is no active market for an equity instrument, the Company establishes the fair value by using valuation techniques. The changes to the fair valuation is recorded under “other operating income” in the consolidated statements of profit or loss and consolidated statements of other comprehensive income (loss).
Shares of Zai Lab: Based on IFRS 9, the Company irrevocably elected to designate this specific investment as a financial asset at fair value through OCI as the participation is not held for trading purposes nor contingent consideration recognized by an acquirer in a business combination. The investment is recorded under “other non-current assets” in consolidated statements of financial position and changes to the fair valuation is recorded under “Fair value gain/(loss) on investments in equity instruments designated as at FVTOCI” in the consolidated statements of profit or loss and consolidated statements of other comprehensive income (loss).
2.14 | Shareholder’s equity |
An equity instrument is any contract that evidences a residual interest in the assets of an entity after deducting all of its liabilities. Equity instruments issued by the Company are recognized at the proceeds received, net of direct issue costs.
The Company has never distributed any dividends to its shareholders. As of December 31, 2023, no profits were available for distribution.
As of January 1, 2021, the Company changed its functional and presentation currency from EUR to USD. Differences resulting from the re-presentation have been presented as translation difference, a component within shareholders’ equity. Share capital, share premium, and other reserves are translated at historic rates prevailing at the date of transaction.
2.15 | Share-based payments |
Equity-settled share-based payments to employees and others providing similar services are measured at the fair value of the equity instruments at the acceptance date. Equity settled share based payments includes expenses related to stock options and restricted stock units granted by the Company.
F-15
The fair value determined at the acceptance date of the equity-settled share-based payments is expensed on a straight-line basis over the vesting period, based on the Company’s estimate of equity instruments that will eventually vest, with a corresponding increase in equity. At the end of each reporting period, the Company revises its estimate of the number of equity instruments expected to vest. The impact of the revision of the original estimates, if any, is recognized in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) such that the cumulative expense reflects the revised estimate, with a corresponding adjustment to the equity-settled share-based payment reserve.
The share-based payment expense is recorded under “Research and development expenses” or “Selling, general & administrative expenses” depending on the nature of the services provided by each beneficiary.
2.16 | Income taxes |
Income tax in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) represents the total of the current tax and deferred tax.
The current tax is based on taxable profit for the year. Taxable profit differs from profit as reported in the consolidated statements of profit or loss and consolidated statements of other comprehensive income (loss) as it excludes items of income or expense that are taxable or deductible in other years and items that are never taxable or deductible. The Company’s liability for current tax is calculated using tax rates that have been enacted or substantively enacted by the end of the reporting period.
Deferred tax is recognized on temporary differences between the carrying amounts of assets and liabilities in the consolidated financial statements and the corresponding tax basis used in the computation of taxable profit. Deferred tax assets are recognized to the extent that it is probable that future taxable profits will be available against which those deductible temporary differences can be utilized. The carrying amount of deferred tax assets is reviewed at the end of each reporting period and reduced to the extent that it is not probable that sufficient taxable profits will be available to allow all or part of the asset to be recovered. Deferred tax assets and liabilities are offset if there is a legally enforceable right to offset.
Deferred tax assets and liabilities are measured at the tax rates that are expected to apply in the period in which the liability is settled or the asset realized, based on tax rates (and tax laws) that have been enacted or substantially enacted by the end of the reporting period.
The Company records uncertain tax positions in accordance with IAS 12 using the 2 step test whereby (1) the Company determines whether it is probable that the tax positions will be accepted by relevant taxing authorities, and (2) for those tax positions that are not probable that a tax authority will accept in full the position, the Company recognizes uncertain tax positions using either the most likely amount or the expected value, depending on specific facts and circumstances.
2.17 | Product net sales |
Revenue from the sale of goods is recognized at an amount that reflects the consideration that the Company expects to be entitled to receive in exchange for transferring goods to a customer, at the time when the customer obtains control of the goods rendered, this means when the customer has the ability to direct the use of the asset. The consideration that is committed in a contract with a customer can include fixed amounts, variable amounts, or both. The amount of the consideration may vary due to discounts, rebates, returns, chargebacks or other similar items. Contingent consideration is included in the transaction price when it is highly probable that the amount of revenue recognized is not subject to future significant reversals.
Product net sales are recognized once we satisfy the performance obligation at a point in time under the revenue recognition criteria in accordance with IFRS 15 Revenue from contracts with customers.
F-16
Revenue arising from the commercial sale of commercial product is presented in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) under “Product net sales”. In accordance with IFRS 15 Revenue from contracts with customers, such revenue is recognized when the product is physically transferred, in accordance with the delivery and acceptance terms agreed with the customer. Payment of the transaction price is payable at the point the customer obtains the legal title to the goods.
The amount of revenue recognized reflects the various types of price reductions or rights of return offered by the Company to its customers. Such price reductions and rights of return qualify as variable consideration under IFRS 15 Revenue from contracts with customers.
Products sold are covered by various Government and State programs (such as Medicare and Medicaid) under which products are sold at a discount. Rebates are granted to healthcare authorities, and under contractual arrangements with certain customers. Some wholesalers are entitled to chargeback incentives based on the selling price to the end customer, under specific contractual arrangements. Rebates, chargebacks and other incentives are recognized in the period in which the underlying sales are recognized as a reduction of product sales.
The significant components of variable consideration are as follows:
Co-payment assistance: We provide co-payment assistance to patients who have commercial insurance and meet certain eligibility requirements. We use the expected-value method for estimating co-payment assistance based on estimates of program redemption using data provided by third-party administrators. Estimates for the co-payment assistance are adjusted quarterly to reflect actual experience. We record an accrued liability for unredeemed co-payment assistance related to products for which control has been transferred to customers.
Chargebacks: Chargebacks are discounts that occur when contracted parties purchase directly from a specialty distributor. Contracted parties, which currently consist primarily of Public Health Service Institutions and federal government entities purchasing via the Federal Supply Schedule, generally purchase the product at a discounted price. The specialty distributor, in turn, charges back the difference between the price initially paid by the specialty distributor and the discounted price paid to the specialty distributor by the contracted parties to the Company. The reserves for chargeback are based on known sales to contracted parties. We establish the reserves for chargebacks in the same period that the related revenue is recognized, resulting in an accrued liability and reduction of product gross sales.
Rebates: We are subject to government mandated rebates for Medicaid Drug Rebate Program, Medicare Part D Prescription Drug Benefit Program, and other government health care programs in the U.S. Rebate amounts are based upon contractual agreements or legal requirements with public sector benefit providers. We use the expected-value method for estimating these rebates. The expected utilization of rebates is estimated based on third-party data from the specialty pharmacies and specialty distributor. Estimates for these rebates are adjusted quarterly to reflect the most recent information. We record an accrued liability and reduction of product sales for unpaid rebates related to products for which control has been transferred to customers.
Medicare Part D Coverage Gap: The Medicare Part D coverage gap is a federal program to subsidize the costs of prescription drugs for Medicare beneficiaries in the U.S., which mandates manufacturers to fund a portion of the Medicare Part D insurance coverage gap for prescription drugs sold to eligible patients. Funding of the coverage gap is generally invoiced and paid in arrears. We estimate the impact of the Medicare Part D coverage gap using the expected-value method based on an amount expected to be incurred for the current quarter’s activity, plus an accrual balance for known prior quarters. Estimates for the impact of the Medicare Part D coverage gap are adjusted quarterly to reflect actual experience. We record an accrued liability for unpaid reserves related to the Medicare Part D coverage gap.
Distributor fees: The specialty distributor provides distribution services to the Company for a fee, based on a contractually determined fixed percentage of sales. As the services being provided by the specialty distributor are not distinct, the recurring service fees paid to specialty distributors are treated as variable consideration and a reduction to the transaction price. We estimate these distributor fees and record such estimates in the same period the related revenue is recognized, resulting in a reduction of product gross sales. We record an accrued liability for unpaid distributor fees.
