UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
|
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
or
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ________________ to __________________.
Commission file number:
(Exact name of registrant as specified in its charter)
|
(State or other jurisdiction of incorporation or organization) |
Not Applicable (I.R.S. Employer Identification No.) |
|
(Address of principal executive offices) |
(Zip Code) |
Registrant’s telephone number, including area code: (
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class |
Trading Symbol(s) |
Name of each exchange on which registered |
||
|
|
|
|
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. YES ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. YES ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer ☐ |
Accelerated filer ☐ |
|
Smaller reporting company |
|
Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). YES
The aggregate market value of the registrant’s voting common shares held by non-affiliates, computed by reference to the closing sales price at which the voting common shares were last sold as of June 30, 2023 (the last business day of the registrant’s most recently completed second fiscal quarter), as reported by The Nasdaq Capital Market on that date, was $
As of March 15, 2024, there were
DOCUMENTS INCORPORATED BY REFERENCE
Part III of this Annual Report on Form 10-K incorporates by reference information (to the extent specific sections are referred to herein) from the registrant’s Proxy Statement for its 2023 Annual General Meeting of Shareholders to be held May 22, 2024.
[page intentionally left blank]
DIAMEDICA THERAPEUTICS INC.
ANNUAL REPORT ON FORM 10-K
FISCAL YEAR ENDED DECEMBER 31, 2023
TABLE OF CONTENTS
| Page | ||
| CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS | 1 | |
| INDUSTRY AND MARKET DATA | 2 | |
| PART I | 3 | |
| Item 1. | Business | 3 |
| Item 1A. | Risk Factors | 29 |
| Item 1B. | Unresolved Staff Comments | 63 |
| Item 1C. | Cybersecurity | 63 |
| Item 2. | Properties | 64 |
| Item 3. | Legal Proceedings | 65 |
| Item 4. | Mine Safety Disclosures | 65 |
| PART II | 66 | |
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | 66 |
| Item 6. | [Reserved] | 76 |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | 77 |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | 85 |
| Item 8. | Financial Statements and Supplementary Data | 86 |
| Item 9. | Changes In and Disagreements With Accountants on Accounting and Financial Disclosure | 108 |
| Item 9A. | Controls and Procedures | 108 |
| Item 9B. | Other Information | 109 |
| Item 9C. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections | 109 |
| PART III | 110 | |
| Item 10. | Directors, Executive Officers and Corporate Governance | 110 |
| Item 11. | Executive Compensation | 110 |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | 110 |
| Item 13. | Certain Relationships and Related Transactions, and Director Independenc | 112 |
| Item 14. | Principal Accountant Fees and Services | 112 |
| PART IV | 113 | |
| Item 15. | Exhibits and Financial Statement Schedules | 113 |
| Item 16. | Form 10-K Summary | 117 |
| SIGNATURES | 118 | |
This annual report on Form 10-K contains certain forward-looking statements that are within the meaning of Section 27A of the United States Securities Act of 1933, as amended, and Section 21E of the United States Securities Exchange Act of 1934, as amended, and are subject to the safe harbor created by those sections. For more information, see “Cautionary Note Regarding Forward-Looking Statements.”
As used in this report, references to “DiaMedica,” the “Company,” “we,” “our” or “us,” unless the context otherwise requires, refer to DiaMedica Therapeutics Inc. and its subsidiaries, all of which are consolidated in DiaMedica’s consolidated financial statements. References in this report to “common shares” mean our voting common shares, no par value per share.
We own various unregistered trademarks and service marks, including our corporate logo. Solely for convenience, the trademarks and trade names in this report are referred to without the ® and ™ symbols, but such references should not be construed as any indicator that the owner of such trademarks and trade names will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend the use or display of other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Statements in this annual report on Form 10-K that are not descriptions of historical facts are forward-looking statements within the meaning of the United States Private Securities Litigation Reform Act of 1995 that are based on management’s current expectations and are subject to risks and uncertainties that could negatively affect our business, operating results, financial condition, prospects and share price. We have attempted to identify forward-looking statements by terminology including “anticipates,” “believes,” “can,” “continue,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “should,” “will,” “would,” the negative of these terms or other comparable terminology, and the use of future dates.
The forward-looking statements in this report are subject to risks and uncertainties and include, among other things:
|
● |
our plans to develop, obtain regulatory approval for and commercialize our DM199 product candidate for the treatment of acute ischemic stroke (AIS) and cardio-renal disease (CRD); |
|
● |
our ability to conduct successful clinical testing of our DM199 product candidate for AIS or CRD and meet certain anticipated or target dates with respect to our clinical studies, including in particular our Phase 2/3 ReMEDy2 clinical trial of DM199 for the treatment of AIS, or ReMEDy2 trial, and anticipated site activations, enrollment and interim analysis timing, especially in the light of the effects of COVID-19, particularly on hospital and medical facility staffing shortages, concerns managing logistics and protocol compliance for participants discharged from the hospital to an intermediate care facility, and competition for research staff and trial subjects due to other pending stroke and stroke related trials; |
|
● |
uncertainties relating to regulatory applications and related filing and approval timelines and the possibility of additional future adverse events associated with or unfavorable results from our ReMEDy2 trial; |
|
● |
the adaptive design of our ReMEDy2 trial, which is intended to enroll approximately 350 participants at up to 100 sites globally, and the possibility that these numbers and other aspects of the study could increase depending upon certain factors, including additional input from the United States Food and Drug Administration (FDA) and results of the interim analysis as determined by the independent data safety monitoring board; |
|
● |
our expectations regarding the perceived benefits of our DM199 product candidate over existing treatment options for AIS and CRD; |
|
● |
the potential size of the markets for our DM199 product candidate for AIS and CRD and our ability to serve those markets and the rate and degree of market acceptance of, and our ability to obtain coverage and adequate reimbursement for, our DM199 product candidate for AIS and CRD both in the United States and internationally; |
|
● |
our ability to partner with and generate revenue from biopharmaceutical or pharmaceutical partners to develop, obtain regulatory approval for and commercialize our DM199 product candidate for AIS and CRD; |
|
● |
the success, cost and timing of our ReMEDy2 trial, as well as our reliance on third parties in connection with our ReMEDy2 trial and any other clinical trials we conduct; |
|
● |
our commercialization, marketing and manufacturing capabilities and strategy; |
|
● |
expectations regarding federal, state and foreign regulatory requirements and developments, such as potential FDA regulation of our DM199 product candidate for AIS and CRD; |
|
● |
our estimates regarding expenses, market opportunity for our product candidates, future revenue, capital requirements, how long our current cash resources will last and need for additional financing; |
|
● |
our expectations regarding our ability to obtain and maintain intellectual property protection for our DM199 product candidate; |
|
● |
expectations regarding competition and our ability to obtain data exclusivity for our DM199 product candidate for AIS and CRD; and |
|
● |
our anticipated use of the net proceeds from our private placements and our ability to obtain additional funding for our operations, including funding necessary to complete planned clinical trials and obtain regulatory approvals for our DM199 product candidate for AIS and CRD. |
These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described under “Part I. Item 1A. Risk Factors” in this report. Moreover, we operate in a very competitive and rapidly-changing environment. New risks emerge from time to time. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this report may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements. Forward-looking statements should not be relied upon as predictions of future events. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee that the future results, levels of activity, performance or events and circumstances reflected in the forward-looking statements will be achieved or occur. Except as required by law, including the securities laws of the United States, we do not intend to update any forward-looking statements to conform these statements to actual results or to changes in our expectations.
INDUSTRY AND MARKET DATA
In addition to the industry, market and competitive position data referenced in this report from our own internal estimates and research, some market data and other statistical information included in this report are based in part upon information obtained from third-party industry publications, research, surveys and studies, none of which we commissioned. Third-party industry publications, research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information.
We are responsible for all of the disclosure in this report, and while we believe that each of the publications, research, surveys and studies included in this report are prepared by reputable sources, we have not independently verified market and industry data from third-party sources. In addition, while we believe our internal company research and estimates are reliable, such research and estimates have not been verified by independent sources. Assumptions and estimates of our and our industry’s future performance are necessarily subject to a high degree of uncertainty and risk due to a variety of factors, including those described in “Part I. Item 1A. Risk Factors.” These and other factors could cause our future performance to differ materially from our assumptions and estimates. See “Cautionary Note Regarding Forward-Looking Statements.”
PART I
|
Item 1. |
Business |
Overview
We are a clinical stage biopharmaceutical company committed to improving the lives of people suffering from serious diseases. DiaMedica’s lead candidate DM199 (rinvecalinase alfa) is the first pharmaceutically active recombinant (synthetic) form of the human tissue kallikrein-1 (KLK1) protein to be clinically studied in patients. KLK1 is an established therapeutic modality in Asia, with human urinary KLK1, for the treatment of acute ischemic stroke and cardio renal disease, including hypertension. We have also produced a potential novel treatment for severe inflammatory diseases, DM300, which is currently in the early preclinical stage of development. Our long-term goal is to use our patented and in-licensed technologies to establish our Company as a leader in the development and commercialization of therapeutic treatments from novel recombinant proteins. Our current focus is on the treatment of acute ischemic stroke (AIS) and currently, to a lesser extent, cardio renal disease (CRD). We plan to advance DM199, our lead drug candidate, through required clinical trials to create shareholder value by establishing its clinical and commercial potential as a therapy for AIS and CRD. In September 2021, the FDA granted Fast Track designation to DM199 for the treatment of AIS where tPA and/or mechanical thrombectomy are not indicated or medically appropriate.
AIS and CRD patients suffer from impaired blood flow in the brain, kidneys, and throughout the body. Many of these patients also exhibit lower than normal levels of endogenous (produced by the body) KLK1 protein, which is produced primarily in the kidneys, pancreas and salivary glands. We believe treatment with DM199 could augment endogenous KLK1 to enhance the function of the kallikrein-kinin system (KKS) to preferentially relax smooth muscle cells in ischemic arteries, thereby vasodilating these arteries and increasing blood flow and oxygen.
We are currently conducting our ReMEDy2 clinical trial of DM199 for the treatment of AIS. Our ReMEDy2 clinical trial is a Phase 2/3, adaptive design, randomized, double-blind, placebo-controlled trial intended to enroll approximately 350 patients at up to 100 sites globally. Patients enrolled in the trial will be treated with either DM199 or placebo within 24 hours of the onset of AIS symptoms. The trial excludes patients treated with tissue plasminogen activator (tPA), a thrombolytic agent intended to dissolve blood clots, and those with large vessel occlusions. The study population is representative of the approximately 80% of AIS patients who do not have treatment options today, primarily due to the limitations on treatment with tPA and/or mechanical thrombectomy. The primary endpoint of the ReMEDy2 trial is physical recovery from stroke as measured by the well-established modified Rankin Scale (mRS) at day 90, specifically recovering to an mRS score of 0-1 (mRS range of 0-6). We believe that our ReMEDy2 trial has the potential to serve as a pivotal registration study of DM199 in this patient population.
We voluntarily paused participant enrollment in the ReMEDy2 trial in May 2022 to investigate three unexpected instances of clinically significant hypotension (low blood pressure) occurring shortly after initiation of the intravenous (IV) dose of DM199. The acutely low blood pressure levels in the three participants recovered back to their baseline blood pressure within minutes after the IV infusion was stopped, and the participants suffered no injuries. On July 6, 2022, we announced that the FDA placed a clinical hold on the investigational new drug application (IND) for our ReMEDy2 trial, and the clinical hold was subsequently lifted in June 2023. In our request for lifting of the clinical hold, we submitted to the FDA in-vitro data supporting that the cause of the hypotensive events was likely related to switching to a new type of IV bag for use in the ReMEDy2 trial, as well as results of an additional in-use, in vitro stability study of all of the materials and equipment used in the IV administration of DM199, which included testing the combination of the IV bag, IV tubing and mechanical infusion pump, to further rule out any other cause of the hypotension events. We also modified the protocol to mitigate the risk of future hypotensive events, including a reduction in the DM199 dose level for the initial IV dose to effectively match the well tolerated IV dose administered in the ReMEDy1 trial.
Concurrently with performing the requested in-use study, we also conducted a Phase 1C open label, single ascending dose (SAD) study of DM199 administered with polyvinyl chloride (PVC) IV bags used in the ReMEDy2 trial. The purpose of the Phase 1C open label SAD study was to confirm, with human data, the DM199 blood concentration levels achieved with the IV dose and further evaluate safety and tolerability. We also included a cohort of hypertensive patients being treated with angiotensin-converting-enzyme inhibitors (ACEi) prior to enrolling. All ACEi patients received the full IV dose at the 0.5 µg/kg level with no instances of hypotension. We believe that these results provide further assurance to potential investigators that ACEi patients may be safely included in the ReMEDy2 trial.
Following in-depth discussions of the ReMEDy2 Phase 2/3 protocol design with global stroke experts, the scientific advisory board and current investigators, we made several important amendments to the protocol subsequent to the June 2023 lifting of the clinical hold. These changes were submitted to the FDA in early October 2023 and we are proceeding with use of the amended protocol as the FDA did not issue any comments during the 30-day review period which ended on November 3, 2023.
We believe DM199 has the potential to treat a variety of diseases where restoring healthy function requires sufficient activity of KLK1 and its system, KKS. Today, forms of KLK1 derived from human urine and the pancreas of pigs are approved and sold in Japan, China and South Korea to treat AIS, hypertension and other related vascular diseases. We believe millions of patients have been treated with these KLK1 therapies, including over 600,000 AIS patients now being treated annually with human urinary-derived KLK1 in China. Over 200 clinical studies in China have found urinary-derived KLK1 effective for increasing blood flow, decreasing ischemia in the penumbra and reducing infarct size. Importantly, human urinary-derived KLK1 has not been shown to increase the risk of severe intracranial hemorrhage. However, there are numerous regulatory, commercial and clinical drawbacks associated with KLK1 derived from these sources which can be overcome by developing a recombinant version of KLK1 such as DM199. We believe higher regulatory standards and potential antibody reactions are the primary reasons why KLK1 derived from these sources are not currently available and used in the United States or Europe. We are not aware of any recombinant version of KLK1 with regulatory approval for human use in any country, nor are we aware of any recombinant version in development other than our drug candidate, DM199.
DM199 Background
Kallikrein-Kinin System
KLK1 is a serine protease, or protein, produced primarily in the kidneys, pancreas and salivary glands. KLK1 plays a critical role in the regulation of local blood flow and vasodilation (the widening of blood vessels, which decreases vascular resistance) in the body, as well as an important role in reducing inflammation and oxidative stress (an imbalance between potentially damaging reactive oxygen species, or free radicals, and antioxidants in the body).
KLK1 is involved in multiple biochemical processes. The most well-characterized activity of KLK1 is the enzymatic cleavage of low molecular weight kininogen (LMWK) to produce Lys-bradykinin (BK)-like peptides, collectively known as kinins, which activate BK receptors (primarily BK2R since the BK1R is typically only activated in pathological situations). As illustrated below, activation of BK receptors by kinins sets in motion metabolic pathways which locally produce nitric oxide, prostaglandins (primarily prostacyclin in endothelial cells),which work through the cyclic guanosine monophosphate (cGMP) and cyclic nucleotides cyclic adenosine monophosphate (cAMP) pathways, to preferentially relax smooth muscle cells and improve blood flow (through vasodilation), potentially protecting tissues and end-organs from ischemic damage. Scientific literature, including publications in Circulation Research, Immunopharmacology and Kidney International, suggests that lower endogenous KLK1 levels in patients are associated with diseases related to vascular disorders, such as stroke, renal diseases and hypertension. DM199, as a protein augmentation therapy, may increase KLK1 levels to properly activate the KKS driving the local production of nitric oxide, prostaglandins and other anti-inflammatory mediators, to promote endothelial health and protect the brain and kidney from damage. By providing additional supply of the KLK1 protein, DM199 treatment could potentially improve blood flow to and reduce inflammation in damaged end-organs, such as the brain and the kidneys, supporting their structural integrity and normal functioning.
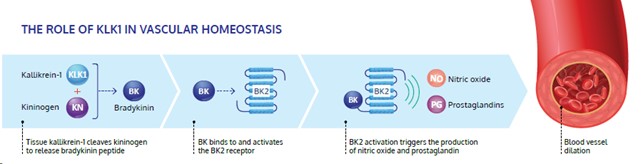
We have conducted numerous internal and third-party analyses to demonstrate that DM199 is structurally and functionally equivalent to KLK1 derived from human urine. Specifically, the amino acid structure of DM199 is nearly identical to the human urine form, and the enzymatic and pharmacokinetic profiles are substantially similar to both human urine and porcine derived KLK1. The physiological effects of DM199 on blood pressure, from our completed studies, is similar to that of human urine and porcine-derived forms of KLK1. We believe that the results of this work suggest that the therapeutic action of DM199 will be the same or potentially better than that of the human urinary and porcine forms of KLK1 marketed in Asia.
We believe DM199 may provide a new treatment with significant benefits over the current standards of care by offering a therapeutic treatment option to a greater number of patients with the potential for fewer side effects.
Summary of Clinical Results
To date, clinical trials have been and/or are being conducted in the United States, Europe and Australia. We believe the clinical data generated to date by DM199 supports the continued development of DM199 as a treatment for AIS and CRD.
|
● |
Our Phase 2 ReMEDy1 trial of DM199 in the treatment of AIS (n=91) met our primary safety and tolerability end points and demonstrated a statistically significant reduction in the number of participants with recurrent ischemic stroke (reported as stroke in evolution or stroke progression by the investigators) in the active treatment group: 0 (0%) participants treated with DM199 vs. 6 (13%) on placebo (p=0.012), with 4 of the 6 resulting in participant death. In a subgroup analysis of participants not receiving mechanical thrombectomy prior to enrollment (n=46), patients treated with DM199 demonstrated a 22% absolute improvement in excellent outcomes (recovering to a National Institutes of Health Stroke Scale (NIHSS) score of 0-1). In participants treated with DM199 (n=25) vs. supportive care and/or tPA (n=21), the results showed that 36% of participants receiving DM199 progressed to a full or nearly full recovery at 90 days (NIHSS: 0-1), compared to 14% of participants in the placebo group. This represents a 22% absolute increase in the proportion of participants achieving a full or nearly full recovery. Additionally, subject deaths decreased from 24% in the placebo group to 12% in the DM199 treatment group, a 50% relative reduction. This subgroup represents the participants most closely aligned with the target treatment population for DM199 in our ReMEDy2 trial. |
|
● |
We conducted our Phase 2 REDUX trial of DM199 in participants with chronic kidney disease (n=84). Most notably, the hypertensive African American cohort demonstrated an over 50% mean reduction in albuminuria in participants with moderate to severe baseline albuminuria and a statistically significant reductions in systolic and diastolic blood pressure levels at the 2µg/kg dose level after three months of treatment. |
In all completed studies, DM199 was shown to be generally safe and well tolerated. The primary adverse events noted in our studies with healthy volunteers included headache, erythema (redness), dizziness, injection site reaction and flushing. The most common adverse events in people with diabetes with or without chronic kidney disease included orthostatic hypotension, local injection site irritation/redness, and diarrhea. The most common adverse events in people with acute ischemic stroke include constipation, oral candidiasis (yeast/fungal infection of mouth) and nausea.
Supporting Data for Use of DM199 (KLK1)
KLK1 derived from human urine was approved in China in 2005. KLK1 derived from the pancreas of pigs has been approved in Japan for several decades. There is one company selling human urine derived KLK1 in China, and we believe human urine derived KLK1 is currently being used to treat over 600,000 AIS patients per year. We believe that approximately 20 companies are marketing porcine KLK1 in Japan, China and South Korea for hypertension, certain chronic kidney and other vascular diseases. We have identified several hundred papers supporting the clinical use of urinary and porcine derived KLK1 from China, Japan and South Korea.
Studies have shown that lower KLK1 levels are also a predictor of stroke recurrence. The red line in the graph below represents patients in the lowest KLK1 quartile who were at the highest risk for recurrence of stroke. (2,478 stroke patients and event free survival over 5 years).
Low KLK1 Levels Are Associated With Stroke Recurrence
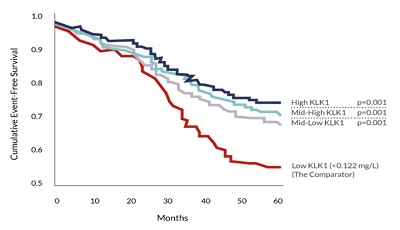
Source: Annals of Neurology (2011) 70:265-73
Our Strategy
Our mission is to improve the lives of people suffering from serious diseases. Our near-term goal is to principally focus on executing our ReMEDy2 Phase 2/3 trial of DM199 in AIS and to finalize plans for the next steps for our CRD program. Key elements of our strategy include:
|
● |
DM199 for AIS – activate additional clinical sites for and enroll participants in our ReMEDy2 Phase 2/3 trial and expand the trial globally to potentially increase enrollment rates; |
|
● |
DM199 for CRD – announce next steps for our CRD program during 2024; |
|
● |
Continue manufacturing process development to support anticipated applications for commercial approval of DM199; and |
|
● |
Identify a strategic partner(s) to assist with future clinical development and commercialization of DM199. |
AIS Background and Disease Pathology
Acute Ischemic Stroke Background
Stroke is characterized by the rapidly developing loss of brain function due to a blockage of blood flow in the brain. As a result, the affected tissues of the brain become inactive and may eventually die. Strokes can be classified into two major categories: AIS and hemorrhagic stroke. AIS is characterized by interruption of the blood supply by a blood clot (ischemia), while a hemorrhagic stroke results from rupture, or bleeding, of a blood vessel in the brain. Risk factors for stroke include, among other things, advanced age, hypertension (high blood pressure), previous stroke or transient ischemic attack (TIA), diabetes, high cholesterol, cigarette smoking, atrial fibrillation, physical inactivity and obesity.
More specifically, with respect to an ischemic stroke, at the site of a blood flow blockage in the brain, there exist two major ischemic zones – the core ischemic zone with nearly complete loss of blood flow (blood flow reduction of 75% to 90%, or more), and the surrounding ischemic penumbra, a rim of mild to moderately ischemic tissue surrounding the core ischemic zone. Within minutes, the significant lack of blood flow in the core ischemic zone deprives these cells of glucose and oxygen which rapidly depletes energy stores and triggers the loss of ion gradients, ultimately leading to neuronal cell death, or apoptosis. The ischemic penumbra zone, however, may remain viable for several hours via collateral arteries that branch from the main occluded artery in the core ischemic zone. Unfortunately, the penumbra is at great risk of delayed tissue damage due to inflammation which may also lead to neuronal cell death. As time goes on, a lack of blood flow in the core ischemic zone (infarct) may lead to fluid buildup (edema) and swelling which creates intracranial pressure. This pressure on the brain leads to tissue compression resulting in additional ischemia. Additional events in AIS include vascular damage to the blood vessel lining or endothelium, loss of structural integrity of brain tissue and blood vessels and inflammation. A stroke can lead to permanent damage with memory loss, speech problems, reading and comprehension difficulties, physical disabilities and emotional/behavioral problems. The long-term costs of stroke are substantial, with many patients requiring extended hospitalization, extended physical therapy or rehabilitation and/or long-term institutional or family care. However, provided the extended window of viability in the penumbra, next generation stroke therapies are being developed to protect valuable brain tissue during the hours to a week after a stroke.
Unmet Medical Need in AIS
According to the World Health Organization, each year 12.2 million people worldwide suffer a stroke, of which 7.6 million are acute ischemic strokes. According to the U.S. Centers for Disease Control and Prevention (CDC) approximately 800,000 people in the U.S. suffer a stroke each year, of which 87% are acute ischemic strokes. We believe that stroke represents an area of significant unmet medical need and a KLK1 therapy (such as DM199) could provide a significant patient benefit, in particular given its proposed treatment window of up to 24 hours after the first sign of symptoms. Currently, the only FDA-approved pharmacological intervention for AIS is tPA, which is approved to be given within 3 hours of symptom onset; however, we understand that based upon supplemental clinical research and common practice, it is typically administered up to 4.5 hours from symptom onset. Treating patients with tPA during this time window can be challenging because it is difficult to determine precisely when symptoms began and a patient must undergo brain imaging before treatment to rule out a hemorrhagic stroke, a ruptured blood vessel causing bleeding within the brain. Mechanical thrombectomy, a procedure which attempts to remove the clot using catheter-based tools, is also available to certain patients. Despite the availability of these treatments, we believe they are relevant to approximately 20% of ischemic stroke patients due to the location of the clot, the elapsed time after the stroke occurred or other safety considerations. Thus, we believe DM199 may offer significant advantages over the current treatment options in that it fills a serious, unmet need for patients who cannot receive tPA or mechanical thrombectomy. Additionally, we believe DM199 may also offer a complementary follow-on treatment for patients who initially receive tPA or mechanical thrombectomy by enabling sustained blood flow improvements to the brain during the critical weeks and months after a stroke, reducing the risk of stroke recurrence.
Acute Ischemic Stroke Treatment Options
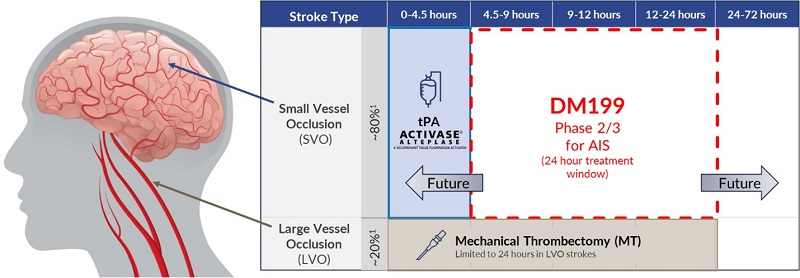
According to the CDC, stroke incidence in the United States and its related effects include:
|
● |
Every year in the United States, approximately 800,000 people experience a stroke (ischemic or hemorrhagic). Approximately 600,000 of these are first events and approximately 25%, or 200,000, are recurrent stroke events. |
|
● |
Approximately one of every 20 deaths in the United States is caused by stroke and is the fifth leading cause of death. On average, someone in the United States has a stroke every 40 seconds and someone dies from a stroke every 3.5 minutes. |
|
● |
Stroke is the leading cause of serious long-term disability and reduces mobility in more than half of stroke survivors aged 65 and over. |
|
● |
Risk of having a first stroke is nearly twice as high for African Americans as for Caucasians, and African Americans have the highest rate of death due to stroke. |
|
● |
Stroke-related costs in the United States came to nearly $53 billion between 2017 and 2018, including the cost of health care services, medications and missed days of work. |
DM199 – Our Novel Solution for the Treatment of AIS
In response to an ischemic stroke, bradykinin 2 receptors (BK2) are significantly upregulated (increased) in the arteries affected by the stroke, the ischemic penumbra. This phenomenon has been observed in animal stroke models, showing a 36-fold increase on the ipsilateral side and a 10-fold increase on the contralateral side (PLOS ONE (2018), 13(6), e0198553.https://doi.org/10.1371/journal.pone.0198553). In these oxygen depleted arteries, the increased BK2 receptors signal the need for BK to bind and restore blood flow to these at-risk arteries in the ischemic penumbra. The treatment with DM199 is intended to increase the body’s production of BK to bind with the BK2 receptors to improve collateral circulation. In binding with the BK2 receptors expressed on endothelial cells (exposed to internal lumen of the artery), DM199, via production of bradykinin, activates the body’s natural physiologic processes and does not need to pass through the blood brain barrier, which is a specialized structure that is difficult for many therapeutic agents to cross.
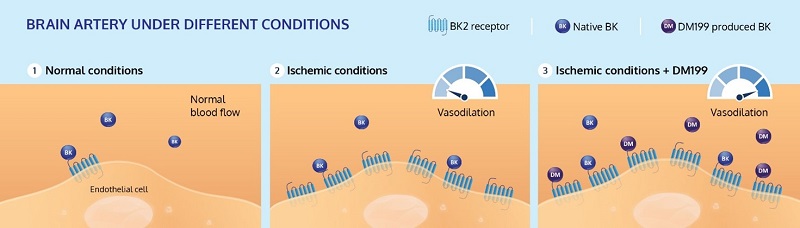
As depicted in the graphic below, we believe the mechanism of action for DM199 (KLK1) has the potential to preserve “at risk” penumbral brain tissue by acutely increasing cerebral blood flow by selectively vasodilating arteries in the ischemic penumbra and increasing collateral blood flow, to improve blood flow and restore oxygen levels rescuing these cerebral tissues.
DM199 Acute Ischemic Stroke: Proposed Mechanism
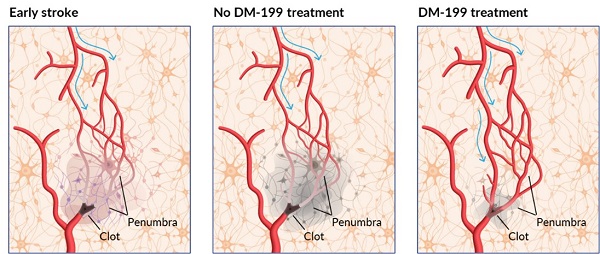
In January 2019, we published a paper titled “Human Tissue Kallikrein in the Treatment of Acute Ischemic Stroke” in a peer reviewed journal (Therapeutic Advances in Neurological Disorders (2019), 12:1-15, .https://doi.org/10.1177/1756286418821918). The paper reviews the scientific literature covering the biochemical role of KLK1 and presents the mechanistic rationale for using KLK1 as an additional pharmacological treatment for AIS. In addition to the biochemical mechanism of KLK1, the review highlights supporting results from human genetics and preclinical animal models of brain ischemia. It also reviews published clinical results for treatment of AIS by a form of KLK1 that is isolated from human urine. This form has been approved for post-stroke treatment of AIS in China and data has been published from clinical trials involving over 4,000 patients. The paper offers a series of testable therapeutic hypotheses for demonstrating the long-term beneficial effect of KLK1 treatment in AIS patients and the reasons for this action.
We are developing DM199 to treat AIS patients with a therapeutic window of up to 24 hours after the first sign of symptoms, well beyond the current window of up to 4.5 hours for tPA, thereby filling a large unmet need for those patients who cannot receive tPA under the currently available treatment window of tPA. This important attribute could potentially make therapy available to the millions of patients worldwide who currently have limited treatment options.
Supporting Data from the Use of Urine-derived KLK1 for the Treatment of AIS in China
In China, Kailikang® is approved and marketed by Techpool Bio-Pharma Inc., a company controlled by Shanghai Pharmaceuticals Holding Co. Ltd. Kailikang has been approved for the treatment of AIS in China. We believe the initial treatment window is up to 48 hours after stroke symptom onset. Based on data from IQVIA real world and health data, other publications and our own internal analysis, we estimate that over 600,000 stroke patients in China were treated in 2022 with Kailikang. More than 50 published clinical studies, covering over 4,000 stroke patients, have demonstrated a beneficial effect of Kailikang treatment in AIS including improvements in standard stroke scores, increased blood flow, reduced infarct size/ischemia in the brain. In a double-blinded, placebo-controlled trial of 446 participants treated with either Kailikang or a placebo with initial treatment administered up to 48 hours after symptom onset showed significantly better scores on the European Stroke Scale and Activities of Daily Living at three weeks post-treatment and after three months using the Barthel Index, (China Journal of Neurology (2007), 40:306–310).
Additionally, a comprehensive meta-analysis covering 24 clinical studies involving 2,433 concluded that human urinary KLK1 appears to ameliorate neurological deficits for patients with AIS and improves long-term outcomes, though a few treated patients suffered from transient hypotension (Journal of Evidence-Based Medicine (2012) 5:31-39, https://doi.org/10.1111/j.1756-5391.2012.01167.x)
Furthermore, in a retrospective study covering 300 consecutive AIS patients, subjects treated with human urinary KLK1 experienced a 6.5% absolute reduction (p=0.009) in recurrent strokes (39% relative) within one year (Brain and Behavior (2018), https://onlinelibrary.wiley.com/doi/pdf/10.1002/brb3.1033).
CRD Background and Disease Pathology
Cardio Renal Disease Background
Cardio-renal syndrome refers to the complex interplay between cardiac and renal dysfunction, where acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other. A key component of this syndrome is hypertension, which serves as both a cause and consequence of cardio-renal interactions. Hypertension contributes to the development and progression of heart and kidney diseases by imposing increased workload on the heart and by causing damage to the kidneys' nephrons, leading to a vicious cycle of worsening heart and kidney function. This relationship underscores the critical need for integrated management strategies that address both cardiac and renal health to effectively treat and prevent the progression of cardio-renal diseases. This integrated approach includes controlling blood pressure, managing fluid and electrolyte balance, and employing therapies that target the underlying mechanisms linking heart and kidney disease, such as the renin-angiotensin-aldosterone system (RAAS) inhibitors.
DM199 – Our Novel Solution for the Treatment of CRD, Including Hypertension
We believe DM199 has the potential to offer meaningful therapeutic benefits for CRD patients. We believe that the KLK1 protein plays a vital role in maintaining normal kidney function, promoting the production of nitric oxide, prostaglandin and other anti-inflammatory mediators which are important for kidney health and integrity. Patients with moderate to severe CRD often excrete abnormally low levels of KLK1 in their urine, leading to the hypothesis that a KLK1 deficit contributes to disease progression.
Additionally, KLK1 is the main source of bradykinin (BK) in resting conditions. BK opposes the prohypertensive renin, angiotensin, aldosterone system (RAAS) by restoring regulation of the epithelial sodium channel (EnaC) and increasing sodium and fluid excretion. DM199 augments low KLK1 levels and promotes natriuresis (excretion of sodium in urine). This regulation of EnaC with DM199 may contribute to lowering blood pressure in hypertensive patients and in particular in patient’s considered to be salt-sensitive.
By providing additional KLK1, we believe DM199 has the potential to:

DM199 treatment is intended to directly replenish KLK1 levels to maintain, or possibly restore, kidney function. Current treatment options, especially ACEi drugs, primarily slow the rate of decline in kidney function but are associated with side effects. Importantly, it is becoming increasingly clear that part of the beneficial effect of ACEi drugs involves preventing the normal breakdown of BK leading to substantial increases in BK levels throughout the body. However, these effects can be unregulated and ACEi drugs therefore can generate excessive BK where it is not needed, potentially leading to side effects such as persistent cough, angioedema (swelling of skin and tissue) and hyperkalemia (abnormally high potassium levels that can lead to cardiac arrest and sudden death). Most importantly, even with the use of ACEi or ARB medications in CRD patients, there remains a high unmet need as a majority of CRD patients still experience a progressive loss of renal function over time. We believe DM199 treatment, either alone or in combination with an ARB, could potentially restore normal KLK1 levels allowing the KKS to perform its normal physiological processes and release BK when and where it is needed, avoiding these side effects.
We intend to clinically evaluate the use of DM199 as a novel therapy for CRD. Protein replacement therapy with DM199, through the activation of the KKS, may complement and balance RAAS, primarily targeted by ACEis and ARBs, and may potentially improve the function of the diseased renal system by improving blood flow and vasodilation, as well as reducing blood pressure.
Our Competition and Current Treatments for Acute Ischemic Stroke and Cardio Renal Disease
The biopharmaceutical industry is highly competitive and characterized by rapidly advancing technologies that focus on rapid development of proprietary drugs. We believe that our DM199 product candidate, development capabilities, experience and scientific knowledge provide us with certain competitive advantages. However, we face significant potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies, academic institutions, governmental agencies and other research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
Many of our competitors, either alone or with their strategic partners, have substantially greater financial, technical and human resources than we do, and experience in obtaining FDA and other regulatory approvals of treatments and commercializing those treatments. Accordingly, our competitors may be more successful than us in obtaining approval for competitive products and achieving widespread market acceptance. Our competitors’ treatments may be more effectively marketed and sold than any products we may commercialize, thus limiting our market share and resulting in a longer period before we can recover the expenses of developing and commercializing our DM199 product candidate.
Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These activities may lead to consolidated efforts that allow for more rapid development of competitive product candidates.
We also compete for staff, development and clinical resources. These competitors may impair our ability to recruit or retain qualified scientific and management personnel, our ability to work with specific advisors, or our ability to work with clinical contract organizations due to conflicts of interest or capacity constraints, and may also delay recruitment of clinical study sites and study volunteers, impeding progress in our development programs.
We expect any products that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, price and the availability of reimbursement from government or other third-party payers. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are viewed as safer, more effective or less expensive than any products that we may develop.
Acute Ischemic Stroke
Currently, there is one approved pharmaceutical treatment for AIS. That treatment is tPA (marketed under the brand name Activase®), and its therapeutic window is limited to up to 4.5 hours after the AIS. There are, however, a number of companies that are actively pursuing a variety of approaches to develop pharmaceutical products for the treatment of AIS including, among others:
|
● |
Stem cells (Athersys, Inc.) |
|
● |
tPA extended treatment window (Genentech / Boehringer Ingelheim) |
|
● |
Tenecteplase (Genentech / Boehringer Ingelheim) |
|
● |
Cerebral edema (Biogen Inc.) |
|
● |
Anti-inflammatory and clot dissolving (Biogen Inc.) |
|
● |
Cell protection and anti-inflammation (ZZ Biotech LLC) |
|
● |
Inhibiting platelet aggregation (Acticor Biotech SAS) |
|
● |
Neuroprotector (Mitsubishi) |
There is a large unmet therapeutic need for AIS treatments that can be administered beyond the 4.5-hour time window of tPA. With this large unmet therapeutic need, there is significant competition to develop new therapeutic options. Currently, the most advanced treatment for AIS uses a medical device for the mechanical removal of blood clots in the large arteries supplying blood to the brain through sophisticated catheter-based approaches, referred to as mechanical thrombectomy. According to published research, use of mechanical thrombectomy is growing and the window of time after a stroke where the procedure can be used is widening. New therapeutic options in development include tissue protection focused therapies (deliverable from hours to days after the stroke) that are intended to preserve and protect brain cells beyond the tPA therapeutic window. The goal is to provide treatment options for the vast majority of AIS patients who do not receive hospital care early enough to qualify for tPA therapy. We believe there is a very significant market opportunity for a drug that has a therapeutic window beyond that of tPA and is able to obtain regulatory approval.
