UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM
(Mark One)
| REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR 12(g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For
the fiscal year ended
OR
OR
Date of event requiring this shell company report: Not applicable
For the transition period from _______ to _______
Commission
file number:
(Exact name of Registrant as specified in its charter)
n/a
(Translation of Registrant’s name into English)
Republic
of
(Jurisdiction of incorporation or organization)
(Address of principal executive offices)
Chairman and Director
Phone:
Email:
c/o CytoMed Therapeutics Limited
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
| Title of class | Trading Symbol | Name of exchange on which registered | ||
Securities registered or to be registered pursuant to Section 12(g) of the Act: None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act: None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or ordinary shares as of the close of the period covered by the annual report: As of December 31, 2023, there were 11,529,328 of the registrant’s ordinary shares, issued and outstanding. As of April 22, 2024 there were ordinary shares outstanding.
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☐ Yes ☒
If
this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section
13 or 15(d) of the Securities Exchange Act of 1934. ☐ Yes ☒
Note – Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 from their obligations under those Sections.
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2)
has been subject to such filing requirements for the past 90 days. ☐ Yes ☒
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule
405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant
was required to submit such files). ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See the definitions of “large accelerated filer”, “accelerated filer” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ | |
| Emerging
growth company |
If
an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant
has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided
pursuant to Section 13(a) of the Exchange Act.
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b) by the registered
public accounting firm that prepared or issued its audit report.
Indicate
by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive based compensation
received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b).
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| U.S. GAAP ☐ | Other ☐ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
☐
Yes ☒
(APPLICABLE ONLY TO ISSUERS INVOLVED IN BANKRUPTCY PROCEEDINGS DURING THE PAST FIVE YEARS)
Indicate by check mark whether the registrant has filed all documents and reports required to be filed by Sections 12, 13 or 15(d) of the Securities Exchange Act of 1934 subsequent to the distribution of securities under a plan confirmed by a court.
☐ Yes ☐ No
TABLE OF CONTENTS
| -i- |
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This annual report contains certain forward-looking statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. Forward-looking statements are based on our beliefs and assumptions and on information currently available to us, and include, without limitation, statements regarding our business, financial condition, strategy, results of operations, certain of our plans, objectives, assumptions, expectations, prospects and beliefs and statements regarding other future events or prospects. Forward-looking statements include all statements that are not historical facts and can be identified by the use of forward-looking terminology such as the words “believe”, “expect”, “plan”, “intend”, “seek”, “anticipate”, “estimate”, “predict”, “potential”, “assume”, “continue”, “may”, “will”, “should”, “could”, “shall”, “risk” or the negative of these terms or similar expressions that are predictions of or indicate future events and future trends.
By their nature, forward-looking statements involve risks and uncertainties because they relate to events and depend on circumstances that may or may not occur in the future. We caution you that forward-looking statements are not guarantees of future performance and that our actual results of operations, financial condition and liquidity, the development of the industry in which we operate and the effect of acquisitions on us may differ materially from those made in or suggested by the forward-looking statements contained in this annual report. In addition, even if our results of operations, financial condition and liquidity, the development of the industry in which we operate and the effect of acquisitions on us are consistent with the forward-looking statements contained in this annual report, those results or developments may not be indicative of results or developments in subsequent periods.
Factors that may cause our actual results to differ materially from those expressed or implied by the forward-looking statements in this annual report include, but are not limited to, the risks described under “Risk Factors”. For example, factors that could cause actual results to vary from projected results include without limitation the following:
| ● | our plans to develop and commercialize our product candidates; | |
| ● | the initiation, timing, progress and results of our current and future pre-clinical studies and clinical trials and our R&D programs; | |
| ● | our expectations regarding the impact of the ongoing COVID-19 pandemic on our business, our industry and the economy; | |
| ● | our estimates regarding expenses, future revenue, capital requirements and needs for additional financing; | |
| ● | our ability to successfully acquire or obtain licenses for additional product candidates on reasonable terms; | |
| ● | our ability to establish and maintain collaborations and/or obtain additional funding; | |
| ● | our ability to obtain regulatory approval for our current and future product candidates from the HSA and/or other relevant regulatory authorities; | |
| ● | our expectations regarding the potential market size and the rate and degree of market acceptance of our current and future product candidates; | |
| ● | our ability to fund our working capital requirements and expectations regarding the sufficiency of our capital resources; | |
| ● | our intellectual property position; | |
| ● | developments in and/or disputes concerning our intellectual property or other proprietary rights; | |
| ● | our expectations regarding government and third-party payor coverage and reimbursement; | |
| ● | our ability to compete in the markets we are in; | |
| ● | the impact of government laws and regulations and liabilities thereunder; | |
| ● | our need to hire additional personnel and our ability to attract and retain such personnel; | |
| ● | developments relating to our competitors and our industry; | |
| ● | we are incorporated in Singapore, and our shareholders may have more difficulty protecting their interests than they would as shareholders of a corporation incorporated in the United States; and | |
| ● | other risk factors as discussed under the section titled “Risk Factors”. |
Forward-looking statements speak only as of the date they are made, and we do not undertake any obligation to update them in light of new information or future developments or to release publicly any revisions to these statements in order to reflect later events or circumstances or to reflect the occurrence of unanticipated events.
