UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission file number:
(Exact name of Registrant as specified in its charter)
The
(Jurisdiction of incorporation or organization)
+49 7071 9883 0
(Address of principal executive offices)
Chief Executive Officer
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
Title of each class |
| Trading Symbol |
| Name of each exchange on which registered |
“ | The |
Securities registered or to be registered pursuant to Section 12(g) of the Act:
None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act:
None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
The number of outstanding shares as of December 31, 2023 was:
Title of Class |
| Number of Shares Outstanding |
Common shares, par value €0.12 per share |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
Yes ☐
Note – Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 from their obligations under those Sections.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
Accelerated Filer ☐ | Non-accelerated Filer ☐ | Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ☐
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
☐ | U.S. GAAP |
☒ |
☐ | Other |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes
TABLE OF CONTENTS
Page | ||
4 | ||
4 | ||
4 | ||
4 | ||
71 | ||
153 | ||
154 | ||
175 | ||
189 | ||
197 | ||
199 | ||
199 | ||
217 | ||
218 | ||
219 | ||
219 | ||
Material Modifications to the Rights of Security Holders and Use of Proceeds | 219 | |
219 | ||
221 | ||
221 | ||
221 | ||
221 | ||
222 | ||
Purchases of Equity Securities by the Issuer and Affiliated Purchasers. | 222 | |
222 | ||
222 | ||
235 | ||
235 | ||
236 | ||
236 | ||
236 | ||
238 | ||
| 1 |
i
PRESENTATION OF FINANCIAL AND OTHER INFORMATION
We report under International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or the IASB. We have made rounding adjustments to some of the figures included in this Annual Report. Accordingly, numerical figures shown as totals in some tables may not be an arithmetic aggregation of the figures that preceded them.
Our financial statements included in this Annual Report are presented in euro and, unless otherwise specified, all monetary amounts are in euro. All references in this Annual Report to “$,” “U.S. dollars” and “dollars” mean U.S. dollars and all references to “€” and “euro” mean euro, unless otherwise noted.
In this Annual Report, unless otherwise indicated, some euro amounts have been translated into U.S. dollars at the rate of $1.105 to €1.00, the official exchange rate quoted as of December 31, 2023 by the European Central Bank.
CERTAIN REFERENCES
Unless otherwise indicated or the context otherwise requires, all references in this Annual Report to “CureVac” or the “Company,” “we,” “our,” “ours,” “ourselves,” “us” or similar terms refer to: (1) on or following the consummation of the Corporate Reorganization, CureVac N.V. together with its subsidiaries, including CureVac SE (formerly known as CureVac AG), and (2) prior to the consummation of the Corporate Reorganization, CureVac AG.
We are a holding company and our sole asset is the capital stock of our wholly owned subsidiaries, including CureVac SE. CureVac N.V. operates and controls all of the business and affairs and consolidates the financial results of CureVac SE. We are incorporated in the Netherlands, and a majority of our outstanding securities are owned by non-U.S. residents. Under the rules of the U.S. Securities and Exchange Commission, or the SEC, we are currently eligible for treatment as a “foreign private issuer.” As a foreign private issuer, we are not required to file periodic reports and financial statements with the SEC as frequently or as promptly as domestic registrants whose securities are registered under the Securities Exchange Act of 1934, as amended, or the Exchange Act.
We own or have rights to various trademarks and trade names, including CureVac® and the CureVac logo, that we use in connection with the operation of our business. This Annual Report may also contain trademarks, service marks and trade names of third parties, which are the property of their respective owners. We do not intend our use or display of other entities’ trademarks, trade names or service marks to imply a relationship with, or endorsement or sponsorship of us by, any other entity. Solely for convenience, the trademarks, trade names and service marks in this Annual Report are referred to without the symbols ® and ™, or SM, but the omission of such references should not be construed as any indication that we will not assert, to the fullest extent under applicable law, our rights or the right of the applicable owner of these trademarks, service marks and trade names.
1
FORWARD-LOOKING STATEMENTS
This Annual Report contains statements that constitute forward-looking statements. Many of the forward-looking statements contained in this Annual Report can be identified by the use of forward-looking words such as “anticipate,” “believe,” “could,” “expect,” “should,” “plan,” “intend,” “estimate” and “potential,” or other similar expressions.
Forward-looking statements appear in a number of places in this Annual Report and include, but are not limited to, statements regarding our intent, belief or current expectations. Forward-looking statements are based on our management’s beliefs and assumptions and on information currently available to our management. Such statements are subject to risks and uncertainties, and actual results may differ materially from those expressed or implied in the forward-looking statements due to various factors, including, but not limited to, those identified under the section entitled “Risk Factors” in this Annual Report. These risks and uncertainties include factors relating to:
| ● | our ability to obtain funding for our operations necessary to complete further development and commercialization of our product candidates; |
| ● | the initiation, timing, progress, results, and cost of our research and development programs and our current and future preclinical studies and clinical trials, including statements regarding the timing of initiation and completion of studies or trials and related preparatory work, the period during which the results of the trials will become available and our research and development programs; |
| ● | the timing of and our ability to obtain and maintain regulatory approval for our product candidates; |
| ● | the ability and willingness of our third-party collaborators to continue research and development activities relating to our product candidates and cost associated with the cancellation of manufacture and supply agreements in the event of termination of our research and development programs; |
| ● | the exercise by the Bill & Melinda Gates Foundation of withdrawal rights; |
| ● | our and our collaborators’ ability to obtain, maintain, defend and enforce our intellectual property protection for our proprietary and collaborative product candidates, and the scope of such protection; |
| ● | the rate and degree of market acceptance of our products; |
| ● | our ability to commercialize our product candidates, if approved; |
| ● | our ability and the potential to successfully manufacture our drug substances and delivery vehicles for preclinical use, for clinical trials and on a larger scale for commercial use, if approved; |
| ● | general economic, political, demographic and business conditions in the United States and Europe; |
| ● | the impact of unstable market and economic conditions such as rising inflation and interest rates and the conflict involving Russia and Ukraine on our business; |
| ● | our ability to implement our growth strategy; |
| ● | our ability to compete and conduct our business in the future; |
| ● | our ability to enroll patients for our clinical trials; |
| ● | the availability of qualified personnel and the ability to retain such personnel; |
| ● | regulatory developments and changes in the United States, Europe and countries outside of Europe including tax matters; |
| ● | our ability to overcome the challenges posed by pandemics (such as COVID-19), to the conduct of our business; |
2
| ● | our ability to implement, maintain and improve effective internal controls; |
| ● | our estimates of our expenses, future revenue and capital requirements and our needs for or ability to obtain additional financing; |
| ● | other factors that may affect our financial condition, liquidity and results of operations; and |
| ● | other risk factors discussed under “Item 3. Key Information — D. Risk Factors.” |
You should read this Annual Report carefully with the understanding that our actual future results may be materially different from and worse than what we expect. If our forward-looking statements prove to be inaccurate, the inaccuracy may be material. Other sections of this Annual Report include additional factors which could adversely impact our business and financial performance. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. Moreover, we operate in an evolving environment. Thus, new risk factors and uncertainties emerge from time to time and it is not possible for our management to predict all risk factors and uncertainties, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. We qualify all of our forward-looking statements by these cautionary statements.
Forward-looking statements speak only as of the date they are made, and we do not undertake any obligation to update them in light of new information or future developments or to release publicly any revisions to these statements in order to reflect later events or circumstances or to reflect the occurrence of unanticipated events or otherwise, except as required by law.
3
PART I
ITEM 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS
Not applicable.
ITEM 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not applicable.
ITEM 3. KEY INFORMATION
Not applicable.
Not applicable.
Summary of Risk Factors
The following is a summary of the risk factors our business faces. The list below is not exhaustive, and investors should read this “Risk Factors” section in full. Some of the risks we face include:
| ● | Interim, “top-line,” and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data. |
| ● | We may face residual business disruption and related risks resulting from inflationary pressures, supply chain issues, labor shortages and increases in commodity prices (including as result of the war in Ukraine) and the lingering effects of the COVID-19 pandemic, which could have a material adverse effect on our business plan or clinical trials. |
| ● | Cyberattacks or other failures in our or our third-party vendors’, contractors’ or consultants’ telecommunications or information technology systems could result in information theft, data corruption and significant disruption of our business operations. |
| ● | If we are unable to obtain, maintain and enforce intellectual property protection for our products or product candidates, or if the scope of our intellectual property protection is not sufficiently broad, our ability to commercialize our product candidates successfully and to compete effectively may be materially adversely affected. |
| ● | We depend on strategic partnerships with other companies to assist in the research, development and commercialization of our platform and product candidates. If our existing or future partners do not perform as expected, if we fail to maintain any of these collaborations or if these collaborations are not successful, our ability to commercialize our product candidates successfully and to generate revenues through technology licensing or otherwise may be materially adversely affected. |
| ● | Clinical drug development involves a lengthy and expensive process with uncertain timelines and uncertain outcomes, and results of earlier studies and trials may not be predictive of future trial results. If clinical trials of our product candidates or production of our product candidates are prolonged or delayed, we may be unable to obtain required regulatory approvals, and therefore be unable to commercialize our product candidates on a timely basis or at all. |
4
| ● | Our proprietary product candidates are still in preclinical or clinical development. We cannot give any assurance that any of our product candidates will receive regulatory approval, and if we are unable to obtain regulatory approval and ultimately commercialize our product candidates or experience significant delays in doing so, our business will be materially harmed. |
| ● | We have no history of commercializing pharmaceutical products, which may make it difficult to evaluate the prospects for our future viability. |
| ● | Our product candidates may cause undesirable side effects that could delay or prevent their regulatory approval, limit the commercial profile of an approved label or result in significant negative consequences following regulatory approval, if any. Significant adverse events may occur during our clinical trials or even after receiving regulatory approval, which could delay or terminate clinical trials, delay or prevent regulatory approval or market acceptance of any of our product candidates. |
| ● | To date, a limited number of products that utilize mRNA as a prophylactic vaccine against COVID-19 have been approved in the United States, Europe and other countries, subject to certain limitations. In addition, no product that utilizes mRNA as a therapeutic vaccine has been approved in the United States or Europe. As such, mRNA drug development has substantial clinical development and regulatory risks due to the novel nature of this new category of medicines. |
| ● | The regulatory approval processes of the FDA and comparable authorities are lengthy, time consuming and inherently unpredictable and if we fail to obtain regulatory approval in any jurisdiction, we will not be able to commercialize our products in that jurisdiction and our business, results of operations, financial condition and prospects, may be materially adversely affected. |
| ● | A potential breakthrough therapy designation by the FDA for a product candidate may not lead to a faster development or regulatory review or approval process, and it would not increase the likelihood that the product candidate will receive marketing approval. |
| ● | The manufacture of mRNA-based medicines is complex and manufacturers often encounter difficulties in production, especially in the field of biologics. If we or any of our third-party manufacturers encounter difficulties, our ability to provide product candidates for clinical trials or products, if approved, to patients or future customers could be delayed or halted. |
| ● | Undetected errors or defects in our production could harm our reputation or expose us to product liability claims. |
| ● | We rely on third parties to conduct our nonclinical and clinical trials and perform other tasks for us. If these third parties do not successfully carry out their contractual duties, meet expected deadlines, or comply with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed. |
| ● | If we or any third-party manufacturer of our product candidates is unable to increase the scale of production of our product candidates, and/or increase the product yield of manufacturing, then our costs to manufacture the product may increase and commercialization may be delayed. |
| ● | At the start of the COVID-19 pandemic, we entered into contractual engagements with third-party manufacturers to have access to additional capacity. The remaining wind-down of these contracts entails risks of financial penalties and litigation. |
| ● | If we fail to comply with our obligations under any license, collaboration or other intellectual property agreements, disagree over contract interpretation, or otherwise experience disruptions to our business relationships with our collaborators or licensors, we could lose intellectual property rights that are necessary to our business. |
| ● | Even if we, or any future collaborators, are able to commercialize any product candidate that we, or they, develop, the successful commercialization of our product candidates will depend in part on the extent to which governmental authorities, private health insurers and other third-party payors provide coverage and adequate reimbursement levels and implement pricing policies favorable for our product candidates. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those products and decrease our ability to generate revenue. |
5
| ● | Some of our product candidates are classified as gene therapies by the FDA and the EMA, and the FDA has indicated that products similar to our product candidates will be reviewed within its Center for Biologics Evaluation and Research, or CBER. Even though our mRNA product candidates are designed to have a different mechanism of action from gene therapies, the association of our product candidates with gene therapies could result in increased regulatory burdens, impair the reputation of our product candidates, or negatively impact our platform or our business. |
Risks Related to Our Financial Position and Need for Additional Capital
We cannot assure you of the adequacy of our capital resources to successfully complete the development and commercialization of our product candidates, and a failure to obtain additional capital, if needed, could force us to delay, limit, reduce or terminate one or more of our product development programs or commercialization efforts.
As of December 31, 2023, we had cash and cash equivalents amounting to €402 million. We believe that we will continue to expend substantial resources for the foreseeable future developing our proprietary product candidates. These expenditures will include costs associated with research and development, conducting preclinical studies and clinical trials, seeking regulatory approvals, as well as launching and commercializing products approved for sale, if any, and costs associated with manufacturing products and maintaining manufacturing facilities. In addition, other unanticipated costs may arise. Because the outcomes of our anticipated clinical trials are highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development and commercialization of our proprietary product candidates.
Our future funding requirements will depend on many factors, including but not limited to:
| ● | the numerous risks and uncertainties associated with developing product candidates and maintaining our mRNA technology platform; |
| ● | the number and characteristics of product candidates that we pursue; |
| ● | the rate of enrollment, progress, cost and outcomes of our clinical trials, which may or may not meet their primary endpoints; |
| ● | the timing of, and cost involved in, conducting nonclinical studies that are regulatory prerequisites to conducting clinical trials of sufficient duration for successful product registration; |
| ● | the cost of manufacturing clinical supply and establishing commercial supply of our product candidates; |
| ● | the costs and timing of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims; |
| ● | the timing of, and the costs involved in, obtaining regulatory approvals for our product candidates if clinical trials are successful; |
| ● | the timing of, and costs involved in, conducting post-approval studies that may be required by regulatory authorities; |
| ● | the cost of commercialization activities for our product candidates, if any of our product candidates are approved for sale, including product manufacturing, marketing and distribution of product candidates generated from our mRNA technology platform and any other product opportunity for which we receive marketing approval in the future; |
| ● | the terms and timing of any collaborative, licensing and other arrangements that we are currently party to or may establish, including any required milestone and royalty payments thereunder and any nondilutive funding that we may receive; |
| ● | the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims, including litigation costs, if any, and the outcome of any such litigation; |
| ● | the timing, receipt, and amount of sales of, or royalties or milestones on, our future products, if any; |
| ● | the costs to recruit and build the organization, including key executives needed to transform to a commercial organization; and |
6
| ● | the costs of operating as a public company, including hiring additional personnel. |
In addition, our operating plan may change as a result of many factors currently unknown to us. As a result of these factors, we may need additional funds sooner than planned. We expect to finance future cash needs primarily through a combination of public or private equity offerings, strategic collaborations, revenues from future product sales, if any, and debt financing. If sufficient funds on acceptable terms are not available when needed, or at all, we could be forced to significantly reduce operating expenses and delay, limit, reduce or terminate one or more of our product development programs or commercialization efforts, which would have a negative impact on our business, prospects, operating results and financial condition.
We have incurred significant losses since our inception. We expect to incur losses for the foreseeable future and may never achieve or maintain profitability.
We have incurred significant losses since our inception. Our consolidated net loss for the years ended December 31, 2023 and 2022 were €260.1 million and €249.1 million, respectively. As of December 31, 2023, our accumulated deficit was €1,566.0 million. We expect to continue to incur losses in the future as we continue our research and development of, and seek regulatory approvals for, our product candidates and maintain and develop new technology platforms, prepare for and begin to commercialize any approved product candidates and add infrastructure and personnel to support our product development efforts and operations as a public company in the United States. We have devoted substantially all of our financial resources and efforts to research and development, including preclinical studies and clinical trials and development of our manufacturing technology. The net losses and negative cash flows from operations incurred to date, together with expected future losses, have had, and likely will continue to have, an adverse effect on our working capital. The amount of future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenue.
Because of the numerous risks and uncertainties associated with biopharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve profitability. For example, our expenses could increase if we are required by the U.S. Food and Drug Administration, or the FDA, the European Medicines Agency, or the EMA, or other regulatory agencies to perform trials in addition to those that we currently expect to perform, or if there are any delays in completing our currently planned clinical trials, the partnering process for our proprietary product candidates or the development of any of our proprietary product candidates.
Our revenue to date has been primarily revenue from the license of our technology platform and from milestone payments for the development of product candidates against targets provided by our collaborators. Our ability to generate revenue and achieve profitability in the future depends in large part on our ability, alone or with our collaborators, to achieve milestones and to successfully complete the development of, obtain the necessary regulatory approvals for, and commercialize, our product candidates and technology platform. This will require us to be successful in a range of challenging activities, including developing product candidates, obtaining regulatory approval for such product candidates, and manufacturing, marketing and selling those product candidates for which we may obtain regulatory approval. We may never succeed in these activities and may never generate revenue from product sales that is significant enough to achieve profitability. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our failure to become or remain profitable could depress our market value and could impair our ability to raise capital, expand our business, develop other product candidates or continue our operations. A decline in the value of our company could also cause you to lose all or part of your investment.
We require substantial financing, which may not be available on acceptable terms, or at all. Raising capital may cause dilution to our shareholders, restrict our operations or require us to relinquish rights to our technology or product candidates.
To the extent that we raise capital through the sale of common shares, convertible securities or other equity securities, the ownership interests of our shareholders may be diluted, and the terms of these securities could include liquidation or other preferences and anti-dilution protections that could adversely affect rights of our common shareholders. In addition, debt financing, if available, may result in fixed payment obligations and may involve agreements that include restrictive covenants that limit our ability to take specific actions, such as incurring debt, making capital expenditures, creating liens, redeeming shares or declaring dividends, that could adversely impact our ability to conduct our business. In addition, securing financing could require a substantial amount of time and attention from our management and may divert a disproportionate amount of their attention away from day-to-day activities, which may adversely affect our management’s ability to oversee the development of our product candidates.
7
If we raise funds through collaborations or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise funds when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market product candidates that we would otherwise prefer to develop and market ourselves.
We cannot be certain that funding will be available on acceptable terms, or at all. If we are unable to raise capital in sufficient amounts or on terms acceptable to us, we may have to significantly delay, scale back or discontinue the development or commercialization of our product candidates or other research and development initiatives. Our current or future license agreements may also be terminated if we are unable to meet the payment or other obligations under the agreements.
We may expend our limited resources to pursue a particular product candidate or indication and fail to capitalize on product candidates or indications that may be more profitable or for which there is a greater likelihood of success.
We have limited financial and managerial resources, and therefore we intend to focus on developing product candidates for specific indications that we believe are most likely to succeed, in terms of both their potential for marketing approval and potential for successful commercialization, if approved. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that may prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs and product candidates for specific indications may not yield any commercially viable product candidates. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through collaboration, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to the product candidate.
We depend on strategic partnerships with other companies to assist in the research, development and commercialization of our platform and product candidates. If our existing or future partners do not perform as expected, if we fail to maintain any of these collaborations or if these collaborations are not successful, our ability to commercialize our product candidates successfully and to generate revenues through technology licensing or otherwise may be materially adversely affected.
We have established strategic partnerships and intend to continue to establish strategic partnerships with third parties to research, develop and commercialize our platform and existing and future product candidates. We have entered into strategic partnerships with Genmab, Acuitas, CRISPR Therapeutics, GlaxoSmithKline Biologicals SA (“GSK”), the Bill & Melinda Gates Foundation, CEPI and Tesla Automation (formerly trading under the name of Tesla Grohmann Automation) and myNEO NV (“myNEO”), among others. For certain of these programs, including our collaborations with Genmab, CRISPR Therapeutics and GSK, we will depend on our partners to design and conduct their clinical studies. As a result, we may not be able to conduct these programs in the manner or on the time schedule we currently contemplate, which may negatively impact our business operations. While we have certain contractual rights to information about preclinical and clinical developments and results under certain of our collaboration agreements, including our agreements with Genmab, CRISPR Therapeutics and GSK, we cannot be certain that clinical trials conducted in connection with such collaboration programs will be conducted in a manner consistent with the best interests of our business. In addition, if any of these partners withdraw support for these programs or proposed products or otherwise impair their development, our business could be negatively affected. Also, our inability to find a partner for any of our product candidates may result in our termination of that specific product candidate program or evaluation of a product candidate in a particular indication. Even if we found a partner for one or more of our product candidates, there is no assurance that upon the approval of one or more of such product candidates we will be able to successfully co-commercialize such products.
8
In addition, our existing licenses and collaboration agreements, including our agreements with Genmab, Acuitas, the Bill & Melinda Gates Foundation, CRISPR Therapeutics, GSK, CEPI and myNEO, impose, and any future licenses, collaborations or other intellectual property agreements we enter into are likely to impose, various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement or other obligations on us. Furthermore, our licenses and collaboration agreements impose, and any future agreement we enter into may also impose, restrictions on our ability to license certain of our intellectual property to third parties or to develop or commercialize certain product candidates or technologies. In spite of our best efforts, our collaborators may conclude that we have breached our obligations under our agreements, in which case, we may be required to pay damages and the collaborator may have the right to terminate the agreement. Any of the foregoing could result in us being unable to develop, manufacture and sell products that are covered by the licensed technology, enable a competitor to gain access to the licensed technology or disrupt our right to receive funding or milestone or royalty payments. See “Item 4. Information on the Company — B. Business Overview — Collaborations.”
Under certain of our collaboration agreements, including our collaborations with Genmab, CRISPR Therapeutics and GSK, we grant our partners an exclusive license to develop and commercialize certain classes of products containing our mRNA technology for specific targets and receive license fees, research and development funding, milestone payments and/or, if a product is approved for marketing, sales royalties in return. Following the discovery and preclinical testing phase, in certain cases, our partners are solely responsible for the further development of the product candidate and therefore exercise full control over its further development and potential commercialization. In certain cases, including under our collaboration with Genmab, we have a limited right to co-commercialize collaboration products. While certain of our existing licenses and collaboration agreements, including our agreements with Genmab, CRISPR Therapeutics and GSK, impose development or commercialization obligations on our collaborators, we cannot be certain that our collaboration partners will allocate sufficient resources or attention to our collaboration programs or that they will progress our collaboration programs consistent with the best interests of our business. Our existing collaborations, and any future collaborations we enter into, therefore, may pose a number of risks, including the following:
| ● | collaborators may have significant discretion in determining the efforts and resources that they will apply to these collaborations; |
| ● | collaborators may not perform their obligations as expected by us or by health authorities, such as the FDA, the EMA or comparable foreign regulatory authorities; |
| ● | collaborators may dissolve, merge, be bought or may otherwise become unwilling to fulfill the initial terms of the collaboration with us; |
| ● | collaborators may fail to perform their obligations under the collaboration agreements or may be slow in performing their obligations; |
| ● | collaborations may be terminated for the convenience of the collaborator and, if terminated, we could be required to raise additional capital to pursue further development or commercialization of the applicable product candidates; |
| ● | collaborators may not pursue development and commercialization of any product candidates that achieve regulatory approval or may elect not to continue or renew development or commercialization programs based on clinical trial results, changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition, that divert resources or create competing priorities or change the actual or perceived competitive situation in a specific indication; |
| ● | collaborators may delay clinical trials, provide insufficient funding for a clinical trial program, stop a clinical trial or abandon a product candidate, repeat or conduct new clinical trials or may require a new formulation of a product candidate for clinical testing; |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates if the collaborators believe that competitive products are more likely to be successfully developed or can be commercialized under terms that are more economically attractive than ours; |
9
| ● | product candidates discovered in collaboration with us may be viewed by our collaborators as competitive with their own product candidates or products, which may cause collaborators to cease to devote resources to the commercialization of our product candidates; |
| ● | a collaborator with marketing and distribution rights to one or more of our product candidates that achieve regulatory approval may not commit sufficient resources to the marketing and distribution of such product or products; |
| ● | disagreements with collaborators, licensors or licensees, including disagreements over proprietary rights, contract interpretation and breach of contract claims, payment obligations or the preferred course of development, might cause delays or termination of the research, development or commercialization of products or product candidates, might lead to additional responsibilities, including financial obligations for us with respect to products or product candidates, or delays or withholding of any payments due or might result in litigation or arbitration, any of which would be time-consuming and expensive, and could limit our ability to execute on our strategies; |
| ● | collaborators may not properly obtain, maintain, enforce or defend our intellectual property or may use our proprietary information in such a way that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation; and |
| ● | collaborators may infringe, misappropriate or otherwise violate the intellectual property of third parties, which may expose us to litigation and potential liability. |
If our collaborations on research and development candidates do not result in the successful development and commercialization of products or if one of our collaborators terminates its agreement with us, we may not receive any future research funding or milestone or royalty payments under the collaboration. If we do not receive the funding we expect under these agreements, our development of our product candidates could be delayed and we may need additional resources to develop our proprietary product candidates. Moreover, our relationships with our partners may divert significant time and effort of our scientific staff and management team and require effective allocation of our resources to multiple internal and collaborative projects. All of the risks relating to product development, regulatory approval and commercialization described in this Annual Report also apply to the activities of our program collaborators.
In the future, we may enter into additional collaborations to fund our development programs or to gain access to sales, marketing or distribution capabilities. Our ability to establish additional strategic alliances will depend, among other things, on our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed strategic alliance and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA, EMA or similar regulatory authorities, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to trial participants, the potential of competing drugs, the existence of uncertainty with respect to our or the proposed collaborator’s ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge, and industry and market conditions generally.
Additionally, subject to its contractual obligations to us, if one of our collaborators is involved in a business combination, the collaborator might deemphasize or terminate the development or commercialization of any product candidate licensed to it by us. If one of our collaborators terminates its agreement with us, we may find it more difficult to attract new collaborators in a timely manner. For more information on our current collaboration agreements, see “Item 4. Information on the Company — B. Business Overview — Collaborations.”
10
We incur significant costs as a result of operating as a public company, and our management is required to devote substantial time to compliance initiatives. We are subject to financial reporting and other requirements for which our accounting and other management systems and resources may not be adequately prepared. We may fail to comply with the rules that apply to public companies, including Section 404 of the Sarbanes-Oxley Act of 2002, which could result in sanctions or other penalties that would harm the business.
As a public company, we incur significant legal, accounting and other expenses. The U.S. federal securities laws, including the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, and rules subsequently implemented by the SEC and the Nasdaq Stock Market LLC, or Nasdaq, have imposed various requirements on public companies, including requirements to file annual and event-driven reports with respect to our business and financial condition, and to establish and maintain effective disclosure and financial controls and corporate governance practices. Our management and other personnel need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations result in substantial legal and financial compliance costs and have made some activities time-consuming and costly. We may not be able to produce reliable financial statements or file these financial statements as part of periodic report in a timely manner with the SEC or comply with Nasdaq listing requirements. In addition, we could make errors in our financial statements that could require us to restate our financial statement.
Pursuant to Section 404 of the Sarbanes-Oxley Act, or Section 404, we are required to furnish a report by our management on our internal control over financial reporting, including the attestation report on internal control over financial reporting issued by our independent registered public accounting firm. To maintain compliance with Section 404, we document and evaluate our internal control over financial reporting, which is both costly and challenging. In this regard, we have needed to continue to dedicate internal resources, have engaged outside consultants, and have adopted a detailed work plan to assess and document the adequacy of internal control over financial reporting. We will continue to implement steps to improve control processes as appropriate, validate through testing that controls are functioning as documented, and implement a reporting and improvement process for internal control over financial reporting. Despite our efforts, there is a risk that in the future neither we nor our independent registered public accounting firm will be able to conclude that our internal control over financial reporting is effective as required by Section 404. This could result in an adverse reaction in the financial markets due to a loss of confidence in the reliability of our financial statements.
Adverse developments affecting financial institutions, companies in the financial service industry or the financial service industry generally, such as actual events or concerns involving liquidity, defaults or non-performance, could adversely affect our operations and liquidity.
Actual events involving limited liquidity, defaults, non-performance or other adverse developments that affect financial institutions or other companies in the financial services industry or the financial services industry generally, or concerns or rumors about any events of these kinds, have in the past and may in the future lead to market-wide liquidity problems, which could adversely affect our operations and liquidity.
Risks Related to the Development, Clinical Testing and Commercialization of Our Product Candidates
Our approach to the discovery and development of product candidates based on mRNA is unproven, and we do not know whether we will be able to successfully develop any products.
We focus on delivering mRNA encoding functional versions of proteins into cells without altering the underlying DNA. Our future success depends on the successful development of this novel therapeutic or vaccine approach. Relatively few mRNA-based product candidates have been tested in animals or humans, and the data underlying the feasibility of developing mRNA-based products are both preliminary and limited. To date, a limited number of products that utilize mRNA as a prophylactic vaccine against COVID-19 have been approved in the United States and Europe, subject to certain limitations. In addition, no product that utilizes mRNA as a therapeutic vaccine has been approved in the United States or Europe. We have not yet succeeded and may not succeed in demonstrating to the FDA or EMA the efficacy and safety of any of our product candidates in clinical trials or in obtaining marketing approval thereafter.
11
Clinical drug development involves a lengthy and expensive process with uncertain timelines and uncertain outcomes, and results of earlier studies and trials may not be predictive of future trial results. If clinical trials of our product candidates or production of our product candidates are prolonged or delayed, we may be unable to obtain required regulatory approvals, and therefore be unable to commercialize our product candidates on a timely basis or at all.
Our business is dependent on the successful development, regulatory approval and commercialization of product candidates based on our technology platform. If we and our collaborators are unable to obtain approval for and effectively commercialize our product candidates, our business would be significantly harmed. Even if we complete the necessary preclinical studies and clinical trials, the marketing approval process is expensive, time-consuming and uncertain, and we may not be able to obtain approvals for the commercialization of any product candidates we may develop.
To obtain the requisite regulatory approvals to market and sell any of our product candidates, we must demonstrate through extensive preclinical studies and clinical trials that our products are safe and effective in humans. Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of our product candidates may not be predictive of the results of later-stage clinical trials. For example, our Phase 2b clinical trial with CV9104, one of our first-generation vaccines based on protamine formulation that was designed to evaluate the investigational mRNA-based cancer vaccine in patients with asymptomatic or minimally symptomatic metastatic castrate resistant prostate cancer, failed to meet the primary endpoint of improving overall survival despite proceeding through preclinical and Phase 1 studies. While we have assessed the results of past trials and these have informed our approach going forward, we can provide no assurance that future clinical trials will not be discontinued or fail to meet their specified endpoints. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and initial clinical trials. A number of companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials. Our future clinical trial results may not be successful.
We are subject to significant regulatory oversight with respect to manufacturing and developing our product candidates. Our manufacturing facilities or the manufacturing facilities of our third-party manufacturers or suppliers may not meet regulatory requirements. Failure to meet GMP requirements set forth in regulations promulgated by the FDA, the EMA and other comparable regulatory authorities could result in significant delays in and costs of our products.
Clinical trials must be conducted in accordance with the FDA, EMA and comparable foreign regulatory authorities’ legal requirements, regulations or guidelines and are subject to oversight by these governmental agencies and Institutional Review Boards, or IRBs, at the medical institutions where the clinical trials are conducted. In addition, clinical trials must be conducted with supplies of our product candidates produced in accordance with current good manufacturing practices, or cGMPs, and other requirements. We depend on medical institutions and clinical research organizations, or CROs, to conduct our clinical trials in compliance with good clinical practice, or GCP, standards. Failure to follow and document adherence to such regulations or other regulatory requirements may lead to significant delays in the availability of a product for our clinical trials, result in the termination of, or a clinical hold being placed on, one or more of our clinical trials, or delay or prevent submission or approval of marketing applications for our product candidates.
To the extent our CROs fail to enroll participants for our clinical trials, fail to conduct the trial in accordance with GCP requirements or are delayed for a significant time in the execution of trials, including achieving full enrollment, we may be affected by increased costs, program delays or both, which may harm our business. Our product candidates, CV2CoV (SARS-CoV-2), CV0501 (modified nucleotides, SARS - CoV - 2), CV0601/0701 (modified nucleotides, SARS-CoV-2), CVSQIV (multivalent seasonal influenza), Flu SV mRNA (modified nucleotides, single antigen seasonal influenza), CV7202 (rabies), CV8102 (melanoma, adenoidcystic carcinoma, squamous cell cancer of skin and head and neck), CVGBM (cancer), Cas9 gene - editing, are in early clinical development. All other of our research programs are in the preclinical development stage.
The completion of clinical trials for our clinical product candidates may be delayed, suspended or terminated as a result of many factors, including but not limited to:
| ● | shortage of materials required for the production of our product candidates including due to inflationary pressures, supply chain issues, labor shortages and increases in commodity prices (including as result of the war in Ukraine) and the lingering effects of COVID-19; |
12
| ● | inability of our manufacturing facilities or the manufacturing facilities of our third-party manufacturers or suppliers to meet regulatory requirements; |
| ● | transfer of manufacturing processes to larger-scale facilities operated by a CMO or by us, and delays or failure by our CMOs or us to make any necessary changes to such manufacturing process; |
| ● | the delay or refusal of regulators or IRBs to authorize us to commence a clinical trial at a prospective trial site and changes in regulatory requirements, policies and guidelines; |
| ● | delays or failure to reach agreement on acceptable terms with prospective CROs and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| ● | the inability to enroll a sufficient number of patients in trials to ensure adequate statistical power to detect statistically significant treatment effects; |
| ● | negative or inconclusive results, which may require us to conduct additional preclinical or clinical trials or to abandon projects that we expect to be promising; |
| ● | safety or tolerability concerns causing us to suspend or terminate a trial if it is determined that the participants are being exposed to unacceptable health risks; |
| ● | regulators or IRBs requiring that we or our investigators suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements or safety concerns, among others; |
| ● | lower than anticipated retention rates of patients and volunteers in clinical trials; |
| ● | our CROs or clinical trial sites failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner, or at all, deviating from the protocol or dropping out of a trial; |
| ● | delays relating to adding new clinical trial sites; |
| ● | difficulty in maintaining contact with patients after treatment, resulting in incomplete data; |
| ● | delays in establishing the appropriate dosage levels; |
| ● | the quality or stability of the product candidate falling below acceptable standards; |
| ● | the inability to produce or obtain sufficient quantities of the product candidate to complete clinical trials on time, or delays in sufficiently developing, characterizing or controlling a manufacturing process suitable for clinical trials; |
| ● | exceeding budgeted costs due to difficulty in accurately predicting costs associated with clinical trials; |
| ● | lack of adequate funding to continue the clinical trial; |
| ● | developments observed in trials conducted by competitors for related technology that raise general FDA or foreign regulatory authority concerns about risk to patients of gene therapy technology; |
| ● | determination that the product will not be producible at the manufacturing stage; and |
13
Disruptions caused by the inflationary pressures, supply chain issues, labor shortages and increases in commodity prices (including result of the war in Ukraine) and the lingering effects of the COVID-19 pandemic may increase the likelihood that we encounter such difficulties or delays in initiating, enrolling, conducting or completing our planned and ongoing clinical trials. We could also encounter delays if a clinical trial is suspended or terminated by us, by the IRBs of the institutions in which such trials are being conducted, by a Data Safety Monitoring Board for such trial or by the FDA or comparable foreign regulatory authorities. Such authorities may impose such a suspension or termination due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or comparable foreign regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. In addition, changes in regulatory requirements and policies may occur, and we may need to amend clinical trial protocols to comply with these changes. Amendments may require us to resubmit our clinical trial protocols to IRBs for reexamination, which may impact the costs, timing or successful completion of a clinical trial.
In addition, preclinical and clinical data are often susceptible to varying interpretations and analyses. Many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials have nonetheless failed to obtain marketing approval for the product candidates. The FDA, the EMA and comparable foreign regulatory authorities have substantial discretion in the approval process and in determining when or whether regulatory approval will be obtained for any of our product candidates. Even if we believe the data collected from clinical trials of our product candidates are promising, such data may not be sufficient to support approval by the FDA, the EMA or any other regulatory authority.
In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, adherence to the dosing regimen and other trial protocols and the rate of dropout among clinical trial participants.
If we are required to conduct additional clinical trials or other testing of our product candidates that we develop beyond the trials and testing that we contemplate, if we are unable to successfully complete clinical trials of our product candidates or other testing, if the results of these trials or tests are unfavorable or are only modestly favorable, or if there are safety concerns associated with our other product candidates, we may:
| ● | be delayed in obtaining marketing approval for our product candidates; |
| ● | not obtain marketing approval at all; |
| ● | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or significant safety warnings, including boxed warnings; |
| ● | be subject to additional post-marketing testing or other requirements; or |
| ● | remove the product from the market after obtaining marketing approval. |
Our product development costs will also increase if we experience delays in testing or receiving marketing approvals and we may be required to obtain additional funds to complete clinical trials. We cannot assure you that our clinical trials will begin as planned or be completed on schedule, if at all, or that we will not need to restructure our trials after they have begun. Significant clinical trial delays also could shorten any periods during which we may have the exclusive right to commercialize our product candidates or allow our competitors to bring products to market before we do, which may harm our business and results of operations. In addition, some of the factors that cause, or lead to, clinical trial delays may ultimately lead to the denial of regulatory approval of our product candidates.
14
Interim, “top-line,” and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data become available and are subject to audit and verification procedures that could result in material changes in the final data.
From time to time, we may publicly disclose preliminary or top-line data from our preclinical studies and clinical trials, which are based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. We also make assumptions, estimations, calculations and conclusions as part of our analyses of data, and we may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the top-line or preliminary results that we report may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Top-line data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, top-line data should be viewed with caution until the final data are available.
From time to time, we may also disclose interim data from our preclinical studies and clinical trials. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available or as patients from our clinical trials continue other treatments for their disease. Adverse differences between preliminary or interim data and final data could significantly harm our business prospects. Further, disclosure of interim data by us or by our competitors could result in volatility in the price of our common stock.
Further, others, including regulatory agencies, may not accept or agree with our assumptions, estimates, calculations, conclusions or analyses or may interpret or weigh the importance of data differently, which could impact the value of the particular program, the approvability or commercialization of the particular product candidate or product and our company in general. In addition, the information we choose to publicly disclose regarding a particular study or clinical trial is based on what is typically extensive information, and you or others may not agree with what we determine is material or otherwise appropriate information to include in our disclosure.
If we encounter difficulties enrolling patients in our clinical trials, our clinical development activities could be delayed and result in increased costs and longer development periods or otherwise be adversely affected.
We will be required to identify and enroll a sufficient number of patients for our planned clinical trials. Trial participant enrollment could be limited in future trials given that many potential participants may be ineligible because of preexisting conditions, medical treatments or other reasons. We may not be able to initiate or continue clinical trials required by the FDA, EMA or other foreign regulatory agencies or any of our other product candidates that we pursue if we are unable to locate and enroll a sufficient number of eligible patients or volunteers to participate in these clinical trials.
Patient enrollment is affected by other factors, including:
| ● | severity of the disease under investigation; |
| ● | design of the clinical trial protocol; |
| ● | size and nature of the patient population; |
| ● | eligibility criteria for the trial in question; |
| ● | perceived risks and benefits of the product candidate under trial; |
| ● | perceived safety and tolerability of the product candidate; |
| ● | proximity and availability of clinical trial sites for prospective patients; |
| ● | availability of competing therapies and clinical trials; |
15
| ● | clinicians’ and patients’ perceptions as to the potential advantages of the drug being studied in relation to other available therapies, including standard-of-care and any new drugs that may be approved for the indications we are investigating; |
| ● | efforts to facilitate timely enrollment in clinical trials; |
| ● | patient referral practices of physicians; and |
| ● | our ability to monitor patients adequately during and after treatment. |
We also may encounter difficulties in identifying and enrolling such patients with a stage of disease appropriate for our ongoing or future clinical trials. In addition, the process of finding and diagnosing patients may prove costly. Our inability to enroll a sufficient number of patients for any of our clinical trials would result in significant delays or may require us to abandon one or more clinical trials.
We may face continued business disruption and related risks resulting from inflationary pressures, supply chain issues, labor shortages and increases in commodity prices (including as result of the war in Ukraine) and the lingering effects of the COVID-19 pandemic, which could have a material adverse effect on our business plan or clinical trials.
The global economy, including credit and financial markets, has experienced extreme volatility and disruptions, including severely diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in unemployment rates, increases in inflation rates, higher interest rates and uncertainty about economic stability. For example, the COVID-19 pandemic resulted in widespread unemployment, economic slowdown and extreme volatility in the capital markets. Similarly, the ongoing military conflict between Russia and Ukraine has created extreme volatility in the global capital markets and is expected to have further global economic consequences, including disruptions of the global supply chain and energy markets. Any such volatility and disruptions may have adverse consequences on us or the third parties on whom we rely. If the equity and credit markets deteriorate, including as a result of political unrest or war, it may make any necessary debt or equity financing more difficult to obtain in a timely manner or on favorable terms, more costly or more dilutive. Increased inflation rates can adversely affect us by increasing our costs, including labor and employee benefit costs. Any significant increases in inflation and related increase in interest rates could have a material adverse effect on our business, results of operations and financial condition.
Moreover, if COVID-19 would restart, we may experience disruptions that could severely impact our business, preclinical studies and clinical trials, including:
| ● | delays in receiving authorization from local regulatory authorities to initiate our planned clinical trials; |
| ● | changes in local regulations as part of a response to the COVID-19 pandemic which may require us to change the ways in which our clinical trials are conducted, which may result in unexpected costs, or to discontinue the clinical trials altogether; |
| ● | diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; |
| ● | interruption of key clinical trial activities, such as clinical trial site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others, or interruption of clinical trial subject visits and study procedures, the occurrence of which could affect the integrity of clinical trial data; |
| ● | risk that participants enrolled in our clinical trials will acquire COVID-19 while the clinical trial is ongoing, which could impact the results of the clinical trial, including by increasing the number of observed adverse events; |
| ● | interruptions in preclinical studies due to restricted or limited operations at our research and development laboratory facility; |
| ● | delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; |
16
| ● | limitations in employee resources that would otherwise be focused on the conduct of our clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people; |
| ● | refusal of the FDA to accept data from clinical trials in affected geographies; and |
| ● | interruption or delays to our sourced discovery and clinical activities. |
These and other disruptions in our operations and the global economy could negatively impact our business, operating results and financial condition.
In addition, quarantines, travel restrictions, shelter-in-place and similar government orders, or the perception that such orders, shutdowns or other restrictions on the conduct of business operations could occur, related to COVID-19 or other infectious diseases could impact personnel at third-party manufacturing facilities upon which we rely, or the availability or cost of materials, which could disrupt the supply chain for our product candidates. We have taken a series of actions aimed at safeguarding our employees and business associates, including regular PCR-based COVID-19 testing, implementing a work-from-home policy for employees except for those related to our production and laboratory operations, and these arrangements may cause reduced productivity of our employees and/or delays or disruptions of our business operations.
Our suppliers, licensors or collaborators could also be disrupted by conditions related to COVID-19, possibly resulting in disruption to our supply chain, clinical trials, partnerships or operations. If our suppliers, licensors, CMOs, CROs or collaborators are unable or fail to fulfill their obligations to us for any reason, our ability to continue meeting clinical supply demand for our product candidates or otherwise advancing development of our product candidates may become impaired.
Our proprietary product candidates are still in preclinical or clinical development. We cannot give any assurance that any of our product candidates will receive regulatory approval, and if we are unable to obtain regulatory approval and ultimately commercialize our product candidates or experience significant delays in doing so, our business will be materially harmed.
Our proprietary product candidates are still in preclinical or clinical development. Although we may receive certain payments from our collaboration partners, including upfront payments, payments for achieving certain development, regulatory or commercial milestones and royalties, our ability to generate revenue from our product candidates’ sales is dependent on receipt of regulatory approval for, and successful commercialization of, such product candidates, which may never occur. Our business and future success is in particular dependent on our ability to develop, either alone or in partnership, successfully, receive regulatory approval for and then successfully commercialize our proprietary product candidates. Each of our product candidates will require additional preclinical and/or clinical development, regulatory approval in multiple jurisdictions, manufacturing supply, substantial investment and significant marketing efforts before we generate any revenue from product sales or royalties. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from applicable regulatory authorities. The success of our product candidates will depend on several factors, including the following:
| ● | successful completion of preclinical and/or clinical studies; |
| ● | negative or inconclusive results from our clinical trials, the clinical trials of our collaborators or the clinical trials of others for product candidates similar to ours, leading to a decision or requirement to conduct additional preclinical testing or clinical trials or abandon a program; |
| ● | successful enrollment of patients in, and completion of, clinical trials; |
| ● | strategic commitment to particular product candidates and indications by us and our collaborators; |
| ● | receipt of regulatory authorizations from applicable regulatory authorities for future clinical trials; |
| ● | receipt of product approvals, including marketing approvals, from applicable regulatory authorities; |
| ● | successful completion of all safety studies required to obtain regulatory approval in the United States, the European Union and other jurisdictions for our product candidates; |
17
| ● | obtaining and maintaining patent and trade secret protection or regulatory exclusivity for our product candidates; |
| ● | launching commercial sales of our product candidates, if and when approved, whether alone or in collaboration with others; |
| ● | acceptance of the product candidates, if and when approved, by patients, the medical community and third-party payors; |
| ● | effectively competing with other therapies; |
| ● | obtaining and maintaining coverage and adequate reimbursement from third-party payors; |
| ● | obtaining, maintaining, enforcing and defending intellectual property and intellectual property-related claims; |
| ● | maintaining a continued acceptable safety and quality profile of the product candidates following approval; and |
| ● | maintaining a continued, sufficient supply of drug substance in acceptable quality. |
If we do not achieve one or more of these factors in a complete and timely manner or at all, we could experience significant delays or an inability to successfully commercialize our product candidates, which would materially adversely affect our business, financial condition, results of operations and prospects and, in case of product candidates, technologies and licenses we have acquired, which may result in a significant impairment of assets.
Although we expect to submit biologics license applications, or BLAs, for our mRNA-based product candidates in the United States, and in the European Union, mRNA-based medicines have been classified as gene therapy medicinal products, other jurisdictions may consider our mRNA-based product candidates to be new drugs, not biologics or gene therapy medicinal products, and require different marketing applications. In addition, we have not previously submitted a BLA to the FDA, or similar regulatory approval filings to comparable foreign authorities, for any product candidate, and we cannot be certain that any of our product candidates will be successful in clinical trials or receive regulatory approval. Further, our product candidates may not receive regulatory approval even if they are successful in clinical trials. If we do not receive regulatory approvals for our product candidates, we may not be able to continue our operations. Even if we successfully obtain regulatory approvals to market one or more of our product candidates, our revenues will be dependent, in part, upon the size of the markets in the territories for which we gain regulatory approval and have commercial rights. If the markets for patient subsets that we are targeting are not as significant as we estimate, we may not generate significant revenues from sales of such products, if approved.
We plan to seek regulatory approval to commercialize our product candidates both in the United States and the EU, and potentially in additional foreign countries. While the scope of regulatory approval is similar in other countries, to obtain separate regulatory approval in many other countries, we must comply with numerous and varying regulatory requirements of such countries regarding safety and efficacy and governing, among other things, clinical trials and commercial sales, pricing and distribution of our product candidates, and we cannot predict success in these jurisdictions.
We have no history of commercializing pharmaceutical products, which may make it difficult to evaluate the prospects for our future viability.
We commenced operations in 2000 and have a long track record of performing clinical trials with multiple product candidates since 2008. Our operations to date have been limited to establishing our company, raising capital, developing our proprietary mRNA technology platform, identifying and testing potential product candidates and conducting clinical trials. We have not yet demonstrated an ability to obtain marketing approvals, manufacture a commercial-scale product, or arrange for a third party to do so on our behalf, or conduct sales and marketing activities necessary for successful product commercialization. Accordingly, you should consider our prospects in light of the costs, uncertainties, delays and difficulties frequently encountered by companies in the early stages of development, especially clinical-stage biopharmaceutical companies such as ours. Any predictions you make about our future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing and commercializing pharmaceutical products.
We may encounter unforeseen expenses, difficulties, complications, delays and other known or unknown factors in achieving our business objectives. We will eventually need to transition from a company with a development focus to a company capable of supporting commercial activities. We may not be successful in such a transition.
18
We may seek to enter into agreements with others to utilize their marketing and distribution capabilities, but we may be unable to enter into agreements on favorable terms, if at all. If we rely on others to commercialize our products if we receive the necessary approvals, our revenues may be lower than if we commercialized these products ourselves. In addition, we may have little or no control over such third parties’ sales efforts. If our partners commit insufficient resources to commercialize our products, and we cannot independently develop necessary marketing capabilities, we may be unable to generate sufficient product revenue to sustain our business.
We expect our financial condition and operating results to continue to fluctuate significantly from quarter to quarter and year to year due to a variety of factors, many of which are beyond our control. Accordingly, you should not rely upon the results of any quarterly or annual periods as indications of future operating performance.
Our product candidates may cause undesirable side effects that could delay or prevent their regulatory approval, limit the commercial profile of an approved label or result in significant negative consequences following regulatory approval, if any. Significant adverse events may occur during our clinical trials or even after receiving regulatory approval, which could delay or terminate clinical trials, delay or prevent regulatory approval or market acceptance of any of our product candidates.
Undesirable side effects that may be caused by our product candidates could cause us, our collaboration partners or the regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, EMA or comparable foreign regulatory authorities. Results of our trials could reveal a high and unacceptable severity and prevalence of side effects. In such an event, our trials could be suspended or terminated and the FDA, EMA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications. The product-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Clinical trials assess a sample of the potential patient population. With a limited number of patients and duration of exposure, rare and severe side effects of our product candidates may only be uncovered with a significantly larger number of patients exposed to the product candidate. If our product candidates receive regulatory approval and we or others identify undesirable side effects caused by such product candidates (or any other similar products) after such approval, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may withdraw or limit their approval of such product candidates and require us to take our approved product(s) off the market; |
| ● | regulatory authorities may require the addition of labeling statements, such as a “boxed” warning or a contraindication, or submission of field alerts to physicians and pharmacies; |
| ● | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
| ● | we may be required to change the way such product candidates are distributed or administered, conduct additional clinical trials or change the labeling of the product candidates; |
| ● | actual or potential drug-related side effects could negatively affect patient recruitment or the ability of enrolled patients to complete a trial for our products or product candidates; |
| ● | market acceptance of our products, if any, by patients and physicians may be reduced and sales of the product, if any, may decrease significantly; |
| ● | regulatory authorities may require a Risk Evaluation and Mitigation Strategy, or a REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools; |
| ● | we may be subject to regulatory investigations and government enforcement actions; |
19
| ● | we may decide or be required to remove such product candidates from clinical trials, or if approved, from the marketplace; |
| ● | we could be sued and potentially held liable for injury caused to individuals exposed to or taking our product candidates; |
| ● | if any product candidates are approved, sales of the product(s) may decrease substantially; and |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product candidates and could substantially increase the costs of commercializing our product candidates, if approved, and therefore could have a material adverse effect on our business, financial condition, results of operations and prospects.
To date, a limited number of products that utilize mRNA as a prophylactic vaccine against COVID-19 have been approved in the United States, Europe and other countries, subject to certain limitations. In addition, no product that utilizes mRNA as a therapeutic vaccine has been approved in the United States or Europe. As such, mRNA drug development has substantial clinical development and regulatory risks due to the novel and unprecedented nature of this new category of medicines.
No product that utilizes mRNA as a therapeutic vaccine has been approved in the United States or Europe. In addition, a limited number of products that utilize mRNA as a prophylactic vaccine against COVID-19 have been approved by the FDA, EMA and other regulatory agencies. Such approvals were provided after parallelized clinical trials, and certain of the products may be subject to ongoing review by the FDA, EMA or other regulatory agencies, and in some cases may be canceled, expire or subject to lengthy renewal. Successful discovery, development and continued market presence of mRNA-based (and other) products by either us or our collaborators is highly uncertain and depends on numerous factors, many of which are beyond our or their control. Our product candidates that appear promising in the early phases of development may fail to advance, experience delays in the clinic or clinical holds, or fail to reach the market or stay in the market for many reasons, including:
| ● | discovery efforts aimed at identifying potential immunotherapies may not be successful; |
| ● | nonclinical or preclinical study results may show product candidates to be less effective than desired or have harmful or problematic side effects; |
| ● | clinical trial results may show the product candidates to be less effective than expected, including a failure to meet one or more endpoints or have unacceptable side effects or toxicities; |
| ● | manufacturing failures, insufficient supply of GMP materials for clinical trials, or higher than expected cost could delay or set back clinical trials, or make our product candidates commercially unattractive; |
| ● | our improvements in the manufacturing processes may not be sufficient to satisfy the clinical or commercial demand of our product candidates or regulatory requirements for clinical trials; |
| ● | changes that we make to optimize our manufacturing, testing or formulating of GMP materials could impact the safety, tolerability and efficacy of our product candidates; |
| ● | pricing or reimbursement issues or other factors could delay clinical trials or make any immunotherapy uneconomical or noncompetitive with other therapies; |
| ● | the failure to timely advance our programs or receive the necessary regulatory approvals, or a delay in receiving such approvals, due to, among other reasons, slow or failure to complete enrollment in clinical trials, withdrawal by trial participants from trials, failure to achieve trial endpoints, additional time requirements for data analysis, data integrity issues, BLA, MAA or the equivalent application, discussions with the FDA or the EMA, a regulatory request for additional nonclinical or clinical data, or safety formulation or manufacturing issues may lead to our inability to obtain sufficient funding; and |
20
| ● | the proprietary rights, products and technologies of our competitors may prevent our immunotherapies from being commercialized. |
Although we expect to submit biologics license applications, or BLAs, for our mRNA-based product candidates in the United States and in the European Union, mRNA-based medicines have been classified as gene therapy medicinal products. Unlike certain gene therapies that irreversibly alter cell DNA and may cause certain side effects, mRNA-based medicines are designed not to irreversibly change cell DNA. Side effects observed in other gene therapies, however, could negatively impact the perception of immunotherapies despite the differences in mechanism. Due to the circumstances surrounding the approval of mRNA-based vaccines against COVID-19, the regulatory pathway for future mRNA products in the United States and other jurisdictions for approval is uncertain. The length of time necessary to complete clinical trials and submit an application for marketing approval by a regulatory authority varies significantly from one pharmaceutical product to the next and may be difficult to predict.
The regulatory approval processes of the FDA and comparable authorities are lengthy, time-consuming and inherently unpredictable and if we fail to obtain regulatory approval in any jurisdiction, we will not be able to commercialize our products in that jurisdiction and our business, results of operations, financial condition and prospects, may be materially adversely affected.
The time required to obtain approval by the FDA, EMA and comparable authorities is unpredictable but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval laws, regulations, policies or the type and amount of clinical data or other information necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. We have not obtained regulatory approval for any product candidate, and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
Our product candidates could fail to receive regulatory approval for many reasons, including the following:
| ● | the FDA, EMA or comparable foreign regulatory authorities may disagree with the design or implementation of our clinical trials; |
| ● | we may be unable to demonstrate to the satisfaction of the FDA, EMA or comparable foreign regulatory authorities that a product candidate is safe and effective for its proposed indication; |
| ● | the designs or our execution of clinical trials might not be considered adequate, or the results of clinical trials may not meet the level of statistical significance required, by the FDA, EMA or comparable foreign regulatory authorities for approval; |
| ● | we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| ● | the FDA, EMA or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| ● | the data collected may not be sufficient to support the submission of a BLA or other submission, or to obtain regulatory approval in the United States, the European Union or elsewhere; |
| ● | the FDA, EMA or comparable foreign regulatory authorities may fail to approve our manufacturing processes or facilities or those of third-party manufacturers with which we contract for clinical and commercial supplies; and |
| ● | the laws, regulations or policies of the FDA, EMA or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data or other regulatory submissions insufficient for approval. |
This lengthy approval process as well as the unpredictability of future clinical trial results may result in our failing to obtain regulatory approval to market any of our product candidates, which would significantly harm our business, results of operations and prospects. The FDA, the EMA and other regulatory authorities have substantial discretion in the approval process and determining when or whether regulatory approval will be obtained for any of our product candidates. Even if we believe the data collected from clinical trials of our product candidates are promising, such data may not be sufficient to support approval by the FDA, the EMA or any other regulatory authority.
21
In order to commercialize our products in more than one jurisdiction, we will be required to obtain separate regulatory approvals in each market and to comply with numerous and varying regulatory requirements. The approval procedures vary from country to country and may require additional testing, administrative review periods, agreements with pricing authorities or other steps. Satisfying these and other regulatory requirements is costly, time-consuming, uncertain and subject to unanticipated delays. In addition, in many countries outside the United States and in particular in many of the Member States of the European Union, a product must undergo health economic assessments to agree on pricing and/or be approved for reimbursement before it can be approved for sale in that country, or before it becomes commercially viable. The FDA and the EMA may come to different conclusions regarding approval of a marketing application. Approval by the FDA or EMA does not ensure approval by regulatory authorities in other countries or jurisdictions, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA or EMA. In addition, our failure to obtain regulatory approval in any country may delay or have negative effects on the process for regulatory approval in other countries. Clinical trials conducted in one country may not be accepted by regulatory authorities in other countries. We may not obtain regulatory approvals on a timely basis, if at all. We may not be able to submit applications for regulatory approvals and may not receive necessary approvals to commercialize our products in any market. We may be required to conduct additional preclinical studies or clinical trials, which would be costly and time-consuming. If we or any future partner are unable to obtain regulatory approval for our product candidates in one or more significant jurisdictions, then the commercial opportunity for our product candidates, and our business, results of operations, financial condition and prospects, may be materially adversely affected.
The regulatory landscape that will govern our product candidates is uncertain. Regulations relating to more established gene therapy and cell therapy products are still developing, and changes in regulatory requirements could result in delays or discontinuation of development of our product candidates or unexpected costs in obtaining regulatory approval.
The regulatory requirements to which our product candidates will be subject are not entirely clear. Even with respect to more established products that fit into the categories of gene therapies or cell therapies, the regulatory landscape is still developing. For example, regulatory requirements governing gene therapy products and cell therapy products have changed frequently and may continue to change in the future. Moreover, there is substantial, and sometimes uncoordinated, overlap in those responsible for regulation of existing gene therapy products and cell therapy products. Although the FDA decides whether individual gene therapy protocols may proceed, the review process and determinations of other reviewing bodies can impede or delay the initiation of a clinical study, even if the FDA has reviewed the study and authorizes its initiation. Conversely, the FDA can place an Investigational New Drug Application, or an IND, on clinical hold even if such other entities have provided a favorable review. Furthermore, gene therapy clinical trials may also require evaluation and assessment by an institutional biosafety committee, or an IBC, a local institutional committee that reviews and oversees basic and clinical research conducted at the institution participating in the clinical trial. The IBC assesses the safety of the research and identifies any potential risk to the public health or the environment, and such assessment may result in some delay before initiation of a clinical trial. In addition, adverse developments in clinical trials of gene therapy products conducted by others may cause the FDA or other regulatory bodies to change the requirements for approval of any of our product candidates.
Complex regulatory environments exist in other jurisdictions in which we might consider seeking regulatory approvals for our product candidates, further complicating the regulatory landscape. For example, in the European Union a special committee called the Committee for Advanced Therapies, or the CAT, was established within the EMA in accordance with Regulation (EC) No 1394/2007 on advanced-therapy medicinal products, or ATMPs, to assess the quality, safety and efficacy of ATMPs, and to follow scientific developments in the field. ATMPs include gene therapy products as well as somatic cell therapy products and tissue-engineered products.
These various regulatory review committees and advisory groups and new or revised guidelines that they promulgate from time to time may lengthen the regulatory review process, require us to perform additional studies, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our product candidates or lead to significant post-approval limitations or restrictions. As the regulatory landscape for our product candidates is new, we may face even more cumbersome and complex regulations than those emerging for gene therapy products and cell therapy products. Furthermore, even if our product candidates obtain required regulatory approvals, such approvals may later be withdrawn as a result of changes in regulations or the interpretation of regulations by applicable regulatory agencies.
Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market could decrease our ability to generate sufficient product sales revenue to maintain our business.
22
Even if we receive regulatory approval for any of our product candidates, we will be subject to ongoing obligations and continued regulatory review, which may materially adversely affect our business, prospects, financial condition and results of operations. We have not previously submitted a BLA to the FDA, or similar regulatory approval filings to comparable foreign authorities, for any product candidate and never received regulatory approval for any of our product candidates. Even if the FDA, EMA or a comparable foreign regulatory authority approves any of our product candidates, the manufacturing processes, labeling, packaging, distribution, product sampling, adverse event reporting, storage, advertising, marketing, promotion and recordkeeping for the product will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with cGMPs and GCPs for any clinical trials that we conduct post-approval, all of which may result in significant expense and limit our ability to commercialize such products. There also are continuing, annual program user fees for any marketed products. Biologic manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
Any regulatory approvals that we receive for our product candidates may also be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing and surveillance to monitor the safety and efficacy of the product. For example, the FDA has the authority to require a REMS as part of a BLA or after approval, which may impose further requirements or restrictions on the distribution or use of an approved product, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements may result in, among other things:
| ● | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory; |
| ● | product recalls; |
| ● | fines, warning letters, untitled letters or holds on clinical trials; |
| ● | refusal by the FDA, EMA or a comparable foreign regulatory authority to approve pending applications or supplements to approved applications, or suspension or revocation of product approvals; |
| ● | requirements to conduct additional clinical trials, change our product labeling or submit additional applications or application supplements; |
| ● | product seizure or detention, or refusal to permit the import or export of products; |
| ● | mandated modification of promotional materials and labeling and the issuance of corrective information; |
| ● | consent decrees, corporate integrity agreements, debarment or exclusion from federal healthcare programs; |
| ● | the issuance of safety alerts, Dear Healthcare Provider letters, press releases and other communications containing warnings or other safety information about the product; or |
| ● | injunctions or the imposition of civil or criminal penalties. |
23
In addition, regulatory policies may change or additional government regulations or legislation may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we fail to comply with existing requirements, are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any regulatory approval that we may have obtained or face regulatory or enforcement actions, which may materially adversely affect our business, prospects, financial condition and results of operations.
In addition, if any of our product candidates is approved, our product labeling, advertising and promotion will be subject to regulatory requirements and continuing regulatory review. The FDA strictly regulates the promotional claims that may be made about prescription products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. If we receive marketing approval for a product candidate, physicians may nevertheless prescribe it to their patients in a manner that is inconsistent with the approved label. If we are found to have promoted such off-label uses, we may become subject to significant liability. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant sanctions. The federal government has levied large civil and criminal fines against companies for alleged improper promotion and has enjoined several companies from engaging in off-label promotion. The FDA has also requested that companies enter into consent decrees or permanent injunctions under which specified promotional conduct is changed or curtailed.
Any government investigation of alleged violations of law could require us to expend significant time and resources in response, and could generate negative publicity. Any failure to comply with ongoing regulatory requirements may significantly and adversely affect our ability to commercialize our product candidates.
Further, the policies of FDA, EMA and other comparable regulatory authorities may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we are slow or unable to adapt to changes in existing requirements or to adopt new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad. It is difficult to predict how current and future legislation, executive actions, and litigation, including the executive orders, will be implemented, and the extent to which they will impact our business, our clinical development, and the FDA’s and other agencies’ ability to exercise their regulatory authority, including the FDA’s pre-approval inspections and timely review of any regulatory filings or applications we submit to the FDA. To the extent any executive actions impose constraints on FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
A breakthrough therapy designation by the FDA for a product candidate may not lead to a faster development or regulatory review or approval process, and it would not increase the likelihood that the product candidate will receive marketing approval.
We may in the future seek a breakthrough therapy designation for one or more product candidates. A breakthrough therapy is defined as a product candidate that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the product candidate may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. For product candidates that have been designated as breakthrough therapies, interaction and communication between the FDA and the sponsor of the trial can help to identify the most efficient path for clinical development while minimizing the number of patients placed in ineffective control regimens. Product candidates designated as breakthrough therapies by the FDA are also eligible for priority review if supported by clinical data at the time of the submission of the BLA.
Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if we believe that one of our product candidates meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. In any event, the receipt of a breakthrough therapy designation for a product candidate may not result in a faster development process, review or approval compared to product candidates considered for approval under conventional FDA procedures and it would not assure ultimate approval by the FDA. In addition, even if one or more of our product candidates qualify as breakthrough therapies, the FDA may later decide that the product candidate no longer meets the conditions for qualification or it may decide that the time period for FDA review or approval will not be shortened.
24
Because we are developing product candidates for the treatment or prevention of diseases in which there is little clinical experience using new technologies, there is increased risk that the FDA, the EMA or other regulatory authorities may not consider the endpoints of our clinical trials to provide clinically meaningful results and that these results may be difficult to analyze.
As we are developing novel treatments and preventative measures for diseases in which we believe there is limited clinical experience with new endpoints and methodologies, there is heightened risk that the FDA, EMA or comparable foreign regulatory bodies may not consider the clinical trial endpoints to provide clinically meaningful results, and the resulting clinical data and results may be more difficult to analyze. It is difficult to determine how long it will take or how much it will cost to obtain regulatory approvals for our product candidates in the United States, the European Union or other jurisdictions, if ever. Further, approvals by one regulatory agency may not be indicative of what other regulatory agencies may require for approval.
During the regulatory review process, we will need to identify success criteria and endpoints such that the FDA, the EMA or other regulatory authorities will be able to determine the clinical efficacy and safety profile of any product candidates we may develop. Because our initial focus is to identify and develop product candidates to treat or prevent diseases in which there is little clinical experience using new technologies, there is heightened risk that the FDA, the EMA or other regulatory authorities may not consider the clinical trial endpoints that we propose to provide clinically meaningful results. In addition, the resulting clinical data and results may be difficult to analyze. Even if the FDA determines that our success criteria is sufficiently validated and clinically meaningful, we may not achieve the prespecified endpoints to a sufficient degree of statistical significance.
This may be a particularly significant risk for many of the genetically defined diseases for which we plan to develop product candidates because many of these diseases have small patient populations, and designing and executing a rigorous clinical trial with appropriate statistical power is more difficult than with diseases that have larger patient populations. Further, even if we do achieve the prespecified criteria, the results may be unpredictable or inconsistent with the results of the non-primary endpoints or other relevant data. The FDA also weighs the benefits of a product against its risks, and the FDA may view the efficacy results in the context of safety as not being supportive of regulatory approval. The EMA and other regulatory authorities may make similar comments with respect to these endpoints and data. Any product candidate we may develop will be based on a novel technology that makes it difficult to predict the time and cost of development and of subsequently obtaining regulatory approval.
We and our collaboration partners have conducted and intend to conduct additional clinical trials for selected product candidates at sites outside the United States, and the FDA may not accept data from trials conducted in such locations or may require additional U.S.-based trials.
We and our collaboration partners have conducted, currently are conducting and intend in the future to conduct, clinical trials outside the United States, particularly in the European Union where we are headquartered. In addition, CureVac and its partner GSK are running the Phase 2 study with CV0601 (monovalent mRNA vaccine candidate encoding the Omicron BA.4-5 variant) and CV0701 (encoding the Omicron BA.4-5 variant as well as the original SARS-CoV-2 virus) in Australia.
Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of this data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be conducted by qualified investigators in accordance with GCPs, and the FDA must be able to validate the trial data through an on-site inspection, if necessary. Generally, the patient population for any clinical trial conducted outside of the United States must be representative of the population for which we intend to seek approval in the United States. There can be no assurance that the FDA will accept data from trials conducted outside of the United States. If the FDA does not accept the data from any clinical trials that we or our collaboration partners conduct outside the United States, it would likely result in the need for additional clinical trials, which would be costly and time-consuming and delay or permanently halt our ability to develop and market these or other product candidates in the United States. In other jurisdictions, for instance, in Japan, there is a similar risk regarding the acceptability of clinical trial data conducted outside of that jurisdiction.
In addition, there are risks inherent in conducting clinical trials in multiple jurisdictions, inside and outside of the United States, such as:
| ● | regulatory and administrative requirements of the jurisdiction where the trial is conducted that could burden or limit our ability to conduct our clinical trials; |
| ● | foreign exchange fluctuations; |
25
| ● | manufacturing, customs, shipment and storage requirements; |
| ● | cultural differences in medical practice and clinical research; and |
| ● | the risk that the patient populations in such trials are not considered representative as compared to the patient population in the target markets where approval is being sought. |
If any of our product candidates receive regulatory approval, the approved products may not achieve broad market acceptance among physicians, patients, the medical community and third-party payors, in which case revenue generated from their sales would be limited.
The commercial success of our product candidates will depend upon their acceptance among physicians, patients and the medical community. The degree of market acceptance of our product candidates will depend on a number of factors, including:
| ● | limitations or warnings contained in the approved labeling for a product candidate; |
| ● | changes in the standard of care for the targeted indications for any of our product candidates; |
| ● | limitations in the approved clinical indications for our product candidates; |
| ● | demonstrated clinical safety and efficacy compared to other products; |
| ● | lack of significant adverse side effects; |
| ● | sales, marketing and distribution support; |
| ● | availability of coverage and extent of reimbursement from managed care plans and other third-party payors; |
| ● | timing of market introduction and perceived effectiveness of competitive products; |
| ● | the degree of cost-effectiveness of our product candidates; |
| ● | availability of alternative therapies at similar or lower cost, including generic and over-the-counter products; |
| ● | whether the product is designated under physician treatment guidelines as a first-line therapy or as a second-or third-line therapy for particular diseases; |
| ● | whether the product can be used effectively with other therapies to achieve higher response rates; |
| ● | adverse publicity about our product candidates or favorable publicity about competitive products; |
| ● | convenience and ease of administration of our products; and |
| ● | potential product liability claims. |
If any of our product candidates are approved, but do not achieve an adequate level of acceptance by physicians, patients and the medical community, we may not generate sufficient revenue from these products, and we may not become or remain profitable. In addition, efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be successful.
26
Our product candidates for which we may seek approval as biologic products may face competition sooner than anticipated.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, the Affordable Care Act, signed into law on March 23, 2010, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or the BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of their product.
To the extent any of our product candidates approved as a biological product under a BLA qualifies for a 12-year period of exclusivity, for which we make no assurances, there is a risk that such exclusivity could be shortened due to congressional action or otherwise, or that the FDA will not consider our product candidates to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. Moreover, the extent to which a biosimilar, once approved, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for nonbiological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
If any approved products are subject to biosimilar competition sooner than we expect, we will face significant pricing pressure and our commercial opportunity will be limited.
Disruptions at the FDA and other government agencies caused by inflation, funding shortages, labor shortages or global health concerns could hinder their ability to hire, retain or deploy key leadership and other personnel, or otherwise prevent new or modified products from being developed, or approved or commercialized in a timely manner or at all, which could negatively impact our business.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, inflation, labor shortages, statutory, regulatory, and policy changes, the FDA’s ability to hire and retain key personnel and accept the payment of user fees, and other events that may otherwise affect the FDA’s ability to perform routine functions. Average review times at the FDA have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable. Disruptions at the FDA and other agencies may also slow the time necessary for our product candidates to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities.
The market opportunities for our product candidates may be smaller than we believe, or we may be unable to successfully identify clinical trial participants.
Our estimates of addressable patient populations are based on our beliefs, and have been derived from a variety of sources, including scientific literature, surveys of clinics, patient foundations or market research, and may prove to be incorrect. Further, new studies may change the estimated incidence or prevalence of these diseases. The number of trial participants may be lower than expected and potential clinical trial participants or patients may not be otherwise amenable to treatment with our product candidates or any products, or new clinical trial participants or patients may become increasingly difficult to identify or gain access to. Even if we obtain significant market share for our product candidates, if approved, because the potential target populations are small, these medicines may never be profitable.
27
If we are not successful in discovering, developing and commercializing additional products, our ability to expand our business and achieve our strategic objectives would be impaired.
An element of our strategy is to discover, develop and commercialize products beyond our current product candidates to treat various conditions and in a variety of therapeutic areas. We intend to do so by investing in our drug discovery efforts, exploring potential strategic alliances for the development of new products and in-licensing technologies. Identifying new product candidates requires substantial technical, financial and human resources. We may fail to identify promising candidates or to successfully develop and commercialize products for many reasons, which would impair our potential for growth.
We could be adversely affected by outbreaks of epidemic, pandemic or other contagious diseases.
In the event of a future epidemic or pandemic, our clinical trials could be paused or delayed due to restrictions (such as quarantines or travel limitations) or reprioritization of resources. Travel limitations could also create challenges and potential delays in our development and production activities, increasing the expense and timelines for producing our products and product candidates.
We utilize third parties to, among other things, manufacture raw materials, components, parts and consumables. If these third parties were to experience delays or disruptions in providing their services in response to an epidemic or pandemic, our supply chain could be disrupted, limiting our ability to manufacture product candidates for our clinical trials, as well as negatively impacting our research and development operations. Such delays or disruptions could adversely impact our strategic collaborators’ ability to fulfill their obligations, which could affect the clinical development or regulatory approvals of product candidates under joint development.
In addition, during a global health crisis, one or more government entities could take actions (such as via the Defense Production Act in the U.S.) that diminish our rights or economic opportunities with respect to our potential products. Our third-party service providers could be impacted by government-imposed restrictions on services they might otherwise offer. Any such action could cause us to experience delays in the development, production, distribution or export of our product candidates and increased expenses.
The increasing use of social media platforms presents risks and challenges.
Social media is increasingly being used to communicate about our research, product candidates, commercial products and the diseases our product candidates are designed to treat. Social media practices in the biopharmaceutical industry continue to evolve and regulations relating to such use are not always clear. This uncertainty creates risk of noncompliance with regulations applicable to our business, resulting in potential regulatory actions against us. For example, subjects may use social media channels to comment on their experience in an ongoing blinded clinical trial or to report an alleged adverse event. When such disclosures occur, we may fail to monitor and comply with applicable adverse event reporting obligations or we may be unable to defend our business in the face of political and market pressures generated by social media due to restrictions on what we may say about our product candidates. There is also a risk of inappropriate disclosure of sensitive information or negative or inaccurate posts or comments about us on any social networking website. If any of these events were to occur or we otherwise fail to comply with applicable regulations, we could incur liability, face regulatory actions or incur other harm to our business.
Risks Related to the Manufacturing of Our Product Candidates
The manufacture of mRNA-based medicines is complex and manufacturers often encounter difficulties in production, especially in the field of biologics. If we or any of our third-party manufacturers encounter difficulties, our ability to provide product candidates for clinical trials or products, if approved, to patients or future customers could be delayed or halted.
The manufacture of mRNA-based medicines is complex and requires significant expertise and capital investment, including the development of advanced manufacturing techniques and analytics. We and our third-party manufacturers must comply with cGMP, regulations and guidelines for the manufacturing of our product candidates used in preclinical studies and clinical trials and, if approved, marketed products. Manufacturers of biotechnology products often encounter difficulties in production, particularly in scaling up and validating initial production. Large-scale mRNA vaccine production requires a high level of (i) equipment to build and run new facilities and (ii) raw materials to produce mRNA and to formulate the drug substance in the required volumes.
Furthermore, if microbial, viral or other contaminations are discovered in our product candidates or in the manufacturing facilities where our product candidates are made, such manufacturing facilities may be closed for an extended period of time to investigate and remedy the contamination. Shortages of raw materials may also extend the period of time required to develop our product candidates.
28
Manufacturing these products requires facilities specifically designed for and validated for this purpose and sophisticated quality assurance and quality control procedures are necessary. Slight deviations anywhere in the manufacturing process, including filling, labeling, packaging, storage and shipping and quality control and testing, may result in lot failures, product recalls or spoilage. When changes are made to the manufacturing process, we may be required to provide preclinical and clinical data showing the comparable identity, strength, quality, purity or potency of the products before and after such changes. The use of biologically derived ingredients can also lead to allegations of harm, including infections or allergic reactions, or closure of product facilities due to possible contamination.
In addition, there are risks associated with large-scale manufacturing for clinical trials or commercial-scale including, among others, cost overruns, potential problems with process scale-up, process reproducibility, stability issues, compliance with good manufacturing practices, lot consistency and timely availability of raw materials. Even if we obtain marketing approval for any of our product candidates, there is no assurance that we or our manufacturers will be able to manufacture the approved product to specifications acceptable to the FDA or other comparable foreign regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential commercial launch of the product or to meet potential future demand. If we or our manufacturers are unable to produce sufficient quantities for clinical trials or for commercialization, our development and commercialization efforts would be impaired, which would have an adverse effect on our business, financial condition, results of operations and growth prospects.
We cannot assure you that any disruptions or other issues relating to the manufacture of any of our product candidates will not occur in the future. Any delay or interruption in the supply of clinical trial supplies could delay the completion of planned clinical trials, increase the costs associated with maintaining clinical trial programs and, depending upon the period of delay, require us to commence new clinical trials at additional expense or terminate clinical trials completely. Any adverse developments affecting clinical or commercial manufacturing of our product candidates or products may result in shipment delays, inventory shortages, lot failures, product withdrawals or recalls or other interruptions in the supply of our product candidates or products. We may also have to take inventory write-offs and incur other charges and expenses for product candidates or products that fail to meet specifications, undertake costly remediation efforts or seek more costly manufacturing alternatives. Accordingly, failures or difficulties faced at any level of our supply chain could delay or impede the development and commercialization of any of our product candidates or products and could have an adverse effect on our business, prospects, financial condition and results of operations.
We and our third-party manufacturers and suppliers could be subject to liabilities, fines, penalties or other sanctions under federal, state, local and foreign environmental, health and safety laws and regulations if we or they fail to comply with such laws or regulations or otherwise incur costs that could have a material adverse effect on our business.
We manufacture and produce mRNA-based active ingredients for our product pipeline. We also currently rely on and expect to continue to rely on third parties for the manufacturing and supply of active pharmaceutical ingredients, or API, and drug products of our product candidates. We and these third parties are subject to various federal, state, local and foreign environmental, health and safety laws and regulations, including those governing laboratory procedures and the generation, handling, labeling, transportation, use, manufacture, storage, treatment and disposal of hazardous materials and wastes and worker health and safety. We do not have control over a manufacturer’s or supplier’s compliance with environmental, health and safety laws and regulations. Liabilities they incur pursuant to these laws and regulations could result in significant costs or, in certain circumstances, an interruption in operations, any of which could adversely affect our business and financial condition.
With respect to any hazardous materials or waste which we are currently, or in the future will be, generating, handling, transporting, using, manufacturing, storing, treating or disposing of, we cannot eliminate the risk of contamination or injury from these materials or waste, including at third-party disposal sites. In the event of such contamination or injury, we could be held liable for any resulting damages and liability. We also could be subject to significant civil or criminal fines and penalties, cessation of operations, investigation or remedial costs or other sanctions for failure to comply with applicable environmental, health and safety laws. In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts or otherwise have a material adverse effect on our business.
At the start of the COVID-19 pandemic, we had entered in contractual engagements with third-party manufacturer to have access to additional capacity. The wind-down of these contracts entail risks of financial penalties and risk of litigation.
29
Undetected errors or defects in our production, misconduct from our business partners and negative media reports could harm our reputation or expose us to product liability claims.
Defects in the cGMP materials we produce may damage the third parties’ businesses we work with and could harm their and our reputation. If that occurs, we may incur significant costs, the attention of our key personnel could be diverted, or other significant problems may arise. We may also be subject to warranty and liability claims for damages related to errors or defects in products made with our cGMP materials. In addition, if we do not meet industry or quality standards, if applicable, such products may be subject to recall. A material liability claim, recall or other occurrence that harms our reputation or decreases market acceptance of such products could harm our business and operating results. In addition, misconduct by our business partners or unfavorable publicity regarding our industry, our business partners or us can lead to reputational damage that adversely affects our business, financial condition and results of operations.
We are subject to operational risks associated with the physical and digital infrastructure at our manufacturing facilities and those of our external service providers.
Our manufacturing facilities incorporate a significant level of automation of equipment with integration of several digital systems to improve efficiency of operations. The digitization of our facilities exposes us to the risk of process equipment malfunctions. These risks include potential system failures or shutdowns due to internal or external factors including, design issues, system compatibility or potential cybersecurity compromises, incidents or breaches. Upgrades or changes to our systems, infrastructure or the software that we implement, use, or upon which our business relies, may result in the introduction of new cybersecurity vulnerabilities and risks.
Our facilities and infrastructure or those of our contract manufacturers or other third-party providers may also be subject to attacks or acts of sabotage by outside actors, contractors or employees. Any disruption in our or our contract manufacturers’ manufacturing capabilities could cause delays in production capacity for our drug substances or product candidates or a shutdown of facilities, could impose additional costs, cause us to fail to meet certain product candidate volume or delivery timing obligations, or may require us to identify, qualify and establish an alternative manufacturing site, the occurrence of which could adversely affect our business, financial condition, results of operations, and prospects.
Risks Related to Our Reliance on Collaborators and Other Third Parties
We rely on third parties to conduct our nonclinical and clinical trials and perform other tasks for us. If these third parties do not successfully carry out their contractual duties, meet expected deadlines, or comply with regulatory requirements, we may not be able to obtain regulatory approval for or commercialize our product candidates and our business could be substantially harmed.
We have relied upon and plan to continue to rely upon third-party CROs to monitor and manage data for our ongoing nonclinical and clinical programs. We rely on these parties for execution of our nonclinical and clinical studies and control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that each of our trials is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities. We and our CROs and other vendors are required to comply with cGMP, GCP, Good Laboratory Practice, or GLP, and other regulations and guidelines enforced by the FDA, the Competent Authorities of the Member States of the European Union and comparable foreign regulatory authorities for all of our product candidates in nonclinical and clinical development. Regulatory authorities enforce these regulations through periodic inspections of study sponsors, principal investigators, trial sites and other contractors. If we or any of our CROs or vendors fail to comply with applicable regulations, the data generated in our nonclinical and clinical trials may be deemed unreliable and the EMA, FDA, or other regulatory authorities may require us to perform additional nonclinical and clinical trials before approving our marketing applications. In addition, even if, for example, the EMA finds our data generated in our nonclinical and clinical trials reliable for approving a marketing application, there is no assurance that other regulatory authorities like the FDA will find such data reliable and sufficient for approving a similar market application. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that all of our clinical trials comply with cGCP regulations. In addition, our clinical trials must be conducted with product produced under cGMP regulations. Our failure to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process.
30
If any of our relationships with these third-party CROs terminates, we may not be able to enter into arrangements with alternative CROs or do so on commercially reasonable terms. In addition, our CROs are not our employees, and except for remedies available to us under our agreements with such CROs, we cannot control whether or not they devote sufficient time and resources to our ongoing nonclinical and clinical programs. If CROs do not successfully carry out their contractual duties or obligations, meet expected deadlines, conduct our studies in accordance with regulatory requirements or our stated study plans and protocols, if they need to be replaced or if the quality or accuracy of the data they obtain is compromised due to the failure to adhere to our protocols, regulatory requirements, or for other reasons, our clinical trials may be extended, delayed, or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. CROs may also generate higher costs than anticipated. As a result, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase, and our ability to generate revenue could be delayed.
Switching or adding additional CROs involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays occur, which can materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter similar challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition, and prospects.
If we or any third-party manufacturer of our product candidates is unable to increase the scale of production of our product candidates, and/or increase the product yield of manufacturing, then our costs to manufacture the product may increase and commercialization may be delayed.
In order to produce sufficient quantities to meet the demand for clinical trials and, if approved, subsequent commercialization of our product candidates in our pipeline or that we may develop, our third-party manufacturers will be required to increase their production and optimize their manufacturing processes while maintaining the quality of the product. The transition to larger-scale and more robust production could prove difficult or costly. Further, any claims in our manufacturing process as a result of scaling up or optimization of the manufacturing, supply and fill and finish process may result in the need to obtain regulatory approvals. If we or our third-party manufacturers are not able to optimize the manufacturing process to increase the product yield for our product candidates or the cGMP production requirement for clinical studies, or if we or our third-party manufacturers are unable to produce increased amounts of our product candidates while maintaining the quality of the product or generally unable to produce the right quality, then we may not be able to meet the demands of clinical trials or market demands, which could decrease our ability to generate profits. Difficulty in achieving commercial scale-up production or production optimization or the need for additional regulatory approvals as a result could have a material adverse impact on our business and results of operations.
Risks Related to Our Intellectual Property Rights
If we are unable to obtain, maintain and enforce intellectual property protection for our products or product candidates, or if the scope of our intellectual property protection is not sufficiently broad, our ability to commercialize our product candidates successfully and to compete effectively may be materially adversely affected.
Our success depends on our ability to obtain and maintain patent and other intellectual property protection in the United States and other countries with respect to our current and future proprietary product candidates. We rely upon a combination of patents, trade secret protection and confidentiality agreements to protect the intellectual property related to our technology, manufacturing processes, products and product candidates. We and our collaborators have primarily sought to protect our proprietary positions by filing patent applications in the United States and abroad related to our proprietary technology, manufacturing processes, and product candidates that are important to our business. Despite our efforts to protect our proprietary rights, unauthorized parties may be able to obtain and use information that we regard as proprietary.
31
The patent prosecution process is expensive and time-consuming, and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner or in all jurisdictions where protection may be commercially advantageous. It is also possible that we may fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. In addition, we or our collaborators, may only pursue, obtain or maintain patent protection in a limited number of countries. There is no assurance that all potentially relevant prior art relating to our patents and patent applications has been found. We may be unaware of prior art that could be used to invalidate or narrow the scope of an issued patent or prevent our pending patent applications from issuing as patents. Because patent applications in the United States, Europe and many other non-U.S. jurisdictions are typically not published until 18 months after filing, or in some cases not at all, and because publications of discoveries in scientific literature lag behind actual discoveries, we cannot be certain that we or our licensors were the first to make the inventions claimed in any of our owned or any in-licensed issued patents or pending patent applications, or that we or our licensors were the first to file for protection of the inventions set forth in our patents or patent applications. As a result, we may not be able to obtain or maintain protection for certain inventions. Even if patents do successfully issue, our owned or in-licensed patents may not adequately protect our intellectual property, provide exclusivity for our products or product candidates, prevent others from designing around our claims or otherwise provide us with a competitive advantage. We cannot offer any assurances about which, if any, patents will issue, the breadth of any such patents or whether any issued patents will be found invalid or unenforceable or will be threatened by third parties. In addition, third parties may challenge the validity, enforceability, ownership, inventorship or scope of any of our patents. Any successful challenge to any of our patents could deprive us of rights necessary for the successful commercialization of any product candidate that we may develop and could impair or eliminate our ability to collect future revenues and royalties with respect to such products or product candidates. If any of our patent applications with respect to our product candidates fail to issue as patents, if their breadth or strength of protection is narrowed or threatened, or if they fail to provide meaningful exclusivity or competitive position, it could dissuade companies from collaborating with us or otherwise adversely affect our competitive position.
The patent position of pharmaceutical companies is generally uncertain because it involves complex legal, scientific and factual considerations for which legal principles remain unsolved. Further, mRNA medicines are a relatively new scientific field and, as the field continues to mature, patent applications are being processed by national patent offices globally. There is uncertainty about which patents will issue, and, if they do, as to when, to whom and with what claims. The standards applied by the United States Patent and Trademark Office, or the USPTO, and foreign patent offices in granting patents are not always applied uniformly or predictably, and can change. Additionally, the laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property rights, particularly those relating to biotechnology, which could make it difficult for us to stop the infringement, misappropriation, or other violation of our patents or other intellectual property, including the unauthorized reproduction of our manufacturing or other know-how or the marketing of competing products in violation of our intellectual property rights generally. Any of these outcomes could impair our ability to prevent competition from third parties, which may have a material adverse effect on our business, financial condition, results of operations and prospects.
Further, the existence of issued patents does not guarantee our right to practice the patented technology or commercialize the patented product candidate. Third parties may have or obtain rights to patents which they may use to prevent or attempt to prevent us from practicing our patented technology or commercializing any of our patented product candidates. If any of these other parties are successful in obtaining valid and enforceable patents, and establishing our infringement of those patents, we could be prevented from selling our products unless we were able to obtain a license under such third-party patents, which may not be available on commercially reasonable terms or at all. In addition, third parties may seek approval to market their own products similar to or otherwise competitive with our products. In these circumstances, we may need to defend or assert our patents, including by filing lawsuits alleging patent infringement. In any of these types of proceedings, a court or agency of competent jurisdiction may find our patents invalid or unenforceable. Our competitors and other third parties may also be able to circumvent our patents by developing similar or alternative product candidates in a non-infringing manner. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
32
In addition, competitors may use our technologies in jurisdictions where we have not obtained or are unable to adequately enforce patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States and Europe. These products may compete with our products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing with us. Proceedings to enforce our patent rights, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or held unenforceable, or interpreted narrowly and our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop, acquire or license.
Our owned and in-licensed patents may be subject to a reservation of rights by one or more third parties. For example, the research resulting in certain of our patents and technology, including patents and technology relating to our yellow fever product candidate, was funded in part by the U.S. government. As a result, the U.S. government has certain rights to such patent rights and technology, which include march-in rights. When new technologies are developed with government funding, in order to secure ownership of such patent rights, the recipient of such funding is required to comply with certain government regulations, including timely disclosing the inventions claimed in such patent rights to the U.S. government and timely electing title to such inventions. Additionally, the U.S. government generally obtains certain rights in any resulting patents, including a nonexclusive license authorizing the government to use the invention or to have others use the invention on its behalf. Accordingly, we have granted the U.S. government a nonexclusive, nontransferable, irrevocable, paid-up license to practice or have practiced for or on behalf of the United States, the inventions described in the patents and patent applications relating to our technology or one or more of our product candidates. If the U.S. government decides to exercise these rights, it is not required to engage us as its contractor in connection with doing so. The government’s rights may also permit it to disclose our confidential information to third parties and to exercise march-in rights to use or allow third parties to use such government-funded technology. The government can exercise its march-in rights if it determines that action is necessary because we fail to achieve practical application of the government-funded technology, or because action is necessary to alleviate health or safety needs, to meet requirements of federal regulations, or to give preference to U.S. industry. In addition, our rights in such inventions may be subject to certain requirements to manufacture products embodying such inventions in the United States. If we fail to comply with those requirements, we could lose our ownership of, or other rights to, any patents subject to such regulations.
In Germany, the German federal government, and the Federal Ministry of Health and downstream authorities, in the event of a national epidemic, have the right to order the use of our owned and in-licensed patents in the interest of the public welfare or the security of the Federal Republic. The German government may issue such an order with respect to our owned or in-licensed patents and we may lose exclusivity with respect to the technologies and product candidates covered by such patents. We would be entitled to compensation in the event a use order is issued with respect to our owned or in-licensed patents; however, such compensation may be less than what we could otherwise receive and any such use order could have a material adverse effect on our competitive position, business, financial condition, results of operations and prospects.
In Russia, the United States and foreign government actions related to Russia’s invasion of Ukraine may limit or prevent filing, prosecution and maintenance of patent applications in Russia. Government actions may also prevent maintenance of issued patents in Russia. These actions could result in abandonment or lapse of our patents or patent applications in Russia, resulting in a partial or complete loss of patent rights in Russia. In addition, a decree was adopted by the Russian government in March 2022, allowing Russian companies and individuals to exploit, without consent or compensation, inventions owned by patentees that have citizenship or nationality in, are registered in, or have predominately primary place of business or profit-making activities in countries that Russia has deemed unfriendly. Consequently, we would not be able to prevent third parties from practicing our inventions in Russia or from selling or importing products made using our inventions in and into Russia. Accordingly, our competitive position may be impaired, and our business, financial condition, results of operations and prospects may be materially adversely affected.
In addition, it is uncertain whether the World Trade Organization, or the WTO, will waive certain intellectual property protections now or in the future on certain technologies related to COVID-19 or other future pandemics. We cannot be certain that any of our current or future product candidates or technologies would not be subject to an intellectual property waiver by the WTO. We also cannot be certain that any of our current or future intellectual property rights, whether patents, trade secrets, or other confidential information would be eliminated, narrowed, or weakened by such a waiver. Given the uncertain future actions by the WTO and other countries and jurisdictions around the world, including the United States, it is unpredictable how our current or future intellectual property rights or how our current or future business would be impacted.
33
Additionally, the research resulting in certain of our patents and technology was funded in part by the German Ministry of Education and Research, or the BMBF. Results of such government-funded research projects must, subject to certain conditions, be made available free of charge for academic research and teaching in Germany and must be published in half-yearly interim reports and a final report following completion of the funded work. Information relating to intellectual property generated, commercial expectations, scientific chances of success and next steps and certain additional information must be disclosed to the German government and must be disclosed to third parties for academic research and teaching upon request under a written confidentiality agreement. The BMBF additionally has, in the case of a special public interest, a nonexclusive and transferable right to use intellectual property generated as part of the funded work. Contracts with third parties relating the exploitation of the results of the funded work must be disclosed to the BMBF and any such contracts with parties outside of the European Union require the prior consent of the BMBF to the extent they deviate from an exploitation plan previously approved by the BMBF. Additionally, if we fail to use or commercialize the results of the funded work we may be required to grant third parties licenses to use such results. In certain scenarios, including if we come under the decisive influence of foreign investors, the funded results are exclusively or predominantly used outside Germany without the prior consent of the BMBF or if we are in breach of our obligations under the grant, the grant funding, including funding already received, can be revoked.
Furthermore, certain of our patents and technology, including patents and technology relating to our rotavirus, malaria, Lassa virus and SARS-CoV-2 projects, were funded in part by grants from nonprofit third parties, including the Bill & Melinda Gates Foundation and CEPI. We are required to fulfill certain contractual obligations with respect to products created using such grant funding, including making certain products available at an affordable price in a list of clearly defined low- and lower-middle income countries and ensuring that certain products are available in geographic regions where there has been an outbreak of an infectious disease at certain reduced economic rates. See “Item 4. Information on the Company — B. Business Overview — Collaborations.”
Furthermore, patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after its effective filing date. Various extensions may be available; however, the life of a patent, and the protection it affords, is limited. Given the amount of time required for the development, testing, regulatory review and approval of new product candidates, our patents protecting such candidates might expire before or shortly after such candidates are commercialized. If we encounter delays in obtaining regulatory approvals, the period of time during which we could market a product under patent protection could be further reduced. Even if patents covering our product candidates are obtained, once such patents expire, we may be vulnerable to competition from similar or biosimilar products. The launch of a similar or biosimilar version of one of our products would likely result in an immediate and substantial reduction in the demand for our product, which could have a material adverse effect on our business, financial condition, results of operations and prospects.
If we fail to comply with our obligations under any license, collaboration or other intellectual property agreements, disagree over contract interpretation, or otherwise experience disruptions to our business relationships with our collaborators or licensors, we could lose intellectual property rights that are necessary to our business.
We rely, in part, on license, collaboration and other intellectual property agreements. These may not provide exclusive rights to use such intellectual property and technology in all relevant fields of use and in all territories in which we may wish to develop or commercialize our product candidates in the future.
In addition, our existing licenses and collaboration agreements, including our agreements with Genmab, Acuitas, GSK, the Bill & Melinda Gates Foundation, CRISPR Therapeutics, CEPI and myNEO, impose, and any future licenses, collaborations or other intellectual property agreements we enter into are likely to impose, various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement or other obligations on us. Our licenses and collaboration agreements, including our agreement with Genmab, impose, and any future agreement we enter into may also impose, restrictions on our ability to license certain of our intellectual property to third parties or to develop or commercialize certain product candidates or technologies. In spite of our best efforts, our licensors, licensees and collaborators may conclude that we have breached our obligations under our agreements, or that we have used the intellectual property licensed to us in an unauthorized manner, in which case, we may be required to pay damages and the licensor, licensee or collaborator may have the right to terminate the agreement. Any of the foregoing could result in us being unable to develop, manufacture and sell products that are covered by the licensed technology, enable a competitor to gain access to the licensed technology or disrupt our right to milestone or royalty payments. We might not have the necessary rights or the financial resources to develop, manufacture or market our current or future product candidates without the rights granted under our licenses, and the loss of sales or potential sales in such product candidates could have a material adverse effect on our business, financial condition, results of operations and prospects.
34
Disputes have arisen and may in the future arise regarding intellectual property subject to licensing, collaboration or other intellectual property agreements, including:
| ● | the scope of rights granted under the license agreement and other interpretation-related issues; |
| ● | the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the license agreement; |
| ● | the sublicensing of patent and other rights under our collaborative development relationships; |
| ● | our diligence obligations under the license agreement and what activities satisfy those diligence obligations; |
| ● | our financial obligations under the license agreement; |
| ● | the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and |
| ● | the priority of invention of patented technology. |
In addition, the agreements under which we currently license intellectual property or technology to or from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or expand the rights of our licensors or partners to the relevant intellectual property or technology, including their ability to use or license such intellectual property or technology to third parties. In addition, such resolution may also increase what we believe to be our financial or other obligations under the relevant agreement. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects. Moreover, if disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
In some circumstances, we may not have the right to control the preparation, filing, prosecution, maintenance, enforcement, and defense of patents and patent applications covering the technology that we license from third parties. We cannot be certain that these patents and applications will be prepared, filed, prosecuted, maintained, enforced, and defended in a manner consistent with the best interests of our business. If our licensors fail to prosecute, maintain, enforce, and defend such intellectual property, or lose rights to such intellectual property, the rights we have licensed and our exclusivity may be reduced or eliminated and our right to develop and commercialize any of our products that are subject to such licensed rights could be adversely affected.
Moreover, our rights to our in-licensed patents and patent applications may depend, in part, on inter-institutional or other operating agreements between the joint owners of such in-licensed patents and patent applications. If one or more of such joint owners breaches such inter-institutional or operating agreements, our rights to such in-licensed patents and patent applications may be adversely affected. In addition, while we cannot currently determine the amount of the royalty obligations we would be required to pay on sales of future products, if any, the amounts may be significant. The amount of our future royalty obligations will depend on the technology and intellectual property we use in products that we successfully develop and commercialize, if any. Therefore, even if we successfully develop and commercialize products, we may be unable to achieve or maintain profitability. In addition, the development of certain of our product candidates is funded by grants that impose certain pricing limitations on such product candidates and limit our ability to commercialize such product candidates and to achieve or maintain profitability. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
If we are unable to successfully obtain rights to required third-party intellectual property rights or maintain the existing intellectual property rights we have on reasonable terms or at all, we may have to abandon development of the relevant program or product candidate and our business, financial condition, results of operations and prospects could suffer.
35
We are and may in the future become involved in lawsuits to protect or enforce our patents, which could be expensive, time-consuming and unsuccessful and could result in a court or administrative body finding our patents to be invalid or unenforceable.
Even if the patent applications we own or license are issued, third parties may infringe our patents. To counter infringement, we have been and may in the future be required to file infringement claims, which can be expensive and time-consuming. If we initiate legal proceedings against a third-party to enforce a patent covering any of our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including novelty, nonobviousness (or inventive step), written description or enablement. In addition, patent validity challenges may, under certain circumstances, be based upon nonstatutory obviousness-type double patenting, which, if successful, could result in a finding that the claims are invalid for obviousness-type double patenting or the loss of patent term if a terminal disclaimer is filed to obviate a finding of obviousness-type double patenting. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld information material to patentability from the USPTO, or made a misleading statement, during prosecution. In an infringement proceeding, a court may decide that one or more of our patents is not valid, is unenforceable or is not infringed, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. Third parties also may raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include reexamination, post-grant review, inter partes review, interference proceedings, derivation proceedings, and equivalent proceedings in foreign jurisdictions (e.g., opposition proceedings, revocation actions, nullity actions or cancellation actions). Such proceedings could result in the revocation or cancellation of, or amendment to, our patents or other intellectual property rights such as utility models, in such a way that they no longer cover our product candidates or provide any competitive advantage.
Litigation is ongoing over the underlying technology to mRNA medicines between many mRNA market participants. It is likely that there will continue to be significant litigation and patent office proceedings in various patent offices relating to patent rights in the mRNA field. For example, one of our manufacturing related U.S. patents was invalidated in an inter partes review proceeding and certain of our European patents relating to RNA-based adjuvants/immunostimulants, RNA-coded antibodies, mRNA vaccination of the elderly, intratumoral (m)RNA treatments, polyvalent mRNA production, optimized in vitro transcription, and mRNA vaccination in combination with an anti-PD1 or anti-PD-L1 antibody have been revoked in European opposition proceedings. Further European patents have been amended after opposition proceedings. For example, European patents related to an mRNA injection solution, mRNA vaccination of newborns and infants, combination of mRNA-based vaccination and agonistic OX40 antibodies and method of RNA analysis have been amended. Further European patents have been maintained after opposition proceedings. One of these patents relates to an mRNA injection solution and the other relates to sequence optimization of a coding sequence. Some of these decisions are currently on appeal and continuation or divisional applications of certain of the maintained, revoked and amended patents have been filed and are currently under examination, although there can be no assurance that any such appeal will be successful or that any such patent applications will issue as patents that provide us with any competitive advantage. Additionally, several of our European and Australian patents relating to intratumoral treatment, mRNA vaccination in combination with an anti-PD1/PD-L1 antibody, prime-boost regimens, lyophilization of RNA, spray-drying of RNA, spray-freeze-drying of RNA, an mRNA comprising a split poly(A) sequence, mRNA production comprising tangential flow filtration, and an improved method for plasmid production for in vitro transcription of mRNA are currently subject to opposition proceedings. The outcome following legal assertions of invalidity and unenforceability is unpredictable. If a third party were to prevail on a legal assertion of invalidity or unenforceability, we could lose part or all of the patent protection on one or more of our product candidates, which could result in our competitors and other third parties using our technology to compete with us. Such a loss of patent protection could have a material adverse impact on our business.
Interference proceedings, or other similar enforcement and revocation proceedings, provoked by third parties or brought by us may be necessary to determine the priority of inventions with respect to our patents or patent applications. We may not be able to prevent, alone or with our licensors, infringement, misappropriation or other violation of our intellectual property rights, particularly in countries where the laws may not protect those rights as fully as in the United States.
In June 2022, we filed a lawsuit against BioNTech SE and its wholly owned subsidiaries, BioNTech Manufacturing GmbH and BioNTech Manufacturing Marburg GmbH (collectively, “BioNTech”), in the Düsseldorf Regional Court, for infringement of one European patent, EP1857122B1, (“EP’122 Patent”), and three utility models, DE202015009961U1, DE202015009974U1, and DE202021003575U1, as a result of the manufacture, use and sale of Comirnaty, BioNTech and Pfizer’s mRNA COVID-19 vaccine.
36
In August 2022, we added European Patent EP3708668B1 (“EP’688 Patent”) to the German lawsuit. In July 2023, we added European Patent EP4023755B1 (“EP’755 Patent”) and two further utility models, DE202021004123U1 and DE 202021004130U1, to the German lawsuit.
In September 2022, BioNTech filed a nullity action in the Federal Patent Court of Germany seeking a declaration that the EP’122 Patent is invalid. In December 2023, the Federal Patent Court in Germany revoked this patent in the first instance. The formal reasons for the decision of the court have not yet been provided. CureVac intends to appeal this decision before the German Federal Court of Justice. Subsequently, the Dusseldorf Court decided to suspend the infringement proceedings until a final decision on validity from the German Federal Court of Justice is made.
In November 2022, BioNTech filed cancellation actions seeking the cancellation of three German utility models DE202015009961U1, DE202015009974U1, and DE202021003575U1 in the German Patent and Trademark Office, and we are defending these actions.In September 2023 the Dusseldorf Court decided to suspend the infringement proceedings for these three utility models until the cancellation division of the German Patent and Trademark Office (DPMA) has reached a decision in the first instance.
In April 2023, BioNTech and Pfizer, among others, filed oppositions against the EP’668 Patent.
In November 2023, BioNTech filed cancellation actions against the two utility models, DE202021004123U1 and DE 202021004130U1 in the German Patent and Trademark Office.
In December 2023 and January 2024, BioNTech and Pfizer, among others, filed oppositions against the EP’755 Patent.
In September 2022, BioNTech and Pfizer filed a declaration of non-infringement and revocation action against the EP’122 Patent and the EP’668 Patent in the Patents Court of the Business and Property Courts of England and Wales. In October 2022, we counterclaimed seeking an order that the EP ‘122 Patent and EP ‘688 Patent are infringed and are valid (the “UK Proceedings”). In February 2024 we added the EP’755 Patent to our infringement claim. The trial is currently scheduled for July 2024.
In July 2022, BioNTech and Pfizer filed a complaint for a declaratory judgment in the U.S. District Court for the District of Massachusetts, seeking a judgment of non-infringement by Comirnaty of U.S. Patent Nos. 11,135,312, 11,149,278 and 11,241,493. Subsequently we filed a motion to have the case transferred to the Eastern District of Virginia, and such motion was granted in May 2023. Subsequently,we filed our counterclaim claiming infringement of ten U.S. patents, U.S. Patent No. 11,135,312; U.S. Patent No. 11,149,278; U.S. Patent No. 11,286,492; U.S. Patent No. 11,345,920; U.S. Patent No. 10,760,070; U.S. Patent No. 11,667,910; U.S. Patent No. 11,241,493; U.S. Patent No. 11,471,525; U.S. Patent No. 11,576,966; and U.S. Patent No. 11,596,686. The jury trial is currently scheduled for January 2025.
