UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
(Mark One)
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number
.
(Exact name of Registrant as specified in its charter and translation of Registrant’s name into English)
(Jurisdiction of incorporation or organization)
Biophytis S.A.
( of principal executive offices)
Chief Executive
Biophytis S.A.
Tel: +
(Name, Telephone, Email and/or Facsimile number and of Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
The | ||
* | The |
*Not for trading, but only in connection with the registration of the American Depositary Shares.
Securities registered or to be registered pursuant to Section 12(g) of the Act. None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act. None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report. Ordinary shares:
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐ Yes ☒
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. ☐ Yes ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ☐ | Accelerated filer ☐ | Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act.
† | The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012. |
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
U.S. GAAP ☐ | Accounting Standards as issued by the International Accounting Standards Board ☒ | Other ☐ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. ☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes
TABLE OF CONTENTS
Page | ||
1 | ||
1 | ||
1 | ||
1 | ||
56 | ||
110 | ||
110 | ||
127 | ||
138 | ||
142 | ||
144 | ||
144 | ||
154 | ||
155 | ||
158 | ||
158 | ||
Material Modifications to the Rights of Security Holders and Use of Proceeds | 158 | |
158 | ||
160 | ||
160 | ||
160 | ||
161 | ||
Purchases of Equity Securities by the Issuer and Affiliated Purchasers | 161 | |
161 | ||
161 | ||
162 | ||
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections | 162 | |
163 | ||
163 | ||
163 | ||
163 | ||
INTRODUCTION
Unless otherwise indicated, “BIOPHYTIS,” “the Company,” “our company,” “we,” “us” and “our” refer to BIOPHYTIS S.A. and its consolidated subsidiaries.
This annual report may contain references to our trademarks and to trademarks belonging to other entities. Solely for convenience, trademarks and trade names referred to in this annual report, including logos, artwork and other visual displays, may appear without the ® or TM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other companies’ trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other company.
Our audited consolidated financial statements have been prepared in accordance with IFRS Accounting Standards, as issued by the International Accounting Standards Board, or IASB. Our financial statements included in this annual report are presented in euros and, unless otherwise specified, all monetary amounts are in euros. All references in this annual report to “$,” “US$,” “U.S.$,” “U.S. dollars,” “dollars” and “USD” mean U.S. dollars and all references to “€” and “euros,” mean euros, unless otherwise noted. Throughout this report, references to ADSs mean American Depositary Shares, or ADSs, or ordinary shares represented by such ADSs, as the case may be.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 20-F (this “annual report”), contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that are based on our management’s beliefs and assumptions and on information currently available to our management. All statements other than present and historical facts and conditions contained in this annual report, including statements regarding our future results of operations and financial positions, business strategy, plans and our objectives for future operations, are forward-looking statements. When used in this annual report, the words “anticipate,” “believe,” “can,” “could,” “estimate,” “expect,” “intend,” “is designed to,” “may,” “might,” “plan,” “potential,” “predict,” “objective,” “should,” or the negative of these and similar expressions identify forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
| ● | the timing, progress and results of clinical trials for our drug candidates, including statements regarding the timing of initiation and completion of clinical trials, dosing of subjects and the period during which the results of the clinical trials will become available and publicly released; |
| ● | the timing, scope or likelihood of regulatory filings and approvals for our drug candidates; |
| ● | our ability to successfully commercialize our drug candidates; |
| ● | potential benefits of the clinical development and commercial experience of our management team; |
| ● | our ability to effectively market any drug candidates that receive regulatory approval, emergency use authorization, or conditional marketing authorization on our own or through third parties; |
| ● | our commercialization, marketing and manufacturing capabilities and strategy; |
| ● | our expectation regarding the safety and efficacy of our drug candidates; |
| ● | the potential clinical utility and benefits of our drug candidates; |
| ● | our ability to advance our drug candidates through various stages of development, especially through pivotal safety and efficacy trials; |
| ● | the likelihood of success and difficulty in ensuring success of clinical investigations; |
| ● | our estimates regarding the potential market opportunity for our drug candidates; |
| ● | developments and projections relating to our competitors or our industry; |
| ● | our ability to become profitable; |
| ● | our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; |
| ● | our ability to secure additional financing when needed on acceptable terms and to continue as a going concern; |
| ● | the impact of government laws and regulations in the United States, France and foreign countries; |
| ● | the implementation of our business model, strategic plans for our business, drug candidates and technology; |
| ● | our intellectual property position; |
| ● | our ability to rely on orphan drug designation for market exclusivity; |
| ● | our ability to attract or retain key employees, advisors or consultants; |
| ● | our expectations regarding the time during which we will be an emerging growth company under the JOBS Act; |
| ● | whether we are classified as a passive foreign investment company (a “PFIC”) for current and future periods; |
| ● | unfavorable conditions in our industry, the global economy or global supply chain, including financial and credit market fluctuations, international trade relations, political turmoil, natural catastrophes, warfare (such as the conflict involving Russia and Ukraine or the Hamas - Israel conflict), and terrorist attacks; and |
| ● | other risks and uncertainties, including those listed in this annual report under the caption “Risk Factors.” |
You should refer to the section of this annual report titled “Item 3.D—Risk Factors” for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this annual report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
You should read this annual report and the documents that we reference in this annual report and have filed as exhibits to this annual report completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements.
This annual report contains market data and industry forecasts that were obtained from industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third-party information. While we believe the market position, market opportunity and market size information included in this annual report is generally reliable, such information is inherently imprecise.
PART I
Item 1. Identity of Director, Senior Management and Advisers.
Not applicable.
Item 2. Offer Statistics and Expected Timetable.
Not applicable.
Item 3. Key Information.
A. | [Reserved]. |
B. | Capitalization and Indebtedness |
Not applicable.
C. | Reasons for the Offer and Use of Proceeds |
Not applicable.
D. | Risk Factors |
Our business is subject to a number of risks and uncertainties that may adversely affect our business, financial condition, results of operations, cash flows, and prospects. These risks are discussed more fully below and include, but are not limited to:
| ● | Risks Related to Our Limited Operating History, Financial Condition, and Capital Requirements |
| o | We are a clinical-stage biotechnology company with no products approved or authorized for sale and a history of losses, which makes it difficult to assess our future prospects and financial results. |
| o | We will need substantial additional financing to achieve our goals, and a failure to obtain this capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our product development or other operations. These factors raise substantial doubt regarding our ability to continue as a going concern. |
| o | We may become dependent on reimbursable financial advances and non-reimbursable subsidies from the French government. |
| o | We have limited resources and may face challenges related to prioritizing drug candidate development. |
| o | Our indebtedness could restrict our operations and make us more vulnerable to adverse economic conditions. |
| o | Our business, financial condition and results of operations could be adversely affected by global or regional health events and related government, private sector and individual responsive actions. |
| ● | Risks Related to Our Business |
| o | Clinical development is lengthy and expensive, and we may not be able to obtain regulatory approval or emergency use authorization for our drug candidates. |
| o | We may have difficulty enrolling patients or finding and retaining investigators that are needed to conduct the clinical studies. |
| o | Our drug candidates could cause undesirable side effects. |
1
| o | Our drug candidates may fail to achieve physician acceptance and patient adoption. |
| o | We rely on third parties for raw materials and to conduct our preclinical studies and clinical trials. |
| o | We face significant competition. |
| o | We will face government restrictions on pricing and reimbursement. |
| o | We will need to establish or secure sales capabilities. |
| o | We may face challenges attracting and retaining senior management and key scientific personnel. |
| o | We may face product liability lawsuits. |
| o | We may not be successful in our existing and future collaborations. |
| o | We may experience significant disruptions to our information technology systems or breaches of data security and/or misconduct by our employees or independent contractors. |
| o | Our employees and independent contractors may engage in misconduct or improper activities. |
| o | We may be unable to comply with environmental laws and regulations. |
| ● | Risks Related to Intellectual Property |
| o | We must protect our intellectual property and proprietary rights. |
| o | We may be unable to resolve disputes concerning the infringement or misappropriation of our proprietary rights or the proprietary rights of others. |
| ● | Risks Related to Government Regulation |
| o | Even if we obtain regulatory approval or authorization for our products, we will remain subject to ongoing regulatory scrutiny. |
| o | Eradication or substantial eradication of COVID-19 could reduce or eliminate the demand for our product, BIO101 (20-hydroxyecdysone) in this indication. |
| o | Regulatory agencies may change the policies and requirements regarding approvals and emergency use authorizations, or revoke emergency use authorizations that the agencies have already issued. |
| o | Our business will be impacted by healthcare legislation and our relationships with investigators, healthcare professionals, consultants, third-party payors, patient organizations and customers. |
| o | We will be impacted by U.S. and foreign anti-corruption and anti-money laundering laws on our business. |
| o | We may be unable to maintain certain tax benefits applicable to French technology companies. |
| o | We will be impacted by U.S. tax laws and regulations on our business. |
2
| ● | Risks Related to the Ownership of the ADSs and Ordinary Shares and Our Status as a Non-U.S. Company with Foreign Private Issuer Status |
| o | The requirements of being a U.S. public company may strain our resources. And if we fail to meet the continued listing requirements of Nasdaq, including the minimum shareholders’ equity requirement of $2.5 million, it could result in a delisting of our securities. |
| o | An active trading market for our ADSs may not continue and the market price of our equity securities may be volatile. |
| o | We may be exposed to foreign exchange risk. |
| o | Investors may be diluted as a result of a significant number of outstanding warrants and convertible debt instruments and future sales of a substantial number of our securities could adversely affect the price of our securities. |
| o | U.S. investors may have difficulty enforcing civil liabilities against our company and directors and senior management and the experts named in this annual report and may not be entitled to a jury trial with respect to claims arising under the deposit agreement. |
| o | Our governing documents and French corporate law may delay or discourage a takeover attempt. |
| o | The ability of ADS holders to exercise voting rights, participate in any future preferential subscription rights, receive dividends, or transfer their ADSs. |
| o | Our status as a foreign private issuer and an “emerging growth company” may be unattractive to certain investors. |
| o | There are risks associated with being characterized a passive foreign investment company. |
| o | We may be unable to maintain effective internal control over financial reporting. |
Risks Related to Our Limited Operating History, Financial Condition, and Capital Requirements
We are a clinical-stage biotechnology company with no products approved or authorized for commercial sale. We have incurred significant losses since our inception and anticipate that we will continue to incur losses for the foreseeable future.
Biotechnology product development is a highly speculative undertaking because it entails substantial upfront capital expenditures and significant risk that any potential drug candidate will not demonstrate adequate effectiveness in the targeted indication or an acceptable safety profile, gain regulatory approval or become commercially viable. We have incurred significant losses since our inception in 2006, and we anticipate that we will continue to incur losses for the foreseeable future, which, together with our limited operating history, may make it difficult to assess our future viability.
We incurred losses of €31.2 million, €24.3 million and €17.6 million ($19.5 million) (translated solely for convenience into dollars at an exchange rate of €1.00-$1.1062, the noon buying rate of the Federal Reserve Bank of New York on December 31, 2023) for the years ended December 31, 2021, 2022 and 2023, respectively. Substantially all of our losses have resulted from expenses incurred in connection with our preclinical and clinical programs and other research and development activities and from general and administrative costs associated with our operations. We expect to continue to incur losses for the foreseeable future, and we anticipate these losses will increase as we continue to develop our drug candidates, conduct clinical trials and pursue research and development activities. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our shareholders’ equity and working capital.
3
We will require substantial additional financing to achieve our goals, and a failure to obtain this capital when needed on acceptable terms, or at all, could force us to delay, limit, reduce or terminate our product development or other operations. These factors raise substantial doubt regarding our ability to continue as a going concern
Since our inception, we have invested a significant portion of our efforts and financial resources on our preclinical studies and clinical trials and other research and development activities. We believe that we will continue to expend substantial resources for the foreseeable future in connection with the preclinical and clinical development of our current drug candidates and the discovery and development of any other drug candidates we may choose to pursue. These expenditures will include costs associated with conducting preclinical studies and clinical trials and obtaining regulatory approvals, emergency use authorizations, or conditional marketing authorization and any expenses associated with commercializing, marketing and selling products approved or authorized for sale that we elect to commercialize ourselves. In addition, other unanticipated costs may arise. Because the outcome of any preclinical study or clinical trial is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development of our current drug candidates or any future drug candidates we may choose to pursue.
As of December 31, 2023, we had capital resources consisting of cash and cash equivalents of € 5.6 million ($ 6.2 million) (translated solely for convenience into dollars at an exchange rate of €1.00-$1.1062, the noon buying rate of the Federal Reserve Bank of New York on December 31, 2023). Since December 31, 2023, we drew down €4 million from our 2021 credit facility with ATLAS Special Opportunities LLC (“ATLAS”).
Our existing capital resources, including our ability to draw down on our credit facility with ATLAS (as described in further detail in “Item 5, Operating and Financial Review and Prospects” of this annual report), will be enough to fund our planned operating expenses into the first quarter of 2025. Accordingly, our current and available capital resources are not sufficient to cover our operating needs for at least the next 12 months. In addition, our current operating plans may change as a result of many factors currently unknown to us, and we may need to seek additional funds even sooner than planned, through public or private equity or debt financings or other sources, such as strategic collaborations. And we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
Our future capital requirements depend on many factors, including:
| ● | the scope, progress, data and costs of researching and developing our current drug candidates and any other drug candidates we may choose to pursue in the future, and conducting preclinical studies and clinical trials; |
| ● | the timing of, and the costs involved in, obtaining regulatory approvals or authorizations for our current drug candidates or any future drug candidates we may choose to pursue; |
| ● | the number and characteristics of any additional drug candidates we develop or acquire; |
| ● | any costs associated with manufacturing our current drug candidates and any future drug candidates; |
| ● | the cost of sourcing purified extracts and a supply chain in sufficient quantity and quality to meet our needs; |
| ● | the cost of commercialization activities associated with any of our current drug candidates or any future drug candidates that are approved or authorized for sale and that we choose to commercialize ourselves, including marketing, sales and distribution costs; |
| ● | our ability to maintain existing, and establish new, strategic collaborations, licensing or other arrangements and the financial terms of any such agreements, including the timing and amount of any future milestone, royalty or other payments due under any such agreement; |
| ● | any product liability or other lawsuits related to any current or future drug candidates that are approved or authorized for sale; |
| ● | the expenses needed to attract, hire and retain skilled personnel; |
| ● | the costs associated with being a public company; |
4
| ● | the costs that become required as a result of modified or revised clinical protocols for our clinical trials; |
| ● | the costs that become required due to necessity of having to perform additional clinical trials; |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing our intellectual property portfolio; and |
| ● | the timing, receipt and amount of sales of any future approved or authorized products, if any. |
Additional funds may not be available when we need them, on terms that are acceptable to us, or at all. If adequate funds are not available to us on a timely basis on terms acceptable to us, we may be required to:
| ● | delay, limit, reduce or terminate preclinical studies, clinical trials or other development activities for our current drug candidates or any future drug candidate; |
| ● | seek corporate partners for our drug candidates when we would otherwise develop our drug candidates on our own, or at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available; |
| ● | delay, limit, reduce or terminate our research and development activities; or |
| ● | delay, limit, reduce or terminate any efforts to establish manufacturing and sales and marketing capabilities or other activities that may be necessary to commercialize our current drug candidates or any future drug candidates. |
These events and conditions indicate that a material uncertainty exists that may cast significant doubt on our ability to continue as a going concern and, therefore, we may be unable to realize our assets and discharge our liabilities in the normal course of business.
We do not expect to realize revenue from sales of products or royalties from licensed products in the foreseeable future, if at all, unless and until our drug candidates are clinically tested, approved or authorized for commercialization, and successfully marketed. To date, we have primarily financed our operations through the sale of debt and equity securities (including our U.S. initial public offering in February 2021), as well as public aid for innovation and reimbursement of the French research tax credit, described elsewhere in this annual report. We will need to seek additional funding in the future and currently intend to do so through collaborations, public or private equity offerings or debt financings, credit or loan facilities, public funding, or a combination of one or more of these funding sources. Our ability to raise additional funds will depend on financial, economic and other factors, many of which are beyond our control. Additional funds may not be available to us on acceptable terms or at all. If we enter into arrangements with collaborators or others, we may be required to relinquish rights to some of our drug candidates that we would otherwise pursue on our own. If we raise additional funds by issuing equity securities, our shareholders will suffer dilution and the terms of any financing may adversely affect the rights of our shareholders. In addition, as a condition to providing additional funds to us, future investors may demand, and may be granted, rights superior to those of existing shareholders. Debt financing, if available, is likely to involve restrictive covenants limiting our flexibility in conducting future business activities, and, in the event of insolvency, debt holders would be repaid before holders of our equity securities received any distribution of our corporate assets.
We have benefited from certain reimbursable financial advances and non-reimbursable subsidies from the French government that if terminated or reduced may restrict our ability to successfully develop, manufacture and commercialize our drug candidates.
We have benefited from certain reimbursable advances and non-reimbursable subsidies from the French government and intend to continue to seek advances and/or subsidies from these agencies in the future in order to accelerate the development of our drug candidates. There is no assurance that these benefits will continue to be available to us in the future. If such benefits and programs were to be terminated or reduced, it could have an adverse effect on our business, operating results and financial condition and could deprive us of financial resources necessary for research and development of our drug candidates. Furthermore, the advances and subsidies are generally subject to contractual conditions, including our compliance with agreed upon preliminary budgets and scientific programs, informing the lender of any deviations from such agreed upon budgets and programs, and our compliance with certain financial ratios to ensure our solvency. In the event that we do not comply with the contractual conditions of the subsidies, we may be required to reimburse the French government for any outstanding payments (€0.9 million as of December 31, 2022) ($1 million) (translated solely for convenience into dollars at an exchange rate of €1.00-$1.1062, the noon buying rate of the Federal
5
Reserve Bank of New York on December 31, 2023) on an accelerated basis and could be liable for any damages incurred by such agencies resulting from the breach of contract.
Due to the significant resources required for the development of our drug candidates, we must prioritize development of certain drug candidates and/or certain disease indications. We may expend our limited resources on candidates or indications that do not yield a successful product and fail to capitalize on drug candidates or indications that may be more profitable or for which there is a greater likelihood of success.
We plan to develop a pipeline of drug candidates to treat age-related diseases and diseases whose progression and symptoms are similar to those associated with aging. Due to the significant resources required for the development of drug candidates, we must focus our attention and resources on specific diseases and disease pathways and decide which drug candidates to pursue and the amount of resources to allocate to each.
Our decisions concerning the allocation of research, development, collaboration, management and financial resources toward particular drug candidates or therapeutic areas may not lead to the development of any viable commercial product and may divert resources away from better opportunities. Similarly, any decision to delay, terminate or collaborate with third parties in respect of certain programs may subsequently prove to be suboptimal and could cause us to miss valuable opportunities. If we make incorrect determinations regarding the viability or market potential of any of our programs, drug candidates, or misread trends in the aging or age-related disease space, or in the biotechnology industry, our business, financial condition and results of operations could be materially adversely affected. As a result, we may fail to capitalize on viable commercial products or profitable market opportunities, be required to forego or delay pursuit of opportunities with other drug candidates or other diseases and disease pathways that may later prove to have greater commercial potential than those we choose to pursue, or relinquish valuable rights to such drug candidates through collaboration, licensing or other royalty arrangements in cases in which it would have been advantageous for us to invest additional resources to retain development and commercialization rights.
Our operating results may fluctuate significantly, which may make our future operating results difficult to predict.
Our operating results may fluctuate significantly, which may make it difficult for us to predict our future operating results. These fluctuations may occur due to a variety of factors, many of which are outside of our control and may be difficult to predict, including:
| ● | the timing and cost of, and level of investment in, research, development and, if approved or authorized, any commercialization activities relating to our drug candidates, which may change from time to time; |
| ● | the timing and status of enrollment for our clinical trials, and availability of medical staff to conduct the clinical trials; |
| ● | the effect of pandemics or endemics (including COVID-19), or emergence of other infectious diseases, on our clinical trials, including government-mandated or -recommended shutdowns, or other restrictions or limitations that are caused by the spread of viruses; |
| ● | the further development and widespread adoption of COVID-19 vaccines and treatment options could reduce or eliminate the demand for our products related to treatment of respiratory failure; |
| ● | the regulatory agencies’ revocation of emergency use authorizations, or conclusion of the public health emergency declaration; |
| ● | the cost of manufacturing our drug candidates, as well as building out our supply chain, which may vary depending on the quantity of production and the terms of our agreements with manufacturers; |
| ● | expenditures that we may incur to acquire, develop or commercialize additional drug candidates; |
| ● | the timing and amount of any future milestone, royalty or other payments due under any collaboration or license agreement; |
| ● | future accounting pronouncements or changes in our accounting policies; |
6
| ● | the timing and success or failure of preclinical studies and clinical trials for our drug candidates and/or redesign, delays and/or change of scope of our preclinical or clinical trials; |
| ● | the timing of receipt of approvals, emergency use authorizations, or conditional marketing authorizations for our drug candidates from regulatory authorities in the United States and internationally; |
| ● | the timing and success of competing drug candidates, or any other change in the competitive landscape of our industry, including consolidation among our competitors or partners; |
| ● | coverage and reimbursement policies with respect to our drug candidates, if approved or authorized for emergency use; and |
| ● | the level of demand for our products, if approved or authorized for emergency use, which may vary significantly over time. |
The cumulative effects of these factors could result in large fluctuations and unpredictability in our annual operating results. As a result, comparing our operating results on a period-to-period basis may not be meaningful. Investors should not rely on our past results as an indication of our future performance.
This variability and unpredictability could also result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any forecasts we may provide to the market, or if the forecasts we provide to the market are below the expectations of analysts or investors, the price of our ordinary shares and ADSs could decline substantially. Such a stock price decline could occur even when we have met any previously publicly stated revenue or earnings guidance we may provide.
Our indebtedness could restrict our operations and make us more vulnerable to adverse economic conditions.
In April 2020, we signed a convertible bond financing of €24 million with ATLAS to continue the development of BIO101 (20-hydroxyecdysone). Pursuant to the terms of the agreement (as amended), ATLAS agreed to subscribe for up to €24 million in convertible bonds, to be issued by us in up to eight tranches of €3 million each. We issued the eighth tranche in December 2021. As of December 31, 2022, all the convertible bonds related to this contract have been converted.
On June 14, 2021, we signed a new convertible bond financing of €32 million with ATLAS. Pursuant to the terms of the agreement, ATLAS agreed to subscribe for up to €32 million in convertible bonds, to be issued by us in up to eight tranches of €4 million each. As of December 2023, the Company issued three tranches for a total of €12 million and 480 ORNANEs, and the outstanding debt as of December 31, 2023 amounted to €1.45 million corresponding to 58 ORNANEs. Since December 31, 2023, the Company issued the fourth tranche of €4 million and 160 ORNANEs as part of its 2021 bond financing agreement with ATLAS. As of the date of this filing and considering the terms and expiration date of the ATLAS agreement as of June 14, 2024, the Company has the capacity to issue no more than two additional tranches for a total amount of €8 million.
On November 19, 2021, we entered into a Subscription Agreement, a Straight Bonds Issue Agreement and a Convertible Bonds Issue Agreement with Kreos Capital VI (UK) Ltd. and Kreos Capital VI (Expert Fund) L.P., which provides for up to €10 million in financing to us. Pursuant to the terms of the agreements, Kreos agreed to subscribe for up to €7.75 million in convertible bonds and for up to €2.25 million in non-convertible bonds, to be issued by us in up to four tranches. The first two tranches were issued on November 22, 2021. Each tranche of non-convertible bonds bears a 10% annual interest rate and must be repaid in 36 monthly installments, with monthly payments commencing in April 2022. Each tranche of convertible bonds bears a 9.5% annual interest rate and must be repaid or converted into shares by 31 March 2025. In connection with the Kreos financing, we issued 2,218,293 warrants giving them the right to purchase 2,218,293 new ordinary shares at an exercise price of €0.56 per share over a seven year period from the issue date. By subscribing to the warrants, Kreos waived its right to exercise the warrants issued to Kreos within the framework of the 2018 loan.
Pursuant to the terms of the agreements with Kreos, we have the right, at any time but with no less than 30 days prior notice to Kreos, to prepay or purchase the bonds, exclusively in full. The prepayment will be equal to (i) the principal amount outstanding, plus (ii) the sum of all interest repayments which would have been paid throughout the remainder of the term of the relevant tranche discounted by 10% per annum.
If we are unable to make the required payments, we may need to refinance all or a portion of our indebtedness, sell assets, delay capital expenditures or seek additional equity. The terms of our existing or future debt agreements may also restrict us from
7
affecting any of these alternatives. Any refinancing of our debt could be at higher interest rates and may require us to comply with more onerous covenants, which could further restrict our business operations. Further, changes in the credit and capital markets, including market disruptions and interest rate fluctuations, may increase the cost of financing, make it more difficult to obtain favorable terms, or restrict our access to these sources of future liquidity. In addition, any failure to make scheduled payments of interest and principal on our outstanding indebtedness would likely result in a reduction of our credit rating, which could harm our ability to incur additional indebtedness on commercially reasonable terms or at all. Our inability to generate sufficient cash flow to satisfy our debt service obligations, or to refinance or restructure our obligations on commercially reasonable terms or at all, could have a material adverse effect on our business, financial condition and results of operations, as well as on our ability to satisfy our obligations in respect of our indebtedness.
Our debt agreements contain restrictions that limit our flexibility in operating our business.
Our Venture Loan Agreement and Bonds Issue Agreement with Kreos Capital V (UK) Ltd., our Subscription Agreement, our Straight Bonds Issue Agreement and our Convertible Bonds Issue Agreement with Kreos Capital VI (UK) Ltd. and Kreos Capital VI (Expert Fund) L.P., and our convertible notes agreements with ATLAS impose certain operating and financial restrictions. These covenants may limit our ability and the ability of our subsidiaries, under certain circumstances, to, among other things:
| ● | incur additional indebtedness; |
| ● | sell or transfer assets; and |
| ● | pay dividends and distributions. |
These agreements also contain certain customary affirmative covenants and events of default, including a change of control.
As a result of the covenants and restrictions contained in our existing debt agreements, we are limited in how we conduct our business, and we may be unable to raise additional debt to compete effectively or to take advantage of new business opportunities. The terms of any future indebtedness we may incur could include more restrictive covenants. We cannot guarantee that we will be able to maintain compliance with these covenants in the future and, if we fail to do so, that we will be able to obtain waivers from Kreos and ATLAS, and/or amend the covenants.
Our failure to comply with the restrictive covenants described above as well as others contained in our future debt instruments from time to time could result in an event of default, which, if not cured or waived, could result in our being required to repay these borrowings before their maturity dates. In addition, any event of default or declaration of acceleration under one debt instrument could also result in an event of default under one or more of our other debt instruments. If we are unable to repay, refinance or restructure our indebtedness under our secured debt, the holders of such debt could proceed against the collateral securing that indebtedness. If we are forced to refinance these borrowings on less favorable terms or if we are unable to repay, refinance or restructure such indebtedness, our financial condition and results of operations could be adversely affected.
Our business has been and could in the future be materially adversely affected by the effects of health pandemics or epidemics, including COVID-19 and its related variants, or emergence of other infectious diseases, and in particular in regions where we or third parties on which we rely have significant manufacturing facilties, concentrations of clinical trial sites or other business operations.
Our business has been and could in the future be materially adversely affected by the effects of health pandemics or epidemics, including COVID-19 and its related variants, or emergence of other infectious diseases. The COVID-19 pandemic prompted severe lifestyle and commercial restrictions aimed at reducing the spread of the disease. Governments imposed quarantines and other restrictions in response to the pandemic. We implemented social distancing and sanitary measures as well as a work-from-home policy for most of our employees during the height of the pandemic. Some of our clinical study sites had to be temporarily closed during the pandemic, and we were forced to revise the protocols and obtain IRB review and approval to continue our SARA INT clinical trial, which has since been completed. Although the COVID-19 pandemic appears to have largely subsided as a result of widespread access to vaccines and other preventative measures, additional variants that are more infectious or deadly may emerge in the future, and any future restrictions implemented in response to COVID-19 or another health pandemic or epidemic, or emergence of other infectious diseases could negatively impact our productivity, disrupt our business and delay our clinical programs and timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions, among other factors. Although we do not currently anticipate any further impacts to our clinical programs from COVID-19 or any other pandemic or endemic, or the
8
emergence of other infectious diseases, these and similar, and perhaps more severe, disruptions in our operations could negatively impact our business operating results and financial condition in the future.
Quarantines, shutdowns and shelter-in-place and similar government orders related to COVID-19 or other infectious diseases, or the perception that such events, orders or other restrictions on the conduct of business operations could occur, could impact personnel at third-party supplier, manufacturing or packaging facilities in European, China or other countries, or the availability or costs of materials, which could disrupt our supply chain. Although we do not currently anticipate any clinical supply issues or concerns for our planned clinical trials, any future restrictions resulting from the COVID-19 pandemic or other health pandemics or epidemics or emergence of other infectious diseases may disrupt our supply chain in the future and delay or limit our ability to obtain sufficient materials for our drug candidates.
In addition, our current clinical trial and planned clinical trials could be affected by a resurgence of COVID-19 (or emergence of new vaccine resistant strains) or emergence of new pandemics, epidemics or other infectious diseases. For example, site initiation and patient enrollment could be impacted by a resurgence of COVID-19 (or emergence of new vaccine resistant strains), and sites conducting potential patient enrollment may not be able or willing to comply with clinical trial protocols whether due to quarantines impeding patient movement or interrupting healthcare services, or due to potential patient concerns regarding interactions with medical facilities or staff. Similarly, our ability to recruit and retain principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 or another virus, may be delayed or disrupted, which could adversely impact our clinical trial operations. Furthermore, when the primary endpoint of one of our studies is a site-based assessment, there is a risk that participants will not be able to or want to undergo this required in person assessment for safety reasons, resulting in a delay to our studies and potentially compromising the timing and results of our study. A resurgence of COVID-19 (or emergence of new vaccine resistant strains) or the emergence of a new pandemic, epidemic or other infectious diseases could also lead to increased costs, due to a prolonged study timeframe, resulting in the need to add study staff and the need to utilize additional technological tools, such as remote monitoring, remote source-data verification and remote audits.
Regulatory authorities may also experience a significantly increased workload, with requirements and demands for short review timelines for COVID-19 studies on the one hand and the need to amend study protocols to address COVID-19-related limitations in study conduct on the other hand. This can prolong review timelines and reduce the availability to run expedited programs, which put a high demand on regulatory staff. There is also a risk that the changes to protocols of ongoing clinical trials (other than for COVID-19 indications) that were made to address restrictions imposed in the context of the coronavirus pandemic will negatively impact the review conducted by the relevant regulatory agencies. In which case, such agencies may consider the data to be insufficient to support acceptance of the data and the statistical plan. For example, changing in-office and in-person checks and visits to phone contacts may not be sufficient for regulatory review. We will not know until we complete our ongoing studies, complete analysis, and submit such data to the regulatory authorities, what, if any, limitations and effects could result.
In addition, the global COVID-19 pandemic has adversely affected, and any future significant outbreak of contagious diseases could similarly adversely affect, the economics and financial markets of many countries, including France and the United States, resulting in an economic downturn that could reduce our ability to access capital, which could negatively affect our liquidity and ability to process our clinical trials and business operations and suppress demand for our future products. Any of these events could have a material adverse effect on our business, financial condition, results of operations or cash flow. In addition, a recession, down-turn or market correction resulting from the COVID-19 pandemic, other pandemic or epidemic, or emergence of other infectious diseases could materially adversely affect the value of our ADSs and ordinary shares.
Risks Related to Our Business
Our business is dependent on the successful development, regulatory approval, emergency use authorization or conditional marketing authorization, manufacture and commercialization of our drug candidates.
We have no products approved or authorized for sale. Our lead drug candidate, BIO101 (20-hydroxyecdysone), is in clinical development and our second drug candidate, BIO201 is still in the preclinical development phase. Our life-cycle extension drug candidates, BIO103 and BIO203, are still in the preclinical development phase.
To secure marketing approval for our lead drug candidates, we will need to meet endpoints satisfactory to the U.S. Food and Drug Administration (“FDA”), and European Medicines Agency (“EMA”), in larger confirmatory clinical trials. The success of our business, including our ability to finance our company and generate any revenue in the future, will primarily depend on the successful development, regulatory approval, emergency use authorization or conditional marketing authorization, and commercialization of drug
9
candidates. It may be many years, if we succeed at all, before we have demonstrated the safety and efficacy of a drug candidate sufficient to warrant approval, emergency use authorization or conditional marketing authorization for commercialization.
In the future, we may also become dependent on other drug candidates that we may develop or acquire. The clinical and commercial success of our current drug candidates and any future drug candidates will depend on a number of factors, including the following:
| ● | our ability to raise any additional required capital on acceptable terms, or at all; |
| ● | our ability to complete Investigational New Drug (“IND”)-enabling studies and successfully submit IND or comparable applications; |
| ● | timely completion of our preclinical studies and clinical trials, which may be significantly slower or cost more than we currently anticipate and will depend substantially upon the performance of third-party contractors; |
| ● | whether we are required by the FDA, EMA or similar regulatory authorities to conduct additional clinical trials or other studies beyond those planned to support the approval, authorization, and commercialization of our drug candidates or any future drug candidates; |
| ● | acceptance of our proposed indications and primary endpoint assessments relating to the proposed indications of our drug candidates by the FDA, the EMA and similar regulatory authorities; |
| ● | our ability to demonstrate to the satisfaction of the FDA, EMA and similar regulatory authorities the safety, efficacy and acceptable risk to benefit profile of our drug candidates or any future drug candidates; |
| ● | the prevalence, duration and severity of potential side effects or other safety issues experienced with our drug candidates or future approved products, if any; |
| ● | the timely receipt of necessary marketing approvals or authorizations from the FDA, EMA and similar regulatory authorities; |
| ● | achieving and maintaining, and, where applicable, ensuring that our third-party contractors achieve and maintain compliance with our contractual obligations and with all regulatory requirements applicable to our drug candidates or any future drug candidates or approved products, if any; |
| ● | the ability of any third parties with whom we contract to manufacture adequate clinical trial and commercial supplies, if approved or authorized, of our current drug candidates or any future drug candidates, remain in good standing with regulatory agencies and develop, validate and maintain commercially viable manufacturing processes that are compliant with applicable requirements including current good manufacturing practices, (“cGMP”); |
| ● | with respect to any approved or authorized drug candidates that we elect to commercialize ourselves, our ability to successfully develop a commercial strategy and thereafter commercialize such drug candidates, whether alone or in collaboration with others; |
| ● | the convenience of our treatment or dosing regimen; |
| ● | our sourcing of purified extracts and a supply chain in sufficient quantity and quality to meet product needs for clinical development and commercialization; |
| ● | acceptance by physicians, payors and patients of the benefits, safety and efficacy of our drug candidates or any future drug candidates, if approved or authorized, including relative to alternative and competing treatments; |
| ● | patient demand for our drug candidates, if approved or authorized; |
| ● | our ability to maintain adequate drug diversion controls for BIO101 (20-hydroxyecdysone), which has a potential for misuse/abuse among body builders and other sportsmen as a result of its intended anabolic effect; |
10
| ● | lifestyle and commercial restrictions as a result of a resurgence of the COVID -19 pandemic or other pandemics or epidemics, or emergence of other infectious diseases; |
| ● | the potential impact of changing government orders in response to upticks in COVID -19 cases or as a result of new pandemics, epidemics, or other infectious diseases, and other limitations on our ability to conduct our business in the ordinary course; |
| ● | prioritization of hospital resources toward any resurgence of the COVID -19 pandemic, other pandemics or epidemics, or emergence of other infectious diseases, which would otherwise be used for clinical studies; |
| ● | the ability of our participants to safely follow clinical trial protocols because of quarantines impeding patient movement or interrupting healthcare services, or due to potential patient concerns regarding interactions with medical facilities or staff as a result of resurgence of the COVID -19 pandemic, other pandemics or epidemics, or emergence of other infectious diseases; |
| ● | the impact, if any, on the data from ongoing studies that have been impacted by the initial and subsequent waves of the COVID-19 pandemic effect, and whether changes that were made to accommodate the pandemic will allow regulatory acceptance of the resulting data or whether the data will be sufficient for regulatory review—the effect of such changes will not be known until we complete ongoing studies, data analysis, and submit the data for regulatory review; |
| ● | our ability to establish and enforce intellectual property rights in and to our current drug candidates and any future drug candidates we may develop; and |
| ● | our ability to avoid third-party patent interference, intellectual property challenges or intellectual property infringement claims. |
These factors, many of which are beyond our control, could cause us to experience significant delays or an inability to obtain regulatory approvals or authorizations, or commercialize or license our drug candidates. Even if regulatory approvals or authorizations are obtained, we may never be able to successfully commercialize or license any of our drug candidates. Accordingly, we cannot provide assurances that we will be able to generate sufficient revenue through the sale of our drug candidates or any future drug candidates we may develop to continue our business or achieve profitability.
We may not be able to obtain regulatory approval, or emergency use authorization, if applicable, for our drug candidates under applicable regulatory requirements. The denial, delay or imposed limitations of or on any such approval or authorization would preclude, delay or limit the commercialization of our drug candidates and adversely impact our potential to generate revenue and/or raise financing, our business and our results of operations.
To gain approval or authorization to market our drug candidates, we must provide the FDA, EMA and other foreign regulatory authorities with clinical data that adequately demonstrate the safety and efficacy of the drug candidate for the intended indication applied for in the applicable regulatory filing. Product development is a long, expensive and uncertain process, and delay or failure can occur at any stage of any of our clinical development programs. A number of companies in the biotechnology and pharmaceutical industries have suffered significant setbacks in clinical trials, even after promising data in preclinical studies or earlier phase clinical trials. These setbacks have been caused by, among other things, new preclinical findings made while clinical trials were underway and safety or efficacy observations made in clinical trials, including previously unreported adverse events. Success in preclinical testing and early phase clinical trials does not ensure that later phase clinical trials will be successful, and the results of clinical trials conducted by other parties may not be indicative of the results in trials we may conduct.
The research, testing, manufacturing, packaging, labeling, approval, authorization, sale, marketing and distribution of drug and biologic products are subject to extensive regulation by the FDA, EMA and other foreign regulatory authorities, and such regulations differ from country to country. We are not permitted to market our investigational drug candidates in the EU, the United States or any other country until they receive the requisite approval or authorization from the applicable regulatory authorities of such jurisdictions.
While the FDA postponed or delayed inspections at certain times since March 10, 2020 as a result of the COVID-19 pandemic, following the latest recommendations from the Centers for Disease Control and Prevention regarding COVID-19, the FDA has resumed normal inspection of domestic and foreign entities and is leveraging technology and resources including remote inspections to facilitate its compliance activities. In the future, global health concerns related to the emergence of new variants or new
11
viruses may prevent the FDA, EMA and other foreign regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, and it could significantly impact the ability of the FDA, EMA or other foreign regulatory authorities to timely review and process regulatory submissions, which could have a material adverse effect on our business.
The FDA, EMA or any foreign regulatory authorities can delay, limit or deny approval or authorization of our drug candidates for many reasons, including:
| ● | our inability to demonstrate to the satisfaction of the agency that a drug candidate is safe and effective for the requested indication; |
| ● | the agency’s disagreement with, or questions on, our trial protocol or the interpretation of data from preclinical studies or clinical trials, including studies impacted by the coronavirus pandemic; |
| ● | the agency’s refusal to accept the data that is produced from modified protocols (e.g., data collected from phone contacts instead of in-office and in-person checks and visits may not be sufficient for regulatory approval or authorization); |
| ● | our inability to demonstrate that the clinical and other benefits of a drug candidate outweigh any safety or other perceived risks; |
| ● | the agency’s requirement for additional preclinical studies or clinical trials; |
| ● | the agency’s non-approval of the formulation, labeling or specifications of a drug candidate; |
| ● | the agency’s failure to approve the manufacturing processes or facilities of third-party manufacturers upon which we rely; |
| ● | our inability to demonstrate to the satisfaction of the agency the sourcing of purified extracts and that our supply chain is in sufficient quantity and quality to meet product specifications; or |
| ● | the potential for regulations and policies of the FDA, EMA or the applicable foreign regulatory agencies relating to drug approval or emergency use authorization to significantly change in a manner rendering our clinical data insufficient for approval or authorization. |
Of the large number of biotechnology and pharmaceutical products in development, only a small percentage successfully complete the applicable regulatory approval or authorization processes and are commercialized.
Even if we eventually complete clinical testing and receive approval or authorization from the FDA, EMA or applicable foreign authorities for any of our drug candidates, the applicable agency may grant approval or authorization contingent on the performance of costly additional clinical trials, which may be required after approval or authorization. The FDA, EMA or the applicable foreign regulatory agency also may approve or authorize our drug candidates for a more limited indication or a narrower patient population than we originally requested, and the applicable agency, may not approve or authorize our drug candidates with the labeling that we believe is necessary or desirable for the successful commercialization of such drug candidates.
Any delay in obtaining, or inability to obtain, applicable regulatory approval or authorization would delay or prevent commercialization of our drug candidates and would materially adversely impact our business and prospects.
Clinical development is a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure or delay can occur at any time during the different phases, or stages, of the clinical trial process. Success in preclinical studies and early clinical trials does not ensure that later clinical trials will be successful. A number of companies in the biotechnology, biopharmaceutical and pharmaceutical industries have suffered significant setbacks in clinical trials, even after positive results in earlier preclinical studies or earlier phase clinical trials. These setbacks have been caused by, among other things, new preclinical findings made while clinical trials were underway and safety or efficacy observations made in clinical trials, including previously unreported adverse events. The results of our preclinical studies or in vivo and in vitro studies provide very limited data in diseases whose physiopathology is not well
12
understood and may not be predictive of the results of study outcomes in human clinical trials. Drug candidates in later stages of clinical trials may fail to show the desired pharmacological properties or safety and efficacy traits despite having progressed through preclinical studies and early phase clinical trials. Notwithstanding any promising results in earlier studies, we cannot be certain that we will not face setbacks and receive less promising results in later studies. Even if we are able to initiate and complete clinical trials, including studies underway during the initial coronavirus pandemic, the safety and efficacy data may not be sufficient to obtain regulatory approval or authorization for our drug candidates.
We may experience delays in obtaining the necessary regulatory authorization for our various clinical programs and initiating other planned studies and trials. Additionally, we cannot be certain that studies or trials for our drug candidates will begin on time, not require redesign, enroll an adequate number of subjects on time or be completed on schedule, if at all. Clinical trials can be delayed or terminated for a variety of reasons, including delays or failures related to:
| ● | the FDA, EMA or comparable foreign regulatory authorities disagreeing as to the design or implementation of our clinical trials; |
| ● | delays in obtaining regulatory approval to commence a trial; |
| ● | reaching agreement on acceptable terms with prospective contract research organizations (“CROs”), and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly from one country to another, notably among different CROs and trial sites; |
| ● | institutional review boards, ethics committee or IRB approval at each trial site; |
| ● | recruiting an adequate number of suitable patients to participate in a trial; |
| ● | having subjects complete a trial or return for post-treatment follow-up; |
| ● | clinical sites deviating from trial protocol or dropping out of a trial; |
| ● | inability to access sites for initiation and patient monitoring and enrollment due to travel or quarantine restrictions imposed by national, federal, state or local governance; |
| ● | addressing subject safety concerns that arise during the course of a trial; |
| ● | adding a sufficient number of clinical trial sites; |
| ● | sourcing of purified extracts and a supply chain in sufficient quantity and quality to meet product needs; |
| ● | supply chain and sourcing may be slow or significantly delayed as the result of pandemic or epidemic restrictions on movement, suspensions of service, and temporary global border closings; or |
| ● | obtaining sufficient product supply of drug candidate for use in preclinical studies, clinical trials, or during industrial scale up from third-party suppliers. |
We may experience numerous adverse or unforeseen events during, or as a result of, preclinical studies and clinical trials that could delay or prevent our ability to receive marketing approval or authorization, or commercialize our drug candidates, including:
| ● | we may receive feedback from regulatory authorities that requires us to modify the design of our clinical trials; |
| ● | clinical trials of our drug candidates may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical trials or abandon drug development programs; |
| ● | patient screening, new patient enrollment, monitoring and data collection may be affected or delayed as a result of restrictions imposed by national, federal, state or local governments due to pandemics, epidemics, or emergence of other infectious diseases; |
13
| ● | the number of patients required for clinical trials of our drug candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials at a higher rate than we anticipate; |
| ● | our third-party contractors may fail to comply with regulatory requirements, fail to maintain adequate quality controls, or be unable to source or provide us with sufficient purified extracts for product supply to conduct and complete preclinical studies or clinical trials of our drug candidates in a timely manner, or at all; |
| ● | we or our investigators might have to suspend or terminate clinical trials of our drug candidates for various reasons, including non-compliance with regulatory requirements, inability to comply with applicable study protocol as a result of a pandemic, epidemic, or other infectious disease, a finding that our drug candidates have undesirable side effects or other unexpected characteristics, or a finding that the participants are being exposed to unacceptable health risks; |
| ● | limitations occurring as a result of public health emergencies; |
| ● | the impact, if any, on the data from ongoing studies that have been impacted by the initial and subsequent waves of the coronavirus pandemic effect and whether changes to accommodate the pandemic will impact regulatory acceptance of the data or whether it will be sufficient for regulatory review, the effect of which will not be known until we complete ongoing studies, data analysis and submit the data for regulatory review; |
| ● | the cost of clinical trials of our drug candidates may be greater than we anticipate; |
| ● | the quality of our drug candidates or other materials necessary to conduct preclinical studies or clinical trials of our drug candidates may be insufficient or inadequate; |
| ● | regulators may revise the requirements for approving or authorizing our drug candidates, or such requirements may not be as we anticipate; and |
| ● | future collaborators may conduct clinical trials in ways they view as advantageous to them but that are suboptimal for us. |
If we are required to conduct additional clinical trials or other testing of our drug candidates beyond those that we currently contemplate, if we are unable to successfully complete clinical trials of our drug candidates or other testing, if the results of these trials or tests are not positive or are only moderately positive or if there are safety concerns, we may:
| ● | incur unplanned costs; |
| ● | be delayed in obtaining marketing approval or authorization for our drug candidates or, in due course, not obtain marketing approval at all; |
| ● | obtain marketing approval in some countries and not in others; |
| ● | obtain marketing approval or authorization for indications or patient populations that are not as broad as intended or desired; |
| ● | obtain marketing approval or authorization with labeling that includes significant use or distribution restrictions or safety warnings, including boxed warnings; |
| ● | be subject to additional post-marketing testing requirements; or |
| ● | have the treatment removed from the market after obtaining marketing approval or authorization. |
We could also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions in which such trials are being conducted, by the Data Safety Monitoring Board (“DSMB”) for such trial or by the FDA, EMA or other regulatory authorities. Such authorities may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA, EMA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or
14
adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
Further, conducting clinical trials in foreign countries presents additional risks that may delay completion of our clinical trials. These risks include the failure of enrolled patients in foreign countries to adhere to the clinical protocol as a result of differences in healthcare services or cultural customs, managing additional administrative burdens associated with foreign regulatory schemes, as well as political and economic risks relevant to such foreign countries, including foreign countries’ enforcement of COVID-19 or other public health - related restrictions on movement and lifestyle.
Principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and may receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, or a regulatory authority concludes that the financial relationship may have affected the interpretation of the trial, the integrity of the data generated at the applicable clinical trial site may be questioned and the utility of the clinical trial itself may be jeopardized, which could result in the delay or rejection of the marketing application we submit. Any such delay or rejection could prevent or delay us from commercializing our current or future drug candidates.
If we experience delays in the completion, or termination, of any preclinical study or clinical trial of our drug candidates, the commercial prospects of our drug candidates may be harmed, and our ability to generate revenues from any of these drug candidates will be delayed or not realized at all. In addition, any delays in completing our clinical trials may increase our costs, slow down our drug candidate development and approval process and jeopardize our ability to commercialize our products and generate revenues. Any of these occurrences may significantly harm our business, financial condition and prospects. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval or authorization of our drug candidates. If one or more of our drug candidates prove to be ineffective, unsafe or commercially unviable, our entire platform and pipeline would have little, if any, value, which would have a material and adverse effect on our business, financial condition, results of operations and prospects.
If we encounter difficulties enrolling patients in our clinical trials, our clinical development activities could be delayed or otherwise adversely affected.
The timely completion of clinical trials in accordance with their protocols depends on, among other things, our ability to enroll a sufficient number of patients who remain in the study until its conclusion. We may experience difficulties in patient enrollment in our clinical trials for a variety of reasons. The enrollment of patients depends on many factors, including:
| ● | the patient eligibility criteria defined in the protocol; |
| ● | the size of the patient population required for analysis of the trial’s primary endpoints; |
| ● | the number of eligible patients in the area where clinical investigation sites are located; |
| ● | the proximity of patients to trial sites; |
| ● | the ability of patients to be assessed in study sites, in the event of lock-downs due to pandemics, epidemics, or emergence of other infectious diseases; |
| ● | the design of the trial; |
| ● | patient enrollment may be delayed due to quarantines impeding patient movement or patient concerns regarding interaction and monitoring with medical facilities and staff; |
| ● | our ability to recruit clinical trial investigators with the appropriate competencies and experience; |
| ● | clinicians’ and patients’ perceptions as to the potential advantages of the drug candidate being studied in relation to other available therapies, including any new drugs that may be approved or authorized for the indications we are investigating; and |
| ● | our ability to obtain and maintain patient consents. |
15
In addition, our clinical trials may compete with other clinical trials for drug candidates that are in the same therapeutic areas as our drug candidates, and this competition will reduce the number and types of patients available to us, because some patients who might have opted to enroll in our trials may instead opt to enroll in a trial being conducted by one of our competitors. Since the number of qualified clinical investigators is limited, we may conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which will reduce the number of patients who are available for our clinical trials in such clinical trial site.
Delays in patient enrollment may result in increased costs or may affect the timing or outcome of the planned clinical trials, which could prevent completion of these trials and adversely affect our ability to advance the development of our drug candidates. The combined effects of high vaccination rates with reduction in patient numbers and mutations in the COVID-19 virus that may decrease its virulence and cause less severe disease, may decrease our ability to complete the study and file for approval.
A resurgence of COVID-19 (or emergence of new vaccine resistant strains), pandemic, endemic, or emergence of other infectious disease, may limit our ability or the investigators’ ability to find and retain medical staffs that are needed to conduct the clinical studies.
The COVID-19 pandemic caused a shortage of labor force, including nurses, clinicians, and other medical staff. This shortage caused medical institutions and other establishments to change their operations to accommodate the shortage, and in many cases, it resulted in increased personnel costs in finding and retaining the staffs that are necessary to conduct the institutions and establishments’ operations. While COVID-19 cases have decreased significantly, due in part to more people being vaccinated, a resurgence of COVID-19 (or emergence of new vaccine resistant strains), or emergence of other pandemic, epidemic, or other infectious diseases, could cause or exacerbate a staffing shortage, our ability to conduct clinical trials may be negatively affected, and we may need to modify, suspend, or stop our clinical trials, or expend greater resource in identifying and retaining the appropriate personnel necessary for the clinical investigations.
Our drug candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval or authorization, limit the commercial profile of an approved or authorized label, or result in significant negative consequences following marketing approval or authorization, if any.
Undesirable side effects caused by our drug candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval or authorization by the FDA, EMA or comparable foreign regulatory authorities. For example, one of our drug products, BIO101 (20-hydroxyecdysone), has been identified as having potential for misuse/abuse of the intended anabolic effect by body builders and sportsmen. Participants in clinical studies with BIO101 (20-hydroxyecdysone) are advised not to allow anyone access to the trial medication and the investigators specifically instruct subjects not to share their medicine. This risk is likely to become more significant after marketing authorization is granted, and the label for the drug, if it becomes approved or authorized, may have warnings and restrictions on the use and distribution of the product.
If unacceptable side effects arise in the development of our drug candidates, we, the FDA, EMA, the IRBs at the institutions in which our studies are conducted, or the DSMB could suspend or terminate our clinical trials or the FDA, EMA or comparable foreign regulatory authorities could order us to cease clinical trials or deny approval or authorization of our drug candidates for any or all targeted indications. Treatment-related side effects could also affect patient recruitment or the ability of enrolled patients to complete any of our clinical trials or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. Failure to recognize or manage the potential side effects of our drug candidates could result in patient injury. Any of these occurrences may harm our business, financial condition and prospects significantly.
If our drug candidates are used in combination with other drugs or treatments, they may interact negatively with those other drugs or treatments. We plan to conduct studies in order to assess the risks of interactions of our drug candidates with other drugs and treatments taken together. However, there can be no guarantee that our drug candidates will not interact negatively with other drugs or treatments not covered by our studies or that such interactions will not be revealed until after the products have been commercialized. These interactions could have adverse, unacceptable or undetected side effects, or could reduce or destroy the effectiveness of our drug candidates, which could diminish the commercial potential of our drug candidates, slow their development and consequently, have a material adverse effect on our business, financial condition and prospects.
16
Even if we successfully advance any of our drug candidates into and through clinical trials, such trials will likely only include a limited number of subjects and limited duration of exposure to our drug candidates. As a result, we cannot be assured that adverse effects of our drug candidates will not be uncovered when a significantly larger number of patients are exposed to the drug candidate. Further, any clinical trials may not be sufficient to determine the effect and safety consequences of taking our drug candidates over a multi-year period. Certain clinical trial protocols that are revised because of public health emergencies may also make it more difficult to identify potential safety concerns early on.
If any of our drug candidates receives marketing approval or authorization, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may withdraw their approval or authorization of the product; |
| ● | we may be required to recall a product or change the way such product is administered to patients; |
| ● | additional restrictions may be imposed on the marketing of the particular product or the manufacturing processes for the product or any component thereof; |
| ● | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or other warnings, including a potential for abuse warning; |
| ● | we may be required to implement a Risk Evaluation and Mitigation Strategy (“REMS”), or create a Medication Guide outlining the risks of such side effects for distribution to patients; |
| ● | we could be sued and held liable for harm caused to patients; |
| ● | the sales of our product may decrease significantly and the product may become less competitive; and |
| ● | our reputation may suffer. |
Any of the foregoing events could prevent us from achieving or maintaining market acceptance of the particular drug candidate, if approved or authorized, and result in the loss of significant revenues to us, which would materially and adversely affect our results of operations and business. In addition, if one or more of our drug candidates prove to be unsafe, our entire platform and pipeline could be affected, which would have a material and adverse effect on our business, financial condition, results of operations and prospects.
Even if our current drug candidates or any future drug candidates obtain regulatory approval or authorization, they may fail to achieve the broad degree of physician and patient adoption and use necessary for commercial success.
Even if one or more of our drug candidates receives the necessary regulatory approvals or authorizations, the commercial success of any of our current or future drug candidates will depend significantly on the broad adoption and use of the resulting product by physicians and patients for approved or authorized indications. Our drug candidates may not be commercially successful. For a variety of reasons, including among other things, competitive factors, pricing or physician preference, reimbursement by insurers, the degree and rate of physician and patient adoption of our current or future drug candidates, if approved or authorized, will depend on a number of factors, including:
| ● | the clinical indications for which the product is approved or authorized and patient demand for approved or authorized products that treat those indications; |
| ● | the safety and efficacy of our product as compared to other available therapies; |
| ● | the feasibility of adhering to heightened drug diversion protocols for drug product BIO101 (20-hydroxyecdysone), which has the potential for misuse/abuse by body builders and other athletes; |
| ● | the availability of coverage and adequate reimbursement from managed care plans, insurers and other healthcare payors for any of our drug candidates that may be approved or authorized; |
17
| ● | acceptance by physicians, operators of clinics and patients of the product as a safe and effective treatment; |
| ● | overcoming any biases physicians or patients may have toward particular therapies for the treatment of approved or authorized indications; |
| ● | public misperception regarding the use of our therapies, or public bias against “anti-aging” companies; |
| ● | any FDA regulatory actions regarding anti-aging claims made in relation to our drugs, if those claims are determined to be unsupported by our approved indications. |
| ● | patient satisfaction with the administration and effectiveness of our drug candidates and overall treatment experience, including, for example, the convenience of any dosing regimen and storage method; |
| ● | the cost of treatment with our drug candidates in relation to alternative treatments and reimbursement levels, if any, and willingness to pay for the product, if approved or authorized, on the part of insurance companies and other third-party payers, physicians and patients; |
| ● | the timing of market introduction of the drug candidate as well as competitive products; |
| ● | the revenue and profitability that our products may offer a physician as compared to alternative therapies; |
| ● | the prevalence and severity of side effects; |
| ● | limitations or warnings contained in the approved or authorized labeling for our products; |
| ● | any regulatory agency’s requirement to undertake a REMS; |
| ● | the effectiveness of our sales, marketing and distribution efforts; |
| ● | COVID-19 may be substantially eradicated prior to our development of a successful therapy in the COVA clinical program by one or more of the vaccines that have been or may in the future be authorized for use, or the therapy produced by the COVA clinical program may not be effective against other or future coronavirus variants, reducing or eliminating the need for this therapy to treat the disease; |
| ● | the SARS-CoV-2 virus could develop resistance to our treatment developed in the COVA clinical program (which is concluding early due to the lack of enrollment), which could affect any long-term demand or sales potential for our potential therapies; |
| ● | adverse publicity about our products, status of ongoing trials, or favorable publicity about competitive products; and |
| ● | potential product liability claims. |
We cannot assure you that our current or future drug candidates, if approved or authorized, will achieve broad market acceptance among physicians and patients. Any failure by our drug candidates that obtain regulatory approval or authorization to achieve market acceptance or commercial success would adversely affect our results of operations.
We rely on third parties to provide the raw materials necessary for our drug candidates and to manufacture preclinical and clinical supplies of our drug candidates and we intend to rely on third parties to produce commercial supplies of any approved or authorized drug candidate. The loss of these suppliers or manufacturers, or their failure to comply with applicable regulatory requirements or to provide us with sufficient quantities at acceptable quality levels or prices, or at all, would materially and adversely affect our business.
We do not have nor do we plan to build or acquire the infrastructure or capability internally to source the raw materials necessary to produce our drug candidates and/or to manufacture our drug candidates on a preclinical, clinical or commercial scale.
18
BIO101 (20-hydroxyecdysone) is a pharmaceutical-grade purification of 20-hydroxyecdysone, which is derived from the Cyamnotis sp or Stemmacantha sp, a plant cultivated in China and used for medicinal purposes in traditional Chinese medicine. There are a limited number of growers of this plant and suppliers of the plant material and we must account for the lead time required to grow sufficient quantities of the plant to meet our needs. At this time, we rely on one supplier for the plant quantities we require for our clinical trials. We have not entered into a long-term supply agreement with this supplier. We have already obtained good manufacturing practices (“GMP”) batches/GMP-compliant batches/batches produced in compliance with GMP of BIO101 (20-hydroxyecdysone) for our clinical trials and we believe we can secure sufficient quantities for our future clinical programs through our current supply chain up to regulatory approval and/or marketing authorization. If our current supplier is unable to provide sufficient quantities of the plant to produce BIO101 (20-hydroxyecdysone) for future clinical trials, our ability to obtain regulatory approval or authorization for BIO101 (20-hydroxyecdysone) would be affected. If we receive regulatory approval or authorization, we will likely need substantial quantities of plants to produce BIO101 (20-hydroxyecdysone) for commercial development. If our current supplier is unable to provide sufficient quantities of the plant to produce BIO101 (20-hydroxyecdysone) and if we are unable to find an alternative source, our ability to commercialize BIO101 (20-hydroxyecdysone) would be impaired. In order to address this issue, we are evaluating alternative methods for producing 20-hydroxyecdysone in order to optimize the supply chain to support our projected commercial needs.
BIO201 is a pharmaceutical-grade purification of norbixin, which is derived from seeds of Bixa orellana L., a plant traditionally used for medicinal purposes in the Amazon and currently used for producing a food color in many countries. Although this plant is more widely available, there are a limited number of suppliers of the plant material that could meet our requirement for quality. At this time, we rely on one supplier for the plant quantities we will require for our MACA clinical program. We have not entered into a long-term supply agreement with this supplier. If our current supplier is unable to provide sufficient supply to produce BIO201 for future clinical trials, our ability to obtain regulatory approval or authorization for BIO201 would be affected. If we receive regulatory approval or authorization, we will likely need substantial quantities of plants to produce BIO201 for commercial development. If our current supplier is unable to provide sufficient quantities of the plant to produce BIO201 and if we are unable to find an alternative source, our ability to commercialize BIO201 would be impaired. In order to address this issue, we are evaluating alternative methods for producing norbixin in order to optimize the supply chain to support our projected commercial needs.
Our contract manufacturing partners are Seqens, an integrated global player in solutions and ingredients for the pharmaceutical and specialty markets headquartered in France, and Eurofins Amatsi Group, an international CMO and Skyepharma, a French pharmaceutical company specializing in the formulation, development and production of pharmaceutical products . We have not entered into a long-term manufacturing agreement with these manufacturers.
The facilities used by our contract manufacturers to manufacture our drug candidates are subject to various regulatory requirements and may be subject to the inspection of the FDA, EMA or other regulatory authorities. We do not control the manufacturing process of, and are completely dependent on, our contract manufacturing partners for compliance with the regulatory requirements, known as cGMPs. If our contract manufacturers cannot successfully manufacture material that conforms to our specifications and the strict regulatory requirements of the FDA, EMA or comparable regulatory authorities in foreign jurisdictions, we may not be able to rely on their manufacturing facilities for the manufacture of our drug candidates. In addition, we have limited control over the ability of our contract manufacturers to maintain adequate quality control, quality assurance and qualified personnel. If the FDA, EMA or a comparable foreign regulatory authority finds these facilities inadequate for the manufacture of our drug candidates or if such facilities are subject to enforcement action in the future or are otherwise inadequate, we may need to find alternative manufacturing facilities, which would significantly impact our ability to develop, obtain regulatory approval or authorization for or market our drug candidates. Any significant delay in, or quality control problems with respect to, the supply of a drug candidate, or the raw material components thereof, for an ongoing study or trial could considerably delay completion of our preclinical studies or future clinical trials, product testing and potential regulatory approval or authorization of our drug candidates.
19
If any of our drug candidates is approved or authorized by the FDA, EMA and/or comparable foreign regulatory authorities and we choose to independently commercialize such drug candidate, we will need to engage manufacturers for the commercial supply of such drug candidates. However, we may be unable to enter into any such agreement or do so on commercially reasonable terms, which could have a material adverse impact upon our business. Moreover, if there is a disruption to one or more of our third-party manufacturers’ or suppliers’ relevant operations, or if we are unable to enter into arrangements for the commercial supply of our drug candidates, we will have no other means of producing our drug candidates until they restore the affected facilities or we or they procure alternative manufacturing facilities or sources of supply. Our ability to progress our preclinical and clinical programs could be materially and adversely impacted if any of the third party suppliers upon which we rely were to experience a significant business challenge, disruption or failure due to issues such as financial difficulties or bankruptcy, issues relating to other customers such as regulatory or quality compliance issues, or other financial, legal, regulatory or reputational issues. Additionally, any damage to or destruction of our third-party manufacturers’ or suppliers’ facilities or equipment may significantly impair our ability to manufacture our drug candidates on a timely basis.
In addition, to manufacture our drug candidates in the quantities that we believe would be required to meet anticipated market demand, our third-party manufacturers would likely need to increase manufacturing capacity and, in some cases, we could be required to secure alternative sources of commercial supply, which could involve significant challenges and could require additional regulatory approvals. If new restrictions are imposed as a result of a COVID-19 resurgence, any new pandemic or epidemic, or emergence of other infectious diseases, we may not be able to develop or scale up manufacturing capacity on a timely basis or have access to logistics or supply channels. In addition, the development of commercial-scale manufacturing capabilities could require us and our third-party manufacturers to invest substantial additional funds and hire and retain the technical personnel who have the necessary manufacturing experience. Neither we nor our third-party manufacturers may successfully complete any required increase to existing manufacturing capacity in a timely manner, or at all. If our manufacturers or we are unable to purchase the raw materials necessary for the manufacture of our drug candidates on acceptable terms, at sufficient quality levels, or in adequate quantities, if at all, the commercial launch of our drug candidates or any future drug candidates would be delayed or there would be a shortage in supply, which would impair our ability to generate revenues from the sale of such drug candidates, if approved.
We rely on third parties in the conduct of all of our preclinical studies and clinical trials and intend to rely on third parties in the conduct of all of our future clinical trials. If these third parties do not successfully carry out their contractual duties, fail to comply with applicable regulatory requirements or meet expected deadlines, we may be unable to obtain regulatory approval for our drug candidates.
We currently do not have the ability to independently conduct preclinical studies that comply with the regulatory requirements known as good laboratory practice (“GLP”) requirements. We also do not currently have the ability to independently conduct any clinical trials. The FDA, EMA and regulatory authorities in other jurisdictions require us to comply with regulations and standards, commonly referred to as good clinical practice (“GCP”) requirements for conducting, monitoring, recording and reporting the results of clinical trials, in order to ensure that the data and results are scientifically credible and accurate and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. We rely on medical institutions, clinical investigators, contract laboratories and other third parties, such as CROs to conduct GLP-compliant preclinical studies and GCP-compliant clinical trials on our drug candidates properly and on time. While we have agreements governing their activities, we control only certain aspects of their activities and have limited influence over their actual performance. The third parties with whom we contract for execution of our GLP-compliant preclinical studies and our GCP-compliant clinical studies play a significant role in the conduct of these studies and trials and the subsequent collection and analysis of data. These third parties are not our employees and, except for restrictions imposed by our contracts with such third parties, we have limited ability to control the amount or timing of resources that they devote to our programs. In addition, third parties may have or adopt their own policies in response to pandemics (such as COVID-19), epidemics, or other infectious diseases that may create delays or service disruptions, including work-from-home policies that lead to reduced workforce productivity. Although we rely on these third parties to conduct our GLP-compliant preclinical studies and GCP-compliant clinical trials, we remain responsible for ensuring that each of our GLP preclinical studies and clinical trials is conducted in accordance with its investigational plan and protocol and applicable laws and regulations, and our reliance on the CROs does not relieve us of our regulatory responsibilities.
20
Many of the third parties with whom we contract may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials or other drug development activities that could harm our competitive position. If the third parties conducting our preclinical studies or our clinical trials do not adequately perform their contractual duties or obligations, experience significant business challenges, disruptions or failures, do not meet expected deadlines, terminate their agreements with us or need to be replaced, or if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to our protocols or to GCPs, or for any other reason, we may need to enter into new arrangements with alternative third parties. This could be difficult, costly or impossible, and our preclinical studies or clinical trials may need to be extended, delayed, terminated or repeated. As a result, we may not be able to obtain regulatory approval or authorization in a timely fashion, or at all, for the applicable drug candidate, our financial results and the commercial prospects for our drug candidates would be harmed, our costs could increase, and our ability to generate revenues could be delayed.
We face significant competition in an environment of rapid technological and scientific change, and our drug candidates, if approved or authorized, will face significant competition and our failure to effectively compete may prevent us from achieving significant market penetration. A number of our competitors have significantly greater resources than we do and we may not be able to successfully compete.
The biotechnology and pharmaceutical industries in particular are characterized by rapidly advancing technologies, intense competition and a strong emphasis on developing proprietary therapeutics. Numerous companies are engaged in the development, patenting, manufacturing and marketing of healthcare products competitive with those that we are developing. We face competition from a number of sources, such as pharmaceutical companies, generic drug companies, biotechnology companies and academic and research institutions, many of which have greater financial resources, marketing capabilities, sales forces, manufacturing capabilities, research and development capabilities, clinical trial expertise, intellectual property portfolios, experience in obtaining patents and regulatory approvals or authorizations for drug candidates and other resources than we do. Some of the companies that offer competing products also have a broad range of other product offerings, large direct sales forces and long-term customer relationships with our target physicians, which could inhibit our market penetration efforts. Mergers and acquisitions in the biotechnology and pharmaceutical industry may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. In addition, certain of our drug candidates, if approved, may compete with other products that treat age-related diseases, including over-the-counter (“OTC”) treatments, for a share of some patients’ discretionary budgets and for physicians’ attention within their clinical practices.
We are aware of other companies seeking to develop treatments to prevent or treat aging-related diseases through various biological pathways. Indeed, the main challenge is to be able to identify the optimal target population given the dynamics in diagnostic criteria. The recent failures, combined with these dynamics, can deter major pharmaceutical companies from re-entering this space.
For COVID-19, many clinical studies have been run to develop medical responses to the virus, but the majority of them have failed to demonstrate any benefit for the patients. In the United States, several products have received emergency use authorizations for specific indications and patient groups such as anti-viral agents, including Paxlovid (nirmatrelvir and ritonavir and molnupiravir), monoclonal antibodies, including Evusheld (tixagevimab co-packaged with cilgavimab and administered together), and anti-inflammatory / immune-suppressing drugs, including anakinra (Kineret). Veklury (remdesivir), baricitinib (Olumiant) and tocilizumab (Actemra) have been approved by the FDA. Moreover, a number of vaccines have been authorized around the globe. In the EU, Veklury (remdesivir), RoActemra (tocilizumab), Kineret (anakinra), Paxlovid (PF-07321332 and ritonavir), Lagevrio (molnupiravir), Regkirona (regdanvimab), Ronapreve (casirivimab/imdevimab), Xevudy (sotrovimab), Regkirona (regdanvimab), Ronapreve (casirivimab/imdevimab and Evusheld (tixagevimab, cilgavimab) have been approved. Age, co-morbidities, heavy smoking, male gender and several ethnic backgrounds are associated with worse outcomes for COVID-19. Our therapeutic approach is aimed at targeting and activating key biological resilience pathways that can protect against and counteract the effects of the multiple biological and environmental stresses, including inflammatory, oxidative, metabolic and viral stresses that lead to age-related diseases.
For DMD, our current focus on non-ambulatory patients with evidence of respiratory deterioration, puts us in a position to become one of the more advanced companies that develop medications for this population. Santhera Therapeutics, has developped Agamree® (vamorolone) targeting all DMD patients from the age of 4. Agamree received marketing authorizations in the US, the UK and Europe in late 2023 and has been launched in Germany in January 2024.
21
For dry AMD, we believe that we will compete with a number of companies that are developing drugs to treat this disease using different technologies (e.g., cellular and gene therapy, integrin regulation, and others), for example, Allegro Ophthalmics, Apellis Pharmaceuticals, Kodiak Sciences, Astellas, Hemera Biosciences, Iveric Bioscience and Roche and Stealth Biotherapeutics.
Certain alternative treatments offered by competitors may be available at lower prices and may offer greater efficacy or better safety profiles. Furthermore, currently approved products could be discovered to have application for treatment of age-related diseases generally, which could give such products significant regulatory and market timing advantages over any of our drug candidates. Our competitors also may obtain FDA, EMA or other regulatory approval for their products more rapidly than we may obtain approval for ours and may obtain orphan product exclusivity from the FDA or EMA for indications our drug candidates are targeting, which could result in our competitors establishing a strong market position before we are able to enter the market. Newly developed systemic or non-systemic treatments that replace existing therapies that are currently only utilized in patients suffering from severe disease may also have lessened side effects or reduced prices compared to current therapies, which make them more attractive for patients suffering from mild to moderate disease. Even if a generic product or an OTC product is less effective than our drug candidates, a less effective generic or OTC product may be more quickly adopted by physicians and patients than our competing drug candidates based upon cost or convenience. For additional information regarding our competition, see the section of this annual report titled “Business—Competition.”
In addition, another party may be successful in producing a more efficacious therapy for COVID-19 or a therapy with a more convenient or preferred route of administration or in producing a therapy in a more timely manner, which may lead to the diversion of funding away from us and toward other companies or lead to decreased demand for our potential therapies. Further, other therapies that are more affordable than our potential therapies may be used to treat COVID-19, including existing generic drugs, which could also hurt the funding of and demand for our potential therapies.
Government restrictions on pricing and reimbursement, as well as other healthcare payor cost-containment initiatives, may negatively impact our ability to generate revenues and become profitable even if we obtain regulatory approval or authorization to market a product.
Our ability to commercialize any products successfully also will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers and other organizations. Government authorities and other third-party payors, such as private health insurers and health maintenance organizations, determine which medications they will cover and establish reimbursement levels. Assuming we obtain coverage for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. Patients who are prescribed medications for the treatment of their conditions, and their prescribing physicians, generally rely on third-party payors to reimburse all or part of the costs associated with their prescription drugs. Patients are unlikely to use our products unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the cost of our products. Therefore, coverage and adequate reimbursement is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are already available or subsequently become available.
Government authorities and other third-party payors are developing increasingly sophisticated methods of controlling healthcare costs, such as by limiting coverage and the amount of reimbursement for particular medications. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices as a condition of coverage, are using restrictive formularies and preferred drug lists to leverage greater discounts in competitive classes, and are challenging the prices charged for medical products.
In the United States, federal programs impose penalties on drug manufacturers in the form of mandatory additional rebates and/or discounts if commercial prices increase at a rate greater than the Consumer Price Index-Urban, and these rebates and/or discounts, which can be substantial, may impact our ability to raise commercial prices. Further, no uniform policy requirement for coverage and reimbursement for drug products exists among third-party payors in the United States. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor. As a result, the coverage determination process is often a time-consuming and costly process that will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage and adequate reimbursement will be applied consistently or obtained in the first instance.
22
In the European Union (“EU”) coverage and reimbursement possibilities for drug products differ from one Member State to another. Each Member State has the ability to set the prices and restrict the range of medicinal products for which their national health insurance systems provide reimbursement. Factors contributing to price changes between Member States depend on different regulatory approaches and instruments used by each Member State to govern the supply and demand of medicinal products. For example, in France, a pharmaceutical company may freely set a price of a drug after obtaining a marketing authorization. However, in order for the product to be reimbursed under the French Social Security scheme, the pharmaceutical company must follow a specific process and submit an application to the French High Authority for Health, or HAS. The opinion issued by the HAS and its subcommittees (Transparency Commission or CT, and the Commission for Economic and Public Health Evaluation or CEESP, if applicable) is then transmitted to the French Economic Committee for Health Products, or CEPS—with which the pharmaceutical company has to negotiate the price of the product and the French National Union of Health Insurance Funds, or UNCAM which fixes the reimbursement rate of medicines covered by statutory health insurance. The final decision on price and reimbursement is issued by the French Minister of Health and can be revised afterwards, for example, depending on the cost/benefit balance of the medicinal product over time. Other EU countries may adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market as well as other price control mechanisms. In view of these differences from one Member State to another, there is still a risk that some EU countries do not allow favorable reimbursements and pricing arrangements.
The continuing efforts of governments, insurance companies, managed care organizations and other payors of healthcare costs to contain or reduce costs of healthcare may negatively affect our commercialization prospects, including:
| ● | our ability to set a price we believe is fair for our products, if approved or authorized; |
| ● | our ability to obtain and maintain market acceptance by the medical community and patients; |
| ● | our ability to generate revenues and achieve profitability; and |
| ● | the availability of capital. |
We cannot be sure that coverage and reimbursement will be available for any potential drug candidate that we may commercialize and, if reimbursement is available, what the level of reimbursement will be. Coverage and reimbursement may impact the demand for, or the price of, any drug candidate for which we obtain marketing approval or authorization. If coverage and reimbursement are not available or reimbursement is available only to limited levels, we may not successfully commercialize any drug candidate for which we obtain marketing approval or authorization.
We expect that additional U.S. state and federal healthcare reform measures, as well as similar measures by non-U.S. governments, will be adopted in the future, any of which could limit the amounts that governments will pay for healthcare products and services, which could result in additional pricing pressure or reduced demand for any drug candidate we develop.