| -ii- |
DEFINITIONS
Unless otherwise stated in this annual report or the context otherwise requires, references to:
| ● | “γδ TCR” are to gamma delta T-cell receptor; | |
| ● | “A*STAR” are to the Agency for Science, Technology and Research of Singapore; | |
| ● | “ACCA” are to the Association of Chartered Certified Accountants; | |
| ● | “ACRA” are to the Accounting and Corporate Regulatory Authority of Singapore; | |
| ● | “ACRA Code” are to the Code of Professional Conduct and Ethics for Public Accountants and Accounting Entities; | |
| ● | “ACTRIS” are to the Advanced Cell Therapy and Research Institute, Singapore; | |
| ● | “Advertisement Regulations” are to the Health Products (Advertisement of Specified Health Products) Regulations 2016 enacted under the HPA (as defined below); | |
| ● | “ANGELICA Trial” are to the Phase I trial to evaluate allogeneic NKG2DL-targeting chimeric antigen receptor-grafted gamma delta T cells (CTM-N2D) in subjects with advanced solid tumours or haematological malignancies; | |
| ● | “ASX” are to the Australian Securities Exchange Ltd; | |
| ● | “ATPL” are to Accelerate Technologies Pte. Ltd. (formerly known as ETPL); | |
| ● | “BAS” are to building automated system; | |
| ● | “BCMA” are to B-cell maturation antigen; | |
| ● | “BMR” are to batch manufacturing records; | |
| ● | “BNM” are to the Central Bank of Malaysia or Bank Negara Malaysia; | |
| ● | “Board” or “Board of Directors” are to the board of directors of the Company; | |
| ● | “CAR” are to chimeric antigen receptor; | |
| ● | “CAR-T” are to chimeric antigen receptor-modified T cells; | |
| ● | “CDCR” are to the Control of Drugs and Cosmetics Regulations 1984 of Malaysia, as amended; |
| -iii- |
| ● | “cGMP” are to current good manufacturing practice, a system for ensuring that products are consistently produced and controlled according to quality standards; | |
| ● | “cGMP Facility” are to the Company’s current good manufacturing practice and processing facility located at 12 Jalan Permas 9/16, Bandar Baru Permas Jaya, 81750 Johor, Malaysia; | |
| ● | “CGTP” are to Cell and Gene Therapy Products; | |
| ● | “Clean Air Regulations” are to the Environmental Quality (Clean Air) Regulations 2014 of Malaysia; | |
| ● | “Clinical Study Agreement” are to the Investigator-Initiated Clinical Study Agreement entered into between the Company and National University Hospital Singapore on March 10, 2023. | |
| ● | “Clinical Trials Regulations” are to the Health Products (Clinical Trials) Regulations of 2016 enacted under the HPA (as defined below), which governs clinical trials of therapeutic products and applicable CTGTP (as defined below) that are not observational trials within Singapore; | |
| ● | “Code” are to the Internal Revenue Code of 1986 of the United States of America, as amended; | |
| ● | “Comptroller” are to the comptroller of income tax in Singapore; | |
| ● | “Contract Manufacturing Organization” or “CMO” are to companies that provide drug development and drug manufacturing services to the companies in the pharmaceutical industry on a contract basis; | |
| ● | “Contract Research Organization” or “CRO” are to companies that provide research support to the companies in pharmaceutical and biotechnology industries on a contract basis; | |
| ● | “COVID-19” are to the worldwide novel coronavirus disease pandemic; | |
| ● | “CRIS” are to the Consortium for Clinical Research and Innovation Singapore, a wholly-owned subsidiary of the Ministry of Health Singapore; | |
| ● | “CRM” are to clinical research materials, which refer to any registered or unregistered therapeutic product, medicinal product, medicinal device, applicable CTGTP or placebo, that is manufactured, imported or supplied for the purpose of being used in clinical research, by way of administration to a trial participant in accordance with the research protocol or for a clinical purpose; | |
| ● | “CRM Regulations” are to the Health Products (Clinical Research Materials) Regulations of 2016 enacted under the HPA (as defined below), which govern the import, manufacture and supply of clinical research materials for use in clinical research studies within Singapore; | |
| ● | “CRS” are to cytokine release syndrome; | |
| ● | “CTA” are to Clinical Trial Authorization, which is required prior to the initiation of a clinical trial of a therapeutic product; | |
| ● | “CTGT” are to cell, tissue and gene therapy; | |
| ● | “CTGTP” are to cell, tissue and gene therapy products; | |
| ● | “CTGTP Regulations” are to the Health Products (Cell, Tissue and Gene Therapy Products) Regulations of 2021 enacted under the HPA (as defined below), which provide for regulating the manufacture, import, supply, presentation, registration, duties, and obligations of manufacturers, importers and other persons carrying out such activities as related to CTGTP in Singapore; | |
| ● | “CTIL” are to Clinical Trial Import License in Malaysia; | |
| ● | “CTM-N2D” are to CAR-gamma delta T cell proprietary product developed by our Group; |
| -iv- |
| ● | “CTM-GDT” are to allogeneic gamma delta T cell proprietary product developed by our Group; | |
| ● | “CTM-MSC” are to allogeneic umbilical cord derived mesenchymal stem cells proprietary product developed by our Group; | |
| ● | “CTX” are to Clinical Trial Exemption in Malaysia; | |
| ● | “CytoMed Malaysia” are to CytoMed Therapeutics (Malaysia) Sdn Bhd, a company incorporated on December 18, 2013 in Malaysia (Company No. 201301044786 (1074609-M)) with its business address at No.12 Jalan Permas 9/16, Bandar Baru Permas Jaya, 81750 Masai, Johor, Malaysia, the manufacturing arm of the Group where a PIC/S GMP laboratory is located and which manufacturing capacity includes stem cells and cancer living medicine; | |
