UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-Q
| QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the quarterly period ended March 31, 2024
OR
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
Commission File Number 0-19311

(Exact name of registrant as specified in its charter)
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |||||||
(617 ) 679-2000
(Address, including zip code, and telephone number, including
area code, of registrant’s principal executive offices)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | |||||||||||||||
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days: Yes x No o
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files): Yes x No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and "emerging growth company" in Rule 12b-2 of the Exchange Act:
| x | Accelerated filer | ☐ | ||||||||||||
| Non-accelerated filer | ☐ | Smaller reporting company | ||||||||||||
| Emerging growth company | ||||||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. o
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No x
The number of shares of the issuer’s Common Stock, $0.0005 par value, outstanding as of April 23, 2024, was 145,596,895 shares.
BIOGEN INC.
FORM 10-Q — Quarterly Report
For the Quarterly Period Ended March 31, 2024
TABLE OF CONTENTS
| Page | ||||||||
PART I — FINANCIAL INFORMATION | ||||||||
| Item 1. | ||||||||
| Item 2. | ||||||||
| Item 3. | ||||||||
| Item 4. | ||||||||
PART II — OTHER INFORMATION | ||||||||
| Item 1. | ||||||||
| Item 1A. | ||||||||
| Item 2. | ||||||||
| Item 5. | ||||||||
| Item 6. | ||||||||
2
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This report contains forward-looking statements that are being made pursuant to the provisions of the Private Securities Litigation Reform Act of 1995 (the Act) with the intention of obtaining the benefits of the “Safe Harbor” provisions of the Act. These forward-looking statements may be accompanied by such words as “aim,” “anticipate,” “believe,” “could,” "contemplate," "continue," “estimate,” “expect,” “forecast,” "goal," “intend,” “may,” “plan,” “potential,” “possible,” "predict," "project", "should," "target," “will,” “would” or the negative of these words or other words and terms of similar meaning. Reference is made in particular to forward-looking statements regarding:
•the anticipated amount, timing and accounting of revenue; contingent, milestone, royalty and other payments under licensing, collaboration, acquisition or divestiture agreements; tax positions and contingencies; collectability of receivables; pre-approval inventory; cost of sales; research and development costs; compensation and other selling, general and administrative expense; amortization of intangible assets; foreign currency exchange risk; estimated fair value of assets and liabilities; and impairment assessments;
•expectations, plans and prospects relating to product approvals, sales, pricing, growth, reimbursement and launch of our marketed and pipeline products;
•the potential impact of increased product competition in the markets in which we compete, including increased competition from new originator therapies, generics, prodrugs and biosimilars of existing products and products approved under abbreviated regulatory pathways, including generic or biosimilar versions of our products or competing products;
•patent terms, patent term extensions, patent office actions and expected availability and periods of regulatory exclusivity;
•our plans and investments in our portfolio as well as implementation of our corporate strategy;
•the execution of our strategic and growth initiatives, including the ultimate success of our acquisition of Reata and our ability to realize the anticipated benefits from the acquisition, including future performance of the SKYCLARYS product and anticipated synergies, as well as the exploration of strategic options for our biosimilars business;
•the drivers for growing our business, including our plans and intention to commit resources relating to discovery, research and development programs and business development opportunities as well as the potential benefits and results of, and the anticipated completion of, certain business development transactions and cost-reduction measures, including our Fit for Growth program;
•the expectations, development plans and anticipated timelines, including costs and timing of potential clinical trials, regulatory filings and approvals, of our products, drug candidates and pipeline programs, including collaborations with third-parties, as well as the potential therapeutic scope of the development and commercialization of our and our collaborators’ pipeline products;
•the timing, outcome and impact of administrative, regulatory, legal and other proceedings related to our patents and other proprietary and intellectual property rights, tax audits, assessments and settlements, pricing matters, sales and promotional practices, product liability, investigations and other matters;
•our ability to finance our operations and business initiatives and obtain funding for such activities;
•adverse safety events involving our marketed or pipeline products, generic or biosimilar versions of our marketed products or any other products from the same class as one of our products;
•the current and potential impacts of geopolitical tensions, acts of war and other large-scale crises, including impacts to our operations, sales and the possible disruptions or delay in our plans to conduct clinical trial activities in areas of geopolitical tension, including regions affected by Russia's invasion of Ukraine and the military conflict in the Middle East;
•the direct and indirect impact of global health outbreaks on our business and operations, including sales, expense, reserves and allowances, the supply chain, manufacturing, research and development costs, clinical trials and employees;
•our use of information systems and data and the potential impacts of any breakdowns, invasions, corruptions, destructions and/or breaches of such systems or those of our business partners;
3
•the potential impact of healthcare reform in the U.S., including the IRA, and measures being taken worldwide designed to reduce healthcare costs and limit the overall level of government expenditures, including the impact of pricing actions and reduced reimbursement for our products;
•our manufacturing capacity, use of third-party contract manufacturing organizations, plans and timing relating to changes in our manufacturing capabilities, activities in new or existing manufacturing facilities and the expected timeline for the gene therapy manufacturing facility in RTP, North Carolina to be operational;
•the impact of the continued uncertainty of the credit and economic conditions in certain countries and our collection of accounts receivable in such countries;
•lease commitments, purchase obligations and the timing and satisfaction of other contractual obligations; and
•the impact of new laws (including tax), regulatory requirements, judicial decisions and accounting standards.
These forward-looking statements involve risks and uncertainties, including those that are described in Item 1A. Risk Factors included in this report and elsewhere in this report, that could cause actual results to differ materially from those reflected in such statements. Because some of these risks and uncertainties cannot be predicted or quantified and some are beyond our control, you should not rely on our forward-looking statements as predictions of future events and you should not place undue reliance on these statements. Moreover, we operate in a very competitive and rapidly changing environment, new risks and uncertainties may emerge from time to time and it is not possible for us to predict all risks nor identify all uncertainties. Forward-looking statements speak only as of the date of this report and are based on information and estimates available to us at this time. Except as required by law, we do not undertake any obligation to publicly update any forward-looking statements, whether as a result of new information, future developments or otherwise. You should read this report with the understanding that our actual future results, performance, events and circumstances might be materially different from what we expect.
NOTE REGARDING COMPANY AND PRODUCT REFERENCES
References in this report to:
•“Biogen,” the “company,” “we,” “us” and “our” refer to Biogen Inc. and its consolidated subsidiaries; and
•“RITUXAN” refers to both RITUXAN (the trade name for rituximab in the U.S., Canada and Japan) and MabThera (the trade name for rituximab outside the U.S., Canada and Japan).
NOTE REGARDING TRADEMARKS
ADUHELM®, AVONEX®, BYOOVIZ®, PLEGRIDY®, QALSODY®, RITUXAN®, RITUXAN HYCELA®, SKYCLARYS®, SPINRAZA®, TECFIDERA®, TYSABRI® and VUMERITY® are registered trademarks of Biogen.
BENEPALI™, FLIXABI™, FUMADERM™, IMRALDI™ and TOFIDENCE™ are trademarks of Biogen.
ACTEMRA®, COLUMVI®, ENBREL®, EYLEA®, FAMPYRA™, GAZYVA®, LEQEMBI®, HUMIRA®, LUCENTIS®, LUNSUMIO®, OCREVUS®, REMICADE®, ZURZUVAE™ and other trademarks referenced in this report are the property of their respective owners.
4
DEFINED TERMS
| 2023 Form 10-K | Annual Report on Form 10-K for the year ended December 31, 2023 | ||||
| 2020 Share Repurchase Program | Board of Directors authorized program to repurchase up to $5.0 billion of our common stock | ||||
AbbVie | AbbVie Inc. | ||||
| Acorda | Acorda Therapeutics, Inc. | ||||
| AI | Artificial Intelligence | ||||
| Alkermes | Alkermes plc | ||||
| ALS | Amyotrophic Lateral Sclerosis | ||||
| AOCI | Accumulated Other Comprehensive Income (Loss) | ||||
| ASU | Accounting Standards Update | ||||
| ATV | Antibody Transport Vehicle | ||||
| BLA | Biologics License Application | ||||
| Blackstone | Blackstone Life Sciences | ||||
| CCPA | California Consumer Privacy Act | ||||
| CHMP | Committee for Medicinal Products for Human Use | ||||
CISA | Cybersecurity and Infrastructure Security Agency | ||||
| CJEU | Court of Justice of the European Union | ||||
| CLE | Cutaneous Lupus Erythematosus | ||||
| CLL | Chronic Lymphocytic Leukemia | ||||
| CLO | Chief Legal Officer | ||||
CODM | Chief Operating Decision Maker | ||||
| Convergence | Convergence Pharmaceuticals Ltd. | ||||
| CRL | Complete Response Letter | ||||
| CROs | Contract Research Organizations | ||||
DEA | Drug Enforcement Agency | ||||
| Denali | Denali Therapeutics Inc. | ||||
District Court | U.S. District Court for the District of Massachusetts | ||||
| DOJ | U.S. Department of Justice | ||||
| EC | European Commission | ||||
| Eisai | Eisai Co., Ltd. | ||||
| EMA | European Medicines Agency | ||||
| EPO | European Patent Office | ||||
| ERM | Enterprise Risk Management | ||||
| E.U. | European Union | ||||
| FA | Friedreich's Ataxia | ||||
| FASB | Financial Accounting Standards Board | ||||
| FCPA | Foreign Corrupt Practices Act | ||||
| FDA | U.S. Food and Drug Administration | ||||
| FDIC | Federal Deposit Insurance Corporation | ||||
| Fit for Growth | Cost saving program initiated in 2023 | ||||
FSS | Federal Supply Schedule | ||||
| Genentech | Genentech, Inc. | ||||
| GILTI | Global Intangible Low Tax Income | ||||
| GloBE | Global Anti-Base Erosion | ||||
| GMP | Good Manufacturing Practices | ||||
5
DEFINED TERMS (continued)
| Humana | Humana Inc. | ||||
| IPR&D | In-process Research and Development | ||||
| Ionis | Ionis Pharmaceuticals Inc. | ||||
| IRA | Inflation Reduction Act of 2022 | ||||
| IT | Information Technology | ||||
| IV | Intravenous | ||||
| LRRK2 | Leucine-Rich Repeat Kinase 2 | ||||
| MAA | Marketing Authorization Application | ||||
| MDD | Major Depressive Disorder | ||||
| MS | Multiple Sclerosis | ||||
| Mylan Ireland | Mylan Ireland Ltd. | ||||
| NCD | National Coverage Decision | ||||
| NDA | New Drug Application | ||||
| Neurimmune | Neurimmune SubOne AG | ||||
| NMPA | National Medicinal Products Administration | ||||
| OECD | Organization for Economic Co-operation and Development | ||||
| OIE | Other (Income) Expense, Net | ||||
| Polpharma | Polpharma Biologics S.A. | ||||
| PPACA | Patient Protection and Affordable Care Act | ||||
| PPD | Postpartum Depression | ||||
| PPMS | Primary Progressive MS | ||||
| PRV | Priority Review Voucher | ||||
| R&D | Research and Development | ||||
| Reata | Reata Pharmaceuticals, Inc. | ||||
| RMS | Relapsing MS | ||||
| RRMS | Relapsing-Remitting MS | ||||
| RTP | Research Triangle Park | ||||
| Sage | Sage Therapeutics, Inc. | ||||
| Samsung Bioepis | Samsung Bioepis Co., Ltd. | ||||
| Samsung BioLogics | Samsung BioLogics Co., Ltd. | ||||
| Sangamo | Sangamo Therapeutics, Inc. | ||||
SEC | U.S. Securities and Exchange Commission | ||||
| SG&A | Selling, General and Administrative | ||||
| SLE | Systemic Lupus Erythematosus | ||||
| SMA | Spinal Muscular Atrophy | ||||
| SMN | Survival Motor Neuron | ||||
| SOD1 | Superoxide Dismutase 1 | ||||
| SWISSMEDIC | Swiss Agency for Therapeutic Products | ||||
| TBA | Technical Boards of Appeal | ||||
| Transition Toll Tax | A one-time mandatory deemed repatriation tax on accumulated foreign subsidiaries' previously untaxed foreign earnings | ||||
| U.K. | United Kingdom | ||||
| U.S. | United States | ||||
| U.S. GAAP | Accounting Principles Generally Accepted in the U.S. | ||||
VA | Veterans Administration | ||||
6
PART I FINANCIAL INFORMATION
BIOGEN INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF INCOME
(unaudited, in millions, except per share amounts)
| For the Three Months Ended March 31, | |||||||||||
| 2024 | 2023 | ||||||||||
| Revenue: | |||||||||||
| Product, net | $ | $ | |||||||||
| Revenue from anti-CD20 therapeutic programs | |||||||||||
| Contract manufacturing, royalty and other revenue | |||||||||||
| Total revenue | |||||||||||
| Cost and expense: | |||||||||||
| Cost of sales, excluding amortization and impairment of acquired intangible assets | |||||||||||
| Research and development | |||||||||||
| Selling, general and administrative | |||||||||||
| Amortization and impairment of acquired intangible assets | |||||||||||
| Collaboration profit sharing/(loss reimbursement) | |||||||||||
| Restructuring charges | |||||||||||
| Other (income) expense, net | |||||||||||
| Total cost and expense | |||||||||||
| Income before income tax (benefit) expense | |||||||||||
| Income tax (benefit) expense | |||||||||||
| Net income | |||||||||||
| Net income (loss) attributable to noncontrolling interests, net of tax | ( | ||||||||||
| Net income attributable to Biogen Inc. | $ | $ | |||||||||
| Net income per share: | |||||||||||
| Basic earnings per share attributable to Biogen Inc. | $ | $ | |||||||||
| Diluted earnings per share attributable to Biogen Inc. | $ | $ | |||||||||
| Weighted-average shares used in calculating: | |||||||||||
| Basic earnings per share attributable to Biogen Inc. | |||||||||||
| Diluted earnings per share attributable to Biogen Inc. | |||||||||||
See accompanying notes to these unaudited condensed consolidated financial statements.
7
BIOGEN INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME
(unaudited, in millions)
| For the Three Months Ended March 31, | |||||||||||
| 2024 | 2023 | ||||||||||
| Net income (loss) attributable to Biogen Inc. | $ | $ | |||||||||
| Other comprehensive income: | |||||||||||
Unrealized gains (losses) on securities available for sale, net of tax | |||||||||||
Unrealized gains (losses) on cash flow hedges, net of tax | ( | ||||||||||
Unrealized gains (losses) on pension benefit obligation, net of tax | ( | ||||||||||
Currency translation adjustment | ( | ||||||||||
| Total other comprehensive income (loss), net of tax | ( | ( | |||||||||
| Comprehensive income (loss) attributable to Biogen Inc. | |||||||||||
| Comprehensive income (loss) attributable to noncontrolling interests, net of tax | ( | ||||||||||
| Comprehensive income (loss) | $ | $ | |||||||||
See accompanying notes to these unaudited condensed consolidated financial statements.
8
BIOGEN INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED BALANCE SHEETS
(unaudited, in millions, except per share amounts)
| As of March 31, 2024 | As of December 31, 2023 | ||||||||||
| ASSETS | |||||||||||
| Current assets: | |||||||||||
| Cash and cash equivalents | $ | $ | |||||||||
Accounts receivable, net of allowance for doubtful accounts of $ | |||||||||||
| Due from anti-CD20 therapeutic programs | |||||||||||
| Inventory | |||||||||||
| Other current assets | |||||||||||
| Total current assets | |||||||||||
| Property, plant and equipment, net | |||||||||||
| Operating lease assets | |||||||||||
| Intangible assets, net | |||||||||||
| Goodwill | |||||||||||
| Deferred tax asset | |||||||||||
| Investments and other assets | |||||||||||
| Total assets | $ | $ | |||||||||
| LIABILITIES AND EQUITY | |||||||||||
| Current liabilities: | |||||||||||
| Current portion of term loan | $ | $ | |||||||||
| Taxes payable | |||||||||||
| Accounts payable | |||||||||||
| Accrued expense and other | |||||||||||
| Total current liabilities | |||||||||||
| Notes payable and term loan | |||||||||||
| Deferred tax liability | |||||||||||
| Long-term operating lease liabilities | |||||||||||
| Other long-term liabilities | |||||||||||
| Total liabilities | |||||||||||
| Commitments, contingencies and guarantees | |||||||||||
| Equity: | |||||||||||
| Biogen Inc. shareholders’ equity: | |||||||||||
Preferred stock, par value $ | |||||||||||
Common stock, par value $ | |||||||||||
| Additional paid-in capital | |||||||||||
| Accumulated other comprehensive income (loss) | ( | ( | |||||||||
| Retained earnings | |||||||||||
| Treasury stock, at cost | ( | ( | |||||||||
| Total equity | |||||||||||
| Total liabilities and equity | $ | $ | |||||||||
See accompanying notes to these unaudited condensed consolidated financial statements.
9
BIOGEN INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOW
(unaudited, in millions)
| For the Three Months Ended March 31, | |||||||||||
| 2024 | 2023 | ||||||||||
| Cash flow from operating activities: | |||||||||||
| Net income | $ | $ | |||||||||
| Adjustments to reconcile net income to net cash flow from operating activities: | |||||||||||
| Depreciation and amortization | |||||||||||
| Excess and obsolescence charges related to inventory | |||||||||||
| Amortization of inventory step-up | |||||||||||
| Share-based compensation | |||||||||||
| Deferred income taxes | ( | ||||||||||
| (Gain) loss on strategic investments | |||||||||||
| Other | |||||||||||
| Changes in operating assets and liabilities, net of effects of business acquired: | |||||||||||
| Accounts receivable | |||||||||||
| Due from anti-CD20 therapeutic programs | |||||||||||
| Inventory | ( | ||||||||||
| Accrued expense and other current liabilities | ( | ( | |||||||||
| Income tax assets and liabilities | ( | ||||||||||
| Other changes in operating assets and liabilities, net | ( | ( | |||||||||
| Net cash flow provided by (used in) operating activities | |||||||||||
| Cash flow from investing activities: | |||||||||||
| Purchases of property, plant and equipment | ( | ( | |||||||||
| Proceeds from sales and maturities of marketable securities | |||||||||||
| Purchases of marketable securities | ( | ||||||||||
| Acquisitions of intangible assets | ( | ( | |||||||||
| Proceeds from sales of strategic investments | |||||||||||
| Other | ( | ( | |||||||||
| Net cash flow provided by (used in) investing activities | ( | ( | |||||||||
| Cash flow from financing activities: | |||||||||||
| Payments related to issuance of stock for share-based compensation arrangements, net | ( | ( | |||||||||
| Repayment of borrowings and premiums paid | ( | ||||||||||
| Net (distribution) contribution to noncontrolling interest | |||||||||||
| Other | |||||||||||
| Net cash flow provided by (used in) financing activities | ( | ( | |||||||||
| Net increase (decrease) in cash and cash equivalents | ( | ||||||||||
| Effect of exchange rate changes on cash and cash equivalents | ( | ||||||||||
| Cash and cash equivalents, beginning of the period | |||||||||||
| Cash and cash equivalents, end of the period | $ | $ | |||||||||
See accompanying notes to these unaudited condensed consolidated financial statements.
10
BIOGEN INC. AND SUBSIDIARIES
CONDENSED CONSOLIDATED STATEMENTS OF EQUITY
(unaudited, in millions)
| March 31, 2024 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Preferred stock | Common stock | Additional paid-in capital | Accumulated other comprehensive loss | Retained earnings | Treasury stock | Total equity | |||||||||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Balance, December 31, 2023 | $ | $ | $ | $ | ( | $ | ( | $ | ( | $ | |||||||||||||||||||||||||||||||||||||||||||||||||
| Net income | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Other comprehensive income (loss), net of tax | — | — | — | — | — | ( | — | — | — | ( | |||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock under stock option and stock purchase plans | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock under stock award plan | — | — | — | ( | — | — | — | — | ( | ||||||||||||||||||||||||||||||||||||||||||||||||||
| Compensation related to share-based payments | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Other | — | — | — | — | ( | — | — | — | — | ( | |||||||||||||||||||||||||||||||||||||||||||||||||
| Balance, March 31, 2024 | $ | $ | $ | $ | ( | $ | ( | $ | ( | $ | |||||||||||||||||||||||||||||||||||||||||||||||||
| December 31, 2023 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Preferred stock | Common stock | Additional paid-in capital | Accumulated other comprehensive loss | Retained earnings | Treasury stock | Total Biogen Inc. shareholders’ equity | Noncontrolling interests | Total equity | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Balance, December 31, 2022 | $ | $ | $ | $ | ( | $ | ( | $ | ( | $ | $ | ( | $ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Net income | — | — | — | — | — | — | — | — | ( | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other comprehensive income (loss), net of tax | — | — | — | — | — | ( | — | — | — | ( | — | ( | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Capital contribution from noncontrolling interest | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock under stock option and stock purchase plans | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock under stock award plan | — | — | — | ( | — | — | — | — | ( | — | ( | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Compensation related to share-based payments | — | — | — | — | — | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other | — | — | — | — | ( | — | — | — | — | ( | — | ( | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Balance, March 31, 2023 | $ | $ | $ | $ | ( | $ | ( | $ | ( | $ | $ | ( | $ | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
See accompanying notes to these unaudited condensed consolidated financial statements.
11
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited)
Note 1: | Summary of Significant Accounting Policies | ||||
References in these notes to "Biogen," the "company," "we," "us" and "our" refer to Biogen Inc. and its consolidated subsidiaries.
Business Overview
Biogen is a global biopharmaceutical company focused on discovering, developing and delivering innovative therapies for people living with serious and complex diseases worldwide. We have a broad portfolio of medicines to treat MS, have introduced the first approved treatment for SMA, co-developed treatments to address a defining pathology of Alzheimer’s disease and launched the first approved treatment to target a genetic cause of ALS. Through our 2023 acquisition of Reata we market the first and only drug approved in the U.S. and the E.U. for the treatment of Friedreich's Ataxia in adults and adolescents aged 16 years and older. We are focused on advancing our pipeline in neurology, specialized immunology and rare diseases. We support our drug discovery and development efforts through internal research and development programs and external collaborations.
Our marketed products include TECFIDERA, VUMERITY, AVONEX, PLEGRIDY, TYSABRI and FAMPYRA for the treatment of MS; SPINRAZA for the treatment of SMA; SKYCLARYS for the treatment of Friedreich's Ataxia; QALSODY for the treatment of ALS; and FUMADERM for the treatment of severe plaque psoriasis.
We also have collaborations with Eisai on the commercialization of LEQEMBI for the treatment of Alzheimer's disease and Sage on the commercialization of ZURZUVAE for the treatment of PPD and we have certain business and financial rights with respect to RITUXAN for the treatment of non-Hodgkin's lymphoma, CLL and other conditions; RITUXAN HYCELA for the treatment of non-Hodgkin's lymphoma and CLL; GAZYVA for the treatment of CLL and follicular lymphoma; OCREVUS for the treatment of PPMS and RMS; LUNSUMIO for the treatment of relapsed or refractory follicular lymphoma; COLUMVI, a bispecific antibody for the treatment of non-Hodgkin's lymphoma; and have the option to add other potential anti-CD20 therapies, pursuant to our collaboration arrangements with Genentech, a wholly-owned member of the Roche Group.
We commercialize a portfolio of biosimilars of advanced biologics including BENEPALI, an etanercept biosimilar referencing ENBREL, IMRALDI, an adalimumab biosimilar referencing HUMIRA, and FLIXABI, an infliximab biosimilar referencing REMICADE, in certain countries in Europe, as well as BYOOVIZ, a ranibizumab biosimilar referencing LUCENTIS, in the U.S. and certain international markets. We also have exclusive rights to commercialize TOFIDENCE, a tocilizumab biosimilar referencing ACTEMRA. We continue to develop potential biosimilar product SB15, a proposed aflibercept biosimilar referencing EYLEA.
For additional information on our collaboration arrangements, please read Note 19, Collaborative and Other Relationships, to these unaudited condensed consolidated financial statements (condensed consolidated financial statements).
Basis of Presentation
In the opinion of management, our condensed consolidated financial statements include all adjustments, consisting of normal recurring accruals, necessary for a fair statement of our financial statements for interim periods in accordance with U.S. GAAP. The information included in this quarterly report on Form 10-Q should be read in conjunction with our audited consolidated financial statements and the accompanying notes included in our 2023 Form 10-K. Our accounting policies are described in the Notes to Consolidated Financial Statements in our 2023 Form 10-K and updated, as necessary, in this report. The year-end condensed consolidated balance sheet data presented for comparative purposes was derived from our audited financial statements, but does not include all disclosures required by U.S. GAAP. The results of operations for the three months ended March 31, 2024, are not necessarily indicative of the operating results for the full year or for any other subsequent interim period.
We operate as one operating segment, focused on discovering, developing and delivering worldwide innovative therapies for people living with serious neurological and neurodegenerative diseases as well as related therapeutic adjacencies.
12
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Consolidation
Our condensed consolidated financial statements reflect our financial statements, those of our wholly-owned subsidiaries and certain variable interest entities where we are the primary beneficiary. For consolidated entities where we own or are exposed to less than 100.0 % of the economics, we record net income (loss) attributable to noncontrolling interests, net of tax in our condensed consolidated statements of income equal to the percentage of the economic or ownership interest retained in such entities by the respective noncontrolling parties. Intercompany balances and transactions are eliminated in consolidation.
In determining whether we are the primary beneficiary of a variable interest entity, we apply a qualitative approach that determines whether we have both (1) the power to direct the economically significant activities of the entity and (2) the obligation to absorb losses of, or the right to receive benefits from, the entity that could potentially be significant to that entity. We continuously assess whether we are the primary beneficiary of a variable interest entity as changes to existing relationships or future transactions may result in us consolidating or deconsolidating one or more of our collaborators or partners. In November 2023 we terminated the Neurimmune Agreement, which resulted in the deconsolidation of our variable interest entity, Neurimmune. For additional information on the deconsolidation of Neurimmune, please read Note 20, Investments in Variable Interest Entities, to these condensed consolidated financial statements.
Use of Estimates
The preparation of our condensed consolidated financial statements requires us to make estimates, judgments and assumptions that may affect the reported amounts of assets, liabilities, equity, revenue and expense and related disclosure of contingent assets and liabilities. On an ongoing basis we evaluate our estimates, judgments and assumptions. We base our estimates on historical experience and on various other assumptions that we believe are reasonable, the results of which form the basis for making judgments about the carrying values of assets, liabilities and equity and the amount of revenue and expense. Actual results may differ from these estimates.
Significant Accounting Policies
New Accounting Pronouncements
From time to time, new accounting pronouncements are issued by the FASB or other standard setting bodies that we adopt as of the specified effective date. Unless otherwise discussed below, we do not believe that the adoption of recently issued standards have had or may have a material impact on our condensed consolidated financial statements or disclosures.
Climate-Related Disclosures
In March 2024 the SEC issued a final rule under SEC Release No. 33-11275, The Enhancement and Standardization of Climate-Related Disclosures for Investors. This new rule will require large accelerated filers to disclose material climate-related risks that are reasonably likely to have a material impact on its business, results of operations or financial condition. The required information about climate-related risks will also include disclosure of material direct greenhouse gas emissions from operations owned or controlled (Scope 1) and/or material indirect greenhouse gas emissions from purchased energy consumed in owned or controlled operations (Scope 2). Additionally, the new rules will require disclosure within the notes to the financial statements of the effects of severe weather events and other natural conditions and information on any climate-related targets or goals, subject to certain materiality thresholds. The final rule, if adopted, includes a phased-in compliance period which will begin phasing in with our annual report for the year ending December 31, 2025.
In April 2024 the SEC voluntarily stayed implementation of the new climate-related disclosure requirements pending judicial review. We are currently evaluating the potential impact that this new rule will have on our company's disclosures.
13
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Segment Reporting
In November 2023 the FASB issued ASU No. 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosure. This standard requires disclosure of significant segment expenses that are regularly provided to the CODM and included within each reported measure of segment profit or loss, an amount and description of its composition for other segment items to reconcile to segment profit or loss and the title and position of the entity's CODM. The amendments in this update also expand the interim segment disclosure requirements. All disclosure requirements under this standard are also required for public entities with a single reportable segment. This standard is effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024. Early adoption is permitted and the amendments in this update are required to be applied on a retrospective basis. We are currently evaluating the potential impact that this new standard will have on our condensed consolidated financial statements and related disclosures.
Note 2: | Acquisitions | ||||
Reata Pharmaceuticals, Inc.
On September 26, 2023, we completed the acquisition of all of the issued and outstanding shares of Reata, a biopharmaceutical company focused on developing therapeutics that regulate cellular metabolism and inflammation in serious neurologic diseases. As a result of this transaction we acquired SKYCLARYS (omaveloxolone), the first and only drug approved in the U.S. and the E.U. for the treatment of Friedreich's Ataxia in adults and adolescents aged 16 years and older, as well as other clinical and preclinical pipeline programs. The acquisition of Reata is expected to complement our global portfolio of neuromuscular and rare disease therapies. The addition of SKYCLARYS is anticipated to provide potential operating synergies with SPINRAZA and QALSODY.
