UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR 12(g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report
For the transition period from to
Commission file number:
(Exact Name of Registrant as Specified in Its Charter)
N/A
(Translation of Registrant’s Name into English)
(Jurisdiction of Incorporation or Organization)
(Address of Principal Executive Offices)
(
Email:
(Name, Telephone, Email and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
Title of Each Class |
| Trading Symbol(s) |
| Name of Each Exchange On Which Registered |
The |
Securities registered or to be registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act:
None
(Title of Class)
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report:
As of December 31, 2023, there were
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐ Yes ☒
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. ☐ Yes ☒
Note - Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 from their obligations under those Sections.
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. ☒
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer ☐ | Accelerated filer ☐ | Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ☐
†The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
International Financial Reporting Standards as issued | Other ☐ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. ☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). ☐ Yes
(APPLICABLE ONLY TO ISSUERS INVOLVED IN BANKRUPTCY PROCEEDINGS DURING THE PAST FIVE YEARS)
Indicate by check mark whether the registrant has filed all documents and reports required to be filed by Sections 12, 13 or 15(d) of the Securities Exchange Act of 1934 subsequent to the distribution of securities under a plan confirmed by a court. ☐ Yes ☐ No
TABLE OF CONTENTS
3
INTRODUCTION
Unless otherwise indicated and except where the context otherwise requires, references in this annual report to:
| ● | “Acrotech” are to Acrotech Biopharma L.L.C.; |
| ● | “ANDA” are to abbreviated new drug application; |
| ● | “CASI”, “us”, “our Company”, “the Company”, “our” and “we” are to (I) CASI Pharmaceuticals, Inc., an exempted company with limited liability incorporated under the laws of the Cayman Islands (“CASI Cayman”) and its subsidiaries after the Redomicile Merger, and (ii) CASI Pharmaceuticals, Inc., a Delaware corporation (“CASI Delaware”) and its subsidiaries prior to the Redomicile Merger, in each case as appropriate based on the context; |
| ● | “CASI China” are to CASI Pharmaceuticals (China) Co., Ltd.; |
| ● | “CASI Wuxi” are to CASI Pharmaceuticals (Wuxi) Co., Ltd.; |
| ● | “CASI Biopharmaceuticals” are to CASI Biopharmaceuticals (WUXI) Co., Ltd; |
| ● | “CASI Hong Kong” are to CASI Pharmaceuticals Co., Limited; |
| ● | “China” or the “PRC” are to the People’s Republic of China, excluding, for the purposes of this annual report only, Hong Kong, Macau and Taiwan; |
| ● | “CDE” are to the China Center for Drug Evaluation; |
| ● | “cGMP” are to current Good Manufacturing Practice; |
| ● | “CTA” are to the Clinical Trial Application; |
| ● | “Companies Act” are to the Companies Act (As Revised) of the Cayman Islands; |
| ● | “EMA” are to the European Medicines Agency; |
| ● | “FDA” are to the U.S. Food and Drug Administration; |
| ● | “IRB” are to institutional review board; |
| ● | “Juventas” are to Juventas Cell Therapy Ltd.; |
| ● | “NMPA” are to the PRC National Medical Products Administration; |
| ● | “ordinary shares” are to our ordinary shares, par value US$0.0001 per share; |
| ● | “PAT” are to Precision Autoimmune Therapeutics, a company established under the laws of China, in which the Company holds an equity investment; |
| ● | “Redomicile Merger” are to a merger between CASI Delaware and CASI Cayman for the purpose of CASI Delaware’s re-domiciliation from the State of Delaware of U.S. to the Cayman Islands, where CASI Delaware merged with and into CASI Cayman with CASI Cayman becoming the surviving entity and the successor issuer; |
| ● | “RMB” and “Renminbi” are to the legal currency of China; |
| ● | “US$,” “U.S. dollars,” “$” and “dollars” are to the legal currency of the United States; and |
| ● | “Wuxi LP” are to Wuxi Huicheng Yuanda Investment Partnership (Limited Partnership) (formerly known as Wuxi Jintou Huicun Investment Enterprise), a limited partnership organized under the laws of the People’s Republic of China. |
4
FORWARD-LOOKING STATEMENTS
This annual report contains forward-looking statements that relate to our current expectations and views of future events. These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from those expressed or implied by the forward-looking statements. These statements are made under the “safe harbor” provisions under Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act and of the U.S. Private Securities Litigations Reform Act of 1995.
You can identify some of these forward-looking statements by words or phrases such as “may,” “will,” “expect,” “anticipate,” “aim,” “estimate,” “intend,” “plan,” “believe,” “is/are likely to,” “potential,” “continue” or other similar expressions. These forward-looking statements include, among others, statements regarding the timing of our commercial launch of products, clinical trials, our cash position and future expenses, and our future revenues. We have based these forward-looking statements largely on our current expectations and projections about future events that we believe may affect our financial condition, results of operations, business strategy and financial needs.
Actual results could differ materially from those currently anticipated due to a number of factors, including: the risk that we may be unable to continue as a going concern as a result of our inability to raise sufficient capital for our operational needs; the possibility that we may be delisted from trading on The Nasdaq Capital Market if we fail to satisfy applicable continued listing standards; the volatility in the market price of our ordinary shares; the risk of substantial dilution of existing shareholders in future share issuances; the difficulty of executing our business strategy on a global basis including China; our inability to enter into strategic partnerships for the development, commercialization, manufacturing and distribution of our proposed product candidates or future candidates; legal or regulatory developments in China that adversely affect our ability to operate in China; our lack of experience in manufacturing products and uncertainty about our resources and capabilities to do so on a clinical or commercial scale; risks relating to the commercialization, if any, of our products and proposed products (such as marketing, safety, regulatory, patent, product liability, supply, competition and other risks); our inability to predict when or if our product candidates will be approved for marketing by the U.S. FDA, EMA, NMPA, or other regulatory authorities; our inability to receive approval for renewal of license of our existing products; the risks relating to the need for additional capital and the uncertainty of securing additional funding on favorable terms; the risks associated with our product candidates, and the risks associated with our other early-stage products under development; the risk that result in preclinical and clinical models are not necessarily indicative of clinical results; uncertainties relating to preclinical and clinical trials, including delays to the commencement of such trials; our ability to protect our intellectual property rights; the lack of success in the clinical development of any of our products and our dependence on third parties; the risks related to our dependence on Juventas to partner with us to co-market CNCT19; risks related to the uncertainty in connection with the ongoing arbitration proceedings between us and Juventas with respect to Juventas’ purported termination of certain CNCT19 license agreements; risks related to our dependence on Juventas to ensure the patent protection and prosecution for CNCT19; risks relating to interests of our largest shareholder and our Chairman and CEO that differ from our other shareholders; and risks related to the success of a new manufacturing facility by CASI Wuxi. Such factors, among others, could have a material adverse effect upon our business, results of operations and financial condition.
You should read this annual report and the documents that we refer to in this annual report and have filed as exhibits to this annual report completely and with the understanding that our actual future results may be materially different from what we expect. Other sections (including “Item 3. Key Information—D. Risk Factors”) of this annual report discuss factors which could adversely impact our business and financial performance. Moreover, we operate in an evolving environment. New risk factors emerge from time to time and it is not possible for our management to predict all risk factors, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. We qualify all of our forward-looking statements by these cautionary statements. We undertake no obligation to publicly release the result of any revision of these forward-looking statements to reflect events or circumstances after the date they are made or to reflect the occurrence of unanticipated events. Additional information about the factors and risks that could affect our business, financial condition and results of operations, are contained in our filings with the U.S. Securities and Exchange Commission (“SEC”), which are available at www.sec.gov.
5
You should not rely upon forward-looking statements as predictions of future events. The forward-looking statements made in this annual report relate only to events or information as of the date on which the statements are made in this annual report. Except as required by law, we undertake no obligation to update or revise publicly any forward-looking statements, whether as a result of new information, future events or otherwise, after the date on which the statements are made or to reflect the occurrence of unanticipated events.
6
PART I
ITEM 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS
Not applicable.
ITEM 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not applicable.
ITEM 3. KEY INFORMATION
Risks and Uncertainties Relating to Doing Business in China
We face various risks and uncertainties related to doing business in China. Our business operations are primarily conducted in China, and we are subject to complex and evolving PRC laws and regulations. For a detailed description of risks related to doing business in China, please refer to risks disclosed under “Item 3. Key Information — D. Risk Factors — Risks Relating to Our Business Operations in China.”
PRC government’s significant authority in regulating our operations and its oversight and control over offerings conducted overseas by, and foreign investment in, China-based issuers could significantly limit or completely hinder our ability to offer or continue to offer securities to investors and cause the value of our securities to significantly decline or become worthless. Implementation of industry-wide regulations, including data security or anti-monopoly related regulations, in this nature could result in a material change in our operations and may cause the value of our securities to significantly decline or become worthless. Risks and uncertainties arising from the legal system in China, including risks and uncertainties regarding the enforcement of laws and quickly evolving rules and regulations in China, could result in a material adverse change in our operations and the value of our ordinary shares. For example, the China’s government has made in recent years statements and regulatory actions to regulate certain market players or to improve its supervision of the market in general, such as those related to data security or anti-monopoly concerns. While we currently do not believe such regulatory actions have materially impacted our business operations, our ability to accept foreign investments, or our ability to maintain listing with the Nasdaq Stock Market, there is no assurance that any new rules or regulations promulgated in the future will not impose additional requirements on us. If any such rules or regulations is adopted, we may be subject to more stringent regulatory scrutinizes for our operation and financing efforts, which may in turn result in more compliance costs and expenses to be incurred by us, delay our investment and financing activities, or otherwise impact our ability to conduct our business, accept foreign investments, or list on a U.S. or other foreign exchange. For more details, see “Item 3. Key Information — D. Risk Factors — Risks Relating to Our Business Operations in China — The legal system in China embodies uncertainties which could impose additional requirements and obligations on our business, and PRC laws, rules, and regulations can evolve quickly with little advance notice, which may materially and adversely affect our business, financial condition, and results of operations.”
Risks Relating to Our Auditor
Our auditor, the independent registered public accounting firm that issues the audit report contained in our annual report, as an auditor of companies that are traded publicly in the United States and a firm registered with the PCAOB, is subject to laws in the United States pursuant to which the PCAOB conducts regular inspections to assess its compliance with the applicable professional standards. Our auditor is located in mainland China, a jurisdiction where the PCAOB was historically unable to conduct inspections and investigations completely before 2022. As a result, we and investors in CASI Delaware’s common stock were deprived of the benefits of such PCAOB inspections. Pursuant to the Holding Foreign Companies Accountable Act, or the HFCAA, if the SEC determines that we have filed audit reports issued by a registered public accounting firm that has not been subject to inspections by the PCAOB for two consecutive years, the SEC will prohibit our securities from being traded on a national securities exchange or in the over-the-counter trading market in the United States.
On December 16, 2021, the PCAOB issued a report to notify the SEC of its determination that the PCAOB was unable to inspect or investigate completely registered public accounting firms headquartered in mainland China and Hong Kong and CASI Delaware’s auditor was subject to that determination. In April 2022, the SEC conclusively listed CASI Delaware as a Commission-Identified Issuer under the HFCAA following the filing of its annual report on Form 10-K for the fiscal year ended December 31, 2021.
7
On December 15, 2022, the PCAOB issued a report that vacated its December 16, 2021 determination and removed mainland China and Hong Kong from the list of jurisdictions where it is unable to inspect or investigate completely registered public accounting firms. For this reason, we do not expect we will be identified as a Commission-Identified Issuer under the HFCAA after we file this annual report for the fiscal year ended December 31, 2023.
Each year in the future, the PCAOB will determine whether it can inspect and investigate completely audit firms in mainland China and Hong Kong, among other jurisdictions. If the PCAOB determines in the future that it no longer has full access to inspect and investigate completely accounting firms in mainland China and Hong Kong and we use an accounting firm headquartered in one of these jurisdictions to issue an audit report on our financial statements filed with the SEC, we would be identified as a Commission-Identified Issuer following the filing of the annual report for the relevant fiscal year. In accordance with the HFCAA, our ordinary shares would be prohibited from being traded on a national securities exchange or in the over-the-counter trading market in the United States if we are identified as a Commission-Identified Issuer for two consecutive years in the future. If our ordinary shares are prohibited from trading in the United States, there is no certainty that we will be able to list on a non-U.S. exchange or that a market for our ordinary shares will develop outside of the United States. A prohibition of being able to trade in the United States would substantially impair your ability to sell or purchase our ordinary shares when you wish to do so, and the risk and uncertainty associated with delisting would have a negative impact on the price of such shares. Also, such a prohibition would significantly affect our ability to raise capital on terms acceptable to us, or at all, which would have a material adverse impact on our business, financial condition, and prospects.
For more details, see “Item 3. Key Information — D. Risk Factors — Risks Relating to Our Auditor.”
Cash and Asset Transfer among the Company and its Subsidiaries
We provide funding to our subsidiaries from time to time through capital contributions or loans, subject to satisfaction of applicable government registration and approval requirements. For the year ended December 31, 2023, we made funding of US$1.0 million through capital contributions to CASI Hong Kong, our newly incorporated Hong Kong subsidiary.
Our subsidiaries may pay dividends and make other distributions to us subject to satisfaction of applicable government filing and approval requirements. Such dividend or other distributions may be subject to limitations and certain tax consequences, a discussion on which is set forth below. For the year ended December 31, 2023, no dividends or other distributions were made by our subsidiaries.
We also pay service fees to our PRC subsidiaries pursuant to certain sales support service agreement and research and development support service agreement. For the year ended December 31 2023, we paid service fees of US$1.1 million to CASI China, one of our PRC subsidiaries. Under PRC tax laws and regulations, earning of our subsidiaries under such agreements are subject to a statutory tax rate of 25%.
In the year ended December 31, 2023, no assets other than cash were transferred through our organization.
All cash transfers among us and our subsidiaries have been eliminated in our consolidated statement of cash flows.
The existing PRC foreign exchange regulations may limit our ability to initiate and complete the cash transfers within our group. Approval from SAFE and PBOC may be required where RMB are to be converted into foreign currencies, including U.S. dollars, and approval from SAFE and PBOC or their branches may be required where RMB are to be remitted out of China. Please see “Item 3. Key Information — D. Risk Factors —Risks Relating to Our Business Operations in China — Governmental control of currency conversion and payments of RMB out of China may limit our ability to utilize our cash balances effectively and affect the value of your investment.”
CASI Delaware and CASI Cayman have never declared or paid dividends on its common stock or any other securities and we do not anticipate paying any dividends on our ordinary shares in the foreseeable future. We may rely on dividends from our subsidiaries in China to pay dividend and other distributions on our ordinary shares. PRC regulations may restrict the ability of our PRC subsidiaries to pay dividends to us. In addition to applicable foreign exchange limitations, under the current regulatory regime in China, a PRC company may pay dividends only out of their accumulated profit, if any, determined in accordance with PRC accounting standards and regulations, and is required to set aside as general reserves at least 10% of its after-tax profit, until the cumulative amount of such reserves reaches 50% of its registered capital, prior to any dividend distribution. In addition, a PRC company shall not distribute any profits in a given year until any losses from prior fiscal years have been offset.
8
Permission and Filing Procedures Required from the PRC Authorities with respect to the Operations of Our PRC Subsidiaries and Future offering in the US
As the date hereof, our PRC subsidiaries have obtained the requisite licenses and permits from the PRC government authorities that are material for our business operations, including, among others, the Business License, the Drug Distribution License, the Drug Manufacturing Permit, the Clinical Trial Application with the NMPA, and the notification filing for international collaborative clinical trial or the application for international collaborative scientific research with the China Human Genetic Resources Administrative Office (“HGRAO”). We also work with our business partners which have obtained the requisite license and permits for their business collaboration with us, including among others the Import Drug Registration for product(s) we promote and distribute in China. Given the uncertainties of interpretation and implementation of relevant laws and regulations and the enforcement practice by relevant government authorities, we may be required to obtain additional permissions or approvals for our business operations. For more details, see “Item 3. Key Information — D. Risk Factors — Risks Relating to Our Business Operations in China — The legal system in China embodies uncertainties which could impose additional requirements and obligations on our business, and PRC laws, rules, and regulations can evolve quickly with little advance notice, which may materially and adversely affect our business, financial condition, and results of operations.”
As the date hereof, we and our PRC subsidiaries (i) are not required to obtain permissions from the China Securities Regulatory Commission, or the CSRC, (ii) are not required to go through cybersecurity review by the Cyberspace Administration of China, or the CAC, and (iii) have not been asked to obtain or were denied such permissions by any PRC authority. On July 7, 2022, the CAC published the Guidelines for Data Export Security Assessment (《数据出境安全评估办法》) (the “Guidelines”), which took effect on September 1, 2022. Pursuant to the Guidelines, the data processor who intends to transfer certain important data or large volume of personal information outside of China shall complete a prior CAC-led data outbound transfer security assessment. However, as the Guidelines has just come into effect, there is no specific enforcement guidelines or interpretation for such security assessment, including what constitutes “important data”, or how to define “outbound transfer”, which results in uncertainties whether our business will be subject to such CAC-led assessment. For the data we accessed through or obtained from clinical trials, we have complied with the laws and regulations then-in-effective, and completed the registration with HGRAO, but it is unclear if we will be required to go through the CAC-led or CAC-involved security assessment or the current HGRAO registration procedure will be changed in the future. We will closely monitor and review any regulatory development and comply with any new approval or license requirement when necessary. If (i) we inadvertently conclude that such permissions or approvals are not required, or (ii) applicable laws, regulations, or interpretations change and we are required to obtain such permissions or approvals in the future, we may have to expend significant time and costs to procure them. If we are unable to do so, on commercially reasonable terms, in a timely manner or otherwise, we may become subject to sanctions imposed by the PRC regulatory authorities, which could include fines and penalties, proceedings against us, and other forms of sanctions, and our ability to conduct our business, invest into China as foreign investments or accept foreign investments, or be listed on a U.S. or other overseas exchange may be restricted, and our business, reputation, financial condition, and results of operations may be materially and adversely affected.
On February 17, 2023, the CSRC released the Trial Administrative Measures of the Overseas Securities Offering and Listing by Domestic Companies (《境内企业境外发行证券和上市管理试行办法》) and five ancillary interpretive guidelines (collectively, the “Overseas Listing Trial Measures”), which apply to overseas offerings and listing by PRC-based companies, or domestic companies, of equity shares, depository receipts, corporate bonds convertible to equity shares, and other equity securities, and came into effect on March 31, 2023. According to the Overseas Listing Trial Measures, (1) domestic companies that seek to offer or list securities overseas, both directly and indirectly, should fulfill the filing procedure and report relevant information to the CSRC, and if a overseas-listed PRC-based issuer issues new securities in the same overseas market after the overseas offering and listing, it is also required to file with the CSRC within three business days after the completion of the issuance; if a domestic company fails to complete the filing procedure or conceals any material fact or falsifies any major content in its filing documents, such domestic company may be subject to administrative penalties, such as order to rectify, warnings, fines, and its controlling shareholders, actual controllers, the person directly in charge and other directly liable persons may also be subject to administrative penalties, such as warnings and fines; (2) if a foreign-incorporated issuer meets both of the following conditions, its overseas offering and listing shall be determined as an indirect overseas offering and listing by a domestic company of the PRC: (i) any of the total assets, net assets, revenues or profits of the domestic operating entities of the issuer in the most recent accounting year accounts for more than 50% of the corresponding line items in the issuer’s audited consolidated financial statements for the same period; and (ii) its major operational activities are carried out in China or its main places of business are located in China, or the senior managers in charge of operation and management of the issuer are mostly Chinese citizens or are domiciled in China; and (3) where a domestic company seeks to indirectly offer and list securities in an overseas market
9
(including issuance of new securities after its overseas offering and listing), the issuer shall designate a major domestic operating entity responsible for all filing procedures with the CSRC.
Furthermore, in case any of the following major events occurs after the overseas offering and listing, the issuer is also required to report the relevant information to the CSRC within three business days of the occurrence and the announcement of the relevant events: (1) change of control; (2) the foreign securities regulatory body or the relevant competent authority has taken such measures as investigation and punishment; (3) conversion of listing status or listing board; and (4) voluntary of compulsory termination of listing. Where there is any material change in the major business and operation of the issuer after overseas offering and listing, and such change does not fall within the scope of filing, the issuer shall, within three business days of the occurrence of such change, submit a special report and a legal opinion issued by a domestic law firm to the CSRC to explain the relevant situation.
As substantially all of our operations are currently based in the PRC, our future offerings and major changes shall be subject to the foregoing filing procedures under the Overseas Listing Trial Measures. We cannot assure you that we could meet such requirements, obtain such permit from the relevant government authorities, or complete such filing in a timely manner or at all. Any failure may significantly limit or completely hinder our ability to continue to offer securities to investors and cause the value of such securities to significantly decline or be worthless. In addition, as the Overseas Listing Trial Measures was recently promulgated, there remains substantial uncertainties as to its interpretation and implementation and how it may impact our ability to raise or utilize fund and business operation.
A. | [Reserved] |
B. | Capitalization and Indebtedness |
Not applicable.
C. | Reasons for the Offer and Use of Proceeds |
Not applicable.
D. | Risk Factors |
Risks Relating to our Financial Position and Need for Additional Capital
We have incurred significant operating losses since inception and anticipate that we will continue to incur operating losses for the foreseeable future and may never achieve or maintain profitability.
To date, we have been engaged primarily in research and development activities. In the years ended December 31, 2021, 2022 and 2023, we had EVOMELA® sales totaling US$30.0 million, US$38.0 million and US$33.9 million, respectively.
We have experienced losses in each year since inception. Through December 31, 2023, we had an accumulated deficit of US$660.8 million. We expect that we will seek to raise capital to continue our operations and, although we have been successfully funded to date through the sales of our equity securities, our capital-raising efforts may not produce the funding needed to sustain our operations. If we are unable to obtain additional funding for operations, we may not be able to continue operations as proposed, requiring us to modify our business plan, curtail various aspects of our operations or cease operations. In any such event, investors may lose a portion or all of their investment.
We expect that our ongoing preclinical, clinical, marketing and corporate activities will result in operating losses for the foreseeable future. In addition, to the extent we rely on others to develop and commercialize our products, our ability to achieve profitability will depend upon the success of these other parties. To support our research and development of certain product candidates, we may seek and rely on cooperative agreements from governmental and other organizations as a source of support. If a cooperative agreement were to be reduced to any substantial extent, it may impair our ability to continue our research and development efforts. To become and remain profitable, we must successfully commercialize one or more product candidates with significant market potential. This will require us to be successful in a range of challenging activities, including completing clinical trials of our candidates, developing commercial scale manufacturing processes, obtaining marketing approval, manufacturing, marketing and selling any current and future
10
product candidates for which we may obtain marketing approval, and satisfying any post-marketing requirements. We may never succeed in any or all of these activities and, even if we do, we may never generate sufficient revenue to achieve profitability.
We may not have sufficient funds to acquire new product candidates or pay milestone payments.
Our growth strategy relies on our in-license of new product candidates from third parties. Our pipeline will be dependent upon the availability of suitable acquisition candidates at favorable prices and upon advantageous terms and conditions. Even if such opportunities are present, we may not be able to successfully identify appropriate acquisition candidates. Moreover, other companies, many of which may have substantially greater financial resources, are competing with us for the right to acquire such product candidates.
If a product candidate is identified, the third parties with whom we seek to cooperate may not select us as a potential partner or we may not be able to enter into arrangements on commercially reasonable terms or at all. Furthermore, the negotiation and completion of collaborative and license arrangements could cause significant diversion of management’s time and resources and potential disruption of our ongoing business.
Our ability to make additional payments in the future for CASI Wuxi is subject to uncertainty, be difficult to accomplish or take longer than expected. We have established a cGMP injectable products manufacturing line in a state-owned industrial facility in Wuxi Huishan Economic Development Zone and it may fail to meet regulatory standard, which may increase our losses.
We have established a cGMP injectable products manufacturing line in a state-owned industrial facility in Wuxi Huishan Economic Development Zone. The injectable products manufacturing line may fail for validation or to meet regulatory standards for authority’s inspection prior to the commercial manufacture. Our ability to establish and operate a manufacturing facility in China may be adversely affected by changes in Chinese laws and regulations such as those related to, among other things, taxation, import and export tariffs, environmental regulations, land use rights, intellectual property, employee benefits and other matters. The success of CASI Wuxi also relies on our ability to make additional payments in the future, which is uncertain. Our plan may require us to obtain additional debt or equity financing, resulting in additional debt obligations, increased interest expense or dilution of equity ownership. If we are unable to establish a new manufacturing facility, purchase equipment, hire an adequate number of experienced personnel to support our manufacturing efforts or implement necessary process improvements, we may be unable to produce commercial materials or meet demand, if any should develop, for our product candidates. Any one of the factors cited above, or a combination of them, could result in unanticipated costs, which could materially and adversely affect our business and planned operations and earnings in China. In addition, our investment plan in connection with the operation of CASI Wuxi has been changed and remains subject to changes due to factors beyond our control, and our ability to make additional payments is subject to uncertainty, see “Risks replating Our Business — The success of CASI Wuxi is subject to uncertainty in our business plan and government regulatory actions. ”
The current capital and credit market conditions may adversely affect our access to capital, cost of capital, and ability to execute our business plan as scheduled.
Access to capital markets is critical to our ability to operate. Traditionally, we have funded our operations by raising capital in the equity markets. Declines and uncertainties in these markets over the past few years have restricted raising new capital in amounts sufficient to conduct our current operations and have affected our ability to continue to expand or fund additional development efforts. We require significant capital for research and development for our product candidates, clinical trials, and marketing activities. Our inability to access the capital markets on favorable terms because of our low stock price, or upon our delisting from the Nasdaq Capital Market if we fails to satisfy a listing requirement, could affect our ability to execute our business plan as scheduled. Moreover, we rely and intend to rely on third parties, including our clinical research organizations, third party manufacturers, and certain other important vendors and consultants. As a result of the current volatile and unpredictable global economic situation, there may be a disruption or delay in the performance of our third-party contractors and suppliers. If such third parties are unable to adequately satisfy their contractual commitments to us in a timely manner, our business could be adversely affected.
We have limited revenue streams and we are uncertain whether additional funding will be available for our future capital needs and commitments. If we cannot raise additional funding, or access the capital markets, we may be unable to complete the development and commercialization of our products and product candidates.
We will require substantial funds in addition to our existing working capital to develop and commercialize our products and product candidates and to otherwise meet our business objectives. We have never generated sufficient revenue during any period since
11
our inception to cover our expenses and have spent, and expect to continue to spend, substantial funds to continue our clinical development programs and commercialization of our products and product candidates. Any one of the following factors, among others, could cause us to require additional funds or otherwise cause our cash requirements in the future to increase materially:
| ● | progress of our clinical trials or correlative studies; |
| ● | results of clinical trials; |
| ● | changes in or terminations of our relationships with strategic partners; |
| ● | changes in the focus, direction, or costs of our research and development programs; |
| ● | competitive and technological advances; |
| ● | establishment and expansion of marketing and sales capabilities; |
| ● | manufacturing; |
| ● | the regulatory approval process; or |
| ● | product launch and distribution. |
On December 31, 2023, we had cash and cash equivalents of US$17.1 million, and short term investments of US$12.0 million. We may continue to seek additional capital through public or private financing or collaborative agreements in 2024 and beyond. Our operations require significant amounts of cash. We may be required to seek additional capital for the future growth and development of our business. We can give no assurance as to the availability of such additional capital or, if available, whether it would be on terms acceptable to us. If we are not successful in obtaining sufficient capital because we are unable to access the capital markets on favorable terms, it could reduce our research and development efforts and materially adversely affect our future growth, results of operations and financial results. There can be no assurance that we would be able to obtain any required financing on a timely basis or at all.
Risks Relating to Our Business
If we or our partners are ultimately unable to obtain regulatory approval for our drug candidates, our business will be substantially harmed.
The time required to obtain approval by the FDA and the NMPA is unpredictable and typically takes many years following the commencement of preclinical studies and clinical trials and depends on numerous factors, including the substantial discretion of the regulatory authorities.
Our drug candidates could be delayed or fail to receive regulatory approval for many reasons, including:
| ● | failure to begin or complete clinical trials due to disagreements with regulatory authorities; |
| ● | delays in subject enrollment or interruptions in clinical trial supplies or investigational product; |
| ● | failure to demonstrate that a drug candidate is safe and effective or that a biologic candidate is safe, pure, and potent for its proposed indication; |
| ● | failure of clinical trial results to meet the level of statistical significance required for approval; |
| ● | reporting or data integrity issues related to our clinical trials; |
| ● | disagreement with our interpretation of data from preclinical studies or clinical trials; |
| ● | changes in approval policies or regulations that render our preclinical and clinical data insufficient for approval or require us to amend our clinical trial protocols; |
| ● | regulatory requests for additional analyses, reports, data, nonclinical studies and clinical trials, or questions regarding interpretations of data and results and the emergence of new information regarding our drug or biologic candidates or other products; |
| ● | regulatory orders new side effect warnings for existing therapies; |
| ● | failure to satisfy regulatory conditions regarding endpoints, patient population, available therapies and other requirements for our clinical trials in order to support marketing approval on an accelerated basis or at all; |
| ● | our failure to conduct a clinical trial in accordance with regulatory requirements or our clinical trial protocols; and |
| ● | clinical sites, investigators or other participants in our clinical trials deviating from a trial protocol, failing to conduct the trial in accordance with regulatory requirements, or dropping out of a trial. |
12
The FDA, NMPA or a comparable regulatory authority may require more information, including additional preclinical, chemical, manufacturing and controls, and/or clinical data, to support approval, which may delay or prevent approval and our commercialization plans, or we may decide to abandon the development program.
Changes in regulatory requirements and guidance may also occur, and we may need to amend clinical trial protocols submitted to applicable regulatory authorities to reflect these changes. Amendments may require us to resubmit clinical trial protocols to IRBs or ethics committees for re-examination, which may impact the costs, timing or successful completion of a clinical trial.
If we or our partners experience delays in the completion of, or the termination of, a clinical trial of any of our product candidates, the commercial prospects of that candidate may be harmed, and our ability to generate product sales revenues from any of those candidates may be delayed. In addition, any delays in completing our clinical trials will increase our costs, slow down our candidate development and approval process, and jeopardize our ability to commence product sales and generate related revenues for that candidate. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
Our success in commercializing these drugs and biologics may be inhibited by a number of factors, including:
| ● | our inability to maintain good collaboration relationship with our partners; |
| ● | our inability to obtain/maintain regulatory approvals; |
| ● | our inability to recruit, train and retain adequate numbers of effective sales and marketing personnel; |
| ● | the inability of sales personnel to obtain access to or educate physicians on the benefits of our products; |
| ● | our lack of experience in manufacturing drugs for commercial sales; |
| ● | our or our partners’ inability to secure widespread acceptance of our products from physicians, healthcare payors, patients and the medical community; |
| ● | our ability to win tenders through the collective tender processes in which we decide to participate; |
| ● | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines; |
| ● | unforeseen costs and expenses associated with creating an independent sales and marketing organization; |
| ● | generic and biosimilar competition; and |
| ● | regulatory exclusivities or patents held by competitors that may inhibit our products’ entry to the market. |
If we decide to rely on third parties to manufacture, sell, market and distribute our products and product candidates, we may not be successful in entering into arrangements with such third parties or may be unable to do so on terms that are favorable to us. In addition, our product revenues and our profitability, if any, may be lower if we rely on third parties for these functions than if we were to market, sell and distribute any products that we develop ourselves. We likely will have little control over such third parties, and any of them may fail to devote the necessary resources and attention to sell and market our products effectively. If we do not establish sales, marketing and distribution capabilities successfully, either on our own or in collaboration with third parties, we will not be successful in commercializing our product candidates, which would adversely affect our business and financial condition.
We are substantially dependent on the commercial success of EVOMELA®, FOLOTYN® and CNCT19. Our approved medical products may fail to achieve and maintain the degree of market acceptance and utilization by physicians, patients, third-party payors, and others in the medical community necessary for commercial success.
| ● | EVOMELA® |
The success of our business is substantially dependent on our ability to successfully commercialize EVOMELA®. On December 3, 2018, we received the NMPA approval for importation, marketing and sales in China for EVOMELA®, and on August 12, 2019, we announced the commercial launch of EVOMELA® in China. We will continue to spend our time, resources and efforts on the commercialization of EVOMELA® in China.
13
Reimbursement and hospital listing may be the most critical market access factors for our commercialization success in China. The National Reimbursement Drug List (the “NRDL”) is updating on an annual basis via a negotiation mechanism. Although participating in the NRDL pricing negotiation is voluntarily, it usually results in significant price discount. The Company has no intention to list EVOMELA® in the NRDL any time before a direct competitor’s compound commercially launch, therefore, our market will be limited given only a small portion of the Chinese population would be able to afford EVOMELA® through self-pay.
The government owned hospitals in China usually restrict the drug use outside the hospital formulary. Therefore, being listed in the hospital formulary is critical. In order to list in the hospital formulary, the Company must participate the provincial level tendering process. Winning the tendering does not guarantee the hospital listing. If we were unable to quickly add EVOMELA® to hospitals’ formulary, doctors and patients will have limited access to EVOMELA® through hospital pharmacies, and the demand for EVOMELA® and the revenues from EVOMELA® will be materially and adversely affected. On the other hand, patients are able to purchase EVOMELA® with a prescription from a physician from pharmacies if the product is not available in the hospital, however, the hospitals do not encourage such activities.
The introduction of a generic melphalan for injection product in China represents a significant business risk for EVOMELA®, potentially eroding its market share in the region. Generic medications, often priced more competitively than their branded counterparts, can quickly attract cost-conscious customers, institutions and healthcare providers, leading to a decrease in sales for the branded product. Furthermore, the situation is compounded by the news that another local company is in the process of registering its injectable melphalan, with a potential market launch in 2025. This impending competition not only threatens to further dilute EVOMELA®'s market presence but also intensifies the pressure on pricing and marketing strategies.
Additionally, we currently rely on a single source for our supply of EVOMELA® which has high risk of supply chain disruption. Early in the COVID-19 pandemic we experienced a disruption to our supply chain for EVOMELA®, and there can be no assurance that restrictions will not be imposed again. If suppliers refuse or are unable to provide products for any reason (including the occurrence of an event like the COVID-19 pandemic that makes delivery impractical), we would have to work with Acrotech, our current supplier, to negotiate an agreement with a substitute supplier, which would likely interrupt further manufacturing of EVOMELA®, cause delays or increase our costs.
| ● | FOLOTYN® |
In July 2023, we entered into a tripartite assignment agreement with Mundipharma International Corporation Limited (“MICL”), Mundipharma Medical Company (“MMCo”) and Acrotech Biopharma Inc. (“Acrotech Inc.”), pursuant to which, MICL’s rights and obligations under that certain License, Development and Commercialization Agreement (as amended and restated) dated as of May 29, 2013 for the commercialization of FOLOTYN® (Pralatrexate) in China, with certain terms of such rights and obligations amended as agreed to by the parties, is assigned to us. We announced the first patient received FOLOTYN® treatment in China on February 15, 2024. We will continue to spend our time, resources and efforts on the commercialization of FOLOTYN® in China.
Securing reimbursement and hospital listings are key to our commercial success in China. The NRDL is updated annually through a negotiation process. While joining the NRDL pricing negotiations is optional, it typically leads to substantial price reductions. Our strategy does not include listing FOLOTYN® on the NRDL until a direct competitor's product is launched commercially. Consequently, our market reach will be restricted as only a limited segment of the Chinese population will be able to afford FOLOTYN® out of pocket.
In China, drugs not listed in a hospital's formulary are often restricted, making formulary inclusion essential. To achieve this, a company must successfully navigate the provincial tender process, though winning does not guarantee formulary listing. If FOLOTYN® is not promptly added to hospital formularies, its availability and demand, as well as revenue, could suffer significantly. While patients can technically obtain FOLOTYN® with a prescription at external pharmacies, hospitals discourage this practice.
We also currently rely on a single source for our supply of FOLOTYN® which has high risk of supply chain disruption. If suppliers refuse or are unable to provide products for any reason (including the occurrence of an event like the COVID-19 pandemic that makes delivery impractical), we would have to work with Acrotech, our current supplier, to negotiate an agreement with a substitute supplier, which would likely interrupt further manufacturing of FOLOTYN®, cause delays or increase our costs.
14
| ● | CNCT19 (Inaticabtagene Autoleucel) |
On November 8, 2023, our partner Juventas, a China-based domestic company engaged in cell therapy, announced market approval for CNCT19 in China from NMPA. The Company acquired in June 2019 worldwide license and commercialization rights to CNCT19 from Juventas pursuant to certain Exclusive License Agreement, which provides that Juventas shall continue to be responsible for the clinical development and regulatory submission and maintenance of CNCT19 regulatory applications and we are responsible for the launch and commercial activities of CNCT19 under the direction of a joint steering committee. In September 2020, the Company and Juventas entered a Supplementary Agreement (together with the Exclusive License Agreement, collectively, “CNCT19 Agreements”), pursuant to which Juventas and the Company will jointly market CNCT19, including, but not limited to, establishing medical teams, developing medical strategies, conducting post-marketing clinical studies, establishing Standardized Cell Therapy Centers, establishing and training providers with respect to cell therapy, testing for cell therapy, and monitoring quality controls (cell collection and transfusion, etc.), and patient management (adverse reactions treatment, patients’ follow-up visits, and establishment of a database). The Company also will reimburse Juventas for a portion of Juventas’ marketing expenses as reviewed and approved by a joint commercial committee to be constituted. The Company will continue to be responsible for recruiting and establishing a sales team to commercialize CNCT19.
The affordability and accessibility of CNCT19, the lowest-priced CAR-T therapy in China, are significant barriers to its widespread adoption in a predominantly self-pay healthcare market. Despite its competitive pricing, the cost of RMB 999,000 may still be prohibitively high for many patients, limited further by the variability in health insurance coverage and the economic disparities across different regions of China. These factors collectively restrict the pool of eligible patients, as high out-of-pocket expenses, insufficient insurance reimbursement, and lack of financial support mechanisms make access to this potentially life-saving treatment challenging. Addressing these issues through strategic pricing, enhanced insurance partnerships, and the establishment of financial aid programs is crucial for improving the accessibility and uptake of CNCT19 among the target patient population. Another risk factor for CNCT19 is its exclusion from the NRDL in the foreseeable future, which poses a considerable barrier to its accessibility. The NRDL in China plays a crucial role in determining which drugs are more affordable to the general population through insurance coverage. Without inclusion in the NRDL, CNCT19's out-of-pocket cost for patients remains high, severely limiting its accessibility to only those who can afford it without financial assistance from public health insurance. This exclusion not only restricts the patient base able to benefit from this advanced therapy but also places CNCT19 at a competitive disadvantage compared to treatments that are covered by the NRDL. Consequently, strategies to mitigate this limitation might include advocating for policy changes, exploring alternative financing models, or implementing patient assistance programs to enhance access despite the lack of NRDL coverage.
The slow acceptance and awareness among doctors of new therapies like CNCT19 present significant risks to its market penetration and sales. Physicians’ hesitation, driven by the need for convincing efficacy, safety data, and familiarity with the treatment, can delay its integration into standard care practices. This challenge is compounded by the necessity for specialized training to administer such advanced therapies. Overcoming these barriers requires targeted educational efforts, dissemination of clinical trial results, and collaboration with key medical opinion leaders to foster a quicker adoption process and enhance CNCT19’s market presence.
The side effects of cell therapy like CNCT19 can elevate the risk of financial compensation claims, impacting the company financially and reputationally. Adverse reactions from patients may lead to increased legal and insurance costs, as well as the need for financial reserves for compensation, posing significant financial risks.
Juventas' limited manufacturing capacity restricts its ability to meet market demand, potentially capping revenue growth and market share. This limitation could result in supply shortages and loss of opportunities to competitors.
Juventas serving as the sole supplier and business partner for CNCT19 introduces a significant risk to the Company’s business strategy. Should Juventas refuse to carry out the Exclusive License Agreement and the Supplementary Agreement, it could severely disrupt the Company’s supply chain, impacting the availability of CNCT19 and, by extension, the Company’s ability to meet market demand. This dependency on a single entity for critical aspects of the business makes us vulnerable to operational and strategic risks, potentially leading to revenue loss, market share decline, and damage to stakeholder relationships.
15
We are involved in arbitration proceedings against Juventas in relation to Juventas’ purported termination of the CNCT19 Agreements.
On March 2, 2024, CASI received a notice from Juventas, which purported to terminate the CNCT19 Agreements based on certain alleged non-performance by CASI of its obligations under the CNCT19 Agreements in relation to the preparation for the commercialization of CNCT19. CASI responded to Juventas’ purported termination notice, noting that Juventas was not entitled to unilaterally terminate the CNCT19 Agreements and further demanding that Juventas cease any conduct that may constitute further breach of the CNCT19 Agreements and execute a written undertaking regarding compliance with the CNCT19 Agreements by March 13, 2024. Juventas did not comply with CASI’s demands. On March 20, 2024, CASI submitted a Notice of Arbitration at the Hong Kong International Arbitration Centre (“HKIAC”) against Juventas pursuant to the CNCT19 Agreements’ dispute resolution clauses, claiming that Juventas’ purported termination was invalid and that Juventas breached the CNCT19 Agreements and seeking, among other things, damages and injunctive reliefs. Together with the Notice of Arbitration, CASI also submitted an application for the appointment of an emergency arbitrator, seeking emergency injunctive reliefs. On the same day, Juventas also submitted a Notice of Arbitration at the HKIAC against CASI, alleging, among other things, that the CNCT19 Agreements were validly terminated and that CASI breached the CNCT19 Agreements. The HKIAC has appointed an emergency arbitrator in accordance with CASI’s application. The arbitration proceedings are ongoing. See “—” “Item 8 — Financial Information — A. Consolidated Statements and Other Financial Information – Legal Proceedings”.
The arbitration proceedings are in their early stages and the Company cannot predict right now the outcome of either of these proceedings. If we do not prevail in either of these proceedings completely or in part, or fail to reach a favorable settlement with Juventas, our plan with respect to the commercialization of CNCT 19 may be delayed or otherwise adversely impacted, which will in turn result in adverse impacts on our results of operations, financial condition and prospects
The success of CASI Wuxi is subject to uncertainty in our business plan and government regulatory actions.
In December 2018, together with our partner Wuxi LP, we established CASI Wuxi, to build and operate a manufacturing facility in the Wuxi Huishan Economic Development Zone in Jiangsu Province, China. We initially held 80% of the equity interests in CASI Wuxi and intended to invest, over time, US$80 million in CASI Wuxi. We have paid US$31 million in cash and transferred selected ANDAs. Wuxi LP held 20% of the equity interest in CASI Wuxi through its investment paid in RMB equivalent of US$20 million in cash in 2019.
In November 2019, CASI Wuxi entered into a lease agreement for the right to use state-owned land in Wuxi for the construction of a manufacturing facility. Pursuant to this agreement, CASI Wuxi intended to invest in land use rights and property, plant and equipment of RMB1 billion by August 2022. Construction of the manufacturing facility began in the fourth quarter of 2020.
Since our business focus has been shifted from ANDAs to the hematology-oncology therapeutic area, a substantial investment in GMP manufacturing facilities does not fit the current business focus. Therefore, in December 2022, we returned the land use right to the local Wuxi government for an amount of RMB 44.42 million, equivalent to the original payment for the land use right. Meanwhile, all construction in progress on the land was disposed. The Company recorded a total disposal loss amounted to US$2.2 million.
Since we failed to meet the land development milestone, the local land administration authority requested CASI Wuxi to pay a land vacancy fee equivalent to 20% of the price for the land use rights according to the PRC Land Administration Law. We paid such fee in the amount of RMB 8.88 million to the local land administration authority in December 2022. Additionally, the Company received a government grant for the land development in April 2020 and November 2021, respectively, in the total amount of RMB 18.9 million. We are currently in negotiation with the local Wuxi government on the further treatment of the grant. The Wuxi government may require the Company to fully or partially return the grant and the Company may incur further losses.
CASI Wuxi is now operating a cGMP injectable products manufacturing line in a state-owned industrial facility in Wuxi Huishan Economic Development Zone. The injectable products manufacturing line may fail for validation or to meet regulatory standards for authority’s inspection prior to the commercial manufacture.
In December 2023, the Company entered into a series of agreements, including a capital reduction agreement, a long term borrowing agreement, and four guarantee agreements, with Wuxi LP, CASI China and CASI Wuxi, pursuant to which, (i) CASI Wuxi will reduce its registered capital and return to Wuxi LP the investment principal made by Wuxi LP in CASI Wuxi in the amount of
16
RMB134.2 million (equivalent to its original investment of US$20 million, the “Investment Principal”), together with certain investment return in the amount of RMB26.2 million to be paid in instalments, and Wuxi LP shall cease to be a shareholder of CASI Wuxi, (ii) Wuxi LP shall reinvest the Investment Principal into a three-year long term borrowing to CASI Wuxi (the “Long term borrowing”), which shall have a non-compounding annual interest rate of 4.05% and can, from the beginning date of the Long term borrowing term till the six month anniversary after the maturity of the Long term borrowing, be partially or fully converted into the equity interest of any subsidiaries of the Company at the conversion date fair value, solely at Wuxi LP’s discretion, and (iii) each of the Company and CASI China will provide irrevocable joint and several liability guarantees on the above-mentioned payment obligations. The term of the Long term borrowing will start on December 25, 2023 and end on December 31, 2026.
The provision allowing Wuxi LP to request immediate repayment of the long term borrowing if CASI Wuxi fails to generate revenue for certain threshold for each of the years during the term, which represents a significant liquidity risk for CASI. If Wuxi LP exercises this option, CASI would be required to allocate a substantial portion of its cash reserves or secure additional financing, which could severely impact its operational cash flow. Converting the investment principal into a three-year long term borrowing introduces uncertainties related to future equity dilution and financial stability.
The existence of counterfeit pharmaceutical products in pharmaceutical markets may compromise our brand and reputation and have a material adverse effect on our business, operations and prospects.
Counterfeit products, including counterfeit pharmaceutical products, are a significant problem, particularly in China. Counterfeit pharmaceuticals are products sold or used for research under the same or similar names, or similar mechanism of action or product class, but which are sold without proper licenses or approvals. Such products may be used for indications or purposes that are not recommended or approved or for which there is no data or inadequate data with regard to safety or efficacy. Such products divert sales from genuine products, often are of lower cost, often are of lower quality (having different ingredients or formulations, for example), and have the potential to damage the reputation for quality and effectiveness of the genuine product. If counterfeit pharmaceuticals illegally sold or used for research result in adverse events or side effects to consumers, we may be associated with any negative publicity resulting from such incidents. Consumers may buy counterfeit pharmaceuticals that are in direct competition with our pharmaceuticals, which could have an adverse impact on our revenues, business and results of operations. In addition, the use of counterfeit products could be used in non-clinical or clinical studies, or could otherwise produce undesirable side effects or adverse events that may be attributed to our products as well, which could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in the delay or denial of regulatory approval by the FDA or other regulatory authorities and potential product liability claims. With respect to China, although the government has combat the counterfeit pharmaceuticals, there is not yet an effective counterfeit pharmaceutical regulation control and enforcement system in China. As a result, we may not be able to prevent third parties from selling or purporting to sell our products in China. The proliferation of counterfeit pharmaceuticals has grown in recent years and may continue to grow in the future. The existence of and any increase in the sales and production of counterfeit pharmaceuticals, or the technological capabilities of counterfeiters, could negatively impact our revenues, brand reputation, business and results of operations.
We face significant competition from other biotechnology and pharmaceutical companies and our business will suffer if we fail to compete effectively.
If competitors were to develop superior drug candidates, our products could be rendered noncompetitive or obsolete, resulting in a material adverse effect to our business. Developments in the biotechnology and pharmaceutical industries are expected to continue at a rapid pace. Success depends upon achieving and maintaining a competitive position in the development of products and technologies. Competition from other biotechnology and pharmaceutical companies can be intense. Many competitors have substantially greater research and development capabilities, marketing, financial and managerial resources and experience in the industry.
The availability of our competitors’ products could limit the demand, and the price we are able to charge, for product candidates we develop. We will not achieve our business plan if the acceptance of our products is inhibited by price competition or reimbursement issues or if physicians switch to other new drug products or choose to reserve our product candidates for use in limited circumstances. The inability to compete with existing or subsequently introduced drug products would have a material adverse impact on our business, financial condition and prospects.
17
We may need new collaborative partners to further develop and commercialize products, and if we enter into such arrangements, we may lose control over the development and approval process.
We may develop and commercialize our product candidates both with and without corporate alliances and partners. Nonetheless, we intend to explore opportunities for new corporate alliances and partners to help us develop, commercialize and market our product candidates. We may grant to our partners certain rights to commercialize any products developed under these agreements, and we may rely on our partners to conduct research and development efforts and clinical trials on, obtain regulatory approvals for, and manufacture and market any products licensed to them. Each individual partner will seek to control the amount and timing of resources devoted to these activities generally. We anticipate obtaining revenues from our strategic partners under such relationships in the form of research and development payments and payments upon achievement of certain milestones. Since we generally expect to obtain a royalty for sales or a percentage of profits of products licensed to third parties, our revenues may be less than if we retained all commercialization rights and marketed products directly. In addition, there is a risk that our corporate partners will pursue alternative technologies or develop competitive products as a means for developing treatments for the diseases targeted by our product candidates.
We may not be successful in establishing any collaborative arrangements. Even if we do establish such collaborations, we may not successfully commercialize any products under or derive any revenues from these arrangements. There is a risk that we will be unable to manage simultaneous collaborations, if any, successfully. With respect to existing and potential future strategic alliances and collaborative arrangements, we will depend on the expertise and dedication of sufficient resources by these outside parties to develop, manufacture, or market products. If a strategic alliance or collaborative partner fails to develop or commercialize a product to which it has rights, we may not recognize any revenues on that particular product.
We must show the safety and efficacy of our product candidates through clinical trials, the results of which are uncertain.
Before obtaining regulatory approvals for the commercial sale of our products, we must demonstrate, through preclinical studies (animal testing) and clinical trials (human testing), that our proposed products are safe and effective for use in each target indication. Testing of our product candidates will be required, and failure can occur at any stage of testing. Clinical trials may not demonstrate sufficient safety and efficacy to obtain the required regulatory approvals or result in marketable products. The failure to adequately demonstrate the safety and efficacy of a product under development could delay or prevent regulatory approval of the potential product.
Clinical trials for the product candidates we are developing may be delayed by many factors, including that potential patients for testing are limited in number. The failure of any clinical trials to meet applicable regulatory standards could cause such trials to be delayed or terminated, which could further delay the commercialization of any of our product candidates. Newly emerging safety risks observed in animal or human studies also can result in delays of ongoing or proposed clinical trials. Any such delays will increase our product development costs. If such delays are significant, they could negatively affect our financial results and the commercial prospects for our products.
Compliance with ongoing post-marketing obligations for our approved products may uncover new safety information that could give rise to a product recall, updated warnings, or other regulatory actions that could have an adverse impact on our business.
After the FDA approves a drug or biologic for marketing, the product’s sponsor must comply with several post-marketing obligations that continue until the product is discontinued. These post-marketing obligations include the reporting of adverse events to the agency within specified timeframes, the submission of product-specific annual reports that include changes in the distribution, manufacturing, and labeling information, and notification when a drug product is found to have significant deviations from its approved manufacturing specifications (among others). Our ongoing compliance with these types of mandatory reporting requirements could result in additional requests for information from the FDA and, depending on the scope of a potential product issue that the FDA may decide to pursue, potentially also result in a request from the agency to conduct a product recall or to strengthen warnings and/or revise other label information about the product. The FDA may also require or request the withdrawal of the product from the market. Any of these post-marketing regulatory actions could materially affect our sales and, therefore, have the potential to adversely affect our business, financial condition, results of operations and cash flows.
18
Undesirable adverse events caused by our medicines and drug candidates could interrupt, delay or halt clinical trials, delay or prevent regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following any regulatory approval.
Undesirable adverse events ("AEs") caused by our medicines and drug candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval, or could result in limitations or withdrawal following approvals. If the conduct or results of our trials or patient experience following approval reveal a high and unacceptable severity or prevalence of AEs, our trials could be suspended or terminated and regulatory authorities could order us to cease further development of, or deny approval of, our drug candidates or require us to cease commercialization following approval.
As is typical in the development of pharmaceutical products, drug-related AEs and serious AEs ("SAEs") have been reported in our clinical trials. Some of these events have led to patient deaths. Drug-related AEs or SAEs could affect patient recruitment or the ability of enrolled subjects to complete the trial and could result in product liability claims. Any of these occurrences may harm our reputation, business, financial condition and prospects significantly. In our periodic and current reports filed with the SEC and our press releases and scientific and medical presentations released from time to time we disclose clinical results for our drug candidates, including the occurrence of AEs and SAEs.
Potential products may subject us to product liability for which insurance may not be available or claims may exceed coverage.
The use of our potential products in clinical trials and the marketing of any pharmaceutical products may expose us to product liability claims. We have obtained a level of liability insurance coverage that we believe is adequate in scope and coverage for our current stage of development. However, our present insurance coverage may not be adequate to protect us from liabilities we might incur. In addition, our existing coverage will not be adequate as we further develop products and, in the future, adequate insurance coverage and indemnification by collaborative partners may not be available in sufficient amounts or at a reasonable cost. If a product liability claim or series of claims are brought against us for uninsured liabilities, or in excess of our insurance coverage, the payment of such liabilities could have a negative effect on our business and financial condition.
If we are unable to obtain both adequate coverage and adequate reimbursement from third-party payers for our products before the competitor’s product launch, our revenues and prospects for profitability will suffer.
Successful commercialization of our products is highly dependent on the extent to which coverage and reimbursement is, and will be, available from third-party payers, including governmental payers and private health insurers. Patients may not be capable of paying for our products themselves and may rely on third-party payers to pay for, or subsidize, the costs of their medications, among other medical costs. If third-party payers do not provide coverage or reimbursement for our products, our revenues and prospects for profitability will suffer. In addition, even if third-party payers provide some coverage or reimbursement for our products, the availability of such coverage or reimbursement for prescription drugs under private health insurance and managed care plans often varies based on the type of contract or plan purchased.
Cybersecurity incidents could impair our ability to conduct business effectively.
Cybersecurity incidents against us or against a third party that has authorized access to our data or networks, failure of our disaster recovery systems, or consequential employee error, could have an adverse effect on our ability to communicate or conduct business, negatively impacting our operations and financial condition. This adverse effect can become particularly acute if those events affect our electronic data processing, transmission, storage, and retrieval systems, or impact the availability, integrity, or confidentiality of our data.
We depend heavily upon IT systems to perform necessary business functions. Our computer systems, networks, and data, like those of other companies, could be subject to cyberattacks and unauthorized access, use, alteration, or destruction. If one or more of these events occurs, it could potentially jeopardize the confidential, proprietary, and other information processed, stored in, and transmitted through our computer systems and networks. Such an attack could cause interruptions or malfunctions in our operations, which could result in financial losses, litigation, regulatory penalties, reputational damage, and increased costs associated with mitigation of damages and remediation. Third parties with which we do business may also be sources of cybersecurity or other technological risk.
19
Policies of extended periods of remote working, whether by us or third parties with which we do business with, could strain technology resources, introduce operational risks and otherwise heighten the risks described above.
Our business depends substantially on the continuing efforts of our senior management, key employees and qualified personnel, and our business operations may be adversely and negatively impacted if we lose their services.
Our future success depends substantially on the continued efforts of our senior management team and key employees. Our employees play key roles in the areas of product development, marketing, sales, and general and administrative functions. Competition for qualified staff or other key employees in the biopharmaceutical industry in China is intense, particularly for individuals with multinational experience. If one or more of our members of senior management or key employees are unable or unwilling to continue their services with us, we might not be able to replace them easily, at an acceptable cost or in a timely manner, if at all.