F-17
Value-based arrangements (VBAs): VBAs are arrangements with third party payers where the Company will pay the third-party payers rebates and other fees on eligible purchases of the Company’s product. In consideration for the rebates and fees paid, the third-party Payers will cover its’ patient purchases made of the Company’s products. The structure of the rebates and fees are largely structured based on volume of product purchased. The rebates and fees paid to will be treated as variable consideration and a reduction to the transaction price. We use the expected-value method for estimating the ultimate rebate and fee paid, which are based on the volume of product sold. We apply the applicable rebate rate against a payer mix factor for the relevant patient populations and to the vials sold in the effective plan year of the rebate to derive a liability recorded. Estimates for these agreements are adjusted quarterly to reflect the most recent information. We record an accrued liability for unpaid value-based agreements.
The estimated amounts described above are recognized in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) within “Product net sales” as a reduction of gross sales, and within “Trade and other payables” in the consolidated statements of financial position. They are subject to regular review and adjustment as appropriate based on the most recent data available to management. Each of the above items require significant estimates, judgement and information obtained from external sources. If management’s estimates differ from actual results, we will record adjustments that would affect product sales in the period of adjustment.
2.18 | Collaborations and license agreements |
The Company has currently two active collaboration and license agreements in scope of IFRS 15:
Zai Lab
For the collaboration agreement with Zai Lab the Company has assessed that there is more than one distinct performance obligation, being the transfer of a license and supply of clinical and commercial product. The Company concluded that these performance obligations are distinct in the context of the contract.
Therefore, the Company assesses to allocate the transaction price to all performance obligations identified. The transaction price of these two agreements is composed of (i) a fixed part, that being an upfront payment in the form of newly issued Zai Lab shares, and a guaranteed, non-creditable, non-refundable payment and (ii) a milestone payment for approval of efgartigimod in the U.S. and the consideration received in return for the supply of clinical and commercial product.
The fixed part of the transaction price, as well as the milestone for approval of efgartigimod in the U.S. has been allocated to the transfer of a license performance obligation. The Company concluded that the license as of the effective date of the contract, being January 2021, has standalone value. As such, the Company concluded that the promise in granting the license to Zai Lab is to provide a right to use the entity’s intellectual property as it exists at the point in time at which the license is granted and therefore, revenue was recognized at a point in time in January 2021.
Under the collaboration agreement, the Company provides clinical and commercial supply to Zai Lab. Company concludes to recognize such sales as revenue given that the Company acts as principal in the transaction as the risk related to inventory is born by the Company until the inventory is transferred to Zai Lab. The revenue related to clinical supply is recorded under line item “Collaboration revenue”. The revenue related to commercial supply is recorded under line item “product net sales” in the Consolidated statements of other comprehensive Income (Loss). The income related to royalties is recorded under line item item “Collaboration revenue”.
AbbVie
For the collaboration agreement with AbbVie the Company has determined that the transfer of license combined with the performance of research and development activities represent one single performance obligation. The Company concluded that the license is not distinct in the context of the contract.
The transaction price is composed of a fixed part, that being an upfront license fee, and a variable part, being milestone payments and cost reimbursements of research and development activities delivered. Milestone payments are
F-18
only included in the transaction price to the extent it is highly probable that a significant reversal in the amount of cumulative revenue recognition will not occur when the uncertainty associate with the variable consideration is subsequently resolved. Management estimates the amount to be included in the transaction price upon achievement of the milestone event. Sales-based milestones and sales-based royalties are a part of the Company’s arrangements but are not yet included in its revenues.
The transaction price has been allocated to the single performance obligation and revenues has been recognized over the estimated service period based on an input model, being the percentage of completion method. The upfront license fee has been fully recognized since 2021 as the performance obligation has been fulfilled at that time. Milestone payments that become highly probable after the performance obligation has been fulfilled are therefore recognized at that point in time.
2.19 | Cost of Sales |
Cost of sales are recognized when the associated revenue from product net sales is recognized. Cost of sales include material, manufacturing costs and other costs attributable to production, including shipping costs, as well as royalties payable on sold products.
3. | Critical accounting estimates and judgments |
In the application of the Company’s accounting policies, which are described above, the Company is required to make judgments, estimates and assumptions about the carrying amounts of assets and liabilities that are not readily apparent from other sources. The estimates and associated assumptions are based on historical experience and other factors that are considered to be relevant. Actual results may differ from these estimates.
The estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimate is revised if the revision affects only that period or in the period of the revision and future periods if the revision affects both current and future periods.
Critical estimates in applying accounting policies
Gross to net adjustments
The product gross sales are subject to various deductions, which are primarily composed of rebates to government agencies, distributors, health insurance companies and managed healthcare organizations. These deductions represent estimates of the related obligations, requiring the use of judgment when estimating the effect of these sales deductions on product gross sales for a reporting period. These adjustments are deducted from product gross sales to arrive at product net sales. The significant components of variable consideration under revenue recognition policy summarizes the nature of these deductions and how the deduction is estimated, see note 2.17. After recording these, product net sales represent the Company’s best estimate of the cash that we expect to ultimately collect. If in future periods the actuals vary from prior period best estimates, this would affect revenue in the period of adjustment.
Please refer to note 14 for the movement over the period and the ending balance of the gross-to-net-accruals.
F-19
4. | Property, plant and equipment |
| IT, office and lab |
| Right-of-use assets |
| Right-of-use assets |
| Leasehold |
| Lease |
| ||||||||
(in thousands of $) | equipment | Buildings | Vehicles | improvements | equipment | Total | ||||||||||||
Cost | ||||||||||||||||||
On January 1, 2021 | $ | | | | | | | |||||||||||
Additions | | | | | — | | ||||||||||||
Disposals | ( | — | — | — | — | ( | ||||||||||||
Currency translation adjustment | | ( | — | | — | ( | ||||||||||||
On December 31, 2021 | | | | | | | ||||||||||||
Additions | | | | — | — | | ||||||||||||
Disposals | ( | — | — | — | — | ( | ||||||||||||
Currency translation adjustment | ( | — | — | — | — | ( | ||||||||||||
On December 31, 2022 | | $ | | $ | | $ | | $ | | $ | | |||||||
Additions | | | | | — | | ||||||||||||
Disposals | ( | — | ( | ( | — | ( | ||||||||||||
On December 31, 2023 | $ | | $ | | $ | | $ | | $ | | $ | | ||||||
Depreciation and impairment | ||||||||||||||||||
On January 1, 2021 | $ | ( | ( | ( | ( | ( | ( | |||||||||||
Depreciation | ( | ( | ( | ( | ( | ( | ||||||||||||
Disposals | | — | — | — | — | | ||||||||||||
Currency translation adjustment | | ( | — | ( | — | | ||||||||||||
On December 31, 2021 | ( | ( | ( | ( | ( | ( | ||||||||||||
Depreciation | ( | ( | ( | ( | ( | ( | ||||||||||||
Disposals | | — | — | — | — | | ||||||||||||
Currency translation adjustment | | | | | — | | ||||||||||||
On December 31, 2022 | ( | $ | ( | $ | ( | $ | ( | $ | ( | $ | ( | |||||||
Depreciation | ( | ( | ( | ( | ( | ( | ||||||||||||
Disposals | | — | | — | — | | ||||||||||||
On December 31, 2023 | $ | ( | $ | ( | $ | ( | $ | ( | $ | ( | $ | ( | ||||||
Carrying Amount | ||||||||||||||||||
On December 31, 2021 | $ | | $ | | $ | | $ | | $ | | $ | | ||||||
On December 31, 2022 | | | | | | | ||||||||||||
On December 31, 2023 | $ | | $ | | $ | | $ | | $ | | $ | | ||||||
Depreciation is recognized as from acquisition date onwards (unless asset is not ready for use) so as to write off the cost or valuation of assets less their residual values over their useful lives, using the straight-line method. Unless revised due to specific changes in the estimated useful life, annual depreciation rates are as follows:
| ● | Office and lab equipment: |
| ● | IT equipment: |
As of December 31, 2023, there are
F-20
| 5. | Intangible assets |
(in thousands of $) |
| Acquired R&D available for use | Acquired In-Process R&D | Software & databases | Other Intangibles | Total | |||||||||
Cost | |||||||||||||||
On January 1, 2021 | $ | — | $ | | $ | | $ | | $ | | |||||
Additions | — | | — | — | | ||||||||||
Translation differences | — | — | ( | — | ( | ||||||||||
On December 31, 2021 | — | | | | | ||||||||||
Additions | — | — | | | | ||||||||||
Disposals | — | — | ( | — | ( | ||||||||||
Derecognition | — | — | — | ( | ( | ||||||||||
On December 31, 2022 | — | | | | | ||||||||||
Additions | | — | — | — | | ||||||||||
Derecognition | — | — | — | ( | ( | ||||||||||
Reclassification | | ( | — | — | — | ||||||||||
On December 31, 2023 | $ | | $ | | $ | | $ | — | $ | | |||||
Amortization and impairment | |||||||||||||||
On January 1, 2021 | $ | — | $ | — | $ | ( | $ | — | $ | ( | |||||
Amortization | ( | — | ( | ||||||||||||
On December 31, 2021 | — | — | ( | — | ( | ||||||||||
Amortization | — | — | ( | ( | ( | ||||||||||
Derecognition | — | — | — | | | ||||||||||
On December 31, 2022 | — | — | ( | — | ( | ||||||||||
Amortization | ( | — | ( | ( | ( | ||||||||||
Derecognition | — | — | — | | | ||||||||||
On December 31, 2023 | $ | ( | $ | — | $ | ( | $ | — | $ | ( | |||||
Carrying Amount | |||||||||||||||
On December 31, 2021 | $ | — | $ | | $ | | $ | | $ | | |||||
On December 31, 2022 | — | | | | | ||||||||||
On December 31, 2023 | $ | | $ | | $ | | $ | — | $ | | |||||
Acquired In-Process R&D is mainly related to the in-licensing of the ENHANZE® drug delivery technology from Halozyme. In line with its accounting policies, the Company has capitalized the upfront payment upon commencement of the in-license agreement in 2019 and the development milestone payments when the respective milestone has been achieved. In June 2023, the Company obtained the FDA approval for VYVGART Hytrulo, which is a subcutaneous product combination of efgartigimod alfa and Halozyme’s ENHANZE® drug delivery technology. Upon this regulatory approval, the has moved from “Acquired In-Process R&D” to “Acquired R&D available for use”.