Under the terms of this acquisition, we paid Reata shareholders $172.50 in cash for each issued and outstanding Reata share, which totaled approximately $6.6 billion. In addition, we agreed to pay approximately $983.9 million in cash for Reata's outstanding equity awards, inclusive of employer taxes, of which approximately $590.5 million was attributable to pre-acquisition services and is therefore reflected as a component of total purchase price paid. Of the $983.9 million paid to Reata's equity award holders, we recognized approximately $393.4 million as compensation attributable to the post-acquisition service period, of which $196.4 million was recognized as a charge to selling, general and administrative expense with the remaining $197.0 million as a charge to research and development expense within our condensed consolidated statements of income for the year ended December 31, 2023. These amounts were associated with the accelerated vesting of stock options and RSUs previously granted to Reata employees and required no future services to vest.
We funded this acquisition through available cash, cash equivalents and marketable securities, supplemented by the issuance of a $1.0 billion term loan under our term loan credit agreement. For additional information on our term loan credit agreement, please read Note 13, Indebtedness, to these condensed consolidated financial statements.
We accounted for this acquisition as a business combination using the acquisition method of accounting in accordance with ASC Topic 805, Business Combinations, and recorded assets acquired and liabilities assumed at their respective fair values as of the acquisition date.
Purchase Price Consideration
Total consideration transferred for the acquisition of Reata is summarized as follows:
| (In millions) | As of September 26, 2023 | |||||||
Cash consideration paid to Reata shareholders(1) | $ | |||||||
Fair value of Reata equity compensation pre-acquisition services and related taxes(2) | ||||||||
| Total consideration | $ | |||||||
(1) Represents cash consideration transferred of $172.50 per outstanding Reata ordinary share based on 38.3 million Reata shares outstanding at closing.
(2) Represents the fair value of Reata stock options and stock units issued to Reata equity award holders and the related taxes attributable to pre-acquisition vesting services.
14
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Preliminary Purchase Price Allocation
The following table summarizes the provisional amounts recognized for assets acquired and liabilities assumed as of the acquisition date, as well as measurement period adjustments made year-to-date to the amounts initially recorded as of the acquisition date on September 26, 2023. The measurement period adjustments summarized below resulted from updates to our valuation assumptions related to the estimated amounts and timing of future cash flows associated with certain intangible assets, updates of our assumptions related to the quantities, selling location and remaining manufacturing and selling costs of acquired inventory, and other assets and liabilities. The related impact to our condensed consolidated statements of income that would have been recognized in previous periods if the adjustments were recognized as of the acquisition date is immaterial.
| (In millions) | Amounts Recognized as of Acquisition Date (as adjusted) March 31, 2024 | |||||||
| Cash and cash equivalents | $ | |||||||
| Accounts receivable | ||||||||
| Inventory | ||||||||
| Other current assets | ||||||||
| Intangible assets: | ||||||||
| Completed technology for SKYCLARYS (U.S.) | ||||||||
| In-process research and development (omaveloxolone) | ||||||||
| Priority review voucher | ||||||||
| Other clinical programs | ||||||||
| Operating lease assets | ||||||||
Accrued expense and other(1) | ( | |||||||
| Debt payable | ( | |||||||
| Contingent payable to Blackstone | ( | |||||||
Deferred tax liability(1) | ( | |||||||
| Operating lease liabilities | ( | |||||||
| Other assets and liabilities, net | ( | |||||||
| Total identifiable net assets | ||||||||
Goodwill(1) | ||||||||
| Total assets acquired and liabilities assumed | $ | |||||||
(1) Includes measurement period adjustments recorded in the first quarter of 2024 that increased accrued expense and other by $4.9 million, deferred tax liability by $4.1 million and goodwill by $9.0 million.
Inventory: Total inventory acquired was approximately $1.3 billion, which reflects a step-up in the fair value of finished goods and work-in-process inventory for SKYCLARYS. The fair value was determined based on the estimated selling price of the inventory, less the remaining manufacturing and selling costs and a normal profit margin on those manufacturing and selling efforts. This fair value step-up adjustment is being amortized to cost of sales within our condensed consolidated statements of income as the inventory is sold, which is expected to be within approximately 3 years from the acquisition date. For the three months ended March 31, 2024, amortization from the fair value step-up adjustment as a result of inventory sold was approximately $44.1 million.
Intangible assets: Intangible assets are comprised of $4.2 billion related to SKYCLARYS commercialization rights in the U.S., $2.3 billion of IPR&D related to the omaveloxolone program outside the U.S., which had not yet received regulatory approval in the E.U. as of the acquisition date, $100.0 million related to a rare pediatric disease priority voucher which may be used to obtain priority review by the FDA for a future regulatory submission or sold to a third party and $40.0 million related to other clinical programs. The estimated fair values of the program related intangible assets were determined using a multi-period excess earnings method, a form of the income approach, utilizing a discount rate of 14.3 % and the estimated fair value of the priority review voucher was based on recent external purchase and sale transactions of similar vouchers.
15
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Our valuation of the SKYCLARYS commercialization rights reflects the assumption that, using an economic consumption model, the related $4.2 billion intangible asset will be amortized over its expected economic life.
Upon SKYCLARYS receiving E.U. regulatory approval in February 2024, we began selling the product in certain countries in Europe, and began amortizing the $2.3 billion IPR&D asset related to the program outside the U.S. over its expected economic life using an economic consumption model.
These fair value measurements were based on significant inputs not observable in the market and thus represent Level 3 fair value measurements.
Leases: We assumed responsibility for a single-tenant, build-to-suit building of approximately 327,400 square feet of office and laboratory space located in Plano, Texas, with an initial lease term of 16 years. We recorded a lease liability of approximately $151.8 million, which represents the net present value of rental expense over the remaining lease term of approximately 15 years, with a corresponding right-of-use asset of approximately $121.2 million, which represents our estimate of the fair value for a market participant of the current rental market in the Dallas, Texas area. Included in our estimate of the market rental rate is the value of any leasehold improvements or tenant allowances related to the building. We do not intend to occupy this building and are evaluating opportunities to sublease the property.
Goodwill: Goodwill was calculated as the excess of the consideration transferred over the net assets recognized and represents the future economic benefits arising from the other assets acquired that could not be individually identified and separately recognized. We recognized goodwill of approximately $473.5 million, which is not deductible for tax purposes. The goodwill recognized from our acquisition of Reata is primarily the result of the deferred tax consequences from the transaction recorded for financial statement purposes.
Acquisition-related expenses: Acquisition-related expense, primarily comprised of regulatory, advisory and legal fees, and other transaction costs, totaled approximately $28.4 million and were recorded within selling, general and administrative expense within our condensed consolidated statements of income for the year ended December 31, 2023.
Assumptions in the Allocations of Purchase Price
The results of operations of Reata, along with the estimated fair values of the assets acquired and liabilities assumed in the Reata acquisition, have been included in our condensed consolidated financial statements since the closing of the Reata acquisition on September 26, 2023.
Our preliminary estimate of the fair value of the specifically identifiable assets acquired and liabilities assumed as of the date of acquisition is subject to the finalization of management's analysis related to certain matters, such as finalizing our assessment of income taxes. The final determination of these fair values will be completed as additional information becomes available but no later than one year from the acquisition date. The final determination may result in asset and liability fair values that are different than the preliminary estimates.
Note 3: | Dispositions | ||||
Sale of Joint Venture Equity Interest in Samsung Bioepis
In April 2022 we completed the sale of our 49.9 % equity interest in Samsung Bioepis to Samsung BioLogics in exchange for total consideration of approximately $2.3 billion. Under the terms of this transaction, we received approximately $1.0 billion in cash at closing, with approximately $1.3 billion in cash to be deferred over two payments. The first deferred payment of $812.5 million was received in April 2023 and the second deferred payment of $437.5 million was received in April 2024.
We elected the fair value option and measured the payments due to us from Samsung BioLogics at fair value. As of March 31, 2024, the estimated fair value of the second deferred payment using a risk-adjusted discount rate of 6.0 % was approximately $436.1 million. This payment has been classified as a Level 3 measurement and is reflected in other current assets within our condensed consolidated balance sheets as of March 31, 2024.
For the three months ended March 31, 2024, we recognized a gain of approximately $6.1 million to reflect the change in fair value related to the second deferred payment due to us. For the three months ended March 31, 2023, we recognized gains of approximately $11.1 million and $6.2 million to reflect the changes in fair value related to
16
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
the first and second deferred payments due to us, respectively. These changes were recorded in other (income) expense, net in our condensed consolidated statements of income.
For additional information on the sale of our equity interest in Samsung Bioepis, please read Note 3, Dispositions, to our consolidated financial statements included in our 2023 Form 10-K.
Note 4: | Restructuring | ||||
2023 Fit for Growth Restructuring Program
In July 2023 we initiated additional cost saving measures as part of our Fit for Growth program to reduce operating costs, while improving operating efficiency and effectiveness. The Fit for Growth program is expected to generate approximately $1.0 billion in gross operating expense savings by the end of 2025, some of which will be reinvested in various initiatives. The Fit for Growth program is currently estimated to include net headcount reductions of approximately 1,000 employees and we expect to incur restructuring charges ranging from approximately $260.0 million to $280.0 million.
Total charges incurred from our 2023 cost saving initiatives are summarized as follows:
| For the Three Months Ended March 31, | |||||||||||||||||||||||
| 2024 | |||||||||||||||||||||||
| (In millions) | Severance Costs | Accelerated Depreciation and Other Costs | Total | ||||||||||||||||||||
| Selling, general and administrative | $ | $ | $ | ||||||||||||||||||||
| Research and development | |||||||||||||||||||||||
| Restructuring charges | |||||||||||||||||||||||
| Total charges | $ | $ | $ | ||||||||||||||||||||
Other Costs: includes costs associated with items such as asset abandonment and write-offs, facility closure costs, pretax gains and losses resulting from the termination of certain leases, employee non-severance expense, consulting fees and other costs.
Reata Integration
Following the close of our Reata acquisition in September 2023, we implemented an integration plan designed to realize operating synergies through cost savings and avoidance. Under this initiative, we estimate we will incur total integration charges ranging from approximately $35.0 million to $40.0 million, related to severance and employment costs, which are expected to be paid by the end of 2024. These amounts were substantially incurred during 2023.
Total charges incurred from our Reata integration are summarized as follows:
| For the Three Months Ended March 31, | |||||||||||||||||||||||
| 2024 | |||||||||||||||||||||||
| (In millions) | Severance Costs | Accelerated Depreciation and Other Costs | Total | ||||||||||||||||||||
| Selling, general and administrative | $ | $ | $ | ||||||||||||||||||||
| Research and development | |||||||||||||||||||||||
| Restructuring charges | |||||||||||||||||||||||
| Total charges | $ | $ | $ | ||||||||||||||||||||
In connection with our acquisition of Reata we assumed responsibility for a single-tenant, build-to-suit building of approximately 327,400 square feet of office and laboratory space located in Plano, Texas, with an initial lease term of 16 years. We do not intend to occupy this building and are evaluating opportunities to sublease the property.
Charges and spending related to workforce reductions from our 2023 Fit for Growth program and Reata Integration are summarized as follows:
17
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Restructuring reserve as of December 31 | $ | $ | ||||||||||||
| Expense | ||||||||||||||
| Payment | ( | ( | ||||||||||||
| Foreign currency and other adjustments | ||||||||||||||
| Restructuring reserve as of March 31 | $ | $ | ||||||||||||
18
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Note 5: | Revenue | ||||
Product Revenue
Revenue by product is summarized as follows:
| For the Three Months Ended March 31, | ||||||||||||||||||||||||||||||||||||||
| 2024 | 2023 | |||||||||||||||||||||||||||||||||||||
| (In millions) | United States | Rest of World | Total | United States | Rest of World | Total | ||||||||||||||||||||||||||||||||
| Multiple Sclerosis: | ||||||||||||||||||||||||||||||||||||||
| TECFIDERA | $ | $ | $ | $ | $ | $ | ||||||||||||||||||||||||||||||||
| VUMERITY | ||||||||||||||||||||||||||||||||||||||
| Total Fumarate | ||||||||||||||||||||||||||||||||||||||
| AVONEX | ||||||||||||||||||||||||||||||||||||||
| PLEGRIDY | ||||||||||||||||||||||||||||||||||||||
| Total Interferon | ||||||||||||||||||||||||||||||||||||||
| TYSABRI | ||||||||||||||||||||||||||||||||||||||
| FAMPYRA | ||||||||||||||||||||||||||||||||||||||
| Subtotal: Multiple Sclerosis | ||||||||||||||||||||||||||||||||||||||
| Rare Disease: | ||||||||||||||||||||||||||||||||||||||
| SPINRAZA | ||||||||||||||||||||||||||||||||||||||
QALSODY(1) | ||||||||||||||||||||||||||||||||||||||
SKYCLARYS(2) | ||||||||||||||||||||||||||||||||||||||
| Subtotal: Rare Disease | ||||||||||||||||||||||||||||||||||||||
| Biosimilars: | ||||||||||||||||||||||||||||||||||||||
| BENEPALI | ||||||||||||||||||||||||||||||||||||||
| IMRALDI | ||||||||||||||||||||||||||||||||||||||
| FLIXABI | ||||||||||||||||||||||||||||||||||||||
BYOOVIZ(3) | ||||||||||||||||||||||||||||||||||||||
| Subtotal: Biosimilars | ||||||||||||||||||||||||||||||||||||||
Other(4) | ||||||||||||||||||||||||||||||||||||||
| Total product revenue | $ | $ | $ | $ | $ | $ | ||||||||||||||||||||||||||||||||
(1) QALSODY became commercially available in the U.S. during the second quarter of 2023.
(2) SKYCLARYS was obtained as part of our acquisition of Reata in September 2023. SKYCLARYS became commercially available in the U.S. during the second quarter of 2023 and we began recognizing revenue from SKYCLARYS in the U.S. during the fourth quarter of 2023, subsequent to our acquisition. SKYCLARYS was approved and became commercially available in the E.U. during the first quarter of 2024.
(3) BYOOVIZ became commercially available in certain international markets in 2023.
(4) Other includes FUMADERM, ADUHELM and ZURZUVAE, which became commercially available in the U.S. during the fourth quarter of 2023.
We recognized revenue from two 25.6 % and 11.7 % of gross product revenue for the three months ended March 31, 2024, and 27.3 % and 7.4 % of gross product revenue for the three months ended March 31, 2023.
19
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
An analysis of the change in reserves for discounts and allowances is summarized as follows:
| (In millions) | Discounts | Contractual Adjustments | Returns | Total | ||||||||||||||||||||||
| Balance, December 31, 2023 | $ | $ | $ | $ | ||||||||||||||||||||||
| Current provisions relating to sales in current year | ||||||||||||||||||||||||||
| Adjustments relating to prior years | ( | ( | ||||||||||||||||||||||||
| Payments/credits relating to sales in current year | ( | ( | ( | ( | ||||||||||||||||||||||
| Payments/credits relating to sales in prior years | ( | ( | ( | ( | ||||||||||||||||||||||
| Balance, March 31, 2024 | $ | $ | $ | $ | ||||||||||||||||||||||
The total reserves above, which are included in our condensed consolidated balance sheets, are summarized as follows:
| (In millions) | As of March 31, 2024 | As of December 31, 2023 | ||||||||||||
| Reduction of accounts receivable | $ | $ | ||||||||||||
| Component of accrued expense and other | ||||||||||||||
| Total revenue-related reserves | $ | $ | ||||||||||||
Revenue from Anti-CD20 Therapeutic Programs
Revenue from anti-CD20 therapeutic programs is summarized in the table below. For the purposes of this footnote, we refer to RITUXAN and RITUXAN HYCELA collectively as RITUXAN.
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Royalty revenue on sales of OCREVUS | $ | $ | ||||||||||||
| Biogen’s share of pre-tax profits in the U.S. for RITUXAN, GAZYVA and LUNSUMIO | ||||||||||||||
| Other revenue from anti-CD20 therapeutic programs | ||||||||||||||
| Total revenue from anti-CD20 therapeutic programs | $ | $ | ||||||||||||
For additional information on our collaboration arrangements with Genentech, please read Note 19, Collaborative and Other Relationships, to our consolidated financial statements included in this report.
Contract Manufacturing, Royalty and Other Revenue
Contract manufacturing, royalty and other revenue is summarized in the table below.
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Contract manufacturing revenue | $ | $ | ||||||||||||
Royalty and other revenue | ( | |||||||||||||
| Total contract manufacturing, royalty and other revenue | $ | $ | ||||||||||||
Contract Manufacturing Revenue
Contract manufacturing revenue primarily reflects amounts earned under contract manufacturing agreements with our strategic customers. During the first quarter of 2023 we began recognizing contract manufacturing revenue for LEQEMBI, upon accelerated approval of LEQEMBI in the U.S. Prior to accelerated approval, our share of contract manufacturing amounts related to LEQEMBI were recognized in research and development expense within our condensed consolidated statements of income.
Royalty and Other Revenue
Royalty and other revenue primarily reflects the royalties we receive from net sales on products related to patents that we have out-licensed, as well as royalty revenue on biosimilar products from our license arrangements with
20
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Samsung Bioepis and our 50.0 % share of LEQEMBI product revenue, net and cost of sales, including royalties, as we are not the principal.
For additional information on our collaboration arrangements with Eisai and our license arrangements with Samsung Bioepis, please read Note 19, Collaborative and Other Relationships, to these condensed consolidated financial statements.
Note 6: | Inventory | ||||
The components of inventory are summarized as follows:
| (In millions) | As of March 31, 2024 | As of December 31, 2023 | ||||||||||||
| Raw materials | $ | $ | ||||||||||||
| Work in process | ||||||||||||||
| Finished goods | ||||||||||||||
| Total inventory | $ | $ | ||||||||||||
| Balance Sheet Classification: | ||||||||||||||
| Inventory | $ | $ | ||||||||||||
| Investments and other assets | ||||||||||||||
| Total inventory | $ | $ | ||||||||||||
Note 7: | Intangible Assets and Goodwill | ||||
Intangible Assets
Intangible assets, net of accumulated amortization, impairment charges and adjustments are summarized as follows:
| As of March 31, 2024 | As of December 31, 2023 | |||||||||||||||||||||||||||||||||||||||||||
| (In millions) | Estimated Life | Cost | Accumulated Amortization | Net | Cost | Accumulated Amortization | Net | |||||||||||||||||||||||||||||||||||||
| Completed technology: | ||||||||||||||||||||||||||||||||||||||||||||
| Acquired and in-licensed rights and patents | $ | $ | ( | $ | $ | $ | ( | $ | ||||||||||||||||||||||||||||||||||||
| Developed technology and other | ( | ( | ||||||||||||||||||||||||||||||||||||||||||
| Total completed technology | ( | ( | ||||||||||||||||||||||||||||||||||||||||||
| In-process research and development | Indefinite until commercialization | — | — | |||||||||||||||||||||||||||||||||||||||||
| Priority review voucher | Indefinite | — | — | |||||||||||||||||||||||||||||||||||||||||
| Trademarks and trade names | Indefinite | — | — | |||||||||||||||||||||||||||||||||||||||||
| Total intangible assets | $ | $ | ( | $ | $ | $ | ( | $ | ||||||||||||||||||||||||||||||||||||
Amortization and Impairments
For the three months ended March 31, 2024, amortization and impairment of acquired intangible assets totaled $78.3 million, compared to $50.2 million in the prior year comparative period. The increase was primarily due to
21
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
amortization for the Reata acquisition acquired intangible assets associated with SKYCLARYS. For the three months ended March 31, 2024 and 2023, we had no impairment charges.
Completed Technology
Completed technology primarily relates to our other marketed products and programs acquired through asset acquisitions, licenses and business combinations. Completed technology intangible assets are amortized over their estimated useful lives, which range between 2 to 31 years, with a remaining weighted average useful life of 12 years for acquired and in-licensed rights and patents and 10 years for developed technology and other. In connection with our acquisition of Reata in September 2023 we acquired SKYCLARYS, a commercially-approved product in the U.S., with an estimated fair value of approximately $4.2 billion, which includes measurement period adjustments. During the first quarter of 2024 SKYCLARYS was approved in the E.U. and became commercially available, which resulted in the reclassification of the related intangible asset, with an estimated fair value of approximately $2.3 billion, from IPR&D to completed technology.
IPR&D Related to Business Combinations
IPR&D represents the fair value assigned to research and development assets that we acquired as part of a business combination and had not yet reached technological feasibility at the date of acquisition. Included in IPR&D balances are adjustments related to foreign currency exchange rate fluctuations. The carrying value associated with our IPR&D assets as of December 31, 2023, related to the IPR&D programs we acquired in connection with our acquisition of Reata in September 2023, with an estimated fair value of approximately $2.3 billion, which includes measurement period adjustments. During the first quarter of 2024 SKYCLARYS was approved in the E.U. and became commercially available, which resulted in the reclassification of the related intangible asset from IPR&D to completed technology.
Priority Review Voucher
In connection with our acquisition of Reata in September 2023 we acquired a rare pediatric disease priority review voucher that may be used to obtain priority review by the FDA for a future regulatory submission or sold to a third party. We recorded the priority review voucher based on its estimated fair value of $100.0 million as an intangible asset. The estimated fair value was based on recent external purchase and sale transactions of similar vouchers.
For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to these condensed consolidated financial statements.
Estimated Future Amortization of Intangible Assets
The estimated future amortization of finite-lived intangible assets for the next five years is expected to be as follows:
| (In millions) | As of March 31, 2024 | |||||||
| 2024 (remaining nine months) | $ | |||||||
| 2025 | ||||||||
| 2026 | ||||||||
| 2027 | ||||||||
| 2028 | ||||||||
| 2029 | ||||||||
Goodwill
The following table provides a roll forward of the changes in our goodwill balance:
| (In millions) | As of March 31, 2024 | |||||||
| Goodwill, December 31, 2023 | $ | |||||||
| Goodwill resulting from Reata acquisition | ||||||||
| Other | ( | |||||||
| Goodwill, March 31, 2024 | $ | |||||||
For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to these condensed consolidated financial statements.
22
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
As of March 31, 2024, we had no accumulated impairment losses related to goodwill. Other includes adjustments related to foreign currency exchange rate fluctuations.
Note 8: | Fair Value Measurements | ||||
The tables below present information about our assets and liabilities that are regularly measured and carried at fair value and indicate the level within the fair value hierarchy of the valuation techniques we utilized to determine such fair value:
| Fair Value Measurements on a Recurring Basis | ||||||||||||||||||||||||||
| As of March 31, 2024 | ||||||||||||||||||||||||||
| (In millions) | Total | Quoted Prices in Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | ||||||||||||||||||||||
| Assets: | ||||||||||||||||||||||||||
| Cash equivalents | $ | $ | $ | $ | ||||||||||||||||||||||
| Marketable equity securities | ||||||||||||||||||||||||||
| Other current assets: | ||||||||||||||||||||||||||
Receivable from Samsung BioLogics(1) | ||||||||||||||||||||||||||
| Derivative contracts | ||||||||||||||||||||||||||
| Other non-current assets: | ||||||||||||||||||||||||||
| Plan assets for deferred compensation | ||||||||||||||||||||||||||
| Total | $ | $ | $ | $ | ||||||||||||||||||||||
| Liabilities: | ||||||||||||||||||||||||||
| Derivative contracts | $ | $ | $ | $ | ||||||||||||||||||||||
| Total | $ | $ | $ | $ | ||||||||||||||||||||||
(1) Represents the fair value of the current payment due from Samsung BioLogics as a result of the sale of our 49.9 % equity interest in Samsung Bioepis to Samsung BioLogics during the second quarter of 2022, for which we elected the fair value option. For additional information on the sale of our equity interest in Samsung Bioepis, please read Note 3, Dispositions, to these condensed consolidated financial statements.
During the third quarter of 2023 we sold all of our marketable debt securities and used the proceeds to partially fund our acquisition of Reata. For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to these condensed consolidated financial statements.
23
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
| Fair Value Measurements on a Recurring Basis | ||||||||||||||||||||||||||
| As of December 31, 2023 | ||||||||||||||||||||||||||
| (In millions) | Total | Quoted Prices in Active Markets (Level 1) | Significant Other Observable Inputs (Level 2) | Significant Unobservable Inputs (Level 3) | ||||||||||||||||||||||
| Assets: | ||||||||||||||||||||||||||
| Cash equivalents | $ | $ | $ | $ | ||||||||||||||||||||||
| Marketable equity securities | ||||||||||||||||||||||||||
| Other current assets: | ||||||||||||||||||||||||||
Receivable from Samsung BioLogics(1) | ||||||||||||||||||||||||||
| Derivative contracts | ||||||||||||||||||||||||||
Other non-current assets: | ||||||||||||||||||||||||||
| Plan assets for deferred compensation | ||||||||||||||||||||||||||
| Total | $ | $ | $ | $ | ||||||||||||||||||||||
| Liabilities: | ||||||||||||||||||||||||||
| Derivative contracts | $ | $ | $ | $ | ||||||||||||||||||||||
| Total | $ | $ | $ | $ | ||||||||||||||||||||||
(1) Represents the fair value of the current payment due from Samsung BioLogics as a result of the sale of our 49.9 % equity interest in Samsung Bioepis to Samsung BioLogics during the second quarter of 2022, for which we elected the fair value option. For additional information on the sale of our equity interest in Samsung Bioepis, please read Note 3, Dispositions, to these condensed consolidated financial statements.
Our marketable equity securities represent investments in publicly traded equity securities. Our ability to liquidate our investments in Denali, Sage and Sangamo may be limited by the size of our interest, the volume of market related activity, our concentrated level of ownership and potential restrictions resulting from our status as a collaborator. Therefore, we may realize significantly less than the current value of such investments.
For additional information on our investments in Denali, Sangamo and Sage common stock, please read Note 19, Collaborative and Other Relationships, to our consolidated financial statements included in our 2023 Form 10-K.
There have been no
For a description of our validation procedures related to prices provided by third-party pricing services and our option pricing valuation model, please read Note 1, Summary of Significant Accounting Policies - Fair Value Measurements, to our consolidated financial statements included in our 2023 Form 10-K.
Level 3 Assets and Liabilities Held at Fair Value
There were no transfers of assets or liabilities into or out of Level 3 as of March 31, 2024 and December 31, 2023.
Financial Instruments Not Carried at Fair Value
Other Financial Instruments
Due to the short-term nature of certain financial instruments, the carrying value reflected in our condensed consolidated balance sheets for current accounts receivable, due from anti-CD20 therapeutic programs, other current assets, accounts payable and accrued expense and other, approximates fair value.
24
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Debt Instruments
The fair and carrying values of our debt instruments, which are Level 2 liabilities, are summarized as follows:
| As of March 31, 2024 | As of December 31, 2023 | |||||||||||||||||||||||||
| (In millions) | Fair Value | Carrying Value | Fair Value | Carrying Value | ||||||||||||||||||||||
| Current portion: | ||||||||||||||||||||||||||
2023 Term Loan 364-day tranche(1) | $ | $ | $ | $ | ||||||||||||||||||||||
2023 Term Loan three-year tranche(1) | ||||||||||||||||||||||||||
| Current portion of notes payable and term loan | ||||||||||||||||||||||||||
| Non-current portion: | ||||||||||||||||||||||||||
2023 Term Loan three-year tranche(1) | ||||||||||||||||||||||||||
| 4.050% Senior Notes due September 15, 2025 | ||||||||||||||||||||||||||
| 2.250% Senior Notes due May 1, 2030 | ||||||||||||||||||||||||||
| 5.200% Senior Notes due September 15, 2045 | ||||||||||||||||||||||||||
| 3.150% Senior Notes due May 1, 2050 | ||||||||||||||||||||||||||
| 3.250% Senior Notes due February 15, 2051 | ||||||||||||||||||||||||||
| Non-current portion of notes payable and term loan | ||||||||||||||||||||||||||
| Total notes payable and term loan | $ | $ | $ | $ | ||||||||||||||||||||||
(1) In connection with our acquisition of Reata we drew $1.0 billion from our 2023 Term Loan, comprised of a $500.0 million floating rate 364-day tranche and a $500.0 million floating rate three-year tranche. For additional information on our 2023 Term Loan, please read Note 13, Indebtedness, to these condensed consolidated financial statements.
The fair values of each of our series of Senior Notes were determined through market, observable and corroborated sources. The changes in the fair values of our Senior Notes as of March 31, 2024, compared to December 31, 2023, are primarily related to increases in U.S. treasury yields partially offset by a decrease in credit spreads used to value our Senior Notes since December 31, 2023. For additional information related to our Senior Notes, please read Note 13, Indebtedness, to our consolidated financial statements included in our 2023 Form 10-K.
Note 9: | Financial Instruments | ||||
The following table summarizes our financial assets with maturities of less than 90 days from the date of purchase included in cash and cash equivalents in our condensed consolidated balance sheets:
| (In millions) | As of March 31, 2024 | As of December 31, 2023 | ||||||||||||
| Money market funds | $ | $ | ||||||||||||
Total | $ | $ | ||||||||||||
The carrying value of our money market funds approximate fair value due to their short-term maturities.