Many of the companies with which we compete for experienced personnel have greater resources than we have and some of these companies may offer more lucrative compensation packages. If any of our key personnel joins a competitor or forms a competing company, we may lose customers, know-how and key professionals and staff members. Even if we enter into employment agreements and non-compete agreements with our employees, certain provisions under these agreements may be deemed invalid or unenforceable under US and PRC laws. Our continued ability to compete effectively depends on our ability to attract new employees and to retain and motivate our existing employees. Since the demand and competition for talent is intense in our industry, we may need to offer higher compensation and other benefits in order to attract and retain key personnel in the future, which could increase our compensation expenses. If we do not succeed in attracting additional highly skilled personnel or retaining or motivating our existing personnel, we may be unable to grow effectively.
Certain our directors and officers may have business interests that may conflict with our interests and those of our shareholders.
Dr. Wei-Wu He, our Chairman and CEO, is the founder and managing partner of Emerging Technology Partners, LLC (“ETP”), a life science focused venture fund, and its related investing entities. Dr. Wei-Wu He, together with ETP, holds 13.7% of the Company’s outstanding shares. To the extent we fail to appropriately deal with any such conflicts of interests, it could negatively impact our reputation and ability to raise additional funds and the willingness of counterparties to do business with us, all of which could have an adverse effect on our business, financial condition, results of operations, and cash flows.
We or the third parties upon whom we rely on may be adversely affected by epidemic outbreaks, earthquakes, tornadoes, hurricanes or other natural disasters, and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
We have the principal executive office in Beijing, China, through which substantially all of our operations are conducted, and an office in Rockville, Maryland, United States. We also rely and intend to rely on third parties, including our clinical research organizations, third party manufacturers, and certain other important vendors and consultants in China and in United States. The occurrence of one or more epidemic outbreaks such as Ebola, Zika, SARS-CoV, COVID-19, pandemic influenza or measles, natural disasters, such as tornadoes, hurricanes, fires, floods, hail storms and earthquakes, unusual weather conditions, terrorist attacks or disruptive political events in regions where we operate our business could adversely affect the operations of the third parties we rely on and our business, results of operations, financial condition and our prospects.
If an epidemic outbreak, natural disaster, power outage or other event occurred that prevented us or the third parties we rely on from using all or a significant portion of our or their offices, damaged critical infrastructure or disrupted operations, it may be difficult, or in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plan we have in place currently are limited and are unlikely to prove adequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which could have a material adverse effect on our business.
20
Risks Relating to Our Reliance on Third Parties
Independent clinical investigators and contract research organizations that we engage to conduct our clinical trials may not devote sufficient time or attention to our clinical trials or be able to repeat their past success.
We depend on independent clinical investigators and contract research organizations (“CROs”) to assist in the conduct of our clinical trials under their agreements with us. The investigators are not our employees, and we cannot control the amount or timing of resources that they devote to our programs. If independent investigators fail to devote sufficient time and resources to our drug development programs, or if their performance is substandard or deviates from regulatory requirements, good clinical practice (“GCPs”), or the protocol, it could delay the approval of our FDA applications and our introduction of new products. The CROs we contract with to assist with the execution of our clinical trials play a significant role in the conduct of the trials and the subsequent collection and analysis of data. Failure of the CROs to meet their obligations, as well as any failure of us or our collaborators to effectively monitor and audit our CROs and clinical trials, could adversely affect clinical development of our products.
We have no currently approved manufacturing capacity and rely on limited suppliers for some of our products.
While we have established a cGMP injectable products manufacturing line in the Wuxi Huishan Economic Development Zone in Jiangsu Province, China, we do not currently have the capacity to manufacture products and we have limited experience in these activities. The manufacturing processes for the pipeline assets which we are developing have not yet been tested at commercial levels, and it may not be possible to manufacture these drug products in a cost-effective manner. If we elect to perform these functions, we will be required to either develop these capacities, or contract with others to perform some or all of these tasks. We may be dependent to a significant extent on corporate partners, licensees, or other entities for manufacturing of our products. If we engage directly in manufacturing, we will require substantial additional funds and personnel and will be required to comply with extensive regulations. We may be unable to develop or contract for these capacities when required to do so in connection with our business.
We depend on our third-party manufacturers to perform their obligations effectively and on a timely basis. These third parties may not meet their obligations and any such non-performance may delay clinical development or submission of products for regulatory approval, or otherwise impair our competitive position. Any significant problem experienced by one of our suppliers could result in a delay or interruption in the supply of materials to us until such supplier resolves the problem or an alternative source of supply is located. Any delay or interruption would likely lead to a delay or interruption of manufacturing operations, which could negatively affect our operations. Although we have identified alternative suppliers for our product candidates, we have not entered into contractual or other arrangements with them. If we needed to use an alternate supplier for any product, we would experience delays while we negotiated an agreement with them for the manufacture of such product. In addition, we may be unable to negotiate manufacturing terms with a new supplier as favorable as the terms we have with our current suppliers.
Problems with any manufacturing processes, including deviations from cGMP, could result in product defects, which could require us to delay shipment of products or recall products previously shipped, as well as regulatory action. In addition, any prolonged interruption in the operations of the manufacturing facilities of one of our sole-source suppliers could result in the cancellation of shipments. A number of factors could cause interruptions, including equipment malfunctions or failures, or damage to a facility due to natural disasters or otherwise. We expect our future manufacturing processes to be highly complex and subject to a lengthy regulatory approval process. Alternative qualified production capacity may not be available on a timely basis or at all. Difficulties or delays in our manufacturing could increase our costs and damage our reputation.
The manufacture of pharmaceutical products can be an expensive, time consuming, and complex process. Manufacturers often encounter difficulties in scaling-up production of new products, including quality control and assurance and shortages of personnel. Delays in formulation and scale-up to commercial quantities could result in additional expense and delays in our clinical trials, regulatory submissions, and commercialization.
Failure of manufacturing facilities producing our product candidates to maintain regulatory approval could delay or otherwise hinder our ability to market our product candidates. Any manufacturer of our product candidates will be subject to applicable cGMP prescribed by the NMPA or other rules and regulations prescribed by the FDA and other foreign regulatory authorities. We and any of our collaborators may be unable to enter into or maintain relationships either domestically or abroad with manufacturers whose facilities and procedures comply or will continue to comply with cGMP and who are able to produce our products in accordance with applicable regulatory standards. Failure by a manufacturer of our products to comply with cGMP could result in significant time delays or our
21
inability to obtain marketing approval or, should we have market approval, for such approval to continue. Changes in our manufacturers could require new product testing and facility compliance inspections. In the U.S., failure to comply with cGMP or other applicable legal requirements can lead to federal seizure of violated products, injunctive actions brought by the federal government, inability to export product, and potential criminal and civil liability on the part of a company and its officers and employees.
If we fail to maintain an effective distribution channel for our medicines, our business and sales could be adversely affected.
We rely on third-party distributors to distribute our approved medicines. Our ability to maintain and grow our business will depend on our ability to maintain an effective distribution channel that ensures the timely delivery of our medicines. However, we have relatively limited control over our distributors, who may fail to distribute our drugs in the manner we contemplate. If price controls or other factors substantially reduce the margins our distributors can obtain through the resale of our medicines to hospitals, medical institutions and sub-distributors, they may terminate their relationship with us. While we believe alternative distributors are readily available, there is a risk that, if the distribution of our medicines is interrupted, our sales volumes and business prospects could be adversely affected.
Risks Related to Extensive Government Regulation
All material aspects of the research, development, manufacturing and commercialization of pharmaceutical products are heavily regulated, and we may face difficulties in complying with or be unable to comply with such regulations, which could have a material adverse effect on our business.
All jurisdictions in which we conduct or intend to conduct our pharmaceutical-industry activities regulate these activities in great depth and detail. We are currently focusing our activities in the major markets of the United States, China and Europe. These geopolitical areas all strictly regulate the pharmaceutical industry, and in doing so they employ broadly similar regulatory strategies, including regulation of product development and approval, manufacturing, and marketing, sales and distribution of products. However, there are differences in the regulatory regimes - some minor, some significant - that make for a more complex and costly regulatory compliance burden for a company like ours that plans to operate in each of these regions.
The process of obtaining regulatory approvals and compliance with appropriate laws and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable requirements at any time during the product development process, approval process, or after approval, may subject us to administrative or judicial sanctions. These sanctions could include a regulator’s refusal to approve pending applications, withdrawal of an approval, license revocation, a clinical hold, voluntary or mandatory product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. The failure to comply with these regulations could have a material adverse effect on our business.
We are subject to certain U.S. healthcare laws, regulation and enforcement; our failure to comply with those laws could have a material adverse effect on our results of operations and financial condition.
We are subject to certain U.S. healthcare laws and regulations and enforcement by the federal government and the states in which we conduct our business. The laws that may affect our ability to operate include, without limitation:
| ● | the federal Anti-Kickback Statute (“AKS”), which governs our business activities, including our marketing practices, educational programs, pricing policies, and relationships with healthcare providers or other entities. The AKS prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. Remuneration has been broadly interpreted to include anything of value, including for example, gifts, discounts, coupons, the furnishing of supplies or equipment, credit arrangements, payments of cash, waivers of payments, ownership interests and providing anything at less than its fair market value. This statute has been broadly interpreted to apply to manufacturer arrangements with prescribers, purchasers and formulary managers, among others; |
22
| ● | the Federal Food, Drug, and Cosmetic Act, and its regulations which prohibit, among other things, the introduction or delivery for introduction into interstate commerce of any food, drug, device, biologic, or cosmetic that is adulterated or misbranded; |
| ● | the Public Health Service Act, which prohibits, among other things, the introduction into interstate commerce of biological product unless a biologics license is in effect for that product; |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent, or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government; |
| ● | federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| ● | Health Insurance Portability and Accountability Act (as amended by the Health Information Technology and Clinical Health Act) and its implementing regulations, which impose certain requirements relating to the privacy, security and transmission of individually identifiable health information; |
| ● | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third-party payer, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; |
| ● | federal and state consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
| ● | federal and state government price reporting laws that require us to calculate and report complex pricing metrics to government programs, where such reported prices may be used in the calculation of reimbursement and/or discounts on our marketed drugs (participation in these programs and compliance with the applicable requirements may subject us to potentially significant discounts on our products, increased infrastructure costs, and could potentially affect our ability to offer certain marketplace discounts); and |
| ● | federal and state financial transparency laws, which generally require certain types of expenditures in the U.S. to be tracked and reported (compliance with such requirements may require investment in infrastructure to ensure that tracking is performed properly, and some of these laws result in the public disclosure of various types of payments and relationships with healthcare providers and healthcare entities, which could potentially have a negative effect on our business and/or increase enforcement scrutiny of our activities). |
In addition, certain marketing practices, including off-label promotion, may also violate certain federal and state healthcare fraud and abuse laws, FDA rules and regulations, as well as false claims laws. If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us, we, or our officers or employees, may be subject to penalties, including administrative civil and criminal penalties, damages, fines, withdrawal of regulatory approval, the curtailment or restructuring of our operations, the exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to sell our products or operate our business and also adversely affect our financial results.
We are subject to certain anti-bribery laws, regulation and enforcement; our failure to comply with those laws could have a material adverse effect on our results of operations and financial condition.
If we fail to comply with applicable anti-bribery laws, our reputation may be harmed and we could be subject to penalties and significant expenses that have a material adverse effect on our business, financial condition and results of operations. We are obligated to adhere to anti-bribery laws in China that prohibit companies and intermediaries from offering payments to government officials to gain or retain business or any other undue advantage. Furthermore, even though our primary business operations are in China, we are also subject to the Foreign Corrupt Practices Act ("FCPA"). The FCPA prohibits us from making inappropriate payments to non-U.S. officials to obtain or retain business. While we have established policies and processes to guarantee compliance with anti-bribery legislation, we cannot ensure that our intermediaries, employees, or agents will abstain from engaging in bribery activities. Failure to comply with anti-bribery regulations could harm our operations and result in severe criminal and civil penalties, including fines, imprisonment, loss of export licenses, suspension of our ability to do business with the government, denial of government reimbursement
23
for our products, and/or exclusion from participating in government healthcare programs. We may need to make additional changes or enhancements to our procedures, policies, and controls, as well as potentially enforce disciplinary measures and personnel changes, any of which may have a substantial negative effect on our business, financial position, operating performance, and liquidity. Furthermore, allegations of violating these laws may adversely impact our organization.
We may not be able to obtain regulatory approval for our drug candidates.
All material aspects of the research, development and commercialization of pharmaceutical products are heavily regulated. Our pharmaceutical-industry activities will be carried out in jurisdictions that impose strict regulations on such activities, particularly in the major markets of China and the United States. These regulatory frameworks cover various aspects of the industry, including product development, approval, manufacturing, marketing, sales, and distribution. Although these jurisdictions employ similar regulatory strategies, there are differences in their regulatory regimes. As a result, compliance with regulations is a more complex and expensive process for our company, which intends to operate in these regions.
Obtaining regulatory approvals and ensuring compliance with laws and regulations is a time-consuming and expensive process. If an applicant fails to comply with applicable requirements at any point during product development or the approval process, or even after approval has been granted, they may face administrative or judicial sanctions. These sanctions may take the form of refusal to approve pending applications, license revocation, clinical holds, product recalls (voluntary or mandatory), product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, requirements for restitution, disgorgement, or other civil or criminal penalties. Noncompliance with regulations could significantly harm our business.
The regulatory approval processes of the FDA, NMPA, EMA and other comparable regulatory authorities are time-consuming and may evolve over time, and if we are ultimately unable to obtain regulatory approval for our drug candidates, our business will be substantially harmed.
The process of obtaining regulatory approval from authorities such as the FDA and NMPA is highly uncertain and dependent on various factors, including the discretion of the regulatory bodies. Typically, such approvals take several years to acquire after the initiation of pre-clinical studies and clinical trials. However, they are typically granted within 12 to 18 months after the completion of regulatory submission of the NDA. Furthermore, regulatory approval policies, regulations, and the type and amount of clinical data required for approval may change during the development of a drug candidate and may differ among jurisdictions.
At present, we have secured IND approvals from the NMPA for two of our drug candidates, namely BI-1206 and CB-5339. Nevertheless, we cannot assure that we will receive regulatory approval for our other drug candidates that are already in existence or any drug candidates we may discover, acquire or license and seek to develop in the future. There are numerous reasons why our drug candidates may fail to obtain regulatory approval from the FDA, NMPA, EMA or other comparable regulatory bodies, including but not limited to:
| ● | Disagreement with the design or implementation of our clinical trials |
| ● | Failure to demonstrate the safety, efficacy, and potency of our drug candidates for their proposed indication |
| ● | Failure of our clinical trial results to meet the required level of statistical significance for approval |
| ● | Failure of our clinical trial process to pass relevant Good Clinical Practice (GCP) inspections |
| ● | Inability to demonstrate that the clinical benefits of a drug candidate outweigh its safety risks |
| ● | Disagreement with our interpretation of data from pre-clinical studies or clinical trials |
| ● | Insufficient data collected from clinical trials to support submissions for regulatory approval |
| ● | Failure of our drug candidates to pass cGMP inspections during regulatory review or production |
24
| ● | Failure of our clinical sites to pass audits by regulatory authorities, potentially invalidating our research data |
| ● | Findings of deficiencies related to our manufacturing processes or facilities of third-party manufacturers with whom we contract for clinical and commercial supplies |
| ● | Changes in approval policies or regulations that render our pre-clinical and clinical data insufficient for approval |
| ● | Inability of our clinical trial process to keep up with scientific or technological advancements required by approval policies or regulations |
Obtaining regulatory approval from the FDA, NMPA, EMA or other comparable regulatory authorities may require additional information, such as more pre-clinical or clinical data, which could delay or prevent approval and our commercialization plans. Even if we were to obtain approval, regulatory authorities may approve our drug candidates for fewer or more limited indications than we requested. They may also grant approval subject to the performance of costly post-marketing clinical trials or approve a drug candidate for an indication that is not desirable for its successful commercialization. Any of these scenarios could have a significant negative impact on the commercial potential of our drug candidates.
Our medicines and any future approved drug candidates will be subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our medicines and drug candidates.
Our medicines and any additional drug candidates that are approved will be subject to ongoing regulatory requirements for manufacturing, labeling, packaging, storage, advertising, promotion, sampling, record-keeping, conduct of post-marketing studies, and submission of safety, efficacy, and other post-marketing information, including requirements in China, both federal and state requirements in the US and requirements in other regions. As such, we and our collaborators will be subject to ongoing review and periodic inspections to assess compliance with applicable post-approval regulations. Additionally, to the extent we want to make certain changes to the approved medicines, product labeling, or manufacturing processes, we will need to submit new applications or supplements to regulatory authorities for approval.
Manufacturers and manufacturers’ facilities are required to comply with extensive FDA, NMPA, EMA or comparable regulatory authority requirements, ensuring that quality control and manufacturing procedures conform to GMP regulations. As such, we and our contract manufacturers are and will be subject to continual review and inspections to assess compliance with GMP and adherence to commitments made in any NDA or BLA, other marketing application, and previous responses to any inspection observations. Accordingly, we and others with whom we work must continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production and quality control. The failure to comply with these requirements could have a material adverse effect on our business.
The regulatory approvals for our medicine and any approvals that we receive for our drug candidates are and may be subject to limitations on the approved indicated uses for which the medicine may be marketed or to the conditions of approval, which could adversely affect the drug’s commercial potential or contain requirements for potentially costly post-marketing testing and surveillance to monitor the safety and efficacy of the drug or drug candidate. The FDA, NMPA, EMA or comparable regulatory authorities may also require a Risk Evaluation and Mitigation Strategy (“REMS”) program or comparable program as a condition of approval of our drug candidates or following approval. In addition, if the FDA, NMPA, EMA or a comparable regulatory authority approves our drug candidates, we will have to comply with requirements including, for example, submissions of safety and other post-marketing information and reports, establishment registration, as well as continued compliance with GMP and GCP for any clinical trials that we conduct post-approval.
The FDA, NMPA, EMA or comparable regulatory authorities may seek to impose a consent decree or withdraw marketing approval if compliance with regulatory requirements is not maintained or if problems occur after the drug reaches the market. Later discovery of previously unknown problems with our medicines or drug candidates or with our drug’s manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of
25
post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| ● | restrictions on the marketing or manufacturing of our medicines, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
| ● | fines, untitled or warning letters, or holds on clinical trials; |
| ● | refusal by the FDA, NMPA, EMA or comparable regulatory authorities to approve pending applications or supplements to approved applications filed by us or suspension or revocation of license approvals or withdrawal of approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of our medicines and drug candidates; and |
| ● | injunctions or the imposition of civil or criminal penalties. |
The FDA, NMPA, EMA and other regulatory authorities strictly regulate the marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for their approved indications and for use in accordance with the provisions of the approved label. The FDA, NMPA, EMA and other regulatory authorities actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. The policies of the FDA, NMPA, EMA and of other regulatory authorities may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our drug candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad, particularly in China, where the regulatory environment is constantly evolving. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any regulatory approval that we may have obtained and we may not achieve or sustain profitability.
In addition, if we obtain accelerated approval or conditional approval of any of our drug candidates, as we have done with the initial approval of EVOMELA® in China, we have been required to conduct a confirmatory study (also called Post Marketing Study, or the “PMS”) to verify the predicted clinical benefit and may also be required to conduct post-marketing safety studies. Other comparable regulatory authorities may have similar requirements. The results from the confirmatory study may not support the clinical benefit, which could result in the approval being withdrawn. While operating under accelerated approval, we will be subject to certain restrictions that we would not be subject to upon receiving regular approval.
Risks Relating to Our Intellectual Property
We depend on patents and other proprietary rights, some of which are uncertain. If we are unable to protect our intellectual property rights our business and competitive position would be harmed.
We have in-licensed rights to a variety of product candidates. Our success, competitive position and future revenues with respect to these product candidates will depend, in part, on our ability to protect our intellectual property. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary rights are covered by valid and enforceable patents or are effectively maintained as trade secrets. We attempt to protect our proprietary position by maintaining trade secrets and by filing U.S. and foreign patent applications related to our in-licensed technology, inventions and improvements that are important to the development of our business. Our failure to do so may adversely affect our business and competitive position.
The patent positions of biotechnology and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. We may not be able to protect our intellectual property rights throughout the world. No consistent policy regarding the breadth of claims allowed in pharmaceutical patents has emerged to date in the U.S. or in many jurisdictions outside of the U.S. Changes in either the patent laws or interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property and therefore we cannot predict with certainty whether any patent applications that we have filed or that we may file in the future will be approved, will cover our products or product candidates or that any resulting patents will be enforced. In addition, third parties may challenge, seek to invalidate, limit the scope of or circumvent any of our patents, once they are issued. Thus, any patents that we own or license from third parties or CASI Wuxi or development partners may not provide any protection against competitors. Any patent applications that we have filed or that we may file in the future, or those we may license from third parties or CASI Wuxi or development partners, may not result in patents being issued. Moreover, disputes
26
between our licensing or joint development partners and us may arise over license scope, or ownership, assignment, inventorship and/or rights to use or commercialize patent or other proprietary rights, which may adversely impact our ability to obtain and protect our proprietary technology and products. Also, patent rights may not provide us with adequate proprietary protection or competitive advantages against competitors with similar technologies or products.
Third parties may initiate legal proceedings alleging infringement of intellectual property rights, the outcome of which would be uncertain and could harm our business.
Third parties may assert patent or other intellectual property infringement claims against us or our licensors arising from the manufacture, use and sale of our current or future product candidates in China or in any other jurisdictions we ultimately commercialize in. The validity of our current or future patents or patent applications or those of our licensors may be challenged in litigation, interference or derivation proceedings, opposition, post grant review, inter parts review, or other similar enforcement and revocation proceedings provoked by third parties or brought by us, as may be necessary to determine the validity of our patents or patent applications or those of our licensors. Our patents could be found invalid, unenforceable, or their scope significantly reduced.
An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
Although China recently adopted changes to its patent law to include patent term extension and an early resolution mechanism for pharmaceutical patent disputes starting in June 2021, key provisions of the law remain unclear and/or subject to implementing regulations. The absence of effective regulatory exclusivity for pharmaceutical products in China could further increase the risk of early generic or biosimilar competition with our medicines in China.
In China, laws on patent term extension, patent linkage, and data exclusivity (referred to as regulatory data protection) are still developing. Therefore, a lower-cost generic drug can emerge onto the market much more quickly. Chinese regulators have set forth a framework for integrating patent linkage and data exclusivity into the Chinese regulatory regime, as well as for establishing a pilot program for patent term extension. The Economic and Trade Agreement Between the United States of America and the People’s Republic of China announced in January 2020 (the “Trade Agreement”) also provides for a mechanism for early resolution of patent disputes and patent term extension systems. To be implemented, this framework will require adoption of legislation and regulations. In October 2020, China adopted amendments to its Patent Law (the “Amended PRC Patent Law”), which became effective on June 1, 2021. The Amended PRC Patent Law contains both patent term extension and a mechanism for early resolution of patent disputes, which may be comparable to patent linkage in the United States. However, the provisions for patent term extension and an early resolution mechanism are unclear and/or remain subject to the approval of implementing regulations that are still in draft form or have not yet been proposed, leading to uncertainty about their scope and implementation.
Until the relevant implementing regulations for patent term extension and an early resolution mechanism in the Amended PRC Patent Law are implemented, and until data exclusivity is adopted and implemented, we may be subject to earlier generic competition.
Risks Relating to Our Ordinary Shares
The market price of our ordinary shares may be highly volatile or may decline regardless of our operating performance.
The volatile price of our securities makes it difficult for investors to predict the value of their investments, to sell shares at a profit at any given time, or to plan purchases and sales in advance. CASI Delaware’s common stock price and our ordinary share price fluctuated from year-to-year and quarter-to-quarter. We expect that the trading price of our ordinary shares is likely to be highly volatile in response to a variety of factors that are beyond our control, such as:
| ● | our ability to maintain regulatory approval for EVOMELA® and FOLOTYN® , and obtain regulatory approval for our other product candidates; |
27
| ● | issues in importation, marketing and sales of EVOMELA® and FOLOTYN®; |
| ● | the outcome of our disputes with Juventas with respect to joint commercialization of CNCT19; |
| ● | the success of CASI Wuxi to build and operate a manufacturing facility in China, and the capability of CASI Wuxi to generate revenue for certain threshold for each of the years during the term of the long term borrowing; |
| ● | the clinical development of CNCT19, BI-1206, CB-5339 and CID-103; |
| ● | publicity regarding actual or potential clinical test results relating to products under development by our competitors or us; |
| ● | initiating, completing or analyzing, or a delay or failure in initiating, completing or analyzing, preclinical or clinical trials or animal trials or the design or results of these trials for products in development; |
| ● | the entry into, or termination of, key agreements, including key commercial partner agreements; |
| ● | the initiation of, material developments in, or conclusion of litigation to enforce or defend any of our intellectual property rights or defend against the intellectual property rights of others; |
| ● | achievement or rejection of regulatory approvals for products in development by our competitors or us; |
| ● | announcements of technological innovations or new commercial products by our competitors or us; |
| ● | developments concerning our collaborations and supply chain; |
| ● | regulatory developments in the United States and foreign countries; |
| ● | economic or other crises and other external factors; |
| ● | the loss of key employees; |
| ● | period-to-period fluctuations in our revenues and other results of operations; |
| ● | changes in financial estimates by securities analysts; or |
| ● | publicity or activity involving possible future acquisitions, strategic investments, partnerships or alliances. |
We will not be able to control many of these factors, and we believe that period-to-period comparisons of our financial results will not necessarily be indicative of our future performance. The valuations of many biotechnology companies without consistent product revenues and earnings are extraordinarily high based on conventional valuation standards, such as price to earnings and price to sales ratios. These trading prices and valuations may not be sustained. In the future, our operating results in a particular period may not meet the expectations of any securities analysts whose attention we may attract, or those of our investors, which may result in a decline in the market price of our ordinary shares. Any negative change in the public’s perception of the prospects of biotechnology companies could depress the price of our securities regardless of our results of operations. These factors may materially and adversely affect the market price of our ordinary shares.
Our largest shareholders, including our directors and officers and investment funds with which they are associated, hold a significant amount of our outstanding ordinary shares and, if they acted together, could influence our management and affairs.
A small number of our shareholders, including our directors and officers and investment funds with which they are associated, hold a significant amount of our outstanding ordinary shares. In addition, our officers and directors and investment funds with which they are associated could determine to make additional purchases of ordinary shares, to the extent permitted by law. In the future, our officers and directors also could be issued ordinary shares as determined by the compensation committee and the board of directors in connection with current or future equity incentive plans.
These shareholders, if they acted together, could significantly influence the vote on all matters requiring approval by our shareholders, including the appointment of directors and the approval of mergers or other business combination transactions. We cannot assure you that our largest shareholder will not seek to influence our business and affairs in a manner that is contrary to the interests of our other shareholders. In addition, the significant concentration of ownership in our ordinary shares may adversely affect the trading price for our ordinary shares because investors often perceive disadvantages in owning stock in companies with significant shareholders.
28
We are a foreign private issuer within the meaning of the rules under the Exchange Act, and as such we are exempt from certain provisions applicable to U.S. domestic public companies.
Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the securities rules and regulations in the United States that are applicable to U.S. domestic issuers, including:
| ● | the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q or current reports on Form 8-K; |
| ● | the sections of the Exchange Act regulating the solicitation of proxies, consents, or authorizations in respect of a security registered under the Exchange Act; |
| ● | the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and |
| ● | the selective disclosure rules by issuers of material nonpublic information under Regulation FD. |
We are required to file an annual report on Form 20-F within four months of the end of each fiscal year. In addition, we intend to publish our results on a quarterly basis as press releases, distributed pursuant to the rules and regulations of the Nasdaq Stock Market. Press releases relating to financial results and material events will also be furnished to the SEC on Form 6-K. However, the information we are required to file with or furnish to the SEC will be less extensive and less timely compared to that required to be filed with the SEC by U.S. domestic issuers. As a result, you may not be afforded the same protections or information that would be made available to you were you investing in a U.S. domestic issuer.
As an exempted company incorporated in the Cayman Islands, we have adopted certain home country practices in relation to corporate governance matters that differ significantly from the Nasdaq’s corporate governance requirements; these practices may afford less protection to shareholders than they would enjoy if we complied fully with the Nasdaq’s corporate governance requirements.
As a Cayman Islands exempted company listed on the Nasdaq Capital Market, we are subject to the Nasdaq listing standards. However, the Nasdaq Stock Market Rules permit a foreign private issuer like us to follow the corporate governance practices of its home country.
Our shareholders may be afforded less protection than they would otherwise enjoy under the Nasdaq listing standards applicable to U.S. domestic issuers given our reliance on the home country practice exception, when and if we decide to rely on it.
Risks Relating to Our Auditor
The PCAOB had historically been unable to inspect our auditor in relation to their audit work performed for our financial statements and the inability of the PCAOB to conduct inspections of our auditor in the past has deprived our investors with the benefits of such inspections.
Our auditor, the independent registered public accounting firm that issued the audit report contained in our annual report and incorporated herein by reference, as an auditor of companies that are traded publicly in the United States and a firm registered with the PCAOB, is subject to laws in the United States pursuant to which the PCAOB conducts regular inspections to assess its compliance with the applicable professional standards. Our auditor is located in mainland China, a jurisdiction where the PCAOB was historically unable to conduct inspections and investigations completely before 2022. As a result, we and investors in CASI Delaware’s common stock were deprived of the benefits of such PCAOB inspections. The inability of the PCAOB to conduct inspections of auditors in China in the past has made it more difficult to evaluate the effectiveness of our independent registered public accounting firm’s audit procedures or quality control procedures as compared to auditors outside of China that are subject to the PCAOB inspections. On December 15, 2022, the PCAOB issued a report that vacated its December 16, 2021 determination and removed mainland China and Hong Kong from the list of jurisdictions where it is unable to inspect or investigate completely registered public accounting firms. However, if the PCAOB determines in the future that it no longer has full access to inspect and investigate completely accounting firms in mainland China and Hong Kong, and we use an accounting firm headquartered in one of these jurisdictions to issue an audit report on our financial statements filed with the SEC, we and investors in our ordinary shares would be deprived of the benefits of such PCAOB inspections again, which
29
could cause investors and potential investors in our ordinary shares to lose confidence in our audit procedures and reported financial information and the quality of our financial statements.
Our ordinary shares will be prohibited from trading in the United States under the Holding Foreign Companies Accountable Act, or the HFCAA, in the future if the PCAOB is unable to inspect or completely investigate auditors located in China. The delisting of our ordinary shares, or the threat of such ordinary shares being delisted, may materially and adversely affect the value of your investment.
Pursuant to the HFCAA, if the SEC determines that we have filed audit reports issued by a registered public accounting firm that has not been subject to inspections by the PCAOB for two consecutive years beginning in 2021 or any year thereafter, the SEC will prohibit our securities from being traded on a national securities exchange or in the over-the-counter trading market in the United States.
On December 16, 2021, the PCAOB issued a report to notify the SEC of its determination that the PCAOB was unable to inspect or investigate completely registered public accounting firms headquartered in mainland China and Hong Kong and our auditor was subject to that determination. In April 2022, the SEC conclusively listed CASI Delaware as a Commission-Identified Issuer under the HFCAA following the filing of its annual report on Form 10-K for the fiscal year ended December 31, 2021.
On December 15, 2022, the PCAOB removed mainland China and Hong Kong from the list of jurisdictions where it is unable to inspect or investigate completely registered public accounting firms. For this reason, we do not expect to be identified as a Commission-Identified Issuer under the HFCAA after we file our annual report on Form 20-F for the fiscal year ended December 31, 2023.
Each year, the PCAOB will determine whether it can inspect and investigate completely audit firms in mainland China and Hong Kong, among other jurisdictions. If the PCAOB determines in the future that it no longer has full access to inspect and investigate completely accounting firms in mainland China and Hong Kong and we use an accounting firm headquartered in one of these jurisdictions to issue an audit report on our financial statements filed with the SEC, we would be identified as a Commission-Identified Issuer following the filing of the annual report for the relevant fiscal year. In accordance with the HFCAA, our ordinary shares would be prohibited from being traded on a national securities exchange or in the over-the-counter trading market in the United States if we are identified as a Commission-Identified Issuer for two consecutive years in the future. If our ordinary shares are prohibited from trading in the United States, there is no certainty that we will be able to list on a non-U.S. exchange or that a market for our securities will develop outside of the United States. A prohibition of being able to trade in the United States would substantially impair your ability to sell or purchase our ordinary shares when you wish to do so, and the risk and uncertainty associated with delisting would have a negative impact on the price of such securities. Also, such a prohibition would significantly affect our ability to raise capital on terms acceptable to us, or at all, which would have a material adverse impact on our business, financial condition, and prospects.
Risks Relating to Our Business Operations in China
We conduct substantially all of our operations in China. Changes in international trade and economic policy by the U.S. and Chinese governments could have a material adverse effect on our business and operations.
We have substantial operations and conduct business in China, and we plan to continue to expand these operations. Therefore, we are subject to risks related to operating in China, which include complex laws or regulatory requirements or unexpected changes to those laws or requirements; other laws and regulatory requirements to which our business activities are subject, such as the Foreign Corrupt Practices Act; changes in the political or economic condition of a specific country or region; fluctuations in the value of foreign currency versus the U.S. dollar; our ability to deploy funds in an efficient manner; tariffs, trade protection measures, import or export licensing requirements, trade embargoes, and sanctions (including those administered by the Office of Foreign Assets Control of the U.S. Department of the Treasury), and other trade barriers; difficulties in attracting and retaining qualified personnel; and cultural differences in the conduct of business. There is currently significant uncertainty about the future relationship between the U.S. and various other countries, including China, with respect to trade policies, treaties, government regulations and tariffs.
Any of the foregoing could harm our business and we cannot anticipate all of the ways in which the current political climate could adversely impact our business.
30
The legal system in China embodies uncertainties which could impose additional requirements and obligations on our business, and PRC laws, rules, and regulations can evolve quickly with little advance notice, which may materially and adversely affect our business, financial condition, and results of operations.
The rules and regulations in China can change quickly with little advance notice
We conduct substantially all of our business through our PRC subsidiaries. Our operations in China are governed by PRC laws and regulations. PRC laws, rules, and regulations evolve rapidly with little advance notice, and the interpretations of laws, regulations, and rules may contain uncertainties. These uncertainties could limit the legal protections available to us. In addition, we cannot predict the effect of future developments in the PRC legal system, including the promulgation of new laws, changes to existing laws or the interpretation or enforcement thereof, or the preemption of local regulations by national laws. Such unpredictability towards our contractual, property (including intellectual property) and procedural rights could adversely affect our business and impede our ability to continue our operations, which may in turn cause the value of our securities to significantly decline or be worthless. From time to time, we may have to resort to judicial and administrative proceedings to enforce our legal rights. Any administrative and court proceedings in China may be protracted, resulting in substantial costs and diversion of resources and management attention. However, since the authorities in China have significant discretion in interpreting and implementing statutory and contractual terms, it may be more difficult to predict the outcome of a judicial or administrative proceeding in China than in other legal systems. Furthermore, the PRC legal system is based, in part, on government policies and internal rules, some of which are not published in a timely manner, or at all, but which may have retroactive effect. As a result, we may not always be aware of any potential violation of these policies and rules.
We may be required to obtain additional permissions or approvals for our business operations and securities offerings
As of the date hereof, our PRC subsidiaries have obtained the requisite licenses and permits from the PRC government authorities that are material for our business operations, including, among others, the Business License, the Drug Distribution License, the Clinical Trial Application with NMPA, and the notification filing for international collaborative clinical trial or the application for international collaborative scientific research with the HGRAO. We also work with our business partners which have obtained the requisite license and permits for their business collaboration with us, including among others the Import Drug Registration for product(s) we promote and distribute in China. Given the uncertainties of interpretation and implementation of relevant laws and regulations and the enforcement practice by relevant government authorities, we may be required to obtain additional licenses, permits, or approvals in the future, failure to obtain which may hinder our ability to carry out our business plan or continue our business operations.
As of the date hereof, we and our PRC subsidiaries (i) are not required to obtain permissions from the CSRC, (ii) are not required to go through cybersecurity review by the CAC, and (iii) have not been asked to obtain or were denied such permissions by any PRC authority. On July 7, 2022, the CAC published the Guidelines for Data Export Security Assessment (《数据出境安全评估办法》) (the “Guidelines”), which took effect on September 1, 2022. Pursuant to the Guidelines, the data processor who intends to transfer certain important data or large volume of personal information outside of China shall complete a prior CAC-led data outbound transfer security assessment. However, as the Guidelines has just come into effect, there is no specific enforcement guidelines or interpretation for such security assessment, including what constitutes “important data,” or how to define “outbound transfer,” which results in uncertainties whether our business will be subject to such CAC-led assessment. For the data we accessed through or obtained from clinical trials, we have complied with the laws and regulations then-in-effect, and completed the registration with HGRAO, but it is unclear if we will be required to go through the CAC-led or CAC-involved security assessment or the current HGRAO registration procedure will be changed in the future. We will closely monitor and review any regulatory development and comply with any new approval or license requirement when necessary. If (i) we inadvertently conclude that such permissions or approvals are not required, or (ii) applicable laws, regulations, or interpretations change and we are required to obtain such permissions or approvals in the future, we may have to expend significant time and costs to procure them. If we are unable to do so, on commercially reasonable terms, in a timely manner or otherwise, we may become subject to sanctions imposed by the PRC regulatory authorities, which could include fines and penalties, proceedings against us, and other forms of sanctions, and our ability to conduct our business, invest into China as foreign investments or accept foreign investments, or be listed on a U.S. or other overseas exchange may be restricted, and our business, reputation, financial condition, and results of operations may be materially and adversely affected.
In addition, new laws and regulations may be enacted from time to time, and PRC laws, rules, and regulations can evolve quickly with little advance notice. Substantial uncertainties exist regarding the interpretation and implementation of current and any future PRC laws and regulations applicable to our businesses. In particular, the PRC government authorities may continue to promulgate
31
new laws, regulations, rules and guidelines with respect to a wide range of issues, such as competition and antitrust, intellectual property, and other matters, which may result in additional obligations imposed on us, and may impact our ability to conduct our business, accept foreign investments, or list on a U.S. or other foreign exchange. Compliance with these laws, regulations, rules, guidelines, and implementations may be costly, and any incompliance or associated inquiries, investigations, and other governmental actions may divert significant management time and attention and our financial resources, bring negative publicity, subject us to liabilities or administrative penalties, or materially and adversely affect our business, financial condition, and results of operations.
The Chinese government may intervene or influence our operations at any time, or may exert more control over offerings conducted overseas and/or foreign investment in China-based issuers
The PRC government authorities may intervene or influence our operations at any time, or may exert more control and strengthen oversight over offerings that are conducted overseas and/or foreign investment in overseas-listed China-based issuers like us. Such actions are beyond our control and could result in a material change in our operations and/or the value of our securities.
On February 17, 2023, the CSRC released the Trial Administrative Measures of the Overseas Securities Offering and Listing by Domestic Companies (《境内企业境外发行证券和上市管理试行办法》) and five ancillary interpretive guidelines (collectively, the “Overseas Listing Trial Measures”), which apply to overseas offerings and listing by PRC-based companies, or domestic companies, of equity shares, depository receipts, corporate bonds convertible to equity shares, and other equity securities, and came into effect on March 31, 2023 . According to the Overseas Listing Trial Measures, (1) domestic companies that seek to offer or list securities overseas, both directly and indirectly, should fulfill the filing procedure and report relevant information to the CSRC, and if a overseas-listed PRC-based issuer issues new securities in the same overseas market after the overseas offering and listing, it is also required to file with the CSRC within three business days after the completion of the issuance; if a domestic company fails to complete the filing procedure or conceals any material fact or falsifies any major content in its filing documents, such domestic company may be subject to administrative penalties, such as order to rectify, warnings, fines, and its controlling shareholders, actual controllers, the person directly in charge and other directly liable persons may also be subject to administrative penalties, such as warnings and fines; (2) if a foreign-incorporated issuer meets both of the following conditions, its overseas offering and listing shall be determined as an indirect overseas offering and listing by a domestic company of the PRC: (i) any of the total assets, net assets, revenues or profits of the domestic operating entities of the issuer in the most recent accounting year accounts for more than 50% of the corresponding line item in the issuer’s audited consolidated financial statements for the same period; and (ii) its major operational activities are carried out in China or its main places of business are located in China, or the senior managers in charge of operation and management of the issuer are mostly Chinese citizens or are domiciled in China; and (3) where a PRC domestic company seeks to indirectly offer and list securities in an overseas market (including issuance of new securities after its overseas offering and listing), the issuer shall designate a major domestic operating entity responsible for all filing procedures with the CSRC.
Furthermore, in case any of the following major events occurs after the overseas offering and listing, the issuer is also required to report the relevant information to the CSRC within three business days of the occurrence and the announcement of the relevant events: (1) change of control; (2) the foreign securities regulatory body or the relevant competent authority has taken such measures as investigation and punishment; (3) conversion of listing status or listing board; and (4) voluntary of compulsory termination of listing. Where there is any material change in the major business and operation of the issuer after overseas offering and listing, and such change does not fall within the scope of filing, the issuer shall, within three business days of the occurrence of such change, submit a special report and a legal opinion issued by a domestic law firm to the CSRC to explain the relevant situation.
As substantially all of our operations are currently based in the PRC, our future offerings and major changes shall be subject to the foregoing filing procedures under the Overseas Listing Trial Measures. We cannot assure you that we could meet such requirements, obtain such permit from the relevant government authorities, or complete such filing in a timely manner or at all. Any failure may significantly limit or completely hinder our ability to continue to offer securities to investors and cause the value of such securities to significantly decline or be worthless. In addition, as the Overseas Listing Trial Measures was recently promulgated, there remains substantial uncertainties as to its interpretation and implementation and how it may impact our ability to raise or utilize fund and business operation.
There is no assurance that any new rules or regulations promulgated in the future will not impose additional requirements on us. If the PRC government authorities later promulgate new rules or implementation of industry-wide regulations directly targeting our operations, it could cause the value of our securities to significantly decline or become worthless. Therefore, investors of our Company and our business face potential uncertainty from actions taken by the PRC government affecting our business.
32
Governmental control of currency conversion and payments of RMB out of China may limit our ability to utilize our cash balances effectively and affect the value of your investment.
Our China subsidiaries have cash and cash equivalents, and short term investments of RMB94.8 million, valued at US$13.4 million in U.S. dollars as of December 31, 2023. On a consolidated basis this balance accounts for 46% of our total cash and cash equivalents and short term investments. The Chinese government imposes controls on the convertibility of RMB into foreign currencies and, in certain cases, the remittance of RMB out of China. Control on payments out of China may restrict the ability of our China subsidiaries to remit RMB to us. Approval from China’s State Administration of Foreign Exchange (“SAFE”) and the People’s Bank of China (“PBOC”) may be required where RMB are to be converted into foreign currencies, including U.S. dollars, and approval from SAFE and the PBOC or their branches may be required where RMB are to be remitted out of China. Specifically, under the existing restrictions, without prior approval from SAFE and the PBOC, the cash balance of our China subsidiaries is not available to us for activities outside of China, including the support of our in-licensing efforts. Furthermore, because repatriation of funds requires the prior approval of SAFE and the PBOC, such repatriation could be delayed, restricted or limited.
Changes in China’s economic, political or social conditions or government policies could have a material adverse effect on our business and operations.
Chinese society and the Chinese economy continue to undergo significant change. Adverse changes in the political and economic policies of the Chinese government could have a material adverse effect on the overall economic growth of China, which could adversely affect our ability to conduct business in China. The Chinese government continues to adjust economic policies to promote economic growth. Some of these measures benefit the overall Chinese economy but may also have a negative effect on us. For example, our financial condition and results of operations in China may be adversely affected by government control over capital investments or changes in tax regulations. As the Chinese pharmaceutical industry grows and evolves, the Chinese government may also implement measures to change the structure of foreign investment in this industry. We are unable to predict the frequency and scope of such policy changes, any of which could materially and adversely affect our liquidity, access to capital and its ability to conduct business in China. Any failure on our part to comply with changing government regulations and policies could result in the loss of our ability to develop and commercialize our product candidates in China.
The Chinese government exerts substantial influence over the manner in which we must conduct our business activities.
The Chinese government has exercised and continues to exercise substantial control over virtually every sector of the Chinese economy through regulation and state ownership. Our ability to operate in China may be harmed by changes in its laws and regulations, including those relating to taxation, import and export tariffs, environmental regulations, land use rights, property, healthcare regulations, and other matters. We believe that our operations in China are in material compliance with all applicable legal and regulatory requirements. However, the central or local governments of the jurisdictions in which we operate may impose new, stricter regulations or interpretations of existing regulations that would require additional expenditures and efforts on our part to ensure our compliance with such regulations or interpretations.
Accordingly, government actions in the future, including any decision not to continue to support recent economic reforms and to return to a more centrally planned economy or regional or local variations in the implementation of economic policies, could have a significant effect on economic conditions in China or particular regions thereof and could require us to divest ourselves of any interest we then hold in Chinese properties, subsidiaries, or joint ventures.
We are subject to the Enterprise Income Tax Law, and we may therefore be subject to PRC income tax on our global income.
Under the PRC Enterprise Income Tax Law and its implementing rules, both of which came into effect on January 1, 2008, enterprises established under the laws of jurisdictions outside of China with “de facto management bodies” located in China may be considered PRC tax resident enterprises for tax purposes and may be subject to the PRC enterprise income tax at the rate of 25% on their global income. “De facto management body” refers to a managing body that exercises substantive and overall management and control over the production and business, personnel, accounting books and assets of an enterprise. The State Administration of Taxation issued the Notice Regarding the Determination of Chinese-Controlled Offshore-Incorporated Enterprises as PRC Tax Resident Enterprises on the basis of de facto management bodies, or Circular 82, on April 22, 2009. Circular 82 provides certain specific criteria for determining whether the “de facto management body” of a Chinese-controlled offshore-incorporated enterprise is located in China.
33
Although Circular 82 only applies to offshore enterprises controlled by PRC enterprises, not those controlled by foreign enterprises or individuals, the determining criteria set forth in Circular 82 may reflect the State Administration of Taxation’s general position on how the “de facto management body” test should be applied in determining the tax resident status of offshore enterprises, regardless of whether they are controlled by PRC enterprises. If we were to be considered a PRC resident enterprise, we would be subject to PRC enterprise income tax at the rate of 25% on our global income. In such case, our profitability and cash flow may be materially reduced as a result of our global income being taxed under the Enterprise Income Tax Law. Although we believe that none of our entities outside of China should be considered a PRC resident enterprise for PRC tax purposes. the tax resident status of an enterprise is subject to determination by the PRC tax authorities and uncertainties remain with respect to the interpretation of the term “de facto management body.”
Chinese regulations relating to investments in offshore companies by China residents may subject our China-resident shareholders, beneficial owners or our China subsidiaries to liability or penalties, limit our ability to inject capital into our China subsidiaries or limit our China subsidiaries’ ability to increase their registered capital or distribute profits to us.
The SAFE promulgated the Circular on Relevant Issues Concerning Foreign Exchange Control on Domestic Residents’ Offshore Investment and Financing and Roundtrip Investment through Special Purpose Vehicles, or SAFE Circular 37, on July 4, 2014, which replaced the former circular commonly known as “SAFE Circular 75” promulgated by SAFE on October 21, 2005. SAFE Circular 37 requires China residents to register with local branches of SAFE in connection with their direct establishment or indirect control of an offshore entity, for the purpose of overseas investment and financing, with such China residents’ legally owned assets or equity interests in domestic enterprises or offshore assets or interests, referred to in SAFE Circular 37 as a “special purpose vehicle.” SAFE Circular 37 further requires amendment to the registration in the event of any significant changes with respect to the special purpose vehicle, such as increase or decrease of capital contributed by China individuals, share transfer or exchange, merger, division or other material event. In the event that a China shareholder holding interests in a special purpose vehicle fails to fulfill the required SAFE registration, the China subsidiaries of that special purpose vehicle may be prohibited from making profit distributions to the offshore parent and from carrying out subsequent cross-border foreign exchange activities, and the special purpose vehicle may be restricted in its ability to contribute additional capital into its China subsidiary. Moreover, failure to comply with the various SAFE registration requirements described above could result in liability under China law for evasion of foreign exchange controls. According to the Notice on Further Simplifying and Improving Policies for the Foreign Exchange Administration of Direct Investment released on February 13, 2015 by SAFE, local banks will examine and handle foreign exchange registration for overseas direct investment, including the initial foreign exchange registration and amendment registration, under SAFE Circular 37 from June 1, 2015.
According to SAFE Circular 37, our shareholders or beneficial owners, who are China residents, are subject to SAFE Circular 37 or other foreign exchange administrative regulations in respect of their investment in our Company. We may not be aware of the identities of all of our shareholders or beneficial owners who are China residents, and we do not know whether they are aware of SAFE Circular 37. We do not have control over our shareholders or beneficial owners and there can be no assurance that all of our China-resident shareholders or beneficial owners will comply with SAFE Circular 37 and subsequent implementation rules, and there is no assurance that the registration under SAFE Circular 37 and any amendment will be completed in a timely manner, or will be completed at all. The failure of our shareholders or beneficial owners who are China residents to register or amend their foreign exchange registrations in a timely manner pursuant to SAFE Circular 37 and subsequent implementation rules, or the failure of future shareholders or beneficial owners who are China residents to comply with the registration procedures set forth in SAFE Circular 37 and subsequent implementation rules, may subject such shareholders or beneficial owners or our China subsidiaries to fines and legal sanctions. Failure to register or comply with relevant requirements may also limit our ability to contribute additional capital to our China subsidiaries and limit our China subsidiaries’ ability to distribute dividends to us. Because a majority of our operating activities take place in and our strategic focus is on China, any such limitations would have a material adverse effect on our business, financial condition and results of operations.
We may be subject to fines and legal sanctions by SAFE or other Chinese government authorities if we or our employees who are China citizens fail to comply with regulations relating to employee share options granted by companies listed on exchanges outside of China to China citizens.
On February 15, 2012, SAFE promulgated the Circular on Relevant Issues Concerning the Foreign Exchange Administration for Domestic Individuals’ Participating in the Share Incentive Plans of Overseas-Listed Companies, or SAFE Circular 7, replacing earlier rules promulgated in 2007. Under SAFE Circular 7, China resident individuals who participate in a share incentive plan of a company that is listed on an overseas exchange are required to register with SAFE and complete certain other procedures. All participants
34
to a plan need to retain a China agent through Chinese subsidiaries of the overseas listed company to handle foreign exchange registration, account opening, funds transfer and remittance and other related matters. An overseas agent should also be designated to handle matters in connection with the exercise or sale of share awards and proceeds transferring for the share incentive plan participants. We believe that our share incentive plans for our China resident employees are in compliance with SAFE Circular 7; however, any failure to comply with these or similar regulations in the future may subject us or our Chinese employees to fines and legal sanctions imposed by SAFE or other government authorities and may prevent us from further granting options under our share incentive plans to our employees. Such events could adversely affect our business operations.
General Risk Factors
We may engage in strategic, commercial and other corporate transactions that could negatively affect our financial condition and prospects.
We may consider strategic, commercial, and other corporate transactions as opportunities present themselves. There are risks associated with such activities. These risks include, among others, incorrectly assessing the quality of a prospective strategic partner, encountering greater than anticipated costs in integration, being unable to profitably deploy assets acquired in the transaction, such as drug candidates, possible dilution to our shareholders, and the loss of key employees due to changes in management. Further, strategic transactions may place additional constraints on our resources by diverting the attention of our management from our business operations. To the extent we issue securities in connection with additional transactions, these transactions and related issuances may have a dilutive effect on existing shareholders. Our financial condition and prospects after an acquisition depend in part on our ability to successfully integrate the operations of the acquired business or technologies. We may be unable to integrate operations successfully or to achieve expected cost savings. Any cost savings which are realized may be offset by losses in revenues or other charges to earnings.
If securities or industry analysts publish inaccurate or unfavorable research about our business, our stock price could decline.
The trading market for our ordinary shares will depend in part on the research and reports that securities or industry analysts publish about us or our business. If one or more of the analysts who may cover us downgrade our securities or publish inaccurate or unfavorable research about our business, the price of our securities would likely decline.
Subsequent resales of our ordinary shares in the public market may cause the market price of our ordinary shares to fall.
The market value of our ordinary shares could decline as a result of sales by investors from time to time, or perceptions that such sales may occur, of a substantial amount of such ordinary shares held by them.
Issuances of additional ordinary shares may cause substantial dilution of existing shareholders.
We may issue additional ordinary shares or other securities that are convertible into or exercisable for such ordinary shares in connection with future acquisitions, future sales of our securities for capital raising purposes, future strategic relationships, or for other business purposes. The future issuance of any additional ordinary shares may create downward pressure on the trading price of our ordinary shares. There can be no assurance that we will not be required to issue additional shares, warrants or other convertible securities in the future in conjunction with any capital raising efforts, including at a price (or exercise prices) below the price at which our ordinary shares are then traded.
ITEM 4. INFORMATION ON THE COMPANY
A. | History and Development of the Company |
CASI Pharmaceuticals, Inc. is a biopharmaceutical company focused on developing and commercializing innovative therapeutics and pharmaceutical products in China, the United States, and throughout the world.
We were incorporated in 1991, and in 2012, with new leadership, we shifted our business strategy to China and has since built an infrastructure in China that includes sales and marketing, medical affairs, regulatory and clinical development and in the foreseeable future, manufacturing. We are focused on acquiring, developing and commercializing products that augment our hematology oncology
35
therapeutic focus as well as other areas of unmet medical need. We are executing our plan to become a biopharmaceutical leader by launching medicines in the greater China market, leveraging our China-based regulatory, clinical and commercial competencies and our global drug development expertise. The majority of our operations and activities are now located in China and are conducted primarily through two of our subsidiaries, CASI China and CASI Wuxi. In China, we have staff currently consists of 235 full-time employees which includes our commercial team of 150 hematology and oncology sales and marketing specialists based in China. We expect our operations in China to continue to grow.
In August 2012, we established CASI China, which is headquartered in Beijing with an office in Shanghai established in 2020. Among its activities, CASI China’s operations oversee the Company’s sales and marketing of EVOMELA® and FOLOTYN®, and the anticipated commercial activities of our pipeline products, technology transfer, local preclinical and clinical operation activities, as well as our NMPA regulatory activities. In addition, CASI China’s operations include business development activities and executive management activities, and our global management decisions are primarily being made out of CASI China where our executive team spends a significant amount of time.
In December 2018, the Company, together with Wuxi LP established CASI Wuxi. CASI Wuxi is part of the long-term strategy to support the Company’s future clinical and commercial manufacturing needs, to manage its supply chain for certain products, and to develop a GMP manufacturing facility in China. In November 2019, CASI Wuxi entered into a lease agreement for the right to use state-owned land in China for the construction of a manufacturing facility. Construction of the manufacturing facility began in the fourth quarter of 2020. Since our business focus has been shifted from ANDAs to the hematology-oncology therapeutic area, a substantial investment in GMP manufacturing facilities does not fit the current business focus. Therefore, in December 2022, we returned the land use right to the Wuxi local government for an amount of RMB 44.42 million, equivalent to the payment for land use right. Meanwhile, all construction in progress on the land was disposed and the Company recorded a disposal loss for the cost of the initial infrastructure development which amounted to RMB 15.8 million. Since we failed to meet the land development milestone, the local land administration authority requested CASI Wuxi to pay a land vacancy fee equivalent to 20% of the price for the land use rights according to the PRC Land Administration Law. We paid such fee in the amount of RMB 8.88 million to the local land administration authority in December 2022. Additionally, the Company received a government grant for the land development in April 2020 and November 2021 respectively, in the total amount of RMB 18.9 million. We are currently in negotiation with the local Wuxi government on the further treatment of the grant. The Wuxi government may require the Company to fully or partially return the grant and the Company may incur further losses.