DM199 Clinical Trials
AIS Phase 2/3 ReMEDy2 Trial
We are currently conducting our ReMEDy2 clinical trial of DM199 for the treatment of AIS. Our ReMEDy2 clinical trial is a Phase 2/3, adaptive design, randomized, double-blind, placebo-controlled trial intended to enroll approximately 350 patients at up to 100 sites globally. Patients enrolled in the trial will be treated with either DM199 or placebo within 24 hours of the onset of AIS symptoms. The trial excludes patients treated with tPA, a thrombolytic agent intended to dissolve blood clots, and those with large vessel occlusions. The study population is representative of the approximately 80% of AIS patients who do not have treatment options today, primarily due to the limitations on treatment with tPA and/or mechanical thrombectomy. We believe that the proposed trial has the potential to serve as a pivotal registration study of DM199 in this patient population.
The primary endpoint of the ReMEDy2 trial is physical recovery from stroke as measured by the well-established modified Rankin Scale (mRS) at day 90. The mRS is a commonly used scale for measuring the degree of disability or dependence in the daily activities of people who have suffered a stroke. Secondary endpoints for the trial will evaluate, among other things, mRS shift (which shows the treatment effect on participants across the full spectrum of stroke severity), participant deaths, the National Institute of Health Stroke Score (NIHSS) and Barthel Index (BI) stroke scales and stroke recurrence. Recurrent strokes represent 25% of all ischemic strokes, often occurring in the first few weeks after an initial stroke and are typically more disabling, costly and fatal than initial strokes.
We voluntarily paused participant enrollment in the ReMEDy2 trial to investigate three unexpected instances of clinically significant hypotension (low blood pressure) occurring shortly after initiation of the intravenous (IV) dose of DM199. The acutely low blood pressure levels in the three participants recovered back to their baseline blood pressure within minutes after the IV infusion was stopped, and the participants suffered no injuries. On July 6, 2022, we announced that the FDA placed a clinical hold on the investigational new drug application (IND) for our ReMEDy2 trial. In September 2022, we submitted to the FDA supporting in-vitro data that the cause of the hypotensive events was likely related to switching to a new type of IV bag for use in the ReMEDy2 trial rather than continue with the type of IV bag used in the prior ReMEDy1 trial, where DM199 was generally safe and well tolerated and no hypotensive episodes were reported. While there were no differences in the compatibility of DM199 with either type of IV bag, we observed significant differences in DM199 binding between the two types of IV bags we believe altered, and unintentionally elevated, the total amount of DM199 being administered to participants in the ReMEDy2 trial and thereby triggering the hypotensive events. We also included in this FDA submission, proposed protocol modifications to further address the mitigation of these events, including a reduction in the DM199 dose level for the initial IV dose to effectively match in the PVC IV bags, the well tolerated IV dose administered in the ReMEDy1 trial. Following review of this analysis, the FDA informed us that they were continuing the clinical hold and requesting, among other items, an additional in-use, in vitro stability study of all of the materials and equipment used in the IV administration of DM199, which included testing the combination of the IV bag, IV tubing and mechanical infusion pump, to further rule out any other cause of the hypotension events. The requested in-use study was completed at an independent laboratory and the results were substantially consistent with our initial stand-alone testing of the IV bags. In May 2023, this additional supporting data was submitted to the FDA in our clinical hold response. In June 2023, the FDA completed review of our clinical hold response and informed us that the clinical hold was fully removed allowing us to begin preparations to resume our ReMEDy2 trial.
Following in-depth discussions of the ReMEDy2 Phase 2/3 protocol design with global stroke experts, the scientific advisory board and current investigators, the Company has made several important amendments to the protocol subsequent to the lifting of the clinical hold. These changes were submitted to the FDA in early October and the Company is proceeding with use of the amended protocol as the FDA did not issue any comments during the 30-day review period which ended on November 3, 2023.
Prior to the clinical hold of our ReMEDy2 trial, we had experienced slower than expected site activations and enrollment in our ReMEDy2 trial and may continue to experience these conditions as we activate additional clinical sites and enrollment participants. We believe this was due primarily to clinical staff shortages resulting from layoffs and employee burnout, the reallocation of clinical nurses to COVID-19 care, particularly during surges in COVID-19 cases, a loss of study coordinators resulting from budget constraints and COVID-19 vaccination requirements and concerns managing logistics and protocol compliance for participants discharged from the hospital to an intermediate care facility. In an effort to mitigate the impact of these factors, we have worked with our contract research organization to develop alternative procedures to support study sites and potential participants as needed. We intend to continue to monitor the results of these efforts or implement additional actions to mitigate the impact of these factors on our ReMEDy2 trial, however no assurances can be provided as to if and when these issues will resolve.
In September 2021, the FDA granted Fast Track designation to DM199 for the treatment of AIS where tPA and/or mechanical thrombectomy are not indicated or medically appropriate. The FDA may grant Fast Track designation to a drug that is intended to treat a serious condition and nonclinical or clinical data demonstrate the potential to address unmet medical need. The FDA provides opportunities for frequent interactions with the review team for a Fast Track product, including end-of-phase 2 meetings with the FDA to discuss study design, extent of safety data required to support approval, dose-response concerns, and use of biomarkers. A Fast Track product may also be eligible for rolling review, where the FDA reviews portions of a marketing application before the sponsor submits the complete application.
Phase 1C Open Label Safety Trial
Concurrently with performing the requested in-use study, we also conducted a Phase 1C open label, single ascending dose (SAD) study of DM199 administered with the PVC IV bags used in the ReMEDy2 trial. The purpose of the study was to confirm, with human data, the DM199 blood concentration levels achieved with the IV dose and further evaluate safety and tolerability. This study was conducted in Australia. The third cohort, which received the 0.50 µg/kg dose level proposed for the ReMEDy2 trial, was dosed in April 2023 with no significant adverse events related to DM199. The pharmacokinetic data, including the DM199 blood concentration levels, for all cohorts was included as supplemental information in our clinical hold response. In investigating the cause of the unexpected instances of hypotension, we noted that all three participants were receiving angiotensin-converting enzyme inhibitor (ACEi) therapy at the time of their enrollment. Given this, we also completed an additional, fourth cohort of hypertensive patients (Part B) being treated with ACEi prior to enrolling. All ACEi patients received the full IV dose at the 0.5 µg/kg level with no instances of hypotension. We believe that these results provide further assurance to potential investigators that ACEi patients may be safely included in the ReMEDy2 trial.
AIS Phase 2 ReMEDy1 Trial
In May 2020, we announced top-line data from our Phase 2 ReMEDy1 trial assessing the safety, tolerability and markers of therapeutic efficacy of DM199 in patients suffering from AIS. We initiated treatment in this trial in February 2018 and completed enrollment in October 2019 with 92 participants. The study drug (DM199 or placebo) was administered as an intravenous (IV) infusion within 24 hours of stroke symptom onset, followed by subcutaneous injections later that day and once every 3 days for 21 days. The trial was designed to measure safety and tolerability along with multiple tests designed to investigate DM199’s therapeutic potential including plasma-based biomarkers and standard functional stroke measures assessed at 90 days post-stroke. Standard functional stroke measurements include the Modified Rankin Scale, National Institutes of Health Stroke Scale, the Barthel Index. The trial met primary safety and tolerability endpoints and was generally safe and well tolerated. In addition, there was a demonstrated therapeutic effect on the rate of severe stroke recurrence inclusive of all participants and there was also a demonstrated therapeutic effect on the physical recoveries of participants that received tPA prior to enrollment but not in participants receiving mechanical thrombectomy prior to enrollment.
Prior to enrollment, 44 of the 91 evaluable participants (48%) received mechanical thrombectomy intervention, a catheter-based treatment intended to physically remove clots and potentially available for patients who have a large vessel occlusion and can be treated within 6 to 24 hours of the onset of stroke symptoms. While approximately 20% of AIS patients are believed to be eligible for a mechanical thrombectomy, currently only about 5% to 10% receive the treatment due to elapsed time post-stroke or unavailability of the therapy at the hospital where the patient presents. DM199 is intended to treat the approximately 80% of AIS patients who are not eligible for either mechanical thrombectomy or tPA. Treatment for these patients is limited to supportive care. Due to the large volume of participants receiving mechanical thrombectomy prior to enrollment in the ReMEDy1 trial, and a disproportionate distribution of these participants between the active treatment and placebo groups, DM199 did not produce a therapeutic effect on physical recoveries in the overall trial analysis.
When participants treated with mechanical thrombectomy are excluded from the trial data set, which represents the group of participants most closely aligned with the target treatment population for DM199 in the ReMEDy2 trial, a positive therapeutic effect on participant physical recoveries was demonstrated. As shown in the table below, when evaluating the participants treated with DM199 (n=25) vs. supportive care and/or tPA (n=21), the results showed that 36% of participants receiving DM199 progressed to a full or nearly full recovery at 90 days (NIHSS: 0-1), compared to 14% of participants in the placebo group. This represents a 22% absolute increase in the proportion of participants achieving a full or nearly full recovery. Additionally, subject deaths decreased from 24% in the placebo group to 12% in the active therapy group, a 50% relative reduction. Note that the number of subjects in these subsets were insufficient for statistical significance.
DM199 vs. Supportive Care and/or tPA
|
NIHSS Outcomes at 90 Days |
||||
|
0-1 |
2-8 |
≥ 9 |
Death |
|
|
Placebo (n=21) |
14% |
57% |
5% |
24% |
|
DM199 (n=25) |
36% |
36% |
16% |
12% |
In addition, in the evaluable participants (n=91), a significant reduction in the number of participants with recurrent ischemic stroke was noted in the active treatment group: 0 (0%) participants treated with DM199 vs. 6 (13%) on placebo (p=0.012), with 4 of the 6 resulting in participant death.
We believe these findings from our Phase 2 ReMEDy1 trial, which are consistent with the use of Kailikang in China, provide a signal that recombinant human KLK1 appears safe and may have promise as a new treatment for physicians who have limited options for the treatment of patients following an AIS.
CRD Phase 2 REDUX Trial
Our REDUX trial was a multi-center, open-label investigation of participants with mild or moderate chronic kidney disease (CKD) (Stage II or III) and albuminuria. The trial was conducted in the United States and included: Cohort 1 enrolled non-diabetic, hypertensive African Americans (AA) with Stage II or III CKD. African Americans are at greater risk for CKD than Caucasians, and those African Americans who have the APOL1 gene mutation are at an even higher risk. Cohort 2 enrolled participants with IgA Nephropathy (IgAN). Cohort 3 enrolled participants with Type 2 diabetes mellitus with CKD, hypertension and albuminuria (DKD). The trial evaluated two dose levels of DM199 within each cohort. Study participants received DM199 by subcutaneous injection twice weekly for 95 days. The primary study endpoints, evaluated after three months of treatment, included safety, tolerability, blood pressure, albuminuria and kidney function, which are evaluated by changes from baseline in estimated glomerular filtration rate, albuminuria, as measured by the urinary albumin to creatinine ratio, and blood pressure in hypertensive participants.
Interim results, issued in November 2021, indicated that after three months of treatment, DM199 was demonstrating clinically meaningful improvements in kidney function in Cohorts 1 and 2, as measured by simultaneously stabilizing estimated glomerular filtration rate (eGFR) and decreasing the urinary albumin-to-creatinine ratio (UACR). Additionally, in participants who were hypertensive (Cohorts 1 and 3), DM199 reduced blood pressure by clinically significant levels and importantly, there was no effect on participants who were not hypertensive (Cohort 2).
DM199 was generally safe and well tolerated across all cohorts. Adverse events (AEs) were generally mild to moderate in severity, with the most common being local injection site irritation, and all resolved without medical intervention.
We completed enrollment in REDUX with a total 84 subjects enrolled, including 24 African American subjects into Cohort 1, 25 subjects with IgAN into Cohort 2 and 35 subjects with Type 2 diabetes in Cohort 3. As of March 31, 2022, all participants had completed their treatment periods.
We plan to disclose additional data related to blood pressure control as part of supporting our plans for our cardio renal program to be disclosed in 2024.
DM199 Safety Summary
Intravenously/subcutaneously administered DM199, in doses ranging from 0.025 μg/kg to 50.0 μg/kg, has been administered to over 250 subjects across 5 completed clinical studies and has been shown to be generally safe and well tolerated. The most frequently reported treatment-emergent adverse events in our Phase 2 ReMEDy1 AIS trial were constipation, oral candidiasis and nausea. These events were predominately mild to moderate in severity. Orthostatic hypotension was determined to be the dose limiting tolerability. There have been 3 reported drug-related serious adverse events (SAEs) in subjects receiving DM199 of transient hypotension; these events were rapidly reversible upon stopping infusion with no long term sequelae (further adverse events).
Potential DM199 Commercial Advantages
Several researchers have studied the structural and functional properties of KLK1. This deep body of knowledge has revealed the potential clinical benefits of KLK1 treatments. Today, forms of KLK1 derived from human urine and the pancreas of pigs are approved and sold in Japan, China and South Korea to treat AIS, CRD, retinopathy, hypertension and related diseases. We are not aware of any recombinant version of KLK1 with regulatory approval for human use in any country, nor any recombinant version in development other than our drug candidate DM199. We believe at least five companies have attempted, unsuccessfully, to create a recombinant version of KLK1.
The growing understanding of the role of KLK1 in human health and its use in Asia as an approved therapeutic highlight two important potential commercial advantages for DM199:
KLK1 treatment is sold in Japan, China and South Korea. Research has shown that patients with low levels of KLK1 are associated with a variety of diseases related to vascular dysfunction, such as AIS, CRD, retinopathy and hypertension. In randomized, controlled clinical trials, human urine and porcine derived KLK1 has demonstrated statistically significant clinical benefits in treating a variety of patients with KLK1 compared to placebo. These efficacy results are further substantiated by established markets in Japan, China and South Korea for pharmaceutical sales of KLK1 derived from human urine and the pancreas of pigs. We estimate that millions of patients have been treated with these forms of KLK1 in Asia. Altogether, we believe this supports a strong market opportunity for a recombinant version of KLK1 such as DM199.
KLK1 treatment has had limited side effects and has been well tolerated in studies to date. KLK1 is naturally produced by the human body; and, therefore, the body’s own control mechanisms act to limit potential side effects. The side effect observed to limit participant tolerability in our clinical trials was orthostatic hypotension, or a sudden drop in blood pressure, which has been primarily seen at doses 10 to 20 times higher than our anticipated therapeutic dose levels. Most recently, clinically significant, transient hypotension (low blood pressure) occurring shortly after initiation of the intravenous (IV) dose of DM199 was experienced by three participants in our ReMEDy2 trial which were the cause of the Company pausing participant enrollment and the FDA placing a clinical hold on the IND for our ReMEDy2 trial. The blood pressure levels of the three participants recovered back to their baseline blood pressure within minutes after the IV infusion was stopped and the participants suffered no injuries. We believe that these events were caused by our switching away from the type of IV bag used in the prior ReMEDy1 trial, where no hypotensive episodes were reported, which resulted in an unintended, elevated dose of DM199 being delivered in the ReMEDy2 trial. We believe that by reducing the dose rate for the IV infusion to a level that matches the effective dose rate in the ReMEDy1 trial, we can manage and/or eliminate the clinically significant hypotensive events.
Moreover, we understand that routine clinical use of KLK1 treatment in Asia has been well-tolerated by patients for several decades. In 2017, we completed a clinical trial comparing the pharmacokinetic profile of DM199 to the human urinary form of KLK1 (Kailikang), which showed DM199, when administered in intravenous form, had a similar pharmacokinetic profile. Further, when DM199 was administered subcutaneously, DM199 demonstrated a longer acting pharmacokinetic profile, superior to the intravenously administered Kailikang and DM199.
In addition, we believe that there are also significant formulation, manufacturing, regulatory and other advantages for recombinant human KLK1 drug candidate DM199:
Potency and Impurity Considerations. KLK1 produced from human urine or the pancreas of pigs presents risks related to preventing impurities, endotoxins and chemical byproducts due to the inherent variability of the isolation and purification process. We believe that this creates the risk of inconsistencies in potency and impurities from one production run to the next. However, we expect to produce a consistent formulation of KLK1 that is free of endotoxins and other impurities.
Cost and Scalability. Large quantities of human urine or pig pancreas must be obtained to derive a small amount of KLK1. This creates potential procurement, cost and logistical challenges to source the necessary raw material, particularly for human urine sourced KLK1. Once sourced, the raw material is processed using chemicals and costly capital equipment and produces a significant amount of byproduct waste. Our novel recombinant manufacturing process utilizes widely available raw materials and can be readily scaled for commercial production. Accordingly, we believe our manufacturing process will have significant cost and scalability advantages.
Regulatory. We are not aware of any attempts by manufacturers of the urine or porcine based KLK1 products to pursue regulatory approvals in the United States. We believe that this is related to challenges presented by using inconsistent and potentially hazardous biomaterials, such as human urine and the pancreas of pigs, and their resulting ability to produce a consistent drug product. Our novel recombinant manufacturing process utilizes widely available raw materials which we believe provides a significant regulatory advantage, particularly in regions such as the United States, Europe and Canada, where safety standards are high. In addition, we believe that DM199 could qualify for 12 years of data exclusivity in the United States under the Biologics Price Competition and Innovation Act of 2009 (BPCIA), which was enacted as part of the Patient Protection and Affordable Care Act (ACA) as amended by the Health Care and Education Reconciliation Act of 2010.
From a strategic perspective, we continue to believe that strategic alternatives with respect to our DM199 product candidate, including licenses and business collaborations, with other regional and global pharmaceutical and biotechnology companies can be important in advancing the clinical development of DM199. Therefore, as a matter of course and from time to time, we engage in discussions with third parties regarding these matters.
Regulatory Approval
Securing regulatory approval for the manufacture and sale of human therapeutic products in the United States, Europe, Canada and other commercial territories is a long and costly process that is controlled by each territory’s national regulatory agency. The national regulatory agency in the United States is the FDA, in Europe it is the European Medicines Agency (EMA), and in Canada it is Health Canada. Other national regulatory agencies have similar regulatory approval requirements, but each national regulatory agency has its own approval processes. Approval in the United States, Europe or Canada does not assure approval by other national regulatory agencies, although often test results from one country may be used in applications for regulatory approval in another country.
Prior to obtaining regulatory approval to market a drug product, every national regulatory agency has a variety of statutes and regulations which govern the principal development activities. These laws require controlled research and testing of products, governmental review, and approval of a submission containing preclinical and clinical data establishing the safety and efficacy of the product for each use sought, as well as approval of manufacturing facilities, including adherence to good manufacturing practices (GMP) during production and storage, and control of marketing activities, including labeling and advertising.
None of our product candidates have been completely developed or tested; and, therefore, we are not yet in a position to seek regulatory approval in any territory to market any of our product candidates.
The clinical testing, manufacturing, labeling, storage, distribution, record keeping, advertising, promotion, import, export and marketing, among other things, of our current or future product candidates, are subject to extensive regulation by governmental authorities in the United States and other countries. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable requirements at any time during the product development process, approval process, or after approval may subject us to a variety of administrative or judicial sanctions, including refusal by the applicable regulatory authority to approve pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters and other types of letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement of profits, or civil or criminal investigations and penalties brought by the FDA and the Department of Justice or other governmental entities.
U.S. Approval Process
In the United States, the FDA is responsible for the drug approval process. The FDA’s mission is to ensure that all medications on the market are safe and effective. The FDA’s approval process examines and thoroughly reviews potential new drugs; only those that are in compliance with the Code of Regulations, 21 CFR 312 and 21 CFR 314 are approved.
The U.S. food and drug regulations require licensing of manufacturing facilities, carefully controlled research and testing of products, governmental review and approval of test results prior to marketing of therapeutic products, and adherence to GMP, as defined by each licensing jurisdiction, during production.
A description of the different stages in the drug approval process in the United States follows.
Stage 1: Preclinical Research. After an experimental drug is discovered, research is conducted to help determine its potential for treating or curing an illness. This is called preclinical research. Animal and/or bench studies are conducted to determine if there are any harmful effects of the drug and to help understand how the drug works. Information from these experiments is submitted to the FDA as part of an IND. The FDA reviews the information in the IND and decides if the drug is safe to study in humans.
Stage 2: Clinical Research. The experimental drug is studied in humans. The studies are known as clinical trials. Clinical trials are carefully designed and controlled experiments in which the experimental drug is administered to patients to test its safety and to determine the effectiveness of an experimental drug. The four general phases of clinical research are described below.
|
● |
Phase 1 Clinical Studies. Phase 1 clinical studies are generally conducted with healthy volunteers who are not taking other medicines; patients with the illness that the drug is intended to treat are not tested at this stage. Ultimately, Phase 1 studies demonstrate how an experimental drug affects the body of a healthy individual. Phase 1 consists of a series of small studies consisting of tens of volunteers. Tests are done on each volunteer throughout the study to see how the person’s body processes, responds to, and is affected by the drug. Low doses and high doses of the drug are usually studied, resulting in the determination of the safe dosage range in volunteers by the end of Phase 1. This information will determine whether the drug proceeds to Phase 2. |
|
● |
Phase 2 Clinical Studies. Phase 2 clinical studies are conducted in order to determine how an experimental drug affects people who have the disease to be treated. Phase 2 usually consists of a limited number of studies that help determine the drug’s short-term safety, side effects, and general effectiveness. The studies in Phase 2 often are controlled investigations involving comparison between the experimental drug and a placebo, or between the experimental drug and an existing drug. Information gathered in Phase 2 studies will determine whether the drug proceeds to Phase 3. |
|
● |
Phase 3 Clinical Studies. Phase 3 clinical studies are expanded controlled and uncontrolled trials that are used to more fully investigate the safety and effectiveness of the drug. These trials differ from Phase 2 trials because a larger number of patients are studied (sometimes in the thousands) and because the studies are usually double blinded, placebo controlled and of longer duration. As well, Phase 3 studies can include patients who have more than one illness and are taking medications in addition to the experimental drug used in the study. Therefore, the patients in Phase 3 studies more closely reflect the general population. The information from Phase 3 forms the basis for most of the drug’s initial labeling, which will guide physicians on how to use the drug. |
|
● |
Phase 4 Clinical Studies. Phase 4 clinical studies are conducted after a drug is approved. Phase 4 studies may be required by the FDA or conducted by companies to more fully understand how their drug compares to other drugs. FDA-required Phase 4 studies often investigate the drug in specific types of patients that may not have been included in the Phase 3 studies and can involve very large numbers of patients to further assess the drug’s safety. |
Stage 3: FDA Review for Approval. Following the completion of Phase 3 clinical studies, the pharmaceutical company prepares an electronic common technical document reporting all clinical nonclinical and chemistry, manufacturing and control studies conducted on the drug that is transmitted to the FDA as a Biologics License Application (BLA). The FDA reviews the information in the BLA to determine if the drug is safe and effective for its intended use. An advisory panel meeting is scheduled for a new drug allowing the FDA to gain feedback from experts. If the FDA determines that the drug is safe and effective, the drug will be approved.
Stage 4: Marketing. After the FDA has approved the experimental drug, the pharmaceutical company can make it available to physicians and their patients. A company also may continue to conduct research to discover new uses for the drug. Each time a new use for a drug is discovered, the drug once again is subject to the entire FDA approval process before it can be marketed for that purpose.
Any FDA approved pharmaceutical products are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA guidance documents, and promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, promoting pharmaceutical products for uses or in patient populations that are not described in the pharmaceutical product’s approved labeling (known as “off-label use”), industry-sponsored scientific and educational activities and promotional activities involving the internet or social media. Failure to comply with FDA requirements is likely to have negative consequences, including adverse publicity, warning or enforcement letters from the FDA or the Federal Trade Commission (FTC), mandated corrective advertising or communications with doctors, product seizures or recalls and state or federal civil or criminal prosecution, injunctions and penalties.
The FDA also may require post-marketing testing, known as Phase 4 testing, risk evaluation and mitigation strategies and surveillance to monitor the effects of an approved product or place conditions on an approval that could restrict the distribution or use of the product.
DM199 may qualify for 12 years of data exclusivity under the BPCIA, which was enacted as part of the ACA, as amended by the Health Care and Education Reconciliation Act of 2010. Under the BPCIA, an application for a biosimilar product (BLA) cannot be submitted to the FDA until four years, or if approved by the FDA, until 12 years, after the original brand product identified as the reference product is approved under a BLA. The BPCIA provides an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. The new law is complex and is only beginning to be interpreted and implemented by the FDA.
European Approval Process
The EMA is roughly parallel to the FDA in terms of the drug approval process and the strict requirements for approval. The EMA was set up in 1995 in an attempt to harmonize, but not replace, the work of existing national medicine regulatory bodies in individual European countries. As with the FDA, the EMA drug review and approval process follows similar stages from preclinical testing through clinical testing in Phase 1, 2, and 3. There are some differences between the FDA and EMA review process, specifically the review process in individual European countries. Such differences may allow certain drug products to be tested in patients at an earlier stage of development.
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the Centers for Medicare and Medicaid Services and other divisions of the U.S. government, including, the Department of Health and Human Services, the Department of Justice and individual U.S. Attorney offices within the Department of Justice, and state and local governments. For example, if a drug product is reimbursed by Medicare, Medicaid, or other federal or state healthcare programs, a company, including its sales, marketing and scientific/educational grant programs, must comply with the federal Food, Drug & Cosmetic Act (FDCA) as it relates to advertising and promotion of drugs, the federal False Claims Act, as amended, the federal Anti-Kickback Statute, as amended, the Physician Payments Sunshine Act, the federal Health Insurance Portability and Accountability Act of 1996 (HIPAA), and similar state laws. If a drug product is reimbursed by Medicare or Medicaid, pricing and rebate programs must comply with, as applicable, the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 (OBRA), and the Medicare Prescription Drug Improvement and Modernization Act of 2003. Among other things, OBRA requires drug manufacturers to pay rebates on prescription drugs to state Medicaid programs and empowers states to negotiate rebates on pharmaceutical prices, which may result in prices for our future products being lower than the prices we might otherwise obtain. Additionally, the ACA substantially changes the way healthcare is financed by both governmental and private insurers. There may continue to be additional proposals relating to the reform of the U.S. healthcare system, in the future, some of which could further limit coverage and reimbursement of drug products. If drug products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements may apply.
Pharmaceutical Coverage, Pricing and Reimbursement
In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of coverage and adequate reimbursement from third-party payers, including government health administrative authorities, managed care providers, private health insurers and other organizations. In the United States, private health insurers and other third-party payers often provide reimbursement for products and services based on the level at which the government (through the Medicare and/or Medicaid programs) provides reimbursement for such treatments. Third-party payers are increasingly examining the medical necessity and cost-effectiveness of medical products and services in addition to their safety and efficacy; and, accordingly, significant uncertainty exists regarding the coverage and reimbursement status of newly approved therapeutics. In particular, in the United States, the European Union and other potentially significant markets for our product candidates, government authorities and third-party payers are increasingly attempting to limit or regulate the price of medical products and services, particularly for new and innovative products and therapies, which has resulted in lower average selling prices. Further, the increased emphasis on managed healthcare in the United States and on country and regional pricing and reimbursement controls in the European Union will put additional pressure on product pricing, reimbursement and usage, which may adversely affect our future product sales and results of operations. These pressures can arise from rules and practices of managed care groups, judicial decisions and governmental laws and regulations related to Medicare, Medicaid and healthcare reform, pharmaceutical reimbursement policies and pricing in general. As a result, coverage and adequate third party reimbursement may not be available for our products to enable us to realize an appropriate return on our investment in research and product development.
The market for our product candidates for which we may receive regulatory approval will depend significantly on access to third-party payers’ drug formularies or lists of medications for which third-party payers provide coverage and reimbursement. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies. Also, third-party payers may refuse to include a particular branded drug in their formularies or may otherwise restrict patient access to a branded drug when a less costly generic equivalent or another alternative is available. In addition, because each third-party payer individually approves coverage and reimbursement levels, obtaining coverage and adequate reimbursement is a time-consuming and costly process. We would be required to provide scientific and clinical support for the use of any product candidate to each third-party payer separately with no assurance that approval would be obtained, and we may need to conduct expensive pharmacoeconomic studies to demonstrate the cost-effectiveness of our product candidates. This process could delay the market acceptance of any of our product candidates for which we may receive approval and could have a negative effect on our future revenues and operating results. We cannot be certain that our product candidates will be considered cost-effective. If we are unable to obtain coverage and adequate payment levels for our product candidates from third-party payers, physicians may limit how much or under what circumstances they will prescribe or administer them and patients may decline to purchase them. This in turn could affect our ability to successfully commercialize our products and impact our profitability, results of operations, financial condition, and future success.
Research and Development
We have devoted substantially all of our efforts to research and development (R&D), which therefore comprises the largest component of our operating costs. Our primary focus over the past approximately 12 years has been our lead product candidate, DM199, which is currently in clinical development for the treatment of AIS.
We expect our R&D expenses will continue to increase in the future as we continue the development and clinical study of our initial product candidate, DM199, in AIS and seek to pursue other indications or expand our product candidate portfolio. The process of conducting the necessary development and clinical research to obtain regulatory approval is costly and time-consuming and we consider the active management and development of our clinical pipeline to be integral to our long-term success. The actual probability of success for each product candidate, clinical indication and preclinical program may be affected by a variety of factors including, among other things, the safety and efficacy data for each product candidate, amounts invested in their respective programs, competition and competitive developments, manufacturing capability and commercial viability.
R&D expenses include:
|
● |
expenses incurred with third party service providers such as contract research organizations and other study support services; |
|
● |
expenses incurred under agreements with clinical trial sites that conduct research activities on our behalf; |
|
● |
laboratory and vendor expenses related to the execution of clinical trials and non-clinical studies; |
|
● |
the cost of acquiring, developing, manufacturing, and distributing clinical trial materials; |
|
● |
employee and consultant-related expenses, which include salaries, benefits, consulting fees, travel and share-based compensation; and |
|
● |
facilities and other expenses, which include direct and allocated expenses for rent and maintenance of facilities, insurance, and other supply costs. |
R&D costs are expensed as incurred. Costs for certain development activities such as clinical trials are recognized based on an evaluation of the progress to completion of specific tasks using information and data provided to us by our vendors and our clinical sites.
We expect that it will be at least three to four years, if ever, before we have any product candidates ready for commercialization.
Manufacturing
We do not own or operate manufacturing facilities for the production of DM199 nor do we have plans to develop our own manufacturing operations in the foreseeable future. We rely on Catalent Pharma Solutions, LLC (Catalent), a contract development and manufacturing organization (CDMO) with proven GMP experience in the manufacturing of recombinant proteins for clinical trials, for procuring all of our required raw materials and producing active pharmaceutical ingredient for our clinical trials. We have licensed certain gene expression technology and we contract with Catalent for the manufacture of DM199 drug substance. We currently employ internal resources and third-party consultants to manage our manufacturing relationship with Catalent.
Sales and Marketing
We have not yet defined our sales, marketing or product distribution strategy for our initial product candidate, DM199, or any future product candidates. We currently expect to partner with a large pharmaceutical company for sales execution. However, our future commercial strategy may include the use of distributors, a contract sales force or the establishment of our own commercial and specialty sales force, as well as similar strategies for regions and territories outside the United States.
Intellectual Property
We view patents and other means of intellectual property protection, including trade secrets, as an important component of our core business. We focus on translating our innovations into intellectual property protecting our proprietary technology from infringement by competitors. To that end, patents are reviewed frequently and continue to be sought in relation to those components or concepts of our preclinical and clinical products to provide protection. Our strategy, where possible, is to file patent applications to protect our product candidates, as well as methods of manufacturing, administering and using a product candidate. Prior art searches of both patent and scientific databases are performed to evaluate novelty, inventiveness and freedom-to-operate. We require all employees, consultants and parties to a collaborative research agreement to execute confidentiality agreements upon the commencement of employment, consulting relationships or a collaboration with us. These agreements require that all confidential information developed or made known during the course of the engagement with us is to be kept confidential. We also maintain agreements with our scientific staff and all parties contracted in a scientific capacity affirming that all inventions resulting from work performed for us, using our property or relating to our business and conceived or completed during the period covered by the agreement are the exclusive property of DiaMedica.
Our DM199 patent portfolio includes four granted U.S. patents, a granted European patent, a granted Canadian patent, and pending applications in Australia, Canada, China, Europe, India, Japan, South Korea, Hong Kong and the United States. Granted or pending claims offer various forms of protection for DM199 including claims to compositions of matter, pharmaceutical compositions, specific formulations and dosing levels and methods for treating a variety of diseases, including stroke, chronic kidney disease and related disorders. These U.S. patents and applications, and their foreign equivalents, are described in more detail below.
Issued patents held by us cover the DM199 composition of matter based on an optimized combination of closely-related isoforms that differ in the extent of glycosylation (process by which sugars are chemically attached to proteins). Issued claims in this patent family cover the most pharmacologically active variants of DM199 and methods of using the same for treating ischemic conditions and these patents are due to expire in 2033. A second patent family includes an issued U.S. patent with claims directed to methods of treating subjects by administering a SC formulation of DM199 or related recombinant kallikrein-1 (KLK1) polypeptides and is predicted to expire in 2033. The pending applications are directed to a range of dose levels and dosing regimens of DM199 that are potentially useful for treating a wide range of diseases including, e.g. pulmonary arterial hypertension, cardiac ischemia, chronic kidney disease, diabetes, stroke and vascular dementia which, if granted, are predicted to expire in 2038.
As previously discussed, we do not own or operate manufacturing facilities for the production of clinical or commercial quantities of DM199. We are contracting with Catalent for the manufacture of DM199. We have licensed certain gene expression technology and we contract with Catalent for the manufacture of DM199. Under the terms of this license, certain milestone and royalty payments may become due and are dependent upon, among other factors, performing clinical trials, obtaining regulatory approvals and ultimately the successful commercialization of a new drug, the outcome and timing of which is uncertain. The royalty term is indefinite but the license agreement may be canceled by us on 90 days’ prior written notice. The license may not be terminated by Catalent unless we fail to make required milestone and royalty payments.
Methods and reagents required for commercial scale manufacture of DM199 are subject to a series of patents issued to Catalent. We license these patents from Catalent, and such license is exclusive as it relates to the production of DM199 or any human KLK1 protein.
We believe that our proprietary technology along with trade secrets and specialized knowledge of the manufacturing process will provide substantial protection from third-party competitors. We also believe that DM199 cannot be easily reverse engineered for the production of a copycat version.
We believe that the most relevant granted patents and applications with composition of matter or method of use claims covering DM199 are listed below, along with their projected expiration dates exclusive of any patent term extension:
|
Patent/Application Number |
Title |
Geography |
Predicted Expiration |
|||
|
DM199 Patent Family |
||||||
|
Issued patents |
||||||
|
US 9,364,521 |
Human Tissue Kallikrein 1 Glycosylation Isoforms |
US |
2033 |
|||
|
US 9,839,678 |
Human Tissue Kallikrein 1 Glycosylation Isoforms |
US |
2033 |
|||
|
CA 2880085 |
Human Tissue Kallikrein 1 Glycosylation Isoforms |
CA |
2033 |
|||
|
EP 2 854 841 |
Human Tissue Kallikrein 1 Glycosylation Isoforms |
Europe |
2033 |
|||
|
US 9,616,015 |
Formulations for Human Tissue Kallikrein-1 for Parenteral Delivery and Related Methods |
US |
2033 |
|||
|
US 11,857,608 |
Dosage Forms of Tissue Kallikrein 1 Application |
US |
2038 |
|||
|
Pending applications |
||||||
|
AU 2018230478 |
Dosage Forms of Tissue Kallikrein 1 |
Australia |
2038 |
|||
|
CA 3054962 |
Dosage Forms of Tissue Kallikrein 1 |
Canada |
2038 |
|||
|
CN 201880016380.4 |
Dosage Forms of Tissue Kallikrein 1 |
China |
2038 |
|||
|
EP 18763243.5 |
Dosage Forms of Tissue Kallikrein 1 |
Europe |
2038 |
|||
|
IN 201917037712 |
Dosage Forms of Tissue Kallikrein 1 |
India |
2038 |
|||
|
JP 2019-548655 |
Dosage Forms of Tissue Kallikrein 1 |
Japan |
2038 |
|||
|
KR 10-2019-7026369 |
Dosage Forms of Tissue Kallikrein 1 |
South Korea |
2038 |
|||
|
HK 62020009783.5 |
Dosage Forms of Tissue Kallikrein 1 |
Hong Kong |
2038 |
|||
|
HK 62020007146.7 |
Dosage Forms of Tissue Kallikrein 1 |
Hong Kong |
2038 |
|||
|
US 18/501,804 |
Dosage Forms of Tissue Kallikrein 1 |
US |
2038 |
|||
|
US 18/295,991 |
Tissue Kallikrein 1 for Treating Chronic Kidney Disease |
US |
||||
|
PCT/US23/65385 |
Tissue Kallikrein 1 for Treating Chronic Kidney Disease |
PCT |
||||
|
DM300 Patent Family |
||||||
|
Issued patents |
||||||
|
11,725,043 |
Ulinastatin Polypeptides |
US |
2041 |
|||
|
Pending applications |
||||||
|
PCT/US2021/021148 |
Ulinastatin Polypeptides |
BR,CA,CN,EP,HK,IN,JP, TW,US |
2041 |
|||
|
PCT/US2022/014095 |
Ulinastatin Polypeptides for Treating Diseases |
CA,CN,EP,JP,US |
2042 |
|||
The base term of a U.S. patent is 20 years from the filing date of the earliest-filed non-provisional patent application from which the patent claims priority. The term of a U.S. patent can be lengthened by patent term adjustment, which compensates the owner of the patent for administrative delays at the U.S. Patent and Trademark Office. In some cases, the term of a U.S. patent is shortened by terminal disclaimer that reduces its term to that of an earlier-expiring patent.