We partially funded our Reata acquisition through available cash, cash equivalents and marketable securities. As of December 31, 2023, we have sold all of our marketable debt securities. For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to these condensed consolidated financial statements.
25
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Our marketable equity securities gains (losses) are recorded in other (income) expense, net in our condensed consolidated statements of income. The following tables summarize our marketable equity securities, classified as available-for-sale:
| As of March 31, 2024 | ||||||||||||||||||||||||||
| (In millions) | Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | ||||||||||||||||||||||
| Marketable equity securities | ||||||||||||||||||||||||||
| Marketable equity securities, current | $ | $ | $ | ( | $ | |||||||||||||||||||||
| Marketable equity securities, non-current | ( | |||||||||||||||||||||||||
| Total marketable equity securities | $ | $ | $ | ( | $ | |||||||||||||||||||||
| As of December 31, 2023 | ||||||||||||||||||||||||||
| (In millions) | Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | ||||||||||||||||||||||
| Marketable equity securities | ||||||||||||||||||||||||||
| Marketable equity securities, current | $ | $ | $ | ( | $ | |||||||||||||||||||||
| Marketable equity securities, non-current | ( | |||||||||||||||||||||||||
| Total marketable equity securities | $ | $ | $ | ( | $ | |||||||||||||||||||||
Proceeds from Marketable Debt Securities
The proceeds from maturities and sales of marketable debt securities and resulting realized gains and losses are summarized as follows:
| (In millions) | For the Three Months Ended March 31, 2023 | |||||||
| Proceeds from maturities and sales | $ | |||||||
| Realized gains | ||||||||
| Realized losses | ||||||||
Realized losses for the three months ended March 31, 2023, primarily relate to sales of U.S. treasuries and corporate bonds.
During the third quarter of 2023 we sold all of our marketable debt securities and used the proceeds to partially fund our acquisition of Reata. For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to these condensed consolidated financial statements.
Strategic Investments
Our strategic investment portfolio includes investments in equity securities of certain biotechnology companies, which are reflected within our disclosures included in Note 8, Fair Value Measurements, to these condensed consolidated financial statements, as well as venture capital funds where the underlying investments are in equity securities of certain biotechnology companies and non-marketable equity securities.
As of March 31, 2024 and December 31, 2023, our strategic investment portfolio was comprised of investments totaling $382.0 million and $460.7 million, respectively, which are included in other current assets and investments and other assets within our condensed consolidated balance sheets.
The decrease in our strategic investment portfolio as of March 31, 2024, was primarily due to the decrease in the fair value of our investments in Sage and Denali common stock. Additionally, during the first quarter of 2024 we sold a portion of our Denali and Sangamo common stock.
For additional information on our strategic investments in Denali, Sangamo and Sage common stock, please read Note 19, Collaborative and Other Relationships, to our consolidated financial statements included in our 2023 Form 10-K.
26
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Note 10: | Derivative Instruments | ||||
Foreign Currency Forward Contracts - Hedging Instruments
Due to the global nature of our operations, portions of our revenue and operating expense are recorded in currencies other than the U.S. dollar. The value of revenue and operating expense measured in U.S. dollars is therefore subject to changes in foreign currency exchange rates. We enter into foreign currency forward contracts and foreign currency options with financial institutions with the primary objective to mitigate the impact of foreign currency exchange rate fluctuations on our international revenue and operating expense.
Foreign currency forward contracts and foreign currency options in effect as of March 31, 2024 and December 31, 2023, had durations of 1 to 9 months and 1 to 12 months, respectively. These contracts have been designated as cash flow hedges and unrealized gains and losses on the portion of these foreign currency forward contracts and foreign currency options that are included in the effectiveness test are reported in AOCI. Realized gains and losses of such contracts and options are recognized in revenue when the sale of product in the currency being hedged is recognized and in operating expense when the expense in the currency being hedged is recorded. We recognize all cash flow hedge reclassifications from AOCI and fair value changes of excluded portions in the same line item in our condensed consolidated statements of income that have been impacted by the hedged item.
The notional amount of foreign currency forward contracts and foreign currency options that were entered into to hedge forecasted revenue and operating expense is summarized as follows:
| Notional Amount | ||||||||||||||
| (In millions) | As of March 31, 2024 | As of December 31, 2023 | ||||||||||||
| Euro | $ | $ | ||||||||||||
| British pound | ||||||||||||||
| Swiss franc | ||||||||||||||
| Canadian dollar | ||||||||||||||
| Total foreign currency forward contracts and options | $ | $ | ||||||||||||
The pre-tax portion of the fair value of these foreign currency forward contracts and foreign currency options that were included in AOCI in total equity is summarized as follows:
| (In millions) | As of March 31, 2024 | As of December 31, 2023 | ||||||||||||
| Unrealized gains | $ | $ | ||||||||||||
| Unrealized (losses) | ( | ( | ||||||||||||
| Net unrealized gains (losses) | $ | ( | $ | ( | ||||||||||
We expect the net unrealized losses of approximately $18.7 million to be settled over the next 9 months, with any amounts in AOCI to be reported as an adjustment to revenue or operating expense. We consider the impact of our and our counterparties’ credit risk on the fair value of the contracts as well as the ability of each party to execute its contractual obligations. As of March 31, 2024 and December 31, 2023, credit risk did not materially change the fair value of our foreign currency forward contracts and forward currency options.
The following table summarizes the effect of foreign currency forward contracts and forward currency options designated as hedging instruments in our condensed consolidated statements of income:
| For the Three Months Ended March 31, | ||||||||||||||||||||||||||||||||
| Net Gains/(Losses) Reclassified from AOCI into Operating Income | Net Gains/(Losses) Recognized in Operating Income | |||||||||||||||||||||||||||||||
| Location | 2024 | 2023 | Location | 2024 | 2023 | |||||||||||||||||||||||||||
| Revenue | $ | $ | Revenue | $ | $ | |||||||||||||||||||||||||||
| Operating expense | ( | ( | Operating expense | ( | ( | |||||||||||||||||||||||||||
27
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Foreign Currency Forward Contracts - Other Derivative Instruments
We also enter into other foreign currency forward contracts, usually with durations of one month or less, to mitigate the foreign currency risk related to certain balance sheet positions. We have not elected hedge accounting for these transactions.
The aggregate notional amount of these outstanding foreign currency forward contracts was $1,308.3 million and $1,301.5 million as of March 31, 2024 and December 31, 2023, respectively. Net losses of $24.3 million and net gains of $1.8 million related to these contracts were recorded as a component of other (income) expense, net for the three months ended March 31, 2024 and 2023, respectively.
Summary of Derivative Instruments
While certain of our derivative instruments are subject to netting arrangements with our counterparties, we do not offset derivative assets and liabilities in our condensed consolidated balance sheets. The amounts in the table below would not be substantially different if the derivative assets and liabilities were offset.
The following table summarizes the fair value and presentation in our condensed consolidated balance sheets of our outstanding derivative instruments, including those designated as hedging instruments:
| (In millions) | Balance Sheet Location | As of March 31, 2024 | As of December 31, 2023 | |||||||||||||||||
| Cash Flow Hedging Instruments: | ||||||||||||||||||||
| Asset derivative instruments | Other current assets | $ | $ | |||||||||||||||||
| Liability derivative instruments | Accrued expense and other | |||||||||||||||||||
| Other Derivative Instruments: | ||||||||||||||||||||
| Asset derivative instruments | Other current assets | |||||||||||||||||||
| Liability derivative instruments | Accrued expense and other | |||||||||||||||||||
Note 11: | Property, Plant and Equipment | ||||
Property, plant and equipment are recorded at historical cost, net of accumulated depreciation. Accumulated depreciation on property, plant and equipment was $2,468.2 million and $2,402.5 million as of March 31, 2024 and December 31, 2023, respectively. For the three months ended March 31, 2024, depreciation expense totaled $69.3 million compared to $62.1 million in the prior year comparative period.
Solothurn, Switzerland Manufacturing Facility
In order to support our future growth and drug development pipeline, we built a large-scale biologics manufacturing facility in Solothurn, Switzerland. This facility includes 393,000 square feet related to a large-scale biologics manufacturing facility, 290,000 square feet of warehouse, utilities and support space and 51,000 square feet of administrative space. As of December 31, 2023, we had approximately $728.8 million capitalized as construction in progress related to this facility. In the second quarter of 2021 a portion of this facility (the first manufacturing suite) received a GMP multi-product license from the SWISSMEDIC and was placed into service. The second manufacturing suite became operational in the first quarter of 2024, resulting in approximately $717.3 million of fixed assets being placed into service. Solothurn has been approved for the manufacture of LEQEMBI by the FDA.
28
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Note 12: | Leases | ||||
6100 Legacy Drive Lease
In connection with our acquisition of Reata we assumed responsibility for a single-tenant, build-to-suit building of approximately 327,400 square feet of office and laboratory space located in Plano, Texas, with an initial lease term of 16 years. We recorded a lease liability of approximately $151.8 million, which represents the net present value of rental expense over the remaining lease term of approximately 15 years, with a corresponding right-of-use asset of approximately $121.2 million, which represents our estimate of the fair value for a market participant of the current rental market in the Dallas, Texas area. Included in our estimate of the market rental rate is the value of any leasehold improvements or tenant allowances related to the building. We do not intend to occupy this building and are evaluating opportunities to sublease the property.
For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to these condensed consolidated financial statements.
Note 13: | Indebtedness | ||||
2023 Term Loan Credit Agreement
In connection with our acquisition of Reata in September 2023 we entered into a $1.5 billion term loan credit agreement (2023 Term Loan). On the closing date of the Reata acquisition we drew $1.0 billion from the 2023 Term Loan, comprised of a $500.0 million floating rate 364-day tranche and a $500.0 million floating rate three-year tranche. The remaining unused commitment of $500.0 million was terminated. As of December 31, 2023, we repaid $350.0 million of the 364--day tranche. The remaining $150.0 million portion of the 364-day tranche was subsequently paid during the first quarter of 2024. Additionally, during the first quarter of 2024 we repaid $250.0 million of the three-year tranche. As of March 31, 2024, we had $250.0 million outstanding under the three-year tranche of the 2023 Term Loan.
Note 14: | Equity | ||||
Share Repurchases
In October 2020 our Board of Directors authorized our 2020 Share Repurchase Program, which is a program to repurchase up to $5.0 billion of our common stock. Our 2020 Share Repurchase Program does not have an expiration date. All share repurchases under our 2020 Share Repurchase Program will be retired. There were no share repurchases of our common stock during the three months ended March 31, 2024 and 2023. Approximately $2.1 billion remained available under our 2020 Share Repurchase Program as of March 31, 2024.
Accumulated Other Comprehensive Income (Loss)
The following tables summarize the changes in AOCI, net of tax by component:
| March 31, 2024 | ||||||||||||||||||||||||||||||||
| (In millions) | Unrealized Gains (Losses) on Securities Available for Sale, Net of Tax | Unrealized Gains (Losses) on Cash Flow Hedges, Net of Tax | Unrealized Gains (Losses) on Pension Benefit Obligation, Net of Tax | Currency Translation Adjustments | Total | |||||||||||||||||||||||||||
| Balance, December 31, 2023 | $ | $ | ( | $ | ( | $ | ( | $ | ( | |||||||||||||||||||||||
| Other comprehensive income (loss) before reclassifications | ( | ( | ( | |||||||||||||||||||||||||||||
| Amounts reclassified from AOCI | ( | ( | ||||||||||||||||||||||||||||||
| Net current period other comprehensive income (loss) | ( | ( | ( | |||||||||||||||||||||||||||||
| Balance, March 31, 2024 | $ | $ | ( | $ | ( | $ | ( | $ | ( | |||||||||||||||||||||||
29
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
| March 31, 2023 | ||||||||||||||||||||||||||||||||
| (In millions) | Unrealized Gains (Losses) on Securities Available for Sale, Net of Tax | Unrealized Gains (Losses) on Cash Flow Hedges, Net of Tax | Unrealized Gains (Losses) on Pension Benefit Obligation, Net of Tax | Currency Translation Adjustments | Total | |||||||||||||||||||||||||||
| Balance, December 31, 2022 | $ | ( | $ | $ | ( | $ | ( | $ | ( | |||||||||||||||||||||||
| Other comprehensive income (loss) before reclassifications | ( | |||||||||||||||||||||||||||||||
| Amounts reclassified from AOCI | ( | ( | ||||||||||||||||||||||||||||||
| Net current period other comprehensive income (loss) | ( | ( | ||||||||||||||||||||||||||||||
| Balance, March 31, 2023 | $ | ( | $ | ( | $ | ( | $ | ( | $ | ( | ||||||||||||||||||||||
The following table summarizes the amounts reclassified from AOCI:
| (In millions) | Amounts Reclassified from AOCI | Income Statement Location | ||||||||||||||||||
| For the Three Months Ended March 31, | ||||||||||||||||||||
| 2024 | 2023 | |||||||||||||||||||
| Gains (losses) on securities available for sale | $ | $ | ( | Other (income) expense | ||||||||||||||||
| Income tax (benefit) expense | ||||||||||||||||||||
| Gains (losses) on cash flow hedges | Revenue | |||||||||||||||||||
| ( | ( | Operating expense | ||||||||||||||||||
| ( | Other (income) expense | |||||||||||||||||||
| ( | ( | Income tax (benefit) expense | ||||||||||||||||||
| Total reclassifications, net of tax | $ | $ | ||||||||||||||||||
Note 15: | Earnings per Share | ||||
Basic and diluted shares outstanding used in our earnings per share calculation are calculated as follows:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Numerator: | ||||||||||||||
| Net income attributable to Biogen Inc. | $ | $ | ||||||||||||
| Denominator: | ||||||||||||||
| Weighted average number of common shares outstanding | ||||||||||||||
| Effect of dilutive securities: | ||||||||||||||
| Time-vested restricted stock units | ||||||||||||||
| Market stock units | ||||||||||||||
| Performance stock units settled in stock | ||||||||||||||
| Dilutive potential common shares | ||||||||||||||
| Shares used in calculating diluted earnings per share | ||||||||||||||
Amounts excluded from the calculation of net income per diluted share because their effects were anti-dilutive were insignificant.
30
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Note 16: | Share-Based Payments | ||||
Share-based Compensation Expense
The following table summarizes share-based compensation expense included in our condensed consolidated statements of income:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
Research and development | $ | $ | ||||||||||||
| Selling, general and administrative | ||||||||||||||
| Subtotal | ||||||||||||||
| Capitalized share-based compensation costs | ( | ( | ||||||||||||
| Share-based compensation expense included in total cost and expense | ||||||||||||||
| Income tax effect | ( | ( | ||||||||||||
| Share-based compensation expense included in net income attributable to Biogen Inc. | $ | $ | ||||||||||||
The following table summarizes share-based compensation expense associated with each of our share-based compensation programs:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Time-vested restricted stock units | $ | $ | ||||||||||||
| Performance stock units settled in stock | ||||||||||||||
| Employee stock purchase plan | ||||||||||||||
| Performance stock units settled in cash | ( | |||||||||||||
| Stock options | ||||||||||||||
| Market stock units | ||||||||||||||
| Subtotal | ||||||||||||||
| Capitalized share-based compensation costs | ( | ( | ||||||||||||
| Share-based compensation expense included in total cost and expense | $ | $ | ||||||||||||
We estimate the fair value of our obligations associated with our performance stock units settled in cash at the end of each reporting period through expected settlement. Cumulative adjustments to these obligations are recognized each quarter to reflect changes in the stock price and estimated outcome of the performance-related conditions.
31
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Note 17: | Income Taxes | ||||
Inflation Reduction Act
In August 2022 the IRA was signed into law in the U.S. The IRA introduced new tax provisions, including a 15.0% corporate alternative minimum tax and a 1.0% excise tax on stock repurchases. The provisions of the IRA are effective for periods after December 31, 2022. The IRA did not result in any material adjustments to our income tax provision or other income tax balances as of March 31, 2024 and December 31, 2023. Preliminary guidance has been issued by the IRS and we expect additional guidance and regulations to be issued in future periods. We will continue to assess its potential impact on our business and results of operations as further information becomes available.
Tax Rate
A reconciliation between the U.S. federal statutory tax rate and our effective tax rate is summarized as follows:
| For the Three Months Ended March 31, | |||||||||||
| 2024 | 2023 | ||||||||||
| Statutory rate | % | % | |||||||||
| State taxes | |||||||||||
| Taxes on foreign earnings | ( | ( | |||||||||
| Tax credits | ( | ( | |||||||||
| Purchased intangible assets | |||||||||||
| GILTI | ( | ||||||||||
| Other, including permanent items | |||||||||||
| Effective tax rate | % | % | |||||||||
Changes in Tax Rate
For the three months ended March 31, 2024, compared to the same period in 2023, the increase in our effective tax rate includes the impact of a resolution of an uncertain tax matter in the prior year related to tax credits and the non-cash tax effects of changes in the value of our equity investments, where we recorded larger unrealized losses in the prior year.
Accounting for Uncertainty in Income Taxes
We and our subsidiaries are routinely examined by various taxing authorities. We file income tax returns in various U.S. states and in U.S. federal and other foreign jurisdictions. With few exceptions, we are no longer subject to U.S. federal tax examination for years before 2019 or state, local or non-U.S. income tax examinations for years before 2013.
The IRS and other national tax authorities routinely examine our intercompany transfer pricing with respect to intellectual property related transactions and it is possible that they may disagree with one or more positions we have taken with respect to such valuations.
It is reasonably possible that we will adjust the value of our uncertain tax positions related to certain transfer pricing, collaboration matters, withholding taxes and other issues as we receive additional information from various taxing authorities, including reaching settlements with such authorities.
We estimate that it is reasonably possible that our gross unrecognized tax benefits, exclusive of interest, could
decrease by up to approximately $25.0 million in the next 12 months as a result of various audit closures, settlements and expiration of the statute of limitations.
32
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Note 18: | Other Consolidated Financial Statement Detail | ||||
Other (Income) Expense, Net
Components of other (income) expense, net, are summarized as follows:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Interest income | $ | ( | $ | ( | ||||||||||
| Interest expense | ||||||||||||||
| (Gains) losses on investments, net | ||||||||||||||
| Foreign exchange (gains) losses, net | ||||||||||||||
| Other, net | ( | |||||||||||||
| Total other (income) expense, net | $ | $ | ||||||||||||
The (gains) losses on investments, net, as reflected in the table above, relate to debt securities, equity securities of certain biotechnology companies, venture capital funds where the underlying investments are in equity securities of certain biotechnology companies and non-marketable equity securities.
The following table summarizes our (gains) losses on investments, net that relate to our equity securities held during the following periods:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Net (gains) losses recognized on equity securities | $ | $ | ||||||||||||
| Less: Net (gains) losses realized on equity securities | ||||||||||||||
| Net unrealized (gains) losses recognized on equity securities | $ | $ | ||||||||||||
The net unrealized losses recognized during the three months ended March 31, 2024, primarily reflect a decrease in the aggregate fair value of our investments in Sage and Denali common stock of approximately $27.7 million, partially offset by an increase in the fair value of Sangamo common stock of approximately $1.4 million.
The net unrealized losses recognized during the three months ended March 31, 2023, primarily reflect a decrease in the aggregate fair value of our investments in Denali, Sangamo and Ionis common stock of approximately $100.0 million, partially offset by an increase in the fair value of Sage common stock of approximately $23.8 million.
Accrued Expense and Other
Accrued expense and other consists of the following:
| (In millions) | As of March 31, 2024 | As of December 31, 2023 | ||||||||||||
| Revenue-related reserves for discounts and allowances | $ | $ | ||||||||||||
| Employee compensation and benefits | ||||||||||||||
| Collaboration expense | ||||||||||||||
| Royalties and licensing fees | ||||||||||||||
| Reata acquisition-related accrued expense | ||||||||||||||
| Other | ||||||||||||||
| Total accrued expense and other | $ | $ | ||||||||||||
Other Long-term Liabilities
Other long-term liabilities were $777.1 million and $781.1 million as of March 31, 2024 and December 31, 2023, respectively, and included accrued income taxes totaling $411.0 million and $403.2 million, respectively.
33
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Note 19: | Collaborative and Other Relationships | ||||
Genentech, Inc. (Roche Group)
We have certain business and financial rights with respect to RITUXAN for the treatment of non-Hodgkin's lymphoma, CLL and other conditions; RITUXAN HYCELA for the treatment of non-Hodgkin's lymphoma and CLL; GAZYVA for the treatment of CLL and follicular lymphoma; OCREVUS for the treatment of PPMS and RMS; LUNSUMIO for the treatment of relapsed or refractory follicular lymphoma; COLUMVI, a bispecific antibody for the treatment of non-Hodgkin's lymphoma; and have the option to add other potential anti-CD20 therapies, pursuant to our collaboration arrangements with Genentech, a wholly-owned member of the Roche Group. For purposes of this footnote, we refer to RITUXAN and RITUXAN HYCELA collectively as RITUXAN.
RITUXAN
Genentech and its affiliates are responsible for the worldwide manufacture of RITUXAN as well as all development and commercialization activities as follows:
•U.S.: We have co-exclusively licensed our rights to develop, commercialize and market RITUXAN in the U.S.
•Canada: We have co-exclusively licensed our rights to develop, commercialize and market RITUXAN in Canada.
GAZYVA
The Roche Group and its sub-licensees maintain sole responsibility for the development, manufacture and commercialization of GAZYVA in the U.S. The level of gross sales of GAZYVA in the U.S. has impacted our percentage of the co-promotion profits for RITUXAN and LUNSUMIO, as summarized in the table below.
OCREVUS
Pursuant to the terms of our collaboration arrangements with Genentech, we receive a tiered royalty on U.S. net sales from 13.5 % and increasing up to 24.0 % if annual net sales exceed $900.0 million. There will be a 50.0 % reduction to these royalties if a biosimilar to OCREVUS is approved in the U.S.
In addition, we receive a gross 3.0 % royalty on net sales of OCREVUS outside the U.S., with the royalty period lasting 11 years from the first commercial sale of OCREVUS on a country-by-country basis.
The commercialization of OCREVUS does not impact the percentage of the co-promotion profits we receive for RITUXAN, LUNSUMIO or GAZYVA. Genentech is solely responsible for development and commercialization of OCREVUS and funding future costs. Genentech cannot develop OCREVUS in CLL, non-Hodgkin's lymphoma or rheumatoid arthritis.
OCREVUS royalty revenue is based on our estimates from third party and market research data of OCREVUS sales occurring during the corresponding period. Differences between actual and estimated royalty revenue will be adjusted for in the period in which they become known, which is generally expected to be the following quarter.
LUNSUMIO (mosunetuzumab)
In January 2022 we exercised our option with Genentech to participate in the joint development and commercialization of LUNSUMIO. Under our collaboration with Genentech, we were responsible for 30.0 % of development costs for LUNSUMIO prior to FDA approval and will be entitled to a tiered share of co-promotion operating profits and losses in the U.S., as summarized in the table below. In addition, we receive low single-digit royalties on sales of LUNSUMIO outside the U.S. In December 2022 LUNSUMIO was granted accelerated approval by the FDA for the treatment of relapsed or refractory follicular lymphoma.
Prior to regulatory approval, we record our share of the expense incurred by the collaboration for the development of anti-CD20 products in research and development expense and pre-commercialization costs within selling, general and administrative expense in our condensed consolidated statements of income. After an anti-CD20 product is approved, we record our share of the development and sales and marketing expense related to that product as a reduction of our share of pre-tax profits in revenue from anti-CD20 therapeutic programs.
34
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
COLUMVI (glofitamab)
In December 2022 we entered into an agreement with Genentech related to the commercialization and sharing of economics for COLUMVI, a bispecific antibody for the treatment of B-cell non-Hodgkin's lymphoma, which was subsequently granted accelerated approval by the FDA in June 2023. Under the terms of this agreement, we will have no payment obligations. Genentech will have sole decision-making rights on the commercialization of COLUMVI within the U.S. and we will receive tiered royalties in the mid-single digit range on net sales of COLUMVI in the U.S. The commercialization of COLUMVI does not impact the percentage of the co-promotion profits we receive for RITUXAN, LUNSUMIO or GAZYVA.
In April 2024 Roche announced that COLUMVI, in combination with chemotherapy GemOx (glofitamab-gxbm), demonstrated a statistically significant improvement in overall survival for people with relapsed or refractory diffuse large B-cell lymphoma.
Profit-sharing Formulas
RITUXAN and LUNSUMIO Profit Share
Our current pretax co-promotion profit-sharing formula for RITUXAN and LUNSUMIO in the U.S. provides for a 30.0 % share on the first $50.0 As a result of the FDA approval of LUNSUMIO our share of the combined annual co-promotion profits for RITUXAN and LUNSUMIO in excess of $50.0
| After LUNSUMIO Approval until the First Threshold Date | % | ||||
| After First Threshold Date until the Second Threshold Date | % | ||||
| After Second Threshold Date | % | ||||
First Threshold Date means the earlier of (i) the first day of the calendar quarter following the date U.S. gross sales of GAZYVA within any consecutive 12-month period have reached $500.0 million or (ii) the first date in any calendar year in which U.S. gross sales of LUNSUMIO have reached $150.0 million.
Second Threshold Date means the later of (i) the first date the gross sales in any calendar year in which U.S. gross sales of LUNSUMIO reach $350.0 million or (ii) January 1 of the calendar year following the calendar year in which the First Threshold Date occurs.
In March 2023 the First Threshold Date was achieved. As a result, beginning in April 2023 the pre-tax profit share for RITUXAN and LUNSUMIO was 35.0 %.
GAZYVA Profit Share
Our current pretax profit-sharing formula for GAZYVA provides for a 35.0 % share on the first $50.0 Our share of annual co-promotion profits in excess of $50.0
| Until Second GAZYVA Threshold Date | % | ||||
| After Second GAZYVA Threshold Date | % | ||||
Second GAZYVA Threshold Date means the first day of the calendar quarter following the date U.S. gross sales of GAZYVA within any consecutive 12-month period have reached $500.0 million. The Second GAZYVA Threshold Date can be achieved regardless of whether GAZYVA has been approved in a non-CLL indication.
In March 2023 the Second GAZYVA Threshold Date was achieved. As a result, beginning in April 2023 the pre-tax profit share for GAZYVA was 35.0 %.
For additional information on our collaboration arrangements with Genentech, please read Note 19, Collaborative and Other Relationships, to our audited consolidated financial statements included in our 2023 Form 10-K.
35
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Eisai Co., Ltd.
During the first quarter of 2023 we accrued a $31.0 million payable to Eisai related to the termination of an agreement whereby Eisai co-promoted or distributed our MS products in certain Asia-Pacific markets and settings. As of December 31, 2023, we paid approximately $16.0 million of the $31.0 million payable. The remaining portion was subsequently paid in January 2024. This termination fee is included in selling, general and administrative expense in our condensed consolidated statements of income for the three months ended March 31, 2023.
LEQEMBI (lecanemab) Collaboration
We have a collaboration agreement with Eisai to jointly develop and commercialize LEQEMBI (lecanemab), an anti-amyloid antibody for the treatment of Alzheimer's disease (the LEQEMBI Collaboration).
Eisai serves as the lead of LEQEMBI development and regulatory submissions globally with both companies co-commercializing and co-promoting the product, and Eisai having final decision-making authority. All costs, including research, development, sales and marketing expense, are shared equally between us and Eisai. We and Eisai co-promote LEQEMBI and share profits and losses equally. We currently manufacture LEQEMBI drug substance and drug product and in March 2022 we extended our supply agreement with Eisai related to LEQEMBI from five years to ten years for the manufacture of LEQEMBI drug substance.
In July 2023 the FDA granted traditional approval of LEQEMBI. Prior to receiving traditional approval, LEQEMBI had been granted accelerated approval by the FDA in January 2023, at which time it became commercially available in the U.S. In September 2023 the Japanese Ministry of Health, Labor and Welfare approved LEQEMBI in Japan, which was subsequently launched in December 2023, and in January 2024 LEQEMBI was approved in China.
Upon commercialization of LEQEMBI, we began recognizing our 50.0 % share of LEQEMBI product revenue, net and cost of sales, including royalties, within other revenue in our condensed consolidated statements of income, as we are not the principal.
Our share of LEQEMBI sales and marketing expense and development expense are recorded within selling, general and administrative expense and research and development expense, respectively, within our condensed consolidated statements of income.
A summary of development and sales and marketing expense related to the LEQEMBI Collaboration is as follows:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Total development expense incurred by the collaboration related to the advancement of LEQEMBI | $ | $ | ||||||||||||
| Biogen's share of the LEQEMBI Collaboration development expense reflected in research and development expense in our condensed consolidated statements of income | ||||||||||||||
Total sales and marketing expense incurred by the LEQEMBI Collaboration | ||||||||||||||
| Biogen's share of the LEQEMBI Collaboration sales and marketing expense reflected in selling, general and administrative expense in our condensed consolidated statements of income | ||||||||||||||
Amounts receivable from Eisai related to the agreements discussed above were approximately $15.9 million and $1.4 million as of March 31, 2024 and December 31, 2023, respectively. Amounts payable to Eisai related to the agreements discussed above were $180.7 million and $118.4 million as of March 31, 2024 and December 31, 2023, respectively.