In November 2023, CASI Wuxi obtained the Drug Manufacturing Permit from local NMPA. In December 2023, the Company entered into a series of agreements, including a capital reduction agreement, a long term borrowing agreement, and four guarantee agreements, with Wuxi LP, CASI China and CASI Wuxi, pursuant to which, (i) CASI Wuxi will reduce its registered capital and return to Wuxi LP the investment principal made by Wuxi LP in CASI Wuxi in the amount of RMB134.2 million (equivalent to its original investment of US$20 million, the “Investment Principal”), together with certain investment return in the amount of RMB26.2 million to be paid in instalments, and Wuxi LP shall cease to be a shareholder of CASI Wuxi, (ii) Wuxi LP shall reinvest the Investment Principal into a three-year long term borrowing to CASI Wuxi (the “Long term borrowing”), which shall have a non-compounding annual interest rate of 4.05% and can, from the beginning date of the Long term borrowing term till the six month anniversary after the maturity of the Long term borrowing, be partially or fully converted into the equity interest of any subsidiaries of the Company at the conversion date fair value, solely at Wuxi LP’s discretion, and (iii) each of the Company and CASI China will provide irrevocable joint and several liability guarantees on the above-mentioned payment obligations. The term of the Long term borrowing will start on December 25, 2023 and end on December 31, 2026. See “Item 3. Key Information—D. Risk Factors — Risks Relating to Our Reliance on Third Parties — The success of CASI Wuxi is subject to uncertainty in our business plan and government regulatory actions.”
CASI Delaware’s common stock traded on the Nasdaq Capital Market under the symbol “CASI.” In May 2022, CASI Delaware completed a 10-to-1 reverse stock split. In March 2023, we completed a redomicile merger, pursuant to which CASI Delaware merged with and into CASI Cayman, with CASI Cayman surviving the merger as the surviving company and successor issuer, and CASI Cayman’s ordinary shares continued trading on the Nasdaq Capital Market under the symbol “CASI.” As described in “Item 10.E. Taxation—United States Federal Income Tax Considerations,” CASI Cayman is treated for U.S. federal income tax purposes as a U.S. corporation, including with respect to any dividends paid by it, which dividends may be subject to U.S. withholding taxes.
Our principal executive offices are located at 1701-1702, China Central Office Tower 1, No. 81 Jianguo Road Chaoyang District, Beijing, 100025, People’s Republic of China. Our telephone number at this address is +86 (10) 6508 6063. Our registered office in the Cayman Islands is located at Maples Corporate Services Limited, P.O. Box 309, Ugland House, Grand Cayman, KY1-1104,
36
Cayman Islands. Our agent for service of process in the United States is located at 9620 Medical Center Drive, Suite 300, Rockville, MD 20850, 240-864-2600.
The SEC maintains an Internet website that contains reports, proxy and information statements, and other information regarding us that filed electronically with the SEC, which can be accessed at http://www.sec.gov. Our annual reports, quarterly results, press release and other SEC filings can also be accessed via our investor relations session at https://www.casipharmaceuticals.com/investor-relations/.
B. | Business Overview |
We have built a fully integrated, world class biopharmaceutical company dedicated to the successful development and commercialization of innovative and other therapeutic products. Our business development strategy is currently focused on acquiring additional targeted drugs and immuno-oncology therapeutics through licensing that will expand our hematology/oncology franchise. We use a market-oriented approach to identify pharmaceutical/biotechnology candidates that we believe to have the potential for gaining widespread market acceptance, either in China or globally, and for which development can be accelerated under our global drug development strategy. In many cases our business development strategy includes direct equity investments in the licensor company. We intend for our pipeline to reflect a diversified and risk-balanced set of assets that include (1) late-stage clinical drug candidates in-licensed for China or global regional rights, (2) proprietary or licensed innovative drug candidates, and (3) select high quality pharmaceuticals that fit our therapeutic focus. We have focused on US/EU approved product candidates, and product candidates with proven targets or product candidates that have reduced clinical risk with a greater emphasis on innovative therapeutics. Although oncology with a focus on hematological malignancies is our principal clinical and commercial target, we are opportunistic about other therapeutic areas that can address unmet medical needs. We will continue to pursue building a robust pipeline of drug candidates for development and commercialization in China as our primary market, and if rights are available for the rest of the world.
We launched our first commercial product, EVOMELA® (Melphalan for Injection) in China in August 2019. In China, EVOMELA® is approved for use as a conditioning treatment prior to stem cell transplantation and as a palliative treatment for patients with multiple myeloma. EVOMELA®, was originally licensed from Spectrum Pharmaceuticals, Inc. (“Spectrum”) and we had a supply agreement with Spectrum to support our application for import drug registration and for commercialization purposes. Spectrum completed the sale of its portfolio of FDA-approved hematology/oncology products including EVOMELA® to Acrotech on March 1, 2019. The original supply agreement with Spectrum was assumed by Acrotech, and Spectrum agreed to continue with a short-term supply agreement for EVOMELA® for the initial commercial product supply in connection with the launch, with the long-term supply assumed by Acrotech.
In July 2023, we entered into a tripartite assignment agreement with MICL, MMCo and Acrotech Inc., pursuant to which, MICL’s rights and obligations under that certain License, Development and Commercialization Agreement (as amended and restated) dated as of May 29, 2013 for the commercialization of FOLOTYN® (Pralatrexate) in China, with certain terms of such rights and obligations amended as agreed to by the parties, is assigned to us. FOLOTYN® is a dihydrofolate reductase inhibitor indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (“PTCL”). This product was approved by both the US FDA and China’s NMPA for PTCL. We also entered into a supply agreement with Acrotech Inc. pursuant to which Acrotech Inc. will supply to the Company FOLOTYN® subject to terms and on the conditions. We announced the first patient received FOLOTYN® treatment in China on February 15, 2024. We will continue to spend our time, resources and efforts on the commercialization of FOLOTYN® in China.
In November 2023, the NMPA granted market approval for Juventas' investigational cell therapy, CNCT19, for the treatment of relapsed and refractory B-cell acute lymphoblastic leukemia (r/r B-ALL) in China. CNCT19 is the first CD19-directed CAR-T product with Chinese independent intellectual property rights, making it a trailblazer in the Chinese biopharmaceutical landscape. It is also the first commercialized cell therapy product in China designed to treat B-ALL.
37
Core Product and Candidates In Hematology/Oncology
EVOMELA® (Melphalan for Injection) - Launched In China
| EVOMELA® (Melphalan for Injection) is an intravenous formulation of melphalan commercialized by Acrotech (formally by Spectrum) in the multiple myeloma treatment setting in the United States, of which we have exclusive greater China rights. The EVOMELA® formulation avoids the use of propylene glycol, which is used as a co-solvent in other formulations of injectable melphalan. The use of Captisol in the EVOMELA® formulation improves the melphalan stability when reconstituted, allowing for longer preparation and infusion times. In August 2019, we launched EVOMELA® in China as its first commercial product. The NMPA required post-marketing study has completed and the clinical study report has been submitted for regulatory review. |
FOLOTYN® (Pralatrexate) - Launched In China
| FOLOTYN® (Pralatrexate) is a dihydrofolate reductase inhibitor indicated for the treatment of patients with relapsed or refractory PTCL. This product was approved by both the FDA and China’s NMPA for PTCL. The Company announced the first patient received FOLOTYN® treatment in China on February 15, 2024. |
CNCT19 (Inaticabtagene Autoleucel)
In November 2023, The NMPA has granted market approval for Juventas' investigational cell therapy, CNCT19, for the treatment of relapsed and refractory B-cell acute lymphoblastic leukemia (r/r B-ALL) in China. In June 2019, the Company acquired worldwide license and commercialization rights to CNCT19 from Juventas, a China-based domestic company engaged in cell therapy. Juventas continues to be responsible for the clinical development and regulatory submission and maintenance of CNCT19 regulatory applications and we are responsible for the launch and commercial activities of CNCT19 under the direction of a joint steering committee.
CNCT19 is an autologous CD19 CAR-T investigative product (CNCT19) being developed by our partner Juventas for which we have exclusive worldwide co-commercial and profit-sharing rights. CNCT19 is being developed as a potential treatment for patients with hematological malignancies which express CD19 including, B-cell acute lymphoblastic leukemia (“B-ALL”) and B-cell non-Hodgkin lymphoma (“B-NHL”). CNCT19 targets CD19, a B-cell surface protein widely expressed during all phases of B-cell development and a validated target for B-cell driven hematological malignancies. CD19 targeted CAR constructs from several different institutions have demonstrated consistently high antitumor efficacy in children and adults with relapsed B-cell acute lymphoblastic leukemia (B-ALL), chronic lymphocytic leukemia (CLL), and B-cell non-Hodgkin lymphoma (B-NHL).
BI-1206 (anti-FcyRIIB antibody)
In October 2020, the Company entered into an exclusive licensing agreement with BioInvent International AB (“BioInvent”) for the development and commercialization of novel anti-FcγRIIB antibody, BI-1206, in China, Taiwan, Hong Kong and Macau. BioInvent is a biotechnology company focused on the discovery and development of first-in-class immune-modulatory antibodies for cancer immunotherapy. BI-1206 is being investigated by BioInvent in a Phase 1/2 trial, in combination with anti-PD1 therapy Keytruda® (pembrolizumab), in patients with solid tumors, and in a Phase 1/2a trial in combination with MabThera® (rituximab) in patients with relapsed/refractory non-Hodgkin lymphoma (NHL).
CTA was approved by NMPA in December 2021 and ethics committee approvals have been received in January of 2022. The Company obtained approval from HGRAO in April 2022. The Company is planning a Phase 1 study of BI-1206 in combination with
38
rituximab with a single agent BI-1206 run in phase in patients with NHL (mantle cell lymphoma, marginal zone lymphoma, and follicular lymphoma) to assess PK, safety and tolerability, select the Recommended Phase 2 Dose and assess early signs of clinical efficacy as part of its development program for BI-1206 in China. The study received regulatory approval from the China Center for Drug Evaluation (“CDE”) in the second quarter of 2022, and the first patient was enrolled and dosed in the third quarter of 2022.
CB-5339 (VCP/p97inhibitor)
CB-5339 is a novel VCP/p97 inhibitor focused on valosin-containing protein (VCP)/p97 as a novel target in protein homeostasis, DNA damage response and other cellular stress pathways for therapeutic use in the treatment of patients with various malignancies. On March 21, 2021, we entered into an exclusive license with Cleave Therapeutics, Inc. (“Cleave”) for the development and commercialization of CB-5339 in mainland China, Hong Kong, Macau and Taiwan. On July 18, 2023, we entered into an assignment agreement with Cleave, pursuant to which we obtained the global intellectual property rights related to CB-5339. CB-5339, an oral second-generation, small molecule VCP/p97 inhibitor, has been evaluated in a Phase 1 clinical trial in patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). We submitted the CB-5339 CTA application for the multiple myeloma indication in March 2022 and received approval from the NMPA in January 2023.
CID 103 (anti-CD38 monoclonal antibody)
CID-103 is a full human IgG1 anti-CD38 monoclonal antibody recognizing a unique epitope that has demonstrated an encouraging preclinical efficacy and safety profile compared to other anti-CD38 monoclonal antibodies, and which we have exclusive global rights. CID-103 is being developed for the treatment of patients with multiple myeloma. The Phase 1 dose escalation and expansion study of CID-103, in patients with previously treated, relapsed or refractory multiple myeloma is closed to further accrual in France and the UK.
Thiotepa
The Company has an exclusive China license and distribution rights to a novel formulation of thiotepa, a chemotherapeutic agent, which has multiple indications including as a conditioning treatment for use prior to certain allogeneic haemopoietic stem cell transplants. Thiotepa has a long history of established use in the hematology/oncology setting. The Company is applying for generic registration and, subject to regulatory and marketing approvals, the Company intends to advance and commercialize this product in China.
License and Distribution Agreements
EVOMELA® License Arrangements with Acrotech
The Company has product rights and perpetual exclusive licenses from Acrotech to develop and commercialize its commercial product EVOMELA® in the greater China region (which includes mainland China, Taiwan, Hong Kong and Macau). The exclusive licenses held by the Company were originally licensed from Spectrum, and Spectrum completed the sale of its portfolio of FDA-approved hematology/oncology products including EVOMELA® to Acrotech on March 1, 2019. On December 3, 2018, the Company received NMPA’s approval for importation, marketing and sales in China and in August 2019 the Company launched commercial sales of EVOMELA® in China. The NMPA required post-marketing study has completed and the clinical study report is being finalized for regulatory submission. As well the Company had similar rights to assets ZEVALIN® (Ibritumomab Tiuxetan) and MARQIBO® (Vincristine Sulfate Liposome Injection). In May 2022, Acrotech and the Company agreed to terminate the license agreement with respect to MARQIBO® and ZEVALIN®.
The Company has established relationships, coupled with supply agreements, to secure the necessary resources to supply the EVOMELA® commercial drug product as well as any clinical trials materials required for our clinical development program. As an import drug product into China, we expect that the future supply of EVOMELA® will continue to be met by our partner Acrotech and its contract manufacturers.
39
FOLOTYN® (Pralatrexate) License Agreement with MICL, MMCo and Acrotech Inc.
On July 31, 2023, the Company entered into a tripartite assignment agreement (the “FOLOTYN® Assignment Agreement”) with MICL, MMCo and Acrotech Inc., pursuant to which, MICL’s rights and obligations under that certain License, Development and Commercialization Agreement (as amended and restated) dated as of May 29, 2013 for the commercialization of FOLOTYN® (Pralatrexate) in China, with certain terms of such rights and obligations amended as agreed to by the parties, is assigned to the Company.
In relation to the FOLOTYN® Assignment Agreement, the Company and MICL entered into a payment agreement (the “Payment Agreement”), pursuant to which the Company will pay MICL a total of US$ 12 million, including (i) a one-time payment of US$ 2 million which will be paid upon completion of the quality audit; (ii) a one time, non-refundable and non-creditable payment of US$ 2 million to be paid when the aggregate net sales of FOLOTYN® in China equals to or exceeds US$ 30,000,000, and (iii) in each calendar quarter, a one-time, non-refundable and non-creditable payment of an amount equal to 10% of net sales of FOLOTYN® in China in the preceding calendar quarter, until an aggregate amount of US$ 8 million is paid in such quarterly instalments. In the event that the Company has not paid the full amount of payment of US$10 million under (ii) and (iii) above (the “Deferred Payments”) on July 31, 2028, the remaining amount shall become immediately due and payable, unless the drug approval in China is not renewed by relevant regulatory authorities. The initial drug approval of FOLOTYN® in China is effective until August 2025 and the renewal of drug license is subject to satisfaction of certain requirements.
Concurrently with the execution of the FOLOTYN® Assignment Agreement and the Payment Agreement, the parties entered into certain other agreements in relation to the commercialization of FOLOTYN®, including a supply agreement entered into by and between the Company and Acrotech Inc., pursuant to which Acrotech Inc. will supply to the Company FOLOTYN® subject to terms and on the conditions set forth therein. The Company is obligated to pay Acrotech Inc. additional $750,000 in three instalments when certain conditions are met.
The Company announced the first patient received FOLOTYN® treatment in China on February 15, 2024. The Company will continue to spend time, resources and efforts on the commercialization of FOLOTYN® in China.
Distribution Agreement with China Resources Pharmaceutical Commercial Group International Trading Co., Ltd. (previously known as China Resources Guokang Pharmaceuticals Co., Ltd.)
In March 2019, the Company entered into a three-year exclusive distribution agreement with China Resources Pharmaceutical Commercial Group International Trading Co., Ltd. (“CRPCGIT”), to appoint CRPCGIT on an exclusive basis as its distributor to distribute EVOMELA® in the territory of China, subject to certain terms and conditions. The Company’s internal marketing and sales team are responsible for commercial activities, including, for example, direct interaction with therapeutic area experts, physicians, hospital centers and the generation of sales. The agreement was renewed in March 2022 for two years, and further extended in February 2024 for an additional three years.
Distribution Agreement with China National Medicines Corporation Ltd.
On December 6, 2023, the Company entered into an exclusive distribution agreement with China National Medicines Corporation Ltd. (“CNMC”) and CASI China. Under the terms of the Agreement, the Company appointed CNMC on an exclusive basis as its sole distributor for the sale of Pralatrexate for Injection (FOLOTYN®) in the territory of China during the term of one year, subject to certain terms and conditions. CNMC may appoint sub-distributors of its choice in furtherance of this goal provided that the Company has been notified in writing and received the due diligence or any other information of the sub-distributor as the Company requests. CASI China is authorized to coordinate and communicate with parties to the Agreement and provide supports for the performance of the Agreement.
Precision Autoimmune Therapeutics Co., Ltd., (previously known as Beijing Tianshi Tongda Pharmaceuticals Technology Co., Ltd)
In May 2022, the Company entered into a Sublicense Agreement (the “PAT Sublicense Agreement”) with PAT, a company established under the laws of China, pursuant to which the Company granted PAT an exclusive (subject to the commercialization and co-marketing rights), perpetual, worldwide license, with the right to freely grant further sublicenses subject to terms and conditions in the PAT Sublicense Agreement, for the investigational anti-CD38 monoclonal antibody TSK011010 licensed and controlled by the
40
Company from Black Belt Therapeutics Limited, in the treatment, prevention and diagnosis of autoimmune diseases, conditions and disorders in humans. Pursuant to the PAT Sublicense Agreement, PAT will make an upfront payment of US$10,000,000 equivalent in two equal instalments upon completion of its first and second financing, respectively, plus potential future payments or reimbursement of development and sales milestones and royalties to the Company.
Also in May 2022, CASI China entered into an agreement for the investment in PAT in the amount of RMB 20.0 million in cash during PAT’s first equity financing.
Juventas License Arrangements
In June 2019, the Company entered into a license agreement for exclusive worldwide license to commercialize an autologous anti-CD19 T-cell therapy product (CNCT19) from Juventas (the “Exclusive License Agreement”). Juventas is a China-based company engaged in cell therapy. The terms of the agreement include RMB 70 million of milestone payments upon the registration of Phase II clinical trial of CNCT19 and sales royalty payments. The milestone was met during the third quarter of 2020, the Company paid the milestone payment of RMB 70 million to Juventas in September 2020, and recognized it as acquired in-process research and development expenses in the consolidated statement of operations and comprehensive loss in 2020.
In September 2020, Juventas and its shareholders (including CASI Biopharmaceuticals, a subsidiary of the Company) agreed to certain terms and conditions required by a new third-party investor to facilitate the Series B financing of Juventas, pursuant to which the Company agreed to amend and supplement the original licensing agreement (the "Supplementary Agreement") by agreeing to pay Juventas certain percentage of net profits generated from commercial sales of CNCT19 in addition to the royalty fee payment calculated as a percentage of net sales. The Supplementary Agreement also specifies a minimum annual target net profit to be distributed to Juventas and certain other terms and obligations. In return, the Company obtained additional equity interests in Juventas.
Under the Supplementary Agreement, Juventas and the Company will jointly market CNCT19, including, but not limited to, establishing medical teams, developing medical strategies, conducting post-marketing clinical studies, establishing Standardized Cell Therapy Centers, establishing and training providers with respect to cell therapy, testing for cell therapy, and monitoring quality controls (cell collection and transfusion, etc.), and patient management (adverse reactions treatment, patients’ follow-up visits, and establishment of a database). The Company also will reimburse Juventas for a portion of Juventas’ marketing expenses as reviewed and approved by a joint commercial committee to be constituted. The Company will continue to be responsible for recruiting and establishing a sales team to commercialize CNCT19.
On October 26, 2021, Juventas completed its Series C financing. Upon the completion of Juventas Series C financing, the Company’s equity ownership in Juventas decreased to 12.01% on a fully diluted basis, with the total fair value of the equity interest amounted to RMB 206 million.
In September 2022, CASI Biopharmaceuticals entered into an Equity Transfer Agreement with Shenzhen Jiadao Gongcheng Equity Investment Fund, LLP (“Jiadao Gongcheng”), pursuant to which CASI Biopharmaceuticals agreed to transfer its equity interest in Juventas to Jiadao Gongcheng in the amount of RMB 240.9 million (approximately US$33.9 million). The transaction was closed in November 2022, and the Exclusive License Agreement is still effective after this equity transfer.
The Company is currently involved in arbitration proceedings against Juventas in relation to Juventas’ purported termination of the CNCT19 Agreements. See “Item 8 — Financial Information — A. Consolidated Statements and Other Financial Information – Legal Proceedings” for further information.
BioInvent International AB
In October 2020, the Company entered into an exclusive licensing agreement with BioInvent for the development and commercialization of novel anti-FcγRIIB antibody, BI-1206, in mainland China, Taiwan, Hong Kong and Macau. BioInvent is a biotechnology company focused on the discovery and development of first-in-class immune-modulatory antibodies for cancer immunotherapy. BI-1206 is being investigated in a Phase 1/2 trial, in combination with anti-PD1 therapy Keytruda® (pembrolizumab), in patients with solid tumors, and in a Phase 1/2a trial in combination with MabThera® (rituximab) in patients with relapsed/refractory non-Hodgkin lymphoma (NHL). CTA was approved by NMPA in December 2021 and ethics committee approvals have been received in January of 2022. The Company obtained approval from HGRAO in April 2022. The Company is planning a Phase 1 study of BI-1206
41
in combination with rituximab with a single agent BI-1206 run in phase in patients with NHL (mantle cell lymphoma, marginal zone lymphoma, and follicular lymphoma) to assess PK, safety and tolerability, select the Recommended Phase 2 Dose and assess early signs of clinical efficacy as part of its development program for BI-1206 in China. The study received regulatory approval from the CDE in the second quarter of 2022, and the first patient was enrolled and dosed in the third quarter of 2022.
Under the terms of the license agreement, BioInvent and CASI will develop BI-1206 in both hematological malignancies and solid tumors, with CASI responsible for commercialization in China and associated markets. CASI made a US$5.9 million upfront payment in November 2020 to BioInvent and will pay up to US$83 million in development and commercial milestone payments plus tiered royalties in the high-single to mid-double-digit range on net sales of BI-1206.
In conjunction with our license agreement entered into with BioInvent, we made a SEK 53.8 million investment in 1.2 million new shares of BioInvent, and 588,000 new warrants, each warrant with a right to subscribe for 1 new share in BioInvent within a period of five years and at a subscription price of SEK 78.50 per share. In the second quarter of 2022, the Company sold 275,000 ordinary shares of BioInvent for US$1.3 million.
As an import drug product into China, we expect that future supply of BI-1206 will be met by our partner BioInvent and its contract manufacturers. For local development in China, we expect that our clinical materials and commercial inventory will be supplied by one or more contract manufacturers.
Black Belt Therapeutics Limited
In April 2019, the Company entered into a license agreement with a newly established, privately held UK Company Black Belt Therapeutics Limited (“Black Belt”) for exclusive worldwide rights to CID-103, an investigational anti-CD38 monoclonal antibody (Mab) (formerly known as TSK011010). In conjunction with the license agreement, CASI invested 2 million euros in Black Belt Tx Ltd. (“Black Belt Tx”), the holding company of Black Belt. In July 2021, Alesta Therapeutics B.V. (“Alesta”) was incorporated as the parent company holding all shares of Black Belt Tx with same ownership structure as Black Belt Tx.
The Company expects that its clinical materials and commercial inventory will be supplied by one or more contract manufacturers with whom the Company has contracted with. Under the terms of the agreement, we obtained global rights to CID-103 for an upfront payment of 5 million euros as well as certain milestone and royalty payments. In June 2021, we achieved the First-Patient-In (FPI) in the Phase 1 dose escalation and expansion study of CID-103, and made US$750,000 milestone payment in June 2021 and €250,000 payment in August 2021 under the terms of the agreement. As mentioned above, in May 2022, the Company entered into the Sublicense Agreement to grant PAT an exclusive, perpetual, worldwide license, for the investigational anti-CD38 monoclonal antibody TSK011010.
Cleave Therapeutics, Inc.
In March 2021, the Company entered into an exclusive license agreement (the “Cleave License Agreement”) with Cleave Therapeutics, Inc. (“Cleave”) for the development and commercialization of CB-5339, an oral novel VCP/p97 inhibitor, in both hematological malignancies and solid tumors, in mainland China, Hong Kong, Macau and Taiwan . Cleave is a clinical-stage biopharmaceutical company focused on valosin-containing protein (VCP)/p97 as a novel target in protein homeostasis, DNA damage response and other cellular stress pathways for therapeutic use in the treatment of patients with cancer.
CB-5339 has been evaluated by Cleave in a Phase 1 clinical trial in patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). Under the terms of the agreement, the Company is responsible for development and commercialization in China and associated markets. The Company paid a US$5.5 million upfront payment to Cleave in 2021 and will pay up to US$74 million in development and commercial milestone payments plus tiered royalties in the high-single to mid-double-digit range on net sales of CB-5339. In conjunction with the license agreement, the Company made a US$5.5 million investment in Cleave through a convertible note.
On July 18, 2023, the Company entered into an assignment agreement with Cleave (the “Cleave Assignment Agreement”), pursuant to which the Company obtained the global intellectual property rights related to CB-5339. The Cleave Assignment Agreement also terminated and superseded the Cleave License Agreement for CB-5339 the Company previously entered into with Cleave. Pursuant to the Cleave Assignment Agreement and partially in exchange for the transfer of the global intellectual property rights for CB-5339 as
42
well as all remaining CB-5339 drug substance and drug product to the Company and a repayment in the amount of USD $1 million to the Company, the Company agreed to the termination of that certain outstanding convertible promissory note issued by Cleave to the Company in 2021 with a principal amount of USD $5.5 million. Cleave is also eligible to receive up to US$66 million in commercial and sales milestone payments, plus a 2.5% royalty on net sales of CB-5339 and any other VCP/p97 inhibitor covered by the Cleave Assignment Agreement, in each case subject to the terms and on the conditions set forth in the Cleave Assignment Agreement.
Riemser Pharma GmbH
In August 2019, the Company entered into an exclusive license and distribution agreement with Riemser Pharma GmbH (“Riemser”), pursuant to which the Company obtained exclusive distribution rights for Thiotepa in China. Under the agreement, Riemser will be responsible for manufacturing and supplying the Company with clinical trials materials and commercial drug product, and costs of clinical trials (if any) for the registration, product launch and commercialization of Thiotepa in China. In January 2020, Riemser was acquired by Esteve Healthcare, S.L. (“ESTEVE”), an international pharmaceutical company headquartered in Barcelona, Spain. In November 2022, the Company entered into an Amendment with Esteve, pursuant to which the Company and Esteve will equally share the costs of clinical trials (if any) for the registration of Thiotepa in China. After the product is launched, the Company will be subject to annual minimum purchase as prescribed in the agreement.
Pharmathen Global BV
In October 2019, the Company entered into an exclusive distribution agreement with Pharmathen Global BV ("Pharmathen") for the development and distribution of Octreotide LAI microsphere in China. Octreotide LAI formulations, which are approved in various European countries, are considered a standard of care for the treatment of acromegaly and the control of symptoms associated with certain neuroendocrine tumors. The Company paid and expensed an upfront payment of 1 million euros in 2019 and milestone payment of 1.5 million euros in 2020 with achievements of certain milestones.
On February 2, 2023, the Company entered into a termination agreement and release with Pharmathen, pursuant to which (i) both parties agreed to terminate the exclusive distribution license agreement in 2019 with respect to product Octreotide LAI, (ii) the Company returned all confidential information provided by Pharmathen and cancelled all regulatory approvals or other registrations it had obtained as an authorized distributor; and (iii) Pharmathen refunded 1.25 million euros to the Company.
Intellectual Property
We generally seek patent protection for our technology and product candidates in China, the United States, Canada and other key markets. The patent position of biopharmaceutical companies generally is highly uncertain and involves complex legal and factual questions. Our success will depend, in part, on whether we can: (i) obtain patents to protect our own products; (ii) obtain licenses to use the technologies of third parties, which may be protected by patents; (iii) protect our trade secrets and know-how; and (iv) operate without infringing the intellectual property and proprietary rights of others.
With regards to our commercial drug EVOMELA® licensed for greater China rights from our partner, we have acquired exclusive licenses to intellectual property to enable us to develop and continue to commercialize EVOMELA® in China.
With regards to our commercial drug FOLOTYN® licensed for China rights from our partner, we have acquired exclusive licenses to intellectual property to enable us to develop and continue to commercialize FOLOTYN® in China.
With regards to CNCT19, we have acquired an exclusive license to intellectual property from our partner Juventas to enable us to co-commercialize CNCT19 in China and well as the rest of the world. Juventas is responsible for prosecuting and maintaining the licensed intellectual property.
With regards to BI-1206, we have acquired an exclusive license to intellectual property and the know-how from our partner BioInvent to enable us to develop and commercialize BI-1206 in our greater China commercial markets. BioInvent is responsible for prosecuting and maintaining the licensed BioInvent intellectual property.
43
With regards to CB-5339, we have acquired an exclusive license to intellectual property and the know-how from our partner Cleave to enable us to develop and commercialize CB-5339 in our greater China commercial markets. Cleave is responsible for prosecuting and maintaining the licensed Cleave intellectual property.
With regards to our in-licensed anti-CD38 antibody candidate CID-103, we have acquired an exclusive worldwide license to patents around CID-103 and other anti-CD38 antibodies, covering multiple pending applications worldwide, directed to the antibodies themselves and treatment methods using the antibodies. We have since filed additional applications, with current pending applications including U.S., Australia, Canada, China, Europe, India, Japan, Korea, New Zealand, Singapore and Hong Kong. We intend to further expand our patent portfolio and in the submission stage of additional applications. The patent term for any patents granted from the earliest of these pending applications will expire in June 2038, assuming all annuities are paid and not considering any term extensions for regulatory approval that might be available.
With regards to the Thiotepa drug candidate, we have acquired exclusive licenses to intellectual property and/or the know-how to enable us to develop and commercialize this drug candidate in the China market.
We hold certain intellectual property in connection with a proprietary aurora kinase inhibitor that we no longer devote resources to. Our intellectual property for this asset remains available for business development partnering.
We currently own a number of registered trademarks and pending trademark applications for CASI, including our corporate logo and product name in China, the United States and other jurisdictions, and we are seeking further trademark protection for CASI, including our corporate logo, product name, and other marks in jurisdictions where available and appropriate.
We review and assess our portfolio on a regular basis to secure protection and to align our intellectual property strategy with our overall business strategy.
Sales and Marketing
We rely on third-party distributors to distribute our approved medicines. For example, we rely on sole third-party distributors to distribute our in-licensed approved medicines in China. Our ability to maintain and grow our business will depend on our ability to maintain an effective distribution channel that ensures the timely delivery of our medicines. We have long-standing business relationship with our sole distributor CRPCGIT for the in-licensed products EVOMELA®, and we newly established business relationship with a sole distributor CNMC for the in-licensed products FOLOTYN®.
Competition
Competition in the pharmaceutical, biotechnology and biopharmaceutical industries is intense and based significantly on scientific and technological factors, the availability of patent and other protection for technology and products, the ability and length of time required to obtain governmental approval for testing, manufacturing and marketing and the ability to commercialize products in a timely fashion. Moreover, the biopharmaceutical industry is characterized by rapidly evolving technology that could result in the technological obsolescence of any products that we develop.
We compete with many specialized biopharmaceutical firms, as well as a growing number of large pharmaceutical companies that are applying biotechnology to their operations. It is probable that the number of companies seeking to develop products and therapies for the treatment of unmet needs in oncology will increase. Many biopharmaceutical companies have focused their development efforts in the human therapeutics area, including oncology and inflammation, and many major pharmaceutical companies have developed or acquired internal biotechnology capabilities or made commercial arrangements with other biopharmaceutical companies. These companies, as well as academic institutions, governmental agencies and private research organizations, also compete with us in recruiting and retaining highly qualified scientific personnel and consultants.
The biopharmaceutical industry has undergone, and is expected to continue to undergo, rapid and significant technological change. Consolidation and competition are expected to intensify as technical advances in each field are achieved and become more widely known. In order to compete effectively, we will be required to continually expand our scientific expertise and technology, identify and retain capable personnel and pursue scientifically feasible and commercially viable opportunities.
44
Our competition will be determined in part by the potential indications for which our product candidates may be developed and ultimately approved by regulatory authorities. The relative speed with which we develop new products, complete clinical trials, obtain regulatory approvals, and complete the other requirements to get a pharmaceutical product on the market are critical factors in gaining a competitive advantage. We may rely on third parties to commercialize our products, and accordingly, the success of these products will depend in significant part on these third parties’ efforts and ability to compete in these markets. The success of any collaboration will depend in part upon our collaborative partners’ own competitive, marketing and strategic considerations, including the relative advantages of alternative products being developed and marketed by our collaborative partners and our competitors.
Many of our existing or potential competitors have substantially greater financial, technical and human resources than we do and may be better equipped to develop, manufacture and market products. In addition, many of these competitors have extensive experience in preclinical testing and human clinical trials and in obtaining regulatory approvals. The existence of competitive products, including products or treatments of which we are not aware, or products or treatments that may be developed in the future, may adversely affect the marketability of products that we may develop. Our competitors’ drugs may be more effective than any drug we may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses of developing our product candidates.
In September 2022, Xi’An Libang Pharmaceuticals Co., Ltd. launched domestically produced injectable melphalan using propylene glycol as co-solvent. The domestic produced injectable melphalan is the direct competitor of the Company’s launched product EVOMELA® and it may adversely affect the marketability of products that we may develop.
CNCT19 faces promotional challenge, primarily due to the saturation of CAR-T clinical trials in China. The abundance of ongoing studies within this therapeutic area creates intense competition for eligible patients, making it increasingly difficult to find a sufficient number of self-paid patients.
FOLOTYN® currently enjoys a unique position in the market without direct competition, providing it a temporary advantage in terms of market share. However, the impending threat from local generic companies in China, which have already filed ANDA applications, looms large. These companies are poised to launch generic versions of FOLOTYN® as soon as its patent expires in China. The entry of generics is expected to lead to significant price reductions and a consequent erosion of FOLOTYN®'s market share, affecting its revenue stream.
Regulations
U.S. Food and Drug Administration (FDA)
Our research, development, testing, manufacture, labeling, sale, marketing, advertising, and distribution of therapeutics in the United States, China and other countries are subject to extensive regulations by federal, state, local and foreign governmental authorities.
In the United States, the FDA regulates the development and commercialization of drugs and biologics. Drugs are subject to regulation under the Federal Food, Drug, and Cosmetic Act (FFDCA), and biological products, in addition to being subject to certain provisions of the FFDCA, are regulated under the Public Health Service Act (PHSA). We believe that the FDA will regulate the products currently being developed by us or our collaborators as drugs or biologics. Both the FFDCA and PHSA and corresponding regulations govern, among other things, the testing, manufacturing, safety, efficacy, labeling, storage, recordkeeping, advertising and other promotion of biologics and drugs, as the case may be.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition to new legislation, FDA regulations and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our product candidates or any future product candidates we may develop. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.
Preparing drug and biologic candidates for regulatory approval is a costly and time-consuming process. Generally, a developer first must conduct preclinical studies in the laboratory and in animal model systems in accordance with applicable FDA requirements, including Good Laboratory Practice regulations, to gain preliminary information on an agent’s effectiveness and to identify any safety
45
problems. The results of these studies, together with manufacturing information and analytical data as well as protocols and detailed descriptions for proposed clinical investigations, are submitted to FDA as a part of an Investigational New Drug Application (IND) for a drug or biologic, which must become effective before human clinical trials of an investigational drug can begin. An IND application will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about issues, such as the conduct of the clinical trials as outlined in the IND application, and places the clinical trial(s) on a clinical hold. In such a case, the IND application sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. We cannot be certain that submission of an IND application will result in the FDA allowing clinical trials to begin.
We or our collaborators must then conduct adequate and well-controlled clinical trials, in accordance with applicable IND regulations, Good Clinical Practices (“GCPs”), and other clinical-trial related regulations, to establish the safety and efficacy of the candidate for each proposed indication. We or our collaborators will be required to select qualified investigators (usually physicians within medical institutions) to supervise the administration of the products, test or otherwise assess patient results, and collect and maintain patient data; monitor the investigations to ensure that they are conducted in accordance with applicable requirements, including the requirements set forth in the general investigational plan and protocols contained in the IND; and comply with applicable reporting and recordkeeping requirements. The study protocol and informed consent information for study subjects in clinical trials must also be approved by an IRB for each institution where the trials will be conducted before the trial can begin, and each IRB must monitor the study until completion. Study subjects must provide informed consent and sign an informed consent form before participating in a clinical trial.
Clinical trials of drugs or biologics are normally done in three phases, although the phases may overlap or be combined. Phase 1 trials usually involve the initial introduction of the investigational candidate into humans to evaluate its short-term safety, dosage tolerance, metabolism, pharmacokinetics and pharmacologic actions, and, if possible, to gain an early indication of its effectiveness. Phase 2 trials normally involve trials in a limited patient population to evaluate dosage tolerance and appropriate dosage, identify possible adverse effects and safety risks, and evaluate preliminarily the efficacy of the candidate for specific target indications. Phase 3 trials are expanded clinical trials with larger numbers of patients which are intended to evaluate the overall benefit-risk relationship of the drug and to gather additional information for proper dosage and labeling of the drug. Phase 3 clinical trials may take several years to complete. Annual progress reports detailing the results of the clinical studies must be submitted to the FDA and IND safety reports must be submitted to the FDA and investigators within 15 calendar days for serious and unexpected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. We or our collaborators, the FDA, or an IRB (with respect to a particular study site) may suspend or terminate clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after receiving initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and are commonly intended to generate additional safety data regarding use of the product in a clinical setting. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of the product or, in certain circumstances, post-approval.
The FDA has various programs, including fast track designation, breakthrough therapy designation, priority review, accelerated approval, and, for regenerative medicine therapies, regenerative medicine advanced therapy designation, which are intended to expedite or simplify the process for the development, and FDA’s review, of drugs and biologics (e.g., granting approval on the basis of surrogate endpoints subject to post-approval trials). Generally, drugs or biologics that may be eligible for one or more of these programs are those intended to treat serious or life-threatening diseases or conditions, those with the potential to address unmet medical needs for those disease or conditions, and/or those that provide a meaningful benefit over existing treatments. Moreover, if a sponsor submits a marketing application for a product intended to treat certain rare pediatric or tropical diseases or for use as a medical countermeasure for a material threat, and that meets other eligibility criteria, upon approval such sponsor may be granted a priority review voucher that can be used for a subsequent application. Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. Furthermore, these programs do not change the standards for approval and may not ultimately expedite the development or approval process.
If clinical trials of a product candidate are completed successfully, the sponsor of the product may seek FDA marketing approval. If the product is classified as a new drug, an applicant must file a New Drug Application (NDA). For biological products, an
46
applicant must file a Biologics License Application (BLA). In each case, FDA must approve the application before the product can be marketed commercially. NDAs and BLAs must include, among other things, detailed information about the product’s chemistry, manufacture, controls, and proposed labeling and the results of preclinical studies and clinical trials. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of a drug, and safety, purity, and potency of a biologic, to the satisfaction of the FDA. A user fee must be paid with the submission of an NDA or BLA (unless a fee waiver applies) in order to support the cost of agency review, which is currently almost US$3 million. FDA usually will inspect the facility or the facilities at which the drug is manufactured and will not approve the product unless the manufacturing and production and testing facilities are in compliance with cGMP regulations. In addition, FDA may also inspect clinical trial sites that generated data for the NDA or BLA as well as us or our collaborators as a clinical trial sponsor.
The testing and approval processes require substantial time and effort, and there can be no assurance that FDA will accept the application for filing or that any approval will be obtained on a timely basis, if at all. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act, the FDA has ten months from the 60 day filing date in which to complete its initial review of a standard application and respond to the applicant. However, the time required by the FDA to review and approve NDAs and BLAs is variable and, to a large extent, beyond our control. Notwithstanding the submission of relevant data, the FDA may ultimately decide that an NDA or BLA does not satisfy its regulatory criteria and deny the approval. In such instance, FDA will issue a Complete Response Letter, describing all the deficiencies that the FDA has identified in an application that must be satisfactorily addressed before it can be approved. A Complete Response Letter may require additional clinical data and/or an additional pivotal Phase 3 clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. Further, even if such additional information is submitted, the FDA may ultimately decide that the application does not satisfy the criteria for approval. The FDA may also refer the application to an appropriate advisory committee, typically a panel of clinicians, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendations of the advisory committee, but the Agency historically has tended to follow such recommendations. In addition, the FDA may condition marketing approval on the conduct of specific post-marketing studies to further evaluate safety and effectiveness or a REMS that may include both special labeling and controls, known as Elements to Assure Safe Use, on the distribution, prescribing, dispensing and use of a drug product. After approval is obtained, a marketed product is subject to continuing regulatory requirements and review relating to cGMP, adverse event reporting, promotion and advertising, and other matters. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Products may be promoted only for the approved indications and consistent with the provisions of the approved label. Discovery of previously unknown problems or failure to comply with the applicable regulatory requirements may result in restrictions on the marketing of a product, mandated labeling changes, or withdrawal of the product from the market, as well as possible civil or criminal sanctions.
Drugs and biological products may be eligible to receive certain regulatory exclusivities upon approval. For example, a drug that constitutes a new chemical entity (i.e., an active moiety that has not been previously approved in another NDA) is entitled to five years of exclusivity during which FDA may not accept an ANDA or 505(b)(2) NDA for filing referencing such chemical entity, unless a “Paragraph IV certification” is made in which case FDA may accept such applications four years after initial approval of the new chemical entity. In addition, three years of exclusivity can be awarded for applications (including supplements) containing the results of new clinical investigations (other than bioavailability studies) conducted by the applicant and essential to the FDA’s approval of new versions or conditions of use of previously approved drug products, such as new indications, delivery mechanisms, dosage forms, strengths, or other conditions of use. A reference biological product is granted twelve years of data exclusivity from the time of first licensure of the product, and the FDA will not accept an application for a biosimilar or interchangeable product based on the reference biological product until four years after the date of first licensure of the reference product. Moreover, a drug or biologic may receive orphan drug designation if intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making the product available in the United States for this type of disease or condition will be recovered from sales of the product in the United States. If a product that has orphan designation subsequently receives the first FDA approval for the disease or condition for which it has such designation, the product is entitled to orphan drug exclusivity, which restricts FDA from approving any other applications to market the same drug for the same indication for seven years from the date of such approval, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity by means of greater effectiveness, greater safety, by providing a major contribution to patient care, or in instances of an inability to assure drug supply.
FDA may approve generic drugs and biological products through abbreviated pathways. Generic drugs may be marketed upon approval of an ANDA, which contains information to show that the proposed product is identical in active ingredient, dosage form,
47
strength, route of administration, labeling, quality, performance characteristics, and intended use, among other things, to a previously approved drug. Approval is generally supported by data from bioequivalence studies, rather than complete preclinical and clinical studies. Biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product are eligible for an abbreviated approval pathway. Although licensure of biosimilar or interchangeable products is generally expected to require less than the full complement of product-specific preclinical and clinical data required for reference products, the FDA has considerable discretion over the kind and amount of scientific evidence required to demonstrate biosimilarity and interchangeability. Under section 610 of the Further Consolidated Appropriations Act, 2020, entitled “Actions for Delays of Generic Drugs and Biological Products”, generic drug and biosimilar developers may sue brand manufacturers, or generic or biosimilar manufacturers, to obtain sufficient quantities of reference product necessary for approval of the developers’ generic or biosimilar product. If a generic drug or biosimilar developer is successful in its suit, the defendant manufacturer would be required to provide sufficient quantities of product on commercially-reasonable, market-based terms and may be required to pay the developer’s reasonable attorney’s fees and costs as well as financial compensation under certain circumstances. While intended to facilitate the timely entry of lower-cost generic and biosimilar products, we cannot determine what effect this new private right of action may have on the development and approval of generic drug and biosimilar products at this time.
The Generic Drug Enforcement Act of 1992 establishes penalties for wrongdoing in connection with the development or submission of an application. In general, the FDA is authorized to temporarily or permanently bar companies and individuals, from submitting or assisting in the submission of applications to FDA, and to temporarily deny approval and suspend applications to market drugs under certain circumstances. FDA’s debarment authority has also been expanded to apply to certain import-related offenses. In addition to debarment, the FDA has numerous enforcement and disciplinary powers, including the authority to withdraw approval of an application or to approve an application under certain circumstances, to suspend the distribution of all drugs approved or developed in connection with certain wrongful conduct, and various civil and criminal penalties. The FDA may also withdraw product approval or take other corrective measures if, among other things, ongoing regulatory requirements are not met or if safety or efficacy questions are raised after the product reaches the market.
Manufacturers and other entities involved in the manufacturing and distribution of approved products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the product. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory requirements, and test each product batch or lot prior to its release. We rely, and expect to continue to rely, on third parties for the production of clinical quantities of our product candidates and any future product candidates we may develop. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production or distribution or may require substantial resources to correct.
Healthcare Regulation
Federal and state healthcare laws in the United States, including fraud and abuse and health information privacy and security laws, also apply to our business. If we fail to comply with those laws, we could face substantial penalties and our business, results of operations, financial condition and prospects could be adversely affected. The laws that may affect our ability to operate include, but are not limited to: the federal Anti-Kickback Statute, which prohibits, among other things, soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; and federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payers that are false or fraudulent. Additionally, we are subject to state law equivalents of each of the above federal laws, which may be broader in scope and apply regardless of whether the payer is a federal healthcare program, and many of which differ from each other in significant ways and may not have the same effect, further complicate compliance efforts.
Numerous federal and state laws, including state security breach notification laws, state health information privacy laws and federal and state consumer protection laws, govern the collection, use and disclosure of personal information. Other countries also have, or are developing, laws governing the collection, use and transmission of personal information. In addition, most healthcare providers who are expected to prescribe our products and from whom we obtain patient health information, are subject to privacy and security requirements under the Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology and Clinical Health Act (HIPAA). Although we are not directly subject to HIPAA, we could be subject to criminal penalties if we obtain
48
and/or disclose individually identifiable health information from a HIPAA-covered entity, including healthcare providers, in a manner that is not authorized or permitted by HIPAA. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing amount of focus on privacy and data protection issues with the potential to affect our business, including recently enacted laws in a majority of states requiring security breach notification. These laws could create liability for us or increase our cost of doing business.
In addition, the Patient Protection and Affordable Care Act, as amended by the Health Care Education Reconciliation Act (PPACA), created a federal requirement under the federal Open Payments program, that requires certain manufacturers to track and report to the Centers for Medicare and Medicaid Services, or CMS, annually certain payments and other transfers of value provided to physicians and teaching hospitals made in the previous calendar year. In addition, there are also an increasing number of state laws that require manufacturers to make reports to states on pricing and marketing information. These laws may affect our sales, marketing, and other promotional activities by imposing administrative and compliance burdens on us. In addition, given the lack of clarity with respect to these laws and their implementation, our reporting actions could be subject to the penalty provisions of the pertinent state and federal authorities.
For those marketed products which are covered in the United States by certain government healthcare programs (e.g., Medicare and Medicaid), we have various obligations, including government price reporting and rebate requirements, which generally require products be offered at substantial rebates/discounts to Medicaid and certain purchasers (including “covered entities” purchasing under the 340B Drug Discount Program). We are also required to discount such products to authorized users of the Federal Supply Schedule of the General Services Administration, under which additional laws and requirements apply. These programs require submission of pricing data and calculation of discounts and rebates pursuant to complex statutory formulas, as well as the entry into government procurement contracts governed by the Federal Acquisition Regulations, and the guidance governing such calculations is not always clear. Compliance with such requirements can require significant investment in personnel, systems and resources, but failure to properly calculate prices, or offer required discounts or rebates could subject us to substantial penalties.
National Medical Products Administration (NMPA, formerly the China Food and Drug Administration)
In the PRC, the NMPA is the authority under the State Administration for Market Regulation (SAMR) that monitors and supervises the administration of pharmaceuticals products, medical appliances and equipment, and cosmetics. We are also subject to regulation and oversight by different levels of the Medical Products Administration and Administration of Market Regulation in China. For clinical-stage product candidates, our development activities in China can follow two purposes: (1) to obtain clinical data to support our global FDA-regulated trials as is the case for our proprietary ENMD 2076, and (2) to obtain clinical data to support local registration with the NMPA. For late-stage product candidates that we in-license for greater China rights, such as EVOMELA®, which has been launched, CID-103, BI-1206 and CB-5339, our development activities in China are to secure clinical trial notification, and marketing approval from the Center of Drug Evaluation (CDE) under the NMPA by conducting import drug registration. The “Law of the PRC on the Administration of Pharmaceuticals,” as last amended on August 26, 2019 and effective as of December 1, 2019, provides the basic legal framework for the administration of the production and sale of pharmaceuticals in China and covers the manufacturing, distributing, packaging, pricing and advertising of pharmaceutical products in China.
We are also subject to other PRC laws and regulations that are applicable to pharmaceutical manufacturers and distributors in general such as “Drug Registration Regulation (DRR)”, which was updated on January 22, 2020 and became effective on July 1, 2020.
The Marketing Authorization Holder System
Pursuant to the amended Law of the PRC on the Administration of Pharmaceuticals, the Marketing Authorization Holder System, previously implemented in a few pilot regions in China, is now implemented nationwide. Companies and research and development institutions can be drug marketing authorization holders after they receive drug approvals. The drug marketing authorization holder are responsible for their products throughout the life cycle, including nonclinical studies, clinical trials, production and distribution, post-market studies, and the monitoring, reporting, and handling of adverse reactions in connection with pharmaceuticals in accordance with the amended law.
The marketing authorization holders may engage contract manufacturers for manufacturing, provided that the contract manufacturers are licensed pharmaceutical manufacturers, and may engage pharmaceutical distribution enterprises with a valid drug distribution license to sell their products. Upon receiving the marketing authorizations from the NMPA, a drug marketing authorization
49
holder may transfer its drug marketing authorization and the transferee should have the capability of quality management, risk prevention and control, and liability compensation to ensure the safety, effectiveness and quality controllability of drugs, and fulfill the obligations of the drug marketing authorization holder.
Product Manufacturing
For the registration of locally manufactured drugs, the drug products need to be manufactured in China through either a self-owned facility or a contract manufacturing organization. The study drug to be used for clinical trials must be manufactured in compliance with NMPA Good Manufacturing Practice (GMP) guidelines. A domestic manufacturer of pharmaceutical products and active pharmaceutical ingredient (API) must obtain the drug manufacturing license to produce pharmaceutical products and API for marketing in China. Pursuant to the newly amended Law of the PRC on the Administration of Pharmaceuticals, the GMP certification has been cancelled, but with its cancellation, drug manufacturing enterprises are still required to strictly comply with GMP requirements. GMP requirements include institution and staff qualifications, production premises and facilities, equipment, raw materials, hygiene conditions, production management, quality controls, product distributions, maintenance of records and manner of handling customer complaints and adverse reaction reports. The drug manufacturing license is valid for five years, and must be renewed at least six months before its expiration date.
In addition, before commencing business, a pharmaceutical manufacturer must also obtain a business license from the Administration of Market Regulation at the local level.
Preclinical Research and Clinical Trials
For an investigational new drug application, a clinical trial approval issued from CDE was historically required to conduct clinical trials. However, since July 24, 2018, the NMPA announced to adopt a negative notification system for clinical trial approvals. In particular, if the applicant does not receive negative comments within 60 days after the CDE accepts the clinical trial application, the applicant can proceed with the clinical trial immediately based on the protocol submitted without waiting to receive an explicit clinical trial approval. Chemical generics, on the other hand, only need to undergo bioequivalent studies upon a filing for record with the NMPA. In order to apply for a clinical trial application approval to support local registration in China, a pharmaceutical company is required to conduct a series of preclinical research including research on chemistry, pharmacology, toxicology and pharmacokinetics of pharmaceuticals.
This preclinical research should be conducted in compliance with the relevant regulatory guidelines issued by the NMPA. In particular, safety evaluation research must be conducted in compliance with China’s Good Laboratory Practice (GLP). To further clarify the GLP certification requirement, the NMPA issued the Administrative Measures for the Certification of Good Laboratory Practices for Non-clinical Laboratory Studies (《药物非临床研究质量管理规范认证管理办法》) on January 19, 2023, which took effect on July 1, 2023, and pursuant to such Administration Measures, the institutions intending to carry out non-clinical safety evaluation studies in China for the purpose of drug registration applications are required to apply for the certification of GLP. The GLP certificate is valid for 5 years. Any entity without such certification must engage a qualified third party to conduct non-clinical studies.
After completion of preclinical studies and obtaining permission to conduct the clinical trial from the NMPA, clinical trials are generally conducted in three sequential phases that may overlap or be combined, known as Phase 1, Phase 2, and Phase 3 clinical trials, and Phase 4 clinical trials may be conducted at the post-marketing surveillance stage, in compliance with China’s Good Clinical Practice (GCP):
Phase 1 – preliminary trial of clinical pharmacology and human safety evaluation studies. The primary objective is to observe the pharmacokinetics and the tolerance level of the human body to the new medicine as a basis for ascertaining the appropriate methods of dosage.
Phase 2 – preliminary exploration on the therapeutic efficacy. The purpose is to assess preliminarily the efficacy and safety of pharmaceutical products on patients with the target indication of the pharmaceutical products and to provide the basis for the design and dosage tests for Phase 3. The dosing and methodology of research in this phase generally adopts double-blind, random methods with limited sample sizes.
50
Phase 3 – confirm the therapeutic efficacy. The objective is to further verify the efficacy and safety of pharmaceutical products on patients within the target indication, to evaluate the benefits and risks and finally to provide sufficient experimentally proven evidence to support the registration application of the pharmaceutical products. In general, the trial should adopt double-blind random methods with sufficient sample sizes.
Phase 4 –assess therapeutic efficacy and adverse reactions post-approval. The purpose is, by conducting a new drug’s post-marketing study, to assess therapeutic efficacy and adverse reactions when the drug is widely used, to evaluate overall benefit-risk relationships of the drug when used among the general population or specific groups and to adjust the administration dose, among others.
In April 2020, the NMPA and the National Health Commission (NHC) released the amended GCP, which took effect on July 1, 2020. The amended GCP is harmonized with the ICH-GCP. Compared to the previous GCP, the amended GCP provides comprehensive and substantive requirements on the design and conduct of clinical trials in China. In particular, the amended GCP enhances the protection for study subjects and tightens the control over bio-samples collected under clinical trials.
Collecting and Using Patients’ Biospecimens and Derived Data
Foreign-invested sponsors that collect and use patients’ biospecimens in clinical trials are required to file with the HGRAO, under the Ministry of Science and Technology, or the MOST. In 2017, the MOST issued the Circular on Optimizing the Administrative Examination and Approval of Human Genetic Resources, which simplified the approval for collecting and using human genetic resources for the purpose of commercializing a drug or medical device in the PRC. In June 2019, the State Council of the PRC issued the Regulation on the Administration of PRC Human Genetic Resources (effective as of July 1, 2019), which formalized the approval requirements pertinent to research collaborations between Chinese and foreign-owned entities.
Pursuant to this new HGR Regulation, a new notification system (as opposed to the advance approval approach originally in place) was put in place for clinical trials using PRC patients’ biospecimens and data at clinical study sites without involving the export of such specimens outside of China. The notification filing must specify the type, quantity, volume size and usage of the biospecimens, among others, with the HGRAO is required before conducting such clinical trials. The collection and use of PRC patients’ biospecimens and data in international basic research collaboration are still subject to the approval of the HGRAO. The notification filing with the HGRAO also applies to access to clinical study data by foreign entities.
In October 2020, the Standing Committee of the NPC promulgated the PRC Biosecurity Law, which took effect on April 15, 2021. The PRC Biosecurity Law, the higher law to the HGR Regulation, reaffirms the regulatory requirements stipulated by the HGR Regulation while potentially increasing the administrative fines significantly in cases where foreign entities are alleged to have collected, preserved or exported Chinese human genetic resources.
To accommodate with the upper-level governing laws’ requirements, the MOST promulgated the Implementing Rules of the Regulation on the Administration of Human Genetic Resources (《人类遗传资源管理条例实施细则》) on May 26, 2023, which took effect on July 1, 2023. The Implementing Rules provide specific requirements on the collection, preservation, utilization and providing human genetic resources out of China.