The term of a U.S. patent may also be eligible for patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act, to account for at least some of the time the drug is under development and regulatory review after the patent is granted. With regard to a drug for which FDA approval is the first permitted marketing of the active ingredient, the Hatch-Waxman Act allows for extension of the term of one U.S. patent that includes at least one claim covering the composition of matter of an FDA-approved drug, an FDA-approved method of treatment using the drug, and/or a method of manufacturing the FDA-approved drug. The extended patent term cannot exceed the shorter of five years beyond the non-extended expiration of the patent or 14 years from the date of the FDA approval of the drug. Some foreign jurisdictions, including Europe and Japan, also have patent term extension provisions, which allow for extension of the term of a patent that covers a drug approved by the applicable foreign regulatory agency. In the future, if and when our pharmaceutical products receive FDA approval, we expect to apply for patent term extension on patents covering those products, their methods of use, and/or methods of manufacture.
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. Companies typically rely on trade secrets to protect aspects of their business that are not amenable to, or that they do not consider appropriate for, patent protection. We protect trade secrets, if any, and know-how by establishing confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and partners. These agreements provide that all confidential information developed or made known during the course of an individual or entity’s relationship with us must be kept confidential during and after the relationship. These agreements also generally provide that all relevant inventions resulting from work performed for us or relating to our business and conceived or completed during the period of employment or assignment, as applicable, shall be our exclusive property. In addition, we take other appropriate precautions, such as physical and technological security measures, to guard against misappropriation of our proprietary information by third parties.
Employees
As of December 31, 2023, we had 19 employees, 18 of whom were full-time and one part-time. We have never had a work stoppage and none of our employees are covered by collective bargaining agreements. We believe our employee relations are good.
Information About Our Executive Officers
The following table sets forth information as of March 15, 2024 regarding each of our current executive officers:
|
Name |
Age |
Positions |
||
|
Rick Pauls |
52 |
President and Chief Executive Officer, Director |
||
|
Lorianne Masuoka, M.D. |
62 |
Chief Medical Officer |
||
|
Scott Kellen |
58 |
Chief Financial Officer and Secretary |
||
|
Dominic Cundari |
73 |
Chief Commercial Officer |
||
|
David Wambeke |
40 |
Chief Business Officer |
||
|
Julie VanOrsdel Daves, MSHS, CCRP |
50 |
Senior Vice President, Clinical Development Operations |
The present principal occupations and recent employment history of each of our executive officers are set forth below.
Rick Pauls was appointed our President and Chief Executive Officer in January 2010. Mr. Pauls has served as a member of our Board of Directors since April 2005 and the Chairman of the Board from April 2008 to July 2014. Prior to joining DiaMedica, Mr. Pauls was the Co-Founder and Managing Director of CentreStone Ventures Inc., a life sciences venture capital fund, from February 2002 until January 2010. Mr. Pauls was an analyst for Centara Corporation, another early stage venture capital fund, from January 2000 until January 2002. From June 1997 until November 1999, Mr. Pauls worked for General Motors Acceptation Corporation specializing in asset-backed securitization and structured finance. Mr. Pauls previously served as an independent member of the board of directors of LED Medical Diagnostics, Inc. Mr. Pauls received his Bachelor of Arts in Economics from the University of Manitoba and his M.B.A. in Finance from the University of North Dakota.
Lorianne Masuoka, M.D. joined DiaMedica as our Chief Medical Officer in January 2024. Prior to joining DiaMedica, Dr. Masuoka served as the Chief Medical Officer of Epygenix Therapeutics, a clinical-stage pharmaceutical company focused on the development of new drugs for the treatment of intractable, rare genetic epilepsies from May 2022 through December 2023. Prior to Epygenix, Dr. Masuoka served as an independent clinical development consultant for several biopharmaceutical companies and as the Chief Medical officer of Marinus Pharmaceuticals From April 2017 through November 2019. Dr. Masuoka served as the Chief Medical Officer or acting Chief Medical Officer at Invivo Therapeutics, from March 2015 through July 2017, Cubist Pharmaceuticals (now Merck) from July 2013 through January 2015 and Nektar Therapeutics from June 2009 through August 2011. Previously, she held various roles of increasing responsibility at FivePrime Therapeutics (now Amgen) and Chiron (now Novartis). In addition to her executive roles, Dr. Masuoka most recently served as a Board member at Pfenex Inc. (now Ligand) and served as a Board member at Opiant Pharmaceuticals (now Indivior). Dr. Masuoka received her medical degree from the University of California, Davis, where she also completed her residency in neurology. She completed her epilepsy fellowship at Yale University and is board certified by the American Boards of Psychiatry and Neurology.
Scott Kellen joined DiaMedica as our Vice President of Finance in January 2018 and was appointed our Chief Financial Officer and Secretary in April 2018. Prior to joining DiaMedica, Mr. Kellen served as Vice President and Chief Financial Officer of Panbela Therapeutics, Inc., formerly known as Sun BioPharma, Inc., a publicly traded clinical stage drug development company, from October 2015 until April 2018. From February 2010 to September 2015, Mr. Kellen served as Chief Financial Officer and Secretary of Kips Bay Medical, Inc., a publicly traded medical device company, and became Chief Operating Officer of Kips Bay in March 2012. From November 2007 to May 2009, Mr. Kellen served as Finance Director of Transoma Medical, Inc. From 2005 to October 2007, Mr. Kellen served as Corporate Controller of ev3 Inc. From March 2003 to April 2005, Mr. Kellen served as Senior Manager, Audit and Advisory Services of Deloitte & Touche, LLP. Altogether, Mr. Kellen has spent more than 25 years in the life sciences industry, focusing on publicly traded early stage and growth companies. Mr. Kellen has a Bachelor of Science degree in Business Administration from the University of South Dakota and is a Certified Public Accountant (inactive).
Dominic Cundari joined DiaMedica as our Chief Commercial Officer in February 2022. Mr. Cundari has over 30 years of pharmaceutical experience in various commercial roles in high growth markets. Prior to joining DiaMedica, Mr. Cundari served as an independent commercial strategy and development consultant for Genentech, a global biotechnology company, since February 2009. From January 1988 to January 2009, Mr. Cundari held a variety of sales and marketing management positions across multiple medical specialties at Genentech. As Senior Director for the Vascular Franchise, Mr. Cundari was responsible for shaping commercial strategies, leading product launches in cardiology, pulmonary and neurology specialties and establishing strategic partnerships with telemedicine companies. Mr. Cundari holds both a Master of Science and Bachelor of Arts in Psychology from Villanova University.
David Wambeke joined DiaMedica as our Chief Business Officer in April 2023. Prior to joining DiaMedica, Mr. Wambeke served as Partner and Managing Director of Investment Banking at Craig-Hallum Capital Group, LLC, a growth focused investment bank. Mr. Wambeke joined Craig-Hallum in May 2007 and was involved in more than one hundred financing and M&A transactions with a focus on the life sciences and biotech industries. Prior to joining Craig-Hallum, Mr. Wambeke was enlisted in the U.S. Army and served as an artilleryman and military police officer. During a deployment in Baghdad, Iraq, in support of Operation Iraqi Freedom, Mr. Wambeke was wounded in combat and awarded the Purple Heart. Mr. Wambeke received a B.S. from the University of Minnesota.
Julie VanOrsdel Daves, MSHS, CCRP joined DiaMedica as our Senior Vice President, Clinical Development Operations in September 2022. Prior to joining DiaMedica, Ms. Daves served as the Vice President and Global Head of Clinical Operations of Sanifit Therapeutics, S.A., a clinical-stage biopharmaceutical company focused on treatments for progressive vascular calcification disorders, from September 2021 through August 2022. Ms. Daves also served as President and Principal Consultant at JVD Pharma Consulting, LLC, a consulting services company specializing in clinical operations and outsourcing, from February 2018 to August 2022. Ms. Daves served as Vice President of Outsourcing and Vendor Management for Edgewise Therapeutics, Inc., a Nasdaq-listed clinical-stage biopharmaceutical company, from May 2021 to August 2021 and served as Edgewise’s Executive Director and Head of Clinical Operations from April 2020 to May 2021. Prior to Edgewise, from February 2018 to April 2020, Ms. Daves served as Senior Director, Clinical Operations & Head of Outsourcing for miRagen Therapeutics, Inc., a development-stage biotechnology company. From February 2016 to February 2018, Ms. Daves served as Global Head and Senior Director of Clinical Vendor Management of Chiltern International Inc., a contract research organization, and from November 2015 to February 2016, she worked as the Director of Clinical Operations. Prior to her time at Chiltern, Ms. Daves worked for Array Biopharma, Inc. as the Director of Clinical Operations & Development Outsourcing from October 2014 to November 2015, as Associate Director of Clinical Outsourcing and Operations from October 2011 to October 2014, and as Clinical Principal Research Manager from January 2011 to October 2011. Ms. Daves worked as a Senior Manager of Clinical Development for BioCryst Pharmaceuticals, Inc. from April 2007 to January 2011, a Study Manager and Senior Clinical Research Associate for Cellgate Pharmaceuticals, Inc., from July 2006 to April 2007, and as a Clinical Project Manager for Inveresk/Charles River Clinical/Kendle from December 2002 to February 2005. Ms. Daves is a certified clinical research professional and received her MSHS in Clinical Research Administration from The George Washington University School of Medicine and Health Sciences and BS in Zoology from North Carolina State University.
Available Information
We are a corporation governed under British Columbia’s Business Corporations Act (BCBCA). Our company was initially incorporated pursuant to The Corporations Act (Manitoba) by articles of incorporation dated January 21, 2000. Our articles were subsequently amended several times, including on April 11, 2016 to continue the Company from The Corporations Act (Manitoba) to the Canada Business Corporations Act (CBCA) and on May 31, 2019, to continue our existence from a corporation incorporated under the CBCA into British Columbia under the BCBCA.
Our registered office is located at 301-1665 Ellis Street, Kelowna, British Columbia, Canada V1Y 2B3 and our principal executive office is located at our wholly owned subsidiary, DiaMedica USA Inc., located at 301 Carlson Parkway, Suite 210, Minneapolis, Minnesota, USA 55305. Our telephone number is 763-496-5454. Our internet website address is http://www.diamedica.com. Information contained on our website does not constitute part of this report.
We make available, free of charge and through our Internet web site, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and any amendments to any such reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, as soon as reasonably practicable after we electronically file such material with, or furnish it to, the Securities and Exchange Commission (SEC). Reports filed with the SEC may be viewed at www.sec.gov.
|
Item 1A. |
Risk Factors |
Below are the material factors known to us that could materially adversely affect our business, operating results, financial condition, prospects or share price. The summary of risk factors is not complete and should be read in conjunction with the more complete and detailed descriptions of risk factors that follow. Risks and uncertainties not currently known to us or that we currently deem to be immaterial also may materially adversely affect our business, operating results, financial condition, prospects or share price.
Risk Factors Summary
Risks Related to Our ReMEDy2 Trial, Future Clinical Trials and DM199 Product Candidate
|
● |
We have had and may continue to have difficulty engaging clinical trial sites for, or enrolling patients in, the trial or we may experience other clinical testing delays or setbacks, which would delay our ability to obtain regulatory approval for DM199 to treat AIS and commercialize it, or partner with a third party to obtain regulatory approval for or commercialization of DM199 to treat AIS, which would substantially harm our business and prospects. |
|
● |
The COVID-19 pandemic adversely impacted hospital and medical facilities, causing, among other things, staffing shortages, which have previously delayed site activations and patient enrollments in our ReMEDy2 trial and could continue to adversely affect the trial. |
|
● |
The adaptive design of our ReMEDy2 trial could result in the trial being required to enroll more patients than anticipated, increasing the time and costs to complete the trial, which may require additional funding that may not be available to us on acceptable terms or at all. |
|
● |
If our ReMEDy2 trial fails to adequately demonstrate the safety and efficacy of DM199 to treat AIS, we will not be able to obtain the regulatory approvals required to market and commercialize DM199 to treat AIS, which would substantially harm our business and prospects. |
|
● |
We may be required to suspend, repeat or terminate our ReMEDy2 trial or future clinical trials if they are deemed not conducted in accordance with regulatory requirements, the results are negative or inconclusive, or the trial is not well designed. |
Risks Related to Our Financial Position and Need for Additional Capital
|
● |
Since we have no revenue from product sales and do not expect any revenue from product sales for at least three or four years, we will likely need additional funding to continue our clinical development activities and other operations, which may not be available to us on acceptable terms, or at all. |
|
● |
We have incurred substantial losses since our inception and expect to continue to incur substantial losses for at least three or four years and may never become profitable, or if achieved, be able to sustain profitability. |
|
● |
Adverse developments with respect to the stability of financial institutions we do business with, or unstable banking, credit and/or capital market conditions generally, or the perception thereof, could adversely affect our ability to access our cash on deposit with financial institutions, obtain additional financing, or meet our liquidity requirements. |
Risks Related to Governmental and Regulatory Compliance and Approvals
|
● |
The regulatory approval process is expensive, time-consuming and uncertain and may prevent us or any future partner or collaborator from obtaining approvals for the commercialization of DM199 or any future product candidate. |
|
● |
Any product candidate for which we or any future partner or collaborator obtains marketing approval could be subject to post-marketing restrictions or recall or withdrawal from the market, and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with the product candidate. |
Risks Related to Our Reliance on Third Parties
|
● |
We rely on third parties to support the planning, execution and/or monitoring of our preclinical and clinical trials, and their failure to perform as required could cause delays in completing our product development and substantially harm to our business. |
|
● |
We rely on contract manufacturers over whom we have limited control. |
|
● |
Future development collaborations are expected to be important to us. |
Risks Related to Intellectual Property
|
● |
We could lose important intellectual property rights that we currently license from a third party if we fail to comply with our obligations under the license agreements under which we license intellectual property rights from this third party or otherwise experience disruptions to our business relationships with our licensor. |
|
● |
We may be unable to adequately protect our technology and enforce our intellectual property rights. |
|
● |
We or a future partner may require additional third-party licenses to effectively develop, manufacture and commercialize DM199, or any future product candidate, and such licenses might not be available on commercially acceptable terms, or at all. |
|
● |
Changes in patent law and its interpretation could diminish the value of our patents. |
|
● |
Intellectual property litigation may be expensive, time consuming and may cause delays in the development, manufacturing and commercialization of DM199 or any future product candidate. |
Risks Related to Human Capital Management
|
● |
We rely heavily on the capabilities and experience of our key executives, clinical personnel and advisors and the loss of any of them could affect our ability to develop DM199 or any future product candidate. |
|
● |
We will likely need to expand our operations and increase the size of our Company and we may experience difficulties in managing our growth. |
Risks Related to the Future Commercialization of DM199 or Any Future Product Candidate
|
● |
The successful commercialization of DM199 or any future product candidate, if approved, will depend on achieving market acceptance and we may not be able to gain sufficient acceptance to generate significant revenue. |
|
● |
If we fail to obtain coverage and adequate reimbursement for DM199 or any future product candidate, its revenue-generating ability will be diminished and there is no assurance that the anticipated market for the product will develop or be sustained. |
|
● |
We or any future partner will likely face competition from other biotechnology and pharmaceutical companies, many of which have substantially greater resources than us. |
|
● |
Our DM199 product candidate may face competition sooner than expected. |
Risks Related to Our Common Shares
|
● |
Our common share price has been volatile and may continue to be volatile. |
|
● |
We do not have a history of a very active trading market for our common shares. |
|
● |
We may issue additional common shares resulting in share ownership dilution. |
|
● |
If there are substantial sales of our common shares or the perception that such sales may occur, the market price of our common shares could decline. |
Risks Related to Our Jurisdiction of Organization
|
● |
We are governed by the corporate laws of British Columbia, which in some cases have a different effect on shareholders than the corporate laws in effect in the United States. |
|
● |
We were classified as a “passive foreign investment company” for 2022 and 2023 and may continue to be in future taxable years, which may have adverse U.S. federal income tax consequences for U.S. shareholders and adversely affect the level of interest in our common shares by U.S. investors. |
Risks Related to Our ReMEDy2 Trial, Future Clinical Trials and DM199 Product Candidate
We have had and may continue to have difficulty engaging clinical trial sites for, or enrolling patients in, our ReMEDy2 trial or we may experience other clinical testing delays or setbacks, which would delay our ability to obtain regulatory approval for DM199 to treat AIS and commercialize it, or partner with a third party to obtain regulatory approval for or commercialization of DM199 to treat AIS, which would substantially harm our business and prospects.
We have had and may continue to have difficulty engaging clinical trial sites for, and enrolling patients in, the ReMEDy2 trial, which could delay the completion of the trial. Concerns regarding the prior clinically significant hypotension events and circumstances surrounding the clinical hold which was lifted in June 2023 may add to these difficulties. In addition, it is possible that we may experience other clinical testing delays or setbacks, which would further delay the completion of the ReMEDy2 trial. Our product development costs will increase if we experience delays in clinical testing. Significant clinical trial delays could not only extend the time period for obtaining regulatory approval of DM199 to treat AIS, but also shorten any periods during which we or a future partner may have the exclusive right to commercialize DM199 to treat AIS or allow our competitors to bring competitive products to market before us, which would impair the ability to successfully commercialize DM199 and may harm our financial condition, results of operations and prospects. The ReMEDy2 trial may be delayed for a number of reasons, including among others:
|
● |
concerns regarding the prior clinically significant hypotension events and circumstances surrounding the clinical hold which was lifted in June 2023; |
|
● |
patients choosing to participate in competing clinical trials or not at all; |
|
● |
scheduling conflicts with participating clinicians and clinical sites; |
|
| ● | complexities in setting up and coordinating with sites that are located outside the United States and additional risks involved in a trial that is being conducted, in part, outside the United States; |
|
● |
suspension or termination of the ReMEDy2 trial by regulators for any reason, including concerns about patient safety or failure of our contract manufacturers to comply with current Good Manufacturing Practices (cGMP) requirements; |
|
● |
any changes to our manufacturing process that may be necessary or desired which affect our ability to produce adequate or timely clinical drug supply; |
|
● |
delays or failure to obtain clinical drug supply of DM199 from contract manufacturers necessary to conduct clinical trials; |
|
● |
our DM199 product candidate demonstrating a lack of safety or efficacy at the planned interim analysis of the ReMEDy2 trial; |
|
● |
patients failing to enroll or complete the ReMEDy2 trial at the rates and within the timelines we expect due to dissatisfaction with the treatment, side effects or other reasons; |
|
● |
clinical investigators not performing the ReMEDy2 trial on their anticipated schedule, dropping out of a trial or employing methods not consistent with the clinical trial protocol and regulatory requirements or other third parties not performing data collection and analysis in a timely or accurate manner; |
|
● |
inspections of our clinical trial sites by regulatory authorities, Institutional Review Boards (IRBs) or ethics committees finding regulatory violations that require us to undertake corrective action, resulting in suspension or termination of one or more sites or the imposition of another clinical hold on the IND for our ReMEDy2 trial; or |
|
● |
public health crises, epidemics or pandemics, such as COVID-19, which may adversely impact our ability to engage and activate clinical trial sites, recruit or enroll subjects for our ReMEDy2 trial or any future trial and obtain the requisite staffing for our ReMEDy2 trial or any future trial. |
Our product development costs may also increase if we need to perform more or larger clinical trials than planned. Additionally, changes in regulatory requirements and policies may occur, and we may need to amend trial protocols or alter our manufacturing processes to reflect these changes. Amendments typically require us to resubmit our trial protocols to regulatory authorities and IRBs or ethics committees for re-examination, which may impact the cost, timing or successful completion of our ReMEDy2 trial. Delays or increased product development costs or any of these events would likely have a material adverse effect on our business, financial condition, and prospects.
The COVID-19 pandemic adversely impacted hospital and medical facilities, causing, among other things, staffing shortages, which have previously delayed site activations and patient enrollments in our ReMEDy2 trial and could continue to adversely affect the trial.
COVID-19 has had, and may continue to have, a severe effect on the clinical trials of many drug candidates, including our ReMEDy2 trial. Prior to the clinical hold of our ReMEDy2 trial, we experienced challenges with engaging and activating clinical trial sites. We believe this was due primarily to clinical staff shortages resulting from layoffs and employee burnout, the reallocation of clinical nurses to COVID-19 care, particularly during surges in COVID-19 cases, a loss of study coordinators resulting from budget constraints and COVID-19 vaccination requirements. Hospital study sites have been especially impacted by these factors. Additionally, prior to the clinical hold of our ReMEDy2 trial, we experienced slower than expected enrollments in the trial due to these factors and patient concerns related to visiting clinical trial sites or being visited by clinical study nurses. In an effort to mitigate the impact of these factors, we have worked with our contract research organization to develop alternative procedures to support study sites and potential participants as needed. We intend to continue to monitor the results of these efforts or implement additional actions to mitigate the impact of these factors on our ReMEDy2 trial. It is also possible that these efforts may draw our employees away from their core responsibilities and create additional expenses, which may adversely affect our business and results of operations. Note however that these efforts may not be effective if patients are unwilling to enroll in our ReMEDy2 trial. We anticipate that COVID-19, and variants of COVID-19, will likely continue to adversely affect our ability to initiate new clinical trial sites and recruit or enroll subjects, and we cannot provide any assurance that we will be able to resolve these issues. Although the severity of the COVID-19 virus has decreased significantly during the past two years, the extent to which COVID-19 may impact our ReMEDy2 trial will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the emergence of new variants, the duration and severity of each variant, and the effectiveness of actions to contain, treat and prevent COVID-19, including the availability, effectiveness and acceptance of vaccines and vaccine booster shots. The resurgence of COVID-19 caused by any new variants in the future or another pandemic could cause us to experience continued and/or additional disruptions that could severely impact our ReMEDy2 trial, as well as our business.
The adaptive design of our ReMEDy2 trial could result in the trial being required to enroll more patients than anticipated, increasing the time and costs to complete the trial, which may require additional funding that may not be available to us on favorable terms or at all.
Our ReMEDy2 trial is currently targeted to enroll approximately 350 patients at up to 100 sites globally. However, with the trial’s adaptive design, it is possible that the number of patients required to complete the trial may increase significantly. If we are required to enroll more patients than currently anticipated, it will increase the time and costs to complete the trial, which may result in a need for additional funding that may not be available to us on acceptable terms, or at all.
If our ReMEDy2 trial fails to adequately demonstrate the safety and efficacy of DM199 to treat AIS, we will not be able to obtain the regulatory approvals required to market and commercialize the product, which would substantially harm our business and prospects.
Before obtaining marketing approval from regulatory authorities for the sale of DM199 to treat AIS, we must demonstrate the safety and efficacy of DM199 to treat AIS to a level acceptable to the FDA or similar regulatory bodies other jurisdictions. Clinical testing is expensive and difficult to design and implement, can take many years to complete and has uncertain outcomes. The outcome of preclinical trials and early clinical trials may not predict the success of later clinical trials, and the interim results of ReMEDy2 may not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in advanced clinical trials due to lack of efficacy or unacceptable safety profiles, including the emergence of undesirable side effects, notwithstanding promising results in earlier trials. We do not know whether our ReMEDy2 trial by itself will demonstrate adequate efficacy and safety to support regulatory approvals to market DM199 to treat AIS in the United States, or in any jurisdiction, or that a second confirmatory trial will be required. A product candidate may fail for safety or efficacy reasons at any stage of the testing process. In addition, the patient populations in our current clinical trial for DM199, and anticipated future clinical trials for DM199, often have co-morbidities that may cause severe illness or death, which may be attributed to DM199 in a manner that negatively affects the safety profile of our DM199 product candidate. If the results of our ReMEDy2 trial are inconclusive with respect to efficacy, if we do not meet our clinical endpoints with statistical significance or if there are unanticipated safety concerns or adverse events that emerge during the ReMEDy2 trial or other clinical trials, such as the events that caused the FDA to place the prior clinical hold on the IND for our ReMEDy2 trial, we may be prevented from or delayed in obtaining marketing approval, and even if we obtain marketing approval, any sales of DM199 for the treatment of AIS may be limited.
We may be required to suspend, repeat or terminate our ReMEDy2 trial or other clinical trials if they are deemed not conducted in accordance with regulatory requirements, the results are negative or inconclusive or the trial is not well designed.
Clinical trials must be conducted in accordance with the FDA’s current Good Clinical Practice (cGCP) requirements, or analogous requirements of applicable foreign regulatory authorities, and designed to provide statistically significant evidence predictive of patient benefit. Clinical trials are subject to oversight by the FDA, other foreign governmental agencies and IRBs or ethics committees at the trial sites where the clinical trials are conducted. In addition, clinical trials must be conducted with product candidates produced in accordance with applicable cGMP. Clinical trials may be suspended by us or by the FDA, other foreign regulatory authorities or by an IRB or ethics committee with respect to a particular clinical trial site, for various reasons, including:
|
● |
deficiencies in the conduct of the clinical trials, including failure to conduct the clinical trial in accordance with regulatory requirements or trial protocols; |
|
● |
deficiencies in the clinical trial operations or trial sites; |
|
● |
unforeseen adverse side effects or the emergence of undue risks to trial subjects; |
|
● |
deficiencies in the trial design necessary to demonstrate efficacy; |
|
● |
the product candidate may not appear to offer benefits over current therapies; or |
|
● |
the quality or stability of the product candidate may fall below acceptable standards. |
The design and implementation of clinical trials is a complex process. As a Company, we have limited experience designing and implementing clinical trials. We may not successfully or cost-effectively design and implement clinical trials that achieve our desired clinical endpoints. A clinical trial that is not well designed or that yields unforeseen adverse side effects or undue risks to trial subjects may delay or even prevent initiation of the trial, can lead to increased difficulty in site activations and enrolling patients, may make it more difficult to obtain regulatory approval for the product candidate on the basis of the trial results or, even if a product candidate is approved, could make it more difficult to commercialize the product successfully or obtain reimbursement from third party payers. Additionally, a trial that is not well designed or that yields unforeseen adverse side effects or undue risks to trial subjects could be delayed and more expensive than it otherwise would have been, or we may incorrectly estimate the costs to complete the clinical trial, which could lead to a shortfall in funding. We can provide no assurance that our ReMEDy2 trial or any other clinical trial conducted by us has been or will be designed and implemented successfully or achieve its desired clinical endpoints.
Our prospects depend on the clinical and commercial success of our DM199 product candidate which is in the clinical stage of development.
We are highly dependent on the success of DM199 and we may not be able to successfully obtain regulatory or marketing approval for, or successfully commercialize, this product candidate. To date, we have expended significant time, resources and effort on the development of DM199, including conducting preclinical and clinical trials, for the treatment of AIS and CRD. DM199 requires significant additional clinical testing and investment prior to seeking marketing approval. A commitment of substantial resources by ourselves and any potential partner or collaborator to continue to conduct the clinical trials for DM199 will be required to obtain required regulatory approvals and successfully commercialize this product candidate. Although we intend to study the use of DM199 to treat multiple diseases, we have no other product candidates in our current clinical development pipeline, with the exception of our new product candidate, DM300, which is in the early, preclinical stage of development and is intended to treat other inflammatory diseases, such as acute pancreatitis. Our ability to generate revenue from product sales and to achieve commercial success with DM199 will depend almost entirely on our ability to demonstrate sufficient safety and efficacy to obtain regulatory approval for DM199. We may fail to complete required clinical trials successfully and not be able to obtain regulatory approvals or commercialize DM199. Competitors may develop alternative products and methodologies to treat the diseases or indications that we are pursuing, thus reducing or eliminating the anticipated competitive advantages of DM199. We do not know whether any of our product development efforts will prove to be effective, meet applicable regulatory standards required to obtain the requisite regulatory approvals, be capable of being manufactured at a reasonable cost or be successfully marketed. DM199 is not expected to be commercially viable for at least three or four years. In addition, although the only significant adverse events that have occurred to date in our clinical trials have been three unexpected instances of transient, clinically significant, hypotension (low blood pressure), it is possible that DM199 may be observed to cause undesirable side effects. Results of early preclinical and clinical research may not be indicative of the results that will be obtained in later stages of clinical research. If regulatory authorities do not approve DM199 for the treatment of AIS or any other indications, or if we fail to maintain regulatory compliance, we would be unable to commercialize DM199 and our business and results of operations would be harmed. If we do succeed in developing viable products from DM199, we will face many potential future obstacles, such as the need to develop or obtain manufacturing, sales and marketing and distribution capabilities.
The clinical success and commercial potential of our DM199 product candidate will depend on a number of factors, many of which are beyond our control.
The clinical success and commercial potential of our DM199 product candidate to treat AIS or any other indication will depend on a number of factors, many of which are beyond our control, including, among others:
|
● |
the timely initiation, continuation and completion of clinical trials, including our Phase 2/3 ReMEDy2 trial and future clinical trials for DM199, which will depend substantially upon requirements for such trials imposed by the FDA and other regulatory agencies and bodies; |
|
● |
our ability to demonstrate the safety and efficacy of DM199 to the satisfaction of the relevant regulatory authorities and/or third-party payers; |
|
● |
whether we are required by the FDA or other regulatory authorities to conduct additional clinical trials, and the scope and nature of such clinical trials, prior to or after approval to market our DM199 product candidate; |
|
● |
the timely receipt of necessary marketing approvals from the FDA and foreign regulatory authorities, as well as achieving adequate pricing and reimbursement determinations; |
|
● |
the ability to successfully commercialize DM199, if approved by the FDA or foreign regulatory authorities, whether alone or in collaboration with others; |
|
● |
our ability and the ability of third-party manufacturers to manufacture the quantities of DM199, with quality attributes necessary to meet regulatory requirements, sufficient to meet anticipated demand and at a cost that allows us or a future partner to achieve profitability; |
|
● |
acceptance of DM199, if approved, as safe and effective by patients and healthcare providers; |
|
● |
the achievement and maintenance of compliance with all regulatory requirements applicable to DM199 by us and our third-party manufacturers and supporting vendors; |
|
● |
the maintenance of an acceptable safety profile of DM199 following any approval; |
|
● |
the availability, perceived advantages, relative cost, relative safety and relative efficacy of alternative and competitive treatments; |
|
● |
our ability to provide approved product with an acceptable patient administration method; |
|
● |
our ability or the ability of a future partner to successfully enforce our intellectual property rights for DM199; and |
|
● |
our ability to avoid or succeed in defending any third-party patent interference or patent infringement claims. |
In addition, because the plastic bags we use in the IV administration of DM199 are made of PVC, certain countries have banned or limited the use of PVC in a manner that, unless we are able to find an alternative, may limit the salability of DM199 in certain countries, thereby decreasing our worldwide market opportunity. No assurance can be provided that we will ever be able to achieve profitability through the sale of, or royalties from, our DM199 product candidate. If we or any future partners or collaborators are not successful in obtaining approval for and commercializing DM199, or are delayed in completing those efforts, our business and operations would be substantially harmed.
Risks Related to Our Financial Position and Need for Additional Capital
Since we currently have no revenue from product sales and do not expect any revenue from product sales for at least three or four years, we will need additional funding to continue our clinical development activities and other operations, which may not be available to us on acceptable terms, or at all.
We expect we will need substantial additional capital to further our R&D activities, planned clinical trials and regulatory activities and to otherwise develop our DM199 product candidate to a point where it may be commercially sold. We expect our current cash resources of $52.9 million in cash, cash equivalents and marketable securities as of December 31, 2023 to be sufficient to allow us to continue our Phase 2/3 trial in patients with AIS and to otherwise fund our planned operations for at least the next 12 months from the date of issuance of the financial statements included in this report. However, the amount and timing of our future funding requirements will depend on many factors, including, among others:
|
● |
the rate of progress in the development of and the conduct of clinical trials with respect to DM199 or any future product candidates; |
|
● |
the timing and results of our ongoing development efforts, including in particular our Phase 2/3 ReMEDy2 trial; |
|
● |
the costs of our development efforts, including the conduct of clinical trials with respect to DM199 or any future product candidates; |
|
● |
the costs associated with identifying additional product candidates and the potential expansion of our current development programs or potential new development programs; |
|
● |
the costs necessary to obtain regulatory approvals for DM199 or any future product candidates; |
|
● |
the costs of developing and validating manufacturing processes for DM199 or any future product candidates; |
|
● |
the costs associated with being a U.S. public reporting company with shares listed on The Nasdaq Capital Market; |
|
● |
the costs we incur in the filing, prosecution, maintenance and defense of our intellectual property; and |
|
● |
the costs related to general and administrative support. |
We may require significant additional funds earlier than we currently expect, and there is no assurance that we will not need or seek additional funding prior to such time. We may elect to raise additional funds even before we need them if circumstances or market conditions for raising additional capital are favorable.
Since our inception, we have financed our operations primarily from public and private sales of equity securities, the exercise of warrants and stock options, interest income on funds available for investment and government grants and tax incentives. We expect to continue this practice for the foreseeable future. We do not have any existing credit facilities under which we could borrow funds. We may seek to raise additional funds through various sources, such as equity and debt financings, or through strategic collaborations and license agreements. We can give no assurances that we will be able to secure additional sources of funds to support our operations, or if such funds are available to us, that such additional financing will be sufficient to meet our needs or on terms acceptable to us. This is particularly true if we experience additional adverse events, if our clinical data is not positive, or economic and market conditions deteriorate.
Although we previously have been successful in obtaining financing through our equity securities offerings, there can be no assurance that we will be able to do so in the future. To the extent we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our shareholders will be diluted. Debt financing, if available, may involve agreements that include conversion discounts or covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through government or other third-party funding, marketing and distribution arrangements or other collaborations or strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. It is possible that financing will not be available or, if available, may not be on favorable terms. The availability of financing could be affected by many factors, including, among others:
|
● |
the results of our clinical trials and other scientific and clinical research; |
|
● |
our ability to obtain regulatory approvals; |
|
● |
market acceptance of DM199 or any future product candidates; |
|
● |
the state of the capital markets generally with particular reference to pharmaceutical, biotechnology and medical companies; |
|
● |
various events outside our control, including without limitation geopolitical events, such as the current war between Russia and Ukraine and the conflict between Israel and Hamas; |
|
● |
the status of strategic alliance agreements; and |
|
● |
other relevant commercial considerations. |
If adequate funding is not available, we may be required to implement cost reduction strategies; delay, reduce or eliminate one or more of our product development programs; relinquish significant rights to DM199 or future product candidates or obtain funds on less favorable terms than we would otherwise accept; and/or divest assets or cease operations through a merger, sale or liquidation of our Company.
We have incurred substantial losses since our inception and expect to continue to incur substantial losses for at least three or four years and may never become profitable, or if achieved, be able to sustain profitability.
We are a clinical stage biopharmaceutical company focused on the development of our DM199 product candidate. Investment in biopharmaceutical product development is highly speculative because it entails substantial upfront financial expenditures and significant risk that a product candidate will fail to prove effective, gain regulatory approval or become commercially viable. Additionally, there has been a general decline in the biotech sector since February 2021, which has further increased the risks associated with investment in biopharmaceutical product development. We do not have any products approved by regulatory authorities and have not generated any revenues from product sales to date, and do not expect to generate any revenue from the sale of products for at least three or four years. We have incurred significant R&D and other administrative expenses related to our ongoing operations and expect to continue to incur such expenses. As a result, we have not been profitable and have incurred significant operating losses in every reporting period since our inception. For the years ended December 31, 2023 and 2022, we incurred a net loss of $19.4 million and $13.7 million, respectively. As of December 31, 2023, we had an accumulated deficit of $115.6 million. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our shareholders’ equity and working capital. We expect to continue to incur substantial operating losses as we continue our R&D activities, planned clinical trials, including our Phase 2/3 ReMEDy2 trial, regulatory activities and other administrative expenses and to support the development of DM199 or any future product candidate to a point where it can be out-licensed or receives required regulatory approvals and may be commercially sold and we begin to recognize future product sales, or receive royalty payments, licensing fees and/or milestone payments sufficient to generate revenues to fund our continuing operations. We expect our operating losses to increase in the near term as we continue development of DM199 and the clinical trials required to seek regulatory approval for DM199, or any future product candidate. We are unable to predict the extent of any future losses or when we will become profitable, if ever. Our failure to become and remain profitable may depress the market price of our common shares and could impair our ability to raise capital, continue to develop DM199, or any future product candidate, expand our business and product offerings or continue our operations. Even if we do achieve profitability, we may not be able to sustain or increase profitability on an ongoing basis.