| ● | “CytoMed Therapeutics”, “CytoMed” or “Company” are to CytoMed Therapeutics Limited, a company incorporated on March 9, 2018 in Singapore (Company Registration Number: 201808327H) with its registered address at 1 Commonwealth Lane, #08-22, Singapore 149544; | |
| ● | “Director” are to the directors of the Company; | |
| ● | “Director General” are to the Director General of Environmental Quality as referred to in the EQA; | |
| ● | “DNA” are to deoxyribonucleic acid, a large complex macromolecule containing the genetic code and which is typically found in the nucleus of a human cell; | |
| ● | “DTC” are to Depository Trust Company; | |
| ● | “ECEG 2016” are to the Ethical Code and Ethical Guidelines (2016 edition) published by the SMC; | |
| ● | “EIS” are to the Employment Insurance System in Malaysia; | |
| ● | “EISA” are to the Employment Insurance System Act 2017 of Malaysia; | |
| ● | “EPFA” are to the Employee Provident Fund Act 1991 of Malaysia; | |
| ● | “ESSA” are to the Employees’ Social Security Act 1969 of Malaysia; | |
| ● | “ETPL” are to Exploit Technologies Pte Ltd (now known as ATPL); | |
| ● | “EQA” are to the Environmental Quality Act 1974 of Malaysia; | |
| ● | “Exchange Act” are to the Securities Exchange Act of 1934 in the United States of America, as amended; | |
| ● | “FDA” are to the United States Food and Drug Administration; | |
| ● | “Federal Reserve System” are to the central bank of the United States of America; | |
| ● | “Financial Institution” is defined in the FX Notices as a person carrying out a financial business regulated under the laws administered by BNM and any person carrying out any other financial business as may be specified by BNM; | |
| ● | “Financial Instrument” is defined in the FX Notices and FSA to include any agreement, including an option, a swap, futures or forward contract, whose market price, value, delivery or payment obligations is derived from, referenced to or based on, but not limited to, securities, commodities, assets, rates (including interest rates or exchange rates) or indices; | |
| ● | “Foreign Exchange Notices” or “FX Notices” are to the notices issued by the BNM under the FSA and IFSA, which local and foreign investors are subject to; |
| -v- |
| ● | “FRS” or “SFRS(I) 9” are to the Singapore Financial Reporting Standard or the Singapore Financial Reporting Standard (International) 9, respectively; | |
| ● | “FSA” are to the Financial Services Act 2013 of Malaysia; | |
| ● | “gamma delta T-iPSCs” are to gamma delta T cells converted into iPSCs; | |
| ● | “GCP” or “Good Clinical Practice” are to good clinical practice; | |
| ● | “GMP” are to good manufacturing practice; | |
| ● | “Group”, “We”, “Us” and “Our” are to the Company and its subsidiaries; | |
| ● | “GST” means goods and services tax imposed pursuant to the Goods and Services Tax Act 1993 of Singapore; | |
| ● | “Guidelines on CGTPs” are to the Guidelines For Registration of Cell and Gene Therapy Products of Malaysia, issued under regulation 29 of the CDCR and effective on January 1, 2021; | |
| ● | “GvHD” are to graft versus host disease; | |
| ● | “HCSA” are to the Healthcare Services Act of Singapore; | |
| ● | “Health Products Act” or “HPA” are to the Health Products Act 2007 of Singapore that regulates the manufacture, import, supply, presentation and advertisement of health products and of active ingredients used in the manufacture of health products within Singapore; | |
| ● | “Health Sciences Authority” or “HSA” are to the statutory board under the Ministry of Health Singapore; | |
| ● | “hESCs” are to human embryonic stem cells; | |
| ● | “HME 2016” are to the Handbook on Medical Ethics (2016 edition) issued by the SMC; | |
| ● | “HPA Order 2021” are to the Health Products Act (Amendment of First Schedule) Order 2021 of Singapore enacted under the HPA, an order in which a new category of health products, namely cell, tissue or gene therapy product, is included in the First Schedule of the HPA; | |
| ● | “hPSCs” are to human pluripotent stem cells; | |
| ● | “HSA Act” are to Health Sciences Authority Act 2001 of Singapore; | |
| ● | “Human Biomedical Research Act” or “HBRA” are to the Human Biomedical Research Act 2015 of Singapore that regulates the conduct of human biomedical research, further regulates certain restricted human biomedical research, and prohibits certain types of human biomedical research; | |
| ● | “IASB” are to International Accounting Standards Board; | |
| ● | “ICA” are to the Industrial Co-ordination Act 1975 of Malaysia, as amended; | |
| ● | “IFRS” are to the international financial reporting standards as adopted by the International Accounting Standards Board; | |
| ● | “IFSA” are to the Islamic Financial Services Act 2013 of Malaysia, as amended; |
| -vi- |
| ● | “IL-2” are to interleukin-2; | |
| ● | “IMCB” are to the Institute of Molecular and Cell Biology, a research institute of A*STAR launched on January 23, 1985 to develop and support the biomedical research and development capabilities in Singapore; | |
| ● | “Incentive Plan” are to the 2023 Equity Incentive Plan of the Company, adopted on January 18, 2023 and effective as of April 18, 2023; | |
| ● | “Income Tax Act” are to the Income Tax Act 1947 of Singapore; | |
| ● | “iPSC” are to induced pluripotent stem cells; | |
| ● | “iPSC-gdNKT” are to iPSC-derived gamma delta natural killer T cell product developed by our Group; | |
| ● | “IRAS” are to the Inland Revenue Authority of Singapore; | |
| ● | “ISAs” are to the International Standards on Auditing; | |
| ● | “ISCA” are to the Institute of Singapore Chartered Accountants; | |