For additional information on our collaboration arrangements with Eisai, please read Note 19, Collaborative and Other Relationships, to our consolidated financial statements included in our 2023 Form 10-K.
UCB
We have a collaboration agreement with UCB, effective November 2003, to jointly develop and commercialize dapirolizumab pegol, an anti-CD40L pegylated Fab, for the potential treatment of SLE and other future agreed indications. Either we or UCB may propose development of dapirolizumab pegol in additional indications. If the parties do not agree to add an indication as an agreed indication to the collaboration, we or UCB may, at the sole expense of the applicable party, pursue development in such excluded indication(s), subject to an opt-in right of the non-pursuing party after proof of clinical activity.
36
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
All costs incurred for agreed indications, including research, development, sales and marketing expense, are shared equally between us and UCB. If marketing approval is obtained, both companies will co-promote dapirolizumab pegol and share profits and losses equally.
A summary of development expense related to the UCB collaboration agreement is as follows:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Total UCB collaboration development expense | $ | $ | ||||||||||||
| Biogen's share of UCB collaboration development expense reflected in research and development expense in our condensed consolidated statements of income | ||||||||||||||
Sage Therapeutics, Inc.
In November 2020 we entered into a global collaboration and license agreement with Sage to jointly develop and commercialize ZURZUVAE (zuranolone) for the treatment of PPD and potential treatment of MDD and BIIB124 (SAGE-324) for the potential treatment of essential tremor with potential in other neurological conditions such as epilepsy.
In August 2023 the FDA approved ZURZUVAE for adults with PPD, pending DEA scheduling, which was completed in October 2023. Upon approval, ZURZUVAE became the first and only oral, once-daily, 14-day treatment that can provide rapid improvements in depressive symptoms by day 15 for women with PPD. ZURZUVAE for PPD became commercially available in the U.S. during the fourth quarter of 2023. Additionally, the FDA issued a CRL for the NDA for zuranolone in the treatment of adults with MDD. The CRL stated that the application did not provide substantial evidence of effectiveness to support the approval of zuranolone for the treatment of MDD and that an additional study or studies would be needed. We and Sage are continuing to seek feedback from the FDA and evaluating next steps.
Under this collaboration, both companies will share equal responsibility and costs for development as well as profits and losses for commercialization in the U.S. Outside of the U.S., we are responsible for development and commercialization, excluding Japan, Taiwan and South Korea, with respect to zuranolone and may pay Sage potential tiered royalties in the high teens to low twenties. During the fourth quarter of 2023 we accrued a milestone payment due to Sage of $75.0 million upon the first commercial sale of ZURZUVAE for PPD in the U.S., which was recorded within intangible assets, net in our condensed consolidated balance sheets, and subsequently paid in January 2024.
For the three months ended March 31, 2024, we recognized net profit-sharing expense of approximately $5.0 million to reflect Sage's 50.0 % share of net collaboration results in the U.S. for ZURZUVAE for PPD, which is recognized in collaboration profit sharing/(loss reimbursement) in our condensed consolidated statements of income.
A summary of development and sales and marketing expense related to the Sage collaboration is as follows:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Total Sage collaboration development expense | $ | $ | ||||||||||||
| Biogen's share of Sage collaboration development expense reflected in research and development expense in our condensed consolidated statements of income | ||||||||||||||
| Total sales and marketing expense incurred by the Sage collaboration | ||||||||||||||
| Biogen's share of Sage collaboration sales and marketing expense reflected in selling, general and administrative expense and collaboration profit sharing/(loss reimbursement) in our condensed consolidated statements of income | ||||||||||||||
Denali Therapeutics Inc.
In August 2020 we entered into a collaboration and license agreement with Denali to co-develop and co-commercialize Denali's small molecule inhibitors of LRRK2 for Parkinson's disease (LRRK2 Collaboration) and also entered into a separate agreement to obtain an exclusive option to license two preclinical programs from Denali's Transport Vehicle platform, including its ATV-enabled anti-amyloid beta program and a second program utilizing its Transport Vehicle technology.
37
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
In April 2023 we exercised our option with Denali to license the ATV-enabled anti-amyloid beta program. In connection with this exercise, we assumed responsibility for all development and commercial activities and associated expenses related to this program. In addition, we made a one-time option exercise payment to Denali and, should certain milestones be achieved, may pay Denali additional development and commercial milestone payments and royalties based on future net sales. Our agreement with Denali was amended in August 2023, whereby certain milestone criteria were changed, while the total amount of development, regulatory and commercial milestones remains the same. In addition, we agreed to waive our option right to the second option program.
Under the LRRK2 Collaboration, both companies share responsibility and costs for global development based on specified percentages as well as profits and losses for commercialization in the U.S. and China. Outside the U.S. and China we are responsible for commercialization and may pay Denali potential tiered royalties.
A summary of development expense related to the Denali collaboration is as follows:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Total Denali collaboration development expense | $ | $ | ||||||||||||
| Biogen's share of Denali collaboration development expense reflected in research and development expense in our condensed consolidated statements of income | ||||||||||||||
Other Research and Discovery Arrangements
These arrangements may include the potential for future milestone payments based on the achievement of certain clinical and commercial development payable over a period of several years.
Other
For the three months ended March 31, 2024, we recorded approximately $7.5 million as research and development expense in our condensed consolidated statements of income related to other research and discovery related arrangements, compared to $0.2 million in the prior year comparative period.
Samsung Bioepis Co., Ltd.
2019 Development and Commercialization Agreement
In December 2019 we completed a transaction with Samsung Bioepis and secured the exclusive rights to commercialize two potential ophthalmology biosimilar products, BYOOVIZ (ranibizumab-nuna), a ranibizumab biosimilar referencing LUCENTIS, and SB15, a proposed aflibercept biosimilar referencing EYLEA, in major markets worldwide, including the U.S., Canada, Europe, Japan and Australia. Samsung Bioepis will be responsible for development and will supply both products to us at a pre-specified gross margin of approximately 45.0 %.
In connection with this transaction, we may also pay Samsung Bioepis up to approximately $180.0 million in additional development, regulatory and sales-based milestones.
We also acquired an option to extend the term of our 2013 commercial agreement for BENEPALI, IMRALDI and FLIXABI by an additional five years , subject to payment of an option exercise fee of $60.0 million by August 2024, and obtained an option to acquire exclusive rights to commercialize these products in China.
2013 Commercial Agreement
We reflect revenue on sales of BENEPALI, IMRALDI and FLIXABI to third parties in product revenue, net in our condensed consolidated statements of income and record the related cost of revenue and sales and marketing expense in our condensed consolidated statements of income to their respective line items when these costs are incurred. Royalty payments to AbbVie on sales of IMRALDI are recognized in cost of sales within our condensed consolidated statements of income.
We share 50.0 % of the profit or loss related to our commercial agreement with Samsung Bioepis, which is recognized in collaboration profit sharing/(loss reimbursement) in our condensed consolidated statements of income. For the three months ended March 31, 2024, we recognized net profit-sharing expense of $60.6 million to reflect Samsung Bioepis' 50.0 % sharing of the net collaboration profits, compared to a net profit-sharing expense of $57.1 million in the prior year comparative period.
38
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Other Services
Simultaneous with the formation of Samsung Bioepis, we also entered into a license agreement with Samsung Bioepis. Under this license agreement, we granted Samsung Bioepis an exclusive license to use, develop, manufacture and commercialize biosimilar products created by Samsung Bioepis using Biogen product-specific technology. In exchange, we receive single digit royalties on biosimilar products developed and commercialized by Samsung Bioepis. Royalty revenue under the license agreement is recognized as a component of contract manufacturing, royalty and other revenue in our condensed consolidated statements of income.
Amounts receivable from Samsung Bioepis related to the agreements discussed above were $18.5 million and $9.9 million as of March 31, 2024 and December 31, 2023, respectively. Amounts payable to Samsung Bioepis related to the agreements discussed above were $35.6 million and $73.7 million as of March 31, 2024 and December 31, 2023, respectively.
Note 20: | Investments in Variable Interest Entities | ||||
Consolidated Variable Interest Entities
Our condensed consolidated financial statements include the financial results of variable interest entities in which we are the primary beneficiary. The following are our significant variable interest entities.
Neurimmune SubOne AG
Beginning in 2007 we consolidated the results of Neurimmune as we determined we were the primary beneficiary because we had the power through the collaboration to direct the activities that most significantly impacted the entity's economic performance and we were required to fund 100.0 % of the research and development costs incurred in support of the collaboration. The collaboration and license agreement with Neurimmune was for the development and commercialization of antibodies for the potential treatment of Alzheimer's disease, including ADUHELM (as amended, the Neurimmune Agreement).
In November 2023 we notified Neurimmune of our decision to terminate the Neurimmune Agreement. Subsequent to the termination, we reconsidered our relationship with Neurimmune and determined that we were no longer the primary beneficiary of the variable interest entity. As a result, we recorded a net gain on the deconsolidation of Neurimmune of approximately $3.0 million, which was recorded in other (income) expense, net within our consolidated statements of income for the year ended December 31, 2023, included in our 2023 Form 10-K.
Unconsolidated Variable Interest Entities
We have relationships with various variable interest entities that we do not consolidate as we lack the power to direct the activities that significantly impact the economic success of these entities. These relationships include investments in certain biotechnology companies and research collaboration agreements.
As of March 31, 2024 and December 31, 2023, the carrying value of our investments in certain biotechnology companies representing potential unconsolidated variable interest entities totaled $23.8 million and $16.4 million, respectively. Our maximum exposure to loss related to these variable interest entities is limited to the carrying value of our investments.
We have also entered into research collaboration agreements with certain variable interest entities where we are required to fund certain development activities. These development activities are included in research and development expense in our condensed consolidated statements of income as they are incurred. We have provided no financing to these variable interest entities other than previous contractually required amounts.
For additional information on our investments in Neurimmune and other variable interest entities, please read Note 20, Investments in Variable Interest Entities, to our consolidated financial statements included in our 2023 Form 10-K.
39
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Note 21: | Litigation | ||||
We are currently involved in various claims, investigations and legal proceedings, including the matters described below. For information as to our accounting policies relating to claims and legal proceedings, including use of estimates and contingencies, please read Note 1, Summary of Significant Accounting Policies, to our consolidated financial statements included in our 2023 Form 10-K.
With respect to some loss contingencies, an estimate of the possible loss or range of loss cannot be made until management has further information, including, for example, (i) which claims, if any, will survive dispositive motion practice; (ii) information to be obtained through discovery; (iii) information as to the parties' damages claims and supporting evidence; (iv) the parties’ legal theories; and (v) the parties' settlement positions. If an estimate of the possible loss or range of loss can be made at this time, it is included in the potential loss contingency description below.
The claims and legal proceedings in which we are involved also include challenges to the scope, validity or enforceability of the patents relating to our products, pipeline or processes and challenges to the scope, validity or enforceability of the patents held by others. These include claims by third parties that we infringe their patents. An adverse outcome in any of these proceedings could result in one or more of the following and have a material impact on our business or consolidated results of operations and financial position: (i) loss of patent protection; (ii) inability to continue to engage in certain activities; and (iii) payment of significant damages, royalties, penalties and/or license fees to third parties.
Loss Contingencies
ADUHELM Securities Litigation
We and certain current and former officers are defendants in two actions, one filed in November 2020 and one in February 2022, related to ADUHELM filed in the District Court. In March 2024 the District Court vacated its dismissal of the action filed in February 2022. Both actions allege violations of federal securities laws under 15 U.S.C. §78j(b) and §78t(a) and 17 C.F.R. §240.10b-5 and seek declarations of the actions as class actions and monetary relief.
Derivative Actions
We and members of the Board of Directors are named as defendants in derivative actions filed by shareholders in February and July 2022, in the District Court. The actions allege violations of federal securities laws under 15 U.S.C. §78n(a) and 17 C.F.R. §240 14.a-9, and breaches of fiduciary duties and waste of corporate assets, and seek declaratory and injunctive relief, monetary relief payable to Biogen, and attorneys’ fees and costs payable to the plaintiffs. The District Court has stayed the case filed in February 2022 and the parties have requested continuation of the stay in the case filed in July 2022.
IMRALDI Patent Litigation
Fresenius Kabi's European Patent 3 145 488 (the EP '488 Patent), which expires in May 2035, is the subject of opposition proceedings in the EPO Technical Boards of Appeal. In June 2022 Fresenius Kabi Deutschland GmbH (Fresenius Kabi) filed a claim for damages and injunctive relief against Biogen France SAS in the Tribunal de Grande Instance de Paris alleging that IMRALDI, the adalimumab biosimilar product of Samsung Bioepis that Biogen commercializes in Europe, infringes the French counterpart of the EP ‘488 Patent. In August 2022 Fresenius Kabi filed a claim for damages and injunctive relief against Biogen GmbH in the Düsseldorf Regional Court, alleging infringement of the German counterpart of the EP '488 Patent, and in March 2024 the court found no infringement and dismissed the case. Fresenius Kabi has appealed to the Higher Regional Court of Düsseldorf.
40
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Litigation with Former Convergence Shareholders
In 2015 Biogen acquired Convergence, a U.K. company. In 2019 Shareholder Representative Services LLC, on behalf of the former shareholders of Convergence, asserted claims of $200.0 million for alleged breaches of the contract pursuant to which we acquired Convergence. In June 2023 Shareholder Representative Services LLC and 24 former shareholders filed suit against us in the High Court of Justice of England and Wales on one of the previously asserted claims, seeking payment of $49.9 million, interest and costs.
Humana Patient Assistance Litigation
In March 2023 the District Court dismissed the previously disclosed action filed against us by Humana in September 2020. Humana had alleged damages related to our providing MS patients with free medications and making charitable contributions to non-profit organizations that assist MS patients and had alleged violations of the federal RICO Act and state laws. In December 2023 Humana appealed to the United States Court of Appeals for the First Circuit and the appeal is pending.
Genentech Litigation
In February 2023 Genentech, Inc. filed suit against us in the U.S. District Court for the Northern District of California, alleging that it is owed royalties on sales of TYSABRI that occurred after the expiration of a patent licensed by Genentech to Biogen, together with interest and costs. The Company estimates that the royalties claimed total approximately $88.3 million.
Bardoxolone Securities Litigation
In March 2024 the United States District Court for the Eastern District of Texas granted final approval of the previously disclosed settlement of litigation filed by putative stockholders of Reata (later acquired by Biogen) alleging violations of the federal securities laws by Reata, certain of its former officers and directors, and certain underwriters under 15 U.S.C. §78j(b) and §78t(a), 17 C.F.R. §240.10b-5, and 15 U.S.C. §§77k, 77l(a)(2) and 77o, and dismissed the action with prejudice.
Lender Dispute
In April 2024, BioPharma Credit PLC, BPCR Limited Partnership, and BioPharma Credit Investments V (Master) LP filed suit against us and Reata Pharmaceuticals, Inc. in the Supreme Court of the State of New York alleging breach of a loan agreement with Reata and seeking payment of approximately $23.2 million, plus interest, costs and attorneys' fees.
Other Matters
Government Investigation
The Company has received subpoenas from the SEC seeking information relating to ADUHELM and its launch. The Company has received a subpoena from the DOJ seeking information relating to our business operations in several foreign countries. The Company is also providing information relating to our business operations in several foreign countries to the SEC.
TYSABRI Biosimilar Patent Matter
In September 2022 we filed an action in the U.S. District Court for the District of Delaware against Sandoz Inc., other Sandoz entities and Polpharma Biologics S.A. under the Biologics Price Competition and Innovation Act, 42 U.S.C. §262, seeking a declaratory judgment of patent infringement.
Annulment Proceedings in the General Court of the European Union relating to TECFIDERA
In November 2020 Mylan Ireland filed an action in the General Court of the European Union to annul the EMA's decision not to validate its applications to market generic versions of TECFIDERA on the grounds that TECFIDERA benefits from regulatory data protection.
Hatch-Waxman Act Litigation relating to VUMERITY Orange-Book Listed Patents
In July 2023 Biogen and Alkermes Pharma Ireland Limited filed patent infringement proceedings relating to VUMERITY Orange-Book listed patents (U.S. Patent Nos. 8,669,281, 9,090,558 and 10,080,733) pursuant to the Drug Price Competition and Patent Term Restoration Act of 1984 (the Hatch-Waxman Act) in the U.S. District Court for the District of Delaware against Zydus Worldwide DMCC.
41
BIOGEN INC. AND SUBSIDIARIES
NOTES TO CONDENSED CONSOLIDATED FINANCIAL STATEMENTS
(unaudited, continued)
Product Liability and Other Legal Proceedings
We are also involved in product liability claims and other legal proceedings incidental to our normal business activities. While the outcome of any of these proceedings cannot be accurately predicted, we do not believe the ultimate resolution of any of these existing matters would have a material adverse effect on our business or financial condition.
Note 22: | Subsequent Events | ||||
Sale of Priority Review Voucher
On April 24, 2024, we entered into a definitive agreement with a third party to sell a rare pediatric disease priority review voucher. In consideration for the PRV we received $103.0 million upon the closing of the PRV purchase.
42
ITEM 2. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following discussion should be read in conjunction with our unaudited condensed consolidated financial statements (condensed consolidated financial statements) and the accompanying notes beginning on page 7 of this quarterly report on Form 10-Q and our audited consolidated financial statements and the accompanying notes included in our 2023 Form 10-K. The results of operations of Reata, along with the estimated fair values of the assets acquired and liabilities assumed in the Reata acquisition, have been included in our condensed consolidated financial statements since the closing of the Reata acquisition on September 26, 2023.
EXECUTIVE SUMMARY
INTRODUCTION
Biogen is a global biopharmaceutical company focused on discovering, developing and delivering innovative therapies for people living with serious and complex diseases worldwide. We have a broad portfolio of medicines to treat MS, have introduced the first approved treatment for SMA, co-developed treatments to address a defining pathology of Alzheimer’s disease and launched the first approved treatment to target a genetic cause of ALS. Through our 2023 acquisition of Reata we market the first and only drug approved in the U.S. and the E.U. for the treatment of Friedreich's Ataxia in adults and adolescents aged 16 years and older. We are focused on advancing our pipeline in neurology, specialized immunology and rare diseases. We support our drug discovery and development efforts through internal research and development programs and external collaborations.
Our marketed products include TECFIDERA, VUMERITY, AVONEX, PLEGRIDY, TYSABRI and FAMPYRA for the treatment of MS; SPINRAZA for the treatment of SMA; SKYCLARYS for the treatment of Friedreich's Ataxia; QALSODY for the treatment of ALS; and FUMADERM for the treatment of severe plaque psoriasis.
We also have collaborations with Eisai on the commercialization of LEQEMBI for the treatment of Alzheimer's disease and Sage on the commercialization of ZURZUVAE for the treatment of PPD and we have certain business and financial rights with respect to RITUXAN for the treatment of non-Hodgkin's lymphoma, CLL and other conditions; RITUXAN HYCELA for the treatment of non-Hodgkin's lymphoma and CLL; GAZYVA for the treatment of CLL and follicular lymphoma; OCREVUS for the treatment of PPMS and RMS; LUNSUMIO for the treatment of relapsed or refractory follicular lymphoma; COLUMVI, a bispecific antibody for the treatment of non-Hodgkin's lymphoma; and have the option to add other potential anti-CD20 therapies, pursuant to our collaboration arrangements with Genentech, a wholly-owned member of the Roche Group.
We commercialize a portfolio of biosimilars of advanced biologics including BENEPALI, an etanercept biosimilar referencing ENBREL, IMRALDI, an adalimumab biosimilar referencing HUMIRA, and FLIXABI, an infliximab biosimilar referencing REMICADE, in certain countries in Europe, as well as BYOOVIZ, a ranibizumab biosimilar referencing LUCENTIS, in the U.S. and certain international markets. We also have exclusive rights to commercialize TOFIDENCE, a tocilizumab biosimilar referencing ACTEMRA. We continue to develop potential biosimilar product SB15, a proposed aflibercept biosimilar referencing EYLEA. We continue to evaluate strategic options for our biosimilars business.
For additional information on our collaboration arrangements, please read Note 19, Collaborative and Other Relationships, to our condensed consolidated financial statements included in this report.
We seek to ensure an uninterrupted supply of medicines to patients around the world. To that end, we continually review our manufacturing capacity, capabilities, processes and facilities. In order to support our future growth and drug development pipeline, we expanded our large molecule production capacity and built a large-scale biologics manufacturing facility in Solothurn, Switzerland. In the second quarter of 2021 a portion of this facility (the first manufacturing suite) received a GMP multi-product license from the SWISSMEDIC and was placed into service. The second manufacturing suite became operational in the first quarter of 2024. Solothurn has been approved for the manufacture of LEQEMBI by the FDA. We believe that the Solothurn facility will support our anticipated near to mid-term needs for the manufacturing of biologic assets. The plant represents a significant increase in our overall manufacturing capacity and is not yet being fully utilized, resulting in our recording of excess capacity charges. If we are unable to fully utilize our manufacturing facilities, we will incur additional excess capacity charges which would have a negative effect on our financial condition and results of operations.
In the longer term, our revenue growth will depend upon the successful clinical development, regulatory approval and launch of new commercial products as well as additional indications for our existing products, our ability to obtain
43
and maintain patents and other rights related to our marketed products, assets originating from our research and development efforts and/or successful execution of external business development opportunities.
BUSINESS ENVIRONMENT
The biopharmaceutical industry and the markets in which we operate are intensely competitive. Many of our competitors are working to develop or have commercialized products similar to those we market or are developing and have considerable experience in undertaking clinical trials and in obtaining regulatory approval to market pharmaceutical products. In addition, the commercialization of certain of our own approved products, products of our collaborators and pipeline product candidates may negatively impact future sales of our existing products.
Our products and revenue streams continue to face increasing competition in many markets from generic versions, prodrugs and biosimilars of existing products as well as products approved under abbreviated regulatory pathways. Such products are likely to be sold at substantially lower prices than branded products. Accordingly, the introduction of such products as well as other lower-priced competing products may significantly reduce both the price that we are able to charge for our products and the volume of products we sell, which will negatively impact our revenue. In addition, in some markets, when a generic or biosimilar version of one of our products is commercialized, it may be automatically substituted for our product and significantly reduce our revenue in a short period of time.
Sales of our products depend, to a significant extent, on the availability and extent of adequate coverage, pricing and reimbursement from government health administration authorities, private health insurers and other organizations. When a new pharmaceutical product is approved, the availability of government and private reimbursement for that product may be uncertain, as is the pricing and amount for which that product will be reimbursed.
Drug prices are under significant scrutiny in the markets in which our products are prescribed, for example the IRA has certain provisions related to drug pricing. We expect drug pricing and other health care costs to continue to be subject to intense political and societal pressures on a global basis.
Our failure to obtain or maintain adequate coverage, pricing or reimbursement for our products could have an adverse effect on our business, reputation, revenue and results of operations, could curtail or eliminate our ability to adequately fund research and development programs for the discovery and commercialization of new products and/or could cause a decline or volatility in our stock price.
In addition to the impact of competition, pricing actions and other measures being taken worldwide designed to reduce healthcare costs and limit the overall level of government expenditures, our sales and operations could also be affected by other risks of doing business internationally, including the impact of public health epidemics on employees, the global economy and the delivery of healthcare treatments, geopolitical events, supply chain disruptions, foreign currency exchange fluctuations, changes in intellectual property legal protections and changes in trade regulations and procedures.
For a detailed discussion on our business environment, please read Item 1. Business, in our 2023 Form 10-K. For additional information on our competition and pricing risks that could negatively impact our product sales, please read Item 1A. Risk Factors included in this report.
TECFIDERA
Multiple TECFIDERA generic entrants are now in North America, Brazil and certain E.U. countries and have deeply discounted prices compared to TECFIDERA. The generic competition for TECFIDERA has significantly reduced our TECFIDERA revenue and we expect that TECFIDERA revenue will continue to decline in the future.
Following a favorable March 2023 decision of the CJEU affirming TECFIDERA's right to regulatory data and marketing protection and the EC determination in May 2023 that TECFIDERA is entitled to an additional year of market protection for its pediatric indication, we believe that TECFIDERA is entitled to regulatory marketing protection in the E.U. until at least February 2, 2025, and are seeking to enforce this protection. As of March 31, 2024, some of the TECFIDERA generics have not yet fully exited some E.U. markets. We are closely monitoring this situation and working to enforce our legal right to market protection. In addition, we will continue to enforce our EP 2 653 873 patent related to TECFIDERA, which expires in 2028.
For additional information, please read Note 21, Litigation, to our condensed consolidated financial statements included in this report.
44
BUSINESS UPDATE REGARDING MACROECONOMIC CONDITIONS AND OTHER DISRUPTIONS
Significant portions of our business are conducted in Europe, Asia and other international geographies. Factors such as global health outbreaks, adverse weather events, geopolitical events, inflation, labor or raw material shortages and other supply chain disruptions could result in product shortages or other difficulties and delays or increased costs in manufacturing our products.
CURRENT ECONOMIC CONDITIONS
Economic conditions remain vulnerable as markets continue to be impacted in part by elevated inflation, higher interest rates, global supply chain uncertainties and risks associated with geopolitical conflicts.
GEOPOLITICAL TENSIONS
Global disputes and interruptions in international relationships, including tariffs, trade protection measures, import or export licensing requirements and the imposition of trade sanctions or similar restrictions by the U.S. or other governments, affect our ability to do business. For example, tensions between the U.S. and China have led to a series of tariffs and sanctions being imposed by the U.S. on imports from China mainland, as well as other business restrictions, with additional restrictive measures being proposed.
We, and the pharmaceutical industry, utilize China-based partners for certain raw materials, ingredients and components for our pharmaceutical products and their delivery devices. Engaging alternative suppliers may involve seeking additional regulatory approvals and be costly in terms of time and resources needed. For example, certain early processes related to our newly acquired SKYCLARYS product rely on a single supplier based in China. We are continuing to evaluate SKYCLARYS' supply chain and prioritizing actions to mitigate risks associated with its manufacturing and our ability to supply patients.
The ongoing geopolitical tensions related to Russia's invasion of Ukraine and the recent military conflict in the Middle East have resulted in global business disruptions and economic volatility.
For example. sanctions and other restrictions have been levied on the government and businesses in Russia. Although we do not have affiliates or employees, in either Russia or Ukraine, we do provide various therapies to patients in Russia through a distributor. In addition, new government sanctions on the export of certain manufacturing materials to Russia may delay or limit our ability to get new products approved. The impact of the conflict on our operations and financial performance remains uncertain and will depend on future developments, including the severity and duration of the conflict between Russia and Ukraine, its impact on regional and global economic conditions and whether the conflict spreads or has effects on countries outside Ukraine and Russia.
We will continue to monitor the ongoing conflict between Russia and Ukraine as well as the military conflict in the Middle East and assess any potential impacts on our business, supply chain, partners or customers, as well as any factors that could have an adverse effect on our results of operations. Revenue generated from sales in Russia and Ukraine represent less than 2.0% of total revenue for the three months ended March 31, 2024 and 2023. Additionally, revenue generated from sales in the broader Middle East region represents less than 2.0% of total revenue for the three months ended March 31, 2024 and 2023.
CLIMATE-RELATED DISCLOSURES
In March 2024 the SEC adopted final rules designed to enhance disclosures related to the impacts of climate-related matters. The final rules require disclosures of material climate-related risks, activities to mitigate or adapt to such risks, information about our board of directors' oversight of climate-related risks and management’s role in managing material climate-related risks and information on any climate-related targets or goals that are material to our business, results of operations or financial condition. In addition, the E.U. and California have enacted similar legislation and regulations. We are currently evaluating the potential impact that these new rules and laws will have on our business.
45
FINANCIAL HIGHLIGHTS
As described below under Results of Operations, our net income and diluted earnings per share attributable to Biogen Inc. for the three months ended March 31, 2024, compared to the three months ended March 31, 2023, reflects the following:
| TOTAL REVENUE | |||||
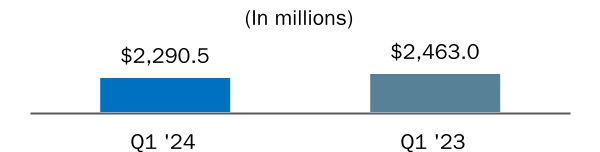
Decreased
$172.5 million or 7.0%
| DILUTED EARNINGS (LOSS) PER SHARE | |||||
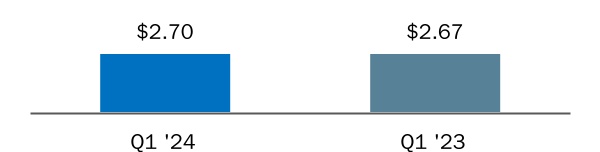
Increased
$0.03 or 1.1%
| PRODUCT REVENUE | |||||
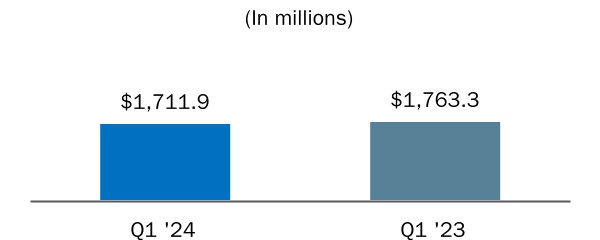
Decreased
$51.4 million or 2.9%
•MS revenue decreased $49.3 million, or 4.4%
•Rare disease revenue decreased $19.4 million, or 4.4%
•The decrease in MS product revenue was primarily due to a decrease in TECFIDERA demand as a result of multiple TECFIDERA generic entrants in North America, Brazil and certain E.U. countries, a decrease in Interferon demand due to competition as patients transition to higher efficacy therapies and a decrease in U.S. TYSABRI revenue primarily driven by increased competition and higher discounts and allowances.