In November 2023, Acuitas filed a Motion to Intervene, a Motion to Sever and Stay and a Complaint for Declaratory Judgement of Inventorship to seek correction of inventorship regarding four United States patents, U.S. Patent No. 11,241,493; U.S. Patent No. 11,471,525; U.S. Patent No. 11,576,966; and U.S. Patent No. 11,596,686 which are part of the litigation against BioNTech and Pfizer. In April 2024, Acuitas' motion was granted, with the judge recommending to stay litigation of all ten patents before the District Court of the Eastern District of Virginia until the Acuitas claim is resolved. CureVac is preparing objections to this recommendation and anticipates a decision within the next two months.
In October 2023, Acuitas filed a Request of Arbitration to resolve a dispute between Acuitas and CureVac regarding the ownership and licensing rights with regard to the patent family WO2021/156267 which is part of the litigation against BioNTech and Pfizer in Germany and the United States. CureVac is defending itself against these claims. The possible outcome of this dispute cannot be predicted at this time.
All of such proceedings are currently pending. In the course of pursuing our case for infringement and defending against the challenges to our patent estate from Pfizer and BioNTech, should we ultimately not be successful, we will be liable for our own and may be liable for BioNTech and Pfizer’s legal costs in at least Germany and the UK.
An unfavorable outcome could also require us to cease using the related technology or attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms.
37
Our defense of litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees.
In many cases, the possibility of appeal exists for any party, and it may be years before final, unappealable rulings are made with respect to these patents in certain jurisdictions. The timing and outcome of these and other proceedings is uncertain and may adversely affect our business if we are not successful in defending the patentability and scope of our pending and issued patent claims. We cannot be certain that any patent will survive or that the claims will remain in the current form. Even if our rights are not directly challenged, disputes could lead to the weakening of our IP rights.
Additionally, an adverse outcome in a litigation or proceeding involving our patents could limit our ability to assert our patents against competitors, affect our ability to receive royalties or other licensing consideration from our licensees, and may curtail or preclude our ability to exclude third parties from making, using and selling similar or competitive products. Any of these occurrences could have a material adverse effect on our business, financial condition, results of operations and prospects.
If we are sued for infringing, misappropriating, or otherwise violating intellectual property rights of third parties, such litigation could be costly and time-consuming and could prevent or delay us from developing or commercializing our product candidates.
Our commercial success depends, in part, on our ability to develop, manufacture, market and sell our product candidates without infringing, misappropriating, or otherwise violating the intellectual property and other proprietary rights of third parties.
There is a substantial amount of intellectual property litigation in the biotechnology and pharmaceutical industries, and we may become party to, or threatened with, litigation or other adversarial proceedings regarding intellectual property rights of third parties with respect to our product candidates, including interference and post-grant proceedings before the USPTO. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the composition, formulation, use or manufacture of our product candidates. Because patent applications can take many years to issue, there may be currently pending patent applications that we may or may not be aware of which may later result in issued patents that our product candidates may be accused of infringing. Additionally, pending patent applications that have been published can, subject to certain limitations, be later amended in a manner that could cover our product candidates or the use of our product candidates. After issuance, the scope of patent claims remains subject to construction based on interpretation of the law, the written disclosure in a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending application may be incorrect. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. Accordingly, third parties may assert infringement claims against us based on intellectual property rights that exist now or arise in the future. The outcome of intellectual property litigation is subject to uncertainties that cannot be adequately quantified in advance. The pharmaceutical and biotechnology industries have produced a significant number of patents, and it may not always be clear to industry participants, including us, which patents cover various types of products or methods of use or manufacture. The scope of protection afforded by a patent is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our product candidates, products or methods either do not infringe the patent claims of the relevant patent or that the patent claims at issue are invalid or unenforceable, and we may not be able to do this. Proving invalidity is difficult. For example, in the United States, proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and the time and attention of our management and scientific personnel could be diverted in pursuing these proceedings, which could significantly harm our business and operating results. In addition, we may not have sufficient resources to bring these actions to a successful conclusion. Some claimants may have substantially greater resources than we do and may be able to sustain the costs of complex intellectual property litigation to a greater degree and for longer periods of time than we could. In addition, patent holding companies that focus solely on extracting royalties and settlements by enforcing patent rights may target us, especially as we gain greater visibility and market exposure as a public company.
Third parties have, and may in the future have, U.S. and non-U.S. issued patents and pending patent applications relating to compounds, methods of manufacturing compounds or methods of use for the treatment of the disease indications for which we are developing our product candidates that may cover our product candidates. For example, we are aware of certain third-party U.S. and non-U.S. issued patents and patent applications, including those of our competitors, that relate to mRNA production, mRNA optimization, modification of mRNA, LNP technology, RNA-based tumor vaccination, LNP formulation, LNP-based mRNA delivery to the eye, lung or liver, mRNA encoding gene-editing enzymes, RNA-encoded antibodies or antigens in LNPs and LNP-formulated RNA that may be construed to cover the LNP-formulated RNA technology used in our vaccines and protein and antibody therapies. We are also aware of certain third-party U.S. and non-U.S. patents and patent applications, including those of our competitors, that
38
relate to coronavirus vaccines, influenza virus vaccines, Respiratory Syncytial Virus (RSV) vaccines and treatments and vaccines against other infectious diseases and we expect such third parties to have filed additional patent applications, which have not yet been published, and to file additional patent applications in the future.
In the event that any of these patent rights were asserted against us, we believe that we have defenses against any such action, including that such patents would not be infringed by our product candidates and/or that such patents are not valid. However, if any such patent rights were to be asserted against us and our defenses to such assertion were unsuccessful, unless we obtain a license to such patents, we could be liable for damages, which could be significant and include treble damages and attorneys’ fees if we are found to willfully infringe such patents, and we could be precluded from commercializing any product candidates that were ultimately held to infringe such patents, any of which could have a material adverse effect on our business, financial condition, results of operations and prospects.
If we are required to obtain a license from any third-party in order to use the infringing technology and continue developing, manufacturing or marketing the infringing product candidate, we may not be able to obtain such required license on commercially reasonable terms or at all. In particular, any of our competitors that control intellectual property that we are found to infringe may be unwilling to provide us a license under any terms. Even if we were able to obtain a license, it could be nonexclusive, thereby giving our competitors access to the same technologies licensed to us; alternatively or additionally it could include terms that impede or destroy our ability to compete successfully in the commercial marketplace. In addition, we could be found liable for monetary damages, including treble damages and attorneys’ fees if we are found to have willfully infringed a patent. Further, if a patent infringement suit is brought against us or our third-party service providers and if we are unable to successfully obtain rights to required third-party intellectual property, we may be required to expend significant time and resources to redesign our product candidates, or to develop or license replacement technology, all of which may not be feasible on a technical or commercial basis, and may delay or require us to abandon our development, manufacturing or sales activities relating to our product candidates. A finding of infringement could prevent us from commercializing our product candidates or force us to cease some of our business operations, which could harm our business. Claims that we have misappropriated the confidential information or trade secrets of third parties could have a similar negative impact on our business. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
Intellectual property litigation and other proceedings could cause us to spend substantial resources and distract our personnel from their normal responsibilities.
Even if resolved in our favor, intellectual property litigation or other legal proceedings relating to our, our licensor’s or other third parties’ intellectual property claims may cause us to incur significant expenses and could distract our personnel from their normal responsibilities. Patent litigation and other proceedings may also absorb significant management time. If not resolved in our favor, litigation may require us to pay any portion of our opponents’ legal fees. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Our competitors or other third parties may be able to sustain the cost of such litigation and proceedings more effectively than we can because of their substantially greater resources. Uncertainties resulting from our participation in patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Furthermore, because of the substantial amount of discovery required in certain jurisdictions in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, the perceived value of our product candidates or intellectual property could be diminished. Accordingly, the market price of our common shares may decline. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our business, financial condition, results of operations and prospects.
39
Changes to the patent law in the United States and other jurisdictions could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, thereby impairing our ability to protect our technologies and product candidates.
As is the case with other biopharmaceutical companies, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves both technological and legal complexity and is therefore costly, time-consuming and inherently uncertain. Changes in either the patent laws or interpretation of the patent laws in the United States could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. For example, the Leahy-Smith America Invents Act, or the America Invents Act, was signed into law on September 16, 2011, and many of the substantive changes became effective on March 16, 2013. The America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations and prospects. Specifically, the America Invents Act reforms United States patent law in part by changing the U.S. patent system from a “first to invent” system to a “first inventor to file” system. Under a “first inventor to file” system, assuming the other requirements for patentability are met, the first inventor to file a patent application generally will be entitled to the patent on an invention regardless of whether another inventor was the first to invent the invention. This will require us to be cognizant going forward of the time from invention to filing of a patent application and be diligent in filing patent applications. Circumstances may arise that could prevent us from promptly filing patent applications on our inventions and allow third parties to file patents claiming our inventions before we are able to do so. The America Invents Act also includes a number of significant changes that affect the way patent applications will be prosecuted and may also affect patent litigation. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by the USPTO administered post-grant proceedings, including reexamination proceedings, inter partes review, post-grant review and derivation proceedings. These adversarial proceedings at the USPTO review patent claims without the presumption of validity afforded to U.S. patents in lawsuits in U.S. federal courts, and use a lower burden of proof than used in litigation in U.S. federal courts. Therefore, it is generally considered easier for a competitor or third party to have a U.S. patent invalidated in a USPTO post-grant review or inter partes review proceeding than in a litigation in a U.S. federal court. One of our manufacturing-related patents has been invalidated in an inter partes proceeding and if any of our other patents are challenged by a third-party in a USPTO proceeding, there is no guarantee that we or our licensors or collaborators will be successful in defending the patent, which would result in a loss or narrowing of the challenged patent right to us.
In addition, the patent positions of companies in the development and commercialization of biologics and pharmaceuticals are particularly uncertain. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. This combination of events has created uncertainty with respect to the validity and enforceability of patents, once obtained. Depending on future actions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways. In addition, the complexity and uncertainty of European patent laws have also increased in recent years. Complying with these laws and regulations could have a material adverse effect on our existing patent portfolio and our ability to protect and enforce our intellectual property in the future.
We may be subject to claims by third parties asserting that our employees, consultants, independent contractors or we have misappropriated their intellectual property, or claiming ownership of what we regard as our own intellectual property and proprietary technology.
Many of our current and former employees, consultants, and independent contractors including our senior management, were previously employed at universities or at other biotechnology or pharmaceutical companies, including some which may be competitors or potential competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these employees, consultants or independent contractors have used or disclosed intellectual property, including trade secrets or other proprietary information, of such individual’s current or former employers, or that patents and applications we have filed to protect inventions of these individuals, even those related to one or more of our product candidates, are rightfully owned by their former or concurrent employer. Litigation may be necessary to defend against such claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel or sustain damages. Such intellectual property rights could be awarded to a third-party, and we could be required to obtain a license from such third-party to commercialize our technology or products. Such a license may not be available on an exclusive basis or on commercially reasonable terms, or at all. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
40
In addition, while we typically require our employees, consultants and independent contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own, or such agreements may be breached or alleged to be ineffective, and the assignment may not be self-executing, which may result in claims by or against us related to the ownership of such intellectual property or may result in such intellectual property becoming assigned to third parties. If we fail in enforcing or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to our senior management and scientific personnel. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
Obtaining and maintaining our patent protection, including patents licensed from third parties, depends on compliance with various procedural, documentary, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for noncompliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and patent applications will be due to be paid to the USPTO and various government patent agencies outside the United States over the lifetime of our patents and patent applications and any patent rights we may own or license in the future. Additionally, the USPTO and various government patent agencies outside the United States require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. In certain cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with rules applicable to the particular jurisdiction. However, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If we or our licensors fail to maintain the patents and patent applications covering or otherwise protecting our product candidates, it could have a material adverse effect on our business. In addition, to the extent that we have responsibility for taking any action related to the prosecution or maintenance of patents or patent applications in-licensed from a third party, any failure on our part to maintain the in-licensed intellectual property could jeopardize our rights under the relevant license and may have a material adverse effect on our business, financial condition, results of operations and prospects.
If we do not obtain patent term extensions and data exclusivity for each of our product candidates, our business may be materially harmed.
Depending upon the timing, duration and specifics of any FDA marketing approval of any product candidates we may develop, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Action of 1984, or Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. In the European Union, a maximum of five and a half years of supplementary protection can be achieved for an active ingredient or combinations of active ingredients of a medicinal product protected by a basic patent, if a valid marketing authorization exists (which must be the first authorization to place the product on the market as a medicinal product) and if the product has not already been the subject of supplementary protection. However, we may not receive an extension because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents, or otherwise failing to satisfy applicable requirements. Moreover, the length of the extension could be less than we request. If we are unable to obtain patent term extension or if the term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and our business, financial condition, results of operations and prospects could be materially harmed.
41
Certain employees and patents are subject to German law.
A significant number of our personnel work in Germany and are subject to German employment law. Inventions which may be the subject of a patent or of protection as a utility model as well as technical improvement proposals for other technical innovations that may not be the subject of a patent or of protection as a utility model made by such employees are subject to the provisions of the German Act on Employees’ Inventions (Gesetz über Arbeitnehmererfindungen), which regulates the ownership of, and compensation for, inventions made by employees. We face the risk that disputes may occur between us and our current or former employees pertaining to the sufficiency of compensation paid by us, allocation of rights to inventions under this act, or alleged nonadherence to the provisions of this act, any of which may be costly to resolve and take up our management’s time and efforts whether we prevail or fail in such dispute. In addition, under the German Act on Employees’ Inventions, certain employees retain rights to patents they invented or co-invented and disclosed to us prior to October 1, 2009 if the employee inventions were not actively claimed by us after notification by the employee inventors. While we believe that all of our current and past German employee inventors have assigned to us their interest in inventions and patents they invented or co-invented, there can be no assurance that all such assignments are fully effective. Therefore, there can be no assurance that present or former employees do not hold rights to intellectual property used by us or that such employees will not demand the registration of intellectual property rights in their name or demand damages pursuant to the German Act on Employees’ Inventions or other applicable laws. Even if we lawfully own all inventions of our employee inventors who are subject to the German Act on Employees’ Inventions, we are required under German law to reasonably compensate such employees for the use of the inventions. If we are required to pay increased compensation or face other disputes under the German Act on Employees’ Inventions, our business, financial condition, results of operations and prospects could be adversely affected.
The German Act on Employees’ Inventions does not generally apply to managing directors, supervisory directors, freelancers or agents who are not employees under German labor law. Unless the German Act on Employees’ Inventions has been referred to in the respective services agreements, inventions and intellectual property rights created by such inventors must be assigned to us by contract. While we believe that all of our managing directors, supervisory directors, freelancers or agents which are not employees have assigned to us their interest in inventions and patents required for our course of business, there can be no assurance that all such assignments are fully effective. If any of our current or past employees, managing directors, supervisory directors, freelancers or agents obtain or retain ownership of any inventions or related intellectual property rights that we believe we own, we may lose valuable intellectual property rights and be required to obtain and maintain licenses from such persons to such inventions or intellectual property rights, which may not be available on commercially reasonable terms or at all, or may be nonexclusive. If we are unable to obtain and maintain a license to any such person’s interest in such inventions or intellectual property rights, we may need to cease the development, manufacture, and commercialization of one or more of our product candidates or the product candidates we may develop. In addition, any loss of exclusivity of our intellectual property rights could limit our ability to stop others from using or commercializing similar or identical technologies and products. Any of the foregoing events could have a material adverse effect on our business, financial condition, results of operations and prospects.
If we are unable to protect the confidentiality of our proprietary information, the value of our technology and products could be materially adversely affected.
In addition to patent protection, we also rely on trade secrets and confidentiality agreements to protect other proprietary information that is not patentable or that we elect not to patent. To maintain the confidentiality of trade secrets and proprietary information, we enter into confidentiality agreements with our employees, consultants, independent contractors, collaborators, CMOs, CROs and others upon the commencement of their relationships with us. These agreements require that all confidential information developed by the individual or entity or made known to the individual or entity by us during the course of the individual’s or entity’s relationship with us be kept confidential and not disclosed to third parties. Our agreements with employees as well as our personnel policies also generally provide that any inventions conceived by the individual in the course of rendering services to us shall be our exclusive property (to the extent not covered by the German Act on Employees’ Inventions) or that we may obtain full rights to such inventions at our election. However, we cannot guarantee that we have entered into such agreements with each party that may have or has had access to our trade secrets or proprietary technology and processes and cannot guarantee that individuals with whom we have these agreements will comply with their terms. We also face the risk that present or former employees could continue to hold rights to intellectual property used by us, may demand the registration of intellectual property rights in their name and demand damages pursuant to the Patent Act. In addition, present or former employees may demand damages due to violation of obligations under the German Act on Employees’ Invention. In the event of unauthorized use or disclosure of our trade secrets or proprietary information, these agreements, even if obtained, may not provide meaningful protection, particularly for our trade secrets.
42
We may not have adequate remedies in the event of unauthorized use or disclosure of our proprietary information in the case of a breach of any such agreements and our trade secrets and other proprietary information could be disclosed to third parties, including our competitors. Many of our partners also collaborate with our competitors and other third parties. The disclosure of our trade secrets to our competitors, or more broadly, would impair our competitive position and may materially harm our business, financial condition, results of operations and prospects. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to maintain trade secret protection could adversely affect our competitive business position. The enforceability of confidentiality agreements may vary from jurisdiction to jurisdiction. Courts outside the United States are sometimes less willing to protect proprietary information, technology and know-how. In addition, others may independently discover or develop substantially equivalent or superior proprietary information and techniques, and the existence of our own trade secrets affords no protection against such independent discovery.
We may not be successful in obtaining necessary intellectual property rights to product candidates for our development pipeline through acquisitions and in-licenses.
Although we intend to develop product candidates through our own internal research, we may need to obtain additional licenses from others to advance our research or allow commercialization of our product candidates and it is possible that we may be unable to obtain additional licenses at a reasonable cost or on reasonable terms, if at all. However, we may be unable to acquire or in-license intellectual property rights relating to, or necessary for, any product candidates from third parties on an exclusive basis or commercially reasonable terms or at all. In that event, we may be unable to develop or commercialize such product candidates. We may also be unable to identify product candidates that we believe are an appropriate strategic fit for our company and intellectual property relating to, or necessary for, such product candidates.
The in-licensing and acquisition of third-party intellectual property is a competitive area, and a number of more established companies are also pursuing strategies to in-license or acquire third-party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, cash resources and greater clinical development and commercialization capabilities. Furthermore, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. In addition, we expect that competition for the in-licensing or acquisition of third-party intellectual property rights for product candidates that are attractive to us may increase in the future, which may mean fewer suitable opportunities for us as well as higher acquisition or licensing costs. We may be unable to in-license or acquire the third-party intellectual property rights for product candidates on terms that would allow us to make an appropriate return on our investment. If we are unable to successfully obtain rights to suitable product candidates, our business, financial condition, results of operations and prospects for growth could suffer.
We may not be able to protect our intellectual property and proprietary rights throughout the world.
Filing, prosecuting, and defending patents on product candidates in all countries throughout the world would be prohibitively expensive, and the laws of foreign countries may not protect our rights to the same extent as the laws of Europe or the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside of Europe and the United States, or from selling or importing products made using our inventions in and into those countries or other jurisdictions. Third parties may use our technologies in jurisdictions where we have not obtained or are unable to adequately enforce patent protection to develop their own products and, further, may export otherwise infringing products to territories where we have patent protection but enforcement is not as strong as that in Europe or the United States. These products may compete with our products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our intellectual property and proprietary rights generally. Proceedings to enforce our intellectual property and proprietary rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property and proprietary rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
43
Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors is forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired, and our business, financial condition, results of operations and prospects may be adversely affected.
If our trademarks and trade names are not adequately protected, we may not be able to build name recognition in our markets of interest and our business, financial condition, results of operations and prospects may be adversely affected.
Our trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names or may be forced to stop using these names or marks which we need for name recognition by potential partners or customers in our markets of interest. During trademark registration proceedings, we may receive rejections. Although we would be given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the USPTO and in comparable agencies in many foreign jurisdictions, third parties are given an opportunity to oppose pending trademark applications and to seek to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our trademarks, and our trademarks may not survive such proceedings. If we are unable to establish name recognition based on our trademarks and trade names, we may not be able to compete effectively and our business, financial condition, results of operations and prospects may be adversely affected.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our proprietary and intellectual property rights is uncertain because such rights offer only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| ● | others may be able to develop products that are similar to, or better than, our product candidates in a way that is not covered by the claims of the patents we license or may own currently or in the future; |
| ● | we, or our licensing partners or current or future collaborators, might not have been the first to make the inventions covered by issued patents or pending patent applications that we license or may own currently or in the future; |
| ● | we, or our licensing partners or current or future collaborators, might not have been the first to file patent applications for certain of our or their inventions; |
| ● | our pending owned or in-licensed patent applications may not lead to issued patents; |
| ● | we may choose not to file a patent for certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property; |
| ● | our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
| ● | it is possible that there are prior public disclosures that could invalidate our or our licensors’ patents; |
| ● | the patents of third parties or pending or future applications of third parties, if issued, may have an adverse effect on our business; |
| ● | any patents that we obtain may not provide us with any competitive advantages or may ultimately be found not to be owned by us, invalid or unenforceable; or |
| ● | we may not develop additional proprietary technologies that are patentable. |
44
Should any of these events occur, they could significantly harm our business, financial conditions, results of operations and prospects.
Risks Related to Our Business and Industry
Our current and future relationships with third-party payors, healthcare professionals and customers in the United States and elsewhere may be subject, directly or indirectly, to applicable anti-kickback, fraud and abuse, false claims, physician payment transparency and other healthcare laws and regulations, which could expose us to significant penalties.
Healthcare providers, physicians and third-party payors in the United States and elsewhere will play a primary role in the recommendation and prescription of any product candidates for which we obtain marketing approval. Our current and future arrangements with healthcare professionals, third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations, including, without limitation, the federal Anti-Kickback Statute and the federal civil False Claims Act, that may constrain the business or financial arrangements and relationships through which we conduct clinical research, sell, market and distribute any products for which we obtain marketing approval. In addition, we may be subject to physician payment transparency laws and patient privacy regulation by the federal government and by the U.S. states and foreign jurisdictions in which we conduct our business. The applicable federal, state and foreign healthcare laws and regulations that may affect our ability to operate include the following:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward, or in return for, either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs, such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. Further, several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of a federal healthcare-covered business, the Anti-Kickback Statute has been violated; |
| ● | federal civil and criminal false claims laws, including, without limitation, the federal civil False Claims Act (that can be enforced through civil whistleblower or qui tam actions), and the civil monetary penalties law, which impose criminal and civil penalties against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. Moreover, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which imposes criminal and civil liability for, among other things, executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
| ● | the U.S. Federal Food, Drug, and Cosmetic Act, which prohibits, among other things, the adulteration or misbranding of drugs, biologics and medical devices; |
| ● | the U.S. Public Health Service Act, or the PHSA, which prohibits, among other things, the introduction into interstate commerce of a biological product unless a biologics license is in effect for that product; |
| ● | the Physician Payments Sunshine Act, created under Section 6002 of the Affordable Care Act, and its implementing regulations, which requires specified manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services, or the CMS, information related to payments or other “transfers of value” made to physicians, which is defined to include doctors, dentists, optometrists, podiatrists and chiropractors, and certain other healthcare providers beginning in 2022, and which requires teaching hospitals and applicable manufacturers to report annually to CMS ownership and investment interests held by physicians and their immediate family members by the 90th day of each calendar year. All such reported information is publicly available; and |
45
| ● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payors, including private insurers; state and foreign laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; and state and foreign laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures. |
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations may involve substantial costs. It is possible that governmental authorities will conclude that our business practices, including our relationships with physicians and other healthcare providers, some of whom may recommend, purchase or prescribe our product candidate, if approved, may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations.
If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, including, without limitation, damages, fines, disgorgement, individual imprisonment, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of noncompliance with these laws and the curtailment or restructuring of our operations, which could have a material adverse effect on our business. If any of the physicians or other healthcare providers or entities with whom we expect to do business is found not to be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from participation in government healthcare programs, which could also materially affect our business.
Even if we, or any future collaborators, are able to commercialize any product candidate that we, or they, develop, the successful commercialization of our product candidates will depend in part on the extent to which governmental authorities, private health insurers and other third-party payors provide coverage and adequate reimbursement levels and implement pricing policies favorable for our product candidates. Failure to obtain or maintain coverage and adequate reimbursement for our product candidates, if approved, could limit our ability to market those products and decrease our ability to generate revenue.
The healthcare industry is acutely focused on cost containment, both in the United States and elsewhere. Government authorities and third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement. The insurance coverage and reimbursement status of newly approved products for orphan diseases is particularly uncertain and failure to obtain or maintain adequate coverage and reimbursement for our product candidates could limit our ability to generate revenue. Third-party payors may not view our product candidates, if approved, as cost-effective, and coverage and reimbursement may not be available to our customers or may not be sufficient to allow our products, if any, to be marketed on a competitive basis. If coverage and reimbursement are not available, or reimbursement is available only to limited levels, we, or any future collaborators, may not be able to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us, or any future collaborators, to establish or maintain pricing sufficient to realize a sufficient return on our or their investments. Cost-control initiatives could also cause us to decrease any price we might establish for our product candidates, which could result in lower than anticipated product revenues. Moreover, eligibility for reimbursement does not imply that any product will be paid for in all cases or at a rate that covers our costs, including our costs related to research, development, manufacture, sale and distribution. Reimbursement rates may vary, by way of example, according to the use of the product and the clinical setting in which it is used. For products administered under the supervision of a physician, obtaining coverage and adequate reimbursement may be particularly difficult because of the higher prices often associated with such drugs. If the prices for our product candidates, if approved, decrease or if governmental and other third-party payors do not provide adequate coverage or reimbursement, our business, prospects, operating results and financial condition will suffer, perhaps materially.
46
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products, including genetic treatments. In the United States, the Centers for Medicare & Medicaid Services, or the CMS, the federal agency responsible for administering the Medicare program, make the principal decisions about coverage and reimbursement for new treatments under Medicare. Private payors tend to follow CMS to a substantial degree. It is difficult to predict what CMS will decide with respect to reimbursement for novel products such as ours. In addition, certain Affordable Care Act marketplace and other private payor plans are required to include coverage for certain preventative services, including vaccinations recommended by the U.S. Centers for Disease Control’s, or the CDC’s, Advisory Committee on Immunization Practices, or ACIP, without cost share obligations (i.e., co-payments, deductibles or co-insurance) for plan members. For Medicare beneficiaries, vaccines may be covered for reimbursement under either the Part B program or Part D depending on several criteria, including the type of vaccine and the beneficiary’s coverage eligibility. If our vaccine candidates, once approved, are reimbursed only under the Part D program, physicians may be less willing to use our products because of the claims adjudication costs and time related to the claims adjudication process and collection of co-payment associated with the Part D program.
Outside the United States, certain countries, including a number of Member States of the European Union, set prices and reimbursement for pharmaceutical products, with limited participation from the marketing authorization holders. We cannot be sure that such prices and reimbursement will be acceptable to us or our collaborators. If the regulatory authorities in these jurisdictions set prices or reimbursement levels that are not commercially attractive for us or our collaborators, our revenues from sales by us or our collaborators, and the potential profitability of our product candidates, in those countries would be negatively affected. Additionally, some countries require approval of the sale price of a product before it can be marketed. In many countries, the pricing review period begins after marketing or product licensing approval is granted. As a result, we might obtain marketing approval for a product in a particular country, but then may experience delays in the reimbursement approval of our product or be subject to price regulations that would delay our commercial launch of the product, possibly for lengthy time periods, which could negatively impact the revenues we are able to generate from the sale of the product in that particular country.
Moreover, an increasing number of countries are taking initiatives to attempt to reduce large budget deficits by focusing cost-cutting efforts on pharmaceuticals for their state-run healthcare systems. These international price control efforts have impacted all regions of the world, but have been most drastic in the European Union. In some countries, in particular, in many Member States of the European Union, we may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our product candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. In addition, publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries.
If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business, financial condition, results of operations or prospects could be materially adversely affected. Cost-control initiatives could cause us, or any future collaborators, to decrease the price we, or they, might establish for products, which could result in lower than anticipated product revenues. An inability to promptly obtain coverage and adequate payment rates from both government-funded and private payors for any of our product candidates for which we, or any future collaborator, obtain marketing approval could significantly harm our operating results, our ability to raise capital needed to commercialize products and our overall financial condition.
Price controls may be imposed in certain markets, which may adversely affect our future profitability.
In some countries, particularly Member States of the European Union, the pricing of prescription drugs is subject to governmental control or control by associations of health insurers. In these countries, pricing negotiations with governmental authorities can take considerable time after receipt of marketing approval for a product. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost-containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various countries and parallel distribution, or arbitrage between low-priced and high-priced countries, can further reduce prices. In some countries, in particular, in many Member States of the European Union, we may be required to conduct a clinical trial or other studies that compare the cost-effectiveness of our product candidates to other available therapies in order to obtain or maintain reimbursement or pricing approval. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business, financial condition, results of operations or prospects could be materially adversely affected.
47
Exchange rate fluctuations or abandonment of the euro currency may materially affect our results of operations and financial condition.
Potential future expense and revenue may be incurred or derived from outside the European Union, particularly the United States. As a result, our business and share price may be affected by fluctuations in foreign exchange rates between the euro and other currencies, particularly the U.S. dollar, which may also have a significant impact on our reported results of operations and cash flows from period to period. In addition, the abandonment of the euro by one or more members of the European Union could lead to the re-introduction of individual currencies in one or more European Union Member States, or in more extreme circumstances, the dissolution of the European Union. The effects on our business of the abandonment of the euro as a currency, the exit of one or more European Union Member States from the European Union (such as Brexit) or a potential dissolution of the European Union, are impossible to predict with certainty, and any such events could have a material adverse effect on our business, financial condition and results of operations.
More generally, shifts in geopolitical balance, political crisis or wars may affect foreign exchange rates between the euro and other currencies, which may also have a significant impact on our reported results of operations and cash flows from period to period.
We could be subject to strict restrictions on the movement of cash and the exchange of foreign currencies.
In some countries, we could be subject to strict restrictions on the movement of cash and the exchange of foreign currencies, which would limit our ability to use this cash across our global operations. This risk could increase as we continue our geographic expansion, and in particular if we seek to expand into emerging markets, which are more likely to impose these restrictions than more established markets.
Current and future legislation may increase the difficulty and cost for us and any collaborators to obtain marketing approval of and commercialize our product candidates and affect the prices we, or they, may obtain.
In the United States and foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system that could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to profitably sell any product candidates for which we obtain marketing approval. We expect that current laws, as well as other healthcare reform measures that may be adopted in the future, may result in additional reductions in Medicare and other healthcare funding, more rigorous coverage criteria, new payment methodologies and in additional downward pressure on the price that we, or any collaborators, may receive for any approved products.
In March 2010, President Obama signed the Affordable Care Act into law. Among the provisions of the Affordable Care Act of potential importance to our business and our product candidates are the following:
| ● | an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription products and biologic products; |
| ● | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program; |
| ● | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for products that are inhaled, infused, instilled, implanted or injected; |
| ● | extension of manufacturers’ Medicaid rebate liability to individuals enrolled in Medicaid managed care organizations; |
| ● | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; |
| ● | a requirement that certain Affordable Care Act marketplace and other private payor plans include coverage for preventative services, including vaccinations recommended by the ACIP without cost share obligations (i.e., co-payments, deductibles or co-insurance) for plan members; |
| ● | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research; |
48
| ● | a new Independent Payment Advisory Board, or an IPAB, which has authority to recommend certain changes to the Medicare program to reduce expenditures by the program that could result in reduced payments for prescription products; and |
| ● | the establishment of the Center for Medicare and Medicaid Innovation within CMS to test innovative payment and service delivery models. |
Since its enactment, there have been judicial and congressional challenges to numerous aspects of the Affordable Care Act. We are continuing to monitor any changes to the Affordable Care Act that, in turn, may potentially impact our business in the future.
Other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include aggregate reductions to Medicare payments to providers of 2% per fiscal year pursuant to the Budget Control Act of 2011 and subsequent laws, which began in 2013 and will remain in effect through 2030, with the exception of a temporary suspension from May 1, 2020 through December 31, 2021. In addition, in January 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several types of providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. New laws may result in additional reductions in Medicare and other healthcare funding, which may materially adversely affect customer demand and affordability for our product candidates, if approved, and, accordingly, the results of our financial operations.
Also, there has been heightened governmental scrutiny recently over the manner in which pharmaceutical companies set prices for their marketed products, which has resulted in several congressional inquiries and proposed federal legislation, as well as state efforts, designed to, among other things, bring more transparency to product pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products.
The policies of the FDA or similar regulatory authorities may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. If we or our collaborators are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or our collaborators are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been obtained and we may not achieve or sustain profitability, which would adversely affect our business.
We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. It is difficult to predict how current and future legislation, executive actions, and litigation, including the executive orders, will be implemented, and the extent to which they will impact our business, our clinical development, and the FDA’s and other agencies’ ability to exercise their regulatory authority, including FDA’s preapproval inspections and timely review of any regulatory filings or applications we submit to the FDA. To the extent any executive actions impose constraints on FDA’s ability to engage in oversight and implementation activities in the normal course, our business may be negatively impacted.
We cannot predict whether future healthcare legislative or policy changes will be implemented at the federal or state level or in countries outside the United States in which we may do business, or the effect any future legislation or regulation will have on us, but we expect there will continue to be legislative and regulatory proposals at the federal and state levels directed at containing or lowering the cost of healthcare.
49
Cyberattacks or other failures in our or our third-party vendors’, contractors’ or consultants’ telecommunications or information technology systems could result in information theft, data corruption and significant disruption of our business operations.
We utilize information technology, or IT, systems and networks and cloud computing services to process, transmit and store electronic information in connection with our business activities. We manage and maintain our applications and data utilizing a combination of on-site systems, managed data centers and cloud-based data centers. We utilize external security and infrastructure vendors to manage our information technology systems and data centers. These applications and data encompass a wide variety of business-critical information, including research and development information, commercial information, and business and financial information. We face a number of risks relative to protecting this critical information, including loss of access risk, inappropriate use or disclosure, inappropriate modification, and the risk of our being unable to adequately monitor, audit and modify our controls over our critical information. This risk extends to the third-party vendors and subcontractors we use to manage this sensitive data. Despite the implementation of cybersecurity measures, given the size and complexity of our internal IT systems and those of our third-party vendors, contractors and consultants, and the increasing amounts of confidential information that they maintain, such IT systems are potentially vulnerable to breakdown or other damage or interruption from service interruptions, system malfunction, natural disasters, terrorism, war, and telecommunication and electrical failures. Such IT systems are additionally vulnerable to cybersecurity breaches from inadvertent or intentional actions by our employees, third-party vendors, contractors, consultants, business partners, and/or other third parties, or from cyberattacks by malicious third parties (including the deployment of harmful malware, ransomware, denial-of-service attacks, social engineering, and other means to affect service reliability and threaten the confidentiality, integrity, and availability of information). These threats pose a risk to the cybersecurity of our systems and networks, the confidentiality and the availability and integrity of our data and these risks apply both to us, and to third parties on whose systems we rely for the conduct of our business.
Cybersecurity threats are persistent and constantly evolving. Such threats have increased in frequency, scope and potential impact in recent years, which increase the difficulty of detecting and successfully defending against them. We may not be able to anticipate all types of cybersecurity threats, and we may not be able to implement preventive measures effective against all such cybersecurity threats. The techniques used by cyber criminals change frequently, may not be recognized until launched, and can originate from a wide variety of sources, including outside groups such as external service providers, organized crime affiliates, terrorist organizations, or hostile foreign governments or agencies. There can be no assurance that we or our third-party service providers, contractors or consultants will be successful in preventing cyberattacks or successfully mitigating their effects. Similarly, there can be no assurance that such third-party service providers, contractors or consultants will be successful in protecting our clinical and other data that is stored on their systems. If the IT systems of our third-party vendors and other contractors and consultants become subject to disruptions or cybersecurity breaches, we may have insufficient recourse against such third parties and we may have to expend significant resources to mitigate the impact of such an event, and to develop and implement protections to prevent future events of this nature from occurring. Any cyberattack or destruction or loss of data could have a material adverse effect on our business, financial condition, results of operations and prospects. For example, if such an event were to occur and cause interruptions in our operations, or those of our third-party vendors and other contractors and consultants, it could result in a material disruption or delay of the development of our product candidates. In addition, we may suffer reputational harm or face litigation or adverse regulatory action as a result of cyberattacks or other data security breaches and may incur significant additional expense to implement further data protection measures. As cybersecurity threats continue to evolve, we may be required to incur material additional expenses in order to enhance our protective measures or to remediate any information security vulnerability.
Our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
We are in the early stages of developing disaster recovery, business continuity plans and document retention plans designed to allow us to be operational despite unforeseen events, including natural disasters such as an earthquake, fire, hurricane, tornado, flood or significant power outage; political crises, such as terrorist attacks, war and other political instability, including the ongoing geopolitical tensions related to Russia’s actions in Ukraine and associated international sanctions in response to such sanctions; or other catastrophic events. Without disaster recovery, business continuity and document retention plans, if we encounter difficulties or disasters with our manufacturing facilities, affiliates, corporate headquarters or those of third parties we rely on, our critical systems, operations and information may not be restored in a timely manner, or at all, and our business activities could be materially disrupted. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which could have a material adverse effect on our business.
50
We may encounter difficulties in managing our growth, which could disrupt our operations.
We may experience growth in the number of our employees and the scope of our operations in the areas of clinical development and regulatory affairs. We are currently constructing a new facility, designed for the development of a cGMP production process on a large industrial scale for market supply. To manage these growth activities, we must continue to implement and improve our managerial, operational and financial systems, expand our facilities and continue to recruit and train additional qualified personnel. Our management may need to devote a significant amount of its attention to managing these activities. Moreover, our growth could require us to relocate to a different geographic area of the country. We may not be able to effectively manage any expansion or relocation of our operations, retain key employees, or identify, recruit and train additional qualified personnel. Our inability to manage the expansion or relocation of our operations effectively may result in weaknesses in our infrastructure, and give rise to operational mistakes, loss of business opportunities, loss of employees and reduced productivity among remaining employees. Our growth could also require significant capital expenditures and may divert financial resources from other projects, such as the development of additional product candidates. If we are unable to effectively manage our expected growth, our expenses may increase more than expected, our ability to generate revenues could be reduced and we may not be able to implement our business strategy, including the successful development and commercialization of our product candidates. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
Engaging in acquisitions, joint ventures or collaborations may increase our capital requirements, dilute our shareholders, cause us to incur debt or assume contingent liabilities, and subject us to other risks. We may not realize the benefits of these acquisitions, joint ventures or collaborations.
We may evaluate various acquisitions and collaborations, including licensing or acquiring complementary products, intellectual property rights, technologies or businesses. Any potential acquisition, joint venture or collaboration may entail numerous risks, including:
| ● | increased operating expenses and cash requirements; |
| ● | the assumption of additional indebtedness or contingent liabilities; |
| ● | assimilation of operations, intellectual property and products of an acquired company, including difficulties associated with integrating new personnel; |
| ● | the diversion of our management’s attention from our existing product programs and initiatives in pursuing such a strategic merger or acquisition; |
| ● | risk in the retention of key employees, the loss of key personnel, and uncertainties in our ability to maintain key business relationships; |
| ● | risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing products or product candidates and regulatory approvals; and |
| ● | our inability to generate revenue from acquired technology or products sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs. |
In addition, if we undertake acquisitions, we may utilize our cash, issue dilutive securities, assume or incur debt obligations, incur large one-time expenses and acquire intangible assets that could result in significant future amortization expense. Moreover, we may not be able to locate suitable acquisition or collaboration opportunities and this inability could impair our ability to grow or obtain access to technology or products that may be important to the development of our business.
51
Our employees, principal investigators and consultants may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading, which could have an adverse effect on the results of our operations.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators and consultants, despite our robust efforts to prevent such misconduct through sponsor oversight. Misconduct by these parties could include intentional failures to comply with FDA regulations or the regulations applicable in the European Union and other jurisdictions, provide accurate information to the FDA, the EMA and other regulatory authorities, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. Such misconduct also could involve the improper use of information obtained in the course of clinical trials or interactions with the FDA or other regulatory authorities, which could result in regulatory sanctions and cause serious harm to our reputation. We have adopted a code of conduct applicable to all of our employees, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from government investigations or other actions or lawsuits stemming from a failure to comply with laws or regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, financial condition, results of operations and prospects, including the imposition of significant fines or other sanctions.
We are subject to stringent privacy laws, information security laws, regulations, policies and contractual obligations related to data privacy and cybersecurity and changes in such laws, regulations, policies and contractual obligations could adversely affect our business, financial condition, results of operations and prospects.
We are subject to data privacy and protection laws and regulations that apply to the collection, transmission, storage and use of personally identifying information, which among other things, impose certain requirements relating to the privacy, security and transmission of personal information. The legislative and regulatory landscape for privacy and data protection continues to evolve in jurisdictions worldwide, and there has been an increasing focus on privacy and data protection issues with the potential to affect our business. Failure to comply with any of these laws and regulations could result in enforcement action against us, including fines, imprisonment of company officials and public censure, claims for damages by affected individuals, damage to our reputation and loss of goodwill, any of which could have a material adverse effect on our business, financial condition, results of operations and prospects. Additionally, if we are unable to properly protect the privacy and security of personal information, including protected health information, we could be found to have breached our contracts.
There are numerous U.S. federal and state laws and regulations related to the privacy and security of personal information. In particular, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, establish privacy and security standards that limit the use and disclosure of individually identifiable health information, or protected health information, and require the implementation of administrative, physical and technological safeguards to protect the privacy of protected health information and ensure the confidentiality, integrity and availability of electronic protected health information. Determining whether protected health information has been handled in compliance with applicable privacy standards and our contractual obligations can be complex and may be subject to changing interpretation. If we fail to comply with applicable privacy laws, including applicable HIPAA privacy and security standards, we could face civil and criminal penalties. The HHS has the discretion to impose penalties without attempting to first resolve violations. HHS enforcement activity can result in financial liability and reputational harm, and responses to such enforcement activity can consume significant internal resources. Even when HIPAA does not apply, failing to take appropriate steps to keep consumers’ personal information secure can constitute unfair acts or practices in or affecting commerce and be construed as a violation of Section 5(a) of the Federal Trade Commission Act, or the FTCA, 15 U.S.C § 45(a). The Federal Trade Commission, or the FTC, expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards and the FTC’s guidance for appropriately securing consumers’ personal information is similar to what is required by the HIPAA Security Rule. In addition, state attorneys general are authorized to bring civil actions seeking either injunctions or damages in response to violations that threaten the privacy of state residents. We cannot be sure how these regulations will be interpreted, enforced or applied to our operations. In addition to the risks associated with enforcement activities and potential contractual liabilities, our ongoing efforts to comply with evolving laws and regulations at the federal and state level may be costly and require ongoing modifications to our policies, procedures and systems.
52
In addition, many states in which we operate have laws that protect the privacy and security of personal information. For example, the California Consumer Privacy Act of 2018, as amended by the California Privacy Rights Act, or the CCPA, increases privacy rights for California residents and imposes obligations on companies that process their personal information. Among other things, the CCPA requires covered companies to provide new disclosures to California consumers and provide such consumers new data protection and privacy rights, including the ability to opt out of certain sales of personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for certain data breaches that result in the loss of personal information. The CCPA also creates a new state agency that will be vested with authority to implement and enforce the CCPA. State laws are changing rapidly and there is discussion in Congress of a new federal data protection and privacy law to which we would become subject if it is enacted.
Internationally, laws, regulations and standards in many jurisdictions apply broadly to the collection, use, retention, security, disclosure, transfer and other processing of personal information. For example, in the European Union, the collection and use of personal data is governed by the provisions of the General Data Protection Regulation, or the GDPR, in addition to other applicable laws and regulations. The GDPR came into effect in May 2018, repealing and replacing the European Union Data Protection Directive, and imposing revised data privacy and security requirements on companies in relation to the processing of personal data of European Union and United Kingdom data subjects. The GDPR, together with national legislation, regulations and guidelines of the European Union Member States and the United Kingdom governing the processing of personal data, impose strict obligations with respect to, and restrictions on, the collection, use, retention, protection, disclosure, transfer and processing of personal data. The GDPR imposes strict rules on the transfer of personal data to countries outside the European Union, including the United States. For example, in 2016, the European Union and United States agreed to a transfer framework for data transferred from the European Union to the United States, called the Privacy Shield, but the Privacy Shield was invalidated in July 2020 by the Court of Justice of the European Union. The standard contractual clauses issued by the European Commission, or the EC, for the transfer of personal data may be similarly invalidated by the Court of Justice of the European Union. On June 4, 2021, the EC adopted new standard contractual clauses, which impose on companies’ additional obligations relating to data transfers, including the obligation to conduct a transfer impact assessment and, depending on a party’s role in the transfer, to implement additional security measures and to update internal privacy practices. If we elect to rely on the new standard contractual clauses for data transfers, we may be required to incur significant time and resources to update our contractual arrangements and to comply with new obligations. If we are unable to implement a valid mechanism for personal data transfers from the EU, we will face increased exposure to regulatory actions, substantial fines and injunctions against processing personal data from the EU. It remains to be seen whether these standard contractual clauses will remain available and whether additional means for lawful data transfers will become available. The GDPR authorizes fines for certain violations of up to 4% of the total global annual turnover of the preceding financial year or €20 million, whichever is greater. Such fines are in addition to any civil litigation claims by data subjects. Separately, Brexit could also lead to further legislative and regulatory changes and increase our compliance costs. As of January 1, 2021, and the expiry of transitional arrangements agreed to between the United Kingdom and the European Union, data processing in the United Kingdom is governed by a United Kingdom version of the GDPR (combining the GDPR and the Data Protection Act 2018), exposing us to two parallel regimes, each of which potentially authorizes similar fines and other potentially divergent enforcement actions for certain violations. On June 28, 2021, the European Commission adopted an adequacy decision in favor of the United Kingdom, enabling data transfers from European Union member states to the United Kingdom without additional safeguards. However, the United Kingdom adequacy decision will automatically expire in June 2025 unless the European Commission reassesses and renews or extends that decision. Other jurisdictions outside the European Union are similarly introducing or enhancing privacy and data security laws, rules and regulations, which could increase our compliance costs and the risks associated with noncompliance. We cannot guarantee that we are, or will be, in compliance with all applicable international regulations as they are enforced now or as they evolve.
It is possible that these laws may be interpreted and applied in a manner that is inconsistent with our practices and our efforts to comply with the evolving data protection rules may be unsuccessful. We must devote significant resources to understanding and complying with this changing landscape. Failure to comply with federal, state and international laws regarding privacy and security of personal information could expose us to penalties under such laws, orders requiring that we change our practices, claims for damages or other liabilities, regulatory investigations and enforcement action, litigation and significant costs for remediation, any of which could adversely affect our business. Even if we are not determined to have violated these laws, government investigations into these issues typically require the expenditure of significant resources and generate negative publicity, which have a material adverse effect on our business, financial condition, results of operations and prospects.
53
We face substantial competition, which may result in others discovering, developing or commercializing products before or more successfully than we do.
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We face and will continue to face competition from third parties that use mRNA, gene editing or gene therapy development platforms and from third parties focused on other therapeutic modalities, such as small molecules, antibodies, biologics and nucleic acid-based therapies. The competition is likely to come from multiple sources, including large and specialty pharmaceutical and biotechnology companies, academic research institutions, government agencies and public and private research institutions.
Many of our potential competitors, alone or with their strategic partners, have substantially greater financial, technical and other resources, such as larger research and development, clinical, marketing and manufacturing organizations. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even greater concentration of resources among a smaller number of competitors. Our commercial opportunity could be reduced or eliminated if competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approvals for their products faster or earlier than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. For example, some of our competitors have already received approval from the FDA and other regulatory agencies for their mRNA-based COVID-19 vaccines. Additionally, technologies developed by our competitors may render our product candidates uneconomical or obsolete, and we may not be successful in marketing our product candidates against competitors’ products. In addition, the availability of our competitors’ products could limit the demand and the prices we are able to charge for any products that we may develop and commercialize.
We depend heavily on our executive officers and managing directors, and the loss of their services would materially harm our business.
Our success depends, and will likely continue to depend, upon our ability to retain the services of our current executive officers, managing directors, principal consultants and other service providers, and our ability to hire new highly qualified personnel. We are highly dependent on the management, development, clinical, financial and business development expertise of our executive officers, managing directors, principal consultants and other service providers. In addition, we have established relationships with universities and research institutions which have historically provided, and continue to provide, us with access to research laboratories, clinical trials, facilities and patients. Our ability to compete in the biotechnology and pharmaceuticals industries depends upon our ability to attract and retain highly qualified managerial, scientific and medical personnel.
In most cases, our personnel may only terminate their employment upon first providing notice. A limited number of agreements provide for at-will termination. If we lose one or more of our executive officers or other key employees, our ability to implement our business strategy successfully could be seriously harmed. Furthermore, replacing executive officers or other key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to develop, gain marketing approval of and commercialize products successfully.
We may be unable to hire, train, retain or motivate these additional key employees on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions.
54
Our employees, independent contractors, consultants, collaborators and contract research organizations may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause significant liability for us and harm our reputation.
We are exposed to the risk that our employees, independent contractors, consultants, collaborators and contract research organizations may engage in fraudulent conduct or other illegal activity. Misconduct by those parties could include intentional, reckless or negligent conduct or disclosure of unauthorized activities to us that violates (i) FDA regulations or similar regulations of comparable non-U.S. regulatory authorities, including those laws requiring the reporting of true, complete and accurate information to such authorities, (ii) manufacturing and clinical trial conduct standards, (iii) federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable non-U.S. regulatory authorities and (iv) laws that require the reporting of financial information or data accurately. Activities subject to these laws also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. It is not always possible to identify and deter misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws, standards or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, contractual damages, reputational harm, diminished profits and future earnings and curtailment of our operations, any of which could have a material adverse effect on our ability to operate our business and our results of operations.
As a result of our geographically diverse operations, we are more susceptible to certain risks.
We have offices and operations in six cities and in five countries. If we are unable to manage the risks of our global operations, including fluctuations in foreign exchange and inflation rates, international hostilities, natural disasters, security breaches, failure to maintain compliance with our clients’ control requirements and multiple legal and regulatory systems, our results of operations and ability to grow could be materially adversely affected.
Changes in our level of taxes, and audits, investigations and tax proceedings, could have a material adverse effect on our results of operations and financial condition.
Although limited in terms of magnitude due to ongoing losses incurred so far, we are subject to income taxes in Germany and the United States. We calculate and provide for income taxes in each tax jurisdiction in which we operate. Tax accounting often involves complex matters and judgment is required in determining our worldwide provision for income taxes and other tax liabilities. We are subject to ongoing tax audits in Germany. In the future, tax authorities may disagree with our judgments or may take increasingly aggressive positions with respect to the judgments we make. We regularly assess the likely outcomes of these audits in order to determine the appropriateness of our tax liabilities. However, our judgments might not be sustained as a result of these audits, and the amounts ultimately paid could be different from the amounts previously recorded. In addition, our effective tax rate in the future could be adversely affected by changes in the mix of earnings in countries with differing statutory tax rates, changes in the valuation of deferred tax assets and liabilities and changes in tax laws. Tax rates in the jurisdictions in which we operate may change as a result of macroeconomic or other factors outside of our control. Increases in the tax rate in any of the jurisdictions in which we operate could have a negative impact on our profitability. In addition, changes in tax laws, treaties or regulations, or their interpretation or enforcement, may be unpredictable, particularly in less developed markets, and could become more stringent, which could materially adversely affect our tax position. Any of these occurrences could have a material adverse effect on our results of operations and financial condition.
On December 20, 2021 the OECD published the Global Anti-Base Erosion Model Rules (the “GloBE Rules”), also known as Pillar II. The GloBE Rules aim to impose a global minimum tax of 15% on multinational enterprises with a revenue in excess of €750 million. On December 22, 2021, the European Commission published a legislative proposal for Pillar II (the “EU Pillar II Directive”). The EU Pillar II Directive is largely aligned with the GloBE Rules. Some deviations were introduced to ensure compliance with EU treaties.
55
On December 15, 2022, the Council of the European Union formally adopted the EU Pillar II Directive. The EU Pillar II Directive aims at consistently implementing among all 27 member states the GloBE Rules. EU Member States will have to transpose the EU Pillar II Directive into their national laws and will have to apply the Pillar II measures in respect of the fiscal years beginning on or after December 31, 2023. The Netherlands has transposed the EU Pillar II Directive into its national legislation with effect from December 31, 2023 pursuant to the Dutch Minimum Tax Act 2024 (Wet minimumbelasting 2024). Germany has transposed the EU Pillar II Directive into its national legislation with effect from December 28, 2023 pursuant to the German Minimum Tax Act.
We do not yet meet the revenue threshold of €750 million for Pillar II but (i) should this threshold be reduced or (ii) should our revenue increase, then the application of Pillar II could have a material adverse effect on our business, financial position and results of operations. We will continue to monitor the impact of Pillar II going forward.
Uninsured losses arising from third-party claims brought against us could result in payment of substantial damages, which would decrease our cash reserves and could harm our profit and cash flow.
Our products are used in applications where the failure to use our products properly or their malfunction could result in serious bodily injury or death. We may not have adequate insurance to cover the payment of any potential claim related to such injuries or deaths. Insurance coverage may not continue to be available to us or, if available, may be at a significantly higher cost.
We are exposed to potential product liability and professional indemnity risks that are inherent in the research, development, manufacturing, marketing and use of pharmaceutical products.
The use of our investigational medicinal products in clinical trials and the sale of any approved products in the future may expose us to liability claims. These claims might be made by patients who use the product, healthcare providers, pharmaceutical companies or others selling such products. Any claims against us, regardless of their merit, could be difficult and costly to defend and could materially adversely affect the market for our product candidates or any prospects for commercialization of our product candidates.
Although the clinical trial process is designed to identify and assess potential side effects, it is always possible that a product, even after regulatory approval, may exhibit unforeseen side effects. If any of our product candidates were to cause adverse side effects during clinical trials or after approval of the product candidate, we may be exposed to substantial liabilities. Physicians and patients may not comply with any warnings that identify known potential adverse effects and patients who should not use our product candidates.
If we cannot maintain our corporate culture, we could lose the innovation, teamwork and passion that we believe contribute to our success.
We invest substantial time and resources in building and maintaining our culture and developing our personnel; however, it may be increasingly difficult to maintain our culture. Recent shifts in workplace and workstyle, increase the risk of our ability to maintain culture. Any failure to preserve our culture could negatively affect our future success, including our ability to retain and recruit personnel and to effectively pursue our strategic plans.
To cover such liability claims, we purchase clinical trial insurance in the conduct of each of our clinical trials. It is possible that our liabilities could exceed our insurance coverage or that our insurance will not cover all situations in which a claim against us could be made. We also intend to expand our insurance coverage to include the sale of commercial products if we receive marketing approval for any of our proprietary products. However, we may not be able to maintain insurance coverage at a reasonable cost or obtain insurance coverage that will be adequate to satisfy any liability that may arise. If a successful product liability claim or series of claims is brought against us for uninsured liabilities or in excess of insured liabilities, our assets may not be sufficient to cover such claims and our business operations could be impaired. Should any of the events described above occur, this could have a material adverse effect on our business, financial condition and results of operations, including, but not limited to:
| ● | decreased demand for our future product candidates; |
| ● | adverse publicity and injury to our reputation; |
| ● | withdrawal of clinical trial participants; |
56
| ● | initiation of investigations by regulators; |
| ● | costs to defend the related litigation; |
| ● | a diversion of management’s time and our resources; |
| ● | compensation in response to a liability claim; |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| ● | loss of revenue; |
| ● | exhaustion of any available insurance and our capital resources; and |
| ● | the inability to commercialize our products or product candidates. |
We could be adversely affected if we are subject to negative publicity. We could also be adversely affected if any of our products or any similar products distributed by other companies prove to be, or are asserted to be, harmful to patients. Any adverse publicity associated with illness or other adverse effects resulting from patients’ use or misuse of our products or any similar products distributed by other companies could have a material adverse impact on our business, financial condition, results of operations or prospects.
Some of our product candidates are classified as gene therapies by the FDA and the EMA, and the FDA has indicated that products similar to our product candidates will be reviewed within its Center for Biologics Evaluation and Research, or CBER. Even though our mRNA product candidates are designed to have a different mechanism of action from gene therapies, the association of our product candidates with gene therapies could result in increased regulatory burdens, impair the reputation of our product candidates or negatively impact our platform or our business.
There have been few approvals of gene therapy products in the United States and other jurisdictions, and there have been well-reported significant adverse events associated with their testing and use. Gene therapy products have the effect of introducing new DNA and potentially irreversibly changing the DNA in a cell. In contrast, mRNA is highly unlikely to localize to the nucleus, integrate into cell DNA or otherwise make any permanent changes to cell DNA. Consequently, we expect that our product candidates will have a different potential side effect profile from gene therapies because they lack risks associated with altering cell DNA irreversibly. Further, we may avail ourselves of ways of mitigating side effects in developing our product candidates to address safety concerns that are not available to all gene therapies, such as lowering the dose of our product candidates during repeat dosing or stopping treatment to potentially ameliorate undesirable side effects.
Regulatory requirements governing gene and cell therapy products have evolved and may continue to change in the future, and the implications for mRNA-based medicines is unknown. For example, the FDA has established the Office of Tissues and Advanced Therapies within CBER to consolidate the review of gene therapy and related products, and convenes the Cellular, Tissue and Gene Therapies Advisory Committee to advise CBER on its review. In the European Union, mRNA has been characterized as a Gene Therapy Medicinal Product. In certain countries, mRNA therapies have not yet been classified or any such classification is not known to us. Specifically, in Japan, the Pharmaceuticals and Medical Devices Agency has not taken a position on the regulatory classification. Notwithstanding the differences between our mRNA product candidates and gene therapies, the classification of some of our mRNA product candidates as gene therapies in the United States, the European Union and potentially other countries could adversely impact our ability to develop our product candidates, and could negatively impact our platform and our business. For instance, a clinical hold on gene therapy products across the field due to risks associated with altering cell DNA irreversibly may apply to our mRNA product candidates irrespective of the mechanistic differences between gene therapies and mRNA.
57
Adverse events reported with respect to gene therapies or genome-editing therapies could adversely impact one or more of our programs. Although our mRNA product candidates are designed not to make any permanent changes to cell DNA, regulatory agencies or others could believe that adverse effects of gene therapy products caused by introducing new DNA and irreversibly changing the DNA in a cell could also be a risk for our mRNA investigational therapies, and as a result, may delay one or more of our trials or impose additional testing for long-term side effects. Any new requirements and guidelines promulgated by regulatory review agencies may have a negative effect on our business by lengthening the regulatory review process, requiring us to perform additional or larger studies, or increasing our development costs, any of which could lead to changes in regulatory positions and interpretations, delay or prevent advancement or approval and commercialization of our product candidates or lead to significant post-approval studies, limitations or restrictions. As we advance our product candidates, we will be required to consult with these regulatory agencies and advisory committees and comply with applicable requirements and guidelines. If we fail to do so, we may be required to delay or discontinue development of some or all of our product candidates.
Risks Related to Our Common Shares
An active trading market for our common shares may not be sustainable. If an active trading market is not maintained, investors may not be able to resell their shares at or above the purchase price and our ability to raise capital in the future may be impaired.
Although our common shares are listed and trade on Nasdaq, an active trading market for our shares may not be maintained. If an active market for our common shares is not maintained, it may be difficult for you to sell shares you purchase without depressing the market price for the shares or at all. An inactive trading market may also impair our ability to raise capital to continue to fund operations by selling shares and may impair our ability to acquire other companies or technologies by using our shares as consideration. In addition to the risks described above, the market price of our common shares may be influenced by many factors, some of which are beyond our control, including:
| ● | the failure of financial analysts to continue to cover our common shares or changes in financial estimates by analysts; |
| ● | actual or anticipated variations in our operating results; |
| ● | changes in financial estimates by financial analysts, or any failure by us to meet or exceed any of these estimates, or changes in the recommendations of any financial analysts that elect to follow our common shares or the shares of our competitors; |
| ● | announcements by us or our competitors of significant contracts or acquisitions; |
| ● | future sales of our shares; and |
| ● | investor perceptions of us and the industries in which we operate. |
These and other factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock. In addition, the stock market in general has from time to time experienced extreme price and volume fluctuations, including in recent months, that have often been unrelated or disproportionate to the operating performance of particular companies affected. These broad market and industry factors may materially harm the market price of our common shares, regardless of our operating performance. In the past, following periods of volatility in the market price of certain companies’ securities, securities class action litigation has been instituted against these companies. This litigation, if instituted against us, could adversely affect our financial condition or results of operations.
Sales of substantial amounts of our common shares in the public market, or the perception that these sales may occur, could cause the market price of our common shares to decline.
Sales of substantial amounts of our common shares in the public market, or the perception that these sales may occur, could cause the market price of our common shares to decline. This could also impair our ability to raise additional capital through the sale of our equity securities. We cannot predict the size of future issuances of our shares or the effect, if any, that future sales and issuances of shares would have on the market price of our common shares.
58
Considerable amounts of common shares are available for issuance under our equity incentive plans, and significant issuances in the future may adversely impact the market price of our common shares.
As of December 31, 2023, we had 386,250,000 authorized common shares, of which 224,305,680 shares were outstanding. In addition, 33,598,301 common shares were reserved for issuance pursuant to our equity incentive plans. The availability of substantial amounts of our common shares resulting from the exercise or settlement of equity awards outstanding under our equity incentive plans, which would be dilutive to existing stockholders, could adversely affect the prevailing market price of our common shares and could impair our ability to raise additional capital through the sale of equity securities.
We have broad discretion in the use of our cash on hand and may invest or spend it in ways with which you do not agree and in ways that may not yield a return on your investment.
As of December 31, 2023, we had cash and cash equivalents amounting to € 402.4 million. Our management will have broad discretion in the use of such cash and could spend it in ways that do not improve our results of operations or enhance the value of our common shares. You will not have the opportunity to influence our decisions on how to use our cash on hand. The failure by our management to apply these funds effectively could result in financial losses that could harm our business, cause the price of our common shares to decline and delay the development of our product candidates. Pending its use, we may invest our cash on hand in a manner that does not produce income or that loses value.
Concentration of ownership by our principal shareholders may conflict with your interest and may prevent you from influencing significant corporate decisions.
As of March 15, 2024, our principal shareholder, dievini Hopp BioTech holding GmbH & Co. KG, or dievini, and its related parties, beneficially owned approximately 37% of our common shares and Kreditanstalt für Wiederaufbau, or KfW, beneficially owns approximately 13% of our common shares. See “Item 7. Major Shareholders and Related Party Transactions.”
In addition, dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) has the right under our articles of association to make a binding nomination for the following number of supervisory directors until dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates as defined by our articles of association and ultimate beneficiaries as defined by our articles of association (individually or collectively) cease to own at least 10% of our issued share capital or an earlier change of control over dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) as defined by our articles of association, which period we refer to as the initial nomination period for dievini:
| ● | four (4) supervisory directors for as long as dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates (as defined by our articles of association) and ultimate beneficiaries (as defined by our articles of association) (individually or collectively) own at least 70% of our issued share capital; |
| ● | three (3) supervisory directors for as long as dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates (as defined by our articles of association) and ultimate beneficiaries (as defined by our articles of association) (individually or collectively) own at least 50% (but less than 70%) of our issued share capital; |
| ● | two (2) supervisory directors for as long as dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates (as defined by our articles of association) and ultimate beneficiaries (as defined by our articles of association) (individually or collectively) own at least 30% (but less than 50%) of our issued share capital; and |
| ● | one (1) supervisory director for as long as dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates (as defined by our articles of association) and ultimate beneficiaries (as defined by our articles of association) (individually or collectively) own at least 10% (but less than 30%) of our issued share capital. |
59
Dievini and Mr. Dietmar Hopp may be able to significantly influence all matters requiring shareholder approval. Even when dievini ceases to own common shares representing a majority of the total voting power, for so long as dievini continues to own a significant percentage of our common shares, dievini will still be able to significantly influence the composition of our supervisory board and the approval of actions requiring shareholder approval. Accordingly, for such period of time, dievini will continue to have significant influence with respect to our management, business plans and policies, including the appointment and removal of our managing directors, decisions on whether to raise future capital and any amending of our organizational documents, which govern the rights attached to our common shares. In particular, for so long as dievini continues to own a significant percentage of common shares, it will be able to cause or prevent a change of control of us or a change in the composition of our supervisory board and could preclude any unsolicited acquisition of us.
In addition, KfW (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) has the right, and has exercised the right under our articles of association, the KfW dievini Shareholders’ Agreement and the ISA to make a binding nomination for one (1) supervisory director until KfW or any KfW affiliates as defined by our articles of association (individually or together with any other KfW affiliate) cease to own at least 10% of our issued share capital, which period we refer to as the initial nomination period for KfW. Certain decisions require, and cannot be taken without, a resolution of our supervisory board that the KfW nominee, and a dievini nominee, have approved. These relate in particular to the location within the European Union of certain of our activities. The KfW dievini Shareholders’ Agreement includes provisions relating to voting together and in a coordinated fashion on certain specified matters as further described under “Item 7. Major Shareholders and Related Party Transactions — B. Related Party Transactions.”
The concentration of ownership and these nomination rights could deprive you of an opportunity to receive a premium for your common shares as part of a sale of us and ultimately might affect the market price of our common shares. In addition, the concentration of voting power and these nomination rights could delay or prevent an acquisition of our company on terms that other shareholders may desire or result in the management of our company in ways with which other shareholders disagree.
We may be required to redeem for cash all, or to facilitate the purchase by a third-party of all, the shares of us held by the Bill & Melinda Gates Foundation as per the date of the ISA if we default under the Global Access Agreement, which could have an adverse impact on us and limit our ability to make distributions to our shareholders.
We entered into a Global Access Agreement with our shareholder, the Bill & Melinda Gates Foundation, in February 2015 pursuant to which we are required to take certain actions to support the Bill & Melinda Gates Foundation’s mission. In the event that we commit a material breach of the Global Access Agreement or certain provisions of the ISA, following a cure period, we may be required to redeem for cash all, or to facilitate the purchase by a third-party of all, the shares of our company held by the Bill & Melinda Gates Foundation as per the date of the ISA at certain terms that may not be favorable to us. If this occurs, cash used for this purpose may adversely affect our liquidity, cause us to reduce expenditures in other areas of our business, or curtail our growth plans. If we do not have sufficient cash on hand to purchase the shares, we would have to seek financing alternatives in order to meet our obligations, and there is no certainty that financing would be available on reasonable terms or at all. For the period that we are unable to redeem the shares held by the Bill & Melinda Gates Foundation or arrange for a third-party to purchase such shares, we will generally not be allowed to pay dividends, redeem the shares of any other shareholder or otherwise make any other distribution to any of our shareholders in connection with their shares. Therefore, meeting this purchase obligation, if necessary, could have a material adverse effect on our business and financial results. For more information on the Bill & Melinda Gates Foundation’s withdrawal rights, see “Item 7. Major Shareholders and Related Party Transactions — B. Related Party Transactions — Investment and Shareholders’ Agreement.”
Being a public company may continue to increase our costs and disrupt the regular operations of our business.
In August 2020, we completed our initial public offering. After the completion of our initial public offering we incurred and expect to continue to incur costs and expenses, including, but not limited to, managing directors’ and supervisory directors’ fees, increased directors and officers insurance, investor relations, and various other costs of a public company.
We also anticipate that we will continue to incur significant costs associated with corporate governance requirements, including requirements under the Sarbanes-Oxley Act of 2002, as amended, or the Sarbanes-Oxley Act, as well as rules implemented by the SEC and Nasdaq. We expect these rules and regulations to continue to increase our legal and financial compliance costs and make some management and corporate governance activities more time-consuming and costly. These rules and regulations may make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. This could have an adverse impact on our ability to retain, recruit and bring on a qualified independent supervisory board.
60
The additional demands associated with being a public company may disrupt regular operations of our business by diverting the attention of some of our senior management team away from revenue producing activities to management and administrative oversight, adversely affecting our ability to attract and complete business opportunities and increasing the difficulty in both retaining professionals and managing and growing our businesses. Any of these effects could harm our business, financial condition and results of operations.
We are a foreign private issuer and, as a result, we are not subject to U.S. proxy rules and are subject to Exchange Act reporting obligations that, to some extent, are more lenient and less frequent than those of a U.S. domestic public company.
We report under the Securities Exchange Act of 1934, as amended, or the Exchange Act, as a non-U.S. company with foreign private issuer status. Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the Exchange Act that are applicable to U.S. domestic public companies, including (i) the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act, (ii) the sections of the Exchange Act requiring insiders to file public reports of their share ownership and trading activities and liability for insiders who profit from trades made in a short period of time and (iii) the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q containing unaudited financial and other specified information, or current reports on Form 8-K, upon the occurrence of specified significant events. In addition, foreign private issuers are not required to file their annual report on Form 20-F until four months after the end of each fiscal year, while U.S. domestic issuers that are accelerated filers are required to file their annual report on Form 10-K within 75 days after the end of each fiscal year. Foreign private issuers are also exempt from the Regulation Fair Disclosure, aimed at preventing issuers from making selective disclosures of material information. As a result of the above, you may not have the same protections afforded to shareholders of companies that are not foreign private issuers.
We may lose our foreign private issuer status which would then require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur significant legal, accounting and other expenses.
We are a foreign private issuer and therefore we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers. If in the future we are not a foreign private issuer as of the last day of the second fiscal quarter in any fiscal year, we would be required to comply with all of the periodic disclosure, current reporting requirements and proxy solicitation rules of the Exchange Act applicable to U.S. domestic issuers. In order to maintain our current status as a foreign private issuer, either (a) a majority of our common shares must be either directly or indirectly owned of record by nonresidents of the United States or (b) (i) a majority of our managing directors, supervisory directors and executive officers may not be United States citizens or residents, (ii) more than 50% of our assets cannot be located in the United States and (iii) our business must be administered principally outside the United States. If we were to lose this status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC and stock exchange rules. The regulatory and compliance costs to us if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be significantly higher than the costs we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time-consuming and costly. These rules and regulations could also make it more difficult for us to attract and retain qualified managing directors and supervisory directors.
61
As a foreign private issuer and as permitted by the listing requirements of Nasdaq, we follow certain home country governance practices rather than the corporate governance requirements of Nasdaq.
We are a foreign private issuer. As a result, in accordance with the listing requirements of Nasdaq, we rely on home country governance requirements and certain exemptions thereunder rather than relying on the corporate governance requirements of Nasdaq. In accordance with Dutch law and generally accepted business practices, our articles of association do not provide quorum requirements generally applicable to general meetings. To this extent, our practice varies from the requirement of Nasdaq Listing Rule 5620(c), which requires an issuer to provide in its bylaws for a generally applicable quorum, and that such quorum may not be less than one-third of the outstanding voting shares. Although we must provide shareholders with an agenda and other relevant documents for the general meeting, Dutch law does not have a regulatory regime for the solicitation of proxies and the solicitation of proxies is not a generally accepted business practice in the Netherlands, thus our practice varies from the requirement of Nasdaq Listing Rule 5620(b). As permitted by the listing requirements of Nasdaq, we have also opted out of the requirements of Nasdaq Listing Rule 5605(d), which requires, among other things, an issuer to have a compensation committee that consists entirely of independent directors, Nasdaq Listing Rule 5605(e), which requires independent director oversight of director nominations, and Nasdaq Listing Rule 5605(b)(1), which requires an issuer to have a majority of independent directors on its board. These rules require that a majority of our supervisory directors must be independent. In addition, we have opted out of shareholder approval requirements, as included in the Nasdaq Listing Rules, for the issuance of securities in connection with certain events such as the acquisition of shares or assets of another company, the establishment of or amendments to equity-based compensation plans for employees, a change of control of our company and certain private placements. To this extent, our practice varies from the requirements of Nasdaq Rule 5635, which generally requires an issuer to obtain shareholder approval for the issuance of securities in connection with such events. Accordingly, you may not have the same protections afforded to shareholders of companies that are subject to these Nasdaq requirements.
Although we do not believe that we were a “passive foreign investment company,” or a PFIC, for U.S. federal income tax purposes for our 2023 taxable year, we may be a PFIC for 2024 or one or more future taxable years. A U.S. holder of common shares may suffer adverse U.S. federal income tax consequences if we are a PFIC for any taxable year.
Under the Internal Revenue Code of 1986, as amended, or the Code, we will generally be a PFIC for any taxable year in which, after the application of certain look-through rules with respect to subsidiaries, either (i) 75% or more of our gross income consists of “passive income,” or (ii) 50% or more of the average quarterly value of our assets consists of assets that produce, or are held for the production of, “passive income.” Passive income generally includes dividends, interest, certain nonactive rents and royalties, and capital gains. The value of a non-U.S. corporation’s goodwill that is associated with activities that produce or are intended to produce active income is generally an active asset for purposes of the asset test unless, for U.S. federal income tax purposes, the non-U.S. corporation is a “controlled foreign corporation,” or a CFC, that is not publicly traded “for the taxable year.” If a non-U.S. corporation is a CFC that is not publicly traded for the taxable year, its PFIC status under the asset test is determined by using the U.S. tax basis of its assets rather than their fair market value and therefore the market value of its goodwill is generally disregarded. Generally, a non-U.S. corporation is a CFC if more than 50% of its shares’ voting power or value is owned, directly, indirectly or constructively, by “United States shareholders” (as defined in Section 951(b) of the Code). Although it is not certain, we may be or may have been a CFC in the 2023 taxable year. However, under Treasury regulations promulgated in 2020, the fair market value of our assets (including goodwill) can be used for purposes of the asset test provided that (i) we are publicly traded on the majority of days during our taxable year or (ii) we would not be a CFC if certain constructive ownership rules were not applied. We believe, and the remainder of this discussion assumes, that we are eligible to use the fair market value of our assets for purposes of the asset test for our 2023 taxable year.
Based on the composition of our income and assets during 2023, we do not believe that we were a PFIC for our 2023 taxable year. However, there can be no assurance that the Internal Revenue Service, or the “IRS,” will agree with our conclusion. Whether we will be a PFIC in 2024 or any future year is uncertain because, among other things, (i) we currently own a substantial amount of passive assets, including cash, (ii) the valuation of our assets that generate nonpassive income for PFIC purposes, including our intangible assets, is uncertain and may vary substantially over time, (iii) the treatment of grants as income for U.S. federal income tax purposes is unclear, and (iv) the composition of our income may vary substantially over time. Accordingly, there can be no assurance that we will not be a PFIC in 2024 or any future taxable year.
62
If we are a PFIC for any taxable year during which a U.S. investor holds common shares, we generally would continue to be treated as a PFIC with respect to that U.S. investor for all succeeding years during which the U.S. investor holds common shares, even if we ceased to meet the threshold requirements for PFIC status. Such a U.S. investor may be subject to adverse U.S. federal income tax consequences, including (i) the treatment of all or a portion of any gain on disposition as ordinary income, (ii) the application of a deferred interest charge on such gain and the receipt of certain dividends and (iii) compliance with certain reporting requirements. There is no assurance that we will provide information that will enable investors to make a qualified electing fund election, also known as a QEF Election, that could mitigate the adverse U.S. federal income tax consequences should we be classified as a PFIC. See “Item 10. Additional Information — E. Taxation — Material U.S. Federal Income Tax Considerations to U.S. Holders.”
Insiders have substantial control over us and could limit your ability to influence the outcome of key transactions, including a change of control.
As of March 15, 2024, our principal shareholders, managing directors, supervisory directors and executive officers and entities affiliated with them own 58% of the outstanding common shares. As a result, these shareholders, if acting together, would be able to influence or control matters requiring approval by our general meeting, including the appointment of managing directors and supervisory directors, changes to our articles of association and approval of mergers or other extraordinary transactions. They may also have interests that differ from yours and may vote in a way with which you disagree and which may be adverse to your interests. The concentration of ownership may have the effect of delaying, preventing or deterring a change of control of our company, could deprive our shareholders of an opportunity to receive a premium for their common shares as part of a sale of our company and might ultimately affect the market price of our common shares.
If securities or industry analysts do not continue to publish research or publish inaccurate or unfavorable research about our business, our share price and trading volume could decline.
The trading market for our common shares depends in part on the research and reports that securities or industry analysts publish about us or our business. If securities or industry analysts do not continue to cover our company, the market price for our common shares would likely be negatively impacted.
In addition, if one or more of the analysts who cover us downgrade our common shares or publish inaccurate or unfavorable research about our business, our share price may decline. If one or more of these analysts cease coverage of our company or fail to publish reports on us regularly, demand for our shares could decrease, which might cause our share price and trading volume to decline.
We do not anticipate paying any cash dividends in the foreseeable future.
We currently intend to retain our future earnings, if any, for the foreseeable future, to fund the development and growth of our business. We do not intend to pay any dividends to holders of our common shares. As a result, capital appreciation in the price of our common shares, if any, will be your only source of gain on an investment in our common shares.
If we do pay dividends, we may need to withhold tax on such dividends payable to holders of our shares in both Germany and the Netherlands.
We do not intend to pay any dividends to holders of our common shares. See “— We do not anticipate paying any cash dividends in the foreseeable future.” However, if we do pay dividends, we may need to withhold tax on such dividends both in Germany and the Netherlands.
63
As an entity incorporated under Dutch law, any dividends distributed by us are subject to Dutch dividend withholding tax on the basis of Dutch domestic law. However, on the basis of the 2012 Convention between the Federal Republic of Germany and the Kingdom of the Netherlands for the avoidance of double taxation with respect to taxes on income, or the “double tax treaty between Germany and the Netherlands,” the Netherlands will be restricted in imposing these taxes if we are also a tax resident of Germany and our effective management is located in Germany, or the “withholding tax restriction.” See also “— We may become taxable in a jurisdiction other than Germany and this may increase the aggregate tax burden on us.” The withholding tax restriction does, however, not apply, and Dutch dividend withholding tax is still required to be withheld from dividends, if and when paid to Dutch resident holders of our common shares (and non-Dutch resident holders of our common shares that have a permanent establishment in the Netherlands to which their shareholding is attributable). As a result, upon a payment of dividends, we will be required to identify our shareholders in order to assess whether there are Dutch residents (or non-Dutch residents with a permanent establishment in the Netherlands to which the common shares are attributable) in respect of which Dutch dividend withholding tax has to be withheld. Such identification may not always be possible in practice. If the identity of our shareholders cannot be determined, withholding of both German and Dutch dividend withholding tax may occur upon a payment of dividends.
Furthermore, the withholding tax restriction referred to above is based on the current reservation made by Germany under the Multilateral Convention to Implement Tax Treaty Related Measures to Prevent Base Erosion and Profit Shifting, or the MLI, with respect to the tie-breaker provision included in Article 4(3) of the double tax treaty between Germany and the Netherlands, or the “MLI tie-breaker reservation.” If Germany changes its MLI tie-breaker reservation, we will not be entitled to any benefits of the double tax treaty between Germany and the Netherlands, including the withholding tax restriction, as long as Germany and the Netherlands do not reach an agreement on our tax residency for purposes of the double tax treaty between Germany and the Netherlands, and, as a result, any dividends distributed by us during the period no such agreement has been reached between Germany and the Netherlands, may be subject to dividend withholding tax both in Germany and the Netherlands.
Our ability to use our net operating loss carryforwards and other tax attributes may be limited.
Our ability to utilize our net operating losses, or NOLs, is currently limited, and may be limited further, under Section 8c of the German Corporation Income Tax Act (Korperschaftsteuergesetz, or KStG) and Section 10a of the German Trade Tax Act (Gewerbesteuergesetz, or GewStG). These limitations apply if a qualified ownership change, as defined by Section 8c KStG, occurs and no exemption is applicable.
Generally, a qualified ownership change occurs if more than 50% of the share capital or the voting rights are directly or indirectly transferred to an acquirer or a party related to the acquirer or a group of acquirers with aligned interests within a period of five years. A qualified ownership change may also occur in case of a transaction comparable to a transfer of shares or voting rights or in case of an increase in capital leading to a respective change in the shareholding.
In the case of such a qualified ownership change, current year tax losses, tax loss carryforwards and interest carryforwards (together ‘tax loss carryforwards’) expire in full. To the extent that the tax loss carryforwards do not exceed the built-in gains (stille Reserven) in the qualifying assets and liabilities taxable in Germany, they may be further utilized despite a qualified ownership change. In case of a qualified ownership change within a group, tax loss carryforwards will be preserved if certain conditions are satisfied. In addition, in case of a qualified ownership change due to a share acquisition in order to restructure business operations, tax loss carryforwards are preserved if certain conditions are met. Further, tax loss carry forwards will be preserved (in the form of a “fortfuhrungsgebundener Verlustvortrag”) in case of a qualified ownership change if the business operations have not been changed and will not be changed within the meaning of Section 8d KStG and a corresponding application is filed.
According to an appeal filed by the fiscal court of Hamburg dated August 29, 2017, Section 8c, paragraph 1, sentence 1 KStG is not in line with the German Constitution. The appeal is still pending. It is unclear when the Federal Constitutional Court will decide this case.
As of December 31, 2023, there are NOLs available for the German entities of CureVac for German corporate income tax purposes of € 1,700 million: € 1,242 million for CureVac SE, € 376 million for CureVac Manufacturing GmbH, € 37 million for CureVac N.V., € 44 million for CureVac RNA Printer GmbH and a NOL for CureVac Netherlands B.V. for Dutch Corporate income tax of € 1 million. For German entities there are NOLs for German trade tax purposes of € 1,685 million: € 1,234 million for CureVac SE, € 370 million for CureVac Manufacturing GmbH, € 37 million for CureVac N.V. and € 44 million for CureVac RNA Printer GmbH available as of December 31, 2023.
64
Future changes in share ownership may also trigger an ownership change and, consequently, a Section 8c KStG or a Section 10a GewStG limitation. Any limitation may result in the expiration of a portion or the complete tax operating loss carryforwards before they can be utilized. As a result, if we earn net taxable income, our ability to use our pre-change net operating loss carryforwards to reduce German income tax may be subject to limitations, which could potentially result in increased future cash tax liability to us.
Shareholders may not be able to exercise preemptive rights and, as a result, may experience substantial dilution upon future issuances of common shares.
In the event of an issuance of common shares or a grant of rights to subscribe for common shares, subject to certain exceptions, each shareholder will have a pro rata preemptive right in proportion to the aggregate nominal value of the common shares held by such holder. These preemptive rights may be restricted or excluded by a resolution of the general meeting or by another corporate body designated by the general meeting. Our management board, subject to approval of our supervisory board, has been authorized, for a period of five years from our initial public offering, to issue shares or grant rights to subscribe for shares up to our authorized share capital from time to time and to limit or exclude preemptive rights in connection therewith. This could cause existing shareholders to experience substantial dilution of their interest in us.
We may become taxable in a jurisdiction other than Germany and this may increase the aggregate tax burden on us.
Since our incorporation, we have had, on a continuous basis, our place of “effective management” in Germany. We will therefore qualify as a tax resident of Germany on the basis of German domestic law. As an entity incorporated under Dutch law, however, we also qualify as a tax resident of the Netherlands on the basis of Dutch domestic law. However, based on our current management structure and the current tax laws of the United States, Germany and the Netherlands, as well as applicable income tax treaties, and current interpretations thereof, we should qualify solely as a tax resident of Germany for the purposes of the double tax treaty between Germany and the Netherlands due to the “effective management” tie-breaker included in Article 4(3) of the double tax treaty between Germany and the Netherlands and the current MLI tie-breaker reservation.
The test of “effective management” is largely a question of fact and degree based on all the circumstances, rather than a question of law. Nevertheless, the relevant case law and OECD guidance suggest that our company is likely to be regarded as having become a German tax resident from incorporation and remaining so if, as our company intends, (i) most meetings of its management board are prepared and held in Germany (and none will be held in the Netherlands) with a majority of managing directors present in Germany for those meetings; (ii) at those meetings there are full discussions of, and decisions are made regarding, the key strategic issues affecting our company and its subsidiaries; (iii) those meetings are properly minuted; (iv) a majority of our managing directors, together with supporting staff, are based in Germany; and (v) our company has permanent staffed office premises in Germany. We may, however, become subject to limited income tax liability in other countries with regard to the income generated in the respective other country, for example, due to the existence of a permanent establishment or a permanent representative in such other country.
The applicable tax laws or interpretations thereof may change, including the MLI tie-breaker reservation. Furthermore, whether we have our place of effective management in Germany and are as such tax resident in Germany is largely a question of fact and degree based on all the circumstances, rather than a question of law, which facts and degree may also change. Changes to applicable laws or interpretations thereof, changes to applicable facts and circumstances (for example, a change of directors or the place where board meetings take place), or changes to applicable income tax treaties, including a change to the MLI tie-breaker reservation, may result in us becoming (also) a tax resident of the Netherlands or another jurisdiction. See “— If we do pay dividends, we may need to withhold tax on such dividends payable to holders of our shares in both Germany and the Netherlands.” As a consequence, our overall effective income tax rate and income tax expense could materially increase, which could have a material adverse effect on our business, results of operations, financial condition and prospects, which could cause our share price and trading volume to decline. In addition, as a consequence, dividends distributed by us, and interest or royalty payments made by us, if any, may become subject to withholding taxes in more than one jurisdiction. The double taxation of income and the double withholding tax on dividends, interest and/or royalties may be reduced or avoided entirely under the double tax treaty between Germany and the Netherlands or under a double tax treaty between the Netherlands and the respective other country.
65
Claims of U.S. civil liabilities may not be enforceable against us.
We are organized and existing under the laws of the Netherlands. As such, under Dutch private international law, the rights and obligations of our shareholders vis-à-vis the company originating from Dutch corporate law and our articles of association, as well as the civil liability of our officers (functionarissen) (including our managing directors, supervisory directors and executive officers) are governed in certain respects by the laws of the Netherlands. Our headquarters is located in Germany.
We are not a resident of the United States and our officers may also not all be residents of the United States. As a result, depending on the subject matter of the action brought against us and/or our officers, United States courts may not have jurisdiction. If a Dutch court has jurisdiction with respect to such action, that court will apply Dutch procedural law and Dutch private international law to determine the law applicable to that action. Depending on the subject matter of the relevant action, a competent Dutch court may apply another law than the laws of the United States.
Also, service of process against non-residents of the United States can in principle (absent, for example, a valid choice of domicile) not be effected in the United States.
Furthermore, most of our assets are located outside the United States. On the date of this Annual Report, (i) there is no treaty in force between the United States and the Netherlands for the reciprocal recognition and enforcement of judgments, other than arbitration awards, in civil and commercial matters and (ii) both the Hague Convention on Choice of Court Agreements (2005) and the Hague Judgments Convention (2019) have entered into force for the Netherlands, but have not entered into force for the United States. Consequently, a judgment rendered by a court in the United States will not automatically be recognized and enforced by the competent Dutch courts. However, if a person has obtained a judgment rendered by a court in the United States that is enforceable under the laws of the United States and files a claim with the competent Dutch court, the Dutch court will in principle give binding effect to that United States judgment if (i) the jurisdiction of the United States court was based on a ground of jurisdiction that is generally acceptable according to international standards, (ii) the judgment by the United States court was rendered in legal proceedings that comply with the Dutch standards of proper administration of justice including sufficient safeguards (behoorlijke rechtspleging), (iii) binding effect of such United States judgment is not contrary to Dutch public order (openbare orde) and (iv) the judgment by the United States court is not incompatible with a decision rendered between the same parties by a Dutch court, or with a previous decision rendered between the same parties by a foreign court in a dispute that concerns the same subject and is based on the same cause, provided that the previous decision qualifies for recognition in the Netherlands. Even if such a United States judgment is given binding effect, a claim based thereon may, however, still be rejected if the United States judgment is not or no longer formally enforceable. Moreover, if the United States judgment is not final (for instance when appeal is possible or pending) a competent Dutch court may postpone recognition until the United States judgment will have become final, refuse recognition under the understanding that recognition can be asked again once the United States judgment will have become final, or impose as a condition for recognition that security is posted.
A competent Dutch court may deny the recognition and enforcement of punitive damages or other awards. Moreover, a competent Dutch court may reduce the amount of damages granted by a United States court and recognize damages only to the extent that they are necessary to compensate actual losses or damages. Thus, United States investors may not be able, or experience difficulty, to enforce a judgment obtained in a United States court against us or our officers.
The United States and Germany currently do not have a treaty providing for the reciprocal recognition and enforcement of judgments, in civil and commercial matters. Consequently, a final judgment for payment or declaratory judgments given by a court in the United States, whether or not predicated solely upon U.S. securities laws, would not automatically be recognized or enforceable in Germany. German courts may deny the recognition and enforcement of a judgment rendered by a U.S. court if they consider the U.S. court not to be competent or the decision to be in violation of German public policy principles. For example, judgments awarding punitive damages are generally not enforceable in Germany. A German court may reduce the amount of damages granted by a U.S. court and recognize damages only to the extent that they are necessary to compensate actual losses or damages.
66
In addition, actions brought in a German court against us, our managing directors, our supervisory directors, our senior management and the experts named herein to enforce liabilities based on U.S. federal securities laws may be subject to certain restrictions. In particular, German courts generally do not award punitive damages. Litigation in Germany is also subject to rules of procedure that differ from the U.S. rules, including with respect to the taking and admissibility of evidence, the conduct of the proceedings and the allocation of costs. German procedural law does not provide for pre-trial discovery of documents, nor does Germany support pre-trial discovery of documents under the 1970 Hague Evidence Convention. Proceedings in Germany would have to be conducted in the German language and all documents submitted to the court would, in principle, have to be translated into German. For these reasons, it may be difficult for a U.S. investor to bring an original action in a German court predicated upon the civil liability provisions of the U.S. federal securities laws against us, our managing directors, our supervisory directors, our senior management and the experts named in this Annual Report.
Based on the lack of a treaty as described above, there can be no assurance that U.S. investors will be able to enforce against us or managing directors, supervisory directors, executive officers or certain experts named herein who are residents of or possessing assets in the Netherlands, Germany, or other countries other than the United States any judgments obtained in U.S. courts in civil and commercial matters, including judgments under the U.S. federal securities laws.
The rights of our shareholders may be different from the rights of shareholders in companies governed by the laws of U.S. jurisdictions and may not protect investors in a similar fashion afforded by incorporation in a U.S. jurisdiction.
We are a public company (naamloze vennootschap) organized under the laws of the Netherlands. Our corporate affairs are governed by our articles of association, the rules of our management board and those of our supervisory board and by the laws governing companies incorporated in the Netherlands. However, there can be no assurance that Dutch law will not change in the future or that it will serve to protect investors in a similar fashion afforded under corporate law principles in the United States, which could adversely affect the rights of investors.
The rights of shareholders and the responsibilities of managing directors and supervisory directors may be different from the rights and obligations of shareholders and directors in companies governed by the laws of U.S. jurisdictions. In the performance of their duties, our managing directors and supervisory directors are required by Dutch law to consider the interests of our company, its shareholders, its employees and other stakeholders, in all cases with due observation of the principles of reasonableness and fairness. It is possible that some of these parties will have interests that are different from, or in addition to, your interests as a shareholder.
For more information on relevant provisions of Dutch corporation law and of our articles of association, see “Item 10. Additional Information — B. Memorandum and Articles of Association.”
The ability for our shareholders to alter the members of our management board or supervisory board may be limited by the Dutch cooling-off period in face of shareholder activism or hostile take-over
Our management board, with the approval of our supervisory board, can invoke a cooling-off period of up to 250 days when shareholders, using their right to have items added to the agenda for a general meeting or their right to request a general meeting, propose an agenda item for our general meeting to dismiss, suspend or appoint one or more managing directors or supervisory directors (or to amend any provision in our articles of association dealing with those matters) or when a public offer for our company is made or announced without our support, provided, in each case, that our management board believes that such proposal or offer materially conflicts with the interests of our company and its business. During a cooling-off period, our general meeting cannot dismiss, suspend or appoint managing directors and supervisory directors (or amend the provisions in our articles of association dealing with those matters) except at the proposal of our management board. During a cooling-off period, our management board must gather all relevant information necessary for a careful decision-making process and at least consult with shareholders representing 3% or more of our issued share capital at the time the cooling-off period was invoked, as well as with our Dutch works council (if we or, under certain circumstances, any of our subsidiaries would have one). Formal statements expressed by these stakeholders during such consultations must be published on our website to the extent these stakeholders have approved that publication. Ultimately one week following the last day of the cooling-off period, our management board must publish a report in respect of its policy and conduct of affairs during the cooling-off period on our website. This report must remain available for inspection by shareholders and others with meeting rights under Dutch law at our office and must be tabled for discussion at the next general meeting. Shareholders representing at least 3% of our issued share capital may request the Enterprise Chamber of the Amsterdam Court of Appeal, or the Enterprise Chamber (Ondernemingskamer), for early termination of the cooling-off period. The Enterprise Chamber must rule in favor of the request if the shareholders can demonstrate that:
67
| ● | our management board, in light of the circumstances at hand when the cooling-off period was invoked, could not reasonably have concluded that the relevant proposal or hostile offer constituted a material conflict with the interests of our company and its business; |
| ● | our management board cannot reasonably believe that a continuation of the cooling-off period would contribute to careful policy-making; or |
| ● | other defensive measures, having the same purpose, nature and scope as the cooling-off period, have been activated during the cooling-off period and have not since been terminated or suspended within a reasonable period at the relevant shareholders’ request (i.e., no “stacking” of defensive measures). |
Provisions of our articles of association or Dutch corporate law might deter acquisition bids for us that might be considered favorable and prevent, delay or frustrate any attempt to replace or remove our managing directors or supervisory directors.
Under Dutch law, various protective measures are possible and permissible within the boundaries set by Dutch law and Dutch case law. In this respect, our general meeting shall authorize our management board, subject to the approval by our supervisory board, to grant a call option to an independent foundation under Dutch law (if and when incorporated), or protective foundation, to acquire preferred shares pursuant to a call option agreement, or the call option agreement, that may be entered into between us and such protective foundation after the later of (a) dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates as defined by our articles of association and ultimate beneficiaries as defined by our articles of association (individually or collectively) no longer holding at least 25% of our issued share capital (or an earlier change of control over dievini, as defined in our articles of association), which we refer to as the initial period, or (b) the termination or expiry of the KfW dievini Shareholders’ Agreement (see “Item 7. Major Shareholders and Related Party Transactions — B. Related Party Transactions — Shareholders’ Agreement Among KfW, dievini and Mr. Hopp” for further information on that agreement), which we refer to as the initial approval period.
This call option, if and when granted, shall be continuous in nature and can be exercised repeatedly on multiple occasions. If the protective foundation, if and when incorporated, would exercise such call option, if and when granted, a number of preferred shares up to 100% of our issued share capital held by others than the protective foundation, minus one share, will be issued to the protective foundation. These preferred shares would then be issued to the protective foundation under the obligation to pay 25% of their nominal value upon issuance. In order for the protective foundation to finance the issue price in relation to the preferred shares, the protective foundation may enter into a finance arrangement with a bank or other financial institution. As an alternative to securing this external financing, subject to applicable restrictions under Dutch law, the call option agreement, if and when entered into, will provide that the protective foundation may request us to provide, or cause our subsidiaries to provide, sufficient funding to the protective foundation to enable it to satisfy the payment obligation (or part thereof) in cash and/or to charge an amount equal to the payment obligation (or part thereof) against our profits and/or reserves in satisfaction of such payment obligation. The articles of association of the protective foundation, if and when incorporated, will provide that it will promote and protect the interests of the company, the business connected with the company and the company’s stakeholders from time to time, and repress possible influences which could threaten the strategy, continuity, independence and/or identity of the company or the business connected with it, to such an extent that it could be considered to be damaging to the aforementioned interests. These influences may include a third-party acquiring a significant percentage of our common shares, the announcement of an unsolicited public offer for our common shares, shareholder activism, other concentration of control over our common shares or any other form of undue pressure on us to alter our strategic policies. The protective foundation, if and when incorporated, shall be structured to operate independently of us.
The voting rights of our shares are based on nominal value and, as we expect our common shares to trade substantially in excess of their nominal value, preferred shares issued at 25% of their nominal value can carry significant voting power for a substantially reduced price compared to the price of our common shares and thus can be used as a defensive measure. These preferred shares, if and when issued, will have both a liquidation and dividend preference over our common shares and will accrue cash dividends at a fixed rate calculated over the amount paid-up on those preferred shares pro rata tempore for the period during which they were outstanding. The protective foundation would be expected to require us to cancel its preferred shares, if and when issued to the protective foundation, once the perceived threat to the company and its stakeholders has been removed or sufficiently mitigated or neutralized. However, subject to the same limitations described above, the protective foundation would, in that case, continue to have the right to exercise the call option in the future in response to a new threat to the interests of us, our business and our stakeholders from time to time.
68
In addition, certain provisions of our articles of association may make it more difficult for a third party to acquire control of us or effect a change in the composition of our management board and supervisory board. These include:
| ● | a provision that our managing directors and supervisory directors are appointed on the basis of a binding nomination, the binding nature of which can only be overruled by a simple majority of votes cast representing at least one-third of our issued share capital; |
| ● | a provision that our managing directors and supervisory directors may only be dismissed by the general meeting by a two-thirds majority of votes cast representing more than 50% of our issued share capital (unless the dismissal is proposed by the supervisory board or, with respect to supervisory directors nominated by dievini or KfW, by dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) during the nomination period for dievini or by KfW (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) during the nomination period for KfW, respectively, in which case a simple majority of the votes would be sufficient); |
| ● | a provision that certain provisions of our articles of association can only be amended with the affirmative vote of (i) during the nomination period for dievini, dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) and (ii) during the nomination period for KfW, KfW (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement); |
| ● | a provision that if a supervisory director is no longer in office or is unable to act, he or she may be replaced temporarily by a person who the supervisory board has designated for that purpose and, where a supervisory director who has been appointed upon a nomination of dievini or KfW, as applicable, is no longer in office or unable to act, such supervisory director may only be temporarily replaced by a person designated for such purposes by dievini or KfW, as applicable. Such person shall become a full member of the supervisory board with the rights of the relevant supervisory director appointed upon a nomination of dievini or KfW, as applicable, as soon as a written designation to that effect has been received by the chairman or vice-chairman of our supervisory board, subject to limitations, under applicable law regarding dievini’s rights under this provision; |
| ● | a provision allowing, among other matters, a former chairman of our supervisory board, a former nominee of dievini, and a former nominee of KfW to jointly take on the supervisory functions, which persons jointly may designate one or more other persons to be charged with the supervision of our company (instead of or together with the former chairman of our supervisory board), as applicable, to supervise our affairs if all of our supervisory directors are removed from office and to appoint others to be charged with the supervision of our affairs, until new supervisory directors are appointed by the general meeting on the basis of a binding nomination discussed above; |
| ● | a provision allowing the management board to temporarily replace a managing director who is no longer in office or unable to act, with another person or persons designated for this purpose by the management board and attributing the management of the company to the supervisory board in case all managing directors are no longer in office or unable to act; and |
| ● | a requirement that certain matters, including an amendment of our articles of association, may only be brought to our shareholders for a vote upon a proposal by our management board with the approval of our supervisory board. |
In addition, Dutch law allows for staggered multiyear terms of our managing directors and supervisory directors, as a result of which only part of our managing directors and supervisory directors may be subject to appointment or reappointment in any one year.
69
We are not obligated to, and do not, comply with all best practice provisions of the Dutch Corporate Governance Code.
We are subject to the Dutch Corporate Governance Code, or the DCGC. The DCGC contains both principles and best practice provisions on corporate governance that regulate relations between the management board, the supervisory board and the general meeting and matters in respect of financial reporting, auditors, disclosure, compliance and enforcement standards. The DCGC is based on a “comply or explain” principle. Accordingly, companies are required to disclose in their annual reports, filed in the Netherlands, whether they comply with the provisions of the DCGC. If they do not comply with those provisions (for example, because of a conflicting Nasdaq requirement), the company is required to give the reasons for such noncompliance. The DCGC applies to Dutch companies listed on a government-recognized stock exchange, whether in the Netherlands or elsewhere, including Nasdaq. We do not comply with all best practice provisions of the DCGC. See “Item 10. Additional Information — B. Memorandum and Articles of Association.” This may affect your rights as a shareholder and you may not have the same level of protection as a shareholder in a Dutch company that fully complies with the DCGC.
We have identified a material weakness in our internal control over financial reporting related to (i) lack of sufficient accounting and supervisory personnel, (ii) lack of appropriate level of technical accounting training; and (iii) lack of consistent performance of controls over financial reporting which, if not remediated appropriately or timely, could result in loss of investor confidence and adversely impact our stock price. If we are unable to remediate the material weakness, or if other material weaknesses are identified, we may not be able to report our financial results accurately, prevent fraud or file our periodic reports as a public company in a timely manner.
Our management is responsible for establishing and maintaining internal control over financial reporting, disclosure controls, and compliance with the other requirements of the Sarbanes-Oxley Act and the rules promulgated by the SEC thereunder. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with international financial reporting standards. A “material weakness” is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
We reported in our Annual Report 20-F for the year ended December 31, 2022, a material weakness in internal control over financial reporting primarily related to an IT system’s functionalities that were not configured to support segregation of duties in the recording of manual journal entries as well as in the authorization of purchase orders.
During 2023, we implemented measures to remediate this material weakness, and even though progress was made to strengthen our control environment, management concluded that at December 31, 2023, there was a material weakness in internal control related to (i) lack of sufficient accounting and supervisory personnel, (ii) lack of appropriate level of technical accounting training; and (iii) lack of consistent performance of controls over financial reporting. As a result, management concluded that our internal control over financial reporting was not effective as of December 31, 2023. While we are working to remediate the weakness as quickly and efficiently as possible, we cannot at this time provide an estimate of the time frame we expect in connection with implementing our plan to remediate this material weakness. These remediation measures may be time consuming, costly and might place significant demands on our financial and operational resources. If we are unable to remediate the material weakness, or are otherwise unable to maintain effective internal control over financial reporting or disclosure controls and procedures, our ability to record, process and report financial information accurately, and to prepare financial statements within required time periods, could be adversely affected, which could subject us to litigation or investigations requiring management resources and payment of legal and other expenses, result in material misstatements in our financial statements that could require a restatement of financial statements, negatively affect investor confidence in our financial statements and adversely impact our stock price.
Notwithstanding the material weakness identified as of December 31, 2023, we have concluded that the financial statements and other financial information included in this Annual Report on Form 20-F, fairly present in all material respects our financial condition, results of operations and cash flows as of, and for, the periods presented.
Our management has and continues to take measures to remediate this material weakness, as discussed in more detail under “Item 15. Controls and Procedures” of this Report and is committed to continue investing significant time and resources and taking actions to remediate the material weakness in training and hiring staff as well as supervisory personnel to further strengthen our internal control over financial reporting. Any failure to maintain or implement required new or improved controls, or any difficulties we encounter in their implementation, could result in additional significant deficiencies or material weaknesses, and result in material misstatements in our financial statements that could result in a restatement of financial statements.
70
Risks Related to ESG
Our ability to effectively monitor and respond to the rapid and ongoing developments and expectations relating to environmental, social and governance, or “ESG,” matters, including related social expectations and concerns, may impose unexpected costs or result in reputational or other harm that could have a material adverse effect on our business, financial condition, cash flows and results of operations and could cause the market value of our common shares to decline.
There are rapid and ongoing developments and changing expectations relating to ESG matters and factors such as the impact of our operations on the environment, corporate governance, management of business ethics, human rights diligence in our supply chain, and human resource development, which may result in increased regulatory, social or other scrutiny on us. Regarding climate risks, we are expected to address climate risks due to our own contribution to climate change (inside-out perspective), risks due to physical effects of climate change as well as transition risks (outside-in perspective), and interactions between both perspectives (“dual materiality”). If we are unable to adequately recognize and respond to such developments and governmental, societal, investor and NGO expectations relating to such ESG matters, we may miss corporate opportunities, become subject to additional scrutiny, incur unexpected costs or experience damage to our reputation or our various brands. If any of these events were to occur, there may be a material adverse effect on our business, financial condition, cash flows and results of operations and the market value of our common shares may decline. We have observed that in addition to the importance of their financial performance, companies are increasingly being judged by their performance on ESG matters. A variety of organizations measure the performance of companies on such ESG topics, and the results of these assessments are widely publicized. We may not comply with some or all of the standards or best practices put forth by such organizations or by governmental or regulatory bodies. In addition, investment in funds that specialize in companies that perform well in such assessments are increasingly popular, and major institutional investors have publicly emphasized the importance of such ESG measures to their investment decisions. In light of investors’ increased focus on ESG matters, there can be no certainty that we will manage such issues successfully. Any failure or perceived failure by us in this regard could have a material adverse effect on our reputation and on our business, share price, financial condition, or results of operations, including the sustainability of our business over time. We are committed to taking actions to manage ESG - related risks. Therefore, in 2023, we initiated a project focused on addressing ESG - related matters based on relevant legal requirements.