Further, the additions to “Acquired R&D available for use” are related to regulatory and sales-based milestones triggered during 2023 related to the in-licensing of the ENHANZE® drug delivery technology from Halozyme. In line with its accounting policies, the Company has capitalized the regulatory and sales-based milestone payments when the respective milestones have been achieved. The “Acquired R&D available for use” are amortized under “Cost of sales” on a straight-line basis over their useful life, being the longer of the patent protection life of the Acquired R&D available for use and patent protection life of the combined product, which is 2036 for VYVGART Hytrulo.
F-21
The Company performs an annual impairment review on the intangible assets. This review did not result in the recognition of an impairment charge for the years ended December 31, 2023, 2022 and 2021.
In the fourth quarter of 2023, the Company utilized the PRV submitted with the sBLA filing for VYVGART Hytrulo for the treatment of CIDP, which resulted in amortization of $
As of December 31, 2023, there are
6. | Other non-current assets |
Other non-current assets consisted of non-current restricted cash and financial assets held at fair value through profit or loss or through OCI.
At December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Non-current restricted cash |
| $ | |
| $ | |
| $ | |
Non-current financial assets held at fair value through profit or loss |
| |
| |
| | |||
Non-current financial assets held at fair value through OCI | | | | ||||||
Total other non-current assets |
| $ | |
| $ | |
| $ | |
Non-current restricted cash on December 31, 2023 was mainly composed of deposit guarantees paid under the lease agreements for the laboratory and offices of the Company.
Non-current financial assets held at fair value through profit or loss is comprised of the profit share in AgomAb Therapeutics NV. In March 2019, the Company entered into a license agreement with AgomAb Therapeutics NV for the use of HGF-mimetic SIMPLE Antibodies™, developed under the Company’s Immunology Innovative Program. In exchange for granting this license, the Company received a profit share in AgomAb Therapeutics NV. Since AgomAb Therapeutics NV is a private company, the valuation of the profit share is based on level 3 assumptions.
In June 2022, AgomAb Therapeutics NV secured €
In October 2023, AgomAb Therapeutics NV secured $
Fair value changes on non-current financial assets with fair value through profit or loss are recognized in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) under “Other operating income”.
As part of the license agreement for the development and commercialization for efgartigimod in Greater China, in 2021 the Company obtained, amongst others,
F-22
election to recognize subsequent changes in fair value through OCI under “Fair value gain/(loss) on investments in equity instruments designated as at FVTOCI”.
The table below illustrates these non-current financials assets at fair value through profit or loss or OCI as of December 31, 2023, 2022 and 2021.
At December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Cost at January 1 |
| $ | |
| $ | |
| $ | |
Additions of the year | — | — | | ||||||
Cost at December 31 | $ | | $ | | $ | | |||
Fair value adjustments at January 1 | $ | ( | $ | ( | $ | | |||
Fair value adjustment of the year through profit or loss | — | | | ||||||
Fair value adjustment of the year through OCI | ( | ( | ( | ||||||
Fair value adjustment at December 31 | $ | ( | $ | ( | $ | ( | |||
Net book value at December 31 |
| $ | |
| $ | |
| $ | |
7. | Inventories |
At December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Raw materials and consumables | $ | |
| $ | |
| $ | | |
Inventories in process | | | | ||||||
Finished goods | | | | ||||||
Total inventories |
| $ | |
| $ | |
| $ | |
The cost of inventories, which is recognized under “Cost of sales” in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss), amounted to $
On December 31, 2023, pre-launch inventory awaiting facility approval amounted to $
As a result of the detection of a latent defect in the second quarter of 2023 in drug substance batches produced in 2022 at one of the facilities awaiting approval, the Company has decreased inventory with an amount of $
8. | Prepaid expenses (Current) |
The current prepaid expenses are composed of prepayments which are details below:
At December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Prepaid inventory | |||||||||
Prepaid research and development expenses | |||||||||
Prepaid advertising expenses | |||||||||
Prepaid software | |||||||||
Other prepaid expenses | |||||||||
Total prepaid expenses |
| $ | |
| $ | |
| $ | |
F-23
9. | Trade and other receivables |
The trade and other receivables are composed of receivables which are detailed below:
At December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Trade receivable | $ | | $ | | $ | | |||
Interest receivable | | | | ||||||
Tax receivables | | | | ||||||
Other receivable | | | | ||||||
Total trade and other receivables |
| $ | |
| $ | |
| $ | |
The carrying amounts of trade and other receivables approximate their respective fair values. On December 31, 2023, 2022 and 2021, we did not have any provision for expected credit losses.
Please also refer to Note 26 for more information on the financial risk management.
10. | Financial assets — current |
These current financial assets relate to term accounts with an initial maturity longer than
At December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Money market funds |
| $ | — |
| $ | |
| $ | |
Term accounts | | | | ||||||
Total current financial assets |
| $ | |
| $ | |
| $ | |
On December 31, 2023, the current financial assets included $
Please also refer to Note 26 for more information on the financial risk management.
11. | Cash and cash equivalents |
At December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Money market funds |
| $ | |
| $ | |
| $ | |
Term accounts | | | | ||||||
Cash and bank balances |
| |
| |
| | |||
Total cash and cash equivalents |
| $ | |
| $ | |
| $ | |
Cash and cash equivalents may comprise of cash and bank balances, saving accounts, term accounts with an original maturity not exceeding 3 months and money market funds that are readily convertible to cash and are subject to an insignificant risk of changes in value.
Cash positions are invested with preferred financial partners, which are mostly considered to be high quality financial institutions with sound credit ratings to reduce credit risk.
F-24
On December 31, 2023, the cash and cash equivalents included $
Please also refer to Note 26 for more information on the financial risk management.
12. | Share capital and share premium |
On December 31, 2023, the Company’s share capital was represented by
Roll forward of number of shares outstanding:
Number of shares outstanding on January 1, 2021 |
| |
Exercise of stock options | | |
Global public offering in Euronext and Nasdaq on February 2, 2021 | | |
Over-allotment option exercised by underwriters on February 4, 2021 | | |
Number of shares outstanding on December 31, 2021 |
| |
Exercise of stock options | | |
Vesting of RSUs | | |
Global public offering in Euronext and Nasdaq on March 23, 2022 | | |
Over-allotment option exercised by underwriters on March 29, 2022 | | |
Number of shares outstanding on December 31, 2022 |
| |
Exercise of stock options | | |
Vesting of RSUs | | |
Global public offering in Nasdaq on July 18, 2023 | | |
Over-allotment option exercised by underwriters on July 19, 2023 | | |
Number of shares outstanding on December 31, 2023 |
| |
On May 2, 2023, at the annual general meeting, the shareholders of the Company approved the authorization to the Board to issue up to a maximum of
On July 18, 2023, argenx SE offered
On December 31, 2023, an amount of €
F-25
13. | Share-based payments |
The Company has an equity incentive plan for the employees, key consultants, board members, senior management and key outside advisors (“key persons”) of the Company and its subsidiaries. In accordance with the terms of the plan, as approved by shareholders, employees may be granted stock options and/or restricted stock units.