| ● | “JOBS Act” are to the Jumpstart Our Business Startups Act of 2012 of the United States, as amended; | |
| ● | “Johor Bahru By-Laws” are to the Trade, License, Business and Industrial (MBJB) By-Laws 2016 of Malaysia; | |
| ● | “JSE” are to the Johannesburg Stock Exchange; | |
| ● | “K562 cells” are to the human myelogenous leukemia cell line; | |
| ● | “K562 Cell License for NK cell expansion” are to the license granted pursuant to the license agreement entered into between the Company and ATPL in December 2020; | |
| ● | “Know-How License” are to the license granted pursuant to the license agreement entered into between Puricell and ATPL in October 2020; | |
| ● | “LMC” are to the Landmark Medical Centre Sdn Bhd, a licensed private hospital located in Johor Bahru, Malaysia; | |
| ● | “Lower-tier PFIC” are to an entity in which a PFIC owns equity interests in; | |
| ● | “MA” are to the Medicines Act 1975 of Singapore; | |
| ● | “Malaysian Ringgit” or “RM” or “Ringgit” are to the currency of Malaysia; | |
| ● | “Management” or our “Management Team” are to our executive officers and Directors; | |
| ● | “Marketing Authorization Application” or “MAA” are to an application submitted by a drug manufacturer seeking marketing authorization, that is permission from a medical regulatory authority to bring a medicinal product to the market; | |
| ● | “Medicare” are to the national health insurance program in the United States of America; | |
| ● | “mDR Convertible Loan” are to the convertible loan provided by mDR Limited to the Company pursuant to the convertible loan agreement dated December 10, 2019, as amended by the supplemental agreements dated December 31, 2021, January 3, 2022 and January 3, 2023, entered into between the Company and mDR Limited. On April 21, 2023, the mDR Convertible Loan was converted into 589,509 Ordinary Shares at approximately U.S.$1.89774 per share pursuant to the terms of the mDR Convertible Loan; | |
| ● | “mGFP-γδ T” are to the gamma delta T cells subjected to electroporation using mRNA encoding mGFP; |
| -vii- |
| ● | “Ministry of Health Malaysia” or “MOH Malaysia” are to the Ministry of Health of the Malaysian Government; | |
| ● | “Ministry of Health Singapore” or “MOH Singapore” are to the Ministry of Health of the Singapore Government; | |
| ● | “MITI” are to the Ministry of Investment Trade and Industry of Malaysia; | |
| ● | “mRNA” are to messenger RNA, a type of RNA that acts as a messenger that is read by ribosomes to build proteins; | |
| ● | “MSCs” are to mesenchymal stem cells; | |
| ● | “Nasdaq” are to The Nasdaq Stock Market LLC; | |
| ● | “Nasdaq Listing Rules” are to the listing requirements of Nasdaq, as amended, modified or supplemented from time to time; | |
| ● | “NDA” are to the new drug application to the HSA and/or other relevant health and regulatory authorities, through which formal proposal is made to the HSA and/or other relevant health and regulatory authorities for approval of a new drug; | |
| ● | “NEOs” are to the Company’s named executive officers, being CHOO Chee Kong, Dr. ZENG Jieming and Dr. TAN Wee Kiat as at the date of this annual report; | |
| ● | “NK cells” are to natural killer cells, a type of immune cell found in the human body; | |
| ● | “NKG2D” are to natural killer group 2D receptor, a type of receptor typically found on the NK cell; | |
| ● | “NKG2DL” are to natural killer group 2D ligands, a type of ligand typically expressed by tumor cells, enabling NK cells to activate and kill tumor cells; | |
| ● | “NKG2Dz-γδT” are to gamma delta T cells grafted with NKG2DL-targeting CAR; | |
| ● | “NKT” are to natural killer T cells; | |
| ● | “NOL” means net operating loss; | |
| ● | “NPCB” are to the National Pharmaceutical Control Bureau, Ministry of Health Malaysia; | |
| ● | “NPRA” are to the National Pharmaceutical Regulatory Authority, Ministry of Health Malaysia; | |
| ● | “Occupational Safety and Health Laws” are to the legislation, regulation and rules in respect of occupational safety and health in Malaysia; | |
| ● | “OECD” are to the Organization for Economic Co-operation and Development; | |
| ● | “Offering” are to the initial public offering of our ordinary shares consummated on April 18, 2023; | |
| ● | “off-the-shelf” are to the manufacturing of the stated cell therapies in quantities, which utilizes either donor blood cells or iPSCs as starting materials, but not the limited patient’s own blood cells and no matching is required between such donor and recipient of the product; | |
| ● | “one-tier system” are to the one-tier corporate tax system in Singapore; | |
| ● | “OSHA” are to Occupational Safety and Health Act 1994 of Malaysia; | |
| ● | “PA 1952” are to the Poisons Act 1952 of Malaysia; |
| -viii- |
| ● | “PAg” are to phosphoantigen, a type of small molecule that stimulates gamma delta T cells expressing Vγ9 and Vδ2 molecules; | |
| ● | “Patent License” are to the license granted pursuant to the license agreement entered into between the Company and ETPL in June 2018, as varied by an addendum entered into in December 2020, second addendum entered into in September 2021, and third addendum entered into effective as of October 18, 2022; | |
| ● | “PCID Regulations” are to the Prevention and Control of Infectious Diseases (Importation and Exportation of Human Remains, Human Tissues and Pathogenic Organisms or Substances) Regulations 2006 of Malaysia, as amended; | |
| ● | “PDPA Malaysia” are to the Personal Data Protection Act 2010 of Malaysia; | |
| ● | “PDPA Singapore” are to the Personal Data Protection Act 2012 of Singapore; | |
| ● | “PFIC” are to passive foreign investment company; | |