If we or our third-party suppliers fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could harm our business.
We are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemicals and biological materials. Our operations also may produce hazardous waste products. We generally anticipate contracting with third parties for the disposal of these materials and wastes. We will not be able to eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from any use by us of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
ITEM 4. INFORMATION ON THE COMPANY
| A. | History and Development of the Company |
On April 7, 2020, CureVac B.V. was incorporated under the laws of the Netherlands and became the holding company of CureVac AG in connection with our initial public offering on August 14, 2020, as part of a corporate reorganization (the “Corporate Reorganization”), in which the legal form of CureVac B.V. was converted from a Dutch private company with limited liability (besloten vennootschap met beperkte aansprakelijkheid) to a Dutch public company (naamloze vennootschap), and the articles of association of CureVac N.V. became effective. Following the Corporate Reorganization, CureVac N.V. became the holding company of CureVac AG. On September 26, 2022, CureVac AG entered into a plan of merger with CureVac Beteiligungsverwaltungs AG, with CureVac SE as the surviving entity and both CureVac AG and CureVac Beteiligungsverwaltungs AG as disappearing entities. The historical consolidated financial statements of CureVac AG included in this Annual Report became part of the historical consolidated financial statements of CureVac N.V. Our legal and commercial name is CureVac N.V.
Our principal executive offices are located at Friedrich-Miescher-Strasse 15, 72076 Tübingen, Germany, and our telephone number at this address is +49 7071 9883 0. Our additional offices are in Wiesbaden (Germany), Louvain La Neuve (Belgium), Amsterdam (Netherlands), Basel (Switzerland), and Boston (Massachusetts, United States).
71
Since August 14, 2020, our common shares are trading on Nasdaq under the symbol “CVAC.” Our agent for service of process in the United States is CureVac Inc., located at 250 Summer St. 3rd Fl., Boston, Massachusetts 02210.
The SEC maintains an internet website that contains reports and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov. Our website can be found at www.curevac.com. The information on our website is not incorporated by reference into this Annual Report, and you should not consider information contained on our website or any websites mentioned in this Annual Report to be part of this Annual Report.
B.Business Overview
Overview
We are a global biopharmaceutical company that is developing a new class of transformative medicines based on messenger ribonucleic acid, or mRNA, which has the potential to improve the lives of people. mRNA plays a central role in cellular biology in the production of proteins in every living cell. Our vision is to revolutionize medicine and open new avenues for developing therapies by enabling the body to make its own drugs. We are pioneers in successfully harnessing mRNAs designed to prevent infections and to treat diseases by mimicking human biology to synthesize the desired proteins. Our technology platform is based on a targeted approach to optimize mRNA constructs that encode functional proteins which either induce a desired immune response or replace defective or missing proteins using the cell’s intrinsic translation machinery. Our current product portfolio includes clinical and preclinical candidates across multiple disease indications in prophylactic vaccines, oncology, and molecular therapy.
Prophylactic Vaccines
In prophylactic vaccines, we are advancing our second-generation mRNA backbone against coronavirus (SARS-CoV-2) and a range of infectious diseases, including seasonal influenza, in collaboration with GSK.
Our improved second-generation mRNA backbone features targeted optimizations designed to improve intracellular mRNA stability and translation for increased and extended protein expression. These optimizations potentially allow for strong and early immune responses at low doses. This supports the development of multivalent vaccines to target spreading COVID-19 variants or different influenza strains as well as combination vaccines against different viral diseases.
COVID-19 Program in collaboration with GSK (second-generation mRNA backbone)
The collaboration on COVID-19 vaccine candidates with GSK, initiated in April 2021, aims to research, develop, and manufacture mRNA vaccines targeting relevant SARS-CoV-2 variants.
On January 5, 2024, CureVac announced positive data from a formal interim analysis in the ongoing Phase 2 clinical study assessing monovalent and bivalent modified COVID-19 vaccine candidates in direct comparison to a licensed bivalent mRNA-based COVID-19 comparator vaccine. The monovalent mRNA vaccine candidate, CV0601, encodes the Omicron BA.4-5 variant; the bivalent candidate, CV0701, encodes the Omicron BA.4-5 variant as well as the original SARS-CoV-2 virus. Both vaccine candidates apply CureVac’s proprietary second-generation mRNA backbone.
Initiation of the Phase 2 study with CV0601 and CV0701 was announced on August 1, 2023. The study is being conducted in Australia and is fully enrolled with 427 healthy adult participants, as announced on November 1, 2023. It assesses the safety and immunogenicity of different single booster doses of CV0601 and CV0701. While the monovalent candidate CV0601 was tested at a single medium dose level, the bivalent candidate CV0701 was tested at low, medium, and high dose levels. All tested dose levels were below those used in mRNA-based COVID-19 vaccines licensed in the U.S. and EU.
Results from the formal interim analysis showed that both candidates exhibited a favorable reactogenicity profile across all tested doses and were generally well tolerated with a lower or similar proportion of participants reporting solicited adverse events when compared to comparator vaccine participants within seven days of dosing.
72
Both candidates produced meaningful immune responses beginning at the lowest tested dose. Interim immunogenicity data showed meaningful titers of neutralizing antibodies for both candidates, which matched or numerically exceeded the titers induced by the licensed comparator vaccine at all tested doses except for the low dose level of CV0701. At the medium dose level tested for CV0601, neutralizing antibody titers against the Omicron BA.4-5 variant on day 29 following the booster vaccination were 5.0 times the pre-boosting titers, numerically exceeding the 3.6-fold ratio generated by the licensed comparator vaccine. At the low, medium, and high dose levels tested for CV0701, neutralizing antibody titers against BA.4-5 on day 29 following the booster vaccination were 2.7-fold, 3.7-fold, and 4.6-fold the titers before the booster, compared to a 3.6-fold ratio for the comparator vaccine.
The Phase 2 study was based on a Phase 1 study with the modified COVID-19 mRNA construct CV0501 also applying our second-generation backbone and encoding the Omicron BA.1 variant. CV0501 was the first vaccine candidate in the COVID-19 program to apply modified mRNA. Vaccination of the first participant in the Phase 1 study with CV0501 was announced on August 18, 2022. The study was conducted at clinical sites in the United States, Australia, and the Philippines to evaluate the safety, reactogenicity, and immunogenicity of a booster vaccination of CV0501 in 180 participants. Positive preliminary data from the Phase 1 study were reported in January and April 2023 and led to the selection of modified mRNA as the preferred technology for further clinical development in the COVID-19 program. The study was completed in August 2023.
The first representative of our joint COVID-19 vaccine program with GSK based on our second-generation backbone was CV2CoV, an unmodified mRNA construct, encoding the original SARS-CoV-2 virus. A Phase 1 clinical study with CV2CoV was announced on March 30, 2022, to evaluate the safety, reactogenicity, and immunogenicity of a booster vaccination of CV2CoV in 98 participants at clinical sites in the United States. The study was completed in March, 2023.
Seasonal Flu Program in collaboration with GSK (second-generation mRNA backbone)
Seasonal flu was disclosed as the first indication from the initial collaboration we started with GSK in July 2020. The collaboration focuses on the development of new products for different targets in the field of infectious diseases, excluding COVID-19.
On April 4, 2024, we announced promising interim data from an ongoing Phase 2 part of the combined Phase 1/2 study of a seasonal influenza vaccine candidate. The multivalent candidate was designed for broad antigen coverage, encoding antigens matched to all four WHO-recommended flu strains. Results from the planned interim analysis showed that the multivalent vaccine candidate boosted antibody titers against all encoded flu strains and across all age groups and tested dose levels, including the lowest tested dose. The vaccine candidate was shown to have an acceptable safety and tolerability profile, with the majority of solicited adverse events reported as either mild or moderate. Among younger and older adults, geometric mean titers generated by the vaccine candidate against influenza A strains numerically exceeded the geometric mean titers of the licensed comparator vaccines consistently across all tested dose levels. For influenza B strains geometric mean titers were lower than those elicited by the licensed comparator vaccines across both age groups and tested dose levels, in line with expectations and other initial mRNA-based clinical flu development programs. Further optimizations to enhance immune responses against influenza B strains will be tested in an additional Phase 2 study.
Initiation of the combined Phase 1/2 for seasonal flu was announced on May 8, 2023. The initial Phase 1 part compared a comprehensive series of multivalent, modified mRNA seasonal flu vaccine candidates with up to eight separate mRNA constructs per candidate, addressing all four WHO-recommended flu strains. Candidates were tested at different dose levels in 270 healthy younger adults (age 18-50). Positive data from an interim analysis of the Phase 1 part was reported on September 12, 2023. Interim safety data showed no safety concerns across all tested dose levels for the multivalent candidates. Immunogenicity of all candidates was assessed in parallel with a licensed seasonal flu vaccine comparator vaccine. The humoral responses observed supported the selection of the preferred vaccine candidate, which is currently being tested in the Phase 2 part of the study.
73
The combined Phase 1/2 study is based on a Phase 1 study with a modified, monovalent influenza vaccine candidate, Flu-SV-mRNA, announced on August 18, 2022. The vaccine candidate expresses an H1N1 hemagglutinin antigen (subtype of influenza A). The Phase 1 dose-escalation study is being conducted in Canada, Spain, and Belgium to evaluate the safety, reactogenicity, and immunogenicity of Flu-SV-mRNA in younger adults aged 18-45 and older adults aged 60-80. Preliminary safety and reactogenicity data in younger adults showed that the monovalent Flu-SV-mRNA candidate was generally well tolerated with no safety concerns observed to date across all tested dose levels. For older adults, a single dose of Flu-SV-mRNA (dose level undisclosed) was assessed for safety and reactogenicity and was also observed to be safe and well tolerated with no grade 3 adverse events in the 32 subjects who were administered the mRNA construct. Immunogenicity of Flu-SV-mRNA was assessed in parallel with a licensed seasonal flu vaccine comparator in both age groups. In younger adults, adjusted geometric mean hemagglutinin inhibition antibody titers elicited by Flu-SV-mRNA increased up to approximately 3.3 times those elicited by the licensed flu vaccine comparator in younger adults. In older adults, adjusted geometric mean hemagglutinin inhibition antibody titers elicited by Flu-SV-mRNA were approximately 2.3 times those elicited by the licensed flu vaccine comparator. In the same age group, the percentage of subjects achieving seroconversion was 89.7% for Flu-SV-mRNA and 56.2% for the licensed flu vaccine comparator.
The first vaccine candidate within the broader infectious disease program applying our second-generation backbone we tested in collaboration with GSK was the flu candidate, CVSQIV, a multivalent vaccine candidate featuring multiple unmodified mRNA constructs to induce immune responses against relevant targets of four different influenza strains. On February 10, 2022, we announced the start of a Phase 1 dose-escalation study in Panama evaluating the safety, reactogenicity, and immunogenicity of CVSQIV. Preliminary safety data reported on April 28, 2022, showed a benign reactogenicity profile across the tested dose groups. The study was completed in September 2022.
Avian Influenza in collaboration with GSK (second-generation mRNA backbone)
Avian influenza was disclosed as the latest program progressing to clinical trials under the initial collaboration we started with GSK in July 2020. The collaboration focuses on the development of new products for different targets in the field of infectious diseases, excluding COVID-19.
On April 24, 2024, the start of the Phase 1 part of a combined Phase 1/2 study of an investigational influenza A (H5N1) pre-pandemic vaccine candidate was announced. The study assesses a monovalent vaccine candidate encoding an influenza A H5-antigen. The H5N1 avian flu virus is considered a potential future pandemic threat, able to sporadically cross species from its original bird host to other animal hosts to humans.
Oncology
In oncology, we plan to build a meaningful portfolio and create long-term value to accelerate growth beyond the recent progress in prophylactic vaccines. Within our oncology strategy, we follow two approaches: (1) the development of off-the-shelf cancer vaccines based on tumor antigens shared across different cancer indications and (2) the development of fully personalized cancer vaccines based on a patient’s individual tumor genomic profile. Developing new cancer vaccine candidates is characterized by similar medical challenges as in infectious diseases, including selection and accessibility of disease-relevant antigens, enhancing antigen-induced immune activation, and triggering immune responses led by a strong induction of tumor-killing T cells.
A key component to deliver on the development of new cancer vaccines is the build-up of a powerful antigen discovery engine. To complement existing in-house expertise to identify and validate promising antigens for mRNA cancer vaccine candidates and gain access to state-of-the-art antigen discovery technologies, we announced a partnership with Belgium-based company myNEO Therapeutics (“myNEO”) on May 25, 2022, and the acquisition of Netherlands-based Frame Cancer Therapeutics (“Frame”) on June 8, 2022.
Together with immunotherapy company myNEO, we aim to identify specific antigens found on the surface of tumors for the development of novel mRNA immunotherapies. myNEO utilizes a broad range of underlying genomic alterations to identify constantly emerging, novel classes of antigens of defined tumor types. Incorporating new ranking methodologies based on tumor cell antigen processing and presentation is expected to allow for selection of antigens with the highest confidence of success for potential clinical testing. On November 22, 2023, both companies announced that CureVac exercised two exclusive options on selected sets of potential cancer vaccine shared antigen targets. The shared antigen targets identified by my NEO within the collaboration demonstrated strong immunogenicity in undisclosed preclinical studies. The most promising targets will be considered for validation for the design of potential mRNA cancer vaccine candidates.
74
With the acquisition of Frame, a private company focused on advanced genomics and bioinformatics, we added sophisticated methods to identify unique neoantigens across different cancer types. The former Frame site was inaugurated as CureVac Netherlands and will further develop the proprietary technology platform, which has the potential to identify a broad panel of neoantigens and tumor-associated antigens that go beyond conventional approaches. This could strongly increase the likelihood of developing highly effective cancer vaccines that activate the human immune system against cancer.
The field of immunotherapy has advanced with the progression of available technologies, such as next-generation sequencing, to extract data from patient samples. Conventional approaches have so far focused on the exome, the protein-coding part of the genome, which represents only about 1.5% of the total genetic information. More recently, breakthrough developments in sequencing capacity have enabled the extraction of vastly larger amounts of data that allows us to utilize the remaining 98.5% of genetic information.
The technologies brought in house with the acquisition of Frame are based on whole-genome-sequencing for every patient sample combined with short as well as long-range RNA sequencing to map the full inventory of genomic changes. More specifically, downstream of the sequencing, a software package integrates all the data to retrieve the exact changes in the DNA of the tumor cells compared to healthy cells. Correlation of this data with changes in the RNA transcription of the tumor results in entirely new and potentially antigenic tumor antigens that we plan to test as targets for a portfolio of new cancer vaccine candidates. These new antigens are not only entirely foreign to the body but are also uniquely expressed in the tumor and not in healthy tissue. In their foreignness, these constructs are expected to raise stronger immune responses than antigens derived from exome-based conventional approaches.
We recently extended our capabilities in oncology with a co-development and licensing agreement with The University of Texas MD Anderson (“M.D. Anderson”) Cancer Center, announced on April 16, 2024. The collaboration creates strong synergies between CureVac’s unique end-to-end capabilities for cancer antigen discovery, mRNA design, and manufacturing and M.D. Anderson’s expertise in cancer antigen discovery and validation, translational drug development, and clinical research. The focus of the collaboration is on the development of differentiated cancer vaccine candidates in selected hematological and solid tumor indications with high unmet medical need. Identification of differentiated cancer antigens via whole genome sequencing, combined with long- and short-read RNA-sequencing and cutting-edge bioinformatics will be followed by joint preclinical validation of the highest-quality cancer antigens and selection of the most promising clinical-lead candidates for conducting initial Phase 1/2 studies in appropriate clinical indications.
Under the collaboration agreement with M.D. Anderson, we are granted an exclusive, fee-bearing, sublicensable license under certain intellectual property to develop, manufacture and commercialize (i) products containing certain RNA-based cancer vaccine candidate(s) developed under the agreement, or M.D. Anderson Cancer Center (MDACC) Program Products, for any and all uses for cancer in humans throughout the world and (ii) certain other products that target one or more antigens identified under the agreement throughout the world and M.D. Anderson is granted an exclusive, fee-bearing, sublicensable license under certain intellectual property jointly created under the agreement to develop, manufacture and commercialize certain non-MDACC Program Products that target one or more antigens identified under the agreement throughout the world.
Further, we are solely responsible for the commercialization and commercial manufacturing of the MDACC Program Products for any and all uses related to cancer in humans worldwide. On a program-by-program basis upon completion of the first Phase I/II or Phase I clinical trial, as applicable, for a program, the parties agree to decide on a commercialization strategy through the joint steering committee, including whether such commercialization will be done by us or a third party via a partnership. On a program-by-program basis, each party will fund a specific percentage of all development costs incurred under the agreement. On a program-by-program basis, we will initially receive a percentage of the commercialization proceeds which is equal to our percentage of development costs, subject to certain assumptions and adjustments.
75
Investigational cancer vaccine CVGBM for surgically resected glioblastoma
To assess the safety and immunogenicity of our second-generation backbone in an oncology setting, we initiated a proof-of-principle Phase 1 study in patients with newly diagnosed surgically resected MGMT-unmethylated glioblastoma or astrocytoma with a molecular signature of glioblastoma on June 20, 2023. The open-label study evaluates the safety and tolerability of CVGBM, a cancer vaccine candidate featuring a single unmodified mRNA encoding eight epitopes derived from known tumor-associated antigens with demonstrated immunogenicity in glioblastoma. On April 24, 2024, we announced that the dose-escalation Part A of the study completed recruitment of all four dose levels. Following review of the safety data, the Data Safety Monitoring Board (DSMB) confirmed no dose limiting toxicities and gave a dose recommendation for 100 µg for the dose-confirmation Part B of the study. Part B is expected to start enrollment mid-2024. A first study data readout is expected in the second half of 2024.
The multiepitope design of CVGBM was supported by preclinical studies assessing the potency of a multiepitope mRNA cancer vaccine construct targeting tumors in a murine melanoma model. The data was presented at the 11th International mRNA Health Conference, hosted by CureVac from October 31 to November 2, 2023, in Berlin, Germany.
The preclinical mRNA construct encoding ten epitopes derived from the murine B.16.F10 melanoma cell line was tested in mice. The study applied three 5 µg doses of LNP-formulated B.16 mRNA, administered intramuscularly at weekly intervals. Data obtained on day 21 confirmed prominent induction of CD8+ and CD4+ T cell responses recognizing epitopes across the full multiepitope construct. Median survival of the animals increased to 30.9 days for treated mice compared to 23.2 days for a group vaccinated with formulated control mRNA.
Strong T cell activation is particularly encouraging and relevant, as the B16-F10 tumor model is characterized as a cytokine deficient “cold” tumor model that exhibits very little immune cell infiltration and resistance to check-point inhibitors. The data suggest that single-agent application of the multiepitope B.16 mRNA construct generated robust T cell activation in the tumor microenvironment, thereby inhibiting tumor growth and extending survival in the applied preclinical model.
The RNA Printer®
In addition to our GMP manufacturing facilities, we are developing a novel downsized, integrated, and automated process for manufacturing of mRNA vaccines and therapeutics, which we refer to as The RNA Printer®. The RNA Printer® is CureVac’s automated end-to-end manufacturing solution for GMP-grade mRNA vaccines and therapeutics and an integral part of our oncology strategy. The highly standardized system is expected to allow for rapid and highly flexible availability of mRNA to screen new targets and transition promising mRNA product candidates more efficiently into the clinic. Designed for small-scale quantities, the automated GMP-grade output of The RNA Printer® is designed to open avenues for personalized mRNA-based cancer therapies. On November 14, 2023, we announced that the system has successfully achieved the first regulatory milestone by obtaining a manufacturing license from the regional authority in Baden-Württemberg for an mRNA in our cancer vaccine development program to support CureVac’s oncology strategy. The second regulatory milestone was achieved on December 12, 2023, when The RNA Printer® received a drug substance framework manufacturing license from the regional authority in Baden-Württemberg. The regulatory review process is ongoing.
Molecular Therapies
Our development efforts for molecular therapy are based on delivering optimized mRNAs to trigger production of antibodies or therapeutic proteins. Using our technology, we can instruct human cells to produce or secrete specific proteins in the nucleus, cytoplasm, cellular organelles, cell membrane. Based on this “healthy” information delivered by mRNA, our cells can produce proteins, which are required to treat the disease caused by missing or inactive proteins.
Our mRNA optimization process, which is a core pillar of our RNA optimizer platform, is designed to increase protein expression with the aim to reach therapeutic levels. In preclinical studies in non-human primates, we have demonstrated that antibodies encoded by mRNA can be produced in hepatocytes very rapidly and can reach therapeutic levels in the blood stream. We are currently advancing undisclosed programs in preclinical studies across eye disorders as well as delivering therapeutic antibodies. Our work in eye disorders is carried out in collaboration with the Schepens Eye Research Institute. We expect to publish data from our collaboration in 2024.
76
Our approach seeks to mitigate clinical and developmental risk across multiple levels to advance and expand our broad product portfolio. We have made advances in utilizing the potential of our technology platform through rational disease selection. We consider a number of factors in our disease selection process including unmet medical need, immune response, duration of expression, dosing requirements, delivery, and targeted tissue types, among other factors. Our programs target the underlying modes of action of the disease that play a critical role in the pathology of the disease. We are initially targeting diseases that require an active immune response (such as prophylactic vaccines and cancer vaccines) and require transient expression of mRNA in tissue types that are more easily accessible. We believe these indications are amenable to localized delivery using a lipid nanoparticle, or LNP, delivery system. Following the encouraging results from our current prophylactic vaccines program in clinical studies and based on our advanced understanding of mRNA biology and immune stimulation control, we have expanded our product portfolio to also target indications that require an immune silent approach (such as protein delivery), given the need for higher doses, repeated dosing and longer expression of the protein. These initial indications are using LNP delivery systems.
We consider our manufacturing process an important part of our strategy that allows us to match our flexible and versatile technology platform with equally flexible and versatile manufacturing setups. In house, we currently operate three GMP-certified suites, with the capacity to supply our clinical programs and support potential early commercialization activities. We are in the process of building a fourth GMP large-scale production facility at CureVac’s headquarters in Tübingen, which is being designed to cover all manufacturing steps from starting material to formulation.
Financial Position
Our capital expenditures for 2023, 2022, and 2021 amounted to € 57.8 million, € 101.6 million, and €135.4 million, respectively. These expenditures were primarily for equipment and intangibles used in our research and development activities, as well as for the development of a GMP production process on a large scale (GMP IV).
To date, our revenues have consisted of upfront licensing payments, product sales, and compensation for research and development services, the majority of which relate to our license and collaboration agreements. For the years ended December 31, 2021, 2022, and 2023, €103.0 million, €67.4 million, and €53.8 million, respectively, or 100%, of our total revenue, in each respective year, was derived through our collaborations. Since our founding in 2000, we have raised €1.94 billion in gross proceeds from equity financings.
The following is a summary of revenue by geographic area. Revenue is attributed to geographic region based on the location of our license and collaboration partner:
| 2021 |
| 2022 |
| 2023 |
| |
North America |
| — | % | — | % | — | % |
Europe |
| 100.0 | % | 100.0 | % | 100.0 | % |
Rest of the World |
| — | % | — | % | — | % |
Intellectual Property Rights
We have built an intellectual property portfolio in the United States, Europe, and other major geographies. As of March 19, 2024, we own approximately 624 issued patents worldwide, including 103 issued U.S. patents, 43 issued European patents (which have been validated in various European countries resulting in a total of approximately 395 national patents in European countries) and including two European patents with unitary effect, one German national patent, and 126 issued patents in other foreign countries, 116 pending U.S. patent applications, 74 pending European patent applications, 311 pending patent applications in other foreign countries, and 18 pending PCT patent applications. Our patent portfolio includes claims relating to our RNA technology platform, our proprietary LNP technology, and our Influenza and SARS-CoV-2 product candidates.
77
Employees
We are led by a team of experts with extensive experience in the biopharmaceutical industry, including in nucleic acid therapy, oncology, rare and infectious diseases, and antibodies. Our management team as well as our supervisory board members have broad expertise in the clinical, regulatory, and commercialization aspects of prophylactic vaccines, oncology and molecular therapy, as well as in drug development, process development, and manufacturing for mRNA therapies. As the result of our organic growth, our workforce has increased from an approximate average workforce of 440 in fiscal 2019 to 1,126 as of December 31, 2023, including 301 employees with advanced scientific degrees.
Our Product Portfolio
Our differentiated mRNA technology platform is designed to address a broad range of diseases across multiple therapeutic areas. Given the strengths of our platform, the broad potential of mRNA-based medicines and our rational approach to disease selection, we have chosen to leverage our platform to initially focus on advancing our product candidates in the areas of prophylactic vaccines, oncology and molecular therapy.
A disease indication may require an approach that triggers an immune response (immune active) or that does not require immune activation (immune silent). Each of the disease indications that we are targeting require different levels of immune activation for the mRNA-based medicines to be effective. For the immune active side of our technology, we focus on mRNA-based medicines in prophylactic vaccines and oncology. For the immune silent side of our technology, we have expanded our preclinical product portfolio to include mRNA therapies based on the expression of therapeutic proteins (including ocular, liver and lung applications).
Our lead programs include:
| ● | Our vaccine candidates CV0601 (monovalent) and CV0701 (bivalent) against SARS-CoV-2, applying our second-generation mRNA backbone and modified mRNA, are being developed in collaboration with GSK. We initiated a Phase 2 study in August 2023, which provided positive interim results in January 2024. |
| ● | Our multivalent vaccine candidate against influenza, applying our second-generation mRNA backbone and modified mRNA, is being developed in collaboration with GSK. We initiated a combined Phase 1/2 study in May 2023 testing a comprehensive series of multivalent vaccine candidates. The preferred candidate selected from the Phase 1 part was advanced to the Phase 2 part in November 2023. The Phase 2 part provided interim results in April 2024. |
| ● | Our cancer vaccine candidate CVGBM is tested in patients with surgically resected Glioblastoma Multiforme. It applies our second-generation mRNA backbone and unmodified mRNA. It encodes eight epitopes derived from known tumor-associated antigens with demonstrated immunogenicity in glioblastoma. A Phase 1 study initiated in June 2023 is expected to provide data in the second half of 2024. |
Our key partnered programs include:
| ● | We have partnered with GSK for the development of COVID-19 vaccine candidates based on our second-generation mRNA backbone and vaccine candidates against other infectious diseases, including seasonal influenza, also based on our second-generation mRNA backbone. |
| ● | We have partnered with the immunotherapy company myNEO to identify specific antigens on the surface of tumors for the development of novel mRNA-based cancer vaccine candidates. The partnership leverages myNEO’s biological datasets and integrated machine learning and bioinformatics platform to identify and validate specific tumor antigens predicted to elicit a strong immune response. |
| ● | We have partnered with CRISPR Therapeutics for the development of novel Cas9 mRNA constructs for use in gene editing therapeutics, with improved properties such as increased potency, decreased duration of expression and reduced potential for immunogenicity. CRISPR Therapeutics has an exclusive license to the improved constructs in three of their in vivo gene editing programs. |
78
| ● | We have a broad strategic partnership with Genmab to leverage our mRNA technology platform to develop up to four mRNA-based novel therapeutic antibodies. This represents the first publicly announced strategic partnership focused on differentiated mRNA-based antibodies. |
| ● | We also have several academic collaborations, including with SERI for target discovery research in mRNA-based eye therapy. |
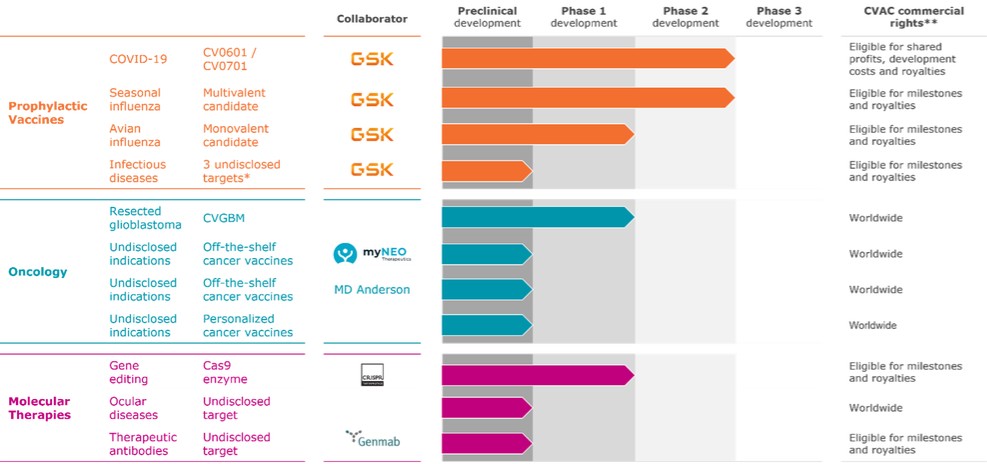
* | Unidentified indication. |
** | For further details on our collaboration agreements, see “Business — Collaborations” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations — Our Collaborations and Related License Agreements.” |
Our Strengths
We are developing a broad portfolio of product candidates currently in preclinical or clinical development stages that we believe position us at the forefront of targeted immune active and immune silent mRNA medicines. Our key strengths include:
| ● | We have a differentiated mRNA technology platform that has the potential to address a wide range of diseases. As the pioneers in the field of mRNA-based medicines, we have a deep understanding of mRNA biology, its interaction with the cellular translation machinery as well as the immune system. We have built our differentiated RNA optimizer platform to incorporate these insights over the past 23 years. We optimize mRNA to preserve critical protein-RNA interactions. Given the potential advantages of the mRNA-based medicines over existing treatment modalities, such as potential for broad application, wide range of activity, flexibility, design versatility, transient expression and a single manufacturing process, we believe that we have the potential to address a broad range of diseases across multiple therapeutic areas. Our technology platform has been validated in clinical and preclinical studies in selected disease indications. |
79
| ● | We have an optimized second-generation mRNA backbone, which catalyzes the expansion of our clinical development pipeline in prophylactic vaccines as well as oncology. Our proprietary second-generation mRNA backbone targets improved intracellular mRNA translation for increased and extended protein expression, resulting in early and strong immune responses. The second-generation mRNA backbone broadly spans unmodified and modified mRNA, as well as monovalent and multivalent vaccine formats to diversify and advance our product development pipeline. We aim to be at the forefront of delivering mRNA-based vaccines against a range of relevant infectious diseases and are executing on a broad mRNA vaccine program in collaboration with GSK. Second-generation backbone-based vaccines are expected to allow for flexible protection against one or more emerging COVID-19 variants and to enable new mRNA vaccines against other infectious diseases, such as influenza, as well as potential combination vaccines against different viruses. Alongside our recent progress in COVID-19 and flu we are also broadening our foundation in oncology and aim to build a meaningful portfolio of cancer vaccine candidates based on the second-generation mRNA backbone in combination with promising new tumor antigens predicted to elicit strong immune responses. |
| ● | We have a broad portfolio of mRNA-based medicines in preclinical and clinical development stages being designed for efficacy, safety and protein expression at relatively low doses. The potential of our technology optimized for immune activation in prophylactic vaccines and oncology has been observed in multiple clinical studies. We are developing our product candidates, have conducted preclinical studies and initiated Phase 1 and Phase 2 trials of vaccine candidates based on our second-generation mRNA backbone for COVID-19 and influenza. In oncology, we plan to build a meaningful portfolio of novel cancer vaccines based on our second-generation mRNA backbone to create long-term value and accelerate growth beyond the recent progress in prophylactic vaccines. A key component to deliver on this strategy is the buildup of a powerful antigen discovery engine. In the immune silent area molecular therapy, our approaches optimized for protein therapies have been evaluated in multiple preclinical disease models. |
| ● | We have a strong clinical development pipeline driven by the technological advantages of our versatile second-generation mRNA backbone. Our development focus in the immune activation area is on our second-generation mRNA backbone. In infectious diseases, we leverage this backbone to advance COVID-19 and flu vaccine candidates jointly developed with GSK. On January 5, 2024, we announced positive data from a formal interim analysis in the ongoing COVID-19 Phase 2 clinical study assessing the monovalent mRNA vaccine candidate, CV0601, encoding the Omicron BA.4-5 variant and the bivalent mRNA candidate, CV0701, encoding the Omicron BA.4-5 variant as well as the original SARS-CoV-2 virus. All tested dose levels were below those used in mRNA-based COVID-19 vaccines licensed in the U.S. and EU. Results from the formal interim analysis showed that both candidates exhibited a favorable reactogenicity profile across all tested doses and were generally well tolerated with a lower or similar proportion of participants reporting solicited adverse events compared to a licensed comparator. Both candidates produced meaningful immune responses beginning at the lowest tested dose with neutralizing antibody titers matching or numerically exceeding the titers induced by the licensed comparator vaccine at all tested doses except for the low dose level of CV0701. In seasonal influenza, we announced promising interim data on April 4, 2024 from the Phase 2 part of the combined Phase 1/2 study of a multivalent candidate, encoding antigens matched to all four WHO-recommended flu strains. Results showed that the multivalent vaccine candidate boosted antibody titers against all encoded flu strains and across all age groups and tested dose levels, and exhibited an acceptable safety and tolerability profile, with the majority of solicited adverse events reported as either mild or moderate. Among younger and older adults, geometric mean titers generated by the vaccine candidate against influenza A strains numerically exceeded the geometric mean titers of the licensed comparator vaccines consistently across all tested dose levels. For influenza B strains geometric mean titers were lower than those elicited by the licensed comparator vaccines across both age groups and tested dose levels. Further optimizations to enhance immune responses against influenza B strains will be tested in an additional Phase 2 study. On April 24, 2024, we announced the start of a combined Phase 1/2 study in avian influenza (H5N1). |
In oncology, we leverage our second-generation mRNA backbone in a Phase 1 study in patients with newly diagnosed resected glioblastoma, initiated in June 2023. The study evaluates the safety and tolerability of CVGBM, a cancer vaccine candidate featuring a single unmodified mRNA encoding eight epitopes derived from known tumor-associated antigens with demonstrated immunogenicity in glioblastoma. On April 24, 2024, we announced that the dose-escalation Part A of the study completed recruitment of all four dose levels. Following review of the safety data, the Data Safety Monitoring Board (DSMB) confirmed no dose limiting toxicities and gave a dose recommendation for 100 µg for the dose-confirmation Part B of the study. Part B is expected to start enrollment toward mid-2024.
80
| ● | We have the ability to target different tissue types based on our delivery systems. We have access to a number of mRNA delivery systems, including third-party and our proprietary systems, which allow us to target distinct tissues in an optimal way. Our current clinical prophylactic vaccine and cancer vaccine programs rely on LNP-based delivery systems administered intramuscularly and providing access to the immune cells. Moreover, LNP-based systems deliver mRNA efficiently to the hepatocytes in the liver, if administered intravenously. Protein expressed in the liver may either restore a specific function in the liver itself or produce secreted proteins for release into circulation. We rely on third-party state-of-the-art LNP delivery systems for our current clinical programs, but we are also developing our own proprietary LNP delivery system. In our proprietary LNP programs, we demonstrated that changes to the ratio and/or composition of the LNP constituents can be applied to fine tune LNP physico-chemical properties and steer immune responses as well as overall biological activity. |
| ● | We have invested in building our in-house manufacturing infrastructure, capabilities and expertise to rapidly, efficiently and cost-effectively produce mRNA-based medicines. We have continued to invest in our manufacturing platform since 2000 and have manufactured thousands of mRNAs. We consider our manufacturing process an important part of our strategy that allows us to continuously improve our technology platform and maintain flexibility in clinical development. Our manufacturing setup allows us to drive innovation and to maintain flexibility as well as to pivot quickly in clinical development and potential commercialization. In house, we currently operate three GMP-certified suites, with the capacity to supply our clinical programs and support potential early commercialization activities. We are in the process of building a fourth GMP large-scale production facility at CureVac’s headquarters in Tübingen, which is being designed to cover all manufacturing steps from starting material to formulation. All our mRNA-based active ingredients for various fields of application originate from a common technology platform and are based on standardized source materials, which enables us to produce mRNA-based medicines using a platform process concept. |
| ● | We have entered into strategic partnerships with leading biopharmaceutical companies and research and nonprofit institutions to expand the applications of our technology platform. We have a history of partnering with leading biopharmaceutical companies such as GSK, myNEO, CRISPR Therapeutics, Genmab, and Bayer. We also have received research grants from the Bill & Melinda Gates Foundation and CEPI for the development of several prophylactic vaccines. Our academic collaborations are focused on identifying and evaluating novel targets in selected therapeutics areas such as our collaboration with the M.D. Anderson Cancer Centre in selected hematological and solid tumor indications with high unmet medical need. These partnerships and collaborations allow us to expand the application of our platform and bring in external expertise and capabilities. |
| ● | We have built an intellectual property portfolio in a variety of markets for our platform and product candidates. As pioneers in the field of mRNA therapies, we have built an intellectual property portfolio in the United States and other major geographies, which is one of the broadest and most diverse intellectual property portfolios in the field. As of March 19, 2024, we own approximately 624 issued patents worldwide, including 103 issued U.S. patents, 43 issued European patents (which have been validated in various European countries resulting in a total of approximately 395 national patents in European countries) and including two European patents with unitary effect, and 126 issued patents in other foreign countries, 116 pending U.S. patent applications, 74 pending European patent applications and 311 pending patent applications in other foreign countries and 18 pending PCT patent applications. These patents include claims relating to our mRNA technology platform, our proprietary LNP technology and other product candidates. We believe our patent applications and other patents are the most cited among mRNA companies’ intellectual property. |
| ● | We have a long history of mRNA research and development and are led by an experienced management team. We are led by veterans of the biopharmaceutical industry with extensive experience in nucleic acid therapy, oncology, rare and infectious diseases and antibodies. Our management team as well as our supervisory board members have broad expertise in the clinical, regulatory and commercialization aspects of oncology, prophylactic vaccines and rare diseases as well as in drug development, process development and manufacturing for mRNA-based medicines. Members of our management team have held senior positions at Novartis, Ipsen, Sanofi, Roche, Roche/Genentech, Merck KGaA, Schering Plough, Pixium Vision and other companies. As of December 31, 2023, our broader team included 294 individuals with advanced scientific degrees working on advancing our mRNA platform. |
81
Our Strategy
Our goal is to build a leading, fully integrated mRNA-based medicines company that can transform the lives of people. The key components of our strategy include:
| ● | Continue to invest in our proprietary technology platform to be the leading mRNA platform company. We intend to invest in our proprietary technology platform to broaden its potential across therapeutic areas, in addition to broadening our pipeline in existing therapeutic areas. We believe our continued investment will enable us to further optimize the three core pillars of our technology platform — protein design, mRNA optimization and mRNA delivery — and to further enhance our treatment approaches by offering higher selectivity, greater protein expression, potential combination therapies and reduced or flexible dosing. We are continuing to build on our deep expertise in mRNA-based medicines based on what we have learned from our current programs to apply to our future programs. |
| ● | Utilize a rational disease selection approach to minimize clinical and commercial risk for our programs and broader platform. Our strategy is to maximize the potential of our technology platform through our rational disease selection approach to clinical development. We are initially targeting diseases that require an active immune response (such as prophylactic vaccines and cancer vaccines) and require transient expression of mRNA in tissue types that are more easily accessible. We are also developing a product portfolio targeting diseases that require an immune silent approach (such as molecular therapy). |
| ● | Rapidly advance our lead product candidates through clinical development and regulatory approval. Our product candidates are currently in preclinical or clinical development stages. In COVID-19, we are currently conducting a Phase 2 clinical study assessing the monovalent mRNA vaccine candidate, CV0601, which encodes the Omicron BA.4-5 variant and the bivalent mRNA candidate, CV0701, which encodes the Omicron BA.4-5 variant as well as the original SARS-CoV-2 virus. The candidates are tested in direct comparison to a licensed bivalent mRNA-based COVID-19 comparator vaccine. Results from a formal interim analysis were announced on January 5, 2024, and showed that both candidates exhibited a favorable reactogenicity profile across all tested doses and were generally well tolerated. Both candidates produced meaningful immune responses beginning at the lowest tested dose. Interim immunogenicity data showed titers of neutralizing antibodies for both candidates, which matched or numerically exceeded the titers induced by the licensed comparator vaccine at all tested doses except for the low dose level of CV0701. In seasonal influenza, within the broader second-generation infectious disease program we are developing in collaboration with GSK, we are conducting a combined Phase 1/2 study. The currently ongoing Phase 2 part of the study assesses a multivalent candidate, which was designed for broad antigen coverage, encoding antigens matched to all four WHO-recommended flu strains. In oncology, we are conducting a Phase 1 study in patients with newly diagnosed surgically resected glioblastoma, initiated in June 2023. The study evaluates the safety and tolerability of CVGBM, a cancer vaccine candidate featuring a single unmodified mRNA encoding eight epitopes derived from known tumor-associated antigens with demonstrated immunogenicity in glioblastoma. A first data readout is expected in the second half of 2024. |
We believe that by initially targeting diseases with high unmet medical need, we will be able to rapidly advance our programs through clinical development. We intend to pursue the appropriate regulatory pathways available to further accelerate our development efforts.
| ● | Continue to invest in our manufacturing capabilities across all manufacturing steps from starting material to formulation to further add scale and flexibility. We believe that our manufacturing capabilities are a key strategic advantage that offer us flexibility, scalability, versatility and reliability in discovery and development. We are currently building our GMP IV facility, which is being designed to cover all manufacturing steps from starting material to formulation. In addition, we are developing a new integrated and automated process for manufacturing of mRNA vaccines and therapeutics, The RNA Printer®. The RNA Printer® is CureVac’s end-to-end solution for integrated and automated manufacturing of small-scale quantities of GMP-grade mRNA vaccines and therapeutics. On April 24, 2024, we announced that the system achieved an important milestone in an ongoing regulatory review process. The system was granted a framework license that significantly broadens its regulatory freedom and flexibility to manufacture different mRNA vaccine candidates in support CureVac’s oncology strategy. The framework license represents an extension of the initial manufacturing license CureVac announced on November 14, 2023. |
82
| ● | Selectively seek strategic partners to develop and commercialize product candidates in certain therapeutic areas and geographies. We plan to continue to seek additional partnerships with other leading biopharmaceutical companies with specialized capabilities, including development and commercialization expertise in selected therapeutic areas and geographies. We may pursue partnerships that allow us to expedite the discovery and development of product candidates, complement our internal development expertise, broaden the breadth of our technology platform, and provide us with non-dilutive financing, while allowing us to retain economic rights to our product candidates that we view as strategically important. Our approach of partnering with biopharmaceutical companies allows us to execute on a broad range of programs simultaneously, while mitigating our drug development risk. |
| ● | Seek strategic acquisitions or in-licenses of technology or assets that are complementary to our programs and technology platform. mRNA-based medicines is an emerging field with ongoing advancements and discoveries. As the pioneers in the field, we have made significant strides in advancing and optimizing our technology platform over the past 23 years. We may seek acquisitions and in-licensing opportunities that can augment our internal expertise, expand our competitive differentiation and further enhance our mRNA technology platform. In June 2022, we announced the acquisition of Frame Cancer Therapeutics, a private company focused on advanced genomics and bioinformatics, to identify both shared and unique neoantigens across different cancer types. The acquisition complements existing in-house expertise to identify and validate promising antigens for mRNA cancer vaccine candidates. The acquired technologies will support the development of a strong antigen discovery engine, which has the potential to identify a broad panel of neoantigens and tumor-associated antigens that go beyond conventional approaches and could strongly increase the likelihood of developing highly effective cancer vaccines that activate the human immune system against cancer. |
| ● | Strengthen and expand our intellectual property portfolio to protect our scientific and technical know-how. We intend to continue to strengthen and expand our intellectual property to protect our advances in scientific and technical know-how. Our intellectual property strategy is focused on covering advancements in our technology platform, manufacturing processes, and product candidates. In addition to patent protection, we also rely on trade secrets and confidentiality agreements to protect other proprietary information that is not patentable or that we elect not to patent. |
Overview of mRNA Therapeutics
The Role of mRNA
mRNA-based medicines represent a novel class of medicine that have the potential to address limitations of conventional treatment modalities. We believe the modular nature of mRNA has the potential for higher efficacy, greater speed and lower cost of production as compared to conventional treatment modalities. mRNA delivery enables direct production of any protein (secreted, membrane and intracellular) in the body and has shown a wide range of activity. The flexible chemical structure of mRNA, utilizing only four nucleotide buildings blocks, allows us to encode for a broad range of proteins with simple sequence changes, offering design versatility, specificity and limited off-target effects. Transient expression of mRNA limits the risk of irreversible changes to the cells’ DNA and allows for flexible dosing based on a patient’s needs as well as the opportunity for repeat dosing. We are leveraging these inherent advantages of mRNA-based medicines in the development of our mRNA technology platform.
We have built an extensive expertise in the fields of mRNA biology, optimization and production. We have continued to invest in developing our technology platform, which we refer to as the RNAoptimizer, over the past 23 years. Our differentiated technology platform is designed to optimize each component of the mRNA-based medicine. Our RNAoptimizer platform is built on three core pillars:
| ● | Protein design: optimizing the specific properties of encoded protein; |
| ● | mRNA optimization: optimizing characteristics such as half-life and translation efficacy of the mRNA molecule; and |
| ● | mRNA delivery: selecting the best-suited delivery system from our diverse portfolio of proprietary and third-party delivery systems. |
83
mRNA is a molecule instructing the translation of genetic information encoded in DNA by cells into proteins, which carry out essential cellular functions. As depicted in the figure below, genetic information stored in DNA is transferred to mRNA in a process called transcription in the cell nucleus. In transcription, double-stranded DNA is temporarily unwound and copied into single-stranded mRNA by the enzyme RNA polymerase. mRNA is then transported to the cytoplasm where it instructs synthesis of proteins through a process called translation. In translation, cellular structures called ribosomes decode mRNA bases in groups of three (called codons) as amino acids. Each codon specifies a particular amino acid, and amino acids are the building blocks of protein molecules which perform distinct functions within the body.
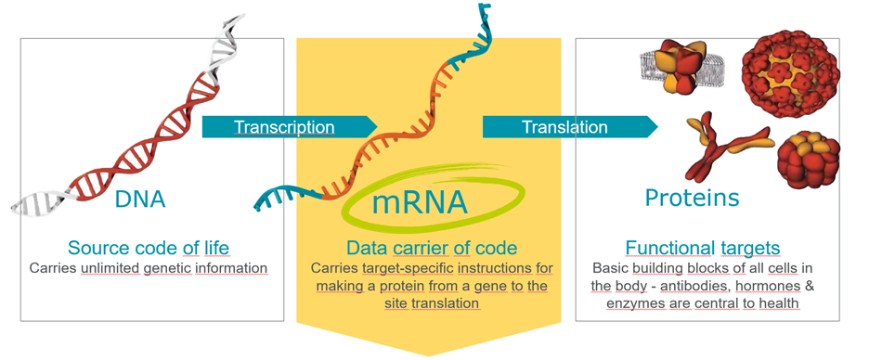
Limitations of Existing Treatment Modalities
There are several existing treatment modalities that seek to address the underlying cause of absent or defective proteins associated with diseases, including protein replacement therapy, gene therapy, gene editing, RNA interference and small molecule therapies. Other treatment modalities seek to harness the immune system, including antibody therapies and traditional prophylactic vaccines. Each of these treatment modalities have certain limitations as discussed below:
| ● | Protein Replacement Therapy: While this approach has been successfully used to treat a subset of protein-based disorders, it is mostly limited to proteins that function outside of the cell. |
| ● | Antibody Therapy: Antibody therapeutics are largely administered intravenously and, being proteins themselves, have applications largely limited to surface molecules. In addition, antibodies have historically faced challenges due to their relatively large size, inadequate pharmacokinetics and tissue accessibility as well as unwanted interactions with the immune system. |
| ● | Gene Therapy: Gene therapy is usually a one-time intervention meant to provide lasting levels of therapeutic protein. While expected to be a one-time treatment, the duration of treatment efficacy is still largely unknown and it may not be amenable to repeat dosing due to neutralizing antibodies against the gene therapy vehicle. In addition, large-scale manufacturing is costly, time consuming and complex. |
| ● | Gene Editing: Despite its promise, gene editing is still in the early-stages of development and has potential risks related to unwanted on and off-target DNA modifications, incomplete targeting or mosaicism that hinder intended modifications. Similar to gene therapies, manufacturing complexities and costs for gene editing are also challenging. |
| ● | RNA Interference: RNA interference has potential in silencing certain genes but has limitations in replacing defective or missing proteins, as well as highly expressed proteins. Most of the current efforts in this treatment modality are focused on genes expressed in the liver, with limited evidence of applications in extra-hepatic tissues. |
| ● | Small Molecule: While small molecules offer advantages over other treatment modalities in terms of biodistribution, tolerability, and delivery, they do not directly address specific gene defects and have a high potential to cause off-target toxicities. |
84
| ● | Traditional Prophylactic Vaccines: While traditional prophylactic vaccines are one of the most successful and cost-effective global health interventions, their complex development and costly production processes create a high barrier to entry, long development cycle and limitation in developing vaccines with high serotype coverage. |
mRNA as a Novel Treatment Modality
mRNA, as the universal template for protein synthesis, can direct the synthesis of any protein in the body. To treat a medical condition, we identify a target protein and encode the information required to synthesize this protein on the mRNA. The mRNA, optimized using our platform, carries this code to give a patient’s body the information to produce its own, custom-tailored protein as medicine.
mRNAs are typically characterized by their rate of translation into protein and their short and predictable, yet steerable half-life. We optimize these mRNA properties for specific therapeutic needs to provide the most efficacious mRNA-based medicine. mRNAs provide the flexibility to deliver medicines that are required for a limited time as well as the opportunity to deliver repeated doses that can be adjusted to patient needs. The development and manufacturing of mRNA-based medicines can also proceed much more quickly than traditional protein-based therapies, including antibodies.
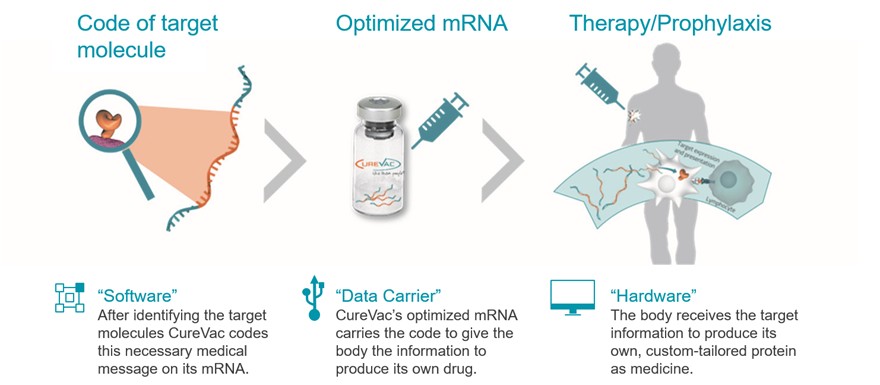
Key potential advantages of mRNA therapies that could position it as a novel treatment modality include:
| ● | Broad application: mRNA has the ability to produce all types of proteins, including secreted, membrane and intracellular proteins. This enables broad applicability across a variety of diseases. |
| ● | Natural biology: mRNAs mimic human biology to produce proteins in the body in contrast to recombinant proteins that are manufactured using processes that are foreign to the body. |
| ● | Wide range of activity: mRNAs can be used to create therapies that can be applied as an agonist, an antagonist or for vaccines. |
| ● | Flexibility: A large number of alternative mRNA candidates can be generated in a short time and tested to optimize both the mRNA and protein format. |
| ● | Design versatility: Therapeutic protein expressed from mRNA in situ can be designed for efficacy without being limited by the constraints which recombinant proteins are subject to. |
| ● | Specificity: mRNA-based medicines encode proteins which offer much higher specificity of interactions compared to small molecule drugs, which limits any potential off-target effects. |
85
| ● | Repeat dosing: mRNA-based medicines can be dosed repeatedly given their low immunogenicity. |
| ● | Transient expression: Short-lived expression of mRNA limits the risk of unforeseen adverse effects of lasting protein expression (as seen in gene therapy and gene editing) and allows for modified dosing schedules adjusted based on a patient’s needs. |
| ● | Manufacturing: mRNA production process is largely independent of the encoded protein as changes to the mRNA sequence do not affect its chemical and physical properties, allowing for higher efficiency, greater speed and lower cost of production. |
Historical Challenges with Developing mRNA Treatments
Using mRNA as a treatment has long been of interest given its potential to address limitations of existing treatment modalities. However, mRNA has historically been limited by the following theoretical and practical hurdles:
| ● | Stability: Naked mRNA is rapidly degraded by RNase enzymes present throughout the body which limits the duration of its therapeutic effect. An effective mRNA would need to be masked from these enzymes. |
| ● | Uptake by cells: Uptake of naked mRNA into cells is relatively inefficient. A more effective mRNA-based medicine would need a delivery system that delivers mRNA efficiently into cells. |
| ● | Expression level: Protein expression levels from synthetic mRNA obtained by in vitro production have been considered too low historically for therapeutic purposes, which underlines the need for an optimized mRNA construct. |
| ● | Immunogenicity: Non-optimized mRNA in the body rapidly activates receptors on immune cells which triggers the innate immune response and can lead to shut down of protein translation in cells. An effective mRNA-based medicine needs to modulate the immune system according to the disease indication being targeted. |
| ● | Tissue targeting: Each indication requires delivery to a specific tissue. An effective mRNA-based medicine would need a delivery system that efficiently delivers mRNA to a specific target tissue with low off-target delivery and toxicity. |
| ● | Manufacturing: mRNA manufacturing technology must be scalable and cost-effective to enable large production for multiple clinical trials and commercialization. |
Our Technology Platform
The therapeutic potential of mRNAs was discovered by our co-founders in 2000. As the pioneers in the field of mRNA, we have built extensive expertise in mRNA biology, optimization and production. We have developed our technology platform, called RNAoptimizer, through continued investments over the past 23 years. We believe that we have created the broadest and most versatile platform to develop optimized mRNA-based medicines that has potential to offer differentiated profile in terms of safety, stability and expression.
Our optimization approach covers three pillars: protein design, mRNA optimization and mRNA delivery. Our approach is based on the extensive data libraries we have generated to date. To improve protein expression from in vitro produced mRNA, we isolated high numbers of human natural mRNAs from different cells and identified elements which stabilize mRNA and improve their interaction with the cellular translation machinery. Our technology platform broadly spans unmodified and modified mRNA, as well as monovalent and multivalent formats. We continue to invest in all levels of optimization to improve the methods we currently employ and continue advancing mRNA-based medicines.
We have a long track record of performing clinical trials with multiple product candidates since 2008. The data generated in these clinical trials has allowed us to better understand the biology of mRNA and to further accelerate development in new therapeutic areas and approaches. We were the first company to demonstrate that mRNA vaccines can induce protective antibody titers in naïve human subjects with a previous version of our current rabies vaccine product candidate.
86
Our product candidates consist of two major components: the protein-coding mRNA and a delivery vehicle. Once we have established delivery capability to a target tissue, we can design new product candidates that vary only in the mRNA component, which we expect will allow for rapid target and development candidate identification. We believe that this will enable our platform to be flexible and scalable as we develop additional product candidates.
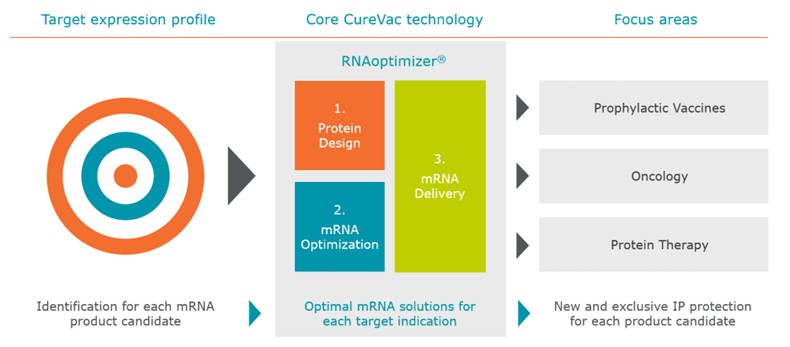
Our process for creating novel mRNA therapies comprises the following three pillars:
| ● | Protein Design: Our goal is to define the amino acid sequence to optimize specific properties of the encoded protein. |
| ● | mRNA Optimization: Our goal is to define the nucleotide sequence of the mRNA encoding the optimized protein to improve the properties of the mRNA molecule. |
| ● | mRNA Delivery: Our goal is to define mRNA encapsulation and delivery to select the optimal formulation for each specific indication and tissue. |
First Pillar: Protein Design
Proteins play a central role in biology, including formation of the structural framework of the body, aiding in intra- and extracellular transport, biological catalysts (such as enzymes), controlling the activity of cells, and enabling signal transduction throughout the body. Accordingly, mutations that alter the function of a protein that plays a critical role inside the body can disrupt normal development and cause disease. Diseases could be caused by low expression, over expression, or abnormal structures for specific proteins.
We target diseases that are caused by these abnormal or missing proteins. Once our team identifies the protein of interest for a specific vaccine or therapeutic target with a defined target product profile, protein design further improves the potential efficacy by adaptation of the amino acid sequence. Protein design is based on modulation of beneficial protein characteristics that are not present in the naturally occurring protein. We have a library of validated protein domains that can be leveraged using a combinatorial approach to optimize the properties of the target protein.
Our protein design process considers multiple factors before the protein is encoded in the mRNA, including half-life, stabilization of tertiary structure, oligomerization, secretion, and immunogenicity. We have the ability to modify each of these parameters while ensuring that these modifications work in harmony with the required function of the target protein.
87
Protein design always depends on the function of the individual protein of interest. The protein can serve as a therapeutic protein without any activation of the immune system or the protein can serve as an antigen with the goal of inducing strong immune responses against it. We employ different optimization strategies to support these distinct functions and requirements. For example, we can enhance certain parameters to extend the half-life or localization of a protein in the case of therapeutic proteins while making sure that RNA sensors remain muted to avoid activation of the immune system. For vaccines, our goal is to induce an optimal immune response mimicking response induced by bacterial or viral infections. Therefore, protein design is always bespoke and multi-factorial to support distinct functions and requirements of the specific target protein.
Below are several specific examples of protein modifications by which we designed a protein’s properties relative to the wild-type protein:
Extended Half-Life of Secreted Protein
This approach relies on the addition of supplementary short domains to the coding sequence of the protein of interest. Although this fusion increases protein size, the additional domains recruit binding proteins already present in blood which promote stabilization of the target protein by preventing proteolytic degradation. To support the efficient persistence of a secreted protein in the bloodstream, we can improve the half-life of this protein by adding specific, endogenous domains. By tailoring the pharmacokinetic profile of secreted proteins, we have the ability to reduce the frequency of dosing, generating a better therapeutic window, and using less material.
For example, wild-type erythropoietin (Epo) is a protein that has a very short half-life of three to four hours in the bloodstream. In a preclinical model, mice were dosed with mouse Epo and protein engineered mouse Epo, both encoded with our optimized mRNA. Dosing with the engineered mouse Epo protein showed an increase in serum titers and pharmacokinetic profile. We were able to increase the half-life and availability of functional Epo in blood from four days to two weeks by fusing endogenous Epo to a selected domain. Notably, both mRNA-encoded Epo proteins showed significantly higher protein expression levels than the injected recombinant Epo, which was cleared from the bloodstream after a single day.
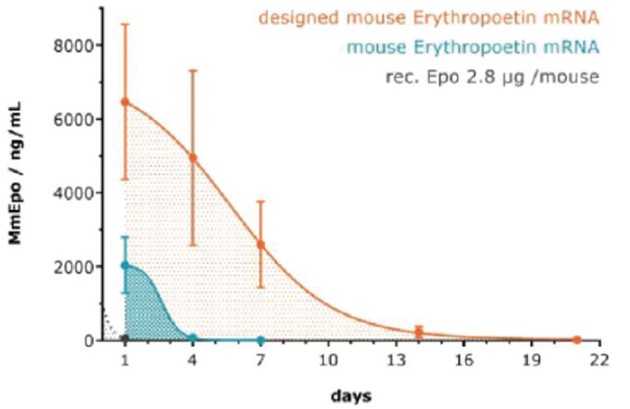
Mice received a single injection in the tail vein of recombinant protein (control) or mRNA encoding proteins. Mice received 2.8 μg of recombinant mouse Epo protein. Wild-type Epo encoded by our optimized mRNA and engineered Epo protein encoded by our optimized mRNA were administered at a dose of 0.4 mg/kg giving rise to relevant serum titers of functional Epo and different pharmacokinetic profiles.
88
Extended Half-Life of Intracellular Protein
Similar approaches can also be applied to intracellular proteins, promoting the half-life of functional target proteins. In the example below, protein variant 1 represents the fusion of a protein of interest with a selected protein domain, while variant 2 represents a construct with a single point mutation within the protein of interest. In contrast to the wild-type protein, both engineered protein variants enabled the detection of protein even one week after mRNA delivery to hepatocyte cells in culture.

Intracellular abundance of engineered protein variants in comparison to unmodified wild-type protein. Protein levels were determined by whole cell Western Blot analysis in human hepatocytes, followed by normalization to signals from a cytosolic loading control and relative to the wild-type protein. Same doses used in wild-type and engineered protein variants.
89
Increased Oligomerization
Protein oligomerization is a process that converts monomers to macromolecular complexes through polymerization. We can engineer protein oligomerization by adding domains capable to perform this process to the target protein. As antigens need to be secreted and build clusters to form virus-like particles, or VLPs, this oligomerization process is beneficial in boosting the immune response.
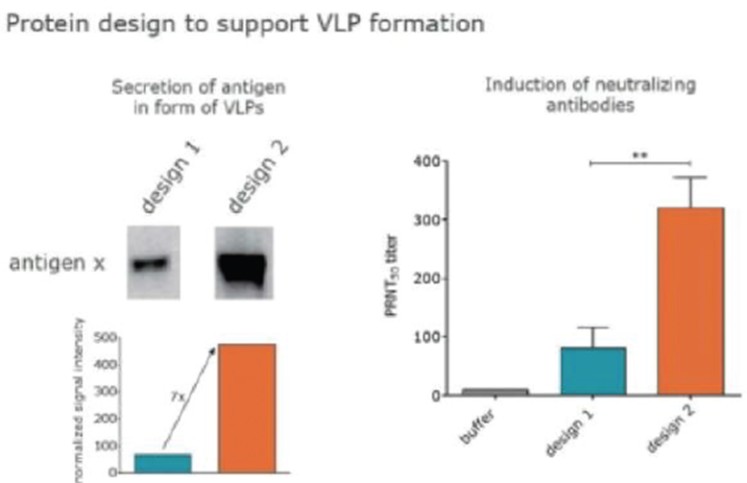
Protein sequence of viral antigen was optimized (design 2) by adding an element promoting secretion and clustering of antigen. In the left-hand side of the graphic, secretion of antigen in form of clusters was confirmed by Western Blot analysis of supernatants from transfected human cells. In the right-hand side of the graphic, vaccination of mice with an mRNA vaccine based on this improved protein design resulted in higher immunogenicity, measured by induction of virus neutralizing antibodies.
Improved Secretion
The potency of secreted target proteins can be improved by using alternative, more powerful signal peptides. These signal peptides are responsible for transporting the target protein from the cytoplasm to the outside of the cell, where the secreted protein fulfills its primary function. We screen large libraries of signal peptides to optimize secretion of any given target protein and in any cell type of choice.
90
For example, we selected a set of 87 verified signal peptides to maximize secretion. These were combined with the novel target protein via automated cloning to enable facile screening and selection of the most potent product candidate. In the figure below, the top hit from this screen increased the secreted protein levels in primary human muscle cells by three-fold relative to the native signal peptide.
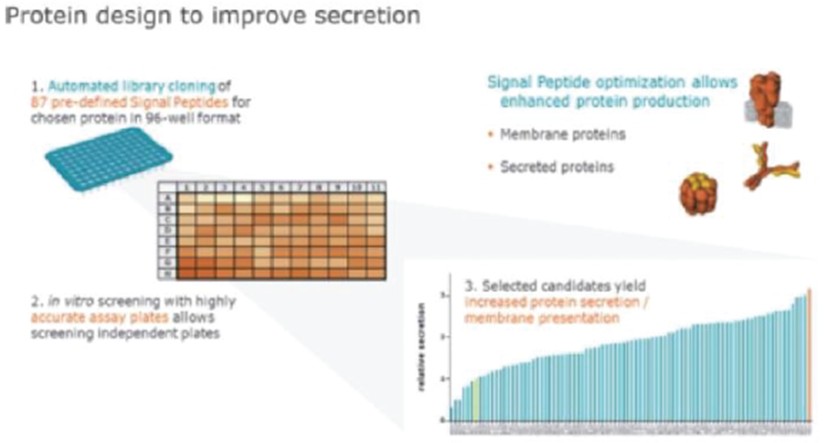
Modified Immunogenicity
If the target protein serves as a therapeutic agent, it is important to curb the protein’s natural immunogenicity. Our protein design process analyzes and replaces immunogenic epitopes, masking immunogenic epitopes and thereby rendering the target protein more immunosilent. In contrast, we also have the ability to improve immunogenicity for certain applications (for example in a cancer vaccine) by protein design. These protein sequence adaptations promote immunogenicity and suppress tumor growth in mouse models, as shown in the below example.
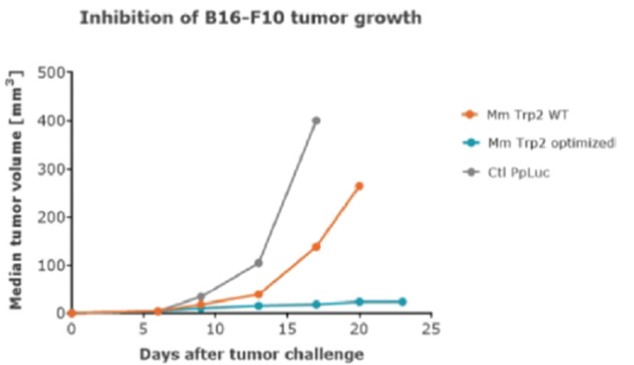
Therapeutic vaccination with mRNA vaccine encoding optimized Trp2 cell antigen inhibited tumor growth in murine melanoma model. Syngeneic mice were challenged subcutaneously with melanoma cells. When tumors were palpable, mice were vaccinated intradermally twice a week with LNP-formulated mRNA encoding either wild-type murine antigen Trp2 or Trp2 designed to improve antigen presentation. Mice vaccinated with LNP-formulated irrelevant mRNA (PpLuc) served as control.
91
Second Pillar: mRNA Optimization
Overview of mRNA Biology
mRNA is a linear polymer comprising four monomers called nucleotides: adenosine (A), guanosine (G), cytidine (C), and uridine (U). The sequence at any mRNA’s center instructing the synthesis of the protein encoded by it is the open reading frame (ORF, also known as coding sequence). The ORF is a continuous stretch of groups of three nucleotides (called codons) that is decoded and translated into protein by the ribosome. The process of translation begins at the first codon of the ORF, always an AUG (the start codon). The start codon signals to the ribosome where to start protein synthesis. The ribosome then progresses along the ORF one codon at a time, adding the amino acid to the protein chain fitting to the codon. A stop codon at the end of the ORF (UAA, UAG, or UGA) signals to the ribosome to terminate protein synthesis. In every cell, hundreds of thousands of mRNAs are translated into hundreds of millions of proteins every day. A typical protein contains 200-600 amino acids; therefore, a typical mRNA coding region ranges from 600-1,800 nucleotides.
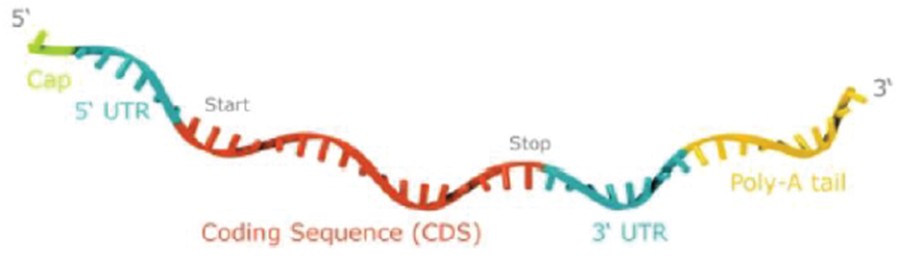
In addition to the coding sequence, mRNAs contain the following elements:
| ● | Untranslated regions, or UTRs — UTRs are sequences that are not translated into protein. The 5’ UTR precedes the start codon, the 3’ UTR follows the stop codon. These regions play important roles in gene expression, including mRNA stability, mRNA localization and translational efficiency via protein-RNA interactions. Some of the elements in the UTRs form characteristic secondary structures that are involved in mRNA regulation. |
| ● | 5’ cap — The cap structure is required to recruit ribosomes and additional proteins involved in translation to the mRNA. |
| ● | 3’ polyadenosine, or poly-A, tail — The 3’ poly-A tail is a long sequence of adenosine nucleotides (often several hundred) at the 3’ end of mRNA. This tail promotes mRNA export from the nucleus and translation and protects mRNA from degradation. In addition, the 3’ end of the mRNA can include a stretch or sequence of nucleotides following the 3’ poly-A tail. |
Our Approach
Our mRNA optimization process is designed to generate the most efficacious mRNA for any particular target and indication by optimizing translation, stability and immunogenicity. Each of these parameters can be modified by changing individual mRNA elements and their interplay guided by the envisaged application. Our mRNA molecule contains six elements that can be optimized to improve the potential efficacy of the mRNA construct. These elements include 5’ cap, 5’ UTR, ORF, 3’ UTR, and 3’ poly-A tail and 3’ end.
Depending on the target and indication, the required pharmacokinetics of protein expression might be different. Some applications may require the highest possible protein expression but only for a limited time, as is the case for gene editing approaches. For other applications, for example some protein replacement therapies, long-lasting protein expression might be key. Peak level and duration of protein expression can be adjusted by the choice or design of enhancer and stabilizing elements in untranslated regions of mRNA. Each of the mRNA elements together in combination with the overall sequence influence the degree of activation of the immune system by any particular mRNA. Therefore, our approach to RNA optimization always considers multiple factors as well as the whole construct to generate the optimal mRNA.
92
UTRs contribute decisively to the potential efficacy of therapeutic mRNAs. Natural mRNAs contain several different 5’ and 3’ UTRs, setting the individual level of translation and stability for each message. We have tapped this natural wealth of regulatory sequences and identified a large set of UTRs that confer translation or mRNA stability via diverse protein-RNA interactions.
Historically, one factor limiting the use of mRNA as a treatment has been the observation that in vitro produced synthetic mRNA activated the innate immune system, resulting in a fast shutdown of protein translation in cells. An effective mRNA therapy would need to evade recognition by the immune system to avoid shut down of protein translation. We have accumulated significant knowledge about the signatures recognized by the innate immune system over the past few years. With the insights we have gained, we are able to avoid signatures activating the immune system in elements at our disposal or eliminate them from mRNA constructs. This is demonstrated by the following example where formulated mRNA was injected intradermally in mice and both B cells and T cells were activated in the draining lymph node. In contrast, unformulated mRNA injected intradermally had limited immunostimulatory capacity.
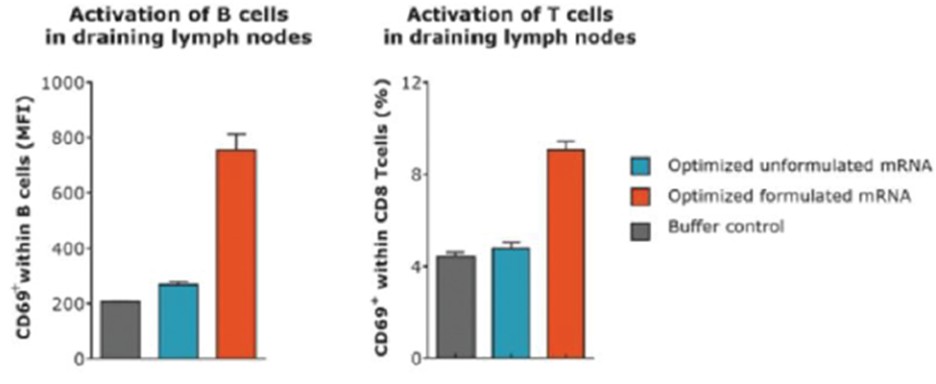
10 μg of mRNA, either free or formulated, was administered intradermally to the back of mice. Twenty-four hours post treatment, draining lymph nodes were isolated, and the activation status of immune cells was analyzed by flow cytometry. A higher CD69 signal indicates activation of the respective immune cells.
Cap Structure
The cap structure influences translation as it recruits the translational machinery, including initiation factors and the ribosome. The cap structure also affects mRNA stability due to its influence on the various proteins recruited to mRNA. Further, the cap structure is a determinant of activation of the innate immune system as different cap structures are differentially recognized by several innate immune sensors. In addition, different cap structures are incorporated during in vitro production of mRNA with different capping efficiency, resulting in varying proportion of mRNA lacking a cap, which is an mRNA species which is recognized by yet other sensors of the innate immune system. Accordingly, there is great potential to improve protein expression and immunosilence in mRNA by optimizing the cap structure. We have access to several cap structures, including those we have developed and commercially available ones.
5’ and 3’ UTRs
We have identified high numbers of naturally occurring 5’ and 3’ UTRs. Using bioinformatics analysis to identify patterns of increased expression, duration of expression, and reduced immunogenicity, we have catalogued more than one million 5’ and 3’ UTRs. From these, we selected a large set of potential enhancer elements (improving the rate of protein expression) and stabilizer elements (improving half-life of protein expression). By running a high throughput combinatorial approach, we identify and create optimized UTR combinations for a specific construct. Further, we have created UTR sub-libraries because we discovered that different UTRs perform differently in various tissue types.
93
Below is an example of the effectiveness of our UTR library to optimize protein expression as part of our collaboration with CRISPR Therapeutics. An open reading frame coding for an optimized Cas9 protein was combined with 83 UTR combinations via automated cloning. This target-specific UTR screening increased Cas9 protein levels in HepG2 cells five-fold compared to an already optimized construct.

To maximize expression of the target protein a set of 83 combinations of untranslated regions (UTR) was selected from screens identifying stable or highly translated endogenous transcripts. These UTR combinations were combined with the target open reading frame (ORF) via automated cloning to enable facile screening and selection of the most potent product candidates. Target-specific UTR screening led to a five-fold increase in protein levels in HepG2 cells compared to an already optimized construct.
Open reading frame (ORF)
The ORF instructs the synthesis of the protein it encodes by the ribosome. The ORF is a continuous stretch of groups of three nucleotides called codons. Ribosomes decode each codon as an amino acid to be added to the nascent protein. Each codon specifies a particular amino acid, however, many amino acids are specified by more than one codon. Due to this multiplicity of codons that specify an amino acid, any protein can be encoded by a myriad of coding sequences differing in their codon composition. These various ORFs differ largely in their properties and for any particular protein a top-performing ORF needs to be identified or designed to make an efficacious mRNA-based medicine. We currently optimize the ORF in a broad, holistic approach that includes multiple parameters taking into account codon optimality. Our algorithms also take into account that, similar to UTRs, different codons are optimal for different tissues. Furthermore, these algorithms also analyze and consider secondary structure. For example, as certain elements are known to drive immune stimulation by secondary structure, our algorithms avoid generation of sequences that may give rise to such immune stimulations.
94
In the following example, protein expressed from our mRNA containing a wild-type coding sequence was abundant in the livers of mice injected intravenously with LNP-encapsulated mRNA. However, protein levels were higher from our mRNA containing a coding sequence engineered for maximal protein expression.
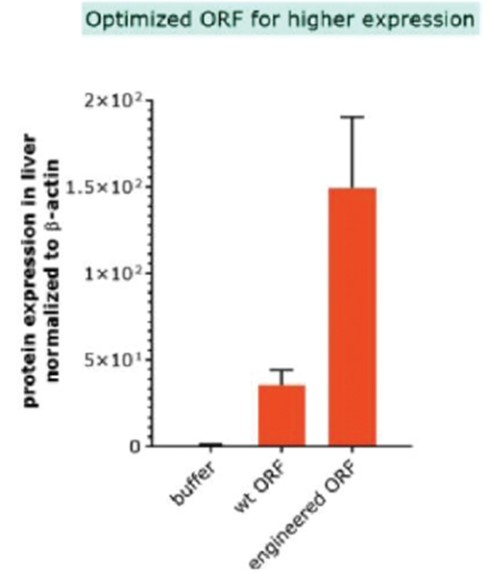
Abundance of a therapeutic protein in mouse liver expressed from an engineered open reading frame (ORF) in comparison to the wild ORF. mRNAs containing ORF variants were formulated in LNPs and injected intravenously into mice (called engineered ORF). Protein levels were determined by Western Blot analysis of liver lysates, followed by normalization to the signal from a loading control.
95
Poly(A) tail and 3’ end
The 3’ end of the mRNA molecule, prone to degradation by nucleases, is another form of optimization. The 3’ end can be sealed using different stabilizing elements, including secondary structure or specific nucleotide sequences, to inhibit RNA nucleases degrading RNA from the 3’ end.
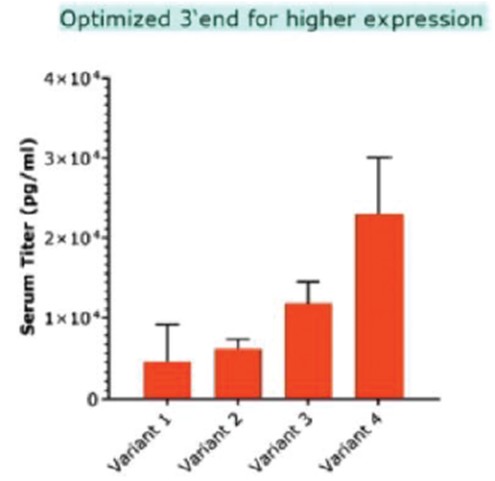
Impact of different mRNA 3’ end on serum levels of a therapeutic protein. mRNAs containing different vector-encoded 3’ end variants were formulated in LNPs and injected intravenously at a dose of 20 μg into female Balb/c mice. Six hours after injection, serum levels of secreted protein were determined by an enzyme-linked immunosorbent assay test, also referred to as ELISA, to measure antibodies in blood.
Finally, we analyze the structure of the optimized mRNA as a whole, including ORF and UTRs, to predict its recognition by RNA sensors and immune activating potential and modify any inappropriate elements.
Third Pillar: mRNA Delivery
The potency of the administered mRNA drug product is the combination of the potential efficacy of the mRNA that encodes the protein and the delivery system that transports the mRNA to the cells. Protein levels are highly correlated with the number of transfected cells which requires optimized delivery systems. While it is possible to deliver mRNA directly into the target tissue without delivery systems in certain cases, the presence of RNA degrading enzymes in blood and interstitial fluids rapidly regrade any extracellular mRNA. Additionally, cell membranes act as a significant barrier to entry of large molecules such as mRNA. These delivery technologies enable us to deliver large quantities of mRNA to the target cells.
We have access to a diverse portfolio of third-party and proprietary delivery systems that allow us to target a range of diseases. Access to this broad range of delivery technologies allows us to select the best-suited technology for development of each of our product candidates. We choose the most suited delivery system based on a number of factors, including immunogenicity, duration of treatment, dose levels, mode of administration and targeted tissue type.
96
The key delivery systems that we currently employ include lipid-based delivery systems. We employ lipid nanoparticles, or LNPs, to deliver our mRNA-based prophylactic and cancer vaccines locally. For protein and antibody therapeutic candidates, we apply LNP-formulated mRNA systemically. We have relied on third-party state-of-the-art LNP delivery systems for our initial clinical programs, and we are developing our proprietary LNP delivery systems for our future clinical programs. With these delivery modalities at hand, we are currently expanding our development pipeline and plan to bring new mRNA therapies to different organs and applications.
Lipid Nanoparticles (LNPs)
A variety of nanoparticles have been developed over the years for use in drug delivery. LNPs represent the most clinically advanced non-viral delivery systems. Encapsulation of the mRNA within LNPs enables delivery to the site of action within the cell. LNPs protect the mRNA from degradation, rapid excretion and liver clearance, enabling higher bioavailability and longer half-life.
LNPs consist of different lipids that form together a lipid nanoparticle with a solid core. The four primary LNP components include cationic lipids, pegylated lipids, phospholipids, and cholesterols. LNPs mimic low-density lipoproteins, which allows them to be taken up by an endogenous cellular transport pathway to deliver the mRNA cargo to cells. When LNPs are injected into biological systems, they attach to natural transport proteins, apolipoproteins, to facilitate the transport of lipids within the bloodstream and throughout the body. Following intravenous administration, the apolipoprotein binding enables efficient transport of the mRNA cargo to the liver. Once internalized in endosomes within cells, the LNPs are designed to escape the endosome and release their mRNA cargo into the cytoplasm, where the mRNA can be translated. Any mRNA and LNP components that do not escape the endosome are typically delivered to lysosomes where they are degraded by the natural process of cellular digestion.
The properties of each LNP system can be customized based on altering each component or overall composition. All of the LNPs we employ in our projects are designed to be biodegradable. We have extensively tested over 40 different delivery solutions and have selected the ones we use based on comparative data for the most efficient LNPs available from third parties for licensure. Having access to these technologies enables us to develop fast powerful solutions for vaccines and protein therapy.
Besides the licensed LNP technology from our partners, we are also developing our own LNP technology. We have established two ionizable lipid families and are developing those LNPs for application in local vaccination and systemic delivery to the liver. We conducted a systematic screening of LNP components and compositions for local vaccination in skin and muscle. Those optimized LNP formulations incorporating our own lipids helped to raise significant levels of immune response comparable with those of approved vaccine formulations.
97
The graphs below demonstrate comparable immunogenicity of CV LNP against a commercial PEG LNP in a vaccination trial with the rabies antigen in mice at same mRNA dose. On the left panel, the virus neutralization titer seven days post the second vaccination is shown, whereas on the right panel the induced T cell response is assessed by EliSpot of peptide library stimulated spleenocytes.
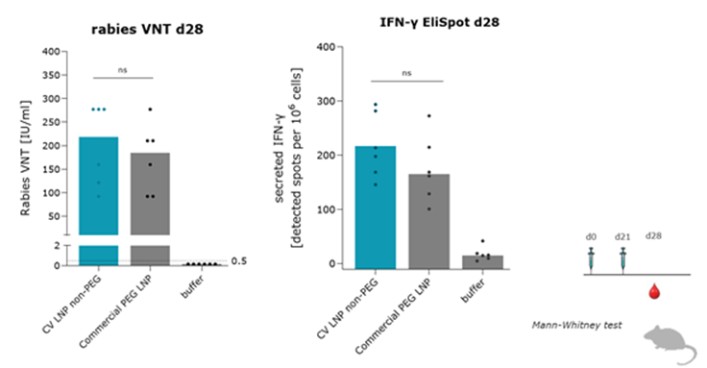
Our Approach to Disease Selection
Our approach seeks to mitigate risk across multiple levels to advance and expand our broad product portfolio. While mRNA is still an emerging treatment modality, we believe that we have made advances towards utilizing the potential of our technology platform through rational disease selection. Our approach for selecting new programs is based on the following key factors:
| ● | Target diseases with high unmet medical needs that are not effectively addressed using the current standard of care. |
| ● | Target areas where the underlying mode of action of the disease is understood or hypothesized which allows us to identify the required protein(s) or antigen(s). |
| ● | Identify areas where mRNA therapies have potential to have differentiated profiles compared to the conventional treatment modalities. |
| ● | Assess the likelihood of being able to address the disease using our technology platform and seek to continuously improve and expand the capabilities of our platform to address an even broader range of diseases. |
| ● | Seek to build on our deep understanding of mRNA biology, data derived from our technology platform and previous clinical and preclinical studies to apply to new indications. |
98
In building our product portfolio, we have considered a number of factors including immune response, duration of expression, dosing requirements, delivery technology, target tissue type, potential for responsiveness to mRNA-based medicine and target disease profile, among other factors. A disease indication may require an mRNA-based medicine that triggers an immune response, or that is immune active, or an mRNA-based medicine that requires no immune activation, or that is immune silent. Each of the disease indications that we are targeting require different levels of immune activation for the mRNA-based medicine to be effective. Our approach is to initially target indications that require an immune active approach (such as prophylactic vaccines or cancer vaccines), given the need for lower doses and transient expression of the antigen. These initial indications are amenable to localized delivery using an LNP delivery system. Based on the clinical validation of our prophylactic vaccines program and our advanced understanding of mRNA biology and immune stimulation modulation, we are also further expanding our product portfolio to target indications that require an immune silent approach (such as protein delivery). Targeting diseases amenable to the immune silent approach requires higher doses and longer expression of the protein, with potential for long-term repeat dosing for chronic diseases.
| ● | Immune active. For indications that require immune stimulation such as prophylactic and therapeutic vaccines, our technology optimizes the combination of mRNA molecules encoding specific antigens and selected delivery modalities to provide the desired immunostimulatory capacity. This allows us to design vaccines with high immunogenic effect. The goal is to induce an immune response against the encoded antigen. The mRNA is taken up by cells, including dendritic cells, at the injection site. Expressed antigens are then presented to the adaptive immune system leading to selective activation of T cells and B cells that recognize these antigens. These activated adaptive immune cells can then recognize and attack similar antigens that are found on tumors or pathogens. |
| ● | Immune silent. For indications that require no immune stimulation such as protein delivery, our technology can also design product candidates to be immunosilent and to express encoded proteins over an extended period of time. These product candidates can be expressed either locally (eye, liver or lung) or systemically, using the liver as a bioreactor for production of the therapeutic proteins (enzymes and antibodies). |
Prophylactic Vaccines
Infectious disease-related proteins, such as viral surface proteins, specific targets for the body’s immune defense system, can be expressed by injected mRNA and then presented to B and differentiated T cells, activating a specific immune response. We believe that our mRNA technology offers a platform for the development and production of prophylactic vaccines against infectious diseases. We believe our mRNA vaccines offer many potential advantages over existing vaccine technologies, including:
| ● | mRNA vaccines mimic several aspects of a natural viral infection and has the potential to offer improved and balanced immune response. |
| ● | mRNAs allow us to encode for specific protein antigens of choice, offering potential for the development against known and yet unidentified pathogenic threats. |
| ● | mRNAs allow production of multivalent vaccines with the potential to either demonstrate a broader efficacy by including additional target pathogens, or to strengthen potential efficacy by better targeting a specific pathogen, for example by adding of immunogenic epitopes, or by addressing different pathogen variants, or both. |
| ● | mRNA vaccines are generally expected to be safer than live or attenuated vaccines since no living virus is injected. As they do not interact with the host-cell DNA, they avoid the potential risk of genomic integration posed by DNA-based vaccines. |
| ● | mRNA binds to pattern recognition receptors and mRNA vaccines are thereby self-adjuvanting, a property which peptide- and protein-based vaccines lack. |
| ● | Rapid speed of development from knowing the sequence of the virus to progressing programs in clinical development given our ability to produce antigens without dedicated cell cultures and fermentation-based manufacturing processes. |
| ● | Commercial-scale production of mRNA is fast, cost-effective and, in contrast to traditional vaccine approaches, does not require cell culture or the use of live pathogens, and as a result, multiple vaccines can be produced in the same plant. |
99
Our current approach to the development of potential prophylactic vaccines is focused on:
| ● | CV0601/CV0701 for SARS-CoV-2: A Phase 2 clinical study was initiated on August 1, 2023, with two modified mRNA COVID-19 vaccine candidates: the monovalent candidate, CV0601, encoding the spike protein of the omicron BA.4-5 variant and the bivalent candidate, CV0701, encoding the spike protein of the omicron BA.4-5 variant as well as the original SARS-CoV-2 strain. The study is being conducted in Australia and fully enrolled with 427 participants. Interim Phase 2 data was announced on January 5, 2024. The data showed that both candidates exhibited a favorable reactogenicity profile across all tested doses and were generally well tolerated with a lower or similar proportion of participants reporting solicited adverse events when compared to comparator vaccine participants. Both candidates produced meaningful immune responses beginning at the lowest tested dose. Interim immunogenicity data showed meaningful titers of neutralizing antibodies for both candidates, which matched or numerically exceeded the titers induced by the licensed comparator vaccine at all tested doses except for the low dose level of CV0701. |
| ● | Multivalent candidate for seasonal influenza: A combined Phase 1/2 clinical study was initiated on May 8, 2023, comparing a comprehensive series of multivalent, modified mRNA seasonal influenza vaccine candidates with up to eight separate mRNA constructs per candidate addressing all four WHO-recommended flu strains. Positive data from an interim analysis of the Phase 1 part was reported on September 12, 2023, and enabled selection of a preferred candidate to be advanced to the Phase 2 part. Promising interim data from the Phase 2 part was announced on April 4, 2024, confirming an acceptable safety profile and boosted antibody titers against all encoded flu strains across age groups and tested dose levels. Geometric mean titers generated by the vaccine candidate against influenza A strains numerically exceeded the geometric mean titers of the licensed comparator vaccines consistently across tested dose levels and age groups. For influenza B strains geometric mean titers were lower than those elicited by the licensed comparator vaccines across age groups and tested dose levels. Targeted optimizations to further improve immune responses against influenza B strains will be tested in an additional Phase 2 study. |
| ● | Monovalent candidate for avian influenza (H5N1): A combined Phase 1/2 clinical study was initiated on April 24, 2024, with a monovalent, modified mRNA vaccine candidate, encoding an influenza A H5 antigen. The Phase 1 part of the study will assess the safety, reactogenicity and immunogenicity of the vaccine candidate in healthy younger and older adults against a placebo control. |
Oncology
mRNA is a versatile platform for cancer vaccine development allowing to encode a wide range of antigens from full length tumor associated antigens to neoepitopes. We are taking multiple approaches in oncology to induce tumor-specific immune responses in patients:
| ● | Novel cancer vaccines: We focus on building a meaningful portfolio of novel cancer vaccine candidates based on differentiated antigen discovery technologies and bioinformatics to target antigens that are overexpressed in tumor tissues with no or little expression on healthy tissues, using our LNP formulations. Within this strategy, we follow two approaches. (1) the development of off-the-shelf cancer vaccines based on tumor antigens shared across different cancer indications and (2) the development of fully personalized cancer vaccines based on a patient’s individual tumor genomic profile. |
| ● | Multi-epitope cancer vaccine candidate CVGBM: A proof-of-principle Phase 1 study was initiated on June 20, 2023 with CVGBM in patients with newly diagnosed surgically resected MGMT-unmethylated glioblastoma or astrocytoma with a molecular signature of glioblastoma. The study evaluates the safety and tolerability of CVGBM, a cancer vaccine candidate featuring a single unmodified mRNA encoding eight epitopes derived from known tumor-associated antigens with demonstrated immunogenicity in glioblastoma. On April 24, 2024, we announced that the dose-escalation Part A of the study completed recruitment of all four dose levels. Following review of the safety data, the Data Safety Monitoring Board (DSMB) confirmed no dose limiting toxicities and gave a dose recommendation for 100 µg for the dose-confirmation Part B of the study. Part B is expected to start enrollment mid-2024. A first study data readout is expected in the second half of 2024. |
100
Molecular Therapy: Deliver mRNA to express the right protein wherever needed
We are seeking to optimize mRNA molecules to trigger production of antibodies. Our antibody work has potential to protect against viruses, bacteria, and toxins and can be applied to many disease indications including cancer, cardiovascular diseases, infectious diseases and autoimmune diseases. In preclinical studies in non-human primates, we have demonstrated that antibodies encoded by mRNA can be produced in hepatocytes very rapidly and can reach the blood stream at relevant therapeutic levels.
With our technology, we can instruct human cells to produce or secrete specific proteins in the nucleus, cytoplasm, cellular organelles or cell membrane. Based on this “healthy” information delivered by mRNA, our cells are designed to produce proteins, which are required to treat the disease caused by missing or inactive proteins.
We believe there are several advantages of our technology applied to development of molecular therapy, including:
| ● | mRNA encoded proteins can function within or outside of cells as well as inside cell membranes, allowing us to address intracellular protein deficiencies that are not addressed by recombinant proteins. |
| ● | mRNAs can enable production of complex proteins that are challenging to make using recombinant technologies due to their folding requirements and complexity. |
| ● | Administered mRNAs encode proteins using natural pathways allowing for post-translational modifications such as glycosylation whereas recombinant proteins use non-human post-translational modifications which may lead to lower effectiveness and increased immunogenicity. |
| ● | mRNA constructs can be optimized to produce proteins that offer desirable pharmacology relative to the wild-type protein, such as increased half-life. |
| ● | mRNA allows for dosing flexibility to meet patient needs without causing irreversible changes to the genome. |
| ● | mRNA can be delivered repeatedly, creating the opportunity to provide long-term benefit for treatment of chronic diseases. |
We currently have collaborations focused on:
| ● | Therapeutic antibodies: We are also developing mRNA therapies to produce antibodies systemically using the liver as a bioreactor for subsequent secretion and systemic distribution of the antibodies to primary organs affected by a disease. Our collaboration with Genmab, a global leader in antibody discovery and design, will allow us to work with novel antibodies produced using our mRNA technology. This partnership represents the first-ever publicly disclosed mRNA antibody-focused deal and will allow us to optimize and manufacture mRNA-encoded antibodies for Genmab. |
| ● | Liver diseases: We are working on mRNA therapies targeted for the liver, including designing and producing intracellular targets such as transcription factors. Our collaboration with the Hannover Medical School aims to design treatments for liver diseases such as NAFLD, NASH and liver cirrhosis, with the potential to treat or reverse liver fibrosis. |
| ● | Eye diseases: We are investigating development of mRNA-based treatments for undisclosed ophthalmic indications. We have a collaboration with SERI for our discovery efforts. |
Our Key Pipeline Candidates
RNA-Based Therapeutics in Oncology
Discovery of new therapeutic cancer vaccine candidates
A key component to the development of new therapeutic cancer vaccine candidates is the build-up of a powerful antigen discovery engine. To gain access to state-of-the-art antigen discovery technologies, we announced a partnership with Belgium-based company myNEO Therapeutics on May 25, 2022, and the acquisition of Netherland-based Frame Cancer Therapeutics on June 8, 2022.
101
Together with immunotherapy company myNEO, we aim to identify specific antigens found on the surface of tumors for the development of novel mRNA immunotherapies. myNEO utilizes a broad range of underlying genomic alterations to identify constantly emerging, novel classes of antigens of defined tumor types. Incorporating new ranking methodologies based on tumor cell antigen processing and presentation is expected to allow for selection of antigens with the highest confidence of success for potential clinical testing.
With the acquisition of Frame Cancer Therapeutics, a private company focused on advanced genomics and bioinformatics, to identify both shared and unique neoantigens across different cancer types, we complement existing in-house expertise to identify and validate promising antigens for mRNA cancer vaccine candidates. Our technology platform has the potential to identify a broad panel of neoantigens and tumor-associated antigens that go beyond conventional approaches and could strongly increase the likelihood of developing highly effective cancer vaccines that activate the human immune system against cancer.
The field of immuno-therapy has advanced with the progression of available technologies to extract data from patient samples, such as next-generation sequencing. Conventional approaches have so far focused on the exome, the protein-coding part of the genome, which represents only about 1.5% of the total genetic information. More recently, breakthrough developments in sequencing capacity have enabled the extraction of vastly larger amounts of data that allows us to utilize the remaining 98.5% of genetic information. The technologies brought in-house with the acquisition of Frame Cancer Therapeutics are based on whole-genome-sequencing for every patient sample and combine it with short as well as long-range RNA sequencing to map the full inventory of genomic changes. More specifically, downstream of the sequencing, a software package integrates all the data to retrieve the exact changes in the DNA of the tumor cells compared to healthy cells. Correlation of this data with changes in the RNA transcription of the tumor results in entirely new and potentially antigenic tumor antigens that we plan to test as targets for a portfolio of new cancer vaccine candidates. These new antigens are not only entirely foreign to the body but are also uniquely expressed in the tumor and not in healthy tissue. In their foreignness, these constructs are expected to raise stronger immune responses than antigens derived from exome-based conventional approaches.
The highly synergistic antigen discovery technologies of Frame Cancer Therapeutics and myNEO are expected to significantly accelerate CureVac’s oncology strategy to build a meaningful portfolio of new cancer vaccine candidates. Within this strategy, we follow two approaches. The first approach assesses tumor antigens shared by different cancer patients for the development of off-the-shelf cancer vaccines. The second approach is tailored to the individual tumor setup of a patient for personalized therapy. We plan to advance new antigens for both approaches based on our second-generation mRNA backbone.
CVGBM
To assess the safety and immunogenicity of our second-generation backbone in an oncology setting, we initiated a proof-of-principle Phase 1 study in patients with newly diagnosed surgically resected MGMT-unmethylated glioblastoma or astrocytoma with a molecular signature of glioblastoma on June 20, 2023. The open-label study evaluates the safety and tolerability of CVGBM, a cancer vaccine candidate featuring a single unmodified mRNA encoding eight epitopes derived from known tumor-associated antigens with demonstrated immunogenicity in glioblastoma. On April 24, 2024, we announced that the dose-escalation Part A of the study completed recruitment of all four dose levels. Following review of the safety data, the Data Safety Monitoring Board (DSMB) confirmed no dose limiting toxicities and gave a dose recommendation for 100 µg for the dose-confirmation Part B of the study. Part B is expected to start enrollment mid-2024. A first study data readout is expected in the second half of 2024.
The multiepitope design of CVGBM was supported by preclinical studies assessing the potency of a multiepitope mRNA cancer vaccine construct targeting tumors in a murine melanoma model. The data was presented at the 11th International mRNA Health Conference, hosted by CureVac from October 31 to November 2, 2023, in Berlin, Germany.
102
The preclinical mRNA construct encoding ten epitopes derived from the murine B.16.F10 melanoma cell line was tested in mice. The study applied three 5 µg doses of LNP-formulated B.16 mRNA, administered intramuscularly at weekly intervals. Data obtained on day 21 can be seen below and confirmed prominent induction of CD8+ and CD4+ T cell responses (left and middle graph) recognizing epitopes across the full multiepitope construct. Median survival (right graph) of the animals increased to 30.9 days for treated mice compared to 23.2 days for a group vaccinated with formulated control mRNA.
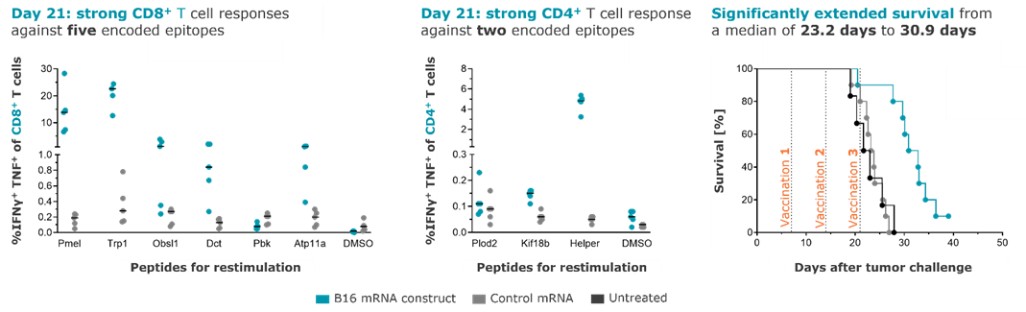
Strong T cell activation is particularly encouraging and relevant, as the B16-F10 tumor model is characterized as a cytokine deficient “cold” tumor model that exhibits very little immune cell infiltration and resistance to check-point inhibitors. The data suggest that single-agent application of the multiepitope B.16 mRNA construct generated robust T cell activation in the tumor microenvironment, thereby inhibiting tumor growth and extending survival in the applied preclinical model.
mRNA-Based Prophylactic Vaccine Programs Jointly Developed With GSK: Second-Generation mRNA Backbone
COVID-19: Disease Overview
Coronaviruses are a family of viruses that can lead to respiratory illness, including Middle East Respiratory Syndrome, or MERS-CoV, and Severe Acute Respiratory Syndrome, or SARS-CoV. Coronaviruses are transmitted between animals and people and can evolve into strains not previously identified in humans. On January 7, 2020, a novel coronavirus (2019-nCoV) was identified as the cause of pneumonia cases and deaths in Wuhan, China and an exponentially increasing number of cases have since then been found worldwide. On March 11, 2020, the World Health Organization (“WHO”) designated COVID-19, the disease caused by the novel coronavirus SARS-CoV-2, an international pandemic. As of January 21, 2024, there have been 774,395,593 confirmed cases of COVID-19 globally, including 7,023,271 deaths, reported to WHO.
Clinical COVID-19 Vaccines Program (second-generation mRNA backbone)
In February 2021, we announced a collaboration with GSK for our COVID-19 vaccine program, which went into effect in April 2021 pursuant to which we are collaborating with GSK to research, develop and manufacture mRNA vaccines based on our second-generation mRNA backbone targeting relevant SARS-CoV-2 variants. The second-generation mRNA backbone can flexibly encode for different COVID-19 variants and features targeted optimizations designed to improve intracellular mRNA stability and translation for increased and extended protein expression. The optimizations potentially allow for strong and early immune responses at low doses, which also supports the development of multivalent vaccines and combination vaccine approaches.
Interim Phase 2 Data of the Monovalent mRNA Vaccine Candidate, CV0601 and Bivalent Vaccine Candidate, CV0701
The Phase 2 study evaluates safety, reactogenicity, and immune responses of single booster doses of the monovalent mRNA vaccine candidate, CV0601, encoding the Omicron BA.4-5 variant and the bivalent candidate, CV0701, encoding the Omicron BA.4-5 variant as well as the original SARS-CoV-2 virus. It is active-controlled, featuring a licensed bivalent mRNA-based COVID-19 comparator vaccine. The study is being conducted in Australia and completion of enrollment at 427 healthy adult participants was announced on November 1, 2023. Interim Phase 2 data were presented on January 5, 2024.
103
Results from the formal interim analysis showed that both vaccine candidates using CureVac’s proprietary second-generation mRNA backbone produced meaningful immune responses and favorable reactogenicity profiles across all tested doses, including the lowest tested dose. All three of the dose levels tested were below those used in mRNA-based COVID-19 vaccines licensed in the U.S. and EU. The candidates were shown to be generally well tolerated with a lower or similar proportion of participants reporting solicited adverse events when compared to comparator vaccine participants within seven days of dosing. Interim immunogenicity data showed meaningful titers of neutralizing antibodies for both candidates at all dose levels.
The graphs below illustrate the reactogenicity (left graph) and immunogenicity (right graph) data reported for the monovalent candidate, CV0601, in comparison to the licensed bivalent comparator vaccine. CV0601 was tested at a medium dose level. The immunogenicity data show that it elicits neutralizing antibody titers against the Omicron BA.4-5 variant on day 29 following the booster vaccination that were 5.0 times the pre-boosting titers, numerically exceeding the 3.6-fold ratio generated by the licensed comparator vaccine. Antibody titers against the original SARS-CoV-2 virus on day 29 following the booster vaccination were 3.1 times the pre-boosting titers, numerically exceeding the 2.8-fold ratio generated by the comparator vaccine.
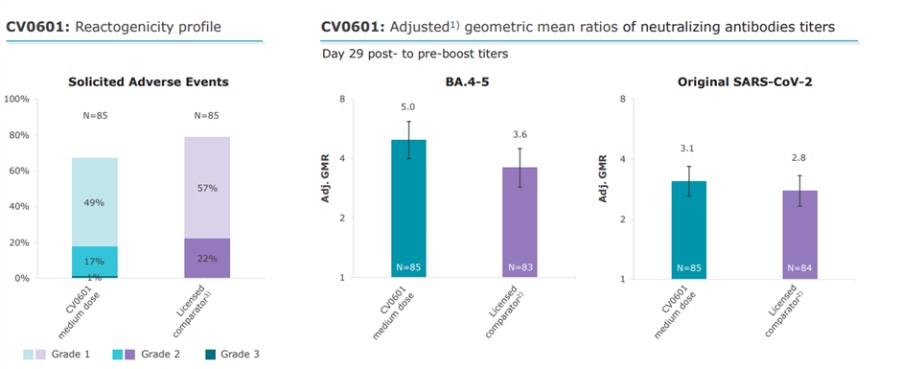
1) GMR and confidence intervals are adjusted for baseline titer, age at baseline (<65 or ≥65) and prior SARS-CoV-2 infection
2) Licensed bivalent, mRNA-based comparator vaccine
104
The bivalent candidate CV0701 was tested at a low, medium, and high dose level. The graphs below illustrate the reported reactogenicity (left graph) and immunogenicity (right graph) data. The immunogenicity data show neutralizing antibody titers against BA.4-5 on day 29 following the booster vaccination that were 2.7-fold, 3.7-fold, and 4.6-fold the titers before the booster, compared to a 3.6-fold ratio of post- to pre-booster titers for the comparator vaccine. Antibody titers against the original SARS-CoV-2 virus on day 29 following the booster vaccination were 2.1-fold, 2.7-fold, and 3.1-fold the titers before the booster, compared to a 2.8-fold ratio of post- to pre-booster titers for the comparator vaccine, matching or numerically exceeded the titers induced by the licensed comparator, except for the low dose level.