Import Drug Registration or Multi Regional Clinical Trials
NMPA regulations allow foreign drug developers to conduct import drug registration or multi regional clinical trials in China for a new drug as part of a global drug development program. An International Multicenter Clinical Trial (IMCT) Application needs to be filed with the CDE for conducting the clinical trials.
In October, 2017, the NMPA released the Decision on Adjusting Items concerning the Administration of Imported Drug Registration, as well as current requirement in DRR, which includes the following key points:
| ● | Phase 1 IMCT is allowed to be conducted in China. The IMCT drug does not need to gain prior approval or have entered into either a Phase 2 or 3 clinical trial in a foreign country before the IMCT could be conducted in China, except for preventive biological products. |
51
| ● | If the IMCT is conducted in China and the local recruitment of patients number allied with CDE, the application for drug marketing authorization can be submitted directly after the completion of the IMCT. |
| ● | With respect to clinical trial and market authorization applications for imported innovative chemical drugs and therapeutic biological products, the marketing authorization in the country or region where the foreign drug manufacturer is located will not be required. |
| ● | With respect to drug applications that have been accepted before the release of this Decision, importation permission can be granted if such applications request exemption of clinical trials for the imported drugs based on the data generated from IMCT and if relevant requirements under the Administrative Measures of the Drug Registration are met. |
The NMPA Decision on IMCT and the application for imported new drugs has streamlined and accelerated the applications for imported new drugs.
In order to apply for an IMCT Application in China, a biopharmaceutical company is required to submit a comprehensive investigation new drug application package filed with foreign regulatory agency, i.e. the FDA in our case, in a format compliant with NMPA guidance.
After obtaining the IMCT permit from the CDE, clinical trials should be conducted in compliance with both the FDA/ICH and NMPA Good Clinical Practice guidelines.
Data derived from IMCT can be used for the marketing authorization applications with the NMPA. When using IMCT data to support marketing authorization applications in China, applicants shall submit completed global clinical trial report, statistical analysis report and database, along with relevant supporting data in accordance with the ICH-CTD (International Conference on Harmonization-Common Technical Document) content and format requirements; subgroup research results summary and comparative analysis shall also be conducted concurrently.
Marketing Authorization Application
After completion of the first 3 phases of clinical trials demonstrating the safety and effectiveness of a pharmaceutical in its targeted indication, a Marketing Authorization Application needs to be filled with the NMPA, which includes research data of chemistry, manufacturing and controls, pre-clinical studies and clinical trial report in order to register the new drug. For imported drugs, the New Drug Registration Application is also known as the Import Drug License Application.
Once a marketing authorization is received, the product can be sold nationwide in China.
Pricing
The government regulates prices for pharmaceuticals (except for narcotic and Type 1 psychotropic drugs) mainly by establishing a price negotiation, consolidated procurement mechanism, and revising medical insurance reimbursement standards. The Chinese government has initiated several rounds of price negotiations with manufacturers of patented drugs, drugs with an exclusive source of supply, and oncology drugs since 2016. The average percentage of price reduction has been over 50%. Once the government agreed with the drug manufacturers on the supply prices, the drugs would be automatically listed in the NRDL and qualified for public hospital purchase.
Reimbursement
Market access in China was effectively transformed from 2016 and 2017 when the first annual negotiations were held for novel drugs to gain coverage under the NRDL. There was initially strong enthusiasm to participate from the majority of market players, with innovative drug makers willing to accept the price cuts demanded by the National Healthcare Security Administration (NHSA) in return for rapid public hospital system access at the earliest stages of a drug’s lifecycle. That enthusiasm has waned as the returns have dwindled, reflecting a combination of overly intense market competition and a natural ceiling on the government’s medical payment capabilities. Statistics shows that this year, over 52% of products eligible for NRDL inclusion decided to refrain from seeking entry.
52
In the eight years since the inception of negotiated reimbursement under China’s Basic Medical Insurance (BMI) system, the NHSA has led 6 rounds of NRDL negotiations, with the latest held on December 13, 2023. During that time, a total of 485 drugs have been newly added to the NRDL, with 88% of those still enjoying market exclusivity, while many more remain in the top-two in terms of market share despite facing generic/biosimilar challenge. It’s clear that despite having a reputation for excessive price pressure, the NRDL continues to play a primary role in drug commercialization in China, and for many remains a sought-after avenue for pharma firms with the right product.
Including traditional Chinese medicines (TCMs), the NRDL now covers 3,088 products following the 2023 updates, including 1,698 western medicines. In 2023, a total of 143 drugs entered the price bidding and negotiation channels, with 121 securing coverage, on average success rate of 84.6%; the overall average price cut required for negotiated/price bidding entry was 61.7%, with the steepest cut at 92% (for Aureole Pharma's inhalable procaterol). In terms of enthusiasm for new drugs to enter the list, 40% of the first-time molecular approvals in 2023 entered the NRDL, down from the high of 69% seen in 2020. For those existing NRDL Listings that renewed in 2023, there was a notable reduction in pricing pressure with 70% maintaining their existing price, and for the remainder on average price cut of only 6.7%.
The NHSA’s NRDL negotiations are focused purely on payment capability at present. Despite being the national payor, the NHSA and the BMI system simply cannot cover the demands of all parties involved. The national government has long advocated for a diversified response to China’s healthcare needs, with the hope that development of the commercial insurance sector can provide support alongside the public system. The private sector contribution has certainly been expanding. According to the NHSA, in 2022, premiums generated in the commercial health insurance sector reached RMB 865.3 billion, while insurers paid out RMB 360 billion in compensation. Chinese citizens have also accumulated ‘life insurance’ product savings of over RMB 1.6 trillion, a form of private pension product that will reduces financial worries, unleashing consumption potential, and promoting economic development. A total of 21 private insurance companies are also involved in supporting the urban and rural major illness insurance schemes, providing reimbursement to over 70 million people in the last ten years.
Hospital Listing
Government hospitals currently represent over 90% of the pharmaceutical market in China. In order for a new drug to be prescribed at a government hospital, it has to be listed in the hospital formulary. The process of entering into the formulary is commonly referred to as “hospital listing”, and typically requires a long lead time. These decisions are made on a hospital-by-hospital basis with timing that can range from every six months to every five years. Some hospitals also have temporary listing procedures that can accelerate timing. Private hospital and non-hospital pharmacies, which represent less than 10% of the drug market in China, do not require a formulary process to sell a drug.
Centralized Procurement and Tenders
Provincial and municipal government agencies will establish a provincial drug procurement agency to operate a mandatory collective tender process for purchases by government hospitals of a medicine included in provincial or local medicine procurement catalogs. The provincial or local medicine procurement catalogs are determined by the provincial drug procurement agency based on the National Essential Drugs List, the NRDL, local hospital formularies, etc. If a new drug has been included in a government hospital formulary, the NRDL or the provincial reimbursement drug list, the relevant hospitals must participate in collective tender processes for the purchase of such new drug. The centralized tender process is in principle conducted once every year in the relevant province or city in China. During the collective tender process, the provincial drug procurement agency will establish a committee consisting of recognized pharmaceutical experts. The committee will assess the bids submitted by the various participating pharmaceutical manufacturers, taking into consideration, among other things, the quality and price of the drug product and the service and reputation of the manufacturer. Only drug products that have been selected in the collective tender processes may be purchased by participating hospitals.
“4+7” Volume-based Drug Procurement and Tenders. In June 2018, the State Council decided to launch a new round of drug pricing and procurement reform. The reform policy aims to lower drug costs for patients, reduce transaction costs for enterprises, regulate drug use of hospitals, and improve the centralized drug procurement and pricing system. This reform is implemented mainly by the NHSA. The NHC supports the reform by introducing policy that encourages purchasing and prescribing of the selected drug. The NMPA is responsible for the quality assurance of the drugs submitted for tenders.
53
The national pilot scheme for centralized volume-based drug procurement and tenders under the reform was launched in November 2018. The selected drugs must pass the GQCE on quality and effectiveness.
The centralized volume-based procurement is open to all approved enterprises that manufacture drugs on the government-set procurement list in China. The NHSA organized nine rounds of volume-based procurement and tenders to this date. On November 6, 2023, the results of the ninth round of the volume-based procurement and tender were announced. All of the 41 listed products were successfully qualified to enter into a supply agreement with the group procurement organization and the average price reduction was 58%.
Regulations on PRC Company Law
The formation, operation and management of enterprises in the PRC are governed by the Company Law of the People’s Republic of China (《中华人民共和国公司法》) (the “Company Law”), which was promulgated by the Standing Committee of the NPC (NPCSC) on December 29, 1993, effective on July 1, 1994, and subsequently amended on December 25, 1999, August 28, 2004, October 27, 2005, December 28, 2013 and October 26, 2018, respectively. The Company Law also applies to foreign-invested companies. On December 29, 2023, the NPCSC further amended the Company Law, and this amendment mainly improves, optimizes and streamlines the provisions with respect to the company’s establishment and dissolution, corporate governance, capitalization management, and accountability of controlling shareholders and management personnel, and further promotes company’s social responsibilities. This amendment will be effective from July 1, 2024 onwards.
In accordance with the current regulatory framework governing foreign-invested enterprises in China, China adopts a pre-access national treatment plus negative list management system for foreign-investments. Pursuant to the provisions of the Law of the People’s Republic of China on Foreign Investment (《中华人民共和国外商投资法》), foreign-invested enterprises engaging in investments in certain industries listed in the Special Administrative Measures for Foreign Investment Entry (Negative List) (2021 Version) (《外商投资准入特别管理措施(负面清单)(2021 年版)》) issued by the NDRC and the Ministry of Commerce on December 27, 2021, are required to adhere to the corresponding pre-investment approval procedures, while, foreign-investments in industries not listed in the Negative List are only required to comply with registration and filing procedures as outlined in the Measures for the Reporting of Foreign Investment Information (《外商投资信息报告办法》), which were issued by the Ministry of Commerce and the State Administration for Market Supervision and Administration on December 30, 2019, and came into effect on January 1, 2020.
Regulations on Overseas Listing and Offering
On February 17, 2023, the CSRC released the Trial Administrative Measures of the Overseas Securities Offering and Listing by Domestic Companies (《境内企业境外发行证券和上市管理试行办法》) and five ancillary interpretive guidelines (collectively, the “Overseas Listing Trial Measures”), which apply to overseas offerings and listing by PRC-based companies, or domestic companies, of equity shares, depository receipts, corporate bonds convertible to equity shares, and other equity securities, and came into effect on March 31, 2023. According to the Overseas Listing Trial Measures, (1) domestic companies that seek to offer or list securities overseas, both directly and indirectly, should fulfill the filing procedure and report relevant information to the CSRC, and if a overseas-listed PRC-based issuer issues new securities in the same overseas market after the overseas offering and listing, it is also required to file with the CSRC within three business days after the completion of the issuance; if a domestic company fails to complete the filing procedure or conceals any material fact or falsifies any major content in its filing documents, such domestic company may be subject to administrative penalties, such as order to rectify, warnings, fines, and its controlling shareholders, actual controllers, the person directly in charge and other directly liable persons may also be subject to administrative penalties, such as warnings and fines; (2) if the a foreign-incorporated issuer meets both of the following conditions, its overseas offering and listing shall be determined as an indirect overseas offering and listing by a domestic company of the PRC: (i) any of the total assets, net assets, revenues or profits of the domestic operating entities of the issuer in the most recent accounting year accounts for more than 50% of the corresponding line item in the issuer’s audited consolidated financial statements for the same period; and (ii) its major operational activities are carried out in China or its main places of business are located in China, or the senior managers in charge of operation and management of the issuer are mostly Chinese citizens or are domiciled in China; and (3) where a PRC domestic company seeks to indirectly offer and list securities in an overseas market (including issuance of new securities after its overseas offering and listing), the issuer shall designate a major domestic operating entity responsible for all filing procedures with the CSRC.
54
Furthermore, in case any of the following major events occurs after the overseas offering and listing, the issuer is also required to report the concrete information to the CSRC within three business days of the occurrence and the announcement of the relevant events: (1) change of control; (2) the foreign securities regulatory body or the relevant competent authority has taken such measures as investigation and punishment; (3) conversion of listing status or listing board; and (4) voluntary of compulsory termination of listing. Where there is any material change in the major business and operation of the issuer after overseas offering and listing, and such change does not fall within the scope of filing, the issuer shall, within three business days of the occurrence of such change, submit a special report and a legal opinion issued by a domestic law firm to the CSRC to explain the relevant situation.
As substantially all of our operations are currently based in the PRC, our future offerings and major changes shall be subject to the foregoing filing procedures under the Overseas Listing Trial Measures. We cannot assure you that we could meet such requirements, obtain such permit from the relevant government authorities, or complete such filing in a timely manner or at all. Any failure may significantly limit or completely hinder our ability to continue to offer securities to investors and cause the value of such securities to significantly decline or be worthless. In addition, as the Overseas Listing Trial Measures was recently promulgated, there remains substantial uncertainties as to its interpretation and implementation and how it may impact our ability to raise or utilize fund and business operation.
On February 24, 2023, the CSRC and other relevant government authorities promulgated the Provisions on Strengthening the Confidentiality and Archives Administration of Overseas Securities Issuance and Listing by Domestic Enterprises (《关于加强境内企业境外发行证券和上市相关保密和档案管理工作的规定》) (the “Provision on Confidentiality”), which became effective on March 31, 2023. Pursuant to the Provision on Confidentiality, where a domestic enterprise provides or publicly discloses documents and materials involving state secrets and working secrets of national government authorities (the “State Confidential Information”) to the relevant securities companies, securities service institutions, overseas regulatory authorities and other entities and individuals, or provides or publicly discloses State Confidential Information through its overseas listing entity, it shall report to the competent governmental department with the examination and approval authority for approval in accordance with the law, and submit to the secrecy administration department of the same level for filing. Domestic enterprises providing accounting archives or copies thereof to entities and individuals concerned such as securities companies, securities service institutions and overseas regulatory authorities shall complete the corresponding procedures pursuant to the relevant national regulations.
Regulations on Foreign Exchange
General Administration of Foreign Exchange
Under the PRC Foreign Currency Administration Rules promulgated by the State Council on January 29, 1996, and last amended on August 5, 2008 and various regulations issued by the SAFE and other relevant PRC government authorities, Renminbi is convertible into other currencies for the purpose of current account items, such as trade related receipts and payments, payment of interest and dividends. The conversion of Renminbi into other currencies and remittance of the converted foreign currency outside China for the purpose of capital account items, such as direct equity investments, loans and repatriation of investment, requires the prior approval from the SAFE or its local branches. Payments for transactions that take place within China must be made in Renminbi. Unless otherwise provided by laws and regulations, PRC companies may repatriate foreign currency payments received from abroad or retain the same abroad. Foreign exchange proceeds under the current accounts may be either retained or sold to a financial institution engaging in settlement and sale of foreign exchange pursuant to relevant PRC rules and regulations. For foreign exchange proceeds under the capital accounts, approval from the SAFE is required for its retention or sale to a financial institution engaging in settlement and sale of foreign exchange, except where such approval is not required under the relevant PRC rules and regulations.
Regulations Relating to Offshore Investment
On July 4, 2014, the SAFE promulgated the Notice of the State Administration of Foreign Exchange on Relevant Issues Concerning Foreign Exchange Administration for Domestic Residents to Engage in Overseas Investment, Financing and Round-Trip Investment via Special Purpose Vehicles, or SAFE Circular 37, which regulates the relevant matters involving foreign exchange registration for round-trip investment. Under SAFE Circular 37, a PRC resident must register with the local SAFE counterpart before contributing assets or equity interests in an offshore special purpose vehicle, that is directly established or indirectly controlled by such PRC resident for the purpose of conducting investment or financing. In addition, following the initial registration, in the event of any major change in respect of the offshore special purpose vehicle, including, among other things, a change of offshore special purpose vehicle’s PRC resident shareholder(s), the name of the offshore special purpose vehicle, terms of operation, or any increase or reduction
55
of the offshore special purpose vehicle’s capital, share transfer or swap, and merger or division, the PRC resident shall complete the change of foreign exchange registration procedures for offshore investment with the local SAFE counterpart. According to the procedural guideline as attached to SAFE Circular 37, the principle of review has been changed to “the domestic individual resident shall only register the offshore special purpose vehicle directly established or controlled (first level).” At the same time, the SAFE has issued the Operation Guidance for the Issues Concerning Foreign Exchange Administration over Round-trip Investment with respect to the procedures for SAFE registration under SAFE Circular 37, which became effective on July 4, 2014, as an attachment to SAFE Circular 37. Under the relevant rules, failure to comply with the registration procedures set out in SAFE Circular 37 may result in restrictions being imposed on the foreign exchange activities of the relevant onshore company, including the payment of dividends and other distributions to its offshore parent or affiliate, and may also subject relevant PRC residents to penalties under PRC foreign exchange administration regulations. PRC residents who hold any shares in the company from time to time are required to register with the SAFE in connection with their investments in the company.
On February 13, 2015, the SAFE promulgated the Notice of the State Administration of Foreign Exchange on Further Simplifying and Improving Foreign Exchange Administration Policy on Direct Investment, or SAFE Notice 13, effective on June 1, 2015, which further amended SAFE Circular 37 by requiring domestic residents to register with qualified banks rather than SAFE or its local counterpart in connection with their establishment or control of an offshore entity established for the purpose of overseas investment or financing.
On March 30, 2015, SAFE promulgated the Circular of the State Administration of Foreign Exchange on Reforming the Administration Measures on Conversion of Foreign Exchange Registered Capital of Foreign-invested Enterprises, or SAFE Circular 19, effective on June 1, 2015, according to which the foreign exchange capital of foreign-invested enterprises must be subject to the Discretional Foreign Exchange Settlement, which refers to the foreign exchange capital in the capital account of a foreign-invested enterprise for which the rights and interests of monetary contribution has been confirmed by the local foreign exchange bureau (or the book-entry registration of monetary contribution by the banks) can be settled at the banks based on the actual operational needs of the foreign-invested enterprise. The proportion of Discretional Foreign Exchange Settlement of the foreign exchange capital of a foreign-invested enterprise is temporarily determined to be 100%. The Renminbi converted from the foreign exchange capital will be kept in a designated account, and if a foreign-invested enterprise needs to make further payment from such account, it still needs to provide supporting documents and go through the review process with the banks.
SAFE issued the Circular of the State Administration of Foreign Exchange on Reforming and Regulating Policies on the Control over Foreign Exchange Settlement of Capital Accounts, or SAFE Circular 16, on June 9, 2016, which became effective on the same day. Pursuant to SAFE Circular 16, enterprises registered in China may also convert their foreign debts from foreign currency to Renminbi on a discretionary basis. SAFE Circular 16 provides an integrated standard for conversion of foreign exchange under capital account items (including but not limited to foreign currency capital and foreign debts) on a discretionary basis which applies to all enterprises registered in China. SAFE Circular 16 reiterates the principle that Renminbi converted from foreign currency-denominated capital of a company may not be directly or indirectly used for purposes beyond its business scope or prohibited by PRC laws or regulations, while such converted Renminbi shall not be provided as loans to its non-affiliated entities. On October 23, 2019, SAFE promulgated the Circular of the State Administration of Foreign Exchange on Further Promoting the Facilitation of Cross-Border Trade and Investment, or SAFE Circular 28, which became effective on the same day. SAFE Circular 28 allows non-investment foreign- invested enterprises to use their capital funds to make equity investments in China as long as such investments do not violate the currently effective Negative List and the target investment projects are genuine and in compliance with laws. In addition, SAFE Circular 28 stipulates that qualified enterprises in certain pilot areas may use their capital income from registered capital, foreign debt, and overseas listing for the purpose of domestic payments without providing authenticity certifications to the relevant banks in advance for those domestic payments.
56
C. | Organizational Structure |
Below set forth is an organization chart of CASI as of March 28, 2024:
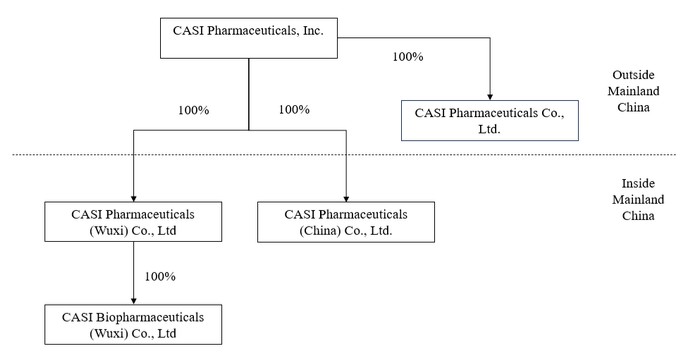
D. | Property, Plant and Equipment |
As of March 28, 2024, our principal executive offices are located in Beijing, China, consisting of approximately 1,300 square meters of leased office space, which serves as the center of our commercial as well as general administration functions. In addition to our principal executive offices, we also leased an office in Shanghai China, consisting of approximately 150 square meters, for regional commercial functions, and in Rockville, Maryland, consisting of approximately 4,100 square feet, for US’s business development and general administration functions. CASI Wuxi leased a workshop and office space of approximately 10,000 square meters. Our offices occupy an aggregate leased area of 11,450 square meters in China and 385 square meters in United States. The lessors of our offices are independent third parties, and we plan to renew these leases from time to time as needed. We believe that our facilities are adequate for our current needs and, should we need additional space, we believe we will be able to obtain additional space on commercially reasonable terms.
CASI Wuxi’s production workshop accounts for 60% of its leased area. While we have established a cGMP injectable products manufacturing line and obtained the Drug Distribution License and the Drug Manufacturing Permit, we are not yet approved by CDE to manufacture products and the manufacturing line has not been put to use.
ITEM 4A. UNRESOLVED STAFF COMMENTS
None.
ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with our consolidated financial statements and the related notes included elsewhere in this annual report on Form 20-F. This discussion may contain forward-looking statements based upon current expectations that involve risks and uncertainties. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of various factors, including those set forth under “Item 3. Key Information—D. Risk Factors” or in other parts of this annual report on Form 20-F.
A. | Operating Results |
Overview
We are a biopharmaceutical company focused on developing and commercializing innovative therapeutics and pharmaceutical products in China, the United States, and throughout the world. We are focused on acquiring, developing and commercializing products
57
that augment our hematology oncology therapeutic focus as well as other areas of unmet medical need. The Company is executing its plan to become a biopharmaceutical leader by launching medicines in the greater China market, leveraging its China-based regulatory, clinical and commercial competencies and its global drug development expertise.
We launched our first commercial product, EVOMELA® (Melphalan for Injection) in China in August 2019. In China, EVOMELA® is approved for use as a conditioning treatment prior to stem cell transplantation and as a palliative treatment for patients with multiple myeloma.
We announced the first patient received FOLOTYN® treatment in China on February 15, 2024. FOLOTYN® (Pralatrexate) is a dihydrofolate reductase inhibitor indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (“PTCL”). This product was approved by both the FDA and China’s NMPA for PTCL.
The other core hematology/oncology assets in our pipeline include:
| ● | CNCT19 is an autologous CD19 CAR-T investigative product (“CNCT19”) being developed by our partner Juventas for which we have exclusive world-wide co-commercial and profit-sharing rights. CNCT19 is being developed as a potential treatment for patients with hematological malignancies which express CD19 including, B-cell acute lymphoblastic leukemia (“B-ALL”) and B-cell non-Hodgkin lymphoma (“B-NHL”). CNCT19 targets CD19, a B-cell surface protein widely expressed during all phases of B-cell development and a validated target for B-cell driven hematological malignancies. CD19 targeted CAR constructs from several different institutions have demonstrated consistently high antitumor efficacy in children and adults with relapsed B-cell acute lymphoblastic leukemia (B-ALL), chronic lymphocytic leukemia (CLL), and B-cell non-Hodgkin lymphoma (B-NHL). In November 2023, the NMPA has granted market approval for Juventas' investigational cell therapy, CNCT19, for the treatment of relapsed and refractory B-cell acute lymphoblastic leukemia (r/r B-ALL) in China. The Company is currently involved in arbitration proceedings against Juventas in relation to Juventas’ purported termination of the CNCT19 Agreements. See “Item 8 — Financial Information — A. Consolidated Statements and Other Financial Information — Legal Proceedings” for further information. |
| ● | BI-1206 In October 2020, the Company entered into an exclusive licensing agreement with BioInvent for the development and commercialization of novel anti-FcγRIIB antibody, BI-1206, in mainland China, Taiwan, Hong Kong and Macau. BioInvent is a biotechnology company focused on the discovery and development of first-in-class immune-modulatory antibodies for cancer immunotherapy. BI-1206 is being investigated in a Phase 1/2 trial, in combination with anti-PD1 therapy Keytruda® (pembrolizumab), in patients with solid tumors, and in a Phase 1/2a trial in combination with MabThera® (rituximab) in patients with relapsed/refractory non-Hodgkin lymphoma (NHL). CTA was approved by NMPA in December 2021 and ethics committee approvals have been received in January of 2022. The Company obtained approval from HGRAO in April 2022. The Company is planning a Phase 1 study of BI-1206 in combination with rituximab with a single agent BI-1206 run in phase in patients with NHL (mantle cell lymphoma, marginal zone lymphoma, and follicular lymphoma) to assess PK, safety and tolerability, select the Recommended Phase 2 Dose and assess early signs of clinical efficacy as part of its development program for BI-1206 in China. The study received regulatory approval from the China Center for Drug Evaluation (“CDE”) in the second quarter of 2022, and the first patient was enrolled and dosed in the third quarter of 2022. |
| ● | CB-5339 is a novel VCP/p97 inhibitor focused on valosin-containing protein (VCP)/p97 as a novel target in protein homeostasis, DNA damage response and other cellular stress pathways for therapeutic use in the treatment of patients with various malignancies. On March 21, 2021, we entered into an exclusive license with Cleave for the development and commercialization of CB-5339 in mainland China, Hong Kong, Macau and Taiwan. On July 18, 2023, we entered into an assignment agreement with Cleave, pursuant to which we obtained the global intellectual property rights related to CB-5339. CB-5339, an oral second-generation, small molecule VCP/p97 inhibitor, has been evaluated in a Phase 1 clinical trial in patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). We submitted the CB-5339 CTA application for the multiple myeloma indication in March 2022 and received approval from the NMPA in January 2023. |
58
| ● | CID-103 is a full human IgG1 anti-CD38 monoclonal antibody recognizing a unique epitope that has demonstrated an encouraging preclinical efficacy and safety profile compared to other anti-CD38 monoclonal antibodies, and which we have exclusive global rights. CID-103 is being developed for the treatment of patients with multiple myeloma. The Phase 1 dose escalation and expansion study of CID-103, in patients with previously treated, relapsed or refractory multiple myeloma is closed to further accrual in France and the UK. |
| ● | Thiotepa is a chemotherapeutic agent, which has multiple indications including use as a conditioning treatment for use prior to certain allogeneic haemopoietic stem cell transplants. Thiotepa has a long history of established use in the hematology/oncology setting. The Company is applying for generic registration and, subject to regulatory and marketing approvals. |
As part of the long-term strategy to support our future clinical and commercial manufacturing needs and to manage our supply chain for certain products, on December 26, 2018, we established CASI Wuxi, between the Company and Wuxi LP, to develop a future GMP manufacturing facility that will be located in the Wuxi Huishan Economic Development Zone in Jiangsu Province, China. In November 2019, CASI Wuxi entered into a lease agreement for the right to use a state-owned land in China for the construction of a manufacturing facility. Pursuant to this agreement, CASI Wuxi intended to invest in land use rights and property, plant and equipment of RMB 1 billion by August 2022. Construction of the manufacturing facility began in the fourth quarter of 2020. Since our business focus has been shifted from ANDAs to the hematology-oncology therapeutic area, a substantial investment in GMP manufacturing facilities does not fit the current business focus. Therefore, in December 2022, we returned the land to the local Wuxi government for an amount of RMB 44.42 million, equivalent to the payment for land use right. Meanwhile, all construction in progress on the land was disposed. The Company recorded a total disposal loss amounted to US$2.2 million. Since we failed to meet the land development milestone, the local land administration authority requested CASI Wuxi to pay a land vacancy fee equivalent to 20% of the price for the land use rights according to the PRC Land Administration Law. We paid such fee in the amount of RMB 8.88 million to the local land administration authority in December 2022. Additionally, the Company received a government grant for the land development in April 2020 and November 2021 respectively, in the total amount of RMB 18.9 million. We are currently in negotiation with the local Wuxi government on the further treatment of the grant. The Wuxi government may require the Company to fully or partially return the grant and the Company may incur further losses.
In November 2023, CASI Wuxi obtained the Drug Manufacture Permit from local NMPA. In December 2023, the Company entered into a series of agreements, including a capital reduction agreement, a long term borrowing agreement, and four guarantee agreements, with Wuxi LP, CASI China and CASI Wuxi, pursuant to which, (i) CASI Wuxi will reduce its registered capital and return to Wuxi LP the investment principal made by Wuxi LP in CASI Wuxi in the amount of RMB134.2 million (equivalent to its original investment of US$20 million, the “Investment Principal”), together with certain investment return in the amount of RMB26.2 million to be paid in instalments, and Wuxi LP shall cease to be a shareholder of CASI Wuxi, (ii) Wuxi LP shall reinvest the Investment Principal into a three-year long term borrowing to CASI Wuxi (the “Long term borrowing”), which shall have a non-compounding annual interest rate of 4.05% and can, from the beginning date of the Long term borrowing term till the six month anniversary after the maturity of the Long term borrowing, be partially or fully converted into the equity interest of any subsidiaries of the Company at the conversion date fair value, solely at Wuxi LP’s discretion, and (iii) each of the Company and CASI China will provide irrevocable joint and several liability guarantees on the above-mentioned payment obligations. The term of the Long term borrowing will start on December 25, 2023 and end on December 31, 2026.
Key Factors Affecting Our Results of Operations
Key factors affecting our results of operations include the following:
Funding for Our Operations
Historically, we funded our operations primarily from financing through the issuance and sale of common stocks in private placement transactions. In recent years, we were also able to fund our operations in part with revenues generated from sales of our successfully commercialized product EVOMELA®. However, with the continuing expansion of our business and our product pipeline, we may require further funding through public or private offerings, debt financing, collaboration, and licensing arrangements or other sources. Any fluctuation in our ability to fund our operations will impact our cash flow plan and our results of operations.
59
Our Ability to Commercialize Our Drug Candidates
Our business and results of operations depend on our ability to commercialize our drug candidates, once and if those candidates are approved for marketing by the respective health authority. Currently, our pipeline consists of drug candidates ranging in development status from pre-clinical to late-stage clinical programs. Although we currently have only one product approved for commercial sale, we expect to generate revenue from sales of other drug candidates after we complete the clinical development, obtain regulatory approval, and successfully commercialize such drug candidates.
Supply and distribution of EVOMELA® and FOLOTYN®
We currently rely on a single source for the supply of both EVOMELA® and FOLOTYN®. The political and economic factors may affect the economies and financial markets of many countries, which may result in a period of economic slowdown or recessions. In such an event, our ability to continue to commercialize and expand distribution of EVOMELA® and FOLOTYN® could be adversely affected if the supplier refuses or is unable to provide products for any reason (including the occurrence of an event that makes delivery impractical). We would have to work with Acrotech to negotiate an agreement with a substitute supplier, which, assuming a substitute supplier was available, would likely interrupt the manufacturing of EVOMELA® and FOLOTYN®, cause supply chain delays and increase costs.
We rely on one single distributor, CRPCGIT for the distribution of EVOMELA® and CNMC for the distribution of FOLOTYN®. Our ability to maintain and grow our business will depend on our ability to maintain an effective distribution channel that ensures the timely delivery of our medicines. However, we have relatively limited control over our distributors, who may fail to distribute our drugs in the manner we contemplate. If price controls or other factors substantially reduce the margins our distributors can obtain through the resale of our medicines to hospitals, medical institutions and sub-distributors, they may terminate their relationship with us. While we believe alternative distributors are readily available, there is a risk that, if the distribution of our medicines is interrupted, our sales volumes and business prospects could be adversely affected.
Key Line Items of Our Results of Operations
Revenues
In the reporting period, we generated revenue primarily from the product sales. We also generated certain revenue from a sublicense agreement with PAT, and an equipment lease with Juventas.
Operating Costs and Expenses
Costs of revenues. Costs of revenues consists primarily of the cost of inventories of EVOMELA® and sales-based royalties related to the sale of EVOMELA®.
Research and Development Expenses. Research and development (R&D) expenses consist primarily of compensation and other expenses related to research and development personnel, research collaborations, costs associated with internal and contract preclinical testing and clinical trials of our product candidates, including the costs of drug substance and drug product, regulatory maintenance costs of ANDAs, facilities expenses, and amortization expense of acquired ANDAs.
General and Administrative Expenses. General and administrative expenses include compensation and other expenses related to executive, finance, business development and administrative personnel, professional services, investor relations and facilities, and amortization expense of acquired license right of FOLOTYN® before its launch.
Selling and Marketing Expenses. Selling and marketing expenses are the direct costs related to the sales of EVOMELA® that was launched in China in August 2019, such as sales force salaries, bonuses, advertising, and other marketing efforts.
60
Taxation
Cayman Islands
We are an exempted company incorporated in the Cayman Islands. The Cayman Islands currently levy no taxes on individuals or corporations based upon profits, income, gains or appreciations and there is no taxation in the nature of inheritance tax or estate duty. There are no other taxes likely to be material to our Company levied by the government of the Cayman Islands save certain stamp duties which may be applicable, from time to time, on certain instruments executed in or brought within the jurisdiction of the Cayman Islands. The Cayman Islands are not party to any double tax treaties that are applicable to any payments made by or to our Company. There are no exchange control regulations or currency restrictions in the Cayman Islands. In addition, the Cayman Islands does not impose withholding tax on dividend payments.
United States
In March 2023, we completed a Redomicile Merger, pursuant to which CASI Delaware merged with and into CASI Cayman, with CASI Cayman surviving the merger. Notwithstanding CASI Cayman’s organization under the laws of the Cayman Islands, pursuant to Section 7874 of the Code, CASI Cayman is treated for U.S. federal income tax purposes as a U.S. corporation, including with respect to any dividends paid by it. The U.S. federal corporate income tax rate is currently 21%.
China
Generally, our PRC subsidiaries and their respective subsidiaries, which are considered PRC resident enterprises under PRC tax law, are subject to enterprise income tax on their worldwide taxable income as determined under PRC tax laws and accounting standards at a rate of 25%. A “high and new technology enterprise” is entitled to a favorable income tax rate of 15% and such qualification is reassessed by relevant governmental authorities every three years. In addition, enterprises of encouraged industries are subject to preferential tax treatment or tax exemption for certain period in certain areas of China.
We are subject to value added tax, or VAT, at a rate of 13% thereafter on the sales of products, at a rate of 6% on the services rendered by us, less any deductible VAT we have already paid or borne. We are also subject to surcharges on VAT payments in accordance with PRC law.
Dividends paid by our wholly foreign-owned subsidiaries in China to us will be subject to a withholding tax rate of 10%, unless they qualify for a special exemption.
If our holding company in the Cayman Islands or any of our subsidiaries outside of China were deemed to be a “resident enterprise” under the PRC Enterprise Income Tax Law, it would be subject to enterprise income tax on its worldwide income at a rate of 25%.
Hong Kong
Our newly incorporated Hong Kong subsidiary is subject to profits tax in Hong Kong at the rate of 8.25%.
Critical Accounting Estimates
The preparation of our financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in our consolidated financial statements and accompanying notes. Actual results could differ materially from those estimates. We consider an accounting estimate to be critical if: (1) the accounting estimate requires us to make assumptions about matters that were highly uncertain at the time the accounting estimate was made, and (2) changes in the estimate that are reasonably likely to occur from period to period, or use of different estimates that we reasonably could have used in the current period, would have a material impact on our financial condition or results of operations. There are other items within our financial statements that require estimation but are not deemed critical, as defined above. Changes in estimates used in these and other items could have a material impact on our financial statements. For a detailed discussion of our significant accounting policies and related judgments, please see “Note 2—Summary of Significant Accounting Policies” to our consolidated
61
financial statements included elsewhere in this annual report. You should read the following description of critical accounting estimates in conjunction with our consolidated financial statements and other disclosures included in this annual report.
Impairment of Long-Lived Assets
Long-lived assets, including property, plant and equipment, right of use (“ROU”) assets and intangible assets subject to amortization, are reviewed for impairment whenever events or changes in circumstances (“triggering events”) indicate that the carrying amount of an asset or asset group may not be recoverable. We identify triggering events and performs impairment testing at asset group level which represents the lowest level for which identifiable cash flows are largely independent of the cash flows of other groups of assets and liabilities. As of December 31, 2023, our asset groups consist of manufacturing asset group and non-manufacturing asset group.
Triggering events include, but are not limited to, significant decrease of market price of the asset group, significant adverse change of an asset group’s use or physical condition, significant adverse changes in the industry conditions, significantly excessive accumulated cost compared with original expectation, expected continuing losses or negative cash flow associated with the use of the asset group, and expected significant early disposal of asset group.
When identifying triggering events for the manufacturing asset group, the assessment of expected operating results associated with the use of asset group and changes in the industry conditions may represent a triggering event required critical estimates and judgments.
If circumstances require an asset group be tested for possible impairment, we first compare undiscounted cash flows expected to be generated by that asset group to its carrying value. If the carrying value of the asset group is not recoverable on an undiscounted cash flow basis, an impairment is recognized to the extent that the carrying value exceeds its fair value. Fair value is determined through various valuation techniques including discounted cash flow models, quoted market values and third-party independent appraisals, as considered necessary.
62
Results of Operations
The following table sets forth a summary of our consolidated results of operations for the periods presented, both in absolute amount and as a percentage of our total net revenue for the periods presented. This information should be read together with our consolidated financial statements and related notes included elsewhere in this annual report.
Years Ended December 31, |
| |||||||||||||
2023 | 2022 |
| 2021 |
|
| |||||||||
| US$ |
| % | US$ |
| % |
| US$ |
| % |
| |||
(in thousands, except for percentages) |
| |||||||||||||
Revenues: |
|
|
|
|
|
|
|
|
|
|
|
| ||
Product sales |
| 33,879 |
| 100.0 | 38,047 |
| 88.3 |
| 30,020 |
| 99.5 |
| ||
Sublicensing revenue from a related party |
| — |
| — | 5,000 |
| 11.6 |
| — |
| — |
| ||
Lease income from a related party |
| — |
| — | 60 |
| 0.1 |
| 148 |
| 0.5 |
| ||
Total revenues |
| 33,879 |
| 100.0 | 43,107 |
| 100.0 |
| 30,168 |
| 100.0 |
| ||
Total costs of revenues |
| 13,827 |
| 40.8 | 15,827 |
| 36.7 |
| 12,557 |
| 41.6 |
| ||
Gross Profit |
| 20,052 |
| 59.2 | 27,280 |
| 63.3 |
| 17,611 |
| 58.4 |
| ||
Operating expenses (income): |
|
|
|
|
|
|
|
| ||||||
Research and development |
| 9,861 |
| 29.1 | 15,996 |
| 37.1 |
| 14,422 |
| 47.8 |
| ||
General and administrative |
| 25,387 |
| 74.9 | 23,449 |
| 54.4 |
| 23,766 |
| 78.8 |
| ||
Selling and marketing |
| 16,450 |
| 48.6 | 14,326 |
| 33.2 |
| 14,705 |
| 48.7 |
| ||
Other operating income | (6,366) | (18.8) | — |
| — | — |
| — | ||||||
Acquired in-process research and development |
| — |
| — | — |
| — |
| 6,555 |
| 21.7 |
| ||
Loss on disposal of long-lived assets |
| — |
| — | 2,058 |
| 4.8 |
| — |
| — |
| ||
Foreign exchange gain | (200) | (0.6) | (3,241) | (7.5) | (321) | (1.1) | ||||||||
Impairment of intangible assets |
| — |
| — | 8,724 |
| 20.2 |
| — |
| — |
| ||
Total operating expenses |
| 45,132 |
| 133.2 | 61,312 |
| 142.2 |
| 59,127 |
| 196.0 |
| ||
Loss from operations |
| (25,080) |
| (74.0) | (34,032) |
| (78.9) |
| (41,516) |
| (137.6) |
| ||
Non-operating income (expense): |
|
|
|
|
|
|
|
|
|
|
|
| ||
Interest income, net |
| 614 |
| 1.8 | 127 |
| 0.3 |
| 321 |
| 1.1 |
| ||
Other income |
| 764 |
| 2.3 | 44 |
| 0.1 |
| 558 |
| 1.8 |
| ||
Change in fair value of investments |
| (581) |
| (1.7) | (8,895) |
| (20.6) |
| 5,660 |
| 18.8 |
| ||
Gain from sale of an equity investment |
| — |
| — | 5,325 |
| 12.4 |
| — |
| — |
| ||
Impairment loss of long-term investments |
| (2,009) |
| (5.9) | — |
| — |
| (865) |
| — |
| ||
Loss before income tax benefit (expense) and share of net loss in an equity investee |
| (26,292) |
| (77.6) | (37,431) |
| (86.8) |
| (35,842) |
| (118.8) |
| ||
Income tax benefit (expense) |
| 81 |
| 0.2 | (1,980) |
| (4.6) |
| — |
| — |
| ||
Share of net loss in equity investee | (48) | (0.1) | (846) | (2.0) |
| — |
| — | ||||||
Net loss |
| (26,259) |
| (77.5) | (40,257) |
| (93.4) |
| (35,842) |
| (118.8) |
| ||
Revenues
Product Sales
Revenue was US$33.9 million for the year ended December 31, 2023, compared to US$38.0 million for the year ended December 31, 2022. Revenues decreased by 11% in the year ended December 31, 2023, as compared to same period in 2022. In 2023, the Company’s business faced a challenging external environment. The decrease of EVOMELA® sales was mainly attributable to the launch of an undifferentiated generic formulation of melphalan for injection product by a Chinese domestic manufacturer.
63
Revenue was US$38.0 million for the year ended December 31, 2022, compared to US$30.0 million for the year ended December 31, 2021. Revenues increased by 27% in the year ended December 31, 2022, as compared to same period in 2021 due to the continued growth in EVOMELA® sales.
Sublicensing revenue
Sublicensing revenue was nil for the year ended December 31, 2023.
In 2022, the Company received an upfront payment of US$5.0 million from PAT and recognized it as sublicensing revenue based on the sublicense agreement entered into between the Company and PAT in May 2022.
Lease Income
Lease income was nil for the year ended December 31, 2023.
Lease income was US$60,000 for the year ended December 31, 2022 compared to US$148,000 for the year ended December 31, 2021. This reduction was attributable to the termination of the leasing agreement upon sale of the underlying assets to Juventas in June 2022 (see Note 20).
Operating Expenses
Costs of Revenues
Costs of revenues were US$13.8 million for the year ended December 31, 2023, as compared to US$15.8 million for the year ended December 31, 2022, representing an decrease of 12.6%. The decrease of costs of revenues primarily resulted from the decreased sales of EVOMELA®. Costs of revenues as a percentage of EVOMELA® sales for 2023 and 2022 were 41% and 42%, respectively, which were stable.
Costs of revenues were US$15.8 million for the year ended December 31, 2022, as compared to US$12.6 million for the year ended December 31, 2021, representing an increase of 26%. The increase of costs of revenues primarily resulted from the increased sales of EVOMELA®. Costs of revenues as a percentage of EVOMELA® sales for 2021 and 2022 were both 42%, which were unchanged.
Research and Development Expenses
Research and development expenses for the year ended December 31, 2023 were US$9.9 million, compared with US$16.0 million for the year ended December 31, 2022. The decrease in R&D expenses primarily due to decrease in CID-103 as we incurred less laboratory tests and decrease in the research and development costs of generic pharmaceuticals in Wuxi manufacturing facility.
Research and development expenses for the year ended December 31, 2022 were US$16.0 million, compared with US$14.4 million for the year ended December 31, 2021. The increase in R&D expenses primarily due to the development of CID-103 and BI-1206.
General and administrative expenses
General and administrative expenses for the year ended December 31, 2023 were US$25.4 million, compared with US$23.4 million for the year ended December 31, 2022. The increase in general and administrative expenses was primarily attributable to incremental share-based compensation expense recognized due to the option modification in May 2023 amounted to US$2.2 million, and increased depreciation expense of US$1.2 million due to the full year depreciation expenses of CASI Wuxi’s leasehold improvement that started to depreciate in August 2022, offset by decrease of land vacancy fee of US$1.3 million in relation to the return of the Wuxi land use right.
64
General and administrative expenses for the year ended December 31, 2022 were US$23.4 million, compared with US$23.8 million for the year ended December 31, 2021. The decrease in general and administrative expenses was primarily due to the decrease in professional fees as we implemented strict cost controls measures in 2022.
Selling and Marketing Expenses
Selling and marketing expenses for the year ended December 31, 2023, were US$16.5 million, compared with US$14.3 million for the year ended December 31, 2022. The increase was primarily due to increased travel and conference expenses incurred for our commercial activities after the Chinese health authority cancelled the stringent COVID-19 controlled measure in December 2022.
Selling and marketing expenses for the year ended December 31, 2022, were US$14.3 million, compared with US$14.7 million for the year ended December 31, 2021. The decrease was primarily due to less travel and conference activities in 2022 resulted from the COVID-19 controlled measure.
Other Operating Income
Other operating income for the year ended December 31, 2023, were US$6.4 million, mainly consisted of a US$4.4 million reimbursement from PAT for certain labor cost and certain preclinical and clinical service incurred in previous years, a US$1.3 million refund from Pharmathen Global BV with respect to the termination of the exclusive distribution license agreement of product Octreotide LAI, and a US$0.6 million reimbursement from ESTEVE for certain costs of clinical trials for the registration of Thiotepa in China.
There was no other operating income for the years ended December 31, 2022 and 2021.
Acquired in-process Research and Development Expenses
There was no acquired in-process R&D expenses for the years ended December 31, 2023 and 2022.
Acquired in-process R&D expenses for the year ended December 31, 2021 were US$6.6 million, consisted of the upfront payment of US$5.5 million to Cleave for the development of CB-5339 and milestone payments to Alesta of US$1.1 million for the development of CID-103.
Loss on disposal of assets
There was no gain or loss on disposal of assets for the years ended December 31, 2023 and 2021.
Loss on disposal of assets for the year ended December 31, 2022 was US$2.1 million. The loss was primarily from the return of the Wuxi land use right. In December 2022, we returned the Wuxi land use right to the local government for an amount equivalent to the payment for the land use right. Meanwhile, all construction in progress on the land was disposed. The Company recorded a total disposal loss of US$2.2 million on these assets.
Foreign exchange gains
Foreign exchange gains for the year ended December 31, 2023 was US$0.2 million compared with US$3.2 million for the year ended December 31, 2022. The foreign exchange gains are primarily comprised of accounts receivable with CRPCGIT and USD denominated cash accounts that are held by our Chinese subsidiaries. The decrease is mainly due to that the exchange rate of RMB against USD fluctuated less than 2022.
Foreign exchange gains for the year ended December 31, 2022 was US$3.2 million compared with US$0.3 million for the year ended December 31, 2021. The foreign exchange gains are primarily comprised of accounts receivable with CRPCGIT and USD denominated cash accounts that are held by our Chinese subsidiaries.
Impairment of intangible assets
There was no impairment of intangible assets for the years ended December 31, 2023 and 2021.
65
For the year ended December 31, 2022, the intangible assets of six ANDAs with a total carrying amount of US$9.7 million were written down to their fair value of US$1.0 million, resulting in an impairment loss of US$8.7 million, which represents the difference between the carrying value of the intangible asset and its fair value. The Company estimated the fair value based on expected selling price.
Non-Operating Items
Interest Income, net
Interest income, net for the year ended December 31, 2023 was US$0.6 million compared with US$0.1 million for the year ended December 31, 2022. The increase in interest income, net was mainly due to our effective operation of funds and increased interest rate for cash and cash equivalents held in the U.S..
Interest income, net for the year ended December 31, 2022 was US$0.1 million compared with US$0.3 million for the year ended December 31, 2021. The decrease in interest income was mainly due to decreased average cash balance in 2022 compared to 2021.
Other income
Other income for the year ended December 31, 2023 was US$0.7 million, compared with US$44,000 for the year ended December 31, 2022. The increase was mainly attributable to a US$0.5 million income recognized from the repayment and termination of the convertible promissory note issued by Cleave to us, and a US$0.3 million government grant.
Other income for the year ended December 31, 2022 was US$44,000, compared with US$558,000 for the year ended December 31, 2021. The decrease of other income was mainly due to US$471,807 recorded in the year ended December 31, 2021 related to the loan to the Company under the Paycheck Protection Program (PPP) that was forgiven in September 2021. The rest of other income was mainly amortized income related to the government grant received from Wuxi’s local government for the construction of a manufacturing facility before the return of the land in December 2022.
Change in fair value of investments
The change in fair value of investments for the years ended December 31, 2023, 2022 and 2021 was a loss of US$0.6 million, a loss of US$8.9 million and a gain of US$5.7 million, respectively. The changes represent gains or losses on the Company’s investments in equity securities and long-term investment. The changes were mainly consisted of the fluctuations in the market price of ordinary shares of two publicly traded companies invested by us; changes in 2022 also include a change in fair value of a convertible loan based on our best estimation by using the discounted cash flow method.
Gain from sale of an equity investment
The Company held an investment in the equity securities of Juventas. In September 2022, CASI Biopharmaceuticals entered into an Equity Transfer Agreement with Jiadao Gongcheng, pursuant to which CASI Biopharmaceuticals agreed to transfer its equity interest in Juventas to Jiadao Gongcheng in the amount of RMB 240.9 million (approximately US$33.9 million). The transaction was closed in November 2022, and the Company recognized a gain of RMB 35.3 million (approximately US$5.3 million), representing the difference between the selling price and the carrying amount of this investment.
Impairment loss of long-term investments
Impairment loss of long-term investments for the year ended December 31, 2023 was US$2.0 million relating to the investment in PAT.
The Company did not record any impairment loss of long-term investments during the year ended December 31, 2022.
Impairment loss of long-term investments for the year ended December 31, 2021 was US$865,000 relating to the investment in a privately held UK Company, Black Belt Tx.
66
Income tax expense
The Company had an income tax benefit of US$81,000, which was attributable to the difference between the income tax provision for the year ended December 31, 2022 and the final tax return.
In relation with gain from the sale of Juventas, CASI Biopharmaceuticals recognized income tax expense of US$2.0 million for the year ended December 31, 2022.
The Company did not record any income tax expenses during the years ended December 31, 2023 and 2021.
Share of net loss in equity investee
In May 2022, CASI China entered into an agreement for the investment in PAT in the amount of RMB 20.0 million (approximately US$3.0 million) in cash during PAT’s first equity financing. CASI China has paid all the consideration in June 2022. Upon consummation of such equity financing, CASI China will hold 15% equity interests of PAT and will hold one of the three board seats. According to the agreement, CASI China began to assume profit or loss of PAT from the date when agreement was signed and recognized losses of US$48,000 and US$0.8 million for the years ended December 31, 2023 and 2022, respectively.
The Company did not record any share of net loss in equity investee during the years ended December 31, 2021.
B. | Liquidity and Capital Resources |
To date, the Company has only two products launched which generated limited revenue stream, and has been engaged in research and development activities for its pipeline products. As a result, the Company has incurred and expect to continue to incur operating losses in 2024 and the foreseeable future.
The Company will require significant additional funding to fund operations beyond the first quarter of 2024 until such time, if ever, it becomes profitable. The Company intends to augment its cash balances by pursuing other forms of capital infusion, including strategic alliances or collaborative development opportunities with organizations that have capabilities and/or products that are complementary to its capabilities and products in order to continue the development of its potential product candidates that they intend to pursue to commercialization. If the Company seeks strategic alliances, licenses, or other alternative arrangements, such as arrangements with collaborative partners or others, to raise further financing, it may need to relinquish rights to certain of its existing product candidates, or products they would otherwise seek to develop or commercialize on its own, or to license the rights to its product candidates on terms that are not favorable to it.
The Company will continue to seek to raise additional capital to fund its commercialization efforts, expansion of its operations, capital expenditure, research and development, and for the acquisition of new product candidates, if any. The Company intends to and is currently actively communicating to explore one or more of the following alternatives to raise additional capital:
| ● | raising bank loans; |
| ● | selling additional equity securities; |
| ● | out-licensing product candidates to one or more corporate partners; |
| ● | in-licensing products to expand revenue streams; |
| ● | completing an outright sale of non-priority assets; and/or |
| ● | engaging in one or more strategic transactions. |
The Company also will continue to manage its cash resources prudently and cost-effectively.
There can be no assurance that adequate additional financing under such arrangements will be available to the Company on terms that we deem acceptable, if at all. If additional funds are raised by issuing equity securities, dilution to existing shareholders may result, or the equity securities may have rights, preferences, or privileges senior to those of the holders of our ordinary shares. If we fail
67
to obtain additional capital when needed, we may be required to delay or scale back our commercialization efforts, advancement of the Acrotech products, or plans for other product candidates, if any.
Since its inception in 1991, the Company has incurred significant losses from operations and, as of December 31, 2023, has incurred an accumulated deficit of US$660.8 million. As of December 31, 2023, the Company had a balance of cash and cash equivalents of US$17.1 million, and short term investments of US$12.0 million. The Company believes that it has sufficient resources to fund its operations at least one year beyond the date that the audited consolidated financial statements are issued.
The following table sets forth a summary of our cash flows for the periods indicated:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
US$ | US$ | US$ | ||||
| (in thousands) | |||||
Summary Consolidated Cash Flow Data |
|
|
|
|
|
|
Net cash used in operating activities |
| (19,967) |
| (21,088) |
| (26,842) |
Net cash provided by (used in) investing activities |
| (9,673) |
| 31,159 |
| (20,691) |
Net cash provided by (used in) financing activities |
| (907) |
| (3,267) |
| 29,642 |
Effect of foreign exchange rate changes on cash and cash equivalents |
| 518 |
| 1,604 |
| (469) |
Net increase (decrease) in cash and cash equivalents |
| (30,029) |
| 8,408 |
| (18,360) |
Cash and cash equivalents at beginning of the year |
| 47,112 |
| 38,704 |
| 57,064 |
Cash and cash equivalents at end of the year |
| 17,083 |
| 47,112 |
| 38,704 |
Operating activities
Our net cash used in operating activities was US$20.0 million in 2023. The decrease of operating cash flow in amount of US$1.1 million compared to 2022 was mainly attributable to decreased research and development expenditures as we incurred less laboratory tests for our pipeline products and generic pharmaceuticals. In 2023, the principal items accounting for the difference between our net cash used in operating activities and our net loss of US$26.3 million, resulted from (i) adjustments for non-cash items totaled US$14.1 million, which mainly consisted of share-based compensation of US$7.2 million, depreciation and amortization expenses of US$3.7 million, and impairment of a long term investment of US$2.0 million, and partially offset by (ii) changes in operating assets and liabilities in the total amount of US$7.8 million.
Our net cash used in operating activities was US$21.1 million in 2022. In 2022, the principal items accounting for the difference between our net cash used in operating activities and our net loss of US$40.3 million, primarily resulted from (i) adjustment for non-cash items, mainly impairment of intangible assets of US$8.7 million, share-based compensation of US$7.0 million, loss on disposal of assets of US$2.1 million, and change in fair value of investments of US$8.9 million, partially offset by gain from disposal of long term investments of US$5.3 million, and partially offset by (ii) changes in operating assets and liabilities, mainly including inventories of US$4.2 million and accounts receivable of US$3.2 million.
Our net cash used in operating activities was US$26.8 million in 2021. In 2021, the principal items accounting for the difference between our net cash used in operating activities and our net loss of US$35.8 million, primarily resulted from (i) adjustment for non-cash or non-operating items, mainly share-based compensation of US$7.8 million and acquired in-process research and development of US$6.6 million, partially offset by change in fair value of investments of US$5.7 million, and partially offset by (ii) changes in operating assets and liabilities, mainly including accounts receivable of US$5.2 million.