| ● | “PHFSA” are to the Private Healthcare Facilities and Services Act 1998 of Malaysia; | |
| ● | “PHMCA” are to the Private Hospitals and Medical Clinics Act 1980 of Singapore; | |
| ● | “PIC/S” are to the pharmaceutical inspection co-operation scheme; | |
| ● | “PTAB” are to the Patent Trial and Appeal Board; | |
| ● | “Puricell” are to Puricell Lab Pte Ltd, a 95% owned subsidiary of the Company; | |
| ● | “QA” are to quality assurance; | |
| ● | “QC” are to quality control; | |
| ● | “R&D” are to research and development; | |
| ● | “REJA” are to the Reciprocal Enforcement of Judgments Act 1958 of Malaysia; | |
| ● | “Resident” is defined in the FX Notices as, among others, a body corporate incorporated or established, or registered with or approved by any authority, in Malaysia; | |
| ● | “RNA” are to ribonucleic acid, a biological macromolecule that functions to convert the genetic information of DNA into proteins; | |
| ● | “Sarbanes-Oxley Act” are to the Sarbanes-Oxley Act of 2002, as amended; | |
| ● | “scFv” are to single-chain variable fragment, a technique used to produce a functional antigen-binding fragment on a cellular level; | |
| ● | “Scientific Advisory Board” or “SAB” are to the Company’s board of scientific advisors which provide the Company with on-going scientific advice; | |
| ● | “Singapore National Healthcare Group Domain Specific Review Boards” are to the independent committee constituted of medical, scientific and nonscientific members who are empowered by the National Healthcare Group Chief Executive Officer, to review research involving patients, staff, premises, or facilities of National Healthcare Group institutions and all other institutions under its oversight. | |
| ● | “SEC” are to the Securities and Exchange Commission of the United States of America; |
| -ix- |
| ● | “Securities Act” are to the Securities Act of 1933 of the United States of America, as amended; | |
| ● | “SFRS(I)” are to the Singapore Financial Reporting Standard (International); | |
| ● | “SGX-ST” are to the Singapore Exchange Securities Trading Limited; | |
| ● | “SIC” are to the Securities Industry Council of Singapore; | |
| ● | “SID” are to the Singapore Institute of Directors; | |
| ● | “Singapore Companies Act” are to the Companies Act 1967 of Singapore; | |
| ● | “Singapore Laboratory” are to the Company’s property located at 1 Commonwealth Lane, #08-22, Singapore 149544, with intended use as an office and research laboratory; | |
| ● | “Singapore Medical Council” or “SMC” are to the statutory board under the Ministry of Health Singapore which regulates the practice of medicine in Singapore; | |
| ● | “Singapore Take-over Code” are to the Singapore Code on Take-overs and Mergers; | |
| ● | “SOCSO” are to the Social Security Organization Fund of Malaysia; | |
| ● | “Stamp Duties Act” are to the Stamp Duties Act 1929 of Singapore; | |
| ● | “TCR” or “TCRs” are to T cell receptors; | |
| ● | “UK” are to the United Kingdom; | |
| ● | “United States” or “U.S.” are to the United States of America; | |
| ● | “VStock” are to V Stock Transfer, LLC, the transfer agent and registrar of the Company; | |
| ● | “U.S. Board of Governors” are to the governing body of the Federal Reserve System in the United States of America; | |
| ● | “U.S. GAAP” are to generally accepted accounting principles in the United States of America; and | |
| ● | “U.S. Holder” are to a person that is, for U.S. federal income tax purposes, a beneficial owner of ordinary shares and a citizen or individual resident of the United States, a corporation, or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state therein or the District of Columbia, or an estate or trust the income of which is subject to U.S. federal income taxation regardless of its source. |
| -x- |
PART I
Unless the context otherwise requires, as used in this annual report, the terms “the Company”, “we”, “us”, and “our” refer to CytoMed Therapeutics Limited and any or all of its subsidiaries. References to our “management” or our “management team” refers to our officers and directors. All references to “$” or “U.S.$” in this annual report refer to U.S. dollars and all references to “S$” are to the currency of Singapore. Unless otherwise noted, all industry and market data in this annual report on Form 20-F (this “annual report”) is presented in U.S. dollars. Unless otherwise noted, all financial and other data related to the Company in this annual report is presented in U.S. dollars.
Please see “Definitions” for defined and capitalized terms used throughout this annual report.
ITEM 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISORS
Not applicable for annual reports on Form 20-F.
ITEM 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not applicable for annual reports on Form 20-F.
ITEM 3. KEY INFORMATION
| A. | [Reserved] |
| B. | Capitalization and Indebtedness |
Not applicable for annual reports on Form 20-F.
| C. | Reasons for the Offer and Use of Proceeds |
Not applicable for annual reports on Form 20-F.
| D. | Risk Factors |
An investment in our ordinary shares involves a high degree of risk. You should carefully consider the following risk factors, as well as all of the other information contained in this annual report, before making an investment decision. The risks described below are not the only ones facing us. The occurrence of any of the following risks, or of additional risks and uncertainties not presently known to us or that we currently believe to be immaterial, could significantly harm our business, financial condition, results of operations and growth prospects. In such case, the trading price of shares of our ordinary shares could decline, and you may lose part or all of your investment.