23
In the event we elect to commercialize any of our drug candidates that receive regulatory approval or authorization, we will need to establish sales capabilities on our own or through third parties. If we are unsuccessful in our efforts, we may not be able to market and sell our drug candidates effectively in the United States, EU and/or other foreign jurisdictions, if approved or authorized, or generate product revenue.
We currently do not have a marketing or sales organization. In order to commercialize our drug candidates in the United States and foreign jurisdictions, we would need to establish marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. If any of our drug candidates receive regulatory approval or authorization and we elect to independently commercialize such drug candidates, we would expect to establish a sales organization with technical expertise and supporting distribution capabilities to commercialize each such drug candidate, which would be expensive and time consuming. We have no prior experience in the marketing, sale and distribution of pharmaceutical products and there are significant risks involved in building and managing a sales organization, including our ability to hire, retain, and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel, and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing and distribution capabilities would adversely impact the commercialization of these products. Alternatively, we may choose to collaborate with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize our drug candidates. If we are not successful in commercializing our drug candidates or any future drug candidates, either on our own or through arrangements with one or more third parties, and are not otherwise able to license these products to third parties, we may not be able to generate any future product revenue and we would incur significant additional losses.
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
As of the date of this annual report, we have 24 full-time employees, 18 of whom are engaged in research and development activities and five of whom are engaged in general and administrative activities. We will need to continue to expand our managerial, operational, finance and other resources in order to manage our operations and clinical trials, continue our development activities and commercialize our drug candidates or any future drug candidates. Our management and personnel, systems and facilities currently in place may not be adequate to support this future growth. Our need to effectively execute our growth strategy requires that we:
| ● | manage our clinical trials effectively; |
| ● | identify, recruit, retain, incentivize and integrate additional employees; |
| ● | manage our internal development and operational efforts effectively while carrying out our contractual obligations to and/or relations with third parties including regulatory agencies and market authorities; |
| ● | continue to improve our operational, financial and management controls, reports systems and procedures; and |
| ● | manage our information technology systems and data security. |
If we fail to attract and retain senior management and key scientific personnel, we may be unable to successfully develop our drug candidates or any future drug candidates, conduct our clinical trials and commercialize our current or any future drug candidates.
We are dependent upon the services of our senior management and the loss of any of these individuals could harm our business. The loss of services of any of our key executive officers or other members of our senior management team, may be disruptive to, or cause uncertainty in, our business and could have a negative impact on our ability to manage and grow our business effectively. Such disruption could have a material adverse impact on our financial performance, financial condition, and the market price of our ordinary shares.
Our success also depends on our ability to continue to attract, retain and motivate highly qualified clinical and scientific personnel. Competition for qualified personnel in the biotechnology and pharmaceuticals field is intense due to the limited number of individuals who possess the skills and experience required by our industry. We will need to hire additional personnel as we expand our clinical development and if we initiate commercial activities. We may not be able to attract and retain quality personnel on acceptable terms, or at all. In addition, to the extent we hire personnel from competitors, we may be subject to allegations that they have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers own their research output.
24
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our current or future drug candidates.
We face an inherent risk of product liability as a result of the clinical testing of our drug candidates and will face an even greater risk if we commercialize any products. For example, we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability, and a breach of warranty. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our drug candidates. Even a successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our current or future drug candidates; |
| ● | injury to our reputation; |
| ● | withdrawal of clinical trial participants; |
| ● | costs to defend the related litigation; |
| ● | a diversion of management’s time and our resources; |
| ● | substantial monetary awards to trial participants or patients; |
| ● | regulatory investigations, product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| ● | loss of revenue; and |
| ● | the inability to commercialize our current or any future drug candidates. |
Our inability to obtain and maintain sufficient product liability insurance at an acceptable cost and scope of coverage to protect against potential product liability claims could prevent or inhibit the commercialization of our current or any future drug candidates we develop. We currently carry product liability insurance covering our clinical trials. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions and deductibles, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient funds to pay such amounts. Moreover, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses. If and when we obtain approval or authorization for marketing any of our drug candidates, we intend to expand our insurance coverage to include the sale of such drug candidate; however, we may be unable to obtain this liability insurance on commercially reasonable terms or at all.
Our existing collaborations as well as additional collaboration arrangements that we may enter into in the future may not be successful, which could adversely affect our ability to develop and commercialize our drug candidates.
We utilize external collaborations and currently maintain several active early-stage research and discovery focused collaborations. We may seek to partner with pharmaceutical laboratories to conduct clinical trials of our drug candidates. We may also seek additional collaboration arrangements for the commercialization, or potentially for the development, of certain of our drug candidates depending on the merits of retaining commercialization rights for ourselves as compared to entering into collaboration arrangements. To the extent that we decide to enter into additional collaboration agreements in the future, we may face significant competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time-consuming to negotiate, document, implement and maintain and challenging to manage. We may not be successful in our efforts to prudently manage our existing collaborations or to enter new ones should we choose to do so. The terms of new collaborations or other arrangements that we may establish may not be favorable to us.
25
The success of our collaboration arrangements will depend heavily on the efforts and activities of our collaborators. Collaborations are subject to numerous risks, which may include risks that:
| ● | collaborators have significant discretion in determining the efforts and resources that they will apply to collaborations; |
| ● | collaborators may not perform their obligations as expected; |
| ● | collaborators may not pursue development and commercialization of our drug candidates or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in their strategic focus due to their acquisition of competitive products or their internal development of competitive products, availability of funding or other external factors, such as a business combination that diverts resources or creates competing priorities; |
| ● | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial, abandon a drug candidate, repeat or conduct new clinical trials or require a new formulation of a drug candidate for clinical testing; |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or drug candidates; |
| ● | a collaborator with marketing, manufacturing and distribution rights to one or more products may not commit sufficient resources to or otherwise not perform satisfactorily in carrying out these activities; |
| ● | we could grant exclusive rights to our collaborators that would prevent us from collaborating with others; |
| ● | collaborators may not properly maintain or defend our intellectual property rights or may use our intellectual property or proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential liability; |
| ● | disputes may arise between us and a collaborator that cause the delay or termination of the research, development or commercialization of our current or future drug candidates or that result in costly litigation or arbitration that diverts management attention and resources; |
| ● | collaborations may be terminated, and, if terminated, this may result in a need for additional capital to pursue further development or commercialization of the applicable current or future drug candidates; |
| ● | collaborators may own or co-own intellectual property covering products that results from our collaborating with them, and in such cases, we would not have the exclusive right to develop or commercialize such intellectual property; |
| ● | disputes may arise with respect to the ownership of any intellectual property developed pursuant to our collaborations; and |
| ● | a collaborator’s sales and marketing activities or other operations may not be in compliance with applicable laws resulting in civil or criminal proceedings. |
26
Significant disruptions of information technology systems, breaches of data security or personnal data breaches could materially adversely affect our business, results of operations and financial condition.
We collect and maintain information in digital form that is necessary to conduct our business, and we are increasingly dependent on information technology systems and infrastructure to operate our business. In the ordinary course of our business, we collect, store and transmit large amounts of confidential information, including intellectual property, proprietary business information and personal information. It is critical that we do so in a secure manner to maintain the confidentiality and integrity of such confidential information. We have established physical, electronic and organizational measures to safeguard and secure our systems to prevent a data compromise, and rely on commercially available systems, software, tools, and monitoring to provide security for our information technology systems and the processing, transmission and storage of digital information. We have also outsourced elements of our information technology infrastructure, and as a result a number of third-party vendors may or could have access to our confidential information. Our internal information technology systems and infrastructure, and those of our current and any future collaborators, contractors and consultants and other third parties on which we rely, are vulnerable to damage from computer viruses, malware, natural disasters, terrorism, war, telecommunication and electrical failures, mishandling, cyber-attacks or cyber-intrusions over the Internet, attachments to emails, persons inside our organization, or persons with access to systems inside and outside our organization.
The risk of a security breach or disruption, particularly through cyber-attacks or cyber-intrusion, including by computer hackers, foreign governments and cyber-terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. In addition, the prevalent use of mobile devices that access confidential information increases the risk of data security breaches, which could lead to the loss of confidential information or other intellectual property. The costs to us to mitigate network security problems, bugs, viruses, worms, malicious software programs and security vulnerabilities could be significant, and while we have implemented security measures to protect our data security and information technology systems, our efforts to address these problems may not be successful, and these problems could result in unexpected interruptions, delays, cessation of service and other harm to our business and our competitive position. If such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our product development programs. For example, the loss or destruction of, or loss of access to, clinical trial data from completed or ongoing or planned clinical trials could result in delays in our regulatory approval or authorization efforts and significantly increase our costs to recover or reproduce the data. Moreover, if a computer security breach affects our systems or results in the unauthorized release of personally identifiable information, our reputation could be materially damaged and we could be subject to regulatory enforcement action undertaken by competent data protection authorities (including financial penalties) and/or claims from private individuals.
In addition, such a breach may require notification to governmental agencies, the media or individuals pursuant to various national, supranational, federal and state privacy and security laws, if applicable, including the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Clinical Health Act of 2009, and its implementing rules and regulations, as well as regulations promulgated by the Federal Trade Commission and state breach notification laws.
Under the data protection laws in the EU, and notably the General Data Protection Regulation (“GDPR”) No. 2016/679, which entered into force on May 25, 2018 and is applicable personal data that we process in relation to our presence in the EU, the offering of products or services to individuals in the EU or the monitoring of the behavior of individuals in the EU, we have also a legal responsibility to report personal data breaches to the competent supervisory authority. The GDPR includes a broad definition and a short deadline for the notification of personal data breaches, which may be difficult to adhere to in practice and requires that we implement robust internal processes. Under the GDPR, we have to report personal data breaches to the competent supervisory authority within 72 hours of the time we become aware of a breach “unless the personal data breach is unlikely to result in a risk to the right and freedoms of natural persons” (Article 33 of the GDPR). In addition, the GDPR requires that we communicate the breach to the data subject if the breach is “likely to result in a high risk to the rights and freedoms of natural persons” (Article 34 of the GDPR). In order to fulfil these requirements, we have to implement specific internal processes to be followed in case of a personal data breach, which will allow us to (a) contain and recover the breach, (b) assess the risk to the data subjects, (c) notify the competent supervisory authority (if required), and possibly communicate the breach to the data subjects, (d) investigate and respond to the breach. The performance of these processes involve substantial costs in resources and time.
27
Finally, as a consequence of the decision by the European Court of Justice issued on July 16, 2020 (known as the “Schrems II decision”), which invalidated the privacy shield for data transfers between the EU and the United States, a reassessment of both data transfers to and storage of EU personal data by our U.S. entities or other U.S. companies was required. As a result of assessment, we implemented additional protective measures in order to ensure the compliance of such transfers, all of which require ongoing monitoring. These additional measures may be required by local data protection authorities, especially since health data is considered as sensitive data pursuant to the GDPR and in any case needs additional protection. They may include the implementation of specific internal processes in order to assess the risks of access by U.S. surveillance authorities or agencies to EU health data pursuant to U.S. laws and impede such access requests where possible. These additional measures, including the ongoing montoring of such measures, incurs substantial cost is resources and time.
Moreover, as we may rely on third parties to process as processor the personal data for which we are a controller—for example, in the context of the manufacturing of our drug candidates or for the conduct of clinical trials, we must contractually ensure that such processors implement strict security measures, maintain applicable certifications and are contractually obliged to comply with appropriate obligations including an obligation to report without undue delay any security incident, in order to allow us to fulfill our own regulatory requirements. In addition, as regards the hosting of health data relating to French patients, it may be necessary to use EU certified health data hosting providers, depending on the purposes for which personal health data is stored, and comply with the specific standards implemented by the French data protection authority where applicable, which may also incur cost in resource and time.
We could also be exposed to a risk of loss or litigation and potential liability for any security breach involving personal data for which we are a controller. The costs of above-mentioned processes together with legal penalties, possible compensation for damages and any resulting lawsuits arising from a breach may be extensive and may have a negative impact on reputation and materially adversely affect our business, results of operations and financial condition.
Our employees and independent contractors, including principal investigators, consultants, commercial collaborators, service providers and other vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could have an adverse effect on our results of operations.
We are exposed to the risk that our employees and independent contractors, including principal investigators, consultants, any future commercial collaborators, service providers and other vendors may engage in misconduct or other illegal activity. Misconduct by these parties could include intentional, reckless and/or negligent conduct or other unauthorized activities that violate the laws and regulations of the FDA, EMA and other similar regulatory authorities, including those laws that require the reporting of true, complete and accurate information to such regulatory authorities; manufacturing standards; healthcare fraud and abuse, data privacy laws and other similar laws; or laws that require the true, complete and accurate reporting of financial information or data. Activities subject to these laws also involve the improper use or misrepresentation of information obtained in the course of clinical trials, the creation of fraudulent data in our preclinical studies or clinical trials, or illegal misappropriation of product, which could result in regulatory sanctions and cause serious harm to our reputation. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. In addition, we are subject to the risk that a person or government could allege such fraud or other misconduct, even if none occurred. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and financial results, including, without limitation, the imposition of significant civil, criminal and administrative penalties, damages, monetary fines, disgorgements, possible exclusion from participation in governmental healthcare programs, individual imprisonment, other sanctions, contractual damages, reputational harm, diminished profits and future earnings and curtailment of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
28
Our business involves the use of hazardous materials and we and our third-party manufacturers and suppliers must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our research and development activities and our third-party manufacturers’ and suppliers’ activities involve the controlled storage, use and disposal of hazardous materials owned by us, including the components of our product and drug candidates and other hazardous compounds. We and any third-party manufacturers and suppliers we engage are subject to numerous federal, state and local environmental, health and safety laws, regulations and permitting requirements, including those governing laboratory procedures; the generation, handling, use, storage, treatment, and disposal of hazardous and regulated materials and wastes; the emission and discharge of hazardous materials into the ground, air and water; and employee health and safety. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also produce hazardous waste. In some cases, these hazardous materials and various wastes resulting from their use are stored at our and our manufacturers’ facilities pending their use and disposal. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination, which could cause an interruption of our commercialization efforts, research and development efforts and business operations, environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products.
Although we believe that the safety procedures utilized by our third-party manufacturers for handling and disposing of these materials generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this is the case or eliminate the risk of accidental contamination or injury from these materials. Under certain environmental laws, we could be held responsible for costs relating to any contamination at our current or past facilities and at third-party facilities. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and state or federal or other applicable authorities may curtail our use of certain materials and/or interrupt our business operations. Furthermore, environmental laws and regulations are complex, change frequently and have tended to become more stringent. We cannot predict the impact of such changes and cannot be certain of our future compliance.
Compliance with applicable environmental laws and regulations may be expensive, and current or future environmental laws and regulations may impair our research, product development and manufacturing efforts. In addition, we cannot entirely eliminate the risk of accidental injury or contamination from these materials or wastes. Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials, this insurance may not provide adequate coverage against potential liabilities. We do not carry specific biological or hazardous waste insurance coverage, and our property, casualty, and general liability insurance policies specifically exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination. Accordingly, in the event of contamination or injury, we could be held liable for damages or be penalized with fines in an amount exceeding our resources, and our clinical trials or regulatory approvals or authorization could be suspended, which could have a material adverse effect on our business, results of operations and financial condition.
Risks Related to Intellectual Property
Our ability to compete may decline if we do not adequately protect our proprietary rights.
Our success depends on obtaining and maintaining proprietary rights to our drug candidates for the treatment of age-related diseases, as well as successfully defending these rights against third-party challenges. We will only be able to protect our drug candidates, and their uses from unauthorized use by third parties to the extent that valid and enforceable patents, or effectively protected trade secrets, cover them. Our ability to obtain patent protection for our drug candidates is uncertain due to a number of factors, including:
| ● | we may not have been the first to make the inventions covered by pending patent applications or issued patents; |
| ● | we may not have been the first to file patent applications for our drug candidates or the compositions we developed or for their uses; |
| ● | others may independently develop identical, similar or alternative products or compositions and uses thereof; |
| ● | our disclosures in patent applications may not be sufficient to meet the statutory requirements for patentability; |
| ● | any or all of our pending patent applications may not result in issued patents; |
29
| ● | we may not seek or obtain patent protection in countries that may eventually provide us a significant business opportunity; |
| ● | any patents issued to us may not provide a basis for commercially viable products, may not provide any competitive advantages, or may be successfully challenged by third parties; |
| ● | our compositions and methods may not be patentable; |
| ● | others may design around our patent claims to produce competitive products which fall outside of the scope of our patents; or |
| ● | others may identify prior art or other bases which could invalidate our patents. |
Even if we have or obtain patents covering our drug candidates or compositions, we may still be barred from making, using and selling our drug candidates or technologies because of the patent rights of others. Others may have filed, and in the future may file, patent applications covering compositions or products that are similar or identical to ours. There are many issued U.S. and foreign patents relating to chemical compounds and therapeutic products, and some of these relate to compounds we intend to commercialize. Numerous U.S. and foreign issued patents and pending patent applications owned by others exist in the allergy treatment field in which we are developing products. These could materially affect our ability to develop our drug candidates or sell our products if approved or authorized. Because patent applications can take many years to issue, there may be currently pending applications unknown to us that may later result in issued patents that our drug candidates or compositions may infringe. These patent applications may have priority over patent applications filed by us.
Obtaining and maintaining a patent portfolio entails significant expense and resources. Part of the expense includes periodic maintenance fees, renewal fees, annuity fees, various other governmental fees on patents and/or applications due in several stages over the lifetime of patents and/or applications, as well as the cost associated with complying with numerous procedural provisions during the patent application process. We may or may not choose to pursue or maintain protection for particular inventions. In addition, there are situations in which failure to make certain payments or noncompliance with certain requirements in the patent process can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If we choose to forgo patent protection or allow a patent application or patent to lapse purposefully or inadvertently, our competitive position could suffer.
In addition, it is unclear at this time what Brexit’s impact will have on our intellectual property rights and the process for obtaining and defending such rights. It is possible that certain intellectual property rights, such as trademarks, granted by the EU will cease being enforceable in the UK absent special arrangements to the contrary. With regard to existing patent rights, the effect of Brexit should be minimal considering enforceable patent rights are specific to the UK, whether arising out of the European Patent Office or directly through the UK patent office.
Legal actions to enforce our proprietary rights (including patents and trademarks) can be expensive and may involve the diversion of significant management time. In addition, these legal actions could be unsuccessful and could also result in the invalidation of our patents or trademarks or a finding that they are unenforceable. We may or may not choose to pursue litigation or other actions against those that have infringed on our patents or trademarks, or used them without authorization, due to the associated expense and time commitment of monitoring these activities. If we fail to protect or to enforce our intellectual property rights successfully, our competitive position could suffer, which could harm our results of operations.
Biotechnology patents and patent applications involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our patent position.
The patent positions of biotechnology companies can be highly uncertain and involve complex legal and factual questions. The interpretation and breadth of claims allowed in some patents covering biotechnology compositions may be uncertain and difficult to determine and are often affected materially by the facts and circumstances that pertain to the patented compositions and the related patent claims. The standards of the United States Patent and Trademark Office (“USPTO”) are sometimes uncertain and could change in the future. Consequently, the issuance and scope of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to reexamination proceedings, post-grant review and/or inter partes review in the USPTO. Foreign patents may be subject also to opposition or comparable proceedings in the corresponding foreign patent office, which could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, reexamination, post-grant review, inter partes review and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
30
In addition, changes in or different interpretations of patent laws in the United States and foreign countries may permit others to use our discoveries or to develop and commercialize our technology and products without providing any compensation to us or may limit the number of patents or claims we can obtain. The laws of some countries do not protect intellectual property rights to the same extent as U.S. laws and those countries may lack adequate rules and procedures for defending our intellectual property rights. This may also result in having the same invention covering differing claims in different countries and provide a different scope of protection in foreign countries.
If we fail to obtain and maintain patent protection and trade secret protection of our drug candidates, we could lose our competitive advantage and competition we face would increase, reducing any potential revenues and adversely affecting our ability to attain or maintain profitability.
Developments in patent law could have a negative impact on our business.
From time to time, the United States Supreme Court, or the Supreme Court, other federal courts, the United States Congress, the USPTO or similar foreign authorities may change the standards of patentability and any such changes could have a negative impact on our business.
In addition, the Leahy-Smith America Invents Act (the “America Invents Act”), which was signed into law in 2011, includes a number of significant changes to U.S. patent law. These changes include a transition from a “first-to-invent” system to a “first-to-file” system, changes to the way issued patents are challenged, and changes to the way patent applications are disputed during the examination process. These changes may favor larger and more established companies that have greater resources to devote to patent application filing and prosecution. The USPTO has developed new and untested regulations and procedures to govern the full implementation of the America Invents Act, and many of the substantive changes to patent law associated with the America Invents Act, and, in particular, the first-to-file provisions, became effective on March 16, 2013. Substantive changes to patent law associated with the America Invents Act may affect our ability to obtain patents, and if obtained, to enforce or defend them. Accordingly, it is not clear what, if any, impact the America Invents Act will have on the cost of prosecuting our patent applications, our ability to obtain patents based on our discoveries and our ability to enforce or defend any patents that may issue from our patent applications, all of which could have a material adverse effect on our business.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to patent protection, because we operate in the highly technical field of development of therapies, we rely in part on trade secret protection in order to protect our proprietary technology and processes. However, trade secrets are difficult to protect. We expect to enter into confidentiality and intellectual property assignment agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keep confidential and not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. These agreements also generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us.
In addition to contractual measures, we try to protect the confidential nature of our proprietary information using physical and technological security measures. Such measures may not, for example, in the case of misappropriation of a trade secret by an employee or third party with authorized access, provide adequate protection for our proprietary information. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. Trade secrets may be independently developed by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, our competitive position could be harmed.
31
We will not seek to protect our intellectual property rights in all jurisdictions throughout the world and we may not be able to adequately enforce our intellectual property rights even in the jurisdictions where we seek protection.
Filing, prosecuting and defending patents on our drug candidates and our trademarks in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States could be less extensive than those in the United States, assuming that rights are obtained in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions or using our trademarks in all countries outside the United States, or from selling or importing products made using our inventions or commercialized under identical or similar trademarks in and into the United States or other jurisdictions. The statutory deadlines for pursuing patent and trademark protection in individual foreign jurisdictions are based on the priority dates of each of our patent and trademark applications.
Competitors may use our technologies or trademarks in jurisdictions where we do not pursue and obtain patent or trademark protection to develop their own products and further, may export otherwise infringing products to territories where we have patent or trademark protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing. Even if we pursue and obtain issued patents and trademarks in particular jurisdictions, our patent claims or other intellectual property rights may not be effective or sufficient to prevent third parties from so competing.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to biopharmaceuticals or biotechnologies. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties, provided that (as a general rule and subject to local laws) the interests of public health so require (e.g., if the treatment is made available to the public in insufficient quantity or quality or at abnormally high prices) and the patent owner is compensated. In addition, many countries limit the enforceability of patents against third parties, including government authorities or government contractors. In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country-by-country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Proceedings to enforce our patent or other intellectual property rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents or other intellectual property at risk of being invalidated or interpreted narrowly, could put our patent or trademark applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. In addition, changes in the law and legal decisions by courts in the United States and foreign countries may affect our ability to obtain adequate protection for our technology and the enforcement of intellectual property. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
32
Third parties may assert ownership or commercial rights to inventions we develop.
Third parties may in the future make claims challenging the inventorship or ownership of our intellectual property. We have written agreements with collaborators that provide for the ownership of intellectual property arising from our collaborations. These agreements provide that we must negotiate certain commercial rights with collaborators with respect to joint inventions or inventions made by our collaborators that arise from the results of the collaboration. In some instances, there may not be adequate written provisions to address clearly the resolution of intellectual property rights that may arise from a collaboration. If we cannot successfully negotiate sufficient ownership and commercial rights to the inventions that result from our use of a third-party collaborator’s materials where required, or if disputes otherwise arise with respect to the intellectual property developed with the use of a collaborator’s samples, we may be limited in our ability to capitalize on the market potential of these inventions. In addition, we may face claims by third parties that our agreements with employees, contractors, or consultants obligating them to assign intellectual property to us are ineffective, or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such inventions. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property, or may lose our exclusive rights in that intellectual property. Either outcome could have an adverse impact on our business.
Our Chief Executive Officer, who is a corporate officer (mandataire social) but not an employee of the Company under French law, is involved in our research and development activities. He has contributed to research results for which we have submitted patent applications in which he is listed as a co-inventor and other inventions that we expect may give rise to patent applications in the future for which we expect he will be included as a co-inventor. Under French intellectual property law, inventors who are employees of a company have legal rights that are typically circumscribed in France by a combination of French labor law and contractual arrangements. Because Mr. Veillet is our CEO, and not an employee, we have entered into an assignment agreement with him, pursuant to which he is entitled to certain payments as consideration for his prior and future contributions to our research projects and inventions. See “Intellectual Property Agreement with Stanislas Veillet” in the “Business” section of this annual report for additional information.
Third parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets.
We employ individuals who were previously employed at universities or other biotechnology companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
A dispute concerning the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be time-consuming and costly, and an unfavorable outcome could harm our business.
There is significant litigation in the biotechnology industry regarding patent and other intellectual property rights. While we are not currently subject to any pending intellectual property litigation, and are not aware of any such threatened litigation, we may be exposed to future litigation by third parties based on claims that our drug candidates, technologies or activities infringe the intellectual property rights of others. If our development activities are found to infringe any such patents, we may have to pay significant damages or seek licenses to such patents. A patentee could prevent us from using the patented drugs or compositions. We may need to resort to litigation to enforce a patent issued to us, to protect our trade secrets, or to determine the scope and validity of third-party proprietary rights. From time to time, we may hire scientific personnel or consultants formerly employed by other companies involved in one or more areas similar to the activities conducted by us. Either we or these individuals may be subject to allegations of trade secret misappropriation or other similar claims as a result of prior affiliations. If we become involved in litigation, it could consume a substantial portion of our managerial and financial resources, regardless of whether we win or lose. We may not be able to afford the costs of litigation. Any adverse ruling or perception of an adverse ruling in defending ourselves against these claims could have a material adverse impact on our cash position and the price of the ADSs. Any legal action against us or our collaborators could lead to:
| ● | payment of damages, potentially treble damages, if we are found to have willfully infringed a party’s patent rights; |
33
| ● | injunctive or other equitable relief that may effectively block our ability to further develop, commercialize, and sell products; or |
| ● | us or our collaborators having to enter into license arrangements that may not be available on commercially acceptable terms, if at all, all of which could have a material adverse impact on our cash position and business and financial condition. As a result, we could be prevented from commercializing current or future drug candidates. |
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts and stop us from commercializing or increase the costs of commercializing our drug candidates, if approved or authorized.
Our success will depend in part on our ability to operate without infringing the intellectual property and proprietary rights of third parties. We cannot assure you that our business, products and methods do not or will not infringe the patents or other intellectual property rights of third parties.
The biotechnology industry is characterized by extensive litigation regarding patents and other intellectual property rights. Other parties may allege that our drug candidates or the use of our technologies infringes patent claims or other intellectual property rights held by them or that we are employing their proprietary technology without authorization. Patent and other types of intellectual property litigation can involve complex factual and legal questions, and their outcome is uncertain. Any claim relating to intellectual property infringement that is successfully asserted against us may require us to pay substantial damages, including treble damages and attorney’s fees if we are found to be willfully infringing another party’s patents, for past use of the asserted intellectual property and royalties and other consideration going forward if we are forced to take a license. In addition, if any such claim were successfully asserted against us and we could not obtain such a license, we may be forced to stop or delay developing, manufacturing, selling or otherwise commercializing products.
Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court, or redesign our products. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, intellectual property litigation or claims could force us to do one or more of the following:
| ● | cease developing, selling or otherwise commercializing our drug candidates; |
| ● | pay substantial damages for past use of the asserted intellectual property; |
| ● | obtain a license from the holder of the asserted intellectual property, which license may not be available on reasonable terms, if at all; |
| ● | harm our reputation and cause potential partners or academic entities to avoid working with us; and |
| ● | in the case of trademark claims, redesign or rename trademarks we own to avoid infringing the intellectual property rights of third parties, which may not be possible and, even if possible, could be costly and time-consuming. |
Any of these risks coming to fruition could have a material adverse effect on our business, results of operations, financial condition and prospects.
34
Issued patents covering our drug candidates could be found invalid or unenforceable if challenged in court.
If we or one of our licensing partners initiated legal proceedings against a third party to enforce a patent covering our drug candidate, the defendant could counterclaim that the patent covering our drug candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for unenforceability assertions include allegations that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review and equivalent proceedings in foreign jurisdictions, e.g., opposition proceedings. Such proceedings could result in revocation or amendment of our patents in such a way that they no longer cover our drug candidates or competitive products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our drug candidates. Such a loss of patent protection would have a material adverse impact on our business.
Risks Related to Government Regulation
Even if we obtain regulatory approval or authorization for a drug candidate, our products will remain subject to regulatory scrutiny.
If our drug candidates are approved or authorized, they may be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing studies, and submission of safety, efficacy, and other post-market information, including both federal and state requirements in the United States and requirements of comparable foreign regulatory authorities.
Manufacturers and manufacturers’ facilities are required to comply with extensive FDA, EMA and comparable foreign regulatory authority requirements, including ensuring that quality control and manufacturing procedures conform to cGMP regulations. As such, we and our contract manufacturers will be subject to continual review and inspections to assess compliance with cGMP and adherence to commitments made in any approved marketing application or authorization. Inspections by regulatory authorities and the potential need for subsequent corrective actions may require additional investment or changes to our manufacturing or suppliers’ manufacturing facilities, and may cause delays, interruptions, or complete stoppage of the manufacturing process. If certain drugs have a potential for misuse/abuse, manufacturers and manufacturers’ facilities must also comply with certain drug diversion regulatory and compliance programs. Accordingly, we and others with whom we work must continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, and quality control.
Given that we expect to have a global supply chain, our supply chain may also be affected by the FDA’s enforcement activity at the U.S. border, such as import detentions, import holds, import refusals, or drug diversion oversight or refusals. Despite our investment in regulatory compliance, the FDA may raise issues with our regulatory compliance, and suppliers outside of our direct control may also fail to adhere to the FDA’s regulatory requirements, in which case our supply chain and business plans may be interrupted. Further import detentions, holds, or refusals may also occur while the FDA attempts to verify the imported products’ compliance with the law. Such detentions, holds, or refusals may affect our supply chain and business plans.
Authorities and policy makers are tightening controls on compliance by suppliers on environmental and social standards. We may be required to further tighten the audit of our suppliers, and to change suppliers in case of non-compliance. Independently, enforcement measures by government authorities such as import bans from suppliers suspected of such non-compliance may impact our supply chain.
35
We will have to comply with requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription drugs and biologics are subject to a variety of legal and regulatory restrictions in the United States and the EU (both at EU and national level, for instance, in France) and must be consistent with the information in the product’s approved or authorized label. As such, we may not promote our products for indications or uses for which they do not have approval or authorization. The holder of an approved or authorized application must submit new or supplemental applications and obtain approval or authorization for certain changes to the approved or authorized product, product labeling, or manufacturing process. We could also be asked to conduct post-marketing clinical trials to verify the safety and efficacy of our products in general or in specific patient subsets. An unsuccessful post-marketing study or failure to complete such a study could result in the withdrawal of marketing approval or authorization. In addition, under European regulation, certain of our drug candidates could be added to the list of drugs subject to additional monitoring and studies. Such list concerns drugs for which there is no experience due to their recent marketing or a lack of data on their long-term use. This classification would lead to additional requirements regarding post-marketing surveillance measures of our products or safety studies, which may require more resources on our end.
If a regulatory authority discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or disagrees with the promotion, marketing or labeling of a product, such regulatory agency may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If we fail to comply with applicable regulatory requirements, a regulatory agency or enforcement authority may, among other things:
| ● | issue warning letters; |
| ● | carry out inspections; |
| ● | seek an injunction or impose administrative, civil or criminal penalties or sanctions; |
| ● | suspend or withdraw regulatory approval or authorization; |
| ● | suspend any of our clinical trials; |
| ● | refuse to approve or authorize pending applications or supplements to approved or authorized applications submitted by us; |
| ● | impose restrictions on our operations, including closing our contract manufacturers’ facilities; |
| ● | seize or detain products, or require a product recall; or |
| ● | refuse product importation, subject the import shipments to scrutiny, or place us or our suppliers on the Import Alert program. |
Any government investigation of alleged violations of law could require us to expend significant time and resources in response, and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize and generate revenue from our products. If regulatory sanctions are applied or if regulatory approval or authorization is withdrawn, the value of our company and our operating results will be adversely affected.
Moreover, the policies of the FDA, EMA and of other regulatory authorities may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval or authorization of our drug candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, in Europe, the United States or elsewhere. For example, Regulation (EU) No 536/2014 on clinical trials on medicinal products for human use was adopted in 2014 and became effective as of January 31, 2022 and could impact the administrative procedure that we will have to follow in order to obtain regulatory approval or authorization for our drug candidates. Depending on the date of our application for clinical trial authorization, we could be required to adapt quickly to the new requirements and procedures resulting from this new regulation, in particular regarding the new required deadlines that will require us to be reactive in the event of additional requests from the authorities. We are also anticipating further guidance and decisions from the European Commission, EMA and national regulators of Member States (such as ANSM for France) as those are involved in the process.
36
In addition, certain policies of the Biden administration in the United States, or the future administration of whichever candidate wins the upcoming 2024 Presidential election, may impact our business and industry. Previously, the Trump administration enacted several executive actions, including the issuance of a number of Executive Orders that restricted the FDA’s ability to engage in routine oversight activities such as implementing rules through rulemaking. The Biden administration rescinded some of the Executive Orders, and has not implemented new Executive Orders that meaningfully restrict the authority of the FDA. However, any presidential administration including the current one may implement new policies and executive actions in the future that could affect the FDA’s ability to exercise its authority. If these executive actions impose restrictions on the FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
Furthermore, new or existing legislation in the United States, and regulations implemented by the FDA, may impact our business and industry. For example, the Consolidated Appropriations Act of 2023 as signed into law by President Biden calls for a number of changes to clinical trial structure and oversight, and calls on the FDA to implement a number of new regulations related to clinical trials that may impact our business or industry. Additionally, FDA regulations and guidance related to advertising of prescription drug products and the modernization and diversification of clinical trial types and data sources may impact our business and industry. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval or authorization that we may have obtained and we may not achieve or sustain profitability.
Eradication or substantial eradication of COVID-19 could reduce or eliminate the demand for our product, BIO101 (20-hydroxyecdysone) in this indication.
Since the outburst of the COVID-19 pandemic in 2020/21, the number of reported cases and hospitalization has decreased significantly. Today, COVID-19 has become a chronic respiratory viral infection, like influenza, and numerous patients still suffer from severe forms and some of them die. With the continued vaccination of elderly people and the lesser activity of new variants, the disease could be largely eradicated or lead to a mild clinical course that only very rarely results in hospitalization, and the demand for our BIO101 (20-hydroxyecdysone) in this indication could be significantly reduced.
If any of our drug candidates obtain regulatory approval, additional competitors could enter the market with generic versions of such drugs, which may result in a material decline in sales of affected products.
Under the Drug Price Competition and Patent Term Restoration Act of 1984 (the “Hatch-Waxman Act”), a pharmaceutical manufacturer may file an abbreviated new drug application (“ANDA”) seeking approval of a generic version of an approved, small molecule innovator product. Under the Hatch-Waxman Act, a manufacturer may also submit a new drug application (“NDA”) under section 505(b)(2) of the Federal Food, Drug, and Cosmetic Act (the “FDCA”) that references the FDA’s prior approval of the innovator product. A 505(b)(2) NDA product may be for a new or improved version of the original innovator product. The Hatch-Waxman Act also provides for certain periods of regulatory exclusivity, which preclude FDA approval (or in some circumstances, FDA filing and review) of an ANDA or 505(b)(2) NDA. In addition to the benefits of regulatory exclusivity, an innovator NDA holder may have patents claiming the active ingredient, product formulation or an approved use of the drug, which would be listed with the product in the FDA publication, “Approved Drug Products with Therapeutic Equivalence Evaluations,” known as the Orange Book. If there are patents listed in the Orange Book for a product, a generic or 505(b)(2) applicant that seeks to market its product before expiration of the patents must include in their applications what is known as a “Paragraph IV” certification, challenging the validity or enforceability of, or claiming non-infringement of, the listed patent or patents. Notice of the certification must be given to the patent owner and NDA holder and if, within 45 days of receiving notice, either the patent owner or NDA holder sues for patent infringement, approval of the ANDA or 505(b)(2) NDA is stayed for up to 30 months.
Accordingly, if any of our drug candidates are approved, competitors could file ANDAs for generic versions of our drug candidates or 505(b)(2) NDAs that reference our small molecule drug products. If there are patents listed for our drug candidates in the Orange Book, those ANDAs and 505(b)(2) NDAs would be required to include a certification as to each listed patent indicating whether the ANDA applicant does or does not intend to challenge the patent. We cannot predict which, if any, patents in our current portfolio or patents we may obtain in the future will be eligible for listing in the Orange Book, how any generic competitor would address such patents, whether we would sue on any such patents, or the outcome of any such suit.
We may not be successful in securing or maintaining proprietary patent protection for products and technologies we develop or license. Moreover, if any of our owned or in-licensed patents that are listed in the Orange Book are successfully challenged by way of a Paragraph IV certification and subsequent litigation, the affected product could immediately face generic competition and its sales would likely decline rapidly and materially.
37
We have received and may seek additional orphan drug designations for certain future drug candidates, but we may be unable to obtain such designations or to maintain the benefits associated with orphan drug designation, including market exclusivity, which may cause our revenue, if any, to be reduced.
We obtained and may pursue orphan drug designation for certain of our future drug candidates. In the European Union, the EMA’s Committee for Orphan Medicinal Products (“COMP”) recommends orphan drug designation to promote the development of products that are intended for the diagnosis, prevention, or treatment of a life-threatening or chronically debilitating condition affecting not more than five in 10,000 persons in the European Union. Additionally, designation is granted for products intended for the diagnosis, prevention, or treatment of a life-threatening, seriously debilitating or serious and chronic condition when, without incentives, it is unlikely that sales of the drug in the European Union would be sufficient to justify the necessary investment in developing the drug or biological product or where there is no satisfactory method of diagnosis, prevention, or treatment, or, if such a method exists, the medicine must be of significant benefit to those affected by the condition. Under the Orphan Drug Act, the FDA may designate a drug or biologic product as an orphan drug if it is intended to treat a rare disease or condition, defined as a patient population of fewer than 200,000 in the United States, or a patient population greater than 200,000 in the United States where there is no reasonable expectation that the cost of developing the drug will be recovered from sales in the United States.
In the European Union, orphan drug designation may entitle a party to financial incentives such as reduction of regulatory fees or fee waivers and ten years of market exclusivity following drug or biological product approval unless a derogation applies. This period may be reduced to six years if the orphan drug designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of market exclusivity. In the United States, orphan drug designation entitles a party to financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages, and application fee waivers. In addition, if a product receives the first FDA approval for the indication for which it has orphan designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity or where the manufacturer is unable to assure sufficient product quantity for the orphan patient population.
We may seek additional orphan drug designations in the future for some of our future drug candidates but FDA or EMA may decline our application. Even if we obtain orphan drug designation, we may not be the first to obtain marketing approval for any particular orphan indication due to the uncertainties associated with developing pharmaceutical products. Further, even if we obtain orphan drug exclusivity for a drug candidate, that exclusivity may not effectively protect the product from competition because different drugs with different active moieties can be approved for the same condition. Orphan drug designations are not in any way indicative of a drug’s likelihood of receiving the final marketing authorization from FDA. The FDA does not evaluate a drug candidate’s safety and effectiveness using the same standard as it would when reviewing a drug candidate’s safety and effectiveness prior to granting final marketing approvals. The FDA may grant orphan drug designations to multiple drugs intended for the same indication. Even after an orphan drug is approved, the EMA or FDA can subsequently approve the same drug with the same active moiety for the same condition if the EMA or FDA concludes that the later drug is clinically superior in that it is safer, more effective, or makes a major contribution to patient care. Orphan drug designation neither shortens the development time or regulatory review time of a drug or biologic nor gives the drug or biologic any advantage in the regulatory review or approval process.
Enacted and future healthcare legislation may increase the difficulty and cost for us to obtain marketing approval or authorization of and commercialize our drug candidates and may affect the prices we may set.
In the United States, the EU and other jurisdictions, there have been, and we expect there will continue to be, a number of legislative and regulatory changes and proposed changes to the healthcare system that could affect our future results of operations. In particular, there have been and continue to be a number of initiatives at the U.S. federal and state levels that seek to reduce healthcare costs and improve the quality of healthcare. For example, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “Affordable Care Act”) was enacted, which substantially changed the way healthcare is financed by both governmental and private insurers. Among the provisions of the Affordable Care Act, those of greatest importance to the pharmaceutical and biotechnology industries include the following:
| ● | an annual, non-deductible fee payable by any entity that manufactures or imports certain branded prescription drugs and biologic agents (other than those designated as orphan drugs), which is apportioned among these entities according to their market share in certain government healthcare programs; |
38
| ● | a Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| ● | requirements to report certain financial arrangements with physicians, teaching hospitals, and other healthcare providers, including reporting “transfers of value” made or distributed to such healthcare providers and reporting ownership or investment interests held by physicians and their immediate family members; |
| ● | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for branded and generic drugs, respectively; |
| ● | a methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; |
| ● | extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
| ● | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability; |
| ● | a Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; and |
| ● | establishment of a Center for Medicare Innovation at the Centers for Medicare & Medicaid Services, or CMS, to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending. |
Since its enactment, there have been judicial and Congressional challenges to certain aspects of the Affordable Care Act, and we expect there will be additional challenges and amendments to the Affordable Care Act in the future.
In addition, other legislative changes have been proposed and adopted in the United States since the Affordable Care Act was enacted. In August 2011, the Budget Control Act of 2011, among other things, led to aggregate reductions of Medicare payments to providers of 2% per fiscal year. These reductions went into effect in April 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2025 unless additional action is taken by Congress. In January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. The Inflation Reduction Act, as passed in 2022, also dramatically revised the Part D benefit for Medicare coverage, eliminating the 5% beneficiary coinsurance requirement above the coverage threshold and capping out-of-pocket costs at $2,000 beginning in 2025. The law also modifies liability for Medicare Part D plans and drug manufacturers, starting in 2025, and reduces Medicare’s liability for spending above the out-of-pocket cap. Medicare’s share of total costs above the spending cap will decrease from 80% to 20% for brand-name drugs and to 40% for generic drugs. Medicare Part D plans’ share of costs will increase from 15% to 60% for both brands and generics above the cap, and drug manufacturers will be required to provide a 20% price discount on brand-name drugs. The legislation also requires manufacturers to provide a 10% discount on brand-name drugs between the deductible and the annual out-of-pocket spending cap, replacing the 70% price discount in the coverage gap phase under the current benefit design.
39
Individual states in the United States have also become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Legally-mandated price controls on payment amounts by third-party payors or other restrictions could harm our business, results of operations, financial condition and prospects. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. This could reduce the ultimate demand for our drug candidates or put pressure on our product pricing. Moreover, payment methodologies may be subject to changes in healthcare legislation and regulatory initiatives. For example, CMS may develop new payment and delivery models, such as bundled payment models. In addition, recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products.
In the EU, similar political, economic and regulatory developments may affect our ability to profitably commercialize our drug candidates, if approved. In addition to continuing pressure on prices and cost containment measures, legislative developments at the EU or at the Member State level may result in significant additional requirements or obstacles that may increase our operating costs. The delivery of healthcare in the EU, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than EU, law and policy. National governments and health service providers have different priorities and approaches to the delivery of health care and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers and payors. Generally, pricing negotiations with governmental authorities can take many months after the receipt of regulatory approval and product launch. In some EU Member States, such as in France, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidates with available therapies in order to obtain favorable reimbursement for the indications sought or pricing approval. Should reimbursement for our drug candidates be unavailable in any country in which we seek reimbursement, or be limited or subject to additional clinical trials, or should pricing be set at unsatisfactory levels, then this might have an impact on our operating results. Coupled with ever-increasing EU and national regulatory burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our drug candidates, restrict or regulate post-approval activities and affect our ability to commercialize our drug candidates, if approved. In markets outside of the United States and EU, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific products and therapies.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action in the United States, the EU or any other jurisdiction. If we or any third parties we may engage are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our drug candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability.
Our business operations and current and future relationships with investigators, healthcare professionals, consultants, third-party payors, patient organizations and customers will be subject to applicable healthcare regulatory laws, which could expose us to penalties.
Our business operations and current and future arrangements with investigators, healthcare professionals, consultants, third-party payors, patient organizations and customers, may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations. These laws may constrain the business or financial arrangements and relationships through which we conduct our operations, including how we research, market, sell and distribute our drug candidates, if approved or authorized. Such laws include:
| ● | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, offering, receiving or providing any remuneration (including any kickback, bribe, or certain rebate), directly or indirectly, overtly or covertly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service, for which payment may be made, in whole or in part, under U.S. federal and state healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
40
| ● | the U.S. federal false claims and civil monetary penalties laws, including the civil False Claims Act, which, among other things, impose criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the U.S. federal government, claims for payment or approval that are false or fraudulent, knowingly making, using or causing to be made or used, a false record or statement material to a false or fraudulent claim, or from knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the U.S. federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
| ● | the U.S. federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which imposes criminal and civil liability for, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, or knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statement, in connection with the delivery of, or payment for, healthcare benefits, items or services; similar to the U.S. federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and its implementing regulations, which also imposes certain obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information without appropriate authorization by covered entities subject to the rule, such as health plans, healthcare clearinghouses and healthcare providers as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information; |
| ● | the FDCA, which prohibits, among other things, the adulteration or misbranding of drugs, biologics and medical devices and the introduction of such products into interstate commerce; |
| ● | the U.S. Public Health Service Act, which prohibits, among other things, the introduction into interstate commerce of a biological product unless a biologics license is in effect for that product; |
| ● | the U.S. Physician Payments Sunshine Act and its implementing regulations, which require certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under Medicare, Medicaid, or the Children’s Health Insurance Program to report annually to the government information related to certain payments and other transfers of value to physicians, teaching hospitals, and other healthcare providers, as well as ownership and investment interests held by the physicians described above and their immediate family members; |
| ● | analogous U.S. state laws and regulations, including: state anti-kickback and false claims laws, which may apply to our business practices, including but not limited to, research, distribution, sales and marketing arrangements and claims involving healthcare items or services reimbursed by any third-party payor, including private insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the U.S. federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws and regulations that require drug manufacturers to file reports relating to pricing and marketing information, which requires tracking gifts and other remuneration and items of value provided to healthcare professionals and entities; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts; and |
41
| ● | similar healthcare laws and regulations in the EU and other jurisdictions, including reporting requirements detailing interactions with and payments to healthcare providers. For example, under French law, the regulation requires strict transparency of the links between the health care industry and other actors such as, but not limited to, health care practioners, and impose reporting on a public record all benefits granted to the various actors involved, in particular health professionals, as well as the existence of agreements concluded with these actors as well as remunerations paid. In addition to financial penalties, any violation of those requirements, such as misleading information or non-publication, could result in additional sanctions that may have harmful effect on the conduct of our business. More generally, as our business activity is heavily regulated and involves a significant interaction with government officials, our dealings with prescriber and authorities are subject to national anti-corruption laws of EU Member States. These laws notably prohibit us and our employees from improperly influencing government officials or commercial parties to obtain or retain business, direct business to any person or gain any advantage and also prohibit our third-party business partner’s representatives and agents from engaging in corruption and bribery. Under these applicable anti-corruption laws, we may be held liable for the acts or the corrupt activities of our third-party business partners, intermediaries, representatives, contractors, channel partners and agents, even if we don’t explicitly authorize or have knowledge of such activities. While we have a formal procedure that defines the process to be used to select our third-party partners, collaborate with them and monitor them in accordance with applicable anti-corruption laws, there is a risk that our third-party partners may act in violation of applicable laws, for which we may be ultimately held responsible. Any violation of applicable anti-corruption laws could result in whistleblower complaints, adverse media coverage, investigations, imposition of significant legal fees, severe criminal, civil and administrative sanctions, suspension or debarment from government contracts, all of which may have an adverse effect on our reputation, business, results of operations and financial condition. In addition, it is possible that as our business grows and evolves, we will become subject to additional compliance requirements, resulting for example from the French Sapin II Act, which requires companies concerned by this regulation to implement a general anti-corruption compliance project under the control of the competent supervisory authority such as staff training, compliance documentation, audits and regular monitoring of commercial relationships. As the EU Commission has stated in one of its reports that the health sector is particularly vulnerable, our business may be subject to increased anti-corruption compliance monitoring. |
Ensuring that our internal operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to significant penalties, including civil, criminal and administrative penalties, damages, fines, exclusion from government-funded healthcare programs, such as Medicare and Medicaid or similar programs in other countries or jurisdictions, disgorgement, individual imprisonment, contractual damages, reputational harm, diminished profits and the curtailment or restructuring of our operations. Further, defending against any such actions can be costly, time-consuming and may require significant personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired.
In addition, considering that our activity involves the processing of personal data, in particular sensitive data such as health data, our business activities are also subject to GDPR and other national data protection laws and guidelines with respect to such data, which implies that we must implement significant and continuous efforts to comply with these data protection regulations, as well as any applicable additional national health care regulations. The GDPR has allowed EU Member States to introduce additional requirements for the processing of health data. This means we must comply with both EU as well as national laws in order to conduct our activities as regards patient data. In particular, our GDPR compliance involves the precise identification of our data processing operations and the risks incurred, the implementation of an organization of our internal processes and the establishment of documentation relating to our compliance. Our GDPR compliance also means being very aware of the fulfilment of our third-party contractors’ obligations and their (own) GPDR compliance, which requires us to impose strict contractual provisions on our third-party contractors as processors or to ensure that they will not use the personal data for other purposes than agreed on. Moreover, the transfer of data from the EU to our U.S. entities or others U.S. companies must (i) have a legal basis in GDPR or other national data protection laws, and (ii) be subject to a valid legal mechanism for the lawful transfer of data, which may have to require some of our third-party contractors who process personal data to take additional privacy and security measures. Non-compliance could lead to severe impact for individuals and cause us to incur potential disruption and expense related to our business processes. Any violations of these laws and regulations could also result in substantial penalties and could materially damage our reputation.
42
Furthermore, following the European Court of Justice’s decision to invalidate the EU—U.S. Privacy Shield as part of the Schrems II decision, any transfer or storage of data from the EU by our U.S. entities, other U.S. companies or contractual counterparties will require the implementation of additional safeguards, which given the current status of regulations, will most certainly require further protection measures in order to ensure an adequate level of protection as defined by the EU and national authorities. In case such additional safeguards do not lead to sufficient protection of personal data, transfers must be suspended or not carried out at all.
We are subject to U.S. and foreign anti-corruption and anti-money laundering laws with respect to our operations and non-compliance with such laws can subject us to criminal and/or civil liability and harm our business.
We are subject to the U.S. Foreign Corrupt Practices Act of 1977, as amended (the “FCPA”), the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, and possibly other state and national anti-bribery and anti-money laundering laws in countries in which we conduct activities. Anti-corruption laws are interpreted broadly and prohibit companies and their employees, agents, third-party intermediaries, joint venture partners and collaborators from authorizing, promising, offering, or providing, directly or indirectly, improper payments or benefits to recipients in the public or private sector. We engage third-party investigators, CROs, and other consultants to design and perform preclinical studies of our drug candidates, and will do the same for any clinical trials. Also, once a drug candidate has been approved, authorized, and commercialized, we may engage third-party intermediaries to promote and sell our products abroad and/or to obtain necessary permits, licenses, and other regulatory approvals or authorizations. We or our third-party intermediaries may have direct or indirect interactions with officials and employees of government agencies or state-owned or affiliated entities. We can be held liable for the corrupt or other illegal activities of these third-party intermediaries, our employees, representatives, contractors, collaborators, partners, and agents, even if we do not explicitly authorize or have actual knowledge of such activities.
Noncompliance with anti-corruption and anti-money laundering laws could subject us to whistleblower complaints, investigations, sanctions, settlements, prosecution, other enforcement actions, disgorgement of profits, significant fines, damages, other civil and criminal penalties or injunctions, suspension and/or debarment from contracting with certain persons, the loss of export privileges, reputational harm, adverse media coverage, and other collateral consequences. If any subpoenas, investigations, or other enforcement actions are launched, or governmental or other sanctions are imposed, or if we do not prevail in any possible civil or criminal litigation, our business, results of operations and financial condition could be materially harmed. In addition, responding to any action will likely result in a materially significant diversion of management’s attention and resources and significant defense and compliance costs and other professional fees. In certain cases, enforcement authorities may even cause us to appoint an independent compliance monitor which can result in added costs and administrative burdens.
Our failure to maintain certain tax benefits applicable to French technology companies may adversely affect our results of operations.
As a French biotechnology company, we have benefited from certain tax advantages, including, for example, the research tax credit (Crédit d’Impôt Recherche), or CIR. The CIR is a French tax credit aimed at stimulating research and development. The CIR can be offset against French corporate income tax due and the portion in excess (if any) may be refunded at the end of a three fiscal-year period (or, sooner, for smaller companies such as ours). The CIR is calculated based on our claimed amount of eligible research and development expenditures in France and represented €4.1 million, €3.3 million and €1.5 million as of December 31, 2021, 2022 and 2023, respectively. The French tax authority with the assistance of the Research and Technology Ministry may audit each research and development program in respect of which a CIR benefit has been claimed and assess whether such program qualifies in its view for the CIR benefit. The French tax authorities may challenge our eligibility to, or our calculation of certain tax reductions and/or deductions in respect of our research and development activities and, should the French tax authorities be successful, we may be liable for additional corporate income tax, and penalties and interest related thereto, or we may not obtain the refunds for which we have applied, which could have a significant impact on our results of operations and future cash flows. Furthermore, if the French Parliament decides to eliminate, or reduce the scope or the rate of, the CIR benefit, either of which it could decide to do at any time, our results of operations could be adversely affected.
43
Future changes to applicable U.S. tax laws and regulations may have an adverse effect on our business, financial condition and results of operations.
In general, changes in laws and policy relating to taxes may have an adverse effect on our business, financial condition and results of operations. For example, at the end of 2017, the U.S. government enacted significant tax reform, with additional guidance from the U.S. Treasury and Internal Revenue Service (the “IRS”) still pending. Changes include, but are not limited to, a federal corporate tax rate decrease to 21% for tax years beginning after December 31, 2017, a reduction to the maximum deduction allowed for net operating losses generated in tax years after December 31, 2017, eliminating carrybacks of net operating losses, and providing for indefinite carryforwards for losses generated in tax years after December 31, 2017. The 2017 legislation remains unclear in many respects and could be subject to potential amendments and technical corrections or even outright changes. Additionally, current tax laws may continue to be subject to interpretations and implementing regulations by the U.S. Treasury and IRS, any of which could mitigate or increase certain adverse effects of prior legislation. In addition, it is unclear how future U.S. federal income tax changes will affect state and local taxation.
Risks Related to the Ownership of the ADSs and Ordinary Shares and Our Status as a Non-U.S. Company with Foreign Private Issuer Status
The requirements of being a U.S. public company may strain our resources, divert management’s attention and affect our ability to attract and retain executive management and qualified board members.
As a U.S. public company, we have and will continue to incur legal, accounting, and other expenses that we did not previously incur. Following our initial public offering of ADSs in the United States, we are now subject to the Exchange Act, including the reporting requirements thereunder, the Sarbanes-Oxley Act of 2002 (the “Sarbanes-Oxley Act”), the Dodd-Frank Wall Street Reform and Consumer Protection Act, the Nasdaq listing requirements and other applicable securities rules and regulations. Compliance with these rules and regulations has and will continue to increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and increase demand on our systems and resources, particularly after we are no longer an “emerging growth company” and/or a foreign private issuer. For example, for so long as we remain a foreign private issuer, we will not be required to file with the SEC quarterly reports with respect to our business and results of operations, which are required to be made by domestic issuers pursuant to the Exchange Act.
Pursuant to Section 404 of the Sarbanes-Oxley Act, we will in the future be required to furnish an attestation report on internal control over financial reporting issued by our independent registered public accounting firm. However, while we remain an emerging growth company, we will not be required to include this attestation report on internal control over financial reporting issued by our independent registered public accounting firm. When our independent registered public accounting firm is required to undertake an assessment of our internal control over financial reporting, the cost of complying with Section 404 will significantly increase and management’s attention may be diverted from other business concerns, which could adversely affect our business and results of operations. We may need to hire more employees in the future or engage outside consultants to comply with these requirements, which will further increase our cost and expense. If we fail to implement the requirements of Section 404 in the required timeframe, we may be subject to sanctions or investigations by regulatory authorities, including the SEC and the Nasdaq. Furthermore, if we are unable to conclude that our internal control over financial reporting is effective, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of the ADSs and our ordinary shares could decline, and we could be subject to sanctions or investigations by regulatory authorities. Failure to implement or maintain effective internal control systems required of public companies could also restrict our future access to the capital markets.
44
Furthermore, if we fail to comply with the Nasdaq continued listing requirements, including their minimum bid requirement, our ADSs could be delisted from Nasdaq, and as a result we and our shareholders could incur material adverse consequences, including a negative impact on our liquidity, our shareholders’ ability to sell shares and our ability to raise capital. On April 24, 2023, we received a deficiency letter from Nasdaq stating that we failed to maintain a minimum of $2,500,000 in stockholders’ equity under Listing Rule 5550(b) as evidenced in the Form 20-F for the year ended December 31, 2022. On August 1, 2023, Nasdaq granted us until October 23, 2023 to regain compliance. On October 26, 2023, we received written correspondence from Nasdaq indicating that we still do not comply with the minimum $2,500,000 stockholders’ equity requirement and that our securities would be delisted unless we requested a hearing. The Company has since appealed the determination and a hearing with the Nasdaq Hearings Panel was held on February 1, 2024 to appeal the determination. Based on the remediation plan we presented, the Panel agreed to extend the grace period until April 23, 2024, thus allowing the Company to continue trading on the Nasdaq and take appropriate actions to increase the level of its shareholders’ equity and regain compliance. These actions included conversion of convertible bonds held by ATLAS funds, at their request, as well as an equity financing. As of the date of this report, compliance has not been achieved yet and planned actions are still in progress.
In parallel, on November 15, 2023, we received written notification from Nasdaq indicating that, based upon a closing bid price of less than $1.00 per share for the Company’s American Depositary Shares (“ADSs”) for the prior 30 consecutive business days period, the Company no longer satisfies Nasdaq Listing Rule 5550(a)(2). Pursuant to this Rule, the applicable grace period to regain compliance is 180 days, or until May 13, 2024. The Company intends to monitor the closing bid price of its ADSs during this grace period and will consider its options in order to regain compliance with the minimum bid price requirement. On March 15, 2024, the Company announced the implementation of a reverse stock split of its shares listed on Euronext Growth, which will result in the allocation of 1 new ordinary share to be issued with a par value of 0.80 euros (the “New Shares ) against 400 old ordinary shares with a par value of 0.002 euros each (the “Old Shares”) and by dividing the number of shares making up the share capital of the Company by 400. The 30-day consolidation period will run from April 2, 2024 to May 3, 2024 (inclusive). At the end of this period, i.e. May 3, 2024, the old shares (ISIN FR0012816825) will be delisted on the Euronext Growth market and the listing of the new shares (ISIN FR001400OLP5) will begin. The amount of capital will be unchanged at the end of the operation and this operation will have no impact on the overall value of the Biophytis securities held in the portfolio by the shareholders, except for fractional shares. If we do not voluntarily change our ADS to share ratio, the reverse stock split of our shares listed on Euronext Growth will be replicated on the ADSs, in order to keep the current ADS-to-share ratio of 1 to 100. However, for our ADSs to trade above the minimum bid price of $1.00 during at least 10 days before May 13, 2024, we will need to adjust our ADS-to-share ratio prior to the reverse stock split effective date of May 3, 2024.
We cannot guarantee that we will continue to comply with the minimum bid price requirement or Nasdaq’s other continued listing requirements. If we fail to satisfy Nasdaq’s conditions for continued listing, our ADSs could be delisted. Delisting from the Nasdaq could have an adverse effect on our business and on the trading of our ADSs. If a delisting of our ADSs were to occur, such securities may trade in the over-the-counter market such as on the OTC Bulletin Board or on the “pink sheets.” The over-the-counter market is generally considered to be a less efficient market, and this could diminish investors’ interest in our ADSs as well as significantly impact the price and liquidity of our ADSs. Any such delisting may also severely complicate trading of our ADSs by our shareholders or prevent them from re-selling their ADSs at/or above the price they paid.
In addition, enhanced legal and regulatory regimes and heightened standards relating to corporate governance and disclosure for public companies result in increased legal and financial compliance costs and make some activities more time consuming. Further, being a U.S. public company and a French public company has and will continue to have an impact on our disclosure of information and requires compliance with two sets of applicable rules. This could result in uncertainty regarding compliance matters and higher costs necessitated by legal analysis of dual legal regimes, ongoing revisions to disclosure and adherence to heightened governance practices.
There was no public market for the ADSs prior to our U.S. initial public offering, and an active market may not continue in which investors can resell their ADSs.
Prior to our U.S. initial public offering, there was no public market for the ADSs. We cannot predict the extent to which an active trading market for the ADSs will develop or be sustained, or how the development of such a market might affect the market price for the ADSs. Investors may not be able to sell their ADSs at or above the price they paid for them. In addition, investors may not be able to successfully withdraw the underlying ordinary shares of the ADSs for the reasons discussed under the risk factor titled “You may not be able to exercise your right to vote the underlying ordinary shares of the ADSs” described below. In connection with any withdrawal of any of our ordinary shares represented by ADSs, the ADSs will be surrendered to the depositary. Unless additional
45
ADSs are issued, the effect of such transactions will be to reduce the number of outstanding ADSs and, if a significant number of transactions are effected, to reduce the liquidity of the ADSs.
The market price of our equity securities may be volatile, and purchasers of our securities could incur substantial losses.
The market price for our securities may be volatile. The stock market in general and the market for biotechnology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may not be able to sell their securities at or above the price paid for the security. The market price for our securities may be influenced by many factors, including:
| ● | actual or anticipated fluctuations in our financial condition and operating results; |
| ● | actual or anticipated changes in our growth rate relative to our competitors; |
| ● | competition from existing products or new products that may emerge; |
| ● | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations, or capital commitments; |
| ● | failure to meet or exceed financial estimates and projections of the investment community or that we provide to the public; |
| ● | issuance of new or updated research or reports by securities analysts; |
| ● | fluctuations in the valuation of companies perceived by investors to be comparable to us; |
| ● | price and volume fluctuations attributable to inconsistent trading volume levels of our securities |
| ● | additions or departures of key management or scientific personnel; |
| ● | lawsuits threatened or filed against us, disputes or other developments related to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies; |
| ● | changes to coverage policies or reimbursement levels by commercial third-party payors and government payors and any announcements relating to coverage policies or reimbursement levels; |
| ● | announcement or expectation of additional debt or equity financing efforts; |
| ● | sales of ADSs or ordinary shares by us, our insiders or our other holders; and |
| ● | general economic and market conditions. |
These and other market and industry factors may cause the market price and demand for our securities to fluctuate substantially, regardless of our actual operating performance, which may limit or prevent investors from readily selling their securities and may otherwise negatively affect the liquidity of the trading market for our securities.
We may be exposed to significant foreign exchange risk. Exchange rate fluctuations may adversely affect the foreign currency value of the ADSs.
We incur portions of our expenses and may in the future derive revenues in currencies other than the euro, in particular, the U.S. dollar. As a result, we are exposed to foreign currency exchange risk as our results of operations and cash flows are subject to fluctuations in foreign currency exchange rates. We currently do not engage in hedging transactions to protect against uncertainty in future exchange rates between particular foreign currencies and the euro. Therefore, for example, an increase in the value of the euro against the U.S. dollar could be expected to have a negative impact on our revenue and earnings growth as U.S. dollar revenue and earnings, if any, would be translated into euros at a reduced value. We cannot predict the impact of foreign currency fluctuations, and foreign currency fluctuations in the future may adversely affect our financial condition, results of operations and cash flows. The
46
ADSs are quoted in U.S. dollars on the Nasdaq Capital Market and our ordinary shares trade in euros on the Euronext Growth Paris. Our financial statements are prepared in euros. Fluctuations in the exchange rate between euros and the U.S. dollar will affect, among other matters, the U.S. dollar value of the ADSs.
If we do not achieve our projected development and commercialization goals in the timeframes we announce and expect, our business will be harmed and the price of our securities could decline as a result.
We sometimes estimate for planning purposes the timing of the accomplishment of various scientific, clinical, regulatory and other product development objectives. These milestones may include our expectations regarding the commencement or completion of scientific studies, clinical trials, the submission of regulatory filings, or commercialization objectives. From time to time, we may publicly announce the expected timing of some of these milestones, such as the completion of an ongoing clinical trial, the initiation of other clinical programs, receipt of marketing approval, authorization, or a commercial launch of a product. The achievement of many of these milestones may be outside of our control. All of these milestones are based on a variety of assumptions which may cause the timing of achievement of the milestones to vary considerably from our estimates, including:
| ● | our available capital resources or capital constraints we experience; |
| ● | the rate of progress, costs and results of our clinical trials and research and development activities, including the extent of scheduling conflicts with participating clinicians and collaborators, and our ability to identify and enroll patients who meet clinical trial eligibility criteria; |
| ● | our receipt of approvals or authorizations by the EMA, FDA and other regulatory authorities and the timing thereof; |
| ● | other actions, decisions or rules issued by regulatory authorities; |
| ● | our ability to access sufficient, reliable and affordable supplies of compounds and raw materials used in the manufacture of our drug candidates; |
| ● | our ability to license and/or generate revenues other than through independent commercialization of our products; |
| ● | the efforts of our collaborators and/or other partners, including licensees, with respect to the commercialization of, in due course, our products; and |
| ● | the securing of, costs related to, and timing issues associated with, product manufacturing as well as sales and marketing activities. |
If we fail to achieve announced milestones in the timeframes we expect, the commercialization of our drug candidates may be delayed, our business and results of operations may be harmed, and the trading price of our securities may decline as a result.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, the price of the ADSs and their trading volume could decline.
The trading market for the ADSs depends in part on the research and reports that securities or industry analysts publish about us or our business. If no or few securities or industry analysts cover our company, the trading price for the ADSs would be negatively impacted. If one or more of the analysts who covers us downgrades our equity securities or publishes incorrect or unfavorable research about our business, the price of our securities would likely decline. If one or more of these analysts ceases coverage of our company or fails to publish reports on us regularly, or downgrades our securities, demand for our securities could decrease, which could cause the price of the ADSs or their trading volume to decline.
We do not currently intend to pay dividends on our ordinary shares and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of our securities. In addition, French law may limit the amount of dividends we are able to distribute.
We have never declared or paid any cash dividends on our ordinary shares and do not currently intend to do so for the foreseeable future. We currently intend to invest our future earnings, if any, to fund our growth. Therefore, you are not likely to receive any dividends on our securities for the foreseeable future and the success of an investment in these securities will depend upon
47
any future appreciation in their value. Consequently, investors may need to sell all or part of their holdings after price appreciation, which may never occur, as the only way to realize any future gains on their investment. There is no guarantee that our securities will appreciate in value or even maintain the price at which investors have purchased them. Investors seeking cash dividends should not purchase our securities.
Further, under French law, the determination of whether we have been sufficiently profitable to pay dividends is made on the basis of our statutory financial statements prepared and presented in accordance with accounting standards applicable in France. Article 34 of our By-laws imposes additional limitations on our ability to declare and pay dividends and there may be taxes imposed on you if we elect to pay a dividend. Therefore, we may be more restricted in our ability to declare dividends than companies not based in France.
In addition, exchange rate fluctuations may affect the amount of euros that we are able to distribute, and the amount in U.S. dollars that our shareholders receive upon the payment of cash dividends or other distributions we declare and pay in euros, if any. These factors could harm the value of the ADSs, and, in turn, the U.S. dollar proceeds that holders receive from the sale of the ADSs.
We have a significant number of outstanding warrants and convertible debt instruments, which may cause significant dilution to our shareholders, have a material adverse impact on the market price of our ordinary shares and make it more difficult for us to raise funds through future equity offerings.
As of February 29th, 2024, we had 1,146,016,581 ordinary shares outstanding. In addition, as of that date we had outstanding warrants to acquire up to 349,762,674 ordinary shares and 126,984,703 free ordinary shares of which 18,369,912 were granted to our two founders on April 14, 2023 and will be delivered to them on April 14, 2024 after a one-year vesting period, and 71,000,000 were granted to them on February 5, 2024 and will be delivered to them on February 5, 2025 after a one-year vesting period. The issuance of ordinary shares upon the exercise of warrants and convertible debt instruments would dilute the percentage ownership interest of all shareholders, might dilute the book value per share of our ordinary shares and would increase the number of our publicly traded shares, which could depress the market price of our ordinary shares.
In addition to the dilutive effects described above, the perceived risk of dilution as a result of the significant number of outstanding warrants and convertible debt may cause our shareholders to be more inclined to sell their shares, which would contribute to a downward movement in the price of our ordinary shares. Moreover, the perceived risk of dilution and the resulting downward pressure on our share price could encourage investors to engage in short sales of our ordinary shares, which could further contribute to price declines in our ordinary shares. The fact that our shareholders, warrant holders and convertible debt holders can sell substantial amounts of our ordinary shares in the public market, whether or not sales have occurred or are occurring could make it more difficult for us to raise additional funds through the sale of equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate, or at all.
Future sales, or the possibility of future sales, of a substantial number of the ADSs or our ordinary shares could adversely affect the price of the ADSs.
Future sales of a substantial number of the ADSs or ordinary shares, or the perception that such sales will occur, could cause a decline in the market price of the ADSs. ADSs sold in our U.S. initial public offering may be resold in the public market without restriction, unless purchased by our affiliates. If ADS holders sell substantial amounts of ADSs in the public market, or the market perceives that such sales may occur, the market price of the ADSs and our ability to raise capital through an issuance of equity securities in the future could be adversely affected.
The rights of shareholders in companies subject to French corporate law differ in material respects from the rights of shareholders of corporations incorporated in the United States.
We are a French company with limited liability. Our corporate affairs are governed by our bylaws and by the laws governing companies incorporated in France. The rights of shareholders and the responsibilities of members of our board of directors are in many ways different from the rights and obligations of shareholders in companies governed by the laws of U.S. jurisdictions. For example, in the performance of its duties, our board of directors is required by French law to consider the interests of our company, rather than solely our shareholders and/or creditors. It is possible that some of these parties will have interests that are different from, or in addition to, yours.
48
U.S. investors may have difficulty enforcing civil liabilities against our company and directors and senior management and the experts named in this annual report.
All of the members of our board of directors and senior management and certain experts named in this annual report are non-residents of the United States, and all or a substantial portion of our assets and the assets of such persons are located outside the United States. As a result, it may not be possible to serve process on such persons or us in the United States or to enforce judgments obtained in U.S. courts against them or us based on civil liability provisions of the securities laws of the United States. Additionally, it may be difficult to assert U.S. securities law claims in actions originally instituted outside of the United States. Foreign courts may refuse to hear a U.S. securities law claim because foreign courts may not be the most appropriate forums in which to bring such a claim. Even if a foreign court agrees to hear a claim, it may determine that the law of the jurisdiction in which the foreign court resides, and not U.S. law, is applicable to the claim. Further, if U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact, which can be a time-consuming and costly process, and certain matters of procedure would still be governed by the law of the jurisdiction in which the foreign court resides. In particular, there is some doubt as to whether French courts would recognize and enforce certain civil liabilities under U.S. securities laws in original actions or judgments of U.S. courts based upon these civil liability provisions. In addition, awards of punitive damages in actions brought in the United States or elsewhere may be unenforceable in France. An award for monetary damages under the U.S. securities laws would be considered punitive if it does not seek to compensate the claimant for loss or damage suffered but is intended to punish the defendant. French law provides that a shareholder, or a group of shareholders, may initiate a legal action to seek indemnification from the directors of a corporation in the corporation’s interest if it fails to bring such legal action itself. If so, any damages awarded by the court are paid to the corporation and any legal fees relating to such action may be borne by the relevant shareholder or the group of shareholders.
The enforceability of any judgment in France will depend on the particular facts of the case as well as the laws and treaties in effect at the time. The United States and France do not currently have a treaty providing for recognition and enforcement of judgments (other than arbitration awards) in civil and commercial matters.
ADSs holders may not be entitled to a jury trial with respect to claims arising under the deposit agreement, which could result in less favorable outcomes to the plaintiff(s) in any such action.
The deposit agreement governing the ADSs representing our ordinary shares provides that, to the fullest extent permitted by law, ADS holders waive the right to a jury trial of any claim they may have against us or the depositary arising out of or relating to our shares, the ADSs or the deposit agreement, including any claim under the U.S. federal securities laws.
If we or the depositary opposed a jury trial demand based on the waiver, the court would determine whether the waiver was enforceable based on the facts and circumstances of that case in accordance with the applicable state and federal law. To our knowledge, the enforceability of a contractual pre-dispute jury trial waiver in connection with claims arising under the federal securities laws has not been finally adjudicated by the United States Supreme Court. However, we believe that a contractual pre-dispute jury trial waiver provision is generally enforceable, including under the laws of the State of New York, which govern the deposit agreement, by a federal or state court in the City of New York, which has non-exclusive jurisdiction over matters arising under the deposit agreement. In determining whether to enforce a contractual pre-dispute jury trial waiver provision, courts will generally consider whether a party knowingly, intelligently and voluntarily waived the right to a jury trial. We believe that this is the case with respect to the deposit agreement and the ADSs. In addition, New York courts will not enforce a jury trial waiver provision in order to bar a viable setoff or counterclaim sounding in fraud or one which is based upon a creditor’s negligence in failing to liquidate collateral upon a guarantor’s demand, or in the case of an intentional tort claim (as opposed to a contract dispute), none of which we believe are applicable in the case of the deposit agreement or the ADSs. It is advisable that you consult legal counsel regarding the jury waiver provision before entering into the deposit agreement.
If you or any other owner or holder of ADSs bring a claim against us or the depositary in connection with matters arising under the deposit agreement or the ADSs, including claims under federal securities laws, you or such other owner or holder may not be entitled to a jury trial with respect to such claims, which may have the effect of limiting and discouraging lawsuits against us and/or the depositary. If a lawsuit is brought against us and/or the depositary under the deposit agreement, it may be heard only by a judge or justice of the applicable trial court, which would be conducted according to different civil procedures and may result in different outcomes than a trial by jury would have had, including results that could be less favorable to the plaintiff(s) in any such action.
49
Nevertheless, if this jury trial waiver provision is not permitted by applicable law, an action could proceed under the terms of the deposit agreement with a jury trial. No condition, stipulation or provision of the deposit agreement or ADSs serves as a waiver by any owner or holder of ADSs or by us or the depositary of compliance with any substantive provision of the U.S. federal securities laws and the rules and regulations promulgated thereunder. By agreeing to the jury trial waiver provision in the deposit agreement, investors will not be deemed to have waived our compliance with or the depositary’s compliance with the federal securities laws and the rules and regulations promulgated thereunder.
Our Articles of Association and By-laws and French corporate law contain provisions that may delay or discourage a takeover attempt.