Adverse developments with respect to the stability of financial institutions we do business with, or unstable banking, credit and/or capital market conditions generally, or the perception thereof, could adversely affect our ability to access our cash on deposit with financial institutions, obtain additional financing, or meet our liquidity requirements.
Potential future disruptions in access to bank deposits or lending commitments due to bank failure, could materially and adversely affect our liquidity, our business, financial condition and stock price. The early 2023 closures of Silicon Valley Bank, Signature Bank and First Republic Bank and their placement into receivership with the Federal Deposit Insurance Corporation (FDIC) created bank-specific and broader financial institution liquidity risk and concerns. Although the depositors at these financial institutions have continued to have access to their funds, even those in excess of the standard FDIC insurance limits, future adverse developments with respect to specific financial institutions or the broader financial services industry may lead to market-wide liquidity shortages. Although we did not have deposits at Silicon Valley Bank, Signature Bank or First Republic Bank, the failure of any bank in which we deposit our funds could reduce the amount of cash we have available for our operations or delay our ability to access such funds. Any such failure may increase the possibility of a sustained deterioration of financial market liquidity, or illiquidity at clearing, cash management and/or custodial financial institutions. In the event we have a commercial relationship with a bank that has failed or is otherwise distressed, we may experience delays or other issues in meeting our financial obligations. If other banks and financial institutions enter receivership or become insolvent in the future in response to financial conditions affecting the banking system and financial markets, our ability to access our cash and cash equivalents and investments may be threatened and could have a material adverse effect on our business and financial condition. In addition, the ability of our suppliers, vendors, and others in which we do business to access their cash and cash equivalents and investments or to obtain any necessary financing to continue their respective businesses could be threatened, which in turn, could harm our business.
Risks Related to Governmental and Regulatory Compliance and Approvals
The regulatory approval process is expensive, time-consuming and uncertain and may prevent us or any future partner or collaborator from obtaining approvals for the commercialization of DM199 or any future product candidate.
The process of obtaining marketing approvals, both in the United States and abroad, is expensive and may take many years, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the product candidate involved. Our DM199 or any future product candidate, and the activities associated with their development and commercialization, including design, research, testing, manufacture, quality control, recordkeeping, labeling, packaging, storage, advertising, promotion, sale, distribution, import, export and reporting of safety and other post-market information, are subject to comprehensive regulation by the FDA, the EMA and other similar foreign regulatory agencies. Failure to obtain marketing approval for DM199 or any future product candidate will prevent us or any future partner or collaborator from commercializing the product candidate. We have only limited experience in filing and supporting the applications necessary to gain marketing approvals and expect to rely on a future partner, collaborator or third-parties to assist us in this process. Securing marketing approval requires the submission of extensive preclinical and clinical data and supporting information to regulatory authorities for each therapeutic indication to establish the product candidate’s safety and efficacy. Securing marketing approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the regulatory authorities. The FDA, EMA or other regulatory authorities may determine that DM199 or any future product candidate may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining marketing approval or prevent or limit its commercial use. One issue of which we are aware is that because the plastic bags we use in the IV administration of DM199 are made of PVC, certain countries have banned or limited the use of PVC in a manner that may limit our ability to conduct the trails in such countries, or in the future in the event we are able to obtain required regulatory approvals, may limit the salability of DM199 in certain countries, thereby decreasing our worldwide market opportunity. Additionally, the regulatory approval process and requirements can change substantially based on amendments to federal regulations, new or amended FDA guidance documents governing the regulatory approval process, and even changes in FDA approval priorities based on the government administration as was recently seen in response to the COVID-19 pandemic. As a result, any marketing approval we ultimately obtain may be limited or subject to restrictions or post-approval commitments that render the approved product not commercially viable. Our or any future partner’s inability to obtain regulatory approval for DM199 or any future product candidate, or if such approval is limited, could substantially harm our business.
Any product candidate for which we or any future partner or collaborator obtains marketing approval could be subject to post-marketing restrictions or recall or withdrawal from the market, and we may be subject to penalties if we fail to comply with regulatory requirements or if we experience unanticipated problems with the product candidate.
The FDA and other federal and state agencies, including the U.S. Department of Justice (DOJ), closely regulate compliance with all requirements governing prescription drug products, including requirements pertaining to marketing and promotion of drugs in accordance with the provisions of the approved labeling and manufacturing of products. The FDA and DOJ impose stringent restrictions on manufacturers’ communications regarding off-label use, , sales and marketing activities, transparency laws, and reimbursement obligations, which restrictions can change substantially based on new and/or amended government interpretations of regulatory priorities, new and/or amended federal regulations, and other external forces. If we do not market our products for their approved indications, we may be subject to enforcement action for off-label marketing. Violations of such requirements may lead to investigations alleging violations of the FDCA and other statutes, including the False Claims Act, the Anti-Kickback Statute, the Sunshine Act and other federal and state health care fraud and abuse laws, as well as state consumer protection laws.
Our or any future partner’s failure to comply with all regulatory requirements, or the later discovery of previously unknown adverse events or other problems with our products, manufacturers or manufacturing processes, may yield various results, including:
|
● |
litigation involving patients using our products; |
|
● |
restrictions on such products, manufacturers or manufacturing processes; |
|
● |
restrictions on the labeling or marketing of a product; |
|
● |
restrictions on product distribution or use; |
|
● |
requirements to conduct post-marketing studies or clinical trials; |
|
● |
warning or untitled letters; |
|
● |
withdrawal of the products from the market; |
|
● |
refusal to approve pending applications or supplements to approved applications that we submit; |
|
● |
recall of products; |
|
● |
fines, restitution or disgorgement of profits or revenues; |
|
● |
suspension or withdrawal of marketing approvals; |
|
● |
damage to relationships with any then current or potential partners; |
|
● |
unfavorable press coverage and damage to our or any future partner’s reputation; |
|
● |
refusal to permit the import or export of our products; |
|
● |
product seizure; or |
|
● |
injunctions or the imposition of civil or criminal penalties. |
Non-compliance by us or any future partner or collaborator with regulatory requirements regarding ongoing safety monitoring, or pharmacovigilance, and with requirements related to the development of products, can also result in significant financial penalties. Similarly, failure to comply with regulatory requirements regarding the protection of personal information can also lead to significant penalties and sanctions.
We may be unable to obtain FDA acceptance of INDs to commence future clinical trials in the United States or on the timelines we expect, and even if we are able to, the FDA may not permit us to proceed in a timely manner.
Prior to commencing additional clinical trials in the United States for DM199 or any future product candidate, we will be required to have an accepted IND for each product candidate and for each targeted indication. In April 2021, we filed, and in May 2021, the FDA accepted, an IND for the Phase 2/3 ReMEDy2 trial in patients with AIS. However, in July 2022, the FDA imposed a clinical hold on the IND under which we are conducting our Phase 2/3 ReMEDy2 trial, which clinical hold was subsequently lifted in June 2023. A submission of an IND may not necessarily result in the FDA allowing further clinical trials to begin and, once begun, issues, such as clinical holds, may arise that will require us to suspend or terminate such clinical trials. Additionally, even if relevant regulatory authorities agree with the design and implementation of the clinical trials set forth in an IND, these regulatory authorities may change their requirements in the future. Failure to obtain acceptance of any future INDs may cause the development of DM199 or any future product candidate to be delayed or terminated, which could materially and adversely affect our business and prospects.
We have received Fast Track designation for DM199 for the treatment of AIS, and we may seek such designation for other uses of DM199 or future product candidates. Fast Track designation may not lead to faster development or a faster FDA review or approval process, and it does not increase the likelihood that DM199 will receive marketing approval in the United States. Further, there is no guarantee we will be able to maintain such designation.
In September 2021, we received Fast Track designation from the FDA for DM199 for the treatment of AIS where tPA and/or mechanical thrombectomy are not indicated or medically appropriate. The FDA may grant Fast Track designation to a drug that is intended to treat a serious condition and nonclinical or clinical data demonstrate the potential to address unmet medical need. The FDA provides opportunities for more frequent interactions with the review team for a Fast Track product, including pre-IND meetings, end-of-phase 1 meetings and end-of-phase 2 meetings with the FDA to discuss study design, extent of safety data required to support approval, dose-response concerns and use of biomarkers. A Fast Track product may also be eligible for rolling review, where the FDA reviews portions of a marketing application before the sponsor submits the complete application.
However, Fast Track designation for DM199 may not result in a faster development process or a faster review or approval compared to products considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA. Any delay in the review process or in the approval of DM199 will delay revenue from potential sales and will increase the capital necessary to fund our development programs and operations. In addition, the FDA may rescind the Fast Track designation for DM199 if the FDA later determines that DM199 no longer meets the qualifying criteria for Fast Track designation.
Current and future legislation may increase the difficulty and cost for us and any future partner or collaborator to obtain marketing approval of and commercialize DM199 or any future product candidate and affect the prices we may obtain.
In the United States and many foreign jurisdictions, there have been a number of legislative and regulatory changes and proposed changes regarding the healthcare system and data privacy that could prevent or delay marketing approval of DM199 or any future product candidate, restrict or regulate post-approval activities and affect our ability to profitably sell DM199 or any future product candidate for which we obtain marketing approval.
Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. For example, the Affordable Care Act (“ACA”) enacted in the United States in 2010, and principally taking effect in 2014, included measures to change health care delivery, decrease the number of individuals without insurance, ensure access to certain basic health care services and contain the rising cost of care. This healthcare reform movement, including the enactment of the ACA, has significantly changed health care financing by both governmental and private insurers in the United States. With respect to pharmaceutical manufacturers, the ACA increased the number of individuals with access to health care coverage, including prescription drug coverage, but it simultaneously imposed, among other things, increased liability for rebates and discounts owed to certain entities and government health care programs, fees for the manufacture or importation of certain branded drugs and transparency reporting requirements under the Physician Payments Sunshine Act. In addition to the ACA, other federal health reform measures have been proposed and adopted in the United States. We expect that the ACA, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and additional downward pressure on the price that we may receive for any product, if approved. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payers.
The U.S. federal government has prioritized and will likely continue to prioritize policies targeting reducing drug prices and healthcare spending and are committed to lowering spending in federal government programs. The Inflation Reduction Act of 2022, which was signed into law on August 16, 2022, includes provisions aimed at lowering prescription drug costs for Medicare patients and reducing the federal government’s spending on prescription drugs by requiring certain prescription drug prices to be negotiated directly with the government, certain rebates to be paid by prescription drug companies, and certain spending caps to be implemented, among other measures. The implementation of cost containment measures or other healthcare reforms may prevent us or a future partner from being able to generate sufficient revenue, attain profitability or even commercialize at all DM199 or any future product candidate.
Future legislation in the United States, Europe or other countries, and/or regulations and policies adopted by the FDA, the EMA or comparable regulatory authorities, may increase the time and cost required for us or any future partners or collaborators to conduct and complete clinical trials of our current or any future product candidates.
The FDA and the European Medicines Agency (EMA) have each established regulations to govern the drug product development and approval process, as have other foreign regulatory authorities. The policies of the FDA, the EMA and other regulatory authorities may change. For example, in December 2016, the 21st Century Cures Act (Cures Act) was signed into law. The Cures Act, among other things, is intended to modernize the regulation of drugs and spur innovation, but not all of its provisions have yet been implemented. Additionally, the EMA issued Annex 1: the Manufacture of Sterile Medicinal Products which was effective August 15, 2023, intended to update standards to reflect change in regulatory and manufacturing environments and to remove ambiguity and inconsistencies in regulations governing the manufacture of sterile medicinal products.. We cannot predict what if any effect the Cures Act, Annex 1 or any existing or future guidance from the FDA, EMA or other regulatory authorities will have on the development of DM199 or any future product candidate.
Risks Related to Our Reliance on Third Parties
We rely and will continue to rely on third parties to support the planning, execution and/or monitoring of our preclinical and clinical trials, and their failure to perform as required could cause delays in completing our product development and substantial harm to our business.
We rely and will continue to rely on third parties to conduct a significant portion of our preclinical and clinical development activities. Preclinical activities include in vivo studies in specific disease models, pharmacology and toxicology studies and assay development. Clinical development activities include trial design, regulatory submissions, clinical site and patient recruitment, clinical trial monitoring, clinical data management and analysis, safety monitoring and project management. If there is any dispute or disruption in our relationship with third parties, or if they are unable to provide quality services in a timely manner and at a feasible cost, including as a result of staffing disruptions, our development programs may face delays. Further, if any of these third parties fail to perform as we expect or if their work fails to meet regulatory requirements, our clinical testing could be delayed, cancelled or rendered ineffective. This happened to us in the past and resulted in us commencing litigation against Pharmaceutical Research Associates Group B.V., which was acquired by ICON plc in July 2021 (PRA Netherlands), as a result of its handling of a double-blinded, placebo-controlled, single-dose and multiple-dose study to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics and proof of concept of DM199 in healthy subjects and in patients with Type 2 diabetes mellitus, as described later in this report, and could happen again.
We rely on contract manufacturers over whom we have limited control. If we are subject to quality, cost or delivery issues with the materials supplied by these or future contract manufacturers, we may be unable to produce adequate supplies of DM199 or any future product candidate, and our clinical and business operations could suffer significant harm.
Completion of our clinical trials and commercialization of our DM199 product candidate and any future product candidate require access to, or development of, facilities to manufacture our product candidates at sufficient yields and, ultimately, assuming approval, at commercial scale. Clinical and commercial drug product must be produced under applicable cGMP regulations. Failure of our CMOs to comply with these regulations may require us to repeat clinical trials, which would delay the regulatory approval process. We rely on CMOs for manufacturing, filling, labeling, packaging, storing and shipping DM199 in compliance with applicable cGMP regulations. The FDA and other regulatory agencies ensure the quality of drug products by carefully monitoring drug manufacturers’ compliance with cGMP regulations.
As a company, we have no direct experience in manufacturing or managing third parties in manufacturing our DM199 product candidate in the volumes that are expected to be necessary to support commercialization, if DM199 is approved. Our efforts to establish these capabilities may not meet our requirements as to scale-up, timeliness, yield, cost or quality in compliance with applicable cGMP regulations. We or any future partner or collaborator or our CMOs may encounter difficulties in production, which may include the following, among others:
|
● |
costs and challenges associated with scale-up and attaining sufficient manufacturing yields; |
|
● |
supply chain issues, including the timely availability and shelf life requirements of raw materials and supplies and the lack of redundant and backup suppliers; |
|
● |
quality control and assurance; |
|
● |
shortages of qualified personnel and capital required to manufacture large quantities of our product candidate; |
|
● |
competing capacity needs at CMOs supporting product development as quantities for supply increase; |
|
● |
establishment of commercial supply capacity through binding supply agreements or to do so on acceptable terms; |
|
● |
compliance with regulatory requirements that vary in each country where a product might be sold; |
|
● |
capacity limitations and scheduling availability in contracted facilities; and |
|
● |
natural disasters, cyberattacks, which could subject us to an increased regulatory burden and increased costs of compliance, or other force majeure events that affect CDMO facilities and possibly limit production or cause loss of product inventory. |
We do not have long-term supply agreements with any of our CMOs and we purchase our required supply on an order-by-order basis. There can be no assurances that our current CMOs or any future CMOs will be able to meet our timetable and requirements for our DM199 product candidate or any future product candidate. If we are unable to arrange for alternative third-party manufacturing sources on commercially reasonable terms or in a timely manner, we may be delayed in the development of DM199 or any future product candidate. Our dependence upon our current CMOs and any future CMOs for the manufacture of our product candidates may adversely affect our ability to develop our product candidates in a timely and competitive basis and, if we or a future partner are able to commercialize our product candidates, may adversely affect our revenues from product sales and significantly harm our business.
Future development collaborations are expected to be important to us. If we are unable to enter into or maintain these collaborations, or if these collaborations are not successful, our business could be adversely affected.
In the future, we intend to seek to collaborate with pharmaceutical and biotechnology companies for the development and/or commercialization of DM199. We face significant competition in seeking appropriate collaborators or partners. Our ability to reach a definitive agreement for any collaboration will depend, among other things, upon our assessment of the collaborator’s or partner’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s or partner’s evaluation of a number of factors. If we are unable to reach agreements with suitable collaborators or partners on a timely basis, on acceptable terms, or at all, we may have to curtail the development of and/or seek alternative means to commercialize our DM199 product candidate resulting in, among other things, reducing or delaying our development program, delaying our potential development schedule or reducing the scope of research activities. If we fail to enter into one or more collaborations and do not have sufficient funds or expertise to undertake the necessary development or commercialization activities, we may not be able to continue or further develop DM199 and our business may be materially and adversely affected.
Future collaborations we may enter into may involve significant risks, including, among others:
|
● |
collaborators may have significant discretion in determining the efforts and resources that they will apply to the collaboration; |
|
● |
collaborators may not perform their obligations as expected; |
|
● |
changes in the collaborators’ strategic focus or available funding, or external factors, such as an acquisition, may divert resources or create competing priorities; |
|
● |
collaborators may delay nonclinical or clinical development, provide insufficient funding for product development of targets selected by us, stop or abandon nonclinical or clinical development for a product candidate, or repeat or conduct new nonclinical and clinical development for a product candidate; |
|
● |
collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates if the collaborators believe that competitive products are more likely to be successfully developed than our products; |
|
● |
product candidates discovered in collaboration with us may be viewed by our future collaborators as competitive with their own product candidates or products, which may cause collaborators to cease to devote resources to the development of our product candidates; |
|
● |
disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of development, might cause delays or termination of the preclinical or clinical development or commercialization of product candidates, might lead to additional responsibilities for us with respect to product candidates, or might result in litigation or arbitration, any of which would be time-consuming and expensive; |
|
● |
collaborators may not properly maintain or defend our intellectual property rights or intellectual property rights licensed to us or may use our proprietary information in such a way as to invite litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation; |
|
● |
collaborators may infringe the intellectual property rights of third parties, which may expose us to litigation and potential liability; and |
|
● |
collaborations may be terminated for the convenience of the collaborator and, if terminated, we could be required to raise additional capital to pursue further development or commercialization of the applicable product candidates. |
If a collaborator terminates its agreement with us, we may find it more difficult to attract new collaborators and the way we are perceived in the business and financial communities could be adversely affected.
If our collaborations do not result in the successful development of DM199, or any future product candidate, development could be delayed, and we may need additional resources to develop DM199 or any future product candidates. All of the risks relating to product development, regulatory approval and commercialization described in this report also apply to the activities of our future collaborators.
Our inability to maintain contractual relationships with physicians could have a negative impact on our research and development.
We maintain contractual relationships with respected physicians in hospitals and universities who assist us in the design of our clinical trials and interpretation of trial results. If we are unable to enter into and maintain these relationships, our ability to develop, obtain required regulatory approvals for, and market our DM199 or any future product candidate could be adversely affected. In addition, it is possible that U.S. federal and state and international laws requiring us to disclose payments or other transfers of value, such as gifts or meals, to surgeons and other healthcare providers could have a chilling effect on the relationships with individuals or entities that may, among other things, want to avoid public scrutiny of their financial relationships with us.
Risks Related to Intellectual Property
We could lose important intellectual property rights that we currently license from a third party if we fail to comply with our obligations under the license agreements under which we license intellectual property rights from this third party or otherwise experience disruptions to our business relationships with our licensor.
We are a party to a license agreement relating to an expression system and cell line for use in the production of DM199 and DM300. We may need to obtain additional licenses from others to advance our R&D activities or allow the commercialization of DM199 or any other product candidates we may identify and pursue. Future license agreements may impose various development, diligence, commercialization and other obligations on us. If any of our current or future in-licenses are terminated, or if the underlying patents fail to provide the intended exclusivity, competitors or other third parties may gain access to technologies that are material to our business, and we may be required to cease our development and commercialization of DM199 or other product candidates that we may identify or to seek alternative manufacturing methods. However, suitable alternatives may not be available or the development of suitable alternatives may result in a significant delay in our commercialization of DM199. Any of the foregoing could have a material adverse effect on our competitive position, business, financial condition, results of operations and prospects.
Moreover, disputes may arise regarding intellectual property subject to a licensing agreement, including, among others:
|
● |
the scope of rights granted under the license agreement and other interpretation-related issues; |
|
● |
the extent to which, our product candidates, technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; |
|
● |
the sublicensing of patent and other rights under our collaborative development relationships; |
|
● |
our diligence obligations under the license agreement and what activities satisfy those diligence obligations; |
|
● |
the inventorship and ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; and |
|
● |
the priority of invention of patented technology. |
In addition, the agreements under which we currently license intellectual property or technology from a third party are complex and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology, or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations, and prospects. Moreover, if disputes over intellectual property that we have in-licensed prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates, which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
We may be unable to adequately protect our technology and enforce our intellectual property rights and our competitors may take advantage of our development efforts or acquired technology and compromise our prospects for marketing and selling DM199 or any future product candidate.
We believe that patents and other proprietary rights are key to our business. Our policy is to file patent applications to protect technology, inventions and improvements that may be important to the development of DM199 or any future product candidate. We also rely upon trade secrets, know-how and continuing technological innovations to develop and maintain our competitive position. We plan to enforce our issued patents and our rights to proprietary information and technology. We review third-party patents and patent applications, both to refine our own patent strategy and to monitor the landscape related to our technology.
Our success depends, in part, on our ability to secure and protect our intellectual property rights and to operate without infringing on the proprietary rights of others or having third parties circumvent the rights owned or licensed by us. We have a number of patents, patent applications and rights to patents related to our compounds, product candidates and technology, but we cannot be certain that they will be enforceable or provide adequate protection or that pending patent applications will result in issued patents.
To the extent that development, manufacturing and testing of our product candidates is performed by third party contractors, such work is performed pursuant to fee for service contracts. Under the contracts, all intellectual property, technology know-how and trade secrets related to our product candidate arising under such agreements are our exclusive property and must be kept confidential by the contractors. It is not possible for us to be certain that we have obtained from the contractors all necessary rights to such technologies. Disputes may arise as to the scope of the contract or possible breach of contract. No assurance can be given that our contracts will be enforceable or would be upheld by a court.
The patent positions of pharmaceutical and biotechnology firms, ourselves included, are uncertain and involve complex questions of law and fact for which important legal issues remain unresolved. Therefore, it is not clear whether our pending patent applications will result in the issuance of patents with commercially meaningful protections or at all, or whether we will develop additional proprietary products which are patentable. Part of our strategy is based on our ability to secure a patent position to protect our technology. There is no assurance that we will be successful in this approach and failure to secure adequate patent protection may have a material adverse effect upon us and our financial condition. Also, we may fail in our attempt to commercialize products using currently patented or licensed technology without having to license additional patents. Moreover, it is not clear whether the patents issued or to be issued will provide us with any competitive advantages or if any such patents will be the target of challenges by third parties, whether the patents of others will interfere with our ability to market our products, or whether third parties will circumvent our patents by means of alternate processes. Furthermore, it is possible for others to develop products that have the same effect as our product candidates or technologies on an independent basis or to design around technologies patented by us. Patent applications relating to or affecting our business may have been filed by pharmaceutical or biotechnology companies or academic institutions. Such applications may conflict with our technologies or patent applications and such conflict could reduce the scope of patent protection that we could otherwise obtain or even lead to the rejection of our patent applications. There is no assurance that we can enter into licensing arrangements on commercially reasonable terms or develop or obtain alternative technology in respect of patents issued to third parties that incidentally cover our products or production technologies. Any inability to secure licenses or alternative technology could result in delays in the introduction of some of our product candidates or even lead to us being prevented from pursuing the development, manufacture or sale of certain products. Moreover, we could potentially incur substantial legal costs in defending legal actions that allege patent infringement, or by initiating patent infringement suits against others. It is not possible for us to be certain that we are the creator of inventions covered by pending patent applications or that we were the first to invent or file patent applications for any such inventions. While we have used commercially reasonable efforts to obtain assignments of intellectual property from all individuals who may have created materials on our behalf (including with respect to inventions covered by our patents and pending patent applications), it is not possible for us to be certain that we have obtained all necessary rights to such materials. No assurance can be given that our patents, or patent applications if issued, would be upheld by a court, or that a competitor’s technology or product would be found to infringe on our patents. Moreover, much of our technology know-how that is not patentable may constitute trade secrets. Therefore, we require our employees, consultants, advisors and collaborators to enter into confidentiality agreements either as stand-alone agreements or as part of their employment or consulting contracts. However, no assurance can be given that such agreements will provide meaningful protection of our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure of confidential information. Also, while we have used commercially reasonable efforts to obtain executed copies of such agreements from all employees, consultants, advisors and collaborators, no assurance can be given that executed copies of all such agreements have been obtained.
We or a future partner may require additional third-party licenses to effectively develop, manufacture and commercialize DM199, or any future product candidate, and such licenses might not be available on commercially acceptable terms, or at all.
A substantial number of patents have already been issued to other biotechnology and pharmaceutical companies. To the extent that valid third-party patent rights cover our product candidates, we or any future collaborator, would be required to seek licenses from the holders of these patents in order to manufacture, use or sell our product candidates, and payments under them would reduce profits from our product candidates. We are currently unable to predict the extent to which we may wish or be required to acquire rights under such patents, the availability and cost of acquiring such rights, and whether a license to such patents will be available on acceptable terms, or at all. There may be patents in the United States or in foreign countries or patents issued in the future that are unavailable to license on acceptable terms. Our inability to obtain such licenses may hinder or eliminate our ability to develop, manufacture and market our product candidates and have a material adverse effect on our business, financial condition, results of operations, and prospects.
Changes in patent law and its interpretation could diminish the value of our patents in general, thereby impairing our ability to protect DM199 or any future product candidate.
As is the case with other biotechnology and pharmaceutical companies, our success is heavily dependent on intellectual property rights, particularly patents. Obtaining and enforcing patents in the biopharmaceutical industry involves technological and legal complexity, and obtaining and enforcing biopharmaceutical patents is costly, time consuming, and inherently uncertain. The U.S. Supreme Court has ruled on several patent cases in recent years, either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our or any licensors’ or collaborators’ ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents once obtained. Depending on decisions by the U.S. Congress, the federal courts, the U.S. Patent and Trademark Office (USPTO) and the European Patent Office (EPO), the laws and regulations governing patents could change in unpredictable ways that would weaken our or any licensors’ or collaborators’ ability to obtain new patents or to enforce existing patents and patents we or any licensors or collaborators may obtain in the future. Changes in either the patent laws or interpretation of the patent laws in the United States or other countries could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents.
Assuming that other requirements for patentability are met, prior to March 2013, in the United States, the first to invent the claimed invention was entitled to the patent, while outside the United States, the first to file a patent application was entitled to the patent. After March 2013, under the Leahy-Smith America Invents Act (the America Invents Act), enacted in September 2011, the United States transitioned to a first inventor to file system in which, assuming that other requirements for patentability are met, the first inventor to file a patent application is entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. A third party that files a patent application in the USPTO after March 2013, but before us could, therefore, be awarded a patent covering an invention of ours even if we had made the invention before it was made by such third party. This will require us to be cognizant of the time from invention to filing of a patent application. Since patent applications in the United States and most other countries are confidential for a period of time after filing or until issuance, we cannot be certain that we or any licensor were the first to either (i) file any patent application related to our product candidates or (ii) invent any of the inventions claimed in our or any licensor’s patents or patent applications.
The America Invents Act also includes a number of significant changes that affect the way patent applications will be prosecuted and also may affect patent litigation. These include allowing third party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent in USPTO-administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. Because of a lower evidentiary standard in USPTO proceedings compared to the evidentiary standard in United States federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. Therefore, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our owned or in-licensed patent applications and the enforcement or defense of our owned or in-licensed issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Intellectual property litigation may be expensive, time consuming and may cause delays in the development, manufacturing and commercialization of DM199 or any future product candidate.
Third parties may claim that we are using their proprietary information without authorization. Third parties may also have or obtain patents and may claim that technologies licensed to or used by us infringe their patents. If we are required to defend patent infringement actions brought by third parties, or if we sue to protect our own patent rights or otherwise to protect our proprietary information and to prevent its disclosure, we may be required to pay substantial litigation costs and managerial attention may be diverted from business operations even if the outcome is in our favor. In addition, any legal action that seeks damages or an injunction to stop us from carrying on our commercial activities relating to the affected technologies could subject us to monetary liability (including treble damages and attorneys’ fees if we are found to have willfully infringed) and require us or any third-party licensors to obtain a license to continue to use the affected technologies. We cannot predict whether we would prevail in any of these types of actions or that any required license would be available on commercially acceptable terms or at all. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources.
Competitors may infringe our patents or other intellectual property. If we were to initiate legal proceedings against a third party to enforce a patent covering our product candidates, the defendant could counterclaim that the patent covering our product candidate is invalid and/or unenforceable. In patent litigation in the United States, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, written description or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. The outcome following legal assertions of invalidity and unenforceability is unpredictable. Moreover, similar challenges may be made by third parties outside the context of litigation, e.g., via administrative proceedings such as post grant or inter partes review in the United States or via oppositions or other similar proceedings in other countries/regions.
Interference or derivation proceedings provoked by third parties or brought by us or declared by the USPTO may be necessary to determine the priority of inventions with respect to our patents or patent applications. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms or at all, or if a non-exclusive license is offered and our competitors gain access to the same technology. Our defense of litigation, validity or enforceability, interference or derivation proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees. In addition, the uncertainties associated with litigation or such other proceedings could have a material adverse effect on our ability to raise the funds necessary to continue our clinical trials, continue our research programs, license necessary technology from third parties, or enter into development partnerships that would help us bring our product candidates to market.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure. There could also be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the market price of our common shares.
Our reliance on third parties may require us to share our trade secrets, which increases the possibility that a competitor will discover them.
Because we rely on third parties to develop and manufacture our DM199 product candidate, we may share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, employment or consulting agreements or other similar agreements with our collaborators, advisors, employees, and consultants prior to beginning research or disclosing proprietary information. These agreements typically restrict the ability of our collaborators, advisors, employees, and consultants to publish data potentially relating to our trade secrets. In the future, we may also conduct joint R&D programs which may require us to share trade secrets under the terms of R&D collaboration or similar agreements. We cannot be certain that our current or any future agreements have been or will be entered into with all relevant parties. Moreover, despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements, independent development or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication. Trade secrets can be difficult to protect. If the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating any trade secrets. A competitor’s discovery of our trade secrets may impair our competitive position and could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Patent terms may be inadequate to protect the competitive position of DM199 or any future product candidate for an adequate amount of time.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. Certain extensions may be available, but the life of a patent, and the protection it affords, is limited. Even if patents covering our product candidates are obtained, once the patent life has expired, we may be open to competition from competitive products, including generics or biosimilars. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our owned and licensed patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may be subject to claims that our employees, consultants or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employees’ former employers or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to be paid to the USPTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and/or applications. We have systems in place to remind us to pay these fees, and we employ an outside firm and rely on our outside counsel to pay these fees due to non-U.S. patent agencies. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might be able to enter the market and this circumstance would have a material adverse effect on our business.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our product candidates in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and may also export infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States. These products may compete with our products and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Many countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of such patent. If we or any of our licensors is forced to grant a license to third parties with respect to any patents relevant to our business, our competitive position may be impaired, and our business, financial condition, results of operations, and prospects may be adversely affected.
Risks Related to Human Capital Management
We rely heavily on the capabilities and experience of our key executives, clinical personnel and advisors and the loss of any of them could affect our ability to develop DM199 or any future product candidate.
We depend heavily on members of our management team and certain other key personnel, including in particular our clinical personnel. We also depend on our clinical collaborators and advisors, all of whom have outside commitments that may limit their availability to us. In addition, we believe that our future success will depend in large part upon our ability to attract and retain highly skilled scientific, managerial, medical, clinical and regulatory personnel, particularly as we continue to expand our activities and seek regulatory approvals for clinical trials and eventually our DM199 product candidate. We enter into agreements with scientific and clinical collaborators and advisors, key opinion leaders and academic partners in the ordinary course of our business. We also enter into agreements with physicians and institutions that will recruit patients into our clinical trials on our behalf in the ordinary course of our business. Notwithstanding these arrangements, we face significant competition for these types of personnel from other companies, research and academic institutions, government entities and other organizations. We cannot predict our success in hiring or retaining the personnel we require for our continued growth. The loss of the services of any of our key executive officers, clinical personnel and advisors could potentially harm our business, operating results or financial condition.
We will likely need to expand our operations and increase the size of our Company and we may experience difficulties in managing our growth.
As we advance our DM199 product candidate through clinical trials, or develop any future product candidates, we expect to increase our product development, scientific, clinical, regulatory and compliance and administrative headcount to manage these programs. In furtherance of these efforts, we recently hired a new Chief Medical Officer and hired a Chief Business Officer during 2023. In addition, to continue to meet our obligations as a U.S. public reporting company, we will likely need to increase our general and administrative capabilities. Our management, personnel and systems currently in place may not be adequate to support this future growth. Our need to effectively manage our operations, growth and various projects requires that we:
|
● |
successfully attract and recruit new employees with the expertise and experience we will require; |
|
● |
manage our clinical programs effectively, which have been and will continue to be conducted at numerous clinical sites; |
|
● |
develop a marketing, distribution and sales infrastructure if we seek to market our products directly; and |
|
● |
continue to improve our operational, manufacturing, quality assurance, financial and management controls, reporting systems and procedures. |
If we are unable to successfully manage this growth and increased complexity of operations, our business may be adversely affected.
Risks Related to the Future Commercialization of DM199 or Any Future Product Candidate
The successful commercialization of DM199 or any future product candidate, if approved, will depend on achieving market acceptance and we may not be able to gain sufficient acceptance to generate significant revenue.
Even if DM199 or any future product candidate is successfully developed and receives regulatory approval, it may not gain market acceptance among physicians, patients, healthcare payers, such as private insurers or governments and other funding parties. The degree of market acceptance for DM199 or any product candidate we develop will depend on a number of factors including, among others:
|
● |
demonstration of sufficient clinical efficacy and safety; |
|
● |
the prevalence and severity of any adverse side effects; |
|
● |
limitations or warnings contained in the product’s approved labeling; |
|
● |
cost-effectiveness and availability of acceptable pricing; |
|
● |
the availability of alternative treatment methods and the superiority of alternative treatment methods; |
|
● |
the effectiveness of marketing and distribution methods and support for the product; and |
|
● |
coverage and reimbursement policies of government and third-party payers to the extent that the product could receive regulatory approval but not be approved for coverage by or receive adequate reimbursement from government and quasi-government agencies or other third-party payers. |
If we fail to obtain coverage and adequate reimbursement for DM199 or any future product candidate, its revenue-generating ability will be diminished and there is no assurance that the anticipated market for the product will develop or be sustained.
Our or any future partner’s ability to successfully commercialize DM199 or any future product candidate will depend, in part, on the extent to which coverage of and adequate reimbursement for such product and related treatments will be available from governmental health payer programs at the federal and state levels, including Medicare and Medicaid, private health insurers, managed care plans and other organizations. No assurance can be given that third-party coverage or adequate reimbursement will be available that will allow us or any future partner to obtain or maintain price levels sufficient for the realization of an appropriate return on our investment in product development. Coverage and adequate reimbursement are critical to new product acceptance by healthcare providers. There is no uniform coverage and reimbursement policy among third-party payers in the United States; however, private third-party payers may follow Medicare coverage and reimbursement policy in setting their own coverage policy and reimbursement rates. Additionally, coverage decisions may depend upon clinical and economic standards that disfavor new drug products when more established or lower cost therapeutic alternatives are or subsequently become available. Even if coverage is obtained for DM199 or any future product candidate, the related reimbursement rates might not be adequate to make the product attractive to providers, or may require patient cost sharing (e.g., copayments and/or deductibles) that patients find unacceptably high. In addition, healthcare reform and controls on healthcare spending may limit coverage of the product and the price we charge and get paid for the product and the volumes thereof that we can sell. Patients are unlikely to use DM199 or any future product candidate unless coverage is provided and reimbursement is adequate to cover a significant portion of its cost.
Outside of the United States, the successful commercialization of DM199 or any future product candidate will depend largely on obtaining and maintaining government coverage, because in many countries, patients are unlikely to use prescription drugs that are not covered by their government healthcare programs. Negotiating coverage and reimbursement with governmental authorities can delay commercialization by 12 months or more. Coverage and reimbursement policies may adversely affect our or a future partner’s ability to sell DM199 or any future product candidate on a profitable basis. In many international markets, governments control the prices of prescription pharmaceuticals, including through the implementation of reference pricing, price cuts, rebates, revenue-related taxes and profit control, and we expect prices of prescription pharmaceuticals to decline over the life of the product or as volumes increase.
We or any future partner will likely face competition from other biotechnology and pharmaceutical companies, many of which have substantially greater resources, and our DM199 product candidate may face competition sooner than expected and our financial condition and operations will suffer if we fail to compete effectively.
Technological competition is intense in the industry in which we operate. Development of new, potentially competitive therapies comes from pharmaceutical companies, biotechnology companies and universities, as well as companies that offer non-pharmaceutical solutions. Many of our competitors have substantially greater financial and technical resources; more extensive R&D capabilities; and greater marketing, distribution, production and human resources than we do. Moreover, competitors may develop products more quickly than us and may obtain regulatory approval for such products more rapidly than we do. Products and processes which are more effective than those that we intend to develop may be developed by our competitors. R&D by others may render our product candidates non-competitive or obsolete.
Our DM199 product candidate may face competition sooner than expected.