RISKS RELATED TO OUR FINANCIAL POSITION
We do not have a long operating history and we do not currently have any product approved for commercial sale.
We are a pre-clinical biopharmaceutical development stage company and we do not have any products approved for commercial sale as of the date of this annual report. We are focused on developing human cells as therapeutics and our technologies are new and unproven. Since our incorporation in 2018, we have invested most of our resources in developing our product candidates, building our intellectual property portfolio, developing our supply chain, conducting business planning, constructing our centralized cGMP Facility, hiring and training staff, raising capital and providing general and administrative support for these operations. Consequently, we do not have a meaningful operating history upon which our business and prospects may be evaluated, and predictions about our future success or viability may not be as accurate as they could be if we had a longer operating history or a history of successfully developing and commercializing drug products. We have not yet demonstrated our ability to overcome many of the risks and uncertainties frequently encountered by companies in the rapidly evolving biopharmaceutical clinical trial industry. If we do not successfully address these risks, our business, financial condition, results of operations and growth prospects will be materially and adversely affected. At present time, we manufacture limited quantities of cells for researchers and institutions on a non-profit, cost recovery basis for the purpose of research.
| 1 |
We have incurred losses since our incorporation and we expect to incur significant losses for the foreseeable future, which raise substantial doubt on our ability to continue as a going concern. Our ability to continue as a going concern is dependent on being able to obtain sufficient additional funding to finance our operations.
Since our incorporation in 2018, we have incurred losses. As of December 31, 2021, 2022 and 2023, we have accumulated losses of S$5.07 million, S$8.20 million and S$12.33 million, respectively. Given that we will continue to invest most of our resources in developing our product candidates, we expect to continue to incur increasing losses for the foreseeable future. In addition, we anticipate a substantial increase in our expenses if, and as, we seek to:
| ● | initiate clinical development of our proprietary product candidates, CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC; | |
| ● | advance additional product candidates to clinical trials, including CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC; | |
| ● | discover and develop additional product candidates; | |
| ● | establish and validate our own clinical-scale and commercial-scale cGMP facilities; | |
| ● | initiate or develop a MAA, or equivalent in the relevant countries, for CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC and/or seek marketing approvals for any of our other product candidates that successfully complete clinical trials; | |
| ● | engage, on an as needed basis, third-party contractors including CROs and CMOs to conduct clinical trials; | |
| ● | maintain, expand and protect our intellectual property portfolio; | |
| ● | acquire or in-license other product candidates and technologies; | |
| ● | incur additional costs associated with operating as a public company; | |
| ● | Explore new partnerships locally, within the region and globally; and | |
| ● | increase our employee headcount and related expenses to support these activities. |
We may not be successful in any or all of these activities and even if we are successful, we may not generate revenues that are significant enough to generate profit. If we seek additional financing to fund our business activities in the future and there remains substantial doubt about our ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding to us on commercially reasonable terms or at all. Further, if we are unable to continue as a going concern, we may have to discontinue operations and liquidate our assets and may receive less than the value at which those assets are carried on our audited financial statements, which would cause our shareholders to lose all or a part of their investment.
We will require additional capital, which, if available, may cause dilution to our shareholders, restrict our operations and/or require us to relinquish rights to our product candidates.
Prior to the Offering, we financed our operations primarily from private equity financing and issuance of convertible loans. As of December 31, 2021, 2022 and 2023, we had cash and cash equivalents of S$2.51 million, S$1.58 million and S$9.00 million, respectively. Our research expenses were S$1.09 million, S$1.52 million and S$1.59 million for the financial year ended December 31, 2021, 2022 and 2023, respectively. During the period from April to June 2021, the Company raised an aggregate of S$4.00 million gross proceeds from the sale of our equities. The Company raised capital in an aggregate of S$1.22 million between September and October 2022. On April 18, 2023, we consummated our Offering resulting in net proceeds of approximately S$10.31 million (U.S.$7.81 million) after deducting underwriting discounts, commissions and offering expenses. We intend to use the proceeds from our Offering to, among other uses, advance CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC through clinical development. Developing pharmaceutical products and conducting pre-clinical studies and clinical trials are expensive. In the future, we may require additional cash funding to continue to execute our strategic plan and fund operations from time to time.
Until and unless we can achieve substantial revenue from our products to offset our expenses, we expect to finance our operations through the proceeds of our Offering, a combination of equity offerings and debt financings, and potentially through additional license and development agreements or strategic partnerships with third parties. We may not be able to obtain sufficient financing or financing on terms which are reasonable to us. In addition, market volatility resulting from the COVID-19 pandemic or other factors could adversely impact our ability to obtain sufficient financing as and when needed. We have no commitments for any additional financing, and will likely be required to raise such financing through the sale of additional securities. If we sell equity or equity-led securities, our current shareholders, may be diluted, and the terms may include liquidation or other preferences that are senior to or otherwise adversely affect the rights of our shareholders. Moreover, if we obtain debt financing, we may need to allocate a significant percentage of our operating cash flow to paying principal and interest on such debt and we may need to comply with operating restrictions, such as limitations on incurring additional debt, which could hinder our future ability to acquire, sell or license intellectual property rights. This consequently could impede our business operations. Furthermore, the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our ordinary shares to decline.