Provisions contained in our Articles of Association and/or French corporate law could make it more difficult for a third party to acquire us, even if doing so might be beneficial to our shareholders. In addition, provisions of our bylaws impose various procedural and other requirements, which could make it more difficult for shareholders to effect certain corporate actions. These provisions include the following:
| ● | under French law, the owner of 90% of voting rights of a public company listed on a regulated market in a Member State of the EU or in a state party to the European Economic Area (“EEA”) Agreement, including France, has the right to force out minority shareholders following a tender offer made to all shareholders; |
| ● | under French law, a non-resident of France as well as any French entity controlled by non-French residents may have to file an administrative notice with French authorities in connection with a direct or indirect investment in us, as defined by administrative rulings; see the section of this annual report titled “Limitations Affecting Shareholders of a French Company”; |
| ● | a merger (i.e., in a French law context, a stock for stock exchange following which our company would be dissolved into the acquiring entity and our shareholders would become shareholders of the acquiring entity) of our company into a company incorporated in the EU would require the approval of our board of directors as well as a two-thirds majority of the votes held by the shareholders present, represented by proxy or voting by mail at the relevant meeting; |
| ● | under French law, a cash merger is treated as a share purchase and would require the consent of each participating shareholder; |
| ● | our shareholders have granted and may grant in the future our board of directors broad authorizations to increase our share capital or to issue additional ordinary shares or other securities, such as warrants, to our shareholders, the public or qualified investors, including as a possible defense following the launching of a tender offer for our shares; |
| ● | our shareholders have preferential subscription rights on a pro rata basis on the issuance by us of any additional securities for cash or a set-off of cash debts, which rights may only be waived by the extraordinary general meeting (by a two-thirds majority vote) of our shareholders or on an individual basis by each shareholder; |
| ● | our board of directors has the right to appoint directors to fill a vacancy created by the resignation or death of a director, for the remaining duration of such director’s term of office, provided that prior to such decision of the board of directors, the number of directors remaining in office exceeds the minimum required by law and our bylaws, and subject to the approval by the shareholders of such appointment at the next shareholders’ meeting, which prevents shareholders from having the sole right to fill vacancies on our board of directors; |
| ● | our board of directors can be convened by our chairman (directly or upon request of our managing director), or, when no board meeting has been held for more than three consecutive months, by directors representing at least one third of the total number of directors; |
| ● | our board of directors meetings can only be regularly held if at least half of the directors attend either physically or by way of videoconference or teleconference enabling the directors’ identification and ensuring their effective participation in the board’s decisions; |
| ● | our shares are nominative or bearer, if the legislation so permits, according to the shareholder’s choice; |
50
| ● | under French law, certain investments in any entity governed by French law relating to certain strategic industries (such as research and development in biotechnologies and activities relating to public health) and activities by individuals or entities not French, not resident in France or controlled by entities not French or not resident in France are subject to prior authorization of the Ministry of Economy; see “Limitations Affecting Shareholders of a French Company”; |
| ● | approval of at least a majority of the votes held by shareholders present, represented by a proxy, or voting by mail at the relevant ordinary shareholders’ general meeting is required to remove directors with or without cause; |
| ● | advance notice is required for nominations to the board of directors or for proposing matters to be acted upon at a shareholders’ meeting, except that a vote to remove and replace a director can be proposed at any shareholders’ meeting without notice; |
| ● | our bylaws can be changed in accordance with applicable laws; |
| ● | the crossing of certain thresholds has to be disclosed and can impose certain obligations; |
| ● | transfers of shares shall comply with applicable insider trading rules and regulations and, in particular, with the Market Abuse Directive and Regulation dated April 16, 2014; and |
| ● | pursuant to French law, our bylaws, including the sections relating to the number of directors and election and removal of a director from office, may only be modified by a resolution adopted by two-thirds of the votes of our shareholders present, represented by a proxy or voting by mail at the meeting. |
You may not be able to exercise your right to vote the ordinary shares underlying your ADSs.
Holders of ADSs may exercise voting rights with respect to the ordinary shares represented by the ADSs only in accordance with the provisions of the deposit agreement. The deposit agreement provides that, upon receipt of notice of any meeting of holders of our ordinary shares, the depositary will fix a record date for the determination of ADS holders who will be entitled to give instructions for the exercise of voting rights. Upon timely receipt of notice from us, if we so request, the depositary will distribute to the holders as of the record date (1) the notice of the meeting or solicitation of consent or proxy sent by us and (2) a statement as to the manner in which instructions may be given by the holders.
Purchasers of ADSs may instruct the depositary of the ADSs to vote the ordinary shares underlying their ADSs. Otherwise, purchasers of ADSs will not be able to exercise voting rights unless they withdraw the ordinary shares underlying the ADSs they hold. However, a holder of ADSs may not know about the meeting far enough in advance to withdraw those ordinary shares. If we ask for a holder of ADSs’ instructions, the depositary, upon timely notice from us, will distribute notice of the upcoming vote and arrange to deliver our voting materials to him or her. We cannot guarantee to any holder of ADSs that he or she will receive the voting materials in time to ensure that he or she can instruct the depositary to vote his or her ordinary shares or to withdraw his or her ordinary shares so that he or she can vote them. If the depositary does not receive timely voting instructions from a holder of ADSs, it may give a proxy to a person designated by us to vote the ordinary shares underlying his or her ADSs. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that a holder of ADSs may not be able to exercise his or her right to vote, and there may be nothing he or she can do if the ordinary shares underlying the ADSs are not voted as he or she requested.
Purchasers of ADSs may not be directly holding our ordinary shares.
A holder of ADSs will not be treated as one of our shareholders and will not have direct shareholder rights. French law governs our shareholder rights. The depositary will be the holder of the ordinary shares underlying ADSs held by ADS holders. The deposit agreement among us, the depositary and the owners and holders of ADSs, sets out ADS holder rights, as well as the rights and obligations of the depositary.
51
The right as a holder of ADSs to participate in any future preferential subscription rights or to elect to receive dividends in shares may be limited, which may cause dilution to ADS holders.
According to French law, if we issue additional securities for cash, current shareholders will have preferential subscription rights for these securities on a pro rata basis unless they waive those rights at an extraordinary meeting of our shareholders (by a two-thirds majority vote) or individually by each shareholder. However, ADS holders in the United States will not be entitled to exercise or sell such rights unless we register the rights and the securities to which the rights relate under the Securities Act of 1933, as amended (“Securities Act”), or an exemption from the registration requirements is available. In addition, the deposit agreement provides that the depositary will not make rights available to ADS holders unless the distribution to ADS holders of both the rights and any related securities are either registered under the Securities Act or exempted from registration under the Securities Act. Further, if we offer holders of our ordinary shares the option to receive dividends in either cash or shares, under the deposit agreement the depositary may require satisfactory assurances from us that extending the offer to holders of ADSs does not require registration of any securities under the Securities Act before making the option available to holders of ADSs. We are under no obligation to file a registration statement with respect to any such rights or securities or to endeavor to cause such a registration statement to be declared effective. Moreover, we may not be able to establish an exemption from registration under the Securities Act. Accordingly, ADS holders may be unable to participate in our rights offerings or to elect to receive dividends in shares and may experience dilution in their holdings. In addition, if the depositary is unable to sell rights that are not exercised or not distributed or if the sale is not lawful or reasonably practicable, it will allow the rights to lapse, in which case you will receive no value for these rights.
ADS holders may be subject to limitations on the transfer of their ADSs and the withdrawal of the underlying ordinary shares.
ADSs, which may be evidenced by ADRs, are transferable on the books of the depositary. However, the depositary may close its books at any time or from time to time when it deems expedient in connection with the performance of its duties. The depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary think it is advisable to do so because of any requirement of law, government or governmental body, or under any provision of the deposit agreement, or for any other reason subject to a holder of ADSs’ right to cancel his or her ADSs and withdraw the underlying ordinary shares. Temporary delays in the cancellation of ADSs and withdrawal of the underlying ordinary shares may arise because the depositary has closed its transfer books or we have closed our transfer books, the transfer of ordinary shares is blocked to permit voting at a shareholders’ meeting or we are paying a dividend on our ordinary shares. In addition, a holder of ADSs may not be able to cancel his or her ADSs and withdraw the underlying ordinary shares when he or she owes money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
As a foreign private issuer, we are exempt from a number of rules under the U.S. securities laws and are permitted to file less information with the SEC than a U.S. company. This may limit the information available to holders of ADSs.
We are a foreign private issuer, as defined in the SEC’s rules and regulations and, consequently, we are not subject to all of the disclosure requirements applicable to public companies organized within the United States. For example, we are exempt from certain rules under the Exchange Act that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act, including the U.S. proxy rules under Section 14 of the Exchange Act. In addition, our officers and directors are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, while we currently make annual and semi-annual filings in France and the United States, we are not required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. public companies and are not required to file quarterly reports on Form 10-Q or current reports on Form 8-K under the Exchange Act. Accordingly, there is less publicly available information concerning our company than there would be if we were not a foreign private issuer.
52
As a foreign private issuer, we are permitted to follow certain home country practices in relation to corporate governance matters that differ significantly from Nasdaq’s corporate governance standards. These practices may afford less protection to shareholders than they would enjoy if we complied fully with the corporate governance standards of Nasdaq.
As a foreign private issuer listed on the Nasdaq Capital Market, we are subject to Nasdaq’s corporate governance standards. However, Nasdaq rules provide that foreign private issuers are permitted to follow home country corporate governance practices in lieu of Nasdaq’s corporate governance standards as long as notification is provided to Nasdaq of the intention to take advantage of such exemptions. We rely on exemptions for foreign private issuers and follow French corporate governance practices in lieu of Nasdaq’s corporate governance standards, to the extent possible. Certain corporate governance practices in France, which is our home country, differ significantly from Nasdaq corporate governance standards. For example, as a French company, neither the corporate laws of France nor our bylaws require a majority of our directors to be independent and we can include non-independent directors as members of our remuneration committee, and our independent directors are not required to hold regularly scheduled meetings at which only independent directors are present.
We are also exempt from provisions set forth in Nasdaq rules which require an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting stock. Consistent with French law, our bylaws provide that a quorum requires the presence of shareholders having at least (1) 20% of the shares entitled to vote in the case of an ordinary shareholders’ general meeting or at an extraordinary shareholders’ general meeting where shareholders are voting on a capital increase by capitalization of reserves, profits or share premium, or (2) 25% of the shares entitled to vote in the case of any other extraordinary shareholders’ general meeting.
As a foreign private issuer, we are required to comply with certain Nasdaq rules and Rule 10A-3 of the Exchange Act, relating to audit committee composition and responsibilities. However, under French law, the audit committee may only have an advisory role and appointment of our statutory auditors, in particular, must be decided by the shareholders at our annual meeting.
Therefore, our shareholders may be afforded less protection than they otherwise would have under Nasdaq’s corporate governance standards applicable to U.S. domestic issuers.
We may lose our foreign private issuer status in the future, which could result in significant additional cost and expense.
While we currently qualify as a foreign private issuer, the determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter and, accordingly, the next determination will be made with respect to us on June 30, 2024. In the future, we would lose our foreign private issuer status if we fail to meet the requirements necessary to maintain our foreign private issuer status as of the relevant determination date. We will remain a foreign private issuer until such time that more than 50% of our outstanding voting securities are held by U.S. residents and any of the following three circumstances applies: (i) the majority of our executive officers or directors are U.S. citizens or residents; (ii) more than 50% of our assets are located in the United States; or (iii) our business is administered principally in the United States.
The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic issuer may be significantly more than costs we incur as a foreign private issuer. If we are not a foreign private issuer, we will be required to file periodic reports and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive in certain respects than the forms available to a foreign private issuer. We would be required under current SEC rules to prepare our financial statements in accordance with U.S. GAAP, rather than IFRS, and modify certain of our policies to comply with corporate governance practices associated with U.S. domestic issuers. Such conversion of our financial statements to U.S. GAAP would involve significant time and cost. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements on U.S. stock exchanges that are available to foreign private issuers such as the ones described herein and exemptions from procedural requirements related to the solicitation of proxies.
53
We are an “emerging growth company” under the JOBS Act and will be able to avail ourselves of reduced disclosure requirements applicable to emerging growth companies, which could make the ADSs less attractive to investors.
We are an “emerging growth company,” as defined in the U.S. Jumpstart Our Business Startups Act of 2012, or the JOBS Act, and we take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies,” including not being required to comply with the auditor attestation requirements of Section 404(b) of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved.
We cannot predict if investors will find the ADSs less attractive because we rely on these exemptions. If some investors find the ADSs less attractive as a result, there may be a less active trading market for the ADSs and the price of the ADSs may be more volatile. We may take advantage of these reporting exemptions until we are no longer an emerging growth company. We will remain an emerging growth company until the earliest of (i) the last day of the fiscal year in which we have total annual gross revenue of $1.235 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the closing date of our U.S. initial public offering (i.e., December 31, 2026); (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; and (iv) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
U.S. holders of ADSs may suffer adverse tax consequences if we are characterized as a passive foreign investment company.
Generally, if, for any taxable year, at least 75% of our gross income is passive income, or at least 50% of the value of our assets is attributable to assets that produce passive income or are held for the production of passive income, including cash, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. For purposes of these tests, passive income includes dividends, interest, and gains from the sale or exchange of investment property and rents and royalties other than rents and royalties which are received from unrelated parties in connection with the active conduct of a trade or business. If we are characterized as a PFIC, U.S. holders of the ADSs may suffer adverse tax consequences, including having gains realized on the sale of the ADSs treated as ordinary income, rather than capital gain, the loss of the preferential rate applicable to dividends received on the ADSs by individuals who are U.S. holders, and having interest charges apply to certain distributions by us and the proceeds of sales of the ADSs. See Item 10.E “Taxation” of this annual report for additional details.
Our status as a PFIC will depend on the composition of our income (including whether we receive certain non-refundable grants or subsidies and whether such amounts and reimbursements of certain refundable research tax credits will constitute gross income for purposes of the PFIC income test) and the composition and value of our assets, which may be determined in large part by reference to the market value of the ADSs, which may be volatile, from time to time. Based on the composition of our gross income, assets, activities, and market capitalization in 2022, and on reasonable assumptions, we believe that we were not a PFIC for our taxable year ending December 31, 2023. However, there can be no assurance that we were not a PFIC for our taxable year ending December 31, 2023, or that the IRS will agree with any position we take regarding our PFIC status for such taxable year. There also can be no assurance that we will not be a PFIC for our current taxable year ending December 31, 2024 or for any future taxable year, because our PFIC status is a factual determination made annually after the end of each taxable year. Our U.S. counsel expresses no opinion and prospective investors should consult their own tax advisors regarding our PFIC status.
Investments in our securities may be subject to prior governmental authorization under the French foreign investment control regime.
Pursuant to the provisions of the French Monetary and Financial Code (code monétaire et financier), any investment by any non-French citizen, any French citizen not residing in France, any non-French entity or any French entity controlled by one of the aforementioned persons or entities that will result in the relevant investor (a) acquiring control of an entity registered in France, (b) acquiring all or part of a business line of an entity registered in France, or (c) for non-EU or non-EEA investors crossing, directly or indirectly, alone or in concert, a 25% threshold of voting rights in an entity registered in France, in each case, conducting activities in certain strategic industries, such as the industry in which we operate, is subject to the prior authorization of the French Ministry of Economy, which authorization may be conditioned on certain undertakings.
The Decree (décret) n°2020-892 dated July 22, 2020 as modified by the Decree (décret) n°2020-1729 dated December 28, 2020 and the Decree (décret) n°2021 1758 dated December 22, 2021, has created, until December 31, 2022, a new 10% threshold of the voting rights for the non-European investments in listed companies, in addition to the 25% abovementioned threshold.
54
The foreign investment control regime described above applies to companies engaged in activities essential to protecting public health as well as biotechnology-related research and development activities.
Therefore, any investor meeting the above criteria willing to acquire all or part of our business with the effect of crossing the applicable share capital thresholds set forth by the French Monetary and Financial Code will have to request this prior governmental authorization before acquiring our ordinary shares or ADSs. We cannot guarantee that such investor will obtain the necessary authorization in due time. The authorization may also be granted subject to conditions that deter a potential purchaser. The existence of such conditions to an investment in our securities could have a negative impact on our ability to raise the funds necessary to our development. In addition, failure to comply with such measures could result in significant consequences for the investor (including the investment to be deemed null and void). Such measures could also delay or discourage a takeover attempt, and we cannot predict whether these measures will result in a lower or more volatile market price of our ADSs or ordinary shares.
We had identified material weaknesses in our internal control over financial reporting as of December 31, 2020, 2021 and 2022 that were remediated as of December 31 2023. Nevertherless a new material weakness was identified in connection with preparing our 2023 consolidated financial statements, relating to a lack of sufficient competent financial reporting and accounting personnel with appropriate understanding of IFRS Accounting Standards to design and implement formal period-end financial reporting controls and procedures and to address complex IFRS Accounting Standards technical accounting issues. We are implementing control procedures to mitigate this risk, but if we are still unable to establish and maintain an effective system of internal control over financial reporting in this matter, we may not be able to accurately report our financial results in a timely manner, which may adversely affect investor confidence in us and materially and adversely affect our business and operating results.
In connection with our fiscal 2020 and 2021 audits, we identified material weaknesses in our internal control over financial reporting related to our failure to correctly apply IFRS 9, “Financial Instruments”, to the fair value assessment of our convertible notes, and its related interpretations and rules with respect to their accounting treatment and IFRS 13, Fair Value Measurement, to the accounting treament of our convertible and non-convertible notes and the valuation of our loan agreement and bonds agreement signed with Kreos on November 19, 2021. A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected and corrected on a timely basis. In response to the material weaknesses described above, our management implemented a remediation plan, which it believes remediates the material weaknesses that had been identified.
In connection with our fiscal 2023 audit, we identified a material weakness in our internal control over financial reporting related to a lack of sufficient competent financial reporting and accounting personnel with appropriate understanding of IFRS Accounting Standards to design and implement formal period-end financial reporting controls and procedures and to address complex IFRS Accounting Standards technical accounting issues. In response to the material weaknesses described above, our management is implementing a remediation plan, which it believes will remediate the material weaknesses that have been identified.
We cannot assure that the measures we have taken to date and may take in the future, will be sufficient to remediate the control deficiencies that led to our material weaknesses in internal control over financial reporting or that we will prevent or avoid potential future material weaknesses. Effective internal controls are necessary for us to provide reliable financial reports. These remediation measures may be time consuming and costly and there is no assurance that these initiatives will ultimately have the intended effects.
If we identify any new material weaknesses in the future, any such newly identified material weaknesses could limit our ability to prevent or detect a misstatement of our accounts or disclosures that could result in a material misstatement of our annual or interim financial statements. In such case, we may be unable to maintain compliance with securities law requirements regarding timely filing of periodic reports in addition to applicable stock exchange listing requirements, investors may lose confidence in our financial reporting and our stock price may decline as a result. We cannot assure you that the measures we have taken to date, or any measures we may take in the future, will be sufficient to avoid potential future material weaknesses.
We must establish and maintain effective internal control over financial reporting, and if we are unable to do so, the accuracy and timeliness of our financial reporting may be adversely affected, which could hurt our business, lessen investor confidence and depress the market price of our securities.
We must establish and maintain effective internal control over financial reporting in order to accurately and timely report our results of operations and financial condition. In addition, as a public company listed in the United States, the Sarbanes-Oxley Act
55
requires, among other things, that we assess the effectiveness of our internal control over financial reporting at the end of each fiscal year.
The rules governing the standards that must be met for our management to assess our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act are complex and require significant documentation, testing and possible remediation. These stringent standards require that our audit committee be advised and regularly updated on management’s review of internal control over financial reporting. This process is time-consuming, costly, and complicated.
Our current controls and any new controls that we develop may become inadequate because of changes in conditions in our business. Further, additional weaknesses in our disclosure controls and internal control over financial reporting may be discovered in the future. Any failure to develop or maintain effective controls or any difficulties encountered in their implementation or improvement could harm our results of operations or cause us to fail to meet our reporting obligations and may result in a restatement of our financial statements for prior periods. Any failure to implement and maintain effective internal control over financial reporting also could adversely affect the results of periodic management evaluations and annual independent registered public accounting firm attestation reports regarding the effectiveness of our internal control over financial reporting that we will eventually be required to include in our periodic reports that will be filed with the SEC. Ineffective disclosure controls and procedures and internal control over financial reporting could also cause investors to lose confidence in our reported financial and other information, which would likely have a negative effect on the trading price of our shares. In addition, if we are unable to continue to meet these requirements, we may not be able to remain listed on the Nasdaq Capital Market.
Our independent registered public accounting firm is not required to formally attest to the effectiveness of our internal control over financial reporting until after we are no longer an emerging growth company as defined in the JOBS Act. At such time, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied with the level at which our internal control over financial reporting is documented, designed, or operating. Any failure to maintain effective disclosure controls and internal control over financial reporting could have an adverse effect on our business and results of operations and could cause a decline in the price of our shares.
Item 4. Information on the Company.
A. | History and Development of the Company |
We were incorporated as a société anonyme (“SA”), on September 27, 2006. We are registered at the Paris Registre du Commerce et des Sociétés under the number 492 002 225. Our principal executive offices are located at Sorbonne University—BC 9, Bâtiment A 4ème étage, 4 place Jussieu 75005 Paris, France and our telephone number is +33 1 44 27 23 00. Our website address is www.biophytis.com. Our agent for service of process in the United States is Puglisi & Associates, 850 Library Avenue, Suite 204, Newark, Delaware 19711. The reference to our website is an inactive textual reference only and the information contained in, or that can be accessed through, our website is not a part of this annual report.
Our actual capital expenditures for the years ended December 31, 2021, 2022 and 2023 amounted to €844 thousand, €412 thousand and €497 thousand, respectively. These capital expenditures primarily consisted of patents rights acquired from our CEO (€270 thousand in 2021, €90 thousand in 2022, €180 thousand in 2023) and rights of use for two scientific equipments (€271 thousand in 2023) recorded in accordance with IFRS 16 Leases. To date, we have expensed all research and development costs as incurred, as we do not currently meet the conditions to capitalize expenditures on drug development activities, as provided in IAS 38 Intangible Assets. Our research and development costs for the years ended December 31, 2021, 2022 and 2023 amounted to €19,665 thousand, €16,059 thousand and €9,503 thousand, respectively. These research and development costs primarily consisted of expenses incurred in connection with the development of our drug candidates such as personnel-related costs, expenses incurred under our agreements with CROs, clinical sites, contract laboratories and costs of acquiring preclinical study and clinical trial materials. We expect our capital expenditures and research and development costs to remain significant as we continue our research and development efforts and advance the clinical development of BIO101 (20-hydroxyecdysone) and BIO201, in the United States, Europe and elsewhere. We anticipate our capital expenditures and research and development costs in 2024 to be financed from our existing cash and cash equivalents, as well as from the funding line of convertible notes set up with ATLAS. For the near future, our investments will mainly remain in France where our research and development facilities are currently located.
The SEC maintains an Internet site that contains reports, proxy information statements and other information regarding issuers that file electronically with the SEC. The address of that site is http://www.sec.gov. Our website address is www.biophytis.com. The reference to our website is an inactive textual reference only and information contained in, or that can be accessed through, our website or any other website cited in this annual report is not part of this annual report.
56
B. | Business Overview |
Overview
We are a clinical-stage biotechnology company focused on the development of therapeutics that are aimed at slowing the degenerative processes associated with aging and improving functional outcomes for patients suffering from age-related diseases. Our goal is to become a leader in the emerging field of aging science by delivering life-changing therapies to the growing number of patients in need. To accomplish this goal, we have assembled an experienced and skilled group of industry professionals, scientists, clinicians and key opinion leaders from leading industry and academic institutions around the world.
A number of degenerative diseases associated with aging have been characterized in the last century, including sarcopenia and AMD. The pathophysiology of these and many other age-related diseases is not yet well understood, and effective treatment options are lacking. The global population of people over the age of 60 is expected to double from approximately 962 million in 2017 to 2.1 billion by 2050, according to estimates from the United Nations’ World Population Prospects: the 2017 Revision. We believe that the need for effective therapeutics for age-related diseases will continue to grow throughout the 21st century. In addition, healthcare costs, including costs associated with treatments and long-term care for age-related diseases associated with this demographic shift, are expected to rise proportionally, as effective treatment options are currently lacking. We believe that developing treatments to slow disease progression and reduce the risk of severe disability associated with age-related diseases is of the utmost importance.
As we age, our physical, respiratory, visual and cognitive performances gradually decline due, in part, to the cumulative deleterious effect of multiple biological and environmental stresses, including current and emerging viral infections, to which we are exposed during our lifetime. The functional decline can be much faster in some individuals as a consequence of, among other things, the degenerative processes affecting specific cells, tissues and organs. Through evolution, cells, tissues and organisms have developed natural means or pathways to counteract and balance the effects of the many stresses they face. This natural ability to compensate for stress and remain functional, called biological resilience, degrades over time. The decline in biological resilience contributes to the acceleration of these degenerative processes and the impairment of functional performances, which, in turn, can lead to severe disability, reduced health-span and ultimately death. This occurs as we age, but can occur at a younger age, when genetic mutations exist, or in the case of infection and inflammation.
Our lead drug candidate, BIO101 (20-hydroxyecdysone), formerly known as Sarconeos (BIO101), is a plant-derived pharmaceutical-grade purified 20-hydroxyecdysone that is an orally administered small molecule.
The initial indication we were seeking approval for is sarcopenia, an age-related degeneration of skeletal muscle, which is characterized by a loss of muscle mass, strength and function in elderly people (adults 65 years of age and older) leading to reduced mobility, or mobility disability, and increased risk of adverse health events and hospitalization, and potential death resulting from falls, fractures, and physical disability. There is currently no approved medication for sarcopenia, which is present in the elderly (greater than 65 years old) with an estimated prevalence range between six to 22% worldwide.
An additional indication we are seeking is to reduce muscle strength loss from GLP-1 agonists in combination with dieting, in adult obese or overweight patients who are treated with semaglutide or liraglutide for weight loss. There are currently no approved therapies for this indication, and it is known that GLP-1 agonists cause an important muscle mass loss as component of the weight loss induced by these treatments.
BIO101 (20-hydroxyecdysone) has also been developed to treat patients with severe respiratory manifestations of COVID-19. We have conducted the COVA study, a global, multicenter, double-blind, placebo-controlled, group-sequential, and adaptive two-part Phase 2-3 study, in patients with SARS-CoV-2 pneumonia. Final results of this study were released on February 2, 2023. The study met its pre-defined primary endpoint demonstrating a statistically significant difference between BIO101 (20-hydroxyecdysone) and placebo in the proportion of patients with respiratory failure or early death at day 28, representing a relative reduction of risk of 44% (p=0.043, Cochran-Mantel-Haenszel test). Moreover, the analysis of time to respiratory failure or early death showed significant differences over 28 days in the Kaplan Meier curves for BIO101 (20-hydroxyecdysone) versus placebo (p=0.022). The pre-specified analysis of time to death over the complete follow-up period over 90 days showed that mortality rate with BIO101 (20-hydroxyecdysone) was reduced compared to placebo in the ITT population (p=0.083) and in the PP population (p=0.038).
Most people infected with the COVID-19 virus and its variants experience mild to moderate respiratory illness and recover without requiring special treatment. Older people, and those with underlying medical problems like cardiovascular disease, diabetes,
57
chronic respiratory disease and cancer are more likely to develop serious illness and to be at risk of respiratory failure. Based on the positive data of COVA Phase 2-3 study, we plan to implement Early Access Programs (EAP) in selected countries and notably to reactivate our EAP in Brazil, as we did initially receive approval for such a program in January 2022. We are also discussing with regulatory authorities in the US and Europe to define market access for BIO101 (20-hydroxyecdysone) in COVID-19 and possibly other respiratory viral infections such as influenza.
We are also developing BIO101 (20-hydroxyecdysone) for DMD, a rare genetic neuromuscular disease in male children and young adults, which is characterized by an accelerated degeneration of muscle and is responsible for a loss of mobility, respiratory failure and cardiomyopathy, leading to premature death. There is currently no cure and limited treatment options for DMD, which affects approximately 2.8 out of 100,000 people worldwide (approximately 20,000 new cases annually worldwide), based on our estimates from publicly available information.
Our second drug candidate, BIO201, formerly known as BIO201, is an orally administered small molecule in development for the treatment of retinopathies. It is a plant-derived pharmaceutical-grade purified norbixin. We have completed preclinical cellular and animal studies of BIO201 for the treatment of retinopathies. While we are still in the early stages of development, we believe that the results from our preclinical studies support continued investigation into whether BIO201 may stimulate biological resilience and protect the retina against phototoxic damage that leads to vision loss. The initial indication we plan to seek approval for is dry AMD, a common eye disorder among people over the age of 50 that affects central vision, impairing functions such as reading, driving, and facial recognition, and has a major impact on quality of life and the ability to live independently. There are currently no approved treatments for dry AMD. Based on our estimates from publicly available information, AMD affects approximately 8.5% of the global population (ages 45 to 85) and is expected to increase over time as the population ages.
We are also exploring BIO201 as a potential treatment for Stargardt disease, which shares many of the characteristics of dry AMD. Stargardt disease is the most common form of inherited macular degeneration that typically develops in childhood and leads to vision loss and, in some cases, blindness.
We hold exclusive commercialization rights through licenses for each of our drug candidates. We currently plan to develop our drug candidates through clinical PoC (typically Phase 2), and then seek licensing and/or partnership opportunities for further clinical development through regulatory approval and commercialization.
We have developed our lead clinical drug candidate BIO101 (20-hydroxyecdysone), our preclinical drug candidate BIO201, and a preclinical pipeline of life-cycle extension products, consisting of BIO103 and BIO203, through a drug discovery platform in collaboration with Sorbonne University in Paris, France based on work with medicinal plants. Plants are major sources of small molecules, called secondary metabolites, which they produce as a defense mechanism to various environmental stresses, including attack from predatory and pathogenic species (e.g., insects, bacteria and fungi). Our drug discovery platform is based on a reverse pharmacology approach that tests a collection of bioactive secondary metabolites along with chemical analogs that we have synthesized in phenotypic screens of various age-related diseases. Our long-term goal is to advance the field of aging science with the continued discovery and development of new drug candidates that treat age-related diseases by stimulating biological resilience pathways that are involved in the aging process and/or age-related diseases.
We have assembled an executive team of scientific, clinical, and business leaders with broad expertise in biotechnology and clinical drug development (see Item 6.A for more information on our directors and senior management).
58
Our Clinical Pipeline
We are developing a portfolio of programs targeting biological resilience pathways that slow the degenerative processes associated with aging and improve functional outcomes for patients suffering from age-related diseases. Our current pipeline of drug candidates is illustrated below.
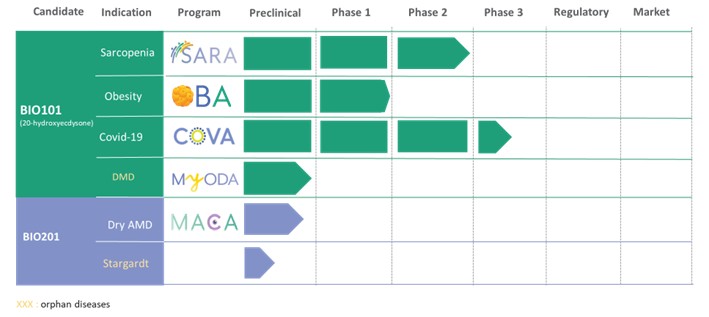
Our Strategy
We are focused on the development of therapeutics that improve functional outcomes for patients suffering from age-related diseases, including severe respiratory failure in patients suffering from respiratory viral infections such as COVID-19 or influenza. Our goal is to build Biophytis into a leading biotechnology company focused on targeting biological resilience pathways that slow the degenerative processes associated with age-related disease progression in order to improve the lives of millions of patients that have limited or no treatment options. We currently plan to develop our drug candidates and then seek licensing and/or partnership opportunities for further clinical development through regulatory approval and commercialization. To achieve our goal, we are pursuing the following strategies:
| ● | Obtain EAPs in Brazil and other selected countries and optimize market access in the US and Europe for BIO101 (20-hydroxyecdysone) for COVID-19 patients at risk of respiratory failure. We reported final analysis of the Phase 2/3 COVA study in hospitalized COVID-19 patients with severe respiratory manifestations in February 2023. The study met its pre-defined primary endpoint demonstrating a statistically significant difference between BIO101 (20-hydroxyecdysone) and placebo in the proportion of patients with respiratory failure or early death at day 28, representing a relative reduction of risk of 44% (p=0.043, Cochran-Mantel-Haenszel test). Based on these results, we are currently reactivating our EAP in Brazil and plan to apply for EAPs in other selected countries. We are also discussing with regulatory authorities in the US and Europe to define market access for BIO101 (20-hydroxyecdysone) in COVID-19 and possibly other respiratory viral infections such as influenza. |
| ● | Demonstrate efficacy of BIO101 (20-hydroxyecdysone) in sarcopenia. We published the topline data for our SARA-INT Phase 2b clinical trial in October 2021. Due to the effect of the pandemic on the patient population, only 45 percent of the study subjects were able to complete the study with end-of-treatment efficacy assessments and the study was underpowered to observe the hypothesized effect size, and the primary and secondary endpoints were not met. However, BIO101 (20-hydroxyecdysone) showed cleanically meaningful improvement in gait speed after 6 months of treatment, which is known to be associated with a reduction in mobility disability and mortality in elderly. Following intensive interactions with the regulatory authorities in Europe (EMA) and the United States (FDA), we have obtained the approval from the FDA and the Belgian regulatory authorities in the second half of 2023 to conduct a Phase 3 clinical, which will be the first Phase 3 trial ever launched in this indication. Additional funding or co-development agreements with pharmaceutical partners will be necessary to be able to conduct this study. |
59
| ● | Initiate clinical development of BIO101 (20-hydroxyecdysone) in obesity. We plan to start a development program with BIO101 (20-hydroxyecdysone) to reduce muscle strength loss from GLP-1 agonists in combination with dieting, in adult obese or overweight patients who are treated with semaglutide or liraglutide for weight loss. We plan to obtain approvals from regulatory authorities and ethics committees / Institutional Review Boards in Europe and the United States to start a proof-of-concept study in the second half of 2024. |
| ● | Initiate clinical development of BIO101 (20-hydroxyecdysone) in DMD. Our efforts are also focused on leveraging our knowledge and the development of BIO101 (20-hydroxyecdysone) in sarcopenia and COVID-19 to commence and advance the clinical development of BIO101 (20-hydroxyecdysone) for the treatment of non-ambulatory DMD patients with signs of respiratory deterioration, independent of genetic mutation and across the disease spectrum. We have already received an IND “may proceed” letter from the FDA in the United States and a CTA approval from FAMHP in Belgium. In the “may proceed” letter from the FDA, the FDA noted that it had significant concerns with the design of the study, and that the results of the study, as originally designed to enroll ambulatory and non-ambulatory patients and measure muscle function deterioration through a composite score, would not be capable of providing interpretable data sufficient to support a marketing application. In its letter, the FDA recommended that we revise the study population and primary endpoint. We have incorporated the agency’s recommendations and have revised the protocol to focus on non-ambulatory patients with signs of respiratory deterioration and changed the primary endpoint to respiratory function. The new protocol and study design will be discussed with Competent Authorities with the objective to start the study in 2024. |
| ● | Advance the development of our second drug candidate, BIO201. We are working on continuing the preclinical development of our second drug candidate, BIO201, for the treatment of retinopathies, with an initial focus on dry AMD. |
| ● | Maintain our presence in the United States to support co-development in Europe and the United States. Our goal is to continue to conduct our clinical trials in this country and, if successful, apply for regulatory approval in both the United States and Europe. We plan to work with patient associations, key experts, regulatory agencies, government and third-party payors and other key constituencies in both regions. |
| ● | Expand our pipeline and explore potential strategic partnerships and alliances to maximize the value of our development programs. We plan to continue to leverage our collaborations with leading scientific and academic institutions in order to pursue new INDs for our existing drug candidates, including BIO101 (20-hydroxyecdysone), BIO103, and BIO201, as well as BIO203. We believe that our drug candidates may be applicable for additional age-related disease research and potential application. We plan to explore the commercial potential of our drug candidates after establishing clinical PoC through Phase 2/3. |
Our Drug Candidates
BIO101 (20-hydroxyecdysone)
Our lead drug candidate, BIO101 (20-hydroxyecdysone), is an orally administered small molecule in development for the treatment of neuromuscular diseases. We have completed preclinical studies and are in various stages of further clinical development for the treatment of neuromuscular diseases. While preclinical studies provide limited data, based on results from our cellular and animal studies, we believe BIO101 (20-hydroxyecdysone) stimulates biological resilience through activation of the MAS Receptor, which may preserve muscle strength, mobility and respiratory function in various age-related conditions.
In addition, MAS activation could potentially counter the deleterious effects of the SARS-CoV -2 infection. Data from models of ALI suggest a further protective role of BIO101 (20-hydroxyecdysone) on the pulmonary tissue. In light of this, we began investigating BIO101 (20-hydroxyecdysone) in the COVA study in patients with severe respiratory manifestations of COVID-19 as an initial potential indication for BIO101 (20-hydroxyecdysone). The enrollment for this study ended April 7, 2022, earlier than planned, due to the inability to recruit sufficient patients in a suitable timeframe as a result of the evolution of the pandemic. It is now less common for patients with COVID-19 disease to be admitted to hospitals for respiratory failure, due to the combined effect of high vaccination rates, high numbers of patients becoming immune through prior infection, the use of new antiviral drugs and the milder symptoms associated with the predominant Omicron variant.
Another indication we are developing is sarcopenia, an age-related degeneration of skeletal muscle, which is characterized by a loss of muscle mass, strength, function and mobility disability, and increased risk of adverse health events and potential death
60
resulting from falls, fractures, and physical disability. There is currently no approved prescription for sarcopenia, which is highly prevalent in the elderly (adults 65 years of age and older) with an estimated prevalence between six to 22% worldwide.
An additional indication we are seeking is to reduce muscle strength loss from GLP-1 agonists in combination with dieting, in adult obese or overweight patients who are treated with semaglutide or liraglutide for weight loss. There are currently no approved therapies for this indication and it is known that GLP-1 agonists cause an important muscle mass loss as component of the weight loss induced by these treatments.
We are also developing BIO101 (20-hydroxyecdysone) for DMD, the most common form of muscular dystrophy in children leading to early mortality. We are focusing on non-ambulatory patients with signs of respiratory deterioration.
History and Development of BIO101 (20-hydroxyecdysone)
In collaboration with Sorbonne University in Paris, France, we began our drug discovery efforts with a class of plant secondary metabolites called phytoecdysteroids, which are produced by plants to protect against insect attack. Phytoecdysteroids are analogs of the insect molting hormones ecdysone, which protects the plants by acting as endocrine disrupters and/or feeding deterrent. Phytoecdysteroids are found in various medicinal plants throughout the world and are used in traditional medicines as tonics or anti-diabetics.
We utilized a reverse pharmacology approach starting with phenotypic screens of a collection of phytoecdysteroids that had been gathered for over 30 years by scientists from Sorbonne University, along with chemical analogs that we have synthesized for their ability to stimulate protein synthesis in muscle cells. We selected 20-hydroxyecdysone for clinical development based on its safety profile, pharmacological activity and potential in maintaining key muscle functions, including mobility and strength. This compound was tested in animal models submitted to different stresses, including metabolic stress (high fat dieting or diabetic models), age-related stress (sarcopenia and disuse models), and genetic-related stress (DMD and Spinal Muscular Atrophy models). We are also testing the compound for infectious-related disease stress (COVID-19). Once pharmacological effects were detected, we identified the molecular target(s) and potential mechanism-of-action.
Potential Mechanism-of-Action
The MAS Receptor, the protective arm of the Renin-Angiotensin System (RAS)
Our preclinical studies demonstrate that BIO101 (20-hydroxyecdysone) activates the MAS Receptor in muscle cells, a key component of the RAS. The RAS is a fundamental endocrine system that is known to control fluid balance and blood pressure, playing a key role in cardio-vascular function. It is also involved in the regulation of smooth, cardiac and skeletal muscle metabolism, and plays a key role in muscle function and mobility in disease states. It is made up of two different arms that counter-regulate each other: (i) the “classical” arm (or ACE / angiotensin-II (Ang-II) / Ang-II receptor type 1 (AT1R) axis), and (ii) the “protective” arm (or ACE2 / angiotensin 1-7 (Ang-1-7) / MAS Receptor axis). Ang-II blood concentration has been shown to be increased with aging and in various neuromuscular diseases, such as sarcopenia and respiratory diseases that are caused by viruses such as SARS-CoV-2. Ang 1-7, the endogenous ligand of the MAS Receptor, opposes the numerous actions of Ang-II on muscle and cardio respiratory functions.
We believe BIO101 (20-hydroxyecdysone), through the activation of the MAS Receptor, triggers two key downstream signaling-pathways: (i) the P13K/AKT/mTOR pathway, or the AKT pathway, which is known to be responsible for increasing protein synthesis, (ii) the AMPK/ACC pathway, or the AMPK pathway, which is known to be involved in stimulating energy production. We have demonstrated that BIO101 (20-hydroxyecdysone) activates major signaling pathways such as the AKT pathway and potentially the AMPK pathway in C2C12 myotubes and human muscle cells through western blot analysis. The AKT pathway and AMPK pathway have all been shown to be impaired in muscle wasting conditions.
61
The potential mechanism-of-action through activation of the MAS Receptor is illustrated in the diagram below:
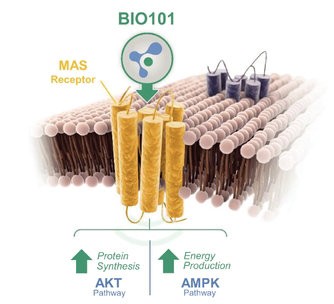
We believe that the AKT and AMPK pathway are potentially the key factors for (i) preserving muscle mass and increasing muscle strength under muscle wasting conditions and (ii) increasing muscle strength and improved endurance, respectively. We have also observed in preclinical studies that activation of the MAS Receptor by BIO101 (20-hydroxyecdysone) shares many common properties with Ang-1-7 at the cellular level. However, BIO101 (20-hydroxyecdysone) did not show an effect on blood pressure or heart rate when compared to enalapril, an angiotensin-converting enzyme, or ACE, inhibitor.
The activation of MAS Receptor is thought to be a key component of the cardio-respiratory function. When it comes to COVID-19, SARS-CoV-2 infection, by down-regulation of ACE2 expression and activity, reduces the conversion of Ang-II to Ang-1-7 resulting in excessive levels of Ang-II. This imbalance between the “classical” and “protective” arms of the RAS due to excessive activation of AT1R and limited activation of MAS Receptor which explain some of the observations in clinical practice reported in COVID-19 patients. Therefore, we believe that restoration of the balance of the RAS, by directly activating MAS Receptor downstream of ACE2, would be a particularly relevant avenue to treat patients infected with SARS-CoV-2.
62
Preclinical proofs of concept
Effect on myocyte differentiation into myotubes (in vitro)
Our preclinical data in C2C12 cell lines and human cell models suggest that BIO101 (20-hydroxyecdysone) enlarges myotubes, the main structural units of muscle, warranting continued research. We believe that this is important for limiting muscle mass loss and increasing muscle strength under muscle wasting conditions. As depicted below, results from an in vitro study demonstrate that human myotubes are larger in muscle cells treated with BIO101 (20-hydroxyecdysone) as compared to untreated control cells.
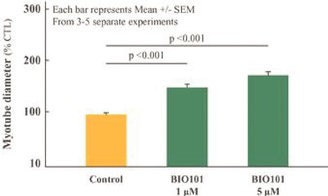
Effect of BIO101 (20-hydroxyecdysone) on mean myotube diameter
We believe BIO101 (20-hydroxyecdysone) directly targets muscle tissue and cells, and improves several key muscle cell functions, including protein synthesis, regeneration and energy production through key signaling pathways that are impaired in muscle wasting conditions, regardless of the disease stage, state of disease progression or severity, and may have the potential to improve muscle function and preserve strength, mobility and respiratory capacity in various neuromuscular diseases, independent of cause (i.e., age-related or genetic) and pathophysiology.
Preclinical Development of BIO101 (20-hydroxyecdysone) in Sarcopenia.
We conducted numerous in vivo experiments in C57Bl/6J mouse models to assess the activity of BIO101 (20-hydroxyecdysone) within the context of aging, specifically studying a high fat diet and immobilization. Key in vivo results are summarized below.
Beneficial effect on mobility in mice. We administered BIO101 (20-hydroxyecdysone) at 50 mg/kg/day or a placebo to “old” mice (22 months old at the beginning of the study) that were fed a high-fat diet over 14 weeks. The mice were exercised on a treadmill and maximum running velocity (Vmax) was recorded after 14 weeks of treatment. Untreated “adult” mice (12 months old at the beginning of the study) were also fed a high-fat diet and exercised similarly to determine a positive control velocity. As shown in the graph below, “old” control mice had a Vmax that was approximately 21% less than “adult” control mice (p<0.001) demonstrating the effects of aging. Further, results showed that “old” mice treated with BIO101 (20-hydroxyecdysone) demonstrated a significant improvement in Vmax as compared to “old” control mice (p<0.01), compensating almost completely for the loss of mobility due to aging. These results were presented in December 2016 at the Society on sarcopenia, Cachexia and Wasting Disorders, or (“SCWD”), conference in Berlin, Germany.
63
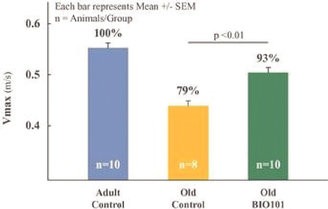
Effect of chronic BIO101 (20-hydroxyecdysone) treatment over
14 weeks on maximum running velocity in old mice
Preservation of muscle strength after immobilization in mice. To model muscle wasting associated with impaired mobility, we immobilized young mice (13 weeks old) and began administering either BIO101 (20-hydroxyecdysone) at 50 mg/kg/day or a placebo control (vehicle). After 14 days, we removed the immobilization and continued administration of BIO101 (20-hydroxyecdysone) for an additional 14 days. The absolute strength of hind limb muscle was recorded at various times over the 28-day period. As shown in the graph below, mice treated with BIO101 (20-hydroxyecdysone) demonstrated a preservation of muscle strength while immobilized compared to vehicle control. We believe these results support continued research to investigate whether BIO101 (20-hydroxyecdysone) could be an effective treatment to preserve muscle function under conditions of disuse or immobility.
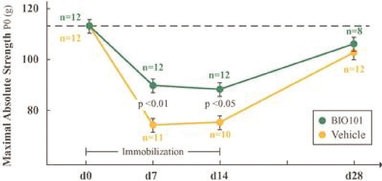
Effect of chronic BIO101 (20-hydroxyecdysone) treatment over 28 days
on maximal absolute strength in hind limb-immobilized mice
64
Preclinical Development of BIO101 (20-hydroxyecdysone) in Obesity.
RAS and Obesity
The renin angiotensin system plays a critical role in the pathogenesis of obesity, notably, the harmful arm of the RAS. Ang II is intimately linked to obesity and its pro-inflammatory effects are involved in obesity-associated co-morbidities. Indeed, Ang II is one of the major pro-inflammatory adipokines produced in obese adipose tissue that may be critical in linking obesity, insulin resistance, and inflammation (Kalupahana et al., 2012; Yvan-Charvet & Quignard-Boulangé, 2011). In humans, plasma Ang II level is correlated with body weight, decreases during weight loss, and is associated with markers of insulin resistance in obese subjects. Conversely, the beneficial effect on obesity and adipose function of Ang-(1–7), which functionally antagonizes angiotensin II through its Mas receptor, has been well elucidated using various rodent models (Santos et al., 2012; Santos et al., 2014). Recent evidence suggests that increasing the activity of the ACE2/Ang-(1-7)/MasR axis in obese animal models leads to significant reductions in body weight (Bruce et al., 2018). Altogether, these results demonstrate that therapeutic strategies/interventions decreasing the production and action of Ang II and/or increasing the protective arm of RAS could be of interest and may improve the clinical conditions in individuals with obesity as described below with BIO101 (20-hydroxyecdysone).
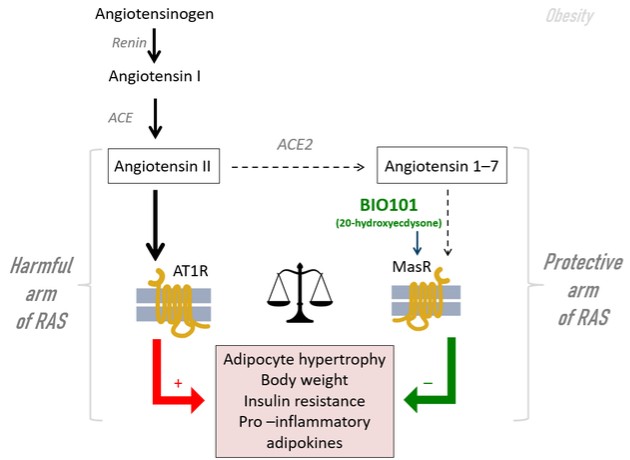
BIO101 (20-hydroxyecdysone) beneficial effects on muscle and on lipid metabolism
BIO101 (20-hydroxyecdysone) activates MasR on the protective arm of renin angiotensin system (Lafont et al. 2021). A clear dose-dependent target engagement was demonstrated on protein synthesis, differentiation in skeletal muscle cells because of the activation of MasR and downstream signaling pathways such as PI3/Akt/mTOR and AMPK (Serova et al. 2023). Biophytis studies demonstrate hypolipidemic effects, showing the protective effect of BIO101 (20-hydroxyecdysone) in mice fed an obesity-inducing high-fat diet (Foucault et al., 2012). BIO101 (20-hydroxyecdysone) food supplementation in these mice prevents the development of adipose tissue by limiting the size of adipocytes and reducing the expression of genes involved in lipid storage, such as lipoprotein lipase and phosphoenolpyruvate carboxykinase (Foucault et al., 2012). Subsequently, this anti-obesity effect of BIO101 (20-hydroxyecdysone) was associated with an overall increase in energy expenditure in these mice, with preferential use of carbohydrate metabolism over oxidation to the detriment of lipogenesis, and a decrease in intestinal absorption of dietary lipids leading to a reduction in their storage in adipose tissue (Foucault et al., 2014).
65
Cross talk between BIO101 (20-hydroxyecdysone) and GLP-1 receptor agonists mechanisms of action: rationale of potential synergism. GLP-1 receptor agonists (GLP-1RA) protect skeletal muscle against obesity-induced muscle atrophy via the SIRT1 pathway (Xiang et al., 2023). In addition, GLP-1RA were shown to have a potent regulatory effect in the RAS biological actions (Mastoor et al. 2022; Seghieri et al. 2018). They display an antagonistic effect on the ACE/AngII/AT1R axis and a positive impact on the ACE2/Ang(1-7)/MasR axis (Yang et al. 2020), indicating that they could play a dual regulatory role on the two arms of the RAS. More specifically, liraglutide activates the ACE2/Ang1-7/Mas axis by upregulating ACE2 and MasR expression. Intracellular transduction of these effects through the PI3K/AKT pathway (Yang et al. 2020) result in increased fatty acid oxidation gene expression, suppression of gluconeogenesis, and inflammation in the liver. This demonstrates that the activation of the RAS protective arm is an attractive target to improve glucose homeostasis, lipid metabolism and energy balance (White et al. 2019). Most interestingly, a SIRT1 activation has been described following MasR activation using Ang1-7 (Olivera Andrade et al. 2014), demonstrating that a cross talk exists between the ACE2/Ang1-7/Mas axis and sirtuins. GLP-1RA’ and BIO101 (20-hydroxyecdysone)’s modes of action as well as the cross talk between the ACE2/Ang1-7/Mas axis and sirtuins are described in the figure below. Because of this cross talk and knowing that both SIRT1 and Akt pathways are promoting muscle cells differentiation and insulin sensitivity while they decrease muscle atrophy, we believe at Biophytis that the combination of the GLP-1RA and BIO101 (20-hydroxyecdysone) could result in synergistic beneficial effects on muscle function and body weight loss in obese patients.
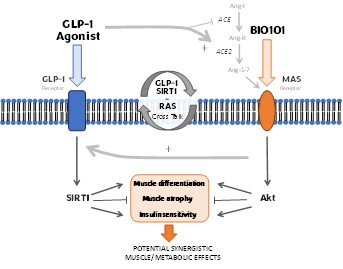
Cross talk between BIO101 (20-hydroxyecdysone) and GLP-1RA mechanisms of action
66
Preclinical Development of BIO101 (20-hydroxyecdysone) in COVID-19
ALI is acute lung injury caused by non-cardiogenic pathogenic factors and may develop to acute respiratory distress syndrome (“ARDS”) in severe cases. One of the important causes of ALI is virus infection that in some cases (including SARS-CoV-2) can deregulate the expression of RAS components by accelerating the imbalance of RAS and the occurrence and development of ALI/ARDS. Of particular interest, BIO101 (20-hydroxyecdysone)’s active principle ingredient (“API”) has shown lung anti-inflammatory and lung protective effects in various in vivo models of ALI known for being associated with severe RAS imbalance. A
2021 preclinical study revealed that BIO101 (20-hydroxyecdysone) daily treatment prevents respiratory function deterioration in SARS-CoV-2-infected mammals and provided a preclinical proof of concept for the concluding phase 2/3 COVA clinical study.
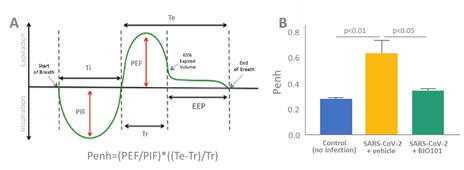
Effect of BIO101 (20-hydroxyecdysone) on SARS-CoV-2 infected hamsters
Enhanced pause (Penh) evaluation after BIO101 IP treatment of Sars-CoV-2-infected hamsters. As demonstrated in (A) above, Penh is a classically used and derived measure of respiratory distress. Penh is derived by assessing several measures of the respiratory response curve (peak expiratory flow of breath (PEF), peak inspiratory flow of breath (PIF), time of expiratory portion of breath (Te) and time required to exhale 65% of breath volume (Tr). EEP: End expiratory Pause. As noted in (B) above, this histogram shows Penh values of control group (not infected with SARS-CoV-2), infected with SARS-CoV-2 and treated with the vehicle (SARS-CoV-2 + vehicle) or infected with the SARS-CoV-2 and treated with BIO101 IP (SARS-CoV-2 + BIO101) with * p <0.05, and ** p <0.01. This data was referenced at at the European Congress of Clinical Microbiology and Infectious Diseases (“ECCMID”) in July 2021.
|
|
|
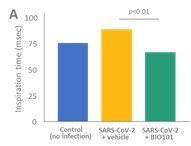
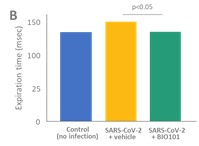
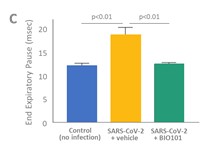
In the graphs above: (A) Inspiration time, (B) expiration time and (C) End Expiratory Pause (EEP) time evaluation after BIO101 IP treatment of SARS-CoV-2 infected hamsters. Histograms show values of control group (not infected with SARS-CoV-2), infected with SARS-CoV-2 and treated with the vehicle (SARS-CoV-2 + vehicle) or infected with the SARS-CoV-2 and treated with BIO101 IP (SARS-CoV-2 + BIO101) with * p <0.05, and ** p <0.01. This data was also referenced at at the ECCMID in July 2021.
Preclinical Development of BIO101 (20-hydroxyecdysone) in DMD
We conducted various in vivo experiments in mdx mice, a commonly used model of DMD. The results from these mdx mice studies were consistent with the results on cellular activity and functional outcomes from both in vitro and in vivo studies of BIO101 (20-hydroxyecdysone) in sarcopenia. We believe these results provide additional support for our belief that BIO101 (20-hydroxyecdysone) has the potential for improving mobility and muscle strength. In addition, we believe these results suggest that BIO101 (20-hydroxyecdysone) may increase respiratory function and decrease fibrosis. Key in vivo results in DMD are summarized below.
67
Improved respiratory function in mice. The loss of respiratory function is a major health issue for later-stage, non-ambulatory patients with DMD. Results have shown that chronic (eight weeks) daily administration of 50/mg/kg of BIO101 (20-hydroxyecdysone) ameliorates the time-dependent degradation of respiratory function observed in C57BL10-mdx mice as compared to C57BL10 control mice. This protective effect on respiratory function is not only associated with breathing parameters as suggested by enhanced pause, or PenH, measurements, but also by an improvement of deep airway structure of the respiratory system shown by FlexiVent experiments, which are a common measurement for in vivo lung function. PenH is calculated as follows: (PIP/PEP) x Pause, where PIP is the maximum change in chamber pressure during inspiration, PEP is the maximum change in chamber pressure during expiration, and Pause equals (TE-TR)/TE, where TE is expiratory time and TR is relaxation time. As shown in the three graphs below, C57BL10-mdx mice treated with BIO101 (20-hydroxyecdysone) exhibited improved respiratory function as measured by resistance, elastance and PenH of the lung. These results were presented in March 2019 at the annual international congress of Myology in Bordeaux, France.
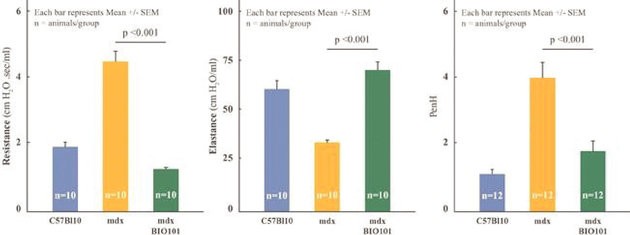
Effect of chronic BIO101 (20-hydroxyecdysone) treatment on resistance, elastance and airway reactivity (PenH).
Improved mobility and muscle strength in mice. We studied the effect of chronic oral administration of 50 mg/kg/day of BIO101 (20-hydroxyecdysone) on mobility and strength over eight weeks in C57BL10-mdx mice. Mobility was measured by running distance and strength was measured by maximum absolute strength (force) in the four-limb grip-test test. Results show that BIO101 (20-hydroxyecdysone) treatment improved mobility in certain animal models, as C57BL10-mdx mice treated with BIO101 (20-hydroxyecdysone) ran 2.4x farther than untreated control C57BL10-mdx mice. Results show that BIO101 (20-hydroxyecdysone) treatment improved muscle strength in animal models, as C57BL10-mdx mice treated with BIO101 (20-hydroxyecdysone) showed an approximate 14% improvement in strength as compared to untreated control C57BL10-mdx mice.
|
|
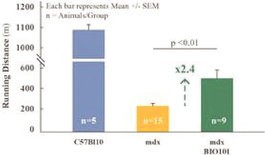
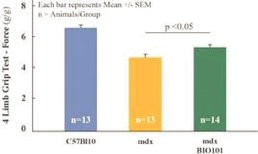
Effect of BIO101 (20-hydroxyecdysone) on mobility (running distance) and muscle strength (four-limb grip-test force).
These in vivo results on muscle functionality (mobility and strength) in mice are consistent with cellular and molecular changes observed in our previous preclinical studies, including (i) improved energy metabolism (mitochondrial respiration and spare respiratory capacity), (ii) improved myoblast differentiation, and (iii) confirmed activation of the AKT Pathway involved in anabolism known for being impaired in DMD muscle. These results were presented in October 2018 at the World Muscle Society, or WMS, conference in Mendoza, Argentina (Dilda et al., 2018).
68
Improved lesion profile in mice. We have observed that BIO101 (20-hydroxyecdysone) treatment may improve the histological (muscular lesion) profile of muscle in mice, consistent with the improvements in physical performance and muscle function (mobility and strength), as mentioned above. We performed histopathological analysis of muscle from C57BL10-control mice, C57BL10-mdx mice and C57BL10-mdx mice treated with BIO101 (20-hydroxyecdysone). Muscles from C57BL10-mdx mice exhibited anisocytosis (atrophy of muscle fibers), as well as chronic inflammation associated with fibrosis as compared to healthy muscles from control mice. Observations of muscle from C57BL10-mdx treated mice showed that chronic administration of BIO101 (20-hydroxyecdysone) decreased anisocytosis and inflammation as compared to muscles from C57BL10-mdx mice. These results were presented in October 2017 at the WMS conference held in Saint Malo, France.
BIO101 (20-hydroxyecdysone) for the treatment of age-related Sarcopenia (the SARA program)
Biophytis is developing BIO101 (20-hydroxyecdysone) to treat age-related degeneration of skeletal muscle. It is a major cause of mobility disability in the elderly, characterized by a loss of muscle mass, strength, balance and the ability to stand and/or walk, resulting in a loss of independence, increased risk of adverse health events and hospitalization, and potential death resulting from falls, fractures, and physical disability.
Sarcopenia was first defined in 1989 and officially classified as a disease in 2016 based on the establishment of a code from the WHO’s International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM), used by physicians, researchers and health systems. There is currently no widely accepted standard of care for sarcopenia, however, to our knowledge, current non-medicinal treatment recommendations primarily focus on moderate physical activity, such as 30 minutes of walking per day or resistance-based (strength) training, as they exert effects on both the nervous and muscular systems that are critical to positive physiological and functional adaptations in older adults, and nutritional intervention. According to the International Clinical Practice Guidelines for Sarcopenia (ICFSR): Screening, Diagnosis and Management (Dent et al., J Nutr Health Aging. 2018;22(10):1148-1161) there is moderate certainty of evidence for the beneficial effects of physical therapy in treating patients with sarcopenia as most of the evidence for physical activity comes from studies of non-sarcopenic older adults or those with mild-moderate sarcopenia. The efficacy of more structured physical activity programs along with certain supplementation (i.e. dietary protein intake and/ or nutrients) for the treatment of sarcopenia is being assessed in various studies, including the SPRINTT trial. However, no consensus on nutritional intervention currently exists.
Phase 1 Clinical Trial (SARA-PK)
We conducted a dose-escalating Phase 1 clinical trial (SARA-PK) to evaluate the safety, PK and PD effects of BIO101 (20-hydroxyecdysone) in 54 healthy adult and elderly subjects. Based on the results of the SARA-PK Phase 1 clinical trial, we chose 175 and 350 mg b.i.d. (twice daily) as the safe, active dosing levels for the SARA-INT Phase 2b clinical trial.
Single Ascending Dose. In the single ascending dose(“SAD”) phase, subjects were dosed once with BIO101 (20-hydroxyecdysone) at a range between 100 to 1,400 mg or placebo. No abnormal clinical vital signs and/or serious adverse events were reported as treatment emergent adverse events, or TEAE. All TEAEs were mild in severity and were resolved by the end of the study. No serious adverse events, or SAEs, were reported in the SAD phase.
Multiple Ascending Dose. The multiple ascending dose(“MAD”) phase was conducted with three selected doses of BIO101 (20-hydroxyecdysone) that were orally administered to 30 patients in total broken into three groups of older adults between 65 and 85 years over 14 days. Each group consisted of eight active and two placebo per dose.
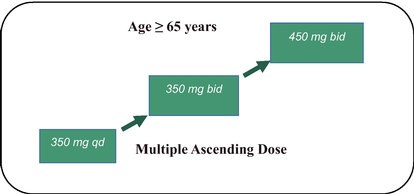
69
No abnormal clinical vital signs and/or adverse events were reported. Study results indicated that several patients experienced TEAEs, the most common were headache and nausea, with one participant reporting an event of food poisoning at the follow-up visit and dizziness postural (vertigo) and are described in the table below. All TEAEs were indicated as mild or moderate and were resolved by the end of the study. No SAEs associated with BIO101 (20-hydroxyecdysone) were reported in the MAD phase.
| No. of treated subjects with TEAE |
|
| ||
Dose | (Type of TEAE) | No. of placebo subjects with TEAE | |||
350 mg q.d. (once daily) | 2 subjects (mainly wound and pain in extremity). | 3 subjects (mainly musculoskeletal and connective tissues (back pain, spasms and stiffness) and nervous system (dizziness and headache)). | |||
350 mg b.i.d. (twice daily) | 7 subjects (mainly gastrointestinal (constipation, diarrhea and bloating), and musculoskeletal and connective tissue (back pain, spasms and stiffness)). | ||||
450 mg b.i.d. (twice daily) | 8 subjects (mainly gastrointestinal (constipation, diarrhea and bloating), musculoskeletal and connective tissue disorders (back pain, spasms and stiffness) and nervous system (dizziness and headache)). |
The pharmacokinetic analysis showed a short half-life between 3 to 4 hours and that the steady state was reached from the second day of administration in the MAD phase. No accumulation of BIO101 (20-hydroxyecdysone) was observed at 350 mg q.d. in the MAD phase (accumulation ratio of 1.14); however, a small accumulation was observed at 350 and 450 mg b.i.d. in the MAD phase (accumulation ratio of 1.31). We determined the optimal dosing of 175 and 350 mg b.i.d. from a PK modeling study.
We also evaluated the effects of BIO101 (20-hydroxyecdysone) on PD markers. Results showed a tendency towards a decreased plasma level in muscle catabolism markers (myoglobin, creatine kinase) and in markers of the RAS (aldosterone and renin). This is consistent with the proposed mechanism-of-action of BIO101 (20-hydroxyecdysone) and is coherent with the activity of BIO101 (20-hydroxyecdysone) on the RAS.
As shown in the graphs below, BIO101 (20-hydroxyecdysone) treatment over 14 days showed (i) a dose-dependent effect on muscle growth and repair, as measured by plasma Procollagen type III N-terminal peptide (PIIINP), a common marker of muscle growth, repair and fibrosis, and (ii) a dose-dependent negative correlation of muscle wasting, as measured by plasma myoglobin, a common marker of muscle catabolism.
|
|
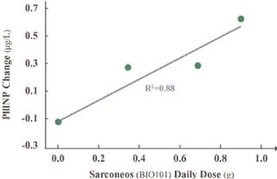
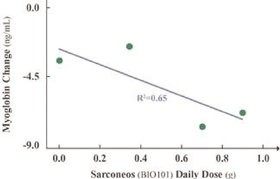
Effect of BIO101 (20-hydroxyecdysone) treatment for 14 days on the evolution of PD markers related to muscle anabolism (PIIINP) and to muscle catabolism (myoglobin)
Results from the SARA-PK Phase 1 clinical trial were released in April 2017 in an oral presentation at the International Conference on Frailty & Sarcopenia Research, in Barcelona, Spain. The results were used to establish the dosing levels for the recently completed SARA-INT Phase 2b clinical trial. Additional dosing studies are recommended by FDA to proceed.
70
Phase 2 Clinical Trial (SARA-OBS and SARA-INT)
| ● | SARA-OBS was an observational study that recruited 218 participants, of whom 185 completed a 6-months follow-up, between April 2017 and April 2019. This study was designed to characterize the target population of elderly patients (65 years old and above), who are at risk for mobility disability. This study was executed in 11 sites in the United States, France, Italy and Belgium. The study was finalized and a preliminary analysis of the SARA-OBS study was presented at the 12th Annual Congress of SCWD in Berlin, Germany in December 2019. The first presentation of the final results was given at the virtual 13th annual congress of SCWD on December 12, 2020. |
| ● | SARA-INT was a global, double-blind, placebo-controlled study, with 233 participants, who received BIO101 (20-hydroxyecdysone) at doses of 175 or 350 mg b.i.d. or placebo for 6 to 9 months. The study was executed in 22 centers in the United States and Belgium. Recruitment was completed in March 2020 with the last patient completing his final on-treatment visit in December 2020. Because of impediments posed by the COVID -19 pandemic, such as the interruption of in-office study visits and other disruptions, only 45 percent of the study subjects were able to complete the study with end-of-treatment efficacy assessments and the study was underpowered to observe the hypothesized effect size, and the primary and secondary endpoints were not met. |
SARA-OBS Study
Objectives and Study Design. The SARA-OBS study aimed to characterize sarcopenia in patients over the age of 65 at risk of mobility disability. The mobility and physical performance of these participants, including body composition was evaluated over a six-month period. This observational phase included two visits, one at the baseline and one at the end of the study, supplemented by a telephone interview at three months to determine whether participants were complaining of a poor physical condition. The SARA-OBS study was designed and structured as a pre-selection for the SARA-INT Phase 2b clinical trial.
Results. Baseline characteristics of the 218 participants were presented in December 2018 at the Society on sarcopenia, Cachexia and Wasting Disorders conference in Maastricht, Netherlands and the virtual 13th annual congress of SCWD on December 12, 2020 and are summarized in the table below. We believe these characteristics are consistent with other clinical trials of sarcopenia patients, including the SPRINTT and LIFE trials.
Age: |
| 79.29 |
BMI: | 29.3 | |
SPPB: |
| 6.12 |
Gait speed: |
| <0.8 m/s |
6-minute walk test: |
| 295.14 meters |
The final results, on the main endpoints, for the 185 completers are:
| Baseline |
| M6 |
| Change |
| P-value | |
400MWT | 0.866 | 0.835 | -0.027 | 0.064 | ||||
SPPB score |
| 6.562 |
| 7.078 |
| 0.439 |
| 0.439 |
6MWT |
| 297.561 |
| 284.841 |
| -16.655 |
| 0.006 |
Chair-stand |
| 1.732 |
| 1.774 |
| 0.007 |
| 0.929 |
Handgrip |
| 23.739 |
| 24.464 |
| 0.957 |
| 0.077 |
400MWT = 400-meters walk-test; SPPB score = Short-Performance Physical Battery; 6MWT = 6-minute walk-test; Chair-stand = the chair-stand component of the SPPB
71
SARA-INT Phase 2b Study
Objectives and Endpoints. The objectives and endpoints of the study are summarized below:
Objectives: |
| · Evaluate the safety and effectiveness of two doses, 175 and 350 mg b.i.d. (twice daily) of BIO101 (20-hydroxyecdysone) administered orally with a meal for 26 weeks against a placebo in participants over 65 at risk of impaired mobility; and · Measure treatment effect on improvement of physical function and on decrease of risk of mobility disability after six-month treatment. |
Primary Endpoint: |
| · The change from baseline in gait speed as measured in the 400MWT. A minimum clinically significant benefit is set at 0.10 meter per second in the mean difference between groups. |
Key Secondary Endpoints: |
| · Change from baseline in the hand grip strength. · Change from baseline and responder analysis on Physical Function domain (PF-10) of the SF-36 questionnaire. · Responder analysis from the 400MWT test with a responder definition of “study participant with an improvement of gait speed at 400MW test greater or equal to 0.1 m/s versus baseline”, at an individual level. |
Other Secondary, Tertiary and Exploratory Endpoints: |
| · Change from baseline of ALM and other parameters of body composition by DEXA; the rate of success to complete 400MW test after a 6-month treatment versus placebo; change from baseline of muscle strength as measured by knee extension and SCPT; change from baseline of the total SPPB score and of the sub-score of the repeated chair stands test; change from baseline using the SarQol, PAT-D, TSD-OC, SF-36 autoself-evaluation questionnaires. · exploratory endpoints: Plasma parameters including safety markers, biomarkers of the RAS (renin, aldosterone), inflammation (IL -6, CRP and hsCRP), and muscle metabolism (PIIINP, myoglobin, creatine kinase MM and creatine kinase MB) and actimetry |
In addition, four pre-defined subgroup analyses were performed:
| ● | a “very low walking speed subpopulation,” defined as having a gait speed< 0.8 m/s in the 4-meter walk test, a component of the SPPB; |
| ● | Subpopulation of participants with a chair stand sub-score of ≤2 of the SPPB; |
| ● | “Subpopulation with sarcopenic obesity” defined by a body fat percentage of > 25% for men and > 35% for women; and |
| ● | Subpopulation of participants who experience a deterioration in their ALM/BMI as measured by the DEXA scan at the end-of-treatment visit compared to the baseline measurement. |
72
These subpopulations represented sarcopenia patients that were at a significantly high risk for deterioration and adverse outcomes.
Trial Design: The trial design is summarized below:
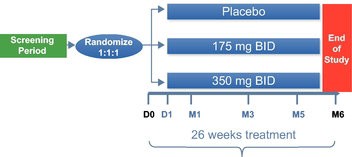
Prospective participants were screened for a period of up to eight weeks prior to inclusion in the trial. The interventional phase was comprised of an inclusion visit (D0) where baseline measurements were taken on the first day and dosing started the following day (D1), a one-month safety visit (M1), a three-month follow-up visit (M3) with safety and reduced measurements in connection with the primary endpoint, a five-month telephone interview (M5), and a final six-month visit (M6) with safety and full measurements. For 50 patients who could not come to the scheduled end of treatment visit at month 6, treatment could be extended to at most 9 months, anticipating that thereafter COVID-19 restrictions would make it possible again for them to come to the site.
A total of 233 elderly patients with sarcopenia at risk of mobility disability were recruited in 22 clinical investigation centers in the United States and Belgium. Recruitment was completed in March 2020. During the first wave of the pandemic, clinical study sites were closed and we revised the protocols to continue our clinical trials. We informed the IRBs that oversee the clinical trials and received approvals for modifications resulting from COVID -19. Despite these and other impediments, we were able to retain most of the participants. A total of 196 participants completed the SARA-INT study, with or without an extension of up to 9 months of treatment. Of those, and due to COVID-19 restrictions, only 106 patients could perform the 400m walk test at the End-of-Study visit (M6/M9), which was the primary endpoint of our study (55% loss of efficacy data). This resulted in the study being underpowered. The last patient completed his final on-treatment visit in December 2020. Top-line results from this study were announced in August 2021, with Clinical Study Report (CSR) finalized in February 2022.
The effect of two doses of BIO101 (20-hydroxyecdysone), 175 mg bid and 350 mg bid, were compared to placebo in the Full Analysis Dataset (FAS) and in the Per-Protocol population (PP, subset of participants that complied to the clinical protocol), as well as in sub-populations of patients.
Results. BIO101 (20-hydroxyecdysone) at the highest dose of 350 mg bid showed an increase of 0.09 m/s in the FAS population and of 0.10 meters per second (m/s) in the PP population compared to placebo in observed data, for the 400-meter walking test (400MWT) in gait speed after 6 months of treatment in observed data. Statistical analyses based on Multiple and Bayesian imputation showed a LS Mean difference at Month 6 of 0.07 m/s in the FAS population (p=0.085) versus Placebo. Minimal Clinically Important Difference (MCID) for the 400MWT in sarcopenia is 0.1 m/s per the study protocol.
However, the increase was not statistically significant, and none of the primary or secondary efficacy endpoints reached statistical significance.
73
Several results from the SARA-INT Phase 2b trial of BIO101 (20-hydroxyecdysone) in sarcopenia were presented at the ICFSR on September 30, 2021. ICFSR is the key international scientific event on frailty and sarcopenia, and is attended by leading researchers, physicians, and personnel from the biotechnology and pharmaceutical companies. The results are provided in graphs.
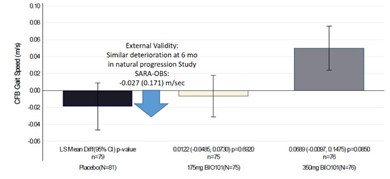
Effect of BIO101 (20-hydroxyecdysone) on the 400 MWT gait speed in the FAS population at Month 6 based on multiple imputation for subjects without data on On-Site Visit
BIO101 (20-hydroxyecdysone) at 350mg bid showed an increase on the 400MWT gait speed in sub-population at higher risk of mobility disability such as slow walkers (LS mean difference of 0.07 m/s versus placebo), obese subgroup (LS mean difference of 0.09 m/s versus Placebo), chair stand sub-score ≤2 of the SPPB (LS mean difference of 0.09 m/s versus placebo, p= 0.004) in the PP population at Month 6.
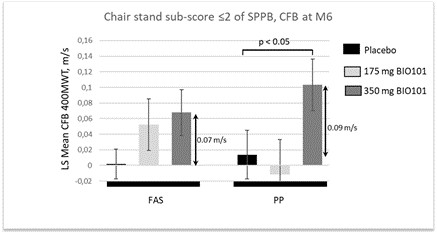
Effect of BIO101 (20-hydroxyecdysone) on the 400MWT gait speed in sub-population with higher risk of mobility disability (chair stand subscore ≤2) at Month 6
The COVID-19 pandemic and its related restrictions had a significant impact on the conduct of the study, with 55% of total participants not allowed to perform their on-site End of Study visit, despite the extension of their treatment period. Only 45 percent of the study subjects were able to complete the end of study assessments at the clinic and thus the study was underpowered to observe the hypothesized effect size, and the primary and secondary endpoints were not met.