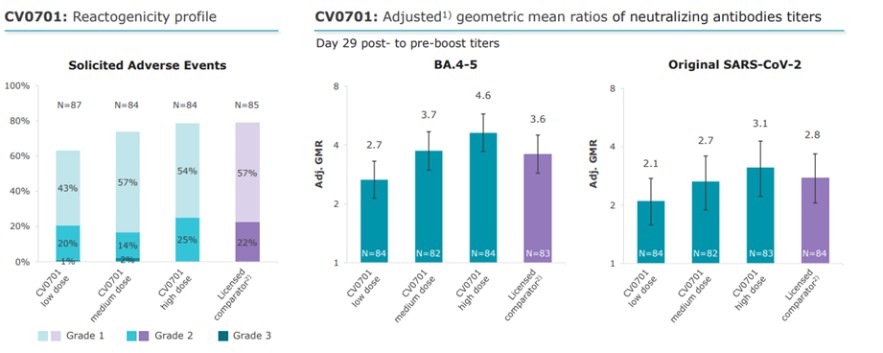
1) GMR and confidence intervals are adjusted for baseline titer, age at baseline (<65 or ≥65) and prior SARS-CoV-2 infection
2) Licensed bivalent, mRNA-based comparator vaccine
Influenza: Disease Overview
Influenza is a highly contagious virus that causes mild to severe respiratory virus that can lead to death. During the 2022-2023 influenza season, influenza was associated with 31 million illnesses, 14 million medical visits, 360,000 hospitalizations and 21,000 deaths in the United States. Preliminary in-season burden estimates for the U.S. in the 2023-2024 flu season showed 32-59 million flu illnesses, 15-27 million flu medical visits, 360,000-730,000 flu hospitalizations and 22,000-65,000 flu deaths. The WHO reports that globally there are around a billion cases of seasonal influenza annually, including three to five million cases of severe influenza, leading to as many as 290,000 to 650,000 deaths.
Influenza viral infections can be prevented by vaccination although there are several limitations associated with current flu vaccines. Flu vaccines are not always effective, primarily because the influenza virus and its associated antigens undergo mutations or changes in its sequence over short periods of time, which is called antigenic drift. Vaccines that are developed for the predominant strain infecting people can be rendered ineffective as the virus mutates as it passes from person to person. The process of developing a standard traditional vaccine typically takes approximately eight months from strain identification to doctor’s office availability, increasing the likelihood that a significant pool of viruses circulating will be poorly recognized by antibodies in vaccinated individuals. Additionally, vaccine efficacy tends to wane over time. For these reasons, vaccination of the target population needs to be repeated every year before the start of the next influenza season, putting a significant burden on the health system. Furthermore, only a part of the population targeted to get the yearly shot is vaccinated each year, leaving many individuals unprotected.
105
Clinical Influenza Vaccines Program (second-generation mRNA backbone)
Influenza is the first indication from the initial collaboration we started with GSK in July 2020, which focuses on the development of new products for different targets in the field of infectious diseases. The jointly developed influenza vaccine candidates are based on our second-generation mRNA backbone, which can flexibly encode for different influenza strains and which features targeted optimizations designed to improve intracellular mRNA stability and translation for increased and extended protein expression. The optimizations potentially allow for strong and early immune responses at low doses, which will also support the state-of-the-art multivalent format of influenza vaccines as well as combination vaccine approaches. The second-generation mRNA backbone differs significantly from the mRNA backbone for our first-generation. The first candidate from the second-generation backbone influenza program was the non-chemically modified mRNA construct CVSQIV, a differentiated multivalent vaccine candidate featuring multiple non-chemically modified mRNA constructs to induce immune responses against relevant targets of four different influenza strains. On February 10, 2022, we announced the start of a Phase 1 dose-escalation study in Panama evaluating the safety, reactogenicity and immunogenicity of CVSQIV in the dose range of 3µg to 28µg. Preliminary safety data reported on April 28, 2022, showed a benign reactogenicity profile across the tested dose groups. Within the jointly developed COVID-19 vaccine program, we also extended our technology platform to chemically modified mRNA constructs to allow for data-driven selection of the best candidate. We announced the start of a Phase 1 dose-escalation study with a chemically modified influenza vaccine candidate, Flu SV mRNA, on August 18, 2022. The candidate is a monovalent candidate. The Phase 1 dose-escalation study is being conducted in Canada, Spain and Belgium to evaluate the safety, reactogenicity and immunogenicity of Flu SV mRNA. On January 6, 2023 we announced that the second-generation mRNA backbone using modified mRNA was selected as the preferred technology for further clinical development in the influenza program.
Flu SV mRNA: Preliminary Phase 1 Data of the Chemically Modified Construct Expressing an H1N1 hemagglutinin antigen
Clinical Influenza Vaccine Programs (second-generation mRNA backbone)
Seasonal influenza and avian influenza (H5N1) are the first two indications from the collaboration we started with GSK in July 2020, which focuses on the development of new products for different targets in the field of infectious diseases. The jointly developed influenza vaccine candidates are based on our second-generation mRNA backbone, which can flexibly encode for different influenza strains, and which features targeted optimizations designed to improve intracellular mRNA stability and translation for increased and extended protein expression. The optimizations potentially allow for strong and early immune responses at low doses, which will also support the state-of-the-art multivalent format of influenza vaccines as well as combination vaccine approaches.
Combined Phase 1/2 Study Start in Avian Influenza
On April 24, 2024, the start of the Phase 1 part of a combined Phase 1/2 study of an investigational influenza A (H5N1) pre-pandemic vaccine candidate was announced. The study assesses a monovalent vaccine candidate encoding an influenza A H5-antigen. The H5N1 avian flu virus is considered a potential future pandemic threat, able to sporadically cross species from its original bird host to other animal hosts to humans.
Interim Phase 2 Data of the Multivalent Seasonal Influenza Vaccine Candidate
A combined Phase 1/2 clinical study was initiated on May 8, 2023. The initial Phase 1 part compared a comprehensive series of multivalent, modified mRNA seasonal flu vaccine candidates with up to eight separate mRNA constructs per candidate, addressing all four WHO-recommended flu strains. Candidates were tested at different dose levels in 270 healthy younger adults (age 18-50). On September 12, 2023, we announced selection of a promising vaccine candidate for continued Phase 2 clinical development based on positive data from an interim analysis of the Phase 1 part. Interim safety data showed no safety concerns across all tested dose levels for the multivalent candidates. Immunogenicity of all candidates was assessed in parallel with a licensed seasonal flu vaccine comparator. The humoral responses observed supported the selection of the preferred vaccine candidate.
106
We announced dosing of the first participant with the preferred vaccine candidate in the Phase 2 part of the study on November 1, 2023. The potentially differentiated, multivalent candidate encodes antigens matched to all WHO-recommended flu strains. In the Phase 2 part, the selected candidate is being tested in younger (age 18-64) and older (age 65-85) adults at different dose levels compared to age-appropriate licensed seasonal flu comparator vaccines. On April 4, 2024, we announced that the selected candidate boosted antibody titers against all encoded flu strains and across all age groups and tested dose levels, including the lowest tested dose. The vaccine candidate was shown to have an acceptable safety and tolerability profile, with the majority of solicited adverse events reported as either grade 1 (mild) or grade 2 (moderate) within seven days of dosing. The results confirm previous findings that the platform elicits strong overall antibody titers at well-tolerated dose levels. Among younger and older adults, geometric mean titers generated by the vaccine candidate against influenza A strains numerically exceeded the geometric mean titers of the licensed comparator vaccines consistently across all tested dose levels. For influenza B strains geometric mean titers were lower than those elicited by the licensed comparator vaccines across both age groups and tested dose levels. Targeted optimizations to further improve immune responses against influenza B strains will be tested in an additional Phase 2 study.
Preliminary Phase 1 Data of Monovalent Seasonal Influenza Construct Flu SV mRNA
On January 6, 2023, the second-generation mRNA backbone using modified mRNA was selected as the preferred technology for further clinical development in the seasonal flu vaccine program. In the Phase 1 study of the monovalent Flu-SV-mRNA, expressing an H1N1 hemagglutinin antigen (subtype of influenza A), five doses ranging from 2 to 54μg with up to 25 subjects per dose cohort were evaluated in younger adults (age 18-45). This dose range takes into account that the candidate is a monovalent construct. Correspondingly, the applied doses reflect doses for a single mRNA construct, which would later be multiplied for a state-of-the-art multivalent flu vaccine. In the younger adult age group, the preliminary safety and reactogenicity data shown below demonstrate that the monovalent Flu-SV-mRNA candidate was generally well tolerated with no safety concerns observed to date across all tested dose levels. A single dose of Flu-SV-mRNA (dose level undisclosed) was assessed for safety and reactogenicity in older adults (age 60-80) and was also observed to be safe and well tolerated with no grade 3 adverse events in the 32 subjects who were administered the mRNA construct.

107
Immunogenicity of the monovalent Flu-SV-mRNA construct was assessed in parallel with a licensed seasonal flu vaccine comparator in both age groups, shown as the orange bar to the right in the figures below. The data illustrate the ratio of post- to pre-boost geometric mean hemagglutinin inhibition antibody titers. In younger adults, Flu-SV-mRNA achieved comparable antibody increases to the licensed vaccine, boosting geometric mean titers at least about 14-fold beginning at the lowest dose of 2µg. In older adults a comparable ratio of post- to pre-boost titers of 14.5 was achieved at the undisclosed dose, which compares to a ratio of 6.2 for the flu vaccine comparator.
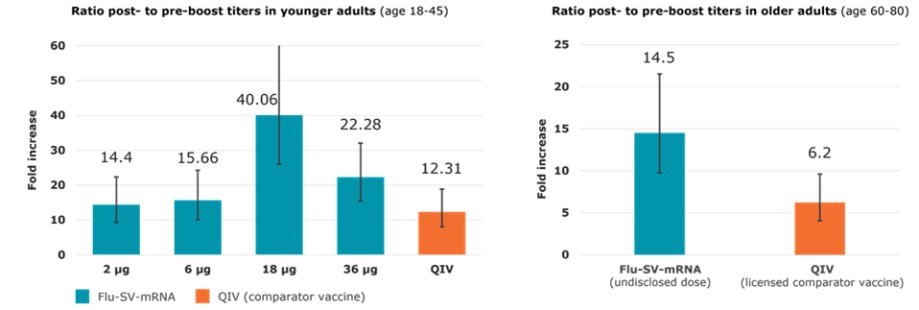
The robust preliminary ratios of post- to pre-boost geometric mean hemagglutinin inhibition antibody titers were shown to result in seroconversion rates that are substantially higher for the modified mRNA candidate. Specifically in the older adults age group, the percentage of subjects achieving seroconversion was 89.7% for Flu-SV-mRNA and 56.2% for the licensed flu vaccine comparator.
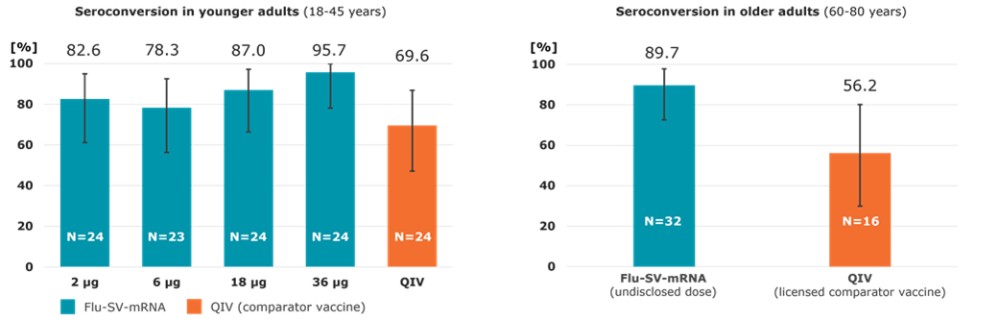
Interim data supported the progression of the modified second-generation mRNA technology for the development of a multivalent mRNA flu vaccine as it is tested in the currently ongoing Phase 2 part of the combined Phase 1/2 study.
RNA-Based Therapeutics in Molecular Therapies
mRNA-based protein supplementation offers a therapeutic approach to compensate for lack of proteins in monogenetic diseases caused by loss-of-function mutations. It offers a potentially curative treatment option, especially in diseases in which the protein is expressed predominantly in organs that can be reached by intravenous delivery (such as the liver). Despite the success of classical enzyme-replacement therapy in several metabolic disorders, this therapeutic approach is not well suited for treatment of diseases caused by the lack of functional intracellular proteins, especially if the proteins are located in or on intracellular compartments. Additionally, as therapeutic proteins are conventionally manufactured by using human, animal or even plant cells, the pharmacological and biochemical properties of such recombinant proteins may differ from endogenously expressed enzymes. Cellular localization, folding and post-translational modifications can especially be critical for the correct function of a therapeutic protein. Delivery of mRNA can overcome these limitations and is likely to result in expression of a functional protein at a physiological cellular location. An example of our rare disease approach is for the potential treatment of hereditary spastic paraplegia, or HSP.
108
Hereditary Spastic Paraplegia
HSP is a group of inherited disorders that are characterized by progressive weakness and spasticity of the legs due to axonal degeneration of the corticospinal tract. Hereditary spastic paraplegia type 5, or SPG5, is caused by autosomal recessive loss-of-function mutations in CYP7B1, a gene encoding for the cytochrome P-450 oxysterol 7-α-hydroxylase, essential for the alternative pathway of bile acid synthesis in the liver. Mutations causing SPG5 lead to decreased enzyme activity of CYP7B1 and accumulation of oxysterols in the serum, the liver and then the central nervous system. The accumulation of hydroxyl cholesterol, or HC, in the brain is what is believed to be the pathologic correlate of this particular disease, which leads to spasms and paraplegia as symptoms. To date, no curative treatment for SPG5 is available. Current clinical treatment strategies for SPG5 are based on the reduction of cholesterol by applying cholesterol-lowering drugs (statins), which consequently lead to a reduction of oxysterols.
Our approach is based on replacement of CYP7B1 by administration of mRNA. We have studied the intravenous application of formulated CYP7B1 mRNA in mice lacking the endogenous Cyp7b1 gene. Comparable to the human situation in SPG5 patients, a drastic increase of these oxysterols was detected in all three compartments (serum, liver and brain) of knockout mice. Using this in vivo model, we were able to demonstrate that a therapeutic approach with mRNA can restore human CYP7B1 protein that exhibits physiological function and eliminates abnormal cholesterol metabolites.
As shown below, we investigated the safety and efficiency of repeated dosing with four consecutive doses of 40 μg LNP-encoded mRNA of CYP7B1 administered intravenously every five days. LNP loaded with a non-translating mRNA were applied as control (vehicle). Prior to the administration, serum samples were taken to determine basal oxysterol levels. Two days after the last injection (17 days of treatment), animals were sacrificed, and serum, liver and brain samples were analyzed. Oxysterol analysis of these samples demonstrated a significant decrease of 25 hydroxy cholesterol, or 25 HC, in the serum and liver. mRNA expression of the human CYP7B1 in the liver led to a reduction of 25 HC in the liver by 8-fold and in serum by approximately 88%. These effects are accompanied by a reduction of the accumulation of 25 HC in the brain by more than 50%. Additionally, repetitive treatment resulted in a significant decrease of 27-HC and 3β-HCA in livers of treated compared to vehicle animals.
In addition, repeat intravenous delivery of CYP7B1 mRNA was found to be well tolerated in this study. Neither the CYP7B1 mRNA nor the restored protein nor the LNP induced liver toxicity. None of the treated animals presented signs of toxic or adverse effects. LNP particles encapsulating non-coding mRNA led to a temporary increase in oxysterol levels (25-HC and 27-HC) in liver and serum in the vehicle group, which is expected given cholesterol is an essential component of LNPs.

Lysosomal Storage Disorders
Lysosomal storage diseases are well-defined, single-gene disorders that are amenable to correction by systemic mRNA therapy.
109
We have conducted preclinical studies in an undisclosed lysosomal storage disease, or LSD, to evaluate LNP delivery of mRNA encoding the deficient enzyme to the liver, production of the enzyme by the liver and subsequent secretion and systemic distribution of the enzyme to the primary organs affected by the disease. In this specific LSD, the enzyme deficiency results in a progressive accumulation of lipid in cellular lysosomes, which ultimately affect the functioning of the heart and kidneys. Enzyme replacement therapy, or ERT, which involves intravenous administration of recombinant enzyme, has been the standard of care for this specific LSD. In contrast to ERTs, our LNP mRNA technology specifically and efficiently targets the liver to naturally produce the missing enzyme, which is subsequently secreted into the bloodstream and distributed to the affected organs. In this specific LSD, the liver is not the target organ, but is used to produce the endogenous native enzyme.
As shown below, LNP delivered mRNA therapy produces a high and homogenous expression of the missing enzyme in the livers of knockout mice (Figure A, brownish stain). The endogenously produced enzyme is then secreted into the bloodstream with a better pharmacokinetic profile than the injected recombinant protein. The enzyme is then taken up by the target organs. In this example, the enzyme is taken up by the heart (Figure B) and kidney (Figure C) and localized into the lysosomes. Our mRNA therapy, through prolonged synthesis and secretion by the liver, led to higher enzyme activity in the organs compared to the infused recombinant enzyme (Figures B and C). This higher enzyme activity leads to a significant and prolonged reduction of accumulated lipids in the organs of mRNA-treated animals (Figure D).
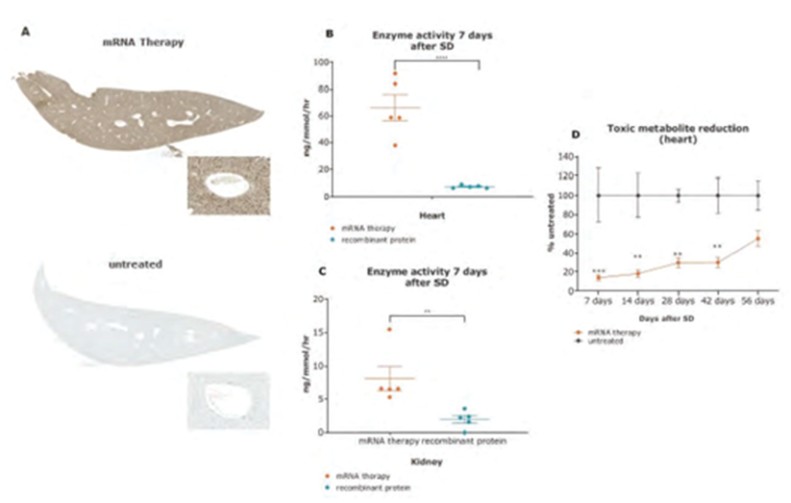
Liver-Specific Metabolic Diseases
We are applying a similar approach to inherited liver-specific metabolic disorders of amino acids, nitrogen and essential nutrients. The goal of these studies is to restore the specific enzyme or protein that is deficient in the liver by LNP-mediated delivery of mRNA to the liver. As such, the target organ for correction is the liver, and secretion and systemic distribution of the enzyme or protein to other organs is not required for a therapeutic effect.
Our ability to optimize mRNA stability and translation, in combination with optimization of the expressed protein, is an important part of our technical expertise. Using a process of mRNA and protein optimization, we believe that we are able to extend the duration of protein expression to meet a defined target product profile.
One example of this technology is the mRNA that we are developing for a metabolic amino acid disorder. In this inherited disorder, a liver-specific intracellular enzyme is deficient resulting in decreased metabolism of the amino acid. As a result, there is a toxic build-up of the amino acid in the blood, which leads to severe consequences for the central nervous system.
110
A single intravenous injection of a liver-targeted LNP formulation containing the therapeutic mRNA leads to a marked decrease in the level of the amino acid in the sera of knockdown animals (Figure A), but also in the brain (Figure B). Several rounds of mRNA and protein optimization were subsequently performed. Improving the mRNA molecular structure during the first round of optimization prolonged the protein and its therapeutic effect (Figure A) compared to the reference mRNA. Protein optimization (Figures C and D) of the expressed target enzyme increased its expression/stability and/or activity in vitro. The combination of both optimization programs resulted in a candidate with improved characteristics.
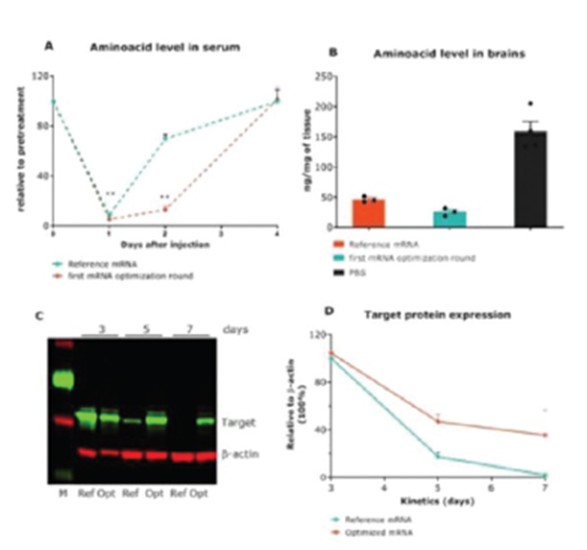
Fibrotic Liver Diseases
According to published literature, chronic liver diseases cause two million deaths a year worldwide. We have shown that the delivery of liver-specific protein factors, which are down regulated in fibrosis, can resolve liver fibrosis, a key pathological feature of NAFLD, NASH, cirrhosis and hepatocellular carcinoma. Protein factor treatment of liver diseases is uniquely suited to mRNA medicines enabling the expression of intracellular proteins. Moreover, we believe that in this particular case, the LNP technology allows us to deliver mRNA almost exclusively to the target cells, hepatocytes.
In a CCL4 chemically induced mouse model of liver fibrosis, we delivered eight doses of LNP-mRNA at an interval of five days at 2 mg/kg. The figure below illustrates the ability of an mRNA-delivered protein factor to reduce collagen, the main fibrotic material deposited in fibrosis, and eliminate activated stellate cells, the source of collagen (stained red). To confirm the potential activity of this mRNA therapy, we obtained similar data in two other unrelated murine models: a diet-induced model and a knockout mouse model of liver fibrosis. These findings offer preclinical proof of concept for this therapeutic concept to treat acute and chronic liver diseases, as well as diseases of other organs.
111
We are currently focused on significantly lowering the dose by employing two parallel strategies – mRNA sequence optimization to improve expression and optimization of the protein sequence for higher and sustained activity.
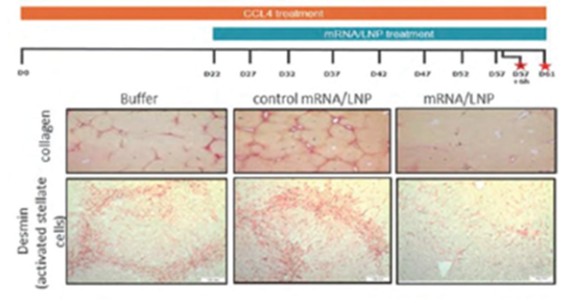
mRNA has the potential to promote expression without inducing an adverse immune response against the encoded protein. We have tested various antibodies using different designs to evaluate our platform’s potential for prophylactic and therapeutic antibodies.
We evaluated the use of mRNA for passive immunization in two indications, rabies and botulism, that can be considered prototypes for anti-pathogen and anti-toxin therapies, respectively. Single injections of mRNA-LNPs were sufficient to establish rapid, strong and long-lasting serum antibody titers in vivo, thereby enabling both prophylactic and therapeutic protection against lethal rabies infection or botulinum intoxication. In both models, the high levels of in vivo serum expression conferred full protection in pre- and post-exposure scenarios.
The left side of the graphic below shows that mice expressing the anti-rabies mAb survived, whereas the majority of control animals which received anti-influenza mAb mRNA succumbed to the rabies infection. The right side of the graphic below shows that mice treated post-intoxication with VNA-BoNTA mRNA or recombinant VNA-BoNTA also survived.
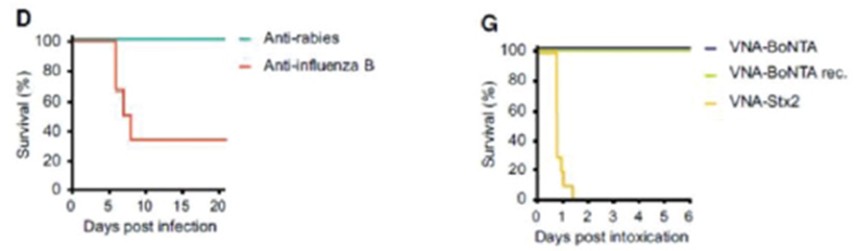
112
In addition, we have demonstrated that mRNA-mediated antibody expression may be effective in the field of cancer immunotherapy, where mAbs are widely used in medical practice. In a preclinical study conducted in mice, we compared the efficacy of rituximab-encoding mRNAs to recombinant rituximab. We inoculated mice intravenously with luciferase expressing Raji lymphoma cells and started treatment with 50 μg of mRNA-LNP encoding rituximab and 200 μg of recombinant rituximab at various time points. mRNA-LNPs coding for an irrelevant antibody were used as further control. Control animals revealed strong tumor cell proliferation and had to be euthanized 17 days after inoculation due to severe symptoms. As shown in the picture below, repeated administration of mRNA-LNP for rituximab strongly decelerated or even abolished tumor cell growth compared to continued tumor growth for recombinant rituximab.
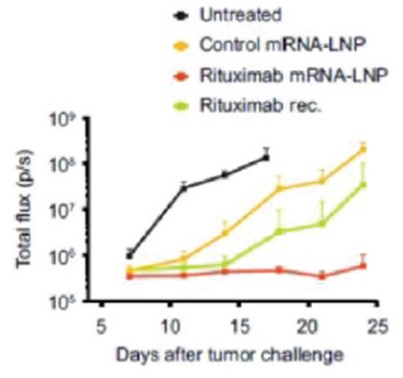
Therapeutic HNF4A mRNA to Attenuate Liver Fibrosis in Mice
On August 25, 2021, we published first preclinical data on the restoration of hepatocyte functions and attenuation of liver fibrosis and cirrhosis in multiple mouse models via an HNF4A encoding mRNA in the Journal of Hepatology. Owing to the extensive amount of data featured in the scientific publication, the following data excerpts represent only a selection of important findings of the preclinical studies. Full data can be accessed via the corresponding publication.
As a first step within the study, existing clinical data was collected that confirmed reduced endogenous HNF4A mRNA levels in patients with liver fibrosis, graded by the Ishak score. In two cohorts from Hannover Medical School, Germany, and Shanghai Zhongshan Hospital, China, liver tissues showed fibrosis stage-dependent reduction in HNF4A mRNA. Subsequently, it was examined whether in vitro synthesized HNF4A mRNA produces functionally active HNF4A protein. The wild-type (non-codon-optimized) or codon-optimized HNF4A mRNA were transfected into the human cervical carcinoma cell line (HeLa) cells as these lack endogenous HNF4A expression. Codon-optimized HNF4A mRNA-transfected cells showed higher HNF4A protein levels than those transfected with wild-type HNF4A mRNA. ZsGreen mRNA-transfected HeLa cells served as a control.
113
Human HNF4A mRNA restoration improves function of fibrotic hepatocytes
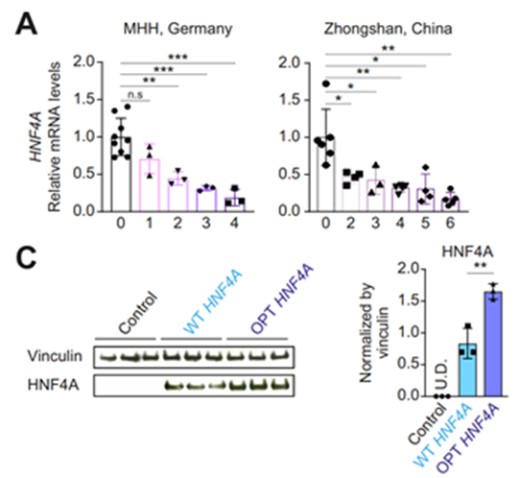
Figure. (A) qPCR analyses of human HNF4A mRNA in patients with fibrosis; 0 (n = 9), 1 (n = 3), 2 (n = 3), 3 (n = 3) and 4 (n = 3) from MHH, Germany and 0 (n = 6), 2 (n = 4) 3 (n = 3), 4 (n = 5), 5 (n = 4), 6 (n = 5) from Zhongshan Hospital, China. (C) Western blot and its quantification for wild-type (non-codon-optimized) and codon-optimized HNF4A mRNA in HeLa cells, 6 hours after HNF4A mRNA delivery.
Efficient Lipid Nanoparticle (LNP)-mediated mRNA delivery into hepatocytes of murine fibrotic livers was established. LNPs interact with apolipoprotein E at the surface of hepatocytes and are subsequently internalized via endocytosis. However, whether LNP-formulated mRNA reaches hepatocytes of fibrotic livers efficiently has remained unknown. LNP carrying mRNA encoding for Photinus pyralis luciferase (Luc/LNP) or ZsGreen (ZsGreen/LNP) were injected into CCl4-induced fibrotic and non-fibrotic control (wild-type) mice i.v. In-vivo imaging revealed robust bioluminescence exclusively in the liver from 8 hours to 120 hours after Luc/LNP injection in both groups (graphic shown below). Co-localization studies showed robust ZsGreen protein expression specifically in hepatocytes of fibrotic and control mice, but not in other hepatic cells. Our results demonstrated that LNP-formulated mRNA technology could specifically and efficiently target hepatocytes in murine fibrotic livers (data not shown below).
114
Targeted delivery of LNP-encapsulated mRNA into hepatocytes of fibrotic livers
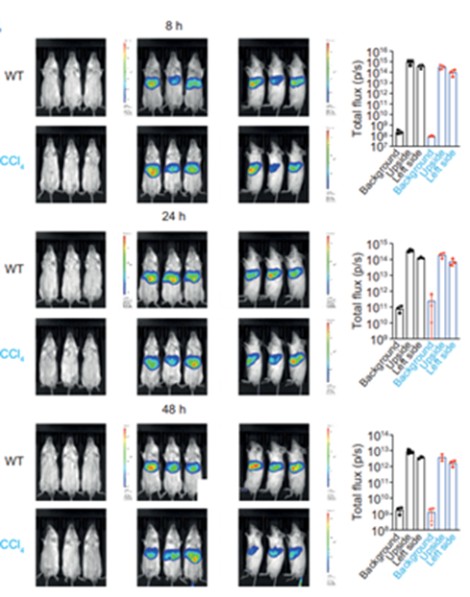
Figure. Successful mRNA delivery into hepatocytes of fibrotic BALB/c mice by LNP. Bioluminescence analyses at 8 h, 24 h and 48 h in wild type control and CCl4-induced fibrotic mice (n = 3 mice per group) injected i.v. with 40µg luciferase mRNA-encapsulated LNP (Luc/LNP).
To address whether systemic administration of HNF4A mRNA inhibits liver fibrosis, mice with toxin- (repeated CCl4 injection, data shown below) or cholestasis-induced fibrosis (induced via 3,5-diethoxycarbonyl-1,4-dihydrocollidine DDC-containing diet, data not shown) were used. HNF4A mRNA levels significantly decreased in fibrotic livers from both models. HNF4A/LNP or ZsGreen/LNP (henceforth referred to as control) were injected i.v. into fibrotic mice at 2 mg/kg per injection. HNF4A protein expression was confirmed in HNF4A/LNP-injected mouse livers and was absent in control mice, since the HNF4A antibody was only specific to human but not mouse HNF4A. HNF4A/LNP-injected mice showed significantly reduced alanine aminotransferase (ALT) and bilirubin indicating improved liver function, and significantly reduced levels of collagen (hydroxyproline assay), suggesting decreased fibrosis in both CCl4 and DDC models. Histological analyses, desmin (profibrogenic activated stellate cell marker) and Sirius red (collagen marker) staining further confirmed reduced fibrosis in CCl4 (Fig. F, N). Our data together provide evidence that HNF4A mRNA delivery attenuates fibrosis in mouse models of toxin as well as cholestasis-induced fibrosis.
115
Therapeutic HNF4A/LNP delivery inhibits toxin induced liver fibrosis
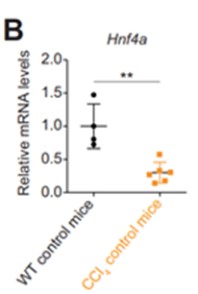

Figure. Therapeutic HNF4A/LNP delivery inhibits toxin induced liver fibrosis. CCl4-induced liver fibrosis (n = 6 mice per CCl4 model). (B) Hnf4a qPCRs after CCl4 injection. (D) Liver function tests for ALT shows reduced injury upon HNF4A/LNP administration. (E) Hydroxyproline assay shows reduced collagen content.
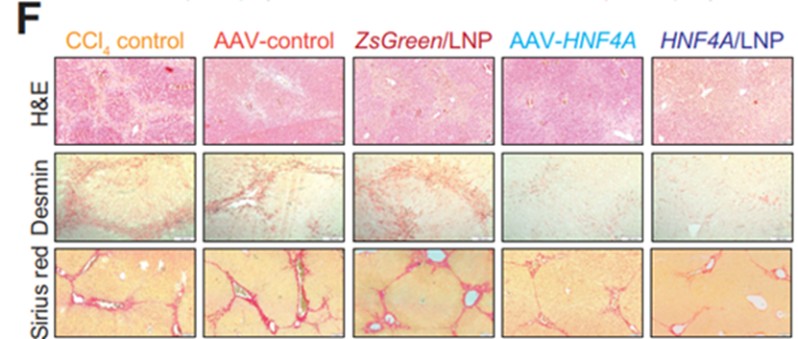
116
Figure. Therapeutic HNF4A/LNP delivery inhibits toxin induced liver fibrosis. CCl4-induced liver fibrosis (n = 6 mice per CCl4 model). (F) Immunohistochemical images of H&E, desmin and Sirius red stainings. Scale bars, 100 lm. (G) Quantification of Sirius red and desmin stainings.
In summary, the preclinical work shown here provides the first evidence that therapeutic mRNA delivery restores intracellular HNF4A transcription factor levels, induces endogenous Hnf4a expression, and improves metabolic activity of targeted hepatocytes. The data show that hepatocyte-specific delivery of HNF4A mRNA in injured livers attenuates fibrosis and cirrhosis in multiple independent mouse models of liver diseases. The shown data represents only a selection of important findings of the comprehensive preclinical studies. Full data can be accessed via the corresponding publication.
Eye Diseases
We are exploring the treatment of eye diseases with mRNA therapy. We have strategic collaborations with SERI for the development of mRNA-based treatments for currently undisclosed eye indications. We believe that the treatment of eye diseases with mRNA therapy represents an excellent opportunity for the mRNA approach for the following reasons:
| ● | Therapeutic protein can be produced directly and locally within the target tissue; |
| ● | Local treatment in the eye requires lower mRNA doses, thereby minimizing systemic exposure; |
| ● | Enables production of endogenous proteins to stop or prevent pathological processes locally in the eye, such as neo-vascularization or apoptosis; |
| ● | Enables expression of multi-domain intracellular or transmembrane proteins in key cells within the eye overcoming limitations of recombinant proteins; |
| ● | No concern with potential side effects typical for viral gene vector; |
| ● | No mRNA construct size restrictions as with viral gene vectors; and |
| ● | The eye is an immune-privileged organ. |
Based on positive preclinical data the agreement and collaboration with SERI moved ahead. We believe that the clinical and research expertise in eye diseases at SERI would allow us to fully leverage our mRNA in the discovery and validation of eye disease targets amenable to mRNA treatment. In collaboration with SERI, a high-priority rare eye condition has been identified for development. Multiple therapeutic targets have been identified for this condition and mRNAs have been generated and are currently being tested in preclinical studies, including models of disease.
117
Significant Agreements
Collaborations
We have entered into various licensing and commercialization agreements, including the following agreements with respect to product candidates:
Collaboration and License Agreements
2020 GlaxoSmithKline Collaboration and License Agreement
In July 2020, we entered into a Collaboration and License Agreement with GSK, which we refer to as the 2020 GSK Agreement, pursuant to which we are collaborating with GSK to research, develop and commercialize prophylactic and therapeutic non-replicating mRNA-based vaccines and antibodies targeting infectious disease pathogens. Under the terms of the 2020 GSK Agreement, we granted GSK a worldwide exclusive, sublicensable (subject to certain conditions) license under certain of our intellectual property relating to vaccines and antibodies encoded by our proprietary mRNA targeting certain selected pathogens, or GSK Program Products, and a non-exclusive license under certain LNP technology to develop, manufacture and commercialize a certain number of such GSK Program Products for use in connection with the infectious diseases targeted under the 2020 GSK Agreement. We additionally granted GSK an exclusive option for a certain period to add additional products in the field of infectious diseases as GSK Program Products. If such additional product targets a coronavirus other than SARS-CoV-2, at our election such product will be developed and commercialized on a cost and profit split basis under the GSK COVID Agreement. GSK is permitted for a certain period to replace any of the GSK Program Products with an alternative product, up to a certain number of times, and to exchange any antigen or antibody for which we have granted GSK a license under LNP technology for an alternative antigen or antibody, up to a certain number of times. In April 2023, GSK exercised its replacement right to replace one product. In the event we obtain rights to any intellectual property controlled by a third party that is useful for the development, manufacture or commercialization of the GSK Program Products, but which is not necessary to obtain freedom to operate with respect to the use or exploitation of our technology or know-how, we must, at GSK’s election, use commercially reasonable efforts to obtain a sublicense to such rights on behalf of GSK. Under the terms of the 2020 GSK Agreement, GSK granted us a royalty-free, non-exclusive license under certain GSK-controlled technology to perform certain development and manufacturing activities under the 2020 GSK Agreement. The 2020 GSK Agreement was amended and restated in April 2021, September 2021, February 2022 and March 2022. Under the September 2021 Amendment, each party also granted the other party a royalty-free, perpetual, worldwide, non-exclusive, sublicensable license under certain inventions created by such party to freely practice, use and exploit such inventions in any field.
For a certain period after the effective date, GSK has the right to reserve up to a certain number of antigens and we and our affiliates will be prohibited from granting any rights to a third party with respect to any such antigen for use in connection with infectious diseases. Under the terms of the 2020 GSK Agreement, GSK and its affiliates and sublicensees and we and our affiliates are additionally prohibited from developing, manufacturing or commercializing, directly or indirectly, any prophylactic or therapeutic mRNA-based vaccine or mRNA-based antibody targeting a pathogen targeted by a GSK Program Product, other than as contemplated under the 2020 GSK Agreement. Such exclusivity obligation will continue on a pathogen-by-pathogen basis for the duration of the 2020 GSK Agreement, so long as such pathogen is targeted by a GSK Program Product. We are additionally prohibited from granting any third party any license under the licensed LNP technology, or using such LNP technology ourselves, in connection with any GSK Program Product for so long as such GSK Program Product is being developed or commercialized under the 2020 GSK Agreement, except as contemplated under the 2020 GSK Agreement. We are additionally prohibited, for the period during which GSK’s option to license additional GSK Program Products remains outstanding, from commercializing or granting any third-party the right to develop or commercialize any prophylactic or therapeutic mRNA-based vaccine or mRNA-based antibody targeting certain pathogens for use in connection with infectious diseases.
118
We and GSK are required to complete certain development activities with respect to the GSK Program Products set forth in various development plans. Among other development responsibilities, we are required to provide clinical supply and will in principle be responsible for sponsoring Phase 1 clinical trials for the GSK Program Products. We and GSK agree to decide whether the products required for clinical studies will be manufactured by us, GSK or jointly. At GSK’s request, we are required to transfer to GSK all know-how necessary for GSK’s development activities under the 2020 GSK Agreement and all know-how necessary for the manufacture of the GSK Program Products. GSK is generally responsible for development activities following completion of Phase 1 clinical trials and is required to use diligent efforts to secure marketing authorization following completion of all necessary clinical trials. GSK is responsible for the commercialization of approved GSK Program Products in all countries other than Austria, Germany and Switzerland and is required to use diligent efforts to commercialize approved GSK Program Products in certain major market countries. At our request, we and GSK will negotiate and agree in good faith to a distribution agreement pursuant to which we will have the exclusive right to commercialize GSK Program Products in Austria, Germany and Switzerland, and we will pay GSK royalties at the rate set out below. We and GSK are required to provide development data to the other party thorough a joint steering committee.
GSK paid us an upfront payment of €120 million and is required to pay us a manufacturing capacity reservation fee of €30 million following a certain regulatory milestone event, which is creditable against future milestone payments. We are eligible to receive up to between €28 million to €45 million in development milestone payments, €32 million to €35 million in regulatory milestone payments and €70 to €100 million in commercial milestone payments, depending on the GSK Program Product. Upon each exercise of its option to add additional products as GSK Program Products, GSK is required to compensate us for certain development costs and pay any accrued milestone payments. If GSK exercises its right to replace a GSK Program Product and if the replacement product was already under development by us, GSK must compensate us for certain development costs and pay any accrued milestone payments. We are eligible to receive tiered royalty payments ranging from a single-digit percentage to a low teens percentage on net sales, subject to certain customary reductions. GSK’s royalty obligations continue on a product-by-product and country-by-country basis until the later of (i) the expiration of the last to expire valid claim covering such product in such country, (ii) the earlier of expiration of regulatory exclusivity for such product in such country or 12 years following the first commercial sale of such product in such country or (iii) ten years following the first commercial sale of such product in such country provided our proprietary know-how is required for such product. In any event, GSK’s royalty obligations with respect to a product will expire in all countries no later than 20 years following the first commercial sale of such product in any country in which GSK is responsible for the commercialization of approved GSK Program Products. GSK is required to compensate us for certain development and regulatory costs we may incur in connection with our performance of our obligations under the 2020 GSK Agreement and we are eligible to receive up to €20,000 in reimbursements for expenses incurred recording or registering the licenses granted under the 2020 GSK Agreement. Under any distribution agreement entered into between us and GSK in connection with our distribution of a GSK Program Product in Austria, Germany and Switzerland, we will be required to purchase supply from GSK and pay GSK a low thirties percentage royalty on net sales. Under the March 2022 amendment, GSK and CureVac agreed on certain changes to the manufacturing strategy for the products developed under the collaboration, as a part of which GSK agreed to reimburse us for certain manufacturing expenses amounting to €42 million.
The term of the 2020 GSK Agreement will continue until the expiration of the last-to-expire royalty term, unless terminated earlier by either party. GSK has the right to terminate the 2020 GSK Agreement in its entirety or on a program-by-program basis for convenience following a certain notice period. We and GSK both have the right to terminate the 2020 GSK Agreement on a program-by-program basis before the first commercial sale of a GSK Program Product under such program in the event of the other party’s material breach following a cure period or after the first commercial sale of a GSK Program Product under such program if the other party fails to make any payments due, commits any willful and material breach of the restrictions on any license granted to such party, commits a material breach of its non-compete obligations or commits a persistent and material breach of its confidentiality obligations following a cure period. We additionally have the right to terminate on a program-by-program basis after the first commercial sale of a GSK Program Product under such program if GSK commits a material breach of its commercialization diligence obligations following a cure period.
119
Upon expiration, the licenses granted to GSK under the 2020 GSK Agreement will become fully paid-up, perpetual and non-exclusive. In the event GSK terminates the 2020 GSK Agreement or a program under the 2020 GSK Agreement for convenience or we terminate a program under the 2020 GSK Agreement for cause, we will have the right to elect to continue with the development and commercialization of such program ourselves. If we decline to continue with the development and commercialization of a terminated program, all licenses granted under the 2020 GSK Agreement will terminate. If we elect to continue with the development and commercialization of a terminated program, all licenses granted by us to GSK will terminate and GSK must grant us an exclusive license under any intellectual property developed under the 2020 GSK Agreement and, at our election, a non-exclusive license under technology, which was used by GSK for the development, manufacture or commercialization of such terminated product. In the case of termination for GSK’s convenience and if we elect to obtain such nonexclusive license, we will be required to pay GSK a single-digit percentage royalty on net sales. In the case of our termination for cause, the grant of rights and transition of the assets from GSK will be subject to a payment to GSK to be mutually agreed by the parties. In the event GSK terminates a program under the 2020 GSK Agreement for cause, GSK will have the right to elect to continue the development and commercialization of such program. If GSK declines to continue with the development and commercialization of a terminated program, all licenses granted under the 2020 GSK Agreement will terminate. If GSK elects to continue development and commercialization, all licenses granted to GSK under the 2020 GSK Agreement will survive termination and all payment obligations will remain in effect except that GSK will have the right to suspend payments until the amount of damages suffered by GSK has been agreed and set off against such payments.
GlaxoSmithKline COVID Collaboration and License Agreement
In April 2021, we entered into a collaboration agreement with GSK, which we refer to as the GSK COVID Agreement, pursuant to which we are collaborating with GSK to research, develop and manufacture next-generation mRNA vaccines targeting the original SARS-CoV-2 strain as well as emerging variants, including multivalent and monovalent approaches, such as our second-generation COVID-19 vaccine candidates, CV0601 or CV0701. These vaccine candidates may either be used to protect unvaccinated individuals or to serve as boosters in the event that SARS-CoV-2 immunity gained from an initial vaccination reduces over time.
Under the terms of the GSK COVID Agreement, we granted GSK a worldwide, exclusive, sublicensable (subject to certain conditions) license under certain of our intellectual property relating to mRNA-based vaccines targeting SARS-CoV-2 and a non-exclusive license under certain LNP technology to develop, manufacture and commercialize certain SARS-CoV-2 pathogen vaccine products, or the GSK COVID Products, for use in connection with the prevention or treatment of diseases caused by the SARS-CoV-2 pathogen. The GSK COVID Products consist of (i) next-generation SARS-CoV-2 pathogen vaccine products (other than CVnCoV), (ii) vaccine products targeting coronaviruses other than SARS-CoV-2 for which GSK exercises its exclusive option pursuant to the 2020 GSK Agreement and where we elect to develop and commercialize such product on a cost and profit split basis under the GSK COVID Agreement and (iii) next-generation SARS-CoV-2 pathogen vaccine products (other than CVnCoV) that also target one or more pathogens that the parties are targeting under the 2020 GSK Agreement, which we refer to as Combination Products. In the event we obtain rights to any intellectual property controlled by a third-party that is useful for the development, manufacture or commercialization of the GSK COVID Products, but which is not necessary to obtain freedom to operate with respect to the use or exploitation of our technology or know-how, we must, at GSK’s election, use commercially reasonable efforts to obtain a sublicense to such rights on behalf of GSK. Under the terms of the GSK COVID Agreement, GSK granted us a royalty-free, non-exclusive license under certain GSK-controlled technology to perform certain development and manufacturing activities under the GSK COVID Agreement. Under the September 2021 Amendment, each party also granted the other party a royalty-free, perpetual, worldwide, non-exclusive, sublicensable license under certain inventions created by such party to freely practice, use and exploit such inventions in any field.
GSK and its affiliates and sublicensees and we and our affiliates are prohibited from, subject to certain exceptions, developing, manufacturing or commercializing, directly or indirectly, any mRNA-based vaccine or mRNA-based antibody products targeting the SARS-CoV-2 pathogen, other than a GSK COVID Product as contemplated under the GSK COVID Agreement or CVnCoV. The exclusivity obligations remain in effect until the expiration or termination of the GSK COVID Agreement.
120
We and GSK are required to complete certain development activities with respect to the GSK COVID Products set forth in various development plans. The GSK COVID Agreement was amended and restated in September 2021, February 2022 and March 2022, as described below, and further amended in August 2022 to reflect conforming updates to the development plans. Pursuant to the amendment in September 2021, we and GSK are required to complete certain development activities with respect to the GSK COVID Products set forth in updated development plans. We and GSK agree to decide whether the GSK COVID Products required for clinical studies will be manufactured by us, GSK or jointly. Once a manufacturing and supply strategy for a given GSK COVID Product has been agreed upon between the parties, we must negotiate and agree in good faith on a commercial supply agreement pursuant to which we may be required to reserve certain manufacturing capacity for GSK. At GSK’s request, we are required to transfer to GSK all know-how necessary for GSK’s development activities under the GSK COVID Agreement and all know-how necessary for the manufacture of the GSK COVID Products. GSK is responsible for the commercialization of GSK COVID Products in all countries other than Austria, Germany and Switzerland and is required to use diligent efforts to commercialize approved GSK COVID Products in certain major market countries. At our request, we and GSK will negotiate and agree in good faith to a distribution agreement pursuant to which we will have the exclusive right to commercialize GSK COVID Products in Austria, Germany and Switzerland. We and GSK are required to provide development data to the other party thorough a joint steering committee.
Under the GSK COVID Agreement, GSK paid us an upfront payment of €75 million. Upon GSK’s exercise of its option to add CVnCoV and boosters for such vaccine as GSK COVID Products, GSK is required to compensate us for certain development costs. We and GSK agreed to equally share all development costs for GSK COVID Products, subject to certain exceptions. We and GSK will share all net profits generated from sales of GSK COVID Products, other than Combination Products, under profit sharing arrangements that in certain cases vary depending upon the GSK COVID Product in question, the time of sale, the number of doses sold and the party to whom the sale is made. We are eligible to receive tiered royalty payments ranging from a sub-teen percentage to a mid-teens percentage on net sales of Combination Products, subject to certain customary reductions. We will pay GSK a high-teen percentage royalty on net sales of all Combination Products in Austria, Germany and Switzerland. All royalty obligations continue on a product-by-product and country-by-country basis until the later of (i) the expiration of the last to expire valid claim covering such product in such country (ii) the earlier of expiration of regulatory exclusivity for such product in such country or 12 years following the first commercial sale of such product in such country, or (iii) 10 years following the first commercial sale of such product in such country provided our proprietary know-how is required for such product. In any event, GSK’s royalty obligations with respect to a product will expire in all countries no later than 20 years following the first commercial sale of such product in the respective party’s territory.
The term of the GSK COVID Agreement will continue until the expiration of all applicable payment obligations, unless terminated earlier by either party. GSK has the right to terminate the GSK COVID Agreement in its entirety for convenience following a certain notice period, and we have the right to opt out of the funding of the development, manufacture and commercialization of a GSK COVID Products on a product-by-product basis. In the event we opt out of funding the development of a GSK COVID Product, GSK can elect to cease the development and commercialization of the relevant product, or to continue it on the terms of the 2020 GSK Agreement. If GSK declines to continue with the development and commercialization, the agreement terminates in connection with that GSK COVID Product and all licenses granted under the GSK COVID Agreement will terminate. We and GSK both have the right to terminate the GSK COVID Agreement before the first commercial sale of a GSK COVID Product in the event of the other party’s material breach following a cure period or after the first commercial sale of a GSK COVID Product if the other party fails to make any payments due, commits any willful and material breach of the restrictions on any license granted to such party, commits a material breach of its non-compete obligations or commits a persistent and material breach of its confidentiality obligations following a cure period. We additionally have the right to terminate after the first commercial sale of a GSK COVID Product if GSK commits a material breach of its commercialization diligence obligations following a cure period.
121
Upon expiration, the licenses granted to GSK under the GSK COVID Agreement will become fully paid-up, perpetual and non-exclusive. In the event GSK terminates the GSK COVID Agreement for convenience or we terminate the GSK COVID Agreement for cause, all licenses granted to GSK under the GSK COVID Agreement will terminate and we will have the right to elect to continue with the development and commercialization of the GSK COVID Products ourselves. If we elect to continue with the development and commercialization of the COVID Products, GSK must grant us an exclusive license under any intellectual property developed under the GSK COVID Agreement and, at our election, a non-exclusive license under technology which was used by GSK for the development, manufacture or commercialization of the GSK COVID Products. In the case of termination for GSK’s convenience and if we elect to obtain such non-exclusive license, we will be required to pay GSK a royalty ranging from a sub-single-digit percentage to a low single-digit percentage on net sales. In the case of our termination for cause, the grant of rights and transition of the assets from GSK will be subject to a payment to GSK to be mutually agreed by the parties. In the event GSK terminates the GSK COVID Agreement for cause, GSK will have the right to elect to continue the development and commercialization of the GSK COVID Products. If GSK declines to continue with the development and commercialization of the GSK COVID Products, all licenses granted under the GSK COVID Agreement will terminate. If GSK elects to continue development and commercialization, all licenses granted to GSK under the GSK COVID Agreement will survive termination, provided that for GSK COVID Products other than Combination Products a one-time payment from GSK to Curevac (in replacement of a continuous profit sharing mechanism) will be mutually agreed upon by the parties. All payment obligations for Combination Products will remain in effect. In each case, GSK will have the right to suspend payments until the amount of damages suffered by GSK has been agreed and set off against such payments.
CureVac-GSK Consortium Agreement
The Federal Republic of Germany, represented by the Vaccine Production Taskforce on behalf of the Federal Ministry of Health, called for tenders relating to pandemic preparedness, which we refer to as the Tender Procedure. The Tender Procedure resulted in framework agreements for the provision to the Federal Republic of Germany of production capacities and, upon demand, the production and supply of mRNA vaccines (referred to as lot 1) and vector- or protein-based vaccines (referred to as lot 2). Because neither we nor GSK were alone in a position to provide the full range of services requested by the Federal Republic of Germany under the Tender Procedure, we established a consortium with GSK (referred to as the CureVac-GSK Consortium) for the purpose of participating in the Tender Procedure, entering into a framework agreement for the provision of production capacities and, upon demand, the production and supply of mRNA vaccines (lot 1), which we refer to as a Pandemic Preparedness Agreement.
The CureVac-GSK Consortium submitted an application and offer under the Tender Procedure for the award of a Pandemic Preparedness Agreement. On April 8, 2022, the Federal Republic of Germany sent a letter confirming that the CureVac-GSK Consortium had been awarded a Pandemic Preparedness Agreement. Following a qualification phase of a maximum of two years from the award date, the Pandemic Preparedness Agreement grants the government access to a manufacturing capacity of 80 million doses of mRNA-based vaccine per year until 2029, subject to extension. Under the contract, after successful achievement of pandemic preparedness by the end of the qualification phase, the contract will enter into a stand-by phase during which the government will pay the CureVac-GSK Consortium an annual stand-by fee. During the stand-by phase, the CureVac-GSK Consortium is required to maintain a manufacturing capacity of 80 million doses of mRNA-based vaccine per year at constant readiness. The Pandemic Preparedness Agreement is subject to termination by the Federal Republic of Germany or the CureVac-GSK Consortium if, among other things, by the end of the qualification phase the CureVac-GSK Consortium does not have an mRNA-based vaccine for which a marketing authorization (which may be temporary) for the German market has been granted. On April 9, 2024, the CureVac-GSK Consortium terminated the Pandemic Preparedness Agreement, effective as of May 31, 2024.
122
Genmab Collaboration and License Agreement
In December 2019, we entered into a Collaboration and License Agreement with Genmab, which we refer to as the Genmab Agreement, to research and develop up to four potential differentiated mRNA-based antibody products, to be selected by Genmab, based on the combination of our proprietary RNAntibody technology with Genmab’s proprietary antibody technology for the treatment of human diseases. The Genmab Agreement was amended in July 2020, December 2020 and June 2021. Pursuant to the Genmab Agreement we granted Genmab an exclusive, worldwide, sublicensable (subject to certain conditions) license under our mRNA technology for the development, manufacture and commercialization of an mRNA antibody product designed to express a certain Genmab proprietary antibody, which we refer to as the Genmab First Program. However, in June 2023 Genmab notified us of its intent to terminate the Genmab First Program under the Genmab Agreement effective in September 2023. Such termination did not terminate the Genmab Agreement in its entirety, but rather only with respect to certain license and exclusivity provisions related to the Genmab First Program. We additionally granted Genmab an exclusive, worldwide, sublicensable license under our mRNA technology for the research and preclinical development of up to four additional mRNA antibody product concepts and an option to obtain an exclusive, worldwide, sublicensable (subject to certain conditions) license to develop, manufacture and commercialize product candidates for up to three of such product concepts. We have the option to share in the costs and profits in connection with the development, manufacture and commercialization of one of the additional mRNA antibody product concepts under predefined terms and conditions.
We may not, directly or indirectly, offer any rights to a third party under the technology we license to Genmab for the additional product concepts and targets being developed under the Genmab Agreement or conduct or participate in the development, manufacture or commercialization of any antibody product that is directed at a target being developed under the Genmab Agreement for certain periods of time.
In partial consideration for entering into the Genmab Agreement, Genmab paid us an upfront fee of $10 million and made a €20 million equity investment. Genmab is additionally required to pay us up to $30 million in option exercise fees. If Genmab exercises any of its options to obtain commercial licenses for the additional mRNA antibody concepts, Genmab would fund all research and would develop and commercialize any resulting product candidates. We are additionally eligible to receive up to between $25 million and $43 million in development milestone payments, $100 million and $125 million in regulatory milestone payments and $150 million and $200 million in commercial milestone payments for each product, depending on the specific product concept. In addition, we are eligible to receive a mid-single-digit to low teens percentage tiered royalty on aggregate net sales of licensed products, on a per product basis and subject to certain customary reductions. Genmab’s royalty obligation continues on a country-by-country and product-by-product basis until the later of the expiration of the last-to-expire valid claim in the licensed patents in such country covering such licensed product, expiration of regulatory exclusivity for such product in such country or 10 years from the date of the first commercial sale of such product. In the event we exercise our right to share in the development, manufacture and commercialization of a product, we must pay Genmab a one-time payment of $3 million and refund any option fee paid by Genmab with respect to such product. As of December 31, 2023, we have received $1 million in development cost reimbursements, and we have not received any reservation, product selection, option exercise or sublicense fees or milestone or royalty payments.
We are required to use commercially reasonable efforts to perform our obligations under the research and development plans established in connection with the Genmab Agreement. Genmab is required to use commercially reasonable efforts to identify and develop each additional product Genmab adds to the development program under the Genmab Agreement, and to further develop each optioned product to marketing authorization and to commercialize each product for which it obtains regulatory approval. We and Genmab are required to make available to the other party all preclinical development data for each program under development under the Genmab Agreement until filing of an IND for such program. Following IND filing for a product, we and Genmab will establish a collaboration committee where Genmab will share the status, progress and results of the development of the respective product.
123
The term of the Genmab Agreement will continue until the expiration of the royalty term, unless terminated earlier by either party. The Genmab Agreement may be terminated upon written notice by either party upon the other party’s material breach or default of any of its obligations following a cure period. Genmab may terminate the Genmab Agreement for convenience after a certain notice period. Upon expiration of the Genmab Agreement, the license rights we granted to Genmab under the Genmab Agreement will become fully paid-up, perpetual and non-exclusive. In the event of termination for our material breach, we will grant Genmab an exclusive (even to us), worldwide and sublicensable license to exploit any product identified prior to termination, subject to Genmab’s continued milestone and royalty obligations. In the event of termination by us for Genmab’s material breach, or Genmab’s termination for convenience, the licenses granted to Genmab will automatically terminate. Additionally, at our request, Genmab will grant us a non-exclusive, royalty-free, sublicensable, perpetual and worldwide license under certain Genmab intellectual property that is created under the Genmab Agreement and that is required to develop, manufacture and commercialize our own mRNA antibody products targeting the collaboration targets under the Genmab Agreement prior to termination. Such license would not include any license to Genmab background intellectual property or the specific products or antibodies developed by Genmab.
Arcturus Development and Option Agreement
In January 2018, we entered into a Development and Option Agreement with Arcturus, which we refer to as the Arcturus Agreement, pursuant to which Arcturus granted us the right to reserve a certain number of targets and an irrevocable offer to obtain a license to a certain number of such reserved targets to develop, manufacture and commercialize products containing Arcturus’s LNP technology (LMD technology) and mRNA constructs intended to express such targets. The Arcturus Agreement expired in July 2023. We did not accept the offer with respect to any targets.
As of December 31, 2023, we made payments totaling $5.6 million to Arcturus reimbursing Arcturus for development costs and in connection with our FTE funding obligations.
Acuitas Development and Option Agreement
In April 2016, we entered into a Development and Option Agreement with Acuitas, which as amended we refer to as the Acuitas Agreement, pursuant to which Acuitas granted us the right to reserve a certain number of vaccine and other targets and an option to obtain a license to a certain number of such reserved targets to develop, manufacture and commercialize products containing Acuitas’s LNP technology and mRNA constructs intended to express such targets. With respect to a certain number of nonexclusive licenses to vaccine targets that we obtain under the Acuitas Agreement, Acuitas additionally granted us an option to exchange each vaccine target licensed under such nonexclusive license for an alternate vaccine target for a certain period. As of December 31, 2023, we have exercised our option to obtain a nonexclusive license to 18 targets, and have not exercised our option to exchange a vaccine target licensed under any nonexclusive license. In connection with an amendment to the Acuitas Agreement dated February 13, 2024, Acuitas also granted us the right to evaluate, and an option to obtain a license to, certain of Acuitas’s LNP technology for personalized cancer vaccine products.
Under the Acuitas Agreement, Acuitas is responsible for the LNP chemistry and formulation and characterization work, and we are responsible for mRNA construct development. Both parties will undertake certain allocated preclinical studies. Each party is required to use diligent efforts to perform its obligations under the work plans established in connection with the Acuitas Agreement. Acuitas is further required to use diligent efforts to manufacture and supply us with certain formulated products. The Acuitas Agreement provides for the establishment of a joint development committee for the discussion of development efforts under the Acuitas Agreement. We are required to reimburse Acuitas for certain costs incurred in connection with development activities and certain FTE costs.
124
We are further required to pay Acuitas annual target reservation and maintenance fees of up to $1.4 million if we reserve the maximum number of targets permitted under the Acuitas Agreement. We are additionally required to pay an option exercise fee ranging from $50,000 to $2 million upon each exercise of our option under the Acuitas Agreement, subject to certain additional fees ranging from $10,000 to $200,000 for the exercise of our option for certain other vaccine targets. We paid Acuitas a $5 million upfront fee in connection with an amendment to the Acuitas Agreement dated July 2020 and, upon each exercise of our option to exchange a vaccine target licensed under any non-exclusive license, we paid an exchange fee of $3 million. We paid Acuitas a $3 million upfront fee in connection with an amendment to the Acuitas Agreement dated December 2020 and paid two annual payments of an additional $250,000 per option to extend certain options. Under each license agreement we enter into in connection with our exercise of our option, we will additionally be required to make low single-digit percentage tiered royalty payments, subject to certain customary reductions, on a country-by-country and a product-by-product basis until the later of the expiration of the last-to-expire licensed patent in such country covering such licensed product, expiration of regulatory exclusivity for such product in such country or 10 years from the date of the first commercial sale of such product in such country. Under each such license we additionally must pay up to between $1.1 million and $9 million in development milestone payments, $1.3 million and $7 million in regulatory milestone payments and $1.3 million and $7 million in commercial milestone payments, depending on whether the license is exclusive or non-exclusive and the number of options exercised to date. As of December 31, 2023, we have exercised our option to obtain a non-exclusive license to 18 targets. As of December 31, 2023, we have paid Acuitas $9.7 million in reservation and option exercise fees, $0.6 million in license maintenance fees and $1.75 million for certain options not yet exercised and have made payments totaling $9.0 million reimbursing Acuitas for development costs and LNP batches and in connection with our FTE funding obligations. Payments made under the license agreements entered into in connection with our exercise of our option under the Acuitas Agreement are described below.
Under the Acuitas Agreement, Acuitas granted us a worldwide, non-exclusive license under its LNP technology for us to perform development activities, and we granted Acuitas a worldwide, non-exclusive license under our mRNA technology solely to enable Acuitas to perform development activities in connection with the Acuitas Agreement.
The Acuitas Agreement will expire in April 2025 unless earlier terminated or extended. Both parties have the right to terminate the Acuitas Agreement in whole or on a program-by-program basis in the event of a material breach by the other party following a cure period. We additionally have the right to terminate the Acuitas Agreement for convenience following a certain notice period or for Acuitas’s change of control. In the event of termination for any reason, Acuitas will transfer all deliverables created under the Acuitas Agreement to us and in the event we terminate for reasons other than for Acuitas’s material breach, we must make any payments owed to Acuitas up to the time of termination. In the event we terminate for Acuitas’s material breach or for Acuitas’s change of control, Acuitas will transfer any technology and provide licenses as reasonably necessary for us to complete work contemplated under the Acuitas Agreement and, in the case of termination for Acuitas’s material breach, Acuitas must refund to us any target reservation and maintenance fees for the remainder of the contract year in which such termination is effective.
Acuitas Non-exclusive License Agreements
For each option we have exercised under the Acuitas Agreement, we have entered into a non-exclusive license agreement with Acuitas with respect to such optioned product, all based on the same form agreement, which we collectively refer to as the Acuitas License Agreements. Under the Acuitas License Agreements, Acuitas grants us a non-exclusive, non-transferable, sublicensable (subject to certain conditions) worldwide license under Acuitas’s LNP technology to develop, manufacture and commercialize licensed products directed to the optioned targets. We may convert the non-exclusive licenses to exclusive licenses subject to certain additional financial obligations. As of December 31, 2023, we have not converted any non-exclusive license to an exclusive license.
125
We must pay Acuitas up to between $1.1 million and $1.6 million in development milestone payments, $1.3 million and $1.8 million in regulatory milestone payments and $1.3 million and $1.8 million in commercial milestone payments under each Acuitas License Agreement upon the occurrence of certain milestone events. We additionally are obligated to pay Acuitas annual fees ranging from $5,000 to $10,000 for any additional protein targeted by a vaccine product licensed under an Acuitas License Agreement after a certain milestone event. We are further required to pay Acuitas a low single-digit tiered percentage royalty on net sales of licensed products, subject to certain potential customary reductions. Our royalty obligations continue under each Acuitas License Agreement on a country-by-country and product-by-product basis until the later of the expiration of the last-to-expire licensed patent claim covering such licensed product in such country, expiration of any regulatory exclusivity period for such product in such country and 10 years following the first commercial sale of such product in such country. As of December 31, 2023, we have made $100,000 in development milestone payments to Acuitas with respect to the license agreement relating to Rabies RAV-G, $1.4 million in development milestone payments (Phase I, Phase II and Phase III milestone payments) to Acuitas with respect to the license agreement relating to the SARS-CoV-2 Spike protein S, $350,000 in development milestone payments to Acuitas with respect to the license agreement relating to the Influenza hemagglutinin (HA) antigen, $1.0 million in development milestone payments to Acuitas with respect to the license agreement relating to CVGBM, and have not made any royalty payments.
Each Acuitas License Agreement will continue on a product-by-product and a country-by-country basis until there are no more payments owed to Acuitas for such product in such country. Either party may terminate an Acuitas License Agreement in the event of a material breach by the other party following a cure period. We additionally have the right to terminate the Acuitas License Agreements for convenience following a certain notice period. Upon expiration of an Acuitas License Agreement, the licenses granted to us under such Acuitas License Agreement will become fully paid-up and will remain in effect. In the event of our termination of an Acuitas License Agreement for Acuitas’s material breach, the rights and licenses granted to us under such agreement will become perpetual and irrevocable. Alternatively, instead of exercising our right to terminate in the event of Acuitas’s material breach, we may elect to instead continue the license but reduce our milestone and royalty payment obligations to Acuitas by a certain percentage. In the event of termination of an Acuitas License Agreement by us for convenience or by Acuitas for our material breach, the licenses granted under such agreement will terminate, except that we will have the right to sell off any remaining inventories of licensed products for a certain period of time.
CRISPR Therapeutics Development and License Agreement
In November 2017, we entered into a Development and License Agreement with CRISPR Therapeutics, which as amended by a first amendment entered into in June 2020 and a second amendment entered into in October 2023, we refer to as the CRISPR Therapeutics Agreement, pursuant to which we will develop novel Cas9 mRNA constructs for use in gene editing therapeutics. Under the terms of the CRISPR Therapeutics Agreement, we granted CRISPR Therapeutics a worldwide, exclusive (even to us), sublicensable (subject to certain conditions) license under certain intellectual property rights that are reasonably necessary or useful to develop, manufacture or commercialize products comprising Cas9 mRNA constructs, and under any patents controlled by us that arise from inventions discovered under the CRISPR Therapeutics Agreement to develop, manufacture and commercialize three of CRISPR Therapeutics’ in vivo gene-editing programs for certain diseases. CRISPR Therapeutics granted us an exclusive (even as to CRISPR Therapeutics), worldwide, cost-free sublicense to manufacture products comprising Cas9 mRNA constructs for CRISPR Therapeutics.
CRISPR Therapeutics has paid us an upfront one-time technology access fee of $3 million and we are eligible to receive up to $28 million in development milestone payments, $52 million in regulatory milestone payments and $260 million in commercial milestone payments, as well as mid single-digit percentage royalties from CRISPR Therapeutics on the net sales of licensed products on a product-by-product and country-by-country basis, subject to certain potential customary reductions. CRISPR Therapeutics’ royalty obligations continue on a product-by-product and country-by-country basis until the later of the date when there are no valid patent claims under our licensed patents covering such licensed product in such country, the date when regulatory exclusivity for such licensed product in such country expires and 10 years following the date of first commercial sale of such licensed product in such country. CRISPR Therapeutics is additionally required to reimburse us for our FTE costs and reasonable out-of-pocket expenses incurred performing development activities under the CRISPR Therapeutics Agreement. In the event CRISPR Therapeutics exercises its right to sublicense under the agreement, CRISPR Therapeutics must pay us a low teens to mid-twenties percentage of any non-royalty sublicense income, depending on the timing of the sublicense and whether the sublicense is granted through an affiliate of CRISPR Therapeutics. As of December 31, 2023, we have received €8.4 million in payments, for the supply of materials and FTE cost, development reimbursements and upfront one-time technology access fee and no milestone, royalty or sublicense fee payments. Additionally, as of December 31, 2023, we have invoiced CRISPR Therapeutics for certain amounts under the agreement in connection with the second amendment, which confirmed the parties’ intention to stop work on two programs under the CRISPR Therapeutics Agreement, terminated all licenses granted by CureVac to CRISPR Therapeutics with respect to such programs and added three new programs.
126
We are required to use commercially reasonable efforts to perform our development obligations under the CRISPR Therapeutics Agreement and to supply certain materials to CRISPR Therapeutics. CRISPR Therapeutics is required to use commercially reasonable efforts to perform its obligations under the development plan and to develop and commercialize licensed products. We and CRISPR are required to keep the other party informed regarding the progress and results of performance of all development activities under the CRISPR Therapeutics Agreement.
The term of the CRISPR Therapeutics Agreement will continue on a product-by-product and country-by-country basis, until the last-to-expire royalty term expires in such country for such product, unless terminated earlier by either party. The CRISPR Therapeutics Agreement may be terminated (i) by CRISPR Therapeutics for convenience following a certain notice period, (ii) by us if CRISPR Therapeutics or any of its affiliates, either directly or indirectly, challenges or assists a third party to challenge the licensed patent rights or in the event CRISPR Therapeutics undergoes a change of control, or (iii) by either party in the event of the other party’s material breach following a cure period (including on a program-by-program basis) or in the event of the other party’s insolvency. Upon expiration, the license granted to CRISPR Therapeutics converts into a fully paid-up, royalty-free, perpetual and irrevocable license. Upon termination, the licenses granted to CRISPR Therapeutics will terminate and, in the case of termination for CRISPR Therapeutics’ material breach or insolvency or for convenience by CRISPR Therapeutics, CRISPR Therapeutics must transfer all Cas9 mRNA constructs and related data to us.
Boehringer Ingelheim Exclusive Collaboration and License Agreement
In August 2014, we entered into an Exclusive Collaboration and License Agreement with Boehringer Ingelheim, which we refer to as the Boehringer Agreement, whereby we granted Boehringer Ingelheim exclusive global rights for development and commercialization of our investigational therapeutic mRNA vaccine BI 1361849 (formerly CV9202) formulated with our protamine technology, and products containing such vaccine for all uses for cancer in humans. We received an upfront payment of €30 million, as well as an option fee payment of €5 million and an additional €7 million in development milestone payments. As of December 31, 2021, we received €7.6 million for the supply of materials and reimbursing us for development costs. In June 2021, Boehringer Ingelheim provided notice of its intention to terminate the Boehringer Agreement, with such termination becoming effective on November 17, 2021.
Bill & Melinda Gates Foundation Partnership
In May 2014, we entered into a contract agreement with the Bill & Melinda Gates Foundation for the development of a vaccine for rotaviruses. Under the terms of the contract grant, as amended by an amendment entered into November 2020, the Bill & Melinda Gates Foundation will provide up to $3.0 million in funding, and we are required to perform certain activities specified in a project collaboration plan. As of December 31, 2023, we have received $3.0 million in funding under the agreement. We own all intellectual property created using contract funding; however, we must make any Bill & Melinda Gates Foundation-funded products available at an affordable price in a list of clearly defined low and lower middle-income countries. The term of the rotavirus agreement expired in June 2022. Our global access commitments survive termination or expiration of the agreement.
In March 2015, the Bill & Melinda Gates Foundation made an equity investment of $40 million to support continued development of our RNA technology platform and the construction of an industrial-scale cGMP production facility, and we entered into the Global Access Commitments Agreement with the Bill & Melinda Gates Foundation in February 2015 pursuant to which we are required to take certain actions to support the Bill & Melinda Gates Foundation’s mission. In particular, we are required to conduct development activities for up to three concurrent projects to be proposed by the Bill & Melinda Gates Foundation, subject to our right to reject proposed projects where we believe there is a reasonable likelihood of a material adverse effect on us. The costs of such projects will be allocated on a project-by-project basis in proportion to the allocation of the expected benefits. All intellectual property developed in connection with such projects will be owned by us.
Under the terms of the Global Access Commitments Agreement, any Bill & Melinda Gates Foundation-funded products will be made available by us at an affordable price in a list of clearly defined low and lower middle-income countries, while we will be able to market such products in developed countries on our own or through licensees. In addition, the new manufacturing facility will have dedicated capacity to focus on products resulting from Bill & Melinda Gates Foundation-related projects for distribution in such low and lower middle-income countries.
127
Our global access commitments are perpetual, however, our obligation to commence new development programs expires in February 2025. In the event that we commit a material breach of the Global Access Agreement, following a cure period, we must grant the Bill & Melinda Gates Foundation a non-exclusive, perpetual, irrevocable, fully paid-up, royalty-free license under any intellectual property controlled by us covering any Bill & Melinda Gates Foundation-funded products to develop, manufacture and commercialize such products in low and lower middle-income countries, and the Bill & Melinda Gates Foundation will have certain withdrawal rights with respect to its equity investment in us. For more information on the Bill & Melinda Gates Foundation’s withdrawal rights, see “Item 7. Major Shareholders and Related Party Transactions — B. Related Party Transactions — Investment and Shareholders’ Agreement.”
In November 2016 in connection with and subject to the terms of the Global Access Agreement, we were awarded a grant for up to $0.9 million in funding from the Bill & Melinda Gates Foundation for the development of a vaccine for picornaviruses. As of December 31, 2023, we have received $0.7 million in funding under the grant agreement. We granted the Bill & Melinda Gates Foundation a non-exclusive, perpetual, irrevocable, worldwide, royalty-free, fully paid-up, sublicensable license to make, use, sell, offer to sell, import, distribute, copy, modify, create derivative works, publicly perform and display any products developed using grant funding; however, in the event we demonstrate to the satisfaction of the Bill & Melinda Gates Foundation that we are able to meet its global access requirements, such license will be modified or terminated. The term of the picornavirus grant expired in June 2022; however, our global access commitments survive. Following the completion of the project, the Bill & Melinda Gates Foundation requested a reimbursement for unspent funds. As of December 31, 2023, we have paid unspent funds of $0.2 million back to the Bill & Melinda Gates Foundation.
In November 2017, also in connection with and subject to the terms of the Global Access Agreement, we were awarded two additional grants for up to $1.9 million and $1.5 million from the Bill & Melinda Gates Foundation for the development of a universal influenza vaccine and a malaria vaccine, respectively. By an amendment entered into November 2020, our grant for the development of a malaria vaccine was increased by an additional $0.8 million. As of December 31, 2023, we have received $1.9 million and $2.2 million, respectively, in funding under each grant agreement. The programs will leverage our advanced RNActive® prophylactic vaccine technology to develop mRNA-based universal influenza and malaria vaccines. The malaria grant agreement expired in December 2022 and the universal influenza grant agreement expired in March 2022; however, our global access commitments survive.
In July 2020, we amended the Global Access Agreement and entered into a Letter Agreement with GSK and the Bill & Melinda Gates Foundation. Pursuant to this letter agreement, the Bill & Melinda Gates Foundation released us of our global access commitments with respect to certain prophylactic and therapeutic vaccines based on our mRNA technology platform to be developed under the 2020 GSK Agreement. This release will remain in effect for a vaccine or medicine only for so long as it is in development or being commercialized under the 2020 GSK Agreement. The letter agreement does not release us from any of our obligations to initiate or continue projects under the Global Access Agreement or related grant agreements and GSK granted to us and the Bill & Melinda Gates Foundation a non-exclusive, royalty-free, perpetual license under intellectual property arising from certain activities under the 2020 GSK Agreement to make vaccines arising from those projects available in low and lower middle-income countries as set forth in the Global Access Agreement.
Coalition for Epidemic Preparedness Innovations Framework Partnering Agreement
In February 2019, we entered into a framework partnership agreement with CEPI, which as amended we refer to as the CEPI Agreement, to develop our RNA Printer using certain intellectual property controlled by us covering the development and manufacture of mRNA products as well as certain additional intellectual property licensed to us. In connection with the CEPI Agreement we have entered into work orders for the preclinical development of a Lassa virus vaccine, a yellow fever vaccine and our rabies virus vaccine. In addition, we entered into a work package for the preclinical development and a Phase 1 clinical trial for our first-generation SARS-CoV-2 vaccine, CVnCoV.
128
We are required to use reasonable efforts to achieve certain development milestones and are responsible for conducting certain clinical trials. We are required to share clinical trial data with CEPI, subject to the terms of specific work packages entered into in connection with the CEPI Agreement. In the event of an infectious disease outbreak, where such outbreak can be addressed by a Lassa virus, SARS-CoV-2 or future vaccine developed under the CEPI Agreement, we must manufacture such vaccine for use in the area affected by the outbreak on economic terms that satisfy CEPI’s equitable access guidelines or otherwise allow CEPI or a third-party to supply such vaccine in the affected area. For the initial term of the CEPI Agreement and for a certain period thereafter, in the event of an outbreak that cannot be addressed by a vaccine already developed under the CEPI Agreement, CEPI may request, and we may agree, that we will develop a product targeted against such outbreak, or we will assist CEPI to develop a candidate product against such outbreak. In the event we decline to enter into such a development agreement, we will grant CEPI the right to develop and stockpile such vaccines under certain of our background intellectual property and intellectual property developed under the CEPI Agreement. We are additionally required to use reasonable efforts, at CEPI’s request, to submit certain optimized antigen nucleotide sequences for up to three specified pathogens in order for CEPI to start its own product development program. We have a right of first refusal to manufacture any pharmaceutical products developed by CEPI using the antigen nucleotide sequences we provide. In certain scenarios, including if we fail to provide Lassa virus, SARS-CoV-2 or future vaccines developed under the CEPI Agreement at prices that comply with CEPI’s equitable access guidelines, we must grant CEPI a license under certain of our background intellectual property and intellectual property developed under the CEPI Agreement to, among other things, develop our automation solution for use in treating such infectious diseases and to develop, manufacture and market such pharmaceutical products for use in geographic areas where there is a disease outbreak.
In connection with a December 2020 amendment to the CEPI Agreement, we agreed to provide CVnCoV to organizations operating under the COVAX Facility, a global collaboration to accelerate the development, production and equitable access to SARS-CoV-2 tests, treatments and vaccines. Under this amendment, we agreed to supply a certain percentage of our total capacity for distribution of CVnCoV to organizations participating in the COVAX Facility.
We are required to grant certain approved manufacturers all necessary rights to use certain of our preexisting intellectual property and intellectual property developed under the CEPI Agreement to further develop our automation solution and manufacture products for the treatment of certain diseases in geographic areas where there is an outbreak on economic terms that satisfy CEPI’s equitable access guidelines. We must provide all necessary commercially reasonable support to such approved manufacturers to facilitate such efforts.
CEPI agreed to contribute up to $34 million in funding for projects undertaken under the CEPI Agreement and an additional $15.3 million in connection with development of CVnCoV. In the event of our commercial use of the pharmaceutical products developed under the CEPI Agreement, other than CVnCoV, we must notify CEPI and agree in good faith how such commercial benefits are to be equitably managed between the parties. As of December 31, 2023, we have received €27.1 million in funding for projects undertaken under the CEPI Agreement.
We solely own all intellectual property developed under the CEPI Agreement but are required to obtain CEPI’s consent prior to exploiting any intellectual property developed under the CEPI Agreement if such exploitation is in conflict with or goes against CEPI’s mission or policies.
The CEPI Agreement terminated in February 2022, except with respect to certain ongoing projects, which are contemplated to be completed in March 2024. Following the completion of the CEPI Agreement, CEPI requested a partial reimbursement of $1.0 million for unspent funds. As of December 31, 2023 we have paid these unspent funds back to CEPI.
Tesla Automation Development and Intellectual Property Agreement
In November 2015, we entered into a development and intellectual property agreement with Tesla Automation, formerly trading under the name of Tesla Grohmann Automation, which we refer to as the Tesla Automation Agreement, pursuant to which Tesla Automation agreed to design, develop and manufacture certain automated manufacturing machines on our behalf. We are obligated to pay Tesla Automation a fee for each machine delivered by Tesla Automation and up to $50 million to $60 million in commercial milestone payments as well as certain development costs under each associated work order. As of December 31, 2023, we have paid Tesla Automation €22 million to €23 million in development costs under various work orders, and we have not paid any fees for machines provided under the Tesla Automation Agreement or made any milestone payments.
129
The parties jointly own any intellectual property developed under the Tesla Automation Agreement, and Tesla Automation granted us a non-exclusive, royalty-free, perpetual, irrevocable as to existing machines, worldwide license to use, sublicense and distribute Tesla Automation background intellectual property that is incorporated into any machine developed under the Tesla Automation Agreement and an exclusive (only with respect to the machines, and until a certain period after the first commercial use of a machine, after which the license shall be non-exclusive), royalty-free, perpetual, irrevocable as to existing machines, worldwide license under Tesla Automation’s interest in any jointly owned intellectual property. We granted Tesla Automation a non-exclusive, nontransferable, no-charge license during the term of the Tesla Automation Agreement under our background intellectual property for Tesla Automation’s performance of its obligations under the Tesla Automation Agreement and a non-exclusive, royalty-free, perpetual, irrevocable as to existing machines, worldwide license under our interest in any jointly owned intellectual property to perform its obligations under the Tesla Automation Agreement and for applications and uses unrelated to the machines developed under the Tesla Automation Agreement.
The Tesla Automation Agreement continues on a machine-by-machine basis until 10 years after the first commercial use of such machine. Either party may terminate any work order entered into in connection with the Tesla Automation Agreement for convenience upon written notice to the other party, and either party may terminate a work order for the other party’s material breach following a cure period, or for the other party’s insolvency. In the event Tesla Automation terminates a work order for convenience or we terminate for Tesla Automation’s material breach or insolvency, Tesla Automation must grant us a non-exclusive, fully paid-up, worldwide, irrevocable, perpetual, transferable and sublicensable license under Tesla Automation background intellectual property and Tesla Automation’s interest in intellectual property developed under the Tesla Automation Agreement for us to complete, either on our own or with another supplier, the work under such terminated work order. In the event we terminate for convenience, we must pay Tesla Automation a termination fee. In the event Tesla Automation terminates for our material breach or insolvency, we must pay Tesla Automation a termination fee and grant Tesla Automation a non-exclusive, fully paid-up, sublicensable, worldwide irrevocable and perpetual license under our background intellectual property and our interest in the intellectual property developed under the Tesla Automation Agreement to manufacture machines relevant to the applicable work order.
Research and Option Agreement with myNEO
On May 12, 2022, we entered into a Research and Option Agreement (“R&O”) with myNEO, pursuant to which, as amended in January 2023, we will collaborate to identify specific antigens found on the surface of tumors for the development of novel mRNA immunotherapies. To achieve this goal, myNEO will leverage its biological datasets, its integrated machine learning and bioinformatics platform to identify and validate specific antigen targets predicted to elicit a strong immune response. Under the R&O, we aim to develop and commercialize at least two new medicinal products for the treatment of non-small cell lung cancer and melanoma (the “Main Indications”) and potentially other indications. We are required to use commercially reasonable efforts to develop at least one product for each of the Main Indications, to file marketing approval applications for such products and commercialize such products in at least one of certain countries. Under the R&O, myNEO will own all intellectual property rights generated solely by myNEO or jointly with us during the first three phases of the R&D plan (the “R&D Project IP”). We received a non-exclusive, royalty-free, non-assignable, sublicensable, worldwide license under certain patents and know-how owned by myNEO and R&D Project IP to the extent required to perform our research and development obligations under the agreement until the completion of a certain phase of the R&D plan. We were also granted with an exclusive option to acquire all of myNEO’s rights under certain R&D Project IP relating to certain target lists, which we exercised on April 12, 2023. In connection with our exercise of such option, we granted myNEO a non-exclusive, royalty-free, perpetual license back to such IP to make, use or sell certain targets in the field of patient-specific vaccines. Under the R&O, myNEO agrees to work exclusively with us to develop and validate shared antigens for the Main Indications until the earlier of the date of the first phase I clinical trial for either Main Indication or 24 months after we exercised our option.
Under the R&O, we paid myNEO an upfront one-time technology access fee of €138,000 and myNEO is eligible to receive up to €17.5 million in research and development milestone payments with respect to the Main Indications, up to €175,000 in research and development milestones payments with respect to indications other than the Main Indications, up to €30 million in commercial milestone payments with respect to the Main Indications and up to €7.5 million in commercial milestone payments with respect to indications other than the Main Indications, as well as low single-digit percentage royalties on the net sales of licensed products in the Main Indications and sub single-digit percentage royalties on the net sales of licensed products for indications other than the Main Indications. Our royalty obligations continue on a product-by-product and country-by-country basis until the earlier of the date when there are no valid patent claims covering such licensed product in such country and 10 years following the date of first commercial sale of such licensed product in such country. On October 9, 2023, we notified myNEO that we reached a research and development milestone where we selected four antigens for further development, which we intend to use in a clinical candidate for the indication head and neck squamous-cell carcinoma, which is an indication other than the Main Indication.
130
The term of the R&O will continue until (i) the expiration of the option period if we do not exercise any of our exclusive options or (ii) the expiry of all applicable royalty payment obligations to myNEO, unless terminated earlier by either party. We have the right to terminate the R&O if certain milestones or if provisions of certain reports are delayed by a certain period, for convenience or in the case of a change of control of myNEO. We and myNEO both have the right to terminate the R&O in the event of the other party’s material breach following a cure period.
Sponsored Collaboration Agreements
Schepens Institute Research Agreement
In March 2019, we entered into a sponsored research agreement, as amended in April 2020, July 2021, September 2021, August 2022, January 2023, June 2023 and February 2024 (as amended, the “Schepens Agreement”), with The Schepens Eye Research Institute, Inc. (“SERI”) and Massachusetts Eye and Ear Infirmary (“MEEI”), pursuant to which SERI and MEEI agreed to perform certain research activities for mRNA-based eye therapy candidates. Under the Schepens Agreement, SERI and MEEI granted us an exclusive option to initiate negotiations for an exclusive or non-exclusive license to SERI’s interest in any inventions developed under the Schepens Agreement. SERI and MEEI additionally granted us an exclusive option to negotiate an exclusive license to certain background intellectual property. Upon the exercise of such option and upon execution of the contemplated license, we would be required to pay SERI a $30,000 upfront payment, up to $0.8 million in development milestone payments and $1.8 million in regulatory milestone payments, and a low single-digit percentage royalty on net sales subject to certain minimum annual payments. We are required to provide $1.7 million in funding to SERI and MEEI in multiple payments during the term of the Schepens Agreement. As of December 31, 2023, we have provided $2.0 million in funding to SERI and MEEI under the Schepens Agreement.
Each party will solely own inventions it solely develops and will jointly own jointly developed inventions. We are responsible for all patent prosecution costs regarding intellectual property developed under the Schepens Agreement, and if we elect not to cover the prosecution costs for such intellectual property, SERI and MEEI will have the right to license such intellectual property to third parties, and we will have no rights in such intellectual property.
The Schepens Agreement continues until June 2024, unless extended by mutual agreement or earlier terminated. Both parties have the right to terminate the Schepens Agreement for the other party’s material breach following a cure period, and SERI has the right to terminate in the event of our insolvency. We additionally have the right to terminate the Schepens Agreement for convenience following a notice period. In the event SERI terminates for our material breach or insolvency or we terminate for convenience, we must reimburse SERI for all costs incurred to date and provide certain additional funding for a three-month period. In the event we terminate for SERI’s material breach, we must reimburse SERI for all noncancellable commitments.
Advance Purchase Agreements
European Commission — COVID-19 Vaccine Candidate
Advance Purchase Agreement for our COVID-19 Vaccine Candidate
On November 30, 2020, we entered into an APA with the EC, acting on behalf and in the name of all Member States of the European Union, which provided for the advance purchase by the Member States of 225 million doses of the vaccine to be allocated among the Member States, and the option to purchase up to an additional 180 million doses. Pursuant to the APA, we received an upfront payment of €450 million. Such upfront payment had to be used solely for the development and commercial supply of CVnCoV. We were required to return any unspent amounts of the upfront payment if, among others, we failed to successfully develop CVnCoV, or if we successfully develop CVnCoV without receiving EU marketing authorization, or if we failed to supply any doses of CVnCoV to any of the Member States by late 2021 (unless we and the EC mutually agree to a later date). In October 2021, we notified the EC of the withdrawal of our regulatory approval application for CVnCoV, which notification automatically terminated the APA. According to the APA, in such case of termination, we would only be required to return any unspent amount of the upfront payment. In the context of the APA, “spent” means either costs incurred or commitments made in connection with the purposes set forth in the APA. On March 8, 2022, we received a letter signed by the EC acknowledging and outlining that we will not be required to return any portion of the upfront payment. Due to the termination of the APA, we will not receive any further payments related to the APA.
131
In other respects, upon the EC’s request, we will transfer any raw materials and/or primary components paid for with the upfront payment that were not used as of the termination date. Additionally, should the EC request, or should we successfully sell, any raw materials and/or primary components, then an applicable portion of such raw materials, primary components or proceeds, as the case may be, will be remitted to the EC. This repayment agreement expired at the end of 2022 and an amount of €4.1 million was paid to the EC as of December 31, 2023.
Pandemic Preparedness Agreement
Federal Republic of Germany
Pandemic Preparedness Agreement with the Federal Republic of Germany
On February 20, 2022, the CureVac-GSK Consortium submitted its best and final offer in the Tender Procedure for the conclusion of framework agreements for the provision of production capacities and, on demand, for the production and supply of mRNA vaccines (lot 1). On April 8, 2022, the CureVac-GSK Consortium received a letter from the Federal Republic of Germany’s counsel confirming that the CureVac-GSK Consortium had been awarded a Pandemic Preparedness Agreement. Pursuant to the Pandemic Preparedness Agreement, the CureVac-GSK Consortium will have to achieve, within a two years’ time frame beginning from the award of the Pandemic Preparedness Agreement, a state in which it is considered qualified to provide manufacturing capacities in Germany for 160 million doses of mRNA vaccine per year, including procurement of the required nonproduct specific manufacturing licenses and insurances and to have achieved “pandemic preparedness,” which means, inter alia, that we maintain the GMP IV facility in a stand-by mode that can be activated for manufacture of a so-called selected vaccine at any time and that the CureVac-GSK Consortium is complying with the material requirements set out in the Pandemic Preparedness Plan (in particular with the requirements regarding the assurance of a supplier network and the availability of the critical supplier products).
If qualification and pandemic preparedness is achieved by the end of the two years’ time frame beginning from the award of the Pandemic Preparedness Agreement (and if the Pandemic Preparedness Agreement is not terminated because the CureVac-GSK Consortium does not have an mRNA-based vaccine for which a marketing authorization for the (at least temporary) placing on the German market has been granted at this time), the CureVac-GSK Consortium will receive a stand-by fee which will be shared between us and GSK in accordance with the agreement governing the CureVac-GSK Consortium. The phase following the qualification phase (stand-by phase) during which pandemic preparedness is to be maintained is for five years, it being understood that this term may be extended by mutual agreement up to three times for a subsequent one-year renewal term.
At any time during the stand-by phase, in case there is a public health emergency, the Federal Republic of Germany may exercise its preferred purchase right and/or its preferred manufacturing right. If the preferred purchase right is exercised the CureVac-GSK Consortium will have to deliver up to 80 million doses of the mRNA vaccine of the CureVac-GSK Consortium, and if the preferred manufacturing right is exercised the CureVac-GSK Consortium will have to act as a contract manufacturer and manufacture a third party’s mRNA vaccine in our GMP IV facility. However, there are strict and narrow requirements to be fulfilled before the Federal Republic of Germany may exercise the preferred manufacturing right.
On April 9, 2024, the CureVac-GSK Consortium notified the Federal Republic of Germany of its termination of the Pandemic Preparedness Agreement.
Intellectual Property
Our commercial success depends in part on our ability to obtain and maintain proprietary or intellectual property protection for our product candidates, and our core technologies and other know-how, defend and enforce our patents, preserve the confidentiality of our trade secrets, operate our business without infringing, misappropriating or otherwise violating the intellectual property or proprietary rights of third parties and prevent third parties from infringing, misappropriating or otherwise violating our proprietary or intellectual property rights. We seek to protect our proprietary and intellectual property position by, among other methods, seeking and maintaining patents in the United States and other major markets. We also rely on trade secrets and know-how to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection, which we generally seek to protect through contractual obligations with third parties.
132
Patents
As of March 19 2024, we own approximately 103 issued U.S. patents, 116 pending U.S. patent applications, 170 issued foreign patents (including 43 European patents, which have been validated in various European countries resulting in a total of approximately 395 national patents in European countries and including two European patents with unitary effect), 410 pending foreign patent applications (including 74 pending European patent applications) and 18 pending Patent Cooperation Treaty, or PCT, patent applications, including several patent families that are jointly owned with third parties. These patents include claims relating to our RNAoptimizer technology platform, and our COVID-19 and Influenza vaccine candidates, and our proprietary LNP technology, as described further below.
RNAoptimizer
As of March 19, 2024, we own 23 issued U.S. patents, 20 pending U.S. patent applications, 92 issued foreign patents, including in Europe, Canada, China, India, Israel, Japan, the Republic of Korea, Singapore, Mexico and Australia, and 90 pending foreign patent applications and three PCT patent applications relating to our RNAoptimizer technology, including patents and patent applications relating to ORF optimization, UTR optimization, novel CAP analogs, protein optimization and formulation. Our RNAoptimizer technology is used in our Influenza and SARS-CoV-2 product candidates. The issued patents are expected to expire between 2025 and 2037, excluding any additional term for patent term adjustments or patent term extensions. If granted, the pending patent applications would be expected to expire between 2022 and 2044, excluding any additional term for patent term adjustments or patent term extensions.
Influenza Vaccine candidates
As of March 19, 2024, we own 11 issued U.S. patents, 17 pending U.S. patent applications, 49 issued foreign patents, including in Europe, Canada, China, India, Israel, Japan, the Republic of Korea, Singapore, Mexico and Australia and 49 pending foreign patent applications and one PCT patent application relating to our Influenza product candidates. The issued patents are expected to expire between 2022 and 2038, excluding any additional term for patent term adjustments or patent term extensions. If granted, the pending patent applications would be expected to expire between 2022 and 2043, excluding any additional term for patent term adjustments or patent term extensions.
COVID-19 Vaccine Candidates
As of March 19, 2024, we own 14 issued U.S. patents, 17 pending U.S. patent applications, 47 issued foreign patents, including in Europe, Canada, China, India, Japan, the Republic of Korea, Singapore, Russia, Mexico and Australia, 80 pending foreign patent applications and one PCT patent application relating to our COVID-19 product candidates. The issued patents are expected to expire between 2022 and 2041, excluding any additional term for patent term adjustments or patent term extensions. If granted, the pending patent applications would be expected to expire between 2022 and 2043, excluding any additional term for patent term adjustments or patent term extensions.
The term of individual patents depends upon the legal term for patents in the countries in which they are obtained. In most countries in which we have filed patent applications, including the United States, the patent term is 20 years from the earliest filing date of a non-provisional patent application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the USPTO in examining and granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier filed patent. The term of a patent that covers a drug or biological product may also be eligible for patent term extension when FDA approval is granted for a portion of the term effectively lost as a result of the FDA regulatory review period, subject to certain limitations and provided statutory and regulatory requirements are met. For more information on patent term extension, see “— Government Regulation — Patent Term Restoration and Extension.”
133
As with other biotechnology and pharmaceutical companies, our ability to maintain and solidify our proprietary and intellectual property position for our product candidates will depend on our success in obtaining effective patent claims and enforcing those claims if granted. However, our owned and licensed pending patent applications, and any patent applications that we may in the future file or license from third parties may not result in the issuance of patents. We also cannot predict the breadth of claims that may be allowed or enforced in our patents. Any issued patents that we may receive in the future may be challenged, invalidated, narrowed, held unenforceable, infringed or circumvented. In addition, because of the extensive time required for clinical development and regulatory review of a product candidate we may develop, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby limiting the protection such patent would afford the respective product and any competitive advantage such patent may provide. See “Item 3. Key Information — D. Risk Factors — Risks Related to Our Intellectual Property Rights.”
Trademarks
As of April 2, 2024, we own trademark registrations or registration applications for CureVac, and the CureVac logo in the United States and in certain foreign jurisdictions including Europe.
Trade Secrets and Proprietary Information
In addition to patents, we rely upon unpatented trade secrets and know-how and continuing technological innovation to develop and maintain our competitive position. However, trade secrets and know-how can be difficult to protect. We seek to protect our proprietary information, in part, by executing confidentiality agreements with our collaborators and scientific advisors, and non-competition, non-solicitation, confidentiality, and invention assignment agreements with our employees, consultants, and independent contractors. We have also executed agreements requiring assignment of inventions with selected scientific advisors and collaborators. The confidentiality agreements we enter into are designed to protect our proprietary information and the agreements or clauses requiring assignment of inventions to us are designed to grant us ownership of technologies that are developed through our relationship with the respective counterparty. We cannot guarantee, however, that we have executed such agreements with all applicable counterparties, such agreements will not be breached, or that these agreements will afford us adequate protection of our intellectual property and proprietary rights. See “Item 3. Key Information — D. Risk Factors — Risks Related to Our Intellectual Property Rights.”
Government Regulation
Government authorities in the United States, at the federal, state and local level, in other countries and jurisdictions and in the European Union, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, post-approval monitoring and reporting, and import and export of pharmaceutical products, including biological products. In addition, some jurisdictions regulate the pricing of pharmaceutical products. The processes for obtaining marketing approvals in the United States and in foreign countries and jurisdictions, along with subsequent compliance with applicable statutes and regulations and other requirements of regulatory authorities, require the expenditure of substantial time and financial resources.
134
Patent Term Restoration and Extension
Depending upon the timing, duration and specifics of FDA approval of product candidates, some of a sponsor’s U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period generally is one-half the time between the effective date of an IND and the submission date of a BLA less any time the sponsor did not act with due diligence during the period, plus the time between the submission date of a BLA and the approval of that application less any time the sponsor did not act with due diligence during the period. Only one patent applicable to an approved biologic product is eligible for the extension, only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended, and the application for the extension must be submitted prior to the expiration of the patent. Moreover, a given patent may only be extended once based on a single product. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. In the future, if and when our products receive FDA approval, we expect to apply for patent term extensions on patents covering those products, however there is no guarantee that the applicable authorities, including the FDA in the United States, will agree with our assessment of whether such extensions should be granted, and if granted, the length of such extensions. For more information regarding the risks related to our intellectual property, see “Item 3. Key Information — D. Risk Factors — Risks Related to Our Intellectual Property Rights.”
Regulation and Procedures Governing Approval of Biological Products in the United States
In the United States, we expect our product candidates will be regulated as biological products, or biologics, under the Public Health Service Act, or PHSA, and the Federal Food, Drug, and Cosmetic Act, or FDCA, and their implementing regulations, and other federal, state, local and foreign statutes and regulations. The failure to comply with the applicable U.S. requirements at any time during the product development process, including during nonclinical testing, clinical testing, the approval process or post-approval process, may subject an applicant to delays in the conduct of a study or regulatory review and approval, and/or to administrative or judicial sanctions and adverse publicity. Sanctions may include, but are not limited to, the U.S. Food and Drug Administration, or FDA’s, refusal to allow an applicant to proceed with clinical testing, refusal to approve pending applications, withdrawal of an approval, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, debarment, disgorgement of profits and civil or criminal investigations and penalties brought by the FDA or the Department of Justice or other governmental entities.
An applicant seeking approval to market and distribute a new biologic in the United States generally must satisfactorily complete each of the following steps:
| ● | nonclinical laboratory tests, animal studies and formulation studies all performed in accordance with applicable regulations, including the FDA’s Good Laboratory Practice, or GLP, regulations; |
| ● | submission to the FDA of an IND application for human clinical testing, which must become effective before human clinical trials may begin; |
| ● | approval by an Institutional Review Board, or IRB, or ethics committee representing each clinical site before each clinical trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials to establish the safety, potency and purity of the product candidate for each proposed indication, in accordance with applicable regulations, including with GCP regulations; |
| ● | after completion of all pivotal clinical trials, preparation and submission to the FDA of a BLA requesting authorization to market the product candidate for one or more proposed indications; |
| ● | satisfactory completion of an FDA advisory committee review, if applicable; |
| ● | a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; |
135
| ● | satisfactory completion of one or more FDA inspections of the manufacturing facility or facilities, including those of third parties, at which the product, or components thereof, are produced to assess compliance with cGMP requirements and to assure that the facilities, methods and controls are adequate to preserve the product’s identity, safety, strength, quality and purity; |
| ● | satisfactory completion of any FDA audits of the clinical study sites to assure compliance with GCPs, and the integrity of clinical data in support of the BLA; |
| ● | payment of user fees and securing FDA approval of the BLA; and |
| ● | compliance with any post-approval requirements, including the potential requirement to implement a REMS and to conduct any post approval studies required by the FDA. |
Nonclinical Studies and Investigational New Drug Application
Before testing any biologic product candidate in humans, the product candidate must undergo nonclinical testing. Nonclinical tests include laboratory evaluations of product chemistry, formulation and stability, as well as animal studies to evaluate the potential for activity and toxicity. The conduct of the nonclinical tests and formulation of the compounds for testing must comply with federal regulations and requirements. The results of the nonclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, are submitted to the FDA as part of an IND or similar application in other jurisdictions. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless within the 30-day time period, the FDA raises concerns or questions about the product or conduct of the proposed clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks, and places the trial on a clinical hold. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns before the clinical trial can begin.
As a result, submission of the IND may result in the FDA not allowing the trial to commence or not be conducted on the terms originally specified by the sponsor in the IND. In addition, the FDA may raise concerns or questions at any time after the IND has become effective, and may impose a clinical hold even after clinical studies have initiated. If the FDA imposes a clinical hold, trials may not recommence without FDA authorization and then only under terms authorized by the FDA. A clinical hold issued by the FDA may therefore delay either a proposed clinical study or cause suspension of an ongoing study, until all outstanding concerns have been adequately addressed and the FDA has notified the company that investigation may proceed. This could cause significant difficulties in completing planned clinical trials in a timely manner.
Human Clinical Trials in Support of a BLA
Clinical trials involve the administration of the investigational product candidate to healthy volunteers or patients with the disease to be treated under the supervision of a qualified principal investigator in accordance with GCP requirements. Clinical trials are conducted under study protocols detailing, among other things, the objectives of the study, inclusion and exclusion criteria, the parameters to be used in monitoring safety, dosing procedures and the effectiveness criteria to be evaluated. A separate protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND.
A sponsor who wishes to conduct a clinical trial outside the United States may, but need not, obtain FDA authorization to conduct the clinical trial under an IND. If a foreign clinical trial is not conducted under an IND, the sponsor may submit data from the clinical trial to the FDA in support of the BLA so long as the clinical trial is well-designed and well-conducted in accordance with GCP, including review and approval by an independent ethics committee, and the FDA is able to validate the study data through an onsite inspection, if necessary.