We believe that DM199 could qualify for 12 years of data exclusivity in the United States under the Biologics Price Competition and Innovation Act of 2009 (BPCIA), which was enacted as part of the ACA. Under the BPCIA, an application for a biosimilar product, or BLA, cannot be submitted to the FDA until four years, or if approved by the FDA, until 12 years, after the original brand product identified as the reference product is approved under a BLA. The BPCIA provides an abbreviated pathway for the approval of biosimilar and interchangeable biological products. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as “interchangeable” based on its similarity to an existing brand product. This law is complex and is only beginning to be interpreted and implemented by the FDA. While it is uncertain when any such processes may be fully adopted by the FDA, any such processes could have a material adverse effect on the future commercial prospects for DM199 or any future product candidate that is a biologic. There is also a risk that the U.S. Congress could repeal or amend the BPCIA to shorten this exclusivity period, potentially creating the opportunity for biosimilar competition sooner than anticipated after the expiration of our patent protection. Moreover, the extent to which a biosimilar, once approved, will be substituted for any reference product in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
Even if, as we expect, our DM199 product candidate is considered to be a reference product eligible for 12 years of exclusivity under the BPCIA, another company could market competing products if the FDA approves a full BLA for such product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity and potency of the products. Moreover, an amendment or repeal of the BPCIA could result in a shorter exclusivity period for our DM199 product candidate, which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
Risks Related to Our Common Shares
Our common share price has been volatile and may continue to be volatile.
Our common shares trade on The Nasdaq Capital Market under the trading symbol “DMAC.” During 2023, the sale price of our common shares ranged from $1.27 to $4.75 per share. A number of factors could influence the volatility in the trading price of our common shares, including changes in the economy or in the financial markets, industry related developments, such as a general decline in the biotech sector, and the impact of material events and changes in our operations, such as our clinical results including the prior clinical hold on the IND for our ReMEDy2 trial, operating results and financial condition. Each of these factors could lead to increased volatility in the market price of our common shares. In addition, the market prices of the securities of our competitors may also lead to fluctuations in the trading price of our common shares.
We do not have a history of a very active trading market for our common shares.
During 2023, the daily trading volume of our common shares ranged from 4,700 shares to 905,600 shares. Although we anticipate a more active trading market for our common shares in the future, we can give no assurance that a more active trading market will develop or be sustained. If we do not have an active trading market for our common shares, it may be difficult for you to sell our common shares at a favorable price or at all.
We may issue additional common shares resulting in share ownership dilution.
Future dilution will likely occur due to anticipated future equity issuances by us. To the extent we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our shareholders will be diluted. In addition, as of December 31, 2023, we had outstanding options to purchase 3,423,103 common shares, deferred stock units representing 196,572 common shares and 927,215 common shares reserved for future issuance in connection with future grants under the DiaMedica Therapeutics Inc. Amended and Restated 2019 Omnibus Incentive Plan and the DiaMedica Therapeutics Inc. 2021 Employment Inducement Incentive Plan and options to purchase 447,910 common shares and deferred stock units representing 17,333 common shares under our prior equity compensation plan. If these or any future outstanding options or deferred stock units are exercised or otherwise converted into our common shares, our shareholders will experience additional dilution.
If there are substantial sales of our common shares or the perception that such sales may occur, the market price of our common shares could decline.
Sales of substantial numbers of our common shares, or the perception that such sales may occur, could cause a decline in the market price of our common shares. Any sales by existing shareholders or holders who exercise their warrants or stock options may have an adverse effect on our ability to raise capital and may adversely affect the market price of our common shares.
We are a “smaller reporting company,” and because we have opted to use the reduced disclosure requirements available to us, certain investors may find investing in our common shares less attractive.
We are currently a “smaller reporting company” under the federal securities laws and, as such, are subject to scaled disclosure requirements afforded to such companies. For example, as a smaller reporting company, we are subject to reduced executive compensation disclosure requirements. Our shareholders and investors may find our common shares less attractive as a result of our status as a “smaller reporting company” and our reliance on the reduced disclosure requirements afforded to these companies. If some of our shareholders or investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and the market price of our common shares may be more volatile.
Risks Related to Our Jurisdiction of Organization
We are governed by the corporate laws of British Columbia, which in some cases have a different effect on shareholders than the corporate laws in effect in the United States.
We are a British Columbia corporation. Our corporate affairs and the rights of holders of our common shares are governed by British Columbia’s Business Corporations Act (BCBCA) and applicable securities laws, which laws may differ from those governing a company formed under the laws of a United States jurisdiction. The provisions under the BCBCA and other relevant laws may affect the rights of shareholders differently than those of a company governed by the laws of a United States jurisdiction and may, together with our Notice of Articles and Articles, have the effect of delaying, deferring or discouraging another party from acquiring control of our Company by means of a tender offer, proxy contest or otherwise, or may affect the price an acquiring party would be willing to offer in such an instance. The material differences between the BCBCA and the Delaware General Corporation Law (DGCL), by way of example, that may be of most interest to shareholders include the following:
|
● |
for material corporate transactions (such as mergers and amalgamations, other extraordinary corporate transactions or amendments to our Notice of Articles), the BCBCA, subject to the provisions of our Articles, generally requires two-thirds majority vote by shareholders; whereas, the DGCL generally only requires a majority vote of shareholders; |
|
● |
under the BCBCA, a holder of 5% or more of our common shares can requisition a special meeting at which any matters that can be voted on at our annual meeting can be considered; whereas, the DGCL does not give this right; |
|
● |
our Articles require two-thirds majority vote by shareholders to pass a resolution for one or more directors to be removed; whereas the DGCL only requires the affirmative vote of a majority of the shareholders; and |
|
● |
our Articles may be amended by resolution of our directors to alter our authorized share structure, including to (a) subdivide or consolidate any of our shares and (b) create additional classes or series of shares; whereas, under the DGCL, a majority vote by shareholders is generally required to amend a corporation’s certificate of incorporation and a separate class vote may be required to authorize alternations to a corporation’s authorized share structure. |
We cannot predict if investors find our common shares less attractive because of these material differences. If some investors find our common shares less attractive as a result, there may be a less active trading market for our common shares and our share price may be more volatile.
We were classified as a “passive foreign investment company” in 2022 and 2023 and may continue to be in future taxable years, which may have adverse U.S. federal income tax consequences for U.S. shareholders and adversely affect the level of interest in our common shares by U.S. investors.
General Rule. For any taxable year in which 75% or more of our gross income is passive income, or at least 50% of the value of our assets (where the value of our total assets is determined based upon the market value of our common shares at the end of each quarter or other measuring period) are held for the production of, or produce, passive income, we would be characterized as a passive foreign investment company (PFIC) for U.S. federal income tax purposes. The percentage of a corporation’s assets that produce or are held for the production of passive income generally is determined based upon the average ratio of passive assets to total assets calculated at the end of each measuring period. Calculation of the value of assets at the end of each measuring period is generally made at the end of each of the four quarters that make up the company’s taxable year, unless an election is made to use an alternative measuring period (such as a week or month). The “weighted average” of those periodic values is then used to determine the value of assets for the passive asset test for the taxable year. In proposed regulations section 1.1297-1(d)(2), a limited exception to the passive asset test valuation rules is provided for the treatment of working capital in order to take into account the short-term cash needs of operating companies. This new rule provides that an amount of cash held in a non-interest bearing account that is held for the present needs of an active trade or business and is no greater than the amount reasonably expected to cover 90 days of operating expenses incurred in the ordinary course of the trade or business of the foreign corporation (for example, accounts payable for ordinary operating expenses or employee compensation) is not treated as a passive asset. The Treasury Department and the IRS indicated that they continue to study the appropriate treatment of working capital for purposes of the passive asset test.
PFIC Status Determination. The tests for determining PFIC status for any taxable year are dependent upon a number of factors, some of which are beyond our control, including the value of our assets, the market price of our common shares, and the amount and type of our gross income. Based on these tests (i) we believe that we were a PFIC for the taxable year ended December 31, 2016, (ii) we do not believe that we were a PFIC for any of the taxable years ended December 31, 2017 through December 31, 2021, and (iii) we believe that we were a PFIC for the taxable years ended December 31, 2022 and December 31, 2023. Our status as a PFIC is a fact-intensive determination made for each taxable year, and we cannot provide any assurance regarding our PFIC status for the taxable year ending December 31, 2024 or for future taxable years. U.S. shareholders who own our common shares for any period during which we are a PFIC (which we believe would currently only be those shareholders that held our common shares in the taxable year ended December 31, 2016, December 31, 2022 or December 31, 2023) will be required to file IRS Form 8621 for each tax year during which they hold our common shares, unless, after we are no longer a PFIC, any such shareholder makes the “purging election” discussed below.
PFIC Consequences. If we are a PFIC for any year during a non-corporate U.S. shareholder’s holding period of our common shares, and the U.S. shareholder does not make a Qualified Electing Fund election (QEF Election) or a “mark-to-market” election, both as described below, then such non-corporate U.S. shareholder generally will be required to treat any gain realized upon a disposition of our common shares, or any so-called “excess distribution” received on our common shares, as ordinary income, rather than as capital gain, and the preferential tax rate applicable to dividends received on our common shares would not be available. This income generally would be allocated over a U.S. shareholder’s holding period with respect to our common shares and the amount allocated to prior years will be subject to tax at the highest tax rate in effect for that year and an interest charge would be imposed on the amount of deferred tax on the income allocated to prior taxable years. Pursuant to the specific provisions of the PFIC rules, a taxpayer may realize gain on the disposition of common shares if the securities are disposed of by a holder whose securities are attributed to the U.S. shareholder, if the securities are pledged as security for a loan, transferred by gift or death, or are subject to certain corporate distributions. Additionally, if we are a PFIC, a U.S. shareholder who acquires our common shares from a decedent would be denied normally available step-up in tax basis for our common shares to fair market value at the date of death but instead would have a tax basis equal to the lower of the fair market value of such common shares or the decedent’s tax basis in such common shares. Proposed regulations, that are not yet effective, address domestic partnerships and S corporations that own stock in a PFIC for which a QEF election or “mark-to-market” election could be made. Currently, only the domestic partnership or S corporation (and not the partners or S corporation shareholders) can make these elections. The proposed regulations would reverse the current rule so that only the partners or S corporation shareholders — not the partnership or S corporation — could make the elections. These proposed regulations would only apply to partnership or S corporation shareholders’ tax years beginning on or after the date they are issued in final form.
QEF Election. A U.S. shareholder may avoid the adverse tax consequences described above by making a timely and effective QEF election. A U.S. shareholder who makes a QEF election generally must report, on a current basis, its share of our ordinary earnings and net capital gains, whether or not we distribute any amounts to our shareholders, and would be required to comply with specified information reporting requirements. Any gain subsequently recognized upon the sale by that U.S. shareholder of the common shares generally would be taxed as capital gain and the denial of the basis step-up at death described above would not apply. The QEF election is available only if the company characterized as a PFIC provides a U.S. shareholder with certain information regarding its earnings and capital gains, as required under applicable U.S. Treasury regulations. We intend to provide all information and documentation that a U.S. shareholder making a QEF election is required to obtain for U.S. federal income tax purposes (e.g., the U.S. shareholder’s pro rata share of ordinary income and net capital gain, and a “PFIC Annual Information Statement” as described in applicable U.S. Treasury regulations).
Mark-to-Market Election. As an alternative to a QEF Election, a U.S. shareholder may also mitigate the adverse tax consequences of PFIC status by timely making a “mark-to-market” election. A U.S. shareholder who makes the mark-to-market election generally must include as ordinary income each year the increase in the fair market value of the common shares and deduct from gross income the decrease in the value of such shares during each of its taxable years. Losses would be allowed only to the extent of the net mark-to-market gain accrued under the election. If a mark-to-market election with respect to our common shares is in effect on the date of a U.S. shareholder’s death, the tax basis of the common shares in the hands of a U.S. shareholder who acquired them from a decedent will be the lesser of the decedent’s tax basis or the fair market value of the common shares. A mark-to-market election may be made and maintained only if our common shares are regularly traded on a qualified exchange, including The Nasdaq Capital Market. Whether our common shares are regularly traded on a qualified exchange is an annual determination based on facts that, in part, are beyond our control. Accordingly, a U.S. shareholder might not be eligible to make a mark-to-market election to mitigate the adverse tax consequences if we are characterized as a PFIC.
Election Tax Risks. Certain economic risks are inherent in making either a QEF Election or a mark-to-market election. If a QEF Election is made, it is possible that earned income will be reported to a U.S. shareholder as taxable income and income taxes will be due and payable on such an amount. A U.S. shareholder of our common shares may pay tax on such “phantom” income, i.e., where income is reported to it pursuant to the QEF Election, but no cash is distributed with respect to such income. There is no assurance that any distribution or profitable sale will ever be made regarding our common shares, so the tax liability may result in a net economic loss. A mark-to-market election may result in significant share price gains in one year causing a significant income tax liability. This gain may be offset in another year by significant losses. If a mark-to-market election is made, this highly variable tax gain or loss may result in substantial and unpredictable changes in taxable income. The amount included in income under a mark-to-market election may be substantially greater than the amount included under a QEF election. Both the QEF and mark-to-market elections are binding on the U.S. shareholder for all subsequent years that the U.S. shareholder owns our shares unless permission to revoke the election is granted by the IRS.
Purging Election. Although we generally will continue to be treated as a PFIC as to any U.S. shareholder if we are a PFIC for any year during a U.S. shareholder’s holding period, if we cease to satisfy the requirements for PFIC classification, the U.S. shareholder may avoid PFIC classification for subsequent years if the U.S. shareholder elects to make a so-called “purging election,” by recognizing income based on the unrealized appreciation in the common shares through the close of the tax year in which we cease to be a PFIC. When a foreign corporation no longer qualifies as a PFIC (due to a change in facts or law), the foreign corporation nonetheless retains its PFIC status with respect to a shareholder unless and until the shareholder makes an election under Code section 1298(b)(1) and regulations section 1.1298–3 (purging election) on IRS Form 8621 attached to the shareholder’s tax return (including an amended return), or requests the consent of the IRS Commissioner to make a late election under Code section 1298(b)(1) and regulations section 1.1298–3(e) (late purging election) on Form 8621-A.
RULES RELATING TO A PFIC ARE VERY COMPLEX. YOU SHOULD CONSULT YOUR TAX ADVISER CONCERNING THE RELATIVE MERITS AND THE ECONOMIC AND TAX IMPACT OF THE PFIC RULES TO YOUR INVESTMENT IN OUR COMMON SHARES AS A NON-ELECTING U.S. SHAREHOLDER, A U.S. SHAREHOLDER MAKING A QEF ELECTION, A U.S. SHAREHOLDER MAKING A MARK-TO-MARKET ELECTION, OR A U.S. SHAREHOLDER MAKING ANY AVAILABLE PURGING ELECTION.
Should we be classified as a PFIC during a U.S. shareholder’s holding period for our common shares, each such U.S. shareholder should consult their own tax advisors with respect to the possibility of making these elections and the U.S. federal income tax consequences of the acquisition, ownership and disposition of our common shares. In addition, the possibility of us being classified as a PFIC may deter certain U.S. investors from purchasing our common shares, which could have an adverse impact on the market price of our common shares and our ability to raise additional financing by selling equity securities, including our common shares.
It may be difficult for non-Canadian shareholders or investors to obtain and enforce judgments against us because of our organization as a British Columbia corporation.
We are a corporation governed under the BCBCA. Two of our directors are residents of Canada, and all or a substantial portion of their assets, and a small portion of our assets, are located outside the United States. Consequently, it may be difficult for holders of our securities who reside in the United States to effect service within the United States upon those directors who are not residents of the United States. It may also be difficult for holders of our securities who reside in the United States to realize in the United States upon judgments of courts of the United States predicated upon our civil liability and the civil liability of our directors, and officers under the United States federal securities laws. Our shareholders and other investors should not assume that British Columbian or Canadian courts (i) would enforce judgments of United States courts obtained in actions against us or such directors, or officers predicated upon the civil liability provisions of the United States federal securities laws or the securities or “blue sky” laws of any state or jurisdiction of the United States, or (ii) would enforce, in original actions, liabilities against us or such directors, or officers predicated upon the United States federal securities laws or any securities or “blue sky” laws of any state or jurisdiction of the United States. In addition, the protections afforded by the securities laws of British Columbia or Canada may not be available to our shareholders or other investors in the United States.
General Risk Factors
We may not achieve our publicly announced milestones according to schedule, or at all.
From time to time, we may announce the timing of certain events we expect to occur, such as the anticipated number of clinical sites and pace of enrollment for our ReMEDy2 trial. These statements are forward-looking and are based on the best estimates of management at the time relating to the occurrence of such events. However, the actual timing of such events may differ significantly from what has been publicly disclosed. The projected timing of events such as the anticipated number of clinical sites and pace of enrollment for our ReMEDy2 trial or the filing of an application to obtain regulatory approval or an announcement of additional clinical trials for a product candidate may ultimately vary from what is publicly disclosed. These variations in timing or events that we anticipate may occur as a result of different factors, including regulatory actions, the nature of the results obtained during a clinical trial or during a research phase, problems with a CMO or contract research organization, health crises, epidemics or pandemics, full or partial clinical holds that may be imposed by the FDA or any other event having the effect of delaying the publicly announced timeline or leading to results that are different from what we expect. We undertake no obligation to update or revise any forward-looking information, whether as a result of new information, future events or otherwise, except as otherwise required by law. Any variation in the timing of previously announced milestones or changes in other events of which we anticipate could have a material adverse effect on our business plan, financial condition or operating results, and the trading price of our common shares.
If securities or industry analysts do not continue to publish research or reports about our business, or publish negative reports about our business, the market price of our common shares and trading volume could decline.
The market price and trading volume for our common shares will depend in part on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. There can be no assurance that analysts will continue to cover us or provide favorable coverage. If one or more of the analysts who cover us downgrade our common shares or negatively change their opinion of our common shares, the market price of our common shares would likely decline. If one or more of these analysts cease coverage of our Company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause the market price of our common shares or trading volume to decline.
We, or our third-party contract research organizations or consultants, may be subject to information technology systems failures, network disruptions, breaches in data security and computer crime and cyber-attacks, which could result in a material disruption of our product candidates' development programs, compromise sensitive information related to our business or prevent us from accessing critical information, potentially exposing us to liability or otherwise adversely affecting our business.
We are increasingly dependent upon information technology systems, infrastructure and data to operate our business. In the ordinary course of business, we collect, store and transmit confidential information (including but not limited to intellectual property, proprietary business information and personal information). It is critical that we do so in a secure manner to maintain the confidentiality and integrity of such confidential information. We also have outsourced elements of our operations to third parties, and as a result we manage a number of third-party consultants who have access to our confidential information.
Information technology system failures, network disruptions, breaches of data security and sophisticated and targeted computer crime and cyber-attacks could disrupt our operations by impeding our drug development programs, including delays in our regulatory efforts, the manufacture or shipment of products, the processing of transactions or reporting of financial results, or by causing an unintentional disclosure of confidential information. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. In the ordinary course of our business, we collect and store sensitive data on our network, including IP, proprietary business information, and personal information of our business partners and employees. Despite our efforts to protect sensitive, confidential or personal data or information, our facilities and systems and those of our third-party service providers may be vulnerable to security breaches, theft, misplaced or lost data, programming and/or human errors that could potentially lead to the compromising of sensitive, confidential or personal data or information, improper use of our systems, software solutions or networks, unauthorized access, use, disclosure, modification or destruction of information, defective products, production downtimes and operational disruptions, which in turn could adversely affect our reputation, competitiveness and results of operations. Although we have been the target of cyber attacks and expect them to continue as cybersecurity threats have been rapidly evolving in sophistication, the aggregate impact of these attacks on our operations and financial condition has not been material to date. In addition, we and the third parties on which we rely may be more susceptible to security breaches and other security incidents due to many of our and their employees working remotely for some portion of time. While management has taken steps to address these concerns by conducting employee training, implementing certain data and system redundancy, hardening and fail-over along with other network security, comprehensive monitoring of our networks and systems, maintenance of backup and protective systems and other internal control measures, there can be no assurance that the measures we have implemented to date would be sufficient in the event of a system failure, loss of data or security breach. As a result, in the event of such a failure, loss of data or security breach, our financial condition and operating results could be adversely affected.
We could be subject to securities class action litigation, which is expensive and could divert management attention.
In the past, securities class action litigation has often been brought against a company following a significant decline or increase in the market price of its securities or certain significant business transactions. We may become involved in this type of litigation in the future, especially if our clinical trial results are not successful or we enter into an agreement for a significant business transaction. If we face such litigation, it could result in substantial costs and a diversion of management’s attention and our resources, which could harm our business. This is particularly true in light of our limited securities litigation insurance coverage.
A variety of risks are associated with operating our business internationally which could materially adversely affect our business.
In the past, we have conducted R&D operations and/or clinical trials in the United States, Canada and Australia. In the future, we expect to conduct certain clinical trials, and plan to seek regulatory approval of DM199, or any future product candidates, outside of the United States. Accordingly, we will be subject to risks related to operating in foreign countries including, among others:
|
● |
differing regulatory requirements for drug approvals; |
|
● |
different standards of care in various countries that could complicate the design of our clinical trials and/or the evaluation of our product candidates; |
|
● |
different reimbursement systems and different competitive drugs indicated to treat the indications for which our product candidates are or will be developed; |
|
● |
different United States and foreign drug import and export rules; |
|
● |
reduced protection for intellectual property rights in certain countries; |
|
● |
withdrawal from, or revision to or unexpected changes in international trade policies or agreements and the imposition or increases in import and export licensing and other compliance requirements, customs duties and tariffs, import and export quotas and other trade restrictions, license obligations, and other non-tariff barriers to trade; |
|
● |
the imposition of U.S. or international sanctions against a country, company, person or entity with whom we do business that would restrict or prohibit continued business with that country, company, person or entity; |
|
● |
economic weakness, including inflation or political instability in particular foreign economies and markets; |
|
● |
compliance with tax, employment, immigration, and labor laws for employees living or traveling abroad; |
|
● |
compliance with the Foreign Corrupt Practices Act and other anti-corruption and anti-bribery laws; |
|
● |
foreign taxes, including withholding of payroll taxes; |
|
● |
foreign currency exchange rate fluctuations, which could result in increased operating expenses and/or reduced revenue, and other obligations incident to doing business in another country; |
|
● |
difficulties in managing and staffing international operations and increases in infrastructure costs, including legal, tax, accounting, and information technology; |
|
● |
workforce uncertainty in countries where labor unrest is more common than in the United States; |
|
● |
production shortages or shipping delays resulting from any events affecting raw material supply or manufacturing capabilities abroad, such as supply chain disruptions, closures and slowdowns caused by COVID-19; |
|
● |
potential liability resulting from development work conducted by foreign partners or collaborators; |
|
● |
transportation delays and interruptions; |
|
● |
business interruptions resulting from natural disasters or geopolitical actions, including war, such as the current war between Russia and Ukraine and the conflict between Israel and Hamas, and terrorism or systems failure, including cybersecurity breaches; and |
|
● |
compliance with evolving and expansive international data privacy laws, such as the European Union General Data Protection Regulation. |
We face the risk of product liability claims, which could exceed our insurance coverage, deplete our cash resources and lead to clinical trial delays.
A risk of product liability claims, and related negative publicity, is inherent in the development of human therapeutics. We are exposed to the risk of product liability claims alleging that use of DM199, or any future product candidate, caused an injury or harm. These claims can arise at any point in the development, testing, manufacture, marketing or sale of a product candidate and may be made directly by patients involved in clinical trials of our product candidate, by consumers, healthcare providers or by individuals, organizations or companies selling our products, if and when approved. Product liability claims can be expensive to defend, even if the product or product candidate did not actually cause the alleged injury or harm, and could lead to clinical trial delays and could negatively impact existing or future collaborations.
Insurance covering product liability claims becomes increasingly expensive as a product candidate moves through the development pipeline to commercialization. To protect against potential product liability risks, we carry product liability insurance coverage at a level we deem appropriate for our stage of development. However, there can be no assurance that such insurance coverage is or will continue to be adequate or available to us at a cost acceptable to us or at all. We may choose or find it necessary under our collaboration agreements to increase our insurance coverage in the future. We may not be able to secure greater or broader product liability insurance coverage on acceptable terms or at reasonable costs when needed. Any liability for damages resulting from a product liability claim could exceed the amount of our coverage, require us to pay a substantial monetary award from our own cash resources, and otherwise have a material adverse effect on our business, financial condition, and results of operations.
If we are unable to maintain product liability insurance required by third parties, certain agreements, such as those with clinical trial sites, contract research organizations and other supporting vendors, would be subject to termination, which could have a material adverse impact on our operations.
Some of our agreements with third parties require, and in the future will likely require, us to maintain product liability insurance in at least certain specified minimum amounts. If we cannot maintain acceptable amounts of coverage on commercially reasonable terms in accordance with the terms set forth in these agreements, the corresponding agreements would be subject to termination, which could have a material adverse impact on our operations.
Our insurance policies are expensive and protect us only from certain business risks, which could leave us exposed to significant uninsured liabilities. Additionally, future fluctuations in insurance cost and availability could adversely affect our operating results or risk management profile.
We hold a number of insurance policies, including, but not limited to, product and general liability insurance, directors’ and officers’ liability insurance, property insurance and workers’ compensation insurance. The costs of maintaining adequate insurance coverage, most notably directors’ and officers’ liability insurance, have increased significantly during the last few years and may continue to do so in the future, thereby adversely affecting our operating results. If such costs increase, we may be forced to accept lower coverage levels and higher deductibles, which, in the event of a claim, could require significant, unplanned expenditures of cash, which could adversely affect our business. Future potential directors and officers could view our directors’ and officers’ liability insurance coverage as limited or even inadequate. Limited directors’ and officers’ liability insurance coverage, or the perception that our directors’ and officers’ liability insurance coverage is inadequate, may make it difficult to attract and retain directors and officers, and we may lose potential independent board members and management candidates to other companies that have more extensive directors’ and officers’ liability insurance coverage. In addition, if any of our current insurance coverages should become unavailable to us or become economically impractical, we would be required to operate our business without indemnity from commercial insurance providers.
Scrutiny and evolving expectations from regulators, investors and other stakeholders with respect to our environmental, social and governance practices may impose additional costs on us or expose us to new or additional risks.
Companies are facing scrutiny from regulators, investors, and other stakeholders related to their environmental, social and governance (ESG) practices and disclosure. For example, during 2022, the SEC proposed new climate disclosure rules, which, if adopted, would require new climate-related disclosure in SEC filings, including certain climate-related metrics and greenhouse gas emissions data, information about climate-related targets and goals, transition plans, if any, and extensive attestation requirements. In addition to requiring companies to quantify and disclose direct emissions data, the new rules also would require disclosure of climate impact arising from the operations and uses by the company’s business partners and contractors and end-users of the company’s products and/or services. We are currently assessing the impact of the new rules, if adopted as proposed, but at this time, we cannot predict the costs of implementation or any potential adverse impacts resulting from the new rules if adopted. However, we may incur increased costs relating to the assessment and disclosure of climate-related risks and increased litigation risks related to disclosures made pursuant to the new rules, either of which could materially and adversely affect our future results of operations and financial condition.
Further, investor advocacy groups, investment funds and influential investors are also increasingly focused on these practices, especially as they relate to the environment, climate change, health and safety, supply chain management, diversity, labor conditions and human rights, both in our own operations and in our supply chain. Increased ESG-related compliance costs could result in material increases to our overall operational costs. Our ESG practices may not meet the standards of all of our stakeholders and advocacy groups may campaign for further changes. A failure, or perceived failure, to adapt to or comply with regulatory requirements or to respond to investor or stakeholder expectations and standards could negatively impact our business and reputation and have a negative impact on the trading price of our common shares.
We no longer qualify as an emerging growth company, and as a result, we now have to comply with increased public company disclosure and compliance requirements, which may have a negative impact on our business and results of operations.
We no longer qualify as an emerging growth company. As such, we are now subject to certain disclosure and compliance requirements that apply to other public companies but did not previously apply to us due to our status as an emerging growth company. While we remain a smaller reporting company and are still subject to certain scaled disclosure requirements, we expect that the loss of emerging growth company status may still increase our legal and financial compliance costs and cause management and other personnel to divert attention from operational and other business matters to devote substantial time to public company reporting requirements, all of which may have a negative impact on our business and results of operations.
Our business or the value of our common shares could be negatively affected as a result of actions by activist shareholders.
We value constructive input from our shareholders, and our Board of Directors and management team are committed to acting in the best interests of our shareholders. However, shareholders may from time to time engage in proxy solicitations, advance shareholder proposals or otherwise attempt to effect changes or acquire control over the Company. Responding to proxy contests and other actions by activist shareholders can be costly and time-consuming, disrupting our operations and diverting the attention of our Board of Directors and senior management from the pursuit of business strategies. In addition, perceived uncertainties as to our future direction, strategy or leadership created as a consequence of activist shareholder initiatives may result in the loss of potential business opportunities, harm our ability to attract new investors, customers, employees, and joint venture partners, and cause our stock price to experience periods of volatility or stagnation.
|
Item 1B. |
Unresolved Staff Comments |
This Item 1B is inapplicable to us as a smaller reporting company.
|
Item 1C. |
Cybersecurity |
Cybersecurity, data privacy, and data protection are critical to our business. In the ordinary course of our business, we collect and store certain confidential information such as information about our employees, contractors, vendors, suppliers, and clinical data. We continue to augment the capabilities of our people, processes, and technologies in order to address our cybersecurity risks. Our cybersecurity risks, and the controls designed to mitigate those risks, are integrated into our overall risk management governance and are reviewed yearly by our Board of Directors.
Risk Management and Strategy
As of December 31, 2023, we have implemented cybersecurity and data protection policies and procedures for assessing, identifying, and managing cybersecurity threats. We take a risk-based approach to cybersecurity, which begins with the identification and evaluation of cybersecurity risks or threats that could affect our operations, finances, legal or regulatory compliance, or reputation. The scope of our evaluation encompasses risks that may be associated with both our internally managed IT systems and key business functions and sensitive data operated or managed by third-party service providers, thereby safeguarding our integrated operations. Risks from cybersecurity threats are regularly evaluated as a part of our broader risk management activities and as a fundamental component of our internal control systems. Our employees receive ongoing cybersecurity awareness trainings, including specific topics related to social engineering and email frauds. We use information technology consultants with significant expertise in cybersecurity related to our industry. We utilize advanced technologies for continuous cybersecurity monitoring across our information technology environment which are designed to prevent, detect, and minimize cybersecurity attacks, as well as alert management of such attacks.
Our IT general controls are firmly established based on recognized industry standards and cover areas such as risk management, data backup, and disaster recovery. We have utilized an outsourced IT services vendor to reduce and monitor security threats and vulnerabilities and respond to all cybersecurity incidents affecting us, including prompt escalation and communication of major security incidents to senior business leadership and our Board of Directors.
Governance
Our Board of Directors is responsible for overseeing our cyber security risk management and strategy, including overseeing management’s responsibility to assess, manage and mitigate risks associated with our business and operational activities, to administer our various compliance programs, in each case including cybersecurity concerns, and to oversee our IT systems, processes and data. Our senior leadership, including our Chief Executive Officer and Chief Financial Officer, regularly meet with and provides periodic briefings to our Board of Directors regarding our cybersecurity risks and activities, including any recent cybersecurity incidents, if any, and related responses, cybersecurity systems testing, and activities of third parties.
Management has implemented risk management policies and procedures, and management is responsible for the day-to-day cybersecurity risk management. Our Chief Financial Officer is responsible for the day-to-day assessment and management of our cybersecurity risks.
Cybersecurity Threat Disclosure
To date, we are not aware of any cybersecurity threats that have materially affected or are reasonably likely to materially affect our Company’s business strategy, results of operations or financial condition.
For further discussion of cybersecurity risks, please see Item 1A, "Risk Factors".
|
Item 2. |
Properties |
Our principal executive offices, together with our research and development operations, are at the office of our wholly owned subsidiary, DiaMedica USA Inc., located at 301 Carlson Parkway, Suite 210, Minneapolis, Minnesota, USA 55305. We lease these premises, which consist of approximately 6,000 square feet, pursuant to a lease that expires in January 2028. We believe that our facilities are adequate for our current needs and that suitable additional space will be available as and when needed on acceptable terms.
|
Item 3. |
Legal Proceedings |
Litigation with Pharmaceutical Research Associates Group B.V.
In March 2013, we entered into a clinical research agreement with Pharmaceutical Research Associates Group B.V., acquired by ICON plc as of July 1, 2021, (PRA Netherlands) to perform a double-blinded, placebo-controlled, single-dose and multiple-dose study to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics and proof of concept of DM199 in healthy subjects and in patients with Type 2 diabetes mellitus. In one arm of this study, we enrolled 36 patients with Type 2 diabetes who were treated with two subcutaneous dose levels of DM199 over a 28-day period. This study achieved its primary endpoint and demonstrated that DM199 was well-tolerated. The secondary endpoints for this study, however, were not met. The secondary efficacy endpoints were confounded due to what we believe were significant execution errors caused by protocol deviations occurring at the clinical study site that were unable to be reconciled. To date, we have been unable to obtain the complete study records from PRA Netherlands necessary to generate a final study report. On November 14, 2017, we initiated litigation with PRA Netherlands in the United States District Court, Southern District of New York. The complaint alleged, among other things, that PRA Netherlands failed to conduct the study in accordance with the study protocol and with generally accepted standards for conducting such clinical studies and that PRA Netherlands further refused to provide us with all data, records and documentation, and/or access thereto, related to the study in accordance with the clinical trial study agreement. The complaint sought to compel PRA Netherlands to comply with the terms of the clinical trial study agreement, including providing full study records and to recover damages.
After several procedural stages, we ceased action against PRA Netherlands in the United States and commenced an action in a Dutch Court, which was subsequently moved to the Netherlands Commercial Court (NCC), which specializes in handling international commercial disputes. On November 23, 2022, we filed a petition requesting leave for a prejudgment attachment of all relevant documents in possession of PRA Netherlands, which was granted on November 28, 2022, by the District Court of Northern Netherlands. A representative of the District Court served PRA Netherlands with the prejudgment attachment on or about December 7 and 8, 2022. The case was formally introduced to the NCC on December 28, 2022 and a hearing by the NCC to determine whether we are entitled to take possession of the records seized was scheduled and held on March 16, 2023. On April 21, 2023, the NCC issued a judgment affirming our ownership of the documents related to the clinical studies performed by PRA Netherlands and seized by the Dutch courts in December 2022. The NCC further ordered PRA Netherlands to allow and tolerate the surrender of the documents. Additionally, the NCC found that we were not in breach of any obligation under the clinical study agreement and PRA Netherlands had no basis to suspend the fulfillment of its obligations under the clinical study agreement to provide us all clinical data and access to perform an audit of the study. On June 15, 2023, PRA Netherlands filed an appeal of this decision and requested a hearing with the NCC. The hearing of this case was conducted on December 7, 2023. On February 7, 2024, the NCC issued a judgement in which they found that, although all data related to the study is the rightful property of DiaMedica, they found that there was an insufficient causal link between PRA Netherlands withholding study data and the damages claimed by us. We have 90 days, or until approximately May 7, 2024, to file an appeal of the decision. We are currently evaluating our options.
From time to time, we may be subject to other various ongoing or threatened legal actions and proceedings, including those that arise in the ordinary course of business, which may include employment matters and breach of contract disputes. Such matters are subject to many uncertainties and to outcomes that are not predictable with assurance and that may not be known for extended periods of time. Other than the PRA matter noted above, we are not currently engaged in or aware of any threatened legal actions.
|
Item 4. |
Mine Safety Disclosures |
Not applicable.
PART II
|
Item 5. |
Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common shares are listed on The Nasdaq Capital Market under the trading symbol “DMAC”.
Number of Record Holders
As of March 15, 2024, we had 25 holders of record of our common shares. This does not include persons whose common shares are in nominee or “street name” accounts through brokers or other nominees.
Dividend Policy
We have never declared or paid cash dividends on our common shares, and currently do not have any plans to do so in the foreseeable future. We expect to retain our future earnings, if any, for use in the operation and expansion of our business. Additionally, we may in the future become subject to contractual restrictions on, or prohibitions against, the payment of dividends. Subject to the foregoing, the payment of cash dividends in the future, if any, will be at the discretion of our Board of Directors and will depend upon such factors as earnings levels, capital requirements, our overall financial condition and any other factors deemed relevant by our Board of Directors. As a result, our shareholders will likely need to sell their common shares to realize a return on their investment and may not be able to sell their shares at or above the price paid for them.
Purchases of Equity Securities by the Company
We did not purchase any common shares or other equity securities of our company during the fourth quarter ended December 31, 2023.
Recent Sales of Unregistered Equity Securities
We did not sell any unregistered equity securities of our company during the fourth quarter ended December 31, 2023.
Exchange Controls
There are no governmental laws, decrees or regulations in Canada that restrict the export or import of capital, including foreign exchange controls, or that affect the remittance of dividends, interest or other payments to non-resident holders of the securities of DiaMedica, other than Canadian withholding tax.