13.1 | Stock Option |
The stock options are granted to key persons of the Company and its subsidiaries. The stock options may be granted to purchase ordinary shares at an exercise price. The stock options have been granted free of charge. Each employee’s stock option converts into
The stock options granted vest, in principle, as follows:
| ● | of the total stock options granted will vest on the first anniversary of the granting of the stock options, and |
| ● | of the total grant on the first day of each month following the first anniversary of the date of grant of the stock options. |
Stock options granted to non-executive directors vest on the third anniversary of the date of grant.
Upon leave of the key persons stock options must be exercised before the later of (i) 90 days after the last working day at argenx, or (ii) March 31 of the 4th year following the date of grant of those stock options, and in any case no later than the expiration date of the option.
In order to prefinance the taxes that are paid upon the grant of stock options, Belgian employees have the ability, in exchange for the taxes due upon the grant of the stock options, to transfer the economic benefits related to part of those stock options to a third party. In the year ending December 31, 2023, the economic benefits of
No other conditions are attached to the stock options.
F-26
The following stock option arrangements were in existence during the current and prior years and which are exercisable at the end of each period presented:
Exercise price | Outstanding | ||||||||
per stock | stock options on | ||||||||
options | December 31, | ||||||||
Expiry date |
| (in $) (1) |
| 2023 |
| 2022 |
| 2021 | |
2022 | $ | | — | — | | ||||
2024 |
| |
| |
| |
| | |
2024 |
| |
| |
| |
| | |
2024 |
| |
| |
| |
| | |
2025 |
| |
| |
| |
| | |
2025 |
| |
| |
| |
| | |
2026 |
| |
| |
| |
| | |
2026 |
| |
| |
| |
| | |
2026 |
| |
| |
| |
| | |
2027 | | | | | |||||
2027 | | | | | |||||
2023 | | — | | | |||||
2028 | | | | | |||||
2023 | | — | | | |||||
2028 | | | | | |||||
2024 | | | | | |||||
2029 | | | | | |||||
2024 | | | | | |||||
2029 | | | | | |||||
2025 | | | | | |||||
2030 | | | | | |||||
2025 | | | | | |||||
2030 | | | | | |||||
2025 | | | | | |||||
2030 | | | | | |||||
2030 | | | | | |||||
2025 | | | | | |||||
2026 | | | | | |||||
2026 | | | | | |||||
2026 | | | | | |||||
2031 | | | | | |||||
2031 | | | | | |||||
2031 | | | | | |||||
2026 | | | | | |||||
2031 | | | | | |||||
2027 | | | | — | |||||
2032 | | | | — | |||||
2027 | | | | — | |||||
2032 | | | | — | |||||
2027 | | | | — | |||||
2032 | | | | — | |||||
2032 | | | | — | |||||
2027 | | | | — | |||||
2025 | | | — | — | |||||
2028 | | | — | — | |||||
2033 | | | — | — | |||||
2028 | | | — | — | |||||
2033 | | | — | — | |||||
2028 | | | — | — | |||||
2033 | | | — | — | |||||
2028-2032 (2) | $ | | | — | — | ||||
|
| | | ||||||
| (1) | Amounts have been converted to USD at the closing rate as of December 31, 2023. |
| (2) | In December 2023, the Company granted stock options for which the Belgian taxed beneficiaries had a 60-day period to choose between a contractual term of or |
F-27
2023 | 2022 | 2021 | |||||||||||||
| Number of |
| Weighted average |
| Number of |
| Weighted average |
| Number of |
| Weighted average | ||||
stock options | exercise price (*) | stock options | exercise price (*) | stock options | exercise price (*) | ||||||||||
Outstanding at January 1 |
| |
| $ | |
| |
| $ | |
| |
| $ | |
Granted |
| |
| |
| |
| |
| |
| | |||
Exercised |
| ( |
| |
| ( |
| |
| ( |
| | |||
Forfeited |
| ( |
| |
| ( |
| |
| ( |
| | |||
Outstanding at December 31 |
| |
| |
| |
| |
| |
| | |||
Exercisable at December 31 |
| |
| $ | |
| |
| $ | |
| |
| $ | |
(*) amounts have been converted to USD at the closing rate of the respective period.
The weighted average share price at the date of exercise of options exercised during the year ended December 31, 2023 was $
|
| Weighted average | ||
remaining | ||||
Outstanding on | contractual life | |||
Exercise price (in $) |
| December 31, 2023 |
| (in years) |
| ||||
| ||||
The fair market value of the stock options has been determined based on the Black and Scholes model using the following unobservable assumptions:
| ● | The expected volatility, determined on the basis of the implied volatility of the share price over the expected life of the option. |
| ● | The expected option life, calculated as the estimated duration until exercise, taking into account the specific features of the plans. |
Below is an overview of the parameters used in relation to the determination of the fair value of the grants during 2023:
Stock options granted in |
| April 2023 |
| July 2023 | October 2023 | December 2023 (1) | |||||||
Number of options granted |
|
| |||||||||||
Average Fair value of options (in $) (*) |
| $ |
| $ | $ | $ | |||||||
Share price (in $) (*) |
| $ |
| $ | $ | $ | |||||||
Exercise price (in $) (*) |
| $ |
| $ | $ | $ | |||||||
Expected volatility |
| % | % | % | % | ||||||||
Average Expected option life (in years) |
|
|
| ||||||||||
Risk‑free interest rate |
| % | % | % | % | ||||||||
Expected dividends |
| — | % | — | % | — | % | — | % | ||||
| (1) | In December 2023, the Company granted a total of |
F-28
| estimate will be reassessed once the acceptance period of 60 days has passed and the beneficiaries will have made a choice between a contractual term of or |
(*) | amounts have been converted to USD at the applicable rate prevailing at the grant date. |
Below is an overview of the parameters used in relation to the determination of the fair value of grants during 2022:
Stock options granted in |
| April 2022 |
| July 2022 | October 2022 |
| December 2022 (1) | ||||||
Number of options granted |
|
| |||||||||||
Average Fair value of options (in $) (*) |
| $ |
| $ | $ | $ | |||||||
Share price (in $) (*) |
| $ |
| $ | $ | $ | |||||||
Exercise price (in $) (*) |
| $ |
| $ | $ | $ | |||||||
Expected volatility |
| % | % | % | % | ||||||||
Average Expected option life (in years) |
|
|
| 4 - 6.50 | |||||||||
Risk‑free interest rate |
| % | % | % | % | ||||||||
Expected dividends |
| — | % | — | % | — | % | — | % | ||||
(*) amounts have been converted to USD at the applicable rate prevailing at the grant date.
(1) In December 2022, the Company granted a total of
Below is an overview of the parameter used in relation to the determination of the fair value of grants during 2021:
Stock options granted in |
| April 2021 |
| July 2021 |
| October 2021 | December 2021 | ||||||
Number of options granted |
|
| |||||||||||
Average Fair value of options (in $) (*) |
| $ |
| $ | $ | $ | |||||||
Share price (in $) (*) |
| $ |
| $ | $ | $ | |||||||
Exercise price (in $) (*) |
| $ | |
| $ | | $ | | $ | | |||
Expected volatility |
| % | % | % | % | ||||||||
Average expected option life (in years) |
|
|
| ||||||||||
Risk‑free interest rate |
| ( | % | ( | % | ( | % | % | |||||
Expected dividends |
| — | % | — | % | — | % | — | % | ||||
(*) amounts have been converted to USD at the closing rate of the respective period.