If we obtain additional financing through licensing or collaboration arrangements with third parties, we may have to relinquish valuable rights to our product candidates, or grant licenses on terms that are not favorable to our business and operations. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
Our efforts to obtain additional financing for our business operations may also divert our time and attention from our day-to-day activities, which may impair or delay the development of our product candidates. In addition, our cash flow requirements may fluctuate as a result of factors currently unknown to us including, but not limited to, any unforeseen costs we may incur as a result of pre-clinical study or clinical trial delays due to the COVID-19 pandemic amongst other causes, and we may need to seek additional financing sooner than anticipated. If we are unable to obtain sufficient financing on a timely basis or at all, we may have to significantly curtail or stop one or more of our research or development programs.
| 2 |
Any acquisitions or strategic collaborations are capital intensive, may dilute our shareholders’ equity interest, cause us to incur debt or assume contingent liabilities and/or subject us to other risks.
From time to time, we may contemplate undertaking various acquisitions and strategic collaborations, including licensing or acquiring complementary drugs, intellectual property rights, technologies or businesses. Any potential acquisition or strategic partnership we may undertake involves several risks and challenges, including, but not limited to:
| ● | increased operating expenditure and cash flow requirements; | |
| ● | taking on clinical trial indebtedness or contingent or unknown liabilities; | |
| ● | assimilation of operations, intellectual property and drugs of an acquired company, including difficulties associated with integrating new personnel; | |
| ● | the diversion of our management’s time and focus from our existing drug programs and initiatives in pursuing such a strategic partnership, merger or acquisition; | |
| ● | retention of key employees, the loss of key personnel, and uncertainties in maintaining crucial business relationships; | |
| ● | risks and uncertainties associated with the other party to such a transaction, including the prospects of that party and their existing drugs or product candidates and regulatory approvals; and | |
| ● | our inability to generate revenue from acquired drugs, intellectual property rights, technologies, and/or businesses sufficient to meet our objectives in undertaking the acquisition or even to offset the associated acquisition and maintenance costs. |
If we undertake acquisitions or strategic partnerships, we may issue securities which will dilute our shareholders’ equity interests, take on debt obligations, incur significant one-off expenditures or acquire intangible assets that could result in substantial future amortization expense. Moreover, we may not identify suitable acquisition or strategic partnership opportunities, and this inability could impair our growth or limit access to technology or drugs that may be important to the development of our business.
Our business and the business and operations of our research partners and/or other third parties with whom we conduct business are subject to the effects of pandemics, health epidemics, and outbreaks of infectious diseases , in regions where we or third parties on which we rely have business operations.
The COVID-19 pandemic and measures taken to mitigate the impact of the pandemic disrupted economic activity and business operations worldwide, including Malaysia, where our primary operations are located. The emergence of one or more pandemics, epidemics, or outbreaks of infectious diseases, including future outbreaks of COVID-19 variants, Respiratory Syncytial Virus (“RSV”), or the flu, could result in similar disruptions.
Our headquarter in Singapore as well as the operations of our cGMP Facility located in Johor, Malaysia, were impacted by the COVID-19 pandemic and may in the future be similarly impacted by future pandemics, epidemics, or outbreaks of infectious disease. For example, as a result of the COVID-19 pandemic, we experienced some delays in the audit and inspection by NPRA of our cGMP Facility, global supply shortages of certain materials that we use for research and cGMP manufacturing, employee turnover/attrition, delays and/or disruptions in achieving license milestones, and delays in commencement of clinical trial and enrollment in our clinical trial.
The emergence of a future pandemic, epidemic, or outbreak of infectious disease may impact the regulatory authorities to which we are subject in our industry, which may, in turn, hamper or delay our clinical development efforts. For instance, the COVID-19 pandemic resulted in a significant increase in the FDA workload, as well as the need to reprioritize the projects under review, and a future pandemic, epidemic, or outbreak of infectious disease may do so again in the future.
In addition, the COVID-19 pandemic has significantly disrupted financial markets around the world and could continue to restrict the level of economic activity, and may limit our ability to access capital, which could adversely impact our liquidity now or in the future. A recession or market correction resulting from the spread of COVID-19 could materially affect our business and the value of our ordinary shares.
| 3 |
We cannot predict the potential future impacts of the emergence of another pandemic, epidemic, or outbreak of infectious disease on us, our research partners, and other third parties with whom we conduct business. We may experience disruptions as a result of a pandemic, epidemic, or outbreak of infectious disease that could severely impact our business, preclinical studies and clinical trials, including but not limited to:
| ● | delay or difficulties in enrolling donors for our clinical trials; | |
| ● | delays or difficulties in enrolling patients for our clinical trials; | |
| ● | delays or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff; | |
| ● | diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; | |
| ● | interruption of key clinical trial activities, such as clinical trial site data monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others or interruption of clinical trial subject visits and study procedures, which may impact the integrity of subject data and clinical study endpoints; | |
| ● | interruption or delays in the operations of the HSA and/or other relevant health and regulatory authorities, which may impact review and approval timelines; | |
| ● | interruption of, or delays in receiving, supplies of our product candidates and other required materials from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in delivery systems; | |
| ● | interruptions in pre-clinical studies due to restricted or limited operations at our cGMP Facility; | |
| ● | increase in cost of operations due to rising logistical and shipping cost; | |
| ● | increase in cost of operations due to rising consumable price; | |
| ● | increase in costs of conducting clinical trials due to additional compliance requirements; | |
| ● | limitations on employee resources that would otherwise be focused on the conduct of our pre-clinical studies and clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people; and | |
| ● | interruption or delays to our discovery and clinical activities, which in turn, could cause us to fail to meet milestone deadlines required to maintain intellectual property licenses upon which we rely. |
The extent of any delays or impacts due to pandemics, epidemics, or outbreaks of infectious disease, or government regulations in response to the foregoing, will depend on future developments that are highly uncertain and cannot be predicted with confidence, but these delays could have a material impact on our business, financial condition, and/or results of operations.