Safety analysis. The proportion of subjects with Treatment-Emergent Adverse Events (TEAEs) was 52 (64.2%), 51 (68.0%), and 44 (59.5%) in the placebo, 175 mg and 350 mg BIO101 groups. The proportion of subjects with serious TEAEs was 9 (11.1%), 10 (13.3%), and 2 (2.7%) in the placebo, 175 mg and 350 mg BIO101 groups. There was no noticeable difference in TEAS, related TEAEs or Serious Adverse Events (SAEs) between treatment groups.
74
A tabular summary of safety data is presented below:
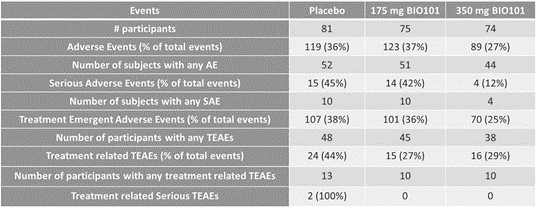
Adverse Events, Serious Adverse Events and Treatment Emergent Adverse Events in the Placebo, 175 mg bid and 350 mg bid groups in the SARA-INT study
Regulatory consultation with FDA and EMA
Upon review of the results, FDA reclassified the scheduled end-of-phase Type B meeting to a Type C meeting. During the meeting, which was held on January 24, 2022, FDA discussed concerns that entering into Phase 3 would be premature, questioning the dose and the sarcopenia condition. We also discussed strategies to further define the proposed population and to refine the proposed indication, and development of other information and data that will assist us in preparing the chemistry, manufacturing, and control information to be submitted to FDA, as well as the regulatory non-clinical plan. Using recent evidence from literature and following discussion with Key External Experts in the sarcopenia indication, we designed a new phase 3 program using Major Mobility Disability (MMD) as the primary endpoint, considered as the earliest occurring of the cascade of hard health-related outcomes of falls, hospitalization, institutionalization and death. MMD is defined as the inability to walk 400 meters within 15 minutes without sitting, help from another person or use of a walker. Individuals who require more than 15 minutes to complete the walk have an extremely slow pace (<0.45 m/sec), which would make their walking capacity of little utility in daily life. This was also the primary endpoint of the two largest recent trials in the field, the SPRINTT and LIFE studies. The primary objective of the Phase 3 trial will be to evaluate the efficacy of BIO101 350mg bid on the hazard of MMD versus placebo. Sarcopenic community-dwelling participants, as defined as having a SPPB score comprised between 3 and 7, a gait speed > 0.8 m/s and an hand grip strength in the dominant hand of > 35.5 kg and >20kg in male and female, will be treated and followed for a minimum of 12 month and up to 36 months. We arranged a scientific advice with EMA and an additional type C meeting with the FDA to discuss the updated design and the planned patient-related outcomes. Overall, EMA agreed with the main parameters of the design of the Phase 3 design: targeted population, primary endpoint, secondary endpoints, dosing duration, whereas the FDA recommended to have a self-reported questionnaire as a co-primary endpoint whilst requiring more pieces of validation on the only available questionnaire specifically developed for the assessment of Quality of Life in sarcopenia. The two competent Authorities commented that the choice of the dose for the Phase 3 program was at our own risk. In 2023, we submitted the Phase 3 protocol to the FDA and in Belgium under the European regulation and both agencies gave the authorization to initiate the study.
Design | Endpoints | Patient Population |
Global, double-blind, randomized, placebo-controlled trial Assessment of efficacy and safety of BIO101 350 mg bid administered orally over at least 52 weeks (up to 156 weeks), as compared to placebo 932 randomized patients planned | Primary endpoint: Time to the first occurrence of Major Mobility Disability (MMD) assessed by the inability to complete the 400-meter walk test within 15 minutes, without sitting, help from another person or use of a walker Secondary endpoints: Handgrip muscle strength, Quality of Life (PRO SarQoL), 4-meter Gait speed from SPPB | Sarcopenic population Age: 65 years old or over living in the community Low mobility measured by Short Performance Physical Battery 3≤(SPPB) ≤7 out of 12 4-meter gait speed ≤ 0.8 m/s Hand grip Strength in the dominant hand <35.5 kg for males, <20 kg for females Reporting a loss of motor function over the last year |
75
Market Opportunity
Sarcopenia (or age-related muscular dystrophy) is a syndrome defined consensually by the European group EWGSOP (The European Working Group on Sarcopenia in Older People), characterized by progressive and generalized loss of skeletal muscle mass and strength associated with an increased risk of adverse events such as disability, poor quality of life and death. Sarcopenia is highly prevalent in adults greater than 60 years of age with an estimated prevalence between 10 to 16% worldwide. It poses a major public health issue and is steadily increasing as the global population ages. The severe sarcopenia of patients aged 65+ that we are targeting represents almost 900,000 US citizens, more than 550,000 in Brazil, 260,000 in France and 1,350,000 in Japan. To date, no pharmacological treatment has yet been approved for either frailty or sarcopenia. Recommendations for the prevention and treatment of frailty and sarcopenia are thus still mainly based on lifestyle interventions, such as nutrition and physical exercise.
If approved by regulatory authorities for commercial use, we believe there is a huge market potential for BIO101 (20-hydroxyecdysone) in severe sarcopenia.
Over the past two decades, other companies have launched multiple clinical development programs to treat sarcopenia, primarily with drug candidates falling in one of two classes: (i) myostatin inhibitors and (ii) selective androgen receptor modulators, or SARMs. Myostatin inhibitors, which primarily aim to increase muscle mass by blocking myostatin (myostatin acts as an essential negative regulator of muscle bulk), have been found to increase muscle mass in early clinical trials. However, they have yet to demonstrate effectiveness on clinically meaningful mobility outcomes (strength and mobility) or safety in larger clinical trials and/or have not progressed through the clinic. Both steroidal and non-steroidal SARMs have been tested as therapeutic agents for several medical conditions, including muscle-wasting diseases, but none have progressed through clinical development mainly due to safety concerns.
Based on our review of publicly available information, currently, neither myostatin inhibitors nor SARMs are being tested in late-stage clinical trials for sarcopenia. Based on our review of research in this area, we believe BIO101 (20-hydroxyecdysone) is currently the most advanced drug candidate being tested in a clinical program for the treatment of sarcopenia and has the potential to improve the vital functional outcomes of mobility disability necessary for regulatory approval. To our knowledge, there is currently no widely accepted standard of care for sarcopenia. Current non-medicinal treatment recommendations primarily focus on moderate physical activity, such as 30 minutes of walking per day or resistance-based (strength) training, as they exert effects on both the nervous and muscular systems that are critical to positive physiological and functional adaptations in older adults, and nutritional intervention. Other potential drug modalities that have been tested in the clinic for sarcopenia have yet to demonstrate effectiveness on clinically meaningful outcomes (strength and mobility) and/or safety in larger clinical trials and/or have not progressed through the clinic. Based on our understanding and discussions with regulatory agencies, including the FDA and EMA, functional mobility endpoints must be achieved in order to obtain marketing approval for sarcopenia.
BIO101 (20-hydroxyecdysone) for the treatment of muscle weakness associated with obesity (the OBA program)
Obesity treatment can lead to loss of muscle mass and function, notably as a consequence of dieting when combined with the recently introduced GLP-1 receptor agonists. Glucagon-like peptide-1 receptor agonist (GLP-1 RA) drugs are very effective drugs that lead to significant weight loss. Up to 40% of the total weight loss comes from muscle, which is a problem as muscle tissue's role is central in controlling metabolism, on top of its motor function, We are seeking to reduce muscle strength loss from GLP-1 agonists (GLP-1As) in combination with dieting, in adult obese or overweight patients who are treated with semaglutide or liraglutide for weight loss. There are currently no approved therapies for this indication and it is known that GLP-1 agonists, in combination with calorie-restricted diet, cause an important lean body mass loss as component of the weight loss induced by these treatments, where muscle mass is a main part of lean body mass. Recently introduced GLP-1 agonists launched for weight loss include semaglutide, tirzepatide and liraglutide. Important declines in lean body mass were reported for semaglutide (5.4 kg compared to placebo, Wilding 2021) and tirzepatide (8.3% of body weight compared to placebo, Jastreboff, 2022). This reduction is deemed to be of clinical relevance especially in patients who have an already compromised muscle strength due to obesity, age or other diseases.
Clinical development
Results obtained from the obese subpopulation in the SARA-INT study in sarcopenic patients support the potential that 20E may be effective in this indication. For example, the gaitspeed from the 400MWT improved nominally statistically significantly in the sarcopenic obesity subgroup in the Per Protocol analysis (see figure below).
76
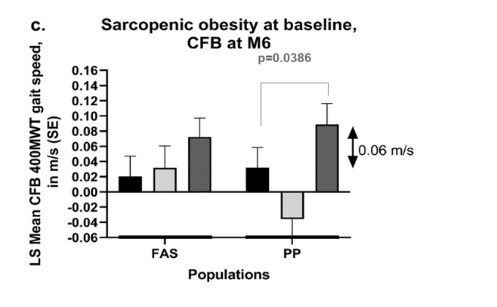
Effect of BIO101 (20-hydroxyecdysone) on the 400MWT gait speed in sub-population with sarcopenic obesity at Month 6
Furthermore, a clear trend towards a dose-response relationship was observed on the change from baseline in handgrip strength in the overall population, although due to the variability these differences were not statistically significant.
Further support comes from a clinical study using Quinolia, a quinoa extract enriched in 20-hydroxyecdysone administered in 58 overweight or obese participants (27 kg/m² ≤ BMI ≤ 38 kg/m²) at a daily consumption corresponding to 37.5 mg of 20-hydroxyecdysone or placebo. Patients followed a weight loss dietary program consisting of a 6-week weight loss (WL) dietary intervention phase followed by a 6-week weight loss maintenance (WLM) phase. Differences in mean hand grip strength between Quinolia® and placebo showed a trend towards an effect when considering the subpopulation of subjects who lost more than 5% of their initial weight during the weight loss phase (p=0.0974). As compared to placebo, Quinolia® induced a statistically significant difference in the android fat mass (p=0.0386) and HOMA-IR index, an index reflecting insulin sensitivity, (p=0.0058) (see figures below; the first timepoint reflects the difference between week 6 and baseline (“S6-S0"); the second timepoint reflects the difference between week 12 and week 6 assessment (“S12-S6”)).
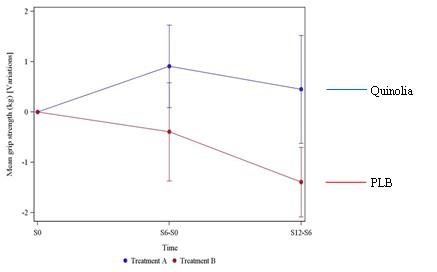
77
Effect of BIO101 (20-hydroxyecdysone) on the Handgrip strength in patients who lost >5% of weight.
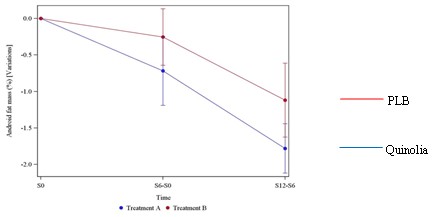
Effect of BIO101 (20-hydroxyecdysone) on the android fat mass.
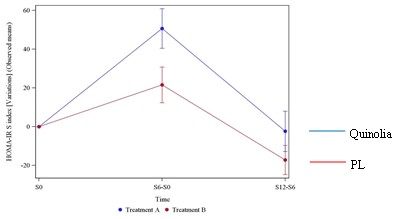
Effect of BIO101 (20-hydroxyecdysone) on the HOMA IR S index.
We are currently designing a Phase 2 study in obese and overweight adult patients who are taking either semaglutide or liraglutide for weight loss and who have a low or poor muscle strength at baseline (ie at the start of the GLP-1A). We are designing our OBA clinical program to specifically to address the following challenges for obese patients:
| o | Muscle weakness and its sequalae due to obesity |
| o | Avoidance of, or decrease in loss of muscle strength in at-risk patients treated with GLP-1A weight-loss drugs plus diet |
| o | Increasing loss of fat mass and/or body weight above that achievable by GLP-1A plus diet |
| o | Improvement of glucose metabolism, insulin sensitivity and/or hypertension |
We propose a Phase 2 placebo-controlled study to obtain pharmacokinetic information on 20-hydroxyecdysone and its main metabolites in this differing patient population which is treated with potentially interacting GLP-1 agonists, and obtain Proof of Concept for 20-hydroxyecdysone efficacy to treat muscle weakness induced by GLP-1As, with the objective to start the study in 2024. This will be a randomized double-blind placebo-controlled study with a 6 month double-blind treatment period in approximately 200-250 subjects. Based on the results of this study we will design a phase 3 program after conducting regulatory consultations (type B meeting with the FDA and Scientific Advice with the EMA).
78
Market Opportunity
Obesity is a serious chronic disease. Nowadays, it’s getting more and more attractions because of new drugs available. 988 million of adults and children are currently living with obesity globally. The global prevalence of obesity has more than tripled since 1975 and the global cost of treating obesity-related complications is expected to rise by over $4 trillion by 2035.
More than 15 million adults only in the US will be treated with an anti-obesity medication at the horizon of 2030, representing 13% penetration into the US adult population. With an estimated market size of $6 billion in 2023 and an estimated average annual growth rate expected around 42%, the addressable market for the treatment of obesity is set to reach $100 billion by 2030.
But obesity treatment can lead to loss of muscle mass and function, notably as a consequence of dieting when combined with the recently introduced GLP-1 receptor agonists. These drugs are very effective drugs that lead to significant weight loss. Up to 40% of the total weight loss comes from muscle, which is a problem as, on top of its motor function, muscle tissue's role is central in controlling metabolism.
BIO101 (20-hydroxyecdysone) is the first oral daily MAS receptor activator and has demonstrated metabolic effects on muscle and fat issues in preclinical studies in obesity. These benefical effects of translate into improved mobility and muscle strength in obese sarcopenic patients, as shown in the SARA-INT phase 2 study. Furthermore, the 20-hydroxyecdysone molecule was already tested in obese patients during hypocaloric dieting in the Quinolia study, showing promising effects on muscle strength and fat mass loss. BIO101 (20-hydroxyecdysone) potential in the treatment of obesity in combination with GLP-1RAs to counteract the undesirable effects on muscle wasting associated with drastic weight-loss was highlighted in Nature Biotechnology (“After obesity drugs’ success, companies rush to preserve skeletal muscle”) on March 05, 2024.
BIO101 (20-hydroxyecdysone) for treatment of severe respiratory manifestation of COVID-19 (the COVA program)
COVID-19 was recognized as a worldwide pandemic by the WHO in March 2020. As of March 2, 2024, approximately 704 million people have been identified as having been infected with the SARS-CoV 2 virus, and more than 7.0 million have died because of COVID 19, as compared to 680 million people infected and 6.8 million deaths as of March 3, 2023. COVID-19 is caused by the SARS-CoV-2 virus. In its severe form, COVID-19 is associated with a plethora of complications, including:
| ● | Acute pneumonia and ARDS; |
| ● | Cardiac injury, including myocarditis and pericarditis; |
| ● | Renal failure; |
| ● | Hepatitis; |
| ● | Vasculitis and thromboembolic events, leading to cardiac and cerebral strokes and pulmonary thromboembolism; |
| ● | Coagulopathy; |
| ● | Muscle injury; and |
| ● | Long-term symptoms such as fatigue, depressive symptoms and respiratory difficulties. |
Ample evidence points towards the membrane-bound ACE2 as the entryway of SARS-CoV -2 into the cells (in a manner similar to the previously described coronavirus-associated severe acute respiratory syndrome (SARS)). Data is emerging that in COVID -19 increased levels of Ang-II are observed and are linked to the severity of the clinical syndrome. Despite the difficulty in measuring Ang -1 -7, some evidence has emerged that the levels of these peptides are indeed decreased in COVID -19 as well.
Our hypothesis has been that by activation of the MAS-receptor, BIO101 (20-hydroxyecdysone) could mitigate some of the downstream effects of the interaction between SARS-CoV-2 and ACE2. Indeed, studies that were conducted in a model of ALI have shown that 20-hydroxyecdysone can mitigate inflammation and reduce the levels of inflammatory markers. We have performed additional studies in animal models of COVID-19, in parallel with the COVA clinical program with the University of Liège in Belgium and other research institutions.
79
SARS-CoV-2 infection, by down-regulation of ACE2 expression and activity, reduces the conversion of Ang-II to Ang-1-7 resulting in excessive levels of Ang-II. Ang-II levels in COVID-19 patients are significantly higher than in non-infected individuals and, more importantly, are linearly associated with viral load and lung injury. Moreover, the plasma levels of Ang-1-7 are significantly lower in COVID-19 patients versus healthy controls and particularly between COVID-19 patients admitted to ICUs compared to those who are not. Because most of SARS-CoV-2 deleterious effects including inflammation, fibrosis, thrombosis, pulmonary damage, point towards an imbalance of the RAS, we strongly believe that acting on the protective arm of RAS via its MAS Receptor downstream of ACE2 could have a beneficial effect in COVID-19-infected patients and, therefore, improve ARDS outcome.
The COVA Study
The COVA study was a global, multicenter, double-blind, placebo-controlled, group-sequential, and adaptive two-part Phase 2-3 study, with a targeted recruitment of a total of 310 hospitalized patients in both parts. The final number of patients was recommended by the DMC based on the blinded second interim analyses and to protect the scientific integrity of the study. We decided to stop enrollment in April 2022 after only 237 patients meeting the study criteria have been enrolled in the trial in France, the United States, Belgium and Brazil, in approximately 35 clinical centers. Progression of the pandemic impacted our ability to enroll the targeted 310 patients in a reasonable timeframe and cost.
The objective of the study was to investigate the efficacy and safety of BIO101 (20-hydroxyecdysone), 350 mg BID in hospitalized COVID-19 patients with hypoxemia, at risk of respiratory failure requiring high flow oxygen or mechanical ventilation, and death. The proportion of patients and time to respiratory failure or early death were studied at 28 days in the primary analysis, corresponding to the maximum treatment period, with follow-up of mortality and safety for at least 90 days. Final results of the study were released on February 2, 2023.
The 233 treated patients (Intent To Treat, ITT population) were 63 years old on average, 64% of the patients were male, recruited in 37 centers in Europe, the United States and Brazil between April 2020 and March 2022, infected with the main SARS-Cov-2 variants. The analysis of the study demographics show that the two groups (treated and placebo) have very comparable characteristics with two caveats: The use of immuno-suppressors is higher in the placebo group and the number of men is lower in the placebo group; both of these data are in favor of the placebo group as compared to the treatment arm.
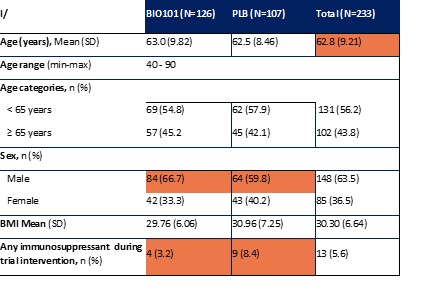
The study met its pre-defined primary endpoint demonstrating a statistically significant difference between BIO101 (20-hydroxyecdysone) and placebo in the proportion of patients with respiratory failure or early death at day 28, representing a relative reduction of risk of 44% (p=0.0426, Cochran-Mantel-Haenszel test).
80
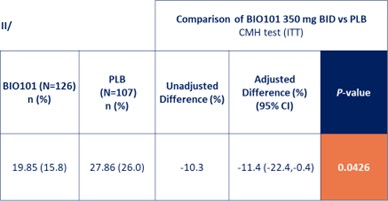
Moreover, the analysis of time to respiratory failure or early death had shown significant differences over 28 days in the Kaplan Meier curves for BIO101 (20-hydroxyecdysone) versus placebo (p=0.022). The pre-specified analysis of time to death over the complete follow-up period over 90 days showed that mortality rate with BIO101 (20-hydroxyecdysone) was reduced compared to placebo in the ITT population (p=0.083) and in the PP population (p=0.038). Post hoc Kaplan-Meier analysis released on November 3, 2022 confirmed the effect of BIO101 (20-hydroxyecdysone) on the primary endpoint and on mortality at day 90.
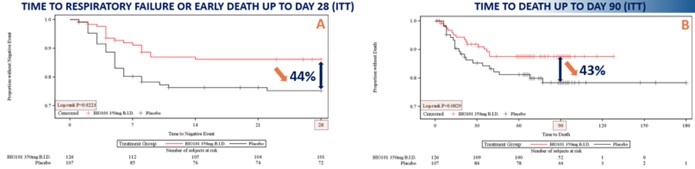
In addition, BIO101 (20-hydroxyecdysone) showed a very good safety profile. The results showed a lower proportion of patients with TEAEs in the BIO101 (20-hydroxyecdysone) group (57%) than in the placebo group (64.4%), in particular a lower frequency of serious TEAEs (25% vs 30.8%) and serious respiratory TEAEs (18.8% vs 26.0%). Frequency of orthostatic hypotension was similar between both groups (7.0% vs 7.7%).
Market opportunity
We believe there is a market opportunity for BIO101 (20-hydroxyecdysone) for the treatment of severe acute respiratory infections associated with COVID-19, subject to successful clinical trials and the FDA’s approval or authorization of BIO101 (20-hydroxyecdysone) for such indication. The viral infections pandemics continue to be a public health issue, in hundreds of countries. As of March 2, 2024, approximately 704 million people have been identified as having been infected with the SARS-CoV 2 virus, and more than 7.0 million have died because of COVID 19.
The Severe Acute Respiratory infection market is driven by 3 main macro-environment factors:
The expanding global population, coupled with factors such as urbanization and international travel, has contributed to the rapid spread of respiratory viruses, creating a substantial patient pool in need of therapeutic interventions.
Governments around the world are implementing strategic initiatives to curb the spread of viral respiratory infections by recognizing the significant impact of these diseases on public health. One of the most effective measures has been the promotion and
81
widespread implementation of vaccination programs aimed at preventing key viral respiratory infections such as influenza, pneumonia, and COVID-19.
The relentless pursuit of innovative solutions has led to the development of novel antiviral treatments, diagnostic tools, and therapeutic approaches, reshaping the landscape of viral respiratory infection management. In recent years, the integration of cutting-edge technologies such as genomics and proteomics has empowered researchers to gain a deeper understanding of viral respiratory pathogens. To our knowledge, although there have been multiple initiatives to develop treatments, treatment regimen has only slightly evolved over the last 18 months. This is summarized in the below scheme:
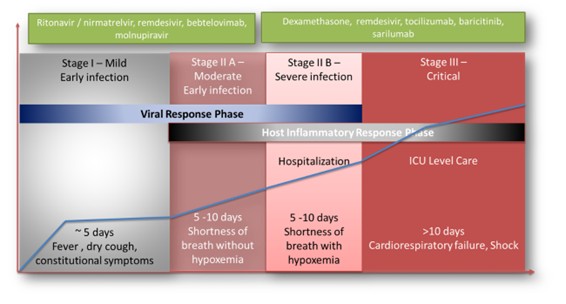
BIO101 (20-hydroxyecdysone) is targeting the hospitalized patients suffering from severe respiratory symptoms, representing a large majority (80%) of in-patients covid19 positive. No treatment specifically targeting the stimulation of respiratory function in hospitalized COVID-19 patients has been approved or recommended for use in United States or in Europe.
BIO101 (20-hydroxyecdysone) for Duchenne Muscular Dystrophy (DMD)
DMD is a rare, genetic neuromuscular disease in male children and young adults, which is characterized by an accelerated degeneration of muscles and is responsible for a loss of mobility, respiratory failure and cardiomyopathy, leading to premature death. DMD is caused by mutations in the dystrophin gene that result in the absence of very low levels of functional dystrophin, a cytoskeletal protein that protects muscle cells. It is the most common form of muscular dystrophy in children, affecting approximately 2.8 out of 100,000 people worldwide (approximately 20,000 new cases annually worldwide), based on our estimates from publicly available information. DMD is caused by mutations in the dystrophin gene that result in the absence or very low levels of functional dystrophin, a cytoskeletal protein that protects muscle cells.
The absence of dystrophin in muscle severely weakens the structural and membrane stability of the muscle fibers. During normal muscle contraction and stretching the muscle fibers become damaged and eventually undergo necrosis (i.e., cell death). In order to compensate for the increased necrosis, muscle tissue regeneration is accelerated. This process soon becomes exhausted and muscle degeneration accelerates as muscle fibers are replaced by fat and connective tissue (fibrosis), resulting in the loss of muscle strength and mobility. DMD evolves according to a very well understood progression with symptoms that are similar to those associated with accelerated aging across all stages. DMD progression can be summarized as follows:
| ● | muscle damage characterized by loss of myofibers, inflammation, and fibrosis beginning at an early age; |
| ● | lower extremity muscle weakness and progressive loss of muscle function beginning in the first few years of life; |
| ● | decline of ambulation and respiratory function after the age of seven; |
82
| ● | total loss of ambulation where the use of a wheelchair is essential in the pre-teenage or early teenage years; |
| ● | progressive loss of upper extremity function during mid to late-teens; and |
| ● | respiratory and/or cardiac failure, resulting in death around the age of 30. |
Our Clinical Development Plans of BIO101 (20-hydroxyecdysone) in DMD (the MYODA program)
We have developed a formulation that is suitable to treat children, especially with swallowing difficulties. We have weight-adjusted the dose range of BIO101 (20-hydroxyecdysone) that we aim to test in the pediatric patient population based on modeling of data from animal studies and the SARA-PK Phase 1 trial in healthy adult and elderly participants. The low end of the dose range is driven by efficacy studies and the upper end of the dose range is driven by safety margins (toxicology and Phase 1). At the low end of the dose range, differences caused by the variance in animal models (i.e., species, age and size) could affect efficacy between animals and humans (both adults and children). At the high end of the dose range, differences in body composition, absorption and metabolism between the age and patient segments could affect safety margins and tolerability. We do not have actual experimental safety PK, PD or efficacy data from clinical testing in a pediatric patient population comprised of developing children (2-12 years), adolescents (12-16 years) or young adults. However, the MYODA clinical study is designed to fill this gap, by testing a range of doses in a dose escalating manner to address these potential differences in safety and efficacy.
We have designed our MYODA clinical program to specifically address the following known challenges in DMD clinical development:
| ● | Currently, DMD programs are very lengthy and may take up to 10 years to finalize. With such a high unmet-need and a situation where young children lose function and experience a much shorter life span, there is a need to utilize fast and robust designs and expedite the development process. |
| ● | A very crowded space, with a lot of competing development programs, which are mostly focusing on ambulatory patients, leading to difficulties in recruitment, while there is very little development that targets non-ambulatory patients—a disease state, where deterioration in respiratory function is becoming a leading cause for mortality. |
In June 2018, we received orphan drug designation from the FDA and EMA for BIO101 (20-hydroxyecdysone) in DMD. In December 2019, we received an IND “may proceed” letter from the FDA (USA) and we received a CTA approval from the FAMHP (Belgium) to start the MYODA study, and to investigate BIO101 (20-hydroxyecdysone) in non-ambulatory patients with signs of respiratory deterioration. In the “may proceed” letter from the FDA, the FDA noted that it had significant concerns with the design of the study, and that the results of the study, as originally designed to enroll ambulatory and non-ambulatory patients and measure muscle function deterioration through a composite score, would not be capable of providing interpretable data sufficient to support a marketing application. In its letter, the FDA recommended that we revise the study population, the primary endpoint and the overall design with the removal of the Part 3. We have incorporated the FDA’s recommendations and deeply revised the protocol to focus on non-ambulatory patients with signs of respiratory deterioration, changed the primary endpoint to respiratory function and now propose a seamless Phase 1/2 design. We propose a Bayesian approach for the analysis of efficacy at the end of the Part 2 (final analysis). This will allow us to have a Proof of Concept on the efficacy of BIO101 in treatment of the respiratory deterioration of non-ambulant DMD patients, with a limited number of participants (48 randomized participants planned). Based on the results, we will seek for a conditional approval or fine-tune the design of a Phase 3 trial. The new design, including the statistical approach, will be discussed with Competent Authorities (Type C meeting with the FDA and Scientific Advice with the EMA) with the objective to start the study in 2024.
All the study participants will be treated for 48 weeks, followed by an open-label extension. Participants who are recruited during Part 1, to the lower dose cohorts, will be moved to a higher dose, once it is cleared to be used. An independent data safety monitoring committee (IDMC) will oversee the study, will review the safety and pharmacokinetics data and allow moving from one dose cohort to the next and will conduct an IA to assess safety and futility.
Because of the high unmet need, we have decided to focus, at this stage, on DMD patients who are non-ambulatory and with evidence of respiratory deterioration. The primary endpoint will be Change from Baseline in Percent Predicted Peak Expiratory Flow (PEF % predictive) at Week 48 (assessed by hospital-based spirometry measurements) and the key secondary endpoint is Change from Baseline in Forced Vital Capacity (FVC % predictive) at Week 48 (assessed by hospital-based spirometry measurements). Additional endpoints include other measures of respiratory function, functional scales, muscle strength and goal-attainment.
83
Market Opportunity
We believe that there is market potential for BIO101 (20-hydroxyecdysone) in DMD, if approved by regulatory authorities for commercial use. DMD is the most common form of genetic muscular dystrophy in children, affecting approximately 2.8 out of 100,000 people worldwide (approximately 20,000 new cases annually worldwide), based on our estimates from publicly available information, resulting in premature death. There is currently no cure for DMD and there are only limited treatment options that aim to control the symptoms and slow the disease progression. In many countries, corticosteroids are the standard drug therapy. However, corticosteroids typically only slow the progression of muscle weakness and delay the loss of ambulation by up to two years, and their benefit for non-ambulatory boys with signs of respiratory deterioration, is not clear. They have also been associated with adverse side effects and are generally not suitable for long-term administration.
There are three targeted therapies (i.e., therapies targeting a specific dystrophin mutation by exon skipping or with stop codons) available on the market (two in the United States and one in Europe). As these therapies each target a specific gene mutation, they can only address the approximately 20% of the overall DMD patient population with those genetic mutations. In addition, there are only a few treatments that are in clinical development that target treatment of non- ambulatory children. There are very few early-stage programs that target treatment of non-ambulatory patients with signs of respiratory deterioration.
In addition to these targeted therapies, gene therapies that are under development aim to introduce a gene coding for a truncated dystrophin protein that could limit immune reactions. These therapies typically suffer from low transfection rates resulting in low levels of dystrophin expression and potential severe immune reactions. This leaves room for combinations of genetic treatments with other disease modifying agents, regardless of the mutation potentially including BIO101. Additional approaches in development include: immune modulators, anti-fibrotic agents and agents that enhance muscle mass and function.
We believe that BIO101 (20-hydroxyecdysone) directly targets muscle tissue and cells, may increase key muscle cell functions that are impaired independent of the genetic mutation that causes the disease, and has the potential to be used complementarily with corticosteroids, current targeted therapies and other gene therapies under development. We also believe that because BIO101 (20-hydroxyecdysone) targets various impaired muscle tissues and cells relevant to muscle strength, mobility and respiratory function, it may have the potential to be used in all stages of DMD progression, including both ambulatory and non-ambulatory patients. Due to the high unmet need, specifically in the population of non-ambulatory patients with signs of respiratory deterioration, we decided to focus on this sub-population, at this stage.
BIO201
Our second drug candidate, BIO201, is an orally administered small molecule in development for the treatment of retinopathies. The initial indication we plan to seek approval for is dry AMD, followed by Stargardt disease.
History and Development of BIO201
Utilizing our expertise in functional screens and assays, we expanded our drug discovery efforts to other age-related diseases, with a focus on retinopathies. Using cellular models developed with the Institute of Vision at Sorbonne University in Paris, we screened a variety of carotenoids and flavonoids for their ability to protect retinal pigment epithelium (“RPE”) cells against the photo-oxidative stress induced by blue light in the presence of A2E, a phototoxic byproduct of the visual pigment cycle. We selected norbixin (an apo-carotenoid) for clinical development based on its pharmacological properties and safety profile in animal models of AMD and Stargardt disease. Next, we identified its molecular target(s) and identified a potential mechanism-of-action.
Potential mechanism-of-action
Inhibition of PPARs
Results from our preclinical studies support continued research to investigate whether BIO201 may protect RPE cells against the photo-oxidative stress induced by blue light in the presence of A2E through transrepression of peroxisome proliferator-activated receptors (“PPARs”). PPARs are nuclear receptors that primarily regulates carbohydrate and lipid metabolism in regenerative tissues only, and inflammatory processes in neuronal tissues, such as the brain or retina. Based on the result from our preclinical studies, we believe that BIO201 potentially counteracts the phototoxic effects of A2E by inhibition of PPARa and PPARg responsible for the anti-oxidative, anti-inflammatory and anti-apoptotic activity observed in the retina. We believe that the mode of action (“MOA”) of BIO201 differs from the MOA of most PPAR activators that are typically associated with known side effects.
84
The potential mechanism-of-action of BIO201 is illustrated in the diagram on the below:
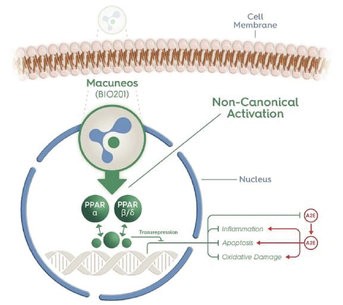
BIO201 is an antagonist of PPAR, involved in protecting retinal cells
Preclinical Development
Proof of concept in cellular models
In collaboration with the Institute of Vision, we used models of primary porcine RPE cell cultures to test the effect of BIO201. We believe this model best preserves functional defense mechanisms against photo-oxidative stress and better represents functioning human RPE cells as compared to existing stable cell lines. We exposed these RPE cells to blue light in the presence of A2E in order to explore the protective effect of BIO201 on RPE cell death.
Increased cell survival. Our preclinical data indicate that BIO201 may protect RPE cells from cell death, in a dose-dependent manner, against the photo-oxidative stress induced by blue light in the presence of A2E. These results were presented in 2016 at the annual meeting of the Association for Research in Vision and Ophthalmology, or ARVO, in Seattle, Washington, and published in PLoSONE (Fontaine et al; 2016).
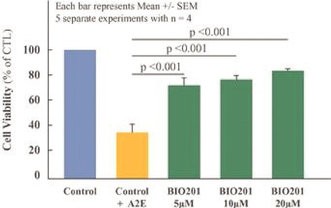
Effect of BIO201 on survival of RPE cells.
85
Proof of concept in animal models
We have observed that BIO201 protects the retina after both oral and intra-vitreal administration in various animal models of AMD and Stargardt disease. The results from the studies, which are summarized below, were presented in 2016 at the annual meeting of the ARVO in Seattle, Washington.
Preservation of visual function in mice. We studied mice in which two genes encoding the proteins involved in the visual pigment cycle (the Abca4 transporter and the retinol dehydrogenase Rdh8) were absent. These animals, called Abca4-/- Rdh8-/-mice, accumulated A2E in their eyes and showed an early loss of electroretinogram amplitude. Our preclinical data suggest that chronic oral administration of BIO201 for three and six months may be effective in protecting the retina, as measured by electroretinography. This is a commonly used way to measure retinal function by looking at the electric signal transport from the retina to the brain. As shown in the figure below, BIO201 treated mice showed a less degraded electroretinogram as compared to the untreated control mice, meaning the treated mice have slower visual function loss. The six-month results were presented in 2018 at the annual meeting of the ARVO in Honolulu, Hawaii and recently published (Fontaine et al. Aging, 2020).
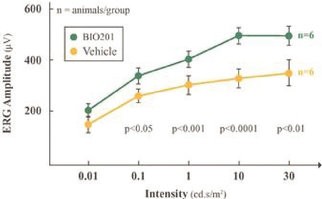
Effects of chronic oral administration of BIO201 on ERG Amplitude in Abca4[ib]-/[ib]- Rdh8[ib]-/[ib]-mice.
Reduced A2E Accumulation in mice. We studied the effect of BIO201 treatment on the accumulation of A2E in the retina of Abca4-/- Rdh8-/- mice. We began a three-month dosing regimen starting on mice that were 2 months of age. We observed that there was significant accumulation of A2E in vehicle Abca4-/- Rdh8-/-mice treated with placebo over three months as compared to control wild type mice at the beginning of the study, confirming a dysfunction of the visual cycle. Results demonstrated that chronic oral administration of BIO201 reduced A2E accumulation in the retina in treated Abca4-/- Rdh8-/-mice by approximately 45% as compared to vehicle control mice, which we believe is a key factor for maintaining visual function (Fontaine et al. PloSOne 2016).
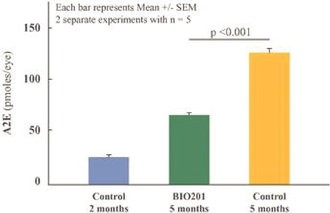
Effects of chronic oral administration of BIO201 on A2E accumulation in Abca4-/—Rdh8-/—mice.
86
Dose-dependent protection of retina integrity in rats. In the classical blue light damage (BLD) rat model using normal albino rats, we observed that intra-peritoneal administration of BIO201 protected the retina in a dose-dependent manner, as measured by the number of remaining layers of photoreceptors. We demonstrated that there was an approximate 90% increase in the number of photoreceptors layers following the maximum dose of 100 µM of BIO201 as compared to the vehicle control. The results were published in PLoSONE (Fontaine et al. 2016).
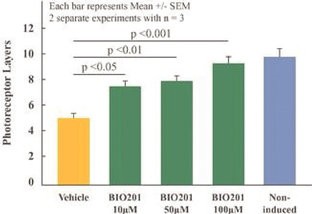
Number of layers of photoreceptors in the blue light damage rat model after intraperitoneal injection of BIO201.
Based on this body of work, we believe that BIO201 may have significant clinical potential for the treatment of retinopathies, including dry AMD and Stargardt disease, and warrants continued investigation.
BIO201 for AMD
AMD is an age-related degeneration of the macula, the central part of the retina. It is one of the leading causes of irreversible vision loss and blindness in the people over the age of 50 worldwide, according to the BrightFocus Foundation’s Age-Related Macular Degeneration: Facts & Figures Fact Sheet. AMD affects the central part of the retina, known as the macula, which is responsible for central vision and its sharpness. There are two types of AMD:
| ● | Dry AMD is a multistage process leading to the progressive loss of vision. Dry AMD affects central vision and impairs many functions affecting quality of life and independent living such as reading, driving, and facial recognition. Early-stage dry AMD is characterized by small drüsen accumulation, which may not cause changes in vision, but as drüsen grow in size and increase in number, they may lead to a dimming or distortion of vision that people find most noticeable when they read. Intermediate stage dry AMD is defined by more abundant and larger drüsen and the appearance of early atrophies. Patients at this stage are at high-risk of advancing into geographic atrophy (“GA”), a late stage form of AMD. Patients in the late stage of AMD may have blind spots in the center of their vision and may lose central vision. The prevalence of dry AMD increases significantly with advancing age. |
| ● | Wet AMD is a late stage form of AMD, which is characterized by abnormal growth of blood vessels from the choroid underneath the macula. This is called choroidal neovascularization. These blood vessels leak blood and fluid into the retina, causing distortion of vision that makes straight lines look wavy, as well as blind spots and loss of central vision. These abnormal blood vessels and their bleeding eventually form a scar, leading to permanent loss of central vision. |
Approximately 85 to 90% of patients with AMD suffer from the dry (atrophic) form of AMD, called dry AMD, according to estimates provided by the American Macular Degeneration Foundation. We believe that photo-oxidative and inflammatory stresses induced by the accumulation of A2E in RPE cells are the main factors responsible for the degenerative process of the retina in diseases such as AMD. We believe the biggest opportunity in treating dry AMD is preventing advancement into the later stages, GA or wet AMD, where vision loss is severe and can lead to visual disability.
87
Clinical Development Plans
We have conducted chronic and acute rodent and non-rodent toxicology studies with BIO201 that we believe will be sufficient to support our IND and clinical trial applications for our MACA clinical development program.
We are currently assessing the relative interest of BIO203 compared to BIO201, before completing the preclinical development and advancing to clinical stage (phase 1 study in healthy volunteers).
Market Opportunity
We believe that there is market potential for BIO201 in dry AMD, if approved by regulatory authorities for commercial use. AMD is one of the leading causes of irreversible vision loss and blindness in people over the age of 50 worldwide, and its prevalence increases with advancing age. Based on our review of publicly available data and to our knowledge, there is currently no approved medication for dry AMD, which represents between 85 to 90% of all AMD cases according to the American Macular Degeneration Foundation, and, based on our estimates from publicly available information, affects approximately 170 million people worldwide, and is expected to increase over time as the population ages.
There are a number of companies currently developing treatments, including anti-complement or neuroprotective agents administered by intraocular injections that may treat or alter the progression of dry AMD. We believe the market for AMD will remain fragmented and will include stand-alone and combination treatments for all stages of the disease and that a large market exists for a drug that could be administered orally rather than by monthly intraocular injections. We will continue to study BIO201 to determine its clinical safety and effectiveness, and to explore the feasibility of oral administration, and to further explain its mode of action.
Preclinical and Discovery Pipeline
Our preclinical pipeline currently consists of BIO201, as well as BIO103 and BIO203, which are chemically synthesized life-cycle extension products for BIO101 (20-hydroxyecdysone) and BIO201, respectively, potentially with better pharmacological properties. We are testing these preclinical drug candidates in preclinical models for multiple age-related diseases. We plan to continue to identify new drug candidates through our drug discovery platform based on our functional assays and reverse pharmacology approach.
Competition
The biotechnology and pharmaceutical industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. While we believe that our expertise in age-related diseases, scientific knowledge and intellectual property portfolio provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions, governmental agencies and public and private research institutions. Not only must we compete with other companies that are focused on neuromuscular diseases and retinopathies, but any drug candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
Many of our competitors may have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
The key competitive factors affecting the success of all of our drug candidates, if approved, are likely to be their efficacy, safety, tolerability, convenience, price and the availability of reimbursement from government and other third-party payors. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA, EMA or other national regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
88
The main competitors for each target indication of our drug candidates include:
| ● | Sarcopenia: We are not currently aware of any approved medications for sarcopenia. Pharmaceutical development of myostatin inhibitors and SARM have been halted, due to lack of evidence of benefit in multiple Phase 2 studies. Therapy development focuses mostly on exercise (including devices that can improve physical function), food supplements and dietary measures. Early stage development of cell therapy and agents that aim to improve muscle function has also started. |
| ● | Obesity: no drugs are approved to alleviate the muscle mass loss caused by GLP-1As in combination with dieting. Several companies have announced they have or will start development drugs which will be added to GLP-1As to either increase weight loss further and/or preserve muscle mass. These include myostatin inhibitors, some of which were previously investigated for sarcopenia but were discontinued for this indication (Regeneron, Roche, Versanis/Lilly), selective androgen receptor modulator (Veru), apelin receptor agonist (BioAge) and mixed activity drugs (ScolaRock). |
| ● | COVID-19: Many clinical studies have been run to develop medical responses to the COVID-19 virus, but the majority of them have failed to demonstrate any benefit for the patients. In the United States, several products have received emergency use authorizations for specific indications and patient groups such as anti-viral agents, including Paxlovid (nirmatrelvir and ritonavir and molnupiravir), monoclonal antibodies, including Evusheld (tixagevimab co-packaged with cilgavimab and administered together), and anti-inflammatory / immune-suppressing drugs, including anakinra (Kineret). Veklury (remdesivir), baricitinib (Olumiant) and tocilizumab (Actemra) have been approved by the FDA. Moreover, a number of vaccines have been authorized around the globe. In the EU, Veklury (remdesivir), RoActemra (tocilizumab), Kineret (anakinra), Paxlovid (PF-07321332 and ritonavir), Lagevrio (molnupiravir), Regkirona (regdanvimab), Ronapreve (casirivimab/imdevimab), Xevudy (sotrovimab), Regkirona (regdanvimab), Ronapreve (casirivimab/imdevimab and Evusheld (tixagevimab, cilgavimab) have been approved. |
| ● | Duchenne Muscular Dystrophy: Corticosteroids are the standard drug therapy for DMD patients in many countries throughout the world, this includes Emflaza (deflazacort, by PTC therapeutics), which was approved by the FDA in 2017, however their benefit for non-ambulatory patients with evidence of respiratory deterioration is limited. To our knowledge, several targeted therapies have been approved to date, which all are treatments that target the genetic mutation: Exondys51 (eteplirsen, by Sarepta), Vyondys53 (golodirsen, by Sarepta) and Amondys 45 (casimersen, by Sarepta) in the United States, and Translarna (ataluren, by PTC therapeutics) in Europe. Several steroid drugs may be prescribed from childhood to slow the progression of the disease, including prednisone, prednisolone or deflazacort. In this category, Santhera Therapeutics, has developped Agamree® (vamorolone) targeting all DMD patients from the age of 4. Agamree® received marketing authorizations in the US, the UK and Europe in late 2023 and has been launched in Germany in January 2024. While many new therapies are in development, most focus on ambulatory children. Only very few candidates, and in early stages, are being developed to treat patients who are non-ambulatory and with signs of respiratory deterioration. |
| ● | Dry Age-Related Macular Degeneration: Based on our review of research in this area, currently there are no approved therapeutic treatments for dry AMD. We believe that a number of other companies are developing drugs that may treat or alter the progression of the disease. Such competitors include, but are not limited to Allegro Ophthalmics, Apellis Pharmaceuticals, Astellas, Hemera Biosciences, Ionis Pharmaceuticals, Ophthotech Corporation, Roche and Stealth Biotherapeutics. |
89
Manufacturing and Supply
We do not own or operate, and currently have no plans to establish any manufacturing facilities. We currently rely, and expect to continue to rely, on third parties for the manufacturing of our drug candidates for both preclinical studies and all phases of clinical trials, as well as for commercial production should any of our drug candidates receive marketing approval for commercialization. We obtain key raw materials for BIO101 (20-hydroxyecdysone) and BIO201 from third-party suppliers. We are developing at the pilot and industrial scales the manufacturing processes and transfer them through agreements to third party European and American Clinical Development Manufacturing Organizations (“CDMOs”). Non GMP and GMP batches are produced in compliance with regulations for preclinical and clinical studies, including in view of the relevant guidelines adopted by the EMA, FDA, ANVISA and other regulatory authorities regarding the COVID-19 context. These batches allowed us to conduct all of our clinical programs. We plan to sign and signed agreements with the same and/or alternative manufacturers for industrial scale-up to submit the regulatory applications for approval and market access, subject to the global COVID-19 pandemic conditions and the impact of the current pandemic on operational capabilities. The manufacturing capacities allowed us to complete the SARA-INT clinical trial in sarcopenia and to conclude COVA phase 2/3 clinical study with BIO101 (20-hydroxyecdysone) in COVID-19, as well as conduct the first two parts of the planned MYODA clinical trial in DMD.
BIO101 (20-hydroxyecdysone)
The API of BIO101 (20-hydroxyecdysone), is a pharmaceutical grade small molecule, 20-hydroxyecdysone (>97% purity of the active molecule). We have produced the API for preclinical and clinical development by purifying the active molecule from Cyanotis sp or Stemmacantha sp, plants cultivated in China and used for medicinal purposes in Traditional Chinese Medicine. We currently rely on one supplier for the quantities of starting material for all our studies. We have not entered into a long-term supply agreement with this supplier for commercial scale up. However, we currently have a supply agreement allowing enough quantities for our ongoing clinical programs, for the manufacturing of validation and registration batches and to treat severe COVID-19 patients around the world, notably in the context of planned EAPs . BIO101 is purified for pharmaceutical use (>97% purity of the active molecule) using proprietary and patented processes, in compliance with GMP for pharmaceuticals, by Seqens our manufacturing partner located in France. We have not entered into a long-term supply agreement with Seqens. The partnership with Seqens has been put in place in July 2023 and manufacturing activities have started based on the robust process developed with our former partner Patheon/Thermofisher. Besides, we have initiated in September 2023 a partnership with Skypharma, based in France, to produce BIO101 (20-hydroxyecdysone) in its final capsule form. We therefore believe that we can secure sufficient quantities to conduct our future clinical trials and early commercial phases. Depending on positive positive outcome of our discussions with regulatory authorities regarding our COVA program, we will have to address significant upscaling of sourcing and manufacturing to support any commercial launch.
We are also evaluating alternative methods for producing BIO101 (20-hydroxyecdysone), such as new chemical synthesis or fermentation, and potential alternative plant sources, to optimize the supply chain.
BIO201
The API of BIO201 is a pharmaceutical grade small molecule norbixin (>97% purity of the active molecule). We have produced the API for preclinical development by chemical conversion into norbixin of the natural molecule bixin, which has been previously purified from seeds of Bixa orellana L., a plant traditionally used for medicinal purposes in the Amazon. At this time, we rely on one supplier of starting material (bixin) and we have not entered yet into a long-term supply agreement with this supplier, as the Company is looking for alternative sources of higher quality. The development of the manufacturing process will start as soon as comparative preclinical studies of BIO203 and BIO201 will indicate the most appropriate API for the development.
Research and Collaboration Agreements with Sorbonne University and Other Academic Research Institutions
Since the inception of the Company, we have entered into several research and collaboration agreements with Sorbonne University and other academic research institutions (i.e., the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Recherche Agronomique, or INRA, Institut National de la Santé et de la Recherche Médicale, or INSERM, and Université Paris Cité) in order to further strengthen our research and development strategies. These agreements are implemented according to the needs of our programs and their objective is to define the terms and conditions of our research (including its financing) and the results of such research.
90
The research and collaboration agreements are generally entered into for an initial fixed term (six to 12 months), and are extended by amendments as long as research is ongoing. The agreements may be terminated by any party to the agreement in the event of a breach by another party that has not been remedied within one month of a notice of the breach.
Pursuant to the terms of the research and collaboration agreements, each of the parties to the agreements remains the owner of intellectual property it owned prior to the time of the agreement, and all parties will have equal ownership of any patents resulting from the research conducted pursuant to such agreements. The parties must jointly agree as to whether the results of research conducted pursuant to the agreement should give rise to the filing of a patent application. In the event that one party does not wish to file a patent application but another party does and agrees to bear alone the cost of such filing, it will have the right to do so and the party who declined to pursue registration of the patent will be required to assign its co-ownership interest of the patent and patent applications to the other party at no charge. For any patent application that is filed, we are responsible for managing the patent application and all intellectual property registrations in France or abroad. In the event that a party desires to assign its co-ownership interest in a patent (except in the event of an assignment between Sorbonne University and CNRS or to one of the inventors within the team dedicated to the research), the other parties to the agreement will have a preemptive right to acquire such party’s co-ownership interest. We have an option to obtain exclusive commercial rights with respect to any products developed through the parties’ research pursuant to the terms of the collaboration agreements (whether patentable or not), which the Company exercised regarding patent families S1 through S10 and patent families MI through MV and still is in a position to exercise regarding ongoing researches and other patent families. The parties may use the results of research conducted pursuant to the agreements for other research purposes, subject to informing the other parties to the agreement if such research is to be carried out in collaboration with third parties.
Pursuant to the terms of the research and collaboration agreements, once a patent is filed, the parties to such agreement enter into (i) a co-ownership agreement providing for the respective rights and obligations of the co-owners of the patents, and (ii) a commercialization/license agreement providing for our right to commercialize products based on the patents in consideration for the payment of royalties to Sorbonne University and/or the other French academic research institutions involved, as applicable, the terms of which will supersede the collaboration agreement. Until these agreements are entered into, the provisions of the collaboration agreements will continue to govern ownership of the results and the rights to commercialize any products developed through such collaborations.
As of the date of this annual report, our historical research and collaboration agreements with Sorbonne University, CNRS and INSERM have expired, but they still produce scientific publications at the international level.
Intellectual Property
We seek to protect and enhance proprietary technology, investments, and improvements that are commercially important to our business by seeking, maintaining and defending patent rights. We also seek to and will continue to rely on regulatory protection afforded through orphan drug designations, data exclusivity, market exclusivity and patent term extensions where available.
Our industrial property protection policy covers our two key fields of innovation: (i) BIO101 (20-hydroxyecdysone) and our life-cycle extension drug candidate, BIO103, for the treatment of neuromuscular disorders, including sarcopenia spinal muscular atrophy (SMA) and DMD, respiratory function impairment resulting from a viral infection and (ii) BIO201 and our life-cycle extension drug candidate, BIO203, for the treatment of retinopathies, including dry AMD.
Current Intellectual Property Portfolio
Our patent portfolio covers 16 patent families, which include a total of 66 co-owned issued patents and a total of 47 co-owned patent applications. We have recently filed other patent applications fully owned by Biophytis that are currently under examination.
The issued patents in our portfolio consist of 13 European patents, 6 U.S. patents, and 32 patents in other jurisdictions, including Australia, Brazil, China, Japan, South Korea and Russia.
The pending patent applications in our portfolio consist of 2 European patent applications, 6 U.S. patent applications, and 42 patent applications pending in other jurisdictions, including notably Australia, Brazil, Canada, China, India, Japan, Mexico, Russia and South Korea.
91
Most of our patents and patent applications are jointly owned by us and Sorbonne, and in some cases together with other academic research institutions (i.e., CNRS, INRA and INSERM). We hold exclusive commercial rights through licenses of each of our drug candidates.
Our drug candidates rely upon one or more patent rights protecting various technologies, including rights related to:
| ● | the use of phytoecdysones in the preparation of a composition to act on metabolic syndrome (Patent family No. S1 “metabolic syndrome”); |
| ● | the use of phytoecdysones in stabilizing weight in overweight or obese subjects after dieting (Patent family No. S2 “weight stabilization”); |
| ● | the use of phytoecdysones to improve muscular quality in obese and/or sarcopenic mammals (Patent family No. S3 “muscular quality”); |
| ● | a process whereby new chemical entities are used in the preparation of medicines (Patent family No. S4 “phytoecdysone analogue”); |
| ● | a process for extracting purified 20-hydroxyecdysone and the therapeutic use of these extracts to improve muscle function or treat cardiovascular disease (Patent family No. S5 “20-hydroxyecdysone; extracts”); |
| ● | the use of 20-hydroxyecdysone components and their derivatives to treat myopathies and other muscular dystrophies (Patent family No. S6 “20-hydroxyecdysone for DMD”); |
| ● | the use of phytoecdysones to prevent loss of muscular strength after immobilization (Patent family No. S7 “Loss of muscle strength”); |
| ● | the use of phytoecdysones in a treatment of neuromuscular disease (Patent family No. S8 “Phytoecdysones in neuromuscular diseases”); |
| ● | the use of phytoecdysones in a treatment of impaired respiratory function (Patent family No. S9 “Phytoecdysones in respiratory diseases); |
| ● | the use of phytoecdysones and their derivatives for use in the treatment of impaired respiratory function during a viral infection (Patent family No.10 “Phytoecdysones in COVID-19 respiratory disease”); |
| ● | the use of phytoecdysones and their derivatives for use in the treatment of inflammatory respiratory diseases (such as asthma) (Patent family No. S11); |
| ● | the use of a composition of bixin and norbixin to protect the skin against sun damage (Patent family No. MI “Photo-protection”); |
| ● | the use of bixin and norbixin compounds to protect the eye against AMD (Patent family No. MII “AMD”); |
| ● | the use of a composition using norbixin in the treatment of AMD (Patent family No. MIII “Composition for protecting retinal epithelial cells”); |
| ● | the use of compounds from the family of flavonoids and anthocyanidins for the treatment, prevention and/or stabilization of AMD and/or Stargardt’s disease, pigmentary retinopathy and/or diabetic retinopathy (Patent family MIV “Use of 3-deoxyanthocyanidins for the treatment of eye diseases”); and |
| ● | The use of compounds targeting the eye and use thereof for treating ocular diseases (Patent family MV “Use of compounds targeting the eye for the treatment of ocular diseases”). |
92
Individual patent terms extend for varying periods of time, depending upon the date of filing of the patent application, the date of patent issuance, and the legal term of patents in the countries in which they are obtained. In most countries in which we file patent applications, including the United States, the patent term is 20 years from the date of filing of the first non-provisional application to which priority is claimed. In certain instances, a patent term can be extended under certain circumstances.
For example, in the United States, the term of a patent that covers an FDA-approved drug may be eligible for a patent term restoration of up to five years to effectively compensate for the patent term lost during the FDA regulatory review process, subject to several limitations discussed below under “Our Intellectual Property Strategy.” Also, in the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office in granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier-filed patent. Similar term extension mechanism may apply for patents filed with the Office Européen des Brevets (European patent office).
Our issued patents and patent applications (if issued) will expire as follows (unless extended):
Patent family No. S1:
| ● | Patent No. FR2924346 expires November 30, 2027. |
| ● | Patent Nos. AU2008332981, CN102231986, BRPI0820455-1, EP2217255, RU2010126625 and US8236359 expire November 19, 2028. |
Patent family No. S2:
| ● | Patent No. FR2982489 expires November 10, 2031. |
| ● | Patent Nos. CN103957727, EP2775859, JP6346094 and JP6462918 expire November 12, 2032. |
Patent family No. S3:
| ● | Patent No. FR2983733 expires December 13, 2031. |
| ● | Patent No. EP2790706 expires December 13, 2032. |
Patent family No. S4:
| ● | Patent No. FR3021318 expires May 20, 2034. |
| ● | Patent Nos. AU2015263121, CN106536539, EP3145942, JP6621217, RU2724329, US9938315 and US10316056 expire May 20, 2035. |
Patent family No. S5:
| ● | Patent No. FR3065644 expires April 28, 2037. |
Patent family No. S6:
| ● | Patent No. FR3065642 expires August 31, 2037. |
Patent family No. S7:
| ● | Patent No. FR3078252 expires February 28, 2038. |
Patent family No. S8:
| ● | Patent No. FR3093640 expires March 15, 2039. |
93
Patent family No. S9:
| ● | Patent No. FR3093641 expires March 15, 2039. |
Patent family No. S10
| ● | Patent No. FR3108504 expires March 30, 2040. |
Patent family No. S11
| ● | Patent No. FR2111920 expires November 10, 2041. |
Patent family No. MI
| ● | Patent Nos. FR2947173 and FR2955767 expire June 25, 2029 |
| ● | Patent Nos. BR1010113-6, EP2445476 and US9173823 expire June 25, 2030 |
Patent family No. MII
| ● | Patent Nos. FR2975008 and FR2996773 expire May 13, 2031. |
| ● | Patent Nos. EP2717891, JP6421306, and JP6432913 expire May 14, 2032. |
Patent family No. MIII
| ● | Patent No. FR3035589 expires April 30, 2035. |
| ● | Patent Nos. EP3288551, JP6660401, MX/a/2017/013918, RU2715889 and US10314804 expire April 28, 2036. |
Patent family No. MIV
| ● | Patent No. FR1554761 expires May 27, 2035. |
| ● | Patent Nos. EP33302463, JP6738412, RU2730854, BR112017025270 and US10513503 expire May 27, 2036. |
Patent family No. MV
| ● | Patent No. FR3105790 expires December 26, 2039 |
| ● | Patent Nos. EP4081507, JP6738412, RU2730854 and US10513503 expire December 26, 2040 |
In China, Patent No. ZL201280066803.6 from Patent family S3 was subject to a motion for invalidation brought by a third party based on several arguments, including the insufficient description of the animal model used in the patent, the novelty of the patent, the extension beyond the application as filed and the inventive step. Under Chinese patent law, the invalidity of a patent may be sought by any person or entity after the grant of the patent. The patent was invalidated in China following oral proceedings before the Court of Revision of the Chinese Patent Office. The arguments in favor of the invalidation by the Court of Revision of the Chinese Patent Office were not considered as relevant objections in the context of the European examination procedure leading to the grant of a European patent on May 8, 2019 (Patent No EP2790706). However, an opposition procedure to the European patent has been started, supposedly by the same opponent as in China (the latter remaining anonymous). The corresponding oral proceedings before the European Opposition Division took place and was won in November in 2021. The patent has thus been granted in Europe.
If patents are issued on our pending patent applications, the resulting patents are projected to expire on dates ranging from 2027 to 2044. However, the actual protection afforded by a patent varies on a product-by-product basis, from country-to-country, and
94
depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory-related extensions, the availability of legal remedies in a particular country, and the validity and enforceability of the patent.
Commercialization/License Agreements
As contemplated by the various research and collaboration agreements, we have entered into two commercialization/license agreements with respect to our patents which are co-owned with Sorbonne University and/or academic research institutions : (i) a commercialization/license agreement, dated January 1, 2016 by and between us and SATT Lutech (acting as agent for CNRS, INRA and Sorbonne University) and CNRS, INRA and Sorbonne University, as amended on April 2, 2019, November 6, 2020 and December 17, 2020, relating to patent families S1 through S10, or the S-Commercialization Agreement, and (ii) a commercialization/license agreement, dated January 1, 2016, by and between us and SATT Lutech (acting as agent for CNRS, INSERM and Sorbonne University) and CNRS, INSERM and Sorbonne University, as amended on December 17, 2020 relating to patent families MI through MV, or the M-Commercialization Agreement.
Unless terminated sooner, these agreements will remain in effect until the expiration or invalidation of the last of the patents covered by such agreement. The terms of the agreements provide that they will automatically terminate upon our termination of activity, wind-up and/or liquidation, a breach of the agreement, or upon a force majeure event (as described in the agreement). In addition, we may terminate these agreements upon 30 days’ notification to SATT Lutech and payment of a penalty equal to three times the annual guaranteed minimum amount, except where termination is justified by the denial of marketing authorizations.
We are required to make certain payments under the S-Commercialization Agreement and M-Commercialization Agreement as follows:
| ● | under the S-Commercialization Agreement, (i) beginning in the year following the first marketing of a product and in any event no later than 2023, we will pay a guaranteed annual minimum amount of €40 thousand, which will be deducted from the amount of royalties due annually (as described below), (ii) for direct commercialization by the us, the agreement provides for annual single-digit royalties based on the net sales of products, distinguishing between sales of nutraceutical and medicinal products, and (iii) for indirect commercialization by a third party, the agreement provides for annual royalties (10-20%) based on income received from licensees, distinguishing (a) between the sales of nutraceutical products (10-20% royalties) and drug products (10-20% royalties or single-digit royalties) and (b) the product development phase (Phase 1, 2 or 3) at the time of the conclusion of the licensing agreement; and |
| ● | under the M-Commercialization Agreement, (i) since 2020, we are paying a guaranteed annual minimum amount of €15 thousand, which will be deducted from the amount of royalties due annually, when applicable (as described below), (ii) beginning in the year following the first marketing of a drug product and in any event no later than 2026, the Company will pay an annual guaranteed minimum amount of €50 thousand, which will be deducted from the amount of royalties due annually (as described below), (iii) for direct commercialization by the us, the agreement provides for annual single-digit royalties based on the net sales of products, distinguishing between sales of nutraceutical and medicinal products, and (iii) for indirect commercialization by a third party, the agreement provides for annual royalties (10-20%) based on income received from licensees, distinguishing (a) between the sales of nutraceutical products (10-20% royalties) and drug products (10-20% or single-digit royalties) and (b) the product development phase (Phase 1, 2 or 3) at the time of the conclusion of the licensing agreement. The payments made under the S-Commercialization Agreement and the M-Commercialization Agreement will end upon termination of these agreements. |
Co-Ownership Agreements
As contemplated by the various research and collaboration agreements, we have entered into 10 co-ownership agreements with Sorbonne University and/or academic research institutions, covering all of our 16 patent families described above (S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, MI, MII, MIII, MIV and MV).
Each of these co-ownership agreements is entered into for a term ending upon expiration or invalidation of the last of the patents covered by such agreement, or, in the case of the co-ownership agreements covering patent families MI, MIII, MIV and MV, until expiration or invalidation of the last of the patents covered by the agreement or as long as the commercialization/license agreement remains in effect. These agreements may be terminated if one of the parties becomes the sole owner of the patents or in the event the parties no longer own the patents. In the event that assignment to a third party is contemplated, the other parties to the agreement will have a preemptive right to acquire such party’s co-ownership share.
95
Intellectual Property Agreement with Stanislas Veillet
Our CEO, who is a corporate officer (mandataire social) but not an employee of the Company under French law, is involved in our research and development activities. He has developed inventions with us for which we have submitted patent applications in which he is listed as a co-inventor and other inventions that we expect may give rise to patent applications in the future for which we expect he will be included as a co-inventor. As an inventor, our CEO has certain rights under French intellectual property law. These rights are distinct from the statutory rights that usually apply to employee inventors under French law. In order to define a framework within which any intellectual property resulting from our CEO’s research and development activities is properly assigned to us, we have entered into an agreement with our CEO, which has been approved by our board of directors, pursuant to which he is entitled to the following payments for his contributions:
| ● | a first lump sum cash payment of €90 thousand to be paid within 30 days of filing of a patent application based on the assigned rights; |
| ● | a second lump sum cash payment of €90 thousand to be paid within 30 days of publication of a patent application based on the assigned rights; and |
| ● | a 6.5% royalty payment with respect to any license income and/or any net sales by us of products manufactured with the patents filed on the basis of the assigned rights. |
These three payments will be capped at €2.1 million on a platform per platform basis, a platform being defined in the agreement as the research and development works which cover the same family of chemical molecules targeting the same molecular receptor or biological pathway for a family of pathologies which are clinically connected.
In the event that a third-party pharmaceutical and/or biotech company acquires 100% of our capital and voting rights, payments will be accelerated, so that the cap (€2.1 million per platform), less any amount previously paid in respect of a platform, will become immediately payable.
The agreement shall remain in effect until no further payments are due. However, the provisions of this agreement will only apply to results generated during the period in which our CEO occupies the position of a corporate officer of the Company or any of its affiliates. Any party to the agreement may, upon material breach of the agreement by the other party, terminate the agreement.
Trademarks
In addition to patent protection, we have trademark protection in many countries for our name (Biophytis) and our drug candidates (in particular, “Ruvembri”). In total, we hold 43 trademarks or trademark applications. None of our trademarks are subject to a third-party license.
Our Intellectual Property Strategy
Our patent policy is to file the first priority application regionally in France, then extend that patent application for international coverage by filing a related international application through the Patent Cooperation Treaty, or PCT. The PCT international application has the potential to be pursued in 142 PCT-contracting countries.
We determine which countries to pursue patent coverage in based on our business strategy. Our business strategy focuses on two main zones in which to pursue patent coverage via the PCT: (1) Europe, and in particular, the major European countries, United States, and Japan because these countries are where most of the main major pharmaceutical companies are concentrated, and (2) the BRIC zone, which is Brazil, Russia, India, and China; and sometimes Canada, Australia and South Korea.
Our objective for this international intellectual property strategy is to secure the earliest patents in these target countries and obtain the broadest and most effective scope of intellectual property protection in these countries. In addition to protecting our innovations by patents, they often have supplemental regulatory data exclusivity in connection with the marketing authorization of our products.
96
Government Regulation
Government authorities in the United States (including federal, state and local authorities) and in other countries, extensively regulate, among other things, the manufacturing, research and clinical development, marketing, labeling and packaging, storage, distribution, post-approval monitoring and reporting, advertising and promotion, pricing, and export and import of pharmaceutical products and active pharmaceutical ingredients, such as those we are developing. The process of obtaining regulatory approvals, authorizations, and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. Government Regulation
In the United States, the FDA regulates drugs under the FDCA and its implementing regulations. FDA approval is required before any new unapproved drug or dosage form, including a new use of a previously approved drug, can be marketed in the United States. Drugs are also subject to other federal, state and local statutes and regulations. If we fail to comply with applicable FDA or other requirements at any time during the drug development process, clinical testing, the approval process or after approval, we may become subject to administrative or judicial sanctions. These sanctions, but are not limited to, could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, untitled letters, product recalls, product seizures, placement on Import Alerts, debarment of personnel, employees or officers, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution.
The process required by the FDA before drug candidates may be marketed in the United States generally involves the following:
| ● | completion of extensive preclinical laboratory tests, preclinical animal studies, and toxicity data, performed in accordance with the GLP regulations; |
| ● | submission to the FDA of an IND, which must become effective before human clinical studies may begin; |
| ● | approval by an independent IRB or ethics committee representing each clinical site before each clinical study may be initiated; |
| ● | performance of several phases of adequate and well-controlled human clinical studies to establish the safety and efficacy of the drug candidate for each proposed indication; |
| ● | preparation of and submission to the FDA of a NDA after completion of all pivotal clinical studies; |
| ● | review of the product application by an FDA advisory committee, where appropriate and if applicable; |
| ● | a determination by the FDA within 60 days of its receipt of an NDA to file the application for review; |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities where the drug candidate is produced to assess compliance with cGMP; and |
| ● | FDA review and approval of an NDA or Biologic License Application, or BLA, prior to any commercial marketing or sale of the drug in the United States. |
An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology and pharmacodynamic characteristics of the product; chemistry, manufacturing and controls information; and any available human data or literature to support the use of the investigational new drug. An IND must become effective before human clinical studies may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions related to the proposed clinical studies within that 30 day window. If the FDA does raise concerns or questions within the 30 days following submission of the IND, the IND may be placed on clinical hold. During this time, the IND sponsor and the FDA must resolve any outstanding concerns or questions before clinical studies can begin. Accordingly, submission of an IND may or may not result in the FDA allowing clinical studies to commence.
97
Clinical Studies
Clinical studies involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical studies are conducted under protocols detailing, among other things, the objectives of the study, and the parameters to be used in monitoring safety and the efficacy criteria to be evaluated. A protocol for each clinical study and any subsequent protocol amendments must be submitted to the FDA as part of the IND. Additionally, approval must also be obtained from each clinical study site’s IRB before the studies may be initiated, and the IRB must monitor the study until completed. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries, such as ClinicalTrials.gov.
The clinical investigation of a drug is generally divided into three or four phases. Although the phases are usually conducted sequentially, they may overlap or be combined.
| ● | Phase 1. The drug is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to evaluate the safety, dosage tolerance, metabolism and pharmacologic actions of the investigational new drug in humans, the side effects associated with increasing doses, and if possible, to gain early evidence on effectiveness. |
| ● | Phase 2. The drug is administered to a limited patient population to evaluate dosage tolerance and determine optimal dosage, identify possible adverse side effects and safety risks and preliminarily evaluate efficacy. |
| ● | Phase 3. The drug is administered to an expanded patient population, generally at geographically dispersed clinical study sites to generate enough data at the recommended dose to evaluate safety , to demonstrate clinical effectiveness, to establish the overall benefit-risk relationship of the investigational product and to provide an adequate basis for product approval. |
| ● | Phase 4. In some cases, the sponsor performs clinical trials in the same condition as the approved product information (same recommended dose, same population, same indication). Such post-approval studies are typically referred to as Phase 4 clinical studies. |
| ● | Post marketing clinical studies: in some cases, the FDA may condition approval of an NDA for a drug candidate on the sponsor’s agreement to conduct additional clinical studies after approval. In other cases, a sponsor may commit to conducting or voluntarily conduct additional clinical studies after approval to gain more information about the drug. |
A confirmatory or pivotal study is a clinical study that adequately meets regulatory agency requirements for the evaluation of a drug candidate’s efficacy and safety such that it can be used to justify the approval of the product. Generally, pivotal studies are Phase 3 studies, but the FDA may accept results from Phase 2 studies if the study design provides a well-controlled and reliable assessment of clinical benefit, particularly in situations where there is an unmet medical need and the results are sufficiently robust. In such cases, the FDA may require post-market studies for safety and efficacy to be conducted for the drug candidate. The FDA may withdraw the approval if the results indicate that the approved drug is not safe or effective.
The FDA, the IRB or the clinical study sponsor may suspend or terminate a clinical study at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Additionally, some clinical studies are overseen by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study. We may also suspend or terminate a clinical study based on evolving business objectives and/or competitive climate.
Submission of an NDA to the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, detailed investigational new drug product information is submitted to the FDA in the form of an NDA requesting approval to market the product for one or more indications. Under federal law, the submission of most NDAs is subject to a substantial application user fee. Applications for orphan drug products are exempted from the NDA application user fees.
98
An NDA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of a product, or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational product to the satisfaction of the FDA.
Once an NDA has been submitted, the FDA’s goal is to review the application within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months after the FDA accepts the application for filing. The review process is often significantly extended by FDA requests for additional information or clarification.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP.
The FDA is required to refer an application for an investigational drug, which has an active ingredient that has not been previously approved, to an advisory committee or explain why such referral was not made. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the investigational product application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions and typically follows such recommendations.
The FDA’s Decision on an NDA
After the FDA evaluates the NDA and conducts inspections of manufacturing facilities, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may require additional clinical data and/or an additional pivotal Phase 3 clinical study(ies), and/or other significant, expensive and time-consuming requirements related to clinical studies, preclinical studies or manufacturing. Even if such additional information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA could also approve the NDA with a REMS to mitigate risks, which could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling, development of adequate controls and specifications or a commitment to conduct one or more post- market studies or clinical studies. Such post-market testing may include post marketing clinical studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. The FDA may have the authority to withdraw its approval if post-market testing fails to verify the approved drug’s clinical benefit, if the applicant does not perform the required testing with due diligence, or if the any other evidence demonstrates the approved drug is not safe or effective, among other reasons. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
Expedited Review, Accelerated-Approval and Emergency Use Authorization Programs
The FDA has various programs, including fast track, priority review, breakthrough therapy, accelerated approval, and regenerative medicine advanced therapy or RMAT designations that are intended to expedite the development and approval of new drugs that address unmet medical needs in the treatment of serious or life-threatening diseases and conditions. To be eligible for a fast track designation, the FDA must determine, based on the request of an applicant, that a product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need. The FDA may review sections of the NDA for a fast-track product on a rolling basis before the complete application is submitted. If the applicant provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable. The applicant pays any required user fees upon submission of the first section of the NDA.
99
The FDA may give a priority review designation to drugs that offer major advances in treatment, or provide a treatment where no adequate therapy exists. A priority review designation means that the goal for the FDA to review an application is six months, rather than the standard review of ten months. These six and ten-month review periods are measured from the “filing” date rather than the receipt date for NDAs for new molecular entities, which typically adds approximately two months to the timeline for review and decision from the date of submission. Products that are eligible for fast-track designation are also likely to be considered appropriate to receive a priority review.
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may be eligible for accelerated approval and may be approved on the basis of adequate and well-controlled clinical studies establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit. This evaluation takes into account the severity, rarity or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require a sponsor of a drug receiving accelerated approval to perform post-marketing studies to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical endpoint, and the drug may be subject to accelerated withdrawal procedures.
Moreover, under the provisions of the Food and Drug Administration Safety and Innovation Act passed in July 2012, a sponsor can request designation of a drug candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development.
Drugs designated as breakthrough therapies are also eligible for priority review and fast track designation. As part of this process, the FDA takes certain actions, such as holding timely meetings and providing advice, intended to expedite the development and review of an application for approval of a breakthrough therapy.
In addition, the 21st Century Cures Act in 2016 made the Regenerative Medicine Advanced Therapy, or RMAT, designation available for investigational drugs that are regenerative medicine therapies intended to treat, modify, reverse, or cure a serious condition, with preliminary clinical evidence indicating that the drug has the potential for addressing unmet medical needs for such condition. The RMAT designation is available for cell therapy, therapeutic tissue engineering products, human cell and tissue products, and combination products that use such therapies or products. The advantages of RMAT designation include those of breakthrough and fast track designations, such as early interactions with the FDA and rolling review of applications, and the drug candidate with the RMAT designation may be eligible for accelerated approval. Requests for RMAT designations should be made with the IND application (if preliminary clinical evidence is available), but no later than the end-of-Phase-2 meeting.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for the FDA review or approval will not be shortened or will be withdrawn.