Investing activities
Net cash used in investing activities was US$9.7 million in 2023, which was primarily attributable to purchase of short term investments of US$51.8 million, and purchase of long lived assets of US$2.2 million, offset by proceeds from sales or maturity of short term investments of US$43.3 million, and proceeds of US$1.0 million from the repayment and termination of the convertible promissory note issued by Cleave to CASI.
68
Net cash provided by investing activities was US$31.2 million in 2022, which was primarily attributable to the sale of the equity investment in Juventas of US$33.9 million.
Net cash used in investing activities was US$20.7 million in 2021, which was primarily attributable to our purchase of property, plant and equipment of US$8.9 million for the Wuxi manufacturing facility; upfront payment of cash to acquire in-process research and development of US$6.6 million, consisting of upfront payment of US$5.5 million to Cleave for the development of CB-5339 and milestone payments to Alesta of US$1.1 million for the development of CID-103, respectively; and payment of cash to acquire equity securities in Cleave of US$5.5 million.
Financing activities
Net cash used in financing activities was US$0.9 million in 2023, which was attributable to the payment of dividends to Wuxi LP of US$0.7 million, and repurchase of our ordinary shares of US$0.3 million, and offset by proceeds from exercise of share options of US$0.1 million.
Net cash used in financing activities was US$3.3 million in 2022, which was attributable to the repurchase of common stock in 2022.
Net cash provided by financing activities was US$29.6 million in 2021, which was attributable to the proceeds from the sale of CASI Delaware’s common stock of US$32.5 million, partially offset by the stock issuance costs of US$2.0 million.
Capital Expenditures
We had capital expenditures of US$8.9 million, US$5.6 million and US$2.2 million in 2021, 2022 and 2023, respectively. In 2023, our capital expenditures were mainly used for purchase of the license right of FOLOTYN®. We intend to fund our future capital expenditures with our existing cash balance.
Contractual Obligations and Commercial Commitments
The following table sets forth our contractual obligations as of December 31, 2023:
Payments Due by Period | ||||||||||||||
| Total |
| 2024 |
| 2025 |
| 2026 |
| 2027 | 2028 |
| Thereafter | ||
(US$ in thousands) | ||||||||||||||
Contractual Obligations: | ||||||||||||||
Lease obligations |
| 2,582 |
| 727 |
| 505 |
| 388 |
| 350 | 350 |
| 262 | |
Long term borrowing, dividends payable and interests payable | 24,192 | 1,508 | 1,883 | 20,801 | — | — | — | |||||||
In conjunction with the FOLOTYN® Assignment Agreement and Payment Agreement entered into in July 2023, in addition to the $2 million upfront payment already paid in 2023, the Company is responsible for certain contingent Deferred Payments up to $10.0 million to MICL and certain contingent payment up to $750,000 to Acrotech Inc. As of December 31, 2023, none of these contingent payments are considered probable based on the uncertainties of the successful renewal of the drug license in China.
In conjunction with various license agreements entered into by the Company, the Company is responsible for certain milestones and royalty payments. The Company has no unrecognized obligations or commitments that are probable to be paid in relation with these certain milestones and royalty payments. Please see Note 21 for details in the consolidated financial statements and notes to consolidated financial statements included in this annual report.
Other than those shown above, we did not have any significant capital and other commitments, long-term obligations, or guarantees as of December 31, 2023.
69
Stock Purchase Warrants
In history, CASI Delaware issued certain shares of common stock with accompanying warrants to certain institutional investors, accredited investors and existing stockholders. All those warrants were equity classified and expired in March 2023.
Off-balance Sheet Arrangements
We have not entered into any material financial guarantees or other commitments to guarantee the payment obligations of any third parties and do not assume credit risk in loans facilitated through our platform. We have not entered into any derivative contracts that are indexed to our shares and classified as shareholder’s equity or that are not reflected in our consolidated financial statements. Furthermore, we do not have any retained or contingent interest in assets transferred to an unconsolidated entity that serves as credit, liquidity or market risk support to such entity. We do not have any variable interest in any unconsolidated entity that provides financing, liquidity, market risk or credit support to us or engages in leasing, hedging or product development services with us.
C. | Research and Development, Patents and Licenses, Etc. |
Our success has benefited from our continuous efforts in building our technologies and protecting our intellectual property, including patents, trademarks, copyrights and trade secrets. See “Item 4. Information on the Company — B. Business Overview — Intellectual Property” for a description on the protection of our intellectual property.
The following table summarizes our research and development expenses by product candidates for the year of 2023:
| Year ended | |
December 31, | ||
2023 | ||
(US$ in thousands) | ||
CID‑103 |
| 3,190 |
BI‑1206 |
| 757 |
ANDAs |
| 605 |
Generic pharmaceuticals |
| 285 |
EVOMELA® |
| 246 |
CB-5339 | 282 | |
Thiotepa |
| 194 |
Others |
| 90 |
Unallocated research and development expenses |
|
|
Labor cost |
| 3,144 |
Depreciation and amortization |
| 1,068 |
Total research and development expenses |
| 9,861 |
D. | Trend Information |
In 2023, the Company’s business faced a challenging external environment, and the launch of an undifferentiated generic formulation of melphalan for injection product by a Chinese domestic manufacture caused decrease of EVOMELA® sales. Other than as disclosed elsewhere in this annual report, we are not aware of any trends, uncertainties, demands, commitments or events since January 1, 2024 that are reasonably likely to have a material adverse effect on our net revenues, income, profitability, liquidity or capital resources, or that caused the disclosed financial information to be not necessarily indicative of future operating results or financial condition.
E. | Critical Accounting Estimates |
See “Item 5. Operating and Financial Review and Prospects — B. Operating Results — Critical Accounting Estimates.”
70
ITEM 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
A. | Directors and Senior Management |
The following table sets forth information regarding our directors and executive officers as of the date of this annual report.
Directors and Officer Appointees | Age | Position/Title | ||
Wei-Wu He, Ph.D. |
| 59 |
| Chairman of the board of directors and CEO |
Y. Alexander Wu, Ph.D. |
| 61 |
| Independent Director |
Zhenbo Su |
| 48 |
| Independent Director |
Thomas Folinsbee |
| 57 |
| Independent Director |
Xuebo Zeng |
| 39 |
| Independent Director |
Wei (Larry) Zhang |
| 64 |
| President and Principal Financial Officer |
Alexander A. Zukiwski, MD |
| 65 |
| Global Chief Medical Officer |
Hai Huang |
| 55 |
| Global Chief Commercial Officer |
Chunhua Wang |
| 52 |
| Chief Operation Officer |
Kun Qian |
| 42 |
| Global Controller |
Wei Gao |
| 42 |
| General Counsel |
Wei-Wu He, Ph.D. Dr. He has served as chairman of the board of directors and CEO of CASI since April 2, 2019. Dr. He served as executive chairman of CASI Delaware from February 23, 2018 to April 2, 2019, as chairman to the board of directors of CASI Delaware from May 2013 to February 23, 2018, and as executive chairman from February 2012 to May 2013. Dr. He has been serving as executive chairman of Human Longevity Inc. (a privately-held biotechnology firm specializing in combining DNA sequencing with machine learning) since July 2019. He also is the founder and general partner of Emerging Technology Partners, LLC, a life sciences focused venture fund established in 2000. Dr. He has been involved in founding or funding over 20 biotech companies throughout his career, some of which went on to be acquired by significantly larger firms. In the earlier part of his career, Dr. He was one of the first few scientists at Human Genome Sciences, and prior to that, was a research fellow at Massachusetts General Hospital and Mayo Clinic. Dr. He is an author to more than 25 research publications and inventor of over 30 issued patents. Dr. He received his Ph.D. from Baylor College of Medicine and MBA from The Wharton School of University of Pennsylvania.
Y. Alexander Wu, Ph.D. Dr. Wu has been a director of CASI since April 2013. From 2006 to 2017, Dr. Wu was co-founder and chief executive officer of Crown Bioscience, Inc., a drug discovery and preclinical research organization in the oncology sector with over 600 employees, which was acquired by JSR for over US$400 million in 2017. Before co-founding Crown Bioscience, Dr. Wu was chief business officer of Starvax International Inc., a biopharmaceutical R&D company focusing on the development of novel therapeutic drugs for the treatment of infectious disease and cancer. Prior to Starvax, he was the head of Asian Operations with Burrill & Company, a life science venture capital and merchant bank. Dr. Wu also co-founded and was chief operating officer of Unimicro Technologies, a life science instrumentation company. He started his career with Hoffmann-La Roche, where he was manager of business development and strategic planning. Dr. Wu obtained his B.S. in biochemistry from Fudan University, China, a M.S. in biochemistry from the University of Illinois, and a Ph.D. in molecular cell biology and MBA from the University of California, Berkeley.
Zhenbo Su. Mr. Su has extensive experiences in the bioscience industry, and he has been a well-known investor in the life-science industry. Mr. Su is currently a partner of Guangzhou Redhill Capital Investment Management Co., Ltd, a leading life-science focused venture capital firm based in Guangzhou, which he co-founded in 2018. Prior to Mr. Su’s efforts in Redhill, he has served as the managing partner of Shenzhen Shared Investment Medical Fund since 2013. Mr. Su has served as an executive director of multiple companies in bioscience industry, such as TCRCure, Genetron, Medprint, Juventus, Hocermed, Polyrey, Pandamed, Light Vision and Meyer FSMP. Before Mr. Su started his career in investment, he was a director at Alcon China, a Novartis company, from 2007 to 2012. Earlier in Mr. Su’s career, he once worked at Johnson & Johnson in medical branch. Mr. Su holds a bachelor’s degree in medicine from Guangdong College of Medicine, a master’s degree in public healthcare policy and medical law from Sun Yat-sen University, and MBA from the University of Chicago.
Thomas Folinsbee. Mr. Folinsbee has over 25 years of experience as a financial and securities professional. He is the founder of Optivest Canada Ltd, a management consulting company focused on business development and investment research mandates for clients in the healthcare industry. Mr. Folinsbee was previously the director of corporate development for 3Sbio Inc. from 2009 to 2019. From 2017 to 2019, Mr. Folinsbee also served as independent director of Bison Capital Acquisition Corporation and was a member of the audit and
71
compensation committees after the merger with Xynomic Pharmaceuticals. From 2011 to 2016, he also worked for Hisanaga Seisakusho Co. Ltd., a Japanese manufacturing company. Before joining 3Sbio Inc., he also worked at Macquarie Equities, BNP Paribas and Optivest Systems Ltd. Mr. Folinsbee graduated in 1990 from McGill University with a bachelor of commerce degree concentrating in finance and international business and is CFA charter holder.
Xuebo Zeng. Mr. Zeng has been an executive director at IDG Capital since 2016, focusing on the investment in drug development, biotechnology, diagnostic devices and medical services. Mr. Zeng started his career in quality control department of Guilin Pharma in 2008 and soon became an investment manager specialized in life sciences investments. Prior to joining IDG Capital, he worked as an investment manager in two other famous private equity firms in China from 2010 to 2016. Mr. Zeng is a member of the board of directors of a number of private and listed companies, including Shanghai Model Organisms and Kelun-Biotech. Mr. Zeng received a bachelor’s degree in pharmacy from Qinghai Nationalities University, China.
Wei (Larry) Zhang. Mr. Zhang joined CASI in September 2018 as president of CASI (Beijing) Pharmaceuticals Co., Ltd., now known as CASI Pharmaceuticals (China) Co., Ltd. (“CASI China”), which is a subsidiary of CASI, and his role expanded to president and principal financial officer of CASI in September 2019. Mr. Zhang has more than 20 years management experience in the healthcare and biopharmaceutical industries in the U.S., Asia Pacific, and China. Prior to joining CASI’s Beijing office, Mr. Zhang was vice president, head of public affairs and corporate responsibility at Novartis Group (China) focusing on the public affairs/public relations strategy including initiating Novartis’ China policy focusing on NMPA new drug approval reform, IP protection, generic quality consistency evaluation and new regulations on biosimilars. From 2011 to 2016, he was chief executive officer of Sandoz Pharmaceutical (China), a Novartis Company. Mr. Zhang has also held executive leadership roles with Bayer Healthcare and Baxter International Corporation in the U.S. and Asia Pacific. He holds a bachelor and master degree in nuclear physics from University of Science & Technology of China, an MBA in marketing/finance from the University of California at Los Angeles (UCLA), and received Ph.D. training in political science from University of Utah.
Alexander A. Zukiwski, MD. Dr. Zukiwski joined CASI in April of 2017 as chief medical officer, and he currently serves as executive vice president and chief medical officer. Prior to joining the Company, Dr. Zukiwski was chief executive officer and chief medical officer of Arno Therapeutics and has been a director of Arno Therapeutics (“Arno”) since 2014. At Arno, his responsibilities included leading the clinical development and regulatory affairs teams to support the company’s pipeline. Prior to joining Arno in 2007, Dr. Zukiwski served as chief medical officer and executive vice president of clinical research at MedImmune LLC (“MedImmune”). Prior to MedImmune, Dr. Zukiwski held several roles of increasing responsibility at Johnson & Johnson’s (“J&J”) medical affairs and clinical development functions at Johnson & Johnson Pharmaceutical Research & Development LLC (“J&JPRD”), Centocor R&D and Ortho Biotech. Before joining J&J, he served in clinical oncology positions at pharmaceutical companies such as Hoffmann-LaRoche, Glaxo Wellcome and Rhone-Poulenc Rorer. Dr. Zukiwski has more than 21 years of experience in global drug development and supported the clinical evaluation and registration of many successful oncology therapeutic agents, including Taxotere®, Xeloda®, Procrit®/Eprex®, Velcade®, Yondelis®, and Doxil®. He previously served as a member of medical advisory board at Gem Pharmaceuticals, LLC and served as a director of Ambit Biosciences Corporation. Dr. Zukiwski holds a bachelor’s degree in pharmacy from the University of Alberta and a Doctor of Medicine degree from the University of Calgary. He conducted his post-graduate training at St. Thomas Hospital Medical Center in Akron, Ohio and the University of Texas MD Anderson Cancer Center.
Hai Huang. Mr. Huang has been global chief commercial officer (executive vice president) of CASI since January 2024. Mr. Huang leads the development of CASI's global commercialization strategy, overseeing the establishment of a global business system, and managing market expansion and strategic business partnerships. Prior to joining us, Mr. Huang served as the chief executive officer of Fosun Kite Biotechnology Co., Ltd. Mr. Huang worked with Pfizer China from 2016 to 2020, acting as general manager of essential business, chief marketing officer/vice president and then chief operating officer of essential business. From 2013 to 2016, Mr. Huang worked with Medtronic as a general manager of Diabetes BU for greater China region. From 2011 to 2013, Mr. Huang worked with Sanofi Aventis as a national sales leader of Diabetes BU. Mr. Huang joined Pfizer in 1997 and worked as sales director till 2010. Mr. Huang currently also holds positions as the Executive Director of the Shanghai CGT Special Committee and a member of the Shanghai Industry-Academia-Research Expert Group Committee. Mr. Huang received the Bi-MBA from Peking University and an undergraduate degree in Biochemistry from Lanzhou University.
Chunhua Wang. Ms. Wang has been chief operation officer of CASI since 2017. Ms. Wang is responsible for the Company’s back-office operation and manufacturing site management and is account for human resources, IT, communication, government affairs, legal, regulatory affairs, R&D, etc. Prior to joining CASI, Ms. Wang was the vice president of Vcanbio from 2015 to 2017. Before she joined Vcanbio, Ms. Wang was the VP at Marsh & McLennan Companies from 2014 to 2015. From 2011 to 2014, Ms. Wang served as the
72
HR director at Schneider Electric SA. Prior her experience in Schneider, Ms. Wang was the vice president at Tycoman Co., Ltd. a medical device company, from 2002 to 2010, and before that, Ms. Wang was the HR director at Tyco Healthcare. Earlier in Ms. Wang’s career, she worked at state-owned enterprises in human resource field. Ms. Wang received the bachelor’s degree in Metallurgy from China Northeastern University, and master’s degree in economics from Renmin University of China.
Kun Qian. Ms. Qian has been CASI’s global controller and vice president since January 2022. She is responsible for all corporate finance functions, including corporate controller, financial planning and analysis, treasury, tax, and regional finance activities. Prior to joining CASI, she served as financial director in Guazi.com from 2021 to 2022. Prior to that, she served as global controller at Wanda Sports Group, an industry group of Wanda Group, from 2016-2020 and completed its initial public offering in Nasdaq in 2019. She worked in international finance department at Weichai Group from 2014 to 2016. Prior to 2014, she was a senior audit manager and spent nearly 10 years with PricewaterhouseCoopers Zhong Tian LLP, Beijing office and PricewaterhouseCoopers, Singapore office. Ms. Qian received a bachelor’s degree in Management from University of International Business and Economics. She is a Certified Public Accountant in the State of New Hampshire and a member of American Institute of Certified Public Accountants. Ms. Qian also qualifies as a Certified Public Accountant in China and a Chartered Professional Accountant in Canada.
Wei Gao. Ms. Gao has been serving as CASI’s general counsel since January 2023. Prior to her current position, Ms. Gao was legal director since December 2020. Prior to joining CASI, Ms. Gao served as legal manager in Medtronic Beijing from 2017 to 2020. Earlier, Ms. Gao was a legal counsel of Syngenta Beijing from 2012 to 2017. From 2006 to 2007, and from 2010 to 2012, Ms. Gao served as legal consultant and senior consultant in Caterpillar. Earlier in her career, Ms. Gao worked as a litigation lawyer specializing international business dispute resolution in Commerce & Finance Law Offices from 2008 to 2010. Ms. Gao started her legal career as a legal assistant in Guo & Partners Attorneys At Law from 2005 to 2006. Ms. Gao received her bachelor’s degree in Economic Law from Jilin University and LL.M. degree in International Business Law from the University of Manchester.
B. | Compensation of Directors and Executive Officers |
For the year ended December 31, 2023, we paid or accrued an aggregate of US$3.7 million in salary and bonus, and other benefits to directors and officers (including a former officer). We have set aside or accrued US$0.3 million to provide pension, retirement or other similar benefits to directors and officers (including a former officer), we also recognized share-based compensation expense of US$3.4 million in relation with our directors and officers (including a former officer). Our China subsidiaries are required by law to make contributions equal to certain percentages of each employee’s salary for his or her pension insurance, medical insurance, unemployment insurance, work-related injury insurance and maternity insurance and other statutory benefits, and a housing provident fund.
Share Incentive Plans and CEO Plan
CASI Cayman succeeded to the interests of CASI Delaware following a redomicile merger pursuant to an agreement and plan of merger dated as of January 31, 2023 (the “Merger Agreement”) between CASI Cayman and CASI Delaware. Pursuant to the Merger Agreement, CASI Delaware merged with and into CASI Cayman, with CASI Cayman surviving the merger and each issued and outstanding shares of CASI Delaware’s common stock being converted into the right to receive one ordinary share of CASI Cayman. In addition, CASI Cayman assumed CASI Delaware’s existing obligations with respect to all outstanding options to purchase shares of CASI Delaware’s common stock and all other outstanding equity awards granted to directors, employees and consultants under CASI Delaware’s 2011 Long-Term Incentive Plan and 2021 Long-Term Incentive Plan, and certain other non-plan stock options to provide for the issuance of an equal number of CASI Cayman’s ordinary shares rather than the common stock of CASI Delaware upon the exercise of the awards, under the same terms and conditions.
The Company has two share incentive plans, the 2011 Long Term Incentive Plan and the 2021 Long Term Incentive Plan (as amended and collectively, the “Share Incentive Plans”). The Share Incentive Plans are adopted to attract and retain the best available personnel, provide additional incentives to employees, directors, officers, and consultants and promote the success of our business. In June 2019, the Company’s stockholders approved an amendment to the 2011 Long-Term Incentive Plan (the “2011 Plan”), increasing the number of shares of common stock reserved for issuance from 2,023,000 to 2,523,000 to be available for grants and awards. In June, 2021, the 2021 Long-Term Incentive Plan (the “2021 Plan”) was approved by the Company’s stockholders. The maximum number of shares of common stock that are available for grants and awards equals to 2,000,000 shares of stock, which includes 1,072,667 shares of common stock remaining under the 2011 Plan as of April 12, 2021. In addition to the Share Incentive Plans, (1) on June 20, 2019, CASI stockholders approved a grant of share options to Dr. He at the 2019 annual meeting, under the terms of which, Dr. He received
73
a share option covering 400,000 shares of common stock, at an exercise price of US$28.50, vesting upon the earlier of (i) the completion of a transformative event by CASI as determined at the discretion of CASI’s compensation committee and (ii) April 2, 2021, the second anniversary of the date of his appointment as CEO of CASI, and (2) on June 15, 2021, the board of directors of CASI approved a grant of share options to Dr. He which consists of 400,000 shares time-based and 400,000 shares performance-based share options (the “CEO Plan”).
On May 12, 2023, the board of directors of the Company adopted certain restated and amended 2011 Long-term Incentive Plan and restated and amended 2021 Long-term Incentive Plan (collectively, the “Amended Plans”), pursuant to which the board of directors of the Company authorized certain downward adjustments to the exercise prices of 3,927,859 options issued to the Company’s directors, executive officers, other management members and employees and outstanding. As of March 21, 2024, there were a total of 188,132 ordinary shares remain available for issuance, and share options in respect of 3,938,564 ordinary shares under the Share Incentive Plans and the CEO Plan are outstanding, the weighted average exercise price of which is US$2.5.
The following paragraphs describe the principal terms of the Share Incentive Plans, which descriptions are subject to the terms of the Share Incentive Plans and are incorporated herein by reference.
Types of Awards. The Share Incentive Plans permit the awards of share options, stock appreciation rights, restricted or unrestricted stock awards, phantom stock, performance awards or any combination of the foregoing.
Unexercised Options. If any award, or portion of an award, under the Share Incentive Plans expires or terminates unexercised, becomes unexercisable or is forfeited or otherwise terminated, surrendered or cancelled as to any shares, or if any ordinary shares are surrendered to the company in connection with any award (whether or not such surrendered shares were acquired pursuant to any award), the shares subject to such award and the surrendered shares shall thereafter be available for further awards under the Share Incentive Plans.
Plan Administration. Our board of directors or a committee of the board of directors (the “administrator”) will administer the Share Incentive Plans. The administrator will have full power and authority to take all other actions necessary to carry out the purpose and intent of the Share Incentive Plans, including, but not limited to, the authority to: (i) determine the eligible persons to whom, and the time or times at which awards will be granted; (ii) determine the types of awards to be granted; (iii) determine the number of shares to be covered by or used for reference purposes for each award; (iv) impose such terms, limitations, restrictions and conditions upon any such award as the administrator deems appropriate, including, but not limited to, whether a share option shall be an incentive share option or a nonqualified share option, any exceptions to nontransferability, any performance goals applicable to awards, any provisions relating to vesting, any circumstances in which the awards would terminate, the period during which awards may be exercised, and the period during which awards will be subject to restrictions; (v) accelerate, extend, or otherwise change the time in which an award may be exercised or becomes payable and to waive or accelerate the lapse, in whole or in part, of any restriction or condition with respect to such award, including, but not limited to, any restriction or condition with respect to the vesting or exercisability of an award due to termination of any participant’s employment or other relationship with our Company or an affiliate; and (vi) establish objectives and conditions, if any, for earning awards and determining whether awards will be paid after the end of a performance period.
Award Agreement. Awards granted under the Share Incentive Plans are evidenced by an award agreement that sets forth terms, conditions and limitations for each award, which may include the term of the award, the provisions applicable in the event of the grantee’s employment or service terminates.
Eligibility. We may grant awards to our employees (including employees-to-be), directors (including directors of a subsidiary or such other entity designated by the administrator) and consultants (including consultants of a subsidiary or such other entity designated by the administrator) of our Company.
Transfer Restrictions. Awards may not be transferred in any manner by the participant other than in accordance with the exceptions provided by the administrator or the relevant award agreement.
Capital Adjustments. In the event of any change in the outstanding ordinary share by reason of any stock dividend, split-up, stock split, recapitalization, reclassification, combination or exchange of shares, merger, consolidation, liquidation or the like, the administrator will provide for a substitution for or adjustment in (i) the number and class of shares of ordinary share subject to outstanding awards, (ii) the exercise price of share options and the base price upon which payments under stock appreciation rights are
74
determined, and (iii) the aggregate number and class of shares of ordinary share for which awards thereafter may be made under the plans.
Modification and Substitution of Awards. Subject to the terms and conditions of Stock Incentive Plans, the administrator may modify the terms of any outstanding awards. However, no modification of an award will, without the consent of the participant, alter or impair any of the participant’s rights or obligations under such award. Awards may, at the discretion of the administrator, be granted under the Share Incentive Plans in substitution for share options and other awards covering capital stock of another corporation which is merged into, consolidated with, or all or a substantial portion of the property or stock of which is acquired by, the company or one of its affiliates. The terms and conditions of the substitute awards so granted may vary from the terms and conditions set forth in the Stock Incentive Plans to such extent as the administrator may deem appropriate in order to conform, in whole or part, to the provisions of the awards in substitution for which they are granted. In the event of (a) a merger or consolidation to which the company is a party, or (b) a sale or exchange of all or substantially all of the company’s ordinary share for cash, securities or other property, the administrator shall take such actions, if any, as it deems necessary or appropriate to prevent the enlargement or diminishment of participants’ rights under the Share Incentive Plans and awards granted thereunder, and may, in its discretion, cause any award granted thereunder to be cancelled in consideration of a cash payment equal to the fair value of the cancelled award, as determined by the administrator in its discretion. The fair value of a share option will be deemed to be equal to the product of (x) the number of shares of ordinary share the share option covers (and has not previously been exercised) and (y) the excess, if any, of the fair market value of a share of ordinary share as of the date of cancellation over the exercise price of the share option.
Foreign Employees. Without amendment of the Share Incentive Plans, the administrator may grant awards to participants who are subject to the laws of foreign countries or jurisdictions on such terms and conditions different from those specified in the plans as may in the judgment of the administrator be necessary or desirable to foster and promote achievement of the purposes of the plans. The administrator may make such modifications, amendments, procedures, sub-plans and the like as may be necessary or advisable to comply with provisions of laws of other countries or jurisdictions in which the company or any of its affiliates operate or have employees.
Termination and Amendment of the Share Incentive Plans. No awards can be granted under the Share Incentive Plans after the 10th anniversary of its effectiveness. The board may terminate, amend or modify the Share Incentive Plans; provided, the board shall not amend or terminate the Plan without approval of (a) CASI’s shareholders to the extent applicable law or regulations or the requirements of the principal exchange or interdealer quotation system on which the ordinary share is listed or quoted, if any, requires shareholder approval of the amendment or termination, and (b) each affected grantee if the amendment or termination would adversely affect the grantee’s rights or obligations under any award granted prior to the date of the amendment or termination.
The following table includes certain information with respect to the value of all unexercised options previously awarded to our directors and executive officers as of March 21, 2024.
|
|
| |||||
Ordinary Shares | Option | Option | |||||
Underlying | Exercise | Expiration | |||||
Name and Principal Position | Options Granted | Price ($) | Date | ||||
Wei-Wu He, Ph.D., | 1,563,499 | $ | 1.93 | 4/6/2025~5/12/2033 | |||
Wei (Larry) Zhang, |
| 330,000 | $ | 1.93 |
| 9/1/2028~5/12/2033 | |
Zukiwski, Alexander Anthony, M.D. |
| 176,000 | $ | 1.93 |
| 4/3/2027~5/12/2033 | |
Hai Huang |
| 250,000 | $ | 6.61 |
| 1/29/2024 | |
Chunhua Wang |
| 155,000 | $ | 1.93 |
| 3/19/2028~5/12/2033 | |
Y. Alexander Wu, Ph.D. |
| * | $ | 1.93 |
| 6/15/2031~5/12/2033 | |
Kun Qian |
| * | $ | 1.93 |
| 3/30/2032~5/12/2033 | |
Wei Gao |
| * | $ | 1.93 |
| 6/18/2031~5/12/2033 | |
Thomas Folinsbee | * | $ | 1.93 | 4/14/2033 | |||
Xuebo Zeng | * | $ | 1.93 | 4/14/2033 | |||
Zhenbo Su | * | $ | 1.93 | 4/14/2033 | |||
* Aggregate number of shares represented by all grants of options to the person account for less than 1% of our total outstanding ordinary shares. | |||||||
75
As of March 21, 2024, other employees as a group hold 1,312,221 options to purchase ordinary shares of our Company, with exercise prices ranging from US$1.93 to US$42.7 per share.
C. | Board Practices |
Board of Directors
Our board of directors consists of five directors. Our currently effective memorandums and articles of association (our “Memorandum and Articles”) provides that the minimum number of directors shall be three and the exact number of directors shall be determined from time to time by our board of directors.
A director is not required to hold any shares in our Company by way of qualification. A director who is in any way, whether directly or indirectly, interested in a contract or transaction or proposed contract or transaction with us is required to declare the nature of his or her interest at a board meeting. Subject to Nasdaq listing rules and disqualification by the chairman of the relevant board meeting, a director may vote in respect of any contract or transaction or proposed contract or transaction, notwithstanding that he or she may be interested therein and if he or she does so, his or her vote shall be counted and he or she may be counted in the quorum at any meeting of directors at which any such contract or transaction or proposed contract or transaction is considered.
Our board of directors may exercise all the powers of our Company to raise or borrow money, and to mortgage or charge its undertaking, property, and assets (present or future) and uncalled capital or any part thereof, and to issue debentures, debenture stock, bonds, or other securities, whether outright or as collateral security for any debt, liability, or obligation of our Company or of any third party.
None of our directors has a service contract with us that provides for benefits upon termination of service.
Committees of the Board of Directors
We have established three committees under the board of directors: an audit committee, a compensation committee and a nominating and corporate governance committee. We have adopted a charter for each of the three committees. Each committee’s members and functions are described below.
Audit Committee. Our audit committee consists of Thomas Folinsbee, Y. Alexander Wu, Ph.D. and Xuebo Zeng. Thomas Folinsbee is the chairperson of the audit committee. Thomas Folinsbee satisfies the criteria of an audit committee financial expert as set forth under the applicable rules of the SEC. Each of Thomas Folinsbee, Y. Alexander Wu, Ph.D. and Xuebo Zeng satisfies the requirements for an “independent director” within the meaning of the Nasdaq listing rules and the criteria for independence set forth in Rule 10A-3 of the Exchange Act.
The audit committee oversees the Company’s accounting and financial reporting processes. The audit committee will be responsible for, among other things:
| ● | appointing the independent auditors and pre-approving all auditing and non-auditing services permitted to be performed by the independent auditors; |
| ● | reviewing with the independent auditors any audit problems or difficulties and management’s response; |
| ● | discussing the annual audited financial statements with management and the independent auditors; |
| ● | reviewing the adequacy and effectiveness of the Company’s accounting and internal control policies and procedures and any steps taken to monitor and control major financial risk exposures; |
| ● | reviewing and approving all proposed related party transactions; |
| ● | meeting separately and periodically with management and the independent auditors; and |
| ● | monitoring compliance with the Company’s code of business conduct and ethics, including reviewing the adequacy and effectiveness of the Company’s procedures to ensure proper compliance. |
76
Compensation Committee. Our compensation committee consists of Y. Alexander Wu, Ph.D., Xuebo Zeng and Zhenbo Su. Y. Alexander Wu, Ph.D. is the chairperson of the compensation committee. Each of Y. Alexander Wu, Ph.D., Xuebo Zeng and Zhenbo Su satisfies the requirements for an “independent director” within the meaning of the Nasdaq listing rules.
The compensation committee will assist the board of directors in reviewing and approving the compensation structure, including all forms of compensation, relating to the Company’s directors and officers. The compensation committee will be responsible for, among other things:
| ● | reviewing and approving, or recommending to the board of directors for its approval, the compensation for the Company’s chief executive officer and other officers; |
| ● | reviewing and recommending to the board of directors for determination with respect to the compensation of the Company’s non-employee directors; |
| ● | reviewing periodically and approving any incentive compensation or equity plans, programs similar arrangements; and |
| ● | selecting compensation consultant, legal counsel or other advisor only after taking into consideration all factors relevant to that person’s independence from management. |
Nominating and Corporate Governance Committee. Our nominating and corporate governance committee consists of Zhenbo Su, Thomas Folinsbee and Xuebo Zeng. Zhenbo Su is the chairperson of the nominating and corporate governance committee. Each of Zhenbo Su, Thomas Folinsbee and Xuebo Zeng satisfies the requirements for an “independent director” within the meaning of the Nasdaq listing rules.
The nominating and corporate governance committee will assist the board of directors in selecting individuals qualified to become directors of the Company and in determining the composition of the board of directors and its committees. The nominating and corporate governance committee will be responsible for, among other things:
| ● | selecting and recommending to the board nominees for election by the shareholders or appointment by the board; |
| ● | reviewing annually with the board the current composition of the board with regards to characteristics such as independence, knowledge, skills, experience and diversity; |
| ● | making recommendations on the frequency and structure of board meetings and monitoring the functioning of the committees of the board; and |
| ● | advising the board periodically with regards to significant developments in the law and practice of corporate governance as well as compliance with applicable laws and regulations, and making recommendations to the board on all matters of corporate governance and on any remedial action to be taken. |
Duties of Directors
Under Cayman Islands law, directors owe fiduciary duties to the company, including a duty of loyalty, a duty to act honestly, and a duty to act in what they consider in good faith to be in the company’s best interests. Directors must also exercise their powers only for a proper purpose. Directors also have a duty to act with skill and care. It was previously considered that a director need not exhibit in the performance of his or her duties a greater degree of skill than may reasonably be expected from a person of his or her knowledge and experience. However, English and Commonwealth courts have moved towards an objective standard with regard to the required skill and care and these authorities are likely to be followed in the Cayman Islands. In fulfilling their duty of care to our Company, our directors must ensure compliance with the Memorandum and Articles, as amended and/or restated from time to time. Our Company has the right to seek damages if a duty owed by any of our directors is breached. A shareholder may in certain circumstances have the right to seek damages in the name of our Company if a duty owed by our directors is breached.
Appointment and Removal of Directors
Our Memorandum and Articles provide that our directors may be appointed by ordinary resolution and removed by ordinary resolution. Our Memorandum and Articles also provide that our directors may, by the affirmative vote of a simple majority of the
77
remaining directors present and voting at a meeting of directors, appoint any person to be a director so as to fill a casual vacancy or as an addition to the existing board of directors.
The office of a director shall be vacated if, amongst other things, such director (a) becomes prohibited by applicable law from being a director; (b) becomes bankrupt or makes any arrangement or composition with his or her creditors, (c) dies or is found to be or becomes of unsound mind, (d) resigns his or her office by notice in writing to the Company, (e) without special leave of absence from the board, is absent from meetings of the board for three consecutive meetings, and the board resolves that his or her office be vacated; or (f) is removed from office pursuant to any other provision of our Memorandum and Articles.
Terms of Directors and Officers
A director shall hold office until such time as he or she resigns his or her office by notice in writing to the Company, is removed from office by ordinary resolution or is otherwise disqualified from acting as a director or removed in accordance with our Memorandum and Articles.
An appointment of a director may be on terms that the director shall automatically retire from office (unless he or she has sooner vacated office) at the next or a subsequent annual general meeting or upon any specified event or after any specified period in a written agreement between our Company and the director, if any; but no such term shall be implied in the absence of express provision. Each director whose term of office expires shall be eligible for re-election at a meeting of the shareholders or re-appointment by the board of directors.
Our officers are appointed by and serve at the discretion of the board of directors.
See “A. Directors and Senior Management” for information on the period during which each of our directors and officers has held office.
Employment Agreements and Indemnification Agreements
Each of our officers is a party to an employment agreement with the Company. Under these agreements, the employment of each officer is for a specified time period, and may be terminated for cause, at any time and without advance notice or compensation, for certain acts of the officer, such as material failure to perform and discharge duties and responsibilities, misconduct that is materially and significantly injurious to the Company, conviction of a felony involving the personal dishonesty or moral turpitude, and material breach of obligations under the employment agreement. The employment may also be terminated without cause upon 30-days advance written notice. The officer may resign at any time with 30-days advance written notice.
Each officer of the Company has agreed that during the employment term and at all times thereafter, such officer shall not, without the written consent of the Company, or except as required by applicable law, disclose to any person, other than a person to whom disclosure is reasonably necessary or appropriate in connection with the performance by the officer of his or her duties as an officer of the Company, any material confidential information obtained by such officer while in the employ of the Company with respect to the businesses of the Company or any of its subsidiaries, including but not limited to, operations, pricing, contractual or personnel data, products, discoveries, improvements, trade secrets, license agreements, marketing information, suppliers, dealers, principles, customers, or methods of distribution, or any other confidential information the disclosure of which the officer knows, or in the exercise of reasonable care should know, will be damaging to the Company.
In addition, each officer of the Company has agreed to be bound by non-competition and non-solicitation restrictions during the term of his or her employment and typically for one year or six months, depending on the nature of the termination, following the last date of employment. Specifically, each officer has agreed not to as an individual, principal, agent, employee, consultant or otherwise, directly or indirectly, or with respect to any company or entity with which the Company has concluded partnership, licensing, joint research and development or other similar business agreements during his employment with the Company, render any services to any firm or company or any division or subsidiary of any firm or company, engaged in the development or commercialization of compounds, analogs or derivatives of those compounds that (a) are of a similar type, that is, small molecules, (b) have more than one mechanism of action and cellular pathway in common with; and (c) are within the same field (i.e. oncology or inflammation) as, those being developed and or commercialized by the Company during the Term (“Competing Company”). In addition, for an additional period of six (6) months after the six-month period set forth above, the officer only may provide services to such a Competing Company if such officer does not
78
work on, or furnish confidential information regarding, any matter related to such compounds defined above. Moreover, for a period of twelve (12) months after the termination of such officer’s employment with the Company, officers shall not take any action, without the prior written consent of the Company, to assist his or her successor employer or any other entity in recruiting or hiring any other employee who was an employee of the Company during such officer’s employment.
The Company has entered into indemnification agreements with each of its director and officer. Under these agreements, the Company agrees to indemnify its directors and officers against certain liabilities and expenses incurred by such persons in connection with claims made by reason of their being a director or officer of the Company.
The Company also has obligations under a Change-in-control Agreement entered by and between CASI Delaware and Dr. Alexander A. Zukiwski, MD, which was later amended. The agreement provides for certain benefits either upon an involuntary termination of employment, other than for cause, or resignation for “good reason,” upon a “Triggering Event.” A Triggering Event includes a merger of the Company with and into an unaffiliated corporation if CASI Delaware is not the surviving corporation or the sale of all or substantially all of CASI Delaware’s assets. “Good reason” generally means any material diminution or change in salary, responsibilities or title; relocation to an office more than 50 miles from Company headquarters or office; a failure to continue health benefits; a failure to pay deferred compensation due under any plan; or the failure to honor any material aspect of the employment agreement. The benefits to be received by Dr. Zukiwski, in the event his employment is terminated after a Triggering Event occurs, include: (i) receipt of a lump sum severance payment equal to Dr. Zukiwski’s then current annual salary and the average of the two prior year’s bonuses; (ii) pro rata current year bonus; (iii) continuation of life, health and disability benefits for twelve months after the termination of employment and (iv) in accordance with the terms of Dr. Zukiwski’s option agreement, all outstanding options would accelerate and become immediately exercisable.
Board Diversity Matrix
Board Diversity Matrix (As of March 21, 2024) | ||||||||
Country of Principal Executive Offices: |
| People’s Republic of China | ||||||
Foreign Private Issuer | Yes | |||||||
Disclosure Prohibited Under Home Country Law | No | |||||||
Total Number of Directors | 5 | |||||||
| Female |
| Male |
| Non-Binary |
| Did Not Disclose Gender | |
Part I: Gender Identity | ||||||||
Directors |
| 0 | 5 | 0 | 0 | |||
Part II: Demographic Background | ||||||||
Underrepresented Individual in Home Country Jurisdiction | 1 | |||||||
LGBTQ+ | 0 | |||||||
D. | Employees |
We had 176, 224 and 243 employees as of December 31, 2021, 2022 and 2023, respectively. The majority of our employees are located in China. The following table sets forth the numbers of our employees categorized by function as of December 31, 2023.
| As of December 31, 2023 | |
Function: |
|
|
Management and General Administration |
| 31 |
Medical, Clinical and Registration |
| 19 |
Commercial |
| 150 |
Manufactory, Quality Control, Research and Development |
| 43 |
Total |
| 243 |
As required by laws and regulations in China, we participate in various employee benefits plans that are organized by municipal and provincial governments, including, among other things, housing fund, pension, medical insurance and unemployment insurance. We are required under PRC law to make contributions to employee benefit plans at specified percentages of the salaries, bonuses and certain allowances of our employees, up to a maximum amount specified by the local government from time to time.
79
We enter into standard employment with confidentiality and non-compete arrangements with our senior management and certain core personnel. These contracts include a standard non-compete covenant that prohibits the employee from competing with us, directly or indirectly, during his or her employment and for one year or six months
We believe that we maintain a good working relationship with our employees. None of our management or employees are represented by labor unions.
E. Share Ownership
Except as specifically noted, the following table sets forth information with respect to the beneficial ownership of our ordinary shares as of March 21, 2024 by:
| ● | each of our directors and executive officers; and |
| ● | each of our principal shareholders who beneficially own 5% or more of our total outstanding shares. |
The calculations in the table below are based on 13,401,375 ordinary shares outstanding as of March 21, 2024.
Beneficial ownership is determined in accordance with the rules and regulations of the SEC. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, we have included shares that the person has the right to acquire within 60 days, including through the exercise of any option, warrant or other right or the conversion of any other security. These shares, however, are not included in the computation of the percentage ownership of any other person.
| Amount and |
|
| ||
Nature of | Percentage of |
| |||
Beneficial | Ordinary Shares |
| |||
Ownership | Outstanding*** |
| |||
Directors and Officer Appointees:** | |||||
Wei-Wu He, Ph.D.(1) |
| 2,957,891 |
| 20.4 | % |
Y. Alexander Wu, Ph.D. (2) |
| * |
| * | |
Zhenbo Su(3) |
| * |
| * | |
Thomas Folinsbee(4) |
| * |
| * | |
Xuebo Zeng(5) |
| * |
| * | |
Wei (Larry) Zhang(6) |
| 198,681 |
| 1.5 | % |
Alexander A. Zukiwski, MD(7) |
| * |
| * | |
Chunhua Wang(8) |
| * |
| * | |
Hai Huang(9) |
| — |
| — | |
Kun Qian(10) |
| * |
| * | |
Wei Gao(11) |
| * |
| * | |
All Directors and Officers as a Group |
| 3,476,749 |
| 23.2 | % |
Principal Shareholders: |
|
|
|
| |
Panacea and affiliated entities(12) | 2,400,000 | 17.8 | % | ||
Emerging Technology Partners, LLC(13) |
| 1,097,341 |
| 8.2 | % |
Sparkle Byte Limited(14) |
| 1,019,852 |
| 7.6 | % |
Wealth Strategy Holding Ltd(15) |
| 908,788 |
| 6.8 | % |
IDG-Accel China and affiliated entities(16) |
| 853,879 |
| 6.4 | % |
* | Less than 1% of our total outstanding shares. |
** | Dr. Alexander A. Zukiwski, MD’s business address is 9620 Medical Center Drive, Suite 300, Rockville, MD 20850. The business address of all other directors and officers of CASI Cayman is 1701-1702, China Central Office Tower 1, No. 81 Jianguo Road Chaoyang District, Beijing, 100025, People’s Republic of China. |
*** None of the Company’s major shareholders have different voting rights than other holders of the Company’s ordinary shares.
80
(1) | Includes: (i) 687,051 ordinary shares held by Dr. He, (ii) 44,107 ordinary shares directly held by Emerging Technology Partners, LLC, (iii) 753,234 ordinary shares beneficially held by ETP Global Fund. L.P., (iv) 300,000 ordinary shares beneficially held by ETP BioHealth III Fund, L.P., (v) 50,000 ordinary shares beneficially owned by Huiying Memorial Foundation, and (vi) 1,123,499 ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. Beneficial ownership is based on the books and records of the Company and based in part on a seventh amendment to Schedule 13D filed on April 18, 2023. Emerging Technology Partners, LLC (“ETP”), a Delaware limited liability company, is the general partner of ETP Global Fund L.P. (“ETP Global”), a Delaware limited partnership. ETP also is the general partner of ETP BioHealth III Fund, L.P. (“ETP BioHealth”), a Delaware limited partnership. Dr. He is founder and managing partner of each of ETP and ETP Global. Huiying Memorial Foundation is a 501(c)(3) private family foundation and Dr. He is a member of the board of trustees and an officer of the Huiying Memorial Foundation. Dr. He does not participate in the investment decisions of the Foundation with respect to CASI’s ordinary shares and disclaims beneficial ownership of CASI’s ordinary shares held by Huiying Memorial Foundation. |
(2) | Represents ordinary shares and ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(3) | Represents ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(4) | Represents ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(5) | Represents ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(6) | Includes: (i) 2,015 ordinary shares and (ii) 196,666 ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(7) | Represents ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(8) | Represents ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(9) | Represents ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(10) | Represents ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(11) | Represents ordinary shares issuable upon exercise of options which are exercisable within 60 days of March 21, 2024. |
(12) | Based on a Form 144 filed on March 15, 2024; a Form 144 filed on March 5, 2024; a Schedule 13G/A filed on January 26, 2024; a Schedule 13G/A filed on November 09, 2023; a Schedule 13G/A filed on June 23, 2023; a Schedule 13G filed on April 10, 2023, and the books and records of the Company, Panacea Innovation Limited, a company incorporated in Cayman Islands, is the sole owner of Panacea Venture Healthcare Fund II GP Company, Ltd., a company incorporated in Cayman Islands, which is the general partner of Panacea Venture Healthcare Fund II, L.P, a partnership organized under the laws of Cayman Islands. James Huang is the sole owner of Panacea Innovation Limited. Each person above mentioned has the shared voting power and dispositive power over the ordinary shares. Pursuant to the abovementioned Schedule 13G/As and Form 144s filed in the years of 2023 and 2024, since the date of our annual report on Form 20-F for the year ended December 31, 2022, Panacea and affiliated entities’ shareholding percentage increased from 9.2% to 17.8%. |
(13) | Includes: (i) 44,107 ordinary shares directly held by Emerging Technology Partners, LLC, (ii) 753,234 ordinary shares beneficially held by ETP Global Fund. L.P., and (iii) 300,000 ordinary shares beneficially held by ETP BioHealth III Fund, L.P. |
(14) | Based solely on a Schedule 13D Amendment filed with the SEC on November 14, 2018, the record owner of these shares is Sparkle Byte Limited. By virtue of holding 100% of the equity interest of Sparkle Byte Limited, Snow Moon Limited may be deemed to have sole voting and dispositive power with respect to these shares. By virtue of holding 100% of the equity interest of Snow Moon Limited, Tianjin Jingran Management Center (Limited Partnership) may be deemed to have sole voting and dispositive power with respect to these shares. By virtue of being the general partner of Tianjin Jingran Management Center (Limited Partnership), He Xie Ai Qi Investment Management (Beijing) Co., Ltd. may be deemed to have sole voting and dispositive power with respect to these shares. By virtue of being the shareholders and/or directors of He Xie Ai Qi Investment Management (Beijing) Co., Ltd., Jianguang Li, Dongliang Lin, Fei Yang and Hugo Shong may be deemed to have shared voting and dispositive power with respect to these ordinary shares. The number of ordinary shares has been adjusted to reflect the reverse stock split in May 2022. |
(15) | Based solely on a Schedule 13G amendment filed on February 19, 2020 by Wealth Strategy Holding Limited (“WSH”), WSH has sole voting power and dispositive power over the shares. By virtue of being the controlling shareholder and/or director of the Reporting Person, Mr. Kung Hung Ka may be deemed to have sole voting and dispositive power with respect to these shares. The number of shares has been adjusted to reflect the reverse stock split in May 2022 and the expiration of certain warrants for purchase of our ordinary shares. |
(16) | Based on a Schedule 13D Amendment filed on February 21, 2023, and the books and records of the Company, the following persons have sole voting and dispositive power and shared voting and dispositive power over the shares: (i) IDG-Accel China Growth Fund III L.P., an exempted Cayman Islands limited partnership (“IDG-Accel Growth”), (ii) IDG-Accel China III |
81
Investors L.P., an exempted Cayman Islands limited partnership (“IDG-Accel Investors”), (iii) IDG-Accel China Growth Fund III Associates L.P., an exempted Cayman Islands limited partnership and the sole general partner of IDG-Accel Growth (“IDG-Accel Associates”), (iv) IDG-Accel China Growth Fund GP III Associates, Ltd., an exempted Cayman Islands limited company (“IDG-Accel GP,” and collectively with IDG-Accel Growth, IDG-Accel Investors and IDG-Accel Associates, “IDG-Accel”), and the sole general partner of each of IDG-Accel Investors and IDG-Accel Associates, (v) Chi Sing Ho, an individual, and director and shareholder of IDG-Accel GP, and (vi) Quan Zhou, an individual, director and shareholder of IDG-Accel GP. The number of ordinary shares has been adjusted to reflect the expiration of certain warrants for purchase of our ordinary shares.]
To our knowledge, as of March 21, 2024, a total of 13,401,375 ordinary shares were held by 72 record holder in the United States. The number of beneficial owners of our ordinary shares in the United States is likely to be much larger than the number of record holders of our ordinary shares in the United States. We are not aware of any arrangement that may, at a subsequent date, result in a change of control of our Company.
F. Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation
Not applicable.
ITEM 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A. | Major Shareholders |
Please refer to “Item 6. Directors, Senior Management and Employees—E. Share Ownership.”
B. | Related Party Transactions |
Employment Agreements and Indemnification Agreements
See “Item 6. Directors, Senior Management and Employees—C. Board Practice—Employment Agreements and Indemnification Agreements.”
Share Incentive Plans
See “Item 6. Directors, Senior Management and Employees—B. Compensation of Directors and Executive Officers—Share Incentive Plans.”
Option Grants
See “Item 6. Directors, Senior Management and Employees—B. Compensation of Directors and Executive Officers—Share Incentive Plans.”
Other Transactions with Entities under Control of Director and Officer
PAT
In May 2022, the Company entered into a Sublicense Agreement (the “PAT Sublicense Agreement”) with PAT, a company established under the laws of China, pursuant to which the Company granted PAT an exclusive (subject to the commercialization and co-marketing rights), perpetual, worldwide license, with the right to freely grant further sublicenses subject to terms and conditions in the PAT Sublicense Agreement, for the investigational anti-CD38 monoclonal antibody TSK011010 licensed and controlled by the Company from Black Belt Therapeutics Limited, in the treatment, prevention and diagnosis of autoimmune diseases, conditions and disorders in humans. Pursuant to the PAT Sublicense Agreement, (i) PAT will make an upfront payment of US$10.0 million equivalent in two equal instalments upon completion of its first and second financing, respectively, plus potential future payments or reimbursement of development and sales milestones and royalties to the Company; (ii) PAT shall reimburse the Company for certain labor cost incurred by the Company related to the assistance for the technology transfer, and certain cost associated with the preclinical and clinical service
82
incurred by the Company. In the fourth quarter of 2022, PAT made the first initial payment of US$5.0 million to CASI China, and CASI China recognized it in revenue upon receipt of the payment.
In the fourth quarter of 2023, the two parties reached an alignment that PAT should reimburse the Company for an amount of US$4.4 million for certain labor cost and certain preclinical and clinical service, the Company recognized this amount in other operating income. The Company received US$3.9 million in 2023 and the remaining balance in amount of US$0.5 million included in receivable from a related party has been collected in January 2024.
Also in May 2022, CASI China entered into an agreement for the investment in PAT in the amount of RMB 20.0 million in cash during PAT’s first equity financing. According to the agreement, CASI China assumes profit or loss of PAT from the date when agreement was signed. For the years ended December 31, 2023 and 2022, CASI China recognized its share of loss of PAT amounted of $48,000 and $0.8 million, respectively. For the year ended December 31, 2023, CASI China also recognized an impairment loss of US$2.0 million relating to the investment in PAT.
Juventas
On July 1, 2019, CASI Delaware entered into a one-year equipment lease with Juventas in the amount of RMB80,000 a month, which is classified as an operating lease. Transactions with Juventas are considered to be related party transactions as Dr. Wei-Wu He is one of the founding shareholders of Juventas. The lease was renewed in July 2020 and June 2021 with the same monthly lease income. During the year ended December 31, 2020, 2021 and 2022, the Company recognized lease income of US$140,000, US$148,000 and US$60,000, respectively. In June 2022, the Company and Juventas entered into a lab equipment transfer contract, pursuant to which the previous leasing agreement was terminated and the Company transferred the equipment to Juventas. Total consideration for the transfer was RMB 900,000 (US$138,000) in cash.
In June 2019, CASI Delaware entered into a license agreement for exclusive worldwide license and commercialization rights to an autologous anti-CD 19 T-cell therapy product (“CNCT19”) from Juventas. In connection with this license agreement, the Company made an upfront equity investment of RMB80 million in Juventas through CASI Biopharmaceuticals, a wholly-owned Chinese subsidiary in lieu of the upfront payment for the license. On September 22, 2020, Juventas and its shareholders (including CASI Biopharmaceuticals) agreed to certain terms and conditions required by a new third-party investor in connection with Juventas’ series B financing. In order to facilitate Juventas’ series B financing, the Company agreed to amend and supplement its original licensing agreement to provide Juventas and the Company with co-marketing and profit-sharing rights for CNCT19. Under the terms of the amended licensing agreement, the Company and Juventas will share a percentage of total net profits, with the Company receiving a tiered percentage increasing up to 50% of the net profit from commercial sales of CNCT19 depending on annual net sales. The amended agreement also specifies a minimum annual target net profit to be distributed to Juventas as a percentage of net profit from commercial sales. In addition, the Company will continue to be obligated to pay Juventas a single digit royalty fee equal to a percentage of net sales that varies by region. In addition, the Company invested in an additional series A plus equity interest in Juventas, resulting in the Company’s equity ownership increasing to 16.45% (post-Juventas series B financing) on a fully diluted basis. The Company is also entitled to appoint a director to Juventas’ board of directors and has a put right upon the occurrence of specified events. In October 2021, Juventas completed its Series C financing, upon which the Company’s equity ownership in Juventas decreased to 12.01% on a fully diluted basis.
In September 2022, CASI Biopharmaceuticals entered into an equity transfer agreement with Shenzhen Jiadao Gongcheng Equity Investment Fund (Limited Partnership) (“Jiadao Gongcheng”), a third party limited partnership enterprise incorporated under the laws of China, pursuant to which CASI Biopharmaceuticals agreed to transfer to Jiadao Gongcheng all of its equity interest in Juventas in the amount of RMB 240.9 million. As of December 31, 2022, the transaction was completed and CASI Biopharmaceuticals received the total payment in full.
March 2021 Underwritten Public Offering Transactions.
On March 24, 2021, CASI Delaware closed an underwritten public offering of 15,853,658 shares of the CASI’s common stock at a price to the public of US$2.05 per share (not adjusted to reflect the reverse stock split completed by CASI Delaware in May 2022, the “Offering”). The gross proceeds to CASI Delaware from the Offering were US$32.5 million before deducting the underwriting discounts and commissions and offering expenses payable by CASI Delaware.
83
ETP BioHealth III Fund L.P. (“ETP BioHealth”), in which Dr. Wei-Wu He is the founder and managing partner of ETP BioHealth’s general partner, purchased shares of common stock in the Offering at the public offering price and on the same terms as the other purchasers in the Offering. ETP BioHealth purchased 3,000,000 shares (not adjusted to reflect the reverse stock split completed by CASI Delaware in May 2022) at the public offering price of US$2.05 per share for a total of US$6.15 million.
C. | Interests of Experts and Counsel |
Not applicable.
ITEM 8. FINANCIAL INFORMATION
| A. | Consolidated Statements and Other Financial Information |
We have appended consolidated financial statements filed as part of this annual report.
Legal Proceedings
CASI is subject in the normal course of business to various legal proceedings in which claims for monetary or other damages may be asserted.