The total share-based payment expense related to stock options recognized in the consolidated statements of profit or loss totaled $
13.2 | Restricted Stock Units (RSUs) |
The RSUs are granted to key persons of the Company and its subsidiaries. The RSUs have been granted free of charge. Each employee’s RSUs converts into
F-29
The following restricted stock units arrangements were in existence during the current and prior years:
2023 | 2022 | 2021 | |||||||||||||
|
| Weighted average |
|
| Weighted average |
|
| Weighted average | |||||||
Number of | Grant Date | Number of | Grant Date | Number of | Grant Date | ||||||||||
RSUs | Fair Value | RSUs | Fair Value | RSUs | Fair Value | ||||||||||
Non-vested units at January 1 |
| | $ | | | $ | | — | $ | — | |||||
Granted |
| | | | | | | ||||||||
Vested |
| ( | ( | — | — | — | |||||||||
Forfeited |
| ( | | ( | | ( | | ||||||||
Non-vested units at December 31 |
| | $ | | | $ | | | $ | | |||||
The total share-based payment expense related to RSUs recognized in the consolidated statements of profit or loss totaled $
14. | Trade and other payables |
At December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Trade payables |
| $ | |
| $ | |
| $ | |
Short‑term employee benefits |
| |
| |
| | |||
Gross-to-net-accruals | | | — | ||||||
Other | | | | ||||||
Total trade and other payables |
| $ | |
| $ | |
| $ | |
The carrying amounts of trade and other payables approximate their respective fair values.
Trade payables correspond primarily to clinical and manufacturing activities and include accrued expenses related to these activities.
Short-term employee benefits include payables and accruals for salaries and bonuses to be paid to the employees of the Company.
F-30
The following table summerizes the movement in the gross-to-net-accruals for the year ended December 31, 2023, 2022:
(in thousands of $) | Rebates and chargebacks | Distribution fees, product returns and other | Total | ||||||
Balance at January 1, 2022 | $ | — | $ | — | $ | — | |||
Current estimate related to the sales made in the current year | | | | ||||||
(Credits or payments related to sales made during the year) | ( | ( | ( | ||||||
Balance at December 31, 2022 | $ | | $ | | $ | | |||
Current estimate related to the sales made in the current year | | | | ||||||
Adjustment for prior year sales | ( | ( | ( | ||||||
(Credits or payments related to sales made during the year) | ( | ( | ( | ||||||
(Credit or payments related to sales made during prior year) | ( | ( | ( | ||||||
Balance at December 31, 2023 | $ | | $ | | $ | |
15. | Product net sales |
Year Ended | |||||||||
December 31, | |||||||||
(in thousands of $) | 2023 | 2022 |
| 2021 | |||||
Product gross sales | $ | - | |||||||
Gross to net adjustment | ( | ( | - | ||||||
Product net sales | $ | - | |||||||
For the twelve months ended December 31, 2023, the product net sales was related to sales of VYVGART and VYVGART SC. For the twelve months ended December 31, 2022, the product net sales was related to sales of VYVGART.
Refer to note 18 for the breakdown of Product net sales by country of sale.
F-31
16. | Collaboration revenue |
The following table summarizes details of collaboration revenues for the year ended December 31, 2023, 2022 and 2021 by collaboration agreement and by category of revenue: upfront payments, milestone payments, research and development service fees and other revenue.
Year Ended | ||||||||||
December 31, | ||||||||||
(in thousands of $) |
| 2023 | 2022 |
| 2021 | |||||
Zai Lab | $ | — | — | | ||||||
J&J |
| — | — | | ||||||
AbbVie |
| — | — | | ||||||
Upfront payments | — | — | | |||||||
Zai Lab | — | — | | |||||||
J&J | — | — | | |||||||
AbbVie | | — | | |||||||
Other | — | | | |||||||
Milestone payments | | | | |||||||
J&J | — | — | | |||||||
Other | — | | | |||||||
Research and development service fees | — | | | |||||||
Zai Lab | | | | |||||||
Other collaboration revenue | | | | |||||||
Total collaboration revenue | $ | | $ | | $ | | ||||
For the years ended December 31, 2023, 2022 and 2021, the collaboration revenue was generated under the agreements with Zai Lab, J&J and AbbVie, each as described below.
Zai Lab
On January 6, 2021, argenx and Zai Lab announced the License agreement for the development and commercialization of efgartigimod in Greater China, granting Zai Lab the exclusive rights to develop and commercialize efgartigimod in Greater China.
Under the terms of the agreement, the Company received $
As stated in the accounting policies regarding this collaboration with Zai Lab, the Company concluded there are
Under the collaboration agreement, the Company provides clinical supply to Zai Lab. The revenue related to clinical supply is recorded under line item “Other revenues” within the collaboration revenue. The income related to
F-32
royalties is recorded as “Other revenues” under “collaboration revenue”. During the year ending December 31, 2023 the first revenue related to commercial supply and related royalties are recognized.
Please refer to Note 2 for the material accounting policies on the remainder elements of the agreement.
AbbVie
In April 2016, the Company entered into a collaboration agreement with AbbVie to develop and commercialize ARGX-115 (ABBV-151).
The Company granted AbbVie an exclusive option, for a specified period following completion of IND enabling studies, to obtain a worldwide, exclusive license to the ARGX-115 (ABBV-151) program to develop and commercialize products.
In October 2023, the Company achieved the second development milestone upon initiation of a non-pivotal Clinical Trial, triggering a $
Subject to the continuing progress of ARGX-115 (ABBV-151) by AbbVie, the Company is eligible to receive future development, regulatory and commercial milestone payments in aggregate amounts of up to $
J&J Innovative Medicines
On June 4, 2021, the Company received a termination notification from Cilag GmbH International, an affiliate of J&J Innovative Medicines (J&J), which results in the termination of the Collaboration Agreement to jointly develop and commercialize cusatuzumab. Following the termination, the Company concluded that it has substantially satisfied the performance obligation, and as a consequence, recorded $
17. | Other operating income |
Year Ended | |||||||||
December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Grants |
| $ | |
| $ | |
| $ | |
Research and development incentives |
| |
| |
| | |||
Payroll tax rebates |
| |
| |
| | |||
Change in fair value on non-current financial assets | — | | | ||||||
Total other operating income |
| $ | |
| $ | |
| $ | |
17.1 | Research and development incentives |
The Company has accounted for a tax incentive following a research and development tax incentive scheme in Belgium according to which the incentive will be refunded after a
17.2 | Payroll tax rebates |
The Company accounted for payroll tax rebates as a reduction in withholding income taxes for its highly qualified personnel employed in its research and development department.
F-33
18. | Segment reporting |
The Company manages its activities and operates as
Following table summarizes the product net sales by country of sales based on the country of the entity that recognizes product net sales:
Year Ended | ||||||
December 31, | ||||||
(in thousands of $) |
| 2023 |
| 2022 | ||
United States | $ | | $ | | ||
Japan | | | ||||
EMEA | | | ||||
China | | — | ||||
Total product net sales |
| $ | |
| $ | |
The Company sells its products through a limited number of distributors and wholesellers.
Collaboration revenue is generated by external customers with their main registered office geographically located as shown in the table below:
Year ended | ||||||||||
December 31, | ||||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 |
| |||
Denmark |
| $ | — |
| $ | |
| $ | |
|
United States |
| |
| — |
| |
| |||
China | | | | |||||||
Other |
| — |
| |
| |
| |||
Total collaboration revenue |
| $ | |
| $ | |
| $ | |
|
The property plant and equipment and intangible assets of the Company are geographically located as shown in the table below:
Non-current assets | |||||||||
At December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Netherlands |
| $ | — |
| $ | — |
| $ | — |
Belgium |
| |
| |
| | |||
United States |
| |
| |
| | |||
Japan |
| |
| |
| | |||
Germany | | — | — | ||||||
Total |
| $ | |
| $ | |
| $ | |
Prior year amounts were updated/recast to match current year presentation
F-34
19. | Research and development expenses |
Year Ended | |||||||||
December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 | 2021 | ||||
Personnel expenses |
| $ | |
| $ | |
| $ | |
External research and development expenses |
| |
| |
| | |||
Materials and consumables |
| |
| |
| | |||
Depreciation and amortization |
| |
| |
| | |||
IT expenses | | | | ||||||
Other expenses |
| |
| |
| | |||
Total Research and development expenses |
| $ | |
| $ | |
| $ | |
20. | Selling, general and administrative expenses |
Year Ended | |||||||||
December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Personnel expenses |
| $ | |
| $ | |
| $ | |
Marketing services |
| | |
| | ||||
Professional fees | | | | ||||||
Supervisory board |
| |
| |
| | |||
Depreciation and amortization | | | | ||||||
IT expenses | | | | ||||||
Other expenses |
| |
| |
| | |||
Total Selling, general and administrative expenses |
| $ | |
| $ | |
| $ | |
21. | Personnel expenses |
The personnel expenses mentioned in notes 19 and 20 above are as follows:
Year Ended | |||||||||
December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Short‑term employee benefits—Salaries |
| $ | |
| $ | |
| $ | |
Short‑term employee benefits—Social Security |
| |
| |
| | |||
Post‑employment benefits |
| |
| |
| | |||
Termination benefits |
| |
| |
| | |||
Share‑based payment |
| |
| |
| | |||
Employer social security contributions stock options |
| |
| |
| | |||
Total personnel expenses |
| $ | |
| $ | |
| $ | |
The post-employment benefits relate to the pension plans the Company has in place for its employees.