Certain Canadian Federal Income Tax Considerations for U.S. Holders
The following is, as of March 15, 2024, a summary of the principal Canadian federal income tax considerations under the Income Tax Act (Canada) (Tax Act) generally applicable to a holder of our common shares who, for purposes of the Tax Act and at all relevant times, is neither resident in Canada nor deemed to be resident in Canada for purposes of the Tax Act and any applicable income tax treaty or convention, and who does not use or hold (and is not deemed to use or hold) common shares in the course of carrying on a business in Canada, deals at arm’s length with us, is not affiliated with us, is not a “specified shareholder” of us (within the meaning of subsection 18(5) of the Tax Act) and holds our common shares as capital property (Holder). A “specified shareholder” for these purposes generally includes a person who (either alone or together with persons with whom that person is not dealing at arm’s length for the purposes of the Tax Act) owns or has the right to acquire or control 25% or more of the common shares determined on a votes or fair market value basis. Generally, common shares will be considered to be capital property to a Holder thereof provided that the Holder does not hold common shares in the course of carrying on a business and such Holder has not acquired them in one or more transactions considered to be an adventure or concern in the nature of trade.
This summary does not apply to a Holder, (i) that is a “financial institution” for purposes of the mark-to-market rules contained in the Tax Act; (ii) that is a “specified financial institution” as defined in the Tax Act; (iii) that holds an interest which is a “tax shelter investment” as defined in the Tax Act; or (iv) that has elected to report its tax results in a functional currency other than Canadian currency. Special rules, which are not discussed in this summary, may apply to a Holder that is an “authorized foreign bank” within the meaning of the Tax Act, a partnership or an insurer carrying on business in Canada and elsewhere. Such Holders should consult their own tax advisors.
This summary is based upon the provisions of the Tax Act (including the regulations (Regulations) thereunder) in force as of March 1, 2023 and our understanding of the current administrative policies and assessing practices of the Canada Revenue Agency (CRA) published in writing by the CRA prior to March 1, 2023. This summary takes into account all specific proposals to amend the Tax Act (and the Regulations) publicly announced by or on behalf of the Minister of Finance (Canada) prior to the date hereof (Tax Proposals) and assumes that the Tax Proposals will be enacted in the form proposed, although no assurance can be given that the Tax Proposals will be enacted in their current form or at all. This summary does not otherwise take into account any changes in law or in the administrative policies or assessing practices of the CRA, whether by legislative, governmental or judicial decision or action. This summary is not exhaustive of all possible Canadian federal income tax considerations and does not take into account other federal or any provincial, territorial or foreign income tax legislation or considerations, which may differ materially from those described in this summary.
This summary is of a general nature only and is not, and is not intended to be, and should not be construed to be, legal or tax advice to any particular Holder, and no representations concerning the tax consequences to any particular Holder are made. Holders should consult their own tax advisors regarding the income tax considerations applicable to them having regard to their particular circumstances.
Dividends
Dividends paid or credited (or deemed to be paid or credited) to a Holder by us are subject to Canadian withholding tax at the rate of 25% unless reduced by the terms of an applicable tax treaty or convention. For example, under the Canada-United States Tax Convention (1980), as amended (US Treaty), the dividend withholding tax rate is generally reduced to 15% (or 5% in the case of a Holder that is a company that beneficially owns at least 10% of our voting shares) in respect of a dividend paid or credited to a Holder beneficially entitled to the dividend who is resident in the United States for purposes of the US Treaty and whose entitlement to the benefits of the US Treaty is not limited by the limitation of benefits provisions of the US Treaty. Holders are urged to consult their own tax advisors to determine their entitlement to relief under the US Treaty or any other applicable tax treaty as well as their ability to claim foreign tax credits with respect to any Canadian withholding tax, based on their particular circumstances.
Disposition of Common Shares
A Holder generally will not be subject to tax under the Tax Act in respect of a capital gain realized on the disposition or deemed disposition of a common share, unless the common share constitutes or is deemed to constitute “taxable Canadian property” to the Holder thereof for purposes of the Tax Act, and the gain is not exempt from tax pursuant to the terms of an applicable tax treaty or convention.
In general, provided the common shares are listed on a “designated stock exchange” (which currently includes The Nasdaq Capital Market) at the date of the disposition, the common shares will only constitute “taxable Canadian property” of a Holder if, at any time within the 60-month period preceding the disposition: (i) such Holder, persons with whom the Holder did not deal at arm’s length, partnerships in which the Holder or a person with whom the Holder did not deal at arm’s length holds a membership interest directly or indirectly through one or more partnerships, or any combination thereof, owned 25% or more of the issued shares of any class or series of the Company’s share capital; and (ii) more than 50% of the fair market value of the common shares was derived directly or indirectly from one or any combination of (A) real or immovable property situated in Canada, (B) Canadian resource properties, (C) timber resource properties, and (D) options in respect of, or interests in, or for civil law rights in, property described in any of subparagraphs (ii)(A) to (C), whether or not the property exists. However, and despite the foregoing, in certain circumstances the common shares may be deemed to be “taxable Canadian property” under the Tax Act.
Holders whose common shares may be “taxable Canadian property” should consult their own tax advisers.
Certain U.S. Federal Income Tax Considerations
The following discussion is generally limited to certain material U.S. federal income tax considerations relating to the purchase, ownership and disposition of our common shares by U.S. Holders (as defined below). This discussion applies to U.S. Holders that hold our common shares as capital assets. This summary is for general information purposes only and does not purport to be a complete analysis or listing of all potential U.S. federal income tax considerations that may apply to a U.S. Holder arising from and relating to the acquisition, ownership, and disposition of our common shares. Accordingly, this summary is not intended to be, and should not be construed as, legal or U.S. federal income tax advice with respect to any U.S. Holder. Although this discussion is generally limited to the U.S. federal income tax considerations to U.S. Holders, the U.S. federal income tax treatment of dividends on and gain on sale or exchange of our common shares by certain “Non-U.S. Holders” (as defined below) is included below at “U.S. Federal Income Taxation of Non-U.S. Holders.”
No legal opinion from U.S. legal counsel or ruling from the Internal Revenue Service (IRS) has been requested, or will be obtained, regarding the U.S. federal income tax consequences of the acquisition, ownership, and disposition of common shares. This summary is not binding on the IRS, and the IRS is not precluded from taking a position that is different from, and contrary to, the positions presented in this summary. In addition, because the guidance on which this summary is based are subject to various interpretations, the IRS and the U.S. courts could disagree with one or more of the positions described in this summary.
This discussion is based on the U.S. Internal Revenue Code of 1986, as amended (Code), U.S. Treasury regulations promulgated thereunder and administrative and judicial interpretations thereof, and the income tax treaty between the United States and Canada (Convention), all as in effect on the date hereof and all of which are subject to change, possibly with retroactive effect. This summary is applicable to U.S. Holders who are residents of the United States for purposes of the Convention and who qualify for the full benefits of the Convention. This summary does not discuss the potential effects, whether adverse or beneficial, of any proposed legislation.
This discussion does not address all of the U.S. federal income tax considerations that may be relevant to specific U.S. Holders in light of their particular circumstances or to U.S. Holders subject to special treatment under U.S. federal income tax law (such as certain financial institutions, insurance companies, broker-dealers and traders in securities or other persons that generally mark their securities to market for U.S. federal income tax purposes, tax-exempt entities, retirement plans, regulated investment companies, real estate investment trusts, certain former citizens or residents of the United States, persons who hold common shares as part of a “straddle,” “hedge,” “conversion transaction,” “synthetic security” or integrated investment, persons that have a “functional currency” other than the U.S. dollar, persons that own (or are deemed to own) 10% or more (by voting power or value) of our common shares, persons that acquire their common shares as part of a compensation arrangement, corporations that accumulate earnings to avoid U.S. federal income tax, partnerships and other pass-through entities, and investors in such pass-through entities). This discussion does not address any U.S. state or local or non-U.S. tax considerations or any U.S. federal estate, gift or alternative minimum tax considerations. In addition, except as specifically set forth below, this summary does not discuss applicable tax reporting requirements.
As used in this discussion, the term “U.S. Holder” means a beneficial owner of common shares that is, for U.S. federal income tax purposes, (1) an individual who is a citizen or resident of the United States, (2) a corporation (or entity treated as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any state thereof, or the District of Columbia, (3) an estate the income of which is subject to U.S. federal income tax regardless of its source or (4) a trust (x) with respect to which a court within the United States is able to exercise primary supervision over its administration and one or more United States persons have the authority to control all of its substantial decisions or (y) that has elected under applicable U.S. Treasury regulations to be treated as a domestic trust for U.S. federal income tax purposes.
If an entity treated as a partnership for U.S. federal income tax purposes holds the common shares, the U.S. federal income tax considerations relating to an investment in the common shares will depend in part upon the status and activities of such entity and the particular partner. Any such entity should consult its own tax advisor regarding the U.S. federal income tax considerations applicable to it and its partners of the purchase, ownership and disposition of the common shares.
Persons holding common shares should consult their own tax advisors as to the particular tax considerations applicable to them relating to the purchase, ownership and disposition of common shares, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Distributions
Subject to the discussion below under “Passive Foreign Investment Company Considerations,” a U.S. Holder that receives a distribution with respect to the common shares generally will be required to include the gross amount of such distribution (before reduction for any Canadian withholding taxes) in gross income as a dividend when actually or constructively received to the extent of the U.S. Holder’s pro rata share of our current and/or accumulated earnings and profits (as determined under U.S. federal income tax principles). To the extent a distribution received by a U.S. Holder is not a dividend because it exceeds the U.S. Holder’s pro rata share of our current and accumulated earnings and profits, it will be treated first as a tax-free return of capital and reduce (but not below zero) the adjusted tax basis of the U.S. Holder’s common shares. To the extent the distribution exceeds the adjusted tax basis of the U.S. Holder’s common shares, the remainder will be taxed as capital gain. However, we cannot provide any assurance that we will maintain or provide earnings and profits determinations in accordance with U.S. federal income tax principles. Therefore, U.S. Holders should expect that a distribution will generally be treated as a dividend even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain under the rules described above.
The U.S. dollar value of any distribution on the common shares made in Canadian dollars generally should be calculated by reference to the exchange rate between the U.S. dollar and the Canadian dollar in effect on the date of receipt (or deemed receipt) of such distribution by the U.S. Holder regardless of whether the Canadian dollars so received are in fact converted into U.S. dollars at that time. If the Canadian dollars received are converted into U.S. dollars on the date of receipt (or deemed receipt), a U.S. Holder generally should not recognize currency gain or loss on such conversion. If the Canadian dollars received are not converted into U.S. dollars on the date of receipt (or deemed receipt), a U.S. Holder generally will have a basis in such Canadian dollars equal to the U.S. dollar value of such Canadian dollars on the date of receipt (or deemed receipt). Any gain or loss on a subsequent conversion or other disposition of such Canadian dollars by such U.S. Holder generally will be treated as ordinary income or loss and generally will be income or loss from sources within the United States for U.S. foreign tax credit purposes. Different rules apply to U.S. Holders who use the accrual method of tax accounting. Each U.S. Holder should consult its own U.S. tax advisors regarding the U.S. federal income tax consequences of receiving, owning, and disposing of foreign currency.
Distributions on the common shares that are treated as dividends generally will constitute income from sources outside the United States for foreign tax credit purposes and generally will constitute “passive category income.” Because we are not a United States corporation, such dividends will not be eligible for the “dividends received” deduction generally allowed to corporate shareholders with respect to dividends received from U.S. corporations. Dividends paid by a “qualified foreign corporation” to a U.S. Holder who is an individual, trust or estate will generally be treated as “qualified dividend income” and are eligible for taxation at a reduced capital gains rate rather than the marginal tax rates generally applicable to ordinary income provided that a holding period requirement (more than 60 days of ownership, without protection from the risk of loss, during the 121-day period beginning 60 days before the ex-dividend date) and certain other requirements are met. However, if we are a PFIC for the taxable year in which the dividend is paid or the preceding taxable year (see discussion below under “Passive Foreign Investment Company Considerations”), we will not be treated as a qualified foreign corporation, and therefore the reduced capital gains tax rate described above will not apply. Each U.S. Holder is advised to consult its own tax advisors regarding the availability of the reduced tax rate on dividends.
If a U.S. Holder is subject to Canadian withholding tax on dividends paid on the holder’s common shares (see discussion above under “Certain Canadian Federal Income Tax Considerations for U.S. Holders—Dividends”), the U.S. Holder may be eligible, subject to a number of complex limitations, to claim a credit against its U.S. federal income tax for the Canadian withholding tax imposed on the dividends. However, if U.S. persons collectively own, directly or indirectly, 50% or more of the voting power or value of our common shares it is possible that a portion of any dividends we pay will be considered U.S. source income in proportion to our U.S. source earnings and profits, which could limit the ability of a U.S. Holder to claim a foreign tax credit for the Canadian withholding taxes imposed in respect of such a dividend, although certain elections may be available under the Code and the Convention to mitigate these effects. A U.S. Holder may claim a deduction for the Canadian withholding tax in lieu of a credit, but only for a year in which the U.S. Holder elects to do so for all creditable foreign income taxes. The rules governing the foreign tax credit are complex. Each U.S. Holder is advised to consult its tax advisor regarding the availability of the foreign tax credit under its particular circumstances.
Sale, Exchange or Other Disposition of Common Shares
Subject to the discussion below under “Passive Foreign Investment Company Considerations,” a U.S. Holder generally will recognize capital gain or loss for U.S. federal income tax purposes upon the sale, exchange or other disposition of common shares. The amount of gain recognized will equal the excess of the amount realized (i.e., the amount of cash plus the fair market value of any property received) over the U.S. Holder’s adjusted tax basis in the common shares sold or exchanged. The amount of loss recognized will equal the excess of the U.S. Holder’s adjusted tax basis in the common shares sold or exchanged over the amount realized. Such capital gain or loss generally will be long-term capital gain or loss if, on the date of sale, exchange or other disposition, the common shares were held by the U.S. Holder for more than one year. Net long-term capital gain derived by a non-corporate U.S. Holder with respect to capital assets is currently subject to tax at reduced rates. The deductibility of a capital loss is subject to limitations. Any gain or loss recognized from the sale, exchange or other disposition of common shares will generally be gain or loss from sources within the United States for U.S. foreign tax credit purposes, except as otherwise provided in an applicable income tax treaty and if an election is properly made under the Code.
Passive Foreign Investment Company Considerations
General Rule. In general, a corporation organized outside the United States will be treated as a PFIC in any taxable year in which either (1) at least 75% of its gross income is “passive income” or (2) at least 50% of the average value of its assets is attributable to assets that produce passive income or are held for the production of passive income. Passive income for this purpose generally includes, among other things, dividends, interest, royalties, rents, and gains from commodities transactions and from the sale or exchange of property that gives rise to passive income. Assets that produce or are held for the production of passive income include cash, even if held as working capital or raised in a public offering, marketable securities and other assets that may produce passive income. The percentage of a corporation’s assets that produce or are held for the production of passive income generally is determined based upon the average ratio of passive assets to total assets calculated at the end of each measuring period. Calculation of the value of assets at the end of each measuring period is generally made at the end of each of the four quarters that make up the company’s taxable year, unless an election is made to use an alternative measuring period (such as a week or month). The “weighted average” of those periodic values is then used to determine the value of assets for the passive asset test for the taxable year. In proposed regulations section 1.1297-1(d)(2), a limited exception to the passive asset test valuation rules is provided for the treatment of working capital in order to take into account the short-term cash needs of operating companies. This working capital rule provides that an amount of cash held in a non-interest bearing account that is held for the present needs of an active trade or business and is no greater than the amount reasonably expected to cover 90 days of operating expenses incurred in the ordinary course of the trade or business of the foreign corporation (for example, accounts payable for ordinary operating expenses or employee compensation) is not treated as a passive asset. The Treasury Department and the IRS indicated that they continue to study the appropriate treatment of working capital for purposes of the passive asset test. In determining whether a foreign corporation is a PFIC, a proportionate share of the items of gross income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) are taken into account.
PFIC Status Determination. Although the tests for determining PFIC status for any taxable year are dependent upon a number of factors, some of which are beyond our control, including the value of our assets, the market price of our common shares, and the amount and type of our gross income, based on those tests: (i) we believe that we were a PFIC for the taxable year ended December 31, 2016, and (ii) we do not believe that we were a PFIC for any of the taxable years ended December 31, 2017 through December 31, 2021, and (iii) we believe that we were a PFIC for the taxable year ended December 31, 2023. Our status as a PFIC is a fact-intensive determination made on an annual basis, and we cannot provide any assurance regarding our PFIC status for the taxable year ending December 31, 2023 or for subsequent taxable years. U.S. Holders who own our common shares for any period during which we are a PFIC will be required to file IRS Form 8621 for each tax year during which they hold our common shares. No opinion of legal counsel or ruling from the IRS concerning our status as a PFIC has been obtained or is currently planned to be requested. However, the determination of our PFIC status is made annually after the close of each taxable year and it is difficult to predict before such determination whether we will be a PFIC for any given taxable year. Even if we determine that we are not a PFIC after the close of a taxable year, there can be no assurance that the IRS will agree with our conclusion. No assurance can be provided regarding our PFIC status, and neither we nor our United States counsel expresses any opinion with respect to our PFIC status.
PFIC Consequences. If we are a PFIC at any time when a non-corporate U.S. Holder owns common shares, and such U.S. Holder does not make a “qualified electing fund” election (QEF election) or a “mark-to-market” election, both as described below, such U.S. Holder will generally be subject to federal tax under the excess distribution rules (described below). Under such rules, additional taxes and interest charges would apply to certain distributions by us or to gain upon dispositions of our common shares. If neither of such elections are made, the excess distribution rules apply to (1) distributions paid during a taxable year that are greater than 125% of the average annual distributions paid in the three preceding taxable years, or, if shorter, the U.S. Holder’s holding period for the common shares, and (2) any gain recognized on a sale, exchange or other disposition (which would include a pledge or transfer by gift or death) of common shares. Under the excess distribution rules, the non-corporate U.S. Holder’s tax liability will be determined by allocating such distribution or gain ratably to each day in the U.S. Holder’s holding period for the common shares. The amount allocated to the current taxable year (i.e., the year in which the distribution occurs or the gain is recognized) and any year prior to the first taxable year in which we were a PFIC during such holding period will be taxed as ordinary income earned in the current taxable year and the preferential tax rate applicable to capital gains or dividends received on our common shares would not be available. The amount allocated to other taxable years (i.e., prior years in which we were a PFIC) will be taxed at the highest marginal rate in effect (for individuals or corporations as applicable) for ordinary income in each such taxable year, and an interest charge, generally applicable to the underpayment of tax, will be added to the tax and the preferential tax rate applicable to capital gains or dividends received on our common shares would not be available. These adverse tax consequences would not apply to a pension or profit-sharing trust or other tax-exempt organization that did not borrow funds or otherwise utilize leverage in connection with its acquisition of our common shares. In addition, if a non-electing U.S. Holder who is an individual dies while owning our common shares, such U.S. Holder's successor generally would not receive a step-up in tax basis with respect to such common shares, but instead would have a tax basis equal to the lower of the fair market value of such common shares or the decedent’s tax basis in such common shares. Newly proposed regulations, that are not yet effective, address domestic partnerships and S corporations that own stock in a PFIC for which a QEF election or "mark-to-market" election could be made. Currently, only the domestic partnership or S corporation (and not the partners or S corporation shareholders) can make these elections. The proposed regulations would reverse the current rule so that only the partners or S corporation shareholders — not the partnership or S corporation — could make the elections. These proposed regulations would only apply to partnership or S corporation shareholders' tax years beginning on or after the date they are issued in final form.
QEF Election. The tax considerations that would apply if we were a PFIC would be different from those described above if a U.S. Holder were able to make a valid QEF election. For each year that we meet the PFIC gross income test or asset test, an electing U.S. Holder would be required to include in gross income its pro rata share of our ordinary income and net capital gains, if any, as determined under U.S. federal income tax principles. The U.S. Holder’s adjusted tax basis in our common shares would be increased by the amount of such inclusions. An actual distribution to the U.S. Holder out of such income generally would not be treated as a dividend and would decrease the U.S. Holder’s adjusted tax basis in our common shares. Gain realized from the sale of our common shares covered by a QEF election would be taxed as a capital gain and the denial of the basis step-up at death described above would not apply. Generally, a QEF election must be made by the U.S. Holder in a timely filed tax return for the first taxable year in which the U.S. Holder held our common shares that includes the close of our taxable year for which we met the PFIC gross income test or asset test. A separate QEF election would need to be made for any of our subsidiaries that are classified as a PFIC. A QEF election is made on IRS Form 8621. U.S. Holders will be eligible to make QEF elections only if we agree to provide U.S. Holders with the information they will need to comply with the QEF rules. In the event we become a PFIC, we intend to provide all information and documentation that a U.S. Holder making a QEF election is required to obtain for U.S. federal income tax purposes (e.g., the U.S. Holder’s pro rata share of ordinary income and net capital gain, and a “PFIC Annual Information Statement” as described in applicable U.S. Treasury regulations).
Mark-to-Market Election. As an alternative to a QEF election, a U.S. Holder may also mitigate the adverse tax consequences of PFIC status by timely making a “mark-to-market” election, provided the U.S. Holder completes and files IRS Form 8621 in accordance with the relevant instructions and related Treasury regulations. A U.S. Holder who makes the mark-to-market election generally must include as ordinary income each year the increase in the fair market value of the common shares and deduct from gross income the decrease in the value of such shares during each of its taxable years, but with losses limited to the amount of previously recognized net gains. The U.S. Holder’s tax basis in the common shares would be adjusted to reflect any income or loss recognized as a result of the mark-to-market election. If a mark-to-market election with respect to our common shares is in effect on the date of a U.S. Holder’s death, the tax basis of the common shares in the hands of a U.S. Holder who acquired them from a decedent will be the lesser of the decedent’s tax basis or the fair market value of the common shares. Any gain from a sale, exchange or other disposition of the common shares in any taxable year in which we are a PFIC (i.e., when we meet the gross income test or asset test described above) would be treated as ordinary income and any loss from a sale, exchange or other disposition would be treated first as an ordinary loss (to the extent of any net mark-to-market gains previously included in income) and thereafter as a capital loss. If we cease to be a PFIC, any gain or loss recognized by a U.S. Holder on the sale or exchange of the common shares would be classified as a capital gain or loss.
A mark-to-market election is available to a U.S. Holder only for “marketable stock.” Generally, stock will be considered marketable stock if it is “regularly traded” on a “qualified exchange” within the meaning of applicable U.S. Treasury regulations. A class of stock is regularly traded during any calendar year during which such class of stock is traded, other than in de minimis quantities, on at least 15 days during each calendar quarter. The common shares should be marketable stock as long as they are listed on The Nasdaq Capital Market and are regularly traded. A mark-to-market election will not apply to the common shares for any taxable year during which we are not a PFIC but will remain in effect with respect to any subsequent taxable year in which we again become a PFIC. Such election will not apply to any subsidiary that we own. Accordingly, a U.S. Holder may continue to be subject to the PFIC rules with respect to any lower-tier PFICs notwithstanding the U.S. Holder’s mark-to-market election. Whether our common shares are regularly traded on a qualified exchange is an annual determination based on facts that, in part, are beyond our control. Accordingly, a U.S. Holder might not be eligible to make a mark-to-market election to mitigate the adverse tax consequences if we are characterized as a PFIC.
Election Tax Risks. Certain economic risks are inherent in making either a QEF Election or a mark-to-market election. If a QEF Election is made, it is possible that earned income will be reported to a U.S. shareholder as taxable income and income taxes will be due and payable on such an amount. A U.S. shareholder of our common shares may pay tax on such “phantom” income, i.e., where income is reported to it pursuant to the QEF Election, but no cash is distributed with respect to such income. There is no assurance that any distribution or profitable sale will ever be made regarding our common shares, so the tax liability may result in a net economic loss. A mark-to-market election may result in significant share price gains in one year causing a significant income tax liability. This gain may be offset in another year by significant losses. If a mark-to-market election is made, this highly variable tax gain or loss may result in substantial and unpredictable changes in taxable income. The amount included in income under a mark-to-market election may be substantially greater than the amount included under a QEF election. Both the QEF and mark-to-market elections are binding on the U.S. shareholder for all subsequent years that the U.S. shareholder owns our shares unless permission to revoke the election is granted by the IRS.
Purging Election. If we are a PFIC at any time when a U.S. Holder holds our common shares, we will generally continue to be treated as a PFIC with respect to the U.S. Holder for all succeeding years during which the U.S. Holder holds our common shares even if we cease to meet the PFIC gross income test or asset test in a subsequent year. However, if we cease to meet these tests, a U.S. Holder can avoid the continuing impact of the PFIC rules by making a special election (a Purging Election) to recognize gain by making a “deemed sale” election with respect to all of the U.S. Holder’s common shares and have such common shares deemed to be sold at their fair market value on the last day of the last taxable year during which we were a PFIC. The shareholder makes a purging election under Code section 1298(b)(1) and regulations section 1.1298–3 on IRS Form 8621 attached to the shareholder’s tax return (including an amended return), or requests the consent of the IRS Commissioner to make a late election under Code section 1298(b)(1) and regulations section 1.1298–3(e) (late purging election) on Form 8621-A. In addition, for a U.S. Holder making such an election, a new holding period would be deemed to begin for our common shares for purposes of the PFIC rules. After the Purging Election, the common shares with respect to which the Purging Election was made will not be treated as shares in a PFIC unless we subsequently again become a PFIC.
Each U.S. person who is a shareholder of a PFIC generally must file an annual report (on IRS Form 8621) with the IRS containing certain information, and the failure to file such report could result in the imposition of penalties on such U.S. person and in the extension of the statute of limitations with respect to federal income tax returns filed by such U.S. person. Should we be classified as a PFIC during a U.S. Holder’s holding period for our common shares, each such U.S. Holder should consult their own tax advisors with respect to the possibility of making these elections and the U.S. federal income tax consequences of the acquisition, ownership and disposition of our common shares. In addition, the possibility of us being classified as a PFIC may deter certain U.S. investors from purchasing our common shares, which could have an adverse impact on the market price of our common shares and our ability to raise additional financing by selling equity securities, including our common shares.
The U.S. federal income tax rules relating to PFICs are very complex. U.S. Holders are urged to consult their own tax advisors with respect to the purchase, ownership and disposition of common shares, the consequences to them of an investment in a PFIC, any elections available with respect to the common shares and the IRS information reporting obligations with respect to the purchase, ownership and disposition of common shares in the event we are considered a PFIC.
Additional Tax on Passive Income
Certain U.S. Holders that are individuals, estates or trusts (other than trusts that are exempt from tax) with adjusted income exceeding certain thresholds, will be subject to a 3.8% tax on all or a portion of their “net investment income,” which includes dividends on the common shares, and net gains from the disposition of the common shares. Further, excess distributions treated as dividends, gains treated as excess distributions, and mark-to-market inclusions and deductions are all included in the calculation of net investment income.
Treasury regulations provide, subject to the election described in the following paragraph, that solely for purposes of this additional tax, that distributions of previously taxed income will be treated as dividends and included in net investment income subject to the additional 3.8% tax. Additionally, to determine the amount of any capital gain from the sale or other taxable disposition of common shares that will be subject to the additional tax on net investment income, a U.S. Holder who has made a QEF election will be required to recalculate its basis in the common shares excluding any QEF election basis adjustments.
Alternatively, a U.S. Holder may make an election which will be effective with respect to all interests in controlled foreign corporations and PFICs that are subject to a QEF election and that are held in that year or acquired in future years. Under this election, a U.S. Holder pays the additional 3.8% tax on QEF election income inclusions and on gains calculated after giving effect to related tax basis adjustments. U.S. Holders that are individuals, estates or trusts should consult their own tax advisors regarding the applicability of this tax to any of their income or gains in respect of the common shares.
U.S. Federal Income Taxation of Non-U.S. Holders
A beneficial owner of our common shares, other than a partnership or entity treated as a partnership for U.S. Federal income tax purposes, that is not a U.S. Holder is referred to herein as a “Non-U.S. Holder”. Non-U.S. Holders generally will not be subject to U.S. federal income tax or withholding tax on dividends received from us with respect to our common shares, unless that income is effectively connected with the Non-U.S. Holder’s conduct of a trade or business in the United States. In general, if the Non-U.S. Holder is entitled to the benefits of certain U.S. income tax treaties with respect to those dividends, that income is taxable only if it is attributable to a permanent establishment maintained by the Non-U.S. Holder in the United States.
Non-U.S. Holders generally will not be subject to U.S. federal income tax or withholding tax on any gain realized upon the sale, exchange or other disposition of our common shares, unless:
|
● |
the gain is effectively connected with the Non-U.S. Holder’s conduct of a trade or business in the United States. In general, if the Non-U.S. Holder is entitled to the benefits of certain income tax treaties with respect to that gain, that gain is taxable only if it is attributable to a permanent establishment maintained by the Non-U.S. Holder in the United States; or |
|
● |
the Non-U.S. Holder is an individual who is present in the United States for 183 days or more during the taxable year of disposition and other conditions are met. |
If the Non-U.S. Holder is engaged in a U.S. trade or business for U.S. federal income tax purposes, the income from the common shares, including dividends and the gain from the sale, exchange or other disposition of the stock, that is effectively connected with the conduct of that trade or business will generally be subject to regular U.S. federal income tax in the same manner as discussed above relating to the general taxation of U.S. Holders. In addition, if you are a corporate Non-U.S. Holder, your earnings and profits that are attributable to the effectively connected income, which are subject to certain adjustments, may be subject to an additional branch profits tax at a rate of 30%, or at a lower rate as may be specified by an applicable U.S. income tax treaty.
Information Reporting with Respect to Foreign Financial Assets
U.S. individuals that own “specified foreign financial assets” (as defined in Section 6038D of the Code) with an aggregate fair market value exceeding certain threshold amounts generally are required to file an information report on IRS Form 8938 with respect to such assets with their tax returns. Significant penalties may apply to persons who fail to comply with these rules. Specified foreign financial assets include not only financial accounts maintained in foreign financial institutions, but also any stock or security issued by a non-U.S. person, such as our common shares, unless held in accounts maintained by certain financial institutions. Upon the issuance of future U.S. Treasury regulations, these information reporting requirements may apply to certain U.S. entities that own specified foreign financial assets. The failure to report information required under the current regulations could result in substantial penalties and in the extension of the statute of limitations with respect to federal income tax returns filed by a U.S. Holder. U.S. Holders should consult their own tax advisors regarding the possible implications of these U.S. Treasury regulations for an investment in our common shares.
Special Reporting Requirements for Transfers to Foreign Corporations
A U.S. Holder that acquires common shares generally will be required to file IRS Form 926 with the IRS if (1) immediately after the acquisition such U.S. Holder, directly or indirectly, owns at least 10% of our common shares, or (2) the amount of cash transferred in exchange for common shares during the 12-month period ending on the date of the acquisition exceeds USD $100,000. Significant penalties may apply for failing to satisfy these filing requirements. U.S. Holders are urged to contact their tax advisors regarding these filing requirements.
Information Reporting and Backup Withholding
Dividends on and proceeds from the sale or other disposition of common shares may be reported to the IRS unless the U.S. Holder establishes a basis for exemption. Backup withholding may apply to amounts subject to reporting if (1) the U.S. holder fails to provide an accurate taxpayer identification number or otherwise establish a basis for exemption, (2) the U.S. Holder is notified by the IRS that backup withholding applies, or (3) the payment is described in certain other categories of persons.
If you sell your common shares through a U.S. office of a broker, the payment of the proceeds is subject to both U.S. backup withholding and information reporting unless you certify that you are a non-U.S. person, under penalties of perjury, or you otherwise establish an exemption. If you sell your common shares through a non-U.S. office of a non-U.S. broker and the sales proceeds are paid to you outside the United States, then information reporting and backup withholding generally will not apply to that payment. However, U.S. information reporting requirements, but not backup withholding, will apply to a payment of sales proceeds, even if that payment is made to you outside the United States, if you sell your common shares through a non-U.S. office of a broker that is a U.S. person or has certain other contacts with the United States, unless you certify that you are a non-U.S. person, under penalty of perjury, or you otherwise establish an exemption.
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules generally will be allowed as a refund or a credit against a U.S. Holder’s U.S. federal income tax liability if the required information is furnished by the U.S. Holder on a timely basis to the IRS.
THE DISCUSSION ABOVE IS A GENERAL SUMMARY. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A U.S. HOLDER. EACH U.S. HOLDER IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT OF AN INVESTMENT IN COMMON SHARES IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
|
Item 6. |
[Reserved] |
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations is based upon accounting principles generally accepted in the United States of America and discusses the financial condition and results of operations for DiaMedica Therapeutics Inc. and our subsidiaries for the years ended December 31, 2023 and 2022.
This discussion should be read in conjunction with our consolidated financial statements and related notes included elsewhere in this report. The following discussion contains forward-looking statements that involve numerous risks and uncertainties. Our actual results could differ materially from the forward-looking statements as a result of these risks and uncertainties. See “Cautionary Note Regarding Forward-Looking Statements” for additional cautionary information.
Business Overview
We are a clinical stage biopharmaceutical company committed to improving the lives of people suffering from serious diseases. Our lead candidate DM199 is the first pharmaceutically active recombinant (synthetic) form of the human tissue kallikrein-1 (KLK1) protein to be clinically studied in patients. KLK1 is an established therapeutic modality in Asia, with human urinary KLK1, for the treatment of acute ischemic stroke and porcine KLK1 for the treatment of cardio renal disease, including hypertension. We have also produced a potential novel treatment for severe inflammatory diseases, DM300, which is currently in the early preclinical stage of development. Our long-term goal is to use our patented and in-licensed technologies to establish our Company as a leader in the development and commercialization of therapeutic treatments from novel recombinant proteins. Our current focus is on the treatment of acute ischemic stroke (AIS) and cardio renal disease (CRD). We plan to advance DM199, our lead drug candidate, through required clinical trials to create shareholder value by establishing its clinical and commercial potential as a therapy for AIS and CRD.
DM199 is a recombinant form of human tissue kallikrein-1 (KLK1). KLK1 is a serine protease (protein) produced primarily in the kidneys, pancreas and salivary glands, which plays a critical role in the regulation of local blood flow and vasodilation (the widening of blood vessels which decreases vascular resistance) in the body, as well as an important role in inflammation and oxidative stress (an imbalance between potentially damaging reactive oxygen species, or free radicals, and antioxidants in your body). We believe DM199 has the potential to treat a variety of diseases where healthy functioning requires sufficient activity of KLK1 and its system, the kallikrein-kinin system (KKS).
Our product development pipeline is as follows:

AIS Phase 2/3 ReMEDy2 Trial
We are currently conducting our ReMEDy2 clinical trial of DM199 for the treatment of AIS. ReMEDy2 clinical trial is a Phase 2/3, adaptive design, randomized, double-blind, placebo-controlled trial intended to enroll approximately 350 patients at up to 100 sites globally. Patients enrolled in the trial will be treated with either DM199 or placebo within 24 hours of the onset of AIS symptoms. The trial excludes patients treated with tissue plasminogen activator (tPA), a thrombolytic agent intended to dissolve blood clots, and those with large vessel occlusions. The study population is representative of the approximately 80% of AIS patients who do not have treatment options today, primarily due to the limitations on treatment with tPA and/or mechanical thrombectomy. The primary endpoint of the ReMEDy2 trial is physical recovery from stroke as measured by the well-established modified Rankin Scale (mRS) at day 90, specifically recovering to an mRS score of 0-1 (mRS range of 0-6). We believe that the proposed trial has the potential to serve as a pivotal registration study of DM199 in this patient population.
We voluntarily paused participant enrollment in the ReMEDy2 trial in May 2022 to investigate three unexpected instances of clinically significant hypotension (low blood pressure) occurring shortly after initiation of the intravenous (IV) dose of DM199. The acutely low blood pressure levels in the three participants recovered back to their baseline blood pressure within minutes after the IV infusion was stopped, and the participants suffered no injuries. On July 6, 2022, we announced that the FDA placed a clinical hold on the investigational new drug application (IND) for our ReMEDy2 trial and the clinical hold was subsequently lifted in June 2023. In our request for lifting of the clinical hold, we submitted to the FDA in-vitro data supporting that the cause of the hypotensive events was likely related to switching to a new type of IV bag for use in the ReMEDy2 trial, as well as results of an additional in-use, in vitro stability study of all of the materials and equipment used in the IV administration of DM199, which included testing the combination of the IV bag, IV tubing and mechanical infusion pump, to further rule out any other cause of the hypotension events. We also modified the protocol to mitigate the risk of future hypotensive events, including a reduction in the DM199 dose level for the initial IV dose to effectively match the well tolerated IV dose administered in the ReMEDy1 trial.
Concurrently with performing the requested in-use study, we also conducted a Phase 1C open label, single ascending dose (SAD) study of DM199 administered with the PVC IV bags used in the ReMEDy2 trial. The purpose of the study was to confirm, with human data, the DM199 blood concentration levels achieved with the IV dose and further evaluate safety and tolerability. We also included a cohort of hypertensive patients being treated with ACEi prior to enrolling. All ACEi patients received the full IV dose at the 0.5 µg/kg level with no instances of hypotension. We believe that these results provide further assurance to potential investigators that ACEi patients may be safely included in the ReMEDy2 trial.