CASI is currently involved in arbitration proceedings against Juventas in relation to Juventas’ purported termination of the CNCT19 Agreements, between the Company and Juventas with respect to the commercialization of Juventas’ cell therapy, Inaticabtagene Autoleucel (CNCT 19). On March 2, 2024, CASI received a notice from Juventas, which purported to terminate the CNCT19 Agreements. CASI responded to Juventas’ purported termination notice, noting that Juventas was not entitled to unilaterally terminate the CNCT19 Agreements and further demanding that Juventas cease any conduct that may constitute further breach of the CNCT19 Agreements and execute a written undertaking regarding compliance with the CNCT19 Agreements by March 13, 2024. Juventas did not comply with CASI’s demands. On March 20, 2024, CASI submitted a Notice of Arbitration at the HKIAC against Juventas pursuant to the CNCT19 Agreements’ dispute resolution clauses, claiming that Juventas’ purported termination was invalid and that Juventas breached the CNCT19 Agreements and seeking, among other things, damages and injunctive reliefs. Together with the Notice of Arbitration, CASI also submitted an application for the appointment of an emergency arbitrator, seeking emergency injunctive reliefs. On the same day, Juventas also submitted a Notice of Arbitration at the HKIAC against CASI, alleging, among other things, that the CNCT19 Agreements were validly terminated and that CASI breached the CNCT19 Agreements. The HKIAC has appointed an emergency arbitrator in accordance with CASI’s application. The arbitration proceedings are ongoing.
Dividend Policy
While there are no restrictions (other than compliance with applicable laws) that limit our ability to pay dividends, CASI Delaware has not paid cash dividends on its common stock, and we have not paid, and do not currently intend to pay cash dividends on CASI Cayman’s ordinary shares in the foreseeable future. Our policy is to retain all earnings, if any, to provide funds for the operation and expansion of our business. Subject to the Amended Articles and certain requirements of Cayman Islands law, our board of directors has discretion on whether to distribute dividends and may consider factors such as our results of operations, financial condition, capital needs and acquisition strategy, among others, in making its determination. In addition, our shareholders may by ordinary resolution declare a dividend, but no dividend may exceed the amount recommended by our directors.
We may rely on dividends from our subsidiaries in China to pay dividend and other distributions on our ordinary shares. PRC regulations may restrict the ability of our PRC subsidiaries to pay dividends to us. Under the current regulatory regime in China, a PRC company may pay dividends only out of their accumulated profit, if any, determined in accordance with PRC accounting standards and regulations, and is required to set aside as general reserves at least 10% of its after-tax profit, until the cumulative amount of such reserves reaches 50% of its registered capital, prior to any dividend distribution. In addition, a PRC company shall not distribute any profits in a given year until any losses from prior fiscal years have been offset.
84
B. | Significant Changes |
Except as disclosed elsewhere in this annual report, we have not experienced any significant changes since the date of our audited consolidated financial statements included in this annual report.
ITEM 9. THE OFFER AND LISTING
| A. | Offering and Listing Details |
See “—C. Markets.”
B. | Plan of Distribution |
Not applicable.
C. | Markets |
CASI Delaware’s common stock was trading on The Nasdaq Capital Market under the symbol “CASI.” In May 2022, CASI Delaware completed a 10-to-1 reverse stock split of its common stock.
In March 2023, CASI Delaware completed a redomicile merger, pursuant to which CASI Delaware merged with and into CASI Cayman, its wholly-owned subsidiary, with CASI Cayman surviving the merger as the surviving company. Pursuant to the redomicile merger, each issued and outstanding share of the common stock of CASI Delaware was converted into the right to receive one ordinary share, par value US$0.0001 each, of CASI Cayman, credited as fully paid. CASI Cayman’s ordinary share continued to trade on the Nasdaq Capital Market under the symbol “CASI.”
D. | Selling Shareholders |
Not applicable.
E. | Dilution |
Not applicable.
F. | Expenses of the Issue |
Not applicable.
ITEM 10. ADDITIONAL INFORMATION
| A. | Share Capital |
Not applicable.
B. | Memorandum and Articles of Association |
The following are summaries of material provisions of our Memorandum and Articles and of the Companies Act, insofar as they relate to the material terms of our ordinary shares.
Objects of Our Company
Under our Memorandum and Articles, the objects of our Company are unrestricted and we have the full power and authority to carry out any object not prohibited by the law of the Cayman Islands.
85
Ordinary Shares
General
All of our issued and outstanding ordinary shares have been issued credited as fully paid and non-assessable. Our ordinary shares are issued in registered form, and are issued when registered in our register of members. We may not issue shares to bearer. Our shareholders who are non-residents of the Cayman Islands may freely hold and transfer their ordinary shares.
Dividends
The holders of our ordinary shares are entitled to such dividends as may be declared by our board of directors, subject to the Companies Act and our Memorandum and Articles, as amended and restated from time to time. In addition, our shareholders may declare dividends by ordinary resolution, but no dividend shall exceed the amount recommended by our board of directors. Under Cayman Islands law, dividends may be declared and paid only out of funds legally available therefor, namely out of either profit or share premium account, provided that in no circumstances may the Company pay a dividend if this would result in the Company being unable to pay its debts as they fall due in the ordinary course of business.
Register of Members
Under the Companies Act, the Company must keep a register of members and there shall be entered therein:
| ● | the names and addresses of the members, together with a statement of the shares held by each member, and such statement shall confirm (i) the amount paid or agreed to be considered as paid, on the shares of each member, (ii) the number and category of shares held by each member, and (iii) whether each relevant category of shares held by a member carries voting rights under the articles of association of the company, and if so, whether such voting rights are conditional; |
| ● | the date on which the name of any person was entered on the register as a member; and |
| ● | the date on which any person ceased to be a member. |
Under Cayman Islands law, the register of members of the Company is prima facie evidence of the matters set out therein (i.e., the register of members will raise a presumption of fact on the matters referred to above unless rebutted) and a member registered in the register of members shall be deemed as a matter of Cayman Islands law to have legal title to the shares as set against its name in the register of members.
Voting Rights
Each holder of ordinary shares is entitled to one vote for each share registered in his name on the register of members on all matters upon which the ordinary shares are entitled to vote on a poll. Voting on any resolution put to the vote at any meeting of shareholders is required to be by way of a poll and not on a show of hands. A poll shall be taken in such manner as the chairperson of the meeting directs, and the result of the poll shall be deemed to be the resolution of the meeting.
A quorum required for a general meeting of shareholders consists of one or more shareholders who hold shares which carry in aggregate not less than one-third of the paid up voting share capital of the Company, present at the meeting. Although not required by the Companies Act or the Memorandum and Articles, the Company expects to hold shareholders’ meetings from time to time and such meetings may be convened by the Company’s board of directors on its own initiative or upon a request to the directors by shareholders holding share which carry in aggregate not less than one-third of all votes attaching to the Company’s issued shares that carry the right to vote at general meetings. A general meeting may also be called by the chairperson of the board of directors of the Company. Advance notice of at least seven calendar days is required for the convening of the Company’s annual general meeting and other shareholders meetings.
An ordinary resolution to be passed by the shareholders requires the affirmative vote of a simple majority of the votes which are cast by those shareholders who, being entitled to do so, vote at a general meeting, while a special resolution requires the affirmative vote of not less than two-thirds of the votes which are cast by those shareholders who, being entitled to do so, vote at a general meeting.
86
Both ordinary resolutions and special resolutions may also be passed by a unanimous written resolution signed by all the shareholders of the Company, as permitted by the Companies Act and the Memorandum and Articles. A special resolution will be required for important matters such as change of name or making changes to the memorandum and articles of association of the Company.
Transfer of Ordinary Shares
Subject to the restrictions of the Memorandum and Articles, as applicable, any of the Company’s shareholders may transfer all or any of his or her ordinary shares by an instrument of transfer in the usual or common form or any other form approved by the Company’s board of directors.
Subject to the Nasdaq listing rules, the Company’s board of directors may, in its absolute discretion, decline to register any transfer of any ordinary share which is not fully paid up or on which the Company has a lien. The Company’s directors may also decline to register any transfer of any ordinary share unless:
| ● | the instrument of transfer is lodged with the Company, accompanied by the certificate for the ordinary shares to which it relates and such other evidence as the Company’s board of directors may reasonably require to show the right of the transferor to make the transfer; |
| ● | the instrument of transfer is in respect of only one class of ordinary shares; |
| ● | the instrument of transfer is properly stamped, if required; |
| ● | in the case of a transfer to joint holders, the number of joint holders to whom the ordinary share is to be transferred does not exceed four; or |
| ● | a fee of such maximum sum as Nasdaq may determine to be payable, or such lesser sum as the Company’s board of directors may from time to time require, is paid to the Company in respect thereof. |
If the Company’s directors refuse to register a transfer they shall, within two calendar months after the date on which the instrument of transfer was lodged, send to each of the transferor and the transferee notice of such refusal, including the relevant reason for such refusal. The registration of transfers may, on 14 days’ notice being given by advertisement in such one or more newspapers or by electronic means, be suspended and the register closed at such times and for such periods as the Company’s board of directors may from time to time determine; provided, however, that the registration of transfers shall not be suspended and the register shall not be closed for more than 30 days in any calendar year.
Liquidation
On a winding up of the Company, if the assets available for distribution among its shareholders shall be more than sufficient to repay the whole of the share capital at the commencement of the winding up, the surplus will be distributed among its shareholders in proportion to the par value of the shares held by them at the commencement of the winding up, subject to a deduction from those shares in respect of which there are monies due, of all monies payable to the Company for unpaid calls or otherwise. If the Company’s assets available for distribution are insufficient to repay all of the paid-up capital, the assets will be distributed so that the losses are borne by its shareholders in proportion to the par value of the shares held by them.
Calls on Ordinary Shares and Forfeiture of Ordinary Shares
The Company’s board of directors may from time to time make calls upon shareholders for any amounts unpaid on their ordinary shares in a notice served to such shareholders at least 14 days prior to the specified time of payment. The ordinary shares that have been called upon and remain unpaid are subject to forfeiture.
Redemption, Repurchase and Surrender of Ordinary Shares
The Company may issue shares on terms that are subject to redemption, at the Company’s option or at the option of the holders, on such terms and in such manner as may be determined before the issue of such shares, by the Company’s board of directors or by an ordinary resolution of the Company’s shareholders. The Company may also repurchase any of its shares provided that the manner and terms of such purchase have been approved by its board of directors or by the Company’s shareholders by ordinary resolution or are
87
otherwise authorized by the Memorandum and Articles. Under the Companies Act, the redemption or repurchase of any share may be paid out of the Company’s profits or out of the proceeds of a fresh issue of shares made for the purpose of such redemption or repurchase, or out of capital (including share premium account and capital redemption reserve) if the Company can, immediately following such payment, pay its debts as they fall due in the ordinary course of business. In addition, under the Companies Act no such share may be redeemed or repurchased (a) unless it is fully paid up, (b) if such redemption or repurchase would result in there being no shares outstanding, or (c) if the company has commenced liquidation. In addition, the Company may accept the surrender of any fully paid share for no consideration.
Variations of Rights of Shares
Whenever the capital of our Company is divided into different classes, the rights attached to any such class of shares (subject to any rights or restrictions for the time being attached to any class) may be materially and adversely varied either with the written consent of the holders of at least two-thirds of the issued shares of that class or with the sanction of a special resolution passed at a separate meeting of the holders of the shares of that class. The rights conferred upon the holders of the shares of any class issued with preferred or other rights shall not, subject to any rights or restrictions for the time being attached to the shares of that class, be deemed to be materially and adversely varied by the creation or issue of further shares ranking pari passu with or subsequent to such existing class of shares or the redemption or purchase of any shares of any class by our company. The rights of the holders of shares shall not be deemed to be materially and adversely varied by the creation or issue of shares with preferred or other rights including, without limitation, the creation of shares with enhanced or weighted voting rights.
Inspection of Books and Records
Holders of the Company’s ordinary shares have no general right under Cayman Islands law to inspect or obtain copies of the Company’s list of shareholders or its corporate records (save for the Memorandum and Articles, special resolutions and the register of mortgages and charges). However, the Company will provide its shareholders with annual audited financial statements.
Changes in Capital
The Company may from time to time by ordinary resolution:
| ● | increase its share capital by new shares of such amount as it thinks expedient; |
| ● | consolidate and divide all or any of its share capital into shares of a larger amount than its existing shares; |
| ● | sub-divide its existing shares, or any of them into shares of a smaller amount that is fixed by the Memorandum and Articles; and |
| ● | cancel any shares which, at the date of the passing of the resolution, have not been taken or agreed to be taken by any person and diminish the amount of its share capital by the amount of the shares so cancelled. |
Subject to the Companies Act and confirmation by the Grand Court of the Cayman Islands on an application by the Company for an order confirming such reduction, the Company may by special resolution reduce its share capital and any capital redemption reserve in any manner authorized by the Companies Act.
Issuance of Additional Shares
Our Memorandum and Articles authorizes the Company’s board of directors to issue additional ordinary shares from time to time as its board of directors shall determine, to the extent of available authorized but unissued shares.
Our Memorandum and Articles authorizes the Company’s board of directors to establish from time to time one or more series of preferred shares and to determine, with respect to any series of preferred shares, the terms and rights of that series, including:
| ● | the designation of the series; |
| ● | the number of shares of the series; |
88
| ● | the dividend rights, dividend rates, conversion rights, voting rights; and |
| ● | the rights and terms of redemption and liquidation preferences. |
The Company’s board of directors may issue preferred shares without action by its shareholders to the extent authorized but unissued. In addition, the issuance of preferred shares may be used as an anti-takeover device without further action on the part of the shareholders. Issuance of these shares may dilute the voting power of holders of ordinary shares.
Appointment and Removal of Directors
Unless otherwise determined by our Company in a general meeting, our Memorandum and Articles provides that our board will consist of not less than three directors. There are no provisions relating to retirement of directors upon reaching any age limit.
The directors have the power to appoint any person as a director either to fill a casual vacancy on the board or as an addition to the existing board. Our shareholders may also appoint any person to be a director by way of ordinary resolution.
An appointment of a director may be on terms that the director shall automatically retire from office (unless he or she has sooner vacated office) at the next or a subsequent annual general meeting or upon any specified event or after any specified period in a written agreement between our Company and the director, if any; but no such term shall be implied in the absence of express provision. Each director whose term of office expires shall be eligible for re-election at a meeting of the shareholders or re-appointment by the board of directors.
A director may be removed with or without cause by ordinary resolution.
In addition, the office of any director shall be vacated if the director (i) becomes prohibited by applicable law from being a director, (ii) becomes bankrupt or makes any arrangement or composition with his creditors, (iii) dies or is found to be or becomes of unsound mind, (iv) resigns his office by notice in writing to the Company; (v) without special leave of absence from the board, is absent from meetings of the board for three consecutive meetings and the board (excluding the absent director) resolves that his office be vacated; or (vi) is removed from office pursuant to any other provision of our Memorandum and Articles.
Proceedings of Board of Directors
Our Memorandum and Articles provide that our business is to be managed and conducted by our board of directors. The quorum necessary for board meetings may be fixed by the board and, unless so fixed at another number, will be a majority of the directors then in office (save that if there is only one director, the quorum shall be one).
A director is not required to hold any shares in our Company by way of qualification. A director who is in any way, whether directly or indirectly, interested in a contract or transaction or proposed contract or transaction with our Company is required to declare the nature of his or her interest at a board meeting. Subject to Nasdaq listing rules and disqualification by the chairman of the relevant board meeting, a director may vote in respect of any contract or transaction or proposed contract or transaction, notwithstanding that he or she may be interested therein and if he or she does so, his or her vote shall be counted and he or she may be counted in the quorum at any meeting of directors at which any such contract or transaction or proposed contract or transaction is considered.
Our Memorandum and Articles provide that the board may exercise all the powers of our Company to raise or borrow money and to mortgage or charge its undertaking, property and assets (present and future) and uncalled capital or any part thereof, to issue debentures, debenture stock, bonds and other securities, whether outright or as collateral security for any debt, liability or obligation of our Company or of any third party.
Exempted Company
The Company is an exempted company duly incorporated with limited liability under the Companies Act. The Companies Act distinguishes between ordinary resident companies and exempted companies. Any company that is registered in the Cayman Islands but conducts business mainly outside of the Cayman Islands, may apply to be registered as an exempted company. The requirements for an exempted company are essentially the same as for an ordinary company except for certain exemptions and privileges, including (a) an
89
exempted company does not have to file an annual return of its shareholders with the Registrar of Companies, (b) an exempted company is not required to open its register of members for inspection, (c) an exempted company does not have to hold an annual general meeting, (d) an exempted company may register by way of continuation in another jurisdiction and be deregistered in the Cayman Islands, (e) an exempted company may obtain an undertaking against the imposition of any future taxation (such undertakings are usually given for 30 years in the first instance), (f) an exempted company may register as a limited duration company and (g) an exempted company may register as a segregated portfolio company.
“Limited liability” means that the liability of each shareholder is limited to the amount unpaid by the shareholder on that shareholder’s shares of the company (except in exceptional circumstances, such as fraud, the establishment of an agency relationship or an illegal or improper purpose or other circumstances in which a court may be prepared to pierce or lift the corporate veil).
Differences in Corporate Law
The Companies Act of the Cayman Islands is derived, to a large extent, from the older Companies Acts of England but does not follow recent United Kingdom statutory enactments, and accordingly there are significant differences between the Companies Act of the Cayman Islands and the current Companies Act of England. In addition, the Companies Act differs from laws applicable to United States corporations and their shareholders. Set forth below is a summary of certain significant differences between the provisions of the Companies Act applicable to us and the comparable provisions of the laws applicable to companies incorporated in the United States and their shareholders.
Mergers and Similar Arrangements. The Companies Act permits mergers and consolidations between Cayman Islands companies and between Cayman Islands companies and non-Cayman Islands companies. For these purposes, (a) “merger” means the merging of two or more constituent companies and the vesting of their undertaking, property and liabilities in one of such companies as the surviving company and (b) a “consolidation” means the combination of two or more constituent companies into a combined company and the vesting of the undertaking, property and liabilities of such companies in the consolidated company. In order to effect such a merger or consolidation, the directors of each constituent company must approve a written plan of merger or consolidation, which must then be authorized by (a) a special resolution of the shareholders of each constituent company, and (b) such other authorization, if any, as may be specified in such constituent company’s articles of association. The written plan of merger or consolidation must be filed with the Registrar of Companies of the Cayman Islands together with a declaration as to the solvency of the consolidated or surviving company, a declaration as to the assets and liabilities of each constituent company and an undertaking that a copy of the certificate of merger or consolidation will be given to the members and creditors of each constituent company and that notification of the merger or consolidation will be published in the Cayman Islands Gazette. Dissenting shareholders have the right to be paid the fair value of their shares (which, if not agreed between the parties, will be determined by the Cayman Islands court) if they complies strictly with the procedures set out in the Companies Act, subject to certain exceptions. Court approval is not required for a merger or consolidation which is effected in compliance with these statutory procedures.
Separate from the statutory provisions relating to mergers and consolidations, the Companies Act also contains statutory provisions that facilitate the reconstruction and amalgamation of companies by way of schemes of arrangement between a company and its members (or any class of them), or between the company and its creditors (or any class of them), provided that the arrangement is approved by (a) 75% in value of the shareholders or class of shareholders, as the case may be, or (b) a majority in number representing 75% in value of the creditors or class of creditors, as the case may be, with whom the arrangement is to be made, that are, in each case, present and voting either in person or by proxy at a meeting, or meetings, convened for that purpose. The convening of the meetings and subsequently the arrangement must be sanctioned by the Grand Court of the Cayman Islands. While a dissenting shareholder has the right to express to the court the view that the transaction ought not to be approved, the Grand Court of the Cayman Islands can be expected to approve the arrangement if it determines that:
| ● | the statutory provisions as to the required majority vote have been met; |
| ● | the shareholders have been fairly represented at the meeting in question and the statutory majority are acting bona fide without coercion of the minority to promote interests adverse to those of the class; |
| ● | the arrangement is such that may be reasonably approved by an intelligent and honest man of that class acting in respect of his interest; and |
| ● | the arrangement is not one that would more properly be sanctioned under some other provision of the Companies Act. |
90
The Companies Act also contains a statutory power of compulsory acquisition which may facilitate the “squeeze out” of dissentient minority shareholder upon a tender offer. When a tender offer is made and accepted by holders of 90% of the shares affected within four months, the offeror may, within a two-month period commencing on the expiration of such four month period, require the holders of the remaining shares to transfer such shares to the offeror on the terms of the offer. An objection can be made to the Grand Court of the Cayman Islands but this is unlikely to succeed in the case of an offer which has been so approved unless there is evidence of fraud, bad faith or collusion.
If an arrangement and reconstruction by way of scheme of arrangement is thus approved and sanctioned, or if a tender offer is made and accepted in accordance with the foregoing statutory procedures, a dissenting shareholder would have no rights comparable to appraisal rights, which would otherwise ordinarily be available to dissenting shareholders of Delaware corporations, providing rights to receive payment in cash for the judicially determined value of the shares.
Shareholders’ Suits. In principle, we will normally be the proper plaintiff to sue for a wrong done to us as a Company, and as a general rule, a derivative action may ordinarily not be brought by a minority shareholder. However, based on English authority, which would in all likelihood be of persuasive authority in the Cayman Islands, the Cayman Islands courts can be expected to follow and apply the common law principles (namely the rule in Foss v. Harbottle and the exceptions thereto) so that a minority shareholder may be permitted to commence a class action against, or derivative actions in the name of, our Company to challenge:
| (a) | an act which is ultra vires or illegal and is therefore incapable of ratification by the shareholders, |
| (b) | an act which constitutes a fraud against the minority where the wrongdoers are themselves in control of our Company, and |
| (c) | an act, although not ultra vires, could only be effected duly if authorized by more than a simple majority vote which has not been obtained. |
Indemnification of Directors and Executive Officers and Limitation of Liability. Cayman Islands law does not limit the extent to which a company’s memorandum and articles of association may provide for indemnification of officers and directors, except to the extent any such provision may be held by the Cayman Islands courts to be contrary to public policy, such as to provide indemnification against civil fraud or the consequences of committing a crime. Our Memorandum and Articles provide that we shall indemnify our officers and directors against all actions, proceedings, costs, charges, expenses, losses, damages or liabilities incurred or sustained by such directors or officers, other than by reason of such person’s dishonesty, willful default or fraud, in or about the conduct of our company’s business or affairs (including as a result of any mistake of judgment) or in the execution or discharge of his duties, powers, authorities or discretions, including, without prejudice to the generality of the foregoing, any costs, expenses, losses or liabilities incurred by such director or officer in defending (whether successfully or otherwise) any civil proceedings concerning our company or its affairs in any court whether in the Cayman Islands or elsewhere. This standard of conduct is generally the same as permitted under the Delaware General Corporation Law for a Delaware corporation.
In addition, we have entered into indemnification agreements with our directors and executive officers that provide such persons with additional indemnification beyond that provided in our Amended Articles.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to our directors, officers or persons controlling us under the foregoing provisions, we have been informed that in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
Directors’ Fiduciary Duties. Under Delaware corporate law, a director of a Delaware corporation has a fiduciary duty to the corporation and its shareholders. This duty has two components: the duty of care and the duty of loyalty. The duty of care requires that a director act in good faith, with the care that an ordinarily prudent person would exercise under similar circumstances. Under this duty, a director must inform himself of, and disclose to shareholders, all material information reasonably available regarding a significant transaction. The duty of loyalty requires that a director acts in a manner he reasonably believes to be in the best interests of the corporation. He must not use his corporate position for personal gain or advantage. This duty prohibits self-dealing by a director and mandates that the best interest of the corporation and its shareholders take precedence over any interest possessed by a director, officer or controlling shareholder and not shared by the shareholders generally. In general, actions of a director are presumed to have been made on an informed basis, in good faith and in the honest belief that the action taken was in the best interests of the corporation. However, this presumption may be rebutted by evidence of a breach of one of the fiduciary duties. Should such evidence be presented concerning
91
a transaction by a director, the director must prove the procedural fairness of the transaction, and that the transaction was of fair value to the corporation.
As a matter of Cayman Islands law, a director of a Cayman Islands company is in the position of a fiduciary with respect to the company and therefore he owes the following duties to the company-a duty to act in good faith in the best interests of the company, a duty not to make a personal profit based on his position as director (unless the company permits him to do so), a duty not to put himself in a position where the interests of the company conflict with his personal interest or his duty to a third party and a duty to exercise powers for the purpose for which such powers were intended. A director of a Cayman Islands company owes to the company a duty to exercise the skill they actually possess and such care and diligence that a reasonably prudent person would exercise in comparable circumstances. It was previously considered that a director need not exhibit in the performance of his duties a greater degree of skill than may reasonably be expected from a person of his knowledge and experience. However, English and Commonwealth courts have moved towards an objective standard with regard to the required skill and care and these authorities are likely to be followed in the Cayman Islands.
Shareholder Action by Written Consent. Under the Delaware General Corporation Law, a corporation may eliminate the right of shareholders to act by written consent by amendment to its certificate of incorporation. Cayman Islands law and our Memorandum and Articles provide that shareholders may approve corporate matters by way of a unanimous written resolution signed by or on behalf of each shareholder who would have been entitled to vote on such matter at a general meeting without a meeting being held.
Shareholder Proposals. Under the Delaware General Corporation Law, a shareholder has the right to put any proposal before the annual meeting of shareholders, provided it complies with the notice provisions in the governing documents. A special meeting may be called by the board of directors or any other person authorized to do so in the governing documents, but shareholders may be precluded from calling special meetings.
The Companies Act provides shareholders with only limited rights to requisition a general meeting, and does not provide shareholders with any right to put any proposal before a general meeting. However, these rights may be provided in a company’s articles of association. Our Memorandum and Articles allow our shareholders holding shares which carry in aggregate not less than one-third of all votes attaching to all issued and outstanding shares of our Company that are entitled to vote at general meetings to requisition a shareholder’s meeting, in which case our directors are required to convene an extraordinary general meeting and to put the resolutions so requisitioned to a vote at such meeting. Other than this right to requisition a shareholders’ meeting, our Memorandum and Articles do not provide our shareholders with any right to put proposals before annual general meetings or extraordinary general meetings not called by such shareholders. As an exempted Cayman Islands company, we are not obliged by law to call shareholders’ annual general meetings .
Cumulative Voting. Under the Delaware General Corporation Law, cumulative voting for elections of directors is not permitted unless the corporation’s certificate of incorporation specifically provides for it. Cumulative voting potentially facilitates the representation of minority shareholders on a board of directors since it permits the minority shareholder to cast all the votes to which the shareholder is entitled on a single director, which increases the shareholder’s voting power with respect to electing such director. There are no prohibitions in relation to cumulative voting under the laws of the Cayman Islands but our Memorandum and Articles do not provide for cumulative voting. As a result, our shareholders are not afforded any less protections or rights on this issue than shareholders of a Delaware corporation.
Removal of Directors. Under the Delaware General Corporation Law, a director of a corporation with a classified board may be removed only for cause with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. Under our Memorandum and Articles, directors may be removed with or without cause, by an ordinary resolution of our shareholders. An appointment of a director may be on terms that the director shall automatically retire from office (unless he or she has sooner vacated office) at the next or a subsequent annual general meeting or upon any specified event or after any specified period in a written agreement between our Company and the director, if any; but no such term shall be implied in the absence of express provision. In addition, the office of any director shall be vacated if the director (i) becomes prohibited by applicable law from being a director, (ii) becomes bankrupt or makes any arrangement or composition with his creditors, (iii) dies or is found to be or becomes of unsound mind, (iv) resigns his office by notice in writing to the Company; (v) without special leave of absence from the board, is absent from meetings of the board for three consecutive meetings and the board (excluding the absent director) resolves that his office be vacated; or (vi) is removed from office pursuant to any other provision of our Memorandum and Articles.
92
Transactions with Interested Shareholders. The Delaware General Corporation Law contains a business combination statute applicable to Delaware corporations whereby, unless the corporation has specifically elected not to be governed by such statute by amendment to its certificate of incorporation, it is prohibited from engaging in certain business combinations with an “interested shareholder” for three years following the date that such person becomes an interested shareholder. An interested shareholder generally is a person or a group who or which owns or owned 15% or more of the target’s outstanding voting share within the past three years. This has the effect of limiting the ability of a potential acquirer to make a two-tiered bid for the target in which all shareholders would not be treated equally. The statute does not apply if, among other things, prior to the date on which such shareholder becomes an interested shareholder, the board of directors approves either the business combination or the transaction which resulted in the person becoming an interested shareholder. This encourages any potential acquirer of a Delaware corporation to negotiate the terms of any acquisition transaction with the target’s board of directors.
Cayman Islands law has no comparable statute. As a result, we cannot avail ourselves of the types of protections afforded by the Delaware business combination statute. However, although Cayman Islands law does not regulate transactions between a company and its significant shareholders, the directors of the company are required to comply with the fiduciary duties which they owe to the company under Cayman Islands law, including the duty to ensure that, in their opinion, any such transactions are bona fide in the best interests of the company and are entered into for a proper purpose and not with the effect of constituting a fraud on the minority shareholders.
Restructuring. A company may present a petition to the Grand Court of the Cayman Islands for the appointment of a restructuring officer on the grounds that the company:
| (a) | is or is likely to become unable to pay its debts; and |
| (b) | intends to present a compromise or arrangement to its creditors (or classes thereof) either pursuant to the Companies Act, the law of a foreign country or by way of a consensual restructuring. |
The Grand Court may, among other things, make an order appointing a restructuring officer upon hearing of such petition, and any restructuring officer so appointed shall have such powers and carry out only such functions as the court may order. At any time (i) after the presentation of a petition for the appointment of a restructuring officer but before an order for the appointment of a restructuring officer has been made, and (ii) when an order for the appointment of a restructuring officer is made, until such order has been discharged, no suit, action or other proceedings (other than criminal proceedings) shall be proceeded with or commenced against the company, no resolution to wind up the company shall be passed, and no winding up petition may be presented against the company, except with the leave of the court and subject to such terms as the court may impose. However, notwithstanding the presentation of a petition for the appointment of a restructuring officer or the appointment of a restructuring officer, a creditor who has security over the whole or part of the assets of the company is entitled to enforce the security without the leave of the court and without reference to the restructuring officer appointed.
Dissolution; Winding up. Under the Delaware General Corporation Law, unless the board of directors approves the proposal to dissolve, dissolution must be approved by shareholders holding 100% of the total voting power of the corporation. Only if the dissolution is initiated by the board of directors may it be approved by a simple majority of the corporation’s outstanding shares. Delaware law allows a Delaware corporation to include in its certificate of incorporation a supermajority voting requirement in connection with dissolutions initiated by the board.
Under Cayman Islands law, a company may be wound up by either an order of the courts of the Cayman Islands or by a special resolution of its members or, if the company is unable to pay its debts as they fall due, by an ordinary resolution of its members. The court has authority to order winding up in a number of specified circumstances including where it is, in the opinion of the court, just and equitable to do so. Under the Companies Act and our Memorandum and Articles, our Company may be dissolved, liquidated or wound up by a special resolution of our shareholders, or by an ordinary resolution on the basis that our Company is unable to pay its debts as they fall due.
Variation of Rights of Shares. Under the Delaware General Corporation Law, a corporation may vary the rights of a class of shares with the approval of a majority of the outstanding shares of such class, unless the certificate of incorporation provides otherwise. Under our Memorandum and Articles, whenever our share capital is divided into different classes, the rights attached to any class may, subject to any rights or restrictions for the time being attached to any class, only be materially and adversely be varied either with the written consent of the holders of at least two-thirds of the issued shares of that class or with the sanction of a special resolution passed
93
at a separate meeting of the holders of the shares of that class. The rights conferred upon the holders of the shares of any class issued with preferred or other rights shall not, subject to any rights or restrictions for the time being attached to the shares of that class, be deemed to be materially and adversely varied by the creation, allotment or issue of further shares ranking pari passu with or subsequent to such existing class of shares or the redemption or purchase of any shares of any class by our company. The rights of the holders of shares shall not be deemed to be materially and adversely varied by the creation or issue of shares with preferred or other rights including, without limitation, the creation of shares with enhanced or weighted voting rights.
Amendment of Governing Documents. Under the Delaware General Corporation Law, a corporation’s governing documents may be amended with the approval of a majority of the outstanding shares entitled to vote, unless the certificate of incorporation provides otherwise. Under the Companies Act, our Memorandum and Articles may only be amended by a special resolution of our shareholders.
Rights of Non-resident or Foreign Shareholders. There are no limitations imposed by our Memorandum and Articles on the rights of non-resident or foreign shareholders to hold or exercise voting rights on our shares. In addition, there are no provisions in our Memorandum and Articles governing the ownership threshold above which shareholder ownership must be disclosed.
C. | Material Contracts |
We have not entered into any material contracts other than in the ordinary course of business and other than those described in “Item 4. Information on the Company,” “Item 7. Major Shareholders and Related Party Transactions — B. Related Party Transactions,” or elsewhere in this annual report on Form 20-F.
D. | Exchange Controls |
The Cayman Islands currently has no exchange control restrictions. See also “Item 4. Information on the Company — B. Business Overview — Regulation — Regulations on Foreign Exchange.”
E. | Taxation |
The following summary of the material Cayman Islands, PRC and U.S. federal income tax consequences of an investment in our ordinary shares is based upon laws and relevant interpretations thereof in effect as of the date of this annual report, all of which are subject to change. This summary does not deal with all possible tax consequences relating to an investment in our ordinary shares, such as the tax consequences under U.S. state and local tax laws or under the tax laws of jurisdictions other than the Cayman Islands, the People’s Republic of China and the United States.
Cayman Islands Taxation
The Cayman Islands currently levies no taxes on individuals or corporations based upon profits, income, gains or appreciation and there is no taxation in the nature of inheritance tax or estate duty. There are no other taxes likely to be material to us levied by the government of the Cayman Islands except for stamp duties which may be applicable on instruments executed in, or after execution brought within the jurisdiction of the Cayman Islands. The Cayman Islands is not party to any double tax treaties that are applicable to any payments made to or by our Company. There are no exchange control regulations or currency restrictions in the Cayman Islands.
Payments of dividends and capital in respect of our ordinary shares will not be subject to taxation in the Cayman Islands and no withholding tax will be required on the payment of a dividend or capital to any holder of our ordinary shares, nor will gains derived from the disposal of our ordinary shares be subject to Cayman Islands income or corporation tax.
People’s Republic of China Taxation
Under the Corporate Income Tax Law of the People’s Republic of China (the “CIT Law”) and its implementation rules, both effective on January 1, 2008, with the CIT Law further being amended on December 29, 2018 and its implementation rules being amended on April 23, 2019, all domestic and foreign investment companies will be subject to a uniform enterprise income tax at the rate of 25% and dividends from PRC enterprises to their foreign shareholders will be subject to a withholding tax at a rate of 10% if the foreign investors are considered as non-resident enterprises without any establishment or place within the PRC or if the dividends payable have no connection with the establishment or place of the foreign investors within the PRC, unless any such foreign investor’s
94
jurisdiction of incorporation has a tax treaty with the PRC that provides for a lower withholding tax rate. In accordance with Caishui (2008) No. 1 issued by the Ministry of Finance, or MOF, and SAT on February 22, 2008, the accumulative undistributed profits of foreign investment companies generated before January 1, 2008, and distributed to foreign investors after year 2008, shall be exempt from withholding tax.
The CIT Law has introduced the concept of “resident enterprises” and corresponding tax liability on resident enterprises’ worldwide income, whilst “non-resident enterprises” without any place or establishment in the PRC are required to pay 10% income tax on their passive incomes from sources within China only. A resident enterprise refers to an enterprise that (i) was established/incorporated within the PRC, or (ii) was established/incorporated under the laws of a foreign jurisdiction but has its “de facto management body” in the PRC. A non-resident enterprise refers to an enterprise which was established/incorporated under the laws of a foreign jurisdiction and does not have its “de facto management body” in the PRC, but has an establishment or place in the PRC, or has China-sourced income even though it does not have any establishment or place in the PRC.
Under the implementation rules of the CIT Law, “de facto management body” is defined as an organization that has material and overall management and control over the business, personnel, accounts and properties of an enterprise. In April 2009, the SAT issued a Notice on Issues Relating to Determination of PRC-Controlled Offshore Enterprises as PRC Resident Enterprises Based on “De Facto Management Body” Test, or SAT Circular No. 82, under which, an offshore enterprise controlled by a PRC enterprise or a PRC enterprise group will be characterized as a “resident enterprise” due to the fact that its “de facto management body” is located within the PRC, if all of the following conditions are met at the same time: (i) the senior management personnel responsible for its daily operations and the place where the senior management departments discharge their responsibilities are located primarily in the PRC, (ii) its finance and human resources related decisions are made by or are subject to the approval of institutions or personnel located in the PRC, (iii) its major assets, books and records, company seals and minutes of its board of directors and shareholder meetings are located or kept in the PRC, and (iv) senior management personnel or 50% or more of the members of its board of directors with voting power of the enterprise reside in the PRC. SAT Circular No. 82 further specifies that the principle of “substance over form” shall be adopted in determining whether the “de facto management body” is located within China.
We currently are not treated as a PRC resident enterprise by the Chinese tax authority and as a result, we have not withheld PRC income taxes from our foreign investors and as a non-resident enterprise, we are subject to PRC withholding tax if we receive dividends directly from our PRC subsidiaries paid by them using funds out of their profits generated on and after January 1, 2008.
Nevertheless, substantially all of our operations are currently based in the PRC. Moreover, substantially all of our management team, who are in charge of finance and human resources related decisions, will perform their duties mainly in the PRC, and over 50% of our board members habitually reside in the PRC. Our main properties, accounting books and records, company seals and minutes of board meetings are maintained in China.
However, the rules regarding the determination of the “de facto management body” are relatively new and whether such rules may apply to us is unclear. Due to lack of further written clarification by the SAT, there is still an uncertainty around the interpretation of each of the four conditions as specified in SAT Circular No. 82 and the principle of “substance over form” and the implementation of SAT Circular No. 82 by Chinese tax authorities in practice. It also remains unclear what percentage of shares of an offshore enterprise must be held by a PRC entity or group in order for the offshore enterprise to be deemed as an offshore enterprise controlled by a PRC enterprise or a PRC enterprise group, and whether shares held by PRC resident individuals are counted pursuant to SAT Circular No. 82.
Due to the lack of clear guidance on the determination of our tax residency under the CIT Law, it remains unclear whether the PRC tax authorities will treat us as a PRC resident enterprise. As a result, we cannot express an opinion as to the likelihood that we will be subject to the tax applicable to resident enterprises or non-resident enterprises under the CIT Law. If the Company treated as a PRC resident enterprise, it will be subject to PRC tax on its worldwide income at the 25% uniform tax rate, but the dividends distributed from its subsidiaries that are or deemed to be PRC resident enterprises should be tax-exempt income. In addition, if the Company is considered a PRC resident enterprise, the dividends paid by it to the non-PRC shareholders may be regarded as income from sources within the PRC pursuant to SAT Circular No. 82, and therefore the non-PRC institutional shareholders may be subject to a 10% withholding tax, and the non-PRC individual shareholders may be subject to a 20% withholding tax unless they are able to claim a lower tax rate pursuant to applicable tax treaties.
95
Furthermore, if the Company is treated as a PRC resident enterprise, there is a possibility that the capital gains realized by its non-PRC shareholders from the transfer of their shares may be regarded as income from sources within the PRC for PRC tax purposes. If such capital gains are taxed in China, the applicable income tax rate would be 10% for non-PRC institutional shareholders, and 20% for non-PRC individual shareholders. If the non-PRC shareholders are U.S. residents that are eligible for PRC-US Tax Treaty benefits, whether capital gains should be taxed in China is unclear.
Pursuant to Paragraph 5 of Article 12 of the PRC-US Tax Treaty, gains from the alienation of shares of a company which is a PRC resident other than those mentioned in paragraph 4 (which refers to shares of a company the property of which consists principally of real property in the PRC) and representing a participation of at least 25% may be taxed in China. Paragraph 6 of Article 12 of the PRC-US Tax Treaty further specifies that “Gains derived by a resident of a Contracting State from the alienation of any property other than that referred to in paragraphs 1 through 5 and arising in the other Contracting State may be taxed in that other Contracting State.” By virtue of this provision, the capital gains realized by U.S. residents may be taxed in the PRC if the capital gains are considered as “arising in” the PRC. Under the CIT Law and its implementing rules, the capital gains from transfer of shares may be considered as “arising in” the PRC if the enterprise whose shares are transferred is “located in” China. If the Company is considered a PRC resident enterprise, and if the Chinese tax authorities take the position that a PRC resident enterprise is deemed to be located in China, the capital gains realized by the U.S. residents from transfer of their shares may be taxed in the PRC depending on how the PRC-US Tax Treaty is interpreted and implemented by the Chinese tax authorities.
United States Federal Income Tax Considerations
The following is a discussion of material U.S. federal income tax consequences of owning and disposing of our ordinary shares. It does not purport to be a comprehensive description of all tax considerations that may be relevant to a particular investor’s decision to own our ordinary shares. This discussion applies only to shareholders that own our ordinary shares as capital assets for U.S. federal income tax purposes. In addition, it does not describe all of the tax consequences that may be relevant in light of a shareholder’s particular circumstances, including alternative minimum tax consequences and tax consequences applicable to shareholders subject to other special rules, such as, but not limited to:
| ● | certain financial institutions; |
| ● | dealers, brokers or traders in securities, commodities or foreign currencies; |
| ● | persons holding our ordinary shares as part of a hedge, straddle, appreciated financial position, conversion transaction or integrated transaction or persons entering into a constructive sale with respect to our ordinary shares; |
| ● | persons whose functional currency for U.S. federal income tax purposes is not the U.S. dollar; |
| ● | entities classified as partnerships or other pass-through entities for U.S. federal income tax purposes; |
| ● | tax-exempt entities, “individual retirement accounts” and “Roth IRAs”; |
| ● | persons that beneficially own five percent or more of our ordinary shares; or |
| ● | persons who acquired our ordinary shares pursuant to the exercise of an employee stock option or otherwise as compensation. |
If an entity that is classified as a partnership for U.S. federal income tax purposes holds our ordinary shares, the U.S. federal income tax treatment of a partner will generally depend on the status of the partner and the activities of the partnership. Partnerships holding our ordinary shares, and partners in such partnerships, should consult their tax advisers as to the U.S. federal income tax consequences of owning and disposing of our ordinary shares.
This discussion is based on the U.S. Internal Revenue Code of 1986, as amended (the “Code”), administrative pronouncements, judicial decisions, and final, temporary and proposed Treasury regulations, all as of the date hereof, any of which is subject to change, possibly with retroactive effect.
96
Pursuant to Section 7874 of the Code, we are treated as a U.S. corporation for U.S. federal income tax purposes and the discussion herein is based on this treatment.
For purposes of this discussion, a “U.S. Holder” is a beneficial owner of our ordinary shares who for U.S. federal income tax purposes is:
| ● | an individual citizen or resident of the United States; |
| ● | a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or the District of Columbia or treated as a U.S. corporation pursuant to section 7874 of the Code; |
| ● | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or |
| ● | a trust if (i) a U.S. court can exercise primary supervision over the trust’s administration and one or more United States persons (as defined in the Code) are authorized to control all substantial decisions of the trust, or (ii) it has a valid election in effect under applicable U.S. Treasury regulations to be treated as a United States person. |
For purposes of this discussion, a “Non-U.S. Holder” is a beneficial owner of our ordinary shares who is neither a U.S. Holder nor a partnership or other pass-through entity for U.S. federal income tax purposes.
This discussion of material U.S. federal income tax consequences is not a complete analysis or description of all potential U.S. federal income tax consequences of acquiring, owning and disposing of our ordinary shares. Shareholders should consult their own tax advisers concerning the U.S. federal, state, local and foreign tax consequences of acquiring, owning and disposing of our ordinary shares in their particular circumstances.
U.S. Holders
Taxation of Distributions. Distributions paid on our ordinary shares, other than certain pro rata distributions of our ordinary shares, will be treated as dividends to the extent paid out of our current or accumulated earnings and profits and will be includable in a U.S. Holder’s income and taxable as ordinary dividend income. If a distribution exceeds our current and accumulated earnings and profits, the excess will be first treated as a tax-free return of capital, to the extent of the U.S. Holder’s tax basis in the ordinary shares. Any remaining excess will be treated as gain from the sale or other taxable disposition of the ordinary shares. Dividends received by a non-corporate U.S. Holder may be eligible to be taxed at reduced rates if certain holding period and other applicable requirements are met. Dividends received by a corporate U.S. Holder may be eligible for the dividends-received deduction if certain holding period requirements and other applicable requirements are met.
Dividends paid on our ordinary shares will be treated as U.S.-source for U.S. federal income tax purposes. As described in “Item 10.E. Taxation—PRC Taxation,” if we were deemed to be a PRC resident enterprise for PRC tax purposes, dividends paid with respect to our ordinary shares might be subject to PRC withholding taxes. For U.S. federal income tax purposes, the amount of a dividend would include any amounts withheld by us in respect of any PRC taxes. U.S. Holders should consult their tax advisers as to whether the rate of any such PRC taxes may be reduced under the provisions of the U.S.-PRC income tax treaty and the creditability of any such PRC taxes in their particular circumstances.
Sale or Other Disposition of Our Ordinary Shares.
Upon the sale or other taxable disposition of our ordinary shares, a U.S. Holder will recognize gain or loss equal to the difference between the amount realized on the sale or other taxable disposition and its tax basis in the ordinary shares. Gain or loss realized on the sale or other disposition of the ordinary shares will be capital gain or loss, and will be long-term capital gain or loss if the U.S. Holder owned the ordinary shares for more than one year. Long-term capital gains recognized by non-corporate taxpayers are currently subject to reduced tax rates. The deductibility of capital losses is subject to limitations.
As described in “Item 10.E. Taxation—PRC Taxation,” if we were deemed to be a PRC resident enterprise for PRC tax purposes, gains from dispositions of our ordinary shares might be subject to PRC tax. In that case, a U.S. Holder’s amount realized
97
would include any amounts paid in respect of PRC taxes. Capital gains realized by a U.S. Holder upon the sale or other taxable disposition of our ordinary shares generally will give rise to U.S.-source gain for foreign tax credit purposes. U.S. Holders should consult their tax advisers as to the creditability of any such PRC taxes in their particular circumstances.
Non-U.S. Holders
Taxation of Distributions. Distributions with respect to our ordinary shares will be treated as paid by a U.S. corporation and generally dividend income to the extent paid from our current or accumulated earnings and profits as determined for U.S. federal income tax purposes. If a distribution exceeds current and accumulated earnings and profits, the excess will be treated first as a return of capital to the extent of the Non-U.S. Holder’s adjusted tax basis in its ordinary shares and thereafter as capital gain from the sale or exchange of such ordinary shares, subject to the tax treatment described below in “—Sale or Other Disposition of Our Ordinary Shares.”
Except as described below, dividends paid to a Non-U.S. Holder are subject to withholding of U.S. federal income tax at a 30% rate or at a lower rate as may be specified by an applicable income tax treaty. In addition, even if a Non-U.S. Holder is eligible for a lower treaty rate, we and other payors will generally be required to withhold at a 30% rate (rather than the lower treaty rate) on dividend payments, unless such Non-U.S. Holder has furnished to us or another payor:
· | a valid IRS Form W-8BEN or W-8BEN-E or an acceptable substitute form upon which such Non-U.S. Holder certifies, under penalties of perjury, its status as a non-U.S. person and entitlement to the lower treaty rate with respect to such payments; or |
· | in the case of payments made outside the United States to an offshore account (generally, an account maintained at an office or branch of a bank or other financial institution at any location outside the United States), other documentary evidence establishing such Non-U.S. Holder’s entitlement to the lower treaty rate in accordance with U.S. Treasury regulations. |
A Non-U.S. Holder that is eligible for a reduced rate of U.S. withholding tax under an income tax treaty may obtain a refund of any excess amounts withheld by filing a refund claim with the IRS.
If dividends paid to a Non-U.S. Holder are “effectively connected” with the conduct of a trade or business within the United States by such Non-U.S. Holder, and, if required by an applicable tax treaty, the dividends are attributable to a permanent establishment that the Non-U.S. Holder maintains in the United States, we and other payors generally are not required to withhold tax from the dividends; provided that the Non-U.S. Holder has furnished to us or another payor a valid IRS Form W-8ECI or an acceptable substitute form upon which the Non-U.S. Holder certifies, under penalties of perjury, that:
· | such Non-U.S. Holder is a non-U.S. person; and |
· | the dividends are effectively connected with the conduct of a trade or business within the United States by such Non-U.S. Holder and are includible in such Non-U.S. Holder’s gross income. |
“Effectively connected” dividends are taxed on a net income basis in the same manner as if a Non-U.S. Holder were a U.S. person, and such dividends if received by a corporate Non-U.S. Holder may, under certain circumstances, be subject to an additional “branch profits tax” at a 30% rate or at a lower applicable treaty rate.
Sale or Other Disposition of Our Ordinary Shares.
A Non-U.S. Holder generally will not be subject to U.S. federal income or withholding tax on gain recognized on a disposition of our ordinary shares unless:
· | the gain is “effectively connected” with the conduct of a trade or business within the United States by such Non-U.S. Holder, and if required by an applicable income tax treaty, the gain is attributable to a permanent establishment that the Non-U.S. Holder maintains in the United States; |
98
· | such Non-U.S. Holder is an individual, is present in the United States for 183 or more days in the taxable year of the disposition and certain other conditions exist; or |
· | (i) we are, or we or CASI Delaware have been, a “United States real property holding corporation” for U.S. federal income tax purposes, (ii) so long as our ordinary shares continues to be regularly traded on an established securities market, such Non-U.S. Holder held, directly or indirectly, at any time during the shorter of the five-year period ending on the date of disposition or such Non-U.S. Holder’s holding period, more than 5% of our ordinary shares or CASI Delaware’s common stock and (iii) such Non-U.S. Holder is not eligible for any treaty exemption. |
“Effectively connected” gains are taxed on a net income basis in the same manner as if a Non-U.S. Holder were a U.S. person. “Effectively connected” gains recognized by a corporate Non-U.S. Holder may also, under certain circumstances, be subject to an additional “branch profits tax” at a 30% rate or at a lower applicable treaty rate. An individual Non-U.S. holder who is subject to U.S. federal income tax because the Non-U.S. Holder was present in the United States for 183 days or more during the year of sale or other disposition of our ordinary shares will generally be subject to a flat 30% tax on the gain derived from such sale or other disposition, which may be offset by U.S. source capital losses (subject to any applicable tax treaty).
We do not believe that we are or that we or CASI Delaware have been, and we do not anticipate becoming, a “United States real property holding corporation” for U.S. federal income tax purposes.
Foreign Account Tax Compliance Act
Under Sections 1471 through 1474 of the Code (such Sections commonly referred to as “FATCA”), a 30% United States federal withholding tax may apply to any dividends paid on our ordinary shares to a “foreign financial institution” (as specifically defined in the Code), unless such foreign financial institution (i) enters into an agreement with the Treasury to report, on an annual basis, information with respect to shares in, and accounts maintained by, the institution held by certain U.S. persons and by certain non-U.S. entities that are wholly or partially owned by U.S. persons and to withhold on certain payments, or (ii) if required under an intergovernmental agreement between the United States and an applicable foreign country, reports such information to its local tax authority, which will exchange such information with the U.S. authorities. Accordingly, the entity through which our ordinary shares are held will affect the determination of whether such withholding is required. Similarly, a 30% United States withholding tax may apply to dividends paid on our ordinary shares to a “non-financial foreign entity” (as specifically defined in the Code) that does not qualify under certain exemptions, unless such entity either (i) certifies to us that such entity does not have any “substantial U.S. owners” or (ii) provides certain information regarding the entity’s “substantial U.S. owners,” which we, or the applicable withholding agent, will in turn provide to the Secretary of the Treasury. If a dividend payment is both subject to withholding under FATCA and subject to the withholding tax discussed above under “—Non-U.S. Holders—Distributions,” an applicable withholding agent may credit the withholding under FATCA against, and therefore reduce, such other withholding tax. We will not pay any additional amounts to holders in respect of any amounts withheld. Holders should consult their own tax advisors regarding these requirements and whether they may be relevant to ownership and disposition of our ordinary shares.
F. | Dividends and Paying Agents |
Not applicable.
G. | Statement by Experts |
Not applicable.
99
H. | Documents on Display |
We are subject to the periodic reporting and other informational requirements of the Exchange Act. Under the Exchange Act, we are required to file reports and other information with the SEC. Specifically, we are required to file annually a Form 20-F no later than four months after the close of each fiscal year. Copies of reports and other information, when so filed, may be inspected without charge and may be obtained at prescribed rates at the public reference facilities maintained by the SEC at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. The public may obtain information regarding the Washington, D.C. Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains a web site at www.sec.gov that contains reports, proxy and information statements, and other information regarding registrants that make electronic filings with the SEC using its EDGAR system. As a foreign private issuer, we are exempt from the rules under the Exchange Act prescribing the furnishing and content of quarterly reports and proxy statements, and officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act.
I.Subsidiary Information
Not applicable.
J. | Annual Report to Security Holders |
Not applicable.
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Inflation
To date, inflation in China has not materially impacted our results of operations. According to the National Bureau of Statistics of China, the year-over-year percent changes in the consumer price index for December 2021, 2022 and 2023 were an increase of 1.5%, an increase of 1.8%, and a decrease of 0.3%, respectively. Although we have not been materially affected by inflation in the past, we can provide no assurance that we will not be affected by higher rates of inflation in China in the future.
Market Risks
Foreign Exchange Risk
Substantially all of our revenues and a major part of our expenses are denominated in RMB. We do not believe that we currently have any significant direct foreign exchange risk and have not used any derivative financial instruments to hedge exposure to such risk. Although our exposure to foreign exchange risks should be limited in general, the value of your investment in our ordinary shares will be affected by the exchange rate between U.S. dollar and Renminbi because the value of our business is effectively denominated in RMB, while our ordinary shares will be traded in U.S. dollars.
The conversion of Renminbi into foreign currencies, including U.S. dollars, is based on rates set by the People’s Bank of China. The Renminbi has fluctuated against the U.S. dollar, at times significantly and unpredictably. It is difficult to predict how market forces or PRC or U.S. government policy may impact the exchange rate between Renminbi and the U.S. dollar in the future.
To the extent that we need to convert U.S. dollars into Renminbi for our operations, appreciation of the Renminbi against the U.S. dollar would have an adverse effect on the RMB amount we receive from the conversion. Conversely, if we decide to convert Renminbi into U.S. dollars for the purpose of making payments for dividends on our ordinary shares or for other business purposes, appreciation of the U.S. dollar against the Renminbi would have a negative effect on the U.S. dollar amounts available to us.
Interest rate risk
We have not been exposed to material risks due to changes in market interest rates, and we have not used any derivative financial instruments to manage our interest risk exposure.
100
We do not expect that the fluctuation of interest rates will have a material impact on our financial condition. However, we cannot provide assurance that we will not be exposed to material risks due to changes in market interest rates in the future.
We may invest our cash in interest-earning instruments. Investments in both fixed rate and floating rate interest earning instruments carry a degree of interest rate risk. Fixed rate securities may have their fair market value adversely impacted due to a rise in interest rates, while floating rate securities may produce less income than expected if interest rates fall.
ITEM 12. DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
A.Debt Securities
Not applicable.
B. | Warrants and Rights |
Not applicable.
C. | Other Securities |
Not applicable.
D. | American Depositary Shares |
Not applicable.
101
PART II.
ITEM 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
None.
ITEM 14. MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS
None.