The average number of full-time equivalents (FTE) employees by function is presented below:
Year Ended | |||||||||
December 31, | |||||||||
Average Number of FTE |
| 2023 |
| 2022 |
| 2021 | |||
Research and development |
| |
| |
| | |||
Selling, general and administrative |
| |
| |
| | |||
| |
| |
| | ||||
F-35
22. | Leases |
The statements of financial position shows the following amounts relating to leases:
Year Ended | |||||||||
December 31, | December 31, | December 31, | |||||||
In thousands of $ |
| 2023 | 2022 | 2021 | |||||
Right-of-use assets |
| ||||||||
Buildings |
| $ | |
| $ | |
| $ | |
Vehicles | | | | ||||||
Equipment | | | | ||||||
| $ | | $ | | $ | | |||
Lease liabilities |
| ||||||||
Current |
| $ | |
| $ | |
| $ | |
Non-current | | | | ||||||
| $ | | $ | | $ | | |||
Additions to the right-of-use assets amounted to $
The table below shows a maturity analysis of the lease liabilities as on December 31, 2023:
(in thousands of $) | Less than 1 year | 1-3 years | 3-5 years | More than 5 years | Total contractual cash flows | Carrying amount | |||||||||||
Lease liabilities | $ | |
| $ | | $ | | $ | | $ | |
| $ | | |||
The consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss) shows the following amounts relating to leases:
Year Ended | |||||||||
December 31, | |||||||||
In thousands of $ |
| 2023 |
| 2022 | 2021 | ||||
Depreciation charges |
| ||||||||
Buildings |
| $ | |
| $ | |
| $ | |
Vehicles | | | | ||||||
Equipment | | | | ||||||
| $ | |
| $ | |
| $ | | |
Interest expense (included in finance cost) | $ | | $ | | $ | | |||
Expense relating to short-term leases | | | | ||||||
Expense relating to leases of low-value assets that are not shown above as short-term leases | | | | ||||||
The total cash outflow for leases in 2023, 2022 and 2021 was $
The Company did not enter into any lease agreement with variable lease payments or residual value guarantees. The Company has leases that include extension options. These options provide flexibility in managing the leased assets and align with the Company’s business needs. The Company exercises judgement in deciding whether it is reasonably certain that the extension options will be exercised.
F-36
23. | Financial result and exchange gains/(losses) |
Year Ended | |||||||||
December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Interest income |
| $ | |
| $ | |
| $ | |
Net gain on cash equivalents & current financial assets held at fair value through profit or loss and cash equivalents |
| |
| |
| | |||
Financial income |
| $ | |
| $ | |
| $ | |
Net loss on cash equivalents & current financial assets held at fair value through profit or loss and cash equivalents | $ | ( | $ | ( | $ | ( | |||
Other financial expense | ( | ( | ( | ||||||
Financial expense | $ | ( | $ | ( | $ | ( | |||
Realized exchange gains/(losses) |
| $ | |
| $ | ( |
| $ | |
Unrealized exchange gains/(losses) | | ( | ( | ||||||
Exchange gains/(losses) |
| $ | |
| $ | ( |
| $ | ( |
The exchange gains of $
24. | Income taxes |
Income taxes recognized in the income statements can be detailed as follows:
Year Ended | |||||||||
December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Current year |
| $ | ( |
| $ | ( |
| $ | ( |
Income tax prior years |
| ( |
| ( |
| | |||
Current tax (expense) / benefit |
| ( |
| ( |
| ( | |||
Originating and reversal of temporary differences | | | | ||||||
Deferred tax (expense) / benefit |
| |
| |
| | |||
Total tax (expense) / benefit | $ | | $ | | $ | ( | |||
F-37
The difference between the provision for income taxes and the amount that would result from applying the Dutch statutory tax rate to income before provision for income taxes is as follows:
Year Ended | |||||||||
December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Loss before taxes |
| $ | |
| $ | |
| $ | |
Income tax (expense)/benefit calculated at the Dutch statutory federal income tax rates for applicable tax years (1) |
| |
| |
| | |||
Effect of intercompany asset deal / transaction | | ( | — | ||||||
Effect of expenses not deductible in determining taxable results |
| ( |
| ( |
| ( | |||
Effect of share based payment expenses that are not deductible in determining taxable results | ( | ( | ( | ||||||
Effect of stock issue expenses that are not taxable in determining taxable results |
| |
| |
| | |||
Effect of concessions |
| |
| |
| | |||
Effect of change of (de)recognition of deferred tax assets on tax losses | ( | ( | ( | ||||||
Effect of different tax rates in jurisdictions in which the company operates |
| ( |
| ( |
| ( | |||
Effect of change of (de)recognition of deferred tax assets |
| ( |
| ( |
| ( | |||
Withholding tax paid | ( | — | ( | ||||||
(Underprovided)/overprovided in prior years | ( | ( | | ||||||
Other | | ( | ( | ||||||
Income tax (expense)/benefit recognized in the consolidated statements of profit or loss |
| $ | |
| $ | |
| $ | ( |
| (1) | Applicable tax rates are |
During 2022, argenx Benelux BV transferred certain pipeline activities to argenx BV through a transfer of assets, (hereafter referred to as “asset deal”), for a total amount of $
The available deferred tax assets relates to argenx US Inc., argenx UK Ltd and argenx Japan KK which are profitable due to the global transfer pricing model of argenx, and the deferred tax liabilities are related to argenx BV. The amount of deferred tax assets and liability by type of temporary difference can be detailed as follow:
At December 31, 2023 | |||||||||
(in thousands of $) |
| Assets | Liabilities |
| Net | ||||
Deferred tax assets / (liabilities) | |||||||||
Accruals and allowances | $ | | $ | — | $ | | |||
Income tax benefit from excess tax deductions related to share-based payments | | — | | ||||||
Profit in inventory | | — | | ||||||
Other tax carryforwards | | — | | ||||||
Property, plant and equipment | | ( | | ||||||
Non-current fixed assets | — | ( | ( | ||||||
Other | | — | | ||||||
Netting by taxable entity | ( | | | ||||||
Net deferred tax assets / (liabilities) |
| $ | |
| $ | ( |
| $ | |
F-38
At December 31, 2022 | |||||||||
(in thousands of $) |
| Assets | Liabilities |
| Net | ||||
Deferred tax assets / (liabilities) | |||||||||
Accruals and allowances | $ | | $ | — | $ | | |||
Income tax benefit from excess tax deductions related to share-based payments | | — | | ||||||
Profit in inventory | | — | | ||||||
R&D capitalized expense | | — | | ||||||
Property, plant and equipment | | ( | | ||||||
Intangible assets | — | ( | ( | ||||||
Non-current fixed assets | — | ( | ( | ||||||
Other | | — | | ||||||
Netting by taxable entity | ( | | — | ||||||
Net deferred tax assets / (liabilities) |
| $ | |
| $ | ( |
| $ | |
At December 31, 2021 | |||||||||
(in thousands of $) |
| Assets | Liabilities |
| Net | ||||
Deferred tax assets / (liabilities) | |||||||||
Accruals and allowances | $ | | $ | — | $ | | |||
Income tax benefit from excess tax deductions related to share-based payments | | — | | ||||||
Profit in inventory | | — | | ||||||
Property, plant and equipment | | ( | ( | ||||||
Intangible assets | — | ( | ( | ||||||
Non-current fixed assets | — | ( | ( | ||||||
Other | | — | | ||||||
Netting by taxable entity | ( | | — | ||||||
Net deferred tax assets / (liabilities) |
| $ | |
| $ | ( |
| $ | |
The change in net deferred taxes recorded in the consolidated statements of financial position can be detailed as follows:
(in thousands of $) |
| Deferred tax assets |
| Deferred tax liabilities | ||
Balance at January 1, 2023 |
| $ | |
| $ | ( |
Recognized in profit or loss | | | ||||
Recognized in equity | | — | ||||
Effects of change in foreign exchange rate | ( | ( | ||||
Balance at December 31, 2023 | $ | | $ | ( | ||
(in thousands of $) |
| Deferred tax assets |
| Deferred tax liabilities | ||
Balance at January 1, 2022 |
| $ | |
| $ | ( |
Recognized in profit or loss | | ( | ||||
Recognized in equity | ( | — | ||||
Effects of change in foreign exchange rate | ( | | ||||
Balance at December 31, 2022 | $ | | $ | ( | ||
F-39
(in thousands of $) |
| Deferred tax assets |
| Deferred tax liabilities | ||
Balance at January 1, 2021 |
| $ | |
| $ | ( |
Recognized in profit or loss | | ( | ||||
Recognized in equity | | — | ||||
Effects of change in foreign exchange rate | | | ||||
Balance at December 31, 2021 | $ | | $ | ( | ||
The unrecognized deferred tax asset on unused tax losses amounts to $
As a company active in research and development in Belgium, we expect to benefit from the innovation income deduction, or IID, in Belgium. The innovation income deduction regime allows net profits attributable to revenue from among others patented products to be taxed at a lower effective tax rate than other revenues. At the end of 2023, 2022 and 2021, we had $
Also, the unrecognized deferred tax asset on the excess depreciations on R&D costs in Belgium amounts to $
Additionally, argenx BV has unrecognized deferred tax asset amounting to $
As of December 31, 2023, the Company had an estimated $
25. | Loss per share |
Year Ended December 31, | |||||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 | |||
Loss for the year |
| $ | ( |
| $ | ( |
| $ | ( |
Weighted average number of shares outstanding |
| |
| |
| | |||
Basic and diluted (loss) per share (in $) |
| $ | ( |
| $ | ( |
| $ | ( |
Earnings/losses per ordinary share are calculated by dividing the loss for the period by the weighted average number of ordinary shares during the year.