•The decrease in rare disease revenue was due to a decrease in rest of world SPINRAZA revenue resulting primarily from the timing of shipments, which we expect to largely normalize throughout the remainder of 2024. Rest of world patient numbers have generally remained stable through the first quarter of 2024. We also saw a modest negative impact from increased competition and the unfavorable impact of foreign currency exchange. The decrease was partially offset by global SKYCLARYS revenue of $78.0 million in the first quarter of 2024.
| TOTAL COST AND EXPENSE | |||||
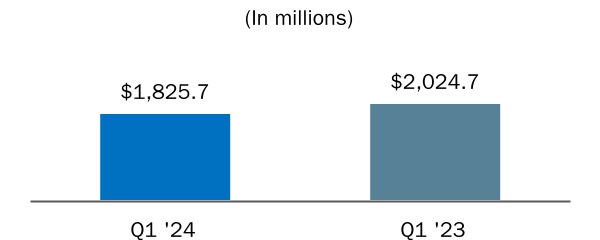
Decreased
$199.0 million or 9.8%
•Cost of sales decreased $120.6 million, or 18.2%
•R&D expense decreased $117.7 million, or 20.6%
•SG&A expense decreased $23.5 million, or 3.9%
•The decrease in cost of sales was primarily due to favorable product mix from decreased contract manufacturing revenue and lower idle capacity charges, offset in part by $44.1 million in amortization costs associated with the acquired SKYCLARYS inventory fair value step-up adjustment.
•The decrease in research and development expense was primarily driven by cost-reduction measures realized in 2024 in connection with our Fit for Growth program and clinical trial close out costs incurred in 2023 related to ADUHELM.
•The decrease in selling, general and administrative expense was primarily due to cost-reduction measures realized in 2024 in connection with our Fit for Growth program, offset by an increase in operational spending on sales and marketing activities in support of LEQEMBI and SKYCLARYS as we continue to expand our U.S. and international product launches.
| FINANCIAL CONDITION, LIQUIDITY AND CAPITAL RESOURCES | |||||
•Cash, cash equivalents and marketable securities totaled approximately $1.1 billion as of March 31, 2024, compared to approximately $1.0 billion as of December 31, 2023.
•We generated $553.2 million of net cash flow from operations for the three months ended March 31, 2024.
46
RECENT DEVELOPMENTS
DEVELOPMENTS IN KEY COLLABORATIVE RELATIONSHIPS
LEQEMBI (lecanemab)
United States
•In March 2024 Eisai completed the submission of LEQEMBI supplemental BLA for monthly IV maintenance dosing for the treatment of early Alzheimer's disease to the FDA.
Rest of World
•In March 2024 Eisai announced that deliberations at the CHMP regarding the MAA in the E.U. for lecanemab have been rescheduled due to procedural reasons at the EMA.
•In January 2024 the NMPA approved LEQEMBI in China, with an expected launch date in 2024.
OTHER KEY DEVELOPMENTS
QALSODY (tofersen)
In February 2024 we announced the CHMP of the EMA adopted a positive opinion recommending a MAA under exceptional circumstances for tofersen, for the treatment of adults with ALS associated with a mutation in the SOD1 gene. If authorized by the EC, tofersen will be the first treatment approved in the E.U. to target a genetic cause of ALS.
SKYCLARYS (omaveloxolone)
In February 2024 the EC approved SKYCLARYS in the E.U. for the treatment of FA in adults and adolescents aged 16 years and older. SKYCLARYS is the first treatment approved within the E.U. for this rare, genetic, progressive neurodegenerative disease.
SALE OF PRIORITY REVIEW VOUCHER
On April 24, 2024, we entered into a definitive agreement with a third party to sell a rare pediatric disease priority review voucher. In consideration for the PRV we received $103.0 million upon the closing of the PRV purchase.
DISCONTINUED PROGRAMS AND STUDIES
ACORDA COLLABORATION
In January 2024 we notified Acorda of our decision to terminate our collaboration and license agreement, effective January 1, 2025. As a result of this termination, Acorda will regain global commercialization rights to FAMPYRA. On April 1, 2024, Acorda filed for bankruptcy protection and announced its intention to sell substantially all its assets to a third party. We are evaluating the impact of these developments on the transition process for the FAMPYRA rights.
47
RESULTS OF OPERATIONS
REVENUE
The following revenue discussion should be read in conjunction with Note 5, Revenue, to our condensed consolidated financial statements included in this report.
Revenue is summarized as follows:
| For the Three Months Ended March 31, | ||||||||||||||||||||||||||||||||||||||
| (In millions, except percentages) | 2024 | 2023 | $ Change | % Change | ||||||||||||||||||||||||||||||||||
| Product revenue: | ||||||||||||||||||||||||||||||||||||||
| United States | $ | 746.1 | 32.6 | % | $ | 701.4 | 28.5 | % | $ | 44.7 | 6.4 | % | ||||||||||||||||||||||||||
| Rest of world | 965.8 | 42.1 | 1,061.9 | 43.1 | (96.1) | (9.0) | ||||||||||||||||||||||||||||||||
| Total product revenue, net | 1,711.9 | 74.7 | 1,763.3 | 71.6 | (51.4) | (2.9) | ||||||||||||||||||||||||||||||||
| Revenue from anti-CD20 therapeutic programs | 394.0 | 17.2 | 399.5 | 16.2 | (5.5) | (1.4) | ||||||||||||||||||||||||||||||||
Contract manufacturing, royalty and other revenue | 184.6 | 8.1 | 300.2 | 12.2 | (115.6) | (38.5) | ||||||||||||||||||||||||||||||||
| Total revenue | $ | 2,290.5 | 100.0 | % | $ | 2,463.0 | 100.0 | % | $ | (172.5) | (7.0) | % | ||||||||||||||||||||||||||
nm Not meaningful
48
PRODUCT REVENUE
Product revenue is summarized as follows:
| For the Three Months Ended March 31, | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2024 | 2023 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| (In millions) | United States | Rest of World | Total | % Total | United States | Rest of World | Total | % Total | $ Change | % Change | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Multiple Sclerosis | $ | 503.2 | $ | 572.7 | $ | 1,075.9 | 62.8 | % | $ | 546.1 | $ | 579.1 | $ | 1,125.2 | 63.8 | % | $ | (49.3) | (4.4) | % | ||||||||||||||||||||||||||||||||||||||||||
| Rare Disease | 225.9 | 198.0 | 423.9 | 24.8 | 146.7 | 296.6 | 443.3 | 25.2 | (19.4) | (4.4) | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Biosimilars | 3.7 | 193.2 | 196.9 | 11.5 | 8.2 | 184.2 | 192.4 | 10.9 | 4.5 | 2.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||
Other(1) | 13.3 | 1.9 | 15.2 | 0.9 | 0.4 | 2.0 | 2.4 | 0.1 | 12.8 | nm | ||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total product revenue, net | $ | 746.1 | $ | 965.8 | $ | 1,711.9 | 100.0 | % | $ | 701.4 | $ | 1,061.9 | $ | 1,763.3 | 100.0 | % | $ | (51.4) | (2.9) | % | ||||||||||||||||||||||||||||||||||||||||||
nm Not meaningful
(1) Other includes FUMADERM, ADUHELM and ZURZUVAE, which became commercially available in the U.S. during the fourth quarter of 2023.
49
| MULTIPLE SCLEROSIS | |||||
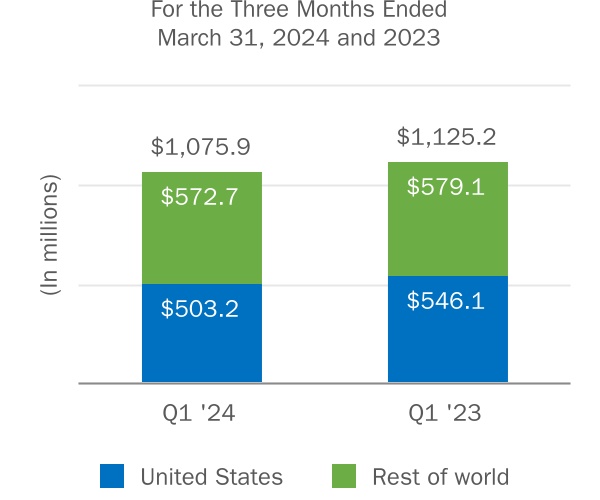
•Global TECFIDERA revenue decreased $20.2 million, from $274.5 million in 2023 to $254.3 million in 2024, or 7.4%, driven by a decrease in demand as a result of multiple TECFIDERA generic entrants in North America, Brazil and certain E.U. countries.
•Global Interferon revenue decreased $2.0 million, from $245.6 million in 2023 to $243.6 million in 2024, or 0.8%, driven by a decrease in demand as patients transition to higher efficacy therapies, partially offset by an favorable channel dynamics in U.S. Interferons.
•Global VUMERITY revenue increased $19.3 million, from $108.2 million in 2023 to $127.5 million in 2024, or 17.8%, primarily due to an increase in global demand and favorable channel dynamics and an increase in pricing in U.S. VUMERITY.
•Global TYSABRI revenue decreased $41.5 million, from $472.8 million in 2023 to $431.3 million in 2024, or 8.8%, primarily due to a decrease in U.S. TYSABRI revenue driven by increased competition and higher discounts and allowances.
In 2024 we expect total MS revenue will continue to decline as a result of increasing competition for many of our MS products in both the U.S. and rest of world markets. We are also aware of a biosimilar entrant of TYSABRI that was approved in the U.S. in August 2023 and the E.U. in September 2023. We believe that future sales of TYSABRI may be adversely affected by the entrance of this biosimilar.
50
| RARE DISEASE | |||||
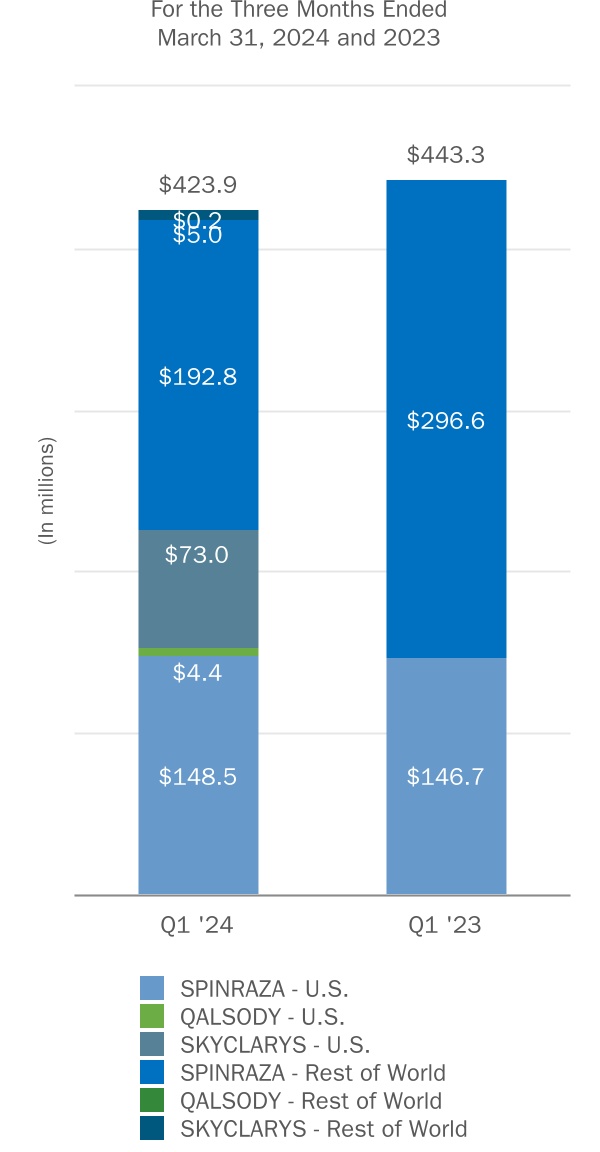
•U.S. SPINRAZA revenue increased $1.8 million, from $146.7 million in 2023 to $148.5 million in 2024, or 1.2%, primarily due to favorable net pricing.
•Rest of world SPINRAZA revenue decreased $103.8 million, from $296.6 million in 2023 to $192.8 million in 2024, or 35.0%. The majority of this year-over-year decline was due to the timing of shipments, which we expect to largely normalize throughout the remainder of 2024. Rest of world patient numbers have generally remained stable through the first quarter of 2024. We also saw a modest negative impact from increased competition and the unfavorable impact of foreign currency exchange.
•Global SKYCLARYS revenue was $78.0 million in 2024, including $73.0 million of U.S. SKYCLARYS revenue, which we began recognizing during the fourth quarter of 2023 subsequent to our acquisition of Reata, and $5.0 million of rest of world SKYCLARYS revenue, which was approved in the E.U. and became commercially available during the first quarter of 2024.
Rare disease revenue includes sales from SPINRAZA, QALSODY, which became commercially available in the U.S. during the second quarter of 2023, and SKYCLARYS, which was obtained as part of our acquisition of Reata in September 2023.
SKYCLARYS became commercially available in the U.S. during the second quarter of 2023 and we began recognizing revenue from SKYCLARYS in the U.S. during the fourth quarter of 2023, subsequent to our acquisition of Reata. In February 2024 the EC approved SKYCLARYS in the E.U. for the treatment of FA in adults and adolescents aged 16 years and older, which became commercially available in the E.U. during the first quarter of 2024.
In 2024 we expect growth in rare disease revenue as we continue to launch SKYCLARYS in the U.S. We expect global SPINRAZA revenue to decrease by low single digits.
51
| BIOSIMILARS | |||||
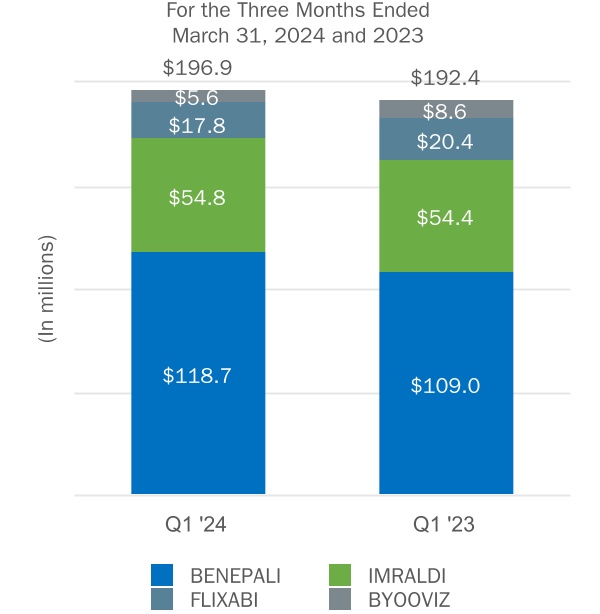
•For the three months ended March 31, 2024, compared to the same period of 2023, the increase in biosimilar revenue was primarily due to channel dynamics, offset in part by a decrease in pricing due to competitive pressures.
Biosimilars revenue includes sales from BENEPALI, IMRALDI, FLIXABI and BYOOVIZ. In 2023 BYOOVIZ became commercially available in certain international markets. During the third quarter of 2023 the FDA approved TOFIDENCE, a tocilizumab biosimilar referencing ACTEMRA, which we expect to become commercially available during 2024.
In 2024 we anticipate modest growth in revenue from our biosimilars business. We continue to work with our third-party contract manufacturers for IMRALDI and BENEPALI to address supply constraints. If not resolved these supply constraints could have an adverse impact on 2024 sales. In addition, one of our contract manufacturers for IMRALDI and BENEPALI entered into a proposed acquisition by a third party, which is expected to close at the end of 2024. We are currently evaluating the impact this will have on our biosimilars business and working to implement mitigation activities.
We continue to evaluate strategic options for our biosimilars business.
REVENUE FROM ANTI-CD20 THERAPEUTIC PROGRAMS
Our share of RITUXAN, including RITUXAN HYCELA, GAZYVA and LUNSUMIO collaboration operating profits in the U.S., royalty revenue on sales of OCREVUS and other revenue from anti-CD20 therapeutic programs are summarized in the table below. For purposes of this discussion, we refer to RITUXAN and RITUXAN HYCELA collectively as RITUXAN.
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Royalty revenue on sales of OCREVUS | $ | 302.7 | $ | 283.6 | ||||||||||
| Biogen’s share of pre-tax profits in the U.S. for RITUXAN, GAZYVA and LUNSUMIO | 87.1 | 112.5 | ||||||||||||
| Other revenue from anti-CD20 therapeutic programs | 4.2 | 3.4 | ||||||||||||
| Total revenue from anti-CD20 therapeutic programs | $ | 394.0 | $ | 399.5 | ||||||||||
ROYALTY REVENUE ON SALES OF OCREVUS
For the three months ended March 31, 2024, compared to the same period in 2023, the increase in royalty revenue on sales of OCREVUS was primarily due to sales growth of OCREVUS in the U.S.
OCREVUS royalty revenue is based on our estimates from third party and market research data of OCREVUS sales occurring during the corresponding period. Differences between actual and estimated royalty revenue will be adjusted for in the period in which they become known, which is generally expected to be the following quarter.
52
BIOGEN'S SHARE OF PRE-TAX PROFITS IN THE U.S. FOR RITUXAN, GAZYVA AND LUNSUMIO
For the three months ended March 31, 2024, compared to the same period in 2023, the decrease in our share of pre-tax profits in the U.S. for RITUXAN, GAZYVA and LUNSUMIO was primarily due to a decrease in sales of RITUXAN in the U.S. resulting from competition from multiple biosimilar products.
Prior to regulatory approval, we record our share of the expense incurred by the collaboration for the development of anti-CD20 products in research and development expense and pre-commercialization costs within selling, general and administrative expense in our condensed consolidated statements of income. After an anti-CD20 product is approved, we record our share of the development and sales and marketing expense related to that product as a reduction of our share of pre-tax profits in revenue from anti-CD20 therapeutic programs.
OTHER REVENUE FROM ANTI-CD20 THERAPEUTIC PROGRAMS
Other revenue from anti-CD20 therapeutic programs consists of our share of pre-tax co-promotion profits from RITUXAN in Canada, royalty revenue on sales of LUNSUMIO outside the U.S. and royalty revenue on net sales of COLUMVI in the U.S., which became commercially available during the second quarter of 2023.
For additional information on our collaboration arrangements with Genentech, including information regarding the pre-tax profit-sharing formula and its impact on future revenue from anti-CD20 therapeutic programs, please read Note 19, Collaborative and Other Relationships, to our condensed consolidated financial statements included in this report.
CONTRACT MANUFACTURING, ROYALTY AND OTHER REVENUE
Contract manufacturing, royalty and other revenue is summarized as follows:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Contract manufacturing revenue | $ | 152.2 | $ | 306.9 | ||||||||||
Royalty and other revenue | 32.4 | (6.7) | ||||||||||||
| Total contract manufacturing, royalty and other revenue | $ | 184.6 | $ | 300.2 | ||||||||||
CONTRACT MANUFACTURING REVENUE
For the three months ended March 31, 2024, compared to the same period in 2023, the decrease in contract manufacturing revenue was primarily driven by higher volumes in 2023 due to the timing of batch production, which includes batches related to LEQEMBI that we began recognizing in the first quarter of 2023 upon the accelerated approval of LEQEMBI in the U.S.
In addition, as part of the 2020 sale of our Hillerød, Denmark manufacturing operations to FUJIFILM, we provided FUJIFILM with certain minimum batch production commitment guarantees, including batches related to our contract manufacturing arrangements. As of December 31, 2023, these batch commitments have been satisfied and we expect that our contract manufacturing revenue will be lower in 2024, compared to 2023, as we are no longer supplying contract manufacturing customers using Hillerød in this manner.
ROYALTY AND OTHER REVENUE
Royalty and other revenue primarily reflects the royalties we receive from net sales on products related to patents that we have out-licensed, as well as royalty revenue on biosimilar products from our license arrangements with Samsung Bioepis and our 50.0% share of LEQEMBI product revenue, net and cost of sales, including royalties, as we are not the principal.
For additional information on our collaboration arrangements with Samsung Bioepis and Eisai, please read Note 19, Collaborative and Other Relationships, to our condensed consolidated financial statements included in this report.
53
RESERVES FOR DISCOUNTS AND ALLOWANCES
Revenue from product sales is recorded net of reserves established for applicable discounts and allowances, including those associated with the implementation of pricing actions in certain international markets where we operate.
Reserves for discounts, contractual adjustments and returns that reduced gross product revenue are summarized as follows:
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Contractual adjustments | $ | 665.7 | $ | 629.9 | ||||||||||
| Discounts | 187.6 | 181.2 | ||||||||||||
| Returns | 10.6 | 4.5 | ||||||||||||
| Total discounts and allowances | $ | 863.9 | $ | 815.6 | ||||||||||
For the three months ended March 31, 2024, reserves for discounts and allowances as a percentage of gross product revenue was 33.2% compared to 31.4% in the prior year comparative period.
CONTRACTUAL ADJUSTMENTS
Contractual adjustments primarily relate to Medicaid and managed care rebates in the U.S., pharmacy rebates, co-payment (copay) assistance, VA, 340B discounts, specialty pharmacy program fees and other government rebates or applicable allowances.
For the three months ended March 31, 2024, compared to the same period in 2023, the increase in contractual adjustments was primarily due to higher Medicaid and government rebates in the U.S., partially offset by lower government rebates in rest of world.
DISCOUNTS
Discounts include trade term discounts and wholesaler incentives.
For the three months ended March 31, 2024, compared to the same period in 2023, the increase in discounts was primarily driven by higher purchase and volume discounts for biosimilars.
RETURNS
Product return reserves are established for returns made by wholesalers. In accordance with contractual terms, wholesalers are permitted to return product for reasons such as damaged or expired product. The majority of wholesaler returns are due to product expiration. Provisions for estimated product returns are recognized in the period the related revenue is recognized, resulting in a reduction to product sales.
For the three months ended March 31, 2024, compared to the same period in 2023, the increase in returns was primarily driven by higher return rates in the U.S.
For additional information on our revenue reserves, please read Note 5, Revenue, to our condensed consolidated financial statements included in this report.
54
COST AND EXPENSE
A summary of total cost and expense is as follows:
| For the Three Months Ended March 31, | ||||||||||||||||||||||||||
| (In millions, except percentages) | 2024 | 2023 | $ Change | % Change | ||||||||||||||||||||||
| Cost of sales, excluding amortization and impairment of acquired intangible assets | $ | 542.2 | $ | 662.8 | $ | (120.6) | (18.2) | % | ||||||||||||||||||
| Research and development | 452.9 | 570.6 | (117.7) | (20.6) | ||||||||||||||||||||||
| Selling, general and administrative | 581.5 | 605.0 | (23.5) | (3.9) | ||||||||||||||||||||||
| Amortization and impairment of acquired intangible assets | 78.3 | 50.2 | 28.1 | 56.0 | ||||||||||||||||||||||
| Collaboration profit sharing/(loss reimbursement) | 65.6 | 57.1 | 8.5 | 14.9 | ||||||||||||||||||||||
| Restructuring charges | 11.5 | 9.6 | 1.9 | 19.8 | ||||||||||||||||||||||
| Other (income) expense, net | 93.7 | 69.4 | 24.3 | 35.0 | ||||||||||||||||||||||
| Total cost and expense | $ | 1,825.7 | $ | 2,024.7 | $ | (199.0) | (9.8) | % | ||||||||||||||||||
55
COST OF SALES, EXCLUDING AMORTIZATION AND IMPAIRMENT OF ACQUIRED INTANGIBLE ASSETS
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions) | 2024 | 2023 | ||||||||||||
| Product | $ | 377.7 | $ | 481.4 | ||||||||||
| Royalty | 164.5 | 181.4 | ||||||||||||
| Total cost of sales | $ | 542.2 | $ | 662.8 | ||||||||||
For the three months ended March 31, 2024, compared to the same period in 2023, the decrease in product cost of sales was primarily due to lower cost of sales associated with contract manufacturing revenue and lower idle capacity charges, offset in part by $44.1 million in SKYCLARYS amortization costs. Contract manufacturing revenue includes LEQEMBI inventory produced for Eisai, beginning in the first quarter of 2023 upon the accelerated approval of LEQEMBI in the U.S. Cost of sales as a percentage of revenue was adversely affected by LEQEMBI batches due to minimal margins.
As a result of our acquisition of Reata in September 2023 we recorded a fair value step-up adjustment related to the acquired inventory of SKYCLARYS of approximately $1.3 billion. This fair value step-up adjustment will be amortized to cost of sales within our condensed consolidated statements of income when the inventory is sold, which is expected to be within approximately 3 years from the acquisition date. For the three months ended March 31, 2024, amortization from the fair value step-up adjustment, associated with SKYCLARYS, as a result of inventory sold was approximately $44.1 million. For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to our condensed consolidated financial statements included in this report.
Write Downs and Other Charges
For the three months ended March 31, 2023, we recorded approximately $44.8 million of aggregate gross idle capacity charges.
For additional information on our collaboration arrangements with Eisai, please read Note 19, Collaborative and Other Relationships, to our condensed consolidated financial statements included in this report.
56
RESEARCH AND DEVELOPMENT
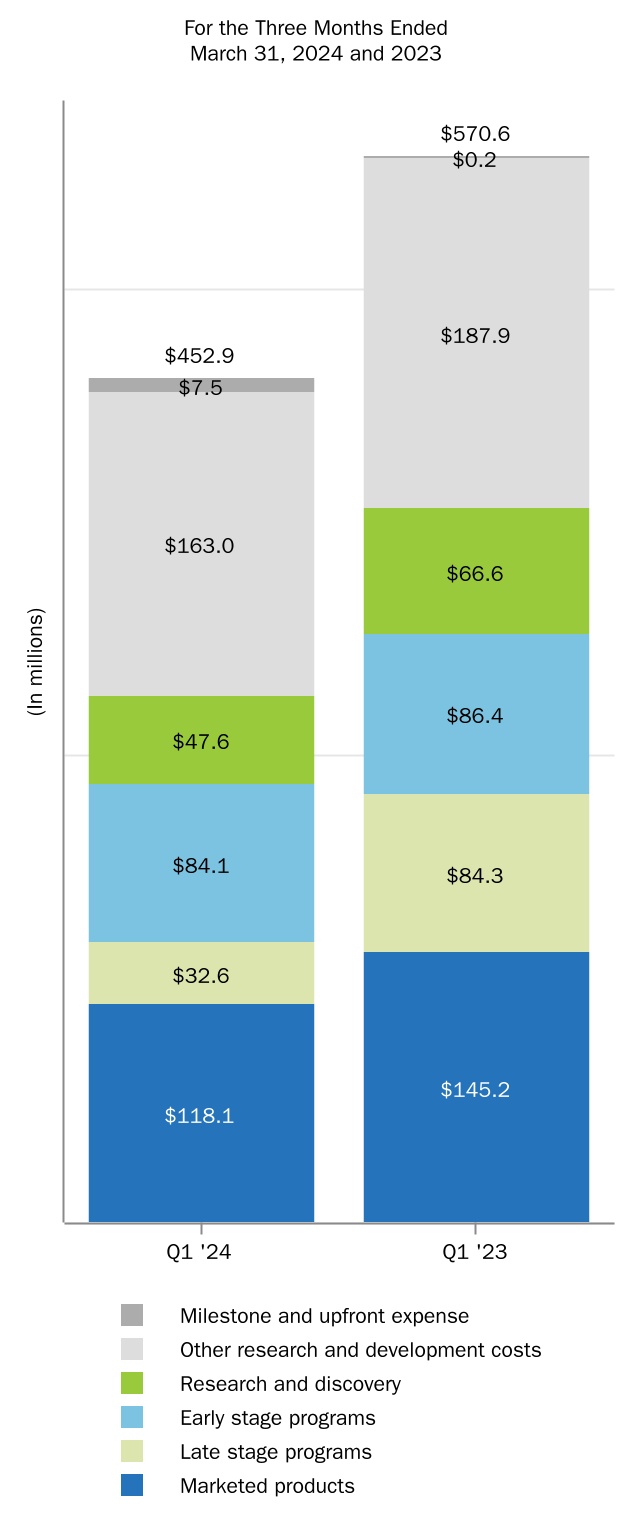
Research and development expense, as a percentage of total revenue, was 19.8% and 23.2% for the three months ended March 31, 2024 and 2023, respectively. For the three months ended March 31, 2024, compared to the same period in 2023, the decrease in research and development was primarily driven by cost-reduction measures realized in 2024 in connection with our Fit for Growth program and clinical trial close out costs incurred in 2023 related to ADUHELM.