ITEM 15. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures
As of December 31, 2023, we carried out an evaluation, under the supervision and with the participation of our management, including our Principal Executive Officer and Principal Financial Officer of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in the Securities Exchange Act of 1934 Rules 13a-15(e) and 15d-15(e)). Our Principal Executive Officer and Principal Financial Officer have concluded that our disclosure controls and procedures are effective to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC and that such information is accumulated and communicated to our management (including our Principal Executive Officer and Principal Financial Officer) to allow timely decisions regarding required disclosures. Based on such evaluation, our Principal Executive Officer and Principal Financial Officer have concluded these disclosure controls and procedures are effective as of December 31, 2023.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Securities Exchange Act Rules 13a-15(f) and 15d-15(f). Our internal control over financial reporting is designed to provide reasonable assurance to our management and board of directors regarding the reliability of financial reporting and the preparation and fair presentation of financial statements for external purposes in accordance with generally accepted accounting principles. Any internal control over financial reporting, no matter how well designed, has inherent limitations. As a result of these inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those internal controls determined to be effective can provide only reasonable assurance with respect to reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Under the supervision and with the participation of our management, including our Principal Executive Officer and Principal Financial Officer, we conducted an assessment of the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control — Integrated Framework 2013. Based on our assessment, we concluded that our internal control over financial reporting was effective as of December 31, 2023.
Changes in Internal Control over Financial Reporting
There have not been any changes in our internal control over financial reporting during the period covered by this annual report on Form 20-F that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 16. RESERVED
ITEM 16A. AUDIT COMMITTEE FINANCIAL EXPERT
Our board of directors has determined that Thomas Folinsbee, the chairperson of our audit committee and an independent director (under the standards set forth in Rule 5605(c)(2) of the Nasdaq Stock Market Rules and Rule 10A-3 under the Exchange Act), is an audit committee financial expert.
102
ITEM 16B. CODE OF ETHICS
Our board of directors adopted a code of business conduct and ethics that applies to our directors, officers and employees, effective in March 21, 2023. We have posted a copy of our code of business conduct and ethics on our website at https://www.casipharmaceuticals.com/about-us/corporate-governance/.
ITEM 16C. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The following table presents aggregate fees for professional services rendered by KPMG and its affiliates (“KPMG”) for the years ended December 31, 2022 and 2023.
KPMG
(US$ in thousands) |
| 2022 |
| 2023 | ||
Audit fees | $ | 755 | $ | 600 | ||
Audit-related fees | $ | — | $ | — | ||
Tax fees | $ | 19 | $ | 5 | ||
All Other Fees | $ | — | $ | — | ||
Total | $ | 774 | $ | 605 | ||
Services rendered by KPMG (for fiscal years 2023 and 2022) in connection with fees presented above were as follows:
Audit Fees
The Company incurred from KPMG audit fees of US$0.6 million in fiscal year 2023, for professional services rendered for the audit of the Company’s annual financial statements included in the Company’s Annual Report on Form 20-F for the fiscal year ended December 31, 2023.
The Company incurred from KPMG audit fees of US$0.8 million in fiscal year 2022, covering professional services rendered for (1) the audit of the Company’s annual financial statements included in the Company’s Annual Report on Form 20-F for the fiscal year ended December 31, 2022 and (2) the reviews of the financial statements included in the CASI Delaware’s quarterly reports on Form 10-Q for the first three quarters of 2022.
Audit-Related Fees
The Company did not incur audit-related fees in fiscal years 2023 or 2022.
Tax Fees
The Company incurred tax consulting service fees of US$5,000 and US$19,000 in fiscal year 2023 and 2022, respectively, for one of the Company’s subsidiary.
All Other Fees
The Company did not incur any other fees in fiscal years 2023 or 2022 from KPMG.
The Audit Committee pre-approves all audit services provided by our independent registered public accounting firm in accordance with the Audit Committee’s pre-approval policy for audit services.
ITEM 16D. EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES
Not applicable.
103
ITEM 16E. PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS
On December 15, 2021, CASI Delaware’s board of directors approved a stock repurchase program for the repurchase of up to $10 million of the Company’s common stock (and no more than 1,250,000 shares of its common stock) through open market purchases in compliance with Rule 10b-18 under the Securities Exchange Act of 1934 and through trading plans established pursuant to Rule 10b5-1 of the Securities Exchange Act (adjusted to reflect the reverse stock split completed in May 2022). The plan was announced by the Company on December 17, 2021.
|
|
| Total Number of |
| Maximum Number | ||||
Total | Shares Purchased as | of Shares that | |||||||
Number of | Average | Part of Publicly | May Yet Be | ||||||
Shares | Price Paid | Announced Plans or | Purchased Under the | ||||||
Purchased | Per Share (US$) | Programs | Plans or Programs | ||||||
January 2023 (from January 3 to January 20) |
| 136,118 | $ | 2.01 |
| 658,129 |
| 591,871 | |
ITEM 16F. CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT
Not applicable.
ITEM 16G. CORPORATE GOVERNANCE
As a Cayman Islands exempted company listed on the Nasdaq Stock Market, we are subject to the Nasdaq listing standards. However, the Nasdaq Stock Market Rules permit a foreign private issuer like us to follow the corporate governance practices of its home country.
Our shareholders may be afforded less protection than they would otherwise enjoy under the Nasdaq listing standards applicable to U.S. domestic issuers given our reliance on the home country practice exception.
See “Item 3. Key Information — D. Risk Factors— Risks Relating to Our Ordinary Shares — As an exempted company incorporated in the Cayman Islands, we have adopted certain home country practices in relation to corporate governance matters that differ significantly from the Nasdaq’s corporate governance requirements; these practices may afford less protection to shareholders than they would enjoy if we complied fully with the Nasdaq’s corporate governance requirements.”
ITEM 16H. MINE SAFETY DISCLOSURE
Not applicable.
ITEM 16I. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
ITEM 16J. INSIDER TRADING POLICIES
Not applicable
ITEM 16K. CYBERSECURITY
Risk Management and Strategy
We have implemented comprehensive cybersecurity risk assessment procedures to ensure effectiveness in cybersecurity management, strategy and governance and reporting cybersecurity risks. We have also integrated cybersecurity risk management into our overall enterprise risk management system. We have ensured that our employees have full access to the basic knowledge and
104
principles of information security, established a sound responding process and disposal mechanism for system security, external attacks and violations, and safeguarded the confidentiality of information and data of the enterprise, employees and customers, making sure information and data can only be obtained and used when necessary. By taking those actions, we have identified the cybersecurity risk management as a core task of our enterprise risk management system and been making efforts to make sure our overall risk assessment can sufficiently cover cybersecurity risks.
We have developed a comprehensive cybersecurity threat defense system to address both internal and external threats. This system encompasses various levels, including network, host and application security and incorporates systematic security capabilities for threat defense, monitoring, analysis, response, deception and countermeasures. We strive to manage cybersecurity risks and protect sensitive information through various means, such as technical safeguards, procedural requirements, an intensive program of monitoring on our corporate network, continuous testing of aspects of our security posture internally, a robust incident response program and regular cybersecurity awareness training for employees. Our IT department regularly monitors the performance of our infrastructure to enable us to respond quickly to potential problems, including potential cybersecurity threats.
As of the date of this annual report, we have not experienced any material cybersecurity incidents or identified any material cybersecurity threats that have affected or are reasonably likely to materially affect us, our business strategy, results of operations or financial condition.
Governance
Our board of directors is responsible for overseeing our cybersecurity risk management. Our board of directors shall (i) maintain oversight of the disclosure related to cybersecurity matters in current reports or periodic reports of our company, (ii) review updates to the status of any material cybersecurity incidents or material risks from cybersecurity threats to our company, and the relevant disclosure issues, if any, presented by our chief executive officer, principal financial officer and cybersecurity officer on a quarterly basis, and (iii) review disclosure concerning cybersecurity matters in our annual report on Form 20-F presented by our chief executive officer, principal financial officer and cybersecurity officer.
At management level, our chief executive officer, principal financial officer and cybersecurity officer are responsible for assessing, identifying and managing material risks from cybersecurity threats to our company and monitoring the prevention, detection, mitigation and remediation of material cybersecurity incident. These officers report to our board of directors on (i) a quarterly basis on updates to the status of any material cybersecurity incidents or material risks from cybersecurity threats to our company, and the relevant disclosure issues, if any, and (ii) on disclosure concerning cybersecurity matters in our annual report on Form 20-F.
105
PART III.
ITEM 17. FINANCIAL STATEMENTS
We have elected to provide consolidated financial statements pursuant to Item 18.
ITEM 18. FINANCIAL STATEMENTS
The consolidated financial statements of CASI Pharmaceuticals, Inc. are included at the end of this annual report.
ITEM 19. EXHIBITS
Exhibit | ||
Number |
| Description of Document |
1.1* | ||
2.1* | ||
2.2 |
| |
4.1 | ||
4.2 | ||
4.3 | ||
4.4 | ||
4.5 | ||
4.6 | ||
4.7 | ||
4.8++ | ||
4.9++ | ||
4.10++ | ||
4.11 |
|
106
4.12++ | ||
4.13 | ||
4.14 | ||
4.15 | ||
4.16 | ||
4.17 | ||
4.18 | ||
4.19 | ||
4.20++ | ||
4.21+ | ||
4.22 | ||
4.23 | ||
4.24+ | ||
4.25+ | ||
4.26++ | ||
4.27++ | ||
4.28++ | ||
4.29*++ |
107
4.30++ | ||
4.31++ | ||
4.32++ | ||
4.33++ | ||
4.34 | ||
4.35++ | ||
4.36++ | ||
4.37*+++ | ||
4.38*+++ | ||
4.39*+++ | ||
4.40*+++ | ||
4.41*+++ | ||
4.42*+++ | ||
4.43*+++ | ||
4.44*+++ | ||
4.45*+++ | ||
4.46*+++ | ||
4.47*+++ |
108
4.48*+++ | ||
8.1* | Significant subsidiaries and consolidated affiliated entities of the Registrant | |
11.1* | ||
12.1* | CEO Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
12.2* | CFO Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 | |
13.1** | CEO Certification Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
13.2** | CFO Certification Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 | |
15.1* | ||
97.1* | ||
101.INS* | Inline XBRL–Instance Document - The instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL Document | |
101.SCH* | Inline XBRL Taxonomy Extension Scheme Document | |
101.CAL* | Inline XBRL Taxonomy Extension Calculation Linkbase Document | |
101.DEF* | Inline XBRL Taxonomy Extension Definition Linkbase Document | |
101.LAB* | Inline XBRL Taxonomy Extension Label Linkbase Document | |
101.PRE* | Inline XBRL Taxonomy Extension Presentation Linkbase Document | |
104* | Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) |
* | Filed with this Annual Report on Form 20-F. |
** | Furnished with this Annual Report on Form 20-F. |
+ | Certain portions of this exhibit have been omitted based upon a request for confidential treatment under 17 C.F.R. section 200.80(b)(4) and 240.24b-2. The confidential portions of this exhibit have been omitted and are marked accordingly. The confidential portions have been filed separately with the Commission pursuant to our confidential treatment request. |
++ | Information in this exhibit identified by brackets is confidential and has been excluded pursuant to Item 601(B)(10)(IV) of Regulation S-K because it (i) is not material and (ii) would likely cause competitive harm to CASI Pharmaceuticals, Inc. if publicly disclosed. |
+++ Information in this exhibit identified by brackets has been redacted because it is not material and is the type that the Company treats as private or confidential.
109
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
CASI Pharmaceuticals, Inc. | ||
By: | /s/ Wei (Larry) Zhang | |
Name: Wei (Larry) Zhang | ||
Title: President and Principal Financial Officer | ||
Date: March 28, 2024 | ||
110
The following consolidated financial statements of CASI Pharmaceuticals, Inc. are included in Item 8:
F-1
Report of Independent Registered Public Accounting Firm
To the Shareholders and Board of Directors
CASI Pharmaceuticals, Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of CASI Pharmaceuticals, Inc. and subsidiaries (the Company) as of December 31, 2023 and 2022, the related consolidated statements of operations and comprehensive loss, shareholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2023, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2023, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of a critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Assessment of impairment triggering events related to the manufacturing asset group
As discussed in Notes 2, 5, 6 and 7 to the consolidated financial statements, as of December 31, 2023, the Company had property, plant and equipment of $9,241, right of use assets of $2,392, and intangible assets subject to amortization of $1,839, a portion of which related to the manufacturing asset group. The Company reviews long-lived assets for impairment whenever events or changes in circumstances (triggering events) indicate that the carrying amount of an asset or asset group may not be recoverable.
We identified the assessment of impairment triggering events related to the manufacturing asset group as a critical audit matter. The assessment of whether expected operating results associated with the use of the asset group and changes in the industry conditions represented a triggering event required a higher degree of subjective auditor judgment.
F-2
The following are the primary procedures we performed to address this critical audit matter. We evaluated the design of certain internal control related to the Company’s triggering events assessment. This included a control related to the Company’s process to identify and evaluate triggering events that indicate the carrying value of the manufacturing asset group may not be recoverable. We evaluated the Company’s identification of triggering events related to the assessment of expected operating results for the manufacturing asset group by comparing the expected operating results assessed by management with historical information and currently available public information. In addition, we evaluated the Company’s assessment of changes in industry conditions by comparing the changes in industry conditions assessed by management to industry outlook using data obtained from publicly available industry and market information.
/s/ |
|
|
|
We have served as the Company’s auditor since 2019. |
|
|
|
| |
March 28, 2024 |
|
F-3
CASI Pharmaceuticals, Inc.
Consolidated Balance Sheets
(In USD thousands, except share and per share data)
| December 31, 2023 |
| December 31, 2022 | |||
ASSETS |
|
|
| |||
Current assets: |
|
|
| |||
Cash and cash equivalents | $ | | $ | | ||
Investment in equity securities, at fair value |
| |
| | ||
Short term investments | | | ||||
Accounts receivable | | | ||||
| — | |||||
Inventories | | | ||||
Prepaid expenses and other |
| |
| | ||
Total current assets |
| |
| | ||
Term deposit, non current | — | | ||||
Property, plant and equipment, net |
| |
| | ||
Intangible assets, net |
| |
| | ||
Long-term investments | | | ||||
Right of use assets | | | ||||
Other assets |
| |
| | ||
Total assets | $ | | $ | | ||
LIABILITIES, REDEEMABLE NONCONTROLLING INTEREST AND SHAREHOLDERS’ EQUITY |
|
|
|
| ||
Current liabilities: |
|
|
|
| ||
Accounts payable | $ | | $ | | ||
Accrued and other current liabilities |
| |
| | ||
Income tax payable |
| — |
| | ||
Total current liabilities |
| |
| | ||
Long term borrowing | | — | ||||
Other liabilities |
| |
| | ||
Total liabilities |
| |
| | ||
Commitments and contingencies (Note 21) |
|
|
|
| ||
Redeemable noncontrolling interest, at redemption value | — | | ||||
Shareholders’ equity: |
|
|
|
| ||
Ordinary shares, $ | ||||||
| ||||||
|
| | ||||
Additional paid-in capital |
| |
| | ||
Treasury shares, at cost: |
| ( |
| ( | ||
Accumulated other comprehensive loss |
| ( |
| ( | ||
Accumulated deficit |
| ( |
| ( | ||
Total shareholders’ equity |
| |
| | ||
Total liabilities, redeemable noncontrolling interest and shareholders’ equity | $ | | $ | | ||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
CASI Pharmaceuticals, Inc.
Consolidated Statements of Operations and Comprehensive Loss
(In USD thousands, except share and per share data)
| Years ended December 31, | ||||||||
| 2023 |
| 2022 |
| 2021 | ||||
Revenues: | |||||||||
Product sales | $ | | $ | | $ | | |||
Sublicensing revenue from a related party | — | | — | ||||||
Lease income from a related party |
| | |
| | ||||
Total revenues | | | | ||||||
Total costs of revenues | | | | ||||||
Gross Profit | | | | ||||||
Operating expenses (income): |
|
|
|
| |||||
Research and development |
| | |
| | ||||
General and administrative | | | | ||||||
Selling and marketing |
| | |
| | ||||
Other operating income | ( | — | — | ||||||
Acquired in-process research and development | — | — | | ||||||
Loss on disposal of long-lived assets | — | | | ||||||
Foreign exchange gain | ( | ( | ( | ||||||
Impairment of intangible assets | — | | — | ||||||
Total operating expenses |
| | |
| | ||||
Loss from operations | ( | ( | ( | ||||||
Non-operating income (expense): | |||||||||
Interest income, net |
| | |
| | ||||
Other income | | | | ||||||
Change in fair value of investments |
| ( | ( |
| | ||||
Gain from sale of an equity investment | — | | — | ||||||
Impairment loss of long-term investments | ( | — | ( | ||||||
Loss before income tax benefit (expense) and share of net loss in an equity investee | ( | ( | ( | ||||||
Income tax benefit (expense) | | ( | — | ||||||
Net loss before share of net loss in an equity investee | ( | ( | ( | ||||||
Share of net loss in an equity investee | ( | ( | — | ||||||
Net loss | ( | ( | ( | ||||||
Less:Loss attributable to redeemable noncontrolling interest | ( | ( | ( | ||||||
Accretion to redeemable noncontrolling interest redemption value | | | | ||||||
Deemed dividends to Wuxi LP | | — | — | ||||||
Net loss attributable to CASI Pharmaceuticals, Inc. | $ | ( | $ | ( | $ | ( | |||
Net loss per share (basic and diluted) | $ | ( | $ | ( | $ | ( | |||
Weighted average number of ordinary shares outstanding (basic and diluted) |
| | |
| | ||||
Comprehensive loss: |
|
| |||||||
Net loss | $ | ( | $ | ( | $ | ( | |||
Foreign currency translation adjustment |
| ( | ( |
| | ||||
Total comprehensive loss | $ | ( | $ | ( | $ | ( | |||
Less: Comprehensive loss attributable to redeemable noncontrolling interest | ( | ( | ( | ||||||
Comprehensive loss attributable to ordinary shareholders | $ | ( | $ | ( | $ | ( | |||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
CASI Pharmaceuticals, Inc.
Consolidated Statements of Shareholders’ Equity
(In USD thousands, except share data)
| Accumulated | |||||||||||||||||||
Additional | Other | |||||||||||||||||||
Ordinary Share | Paid-in | Treasury | Comprehensive | Accumulated | ||||||||||||||||
| Shares |
| Amount |
| Capital |
| Shares |
| Income (Loss) |
| Deficit |
| Total | |||||||
Balance at December 31, 2020 |
| | $ | | $ | | $ | ( | $ | | $ | ( | $ | | ||||||
Issuance of ordinary shares pursuant to financing agreements |
| |
| |
| |
| |
| |
| |
| | ||||||
Stock issuance costs |
| |
| |
| ( |
| |
| |
| |
| ( | ||||||
Share-based compensation expense, net of forfeitures |
| |
| |
| |
| |
| |
| |
| | ||||||
Foreign currency translation adjustment |
| |
| |
| — |
| |
| |
| |
| | ||||||
Net loss attributable to CASI Pharmaceuticals, Inc. |
| |
| |
| ( |
| |
| |
| ( |
| ( | ||||||
Balance at December 31, 2021 | | $ | $ | | $ | ( | $ | | $ | ( | $ | | ||||||||
Repurchase and retirement of ordinary shares | ( | | ( | ( | | | ( | |||||||||||||
Share-based compensation expense, net of forfeitures | | | | | | | | |||||||||||||
Foreign currency translation adjustment | | | — | | ( | | ( | |||||||||||||
Net loss attributable to CASI Pharmaceuticals, Inc. | | | ( | | | ( | ( | |||||||||||||
Balance at December 31, 2022 |
| | $ | | | $ | ( | $ | ( | $ | ( | $ | | |||||||
Repurchase of ordinary shares | ( | | ( | ( | | | ( | |||||||||||||
Issuance of ordinary shares for options exercised | | | | | | | | |||||||||||||
Share-based compensation expense, net of forfeitures | | | | | | | | |||||||||||||
Foreign currency translation adjustment | | | — | | ( | | ( | |||||||||||||
Net loss attributable to CASI Pharmaceuticals, Inc. | | | ( | | | ( | ( | |||||||||||||
Balance at December 31, 2023 |
| | $ | | | $ | ( | $ | ( | $ | ( | $ | | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
CASI Pharmaceuticals, Inc.
Consolidated Statements of Cash Flows
(In USD thousands)
| Years Ended December 31, | ||||||||
2023 | 2022 |
| 2021 | ||||||
CASH FLOWS FROM OPERATING ACTIVITIES |
|
|
| ||||||
Net loss | $ | ( | $ | ( | $ | ( | |||
Adjustments to reconcile net loss to net cash used in operating activities: |
|
| |||||||
Depreciation for property, plant and equipment |
| | | | |||||
Amortization of intangible assets |
| | | | |||||
Reduction in the carrying amount of the right-of-use assets | | | | ||||||
Loss on disposal of long-lived assets |
| — | | | |||||
Impairment of intangible assets | — | | — | ||||||
Share-based compensation expense |
| | | | |||||
Acquired in-process research and development |
| — | — | | |||||
Government grant as a result of loan forgiveness | — | — | ( | ||||||
Change in fair value of investments |
| | | ( | |||||
Share of net loss of an equity investee | | | — | ||||||
Gain from extinguishment of investment in a convertible loan | ( | — | — | ||||||
Gain from sale of an equity investment | — | ( | — | ||||||
Impairment loss of long-term investments | | — | | ||||||
Changes in operating assets and liabilities: |
|
| |||||||
Accounts receivable | | ( | ( | ||||||
Receivable from a related party | ( | — | — | ||||||
Inventories | ( | ( | ( | ||||||
Prepaid expenses and other |
| | ( | ( | |||||
Accounts payable |
| | | | |||||
Accrued liabilities and other liabilities |
| ( | ( | | |||||
Income tax payable | ( | | — | ||||||
Net cash used in operating activities |
| ( | ( | ( | |||||
CASH FLOWS FROM INVESTING ACTIVITIES |
|
|
| ||||||
Proceeds from disposal of property and equipment |
| | | | |||||
Purchases of property, plant and equipment | ( | ( | ( | ||||||
Proceeds from sales of equity securities in MaxCyte and BioInvent | — | | — | ||||||
Receipt from sales of equity investment in Juventas | — | | — | ||||||
Cash paid to acquire equity investment in Precision Autoimmune Therapeutics Co., Ltd. | — | ( | — | ||||||
Purchase of short term investments and term deposits | ( | ( | — | ||||||
Proceeds from sales or maturity of short term investments and term deposits | | — | — | ||||||
Receipt from government for return of Wuxi land use right | — | | — | ||||||
Purchase of intangible asset | ( | — | — | ||||||
Proceeds from extinguishment of investment in a convertible loan | | — | — | ||||||
Cash paid to acquire in-process research and development | — | — | ( | ||||||
Cash paid to acquire convertible loan in Black Belt Tx Limited | — | — | ( | ||||||
Receipt of repayment of Black Belt convertible note | — | — | | ||||||
Cash paid to acquire convertible loan in Alesta Tx | — | — | ( | ||||||
Cash paid to acquire convertible loan in Cleave | — | — | ( | ||||||
Receipt of government grants related to land use right | — | — | | ||||||
Net cash provided by (used in) investing activities |
| ( | | ( | |||||
CASH FLOWS FROM FINANCING ACTIVITIES |
| ||||||||
Proceeds from bank borrowings | — | | | ||||||
Repayment of bank borrowings | — | ( | ( | ||||||
Dividend payment to Wuxi LP | ( | — | — | ||||||
Repurchase of ordinary shares | ( | ( | — | ||||||
Stock issuance costs |
| — | — | ( | |||||
Proceeds from sale of ordinary shares |
| — | — | | |||||
Proceeds from exercise of share options |
| | — | — | |||||
Net cash provided by (used in) financing activities |
| ( | ( | | |||||
Effect of exchange rate change on cash and cash equivalents |
| | | ( | |||||
Net increase (decrease) in cash and cash equivalents | ( | | ( | ||||||
Cash and cash equivalents at beginning of year | | | | ||||||
Cash and cash equivalents at end of year | | | | ||||||
Supplemental disclosure of cash flow information: | |||||||||
Interest paid | — | | | ||||||
Income taxes paid | | — | — | ||||||
|
| ||||||||
Non-cash investing and financing activities: | |||||||||
Payables related to property, plant and equipment | | | | ||||||
Exchange of redeemable noncontrolling interest to long term borrowing (Note 12) | | — | — | ||||||
Government grant as a result of loan forgiveness | — | — | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-7
CASI Pharmaceuticals, Inc.
Notes to Consolidated Financial Statements
1. DESCRIPTION OF BUSINESS
Nature of Operations and Organization
CASI Pharmaceuticals, Inc. (“CASI” or the “Company”) (Nasdaq: CASI) is a biopharmaceutical company focused on developing and commercializing innovative therapeutics and pharmaceutical products in China, the United States, and throughout the world. The Company was incorporated in 1991, and in 2012, with new leadership, the Company shifted its business strategy to China and has since built an infrastructure in China that includes sales and marketing, medical affairs, regulatory and clinical development and in the foreseeable future, manufacturing.
The Company is focused on acquiring, developing and commercializing products that augment our hematology oncology therapeutic focus as well as other areas of unmet medical need. The Company is executing its plan to become a biopharmaceutical leader by launching medicines in the greater China market, leveraging its China-based regulatory, clinical and commercial competencies and its global drug development expertise. The majority of the Company’s operations and activities are now located in China and are conducted primarily through two subsidiaries: (i) CASI Pharmaceuticals (China) Co., Ltd. (“CASI China”), which is wholly owned and is located in Beijing, China, and (ii) CASI Pharmaceuticals (Wuxi) Co., Ltd. (“CASI Wuxi”), which is located in Wuxi, China. CASI China is primarily responsible for the day-to-day operations, and oversee the Company’s commercial activities throughout China. CASI Wuxi is part of the long-term strategy to support the Company’s future clinical and commercial manufacturing needs, to manage its supply chain for certain products, and to develop a GMP manufacturing facility in China.
On January 31, 2023, CASI Pharmaceuticals, Inc., the Delaware corporation (“CASI Delaware”) and CASI Pharmaceuticals Holdings, Inc., an exempted company incorporated under the laws of the Cayman Islands and a wholly owned subsidiary of the Company (“CASI Cayman”) entered into a definitive agreement and plan of merger (the “Merger Agreement”) related to a proposed merger transaction. The Merger Agreement provides that, upon the terms and subject to the conditions set forth therein, CASI Delaware will merge with and into CASI Cayman (the “Redomicile Merger”), with CASI Cayman surviving and changing its name to CASI Pharmaceuticals, Inc. Following the Redomicile Merger, CASI Cayman, together with its subsidiaries, owns and continues to conduct the Company’s business in substantially the same manner as is currently being conducted by the Company and its subsidiaries.
The Merger Agreement and the Redomicile Merger were approved by the stockholders of CASI Delaware at a special meeting of stockholders held on March 20, 2023. The Merger Agreement was filed with CASI Cayman’s Registration Statement on Form F-4 filed with the Securities and Exchange Commission (the “SEC”) on January 31, 2023 (the “Registration Statement”) and CASI Cayman’s prospectus filed with the SEC on February 14, 2023 (the “Prospectus”). The Merger Agreement and Redomicile Merger is described in details in CASI Cayman’s proxy statement/prospectus filed with the SEC on January 31, 2023.
On March 21, 2023, CASI Delaware and CASI Cayman completed the Redomicile Merger. Each issued and outstanding stock of CASI Delaware’s common stock was converted to one ordinary share of CASI Cayman. The consolidated financial statements of CASI Cayman represents the continuation of the financial statements of CASI Delaware, reflecting the assets and liabilities, accumulated deficit, and other equity balances of CASI Delaware before the Redomicile Merger. The equity structure is restated using the exchange ratio established in the Merger Agreement to reflect the number of shares of CASI Cayman.
Business Overview
The Company has built a fully integrated, world class biopharmaceutical company dedicated to the successful development and commercialization of innovative and other therapeutic products. Its business development strategy is currently focused on acquiring additional targeted drugs and immuno-oncology therapeutics through licensing that will expand its hematology/oncology franchise. The Company uses a market-oriented approach to identify pharmaceutical/biotechnology candidates that it believes to have the potential for gaining widespread market acceptance, either globally or in China, and for which development can be accelerated under its global drug development strategy. In many cases its business development strategy includes direct equity investments in the licensor company. The Company intends for its pipeline to reflect a diversified and risk-balanced set of assets that include (1) late-stage clinical drug candidates in-licensed for China or global regional rights, (2) proprietary or licensed innovative drug candidates, and (3) select high quality
F-8
pharmaceuticals that fit its therapeutic focus. The Company has focused on US/EU approved product candidates, and product candidates with proven targets or product candidates that have reduced clinical risk with a greater emphasis on innovative therapeutics. Although oncology with a focus on hematological malignancies is its principal clinical and commercial target, the Company is opportunistic about other therapeutic areas that can address unmet medical needs. The Company will continue to pursue building a robust pipeline of drug candidates for development and commercialization in China as its primary market, and if rights are available for the rest of the world.
The Company believes its China operations offer a significant market and growth potential due to the extraordinary increase in demand for high quality medicines coupled with regulatory reforms in China that facilitate the entry of new pharmaceutical products into the country. The Company will continue to in-license clinical-stage and late-stage drug candidates, and leverage its cross-border operations and expertise, and hope to be the partner of choice to provide access to the China market. The Company expects the implementation of its plans will include leveraging its resources and expertise in both the U.S. and China so that the Company can maximize regulatory, development and clinical strategies in both countries.
The Company launched its first commercial product, EVOMELA® (Melphalan for Injection) in China in August 2019. In China, EVOMELA® is approved for use as a conditioning treatment prior to stem cell transplantation and as a palliative treatment for patients with multiple myeloma. EVOMELA®, was originally licensed from Spectrum Pharmaceuticals, Inc. (“Spectrum”) and it had a supply agreement with Spectrum to support its application for import drug registration and for commercialization purposes. Spectrum completed the sale of its portfolio of FDA-approved hematology/oncology products including EVOMELA® to Acrotech Biopharma L.L.C. (“Acrotech”) on March 1, 2019. The original supply agreement with Spectrum was assumed by Acrotech.
On July 31, 2023, the Company entered into a tripartite assignment agreement (the “FOLOTYN® Assignment Agreement”) with Mundipharma International Corporation Limited (“MICL”), Mundipharma Medical Company (“MMCo”) and Acrotech Biopharma Inc. (“Acrotech Inc.”), pursuant to which, MICL’s rights and obligations under that certain License, Development and Commercialization Agreement (as amended and restated) dated as of May 29, 2013 for the commercialization of FOLOTYN® (Pralatrexate) in China, with certain terms of such rights and obligations amended as agreed to by the parties, is assigned to the Company. FOLOTYN® (Pralatrexate) is a dihydrofolate reductase inhibitor indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (“PTCL”). This product was approved by both the FDA and China’s NMPA for PTCL. The Company announced the first patient received FOLOTYN® treatment in China on February 15, 2024.
The other core hematology/oncology assets in the Company’s pipeline include:
· | CNCT19 is an autologous CD19 CAR-T investigative product (“CNCT19”) being developed by the Company’s partner Juventas for which it has exclusive worldwide co-commercial and profit-sharing rights. CNCT19 is being developed as a potential treatment for patients with hematological malignancies which express CD19 including, B-cell acute lymphoblastic leukemia (“B-ALL”) and B-cell non-Hodgkin lymphoma (“B-NHL”). CNCT19 targets CD19, a B-cell surface protein widely expressed during all phases of B-cell development and a validated target for B-cell driven hematological malignancies. CD19 targeted CAR constructs from several different institutions have demonstrated consistently high antitumor efficacy in children and adults with relapsed B-cell acute lymphoblastic leukemia (B-ALL), chronic lymphocytic leukemia (CLL), and B-cell non-Hodgkin lymphoma (B-NHL). In November 2023, The NMPA has granted market approval for Juventas' investigational cell therapy, CNCT19 (Inaticabtagene Autoleucel), for the treatment of relapsed and refractory B-cell acute lymphoblastic leukemia (r/r B-ALL) in China. |
· | BI-1206 In October 2020, the Company entered into an exclusive licensing agreement with BioInvent International AB (“BioInvent”) for the development and commercialization of novel anti-FcγRIIB antibody, BI-1206, in Mainland China, Taiwan, Hong Kong and Macau. BioInvent is a biotechnology company focused on the discovery and development of first-in-class immune-modulatory antibodies for cancer immunotherapy. BI-1206 is being investigated in a Phase 1/2 trial, in combination with anti-PD1 therapy Keytruda® (pembrolizumab), in patients with solid tumors, and in a Phase 1/2a trial in combination with MabThera® (rituximab) in patients with relapsed/refractory non-Hodgkin lymphoma (NHL). CTA was approved by China NMPA in December 2021 and ethics committee approvals have been received in January of 2022. The Company obtained approval from China Human Genetic Resources Administrative Office (“HGRAO”) in April 2022. The Company is planning a Phase 1 study of BI-1206 in combination with rituximab with a single agent BI-1206 run in phase in patients with NHL (mantle cell lymphoma, marginal zone lymphoma, and follicular lymphoma) to assess PK, safety and tolerability, select the Recommended Phase 2 Dose and assess early signs of clinical efficacy as part of its |
F-9
development program for BI-1206 in China. The study received regulatory approval from the China Center for Drug Evaluation (“CDE”) in the second quarter of 2022, and the first patient was enrolled and dosed in the third quarter of 2022.
· | CB-5339 is a novel VCP/p97 inhibitor focused on valosin-containing protein (VCP)/p97 as a novel target in protein homeostasis, DNA damage response and other cellular stress pathways for therapeutic use in the treatment of patients with various malignancies. The Company entered into an exclusive license on March 21, 2021 with Cleave Therapeutics, Inc. (“Cleave”) for the development and commercialization of CB-5339 in Mainland China, Hong Kong, Macau and Taiwan. CB-5339, an oral second-generation, small molecule VCP/p97 inhibitor, has been evaluated in a Phase 1 clinical trial in patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). The Company submitted the CB-5339 CTA application for the multiple myeloma indication in March 2022 and received approval from the NMPA in January 2023. |
· | CID-103 is a full human IgG1 anti-CD38 monoclonal antibody recognizing a unique epitope that has demonstrated an encouraging preclinical efficacy and safety profile compared to other anti-CD38 monoclonal antibodies, and which the Company has exclusive global rights. CID-103 is being developed for the treatment of patients with multiple myeloma. The Phase 1 dose escalation and expansion study of CID-103, in patients with previously treated, relapsed or refractory multiple myeloma is closed to further accrual in France and the UK. |
· | Thiotepa is a chemotherapeutic agent, which has multiple indications including use as a conditioning treatment for use prior to certain allogeneic haemopoietic stem cell transplants. Thiotepa has a long history of established use in the hematology/oncology setting. The Company is applying for generic registration and, subject to regulatory and marketing approvals. |
Liquidity and Capital Resources
Since its inception in 1991, the Company has incurred significant losses from operations and, as of December 31, 2023, has incurred an accumulated deficit of $
The Company believes that it has sufficient resources to fund its operations at least one year beyond the date that the consolidated financial statements are issued. As of December 31, 2023, the Company had a balance of cash and cash equivalents of $
License and Distribution Agreements
EVOMELA® License Arrangements with Acrotech
The Company has product rights and perpetual exclusive licenses from Acrotech Biopharma L.L.C. to develop and commercialize its commercial product EVOMELA® (Melphalan Hydrochloride For Injection) in the greater China region (which includes Mainland China, Taiwan, Hong Kong and Macau). As well the Company had similar rights to assets ZEVALIN® (Ibritumomab Tiuxetan) and MARQIBO® (Vincristine Sulfate Liposome Injection). The exclusive licenses held by the Company were originally licensed from Spectrum Pharmaceuticals Cayman, LP Inc. (“Spectrum”), and Spectrum completed the sale of its portfolio of FDA-approved hematology/oncology products including EVOMELA® to Acrotech on March 1, 2019. On December 3, 2018, the Company received NMPA’s approval for importation, marketing and sales in China and in August 2019 the Company launched commercial sales EVOMELA® in China. The NMPA required EVOMELA® post-marketing study has been completed and the clinical study report has been submitted to CDE for review and comment. In May 2022, Acrotech and the Company agreed to terminate the license agreement with respect to MARQIBO® and ZEVALIN®.
F-10
FOLOTYN® (Pralatrexate) License Agreement with MICL, MMCo and Acrotech Inc.
On July 31, 2023, the Company entered into a tripartite assignment agreement (the “FOLOTYN® Assignment Agreement”) with MICL, MMCo and Acrotech Inc., pursuant to which, MICL’s rights and obligations under that certain License, Development and Commercialization Agreement (as amended and restated) dated as of May 29, 2013 for the commercialization of FOLOTYN® (Pralatrexate) in China, with certain terms of such rights and obligations amended as agreed to by the parties, is assigned to the Company.
In relation to the FOLOTYN® Assignment Agreement, the Company and MICL entered into a payment agreement (the “Payment Agreement”), pursuant to which the Company will pay MICL a total of US$
Concurrently with the execution of the FOLOTYN® Assignment Agreement and the Payment Agreement, the parties entered into certain other agreements in relation to the commercialization of FOLOTYN®, including a supply agreement entered into by and between the Company and Acrotech Inc., pursuant to which Acrotech Inc. will supply to the Company FOLOTYN® subject to terms and on the conditions set forth therein. The Company is obligated to pay Acrotech Inc. additional $
The Company announced the first patient received FOLOTYN® treatment in China on February 15, 2024. The Company will continue to spend time, resources and efforts on the commercialization of FOLOTYN® in China.
China Resources Pharmaceutical Commercial Group International Trading Co., Ltd. (previously known as China Resources Guokang Pharmaceuticals Co., Ltd.)
In March 2019, the Company entered into a
China National Medicines Corporation Ltd.
On December 6, 2023, the Company entered into an Exclusive Distribution Agreement (the “Agreement”) with China National Medicines Corporation Ltd. (“CNMC”) and CASI China. Under the terms of the Agreement, the Company appointed CNMC on an exclusive basis as its sole distributor for the sale of Pralatrexate for Injection (Folotyn®) in the territory of the People’s Republic of China (excluding Hong Kong, Taiwan and Macau) (the “Territory”) during the term of
Precision Autoimmune Therapeutics Co., Ltd., (previously known as Beijing Tianshi Tongda Pharmaceuticals Technology Co., Ltd)
In May 2022, the Company entered into a Sublicense Agreement (the “Sublicense Agreement”) with Precision Autoimmune Therapeutics Co., Ltd. (“PAT”), a company established under the laws of China, pursuant to which the Company granted PAT an exclusive (subject to the commercialization and co-marketing rights), perpetual, worldwide license, with the right to freely grant further sublicenses subject to terms and conditions in the Sublicense Agreement, for the investigational anti-CD38 monoclonal antibody TSK011010 licensed and controlled by the Company from Black Belt Therapeutics Limited, in the treatment, prevention and diagnosis
F-11
of autoimmune diseases, conditions and disorders in humans. Pursuant to the Sublicense Agreement, PAT will make an upfront payment of $
Also in May 2022, CASI Pharmaceuticals (China) Co., Ltd. (“CASI China”) entered into an agreement for the investment in PAT in the amount of RMB
Juventas Cell Therapy Ltd. (“Juventas”)
In June 2019, the Company entered into a license agreement for exclusive worldwide license to commercialize an autologous anti-CD19 T-cell therapy product (CNCT19) from Juventas (the “Exclusive License Agreement”). Juventas is a China-based company engaged in cell therapy. The terms of the agreement include RMB
In September 2020, Juventas and its shareholders (including CASI Biopharmaceuticals (Wuxi) Co., Ltd (“CASI Biopharmaceuticals”), a subsidiary of the Company, ) agreed to certain terms and conditions required by a new third-party investor to facilitate the Series B financing of Juventas, pursuant to which the Company agreed to amend and supplement the original “licensing agreement (the "Supplementary Agreement") by agreeing to pay Juventas certain percentage of net profits generated from commercial sales of CNCT19 in addition to the royalty fee payment calculated as a percentage of net sales. The Supplementary Agreement also specifies a minimum annual target net profit to be distributed to Juventas and certain other terms and obligations. In return, the Company obtained additional equity interests in Juventas.
Under the Supplementary Agreement, Juventas and the Company will jointly market CNCT19, including, but not limited to, establishing medical teams, developing medical strategies, conducting post-marketing clinical studies, establishing Standardized Cell Therapy Centers, establishing and training providers with respect to cell therapy, testing for cell therapy, and monitoring quality controls (cell collection and transfusion, etc.), and patient management (adverse reactions treatment, patients’ follow-up visits, and establishment of a database). The Company also will reimburse Juventas for a portion of Juventas’ marketing expenses as reviewed and approved by a joint commercial committee to be constituted. The Company will continue to be responsible for recruiting and establishing a sales team to commercialize CNCT19.
In September 2022, CASI Biopharmaceuticals entered into an Equity Transfer Agreement to transfer its equity interest in Juventas in the amount of RMB
On March 2, 2024, CASI received a notice from Juventas, which purported to terminate the CNCT19 Agreements. CASI responded to Juventas’ purported termination notice, noting that Juventas was not entitled to unilaterally terminate the CNCT19 Agreements and further demanding that Juventas cease any conduct that may constitute further breach of the CNCT19 Agreements and execute a written undertaking regarding compliance with the CNCT19 Agreements by March 13, 2024. Juventas did not comply with CASI’s demands. On March 20, 2024, CASI submitted a Notice of Arbitration at the Hong Kong International Arbitration Centre (“HKIAC”) against Juventas pursuant to the CNCT19 Agreements’ dispute resolution clauses, claiming that Juventas’ purported termination was invalid and that Juventas breached the CNCT19 Agreements and seeking, among other things, damages and injunctive reliefs. Together with the Notice of Arbitration, CASI also submitted an application for the appointment of an emergency arbitrator, seeking emergency injunctive reliefs. On the same day, Juventas also submitted a Notice of Arbitration at the HKIAC against CASI, alleging, among other things, that the CNCT19 Agreements were validly terminated and that CASI breached the CNCT19 Agreements. The HKIAC has appointed an emergency arbitrator in accordance with CASI’s application. The arbitration proceedings are in their early stages and the Company cannot predict right now the outcome of either of these proceedings. If we do not prevail in either of these proceedings completely or in part, or fail to reach a favorable settlement with Juventas, our plan with respect to the commercialization of CNCT 19 may be delayed or otherwise adversely impacted, which may in turn result in adverse impacts on our results of operations, financial condition and prospects.
F-12
BioInvent International AB
In October 2020, the Company entered into an exclusive licensing agreement with BioInvent International AB (“BioInvent”) for the development and commercialization of novel anti-FcγRIIB antibody, BI-1206, in Mainland China, Taiwan, Hong Kong and Macau. BioInvent is a biotechnology company focused on the discovery and development of first-in-class immune-modulatory antibodies for cancer immunotherapy. BI-1206 is being investigated in a Phase 1/2 trial, in combination with anti-PD1 therapy Keytruda® (pembrolizumab), in patients with solid tumors, and in a Phase 1/2a trial in combination with MabThera® (rituximab) in patients with relapsed/refractory non-Hodgkin lymphoma (NHL). The CASI Clinical Trial Application (CTA) was approved by China National Medical Products Administration (NMPA) in December 2021 and ethics committee approvals have been received in January of 2022. The Company obtained approval from HGRAO in April 2022. The Company is planning a Phase 1 study of BI-1206 in combination with rituximab with a single agent BI-1206 run in phase in patients with NHL (mantle cell lymphoma, marginal zone lymphoma, and follicular lymphoma) to assess PK, safety and tolerability, select the Recommended Phase 2 Dose and assess early signs of clinical efficacy as part of its development program for BI-1206 in China. The study received regulatory approval from the China Center for Drug Evaluation (“CDE”) in the second quarter of 2022, and the first-patient dosing was achieved in the third quarter of 2022.
Under the terms of the agreement, BioInvent and CASI will develop BI-1206 in both hematological malignancies and solid tumors, with CASI responsible for commercialization in China and associated markets. CASI made a $
In conjunction with the license agreement entered into with BioInvent, the Company made a SEK
Black Belt Therapeutics Limited
In April 2019, the Company entered into a license agreement with Black Belt Therapeutics Limited (“Black Belt”) for exclusive worldwide rights to CID-103, an investigational anti-CD38 monoclonal antibody (Mab) (formerly known as TSK011010). The Company expects that its clinical materials and commercial inventory will be supplied by one or more contract manufacturers with whom the Company has contracted with. Under the terms of the agreement, CASI obtained global rights to CID-103 for an upfront payment of
Cleave Therapeutics, Inc.
In March 2021, the Company entered into an exclusive license with Cleave Therapeutics, Inc. (“Cleave”) for the development and commercialization of CB-5339, an oral novel VCP/p97 inhibitor, in both hematological malignancies and solid tumors, in Mainland China, Hong Kong, Macau and Taiwan. Cleave is a clinical-stage biopharmaceutical company focused on valosin-containing protein (VCP)/p97 as a novel target in protein homeostasis, DNA damage response and other cellular stress pathways for therapeutic use in the treatment of patients with cancer. Cleave and the Company will develop CB-5339 in both hematological malignancies and solid tumors, with CASI responsible for development and commercialization in China and associated markets. The Company paid a $
F-13
CB-5339 has been evaluated by Cleave in a Phase 1 clinical trial in patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). Because CB-5339 has not yet reached technological feasibility and has no alternative future uses, the Company expensed the $
On July 18, 2023, the Company entered into an assignment agreement (the “Cleave Assignment Agreement”) with Cleave, pursuant to which the Company obtained the global intellectual property rights related to CB-5339. The Assignment Agreement terminates and supersedes the License Agreement for CB-5339 the Company previously entered into with Cleave. Pursuant to the Assignment Agreement and partially in exchange for the transfer of the global intellectual property rights for CB-5339 as well as all remaining CB-5339 drug substance and drug product to the Company and a repayment in the amount of USD $
Pharmathen Global BV
On October 29, 2019, the Company entered into an exclusive distribution agreement with Pharmathen Global BV (“Pharmathen”) for the development and distribution of octreotide long acting injectable (Octreotide LAI) microsphere in China. Octreotide LAI formulations, which are approved in various European countries, are considered a standard of care for the treatment of acromegaly and the control of symptoms associated with certain neuroendocrine tumors. The Company paid and expensed an upfront payment of
Riemser Pharma GmbH
In August 2019, the Company entered into a distribution agreement in China with Riemser Pharma GmbH (“Riemser”) to a novel formulation of thiotepa, a chemotherapeutic agent, which has multiple potential indications including use as a conditioning treatment for use prior to allogenic hematopoietic stem cell transplantation. Thiotepa has a long history of established use in the hematology/oncology setting. Pursuant to the distribution agreement, CASI obtained the exclusive distribution right of the products in China, and Riemser will be responsible for manufacturing and supplying CASI with clinical materials and commercial inventory. The Company is applying for generic registration and, subject to regulatory and marketing approvals, the Company intends to advance and commercialize this product in China. In January 2020, Riemser was acquired by Esteve Healthcare, S.L. (“ESTEVE”), an international pharmaceutical company headquartered in Barcelona, Spain. In November 2022, the Company entered into an Amendment with Esteve, pursuant to which the Company and Esteve will equally share the costs of clinical trials (if any) for the registration of Thiotepa in China. After the product is launched, the Company will be subject to annual minimum purchase as prescribed in the agreement.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation
The accompanying consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). The Company’s Redomicile Merger was accounted for as a legal reorganization with no change in ultimate ownership interest immediately before and after the transaction. Accordingly, all assets and liabilities will be recorded at historical cost as an exchange between entities under common control, and the consolidated assets and liabilities of CASI Cayman will be the same as those of the Company immediately prior to the Redomicile Merger.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. The Company’s significant
F-14
accounting estimates relate to recoverability of property, plant and equipment, right of use assets, intangible assets and long-term investments, valuation allowance for deferred tax assets, share-based arrangements and fair value of investments. Management bases its estimates on historical experience and on various other assumptions that it believes are reasonable under the circumstances. Actual results may differ from those estimates, and such differences may be material to the consolidated financial statements.
Consolidation
The accompanying consolidated financial statements include the accounts of the Company and its subsidiaries, in which CASI, directly or indirectly, has a controlling financial interest.
These subsidiaries include CASI China, CASI Wuxi, CASI Biopharmaceuticals, CASI Pharmaceuticals (Hainan) Co., Ltd. (“CASI Hainan”), ZhongBio (Beijing) Tech Co. Ltd. (“ZhongBio”) and CASI Pharmaceuticals Co., Ltd., a subsidiary incorporated in Hong Kong in 2023. ZhongBio was dissolved in January 2023, and CASI Hainan was dissolved in May 2023.
All inter-company balances and transactions have been eliminated in consolidation. The Company currently operates in
Foreign Currency Translation and Transactions
The accompanying consolidated financial statements of the Company are reported in US dollars. The financial position and results of operations of the Company’s subsidiaries in the PRC are measured using the Renminbi (RMB), which is the local and functional currency of these entities. Assets and liabilities of the Company’s PRC subsidiaries are translated into US$ using the exchange rates in effect at the consolidated balance sheet date. The revenues and expenses of these entities are translated into US$ at the weighted average exchange rates for the period. The resulting translation gains (losses) are recorded in accumulated other comprehensive loss as a component of shareholders’ equity.
Transactions denominated in foreign currencies are remeasured into the functional currency at the exchange rates prevailing on the transaction dates. Foreign currency denominated financial assets and liabilities are remeasured at the exchange rates prevailing at the balance sheet date. Net gains or losses resulting from foreign currency denominated transactions are recorded in foreign exchange gain (losses) in the consolidated statements of operations and comprehensive loss.
Segment Reporting
In accordance with ASC 280, Segment Reporting, the Company’s chief operating decision maker, the Chief Executive Officer, reviews the consolidated results when making decisions about allocating resources and assessing performance of the Company as a whole and hence, the Company has only
Revenue Recognition
Product sales and sublicensing revenue recognized in the consolidated statements of operations and comprehensive loss are considered revenue from contracts with customers and, accordingly, the Company recognizes revenue using the following steps:
| ● | Identification of the contract, or contracts, with a customer; |
| ● | Identification of the performance obligations in the contract; |
| ● | Determination of the transaction price, including the identification and estimation of variable consideration; |
| ● | Allocation of the transaction price to the performance obligations in the contract; and |
| ● | Recognition of revenue when the Company satisfies a performance obligation. |
F-15
The Company recognizes revenue on sales of EVOMELA® when the control of the product is transferred to the distributor, which occurs upon delivery of the product to the carrier appointed by the distributor, in an amount that reflects the consideration to which the Company expects to be entitled to in exchange for the product, excluding amounts collected on behalf of third parties (e.g. value-added taxes). Payment terms for these sales are due within 90 days. The arrangement does not include any variable consideration. The Company recognizes accounts receivable when it recognizes revenues as its right to consideration is unconditional and only the passage of time is required before payment of that consideration is due.
The costs of assurance type warranties that provide the customer the right to exchange purchased product that does not meet appropriate quality standards are recognized when they are probable and are reasonably estimable. There was no product exchange during the years ended December 31, 2023, 2022 and 2021. As of December 31, 2023, 2022 and 2021, the Company did not incur, and therefore did not defer, any material costs to obtain or fulfill contracts. The Company did not have any contract assets or contract liabilities as of December 31, 2023 and 2022.
The Company recognizes revenue from non-refundable upfront payments and milestone payments on sublicense arrangements when the license that represents a functional intellectual property is transferred to the customer and the customer is able to use and benefit from the license. When certain upfront payments or milestone payments is considered as constrained variable considerations as they are contingent upon achievement of certain milestones that are not within the control of the Company, they are excluded from the transaction price before the constraints are resolved.
Concentrations Risks
Cash Concentration Risk
The Company maintains its U.S., Hong Kong dollar and RMB cash in bank deposit accounts, which, at times, may exceed regulated insured limits. The Company believes it is not exposed to significant credit risk on cash and cash equivalents.
Vendor Concentration Risk
The Company has a sole supplier, Acrotech, for its EVOMELA® and FOLOTYN® products. The Company’s ability to select other providers of EVOMELA® and FOLOTYN® is limited by FDA regulations.
Geographic Concentration Risk
The Company revenue is solely generated in mainland China.
Accounts Receivable and Credit Concentration
CRPCGIT is the sole customer of the Company's EVOMELA® product sales in China. All consolidated product sales for the years ended December 31, 2023, 2022 and 2021 were generated from sales to CRPCGIT in China, and all the Company’s accounts receivable balance as of December 31, 2023 and 2022 were due from CRPCGIT.
The Company extends credit to CRPCGIT on an unsecured basis and does not believe there is significant credit risk.
Fair Value of Financial Instruments
Fair value is the price that would be received from the sale of an asset or paid to transfer a liability assuming an orderly transaction in the most advantageous market at the measurement date. U.S. GAAP establishes a hierarchical disclosure framework which prioritizes and ranks the level of observability of inputs used in measuring fair value. These tiers include:
| ● | Level 1—Quoted prices (unadjusted) in active markets that are accessible at the measurement date for identical assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs. |
| ● | Level 2—Observable market-based inputs other than quoted prices in active markets for identical assets or liabilities. |
F-16
Level 3—Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
See Note 3 and Note 19 for additional fair value disclosures.
Cash and Cash Equivalents
Cash and cash equivalents include cash and highly liquid investments with original maturities of less than 90 days that are readily convertible to known amounts of cash. As of December 31, 2023, the Company had $
Short Term Investments
The Company’s short term investments represent investments in financial instruments with a variable interest rate and term deposits with original maturities of more than 90 days that are readily convertible to known amounts of cash. The Company elected the fair value method at the date of initial recognition, and carried these short term investments subsequently at fair value. Changes in fair values are reflected in the consolidated statements of comprehensive income. As of December 31, 2023, the Company held its short term investments solely in financial institutions in mainland China. The aggregate cost and fair value are as follows:
December 31 | ||||||
(In thousands) |
| 2023 |
| 2022 | ||
Aggregate cost basis | $ | |
| $ | | |
Gross unrealized gain |
| |
| — | ||
Aggregate fair value | $ | | $ | | ||
Allowance for Credit Losses
The Company adopted Accounting Standards Update (“ASU”) No. 2016-13, Financial Instruments — Credit Losses (Topic 326) (“ASU 2016-13”) and subsequent amendments for the year beginning January 1, 2023.
The Company estimates the allowance for credit losses based on the historical loss rates from historical observation period, and adjusted to reflect the effects of current and future economic conditions over reasonable and supportable forecast period.
Inventories
Inventories consist of EVOMELA® and FOLOTYN® finished goods and are stated at the lower of cost or net realizable value. Cost is determined using a first-in, first-out method. Net realizable value is the estimated selling prices in the ordinary course of business, less reasonably predictable costs of completion, disposal, and transportation. Adjustments are recorded to write down the carrying amount of any obsolete and excess inventory to its estimated net realizable value based on historical and forecasted demand.
Research and Development Expenses
Research and development expenses consist primarily of compensation and other expenses related to research and development personnel, research collaborations, costs associated with pre-clinical testing and clinical trials of the Company’s product candidates, including the costs of manufacturing drug substance and drug product, regulatory maintenance costs, and facilities expenses, along with the amortization of acquired ANDAs. Research and development costs are expensed as incurred.
Property, Plant and Equipment
Property, plant and equipment are stated at cost, less accumulated depreciation and impairment, if any.
F-17
Costs incurred in the construction of property, plant and equipment, including down payments and progress payments, are initially capitalized as construction-in-progress and transferred into their respective asset categories when the assets are ready for their intended use, at which time depreciation commences. Office equipment and furniture are depreciated over their estimated useful lives of
Costs of Revenues
Costs of revenues consist primarily of the cost of inventories of EVOMELA® and sales-based royalties related to the sale of EVOMELA®.
Investments
The Company’s investments mainly consist of investments in equity securities with readily determinable fair value, equity securities without readily determinable fair value, investments measured using fair value option, and investment of equity method.
Investment in equity securities with readily determinable fair value are measured at fair values, and any changes in fair value are recognized in earnings. Where the fair value of an investment in equity securities is not readily determinable, the Company recognizes such investment in long-term investments, and uses the measurement alternative of cost minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for the identical or a similar investment of the same issuer.