As the Company reported a net loss in 2023, 2022 and 2021, stock options and RSUs have an anti-dilutive effect rather than a dilutive effect. As such, there is no difference between basic and diluted loss per ordinary share.
26. | Financial risk management |
The financial risks are managed centrally. The Company coordinates the access to national and international financial markets and considers and manages continuously the financial risks concerning the Company’s activities. These relate to the financial markets risk, credit risk, liquidity risk and currency risk. There are no other important risks,
F-40
such as interest rate risk on borrowings, as the Company has no financial debt. The Company does not buy or trade financial instruments for speculative purposes.
Categories of financial assets and liabilities:
Measurement category | Carrying amount | ||||||||||
At December 31, | |||||||||||
(in thousands of $) |
|
| 2023 | 2022 |
| 2021 | |||||
Financial assets — non-current |
| FVTPL |
| $ | | $ | |
| $ | | |
Financial assets — non-current | FVTOCI | | | | |||||||
Research and development incentive receivables — non-current |
| Amortised cost | | |
| | |||||
Restricted cash — non-current |
| Amortised cost | | |
| | |||||
Trade and other receivables | Amortised cost | | |
| | ||||||
Financial assets—current | FVTPL | — | | | |||||||
Financial assets—current | Amortised cost | | | | |||||||
Research and development incentive receivables — current | Amortised cost | | | — | |||||||
Cash and bank balances |
| Amortised cost | | |
| | |||||
Cash equivalents | FVTPL | | | | |||||||
Cash equivalents | Amortised cost | | | | |||||||
Trade and other payables | Amortised cost | | |
| | ||||||
The carrying amounts of trade and other payables and trade and other receivables are considered to be the same as their fair values, due to their short-term nature.
Financial assets held at fair value through profit or loss or OCI
Financial assets held at fair value through profit or loss or OCI consisted of equity instruments of listed and non-listed companies and money market funds.
The Company has no restrictions on the sale of these equity instruments and the assets are not pledged under any of its liabilities. These instruments are classified as financial assets held at fair value through profit or loss or OCI which qualify for:
| ● | Level 1 fair value measurement with respect to current financial assets and cash equivalents based upon the closing price (net asset value) of such securities at each reporting date. |
| ● | Level 3 fair value measurement with respect to non-current financial assets. |
The market price of these financial instruments might face fluctuations and might be affected by a variety of factors, such as the global economic situation. Current financial assets and cash equivalents include collective investment funds nominated in € and $ of which the underlying investments include bonds and other international debt securities. Based on the weighted average maturity of the underlying instruments, amongst others, these investments are either classified as current financial assets or cash equivalents.
The maximum exposure to credit risk is the carrying amount at reporting date.
F-41
The Company carried the following assets at fair value on December 31, 2023, 2022 and 2021 respectively:
At December 31, 2023 | |||||||||
(in thousands of $) |
| Level 1 |
| Level 2 |
| Level 3 | |||
Non-current financial assets |
| $ | |
| $ | — |
| $ | |
Cash and cash equivalents | | — | — | ||||||
Assets carried at fair value |
| $ | |
| $ | — |
| $ | |
At December 31, 2022 | |||||||||
(in thousands of $) |
| Level 1 |
| Level 2 |
| Level 3 | |||
Non-current financial assets |
| $ | |
| $ | — |
| $ | |
Current financial assets |
| |
| — |
| — | |||
Cash and cash equivalents | | — | — | ||||||
Assets carried at fair value |
| $ | |
| $ | — |
| $ | |
At December 31, 2021 | |||||||||
(in thousands of $) |
| Level 1 |
| Level 2 |
| Level 3 | |||
Non-current financial assets |
| $ | |
| $ | — |
| $ | |
Current financial assets |
| |
| — |
| — | |||
Cash and cash equivalents | | — | — | ||||||
Assets carried at fair value |
| $ | |
| $ | — |
| $ | |
During the disclosed calendar year,
Non-current financial assets – Level 3
In March 2019, the Company entered into a license agreement with AgomAb Therapeutics NV for the use of HGF-mimetic SIMPLE Antibodies™, developed under the Company’s Immunology Innovative Program. In exchange for granting this license, the Company received a profit share in AgomAb Therapeutics NV.
In March 2021, AgomAb Therapeutics NV secured $
In June 2022, AgomAb Therapeutics NV secured €
In October 2023, AgomAb Therapeutics NV secured $
Non-current financial assets – Level 1
In January 2021, as part of the license agreement for the development and commercialization for efgartigimod in Greater China (see note 16 for further information), the Company obtained, amongst others,
F-42
Capital risk
The Company manages its capital to ensure that it will be able to continue as a going concern. The capital structure of the Company consists of equity attributed to the holders of equity instruments of the Company, such as capital, reserves and accumulated losses as mentioned in the consolidated statements of changes in equity. The Company makes the necessary adjustments in the light of changes in the economic circumstances, risks associated to the different assets and the projected cash needs of the current and projected research activities. On December 31, 2023, cash and cash equivalents amounted to $
Credit risk
Credit risk refers to the risk that a counterparty will default on its contractual obligations resulting in financial loss to the Company. The Company has adopted a policy of only dealing with creditworthy counterparties and obtaining sufficient collateral, where appropriate, as a means of mitigating the risk of financial loss from defaults. Concentrations in credit risk are determined based on an analysis of counterparties and their importance on the overall outstanding contractual obligations at year-end.
The Company has a limited number of collaboration and license partners and therefore has a significant concentration of credit risk. However, it has policies in place to ensure that credit exposure is kept to a minimum and significant concentrations of credit exposure are only granted for short periods of time to high credit quality collaboration partners.
The Company applied the IFRS 9 simplified approach to measuring expected credit losses which uses a lifetime expected loss allowance for all receivables. To measure the expected credit losses, receivables have been grouped based on credit risk characteristics and the days past due. The provision for expected credit losses was not significant given that there have been no credit losses over the last three years and the high quality nature of the Company’s customers.