Prior to the clinical hold of our ReMEDy2 trial, we had experienced slower than expected site activations and enrollment in our ReMEDy2 trial and may continue to experience these conditions as we activate additional clinical sites and enroll participants. We believe this was due primarily to clinical staff shortages resulting from layoffs and employee burnout, the reallocation of clinical nurses to COVID-19 care, particularly during surges in COVID-19 cases, a loss of study coordinators resulting from budget constraints and COVID-19 vaccination requirements and concerns managing logistics and protocol compliance for participants discharged from the hospital to an intermediate care facility. In an effort to mitigate the impact of these factors, we have worked with our contract research organization to develop alternative procedures to support study sites and potential participants as needed. We intend to continue to monitor the results of these efforts or implement additional actions to mitigate the impact of these factors on our ReMEDy2 trial, however no assurances can be provided as to if and when these issues will resolve.
Cardio Rental Program
We plan to disclose additional data related to blood pressure control as part of supporting our plans for our cardio renal program, which we expect to disclose in 2024.
DM300
We have also produced a potential novel treatment for severe inflammatory diseases, DM300, which is currently in the early preclinical stage of development.
Financial Overview
We have not generated any revenues from product sales. Since our inception, we have financed our operations from public and private sales of equity, the exercise of warrants and stock options, interest income on funds available for investment and government grants. We have incurred losses in each year since our inception. Our net losses were $19.4 million and $13.7 million for the years ended December 31, 2023 and 2022, respectively. As of December 31, 2023, we had an accumulated deficit of $115.6 million. Substantially all of our operating losses resulted from expenses incurred in connection with our product candidate development programs, our primary R&D activities, and general and administrative (G&A) support costs associated with our operations and status as a publicly listed company.
We expect to continue to incur significant expenses and increased operating losses for at least the next several years. We anticipate that our quarterly expenses will increase relative to recent prior periods as we expand our ReMEDy2 trial globally and enrollment increases. Our efforts to expand our team to provide support for our operations and maintaining, expanding and protecting our intellectual property portfolio will also likely contribute to such increases.
While we expect our rate of future negative cash flow per month will generally increase as we globally expand our ReMEDy2 trial, we expect our current cash resources will be sufficient to allow us to continue our ReMEDy2 trial and otherwise fund our planned operations for at least the next 12 months from the date of issuance of the consolidated financial statements included in this report. However, the amount and timing of our future funding requirements will depend on many factors, including the timing and results of our ongoing development efforts, and the global expansion of our ReMEDy2 trial, specifically the rate of site activations and enrollment, the ongoing effects on our trial of COVID-19, including site staffing shortages, and competition for research staff due to other neurologic trials. Other factors, such as the potential expansion of our current and new development programs, and operating expenses incurred in connection with such activities may also contribute to fluctuations in the amount and timing of our future funding requirements. We may require significant additional funds earlier than we currently expect and there is no assurance that we will not need or seek additional funding prior to such time. We may elect to raise additional funds even before we need them if market conditions for raising additional capital are favorable.
Components of Our Results of Operations
Research and Development Expenses
We incurred R&D expenses of $13.1 million and $7.8 million for the years ended December 31, 2023 and 2022, respectively. R&D expenses consist primarily of fees paid to external service providers such as contract research organizations; clinical support services; clinical development including clinical site costs; outside nursing services and laboratory testing; and preclinical trials; fees paid to our contract manufacturing and development organizations and outside laboratories for development of DM199 and related manufacturing processes; costs for production runs of DM199; salaries, benefits, share-based compensation; and other personnel costs. Over the past approximately ten years, our R&D efforts have been primarily focused on developing DM199. At this time, due to the risks inherent in the clinical development process and the clinical stage of our product development programs, we are unable to estimate with any certainty the costs we will incur in developing DM199 through marketing approval or any of our preclinical development programs. The process of conducting clinical studies necessary to obtain regulatory approval and manufacturing scale-up to support expanded development and potential future commercialization is costly and time consuming. Any failure by us or delay in completing clinical studies, manufacturing scale-up or in obtaining regulatory approvals could lead to increased R&D expenses and, in turn, have a material adverse effect on our results of operations.
General and Administrative Expenses
We incurred G&A expenses of $8.2 million and $6.2 million for the years ended December 31, 2023 and 2022, respectively. G&A expenses consist primarily of salaries and related benefits, including share-based compensation related to our executive, finance, business development and support functions. G&A expenses also include insurance, including directors and officers liability coverage, rent and utilities, travel expenses, patent costs, and professional fees, including for auditing, tax and legal.
Other Income, Net
Other income, net consists primarily of interest income earned on marketable securities.
Critical Accounting Policies and Estimates
Management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these consolidated financial statements requires us to make estimates and assumptions for the reported amounts of assets, liabilities, revenue, expenses and related disclosures. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions and any such differences may be material.
While our significant accounting policies are more fully described in Note 3 to our consolidated financial statements included elsewhere in this report, we believe the following discussion addresses our most critical accounting policies, which are those that are most important to the portrayal of our financial condition and results of operations and require our most difficult, subjective and complex judgments.
Research and Development Costs
R&D costs include expenses incurred in the conduct of human clinical trials such as fees paid to external service providers such as contract research organizations; clinical support services; clinical development including clinical site costs; outside nursing services and laboratory testing. R&D costs also include non-clinical research studies; fees paid to contract manufacturing and development organizations and outside laboratories for the development of DM199 and related manufacturing processes; and costs to produce sufficient amounts of the DM199 compound for use in our clinical studies; consulting resources with specialized expertise related to execution of our development plan for our DM199 product candidate; and personnel costs, including salaries, benefits and share-based compensation.
We charge R&D costs to expense when incurred. Our human clinical trials are performed at clinical trial sites and are generally administered by us with assistance from contract research organizations (CROs), and include outside service providers such as outside nursing services, testing laboratories and data coordination and collection. Costs of setting up clinical trial sites are accrued upon execution of the trial agreement. Expenses related to the performance of clinical trials are accrued based on contracted amounts and the achievement of agreed upon milestones, such as participant enrollment, participant follow-up, etc. While we utilize electronic data capture systems to facilitate the transmission and capture of clinical trial activity, such information is often incomplete or delayed. Therefore we are required to estimate levels of performance under each significant contract, including, among other things, the extent of participant enrollment, the extent of supporting services performed and other activities through communications with the clinical trial sites, CROs and supporting vendors and adjust the estimates, if required, on a quarterly basis so that clinical expenses reflect the actual work performed at each clinical trial site and by each CRO or supporting vendor.
Share-based Compensation
We account for all share-based compensation awards using a fair value method. The cost of employee and non-employee services received in exchange for awards of equity instruments is measured and recognized based on the estimated grant date fair value of those awards. Compensation cost is recognized ratably using the straight-line attribution method over the vesting period, which is considered to be the requisite service period. We record forfeitures in the periods in which they occur.
The fair value of share-based awards is estimated using the Black-Scholes option pricing model. The determination of the fair value of share-based awards is affected by our common share price, as well as assumptions regarding a number of complex and subjective variables. Risk-free interest rates are based upon United States Government securities rates appropriate for the expected term of each award. Expected volatility rates are based on the historical volatility equal to the expected term of the option. The assumed dividend yield is zero, as we do not expect to declare any dividends in the foreseeable future. The expected term of options is estimated considering the vesting period at the grant date, the life of the option and the average length of time similar grants have remained outstanding in the past.
The assumptions used in calculating the fair value under the Black-Scholes option valuation model are set forth in the following table for options issued by us for the years ended December 31, 2023 and 2022:
|
2023 |
2022 |
|||||||||
|
Common share fair value |
$1.57 | – | $3.24 | $1.47 | – | $3.88 | ||||
|
Risk-free interest rate |
3.5 | – | 4.6% | 1.4 | – | 3.6% | ||||
|
Expected dividend yield |
0% | 0% | ||||||||
|
Expected option life (in years) |
5.0 | – | 5.7 | 5.0 | – | 5.6 | ||||
|
Expected stock price volatility |
101.7 | – | 108.1% | 102.1 | – | 104.0% | ||||
Results of Operations
Comparison of the Years Ended December 31, 2023 and 2022
The following table summarizes our results of operations for the years ended December 31, 2023 and 2022 (in thousands):
|
Year Ended December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Research and development expense |
$ | 13,110 | $ | 7,839 | ||||
|
General and administrative expense |
8,157 | 6,162 | ||||||
|
Other income, net |
(1,929 | ) | (353 | ) | ||||
Research and Development Expenses
R&D expenses increased to $13.1 million for the year ended December 31, 2023, up from $7.8 million in the prior year. The increase was driven principally by costs incurred for the in-use studies performed to address the recently lifted clinical hold on our ReMEDy2 AIS trial, costs incurred for the Phase 1C study and increased manufacturing and process development costs for DM199. Also contributing to the increase were higher personnel costs, including non-cash share-based compensation, associated with expanding the clinical team. We expect our R&D expenses to increase moderately as we globally expand the ReMEDy2 trial. The increases will be moderated by the completion of the REDUX and Phase 1C trials during 2023.
General and Administrative Expenses
G&A expenses were $8.2 million and $6.2 million for the year ended December 31, 2023 and 2022, respectively. This increase was primarily driven by increased legal fees incurred in connection with our lawsuit against PRA Netherlands and increased personnel costs incurred in conjunction with expanding our team. Increased costs for patent prosecution and non-cash share-based compensation also contributed to the increase. We expect that G&A expenses will remain steady or decline slightly as compared to prior periods.
Other Income, Net
Other income, net, was $1.9 million for the year ended December 31, 2023 compared to $0.4 million for 2022. This increase was driven by increased interest income recognized during 2023 as compared to 2022, related to both higher interest rates and increased marketable securities balances during 2023.
Liquidity and Capital Resources
The following tables summarize our liquidity and capital resources as of December 31, 2023 and 2022 and cash flows for each of the years ended December 31, 2023 and 2022, and are intended to supplement the more detailed discussion that follows (in thousands):
|
Liquidity and Capital Resources |
December 31, 2023 |
December 31, 2022 |
||||||
|
Cash, cash equivalents and marketable securities |
$ | 52,895 | $ | 33,502 | ||||
|
Total assets |
54,160 | 34,395 | ||||||
|
Total current liabilities |
2,786 | 2,168 | ||||||
|
Total shareholders’ equity |
51,057 | 31,827 | ||||||
|
Working capital |
50,889 | 31,667 | ||||||
|
Year Ended December 31, |
||||||||
|
Cash Flow Data |
2023 |
2022 |
||||||
|
Cash flow provided by (used in): |
||||||||
|
Operating activities |
$ | (18,728 | ) | $ | (11,511 | ) | ||
|
Investing activities |
(18,299 | ) | 11,538 | |||||
|
Financing activities |
36,842 | (6 | ) | |||||
|
Net increase (decrease) in cash and cash equivalents |
$ | (185 | ) | $ | 21 | |||
Liquidity and Capital Resources
We had cash, cash equivalents and marketable securities of $52.9 million, current liabilities of $2.8 million and working capital of $50.9 million as of December 31, 2023, compared to $33.5 million in cash, cash equivalents and marketable securities, $2.2 million in current liabilities and $31.7 million in working capital as of December 31, 2022. The increases in our combined cash, cash equivalents and marketable securities and in our working capital were due primarily to the net proceeds received from our April and June 2023 private placements, partially offset by cash used to fund our operations.
Cash Flows
Operating Activities
Net cash used in operating activities for the year ended December 31, 2023 was $18.7 million compared to $11.5 million for the year ended December 31, 2022. The increase in cash used in operating activities is driven primarily by our higher net loss and increased amortization of discounts on purchased marketable securities, partially offset by non-cash share-based compensation and the effects of the changes in operating assets and liabilities during 2023.
Investing Activities
Investing activities consist primarily of purchases and maturities of marketable securities. Net cash used in investing activities was $18.3 million for the year ended December 31, 2023 compared to net cash provided by investing activities of $11.5 million for the year ended December 31, 2022. This change resulted primarily from the timing of maturities and, in the current year, investments of the net proceeds from our June 2023 private placement.
Financing Activities
Net cash provided by financing activities was $36.8 million for the year ended December 31, 2023 consisting primarily of net proceeds from the sale of common shares in our April and June 2023 private placements. For the year ended December 31, 2022, net cash used in financing activities of $6,000 was comprised entirely of principal payments on finance lease obligations.
Capital Requirements
Since our inception, we have incurred losses while advancing the R&D of our DM199 product candidate. We have not generated any revenues from product sales and do not expect to do so for at least three to four years. We do not know when or if, we will generate any revenues from product sales or out-licensing of our DM199 product candidate or any future product candidate. We do not expect to generate any revenue from product sales unless and until we obtain regulatory approval. We expect to continue to incur substantial operating losses until such time as any future product sales, licensing fees, milestone payments and/or royalty payments are sufficient to generate revenues to fund our continuing operations. We expect our operating losses to increase as compared to prior periods as we continue the research, development and clinical studies of, and seek regulatory approval for, our DM199 product candidate, including, in particular, the resumption and global expansion of our ReMEDy2 trial. In the long-term, subject to obtaining regulatory approval of our DM199 product candidate, or any future product candidate, and in the absence of the assistance of a strategic partner, we expect to incur significant commercialization expenses for product sales, marketing, manufacturing and distribution.
Accordingly, and notwithstanding the completion of our April and June 2023 private placements from which we received aggregate net proceeds of $36.8 million, we expect we will need substantial additional capital to further our R&D activities, current and anticipated future clinical studies, regulatory activities and otherwise develop our product candidate, DM199, or any future product candidate, to a point where the product candidate may be out-licensed or commercially sold. Although we are striving to achieve these plans, there is no assurance that these and other strategies will be achieved or that additional funding will be obtained on favorable terms or at all. We expect our rate of future negative cash flow per month will vary depending on our clinical activities and the timing of expenses incurred and will increase as we resume and globally expand our ReMEDy2 trial. We expect our current cash resources will be sufficient to continue our ReMEDy2 trial and otherwise fund our planned operations for at least the next twelve months from the date of issuance of the consolidated financial statements included in this report. However, the amount and timing of our future funding requirements will depend on many factors, including the timing and results of our ongoing development efforts, and specifically our ReMEDy2 trial, the rate of site activation and enrollment in such trial, the effects on such trial of COVID-19, site staffing shortages, competition for research staff and trial subjects due to other stroke trials, and other factors, as well as the potential expansion of our current and potential new development programs, and operating expenses incurred in connection with such activities. We may require significant additional funds earlier than we currently expect and there is no assurance that we will not need or seek additional funding prior to such time, especially if market conditions for raising additional capital are favorable.
Historically, we have financed our operations primarily from sales of equity securities and the exercise of warrants and stock options, and we expect to continue this practice for the foreseeable future. Our most recent equity financing was our June 2023 private placement in which we issued and sold an aggregate of 11,011,406 common shares pursuant to a securities purchase agreement at a purchase price of $3.40 per share to accredited investors, or $3.91 per share in the case of our participating directors and officers. As a result of the offering, we received gross proceeds of $37.5 million, which resulted in net proceeds to us of approximately $36.1 million, after deducting offering expenses. We do not have any existing credit facilities under which we could borrow funds. We may seek to raise additional funds through various sources, such as equity or debt financings, or through strategic collaborations and license agreements. We can give no assurances that we will be able to secure additional sources of funds to support our operations, or if such funds are available to us, that such additional financing will be sufficient to meet our needs or on terms acceptable to us. This is particularly true if our clinical data is not positive or economic and market conditions deteriorate.
To the extent we raise additional capital through the sale of equity or convertible debt securities, the ownership interests of our shareholders will be diluted. Debt financing, if available, may involve agreements that include conversion discounts, pledging our intellectual property as collateral or covenants limiting or restricting our ability to take specific actions, such as incurring additional debt or making capital expenditures. If we raise additional funds through government or other third-party funding, marketing and distribution arrangements or other collaborations, or strategic alliances or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. The availability of financing will be affected by our clinical data and other results of scientific and clinical research; the ability to attain regulatory approvals and other regulatory actions; market acceptance of our product candidates; the state of the capital markets generally with particular reference to pharmaceutical, biotechnology and medical companies; the status of strategic alliance agreements; and other relevant commercial considerations.
If adequate funding is not available when needed, we may be required to scale back our operations by taking actions that may include, among other things, implementing cost reduction strategies, such as reducing use of outside professional service providers, reducing the number of our employees or employee compensation, modifying or delaying the development of our DM199 product candidate; licensing to third parties the rights to commercialize our DM199 product candidate for AIS, CRD or other indications that we would otherwise seek to pursue, or otherwise relinquishing significant rights to our technologies, future revenue streams, research programs or product candidates or granting licenses on terms that may not be favorable to us; and/or divesting assets or ceasing operations through a merger, sale, or liquidation of our company.
Commitments and Contingencies
In the normal course of business, we incur obligations to make future payments as we execute our business plan. These obligations may relate to preclinical or clinical studies, manufacturing or manufacturing process development and other related or supporting activities. Currently, these obligations include costs to be incurred with contract research organizations, central laboratory and pharmacy services, clinical study sites, home nursing services, various other vendors supporting the performance of our clinical trials and contract manufacturing and development organizations. The contracts we enter into with these vendors and the commitments within these contracts are subject to significant variability based upon the actual activities/services performed by each vendor. As a result, the ultimate amounts due may be materially different as these obligations are affected by, among other factors, the number and pace of clinical study sites activated, the number of participants enrolled, the amount of time to complete trial enrollment and the time required to finalize, analyze and report our clinical trial results. Clinical research agreements are generally cancelable upon up to 60-90 days’ notice, with our obligation limited to costs incurred up to that date, including any non-cancelable costs. Cancelation terms for product manufacturing and process development contracts vary and are generally dependent upon timelines for sourcing research materials and reserving laboratory time. As of December 31, 2023, we estimate that our outstanding commitments, including such cancellable contracts, are approximately $15.3 million over the next 12 months and approximately $12.5 million in the following 12 months.
As of December 31, 2023, we had future operating lease obligation totaling approximately $396,000 over the remainder of the lease, of which approximately $80,000 is due over the next 12 months.
We have entered into a license agreement with Catalent Pharma Solutions, LLC (Catalent) whereby we have licensed certain gene expression technology and we contract with Catalent for the manufacture of DM199. Under the terms of this license, certain milestone and royalty payments may become due under this agreement and are dependent upon, among other factors, clinical trials, regulatory approvals and ultimately the successful development of a new drug, the outcome and timing of which is uncertain As of December 31, 2023, one milestone payment obligation remains which is tied to the first commercial sale. Following the launch of our first product, we will also incur a royalty of less than 1% on net sales. The royalty term is indefinite but the license agreement may be canceled by us on 90 days’ prior written notice. The license may not be terminated by Catalent unless we fail to make required milestone and royalty payments.
|
Item 7A. |
Quantitative and Qualitative Disclosures About Market Risk |
This Item 7A is inapplicable to DiaMedica as a smaller reporting company and has been omitted pursuant to Item 305(e) of SEC Regulation S-K.
|
Item 8. |
Financial Statements and Supplementary Data |
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page | |
| Report of Independent Registered Public Accounting Firm (PCAOB Firm ID |
87 |
| Consolidated Balance Sheets as of December 31, 2023 and 2022 | 88 |
| Consolidated Statements of Operations and Comprehensive Loss for the years ended December 31, 2023 and 2022 | 89 |
| Consolidated Statements of Shareholders’ Equity for the years ended December 31, 2023 and 2022 | 90 |
| Consolidated Statements of Cash Flows for the years ended December 31, 2023 and 2022 | 91 |
| Notes to Consolidated Financial Statements | 92-107 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Shareholders and Board of Directors of DiaMedica Therapeutics Inc.:
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of DiaMedica Therapeutics Inc. and Subsidiaries (the “Company”) as of December 31, 2023 and 2022, the related consolidated statements of operations and comprehensive loss, shareholders’ equity and cash flows for the years then ended, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved or are especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
/s/
We have served as the Company’s auditor since 2018.
March 19, 2024
DiaMedica Therapeutics Inc.
Consolidated Balance Sheets
(In thousands, except share amounts)
|
December 31, 2023 |
December 31, 2022 |
|||||||
|
ASSETS |
||||||||
|
Current assets: |
||||||||
|
Cash and cash equivalents |
$ | $ | ||||||
|
Marketable securities |
||||||||
|
Prepaid expenses and other assets |
||||||||
|
Amounts receivable |
||||||||
|
Total current assets |
||||||||
|
Non-current assets: |
||||||||
|
Operating lease right-of-use asset |
||||||||
|
Property and equipment, net |
||||||||
|
Total non-current assets |
||||||||
|
Total assets |
$ | $ | ||||||
|
LIABILITIES AND EQUITY |
||||||||
|
Current liabilities: |
||||||||
|
Accounts payable |
$ | $ | ||||||
|
Accrued liabilities |
||||||||
|
Finance lease obligation |
||||||||
|
Operating lease obligation |
||||||||
|
Total current liabilities |
||||||||
|
Non-current liabilities: |
||||||||
|
Finance lease obligation, non-current |
||||||||
|
Operating lease obligation, non-current |
||||||||
|
Total non-current liabilities |
||||||||
|
Commitments and contingencies (Note 10) |
|
|
||||||
|
Shareholders’ equity: |
||||||||
|
Common shares, par value; authorized; |
||||||||
|
Paid-in capital |
||||||||
|
Accumulated other comprehensive income (loss) |
( |
) | ||||||
|
Accumulated deficit |
( |
) | ( |
) | ||||
|
Total shareholders’ equity |
||||||||
|
Total liabilities and shareholders’ equity |
$ | $ | ||||||
See accompanying notes to consolidated financial statements.
DiaMedica Therapeutics Inc.
Consolidated Statements of Operations and Comprehensive Loss
(In thousands, except share and per share amounts)
|
Year Ended December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Operating expenses: |
||||||||
|
Research and development |
$ | $ | ||||||
|
General and administrative |
||||||||
|
Total operating expenses |
||||||||
|
Operating loss |
( |
) | ( |
) | ||||
|
Other income: |
||||||||
|
Other income, net |
||||||||
|
Total other income, net |
||||||||
|
Loss before income tax expense |
( |
) | ( |
) | ||||
|
Income tax expense |
( |
) | ( |
) | ||||
|
Net loss |
( |
) | ( |
) | ||||
|
Other comprehensive income (loss) |
||||||||
|
Unrealized gain (loss) on marketable securities |
( |
) | ||||||
|
Net loss and comprehensive loss |
$ | ( |
) | $ | ( |
) | ||
|
Basic and diluted net loss per share |
$ | ( |
) | $ | ( |
) | ||
|
Weighted average shares outstanding – basic and diluted |
||||||||
See accompanying notes to consolidated financial statements.
DiaMedica Therapeutics Inc.
Consolidated Statements of Shareholders’ Equity
(In thousands, except share amounts)
|
Common Shares |
Paid-In Capital |
Accumulated Other Comprehensive Income (Loss) |
Accumulated Deficit |
Total Shareholders’ Equity |
||||||||||||||||
|
Balances at December 31, 2021 |
$ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||
|
Share-based compensation expense |
— | |||||||||||||||||||
|
Unrealized loss on marketable securities |
— | — | ( |
) | ( |
) | ||||||||||||||
|
Net loss |
— | — | ( |
) | ( |
) | ||||||||||||||
|
Balances at December 31, 2022 |
$ | $ | ( |
) | $ | ( |
) | $ | ||||||||||||
|
Issuance of common shares, net of offering costs of $ |
||||||||||||||||||||
|
Issuance of common shares in settlement of deferred stock units |
— | — | — | — | ||||||||||||||||
|
Issuance of common shares in settlement of restricted stock units |
||||||||||||||||||||
|
Share-based compensation expense |
— | — | — | |||||||||||||||||
|
Unrealized gain on marketable securities |
— | — | ||||||||||||||||||
|
Net loss |
— | ( |
) | ( |
) | |||||||||||||||
|
Balances at December 31, 2023 |
$ | $ | $ | ( |
) | $ | ||||||||||||||
See accompanying notes to consolidated financial statements.
DiaMedica Therapeutics Inc.
Consolidated Statements of Cash Flows
(In thousands, except share amounts)
|
Year Ended December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Cash flows from operating activities: |
||||||||
|
Net loss |
$ | ( |
) |
$ | ( |
) |
||
|
Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
|
Share-based compensation |
||||||||
|
Amortization of discounts on marketable securities |
( |
) | ( |
) | ||||
|
Non-cash lease expense |
||||||||
|
Depreciation |
||||||||
|
Changes in operating assets and liabilities: |
||||||||
|
Amounts receivable |
( |
) | ||||||
|
Prepaid expenses and other assets |
( |
) |
( |
) |
||||
|
Accounts payable |
||||||||
|
Accrued liabilities |
||||||||
|
Net cash used in operating activities |
( |
) | ( |
) | ||||
|
Cash flows from investing activities: |
||||||||
|
Purchase of marketable securities |
( |
) |
( |
) |
||||
|
Maturities of marketable securities |
||||||||
|
Purchase of property and equipment |
( |
) |
( |
) |
||||
|
Net cash provided by (used in) investing activities |
( |
) | ||||||
|
Cash flows from financing activities: |
||||||||
|
Proceeds from issuance of common shares, net of offering costs |
||||||||
|
Principal payments on finance lease obligations |
( |
) |
( |
) |
||||
|
Net cash provided by (used in) financing activities |
( |
) | ||||||
|
Net increase (decrease) in cash and cash equivalents |
( |
) | ||||||
|
Cash and cash equivalents at beginning of period |
||||||||
|
Cash and cash equivalents at end of period |
$ | $ | ||||||
|
Supplemental disclosure of cash flow information: |
||||||||
|
Cash paid for income taxes |
$ | $ | ||||||
|
Assets acquired under operating lease |
$ | $ | ||||||
See accompanying notes to consolidated financial statements.
DiaMedica Therapeutics Inc.
Notes to Consolidated Financial Statements
|
1. |
Business |
DiaMedica Therapeutics Inc. and its wholly owned subsidiaries, DiaMedica USA Inc. and DiaMedica Australia Pty Ltd. (collectively, we, us, our, DiaMedica and the Company), exist for the primary purpose of advancing the clinical and commercial development of our proprietary recombinant KLK1 protein called DM199, for the treatment of neurological and cardio-renal diseases. Currently, our primary focus is on developing DM199, a recombinant form of the human tissue kallikrein-1 (KLK1) protein, for the treatment of acute ischemic stroke (AIS) and cardio-renal disease (CRD). Our parent company is governed under British Columbia’s Business Corporations Act, and our common shares are publicly traded on The Nasdaq Capital Market under the symbol “DMAC.”
|
2. |
Risks and Uncertainties |
DiaMedica operates in a highly regulated and competitive environment. The development, manufacturing and marketing of pharmaceutical products require approval from, and are subject to ongoing oversight by, the United States Food and Drug Administration (FDA) in the United States, the European Medicines Agency (EMA) in the European Union and comparable agencies in other countries. We are in the clinical stage of development of our initial product candidate, DM199, for the treatment of AIS and CRD. We have not completed the development of any product candidate and do not generate any revenues from the commercial sale of any product candidate. DM199 requires significant additional clinical testing and investment prior to seeking marketing approval and is not expected to be commercially available for at least three years, if at all.
On July 6, 2022, we announced that the FDA placed a clinical hold on the investigational new drug application (IND) for our Phase 2/3 ReMEDy2 trial. The clinical hold was issued following us voluntarily pausing participant enrollment in the trial to investigate three unexpected instances of clinically significant hypotension (low blood pressure) occurring shortly after initiation of the intravenous (IV) dose of DM199. In September 2022, we submitted our analysis of the events leading to and causing the hypotensive events, and proposed protocol modifications to address the mitigation of these events for future trial participants. Following review of this analysis, the FDA informed us that they were continuing the clinical hold and requesting, among other items, an additional in-use in vitro stability study of the IV administration of DM199, which includes testing the combination of the IV bag, IV tubing and mechanical infusion pump, to further rule out any other cause of the hypotension events. The requested in-use study was completed at an independent laboratory and the results were substantially consistent with our earlier testing of the IV bags. In May 2023, these additional supporting data were submitted to the FDA in our clinical hold response. In June 2023, the FDA completed review of our clinical hold response and informed us that the clinical hold was removed allowing us to resume our Phase 2/3 ReMEDy2 trial.
Prior to the clinical hold of our ReMEDy2 trial, we had experienced slower than expected site activations and enrollment in our ReMEDy2 trial and may continue to experience these conditions as we activate additional clinical sites and enroll participants. We believe this was due primarily to clinical staff shortages resulting from layoffs and employee burnout, the reallocation of clinical nurses to COVID-19 care, particularly during surges in COVID-19 cases, a loss of study coordinators resulting from budget constraints and COVID-19 vaccination requirements and concerns managing logistics and protocol compliance for participants discharged from the hospital to an intermediate care facility. In an effort to mitigate the impact of these factors, we have worked with our contract research organization to develop alternative procedures to support study sites and potential participants as needed. We intend to continue to monitor the results of these efforts or implement additional actions to mitigate the impact of these factors on our ReMEDy2 trial, however no assurances can be provided as to if and when these issues will resolve.
Our future success is dependent upon the success of our development efforts, our ability to demonstrate clinical progress for our DM199 product candidate in the United States or other markets, our ability, or the ability of any future partner, to obtain required governmental approvals of our product candidate, our ability to license or market and sell our DM199 product candidate and our ability to obtain additional financing to fund these efforts.
As of December 31, 2023, we have incurred losses of $
Our principal source of cash has been net proceeds from the issuance of equity securities. Although we have previously been successful in obtaining financing through equity securities offerings, there is no assurance that we will be able to do so in the future. This is particularly true if our clinical data is not positive or if economic and market conditions deteriorate.
We expect that we will need substantial additional capital to further our research and development activities, complete the required clinical studies, regulatory activities and manufacturing development for our product candidate, DM199, or any future product candidates, to a point where they may be licensed or commercially sold. We expect our current cash, cash equivalents and marketable securities to continue our ReMEDy2 trial and otherwise fund our planned operations for at least the next 12 months from the date of issuance of these consolidated financial statements. The amount and timing of our future funding requirements will depend on many factors, including timing and results of our ongoing development efforts, including our current ReMEDy2 trial and the rate of site activation and enrollment in the study, the potential expansion of our current development programs, potential new development programs, the effects of COVID-19, staffing shortages and other factors on our clinical trials and our operating expenses. We may require significant additional funds earlier than we currently expect and there is no assurance that we will not need or seek additional funding prior to such time, especially if market conditions for raising capital are favorable.
|
3. |
Summary of Significant Accounting Policies |
Basis of consolidation
The accompanying consolidated financial statements include the assets, liabilities and expenses of DiaMedica Therapeutics Inc., and our wholly-owned subsidiaries, DiaMedica USA, Inc. and DiaMedica Australia Pty Ltd. All significant intercompany transactions and balances have been eliminated in consolidation.
Functional currency
Use of estimates
Cash and cash equivalents
Marketable securities
The Company’s marketable securities may consist of obligations of the United States government and its agencies, bank certificates of deposit and/or investment grade corporate obligations, which are classified as available-for-sale. Marketable securities which mature within 12 months from their date of purchase are included in current assets. Securities are valued based on market prices for similar assets using third party certified pricing sources. Available-for-sale securities are carried at fair value. The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity. Such amortization or accretion is included in interest income. Realized gains and losses, if any, are calculated on the specific identification method and are included in other income in the consolidated statements of operations.
We conduct periodic reviews to identify and evaluate each available-for-sale debt security that is in an unrealized loss position in order to determine whether an other-than-temporary impairment exists. An unrealized loss exists when the current fair value of an individual security is less than its amortized cost basis. Declines in fair value considered to be temporary and caused by noncredit-related factors, are recorded in accumulated other comprehensive loss, which is a separate component of shareholders’ equity. Declines in fair value that are other than temporary or caused by credit-related factors, are recorded within earnings as an impairment loss. There were no other-than-temporary unrealized losses as of December 31, 2023.
Concentration of credit risk
Financial instruments that potentially expose the Company to concentration of credit risk consist primarily of cash, cash equivalents and marketable securities. The Company maintains its cash balances primarily with two financial institutions. These balances generally exceed federally insured limits. The Company has not experienced any losses in such accounts and believes it is not exposed to any significant credit risk in cash and cash equivalents. The Company believes that the credit risk related to marketable securities is limited due to the adherence to an investment policy focused on the preservation of principal.
Fair value measurements
Under the authoritative guidance for fair value measurements, fair value is defined as the exit price, or the amount that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants as of the measurement date. The authoritative guidance also establishes a hierarchy for inputs used in measuring fair value that maximizes the use of observable inputs and minimizes the use of unobservable inputs by requiring that the most observable inputs be used when available. Observable inputs are inputs market participants would use in valuing the asset or liability developed based on market data obtained from sources independent of the Company. Unobservable inputs are inputs that reflect the Company’s assumptions about the factors market participants would use in valuing the asset or liability developed based upon the best information available in the circumstances. The categorization of financial assets and financial liabilities within the valuation hierarchy is based upon the lowest level of input that is significant to the fair value measurement.
The hierarchy is broken down into three levels defined as follows:
Level 1 Inputs — quoted prices in active markets for identical assets and liabilities
Level 2 Inputs — observable inputs other than quoted prices in active markets for identical assets and liabilities
Level 3 Inputs — unobservable inputs
As of December 31, 2023, the Company believes that the carrying amounts of its other financial instruments, including amounts receivable, accounts payable and accrued liabilities, approximate their fair value due to the short-term maturities of these instruments. See Note 4, titled “Marketable Securities” for additional information.
Long-lived assets
Leases
We determine if an arrangement is a lease at inception. We have made a policy election to not separate lease and non-lease components for our real estate leases to the extent they are fixed. Non-lease components that are not fixed are expensed as incurred as variable lease expense. Our facility lease includes variable non-lease components, such as common-area maintenance costs. Our operating lease is included in operating lease right-of-use (“ROU”) asset and operating lease obligations on our consolidated balance sheets. Our operating lease ROU asset represents our right to use an underlying asset for the lease term and operating lease liabilities represent our obligation to make lease payments arising from the lease. The operating lease ROU asset and operating lease obligation are recognized based on the present value of lease payments over the lease term. The lease does not provide an implicit rate and, due to the lack of a commercially salable product, we are generally considered unable to obtain commercial credit. Therefore, considering the quoted rates for the lowest investment-grade debt and the interest rates implicit in recent financing leases, we estimated our incremental borrowing rate. The operating lease ROU asset excludes lease incentives. Our lease includes an option to extend or terminate the lease; lease terms are only adjusted for these options when it is reasonably certain that we will exercise such options to extend or terminate the lease. Lease expense is recognized on a straight-line basis over the lease term.
Assumptions made by us at the commencement date are re-evaluated upon occurrence of certain events, including a lease modification. A lease modification results in a separate contract when the modification grants the lessee an additional right of use not included in the original lease and when lease payments increase commensurate with the standalone price for the additional right of use. When a lease modification results in a separate contract, it is accounted for in the same manner as a new lease.
Research and development costs
Research and development (R&D) costs include expenses incurred in the conduct of human clinical trials such as fees paid to external service providers such as contract research organizations; clinical support services; clinical development including clinical site costs; outside nursing services and laboratory testing. R&D costs also include non-clinical research studies; fees paid to contract manufacturing and development organizations and outside laboratories for the development of DM199 and related manufacturing processes; and costs to produce sufficient amounts of the DM199 compound for use in our clinical studies; consulting resources with specialized expertise related to execution of our development plan for our DM199 product candidate; and personnel costs, including salaries, benefits and share-based compensation.
We charge research and development costs, including clinical trial costs, to expense when incurred. Our human clinical trials are performed at clinical trial sites and are administered jointly by us with assistance from various contract research organizations. Costs of setting up clinical trial sites are accrued upon execution of the study agreement. Expenses related to the performance of clinical trials are recorded or accrued based on actual invoices received and estimates of work completed to date by clinical trial sites, contract research organizations and outside vendors that assist with management and performance of the trials, and those that manufacture the investigational product. While we utilize electronic data capture systems to facilitate the transmission and capture of clinical trial activity, such information is often incomplete or delayed. Therefore we are required to estimate the levels of performance under each significant contract, including, among other things, the extent of participant enrollment, the extent of supporting services performed and other activities through communications with the clinical trial sites, CROs and supporting vendors and adjust the estimates, if required, on a quarterly basis so that clinical expenses reflect the actual work performed at each clinical trial site and by each CRO or supporting vendor. Additionally, actual costs may be charged to us and are recognized as the tasks are completed by the clinical trial site. Accrued R&D costs may be subject to revisions as clinical trials, non-clinical research and DM199 development programs progress and any revisions are recorded in the period in which the facts that give rise to the revisions become known.
Patent costs
Costs associated with applying for, prosecuting and maintaining patents are expensed as incurred given the uncertainty of patent approval and, if approved, the resulting probable future economic benefit to the Company. Patent-related costs, consisting primarily of legal expenses and filing/maintenance fees, are included in general and administrative costs and were $
Share-based compensation
The cost of employee and non-employee services received in exchange for awards of equity instruments is measured and recognized based on the estimated grant date fair value of those awards. Compensation cost is recognized ratably using the straight-line attribution method over the vesting period, which is considered to be the requisite service period. We record forfeitures in the periods in which they occur.
The fair value of option awards is estimated at the date of grant using the Black-Scholes option pricing model. The determination of the fair value of share-based awards is affected by our share price, as well as assumptions regarding a number of complex and subjective variables. Risk free interest rates are based upon United States Government bond rates appropriate for the expected term of each award. Expected volatility rates are based on the historical volatility over a period equal to the expected term of the option. The assumed dividend yield is zero, as we do not expect to declare any dividends in the foreseeable future. The expected term of options is estimated considering the vesting period at the grant date, the life of the option and the average length of time similar grants have remained outstanding in the past.