100
With the previous declaration of COVID-19 as a worldwide pandemic and public health emergency, several programs have been utilized, to expedite review of medications. These include:
| ● | EUA: Authority allows the FDA to help strengthen the nation’s public health protections against chemical, biological, radiological and nuclear threats by facilitating the availability and use of medical countermeasures needed during public health emergencies. Under section 564 of the Federal Food, Drug and Cosmetic Act, or FD&C Act, the FDA Commissioner may allow unapproved medical products, or unapproved uses of approved medical products, to be used in an emergency to diagnose, treat, or prevent serious or life-threatening diseases or conditions caused by chemical, biological, radiological and nuclear (CBRN) threat agents when there are no adequate, approved, and available alternatives. The EUA allows temporary use of the medical product, based on efficacy data, which is usually not sufficient on its own for approval. For example, A few anti-viral agents (including Paxlovid (nirmatrelvir and ritonavir), and molnupiravir) as well as monoclonal antibodies REGEN-COV (Casirivimab and Imdevimab), Bebtelovimab, (sotrovimab and Evusheld (tixagevimab co-packaged with cilgavimab and administered together)) have received emergency use authorizations for certain patient populations and indications. An EUA may be revoked at the conclusion of a public health emergency, and there may be certain limitations to its uses, such as label statements specifying that the product only has an EUA, and that it has not received the FDA’s clearance or approval. While some EUAs available due to COVID-19 remain in place in limited circumstances, these pathways could be terminated at any time by declaration by the Commissioner or the Secretary of the Department of Health and Human Services. |
Post-Approval Requirements
Drugs marketed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims are subject to prior FDA review and approval. There also are continuing, annual user fee requirements.
Manufacturers are subject to periodic unannounced inspections by FDA and state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon us and any third- party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
Discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post- market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, but are not limited to:
| ● | restrictions on the marketing or manufacturing of the product; |
| ● | complete withdrawal of the product from the market or product recalls; |
| ● | fines, Form 483 observations, warning letters, untitle letters, or holds on post-approval clinical studies; |
| ● | refusal of the FDA to approve pending NDAs or supplements to approved NDAs or suspension or revocation of product approvals; |
101
| ● | product seizure or detention, or refusal to permit the import or export of products; and/or |
| ● | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications, in accordance with the provisions of the approved label, and materials may not be false or misleading. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses and false or misleading advertising, and a company that is found to have improperly marketed its products may be subject to significant liability.
It is expected that, with respect to COVID-19-related EUA programs, data collection post-approval will be required, either in the form of a clinical trial, or by other methods (e.g. real-world data). Several anti-viral agents, for example Paxlovid (nirmatrelvir and ritonavir), and molnupiravir) as well as monoclonal antibodies (sotrovimab and Evusheld (tixagevimab co-packaged with cilgavimab and administered together)) have already received emergency use authorizations in the United States for specific indications and patient groups. These products are reporting post-approval data and are undergoing additional clinical study, and other products with EUAs will likely require similar reporting to retain their authorizations under the FDA’s current approach.
Orphan Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan designation to a drug intended to treat a rare disease or condition, defined as a disease or condition with a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 individuals in the United States and when there is no reasonable expectation that the cost of developing and making available the drug in the United States will be recovered from sales in the United States for that drug. Orphan drug designation must be requested before submitting a BLA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.
If a product that has orphan drug designation subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan product marketing exclusivity, which means that the FDA may not approve any other applications, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or if the FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition.
Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
A designated orphan drug may not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or, as noted above, if the second applicant demonstrates that its product is clinically superior to the approved product with orphan exclusivity or the manufacturer of the approved product is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
102
Hatch-Waxman Amendments and Exclusivity
Section 505 of the FDCA describes three types of marketing applications that may be submitted to the FDA to request marketing authorization for a new drug. A Section 505(b)(1) NDA is an application that contains full reports of investigations of safety and efficacy. A 505(b)(2) NDA is an application that contains full reports of investigations of safety and efficacy but where at least some of the information required for approval comes from investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. This regulatory pathway enables the applicant to rely, in part, on the FDA’s prior findings of safety and efficacy for an existing product, or published literature, in support of its application. Section 505(j) establishes an abbreviated approval process for a generic version of approved drug products through the submission of an ANDA. An ANDA provides for marketing of a generic drug product that has the same active ingredients, dosage form, strength, route of administration, labeling, performance characteristics and intended use, among other things, to a previously approved product. ANDAs are termed “abbreviated” because they are generally not required to include preclinical (animal) and clinical (human) data to establish safety and efficacy. Instead, generic applicants must scientifically demonstrate that their product is bioequivalent to, or performs in the same manner as, the innovator drug through in vitro, in vivo or other testing. The generic version must deliver the same amount of active ingredients into a subject’s bloodstream in the same amount of time as the innovator drug and can often be substituted by pharmacists under prescriptions written for the reference listed drug. In seeking approval for a drug through an NDA, applicants are required to list with the FDA each patent with claims that cover the applicant’s drug or a method of using the drug. Upon approval of a drug, each of the patents listed in the application for the drug is then published in the Orange Book. Drugs listed in the Orange Book can, in turn, be cited by potential competitors in support of approval of an ANDA or 505(b)(2) NDA.
Upon submission of an ANDA or a 505(b)(2) NDA, an applicant must certify to the FDA that (1) no patent information on the drug product that is the subject of the application has been submitted to the FDA; (2) such patent has expired; (3) the date on which such patent expires; or (4) such patent is invalid or will not be infringed upon by the manufacture, use or sale of the drug product for which the application is submitted. Generally, the ANDA or 505(b)(2) NDA cannot be approved until all listed patents have expired, except where the ANDA or 505(b)(2) NDA applicant challenges a listed patent through the last type of certification, also known as a paragraph IV certification. If the applicant does not challenge the listed patents, or indicates that it is not seeking approval of a patented method of use, the ANDA or 505(b)(2) NDA application will not be approved until all of the listed patents claiming the referenced product have expired.
If the ANDA or 505(b)(2) NDA applicant has provided a Paragraph IV certification to the FDA, the applicant must send notice of the Paragraph IV certification to the NDA and patent holders once the application has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the paragraph IV certification. If the paragraph IV certification is challenged by an NDA holder or the patent owner(s) asserts a patent challenge to the paragraph IV certification, the FDA may not approve that application until the earlier of 30 months from the receipt of the notice of the paragraph IV certification, the expiration of the patent, when the infringement case concerning each such patent was favorably decided in the applicant’s favor or settled, or such shorter or longer period as may be ordered by a court. This prohibition is generally referred to as the 30-month stay. In instances where an ANDA or 505(b)(2) NDA applicant files a paragraph IV certification, the NDA holder or patent owner(s) regularly take action to trigger the 30-month stay, recognizing that the related patent litigation may take many months or years to resolve.
The FDA also cannot approve an ANDA or 505(b)(2) application until all applicable non-patent exclusivities listed in the Orange Book for the branded reference drug have expired. For example, a pharmaceutical manufacturer may obtain five years of non-patent exclusivity upon NDA approval of a new chemical entity, or NCE, which is a drug containing an active moiety that has not been approved by the FDA in any other NDA. An “active moiety” is defined as the molecule responsible for the drug substance’s physiological or pharmacologic action. During that five-year exclusivity period, the FDA cannot accept for filing (and therefore cannot approve) any ANDA seeking approval of a generic version of that drug or any 505(b)(2) NDA that relies on the FDA’s approval of the drug, provided that the FDA may accept an ANDA four years into the NCE exclusivity period if the ANDA applicant also files a Paragraph IV certification.
A drug, including one approved under Section 505(b)(2), may obtain a three-year period of exclusivity for a particular condition of approval, or change to a marketed product, such as a new formulation for a previously approved product, if one or more new clinical studies (other than bioavailability or bioequivalence studies) was essential to the approval of the application and was conducted/sponsored by the applicant. Should this occur, the FDA would be precluded from approving any ANDA or 505(b)(2) application for the protected modification until after that three-year exclusivity period has run. However, unlike NCE exclusivity, the FDA can accept an application and begin the review process during the exclusivity period.
103
Other Healthcare Laws and Compliance Requirements
U.S. pharmaceutical companies are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which they conduct their business. Such laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security and physician sunshine laws and regulations. If pharmaceutical company operations are found to be in violation of any of such laws or any other applicable governmental regulations, these companies may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, the curtailment or restructuring of operations, exclusion from participation in federal and state healthcare programs and individual imprisonment.
Coverage and Reimbursement
Sales of any product depend, in part, on the extent to which such product will be covered by third-party payors, such as federal, state and foreign government healthcare programs, commercial insurance and managed healthcare organizations and the level of third-party reimbursement for such product. Third-party payor decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. These third-party payors often reduce reimbursements for medical products, drugs and services. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit sales of any product. Decreases in third-party reimbursement for any product or a decision by a third-party payor not to cover a product could reduce physician usage and patient demand for the product and also has a material adverse effect on sales. Even after the FDA approves a product, for example, the failure to obtain third-party payor coverage may impose a material adverse effect on sales. As federal and state governments continue to promulgate new policies and regulations, these policies and regulations may also impose a material adverse effect on sales. These laws and regulations may restrict, prohibit, or preventing us from implementing a wide range of pricing, discounting, marketing, promotion, sales commission, incentive programs, and other business activities. No uniform policy of coverage and reimbursement among third-party payors exists in the United States. Finally, although payors often rely upon Medicare coverage policy establishing their coverage and reimbursement policies. Instead, each payor makes independent and separate decisions regarding the extent of coverage and amount of reimbursement to be provided.
Healthcare Reform
In March 2010, former President Obama signed the Affordable Care Act, which substantially changed the way healthcare is financed by both governmental and private insurers in the United States, and significantly affected the pharmaceutical industry. The Affordable Care Act contains a number of provisions, including those governing enrollments in federal healthcare programs, reimbursement adjustments and fraud and abuse changes. Additionally, the Affordable Care Act increases the minimum level of Medicaid rebates payable by manufacturers of brand name drugs from 15.1% to 23.1%; requires collection of rebates for drugs paid by Medicaid managed care organizations; requires manufacturers to participate in a coverage gap discount program, under which they must agree to offer 50% point-of- sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and imposes a non-deductible annual fee on pharmaceutical manufacturers or importers who sell “branded prescription drugs” to specified federal government programs.
Since its enactment, there have been judicial and Congressional challenges to certain aspects of the Affordable Care Act, and we expect there will be additional challenges and amendments to the Affordable Care Act in the future. Other legislative changes have been proposed and adopted since the Affordable Care Act was enacted, including aggregate reductions of Medicare payments to providers of 2% per fiscal year for certain Medicare providers and suppliers, and further reduced payments to several types of Medicare providers.
Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed bills designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. Individual states in the United States have also become increasingly active in implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, proposing to encourage importation from other countries and bulk purchasing.
104
CARES Act
In March 2020, the United States Congress passed the Coronavirus Aid, Relief, and Economic Security, or CARES, Act, a $2 trillion relief package created in response to the ongoing COVID-19 pandemic in the United States. Although Congress directed a significant portion of CARES Act aid to health care providers and institutions that serve on the “front line” of the COVID-19 crisis, Congress also allocated $940 million to the NIH, the U.S. government’s primary agency responsible for biomedical research. Additionally, for companies that engage in research studies that involve routine costs payable by federal health care programs, such as Medicare and Medicaid, the CARES Act includes a number of measures designed to ease restrictions, enhance coverage, or even accelerate reimbursement for those routine costs in limited circumstances. Although CARES Act financial aid and easing of restrictions are specifically intended to address the COVID-19 emergency and are thus generally temporary, the sheer size and breadth of relief opportunities afforded under the law could positively impact life science and biotechnology companies’ growth in the long term (i.e., even beyond the pandemic), particularly for early stage companies engaged in COVID-19 related research.
Foreign Corrupt Practices Act
Our business activities may be subject to the FCPA and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we operate. The FCPA generally prohibits offering, promising, giving, or authorizing others to give anything of value, either directly or indirectly, to a non-U.S. government official in order to influence official action, or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise and maintain an adequate system of internal accounting controls. Our business is heavily regulated and therefore involves significant interaction with public officials, including officials of non-U.S. governments. Additionally, in many other countries, the health care providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are government entities; therefore, our dealings with these prescribers and purchasers are subject to regulation under the FCPA. There is no certainty that all of our employees, agents, suppliers, manufacturers, contractors, or collaborators, or those of our affiliates, will comply with all applicable laws and regulations, particularly given the high level of complexity of these laws. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers, or our employees, the closing down of facilities, including those of our suppliers and manufacturers, requirements to obtain export licenses, cessation of business activities in sanctioned countries, implementation of compliance programs, and prohibitions on the conduct of our business. Any such violations could include prohibitions on our ability to offer our products in one or more countries as well as difficulties in manufacturing or continuing to develop our products, and could materially damage our reputation, our brand, our international expansion efforts, our ability to attract and retain employees, and our business, prospects, operating results, and financial condition.
European Union Drug Development
In the European Union, our drug candidates may also be subject to extensive regulatory requirements. As in the United States, medicinal products can only be marketed if a marketing authorization from the competent regulatory agencies has been obtained.
Similar to the United States, the various phases of preclinical and clinical development in the European Union should be performed in compliance with the EU regulations and directives, the national regulations and the international standards for GMP, GLP and GCP.
Clinical trials are currently governed by EU Clinical Trials Regulation No 536/2014/EC adopted on 16 April 2014 and repealing Directive 2001/20/EC that set out common rules for the control and authorization of clinical trials in the European Union, as well as by the Good Clinical Practice (GCP) Directive 2005/28/EC and the ICH guidelines on GMP and GCP
Before a clinical trial can be initiated it must be approved in each of the EU Member States where the trial is to be conducted by two distinct bodies: the National Competent Authority, or NCA, and one national Ethics Committee, or ECs. All suspected unexpected serious adverse reactions, or SUSARs, to the investigated drug that occur during the clinical trial have to be reported to the NCA and ECs of the Member State where they occurred. The Regulation also imposes Drug Safety Update Report (DSUR) to be submitted annually to both the Competent authorities and EC and public transparency rules.
105
European Union Drug Review and Approval
In the EEA (which is comprised of the 27 (after the Brexit) Member States of the European Union plus Norway, Iceland and Liechtenstein), medicinal products can only be commercialized after obtaining a Marketing Authorization, or MA. MAs may be granted either centrally (Community MA) or nationally (National MA).
The Community MA is issued centrally by the European Commission after the CHMP of the EMA issued a positive opinion based on a MA dossier has been evaluated through the Centralized Procedure. This single Community MA is valid throughout the entire territory of the EU and will be recognized in the other EEA Member States Norway, Iceland and Liechtenstein which issue their own national MA. The Centralized Procedure is mandatory for certain types of products such as orphan medicinal products and medicinal products containing a new active substance indicated for the treatment of neurodegenerative disorders, of cancers or diabetes. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EU, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the European UnionThe European Commission may also grant a “conditional marketing authorization” prior to obtaining the comprehensive clinical data required for an application for a full marketing authorization. Such conditional marketing authorizations may be granted for product candidates (including medicines designated as orphan medical products), if:
| ● | the risk-benefit balance of the product candidate is positive; |
| ● | it is likely that the applicant will be in a position to provide the required comprehensive clinical trial data; |
| ● | the product fulfills an unmet medical need; and |
| ● | the benefit to public health of the immediate availability on the market of the medicinal product concerned outweighs the risk inherent in the fact that additional data are still required. |
A conditional marketing authorization may contain specific obligations to be fulfilled by the marketing authorization holder including obligations with respect to the completion of ongoing or new studies, and with respect to the collection of pharmacoviligance data. Conditional marketing authorizations are valid for one year, and may be renewed annually, if the risk-benefit balance remains positive, and after an assessment of the need for additional or modified conditions and/or specific obligations.
National MAs are issued nationally by the competent authorities of the Member States of the EEA and only cover their respective territory. National MAs are available for products not falling within the mandatory scope of the Centralized Procedure.
Under the above-described procedures, before granting the MA, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
Pursuant to Regulation (EC) No. 1901/2006, or the “Pediatric Regulation”, all applications for marketing authorization for new medicines, as well as all applications for new indications of approved medicines, must include to be valid, in addition to the particulars and documents referred to in Directive 2001/83/EC, the results of all studies performed and details of all information collected in compliance with a pediatric investigation plan agreed between regulatory authorities and the MA holder, unless the medicine is exempt because of a deferral or waiver of the EMA.
Before the EMA is able to begin its assessment of a Community MA application, it will validate that the applicant has complied with the agreed pediatric investigation plan. Products that are granted a MA on the basis of the pediatric clinical trials conducted in accordance with the Pediatric Investigation Plan, or PIP, are eligible for a six-month extension of the protection under a supplementary protection certificate (if any is in effect at the time of approval) or, in the case of orphan medicinal products, a two-year extension of the orphan market exclusivity. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
106
Orphan Drugs
In the European Union, Regulation (EC) No 141/2000 of the European Parliament and of the Council of December 16, 1999 on orphan medicinal products, as amended, states that a drug shall be designated as an orphan drug if its sponsor can establish that the three following cumulative conditions are met:
| ● | the product is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition; |
| ● | the prevalence of the conditions is not more than five in ten thousand persons in the European Union when the application is made, or that it is intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely that the marketing of the drug in the European Union would generate sufficient return to justify the necessary investment; and |
| ● | that there is no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized in the European Union or, if such method exists, that the drug will be of significant benefit to those affected by that condition. |
Pursuant to Regulation (EC) No. 847/2000 of April 27, 2000 laying down the provisions for implementation of the criteria for designation of a medicinal product as an orphan medicinal product and definitions of the concepts “similar medicinal product” and “clinical superiority”, an application for the designation of a drug as an orphan drug must be submitted at any stage of development of the drug before filing of a MA application.
The European Union offers incentives to encourage the development of designated orphan medicines (protocol assistance, fee reductions, etc.) and provides opportunities for market exclusivity.
Pursuant to abovementioned Regulation (EC) No. 141/2000, If a Community MA in respect of an orphan drug is granted, regulatory authorities will not, for a period of usually ten years, accept another application for a MA, or grant a MA or accept an application to extend an existing MA, for the same therapeutic indication, in respect of a similar drug (Market exclusivity). This period may however be reduced to six years if, at the end of the fifth year, it is established, in respect of the drug concerned, that the above-mentioned criteria for orphan drug designation are no longer met, in other words, when it is shown on the basis of available evidence that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Pursuant to Regulation No. 1901/2006, for orphan medicinal products, instead of an extension of the supplementary protection certificate, the ten-year period of orphan market exclusivity should be extended to 12 years if the requirement for data on use in the pediatric population is fully met (i.e. when the request contains the results of all studies carried out under the approved PIP and when the declaration attesting the conformity of the request to this PIP is included in the MA).
Notwithstanding the foregoing, a MA may be granted, for the same therapeutic indication, to a similar drug if:
| ● | the holder of the MA for the original orphan drug has given its consent to the second applicant; |
| ● | the holder of the MA for the original orphan drug is unable to supply sufficient quantities of the drug; or |
| ● | the second applicant can establish in the application that the second drug, although similar to the orphan drug already authorized, is safer, more effective or otherwise clinically superior. |
The abovementioned Regulation (EC) No. 141/2000 provides for other incentives regarding orphan medicinal products.
Post-Approval Controls
The holder of a MA must comply with EU requirements applicable to manufacturing, marketing, promotion and sale of medicinal products. In particular, the holder of the MA must establish and maintain a pharmacovigilance system and appoint a Qualified Person Responsible for Pharmacovigilance, or QPPV, who is responsible for oversight of that system and who will reside and operate in the EU. Key obligations include safety expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
107
All new application must include a risk management plan, or RMP, to submit to the EMA, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the MA. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies. RMPs and PSURs are routinely available to third parties requesting access, subject to limited redactions. All advertising and promotional activities for the product must be consistent with the approved summary of product characteristics, and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription medicines is also prohibited in the European Union. Although general requirements for advertising and promotion of medicinal products are established under EU directives, the details are governed by regulations in each EU Member State and can differ from one country to another.
Reimbursement
The European Union provides options for its Member States to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A Member State may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. For example, in France, effective market access will be supported by agreements with hospitals and products may be reimbursed by the Social Security Fund. The price of medicines covered by national health insurance is negotiated with the French Economic Committee for Health Products, or CEPS. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our drug candidates.
Historically, products launched in the European Union do not follow price structures of the United States and generally prices tend to be significantly lower.
Other European Regulatory Matters
French Regulatory Framework for Clinical Development
The EU Regulation No 536/2014/EC adopted on 16 April 2014 requiring prior favorable opinion from an ethics committee before starting any clinical trial applies to all EU Member States including France.
In France, parties to a CTA must use a CTA template (“unique agreement” or convention unique) to organize the conduct of interventional clinical trials with commercial purpose, as well as specific template exhibits to this agreement. The use of the unique agreement template is mandatory if the research takes place in a public health establishment, institution (“maison de sante”), or centre (“centre de sante”) in France. Once concluded, the CTA is communicated for information by the sponsor to the French national board of physicians (Ordre national des médecins) without delay.
The processing of personal data, including health data, collected during clinical trials has to comply with the Regulation (EU) 2016/679 of the European Parliament and of the Council of April 27, 2016 and Law No 2018-493 of June 20, 2018 on the protection of personal data, implementing the Regulation (EU) 2016/679 requirements, as well as the guidelines of the French data protection authority, the Commission Nationale de l’Informatique et des Libertés, or CNIL. Regarding automatic processing operations for the purpose of research or clinical studies, formalities have to be completed before the CNIL, so as to obtain the authorization to process personal data. However, there are simplified standards.
French Law No. 2011-2012 of December 29, 2011, or Loi Bertrand, aimed at strengthening the health safety of medicinal and health products, as amended (and its implementing decrees), introduced into French law provisions regarding transparency of fees received by some healthcare professionals from health product industries, i.e. companies manufacturing or marketing health products (Article L.1453-1 of the French Public Health Code). The French Decree No. 2016-1939 of December 28, 2016 clarifies that companies manufacturing or marketing health care products (medicinal products, medical devices, etc.) in France shall publicly disclose (mainly on a specific public website available at: https://www.entreprises-transparence.sante.gouv.fr) the advantages and fees paid to healthcare professionals amounting to €10 or above, as well as the agreements concluded with the latter, along with detailed information about each agreement (the precise subject matter of the agreement, the date of signature of the agreement, its end date, the total amount paid to the healthcare professional, etc.). Another declaration must also be filed to the competent healthcare professional body. Several decrees have further extended the scope of these declarations. For instance, under Decree No. 2019-1530 of December 30, 2019, companies will also have to disclose agreements concluded with persons who present one or more health products in the media or social networks in such a way as to influence the public.
108
Law No. 2011-2012 also reinforced the French anti-gift rules. Further to subsequent modifications in 2017 and 2019, new Articles L. 1453-3 et seq. of the French Public Health Code amended the Anti-Gift regime and expanded the scope of the general prohibition of payments from pharmaceutical and device manufacturers to healthcare professionals to broadly cover any company manufacturing or marketing health products, regardless of whether or not payment for the products is reimbursed under the French social security system (new Articles L. 1453-3 et seq. of the French Public Health Code). Derogation must be submitted to the relevant healthcare professional body. Moreover, the penalties incurred for non-compliance with the requirements of the Anti-Gift regime by a healthcare company may lead to a fine of up to €750,000.
Early Access Program
The Early Access Program (EAP) is governed by the French Public Health Law art. L5121-12 in France. This an exceptional, temporary regulatory disposition which allows to make unapproved drugs or unapproved use of approved drugs available to the patients waiting for the granting of its MA. This regulatory disposition is considered when the following criteria are met:
- | the disease is rare or serious |
- | there is an unmet need |
- | the treatment could not be deferred |
- | the benefit/risk ratio of the drug is presumed positive in the sought indication |
- | the drug is presumed to be an innovative drug |
The authorization of an EAP in France is granted by the HAS (Haute Autorité de Santé) taking into account the opinion of the French competent authorities (ANSM) in charge of evaluating the benefit/risk ratio of the concerned drug in the seek EAP indication. The company who is seeking for an EAP should submit a dossier containing quality, non-clinical and clinical data, a commitment to apply for a MA within 2 years of the EAP authorization and a protocol for the collection of patient’s clinical safety and efficacy data. This data collection is maintained until the end of the EAP and periodic reports should be submitted to the competent authorities. When approved, the EAP is valid until the effective marketing of the drug unless the above mentioned eligibility criteria are not met anymore, the company does not comply with its commitment to apply for a MA, in case of public health reasons, non-compliance with the obligation to collect patients clinical data, if the benefit/risk ratio is no more presumed positive or in case the outcome of the MA dossier assessment is negative.
French Pharmaceutical Company Status
In France, there is a regulated status of pharmaceutical establishment and operating company, which allows us to manufacture, store, import, distribute and market drugs. Obtaining a pharmaceutical establishment license, either as a distributor or as a manufacturer requires the submission of an application file to the ANSM. The application package will vary depending on the type of application (distribution license or manufacturing license). The ANSM grants such license after verifying that the company has adequate premises, the necessary personnel and adequate procedures to carry out the proposed pharmaceutical activities.
109
C.Organizational Structure
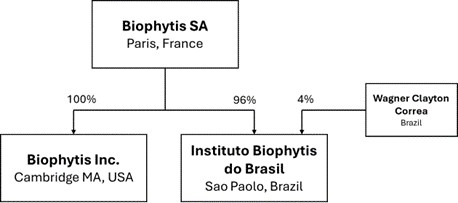
D. | Property, Plants and Equipment |
We lease approximately 504 square meters of office space at Sorbonne University—BC 9, Bâtiment A 4ème étage, 4 place Jussieu, 75005 Paris, France for research and development and administrative activities. The lease agreement (convention d’occupation du domaine public) provides for a one-year renewable term. We incurred annual lease expense of €200 thousand for the year 2023. We believe that our existing facility is adequate to meet our current needs, and that suitable additional alternative spaces will be available in the future on commercially reasonable terms.
Item 4A. Unresolved Staff Comments.
Not applicable.
Item 5. Operating and Financial Review and Prospects.
Overview
We are a clinical-stage biotechnology company specialized in the development of therapeutics that are aimed at slowing the degenerative processes associated with aging and improving functional outcomes for patients suffering from age-related diseases, including severe respiratory failure in patients suffering from COVID-19. BIO101 (20-hydroxyecdysone), our leading drug candidate, is a small molecule, administered orally, being developed as a treatment for sarcopenia (Phase 2 clinical trial SARA-INT completed in the United States and Europe), as well as for severe respiratory manifestations of COVID-19 (Phase 2-3 study COVA completed in Europe, Latin America, and the United States). A pediatric formulation of BIO101 (20-hydroxyecdysone) is being developed for the treatment of Duchenne Muscular Dystrophy (DMD).
Financial Operations Overview
The following discussion sets forth certain components of our statements of operations as well as factors that impact those items.
Revenues
To date we have not generated any revenues from product sales, or otherwise, and we do not expect to recognize any revenue from the sale of products, even if we obtain regulatory approval for the products, in the near term. Our ability to generate revenue in the future will depend almost entirely on our ability to successfully develop, obtain regulatory approval for and then commercialize our drug candidates.
110
Research and Development Expenses
Our research and development expenses consist primarily of costs incurred in connection with the development of our drug candidates, including:
| ● | personnel-related costs, such as salaries, bonuses, benefits, travel and other related expenses, including share-based compensation; |
| ● | expenses incurred under our agreements with CROs, clinical sites, contract laboratories, medical institutions and consultants that plan and conduct our preclinical studies and clinical trials; |
| ● | costs associated with regulatory filings; |
| ● | costs of acquiring preclinical study and clinical trial materials, and costs associated with preclinical development formulation and process development; |
| ● | depreciation, maintenance and other facility-related expenses; and |
| ● | as an offset of our research and development expenses, the CIR, which is a French tax credit dedicated to R&D. |
To date, we have expensed all research and development costs as incurred, as we do not currently meet the conditions to capitalize expenditures on drug development activities, as provided in IAS 38 Intangible Assets.
Clinical development expenses for our drug candidates are a significant component of our current research and development expenses as we advance our drug candidates into and through clinical trials. Drug candidates in later stage clinical development generally have higher research and development costs than those in earlier stages of development, primarily due to increased size and duration of the clinical trials. We recognize costs for each grant project, preclinical study or clinical trial that we conduct based on our evaluation of the progress to completion, using information and data provided to us by our research and development vendors and clinical sites.
We expect our research and development expenses to increase for the foreseeable future as we progress our drug candidates into and through clinical trials. Furthermore, to the extent we undertake to commercialize any drug candidates approved or authorized for any indication for sale, our expenses will likely increase even more. The process of conducting the necessary clinical research to obtain regulatory approval or authorization of a drug candidate is costly and time consuming, and we will require additional funding to fund our continuing operations. The probability that any of our drug candidates receives regulatory approval or authorization and eventually is able to generate revenue depends on a variety of factors, including the quality of our drug candidates, early clinical data, investment in our clinical program and further clinical validation, competition, manufacturing capability and commercial viability. We may never succeed in obtaining regulatory approval or authorization for any of our drug candidates. As a result of these uncertainties, we are unable to determine the duration and completion costs of our research and development projects or if, when and to what extent we will generate revenue from the commercialization and sale of any of our drug candidates, if approved or authorized.
General and Administrative Expenses
General and administrative expenses include personnel costs, costs for outside professional services and other allocated expenses. Personnel costs consist of salaries, bonuses, benefits, travel and share-based compensation. Outside professional services consist of legal, accounting and audit services, commercial evaluation and strategy services, and other consulting services. We expect general and administrative expenses to increase in the near future with the expansion of our staff and management team to include new personnel responsible for finance, legal, information technology and later, sales and business development functions. We also expect to incur additional general and administrative costs as a result of operating as a U.S. public company, including expenses related to compliance with the rules and regulations of the SEC and those of any national securities exchange on which our securities are traded, additional insurance expense, investor relations activities and other administrative and professional services. We also expect to incur additional expenses related to in-licenses, acquisitions or similar transactions that we may pursue as part of our strategy, including legal, accounting and audit services and other consulting fees.
111
Net Financial Income (Expense)
Net financial income (expense) includes amortized cost of the reimbursable advances, amortized cost of non-convertible bonds and of the debt component of the convertible notes issued to Kreos Capital, change in fair value on derivative financial instruments related to the conversion option of the convertible notes issued to Kreos Capital and related to the bonds issued to Kreos Capital, fair value adjustments on convertible notes issued to ATLAS, other financial income and expense, the net financial income related to NEGMA returning to us cash settlement related to contractual terms in relation with the NEGMA litigation in 2022 (as described in further detail in the paragraph “Results of Operations” below and in Note 17 to the audited consolidated financial statements), and foreign exchange gains and losses.
COVID19 Impact
Our clinical studies have been impacted by the COVID-19 pandemic. Our SARA-INT trial in sarcopenia was impacted by the emergence of COVID-19 and lockdowns in Belgium and several American states (California and New York in particular), and we had to adapt our SARA-INT protocol in order to ensure the continuity of the trial, in particular by closing all on-site activities, replacing them by phone calls, organizing Investigational Product delivery to patients’ homes, and expanding the treatment from six to nine months for some patients. Despite these interruptions of in-office study visits and other disruptions, we were able to retain most of the study participants, and a total of 203 participants completed the SARA-INT study. The last patient completed his final on-treatment visit in December 2020. However only 106 patients could perform the 400m walk test, which was the primary endpoint of our study.
Patient recruitment for our COVA trial to treat COVID19 patients has ended earlier than planned as a result of the evolution of the pandemic. In, April 2022, due to a lack of subjects meeting enrollment criteria, we have decided to stop enrollment in the COVA study with immediate effect. Final results of the study were communicated on February 2, 2023
The JOBS Act
As an “emerging growth company” under the JOBS Act, we can take advantage of an extended transition period for complying with new or revised accounting standards. This allows an “emerging growth company” to delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We will not take advantage of the extended transition period provided under Section 7(a)(2)(B) of the Securities Act for complying with new or revised accounting standards. Since IFRS makes no distinction between public and private companies for purposes of compliance with new or revised accounting standards, the requirements for our compliance as a private company and as a public company are the same. In addition, subject to certain conditions, as an “emerging growth company”, we intend to rely on certain of these exemptions including, without limitation, the exemptions from providing an auditor’s attestation report on our system of internal controls over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act. We will remain an “emerging growth company” until the earliest of: (1) the last day of the fiscal year in which we have total annual gross revenues of $1.235 billion or more; (2) the last day of the fiscal year following the fifth anniversary of the closing date of our U.S. initial public offering; (3) the date on which we have issued more than $1 billion in non-convertible debt during the previous three years; and (4) the date on which we are deemed to be a large accelerated filer under the rules of the SEC.
112
A. | Operating Results |
Comparison for the years ended December 31, 2022 and 2023
| December 31, |
| December 31, | |
(Amounts in thousands of euros) | 2022 | 2023 | ||
Revenue |
| — |
| — |
Costs of sales |
| — |
| — |
Gross margin |
| — |
| — |
Research and development expenses |
| (16,034) |
| (8,845) |
General and administrative expenses |
| (7,237) |
| (5,488) |
Operating loss |
| (23,272) |
| (14,333) |
Financial expenses |
| (2,564) |
| (1,633) |
Financial income |
| 983 |
| 269 |
Change in fair value of financial instruments |
| 637 |
| (1,330) |
Net financial expense |
| (944) |
| (2,694) |
Loss before taxes |
| (24,216) |
| (17,026) |
Income taxes benefit (expenses) |
| — |
| — |
Net loss |
| (24,216) |
| (17,026) |
Research and Development Expenses
Research and development expenses may be summarized as follows for the years ended December 31, 2022 and 2023.
| December 31, |
| December 31, | |
(Amounts in thousands of euros) | 2022 | 2023 | ||
Personnel expenses |
| (6,179) |
| (3,993) |
Purchases and external expenses |
| (12,991) |
| (6,378) |
Other |
| (285) |
| (35) |
Research and development expenses |
| (19,455) |
| (10,406) |
Research tax credit |
| 3,413 |
| 1,561 |
Subsidies |
| 7 |
| |
Research tax credit and Subsidies |
| 3,420 |
| 1,561 |
Research and development, net |
| (16,034) |
| (8,845) |
Personnel costs, including stock-based payments for engineers and research personnel, were €(6,179) thousand and €(3,993) thousand for the years ended December 31, 2022 and 2023, respectively. The decrease in personnel expenses in 2023 compared to 2022 was related to share-based payment of €560 thousand in 2023 compared to €3,281 thousand in 2022.
Purchases and external expenses related to our research activity were €(12,991) thousand and €(6,378) thousand for the years ended December 31, 2022 and 2023, respectively. The decrease in purchases and external expenses related to our studies and research costs is essentially linked to the finalization of clinical trials of the COVA and SARA programs in the second half of 2022. Residual costs linked to clinical development were recognized in 2023, however the majority of R&D expenses over the year concerned various preclinical work on the various programs of the Company and operations relating to the production of BIO101 (20-hydroxyecdysone).
We have benefited from the Research Tax Credit (CIR) since our incorporation. The CIR amounted to €3,274 thousand and €1,561 thousand for the years ended December 31, 2022 and 2023, respectively. In December 2022 and 2023, a portion of the CIR receivables for 2022 and for 2023 were prefinanced by the fonds commun de titrisation Predirec Innovation with Neftys Conseil Sarl, or Neftys, as arranger, in an amount of € 2,167 thousand and €1,098 thousand, respectively.
For the year ended December 31, 2023 no subsidy was granted to the Company.
113
General and Administrative Expenses
General and Administrative expenses may be summarized as follows for the years ended December 31, 2022 and 2023.
December 31, | December 31, | |||
(Amounts in thousands of euros) |
| 2022 |
| 2023 |
Personnel costs |
| (4,110) |
| (1,570) |
Purchases and external expenses |
| (2,928) |
| (3,427) |
Other |
| (199) |
| (491) |
General and administrative expenses |
| (7,237) |
| (5,488) |
Personnel costs, including share-based payments, for general management and administrative staff were €(4,110) thousand and €(1,570) thousand for the years ended December 31, 2022 and 2023, respectively. The decrease in personnel expenses in 2023 compared to 2022 was related to share-based payment of €251 thousand in 2023 compared to €2,285 thousand in 2022.
Purchases and external expenses were €(2,928) thousand and €(3,427) thousand for the years ended December 31, 2022 and 2023, respectively. These expenses consisted primarily of administrative expenses associated with being a public listed company in France and in the United States since February 2021, accounting and audit fees, insurance and legal fees. The €499 thousand increase is mainly linked to legal and financial consulting fees.
Net Financial Expense
Net financial expense may be summarized as follows for the years ended December 31, 2022 and 2023.
| December 31, |
| December 31, | |
(Amounts in thousands of euros) | 2022 | 2023 | ||
Interest and amortized cost on Kreos financing contract |
| (1,597) |
| (1,094) |
Change in fair value of convertible bonds and derivative liabilities |
| 637 |
| (1,330) |
Provision for Negma litigation risks | (75) | — | ||
Other financial expenses |
| (31) |
| (157) |
Expenses relating to the issue of convertible bonds |
| (820) |
| (330) |
Net financial income related to the repayment of penalties by Negma |
| 990 |
| — |
Other financial income |
| (17) |
| 174 |
Foreign exchange gains (losses) |
| (31) |
| 43 |
Total financial income and expense |
| (944) |
| (2,694) |
On September 8, 2022, as part of the Negma litigation, the Paris Court of Appeal partially overturned the 2021 judgment of the Execution Judge of the Paris Court. Negma Group Ltd was ordered to return to Biophytis the sum of €1,000 thousand. This indemnity was recorded as a financial income.
ATLAS contracts
In April 2020, we signed a new convertible bond financing of €24 million with ATLAS (the “2020 ATLAS Contract”) to continue the development of BIO101 (20-hydroxyecdysone). We issued three tranches of €3 million in 2020, and five additional tranches of €3 million in 2021. As of December 31, 2022, all of the convertible notes of the 2020 ATLAS Contract had been converted.
In June 2021, we signed a new convertible bond financing of up to €32 million (8 tranches with a nominal value of €4 million each) with ATLAS (the “2021 ATLAS Contract”) to continue the development of BIO101 (20-hydroxyecdysone) through the issuance of multiple convertible notes. As of December 31, 2022 and 2023, we have drawn down respectively €10 million and €12 million from our 2021 credit facility with ATLAS, corresponding to the first three tranches. Since December 31, 2023, the Company issued the fourth tranche of €4 million and 160 ORNANEs as part of its 2021 bond financing agreement with ATLAS. As of the date of this filing and considering the terms and expiration date of the ATLAS agreement as of June 14, 2024, the Company has the capacity to issue no more than two additional tranches for a total amount of €8 million.
During the years ended December 31, 2023 and December 31, 2022, the Company recorded in the statement of operations changes in fair value of the convertible notes issued to Atlas for €(1,342) thousand and €(675) thousand, respectively.
114
Kreos agreements
On September 10, 2018, the Company entered into a “venture loan agreement” with Kreos serving as a framework agreement organizing the issuance of a bond loan of an amount of up to €10 million for through the issue of four tranches of 2.5 million euros each, the first tranche being accompanied by share subscription warrants. All tranches were issued over the 2018 and 2019 financial years, for a total amount of 10 million euros. Each tranche bore interest at 10% per year. All non-convertible loan tranches issued were repayable in 36 monthly installments from April 2019. As of December 31, 2022, the financing was fully repaid. In accordance with IFRS 9, the non-convertible debt component related to the 2018 Kreos venture loan agreement was measured according to the amortized cost method, which is nil as of December 31, 2022, as the financing has been fully repaid.
On November 19, 2021, the Company entered into a new “venture loan agreement” with Kreos serving as a framework agreement organizing the issuance of a bond loan for an amount of up to €10 million through the issuance of €7.75 million in non-convertible bonds (“Straight bonds”), the issuance of €2.25 million in convertible bonds (“Convertible bonds”), and the issuance of Biophytis share subscription warrants. The issuance of the first tranche is conditioned to the subscription of the warrants previously mentioned. The loan contract comprising four tranches was partially drawn down by the Company during the 2021 financial year for a total amount of 6.2 million euros.
The Company determined that in accordance with IFRS9 and IAS 32, the conversion options from the convertible bonds and the warrants qualify as derivative instruments and are accounted for as financial liabilities, at amortized cost, based on an average effective interest rate of 26.37% for the non-convertible tranches, and 22.85% for the convertible tranches.
The instruments presented as a derivative are accounted for at fair value, with changes in fair value recorded in the income statement. Fair value is estimated using a binomial valuation model for convertible bonds, and a Black Scholes valuation model for warrants.
During the year ended December 31, 2022, the Company recognized €(1,425) thousand of interest expense and amortized costs related to Kreos loan agreements, and the change in fair value of the derivative instruments related to the conversion option and the warrants amounted to €1,312 thousand.
During the year ended December 31, 2023, the Company recognized €(1,094) thousand of interest expense and amortized costs related to Kreos loan agreements, and the change in fair value of the derivative instruments related to the conversion option and the warrants amounted to €12 thousand.
Comparison for the years ended December 31, 2021 and 2022
| December 31, |
| December 31, | |
(Amounts in thousands of euros) |
| 2021 |
| 2022 |
Revenue |
| — |
| — |
Costs of sales |
| — |
| — |
Gross margin |
| — |
| — |
Research and development expenses |
| (19,665) |
| (16,034) |
General and administrative expenses |
| (7,150) |
| (7,237) |
Operating loss |
| (26,815) |
| (23,272) |
Financial expenses |
| (2,517) |
| (2,564) |
Financial income |
| 24 |
| 983 |
Change in fair value of financial instruments |
| (1,856) |
| 637 |
Net financial expense |
| (4,349) |
| (944) |
Loss before taxes |
| (31,164) |
| (24,216) |
Income taxes benefit (expenses) |
| — |
| — |
Net loss |
| (31,164) |
| (24,216) |
115
Research and Development Expenses
Research and development expenses may be summarized as follows for the years ended December 31, 2021 and 2022.
December 31, | December 31, | |||
(Amounts in thousands of euros) |
| 2021 |
| 2022 |
Personnel expenses | (4,392) | (6,179) | ||
Purchases and external expenses |
| (19,345) |
| (12,991) |
Other |
| (264) |
| (285) |
Research and development expenses |
| (24,001) |
| (19,455) |
Research tax credit |
| 4,080 |
| 3,413 |
Subsidies |
| 256 |
| 7 |
Research tax credit and Subsidies |
| 4,336 |
| 3,420 |
Research and development, net |
| (19,665) |
| (16,034) |
Personnel costs, including stock-based payments for engineers and research personnel, were €(4,392) thousand and €(6,179) thousand for the years ended December 31, 2021 and 2022, respectively. The increase in personnel expenses in 2022 compared to 2021 was related to the full year compensation expense of recruitments made during 2021, the reinforcement of the regulatory department within the framework of the COVA clinical study and expenses relating to share-based payment of €(3,281) thousand in 2022 compared to €(2,125) thousand in 2021.
Purchases and external expenses related to our research activity were €(19,345) thousand and €(13,016) thousand for the years ended December 31, 2021 and 2022, respectively. The decrease in purchases and external expenses related to our studies and research costs was primarily related to the end of our COVA Phase 2-3 study as well as to the costs of preliminary meetings with public regulatory authorities in the context of the pursuit of our post-phase 2 SARA-INT study. These expenses consisted primarily of the cost of CROs in conducting clinical trials and non-clinical studies, as well as the costs of CDMOs for the manufacturing scaling-up of Sarconeos (BIO101) in preparation of a filing with Regulatory Authorities.
We have benefited from the Research Tax Credit (CIR) since our incorporation. The CIR amounted to €4,080 thousand and €3,274 thousand for the years ended December 31, 2021 and 2022, respectively. In December 2021 and 2022, a portion of the CIR receivable for 2021 and for 2022 were prefinanced by the fonds commun de titrisation Predirec Innovation with Neftys Conseil Sarl as arranger, or Neftys. The portion of the CIR 2021 and CIR 2022 receivables were prefinanced by Neftys for amounts of €3,450 thousand and €2,167 thousand, as of December 31, 2021 and 2022, respectively.
As part of the BPI France conditional advance for the “BIO 201” project, the Company was entitled to receive a grant of €380 thousand, of which €202 thousand was recognized as a subsidy in 2021 since 53% of the budget of research and development expenses were incurred at the closing date. No additional expenses were incurred in 2022.
For the year ended December 31, 2022 no subsidy was granted to the Company.
General and Administrative Expenses
General and Administrative expenses may be summarized as follows for the years ended December 31, 2021 and 2022.
December 31, | December 31, | |||
(Amounts in thousands of euros) |
| 2021 |
| 2022 |
Personnel costs |
| (3,107) |
| (4,110) |
Purchases and external expenses |
| (3,991) |
| (2,928) |
Other |
| (52) |
| (199) |
General and administrative expenses |
| (7,150) |
| (7,237) |
Personnel costs, including share-based payments, for general management and administrative staff were €(3,107) thousand and €(4,110) thousand for the years ended December 31, 2021 and 2022, respectively. This increase was mainly due to the replacement of the finance staff, as well as to the impact of the stock-based compensation expense related to Founders’ warrants and free shares granted in 2020, in 2021 and in 2022.
116
Purchases and external expenses were €(3,991) thousand and €(2,945) thousand for the years ended December 31, 2021 and 2022, respectively. These expenses consisted primarily of administrative expenses associated with being a public listed company in France and in the United States since February 2021, accounting and audit fees, insurance and legal fees. The €1,046 thousand decrease is mainly linked to a reduction of legal and financial consulting fees.
Net Financial Expense
Net financial expense may be summarized as follows for the years ended December 31, 2021 and 2022.
| December 31, |
| December 31, | |
(Amounts in thousands of euros) |
| 2021 |
| 2022 |
Financial interest of the non-convertible bonds and convertible notes issued and amortized cost of the non-convertible to Kreos |
| (545) |
| (1,597) |
Change in fair value of convertible notes issued to ATLAS and KREOS derivative instruments |
| (1,856) |
| 637 |
NEGMA financial indemnity |
| (1,695) |
| 990 |
Accrual Negma litigation provision | — | (75) | ||
Other financial expenses |
| (166) |
| (31) |
Transaction costs related to the issuance of convertible notes |
| (125) |
| (820) |
Net financial income related to NEGMA returning to Biophytis Damages paid |
| 20 |
| — |
Other financial income |
| 4 |
| (17) |
Foreign exchange gains (losses) |
| 14 |
| (31) |
Net financial expense |
| (4,349) |
| (944) |
Net financial expense was €(4,349) thousand and €(944) thousand for the years ended December 31, 2021 and 2022, respectively.
During the year ended December 31, 2022, the change in fair value of convertible notes and derivative instruments was related to (i) the change in fair value of the ORNANE issued to Atlas for €(675) thousand and (ii) the change in fair value of the derivative instruments for €1,312 thousand.
During the year ended December 31, 2021, the change in fair value of convertible notes and derivative instruments was related to (i) the change in fair value of the ORNANE issued to Negma for €1,307 thousand, (ii) the change in fair value of the ORNANE issued to Atlas for (€3,017) thousand and (iii) the change in fair value of the derivative instruments for (€150) thousand.
On July 16, 2021, the Paris Court of Justice assigned Biophytis a penalty payment of €1,500 thousand in favor of Negma Group Ltd, following a judgment by the Paris Commercial Court.
On September 8, 2022, the Paris Court of Appeal partially overturned the judgment of the Execution Judge of the Paris Court. Negma Group Ltd was ordered to return to Biophytis the sum of €1,000 thousand. This indemnity was recorded as a financial income.
ATLAS contracts
In April 2020, we signed a new convertible bond financing of €24 million with ATLAS (the “2020 ATLAS Contract”) to continue the development of Sarconeos (BIO101). We issued a first tranche of €3 million on April 29, 2020, a second tranche of €3 million on June 19, 2020, and a third tranche of €3 million on August 28, 2020.
In 2020, 330 convertible notes (nominal value of €25 thousand each) were converted and the remaining 30 notes were redeemed in cash. As of December 31, 2020, there were no outstanding convertible notes issued to ATLAS.
On May 27, 2021, we issued a fourth and fifth tranche of €3 million each. On September 20, 2021, we issued a sixth and seventh tranche of €3 million each. On December 20, 2021, we issued an eighth of €3 million.
As of December 31, 2022, all of the convertible notes of the 2020 ATLAS Contract had been converted. Pursuant to the 2020 ATLAS Contract, all ORNANEs have been issued to ATLAS.
117
In June 2021, we signed a new convertible bond financing of up to €32 million (8 tranches with a nominal value of €4 million each) with ATLAS (the “2021 ATLAS Contract”) to continue the development of Sarconeos (BIO 101) through the issuance of multiple convertible notes. As of December 31, 2021, no convertible notes have been issued as part of this contract. As of December 31, 2022, we have drawn down €10 million from our 2021 credit facility with ATLAS. As of the date of this filing and considering the terms and expiration date of the ATLAS agreement as of June 14, 2024, the Company has the capacity to issue no more than two additional tranches for a total amount of €8 million.
During the years ended December 31, 2022 and December 31, 2021, the Company recorded in the statement of operations changes in fair value of the convertible notes issued to Atlas for €(675) thousand and (€3,017) thousand, respectively.
Kreos agreements
In accordance with IFRS 9, the non-convertible debt component related to the 2018 Kreos venture loan agreement is measured according to the amortized cost method, which is nill as of December 31, 2022, as the financing has been fully repaid, and to €0.9 million as of December 31, 2021.
On November 19, 2021, the Company entered into a “venture loan agreement” with Kreos in lieu of a framework agreement organizing the issuance of a bond loan for an amount of up to €10 million through the issuance of €7.75 million in non-convertible bonds (“Straight bonds”), the issuance of €2.25 million in convertible bonds (“Convertible bonds”), and the issuance of Biophytis share subscription warrants. The issuance of the first tranche is conditioned to the subscription of the warrants previously mentioned.
The loan agreement comprises four tranches of respectively €2.5 million (A), €3.0 million (B), €2.5 million (C) and €2.0 million (D). The first two tranches (A and B) were drawn upon signature of the contract on November 19, 2021, and the third tranche (C) limited to €677 thousand was drawn as of December 31, 2021. The first two Tranche (A and B) include straight bonds for a nominal amount of €3,250 thousand, and convertible bonds for €2,250 thousand.
The Company determined that in accordance with IFRS9 and IAS 32, the conversion options from the convertible bonds and the warrants qualify as derivative instruments and are accounted for as financial liabilities.
The amount of cash of €5 million, received on November 19, 2021 (excluding transaction costs) corresponds to the estimated fair value of the instruments put in place on the date the funds were drawn: financial debt for tranches A and B for €(4,3) million (convertible and non-convertible), liability derivatives for premiums received on options sold for €(1,2) million (€464 thousand for conversion options and €710 thousand for BSAs issued), and financial compensation of €48 thousand for the 2018 BSAs bought back by Biophytis from KREOS.
Regarding the third tranche (C) of the straight bond issued in December 2021 for €677 thousand, as the drawdown conditions were fulfilled outside the framework of the contract, the company analyzed the drawdown of the third tranche (C) as a new loan contract, with Kreos Capital VI UK. As such, the third tranche (C) is recognized for its fair value on the balance sheet, estimated on the basis of the financing rate deducted from the Kreos VI financing. The entry value of the liabilities of the Tranche C leads to the recognition of a day one gain of €98 thousand. Given the unobservable nature of the market rate, the day one gain is deferred on the Company’s balance sheet and recorded as financial liabilities.
The instruments presented as financial debt is accounted for at amortized cost, based on an average effective interest rate of 26.37% for the non-convertible tranches, and 22.85% for the convertible tranches.
The instruments presented as a derivative is accounted for at fair value, with changes in fair value recorded in the income statement. Fair value is estimated using a binomial valuation model for convertible bonds, and a Black Scholes valuation model for BSAs.
During the year ended December 31, 2022, the Company recognized €(1,425) thousand (545) thousand in 2021) of interest expense and amortized costs related to Kreos loan agreements.
During the year ended December 31, 2022, the change in fair value of the derivative instruments related to the conversion option and the warrants amounted to €1,312 thousand compared to €(150) thousand during the year ended December 2021.
118
B. | Liquidity and Capital Resources |
Our operations have been financed primarily by capital contributions from our founders, capital increases carried out between 2006 and 2023, convertible debt instruments with warrants, non-convertible bonds and net proceeds from the initial public offering of our ordinary shares on the Euronext Growth Market in France in 2015 and, into 2021, from the proceeds from our U.S initial public offering. Our primary uses of capital are, and we expect will continue to be, third-party expenses associated with the planning and conduct of preclinical studies and clinical trials, costs of process development services and manufacturing of our drug candidates, and compensation-related expenses.
We do not expect to generate significant revenue from product sales unless and until we out-license one or more drug candidates or we obtain regulatory approval or authorization for and commercialize our current or any future drug candidates, either directly or through others. We anticipate that we will continue to generate losses for the foreseeable future, and we expect our losses to increase as we continue the development of and seek regulatory approvals and authorizations for our drug candidates and begin to commercialize any approved or authorized products.
We are subject to numerous risks applicable to the development of new products, and we may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may harm our business. We expect to incur additional costs associated with operating as a public company in the United States and we anticipate that we will need substantial additional funding in connection with our continuing operations.
Our future funding requirements will depend on many factors, including the following:
| ● | the scope, rate of progress, results and cost of our preclinical studies and clinical trials and other related activities; |
| ● | the cost of formulation, development, manufacturing of clinical supplies and establishing commercial supplies of our drug candidates and any other drug candidates that we may develop, in-license or acquire; |
| ● | the cost, timing and outcomes of pursuing regulatory approvals and authorizations; |
| ● | the cost and timing of establishing administrative, sales, marketing and distribution capabilities, to the extent we undertake to commercialize our products directly; |
| ● | the terms and timing of any collaborative, licensing and other arrangements that we may establish, including any required milestone and royalty payments thereunder; and |
| ● | the emergence of competing technologies and their achieving commercial success before we do or other adverse market developments. |
Our ability to achieve and maintain profitability will depend upon the successful development, regulatory approval, authorization, and commercialization of our drug candidates and achieving a level of revenues adequate to support our cost structure. We may never achieve profitability, and unless and until we do, we will continue to need to raise additional capital. If we need to raise additional capital to fund our operations and complete our ongoing and planned clinical trials, funding may not be available to us on acceptable terms, or at all.
We plan to continue to fund our operations and capital funding needs through a combination of equity offerings, debts and collaborations. The sale of additional equity would result in additional dilution to our shareholders. The incurrence of debt financing would result in debt service obligations and the instruments governing such debt could provide for operating and financing covenants that would restrict our operations. If we are not able to secure adequate additional funding, we may be forced to make reductions in spending, extend payment terms with suppliers, sell assets where possible or suspend or curtail planned programs. In addition, lack of funding would limit any strategic initiatives to in-license or acquire additional drug candidates or programs.
As of December 31, 2023, we had capital resources consisting of cash, cash equivalents of €5.6 million ($6.2 million, translated solely for convenience into dollars at an exchange rate of €1.00 – $1.1062, the noon buying rate of the Federal Reserve Bank of New York on December 31, 2023). Cash in excess of immediate requirements is invested in accordance with our investment policy, primarily with a view to liquidity and capital preservation. Currently, our funds are held in bank accounts and fixed bank deposits primarily in France.
119
In June 2021, we signed a new convertible bond financing of up to €32 million (8 tranches with a nominal value of €4 million each) with ATLAS (the “2021 ATLAS Contract”) to continue the development of BIO101 (20-hydroxyecdysone) through the issuance of multiple convertible notes. As of December 31, 2023, we have drawn down €12 million from our 2021 credit facility with ATLAS, corresponding to the first three tranches. Since December 31, 2023, the Company issued the fourth tranche of €4 million and 160 ORNANEs as part of its 2021 bond financing agreement with ATLAS. As of the date of this filing and considering the terms and expiration date of the ATLAS agreement as of June 14, 2024, the Company has the capacity to issue no more than two additional tranches for a total amount of €8 million.
We expect that our existing capital resources as adjusted by the effect of those events, and including our ability to draw down on our credit facility with ATLAS will be sufficient to fund our current operations into the first quarter of 2025. However, this estimate is based on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect. As of the date of the filing, our available cash is not projected to be sufficient to support our operating plan for at least the next 12 months. As such, there is substantial doubt regarding our ability to continue as a going concern. We intend to seek additional capital to pursue preclinical and clinical activities, obtain regulatory approval and authorization for, and commercialize our drug candidates. Notably during 2024, we may conduct equity financing transactions on Euronext Growth or Nasdaq, enter into new debt financing agreements or enter into partnership or licensing agreements for our R&D programs that could provide additional non-dilutive financial resources or reduce our costs.
We cannot guarantee that we will be able to obtain the necessary financing to meet our needs or to obtain funds at attractive terms and conditions, including as a result of disruptions to the global financial markets resulting from geopolitical instability, macroeconomic conditions, global health crises, or other factors.
If we are not successful in our financing objectives, we could have to scale back our operations, notably by delaying or reducing the scope of our research and development efforts or obtain financing through arrangements with collaborators or others that may require us to relinquish rights to our product candidates that we might otherwise seek to develop or commercialize independently.
Our financial statements have been prepared on a going concern basis assuming that we will be successful in our financing objectives. As such, no adjustments have been made to the financial statements relating to the recoverability and classification of the asset carrying amounts or classification of liabilities that might be necessary should we not be able to continue as a going concern.
Cash Flows
Year Ended | ||||||
December 31, | ||||||
(Amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Net cash (used in) provided by: |
|
|
|
|
|
|
Operating activities |
| (23,795) |
| (18,988) |
| (12,873) |
Investing activities |
| 12,160 |
| (17) |
| (662) |
Financing activities |
| 29,715 |
| 6,134 |
| 7,027 |
Effect of exchange rate changes on cash and cash equivalents |
| (1) |
| (3) |
| (9) |
Net increase (decrease) in cash and cash equivalents |
| 18,079 |
| (12,873) |
| (5,485) |
Operating Activities
Net cash used in operating activities was €(23,795) thousand, €(18,988) thousand and €(12,873) for the years ended December 31, 2021, 2022 and 2023, respectively. The decrease in net cash used from 2021 to 2022 is mainly related to expenses incurred for the SARA-INT clinical program and the launch of the COVA program in 2021. The decrease in net cash used from 2022 to 2023 is mainly related to the end of the COVA and SARA-INT studies.
Investing Activities
Net cash provided by (used in) investing activities was €12,160 thousand, €(17) thousand and €370 thousand for the years ended December 31, 2021, 2022 and 2023, respectively.
Investing activities include to the purchase of office and laboratory equipment, generally of low value.
120
In 2020, we purchased short-term deposits for €12,500 thousand, classified as other current financial assets in accordance with IAS 7. In 2021, we sold short-term deposits for €12,500 thousand, classified as other current financial assets in accordance with IAS 7.
Financing Activities
Net cash provided by financing activities was €29,715 thousand, €8,267 thousand and €7,027 thousand for the years ended December 31, 2021, 2022 and 2023, respectively.
Net proceeds from capital increases linked with equity financing amounted to €14,485 thousand in 2021, nil in 2022 and €5,073 thousand in 2023.
Proceeds from the subscription of warrants and Founders’ warrants amounted to €742 thousand in 2021 and were non-significant in 2022 and 2023.
In 2021, we issued five tranches of convertible notes to ATLAS for a total of €15 million. In 2022, we issued the first, second and a half of the third tranche of convertible notes to ATLAS for a total of €10 million, corresponding to a net cash receipt of €9.6 million. In 2023, we issued half a tranche of convertible notes to ATLAS for €2 million, corresponding to a net cash receipt of €1.9 million.
We repaid the Kreos non-convertible bonds in an amount of €1,722, thousand, €2,505 thousand and €3,550 thousand for 2023, 2022 and 2021, respectively. These amounts include interests of €450 thousand, €662 thousand and €562 thousand for 2023, 2022 and 2021, respectively.
We received proceeds from subsidies of €204 thousand in 2022 compared to €400 thousand in 2021. We repaid €220 thousand, €224 thousand and €279 thousand of conditional advances, for 2023, 2022 and 2021, respectively.
We received payment of a portion of our research tax credit (CIR) receivable for 2021, 2022 and 2023, either by way of prefinancing by Neftys or directly from the French State, in an amount of €3,011 thousand, €1,834 thousand and €3,138 thousand respectively. We repaid the prefinanced portion of our CIR for an amount of €2,252 thousand in 2021, €3,450 thousand in 2022 and Nil in 2023.
On September 8, 2022, the Paris Court of Appeal partially overturned the 2021 judgment of the Execution Judge of the Paris Court in the Negma litigation. Negma Group Ltd was ordered to return to Biophytis the sum of €1,000 thousand, previously paid as part of the total €1,675 thousand paid to Negma in 2021.
Cash and Funding Sources
Research Tax Credit (CIR)
We have benefited from the CIR since our incorporation. The CIR is usually payable by the French government in the year following its recognition when there is no taxable net income to be offset if certain business size criteria are met.
The CIR for the years 2023, 2022 and 2021 amounted to €1,561 thousand, €3,364 thousand and €4,080 thousand, respectively. The CIR for 2021 has been repaid in 2022 and the CIR 2022 has been repaid in 2023. The CIR for 2023 has been partly prefinanced by Neftys and is expected to be repaid end 2024.
Reimbursable Advances
A reimbursable advance was granted to us by BPI France on February 4, 2015. This was a non-interest-bearing reimbursable advance of €260 thousand for the “in vitro, in vivo, and pharmacokinetic characterization of a candidate drug.” Following the successful completion of the project and the extension of the repayment terms granted by BPI France, this advance is being repaid by means of quarterly payments made between June 30, 2017 and March 31, 2022. The payment schedule has been postponed by six months automatically by BPI France as part of the financial support measures for companies in the management of the COVID19 crisis. As a result, the last payment will occur in June 30, 2024.
121
A reimbursable advance was granted to us by BPI France on November 28, 2016. This is a non-interest-bearing reimbursable advance of €1,100 thousand for the production of clinical batches, in the preclinical regulatory phase and clinical Phase 1 of BIO101 (20-hydroxyecdysone), for the treatment of sarcopenic obesity. The agreement with BPI France provides that the advance would be paid to us in two tranches, with the first of €600 thousand paid at the signing date of the agreement, and the second €500 thousand to be paid at the end of the program. We received €500 thousand during the year ended December 31, 2018 related to the second tranche. Following the successful completion of the project, this advance is being repaid by means of quarterly payments made between December 31, 2018 and September 30, 2023. The payment schedule has been postponed by six months automatically by BPI France as part of the financial support measures for companies in the management of the COVID-19 crisis. As a result, the last payment will occur in March 31, 2024.
On June 3, 2019, we entered into a collaboration agreement with the French Muscular Dystrophy Association (AFM-Telethon), pursuant to which AFM-Telethon has provided funding of €400,000 to us. This is a non-interest-bearing reimbursable advance of €400,000 for certain preclinical studies and preparations for our MYODA program. Under the terms of the agreement, subject to regulatory approval to conduct the MYODA clinical trial in Europe and conclusive results from the collaboration, we will submit to AFM-Telethon a new research project for further collaboration on the clinical development of BIO101 (20-hydroxyecdysone) in DMD. If funding for the new research project is approved by AFM-Telethon, we will negotiate in good faith the terms of a new collaboration agreement with AFM-Telethon. If entered into, the new collaboration agreement will grant certain rights to AFM-Telethon that may, in the event we later decide to abandon or not pursue the development of BIO101 (20-hydroxyecdysone), entitle AFM-Telethon to continue the development and/or commercialization of BIO101 (20-hydroxyecdysone) and/or any pharmaceutical product derived from BIO101 (20-hydroxyecdysone) for the purpose of guaranteeing the access of such products to DMD patients. The advance will be repaid upon our obtaining authorization to commence a Phase 3 clinical trial of BIO101 (20-hydroxyecdysone) for the treatment of DMD. In addition, we will be required to repay the advance if we are unable to come to an agreement with AFM-Telethon on further funding of our MYODA clinical program or we materially breach the agreement and AFM-Telethon requests reimbursement.
On August 23, 2019, we entered into an agreement with BPI France for an interest-free conditional advance of €600 thousand payable in milestone installments for its MACA program of BIO201 in dry Age-Related Macular Degeneration (AMD). The proceeds were subject to financial conditions that have been met in April 2021. The Company received €400 thousand in April 2021 in connection with this agreement. The balance of €200 thousand will be received once the Company finalizes the program. The repayment of this conditional advance is subject to the successful completion of the project: in case of technical and economic failure, a minimum repayment of €240 thousand is due at the end of the project timeline (36 months after first conditional advance received) and in case of successful completion, repayment over a 5-year period will commence in September 2022. As part of this agreement, the Company was entitled to receive a grant of €380 thousand, of which €260 thousand was received in April 2021. As of December 31, 2021, we recognized €202 thousand as subsidies since 53% of the budget of research and development expenses was incurred on that project at the closing date. As of December 31, 2022 and 2023, no research and development expenses has been incurred. Therefore, no additional subsidies has been recognized.
Non-convertible bonds issued to Kreos (2018 Kreos contract)
In September 2018, we entered into a venture loan agreement and bonds issue agreement with Kreos providing for up to €10 million in financing to us. Pursuant to the terms of the agreements, Kreos agreed to subscribe for up to €10 million in non-convertible bonds, to be issued by us in up to four tranches of €2.5 million each, with a warrant to purchase 442,477 ordinary shares attached to the first tranche. As required under the terms of the agreements, we pledged a security interest in our assets for the benefit of Kreos. We also granted a security over the business as a going concern (nantissement de fonds de commerce), including a portion of our patents, to Kreos.
Each tranche of non-convertible bonds bears a 10% annual interest rate and must be repaid in 36 monthly installments of €320,004 per month commencing in April 2019. The first and second tranches were issued to Kreos on September 10, 2018. The third tranche was issued to Kreos on December 17, 2018. The final tranche was issued on March 1, 2019.
In connection with the first tranche, we issued 442,477 warrants to Kreos giving them the right to purchase 442,477 new ordinary shares at an exercise price of €2.67 per share over a 7-year period from the issue date.
Pursuant to the terms of the agreements, we have the right, at any time but with no less than 30 days prior notice to Kreos, to prepay or purchase the bonds, exclusively in full. The prepayment will be equal to (i) the principal amount outstanding, plus (ii) the
122
sum of all interest repayments which would have been paid throughout the remainder of the term of the relevant tranche discounted by 10% per annum.
As of December 31, 2022, the financing was fully repaid.
Non-convertible bonds and convertible notes issued to Kreos (2021 Kreos contract)
On November 19, 2021, we signed a new venture loan agreement and bonds issue agreement that could provide for up to €10 million in financing to us through the issuance by us to Kreos of non-convertible bonds for €7.75 million (straight bonds) and convertible notes of €2.25 million, plus the issuance of attached warrants to the first tranche. The loan agreement includes four tranches of respectively €2.5 million, €3.0 million, €2.5 million and €2.0 million. The two first tranches were drawn upon signing of the contract on November 19, 2021, the third tranche limited to €676 thousand was drawn up before December 31, 2021. The last tranch was not drawn up.
Non-convertible bonds bears a 10% annual interest rate and must be repaid in cash in 36 monthly installments commencing on April 1, 2022. Convertible notes bears a 9.5% annual interest rate.
We must repay the convertible bonds at their principal amount at the latest March 31, 2025, unless they are converted prior to that time into shares, at the option of Kreos Capital, at a fixed conversion price of €0,648.
We issued for the benefit of Kreos Capital 2,218,293 warrants giving the right to subscribe to new Biophytis ordinary shares, on the basis of one share for one warrant. The warrants may be exercised over a 7-year period after being issued. The exercise price of the share warrants has been set at €0.56.
By subscribing to the BSAs, Kreos Capital has expressly waived the right to exercise the 2018 BSAs as held following their detachment from the non-convertible bonds subscribed on September 10, 2018 within the framework of the 2018 loan structure.
As required under the terms of the venture loan agreement, we pledged a security interest in our assets for the benefit of Kreos. We also granted a security interest in the business as a going concern (nantissement de fonds de commerce), including a portion of the Company’s patents, to Kreos.
Pursuant to the terms of the agreements, we have the right, at any time but with no less than 30 days prior notice to Kreos, to prepay or purchase the bonds, exclusively in full. The prepayment will be equal to (i) the principal amount outstanding, plus (ii) the sum of all interest repayments which would have been paid throughout the remainder of the term of the relevant tranche discounted by 10% per annum.
Pursuant to the terms of the agreements, in the event conversion occurs on the repayment date, Kreos shall repay to Biophytis, upon issuance of the conversion shares, an amount equal to 10% of the total interest paid by Biophytis. In case of a partial conversion upon that date, the amount shall be reduced accordingly.
Convertible notes issued to ATLAS
In April 2020, we signed a convertible note financing of €24 million from ATLAS to continue the development of BIO101 (20-hydroxyecdysone).
The 960 3-year note warrants require their holder to exercise them, at our request, in tranches of 120 warrants each. Each warrant grants its holder the right to one ORNANE. Note warrants may not be transferred and will not be subject to a request for admission to trading on the Euronext Growth market.
The ORNANE have a par value of €25,000 and are issued at a subscription price of 97% of the nominal value. They do not bear interest and have a 24-month maturity from issuance. Holders of ORNANE may request at any time to convert them during their maturity period, and at that time, we will be able to redeem the ORNANE in cash. At the end of the maturity period, and if the ORNANE have not yet been converted or redeemed, the holder will have to convert them.
ORNANE may be transferred by their holders only to Affiliates and will not be subject to a request for admission to trading on the Euronext Growth market.
123
We issued the eighth and last tranche of €3 million in December 2021. As of December 31, 2021, there are 224 outstanding convertible notes issued to ATLAS. A commitment fee of €375 thousand was withheld from the proceeds received for the first tranche. Other issuance costs were incurred by us for approximately €66 thousand (€16 thousand for the first tranche, €23 thousand for the second tranche and €27 thousand for the third tranche).
As of December 31, 2021, 376 convertible notes had been converted resulting in the issuance of 16,379,256 ordinary shares within the frame of ATLAS 2020 contract.
As of December 31, 2022, the remaining 224 convertible notes had been converted resulting in the issuance of 29,179,474 ordinary shares within the frame of ATLAS 2020 contract.
On June 14, 2021, we signed a new convertible bond financing of €32 million with ATLAS. Pursuant to the terms of the agreement, ATLAS agreed to subscribe for up to €32 million in convertible bonds, to be issued by us in up to eight tranches of €4 million each. We issued the first, second and half of the third tranche for €10 million in 2022 correponding to a net amount of €9.6 million. As of December 31, 2022, there were 228 outstanding convertible notes issued to ATLAS, amounting to €5.7 million. We issued the second half of the third tranche for €2 million in 2023. As of December 31, 2023, there are 58 outstanding convertible notes issued to ATLAS, amounting to €1.45 million, subsequent to the conversion of 250 notes during the year 2023 for a total amount of €6.3 million. Since December 31, 2023, the Company issued the fourth tranche of €4 million and 160 ORNANEs as part of its 2021 bond financing agreement with ATLAS. As of the date of this filing and considering the terms and expiration date of the ATLAS agreement as of June 14, 2024, the Company has the capacity to issue no more than two additional tranches for a total amount of €8 million.
Public Offering of Share Subscription Warrants
On April 3, 2020, we decided to launch a public offering of share subscription warrants. Upon completion of the public offering, we issued 7,475,708 share subscription warrants, after full exercise of the extension clause. The subscription price was €0.06 per warrant. The warrants can be exercised for a period of 5 years from April 30, 2020, at an exercise price of €0.27 per new share. Each warrant gives its holder the right to subscribe to one new Biophytis share. In 2021, 2022 and 2023 warrants were exercised for €302 thousand, €6 thousand and €3 thousand, respectively.
As of December 2021, the subscription and the exercise of the investors warrants by our CEO was settled by the remaining amount of €630 thousand due our CEO as part of the Intellectual Property agreement (€177 thousand for the subscription of warrants and €453 thousand for the exercise of warrants).
Capital increase in the form of a private placement on Euronext
On May 11, 2023, the Company announced a new financing in the form of a private placement with professional investors combined with a public offering to individual investors, for a gross amount of 2.3 million euros. The operation was implemented and carried out pursuant to the 2nd and 4th resolution of the combined general meeting of shareholders of the Company dated April 17, 2023. A total of 103,717,811 new ordinary shares, representing 32% of the capital share of the Company before the operation were issued at a price of 0.0222 euros per share, showing a discount of 25% compared to the average price weighted by the volumes of the Biophytis share of the 5 trading sessions preceding the operation and representing a nominal amount of 1,037 thousand euros and a total issue premium of 1,265 thousand euros. The admission of the new shares to Euronext Growth Paris under the ISIN code FR0012816825 ALBPS took place on May 15, 2023 at the opening of the market and these shares are assimilated to existing shares and carry immediate dividend rights.
Capital increase in the form of a registered direct offering on the Nasdaq Capital Market
On July 19, 2023, the Company announced a registered direct offering for a gross amount of $3.8 million, equivalent to €3.4 million. This transaction, closed on July 21, consisted of the purchase and sale of 1,333,334 units, each consisting of one (1) American Depositary Share (“ADS”) or one (1) pre-funded warrant entitling itself to one (1) ADS (the “Pre-Funded Warrants”), and one (1) warrant (the “Ordinary Warrant”) entitling the holder to one (1) ADS, at a purchase price of $2.85 per unit with an ADS and $2.84 per unit with a pre-funded warrant. Each ADS represents the right to receive one hundred new ordinary shares of the Company, with a par value of €0.01 per share.
124
The ADSs and Pre-Funded Warrants were offered and sold in a direct offering registered pursuant to a “shelf” registration statement on Form F-3 (File No. 333-271385) filed with the Securities and Exchange Commission United States Exchange Commission (the “SEC”) on April 21, 2023 and declared effective by the SEC on May 1, 2023. The ordinary warrants were issued in a concurrent private placement.
The issue price of the ordinary shares underlying the ADSs represented a premium of 2% compared to the volume-weighted average price (VWAP) of the Company’s ordinary shares on the Euronext Growth Paris market during the 15 trading sessions preceding the determination of the issue price on July 18, 2023 and a discount of 21% compared to the VWAP including 23% of the theoretical value of a subscription warrant, the value of which per warrant was €0.013.
The issue of the 50,500,000 new ordinary shares underlying the ADSs resulted in an immediate capital increase of 1,278 thousand euros, share premium included.
Each pre-funded warrant, giving right to one (1) ADS, was subscribed for at a price of $2.84 and their exercise price amounted to $0.01 per ADS, i.e. a total amount received by the Company of 2,090 thousand euros. All of the pre-funded warrants, representing 82,833,400 new ordinary shares, were exercised during the financial year. The ordinary warrants, representing an additional 133,333,400 potential new ordinary shares, have an exercise price of $3 per ADS and are exercisable upon issuance and will expire three years after issuance. As of December 31, 2023, no ordinary warrants have yet been exercised.
Capital increase with maintenance of preferential subscription rights on Euronext
On November 20, 2023, the Company issued 210,733,954 Shares with Redeemable Share Subscription Warrants (ABSAR), of which the amount, share premium included, amounts to approximately €1.96 million. The issue price of the ABSARs was 0.0093 euros. The BSARs may be exercised at any time until December 31, 2026, one (1) BSAR giving the right to subscribe to one (1) new ordinary share upon payment of an exercise price of 0.012 euros. As of December 31, 2023, 2,477,006 BSARs were exercised.