136
Further, each clinical trial must be reviewed and approved by an IRB either centrally or individually at each institution at which the clinical trial will be conducted. The IRB will consider, among other things, clinical trial design, patient informed consent, ethical factors and the safety of human subjects. An IRB must operate in compliance with FDA regulations. The FDA, IRB, or the clinical trial sponsor may suspend or discontinue a clinical trial at any time for various reasons, including a finding that the clinical trial is not being conducted in accordance with FDA requirements, the trial is unlikely to meet its stated objectives or that the subjects or patients are being exposed to an unacceptable health risk. Clinical testing also must satisfy extensive GCP rules, including the requirements for informed consent. The IRB also approves the form and content of the informed consent that must be signed by each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group may recommend continuation of the study as planned, changes in study conduct, or cessation of the study at designated check points based on access to certain data from the study.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined. Additional studies may be required after approval.
| ● | Phase 1 clinical trials are initially conducted in a limited population to test the product candidate for safety, including adverse effects, dose tolerance, absorption, metabolism, distribution, excretion and pharmacodynamics in healthy humans or, on occasion, in patients, such as in the case of some products for severe or life-threatening diseases as cancer, especially when the product may be too inherently toxic to ethically administer to healthy volunteers. |
| ● | Phase 2 clinical trials are generally conducted in a limited patient population to identify possible adverse effects and safety risks, preliminarily evaluate the efficacy of the product candidate for specific targeted indications and determine dose tolerance and optimal dosage. Multiple Phase 2 clinical trials may be conducted by the sponsor to obtain information prior to beginning larger Phase 3 clinical trials. |
| ● | Phase 3 clinical trials proceed if the Phase 2 clinical trials demonstrate that a certain dose or dose range of the product candidate is potentially effective and has an acceptable safety profile. Phase 3 clinical trials are undertaken within an expanded patient population, often at geographically dispersed clinical trial sites, to gather additional information about safety and effectiveness necessary to evaluate the overall benefit-risk relationship of the investigational product and to provide an adequate basis for physician labeling and product approval. |
In some cases, the FDA may approve a BLA for a product candidate but require the sponsor to conduct additional clinical trials to further assess the product candidate’s safety and effectiveness after approval. Such post-approval trials are typically referred to as Phase 4 clinical trials, or Phase 4. These studies may be used to gain additional experience from the treatment of patients in the intended therapeutic indication and to document a clinical benefit in the case of biologics approved under accelerated approval regulations. Failure to exhibit due diligence with regard to conducting required Phase 4 clinical trials could result in withdrawal of approval for products. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the biological characteristics of the product candidate.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected adverse events, any findings from other trials, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the biological candidate has been associated with unexpected serious harm to patients.
137
There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries. Sponsors of clinical trials of FDA-regulated products, including biologics, are required to register and disclose certain clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, trial sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to discuss the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed until the new product or new indication being studied has been approved.
Review and Approval of a BLA
The results of product candidate development, nonclinical testing and clinical trials, including negative or ambiguous results as well as positive findings, are submitted to the FDA as part of a BLA requesting a license to market the product for one or more indications. The BLA must contain extensive chemistry manufacturing and controls information and detailed information on the composition of the product and proposed labeling. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by investigators. The submission of a BLA requires payment of a substantial user fee to the FDA, and the sponsor of an approved BLA is also subject to annual program fees. The FDA adjusts the Prescription Drug User Fee Act, or PDUFA, user fees on an annual basis. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
In addition, under the Pediatric Research Equity Act, or PREA, a BLA or supplement to a BLA must contain data to assess the safety and effectiveness of the biological product candidate for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The Food and Drug Administration Safety and Innovation Act, or FDASIA, requires that a sponsor who is planning to submit a marketing application for a drug or biological product that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration submit an initial Pediatric Study Plan, or PSP, within sixty days after an end-of-Phase 2 meeting or as may be agreed between the sponsor and FDA. Unless otherwise required by regulation, PREA does not apply to any biological product for an indication for which orphan designation has been granted.
The FDA has 60 days after submission of the application to conduct an initial review to determine whether the BLA is sufficient to accept for filing based on the agency’s threshold determination that it is substantially complete so as to permit substantive review. Once the submission has been accepted for filing, the FDA begins an in-depth review of the application. Under the goals and policies agreed to by the FDA under PDUFA, the FDA aims to complete its initial review of a standard application and respond to the applicant within 10 months of the 60-day filing date, and for a priority review application within six months. The FDA does not always meet its PDUFA goal dates for standard and priority BLAs, and its review goals are subject to change from time to time. The review process may often be significantly extended by FDA requests for additional information or clarification. The review process and the PDUFA goal date may also be extended by three months if the FDA requests or if the applicant otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
The FDA reviews a BLA to determine, among other things, whether the proposed product is safe and potent, or effective, for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with cGMP requirements to assure and preserve the product’s identity, safety, strength, quality, potency and purity. The FDA may convene an advisory committee to provide clinical insight on application review questions. Before approving a BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with cGCP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
138
On the basis of the FDA’s evaluation of the application and accompanying information, including the results of the inspection of the manufacturing facilities and any FDA audits of clinical trial sites to assure compliance with GCPs, the FDA may issue an approval letter or a complete response letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. If the application is not approved, the FDA may issue a complete response letter indicating that the review cycle is complete and the application is not ready for approval. A complete response letter will describe the deficiencies that must be addressed in order to secure final approval of the application, and when possible, will outline recommended actions the sponsor might take to obtain approval of the application. If a complete response letter is issued, the applicant may either resubmit the BLA, addressing all of the deficiencies identified in the letter, or withdraw the application. The FDA may also request additional information or clarification.
Sponsors that receive a complete response letter who elect to address the deficiencies may submit to the FDA information that represents a complete response to the issues identified by the FDA in the response letter. Such resubmissions are classified under PDUFA as either Class 1 or Class 2, based on the information submitted by an applicant in response to an action letter. Under the goals and policies agreed to by the FDA under PDUFA, the FDA aims to review and act on a Class 1 resubmission with two months of receipt and, with respect to a Class 2 resubmission, within six months of receipt. The FDA will not approve an application until issues identified in the complete response letter have been addressed. The FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product.
If the FDA approves a new product, it may limit the approved indications for use of the product, or limit the approval to specific dosages. It may also require development of adequate controls or specifications and that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may call for post-approval studies, including Phase 4 clinical trials, to further assess the product’s safety after approval and may limit further marketing of the product based on the results of these post-marketing studies. The agency may also require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, including REMS, to help ensure that the benefits of the product outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, or ETASU. ETASU can include, but are not limited to, special training or certification for prescribing or dispensing, dispensing only under certain circumstances, special monitoring and the use of patent registries. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS; the FDA will not approve the BLA without a REMS, if required. The FDA may prevent or limit further marketing of a product based on the results of post-market studies or surveillance programs. After approval, many types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval. Once approved, the FDA may withdraw the product approval if compliance with pre-and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
Expedited Development and Review Programs
The FDA has a number of programs intended to expedite the development and/or review of new products intended for serious or life-threatening diseases or conditions. These programs include fast track designation, breakthrough therapy designation, priority review and accelerated approval.
The FDA may issue a fast track designation to a product candidate if it is intended, whether alone or in combination with one or more other products, for the treatment of a serious or life-threatening disease or condition, and it demonstrates the potential to address unmet medical needs for such a disease or condition. Fast track designation applies to the combination of the product and the specific indication for which it is being studied. The sponsor of a new biologic may request that the FDA designate the biologic as a fast track product at any time during the clinical development of the product. For fast track products, sponsors may have greater interactions with the FDA during product development. A fast track product may also be eligible for rolling review, where the FDA may consider for review sections of the BLA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the BLA, the FDA agrees to accept sections of the BLA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the BLA. However, the FDA’s PDUFA goal for reviewing a BLA fast track application does not begin until the last section of the application is submitted. Fast track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
139
A product may be designated as a breakthrough therapy if it is intended, either alone or in combination with one or more other products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. The FDA may take certain actions with respect to breakthrough therapies, including holding meetings with the sponsor throughout the development process; providing timely advice to the product sponsor regarding development and approval; involving more senior staff in the review process; assigning a cross-disciplinary project lead for the review team; and taking other steps to facilitate the design of clinical trials in an efficient manner. The designation includes all of the fast track program features, as well as more intensive FDA interaction and guidance beginning as early as Phase 1 and an organizational commitment to expedite the development and review of the product, including involvement of senior managers.
Any marketing application for a biologic submitted to the FDA for approval, including a product with a fast track designation and/or breakthrough therapy designation, may be eligible for other types of FDA programs intended to expedite the FDA review and approval process, such as priority review and accelerated approval. The FDA may designate a product for priority review if it is a product that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. The FDA determines, on a case-by-case basis, whether the proposed product represents a significant improvement when compared with other available therapies. Significant improvement may be illustrated by evidence of increased effectiveness in the treatment of a condition, elimination or substantial reduction of a treatment-limiting product reaction, documented enhancement of patient compliance that may lead to improvement in serious outcomes and evidence of safety and effectiveness in a new subpopulation. A priority review is intended to direct overall attention and resources to the evaluation of such applications, and to shorten the FDA’s goal for taking action on an original BLA from 10 months to six months from the 60-day filing date.
The FDA may grant accelerated approval to a product for a serious or life-threatening condition that provides meaningful therapeutic advantage to patients over existing treatments based upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a condition when the product has an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, or IMM, and that is reasonably likely to predict an effect on IMM or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Products granted accelerated approval must meet the same statutory standards for safety and effectiveness as those granted traditional approval.
For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign, or other measure that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. An intermediate clinical endpoint is a measurement of a therapeutic effect that is considered reasonably likely to predict the clinical benefit of a product, such as an effect on IMM. The FDA has stated that although it has limited experience with accelerated approvals based on intermediate clinical endpoints, such endpoints generally may support accelerated approval where the therapeutic effect measured by the endpoint is not itself a clinical benefit and basis for traditional approval, if there is a basis for concluding that the therapeutic effect is reasonably likely to predict the ultimate clinical benefit of a product.
The accelerated approval pathway is most often used in settings in which the course of a disease is long and an extended period of time is required to measure the intended clinical benefit of a product. Thus, accelerated approval has been used extensively in the development and approval of products for treatment of a variety of cancers in which the goal of therapy is generally to improve survival or decrease morbidity and the duration of the typical disease course requires lengthy and sometimes large trials to demonstrate a clinical or survival benefit.
The accelerated approval pathway is usually contingent on a sponsor’s agreement to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the product’s clinical benefit. As a result, a product candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of Phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or to confirm a clinical benefit during post-marketing studies, may lead the FDA to withdraw the product from the market on an expedited basis. All promotional materials for product candidates approved under accelerated regulations are subject to prior review by the FDA.
Fast track designation, priority review, accelerated approval, and breakthrough therapy designation may expedite the development or approval process, but do not change the standards for approval. Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
140
Post-Approval Regulation
If regulatory approval for marketing of a product or for a new indication for an existing product is obtained, the sponsor will be required to comply with rigorous and extensive post-approval regulatory requirements as well as any post-approval requirements that the FDA has imposed on the particular product as part of the approval process. The sponsor will be required, among other things, to report certain adverse reactions and production problems to the FDA, provide updated safety and efficacy information and comply with requirements concerning advertising, promotional labeling, product sampling and distribution. Manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMP regulations, which impose certain procedural and documentation requirements upon manufacturers. Accordingly, the BLA-holder and its third-party manufacturers must continue to expend time, money and effort in the areas of production and quality control to maintain compliance with cGMP regulations and other regulatory requirements. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon us and any third-party manufacturers that we or our partners may decide to use. In addition, changes to the manufacturing process or facility are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented, and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market study requirements or clinical trial requirements to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| ● | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| ● | fines, untitled letters or warning letters or holds on post-approval clinical trials; |
| ● | adverse publicity; |
| ● | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| ● | product seizure or detention, or refusal to permit the import or export of products; or |
| ● | injunctions, fines, debarment, disgorgement of profits or the imposition of civil or criminal penalties. |
The FDA closely regulates marketing, labeling, advertising and promotion of products that are placed on the market. Pharmaceutical products may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict marketing authorization holders’ communications on the subject of off-label use of their products.
141
Orphan Drug Designation
Orphan drug designation in the United States is designed to encourage sponsors to develop products intended for rare diseases or conditions. In the United States, a rare disease or condition is statutorily defined as a disease or condition that affects fewer than 200,000 individuals in the United States or that affects more than 200,000 individuals in the United States but for which there is no reasonable expectation that the cost of developing and making available the product for the disease or condition will be recovered from sales of the product in the United States.
Orphan drug designation qualifies a company for certain financial incentives, including tax advantages, waiver of the BLA application user fee and, if the product receives the first FDA approval for the indication for which it has orphan designation, market exclusivity. Orphan product exclusivity means that the FDA may not approve any other applications, including a full BLA, to market the same biologic for the same indication for seven years from the approval of the BLA, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or if the FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. An application for designation as an orphan product can be made any time prior to the filing of an application for approval to market the product.
In addition, a sponsor of a product that is otherwise the same product as an already approved orphan drug may seek and obtain orphan drug designation for the subsequent product for the same rare disease or condition if it can present a plausible hypothesis that its product may be clinically superior to the first product. More than one sponsor may receive orphan drug designation for the same product for the same rare disease or condition, but each sponsor seeking orphan drug designation must file a complete request for designation.
The period of exclusivity begins on the date that the marketing application is approved by the FDA and applies only to the indication for which the product has been designated. The FDA may approve a second application for the same product for a different use or a second application for a clinically superior version of the product for the same use. The FDA cannot, however, approve the same product made by another manufacturer for the same indication during the market exclusivity period unless it has the consent of the sponsor, the manufacturer makes a showing of clinical superiority over the product with orphan exclusivity, or the sponsor is unable to provide sufficient quantities.
An orphan-designated product may not receive orphan exclusivity if it is approved for a use that is broader than the indication for which it received orphan designation. In addition, orphan exclusivity in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
Biosimilars and Exclusivity
The BPCIA (under the Affordable Care Act) created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. The FDA has issued several guidance documents outlining an approach to review and approve of biosimilars.
Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity and potency, can be shown through analytical studies, animal studies, and a clinical study or studies. Interchangeability requires that a product is biosimilar to the reference product, and the product must demonstrate that it can be expected to produce the same clinical results as the reference product in any given patient and, for products that are administered multiple times to an individual, the biologic and the reference biologic may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biologic. However, complexities associated with the larger, and often more complex, structures of biological products, as well as the processes by which such products are manufactured, pose significant hurdles to implementation of the abbreviated approval pathway that are still being worked out by the FDA.
142
Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a full BLA for the competing product containing that applicant’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of its product. The BPCIA also created certain exclusivity periods for biosimilars approved as interchangeable products. At this juncture, it is unclear whether products deemed “interchangeable” by the FDA will, in fact, be readily substituted by pharmacies, which are governed by state pharmacy law.
A biological product can also obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study.
The BPCIA is complex and continues to be interpreted and implemented by the FDA. In addition, government proposals have sought to reduce the 12-year reference product exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. As a result, the ultimate impact, implementation, and impact of the BPCIA is subject to significant uncertainty.
Regulation and Procedures Governing Approval of Medicinal Products in the European Union
In order to market any product outside the United States, a company also must comply with numerous and varying regulatory requirements of other countries and jurisdictions regarding quality, safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of products. Whether or not it obtains FDA approval for a product, an applicant will need to obtain the necessary approvals by the comparable foreign regulatory authorities before it can initiate clinical trials or marketing of the product in those countries or jurisdictions. Specifically, the process governing approval of medicinal products in the European Union generally follows the same lines as in the United States. It entails satisfactory completion of pharmaceutical development, nonclinical studies and adequate and well-controlled clinical trials to establish the safety and efficacy of the medicinal product for each proposed indication.
Clinical Trial Approval
On January 31, 2022, the new Clinical Trial Regulation (EU) No 536/2014 became effective, replacing Clinical Trials Directive 2001/20/EC (which can still be applied for new clinical studies during a transition period until January 2023). This new legislation, which is directly applicable in all Member States, aims at simplifying and streamlining the approval of clinical trials in the European Union by allowing for a streamlined application procedure via a single-entry point and strictly defined deadlines for the assessment of clinical trial applications. The Clinical Trial Regulation harmonizes the assessment and supervision processes for clinical trials throughout the European Union, via a Clinical Trials Information System, or CTIS, which contains the centralized European Union portal and database for clinical trials foreseen by the Regulation. The EMA sets up and maintains CTIS, in collaboration with the competent national authority of each European Union Member Sate and the EC.
143
Marketing Authorization
To obtain a marketing authorization for a product under the European Union regulatory system, an applicant must obtain Marketing Authorization, or MA. There are two types of MAs:
| ● | The Community MA, which is issued by the EC through the Centralized Procedure, based on the opinion of the Committee for Medicinal Products for Human Use, or CHMP, of the EMA, and which is valid throughout the entire territory of the EEA. The Centralized Procedure is mandatory for certain types of products, such as biotechnology medicinal products, orphan medicinal products, advanced therapy medicinal products (such as gene therapies, somatic cell therapies and tissue engineered products), and medicinal products that contain a new active substance indicated for the treatment of AIDS, cancer, neurodegenerative disorders, diabetes, auto-immune and viral diseases. The Centralized Procedure is optional for products containing a new active substance not yet authorized in the EEA, or for products that constitute a significant therapeutic, scientific or technical innovation or which are in the interest of public health in the EU. Under the Centralized Procedure the maximum time frame for the evaluation of a marketing authorization application is 210 days (excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP). Accelerated evaluation might be granted by the CHMP in exceptional cases, when the authorization of a medicinal product is of major interest from the point of view of public health and in particular from the viewpoint of therapeutic innovation. Under the accelerated procedure the standard 210-day review period is reduced to 150 days. |
| ● | National MAs, which are issued by the competent authorities of the Member States of the EEA and only cover their respective territory, are available for products not falling within the mandatory scope of the Centralized Procedure. Where a product has already been authorized for marketing in a Member State of the EEA, this National MA can be recognized in other Member States through the Mutual Recognition Procedure. If the product has not received a National MA in any Member State at the time of application, it can be approved simultaneously in various Member States through the Decentralized Procedure. |
An MA may be granted only to an applicant established in the European Union. Regulation 1901/2006 on Medicinal Products for Pediatric Use provides that prior to obtaining a marketing authorization in the European Union in the centralized procedure, an applicant must demonstrate compliance with all measures included in an EMA-approved Pediatric Investigation Plan covering all subsets of the pediatric population, unless the EMA has granted a product-specific waiver, class waiver, or a deferral for one or more of the measures included in the Pediatric Investigation Plan.
The European Union also provides opportunities for market exclusivity. For example, in the European Union, upon receiving marketing authorization, new chemical entities generally receive eight years of data exclusivity and an additional two years of market exclusivity. If granted, data exclusivity prevents regulatory authorities in the European Union from referencing the innovator’s data to assess a generic or biosimilar application. During the additional two-year period of market exclusivity, a generic or biosimilar marketing authorization can be submitted, and the innovator’s data may be referenced, but no generic or biosimilar product can be marketed until the expiration of the market exclusivity. However, there is no guarantee that a product will be considered by the European Union’s regulatory authorities to be a new chemical entity, and products may not qualify for data exclusivity.
Periods of Authorization and Renewals
A marketing authorization is valid for five years, in principle, and it may be renewed after five years on the basis of a reevaluation of the risk-benefit balance by the EMA or by the competent authority of the authorizing Member State. To that end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least nine months before the marketing authorization ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the EC or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal period. Any authorization that is not followed by the placement of the product on the European Union market (in the case of the centralized procedure) or on the market of the authorizing Member State within three years after authorization ceases to be valid.
144
Regulatory Requirements After Marketing Authorization
Following approval, the holder of the marketing authorization is required to comply with a range of requirements applicable to the manufacturing, marketing, promotion and sale of the medicinal product. These include compliance with the European Union’s stringent pharmacovigilance or safety reporting rules, pursuant to which post-authorization studies and additional monitoring obligations can be imposed. In addition, the manufacturing of authorized products, for which a separate manufacturer’s license is mandatory, must also be conducted in strict compliance with the EMA’s cGMP requirements and comparable requirements of other regulatory bodies in the European Union, which mandate the methods, facilities and controls used in manufacturing, processing and packing of products to assure their safety and identity. Finally, the marketing and promotion of authorized products, including industry-sponsored continuing medical education and advertising directed toward the prescribers of products and/or the general public, are strictly regulated in the European Union under Directive 2001/83.
Orphan Drug Designation and Exclusivity
Regulation 141/2000 and Regulation 847/2000 provide that a product can be designated as an orphan drug by the EC if its sponsor can establish: that the product is intended for the diagnosis, prevention or treatment of (1) a life-threatening or chronically debilitating condition affecting not more than five in ten thousand persons in the European Union when the application is made, or (2) a life-threatening, seriously debilitating or serious and chronic condition in the European Union and that without incentives it is unlikely that the marketing of the product in the European Union would generate sufficient return to justify the necessary investment. For either of these conditions, the applicant must demonstrate that there exists no satisfactory method of diagnosis, prevention, or treatment of the condition in question that has been authorized in the European Union or, if such method exists, the product has to be of significant benefit compared to products available for the condition.
An orphan drug designation provides a number of benefits, including fee reductions, regulatory assistance and the possibility to apply for a centralized European Union marketing authorization. Orphan drugs also benefit from a 10-year period of market exclusivity. During this market exclusivity period, neither the EMA nor the EC or the Member States can accept an application or grant a marketing authorization for a “similar medicinal product.” A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. The market exclusivity period for the authorized therapeutic indication may, however, be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan drug designation because, for example, the product is sufficiently profitable not to justify market exclusivity. Additionally, marketing authorization may be granted to a similar product for the same indication at any time if the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior, the applicant consents to a second orphan medicinal product application, or applicant cannot supply enough orphan medicinal product.
Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any product candidates for which we may obtain regulatory approval. Even if our product candidates are approved for marketing, sales of such product candidates will depend, in part, on the extent to which third-party payors, including government health programs in the United States (such as Medicare and Medicaid), commercial health insurers, and managed care organizations, provide coverage and establish adequate reimbursement levels for such product candidates. In the United States, the Member States of the European Union and markets in other countries, patients who are prescribed treatments for their conditions and providers performing the prescribed services generally rely on third-party payors to reimburse all or part of the associated healthcare costs. Reimbursement rules and levels are not harmonized in the European Union and therefore differ from Member State to Member State. Patients are unlikely to use any product candidates we may develop unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of such product candidates. The process for determining whether a payor will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Third-party payors are increasingly challenging the price and examining the medical necessity and cost effectiveness of medical products and services and imposing controls to manage costs.
145
In order to secure coverage and reimbursement for any product that might be approved for sale, a company may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of the product, and the cost of these studies would be in addition to the costs required to obtain FDA or other comparable marketing approvals. Even after pharmacogenomic studies are conducted, product candidates may not be considered medically necessary or cost effective. A decision by a third-party payor not to cover any product candidates we may develop could reduce physician utilization of such product candidates once approved and have a material adverse effect on our sales, results of operations and financial condition. Additionally, a payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. For example, the payor may require co-payments that patients find unacceptably high. Further, one payor’s determination to provide coverage for a product does not assure that such coverage will continue or that other payors will also provide coverage and reimbursement for the product, and the level of coverage and reimbursement can differ significantly from payor to payor. Third-party reimbursement and coverage may not be adequate to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. The insurance coverage and reimbursement status of newly approved products for orphan diseases is particularly uncertain, and failure to obtain or maintain adequate coverage and reimbursement for any such product candidates could limit a company’s ability to generate revenue.
The containment of healthcare costs also has become a priority of federal, state and foreign governments as well as other third-party payors such as statutory health insurance funds and the prices of pharmaceuticals have been a focus in this effort. Governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit a company’s revenue from the sale of any approved products. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which a company or its collaborators receive marketing approval, less favorable coverage policies and reimbursement rates may be implemented or coverage may be ended in the future.
Outside the United States, we will face challenges in ensuring obtaining adequate coverage and payment for any product candidates we may develop. Pricing of prescription pharmaceuticals is subject to governmental control in many countries. Pricing negotiations with governmental authorities or other third-party payors such as statutory health insurance funds can extend well beyond the receipt of regulatory marketing approval for a product and may require us to conduct a clinical trial that compares the cost effectiveness of any product candidates we may develop to other available therapies. The conduct of such a clinical trial could be expensive and result in delays in our commercialization efforts.
In the European Union, pricing and reimbursement schemes vary widely from country to country. Some countries provide that products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost effectiveness of a particular product candidate to currently available therapies (so-called health technology assessments) in order to obtain reimbursement or pricing approval. For example, the European Union provides options for its Member States to restrict the range of products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. European Union Member States may approve a specific price for a product or may instead adopt a system of direct or indirect controls on the profitability of the company placing the product on the market. Other Member States allow companies to fix their own prices for products, but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. Recently, many countries in the European Union have increased the amount of discounts required on pharmaceuticals and these efforts could continue as countries attempt to manage healthcare expenditures, especially in light of the severe fiscal and debt crises experienced by many countries in the European Union. The downward pressure on healthcare costs in general, particularly prescription products, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various European Union Member States and parallel trade (arbitrage between low-priced and high-priced Member States) can further reduce prices. Special pricing and reimbursement rules may apply to orphan drugs. Inclusion of orphan drugs in reimbursement systems tend to focus on the medical usefulness, need, quality and economic benefits to patients and the healthcare system as for any product. Acceptance of any medicinal product for reimbursement may come with cost, use and often volume restrictions, which again can vary by country. In addition, results based rules of reimbursement may apply. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products, if approved in those countries.
146
Healthcare Laws and Regulations
Healthcare providers and third-party payors play a primary role in the recommendation and prescription of pharmaceutical products that are granted marketing approval. Our current and future arrangements with providers, researchers, consultants, third-party payors and customers are subject to broadly applicable federal and state fraud and abuse, anti-kickback, false claims, physician payment transparency and other healthcare laws and regulations that may constrain our business and/or financial arrangements. Restrictions under applicable federal and state healthcare laws and regulations include, without limitation, the following:
| ● | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering, or paying remuneration, directly or indirectly, in cash or in-kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or a specific intent to violate it in order to have committed a violation; |
| ● | the federal civil and criminal false claims laws, including the civil False Claims Act, and civil monetary penalties laws, which prohibit individuals or entities from, among other things, knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false, fictitious, or fraudulent or knowingly making, using, or causing to be made or used a false record or statement to avoid, decrease, or conceal an obligation to pay money to the federal government. Moreover, the government may assert that a claim that includes items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the civil False Claims Act; |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal laws that prohibit, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or a specific intent to violate it in order to have committed a violation; |
| ● | the federal transparency requirements known as the federal Physician Payments Sunshine Act, under the Affordable Care Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies to report annually to the CMS, within the HHS, information related to payments and other transfers of value made by that entity to physicians (as defined by statute), certain other healthcare providers beginning in 2022, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; |
| ● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; |
| ● | federal government price reporting laws, which require us to calculate and report complex pricing metrics to government programs and which may be used in the calculation of reimbursement and/or discounts on marketed products; |
| ● | the Foreign Corrupt Practices Act, a U.S. law which regulates certain financial relationships with foreign government officials (which could include, for example, certain medical professionals); and |
| ● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may apply to healthcare items or services that are reimbursed by non-governmental third-party payors, including private insurers; and some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government in addition to requiring pharmaceutical manufacturers to report information related to payments to physicians and other healthcare providers or marketing expenditures and pricing information. |
Violations of these laws can subject us to criminal, civil and administrative sanctions including monetary penalties, damages, fines, disgorgement, individual imprisonment and exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, additional reporting requirements and oversight if we become subject to a corporate integrity agreement or similar agreement to resolve allegations of noncompliance with these laws, reputational harm, and we may be required to curtail or restructure our operations. Moreover, we expect that there will continue to be federal and state laws and regulations, proposed and implemented, that could impact our future operations and business.
147
Data Privacy and Cybersecurity Laws
We may be subject to, or our marketing activities may be limited by, HIPAA and its implementing regulations, which established uniform standards for certain covered entities (healthcare providers, health plans and healthcare clearinghouses) governing the conduct of certain electronic healthcare transactions and protecting the security and privacy of protected health information, including, among other requirements, mandatory contractual terms and technical safeguards to protect the privacy, security and transmission of protected health information and notification to affected individuals and regulatory authorities in the event of certain breaches of security of protected health information. The American Recovery and Reinvestment Act of 2009, commonly referred to as the economic stimulus package, included sweeping expansion of HIPAA’s privacy and security standards called the Health Information Technology for Economic and Clinical Health Act, or HITECH, which became effective on February 17, 2010. Among other things, the HITECH makes HIPAA’s privacy and security standards directly applicable to business associates, or independent contractors or agents of covered entities, that receive or obtain protected health information in connection with providing a service on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions.
Even when HIPAA does not apply, failing to take appropriate steps to keep consumers’ personal information secure can constitute unfair acts or practices in or affecting commerce and be construed as a violation of Section 5(a) of the Federal Trade Commission Act, or the FTCA, 15 U.S.C § 45(a). The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards and the FTC’s guidance for appropriately securing consumers’ personal information is similar to what is required by the HIPAA Security Rule.
State laws may be more stringent, broader in scope or offer greater individual rights with respect to protected health information, or PHI, than HIPAA, and state laws may differ from each other, which may complicate compliance efforts. By way of example, the California Consumer Privacy Act as amended by the California Privacy Rights Act, or CCPA, gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing, and receive detailed information about how their personal information is used. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA may increase our compliance costs and potential liability. The CCPA also creates a new state agency that will be vested with authority to implement and enforce the CCPA. In addition, other states may choose to adopt more stringent privacy legislation, which could increase our potential liability and compliance costs and adversely affect our business.
In the European Union, we may be subject to strict data protection regulations, in particular with regard to health data of individuals pursuant to Art. 4 Nr. 15 of the GDPR, effective since May 25, 2018. The GDPR, together with national legislation, regulations and guidelines of the European Union Member States and the United Kingdom governing the processing of personal data, impose strict obligations with respect to, and restrictions on, the collection, use, retention, protection, disclosure, transfer and processing of personal data. In particular, the GDPR includes obligations and restrictions concerning the consent and rights of data subjects, the transfer of personal data to countries outside the European Union, security breach notifications, and other requirements concerning the security and confidentiality of personal data. For example, in 2016, the European Union and United States agreed to a transfer framework for data transferred from the European Union to the United States, called the Privacy Shield, but the Privacy Shield was invalidated in July 2020 by the Court of Justice of the European Union. The standard contractual clauses issued by the European Commission for the transfer of personal data may be similarly invalidated by the Court of Justice of the European Union. On June 4, 2021, the European Commission adopted new standard contractual clauses, which impose on companies additional obligations relating to data transfers, including the obligation to conduct a transfer impact assessment and, depending on a party’s role in the transfer, to implement additional security measures and to update internal privacy practices. It remains to be seen whether these standard contractual clauses will remain available and whether additional means for lawful data transfers will become available. The GDPR imposes special requirements concerning the protection of special categories of personal data which include health and genetic information of data subjects. These special categories of data may only be processed under certain circumstances, including if the data subject consented to such processing or if (i) processing is necessary in order to protect vital interests of the data subject or of another natural person, insofar as the data subject is unable to provide consent for physical or legal reasons; (ii) the data concerned have manifestly been made public by the data subject; (iii) processing is necessary in order to assert, exercise or defend legal claims; or (iv) processing is necessary for the purposes of scientific research and to ensure that any additional requirements under applicable data protection laws, including national legislation, regulations and guidelines, are met.
148
Therefore, we may be subject to and our marketing activities may be limited by the regulations regarding the data protection of individuals according to the GDPR, the German Federal Data Protection Act and other applicable data protection laws. These regulations could also restrict the transfer of data from European Union member states to the United States. The general transfer of personal data outside of the European Union is prohibited unless the conditions laid out in Art. 44 et. seq. of the GDPR are fulfilled and an adequate level of data protection can be ensured. Currently the United States is not considered to be a country with an adequate level of data protection, and further contractual arrangements must be adopted to permit the international transfer of personal data to the United States. European data protection authorities may interpret the GDPR and national laws differently and impose additional requirements, which contributes to the complexity of processing personal data in or from the European Union. Guidance on implementation and compliance practices is regularly updated or otherwise revised. The GDPR has increased our responsibility and liability in relation to personal data that we process and we may be required to put in place additional mechanisms ensuring compliance with the relevant data protection regimes. Separately, Brexit could also lead to further legislative and regulatory changes and increase our compliance costs. As of January 1, 2021, and the expiry of transitional arrangements agreed to between the United Kingdom and the European Union, data processing in the United Kingdom is governed by a United Kingdom version of the GDPR (combining the GDPR and the Data Protection Act 2018), exposing us to two parallel regimes, each of which potentially authorizes similar fines and other potentially divergent enforcement actions for certain violations. On June 28, 2021, the European Commission adopted an adequacy decision in favor of the United Kingdom, enabling data transfers from European Union member states to the United Kingdom without additional safeguards. However, the United Kingdom adequacy decision will automatically expire in June 2025 unless the European Commission re-assesses and renews or extends that decision. For more information regarding the risks related to data security and privacy, see “Risk Factors — Risks Related to Our Business and Industry.”
Competition
We participate in an industry that is characterized by a rapidly growing understanding of disease biology, quickly changing technologies, strong emphasis on proprietary products, and a multitude of companies involved in the creation, development and commercialization of novel therapeutics. These companies are highly sophisticated and often collaborate strategically with each other.
We are developing a broad portfolio of product candidates that, coupled with our capabilities across mRNA technology, development and manufacturing, we believe position us at the forefront of targeted immune active and immune silent mRNA-based medicines. However, we compete with a wide range of pharmaceutical companies, biotechnology companies, academic institutions and other research organizations for novel therapeutic targets, new technologies, talent, financial resources, intellectual property rights and collaboration opportunities. As such, many of our competitors and potential competitors have substantially greater scientific, research and product development capabilities as well as greater financial, manufacturing, marketing and human resources than we do. In addition, there is intense competition to establish clinical trial sites and register patients for clinical trials. Many specialized biotechnology firms have formed collaborations with large, established companies to support the research, development and commercialization of products that may be competitive with ours. Accordingly, our competitors may be more successful than we may be in developing, commercializing and achieving widespread market acceptance. In addition, our competitors’ products may be more effective or more effectively marketed and sold than any treatment we or our development partners may commercialize and may render our product candidates obsolete or noncompetitive before we can recover the expenses related to developing and commercializing any of our product candidates.
There are additional companies that are working on potential mRNA medicines. Companies with clinical programs with mRNA include BioNTech/Pfizer, Moderna, eTheRNA Immunotherapies, Translate Bio, GlaxoSmithKline Sanofi, AstraZeneca, Merck & Co. and Arcturus Therapeutics and those programs include, Ethris and Genevant Sciences. In COVID-19, our monovalent vaccine candidate, CV0601, and in seasonal influenza our multivalent vaccine candidate are currently the main focus of other pharmaceutical companies, some with more considerable capital resources than ours. For example, a limited number of products that utilize mRNA as a prophylactic vaccine against COVID-19 have been approved by the FDA, EMA and other regulatory agencies.. For influenza, no mRNA-based vaccine has been approved so far with the current standard-of-care being well-established protein-based vaccines. Thus, we expect intense competition for our vaccine candidates from other pharmaceutical companies not limited to the field of mRNA medicines. In addition, the oncology therapeutics landscape in general is highly competitive and includes large and specialty pharmaceutical and biotechnology companies, academic research institutions and governmental agencies and public and private research institutions. It includes both competition from marketed therapies as well as potential new therapeutics in development. We may compete with products with different mechanisms of action as well as against established standards of care. We expect our intratumoral immunotherapy candidates for the treatment of solid tumors to face direct competition from companies such as Moderna and BioNTech in collaboration with Sanofi in addition to several non-mRNA-based approaches.
149
C. | Organizational Structure |
We are a holding company, and our sole asset is the capital stock of our wholly owned subsidiaries. Our major subsidiaries are listed under Exhibit 8.1 filed herewith.
D. | Property, Plant and Equipment |
Our Manufacturing Platform
We are an integrated biopharmaceutical company with in-house manufacturing capabilities and expertise. We consider our manufacturing process an important part of our strategy that allows us to continuously improve our technology platform and maintain flexibility in clinical development and allow us to manufacture potential future commercial products. The close interaction of our technical development and research teams enables us to rapidly implement innovations and robustness to the manufacturing process. We control the critical steps of manufacturing in-house and also collaborate with manufacturing organization partners. Both of which allow us to drive innovation and to maintain flexibility, and in turn allows us to pivot quickly in pandemic settings.
All of our mRNA-based active ingredients for various fields of application originate from a common technology platform and are based on identical source materials. This enables us to produce all mRNA therapies using a platform process concept. Given the differences in the encoded protein only require alterations of the sequence of the mRNA molecule, leaving its physicochemical characteristics largely unaffected, we can use the same mRNA production strategy applying the same unit operations for diverse products. This allows us to save time and reduce costs compared to other manufacturing processes. Our approach supports a seamless production concept based on our experience and know-how in mRNA manufacturing.
Our GMP Manufacturing Facilities
150
We have continued to invest significantly in building and expanding our manufacturing capabilities since 2006. We currently have the capacity to produce early and late-stage clinical trial RNA material and potential future commercial lots. Since 2006, we have manufactured thousands of mRNA constructs, from high throughput and small amounts for discovery and preclinical development to GMP compliant products.
We are currently operating three GMP-certified suites. Our GMP I and II facilities were designed to run up to 14 different products in parallel, using a lab-scale process. The facilities cover all steps from starting material pDNA, through mRNA manufacturing to formulated bulk. Our GMP I and II facilities are dedicated to providing supplies for early clinical development (Phase 1 and 2), with capacity to produce multiple batches per year. In 2019, we expanded our production capacity to meet the increasing demands for clinical studies and potential future initial commercial supply by adding a GMP III facility. Our GMP III facility allows us to achieve additional scale and reduce manufacturing process time. This facility focuses on the production of mRNA, and formulated bulk. We currently use our GMP/II facility and CMOs for starting material plasmid DNA (pDNA). The GMP III facility is intended to provide supply for early to late-stage clinical studies and initial potential market supply and is based on a new scalable process design compared to our GMP I and II facilities. We are currently in the process of certifying a GMP IV facility (shown in the picture below), which is being designed to cover all manufacturing steps from starting material (pDNA) to formulation.

GMP IV facility
151
The RNA Printer
In addition to our GMP I-IV manufacturing facilities, we are operating an integrated and highly automated production unit, The RNA Printer®. The RNA Printer® is a GMP-grade end-to-end system that is being designed to downscale, accelerate and automate the manufacturing process. The fully synthetic production process allows us to have rapid manufacturing of products and offer reproducibility and high standardization. It includes automated cleaning and sanitization in place procedures and continuous process verification. The current setup covers DNA and RNA production for automated downstream and upstream production up to drug substance. We have obtained a Manufacturing License for a mRNA in the cancer vaccine development program to support CureVac’s oncology strategy from the regional authority in Baden-Württemberg as announced in November 2023. In April 2024, we announced that the system was also granted a Framework License from the same authority that significantly broadens its regulatory freedom and flexibility to manufacture different mRNA vaccine candidates. The regulatory approval process for the formulation Module is ongoing.

The RNA Printer®
The key characteristics of The RNA Printer® are rapid throughput, easy operator access to equipment, sophisticated precision control software and data capture, and the small footprint that allows for easy decentralization. We view The RNA Printer® as complementary to our manufacturing strategy. With its modular design and decentralized concept, we believe The RNA Printer® could be used to facilitate broad access to mRNA technology and enable mRNA product developments, e.g. for patient access to advanced and personalized mRNA-based therapies in oncology.
Our objective is to cover the entire production stream, and we believe efficient accompanying analytics will help to rapidly produce high-quality material. All data generated during production would be collected to further improve production processes and product development.
Facilities
Our headquarters are in Tübingen, Germany, Friedrich-Miescher-Strasse 15, where we occupy approximately 123,000 square feet of office and laboratory space under a sublease agreement entered into with CureVac Real Estate GmbH that started on June 6, 2018. The fixed-term 15-year lease payment period began on March 1, 2020. We also occupy approximately 53,000 square feet of additional office and laboratory space in Tübingen, Germany, Paul-Ehrlich-Strasse 15, under sublease agreements also entered into with CureVac Real Estate GmbH, that started on February 1, 2018.
152
Since 2006, we have operated a manufacturing facility in Tübingen, Germany, the first worldwide GMP-compliant mRNA production plant with two multi-product suites (GMP I and II). These facilities contain approximately 16,145 square feet of laboratory space, including 2,800 square feet of GMP facilities, and are dedicated to providing supplies for early clinical development (Phases 1 and 2 of clinical trials). In addition, we have established a third in-house production suit (GMP III) with an upscaled manufacturing process, which was certified in December 2019. We currently occupy 2,800 square feet of GMP III facility for the production of mRNA. Our GMP III facility is intended to provide supplies for our late-stage clinical studies and anticipated early market supply. These manufacturing facilities are located in Tübingen, Germany, Paul-Ehrlich-Strasse 15 and are leased via the abovementioned sublease agreements entered into with CureVacManufacturing GmbH.
We have also constructed a new CureVac owned manufacturing facility, for a GMP production process at large scale, from starting material to formulation(GMP IV). This facility is expected to be approximately 128,000 square feet. The expected cost of completion is €171.1 million, and as of December 31, 2023, we have spent €170.6 million on completing the GMP IV facility.
In addition, we lease land and small- to mid-scale buildings for our offices and laboratories in Tübingen approximately 111,000 sq. feet. We occupy approximately 27,000 square feet of additional offices and laboratories in Tübingen, Germany, Rosentalstrasse 15 for research and development (or Technology Department). The agreement started on March 1, 2022.
We lease an aggregate of approximately 337,300 square feet, in Germany, Europe and the United States. The following table summarizes information with respect to the principal facilities leased by us:
Area | ||
Location |
| (Approximate Sq. Feet) |
Germany: | ||
Tübingen |
| 287,000 |
Frankfurt am Main |
| 8,600 |
Wiesbaden | 14,800 | |
Total: |
| 310,400 |
Belgium: | ||
Louvain La Neuve |
| 5,200 |
Total |
| 5,200 |
Netherlands: |
| |
Amsterdam | 8,800 | |
Total: | 8,800 | |
United States: | ||
Boston | 12,900 | |
Total: | 12,900 | |
Total | 337,300 |
Our leases expire on various dates from 2021 to 2035. The lease in Boston, United States, is held by our U.S. subsidiary, CureVac Inc. The Lease in Louvain La Neuve, Belgium, is held by our Belgian subsidiary, CureVac Belgium SA. The lease in Amsterdam, Netherlands, is held by our Dutch subsidiary, CureVac Netherlands B.V.
Environmental Issues
To the best of our knowledge, currently there are no foreign, federal, state or local environmental laws, rules or regulations that will materially affect our results of operations or our position with respect to our competitors. However, we can provide no assurance of the effect that any possible future environmental laws will have on our operating results.
ITEM 4A. UNRESOLVED STAFF COMMENTS
Not applicable.
153
ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
A. Operating Results
The following discussion of our financial condition and results of operations should be read in conjunction with CureVac’s audited consolidated financial statements as of December 31, 2022 and 2023, and for the years ended December 31, 2021, 2022 and 2023, and the notes thereto, included elsewhere in this Annual Report. Financial information presented in the consolidated financial statements for periods prior to the completion of our corporate reorganization is that of CureVac AG (currently CureVac SE), our wholly owned subsidiary. The consolidated financial statements of CureVac N.V. are a continuation of the historical consolidated financial statements of CureVac AG (currently CureVac SE). CureVac AG (currently CureVac SE) was acquired by CureVac B.V., which subsequently converted into CureVac N.V. on August 14, 2020, as part of our corporate reorganization. CureVac B.V. had no assets, liabilities or contingent liabilities until the completion of our corporate reorganization. Following the corporate reorganization, CureVac N.V. became the holding company of CureVac AG (currently CureVac SE) and the historical consolidated financial statements of CureVac AG (currently CureVac SE) included in this Annual Report became the historical consolidated financial statements of CureVac N.V. The following discussion is based on our financial information prepared in accordance with IFRS as issued by the IASB, which may differ in material respects from generally accepted accounting principles in the United States and other jurisdictions. The following discussion includes forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of many factors, including but not limited to those described under “Item 3. Key Information — D. Risk Factors” and elsewhere in this Annual Report.
Key Factors Affecting Our Results of Operations
We believe that the most significant factors affecting our results of operations include:
Research and Development Expenses
Our ability to successfully pioneer a robust mRNA technology platform and develop innovative product candidates will be the primary factor affecting our future growth and development. Our approach to the discovery and development of product candidates based on mRNA technology is still being demonstrated. As such, we do not know whether we will be able to successfully develop any products. Developing novel product candidates requires a significant investment of resources over a prolonged period of time, and a core part of our strategy is to continue making sustained investments in this area. We have chosen to leverage our platform to initially focus on advancing our product candidates in the areas of prophylactic vaccines, oncology and molecular therapy.
For more information on our proprietary technology and clinical development pipeline, see “Item 4. Information on the Company — B. Business Overview — Our Product Portfolio.”
154
All of the product candidates are still in development, and we have incurred and will continue to incur significant research and development costs for preclinical studies and clinical trials. We expect that our research and development expenses will constitute the most substantial part of our expenses in future periods in line with the advance and expansion of the development of our product candidates. Due to our accelerated efforts to develop our first-generation backbone COVID-19 vaccine candidate, CVnCoV, we incurred research and development expenses that significantly exceeded our historical levels of research and developments expenses. Additionally, our October 2021 notification to the European Commission, or EC, of the withdrawal of our regulatory approval application for CVnCoV resulted in our recognition of several expenses, which have contributed to our increased expense levels, but which we do not expect to recur in future periods. In April 2021, we entered into a collaboration agreement with GSK for the development of a broad COVID-19 vaccine program based on our second-generation backbone. CV2CoV, a non-chemically modified mRNA, encoding the prefusion stabilized full-length spike protein of the SARS-CoV-2 virus, and formulated within LNPs, is the first representative of our COVID-19 vaccine program based on the second-generation backbone and presently in the Phase 1 clinical trial, as announced on March 30, 2022. Within this COVID-19 vaccine program, we plan to extend our technology platform also to chemically modified mRNA constructs to allow for data-driven selection of the best candidate. We expect to incur significant expenses related to such second-generation backbone vaccine candidates. But, as we and GSK agreed to equally share the development costs for GSK COVID Products, our current level of research and development expenses will not continue to increase in the level as it did from 2020 to 2021. Once we conclude our research and development efforts related to a selected second-generation backbone vaccine candidate, we expect that our research and development expenses shall be consistent with our past trends before the COVID-19 pandemic, but we may find it necessary to continue such current trend with respect to our research and developments expenses or we may continue to increase further our research and development expenses. For example, we may continue to increase our research and development expenses for future research and development related to the next generation backbone for our COVID-19 vaccine candidates, such as for our second-generation backbone COVID-19 vaccine candidates or may pursue new indications with our technology platform.
We have historically funded the research and development expenses primarily through public offerings of our common stock, private placements of equity securities, convertible loans, grants from government agencies and similar bodies and payments for collaborative research and development services with strategic partners. In addition, we signed an advance purchase agreement, or APA, with the EC that provided substantial support for our efforts to advance our first-generation backbone vaccine candidate, CVnCoV. In October 2021, we notified the European Commission of the withdrawal of our regulatory approval application for CVnCoV, which automatically terminated the APA.
Our and Our Collaborators’ Ability to Commercialize Our Product Candidates
Our ability to generate revenue from our product candidates depends on our and our collaborators’ ability to successfully advance clinical trials for our product candidates and receive regulatory approval, particularly in the United States, Europe, and other major markets.
We believe that our broad portfolio of product candidates with both novel and validated targets enhances the likelihood that our research and development efforts will yield successful product candidates. Nonetheless, we cannot be certain if any of our product candidates will receive regulatory approvals. Even if such approvals are granted, we will thereafter need to maintain manufacturing and supply arrangements and engage in extensive marketing prior to generating any revenue from such products, and the ultimate commercial success of our products will depend on their acceptance by patients, the medical community and third-party payors and their ability to compete effectively with other therapies on the market. See “Item 3. Key Information — D. Risk Factors — Risks Related to the Development, Clinical Testing and Commercialization of Our Product Candidates.”
The competitive environment is also an important factor with the commercial success of our product candidates, and our ability to successfully commercialize a product candidate will depend on whether there are competing product candidates being developed or already marketed by other companies.
155
We currently do not have any product candidates that have received regulatory approval. As such, we have not incurred any material commercialization expenses in connection with an approved product candidate. In February 2021, we initiated a rolling submission for our first generation COVID-19 vaccine candidate, CVnCoV, with the EMA, which was designed to allow the EMA to assess CVnCoV’s compliance with standards for vaccine efficacy, safety and pharmaceutical quality as a prerequisite for a formal market authorization application. Later in 2021, EMA informed us that the EMA would not start reviewing our submission for CVnCoV before the beginning of 2022. As a result, we estimated that the earliest possible approval of CVnCoV would come in the second quarter of 2022. Data on the efficacy of CVnCoV was generated and published in June 2021. This efficacy data did not live up to our pre-trial expectations and fell behind the efficacy of competing COVID-19 vaccine products. The application for the marketing authorization for CVnCoV was withdrawn in early October 2021, as a necessary reaction to the efficacy data as well as the concerns and uncertainties resulting from such data on the granting of a marketing authorization and the expected concerns of prescribers and patients about using a COVID-19 vaccine with a lower efficacy compared to the vaccines already available on the market. After the withdrawal of the application for a marketing authorization for CVnCoV, we have focused our efforts on second-generation mRNA vaccines. The decision is aligned with the evolving dynamics of the pandemic response toward greater need for differentiated vaccines with the gradual transition from an acute pandemic to an endemic SARS-CoV-2 environment. In connection with the regulatory approval process, and in preparation for the commercialization of a second-generation COVID-19 vaccine, we expect our expenses related to commercialization to significantly decrease in the short-term due to our past commercialization efforts for CVnCoV. However, we expect that our expenses related to commercialization will significantly increase in the long term if a second-generation COVID-19 vaccine candidate reaches late clinical stages, but we expect that this increase in expenses will be mitigated by the GSK COVID Agreement, as described below. As part of the commercialization process of CVnCoV, we also entered into strategic partnerships with Bayer for the development, production and distribution of CVnCoV. In addition, pursuant to a preliminary agreement regarding the secondary manufacturing of CVnCoV we entered into with GSK, GSK would have supported the secondary manufacturing of up to 100 million doses of CVnCoV in 2021. Additionally, we also set up an integrated European manufacturing and supply network. After our decision to withdraw CVnCoV from the regulatory approval process, we have restructured this network to focus our efforts on the second-generation mRNA vaccine.
Our Collaborations, Related License Agreements and Advance Purchase Agreements
Our results of operations have been, and we expect them to continue to be, affected by our contractual collaborations with third parties for the development and commercialization of certain of our product candidates. In addition, our future results of operation may be affected by future advance purchase agreements for our COVID-19 vaccine candidates. To date, our revenues have been recognized pursuant to license and collaboration agreements, which include upfront payments for licenses or options to obtain licenses, milestone payments, payments for product sales and payments for research and development services. Grants from government agencies or similar bodies are recognized as other operating income or as a reduction to depreciation and amortization expense recognized from assets purchased under the associated arrangements.
We have entered into strategic collaborations and license agreements with third parties. In addition, on November 30, 2020, we entered into an advance purchase agreement, or APA, with the European Commission, or EC, which provided for the advance purchase by the commission of our first-generation vaccine candidate, CVnCoV. In October 2021, we notified the EC of the withdrawal of our regulatory approval application for CVnCoV, which automatically terminated the APA. As part of our business development strategy, we aim to increase the number of our strategic collaborations in order to derive further value from our platform and more fully exploit the potential of our collaboration and license agreements.
On April 8, 2022, we received a letter from the Federal Republic of Germany’s counsel confirming that the CureVac-GSK Consortium, was awarded with the Pandemic Preparedness Agreement. On April 9, 2024, the CureVac-GSK Consortium notified the Federal Republic of Germany of its termination of the Pandemic Preparedness Agreement, effective as of May 31, 2024. For further details on our Pandemic Preparedness Agreement, see “Item 4. Information on the Company — B. Business Overview — Pandemic Preparedness Agreement”.
Certain key terms of our current material collaboration and license agreements, as well as our advance purchase agreement with the EC are summarized below. For further details on our collaboration agreements, see “Item 4. Information on the Company — B. Business Overview — Collaborations” and “Item 4. Information on the Company — B. Business Overview — Advance Purchase Agreements,” respectively.
156
GlaxoSmithKline
In July 2020, we entered into a Collaboration and License Agreement with GSK, which we refer to as the 2020 GSK Agreement, pursuant to which we are collaborating with GSK to research, develop and commercialize prophylactic and therapeutic non-replicating mRNA-based vaccines and antibodies targeting infectious disease pathogens. The 2020 GSK Agreement was amended and restated in April 2021, September 2021, February 2022 and March 2022.
GSK paid us an upfront payment of €120 million and is required to pay us a manufacturing capacity reservation fee of €30 million following a certain regulatory milestone event, which is creditable against future milestone payments. We are eligible to receive up to between €28 million to €45 million in development milestone payments, €32 million to €35 million in regulatory milestone payments and €70 million to €100 million in commercial milestone payments, depending on the product. Under the 2020 GSK Agreement, we granted GSK an exclusive option to add additional products in the field of infectious diseases to the license granted under the 2020 GSK Agreement and, upon each exercise of such option, GSK is required to compensate us for certain development costs and pay any accrued milestone payments. Additionally, GSK has the right to replace products licensed under the 2020 GSK Agreement and, if the replacement product was already under development by us, GSK must compensate us for certain development costs and pay any accrued milestone payments. We are additionally eligible to receive tiered royalty payments ranging from single-digit to low - teens percentages on net sales, subject to certain customary reductions. GSK is required to compensate us for certain development and regulatory costs we may incur in connection with performing our obligations under the 2020 GSK Agreement, and we are eligible to receive up to €20,000 in reimbursements for expenses incurred by recording or registering the licenses granted under the 2020 GSK Agreement. We retain the right to commercialize products developed under the 2020 GSK Agreement in Germany, Austria and Switzerland, as GSK’s exclusive distributor in these markets. Under any such distribution agreement to be entered into between us and GSK, we will be required to purchase supply from GSK and pay GSK a low thirties percentage royalty on net sales. Pursuant to the amendment in September 2021, we and GSK are required to complete certain development activities set forth in updated development plans. We and GSK agree to decide whether the products required for clinical studies will be manufactured by us, GSK or jointly.
Additionally, in April 2021, we entered into a new collaboration agreement with GSK, which we refer to as the GSK COVID Agreement, pursuant to which we are collaborating with GSK to research, develop and manufacture next-generation mRNA vaccines targeting the original SARS-CoV-2 strain as well as emerging variants, including multivalent and monovalent approaches, such as our second-generation COVID 19 vaccine candidates, CV0601 and CV0701. These vaccine candidates may either be used to protect unvaccinated individuals or to serve as boosters in the event that SARS-CoV-2 immunity gained from an initial vaccination reduces over time. The GSK COVID Agreement was amended and restated in September 2021, February 2022 and March 2022, and further amended to update the research and development plan in August 2022. Pursuant to the amendment in September 2021, we and GSK are required to complete certain development activities with respect to the GSK COVID Products set forth in updated development plans. We and GSK agree to decide whether the GSK COVID Products required for clinical studies will be manufactured by us, GSK or jointly.
Under the GSK COVID Agreement, GSK paid us an upfront payment of €75 million. We and GSK agreed to equally share all development costs for GSK COVID Products, subject to certain exceptions. We and GSK will share all net profits generated from sales of GSK COVID Products, other than Combination Products (as defined therein), under profit sharing arrangements that in certain cases vary depending upon the GSK COVID Product in question, the time of sale, the number of doses sold and the party to whom the sale is made. We are eligible to receive tiered royalty payments ranging from sub-teen to mid-teen percentages on net sales of Combination Products, subject to certain customary reductions. Under the GSK COVID Agreement we have the right to commercialize GSK COVID Products in Austria, Germany and Switzerland and if we exercise such right, our sales of GSK COVID Products, other than Combination Products will be subject to the profit share and we will be required to pay GSK a high-teen percentage royalty on net sales of all Combination Products in such countries.
157
Genmab
In December 2019, we entered into a Collaboration and License Agreement, which we refer to as the Genmab Agreement, with Genmab to research and develop up to four potential differentiated mRNA-based antibody products, to be selected by Genmab, based on the combination of our proprietary RNAntibody technology with Genmab’s proprietary antibody technology for the treatment of human diseases. We collaborated on research to identify an initial product candidate designed to express a certain Genmab proprietary antibody, and we contributed a portion of the overall costs for the development of such product candidate. In June 2023, Genmab notified us of its intent to terminate the Genmab First Program under the Genmab Agreement effective in September 2023. Such termination did not terminate the Genmab Agreement in its entirety, but rather only with respect to certain license and exclusivity provisions related to the Genmab First Program. Under the Genmab Agreement, we further grant Genmab a license for the preclinical development of up to four additional mRNA antibody product concepts and options to obtain commercial licenses under our mRNA technology to develop, manufacture and commercialize product candidates for up to three of such product concepts.
Under the terms of the Genmab Agreement, Genmab paid us a $10 million upfront fee and made a €20 million equity investment in March 2020. Genmab is additionally required to pay us up to $30 million in option exercise fees. If Genmab exercises any of its options to obtain commercial licenses for the additional mRNA antibody concepts, Genmab would fund all research and would develop and commercialize any resulting product candidates. We are additionally eligible to receive up to between $25 million and $43 million in development milestone payments, $100 million and $125 million in regulatory milestone payments and $150 million and $200 million in commercial milestone payments for each product, depending on the specific product concept. In addition, we are eligible to receive a mid single-digit to low teens percentage tiered royalty on aggregate net sales of licensed products, on a per-product basis and subject to certain customary reductions. We retain an option to participate in development and commercialization of one of the potential additional mRNA antibody product concepts under predefined terms and conditions. In the event we exercise such right, we must pay Genmab a one-time payment of $3 million and refund any option fee paid by Genmab with respect to such product. As of December 31, 2023, we have received $1 million in development cost reimbursements and we have not received any reservation, product selection, option exercise or sublicense fees or milestone or royalty payments.
Arcturus
In January 2018, we entered into a Development and Option Agreement, which we refer to as the Arcturus Agreement, with Arcturus, which provided us with access to Arcturus LNP formulation technology which we used in combination with our mRNA technology. Such agreement expired in July 2023. As of December 31, 2023, we made payments totaling $5.6 million to Arcturus reimbursing Arcturus for development costs and in connection with our FTE funding obligations.
Acuitas
In April 2016, we entered into a Development and Option Agreement, which as amended we refer to as the Acuitas Agreement, with Acuitas, which provides us with access to Acuitas LNP formulation technology that we use in combination with our mRNA technology. We are required to pay Acuitas annual target reservation and maintenance fees of up to $1.4 million if we reserve the maximum number of targets permitted under the Acuitas Agreement and to reimburse Acuitas for certain costs incurred in connection with development activities and certain FTE costs. We are additionally required to pay an option exercise fee ranging from $50,000 to $2 million upon each exercise of our option to obtain a license for further development and commercialization with respect to a selected target, subject to certain additional fees ranging from $10,000 to $200,000 for the exercise of our option for certain other vaccine targets. We paid Acuitas a $5 million upfront fee in connection with an amendment to the Acuitas Agreement dated July 2020 and, upon each exercise of our option to exchange a vaccine target licensed under any non-exclusive license, we are required to pay an exchange fee of $3 million. We additionally paid Acuitas a $3 million upfront fee in connection with an amendment to the Acuitas Agreement dated December 2020 and paid two annual payments of an additional $250,000 per option to extend certain options. Under each license agreement in connection with our exercise of our option, we will additionally be required to make low single-digit percentage tiered royalty payments and must pay up to between $1.1 million and $9 million in development milestone payments, $1.3 million and $7 million in regulatory milestone payments and $1.3 million and $7 million in commercial milestone payments, depending on whether the license is exclusive or non-exclusive and the number of options exercised to date. As of December 31, 2023, we have exercised our option to obtain a non-exclusive license to 18 targets, subject to customary closing conditions. As of December 31, 2023, we have paid Acuitas $9.7 million in reservation and option exercise fees, $0.6 million in license maintenance fees and $1.75 million for certain options not yet exercised and have made payments totaling $9.0 million reimbursing Acuitas for development costs and LNP batches and in connection with our FTE funding obligations.
158
For each option that we have exercised under the Acuitas Agreement, we have entered into a non-exclusive license agreement with Acuitas with respect to such optioned target, all based on the same form agreement, which we refer to as the Acuitas License Agreements. We are required to pay Acuitas up to between $1.1 million and $1.6 million in development milestone payments, $1.3 million and $1.8 million in regulatory milestone payments and between $1.3 million and $1.8 million in commercial milestone payments under each Acuitas License Agreement. We must pay Acuitas annual fees ranging from $5,000 to $10,000 for any additional protein targeted by a vaccine product licensed under each Acuitas License Agreement after a certain milestone event. Additionally, we are obligated to pay Acuitas a low single-digit percentage royalty on net sales of licensed products. As of December 31, 2023, we have made $100,000 in development milestone payments to Acuitas with respect to the license agreement relating to Rabies RAV-G, $1.4 million in development milestone payments (Phase I, Phase II and Phase III milestone payments) to Acuitas with respect to the license agreement relating to the SARS-CoV-2 Spike protein S, $350,000 in development milestone payments to Acuitas with respect to the license agreement relating to the Influenza hemagglutinin (HA) antigen, $1.0 million in development milestone payments to Acuitas with respect to the license agreement relating to CVGBM, and have not made any royalty payments.
CRISPR Therapeutics
In November 2017, we entered into a Development and License Agreement with CRISPR Therapeutics, which, as amended by the first amendment entered into in June 2020 and the second amendment entered into in October 2023, we refer to as the CRISPR Therapeutics Agreement, pursuant to which we will develop novel Cas9 mRNA constructs for use in gene editing therapeutics. Under the CRISPR Therapeutics Agreement, we granted CRISPR Therapeutics an exclusive worldwide license to use our improved Cas9 constructs for the development and commercialization of three of its in vivo gene-editing programs for certain diseases.
CRISPR Therapeutics has paid us an upfront one-time technology access fee of $3 million and we are eligible to receive up to $28 million in development milestone payments, $52 million in regulatory milestone payments and $260 million in commercial milestone payments, as well as mid-single-digit percentage royalties from CRISPR Therapeutics on the net sales of licensed products on a product-by-product and country-by-country basis, subject to certain potential customary reductions. Additionally, CRISPR Therapeutics will make payments to us for services provided by us in conjunction with research programs under the CRISPR Therapeutics Agreement. In the event CRISPR Therapeutics exercises its right to sublicense under the CRISPR Therapeutics Agreement, CRISPR Therapeutics must pay us a low-teens to mid-twenties percentage of any non-royalty sublicense income, depending on the timing of the sublicense and whether the sublicense is granted through an affiliate of CRISPR Therapeutics. As of December 31, 2023, we have received €8.4 million in payments for the supply of materials and FTE cost, development reimbursements and upfront one-time technology access fee and no milestone, royalty or sublicense fee payments. Additionally, as of December 31, 2023, we have invoiced CRISPR Therapeutics for certain amounts under the agreement in connection with the second amendment, which confirmed the parties’ intention to stop work on two programs under the CRISPR Therapeutics Agreement and added three new programs.
Bill & Melinda Gates Foundation
In May 2014, we were awarded a contract from the Bill & Melinda Gates Foundation for the development of a vaccine for rotaviruses, as amended in November 2020, for up to $3.0 million in funding. As of December 31, 2023, we have received $3.0 million in funding under the agreement. In March 2015, the Bill & Melinda Gates Foundation made an equity investment of $40 million to support continued development of our RNA technology platform and the construction of an industrial-scale cGMP production facility. We entered into a Global Access Commitments Agreement with the Bill & Melinda Gates Foundation in February 2015 pursuant to which we are required to take certain actions to support the Bill & Melinda Gates Foundation mission. In connection with the investment by the Bill & Melinda Gates Foundation, we are required to conduct development activities for up to three concurrent projects to be proposed by the Bill & Melinda Gates Foundation. The costs of such projects will be allocated on a project-by-project basis in proportion to the allocation of the expected benefits.
159
In November 2016, in connection with the Global Access Commitments Agreement, we were awarded a grant for up to $0.9 million in funding from the Bill & Melinda Gates Foundation for the development of a vaccine for picornaviruses. As of December 31, 2023, we have received $0.7 million in funding under the picornaviruses grant agreement. The term of the picornavirus grant expired in June 2022; however, our global access commitments survive. Following the completion of the project, the Bill & Melinda Gates Foundation requested a reimbursement for unspent funds. As of December 31, 2023, we have paid unspent funds of $0.2 million back to the Bill & Melinda Gates Foundation. In November 2017, we were awarded two additional grants each for up to $1.9 million and $1.5 million in funding from the Bill & Melinda Gates Foundation for the development of a universal influenza and a malaria vaccine, respectively. By an amendment entered into November 2020, our grant for the development of a malaria vaccine was increased by an additional $0.8 million. As of December 31, 2023, we have received $1.9 million and $2.2 million, respectively, in funding under each grant agreement. The malaria grant agreement expired in December 2022 and the universal influenza grant agreement expired in March 2022; however, our global access commitments survive.
Coalition for Epidemic Preparedness Innovations
In February 2019, we entered into a framework partnership agreement, which as amended we refer to as the CEPI Agreement, with the Coalition for Epidemic Preparedness, or CEPI, to develop our RNA Printer using certain intellectual property controlled by us covering the development and manufacture of mRNA products, as well as certain additional intellectual property licensed to us. In connection with the CEPI Agreement we have entered into work orders for the preclinical development of a Lassa virus vaccine, a yellow fever vaccine and our rabies virus vaccine. In addition, we entered into a work package for the preclinical development and a Phase 1 clinical trial for our first-generation COVID-19 vaccine candidate, CVnCoV. The CEPI Agreement terminated in February 2022, except with respect to certain ongoing projects, which are contemplated to be completed in March 2024. CEPI agreed to contribute up to $34 million in funding for projects undertaken under the CEPI Agreement and an additional $15.3 million in connection with development of CVnCoV. As of December 31, 2023, we have received €27.1 million in funding for projects undertaken under the CEPI Agreement. Following the completion of the CEPI Agreement, CEPI requested a partial reimbursement of $1.0 million for unspent funds. As of December 31, 2023 we have paid these unspent funds back to CEPI.
Tesla Automation
In November 2015, we entered into a development and intellectual property agreement with Tesla Automation, formerly trading under the name of Tesla Grohmann Automation, which we refer to as the Tesla Automation Agreement, pursuant to which Tesla Automation agreed to design, develop and manufacture certain automated manufacturing machines on our behalf. We are obligated to pay Tesla Automation a fee for each machine delivered by Tesla Automation and up to $50 million to $60 million in commercial milestone payments as well as certain development costs under each associated work order. As of December 31, 2023, we have paid Tesla Automation €22 million to €23 million in development costs under various work orders, and we have not paid any fees for machines provided under the Tesla Automation Agreement or made any milestone payments.
160
Research and Option Agreement with myNEO
On May 12, 2022, we entered into a Research and Option Agreement (“R&O”) with myNEO, pursuant to which, as amended in January 2023, we will both collaborate to identify specific antigens found on the surface of tumors for the development of novel mRNA immunotherapies. To achieve this goal, myNEO will leverage its biological datasets, its integrated machine learning and bioinformatics platform to identify and validate specific antigen targets predicted to elicit a strong immune response. Under the R&O, we aim to develop and commercialize at least two new medicinal products for the treatment of non-small cell lung cancer and melanoma (the “Main Indications”) and potentially other indications. We are required to use commercially reasonable efforts to develop at least one product for each of the Main Indications, to file marketing approval applications for such products and commercialize such products in at least one of certain countries. Under the R&O, myNEO will own all intellectual property rights generated solely by myNEO or jointly with us during the first three phases of the R&D plan (the “R&D Project IP”). We receive a non-exclusive, royalty-free, non-assignable, sublicensable, worldwide license under certain patents and know-how owned by myNEO and R&D Project IP to the extent required to perform our research and development obligations under the agreement until the completion of a certain phase of the R&D plan. We were also granted an exclusive option to acquire all of myNEO’s rights under certain R&D Project IP relating to certain target lists, which we exercised on April 12, 2023. myNEO receives a non-exclusive, royalty-free, perpetual license back to such IP to make, use or sell certain targets in the field of patient-specific vaccines. Under the R&O, myNEO agrees to work exclusively with us to develop and validate shared antigens for the Main Indications until the earlier of the date of the first phase I clinical trial for either Main Indication or 24 months after we exercised our option. On October 9, 2023, we notified myNEO that we reached a research and development milestone where we selected four antigens for further development, which we intend to use in a clinical candidate for the indication head and neck squamous-cell carcinoma, which is an indication other than the Main Indication.
Under the R&O, we paid myNEO an upfront one-time technology access fee of €138,000 and myNEO is eligible to receive up to €17.5 million in research and development milestone payments with respect to the Main Indications, up to €175,000 in research and development milestone payments with respect to indications other than the Main Indications, up to €30 million in commercial milestone payments with respect to the Main Indications and up to €7.5 million in commercial milestone payments with respect to indications other than the Main Indications, as well as low single-digit percentage royalties on the net sales of licensed products in the Main Indications and sub single-digit percentage royalties on the net sales of licensed products for indications other than the Main Indications. Our royalty obligations continue on a product-by-product and country-by-country basis until the earlier of the date when there are no valid patent claims covering such licensed product in such country and 10 years following the date of first commercial sale of such licensed product in such country.
Advance Purchase Agreement for our First-Generation COVID-19 Vaccine Candidate
On November 30, 2020, we entered into an APA with the EC, acting on behalf and in the name of all Member States of the European Union, which provided for the advance purchase by the Member States of 225 million doses of the vaccine to be allocated among the Member States and the option to purchase up to an additional 180 million doses. Pursuant to the APA, we received an upfront payment of €450 million. Such upfront payment had to be used solely for the development and commercial supply of CVnCoV. We are required to return any unspent amounts of the upfront payment if, among others, we fail to successfully develop CVnCoV or if we successfully develop CVnCoV, but we do not receive EU marketing authorization or fail to supply any doses of CVnCoV to any of the Member States by late 2021, unless we and the EC mutually agree to a later date. In October 2021, we notified the EC of the withdrawal of our regulatory approval application for CVnCoV, which notification automatically terminated the APA. According to the APA, in such case of termination, CureVac would only return the unspent amount of the upfront payment. In the context of the APA, “spent” means either costs incurred or commitments made in connection with the purposes set forth in the APA. On March 8, 2022, we received a letter signed by the EC acknowledging and outlining that we will not be required to return any portion of the upfront payment. Due to the termination of the APA, we will not receive any further payments related to the APA.
In other respects, upon the EC’s request, we will transfer any raw materials and/or primary components paid for with the upfront payment that were not used as of the termination date. Additionally, should the EC request, or should we successfully sell, any raw materials and/or primary components, then an applicable portion of such raw materials, primary components or proceeds, as the case may be, will be remitted to the EC. This repayment agreement expired at the end of 2022 and an amount of €4.1 million was paid to the EC as of December 31, 2023.
161
Acquisition of Frame Pharmaceuticals
On June 8, 2022, we entered into a Share Purchase Agreement (“SPA”), to acquire all of the issued and outstanding shares of Frame Pharmaceuticals B.V., domiciled in Amsterdam, the Netherlands, a private company with limited liability (besloten vennootschap met beperkte aansprakelijkheid), organized and existing under the laws of the Netherlands, focused on advanced genomics and bioinformatics to identify both unique and shared neoantigens across different cancer types. Under the SPA, the total consideration for the purchase was €34 million, conditioned on certain development milestone payments, as described therein. This acquisition serves to complement and strengthen our discovery capabilities to identify and validate promising neoantigens for our mRNA cancer vaccine programs and could strongly increase the likelihood of developing highly effective cancer vaccines for patients.
Results of Operations
Year Ended December 31, 2022 Compared to Year Ended December 31, 2023
We have based the following discussion of our financial condition and results of operations on our audited consolidated financial statements as of and for the years ended December 31, 2022 and 2023, and the notes thereto, included elsewhere in this Annual Report and which have been retrospectively adjusted to reflect the impact of the share split resulting from the Corporate Reorganization.
The following is a discussion of our consolidated results of operations for each of the years ended December 31, 2022 and December 31, 2023. This information is derived from our accompanying consolidated financial statements prepared in accordance with IFRS as issued by IASB.
The following table summarizes our results of operations for the fiscal year ended December 31, 2022 and 2023:
| For Years Ended | |||
December 31, | ||||
2022 | 2023 | |||
(in thousands of euros, except | ||||
per share data) | ||||
Statement of Operations and Comprehensive Income (Loss) Data: | ||||
Revenue |
| 67,420 |
| 53,758 |
Cost of sales |
| (183,993) |
| (124,366) |
Selling and distribution expenses |
| (2,817) |
| (3,912) |
Research and development expenses |
| (62,550) |
| (115,724) |
General and administrative expenses |
| (104,178) |
| (91,758) |
Income from release of governmental contract liabilities |
| — |
| — |
Other operating income |
| 37,932 |
| 9,151 |
Other operating expenses |
| (1,271) |
| (1,356) |
Operating loss |
| (249,457) |
| (274,207) |
Finance income |
| 4,009 |
| 16,731 |
Finance expenses |
| (3,707) |
| (2,493) |
Loss before income tax |
| (249,155) |
| (259,969) |
Income tax benefit |
| 126 |
| (198) |
Net loss |
| (249,029) |
| (260,167) |
Other comprehensive income/loss: |
|
|
|
|
Items that may be subsequently reclassified to profit or loss |
|
|
|
|
Foreign currency adjustments |
| (105) |
| 72 |
Total comprehensive loss |
| (249,134) |
| (260,095) |
Net loss per share (basic and diluted) |
| (1.32) | (1.18) | |
Revenue
Revenue was €53.8 million for the year ended December 31, 2023, representing a decrease of €13.7 million, or 20%, from €67.4 million for the year ended December 31, 2022. The decrease was primarily driven by lower revenues recognized from collaborations with GSK due to changes in the timeline of certain projects. For the full year ended December 31, 2023 and 2022, €47.1 million and €62.3 million, respectively, in revenue was recognized through the collaborations with GSK.
162
Cost of Sales
Cost of sales was €124.4 million for the year ended December 31, 2023, representing a decrease of €59.6 million, or 32%, from €184.0 million for the year ended December 31, 2022. The decrease was primarily attributable to decrease in write-offs of raw materials, which were in prior periods procured for manufacturing products to sell to GSK and due to a change of contract termination provisions. Additionally, prior year period was impacted by a higher impairment of CMO equipment identified for sale. Personnel expenses increased mainly due to increased workforce. We recognize in Cost of Sales costs related to all R&D-services that we provide to GSK and all other collaboration partners as well as costs from set-up and quality assurance activities for our production processes, including those relating to pharmaceutical products which are under development in our collaboration agreements and for which we have not yet generated revenues.
| For the Years Ended | |||
December 31, | ||||
2022 | 2023 | |||
(in thousands of euros) | ||||
Personnel |
| (27,185) |
| (37,734) |
Materials |
| (88,891) |
| (59,641) |
Third-party services |
| (32,331) |
| (9,398) |
Maintenance and lease |
| (2,425) |
| (2,672) |
Amortization and depreciation |
| (6,295) |
| (4,850) |
Impairment of equipment |
| (24,948) |
| (8,085) |
Other |
| (1,918) |
| (1,986) |
Total |
| (183,993) |
| (124,366) |
Selling and Distribution Expenses
Selling and distribution expenses were €3.9 million for the year ended December 31, 2023, representing an increase of €1.1 million, or 39%, from €2.8 million in the year ended December 31, 2022. The increase was primarily attributable to higher personnel expenses related to the satellite offices in Belgium and the United States.
| For the Years Ended | |||
| December 31, | |||
2022 | 2023 | |||
(in thousands of euros) | ||||
Personnel | (2,029) |
| (3,425) | |
Maintenance and lease | (35) |
| (45) | |
Amortization and depreciation | (336) |
| (18) | |
Other | (417) |
| (424) | |
Total | (2,817) |
| (3,912) | |
163
Research and Development Expenses
Research and development costs were €115.7 million for the year ended December 31, 2023, representing an increase of €53.1 million, or 85%, from €62.6 million in the year ended December 31, 2022. The increase was primarily attributable to prior year impacts caused by (i) the reversal of provision for onerous contracts in the amount of €38.5 million as a result of more participants leaving the clinical trials, prior to completion, than originally estimated; and (ii) renegotiations of contracts with CROs. Additionally, in the prior year period, GSK took over the Group’s committed capacity at a CMO which resulted in a reduction in the estimated contract termination provisions in the amount of €25.1 million The net effect of these two events resulted in an overall gain within the Third-party services category. Personnel expenses increased mainly due to increased workforce and the acquisition of Frame Pharmaceuticals. Additionally, share-based payment expense was higher compared to prior year period. Materials decreased due to prior years impact of further write-off and scrap related to our first generation COVID-program, CVnCoV.
| For the Years Ended | |||
December 31, | ||||
2022 | 2023 | |||
(in thousands of euros) | ||||
Materials |
| (32,982) |
| (19,126) |
Personnel |
| (33,944) |
| (43,267) |
Amortization and depreciation |
| (8,650) |
| (8,539) |
Patents and fees to register a legal right |
| (3,813) |
| (6,666) |
Third-party services |
| 20,499 |
| (28,587) |
Maintenance and lease |
| (1,069) |
| (7,287) |
Other |
| (2,591) |
| (2,252) |
Total |
| (62,550) |
| (115,724) |
The following table reflects our research and development costs for each of our programs for the year ended December 31, 2022 and 2023:
| For the Years Ended | |||
December 31, | ||||
2022 | 2023 | |||
(in thousands of euros) | ||||
Key Programs (CV8102, CVGBM, SARS-CoV-2 and CVnCoV) | ||||
CV8102 |
| (2,781) |
| (1,773) |
CVGBM |
| (3,062) |
| (4,821) |
Second Generation COVID (CV0601, CV0701 and CV0501) |
| (24,983) |
| (9,233) |
CVnCoV |
| 39,458 |
| 4,002 |
Other Research and Development Programs |
| (11,106) |
| (19,848) |
Unallocated costs(1) |
| (60,076) |
| (84,051) |
Total |
| (62,550) |
| (115,724) |
| (1) | Unallocated costs primarily consist of costs associated with personnel expenses, patents and fees to register a legal right, amortization and depreciation, maintenance and lease expenses, certain third-party service expenses and certain material expenses. |
We expect that our research and development expenses will constitute the most substantial part of our expenses in future periods in line with the advance and expansion of the development of our product candidates.
Considering that, our research and development expenses primarily relate to the following key programs:
| ● | For SARS-CoV-2, we are developing our modified mRNA vaccine candidates CV0601 (monovalent) and CV0701 (bivalent) in collaboration with GSK. While CV0601, encodes the Omicron BA.4-5 variant; CV0701, encodes the Omicron BA.4-5 variant as well as the original SARS-CoV-2 virus. Both candidates are currently tested in a Phase 2 study, comparing both candidates to a licensed bivalent mRNA-based COVID-19 comparator vaccine initiated on August 1, 2023. Positive data from a formal interim analysis were announced on January 5, 2024. Both vaccine candidates, CV0601 and CV0701, apply CureVac’s proprietary second-generation mRNA backbone. |
164
| ● | In our oncology therapeutic area, we work on novel cancer vaccine candidates based on differentiated antigen discovery technologies and bioinformatics to target antigens that are overexpressed in tumor tissues with no or little expression in healthy tissues. Within this strategy, we follow two approaches: (1) the development of off-the-shelf cancer vaccines based on tumor antigens shared across different cancer indications and (2) the development of fully personalized cancer vaccines based on a patient’s individual tumor genomic profile. We plan to advance new antigens for both approaches based on our second-generation mRNA backbone. |
| ● | Our oncology program, CVGBM, is a single mRNA construct based on our second-generation mRNA backbone, encoding eight epitopes from known tumor associated antigens with demonstrated relevance in glioblastoma. It is currently being tested in a Phase 1 study to assess safety and tolerability as a monotherapy in patients with newly diagnosed and surgically resected MGMT-unmethylated glioblastoma or astrocytoma with a molecular signature of glioblastoma. The study consists of two parts, a dose-escalation part (Part A) and a dose-expansion part (Part B). In the currently ongoing Part A, patients receive a total of seven intramuscular administrations of CVGBM at escalating doses in the range of 12µg to 100µg. |
| ● | Our oncology program, CV8102, has completed a Phase 1 dose escalating clinical trial for four types of solid tumors as a monotherapy and in combination with anti-PD-1 and an expansion of the Phase 1 study to confirm the safety, tolerability and efficacy of CV8102 at a 600μg dose. In the context of our strategic focus on the development of novel mRNA-based cancer vaccines, current and upcoming clinical developments will provide the basis for any potential integration of CV8102 into this priority program as a strong immune-modulatory adjunct. Clinical development of CV8102 will only be considered in combination with a defined mRNA cancer vaccine. |
General and Administrative Expenses
General and administrative expenses were €91.8 million for the year ended December 31, 2023, representing a decrease of €12.4 million, or 12%, from €104.2 million in the year ended December 31, 2022. The decrease was primarily attributable to less personnel expenses due to lower workforce in the corporate service functions and due to lower other expenses.
| For the Years Ended | |||
December 31, | ||||
2022 | 2023 | |||
(in thousands of euros) | ||||
Personnel |
| (36,765) |
| (28,996) |
Maintenance and lease costs |
| (5,853) |
| (5,353) |
Third-party services |
| (27,669) |
| (29,920) |
Legal and other professional services |
| (10,394) |
| (10,160) |
Amortization and depreciation |
| (11,360) |
| (13,821) |
Other |
| (12,137) |
| (3,508) |
Total |
| (104,178) |
| (91,758) |
Other Operating Income
Other operating income was €9.2 million in the year ended December 31, 2023, representing a decrease of €28.8 million or 76%, from €37.9 million for the year ended December 31, 2022.
The decrease was primarily attributable to the impacts of the amendments to the 2020 GSK Agreement and the GSK COVID Agreement, under which CureVac was entitled to further compensation by GSK for set-up activities undertaken by CureVac (€20.5 million) and for reimbursement of prepayments (€12.0 million) in prior year period.
In January 2024, we have been granted the positive research allowance notice by the German Federal Ministry of Education and Research for the years 2020-2026. The Group recognized EUR €3.1 million as other operating income for the year ended December 31, 2023.
165
Other Operating Expense
Other operating expense was €1.4 million in the year ended December 31, 2023 representing an increase of €0.1 million, or 7%, from €1.3 million for the year ended December 31, 2022. Other operating expense related primarily to compensation expense of our supervisory board.
Finance Income
Finance income was €16.7 million for the year ended December 31, 2023, representing an increase of €12.7 million, or 317%, from €4.0 million for the year ended December 31, 2022. The increase was primarily attributable to interest income on cash investments.
Finance Expenses
Finance expenses were €2.5 million for the year ended December 31, 2023, representing a decrease of €1.2 million, or 33%, from €3.7 million for the year ended December 31, 2022. The decrease was mainly related to prior year impacted by negative interest on cash.
Income Tax Benefit (Expense)
An income tax expense of €0.2 million was generated for the year ended December 31, 2023, representing an increase of €0.3 million, from an income tax benefit of €0.1 million generated for the year ended December 31, 2022. The increase to an expense was primarily attributable to income tax expense of CureVac Belgium S.A. and release of DTA at CureVac Corporate Services GmbH.
Year Ended December 31, 2021 Compared to Year Ended December 31, 2022
We have based the following discussion of our financial condition and results of operations on our audited consolidated financial statements for the years ended December 31, 2021 and 2022, and the notes thereto, included elsewhere in this Annual Report and which have been retrospectively adjusted to reflect the impact of the share split resulting from the Corporate Reorganization.
The following is a discussion of our consolidated results of operations for each of the years ended December 31, 2021 and December 31, 2022. This information is derived from our accompanying consolidated financial statements prepared in accordance with IFRS as issued by IASB.
166
The following table summarizes our results of operations for the fiscal year ended December 31, 2021 and 2022:
For Years Ended | ||||
December 31, | ||||
| 2021 |
| 2022 | |
(in thousands of euros, except | ||||
per share data) | ||||
Statement of Operations and Comprehensive Income (Loss) Data: |
| |||
Revenue |
| 102,990 |
| 67,420 |
Cost of sales |
| (238,195) |
| (183,993) |
Selling and distribution expenses |
| (1,743) |
| (2,817) |
Research and development expenses |
| (815,907) |
| (62,550) |
General and administrative expenses |
| (100,402) |
| (104,178) |
Income from release of governmental contract liabilities | 574,502 | — | ||
Other operating income |
| 67,702 |
| 37,932 |
Other operating expenses |
| (1,210) |
| (1,271) |
Operating loss |
| (412,263) |
| (249,457) |
Finance income |
| 10,103 |
| 4,009 |
Finance expenses |
| (10,338) |
| (3,707) |
Loss before income tax |
| (412,498) |
| (249,155) |
Income tax benefit |
| 782 |
| 126 |
Net loss |
| (411,716) |
| (249,029) |
Other comprehensive income/loss: |
|
| ||
Items that may be subsequently reclassified to profit or loss |
|
| ||
Foreign currency adjustments |
| (91) |
| (105) |
Total comprehensive loss |
| (411,807) |
| (249,134) |
Net loss per share (basic and diluted) |
| (2.21) |
| (1.32) |
Revenue
Revenue was €67.4 million for the year ended December 31, 2022, representing a decrease of €35.6 million, or 35%, from €103.0 million for the year ended December 31, 2021. The decrease was primarily driven by the termination of the Boehringer Agreement in 2021, which led to a recognition of the remaining contract liability, option fee payment and development milestone in revenue amounting to €26.0 million for the full year ended December 31, 2021. For the full year ended December 31, 2022 and 2021, €62.3 million and €74.3 million, respectively, in revenue was recognized under the collaboration agreements with GSK, for both the 2020 GSK Agreement and the GSK COVID Agreement.
167
Cost of Sales
Cost of sales was €184.0 million for the year ended December 31, 2022, representing a decrease of €54.2 million, or 23%, from €238.2 million for the year ended December 31, 2021. The decrease was primarily attributable to less expenses on CMO services (fiscal year 2021 was highly impacted by significant expenses for the set-up of a CMO network), partially offset by increased write-offs and scrap of raw materials amounting to €80.0 million in 2022, which were procured for manufacturing of products to sell to GSK that are no longer expected to be sold to them or were determined to be excess materials on hand. We recognize in Cost of Sales costs related to all R&D-services that we provide to GSK and all other collaboration partners as well as costs from set-up and quality assurance activities for our production processes, including those relating to pharmaceutical products which are under development in our collaboration agreements and for which we have not yet generated revenues.
For the Years Ended | ||||
December 31, | ||||
| 2021 |
| 2022 | |
(in thousands of euros) | ||||
Personnel |
| (22,159) |
| (27,185) |
Materials |
| (46,250) |
| (88,891) |
Third-party services |
| (139,975) |
| (32,331) |
Maintenance and lease |
| (2,874) |
| (2,425) |
Amortization, depreciation and derecognition |
| (3,992) |
| (6,295) |
Impairment of equipment | (22,810) | (24,948) | ||
Other |
| (135) |
| (1,918) |
Total |
| (238,195) |
| (183,993) |
Selling and Distribution Expenses
Selling and distribution expenses were €2.8 million for the year ended December 31, 2022, representing an increase of €1.1 million, or 65%, from €1.7 million in the year ended December 31, 2021. The increase was primarily attributable to higher personnel expenses due to an increase in headcount.
For the Years Ended | ||||
December 31, | ||||
| 2021 |
| 2022 | |
(in thousands of euros) | ||||
Personnel |
| (1,369) |
| (2,029) |
Amortization and depreciation |
| (86) |
| (371) |
Other |
| (288) |
| (417) |
Total |
| (1,743) |
| (2,817) |
168
Research and Development Expenses
Research and development costs were €62.6 million for the year ended December 31, 2022, representing a decrease of €753.3 million, or 92%, from €815.9 million in the year ended December 31, 2021. The decrease was primarily attributable to significantly lower research and development costs. Fiscal year 2021 was highly impacted from the start of our Phase 2/3 clinical trial for CVnCoV. As of December 2021, we had recognized a provision for all remaining costs related to the CVnCoV clinical trials. During 2022, we were able to renegotiate the existing contracts such that our estimated remaining costs decreased, and more participants exited the trials earlier than originally estimated. As a result of these changes, we recognized a gain from the reversal of €38.5 million of the provision. Additionally, during this period, we recognized a net gain of €25.1 million for a change of estimate in the contract termination provisions as a result of GSK taking over our committed capacity at a CMO. As a result of these gains, we experienced a net gain in third-party services costs.
For the Years Ended | ||||
December 31, | ||||
| 2021 |
| 2022 | |
(in thousands of euros) | ||||
Materials |
| (232,292) |
| (32,982) |
Personnel |
| (33,733) |
| (33,944) |
Amortization and depreciation |
| (4,259) |
| (8,650) |
Patents and fees to register a legal right |
| (11,157) |
| (3,813) |
Third-party services |
| (531,827) |
| 20,499 |
Maintenance and lease |
| (347) |
| (1,069) |
Other |
| (2,292) |
| (2,591) |
Total |
| (815,907) |
| (62,550) |
The following table reflects our research and development costs for each of our programs for the year ended December 31, 2021 and 2022:
For the Years Ended | ||||
December 31, | ||||
| 2021 |
| 2022 | |
(in thousands of euros) | ||||
Key Programs (CV8102, CV7202, CV2CoV and CVnCoV) |
|
| ||
CV8102 |
| (6,591) |
| (2,781) |
CV7202 |
| (518) |
| (219) |
CV2CoV | (5,782) | (24,983) | ||
CVnCoV |
| (753,627) |
| 39,458 |
Other Research and Development Programs |
| (4,610) |
| (13,949) |
Unallocated costs(1) |
| (44,779) |
| (60,076) |
Total |
| (815,907) |
| (62,550) |
(1) | Unallocated costs primarily consist of costs associated with personnel expenses, patents and fees to register a legal right, amortization and depreciation, maintenance and lease expenses, certain third-party service expenses and certain material expenses. |
169
We expect that our research and development expenses will constitute the most substantial part of our expenses in future periods in line with the advance and expansion of the development of our product candidates. Considering that, our research and development expenses primarily relate to the following key programs:
| ● | Our modified mRNA vaccine candidate against SARS-CoV-2, CV0501, a monovalent construct, developed in collaboration with GSK. A Phase 1 study was initiated in August 2022. We reported positive preliminary data in early 2023. A Phase 2 clinical study, expected to start later in 2023, will assess monovalent and/or bivalent vaccine candidates designed to target clinically relevant variants. Novel cancer vaccine candidates based on differentiated antigen discovery technologies and bioinformatics to target antigens that are overexpressed in tumor tissues with no or little expression on healthy tissues. Within this strategy, we follow two approaches. The first approach assesses tumor antigens shared by different cancer patients for the development of off-the-shelf cancer vaccines. The second approach is tailored to the individual tumor setup of a patient for personalized therapy. We plan to advance new antigens for both approaches based on our second-generation mRNA backbone. To assess the safety and immunogenicity of our second-generation backbone in an oncology setting, we expect to initiate a proof-of-principle study in the second quarter of 2023, assessing an mRNA construct encoding eight epitopes from tumor associated antigens in patients with surgically resected Glioblastoma Multiforme. |
| ● | Our oncology program, CV8102, which is currently in a Phase 1 dose escalating clinical trial for four types of solid tumors as a monotherapy and in combination with anti-PD1 and an expansion of the Phase 1 study to evaluate the safety, tolerability and efficacy of CV8102 at a 600μg dose in patients with PD-1 refractory melanoma – also as a monotherapy and in combinations with anti-PD-1 antibodies. |
| ● | Our vaccine program, CV7202, which is currently in a Phase 1 clinical trial as a vaccine candidate for rabies. |
General and Administrative Expenses
General and administrative expenses were €104.2 million for the year ended December 31, 2022, representing an increase of €3.8 million, or 4%, from €100.4 million in the year ended December 31, 2021. The increase was primarily attributable to increased amortization and depreciation expenses due to increased depreciation expense of right-of-use assets.
For the Years Ended | ||||
December 31, | ||||
| 2021 |
| 2022 | |
(in thousands of euros) | ||||
Personnel |
| (37,393) |
| (36,765) |
Maintenance and lease costs |
| (4,306) |
| (5,853) |
Third-party services |
| (28,875) |
| (27,669) |
Legal and other professional services |
| (9,230) |
| (10,394) |
Amortization and depreciation |
| (8,895) |
| (11,360) |
Other |
| (11,703) |
| (12,137) |
Total |
| (100,402) |
| (104,178) |
Income from release of governmental contract liabilities
Due to the withdrawal of the EMA regulatory approval application for CVnCoV in October 2021, CureVac recorded in 2021 an “Income from release of governmental contract liabilities” amounting to €574.5 million (refer to the “Year Ended December 31, 2020 Compared to Year Ended December 31, 2021” section for additional information regarding this prior year event). There was no such event occurring in 2022.
Other Operating Income
Other operating income was €37.9 million in the year ended December 31, 2022, representing a decrease of €29.8 million or 44%, from €67.7 million for the year ended December 31, 2021.
170
In March 2022, CureVac SE (as the surviving entity from the merger of CureVac AG and CureVac Beteiligungsverwaltungs AG) and GlaxoSmithKline Biologicals SA amended and restated the 2020 GSK Agreement and the GSK COVID Agreement in connection with GSK entering into a direct Agreement with Novartis relating to the use of Novartis as CMO at the same time as CureVac exits its CMO agreement with Novartis and is released from its pre-existing capacity commitments under the agreement. As a result, the Company avoided an outflow of resources. Additionally, under the restated agreement, CureVac was entitled to further compensation by GSK. The compensation mainly consists of consideration for set-up activities undertaken by CureVac (€20.5 million) and for reimbursement of prepayments (€12.0 million).
In 2021 the other operating income was primarily attributable to amounts recognized from grants from government agencies and similar bodies, primarily the German Federal Ministry of Education and Research, or BMBF.
Other Operating Expense
Other operating expense was €1.3 million in the year ended December 31, 2022 representing an increase of €0.1 million, or 8%, from €1.2 million for the year ended December 31, 2021. Other operating expense related primarily to compensation expense of our supervisory board.
Finance Income
Finance income was €4.0 million for the year ended December 31, 2022, representing an decrease of €6.1 million, or 60%, from €10.1 million for the year ended December 31, 2021. The decrease was mainly attributable to less foreign exchange gains partially compensated by positive interest on cash investments.
Finance Expenses
Finance expenses were €3.7 million for the year ended December 31, 2022, representing an decrease of €6.6 million, or 64%, from €10.3 million for the year ended December 31, 2021. The decrease was mainly related to less foreign exchange losses and less negative interest on cash.
Income Tax Benefit (Expense)
An income tax benefit of €0.1 million for the year ended December 31, 2022, representing a decrease of €0.7 million, or 88%, from €0.8 million for the year ended December 31, 2021. The change was primarily attributable to decreased deferred tax benefits.
B. Liquidity and Capital
Resources
Overview
Since inception, we have incurred significant operating losses. For the year ended December 31, 2022 and 2023, we incurred net losses of €249.0 million and €260.2 million, respectively. To date, we have financed our operations primarily through the IPO in August 2020, follow-on public offerings, private placements of equity securities, issuance of convertible debt, grants from government agencies and similar bodies and payments for collaborative research and development services. Our cash and cash equivalents as of December 31, 2023, were €402.5 million. Our primary cash needs are to fund our non-clinical and clinical development programs, for working capital requirements and for capital expenditures. The expected cost of completion for GMP IV is €171.1 million, and as of December 31, 2023, we have spent €170.6 million on completing the GMP IV facility. We believe that our existing cash, cash equivalents and short-term investments will enable us to fund our operating expenses and capital expenditure requirements at least through the fourth quarter of 2025. We have based this estimate on assumptions that may prove to be wrong, and we could exhaust our available capital resources sooner than we expect.
In February 2023, we sold an additional 27,027,028 common shares in an underwritten public offering at an offering price of $9.25 per share raising $234.2 million in net proceeds, after deducting underwriting discounts and commissions and offering expenses payable by us.
171
In September 2021, we entered into a sales agreement, the Open Sale Agreement, with Jefferies LLC and SVB Leerink LLC, as sales agents, to establish an at-the-market offering (ATM) program, pursuant to which we may sell, from time to time, ordinary shares for aggregate gross proceeds of up to $600 million. As of December 31, 2023, we had issued 8,656,711 common shares through the ATM program, resulting in $87 million in gross proceeds to us. Following these issuances, the remaining value authorized for sale under the at-the-market program is $513 million.
Our financial condition and liquidity is and will continue to be influenced by a variety of factors, including our ability to generate cash flows from our operations, future indebtedness and the interest we are obligated to pay on this indebtedness, the availability of public and private debt and equity financing, changes in exchange rates which will impact our generation of cash flows from operations when measured in euros and our capital expenditure requirements, which are described in more detail at “Item 3. Key Information — D. Property, Plant and Equipment.”
The following table summarizes our contractual obligations as of December 31, 2023, and the effects, including estimated interest payments, that such obligations are expected to have on our liquidity and cash flows in future periods:
Total | ||||||||||||||
| (in thousands of euros) |
| 2024 |
| 2025 |
| 2026 |
| 2027 |
| 2028 |
| Thereafter | |
Contractual commitments |
| 3,269 |
| 3,269 |
| — |
| — |
| — |
| — |
| — |
Lease liabilities |
| 53,656 |
| 7,254 |
| 7,096 |
| 5,977 |
| 5,633 |
| 4,875 |
| 22,821 |
Total |
| 56,925 |
| 10,523 |
| 7,096 |
| 5,977 |
| 5,633 |
| 4,875 |
| 22,821 |
We have entered into various agreements with collaborators, including licensing agreements. These agreements provide for us to make milestone and royalty payments that are conditional on the achievement of certain development, regulatory and commercial milestones and certain of these agreements provide us an option to obtain further licenses which could additionally require us to make such milestone and royalty payments. As of December 31, 2023, the aggregate amount of such potential milestone payments, including those relating to licenses acquired from exercised options, under all such collaboration agreements, was up to $192.4 million. The timing of these payments, and whether they become due, is conditional on achieving the applicable milestones.
We have not included milestone or royalty payments or other contractual payment obligations in the table above if the timing and amount of such obligations are unknown or uncertain.
European Investment Bank Loan
In June 2020, we signed a financing arrangement with the European Investment Bank, or EIB, under which EIB agreed to provide us with a line of credit in an amount of up to €75 million for the partial financing of our clinical developments and large-scale production of our infectious diseases vaccine candidates including our vaccine against SARS-CoV-2, or the Investment, provided that the amount of financing does not exceed 50% of the cost of the Investment.
As of December 31, 2020, we had drawn €25 million under the first of the three tranches. In November 2020, a land charge (mortgage) amounting to €75 million was registered in favor of the EIB to secure the loan.
In November 2021, we issued a prepayment request and cancellation notice to the EIB, pursuant to which we voluntarily repaid the €25 million in principal in addition to accrued interest in December 2021 and canceled the remaining €50 million available under the EIB loan. The mortgage was deleted from the land register in 2022.
172
BMBF Grant
We received from BMBF a grant to support the development of our first-generation COVID-19 vaccine candidate, CVnCoV of up to €252 million. In July 2020, we had applied to that grant as part of a special program to accelerate the research and development of urgently needed vaccines against SARS-CoV-2. In addition to the further development of our first-generation COVID-19 vaccine candidate, CVnCoV, against COVID-19, the grant was used for the rapid expansion of the vaccine production. Payments were contingent on reaching predefined milestones. Amounts incurred in 2020 and 2021 were eligible for reimbursement through the grant. Due to the withdrawal of the regulatory approval application for CVnCoV, we were not able to reach all predefined milestones for 2021 under the grant. We received funding of €103 million in 2020 and funding of €93 million in 2021. As of December 31, 2021, we had drawn €196 million of the grant. In November 2021, we notified BMG of our inability to supply CVnCoV, thereby triggering automatic termination of the supply arrangement.
Advance Purchase Agreement for our First-Generation COVID-19 Vaccine Candidate
On November 30, 2020, we entered into an APA with the EC, acting on behalf and in the name of all Member States of the European Union, which provides for the advance purchase by the Member States of 225 million doses of the vaccine to be allocated among the Member States, and the option to purchase up to an additional 180 million doses. Pursuant to the APA, we received an upfront payment of €450 million. Such upfront payment had to be used solely for the development and commercial supply of CVnCoV. We are required to return any unspent amounts of the upfront payment if, among others, we fail to successfully develop CVnCoV or if we successfully develop CVnCoV, but we do not receive EU marketing authorization or fail to supply any doses of CVnCoV to any of the Member States by late 2021, unless we and the EC mutually agree to a later date. In October 2021, we notified the EC of the withdrawal of our regulatory approval application for CVnCoV, which notification automatically terminated the APA. According to the APA, in such case of termination, we would only return the unspent amount of the upfront payment. In the context of the APA, “spent” means either costs occurred, or commitments made in relation to the purpose as set out in the APA. We were able to demonstrate that the upfront payment was used in accordance with the contract and no repayment was required.
Comparative Cash Flows
Comparison of the years ended December 31, 2022 and 2023
For the Year Ended | ||||
December 31, | ||||
| 2022 |
| 2023 | |
(in thousands of euros) | ||||
Net cash flow provided by (used in): | ||||
Operating activities |
| (286,177) |
| (267,887) |
Investing activities |
| (93,499) |
| (55,200) |
Financing activities |
| 63,173 |
| 230,893 |
Effect of currency translation gains on cash and cash equivalents |
| 836 |
| (1,151) |
Overall cash inflow/(outflow) |
| (315,667) |
| (93,345) |
Operating Activities
Net cash used in operating activities for the year ended December 31, 2023, was €267.9 million as compared to net cash used by operating activities of €286.2 million for the year ended December 31, 2022. The decrease in net cash used in operating activities was primarily attributable to less payments for service agreements with contract manufacturing organizations and raw material suppliers.
Investing Activities
Net cash used in investing activities for the year ended December 31, 2023, was €55.2 million as compared to net cash used in investing activities of €93.5 million for the year ended December 31, 2022. The change in cash flows used in investing activities was primarily attributable to decreased purchases of property, plant and equipment for manufacturing facilities and intangible assets.
173
Financing Activities
Net cash provided by financing activities was €230.9 million for the year ended December 31, 2023, as compared to €63.2 million for the year ended December 31, 2022. The increase in cash flows provided by financing activities was mainly attributable to the raising of cash in the follow-on underwritten public offering conducted in February 2023.
Comparison of the years ended December 31, 2021 and 2022
For the Year Ended | ||||
December 31, | ||||
| 2021 |
| 2022 | |
(in thousands of euros) | ||||
Net cash flow provided by (used in): | ||||
Operating activities |
| (733,128) |
| (286,177) |
Investing activities |
| (127,901) |
| (93,499) |
Financing activities |
| 344,964 |
| 63,173 |
Effect of currency translation gains on cash and cash equivalents |
| 4,936 |
| 836 |
Overall cash inflow/(outflow) |
| (511,129) |
| (315,667) |
Operating Activities
Net cash used in operating activities for the year ended December 31, 2022, was €286.2 million as compared to net cash provided by operating activities of €733.1 million for the year ended December 31, 2021. The decrease in net cash used in operating activities was primarily attributable to less payments for service agreements with contract research organizations and contract manufacturing organizations, including related settlements which were driving the cash outflows in the year ended December 31, 2021.
Investing Activities
Net cash used in investing activities for the year ended December 31, 2022, was €93.5 million as compared to net cash used in investing activities of €127.9 million for the year ended December 31, 2021. The change in cash flows used in investing activities was primarily attributable to decreased purchases of property, plant and equipment for use in contract manufacturing facilities.
Financing Activities
Net cash provided by financing activities was €63.2 million for the year ended December 31, 2022, as compared to €345.0 million for the year ended December 31, 2021. The decrease in cash flows provided by financing activities was mainly attributable to the proceeds in 2022 from the at-the-market offering program being lesser than those from the follow-on public offering, which closed in February 2021.
C. Research and Development, Patents and Licenses, etc.
Research and development expenses consist primarily of costs incurred for our research and preclinical and clinical development activities, including our product discovery efforts and certain activities relating to the design of GMP-manufacturing facilities. Research and development expenses contain wages and salaries, share-based compensation, fringe benefits and other personnel costs, the costs of clinical testing and the associated clinical production costs, research material production costs, fees for contractual partners, consultants and other third parties, fees to register legal rights, amortization of licensed software and intellectual property as well as costs for plant and facilities. Research and development expenses contain costs for independent research and development work as well as work carried out in the context of collaboration and licensing agreements; such expenses include all costs related to research and development services delivered under our collaboration arrangements. Additionally, prior to initial regulatory approval, if any, costs relating to production of products are expensed as research and development expenses in the period incurred. If pre-launch products are sold, the respective product gross margin may be higher compared to the expected recurring margin as the underlying costs will not be included in cost of sales as they will have been recognized in research and development expense in the period incurred.
174
We also have partnered programs as further described under “Item 4. Information on the Company — B. Business Overview — Collaborations” and “Item 4. Information on the Company — B. Business Overview — Advance Purchase Agreements,” for which we incur additional expenses. In addition, our research and development expenses relate to our preclinical studies of further product candidates and discovery activities. These expenses mainly consist of salaries, share based-compensation, costs for production of preclinical compounds and costs paid to contract research organizations.
We expense research and development expenses as incurred. We recognize costs for certain development activities, such as preclinical studies and clinical trials, based on an evaluation of the progress to completion of specific tasks. We use information provided to us by our vendors such as patient enrollment or clinical site activations for services received and efforts expended. We expect research and development costs, including manufacturing, to support these activities, to increase significantly for the foreseeable future as our current development programs progress and new programs are added.