EARLY STAGE PROGRAMS
Q1 2024 vs. Q1 2023
The decrease in early stage programs was driven by a decrease in costs associated with:
•discontinuation of BIIB131 for the treatment of acute ischemic stroke; and
•discontinuation of BIIB135 for the treatment of MS.
The decrease was partially offset by an increase in costs associated with:
•development of cemdomespib for the treatment of diabetic neuropathic pain; and
•development of BIIB080 for the treatment of Alzheimer's disease.
LATE STAGE PROGRAMS
Q1 2024 vs. Q1 2023
The decrease in late stage programs was driven by a decrease in costs associated with:
•advancement of ZURZUVAE from late stage to marketed upon the approval of ZURZUVAE for PPD in the U.S.;
•advancement of QALSODY from late stage to marketed upon the accelerated approval of QALSODY in the U.S.;
•discontinuation of BIIB093 for large hemispheric infarction; and
•advancement of TOFIDENCE, a tocilizumab biosimilar referencing ACTEMRA, from late stage to marketed upon the approval of TOFIDENCE in the U.S.
MARKETED PROGRAMS
Q1 2024 vs. Q1 2023
The decrease in marketed programs was driven by a decrease in costs associated with:
•discontinuation of ADUHELM for the treatment of Alzheimer's disease; and
•decreased spend in LEQEMBI due to the timing of clinical spend.
The decrease was partially offset by an increase in costs associated with:
•advancement of QALSODY from late stage to marketed upon the accelerated approval of QALSODY in the U.S.; and
•increased spend in SKYCLARYS as a result of our acquisition of Reata in September 2023.
57
Research and development expense is reported above based on the following classifications. The development stage reported is based upon the program status when incurred. Therefore, the same program could be reflected in different development stages in the same year. For several of our programs, the research and development activities are part of our collaborative and other relationships. Our costs reflect our share of the total costs incurred.
•Research and discovery: represents costs incurred to support our discovery research and translational science efforts.
•Early stage programs: are programs in Phase 1 or Phase 2 development.
•Late stage programs: are programs in Phase 3 development or in registration stage.
•Marketed products: includes costs associated with product lifecycle management activities including, if applicable, costs associated with the development of new indications for existing products.
•Other research and development costs: A significant amount of our research and development costs consist of indirect costs incurred in support of overall research and development activities and non-specific programs, including activities that benefit multiple programs, such as management costs, as well as depreciation, information technology and facility-based expenses. These costs are considered other research and development costs in the table above and are not allocated to a specific program or stage.
Excluding any milestone and upfront payments, we expect our core research and development expense to decrease in 2024, while continuing to invest in our pipeline. This is primarily due to the continued realization of our cost savings initiatives. We intend to continue committing significant resources to targeted research and development opportunities where there is a significant unmet need and where a drug candidate has the potential to be highly differentiated.
SELLING, GENERAL AND ADMINISTRATIVE
For the three months ended March 31, 2024, compared to the same period in 2023, selling, general and administrative expense decreased by approximately 3.9% primarily due to cost-reduction measures realized in 2024 in connection with our Fit for Growth program, offset by an increase in operational spending on sales and marketing activities in support of LEQEMBI and SKYCLARYS as we continue to expand our U.S. and international product launches. For the three months ended March 31, 2023, selling, general and administrative expense included a $31.0 million obligation to Eisai related to the termination of the co-promotion agreement for our MS products in Japan during the first quarter of 2023. General and administrative expense decreased by approximately $8.8 million, or 5.1%, in the first quarter of 2024, compared to the same period in 2023, due to cost-reduction measures. Excluding exit-related costs and acquisition-related expenses incurred during the first quarter of 2024, general and administrative expense decreased by approximately $19.3 million, or 11.3%.
For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to our condensed consolidated financial statements included in this report. For additional information on our collaboration arrangements with Eisai, please read Note 19, Collaborative and Other Relationships, to our condensed consolidated financial statements included in this report.
AMORTIZATION AND IMPAIRMENT OF ACQUIRED INTANGIBLE ASSETS
Our amortization expense is based on the economic consumption and impairment of intangible assets. Our most significant amortizable intangible assets are related to TYSABRI, AVONEX, SPINRAZA, VUMERITY and SKYCLARYS, which was obtained as part of our acquisition of Reata in September 2023. For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to our condensed consolidated financial statements included in this report.
For the three months ended March 31, 2024, compared to the same period in 2023, the increase in amortization and impairment of acquired intangible assets was primarily due to amortization for the Reata acquisition acquired intangible assets associated with SKYCLARYS. For the three months ended March 31, 2024 and 2023, we had no impairment charges.
For additional information on the amortization and impairment of our acquired intangible assets, please read Note 7, Intangible Assets and Goodwill, to our condensed consolidated financial statements included in this report.
58
COLLABORATION PROFIT SHARING/(LOSS REIMBURSEMENT)
Collaboration profit sharing/(loss reimbursement) primarily includes Samsung Bioepis' 50.0% share of the profit or loss related to our biosimilars 2013 commercial agreement with Samsung Bioepis. In the third quarter of 2023 we began recognizing collaboration profit sharing/(loss reimbursement) related to Sage's 50.0% share of income and expense in the U.S. related to ZURZUVAE for PPD.
For the three months ended March 31, 2024, we recognized net profit-sharing expense of $60.6 million to reflect Samsung Bioepis' 50.0% share of the net collaboration profits, compared to a net profit-sharing expense of $57.1 million in the prior year comparative period.
For the three months ended March 31, 2024, we recognized net profit-sharing expense of approximately $5.0 million to reflect Sage's 50.0% share of net collaboration results in the U.S. for ZURZUVAE for PPD.
For additional information on our collaboration and license arrangements with Samsung Bioepis and Sage, please read Note 19, Collaborative and Other Relationships, to our condensed consolidated financial statements included in this report.
RESTRUCTURING CHARGES
2023 FIT FOR GROWTH RESTRUCTURING PROGRAM
In July 2023 we initiated additional cost saving measures as part of our Fit for Growth program to reduce operating costs, while improving operating efficiency and effectiveness. The Fit for Growth program is expected to generate approximately $1.0 billion in gross operating expense savings by the end of 2025, some of which will be reinvested in various initiatives. The Fit for Growth program is currently estimated to include net headcount reductions of approximately 1,000 employees and we expect to incur restructuring charges ranging from approximately $260.0 million to $280.0 million.
Total charges incurred from our 2023 cost saving initiatives are summarized as follows:
| For the Three Months Ended March 31, 2024 | ||||||||||||||||||||
| (In millions) | Severance Costs | Accelerated Depreciation and Other Costs | Total | |||||||||||||||||
| Selling, general and administrative | $ | — | $ | 1.4 | $ | 1.4 | ||||||||||||||
| Research and development | — | 4.9 | 4.9 | |||||||||||||||||
| Restructuring charges | 9.3 | — | 9.3 | |||||||||||||||||
| Total charges | $ | 9.3 | $ | 6.3 | $ | 15.6 | ||||||||||||||
Other Costs: includes costs associated with items such as asset abandonment and write-offs, facility closure costs, pretax gains and losses resulting from the termination of certain leases, employee non-severance expense, consulting fees and other costs.
REATA INTEGRATION
Following the close of our Reata acquisition in September 2023, we implemented an integration plan designed to realize operating synergies through cost savings and avoidance. Under this initiative, we estimate we will incur total integration charges ranging from approximately $35.0 million to $40.0 million, related to severance and employment costs, which are expected to be paid by the end of 2024. These amounts were substantially incurred during 2023.
59
Total charges incurred from our Reata integration are summarized as follows:
| For the three months ended March 31, 2024 | |||||||||||||||||||||||
| (In millions) | Severance Costs | Accelerated Depreciation and Other Costs | Total | ||||||||||||||||||||
| Selling, general and administrative | $ | — | $ | 1.8 | $ | 1.8 | |||||||||||||||||
| Research and development | — | 2.7 | 2.7 | ||||||||||||||||||||
| Restructuring charges | 2.2 | — | 2.2 | ||||||||||||||||||||
| Total charges | $ | 2.2 | $ | 4.5 | $ | 6.7 | |||||||||||||||||
In connection with our acquisition of Reata we assumed responsibility for a single-tenant, build-to-suit building of approximately 327,400 square feet of office and laboratory space located in Plano, Texas, with an initial lease term of 16 years. We do not intend to occupy this building and are evaluating opportunities to sublease the property.
For additional information on our cost saving initiatives, please read Note 4, Restructuring, to our condensed consolidated financial statements included in this report.
OTHER (INCOME) EXPENSE, NET
For the three months ended March 31, 2024, compared to the same period in 2023, the change in other (income) expense, net primarily reflects lower interest income driven by lower cash balances in 2024, compared to the same period in 2023, partially offset by a decrease in our net unrealized losses on our holdings in equity securities.
NET (GAINS) LOSSES IN EQUITY SECURITIES
For the three months ended March 31, 2024, net unrealized and realized losses on our holdings in equity securities were approximately $25.7 million and $4.9 million, respectively, compared to net unrealized and realized losses of approximately $76.5 million and $1.6 million, respectively, in the prior year comparative period.
•The net unrealized losses recognized during the three months ended March 31, 2024, primarily reflect a decrease in the aggregate fair value of our investments in Sage and Denali common stock of approximately $27.7 million, partially offset by an increase in the fair value of Sangamo common stock of approximately $1.4 million.
•The net unrealized losses recognized during the three months ended March 31, 2023, primarily reflect a decrease in the aggregate fair value of our investments in Denali, Sangamo and Ionis common stock of approximately $100.0 million, partially offset by an increase in the fair value of Sage common stock of approximately $23.8 million.
INCOME TAX PROVISION
| For the Three Months Ended March 31, | ||||||||||||||
| (In millions, except percentages) | 2024 | 2023 | ||||||||||||
| Income before income tax (benefit) expense | $ | 464.8 | $ | 438.3 | ||||||||||
| Income tax (benefit) expense | 71.4 | 50.7 | ||||||||||||
| Effective tax rate | 15.4 | % | 11.6 | % | ||||||||||
Our effective tax rate fluctuates from year to year due to the global nature of our operations. The factors that most significantly impact our effective tax rate include changes in tax laws, variability in the allocation of our taxable earnings among multiple jurisdictions, the amount and characterization of our research and development expense, the levels of certain deductions and credits, acquisitions and licensing transactions.
For the three months ended March 31, 2024, compared to the same period in 2023, the increase in our effective tax rate includes the impact of a resolution of an uncertain tax matter in the prior year related to tax credits and the non-cash tax effects of changes in the value of our equity investments, where we recorded larger unrealized losses in the prior year.
PILLAR TWO
The OECD has issued model rules, which generally provide for a jurisdictional minimum effective tax rate of 15.0%. Various countries have or are in the process of enacting legislation intended to implement the principles effective
60
January 1, 2024. Our income tax provision for the three months ended March 31, 2024, reflects currently enacted legislation and guidance related to the OECD model rules.
For additional information on our income taxes, please read Note 17, Income Taxes, to our condensed consolidated financial statements included in this report.
FINANCIAL CONDITION, LIQUIDITY AND CAPITAL RESOURCES
Our financial condition is summarized as follows:
| (In millions, except percentages) | As of March 31, 2024 | As of December 31, 2023 | $ Change | % Change | ||||||||||||||||||||||
| Financial assets: | ||||||||||||||||||||||||||
| Cash and cash equivalents | $ | 1,074.4 | $ | 1,049.9 | $ | 24.5 | 2.3 | % | ||||||||||||||||||
| Marketable securities — current | — | — | — | — | ||||||||||||||||||||||
| Marketable securities — non-current | — | — | — | — | ||||||||||||||||||||||
| Total cash, cash equivalents and marketable securities | $ | 1,074.4 | $ | 1,049.9 | $ | 24.5 | 2.3 | % | ||||||||||||||||||
| Borrowings: | ||||||||||||||||||||||||||
Current portion of term loan | $ | 250.0 | $ | 150.0 | $ | 100.0 | 66.7 | % | ||||||||||||||||||
Notes payable and term loan | 6,290.1 | 6,788.2 | (498.1) | (7.3) | ||||||||||||||||||||||
| Total borrowings | $ | 6,540.1 | $ | 6,938.2 | $ | (398.1) | (5.7) | % | ||||||||||||||||||
| Working capital: | ||||||||||||||||||||||||||
| Current assets | $ | 6,756.2 | $ | 6,859.3 | $ | (103.1) | (1.5) | % | ||||||||||||||||||
| Current liabilities | (3,222.8) | (3,434.3) | 211.5 | (6.2) | ||||||||||||||||||||||
| Total working capital | $ | 3,533.4 | $ | 3,425.0 | $ | 108.4 | 3.2 | % | ||||||||||||||||||
OVERVIEW
We have historically financed and expect to continue to fund our operating and capital expenditures primarily through cash flow earned through our operations, as well as our existing cash resources. We believe that generic and biosimilar competition for many of our key products, the continued overall decline of our MS business and our investments in the launch of key new products and the development of our pipeline will have a significant adverse impact on our future cash flow from operations.
We believe that our existing funds, when combined with cash generated from operations and our access to additional financing resources, if needed, are sufficient to satisfy our operating, working capital, strategic alliance, milestone payment, capital expenditure and debt service requirements for the foreseeable future. In addition, we may choose to opportunistically return cash to shareholders and pursue other business initiatives, including acquisition and licensing activities. We may also seek additional funding through a combination of new collaborative agreements, strategic alliances and additional equity and debt financings or from other sources should we identify a significant new opportunity.
In April 2024 we received the remaining $437.5 million payment from Samsung BioLogics related to the sale of Samsung Bioepis. Additionally, in 2024 we plan to repay the remaining $250.0 million balance of our 2023 Term Loan three-year tranche.
For additional information on certain risks that could negatively impact our financial position or future results of operations, please read Item 1A. Risk Factors and Item 3. Quantitative and Qualitative Disclosures About Market Risk included in this report.
LIQUIDITY
WORKING CAPITAL
Working capital is defined as current assets less current liabilities. Our working capital was $3.5 billion and $3.4 billion as of March 31, 2024 and December 31, 2023, respectively. The change in working capital reflects a decrease in total current assets of approximately $103.1 million and a decrease in total current liabilities of approximately $211.5 million. The changes in total current assets and total current liabilities were primarily driven by the following:
61
CURRENT ASSETS
•$24.5 million increase in cash, cash equivalents and current marketable securities;
•$59.6 million decrease in accounts receivable, net related to our ongoing operations; and
•$16.7 million decrease in other current assets partially due to the sale of a portion of our short-term strategic investments.
CURRENT LIABILITIES
•$269.0 million decrease in accrued expense and other primarily due to the timing of our annual incentive compensation payment; and
•$100.0 million increase in current portion of debt due to the reclassification of $250.0 million from long-term to short-term related to the three-year tranche of our 2023 Term Loan, partially offset by the repayment of $150.0 million related to the 365-day tranche of our 2023 Term Loan.
For additional information on the sale of our equity interest in Samsung Bioepis, please read Note 3, Dispositions, to our condensed consolidated financial statements included in this report.
CASH, CASH EQUIVALENTS AND MARKETABLE SECURITIES
As of March 31, 2024, we had cash, cash equivalents and marketable securities totaling approximately $1.1 billion compared to approximately $1.0 billion as of December 31, 2023. The increase in the balance was primarily due to cash flows from operations, partially offset by $400.0 million of cash used for the repayment of our 2023 Term Loan.
Until required for another use in our business, we typically invest our cash reserves in bank deposits, certificates of deposit, commercial paper, corporate notes, U.S. and foreign government instruments, overnight reverse repurchase agreements and other interest-bearing marketable debt instruments in accordance with our investment policy. It is our policy to mitigate credit risk in our cash reserves and marketable securities by maintaining a well-diversified portfolio that limits the amount of exposure as to institution, maturity and investment type. We have experienced no significant limitations in our liquidity resulting from uncertainties in the banking sector.
The following table summarizes the fair value of our significant common stock investments in our strategic investment portfolio:
| (In millions) | March 31, 2024 | December 31, 2023 | ||||||||||||
| Denali | $ | 205.4 | $ | 273.6 | ||||||||||
| Sage | 117.0 | 135.3 | ||||||||||||
| Sangamo | 7.1 | 7.9 | ||||||||||||
| Total | $ | 329.5 | $ | 416.8 | ||||||||||
Our ability to liquidate our investments in Denali, Sage and Sangamo may be limited by the size of our interest, the volume of market related activity, our concentrated level of ownership and potential restrictions resulting from our status as a collaborator. Therefore, we may realize significantly less than the current value of such investments.
For additional information on our collaboration arrangements, please read Note 19, Collaborative and Other Relationships, to our condensed consolidated financial statements included in this report.
CASH FLOW
The following table summarizes our cash flow activity:
| For the Three Months Ended March 31, | ||||||||||||||||||||
| (In millions, except percentages) | 2024 | 2023 | % Change | |||||||||||||||||
| Net cash flow provided by (used in) operating activities | $ | 553.2 | $ | 455.3 | 21.5 | % | ||||||||||||||
| Net cash flow provided by (used in) investing activities | (66.0) | (953.0) | (93.1) | |||||||||||||||||
| Net cash flow provided by (used in) financing activities | (439.6) | (43.4) | 912.9 | |||||||||||||||||
62
OPERATING ACTIVITIES
Operating cash flow is derived by adjusting our net income for:
•non-cash operating items such as depreciation and amortization, impairment charges, unrealized (gain) loss on strategic investments and share-based compensation;
•changes in operating assets and liabilities, which reflect timing differences between the receipt and payment of cash associated with transactions and when they are recognized in results of operations; and
•(gains) losses on the disposal of assets, deferred income taxes, changes in the fair value of contingent payments associated with our acquisitions of businesses and acquired IPR&D.
For the three months ended March 31, 2024, compared to the same period in 2023, the increase in net cash flow provided by operating activities was primarily due to lower employee-benefit payments made during the first quarter of 2024, as compared to the same period in 2023, timing of payments and changes in non-cash adjustments to net income. The increase was partially offset by an unfavorable change in inventory.
INVESTING ACTIVITIES
For the three months ended March 31, 2024, compared to the same period in 2023, the decrease in net cash flow used in investing activities was primarily due to higher net purchases of marketable securities in 2023.
FINANCING ACTIVITIES
For the three months ended March 31, 2024, compared to the same period in 2023, the increase in net cash flow used in financing activities was primarily due to the repayment of our 2023 Term Loan for $400.0 million during the first quarter of 2024.
For additional information on our acquisition of Reata, please read Note 2, Acquisitions, to our condensed consolidated financial statements included in this report.
CAPITAL RESOURCES
DEBT AND CREDIT FACILITIES
LONG-TERM DEBT AND TERM LOAN CREDIT AGREEMENTS
Our long-term obligations primarily consist of long-term debt related to our Senior Notes with final maturity dates ranging between 2025 and 2051. As of March 31, 2024, our outstanding balance related to long-term debt was $6,290.1 million.
In connection with our acquisition of Reata in September 2023 we entered into a $1.5 billion term loan credit agreement (2023 Term Loan). On the closing date of the Reata acquisition we drew $1.0 billion from the 2023 Term Loan, comprised of a $500.0 million floating rate 364-day tranche and a $500.0 million floating rate three-year tranche. The remaining unused commitment of $500.0 million was terminated. As of December 31, 2023, we repaid $350.0 million of the 364--day tranche. The remaining $150.0 million portion of the 364-day tranche was subsequently paid during the first quarter of 2024. Additionally, during the first quarter of 2024 we repaid $250.0 million of the three-year tranche. As of March 31, 2024, we had $250.0 million outstanding under the three-year tranche of the 2023 Term Loan.
2020 REVOLVING CREDIT FACILITY
In January 2020 we entered into a $1.0 billion, five-year senior unsecured revolving credit facility under which we are permitted to draw funds for working capital and general corporate purposes. The terms of the revolving credit facility include a financial covenant that requires us not to exceed a maximum consolidated leverage ratio. As of March 31, 2024 and December 31, 2023, we had no outstanding borrowings and were in compliance with all covenants under this facility.
For a summary of the fair and carrying values of our outstanding borrowings as of March 31, 2024 and December 31, 2023, please read Note 8, Fair Value Measurements, to our condensed consolidated financial statements included in this report.
For additional information on our Senior Notes and credit facility please read, Note 13, Indebtedness, to our consolidated financial statements included in our 2023 Form 10-K.
63
SHARE REPURCHASE PROGRAMS
In October 2020 our Board of Directors authorized our 2020 Share Repurchase Program, which is a program to repurchase up to $5.0 billion of our common stock. Our 2020 Share Repurchase Program does not have an expiration date. All share repurchases under our 2020 Share Repurchase Program will be retired. There were no share repurchases of our common stock during the three months ended March 31, 2024 and 2023. Approximately $2.1 billion remained available under our 2020 Share Repurchase Program as of March 31, 2024.
CONTRACTUAL OBLIGATIONS AND OFF-BALANCE SHEET ARRANGEMENTS
CONTRACTUAL OBLIGATIONS
Our contractual obligations primarily consist of our obligations under non-cancellable operating leases, long-term debt obligations and defined benefit and other purchase obligations, excluding amounts related to uncertain tax positions, funding commitments, contingent development, regulatory and commercial milestone payments and contingent payments, as described below.
In addition, certain of our collaboration and licensing arrangements include royalty payment obligations. For additional information on our royalty payments please read, Note 22, Commitments and Contingencies, to our consolidated financial statements included in our 2023 Form 10-K.
There have been no material changes in our contractual obligations since December 31, 2023.
CONTINGENT DEVELOPMENT, REGULATORY AND COMMERCIAL MILESTONE PAYMENTS
Based on our development plans as of March 31, 2024, we could trigger potential future milestone payments to third parties of up to approximately $5.1 billion, including approximately $1.0 billion in development milestones, approximately $0.4 billion in regulatory milestones and approximately $3.7 billion in commercial milestones, as part of our various collaborations, including licensing and development programs. Payments under these agreements generally become due and payable upon achievement of certain development, regulatory or commercial milestones. Because the achievement of these milestones was not considered probable as of March 31, 2024, such contingencies have not been recorded in our financial statements. Amounts related to contingent milestone payments are not considered contractual obligations as they are contingent on the successful achievement of certain development, regulatory or commercial milestones.
If certain clinical and commercial milestones are met, we may pay up to approximately $82.5 million in milestones in 2024 under our current agreements. This excludes potential opt-in payments.
OTHER FUNDING COMMITMENTS
As of March 31, 2024, we have several ongoing clinical studies in various clinical trial stages. Our most significant clinical trial expenditures are to CROs. The contracts with CROs are generally cancellable, with notice, at our option. We recorded accrued expense of approximately $40.6 million in our condensed consolidated balance sheets for expenditures incurred by CROs as of March 31, 2024. We have approximately $607.3 million in cancellable future commitments based on existing CRO contracts as of March 31, 2024.
TAX RELATED OBLIGATIONS
We exclude liabilities pertaining to uncertain tax positions from our summary of contractual obligations as we cannot make a reliable estimate of the period of cash settlement with the respective taxing authorities. As of March 31, 2024, we have approximately $160.4 million of liabilities associated with uncertain tax positions.
As of March 31, 2024 and December 31, 2023, we have accrued income tax liabilities of approximately $421.3 million and $419.5 million, respectively, under the Transition Toll Tax. Of the amounts accrued as of March 31, 2024, approximately $187.3 million is expected to be paid within one year. The Transition Toll Tax is being paid in installments over an eight-year period, which started in 2018, and will not accrue interest.
NEW ACCOUNTING STANDARDS
For a discussion of new accounting standards please read Note 1, Summary of Significant Accounting Policies, to our condensed consolidated financial statements included in this report.
64
CRITICAL ACCOUNTING ESTIMATES
The preparation of our condensed consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the U.S., requires us to make estimates, judgments and assumptions that may affect the reported amounts of assets, liabilities, equity, revenue and expense and related disclosure of contingent assets and liabilities. On an ongoing basis we evaluate our estimates, judgments and assumptions. We base our estimates on historical experience and on various other assumptions that we believe are reasonable, the results of which form the basis for making judgments about the carrying values of assets, liabilities and equity and the amount of revenue and expense. Actual results may differ from these estimates.
There have been no material changes to our critical accounting estimates since our 2023 Form 10-K. For a discussion of our other critical accounting estimates, please read Part II, Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations in our 2023 Form 10-K.
ITEM 3. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
We are subject to certain risks that may affect our results of operations, cash flow and fair values of assets and liabilities, including volatility in foreign currency exchange rates, interest rate movements and equity price exposure as well as changes in economic conditions in the markets in which we operate as a result of the conflict between Russia and Ukraine and the military conflict in the Middle East. We manage the impact of foreign currency exchange rates and interest rates through various financial instruments, including derivative instruments such as foreign currency forward contracts, foreign currency options, interest rate lock contracts and interest rate swap contracts. We do not enter into financial instruments for trading or speculative purposes. The counterparties to these contracts are major financial institutions, and there is no significant concentration of exposure with any one counterparty.
FOREIGN CURRENCY EXCHANGE RISK
Our results of operations are subject to foreign currency exchange rate fluctuations due to the global nature of our operations. As a result, our consolidated financial position, results of operations and cash flow can be affected by market fluctuations in foreign currency exchange rates, primarily with respect to the Euro, British pound sterling, Canadian dollar and Swiss franc.
While the financial results of our global activities are reported in U.S. dollars, the functional currency for most of our foreign subsidiaries is their respective local currency. Fluctuations in the foreign currency exchange rates of the countries in which we do business will affect our operating results, often in ways that are difficult to predict. In particular, as the U.S. dollar strengthens versus other currencies, the value of the non-U.S. revenue will decline when reported in U.S. dollars. The impact to net income as a result of a strengthening U.S. dollar will be partially mitigated by the value of non-U.S. expense, which will also decline when reported in U.S. dollars. As the U.S. dollar weakens versus other currencies, the value of the non-U.S. revenue and expense will increase when reported in U.S. dollars.
We have established revenue and operating expense hedging and balance sheet risk management programs to protect against volatility of future foreign currency cash flow and changes in fair value caused by volatility in foreign currency exchange rates.
During the second quarter of 2018 the International Practices Task Force of the Center for Audit Quality categorized Argentina as a country with a projected three-year cumulative inflation rate greater than 100.0%, which indicated that Argentina's economy is highly inflationary. This categorization did not have a material impact on our results of operations or financial position as of March 31, 2024, and is not expected to have a material impact on our results of operations or financial position in the future. In December 2023 the Argentinian Peso experienced a substantial devaluation following a presidential election. The devaluation resulted in a $16.0 million charge recorded during the fourth quarter of 2023 in other (income) expense, net within our consolidated statements of income for the year ended December 31, 2023, included in our 2023 Form 10-K.
REVENUE AND OPERATING EXPENSE HEDGING PROGRAM
Our foreign currency hedging program is designed to mitigate, over time, a portion of the impact resulting from volatility in exchange rate changes on revenue and operating expense. We use foreign currency forward contracts and foreign currency options to manage foreign currency risk, with the majority of our forward contracts and options used to hedge certain forecasted revenue and operating expense transactions denominated in foreign currencies in the next 9 months. We do not engage in currency speculation. For a more detailed disclosure of our revenue and operating expense hedging program, please read Note 10, Derivative Instruments, to our condensed consolidated financial statements included in this report.
65
Our ability to mitigate the impact of foreign currency exchange rate changes on revenue and net income diminishes as significant foreign currency exchange rate fluctuations are sustained over extended periods of time. In particular, devaluation or significant deterioration of foreign currency exchange rates are difficult to mitigate and likely to negatively impact earnings. The cash flow from these contracts are reported as operating activities in our condensed consolidated statements of cash flow.
BALANCE SHEET RISK MANAGEMENT HEDGING PROGRAM
We also use forward contracts to mitigate the foreign currency exposure related to certain balance sheet items. The primary objective of our balance sheet risk management program is to mitigate the exposure of foreign currency denominated net monetary assets and liabilities of foreign affiliates. In these instances, we principally utilize currency forward contracts. We have not elected hedge accounting for the balance sheet related items. The cash flow from these contracts are reported as operating activities in our condensed consolidated statements of cash flow.
The following quantitative information includes the impact of currency movements on forward contracts used in our revenue, operating expense and balance sheet hedging programs. As of March 31, 2024 and December 31, 2023, a hypothetical adverse 10.0% movement in foreign currency exchange rates compared to the U.S. dollar across all maturities would result in a hypothetical decrease in the fair value of forward contracts of approximately $246.5 million and $249.4 million, respectively. The estimated fair value change was determined by measuring the impact of the hypothetical exchange rate movement on outstanding forward contracts. Our use of this methodology to quantify the market risk of such instruments is subject to assumptions and actual impact could be significantly different. The quantitative information about market risk is limited because it does not take into account all foreign currency operating transactions.
CREDIT RISK
Financial instruments that potentially subject us to concentrations of credit risk include cash and cash equivalents, investments, derivatives and accounts receivable. We attempt to minimize the risks related to cash and cash equivalents and investments by investing in a broad and diverse range of financial instruments. We have established guidelines related to credit ratings and maturities intended to safeguard principal balances and maintain liquidity. Our investment portfolio is maintained in accordance with our investment policy, which defines allowable investments, specifies credit quality standards and limits the credit exposure of any single issuer. We minimize credit risk resulting from derivative instruments by choosing only highly rated financial institutions as counterparties.