Income taxes
Income taxes are accounted for under the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the consolidated financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carry-forwards. Deferred tax assets and liabilities are measured using enacted rates, for each of the jurisdictions in which the Company operates, expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in operations in the period that includes the enactment date. Valuation allowances are established when necessary to reduce deferred tax assets to the amount that is more likely than not to be realized. The Company has provided a full valuation allowance against the gross deferred tax assets as of December 31, 2023 and 2022. See Note 14, “Income Taxes” for additional information. The Company’s policy is to classify interest and penalties related to income taxes as income tax expense.
Net loss per share
We compute net loss per share by dividing our net loss (the numerator) by the weighted-average number of common shares outstanding (the denominator) during the period. Shares issued during the period and shares reacquired during the period, if any, are weighted for the portion of the period that they were outstanding. The computation of diluted earnings per share, or EPS, is similar to the computation of basic EPS except that the denominator is increased to include the number of additional common shares that would have been outstanding if the dilutive potential common shares had been issued. Our diluted EPS is the same as basic EPS due to the exclusion of common share equivalents as their effect would be anti-dilutive.
The following table summarizes our calculation of net loss per common share for the periods presented (in thousands, except share and per share data):
|
Year Ended December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Net loss |
$ | ( |
) | $ | ( |
) | ||
|
Weighted average shares outstanding—basic and diluted |
||||||||
|
Basic and diluted net loss per share |
$ | ( |
) | $ | ( |
) | ||
The following outstanding potential common shares were not included in the diluted net loss per share calculations as their effects were not dilutive:
|
Year Ended December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Employee and non-employee stock options |
||||||||
|
Common shares issuable under common share purchase warrants |
||||||||
|
Common shares issuable upon settlement of deferred stock units |
||||||||
Recent Accounting Pronouncements
Recently Adopted Accounting Pronouncements
In June 2016, the Financial Accounting Standards Board issued ASU No. 2016-13, Financial Instruments – Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments, which requires the measurement and recognition of expected credit losses for financial assets held at amortized cost. This ASU replaces the existing incurred loss impairment model with an expected loss model. It also eliminates the concept of other-than-temporary impairment and requires credit losses related to available-for-sale debt securities to be recorded through an allowance for credit losses rather than as a reduction in the amortized cost basis of the securities. These changes will result in earlier recognition of credit losses. The standard was effective for smaller reporting companies in fiscal years beginning after December 15, 2022 with early adoption permitted for all periods beginning after December 15, 2018. We adopted ASU No. 2016-13 on January 1, 2023, which did not have an impact on our consolidated financial statements.
|
4. |
Marketable Securities |
The available-for-sale marketable securities are primarily comprised of investments in commercial paper, corporate bonds and government securities and consist of the following, measured at fair value on a recurring basis (in thousands):
| Fair Value Measurements as of December 31, 2023
Using Inputs Considered as |
||||||||||||||||
|
Fair Value |
Level 1 |
Level 2 |
Level 3 |
|||||||||||||
|
Commercial paper and corporate bonds |
$ | $ | $ | $ | ||||||||||||
|
Government securities |
||||||||||||||||
|
Total marketable securities |
$ | $ | $ | $ | ||||||||||||
|
Fair Value Measurements as of December 31, 2022 Using Inputs Considered as |
||||||||||||||||
|
Fair Value |
Level 1 |
Level 2 |
Level 3 |
|||||||||||||
|
Commercial paper and corporate bonds |
$ | $ | $ | $ | ||||||||||||
|
Government securities |
||||||||||||||||
|
Total marketable securities |
$ | $ | $ | $ | ||||||||||||
Accrued interest receivable on available-for-sale securities was $
There were no transfers of assets between Level 1 and Level 2 of the fair value measurement hierarchy during the year ended December 31, 2023.
Under the terms of the Company’s investment policy, purchases of marketable securities are limited to investment grade governmental and corporate obligations and bank certificates of deposit with a primary objective of principal preservation. Maturities of individual securities are less than one year, and the amortized cost of all securities approximated fair value as of December 31, 2023 and 2022.
|
5. |
Amounts Receivable |
Amounts receivable consisted of the following (in thousands):
|
December 31, 2023 |
December 31, 2022 |
|||||||
|
Accrued interest receivable on marketable securities |
$ | $ | ||||||
|
Other |
||||||||
|
Total amounts receivable |
$ | $ | ||||||
|
6. |
Prepaid Expenses and Other Assets |
Prepaid expenses and other assets consisted of the following (in thousands):
|
December 31, 2023 |
December 31, 2022 |
|||||||
|
Advances to vendors |
$ | $ | ||||||
|
Prepaid expenses |
||||||||
|
Total prepaid expenses and other assets |
$ | $ | ||||||
We periodically advance funds to vendors engaged to support the performance of our clinical trials and related supporting activities. The funds advanced are held, interest free, for varying periods of time and may be recovered by the Company through partial reductions of ongoing invoices, application against final study/project invoices or refunded upon completion of services to be provided. Deposits are classified as current or non-current based upon their expected recovery time.
|
7. |
Property and Equipment |
Property and equipment consisted of the following (in thousands):
|
December 31, 2023 |
December 31, 2022 |
|||||||
|
Furniture and equipment |
$ | $ | ||||||
|
Computer equipment |
||||||||
|
Leasehold Improvements |
||||||||
|
Less accumulated depreciation |
( |
) | ( |
) | ||||
|
Property and equipment, net |
$ | $ | ||||||
Depreciation expense was $
|
8. |
Accrued Liabilities |
Accrued liabilities consisted of the following (in thousands):
|
December 31, 2023 |
December 31, 2022 |
|||||||
|
Accrued compensation |
||||||||
|
Accrued research and other professional fees |
||||||||
|
Accrued clinical trial costs |
||||||||
|
Accrued other liabilities |
||||||||
|
Total accrued liabilities |
$ | $ | ||||||
|
9. |
Operating Lease |
In June 2022, we entered into an agreement to lease approximately
Our operating lease costs were $
Maturities of our operating lease obligation are as follows as of December 31, 2023 (in thousands):
|
2024 |
||||
|
2025 |
||||
|
2026 |
||||
|
2027 |
||||
|
2028 |
||||
|
Total lease payments |
$ | |||
|
Less interest portion |
( |
) | ||
|
Present value of lease obligation |
$ |
Former office lease
We leased certain office space under a non-cancelable operating lease that terminated on August 31, 2022, and we did not renew it. This lease included lease (e.g., fixed rent) and non-lease components (e.g., common-area and other maintenance costs). The right-of-use asset for this lease was fully amortized as of August 31, 2022.
|
10. |
Commitments and Contingencies |
Clinical trials and product development
In the normal course of business, we incur obligations to make future payments as we execute our business plan. These obligations may relate to preclinical or clinical studies, manufacturing or manufacturing process development and other related or supporting activities. Currently, these obligations include costs to be incurred with contract research organizations, central laboratory and pharmacy services, clinical study sites, home nursing services, various other vendors supporting the performance of our clinical trials and contract manufacturing and development organizations. The contracts we enter into with these vendors and the commitments within these contracts are subject to significant variability based upon the actual activities/services performed by each vendor. As a result, the ultimate amounts due may be materially different as these obligations are affected by, among other factors, the number and pace of clinical study sites activated, the number of participants enrolled, the amount of time to complete trial enrollment and the time required to finalize, analyze and report our clinical trial results. Clinical research agreements are generally cancelable upon up to 60-90 days’ notice, with our obligation limited to costs incurred up to that date, including any non-cancelable costs. Cancelation terms for product manufacturing and process development contracts vary and are generally dependent upon timelines for sourcing research materials and reserving laboratory time. As of December 31, 2023, we estimate that our outstanding commitments, including such cancellable contracts, are approximately $
Technology license
We have entered into a license agreement with Catalent Pharma Solutions, LLC (Catalent) whereby we have licensed certain gene expression technology and we contract with Catalent for the manufacture of DM199. Under the terms of this license, certain milestone and royalty payments may become due under this agreement and are dependent upon, among other factors, clinical trials, regulatory approvals and ultimately the successful development of a new drug, the outcome and timing of which is uncertain. As of December 31, 2023, one milestone payment obligation remains which is due upon our first regulatory approval of DM199 for commercial sale. Following the launch of our first product, we will also incur a royalty of less than 1% on net sales. The royalty term is indefinite but the license agreement may be canceled by us on 90 days’ prior written notice. The license may not be terminated by Catalent unless we fail to make required milestone and royalty payments.
Indemnification of directors and officers
The Company, as permitted under laws of the BCBCA and in accordance with the Company’s Articles and indemnification agreements, will indemnify and advance expenses to its directors and officers to the fullest extent permitted by law and may choose to indemnify other employees or agents from time to time. The Company has secured insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in connection with their services to the Company. As of December 31, 2023, there was no pending litigation or proceeding involving any director or officer of the Company as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification. Insofar as indemnification for liabilities arising under the United States Securities Act of 1933, as amended (Securities Act) may be permitted to directors, officers and controlling persons of the Company, the Company has been advised that, in the opinion of the United States Securities and Exchange Commission (SEC), such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable. The Company believes the fair value of these indemnification agreements is minimal. Accordingly, the Company had recorded any liabilities for these obligations as of December 31, 2023 or 2022.
|
11. |
Shareholders’ Equity |
Authorized capital stock
DiaMedica has authorized share capital of an unlimited number of common voting shares, and the shares do not have a stated par value. Common shareholders are entitled to receive dividends as declared by the Company, if any, and are entitled to one vote per share at the Company’s annual general meeting and any extraordinary or special general meeting.
Equity issued during the year ended December 31, 2023
On April 10, 2023, in conjunction with his appointment as Chief Business Officer of DiaMedica, David Wambeke purchased
On June 21, 2023, we issued and sold an aggregate
In connection with the June 2023 private placement, we entered into a registration rights agreement (Registration Rights Agreement) with the investors pursuant to which we agreed to file with the United States Securities and Exchange Commission (SEC) a registration statement registering the resale of the shares sold in the June 2023 private placement (Resale Registration Statement). The Resale Registration Statement was filed with the SEC on June 30, 2023 and declared effective by the SEC on July 7, 2023. Under the terms of the Registration Rights Agreement, we agreed to keep the Resale Registration Statement effective at all times until the shares are no longer considered “Registrable Securities” under the Registration Rights Agreement and if we fail to keep the Resale Registration Statement effective, subject to certain permitted exceptions, we will be required to pay liquidated damages to the investors in an amount of up to
During the year ended December 31, 2023,
Equity issued during the year ended December 31, 2022
During the year ended December 31, 2022, we did issue any common shares or other equity securities, other than stock options and deferred stock units.
Shares reserved
Common shares reserved for future issuance are as follows:
|
December 31, 2023 |
||||
|
Employee and non-employee stock options |
||||
|
Common shares issuable upon settlement of deferred stock units |
||||
|
Common shares issuable under common share purchase warrants |
||||
|
Shares available for grant under the Amended and Restated 2019 Omnibus Incentive Plan |
||||
|
Shares available for grant under the 2021 Employment Inducement Incentive Plan |
||||
|
Total |
||||
|
12. |
Share-Based Compensation |
Amended and Restated 2019 Omnibus Incentive Plan
The DiaMedica Therapeutics Inc. Amended and Restated 2019 Omnibus Incentive Plan (the 2019 Plan) was adopted by the Board of Directors (Board) on March 10, 2022 and approved by our shareholders at our 2022 Annual General Meeting of Shareholders held on May 18, 2022.
The 2019 Plan permits the Board, or a committee or subcommittee thereof, to grant to the Company’s eligible employees, non-employee directors and certain consultants non-statutory and incentive stock options, stock appreciation rights, restricted stock awards, restricted stock units (RSUs), deferred stock units (DSUs), performance awards, non-employee director awards and other stock-based awards. We grant options to purchase common shares under the 2019 Plan at no less than the fair market value of the underlying common shares as of the date of grant. Options granted to employees and non-employee directors have a maximum term of years and generally vest over to years. Options granted to non-employees have a maximum term of years and generally vest over year. Subject to adjustment as provided in the 2019 Plan, the maximum number of the Company’s common shares authorized for issuance under the 2019 Plan is
2021 Employment Inducement Incentive Plan
On December 3, 2021, the Board adopted the DiaMedica Therapeutics Inc. 2021 Employment Inducement Incentive Plan (Inducement Plan) to facilitate the granting of equity awards as an inducement material to new employees joining the Company. The Inducement Plan was adopted without shareholder approval pursuant to Nasdaq Listing Rule 5635(c)(4) and is administered by the Compensation Committee of the Board of Directors. The Board reserved
Prior Stock Option Plan
The DiaMedica Therapeutics Inc. Stock Option Plan, Amended and Restated November 6, 2018 (Prior Plan), was terminated by the Board of Directors in conjunction with the shareholder approval of the 2019 Plan. Awards outstanding under the Prior Plan remain outstanding in accordance with and pursuant to the terms thereof. Options granted under the Prior Plan have terms similar to those used under the 2019 Plan. As of December 31, 2023, options to purchase an aggregate of
Prior Deferred Stock Unit Plan
The DiaMedica Therapeutics Inc. Amended and Restated Deferred Stock Unit Plan (Prior DSU Plan) was terminated by the Board of Directors in conjunction with the shareholder approval of the 2019 Plan. Awards outstanding under the Prior DSU Plan remain outstanding in accordance with and pursuant to the terms thereof. As of December 31, 2023, there were
Share-based compensation expense for each of the periods presented is as follows (in thousands):
|
December 31, 2023 |
December 31, 2022 |
|||||||
|
Research and development |
$ | $ | ||||||
|
General and administrative |
||||||||
|
Total share-based compensation |
$ | $ | ||||||
We recognize share-based compensation based on the fair value of each award as estimated using the Black-Scholes option valuation model. Ultimately, the actual expense recognized over the vesting period will only be for those shares that actually vest.
A summary of option activity is as follows (in thousands except share and per share amounts):
|
Shares Underlying Options |
Weighted Average Exercise Price Per Share |
Aggregate Intrinsic Value |
||||||||||
|
Balances as of December 31, 2021 |
$ | $ | ||||||||||
|
Granted |
||||||||||||
|
Exercised |
||||||||||||
|
Expired/cancelled |
( |
) | ||||||||||
|
Forfeited |
( |
) | ||||||||||
|
Balances as of December 31, 2022 |
$ | $ | ||||||||||
|
Granted |
||||||||||||
|
Exercised |
||||||||||||
|
Expired/cancelled |
( |
) | ||||||||||
|
Forfeited |
( |
) | ||||||||||
|
Balances as of December 31, 2023 |
$ | $ | ||||||||||
A summary of the status of our unvested shares underlying options during the year ended and as of December 31, 2023 is as follows:
|
Shares Underlying Options |
Weighted Average Grant Date Fair Value Per Share |
|||||||
|
Unvested as of December 31, 2022 |
$ | |||||||
|
Granted |
||||||||
|
Vested |
( |
) | ||||||
|
Forfeited |
( |
) | ||||||
|
Unvested as of December 31, 2023 |
$ | |||||||
Information about stock options outstanding, vested and expected to vest as of December 31, 2023, is as follows:
|
Outstanding, Vested and Expected to Vest |
Options Vested and Exercisable |
|||||||||||||||||||||
|
Per Share Exercise Price |
Shares |
Weighted Average Remaining Contractual Life (Years) |
Weighted Average Exercise Price |
Options Exercisable |
Weighted Average Remaining Contractual Life (Years) |
|||||||||||||||||
| $ |
- | $ |
$ | |||||||||||||||||||
| $ |
- | $ |
||||||||||||||||||||
| $ |
- | $ |
||||||||||||||||||||
| $ |
- | $ |
||||||||||||||||||||
| $ |
- | $ |
||||||||||||||||||||
| $ | ||||||||||||||||||||||
The cumulative grant date fair value of employee options vested during the years ended December 31, 2023 and 2022 was $
As of December 31, 2023, total compensation expense related to unvested employee stock options not yet recognized was $
The assumptions used in calculating the fair value under the Black-Scholes option valuation model are set forth in the following table for options issued by the Company for the years ended December 31, 2023 and 2022:
|
2023 |
2022 |
|||||||||
|
Common share fair value |
$ |
– | $ |
$ |
– | $ |
||||
|
Risk-free interest rate |
– | – | ||||||||
|
Expected dividend yield |
||||||||||
|
Expected option life (years) |
– | – | ||||||||
|
Expected stock price volatility |
– | – |
Deferred Stock Units and Restricted Stock Units
Under our non-employee director compensation program, non-employee directors may elect to receive RSUs or DSUs in lieu of all or a portion of the annual cash retainers payable to such director. Each RSU or DSU represents the right to receive one share of our common stock. These recipients receive a number of RSUs or DSUs equal to the amount of the elected portion of the annual cash retainers divided by the 10-trading day average closing sale price of the common stock as determined on the third (3rd) business day prior to the anticipated grant date of the award. Vesting for these annual RSU and DSU grants is quarterly over one year, conditioned on continuous service. The cost of the RSUs and DSUs is measured and recognized based on the fair market value of our common shares on the date of grant. RSUs will be settled immediately upon vesting and DSU awards will be settled following a separation from service by such director.
There were approximately
13. Employee Benefit Plan
We maintain an employee 401(k) retirement savings plan (401(k) Plan). The 401(k) Plan provides eligible employees with an opportunity to make tax-deferred contributions into a long-term investment and savings program. All employees over the age of 21 may elect to participate in the 401(k) Plan beginning on their hire date. The 401(k) Plan allows eligible employees to contribute a portion of their annual compensation, subject only to maximum limits required by law. We contribute an amount up to
We have recorded contribution expenses of $
14. Income Taxes
The Company has incurred net operating losses since inception. The Company has not reflected the benefit of net operating loss carryforwards in the accompanying consolidated financial statements and has established a full valuation allowance against its deferred tax assets.
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes as well as operating losses and tax credit carryforwards.
The significant components of our deferred tax assets and liabilities are as follows (in thousands):
|
December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Deferred tax assets (liabilities): |
||||||||
|
Non-capital losses carried forward |
$ | $ | ||||||
|
Research and development expenditures |
||||||||
|
Share issue costs |
||||||||
|
Patents and other |
||||||||
|
Accruals |
||||||||
|
Share-based compensation |
||||||||
|
Property and equipment |
( |
) | ( |
) | ||||
|
Total deferred tax asset, net |
||||||||
|
Valuation allowance |
( |
) | ( |
) | ||||
|
Net deferred tax asset |
$ | $ | ||||||
Realization of the future tax benefits is dependent on our ability to generate sufficient taxable income within the carryforward period. Because of our history of operating losses, management believes that the deferred tax assets arising from the above-mentioned future tax benefits are currently not likely to be realized and, accordingly, we have provided a full valuation allowance.
The reconciliation of the Canadian statutory income tax rate applied to the net loss for the year to the income tax expense is as follows (in thousands):
|
December 31, |
||||||||
|
2023 |
2022 |
|||||||
|
Statutory income tax rate |
% | % | ||||||
|
Income tax recovery based on statutory rate |
$ | ( |
) | $ | ( |
) | ||
|
Share-based compensation |
||||||||
|
Prior-year true-ups |
( |
) | ( |
) | ||||
|
Share issuance costs |
( |
) | ||||||
|
Other |
||||||||
|
Change in valuation allowance |
||||||||
|
Income tax expense |
$ | $ | ||||||
Net operating losses and tax credit carryforwards as of December 31, 2023, are as follows:
|
Amount (In thousands) |
Expiration Years |
||||
|
Non-capital income tax losses, net |
$ |
Beginning 2026 |
|||
|
Research and development expense carry forwards |
Indefinitely |
||||
|
Tax credits |
Beginning 2024 |
||||
The Company is subject to taxation in Canada, the United States and Australia. Tax returns, since the inception of DiaMedica Therapeutics Inc., are subject to examinations by Canadian tax authorities and may change upon examination. Tax returns of DiaMedica USA, Inc., since its inception in 2012 and thereafter, are subject to examination by the U.S. federal and state tax authorities. Tax returns of DiaMedica Therapeutics Australia Pty Ltd., since its inception in 2016 and thereafter, are subject to examination by the Australian tax authorities.
Item 9. Changes In and Disagreements With Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the United States Securities Exchange Act of 1934, as amended (Exchange Act)) that are designed to provide reasonable assurance that information required to be disclosed by us in the reports we file or submit under the Exchange Act, is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure. Our management evaluated, with the participation of its Chief Executive Officer and its Chief Financial Officer, the effectiveness of the design and operation of our disclosure controls and procedures as of the end of the period covered in this report. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures were effective as of the end of such period to provide reasonable assurance that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure.
Management’s Report on Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Rule 13a-15(f) under the Exchange Act.
Our management conducted an assessment of the effectiveness of our internal control over financial reporting based on the criteria set forth in “Internal Control-Integrated Framework (2013)” issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on this assessment, management concluded that, as of December 31, 2023, our internal control over financial reporting was effective.
This annual report on Form 10-K does not include an attestation report of our independent registered public accounting firm on internal control over financial reporting due to an exemption established by the JOBS Act for “smaller reporting companies.”
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting that occurred during the fourth quarter ended December 31, 2023 that has materially affected or is reasonably likely to materially affect our internal control over financial reporting.
Item 9B. Other Information
Rule 10b5-1 Plan and Non-Rule 10b5-1 Trading Arrangement Adoptions, Terminations, and Modifications
During the three months ended December 31, 2023, of our directors or “officers” (as defined in Rule 16a-1(f) under the Exchange Act) adopted or terminated a “Rule 10b5-1 trading arrangement” or “non-Rule 10b5-1 trading arrangement,” as each term is defined in Item 408(a) and 408(c) respectively of SEC Regulation S-K.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
Directors
The information in the “Voting Proposal One – Election of Directors” section of our definitive proxy statement to be filed with the SEC with respect to our next annual general meeting of shareholders, which involves the election of directors, is incorporated in this annual report on Form 10-K by reference.
Executive Officers
Information concerning our executive officers is included in this annual report on Form 10-K under Item 1 of Part I under “Information About Our Executive Officers.”
Code of Ethics
We have adopted a code of business conduct and ethics applicable to all of our directors, officers and employees, in accordance with Section 406 of the Sarbanes-Oxley Act, the rules of the SEC promulgated thereunder, and the Nasdaq Listing Rules. In the event that any changes are made or any waivers from the provisions of the code of business conduct and ethics are made, these events would be disclosed on our website or in a report on Form 8-K within four business days of such event. The code of business conduct and ethics is posted on our website at www.diamedica.com. Copies of the code of business conduct and ethics will be provided free of charge upon written request directed to Investor Relations, DiaMedica Therapeutics Inc., 301 Carlson Parkway, Suite 210, Minneapolis, Minnesota 55305.
Changes to Nomination Procedures
During the fourth quarter of fiscal 2023, we made no material changes to the procedures by which shareholders may recommend nominees to our Board of Directors.
Audit Committee Matters
The information in the “Corporate Governance—Audit Committee” section of our definitive proxy statement to be filed with the SEC with respect to our next annual general meeting of shareholders, which involves the election of directors, is incorporated in this annual report on Form 10-K by reference.
Item 11. Executive Compensation
The information in the “Director Compensation” and “Executive Compensation” sections of our definitive proxy statement to be filed with the SEC with respect to our next annual general meeting of shareholders, which involves the election of directors, is incorporated in this annual report on Form 10-K by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
Stock Ownership
The information in the “Stock Ownership—Security Ownership of Significant Beneficial Owners” and “Stock Ownership—Security Ownership of Management” sections of our definitive proxy statement to be filed with the SEC with respect to our next annual general meeting of shareholders, which involves the election of directors, is incorporated in this annual report on Form 10-K by reference.
Securities Authorized for Issuance under Equity Compensation Plans
The following table summarizes outstanding options and other awards under our equity compensation plans as of December 31, 2023. Our equity compensation plans as of December 31, 2022 were the DiaMedica Therapeutics Inc. Amended and Restated 2019 Omnibus Incentive Plan (2019 Plan), the DiaMedica Therapeutics Inc. Stock Option Plan, Amended and Restated November 6, 2018 (Prior Plan), the DiaMedica Therapeutics Inc. Amended and Restated Deferred Share Unit Plan (DSU Plan) and the DiaMedica Therapeutics Inc. 2021 Employment Inducement Incentive Plan (Inducement Plan).
Equity Compensation Plan Information
|
(a) |
(b) |
(c) |
||||||||||
|
Plan Category |
Number of Securities to be Issued Upon Exercise of Outstanding Options, Warrants and Rights |
Weighted-Average Exercise Price of Outstanding Options, Warrants and Rights |
Number of Securities Remaining Available for Future Issuance Under Equity Compensation Plans (excluding securities reflected in column (a)) |
|||||||||
|
Equity compensation plans approved by security holders |
3,479,918 | (1) | $ | 4.14 | (2) | 927,215 | ||||||
|
Equity compensation plans not approved by security holders |
605,000 | $ | 2.52 | 395,000 | (3) | |||||||
|
Total |
4,084,918 | $ | 5.10 | (2) | 1,322,215 | (4) | ||||||
|
(1) |
Amount includes 2,818,103 common shares issuable upon the exercise of stock options and 196,572 common shares issuable upon the settlement of DSU awards outstanding under the 2019 Plan, 447,910 common shares issuable upon the exercise of stock options under the Prior Plan and 17,333 common shares issuable under the DSU Plan. |
|
(2) |
Not included in the weighted-average exercise price calculation are 196,572 deferred stock unit awards under the 2019 Plan and 17,333 deferred stock unit awards under the DSU Plan. |
|
(3) |
On December 3, 2021, the Board adopted Inducement Plan to facilitate the granting of equity awards as an inducement material to new employees joining the Company. The Inducement Plan was adopted without shareholder approval pursuant to Nasdaq Listing Rule 5635(c)(4) and is administered by the Compensation Committee of the Board of Directors. The Board reserved 1,000,000 common shares of the Company for issuance under the Inducement Plan, which permits the grant of non-statutory options, stock appreciation rights, restricted stock awards, restricted stock units, performance awards and other stock-based awards, to eligible recipients. The only persons eligible to receive awards under the Inducement Plan are individuals who are new employees and satisfy the standards for inducement grants under Nasdaq Listing Rule 5635(c)(4) or 5635(c)(3), as applicable. Also on December 3, 2021, the Compensation Committee adopted a form of notice of option grant and option award agreement for use under the Inducement Plan, which contains terms substantially identical to the form of notice of option grant and option award agreement for use under the shareholder-approved 2019 Plan. The Inducement Plan has a term of 10 years. The share reserve under the Inducement Plan may be increased at the discretion of and approval by the Board. As of December 31, 2023, 605,000 option awards had been granted under the Inducement Plan. |
|
(4) |
Amount includes 927,215 shares remaining available for future issuance under the 2019 Plan and 395,000 remaining available for future issuance under the Inducement Plan. |
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information in the “Related Person Relationships and Transactions” and “Corporate Governance—Director Independence” sections of our definitive proxy statement to be filed with the SEC with respect to our next annual general meeting of shareholders, which involves the election of directors, is incorporated in this annual report on Form 10-K by reference.
Item 14. Principal Accountant Fees and Services
The information in the “Voting Proposal Two—Appointment of Baker Tilly US, LLP as our Independent Registered Public Accounting Firm and Authorization to Fix Remuneration” section of our definitive proxy statement to be filed with the SEC with respect to our next annual general meeting of shareholders, which involves the election of directors, is incorporated in this annual report on Form 10-K by reference.
PART IV
Item 15. Exhibits and Financial Statement Schedules
Financial Statements
Our consolidated financial statements are included in “Part II, Item 8. Financial Statements and Supplementary Data.”
Financial Statement Schedules
All financial statement schedules are omitted because they are inapplicable since we are a smaller reporting company.
Exhibits
The exhibits being filed or furnished with this report are listed below, along with an indication as to each management contract or compensatory plan or arrangement.
A copy of any of the exhibits listed or referred to herein will be furnished at a reasonable cost to any person who is a shareholder upon receipt from any such person of a written request for any such exhibit. Such request should be sent to: Mr. Scott Kellen, Chief Financial Officer and Corporate Secretary, DiaMedica Therapeutics Inc., 301 Carlson Parkway, Suite 210, Minneapolis, Minnesota 55305, Attn: Shareholder Information.
|
Item No. |
Item |
Method of Filing |
|
3.1 |
Notice of Articles of DiaMedica Therapeutics Inc. dated May 31, 2019 |
Incorporated by reference to Exhibit 3.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on June 4, 2019 (File No. 001-36291) |
|
3.2 |
Amended and Restated Articles of DiaMedica Therapeutics Inc. Effective May 17, 2023 |
Incorporated by reference to Exhibit 3.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on May 18, 2023 (File No. 001-36291) |
|
4.1 |
Description of Securities Registered Pursuant to Section 12 of the Securities Exchange Act of 1934 |
Filed herewith |
|
4.2 |
Specimen Certificate representing Voting Common Shares of DiaMedica Therapeutics Inc. |
Incorporated by reference to Exhibit 4.2 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on June 4, 2019 (File No. 001-36291) |
|
4.3 |
Incorporated by reference to Exhibit 4.5 to DiaMedica’s Registration Statement on Form S-3 as filed with the Securities and Exchange Commission on October 5, 2021 (File No. 333-260066) |
| Item No. | Item | Method of Filing |
|
4.4 |
Incorporated by reference to Exhibit 4.6 to DiaMedica’s Registration Statement on Form S-3 as filed with the Securities and Exchange Commission on June 30, 2023 (File No. 333-273068) |
|
|
10.1# |
DiaMedica Therapeutics Inc. Amended and Restated 2019 Omnibus Incentive Plan |
Incorporated by reference to Exhibit 10.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on May 19, 2022 (File No. 001-36291) |
|
10.2# |
Form of Option Award Agreement under the DiaMedica Therapeutics Inc. 2019 Omnibus Incentive Plan |
Incorporated by reference to Exhibit 10.2 to DiaMedica’s Annual Report on Form 10-K for the year ended December 31, 2021 (File No. 001-36291) |
|
10.3# |
Incorporated by reference to Exhibit 10.3 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on June 21, 2019 (File No. 001-36291) |
|
|
10.4# |
Incorporated by reference to Exhibit 10.1 to DiaMedica’s Quarterly Report on Form 10-Q for the quarterly period ended June 30, 2020 (File No. 001-36291) |
|
|
10.5# |
DiaMedica Therapeutics Inc. 2021 Employment Inducement Incentive Plan |
Incorporated by reference to Exhibit 10.5 to DiaMedica’s Annual Report on Form 10-K for the year ended December 31, 2021 (File No. 001-36291) |
|
10.6# |
Incorporated by reference to Exhibit 10.6 to DiaMedica’s Annual Report on Form 10-K for the year ended December 31, 2021 (File No. 001-36291) |
|
|
10.7# |
DiaMedica Therapeutics Inc. Stock Option Plan Amended and Restated November 6, 2018 |
Incorporated by reference to Exhibit 10.1 to DiaMedica’s Registration Statement on Form S-1 as filed with the Securities and Exchange Commission on November 9, 2018 (File No. 333-228313) |
|
10.8# |
Incorporated by reference to Exhibit 10.3 to DiaMedica’s Registration Statement on Form S-1 as filed with the Securities and Exchange Commission on November 9, 2018 (File No. 333-228313) |
|
|
10.9# |
Incorporated by reference to Exhibit 10.2 to DiaMedica’s Registration Statement on Form S-1 as filed with the Securities and Exchange Commission on November 9, 2018 (File No. 333-228313) |
| Item No. | Item | Method of Filing |
|
10.10# |
DiaMedica Therapeutics Inc. Amended and Restated Deferred Share Unit Plan |
Incorporated by reference to Exhibit 10.4 to DiaMedica’s Registration Statement on Form S-1 as filed with the Securities and Exchange Commission on November 9, 2018 (File No. 333-228313) |
|
10.11# |
Incorporated by reference to Exhibit 10.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on June 21, 2019 (File No. 001-36291) |
|
|
10.12# |
Form of Indemnification Agreement between DiaMedica Therapeutics Inc. and Each Director and Officer |
Incorporated by reference to Exhibit 10.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on June 4, 2019 (File No. 001-36291) |
|
10.13# |
Employment Agreement effective as of September 12, 2018 between DiaMedica USA, Inc. and Rick Pauls |
Incorporated by reference to Exhibit 10.6 to DiaMedica’s Registration Statement on Form S-1 as filed with the Securities and Exchange Commission on November 9, 2018 (File No. 333-228313) |
|
10.14# |
Employment Agreement effective as of September 12, 2018 between DiaMedica USA, Inc. and Scott Kellen |
Incorporated by reference to Exhibit 10.7 to DiaMedica’s Annual Report on Form 10-K for the year ended December 31, 2018 (File No. 001-36291) |
|
10.15# |
Employment Agreement effective as of January 3, 2022 between DiaMedica USA, Inc. and Kirsten Gruis |
Incorporated by reference to Exhibit 10.17 to DiaMedica’s Annual Report on Form 10-K for the year ended December 31, 2022 (File No. 001-36291) |
|
10.16# |
Incorporated by reference to Exhibit 10.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on September 5, 2023 (SEC File No. 001-36291) |
|
|
10.17# |
Incorporated by reference to Exhibit 10.2 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on September 5, 2023 (SEC File No. 001-36291) |
|
|
10.18 |
Incorporated by reference to Exhibit 10.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on June 29, 2022 (File No. 001-36291) |
|
|
10.19 |
Lease Guaranty Agreement dated June 22, 2022 by DiaMedica Therapeutics Inc. |
Incorporated by reference to Exhibit 10.2 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on June 29, 2022 (File No. 001-36291) |
| Item No. | Item | Method of Filing |
|
10.20(1) |
Incorporated by reference to Exhibit 10.12 to DiaMedica’s Registration Statement on Form S-1 as filed with the Securities and Exchange Commission on November 9, 2018 (File No. 333-228313) |
|
|
10.21 |
Incorporated by reference to Exhibit 10.13 to DiaMedica’s Registration Statement on Form S-1 as filed with the Securities and Exchange Commission on November 9, 2018 (File No. 333-228313) |
|
|
10.22 |
Incorporated by reference to Exhibit 10.19 to DiaMedica’s Annual Report on Form 10-K for the year ended December 31, 2019 (File No. 001-36291) |
|
|
10.23 |
Filed herewith |
|
|
10.24 |
Incorporated by reference to Exhibit 10.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on September 27, 2021 (File No. 001-36291) |
|
|
10.25# |
Incorporated by reference to Exhibit 10.1 to DiaMedica’s Current Report on Form 8-K as filed with the Securities and Exchange Commission on June 21, 2023 (SEC File No. 001-36291) |
|
|
21.1 |
Incorporated by reference to Exhibit 21.1 to DiaMedica’s Annual Report on Form 10-K for the year ended December 31, 2019 (File No. 001-36291) |
|
|
23.1 |
Filed herewith |
|
|
31.1 |
Filed herewith |
|
|
31.2 |
Filed herewith |
| Item No. | Item | Method of Filing |
|
32.1 |
Furnished herewith |
|
|
32.2 |
Furnished herewith |
|
|
97.1 |
Filed herewith |
|
|
101 |
The following materials from DiaMedica Therapeutics Inc.’s Annual Report on Form 10-K for the year ended December 31, 2023, formatted in Inline XBRL (Extensible Business Reporting Language): (i) the Consolidated Balance Sheets, (ii) the Consolidated Statements of Operations, (iii) the Consolidated Statements of Comprehensive Income (Loss), (iv) the Consolidated Statements of Equity, (v) the Consolidated Statements of Cash Flows, and (vi) Notes to Consolidated Financial Statements |
Filed herewith |
|
104 |
Cover Page Interactive Data File |
Embedded within the Inline XBRL document |
|
# |
Indicates a management contract or compensatory plan or arrangement. |
|
(1) |
Portions of this exhibit have been redacted and are subject to an order granting confidential treatment under Rule 406 of the United States Securities Act of 1933, as amended (File No. 333-228313, CF #36833). The redacted material was filed separately with the Securities and Exchange Commission. |
Item 16. Form 10-K Summary
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
DIAMEDICA THERAPEUTICS INC. |
|
|
|
|
|
|
|
|
|
|
Date: March 19, 2024 |
By: |
/s/ Rick Pauls |
|
|
|
Rick Pauls |
|
|
|
President and Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
Name |
Title |
Date |
||
|
/s/ Rick Pauls |
President, Chief Executive Officer and Director |
March 19, 2024 |
||
| Rick Pauls | (principal executive officer) | |||
|
/s/ Scott Kellen |
Chief Financial Officer and Secretary |
March 19, 2024 |
||
| Scott Kellen | (principal financial and accounting officer) | |||
|
/s/ Richard Pilnik |
Chairman of the Board |
March 19, 2024 |
||
| Richard Pilnik | ||||
|
/s/ Michael Giuffre, M.D. |
Director |
March 19, 2024 |
||
| Michael Giuffre, M.D. | ||||
|
/s/ Richard Kuntz |
Director |
March 19, 2024 |
||
| Richard Kuntz | ||||
|
/s/ Tanya N. Lewis |
Director |
March 19, 2024 |
||
| Tanya N. Lewis | ||||
|
/s/ James Parsons |
Director |
March 19, 2024 |
||
| James Parsons | ||||
|
/s/ Charles P. Semba, M.D. |
Director |
March 19, 2024 |
||
| Charles P. Semba, M.D. |
[page intentionally left blank]