D. Trend Information
For a discussion of trend information, see “Item 5. Operating and Financial Review and Prospects.”
E. Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements that have or are reasonably likely to have a current or future effect on our financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources, except for those noncancellable contractual obligations from certain of our arrangements with contract manufacturing organizations disclosed in “Item 5. Operating and Financial Review and Prospects — B. Liquidity and Capital Resources.”
F. Safe Harbor
See “Forward-Looking Statements.”
G. Critical Accounting Policies and Estimates
Our consolidated financial statements are prepared in accordance with IFRS as issued by the IASB. Some of the accounting methods and policies used in preparing the financial statements under IFRS are based on complex and subjective assessments by our management or on estimates based on past experience and assumptions deemed realistic and reasonable based on the circumstances concerned. The actual value of our assets, liabilities and shareholders’ equity and of our earnings could differ from the value derived from these estimates if conditions changed and these changes had an impact on the assumptions adopted.
Our significant accounting policies that we believe to be critical to the judgments and estimates used in the preparation of our financial statements are included in “note 2 — Significant accounting policies” and “note 10 — Share-based payments” to our consolidated financial statements included elsewhere in this Annual Report.
ITEM 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
A. Directors and Senior Management
Board Structure
We have a two-tier board structure consisting of a management board (bestuur) and a separate supervisory board (raad van commissarissen). There are no family relationships among any of our managing directors and supervisory directors.
175
Management Board
Our management board is composed of four members, who we refer to as our managing directors and who we consider to be our executive officers. Each managing director of CureVac N.V. holds office for the term set by our general meeting (as set forth in the table below), except in the case of his or her earlier death, resignation or removal. Our managing directors do not have a retirement age requirement under our articles of association.
Our managing directors are responsible for the management and representation of our company. Our senior management has an average of 25 years of experience in the biopharmaceutical industry.
The following table lists our current managing directors who are also executive officers, as well as their ages, term served, the year of expiration of their term as managing directors of CureVac N.V. and position:
Year in which | ||||||||
Name |
| Age |
| Term Served |
| Term Expires |
| Position |
Alexander Zehnder, MD, MBA(1) |
| 54 |
| 4/2023 – Present |
| 2026 |
| Chief Executive Officer |
Pierre Kemula, B.Sc. |
| 50 |
| 11/2016 – Present |
| 2024(3) |
| Chief Financial Officer |
Myriam Mendila, MD(2) | 58 | 2/2023 – Present | 2026 | Chief Development Officer | ||||
Malte Greune, Ph.D | 59 | 7/2021 – Present | 2024 | Chief Operating Officer |
| (1) | Dr. Zehnder was formally appointed as a managing director of CureVac N.V. on March 28, 2023. |
| (2) | Dr. Mendila joined the executive team as Chief Development Officer on February 1, 2023. She was formally appointed as a managing director of CureVac N.V. on March 28, 2023. |
| (3) | Following the lapse of his term in office as managing director on October 31, 2023 and with effect from November 1, 2023, Pierre Kemula was appointed as a managing director in the event of absence or inability to act of a managing director until the end of the AGM 2024 and his date of initial appointment includes the term of office served as managing director of CureVac SE (at that time named: CureVac AG). |
The above table does not include Dr. Antony Blanc and Dr. Igor Splawski, who each resigned as a managing director of CureVac N.V. on November 30, 2023 and July 14, 2023, respectively.
The following is a brief summary of the prior business experience and principal business activities performed outside of CureVac of our managing directors. Unless otherwise indicated, the current business addresses for each managing director is Friedrich-Miescher-Strasse 15, 72076 Tübingen, Germany.
Alexander Zehnder, MD, MBA became our chief executive officer in April 2023. Dr. Zehnder earned his degree as a Medical Doctor from the University of Bern, Switzerland, and completed an MBA at IMD Business School in Lausanne, Switzerland. He has held roles of increasing complexity and responsibility in the pharmaceutical industry for more than 20 years, across multiple business units and functional areas in Europe, the United States and Japan. Prior to joining CureVac, Dr. Zehnder was the Global Head of Oncology at Sanofi and previously held leadership positions at Sanofi at both the headquarter and country level since 2014. Prior to Sanofi, Dr. Zehnder worked at Roche/Genentech where he served as Vice President, Global Product Strategy and Global Franchise Head Avastin, the company’s blockbuster oncology drug.
Pierre Kemula, B.Sc. has been our chief financial officer since 2016. Previously, he was the chief financial officer of Pixium Vision from 2014 until 2016, where he successfully contributed to the listing of the company on Euronext in Paris, and Vice President of Corporate Finance, Treasury and Financial Markets, as well as Director of Investor Relations, Vice President of Investor Relations and Investor Relations Officer at lpsen from 2008 until 2014. Earlier in his career, Mr. Kemula worked with major strategy consulting firms (Roland Berger, Bossard Consultants and Gemini Consulting). He holds a Bachelor of Science in Management Sciences from the London School of Economics, or LSE, in the United Kingdom.
176
Myriam Mendila, MD has been our chief development officer since February 2023. She has more than 20 years of global experience in product development, medical affairs, pharmacovigilance and healthcare compliance as well as global product strategy, including commercial strategy at Roche, Genentech and Novartis. Over the last 5 years, she has held the position of Worldwide Head of Medical Affairs and Chief Medical Officer Oncology at Novartis Pharma AG, Switzerland where she drives and oversees the development and cross-functional execution of the long-range global medical affairs vision and strategy for the Novartis oncology portfolio. Myriam earned her medical degree and subsequently doctoral degree from the Medical University of Hanover, Germany.
Malte Greune, Ph.D. has been our chief operating officer since July 2021. Dr. Greune joins CureVac from Sanofi-Aventis Deutschland GmbH, where he held various management positions for almost ten years. As General Manager and Vice President Cartridges, Devices & Insulin Technology Group, he was responsible for several manufacturing sites in Frankfurt. Under his leadership, six isolator filling lines for insulins, oncology drugs and biologics were set up including one for a COVID-19 vaccine. Prior to his position as Head of Diabetes, Oncology and Devices at Sanofi, he worked as the Senior Vice President of Animal Health Manufacturing for the Merck Manufacturing Division, USA, where he led an international network of 28 sites, including 18 integrated vaccine sites. Furthermore, he held various leadership roles at the pharmaceutical companies Schering-Plough and Intervet International B.V. Dr. Greune started his career at Hoechst AG in Corporate Planning. Dr. Greune received his Ph.D. in Economics from the University of Cologne, Germany, graduated from the University of Trier, Germany, and completed a Master of Business Administration at Clark University in Worcester, USA.
Supervisory Board
Our supervisory board is composed of seven current members, who we refer to as our supervisory directors. Each supervisory director holds office for the term set by our general meeting (as set forth in the table below), except in the case of his or her earlier death, resignation or removal. Our supervisory directors do not have a retirement age requirement under our articles of association.
The following table sets forth the names and functions of our current supervisory directors, their ages, term served and the year of expiration of their term as supervisory director of CureVac N.V.
Year in which | ||||||||
Name |
| Age |
| Term Served |
| Term Expires |
| Functions |
Baron Jean Stéphenne, MSc, MBA |
| 74 |
| 8/2015 – Present |
| 2024 |
| Chairman and Supervisory Director |
Mathias Hothum, Ph.D. |
| 57 |
| 8/2015 – Present |
| 2024 |
| Supervisory Director |
Craig A. Tooman, MBA |
| 58 |
| 6/2019 – Present |
| 2025 |
| Supervisory Director |
Viola Bronsema, Ph.D. |
| 61 |
| 8/2020 – Present |
| 2024 |
| Supervisory Director |
Debra Barker, MD |
| 61 |
| 6/2022 – Present |
| 2025 |
| Supervisory Director |
Klaus Schollmeier Ph.D. | 67 | 6/2022 – Present | 2025 | Supervisory Director | ||||
Michael Brosnan BSc.(1) |
| 69 |
| 6/2023 – Present |
| 2026 |
| Supervisory Director |
(1) | Mr. Brosnan joined the Supervisory Board on June 19, 2023. |
The above table does not include Dr. Hans Christoph Tanner and Dr. Ralf Clemens, who each resigned from the Supervisory Board on June 19, 2023 and September 30, 2023, respectively.
The following is a brief summary of the prior business experience and principal business activities performed outside of CureVac of our supervisory directors. Unless otherwise indicated, the current business addresses for each of our supervisory directors is Friedrich-Miescher-Strasse 15, 72076 Tübingen, Germany.
Baron Jean Stéphenne, MSc, MBA has served as a supervisory director since 2015. Mr. Stéphenne was the CEO of GSK Biologicals from 1989 until 2012 and the President of GSK Biologicals from 2002 until 2012, where he was instrumental in building one of the world’s leading vaccine companies. In 1974, Mr. Stéphenne joined SmithKline-Rit, as engineer in biology in research and development. He also served as the President of UWE (Union Wallonne des Entreprises) from 1997 until 2000. Mr. Stéphenne was the chairman of BESIX Group S.A./N.V. and TiGenix N.V., IBA Wallonia Foreign Trade and Investment Agency, Henogen S.A., Aseptic Technologies and Bone Therapeutics (now BioSenic SA). He was also a director of Fortis bank, GBL and Bone Therapeutics.
177
Mathias Hothum, Ph.D. has served as a supervisory director since 2015. Dr. Hothum is the managing director of dievini Hopp BioTech holding GmbH & Co. KG, or dievini. dievini manages the biotech investments of SAP co-founder Dietmar Hopp. For the past 25 years, Dr. Hothum has worked as a health economist in the healthcare, health services and life sciences sectors. Dr. Hothum specializes in financing, pricing, reimbursement and in the evaluation of mid-sized companies as well as of publicly owned/market-listed companies. He is the owner and founder of HMM-Consulting. Furthermore, Dr. Hothum serves as a supervisory director of a few biotech companies, including Heidelberg Pharma AG, Apogenix AG, Weinheim 216 GmbH, Novaliq GmbH, Molecular Health GmbH and Joimax GmbH. He received his Ph.D. in economics from the University of Magdeburg and degree in economics from the University of Mannheim.
Craig A. Tooman, MBA has served as a supervisory director since 2019. Mr. Tooman has experience in the biopharmaceutical industry spanning more than 30 years, including more than 15 years of such experience as the Chief Executive Officer and Chief Financial Officer at several public companies. Mr. Tooman currently serves as the President, Chief Executive Officer and as a member of the board of directors of Silence Therapeutics. He was previously the Chief Financial Officer of Silence. Prior to joining Silence, from September 2019 to January 2021, he served as CFO and COO at Vyome Therapeutics, Inc. and prior to his tenure at Vyome, from November 2013 to July 2019, Mr. Tooman served as CFO, and then subsequently as CEO and Board Director of Aratana Therapeutics, where he successfully negotiated a merger with Elanco. Before Aratana, from 2005 to 2010, Mr. Tooman served as the CFO of Enzon Pharmaceuticals until its acquisition by Sigma Tau, and prior to that led the $1.1 billion M&A initiative and integration of ILEX Oncology and Genzyme Corporation. Mr. Tooman has also held key positions at Pharmacia, and Upjohn. Mr. Tooman also serves on the Board of Directors of Ondine Biomedical Inc. Mr. Tooman earned his MBA in finance from the University of Chicago and a Bachelor of Arts degree in economics from Kalamazoo College.
Viola Bronsema, Ph.D. has served as a supervisory director since August 2020. Dr. Bronsema has been Secretary General and CEO of BIO Deutschland, Germany’s Biotechnology Industry Association, since 2006. With its 390 corporate members, the sector association represents the interests of Germany’s biotechnology industry nationally and internationally. Currently, she is member of the Advisory Boards of the German Federal Government (Bioeconomy Advisory Board, Council for technological Sovereignty), and one of the oldest German economic policy associations (WPCD e.V.). Previously, Dr. Bronsema was Head of Communications of Roche Diagnostics GmbH and of Roche Diagnostics Europe, Middle East, Africa, and before that, of Lilly Pharma Holding GmbH. She earned her Ph.D. at the Centre for Molecular Biology (ZMBH) at the University of Heidelberg, Germany.
Debra Barker, MD has served as a supervisory director since June 2022. Dr. Barker is a seasoned pharmaceutical executive with more than 25 years of experience in drug development and commercialization from Novartis, Roche, SmithKline Beecham and Knall in Europe, the Americas and Asia. She currently serves as a Non-Executive Director for two additional public companies including BergenBio ASA, a Norwegian Oncology company, and Destiny Pharma PLC, a late-stage British Anti-infective company. Previously, Dr. Barker led as Chief Medical and Development Officer at Polyphor LTD for two years. From 2006 to 2017 she held various leadership positions at Novartis, where she most recently served as Vice President, Medical Affairs Franchise Head for Ophthalmology as well as interim Franchise Head for Neuroscience. In addition, she held the role of Regional Medical Director, Asia Pacific for Novartis where she led teams to develop and execute medical communication, clinical trial and opinion leader development activities across the franchises. Dr. Barker holds a degree in pharmaceutical medicine and received a master’s degree in immunology from King’s College in London and a medical degree from Queens’ College, Cambridge, UK.
Klaus Schollmeier, Ph.D has served as a supervisory director since June 2022. Dr. Schollmeier is an advisor to the Pharma/Biotech industry. He was Chief Executive Officer of SuppreMol in Munich from 2013 until 2015, when the company was sold to Baxalta. From 2004 to 2011, he served as CEO of Santhera in Basel, and as Chairman of the Board of Santhera until 2013. Dr. Schollmeier joined Graffinity Pharmaceuticals AG in Heidelberg, Germany as CEO in 2003 and merged the company with MyoContract AG, Basel in 2004 to form Santhera. Prior to joining the biotechnology industry in 2003, he was managing director of the healthcare/biotechnology group at ING-BHF Bank for ING Group Europe. Before that, he spent 16 years in the pharmaceutical industry at BASF, Knoll and Abbott. Dr. Schollmeier is member of the board and chairman of several biotech companies including Tacalyx (Germany), Modra Pharmaceuticals (Netherlands), Affiris Pharma (Austria) and Eternygen (Germany).He holds a Ph.D. in biology from the University of Düsseldorf, Germany, and is currently an adjunct research associate professor at the Boston University Medical School, Massachusetts.
178
Michael Brosnan, BSc has served as a supervisory director since June 2023. Mr. Brosnan currently serves as chairman of the audit committee and member of the supervisory board at MorphoSys AG, a biotechnology company. He also is chairman of the audit committee and member of the supervisory board of Daimler Truck AG. Until his retirement in 2019, he was the CFO of Fresenius Medical Care (“FMC”), headquartered in Germany, a worldwide leader in dialysis products and services. FMC is dual listed in New York and Frankfurt. Prior to taking over management roles in financial departments at FMC in 2003, he was an audit partner at KPMG. Mr. Brosnan holds a degree in business administration and accounting from Northeastern University, Boston.
Diversity
The table below provides certain information regarding the diversity of our supervisory board members as of the date of this Annual Report.
Supervisory Board Diversity Matrix | ||||
Country of Principal Executive Officers | Germany | |||
Foreign Private Issuer | Yes | |||
Disclosure Prohibited under Home Country Law | Yes | |||
Total Number of Supervisory Board Members | 7 | |||
Female | Male | Non-Binary | Did not Disclose | |
Part I: Gender Identity | ||||
Supervisory Board Members | 2 | 5 | - | - |
Part II: Demographic Background | ||||
Underrepresented individual in Germany | - | |||
LGBTQ+ | - | |||
Did Not Disclose Demographic Background | - | |||
We have diversity objectives with respect to the composition of our management board and supervisory board as part of our diversity policy. We are committed to supporting, valuing and leveraging diversity and we recognize and welcome diversity with respect to gender, age, race, ethnicity, nationality, sexual orientation and other important cultural differences. In its evaluation of new candidates, the nomination and corporate governance committee may consider race or ethnicity, nationality, gender, sexual orientation, age, background, education skills and experience, as well as the restrictions, requirements and recommendations concerning those matters under applicable law, Nasdaq rules or best practice provisions of the Dutch Corporate Governance Code in relation to the full our management board, supervisory board and/or individual directors or director candidates. However, we also believe that there is a fine line between diversity and unintentional discrimination. For that reason, the importance of diversity, in and of itself, should not set aside the overriding principle that someone should be recommended, nominated and appointed for being “the right person for the job.”
Although we have not yet set specific targets with respect to certain elements of diversity (other than with regard to gender as required by section 2:166(2) of the Dutch Civil Code), we believe that it is important for the management board, supervisory board and our senior management to represent a diverse composite mix of personal backgrounds, experiences, qualifications, knowledge, abilities and viewpoints, consistent with the principles outlined above. We also seek to combine the skills and experience of long-standing members of the management board, supervisory board and our senior management with the fresh perspectives, insights, skills and experiences of new candidates from time to time. To further increase the range of viewpoints, perspectives, talents and experience, we strive for a mix of experience among the management board, supervisory board and its senior management, but we have not set a specific target in this respect.
Three of our managing directors are male and one is female. We believe that we are on a trajectory to achieve our diversity objectives with respect to the management board.
179
Five of our supervisory directors are male and two are female. We believe that we are on a trajectory to achieve our diversity objectives with respect to the supervisory board.
In connection with our diversity objectives, we have defined our senior management as employees at the vice president position and above. Seven out of our thirty-four senior management members are female. We believe that we are on a trajectory to achieve our diversity objectives with respect to our senior management.
We have taken the following activities and measures for a more balanced gender ratio within the organization:
| ● | establishing policies that support equal opportunities for employees, senior management and applicants for employment; |
| ● | encouraging and enforcing respectful communication and cooperation among all employees and senior management; |
| ● | fostering a corporate culture where employees and senior management are treated with dignity, respect and understanding; |
| ● | actively encouraging employees and officers who feel that they have been subjected to discrimination or harassment to report this to their supervisor, to our human resources department or via our Speak-Up tool; and |
| ● | regularly review our corporate policies to ensure equal treatment. |
Given the gender ratios both in the management board, supervisory board and as well as senior management, we are satisfied overall with our efforts towards improving gender diversity and we believe that our activities, training, mentoring and programs will continue to enhance our diversity objectives.
B. Compensation
Remuneration and Other Benefits to Supervisory and Managing Directors as of December 31, 2023
As a foreign private issuer, in accordance with Nasdaq listing requirements, we must comply with home country compensation requirements and certain exemptions thereunder rather than complying with Nasdaq compensation requirements. Dutch law does not provide for limitations with respect to the aggregate annual compensation paid to our managing directors or supervisory directors, provided that such compensation is consistent with our compensation policy. Such compensation policy requires approval by the general meeting. The supervisory board, in conjunction with our compensation committee, determines the remuneration of individual managing directors with due observance of the compensation policy. A proposal with respect to remuneration schemes in the form of shares or rights to shares in which managing directors may participate is subject to approval by our general meeting. Such a proposal must set out at least the maximum number of shares or rights to subscribe for shares to be granted to the managing directors and the criteria for granting or amendment. The compensation for our supervisory directors is set by the general meeting.
Our compensation policy authorizes our supervisory board to determine the amount, level and structure of the compensation packages of our managing directors at the recommendation of our compensation committee. These compensation packages may consist of a mix of fixed and variable compensation components, including base salary, short-term incentives, long-term incentives, fringe benefits, severance pay and pension arrangements, as determined by our supervisory board.
We did not provide pension, retirement or similar benefits to our managing directors and supervisory directors in the year ended December 31, 2023.
180
Supervisory Board
Compensation of Supervisory Directors
For the year ended December 31, 2023, the cash compensation paid to our supervisory directors for services in all capacities was €673,742. The following table sets forth the aggregate compensation and benefits provided to our supervisory board members for services in the year ended December 31, 2023.
Cash | Total | |||
Compensation |
| Compensation | ||
Name |
| (€) |
| (€)(4) |
Baron Jean Stéphenne, MSc, MBA | 123,750 | 194,500 | ||
Ralf Clemens, MD, PhD (1) |
| 41,250 |
| 94,400 |
Mathias Hothum, PhD |
| 96,250 |
| 128,598 |
Hans Christoph Tanner, PhD (2) |
| 45,216 |
| 70,942 |
Craig A. Tooman, MBA |
| 96,250 |
| 116,741 |
Viola Bronsema, PhD |
| 68,750 |
| 68,750 |
Debra Barker, MD |
| 82,500 | 96,140 | |
Klaus Schollmeier PhD | 68,750 | 73,002 | ||
Michael Brosnan BSc. (3) | 51,026 | 51,026 |
(1) | Dr. Clemens resigned from the Supervisory Board of CureVac N.V. on September 30, 2023. |
(2) | Dr. Tanner resigned from the Supervisory Board of CureVac N.V. on June 19, 2023. |
(3) | Mr. Brosnan joined the Supervisory Board of CureVac on June 19, 2023. |
(4) | This column represents the cash compensation and value of shares delivered in 2023 and does not include RSUs held by certain of the supervisory board members, as described in the share ownership section below. |
Management Board
Compensation of Managing Directors
For the year ended December 31, 2023, the aggregate compensation accrued or paid to our managing directors for services in all capacities was €5,627,583. The following table sets forth the compensation and benefits provided to our management board for services in the year ended December 31, 2023.
All other | Total | |||||||
Salary |
| Bonus(1) |
| Compensation(2) |
| Compensation(3) | ||
Name* |
| (€) |
| (€) |
| (€) |
| (€) |
Alexander Zehnder, MD, MBA (4) |
| 500,000 |
| 250,000 |
| 68,831 |
| 818,831 |
Franz-Werner Haas, LLD, LLM (5) |
| 107,500 |
| 59,043 |
| 880,000 |
| 1,046,543 |
Pierre Kemula, B.Sc |
| 355,250 |
| 179,375 |
| 119,550 |
| 654,175 |
Antony Blanc, Ph.D. (6) |
| 313,834 |
| 175,875 |
| 557,607 |
| 1,047,316 |
Igor Splawski, Ph.D., MSc (7) |
| 176,556 |
| 89,803 |
| 573,115 |
| 839,474 |
Myriam Mendila, MD (8) | 359,239 | 184,175 | 38,381 | 581,795 | ||||
Malte Greune Ph.D. |
| 355,250 |
| 179,375 |
| 66,111 |
| 600,736 |
Senta Ulrike Gnad-Vogt MD (9) | 22,312 | 10,041 | 6,360 | 38,713 |
| (1) | This amount represents the annual variable amount accrued in full in 2023 for the year 2023 based on a percentage of yearly gross remuneration for reaching certain targets agreed upon with the supervisory board, as described further below. These amounts may differ from actual amounts that will be paid in 2024 as the Compensation Committee assesses the performance of the management team. |
| (2) | All other compensation includes other monetary benefits and contributions to social security insurance, if any. |
| (3) | This column does not include the virtual shares, options or RSUs held by certain of the management board members, as described in the “Virtual Share Ownership of Managing Directors” table below. |
| (4) | Dr. Zehnder was formally appointed as a managing director of CureVac N.V. on March 28, 2023. |
| (5) | Dr. Haas resigned as a managing director of CureVac N.V. on March 31, 2023. |
181
| (6) | Dr. Blanc resigned as a managing director of CureVac N.V. on November 30, 2023. |
| (7) | Dr. Splawski resigned as a managing director of CureVac N.V. on July 14, 2023. Amounts in USD (Conv.rate 1,1058). |
| (8) | Dr. Mendila joined the executive team as Chief Development Officer on February 1, 2023. She was formally appointed as a managing director of CureVac N.V. on March 28, 2023. |
| (9) | Dr. Gnad-Vogt served as our interim Chief Development Officer from July 1, 2022 until January 31, 2023. |
Bonus Plan
We maintain and implement a management bonus plan for our managing directors. Under the management bonus plan, we provide a variable bonus payment as a component of management compensation that ranges from 50% to 55% of the individual’s annual base salary, depending on management level. We agree upon the respective individual target bonus amount with each employee on an individual contractual basis. The annual performance review is used to measure the achievement of objectives. The Supervisory Board finally decides on the goal achievement of the Managing Directors. The bonus payment is calculated based on the individual’s degree of target bonus achievement, which is then calculated as a percentage of annual base salary, and is generally paid out in March of the following year. The bonus is calculated on a pro rata basis if the individual joins or leaves CureVac during the year.
Equity Incentive Plans
We maintain a virtual share plan (the “Prior VSOP”) for members of the management board and the supervisory board. As of December 31, 2023, there are 5,603,155 virtual shares outstanding and no awards are available for issuance under the Prior VSOP. Ten percent (10%) of each award under the Prior VSOP became exercisable upon expiration of the 180-day lock-up period following the closing of CureVac’s IPO, which occurred on February 9, 2021. The exercise case “Liquidity after IPO” which relates to certain minimum trading volumes of the CureVac N.V. shares and liquidity levels being reached led to a second 10% portion of the (vested) virtual shares becoming exercisable on the first anniversary after the IPO on August 14, 2021, a third 10% portion of the (vested) virtual shares becoming exercisable on the second anniversary of the IPO on August 14, 2022 and a fourth 10% portion of the (vested) virtual shares becoming exercisable on the third anniversary of the IPO on August 14, 2023. The remaining portion of each award may be exercised (in whole or in part) upon the occurrence of certain defined triggering events, including, but not limited to, drug approval or the sale by a majority shareholder of 5% of our outstanding shares, in each case subject to the terms and conditions of the Prior VSOP. All rights under the Prior VSOP will terminate following the ninth calendar year of our listing on Nasdaq. The Prior VSOP was restructured upon the completion of our Corporate Reorganization. Following this restructuring, upon vesting of virtual shares, the holder will be able to exchange his or her virtual shares (in whole or in part) for cash or common shares of CureVac N.V. (instead of shares of CureVac SE) on a 1 to 133.0778 basis.
Due to the increase in value of CureVac prior to our Corporate Reorganization, we modified our incentive program to allow members of the supervisory board, management board and other employees to participate in the value-increased business based on CureVac’s valuation at the time of the Corporate Reorganization subject to the occurrence of certain enumerated exercise cases reflecting such value-increase (the “New VSOP”). Each virtual share awarded under the New VSOP tracked one underlying series A share of CureVac SE (formerly CureVac AG). The New VSOP provides a cash-claim against CureVac in the amount of the positive difference between the value of CureVac per virtual share at the grant date (as determined by CureVac when the New VSOP was established) and the value per virtual share at the time of exercise of such virtual share (such value to be derived from the valuation of CureVac in the relevant triggering event), subject to CureVac’s discretion to provide tradable shares against payment of the value of CureVac per virtual share at the grant date. Such awards provided under the New VSOP have a term of ten years from the date of grant and vest over four years, where 25% vest after the first anniversary of the individual’s hire date and the remainder vesting monthly. These virtual shares were assumed by CureVac N.V. upon the completion of our Corporate Reorganization, and were converted into options, exercisable for common shares of CureVac N.V. on a 1 to 133.0778 basis. Following this conversion, subject to the same vesting, exercise and expiration terms discussed above, these option awards are governed by the Plan (as defined below).
182
In connection with our initial public offering, we established the Long Term Incentive Plan “LTIP” (the “Plan”) pursuant to which we may grant options, restricted stock, restricted stock units, share appreciation rights and other equity and equity-based awards to our employees, managing directors and supervisory directors, consultants or other advisors. As of December 31, 2023, there are 7,391,361 awards outstanding and 26,206,940 shares remained available for issuance under the Plan. The maximum number of common shares underlying awards granted pursuant to the Plan, including the awards granted under the New VSOP and the common shares underlying outstanding awards under the Prior VSOP, will not exceed an equivalent of 15% of our issued share capital from time to time. The Plan is administered jointly by our management board, supervisory board and compensation committee. Awards under the Plan may be granted to our employees, managing directors and supervisory directors, consultants or other advisors. Awards under the Plan may be conditioned upon the achievement or satisfaction of certain performance criteria. The vesting conditions for awards under the Plan are determined by the Committee and will be set forth in the applicable award documentation. The Plan provides for special provisions for good leavers and bad leavers as well as for a change in control of CureVac N.V.
C. Board Practices
Committees
Audit Committee
The audit committee consists of Michael Brosnan (as chairman), Craig A. Tooman and Klaus Schollmeier. The audit committee assists the supervisory board in overseeing our accounting and financial reporting processes and the audits of our financial statements. In addition, the audit committee is responsible for the appointment, compensation, retention and oversight of the work of our independent registered public accounting firm. Our supervisory board has determined that Michael Brosnan, Craig A. Tooman and Klaus Schollmeier satisfy the “independence” requirements set forth in Rule 10A-3 under the Exchange Act and qualifies as an “audit committee financial expert,” as such term is defined in the rules of the SEC. The composition of our audit committee is consistent with the best practice provisions of the DCGC.
The audit committee is governed by a charter that complies with applicable Nasdaq rules, which charter has been posted on our website.
Compensation Committee
The compensation committee consists of Craig A. Tooman (as chairman), Michael Brosnan, Mathias Hothum and Viola Bronsema. The compensation committee assists the supervisory board in determining compensation for our management and supervisory board members.
The composition of our compensation committee deviates from the best practice provisions of the DCGC, because half of its members are not independent within the meaning of the DCGC because of their affiliation with dievini. Under SEC and Nasdaq rules, there are heightened independence standards for members of the Committee, including a prohibition against the receipt of any compensation from us other than standard director fees. As permitted by the listing requirements of Nasdaq, we opted out of Nasdaq Listing Rule 5605(d), which requires that a compensation committee consist entirely of independent supervisory directors.
The compensation committee is governed by a charter that complies with applicable Nasdaq rules, which has been posted on our website.
Nomination and Corporate Governance Committee
The nomination and corporate governance committee consists of Craig Tooman (as chairman), Mathias Hothum, Michael Brosnan and Viola Bronsema. The nomination and corporate governance committee assists our supervisory board in identifying individuals qualified to become our managing directors or supervisory directors consistent with criteria established by us and in developing our code of business conduct and ethics. The composition of our nomination and corporate governance committee deviates from the best practice provisions of the DCGC, because more than half of its members are not independent within the meaning of the DCGC because of their affiliation with dievini or KfW. As permitted by the listing requirements of Nasdaq, we opted out of Nasdaq Listing Rule 5605(e), which requires independent director oversight of director nominations.
183
The nominating and corporate governance committee is governed by a charter that complies with applicable Nasdaq rules, which has been posted on our website.
Special Committee
Resolutions of our supervisory board to approve a resolution of our management board to exclude or limit pre-emption rights (except in connection with the ordinary operation of our equity incentive plans), or to issue shares against non-cash contribution, require approval by a special committee consisting of one supervisory director nominated by dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement, as defined below) (during the initial nomination period for dievini), one supervisory director nominated by KfW (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) (during the initial nomination period for KfW) and, if applicable, one supervisory director nominated by a nomination concert. In this special committee, the affirmative votes of one supervisory director nominated by dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) (during the initial nomination period for dievini) and the supervisory director nominated by KfW (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) (during the initial nomination period for KfW) are required. See “Item 16G. Corporate Governance — Duties of Managing and Supervisory Directors.”
Service Agreements
Consulting Agreement with Craig Tooman
With the approval of the supervisory board, we entered into a consulting agreement with Craig Tooman, a current member of our supervisory board in January 2023 (the “Tooman Consulting Agreement”), whereby Mr. Tooman agreed to provide consulting services and to advise on the strategic and operative planning of CureVac´s organization. The Tooman Consulting Agreement was concluded for a term of one year, payment of certain travel and out-of-pocket expenses in addition to his consulting fee and restrictive covenants (including covenants related to confidentiality and proprietary information).
Consulting Agreement with Debra Barker
With the approval of the supervisory board, we entered into a consulting agreement with Debra Barker, a current member of our supervisory board in January 2023 (the “Barker Consulting Agreement”), whereby Ms. Barker agreed to provide consulting services and to advise on the strategic and operative planning of the fast-track build-up of state-of-the-art oncology clinical pipeline. The Barker Consulting Agreement was concluded for a term of one year, payment of certain travel and out-of-pocket expenses in addition to her consulting fee and restrictive covenants (including covenants related to confidentiality and proprietary information).
Consulting Agreement with Ralf Clemens
With the approval of the supervisory board, we entered into a consulting agreement with Dr. Clemens, a former member of our supervisory directors in March 2013 (the “Clemens Consulting Agreement”), whereby Dr. Clemens agreed to provide consulting services and agreed to act as a member of our scientific advisory board for an indefinite period. The Clemens Consulting Agreement provides for a notice of termination period of four weeks, payment of certain travel and out-of-pocket expenses in addition to his consulting fee and restrictive covenants (including covenants related to confidentiality and proprietary information). In addition, we entered with the approval of the supervisory board into a consulting agreement with GRID EUROPE in June 2023 (the “GRID Consulting Agreement”). The consulting services were provided by Dr. Clemens. The Grid Consulting Agreement included the information of the Scientific Committee of the Supervisory Board on latest mRNA based clinical activities and global policies. The Grid Consulting Agreement was concluded for a term of six months, payment of certain travel and out-of-pocket expenses in addition to his consulting fee and restrictive covenants (including covenants related to confidentiality and proprietary information).
184
Management Board Service Contracts
We entered into management board service agreements (the “Management Contracts”) with the following current or former managing directors: Dr. Gnad-Vogt, Dr. Haas, Mr. Kemula, Dr. Splawski, Dr. Greune, Dr. Blanc, Dr. Mendila and Dr. Zehnder. The Management Contracts generally provide for an initial term of three years, a 12-month notice period thereafter, a base salary, certain administrative allowances, an annual variable payment expressed as a percentage of annual base salary that is dependent on the achievement of certain objectives agreed to by the supervisory board and a severance payment upon termination of the Management Contracts. The supervisory board is entitled to grant managing directors additional compensation at its discretion. The managing directors are also eligible to participate in a virtual stock plan or equivalent plan that is established in a manner substantially similar to other of the executive officers.
The Management Contracts provide for the following restrictive covenants: (i) a non-compete during employment and for 12 months after termination; (ii) a non-solicit of employees during employment and for two years after termination; and (iii) a perpetual confidentiality covenant. Under the Management Contracts, we are obligated to pay the managing directors compensation for the duration of their post-employment non-compete period in monthly installments that are equal to half of the total compensation they received prior to their termination.
Employment Contract with Senta Ulrike Gnad-Vogt
We entered into an employment contract with Dr. Gnad-Vogt in July 2011, as amended in September 2019, (the “Gnad-Vogt Employment Contract”), whereby Dr. Gnad-Vogt is employed for an indefinite period of time. The Gnad-Vogt Employment Contract provides for a notice of termination period of six months, which can be initiated in writing by either party, and restrictive covenants, including non-compete and non-solicitation covenants and a covenant related to confidentiality. Dr. Gnad-Vogt resigned as our interim chief development officer on July 31, 2021. Dr. Gnad-Vogt also served as our interim Chief Development Officer from July 1, 2022 until January 31, 2023.
Employment Contract with Franz-Werner Haas
We entered into an addendum to the employment contract with Franz-Werner Haas in April 2022 (the “Haas Addendum”) pursuant to which Dr. Haas agreed to extend his service on the management board until March 31, 2023. Under Dr. Haas’s addendum, we awarded him 36,000 RSUs under the Plan on April 1, 2022, which vested immediately and became exercisable on or after January 1, 2023. The Haas Addendum also provides that Dr. Haas be entitled to the reimbursement of certain costs. On March 31, 2023, Dr. Haas stepped down as CureVac’s Chief Executive Officer.
Employment Contract with Pierre Kemula
We entered into an addendum to the employment contract with Pierre Kemula in April 2022 (the “Kemula Addendum”) pursuant to which Mr. Kemula agreed to an indefinite extension of his service on the management board, subject to a notice period of 9 months, but in any case not before October 4, 2024. The Kemula Addendum also provides that Mr. Kemula be entitled to the reimbursement of certain administrative costs.
Employment Contract with Igor Splawski
We entered into an addendum to the employment contract with Igor Splawski in April 2022 (the “1st Splawski Addendum”) pursuant to which Dr. Splawski is entitled to the reimbursement of certain administrative costs.
We entered into another addendum to the employment contract with Igor Splawski in July 2022 (the “2nd Splawski Addendum”) pursuant to which Dr. Splawksi agreed to relocate his place of work to the U.S.
On July 14, 2023, Dr. Splawski stepped down as CureVac’s Chief Scientific Officer.
185
Employment Contract with Malte Greune
We entered into an addendum to the employment contract with Malte Greune in April 2022 (the “Greune Addendum”) pursuant to which Dr. Greune agreed to an indefinite extension of his service on the management board, subject to a notice period of 12 months, but in any case not before July 31, 2024. The Greune Addendum also provides that Dr. Greune be entitled to the reimbursement of certain administrative costs.
Employment Contract with Antony Blanc
We entered into an addendum to the employment contract with Antony Blanc in April 2022 (the “Blanc Addendum”) pursuant to which Dr. Blanc agreed to an indefinite extension of his service on the management board, subject to a notice period of 12 months, but in any case not before November 30, 2023. The Blanc Addendum also provides that Dr. Blanc be entitled to the reimbursement of certain administrative costs. Dr. Blanc stepped down as CureVac’s Chief Business Officer on November 30, 2023.
Employment Contract with Myriam Mendila
We entered into an employment contract with Myriam Mendila in June 2022 (the “Mendila Employment Contract”), pursuant to which Dr. Mendila agreed to serve on the management board with an indefinite term, subject to a notice period of 12 months. The Mendila Employment Contract also provides that Dr. Mendila be entitled to the reimbursement of certain administrative costs.
Employment Contact with Alexander Zehnder
We entered into a future service agreement with Alexander Zehnder on January 6, 2023, and Dr. Zehnder became our CEO on April 1, 2023. On February 28, 2023, we entered into an addendum to the future service agreement with Dr. Zehnder (the “Zehnder addendum”) pursuant to which Dr. Zehnder agreed to an earlier start date into his new role already as of March 1, 2023.
D. Employees
As of December 31, 2023, we employed 1,172 total employees worldwide, 999 of whom were full-time, 294 of whom hold Ph.D. or M.D. degrees, 466 of whom were engaged directly or indirectly in production, 523 of whom were engaged in research and development activities, 48 of whom were engaged in clinical and regulatory activities, 15 of whom were engaged in marketing and sales activities, and 120 of whom were engaged in management, business development or marketing, finance, human resources or administrative support. Of our 1,172 total employees, 1,129 work in Germany, 13 work in Belgium, 22 work in the Netherlands and 8 work in the United States. We are not subject to collective bargaining agreements or similar labor contracts and have a workers’ council established since November 2021.
E. Share Ownership
Share Ownership of Supervisory Directors
The following table sets forth the share ownership of our supervisory directors as of March 15, 2024.
Percentage of | ||||||
Number of |
| Shares |
| |||
Name |
| Shares |
| Outstanding |
| Voting Rights |
Baron Jean Stéphenne, MSc, MBA | 16,758 | 0.01 | % | 16,758 | ||
Mathias Hothum, Ph.D. |
| 106,349 |
| 0.05 | % | 106,349 |
Craig A. Tooman, MBA |
| 9,653 |
| — | 9,653 | |
Viola Bronsema, Ph.D. |
| 0 |
| — |
| 0 |
Debra Barker, MD. |
| 7,125 |
| — |
| 7,125 |
Klaus Schollmeier, Ph.D. | 5,032 | — | 5,032 | |||
Michael Brosnan BSc. (1) | 4,186 | — |
| 4,186 |
(1)Mr. Brosnan joined the Supervisory Board of CureVac on June 19, 2023.
186
Restricted Stock Unit Ownership of Supervisory Directors
The following table sets forth the restricted stock unit (RSU) ownership of our supervisory directors as of March 15, 2024.
Name |
| Number of RSUs |
| Title |
| Vesting Date |
Baron Jean Stéphenne | 2,370 | CureVac LTIP 2022 RSUs | Dec 31, 2024 | |||
Baron Jean Stéphenne | 13,481 | CureVac LTIP 2023 RSUs | 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 | |||
Mathias Hothum, Ph.D. |
| 1,843 |
| CureVac LTIP 2022 RSUs |
| Dec 31, 2024 |
Mathias Hothum, Ph.D. |
| 10,485 |
| CureVac LTIP 2023 RSUs |
| 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 |
Craig A. Tooman, MBA |
| 1,521 |
| CureVac LTIP 2022 RSUs |
| Dec 31, 2024 |
Craig A. Tooman, MBA | 10,485 | CureVac LTIP 2023 RSUs | 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 | |||
Viola Bronsema, Ph.D. | 0 | — | ||||
Debra Barker, MD | 1,317 | CureVac LTIP 2022 RSUs | 1/2 on Dec 31, 2024 | |||
Debra Barker, MD | 8,987 | CureVac LTIP 2023 RSUs | 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 | |||
Klaus Schollmeier, Ph.D. | 695 | CureVac LTIP 2022 RSUs | Dec 31, 2024 | |||
Klaus Schollmeier, Ph.D. | 7,490 | CureVac LTIP 2023 RSUs | 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 | |||
Michael Brosnan BSc.(1) | 8,373 | CureVac LTIP 2023 RSUs | 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 |
(1)Mr. Brosnan joined the Supervisory Board of CureVac on June 19, 2023.
RSU awards which have vested and have been delivered as shares are not included in the table above. The RSUs included in the table above which have not yet vested or been settled (delivered as shares) are not included in the fully diluted shares outstanding.
Share Ownership of Managing Directors
The following table sets forth the share ownership of our managing directors as of March 15, 2024.
Percentage | ||||||
Number of |
| of Shares |
| |||
Name* |
| Shares |
| Outstanding |
| Voting Rights |
Alexander Zehnder, MD, MBA (1) | 56,374 | 0.03 | % | 56,374 | ||
Pierre Kemula, B.Sc |
| 79,013 |
| 0.04 | % | 79,013 |
Myriam Mendila, MD (2) | 4,752 | — | 4,752 | |||
Malte Greune, Ph.D |
| 8,523 |
| — | 8,523 |
| (2) | Dr. Mendila joined the executive team as Chief Development Officer on February 1, 2023. She was formally appointed as a managing director of CureVac N.V. on March 28, 2023. |
Option Ownership of Managing Directors
The following table sets forth the option ownership of our managing directors as of March 15, 2024.
Exercise | ||||||||
Number of |
|
| Price |
| ||||
Name |
| Options |
| Title |
| (in USD) |
| Expiration Date |
Alexander Zehnder (1) | 144,379 | LTIP Option Award 2023 | 6.66 | April 1, 2033 | ||||
Pierre Kemula |
| 25,000 |
| LTIP Option Award 2022 |
| 19.35 |
| March 1, 2032 |
Myriam Mendila (2) | — | — | — | — | ||||
Malte Greune | 20,000 | LTIP Option Award 2021 | 84.03 | July 1, 2025 | ||||
| 25,000 |
| LTIP Option Award 2022 |
| 19.35 |
| March 1, 2032 |
| (1) | Dr. Zehnder was formally appointed as a managing director of CureVac N.V. on March 28, 2023. |
| (2) | Dr. Mendila joined the executive team as Chief Development Officer on February 1, 2023. She was formally appointed as a managing director of CureVac N.V. on March 28, 2023. |
187
Restricted Stock Unit Ownership of Managing Directors
The following table sets forth the restricted stock unit (RSU) ownership of our managing directors as of March 15, 2024.
Name |
| Number of RSUs |
| Title |
| Vesting Date |
Alexander Zehnder (1) | 27,233 | CureVac LTIP | 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 | |||
Pierre Kemula |
| 3,203 |
| CureVac LTIP |
| Dec 31, 2024 |
| 19,063 |
| CureVac LTIP |
| 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 | |
Myriam Mendila (2) | 19,472 | CureVac LTIP | 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 | |||
Malte Greune | 3,203 | CureVac LTIP | Dec 31, 2024 | |||
5,000 | CureVac LTIP | 1/2 on Dec 31, 2022 & 1/2 on Dec 31, 2023 | ||||
19,063 | CureVac LTIP | 1/2 on Dec 31, 2024 & 1/2 on Dec 31, 2025 | ||||
| 25,000 |
| CureVac LTIP |
| 1/33 on Dec 31, 2023,1/3 on Dec 31, 2024 & 1/3 on Dec 31, 2025 |
| (1) | Dr. Zehnder was formally appointed as a managing director of CureVac N.V. on March 28, 2023. |
| (2) | Dr. Mendila joined the executive team as Chief Development Officer on February 1, 2023. She was formally appointed as a managing director of CureVac N.V. on March 28, 2023. |
Please note: RSU awards which have vested and have been delivered as shares are not included in the table above. The RSUs included in the table above which have not yet vested or been settled (delivered as shares) are not included in the fully diluted shares outstanding.
Virtual Share, or VS, Ownership of Management Board Members from VSOP Programs as of March 15, 2024.
Start of | ||||||||||||||||
Vesting | Grant date | |||||||||||||||
Max | Period | (Date of | ||||||||||||||
VS Points | Vested | (Only for New | Allocation | Vesting | ||||||||||||
Name |
| Program |
| Granted |
| Points |
| Participants) |
| Letter) |
| Period |
| VSOP Plan |
| Valid until |
Pierre Kemula |
| VS |
| 465,779 |
| 465,779 |
| 01.10.2016 |
| 18.04.2019 |
| 36 | Prior VSOP | Aug 13, 2029 |
F. Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation
Not applicable.
188
ITEM 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A. Major Shareholders
The following table presents information relating to the beneficial ownership of our common shares as of March 15, 2024 by:
| ● | each person, or group of affiliated persons, known by us to own beneficially 5% or more of our outstanding common shares; |
| ● | each managing director and supervisory director; and |
| ● | all managing directors and supervisory directors as a group. |
The number of common shares beneficially owned by each entity, person, supervisory director or managing director is determined in accordance with the rules of the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under such rules, beneficial ownership includes any common shares over which the individual has sole or shared voting power or investment power as well as any common shares that the individual has the right to acquire within 60 days of March 15, 2024, through the exercise of any option, warrant or other right. Except as otherwise indicated, and subject to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all common shares held by that person. The table is based on information supplied by our managing directors and supervisory directors and by Schedules 13D and Schedules 13G, if any, filed with the SEC.
The percentage of outstanding common shares is computed on the basis of 224,305,680 common shares outstanding as of March 15, 2024. Common shares that a person has the right to acquire within 60 days of March 15, 2024 are also deemed outstanding for purposes of computing the percentage ownership of the person holding such rights, but are not deemed outstanding for purposes of computing the percentage ownership of any other person, except with respect to the percentage ownership of all members of the supervisory board and management board as a group. Unless otherwise indicated below, the address for each beneficial owner is CureVac SE, Friedrich-Miescher-Strasse 15, 72076 Tübingen, Germany.
Shares Beneficially Owned | |||||
Shareholder |
| Number |
| Percentage | |
5% Shareholders: |
|
| |||
Dietmar Hopp(1) |
| 83,880,935 | (2) | 37.4 | % |
Kreditanstalt für Wiederaufbau(2) |
| 29,871,441 |
| 13.3 | % |
Glaxo Group Limited |
| 16,591,937 |
| 7.4 | % |
|
| ||||
Managing Directors: |
|
| |||
Alexander Zehnder, Ph.D. |
| 56,374 |
| * | |
Pierre Kemula, BSc. |
| 79,013 |
| * | % |
Myriam Mendila, MD |
| 4,752 |
| * | |
Malte Greune, Ph.D. | 8,523 | * | % | ||
Supervisory Directors: |
|
| |||
Mathias Hothum, Ph.D.(4) |
| 70,288,109 |
| 31.3 | % |
Baron Jean Stephenne, MSc, MBA |
| 16,758 |
| * | |
Michael Brosnan, BSc. | 4,186 | * | % | ||
Debra Barker, MD |
| 7,125 |
| * | % |
Viola Bronsema, MD |
| — |
| — | |
Klaus Schollmeier, Ph.D. |
| 5,032 |
| * | |
Craig A. Tooman, MBA |
| 9,653 |
| * | % |
All Managing Directors and Supervisory Directors as a Group: |
| 70,475,339 |
| 31.4 | % |
* | Represents beneficial ownership of less than 1%. |
189
| (1) | This information is based on a Schedule 13D filed with the SEC on February 24, 2023 by dievini Hopp BioTech holding GmbH & Co. KG (“dievini”), DH-LT-Investments GmbH (“DH-LT-Investments”), DH-Capital GmbH & Co. KG (“DH-Capital”), OH Beteiligungen GmbH & Co. KG (“OH Beteiligungen”), Dietmar Hopp, Oliver Hopp, Daniel Hopp, DHFS II Holding GmbH & Co. KG (“DHFS II Holding”) and Zweite DH Verwaltungs GmbH (“Zweite DH”). dievini is the record holder of 70,181,760 shares and DH-LT Investments is the record holder of 10,102,286 shares. Mr. Hopp is the record holder of 158,700 shares and is the sole shareholder of a company that is the record holder of 33,517 shares. Zweite DH is the record holder of 3,404,672 shares. DH-Capital and OH Beteiligungen, are collectively the holders of 100% of the limited partner interest in dievini. DH-Capital and OH Beteiligungen each hold a 50% limited partner interest in dievini and therefore, control the voting and dispositive decisions of dievini together and may be deemed to beneficially own the shares held by dievini. Dietmar Hopp, Daniel Hopp and Oliver Hopp are the ultimate controlling persons of dievini, DH-Capital and OH Beteiligungen, and control the voting and investment decisions of the ultimate parent company of dievini and therefore, may be deemed to beneficially own the shares held by dievini by virtue of their status as controlling persons of dievini. The sole general partner of dievini with the authorization to represent are dievini Verwaltungs GmbH and Dr. Hothum; however, 100% of the shares of dievini Verwaltungs GmbH are held by dievini so dievini Verwaltungs GmbH is not considered to have control over dievini. The managing directors of dievini Verwaltungs GmbH are Dietmar Hopp and Dr. Hothum. Voting and dispositive decisions made within dievini Verwaltungs GmbH regarding the securities held by dievini are made by at least two managing directors acting together; however, Dietmar Hopp is entitled to represent dievini Verwaltungs GmbH solely. Therefore, in their capacity as managing directors, Dietmar Hopp and Dr. Hothum share voting and dispositive power over the shares held by dievini, and may be deemed to beneficially own such shares held by dievini; however, each of Dietmar Hopp and Dr. Hothum disclaims beneficial ownership of the shares held by dievini except to the extent of their pecuniary interests therein. The address for dievini, DH-LT Investments and Dietmar Hopp is c/o dievini Hopp BioTech holding GmbH & Co. KG, Johann-Jakob-Astor Str. 57, 69190 Walldorf, Germany, the address for DH-Capital and OH Beteilungen is Heidelberger Straße 43, 69168 Wiesloch, Germany, the address for Oliver Hopp, Daniel Hopp, DHFS II Holding and Zweite DH is Johann-Jakob-Astor Straße 57, 69190 Walldorf, Germany. |
| (2) | This amount includes the amount of 672,875 shares which was delivered to the Company to fulfil dievini’s prior VSOP obligation end of March 2024. As of March 31, 2024 the amount has to be reduced by 672,875. |
| (3) | This information is based on a Schedule 13D filed with the SEC on February 24, 2023 by KfW, or Kreditanstalt für Wiederaufbau, an institution (Anstalt des öffentlichen Rechts) organized under public law of the Federal Republic of Germany. The Federal Republic of Germany holds 80% of KfW’s subscribed capital, and the German federal states (Länder) hold the remaining 20%. The address for KfW is Palmengartenstrasse 5-9, 60325 Frankfurt am Main, Germany. |
| (4) | Includes 70,181,760 shares held by dievini Hopp BioTech holding GmbH & Co. KG that may also be deemed beneficially owned by Dr. Mathias Hothum, 95,504 shares held by MH-LT-Investments GmbH and 10,845 shares held by Dr. Mathias Hothum. |
Each of our shareholders is entitled to one vote per common share. None of the holders of our shares will have different voting rights from other holders of shares. We are not aware of any arrangement that may, at a subsequent date, result in a change of control of our company.
As a number of our shares are held in book-entry form, we are not aware of the identity of all of our shareholders.
B. Related Party Transactions
The following is a description of related party transactions we have entered into since January 1, 2021, with any of our management and supervisory directors and the holders of more than 5% of our common shares.
dievini Hopp BioTech holding GmbH & Co. KG, Walldorf
As of December 31, 2023, dievini holds the majority of our capital stock and is the controlling shareholder. Molecular Health GmbH, or Molecular Health, is a subsidiary of dievini. In December 2017, we concluded a contract with Molecular Health, according to which Molecular Health provides services in conjunction with the Modeling of the biological and clinical effects of Toll-like receptor 7 and 8 agonists in cancer and immune cells. In fiscal years 2021, 2022 and 2023, payments to Molecular Health with respect to research and development amounted to €0. In 2023 a total of EUR 1k (2022: EUR 58k) was paid to dievini Hopp BioTech Holding GmbH & Co. KG as a reimbursement of travel cost.
190
Convertible Loans with Mr. Hopp
We entered into a convertible loan agreement on May 3, 2019 with Mr. Dietmar Hopp, managing director of dievini, under which Mr. Hopp disbursed to us the amount of EUR 50,000k, or Convertible Loan I. On October 24, 2019, we entered into an additional convertible loan agreement with Mr. Hopp, as amended, under which we have the right to call for disbursements in two tranches of EUR 20,000k and a final tranche of €23,927k, until December 31, 2021, or Convertible Loan II, and together with Convertible Loan I, or the Convertible Loans. The Convertible Loans bear an interest rate of 8.00% per annum. We repaid the Convertible Loans on August 7, 2020, and as of December 31, 2020, no Convertible Loans were outstanding.
Immatics Biotechnologies GmbH
In August 2023, a purchase agreement between CureVac Manufacturing GmbH and Immatics Biotechnologies GmbH was entered into. CureVac purchased technical equipment of containers which CureVac will use as a facility in the future. dievini held 16,7% of the shares in Immatics N.V. (Immatics) as of December 31st, 2023 per the Form 20-F of Immatics filed in March 2024. CureVac paid EUR 178k under this agreement.
BePharBel Manufacturing S.A.
In December 2020, CureVac Manufacturing GmbH (formerly CureVac Real Estate GmbH) and BePharBel Manufacturing S.A., entered into a commercial supply agreement to develop and manufacture the diluent that was expected to be used to dilute the Group’s first concentrated COVID-19 vaccine candidate, CVnCoV. Pursuant to the terms of the agreement, it was intended that BePharBel Manufacturing would manufacture and deliver to CureVac Manufacturing GmbH a low seven figure amount of commercial batches of diluent per year, in 2021 and 2022. Following the withdrawal of the CVnCoV in October 2021, CureVac Manufacturing GmbH terminated the commercial and supply agreement with BePharBel and entered into negotiations on a structured and rapid wind-down of the ordered production. The Parties agreed on a settlement in May 2022 of all claims resulting from the commercial and supply agreement for an amount of EUR 3,900k. In total an amount of EUR 4,016k was paid. Baron Jean Stéphenne, our supervisory board member, holds directly and indirectly 15.61% of BePharBel Manufacturing’s equity and is a director of BePharBel Manufacturing, and Baron Jean Stéphenne’s son, Vincent Stéphenne, holds 1.43% of BePharBel Manufacturing’s equity and is a managing director of BePharBel Manufacturing.
Rittershaus law firm, Mannheim
A consulting agreement dated December 15, 2005, was in place for an indefinite term with the law firm Rittershaus Rechtsanwälte Partnerschaftsgesellschaft mbB, Mannheim (Rittershaus). The agreement was replaced by a new consulting agreement dated January 1, 2015.
The agreement can be terminated without notice by us and with notice of three months to the end of the quarter by Rittershaus. In fiscal years 2021, 2022 and 2023, consulting fees of EUR 757k, EUR 518k and EUR 212k were paid to Rittershaus, respectively. Prof. Dr. Christof Hettich, who was one of the managing directors of dievini until June 2022, is a partner of Rittershaus.
Dr. Ingmar Hörr
In June 2018, an advisory agreement was implemented between Dr. Ingmar Hoerr and CureVac. Dr. Hoerr received EUR 0k, EUR 0k and EUR 0k for consulting services in fiscal years 2021, 2022 and 2023, respectively. The advisory agreement with Dr. Ingmar Hoerr was terminated in March 2020. Due to the exercise of the Prior VSOP award in December 2022, CureVac had a receivable position of EUR 573k as per year-end 2022 for the income tax and social security liability. CureVac has received the money in February 2023.
191
Antony Blanc
In July 2020, a consulting agreement was implemented between CureVac AG (currently CureVac SE) and Clarentis SRL, which is wholly owned by Antony Blanc, our chief business officer. The consulting agreement was further amended in September 2020. The consulting agreement was terminated in February 2021 upon Antony Blanc’s appointment as our chief business officer. The consulting agreement provided for payments that were contingent upon the achievement of certain milestones. Clarentis SRL received EUR 150k and EUR100k in fiscal years 2020 and 2021, respectively. In addition to his Management Board position at CureVac NV, Antony Blanc also took over the role as Management Director at CureVac Belgium SA. He executes this function by using Clarentis SRL. As it relates to these services in 2023, CureVac paid EUR 85k in 2023 (2022: EUR 69k, 2021: EUR 0k). The amounts invoiced for these function/services are offset/deducted from his base compensation for his function on the Board of Management of CureVac N.V. As Antony Blanc has left the company as of November 30, 2023, CureVac and Antony Blanc signed a settlement agreement as of September 26, 2023. Under this agreement CureVac incurred cost of EUR 107k for Clarentis SRL entity in 2023.
Dr. Mariola Fotin-Mleczek
Due to the exercise of the Prior VSOP award in December 2022, CureVac had a receivable position of EUR 131k as per year-end for the income tax and social security liability. CureVac has received the money in February 2023.
In 2022, a consulting agreement between CureVac N.V. and Mariola Fotin-Mleczek was made. In 2022 and 2023, a total of EUR 20k and EUR 2k were paid, respectively.
Dr. Florian von der Mülbe
Due to the exercise of the Prior VSOP award in December 2022, CureVac had a receivable position of EUR 559k as per year-end 2022 for the income tax and social security liability. CureVac has received the money in February 2023.
Consulting agreements between CureVac Printer GmbH and Florian von der Mülbe related to the years 2022 to 2024 have been executed. In 2023 CureVac paid EUR 146k under such agreement related to 2022.
Franz-Werner Haas
In Q1 2023, a consulting agreement between CureVac SE and Franz-Werner Haas was executed. In 2023 CureVac paid EUR 65k under this agreement. The payment was partially compensated by Franz-Werner Haas taking over his former company car for EUR 40k.
Alexander Zehnder
In Q1 2023, a first addendum to the future service agreement was entered into to ensure a smooth transition from CEO Franz-Werner Haas to the new CEO Alexander Zehnder. Total compensation amounted to EUR 51k during the month of March.
Barker BioMedical GmbH
In Q1 2023, a consulting agreement between CureVac SE and Barker BioMedical GmbH was executed. Barker BioMedical GmBH is a wholly owned consulting company of Debra Barker, Supervisory Board member of CureVac N.V. In 2023 CureVac paid EUR 14k under this agreement.
Craig Tooman
In Q1 2023, a consulting agreement between CureVac SE and Craig Tooman was executed. In 2023, CureVac paid EUR 5k under this agreement.
Ralf Clemens
In Q3 2023, a consulting agreement between CureVac N.V. and GRID EUROPE was executed. GRID EUROPE is a wholly owned consulting company of Ralf Clemens, who was a member of the supervisory board up to September 30, 2023. CureVac incurred EUR 23k under this agreement in 2023.
192
2020 Private Investment
On July 17, 2020, we entered into a binding agreement with KfW, Glaxo Group Limited, or GSK, QIA and other investors, pursuant to which we agreed to issue new Series B shares in CureVac AG (currently CureVac SE), representing approximately 36% of the shares in CureVac SE in exchange for an aggregate investment of €560 million.
According to the mandate of KfW by the Federal Republic of Germany pursuant to and in accordance with Article 2 paragraph 4 of the KfW Law (Zuweisungsgeschäft) KfW, acquired approximately 19% shareholding in CureVac AG (currently CureVac SE), for an aggregate investment of €300 million. In addition, GSK acquired approximately a 9% shareholding in CureVac SE for an investment of €150 million and QIA acquired approximately 4% shareholding in CureVac SE for an investment of €60 million. We refer to the investment of KfW as the KfW Investment and to the investment by GSK as the GSK Investment. In addition, the other several shareholders purchased an aggregate 3% shareholding in CureVac SE for an aggregate investment of €50 million. As part of our Corporate Reorganization, outstanding shares of all series in CureVac SE have been exchanged for common shares in CureVac B.V., which subsequently were converted into shares of CureVac N.V.
As part of the 2020 Private Investment those investors who have subscribed for our Series A, B and C shares, or the pre-IPO shareholders, became parties to an Investment and Shareholders’ Agreement, as amended, pursuant to which certain of our pre-IPO shareholders holding at least 10% of our shares have entered into a Registration Right Agreement, as further described below. In addition, KfW has become a party to a Relationship Agreement and entered into a separate Shareholders’ Agreement with dievini and Mr. Hopp as further described below. As part of the GSK Investment, we also entered into a collaboration agreement with GSK pursuant to which we are collaborating with GSK to research, develop and commercialize prophylactic and therapeutic non-replicating mRNA-based vaccines and antibodies targeting infectious disease pathogens. For further information regarding our collaboration agreement with GSK please refer to “Item 4. Information on the Company — B. Business Overview — Collaborations.”
We are obligated to use the funds raised in the 2020 Private Investment solely to fund the (i) development of our proprietary pipeline, including earlier stage assets currently in preclinical development, (ii) research and development activities to expand our mRNA platform technology, in particular with respect to our vaccine candidate against SARS-CoV-2 and other infectious diseases and (iii) manufacturing capacities for mRNA-based drug product candidates and future approved products.
Shareholders’ Agreement among KfW, dievini, DH-LT Investments GmbH and Mr. Hopp
In connection with the KfW Investment, KfW, dievini and Mr. Hopp entered into a shareholders’ agreement on June 16, 2020, or the KfW dievini Shareholders’ Agreement, agreeing to certain transfer restrictions and rights of first refusal relating to their interests in our company, nomination rights as provided elsewhere in this Annual Report, and a voting agreement relating to certain specified actions. In particular, dievini and Mr. Hopp agree to vote a specified number of their shares as directed by KfW on certain specified actions, subject to certain exceptions. These specified actions include, inter alia: (1) transferring the tax domicile of CureVac N.V. and/or the approval of the transfer of the corporate or administrative seat of CureVac AG (currently CureVac SE); (2) relocating or ceasing activities in specified areas to a state outside the European Union to the extent (in particular in the area of the development of vaccines) material for the protection of the health of the population of the European Union; (3) entering into material mergers and acquisitions; and (4) amendments to the articles of association of CureVac AG (currently CureVac SE) which would affect the foregoing matters. Under the terms of KfW dievini Shareholders’ Agreement, Mr. Hopp agreed to purchase an aggregate of €100 million of our common shares in a private placement that took place with our initial public offering, in August 2020, at a price per share equal to our initial public offering price concurrently. Mr. Hopp effected this purchase through DH-LT-Investments GmbH, an affiliated entity. In connection with such concurrent private placement DH-LT-Investments GmbH has become a party to the KfW dievini Shareholders’ Agreement. The KfW dievini Shareholders’ Agreement has been extended to December 31, 2024. The Shareholders’ Agreement may be terminated after December 31, 2024, by either party subject to six months’ notice prior the end of the applicable calendar year. In addition, the Shareholders’ Agreement shall automatically terminate if KfW sells all or a part of its interest in the Issuer to a third party, subject to certain exceptions.
Investment and Shareholders’ Agreement
We and our pre-IPO shareholders, entered into an investment and shareholders’ agreement July 17, 2020, or the ISA. The ISA provides for certain particular shareholders’ rights and also envisages restrictions on the shareholders party thereto, including the obligation to enter into a registration rights agreement, restrictions on transfer, as well as certain tag-along rights, drag-along rights, demand rights, rights of first offer and rights of first refusal.
193
Upon the listing of our shares on Nasdaq, only certain limited provisions of the ISA survived the Corporate Reorganization. Pursuant to such surviving provisions, we and/or our pre-IPO shareholders are subject to certain obligations, as listed below.
| ● | We agreed to prepare and provide KfW our interim financials not later than 30 days after the end of each quarter. In addition, not later than 30 days prior to the start of each fiscal year we agreed to prepare and provide KfW our operating plans that shall include certain projections regarding our financials, our business plan relating to the succeeding fiscal year including our development plans, financial and investment plans, budgeted and projected figures and other information and forecasts. Our supervisory board needs to approve the planning by a simple majority vote. In addition, subject to certain limitations, the member of our supervisory board designated (nominiert) by KfW shall, to the extent not prohibited by any mandatory law and/or Nasdaq rules, be entitled to pass on and discuss any information received in his or her capacity as a member of the supervisory board with KfW and certain governmental agencies and offices of the federal government of the federal republic of Germany, however, restricted to the extent required for KfW and any of the aforementioned institutions to comply with their respective obligations. We are obligated to also provide to KfW upon its request such information that is reasonably requested by KfW for the management and the controlling of KfW’s shareholding in us in order for KfW and certain other institutions as of the Federal Republic of Germany to comply with their respective obligations; |
| ● | KfW has the right to designate (nominieren) one member of our supervisory board for appointment by the general meeting (and to prompt the recall of such member of the supervisory board at its sole discretion) as long as KfW’s shareholding in our share capital is at least 10% in accordance with the arrangements included in our articles of association. Furthermore, our pre-IPO shareholders shall, to the extent legally permissible, ensure that any established advisory board or any other comparable panel of our subsidiaries or affiliate shall also provide KfW the right, upon its discretion, to include a member to be designated (nominieren); |
| ● | We need to reasonably cooperate with each of our pre-IPO shareholders to provide them with information that is mandatory for their tax obligations; |
| ● | If we fail to withhold taxes on certain amounts paid to a pre-IPO shareholder, its affiliates and certain related persons of such pre-IPO shareholder (excluding a shareholder that was not our shareholder prior to the 2020 Private Investment), and such certain amounts are reimbursable or creditable to such pre-IPO shareholder, its affiliates and certain related persons of such pre-IPO shareholder, then such pre-IPO shareholder needs to use his or her best efforts in order to obtain a credit or reimbursement from the applicable tax authority and such credit or reimbursement shall be paid to us; |
| ● | We are obligated to use the funds raised in the 2020 Private Investment solely to fund the (i) development of our proprietary pipeline, including earlier stage assets currently in preclinical development, (ii) research and development activities to expand our mRNA platform technology, in particular with respect to our vaccine candidate against SARS-CoV-2 and other infectious diseases and (iii) manufacturing capacities for mRNA-based drug product candidates and future approved products; |
194
| ● | We entered into a Global Access Agreement with the Bill & Melinda Gates Foundation in February 2015 pursuant to which we are required to take certain actions to support the Bill & Melinda Gates Foundation’s mission. We may be required to redeem all, or to facilitate the purchase by a third party of all, the shares held in us by the Bill & Melinda Gates Foundation as per the date of the ISA on certain terms that may not be favorable to us, if we receive from the Bill & Melinda Gates Foundation a notification that we have (a) committed a material breach of certain commitments under the Global Access Agreement, (b) used the funds received from the Bill & Melinda Gates Foundation for purposes other than those described in the Global Access Agreement, including not for the (i) finance of our facility to be used inter alia to manufacture vaccines and drugs in support of the Bill & Melinda Gates Foundation’s charitable purpose, (ii) continued development of technology for prophylactic and therapeutic mRNA vaccines and drugs against infectious diseases and vaccine adjuvants, comprised of long, non-coding RNA molecules and formulation/delivery technology necessary to develop the mRNA vaccines and drugs, or the Platform Technology, (iii) the use of the Platform Technology to advance vaccine and drug candidates in support of the Bill & Melinda Gates Foundation charitable purpose and/or (c) failed to comply with certain U.S. regulatory and tax obligations, or together the BMGF Default, and the BMGF Default continues to exist following a cure period. In addition, if we are required but fail to purchase all of the shares held in us by the Bill & Melinda Gates Foundation as per the date of the ISA, we shall not be allowed to pay dividends, redeem the shares of any other shareholder (other than repurchases at cost of shares from our employees, officers, directors, consultants or other persons performing services for us pursuant to agreements under which we have the option to repurchase our shares upon the occurrence of the termination of employment or service) or otherwise make any other distribution to any of our shareholders in connection with their shares. In addition, if within 12 months after such redemption or sale, we close an underwritten public financing or a change of control occurs and the valuation used for such underwritten public financing or a change of control, as the case may be, is in excess of 200% of the valuation used for the redemption or the sale of the shares held by the Bill & Melinda Gates Foundation, we will need to pay the Bill & Melinda Gates Foundation compensation equal to the excess of what it would have received in such transaction if it still held its shares at the time of such underwritten public financing or a change of control over what it received in the sale or redemption of its shares had the BMGF Default not occurred. For further information regarding the Global Access Agreement see “Item 4. Information on the Company — B. Business Overview — Collaborations — Bill & Melinda Gates Foundation Partnership;” |
| ● | Upon the consummation of our initial public offering, we agreed to file, and filed, an application for continuation of tax book value (Antrag auf Buchwertfortführung acc. to Sec. 21 par. 1 second and third sentences of the German Tax Conversion Act — UmwStG) regarding the shares of CureVac AG (currently CureVac SE) that has been transferred as part of our initial public offering, as set out above, with the respectively competent tax authorities within four (4) months after the date of the ISA at the latest; and |
| ● | We agreed to provide pre-IPO shareholders holding at least 10% of our share capital certain information required to facilitate the disposition of our shares held by any such pre-IPO shareholder, or in some circumstances to provide information that we intend to file with the SEC and in such case we may be required to take into account the input of such pre-IPO shareholder holding prior to the filing with the SEC. In addition, we agreed that upon a written request of such a pre-IPO shareholder, we will add such pre-IPO shareholder holding to our liability insurance as a named insured in connection with the consummation of a registered offering, but any related premium shall be borne by such pre-IPO shareholder holding at least 10% of our share capital; and dievini and certain other limited pre-IPO shareholders agreed to bear the economics with respect to the Prior VSOP for a total of up to 60,175 participation rights corresponding with 10% of our share capital as of February 1, 2015, in the amount of altogether €601,750,000. In case of an exercise event under the Prior VSOP, dievini and such other limited pre-IPO shareholders agreed to transfer common shares up to a pre-determined amount to allow us to fulfill any claims of the beneficiaries under the Prior VSOP. |
195
Registration Rights Agreement
We entered into a registration rights agreement upon the consummation off our initial public offering, pursuant to which each holder of at least 10% of our common shares and certain other holders of our common shares is entitled to various rights with respect to the registration of their common shares under the Securities Act. Each holder party to the registration rights agreement or an Eligible Shareholder shall continue to have various rights with respect to the registration of its common shares under the Securities Act, until 90 days after such shareholder, including its affiliates, cease to hold at least 10% of our common shares, or any other future class of shares. The registration of these common shares under the Securities Act would result in these common shares becoming freely tradable without restriction under the Securities Act immediately upon the effectiveness of the registration, except for shares purchased by affiliates. We are not required to register such common shares if an exemption from the registration requirements of the U.S. Securities Act is available with respect to the number of our common shares desired to be sold.
Form F-1 Registration Statement
As of February 9, 2021, any Eligible Shareholder or group of Eligible Shareholders, is entitled to demand in writing that we effect the registration under the Securities Act of the sale or other transfer of such shareholder or shareholders’ common shares, provided that we are not required to effect more than three such registrations and that the aggregate anticipated offering price from the sale of such shares equals at least $35 million, subject to certain exemptions.
Form F-3 Registration Statement
As of August 13, 2021, each Eligible Shareholder or group of Eligible Shareholders may request in writing, not more than three times within 12-month period, that we effect a registration of the sale or other transfer of such shares, provided that the aggregate anticipated offering price from the sale of such shares equals at least $15 million, subject to certain exemptions. We will not be obligated to file a registration statement on Form F-3 in certain customary cases, subject to certain exemptions.
Piggyback Registration Rights
The registration rights agreement provides our Eligible Shareholders with “piggy back” registration rights in the event that we determine to register the sale of any of our securities. With respect to such registration rights, we are committed to use our reasonable best efforts to include in a registration statement a prospectus relating to the resale of certain securities held by certain of our Eligible Shareholders.
Relationship Agreement
In connection with the KfW Investment, KfW, dievini and CureVac B.V. entered into a relationship agreement on July 17, 2020, or the KfW dievini Relationship Agreement. Pursuant the KfW dievini Relationship Agreement the parties provide for our agreed form of the articles of association, the supervisory board rules and the management board rules. In addition, the KfW dievini Relationship Agreement establishes that if at any time during the effectiveness of the KfW dievini Shareholders’ Agreement any of our shareholders other than KfW or dievini is granted a nomination right to our supervisory board or the supervisory board of any of our subsidiaries, then all of the parties to the KfW dievini Relationship Agreement shall use best efforts to exercise their shareholder rights to ensure that KfW and dievini shall each be given a nomination right as well. We also agreed not to procure, not to propose or implement during the terms of the KfW dievini Shareholders’ Agreement and the ISA, any amendment to the corporate documents of CureVac AG (currently CureVac SE) or CureVac B.V. or CureVac N.V., which would violate or not observe the Relationship Agreement, the ISA or other agreements concluded with us and KfW in connection with KfW’s investment.
Indemnification Agreements
Our articles of association require us to indemnify our current and former managing directors and supervisory directors to the fullest extent permitted by law, subject to certain exceptions. We entered into indemnification agreements with all our managing directors and supervisory directors and may enter into additional indemnification agreements with future managing directors and supervisory directors.
196
Employment Agreements
Certain of our managing directors and supervisory directors have entered into service agreements with us as discussed in more detail within “Item 6. Directors, Senior Management and Employees — C. Board Practices — Service Agreements.”
Transfer of Shares from Prior Shareholders
As of March 10, 2021, October 18, 2021, and December 12, 2022, the holders of the virtual shares under the Prior VSOP plan exercised all of their exercisable virtual shares. See note 10 to our audited consolidated financial statements contained elsewhere in this Annual Report for further information on the exercise of the virtual shares.
C. Interests of Experts and Counsel
Not applicable.
ITEM 8. FINANCIAL INFORMATION
A. Consolidated Statements and Other Financial Information
Financial Statements
See “Item 18. Financial Statements,” which contains our audited financial statements prepared in accordance with IFRS.
Legal Proceedings
From time to time, we are subject to various legal proceedings and claims that arise in the ordinary course of our business activities including proceedings initiated by former employees and directors. Although the results of litigation and claims cannot be predicted with certainty, as of the date of this Annual Report, we do not believe such claims and proceedings, would individually or in the aggregate be reasonably expected to have a material adverse effect on our business. If and to the extent necessary, we have formed appropriate accruals.
In connection with our adjustment of our external European manufacturing network after the withdrawal of our EMA dossier for our vaccine candidate, CvnCoV, we are involved in disputes with three former contract manufacturers.
In February 2022, we were served with a request for arbitration filed with the German Arbitration Institute by Wacker Biotech B.V. seeking payments based on a terminated agreement with us. In April 2022, Celonic Deutschland GmbH & Co.KG initiated arbitration proceedings according to the procedural rules of the German Arbitration Institute against us also requesting payments based on a terminated agreement. The agreements with Wacker and Celonic were terminated in connection with the withdrawal of our EMA dossier for our drug candidate CVnCoV. We have defended ourselves vigorously against Wacker’s and Celonic’s claims in written submission and the oral hearings. The taking of evidence has concluded in both proceedings, and we are awaiting the final awards of the arbitration tribunals.
In August 2022, Rentschler Biopharma SE initiated arbitration proceedings according to the procedural rules of the German Arbitration Institute against us. Rentschler's claims are based on the assumption that certain agreements between Rentschler and CureVac Manufacturing GmbH (then: CureVac Real Estate GmbH) have been terminated due to the withdrawal of our EMA dossier for CVnCoV or alternatively that CureVac would have been obliged to terminate these agreements. CureVac's position always was that the agreements had ended upon expiration of their term. We did not believe that there was a legal basis for Rentschler's claims and therefore defended ourselves vigorously against these claims in written submissions and the oral hearing in October 2023. In its final award rendered in April 2024, the arbitration tribunal followed CureVac's arguments and dismissed Rentschler's claims in their entirety.
In addition, we are involved in several intellectual property related legal proceedings.
197
In June 2022, we filed a lawsuit against BioNTech in the Düsseldorf Regional Court for infringement of several of CureVac’s patents and utility models as a result of the manufacture, use and sale of Comirnaty. Additionally, certain of our patents and utility models are part of infringement claims in Germany filed by BioNTech. For all patents and utility models parallel invalidity claims (nullity action, oppositions and cancellation actions) were filed by BioNTech. In December 2023, the Federal Patent Court in Germany revoked one patent which is part of the infringement claim. The formal reasons for the decision of the court have not yet been provided. CureVac intends to appeal this decision before the German Federal Court of Justice. The proceedings before the Düsseldorf Regional Court regarding the remaining intellectual property rights are still ongoing and will be decided individually.
In November 2022, BioNTech filed cancellation actions seeking the cancellation of three of CureVac’s German utility models in the German Patent and Trademark Office. In November 2023, BioNTech filed cancellation actions against two additional utility models in the German Patent and Trademark Office. In December 2023, BioNTech filed an opposition against a European patent at the European Patent Office. CureVac is defending against these actions.
In September 2022, BioNTech and Pfizer filed a declaration of non-infringement and revocation action against two of CureVac’s patents in the Patents Court of the Business and Property Courts of England and Wales. We counterclaimed that such patents are valid and infringed by Comirnaty and subsequently added a third patent to our counterclaim.
In July 2022, BioNTech and Pfizer filed a complaint in the U.S. District Court for the District of Massachusetts seeking declaratory judgment of non-infringement of several of CureVac’s patents by Comirnaty. Subsequently we successfully filed a motion to have the case transferred to the Eastern District of Virginia. In May 2023, CureVac filed a counterclaim alleging infringement of several of our U.S. patents by the manufacture and sale of Comirnaty. Pfizer and BioNTech filed a counterclaim alleging non-infringement and validity of the asserted rights. In November 2023, Acuitas filed a motion to intervene and a motion to sever and stay in the U.S. proceedings regarding four U.S. patents. Simultaneously, Acuitas filed a separate case in the Eastern District of Virginia (Norfolk) claiming co-inventorship for the same patents.
In October 2023, Acuitas initiated arbitration proceedings against CureVac. Acuitas requests a declaration of the tribunal that Acuitas co-owns a patent family which is part of CureVac’s litigation against BioNTech and Pfizer in Germany and the United States and asserts consequential claims. CureVac will defend itself against these claims. The possible outcome of this dispute cannot be predicted at this time.
For more information, see also “Item 3. Key Information — D. Risk Factors — Risks Related to Our Intellectual Property Rights.”
Legal disputes, arbitration and litigation are inherently unpredictable. While we do not believe that any of these matters will have a material adverse effect on our financial position, we could incur judgments, enter into settlements or revise our expectations regarding the outcome of matters, which could have a material adverse effect on our financial condition or on our results of operations and/or our cash flows in the period in which any such amounts are accrued or paid. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Dividends and Dividend Policy
We have never paid or declared any cash dividends on our common shares, and we do not anticipate paying any cash dividends on our common shares in the foreseeable future. We intend to retain all available funds and any future earnings to fund the development and expansion of our business. Under Dutch law, we may only pay dividends to the extent our shareholders’ equity (eigen vermogen) exceeds the sum of the paid-in and called-up share capital plus the reserves required to be maintained by Dutch law or by our articles of association and (if it concerns a distribution of profits) after adoption of the annual accounts by the general meeting from which it appears that such dividend distribution is allowed. Subject to such restrictions, any future determination to pay dividends will be at the discretion of our management board with the approval of our supervisory board and will depend upon a number of factors, including our results of operations, financial condition, future prospects, contractual restrictions, restrictions imposed by applicable law and other factors our management board and supervisory board deem relevant.
198
Under our articles of association, our management board may decide that all or part of the profits are added to our reserves. Before reservation of any profit, to the extent that preferred shares have been canceled and preferred distributions on those canceled shares are outstanding, the profits are first to be used to satisfy the outstanding claim to those who held those preferred shares at the moment of such cancellation becoming effective and subsequently if any preferred shares are outstanding, a dividend is paid out of the remaining profit on the preferred shares in accordance with our articles of association. This dividend, or preferred dividend, shall be calculated on the basis of a fixed rate over the amount paid-up on the outstanding preferred shares pro rata tempore for the period during which they were outstanding during the financial year concerned, and shall include any arrears in payment of prior years’ preferred dividends (if any). The remaining profit will be at the disposal of the general meeting at the proposal of the management board for distribution on the common shares, subject to restrictions of Dutch law and approval by our supervisory board of such proposal of our management board. Our management board is permitted, subject to certain requirements, to declare interim dividends without the approval of the general meeting, but only with the approval of the supervisory board. Dividends and other distributions shall be made payable not later than the date determined by the management board. Claims to dividends and other distributions not made within five years from the date that such dividends or distributions became payable will lapse and any such amounts will be considered to have been forfeited to us (verjaring).
B. Significant Changes
None.
ITEM 9. THE OFFER AND LISTING
A. Offering and Listing Details
Our common shares, with a par value of €0.12 per share, have traded on Nasdaq under the symbol “CVAC” since August 14, 2020.
B. Plan of Distribution
Not applicable.
C. Markets
For a description of our publicly traded common shares, see “Item 9. The Offering and Listing — A. Offer and Listing Details.”
D. Selling Shareholders
Not applicable.
E. Dilution
Not applicable.
F. Expenses of the Issue
Not applicable.
ITEM 10. ADDITIONAL INFORMATION
A. Share Capital
Not applicable.
199
B. Memorandum and Articles of Association
Our shareholders adopted the Articles of Association included as Exhibit 3.1 to our revised registration statement on Form F-1 (File No. 333-240076) filed with the SEC on August 7, 2020.
We incorporate by reference into this Annual Report on Form 20-F the description of our Articles of Association effective upon the closing of our last public offering contained in our Registration Statement on Form F-3 (File No. 333-259613), under “Description of Share Capital and Articles of Association” filed with the SEC on September 17, 2021. Such description sets forth a summary of certain provisions of our articles of association, and certain descriptions of applicable German and Dutch law, each as currently in effect.
C. Material Contracts
See “Item 4. Information on the Company — B. Business Overview — Significant Agreements.”
D. Exchange Controls
Under Dutch law, there are no exchange controls applicable to the transfer to persons outside of the Netherlands of dividends or other distributions with respect to, or of the proceeds from the sale of, shares of a Dutch company, subject to applicable restrictions under sanctions and measures, including those concerning export control, pursuant to European Union regulations, the Sanctions Act 1977 (Sanctiewet 1977) or other legislation, applicable anti-boycott regulations and similar rules. There are no special restrictions in the articles of association or Dutch law that limit the right of shareholders who are not citizens or residents of the Netherlands to hold or vote shares.
E. Taxation
The following summary contains a description of certain Dutch, German and U.S. federal income tax consequences of the acquisition, ownership and disposition of common shares, but it does not purport to be a comprehensive description of all the tax considerations that may be relevant to a decision to purchase common shares. The summary is based upon the tax laws of the Netherlands and regulations thereunder, the tax laws of Germany and regulations thereunder and the tax laws of the United States and regulations thereunder as of the date hereof, which are subject to change. You should consult your tax advisor regarding the applicable tax consequences to you of investing in our common shares.
Material Dutch Tax Considerations
General
The following is a general summary of certain material Dutch tax consequences of the acquisition, holding and disposal of our common shares. This summary does not purport to set forth all possible tax considerations or consequences that may be relevant to a holder or prospective holder of our common shares and does not purport to deal with the tax consequences applicable to all categories of investors, some of which (such as trusts or similar arrangements) may be subject to special rules. In view of its general nature, it should be treated with corresponding caution.
This summary is based on the tax laws of the Netherlands, published regulations thereunder and published authoritative case law, all as in effect on the date hereof, including, for the avoidance of doubt, the tax rates applicable on the date hereof, and all of which are subject to change, possibly with retroactive effect. Any such change may invalidate the contents of this summary, which will not be updated to reflect such change. Where the summary refers to “the Netherlands” or “Dutch” it refers only to the part of the Kingdom of the Netherlands located in Europe.
This discussion is for general information purposes and is not Dutch tax advice or a complete description of all Dutch tax consequences relating to the acquisition, holding and disposal of our common shares. Holders or prospective holders of our common shares should consult their own tax advisor regarding the Dutch tax consequences relating to the acquisition, holding and disposal of our common shares in light of their particular circumstances.
200
Please note that this section does not set forth the Dutch tax considerations for:
| (i) | holders of common shares if such holders have a substantial interest (aanmerkelijk belang) or deemed substantial interest (fictief aanmerkelijk belang) under the Dutch Income Tax Act 2001 (Wet inkomstenbelasting 2001). Generally, a holder of securities in a company is considered to hold a substantial interest in such company if such holder alone or, in the case of individuals, together with such holder’s partner for Dutch income tax purposes, or any relatives by blood or marriage in the direct line (including foster children), directly or indirectly holds (i) an interest of 5% or more of the total issued and outstanding capital of that company or of 5% or more of the issued and outstanding capital of a certain class of shares of that company; or (ii) rights to acquire, directly or indirectly, such interest; or (iii) certain profit sharing rights in that company that relate to 5% or more of the company’s annual profits and/or to 5% or more of the company’s liquidation proceeds. A deemed substantial interest exists if a substantial interest (or part thereof) in a company has been disposed of, or is deemed to have been disposed of, on a non-recognition basis; |
| (ii) | holders of common shares for which the common shares qualify or qualified as a participation (deelneming) for purposes of the Dutch Corporate Income Tax Act 1969 (Wet op de vennootschapsbelasting 1969). Generally, a holder’s shareholding of 5% or more in a company’s nominal paid-up share capital qualifies as a participation. A holder may also have a participation if (a) such holder does not have a shareholding of 5% or more but a related entity (statutorily defined term) has a participation or (b) the company in which the shares are held is a related entity (statutorily defined term); |
| (iii) | holder of common shares which are or who are entitled to the dividend withholding tax exemption (inhoudingsvrijstelling) with respect to any income (opbrengst) derived from the common shares (as defined in Article 4 of the Dutch Dividend Withholding Tax Act 1965 (Wet op de dividendbelasting 1965). Generally, a holder of common shares may be entitled or required to apply, subject to certain other requirements, the dividend withholding tax exemption if it is an entity and holds and interest of 5% or more in our nominal paid-up share capital; |
| (iv) | pension funds, investment institutions (fiscale beleggingsinstellingen), tax exempt investment institutions (vrijgestelde beleggingsinstellingen) (each as defined in the Dutch Corporate Income Tax Act 1969) and other entities that are, in whole or in part, not subject to or exempt from Dutch corporate income tax, entities that have a function comparable to an investment institution or a tax exempt investment institution, as well as entities that are exempt from corporate income tax in their country of residence, such country of residence being another state of the European Union, Norway, Liechtenstein, Iceland or any other state with which the Netherlands has agreed to exchange information in line with international standards; and |
| (v) | holders of common shares who are individuals and for whom the common shares or any benefit derived from the common shares are a remuneration or deemed to be a remuneration for (employment) activities performed by such holders or certain individuals related to such holders (as defined in the Dutch Income Tax Act 2001). |
Dividend Withholding Tax
Dividends distributed by us generally are subject to Dutch dividend withholding tax at a rate of 15%. Generally, we are responsible for the withholding of such dividend withholding tax at source; the Dutch dividend withholding tax is for the account of the holder of our common shares.
The expression “dividends distributed” includes, among other things:
| ● | distributions in cash or in kind, deemed and constructive distributions and repayments of paid-in capital not recognized for Dutch dividend withholding tax purposes; |
| ● | liquidation proceeds, proceeds from the redemption of common shares, or proceeds from the repurchase of common shares (other than as temporary portfolio investment; tijdelijke belegging) by us or one of our subsidiaries or other affiliated entities to the extent such proceeds exceed the average paid-in capital of those common shares as recognized for Dutch dividend withholding tax purposes; |
| ● | an amount equal to the par value of common shares issued or an increase of the par value of common shares, to the extent that it does not appear that a contribution, recognized for Dutch dividend withholding tax purposes, has been made or will be made; and |
201
| ● | partial repayment of the paid-in capital, recognized for Dutch dividend withholding tax purposes, if and to the extent that we have net profits (zuivere winst), unless (i) the general meeting has resolved in advance to make such repayment and (ii) the par value of the common shares concerned has been reduced by an equal amount by way of an amendment of our articles of association. The term “net profits” includes anticipated profits that have yet to be realized. |
Corporate legal entities that are resident or deemed to be resident of the Netherlands for Dutch corporate income tax purposes (“Dutch Resident Entities”) generally are entitled to an exemption from, or a credit for, any Dutch dividend withholding tax against their Dutch corporate income tax liability. The credit in any given year is, however, limited to the amount of Dutch corporate income tax payable in respect of the relevant year with an indefinite carry forward of any excess amount. Individuals who are resident or deemed to be resident of the Netherlands for Dutch personal income tax purposes (“Dutch Resident Individuals”) generally are entitled to a credit for any Dutch dividend withholding tax against their Dutch personal income tax liability and to a refund of any residual Dutch dividend withholding tax. The above generally also applies to holders of our common shares that are neither resident nor deemed to be resident of the Netherlands (“Non-Resident Holders”) if the common shares are attributable to a Dutch permanent establishment of such Non-Resident Holder.
A holder of common shares resident of a country other than the Netherlands may, depending on such holder’s specific circumstances, be entitled to exemptions from, reduction of, or full or partial refund of, Dutch dividend withholding tax under Dutch domestic tax law, EU law, or treaties for the avoidance of double taxation in effect between the Netherlands and such other country.
Dividend Stripping
According to Dutch domestic anti-dividend stripping rules, no credit against Dutch tax, exemption from, reduction, or refund of Dutch dividend withholding tax will be granted if the recipient of the dividends we paid is not considered the beneficial owner (uiteindelijk gerechtigde; as described in the Dutch Dividend Withholding Tax Act 1965) of those dividends. This legislation generally targets situations in which a shareholder retains its economic interest in shares but reduces the withholding tax costs on dividends by a transaction with another party. It is not required that the recipient of the dividends is aware that a dividend stripping transaction took place for these rules to apply. The Dutch State Secretary of Finance takes the position that the definition of beneficial ownership introduced by this legislation will also be applied in the context of a double taxation convention. As from January 1, 2024, more stringent rules apply to the setoff, exemption from, and reduction or refund of Dutch dividend withholding tax to address situations where a claim for setoff, exemption, reduction or refund may align with the letter of Dutch tax law or a double taxation convention but goes against the underlying intention or spirit of the dividend stripping rules, as perceived by the legislator. The burden of proof with respect to beneficial ownership of dividends distributed by us rests on the Dutch tax authorities. If, however, a shareholder would receive dividends, including dividends on the common shares, in a calendar year in respect of which an aggregate amount of €1,000 in Dutch dividend withholding tax would otherwise be due based on the rate of 15%, the burden of proof with respect to beneficial ownership of such dividends lies with the shareholder. Furthermore, for shares traded on a regulated market, including the common shares, it has been codified that the record date is used when determining the person who is entitled to the dividend.
Conditional withholding tax on dividends
In addition to the regular Dutch dividend withholding tax as described above, a Dutch conditional withholding tax will be imposed on dividends distributed by us to entities related (gelieerd) to us (within the meaning of the Dutch Withholding Tax Act 2021; Wet bronbelasting 2021), if such related entity:
| (i) | is considered to be resident (gevestigd) in a jurisdiction that is listed in the yearly updated Dutch Regulation on low-taxing states and non-cooperative jurisdictions for tax purposes (Regeling laagbelastende staten en niet-coöperatieve rechtsgebieden voor belastingdoeleinden) (a “Listed Jurisdiction”); or |
| (ii) | has a permanent establishment located in a Listed Jurisdiction to which the common shares are attributable; or |
| (iii) | holds the common shares for the main purpose or one of the main purposes to avoid taxation for another person or entity and there is an artificial arrangement or transaction or a series of artificial arrangements or transactions; or |
| (iv) | is not considered to be the beneficial owner of the common shares in its jurisdiction of residence because such jurisdiction treats another entity as the beneficial owner of the common shares (a hybrid mismatch); or |
202
| (v) | is not resident in any jurisdiction (also a hybrid mismatch); or |
| (vi) | is a reverse hybrid (within the meaning of Article 2(12) of the Dutch Corporate Income Tax Act 1969), if and to the extent (x) there is a participant in the reverse hybrid which is related (gelieerd) to the reverse hybrid, (y) the jurisdiction of residence of such participant treats the reverse hybrid as transparent for tax purposes and (z) such participant would have been subject to the Dutch conditional withholding tax in respect of dividends distributed by us without the interposition of the reverse hybrid, all within the meaning of the Dutch Withholding Tax Act 2021. |
The Dutch conditional withholding tax on dividends will be imposed at the highest Dutch corporate income tax rate in effect at the time of the distribution 2024: 25.8%). The Dutch conditional withholding tax on dividends will be reduced, but not below zero, by any regular Dutch dividend withholding tax withheld in respect of the same dividend distribution. As such, based on the currently applicable rates, the overall effective tax rate of withholding the regular Dutch dividend withholding tax (as described above) and the Dutch conditional withholding tax on dividends will not exceed the highest corporate income tax rate in effect at the time of the distribution (2024: 25.8%).
Dual Tax Residency
We are incorporated under the laws of the Netherlands, and we are therefore a Dutch tax resident for Dutch domestic tax law purposes, including the Dutch Dividend Withholding Tax Act 1969. We are also treated as a German tax resident for German domestic tax law purposes, since our place of effective management is located in Germany. Based on the so-called tie-breaker provision (the “Tie-Breaker Provision”) included in Section 4(3) of the 2012 Convention between the Federal Republic of Germany and the Kingdom of the Netherlands for the avoidance of double taxation with respect to taxes on income (the “double tax treaty between Germany and the Netherlands”) as in effect on the date hereof, our tax residence in either the Netherlands or Germany for the purposes of the double tax treaty between Germany and the Netherlands should be determined on our place of effective management. As long as our place of effective management is in Germany, and the Tie-Breaker Provision and the reservation made by Germany with respect to the Tie-Breaker Provision as part of the Multilateral Convention to Implement Tax Treaty Related Measures to Prevent Base Erosion and Profit Shifting are not changed, we should exclusively be a tax resident of Germany for purposes of the double tax treaty between Germany and the Netherlands. As a consequence, the Netherlands will be restricted from imposing Dutch dividend withholding tax on dividends distributed by us pursuant to Section 10(5) of the double tax treaty between Germany and the Netherlands, except for dividends distributed to Dutch Resident Entities, Dutch Resident Individuals and Non-Resident Holders if the common shares are attributable to a permanent establishment in the Netherlands of such Non-Resident Holder.
Taxes on Income and Capital Gains
Dutch Resident Entities
Generally speaking, if the holder of common shares is a Dutch Resident Entity, any benefits derived or deemed to be derived from the common shares or any capital gains realized on the disposal or deemed disposal of the common shares is subject to Dutch corporate income tax at a rate of 19% with respect to taxable profits up to €200,000 and 25.8% with respect to taxable profits in excess of that amount (rates and brackets for 2024).
Dutch Resident Individuals
If the holder of common shares is a Dutch Resident Individual, any benefits derived or deemed to be derived from the common shares or any capital gains realized on the disposal or deemed disposal of the common shares is taxable at the progressive Dutch income tax rates (with a maximum of 49.5% in 2024), if:
(i) | the common shares are attributable to an enterprise from which the holder of common shares derives a share of the profit, whether as an entrepreneur (ondernemer) or as a person who has a co-entitlement to the net worth (medegerechtigd tot het vermogen) of such enterprise without being a shareholder (as defined in the Dutch Income Tax Act 2001); or |
(ii) | the holder of common shares is considered to perform activities with respect to the common shares that go beyond ordinary asset management (normaal, actief vermogensbeheer) or otherwise derives benefits from the common shares that are taxable as benefits from miscellaneous activities (resultaat uit overige werkzaamheden). |
203
Taxation of savings and investments
If the above-mentioned conditions (i) and (ii) do not apply to the Dutch Resident Individual, the common shares will be subject to an annual Dutch income tax under the regime for savings and investments (inkomen uit sparen en beleggen). Taxation only occurs insofar the Dutch Resident Individual’s net investment assets for the year exceed a statutory threshold (heffingvrij vermogen). The net investment assets for the year are the fair market value of the investment assets less the fair market value of the liabilities on January 1 of the relevant calendar year (reference date; peildatum). Actual income or capital gains realized in respect of the common shares are as such not subject to Dutch income tax.
The Dutch Resident Individual’s assets and liabilities taxed under this regime, including the common shares, are allocated over the following three categories: (a) bank savings (banktegoeden), (b) other investments (overige bezittingen), including the common shares, and (c) liabilities (schulden). The taxable benefit for the year (voordeel uit sparen en beleggen) is equal to the product of (x) the total deemed return divided by the sum of bank savings, other investments and liabilities and (b) the sum of bank savings, other investments and liabilities minus the statutory threshold, and is taxed at a flat rate of 36% (rate for 2024).
The deemed return applicable to other investments, including the common shares, is set at 6.04% for the calendar year 2024. Transactions in the three-month period before and after January 1 of the relevant calendar year implemented to arbitrate between the deemed return percentages applicable to bank savings, other investments and liabilities will for this purpose be ignored if the holder of common shares cannot sufficiently demonstrate that such transactions are implemented for other than tax reasons.
The current Dutch income tax regime for savings and investments was implemented in Dutch tax law following the decision of the Dutch Supreme Court (Hoge Raad) of 24 December 2021 (ECLI:NL:2021:1963) (the “Decision”). In the Decision, the Dutch Supreme Court ruled that the (old) system of taxation for savings and investments based on a deemed return may under specific circumstances contravene with Section 1 of the First Protocol to the European Convention on Human Rights in combination with Section 14 of the European Convention on Human Rights (the “EC-Human Rights”). A new court procedure is pending before the Dutch Supreme Court questioning whether the current tax system for savings and investments is in line with the Decision. On 18 September 2023 (ECLI:NL:PHR:2023:655) the Attorney General Wattel concluded that the new tax system is not in line with the Decision, except for the taxation of bank savings, as the system is, in short, still based on a deemed return rather than actual returns, and as a result, the regime contravenes with the EC-Human Rights. The decision of the Dutch Supreme Court is expected mid-2024. In addition, on 8 September 2023, the former cabinet published a law proposal for a new tax system for savings and investments on the basis of actual returns according to an asset accumulation system, the ‘Actual Return Box 3 Act’ (Wet werkelijk rendement box 3). The proposed system is expected to come into effect on 1 January 2027 at the earliest. However, it is up to the new cabinet to submit a final law proposal to the Dutch parliament.
Holders of common shares are advised to consult their own tax advisor to ensure that the tax in respect of the common shares is levied in accordance with the applicable Dutch tax rules at the relevant time.
Non-residents of the Netherlands
A holder of common shares that is neither a Dutch Resident Entity nor a Dutch Resident Individual will not be subject to Dutch (corporate) income tax in respect of income derived or deemed to be derived from the common shares or in respect of capital gains realized on the disposal or deemed disposal of the common shares, provided that:
| (i) | such holder does not have an interest in an enterprise or deemed enterprise (as defined in the Dutch Income Tax Act 2001 and the Dutch Corporate Income Tax Act 1969) which, in whole or in part, is either effectively managed in the Netherlands or carried on through a permanent establishment, a deemed permanent establishment or a permanent representative in the Netherlands and to which enterprise or part of an enterprise the common shares are attributable; and |
| (ii) | in the event the holder is an individual, such holder does not carry out any activities in the Netherlands with respect to the common shares that go beyond ordinary asset management and does not otherwise derive benefits from the common shares that are taxable as benefits from miscellaneous activities in the Netherlands. |
204
Gift and Inheritance Taxes
Residents of the Netherlands
Gift or inheritance taxes will arise in the Netherlands with respect to a transfer of common shares by way of a gift by, or on the death of, a holder of such common shares who is resident or deemed resident of the Netherlands at the time of the gift or the holder’s death.
Non-residents of the Netherlands
No gift or inheritance taxes will arise in the Netherlands with respect to a transfer of common shares by way of gift by, or on the death of, a holder of common shares who is neither resident nor deemed to be resident of the Netherlands, unless:
| (i) | in the case of a gift of common shares by an individual who at the date of the gift was neither resident nor deemed to be resident of the Netherlands, such individual dies within 180 days after the date of the gift, while being resident or deemed to be resident of the Netherlands; or |
| (ii) | in the case of a gift of common shares is made under a condition precedent, the holder of common shares is resident or is deemed to be resident of the Netherlands at the time the condition is fulfilled; or |
| (iii) | the transfer is otherwise construed as a gift or inheritance made by, or on behalf of, a person who, at the time of the gift or death, is or is deemed to be resident of the Netherlands. |
For purposes of Dutch gift and inheritance taxes, among others, a person that holds the Dutch nationality will be deemed to be resident of the Netherlands if such person has been resident in the Netherlands at any time during the ten (10) years preceding the date of the gift or such person’s death. Additionally, for purposes of Dutch gift tax, amongst others, a person not holding the Dutch nationality will be deemed to be resident of the Netherlands if such person has been resident in the Netherlands at any time during the twelve (12) months preceding the date of the gift. Applicable tax treaties may override deemed residency.
Value Added Tax (VAT)
No Dutch VAT will be payable by a holder of common shares in respect of any payment in consideration for the holding or disposal of the common shares.
Stamp Duties
No Dutch documentation taxes (commonly referred to as stamp duties) will be payable by a holder of common shares in respect of any payment in consideration for the holding or disposal of the common shares.
Material German Tax Considerations
The following section is a description of the material German tax considerations that become relevant when purchasing, holding or transferring the company’s shares. The section is based on the German tax law applicable as of the date of this Annual Report. It should be noted that the law may change following the issuance of this Annual Report and that such changes may have retroactive effect.
The material German tax principles of purchasing, owning and transferring of shares are set forth in the following. This section does not purport to be a comprehensive or complete analysis or listing of all potential tax effects of the purchase, ownership or disposition of shares and does not set forth all tax considerations that may be relevant to a particular person’s decision to acquire common shares.
205
This section is intended as general information only and does not purport to be a comprehensive or complete description of all potential German tax effects of the acquisition, ownership or transfer of ordinary shares and does not set forth all German tax considerations that may be relevant to a particular person’s decision to acquire ordinary shares. It does not constitute particular German tax advice and potential purchasers of CureVac’s ordinary shares are urged to consult their own tax advisors regarding the tax consequences of the acquisition, ownership and transfer of ordinary shares in light of their particular circumstances with regard to the application of German tax law to their particular situations, in particular with respect to the procedure to be complied with to obtain a relief of withholding tax on dividends and on capital gains (Kapitalertragsteuer) and with respect to the influence of double tax treaty provisions, as well as any tax consequences arising under the laws of any state, local or other non-German jurisdiction. For German tax purposes, a shareholder may include an individual who or an entity that does not have the legal title to the ordinary shares, but to whom nevertheless the ordinary shares are attributed, based either on such individual or entity owning a beneficial interest in the ordinary shares or based on specific statutory provisions.
All of the following is subject to change. Such changes could apply retroactively and could affect the consequences set forth below. This section does not refer to any U.S. Foreign Account Tax Compliance Act aspects (FACTA).
CureVac’s Tax Residency Status
CureVac has its statutory seat in the Netherlands and its sole place of management in Germany and is therefore tax resident in Germany (for purposes of the German-Dutch tax treaty). Thus, CureVac qualifies as a corporation subject to German unlimited liability for corporate income tax purposes. However, because CureVac’s tax residency depends on future facts regarding its place of management the German unlimited liability for corporate income tax purposes may change in the future.
Dividends Tax
Withholding Tax on Dividends
Dividends distributed from a company to its shareholders are subject to withholding tax, subject to certain exemptions (for example, repayments of capital from the tax equity account (steuerliches Einlagekonto)), as described in the following. The withholding tax rate is 25% plus 5.5% solidarity surcharge (Solidaritätszuschlag) thereon (in total 26.375%) of the gross dividend approved by the ordinary shareholders’ meeting. Withholding tax is to be withheld and passed on for the account of the shareholders by a domestic branch of a domestic or foreign credit or financial services institution (Kredit-und Finanzdienstleistungsinstitut), by the domestic securities trading company (inländisches Wertpapierhandelsunternehmen) or a domestic securities trading bank (inländische Wertpapierhandelsbank) which keeps and administers the shares and disburses or credits the dividends or disburses the dividends to a foreign agent, or by the securities custodian bank (Wertpapiersammelbank) to which the shares were entrusted for collective custody if the dividends are distributed to a foreign agent by such securities custodian bank (which is referred to as the “Dividend Paying Agent”). In case the shares are not held in collective deposit with a Dividend Paying Agent, the company is responsible for withholding and remitting the tax to the competent tax office.
Such withholding tax is levied and withheld irrespective of whether, and to what extent, the dividend distribution is taxable at the level of the shareholder, and whether the shareholder is a person residing in Germany or in a foreign country.