Material cash requirements
The following table discloses aggregate information about material contractual obligations and periods in which payments were due as of December 31, 2023 :
(amounts in thousands of euros) |
| 2024 |
| Thereafter |
| Total |
Conditional advances |
| 196 |
| 686 |
| 882 |
Convertible and Non-convertible bonds |
| 3,560 |
| 2,425 |
| 5,985 |
Lease obligations |
| 54 |
| 136 |
| 190 |
Financial liabilities related to the prefinancing of a portion of the research tax credit receivables |
| 1,213 |
|
| 1,213 | |
Total |
| 5,023 |
| 3,247 |
| 8,270 |
The commitment amounts in the table above are associated with contracts that are enforceable and legally binding and that specify all significant terms, including interest on long-term debt, fixed or minimum services to be used, fixed, minimum or variable price provisions, and the approximate timing of the actions under the contracts. The table does not include obligations under agreements that we can cancel without a significant penalty. Future events could cause actual payments to differ from these estimates.
The company benefits from conditional advances (as described in further detail in the Note 13.1 to the audited consolidated financial statements), with a repayment horizon up to 2029.
Bonds represent a €5.9 million cash requirement as of December 31, 2023 and are related to the Kreos and Atlas loan agreements. The sum that is due in 2024 is related to the Kreos non-convertible loan that is repaid on a monthly basis. The sum in subsequent years is related to the Kreos convertible loan, repayable in April 2025 if not converted beforehand as well as to the Atlas convertible loan that may be repaid in cash at the Company’s choice, the maturity date of each bon being two years after issuance).
Leases represent a €0.2 million cash requirements as of December 31, 2023 with a repayment horizon up to 2027. Most of this cash requirement is linked to the Sorbonne lease agreement accounting for €56 thousand of the total lease obligations amount.
125
Commitments provided as part of license agreements
We have signed several agreements to license industrial property to further our research and developments efforts with royalties due to the counterparties that are variable starting the year after the first marketing of a product and royalty arrangements. However, there are certain guaranteed annual minimum amounts due starting in various future years. These guaranteed annual minimum amounts are shown in the table above. Other than these minimum guaranteed amounts (as further described below), amounts of royalties to be paid after 2024 cannot be determined precisely.
The following table discloses the commitments given as part of the licensing agreements mentioned above:
Agreements for the exploitation of industrial property |
| Commitments given |
MACULIA commercialization agreement—SATT Lutech Agreement of January 1, 2016, as amended on December 17, 2020 |
| This agreement covers the MI through MIV patent families. The contractual structure of the consideration payable by us is as follows: firstly, in the year following the first marketing of a nutraceutical product and in any event no later than in 2020, we will pay an annual guaranteed minimum amount of €15 thousand. In the same way, we will pay a guaranteed minimum amount of €50 thousand in the event of marketing of a drug product and in any event no later than from 2026. These amounts will be deducted from the amount of royalties effectively due annually to SATT Lutech. For direct exploitation, the agreement also provides for an annual royalty of a figure based on net sales of products, distinguishing between sales of nutraceutical and medicinal drugs. For indirect exploitation, it also provides for annual double-digit royalties based on income received from licensees, distinguishing (i) between the sales of nutraceuticals (double-digit royalties) and drug products (one or two-digit royalties) and (ii) the product development phase of these products (Phase 1, 2 or 3) at the time of conclusion of the licensing agreement. The royalty payments will end upon termination of the agreement. |
Off-Balance Sheet Arrangements
We do not have variable interests in variable interest entities or any off-balance sheet arrangements as defined under the SEC rules, such as relationships with unconsolidated entities or financial partnerships, which are often referred to as structured finance or special purpose entities, established for the purpose of facilitating financing transactions that are not required to be reflected on our statements of financial position.
C.Research and Development
For a discussion of our research and development activities, see “Item 4.B—Business Overview” and “Item 5.A—Operating Results.”
D.Trend Information
For a discussion of trends, see “Item 4.B—Business Overview,” “Item 5.1—Operating Results” and “Item 5.B—Liquidity and Capital Resources.”
E.Critical Accounting Estimates
Our audited consolidated financial statements have been prepared in accordance with IFRS Accounting Standards, as issued by the IASB. Some of the accounting methods and policies used in preparing our financial statements under IFRS Accounting Standards are based on complex and subjective assessments by our management or on estimates based on past experience and assumptions deemed realistic and reasonable based on the circumstances. The actual value of our assets, liabilities and shareholders’ equity and of our losses could differ from the value derived from these estimates if conditions change and these changes have an impact on the assumptions adopted. We believe that the most significant management judgments and assumptions in the preparation of our financial statements are described in Note 3.1 to our audited consolidated financial statements.
126
Item 6. Directors, Senior Management and Employees.
A.Directors and Senior Management
The following table presents information about our officers and directors as of the date of this annual report.
NAME |
| AGE |
| POSITION |
Executive Officers |
|
|
|
|
Stanislas Veillet |
| 58 |
| Chairman of the Board, Chief Executive Officer and Director |
Nicolas Fellmann | 55 | Chief Financial Officer | ||
Chiara Baccelli |
| 48 |
| Chief Pharmaceutical Operations Officer & Quality Assurance Director |
Edouard Bieth |
| 44 |
| Chief Business Officer |
Pierre J. Dilda |
| 54 |
| Chief Scientific Officer |
Waly Dioh |
| 55 |
| Chief Clinical Operations Officer |
Rob Van Maanen | 54 | Chief Medical Officer | ||
Non-Employee Directors |
|
|
| |
Claude Allary | 69 | Director | ||
Nadine Coulm |
| 61 |
| Director |
Jean Mariani |
| 74 |
| Director |
There are no family relationships among any of our executive officers or directors. Unless otherwise indicated, the current business addresses for our executive officers and directors is Sorbonne University—BC 9, Bâtiment A 4ème étage, 4 place Jussieu 75005 Paris, France.
Biographies
Executive Officers
Stanislas Veillet is the co-founder of Biophytis. He has served as our President since the Company’s inception and served as Chief Executive Officer (Directeur Général) and chairman of our board since May 2015. He began his career in Brazil as a researcher at the Centre de coopération international en recherche agronomique pour le développement, or CIRAD from 1989 to 1993, before obtaining a Ph.D. in Genetics. From 1994 to 2001, Mr. Veillet managed a biotechnology laboratory for the Cargill Group, then Pharmacia-Monsanto, to develop a high throughput platform for whole genome genotyping. From 2002 to 2006, he managed the Life Sciences Department of the Danone Group, where he developed several products, including Danacol and Danaten for the prevention of cardiovascular diseases. Mr. Veillet has a degree in Engineering and a Ph.D. in Genetics from AgroParisTech. Mr. Veillet is also a member of the board of directors and chairman of the compensation committee of Drone Volt S.A.
Nicolas Fellmann joined Biophytis in May 2023 as Chief Financial Officer. From 2014 to 2023, Mr. Fellmann served as Chief Financial Officer of Onxeo (today renamed Valerio Therapeutics), a French biotechnology company developing drugs in oncology and listed both on Euronext and the Nasdaq Copenhagen. From 2006 to 2014, he was Chief Financial Officer of BioAlliance Pharma, a French specialty pharmaceutical company developing and selling supportive care products in oncology. In these two companies, Mr. Fellmann raised more than €170 million, managed two M&A transactions and several out-licensing deals in Europe, the United States and Asia. From 1995 to 2006, Mr. Fellmann worked at Pfizer France in positions with increasing responsibilities, notably as Director Treasury Tax and Audit, responsible for the management of financial risks for the French affiliates. Prior experiences include Ernst & Young and Total. Mr. Fellmann received an MBA from EM Lyon business school (1990).
Chiara Baccelli has served as our Chief Pharmaceutical Operations Officer & Quality Assurance Director since January 2023. She holds more than 20 years of experience in the development and production of pharmaceutical products. Prior to joining us, she held different roles around the development of innovative products, from CMC to Quality Assurance and Control, Industrialization and Regulatory Affairs and worked for UCB in Belgium, for the French group Delpharm, the company Bioprojet and also for the consulting firm IDD in France in collaboration with large pharmaceutical groups such as Boehringer Ingelheim, Ipsen, Sanofi, Nemera, Harmony Bioscience. Ms. Baccelli holds a PharmD from the University of Pisa in Italy, a PhD in Pharmaceutical and Biomedical Sciences from the Catholic University of Leuven in Belgium and a MBA in Innovation and Strategy from IRIIG in Lyon.
127
Edouard Bieth has served as our Chief Business Officer since September 2023. Prior to joining us, Mr. Bieth was General Manager France & BeNeLux of Tillotts Pharma’s commercial subsidiary. During his career, he has also worked for various industry players, including AstraZeneca, Servier and Menarini, where he held senior management positions. With over 18 years’ experience in the pharmaceutical industry, Mr. Bieth has developed strong expertise in market access and sales and marketing strategy, both globally and locally. He has a Master’s degree in biology and pharmacology of ageing and trained in sales and marketing strategy at HEC Paris and in finance at the London Business School.
Pierre J. Dilda has served as our Chief Scientific Officer since October 2019 and previously served as our Vice President of Research from 2015 to 2019. Before joining us, he was Senior Research Fellow at the Lowy Cancer Research Center at the University of New South Wales (UNSW) in Sydney, Australia, from 2006 to 2015, where he was responsible for advancing several cancer therapeutics. Dr. Dilda holds a bachelor’s degree in biochemistry and a Masters in biochemistry and immunology from the University of Paris VII (Denis Diderot), Faculty of Sciences, Paris, France, and a Masters in physiology and physiopathology and a PhD in pharmacology from the University of Paris V, Faculty of Medicine, Paris, France.
Waly Dioh has served as our Chief Clinical Operating Officer since October 2019 and previously served as Vice President of Clinical Development from October 2015 to October 2019 and as our Director of Research and Development from October 2006 to October 2015. Previously, Mr. Dioh worked at Monsanto Company, initially in France and then in the United States. Mr. Dioh received a DUES in natural sciences from Dakar University in Senegal, a masters in biology/plant pathology from Pierre and Marie Curie University Paris VI in Paris, France, a PhD in plant pathology from his doctorate from the University of Paris XI, Orsay in Paris, France and an MBA from the ESLSCA Business School in Paris, France.
Rob Van Maanen has served as our Chief Medical Officer since September 2021. Prior to joining us, he served as Chief Medical Officer for Khondrion, a dutch clinical-stage company discovering and developing therapies targeting orphan inherited mitochondtrial diseases. Before that he was Senior Medical Director at Astellas in Leiden, the Netherlands. Dr. Van Maanen holds an MBA from University of Amsterdam (NL), as well as medical licences in the UK (as specialist in the Pharmaceutical Medicine) and the Netherlands. He is an expert in global drug development, medical affairs, and pharmacovigilance with more than 20 years of experience in both large pharmaceutical companies and small biotechs.
Non-Employee Directors
Claude Allary has served as a director since July 2021. Mr. Allary has been working in health industries for the past 40 years. He is currently a Senior Advisor for Bionest Partners, which he co-founded in 2002. He is also a co-founder and Director of Institut Colisée, a think tank for the betterment of human relationship in companies and organizations. Mr. Allary graduated from ESSEC, a French Business School and holds a Ph.D. in Management Sciences from Paris II University.
Nadine Coulm has served as a director since May 2015.She has over 30 years of experience in Corporate Finance, with a focus on Investor Relations and Financing. Ms. Coulm served as the Vice President of Investor Relations and Financing for the Korian Group, which provides long-term care to the elderly, from March 2017 to August 2019. Previously, she served as the Vice President Financing and Investor Relations for FNAC Group, a consumer electronics company, from January 2013 to March 2017. From November 2006 to November 2011, she served as Vice President of Fianancial Communication and Investor Relations at Casino Group. From 1988 to 2006, she held various positions at Danone Group.. Ms. Coulm received an MBA in Finance from HEC Paris.
Jean Mariani has served as a director since October 2019. Dr. Mariani was employed by the Company from October 2017 to September 2019. Since October 2019, Dr. Mariani has served as president Successful Life SAS. He has served as a Professor Emeritus at the faculty of Medecine of Sorbonne University since October 2017. He has served as director of the team Brain Development Aging and Repair in the UMR UPMC-CNRS 8256 (Research laboratory) since 2014. He has been director of the UMR UPMC-CNRS 7102 (Research laboratory) from 2001 to 2013. He has been director of the University Hospital Department FAST (Fight Ageing and Stress) from 2013 to 2018 and of the Institute of Longevity Charles Foix since 2008. He has been a professor and hospital practitioner since September 2005. He was member of the Scientific Council of the Faculty of Medicine Pierre et Marie Curie from 2011 to 2015. Dr. Mariani has been a member of the Scientific Council of the Ataxia Telangectasia Fund since 1997 and president of the Society for Research on Cerebellum and Ataxia since 2012. Dr. Mariani holds an MD and a DSc in Biochemistry. Dr. Mariani has been credited with 241 scientific articles and 25 book chapters.
128
Board Diversity
The table below provides certain information regarding the diversity of our board of directors as of the date of this annual report.
Board Diversity Matrix 2023 | ||||||||
Country of Principal Executive Offices: |
| France | ||||||
Foreign Private Issuer |
| Yes | ||||||
Disclosure Prohibited under Home Country Law |
| Yes | ||||||
Total Number of Directors |
| 4 | ||||||
| Female |
| Male |
| Non- |
| Did Not | |||
Part I: Gender Identity | ||||||||||
Directors |
| 1 |
| 3 |
| — |
| — | ||
Part II: Demographic Background | ||||||||||
Underrepresented Individual in Home Country Jurisdiction | — | |||||||||
LGBTQ+ | — | |||||||||
Did Not Disclose Demographic Background | — |
Board Diversity Matrix 2022 | ||||||||
Country of Principal Executive Offices: |
| France | ||||||
Foreign Private Issuer |
| Yes | ||||||
Disclosure Prohibited under Home Country Law |
| Yes | ||||||
Total Number of Directors |
| 5 | ||||||
|
| Female |
| Male |
| Non- Binary |
| Did Not |
Part I: Gender Identity |
| |||||||
Directors |
| 1 |
| 4 |
| — |
| — |
Part II: Demographic Background |
| |||||||
Underrepresented Individual in Home Country Jurisdiction |
| — | ||||||
LGBTQ+ |
| — | ||||||
Did Not Disclose Demographic Background |
| — | ||||||
B.Compensation
The aggregate compensation paid and benefits in kind granted by us to our current executive officers and directors, including share-based compensation, for the year ended December 31, 2023, was €2,811 thousand. For the year ended December 31, 2023, we allocated €237 thousand to be accrued to provide retirement indemnity to our directors or executive officers, except to the extent required by French law.
There are no agreements or contracts between us (or our subsidiaries) and any of our directors or executive officers that provide for benefits to the directors upon termination of service or to the executive officers upon termination of employment.
In application of the Exchange Act Rule 10D-1 and amendments to existing rules and forms, and in response to the requirements of the final clawback rules adopted by the SEC on June 9, 2023, the Company adopted a Clawback Policy applicable to any incentive-based compensation that is granted, earned or vested based wholly or in part on the attainment of any financial reporting measure on or after the effective date of October 2, 2023. This Clawback Policy is attached as Exhibit 97.1.
129
Director Compensation
The following table sets forth the total compensation paid to our non-employee directors for service on our board of directors during the year ended December 31, 2023.
Total | ||
Compensation(1) | ||
Name |
| (K€) |
Stanislas Veillet |
| 25 |
Claude Allary | 25 | |
Nadine Coulm |
| 25 |
Jean Mariani |
| 25 |
| (1) | Represents meeting attendance fees paid to or earned by directors. |
Chief Executive Officer Compensation
The following table sets forth information regarding compensation paid to or earned by our Chief Executive Officer during the year ended December 31, 2023.
Amounts | ||
Paid | ||
| or Earned | |
Nature of Compensation |
| (K€) |
Fixed remuneration(1) |
| 250 |
Variable annual remuneration(2) |
| 45 |
Benefits in kind(3) |
| 29 |
Total |
| 324 |
| (1) | Mr. Veillet receives a fixed annual remuneration of €250,000 payable over 12 months. |
| (2) | Mr. Veillet was entitled to receive variable annual remuneration of up to €75,000 for the year ended December 31, 2023, based on satisfaction of the following 2021 annual targets: (i) Get approval to start and partner SARA phase 3 study H1, (ii) Raise €16 million on Nasdaq and/or Euronext in 2023, (iii) Get COVA EAP approval in Brazil and France H1, (iv) File for conditional marketing authorization in COVID-19 and generate revenues of €10 million H2. Based on an overall achievement of 60%, the Compensation and Governance Committee determined that Mr. Veillet was entitled to receive €45,000, which amount was paid to him in March 2024. |
| (3) | Mr. Veillet benefits from a “GSC” private unemployment insurance policy. In France, directors and officers do not have employee status and are not covered by the legal unemployment regime. “GSC” enables directors and officers to receive income in the event of unemployment. |
Mr. Veillet is also entitled to receive reimbursement of expenses incurred within the context of performing his duties as Chairman and Chief Executive Officer.
Employment Agreements with Executive Officers and Change of Control Severance Benefits
We have entered into employment agreements with our executive officers, except for our CEO who is a corporate officer (mandataire social) and does not have an employment contract. Each of our executive officers is employed for a continuous term unless either we or the executive officer gives prior notice to terminate such employment. We may terminate the employment of our executive officers for just cause (cause réelle et sérieuse), at any time, with the notice and indemnification requirements provided by French law and the applicable collective bargaining agreement. An executive officer may terminate his or her employment at any time with the prior written notice period provided by French law and the applicable collective bargaining agreement.
Each executive officer has agreed to maintain the confidentiality of any confidential information, both during and after the employment agreement expires or is earlier terminated. In addition, all executive officers have agreed to be bound by a non-solicitation covenant that prohibits each executive officer from soliciting our customers, or soliciting or hiring our executive
130
employees and those of our employees working in the same team as our executive officer, during his or her employment and for one year after the termination of his or her employment. In addition, our executive employees (other than René Lafont), are bound by a non-compete covenant that prohibits each executive officer from competing with us, directly or indirectly, during his or her employment and for six months after the termination of his or her employment.
In accordance with statutory provisions, Mr. Veillet may be freely removed from his position as Chairman and/or Chief Executive Officer by the board of directors. As director, he may be removed by decision of the shareholders. When the Chief Executive Officer does not hold the position of Chairman of the board of directors, he may be entitled to receive an indemnity in the event that he is removed without just cause. Mr. Veillet benefits from a “GSC” private unemployment insurance policy, the cost of which is borne by the Company as a benefit in kind.
Limitations on Liability and Indemnification Matters
Under French law, provisions of by-laws that limit the liability of directors are prohibited. However, French law allows sociétés anonymes to contract for and maintain liability insurance against civil liabilities incurred by any of their directors and officers involved in a third-party action, provided that they acted in good faith and within their capacities as directors or officers of the company. Criminal liability cannot be indemnified under French law, whether directly by the company or through liability insurance.
We expect to maintain customary liability insurance coverage for our directors and executive officers, including insurance against liability under the Securities Act, and we intend to enter into agreements with our directors and executive officers to provide contractual indemnification. With certain exceptions and subject to limitations on indemnification under French law, these agreements will provide for indemnification for damages and expenses including, among other things, attorneys’ fees, judgments, fines and settlement amounts incurred by any of these individuals in any action or proceeding arising out of his or her actions in that capacity. We believe that this insurance and these agreements are necessary to attract qualified directors and executive officers.
These agreements may discourage shareholders from bringing a lawsuit against our directors and executive officers for breach of their fiduciary duty. These provisions also may have the effect of reducing the likelihood of derivative litigation against directors and executive officers, even though such an action, if successful, might otherwise benefit us and our shareholders. Furthermore, a shareholder’s investment may be adversely affected to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these insurance agreements.
Certain of our non-employee directors may, through their relationships with their employers or partnerships, be insured against certain liabilities in their capacity as members of our board of directors.
Equity Incentives
We believe our ability to grant equity incentives is a valuable and necessary compensation tool that allows us to attract and retain the best available personnel for positions of substantial responsibility, provides additional incentives to employees and promotes the success of our business. Due to French corporate law and tax considerations, we have historically granted three different types of equity incentive instruments to our directors, executive officers, employees and other service providers, including:
| ● | founders’ share warrants (otherwise known as bons de souscription de parts de créateurs d’entreprise (“BSPCE”)), which are granted to our officers, directors and employees; |
| ● | share warrants (otherwise known as bons de souscription d’actions (“BSA”)), which have historically only been granted to non-employee directors; and |
| ● | free shares (otherwise known as actions gratuities (“AGA”)), which have historically only been granted to our officers and employees. |
Our board of directors’ authority to grant these equity incentive instruments and the aggregate amount authorized to be granted under these instruments must be approved by a two-thirds majority of the votes by our shareholders present, represented or voting by authorized means, at the relevant extraordinary shareholders’ meeting. Once approved by our shareholders, our board of directors can grant share warrants (BSA) or founder’s share warrants (BSPCE) for up to 18 months from the date of the applicable shareholders’ approval or free shares (AGA) for up to 38 months from the date of the applicable shareholders’ approval. The authority of our board of directors to grant equity incentives may be extended or increased only by receipt of the requisite shareholder approval
131
at an extraordinary shareholders’ meeting. As a result, we typically request that our shareholders authorize new pools of equity incentive instruments at every annual shareholders’ meeting.
All vested shares underlying warrants must be exercised within exercise periods set forth in the grant documents. In the event of certain changes in our share capital structure, such as a consolidation or share split or dividend, French law and applicable grant documentation provide for appropriate adjustments to the numbers of shares issuable and/or the exercise price of the outstanding warrants.
As of February 29, 2024, founders’ share warrants and share warrants granted pursuant to equity incentive awards were outstanding allowing for the issuance or purchase of an aggregate of 5,276,436 ordinary shares (assuming that such instruments’ vesting conditions are met) at a weighted average exercise price of €0.45 per ordinary share. As of February 29, 2024, there were 126,964,703 free shares granted pursuant to equity incentive awards outstanding.
Founder’s Share Warrants (BSPCE)
Employee warrants may only be issued by growth companies meeting certain criteria. Most significantly, the issuer must have been registered for less than 15 years and 25% of the issuer’s share capital must have been continuously held since the company’s formation by natural persons or by holding companies, of which 75% of such holding company’s share capital is held by natural persons. The calculation of such threshold does not include venture capital mutual investment funds (fonds commun de placement à risques), specialized professional funds (fonds professionnels spécialisés), private equity funds (fonds professionnels de capital investissement), local investment funds (fonds d’investissement de proximité) and innovation-focused mutual funds (fonds commun de placement dans l’innovation).
As the Company was registered more than 15 years ago, the Company does no longer fulfil the conditions required to issue founders’ share warrants.
132
Founder’s share warrants have traditionally been granted to certain of our employees and/or officers who were French tax residents because the warrants carry favorable tax and social security treatment for French tax residents. Since French law n°2019-486 of May 22, 2019 relating to the growth and transformation of companies, we may grant founder’s share warrants to our directors. Similar to options, founder’s share warrants entitle a holder to exercise the warrant for the underlying vested shares at an exercise price per share determined by our board of directors and at least equal to the fair market value of an ordinary share on the date of grant. However, unlike options, the exercise price per share is fixed as of the date of implementation of the plans pursuant to which the warrants may be granted, rather than as of the date of grant of the individual warrants. Founder’s share warrants may only be exercised if, at the exercise date, the employee is employed by us. The table below summarizes our outstanding founder’s share warrants to employees/directors employed by us as of February 29, 2024.
Number of | ||||||||||||||||||
ordinary | ||||||||||||||||||
| shares |
|
|
| Purchase |
|
| Number |
| Founders’ | ||||||||
| underlying | Date of | Date of |
| Price | Exercise |
| of Shares |
| Warrants | ||||||||
| Founders’ | General | Board | per share | Start Date | Expiration | Price | subscribed | outstanding as | |||||||||
Name |
| warrants |
| Meeting |
| Meeting |
| (€) |
| for Exercise |
| Date |
| (€) |
| to date |
| of 2/29/2024 |
Stanislas Veillet |
| 940,249 | (1) | 08/08/2019 |
| 04/03/2020 |
| — |
| 04/08/2020 |
| 04/08/2026 |
| 0.27 |
| 313,417 |
| 626,832 |
Pierre Dilda | 50,424 | (1) | 08/08/2019 | 04/03/2020 | — | 04/08/2020 | 04/08/2026 | 0.27 | — | 50,424 | ||||||||
| 100,848 | (2) | 05/28/2020 |
| 12/22/2020 |
| — |
| 12/22/2020 |
| 12/22/2026 |
| 0.47 |
| — |
| 100,848 | |
| 267,394 | (3) | 05/10/2021 |
| 09/15/2021 |
| — |
| 09/15/2021 |
| 09/15/2027 |
| 0.73 |
| — |
| 267,394 | |
Waly Dioh |
| 79,201 | (1) | 08/08/2019 |
| 04/03/2020 |
| — |
| 04/08/2020 |
| 04/08/2026 |
| 0.27 |
| — |
| 79,201 |
| 158,401 | (2) | 05/28/2020 |
| 12/22/2020 |
| — |
| 12/22/2020 |
| 12/22/2026 |
| 0.47 |
| — |
| 158,401 | |
| 419,994 | (3) | 05/10/2021 |
| 09/15/2021 |
| — |
| 09/15/2021 |
| 09/15/2027 |
| 0.73 |
| — |
| 419,994 | |
Nadine Coulm |
| 103,946 | (1) | 08/08/2019 |
| 04/03/2020 |
| — |
| 04/08/2020 |
| 04/08/2026 |
| 0.27 |
| — |
| 103,946 |
| 207,892 | (2) | 05/28/2020 |
| 12/22/2020 |
| — |
| 12/22/2020 |
| 12/22/2026 |
| 0.47 |
| — |
| 207,892 | |
551,218 | (3) | 05/10/2021 | 09/15/2021 | — | 09/15/2021 | 09/15/2027 | 0.73 | — | 551,218 | |||||||||
Jean Mariani | 103,946 | (1) | 08/08/2019 | 04/03/2020 | — | 04/08/2020 | 04/08/2026 | 0.27 | — | 103,946 | ||||||||
| 207,892 | (2) | 05/28/2020 |
| 12/22/2020 |
| — |
| 12/22/2020 |
| 12/22/2026 |
| 0.47 |
| — |
| 207,892 | |
| 551,218 | (3) | 05/10/2021 |
| 09/15/2021 |
| — |
| 09/15/2021 |
| 09/15/2027 |
| 0.73 |
| — |
| 551,218 | |
Claude Allary | 551,218 | (3) | 05/10/2021 | 09/15/2021 | — | 09/15/2021 | 09/15/2027 | 0.73 | — | 551,218 | ||||||||
Rob Van Maanen | 267,394 | (3) | 05/10/2021 | 09/15/2021 | — | 09/15/2021 | 09/15/2027 | 0.73 | — | 267,394 |
| (1) | These founder’s share warrants are exercisable for (i) 33.33% between the grant date and the second anniversary of the grant date, (ii) for 66.66% between the second anniversary of the grant date and the fourth anniversary of the grant date and (iii) in full beginning on the fourth anniversary of the grant date. |
| (2) | These founder’s share warrants are exercisable for (i) 33.33% between the grant date and the second anniversary of the grant date, (ii) for 66.66% between the second anniversary of the grant date and the fourth anniversary of the grant date and (iii) in full beginning on the fourth anniversary of the grant date. |
| (3) | These founder’s share warrants are exercisable for (i) 33.33% between the grant date and the first anniversary of the grant date, (ii) for 66.66% between the first anniversary of the grant date and the second anniversary of the grant date and (iii) in full beginning on the second anniversary of the grant date. |
Share Warrants (BSA)
Similar to options, share warrants entitle a holder to exercise the warrant for the underlying vested shares at an exercise price per share determined by our board of directors. However, unlike options, the exercise price per share is fixed as of the date of
133
implementation of the plans pursuant to which the warrants may be granted, rather than as of the date of grant of the individual warrants. The table below summarizes our outstanding share warrants as of February 29, 2024.
Number of | ||||||||||||||||||
| ordinary |
|
|
| Purchase |
| Exercise |
| Number of |
| Warrants | |||||||
| shares | Date of | Date of |
| Price |
| Price per |
| Shares |
| outstanding | |||||||
| underlying | General | Board | per share | Start Date | Expiration | share | subscribed to | as of | |||||||||
Name |
| share warrants |
| Meeting |
| Meeting |
| (€) |
| for Exercise |
| Date |
| (€) |
| date |
| 2/29/2024 |
Nadine Coulm |
| 27,956 | (1) | 08/08/2019 |
| 03/04/2020 |
| 0.06 |
| 04/30/2020 |
| 04/30/2025 |
| 0.27 |
| 535 |
| 27,956 |
99,619 | (1) | 05/10/2021 | 02/28/2022 | 0.0048 | 06/17/2022 | 06/17/2026 | 0.0967 | — | 99,619 | |||||||||
251,543 | (2) | 06/21/2022 | 02/15/2023 | 0.0027 | 04/14/2023 | 04/14/2027 | 0.0544 | — | 251,543 | |||||||||
Jean Mariani |
| 25,566 | (1) | 08/08/2019 |
| 03/04/2020 |
| 0.06 |
| 04/30/2020 |
| 04/30/2025 |
| 0.27 |
| — |
| 25,566 |
99,619 | (1) | 05/10/2021 | 02/28/2022 | 0.0048 | 06/17/2022 | 06/17/2026 | 0.0967 | — | 99,619 | |||||||||
172,594 | (2) | 06/21/2022 | 02/15/2023 | 0.0027 | 04/14/2023 | 04/14/2027 | 0.0544 | — | 172,594 | |||||||||
Claude Allari | 99,619 | (1) | 05/10/2021 | 02/28/2022 | 0.0048 | 06/17/2022 | 06/17/2026 | 0.0967 | — | 99,619 | ||||||||
251,543 | (2) | 06/21/2022 | 02/15/2023 | 0.0027 | 04/14/2023 | 04/14/2027 | 0.0544 | — | 251,543 | |||||||||
Stanislas Veillet |
| 2,935,701 | (1) | 08/08/2019 |
| 03/04/2020 |
| 0.06 |
| 04/30/2020 |
| 04/30/2025 |
| 0.27 |
| 2,853,201 |
| 82,500 |
Pierre Dilda |
| 20,000 | (1) | 08/08/2019 |
| 03/04/2020 |
| 0.06 |
| 04/30/2020 |
| 04/30/2025 |
| 0.27 |
| 2,000 |
| 18,000 |
Waly Dioh | 26,428 | (1) | 08/08/2019 | 03/04/2020 | 0.06 | 04/30/2020 | 04/30/2025 | 0.27 | — | 26,428 |
| (1) | These share warrants are exercisable in full, beginning on the subscription date. |
| (2) | These warrants are exercisable as follows: 33.33% beginning on the subscription date, 66.66% beginning one year after the subscription date and 100% beginning two years after the subscription date. |
Free Shares (AGA)
Under our free share plan, adopted by our board of directors on February 15, 2023 we have granted free shares (actions gratuites) to certain of our officers and employees.
Free shares (actions gratuites) may be granted to any individual employed by us or by any affiliated company. Free shares may also be granted to our Chairman and Chief Executive Officer. However, no free share may be granted to a beneficiary holding more than 10% of our share capital or to a beneficiary who would hold more than 10% of our share capital as a result of such grant. In addition, under French law, the maximum number of shares that may be granted may not exceed 10% of the share capital as of the date of grant of such free shares (30% if the allocation benefits all employees).
134
The table below summarizes our outstanding free shares as of February 29/2024.
Number of | ||||||||||||||
ordinary shares | Free Shares | |||||||||||||
underlying | Date of General | Date of Board | Date of | Duration of | outstanding as | |||||||||
Name |
| Free Shares |
| Meeting |
| Meeting |
| Grant Date |
| acquisition |
| holding period |
| of 2/29/2024 |
Stanislas Veillet | 13,812,813 | 06/21/2022 | 02/15/2023 | 04/14/2023 | 04/14/2024 | 1 year | 13,812,813 | |||||||
54,000,000 | 04/17/2023 | 01/23/2024 | 02/05/2024 | 02/05/2025 | 1 year | 54,000,000 | ||||||||
René Lafont |
| 4,557,099 |
| 06/21/2022 |
| 02/15/2023 |
| 04/14/2023 |
| 04/14/2024 |
| 1 year |
| 4,557,099 |
17,000,000 | 04/17/2023 | 01/23/2024 | 02/05/2024 | 02/05/2025 | 1 year | 17,000,000 | ||||||||
Nicolas Fellmann | 10,000,000 | 04/17/2023 | 01/23/2024 | 02/05/2024 | 02/05/2025 | 1 year | 10,000,000 | |||||||
Edouard Bieth | 6,000,000 | 04/17/2023 | 01/23/2024 | 02/05/2024 | 02/05/2025 | 1 year | 6,000,000 | |||||||
Waly Dioh |
| 119,345 |
| 06/21/2022 |
| 02/15/2023 |
| 04/14/2023 |
| 04/14/2024 |
| 1 year |
| 119,345 |
3,500,000 | 04/17/2023 | 01/23/2024 | 02/05/2024 | 02/05/2025 | 1 year | 3,500,000 | ||||||||
Pierre Dilda |
| 75,983 |
| 06/21/2022 |
| 02/15/2023 |
| 04/14/2023 |
| 04/14/2024 |
| 1 year |
| 75,983 |
3,500,000 | 04/17/2023 | 01/23/2024 | 02/05/2024 | 02/05/2025 | 1 year | 3,500,000 | ||||||||
Chiara Baccelli |
| 75,983 |
| 06/21/2022 |
| 02/15/2023 |
| 04/14/2023 |
| 04/14/2024 |
| 1 year |
| 75,983 |
3,500,000 | 04/17/2023 | 01/23/2024 | 02/05/2024 | 02/05/2025 | 1 year | 3,500,000 | ||||||||
Rob Van Maanen |
| 75,983 |
| 06/21/2022 |
| 02/15/2023 |
| 04/14/2023 |
| 04/14/2024 |
| 1 year |
| 75,983 |
3,500,000 | 04/17/2023 | 01/23/2024 | 02/05/2024 | 02/05/2025 | 1 year | 3,500,000 |
None of the free share recipients held more than 10% of our share capital prior to any grant, no allocation of free shares resulted in any of the free share recipients holding more than 10% of the share capital.
C.Board Practices
Board Composition
We currently have five directors.
Under French law and our Articles of Association, our board of directors must be comprised of between three and 18 members. Within this limit, the number of directors is determined by our shareholders. Directors are elected, re-elected and may be removed at a shareholders’ general meeting with a simple majority vote of our shareholders. Pursuant to our by-laws, our directors are elected for three-year terms. In accordance with French law, our by-laws also provide that our directors may be removed with or without cause by the affirmative vote of the holders of at least a majority of the votes of the shareholders present, represented by a proxy or voting by mail at the relevant ordinary shareholders’ meeting, and that any vacancy on our board of directors resulting from the death or resignation of a director, provided there are at least three directors remaining, may be filled by vote of a majority of our directors then in office provided that there has been no shareholders meeting since such death or resignation. Directors chosen or appointed to fill a vacancy shall be elected by the board of directors for the remaining duration of the current term of the replaced director. The appointment must then be ratified at the next shareholders’ general meeting. In the event the board of directors would be composed of less than three directors as a result of a vacancy, the remaining directors shall immediately convene a shareholders’ general meeting to elect one or several new directors so there are at least three directors serving on the board of directors, in accordance with French law.
The following table sets forth the names of our directors, the years of their initial appointment as directors and the expiration dates of their current term.
Year of | Term | |||||
Current | Initial | Expiration | ||||
Name |
| Position |
| Appointment |
| Year |
Stanislas Veillet |
| Chairman |
| 2015 |
| 2024 |
Claude Allary | Director | 2021 | 2026 | |||
Nadine Coulm |
| Director |
| 2015 |
| 2024 |
Jean Mariani |
| Director |
| 2019 |
| 2023 |
135
Director Independence
As a foreign private issuer, under the listing requirements and rules of Nasdaq, we are not required to have independent directors on our board of directors, except with respect to our audit committee, for which Nasdaq listing requirements permit specified phase-in schedules. Nevertheless, our board of directors has undertaken a review of the independence of the directors and considered whether any director has a material relationship with us that could compromise his or her ability to exercise independent judgment in carrying out his or her responsibilities. Based upon information requested from, and provided by, each director concerning such director’s background, employment and affiliations, including family relationships, our board of directors determined that all of our directors, except for Mr. Veillet and Mr. Mariani, qualify as “independent directors” as defined under applicable rules of Nasdaq and the independence requirements contemplated by Rule 10A-3 of the Exchange Act. In making these determinations, our board of directors considered the current and prior relationships that each non-employee director has with our company and all other facts and circumstances that our board of directors deemed relevant in determining their independence, including the beneficial ownership of our ordinary shares by each non-employee director and his or her affiliated entities (if any).
Our board of directors also determined that, except for Stanislas Veillet and Jean Mariani, all of our directors qualify as “independent directors” as defined by the Corporate Governance Code (Code de Gouvernement d’Entreprise) for small and mid-cap companies as published in September 2016 by MiddleNext and validated as a reference code by the French Financial Markets Authority (Autorité des Marchés Financiers).
Board Committees
The board of directors has established an audit committee and a compensation and governance committee, which operate pursuant to rules of procedure adopted by our board of directors. The board of directors has also established a scientific committee, which is responsible for analyzing and reviewing our clinical and regulatory strategy. Subject to available exemptions, the composition and functioning of all of our committees will comply with all applicable requirements of the French Commercial Code, the by-laws, the Exchange Act, Nasdaq and SEC rules and regulations.
In accordance with French law, committees of our board of directors only have an advisory role and can only make recommendations to our board of directors based on their area of competence. As a result, all decisions will be made by our board of directors taking into account non-binding recommendations of the relevant board committee.
Audit Committee
The Audit Committee consists of at least two members appointed by our board of directors. The members of the Audit Committee may or may not be directors or shareholders of the Company; provided, however, that as far as possible, the members of the Audit Committee consists of independent members and, in any event, the Audit Committee must include at least one independent director. The Chairperson of the Audit Committee is appointed by our board of directors for the duration of his or her mandate as a board member.
The current members of our Audit Committee are Nadine Coulm (Chairwoman) and Claude Allary, both independent directors. We intend to rely on the exemption available to foreign private issuers for the requirement that an audit committee be comprised of at least three members, although we may, in the future, look to expand this committee.
The duration of the mandates of the members of the Audit Committee is three years, ending at the first board meeting held after the Ordinary General Meeting called to approve the financial statements. The mandates of the members of the Audit Committee are renewable.
The Audit Committee is responsible for assisting the board of directors in:
| ● | ensuring the truthfulness of the financial statements, the quality of internal controls and the quality and relevance of the financial information provided; |
| ● | assessing the existence and relevance of the financial control and internal audit procedures; |
| ● | assessing the relevance of the Company’s accounting policies; |
136
| ● | examining the accounts of the Company, as well as the information issued before their submission to the board of directors; |
| ● | examining the changes and adaptations of accounting principles and rules used in the context of drawing up of financial statements, as well as their relevance; |
| ● | examining the candidates proposed to the positions of statutory auditor or substitute auditor, or proposing the appointment of the auditors; |
| ● | guaranteeing the independence and competence of auditors and ensuring the proper performance of their duties; and |
| ● | examining the significant risks for the Company and notably the off-balance-sheet risks and commitments. |
In this capacity, the Audit Committee issues opinions, proposals and recommendations to our board of directors and regularly reports to it on its work.
The Audit Committee meets as often as it considers necessary, but at least twice a year before the meeting of the board of directors at which the annual and interim financial statements of the Company are reviewed.
Compensation and Governance Committee
The Compensation and Governance Committee consists of at least two members, appointed by our board of directors. The members of the Compensation and Governance Committee may or may not be directors or shareholders of the company; provided, however, that the Compensation and Governance Committee must include at least one independent director. No member of the board of directors exercising management functions within the Company may be a member of the Compensation and Governance Committee. The Chairman of the Compensation and Governance Committee is appointed by the board of directors of the Company for the duration of his or her mandate as Committee member.
The current members of our Compensation and Governance Committee are Dimitri Batsis (Chairman) and Nadine Coulm, both independent directors.
The duration of the mandates of the members of the Compensation and Governance Committee is three years, ending at the first meeting of the board of directors held after the Ordinary General Meeting called to approve the financial statements. The mandate of the members of the Compensation and Governance Committee is renewable.
The Compensation and Governance Committee is responsible for:
| ● | making recommendations to the board of directors (i) on remuneration (fixed and variable) of company officers and key executives and notably contributing to the review of remuneration procedures, setting objectives and bonuses for objectives reached and incentives for the company’s officers; (ii) the recruitment, training, development, retention of employees with remuneration programs; and (iii) the shareholder policy and incentive tools for managers and employees, taking into account the objectives of the Company and individual and collective performance, including the fixing and/or modification of the conditions for the award or exercise of securities granted to the officers or of the employees, and, where appropriate, the achievement of objectives permitting the exercise of the said securities, as provided under the terms and conditions of the said securities; |
| ● | participating in the implementation of the Company’s governing bodies; |
| ● | identifying, assessing and proposing the appointment of independent directors with a view to the good governance of the Company; and |
| ● | pronouncing on any other issue relating to human resources which it considers appropriate or which is referred to it by the board of directors. |
The Compensation and Governance Committee has only consultative powers. The Compensation and Governance Committee reports on its mission to the board of directors and communicates its recommendations, specifications, and opinions.
137
The Compensation and Governance Committee meets as often as it considers necessary, but at least twice a year.
D.Employees
As of December 31, 2023, we had 22 employees, all of whom are full-time, 17 of whom are engaged in research and development activities and 5 of whom are engaged in general and administrative activities. As of the date of this annual report, 100% of our employees are located in France. None of our employees are subject to a collective bargaining agreement. We consider our relationship with our employees to be good. France-based employees are subject to the national collective bargaining agreement for the pharmaceutical industry (the convention collective nationale de l’industrie pharmaceutique).
E.Share Ownership
For information regarding the share ownership of our directors and senior management, see “Item 6.B—Compensation” and “Item 7.A—Major Shareholders.”
F.Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation
None
Item 7. Major Shareholders and Related Party Transactions.
A.Major Shareholders
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as of February 29, 2024 for:
| ● | each beneficial owner of more than 5% of our outstanding ordinary shares; |
| ● | each of our directors and executive officers; and |
| ● | all of our directors and executive officers as a group. |
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include ordinary shares that can be acquired within 60 days of February 29, 2024. The percentage ownership information shown in the table is based upon 1,146,016,582 ordinary shares outstanding as of February 29, 2024.
Except as otherwise indicated, all of the shares reflected in the table are ordinary shares and all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
138
In computing the number of ordinary shares beneficially owned by a person and the percentage ownership of that person, we deemed outstanding ordinary shares subject to options and warrants held by that person that are immediately exercisable or exercisable within 60 days of February 29, 2024. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. Beneficial ownership representing less than 1% is denoted with an asterisk (*). The information in the table below is based on information known to us or ascertained by us from public filings made by the shareholders. Except as otherwise indicated in the table below, addresses of the directors, executive officers and named beneficial owners are in care of Biophytis S.A., Sorbonne University—BC 9, Bâtiment A 4ème étage, 4 place Jussieu 75005 Paris, France.
Number of Ordinary |
| ||||
Owner |
| Shares |
| Percentage |
|
Directors and Executive Officers: |
|
|
|
| |
Stanislas Veillet(1) |
| 33,484,803 |
| 2.86 | % |
Claude Allary (2) | 902,380 | * | |||
Nadine Coulm (3) | 1,243,034 | * | |||
Jean Mariani (4) |
| 1,162,835 |
| * | |
Pierre Dilda (5) |
| 560,974 |
| * | |
Waly Dioh (6) |
| 904,272 |
| * | |
Rob Van Maanen (7) |
| 343,917 |
| * | |
Chiara Baccelli (8) |
| 75,983 |
| * | |
Nicolas Fellmann | — | * | |||
Edouard Bieth | — | * | |||
All directors, executive officers and key employees as a group (15 persons) (9) |
| 4,703,019 |
| * |
* | Represents beneficial ownership of less than 1%. |
(1) | The shares beneficially owned by Mr. Veillet include 709,332 shares issuable upon the exercise of warrants that are currently exercisable or exercisable within 60 days of February 29, 2024 and 13,812,813 free shares that will be definitely acquired within 60 days of February 29, 2024. |
(2) | The shares beneficially owned by Mr. Allary include 902,380 shares issuable upon the exercise of warrants that are currently exercisable or exercisable within 60 days of February 29, 2024 . |
(3) | The shares beneficially owned by Mrs. Coulm include 1,241,784 shares issuable upon the exercise of warrants that are currently exercisable or exercisable within 60 days of February 29, 2024 . |
(4) | The shares beneficially owned by Mr. Mariani include 1,162,835 shares issuable upon the exercise of warrants that are currently exercisable or exercisable within 60 days of February 29, 2024 . |
(5) | The shares beneficially owned by Mr. Dilda include 418,666 shares issuable upon the exercise of warrants that are currently exercisable or exercisable within 60 days of February 29, 2024 and 75,983 free shares that will be definitely acquired within 60 days of February 29, 2024. |
(6) | The shares beneficially owned by Mr. Dioh include 657,956 shares issuable upon the exercise of warrants that are currently exercisable or exercisable within 60 days of February 29, 2024 and 119,345 free shares that will be definitely acquired within 60 days of February 29, 2024. |
(7) | The shares beneficially owned by Mr. Van Maanen include 267,934 shares issuable upon the exercise of warrants that are currently exercisable or exercisable within 60 days of February 29, 2024 and 75,983 free shares that will be definitely acquired within 60 days of February 29, 2024. |
(8) | The shares beneficially owned by Mrs. Baccelli include 0 shares issuable upon the exercise of warrants that are currently exercisable or exercisable within 60 days of February 29, 2024 and 75,983 free shares that will be definitely acquired within 60 days of February 29, 2024. |
(9) | The shares beneficially owned by our officers and directors as a group include an aggregate of 4,703,019 shares issuable upon acquisition of free shares within 60 days of February 29, 2024. |
139
Each of our shareholders is entitled to one vote per ordinary share. None of the holders of our shares has different voting rights from other holders of shares. We are not aware of any arrangement that may, at a subsequent date, result in a change of control of our company.
As of February 29, 2024, assuming that all of our ordinary shares represented by ADSs are held by residents of the United States, we estimate that approximately 9% of our outstanding ordinary shares were held in the United States by one registered holder of record, BNY Mellon, our ADS depositary. The actual number of holders is greater than these numbers of record holders and includes beneficial owners whose ADSs are held in street name by brokers and other nominees. This number of holders of record also does not include holders whose shares may be held in trust by other entities.
B. | Related Party Transactions |
Since January 1, 2023, we have engaged in the following transactions with our directors, executive officers and holders of more than 5% of our outstanding voting securities and their affiliates, which we refer to as our related parties.
Transactions with Our Affiliates, Principal Shareholders, Directors and Executive Officers
Intellectual Property Agreement with Stanislas Veillet
Our CEO, who is a corporate officer (mandataire social) but not an employee of the Company under French law, is involved in our research and development activities. He has developed inventions with us for which we have submitted patent applications in which he is listed as a co-inventor and other inventions that we expect may give rise to patent applications in the future for which we expect he will be included as a co-inventor. As an inventor, our CEO has certain rights under French intellectual property law. These rights are distinct from the statutory rights that usually apply to employee inventors under French law. In order to define a framework within which any intellectual property resulting from our CEO’s research and development activities is properly assigned to us, we entered into an agreement on May 22, 2019 and into an amendment agreement to this agreement on April 6, 2020, both of which were approved by our board of directors. Pursuant to this agreement (as amended), our CEO is entitled to the following payments for his contributions:
| ● | a first lump sum cash payment of €90 thousand to be paid within 30 days of filing of a patent application based on the assigned rights; |
| ● | a second lump sum cash payment of €90 thousand, to be paid within 30 days of publication of a patent application based on the assigned rights; and |
| ● | a 6.5% royalty payment with respect to any license income and/or any net sales by us of products manufactured with the patents filed on the basis of the assigned rights. |
These three payments will be capped at €2.1 million on a platform per platform basis, a platform being defined in the agreement as the research and development works which cover the same family of chemical molecules targeting the same molecular receptor or biological pathway for a family of pathologies which are clinically connected.
In the event that a third party pharmaceutical and/or biotech company acquires 100% of our capital and voting rights, payments will be accelerated, so that the cap (€2.1 million per platform), less any amount previously paid in respect of a platform, will become immediately payable.
The agreement shall remain in effect until no further payments are due. However, the provisions of this agreement will only apply to results generated during the period in which our CEO occupies the position of a corporate officer of the Company or any of its affiliates. Any party to the agreement may, upon material breach of the agreement by the other party, terminate the agreement.
As part of the Intellectual Property agreement signed with our CEO and its amendment, the total patents rights acquired from our CEO amounted to €1530 thousand. Of this amount, €270 thousand, €90 thousand and €90 thousand were paid to the Company’s CEO in 2021, 2022 and 2023, respectively.
140
Agreements with Biophytis, Inc.
We have entered into a current account advance agreement with Biophytis, Inc., dated November 9, 2015, which provides for certain cash advances to be made to Biophytis, Inc. by us. The amounts advanced to Biophytis, Inc. under this agreement bear interest from the date such advances are made at the quarterly average effective rate of floating-rate loans with an initial maturity of more than two years, as used by credit institutions and published by the Banque de France. Biophytis, Inc. undertakes to reimburse us for the sums borrowed at any time, subject to budget constraints and immediately upon ceasing to be under our direct or indirect control. However, no repayment schedule has been set. Since January 1, 2023, the largest amount owed by Biophytis, Inc. to us under this agreement was € 1,388 thousand. The outstanding amount owed by Biophytis, Inc. to us as of February 29, 2024 is € 1,388 thousand.
We are also party to a debt compensation agreement with Biophytis, Inc., dated March 14, 2017, with retroactive effect as of January 1, 2017. This agreement provides that in exchange for services rendered to Biophytis, Inc., Biophytis, Inc. will pay us the amounts we invoice to them and that amounts billed to Biophytis, Inc. will bear interest at the quarterly average effective rate of floating-rate loans with an initial maturity of more than two years, as used by credit institutions and published by the Bank of France. Since January 1, 2023, the largest amount owed by us to Biophytis, Inc. was nil. The outstanding amount owed by us to Biophytis, Inc. as of February 29, 2024 is nil.
On March 22, 2019, we also entered into a services agreement with Biophytis, Inc., effective as of January 1, 2019. Pursuant to the terms of the agreement, Biophytis, Inc. has agreed to provide certain clinical and regulatory assistance to us (including supporting our clinical development efforts, assisting with the preparation and submission of regulatory and clinical documents to the various regulatory agencies and interacting with those agencies, and assisting with the preparation of other scientific communications) and certain financial and communication services (including financial and accounting support and investor relations services). In consideration for their services, we have agreed to reimburse Biophytis, Inc. for all of their direct and indirect costs and expenses in providing the services plus a 5% margin. The agreement is effective for one year and may be renewed for subsequent one year periods. On June 7, 2019, this agreement was amended to expand the financial services to be provided to us by Biophytis, Inc. under the agreement. Since January 1, 2023, the largest amount owed by us to Biophytis, Inc. was nil. The outstanding amount owed by us to Biophytis, Inc. as of February 29, 2024 is nil.
Agreements with Biophytis Instituto Do Brasil Serviços, Comércio, Importação E Exportação de Alimentos Ltda.
Since 2009, we have entered into several loan contracts providing for advances to Biophytis Instituto Do Brasil Serviços, Comércio, Importacao E Exportaçao de Alimentos Ltda, or Biophytis Brazil. We own 94.6% of Biophytis Brazil’s share capital and voting rights. Biophytis Brazil’s other shareholder is M. Wayne Clayton Correa, manager of Biophytis Brazil. Since January 1, 2023, the largest aggregate amount outstanding under these loan contracts was € 609 thousand. The outstanding amount owed by Biophytis Brazil to us as of February 29, 2024 was € 609 thousand. The terms of these loan contracts do not provide for interest or penalty in the event of default or late repayment. If Biophytis Brazil fails to pay the principal of the loan at the maturity date, we may extend the loan for a new term as agreed with Biophytis Brazil.
We have entered into a current account advance agreement with Biophytis Brazil dated December 28, 2020, with retroactive effect as of January 1, 2020, which provides for certain cash advances to be made to Biophytis Brazil by us. The amounts advanced to Biophytis Brazil under this agreement bear interest from the date such advances are made at the quarterly average effective rate of floating-rate loans with an initial maturity of more than two years, as used by credit institutions and published by the Banque de France. Biophytis Brazil undertakes to reimburse us for the sums borrowed at any time, subject to budget constraints and immediately upon ceasing to be under our direct or indirect control. However, no repayment schedule has been set. Since January 1, 2023, the largest aggregate amount outstanding under this agreement was nil. The outstanding amount owed by Biophytis Brazil to us as of February 29, 2024 was nil.
We are also party to a debt compensation agreement with Biophytis Brazil, dated December 28, 2020, with retroactive effect as of July 1, 2020. This agreement provides that in exchange for services rendered to Biophytis Brazil, Biophytis Brazil will pay us the amounts we invoice to them, as soon as its financial resources allow it reasonably, and that amounts billed to Biophytis Brazil will bear interest at the quarterly average effective rate of floating-rate loans with an initial maturity of more than two years, as used by credit institutions and published by the Bank of France. Since January 1, 2023, the largest aggregate amount outstanding under this agreement was nil. The outstanding amount owed by Biophytis Brazil to us as of February 29, 2024 was nil.
141
On December 28, 2020, we also entered into a services agreement with Biophytis Brazil, with retroactive effect as of July 1, 2020. Pursuant to the terms of the agreement, Biophytis Brazil has agreed to provide certain clinical and regulatory assistance to us (including supporting our clinical development efforts, assisting with the preparation and submission of regulatory and clinical documents to the various regulatory agencies and interacting with those agencies, and assisting with the preparation of other scientific communications). In consideration for their services, we have agreed to reimburse Biophytis Brazil for all of their direct and indirect costs and expenses in providing the services plus a 5% margin. The agreement is effective for one year and will be renewed by tacit agreement for subsequent one year periods. Since January 1, 2023, the largest aggregate amount outstanding under this agreement was nil. The outstanding amount owed by us to Biophytis Brazil as of February 29, 2024 was nil.
Services Agreement with Successful Life
On October 1, 2019, we entered into a services agreement with Successful Life SAS in which Jean Mariani, its legal representative, has a controlling interest. This agreement was entered into for a period of one year and was renewed by written amendment dated October 1, 2020 for an additional period of one year, tacitly renewable. This agreement has been terminated and a new agreement has been signed for a period of one year tacitly renewable with effect as from January 1, 2021 following the March 9, 2021 Board decision.The contract runs by tacit renewal for one additional year in 2023. This services agreement provides for the scientific and strategic advice in relation to the biology of aging. The agreement provides for a fixed remuneration of €450 per day within the cap of €32,400 per year and reimbursement of costs and expenses upon presentation of supporting documentation.
In addition, on July 7, 2021, we signed a second agreement with Successful Life SAS by which Jean Mariani is to serve as interim Chief Medical officer until September 8, 2021, as approved by the July 7, 2021 Board decision, for a fixed remuneration of €15,000 per month. This agreement was extended on August 31 until September 30 2022 and was terminated on that date.
Indemnification Agreement
We have entered into nominative indemnification agreements with our directors and executive officers, pursuant to which we have undertaken to provide, in the event of claims, indemnification against any and all losses of and the advancement of expenses to, our directors and executive officers who become a party to or are threatened to be made a party to any claim.
Director and Executive Officer Compensation
See “Item 6.B—Compensation” of this annual report for information regarding compensation of directors and executive officers.
Related Person Transaction Policy
Under French law, transactions between a company and its general managers, directors, shareholders holding more than 10% of the voting rights of the company and any company controlling a shareholder holding more than 10% of the voting rights of the company, other than transactions in the ordinary course of business and at arm’s length, must be (i) approved by the board of directors of the company prior to entering into the transaction, (ii) reported to the statutory auditors who must then prepare a report on such transaction, and (iii) ratified by the company’s shareholders at the annual general meeting.
C.Interests of Experts and Counsel
Not applicable.
Item 8. Financial Information.
A.Consolidated Statements and Other Financial Information
Consolidated Financial Statements
Our consolidated financial statements are appended at the end of this annual report, starting at page F-1, and are incorporated by reference herein.
142
Legal Proceedings
From time to time, we may be involved in various claims and legal proceedings relating to claims arising out of our operations, including those described in Note 14 of our consolidated financial statements for the year ended December 31, 2023 appended to this annual report.
On August 21, 2019, the Company signed an agreement with Negma Group Limited (Negma Group) providing for up to €24 million in financing of the Company through the issuance of several tranches of convertible bonds with warrants (ORNANEBSA).
In accordance with this agreement, the Board of Directors decided to issue the following convertible bonds and warrants during the financial year ended December 31, 2019:
| ● | a first tranche on August 21, 2019 of 300 ORNANE plus a commitment fee of 30 ORNANE, generating gross proceeds for the Company of 3 million euros, accompanied by share subscription warrants giving right to 585,936 shares (BSAT1); |
| ● | a second tranche on December 27, 2019 of 300 ORNANE, 50% of which were paid by Negma Group as of December 31, 2019, generating gross proceeds for the Company of €1.5 million, accompanied by BSAs giving right to 694,444 shares ( BSAT2). |
On April 6, 2020, the Company unilaterally terminated the contract with Negma Group. Following this termination, Negma Group took various litigation steps, both in summary proceedings and on the merits. On the summary aspect, ruling on an order of May 7, 2020, the Paris Court of Appeal, by a judgment of November 18, 2020, overturned the said order, which had partially granted the requests of Negma Group, and condemned this last to return to Biophytis the €378k and the 2,050,000 shares that Biophytis had been forced to deliver to it. It also ordered Negma Group to pay additional penalties to Biophytis for an amount of €41,000 (recognized in financial income for the 2020 financial year).
On June 16, 2020, Negma Group summoned Biophytis before the Paris Commercial Court in order to obtain on the merits what the order of May 7, 2020 refused it. Following a judgment of March 16, 2021, the Paris Commercial Court ordered Biophytis to pay Negma Group a principal amount of €910 thousand in contractual penalties and to deliver €7,000 thousand to Negma Group. Biophytis has appealed to the Paris Court of Appeal against the judgment of the Paris Commercial Court of March 16, 2021. Under the terms of a judgment of July 16, 2021, the execution judge of the Paris Judicial Court partially granted Negam Group and sentenced Biophytis to a penalty of €1,500 thousand for non-execution of the judgment of the Paris Commercial Court of March 16, 2021, which was reduced to €500 thousand by a judgment of the Paris Court of Appeal of September 8, 2022. Biophytis has fulfilled all of the obligations imposed on it under the above-mentioned judgments and delivered to Negma Group 2,050,000 shares held in treasury in July 2021 and 4,950,000 new shares in August 2021. Following a judgment of January 17, 2023, the Paris Court of Appeal confirmed the judgment of the Paris Commercial Court of March 16, 2021. The Paris Court of Appeal adopted a reasoning similar to that of the Paris Commercial Court, contenting itself with a analysis of the Contract at the stage of its conclusion and did not take into account the execution of the said Contract by Negma Group.
On the criminal aspect, an investigating judge was appointed on March 6, 2023.
Biophytis filed a cassation appeal against the judgment of January 17, 2023 on May 10, 2023.
At the same time, on March 29, 2023, Negma Group sued Biophytis before the Paris Commercial Court. Based on the judgment of the Paris Court of Appeal of January 17, 2023, Negma Group requested that Biophytis be ordered to compensate it for the alleged material and image damage resulting from the termination of the ORNANEBSA contract. On September 6, 2023, Biophytis requested that the stay of proceedings be pronounced pending the outcome of the cassation appeal against the judgment of the Paris Court of Appeal of January 17, 2023. By a judgment of February 9, 2024, the Paris Commercial Court granted Biophytis’ requests and suspended the proceedings.
On March 6, 2024, Negma Group summoned Biophytis before the First President of the Paris Court of Appeal for the purpose of being authorized to appeal against the decision to stay proceedings rendered on February 9, 2024 by the Court of commerce of Paris.
143
Other than the legal proceeding described above, we are not currently a party to any legal proceedings that, in the opinion of our management, are likely to have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Dividend Distribution Policy
We have never declared or paid any cash dividends on our ordinary shares. We do not anticipate paying cash dividends on our equity securities in the foreseeable future and intend to retain all available funds and any future earnings for use in the operation and expansion of our business.
Subject to the requirements of French law and our by-laws, dividends may only be distributed from our distributable profits, plus any amounts held in our reserves other than those reserves that are specifically required by law. Article 34 of our By-laws imposes additional limitations on our ability to declare and pay dividends and there may be taxes imposed on you if we elect to pay a dividend. Dividend distributions, if any, will be made in euros and converted into U.S. dollars with respect to the ADSs, as provided in the deposit agreement.
B.Significant Changes
Not applicable.
Item 9. The Offer and Listing.
A.Offer and Listing Details
Our ADS have been listed on the Nasdaq Capital Market under the symbol “BPTS” since February 10, 2021. Prior to that date, there was no public trading market for ADSs. Our ordinary shares have been trading on Euronext Growth Paris under the symbol “ALBPS” since July 13, 2015. Prior to that date, there was no public trading market for our ordinary shares.
B.Plan of Distribution
Not applicable.
C.Markets
Our ADSs have been listed on the Nasdaq Capital Market under the symbol “BPTS” since February 10, 2021 and our ordinary shares have been trading on Euronext Growth Paris under the symbol “ALBPS” since July 13, 2015.
D.Selling Shareholders
Not applicable.
E.Dilution
Not applicable.
F.Expenses of the Issue
Not applicable.
Item 10. Additional Information.
A.Share Capital
Not applicable.
144
B.Memorandum and Articles of Association
The information set forth in Exhibit [2.3] to this annual report is incorporated herein by reference.
C.Material Contracts
We entered into an underwriting agreement with H.C. Wainwright & Co., or H.C. Wainwright, as underwriter, on February 9, 2021, with respect to the ADSs sold in our U.S. initial public offering. We have agreed to indemnify H. C. Wainwright against certain liabilities, including liabilities under the Securities Act, and to contribute to payments H.C. Wainwright may be required to make in respect of such liabilities.
For additional information regarding our material contracts, please see “Item 4—Information on the Company”, “Item 6—Directors, Senior Management and Employees,” and “Item 7.B—Related Party Transactions” of this annual report.
D.Exchange Controls
Under current French foreign exchange control regulations there are no limitations on the amount of cash payments that we may remit to residents of foreign countries. Laws and regulations concerning foreign exchange controls do, however, require that all payments or transfers of funds made by a French resident to a non-resident such as dividend payments be handled by an accredited intermediary. All registered banks and substantially all credit institutions in France are accredited intermediaries.
E.Taxation
Material U.S. Federal Income Tax Considerations
The following is a summary of certain material U.S. federal income tax considerations relating to the acquisition, ownership and disposition of ADSs by a U.S. holder (as defined below). This summary addresses such U.S. federal income tax considerations only for U.S. holders that are initial purchasers of the ADSs and that will hold such ADSs as capital assets within the meaning of Section 1221 of the U.S. Internal Revenue Code of 1986, as amended (the “Code”). This summary does not address all U.S. federal income tax matters that may be relevant to a particular U.S. holder, including U.S. federal estate, gift, or alternative minimum tax considerations, or any U.S. state, local, or non-U.S. tax considerations of the acquisition, ownership and disposition of the ADSs. This summary also does not address tax considerations applicable to a U.S. holder of ADSs that may be subject to special tax rules including, without limitation, the following:
| ● | banks, financial institutions or insurance companies; |
| ● | brokers, dealers or traders in securities, currencies, commodities, or notional principal contracts; |
| ● | tax-exempt entities or organizations, including an “individual retirement account” or “Roth IRA” as defined in Section 408 or 408A of the Code (as defined below), respectively; |
| ● | an entity subject to special tax rules prescribed pursuant to Section 7874 of the Code (as defined below); |
| ● | real estate investment trusts, regulated investment companies or grantor trusts; |
| ● | persons that hold the ADSs as part of a “hedging,” “integrated,” “wash sale” or “conversion” transaction or as a position in a “straddle” for U.S. federal income tax purposes; |
| ● | S corporations; |
| ● | certain former citizens or long term residents of the United States; |
| ● | persons subject to Section 451(b) of the Code; |
| ● | persons that received ADSs as compensation for the performance of services; |
145
| ● | persons acquiring ADSs in connection with a trade or business conducted outside of the United States, including a permanent establishment in France; |
| ● | holders that own directly, indirectly, or through attribution 10% or more of the voting power or value of the ADSs and shares or, in the case of the discussion of French tax consequences, 5% or more of the voting stock or our share capital; and |
| ● | holders that have a “functional currency” other than the U.S. dollar. |
For the purposes of this description, a “U.S. holder” is a beneficial owner of ADSs that is (or is treated as), for U.S. federal income tax purposes:
| ● | an individual who is a citizen or resident of the United States; |
| ● | a domestic corporation; or |
| ● | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or a trust, if a court within the United States is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of the substantial decisions of such trust, or if such trust has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person. |
If a partnership (or any other entity treated as a partnership for U.S. federal income tax purposes) holds ADSs, the U.S. federal income tax consequences relating to an investment in the ADSs will depend in part upon the status of the partner and the activities of the partnership. Such a partner or partnership should consult its tax advisor regarding the U.S. federal income tax considerations of acquiring, owning and disposing of the ADSs in its particular circumstances.
The discussion in this section is based in part upon the representations of the depositary and the assumption that each obligation in the deposit agreement and any related agreement will be performed in accordance with its terms. In general, and taking into account the earlier assumptions, for U.S. federal income tax purposes, a U.S. holder holding ADSs should be treated as the owner of the ordinary shares represented by the ADSs. Accordingly, no gain or loss should be recognized upon an exchange of ADSs for ordinary shares. The U.S. Treasury has expressed concerns that intermediaries in the chain of ownership between the holder of an ADS and the issuer of the security underlying the ADS may be taking actions that are inconsistent with the beneficial ownership of the underlying security. Accordingly, the creditability of foreign taxes, if any, as described below, could be affected by actions taken by intermediaries in the chain of ownership between the holders of ADSs and our company if as a result of such actions the holders of ADSs are not properly treated as beneficial owners of the underlying ordinary shares.
This summary is based on the Code, final, proposed and temporary U.S. Treasury regulations promulgated thereunder and administrative and judicial interpretations thereof, in each case as in effect and available on the date hereof. All the foregoing is subject to change, which change could apply retroactively, and to differing interpretations, all of which could affect the tax considerations described below. There can be no assurances that the IRS will not take a position concerning the tax consequences of the acquisition, ownership and disposition of the ADSs or that such a position would not be sustained by a court. Holders should consult their tax advisers concerning the U.S. federal, state, local and non-U.S. tax consequences of acquiring, owning and disposing of the ADSs in their particular circumstances.
146
PERSONS CONSIDERING AN INVESTMENT IN THE ADSs SHOULD CONSULT THEIR TAX ADVISORS AS TO THE PARTICULAR TAX CONSEQUENCES APPLICABLE TO THEM RELATING TO THE ACQUISITION, OWNERSHIP AND DISPOSITION OF THE ADSs, INCLUDING THE APPLICABILITY OF U.S. FEDERAL, STATE AND LOCAL TAX LAWS AND OTHER NON-U.S. TAX LAWS.
Distributions. Subject to the discussion under “—Passive Foreign Investment Company Considerations,” below, the gross amount of any distribution (including any amounts withheld in respect of foreign tax) actually or constructively received by a U.S. holder with respect to the ADSs will be taxable to the U.S. holder as a dividend to the extent of the U.S. holder’s pro rata share of our current and accumulated earnings and profits as determined under U.S. federal income tax principles. Distributions in excess of earnings and profits will be non-taxable to the U.S. holder to the extent of, and will be applied against and reduce, the U.S. holder’s adjusted tax basis in the ADSs. Distributions in excess of earnings and profits and such adjusted tax basis generally will be taxable to the U.S. holder as either long-term or short-term capital gain depending upon whether the U.S. holder has held the ADSs for more than one year as of the time such distribution is received. However, since we do not calculate our earnings and profits under U.S. federal income tax principles, it is expected that any distribution will be reported as a dividend, even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain under the rules described above. Non-corporate U.S. holders may qualify for the preferential rates of taxation with respect to dividends on ADSs applicable to long-term capital gains (i.e., gains from the sale of capital assets held for more than one year) applicable to “qualified dividend income” (as discussed below) if we are a “qualified foreign corporation” and certain other requirements (discussed below) are met. A non-U.S. corporation (other than a corporation that is classified as a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation (a) if it is eligible for the benefits of a comprehensive tax treaty with the United States which the Secretary of Treasury of the United States determines is satisfactory for purposes of this provision and which includes an exchange of information provision, or (b) with respect to any dividend it pays on ADSs which are readily tradable on an established securities market in the United States. The Company, which is incorporated under the laws of France, believes that it qualifies as a resident of France for purposes of, and is eligible for the benefits of, the Convention between the Government of the United States of America and the Government of the French Republic for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income and Capital, signed on August 31, 1994, as amended and currently in force (the “U.S.-France Tax Treaty”), although there can be no assurance in this regard. Further, the IRS has determined that the U.S.-France Tax Treaty is satisfactory for purposes of the qualified dividend income rules and that it includes an exchange-of-information program. The ADSs are listed on the Nasdaq Capital Market, which is an established securities market in the United States. There can, however, be no assurance that the ADSs will be considered readily tradable on an established securities market in the United States in later years. Therefore, subject to the discussion under “—Passive Foreign Investment Company Considerations,” below, dividends on ADSs generally will be “qualified dividend income” in the hands of individual U.S. holders, provided that a holding period requirement (more than 60 days of ownership, without protection from the risk of loss, during the 121-day period beginning 60 days before the ex-dividend date) and certain other requirements are met. The dividends will not be eligible for the dividends-received deduction generally allowed to corporate U.S. holders.
A U.S. holder generally may claim the amount of any French withholding tax as either a deduction from gross income or a credit against its U.S. federal income tax liability. However, the foreign tax credit is subject to numerous complex limitations that must be determined and applied on an individual basis. Generally, the credit cannot exceed the proportionate share of a U.S. holder’s U.S. federal income tax liability that such U.S. holder’s taxable income bears to such U.S. holder’s worldwide taxable income. In applying this limitation, a U.S. holder’s various items of income and deduction must be classified, under complex rules, as either “foreign source” or “U.S. source.” In addition, this limitation is calculated separately with respect to specific categories of income. The amount of a distribution with respect to the ADSs that is treated as a “dividend” may be lower for U.S. federal income tax purposes than it is for French income tax purposes, potentially resulting in a reduced foreign tax credit for the U.S. holder. Each U.S. holder should consult its tax advisor regarding the foreign tax credit rules.
In general, the amount of a distribution paid to a U.S. holder in a foreign currency will be the dollar value of the foreign currency calculated by reference to the spot exchange rate on the day the depositary receives the distribution, regardless of whether the foreign currency is converted into U.S. dollars at that time. Any foreign currency gain or loss a U.S. holder realizes on a subsequent conversion of foreign currency into U.S. dollars will be U.S. source ordinary income or loss. If dividends received in a foreign currency are converted into U.S. dollars on the day they are received, a U.S. holder should not be required to recognize foreign currency gain or loss in respect of the dividend.
147
Sale, Exchange or Other Taxable Disposition of the ADSs. A U.S. holder generally will recognize gain or loss for U.S. federal income tax purposes upon the sale, exchange or other taxable disposition of ADSs in an amount equal to the difference between the U.S. dollar value of the amount realized from such sale, exchange or other taxable disposition and the U.S. holder’s tax basis in those ADSs, determined in U.S. dollars. Subject to the discussion under “—Passive Foreign Investment Company Considerations” below, this gain or loss generally will be a capital gain or loss. The adjusted tax basis in the ADSs generally will be equal to the cost of such ADSs. Capital gain from the sale, exchange or other taxable disposition of ADSs of a non-corporate U.S. holder generally is eligible for a preferential rate of taxation applicable to capital gains, if the non-corporate U.S. holder’s holding period determined at the time of such sale, exchange or other taxable disposition for such ADSs exceeds one year (i.e., such gain is long-term capital gain). The deductibility of capital losses for U.S. federal income tax purposes is subject to limitations. Any such gain or loss that a U.S. holder recognizes generally will be treated as U.S. source gain or loss for foreign tax credit limitation purposes.
For a cash basis taxpayer, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the settlement date of the purchase or sale. In that case, no foreign currency exchange gain or loss will result from currency fluctuations between the trade date and the settlement date of such a purchase or sale. An accrual basis taxpayer may elect the same treatment required of cash basis taxpayers with respect to purchases and sales of the ADSs that are traded on an established securities market, provided the election is applied consistently from year to year. Such election may not be changed without the consent of the IRS. For an accrual basis taxpayer who does not make such election, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the trade date of the purchase or sale. Such an accrual basis taxpayer may recognize exchange gain or loss based on currency fluctuations between the trade date and the settlement date. Any foreign currency gain or loss a U.S. Holder realizes will be U.S. source ordinary income or loss.
Medicare Tax. Certain U.S. holders that are individuals, estates or trusts are subject to a 3.8% tax on all or a portion of their “net investment income,” which may include all or a portion of their dividend income and net gains from the disposition of ADSs. Each U.S. holder that is an individual, estate or trust is urged to consult its tax advisor regarding the applicability of such tax to its income and gains in respect of its investment in the ADSs.
Passive Foreign Investment Company Considerations. If we are classified as a PFIC in any taxable year, a U.S. holder will be subject to special rules generally intended to reduce or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. holder could derive from investing in a non-U.S. company that does not distribute all of its earnings on a current basis.
PFIC Tests. We will be classified as a PFIC for U.S. federal income tax purposes in any taxable year in which, after applying certain look-through rules with respect to the income and assets of our subsidiaries, either: (i) at least 75% of our gross income is “passive income” (the “PFIC Income Test”), or (ii) at least 50% of the average quarterly value of our total gross assets (which would generally be measured by the fair market value of our assets, and for which purpose the total value of our assets may be determined in part by the market value of the ADSs and our ordinary shares, which are subject to change) is attributable to assets that produce “passive income” or are held for the production of “passive income” (the “PFIC Asset Test”).
Passive income for purposes of the PFIC Income Test and PFIC Asset Test generally includes dividends, interest, royalties, rents, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income, and amounts derived by reason of the temporary investment of funds raised in offerings of the ADSs. If a non-U.S. corporation owns directly or indirectly at least 25% by value of the stock of another corporation, the non-U.S. corporation is treated for purposes of the PFIC Income Test and the PFIC Asset Test as owning its proportionate share of the assets of the other corporation and as receiving directly its proportionate share of the other corporation’s income. If we are classified as a PFIC in any year with respect to which a U.S. holder owns the ADSs, we will continue to be treated as a PFIC with respect to such U.S. holder in all succeeding years during which the U.S. holder owns the ADSs, regardless of whether we continue to meet the PFIC Income Test and/or the PFIC Asset Test.
For purposes of the PFIC Asset Test, the market value of our assets may be determined in large part by reference to the market price of the ADSs and our ordinary shares, which is likely to fluctuate. In addition, whether we meet PFIC Income Test for any taxable year may depend on whether we receive certain non-refundable grants or subsidies and whether such amounts and reimbursements of certain refundable research tax credits constitute gross income for purposes of PFIC Income Test. Based on the composition of our gross income, assets, activities, and market capitalization in 2023, and on reasonable assumptions, we believe that we were not a PFIC for our taxable year ending December 31, 2023. However, there can be no assurance that we were not a PFIC for our taxable year ending December 31, 2023, or that the IRS will agree with any position we take regarding our PFIC status for such taxable year. There also can be no assurance that we will not be a PFIC for our current taxable year ending December 31, 2024 or for any future taxable year, because our PFIC status is a factual determination made annually after the end of each taxable year. Our U.S. counsel expresses no opinion , and prospective investors should consult their own tax advisors,regarding our PFIC status.