We operate in certain countries where weakness in economic conditions, including the effects of the conflict between Russia and Ukraine and the military conflict in the Middle East, can result in extended collection periods. We continue to monitor these conditions, including the volatility associated with international economies and the relevant financial markets, and assess their possible impact on our business. To date, we have not experienced any significant losses with respect to the collection of our accounts receivable.
We believe that our allowance for doubtful accounts was adequate as of March 31, 2024 and December 31, 2023.
EQUITY PRICE RISK
Our strategic investment portfolio includes investments in equity securities of certain biotechnology companies. While we are holding such securities, we are subject to equity price risk, and this may increase the volatility of our income in future periods due to changes in the fair value of equity investments. We may sell such equity securities based on our business considerations, which may include limiting our price risk.
Changes in the fair value of these equity securities are impacted by the volatility of the stock market and changes in general economic conditions, among other factors. The potential change in fair value for equity price sensitive instruments has been assessed on a hypothetical 10.0% adverse movement. As of March 31, 2024 and December 31, 2023, a hypothetical adverse 10.0% movement would result in a hypothetical decrease in fair value of approximately $33.0 million and $41.7 million, respectively.
66
ITEM 4. CONTROLS AND PROCEDURES
DISCLOSURE CONTROLS AND PROCEDURES AND INTERNAL CONTROL OVER FINANCIAL REPORTING
CONTROLS AND PROCEDURES
We have carried out an evaluation, under the supervision and with the participation of our management, including our principal executive officer and principal financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in Rules 13a-15(e) or 15d-15(e) under the Securities Exchange Act of 1934, as amended), as of March 31, 2024. Based upon that evaluation, our principal executive officer and principal financial officer concluded that, as of the end of the period covered by this report, our disclosure controls and procedures are effective in ensuring that:
(a) the information required to be disclosed by us in the reports that we file or submit under the Securities Exchange Act is recorded, processed, summarized and reported within the time periods specified in the U.S. Securities and Exchange Commission’s rules and forms; and
(b) such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure.
In designing and evaluating our disclosure controls and procedures, our management recognized that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and our management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
CHANGES IN INTERNAL CONTROL OVER FINANCIAL REPORTING
There were no changes in our internal control over financial reporting during the quarter ended March 31, 2024, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
67
PART II OTHER INFORMATION
ITEM 1. LEGAL PROCEEDINGS
For a discussion of legal proceedings as of March 31, 2024, please read Note 21, Litigation, to our condensed consolidated financial statements included in this report, which is incorporated into this item by reference.
ITEM 1A. RISK FACTORS
Risks Related to Our Business
We are substantially dependent on revenue from our products.
Our revenue depends upon continued sales of our products as well as the financial rights we have in our anti-CD20 therapeutic programs. A significant portion of our revenue is concentrated on sales of our products in increasingly competitive markets. Any of the following negative developments relating to any of our products or any of our anti-CD20 therapeutic programs may adversely affect our revenue and results of operations or could cause a decline in our stock price:
•the introduction, greater acceptance or more favorable reimbursement of competing products, including new originator therapies, generics, prodrugs and biosimilars of existing products and products approved under abbreviated regulatory pathways;
•safety or efficacy issues;
•limitations and additional pressures on product pricing or price increases, including those relating to inflation and those resulting from governmental or regulatory requirements, including those relating to any future potential drug price negotiation under the IRA; increased competition, including from generic or biosimilar versions of our products; or changes in, or implementation of, reimbursement policies and practices of payors and other third-parties;
•adverse legal, administrative, geopolitical events, regulatory or legislative developments; or
•our ability to maintain a positive reputation among patients, healthcare providers and others, which may be impacted by our pricing and reimbursement decisions.
LEQEMBI is in the early stages of commercial launch in the U.S. and certain international markets and SKYCLARYS is in the early stages of commercial launch in the U.S. and certain European markets. In addition to risks associated with new product launches and the other factors described in these Risk Factors, Biogen’s and Eisai’s ability to successfully commercialize LEQEMBI and our ability to successfully commercialize SKYCLARYS may be adversely affected due to:
•Eisai’s ability to obtain and maintain adequate reimbursement for LEQEMBI;
•the effectiveness of Eisai's and Biogen’s commercial strategy for marketing LEQEMBI;
•requirements such as participation in a registry and the use of imaging or other diagnostics for LEQEMBI;
•our ability to obtain approval in other markets;
•the approval of other new products for the same or similar indications;
•Eisai’s and Biogen’s ability to maintain a positive reputation among patients, healthcare providers and others in the Alzheimer’s disease community, which may be impacted by pricing and reimbursement decisions relating to LEQEMBI, which are made by Eisai and/or third parties;
•Biogen's ability to obtain and maintain adequate reimbursement for SKYCLARYS; and
•the effectiveness of Biogen's commercial strategy for marketing SKYCLARYS.
Our long-term success depends upon the successful development of new products and additional indications for our existing products.
Our long-term success will depend upon the successful development of new products from our research and development activities or our licenses or acquisitions from third parties, as well as additional indications for our existing products.
Product development is very expensive and involves a high degree of uncertainty and risk and may not be successful. Only a small number of research and development programs result in the commercialization of a product. It is difficult to predict the success and the time and cost of product development of novel approaches for the treatment
68
of diseases. The development of novel approaches for the treatment of diseases, including development efforts in new modalities such as those based on the antisense oligonucleotide platform and gene therapy, may present additional challenges and risks, including obtaining approval from regulatory authorities that have limited experience with the development of such therapies. For example, we are currently seeking approval of LEQEMBI in Europe and the approval of a subcutaneous formulation of LEQEMBI in the U.S. and any delays or challenges may impact our ability to realize the anticipated benefits from LEQEMBI.
Clinical trial data are subject to differing interpretations and even if we view data as sufficient to support the safety, effectiveness and/or approval of an investigational therapy, regulatory authorities may disagree and may require additional data, limit the scope of the approval or deny approval altogether. Furthermore, the approval of a product candidate by one regulatory agency does not mean that other regulatory agencies will also approve such product candidate.
Success in preclinical work or early-stage clinical trials does not ensure that later stage or larger scale clinical trials will be successful. Clinical trials may indicate that our product candidates lack efficacy, have harmful side effects, result in unexpected adverse events or raise other concerns that may significantly reduce or delay the likelihood of regulatory approval. This may result in terminated programs, significant restrictions on use and safety warnings in an approved label, adverse placement within the treatment paradigm or significant reduction in the commercial potential of the product candidate.
Even if we could successfully develop new products or indications, we may make a strategic decision to discontinue development of a product candidate or indication if, for example, we believe commercialization will be difficult relative to the standard of care or we prioritize other opportunities in our pipeline.
Sales of new products or products with additional indications may not meet investor expectations.
If we fail to compete effectively, our business and market position would suffer.
The biopharmaceutical industry and the markets in which we operate are intensely competitive. We compete in the marketing and sale of our products, the development of new products and processes, the acquisition of rights to new products with commercial potential and the hiring and retention of personnel. We compete with biotechnology and pharmaceutical companies that have a greater number of products on the market and in the product pipeline, substantially greater financial, marketing, research and development and other resources and other technological or competitive advantages.
Our products continue to face increasing competition from the introduction of new originator therapies, generics, prodrugs and biosimilars of existing products and products approved under abbreviated regulatory pathways. Some of these products are likely to be sold at substantially lower prices than our branded products. The introduction of such products as well as other lower-priced competing products has reduced, and may in the future, significantly reduce both the price that we are able to charge for our products and the volume of products we sell, which will negatively impact our revenue. For instance, demand and price for TECFIDERA declined significantly as a result of multiple TECFIDERA generic entrants entering the U.S. market in 2020. In addition, in some markets, when a generic or biosimilar version of one of our products is commercialized, it may be automatically substituted for our product and significantly reduce our revenue in a short period of time.
Our ability to compete, maintain and grow our business may also be adversely affected due to a number of factors, including:
•the introduction of other products, including products that may be more efficacious, safer, less expensive or more convenient alternatives to our products, including our own products and products of our collaborators;
•the off-label use by physicians of therapies indicated for other conditions to treat patients;
•patient dynamics, including the size of the patient population and our ability to identify, attract and maintain new and current patients to our therapies;
•the reluctance of physicians to prescribe, and patients to use, our products without additional data on the efficacy and safety of such products;
•damage to physician and patient confidence in any of our products, generic or biosimilars of our products or any other product from the same class as one of our products, or to our sales and reputation as a result of label changes, pricing and reimbursement decisions or adverse experiences or events that may occur with patients treated with our products or generic or biosimilars of our products;
•inability to obtain and maintain appropriate pricing and adequate reimbursement for our products compared to our competitors in key markets; or
69
•our ability to obtain and maintain patent, data or market exclusivity for our products.
Our business may be adversely affected if we do not successfully execute or realize the anticipated benefits of our strategic and growth initiatives.
The successful execution of our strategic and growth initiatives may depend upon internal development projects, commercial initiatives and external opportunities, which may include the acquisition and in-licensing of products, technologies, companies, the entry into strategic alliances and collaborations or our Fit for Growth program, as well as our ability to execute on previously-announced initiatives such as the exploration of strategic options for our biosimilars business.
While we believe we have a number of promising programs in our pipeline, failure or delay of internal development projects to advance or difficulties in executing on our commercial initiatives could impact our current and future growth, resulting in additional reliance on external development opportunities for growth.
Supporting the further development of our existing products and potential new products in our pipeline will require significant capital expenditures and management resources, including investments in research and development, sales and marketing, manufacturing capabilities and other areas of our business. We have made, and may continue to make, significant operating and capital expenditures for potential new products prior to regulatory approval with no assurance that such investment will be recouped, which may adversely affect our financial condition, business and operations.
The availability of high quality, fairly valued external product development is limited and the opportunity for their acquisition is highly competitive. As such, we are not certain that we will be able to identify suitable candidates for acquisition or if we will be able to reach agreement to make any such acquisition if suitable candidates are identified.
We may fail to initiate or complete transactions for many reasons, including failure to obtain regulatory or other approvals as well as a result of disputes or litigation. Furthermore, we may not be able to achieve the full strategic and financial benefits expected to result from transactions, or the benefits may be delayed or not occur at all. We may also face additional costs or liabilities in completed transactions that were not contemplated prior to completion.
Any failure in the execution of a transaction, in the integration of an acquired asset or business or in achieving expected synergies could result in slower growth, higher than expected costs, the recording of asset impairment charges and other actions which could adversely affect our business, financial condition and results of operations. For example, we recently acquired Reata and are in the process of integrating Reata into our Company. The ultimate success of our acquisition of Reata and our ability to realize the anticipated benefits from the acquisition, including the SKYCLARYS product and anticipated synergies, depends on, among other things, how effective we are in integrating the Biogen and Reata operations.
We face risks associated with our Fit for Growth program that may impair our ability to achieve anticipated savings and operational efficiencies or that may otherwise harm our business. These risks include delays in implementation of cost optimization actions, loss of workforce capabilities, higher than anticipated separation expenses, litigation and the failure to meet financial and operational targets. In addition, the calculation of the anticipated cost savings and other benefits resulting from our Fit for Growth program are subject to many estimates and assumptions. These estimates and assumptions are subject to significant business, economic, competitive and other uncertainties and contingencies, many of which are beyond our control. if these estimates and assumptions are incorrect or if we experience delays or unforeseen events, our business and financial results could be adversely affected.
Sales of our products depend, to a significant extent, on adequate coverage, pricing and reimbursement from third-party payors, which are subject to increasing and intense pressure from political, social, competitive and other sources. Our inability to obtain and maintain adequate coverage, or a reduction in pricing or reimbursement, could have an adverse effect on our business, reputation, revenue and results of operations.
Sales of our products depend, to a significant extent, on adequate coverage, pricing and reimbursement from third-party payors. When a new pharmaceutical product is approved, the availability of government and private reimbursement for that product, diagnosis of the condition it treats and the cost to administer it may be uncertain, as is the pricing and amount for which that product will be reimbursed.
Pricing and reimbursement for our products may be adversely affected by a number of factors, including:
•changes in, and implementation of, federal, state or foreign government regulations or private third-party payors’ reimbursement policies;
•pressure by employers on private health insurance plans to reduce costs;
70
•consolidation and increasing assertiveness of payors seeking price discounts or rebates in connection with the placement of our products on their formularies and, in some cases, the imposition of restrictions on access or coverage of particular drugs or pricing determined based on perceived value;
•our ability to receive reimbursement for our products or our ability to receive comparable reimbursement to that of competing products; and
•our value-based contracting program pursuant to which we aim to tie the pricing of our products to their clinical values by either aligning price to patient outcomes or adjusting price for patients who discontinue therapy for any reason, including efficacy or tolerability concerns.
Our ability to set the price for our products varies significantly from country to country and, as a result, so can the price of our products. Governments may use a variety of cost-containment measures to control the cost of products, including price cuts, mandatory rebates, value-based pricing and reference pricing (i.e., referencing prices in other countries and using those reference prices to set a price). Drug prices are under significant scrutiny in the markets in which our products are prescribed; for example the IRA has certain provisions related to drug pricing. We expect drug pricing and other health care costs to continue to be subject to intense political and societal pressures on a global basis. Certain countries set prices by reference to the prices in other countries where our products are marketed. Our inability to obtain and maintain adequate prices in a particular country may not only limit the revenue from our products within that country but may also adversely affect our ability to secure acceptable prices in existing and potential new markets, which may limit market growth and result in reductions in revenue. This may create the opportunity for third-party cross-border trade or influence our decision to sell or not to sell a product, thus adversely affecting our geographic expansion plans and revenue. Additionally, in certain jurisdictions governmental health agencies may adjust, retroactively and/or prospectively, reimbursement rates for our products. Reimbursement for our products by governments, including the timing of any reimbursements, may also be affected by budgetary or political constraints, particularly in challenging economic environments. Government agencies often do not set their own budgets and therefore, have limited control over the amount of money they can spend. In addition, these agencies experience political pressure that may dictate the manner in which they spend money. There can be no assurance that the economic, budgeting or political issues will not worsen and adversely impact sales or reimbursements of our products.
Competition from current and future competitors may negatively impact our ability to maintain pricing and our market share. New products marketed by our competitors could cause our revenue to decrease due to potential price reductions and lower sales volumes. Additionally, the introduction of generic or biosimilar versions of our products, follow-on products, prodrugs or products approved under abbreviated regulatory pathways may significantly reduce the price that we are able to charge for our products and the volume of products we sell.
Many payors continue to adopt benefit plan changes that shift a greater portion of prescription costs to patients, including more limited benefit plan designs, higher patient co-pay or co-insurance obligations and limitations on patients' use of commercial manufacturer co-pay payment assistance programs (including through co-pay accumulator adjustment or maximization programs). Significant consolidation in the health insurance industry has resulted in a few large insurers and pharmacy benefit managers exerting greater pressure in pricing and usage negotiations with drug manufacturers, significantly increasing discounts and rebates required of manufacturers and limiting patient access and usage. Further consolidation among insurers, pharmacy benefit managers and other payors would increase the negotiating leverage such entities have over us and other drug manufacturers. Additional discounts, rebates, coverage or plan changes, restrictions or exclusions as described above could have a material adverse effect on sales of our affected products.
Our failure to obtain or maintain adequate coverage, pricing or reimbursement for our products could have an adverse effect on our business, reputation, revenue and results of operations.
We depend on relationships with collaborators and other third-parties for revenue, and for the development, regulatory approval, commercialization and marketing of certain of our products and product candidates, which are outside of our full control.
We rely on a number of collaborative and other third-party relationships for revenue and the development, regulatory approval, commercialization and marketing of certain of our products and product candidates. We also outsource certain aspects of our regulatory affairs and clinical development relating to our products and product candidates to third-parties. Reliance on third-parties subjects us to a number of risks, including:
•we may be unable to control the resources our collaborators or third-parties devote to our programs, products or product candidates, which may affect our ability to achieve development goals or milestones;
71
•disputes may arise under an agreement, including with respect to the achievement and payment of milestones, payment of development or commercial costs, ownership of rights to technology developed, and the underlying agreement may fail to provide us with significant protection or may fail to be effectively enforced if the collaborators or third-parties fail to perform;
•the interests of our collaborators or third-parties may not always be aligned with our interests, and such parties may not pursue regulatory approvals or market a product in the same manner or to the same extent that we would, which could adversely affect our revenue, or may adopt tax strategies that could have an adverse effect on our business, results of operations or financial condition;
•third-party relationships require the parties to cooperate, and failure to do so effectively could adversely affect product sales or the clinical development or regulatory approvals of product candidates under joint control, could result in termination of the research, development or commercialization of product candidates or could result in litigation or arbitration;
•any failure on the part of our collaborators or third-parties to comply with applicable laws, including tax laws, regulatory requirements and/or applicable contractual obligations or to fulfill any responsibilities they may have to protect and enforce any intellectual property rights underlying our products could have an adverse effect on our revenue or reputation as well as involve us in possible legal proceedings; and
•any improper conduct or actions on the part of our collaborators or third-parties could subject us to civil or criminal investigations and monetary and injunctive penalties, require management attention, impact the accuracy and timing of our financial reporting and/or adversely impact our ability to conduct business, our operating results and our reputation.
Given these risks, there is considerable uncertainty regarding the success of our current and future collaborative efforts. If these efforts fail, our product development or commercialization of new products could be delayed, revenue from products could decline and/or we may not realize the anticipated benefits of these arrangements.
Our results of operations may be adversely affected by current and potential future healthcare reforms.
In the U.S., federal and state legislatures, health agencies and third-party payors continue to focus on containing the cost of health care. Legislative and regulatory proposals, enactments to reform health care insurance programs (including those contained in the IRA) and increasing pressure from social sources could significantly influence the manner in which our products are prescribed, purchased and reimbursed. For example, provisions of the PPACA have resulted in changes in the way health care is paid for by both governmental and private insurers, including increased rebates owed by manufacturers under the Medicaid Drug Rebate Program, annual fees and taxes on manufacturers of certain branded prescription drugs, the requirement that manufacturers participate in a discount program for certain outpatient drugs under Medicare Part D and the expansion of the number of hospitals eligible for discounts under Section 340B of the Public Health Service Act. These changes have had and are expected to continue to have a significant impact on our business.
We may face uncertainties as a result of efforts to repeal, substantially modify or invalidate some or all of the provisions of the PPACA. There is no assurance that the PPACA, as currently enacted or as amended in the future, will not adversely affect our business and financial results, and we cannot predict how future federal or state legislative or administrative changes relating to healthcare reform will affect our business.
There is substantial public attention on the costs of prescription drugs and we expect drug pricing and other health care costs to continue to be subject to intense political and societal pressures on a global basis. In addition, there have been (including elements of the IRA), and are expected to continue to be, legislative proposals to address prescription drug pricing. Some of these proposals could have significant effects on our business, including an executive order issued in September 2020 to test a “most favored nation” model for Part B and Part D drugs that tie reimbursement rates to international drug pricing metrics. These actions and the uncertainty about the future of the PPACA and healthcare laws may put downward pressure on pharmaceutical pricing and increase our regulatory burdens and operating costs.
There is also significant economic pressure on state budgets, that may result in states increasingly seeking to achieve budget savings through mechanisms that limit coverage or payment for our drugs. In recent years, some states have considered legislation and ballot initiatives that would control the prices of drugs, including laws to allow importation of pharmaceutical products from lower cost jurisdictions outside the U.S. and laws intended to impose price controls on state drug purchases. State Medicaid programs are requesting manufacturers to pay supplemental rebates and requiring prior authorization by the state program for use of any drug for which supplemental rebates are not being paid. Government efforts to reduce Medicaid expense may lead to increased use of managed care organizations by Medicaid programs. This may result in managed care organizations influencing prescription
72
decisions for a larger segment of the population and a corresponding limitation on prices and reimbursement for our products.
In the E.U. and some other international markets, the government provides health care at low cost to consumers and regulates pharmaceutical prices, patient eligibility or reimbursement levels to control costs for the government-sponsored health care system. Many countries have announced or implemented measures, and may in the future implement new or additional measures, to reduce health care costs to limit the overall level of government expenditures. These measures vary by country and may include, among other things, patient access restrictions, suspensions on price increases, prospective and possible retroactive price reductions and other recoupments and increased mandatory discounts or rebates, recoveries of past price increases and greater importation of drugs from lower-cost countries. These measures have negatively impacted our revenue and may continue to adversely affect our revenue and results of operations in the future.
Our success in commercializing biosimilars is subject to risks and uncertainties inherent in the development, manufacture and commercialization of biosimilars. If we are unsuccessful in such activities, our business may be adversely affected.
The development, manufacture and commercialization of biosimilar products require specialized expertise and are very costly and subject to complex regulation. Our success in commercializing biosimilars is subject to a number of risks, including:
•Reliance on Third-Parties. We are dependent, in part, on the efforts of collaboration partners and other third-parties over whom we have limited or no control in the development and manufacturing of biosimilars products. For example, a recently announced potential acquisition of a contract development and manufacturing organization by a third party may impact its operational, strategic or financial risk. If these third-parties fail to perform successfully, or reduce their third party manufacturing production, our biosimilar product development or commercialization of biosimilar products could be delayed, revenue from biosimilar products could decline and/or we may not realize the anticipated benefits of these arrangements;
•Regulatory Compliance. Biosimilar products may face regulatory hurdles or delays due to the evolving and uncertain regulatory and commercial pathway of biosimilars products in certain jurisdictions;
•Ability to Provide Adequate Supply. Manufacturing biosimilars is complex. If we encounter any manufacturing or supply chain difficulties we may be unable to meet demand. We are dependent on a third-party for the manufacture of our biosimilar products and such third-party may not perform its obligations in a timely and cost-effective manner or in compliance with applicable regulations and may be unable or unwilling to increase production capacity commensurate with demand for our existing or future biosimilar products;
•Intellectual Property and Regulatory Challenges. Biosimilar products may face extensive intellectual property clearances and infringement litigation, injunctions or regulatory challenges, which could prevent the commercial launch of a product or delay it for many years or result in imposition of monetary damages, penalties or other civil sanctions and damage our reputation;
•Failure to Gain Market and Patient Acceptance. Market success of biosimilar products will be adversely affected if patients, physicians and/or payors do not accept biosimilar products as safe and efficacious products offering a more competitive price or other benefit over existing therapies; and
•Competitive Challenges. Biosimilar products face significant competition, including from innovator products and biosimilar products offered by other companies that may receive greater acceptance or more favorable reimbursement. Local tendering processes may restrict biosimilar products from being marketed and sold in some jurisdictions. The number of competitors in a jurisdiction, the timing of approval and the ability to market biosimilar products successfully in a timely and cost-effective manner are additional factors that may impact our success in this business area. The decision to explore strategic options related to our biosimilars business could adversely affect our operations related to our biosimilars business.
Risks Related to Intellectual Property
If we are unable to obtain and maintain adequate protection for our data, intellectual property and other proprietary rights, our business may be harmed.
Our success, including our long-term viability and growth, depends, in part, on our ability to obtain and defend patent and other intellectual property rights, including certain regulatory forms of exclusivity, that are important to the commercialization of our products and product candidates. Patent protection and/or regulatory exclusivity in the U.S. and other important markets remains uncertain and depends, in part, upon decisions of the patent offices, courts, administrative bodies and lawmakers in these countries. We may fail to obtain, defend or preserve patent and other
73
intellectual property rights, including certain regulatory forms of exclusivity, or the protection we obtain may not be of sufficient breadth and degree to protect our commercial interests in all countries where we conduct business, which could result in financial, business or reputational harm to us or could cause a decline or volatility in our stock price. In addition, settlements of such proceedings often result in reducing the period of exclusivity and other protections, resulting in a reduction in revenue from affected products.
In many markets, including the U.S., manufacturers may be allowed to rely on the safety and efficacy data of the innovator's product and do not need to conduct clinical trials before marketing a competing version of a product after there is no longer patent or regulatory exclusivity. In such cases, manufacturers often charge significantly lower prices and a major portion of the company's revenue may be reduced in a short period of time. In addition, manufacturers of generics and biosimilars may choose to launch or attempt to launch their products before the expiration of our patent or other intellectual property protections.
Furthermore, our products may be determined to infringe patents or other intellectual property rights held by third-parties. Legal proceedings, administrative challenges or other types of proceedings are and may in the future be necessary to determine the validity, scope or non-infringement of certain patent rights claimed by third-parties to be pertinent to the manufacture, use or sale of our products. Legal proceedings may also be necessary to determine the rights, obligations and payments claimed during and after the expiration of intellectual property license agreements we have entered with third parties. Such proceedings are unpredictable and are often protracted and expensive. Negative outcomes of such proceedings could hinder or prevent us from manufacturing and marketing our products, require us to seek a license for the infringed product or technology or result in the assessment of significant monetary damages against us that may exceed amounts, if any, accrued in our financial statements. A failure to obtain necessary licenses for an infringed product or technology could prevent us from manufacturing or selling our products. Furthermore, payments under any licenses that we are able to obtain could reduce our profits from the covered products and services. Any of these circumstances could result in financial, business or reputational harm to us or could cause a decline or volatility in our stock price.
Risks Related to Development, Clinical Testing and Regulation of Our Products and Product Candidates
Successful preclinical work or early stage clinical trials does not ensure success in later stage trials, regulatory approval or commercial viability of a product.
Positive results in a clinical trial may not be replicated in subsequent or confirmatory trials. Additionally, success in preclinical work or early stage clinical trials does not ensure that later stage or larger scale clinical trials will be successful or that regulatory approval will be obtained. Even if later stage clinical trials are successful, regulatory authorities may delay or decline approval of our product candidates. Regulatory authorities may disagree with our view of the data, require additional studies, disagree with our trial design or endpoints or not approve adequate reimbursement. Regulatory authorities may also fail to approve the facilities or processes used to manufacture a product candidate, our dosing or delivery methods or companion devices. Regulatory authorities may grant marketing approval that is more restricted than anticipated, including limiting indications to narrow patient populations and the imposition of safety monitoring, educational requirements, requiring confirmatory trials and risk evaluation and mitigation strategies. The occurrence of any of these events could result in significant costs and expense, have an adverse effect on our business, financial condition and results of operations and/or cause our stock price to decline or experience periods of volatility.
Clinical trials and the development of biopharmaceutical products is a lengthy and complex process. If we fail to adequately manage our clinical activities, our clinical trials or potential regulatory approvals may be delayed or denied.
Conducting clinical trials is a complex, time-consuming and expensive process. Our ability to complete clinical trials in a timely fashion depends on a number of key factors, including protocol design, regulatory and institutional review board approval, patient enrollment rates and compliance with current Good Clinical Practices. If we or our third-party clinical trial providers or third-party CROs do not successfully carry out these clinical activities, our clinical trials or the potential regulatory approval of a product candidate may be delayed or denied.
We have opened clinical trial sites and are enrolling patients in a number of countries where our experience is limited. In most cases, we use the services of third-parties to carry out our clinical trial related activities and rely on such parties to accurately report their results. Our reliance on third-parties for these activities may impact our ability to control the timing, conduct, expense and quality of our clinical trials. One CRO has responsibility for a substantial portion of our activities and reporting related to our clinical trials and if such CRO does not adequately perform, many of our trials may be affected, including adversely affecting our expenses associated with such trials. We may need to replace our CROs, which may result in the delay of the affected trials or otherwise adversely affect our efforts to obtain regulatory approvals and commercialize our product candidates.
74
Adverse safety events or restrictions on use and safety warnings for our products can negatively affect our business, product sales and stock price.
Adverse safety events involving our marketed products, generic or biosimilar versions of our marketed products or products from the same class as one of our products may have a negative impact on our business. Discovery of safety issues with our products could create product liability and could cause additional regulatory scrutiny and requirements for additional labeling or safety monitoring, withdrawal of products from the market and/or the imposition of fines or criminal penalties. Adverse safety events may also damage physician, patient and/or investor confidence in our products and our reputation. Any of these could result in adverse impacts on our results of operations.
Regulatory authorities are making greater amounts of stand-alone safety information directly available to the public through periodic safety update reports, patient registries and other reporting requirements. The reporting of adverse safety events involving our products or products similar to ours and public rumors about such events may increase claims against us and may also cause our product sales to decline or our stock price to experience periods of volatility.
Restrictions on use or safety warnings that may be required to be included in the label of our products may significantly reduce expected revenue for those products and require significant expense and management time.
Risks Related to Our Operations
A breakdown or breach of our information systems could subject us to liability or interrupt the operation of our business.