For equity investments measured at fair value with changes in fair value recorded in earnings, the Company does not assess whether those securities are impaired. For equity investments without readily determinable fair value, at each reporting period, the Company makes a qualitative assessment considering impairment indicators to evaluate whether the investment is impaired. Impairment indicators that the Company considers include, but are not limited to, (i) the deterioration of earnings performance, credit rating, asset quality, or business prospects of the investee, (ii) a significant adverse change in the regulatory, economic, or technological environment of the investee, (iii) a significant adverse change in the general market condition of either the geographic area or the industry in which the investee operates. If a qualitative assessment indicates that the investment is impaired, the Company has to estimate the investment’s fair value and if the fair value is less than the investment’s carrying value, the Company recognizes an impairment loss in non-operating expenses equal to the difference between the carrying value and fair value.
Dividend income is recognized in other income when earned.
The Company elected to use fair value option to account for its investment in Cleave (see Note 3) as permitted under Accounting Standards Codification (“ASC”) 825, Financial Instruments (“ASC 825”), which then refers to ASC 820, Fair Value Measurement (“ASC 820”) to provide the fair value framework for valuing such investments. In accordance with ASC 820, the Company records such investment at fair value, with changes in fair value recorded in change in fair value of investments in the consolidated statements of operations and comprehensive loss.
For investments in common stock or in-substance common stock of entities over which the Company can exercise significant influence but does not own a majority equity interest or control, the equity method is applied, and the Company adjusts the carrying amount of an investment and recognizes investment income or loss for the Company’s share of the earnings or loss of the investee after the date of investment.
Leases
At contract inception, the Company determines whether an arrangement is or contains a lease and whether the lease should be classified as an operating or a financing lease. A contract is or contains a lease if the contract conveys the right to control the use of the identified asset for a period of time in exchange for consideration. Control is determined based on the right to obtain substantially all of the economic benefits from use of the identified asset and the right to direct the use of the identified asset. Right of use (“ROU”) assets
F-18
for operating leases represent the right to use an underlying asset for the lease term, and operating lease liabilities represent the obligation to make lease payments.
ROU assets and lease liabilities are recognized upon lease commencement for operating leases based on the present value of lease payments over the lease term. As the rate implicit in the lease cannot be readily determined, the Company uses incremental borrowing rate at the lease commencement date in determining the imputed interest and present value of lease payments. The incremental borrowing rate was determined based on the rate of interest that the Company would have to borrow an amount equal to the lease payments on a collateralized basis over a similar term. The incremental borrowing rate is primarily influenced by the risk-free interest rate of China and the US, the Company’s credit rating and lease term, and is updated for measurement of new lease liabilities.
For operating leases, the Company recognizes a single lease cost on a straight-line basis over the remaining lease term.
The Company has elected not to recognize ROU assets or lease liabilities for leases with an initial term of 12 months or less; the Company recognizes lease expense for these leases on a straight-line basis over the lease term. In addition, the Company has elected not to separate non-lease components (e.g., common area maintenance fees) from the lease components.
Land use rights acquired are recognized in right-of-use assets if they meet the definition of lease.
As of December 31, 2023, all of the Company’s ROU assets are located in the mainland China.
Impairment of Long-Lived Assets
Long-lived assets, including property, plant and equipment, right of use (“ROU”) assets and intangible assets subject to amortization, are reviewed for impairment whenever events or changes in circumstances (“triggering events”) indicate that the carrying amount of an asset or asset group may not be recoverable. The Company identifies triggering events and performs impairment testing at asset group level which represents the lowest level for which identifiable cash flows are largely independent of the cash flows of other groups of assets and liabilities. As of December 31, 2023, the Company’s asset groups consist of manufacturing asset group and non-manufacturing asset group.
Triggering events include, but are not limited to, significant decrease of market price of the asset group, significant adverse change of an asset group’s use or physical condition, significant adverse changes in the industry conditions, significantly excessive accumulated cost compared with original expectation, expected continuing losses or negative cash flow associated with the use of the asset group, and expected significant early disposal of asset group.
If circumstances require an asset or asset group be tested for possible impairment, the Company first compares undiscounted cash flows expected to be generated by that asset or asset group to its carrying value. If the carrying value of the long-lived asset or asset group is not recoverable on an undiscounted cash flow basis, an impairment is recognized to the extent that the carrying value exceeds its fair value. Fair value is determined through various valuation techniques including discounted cash flow models, quoted market values and third-party independent appraisals, as considered necessary.
Acquired In-Process Research and Development Expense
The Company has acquired rights to develop and commercialize product candidates. Upfront payments that relate to the acquisition of a new drug compound, as well as pre-commercial milestone payments, are immediately expensed as acquired in-process research and development in the period in which they are incurred, provided that the new drug compound did not also include processes or activities that would constitute a “business” as defined under U.S. GAAP, the drug has not achieved regulatory approval for marketing and, absent obtaining such approval, has no established alternative future use.
The Company also pays contingent development milestone payments in accordance with agreements (see Note 1). The Company recognizes development milestone payments as acquired in-process research and development expenses when the milestones are reached.
F-19
Share-Based Compensation
The Company records compensation expense associated with service and performance-based share options in accordance with provisions of authoritative guidance. The estimated fair value of solely service-based awards that have a graded vesting schedule is measured on the grant date and is generally recognized on a straight-line basis over the requisite service period for the entire award, provided that the cumulative amount of compensation cost recognized at any date at least equals the portion of the grant date fair value of such award that is vested at that date. The estimated fair value of performance-based awards is measured on the grant date and is recognized when it is determined that it is probable that the performance condition will be achieved at tranche-by-tranche basis. If the required vesting conditions are not met resulting in the forfeiture of the share-based awards, previously recognized compensation expense relating to those awards are reversed as occurred.
When there is a modification of an award, the accounting for the modification depends on the likelihood, at the date of the modification that the original award would have vested under its original terms. If, at the date of the modification, it is probable that the original award would have vested under its original terms, the cumulative compensation cost to be recognized equals the grant-date fair value of the original award plus the incremental value of the award given in the modification. In calculating the incremental compensation cost of a modification, the fair value of the modified award is compared to the fair value of the original award measured immediately before its terms or conditions are modified. The Company recognizes share-based compensation over the vesting periods of the modified awards, which comprises, (i) the amortization of the incremental portion of share-based compensation over the remaining vesting term and (ii) any unrecognized compensation cost of the original award over the original term. If, at the date of the modification, it is not probable that the original award would have vested under its original terms, the fair value of the modified awards on the modification date will be used for recognizing compensation cost when it becomes probable.
Grant date and modification date fair value was determined using an option pricing model which is affected by the fair value of underlying ordinary shares as well as assumptions regarding a number of complex and subjective variables, such as expected volatility, expected term of options, risk-free rate, and expected dividend yield.
Government Grants
Government grants are recognized when there is reasonable assurance that the Company will comply with required conditions and the grants will be received. Government grants related to assets are presented as deferred income that is recognized on a systematic basis over the useful life of the asset.
Income Taxes
Income tax expense is recognized using the asset and liability method. Deferred tax assets and liabilities are determined based on the difference between the financial statement and tax bases of assets and liabilities and operating loss and tax credit carryforwards as measured by the enacted tax rates that will be in effect when these differences reverse. A valuation allowance is provided to reduce the amount of deferred income tax assets if it is considered more likely than not that some portion or all of the deferred income tax assets will not be realized.
The Company recognizes in its consolidated financial statements the impact of a tax position if a tax return position or future tax position is “more likely-than-not” to be sustained upon examination, based on the technical merits of the position. Tax positions that meet the “more-likely-than-not” recognition threshold are measured at the largest amount of tax benefit that has a greater than fifty percent likelihood of being realized upon settlement. The Company recognizes interest and penalties related to uncertain tax positions, if any, in income tax expense.
Net Loss Per Share
Net loss per share (basic and diluted) was computed by dividing net loss attributable to ordinary shareholders by the weighted average number of ordinary shares outstanding.
F-20
New Accounting Pronouncements
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments — Credit Losses (Topic 326) (“ASU 2016-13”) and subsequent amendments to the initial guidance including ASU No. 2018-19, ASU No. 2019-04, and ASU No. 2019-05 (collectively, “Topic 326”). Topic 326 requires entities to measure all expected credit losses for financial assets held at the reporting date based on historical experience, current conditions, and reasonable and supportable forecasts. This replaces the existing incurred loss model and is applicable to the measurement of credit losses on financial assets measured at amortized cost. This standard is effective for public business entities, excluding entities eligible to be smaller reporting companies for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. For all other entities, this standard is effective for annual and interim periods beginning after December 15, 2022 and early adoption is permitted for annual and interim periods beginning after December 15, 2018. As CASI Delaware was a smaller reporting company and was not required to adopt it for annual and interim periods before December 15, 2022, the Company adopted ASU 2016-13 and subsequent amendments for the year beginning January 1, 2023. The adoption of ASU 2016-13 and subsequent amendments does not have a material impact on the Company’s consolidated financial statements.
In November 2021, the FASB issued ASU No. 2021-10, Government Assistance (Topic 832): Disclosures by Business Entities about Government Assistance. This update requires certain annual disclosures about transactions with a government that are accounted for by applying a grant or contribution accounting model by analogy. This update is effective for annual periods beginning after December 15, 2021, and early application is permitted. This guidance should be applied either prospectively to all transactions that are reflected in financial statements at the date of initial application and new transactions that are entered into after the date of initial application or retrospectively to those transactions. The Company adopted ASU 2021-10 for the annual period from January 1, 2022 and retrospectively disclosed such transactions in Note 11.
In June 2022, the FASB issued ASU 2022-03, Fair Value Measurement (Topic 820): Fair Value Measurement of Equity Securities Subject to Contractual Sale Restrictions, which aims to (1) clarify the guidance in Topic 820, Fair Value Measurement, when measuring the fair value of an equity security subject to contractual restrictions that prohibit the sale of an equity security, (2) to amend a related illustrative example, and (3) to introduce new disclosure requirements for equity securities subject to contractual sale restrictions that are measured at fair value in accordance with Topic 820. The amendments in this Update clarify that a contractual restriction on the sale of an equity security is not considered part of the unit of account of the equity security and, therefore, is not considered in measuring fair value. The amendments also clarify that an entity cannot, as a separate unit of account, recognize and measure a contractual sale restriction. The amendments in this Update also require the following disclosures for equity securities subject to contractual sale restrictions: (1) the fair value of equity securities subject to contractual sale restrictions reflected in the balance sheet, (2) the nature and remaining duration of the restriction(s) and (3) the circumstances that could cause a lapse in the restriction(s). The amendments in this Update are effective for public business entities for fiscal years beginning after December 15, 2023. The Company plans to adopt ASU 2022-03 for the year beginning January 1, 2024 and does not believe there is material impact to the consolidated financial statements other than disclosures required above.
In November 2023, the FASB issued ASU 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures. The amendments in ASU 2023-07 improve reportable segment disclosure requirements through enhanced disclosures about significant segment expenses. The amendments introduce a new requirement to disclose significant segment expenses regularly provided to the chief operating decision maker (CODM), extends certain annual disclosures to interim periods, clarifies single reportable segment entities must apply ASC 280 in its entirety, permits more than one measure of segment profit or loss to be reported under certain conditions, and requires disclosure of the title and position of the CODM. ASU 2023-07 is effective for fiscal years beginning after December 15, 2023, and interim periods within fiscal years beginning after December 15, 2024. Early adoption is permitted. The Company will adopt ASU 2023-07 for the year beginning January 1, 2024 and does not believe there is material impact to the consolidated financial statements other than disclosures for single reportable segment entities required above.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures. The amendments in ASU 2023-09 address investor requests for more transparency about income tax information through improvements to income tax disclosures primarily related to the rate reconciliation and income taxes paid information. This ASU also includes certain other amendments to improve the effectiveness of income tax disclosures. For public business entities, the amendments in this ASU are effective for annual periods beginning after December 15, 2024. Early adoption is permitted for annual financial statements that have not yet been issued or made available for issuance. The amendments in this ASU should be applied on a prospective basis. Retrospective application is permitted. The Company plans to adopt ASU 2023-09 for the year beginning January 1, 2025 is currently evaluating the impact of this new guidance on its consolidated financial statements.
F-21
There are no other recently issued accounting pronouncements that are expected to have a material effect on the Company’s financial position, results of operations or cash flows.
3. INVESTMENT IN EQUITY SECURITIES, AT FAIR VALUE AND LONG-TERM INVESTMENTS
Investment in equity securities, at fair value
MaxCyte Inc.
The Company had an equity investment in the common stock of MaxCyte, a publicly traded company. The fair value of this security was measured using its quoted market price, a Level 1 input, and was $
BioInvent International AB – ordinary shares
In October 2020, in conjunction with its license agreement entered into with BioInvent (see Note 1), a publicly traded company, CASI made a $
The fair value of the ordinary shares was measured using its quoted market price, a Level 1 input (see Note 19). The Company recognized losses of $
The following table summarizes the Company’s investments in equity securities with readily determinable fair value as of December 31, 2023 and 2022, respectively:
Gross | |||||||||
(In thousands) | unrealized | Aggregate fair | |||||||
As of December 31, 2023 |
| Cost |
| losses |
| value | |||
BioInvent - ordinary shares | $ | | $ | ( | $ | | |||
Gross | |||||||||
(In thousands) | unrealized | Aggregate fair | |||||||
As of December 31, 2022 |
| Cost |
| losses |
| value | |||
BioInvent - ordinary shares | $ | | $ | ( | $ | | |||
F-22
Long-term investments
Long-term investments include long term investments measured at fair value or measurement alternative, and an equity method investment.
Long-term investments measured at fair value or measurement alternative
Long-term investments measured at fair value or measurement alternative as of December 31, 2023 and 2022 consisted of the following:
Gross | Foreign | ||||||||||||||
Gross | unrealized | currency | |||||||||||||
As of December 31, 2023 | unrealized | losses (including | translation | Carrying | |||||||||||
(In thousands) |
| Cost |
| gains |
| impairment) |
| adjustment |
| amount | |||||
Investments measured at fair value | |||||||||||||||
Available-for-sale debt securities: |
|
|
|
|
|
|
|
| |||||||
Alesta Therapeutics B.V. - convertible loan | $ | | $ | | $ | — | $ | — | $ | | |||||
Securities measured at fair value: | |||||||||||||||
BioInvent International AB - warrants | | — | ( | — | | ||||||||||
Investments in equity securities using measurement alternative |
|
|
|
|
|
|
|
|
| ||||||
Alesta Therapeutics B.V. - equity interests |
| |
| — |
| ( |
| — |
| | |||||
Total | $ | | $ | | $ | ( | $ | — | $ | | |||||
Gross | Foreign | ||||||||||||||
Gross | unrealized | currency | |||||||||||||
As of December 31, 2022 | unrealized | losses (including | translation | Carrying | |||||||||||
(In thousands) |
| Cost |
| gains |
| impairment) |
| adjustment |
| amount | |||||
Investments measured at fair value | |||||||||||||||
Available-for-sale debt securities: |
|
|
|
|
|
|
|
| |||||||
Alesta Therapeutics B.V. - convertible loan | $ | | $ | | $ | — | $ | — | $ | | |||||
Securities measured at fair value: | |||||||||||||||
BioInvent International AB - warrants | | — | ( | — | | ||||||||||
Cleave Therapeutics, Inc. - convertible loan | | — | ( | — | | ||||||||||
Investments in equity securities using measurement alternative |
|
|
|
|
| ||||||||||
Alesta Therapeutics B.V. - equity interests |
| |
| — |
| ( |
| — |
| | |||||
Total | $ | | $ | | $ | ( | $ | — | $ | | |||||
Juventas
In June 2019, in conjunction with its license agreement entered into with Juventas (see Note 1), the Company, through CASI Biopharmaceuticals, made an RMB
In September 2020, in conjunction with the Supplementary Agreement entered into with Juventas (see Note 1), the Company obtained additional Series A plus equity interests in Juventas with substantive liquidation preference over Juventas’ founding shareholders, resulting in the Company's equity ownership increasing to
F-23
shareholders at RMB
Since the equity interests with substantive liquidation preference is not in-substance common stock, the investment in the additional equity interests of Juventas was accounted for as an investment in equity securities at transaction date fair value with a corresponding credit to other liabilities. The profit-sharing liability represents the Company’s obligation to pay an increased share of future profits pursuant to the Supplementary Agreement (see Note 1) which was conveyed by the Company in exchange for the additional equity interests in Juventas. The Company views this as a payment from a vendor that should reduce cost of revenues over the period of royalty payments. The long-term liability will be derecognized as payments are made on a systematic and rational basis representing the pattern in which the Company expects to settle the profit-sharing payment during the commercialization period of CNCT19.
The investments are measured using the measurement alternative at its cost, minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for the identical or a similar investment of the same issuer, as the fair value of the equity securities of Juventas is not readily determinable.
In October 2021, Juventas completed its Series C financing through which it raised capital of RMB
In September 2022, CASI Biopharmaceuticals entered into an equity transfer agreement (the “Equity Transfer Agreement”) with Shenzhen Jiadao Gongcheng Equity Investment Fund (Limited Partnership) (“Jiadao Gongcheng”), a third party limited partnership enterprise incorporated under the laws of China, pursuant to which CASI Biopharmaceuticals agreed to transfer to Jiadao Gongcheng all of its equity interest in Juventas in the amount of RMB
BioInvent International AB – warrants
The fair value of the warrants of BioInvent was measured using observable market-based inputs other than quoted prices in active markets for identical assets, level 2 inputs (see Note 19). The Company uses the Black-Scholes-Merton valuation model to estimate the fair value of warrants. The fair value of the warrants was $
Alesta Therapeutics B.V. (previously Black Belt Tx Limited)
In April 2019, in conjunction with its license agreement the Company entered into with Black Belt (see Note 1), the Company made a
In July 2021, Alesta Therapeutics B.V. (“Alesta Tx”) was incorporated as the parent company holding all shares of Black Belt Tx with same ownership structure as Black Belt Tx. CASI obtained
F-24
ownership in Alesta Tx was diluted from
In July 2021, the Company entered into a
Cleave Therapeutics, Inc.
In March 2021, in conjunction with its license agreement entered into with Cleave (see Note 1), CASI made a $
In the event that Cleave, on or prior to the maturity date, completes an equity financing round of preferred stock of at least $
On July 18, 2023, the Company entered into an assignment agreement with Cleave, pursuant to which the Company obtained a repayment in the amount of USD $
Equity method investment
Investment in Precision Autoimmune Therapeutics Co., Ltd., (“PAT”)
In May 2022, CASI China entered into an agreement for the investment in PAT in the amount of RMB
The investment is accounted for under the equity method as CASI China does not control the investee but has the ability to exercise significant influence over the operating and financial policies of the investee through its board representation. The Company recognized its share of loss of the equity method investment net of income tax, in the amount of $
4. INVENTORIES
The Company’s inventories consist of finished goods of EVOMELA® and FOLOTYN®. As of December 31, 2023 and 2022, the Company had balance of inventories in the aggregate of $
F-25
5. LEASES
Right of use (“ROU”) assets and liabilities for operating leases are recognized at commencement date based on the present value of lease payments over the lease term. Rent expense is recognized on a straight-line basis over the lease term.
Operating lease liabilities are included in accrued and other current liabilities and other liabilities (noncurrent) in the consolidated balance sheets as of December 31, 2023 and 2022. As of December 31, 2023 and 2022, the Company did not have any finance leases.
In November 2019, CASI Wuxi entered into a
Rent expense for the years ended December 31, 2023, 2022 and 2021 was $
Right of use assets and liabilities as of December 31, 2023 and 2022 were classified on the consolidated balance sheets as follows:
| December 31, | December 31, | ||||
(In thousands) |
| 2023 |
| 2022 | ||
Right of use assets | $ | | $ | | ||
$ | | $ | | |||
| |
| | |||
Total lease liabilities | $ | | $ | | ||
Supplemental cash flow information related to leases was as follows:
| Year Ended December 31, | ||||||||
(In thousands) |
| 2023 |
| 2022 |
| 2020 | |||
Cash paid for amounts included in the measurement of lease liabilities: |
|
|
| ||||||
Operating cash flows | $ | | $ | | $ | | |||
Right of use assets obtained in exchange for lease obligations | $ | | $ | | $ | | |||
Increase of right of use assets from remeasurement | $ | | $ | — | $ | — | |||
All of the Company’s existing leases as of December 31, 2023 and 2022 are classified as operating leases. As of December 31, 2023 and 2022, the Company had
F-26
that the Company would have to pay to borrow, on a collateralized basis for a similar term, an amount equal to the lease payments in a similar economic environment.
A maturity analysis representing the future undiscounted cash flow of the Company’s operating leases liabilities as of December 31, 2023 is as follows:
(In thousands) |
| ||
2024 | $ | | |
2025 |
| | |
2026 |
| | |
2027 | | ||
2028 | | ||
Thereafter |
| | |
Total |
| | |
Discount factor |
| ( | |
Lease liability |
| | |
Amounts due within 12 months |
| | |
Non-current lease liability | $ | |
6. PROPERTY, PLANT AND EQUIPMENT
The Company’s property, plant and equipment (“PP&E”) mainly includes leasehold improvements, machinery and lab equipment, office equipment and furniture, and construction in progress (“CIP”).
Leasehold improvements are stated at cost and are amortized over the shorter of their useful lives or the lease term. Machinery and lab equipment are stated at cost and are depreciated over their estimated useful lives of
CASI Wuxi entered into a lease agreement of a state-owned land in 2019, and a series of contracts for construction and equipment since 2020. In December 2022, the Company returned the land use right to the Wuxi local government (Note 5). Meanwhile, all construction in progress on the land was disposed. The Company recorded a disposal loss for the cost of the initial infrastructure development.
In the third quarter of 2022, CASI Wuxi obtained the Drug Distribution License, and in the fourth quarter of 2023, CASI Wuxi obtained the Drug Manufacturing Permit for its manufacturing line in the leased building in Wuxi, which means that certain related assets are ready for their respective usage. The Company started to depreciated those assets based on their estimated useful lives.
Property, plant and equipment consist of the following:
(In thousands) | December 31, | |||||
| 2023 |
| 2022 | |||
Leasehold improvements |
| |
| | ||
Machinery and equipment | | | ||||
Office equipment and furniture | | | ||||
Construction in progress | | | ||||
Property, plant and equipment, gross |
| |
| | ||
Accumulated depreciation | ( | ( | ||||
Property, plant and equipment, net | $ | | $ | | ||
F-27
Depreciation expense were $
7. INTANGIBLE ASSETS
As of December 31, 2023, intangible assets mainly include the license right of FOLOTYN®, as well as cloud computing or office administration software. These intangible assets were originally recorded at fair values and are stated net of accumulated amortization and impairment, if any.
As mentioned in Note 1, the Company obtained the license of FOLOTYN® in 2023 under the FOLOTYN® Assignment Agreement and Payment Agreement which includes certain Deferred Payments. Considering the uncerntainties related to the renewal of the drug approval in China, as of December 31, 2023, the Company believes it is not probable that the Deferred Payments will be paid. The Company recorded the intangible asset (License of FOLOTYN®) in amount of US$
Other intangible assets are being amortized over their useful lives of
The Company used to own
In November 23, 2023, the Company applied for withdrawal of the ANDAs, and FDA approved the withdrawal. The Company derecognized the ANDAs.
F-28
Intangible assets at December 31, 2023 and 2022 consists of the following:
(In thousands) | ||||||||||||
Asset as of December 31, 2023 |
| Purchase Price |
| Accumulated Amortization |
| Impairment |
| Carrying Amount | ||||
License of FOLOTYN® | | ( | — | | ||||||||
Others | | ( | — | | ||||||||
Total | $ | | $ | ( | $ | — | $ | | ||||
(In thousands) | ||||||||||||
Asset as of December 31, 2022 |
| Purchase Price |
| Accumulated Amortization |
| Impairment |
| Carrying Amount | ||||
ANDAs | $ | | $ | ( |
| $ | ( |
| $ | | ||
Others | | ( | — | | ||||||||
Total | $ | | $ | ( |
| $ | ( |
| $ | | ||
The changes in intangible assets for the years ended December 31, 2023, 2022 and 2021 are as follows:
(In thousands) | 2023 | 2022 | 2021 | ||||||
Balance at the beginning of the year |
| $ | |
| $ | |
| $ | |
Addition |
| | |
| — | ||||
Amortization expense |
| ( | ( |
| ( | ||||
Impairment | — | ( | — | ||||||
Foreign currency translation adjustment |
| ( | ( |
| | ||||
Balance at the ending of the year | $ | | $ | | $ | | |||
Expected future amortization expense is as follows as of December 31, 2023:
(In thousands) | |||
2024 |
| $ | |
2025 |
| | |
2026 |
| | |
2027 |
| | |
2028 |
| | |
2029 and thereafter |
| |
F-29
8. ACCRUED AND OTHER CURRENT LIABILITIES, AND OTHER LIABILITIES
Year Ended December 31, | ||||||
(In thousands) |
| 2023 |
| 2022 | ||
Accrued and other current liabilities: | ||||||
Payroll and welfare payable | $ | | $ | | ||
Grants related to land use right | | | ||||
Payable to sales and marketing services |
| | | |||
Payable to professional consulting service | | | ||||
Payable to clinical study service |
| | | |||
Dividends and interest payable to Wuxi LP, current (Note 12) | | — | ||||
| | |||||
Value-added tax and other tax payable | |
| | |||
Payables related to property, plant and equipment | |
| | |||
Other | | | ||||
$ | $ | |||||
Other Liabilities | ||||||
Profit-sharing liability to Juventas (Note 3) | $ | | $ | | ||
Dividends and interest payable to Wuxi LP, non current (Note 12) | | — | ||||
| | |||||
$ | | $ | | |||
9. SHORT TERM BORROWINGS
In November 2020, Beijing Branch of China CITIC Bank Corporation Limited approved a guaranteed line of credit (“Bank Borrowings”) to the Company with maximum borrowings of RMB
In May 2022, the Company entered into a Business Loan Agreement (together with related security agreements and promissory note, the “Agreement”) with East West Bank (“EWB”). Under the Agreement, EWB made available to the Company a revolving line of credit up to a maximum of $
In June 2022, the Company’s subsidiary, CASI China entered into a Loan Agreement with China International Trust and Investment Corporation Bank (“CITIC Bank”). Under the Loan Agreement, CITIC Bank made available to CASI China a guaranteed line of credit up to a maximum of RMB
10. NOTES PAYABLE
On April 27, 2020, M&T Bank approved a $
F-30
evidenced by a promissory note to M&T Bank as lender and dated April 29, 2020, has a term of
11. GRANTS
In November 2019, CASI Wuxi entered into a
In April 2020, CASI Wuxi received RMB
The Company recognized
12. REDEEMABLE NONCONTROLLING INTEREST AND LONG TERM BORROWING
On December 26, 2018, the Company, together with Wuxi Huicheng Yuanda Investment Partnership (Limited Partnership) (“Wuxi LP”) established CASI Wuxi to build and operate a manufacturing facility in the Wuxi Huishan Economic Development Zone in Jiangsu Province, China. The Company holds
Pursuant to the investment contract between the Company and Wuxi LP and Articles of Association of CASI Wuxi, the Company has the call option to purchase the
The investment of Wuxi LP in CASI Wuxi is treated as redeemable noncontrolling interest and is classified outside of permanent equity on the consolidated balance sheets because (1) the noncontrolling interest is not mandatorily redeemable financial instruments, and (2) it is redeemable at the option of the holder, or upon the occurrence of an event that is not solely within the control of the Company. The Company initially recorded the redeemable noncontrolling interest at its fair value of $
F-31
value, assuming the noncontrolling interest is redeemable at the balance sheet date. Accretion of the carrying amount of redeemable noncontrolling interest to the redemption value is recorded in additional paid-in capital.
In December 2023, the Company entered into a series of agreements, including a capital reduction agreement, a long term borrowing agreement, and
The provision related to the Long term borrowing includes certain financial and non-financial covenants, including certain revenue threshold generated by CASI Wuxi for each of the years from 2024 to 2028. If CASI Wuxi were to breach the covenants, the remaining balance of Dividends Payable and Long term borrowing become payable on demand.
Changes in redeemable noncontrolling interest during the years ended December 31, 2023, 2022 and 2021 are as follows:
Year Ended December 31, | |||||||||
(In thousands) |
| 2023 |
| 2022 |
| 2021 | |||
Balance at beginning of year | $ | | $ | |
| $ | | ||
Share of CASI Wuxi net loss | ( | ( | ( | ||||||
Accretion of redeemable noncontrolling interest | | | | ||||||
Foreign currency translation adjustment | ( | ( | | ||||||
Redemption of Wuxi NCI | ( | — | — | ||||||
Balance at end of year | $ | — | $ | |
| $ | | ||
F-32
13. SHAREHOLDERS’ EQUITY
In March 2023, the Company completed a redomicile merger, pursuant to which CASI Delaware merged with and into CASI Cayman, with CASI Cayman surviving the merger as the surviving company and successor issuer, and CASI Cayman’s ordinary shares continued trading on the Nasdaq Capital Market under the symbol “CASI.” CASI Cayman had
Reverse Stock Split
On June 1, 2022, CASI Delaware effectuated a reverse stock split of its Common Stock (the “Reverse Stock Split”) pursuant to an amendment to its Amended and Restated Certificate of Incorporation filed on May 26, 2022. Trading of the Common Stock on a reverse stock split-adjusted basis began at the opening of trading on the Nasdaq Capital Market on June 2, 2022. After the reverse stock split, each
Stock Repurchase Program
On December 15, 2021, the board of directors of CASI Delaware approved a stock repurchase program for the repurchase of up to $
March 2021 Underwritten Public Offering
On March 24, 2021, CASI Delaware closed an underwritten public offering of
CASI Delaware is using the net proceeds of this offering for working capital and general corporate purposes, which include, but are not limited to advancing the Company’s product portfolio, acquiring the rights to new product candidates and general and administrative expenses.
Common Stock Sales Agreements
On October 29, 2021, CASI Delaware has entered into a Common Stock sales agreement (“Stock Sales Agreement”), with H.C. Wainwright & Co., LLC, relating to shares of Common Stock of the Company. In accordance with the terms of the sales agreement,
F-33
CASI Delaware may offer and sell shares of Common Stock in “at-the-market” transactions, subject to compliance with the terms and conditions of the Stock Sales Agreement, with an aggregate offering price of not more than $
Stock Purchase Warrants
In history, CASI Delaware issued shares of its common stock with accompanying warrants to certain institutional investors, accredited investors and existing shareholders.
Stock purchase warrants activity for the years ended December 31, 2023, 2022 and 2021 is as follows:
Number of | Weighted Average | ||||
| Warrants |
| Exercise Price | ||
Outstanding at December 31, 2020 |
| | $ | | |
Expired |
| ( | $ | | |
Outstanding at December 31, 2021 |
| | $ | | |
Outstanding at December 31, 2022 | $ | ||||
Expired | ( | $ | |||
Outstanding at December 31, 2023 | — | $ | — | ||
14. COSTS OF REVENUES
Costs of revenues consists primarily of the cost of inventories of EVOMELA® and sales-based royalties related to the sale of EVOMELA®. The Company is obligated to pay certain percentage of the Company’s revenue from EVOMELA® sales as royalties for a period of
15. NET LOSS PER SHARE
Net loss per share (basic and diluted) was computed by dividing net loss attributable to ordinary shareholders, considering the accretions to redemption value of the redeemable noncontrolling interest, by the weighted average number of shares of ordinary shares outstanding. As of December 31, 2023, 2022, and 2021, outstanding share options totaling
The following table sets forth the basic and diluted net loss per share computation and provides a reconciliation of the numerator and denominator for the periods presented:
| Year Ended December 31, | ||||||||
(In thousands, except share and per share data) |
| 2023 |
| 2022 |
| 2021 | |||
Numerator: |
|
| |||||||
Net loss attributable to CASI Pharmaceuticals, Inc. | $ | ( | $ | ( | $ | ( | |||
Denominator: |
|
|
| ||||||
Weighted average number of ordinary shares |
| |
| |
| | |||
Denominator for basic and diluted net loss per share calculation |
| |
| |
| | |||
Net loss per share |
|
|
| ||||||
— Basic and diluted | $ | ( | $ | ( | $ | ( | |||
16. EMPLOYEE BENEFIT PLAN
The Company sponsors the CASI Pharmaceuticals, Inc. 401(k) Plan and Trust. The plan covers substantially all U.S. employees and enables participants to contribute a portion of salary and wages on a tax-deferred basis. Contributions to the plan by the Company
F-34
are discretionary. Contributions by the Company totaled $
Full time employees of the Company in the PRC participate in a government mandated defined contribution plan, pursuant to which certain pension benefits, medical care, employee housing fund and other welfare benefits are provided to employees. Chinese labor regulations require that the PRC subsidiaries of the Company make contributions to the government for these benefits based on certain percentages of the employees’ salaries. The Company has no legal obligation for the benefits beyond the contributions made. The total amounts for such employee benefits, which were expensed as incurred, were $
17. SHARE-BASED COMPENSATION
In March 2023, the Company completed a redomicile merger, pursuant to which CASI Delaware merged with and into CASI Cayman, with CASI Cayman surviving the merger as the surviving company and successor issuer. As a result, CASI Cayman succeeded CASI Delaware’s share compensation plans. CASI Delaware has adopted various share compensation plans for executive, management and staff of the Company, as well as outside directors and consultants.
On June 15, 2021, the 2021 Long-Term Incentive Plan (the “2021 Plan”) was approved by CASI Delaware’s shareholders. The maximum number of shares of common stock that are available for grants and awards equals to
In addition, CASI Delaware also granted share options to Dr. He, its Chairman and CEO. On June 15, 2021, the Board approved a grant of share options to Dr. He which consists of
On May 12, 2023, the board of directors of the Company adopted certain restated and amended 2011 Long-term Incentive Plan and restated and amended 2021 Long term Incentive Plan, pursuant to which the board of directors of the Company authorized certain downward adjustments to the exercise prices of
The share-based compensation expenses are recorded as components of general and administrative expense, selling and marketing expense, and research and development expense, as follows:
Year Ended December 31, | ||||||||||||||
(In thousands) |
| 2023 |
| 2022 |
| 2021 | ||||||||
Research and development | $ | | $ | 624 | $ | 361 | ||||||||
Sales and marketing | | 174 | 449 | |||||||||||
General and administrative |
| |
| 6,212 |
| 6,960 | ||||||||
Total | $ | | $ | 7,010 | $ | 7,770 | ||||||||
Compensation expense related to share options with service conditions is recognized over the requisite service period, which is generally the option vesting term of up to
The Company uses the Black-Scholes-Merton valuation model to estimate the fair value of service based and performance-based share options granted to employees. Such model requires the input of highly subjective assumptions, and changes in the assumptions used can materially affect the grant date fair value of an award.
F-35
Expected Volatility—Volatility is a measure of the amount by which a financial variable such as a share price has fluctuated (historical volatility) or is expected to fluctuate (expected volatility) during a period. The Company uses the historical volatility based on the daily price observations of its ordinary share during the period immediately preceding the share-based award grant that is equal in length to the award’s expected term. The Company believes that historical volatility represents the best estimate of future long term volatility.
Risk-Free Interest Rate—This is the average interest rate consistent with the yield available on a U.S. Treasury note (with a term equal to the expected term of the underlying grants) at the date the option was granted.
Expected Term of Options—This is the period of time that the options granted are expected to remain outstanding. The Company uses a simplified method for estimating the expected term of service based awards granted. For performance based awards, the expected term is based on the derived service period.
Expected Dividend Yield—The Company has never declared or paid dividends on its ordinary share and does not anticipate paying any dividends in the foreseeable future. As such, the dividend yield percentage is assumed to be .
The weighted average grant date fair value is $
Year Ended December 31, | ||||||||||||
| 2023 |
| 2022 |
| 2021 |
| ||||||
Range of expected volatility |
| % | % | % | ||||||||
Range of risk free interest rate | % | % | % | |||||||||
Weighted average expected option life (in years) |
| |||||||||||
Expected dividend yield |
| | % | | % | | % | |||||
In terms of the Modification in 2023, the weighted average modification date fair value is $
F-36
A summary of the Company’s share option plans and changes in options outstanding under the plans during the years ended December 31, 2023, 2022 and 2021 is as follows:
Weighted Average | Weighted Average Remaining | |||||||||
| Number of Options |
| Exercise Price |
| Contractual Term In Years |
| Aggregate Intrinsic Value | |||
Outstanding at December 31, 2020 | $ | | ||||||||
Exercised |
| — | $ | — | ||||||
Granted | | $ | | |||||||
Expired |
| ( | $ | | ||||||
Forfeited | ( | $ | | |||||||
Outstanding at December 31, 2021 | $ | | ||||||||
Exercised | — | $ | — | |||||||
Granted | $ | | ||||||||
Expired | ( | $ | | |||||||
Forfeited | ( | $ | | |||||||
Outstanding at December 31, 2022 | $ | | ||||||||
Exercised | ( | $ | | |||||||
Granted | | $ | | |||||||
Expired | ( | $ | | |||||||
Forfeited | ( | $ | | |||||||
Outstanding at December 31, 2023 | $ | | | |||||||
Vested and expected to vest at December 31, 2023 | $ | | | |||||||
Exercisable at December 31, 2023 | $ | | | |||||||
The aggregate intrinsic value is calculated as the difference between (i) the closing price of the ordinary share at December 31, 2023 and (ii) the exercise price of the underlying awards, multiplied by the number of options that had an exercise price less than the closing price on the last trading day of the year. Cash received from option exercises under all share-based payment arrangements for the years ended December 31, 2023, 2022 and 2021 was $
The following summarizes information about share options that are outstanding at December 31, 2023:
| Options Outstanding |
| Options Exercisable | |||||||||
| Weighted | |||||||||||
| Average |
| Weighted |
| Weighted | |||||||
| Number |
| Remaining |
| Average |
| Number |
| Average | |||
Range of |
| Outstanding at |
| Contractual |
| Exercise |
| Exercisable at |
| Exercise | ||
Exercise Prices |
| December 31, 2023 |
| Life in Years |
| Price |
| December 31, 2023 |
| Price | ||
$ | |
| $ | | $ | |||||||
$ | |
| $ | | $ | |||||||
$ | | $ | | $ | ||||||||
$ | |
| $ | | $ | |||||||
|
| $ | | $ | ||||||||
As of December 31, 2023, there was $
F-37
18. INCOME TAXES
The Company is subject to U.S. federal tax rate of
For financial reporting purposes, loss before income taxes includes the following components:
(In thousands) |
| 2023 |
| 2022 |
| 2021 | |||
Domestic - United States | $ | | $ | ( | $ | ( | |||
Foreign - mainland China and Hong Kong |
| ( |
| ( |
| ( | |||
Total | $ | ( | $ | ( | $ | ( | |||
Significant components of the Company’s deferred income tax assets and liabilities as of December 31, 2023 and 2022 are as follows:
December 31, | ||||||
(In thousands) |
| 2023 |
| 2022 | ||
Deferred income tax assets: |
|
|
|
| ||
Net operating loss carryforwards | $ | | $ | | ||
Research and development credit carryforwards |
| |
| | ||
Intangible assets |
| |
| | ||
Share-based compensation |
| |
| | ||
Change in fair value of investments | | | ||||
Capitalized R&D amortization | | | ||||
Lease liability | | | ||||
Impairment loss of long-term investments | | | ||||
Others |
| |
| | ||
Valuation allowance for deferred income tax assets |
| ( |
| ( | ||
$ | | $ | | |||
Deferred income tax liabilities: | ||||||
Deferred Royalty Income | — | ( | ||||
Right of use asset | ( | ( | ||||
Others | ( | ( | ||||
$ | ( | $ | ( | |||
The Company has U.S. federal and state net operating loss (NOL) carryforwards of $
Under the provisions of the US Internal Revenue Code, the NOL and tax credit carryforwards are subject to review and possible adjustment by the Internal Revenue Service and state tax authorities. NOL and tax credit carryforwards may become subject to an annual limitation in the event of certain cumulative changes in the ownership interest of significant shareholders over a three-year period in excess of 50%, as defined under Sections 382 and 383 of the Internal Revenue Code of 1986, respectively, as well as similar state tax provisions. This could limit the amount of tax attributes that the Company can utilize annually to offset future taxable income or tax liabilities. The amount of the annual limitation, if any, will be determined based on the value of the Company immediately prior to the ownership change. Subsequent ownership changes may further affect the limitation in future years. For financial reporting purposes, a
F-38
A reconciliation of the provision for income taxes to the federal statutory rate is as follows:
(In thousands) |
| 2023 |
| 2022 |
| 2021 | |||
Tax benefit at statutory rate of | $ | ( | $ | ( | $ | ( | |||
Attribute expiration |
| |
| |
| | |||
Tax rate differential | ( | ( | ( | ||||||
Change in applicable tax rates (1) |
| — |
| — |
| | |||
Nondeductible expenses | | | | ||||||
Research and development bonus deduction | ( | — | — | ||||||
Tax shortfalls from share options | | — | — | ||||||
Others |
| ( |
| ( |
| | |||
Change in valuation allowance (2) |
| ( |
| |
| ( | |||
$ | ( | $ | | $ | | ||||
Note (1) Change in applicable tax rates reflects the reduction of the Company’s deferred tax assets related to the change in the Company’s activities in and the tax laws of certain states.
Note (2) Change in valuation allowance is as follows:
(In thousands) |
| 2023 |
| 2022 |
| 2021 | |||
Balance at January 1 | $ | | $ | | $ | | |||
Additions (Reversals) | | | ( | ||||||
Decrease from attribute expiration | ( | ( | ( | ||||||
Foreign currency translation adjustment | ( | — | — | ||||||
Balance at December 31 | $ | | $ | | $ | | |||
A reconciliation of the beginning and ending amount of gross unrecognized tax benefits is as follows:
(In thousands) |
| 2023 |
| 2022 |
| 2021 | |||
Unrecognized tax benefits balance at January 1 | $ | | $ | | $ | | |||
Reductions for tax positions of prior periods |
| ( |
| ( |
| ( | |||
Additions for tax positions of current period |
|
|
| | |||||
Unrecognized tax benefits balance at December 31 | $ | | $ | | $ | ||||
The Company had $
The Company and each of its PRC subsidiaries file income tax returns in the United States and the PRC, respectively. Due to the existence of tax attribute carryforwards (which are currently offset by a full valuation allowance), all of the Company’s tax returns since 1999 are open to examination by the taxing authorities. According to the PRC Tax Administration and Collection Law, the statute of limitations is three years if the underpayment of taxes is due to computational errors made by the taxpayer or the withholding agent. The statute of limitations is extended to five years under special circumstances where the underpayment of taxes is more than RMB
F-39
19. FAIR VALUE MEASUREMENTS
Financial instruments of the Company primarily consist of cash and cash equivalents, investment in equity securities, short-term investments, accounts receivable, receivable from a related party, term deposit, prepaid expenses and other, long-term investments, accounts payable, accrued and other current liabilities, and long term borrowing. As of December 31, 2023 and 2022, the carrying amount of cash and cash equivalents, accounts receivable, receivable from a related party, prepaid expenses and other, accounts payable and accrued and other current liabilities are carried at cost which approximates their fair values due to the short-term nature of the instruments, the carrying amount of long term borrowing approximates its fair value as interest rate is comparable to the prevailing interest rate in the market.
Financial Assets and Liabilities Measured at Fair Value on a Recurring Basis
The Company evaluates financial assets and liabilities subject to fair value measurements on a recurring basis to determine the appropriate level at which to classify them each reporting period. This determination requires the Company to make subjective judgments as to the significance of inputs used in determining fair value and where such inputs lie within the hierarchy.
The Company has an equity investment in the common stock of a publicly traded company. The Company’s investments in these equity securities are carried at their estimated fair value, with changes in fair value reported in the consolidated statement of operations and comprehensive loss each reporting period (see Note 3). The fair value of the common stock is based on quoted market price for the investees’ common stock, a Level 1 input.
The Company has an equity investment in the warrants of a publicly traded company. The Company’s investment is carried at its estimated fair value, with changes in fair value reported in the consolidated statement of operations and comprehensive loss each reporting period (see Note 3). The fair value of the warrants was measured using observable market-based inputs other than quoted prices in active markets for identical assets, level 2 inputs. The Company uses the Black-Scholes-Merton valuation model to estimate the fair value of warrants. Option valuation models, including Black-Scholes-Merton, require the input of highly subjective assumptions, and changes in the assumptions used can materially affect the fair value determination of a warrant.
The Company has investments in the convertible debt of a private company. The Company’s investment is carried at its estimated fair value, with changes in fair value reported in the consolidated statement of operations and comprehensive loss each reporting period (see Note 3) using Level 3 input.
The following tables present the Company’s financial assets accounted for at fair value on a recurring basis as of December 31, 2023 and 2022, by level within the fair value hierarchy:
(In thousands) | Fair Value at | |||||||||||
Description |
| December 31, 2023 |
| Level 1 |
| Level 2 |
| Level 3 | ||||
Investment in equity securities, at fair value: | $ | | $ | | $ | — | $ | — | ||||
Short term investments: | $ | | $ | — | $ | | $ | — | ||||
Long term investments: | ||||||||||||
Investment in warrants - Designated as investment measured at FVTPL | $ | | $ | — | $ | | $ | — | ||||
Investment in convertible loan - AFS | $ | | $ | — | $ | — | $ | | ||||
F-40
(In thousands) |
| Fair Value at | ||||||||||
Description |
| December 31, 2022 |
| Level 1 |
| Level 2 |
| Level 3 | ||||
Investment in equity securities, at fair value: | $ | | $ | | $ | | $ | | ||||
Short term investments: | $ | | $ | | $ | | $ | | ||||
Term deposit, non current: | $ | | $ | | $ | | $ | | ||||
Long term investments: | ||||||||||||
Investment in warrants - Designated as investment measured at FVTPL | $ | | $ | | $ | | $ | |||||
Investment in convertible loan - AFS | $ | | $ | | $ | | $ | | ||||
Investment in convertible loan - Designated as investment measured at FVTPL | $ | | $ | | $ | | $ | | ||||
Financial Assets and Liabilities Measured at Fair Value on a Non-Recurring Basis
The Company reduces the carrying amount of equity method investments to its fair value when an impairment is determined to be other-than-temporary. In 2023, due to PAT’s slow business progress and delay of financing, the Company performed an impairment test using discounted cash flow approach. As the projected cash flow of the investment is negative, the Group recognized full impairment for the investment.
The Company measures equity investments without readily determinable fair values at its cost, minus impairment, if any, plus or minus changes resulting from observable transactions of identical or similar securities of the same issuer.
On October 23, 2021, the Company remeasured the investments in equity securities in Juventas to the fair value. The Company estimated the fair value of these securities based on the transaction price of similar securities issued by the investee. In the fourth quarter of 2022, the Company completed the sales of the equity interests of Juventas (see Note 3).
Quantitative Information about Level 3 Fair Value Measurements | |||||||||
Fair Value at | |||||||||
| October 23, 2021 |
|
|
| |||||
Description |
| (remeasurement date) |
| Valuation Techniques |
| Unobservable Input |
| Average/Median | |
Investment in equity securities using measurement alternative | $ | |
| Market approach |
| Expected volatility |
| ||
On June 30, 2021, the Company remeasured the investment in equity securities in Alesta to the fair value of $
Non-Financial Assets and Liabilities Measured at Fair Value on a Recurring Basis
The Company has
Non-Financial Assets and Liabilities Measured at Fair Value on a Non-Recurring Basis
On December 31, 2022, the intangible assets of six ANDAs with a total carrying amount of $
20. RELATED PARTY TRANSACTIONS
PAT. In May 2022, the Company entered into a Sublicense Agreement (the “Sublicense Agreement”) with PAT, a company established under the laws of China, pursuant to which the Company granted PAT an exclusive (subject to the commercialization and co-marketing rights), perpetual, worldwide license, with the right to freely grant further sublicenses subject to terms and conditions in
F-41
the Sublicense Agreement, for the investigational anti-CD38 monoclonal antibody TSK011010 licensed and controlled by the Company from Black Belt Therapeutics Limited, in the treatment, prevention and diagnosis of autoimmune diseases, conditions and disorders in humans. Pursuant to the Sublicense Agreement, PAT shall make an upfront payment of $
In the fourth quarter of 2023, the two parties reached an alignment that PAT should reimburse the Company for an amount of US$
Juventas. On July 1, 2019 the Company entered into a
March 2021 Underwritten Public Offering Transactions. On March 24, 2021, the Company closed an underwritten public offering of
ETP BioHealth III Fund LP (“ETP BioHealth”), in which CASI's Chairman and CEO is the founder and managing partner of ETP BioHealth’s general partner (Emerging Technology Partners, LLC (“ETP”)), purchased shares of common stock in the Offering at the public offering price and on the same terms as the other purchasers in the Offering. ETP BioHealth purchased
21. COMMITMENTS AND CONTINGENCIES
In conjunction with the Cleave assignment agreement entered into on July 18, 2023 (see Note 1), the Company is responsible for certain milestone and royalty payments. As of December 31, 2023,
In conjunction with the BioInvent agreement entered into during 2020 (see Note 1), the Company is responsible for certain milestone and royalty payments. As of December 31, 2023,
In conjunction with the Black Belt agreement entered into during 2019 (see Note 1), the Company is responsible for certain milestone and royalty payments. In June 2021, the Company achieved the First-Patient-In (FPI) in the Phase 1 dose escalation and expansion study of CID-103, and made $
In conjunction with the Pharmathen agreement entered into during 2019 (see Note 1), the Company is responsible for
In conjunction with the FOLOTYN® Assignment Agreement and Payment Agreement entered into in July 2023 (see Note 1), in addition to the $
The Company is subject in the normal course of business to various legal proceedings in which claims for monetary or other damages may be asserted. Management does not believe such legal proceedings, unless otherwise disclosed herein, are material.
F-42
22. RESTRICTED NET ASSETS
Relevant PRC law and regulations permit payment of dividends by PRC-based operating entities only out of their retained earnings, if any, as determined in accordance with PRC accounting standards and regulations. In addition, a PRC-based operating entity is required to annually appropriate 10% of net after-tax income to the statutory surplus reserve fund prior to payment of any dividends, unless such reserve funds have reached 50% of the entity’s registered capital. As a result of these and other restrictions under PRC law and regulations, PRC-based operating entities are restricted in their ability to transfer a portion of their net assets to the Company either in the form of dividends, loans or advances. The Company may in the future require additional cash resources from PRC-based operating entities due to changes in business conditions, to fund future acquisitions and development, or to declare and pay dividends to or distribution to its shareholders. As of December 31, 2023, the Group had restricted net assets in the amount of $
23. SUBSEQUENT EVENTS
On March 2, 2024, CASI received a notice from Juventas, which purported to terminate the CNCT19 Agreements. The Company is currently involved in arbitration proceedings against Juventas. See “Note 1 – License and Distribution Agreements” for further information.
F-43
24. CONDENSED FINANCIAL INFORMATION OF REGISTRANT
a) Condensed Balance Sheets (In thousands)
| December 31, 2023 |
| December 31, 2022 | |||
ASSETS |
|
|
|
| ||
Current assets: |
|
|
|
| ||
Cash and cash equivalents | $ | | $ | | ||
Investment in equity securities, at fair value |
| |
| | ||
Accounts receivable, net |
| |
| | ||
| — | |||||
Inventories |
| |
| | ||
Prepaid expenses and other |
| |
| | ||
Total current assets |
| |
| | ||
Intercompany receivables |
| |
| | ||
Property, plant and equipment, net |
| |
| | ||
Intangible assets, net |
| |
| | ||
Long-term investments |
| |
| | ||
Investment in subsidiaries |
| |
| | ||
Total assets | $ | | $ | | ||
LIABILITIES AND SHAREHOLDERS’ EQUITY |
|
|
|
| ||
Current liabilities: |
|
|
|
| ||
Accounts payable | $ | | $ | | ||
Accrued and other current liabilities |
| |
| | ||
Intercompany payables |
| |
| | ||
Total current liabilities |
| |
| | ||
Other liabilities |
| |
| | ||
Total liabilities |
| |
| | ||
Shareholders’ equity: |
|
|
|
| ||
Ordinary shares |
| |
| | ||
Additional paid-in capital |
| |
| | ||
Treasury shares |
| ( |
| ( | ||
Accumulated other comprehensive loss | ( | ( | ||||
Accumulated deficit |
| ( |
| ( | ||
Total shareholders’ equity |
| |
| | ||
Total liabilities and shareholders' equity | $ | | $ | | ||
F-44
b) Condensed Statements of Operations and Comprehensive Loss (In thousands)
| Years ended December 31, | ||||||||
| 2023 |
| 2022 |
| 2021 | ||||
Total revenues |
| |
| |
| | |||
Total costs of revenues |
| |
| |
| | |||
Gross Profit |
| |
| |
| | |||
Operating expenses: |
|
|
|
|
|
| |||
Intercompany expenses |
| |
| |
| | |||
Other operating expenses | | | | ||||||
Total operating expenses |
| |
| |
| | |||
Loss from operations |
| ( |
| ( |
| ( | |||
Non-operating income (expense): |
|
|
|
|
|
| |||
Share of loss of subsidiaries |
| ( |
| ( |
| ( | |||
Other non-operating income (expenses) | | ( | | ||||||
Loss before income tax expense |
| ( |
| ( |
| ( | |||
Income tax expense | — | — | — | ||||||
Net loss |
| ( |
| ( |
| ( | |||
F-45
c) Condensed Statements of Cash Flows (In thousands)
| Years Ended December 31, | |||||||
| 2023 |
| 2022 |
| 2021 | |||
CASH FLOWS FROM OPERATING ACTIVITIES |
|
|
|
|
|
| ||
Net cash provided by (used in) operating activities | $ | |
| $ | ( |
| $ | ( |
CASH FLOWS FROM INVESTING ACTIVITIES |
|
|
|
|
|
| ||
Proceeds from sales of equity securities in MaxCyte and BioInvent |
| — |
| |
| — | ||
Proceeds from disposal of property and equipment |
| — |
| — |
| | ||
Proceeds from extinguishment of investment in a convertible loan | | — | — | |||||
Purchases of property, plant and equipment |
| ( |
| — |
| — | ||
Purchases of intangible asset | ( | — | — | |||||
Cash paid to acquire in-process research and development |
| — |
| — |
| ( | ||
Cash paid to acquire convertible loan in Black Belt Tx Limited |
| — |
| — |
| ( | ||
Receipt of repayment of Black Belt Tx convertible note |
| — |
| — |
| | ||
Cash paid to acquire convertible loan in Alesta Tx |
| — |
| — |
| ( | ||
Cash paid to acquire convertible loan in Cleave |
| — |
| — |
| ( | ||
Investment in subsidiaries |
| ( |
| — |
| ( | ||
Net cash provided by (used in) investing activities |
| ( |
| |
| ( | ||
CASH FLOWS FROM FINANCING ACTIVITIES |
|
|
|
|
|
| ||
Proceeds from bank borrowings |
| — |
| |
| — | ||
Repayment of bank borrowings |
| — |
| ( |
| — | ||
Repurchase of ordinary share |
| ( |
| ( |
| — | ||
Stock issuance costs |
| — |
| — |
| ( | ||
Proceeds from sale of ordinary share |
| — |
| — |
| | ||
Proceeds from exercise of share options |
| |
| — |
| — | ||
Net cash provided by (used in) provided by financing activities |
| ( |
| ( |
| | ||
Net increase (decrease) in cash and cash equivalents |
| |
| ( |
| ( | ||
Cash and cash equivalents at beginning of year |
| |
| |
| | ||
Cash and cash equivalents at end of year | $ | | $ | | $ | | ||
Note: For the presentation of the condensed financial information, the Company records its investment in subsidiaries under the equity method of accounting as prescribed in ASC 323, “Investments-Equity Method and Joint Ventures”. Such investments are presented on the condensed balance sheets as “Investment in subsidiaries” and the subsidiaries income (loss) as “Share of income (loss) of subsidiaries” on the condensed statements of operations and comprehensive loss. The condensed financial information should be read in conjunction with the Company’s consolidated financial statements. As of December 31, 2023 and 2022, there were no material contingencies, significant provisions of long-term obligations, mandatory dividend or redemption requirements of redeemable shares or guarantees of the Company, except for those, which have been separately disclosed in the consolidated financial statements.
F-46