148
If we are a PFIC, and you are a U.S. holder that does not make one of the elections described below, a special tax regime will apply to both (a) any gain realized on the sale, exchange, or other taxable disposition of ADSs and (b) any “excess distribution” by us to you (generally, your ratable portion of distributions in any year that are greater than 125% of the average annual distribution received by you in the shorter of the three preceding years or your holding period for the ADSs), unless you elect to treat us as a “qualified electing fund” (a “QEF”) or make a “mark-to-market” election, each as discussed below. Under this regime, any excess distribution and realized gain will be treated as ordinary income and will be subject to tax as if (a) the excess distribution or gain had been realized ratably over your holding period, (b) the amount deemed realized in each year had been subject to tax in each year of that holding period at the highest marginal rate for such year (other than income allocated to the current period or any taxable period before we became a PFIC, which would be subject to tax at the U.S. holder’s regular ordinary income rate for the current year and would not be subject to the interest charge discussed below), and (c) the interest charge generally applicable to underpayments of tax had been imposed on the taxes deemed to have been payable in those years. In addition, dividend distributions made to you will not qualify for the lower rates of taxation applicable to “qualified dividend income” discussed above under “—Distributions.”
If we are determined to be a PFIC, the general tax treatment for U.S. holders described in this section would apply to indirect distributions and gains deemed to be realized by U.S. holders in respect of any of our subsidiaries that also may be determined to be PFICs.
If a U.S. holder owns ADSs during any taxable year in which we are a PFIC, the U.S. holder generally will be required to file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund) with respect to the Company, generally with the U.S. holder’s federal income tax return for that year.
PFIC Elections. Certain elections may alleviate some of the adverse consequences of PFIC status and would result in an alternative treatment of the ADSs.
A U.S. holder may make a “mark-to-market” election with respect to its ADSs if the ADSs meet certain minimum trading requirements, as described below. If a U.S. holder makes a mark-to-market election, the U.S. holder generally will recognize as ordinary income any excess of the fair market value of the ADSs at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ADSs over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). If a U.S. holder makes a mark-to-market election, the U.S. holder’s tax basis in the ADSs will be adjusted to reflect these income or loss amounts. Any gain recognized on the sale, exchange, or other taxable disposition of ADSs in a taxable year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). The mark-to-market election is available only if we are a PFIC and the ADSs are “regularly traded” on a “qualified exchange.” The ADSs will be treated as “regularly traded” in any calendar year in which more than a de minimis quantity of the ADSs are traded on a qualified exchange on at least 15 days during each calendar quarter (subject to the rule that trades that have as one of their principal purposes the meeting of the trading requirement as disregarded). The Nasdaq Capital Market is a qualified exchange for this purpose and, consequently, if the ADSs are regularly traded, the mark-to-market election should be available to a U.S. holder.
As an alternative to making a mark-to-market election, the excess distribution rules may be avoided if a U.S. holder makes a QEF election effective beginning with the first taxable year in the U.S. holder’s holding period in which we are treated as a PFIC with respect to such U.S. holder. A U.S. holder that makes a QEF election with respect to a PFIC is required to include in income its pro rata share of the PFIC’s ordinary earnings and net capital gain as ordinary income and capital gain, respectively, subject to a separate election to defer payment of taxes, which deferral is subject to an interest charge.
In general, a U.S. holder makes a QEF election by attaching a completed IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund) to a timely filed (taking into account any extensions) U.S. federal income tax return for the taxable year beginning with which the QEF election is to be effective. In certain circumstances, a U.S. holder may be able to make a retroactive QEF election. A QEF election can be revoked only with the consent of the IRS. In order for a U.S. holder to make a valid QEF election, the corporation must annually provide or make available to the U.S. holder certain information.
We do not currently intend to provide the information necessary for U.S. holders to make or maintain QEF elections if we are treated as a PFIC for any taxable year. U.S. holders should consult their tax advisors to determine whether any of these elections would be available and if so, what the consequences of the alternative treatments would be in their particular circumstances.
149
The U.S. federal income tax rules relating to PFICs are complex. U.S. HOLDERS are urged to consult their tax advisers with respect to the consequences of THE acquisition, ownership and disposition of the ADSs, the consequences to them of an investment in a PFIC, any elections available with respect to the ADSs and the IRS information reporting obligations with respect to the acquisition, ownership and disposition of the ADSs.
Backup Withholding and Information Reporting. U.S. holders generally will be subject to information reporting requirements with respect to dividends on the ADSs and on the proceeds from the sale, exchange or other taxable disposition of the ADSs that are paid within the United States or through U.S.-related financial intermediaries, unless the U.S. holder is an “exempt recipient.” In addition, U.S. holders may be subject to backup withholding on such payments, unless the U.S. holder provides a taxpayer identification number and a duly executed IRS Form W-9 or otherwise establishes an exemption from backup withholding. Backup withholding is not an additional tax, and the amount of any backup withholding should be allowed as a credit against a U.S. holder’s U.S. federal income tax liability and may entitle such holder to a refund, provided that the required information is timely furnished to the IRS. U.S. holders are urged to consult their tax advisors regarding the application of the information reporting and backup withholding rules to its particular circumstances.
Certain Reporting Requirements With Respect to Payments of Offer Price. U.S. holders paying more than U.S. $100,000 for the ADSs generally may be required to file IRS Form 926 reporting the payment of the Offer Price for the ADSs to us. Substantial penalties may be imposed upon a U.S. holder that fails to comply. Each U.S. holder should consult its own tax advisor as to the possible obligation to file IRS Form 926.
Foreign Asset Reporting. Certain individual U.S. holders are required to report information relating to an interest in the ADSs, subject to certain exceptions (including an exception for shares held in accounts maintained by U.S. financial institutions) by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their federal income tax return. In addition, U.S. holders should consider their possible obligation to file FinCEN Form 114 (Foreign Bank and Financial Accounts Report) with the U.S. Treasury, as a result of holding the ADSs. Moreover, U.S. holders who paid us more than U.S. $100,000 for the ADSs generally may be required to file IRS Form 926 to report such payment and substantial penalties may be imposed upon a U.S. holder that fails to comply. U.S. holders are urged to consult their tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of the ADSs.
Material French Tax Considerations
The following describes the material French income tax consequences to U.S. holders of purchasing, owning and disposing of the ADSs. This discussion does not purport to be a complete analysis or listing of all potential tax effects of the acquisition, ownership or disposition of the ADSs to any particular investor, and does not discuss tax considerations that arise from rules of general application or that are generally assumed to be known by investors. All of the following is subject to change. Such changes could apply retroactively and could affect the consequences described below.
The description of the French income tax and wealth tax consequences set forth below is based on the Convention Between the Government of the United States of America and the Government of the French Republic for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income and Capital of August 31, 1994, or the Treaty, which came into force on December 30, 1995 (as amended by any subsequent protocols, including the protocol of January 13, 2009), and the tax guidelines issued by the French tax authorities in force as of the date of this annual report.
This discussion applies only to investors that are entitled to Treaty benefits under the “Limitation on Benefits” provision contained in the Treaty.
In 2011, France introduced a comprehensive set of new tax rules applicable to French assets that are held by or in foreign trusts. These rules provide inter alia for the inclusion of trust assets in the settlor’s net assets for the purpose of applying the French wealth tax, for the application of French gift and death duties to French assets held in trust, for a specific tax on capital on the French assets of foreign trusts not already subject to the French wealth tax and for a number of French tax reporting and disclosure obligations. The following discussion does not address the French tax consequences applicable to securities (including ADSs) held in trusts. If ADSs are held in trust, the grantor, trustee and beneficiary are urged to consult their own tax advisor regarding the specific tax consequences of acquiring, owning and disposing of securities (including ADSs).
150
U.S. holders are urged to consult their own tax advisors regarding the tax consequences of the purchase, ownership and disposition of securities in light of their particular circumstances, especially with regard to the “Limitations on Benefits” provision.
Estate and Gift Taxes and Transfer Taxes
In general, a transfer of securities by gift or by reason of death of a U.S. holder that would otherwise be subject to French gift or inheritance tax, respectively, will not be subject to such French tax by reason of the Convention between the Government of the United States of America and the Government of the French Republic for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Estates, Inheritances and Gifts, dated November 24, 1978, unless (i) the donor or the transferor is domiciled in France at the time of making the gift or at the time of his or her death, or (ii) the securities were used in, or held for use in, the conduct of a business through a permanent establishment or a fixed base in France.
Pursuant to Article 235 ter ZD of the Code général des impôts (French Tax Code, or FTC), purchases of shares or ADSs of a French company listed on a regulated market of the European Union or on a foreign regulated market formally acknowledged by the AMF are subject to a 0.3% French tax on financial transactions, or the TFT, provided that the issuer’s market capitalization exceeds €1 billion as of December 1 of the year preceding the taxation year.
A list of relevant French companies whose market capitalization exceeds €1 billion as of December 1 of the year preceding the taxation year within the meaning of Article 235 ter ZD of the FTC used to be published annually by the French Ministry of Economy. It is now published by the French tax authorities, and could be amended at any time. Pursuant to Regulations BOI-ANNX-000467- 29/12/2021 issued on December 29, 2021, we are currently not included in such list. Such list may be updated from time to time, or may not be published anymore in the future.
As a result, neither the ADSs nor the ordinary shares are currently within the scope of the TFT. However, following our U.S. initial public offering, purchases of our securities may be subject to TFT, provided that our market capitalization exceeds €1 billion.
In the case where Article 235 ter ZD of the FTC is not applicable, transfers of shares issued by a listed French company are subject to uncapped registration duties at the rate of 0.1% if the transfer is evidenced by a written statement (“acte”) executed either in France or outside France. Although there is no case law or official guidelines published by the French tax authorities on this point, transfers of ADSs should remain outside of the scope of the aforementioned 0.1% registration duties.
Tax on Sale or Other Disposition
As a matter of principle, under French tax law, a U.S. holder should not be subject to any French tax on any capital gain from the sale, exchange, repurchase or redemption by us of ordinary shares or ADSs, provided such U.S. holder is not a French tax resident for French tax purposes and has not held more than 25% of our dividend rights, known as “droits aux benefices sociaux,” at any time during the preceding five years, either directly or indirectly, and, as relates to individuals, alone or with relatives (as an exception, a U.S holder resident, established or incorporated in a non-cooperative state or territory as defined in Article 238-0 A of the FTC should be subject to a 75% withholding tax in France on any such capital gain, regardless of the fraction of the dividend rights it holds).
Under application of the Treaty, a U.S. holder who is a U.S. resident for purposes of the Treaty and entitled to Treaty benefit will not be subject to French tax on any such capital gain unless the ordinary shares or the ADSs form part of the business property of a permanent establishment or fixed base that the U.S. holder has in France. U.S. holders who own ordinary shares or ADSs through U.S. partnerships that are not resident for Treaty purposes are advised to consult their own tax advisors regarding their French tax treatment and their eligibility for Treaty benefits in light of their own particular circumstances. A U.S. holder that is not a U.S. resident for Treaty purposes or is not entitled to Treaty benefit (and in both cases is not resident, established or incorporated in a non-cooperative State or territory as defined in Article 238-0 A of the FTC) and has held more than 25% of our dividend rights, known as “droits aux benefices sociaux,” at any time during the preceding five years, either directly or indirectly, and, as relates to individuals, alone or with relatives will be subject to a levy in France at the rate (i) of 12.8% for individuals, and (ii) corresponding to the standard corporate income tax set forth in Article 219-I of the FTC legal persons (i.e., 25% for financial years beginning on or after January 1, 2022).
151
Taxation of Dividends
Dividends paid by a French corporation to non-residents of France are generally subject to French withholding tax at a rate (i) aligned on the standard corporate income tax rate set forth in Article 219-I of the FTC for financial years beginning January 1, 2022, for payments benefitting legal persons who are not French tax residents (i.e. 25% for financial years beginning on or after January 1, 2022), and (ii) equal to 12.8% for payments benefitting individuals who are not French tax residents. Dividends paid by a French corporation in a non-cooperative State or territory, as defined in Article 238-0 A of the FTC, will generally be subject to French withholding tax at a rate of 75%. However, eligible U.S. holders entitled to Treaty benefits under the “Limitation on Benefits” provision contained in the Treaty who are U.S. residents, as defined pursuant to the provisions of the Treaty, will not be subject to the above-mentioned withholding tax rates, but may be subject to the withholding tax at a reduced rate (as described below).
Under the Treaty, the rate of French withholding tax on dividends paid to an eligible U.S. holder who is a U.S. resident as defined pursuant to the provisions of the Treaty and whose ownership of the ordinary shares or ADSs is not effectively connected with a permanent establishment or fixed base that such U.S. holder has in France, is generally reduced to 15%, or to 5% if such U.S. holder is a corporation and owns directly or indirectly at least 10% of the share capital of the issuer; such U.S. holder may claim a refund from the French tax authorities of the amount withheld in excess of the Treaty rates of 15% or 5%, if any.
For U.S. holders that are not individuals but are U.S. residents, as defined pursuant to the provisions of the Treaty, the requirements for eligibility for Treaty benefits, including the reduced 5% or 15% withholding tax rates contained in the “Limitation on Benefits” provision of the Treaty, are complex, and certain technical changes were made to these requirements by the protocol of January 13, 2009. U.S. holders are advised to consult their own tax advisors regarding their eligibility for Treaty benefits in light of their own particular circumstances. Dividends paid to an eligible U.S. holder may immediately be subject to the reduced rates of 5% or 15% provided that:
| ● | such holder establishes before the date of payment that it is a U.S. resident under the Treaty by completing and providing the depositary with a treaty form (Form 5000); or |
| ● | the depositary or other financial institution managing the securities account in the United States of such holder provides the French paying agent with a document listing certain information about the U.S. holder and its ordinary shares or ADSs and a certificate whereby the financial institution managing the U.S. holder’s securities account in the United States takes full responsibility for the accuracy of the information provided in the document. |
Otherwise, dividends paid to a U.S. holder will be subject to French withholding tax at the rate of 12.8%, 25%, or 75% if paid in a non-cooperative State or territory (as defined in Article 238-0 A of the FTC), and may then be reduced at a later date to 5% or 15%, provided that such holder duly completes and provides the French tax authorities with the treaty forms Form 5000 and Form 5001 before December 31 of the second calendar year following the year during which the dividend is paid.
Certain qualifying pension funds and certain other tax-exempt entities are subject to the same general filing requirements as other U.S. holders except that they may have to supply additional documentation evidencing their entitlement to these benefits.
Form 5000 and Form 5001, together with instructions, will be provided by the depositary to all U.S. holders registered with the depositary. The depositary will arrange for the filing with the French tax authorities of all such forms properly completed and executed by U.S. holders of ordinary shares or ADSs and returned to the depositary in sufficient time so that they may be filed with the French tax authorities before the distribution in order to immediately obtain a reduced withholding tax rate. Otherwise, the depositary must withhold tax at the full rate of 12.8%, 30% or 75% as applicable. In that case, the U.S. holders may claim a refund from the French tax authorities of the excess withholding tax, if any.
152
Wealth Tax
The French wealth tax (impôt de solidarité sur la fortune) has been repealed by the finance bill for 2018 (loi de finances pour 2018), dated December 30, 2017. The French wealth tax used to apply only to individuals and did not generally apply to securities held by an eligible U.S. holder who is a U.S. resident, as defined pursuant to the provisions of the Treaty, provided that such U.S. holder does not own directly or indirectly more than 25% of the issuer’s financial rights and that the securities did not form part of the business property of a permanent establishment or fixed base in France. It has been replaced by a new real estate wealth tax (impôt sur la fortune immobilière) as from January 1, 2018. The scope of such new tax is narrowed to real estate assets (and certain assets deemed to be real estate assets) or rights, directly or indirectly through one or more legal entities and whose net taxable assets amount to at least €1,300,000. Our securities owned by a U.S. Holder should not fall within the scope of the new real estate wealth tax provided that such U.S. Holder does not own directly or indirectly a shareholding exceeding 10% of the financial rights and voting rights of the company.
THE DISCUSSION ABOVE IS A SUMMARY OF THE MATERIAL U.S. FEDERAL AND FRENCH INCOME TAX CONSEQUENCES OF AN INVESTMENT IN THE ADSs OR ORDINARY SHARES AND IS BASED UPON LAWS AND RELEVANT INTERPRETATIONS THEREOF IN EFFECT AS OF THE DATE OF THIS ANNUAL REPORT, ALL OF WHICH ARE SUBJECT TO CHANGE, POSSIBLY WITH RETROACTIVE EFFECT. EACH PROSPECTIVE INVESTOR IS URGED TO CONSULT ITS TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT OF AN INVESTMENT IN ADSs OR ORDINARY SHARES IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
F.Dividends and Paying Agents
Not applicable.
G.Statement by Experts
Not applicable.
H.Documents on Display
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and file reports with the SEC under those requirements. Such reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an Annual Report on Form 20-F each year containing financial statements that have been examined and reported on, with and opinion expressed by an independent registered public accounting firm.
We maintain a corporate website at www.biophytis.com. We intend to post our annual report on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this annual report. We have included our website address in this annual report solely as an inactive textual reference.
The Securities and Exchange Commission maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as Biophytis S.A., that file electronically with the SEC.
With respect to references made in this annual report to any contract or other document of our company, such references are not necessarily complete and you should refer to the exhibits attached or incorporated by reference to this annual report for copies of the actual contract or document.
I.Subsidiary Information
Not required.
153
J.Annual Report to Security Holders
If we are required to provide an annual report to security holders in response to the requirements of Form 6-K, we will submit the annual report to security holders in electronic format in accordance with the EDGAR Filer Manual.
Item 11. Quantitative and Qualitative Disclosures About Market Risk.
We are exposed to a variety of financial risks: market risk (including interest rate risk and foreign exchange risk), credit risk and liquidity risk. Our overall risk management program focuses on preservation of capital given the unpredictability of financial markets. For additional information on general risk factors, please see the section of this annual rport titled “Item 3.D—Risk Factors.”
Market risk
Interest rate risk
Interest rate risk reflects the Company’s exposure to fluctuations in interest rates in the market.
Changes in interest rate could affect returns achieved on cash and fixed-term deposits but this risk is not considered material given the current low returns on deposits held by the Company.
Change in interest rate could affect the consolidated statement of profit or loss for financial liabilities but this risk is considered as not significant given the implementation by the Company of debts bearing fixed interest rate.
Foreign exchange risk
The major risks linked to foreign exchange rate are considered not significant due to the low level of activity of its foreign subsidiaries.
The Company currently does not use hedging instruments to protect its activity from exchange rate fluctuations. However, any major development in its activity may result in an increase of its exposure to exchange rate risk. Should such increase materialize, the Company may consider adopting an appropriate policy to hedge such risks.
Equity risk
The Company entered into ORNANE agreement with Negma and Atlas, loan agreement and bonds issue agreement with Kreos providing financing through the issuance of multiple tranches of convertible notes eventually with attached warrants. As part of these agreements, the Company is exposed to changes in the market price of its own shares.
Credit risk
Credit risk is linked to deposits with banks and financial institutions.
The Company seeks to minimize the risk related to banks and financial institutions by placing cash deposits with highly rated financial institutions. The maximum level of the credit risk corresponds to the book value of the financial assets. As outstanding receivables consist primarily of Research Tax Credit “CIR” granted by the French government, the Company does not carry significant credit risk.
Liquidity risk
Since our inception, we have funded our operations and growth by strengthening our shareholders’ equity through capital increases (including the capital increase realized during its French IPO in July 2015), bank loans and notes, and obtaining public aid for innovation and reimbursement of CIR receivables, including the prefinancing arrangement initiated in 2019.
Significant research and development expenses have been incurred since inception generating negative cash flows from operating activities of €23,795 thousand, €18,988 and €12,873 thousand for the years ended December 31, 2021, 2022 and 2023, respectively.
154
The Financial Statements have been approved on a going concern basis by the Board of Directors. As of the date of authorization of these financial statements, our available cash and our ORNANE financing line are not projected to be sufficient to support our operating plan for at least the next 12 months. These events and conditions indicate that a material uncertainty exists that may cast significant doubt on the Company’s ability to continue as a going concern and, therefore, the Company may be unable to realize its assets and discharge its liabilities in the normal course of business (refer to Item 18 note 3.1).
The Company will continue to have major funding requirements in the future to support the development of its drug candidates. The precise extent of funding required is difficult to predict accurately and will depend in part on factors outside the Company’s control. Areas subject to significant uncertainty include, but are not limited to:
| ● | The Company’s ability to conduct successful clinical trials, including the capacity to recruit patients in a timely-manner for the Company’s clinical trials; |
| ● | the change in the regulatory landscape; and |
| ● | the approval for other drugs on the market that may potentially reduce the attractiveness for the Company’s drug candidates. |
Should the Company find itself unable to finance its own growth through partnership agreements, the Company would be dependent on other sources of financing, including equity and/or debt funding or research grants.
Item 12. Description of Securities Other than Equity Securities.
A.Debt Securities
Not applicable.
B.Warrants and Rights
Not applicable.
C.Other Securities
Not applicable.
D.American Depositary Shares
The Bank of New York Mellon, as depositary, registers and deliver ADSs. Each ADS represents 100 ordinary shares (or a right to receive 100 ordinary shares) deposited with Societe Generale, as custodian for the depositary in France. Each ADS will also represent any other securities, cash or other property which may be held by the depositary. The deposited shares together with any other securities, cash or other property held by the depositary are referred to as the deposited securities. The depositary’s office at which the ADSs will be administered and its principal executive office are located at 240 Greenwich Street, New York, New York 10286.
A deposit agreement among us, the depositary and the ADS holders sets out the ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADSs. A copy of the deposit agreement is incorporated by reference as an exhibit to this annual report.
155
Fees and Charges
Pursuant to the terms of the deposit agreement, the holders of ADSs will be required to pay the following fees:
Persons depositing or withdrawing ordinary shares or ADS holders must pay: |
| For: |
$5.00 (or less) per 100 ADSs (or portion of 100 ADSs) |
| · Issuance of ADSs, including issuances resulting from a distribution of ordinary shares or rights or other property · Cancellation of ADSs for the purpose of withdrawal, including if the deposit agreement terminates
|
$.05 (or less) per ADS |
| · Any cash distribution to ADS holders
|
A fee equivalent to the fee that would be payable if securities distributed to you had been ordinary shares and the ordinary shares had been deposited for issuance of ADSs |
| · Distribution of securities distributed to holders of deposited securities (including rights) that are distributed by the depositary to ADS holders
|
$.05 (or less) per ADS per calendar year |
| · Depositary services
|
Registration or transfer fees |
| · Transfer and registration of ordinary shares on our share register to or from the name of the depositary or its agent when you deposit or withdraw ordinary shares
|
Expenses of the depositary |
| · Cable (including SWIFT) and facsimile transmissions (when expressly provided in the deposit agreement) · Converting foreign currency to U.S. dollars
|
Taxes and other governmental charges the depositary or the custodian has to pay on any ADSs or ordinary shares underlying ADSs, such as stock transfer taxes, stamp duty or withholding taxes
|
| · As necessary |
Any charges incurred by the depositary or its agents for servicing the deposited securities |
| · As necessary |
The depositary collects its fees for delivery and surrender of ADSs directly from investors depositing shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by deduction from cash distributions or by directly billing investors or by charging the book-entry system accounts of participants acting for them. The depositary may collect any of its fees by deduction from any cash distribution payable (or by selling a portion of securities or other property distributable) to ADS holders that are obligated to pay those fees. The depositary may generally refuse to provide fee-attracting services until its fees for those services are paid.
From time to time, the depositary may make payments to us to reimburse us for costs and expenses generally arising out of establishment and maintenance of the ADS program, waive fees and expenses for services provided to us by the depositary or share revenue from the fees collected from ADS holders. In performing its duties under the deposit agreement, the depositary may use brokers, dealers, foreign currency dealers or other service providers that are owned by or affiliated with the depositary and that may earn or share fees, spreads or commissions.
156
The depositary may convert currency itself or through any of its affiliates, or the custodian or we may convert currency and pay U.S. dollars to the depository. Where the depository converts currency itself or through any of its affiliates, the depository acts as principal for its own account and not as agent, advisor, broker or fiduciary on behalf of any other person and earns revenue, including, without limitation, transaction spreads, that it will retain for its own account. The revenue is based on, among other things, the difference between the exchange rate assigned to the currency conversion made under the deposit agreement and the rate that the depositary or its affiliate receives when buying or selling foreign currency for its own account. The depositary makes no representation that the exchange rate used or obtained by it or its affiliates in any currency conversion under the deposit agreement will be the most favorable rate that could be obtained at the time or that the method by which that rate will be determined will be the most favorable to ADS holders, subject to the depositary’s obligation under to act without negligence or bad faith. The methodology used to determine exchange rates used in currency made by the depositary is available upon request. Where the custodian converts currency, the custodian has no obligation to obtain the most favorable rate that could be obtained at the time or to ensure that the method by which that rate will be determined will be the most favorable to ADS holders, and the depositary makes no representation that the rate is the most favorable rate and will not be liable for any direct or indirect losses associated with the rate. In certain instances, the depositary may receive dividends or other distributions in U.S. dollars that represent the proceeds of a conversion of foreign currency or translation from foreign currency at a rate that was obtained or determined by us and, in such cases, the depositary will not engage in, or be responsible for, any foreign currency transactions and neither it nor we make any representation that the rate obtained or determined by us is the most favorable rate and neither it nor we will be liable for any direct or indirect losses associated with the rate.
Payment of Taxes
ADS Holders are responsible for any taxes or other governmental charges payable on the ADSs or on the deposited securities represented by any of the ADSs. The depositary may refuse to register any transfer of the ADSs or allow you to withdraw the deposited securities represented by the ADSs until those taxes or other charges are paid. It may apply payments owed to you or sell deposited securities represented by the ADSs to pay any taxes owed and you will remain liable for any deficiency. If the depositary sells deposited securities, it will, if appropriate, reduce the number of ADSs to reflect the sale and pay to ADS holders any proceeds, or send to ADS holders any property, remaining after it has paid the taxes.
157
PART II
Item 13. Defaults, Dividend Arrearages and Delinquencies.
Not applicable.
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds.
Not applicable.
Item 15. Controls and Procedures.
A.Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the Securities and Exchange Commission. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed in our reports filed or submitted under the Exchange Act is accumulated and communicated to management, including our chief executive officer (principal executive officer) and chief financial officer. We carried out an evaluation, under the supervision and with the participation of our management, including our chief executive officer (principal executive officer) and chief financial officer (principal financial officer), of the effectiveness of the design and operation of our disclosure controls and procedures as of December 31, 2023 in connection with the preparation of this annual report. Based on that evaluation, our chief executive officer (principal executive officer) and chief financial officer (principal financial officer) concluded that our disclosure controls and procedures were not effective as of December 31, 2023 due to a material weakness in our internal control over financial reporting as described below.
B.Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f) and 15d-15(f). Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with generally accepted accounting principles.
Under the supervision and with the participation of our management, including our Chief Executive Officer (principal executive officer) and Chief Financial Officer (principal financial officer), we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2023 based on the guidelines established in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
In connection with preparing our 2023 consolidated financial statements, a material weakness in our internal control over financial reporting was identified relating to a lack of sufficient competent financial reporting and accounting personnel with appropriate understanding of IFRS Accounting Standards to design and implement formal period-end financial reporting controls and procedures and to address complex IFRS Accounting Standards technical accounting issues. This material weakness resulted in significant misstatements in aggregate, identified by our external auditors in connection with their audit of the 2023 consolidated financial statements.
Based on our assessment as of December 31, 2023, our management, including our chief executive officer (principal executive officer) and chief financial officer (principal financial officer), believes that our internal control over financial reporting was not effective as of December 31, 2023, because of the material weakness described above.
Notwithstanding this material weakness and management’s assessment that internal control over financial reporting was ineffective as of December 31, 2023, our management, including our chief executive officer (principal executive officer) and chief
158
financial officer (principal financial officer), believes that the consolidated financial statements contained in this annual report as of December 31, 2023 present fairly, in all material respects, our financial position, results of operations and cash flows for the periods presented in conformity with IFRS Accounting Standards.
Management’s Plan for Remediation
We are in the process of remediating this identified material weakness and we have taken and will continue to take actions to eventually eliminate it. We have strengthened our financial control and our accounting team by hiring a new financial controller with knowledge in both French GAAP and IFRS Accounting Standards, who joined the Company mid-February. We are also working with financial experts knowledgeable in IFRS Accounting Standards to secure the application of appropriate accounting treatments in our consolidated accounts. Besides, we will strengthen our closing control checklist with reinforced controls of data provided by external providers, such as our accounting advisors or our consultant in charge of preparing our research tax credit calculation.
C.Attestation Report of the Registered Public Accounting Firm
Because we qualify as an emerging growth company under the JOBS Act, this annual report does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting as required by Section 404(b) of the Sarbanes Oxley Act of 2002.
D.Changes in Internal Control Over Financial Reporting
In our annual report on Form 20-F (as amended) for the fiscal year ended December 31, 2022, we reported the continued existence of material weaknesses, previously disclosed in Item 15.B of our annual report on Form 20-F (as amended) for the fiscal year ended December 31, 2021, related to our internal control over the application of IFRS 9 “Financial Instruments”, and their related interpretations and rules, and IFRS 13 “Fair Value Measurement” to the accounting treatment and fair value assessment of our convertible notes, non-convertible notes, and warrants. The deficiencies, which aggregate to material weaknesses, were the result of:
| ● | A lack of sufficient formalized and documented policies and procedures to monitor the accounting treatment of complex financial instruments. |
| ● | A lack of sufficient technical resources and expertise within the Company to evaluate the accounting treatment for complex financial instruments, including ensuring that all provisions of such instruments are incorporated into the accounting analysis and the fair value assessment, to develop the required complex valuation models, and to oversee third party experts engaged to assist with such accounting analysis and creation of valuation models. |
These material weaknesses were remediated during the year 2023, which included:
| ● | Updating our policies and procedures and redesigning controls to include evaluation of existing accounting for financial instruments at each reporting period. |
| ● | Providing additional training to staff and management on accounting for complex financial instruments. |
| ● | Improving the design of control procedures over review of qualified third-party specialist analyses. |
| ● | Implementing such control procedures. |
In order to comply with Section 404(a) of the Sarbanes Oxley Act of 2002, in addition to implementing our remediation plan described above, we have also established a risk matrix for our operations, and implemented internal control procedures to mitigate the risks moving forward, as follows:
159
We identified and analyzed risks associated with financial and operational processes and implemented new and/or improved control activities to mitigate these risks. We developed written standard operating procedures for major processes and training programs to inform employees regarding SOX best practices and newly implemented internal controls. We also
| ● | put into service a new purchasing system that has improved controls over our procurement process, including vendor management, automatic approval workflow for purchase orders and invoices, automatic matching of orders and invoices, and improved financial and supplier reports; |
| ● | Supported the team by external advisors with subject matter expertise to provide assistance with all elements related to complex financial reporting elements. |
We will continue to put into place additional improvements and/or corrective actions to control activities and procedures, and expanding our corporate training programs going forward, as well as implement a plan to test key controls during 2024 to help ensure the effectiveness of internal controls over financial reporting and processes.
If we identify any new material weaknesses in the future, any such newly identified material weaknesses could limit our ability to prevent or detect a misstatement of our accounts or disclosures that could result in a material misstatement of our annual or interim financial statements. In such case, we may be unable to maintain compliance with securities law requirements regarding timely filing of periodic reports in addition to applicable stock exchange listing requirements, investors may lose confidence in our financial reporting and our stock price may decline as a result. We cannot assure you that the measures we have taken to date, or any measures we may take in the future, will be sufficient to avoid potential future material weaknesses.
Item 16A. Audit Committee Financial Expert.
Our board of directors has determined that Nadine Coulm is an “audit committee financial expert” as defined by the rules and regulations of the Securities and Exchange Commission and has the requisite financial sophistication under the applicable rules and regulations of the Nasdaq Stock Market. Ms. Coulm is also independent as such term is defined in Rule 10A-3 under the Exchange Act and under the listing standards of the Nasdaq Stock Market.
Item 16B. Code of Ethics.
We have adopted a Code of Business Conduct and Ethics (“Code of Conduct”), applicable to all of our employees, executive officers and directors. The Code of Conduct is available on our website at www.biophytis.com. The board of directors is responsible for administering the Code of Conduct, but has delegated day-to-day responsibility for administering and interpreting the Code of Conduct to our Chief Financial Officer, who has been appointed Compliance Officer under the Code of Conduct. Any waivers of the Code of Conduct for employees, executive officers and directors must be approved by the board of directors and promptly disclosed to our shareholders. We expect that any amendments to the Code of Conduct, or any waivers of its requirements, will be disclosed on our website.
Item 16C. Principal Accountant Fees and Services.
KPMG served as our independent registered public accounting firm for 2022 and 2023, and they billed the following fees to us for professional services in each of those fiscal years:
Year Ended December 31, | ||||
| 2022 |
| 2023 | |
(in € thousands) | ||||
Audit Fees |
| 341 |
| 303 |
Audit-Related Fees |
| 1 |
| — |
Tax Fees |
| — |
| — |
Other Fees |
| — |
| — |
Total |
| 342 |
| 303 |
“Audit Fees” are the aggregate fees billed for the audit of our annual financial statements. This category also includes services provided by KPMG, such as consents and review of documents filed with the SEC.
160
“Audit-Related Fees” are the aggregate fees billed for assurance and related services that are reasonably related to the performance of the audit and are not reported under Audit Fees.
“Tax Fees” are the aggregate fees billed for professional services rendered by KPMG for tax compliance, tax advice and tax planning related services.
“Other Fees” are any additional amounts billed for products and services provided by KPMG.
Audit and Non-Audit Services Pre-Approval Policy
The audit committee has responsibility for reviewing the candidates for the position of (and proposing in some cases their appointment), setting compensation of and overseeing the work of the independent registered public accounting firm. The audit committee ensures the independence and competence of the independent registered public accounting firm. Unless a type of service to be provided by our independent registered public accounting firm has received general pre-approval from the audit committee, it requires specific pre-approval by the audit committee. The payment for any proposed services in excess of pre-approved cost levels requires specific pre-approval by the audit committee.
Item 16D. Exemptions from the Listing Standards for Audit Committees.
Not applicable.
Item 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
Not applicable.
Item 16F. Change in Registrant’s Certifying Accountant.
Not applicable.
Item 16G. Corporate Governance.
As a French société anonyme, we are subject to various corporate governance requirements under French law. In addition, as a foreign private issuer listed on the Nasdaq Capital Market, we are subject to Nasdaq’s corporate governance listing standards. However, Nasdaq’s listing standards provide that foreign private issuers are permitted to follow home country corporate governance practices in lieu of Nasdaq’s rules, with certain exceptions. Certain corporate governance practices in France may differ significantly from corporate governance listing standards. For example, neither the corporate laws of France nor our bylaws require that (i) a majority of our directors be independent, (ii) our compensation committee include only independent directors, or (iii) our independent directors hold regularly scheduled meetings at which only independent directors are present. Other than as set forth below, we currently intend to comply with the corporate governance listing standards of Nasdaq to the extent possible under French law. However, we may choose to change such practices to follow home country practice in the future.
As a foreign private issuer, we are required to comply with Rule 10A-3 of the Exchange Act relating to audit committee composition and responsibilities. Rule 10A-3 of the Exchange Act provides that the audit committee must have direct responsibility for the nomination, compensation and choice of our auditors, as well as control over the performance of their duties, management of complaints made, and selection of consultants. However, if the laws of a foreign private issuer’s home country require that any such matter be approved by the board of directors or the shareholders, the audit committee’s responsibilities or powers with respect to such matter may instead be advisory. Under French law, the audit committee may only have an advisory role and appointment of our statutory auditors, in particular, must be decided by the shareholders at our annual meeting.
161
In addition, Nasdaq rules require that a listed company specify that the quorum for any meeting of the holders of common stock be at least 331/3% of the outstanding shares of the company’s common voting stock. Consistent with French law, our by-laws provide and will continue to provide that a quorum requires the presence of shareholders having at least (1) 20% of the shares entitled to vote in the case of an ordinary shareholders’ general meeting or at an extraordinary shareholders’ general meeting where shareholders are voting on a capital increase by capitalization of reserves, profits or share premium, or (2) 25% of the shares entitled to vote in the case of any other extraordinary shareholders’ general meeting. If a quorum is not present, the meeting is adjourned. There is no quorum requirement when an ordinary general meeting is reconvened, but the reconvened meeting may consider only questions which were on the agenda of the adjourned meeting. When an extraordinary general meeting is reconvened, the quorum required is 20% of the shares entitled to vote, except where the reconvened meeting is considering capital increases through capitalization of reserves, profits or share premium. For these matters, no quorum is required at the reconvened meeting. If a quorum is not present at a reconvened meeting requiring a quorum, then the meeting may be adjourned for a maximum of two months.
See the section of this annual report titled “Description of Share Capital—Key Provisions of Our By-laws and French Law Affecting Our Ordinary Shares.”
Item 16H. Mine Safety Disclosure.
Not applicable.
Item 16I. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections.
Not applicable.
162
Item 16K. Cybersecurity
[We believe that cybersecurity is fundamental in our operations and, as such, we are committed to maintaining robust governance and oversight of cybersecurity risks and to implementing comprehensive processes and procedures for identifying, assessing, and managing material risks from cybersecurity threats as part of our broader risk management system and processes. Our cybersecurity risk management strategy prioritizes detection, analysis and response to known, anticipated or unexpected threats; effective management of security risks; and resiliency against incidents. With the ever-changing cybersecurity landscape and continual emergence of new cybersecurity threats, our management team ensure that adequate resources are devoted to cybersecurity risk management and the technologies, processes and people that support it. Our IT operations are managed by a specialized third-party company, which provides a dedicated team with multiple competencies in charge of ensuring that our systems and software are efficient and up to date, including notably our firewall and IT access management system. This team also manages the backup of our files outside of the Company premises and recovery tests are regularly performed to ensure quick availability of data in case of an incident on our servers. The IT team maintains awareness about security risks throughout the Company by sending messages to all employees about current threats and good cybersecutity practices.
As we do not have a dedicated board committee solely focused on cybersecurity, our management team has oversight responsibility for risks and incidents relating to cybersecurity threats, including compliance with disclosure requirements, cooperation with law enforcement, and related effects on financial and other risks, and it reports any material findings and recommendations, as appropriate, to our board of directors for consideration.
We continue to invest in our cybersecurity systems and to enhance our internal controls and processes. Our business strategy, results of operations and financial condition have not been materially affected by risks from cybersecurity threats, including as a result of previously identified cybersecurity incidents, but we cannot provide assurance that they will not be materially affected in the future by such risks or any future material incidents. While we have dedicated appropriate resources to identifying, assessing, and managing material risks from cybersecurity threats, our efforts may not be adequate, may fail to accurately assess the severity of an incident, may not be sufficient to prevent or limit harm, or may fail to sufficiently remediate an incident in a timely fashion, any of which could harm our business, reputation, results of operations and financial condition. ]
PART III
Item 17. Financial Statements.
See response to Item 18.
Item 18. Financial Statements.
See pages F-1 through F-[53] of this annual report.
Item 19. Exhibits.
Exhibit No. |
| Description of Exhibit |
1.1 |
2.1 | ||
2.2 | ||
2.3 | ||
163
4.1 | ||
4.2 | ||
4.3† | ||
4.4† | ||
4.5† | ||
4.6† | ||
4.7† | ||
4.8† | ||
4.9† |
| |
|
|
|
4.10† |
| |
|
|
|
4.11† |
| |
|
|
|
4.12† |
| |
|
|
|
164
4.13† |
| |
|
|
|
4.14 |
| |
|
|
|
4.15† |
| |
|
|
|
4.16† |
| |
|
|
|
4.17† |
| |
|
|
|
4.18† |
| |
|
|
|
4.19† |
| |
|
|
|
4.20† |
| |
4.21† |
| |
|
|
|
4.22† |
| |
|
|
|
4.23 |
| |
|
|
|
4.24 |
| |
|
|
|
4.25 |
| |
|
|
|
165
4.26† |
| |
|
|
|
4.27 |
| |
|
|
|
4.28 |
| |
|
|
|
4.29 |
| |
|
|
|
4.30 |
| |
|
|
|
4.31 |
| |
|
|
|
4.32 |
| |
|
|
|
4.33 |
| |
|
|
|
4.34† |
| |
|
|
|
4.35 |
| |
4.36 |
| |
|
|
|
4.37† |
| |
|
|
|
4.38† |
| |
4.39 |
| |
166
4.40 | ||
4.41 | ||
4.42 | ||
4.43 | ||
4.44 | ||
4.45 | ||
4.46 | IP Pledge Agreement between Biophytis S.A. and Kreos Capital VI (UK) Ltd dated November 19, 2021 | |
4.39 |
| |
|
|
|
8.1 |
| |
|
|
|
12.1 |
| |
|
|
|
12.2 |
| |
|
|
|
13.1 |
| |
|
|
|
13.2 |
| |
15.1* | Consent of KPMG S.A,, the independent registered public accounting firm. | |
15.2* | Consent from Ernst & Young et Autres, the independent registered public accounting firm | |
97.1* | ||
101.INS XBRL Instance Document* | ||
|
|
|
101.SCH XBRL Taxonomy Extension Schema Document** | ||
|
|
|
101.CAL XBRL Taxonomy Extension Calculation Linkbase Document** | ||
|
|
|
101.DEF XBRL Taxonomy Extension Definition Linkbase Document** | ||
|
|
|
101.LAB XBRL Taxonomy Extension Label Linkbase Document** | ||
|
|
|
101.PRE XBRL Taxonomy Presentation Linkbase Document** | ||
† | Confidential portions of the exhibit have been omitted. |
* | Filed herewith. |
167
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
BIOPHYTIS S.A. | ||
| ||
By: | /s/ Stanislas Veillet | |
Stanislas Veillet | ||
Chief Executive Officer and Chairman |
Date: April 8, 2024
169
BIOPHYTIS S.A.
Index to the Financial Statements
Auditor Firm Id: | Auditor Name: | Auditor Location: |
Auditor Firm Id: | Auditor Name: | Auditor Location: |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors
Biophytis S.A.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated statements of financial position of Biophytis S.A. and subsidiaries (the Company) as of December 31, 2023 and 2022, the related consolidated statements of profit or loss, consolidated statements of profit or loss and other comprehensive profit or loss, statements of changes in consolidated shareholders’ equity, and statements of consolidated cash flows for each of the years in the two-year period ended December 31, 2023 and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022 and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2023 in conformity with IFRS Accounting Standards as issued by the International Accounting Standards Board.
Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 3.1 to the consolidated financial statements, the Company has incurred operating losses and negative cash flows from operations since inception and current cash and cash equivalents, complemented with the ORNANE financing line, are not sufficient for at least the next twelve months. These matters raise substantial doubt about the ability of the Company to continue as a going concern. Management’s plans in regard to these matters are also described in Note 3.1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
We have served as the Company’s auditor since 2022.
Paris La Défense, France
April 8, 2024
/s/ KPMG S.A.
Cédric Adens
Partner
F-2
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of Biophytis S.A.,
Opinion on the Financial Statements
We have audited the accompanying statement of consolidated financial position of Biophytis S.A. (the Company) as of December 31, 2021, the related consolidated statement of profit or loss, consolidated statement of profit or loss and other comprehensive profit or loss, statement of changes in consolidated shareholders’ equity and statement of consolidated cash flows for the year ended December 31, 2021, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2021, and the results of its operations and its cash flows for the year ended December 31, 2021, in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board (“IFRS”) and in accordance with International Financial Reporting Standards as endorsed by the European Union.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/ Ernst & Young et Autres
We have served as the Company’s auditors from 2016 to 2022.
Paris-La Défense, France
April 21, 2022, except for Note 13.2.3, as to which the date is January 30, 2023
F-3
CONSOLIDATED FINANCIAL STATEMENTS PREPARED IN ACCORDANCE WITH IFRS FOR THE YEAR ENDED DECEMBER 31 2023
CONSOLIDATED STATEMENT OF FINANCIAL POSITION
AS OF DECEMBER 31, | ||||||||
(amounts in thousands of euros) |
| NOTES |
| 2021 |
| 2022 |
| 2023 |
ASSETS | ||||||||
Patents and software | 4 |
| | | ||||
Property, plant and equipment | 5 |
| | | | |||
Other non-current financial assets | 6, 10 |
| | | | |||
Total non-current assets |
| | | | ||||
|
|
| ||||||
Other receivables and prepaid expenses | 8, 10 |
| | | | |||
Other current financial assets | 7 |
| | | | |||
Cash and cash equivalents | 9, 10 |
| | | | |||
Total current assets |
| | | | ||||
|
|
| ||||||
TOTAL ASSETS |
| | | | ||||
|
|
| ||||||
|
|
| ||||||
LIABILITIES AND SHAREHOLDERS’ EQUITY (DEFICIT) |
|
|
| |||||
Shareholder’ equity |
|
|
| |||||
Share capital | 11 |
| | | | |||
Premiums related to the share capital | 11 |
| |
| ( |
| | |
Treasury shares | 11 |
| ( | ( | ( | |||
Foreign currency translation adjustment | ( | ( | ( | |||||
Reserves - attributable to the owners of the parent |
| ( | ( | ( | ||||
Net loss - attributable to the owners of the parent |
| ( | ( | ( | ||||
Shareholder equity - attributable to the owners of the parent |
| | ( | ( | ||||
Non-controlling interests |
| ( | ( | ( | ||||
Total shareholder equity (deficit) |
| | ( | ( | ||||
|
|
| ||||||
Liabilities | ||||||||
Employee benefit obligations | 14 | | | | ||||
Non-current financial liabilities | 10, 13 |
| | | | |||
Non-current derivative financial instruments | 13 | | — | — | ||||
Total non-current liabilities |
| | | | ||||
|
|
| ||||||
Current financial liabilities | 10, 13 |
| | | | |||
Provisions | 15 |
| — | | | |||
Trade payable | 10, 16.1 |
| | | | |||
Tax and social liabilities | 16.2 |
| | | | |||
Current derivative financial liabilities | 13 | | | | ||||
Other creditors and miscellaneous liabilities | 16.3 |
| | | | |||
Total current liabilities |
| | | | ||||
|
|
| ||||||
TOTAL LIABILITIES AND SHAREHOLDERS’ EQUITY (DEFICIT) |
| | | | ||||
The accompanying Notes form an integral part of these consolidated financial statements
F-4
CONSOLIDATED STATEMENT OF PROFIT OR LOSS
FOR THE YEARS ENDED DECEMBER 31, | ||||||||
|
| 2021 |
| 2022 |
| 2023 | ||
(Amounts in thousands of euros, except share and per share data) |
| NOTES |
| 12 months |
| 12 months |
| 12 months |
Revenues |
|
| — | — | — | |||
Cost of sales |
|
| — | — | — | |||
Gross margin |
|
| — | — | — | |||
Research and development expenses, net |
| 17.1 | ( | ( | ( | |||
General and administrative expenses |
| 17.2 | ( | ( | ( | |||
Operating loss |
| ( | ( | ( | ||||
Financial expenses |
| ( | ( | ( | ||||
Financial income |
| | | | ||||
Change in fair value of derivative liabilities and convertible bonds |
| ( | | ( | ||||
Net financial expense |
| 18 | ( | ( | ( | |||
Loss before taxes |
| ( | ( | ( | ||||
Income taxes benefit |
| — | — | — | ||||
Net loss |
| ( | ( | ( | ||||
Attributable to the owners of the parent |
| ( | ( | ( | ||||
Non-controlling interests |
| ( | — | — | ||||
Basic and diluted weighted average number of shares outstanding |
| | | | ||||
Basic loss per share (€/share) |
| 20 | ( | ( | ( | |||
Diluted loss per share (€/share) |
| 20 | ( | ( | ( | |||
The accompanying Notes form an integral part of these consolidated financial statements
F-5
CONSOLIDATED STATEMENT OF PROFIT OR LOSS AND OTHER COMPREHENSIVE PROFIT OR LOSS
FOR THE YEARS ENDED DECEMBER 31, | ||||||
| 2021 |
| 2022 |
| 2023 | |
(amounts in thousands of euros) |
| 12 months |
| 12 months |
| 12 months |
Net loss for the year |
| ( | ( |
| ( | |
Items that will not be reclassified to profit or loss |
|
| ||||
Actuarial gains and losses |
| | |
| | |
Items that will be reclassified to profit or loss |
|
| ||||
Foreign currency translation adjustment |
| — | |
| ( | |
Other comprehensive income items |
| | |
| | |
Total other comprehensive profit or loss |
| ( | ( |
| ( | |
(17,026) | ||||||
Attributable to the owners of the parent |
| ( | ( |
| ( | |
Non-controlling interests |
| ( | — |
| — | |
The accompanying Notes form an integral part of these consolidated financial statements
F-6
Statement of changes in consolidated shareholders’ equity
Impact of | ||||||||||||||||||||||
separate | Shareholder | |||||||||||||||||||||
accounting for | equity - | |||||||||||||||||||||
Additional | Reserves | Share- | convertible and | attributable | Non- | |||||||||||||||||
paid-in | and | Conversion | based | non-convertible | Own | to Biophytis | controlling | Shareholder | ||||||||||||||
(amounts in thousands of euros, except share data) |
| NOTES |
| Capital |
| capital |
| income |
| reserve |
| payment |
| bonds |
| shares |
| shareholders |
| interests |
| equity |
As of December 31, 2020 |
| | | ( | ( | | | ( | | ( | | |||||||||||
Net loss for the period |
| — | — | ( | — | — | — | — | ( | ( | ( | |||||||||||
Other comprehensive profit or loss |
| — | — | | — | — | — | — | | — | | |||||||||||
Total other comprehensive profit or loss |
| — | — | ( | — | — | — | — | ( | ( | ( | |||||||||||
Conversion of convertible notes | | | — | — | — | — | — | | — | | ||||||||||||
Share capital increase | | | — |
| — | — | — | — | | — | | |||||||||||
Exercise of warrants | | | — |
| — | — | — | — | | — | | |||||||||||
Cancellation of 2018 Kreos warrants | — | — | — | — | — | ( | — | ( | — | ( | ||||||||||||
Biophytis shares delivered to Negma | — | — | | — | — | — | — | | — | | ||||||||||||
Allocation of premiums to retained earnings | — | ( | | — | — | — | — | — | — | — | ||||||||||||
Treasury shares net movements | — | — | — | — | — | — | ( | ( | — | ( | ||||||||||||
Gains and losses, net related to treasury shares | — | — | | — | — | — | — | | — | | ||||||||||||
Equity settled share-based payments | — | — | — |
| — | | — | — | | — | | |||||||||||
Costs incurred in relation to equity transactions | — | ( | — |
| — | — | — | — | ( | — | ( | |||||||||||
On December 31, 2021 |
|
|
| |
| |
| ( | ( |
| |
| |
| ( |
| |
| ( |
| | |
Net loss for the period |
|
|
| — |
| — |
| ( | — |
| — |
| — |
| — |
| ( |
| — |
| ( | |
Other comprehensive profit or loss |
|
|
| — |
| — |
| |
| |
| — |
| — |
| — |
| |
| — |
| |
Total other comprehensive profit or loss |
|
|
| — |
| — |
| ( | |
| — |
| — |
| — |
| ( |
| — |
| ( | |
Bond conversions |
|
| |
| ( |
| — |
| — |
| — |
| — |
| — |
| |
| — |
| | |
Exercise of ‘BSA’ warrants, ‘BSPCE’ warrants and AGAs |
|
| |
| ( |
| — |
| — |
| — |
| — |
| — |
| |
| — |
| | |
Net own share transactions |
|
| — |
| — |
| — | — |
| — |
| — |
| |
| |
| — |
| | ||
Allocation of retained earnings to premium |
|
|
| — |
| ( |
| |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
Gains and losses, net related to treasury shares |
|
|
| — |
| — |
| ( |
| — |
| — | — |
| — |
| ( |
| — |
| ( | |
Equity settled share-based payments |
|
| — |
| — |
| — |
| — |
| |
| — |
| — |
| |
| — |
| | |
On December 31, 2022 | | ( | ( | ( | | | ( | ( | ( | |||||||||||||
Net loss for the period | — | — | ( | — | — | — | — | ( | — | ( | ||||||||||||
Other comprehensive profit or loss | — | — | | ( | — | — | — | | — | | ||||||||||||
Total other comprehensive profit or loss | — | — | ( | ( | — | — | — | — | ( | |||||||||||||
Bond conversions | 11 | | ( | — | — | — | — | — | | — | | |||||||||||
Capital increase | 11 | | | — | — | — | — | — | | — | | |||||||||||
Exercise of ‘BSA’ warrants and ‘BSPCE’ warrants | 11 | | | — | — | — | — | — | | — | | |||||||||||
Capital reduction | 11 | ( | — | | — | — | — | — | — | — | — | |||||||||||
Net own share transactions | — | — | — | — | — | — | | | — | | ||||||||||||
Net gains and losses on own shares | — | — | ( | — | — | — | — | ( | — | ( | ||||||||||||
Allocation of retained earnings to premiums | 11 | — | | ( | — | — | — | — | — | — | — | |||||||||||
Share-based payments | 12.4 | — | — | — | — | | — | — | | — | | |||||||||||
Other changes affecting shareholder equity | — | — | | — | — | — | — | | — | | ||||||||||||
Expenses related to capital increases | 11 | — | ( | — | — | — | — | — | ( | — | ( | |||||||||||
On December 31, 2023 | | | ( | ( | | | ( | ( | ( | ( |
The accompanying Notes form an integral part of these consolidated financial statements
F-7
STATEMENT OF CONSOLIDATED CASH FLOWS
FOR THE YEARS ENDED DECEMBER 31, | ||||||||
2021 | 2022 | 2023 | ||||||
(amounts in thousands of euros) |
| NOTES |
| 12 months |
| 12 months |
| 12 months |
Cash flow used in operating activities |
|
|
|
|
|
| ||
Net loss |
|
| ( | ( |
| ( | ||
Amortization and depreciation of intangible and tangible assets |
| 4, 5 |
| | |
| | |
Additions of provisions, net of reversals |
| 14, 15 |
| | ( |
| ( | |
Expenses associated with share-based payments |
| 12.4 |
| | |
| | |
Financial interest |
|
| | |
| | ||
Amortization of the day one Loss | 13.2 | | — | — | ||||
Change in fair value of financial instruments |
| 13.2 |
| | ( |
| | |
Interests on investment accounts |
|
|
| ( | ( |
| — | |
Net financial indemnities Negma |
| 13.2 |
| | ( |
| — | |
Unwinding of conditional advances and other financial expenses |
| 13.1 |
| | |
| | |
Amortized cost of non-convertible bonds and debt component of the convertible notes |
| 13.2 |
| | |
| | |
Cash flow used in operating activities before changes in working capital |
|
|
| ( | ( |
| ( | |
(-) Change in working capital (net of depreciation of trade receivables and inventories) |
|
|
| | ( |
| ( | |
(Increase) decrease in Other non-current financial assets |
|
|
| ( | — |
| | |
(Increase) decrease in other receivables |
|
|
| | ( |
| | |
Increase (decrease) in trade accounts payable |
|
|
| | ( |
| ( | |
Increase (decrease) in tax and social security liabilities |
| ( | ( | ( | ||||
Increase (decrease) in other creditors and accrued liabilities |
|
|
| ( | ( |
| | |
|
|
|
| |||||
Cash flow used in operating activities |
|
|
| ( | ( |
| ( | |
|
|
|
|
|
|
|
| |
Cash flow used in investment operations |
|
|
|
|
|
|
| |
Acquisition of intangible and tangible assets |
| 4, 5 |
| ( | ( |
| ( | |
Interest on investment account |
|
|
| | — |
| — | |
Purchase of term deposits classified as other current financial assets | 7 | — | | — | ||||
Decrease (increase) in short term deposits accounts | | | | |||||
Cash flow used in investment operations |
|
|
| | ( |
| | |
|
|
|
|
|
|
|
| |
Cash flow from/used in to financing operations |
|
|
|
|
|
|
| |
Proceeds from share capital increase | 11 | | — | | ||||
Costs paid in relation to equity transactions |
| 11 |
| ( | — |
| ( | |
Net financial indemnity received from/ (paid to) Negma |
| 12.2 |
| ( | |
| — | |
Exercise of warrants (BSA) and founders’ warrants (BSPCE) |
| 12 |
| | |
| | |
Proceeds from research tax credit prefinancing, net of guarantee deposit |
| 13.3 |
| | |
| | |
Reimbursement of the prefinanced CIR receivables, net of guarantee deposit | 12 | ( | ( | — | ||||
Proceeds from conditional advances |
| 13.1 |
| | |
| — | |
Repayment of conditional advances | ( | ( | ( | |||||
Proceeds from subsidies |
|
|
| — | |
| — | |
Financial interest paid | ( | ( | ( | |||||
Conversion settled with cash payment |
| 13.2 |
| ( | — |
| — | |
Proceeds from the issuance of convertible notes and non-convertible bonds |
| 13.2 |
| | |
| | |
Repayment non-convertible bonds | 13.2 | ( | ( | ( | ||||
Costs incurred in relation to the issuance of convertible notes and non-convertible bonds | 13.2 | ( | — | ( | ||||
Repayment of lease obligations |
| 13.3 |
| ( | ( |
| ( | |
Cash flow used in financing operations |
|
|
| | |
| | |
|
|
|
| |||||
Net effect of exchange rate changes on cash and cash equivalents |
|
|
| ( | ( |
| ( | |
|
|
|
|
| ||||
Increase (decrease) in cash and cash equivalents |
|
|
| | ( |
| ( | |
|
|
|
|
|
|
|
| |
Cash and cash equivalents at the beginning of the period |
| | | | ||||
Cash and cash equivalents at the end of the period |
| | | |||||
The accompanying Notes form an integral part of these consolidated financial statements
F-8
Notes to the consolidated financial statements
(Unless otherwise indicated, the consolidated financial statements are presented in thousands of euros. Certain amounts may be rounded up for the purpose of calculating the financial information contained in the consolidated financial statements. As a result, the totals in some tables may differ slightly to the sum of the preceding figures).
Note 1: General information about the Company
Founded in September 2006, Biophytis SA is a clinical-stage biotechnology company specializing in the development of treatments aimed at slowing down the degenerative processes associated with aging and improving functional outcomes for patients suffering from age-related diseases.
Biophytis is a limited company (société anonyme) subject to French law, with its registered office at 14, avenue de l’Opéra, 75001 Paris, France (Company registration number: 492 002 225 RCS PARIS).
The Company’s standard shares are listed on Euronext Growth Paris (Mnemo: ALBPS-ISIN: FR0012816825). The ADSs (American Depositary Shares) have been listed on the Nasdaq Capital Market since February 10, 2021 under the symbol “BPTS”.
Biophytis and its subsidiaries are hereinafter referred to as “Biophytis” or the “Company”.
The consolidated financial statements of Biophytis for the year ended December 31, 2023, or the “Financial Statements”, were prepared under the responsibility of the Company’s management and were approved and authorized for issue by the Company’s Board of Directors on April 8, 2024. The accounts will also be submitted to the Annual General Meeting for approval.
Note 2: Notable events
2.1.Research and development activity
During the 2023 financial year, the Company continued the development of its main clinical and preclinical programs involving BIO101 (20-hydroxyecdysone), formerly known as Sarconeos (BIO101).
2.1.1.COVA program for severe forms of COVID-19
In early February 2023, Biophytis announced the final results of its COVA phase 2-3 clinical trial, including the data from the
On the strength of these results, the Company has begun the regulatory formalities that will allow BIO101 (20-hydroxyecdysone) to be deployed as quickly as possible for hospitalized patients with severe COVID-19 at risk of respiratory failure and death. The Company has pursued a multi-pronged strategy for this:
| Application for Conditional Marketing Authorization (cMA) for Europe and Emergency Use Authorization (EUA) in the United States. The Company has requested a pre-submission meeting with a view to applying for conditional marketing authorization in Europe from the EMA, and in the USA from the FDA in view of the health emergency. The company announced on August 16, 2023 that it had received responses from the European Medicines Agency (EMA) and the Food and Drug Administration (FDA) with the recommendation to seek Scientific Opinions from the relevant agencies on the proposed clinical and regulatory development plan of BIO101 (20-hydroxyecdysone) for severe forms of COVID-19 up to full MA. Responses are expected for the first half of 2024. |
F-9
| Applications for early access programs in key countries. In France, this application was made in May. The early access program in France will be run in partnership with Intsel Chimos, a pharmaceutical company based in Saint-Cloud, France, which specializes in importing, distributing and exploiting innovative drugs to treat patients with rare and/or serious diseases that have reached a treatment impasse. On September 19, Biophytis announced that it had received a reply from the French National Health Authority (HAS) asking it to complete the application file by providing certain results from pharmaceutical studies underway with its industrial partner Sequens, as well as additional data and scientific arguments relating to the phase 2-3 COVA clinical trial. The HAS application will be re-submitted including these different elements in 2024, depending on the EMA’s responses concerning the possibility of applying for conditional marketing authorization. In Brazil, an early access program has already been approved in 2022 to treat critically ill COVID-19 patients in intensive care units (ICUs), but was suspended pending the results of the COVA study. This program is currently being reactivated in response to the positive results obtained. |
On the preclinical front, the Company entered into a partnership with the University of Liège at the end of 2023 to carry out a number of research projects, notably concerning the treatment of respiratory failure caused by the Influenza virus. Given its original operational mode on the renin-angiotensin system, BIO101 (20-hydroxyecdysone) could be used to treat severe forms of the main viral respiratory diseases. These pathologies, where medical needs remain unsatisfied, represent a significant potential for Biophytis.
2.1.2.SARA program for sarcopenia
In May 2023, the Company submitted an authorization application via the EMA (European Medicines Agency) European portal to launch SARA-31, the first Phase 3 study ever launched for sarcopenia. A similar application was submitted to the FDA (Food and Drug Administration) at the beginning of July to launch the same study in the United States.
The launch of the Phase 3 program follows encouraging results from the SARA-INT Phase 2b study and interactions with health authorities in 2022. On August 8, 2023, Biophytis announced that it had received a positive response from the Belgian authorities to carry out its SARA-31 program. A positive response from the FDA to conduct the study in the United States was also received and announced by the Company on September 11, 2023. The actual start of the study is scheduled for 2024, and will depend on the conclusion of partnership agreements and the Company’s financial resources.
2.1.3.MYODA program for Duchenne muscular dystrophy
At the Clinical and Scientific Conference organized by the Muscular Dystrophy Association (MDA) between March 19 to 22, 2023 in Dallas, Texas, the Company shared new information concerning its MYODA program for Duchenne Muscular Dystrophy (DMD) in the form of posters, for which a clinical development plan is in preparation, and the therapeutic potential of BIO101 (20-hydroxyecdysone) in rare neuromuscular diseases such as Spinal Muscular Atrophy (SMA), for which promising preclinical results have been obtained, particularly in combination with gene therapy. In Duchenne Muscular Dystrophy, the Company has reviewed its clinical development protocol after interactions with the FDA and plans to start a phase 1/2 clinical study in 2024.
2.2.Financing
During the financial year, the Company carried out several financing transactions for a total gross amount of approximately
2.2.1.Capital increase through private placement on Euronext
On May 11, 2023, the Company announced a new financing operation in the form of a private placements by professional investors combined with a public proposal to private individuals, for a gross amount of
F-10
on Euronext Growth Paris under the ISIN code FR0012816825 ALBPS for the opening of trading on May 15, 2023, and are assimilated to existing shares with immediate dividend rights.
2.2.2.Registered direct offering on the Nasdaq Capital Market
On July 19, 2023, the Company announced a registered direct offering for a gross amount of $
The ADSs and pre-funded warrants were offered and sold in a registered direct offering pursuant to a shelf registration statement on Form F-3 (File No. 333-271385) filed with the U.S. Securities and Exchange Commission (the “SEC”) on April 21, 2023 and declared effective by the SEC on May 1st, 2023. The ordinary warrants were issued within the framework of a concurrent private placement.
The issue price of the ordinary shares underlying the ADSs represented a
The issue of the
Each pre-funded warrant, giving entitlement to
The accounting impacts of this transaction are presented in note 11.
2.2.3.Capital increase with preferential subscription rights on Euronext
On November 20, 2023, the Company announced the successful completion of its capital increase with shareholders’ preferential subscription rights (DPS), through the issue of
The BSARs attached to each new share were listed on a separate pricing line under the ISIN code: FR001400LN79. They may be exercised at any time up to December 31, 2026, with
The accounting impacts of this transaction are presented in note 11.
2.2.4.Convertible bond issues
The financing transactions described above enabled Biophytis to limit its need for bond financing via the convertible bond issue agreement with Atlas. This contract covers the issue of a maximum of
In the first half of 2023, the Company issued
F-11
At the end of 2023, the Company also announced the issue of the fourth tranche of 160 ORNANE bonds, of which the first half was effectively cashed in early January 2024 and the second half was issued in February 2024. The net amount received was
2.2.5. Prefinancing of the French research tax credit (CIR)
During December 2023, the company pre-financed part of its CIR for the year 2023 for an amount of
2.3.Post-balance sheet events
2.3.1.Issue and conversion of convertible bonds
In
Since December 31, 2023, the company has carried out, upon ATLAS’ request, the conversion of
2.3.2.Reverse stock split of Biophytis shares listed on Euronext Growth
On March 15, 2024, the Company announced the implementation of a reverse stock split of its shares listed on Euronext Growth, which will result in the allocation of 1 new ordinary share to be issued with a par value of
2.3.3.New OBA program in obesity
On April 8, 2024, the Company announced the launch of a new clinical development program named OBA, with BIO101 (20-hydroxyecdysone) as a potential treatment for obesity in combination with GLP-1 receptor agonists. Treatment of obesity may result in loss of muscle mass and function, particularly following a diet combined with recently introduced GLP-1 receptor agonists. BIO101 (20-hydroxyecdysone) is the first MAS receptor activator, administered daily orally, that has demonstrated metabolic effects on muscle and fat mass in preclinical obesity studies. These beneficial effects of BIO101 (20-hydroxyecdysone) translate into improved mobility and muscle strength in sarcopenic obese patients, as shown in the SARA-INT phase 2 study. In addition, the molecule 20-hydroxyecdysone has already been tested in obese patients during a low-calorie diet in the Quinolia study, showing promising effects on muscle strength and fat loss. The Company plans to start the phase 2 OBA clinical study in mid-2024, after obtaining regulatory authorizations, and the first patients are expected to be treated during the second half of 2024. BIO101 (20-hydroxyecdysone) will be evaluated in obese patients treated by GLP-1 RA and following a hypocaloric diet. The first efficacy results should be available in 2025.
F-12
Note 3: Accounting principles, rules and methods
3.1.Principles used in preparing the Financial Statements
Unless otherwise indicated, the consolidated financial statements are presented in thousands of euros. Certain amounts may be rounded up for the purpose of calculating the financial information contained in the consolidated financial statements. As a result, the totals in some tables may not correspond exactly to the sum of the preceding figures.
Statement of compliance
The consolidated financial statements for the year ended December 31, 2023 have been prepared in accordance with IFRS Accounting Standards issued by the International Accounting Standards Board (IASB), in compliance with the international standards as published by the IASB on December 31, 2023, and with the international standards as adopted by the European Union on December 31, 2023.
The reference system adopted by the European Commission can be consulted on the following website: https://eur-lex.europa.eu/legal-content/FR/TXT/?uri=LEGISSUM%3Al26040
The accounting principles and methods applied for the consolidated financial statements for the year ended December 31, 2023 are identical to those used in the consolidated financial statements for the years ended December 31, 2022 and December 31, 2021, and comply with the IFRS Accounting Standards, amendments and interpretations as adopted by the European Union and the IASB, mandatory for financial years beginning on or after January 1, 2023 (and which had not been applied early by the Group), namely:
Standard |
| Name |
IFRS 17 and amendments to IFRS 17 | Insurance contract including amendments published on 06/25/20. First adoption of IFRS 17 and IFRS 9 - Comparative information | |
Amendments to IAS 8 | Definition of accounting estimates | |
Amendments to IAS 1 and Statement Practice 2 | Disclosure of accounting policies | |
Amendments to IAS 12 | Deferred taxes on assets and liabilities arising from the same transaction | |
Amendments to IAS 12 | International tax reform - Pillar 2 rule model |
The application of these standards, amendments and interpretations has no material impact on the Group’s consolidated financial statements. It is specified that the Company is not affected by the amendments to IAS 12 presented above.
In addition, the other standards, amendments or interpretations published respectively by the IASB and the IFRIC (International Financial Reporting Interpretations Committee) and adopted by the European Union on December 31, 2023 but whose mandatory application is subsequent to the financial year beginning January 1, 2023 have not been applied early by the Group: amendments to IFRS 16 (lease liabilities relating to a sale and leaseback), amendments to IAS 1 (classification of liabilities as current and non-current), and amendments to IAS 21 (absence of exchangeability).
Going concern
Our financial statements have been prepared on a going concern basis assuming that we will be successful in our financing objectives. As such, no adjustments have been made to the financial statements relating to the recoverability and classification of the asset carrying amounts or classification of liabilities that might be necessary should we not be able to continue as a going concern.
The Company has incurred operating losses and negative cash flows from operations since inception due to the innovative nature of the product candidates it is developing, which necessitates a research and development phase spanning multiple years. The Company does not expect to generate revenue from product sales in the near future.
We estimate our existing capital resources consisting of cash and cash equivalents €
F-13
to be wrong, and we could use our capital resources sooner than we currently expect. As of the date of authorization of these financial statements, our available cash and our ORNANE financing line are not projected to be sufficient to support our operating plan for at least the next 12 months. These events and conditions indicate that a material uncertainty exists that may cast significant doubt on the Company’s ability to continue as a going concern and, therefore, the Company may be unable to realize its assets and discharge its liabilities in the normal course of business.
We intend to seek additional capital to pursue preclinical and clinical activities, obtain regulatory approval and authorization for, and commercialize our drug candidates. Notably during 2024, we may conduct equity financing transactions on Euronext Growth or Nasdaq, enter into new debt financing agreements or enter into partnership or licensing agreements for our R&D programs that could provide additional non-dilutive financial resources or reduce our costs.
We cannot guarantee that we will be able to obtain the necessary financing to meet our needs or to obtain funds at attractive terms and conditions, including as a result of disruptions to the global financial markets resulting from geopolitical instability, macroeconomic conditions, global health crises, or other factors.
If we are not successful in our financing objectives, we could have to scale back our operations, notably by delaying or reducing the scope of our research and development efforts or obtain financing through arrangements with collaborators or others that may require us to relinquish rights to our product candidates that we might otherwise seek to develop or commercialize independently.
Use of judgments and estimates
The preparation of financial statements requires that the management makes reasonable estimates and assumptions based on the information available at the date that the financial statements are finalized. These estimates and assumptions may affect the values of assets, liabilities and expenses given in the financial statements, and the disclosure of contingent assets and liabilities when the financial statements are reviewed.
These estimates are based on the going concern assumption and are prepared using the information available at the time of preparation. They are continuously assessed on the basis of past experience and various other factors deemed reasonable and form the basis for assessments of the accountable values of assets and liabilities. Estimates may be revised if the circumstances on which they were based change or if new information becomes available. Actual results could differ materially from these estimates depending on different assumptions or conditions.
The main judgments and estimates made by the Company’s management relate in particular to:
| ● | The definition of the fair value of share-based payments, including stock subscription warrants (“BSA”), warrants to purchase shares in business creators (“BSPCE”) and bonus shares (“AGA”) granted to employees, directors and external service providers. This is based on the Black & Scholes option pricing model, which takes into account assumptions on complex and subjective variables. These variables include the value of the shares, the expected volatility of the share value over the life of the instrument, and the current and future behavior of the holders of these instruments. There is a high inherent risk of subjectivity when using an option pricing model to measure the fair value of share-based payments in accordance with IFRS 2 Share-based Payment standard. The valuation assumptions used are presented in Note 12. |
| ● | The definition of the fair value of convertible bonds and non-convertible bonds issued to Kreos with attached stock warrants. The determination of the fair value of stock warrants for the benefit of Kreos is based on the Black & Scholes model. This model takes as input observable variables such as the value of the company's shares and the risk-free interest rate, but also unobservable variables such as the volatility of the share price. There is a high inherent risk of subjectivity arising from the use of an option pricing model to define the fair value of derivative liabilities and equity instruments in accordance with IAS 32 Financial Instruments - Presentation ("IAS 32") and IFRS 9. The convertible bond is valued using the “One-factor equity convertible model”. This model takes as input observable variables such as the value of the company's shares and the risk-free interest rate, but also unobservable variables such as the volatility of the share price and the credit spread of the company. business. The fair value of the debt component of convertible bonds was determined by discounting future cash flows at a market rate (unobservable data). The valuation assumptions used are presented in Note 13.2. The fair value of the conversion option is deducted by subtracting the fair value of the convertible bond from the fair value of the debt component. The credit |
F-14
| spread was determined by equalizing the sum of the fair value of the instruments (Warrants and Convertible Bond) on the issue date to the amount of cash received. |
| ● | The definition of the fair value of Negma and Atlas bonds convertible into ordinary shares and/or redeemable in cash. This is based on the binomial option pricing model and the Longstaff Schwartz model, respectively, which take into account unobservable assumptions and variables , such as stock volatility and issuer credit spread. These variables include the value of the Company’s securities, the expected volatility of the share price over the expected life of the instrument, and the present and future behavior of the Company and the holders of these instruments. There is an inherent high risk of subjectivity arising from the use of an option pricing model to define the fair value of convertible bonds in accordance with IFRS 9 and IAS 32. The valuation assumptions used are presented in Note 13.2. |
| ● | The determination of the amount of deferred tax assets that can be recognized in the financial statements. This requires management to make estimates both of the period over which losses carried forward will be used up, and of the level of future taxable profits, in the light of tax management strategies. The accounting principles applied by the Company regarding the recognition of deferred tax assets are specified in Note 3.19. |
3.2.Consolidation scope and method
The scope of consolidation included the following companies on December 31, 2023:
Biophytis,
| ● | Instituto Biophytis Do Brasil, a |
| ● | Biophytis Inc., a |
As the Company controls its two subsidiaries, they are fully consolidated.
Group companies close their accounts on December 31 of each year. Intra-group transactions and balances are eliminated. Subsidiary financial statements are prepared for the same reference period as those of the parent company, using consistent accounting policies.
3.3.Foreign currency translation
For each entity, Group entities determine a functional currency, and items included in the financial statements of each entity will be measured using that functional currency.
The Company’s financial statements are drawn up in euros (€), which is its presentation currency.
3.3.1.Accounting for foreign currency transactions
Transactions in foreign currencies are translated into the company’s functional currency of each entity at the exchange rate prevailing on the transaction date. Monetary assets and liabilities denominated in foreign currencies at the balance sheet date are translated into the functional currency using the exchange rate for that date.
Gains and losses arising from the translation of monetary items correspond to the difference between the amortized cost denominated in the functional currency at the start of the period, adjusted for the impact of the effective interest rate and payments over the period, and the amortized cost denominated in the foreign currency translated at the exchange rate on the balance sheet date.
3.3.2.Translation of foreign subsidiary financial statements
The financial statements of entities whose functional currency is not the euro are translated as follows:
| ● | Assets and liabilities are translated at the year-end rate; |
F-15
| ● | income statement items are translated using the average exchange rate for the period, as long as this rate is not altered by significant changes in exchange rates; and |
| ● | Equity items are translated at the historical rate. |
Exchange differences arising on translation for consolidation purposes are recognized in other elements of comprehensive income and stored in shareholder equity under “Translation reserve”.