We are increasingly dependent upon information systems and data to operate our business. Changes in how we operate have caused us to modify our business practices in ways that heighten this dependence, including changing the requirement that most of our office-based employees in the U.S. and our other key markets work from the office, with many of our employees now working in hybrid or full-remote positions. As a result, we are increasingly dependent upon our information systems to operate our business and our ability to effectively manage our business depends on the security, reliability and adequacy of our information systems and data, which includes use of cloud technologies, including Software as a Service (SaaS), Platform as a Service (PaaS) and Infrastructure as a Service (IaaS). Breakdowns, invasions, corruptions, destructions and/or breaches, which impact may include, but not limited to, comprising the capacity, reliability or security of our information systems or those of our business partners, including our cloud technologies, and/or unauthorized access to our data and information could subject us to significant liability, negatively impact our business operations, and/or require replacement of technology and/or sizeable ransom payments. Our information systems, including our cloud technologies, continue to increase in multitude and complexity, increasing our vulnerability when breakdowns, malicious intrusions and random attacks occur. Data privacy or security breaches also pose a risk that sensitive data, including intellectual property, trade secrets or personal information belonging to us, patients, customers or other business partners, may be exposed to unauthorized persons or to the public.
Cybersecurity threats and incidents are increasing in their frequency, sophistication and intensity, and are becoming increasingly difficult to detect, particularly when they impact vendors, customers or suppliers, and other companies in our supply chain. Cybersecurity threats and incidents are often carried out by motivated, well-resourced, skilled and persistent actors, including nation states, organized crime groups, “hacktivists” and may include or target employees or contractors acting with careless or malicious intent. Recent developments in the threat landscape include use of AI and machine learning, as well as an increased number of cyber extortion attacks, with higher financial ransom demand amounts and increasing sophistication and variety of ransomware techniques and methodology. Geopolitical instability, including that related to Russia's invasion of Ukraine or the conflict in the Middle East, may increase the risk of cybersecurity threats. Cybersecurity threats or incidents may include deployment of harmful malware and key loggers, ransomware, a denial-of-service attack, a malicious website, the use of social engineering and other means to affect the confidentiality, integrity and availability of our information systems and data. Cybersecurity threats and incidents also include manufacturing, hardware or software supply chain attacks, which could cause a delay in the manufacturing of products or products produced for contract manufacturing or lead to a data privacy or security breach. Our key business partners face similar risks and any security breach of their systems could adversely affect our security posture. In addition, our increased use of cloud technologies heightens these and other operational risks, and any failure by cloud or other technology service providers to adequately safeguard their systems and prevent cyber-attacks could disrupt our operations and result in misappropriation, corruption or loss of confidential or propriety information.
While we continue to build and improve our systems and infrastructure, including our business continuity plans, there can be no assurance that our efforts will prevent cybersecurity threats or incidents in our systems and any such
75
incidents could materially adversely affect our business and operations and/or result in the loss of critical or sensitive information, which could result in material financial, legal, operational or reputational harm to us, loss of competitive advantage or loss of consumer confidence. Our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyber-attacks and other related breaches.
Regulations continue to change as regulators worldwide consider new rules. For example, the SEC has adopted additional disclosure rules regarding cyber security risk management, strategy, governance and incident reporting by public companies. These new regulations or other regulations being considered in Europe and around the world may impact the manner in which we operate.
Regulators are imposing new data privacy and security requirements, including new and greater monetary fines for privacy violations. For example, the E.U.’s General Data Protection Regulation established regulations regarding the handling of personal data, and provides an enforcement authority and imposes large penalties for noncompliance. New U.S. data privacy and security laws, such as the CCPA, and others that may be passed, similarly introduce requirements with respect to personal information, and non-compliance with the CCPA may result in liability through private actions (subject to statutorily defined damages in the event of certain data breaches) and enforcement. Failure to comply with these current and future laws, policies, industry standards or legal obligations or any security incident resulting in the unauthorized access to, or acquisition, release or transfer of personal information may result in governmental enforcement actions, litigation, fines and penalties or adverse publicity and could cause our customers to lose trust in us, which could have a material adverse effect on our business and results of operations.
Manufacturing issues could substantially increase our costs, limit supply of our products and/or reduce our revenue.
The process of manufacturing our products is complex, highly regulated and subject to numerous risks, including:
•Risks of Reliance on Third-Parties and Single Source Providers. We rely on third-party suppliers and manufacturers for many aspects of our manufacturing process for our products and product candidates. In some cases, due to the unique manner in which our products are manufactured, we rely on single source providers of raw materials and manufacturing supplies. These third-parties are independent entities subject to their own unique operational, strategic and financial risks that are outside of our control. For example, a recently announced potential acquisition of a contract development and manufacturing organization by a third party may impact its operational, strategic or financial risk. These third-parties may not perform their obligations in a timely and cost-effective manner or in compliance with applicable regulations, and they may be unable or unwilling to increase production capacity commensurate with demand for our existing or future products. Finding alternative providers could take a significant amount of time and involve significant expense due to the specialized nature of the services and the need to obtain regulatory approval of any significant changes to our suppliers or manufacturing methods. We cannot be certain that we could reach agreement with alternative providers or that the FDA or other regulatory authorities would approve our use of such alternatives.
•Global Bulk Supply Risks. We rely on our manufacturing facilities for the production of drug substance for our large molecule products and product candidates. Our global bulk supply of these products and product candidates depends on the uninterrupted and efficient operation of these facilities, which could be adversely affected by equipment failures, labor or raw material shortages, geopolitical instability, public health epidemics, natural disasters, power failures, cyber-attacks and many other factors.
•Risks Relating to Compliance with current GMP (cGMP). We and our third-party providers are generally required to maintain compliance with cGMP and other stringent requirements and are subject to inspections by the FDA and other regulatory authorities to confirm compliance. Any delay, interruption or other issues that arise in the manufacture, fill-finish, packaging or storage of our products as a result of a failure of our facilities or operations or those of third-parties to receive regulatory approval or pass any regulatory agency inspection could significantly impair our ability to develop and commercialize our products. Significant noncompliance could also result in the imposition of monetary penalties or other civil or criminal sanctions and damage our reputation.
•Risk of Product Loss. The manufacturing process for our products is extremely susceptible to product loss due to contamination, oxidation, equipment failure or improper installation or operation of equipment or vendor or operator error. Even minor deviations from normal manufacturing processes could result in reduced production yields, product defects and other supply disruptions. If microbial, viral or other contaminations are discovered in our products or manufacturing facilities, we may need to close our manufacturing facilities for an extended period of time to investigate and remediate the contaminant.
Any adverse developments affecting our manufacturing operations or the operations of our third-party suppliers and manufacturers may result in shipment delays, inventory shortages, lot failures, product withdrawals or recalls or other interruptions in the commercial supply of our products.
76
Furthermore, factors such as geopolitical events, global health outbreaks, weather events, labor or raw material shortages and other supply chain disruptions could result in difficulties and delays in manufacturing our products, which could have an adverse impact on our results in operations or result in product shortages. We may also have to take inventory write-offs and incur other charges and expense for products that fail to meet specifications, undertake costly remediation efforts or seek more costly manufacturing alternatives. Such developments could increase our manufacturing costs, cause us to lose revenue or market share as patients and physicians turn to competing therapeutics, diminish our profitability or damage our reputation.
In addition, although we have business continuity plans to reduce the potential for manufacturing disruptions or delays and reduce the severity of a disruptive event, there is no guarantee that these plans will be adequate, which could adversely affect our business and operations.
Management, personnel and other organizational changes may disrupt our operations, and we may have difficulty retaining personnel or attracting and retaining qualified replacements on a timely basis for the management and other personnel who may leave the Company.
Changes in management, other personnel and our overall retention rate may disrupt our business, and any such disruption could adversely affect our operations, programs, growth, financial condition or results of operations. New members of management may have different perspectives on programs and opportunities for our business, which may cause us to focus on new opportunities or reduce or change emphasis on our existing programs.
Our success is dependent upon our ability to attract and retain qualified management and other personnel in a highly competitive environment. Qualified individuals are in high demand, and we may incur significant costs to attract or retain them. We may face difficulty in attracting and retaining talent for a number of reasons, including management changes, integration related to the Reata acquisition, the underperformance or discontinuation of one or more marketed, pre-clinical or clinical programs, recruitment by competitors or changes in the overall labor market. In addition, changes in our organizational structure or in our flexible working arrangements could impact employees' productivity and morale as well as our ability to attract, retain and motivate employees. We cannot ensure that we will be able to hire or retain the personnel necessary for our operations or that the loss of any personnel will not have a material impact on our financial condition and results of operations.
If we fail to comply with the extensive legal and regulatory requirements affecting the health care industry, we could face increased costs, penalties and a loss of business.
Our activities, and the activities of our collaborators, distributors and other third-party providers, are subject to extensive government regulation and oversight in the U.S. and in foreign jurisdictions, and are subject to change and evolving interpretations, which could require us to incur substantial costs associated with compliance or to alter one or more of our business practices. The FDA and comparable foreign agencies directly regulate many of our most critical business activities, including the conduct of preclinical and clinical studies, product manufacturing, advertising and promotion, product distribution, adverse event reporting, product risk management and our compliance with good practice quality guidelines and regulations. Our interactions with physicians and other health care providers that prescribe or purchase our products are also subject to laws and government regulation designed to prevent fraud and abuse in the sale and use of products and place significant restrictions on the marketing practices of health care companies. Health care companies are facing heightened scrutiny of their relationships with health care providers and have been the target of lawsuits and investigations alleging violations of laws and government regulation, including claims asserting submission of incorrect pricing information, impermissible off-label promotion of pharmaceutical products, payments intended to influence the referral of health care business, submission of false claims for government reimbursement, antitrust violations or violations related to environmental matters. There is also enhanced scrutiny of company-sponsored patient assistance programs, including testing, insurance premium and co-pay assistance programs and donations to third-party charities that provide such assistance. The U.S. government has challenged some of our donations to third-party charities that provide patient assistance. If we, or our vendors or donation recipients, are found to fail to comply with relevant laws, regulations or government guidance in the operation of these or other patient assistance programs, we could be subject to significant fines or penalties. Risks relating to compliance with laws and regulations may be heightened as we continue to expand our global operations and enter new therapeutic areas with different patient populations, which may have different product distribution methods, marketing programs or patient assistance programs from those we currently utilize or support.
Conditions and regulations governing the health care industry are subject to change, with possible retroactive effect, including:
•new laws, regulations or judicial decisions, or new interpretations of existing laws, regulations or judicial decisions, related to health care availability, pricing or marketing practices, compliance with employment
77
practices, method of delivery, payment for health care products and services, compliance with health information and data privacy and security laws and regulations, tracking and reporting payments and other transfers of value made to physicians and teaching hospitals, extensive anti-bribery and anti-corruption prohibitions, product serialization and labeling requirements and used product take-back requirements;
•changes in the FDA and foreign regulatory approval processes or perspectives that may delay or prevent the approval of new products and result in lost market opportunity;
•government shutdowns or relocations may result in delays to the review and approval process, slowing the time necessary for new drug candidates to be reviewed and/or approved, which may adversely affect our business;
•requirements that provide for increased transparency of clinical trial results and quality data, such as the EMA's clinical transparency policy, which could impact our ability to protect trade secrets and competitively-sensitive information contained in approval applications or could be misinterpreted leading to reputational damage, misperception or legal action, which could harm our business; and
•changes in FDA and foreign regulations that may require additional safety monitoring, labeling changes, restrictions on product distribution or use or other measures after the introduction of our products to market, which could increase our costs of doing business, adversely affect the future permitted uses of approved products or otherwise adversely affect the market for our products.
Violations of governmental regulation may be punishable by criminal and civil sanctions, including fines and civil monetary penalties and exclusion from participation in government programs, including Medicare and Medicaid, as well as against executives overseeing our business. We could also be required to repay amounts we received from government payors or pay additional rebates and interest if we are found to have miscalculated the pricing information we submitted to the government. In addition, legal proceedings and investigations are inherently unpredictable, and large judgments or settlements sometimes occur. While we believe that we have appropriate compliance controls, policies and procedures in place to comply with the laws or regulations of the jurisdictions in which we operate, there is a risk that acts committed by our employees, agents, distributors, collaborators or third-party providers might violate such laws or regulations. Whether or not we have complied with the law, an investigation or litigation related to alleged unlawful conduct could increase our expense, damage our reputation, divert management time and attention and adversely affect our business.
Our sales and operations are subject to the risks of doing business internationally.
We are increasing our presence in international markets, subjecting us to many risks that could adversely affect our business and revenue. There is no guarantee that our efforts and strategies to expand sales in international markets will succeed. Emerging market countries may be especially vulnerable to periods of global and local political, legal, regulatory and financial instability and may have a higher incidence of corruption and fraudulent business practices. Certain countries may require local clinical trial data as part of the drug registration process in addition to global clinical trials, which can add to overall drug development and registration timelines. We may also be required to increase our reliance on third-party agents or distributors and unfamiliar operations and arrangements previously utilized by companies we collaborate with or acquire in emerging markets.
Our sales and operations are subject to the risks of doing business internationally, including:
•the impact of public health epidemics on the global economy and the delivery of healthcare treatments;
•less favorable intellectual property or other applicable laws;
•the inability to obtain necessary foreign regulatory approvals of products in a timely manner;
•limitations and additional pressures on our ability to obtain and maintain product pricing, reimbursement or receive price increases, including those resulting from governmental or regulatory requirements;
•increased cost of goods due to factors such as inflation and supply chain disruptions;
•additional complexity in manufacturing or conducting clinical research internationally, including materials manufactured in China or working with CROs in China;
•delays in clinical trials relating to geopolitical instability related to Russia's invasion of Ukraine and the military conflict in the Middle East;
•the inability to successfully complete subsequent or confirmatory clinical trials in countries where our experience is limited;
78
•longer payment and reimbursement cycles and uncertainties regarding the collectability of accounts receivable;
•fluctuations in foreign currency exchange rates that may adversely impact our revenue, net income and value of certain of our investments;
•the imposition of governmental controls;
•diverse data privacy and protection requirements;
•increasingly complex standards for complying with foreign laws and regulations that may differ substantially from country to country and may conflict with corresponding U.S. laws and regulations;
•the far-reaching anti-bribery and anti-corruption legislation in the U.K., including the U.K. Bribery Act 2010, and elsewhere and escalation of investigations and prosecutions pursuant to such laws;
•compliance with complex import and export control laws;
•changes in tax laws; and
•the imposition of tariffs or embargoes and other trade restrictions.
In addition, our international operations are subject to regulation under U.S. law. For example, the U.S. FCPA prohibits U.S. companies and their representatives from paying, offering to pay, promising to pay or authorizing the payment of anything of value to any foreign government official, government staff member, political party or political candidate for the purpose of obtaining or retaining business or to otherwise obtain favorable treatment or influence a person working in an official capacity. In many countries, the health care professionals we regularly interact with may meet the FCPA's definition of a foreign government official. Failure to comply with domestic or foreign laws could result in various adverse consequences, including possible delay in approval or refusal to approve a product, recalls, seizures or withdrawal of an approved product from the market, disruption in the supply or availability of our products or suspension of export or import privileges, the imposition of civil or criminal sanctions, the prosecution of executives overseeing our international operations and damage to our reputation. Any significant impairment of our ability to sell products outside of the U.S. could adversely impact our business and financial results. In addition, while we believe that we have appropriate compliance controls, policies and procedures in place to comply with the FCPA, there is a risk that acts committed by our employees, agents, distributors, collaborators or third-party providers might violate the FCPA and we might be held responsible. If our employees, agents, distributors, collaborators or third-party providers are found to have engaged in such practices, we could suffer severe penalties and may be subject to other liabilities, which could negatively affect our business, operating results and financial condition.
We built a large-scale biologics manufacturing facility and are building a gene therapy manufacturing facility, which will result in the incurrence of significant investment with no assurance that such investment will be recouped.
In order to support our future growth and drug development pipeline, we have expanded our large molecule production capacity by building a large-scale biologics manufacturing facility in Solothurn, Switzerland with no assurance that the additional capacity will be required or this investment will be recouped.
Although the Solothurn facility was approved by the FDA for LEQEMBI, there can be no assurance that the regulatory authorities will approve the Solothurn facility for the manufacturing of other products.
Additionally, we are building a new gene therapy manufacturing facility in RTP, North Carolina with no assurance that this investment will be fully utilized. If we are unable to fully utilize this gene therapy manufacturing facility, charges from excess capacity may occur and would have a negative effect on our financial condition and results of operations.
If we are unable to fully utilize our manufacturing facilities, our business may be harmed. Charges resulting from excess capacity may continue to occur and would have a negative effect on our financial condition and results of operations.
The illegal distribution and sale by third-parties of counterfeit or unfit versions of our products or stolen products could have a negative impact on our reputation and business.
Third-parties might illegally distribute and sell counterfeit or unfit versions of our products, which do not meet our rigorous manufacturing, distribution and testing standards. A patient who receives a counterfeit or unfit drug may be at risk for a number of dangerous health consequences. Our reputation and business could suffer harm as a result of counterfeit or unfit drugs sold under our brand name. Inventory that is stolen from warehouses, plants or while in-transit, and that is subsequently improperly stored and sold through unauthorized channels, could adversely impact patient safety, our reputation and our business.
79
The increasing use of social media platforms and artificial intelligence based software presents new risks and challenges.
Social media is increasingly being used to communicate about our products and the diseases our therapies are designed to treat. Social media practices in the biopharmaceutical industry continue to evolve and regulations relating to such use are not always clear and create uncertainty and risk of noncompliance with regulations applicable to our business. For example, patients may use social media channels to comment on the effectiveness of a product or to report an alleged adverse event. When such disclosures occur, there is a risk that we fail to monitor and comply with applicable adverse event reporting obligations or we may not be able to defend the company or the public's legitimate interests in the face of the political and market pressures generated by social media due to restrictions on what we may say about our products. There is also a risk of inappropriate disclosure of sensitive information or negative or inaccurate posts or comments about us on social media. We may also encounter criticism on social media regarding our company, management, product candidates or products. The immediacy of social media precludes us from having real-time control over postings made regarding us via social media, whether matters of fact or opinion. Our reputation could be damaged by negative publicity or if adverse information concerning us is posted on social media platforms or similar mediums, which we may not be able to reverse. If any of these events were to occur or we otherwise fail to comply with applicable regulations, we could incur liability, face restrictive regulatory actions or incur other harm to our business. Additionally, the use of AI based software is increasingly being used in the biopharmaceutical industry. Use of AI based software may lead to the release of confidential proprietary information which may impact our ability to realize the benefit of our intellectual property.
Risks Related to Holding Our Common Stock
Our operating results are subject to significant fluctuations.
Our quarterly revenue, expense and net income (loss) have fluctuated in the past and are likely to fluctuate significantly in the future due to the risks described in these Risk Factors as well as the timing of charges and expense that we may take. We have recorded, or may be required to record, charges that include:
•the cost of restructurings or other initiatives to streamline our operations and reallocate resources;
•the costs associated with decisions to terminate research and development programs;
•impairments with respect to investments, fixed assets and long-lived assets, including IPR&D and other intangible assets;
•inventory write-downs for failed quality specifications, charges for excess capacity, charges for excess or obsolete inventory and charges for inventory write-downs relating to product suspensions, expirations or recalls;
•changes in the fair value of contingent consideration or our equity investments;
•bad debt expense and increased bad debt reserves;
•outcomes of litigation and other legal or administrative proceedings, regulatory matters and tax matters;
•payments in connection with acquisitions, divestitures and other business development activities and under license and collaboration agreements;
•failure to meet certain contractual commitments; and
•the impact of public health epidemics, on employees, the global economy and the delivery of healthcare treatments.
Our revenue and certain assets and liabilities are also subject to foreign currency exchange rate fluctuations due to the global nature of our operations. Our efforts to mitigate the impact of fluctuating currency exchange rates may not be successful. As a result, currency fluctuations among our reporting currency, the U.S. dollar, and other currencies in which we do business will affect our operating results, often in unpredictable ways. Our net income may also fluctuate due to the impact of charges we may be required to take with respect to foreign currency hedge transactions. In particular, we may incur higher than expected charges from early termination of a hedge relationship.
Our operating results during any one period do not necessarily suggest the anticipated results of future periods.
Our investments in properties may not be fully realized.
We own or lease real estate primarily consisting of buildings that contain research laboratories, office space and manufacturing operations. We may decide to consolidate or co-locate certain aspects of our business operations or dispose of one or more of our properties, some of which may be located in markets that are experiencing high vacancy rates and decreasing property values. If we determine that the fair value of any of our owned properties is
80
lower than their book value, we may not realize the full investment in these properties and incur significant impairment charges or additional depreciation when the expected useful lives of certain assets have been shortened due to the anticipated closing of facilities. If we decide to fully or partially vacate a property, we may incur significant cost, including facility closing costs, employee separation and retention expense, lease termination fees, rent expense in excess of sublease income and impairment of leasehold improvements and accelerated depreciation of assets. Any of these events may have an adverse impact on our results of operations.
We may not be able to access the capital and credit markets on terms that are favorable to us.
We may seek access to the capital and credit markets to supplement our existing funds and cash generated from operations for working capital, capital expenditure and debt service requirements and other business initiatives. The capital and credit markets are experiencing, and have in the past experienced, extreme volatility and disruption, which leads to uncertainty and liquidity issues for both borrowers and investors. In the event of adverse market conditions, we may be unable to obtain capital or credit market financing on favorable terms which could significantly increase our financing costs. Changes in credit ratings issued by nationally recognized credit rating agencies could also adversely affect our cost of financing and the market price of our securities.
Our indebtedness could adversely affect our business and limit our ability to plan for or respond to changes in our business.
Our indebtedness, together with our significant contingent liabilities, including milestone and royalty payment obligations, could have important consequences to our business; for example, such obligations could:
•increase our vulnerability to general adverse economic and industry conditions;
•limit our ability to access capital markets and incur additional debt in the future;
•require us to dedicate a substantial portion of our cash flow from operations to payments on our indebtedness, thereby reducing the availability of our cash flow for other purposes, including business development, research and development and mergers and acquisitions; and
•limit our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate, thereby placing us at a disadvantage compared to our competitors that have less debt.
Our investment portfolio is subject to market, interest and credit risk that may reduce its value.
We maintain a portfolio of marketable securities for investment of our cash as well as investments in equity securities of certain biotechnology companies. Changes in the value of our investment portfolio could adversely affect our earnings. The value of our investments may decline due to, among other things, increases in interest rates, downgrades of the bonds and other securities in our portfolio, negative company-specific news, biotechnology market sentiment, instability in the global financial markets that reduces the liquidity of securities in our portfolio, declines in the value of collateral underlying the securities in our portfolio and other factors. Each of these events may cause us to record charges to reduce the carrying value of our investment portfolio or sell investments for less than our acquisition cost. Although we attempt to mitigate these risks through diversification of our investments and continuous monitoring of our portfolio's overall risk profile, the value of our investments may nevertheless decline.
There can be no assurance that we will continue to repurchase shares or that we will repurchase shares at favorable prices.
From time to time our Board of Directors authorizes share repurchase programs. The amount and timing of share repurchases are subject to capital availability and our determination that share repurchases are in the best interest of our shareholders and are in compliance with all respective laws and our applicable agreements. Our ability to repurchase shares will depend upon, among other factors, our cash balances and potential future capital requirements for strategic transactions, our results of operations, our financial condition and other factors beyond our control that we may deem relevant. Additionally, the recently enacted IRA includes an excise tax on share repurchases, which will increase the cost of share repurchases. A reduction in repurchases under, or the completion of, our share repurchase programs could have a negative effect on our stock price. We can provide no assurance that we will repurchase shares at favorable prices, if at all.
Some of our collaboration agreements contain change in control provisions that may discourage a third-party from attempting to acquire us.
Some of our collaboration agreements include change in control provisions that could reduce the potential acquisition price an acquirer is willing to pay or discourage a takeover attempt that could be viewed as beneficial to shareholders. Upon a change in control, some of these provisions could trigger reduced milestone, profit or royalty
81
payments to us or give our collaboration partner rights to terminate our collaboration agreement, acquire operational control or force the purchase or sale of the programs that are the subject of the collaboration.
General Risk Factors
Our effective tax rate fluctuates, and we may incur obligations in tax jurisdictions in excess of accrued amounts.
As a global biopharmaceutical company, we are subject to taxation in numerous countries, states and other jurisdictions. As a result, our effective tax rate is derived from a combination of applicable tax rates, including withholding taxes, in the various places that we operate. In preparing our financial statements, we estimate the amount of tax that will become payable in each of such places. Our effective tax rate may be different than experienced in the past or our current expectations due to many factors, including changes in the mix of our profitability from country to country, the results of examinations and audits of our tax filings, adjustments to the value of our uncertain tax positions, interpretations by tax authorities or other bodies with jurisdiction, the result of tax cases, changes in accounting for income taxes and changes in tax laws and regulations either prospectively or retrospectively and the effects of the integration of Reata.
Our inability to secure or sustain acceptable arrangements with tax authorities and future changes in the tax laws, among other things, may result in tax obligations in excess of amounts accrued in our financial statements.
The enactment of some or all of the recommendations set forth or that may be forthcoming in the OECD’s project on “Base Erosion and Profit Shifting” by tax authorities and economic blocs in the countries in which we operate, could unfavorably impact our effective tax rate. These initiatives focus on common international principles for the entitlement to taxation of global corporate profits and minimum global tax rates. Many countries have or are in the process of enacting legislation intended to implement the OECD GloBE Model Rules effective on January 1, 2024. The impact on the Company will depend on the timing of implementation, the exact nature of each country's GloBE legislation, guidance and regulations thereon and their application by tax authorities either prospectively or retrospectively.
Our business involves environmental risks, which include the cost of compliance and the risk of contamination or injury.
Our business and the business of several of our strategic partners involve the controlled use of hazardous materials, chemicals, biologics and radioactive compounds which make us subject to changing and evolving rules and interpretations, which could require us to incur substantial costs associated with compliance or to alter one or more of our business practices. Although we believe that our safety procedures for handling and disposing of such materials comply with state, federal and foreign standards, there will always be the risk of accidental contamination or injury. If we were to become liable for an accident, or if we were to suffer an extended facility shutdown, we could incur significant costs, damages and penalties that could harm our business. Manufacturing of our products and product candidates also requires permits from government agencies for water supply and wastewater discharge. If we do not obtain appropriate permits, including permits for sufficient quantities of water and wastewater, we could incur significant costs and limits on our manufacturing volumes that could harm our business. Additionally, regulators have passed new environmental disclosure rules. For example, the SEC, the E.U. and California have implemented new climate disclosure rules that will generally require additional disclosure. Additionally, other regulators are considering environmental disclosure rules. These new rules collectively will impose additional disclosure requirements elating to climate-related risks and emissions disclosures. We expect to be subject to these new laws, which impose extensive reporting obligations about greenhouse gas emissions and climate-related financial risks. These recently enacted and proposed regulations may require us to incur compliance and disclosure costs and will likely require substantial management attention.
82
ITEM 2. UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS
ISSUER PURCHASES OF EQUITY SECURITIES
The following table summarizes our common stock repurchase activity under our 2020 Share Repurchase Program during the first quarter of 2024:
| Period | Total Number of Shares Purchased (#) | Average Price Paid per Share ($) | Total Number of Shares Purchased as Part of Publicly Announced Programs (#) | Approximate Dollar Value of Shares That May Yet Be Purchased Under Our Programs ($ in millions) | ||||||||||||||||||||||
| January 2024 | — | $ | — | — | $ | 2,050.0 | ||||||||||||||||||||
| February 2024 | — | $ | — | — | $ | 2,050.0 | ||||||||||||||||||||
| March 2024 | — | $ | — | — | $ | 2,050.0 | ||||||||||||||||||||
Total(1) | — | $ | — | |||||||||||||||||||||||
(1) There were no share repurchases during the first quarter of 2024.
In October 2020 our Board of Directors authorized our 2020 Share Repurchase Program, which is a program to repurchase up to $5.0 billion of our common stock. Our 2020 Share Repurchase Program does not have an expiration date. All share repurchases under our 2020 Share Repurchase Program will be retired. There were no share repurchases of our common stock during the three months ended March 31, 2024 and 2023. Approximately $2.1 billion remained available under our 2020 Share Repurchase Program as of March 31, 2024.
ITEM 5. OTHER INFORMATION
TRADING ARRANGEMENTS
There were no terminated
83
ITEM 6. EXHIBITS
The exhibits listed below are filed or furnished as part of this Quarterly Report on Form 10-Q.
EXHIBIT INDEX
Exhibit Number | Description of Exhibit | |||||||
| 31.1+ | ||||||||
| 31.2+ | ||||||||
| 32.1++ | ||||||||
| 101++ | The following materials from Biogen Inc.’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2024, formatted in iXBRL (Inline Extensible Business Reporting Language): (i) the Condensed Consolidated Statements of Income, (ii) the Condensed Consolidated Statements of Comprehensive Income, (iii) the Condensed Consolidated Balance Sheets, (iv) the Condensed Consolidated Statements of Cash Flow, (v) the Condensed Consolidated Statements of Equity and (vi) Notes to Condensed Consolidated Financial Statements. | |||||||
| 104++ | The cover page from this Quarterly Report on Form 10-Q for the quarter ended March 31, 2024, formatted in Inline XBRL. | |||||||
+ Filed herewith
++ Furnished herewith
84
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| BIOGEN INC. | ||
| /s/ Michael R. McDonnell | ||
| Michael R. McDonnell | ||
| Chief Financial Officer | ||
| (principal financial officer) | ||
April 24, 2024
85