Cash and cash equivalents and current financial assets are invested with several highly reputable banks and financial institutions. The Company holds its cash and cash equivalents mainly with different banks which are independently rated with a minimum rating of ‘A-’. The Company also holds cash equivalents in the form of money market funds with a recommended investment horizon of
Liquidity risk
The Company manages liquidity risk by maintaining adequate reserves, by continuously monitoring forecast and actual cash flows, and by matching the maturity profiles of financial assets and liabilities.
The Company’s main sources of cash inflows are obtained through sale of commercial product, capital increases and collaboration agreements. This cash is invested in savings accounts, term accounts and short term investment funds in the form of money market funds. These money market funds represent the majority of the Company’s available sources of liquidity. Since all of these are immediately tradable and convertible in cash they have a no impact on the liquidity risk.
Interest rate risk
The only variable interest-bearing financial instruments are cash and cash equivalents and current financial assets. Changes in interest rates may cause variations in interest income and expense resulting from short-term interest-bearing assets. Interest rate cuts may have a negative impact on the interest income of the Company.
F-43
For the year ended December 31, 2023, if applicable interest rates would increase/decrease by
Foreign exchange risk
The Company undertakes transactions denominated in foreign currencies, causing exposures to exchange rate fluctuations. The Company is mainly exposed to the Euro, Japanese yen, British pound and Swiss franc. To limit this risk, the Company attempts to align incoming and outgoing cash flows in currencies other than USD.
The net exposure to exchange differences of the monetary assets (being cash, cash equivalents and current financial assets) of the Company at the end of the reporting period are as follows:
At December 31, | ||||||
(in thousands of $) |
| 2023 |
| 2022 |
| 2021 |
EUR |
| |
| |
| |
JPY | | | | |||
GBP |
| |
| |
| |
CHF | | | | |||
CAD | | | — | |||
SEK | | | — | |||
DKK |
| |
| | — | |
On December 31, 2023, if the EUR would have versus the USD by
27. | Related party transactions |
27.1 | Relationship and transactions with joint venture entity |
In July 2022, the Company entered into a joint venture agreement with the University of Colorado Anschutz Medical Campus and UCHealth and created a separate legal entity, OncoVerity, Inc., which is focused on optimizing and advancing the development of cusatuzumab, a novel anti-CD70 antibody, in AML. The Company contributed $
27.2 | Relationship and transactions with subsidiaries |
See note 31 for an overview of the consolidated companies of the group, which are all wholly-owned subsidiaries of argenx SE.
Balances and transactions between the Company and its subsidiaries, which are related parties of the Company, have been eliminated on consolidation and are not disclosed in this note.
27.3 | Relationship and transactions with key personnel |
The Company’s key management personnel consists of the members of the management team and the members of the board of directors.
F-44
Remuneration of key management personnel
On December 31, 2023, the senior management consisted of 8 members: Chief Executive Officer, Chief Operating Officer, Chief Financial Officer, Chief Scientific Officer, General Counsel, Chief Medical Officer, Vice President Corporate Development and Strategy and Global Head of Quality Assurance. They provide their services on a full-time basis.
On December 31, 2023, the board of directors consisted of 9 members: Peter Verhaeghe, Don deBethizy, Pamela M. Klein, A.A. Rosenberg, James M. Daly, Camilla Sylvest, Ana Cespedes, Steve Krognes and Tim Van Hauwermeiren.
Only the Chief Executive Officer is a member of both the senior management team and the board of directors. The Chief Executive Officer does not receive any remuneration for his board membership, as this is part of his total remuneration package in his capacity as member of the senior management team.
The remuneration package of the members of key management personnel comprises:
| Year Ended December 31, | ||||||||
(in thousands of $, except for the number of stock options & RSUs) |
| 2023 |
| 2022 |
| 2021 | |||
Remuneration of key management personnel |
|
|
| ||||||
Short-term benefits for senior management members as a group |
|
|
| ||||||
Gross salary | $ | | $ | | $ | | |||
Variable pay | | | | ||||||
Employer social security | | | | ||||||
Other short term benefits | | | | ||||||
Termination Benefits | — | — | | ||||||
Post-employment benefits for senior management members as a group | | | | ||||||
Cost of stock options granted in the year for senior management members as a group | | | | ||||||
Cost of restricted stock units granted in the year for senior management members as a group | | | | ||||||
Employer social security cost related to stock options | ( | | | ||||||
Total benefits for key management personnel | | | | ||||||
Numbers of stock options granted in the year | |||||||||
Senior Management as a group | |||||||||
Numbers of restricted stock units granted in the year | |||||||||
Senior Management as a group | | | | ||||||
Remuneration of non-executive directors | |||||||||
Board fees and other short-term benefits for non-executive directors | | | | ||||||
Cost of stock options granted in the year for non-executive directors | | | | ||||||
Cost of restricted stock units granted in the year for non-executive directors | | | | ||||||
Total benefits for non-executive board members | $ | | $ | | $ | | |||
Numbers of stock options granted in the year | |||||||||
Non-executive directors | | | | ||||||
Numbers of restricted stock units granted in the year | |||||||||
Non-executive directors | | | | ||||||
F-45
Other
No loans, quasi-loans or other guarantees were given by the Company or any of its subsidiaries to members of the board of directors or the senior management. We have not entered into transactions with the Company’s key management personnel, other than as described above with respect to remuneration arrangements relating to the exercise of their mandates as members of the senior management and the board of directors.
28. | Contingencies |
The Company is currently not facing any outstanding claims or litigations that may have a significant adverse impact on the Company’s consolidated financial position.
29. | Commitments |
At balance sheet date, there were
In February 2019, and as amended in September 2020, the Company entered into a global collaboration and license agreement with Halozyme Therapeutics, Inc. Under the terms of the agreement, the Company will pay up to $
The Company’s manufacturing commitments with Lonza, its drug substance manufacturing contractor, relate to the ongoing execution of the biologic license application (BLA) services for efgartigimod and its manufacturing activities related to the potential future commercialisation. In December 2018, the Company signed its first commercial supply agreement with Lonza related to the reservation of commercial drug substance supply capacity for efgartigimod. In the aggregate, the Company has outstanding commitments for efgartigimod under the commercial supply agreements of $
During 2022, Company signed an agreement with Fujifilm, for activities relating to the large-scale manufacturing of efgartigimod drug substance. In the aggregate, the Company has outstanding commitments for efgartigimod under the commercial supply agreement of $
As of December 31, 2023, the Company had a line of credit totalling to $
30. | Audit fees |
The following auditors’ fees were expensed in the consolidated statements of profit or loss and the consolidated statements of other comprehensive income (loss):
Year Ended December 31, | |||||||||
Fees |
| 2023 |
| 2022 |
| 2021 | |||
in thousands of $ | |||||||||
Audit fees (1) |
| $ | |
| $ | |
| $ | |
Audit-related fees |
| |
| |
| | |||
Tax fees (2) |
| — |
| — |
| | |||
Total |
| $ | |
| $ | |
| $ | |
| (1) | Auditor services performed by Deloitte Accountants B.V. as the external auditor referred to in Section 1 of the Dutch Accounting Firms Oversight Act (Wta) as well as by the Deloitte network. |
| (2) | Tax services performed by the Deloitte network. |
F-46
31. | Overview of consolidation scope |
The parent company argenx SE is domiciled in the Netherlands. The Company, argenx SE, has
Name |
| Country |
| Participation |
|
argenx SE | The Netherlands | | % | ||
argenx BV |
| Belgium |
| | % |
argenx Benelux BV | Belgium | | % | ||
argenx US, Inc. |
| USA |
| | % |
argenx Switzerland, SA | Switzerland | | % | ||
argenx Japan KK |
| Japan |
| | % |
argenx France SAS | France | | % | ||
argenx Germany GmbH | Germany | | % | ||
argenx Canada Inc. | Canada | | % | ||
argenx UK Ltd. | United Kingdom | | % | ||
argenx Netherlands Services B.V. | The Netherlands | | % | ||
argenx Italy S.r.l. | Italy | | % | ||
argenx Spain S.L. | Spain | | % |
32. | Events after the balance sheet date |
No events have occurred after the balance sheet date that could have a material impact on the consolidated financial statements.
F-47