The exchange rates used to prepare the consolidated financial statements are as follows:
Closing rate | Average rate | |||||||||||
EXCHANGE RATES | AS OF DECEMBER 31, | DECEMBER 31 | ||||||||||
(currency for €1) |
| 2021 |
| 2022 |
| 2023 |
| 2021 |
| 2022 |
| 2023 |
BRL |
| | | | | |
| | ||||
USD |
| | |
| |
| | |
| | ||
3.4.Intangible assets
3.4.1.Research and development costs
Research costs are accounted for as incurred. Costs incurred on development projects are included as intangible assets when the following criteria are met, in application of IAS 38:
| ● | It is technically possible to complete the intangible asset such that it can be available for use or sale; |
| ● | Management plans to complete, use or sell the intangible asset; |
| ● | The intangible asset can be used or sold; |
| ● | It can be demonstrated that the intangible asset is likely to generate future economic benefits; |
| ● | Adequate technical, financial and other resources are available for the full development, use or sale of the intangible asset; |
| ● | The expenditure attributable to the intangible asset during its development can be reliably measured. |
In the opinion of the Company’s management, and due to the uncertainties inherent in the development of the Company’s drug candidates, the criteria required for development costs to be recognized as an asset, as defined by IAS 38, “Intangible Assets”, are not met.
3.4.2.Patents and software
Costs relating to the acquisition of patents and software are capitalized on the basis of the costs incurred to acquire the patents and software concerned.
3.4.3.Amortization period and expense
Where intangible assets have a finite useful life, amortization is calculated on a straight-line basis over this period, i.e.:
Items |
| Amortization period |
Development costs | Estimated useful life of the project | |
Purchased patents | Estimated useful life of patents | |
Metabrain | ||
Iris Pharma | ||
Stanislas Veillet (BIO101) |
F-16
Software |
Amortization of intangible assets is accounted for in the consolidated income statement under:
| ● | “General and administrative expenses” for amortization of software; and |
| ● | “Research and development costs” for amortization of patents |
The value of intangible assets is tested whenever there is an impairment indicator. Quantitative and qualitative indicators are reviewed at each balance sheet date, the main ones being those relating to the development of the R&D portfolio, pharmacovigilance, patent disputes and the arrival of competing products. If there is any internal or external indication of impairment, Biophytis assesses the asset’s recoverable value. The test consists of comparing the net book value of these assets with their recoverable value. When the book value of an asset exceeds its recoverable value, an impairment loss is included to account for the difference.
3.5.Property, plant and equipment
Property, plant and equipment are valued at acquisition cost (purchase price plus incidental expenses) or at the Company’s production cost.
Assets are depreciated on a straight-line basis over their estimated useful lives:
Items |
| Amortization period |
General fixtures and fittings |
| |
Plant, machinery and equipment |
| |
Office and computer equipment |
| |
Furniture |
| |
Transport equipment |
|
Amortization of property, plant and equipment is accounted for in the consolidated income statement under :
| ● | “General and administrative expenses” for the amortization of fixtures and fittings, office and computer equipment and furniture; and |
| ● | “Research and development costs” for the amortization of laboratory equipment. |
3.6.Lease contracts
Items financed by leases as defined by the IFRS 16 standard concerning leases that do not meet the accounting exemption criteria for the lessees (leases of “low-value” assets and short-term leases of less than 12 months) are accounted for as assets in the financial position statement. The corresponding debt is recorded under “Liabilities”.
Payments made for leases that meet the exemption criteria are accounted for as expenses in the income statement on a straight-line basis over the term of the contract,.
Rights of use are amortized on a straight-line basis over the lease term.
3.7.Recoverable value of non-current assets
Amortized assets are tested for impairment whenever there is an internal or external indication that an asset may be impaired. Impairment indicators include the following:
| ● | Mixed or negative results from preclinical and clinical trials; |
F-17
| ● | Significant delays or non-compliance with clinical trial development schedules. |
3.8.Financial assets
In accordance with IFRS 9, the Company’s financial assets are classified in two categories based on their nature and holding intention:
| ● | Financial assets at fair value through profit or loss; and |
| ● | Financial assets at amortized cost. |
All financial assets are initially accounted for at fair value plus acquisition costs. All purchases and sales of financial assets are accounted for on the settlement date.
Financial assets are removed from the accounts when the rights to receive cash flows from them expire, or when they have been sold and the Company has transferred the majority of the risks and benefits of ownership.
Financial assets linked to guarantee deposits and the corresponding financial liabilities are presented separately in accordance with IAS 32.
3.8.1.Financial assets at fair value through profit or loss
Financial assets at fair value through profit or loss are made up of cash and cash equivalents.
Gains or losses arising from changes in the value of “financial assets at fair value through profit or loss” are presented under “financial income” in the income statement for the period in which they occur.
Other assets may also be voluntarily classified in this category if the criteria are met, in accordance with IFRS 9.
3.8.2.Financial assets at amortized cost
Financial assets at amortized cost are largely made up of non-current financial assets and other loans and receivables. They are valued at their amortized cost using the effective interest rate method, adjusted for expected credit losses.
3.8.3.Impairment of financial assets at amortized cost
A financial asset is impaired using the expected loss method, taking into account any default during the asset’s holding period. Expected losses are accounted for in the financial position statement. Impairment is accounted for in the consolidated income statement.
3.9.Cash and cash equivalents
Cash and cash equivalents accounted for in the statement of financial position are made up of available cash at bank and in hand as well as short-term deposits with initial maturities of less than three months.
Cash equivalents are readily convertible to a known amount of cash and are subject to an insignificant risk of change in value. They are held for the purpose of meeting short-term cash commitments. They are valued at fair value, with changes in value accounted for as “financial income”.
3.10.Fair value of financial instruments
Borrowings (excluding derivatives and convertible bonds) are initially accounted for at fair value less any transaction costs, and subsequently valued at their amortized cost using the effective interest method.
F-18
The convertible bonds issued have been valued at fair value through profit or loss in accordance with IFRS 9 and the related costs, if applicable, are directly recorded as financial expenses.
The fair value of trade receivables and payables is equivalent to their balance sheet value, given the very short payment terms of these receivables. The same applies to other current receivables and payables.
The Company has defined three categories of financial instruments based on their valuation methods, and uses this classification to present some of the disclosures required by IFRS 7 Financial instruments - disclosures:
| ● | Level 1: financial instruments listed on an active market; |
| ● | Level 2: financial instruments whose valuation methods are based on observable data; |
| ● | Level 3: financial instruments whose valuation methods are based in whole or in part on unobservable data. Unobservable data is defined as data the value of which is based on assumptions or correlations that are neither based on observable market transaction prices for the same instrument, nor on observable market data at the valuation date. |
Financial instruments held by the Company and accounted for at fair value through profit or loss are the derivatives and convertible bonds issued to Kreos and Atlas (see Note 12.2), which are classified as level 3.
3.11.Liquidity contract
As part of its listing on the Euronext Growth Paris market, the Company has signed a liquidity contract with a specialist institution to limit the intra-day volatility of Biophytis shares by taking buy and sell positions on the Company’s shares. Shares acquired under this contract are accounted for as treasury shares at their acquisition cost. Gains and losses on the sale of treasury shares are accounted for in shareholder equity. The cash reserve related to the liquidity contract is shown under “Other non-current financial assets”.
The accounting treatment relating to the liquidity contract is presented in note 11.
3.12.Public subsidies
3.12.1.Repayable advances
The Company benefits from repayable advances. Details of these aids are provided in Note 13.1.
They are accounted for in accordance with IAS 20 Accounting for Government Grants and Disclosure of Government Assistance. Financial advances granted at below-market interest rates are valued at amortized cost in accordance with IFRS 9 :
| ● | The interest rate benefit is determined using a discount rate corresponding to a market rate on the grant award date. The amount resulting from the rate advantage obtained on the award of repayable advances is treated as a subsidy recorded as income in the comprehensive income statement; and |
| ● | The financial cost of repayable advances, calculated at the market rate, is then recorded as a financial expense. |
Subsidies corresponding to the rate advantage are shown as a reduction in the “Research and development” category.
These advances are recorded under “Non-current borrowings” or “Current borrowings”, depending on their maturity. If the project fails, the waiver is recorded as a grant.
3.12.2.Grants
Government grants are accounted for when there is reasonable assurance that the entity will comply with the applicable conditions and that the grant will be received.
F-19
Operating subsidies are deducted from research and development expenses.
3.12.3.Research tax credit
The Company benefits from certain provisions of the French General Tax Code relating to research tax credits.
The Company benefits from research tax credits for specific projects (“crédit d’impôt recherche”, or “CIR”), granted to companies based in France to encourage scientific and technical research. Companies whose expenses meet the required criteria receive a tax credit which (i) may be deducted from the income tax due for the year in which it was granted, as well as for the three following years, or (ii) in certain circumstances, may also be refunded to the Company for its excess share.
If a company meets certain criteria in terms of sales, staff or assets that enable it to be considered a small or medium-sized enterprise as defined by the European Union, it can apply for immediate repayment of the research tax credit. Biophytis meets these criteria.
The Company considers the research tax credit granted by the French government to be a public subsidy, since it is received independently of the Company’s tax payments. The Company accounts for this receivable in other current receivables, given the expected repayment period. Research tax credits are deducted from research and development expenses in the consolidated income statement.
The research tax credit is subject to audit by the French tax authorities.
3.13.Other receivables
Other receivables include the nominal value of the research tax credit, which is recorded when the eligible expenses giving rise to the research tax credit have been incurred.
3.14.Capital
Classification as equity depends on a specific analysis of the characteristics of each instrument issued. The Company’s standard shares are classified as equity.
Incidental costs directly attributable to the share issue are deducted from shareholder equity, net of tax.
3.15.Share-based payments
Since its creation, the Company has set up several equity-based compensation plans in the form of “stock subscription warrants” (“BSA”), “business creator share subscription warrants” (“BSPCE”) or “bonus shares” (“AGA”) awarded to employees and Board members.
In application of IFRS 2 Share-based payment, the cost of equity-based transactions is accounted for over the period in which the rights to benefit from the equity instruments are attributed to the recipient.
The fair value of warrants granted to employees is determined by applying the Black-Scholes option pricing model. The same applies to options granted to other individuals providing similar services, as their market value cannot be defined.
All the assumptions used to determine the fair value of the programs are described in Note 11.
3.16.Employment benefit obligations
The Company’s French employees are entitled to the pension benefits set out by French law, including:
| ● | A retirement indemnity paid by the Company on retirement (defined benefit plan); and |
F-20
| ● | The payment of retirement pensions by Social Security organizations, which are financed by contributions from companies and employees (defined-contribution plans). |
Pension plans, similar benefits and other employee benefits that are analyzed as defined benefit plans (plans under which the Company undertakes to guarantee a defined amount or level of benefits) are accounted for in the consolidated statement of financial position on the basis of an actuarial valuation of the obligations at the accounts closing date, less the fair value of the related program assets dedicated to them.
This valuation is based on the projected unit credit method, taking into account staff turnover and mortality estimations. Actuarial gains and losses, if any, are accounted for in “Other comprehensive income”.
Company payments for defined contribution plans are accounted for in the income statement for the period to which they relate.
3.17.Provisions
A provision is constituted if, as a result of past events, the Company has a present legal or implicit obligation, the value of which can be reliably estimated, and it is probable that an outflow of economic benefits will be required to settle the obligation.
The amount recorded as a provision corresponds to the best estimate of the expenditure required to settle the present obligation on the balance sheet date.
3.18.Financial liabilities
Financial liabilities are classified in two categories and include:
| ● | Financial liabilities accounted for at amortized cost and, |
| ● | Financial liabilities accounted for at fair value through profit or loss. |
3.18.1.Financial liabilities accounted for at amortized cost
Borrowings and other financial liabilities, such as repayable advances, are accounted for at amortized cost, calculated using the effective interest rate. The current portion of borrowings is shown under “Current borrowings”.
The accounting treatment of non-convertible bonds and convertible bonds issued by the Company is detailed in Note 13.2.
3.18.2.Financial liabilities at fair value through profit or loss
The Company issued Kreos with non-convertible bonds and convertible bonds. This financial instrument is made up of several components valued at their fair value through profit or loss in accordance with IFRS 9: a derivative liability linked to the convertible bond conversion option and a derivative liability linked to the warrants. It also includes a component relating to non-convertible bonds valued at amortized cost
The Company also issued Atlas with bonds that were convertible into ordinary shares, with warrants attached. This financial instrument is made up of: a hybrid component linked to convertible bonds (valued at their fair value through profit or loss in accordance with IFRS 9) and an equity instrument linked to warrants (valued at their fair value on the issue date in equity instruments in accordance with IAS 32).
Transaction costs are accounted for in financial expenses at the date of issue of the convertible bonds.
The accounting treatment of these compound financial instruments is detailed in Note 13.2.
F-21
3.19.Corporate income tax
The taxable assets and liabilities for the current and previous years are valued at the amount expected to be recovered from or paid to the tax authorities.
The tax rates and regulations used to determine these amounts are those that are fully or at least substantially legally valid on the financial year end date.
Deferred taxes are recorded, using the liability method, on all temporary differences between the tax base of assets and liabilities and their carrying amount in the financial statements at the financial year end date, as well as on any deficits to be carried forwards.
Deferred tax assets are recorded as tax losses to be carried forwards when it is probable that future taxable profits will be available against which the unused tax losses can be utilized. Determining the amount of deferred tax assets to be recorded requires management to make estimates both of the period over which losses carried forward will be used, and of the level of future taxable profits, in the light of tax management strategies.
3.20.Segment information
The Company operates in a single business sector: the development of drug candidates for the treatment of degenerative diseases and the improvement of muscular and visual functions for patients suffering from age-related diseases.
The assets, liabilities and operating loss presented in the financial statements relate to the parent company’s activities in France. Most research and development and administrative costs are incurred in France and, since 2018, in the United States.
3.21.Earnings per share
Basic earnings per share are calculated by dividing profit or loss attributable to Biophytis equity holders by the weighted average number of ordinary shares outstanding for the period.
Diluted earnings per share are determined by adjusting the earnings attributable to Biophytis shareholders and the weighted average number of ordinary shares outstanding to allow for the effects of all dilutive potential ordinary shares.
If the inclusion of instruments giving deferred rights to the capital (BSA, BSPCE, AGA and convertible bonds) generates an anti-dilutive effect, these instruments are not taken into account.
F-22
Note 4: Intangible assets
(amounts in thousands of euros) |
| Patents |
| Software |
| Total |
GROSS VALUES |
|
|
|
|
|
|
Statement of financial position for December 31, 2021 |
| |
| |
| |
Acquisition |
| |
| — |
| |
Transfer |
| ( |
| |
| ( |
Statement of financial position for December 31, 2022 |
| |
| |
| |
Acquisition |
| |
| — |
| — |
Transfer |
| |
| |
| |
Statement of financial position for Sunday, December 31, 2023 |
| |
| |
| |
AMORTIZATION |
|
|
|
|
|
|
Statement of financial position for December 31, 2021 |
| |
| |
| |
Increase |
| |
| — |
| |
Decrease |
| |
| |
| |
Statement of financial position for December 31, 2022 |
| |
| |
| |
Increase |
| |
| — |
| |
Decrease |
| |
| |
| |
Statement of financial position for December 31, 2023 |
| |
| |
| |
NET BOOK VALUES |
|
|
|
|
|
|
On December 31, 2021 |
| |
| — |
| |
On December 31, 2022 |
| |
| — |
| |
On December 31, 2023 |
| |
| — |
| |
The Company has not identified any indicators of loss of value leading it to carry out impairment tests as of December 31, 2023 in accordance with IAS 36
The Company holds patent co-ownership shares with public-sector partners.
As part of the intellectual property agreement signed with the Company’s Chief Executive Officer (see Note 21.2), the total patent rights acquired from the Company’s Chief Executive Officer on December 31, 2023 amounted to
F-23
Note 5: Property, plant and equipment
|
|
|
| Office |
|
| ||||||
Equipment | Fixtures | equipment, | ||||||||||
Equipment | and tools | and | computers, | Buildings | ||||||||
(amounts in thousands of euros) |
| and tools | (rights of use) | fittings | furniture | (rights of use) | Total | |||||
GROSS VALUES |
|
|
|
| ||||||||
Statement of financial position for December 31, 2021 |
| |
| |
| |
| |
| | | |
Acquisition |
| |
| |
| |
| |
| — | | |
Transfer | ( | — | — | — | — | ( | ||||||
Exchange Rate impact |
| — |
| — |
| |
| |
| — | | |
Statement of financial position for December 31, 2022 |
| |
| |
| |
| |
| | | |
Acquisition |
| |
| — |
| |
| |
| — | | |
Disposal | — | ( | — | — | ( | ( | ||||||
Exchange Rate impact |
| — |
| — |
| |
| |
| — | | |
Statement of financial position for December 31, 2023 |
| |
| |
| |
| |
| — | | |
|
|
|
|
| ||||||||
AMORTIZATION |
|
|
|
|
|
|
|
|
|
| ||
Statement of financial position for December 31, 2021 |
| |
| |
| |
| |
| | | |
Increase |
| |
| |
| |
| |
| | | |
Decrease | ( | — | — | — | — | ( | ||||||
Exchange Rate impact |
| — |
| — |
| — |
| — |
| — | — | |
Statement of financial position for December 31, 2022 |
| |
| |
| |
| |
| | | |
Increase |
| |
| |
| |
| |
| | | |
Decrease |
| — |
| ( |
| — |
| — |
| ( | ( | |
Exchange Rate impact | — | — | — | — | — | — | ||||||
Statement of financial position for December 31, 2023 |
| |
| |
| |
| |
| — | | |
NET BOOK VALUES |
|
|
|
|
|
|
|
|
|
| ||
On December 31, 2021 |
| |
| — |
| |
| |
| | | |
On December 31, 2022 |
| |
| |
| |
| |
| | ||
On December 31, 2023 |
| |
| |
| |
| |
| — | |
Building lease payments correspond to the rent paid to the Sorbonne Université for the Company’s Paris premises. The contract is concluded on an annual basis and has been renewed in December 2023 for one year. As this contract is less than one year, the right of use has not been recognized in the consolidated accounts, in accordance with IFRS 16.18.
Note 6: Other non-current financial assets
AS OF DECEMBER 31, | ||||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Liquidity contract - cash balance | | | | |||
Security deposit for non-convertible bonds (“Kreos contract 2018”) | | | | |||
Security deposit for the “Kreos contract 2021” loan agreement (see Note 12.2.3) | — | — | — | |||
Other security deposits |
| | |
| — | |
Total other non-current financial assets |
| | |
| | |
F-24
Note 7: Other current financial assets
AS OF DECEMBER 31, | ||||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Deductions in connection with the pre-financing of the CIR (Research Tax Credit) by Neftys (cf. Note 13.3). |
| | |
| | |
Guarantee deposit related to the non-convertible bonds (Kreos 2018 contract) |
| | — |
| — | |
Total other current financial assets |
| | |
| | |
In accordance with IAS 7, term deposits have been classed as current financial assets.
Note 8: Other receivables and prepaid expenses
| AS OF DECEMBER 31, | |||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Research tax credit (CIR) |
| | |
| | |
Value added tax |
| | |
| | |
Prepaid expenses |
| | |
| | |
Trade payables - prepayments and trade debtors |
| | |
| | |
Miscellaneous |
| | |
| — | |
Total other receivables and prepaid expenses |
| | |
| | |
The “research tax credit (CIR)” item corresponds to the French CIR receivable for the 2023 financial year (vs
Note 9: Cash and cash equivalents
Cash and cash equivalents break down:
AS OF DECEMBER 31, | ||||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Cash |
| | |
| | |
Cash equivalents |
| | |
| | |
Total cash and cash equivalents |
| | |
| | |
Cash equivalents correspond to term deposits complying with the provisions of IAS 7.6 and IAS 7.7, i.e. short-term, liquid investments that can be drawn down rapidly.
F-25
Note 10: Financial assets and liabilities and their impact on income
The Company’s assets and liabilities are valued as follows for the years ending December 31, 2022 and December 31, 2023, respectively:
| Value - IFRS 9 statement of | |||||||
AS OF DECEMBER 31, 2022 | financial position | |||||||
Statement of | Fair value | |||||||
financial | Fair | through profit | ||||||
(amounts in thousands of euros) |
| position value |
| value |
| or loss |
| Amortized cost |
Non-current financial assets (excluding deferred losses) | |
| |
|
| | ||
Other receivables (excluding prepaid expenses) | |
| |
| — |
| | |
Current financial assets (excluding deferred losses) | |
| |
| — |
| | |
Cash and cash equivalents | |
| |
| |
| | |
Total assets | |
| |
| |
| | |
Non-current borrowing | |
| |
| — |
| | |
Non-current derivative liabilities | — | — | — | — | ||||
Current borrowings | |
| |
| |
| | |
Current derivative liabilities | | | | — | ||||
Trade accounts payable | |
| |
| — |
| | |
Tax and social security liabilities | | | — | | ||||
Other creditors and accrued liabilities | | | — | | ||||
Total liabilities | |
| |
| |
| | |
|
| Value - IFRS 9 statement of | ||||||
AS OF DECEMBER 31, 2023 | financial position | |||||||
Statement of | Fair value | |||||||
financial |
|
| through profit |
| ||||
(amounts in thousands of euros) |
| position value | Fair value | or loss | Amortized cost | |||
Non-current financial assets (excluding deferred losses) |
| |
| |
| — | | |
Other receivables (excluding prepaid expenses) |
| — |
| — |
| — | — | |
Current financial assets (excluding deferred losses) |
| — |
| — |
| — | — | |
Cash and cash equivalents |
| |
| |
| | | |
Total assets |
| |
| |
| | | |
Non-current borrowing |
| ( |
| ( |
| — | ( | |
Non-current derivative liabilities | — | — | — | — | ||||
Current borrowings |
| ( |
| ( |
| ( | ( | |
Current derivative liabilities | ( | ( | ( | — | ||||
Trade accounts payable |
| ( |
| ( |
| — | ( | |
Tax and social security liabilities | ( | ( | — | ( | ||||
Other creditors and accrued liabilities | ( | ( | — | ( | ||||
Total liabilities |
| ( |
| ( |
| ( | ( | |
F-26
The impact of the Company’s financial assets and liabilities on the consolidated income statement for the years ended December 31, 2022 and December 31, 2023:
| AS OF DECEMBER 31, | |||||||||||
2021 |
| 2022 | 2023 | |||||||||
|
| Change in fair |
|
| Change in fair | Change in fair | ||||||
(amounts in thousands of euros) | Interest | value | Interests |
| value |
| Interests |
| value | |||
Liabilities |
|
|
|
|
|
|
|
| ||||
Derivative liabilities | — | ( | — | | — | | ||||||
Liabilities valued at fair value: bonds |
| — | ( | — |
| |
| — |
| ( | ||
Liabilities valued at amortized cost: non-convertible bonds and debt component of convertible bonds |
| ( | — | ( |
| — |
| |
| — | ||
Liabilities valued at amortized cost: advances |
| ( | — | ( |
| — |
| |
| — | ||
Note 11: Capital and premiums
| December 31, | ||||||||
2021 |
| 2022 |
| 2023 | |||||
Share capital (in thousand of euros) |
| |
| |
| | |||
Number of outstanding shares |
| |
| |
| | |||
Nominal value per share (in euros) | € | | € | | € | | |||
On December 31, 2023, the Company’s share capital stood at
On April 17, 2023, the Board of Directors decided on an initial capital reduction motivated by losses, for a total amount of €
Capital movements for the 2023 financial year were as follows:
Nominal amount | |||||
| Number of shares |
| (in thousands of euros) | ||
Capital on December 31, 2022 | |||||
May 11, 2023 private placement (1) | |||||
July 18, 2023 private placement (2) | |||||
Exercise of pre-funded share subscription warrants (2) | | | |||
November 20, 2023 capital increase (3) | |||||
Conversion of convertible bonds (4) | | | |||
Exercise of share warrants (5) | | | |||
Definitive acquisition of bonus shares (6) | | | |||
Total impact of nominal reduction | — | | |||
Capital on December 31, 2023 |
| (1) | Capital increase by private placement combined with a public proposal, for a net amount of |
F-27
| (2) | Capital increase by registered direct placement for a gross amount of $ |
| (3) | Capital increase with shareholders’ pre-emptive rights maintained, through the issue of |
| (4) |
| (5) | Following the exercise of warrants during the period, share capital was increased by |
| (6) | |
Changes in capital for the year ended December 31, 2022:
During the year ended December 31, 2022,
Following the exercise of warrants and acquisition of free shares during the period, the share capital was increased by €
In total, the share capital was increased by €
Changes in capital for the year ended December 31, 2021:
On February 12, 2021, Biophytis announced the closing of the ADS Offering. The gross proceeds from the Offering were $
On July 30, 2021,
During the year ended December 31, 2021,
The costs incurred during the period by the Company in connection with the Initial Public Offering on Nasdaq Capital Market in February 2021 were recognized as a reduction from shareholders’ equity for €
F-28
Following the exercise of warrants during the period, the share capital was increased by €
Own shares
Under the liquidity contract signed with Invest Securities, the Company held
Share premium
The Annual General Meeting of April 17, 2023 decided to increase the “share premium” account by
Distribution of dividends
The Company did
Note 12: Share subscription warrants (BSA), Founders share subscription warrants (BSPCE) and Free shares (AGA)
12.1 Share subscription warrants
Changes in the number of warrants outstanding over the 2022 and 2023 financial years are analyzed as follows:
|
|
|
|
|
|
| Maximum | |||||||
number of | ||||||||||||||
Number of warrants outstanding | shares that | |||||||||||||
Allocation | may be | |||||||||||||
Type | date | 12/31/2021 | Allocated | Exercised | Expired | 12/31/2022 |
| subscribed | ||||||
BSA2018 |
| 9/10/2018 |
| |
| — |
| — |
| — |
| |
| |
BSA2020 | 4/7/2020 | | — | ( | — | | | |||||||
BSA2021 | 6/17/2022 | — | | — | — | | | |||||||
Total |
|
| |
| |
| ( |
| — |
| |
| | |
|
|
|
|
|
|
| Maximum | |||||||
number of | ||||||||||||||
Number of warrants outstanding | shares that | |||||||||||||
Allocation | may be | |||||||||||||
Type | date | 12/31/2022 | Allocated | Exercised | Expired | 12/31/2023 | subscribed | |||||||
BSA2018 |
| 9/10/2018 |
| |
| — |
| — |
| — |
| |
| |
BSA2020 | 4/7/2020 | | — | ( | — | | | |||||||
BSA2021 | 6/17/2022 | | — | — | — | | | |||||||
BSA2022 | 4/14/2023 | — | | — | — | | | |||||||
Pre-funded warrants2023-07 | 7/18/2023 | — | | ( | — | — | — | |||||||
BSA2023-07 | 7/18/2023 | — | | — | — | | | |||||||
BSAR2023-11 | 11/17/2023 | — | | ( | — | | | |||||||
Total |
| |
| |
| ( |
| — |
| |
| | ||
On June 17, 2022, the Company allocated
F-29
On April 14, 2023, the Company allocated
As part of the July 2023 registered direct proposal, the Company issued
The Company issued
12.2. Business creator share subscription warrants (“BSPCE”)
Plan features | Assumptions | ||||||||||||||||
Initial IFRS2 total | |||||||||||||||||
valuation | |||||||||||||||||
Allocation | Total number of | Risk- | (Black& Scholes) (in | ||||||||||||||
Type |
| date |
| warrants allocated |
| Maturity date |
| Expected term |
| Exercise price |
| Volatility |
| free rate | thousands of euros) | ||
BSPCE2019-1 |
| 4/3/2020 |
| |
| 4/3/2026 |
| 2 years | € | |
| | % | ( | % | | |
BSPCE2019-2 | 4/3/2020 | | 4/3/2026 | 4 years | € | | | % | ( | % | | ||||||
BSPCE2020-1 | 12/22/2020 | | 12/22/2026 | 2 years | € | | | % | ( | % | | ||||||
BSPCE2020-2 | 12/22/2020 | | 12/22/2026 | 4 years | € | | | % | ( | % | | ||||||
BSPCE2021-1 | 9/15/2021 | | 9/15/2027 | 1 year | € | | | % | ( | % | | ||||||
BSPCE2021-2 |
| 9/15/2021 |
| |
| 9/15/2027 |
| 2 years | € | |
| | % | ( | % | | |
The change in the number of BSPCEs outstanding over the 2022 and 2023 financial years can be analyzed as follows:
Number of warrants outstanding | Maximum | |||||||||||||
number of | ||||||||||||||
Allocation | shares that may | |||||||||||||
Type |
| date |
| 12/31/2021 |
| Allocated |
| Exercised |
| Expired |
| 12/31/2022 |
| be subscribed |
BSPCE2019-1 |
| 04/03/2020 |
| |
| — |
| ( |
| ( |
| |
| |
BSPCE2020-2 | 04/03/2020v | | — | ( | | | ||||||||
BSPCE2021-1 | 12/22/2020 | | — | ( | | | ||||||||
BSPCE2021-2 | 12/22/2020 | | — | ( | | | ||||||||
BSPCE2019-1 | 9/15/2021 | | — | ( | | | ||||||||
BSPCE2019-2 | 9/15/2021 | | — | ( | | | ||||||||
Total |
|
|
| |
| — |
| ( |
| ( |
| |
| |
F-30
Number of warrants outstanding | Maximum | |||||||||||||
number of | ||||||||||||||
Allocation | shares that may | |||||||||||||
Type |
| date |
| 12/31/2022 |
| Allocated |
| Exercised |
| Expired |
| 12/31/2023 |
| be subscribed |
BSPCE2019-1 | 04/03/2020 | | — | — | ( | | | |||||||
BSPCE2020-2 |
| 04/03/2020v |
| |
| — |
| — |
| ( |
| |
| |
BSPCE2021-1 |
| 12/22/2020 |
| |
| — |
| — |
| ( |
| |
| |
BSPCE2021-2 |
| 12/22/2020 |
| |
| — |
| — |
| ( |
| |
| |
BSPCE2019-1 |
| 9/15/2021 |
| |
| — |
| — |
| ( |
| |
| |
BSPCE2019-2 |
| 9/15/2021 |
| |
| — |
| — |
| ( |
| |
| |
Total |
|
| |
| — |
| — | ( |
| |
| | ||
The vesting period for the BSPCE plans is as follows:
Type |
| Vesting period |
BSPCE2017-1 |
| 1/3 to 07/21/2017 1/3 to 07/21/2018 1/3 to 07/21/2019 |
BSPCE2017-2 |
| 1/3 to 07/21/2017 1/3 to 07/21/2018 1/3 to 07/21/2019 |
BSPCE2019-1 |
| 1/3 to 4/10/2020 1/3 to 4/10/2022 1/3 to 4/10/2024 |
BSPCE2019-2 |
| 1/3 to 4/10/2020 1/3 to 4/10/2022 1/3 to 4/10/2024 |
BSPCE2020-1 |
| 1/3 to 12/22/2020 1/3 to 12/22/2022 1/3 to 12/22/2024 |
BSPCE2020-2 | 1/3 to 12/22/2020 1/3 to 12/22/2022 1/3 to 12/22/2024 | |
BSPCE2021-1 | 1/3 to 9/15/2021 1/3 to 9/15/2022 1/3 to 9/15/2023 | |
BSPCE2021-2 |
| 1/3 to 9/15/2021 1/3 to 9/15/2022 1/3 to 9/15/2023 |
The parameters used to value the BSPCEs issued in 2022 and 2023 are as follows:
|
| Plan features |
| Assumptions | |||||||||||||
Total |
|
|
|
| Initial IFRS2 total | ||||||||||||
number of | valuation (Black & | ||||||||||||||||
Allocation | warrants | Expected | Exercise | Scholes) (in | |||||||||||||
Type | date | allocated | Maturity date | term | price | Volatility | Risk-free rate | thousands of euros) | |||||||||
BSPCE2019-1 |
| 4/3/2020 |
| |
| 4/3/2026 | 2 years | € | |
| | % | ( | % | | ||
BSPCE2019-2 |
| 4/3/2020 |
| |
| 4/3/2026 | 4 years | € | |
| | % | ( | % | | ||
BSPCE2020-1 |
| 12/22/2020 |
| |
| 12/22/2026 | 2 years | € | |
| | % | ( | % | | ||
BSPCE2020-2 |
| 12/22/2020 |
| |
| 12/22/2026 | 4 years | € | |
| | % | ( | % | | ||
BSPCE2021-1 |
| 9/15/2021 |
| |
| 9/15/2027 | 1 year | € | |
| | % | ( | % | | ||
BSPCE2021-2 |
| 9/15/2021 |
| |
| 9/15/2027 | 2 years | € | |
| | % | ( | % | | ||
12.3. Allocation of bonus shares (“AGA”)
Plan features | Assumptions | |||||||||||||
Total initial | ||||||||||||||
Total number | IFRS2 valuation | |||||||||||||
of bonus | (Black & Scholes) | |||||||||||||
Allocation | shares | Maturity | Exercise | (in thousands | ||||||||||
Type |
| date |
| allocated |
| date |
| price |
| Volatility |
| Risk-free rate |
| of euros) |
AGA2021-2 | 4/25/2021 | | N/A | N/A | N/A | N/A | | |||||||
AGA2022 |
| 4/14/2023 |
| |
| N/A |
| N/A |
| N/A |
| N/A |
| |
Total |
|
| |
|
|
|
|
| | |||||
F-31
The change in the number of AGAs under acquisition over the 2022 and 2023 financial years can be analyzed as follows:
Number of bonus shares under acquisition | Maximum | |||||||||||||
number of | ||||||||||||||
Allocation | shares that may | |||||||||||||
Type |
| date |
| 12/31/2021 |
| Allocated |
| Acquired |
| Expired |
| 12/31/2022 |
| be subscribed |
AGA2020 |
| 12/22/2020 |
| |
| — |
| ( |
| — |
| — |
| — |
AGA2021-1 | 9/15/2021 | | — | ( | — | — | — | |||||||
AGA2021-2 | 4/25/2023 | — | | — | — | | | |||||||
Total |
| |
| ( |
| ( |
| — |
| |
| | ||
Number of bonus shares under acquisition | Maximum | |||||||||||||
number of | ||||||||||||||
Allocation | shares that may be | |||||||||||||
Type |
| date |
| December 12, 2021 |
| Allocated |
| Acquired |
| Expired |
| December 31, 2023 |
| acquired |
AGA2021-2 | 4/25/2021 | | — | ( | ( | — | — | |||||||
AGA2022 | 4/14/2023 | — | | — | ( | | | |||||||
Total | | ( | ( | | | |||||||||
On April 14, 2023, the Company allocated
12.4. Share-based payment expenses as accounted for on December 31, 2022 and December 31, 2023
| DECEMBER 31, 2022 |
| DECEMBER 31, 2023 | |||||||||||||
| Cumulative |
|
|
| Cumulative |
|
| |||||||||
Probabilized | expense at | Expense | Accumulated | expense at | Expense | Accumulated | ||||||||||
in thousands | cost of plan | beginning of | for the | cost of plan | Probablilized cost | beginning of | for the | expense to | ||||||||
of euros | to date | financial year | period | to date | of the date | financial year | period | date | ||||||||
BSA2021 |
| |
| — |
| |
| |
| |
| |
| — |
| |
BSA2022 |
| — |
| — |
| — |
| — |
| |
| — |
| |
| |
BSPCE2019--1 |
| |
| |
| |
| |
| |
| |
| — |
| |
BSPCE2019-2 |
| |
| |
| |
| |
| |
| |
| |
| |
BSPCE2020-1 |
| |
| |
| |
| |
| |
| |
| — |
| |
BSPCE2020-2 |
| |
| |
| |
| |
| |
| |
| |
| |
BSPCE2021-1 |
| |
| |
| |
| |
| |
| |
| — |
| |
BSPCE2021-2 | | | | | | | | | ||||||||
AGA2020-1 | | | | | | | — | | ||||||||
AGA2021-1 | | | | | | | — | | ||||||||
AGA2021-2 |
| |
| — |
| |
| |
| |
| |
| |
| |
AGA2022 | — | — | — | — | | — | | | ||||||||
Sub-total | | | ||||||||||||||
Social contribution (1) | | | ||||||||||||||
Total |
|
|
| |
|
|
|
| |
| ||||||
F-32
| DECEMBER 31, 2021 | |||||||
Cumulative | ||||||||
Probable | expenses - | Expense | ||||||
cost of | beginning | for the | Cumulative expense | |||||
Type |
| the plan |
| of period |
| period |
| to date |
Warrants 2021 | |
| |
| — |
| | |
Founders’ warrants2017-1 | |
| |
| — |
| | |
Founders’ warrants2017-2 | |
| |
| — |
| | |
Founders’ warrants2019-1 | |
| |
| |
| | |
Founders’ warrants2019-2 | |
| |
| |
| | |
Founders’ warrants2020-1 | |
| |
| |
| | |
Founders’ warrants2020-2 | |
| |
| |
| | |
Founders' warrants2021-1 | |
| — |
| |
| | |
Founders' warrants2021-2 | |
| — |
| |
| | |
Free shares 2020 | |
| |
| |
| | |
Free shares 2021-1 | |
| — |
| |
| | |
Free shares 2021-2 | — |
| — |
| — |
| — | |
Sub-total |
|
|
|
| |
|
| |
Social contribution (1) |
|
|
|
| |
|
| |
Total |
|
|
|
| |
|
| |
Bonus shares are subject to an additional social security contribution payable on allocation of the bonus shares at the end of the vesting period. It is accounted for on a straight-line basis over the vesting period and revalued in line with the Company’s share price at the end of each financial year. This social security contribution, recorded under social security and other social bodies liabilities (see Note 15.2), amounted to
Note 13: Borrowings and financial liabilities
AS OF DECEMBER 31, | ||||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Repayable advances |
| | |
| | |
Non-convertible bonds |
| | |
| | |
Convertible bonds |
| | |
| | |
Non-current lease obligations | | | | |||
Non-current borrowing |
| | |
| | |
Non-current derivative liabilities |
| | — |
| — | |
AS OF DECEMBER 31, | ||||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Repayable advances | | | | |||
Non-convertible bonds | | | | |||
Convertible bonds | | | | |||
Debt relating to pre-financing of part of CIR (Research Tax Credit) receivables | | | | |||
Payables on current rental obligations | | | | |||
Accrued interest payable | — | — | | |||
Current borrowings |
| | |
| | |
Current derivative liabilities | | |
| | ||
F-33
Breakdown of borrowings by maturity, at repayment value
Borrowing maturities break down:
| AS OF |
|
|
| ||||
DECEMBER 31, | Current | Non-current | ||||||
(amounts in thousands of euros) | 2023 | < 1 year | 1 to 5 years | > 5 years | ||||
Repayable advances |
| |
| |
| |
| — |
Non-convertible bonds |
| |
| |
| |
| — |
Convertible bonds |
| |
| |
| | — | |
Debts on leasing obligations | | | | — | ||||
Debt relating to pre-financing of part of CIR (Research Tax Credit) receivables |
| |
| |
| — |
| — |
Accrued interest payable | | | — | — | ||||
Total borrowings |
| |
| |
| |
| — |
Derivative liabilities | | — | — | — | ||||
AS OF | ||||||||
DECEMBER 31, |
| Current |
| Non-current | ||||
(amounts in thousands of euros) |
| 2022 | < 1 year |
| 1 to 5 years |
| > 5 years | |
Repayable advances | | | | — | ||||
Non-convertible bonds |
| |
| |
| |
| — |
Convertible bonds |
| |
| |
| |
| — |
Debts on leasing obligations |
| |
| |
| |
| — |
Debt relating to pre-financing of part of CIR (Research Tax Credit) receivables |
| |
| |
| — |
| — |
Total borrowings |
| |
| |
| |
| — |
Derivative liabilities |
| |
| |
| — |
| — |
| AS OF |
|
|
| ||||
DECEMBER 31, | Current | Non-current | ||||||
(amounts in thousands of euros) | 2021 | < 1 year | 1 to 5 years | > 5 years | ||||
Repayable advances |
| |
| |
| |
| |
Non-convertible bonds |
| |
| |
| |
| — |
Convertible bonds |
| |
| |
| | — | |
Debts on leasing obligations | | | | — | ||||
Debt relating to pre-financing of part of CIR (Research Tax Credit) receivables |
| |
| |
|
| — | |
Total borrowings |
| |
| |
| |
| |
Derivative liabilities | | | | — | ||||
F-34
13.1. Repayable advances
The table below shows changes in repayable advances:
(in thousands of euros) |
| BPI - Sarcob |
| BPI – BIO 101 |
| AFM – Telethon |
| BPI – BIO 201 | Total | |
December 31, 2021 |
| |
| |
| |
| |
| |
(+) Cash inflow |
| — |
| — |
| — |
| — | — | |
(-) Repayment |
| ( |
| ( |
| — |
| — | ( | |
Grants |
| — |
| — |
| — |
| — | — | |
Financial expenses |
| |
| |
| |
| | | |
Other | | — | ( | — | ( | |||||
On December 31, 2022 |
| — |
| |
| |
| | | |
(+) Cash inflow |
| — |
| — |
| — |
| — | — | |
(-) Repayment |
| — |
| ( |
| — |
| — | ( | |
Grants |
| — |
| — |
| — |
| — | — | |
Financial expenses |
| — |
| |
| |
| | | |
Others | — | — | — | — | — | |||||
On December 31, 2023 |
| — |
| |
| |
| | |
Breakdown of repayable advances by maturity at repayment value
(in thousands of euros) |
| BPI -Sarcob |
| BPI - BIO 101 |
| AFM – Telethon |
| BPI - BIO 201 |
| Total |
On December 31, 2022 |
| — |
| |
| |
| |
| |
Current portion | — | | | — | ||||||
One to 5 years | — | | | | ||||||
Over 5 years | — | — | — | — | — |
(in thousands of euros) |
| BPI -Sarcob |
| BPI – BIO 101 |
| AFM – Telethon |
| BPI – BIO 201 |
| Total |
On December 31, 2023 |
| — |
| |
| |
| |
| |
Current portion |
| — |
| |
| |
| |
| |
One to 5 years |
| — |
| — |
| |
| |
| |
Over 5 years |
| — |
| — |
| — |
| — |
| — |
| ● | BPI France repayable advance - “BIO 101” project |
Under a contract signed with BPI France on November 28, 2016, the Company received a non-interest-bearing recoverable advance of €
| ● | Collaboration agreement with AFM-Téléthon - “BIO 101” project |
Biophytis signed a collaboration agreement with AFM-Téléthon on June 3, 2019 for the development of BIO101 (20-hydroxyecdysone) for the treatment of Duchenne Muscular Dystrophy (DMD) as part of the MYODA program. The Company received
| ● | BPI France repayable advance - “BIO 201” project |
On August 23, 2019, the Company entered into an agreement with BPI France for a conditional interest-free advance of
F-35
Repayment of this advance depends on the successful completion of the project:
| - | in the event of technical and economic failure, a minimum repayment of € |
| - | in the event of technical and economic success, reimbursement is scheduled over a |
Under this agreement, the Company was entitled to receive a grant of €
This project was suspended in 2023 due to limited financial resources which require establishing priorities for financing R&D programs. Although this temporary shutdown can be considered a failure, the Company intends to negotiate a postponement of the end date of the program.
In accordance with IFRS, the fact that the repayable advances received by the Company do not bear annual interest means that the Company has benefited from interest-free loans, i.e. financing conditions that are more favorable than market conditions. The difference between the amount of the advance at historical cost and the amount of the advance discounted at a market rate has been accounted for as a subsidy received from the State.
13.2 Convertible and non-convertible bonds
13.2.1 ATLAS convertible bond issue - Atlas 2020 contract
(amounts in thousands of euros) |
| 2020 ORNANE ATLAS |
On December 31, 2021 - Convertible bonds - Current |
| |
(+) Net cash inflow |
| — |
(+) Change in fair value of debt |
| ( |
(-) Conversion |
| ( |
On December 31, 2022 - Convertible bonds - Current |
| — |
On April 2020, the Company signed a convertible bond financing program with Atlas for up to €
On December 31, 2022, all the convertible bonds associated with this contract had been converted.
Accounting treatment
The Company determined that it could not reliably estimate the fair value of the conversion option embedded in the convertible bonds separately, and therefore concluded that the entire hybrid contract should be valued at fair value through profit or loss until settlement. Fair value was evaluated using a binomial valuation model. As the expected maturity of the bonds is short, the “Day one loss” (including repayment premium and/or issue premium) was immediately accounted for in the income statement.
F-36
13.2.2 ATLAS convertible bond issue - Atlas 2021 contract
| 2021 ORNANE | |
(amounts in thousands of euros) |
| ATLAS |
On December 31, 2021 - Convertible bonds - Current | — | |
(+) Net cash inflow (1) | | |
(+) Change in fair value of debt | | |
(-) Conversion | ( | |
On December 31, 2022 - Convertible bonds - Current |
| |
(+) Net cash inflow (2) | | |
(+) Change in fair value of debt |
| |
(-) Conversion | ( | |
On December 31, 2023 - Convertible bonds - Current |
| |
In June 2021, the Company arranged up to €
The contract imposes certain operational and financial restrictions. These covenants may limit the ability of the parent company and its subsidiaries, in certain circumstances, to, among other things, incur additional debt, create or incur privileges, sell or transfer assets and pay out dividends. On December 31, 2023, these covenants have been met. The contract also contains certain customary covenants and default situations, including changes to the company’s controlling interests.
The ORNANE bonds have a par value of
The holder will be able to request the conversion of the ORNANE bonds at any time in accordance with the conversion parity determined by the following formula: N = CA / CP, where
| ● | “N” is the number of shares resulting from the conversion, |
| ● | “CA” is the nominal value of the ORNANE bonds (i.e. |
| ● | “CP” is the conversion price (i.e. |
On the date of the conversion request, the Company will have the option of redeeming the ORNANE in cash in accordance with the following formula: V = CA / CP x CPr, where
| ● | “V” is the amount to be reimbursed to the bearer. |
| ● | “CPr” is the revised price, corresponding to the lower of (i) the volume-weighted average price over the |
F-37
Accounting treatment
The Company determined that it could not reliably estimate the fair value of the conversion option embedded in the convertible bonds separately, and therefore concluded that the entire hybrid contract should be valued at fair value through profit or loss until settlement. Fair value is evaluated using a Longstaff Schwartz valuation model. As the expected maturity of the bonds is short, the “Day one loss” (including repayment premium and/or issue premium) will be immediately accounted for in the income statement.
During the 2022 financial year, the Company issued
During the 2023 financial year, the Company issued
In addition, during the month of December 2023, the company decided to draw the 4th tranche for an amount of 4 million euros (corresponding to 160 ORNANE), this sum to be paid in 2 installments: 2 million euros at the beginning of January 2024 and 2 million euros in mid-February 2024. The Company determined that it could not reliably estimate separately the fair value of the conversion option embedded in the convertible bonds and therefore concluded that the entire hybrid contract should be measured at fair value through the income statement until settlement. Fair value is assessed using a binomial valuation model. As the expected maturity of the bonds is short, the loss on the issue date (“Day one loss”) (including the redemption premium and/or the issue premium) is immediately recognized in profit or loss.
The table below summarizes the main data used to evaluate the fair value of the convertible bonds:
Conversion option |
| Tranche 2 | Tranche 3 | Tranche 4 | |||||||||||
On issue | On issue | ||||||||||||||
ATLAS 2021 | (21/06/2022) | 12/31/2023 |
| (28/10/2022) |
| 12/31/2023 |
| 12/31/2023 |
| ||||||
Number of bonds outstanding | |
| | | | | |||||||||
Share price | | € | | € | |
| € | | € | | € | ||||
Volatility |
| | % | | % |
| | % | | % |
| | % | ||
Risk-free rate |
| % | NA |
| % | | % |
| | % | |||||
Value of bond issue (in K€) |
| |
| — |
| |
| |
| |
| ||||
The sensitivity analysis of the degree of valuation of convertible bonds as impacted by the change in assumptions has not been presented because the impacts are negligible.
F-38
13.2.3 KREOS convertible and non-convertible bonds
KREOS |
|
|
| |||||||||||
contract | KREOS | KREOS | KREOS | |||||||||||
2018 | contract 2021 | contract | KREOS | 2021 | ||||||||||
Non- | Non- | 2021 | loan | security | KREOS | |||||||||
convertible | convertible | Convertible | Derivative | buyback | 2021 day | |||||||||
(amounts in thousands of euros) |
| bonds |
| bonds |
| bonds |
| contract |
| 2018 |
| one gain |
| Total |
On December 31, 2021 | | | | | ( | | | |||||||
(+) Gross cash inflow |
| — | — | — | — | — | — | — | ||||||
(+) Security deposit |
| — | — | — | — | — | — | — | ||||||
(-) Expenses charged to bond issue | — | — | — | — | — | — | — | |||||||
(+) Change in fair value of debt (1) |
| — | — | — | ( | — | — | ( | ||||||
(-) Bifurcation of the conversion option recognized as a derivative liability | — | — | — | — | — | — | — | |||||||
(+/-) Impact of amortized cost |
| | | | — | — | ( | | ||||||
(-) Repayment |
| ( | ( | — | — | — | — | ( | ||||||
On December 31, 2022 |
| — | | | | ( | | | ||||||
(+) Change in fair value of debt | — | — | — | | — | — | | |||||||
(+/-) Impact of amortized cost | — | | | — | — | — | | |||||||
(-) Repayment | — | ( | — | — | — | ( | ( | |||||||
On December 31, 2023 | — | | | | ( | | |
| (1) | Decrease in value per option: € |
| ● | Issue of non-convertible bonds to Kreos - 2018 Contract |
On September 10, 2018, the Company entered into a venture loan agreement with Kreos in the form of a framework agreement organizing the issue of a bond loan of up to €
A security deposit totaling €
The warrants issued to Kreos under the first installment give the right to subscribe for
In accordance with IFRS 9, the non-convertible debt component was initially accounted for at fair value and subsequently evaluated at amortized cost. The effective interest rate after recording the warrants as a reduction in debt was
| ● | Issue of non-convertible bonds and convertible bonds to Kreos - 2021 Contract |
On November 19, 2021, the Company signed a new venture loan agreement and a bond issue agreement that could provide up to
The four-installment loan agreement was partially drawn down by the Company during the 2021 financial year for a total amount of
The non-convertible bonds bear interest at an annual rate of
F-39
Convertible bonds bear interest at an annual rate of
The Company has also issued
The loan agreement provides for the pledge to Kreos of the Company’s goodwill, bank account balances and intellectual property rights. It also imposes certain operational and financial restrictions. These covenants may limit the ability of the company and its subsidiaries, in certain circumstances, to, among other things, incur additional debt, sell or transfer assets and pay out dividends. This contract also contains certain customary covenants and default situations, including changes to the company’s controlling interests. As of December 31, 2023, these covenants are respected.
Accounting treatment for hybrid financing
Analysis of the specifications of the hybrid contract with reference to IFRS9 and IAS32 criteria led to the need to account for the conversion options and BSA warrants as derivative instruments separate from the host contract (no equity component insofar as these options do not in all circumstances result in the delivery of a fixed number of shares at a fixed price).
The cash amount of
With regard to installment (C) of the ordinary bond issued in December 2021 for
In accordance with IAS
F-40
The table below summarizes the valuation of this derivative on December 31, 2023:
| As of December 31, |
| ||||||||
Fair value of derivative liabilities KREOS 2021 | 2021 | 2022 | 2023 | |||||||
Number of bonds outstanding |
| | | | ||||||
Number of shares available for subscription |
| | | | ||||||
Share price | € | € | € | |||||||
Exercise price | € | € | € | |||||||
Volatility over 12 months |
| | % | | % | | % | |||
Risk-free rate |
| — | % | | % | | % | |||
Credit spread |
| | % | | % | | % | |||
Fair value of derivative instrument (in K€) |
| ( | — | — | ||||||
Change in fair value of derivative liability over the period (in K€) | ( | | — | |||||||
The table below summarizes the accounting procedure for derivatives:
| As December 31, |
| ||||||||
BSA – KREOS 2021 Derivative instruments | 2021 | 2022 | 2023 | |||||||
Number of BSA warrants outstanding |
| |
| | | |||||
Exercise price per share | € | | € | | € | | ||||
Maturity |
|
| ||||||||
Volatility |
| | % | | % |
| | % | ||
Risk-free rate |
| — | % | | % |
| | % | ||
Fair value of BSA 2021 issued to KREOS (in K€) |
| ( | ( |
| ( | |||||
Change in fair value of derivative instrument (in K€) |
| ( | |
| | |||||
The table below summarizes the sensitivity analysis of the valuation of financial instruments impacted by the change in assumptions:
| On December 31, 2023 | |||||
Unconverted | Convertible | Bifurcated | ||||
Sensitivity analysis | installments |
| installments |
| derivatives | |
Value of instruments (in K€) | | | | |||
Impact of a | — | — | — | |||
Impact of a | — |
| — |
| — | |
Impact of a | ( |
| ( |
| — | |
Impact of a | |
| |
| — | |
Impact of a | ( |
| ( |
| — | |
Impact of a | |
| |
| — | |
Impact of a | — |
| — |
| — | |
Impact of a | — |
| — |
| — | |
13.3 Research Tax Credit (CIR) pre-financing debt
Part of the CIR 2022 and 2023 receivables was pre-financed by the PREDIREC INNOVATION 3 securitization fund, with Neftys Conseil SARL as arranger. As a result, the Company has recorded:
| ● | a liability for the amount due to NEFTYS on receipt of the CIR; |
| ● | a financial asset for amounts drawn by NEFTYS from assigned receivables (considered as a security deposit, see Note 7), and |
| ● | a current asset in the form of the French government research tax credit (crédit d’impôt recherche - CIR). |
In accordance with IFRS 9, the financial debt owed to NEFTYS has been determined using the amortized cost method.
F-41
Statement of changes in financial liabilities
Reclassification | ||||||||||||||||||||||||
New | Change in | of the day- | ||||||||||||||||||||||
Impact of | financial debt | fair Value | Loan | Transfer between | one gain as a | |||||||||||||||||||
amortized | related to lease | through | charges | Conversion | non-current and | financial | ||||||||||||||||||
(amounts in thousands of euros) |
| 12/31/2022 |
| Pay-ment |
| Repay-ment |
| cost |
| obligations |
| profit or loss |
| and interest |
| to equity |
| Holdback |
| current liabilities |
| asset |
| 12/31/2023 |
Repayable advances | | — | — | | — | — | — | — | — | — | — | | ||||||||||||
Non-convertible bonds | | — | — | | — | — | — | — | — | — | ( | | ||||||||||||
Convertible bonds | | — | — | | — | — | — | — | — | — | — | | ||||||||||||
Non-current lease obligations | | — | — | — | — | — | — | — | — | — | ( | | ||||||||||||
Non-current borrowing |
|
| — |
| — |
| |
| — |
| — |
| — |
| — |
| — |
| — |
| ( |
| | |
Non-current derivative liabilities |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
Repayable advances |
| |
| — |
| ( |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| |
Non-convertible bonds |
| |
| — |
| ( |
| |
| — |
| — |
| — |
| — |
| — |
| — |
| |
| |
Convertible bonds |
| |
| |
| — |
| — |
| — |
| |
| — |
| ( |
| — |
| — |
| — |
| |
Accrual Interests to pay |
| — |
| — |
| — |
| — |
| — |
| — |
| |
| — |
| — |
| — |
| — |
| |
CIR pre-financing debt |
| |
| |
| ( |
| — |
| — |
| — |
| |
| — |
| — |
| |
| — |
| |
Payables on current rental obligations |
| |
| — |
| ( |
| — |
| — |
| — |
| — |
| — |
| — |
| — |
| |
| |
Current borrowings |
|
| |
| ( |
| |
| — |
| |
| |
| ( |
| — |
| |
| |
| | |
Current derivative liabilities | | — | — | — | — | ( | — | — | — | — | — | |
Transfer | ||||||||||||||||||||||
New | Change | between | ||||||||||||||||||||
Financial | in fair | non- | ||||||||||||||||||||
debt | value | Loan | current | |||||||||||||||||||
Impact of | related to | through | changes | and | ||||||||||||||||||
amortized | lease | profit | and | Conversion | current | |||||||||||||||||
(amounts in thousands of euros) |
| 12/31/2021 |
| Payment |
| Repayment |
| cost |
| obligations |
| or loss |
| interest |
| Conversion to equity |
| Holdback |
| liabilities |
| 12/31/2022 |
Repayable advances | — | — | ( | | — | — | — | — | ( | | ||||||||||||
Non-convertible bonds | | — | — | — | | — | — | — | — | ( | | |||||||||||
Convertible bonds | | — | — | — | | — | — | — | — | — | | |||||||||||
Non-current lease obligations | | — | — | — | — | | — | — | — | ( | | |||||||||||
Non-current borrowing | | — | — | ( | | | — | — | — | ( | | |||||||||||
Non-current derivative liabilities |
| |
| — |
| — |
| — |
| — |
| ( |
| — |
| — |
| — |
| — |
| — |
Repayable advances |
| |
| — |
| — |
| |
| — |
| — |
| — |
| — |
| — |
| |
| |
Non-convertible bonds |
| |
| — |
| ( |
| |
| — |
| — |
| — |
| — |
| — |
| |
| |
Convertible bonds |
| |
| |
| — |
| — |
| — |
| |
| — |
| ( |
| — |
| — |
| |
CIR pre-financing debt |
| |
| |
| ( |
| |
| — |
| — |
| |
| — |
| |
| — |
| |
Payables on current rental obligations |
| |
| — |
| ( |
| — |
| |
| — |
| — |
| — |
| — |
| |
| |
Current borrowings |
| |
| |
| ( |
| |
| |
| |
| |
| ( |
| |
| |
| |
Current derivative liabilities |
| |
| — |
| — |
| — |
| — |
| ( |
| — |
| — |
| — |
| — |
| |
F-42
Note 14: Employee benefit obligation
Employee benefit obligation correspond to retirement indemnities, valued on the basis of the clauses set out in the applicable collective bargaining agreement. This commitment concerns only employees subject to French law.
The amended social security financing law 2023-270 for 2023, promulgated on April 14, 2023, modified the general pension system in France. Its main measures concern the gradual increase in the legal retirement age from
The main actuarial assumptions are as follows:
AS OF DECEMBER 31, | ||||||
ACTUARIAL ASSUMPTIONS |
| 2021 | 2022 |
| 2023 | |
Voluntary retirement | ||||||
Retirement age | between the ages of | |||||
Collective bargaining agreements | Pharmaceutical industry | |||||
Discount rate (IBOXX Corporates AA) | ||||||
Mortality table | INSEE 2017 | INSEE: TH/TF 2016-2018 | INSEE 2018 | |||
Salary increase rate | ||||||
Turnover rate | Medium | Medium | Medium | |||
Social security contribution rates for Executives | ||||||
Movements in the provision for retirement commitments were as follows:
(amounts in thousands of euros) |
| Retirement benefits |
On December 31, 2021 |
| |
Past service cost |
| |
Financial costs |
| |
Actuarial gains and losses |
| ( |
On December 31, 2022 |
| |
Past service cost |
| |
Financial costs |
| |
Actuarial gains and losses |
| ( |
On December 31, 2023 |
| |
Note 15: Provisions
Reversals | Reversals | |||||||||
(amounts in thousands of euros) |
| 12/31/2021 |
| Endowments |
| (used) |
| (unused) |
| 12/31/2022 |
Provision for litigation |
| — |
| |
| — |
| — |
| |
Provision for contingencies |
| — |
| — |
| — |
| — |
| — |
Total provisions |
| — |
| |
| — |
| — |
| |
In the financial year ended December 31, 2022, the Company set aside a provision of
Reversals | Reversals | |||||||||
(amounts in thousands of euros) | 12/31/2022 | Endowments | (used) | (unused) | 12/31/2023 | |||||
Provision for litigation |
| |
| |
|
| ( |
| | |
Provision for contingencies |
| — |
| — |
| — |
| — |
| — |
Total provisions | |
| |
|
| ( |
| |
F-43
As part of the dispute with Negma Group, the Company is the subject of various requests for compensation but has not concluded that it is necessary to recognize provisions for risks due to the stay of ruling pronounced by the Commercial Court on February 9, 2024 and the existence of criminal proceedings against Negma
Note 16: Other current liabilities
16.1 Trade payables
| AS OF DECEMBER 31, | |||||
(amounts in thousands of euros) | 2021 | 2022 |
| 2023 | ||
Suppliers - research and development |
| |
| |
| |
Suppliers - general and administrative expenses |
| |
| |
| |
Total trade payables |
| |
| |
| |
The decrease in debt to research and development suppliers between December 31, 2022 and December 31, 2023 is mainly due to the end of the COVA phase 2/3 and SARA-INT phase 2 clinical trials, this impact being partially offset by industrialization costs for BIO101 (20-hydroxyecdysone) as well as preclinical studies initiated in 2023.
16.2 Tax and social security liabilities
| AS OF DECEMBER 31, | |||||
(amounts in thousands of euros) | 2021 | 2022 |
| 2023 | ||
Personnel and related accounts |
| |
| |
| |
Social security and other social organizations |
| |
| |
| |
Other taxes and levies |
| |
| |
| ( |
Total tax and social security liabilities |
| |
| |
| |
16.3 Other creditors and accrued liabilities
AS OF DECEMBER 31, | ||||||
(amounts in thousands of euros) |
| 2021 | 2022 |
| 2023 | |
Directors’ salaries | |
| |
| | |
Deferred income | | | | |||
Other | |
| |
| | |
Total other creditors and accrued liabilities | |
| |
| | |
As part of the “BIO 201” repayable advance project from BPI France, the Company has also received a grant of €
F-44
Note 17: Operating expenses by function
17.1 Research and development costs
FOR THE YEAR ENDED DECEMBER 31, | ||||||
(amounts in thousands of euros) |
| 2021 | 2022 |
| 2023 | |
Personnel expenses | ( | ( |
| ( | ||
Other purchases and external charges | ( | ( |
| ( | ||
Miscellaneous | ( | ( |
| ( | ||
Research and development costs | ( | ( |
| ( | ||
Research tax credit (CIR) | | |
| | ||
Grants | | |
| |||
Subsidies and CIR | | |
| | ||
Research and development costs, net | ( | ( |
| ( | ||
The decrease in research personnel expenses in 2023 compared to 2022 stems mainly from the impact of share-based payments, which accounted for
The increase in personnel costs in 2022 compared to 2021 is due to the full year of recruitment in 2021, to the increase in the number of statutory employees, and to the impact of the share-based compensation expense related to the BSPCEs and bonus shares granted at the end of 2021 and 2022 (€
The decrease in R&D purchases and external expenses in 2023 compared to 2022 is mainly due to the completion of clinical trials for the COVA and SARA programs in the second half of 2022. Residual costs relating to clinical development were recognized in 2023, but the bulk of R&D expenditure over the year concerned various preclinical work on the Company’s different programs and operations relating to the production of BIO101 (20-hydroxyecdysone).
The decrease in external purchases and expenses in 2022 compared to 2021 is mainly related to the end of our COVA phase 2-3 study as well as the costs of preliminary meetings with public regulatory bodies as part of the continuation of our study. post-phase 2 SARA-INT. These expenses consisted primarily of contract research organization (CRO) costs for conducting clinical trials and non-clinical studies, as well as contract development and manufacturing organization (CDMO) costs for the manufacturing scale of Sarconeos (BIO101) for filing with regulatory authorities.
As part of the BPI France conditional advance “BIO 201” project, the Company was entitled to receive a grant of €
17.2 General and administrative expenses
FOR THE YEAR ENDED DECEMBER 31, | ||||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Personnel expenses | ( | ( |
| ( | ||
Other purchases and external charges | ( | ( |
| ( | ||
Miscellaneous | ( | ( |
| ( | ||
General and administrative expenses | ( | ( |
| ( | ||
The drop in personnel expenses for general management and administrative staff in 2023 compared to 2022 stems mainly from the impact of share-based compensation, which accounted for
Between 2021 and 2022, personnel expenses, including share-based compensation, for senior management and administrative staff increased by €
F-45
Other external purchases and expenses mainly comprise administrative expenses related to the French and United States stock market listing operations, accounting and auditing fees, insurance and legal fees.
F-46
Note 18: Net financial income and expense
FOR THE YEAR ENDED DECEMBER 31, | ||||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Interest and amortized cost on Kreos financing contract (1) | ( | ( |
| ( | ||
Change in fair value of convertible bonds and derivative liabilities (2) | ( | |
| ( | ||
Negma financial indemnities (3) | ( | — |
| — | ||
Provision for Negma litigation risks | — | ( |
| — | ||
Other financial expenses | ( | ( | ( | |||
Expenses relating to the issue of convertible bonds | ( | ( | ( | |||
Net financial income related to the repayment of penalties by Negma (4) | | |
| — | ||
Other financial income | | ( |
| | ||
Foreign exchange gains (losses) | | ( |
| | ||
Total financial income and expense | ( | ( |
| ( | ||
| (1) | See Note 13.2 Convertible and non-convertible bonds |
| (2) | In the financial year ended December 31, 2023, the change in fair value of convertible bonds and derivative liabilities was essentially related to the change in fair value of the ORNANE bonds issued to ATLAS for ( |
| (3) | In the financial year ended December 31, 2021, the financial indemnities paid to Negma is comprised of the fine for non-performance imposed by the Judgment € |
| (4) | On September 8, 2022, the Paris Court of Appeal partially overturned the decision of the Paris tribunal enforcement judge in the Negma litigation. Negma Group Ltd was ordered to repay the Company the sum of € |
Note 19: Income tax
The total amount of tax losses on December 31, 2023 is estimated at €
| ● | French tax losses carried forward indefinitely for a total of € |
| ● | Tax losses of the US subsidiary for a total of € |
| o | € |
| o | € |
| o | € |
| o | € |
| ● | Tax losses of the Brazilian subsidiary for a total of € |
F-47
The tax rate applicable to:
| ● | Biophytis: the current French rate of |
| ● | Biophytis Inc.: the current U.S. rate of |
| ● | Instituto Biophytis Do Brasil: the current Brazil rate of |
Reconciliation of theoretical and effective taxes
FOR THE YEAR ENDED DECEMBER 31, | |||||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 | |
Net loss |
| ( | ( |
| ( | ||
Consolidated tax |
| — | — |
| — | ||
Loss before tax |
| ( | ( |
| ( | ||
Current tax rate in France |
| | % | | % | | % |
Theoretical tax at current rate in France |
| | |
| | ||
Permanent differences |
| | |
| | ||
Share - based payments |
| ( | ( |
| ( | ||
Unused tax losses adjusted for deferred taxes |
| ( | ( |
| ( | ||
Tax rate differences |
| — | ( |
| — | ||
Group income tax (expense)/ income |
| — | — |
| — | ||
Effective tax rate |
| | % | | % | | % |
Permanent differences include the impact of the research tax credit (non-taxable operating income).
Nature of deferred taxes
| AS OF DECEMBER 31, | |||||
(amounts in thousands of euros) | 2021 |
| 2022 | 2023 | ||
Temporary shifts |
| | |
| | |
Tax loss carry-forwards |
| | |
| ||
Total deferred tax assets |
| | |
| ||
— | — | - | ||||
Temporary shifts |
| ( | ( |
| ( | |
Total deferred tax liabilities |
| ( | ( |
| ( | |
— | — | - | ||||
Total net deferred tax items |
| | |
| ||
Unrecognized deferred taxes |
| ( | ( |
| ( | |
Total net of deferred taxes |
| — | — |
| — | |
F-48
Note 20: Loss per share
| FOR THE YEAR ENDED DECEMBER 31, | |||||
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Weighted average number of shares outstanding |
| | |
| | |
Own shares |
| | |
| | |
Weighted average number of shares outstanding (excluding treasury shares) |
| | |
| | |
| ||||||
Net loss for the fiscal year |
| ( | ( |
| ( | |
Basic loss per share (€/share) |
| ( | ( |
| ( | |
Diluted loss per share (€/share) |
| ( | ( |
| ( | |
The accounting of instruments giving deferred rights to the capital (BSA, BSPCE, AGA, convertible bonds) has an anti-dilutive effect for the years presented. They are therefore not taken into account when calculating diluted earnings (see notes 11 and 12.1).
On December 31, 2023, there were outstanding BSAs entitling their holders to acquire up to
As of December 31, 2022, there was outstanding warrants to acquire up to
Note 21: Related parties
21.1 Compensation paid to corporate officers and management
(amounts in thousands of euros) |
| 2021 |
| 2022 |
| 2023 |
Fixed compensation payable |
| | |
| | |
Variable compensation payable |
| | |
| | |
Benefits in kind |
| | |
| | |
Directors’ fees |
| | |
| | |
Share-based payments |
| | |
| | |
Consulting fees | | | | |||
Total executive compensation |
| | |
| |
No post-employment benefits have been granted to the Chief Executive Officer or other corporate officers.
21.2 Intellectual property agreement signed with the Company’s Chief Executive Officer
The Company’s Chief Executive Officer, who is not an employee, is involved in the Company’s research and development activities. In collaboration with the Company, he has developed inventions for which the Company has submitted patent applications in which he is listed as co-inventor, and other inventions which may give rise to new patent applications in the future and for which he will be listed as co-inventor.
As an inventor, the Chief Executive Officer has certain rights under French intellectual property law. These rights are distinct from the legal rights that usually apply to salaried inventors under French law.
F-49
In order to define a framework under which any intellectual property rights arising from the CEO’s research and development activities would be assigned to the Company, the Company and the CEO entered into an agreement in May 2019, approved by the Board of Directors on May 13, 2019, under which the CEO will be entitled to the following payments for his contributions:
| a) | a first lump-sum cash payment of |
| b) | a second lump-sum cash payment of |
| c) | a royalty of |
The total amount resulting from the combination of the
In the event of a third-party pharmaceutical and/or biotechnology company acquiring
Following signature of the Transfer Agreement, an amount of €
In April 2020, the company amended the intellectual property agreement signed with the company’s CEO to take into account
Since the inception of this agreement, the Company has acquired rights to use patents from the Company’s Chief Executive Officer for a total of €
21.3 Consulting contract with Successful Life
On January 1, 2021, we entered into a service agreement with Successful Life SAS, owned by Jean Mariani, a director of the Company. This agreement, for an initial term of
In addition, on July 7, 2021, we signed a second agreement with Successful Life SAS by which Jean Mariani is to serve as interim Chief Medical officer until September 8, 2021, as approved by the July 7, 2021 Board decision, for a fixed remuneration of €
21.4 Indemnification agreements with the directors of the Company
During the 2021 financial year, following approval of the combined general meeting of May 10, 2021, the Company signed compensation agreements with its directors, ensuring the latter are covered by an insurance policy and compensation in cases of personal liability actions against them in relation to the exercise of their corporate mandate.
F-50
Note 22: Off-balance sheet commitments
22.1 Financial debt commitments
Residual | ||||||
amount on | ||||||
Borrowing |
| Commitments given |
| Nominal |
| 12/31/2023 |
BPI France repayable advance - “BIO 101” project |
| The agreement provides for an annual repayment starting on January 1, 2018 and no later than March 31 of each year until September 30, 2023 corresponding to : |
| |
| |
Kreos 2021 |
| In accordance with the terms of the subprime loan agreements signed with Kreos on September 10, 2018 (see note 12.2.3) and November 19, 2021 (see note 12.2.3), the Company has pledged a security interest in the Company's assets for the benefit of Kreos. The Company has also granted a security interest in the operating business, including part of the Company's patents, to Kreos. |
| N/A |
| N/A |
22.2 Commitments given in respect of the use of industrial property
Agreements on the use of |
| |
industrial property |
| Commitments given |
MACULIA commercialization contract - SATT Lutech Agreements of January 1, 2016 modified by the amendment of December 17, 2020. |
| This contract covers patent families from M1 to M4. The consideration payable by the Company is as follows: firstly, in the year following the first product launch, and in any event no later than 2020, the Company will pay a minimum guaranteed amount of € |
Note 23: Financial risk management and assessment
Biophytis may be exposed to various types of financial risk, including market risk, liquidity risk and credit risk. Biophytis implements simple measures proportionate to its size to minimize the potentially adverse effects of these risks on financial performance.
Biophytis’ policy is not to underwrite financial instruments for speculative purposes.
23.1 Market risk
Interest rate risk
Interest rate risk represents the Company’s exposure to variations in market interest rates.
Changes in interest rates could affect returns on cash and term deposits. Nevertheless, this risk is considered insignificant given the current low yields on term deposits held by the Company.
Foreign exchange risk
The main risks relating to foreign exchange impacts are considered insignificant due to the low level of activity of our foreign subsidiaries.
F-51
At the present stage of its development, the Company has not taken any hedging measures to protect its business against exchange rate fluctuations. On the other hand, the Company cannot rule out the possibility that a significant increase in its business may result in greater exposure to foreign exchange risk. The Company will then consider the use of an appropriate hedging policy to cover these risks.
Equity risk
The Company has signed agreements with Atlas and Kreos, providing for financing through the issue of several installments of convertible bonds, with warrants where applicable. Under these agreements, the Company is exposed to variations in the market price of its own shares.
23.2 Credit risk
Credit risk is associated with deposits with banks and financial institutions.
The Company seeks to minimize its exposure to banks and financial institutions by placing term deposits with first-class financial institutions. The maximum level of credit risk corresponds to the carrying amount of financial assets. As outstanding receivables mainly comprise research tax credits granted by the French government, the Company is not exposed to any significant credit risk.
23.3 Liquidity risk
Since its creation, the Company has financed its business and growth by strengthening its equity through successive capital increases (including its initial public flotation in July 2015), bank loans and bonds, public grants for innovation and pre-financing of CIR receivables.
Significant research and development expenditure has been incurred since the start of the Company’s operations, generating negative cash flow from operating activities to date of €
In addition, the Company has contracted debts, notably in connection with convertible or non-convertible bond financing, presented in note 13.2 and summarized in the table below:
Year ended |
|
|
|
| ||||||
December 31, 2023 | 2024 | 2025 / 2026 | 2027 / 2028 | More than 5 | ||||||
Amounts in K€ |
| Total |
| Less than 1 year |
| between 1 and 3 years |
| between 3 and 5 years |
| years |
Non-convertible bonds issued to Kreos |
| |
| |
| |
| — |
| — |
Repayable advances |
| |
| |
| |
| — |
| — |
Leasing obligations |
| |
| |
| |
| — |
| — |
Convertible bonds issued to Kreos |
| |
| — |
| |
| — |
| — |
Convertible bonds issued to ATLAS | | | — | — | — | |||||
Financial debts related to CIR pre-financing | | | — | — | — | |||||
Accrual interests to pay | | | — | — | — | |||||
Derivative liabilities | | | — | — | — | |||||
Total |
| |
| |
| |
| — |
| — |
The going concern assumption has been adopted by the Board of Directors As of the date of authorization of these financial statements, our available cash and our ORNANE financing line are not projected to be sufficient to support our operating plan for at least the next 12 months. These events and conditions indicate that a material uncertainty exists that may cast significant doubt on the Company’s ability to continue as a going concern and, therefore, the Company may be unable to realize its assets and discharge its liabilities in the normal course of business (see note 3.1).
F-52
The Company will continue to have significant financing needs in the future to support the development of its drug candidates. The precise extent of financing required is difficult to estimate accurately, and will depend in part on factors beyond the Company’s control. Areas of significant uncertainty include, but are not limited to:
| ● | Its ability to conduct successful clinical trials, including the ability to recruit patients for our clinical trials in a timely manner; |
| ● | Changes in the regulatory environment; and |
| ● | Approval of other drugs on the market that could potentially reduce the attractiveness of its drug candidates. |
If the Company were unable to finance its own growth through partnership agreements, it would be dependent on other sources of financing, including raising capital or seeking grants.
F-53