In the case of dividends distributed to a company within the meaning of Art. 2 of the amended EU Directive 2011/96/EU of the Council of November 30, 2011, or the EU Parent Subsidiary Directive, domiciled in another Member State of the European Union, an exemption from withholding tax will be granted upon request if further prerequisites are satisfied (Freistellung im Steuerabzugsverfahren). This also applies to dividends distributed to a permanent establishment located in another Member State of the European Union of such a parent company or of a parent company tax resident in Germany if the participation in the company is effectively connected with this permanent establishment. The key prerequisite for the application of the EU Parent Subsidiary Directive is that the shareholder has held a direct participation in the share capital of the company of at least 10% for at least one year.
206
The withholding tax on distributions to other foreign resident shareholders is reduced in accordance with a double taxation treaty (e.g., to 15%, 5% or 0% depending on certain prerequisites) if Germany has concluded such double taxation treaty with the country of residence of the shareholder and if the shareholder does not hold his shares either as part of the assets of a permanent establishment or a fixed place of business in Germany or as business assets for which a permanent representative has been appointed in Germany. The reduction of the withholding tax is procedurally granted in such a manner that the difference between the total amount withheld, including the solidarity surcharge, and the tax liability determined on the basis of the tax rate set forth in the applicable double taxation treaty (15% unless further qualifications are met) is refunded by the German tax administration upon request (Federal Central Office for Taxes (Bundeszentralamt für Steuern).
In the case of dividends received by corporations whose statutory seat and effective place of management are not located in Germany and who are therefore not tax resident in Germany, two-fifths of the withholding tax deducted and remitted are refunded without the need to fulfill all prerequisites required for such refund under the EU Parent Subsidiary Directive or under a double taxation treaty or if no double taxation treaty has been concluded between the state of residence of the shareholder. However, such refund is subject to the German anti-directive/treaty shopping provision of Section 50d paragraph 3 of the German Income Tax Act (Einkommensteuergesetz).
In order to receive a refund pursuant to a double taxation treaty or the aforementioned option for foreign corporations, the shareholder has to electronically submit a n application via the Federal Central Tax Office’s online portal (www.elster.de/bportal) and upload certain documents during the application, i.a. a withholding tax certificate (Kapitalertragsteuerbescheinigung) issued by the institution that withheld the tax.
The exemption from withholding tax in accordance with the EU Parent Subsidiary Directive or a double tax treaty and the aforementioned options for a refund of the withholding tax (with or without protection under a double taxation treaty) depend on whether certain additional prerequisites (in particular so-called substance requirements) are fulfilled. The applicable withholding tax relief will only be granted if the preconditions of the German anti avoidance rules (so called Directive Override or Treaty Override), in particular Section 50d, paragraph 3, German Income Tax Act (Einkommensteuergesetz) are fulfilled.
The aforementioned reductions of (or exemptions from) withholding tax are further restricted if (i) the applicable double taxation treaty provides for a tax reduction resulting in an applicable tax rate of less than 15% and (ii) the shareholder is not a corporation that directly holds at least 10% in the equity capital of the company and is subject to tax on its income and profits in its state of residence without being exempt. In this case, the reduction of (or exemption from) withholding tax is subject to the following three cumulative prerequisites: (i) the shareholder must qualify as beneficial owner of the shares in the company for a minimum holding period of 45 consecutive days occurring within a period of 45 days prior and 45 days after the due date of the dividends, (ii) the shareholder has to bear at least 70% of the change in value risk related to the shares in the company during the minimum holding period without being directly or indirectly hedged and (iii) the shareholder must not be required to fully or largely compensate directly or indirectly the dividends to third parties. However, these further prerequisites do not apply if the shareholder has been the beneficial owner of the shares in the company for at least one uninterrupted year upon receipt of the dividends.
Absent the fulfillment of all of the three prerequisites, three-fifths of the withholding tax imposed on the dividends must not be credited against the shareholder’s (corporate) income tax liability, but may, upon application, be deducted from the shareholder’s tax base for the relevant assessment period. A shareholder that has received gross dividends without any deduction of withholding tax due to a tax exemption without qualifying for a full tax credit has to notify the competent local tax office accordingly and has to make a payment in the amount of the omitted withholding tax deduction. The special rules on the restriction of withholding tax credit do not apply to a shareholder whose overall dividend earnings within an assessment period do not exceed €20,000 or that has been the beneficial owner of the shares in the company for at least one uninterrupted year upon receipt of the dividends.
207
For individual or corporate shareholders tax resident outside Germany not holding the shares through a permanent establishment (Betriebsstätte) in Germany or as business assets (Betriebsvermögen) for which a permanent representative (ständiger Vertreter) has been appointed in Germany, the remaining and paid withholding tax (if any) is final (i.e., not refundable) and settles the shareholder’s limited tax liability in Germany. For individual or corporate shareholders tax resident in Germany (that are, for example, shareholders whose residence, domicile, registered office or place of management is located in Germany) holding their shares as business assets, as well as for shareholders tax resident outside of Germany holding their shares through a permanent establishment in Germany or as business assets for which a permanent representative has been appointed in Germany, the withholding tax withheld (including solidarity surcharge) can be credited against the shareholder’s personal income tax or corporate income tax liability in Germany. Any withholding tax (including solidarity surcharge) in excess of such tax liability is refunded. For individual shareholders tax resident in Germany holding the company’s shares as private assets, the withholding tax is a final tax (Abgeltungsteuer), subject to the exceptions described in the following section. Taxation of Dividend Income of Shareholders Tax Resident in Germany Holding the Company’s Shares as Private Assets
For individual shareholders (individuals) resident in Germany holding the company’s shares as private assets, dividends are subject to a flat tax rate which is satisfied by the withholding tax actually withheld (Abgeltungsteuer). Accordingly, dividend income will be taxed at a flat tax rate of 25% plus 5.5% solidarity surcharge thereon (in total 26.375%) and church tax (Kirchensteuer) in case the shareholder is subject to church tax because of his individual circumstances. An automatic procedure for deduction of church tax by way of withholding will apply to shareholders being subject to church tax unless the shareholder has filed a blocking notice (Sperrvermerk) with the German Federal Tax Office (details related to the computation of the concrete tax rate including church tax are to be discussed with the individual tax adviser of the relevant shareholder). Except for an annual lump sum savings allowance (Sparer-Pauschbetrag) of up to € 1.000 (for individual filers) or up to € 2.000 (for married couples and for partners in accordance with the registered partnership law (Gesetz über die Eingetragene Lebenspartnerschaft) filing jointly), private individual shareholders will not be entitled to deduct expenses incurred in connection with the capital investment from their dividend income.
The income tax owed for the dividend income is satisfied by the withholding tax withheld by the Dividend Paying Agent. However, if the flat tax results in a higher tax burden as opposed to the private shareholder’s individual tax rate, the private shareholder can opt for taxation at his individual personal income tax rate. In that case, the final withholding tax will be credited against the income tax. However, private shareholders are nevertheless not entitled to deduct expenses incurred in connection with the capital investment from their income. The option can be exercised only for all capital income from capital investments received in the relevant assessment period uniformly, and married couples as well as partners in accordance with the registered partnership law filing jointly may only jointly exercise the option.
Exceptions from the flat tax rate (satisfied by withholding at source) (Abgeltungsteuer) may apply — that is, only upon application — for shareholders who have a shareholding of at least 25% in CureVac or for shareholders who have a shareholding of at least 1% in CureVac and work for the company in a professional capacity. In such a case, the same rules apply as for sole proprietors holding the shares as business assets. See “— Taxation of Dividend Income of Shareholders Tax Resident in Germany Holding the Company’s shares as Business Assets — Sole Proprietors.” Further, the flat rate tax does not apply if and to the extent dividends reduced CureVac taxable income.
Taxation of Dividend Income of Shareholders Tax Resident in Germany Holding the Company’s shares as Business Assets
If a shareholder holds the company’s shares as business assets, the taxation of the dividend income depends on whether the respective shareholder is a corporation, a sole proprietor or a partnership.
208
Corporations
Dividend income of corporate shareholders is exempt from corporate income tax, provided that the incorporated entity holds a direct participation of at least 10% in the share capital of a company at the beginning of the calendar year in which the dividends are paid. The acquisition of a participation of at least 10% in the course of a calendar year is deemed to have occurred at the beginning of such calendar year for the purpose of this rule. Participations in the share capital of the company which a corporate shareholder holds through a partnership, including co-entrepreneurships (Mitunternehmerschaften), are attributable to such corporate shareholder only on a pro rata basis at the ratio of the interest share of the corporate shareholder in the assets of the relevant partnership. However, 5% of the tax-exempt dividends are deemed to be non-deductible business expenses for tax purposes and therefore are subject to corporate income tax (plus solidarity surcharge) and trade tax, i.e., tax exemption of 95%. Business expenses incurred in connection with the dividends received are entirely tax-deductible. The participation exemption does not apply if and to the extent dividends reduced CureVac’s taxable income.
For trade tax purposes the entire dividend income is subject to trade tax (i.e., the tax-exempt dividends must be added back when determining the trade taxable income), unless the corporation shareholder holds at least 15% of the company’s registered share capital at the beginning of the relevant tax assessment period (Erhebungszeitraum). In case of an indirect participation via a partnership please refer to the section “Partnerships” below.
If the shareholding is below 10% in the share capital, dividends are taxable at the applicable corporate income tax rate of 15% plus 5.5% solidarity surcharge thereon and trade tax (the rate of which depends on the municipalities the corporate shareholder resides in).
Special regulations apply which abolish the 95% tax exemption if the company’s shares are held as trading portfolio assets in the meaning of Section 340e of the German commercial code (Handelsgesetzbuch) by (i) a credit institution (Kreditinstitut), (ii) a financial service institution (Finanzdienstleistungsinstitut) or (iii) a financial enterprise within the meaning of the German Banking Act (Kreditwesengesetz), in cases where more than 50% of the shares of such financial enterprise are held directly or indirectly by a credit institution or a financial service institution, as well as by a life insurance company, a health insurance company or a pension fund in case the shares are attributable to the capital investments, resulting in fully taxable income.
Sole Proprietors
For sole proprietors (individuals) resident in Germany holding shares as business assets dividends are subject to the partial income rule (Teileinkünfteverfahren). Accordingly, only (i) 60% of the dividend income will be taxed at his/her individual personal income tax rate plus 5.5% solidarity surcharge thereon and church tax (if applicable) and (ii) 60% of the business expenses related to the dividend income are deductible for tax purposes. In addition, the dividend income is entirely subject to trade tax if the shares are held as business assets of a permanent establishment in Germany within the meaning of the German Trade Tax Act (Gewerbesteuergesetz), unless the shareholder holds at least 15% of the company’s registered share capital at the beginning of the relevant assessment period.
However, if a partner is an individual, depending on the applicable municipal trade tax rate and the individual tax situation, the trade tax paid at the level of the partnership is credited against the partner’s personal income tax liability up to four times of the trade measurement amount (Gewerbesteuer-Messbetrag) depending on the applicable municipal trade tax levy rate and the personal tax situation.
Partnerships
In case shares are held by a partnership, the partnership itself is not subject to corporate income tax or personal income tax. In this regard, corporate income tax or personal income tax (and church tax, if applicable) as well as solidarity surcharge, are levied only at the level of the partner with respect to their relevant part of the profit and depending on their individual circumstances.
If the partner is a corporation, the dividend income will be subject to corporate income tax plus solidarity surcharge. See “— Corporations.”
If the partner is a sole proprietor, the dividend income will be subject to the partial income rule. See “— Sole Proprietors.”
209
If the partner is a private individual, the dividend income will be subject to the flat tax rate. See “Taxation of Dividend Income of Shareholders Tax Resident in Germany Holding the Company’s Shares as Private Assets” above; unless the partnership is a (operative or deemed) commercial partnership in which case the partial income rule applies.
If a partner is an individual, depending on the applicable municipal trade tax rate and the individual tax situation, the trade tax paid at the level of the partnership is partly or entirely be credited against the partner’s personal income tax liability.
In case the partnership is a (operative or deemed) commercial partnership with its place of management in Germany, the dividend income is subject to German trade tax at the level of the partnership, unless the partnership holds at least 15% of a company’s registered share capital at the beginning of the relevant assessment period, in which case the dividend income is exempt from trade tax.
Taxation of Dividend Income of Shareholders Tax Resident Outside of Germany
For foreign individual or corporate shareholders tax resident outside of Germany not holding the shares through a permanent establishment in Germany or as business assets for which a permanent representative has been appointed in Germany, the deducted withholding tax (possibly reduced by way of a tax relief under a double tax treaty or domestic tax law, such as in connection with the EU Parent Subsidiary Directive) is final (that is, not refundable) and settles the shareholder’s limited tax liability in Germany, unless the shareholder is entitled to apply for a withholding tax refund or exemption.
In contrast, individual or corporate shareholders tax resident outside of Germany holding the company’s shares through a permanent establishment in Germany or as business assets for which a permanent representative has been appointed in Germany are subject to the same rules as applicable (and described above) to shareholders resident in Germany holding the shares as business assets. The withholding tax withheld (including solidarity surcharge) is credited against the shareholder’s personal income tax or corporate income tax liability in Germany.
Taxation of Capital Gains
Withholding Tax on Capital Gains
Capital gains realized on the disposal of shares are only subject to withholding tax if a German branch of a German or foreign credit or financial institution, a German securities trading company or a German securities trading bank stores or administrates or carries out the sale of the shares and pays or credits the capital gains. In those cases, the institution (and not the company) is required to deduct the withholding tax at the time of payment for the account of the shareholder and has to pay the withholding tax to the competent tax authority.
In the case that the shares in CureVac N.V. are held (i) as business assets by a sole proprietor, a partnership or a corporation and such shares are attributable to a German business or (ii) in the case of a corporation being subject to unlimited corporate income tax liability in Germany, the capital gains are not subject to withholding tax. In the case of clause (i), the withholding tax exemption is subject to the condition that the paying agent has been notified by the beneficiary (Gläubiger) that the capital gains are exempt from withholding tax. The respective notification has to be filed by using the officially prescribed form.
Taxation of Capital Gains Realized by Shareholders Tax Resident in Germany Holding Shares as Private Assets
For individual shareholders (individuals) resident in Germany holding shares as private assets, capital gains realized on the disposal of shares are subject to final withholding tax. Accordingly, capital gains will be taxed at a flat tax rate of 25% plus a 5.5% solidarity surcharge thereon (in total 26.375%) and church tax, in case the shareholder is subject to church tax because of his individual circumstances. An automatic procedure for deduction of church tax by way of withholding will apply to shareholders being subject to church tax unless the shareholder has filed a blocking notice (Sperrvermerk) with the German Federal Tax Office (details related to the computation of the concrete tax rate including church tax are to be discussed with the individual tax adviser of the relevant shareholder). The taxable capital gain is calculated by deducting the acquisition costs of the shares and the expenses directly related to the disposal from the proceeds of the disposal. Apart from that, except for an annual lump sum savings allowance (Sparer-Pauschbetrag) of up to €1,000 (for individual filers) or up to €2,000 (for married couples and for partners in accordance with the registered partnership law (Gesetz über die Eingetragene Lebenspartnerschaft) filing jointly), private individual shareholders will not be entitled to deduct expenses incurred in connection with the capital investment from their capital gain.
210
In case the flat tax results in a higher tax burden as opposed to the private shareholder’s individual tax rate, the private shareholder can opt for taxation at his or her individual personal income tax rate. In that case, the withholding tax (including solidarity surcharge) withheld will be credited against the income tax. However, the private shareholders are nevertheless not entitled to deduct expenses incurred in connection with the capital investment from their income. The option can be exercised only for all capital income from capital investments received in the relevant assessment period uniformly, and married couples as well as for partners in accordance with the registered partnership law filing jointly may only jointly exercise the option.
Capital losses arising from the sale of the shares can only be offset against other capital gains resulting from the disposition of the shares or shares in other stock corporations during the same calendar year. Offsetting of overall losses with other income (such as business or rental income) and other capital income is not possible. Such losses are to be carried forward and to be offset against positive capital gains deriving from the sale of shares in stock corporations in future years. In case of a derecognition or transfer of worthless shares (or other capital assets), the utilization of such loss is further restricted and can only be offset up to the amount of €20,000 per calendar year.
The final withholding tax will not apply if the seller of the shares or, in the case of gratuitous transfer, its legal predecessor has held, directly or indirectly, at least 1% of the company’s registered share capital at any time during the five years prior to the disposal. In that case capital gains are subject to the partial income rule. Accordingly, only (i) 60% of the capital gains will be taxed at his individual personal income tax rate plus a 5.5% solidarity surcharge thereon and church tax (if applicable) and (ii) 60% of the business expenses related to the capital gains are deductible for tax purposes. The withholding tax withheld (including solidarity surcharge) will be credited against the shareholder’s personal income tax liability in Germany.
Taxation of Capital Gains Realized by Shareholders Tax Resident in Germany Holding the Company’s Shares as Business Assets
If a shareholder holds shares as business assets, the taxation of capital gains realized on the disposal of such shares depends on whether the respective shareholder is a corporation, a sole proprietor or a partnership:
Corporations
Capital gains realized on the disposal of shares by a corporate shareholder are generally exempt from corporate income tax and trade tax. However, 5% of the tax-exempt capital gains are deemed to be non-deductible business expenses for tax purposes and therefore are subject to corporate income tax (plus solidarity surcharge) and trade tax, i.e., tax exemption of 95%. Business expenses incurred in connection with the capital gains are entirely tax-deductible.
Capital losses incurred upon the disposal of shares or other impairments of the share value are not tax deductible. A reduction of profit is also defined as any losses incurred in connection with a loan or security in the event the loan or the security is granted by a shareholder or by a related party thereto or by a third person with the right of recourse against the aforementioned persons, and the shareholder holds directly or indirectly more than 25% of the company’s registered share capital.
Special regulations apply if the shares are held as trading portfolio assets by a credit institution, a financial service institution or a financial enterprise within the meaning of the German Banking Act (Kreditwesengesetz) as well as by a life insurance company, a health insurance company or a pension fund. See “— Taxation of Dividend Income of Shareholders Tax Resident in Germany Holding the Company’s shares as Business Assets — Corporations.”
Sole Proprietors
If the shares are held by a sole proprietor, capital gains realized on the disposal of the shares are subject to the partial income rule. Accordingly, only (i) 60% of the capital gains will be taxed at his/her individual personal income tax rate plus a 5.5% solidarity surcharge thereon and church tax (if applicable) and (ii) 60% of the business expenses related to the dividend income are deductible for tax purposes. In addition, 60% of the capital gains are subject to trade tax if the shares are held as business assets of a permanent establishment in Germany within the meaning of the German Trade Tax Act (Gewerbesteuergesetz). The trade tax levied, depending on the applicable municipal trade tax rate and the individual tax situation, is partly or entirely credited against the shareholder’s personal income tax liability.
211
Partnerships
In case the shares are held by a partnership, the partnership itself is not subject to corporate income tax or personal income tax as well as a solidarity surcharge (and church tax) since partnerships qualify as transparent for German tax purposes. In this regard, corporate income tax or personal income tax as well as a solidarity surcharge (and church tax, if applicable), are levied only at the level of the partner with respect to their relevant part of the profit and depending on their individual circumstances.
If the partner is a corporation, the capital gains will be subject to corporate income tax plus a solidarity surcharge. See “— Corporations.” Trade tax will be levied additionally at the level of the partner insofar as the relevant profit of the partnership is not subject to trade tax at the level of the partnership. However, with respect to both corporate income and trade tax, the 95% exemption rule as described above applies. With regard to corporate partners, special regulations apply if they are held as trading portfolio assets by credit institutions, financial service institutions or financial enterprises within the meaning of the German Banking Act or life insurance companies, health insurance companies or pension funds, as described above.
If the partner is a sole proprietor (individual), the capital gains are subject to the partial income rule. See “— Sole Proprietors.”
In addition, if the partnership is liable to trade tax, 60% of the capital gains are subject to trade tax at the level of the partnership, to the extent the partners are individuals, and 5% of the capital gains are subject to trade tax, to the extent the partners are corporations. However, if a partner is an individual, depending on the applicable municipal trade tax rate and the individual tax situation, the trade tax paid at the level of the partnership is credited against the partner’s personal income tax liability up to four times of the trade tax measurement amount (Gewerbesteuer-Messbetrag) depending on the applicable municipal trade tax levy rate and the personal tax situation.
A new tax regime enables certain partnerships to be taxed as a corporation and their partners to be taxed like shareholders of a corporation. The election is available for fiscal years ending in 2022 or later years. See “— Corporations.”
Taxation of Capital Gains Realized by Shareholders Tax Resident Outside of Germany
Capital gains realized on the disposal of the shares by a shareholder tax resident outside of Germany are subject to German taxation provided that (i) the company’s shares are held as business assets of a permanent establishment or as business assets for which a permanent representative has been appointed in Germany, or (ii) the shareholder or, in case of a gratuitous transfer, its legal predecessor has held, directly or indirectly, at least 1% of the company’s shares capital at any time during a five-year period prior to the disposal. In these cases, capital gains are generally subject to the same rules as described above for shareholders resident in Germany. However, except for the cases referred to in (i) above, most double tax treaties concluded by Germany provide for a full exemption from German taxation unless the company is considered a real estate holding entity for treaty purposes. Further, in case of non-German corporations, the participation exemption applies in full resulting in a tax exemption of 100% (i.e., no deemed non-tax-deductible business expenses).
Inheritance and Gift Tax
The transfer of the company’s shares to another person by way of succession or donation is subject to German inheritance and gift tax (Erbschaft-und Schenkungsteuer) if:
| (i) | the decedent, the donor, the heir, the donee or any other beneficiary has his/her/its residence, domicile, registered office or place of management in Germany at the time of the transfer, or is a German citizen who has not stayed abroad for more than five consecutive years without having a residence in Germany; or |
| (ii) | (irrespective of the personal circumstances) the shares are held by the decedent or donor as business assets for which a permanent establishment in Germany is maintained or a permanent representative is appointed in Germany; or |
| (iii) | (irrespective of the personal circumstances) at least 10% of the shares are held, directly or indirectly by, the decedent or person making the gift, himself or together with a related party in terms of Section 1, paragraph 2 of the Foreign Tax Act (Außensteuergesetz). |
212
Special regulations apply to qualified German citizens who maintain neither a residence nor their domicile in Germany but in a low tax jurisdiction, and to former German citizens, also resulting in inheritance and gift tax. The few double tax treaties on inheritance and gift tax which Germany has entered into provide that German inheritance and gift tax is levied only in the case of (i) and, with certain restrictions, in the case of (ii).
Other Taxes
No German capital transfer tax (Kapitalverkehrsteuer), value added tax (Umsatzsteuer), stamp duty (Stempelgebühr) or similar taxes are levied when acquiring, holding or transferring the company’s shares. No value added tax will be levied unless the shareholder validly opts for it. Net wealth tax (Vermögensteuer) is currently not levied in Germany.
On January 22, 2013, the Council of the European Union approved the resolution of the ministers of finance from 11 European Union member states (including Germany) to introduce a Financial Transaction Tax, or FTT, within the framework of enhanced cooperation. On February 14, 2013, the EC published a proposal for a Council Directive implementing enhanced cooperation in the area of financial transaction tax. The plan focuses on levying a tax of 0.1% (0.01% for derivatives) on the purchase and sale of financial instruments.
A joint statement issued by 10 of the 11 participating European Union member states in October 2016 reaffirmed the intention to introduce FTT. However, at the moment not many details are available. Thus, it is not known to what extent the elements of the European Commission’s proposal outlined in the preceding paragraph will be followed in relation to the taxation of shares. The FTT proposal remains subject to negotiation between the participating member states and is subject to political discussion. It may, therefore, be altered prior to the implementation, the timing of which remains unclear. Additional European Union member states may decide to participate.
Prospective holders of the shares are advised to seek their own professional advice in relation to FTT.
Material U.S. Federal Income Tax Considerations to U.S. Holders
The following is a description of the material U.S. federal income tax consequences to the U.S. Holders, as defined below, of owning and disposing of our common shares. It does not describe all tax considerations that may be relevant to a particular person’s decision to invest in common shares.
This discussion applies only to a U.S. Holder that holds common shares as capital assets for U.S. federal income tax purposes. In addition, it does not describe all of the U.S. federal income tax consequences that may be relevant in light of the U.S. Holder’s particular circumstances, including alternative minimum tax consequences, the potential application of the provisions of the Code known as the Medicare contribution tax and tax consequences applicable to U.S. Holders subject to special rules, such as:
| ● | certain financial institutions; |
| ● | dealers or traders in securities who use a mark-to-market method of tax accounting; |
| ● | persons holding common shares as part of a hedging transaction, straddle, wash sale, conversion transaction or other integrated transaction or persons entering into a constructive sale with respect to the common shares; |
| ● | persons whose functional currency for U.S. federal income tax purposes is not the U.S. dollar; |
| ● | entities classified as partnerships for U.S. federal income tax purposes; |
| ● | tax-exempt entities, including an “individual retirement account” or “Roth IRA”; |
| ● | persons that own or are deemed to own ten percent or more of our common shares (by vote or value); or |
| ● | persons holding common shares in connection with a trade or business conducted outside of the United States. |
213
If an entity that is classified as a partnership for U.S. federal income tax purposes holds common shares, the U.S. federal income tax treatment of a partner will generally depend on the status of the partner and the activities of the partnership. Partnerships holding common shares and partners in such partnerships should consult their tax advisers as to the particular U.S. federal income tax consequences of owning and disposing of the common shares.
This discussion is based on the Code, administrative pronouncements, judicial decisions, final, temporary and proposed Treasury regulations, and the income tax treaty between the Federal Republic of Germany and the United States, or the Treaty, all as of the date hereof, any of which is subject to change or differing interpretations, possibly with retroactive effect.
A “U.S. Holder” is a holder who, for U.S. federal income tax purposes, is a beneficial owner of our common shares, who is eligible for the benefits of the Treaty and who is:
| ● | an individual who is a citizen or resident of the United States; |
| ● | a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or the District of Columbia; or |
| ● | an estate or trust, the income of which is subject to U.S. federal income taxation regardless of its source. |
Treasury regulations that apply to taxable years beginning on or after December 28, 2021, may in some circumstances prohibit a U.S. person from claiming a foreign tax credit with respect to certain non-U.S. taxes that are not creditable under applicable income tax treaties. Accordingly, U.S. investors that are not eligible for Treaty benefits should consult their tax advisers regarding the creditability or deductibility of any non-U.S. taxes imposed on them. This discussion does not apply to investors in this special situation. U.S. Holders should consult their tax advisers concerning the U.S. federal, state, local and non-U.S. tax consequences of owning and disposing of our common shares in their particular circumstances.
Taxation of Distributions
As discussed above under “Item 8. Financial Information — A. Consolidated statements and other financial information — Dividends and dividend policy” we have never paid or declared any cash dividends on our common shares, and we do not anticipate paying any cash dividends on our common shares in the foreseeable future. In the event that we do make distributions of cash or other property, subject to the passive foreign investment company rules described below, distributions paid on common shares, other than certain pro rata distributions of common shares, will generally be treated as dividends to the extent paid out of our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). Because we do not maintain calculations of our earnings and profits under U.S. federal income tax principles, we expect that distributions generally will be reported to U.S. Holders as dividends. For so long as our common shares are listed on the Nasdaq or another established securities market in the United States or we are eligible for benefits under the Treaty, dividends paid to certain non-corporate U.S. Holders may be eligible for taxation as “qualified dividend income” and therefore, subject to applicable limitations, taxable at rates not in excess of the long-term capital gain rate applicable to such U.S. Holders. U.S. Holders should consult their tax advisers regarding the availability of the reduced tax rate on dividends in their particular circumstances. The amount of a dividend will include any amounts withheld by us in respect of German income taxes. The amount of the dividend will be treated as foreign-source dividend income to U.S. Holders and will not be eligible for the dividends-received deduction generally available to U.S. corporations under the Code. Dividends will be included in a U.S. Holder’s income on the date of the U.S. Holder’s receipt of the dividend. The amount of any dividend income paid in euros will be the U.S. dollar amount calculated by reference to the exchange rate in effect on the date of actual or constructive receipt, regardless of whether the payment is in fact converted into U.S. dollars at that time. If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder should not be required to recognize foreign currency gain or loss in respect of the dividend income. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt.
Subject to applicable limitations, some of which vary depending upon the U.S. Holder’s particular circumstances, German income taxes withheld from dividends on common shares at a rate not exceeding the rate provided by the Treaty will be creditable against the U.S. Holder’s U.S. federal income tax liability.
214
German taxes withheld in excess of the rate applicable under the Treaty will not be eligible for credit against a U.S. Holder’s federal income tax liability. The rules governing foreign tax credits are complex, and U.S. Holders should consult their tax advisers regarding the creditability of foreign taxes in their particular circumstances. In lieu of claiming a foreign tax credit, U.S. Holders may, at their election, deduct foreign taxes, including any German income tax, in computing their taxable income, subject to generally applicable limitations under U.S. law. An election to deduct foreign taxes instead of claiming foreign tax credits applies to all foreign taxes paid or accrued in the taxable year.
Sale or Other Disposition of Common Shares
Subject to the passive foreign investment company rules described below, gain or loss realized on the sale or other disposition of common shares will be capital gain or loss, and will be long-term capital gain or loss if the U.S. Holder held the common shares for more than one year. The amount of the gain or loss will equal the difference between the U.S. Holder’s tax basis in the common shares disposed of and the amount realized on the disposition, in each case as determined in U.S. dollars. This gain or loss will generally be U.S.-source gain or loss for foreign tax credit purposes. The deductibility of capital losses is subject to various limitations. The Treaty generally exempts a U.S. Holder from German tax on capital gains realized on the sale or other disposition of common shares and, accordingly, no such tax will be creditable against the U.S. Holder’s U.S. federal income tax liability.
Passive Foreign Investment Company Rules
Under the Code, we will generally be a “passive foreign investment company,” or a PFIC, for any taxable year in which, after the application of certain “look-through” rules with respect to subsidiaries, either (i) 75% or more of our gross income consists of “passive income,” or (ii) 50% or more of the average quarterly value of our assets consist of assets that produce, or are held for the production of, “passive income.” For purposes of the above calculations, we will be treated as if we hold our proportionate share of the assets of, and receive directly our proportionate share of the income of, any other corporation in which we directly or indirectly own at least 25%, by value, of the shares of such corporation. Passive income generally includes dividends, interest, rents, certain non-active royalties and capital gains. The value of a non-U.S. corporation’s goodwill that is associated with activities that produce, or are intended to produce, active income is generally an active asset for purposes of the asset test unless, for U.S. federal income tax purposes, the non-U.S. corporation is a “controlled foreign corporation,” or CFC, that is not publicly traded “for the taxable year.” If a non-U.S. corporation is a CFC that is not publicly traded for the taxable year, its PFIC status under the asset test is determined by using the U.S. tax basis of its assets rather than their fair market value and therefore the market value of its goodwill is generally disregarded. Generally, a non-U.S. corporation is a CFC if more than 50% of its shares’ voting power or value is owned, directly, indirectly or constructively, by “United States shareholders” (as defined in Section 951(b) of the Code). Although it is not certain, we may be or may have been a CFC in our 2023 taxable year. However, under Treasury regulations promulgated in 2020, the fair market value of our assets (including goodwill) can be used for purposes of the asset test provided that (i) we are publicly traded on the majority of days during our taxable year or (ii) we would not be a CFC if certain constructive ownership rules were not applied. We believe, and the remainder of this discussion assumes, that we are eligible to use the fair market value of our assets for purposes of the asset test, for our 2023 taxable year.
Based on the composition of our income and assets during 2023, we do not believe that we were a PFIC for our 2023 taxable year. However, there can be no assurance that the IRS will agree with our conclusion. Whether we will be a PFIC in 2024 or any future year is uncertain because, among other things, (i) we currently own a substantial amount of passive assets, including cash, (ii) the valuation of our assets that generate non-passive income for PFIC purposes, including our intangible assets, is uncertain and may vary substantially over time, (iii) the treatment of grants as income for U.S. federal income tax purposes is unclear, and (iv) the composition of our income may vary substantially over time. Accordingly, there can be no assurance that we will not be a PFIC in 2024 or any future taxable year. If we are a PFIC for any year during which a U.S. Holder holds common shares, we generally would continue to be treated as a PFIC with respect to that U.S. Holder for all succeeding years during which the U.S. Holder holds common shares, even if we ceased to meet the threshold requirements for PFIC status. In addition, we may, directly or indirectly, have held or hold equity interests in other PFICs, collectively referred to as Lower-Tier PFICs. Under attribution rules, if we are a PFIC, U.S. Holders will be deemed to own their proportionate shares of the stock of Lower-Tier PFICs and will be subject to U.S. federal income tax according to the rules described in the following paragraphs on (i) certain distributions by a Lower-Tier PFIC and (ii) a disposition of shares of a Lower-Tier PFIC, in each case as if the U.S. Holder held such shares directly, even though holders have not received the proceeds of those distributions or dispositions directly. U.S. Holders should consult their tax advisors about the consequences to them of our investment in one or more Lower-Tier PFICs.
215
If we were a PFIC for any taxable year during which a U.S. Holder held common shares (assuming such U.S. Holder has not made a timely mark-to-market election, as described below), gain recognized by a U.S. Holder on a sale or other disposition (including certain pledges) of the common shares would be allocated ratably over the U.S. Holder’s holding period for the common shares. The amounts allocated to the taxable year of the sale or other disposition and to any year before we became a PFIC would be taxed as ordinary income. The amount allocated to each other taxable year would be subject to tax at the highest rate in effect for individuals or corporations, as appropriate, for that taxable year, and an interest charge would be imposed on the amount allocated to that taxable year. Further, to the extent that any distribution received by a U.S. Holder on its common shares exceeds 125% of the average of the annual distributions on the common shares received during the preceding three years or the U.S. Holder’s holding period, whichever is shorter, that distribution would be subject to taxation in the same manner as gain, described immediately above.
A U.S. Holder can avoid certain of the adverse rules described above by making a mark-to-market election with respect to its common shares, provided that the common shares are “marketable.” Common shares will be marketable if they are “regularly traded” on a “qualified exchange” or other market within the meaning of applicable Treasury regulations. If a U.S. Holder makes the mark-to-market election, it generally will recognize as ordinary income any excess of the fair market value of the common shares at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the common shares over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). If a U.S. Holder makes the election, the U.S. Holder’s tax basis in our common shares will be adjusted to reflect the income or loss amounts recognized. Any gain recognized on the sale or other disposition of common shares in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). A mark-to-market election generally cannot be made for equity interests in any Lower-Tier PFIC unless shares of such Lower-Tier PFIC are themselves “marketable.” As a result, if a U.S. Holder makes a mark-to-market election with respect to our common shares, the U.S. Holder would nevertheless be subject to the PFIC rules described above with respect to its indirect interest in any Lower-Tier PFIC unless the U.S. Holder makes a qualified electing fund election, also known as a QEF Election, with respect to such Lower-Tier PFIC, as discussed below.
In addition, in order to avoid the application of the foregoing rules, a U.S. Holder that owns stock in a PFIC for U.S. federal income tax purposes may make a QEF Election with respect to such PFIC if the PFIC provides the information necessary for such election to be made. If a U.S. Holder makes a QEF Election with respect to a PFIC, the U.S. Holder will be currently taxable on its pro rata share of the PFIC’s ordinary earnings and net capital gain (at ordinary income and capital gain rates, respectively) for each taxable year that the entity is classified as a PFIC and will not be required to include such amounts in income when actually distributed by the PFIC. There is no assurance that we will provide information necessary for U.S. Holders to make QEF Elections. A QEF Election with respect to us will not apply to any Lower-tier PFIC. If we determine that any of our subsidiaries is a Lower-Tier PFIC for any taxable year, there is no assurance that we will provide information necessary for U.S. Holders to make a QEF Election with respect to such Lower-Tier PFIC.
In addition, if we were a PFIC or, with respect to a particular U.S. Holder, were treated as a PFIC for the taxable year in which we paid a dividend or for the prior taxable year, the preferential dividend rates discussed above with respect to dividends paid to certain non-corporate U.S. Holders would not apply.
If a U.S. Holder owns common shares during any year in which we are a PFIC, the U.S. Holder generally must file annual reports, containing such information as the U.S. Treasury may require on IRS Form 8621 (or any successor form) with respect to us, generally with the U.S. Holder’s federal income tax return for that year.
U.S. Holders should consult their tax advisers concerning our potential PFIC status and the potential application of the PFIC rules.
Information Reporting and Backup Withholding
Payments of dividends and sales proceeds that are made within the United States or through certain U.S.-related financial intermediaries generally are subject to information reporting, and may be subject to backup withholding, unless (i) the U.S. Holder is a corporation or other exempt recipient, or (ii) in the case of backup withholding, the U.S. Holder provides a correct taxpayer identification number and certifies that it is not subject to backup withholding.
The amount of any backup withholding from a payment to a U.S. Holder will be allowed as a credit against the U.S. Holder’s U.S. federal income tax liability and may entitle it to a refund, provided that the required information is timely furnished to the IRS.
216
F. Dividends and Paying Agents
Not applicable.
G. Statement by Experts
Not applicable.
H. Documents on Display
We are subject to the informational requirements of the Exchange Act. Accordingly, are required to file reports and other information with the SEC, including annual reports on Form 20-F and reports on Form 6-K. The SEC maintains an Internet website that contains reports and other information about issuers, like us, that file electronically with the SEC. The address of that website is www.sec.gov.
I. Subsidiary Information
Not applicable.
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
In the ordinary course of our business activities, we are exposed to various market risks that are beyond our control, including fluctuations in foreign exchange rates, which may have an adverse effect on the value of our financial assets and liabilities, future cash flows and profit. As a result of these market risks, we could suffer a loss due to adverse changes in foreign exchange rates in the countries in which we operate. Our policy with respect to these market risks is to assess the potential of experiencing losses and the consolidated impact thereof and to mitigate these market risks. We are not currently exposed to significant interest rate risk because we do not currently hold long-term debt that is exposed to market rates. See note 16 to our financial statements contained elsewhere in this Annual Report for further information on our risk management policies and exposure to market risks.
Credit Risk
Our credit risk arises primarily from cash and cash equivalents and other financial assets, including deposits with banks and financial institutions, as well as credit exposures to customers, including outstanding receivables and contract assets. These financial instruments approximate fair value due to short-term maturities. We maintain our cash and cash equivalents and short-term investments with high credit quality financial institutions. We believe that our credit policies reflect normal industry terms and business risk.
Foreign Currency Risk
Foreign currency risk is the risk that the fair value or future cash flows of an exposure will fluctuate because of changes in foreign exchange rates. Our exposure to the risk of changes in foreign exchange rates relates primarily to our operating activities (when revenue or expense is denominated in a foreign currency) and the amounts held as cash and cash equivalents. Our consolidated financial statements are reported in euros. We generate a significant portion of our revenue and incur a significant portion of our expenditures in certain non-euro currencies, principally U.S. dollars. We are exposed to fluctuations in foreign currency exchange rates primarily on revenue generated from sales in these foreign currencies. Our results of operations can be affected if the U.S. dollar appreciates or depreciates against the euro. As of December 31, 2023, if the euro had weakened 10% against the U.S. dollar with all other variables held constant, pre-tax loss for the year would have been €8.0 million (2022: €8.6 million) lower and post-tax loss would have been €5.6 million lower (2022: €6.1 million). Conversely, if the euro had strengthened 10% against the U.S. dollar with all other variables held constant, pre-tax loss would have been €6.6 million (2022: €7.1 million) higher and post-tax loss would have been €4.6 million (2022: €5.0 million) higher. The effects on pre- and post-tax loss and (accumulated) other comprehensive income due to fact that our subsidiary CureVac Inc.’s functional currency is the U.S. dollar would still have been immaterial at December 31, 2022 and 2023.
217
To the extent that we need to convert U.S. dollars into foreign currencies for our operations, appreciation of such foreign currencies against the U.S. dollar would adversely affect the amount of such foreign currencies we receive from the conversion. Sensitivity analysis is used as a primary tool in evaluating the effects of changes in foreign currency exchange rates on our business operations. The analysis quantifies the impact of potential changes in these rates on our earnings, cash flows and fair values of assets and liabilities during the forecast period, most commonly within a one-year period. The ranges of changes used for the purpose of this analysis reflect our view of changes that are reasonably possible over the forecast period. Fair values are the present value of projected future cash flows based on market rates and chosen prices.
Interest Rate Risk
Interest rate risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market interest rates. Our exposure to the risk of changes in market interest rates relates primarily to our cash and cash equivalents with floating interest rates. Due to persistent low interest rates we may be exposed to the risk of being charged negative interest rates on our bank deposits. If interest rates as of December 31, 2022 and 2023, had been 1% higher while all other variables had remained the same, the net loss for the year (before and after tax) would have been €4.0 million (2022: €5.0 million) lower, because the higher interest income would have been generated from floating rates on invested cash and cash equivalents.
ITEM 12. DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
A. Debt Securities
Not applicable.
B. Warrants and Rights
Not applicable.
C. Other Securities
Not applicable.
D. American Depositary Shares
Not applicable.
218
PART II
ITEM 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
A. Defaults
No matters to report.
B. Arrears and Delinquencies
No matters to report.
ITEM 14. MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS
A. Material Modifications to Instruments
Not applicable.
B. Material Modifications to Rights
Not applicable.
C. Withdrawal or Submission of Assets
Not applicable.
D. Changes in Trustees or Paying Agents
Not applicable.
E. Use of Proceeds
None.
ITEM 15. CONTROLS AND PROCEDURES
A. Disclosure Controls and Procedures
As required by Rules 13a-15(e) and 15d-15(e) under the Exchange Act, our management, including our Chief Executive Officer (CEO) and our Chief Financial Officer (CFO), has performed an evaluation of the effectiveness of our disclosure controls and procedures as of December 31, 2023. Disclosure controls and procedures refer to controls and other procedures designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. Disclosure controls and procedures include, without limitations, controls and procedures designed to ensure that information required to be disclosed by us in our reports that we file or submit under the Exchange Act is accumulated and communicated to management, including our principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding our required disclosures.
Based on the foregoing, our Chief Executive Officer and our Chief Financial Officer, together with our other members of management, have concluded that, as of December 31, 2023, due to a material weakness in internal control over financial reporting described below, our disclosure controls and procedures were not effective.
219
B. Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act). Internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with IFRS as issued by IASB and includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of our Company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with IFRS as issued by IASB, and that receipts and expenditures of our Company are being made only in accordance with authorizations of management and directors of our Company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our Company’s assets that could have a material effect on the financial statements. A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
Any internal control system, no matter how well designed, has inherent limitations, including the possibility of human error and the circumvention or overriding of the controls and procedures, which may not prevent or detect misstatements.
Our management has assessed the effectiveness of our internal control over financial reporting as of December 31, 2023. In making this assessment, our management used the updated criteria set forth in 2013 by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in “Internal Control – Integrated Framework”.
Our management has concluded, based on its assessment, that our internal control over financial reporting was not effective as of December 31, 2023 due to a material weakness resulting from (i) lack of sufficient accounting and supervisory personnel, (ii) lack of appropriate level of technical accounting training; and (iii) lack of consistent performance of controls over financial reporting.
Notwithstanding the material weakness identified as of December 31, 2023, our management has concluded that the consolidated financial statements included in this Annual Report on Form 20-F, present fairly, in all material respects, our financial position, results of operations and cash flows for the periods presented in conformity with IFRS as issued by IASB.
Remediation plan
As previously disclosed, we reported in our Annual Report 20-F for the year ended December 31, 2022, a material weakness in internal control over financial reporting primarily related to an IT system’s functionalities that were not configured to support segregation of duties in the recording of manual journal entries as well as in the authorization of purchase orders. During 2023, we implemented measures to successfully remediate this material weakness.
Management’s planned remediation measures for the material weakness identified as of December 31, 2023 include (i) training and hiring additional qualified staff; (ii) implementing, streamlining, and/or replacing certain IT systems to increase automation in accounting processes and procedures; and (iii) engaging third parties, as required, to assist with technical expertise, integration and streamlining of business controls.
Management believes the foregoing plans will effectively remediate the deficiencies constituting the material weakness. However, there is no assurance as to when such remediation will be successful. As the remediation plans are implemented, management may take additional measures or modify the plan described above. See “Item 3. Key Information — D. Risk Factors — We have identified a material weakness in our internal control related (i) lack of sufficient accounting and supervisory personnel, (ii) lack of appropriate level of technical accounting training; and (iii) lack of consistent performance of controls over financial reporting which, if not remediated appropriately or timely, could result in loss of investor confidence and adversely impact our stock price. If we are unable to remediate the material weakness, or if other material weaknesses are identified, we may not be able to report our financial results accurately, prevent fraud or file our periodic reports as a public company in a timely manner.”
220
C. Attestation Report of the Registered Public Accounting Firm
KPMG AG Wirtschaftsprüfungsgesellschaft, an independent registered public accounting firm who audited the consolidated financial statements as of and for the year ended December 31, 2023, has also audited the effectiveness of the Company’s internal control over financial reporting as of December 31, 2023. Their reports are included in this Annual Report on Form 20-F beginning on page F-3.
D. Changes in Internal Control over Financial Reporting
Other than the changes described above there were no changes to our internal control over financial reporting (as defined in Rule 13a-15(f) and 15d-15(f) under the Exchange Act of 1934), which occurred during the period covered by this Form 20-F that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 16. [Reserved]
ITEM 16A. Audit Committee Financial Expert
Our supervisory board has determined that Michael Brosnan, Craig A. Tooman, Klaus Schollmeier, Hans Christoph Tanner and Ralf Clemens satisfy the “independence” requirements set forth in Rule 10A-3 under the Exchange Act and each qualifies as an “audit committee financial expert,” as such term is defined in the rules of the SEC. Hans Christoph Tanner resigned from the Supervisory Board on June 19, 2023 and Ralf Clemens resigned from the Supervisory Board on September 30, 2023. For more information see “Item 6. Directors, Senior Management and Employees — C. Board Practices — Committees — Audit Committee.”
ITEM 16B. Code of Conduct
We adopted a written code of business conduct and ethics, or code of conduct, which outlines the principles of legal and ethical business conduct under which we do business, and is a code of ethics as defined in Item 16B of Form 20-F promulgated by the SEC. The code of conduct applies to all of our management board and supervisory directors and employees. The full text of the code of conduct is available on our website at www.curevac.com. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report and is not incorporated by reference herein. If we make any amendment to the code of conduct or grant any waivers, including any implicit waiver, from a provision of the code of conduct, we will disclose the nature of such amendment or waiver on our website to the extent required by the rules and regulations of the SEC.
In addition, we have implemented a compliance management policy which describes the compliance management system implemented at CureVac SE, which is designed to ensure compliance with all legal requirements, while at the same time implementing high ethical standards that are mandatory for both management and each employee. The overall responsibility for the compliance management system lies with the management board, which reports regularly to the audit committee. In the performance of its compliance responsibilities, the management board has delegated the corresponding tasks to various functions at CureVac SE.
ITEM 16C. Principal Accountant Fees and Services
Our financial statements have been prepared in accordance with IFRS as issued by the IASB and are audited by KPMG. KPMG has acted as our independent registered public accounting firm registered with the Public Company Accounting Oversight Board in the United States.
KPMG has served as our independent registered public accounting firm for the year ended December 31, 2023, for which audited financial statements appear in this Annual Report. EY GmbH & Co. KG Wirtschaftsprüfungsgesellschaft (“EY”) served as our independent registered public accounting firm for the years ended December 31, 2021 and 2022, for which audited financial statements appear in this Annual Report.
Audit Fees
We incurred fees of €1.2 million to KPMG for audit services for fiscal 2023 and €1.8 million to EY for audit services for fiscal 2022, respectively, including fees associated with the annual audit, consultations on various accounting issues, performance of local statutory audits and comfort letters and review of offering documents filed with the SEC.
221
Audit-Related Fees
No audit-related services fees were incurred for fiscal 2023 and 2022.
Tax Fees
We incurred €0.1 million to KPMG for tax fees for fiscal 2023 and €0.5 million for tax fees to EY for fiscal 2022, including fees associated with tax compliance, tax advice and tax planning services for fiscal 2023 and 2022, respectively.
All Other Fees
No fees for services other than those categorized in Audit Fees, Audit-Related Fees and Tax Fees described above incurred to KPMG in 2023.We incurred €0.1 million for services other than those categorized in Audit Fees, Audit-Related Fees and Tax Fees described above to EY in 2022.
Pre-Approval Policies and Procedures
The Audit Committee has adopted policies and procedures relating to the approval of all audit and non-audit services that are to be performed by our independent registered public accounting firm. These policies generally provide that we will not engage our independent registered public accounting firm to render audit or non-audit services unless the service is specifically approved in advance by the Audit Committee or the engagement is entered into pursuant to the pre-approval procedure described below.
From time to time, the Audit Committee may pre-approve specified types of services that are expected to be provided to us by our independent registered public accounting firm during the next 12 months. Any such pre-approval is detailed as to the particular service or type of services to be provided and is also generally subject to a maximum dollar amount. In fiscal 2023, our Audit Committee approved all of the services provided by KPMG.
ITEM 16D. Exemptions from the Listing Standards for Audit Committees
None.
ITEM 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None.
ITEM 16F. Change in Registrant’s Certifying Accountant
None.
ITEM 16G. Corporate Governance
The following comparison between Dutch corporate law, which applies to us, and Delaware corporation law, the law under which many publicly listed corporations in the United States are incorporated, discusses additional matters not otherwise described in this Annual Report. Although we believe this summary is materially accurate, the summary is subject to Dutch law, including Book 2 of the Dutch Civil Code and the DCGC and Delaware corporation law, including the Delaware General Corporation Law.
Duties of Managing and Supervisory Directors
The Netherlands. In the Netherlands, a listed company typically has a two-tier board structure with a management board comprised of the managing directors (executive directors) and a supervisory board comprised of the supervisory directors (non-executive directors). We have a two-tier board structure consisting of our management board (bestuur) and a separate supervisory board (raad van commissarissen).
222
Under Dutch law, the management board is charged with the management of the company, subject to the restrictions contained in our articles of association, and the supervisory board is charged with the supervision of the policy of the management board and the general course of affairs of the company and of the business connected with it. The managing directors may divide their tasks among themselves in or pursuant to the internal rules applicable to the management board. Each managing director and supervisory director has a statutory duty to act in the corporate interest of the company and its business. Under Dutch law, the corporate interest extends to the interests of all corporate stakeholders, such as shareholders, creditors, employees, customers and suppliers. The duty to act in the corporate interest of the company also applies in the event of a proposed sale or breakup of the company, provided that the circumstances generally dictate how such duty is to be applied and how the respective interests of various groups of stakeholders should be weighed. Any resolution of the management board regarding a material change in our identity or character requires approval of the general meeting. In addition, during the initial approval period, the following additional resolutions of the management board will require approval of the general meeting and our supervisory board:
| ● | transferring the tax domicile of CureVac N.V. and/or the approval of the transfer of the corporate or administrative seat of CureVac SE; |
| ● | relocating or ceasing (including by way of disposal, demerger or similar transactions) activities in specified areas in or to a state outside the European Union, except to the extent our supervisory board considers such activities (in particular in the area of the development of vaccines) not to be material for the protection of the health of the population of the European Union; |
| ● | entering into mergers, demergers and similar reorganizations and entering into acquisitions or businesses or participations, except to the extent our supervisory board considers such transactions not to be material; |
| ● | amendments to the articles of association of CureVac SE which would affect these approval rights during the initial approval period; and |
| ● | the exercise of voting rights in CureVac SE approving, directing or causing any of the foregoing matters. |
Under our articles of association, the approval of our supervisory board is also required for resolutions of the management board, including concerning the following matters:
| ● | the making of certain proposals to the general meeting; |
| ● | the issue of shares or the granting of rights to subscribe for shares; |
| ● | the limitation or exclusion of pre-emption rights; |
| ● | the establishment of new activities of us or our direct or indirect subsidiaries in the areas of research, development, production and administration and/or the approval to establish activities of CureVac SE or its subsidiaries in these areas, in each case in a state outside the European Union; |
| ● | the acquisition of shares by us in our own capital; |
| ● | the drawing up or amendment of our management board rules; |
| ● | the performance of legal acts relating to non-cash contributions on shares; |
| ● | material changes to the identity or the character of the company or its business; |
| ● | the charging of amounts to be paid up on shares against the company’s reserves; |
| ● | the making of an interim distribution; |
| ● | designating a current or former officer or employee as indemnitees under our articles of association; |
223
| ● | the stipulation of additional terms, conditions and restrictions in relation to the indemnification offered under our articles of association; |
| ● | and such other resolutions as the supervisory board shall have specified in a resolution to that effect and notified to the management board. |
Under the internal rules applicable to our management board, certain additional resolutions are subject to the approval of our supervisory board.
Under the internal rules applicable to our supervisory board, resolutions of our supervisory board to approve a resolution of our management board to exclude or limit pre-emption rights (except in connection with the ordinary operation of our equity incentive plans) or to issue shares against non-cash contribution, shall require the approval of a special committee consisting of one supervisory director nominated by dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) (during the initial nomination period for dievini), the supervisory director nominated by KfW (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) (during the initial nomination period for KfW) and, if applicable, one supervisory director nominated by a nomination concert. In this special committee, the affirmative votes of one supervisory director nominated by dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) (during the initial nomination period for dievini) and the supervisory director nominated by KfW (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) (during the initial nomination period for KfW) shall be required. Similarly, the affirmative votes of at least one supervisory director nominated by dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) (during the initial nomination period for dievini) and the supervisory director nominated by KfW (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) (during the initial nomination period for KfW) shall be required for certain resolutions of the supervisory board specified by our articles of association and the internal rules applicable to our supervisory board.
The absence of the approval of the supervisory board shall result in the relevant resolution being null and void but shall not affect the powers of representation of the management board or of the managing directors.
Our management board is entitled to represent us. The power to represent us also vests in the chief executive officer individually, as well as in any other two managing directors acting jointly.
Delaware. The board of directors bears the ultimate responsibility for managing the business and affairs of a corporation. In discharging this function, directors of a Delaware corporation owe fiduciary duties of care and loyalty to the corporation and to its stockholders. Delaware courts have decided that the directors of a Delaware corporation are required to exercise informed business judgment in the performance of their duties. Informed business judgment means that the directors have informed themselves of all material information reasonably available to them. Delaware courts have also imposed a heightened standard of conduct upon directors of a Delaware corporation who take any action designed to defeat a threatened change in control of the corporation. In addition, under Delaware law, when the board of directors of a Delaware corporation approves the sale or breakup of a corporation, the board of directors may, in certain circumstances, have a duty to obtain the highest value reasonably available to the stockholders.
Director Terms
The Netherlands. The DCGC provides the following best practice recommendations on the terms for tenure of managing directors and supervisory directors:
| ● | Managing directors should be appointed for a maximum period of four years, without limiting the number of consecutive terms managing directors may serve. |
| ● | Supervisory directors should be appointed for two consecutive periods of no more than four years. Thereafter, supervisory directors may be reappointed for a maximum of two consecutive periods of no more than two years, provided that any reappointment after an eight-year term of office should be disclosed in the company’s annual report. |
224
The general meeting shall at all times be entitled to suspend or dismiss a managing director or supervisory director. Under our articles of association, the general meeting may only adopt a resolution to suspend or dismiss such director by at least a two-thirds majority of the votes cast, provided that such majority represents more than half of the issued share capital, unless the resolution is passed at the proposal of the supervisory board or, with respect to supervisory directors nominated by dievini or KfW, by dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) during the nomination period for dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) or by KfW (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) during the nomination period for KfW, respectively, in which case a simple majority of the votes cast is sufficient. In addition, the supervisory board may at any time suspend a managing director. A suspension by the supervisory board can at any time be lifted by the general meeting. If a managing director is suspended and the general meeting does not resolve to dismiss him or her within three months from the date of such suspension, the suspension shall lapse.
Delaware. The Delaware General Corporation Law generally provides for a one-year term for directors, but permits directorships to be divided into up to three classes with up to three-year terms, with the years for each class expiring in different years, if permitted by the certificate of incorporation, an initial bylaw or a bylaw adopted by the stockholders. A director elected to serve a term on a “classified” board may not be removed by stockholders without cause. There is no limit in the number of terms a director may serve.
Director Vacancies
The Netherlands. Under Dutch law, managing directors and supervisory directors of a company like ours are appointed and reappointed by the general meeting. Under our articles of association, managing directors and supervisory directors are appointed by the general meeting upon the binding nomination by our supervisory board. During the periods specified below, dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement), KfW (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement), and any nomination concert have the right to make a binding nomination for one or more supervisory directors as specified below:
| ● | during the initial nomination period for dievini, dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) will have the right under our articles of association to make a binding nomination for the following number of supervisory directors: |
| ● | four (4) supervisory directors for as long as dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates, as defined by our articles of association, and ultimate beneficiaries, as defined by our articles of association (individually or collectively) owns at least 70% of our issued share capital; |
| ● | three (3) supervisory directors for as long as dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates, as defined by our articles of association, and ultimate beneficiaries, as defined by our articles of association (individually or collectively) owns at least 50% (but less than 70%) of our issued share capital; |
| ● | two (2) supervisory directors for as long as dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates, as defined by our articles of association, and ultimate beneficiaries, as defined by our articles of association (individually or collectively) owns at least 30% (but less than 50%) of our issued share capital; and |
| ● | one (1) supervisory director for as long as dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) and its affiliates, as defined by our articles of association, and ultimate beneficiaries, as defined by our articles of association (individually or collectively) owns at least 10% (but less than 30%) of our issued share capital. |
| ● | during the initial nomination period for KfW, KfW (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) will have the right under our articles of association to make a binding nomination for one supervisory director; and |
225
| ● | at any time, each nomination concert (excluding dievini, its affiliates and its ultimate beneficiaries and excluding KfW and its affiliates for as long as dievini and KfW, respectively, have the nomination rights discussed above) will have the right under our articles of association to make a binding nomination for one supervisory director for each 20% of our issued share capital represented by that nomination concert, provided such nominee is independent from the nomination concert and CureVac N.V. under the DCGC and applicable U.S. securities laws and Nasdaq rules. |
The general meeting may at all times overrule the binding nomination by a resolution adopted by a simple majority of the votes cast, provided that such majority represents at least one-third of the issued share capital. If the general meeting overrules the binding nature of a binding nomination, a new nomination shall be prepared by whoever made the overruled nomination.
If dievini, KfW and/or a nomination concert loses the right to nominate one or more of our supervisory directors, as applicable, the supervisory director(s) so nominated must promptly resign. A supervisory director nominated by a nomination concert must also promptly resign once that supervisory director is no longer independent from the nomination concert or our company.
Under Dutch law, when nominating a person for appointment or reappointment as a supervisory director, the nomination must be supported by reasons (if it concerns a reappointment, past performance must be taken into consideration) and the following information about such person must be provided: (i) age and profession; (ii) the aggregate nominal value of the shares held in the company’s capital; (iii) present and past positions, to the extent relevant for the performance of the tasks of a supervisory director; and (iv) the name of each entity where such person already holds a position as supervisory director or non-executive director (in case of multiple entities within the same group, the name of the group shall suffice).
Delaware. The Delaware General Corporation Law provides that vacancies and newly created directorships may be filled by a majority of the directors then in office (even though less than a quorum) unless (i) otherwise provided in the certificate of incorporation or bylaws of the corporation or (ii) the certificate of incorporation directs that a particular class of stock is to elect such director, in which case any other directors elected by such class, or a sole remaining director elected by such class, will fill such vacancy.
Conflict-of-Interest Transactions
The Netherlands. Under Dutch law and our articles of association, our managing directors and supervisory directors shall not take part in any discussion or decision-making that involves a subject or transaction in relation to which he or she has a direct or indirect personal conflict of interest with us. Such a conflict of interest would generally arise if the managing director or supervisory director concerned is unable to serve our interests and the business connected with it with the required level of integrity and objectivity due to the existence of the conflicting personal interest. Our articles of association provide that if as a result of conflicts of interests no resolution of the management board can be adopted, the resolution may be passed by the supervisory board and that, if as a result of conflicts of interests no resolution of the supervisory board can be adopted, the resolution may nonetheless be adopted by the supervisory board as if none of the supervisory directors had a conflict of interest. In that case, each supervisory director is entitled to participate in the discussion and decision-making process and to cast a vote.
The DCGC provides the following best practice recommendations in relation to conflicts of interests in respect of managing directors or supervisory directors:
| ● | A managing director should report any potential conflict of interest in a transaction that is of material significance to the company and/or to such person to the chairman of the supervisory board and to the other members of the management board without delay. The managing director should provide all relevant information in that regard, including the information relevant to the situation concerning his or her spouse, registered partner or other life companion, foster child and relatives by blood or marriage up to the second degree; |
| ● | A supervisory director should report any conflict of interest or potential conflict of interest in a transaction that is of material significance to the company and/or to such person to the chairman of the supervisory board without delay and should provide all relevant information in that regard, including the relevant information pertaining to his or her spouse, registered partner or other life companion, foster child and relatives by blood or marriage up to the second degree. If the chairman of the supervisory board has a conflict of interest or potential conflict of interest, he or she should report this to the vice-chairman of the supervisory board without delay; |
226
| ● | The supervisory board should decide, outside the presence of the managing director or supervisory director concerned, whether there is a conflict of interest; |
| ● | All transactions in which there are conflicts of interest with managing directors or supervisory directors should be agreed on terms that are customary in the market; and |
| ● | Decisions to enter into transactions in which there are conflicts of interest with managing directors or supervisory directors that are of material significance to the company and/or to the relevant managing directors or supervisory directors should require the approval of the supervisory board. Such transactions should be published in the annual report, together with a description of the conflict of interest and a declaration that the relevant best practice provisions of the DCGC have been complied with. |
Delaware. The Delaware General Corporation Law generally permits transactions involving a Delaware corporation and an interested director of that corporation if:
| ● | the material facts as to the director’s relationship or interest are disclosed and a majority of disinterested directors consent; |
| ● | the material facts are disclosed as to the director’s relationship or interest and a majority of shares entitled to vote thereon consent; or |
| ● | the transaction is fair to the corporation at the time it is authorized by the board of directors, a committee of the board of directors or the stockholders. |
Proxy Voting by Directors
The Netherlands. An absent managing director may issue a proxy for a specific management board meeting but only to another managing director in writing or by electronic means. An absent supervisory director may issue a proxy for a specific supervisory board meeting but only to another supervisory director in writing or by electronic means.
Delaware. A director of a Delaware corporation may not issue a proxy representing the director’s voting rights as a director.
Shareholder Rights
Voting Rights
The Netherlands. In accordance with Dutch law and our articles of association, each issued common share confers the right to cast one vote at the general meeting. Each holder of shares may cast as many votes as it holds shares. No votes may be cast on shares that are held by us or our direct or indirect subsidiaries or on shares for which we or our subsidiaries hold depository receipts. Nonetheless, the holders of a right of use and enjoyment (vruchtgebruik) and the holders of a right of pledge (pandrecht) in respect of shares held by us or our subsidiaries in our share capital are not excluded from the right to vote on such shares, if the right of use and enjoyment (vruchtgebruik) or the right of pledge (pandrecht) was granted prior to the time such shares were acquired by us or any of our subsidiaries. Neither we nor any of our subsidiaries may cast votes in respect of a share on which we or such subsidiary holds a right of use and enjoyment (vruchtgebruik) or a right of pledge (pandrecht). Shares which are not entitled to voting rights pursuant to the preceding sentences will not be taken into account for the purpose of determining the number of shareholders that vote and that are present or represented, or the amount of the share capital that is provided or that is represented at a general meeting.
In accordance with our articles of association, for each general meeting, the management board may determine that a record date will be applied in order to establish which shareholders are entitled to attend and vote at the general meeting. Such record date shall be the 28th day prior to the day of the general meeting. The record date and the manner in which shareholders can register and exercise their rights will be set out in the notice of the meeting which must be published in a Dutch daily newspaper with national distribution at least 15 days prior to the meeting (and such notice may therefore be published after the record date for such meeting). Under our articles of association, shareholders and others with meeting rights under Dutch law must notify us in writing or by electronic means of their identity and intention to attend the general meeting. This notice must be received by us ultimately on the seventh day prior to the general meeting, unless indicated otherwise when such meeting is convened.
227
Delaware. Under the Delaware General Corporation Law, each stockholder is entitled to one vote per share of stock, unless the certificate of incorporation provides otherwise. In addition, the certificate of incorporation may provide for cumulative voting at all elections of directors of the corporation, or at elections held under specified circumstances. Either the certificate of incorporation or the bylaws may specify the number of shares and/or the amount of other securities that must be represented at a meeting in order to constitute a quorum, but in no event will a quorum consist of less than one-third of the shares entitled to vote at a meeting.
Stockholders as of the record date for the meeting are entitled to vote at the meeting, and the board of directors may fix a record date that is no more than 60 nor less than 10 days before the date of the meeting, and if no record date is set then the record date is the close of business on the day next preceding the day on which notice is given, or if notice is waived then the record date is the close of business on the day next preceding the day on which the meeting is held. The determination of the stockholders of record entitled to notice or to vote at a meeting of stockholders shall apply to any adjournment of the meeting, but the board of directors may fix a new record date for the adjourned meeting.
Shareholder Proposals
The Netherlands. Pursuant to our articles of association, extraordinary general meetings will be held whenever required under Dutch law or whenever our management board or supervisory board deems such to be appropriate or necessary. Pursuant to Dutch law, one or more shareholders or others with meeting rights under Dutch law representing at least one-tenth of the issued share capital may request us to convene a general meeting, setting out in detail the matters to be discussed. If we have not taken the steps necessary to ensure that such meeting can be held within six weeks after the request, the requesting party or parties may, on their application, be authorized by the competent Dutch court in preliminary relief proceedings to convene a general meeting.
Also, the agenda for a general meeting shall include such items requested by one or more shareholders, and others entitled to attend general meetings, representing at least 3% of the issued share capital, except where the articles of association state a lower percentage. Our articles of association do not state such lower percentage. Requests must be made in writing or by electronic means and received by us at least 60 days before the day of the meeting.
In accordance with the DCGC and our articles of association, a shareholder shall exercise the right of putting an item on the agenda only after consulting the management board in that respect. If one or more shareholders intend to request that an item be put on the agenda that may result in a change in the company’s strategy (for example, the removal of managing directors or supervisory directors), the management board must be given the opportunity to invoke a reasonable period to respond to such intention. Such period shall not exceed 180 days (or such other period as may be stipulated for such purpose by Dutch law and/or the DCGC from time to time). If invoked, the management board must use such response period for further deliberation and constructive consultation, in any event with the shareholders(s) concerned, and shall explore the alternatives. At the end of the response time, the management board shall report on this consultation and the exploration of alternatives to the general meeting. This shall be supervised by our supervisory board. The response period may be invoked only once for any given general meeting and shall not apply: (a) in respect of a matter for which a response period has been previously invoked; or (b) if a shareholder holds at least 75% of the company’s issued share capital as a consequence of a successful public bid. The response period may also be invoked in response to shareholders or others with meeting rights under Dutch law requesting that a general meeting be convened, as described above.
228
Moreover, our management board, with the approval of our supervisory board, can invoke a cooling-off period of up to 250 days when shareholders, using their right to have items added to the agenda for a general meeting or their right to request a general meeting, propose an agenda item for our general meeting to dismiss, suspend or appoint one or more managing directors or supervisory directors (or to amend any provision in our articles of association dealing with those matters) or when a public offer for our company is made or announced without our support, provided, in each case, that our management board believes that such proposal or offer materially conflicts with the interests of our company and its business. During a cooling-off period, our general meeting cannot dismiss, suspend or appoint managing directors and supervisory directors (or amend the provisions in our articles of association dealing with those matters) except at the proposal of our management board. During a cooling-off period, our management board must gather all relevant information necessary for a careful decision-making process and at least consult with shareholders representing 3% or more of our issued share capital at the time the cooling-off period was invoked, as well as with our Dutch works council (if we or, under certain circumstances, any of our subsidiaries would have one). Formal statements expressed by these stakeholders during such consultations must be published on our website to the extent these stakeholders have approved that publication. Ultimately one week following the last day of the cooling-off period, our management board must publish a report in respect of its policy and conduct of affairs during the cooling-off period on our website. This report must remain available for inspection by shareholders and others with meeting rights under Dutch law at our office and must be tabled for discussion at the next general meeting. Shareholders representing at least 3% of our issued share capital may request the Enterprise Chamber for early termination of the cooling-off period. The Enterprise Chamber must rule in favor of the request if the shareholders can demonstrate that:
| ● | our management board, in light of the circumstances at hand when the cooling-off period was invoked, could not reasonably have concluded that the relevant proposal or hostile offer constituted a material conflict with the interests of our company and its business; |
| ● | our management board cannot reasonably believe that a continuation of the cooling-off period would contribute to careful policy-making; or |
| ● | other defensive measures, having the same purpose, nature and scope as the cooling-off period, have been activated during the cooling-off period and have not since been terminated or suspended within a reasonable period at the relevant shareholders’ request (i.e., no ‘stacking’ of defensive measures). |
Delaware. Delaware law does not specifically grant stockholders the right to bring business before an annual or special meeting. However, if a Delaware corporation is subject to the SEC’s proxy rules, a stockholder who owns at least $2,000 in market value, or 1% of the corporation’s securities entitled to vote, may propose a matter for a vote at an annual or special meeting in accordance with those rules.
Action by Written Consent
The Netherlands. Under Dutch law, shareholders’ resolutions may be adopted in writing without holding a meeting of shareholders, provided that (i) the articles of association allow such action by written consent, (ii) the company has not issued bearer shares or, with its cooperation, depository receipts for shares in its capital, and (iii) the resolution is adopted unanimously by all shareholders that are entitled to vote. Although our articles of association allow for shareholders’ resolutions to be adopted in writing, the requirement of unanimity renders the adoption of shareholder resolutions without holding a meeting not feasible for us as a publicly traded company.
Delaware. Although permitted by Delaware law, publicly listed companies do not typically permit stockholders of a corporation to take action by written consent.
Appraisal Rights
The Netherlands. Subject to certain exceptions, Dutch law does not recognize the concept of appraisal or dissenters’ rights. However, Dutch law does provide for squeeze-out procedures. Also, Dutch law provides for cash exit rights in certain situations for dissenting shareholders of a company organized under Dutch law entering into certain types of mergers. In those situations, a dissenting shareholder may file a claim with the Dutch company for compensation. Such compensation shall then be determined by one or more independent experts. The shares of such shareholder that are subject to such claim will cease to exist as of the moment of entry into effect of the merger.
229
Delaware. The Delaware General Corporation Law provides for stockholder appraisal rights, or the right to demand payment in cash of the judicially determined fair value of the stockholder’s shares, in connection with certain mergers and consolidations.
Shareholder Suits
The Netherlands. In the event a third-party is liable to a Dutch company, only the company itself can bring a civil action against that party. The individual shareholders do not have the right to bring an action on behalf of the company. Only in the event that the cause for the liability of a third-party to the company also constitutes a tortious act directly against a shareholder does that shareholder have an individual right of action against such third-party in its own name. Dutch law provides for the possibility to initiate such actions collectively, in which a foundation or an association can act as a class representative and has standing to commence proceedings and claim damages if certain criteria are met. The court will first determine if those criteria are met. If so, the case will go forward as a class action on the merits after a period allowing class members to opt out from the case has lapsed. All members of the class who are residents of the Netherlands and who did not opt out will be bound to the outcome of the case. Residents of other countries must actively opt in in order to be able to benefit from the class action. The defendant is not required to file defenses on the merits prior to the merits phase having commenced. It is possible for the parties to reach a settlement during the merits phase. Such a settlement can be approved by the court, which approval will then bind the members of the class, subject to a second opt out. This new regime applies to claims brought after January 1, 2020, and which relate to certain events that occurred prior to that date. For other matters, the old Dutch class actions regime will apply. Under the old regime, no monetary damages can be sought. Also, a judgment rendered under the old regime will not bind individual class members. Even though Dutch law does not provide for derivative suits, directors and officers can still be subject to liability under U.S. securities laws.
Delaware. Under the Delaware General Corporation Law, a stockholder may bring a derivative action on behalf of the corporation to enforce the rights of the corporation. An individual also may commence a class action suit on behalf of himself and other similarly situated stockholders where the requirements for maintaining a class action under Delaware law have been met. A person may institute and maintain such a suit only if that person was a stockholder at the time of the transaction which is the subject of the suit. In addition, under Delaware case law, the plaintiff normally must be a stockholder at the time of the transaction that is the subject of the suit and throughout the duration of the derivative suit. Delaware law also requires that the derivative plaintiff make a demand on the directors of the corporation to assert the corporate claim before the suit may be prosecuted by the derivative plaintiff in court, unless such a demand would be futile.
Repurchase of Shares
The Netherlands. Under Dutch law, when issuing shares, a public company such as ours may not subscribe for newly issued shares in its own capital. Such company may, however, subject to certain restrictions of Dutch law and its articles of association, acquire shares in its own capital. A listed public company such as ours may acquire fully paid shares in its own capital at any time for no valuable consideration. Furthermore, subject to certain provisions of Dutch law and its articles of association, such company may repurchase fully paid shares in its own capital if (i) the company’s shareholders’ equity less the payment required to make the acquisition does not fall below the sum of paid-in and called-up share capital plus any reserves required by Dutch law or its articles of association and (ii) the aggregate nominal value of shares of the company which the company acquires, holds or on which the company holds a pledge (pandrecht) or which are held by a subsidiary of the company, would not exceed 50% of its then-current issued share capital. Such company may only acquire its own shares if its general meeting has granted the management board the authority to effect such acquisitions.
An acquisition of common shares for a consideration must be authorized by our general meeting. Such authorization may be granted for a maximum period of 18 months and must specify the number of common shares that may be acquired, the manner in which common shares may be acquired and the price limits within which common shares may be acquired. The actual acquisition may only be effected pursuant to a resolution of our management board, with the approval of our supervisory board. Our management board, subject to approval of our supervisory board, has been authorized, for a period of 18 months following the completion of our initial public offering, to cause the repurchase of common shares by us of up to 20% of our issued share capital, for a price per share not exceeding 110% of the average market price of our common shares on Nasdaq (such average market price being the average of the closing prices on each of the five consecutive trading days preceding the date the acquisition is agreed upon by us). These shares may be used to deliver shares underlying awards granted pursuant to our equity-based compensation plans.
230
Our management board, subject to approval by our supervisory board, has also been authorized for a period of 18 months following the completion of our initial public offering to cause the repurchase of preferred shares, for a price which is higher than nil and does not exceed the nominal value thereof. No authorization of the general meeting is required if fully paid common shares are acquired by us with the intention of transferring such common shares to our employees under an applicable employee share purchase plan.
Delaware. Under the Delaware General Corporation Law, a corporation may purchase or redeem its own shares unless the capital of the corporation is impaired or the purchase or redemption would cause an impairment of the capital of the corporation. A Delaware corporation may, however, purchase or redeem out of capital any of its preferred shares or, if no preferred shares are outstanding, any of its own shares if such shares will be retired upon acquisition and the capital of the corporation will be reduced in accordance with specified limitations.
Anti-Takeover Provisions
The Netherlands. Under Dutch law, various protective measures are possible and permissible within the boundaries set by Dutch law and Dutch case law. In this respect, certain provisions of our articles of association may make it more difficult for a third-party to acquire control of us or effect a change in our management board and supervisory board. These provisions include:
| ● | the authorization of a class of preferred shares that, after the expiration of the later of the initial period or the initial approval period, may be issued to a protective foundation pursuant to a call option to that effect, see “Item 3. Key Information — D. Risk Factors — Provisions of our articles of association or Dutch corporate law might deter acquisition bids for us that might be considered favorable and prevent, delay or frustrate any attempt to replace or remove the members of our managing directors or supervisory directors; |
| ● | a provision that our managing directors and supervisory directors are appointed on the basis of a binding nomination, the binding nature of which can only be overruled by a simple majority of votes cast representing at least one-third of our issued share capital; |
| ● | a provision that our managing directors and supervisory directors may only be dismissed by the general meeting by a two-thirds majority of votes cast representing more than 50% of our issued share capital (unless the dismissal is proposed by the supervisory board or, with respect to supervisory directors nominated by dievini or KfW, by dievini (or its legal successor) or permitted assigns under the KfW dievini Shareholders’ Agreement during the nomination period for dievini or by KfW (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) during the nomination period for KfW, respectively, in which case a simple majority of the votes would be sufficient); |
| ● | a provision that where a supervisory director is no longer in office or is unable to act, he or she may be replaced temporarily by a person whom the supervisory board has designated for that purpose and, where a supervisory director who has been appointed upon a nomination of dievini or KfW, as applicable, is no longer in office or unable to act, such supervisory director may only be temporarily replaced by a person designated for such purposes by dievini or KfW, as applicable. Such person shall become a full member of the supervisory board with the rights of the relevant supervisory director appointed upon a nomination of dievini or KfW, as applicable, as soon as a written designation to that effect has been received by the chairman or vice-chairman of our supervisory board, subject to limitations, under applicable law regarding dievini’s rights under this provision; |
| ● | a provision allowing, among other matters, a former chairman of our supervisory board, a former nominee of dievini, and a former nominee of KfW, to jointly take on the supervisory functions, which persons jointly may designate one or more other persons to be charged with the supervision of our company (instead of or together with the former chairman of our supervisory board), as applicable, to manage our affairs if all of our managing directors and supervisory directors are removed from office and to appoint others to be charged with the management and supervision of our affairs, until new managing directors and supervisory directors are appointed by the general meeting on the basis of a binding nomination discussed above; |
| ● | a provision allowing the management board to temporarily replace a managing director who is no longer in office or unable to act, by another person or persons designated for this purpose by the management board and attributing the management of the company to the supervisory board in case all managing directors are no longer in office or unable to act; |
231
| ● | a provision that certain provisions of our articles of association can only be amended with the affirmative vote of (i) during the nomination period for dievini, dievini (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement) and (ii) during the nomination period for KfW, KfW (or its legal successors or permitted assigns under the KfW dievini Shareholders’ Agreement); and |
| ● | a requirement that certain matters, including an amendment of our articles of association, may only be brought to our shareholders for a vote upon a proposal by our management board. |
In addition, Dutch law allows for staggered multi-year terms of our managing directors and supervisory directors, as a result of which only part of our managing directors and supervisory directors may be subject to appointment or reappointment in any one year.
Furthermore, our management board may, under certain circumstances invoke a reasonable period of up to 180 days to respond to certain shareholder proposals or a statutory cooling-off period of up to 250 days to respond to certain shareholder proposals or a hostile bid. See above under “Shareholder Proposals.”
Delaware. In addition to other aspects of Delaware law governing fiduciary duties of directors during a potential takeover, the Delaware General Corporation Law also contains a business combination statute that protects Delaware companies from hostile takeovers and from actions following the takeover by prohibiting some transactions once an acquirer has gained a significant holding in the corporation.
Section 203 of the Delaware General Corporation Law prohibits “business combinations,” including mergers, sales and leases of assets, issuances of securities and similar transactions by a corporation or a subsidiary with an interested stockholder that beneficially owns 15% or more of a corporation’s voting stock, within three years after the person becomes an interested stockholder, unless:
| ● | the transaction that will cause the person to become an interested stockholder is approved by the board of directors of the target prior to the transactions; |
| ● | after the completion of the transaction in which the person becomes an interested stockholder, the interested stockholder holds at least 85% of the voting stock of the corporation not including shares owned by persons who are directors and officers of interested stockholders and shares owned by specified employee benefit plans; or |
| ● | after the person becomes an interested stockholder, the business combination is approved by the board of directors of the corporation and holders of at least 66.67% of the outstanding voting stock, excluding shares held by the interested stockholder. |
A Delaware corporation may elect not to be governed by Section 203 by a provision contained in the original certificate of incorporation of the corporation or an amendment to the original certificate of incorporation or to the bylaws of the company, which amendment must be approved by a majority of the shares entitled to vote and may not be further amended by the board of directors of the corporation. Such an amendment is not effective until 12 months following its adoption.
Inspection of Books and Records
The Netherlands. The management board and the supervisory board provide the general meeting, within a reasonable amount of time, all information that the shareholders require for the exercise of their powers, unless this would be contrary to an overriding interest of our company. If the management board or supervisory board invokes such an overriding interest, it must give reasons.
Delaware. Under the Delaware General Corporation Law, any stockholder may inspect for any proper purpose certain of the corporation’s books and records during the corporation’s usual hours of business.
232
Dismissal of Directors
The Netherlands. Under our articles of association, the general meeting shall at all times be entitled to dismiss a managing director or supervisory director. The general meeting may only adopt a resolution to suspend or dismiss a managing director or supervisory director by at least a two-thirds majority of the votes cast, provided that such majority represents more than half of the issued share capital, unless the proposal was made by the supervisory board or, with respect to supervisory directors nominated by dievini or KfW, by dievini (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) during the nomination period for dievini or by KfW (or its legal successor or permitted assigns under the KfW dievini Shareholders’ Agreement) during the nomination period for KfW, respectively, in which latter case a simple majority is sufficient.
Delaware. Under the Delaware General Corporation Law, any director or the entire board of directors may be removed, with or without cause, by the holders of a majority of the shares then entitled to vote at an election of directors, except (i) unless the certificate of incorporation provides otherwise, in the case of a corporation whose board is classified, stockholders may effect such removal only for cause, or (ii) in the case of a corporation having cumulative voting, if less than the entire board is to be removed, no director may be removed without cause if the votes cast against his removal would be sufficient to elect him if then cumulatively voted at an election of the entire board of directors, or, if there are classes of directors, at an election of the class of directors of which he or she is a part.
Issuance of Shares
The Netherlands. Under Dutch law, a company’s general meeting is the corporate body authorized to resolve on the issuance of shares and the granting of rights to subscribe for shares. The general meeting can delegate such authority to another corporate body of the company, such as the management board, for a period not exceeding five years; this authorization may only be extended from time to time for a maximum period of five years. In order for a resolution of the general meeting on an issuance or an authorization as discussed in the previous sentence to be valid, a prior or simultaneous approval shall be required from each meeting of holders of a certain class of shares whose rights are prejudiced by the issuance.
Our management board, subject to approval of our supervisory board, has been authorized, for a period of five years from the completion of our initial public offering, to issue shares or grant rights to subscribe for shares up to our authorized share capital from time to time. We may not subscribe for our own shares on issue.
Delaware. All creation of shares require the board of directors to adopt a resolution or resolutions, pursuant to authority expressly vested in the board of directors by the provisions of the company’s certificate of incorporation.
Preemptive Rights
The Netherlands. Under Dutch law, in the event of an issuance of common shares, each shareholder will have a pro rata preemptive right in proportion to the aggregate nominal value of the common shares held by such holder (with the exception of common shares to be issued to employees or common shares issued against a contribution other than in cash or pursuant to the exercise of a previously acquired right to subscribe for shares). Our preferred shares carry no preemptive rights. Under our articles of association, the preemptive rights in respect of newly issued common shares may be restricted or excluded by a resolution of the general meeting. Another corporate body, such as the management board, may restrict or exclude the preemptive rights in respect of newly issued common shares if it has been designated as the authorized body to do so by the general meeting. Such designation can be granted for a period not exceeding five years. A resolution of the general meeting to restrict or exclude the preemptive rights or to designate another corporate body as the authorized body to do so requires a majority of not less than two-thirds of the votes cast, if less than one-half of our issued share capital is represented at the meeting. Our management board, subject to approval of our supervisory board, has been authorized, for a period of five years from the completion of our initial public offering, to limit or exclude preemptive rights in relation to an issuance of shares or a grant of rights to subscribe for shares that the management board is authorized to resolve upon. See “—Issuance of Shares.”
Delaware. Under the Delaware General Corporation Law, stockholders have no preemptive rights to subscribe for additional issues of stock or to any security convertible into such stock unless, and to the extent that, such rights are expressly provided for in the certificate of incorporation.
233
Dividends
The Netherlands. Dutch law provides that dividends (if it concerns a distribution of profits) may be distributed after adoption of the annual accounts by the general meeting from which it appears that such dividend distribution is allowed. Moreover, dividends may be distributed, whether as a distribution of profits or of freely distributable reserves, only to the extent the shareholders’ equity exceeds the amount of the paid-in and called-up issued share capital and the reserves that must be maintained under the law or the articles of association. Interim dividends may be declared as provided in the articles of association and may be distributed to the extent that the shareholders’ equity exceeds the amount of the paid-in and called-up issued share capital plus any reserves as described above as apparent from our interim financial statements prepared under Dutch law.
Under our articles of association, our management board, with the approval of our supervisory board, may decide that all or part of the profits are carried to reserves. Before reservation of any profit, to the extent that preferred shares have been canceled and preferred distributions on those canceled shares are outstanding, the profits are first to be used to satisfy the outstanding claim to those who held those preferred shares at the moment of such cancellation becoming effective and subsequently if any preferred shares are outstanding, the preferred dividend is paid out of the remaining profit on the preferred shares in accordance with our articles of association. The remaining profit will be at the disposal of the general meeting at the proposal of the management board for distribution on the common shares, subject to restrictions of Dutch law and approval by our supervisory board of such proposal of our management board. Our management board is permitted, subject to certain requirements, to declare interim dividends without the approval of the general meeting, but only with the approval of the supervisory board. Dividends and other distributions shall be made payable not later than the date determined by the management board. Claims to dividends and other distributions not made within five years from the date that such dividends or distributions became payable will lapse and any such amounts will be considered to have been forfeited to us (verjaring).
Delaware. Under the Delaware General Corporation Law, a Delaware corporation may pay dividends out of its surplus (the excess of net assets over capital), or in case there is no surplus, out of its net profits for the fiscal year in which the dividend is declared and/or the preceding fiscal year (provided that the amount of the capital of the corporation is not less than the aggregate amount of the capital represented by the issued and outstanding stock of all classes having a preference upon the distribution of assets). In determining the amount of surplus of a Delaware corporation, the assets of the corporation, including stock of subsidiaries owned by the corporation, must be valued at their fair market value as determined by the board of directors, without regard to their historical book value. Dividends may be paid in the form of common stock, property or cash.
Shareholder Vote on Certain Reorganizations
The Netherlands. Under Dutch law, the general meeting must approve resolutions of the management board relating to a significant change in the identity or the character of the company or the business of the company, which includes:
| ● | a transfer of the business or virtually the entire business to a third party; |
| ● | the entry into or termination of a long-term cooperation of the company or a subsidiary with another legal entity or company or as a fully liable partner in a limited partnership or general partnership, if such cooperation or termination is of a far-reaching significance for the company; and |
| ● | the acquisition or divestment by the company or a subsidiary of a participating interest in the capital of a company having a value of at least one-third of the amount of its assets according to its balance sheet and explanatory notes or, if the company prepares a consolidated balance sheet, according to its consolidated balance sheet and explanatory notes in the last adopted annual accounts of the company. |
Delaware. Under the Delaware General Corporation Law, the vote of a majority of the outstanding shares of capital stock entitled to vote thereon generally is necessary to approve a merger or consolidation or the sale of all or substantially all of the assets of a corporation. The Delaware General Corporation Law permits a corporation to include in its certificate of incorporation a provision requiring for any corporate action the vote of a larger portion of the stock or of any class or series of stock than would otherwise be required.
234
Under the Delaware General Corporation Law, no vote of the stockholders of a surviving corporation to a merger is needed, however, unless required by the certificate of incorporation, if (i) the agreement of merger does not amend in any respect the certificate of incorporation of the surviving corporation, (ii) the shares of stock of the surviving corporation are not changed in the merger and (iii) the number of shares of common stock of the surviving corporation into which any other shares, securities or obligations to be issued in the merger may be converted does not exceed 20% of the surviving corporation’s common stock outstanding immediately prior to the effective date of the merger. In addition, stockholders may not be entitled to vote in certain mergers with other corporations that own 90% or more of the outstanding shares of each class of stock of such corporation, but the stockholders will be entitled to appraisal rights.
Remuneration of Managing Directors and Supervisory Directors
The Netherlands. The supervisory board determines the remuneration of individual managing directors with due observance of the compensation policy at the recommendation of our compensation committee. A proposal with respect to remuneration schemes in the form of shares or rights to shares in which managing directors may participate is subject to approval by our general meeting. Such a proposal must set out at least the maximum number of shares or rights to subscribe for shares to be granted to the managing directors and the criteria for granting or amendment. The compensation for our supervisory directors is set by the general meeting.
Delaware. Under the Delaware General Corporation Law, the stockholders do not generally have the right to approve the compensation policy for directors or the senior management of the corporation, although certain aspects of the compensation policy may be subject to stockholder vote due to the provisions of U.S. federal securities and tax law.
ITEM 16H. Mine Safety Disclosure
Not applicable.
ITEM 16K. Cybersecurity.
Protecting the security and integrity of CureVac’s IT systems and safeguarding the privacy of our business partners, patients and employees along with our intellectual property is a key priority for us. Cybersecurity risks are among the core risks evaluated through our enterprise risk management program.
Our IT security team is responsible for assessing and maintaining our cybersecurity risk management program. Our Head of IT oversees our cybersecurity program and is supported by our Manager of Information Security, the Senior Director of Infrastructure & Operations, the IT Leadership Team and other business leaders. Our cybersecurity program is designed to enable the Company to respond to the threat of security breaches and cyberattacks and to protect and preserve the confidentiality, integrity, and continued availability of information assets processed by the Company. Our Manager of Information Security and his team are experienced subject matter experts in the field of information security. The Senior Director of Infrastructure & Operations has over 25 years of international experience in information security, IT strategy and IT management, including security consulting. Similarly, our Head of IT has over 25 years of experience in various senior international IT leadership roles. Our internal IT security team is augmented by third parties, who provide additional information security expertise.
To address cybersecurity threats and in an effort to prevent IT system interruptions, we are developing enterprise-wide policies, procedures, and practices based on and audited against industry standards, including the ISO 27001 Standard and “BSI-Grundschutz”. CureVac’s systems are regularly patched and updated with supported software releases. We conduct regular internal vulnerability analyses (including simulated hacking) as well as external penetration testing utilizing third parties to verify and ensure the effectiveness of our cybersecurity controls. Our employees are required to participate in our cybersecurity awareness program, which includes access to specific cybersecurity and information security training sessions and content and periodic phishing attack simulations.
CureVac requires employees to report IT security incidents to the IT security team via CureVac’s internal service management platform. Additionally, we have implemented a 24/7/365 Cyber Security Operations Center (CSOC) operated by an external third party. The CSOC provides active monitoring and is responsible for investigating any security incidents and alerts on our platforms.
Where appropriate, threats or incidents are escalated to our Head of IT, who informs our CEO, management board and/or Data Protection Officer of any such threats or incidents. Any breaches related to personal data are managed by the Data Protection Officer in consultation with CureVac’s IT security team.
235
The Head of IT updates the status of the cybersecurity program quarterly within the enterprise risk management program. The management board provides oversight over the enterprise risk management program, reviews the risk portfolio on a quarterly basis and is responsible for ensuring that we have implemented an appropriate and effective risk management system and process. The supervisory board reviews the enterprise risk management program on an annual basis. In addition, the Head of IT provides regular updates on the status of the cybersecurity program, and emerging cybersecurity threats and incidents to the management board. At least once each year, the Audit Committee discusses our approach to cybersecurity risk management with the Head of IT and, as necessary, to the full supervisory board, based on management’s assessment of risk.
In 2023, we did not identify any cybersecurity threats or incidents that have materially affected or are reasonably likely to materially affect our business strategy, results of operations, or financial condition. However, despite our efforts, we cannot eliminate all risks from cybersecurity threats or incidents or provide assurances that we have not experienced an undetected cybersecurity incident. For more information about these risks, please see “Risk Factors–Risks Related to Our Business and Industry” in this Annual Report on Form 20-F.
PART III
ITEM 17. Financial Statements
We have responded to Item 18 in lieu of this item.
ITEM 18. Financial Statements
Our audited consolidated financial statements are included in this Annual Report beginning at Page F-1.
236
SIGNATURE
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this Annual Report on its behalf.
CureVac N.V. | |||
| By: | /s/ Alexander Zehnder | |
Name: Alexander Zehnder | |||
Title: Chief Executive Officer | |||
Date: April 24, 2024 | |||
237
Exhibit no. |
| Description |
|---|---|---|
4.10 | ||
4.11 | ||
4.12 | ||
4.13 | ||
4.14 | ||
4.15 | ||
4.16 | ||
4.17 | ||
4.18 | ||
4.19 | ||
4.20 | ||
4.21 | ||
4.22 | ||
4.23 | ||
4.24 | ||
239
Exhibit no. |
| Description |
|---|---|---|
4.25 | ||
4.26 | ||
4.27 | ||
4.28 | ||
4.29 | ||
4.30 | ||
4.31 | ||
4.32 | ||
4.33 | ||
4.34 | ||
4.35 | ||
4.36 | ||
4.37 | ||
4.38 | ||
240
Exhibit no. |
| Description |
|---|---|---|
4.39 | ||
4.40 | ||
4.41 | ||
4.42 | ||
4.43 | ||
4.44 | ||
4.45 | ||
4.46 | ||
4.47 | ||
4.48 | ||
4.49 | ||
4.50* | CureVac N.V. Long-Term Incentive Plan (Amended and Restated as of 2024). | |
4.51* | ||
4.52* | ||
4.53* | ||
4.54* | ||
241
Exhibit no. |
| Description |
|---|---|---|
4.55* | ||
8.1* | ||
12.1** | Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
12.2** | Certification Pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | |
13.1** | ||
13.2** | ||
15.1* | ||
15.2* | ||
97.1* | ||
101.INS** | Inline XBRL Instance Document | |
101.SCH** | Inline XBRL Taxonomy Extension Schema Document | |
101.CAL** | Inline XBRL Taxonomy Extension Calculation Linkbase Document | |
101.DEF** | Inline XBRL Taxonomy Extension Definition Linkbase Document | |
101.LAB** | Inline XBRL Taxonomy Extension Label Linkbase Document | |
101.PRE** | Inline XBRL Taxonomy Extension Presentation Linkbase Document | |
104** | Inline Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) |
† | Certain information has been excluded from the exhibit because it both (i) is not material and (ii) would likely cause competitive harm to the Registrant if publicly disclosed. |
* | Filed with this Annual Report on Form 20-F. |
** | In accordance with Rule 402 of Regulation S-T, the information in these exhibits shall not be deemed to be “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, and shall not be incorporated by reference into any registration statement or other document filed under the Securities Act or the Exchange Act, except as shall be expressly set forth by specific reference in such filing. |
242

CureVac N.V.
Consolidated Financial
Statements
As of December 31, 2023 and 2022
and for the years ended December 31, 2023, 2022 and 2021
F-1
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Audited Annual Consolidated Financial Statements |
| Page |
Reports of Independent Registered Public Accounting Firm (PCAOB ID | F-3 | |
Report of Independent Registered Public Accounting Firm (PCAOB ID 01251) | F-7 | |
F-8 | ||
Consolidated Statements of Financial Position as of December 31, 2023 and 2022 | F-9 | |
F-10 | ||
Consolidated Statements of Cash Flows for the Fiscal Years ended December 31, 2023, 2022 and 2021 | F-11 | |
F-12 |
F-2
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors
CureVac N.V.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated statement of financial position of CureVac N.V. and subsidiaries (the Company) as of December 31, 2023, the related consolidated statements of operations and comprehensive income (loss), changes in shareholders’ equity, and cash flows for the year ended December 31, 2023, and the related notes (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023, and the results of its operations and its cash flows for the year ended December 31, 2023, in conformity with International Financial Reporting Standards (IFRS) as issued by the International Accounting Standards Board.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated April 24, 2024 expressed an adverse opinion on the effectiveness of the Company’s internal control over financial reporting.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audit included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
F-3
Identification of distinct performance obligations and determining the over-time revenue recognition method under the GSK collaboration agreements
As discussed in Notes 2 and 3.1 to the consolidated financial statements, the Company entered into collaboration and license agreements with GlaxoSmithKline Biologicals SA (“GSK”) in July 2020, as amended and restated through March 2022. Under these arrangements, the Company recognized revenue in 2023 of EUR 38.3 million. These agreements contain multiple performance obligations and include licenses for the Company’s technology or programs, research and development services, and services or obligations in connection with participation in research or steering committees.
As part of our initial audit, we identified the evaluation of the distinct performance obligations identified by the Company and the determination of the appropriate method for measuring progress as a critical audit matter. Complex auditor judgment was required in evaluating the terms and conditions in the agreements to assess the identification of distinct performance obligations and to assess the most appropriate method to measure progress towards complete satisfaction of the identified performance obligations.
The following are the primary procedures we performed to address this critical audit matter. We evaluated the design of a certain internal control related to the identification of distinct performance obligations and the determination of the appropriate method to measure progress. We obtained and read the amended and restated collaboration and license agreements and evaluated the terms and conditions of the agreements to assess that the performance obligations within the agreements were completely and accurately identified in accordance with the relevant accounting guidance, and an appropriate measure of progress has been selected that best depicts the transfer of control to the customer.
Provisions for contract termination costs
As discussed in Note 2 to the consolidated financial statements, the Company recognizes provisions for contract termination costs when it is probable that an outflow of resources will be required to settle a present obligation and when the outflow can be reliably estimated. As described in Note 12 to the consolidated financial statements, as of December 31, 2023, the Company recorded provisions in the amount of EUR 37.4 million for the estimated costs that are expected to be required to settle the termination of arrangements that had been concluded with contract manufacturing organizations (CMOs) in prior years.
We have identified the evaluation of the provisions for terminated CMO contracts as a critical audit matter as the assessment required significant auditor judgment, due to the significant measurement uncertainty in the amount that will ultimately be required to be paid to release the Company from its remaining obligations under the CMO contracts, in particular considering the impact of future arbitration decisions.
The following are the primary procedures we performed to address this critical audit matter. We evaluated the design and tested the operating effectiveness of a certain internal control related to the evaluation of legal proceedings, including the evaluation of information from external and internal legal counsels. We evaluated the competence of the external legal counsel of the Company. We assessed the provisions by: (1) Inspecting confirmation letters received directly from the Company’s external legal counsels that assessed the likelihood of loss and the amounts that would be paid in the event of loss of the arbitration proceedings, and comparing these assessments and estimates to those made by the Company; (2) Inquiring of the Company’s in-house legal counsel; and (3) Inspecting the latest correspondence between the Company and the CMOs.
/s/
We have served as the Company’s auditor since 2023.
April 24, 2024
F-4
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors
CureVac N.V.:
Opinion on Internal Control Over Financial Reporting
We have audited CureVac N.V.’s and subsidiaries (the Company) internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. In our opinion, because of the effect of the material weakness, described below, on the achievement of the objectives of the control criteria, the Company has not maintained effective internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated statement of financial position of the Company as of December 31, 2023, the related consolidated statements of operations and comprehensive income (loss), changes in shareholders’ equity, and cash flows for the year ended December 31, 2023, and the related notes (collectively, the consolidated financial statements), and our report dated April 24, 2024 expressed an unqualified opinion on those consolidated financial statements.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis. A material weakness related to (i) lack of sufficient accounting and supervisory personnel, (ii) lack of appropriate level of technical accounting training; and (iii) lack of consistent performance of controls over financial reporting has been identified and included in management’s assessment. The material weakness was considered in determining the nature, timing, and extent of audit tests applied in our audit of the 2023 consolidated financial statements, and this report does not affect our report on those consolidated financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Annual Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
F-5
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ KPMG AG Wirtschaftsprüfungsgesellschaft
Mannheim, Germany
April 24, 2024
F-6
Report of Independent Registered Public Accounting Firm
To the Shareholders and the Audit Committee of CureVac N.V.
Opinion on the Financial Statements
We have audited the accompanying consolidated statement of financial position of CureVac N.V. (the Company) as of December 31, 2022, the related consolidated statements of operations and other comprehensive income (loss), changes in shareholders’ equity and cash flows for each of the two years in the period ended December 31, 2022, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2022, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2022, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ EY GmbH & Co. KG Wirtschaftsprüfungsgesellschaft
We served as the Company’s auditor from 2015 to 2023.
Stuttgart, Germany
April 25, 2023
F-7
CureVac N.V.
Consolidated Statements of Operations and
Other Comprehensive Income (Loss)
|
|
| Year ended December 31, | |||||
(in thousands of EUR, except per share amounts) |
| Note |
| 2021 |
| 2022 |
| 2023 |
Revenue | 3.1 | |
| | | |||
Cost of sales | 3.2 | ( |
| ( | ( | |||
Selling and distribution expenses | 3.3 | ( |
| ( | ( | |||
Research and development expenses | 3.4 | ( |
| ( | ( | |||
General and administrative expenses | 3.5 | ( |
| ( | ( | |||
Income from release of governmental contract liabilities | 3.7 | |
| — | — | |||
Other operating income | 3.8 | |
| | | |||
Other operating expenses | ( |
| ( | ( | ||||
Operating loss |
| ( |
| ( | ( | |||
Finance income | 15 | |
| | | |||
Finance expenses | 15 | ( |
| ( | ( | |||
Loss before income tax | ( |
| ( | ( | ||||
Income tax benefit/ (expense) | 13 | |
| | ( | |||
Net loss for the period |
| ( |
| ( | ( | |||
Other comprehensive income: |
|
| ||||||
Items that may be subsequently reclassified to profit or loss |
|
| ||||||
Foreign currency adjustments |
| ( |
| ( | ||||
Total comprehensive loss for the period |
| ( |
| ( | ( | |||
Net loss per share (basic and diluted) | 14 | ( | ( | ( | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-8
CureVac N.V.
Consolidated Statements of Financial Position
| December 31, | December 31, | ||||
(in thousands of EUR) |
| Note |
| 2022 |
| 2023 |
Assets |
|
|
| |||
Non-current assets |
|
|
| |||
Intangible assets and goodwill | 4.1 |
| | | ||
Property, plant and equipment | 4.1 |
| | | ||
Right-of-use assets | 4.2 |
| | | ||
Other assets |
| | | |||
Deferred tax assets | 13 |
| | | ||
Total non-current assets |
|
| | | ||
Current assets |
|
| ||||
Assets held for sale | 5 | | | |||
Inventories | 6 |
| | | ||
Trade receivables |
| | | |||
Contract assets | 3.1 |
| | | ||
Other financial assets | 7 |
| | | ||
Prepaid expenses and other assets | 8 |
| | | ||
Current tax assets | 13 | | | |||
Cash and cash equivalents |
|
| | | ||
Total current assets |
|
| | | ||
Total assets |
|
| | | ||
Equity and liabilities |
|
| ||||
Equity |
| |||||
Issued capital | 9 |
| | | ||
Capital reserve |
|
| | | ||
Treasury Shares |
|
| ( | — | ||
Accumulated deficit |
|
| ( | ( | ||
Other comprehensive income |
| ( | ( | |||
Total equity |
|
| ||||
Non-current liabilities |
| |||||
Lease liabilities | 4.2 |
| | | ||
Contract liabilities | 3.1 | | | |||
Provisions | 12 |
| | — | ||
Other liabilities | 12 |
| | — | ||
Total non-current liabilities |
|
| | | ||
Current liabilities |
|
| ||||
Lease liabilities | 4.2 |
| | | ||
Trade and other payables | 11 |
| | | ||
Provisions | 12 | | | |||
Other liabilities | 12 |
| | | ||
Income taxes payable | 13 |
| | | ||
Contract liabilities | 3.1 |
| | | ||
Total current liabilities |
| | | |||
Total liabilities |
| | | |||
Total equity and liabilities |
|
| | |
The accompanying notes are an integral part of these consolidated financial statements.
F-9
CureVac N.V.
Consolidated Statements of Changes in Shareholders’ Equity
|
|
|
|
| Currency |
| ||||||
Issued | Capital | Treasury | Accumulated | translation | Total | |||||||
(in thousands of EUR) | capital | reserve |
| Shares | deficit | reserve | equity | |||||
Balance as of January 1, 2021 |
| |
| |
| — | ( |
| |
| | |
Net loss |
| — |
| — |
| — | ( |
| — |
| ( | |
Other comprehensive income (loss) |
| — |
| — |
| — | — |
| ( |
| ( | |
Total comprehensive income (loss) |
| — |
| — |
| — | ( |
| ( |
| ( | |
Share-based payment expense | — |
| |
| — | — |
| — |
| | ||
Exercise of options | | | — | — | — | | ||||||
Issuance of share capital (net of transaction costs) | |
| |
| — | — |
| — |
| | ||
Repurchase of common shares | — | ( | ( | — | — | ( | ||||||
Balance as of December 31, 2021 |
| |
| |
| ( | ( |
| ( |
| |
|
|
|
|
| Currency |
| ||||||
Issued | Capital | Treasury | Accumulated | translation | Total | |||||||
(in thousands of EUR) | capital | reserves |
| Shares | deficit | reserve | equity | |||||
Balance as of January 1, 2022 |
| |
| |
| ( | ( |
| ( |
| | |
Net loss |
| — |
| — |
| — | ( |
| — |
| ( | |
Other comprehensive income (loss) |
| — |
| — |
| — | — |
| ( |
| ( | |
Total comprehensive income (loss) |
| — |
| — |
| — | ( |
| ( |
| ( | |
Share-based payment expense |
| — |
| |
| — | — |
| — |
| | |
Issuance of share capital (net of transaction costs) |
| |
| |
| — | — |
| — |
| | |
Share issuances and contingent consideration from business combination | | | — | — | — | | ||||||
Exercise of options / Settlement of share-based payment awards | | ( | | — | — | | ||||||
Balance as of December 31, 2022 |
| |
| |
| ( | ( |
| ( |
| |
| Currency | |||||||||||
Issued | Capital | Treasury | Accumulated | translation | Total | |||||||
(in thousands of EUR) |
| capital |
| reserves |
| Shares |
| deficit |
| reserve |
| equity |
Balance as of January 1, 2023 |
| |
| | ( |
| ( |
| ( |
| | |
Net loss | — |
| — | — |
| ( |
| — |
| ( | ||
Other comprehensive income (loss) | — |
| — | — |
| — |
| |
| | ||
Total comprehensive income (loss) | — |
| — | — |
| ( |
| |
| ( | ||
Share-based payment expense | — |
| | — |
| — |
| — |
| | ||
Issuance of share capital (net of transaction costs) | | | — | — | — | | ||||||
Settlement of share-based payment awards | | ( | | — | — | | ||||||
Balance as of December 31, 2023 |
| |
| | — |
| ( |
| ( |
| |
The accompanying notes are an integral part of these consolidated financial statements.
F-10
CureVac N.V.
Consolidated Statements of Cash Flows
|
|
| Year ended December 31, | |||||
(in thousands of EUR) |
| Note |
| 2021 |
| 2022 |
| 2023 |
Operating activities | ||||||||
Loss before income tax | ( |
| ( | ( | ||||
Adjustments to reconcile loss before tax to net cash flows |
| |||||||
Finance income | ( |
| ( | ( | ||||
Finance expense | |
| | | ||||
Depreciation and amortization | 4.1 | |
| | | |||
Impairment of property, plant and equipment | 4.1 | |
| | | |||
Loss on disposal of fixed assets | 4.1 | |
| | | |||
Impairment of assets held for sale | 5 | — | | | ||||
Impairment of inventory | 6 | |
| | | |||
Share-based payment expense | | | | |||||
Income from release of governmental contract liabilities | 3.7 | ( | — | — | ||||
Changes of provisions | 12 | — | ( | ( | ||||
Working capital changes |
| |||||||
Decrease / (increase) in asset held for sale | — | — | | |||||
Decrease / (increase) in trade receivables and contract assets | ( |
| | ( | ||||
Decrease / (increase) in inventory | 6 | ( |
| ( | ( | |||
Decrease / (increase) in prepaid expenses and other assets | ( |
| | | ||||
Receipts from grants from government agencies and similar bodies | |
| — | — | ||||
(Decrease) / increase in trade and other payables and contract liabilities | |
| ( | ( | ||||
(Decrease) / increase in other current financial and other liabilities | ( | — | — | |||||
Decrease / (increase) in deferred taxes | ( |
| | ( | ||||
Income taxes paid | ( |
| ( | ( | ||||
Interest received | |
| | | ||||
Interest paid | ( |
| ( | ( | ||||
Net cash flow provided by (used in) operating activities | ( | ( | ( | |||||
Investing activities |
| |||||||
Purchase of property, plant and equipment | ( |
| ( | ( | ||||
Purchase of intangible assets | ( |
| ( | ( | ||||
Acquisition of subsidiary, net of cash acquired | 20 | — | ( | — | ||||
Net cash flow provided by (used in) investing activities | ( | ( | ( | |||||
Financing activities |
| |||||||
Payments on lease obligations | ( | ( | ( | |||||
Proceeds from the issuance of shares (net of transaction costs) | |
| — | | ||||
Payment on / proceeds from treasury shares/exercise of options | ( |
| | | ||||
Proceeds from at-the-market offering program (net of transaction costs) | — | | — | |||||
Repayment of the EIB loan | ( | — | — | |||||
Net cash flow provided by financing activities | |
| | | ||||
Net increase (decrease) in cash and cash equivalents | ( |
| ( | ( | ||||
Effect of currency translation gains on cash and cash equivalents | |
| | ( | ||||
Cash and cash equivalents, beginning of period | |
| | | ||||
Cash and cash equivalents, end of period | |
| | | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-11
1. Corporate Information
CureVac N.V. (CureVac or CV or the Company) is the parent company of CureVac Group (Group) and, along with its subsidiaries, is a global biopharmaceutical company developing a new class of transformative medicines based on the messenger ribonucleic acid (mRNA) that has the potential to improve the lives of people.
With in - house manufacturing capabilities and expertise we are an integrated biopharmaceutical company. We consider our manufacturing process an important part of our strategy that allows us to continuously improve our technology platform and maintain flexibility in clinical development and allow us to manufacture potential future commercial products. The close interaction of our technical development and research teams enables us to rapidly implement innovations and robustness to the manufacturing process.
We were incorporated pursuant to the laws of the Netherlands as CureVac B.V. on April 7, 2020 to become a holding company for CureVac AG prior to our initial public offering. Pursuant to the terms of a corporate reorganization (the “Corporate Reorganization”), all of the outstanding shares in CureVac AG were contributed and transferred to CureVac B.V. in a capital increase in exchange for common shares of CureVac B.V. and, as a result, CureVac AG became a wholly-owned subsidiary of CureVac B.V. and then current shareholders of CureVac AG became the shareholders of CureVac B.V. Immediately following such exchange, and prior to the listing of our common shares on Nasdaq, we converted into a public company (naamloze vennootschap) under Dutch law pursuant to a Dutch notarial deed of amendment and conversion, following which our legal name became CureVac N.V. As part of our Corporate Reorganization, outstanding shares of all series in CureVac AG were exchanged for common shares in CureVac N.V.
We are registered in the commercial register at the Netherlands Chamber of Commerce under company number 77798031 (RSIN 861149336). Our principal executive offices are located at Friedrich-Miescher-Strasse 15, 72076 Tübingen, Germany.
During fiscal 2023, dievini Hopp BioTech holding GmbH & Co. KG (dievini), which is an investment company dedicated to the support of companies in health and life sciences, was the largest shareholder of CureVac. Together with its related parties, dievini held shares and voting rights in CureVac between appr.
2. Accounting Policies
Basis of preparation
The consolidated financial statements of the Group have been prepared in accordance with International Financial Reporting Standards (IFRS) as issued by the International Accounting Standards Board (IASB) and were authorized by the Management Board for presentation to the Supervisory Board on April 23, 2024. The Group’s consolidated financial statements are presented in Euros (EUR), which is also the parent company’s functional currency. Unless otherwise stated, the numbers are rounded to thousands of Euros, except per share amounts.Due to rounding, differences may arise when individual amounts or percentages are added together.
The Group has prepared the consolidated financial statements on the basis that it will continue to operate as a going concern. No commercial products have been marketed yet as CureVac is still in the development phase. Hence, the Group is raising capital to perform research & development activities and to advance its technologies up to a successful development and regulatory approval of its products. To reach its development and commercialization objectives, the Group will seek additional funding through collaboration or licensing agreements or other financing activities. While the Group’s business projections could be unfavorably affected by being unable to obtain funding or enter into collaboration or license agreements on acceptable terms, CureVac’s cash and cash equivalents will be sufficient to fund operating expenses and capital expenditure requirements for at least 12 months from the issuance date of the financial statements. This anticipates, that for the foreseeable future the Group will continue to operate. Thus, no adjustments to the consolidated financial statements have been made related to the Group not being able to continue as a going concern.
Details of the Group’s accounting policies, including changes thereto are described below. The accounting policies have been consistently applied to all years presented unless otherwise stated.
F-12
Basis of consolidation
Subsidiaries are entities controlled by the Group. The Group “controls” an entity when it is exposed, or has rights, to variable returns from its involvement with the investee and has the ability to affect those returns through its power over the investee.
The consolidated financial statements include the Company’s wholly-owned subsidiaries CureVac SE (until September 26, 2022: CureVac AG, Tuebingen, Germany), CureVac Inc. (Boston, Massachusetts, USA), CureVac Manufacturing GmbH (prior years: CureVac Real Estate GmbH, Tuebingen, Germany), CureVac Corporate Services GmbH (Tuebingen, Germany), CureVac RNA Printer GmbH (Tuebingen, Germany), CureVac Swiss AG (Basel, Switzerland – incorporated in 2021) and CureVac Belgium SA (Ottignies-Louvain-la-Neuve, Belgium – incorporated in 2022). Effective July 1, 2022, we acquired Frame Pharmaceuticals B.V., Amsterdam, Netherlands (Frame Pharmaceuticals), which was renamed to CureVac Netherlands B.V.; refer to Note 20 for additional information about this business combination.
The fiscal year of all Group entities corresponds to the calendar year ending December 31.
Material accounting policies
Business combinations and goodwill
The Group determines that it has acquired a business when the acquired set of activities and assets include an input and a substantive process that together significantly contribute to the ability to create outputs.
Any contingent consideration to be transferred by the acquirer is recognized at fair value at the acquisition date. Contingent consideration classified as equity is not remeasured and its subsequent settlement is accounted for within equity. Contingent consideration classified as an asset or liability that is a financial instrument and within the scope of IFRS 9 Financial Instruments, is measured at fair value with the changes in fair value recognized in the statement of operations in accordance with IFRS 9. Other contingent consideration that is not within the scope of IFRS 9 is measured at fair value at each reporting date with changes in fair value recognized in the statement of operations.
Goodwill is initially measured at cost (being the excess of the aggregate of the consideration transferred over the fair value of net identifiable assets acquired and liabilities assumed).
After initial recognition, goodwill is measured at cost less any accumulated impairment losses.
Revenue recognition
Revenue from the sale of products and services is recognized when the Group transfers control to the customer. Control generally transfers when the customer gains the ability to direct the use of and obtain substantially all of the remaining benefits from the good or service. If the contract contains more than one performance obligation, the consideration which the Group expects to receive is allocated to each of the performance obligations, using the relative stand-along selling price method. Revenue is recognized at the amount of consideration that the Group is expected to receive in exchange for these goods or services.
The Group primarily generates revenue from its licensing and development agreements with its customers, which include collaboration partners for the development of mRNA medicines against a variety of targets in diseases and conditions. These arrangements contain multiple contractual promises, including (i) licenses, or options to obtain licenses to the Group’s mRNA technology, as well as related research and development technology services, (ii) delivery of products, and (iii) research and development services. Such arrangements provide for various types of payments to the Group, including upfront fees, funding of research and development services, payment for delivered products, development, regulatory and commercial milestone payments, license fees, and royalties on product sales, all of which may be satisfied at different points in time. Outlicensing agreements may be entered into with or without any further significant contractual obligations.
Goods or services promised in collaborative arrangements are accounted for as separate performance obligations if such promises are distinct (i.e., if the customer can benefit from the good or service on its own or together with other resources readily available to it and if the promise is separately identifiable from other promises in the contract).
F-13
In determining whether contractual promises are separately identifiable, the Group considers whether:
| ● | It provides a significant service of integrating the goods or services with other goods or services that represent the combined output or outputs for which the other party has contracted. |
| ● | One or more of the goods or services significantly modifies or customizes one or more of the other goods or services promised in the agreement. |
| ● | The goods or services the Group promised to transfer or to provide are highly interdependent or highly interrelated. |
Based on these criteria, management evaluates whether the intellectual property (IP) licenses granted, and to which further research and development activities may apply under the terms of a collaboration agreement, are distinct from the unperformed obligations to the collaboration partner, considering the relevant facts and circumstances of each arrangement. Factors considered in this determination include the nature of the IP license, the stage of development of the IP license granted, the research capabilities of the partner, and the availability of mRNA technology research expertise in the general marketplace.
When an IP license is not considered to be distinct from research services, the Group generally recognizes revenue, including any upfront payment, attributable to the license on a straight-line basis, which reflects the performance of services by the Group towards satisfaction of the obligation, over the contractual or estimated performance period, which is typically from the effective date of the related collaboration agreement through to the estimated date of Marketing Authorization Application (MAA) or Biologics License Application (BLA) for a product developed under the agreement. The determination of the estimated date of MAA or BLA is a significant estimate given the uncertainty inherent in developing innovative pharmaceutical products and is based upon development plans with the customer, which are subject to change, clinical trials, and approval of regulatory authorities Changes in the estimated date of market entry could have a material impact on the amount and timing of revenue the Group records in future periods.
Sometimes an IP license is granted together with research and development services relating to the technology. In this case, the services are not considered distinct from the licenses and form a bundle for which revenue is recognized on a straight line basis over the time of services, as the services are determined predominant.
When an IP license is considered to be distinct, the Group determines whether it provides the customer with either (1) a right to access the IP throughout the license period (for which revenue is recognized over the license period) or (2) a right to use the IP as it exists at the point in time that the license is granted (for which revenue is recognized at a point in time where the customer can first use and benefit from the license).
Some of the collaboration arrangements include rights and obligations that are considered outside the scope of a customer-vendor relationship as they are more akin to the activities that joint operators perform. CureVac decided that the nature of these types of activities is best reflected by applying joint arrangements accounting which, as the main effect, usually leads to revenue recognition at a later point in time.
If the transaction price in an agreement includes a variable amount, the Group estimates the amount of consideration to which the Group will be entitled in exchange for transferring the goods to the customer. At contract inception, the variable consideration is estimated based on the most likely amount of consideration expected from the transaction and constrained until it is highly probable that a significant revenue reversal in the amount of cumulative revenue recognized will not occur when the associated uncertainty with respect to the variable consideration is subsequently resolved. The estimated deferred contract liability is updated at each reporting date to reflect the current facts and circumstances.
Collaboration agreements may also provide a customer with the option to acquire additional goods or services. In these cases, the Group determines as to whether the customer was granted a material right.
F-14
Product sales related to collaboration agreements include RNA products and are recognized over time as goods are produced because such goods have no alternative use. Otherwise, revenue for product sales is recognized at a point in time. In 2023 revenue for produced LNP batches were recognized on a point-in-time basis. No product revenue was recognized in 2022 and 2021. Revenue from certain research and development services, delivered as a distinct performance obligation under the collaboration agreements, are recognized over time as the services are provided.
A receivable is recognized when the consideration is unconditional and only the passage of time is required before payment is due. Amounts received prior to satisfying the above revenue recognition criteria are recorded as contract liability in the statements of financial position.
The Group may present the following contract balances:
| ● | Contract assets — Represents the Group’s right to consideration in exchange for goods or services that the Group has transferred to the customer when that right is conditioned on something other than the passage of time |
| ● | Trade receivables — Represents the Group’s right to an amount of consideration that is unconditional (i.e., only the passage of time is required before payment of the consideration is due) |
| ● | Contract liabilities — Represents the Group’s obligations to transfer goods or services to a customer for which the Group has received consideration (or consideration is due) from the customer |
The Group recognizes revenue from contracts with customers relating to its core business. All other operating proceeds are presented as other operating income in the statements of operations.
Grants from government agencies and similar bodies
The Group receives grants from government agencies and similar bodies for the active participation in specific research and development projects. Each grant agreement is assessed to determine whether there are elements of the supply of products from the group to the grantor that are recognized separately from the grant. For the supply of products, the standalone selling price is determined by reference to observed prices with other customers. The grants are recognized when there is reasonable assurance that the grant will be received and all grant conditions will be met. If grant funds are received prior to qualifying expenses being incurred or assets purchased, they are recorded as a liability in other liabilities. If the funds reimburse expenses, the liability is amortized into other operating income on a systematic basis over the period in which the corresponding expenses are incurred. If the funds reimburse purchased assets, the liability is reduced with a corresponding amount deducted from the asset’s carrying amount upon recording of the qualified asset. According to the terms of the grants, grantors generally have the right to audit qualifying expenses submitted by the Group.
Financial instruments
A financial instrument is any contract that gives rise to a financial asset of one entity and a financial liability or equity instrument of another entity.
i) Financial assets
Initial recognition and measurement
Financial assets are classified, at initial recognition, as subsequently measured at amortized cost, fair value through other comprehensive income (OCI), and fair value through profit or loss.
The classification of financial assets at initial recognition depends on the financial asset’s contractual cash flow characteristics and the Group’s business model for managing them. The Group initially measures a financial asset at its fair value plus, in the case of a financial asset not at fair value through profit or loss, transaction costs. Trade receivables that do not contain a significant financing component are measured at the transaction price determined under IFRS 15.
F-15
Subsequent measurement
For purposes of subsequent measurement, financial assets are classified into four categories:
| ● | financial assets at amortized cost (debt instruments); |
| ● | financial assets at fair value through other comprehensive income with recycling of cumulative gains and losses (debt instruments); |
| ● | financial assets designated at fair value through other comprehensive income with no recycling of cumulative gains and losses upon derecognition (equity instruments); or |
| ● | financial assets at fair value through profit or loss. |
In fiscal 2021, 2022, and 2023, the Group only had the following financial assets to be measured at amortized cost:
| ● | Cash and cash equivalents |
| ● | Trade receivables |
| ● | Other financial assets |
Financial assets at amortized cost are subsequently measured using the effective interest (EIR) method and are subject to impairment. Gains and losses are recognized in the statement of operations when the asset is derecognized, modified, or impaired.
Impairment of financial assets
An allowance for expected credit losses (ECLs) is recognized for all debt instruments not held at fair value through profit or loss including contract assets.
For credit exposures for which there has not been a significant increase in credit risk since initial recognition, ECLs are provided for credit losses that result from default events that are possible within the next 12- months (a 12- month ECL). For those credit exposures for which there has been a significant increase in credit risk since initial recognition, a loss allowance is required for credit losses expected over the remaining life of the exposure, irrespective of the timing of the default (a lifetime ECL).
For financial assets being due more than 30 days, a significant increase in credit risk is assumed.
For trade receivables and contract assets, the Group applies a simplified approach in calculating ECLs. Therefore, the Group does not track changes in credit risk but instead recognizes a loss allowance based on lifetime ECLs at each reporting date.
The Group considers a financial asset in default when contractual payments are 180 days past due. However, in certain cases, the Group may also consider a financial asset to be in default when internal or external information indicates that the Group is unlikely to receive the outstanding contractual amounts in full before taking into account any credit enhancements held by the Group. A financial asset is written off when there is no reasonable expectation of recovering the contractual cash flows.
ii) Financial liabilities
The Group’s material financial liabilities consists of lease liabilities and trade payables.
Trade payables at initial recognition are accounted for at fair value and subsequently at amortized costs. The material accounting policy information regarding accounting of leases is described below.
F-16
Acquired intangible assets
Acquired intangible assets are mainly comprised of software and licenses. Regarding the acquired intangible assets (i.e., technology and goodwill) in the business combination with Frame Pharmaceuticals, refer to Note 20.
The estimated useful lives for each intangible asset class are as follows:
Software |
| |
Licenses | ||
Technology |
With the exception of goodwill, the Group does not have any intangible assets with indefinite useful lives.
Research and development costs
Research costs are expensed as incurred. Since our own development projects are mostly subject to regulatory approval and all studies are at an early phase, where the outcome of reaching the next phases is uncertain, the conditions for the capitalization of expenditures incurred prior to approval are not met.
Property, plant and equipment
Property, plant and equipment are stated at cost less accumulated depreciation and accumulated impairments. These costs also comprise the costs for replacement parts, which are recognized at the time they are incurred, providing they meet the recognition criteria. All other repair and maintenance costs are expensed as incurred. Depreciation is recognized on a straight-line basis over the estimated useful lives as follows:
Leasehold improvements |
| |
Technical equipment and machines: |
| |
Other equipment, furniture and fixtures: |
|
The estimated useful lives are reviewed regularly and revised if necessary. The depreciation method remained unchanged from 2021 through 2023. The residual values of the assets are generally considered to be
Impairment of non-financial non-current assets
At each reporting date, the Group assesses whether there is an indication that a non-financial asset may be impaired. If there is any indication of impairment (triggering event) or if an annual impairment test is required, the Group estimates the recoverable amount of the asset. The recoverable amount of an asset is the higher of the asset’s or CGU’s fair value less costs of disposal and its value-in-use. It is determined for an individual asset unless the asset does not generate cash inflows that are largely independent of those from other assets or groups of assets, in which case it is determined at the level of the cash-generating unit (CGU). If the carrying amount of an asset or CGU exceeds its recoverable amount, the asset or CGU is impaired and written down to its recoverable amount. In determining fair value less costs of disposal, recent market transactions are taken into account. If no such transactions can be identified, an appropriate valuation model is used.
In principle, the recoverable amount has to be determined for each individual asset where such asset generates cash flows that are largely independent of those generated by other assets. Where it is not possible, the recoverable amount has been determined on the basis of a group of assets representing a cash generating unit.
F-17
In assessing whether the cash flows are largely independent, CureVac considers various factors, including how management manages the company activities and how the CureVac companies are linked in business operations. As management decisions are centralized on group level and the CureVac companies are closely linked in business operations, the Group operates as one cash-generating unit. Therefore, for the purpose of annual goodwill impairment testing in 2022 and 2023, goodwill was allocated at Group level. The recoverable amount for the Company’s cash-generating unit based on fair values less costs of disposal using market capitalization derived from public quotations of CureVac stock. Management believes that no reasonably foreseeable change in the price of CureVac stock would cause the carrying amount of our one cash-generating unit to exceed its recoverable amount.Impairment losses of continuing operations are recognized in the statement of operations in expense categories consistent with the function of the impaired asset.
In fiscal year 2021, impairment of EUR
Beginning in year 2022, goodwill is tested for impairment annually as of December 31 and when circumstances indicate that the carrying value may be impaired.
For 2023, impairment was evaluated for goodwill by assessing the recoverable amount on Group level. Impairment losses relating to goodwill cannot be reversed in future periods.
There was
Non-current assets held for sale
The Group classifies non-current assets as held for sale if their carrying amount is expected to be recovered principally through a sale transaction rather than through continuing use. Non-current assets classified as held for sale are measured at the lower of their carrying amount and fair value less costs to sell. Costs to sell are the incremental costs directly attributable to the disposal of an asset, excluding finance costs and income tax expenses. The criteria for held for sale classification is regarded as met only when the sale is highly probable, and the asset is available for immediate sale in its present condition. Actions required to complete the sale indicate that it is unlikely that significant changes to the sale will be made or that the decision to sell will be withdrawn. Management is committed to the plan to sell the assets. Property, plant and equipment are not depreciated or amortized once classified as held for sale.
Right-of-use assets
The Group recognizes right-of-use assets at the commencement date of the lease (i.e., the date the underlying asset is available for use). Right-of-use assets are measured at cost, less any accumulated depreciation and impairment losses, and adjusted for any remeasurement of lease liabilities. The cost of right-of-use assets includes the amount of lease liabilities recognized, initial direct costs incurred, and lease payments made at or before the commencement date less any lease incentives received as well as any estimated costs to be incurred by the lessee for dismantling and removing the underlying asset. Unless the Group is reasonably certain to obtain ownership of the leased asset at the end of the lease term, the recognized right-of-use assets are depreciated on a straight-line basis over the shorter of its estimated useful life, indicated below, and the lease term. Right-of-use assets are subject to impairment. Refer to the section above “Impairment of non-financial assets”.
Land, Buildings and supply installations |
| |
Vehicles |
| |
Other equipment |
|
Lease liabilities
At the commencement date of the lease, the Group recognizes lease liabilities measured at the present value of lease payments to be made over the lease term. The lease payments include fixed payments (including in- substance fixed payments) less any lease incentives receivable and amounts expected to be paid under residual value guarantees. The lease payments also include the exercise price of a purchase option reasonably certain to be exercised by the Group and payments of penalties for terminating a lease, if the lease term reflects the Group exercising the option to terminate.
F-18
In calculating the present value of lease payments, the Group uses the incremental borrowing rate at the lease commencement date if the interest rate implicit in the lease is not readily determinable. After the commencement date, the amount of lease liabilities is increased to reflect the accretion of interest and reduced for the lease payments made. In addition, the carrying amount of lease liabilities is remeasured if there is a modification, a change in the lease term, a change in the in-substance fixed lease payments, or a change in the assessment to purchase the underlying asset. When the lease liability is remeasured, a corresponding adjustment is made to the carrying amount for the right-of-use asset or is recorded in profit or loss if the carrying amount of the right-of-use asset has been reduced to zero.
Short-term leases and leases of low-value assets
The Group applies the short-term lease recognition exemption to its short-term leases (i.e., leases that have a lease term of 12 months or less from the commencement date and do not contain a purchase option). It also applies the lease of low-value assets recognition exemption. Lease payments on short-term leases and leases of low-value assets are recognized as expenses on a straight-line basis over the lease term.
Inventories
Inventories are measured at the lower of cost and net realizable value. Net realizable value is the estimated selling price in the ordinary course of business, less estimated costs of completion and the estimated costs necessary to make the sale. Inventories are comprised of raw materials, work in progress, and finished goods.
Costs incurred in bringing each product to its present location and condition are accounted for, as follows:
| ● | Raw materials: purchase cost on a first-in/first-out basis |
| ● | Finished goods and work in progress: cost of direct materials and labor and a proportion of manufacturing overhead based on normal operating capacity, but excluding borrowing costs |
Cash and cash equivalents
Cash and cash equivalents include cash on hand, bank balances on-demand, and short-term deposits with original maturities of three months or less.
Onerous contracts
An onerous contract is a contract under which the unavoidable costs (i.e., the costs that the Group cannot avoid because it has the contract) of meeting the obligations under the contract exceed the economic benefits expected to be received under it. The unavoidable costs under a contract reflect the least net cost of exiting from the contract, which is the lower of the cost of fulfilling it and any compensation or penalties arising from failure to fulfil it. The cost of fulfilling a contract comprises the costs that relate directly to the contract (i.e., both incremental costs and an allocation of costs directly related to contract activities).
If the Group has a contract that is onerous, the present obligation under the contract is recognized and measured as a provision. However, before a separate provision for an onerous contract is established, the Group recognizes any impairment loss that has occurred on assets dedicated to that contract.
Share-based payment awards
The Group operates several share-based payment programs.
An equity-settled share-based payment award is accounted for by recognizing the related expense over the vesting period of the award, with a corresponding increase recorded in equity. The expense is based on the fair value determined at the grant date of the award and the number of awards expected to vest. The fair value remains unchanged after grant date. Once the award has vested, there is no reversal of expense related to the award.
F-19
When a share-based payment award provides for different ways of settlement (i.e. cash versus shares) depending on the occurrence of contingent events, CureVac has the right to choose the manner of settlement and accounts the award based on this.
The related share-based payment expense is recorded in the functional cost category to which the award recipient’s costs are classified.
Income Taxes
Income taxes comprise current and deferred tax. Current and deferred taxes are recognized in profit or loss except to the extent that it relates to items recognized directly in equity or in other comprehensive loss.
Current tax is the expected tax payable or receivable on the taxable income or loss for the year, using tax rates enacted or substantively enacted at the reporting date, and adjustments to taxes payable in respect of previous years.
Tax loss carryforwards
Tax loss carryforwards are examined by the German taxation authorities and may be adjusted. Furthermore, significant changes in the shareholder and company structure can lead to a reduction in the loss carryforwards under the current provisions of German tax law, which can be used to calculate the annual amount for offsetting against the future taxable income.
CureVac assesses whether it is likely that a tax authority will accept an uncertain tax treatment. The amount of the expected tax liability or tax receivable reflects the amount, which represents the best estimation taken into account tax uncertainties. Based on management’s estimation, no provisions for uncertain tax treatments need to be accounted for.
Deferred income taxes
Deferred tax assets are recognized for deductible temporary differences, the carry forward of unused tax credits and any unused tax losses to the extent that it is probable that future taxable income will allow the deferred tax asset to be realized.
The carrying amount of deferred tax assets is reviewed at each reporting date and reduced to the extent that it is no longer probable that sufficient taxable profit will be available to allow all or part of the deferred tax asset to be utilized. Unrecognized deferred tax assets are re-assessed at each reporting date and are recognized to the extent that it has become probable that future taxable profits will allow the deferred tax asset to be recovered.
Segment Reporting
An operating segment is defined as a component of an entity for which discrete financial information is available and whose operating results are regularly reviewed by our Management Board as the Chief Operating Decision Maker (CODM). The Group operates as a single segment dedicated to the discovery and development of biotechnological applications and the CODM makes decisions about allocating resources and assessing performance based on the Group as a whole. Accordingly, the Group has determined it operates in
Significant accounting judgments, estimates and assumptions
The preparation of financial statements in conformity with IFRS requires management to make judgments, estimates, and assumptions that affect the reported amounts in the financial statements. Management continually evaluates its judgments and estimates in relation to assets, liabilities, contingent liabilities, uncertain tax position, revenues, and expenses. Management bases its judgments and estimates on historical experience and other various factors, which it believes to be reasonable under the circumstances, the result of which forms the basis of the carrying values of assets and liabilities that are not readily apparent from other sources. CureVac assesses whether it is likely that a tax authority will accept an uncertain tax treatment. Based on management’s estimation, no provisions for uncertain tax treatments need to be accounted for. Actual results may differ from these estimates under different assumptions and conditions and may materially affect the financial results or the financial position reported in future periods.
F-20
Judgments
In the process of applying the accounting policies, management has made the following judgments, which have the most significant effect on the amounts recognized in the consolidated financial statements.
Revenue recognition and collaboration agreements
The Group applied the following judgments in determining the amount and timing of revenue from collaboration agreements:
| ● | Identification and determination of the nature of performance obligations in collaboration and license agreements. |
The Group generates revenues from collaboration and license agreements under which the Group grants licenses to use, research, develop, manufacture, and commercialize candidates and products. As these agreements comprise several promises, it must be assessed whether these promises are capable of being distinct within the context of the contract. If these promises are not distinct, they are combined until the bundle of promised goods and services is distinct. For some agreements, this results in the Group accounting for all goods and services promised in a collaboration and license agreement as a single performance obligation with a single measure of progress.
For these combined performance obligations, it must be assessed which of these promises is the predominant promise to determine the nature of the performance obligation. The Group determined that the research and development technology services for the grant of the license is the predominant promise within the (combined) performance obligation.
If the Group shares the risks and benefits of a development project in such a way that the nature of the arrangement is not considered to be purely a customer-vendor-relationship, activities that are determined to be outside of a customer-vendor-relationship are accounted for in consideration of the principles of a joint operation accounting.
As a result, the promise of the research and development technology services for granting a license is accounted for as a performance obligation satisfied over time as the Group’s customer simultaneously receives and consumes the benefits from the Group’s performance.
Estimates
Revenue recognition and collaboration agreements
| • | Estimation of variable consideration and assessment of the constraint when determining the amount of revenue of which to defer recognition |
The Group’s collaboration and license agreements comprise variable considerations which are contingent on the occurrence or non-occurrence of a future event (i.e., reaching a certain milestone). When determining the deferral of revenue in a collaboration and license agreement, the Group is required to estimate the amount of consideration to which it will be entitled in exchange for transferring the promised goods or services to the customer.
As there are usually only two possible outcomes (i.e., a milestone is reached or not), the Group has assessed that the method of the most likely amount is the best method to predict the amount of consideration to which the Group will be entitled.
The most likely amount of these milestone payments (i.e., the full milestone payment) is only included in the transaction price if the occurrence of reaching a future milestone is highly probable. The Group has assessed that the likelihood of achieving the respective milestone decreases depending on how far the expected date of achieving the milestone lies in the future.
The Group has concluded that future milestone payments are fully constrained at each of the fiscal years. Future milestone payments would become unconstrained at the satisfaction of the milestone event, specifically a development event, regulatory approval, or achievement of a sales milestone.
F-21
•Estimation of determination of date of market authorization application for revenue release over-time
A significant amount of judgment given the uncertainty inherent in developing innovative pharmaceutical products is being used in the determination of the estimated date of the Marketing Authorization Application or Biologics License Applications. The group regularly reviews the timelines and changes are reflected in the release of revenue amount when they become effective.
Accounting for non - current assets held for sale
Non-current assets are classified as asset held for sale if their carrying amount will be recovered through a sale transaction rather than through continued use; the carrying value of such assets is measured at the lower of their previous carrying value and their fair value less costs to sell. In evaluating whether the criterion, for classification as assets held for sale, of a sale being highly probable, the Group considered that a plan to sell the assets was committed to and an active program to locate a buyer was initiated with an equipment reseller. By estimating the fair value less costs to sell the Group considered various indicative prices of expected auction proceeds quoted by the equipment reseller. Additionally, the Group estimated, whether the plan to sell the assets could be completed within one year from the date of classification and whether actions required to complete the plan indicate that it is unlikely that significant changes to the plan will be made or that the plan will be withdrawn.
Accounting for share-based payments
The Group has multiple share-based payment programs. Significant estimates are included in the determination of the grant date fair value of the awards.
Accounting for onerous contract provisions
The Group has entered into binding legal agreements for the supply of services by contract research organizations (CROs) to the Group for clinical trials of its first-generation COVID-19 vaccine candidate, CVnCoV. Such services are generally associated with the ongoing monitoring and care for enrolled participants in the clinical trials. Due to the discontinuation of the CVnCoV program, the remaining services, which the Group is obligated to procure, do not have a value for the Group anymore. In estimating the cost of the remaining services, judgment is required, particularly in estimating the number of participants completing the clinical trials, when measuring provisions for such contracts with clinical research organizations.
Accounting for contract termination provisions
Contract termination provisions are established under certain conditions in the case of legal risks. Settlement and legal proceedings often raise complex issues and are subject to many uncertainties and complexities including, but not limited to, the facts and circumstances of each particular case. The outcome of any current or future proceedings cannot normally be predicted. The Group considers the need for accounting measures in respect of pending or future settlements for terminated contracts on the basis of the information available to its legal department and in close consultation with legal counsel acting for the Group. Where it is more likely than not that such settlement will result in an outflow of resources that is already reasonably estimable, a provision for settling terminated contracts is recorded in the amount of the present value of the expected cash outflows.
Current income tax
CureVac assesses whether it is likely that a tax authority will accept an uncertain tax treatment. Based on management’s estimation, no provisions for uncertain tax treatments need to be accounted for. The risk for the assessment regarding i) the deferral of upfront payments, ii) the cooperation between CureVac and GlaxoSmithKline Biologicals SA (GSK), and iii) the transfer pricing setup not to be accepted by the tax authorities is estimated to be between
Changes in accounting policies and disclosures
New and amended standards and interpretations
The Group has not early adopted any standards, interpretations, or amendments that have been issued but are not yet effective.
F-22
Standards issued but not yet effective
The following amendments will be adopted effective January 1, 2024, or at a later effective date:
| ● | Amendments to IAS 1: Classification of Liabilities as Current or Non-current and Non-current Liabilities with Covenants |
| ● | Amendments to IAS 7 and IFRS 7: Supplier Finance Arrangements |
| ● | Amendments to IFRS 16: Lease Liability in a Sale and Leaseback |
| ● | Amendments to IAS 21: Lack of Exchangeability |
CureVac does not expect these standards to have an impact on the Group´s consolidated financial statements.
3. Notes to the consolidated financial statements
3.1 Revenue from contract with customers
The Group recognized the following revenues in 2021, 2022 and 2023:
| December 31 | |||||
2021 |
| 2022 |
| 2023 | ||
EUR k | EUR k | EUR k | ||||
Belgium |
|
|
| |||
GSK | | | | |||
Germany | ||||||
Boehringer Ingelheim | | — | — | |||
Netherlands | ||||||
Genmab | | | | |||
Switzerland | ||||||
CRISPR | | | | |||
Others | ||||||
Others | — | — | | |||
Total | | | | |||
During the fiscal year ended December 31, 2023, the Company recognized revenues over-time (i) EUR
F-23
GlaxoSmithKline
In July 2020, the Group entered a collaboration with GSK for the research, development, manufacture and commercialization of mRNA-based vaccines and monoclonal antibodies targeting infectious disease pathogens. In addition to an equity investment of EUR
Additionally, in April 2021, the Group entered into a new collaboration agreement with GSK, which we refer to as the GSK COVID Agreement, pursuant to which we are collaborating with GSK to research, develop and manufacture next-generation mRNA vaccines targeting the original SARS-CoV-2 strain as well as emerging variants, including multivalent and monovalent approaches (“GSK COVID Products”), such as the CureVac’s second-generation COVID-19 vaccine candidate, CV2CoV. These vaccine candidates may either be used to protect unvaccinated individuals or to serve as boosters in the event that SARS-CoV-2 immunity gained from an initial vaccination reduces over time. The GSK COVID Agreement was amended and restated in September 2021. Pursuant to the amendment in September 2021, CureVac and GSK are required to complete certain development activities with respect to the GSK COVID Products set forth in updated development plans. CureVac and GSK agree to decide whether the GSK COVID Products required for clinical studies will be manufactured by CureVac, GSK or jointly.
Under the GSK COVID Agreement, GSK has paid CureVac an upfront payment of EUR
F-24
Boehringer Ingelheim
In August 2014, the Group entered into an Exclusive Collaboration and License Agreement, which it refers to as the Boehringer Agreement, with Boehringer Ingelheim, whereby it granted Boehringer Ingelheim exclusive global rights for development and commercialization of its investigational therapeutic mRNA vaccine BI 1361849 (formerly CV9202) formulated with a legacy protamine technology. The Group received, in 2014, an upfront payment of EUR
CRISPR Therapeutics Development and License Agreement
In November 2017, we entered into a Development and License Agreement with CRISPR Therapeutics, which, as amended by amendments entered into in June 2020 and in October 2023, we refer to as the CRISPR Therapeutics Agreement, pursuant to which we will develop novel Cas9 mRNA constructs for use in gene editing therapeutics. CureVac and CRISPR confirmed to stop working on two programs under the CRISPR Therapeutics Agreement and added three new programs. CRISPR Therapeutics has paid us an upfront one-time technology access fee of USD
Genmab Collaboration and License Agreement
In December 2019, the Group entered into a Collaboration and License Agreement with Genmab, which we refer to as the Genmab Agreement, to research and develop up to four potential differentiated mRNA-based antibody products, to be selected by Genmab, based on the combination of our proprietary RNAntibody technology with Genmab’s proprietary antibody technology for the treatment of human diseases.
In June 2023, Genmab notified the Group of its intent to terminate the Genmab First Program under the Genmab Agreement effective in September 2023. Such termination did not terminate the Genmab Agreement in its entirety, but rather only with respect to certain license and exclusivity provisions related to the Genmab First Program. Under the Genmab Agreement, the Group further grants Genmab a license for the preclinical development of up to four additional mRNA antibody product concepts and options to obtain commercial licenses under CureVac’s mRNA technology to develop, manufacture and commercialize product candidates for up to three of such product concepts.
In partial consideration for entering into the Genmab Agreement, in 2019 Genmab made a USD
F-25
The Group has received upfront payments which were initially deferred and are subsequently recognized as revenue as the Group renders services over the performance period or upon termination of the agreement, when no services are provided anymore. Below is a summary of such payments and the related revenues recognized over-time:
Upfront and milestone payments included | ||||||||||||
| Upfront and milestone payments |
| in contract liabilities at |
| Revenue recognized from upfront and milestone payments | |||||||
Customer | December 31, 2022 | December 31, 2023 | 2021 | 2022 | 2023 | |||||||
(in k) | EUR k | EUR k | EUR k | |||||||||
GSK |
| EUR |
| |
| |
| |
| |
| |
Boehringer Ingelheim |
| EUR |
| — |
| — |
| |
| — |
| — |
Genmab |
| USD |
| |
| |
| |
| |
| |
CRISPR |
| USD |
| |
| |
| |
| |
| |
Total |
|
| |
| |
| |
| |
| | |
*Translated at the currency exchange rate prevailing on the transaction date
Contract balances:
| December 31, |
| December 31, | |
2022 | 2023 | |||
EUR k | EUR k | |||
Trade receivables |
| |
| |
Contract assets |
| |
| |
Contract liabilities |
| |
| |
Contract liabilities include advances received from the Group’s license and collaboration agreements.
Contract liabilities allocated to the remaining performance obligations (unsatisfied or partially unsatisfied) as at year-end are as follows:
Year ended December 31, | ||||
| 2022 |
| 2023 | |
EUR k | EUR k | |||
Within one year |
| | | |
More than one year |
| | | |
Total |
| | | |
Trade receivables are non-interest bearing and are generally settled within 30 to 45 days.
As of December 31, 2023, the Group had
F-26
The nature of expenses recognized in the functional categories of the statement of operations are as follows:
3.2 Cost of sales
The cost of sales consists of the following:
| 2021 |
| 2022 |
| 2023 | |
EUR k | EUR k | EUR k | ||||
Personnel |
| ( |
| ( | ( | |
Materials |
| ( |
| ( | ( | |
Third-party services |
| ( |
| ( | ( | |
Maintenance and lease |
| ( |
| ( | ( | |
Amortization and depreciation |
| ( |
| ( | ( | |
Impairment of equipment |
| ( |
| ( | ( | |
Other | ( | ( | ( | |||
Total |
| ( |
| ( | ( |
During the year ended December 31, 2023, cost of sales mainly decreased compared to the same period of 2022 due to higher material costs in the prior year, which were driven by write-offs of raw materials originally procured for the manufacturing of products intended to be sold to GSK. However, these raw materials were no longer expected to be sold to them (refer to Note 6 for further information). Additionally, in the prior year, the Company recognized a higher impairment of assets held for sale (refer to Note 5 for further information). Personnel expenses increased mainly due to increased workforce in the manufacturing organization.
3.3 Selling and distribution expenses
Selling and distribution expenses consist of the following:
| 2021 |
| 2022 |
| 2023 | |
EUR k | EUR k | EUR k | ||||
Personnel |
| ( |
| ( | ( | |
Maintenance and lease |
| ( |
| ( | ( | |
Amortization and depreciation |
| ( |
| ( | ( | |
Other |
| ( |
| ( | ( | |
Total |
| ( |
| ( | ( |
During the year ended December 31, 2023, selling and distribution expenses increased compared to the same period of 2022. The increase was primarily attributable to higher personnel expenses mainly related to the satellite offices in Belgium and the US.
3.4 Research and development (R&D) expenses
R&D expenses consist of the following:
| 2021 |
| 2022 |
| 2023 | |
EUR k | EUR k | EUR k | ||||
Materials |
| ( |
| ( | ( | |
Personnel |
| ( |
| ( | ( | |
Amortization and depreciation |
| ( |
| ( | ( | |
Patents and fees to register a legal right |
| ( |
| ( | ( | |
Third-party services |
| ( |
| | ( | |
Maintenance and lease |
| ( |
| ( | ( | |
Other |
| ( |
| ( | ( | |
Total |
| ( |
| ( | ( |
During the year ended December 31, 2023, research and development expenses increased significantly in comparison to the same period of 2022, as the prior period was largely impacted by the reversal of provision for onerous contracts in the amount of EUR
F-27
Since inception through December 31, 2023, the Group had no development expenditures which met the requirements for capitalization and thus none have been capitalized. In 2021, according to the terms and conditions of the grant from the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, or BMBF), the Group earned income (recognized in other operating income) for certain eligible expenses incurred for the COVID-19 vaccine development; refer to Note 3.7 for more information on amounts recognized from this grant in the year ended December 31, 2021.
Personnel expenses increased mainly due to increased workforce and the acquisition of Frame Pharmaceuticals. Additionally, share-based payment expense was higher compared to prior year period (refer to Note 10 for further details).
3.5 General and administrative expenses
General and administrative expenses include the following:
2021 | 2022 | 2023 | ||||
| EUR k |
| EUR k |
| EUR k | |
Personnel |
| ( |
| ( | ( | |
Maintenance and lease |
| ( |
| ( | ( | |
Third-party services |
| ( |
| ( | ( | |
Legal and other professional services |
| ( |
| ( | ( | |
Amortization and depreciation |
| ( |
| ( | ( | |
Other |
| ( |
| ( | ( | |
Total |
| ( |
| ( | ( |
During the year ended December 31, 2023, general and administrative expenses decreased in comparison to the same period of 2022. The decrease was primarily attributable to less personnel expenses due to lower workforce in the corporate service functions. Others include mainly expenses for D&O insurance and allocations.
3.6 Expenses by nature
The nature of the expenses is as follows:
| 2021 |
| 2022 |
| 2023 | |
EUR k | EUR k | EUR k | ||||
Personnel |
| ( |
| ( |
| ( |
Materials |
| ( |
| ( |
| ( |
Third-party services |
| ( |
| ( |
| ( |
Maintenance and lease |
| ( |
| ( |
| ( |
Amortization and depreciation |
| ( |
| ( |
| ( |
Impairment of equipment |
| ( |
| ( |
| ( |
Patents and fees to register a legal right |
| ( |
| ( |
| ( |
Legal and other professional services |
| ( |
| ( |
| ( |
Other |
| ( |
| ( |
| ( |
Total |
| ( |
| ( |
| ( |
We refer to Notes 3.2 - 3.5 for additional information.
F-28
3.7. Income from release of governmental contract liabilities
Due to the withdrawal of the EMA regulatory approval application for CVnCoV, in October 2021, CureVac recorded in 2021 “Income from release of governmental contract liabilities” amounting to EUR
Advance Purchase Agreement with European Commission
On November 30, 2020, CureVac entered into an Advance Purchase Agreement (APA) with the European Commission (EC), acting on behalf and in the name of all Member States of the European Union. The APA provided for the advance purchase by the Member States of
The second up-front payment would have had been due after the submission of the interim data package to the EMA in view of obtaining EC marketing authorization for CVnCoV. The up-front payments were designed to support the development and prepare the commercial supply of the vaccine.
In October 2021, we notified the EC of the withdrawal of our regulatory approval application for CVnCoV, which notification automatically terminated the APA. According to the APA, in such case of termination, CureVac would return the unspent amount of the up-front payment. In the context of the APA, “spent” means either costs occurred, or commitments made in relation to the purpose as set out in the APA. CureVac demonstrated that the up-front payment was used in accordance with the contract and
As described above, CureVac recognized the consideration related to its delivery obligations, existing at the outset of the arrangement, as contract liabilities. Upon the automatic termination of the APA, the ability for CureVac to satisfy the contractual performance obligations of the arrangement ceased and the EC ceased to have the ability to exercise its rights for performance by the CureVac. As such, the substance of the arrangement changed from a revenue contract to that of a government grant. Due to the material magnitude of the amount, its non-recurring nature and to better enable comparability to past performance and predictability of future performance, CureVac recognized the EUR
Additionally, CureVac was required to transfer, upon EC’s request any raw material and primary components paid for with the up-front payment and not used as of the termination date. Should the EC request any raw material and primary components or should CureVac successfully sell some of these, an applicable portion of raw material, primary components or proceeds would be remitted to the EC. This agreement expired at the end of 2022 and an amount of EUR
German Federal Ministry of Education and Research
In 2020, the Company announced that the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung), or BMBF, a German government-related entity,had established a grant to support the development and production of its COVID-19 vaccine candidates. In July 2020, CureVac applied for this grant as part of a special program to accelerate the research and development of urgently needed vaccines against SARS-CoV-2. The grant amounted up to EUR
The Company reached all the predefined milestones for 2020. Due to the withdrawal of the EMA regulatory approval application for CVnCoV in October 2021, CureVac was not able to reach all predefined milestones in 2021. From 2020 to December 2021, CureVac received a total of EUR
F-29
Consistent with the rationale and treatment described above under the APA with the EC, the substance of the supply component of the BMBF arrangement changed from a revenue contract to that of a government grant and thus, consistent with the presentation of the contract liabilities under the APA, CureVac recognized the EUR
3.8 Other operating income
Other operating income relates to:
2021 | 2022 | 2023 | ||||
| EUR k |
| EUR k |
| EUR k | |
Compensation for CMO/Materials transfer | — | | | |||
Reimbursement claim | — | | | |||
Sale of equipment | — | | | |||
Grants and other reimbursements from government agencies and similar bodies | | | | |||
Other |
| |
| | | |
Total |
| |
| | |
In March 2022, CureVac AG and GSK amended and restated the 2020 GSK agreement and the GSK COVID Agreement in connection with GSK entering into a direct agreement with Novartis for use of Novartis as a CMO at the same time as CureVac exited its CMO agreement with Novartis. Additionally, under the restated agreement, CureVac is entitled to further compensation by GSK. The compensation mainly consists of consideration for set-up activities undertaken by CureVac (EUR
In 2023, 2022 and 2021 income from grants with government agencies and similar bodies resulted from the following:
German Federal Ministry of Education and Research
As discussed in Note 3.7, in 2020 the Company received a grant from BMBF to support the development of its COVID-19 vaccine candidate for which it was determined the arrangement contained two components: a grant component (in the scope of IAS 20) and a supply component (in the scope of IFRS 15). The Group recognized grant income of EUR
In January 2024, CureVac has been granted the positive research allowance notice by the German Federal Ministry of Education and Research for the years 2020 - 2026. The research allowance is an instrument that provides tax relief for research expenditure by companies subject to tax in Germany - regardless of size, legal form and sector - and is intended to create incentives to invest in R&D. During the year ended December 31, 2023 the Group recognized EUR
Coalition for Epidemic Preparedness Innovations
The Coalition for Epidemic Preparedness Innovations (CEPI) is an innovative partnership between public, private, philanthropic, and civil organizations, launched at the World Economic Forum in Davos in 2017, to develop vaccines to stop future epidemics. CEPI’s priority diseases include Ebola virus, Lassa virus, Middle East Respiratory Syndrome coronavirus, Nipah virus, Rift Valley Fever and Chikungunya virus. CEPI also invests in platform technologies that can be used for rapid vaccine and immunoprophylactic development against unknown pathogens (i.e., Disease X).
In February 2019, CureVac entered into a partnership agreement worth up to USD
F-30
CureVac is required to use reasonable efforts to achieve certain development milestones and is responsible for conducting certain clinical trials. In the event of an infectious disease outbreak, where such outbreak can be addressed by a Lassa virus, SARS-CoV-2 or future vaccine developed under the agreement, CureVac must manufacture such vaccine for use in the area affected by the outbreak on economic terms that satisfy CEPI’s equitable access guidelines or otherwise allow CEPI or a third party to supply such vaccine in the affected area.
CureVac is required to grant certain approved manufacturers all necessary rights to use certain of CureVac’s pre-existing IP and IP developed under the CEPI Agreement to further develop CureVac’s automation solution and manufacture products for the treatment of certain diseases in geographic areas where there is an outbreak on economic terms that satisfy CEPI’s equitable access guidelines. CureVac must provide all necessary commercially reasonable support to such approved manufacturers to facilitate such efforts.
CureVac solely owns all IP developed under the CEPI Agreement but is required to obtain CEPI’s consent prior to exploiting any IP developed under the CEPI Agreement if such exploitation is in conflict with or goes against CEPI´s mission or policies.
In the event that CEPI terminates the agreement, CureVac will grant CEPI a license under CureVac’s background IP and IP developed under the agreement to, among other things, develop and use CureVac’s RNA Printer for use in treating certain infectious diseases and to manufacture products developed under the agreement.
In January 2020, CureVac and CEPI entered a collaboration to develop a vaccine against the new coronavirus SARS-CoV-2. The aim of the cooperation is to safely advance vaccine candidates into clinical testing as quickly as possible. The agreement builds upon the existing partnership between CureVac and CEPI to develop a rapid-response vaccine platform and included additional initial funding of up to USD
During the year ended December 31, 2023, CureVac recognized the reimbursement of approved expenses of EUR
As of December 31, 2023, EUR
Bill & Melinda Gates Foundation (BMGF)
BMGF finances, in the form of grants, various programs that CureVac operates for the development of vaccines, hence promoting and accelerating the development of CureVac’s technology platform. Through its equity investment, BMGF supports mainly the development of CureVac’s technology platform including the construction of a production plant in accordance with the GMP (Good Manufacturing Practice) standard on an industrial scale.
In 2015, CureVac entered into a Global Access Commitments Agreement with the Bill & Melinda Gates Foundation pursuant to which the Company is required to take certain actions to support the Bill & Melinda Gates Foundation’s mission.
In November 2016, in connection with the Global Access Agreement, CureVac received a grant of USD
During the year ended December 31, 2023 CureVac recognized EUR
F-31
As of December 31, 2023, EUR
4. Fixed Assets
4.1 Development of intangible assets and property, plant and equipment
The development of intangible assets and property, plant and equipment for the years ended December 31, 2023 and 2022 were as follows:
Intangible assets
| Advance |
| ||||||||||
(in thousands of EUR) |
| Software |
| Licenses |
| Technology |
| Goodwill |
| payments |
| Total |
Acquisition costs |
|
|
|
|
| |||||||
As of January 1, 2022 |
| | |
| — | — | |
| | |||
Additions |
| | |
| | | — |
| | |||
Disposals |
| ( | ( |
| — | — | ( |
| ( | |||
Reclassifications | | — | — | — | ( | — | ||||||
Currency translation | | — | — | — | — | | ||||||
As of December 31, 2022 |
| | |
| | | |
| | |||
Accumulated amortization and impairment charges |
|
|
| |||||||||
As of January 1, 2022 |
| | |
| — | — | — |
| | |||
Amortization |
| | |
| | — | — |
| | |||
Disposals | ( | ( | — | — | — | ( | ||||||
Currency translation | | — |
| — | — | — |
| | ||||
As of December 31, 2022 |
| | |
| | — | — |
| | |||
Acquisition costs | ||||||||||||
As of January 1, 2023 | | | | | | | ||||||
Additions | | | — | — | — | | ||||||
Disposals | ( | ( | — | — | ( | ( | ||||||
Reclassifications | ( | | — | — | — | — | ||||||
Currency translation | — | — | — | — | — | — | ||||||
As of December 31, 2023 | | | | | — | | ||||||
Accumulated amortization and impairment charges | ||||||||||||
As of January 1, 2023 | | | | — | — | | ||||||
Amortization | | | | — | — | | ||||||
Disposals | ( | ( | — | — | — | ( | ||||||
Reclassifications | ( | | — | — | — | — | ||||||
Currency translation | — | — | — | — | — | — | ||||||
As of December 31, 2023 | | | | — | — | | ||||||
Carrying amount |
|
| ||||||||||
As of January 1, 2022 |
| | |
| — | — | |
| | |||
As of December 31, 2022 |
| | |
| | | |
| | |||
As of December 31, 2023 |
| | | | | — |
| |
As the Group operates as one cash-generating unit, for the purpose of impairment testing in 2023, goodwill was allocated at Group level. For the impairment test the market capitalization was compared to the Group’s equity; the Goodwill was covered by the exceeding amount. The market capitalization derived from public quotations of CureVac stock (Nasdaq Dec 29, 2023: USD
F-32
Property, plant and equipment
Other | ||||||||||
equipment, | ||||||||||
Technical | furniture | Assets | ||||||||
| equipment |
| and |
| under | |||||
(in thousands of EUR) |
| Buildings |
| and machines |
| fixtures |
| construction |
| Total |
Acquisition costs |
|
|
|
|
|
|
|
|
|
|
As of January 1, 2022 |
| |
| |
| |
| |
| |
Additions |
| |
| |
| |
| |
| |
Assets held for sale | — | ( | — | ( | ( | |||||
Disposals |
| ( |
| ( |
| ( |
| ( |
| ( |
Reclassifications |
| — |
| |
| — |
| ( |
| — |
Currency translation |
| — |
| — |
| |
| — |
| |
As of December 31, 2022 |
| |
| |
| |
| |
| |
Accumulated depreciation and impairment charges |
|
|
|
|
| |||||
As of January 1, 2022 |
| |
| |
| |
| |
| |
Depreciation |
| |
| |
| |
| — |
| |
Impairment | — | | — | | | |||||
Disposals |
| ( |
| ( |
| ( |
| ( |
| ( |
Currency translation |
| — |
| — |
| |
| — |
| |
As of December 31, 2022 |
| |
| |
| |
| |
| |
Acquisition costs | ||||||||||
As of January 1, 2023 | | | | | | |||||
Additions | | | | | | |||||
Disposals | ( | ( | ( | ( | ( | |||||
Reclassifications | | | ( | ( | | |||||
Currency translation | — | — | ( | — | ( | |||||
As of December 31, 2023 | | | | | | |||||
Accumulated depreciation and impairment charges | ||||||||||
As of January 1, 2023 | | | | | | |||||
Depreciation | | | | — | | |||||
Impairment | — | — | — | | | |||||
Disposals | ( | ( | ( | — | ( | |||||
Currency translation | — | — | ( | — | ( | |||||
As of December 31, 2023 | | | | | | |||||
Carrying amount |
|
|
|
|
| |||||
As of January 1, 2022 |
| |
| |
| |
| |
| |
As of December 31, 2022 |
| |
| |
| |
| |
| |
As of December 31, 2023 |
| |
| |
| |
| |
| |
In fiscal 2023, impairments of EUR
The Group capitalized EUR
F-33
4.2 Right-of-use assets and lease liabilities
Set out below, are the carrying amounts of the Group’s right-of-use assets and the movements during the period:
Right-of-use assets | ||||||||
Land and | Other | |||||||
Buildings | Vehicles | equipment | Total | |||||
| EURk |
| EURk |
| EURk |
| EURk | |
As of January 1, 2023 |
| | | | | |||
Additions (new leases and reassessment of existing leases) |
| | | — | | |||
Depreciation expense |
| ( | ( | ( | ( | |||
Impairment | ( | — | — | ( | ||||
Foreign currency translation |
| ( | — | — | ( | |||
As of December 31, 2023 |
| | | | | |||
The most significant lease agreements that have been initiated relate to several buildings in Tübingen, a building in Wiesbaden and Frankfurt am Main, a building in Amsterdam/Netherlands and a building in Boston/USA.
The additions mainly relate to
Below are the carrying amounts of lease liabilities and the movements during the period:
| EUR k | |
As of January 1, 2023 |
| |
Additions (new leases and reassessment of existing leases) |
| |
Accretion of interest |
| |
Payments |
| ( |
Foreign currency translation |
| ( |
As of December 31, 2023 |
| |
Current |
| |
Non-current |
| |
A maturity analysis of lease liabilities is disclosed in Note 15.
The following are the amounts recognized in the statement of operations:
2021 | 2022 | 2023 | ||||
| EUR k |
| EUR k |
| EUR k | |
Depreciation expense of right-of-use assets |
| ( | ( | ( | ||
Impairment expense / reversal of impairment |
| — | ( | ( | ||
Interest expense on lease liabilities | ( | ( | ( | |||
Expense relating to short-term leases (included in cost of sales) |
| ( | ( | ( | ||
Expense relating to leases of low-value assets (included in general and administrative expenses) |
| ( | ( | ( | ||
Income from sub-leasing right-of-use assets presented in “other operating income” | — | — | |
F-34
Set out below, are the carrying amounts of the Group’s right-of-use assets and the movements of prior period:
Right-of-use assets | ||||||||
Land and | Other | |||||||
Buildings | Vehicles | equipment | Total | |||||
| EURk |
| EURk |
| EURk |
| EURk | |
As of January 1, 2022 |
| |
| |
| |
| |
Additions |
| |
| |
| |
| |
Depreciation expense |
| ( |
| ( |
| ( |
| ( |
Impairment | ( | — | — | ( | ||||
Foreign currency translation |
| |
| — |
| — |
| |
As of December 31, 2022 |
| |
| |
| |
| |
Below are the carrying amounts of lease liabilities and the movements during the period 2022:
| EUR k | |
As of January 1, 2022 |
| |
Additions |
| |
Accretion of interest |
| |
Payments |
| ( |
Foreign currency translation |
| |
As of December 31, 2022 |
| |
Current |
| |
Non-current |
| |
A maturity analysis of lease liabilities is disclosed in Note 15.
There are no commitments for leases not yet commenced as of December 31, 2023. Commitments for leases not yet commenced as of December 31, 2022, related to two lease agreements of buildings in Tübingen, Germany that have been signed in 2021 and one lease agreement of a building in Wiesbaden, Germany that has been signed in 2022.
Optional lease payments from using extension options not included in the measurement of the lease liability exist in a gross amount of EUR
5. Assets held for sale
In 2022, Management decided to dispose of certain equipment which had been procured for CMO activities (CMO Equipment) but that was no longer planned to be used by the Company. An external service-provider was appointed on June 14, 2022 to organize the sale of the CMO Equipment. As of December 31, 2022, the CMO Equipment identified for sale had a gross book value of EUR
During fiscal year 2023 assets with a net book value of EUR
F-35
6. Inventories
The inventories include only raw materials and supplies amounting to EUR
Timelines related to the Pandemic Preparedness Agreement were ambitious with the aim of providing acute pandemic preparedness in Germany in the context of emerging COVID-19 variants. In 2023, CureVac identified a risk that necessary regulatory approvals needed to meet these timelines may not be achieved within the contractually agreed timeframe. After consultation with ZEPAI, CureVac applied for a timeline extension as foreseen under the agreement to avoid procurement of further substantial amounts of raw material at risk. As of December 31, 2023, it was considered unlikely that the extension would be granted. Additionally, a rapidly changing epidemiological environment did no longer prompt an acute pandemic threat. Accordingly, inventories which had been stockpiled as required by the PPA, were written down to their net realizable value. Ultimately, the extension was not granted, whereupon CureVac and GSK jointly decided to terminate the agreement. For more detailed information we refer to Note 21. In total, inventory in the amount of EUR
The expense recognized related to the write-offs, scraps and consumption are mainly included in ‘Cost of Sales’. In the year 2022 EUR
7. Other financial assets
Other financial assets as of December 31, 2023 amounted to EUR
8. Prepaid expenses and other current assets
Prepaid expenses and other current assets of EUR
9. Equity
Overview
According to the Company’s articles of association, the Company’s authorized shares are divided into
All payments received from shareholders in excess of the nominal value of the shares issued and net of transaction costs are recognized in capital reserves. Capital reserves also consists of recognition of share-based payments and the equity components of convertible loans. The Company may only make distributions, whether a distribution of profits or of freely distributable reserves, to shareholders to the extent shareholders’ equity exceeds the sum of the paid-in and called-up share capital plus any reserves required by Dutch law or by the Company’s articles of association.
F-36
The number of shares issued and outstanding in fiscal 2023 are as follows:
Common shares issued and outstanding at December 31, 2020 |
| |
Follow-on public offering, incl. Greenshoe | ||
Share option exercises | ||
Common shares issued and outstanding at December 31, 2021 | ||
Shares issued as part of the at-the-market offering program | | |
Shares issued to former shareholders of Frame Pharmaceuticals | | |
Shares issued for LTIP option exercises and RSU deliveries | | |
Common shares issued and outstanding at December 31, 2022 | | |
Shares issued as part of the at-the-market offering program | | |
Shares issued as part of the public offering | | |
Shares issued for LTIP option exercises and RSU releases | | |
Common shares issued and outstanding at December 31, 2023 | |
Refer to Note 21 for additional information on share transactions occurring after December 31, 2023. The share transactions which occurred in 2021, 2022 and 2023 are as described below.
Follow-on public offering
In February 2021, the Company completed a follow-on public offering whereby it sold
At-the-market offering
On September 17, 2021, CureVac filed a prospectus for an “at-the-market” (ATM) offering program to raise additional cash of up to USD
Follow-on public offering 2023
In February 2023, the Company completed a follow - on public offering whereby it sold
Frame Pharmaceuticals acquisition
On June 8, 2022, CureVac entered into a Share Purchase Agreement (SPA) to acquire all of the issued and outstanding shares of Frame Pharmaceuticals B.V., a research company focused on advanced genomics and bioinformatics, based in Amsterdam, Netherlands. Under the SPA, the total consideration for the the purchase was up to EUR
F-37
Exercises of share options under the prior VSOP plan
The IPO in August 2020 triggered an exercise event under the set terms of the prior VSOP plan (see Note 10). In March 2021, CureVac received
A third triggering event, again “liquidity after IPO”, was met on the second anniversary of the IPO. In December 2022, CureVac received
Exercises of share options under the new VSOP plan
Participants of the new VSOP plan (see Note 10) were able to continue to exercise their options throughout the year of 2023. In 2023
10. Share-based payments
During the years ended December 31, 2023, 2022, and 2021, the Group operated the following share-based plans for members of management and other key employees of the Group, as well as members of the supervisory board:
| ● | Prior VSOP |
| ● | New VSOP — for US employees (from 2019 onwards) |
| ● | LTIP Stock Options |
| ● | LTIP RSUs (from 2021 onwards) |
All programs were accounted for as equity-settled share-based payment awards.
Measurement of the grant date fair value is based on valuation techniques appropriate in the circumstances, such as Black Scholes option pricing models or a Monte Carlo simulation. Expected volatility, a key input to such models, was based on an evaluation of the historical volatilities of comparable listed biotech-companies over the historical period commensurate with the expected option life. Regarding the expected option life of the stock option programs, this was based on the assumptions that the beneficiary would exercise his option in equal installments from the date of the first time possible (taking into account lock-up and potential trading windows restrictions) until maturity. The risk-free interest was derived from German or US-Government bonds, as appropriate.
The expense recognized for share-based payments during the years ended December 31 is as follows:
| 2021 |
| 2022 |
| 2023 | |
EUR k | EUR k | EUR k | ||||
Prior VSOP |
| ( |
| ( |
| ( |
New VSOP |
| ( |
| |
| ( |
LTIP Stock Options |
| ( |
| ( |
| ( |
LTIP RSUs |
| ( |
| ( |
| ( |
RSU Supervisory board |
| ( |
| ( |
| ( |
Total |
| ( |
| ( |
| ( |
F-38
Prior VSOP
The development of the virtual shares in the Prior VSOP program granted to management and key employees was as follows:
| 2021 |
| 2022 |
| 2023 | |
Outstanding at the beginning of the period | | | | |||
Granted during the period |
| — | — | — | ||
Forfeited during the period |
| — | ( | ( | ||
Exercised during the period | ( | ( | — | |||
Outstanding at the end of the period |
| | | | ||
Thereof vested |
| | | | ||
Thereof exercisable |
|
The IPO on August 14, 2020, triggered the right to exercise
A second
A third
A fourth
Expense recognized in the statement of operations and other comprehensive income (loss)
The expense recognized for this share-based payment plan during the years ended December 31 is as follows:
2021 | 2022 | 2023 | ||||
| EUR k |
| EUR k |
| EUR k | |
Cost of Sales | — | — | ( | |||
Selling and distribution expenses | ( | ( | ( | |||
Research and development expenses |
| ( |
| ( |
| |
General and administrative expenses |
| ( |
| ( |
| ( |
Total |
| ( |
| ( |
| ( |
F-39
New VSOP
The number of awards in the New VSOP program granted to key employees developed as follows:
| 2021 |
| 2022 |
| 2023 | |
Outstanding at the beginning of the period | |
| |
| | |
Granted during the period | — |
| — |
| — | |
Forfeited during the period | ( | — | ||||
Exercised during the period | ( | ( | ( | |||
Outstanding at the end of the period |
| | | | ||
Thereof vested | | | | |||
Thereof exercisable |
| | | — |
The remaining life of the option awards as of December 31, 2023 is between
In 2021 multiple exercises happened throughout the year. In total
In 2023, a number of exercises were carried out throughout the year. In total,
Expense recognized in the statement of operations and other comprehensive income (loss)
The expense recognized for employee services received during the years ended December 31, 2023, 2022, and 2021 is shown in the following table:
| 2021 |
| 2022 |
| 2023 | |
EUR k | EUR k |
| EUR k | |||
Research and development expenses |
| ( | | ( | ||
Selling and distribution expenses |
| ( | | — | ||
General and administrative expenses |
| ( | | | ||
Total |
| ( | | ( |
Long-Term Incentive Plan (LTIP) - Options
On November 16, 2020, CureVac granted
Options granted to the former CSO have an exercise price of EUR
F-40
For the grant to the former CSO, a Monte Carlo simulation has been used to measure the fair value at the relevant grant date. The inputs used in the measurement of the fair value at grant date were as follows:
Weighted average fair value per option |
| EUR | |
Weighted average share price (10-days VWAP before grant date) |
| EUR | |
Exercise price (USD |
| EUR | |
Expected volatility (%) |
| | % |
Expected life (years) |
| | |
Risk-free interest rate (%) |
| % |
At December 31, 2022,
Options granted to the former CBO / CCO have been granted in 3 tranches vesting over to
For the grant to the former CBO/CCO, a Monte Carlo simulation has been used to measure the fair value at the relevant grant date. The inputs used in the measurement of the fair value at grant date were as follows:
First tranche:
Weighted average fair value per option |
| EUR | |
Weighted average share price (actual 10-days VWAP before grant date, USD |
| EUR | |
Exercise price (USD |
| EUR | |
Expected volatility (%) |
| | % |
Expected life (years) |
| | |
Risk-free interest rate (%) |
| % |
Second tranche:
Weighted average fair value per option |
| EUR | |
Weighted average share price (estimated by Monte Carlo simulation to be USD |
| EUR | |
Exercise price (estimated by Monte Carlo simulation to be USD |
| EUR | |
Expected volatility (%) |
| | % |
Expected life (years) |
| | |
Risk-free interest rate (%) |
| % |
Third tranche:
Weighted average fair value per option |
| EUR | |
Weighted average share price (estimated by Monte Carlo simulation to be USD |
| EUR | |
Exercise price (estimated by Monte Carlo simulation to be USD |
| EUR | |
Expected volatility (%) |
| | % |
Expected life (years) |
| | |
Risk-free interest rate (%) |
| % |
As the former CBO/CCO left the Company as of November 30, 2023, all remaining unvested awards were subject to accelerated vesting.
F-41
On March 1, 2021, CureVac granted
For the grant to the key employee, a Monte Carlo simulation has been used to measure the fair value at the relevant grant date. The inputs used in the measurement of the fair value at grant date were as follows:
Weighted average fair value per option |
| EUR | |
Weighted average share price (30-days VWAP after grant date) |
| EUR | |
Exercise price (USD |
| EUR | |
Expected volatility (%) |
| | % |
Expected life (years) |
| | |
Risk-free interest rate (%) |
| % |
On July 1, 2021, CureVac granted
Options granted to the COO have an exercise price of EUR
For the grant to the COO, a Monte Carlo simulation has been used to measure the fair value at the relevant grant date. The inputs used in the measurement of the fair value at grant date were as follows:
Weighted average fair value per option |
| EUR | |
Weighted average share price (20-day VWAP before grant date) |
| EUR | |
Exercise price (USD |
| EUR | |
Expected volatility (%) | | % | |
Expected life (years) |
| ||
Risk-free interest rate (%) | % |
Options granted to the former CDO have an exercise price: EUR
For the grant to the former CDO, a Monte Carlo simulation has been used to measure the fair value at the relevant grant date. The inputs used in the measurement of the fair value at grant date were as follows:
Weighted average fair value per option |
| EUR | |
Weighted average share price (20-days VWAP before grant date) |
| EUR | |
Exercise price (USD |
| EUR | |
Expected volatility (%) | | % | |
Expected life (years) |
| ||
Risk-free interest rate (%) | % |
The former CDO has since left the company and, under the terms of his LTIP agreement, his options had expired as of December 31, 2022.
F-42
On January 1, 2022, CureVac granted
For the grant to the key employee, a Monte Carlo simulation has been used to measure the fair value at the relevant grant date. The inputs used in the measurement of the fair value at grant date were as follows:
Weighted average fair value per option |
| EUR | |
Weighted average share price (30-days VWAP after grant date) |
| EUR | |
Exercise price (USD |
| EUR | |
Expected volatility (%) |
| | % |
Expected life (years) |
| | |
Risk-free interest rate (%) |
| % |
The key employee has left the company and, under the terms of his LTIP agreement, his unvested options have been forfeited as of January 31, 2024.
On March 1, 2022, CureVac granted
The options granted to the management board have an exercise price of USD
For the grants to the management board, a Monte Carlo simulation has been used to measure the fair value at the grant date. The inputs used in the measurement of the fair value at grant date were as follows:
Weighted average fair value per option |
| EUR | |
Weighted average share price (10 - day VWAP before grant date) |
| EUR | |
Exercise price (USD |
| EUR | |
Expected volatility (%) |
| % | |
Expected life (years) |
| | |
Risk-free interest rate (%) |
| % |
As the former CEO, former CSO and former CBO/CCO left the Group, in 2023, all of their remaining unvested awards were subject to accelerated vesting. As of December 31, 2023 none of these options had been exercised.
On April 1, 2022, CureVac granted
F-43
For the grant to the key employee, a Monte Carlo simulation has been used to measure the fair value at the grant date. The inputs used in the measurement of the fair value at grant date were as follows:
Weighted average fair value per option |
| EUR |
|
Weighted average share price (10-days VWAP before grant date) | EUR |
| |
Exercise price (USD |
| EUR | |
Expected volatility (%) |
| | % |
Expected life (years) |
| | |
Risk-free interest rate (%) |
| | % |
On April 1, 2023, CureVac granted
For the grant to the CEO, a Monte Carlo simulation has been used to measure the fair value at the grant date. The inputs used in the measurement of the fair value at grant date were as follows:
Weighted average fair value per option |
| EUR | |
Weighted average share price (10-day VWAP before grant date) |
| EUR | |
Exercise price (USD |
| EUR | |
Expected volatility (%) |
| | % |
Expected life (years) |
| | |
Risk-free interest rate (%) |
| | % |
The expense recognized for employee services received under the LTIP – options during the year ended December 31, 2023, in an amount of EUR
Long-Term Incentive Plan (LTIP) - Restricted Stock Units (RSUs)
Restricted Stock Units (RSUs)
In 2021, as part of the LTIP program, the group awarded RSUs (restricted stock units) to senior executives as well as supervisory board members.
On June 24, 2021, the group awarded
In addition, on July 1, 2021, the group also awarded
In 2022, as part of the LTIP program, the group awarded RSUs (RSU Award 2022) to senior executives as well as supervisory board members.
On June 22, 2022 the group awarded
In addition, on January 1, 2022, the group awarded
F-44
On January 31, 2022, the group also awarded
On July 1, 2022, the group awarded
On March 31, 2023, the Group awarded
On September 30, 2023 the group also awarded
As the former CEO, former CSO and former CBO/CCO left the Group in 2023, all of their remaining unvested awards were subject to accelerated vesting. As a Supervisory Board member left the Group as of June 19, 2023, all remaining unvested awards were subject to accelerated vesting.
Expenses for employer taxes arising upon the delivery of RSUs are recognized in profit or loss.
The related RSU expense is recorded in the functional cost category to which the award recipient’s costs are classified.
| 2021 |
| 2022 |
| 2023 | |
EUR k | EUR k | EUR k | ||||
Cost of Sales | — | — | ( | |||
Research and development expenses |
| ( |
| ( |
| ( |
Selling and distribution expenses |
| ( |
| ( |
| ( |
General and administrative expenses |
| ( |
| ( |
| ( |
Total |
| ( |
| ( |
| ( |
11. Trade and other payables
Trade payables and other payables are all due within one year and include the following:
2022 | 2023 | |||
| EUR k |
| EUR k | |
Trade payables |
| |
| |
Miscellaneous liabilities |
| |
| |
Total |
| |
| |
Trade and other payables are all due within one year. The prior year was highly impacted by received invoices of raw material suppliers before December 31, 2022. Thus, during the year ended December 31, 2023, Trade and other payables decreased by EUR
F-45
12. Other liabilities and provisions
Provisions include the following:
| 2022 |
| 2023 | |
| EUR k |
| EUR k | |
Contract termination provisions |
| |
| — |
Provisions (non-current) |
| |
| — |
Provision for onerous contracts |
| |
| |
Contract termination provisions |
| — |
| |
Provisions (current) |
| |
| |
Total Provisions (current & non-current) |
| |
| |
Below are movements during the period:
| EUR k | |
As of January 1, 2023 |
| |
additions |
| |
used (amounts charged against the provision) |
| ( |
unused amounts reversed |
| ( |
As of December 31, 2023 |
| |
Current |
| |
Non-current |
| — |
The provision for onerous contracts relates to CRO agreements and are expected to be settled within one year from December 31, 2023. All amounts recognized as of December 31, 2022 arose during 2021. During the year ended December 31, 2023, EUR
Contract termination provisions relate to amounts which the Company expects to pay out to settle its obligations under certain CMO contracts which it has terminated. All amounts recognized as of December 31, 2023 arose during 2021 and 2022 except for EUR
F-46
Other liabilities include the following:
2022 | 2023 | |||
| EUR k |
| EUR k | |
Deferred Tax Liability | | — | ||
Other liabilities (non-current) | | — | ||
Personnel accrued liabilities (e.g. bonus, vacation) |
| |
| |
Grants from government agencies and similar bodies |
| |
| |
Outstanding invoices |
| |
| |
Professional fees |
| |
| |
Other taxes | | | ||
Other | | | ||
Other liabilities (current) |
| |
| |
Total other liabilities (current & non-current) |
| |
| |
13. Income tax
In fiscal 2023, the Group recorded a consolidated income tax expense of EUR
A reconciliation between actual income taxes and the expected tax benefit from the loss before tax multiplied by the Group’s applicable tax rate of
2021 | 2022 | 2023 | ||||
| EUR k |
| EUR k |
| EUR k | |
Loss before tax |
| ( | ( | ( | ||
Expected tax benefit (based on statutory tax rate of |
| | | | ||
Recognition of tax loss carryforwards recognized in prior years | — | | — | |||
Effects from differences between Group and local tax rates |
| ( | ( | ( | ||
Non-recognition of tax loss carryforwards |
| ( | ( | ( | ||
Non-recognition of deferred tax assets |
| ( | ( | — | ||
Recognition of deferred tax assets | — | — | | |||
Non-deductible expenses for tax purposes |
| — | ( | ( | ||
Additions for local trade taxes |
| ( | ( | — | ||
Other non-deductible expenses |
| ( | — | — | ||
Other effects |
| | ( | | ||
Effective tax benefit / (expense) |
| | | ( |
F-47
Deferred tax assets and deferred tax liabilities consist of the following:
| January 01, 2023 |
| Recognized in |
| Acquired in |
| December 31, 2023 | |||||||
| Net balance |
| profit and loss |
| Equity |
| Business combination |
| Net balance |
| DTA |
| DTL | |
EUR k | EUR k | EUR k | EUR k | EUR k | EUR k | EUR k | ||||||||
Non-current assets | ||||||||||||||
Intangible assets | ( | |
| — | — | ( |
| — |
| ( | ||||
Property, plant and equipment | ( | ( |
| — | — | ( |
| — |
| ( | ||||
Right-of-use assets | ( | |
| — | — | ( |
| — |
| ( | ||||
Other assets | ( | |
| — | — | — |
| — |
| — | ||||
Current assets |
|
|
|
|
|
|
|
|
|
| ||||
Inventories | — | |
| — | — | |
| |
| — | ||||
Trade receivables | ( | |
| — | — | |
| |
| — | ||||
Prepaid expenses and other assets | | — |
| — | — | |
| |
| — | ||||
Cash and cash equivalents | ( | |
| — | — | ( |
| |
| ( | ||||
Non - current liabilities |
|
|
|
|
|
|
|
|
|
| ||||
Lease liabilities (Non-current ) | | ( |
| — | — | |
| |
| — | ||||
Other liabilities (Non-current) | | ( |
| — | — | ( |
| — |
| ( | ||||
Current liabilities |
|
|
|
|
|
|
|
|
|
| ||||
Lease liabilities (Current) | | ( |
| — | — | |
| |
| — | ||||
Trade and other payables | ( | |
| — | — | ( |
| — |
| ( | ||||
Other liabilities (Current) | | ( |
| — | — | ( |
| |
| ( | ||||
Tax losses carried forward | | |
| — | — | |
| |
| — | ||||
Share-based payments | | ( |
| — | — | |
| |
| — | ||||
Netting | — | — |
| — | — | — |
| ( |
| | ||||
Deferred Taxes Total | | ( |
| — | — | |
| |
| ( | ||||
F-48
January 01,2022 | Recognized in | Acquired in | December 31, 2022 | |||||||||||||
| Net balance |
| profit and loss |
|
| Equity |
| Business combination |
|
| Net balance |
| DTA |
| DTL | |
EUR k | EUR k | EUR k | EUR k | EUR k | EUR k | EUR k | ||||||||||
Non-current assets | ||||||||||||||||
Intangible assets | — |
| — | — | ( | ( | — | ( | ||||||||
Property, plant and equipment | ( |
| ( | — | — | ( | | ( | ||||||||
Right-of-use assets | ( |
| ( | — | — | ( | — | ( | ||||||||
Other assets | — |
| ( | — | — | ( | — | ( | ||||||||
Current assets | ||||||||||||||||
Inventories | ( | | — | — | — | — | — | |||||||||
Trade receivables | |
| ( | — | — | ( | — | ( | ||||||||
Prepaid expenses and other assets | ( |
| | — | — | | | — | ||||||||
Cash and cash equivalents | ( |
| ( | — | — | ( | — | ( | ||||||||
Non - current liabilities | ||||||||||||||||
Lease liabilities (Non-current) | |
| | — | — | | | — | ||||||||
Other liabilities (Non-current) | ( |
| | — | — | | | ( | ||||||||
Current liabilities | ||||||||||||||||
Lease liabilities (Current) | |
| | — | — | | | — | ||||||||
Trade and other payables | ( | ( | — | — | ( | — | ( | |||||||||
Other liabilities (Current) | |
| | — | — | | | ( | ||||||||
Tax losses carried forward | | ( | — | | | | — | |||||||||
Share-based payments | | | ( | — | | | — | |||||||||
Netting | — | — | — | — | — | ( | | |||||||||
Deferred Taxes Total | |
| | ( | ( | | | ( | ||||||||
Deferred Tax Assets for CureVac Corporate Services, which incurred a tax loss in the current or prior year, are recognized in the amount of EUR
The following unused tax losses for which
Tax loss carryforwards |
| 2021 |
| 2022 |
| 2023 |
EUR k | EUR k | EUR k | ||||
Unused tax losses for corporate income tax |
| |
| |
| |
Unused tax losses for trade tax | |
| |
| | |
Unused interest carryforward ("Zinsschranke") |
| | — | — |
CureVac has tax losses in Germany and Netherlands that are available indefinitely for offsetting against future taxable profits of the companies in which the losses arose. In Germany and Netherlands tax losses carryforwards are unlimited.
DTA’s for temporary differences in the amount of EUR
Pillar Two legislation has been enacted or substantively enacted in certain jurisdictions in which the Group operates. However, this legislation does not apply to the Group as its consolidated revenue is lower than EUR 750 million.
Current tax assets of EUR
14. Earnings per share
Earnings per share is calculated by dividing the consolidated net loss of CureVac by the weighted average number of shares outstanding in the fiscal period.
F-49
The weighted average number of shares outstanding in fiscal 2021, 2022 and 2023 was
Diluted earnings per share is calculated using CureVac’s weighted-average outstanding common shares including the dilutive effect of share-based payment awards as determined under the treasury stock method. In periods when CureVac has a net loss, share-based payment awards are excluded from the calculation of earnings per share as their inclusion would have an antidilutive effect. Share options and RSUs of 1.217.526 and
15. Disclosure of financial instruments and management of financial risks
General information
CureVac is exposed to certain financial risks with respect to its assets and liabilities and the transactions associated with its business model. These risks generally relate to credit risks, liquidity risks and market risks (including currency risk, interest rate risk and price risk).
The aim of risk management is to limit the potential negative impact on expected cash flows and take advantage of any opportunities that arise. As a result, the management of CureVac assesses at least once a year whether risks have changed and whether the measures in place to limit risk are still sufficient.
Finance Income / Finance Expenses
During the year ended December 31, 2023, finance income was EUR
During the year ended December 31, 2023, finance expenses were EUR
Credit risk
Credit risk is managed by CureVac’s finance department. Credit risk arises from cash and cash equivalents and other financial assets, including deposits with banks and financial institutions, as well as credit exposures to customers, including outstanding receivables and contract assets.
CureVac is exposed to bank default and concentration risk as its cash is concentrated at few financial institutions. Management distributed the cash to decrease concentration risk as of December 31, 2023, deciding to allocate
CureVac is also exposed to a credit risk for its receivables and contract assets. The risk of default is considered to be low because the structure of customers consists of reputable collaborating parties and government grantors. Receivables management and financial accounting incorporates monitoring of payments received and any overdue receivables. The risk of counterparty nonperformance is not material.
The carrying amount of other financial assets recognized determines the maximum theoretical credit risk. As of the end of fiscal 2023, available funds are deposited at three reputable financial institutions.
As of December 31, 2023, the loss allowance for the “expected credit losses” totaled to EUR
F-50
Liquidity risk / Capital management
For the purpose of CureVac’s capital management, capital includes share capital and all other equity reserves attributable to the equity holders. The primary objective of CureVac’s capital management is to maximize the shareholder value through investment in the development activities of the Group.
Based on its business as an active research group, CureVac has historically relied almost exclusively on equity funding by its shareholders and lenders as a means of financing itself prior to successful development and sales of a marketable product.
The Group’s finance department reviews the total amount of cash of the Group on a weekly basis. As part of this review, the finance department considers the total cash and cash equivalents, the cash outflow, currency translation differences and refinancing activities. The Group monitors cash using a burn rate. The cash burn rate is defined as the average monthly net cash flow from operating and investing activities during a financial year.
In meeting its financing objectives, the Group negotiates and enters into research cooperation agreements. In general, the aim is to maximize the financial resources available for further research and development projects.
CureVac is not subject to externally imposed capital requirements. However, certain grant funds received may be required to be returned if qualifying costs are not incurred or are not incurred in accordance with the grant terms (see also Note 3.7. and Note 3.8.).
As described in Note 9, the Group has an at-the-market offering program through which, from time to time, may be able to raise additional capital through the issuance of common shares.
No changes were made in the objectives, policies or processes for managing cash during the years ended December 31, 2023 and 2022.
In order to safeguard liquidity, the Group invests funds not required immediately for operating purposes in call-deposit accounts with original maturities up to three months. Liquidity risks are therefore expected to be low. The Group does not enter into trading of financial instruments and monitors its risk of a shortage of funds using a liquidity planning tool.
Historically, CureVac has relied on financing from shareholders, grant income and collaborators in order to ensure sufficient liquidity. Lack of external financial support could pose a risk of going concern. The liquidity management of CureVac ensures the availability of cash and cash equivalents for operational activities and further investments through appropriate budget planning.
Ultimately, the responsibility for liquidity risk management lies with management, who has established an appropriate approach to managing short-, medium- and long-term financing and liquidity requirements. CureVac manages liquidity risks by holding appropriate reserves, as well as by monitoring forecasted and actual cash flows and reconciling the maturity profiles of financial assets and liabilities.
The table below summarizes the maturity profile of the Group’s liabilities based on contractual undiscounted payments:
<1 year | 1 to 5 years | > 5 years | Total | |||||
2023 |
| EUR k |
| EUR k |
| EUR k |
| EUR k |
Contractual commitments | ( |
| — |
| — |
| ( | |
Lease liabilities (Note 4.2) | ( |
| ( |
| ( |
| ( | |
Other liabilities and provisions (Note 12) | ( |
| — |
| — |
| ( | |
Trade and other payables (Note 11) | ( | — | — | ( | ||||
Total |
| ( |
| ( |
| ( |
| ( |
F-51
< 1 year | 1 to 5years | > 5 years | Total | |||||
2022 |
| EUR k |
| EUR k |
| EUR k |
| EUR k |
Contractual commitments |
| ( |
| ( |
| — |
| ( |
Lease liabilities (Note 4.2) |
| ( |
| ( |
| ( |
| ( |
Other liabilities and provisions (Note 12) |
| ( |
| ( |
| ( |
| ( |
Trade and other payables (Note 11) |
| ( |
| — |
| — |
| ( |
Total |
| ( |
| ( |
| ( |
| ( |
Foreign currency risk
Foreign currency risk is the risk that the fair value or future cash flows of an exposure will fluctuate because of changes in foreign exchange rates. CureVac’s exposure to the risk of changes in foreign exchange rates relates primarily to the Group’s operating activities (when revenue or expense is denominated in a foreign currency) and the amounts held as cash and cash equivalents.
For all CureVac entities except of CureVac Inc. and CureVac Swiss AG the functional currency is EUR. The functional currency of CureVac Inc. is the USD and of CureVac Swiss AG the CHF. CureVac SE’s exposure in foreign currency at the end of 2023 and 2022 is as follows:
2023 (in thousands) |
| ||||
Cash and cash equivalents |
| EUR | USD | ||
EUR | CHF | ||||
Trade and other receivables |
| | EUR | | USD |
Total monetary assets in foreign currency |
| | EUR | ||
Trade and other payables |
| | EUR | | USD |
| | EUR | | GBP | |
| | EUR | | CHF | |
Total monetary liabilities in foreign currency |
| | EUR |
| |
2022 (in thousands) | |||||
Cash and cash equivalents |
| | EUR | | USD |
Trade and other receivables |
| — | EUR | — | USD |
Total monetary assets in foreign currency |
| | EUR | | USD |
Trade and other payables |
| | EUR | | USD |
| | EUR | | GBP | |
| EUR | | CHF | ||
Total monetary liabilities in foreign currency |
| | EUR |
| |
As shown in the tables above, CureVac N.V. is exposed to a currency risk only in relation to the USD. Therefore, a foreign currency sensitivity analysis is only presented in respect to the net exposure in USD at fiscal year ends. CureVac’s net exposure in USD is the difference between monetary assets in USD and monetary liabilities in USD and developed as follows:
Net exposure in USD
2022 (1 EUR= |
| 2023 (1 EUR = |
|
EUR | EUR |
At December 31, 2023, if the EUR had weakened and conversely, had strengthened,
| Effect on pre-tax profit or loss |
| Effect on post-tax profit or loss | |||||
| Strengthening |
| Weakening |
| Strengthening |
| Weakening | |
31.12.2023 EUR against USD | |
| ( | |
| ( | ||
31.12.2022 EUR against USD | |
| ( | |
| ( | ||
CureVac did not have derivatives in 2023 and 2022.
F-52
Interest rate risk
Interest rate risk is the risk that the fair value or future cash flows of a financial instrument will fluctuate because of changes in market interest rates. CureVac’s exposure to the risk of changes in market interest rates relates primarily to CureVac’s cash and cash equivalents with floating interest rates.
If interest rates as of December 31, 2023 had been
Fair value measurement
All assets and liabilities for which fair value is measured or disclosed in the financial statements are categorized with the fair value hierarchy, described as follows, based on the lowest level input that is significant to the fair value measurement as a whole:
| ● | Level 1 — Inputs use quoted prices in active markets for identical assets or liabilities |
| ● | Level 2 — Inputs are inputs, other than quoted prices included in Level 1, which are directly or indirectly observable |
| ● | Level 3 — Inputs for the asset or liability that are not based on observable market data (unobservable inputs) |
All financial instruments are measured at amortized cost at December 31, 2023 and December 31, 2022.
The group measures assets held for sale at fair value less cost of disposals. For all short term liabilities and receivables the book value approximates the fair value.
16. Notes to the consolidated statements of cash flows
Changes in liabilities arising from financing activities
Foreign | ||||||||||||||||||
| January 1, |
|
|
|
| Other |
| Accrued |
| Paid |
| Exchange |
| December 31, | ||||
in thousands of EUR | 2023 |
| Cash flows | Reclassification | Disposals | Changes | interest | Interest | Movements | 2023 | ||||||||
Payment of lease liabilities (Note 4.2) | | ( | — | ( | | — | | ( | | |||||||||
Total liabilities from financing activities |
| |
| ( | — | ( | | — | | ( |
| |
Foreign | ||||||||||||||||||
January 1, | Other | Accrued | Paid | Exchange | December 31, | |||||||||||||
in thousands of EUR |
| 2022 |
| Cash flows |
| Reclassification |
| Disposals |
| Changes |
| interest |
| Interest |
| Movements |
| 2022 |
Payment of lease liabilities (Note 4. 2) |
| | ( | — | — | | — | | | | ||||||||
Total liabilities from financing activities |
| |
| ( | — | — | | — | | |
| |
The cash flow includes an interest component which is presented separately.
17. Commitments and contingencies
No material contingent liabilities resulting from claims and legal proceedings exist as of December 31, 2023. Refer to Note 12 for provisions recognized for contract terminations. For contractual commitments, refer to Note 15.
F-53
18. Remuneration of the Company’s key management personnel
Total remuneration of key management personnel
Remuneration of the Company’s key management personnel was as follows :
| 2021 |
| 2022 |
| 2023 | |||||||
Management |
| Supervisory | Management |
| Supervisory | Management |
| Supervisory | ||||
Remuneration of key management | Board | Board | Board | Board | Board | Board | ||||||
EUR k | EUR k | EUR k | EUR k | EUR k | EUR k | |||||||
Short-term benefits |
| |
| | ||||||||
Termination benefits | — | — | — | — | | — | ||||||
Share-based payments |
|
|
| |
| | ||||||
Total |
|
|
| |
| | ||||||
Out of the total remuneration, for Management Board EUR
In 2023,
19. Related party disclosures
Parent and ultimate controlling party
As disclosed in Note 1, during fiscal 2023, dievini Hopp BioTech holding GmbH & Co. KG (dievini), which is an investment company dedicated to the support of companies in health and life sciences, was the largest shareholder of CureVac. Together with its related parties, dievini has held shares and voting rights in CureVac between approximately 37-43 % during that period. dievini is thus the de facto parent of the Group. Dietmar Hopp, Daniel Hopp and Oliver Hopp are the ultimate controlling persons (of the main shareholders) of dievini, and, therefore, control the voting and investment decisions of dievini.
Transfer of shares from the former major shareholders
As discussed in Note 10, due to certain virtual share exercises under the Prior VSOP during the year 2023:
Dietmar Hopp
During 2019, Dietmar Hopp, principal of dievini Hopp BioTech holding GmbH & Co. KG (dievini), the majority shareholder of the Group, granted
Immatics Biotechnologies GmbH
In August 2023, a purchase agreement between CureVac Manufacturing GmbH and Immatics Biotechnologies GmbH was entered into. CureVac purchased technical equipment of containers which CureVac will use as a facility in the future. dievini held
F-54
Key management personnel compensation
Referring to key management personnel compensation we refer to Note 18.
Key management personnel transactions
Franz-Werner Haas
In Q1 2023, a consulting agreement between CureVac SE and Franz-Werner Haas was executed. In 2023 CureVac paid EUR
Alexander Zehnder
In Q1 2023, a first addendum to the future service agreement was entered into to ensure a smooth transition from CEO Franz-Werner Haas to the new CEO Alexander Zehnder. Total compensation amounted to EUR
Antony Blanc
In 2020, a consulting agreement between CureVac AG and Clarentis SRL was made. Clarentis SRL is a wholly owned consulting company of Antony Blanc, PhD, the CBO of CureVac. After the transition of Antony Blanc to the Management Board in February 2021, the contract was no longer active, and no new orders were placed. In Q3 2021, a milestone payment, which related to the submission of the EMA dossier for CVnCoV and which amounted to EUR
Dr. Ingmar Hörr
Due to the exercise of the Prior VSOP award in December 2022, CureVac had a receivable position of EUR
Florian von der Mülbe
Due to the exercise of the Prior VSOP award in December 2022, CureVac had a receivable position of EUR
Consulting agreements between CureVac Printer GmbH and Florian von der Mülbe related to the years 2022 to 2024 have been executed. In 2023 CureVac paid EUR
Mariola Fotin-Mleczek
In 2022, a consulting agreement between CureVac and Mariola Fotin-Mleczek was made. In 2023 a total amount of EUR
Due to the exercise of the Prior VSOP award in December 2022, CureVac had a receivable position of EUR
F-55
Barker BioMedical GmbH
In Q1 2023, a consulting agreement between CureVac SE and Barker BioMedical GmbH was executed. Barker BioMedical GmBH is a wholly owned consulting company of Debra Barker, Supervisory Board member of CureVac. In 2023 CureVac paid EUR
Craig Tooman
In Q1 2023, a consulting agreement between CureVac SE and Craig Tooman was executed. In 2023, CureVac paid EUR
Ralf Clemens
In Q3 2023, a consulting agreement between CureVac and GRID EUROPE was executed. GRID EUROPE is a wholly owned consulting company of Ralf Clemens, who was a member of the supervisory board up to September 30, 2023. CureVac incurred EUR
Indemnification Agreements
The Company’s articles of association require it to indemnify its current and former managing directors and supervisory directors in relation to acts or omissions in the performance of their duties to the fullest extent permitted by law, subject to certain exceptions. We entered into indemnification agreements with all our managing directors and supervisory directors.
Other related party transactions
Rittershaus Rechtsanwaelte
Since December 15, 2005, a consultant agreement is in place for an indefinite term with Rittershaus. The agreement can be terminated without notice by CureVac and with notice of
BePharBel Manufacturing S.A.
In December 2020, CureVac Manufacturing GmbH (formerly CureVac Real Estate GmbH) and BePharBel Manufacturing S.A., entered into a commercial supply agreement to develop and manufacture the diluent that was expected to be used to dilute the Group’s first concentrated COVID-19 vaccine candidate, CVnCoV, to the amount specified by each dose level. Pursuant to the terms of the agreement, it was intended that BePharBel Manufacturing would manufacture and deliver to CureVac Manufacturing GmbH a low seven figure amount of commercial batches of diluent per year, in 2021 and 2022. Following the withdrawal of the CVnCoV in October 2021 due to COVID-19 virus drift, WHO COVID vaccine efficiency recommendation and market expectations, CureVac Manufacturing GmbH terminated the commercial and supply agreement with BePharBel and entered into negotiations on a structured and rapid wind-down of the ordered production. The Parties agreed on a settlement in May 2022 of all claims resulting from the commercial and supply agreement for an amount of EUR
F-56
20. Business Combinations
Business Combination and Goodwill – Frame Acquisition
Effective July 1, 2022 (‘closing date’), CureVac acquired all shares of Frame Pharmaceuticals B.V., Amsterdam, Netherlands (‘Frame Pharmaceuticals’ or ‘Frame’). Frame Pharmaceuticals focuses on the development of a proprietary platform enabling the identification of structural changes within the cancer genome and has strong competencies in antigen discovery as well as validation for personalized cancer vaccines. In July 2022, Frame Pharmaceuticals was renamed CureVac Netherlands B.V.
In the purchase price agreement (‘SPA’) dated June 8, 2022, total consideration of up to EUR
The total consideration is split into
| ● | Issuance and transfer of |
| ● | Issuance and transfer of further |
| ● | Payment of EUR |
Payment of the remaining
Consequently, the total consideration transferred for the business combination was determined to be EUR
| ● | Issuance and transfer of |
| ● | Payment of EUR |
| ● | Contingent consideration, classified as equity, with a fair value of EUR |
The contingent consideration of EUR
In addition,
F-57
The final fair values in accordance with IFRS 3 of the identifiable net assets as of the date of acquisition were as follows:
in EUR thousands |
| Fair value recognized on acquisition |
Non-current assets |
| |
Property, plant and equipment |
| |
Right-of-use assets |
| |
Intangible asset (Technology) |
| |
Current assets |
| |
Trade and other receivables |
| |
Cash and cash equivalents |
| |
Total assets |
| |
Non-current liabilities |
| |
Lease liabilities |
| |
Deferred tax liabilities (net of deferred tax assets) |
| |
Current liabilities |
| |
Lease liabilities |
| |
Accounts Payables |
| |
Other current liabilities |
| |
Total liabilities |
| |
Net assets acquired |
| |
The technology intangible asset consists of a bioinformatical platform for cancer antigen discovery and validation for off-the-shelf and personalized cancer vaccines. The fair value of the technology of EUR
Goodwill was recognized as a result of the acquisition as follows:
in EURk |
|
|
Consideration transferred |
| |
Net Assets acquired |
| ( |
Goodwill |
| |
The goodwill is mainly attributable to the synergies and an assembled workforce as well as the strategic benefits to the Group. The goodwill is not deductible for tax purposes.
Transactions costs in relation to the business combination amounting to EUR
The consolidated statements of profit or loss include the results of Frame Pharmaceuticals (meanwhile: CureVac Netherlands B.V.) since the acquisition date. From the date of acquisition through December 31, 2022 Frame Pharmaceuticals did not have any significant impact on the operating income or revenues of the Group. The same applies if the transaction had occurred at the beginning of the reporting period.
21. Subsequent events
In January 2024, positive interim data from the ongoing Phase 2 clinical program assessing monovalent and bivalent modified vaccine candidates against COVID-19 in comparison with a licensed bivalent mRNA-based comparator vaccine were published. The tested vaccine candidates are being developed in collaboration with GSK. Results from the interim analysis showed that both vaccine candidates using CureVac’s proprietary second-generation mRNA backbone produced meaningful immune responses and favorable tolerability profiles across all tested doses, including the lowest tested dose. All three tested dose levels were below those used in mRNA-based COVID-19 vaccines licensed in the U.S. and EU.
F-58
In March 2024, the beneficiaries of the Prior VSOP plan declared their intent to exercise their exercisable shares. The shares became exercisable on the third anniversary after IPO (August 14, 2023), because certain minimum trading volumes of the CureVac N.V. shares and liquidity levels had been reached.
In March 2024, the Company started a voluntary leaver program intended to align the workforce to the Company’s business scope and priorities. The business goal is to reduce 150 positions by year-end, including natural attrition. Unnecessary residual pandemic infrastructure is being adapted to significantly increase efficiency and performance while maintaining a strong focus on innovation and R&D activities. For this program we anticipate costs in a range between EUR
In April 2024, promising interim data from the ongoing Phase 2 part of the combined Phase 1/2 study of a seasonal influenza vaccine candidate were reported. The multivalent candidate was designed for broad antigen coverage, encoding antigens matched to all four WHO-recommended flu strains. Results from the planned interim analysis showed that the multivalent vaccine candidate boosted antibody titers against all encoded flu strains and across all age groups and tested dose levels, including the lowest tested dose. The vaccine candidate was shown to have an acceptable safety and tolerability profile, with the majority of solicited adverse events reported as either grade 1 (mild) or grade 2 (moderate) within seven days of dosing. Among younger and older adults, geometric mean titers generated by the vaccine candidate against influenza A strains numerically exceeded the geometric mean titers of the licensed comparator vaccines consistently across all tested dose levels. For influenza B strains geometric mean titers were lower than those elicited by the licensed comparator vaccines across both age groups and tested dose levels. Targeted optimizations to further improve immune responses against influenza B strains will be assessed in an additional Phase 2 study.
In April 2024, the Company entered into a co-development and licensing agreement with The University of Texas MD Anderson Cancer Center for the development of novel mRNA-based cancer vaccines. The collaboration creates strong synergies between CureVac’s unique end-to-end capabilities for cancer antigen discovery, mRNA design, and manufacturing and MD Anderson’s expertise in cancer antigen discovery and validation, translational drug development, and clinical research. The collaboration will focus on the development of differentiated cancer vaccine candidates in selected hematological and solid tumor indications with high unmet medical need. Both parties will contribute to the identification of differentiated cancer antigens based on whole genome sequencing, combined with long- and short-read RNA sequencing and cutting-edge bioinformatics followed by joint preclinical validation of the highest-quality cancer antigens and selection of the most promising clinical-lead candidates for conducting initial Phase 1/2 studies in appropriate clinical indications.
In 2022, Curevac and GSK entered into a Pandemic Preparedness Agreement with the Federal Republic of Germany. Timelines related to the Pandemic Preparedness Agreement were ambitious with the aim of providing acute pandemic preparedness in Germany in the context of emerging COVID-19 variants. In 2023, CureVac identified a risk that necessary regulatory approvals needed to meet these timelines may not be achieved within the contractually agreed timeframe. After consultation with ZEPAI, CureVac applied for a timeline extension as foreseen under the agreement to avoid procurement of further substantial amounts of raw material at risk. As of December 31, 2023, it was considered unlikely that the extension would be granted. Additionally, a rapidly changing epidemiological environment did no longer prompt an acute pandemic threat. Accordingly, inventories which had been stockpiled as required by the PPA, were written down to their net realizable value. Ultimately, the extension was not granted, whereupon CureVac and GSK jointly decided to terminate the agreement. Despite the termination, CureVac’s GMP IV manufacturing plant for the large-scale production of mRNA-based vaccines will be completed. Contingent upon regulatory approval, the facility is expected to be certified mid-2024.
F-59
In April 2024, the Company announced the start of the Phase 1 part of a combined Phase 1/2 study of an investigational influenza A (H5N1) pre-pandemic vaccine candidate developed in collaboration with GSK. The H5N1 avian influenza virus is considered a potential future pandemic threat, able to cross species from its original bird host to humans. The monovalent vaccine candidate is based on CureVac’s proprietary second-generation mRNA backbone and encodes an influenza A H5-antigen. Avian influenza represents latest program progressing to clinical trials under broad infectious disease collaboration agreement with GSK.
In April 2024, the Company had the first ruling in one of its CMO arbitration. In August 2022, Rentschler Biopharma SE initiated arbitration proceedings according to the procedural rules of the German Arbitration Institute against the Company. Rentschler’s claims are based on the assumption that certain agreements between Rentschler and CureVac Manufacturing GmbH (then: CureVac Real Estate GmbH) have been terminated due to the withdrawal of the EMA dossier for CVnCoV or alternatively that CureVac would have been obliged to terminate these agreements. CureVac’s position always was that the agreements had ended upon expiration of their term. We did not believe that there was a legal basis for Rentschler’s claims and therefore defended ourselves against these claims in written submissions and the oral hearing in October 2023. In its final award rendered, the arbitration tribunal followed CureVac’s arguments and dismissed Rentschler’s claims in their entirety. The totality of Rentschler Biopharma SE related provision was reversed as of December 31, 2023.
F-60