RISKS RELATED TO OUR BUSINESS IN THE BIOPHARMACEUTICAL INDUSTRY
Our business depends upon the success of our CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC product candidates in obtaining regulatory approval and being commercialized.
The success of our business depends on our ability to utilize our proprietary technologies to manufacture our product candidates obtain regulatory approval for such product candidates, and commercialize the approved products. Our product candidates have not yet been evaluated in humans and may never become commercialized. All our product candidates developed from our proprietary technology platforms will require significant additional clinical and non-clinical development, review and approval by the relevant health and regulatory authorities in the various jurisdictions, substantial capital and time investment, access to sufficient commercial manufacturing capacity and significant marketing efforts before they can be successfully commercialized. If any of our product candidates encounter safety or efficacy issues, developmental delays or regulatory issues or other roadblocks, it could impact the development plans for our other product candidates.
| 4 |
Our research on utilizing CTM-N2D, CTM-GDT and iPSC-gdNKT to treat cancer; and CTM-MSC to treat degenerative diseases is novel, and we must overcome significant challenges such as obtaining required regulatory approvals in order to develop, commercialize and manufacture our product candidates.
Cell-based therapies are still considered novel and a new medical science in South-east Asia. The CTGTP Regulations in Singapore became effective on March 1, 2021. Malaysia’s CGTP regulations became effective on January 1, 2021. It can be expected that such regulations will be updated as more experience and expertise are gained by regulators, clinicians and the scientific community. Although future regulations may hinder or be detrimental to cell-based therapies, the medical community generally considers that such cellular “living” medicine has the potential to enhance patient treatment modalities for many diseases. Reflecting this, Singapore set up a national agency in April 2020 known as ACTRIS comprised of, inter alia, all the national universities, hospitals, research institutions and funding agencies in Singapore.
Our R&D efforts are focused on utilizing CTM-N2D, iPSC-gdNKT and CTM-GDT as immunotherapies for cancers, and CTM-MSC for degenerative diseases. To date, HSA has approved only two cell-based cancer therapy for commercialization (i.e. Kymriah® by Novartis (Singapore) Pte Ltd, Yescarta by Gilead Sciences Singapore Pte Ltd, and SpheChon by Labcorp) and as far as we are aware, no gamma delta T cell-based therapy has been approved for commercial use by HSA and/or any other relevant regulatory authority in any of the key markets. The processes and requirements imposed by HSA and/or other relevant regulatory authorities in multiple jurisdictions may cause delays and additional costs in obtaining approvals for marketing authorization for our product candidates. Because our CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC products are novel, and cell-based therapies are relatively new, regulatory agencies may lack the necessary experience to assess our product candidates. As such, this may increase the regulatory review process and period, including the time it takes for HSA and/or other relevant regulatory authorities to review our clinical trial application if and when submitted, such as the review of the Phase I clinical trial as described in paragraph “Development Status of CTM-N2D Therapy” in the section entitled “Business” which was submitted to the regulatory authorities in respect of CTM-N2D for the treatment of relapsed or refractory solid tumors of different types. Such regulatory delays may thereby increase our development costs and cause further delays in the commercialization of our CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC products. Additionally, commercializing novel immuno-oncology therapies will create significant challenges for us, including but not limited to:
| ● | educating medical personnel regarding the potential side-effect profile of our cell products and, as the clinical program progresses, on observed side effects with the immune-oncology therapy; | |
| ● | training a sufficient number of medical personnel on how to properly handle and administer our cell products, especially in our planned solid tumor trial wherein the cells are given through a procedure by trained medical doctors; | |
| ● | enrolling sufficient numbers of patients in clinical trials; | |
| ● | developing a reliable and safe and an effective means of manufacturing our cell products; | |
| ● | manufacturing our cells on a large scale and in a cost-effective manner; | |
| ● | sourcing starting material suitable for clinical and commercial manufacturing; and | |
| ● | establishing sales and marketing capabilities, as well as developing a manufacturing process and distribution network to support the commercialization of any approved products. |
We must be able to overcome these challenges in order for us to develop, manufacture and commercialize our product candidates utilizing CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC in each geographical market. As such, we remain open to exploring the possibility of conducting clinical trials in other countries.
Our pre-clinical studies may experience delays or may not progress to clinical trials, which would adversely affect our ability to obtain regulatory approvals or commercialize these programs on a timely basis or at all.
In order to obtain HSA and/or other relevant regulatory authority approval to market a new biological product we must demonstrate proof of safety, purity and potency or efficacy in humans. We will have to conduct adequate and well-controlled clinical trials in order to meet high safety standards and requirements. We cannot guarantee the timely completion or successful outcome of our pre-clinical testing and studies and cannot determine whether the regulatory authority will accept our proposed clinical programs or if the outcome of our pre-clinical testing and studies will ultimately support the further development of our programs. As a result, we cannot be certain that we can submit any clinical trial or similar applications for our pre-clinical programs on the timelines we expect, if at all, and we cannot be sure that submission of clinical trials or similar applications will result in the regulatory authority or other regulatory authorities allowing clinical trials to begin.
| 5 |
Conducting pre-clinical testing is a prolonged, time-consuming and costly process. The period of pre-clinical testing may vary substantially depending on the type, complexity and novelty of the program, and may often take several years or more for each program. Any delays in pre-clinical testing and studies conducted by us or potential future partners may cause us to incur additional operating expenses. The commencement and rate of completion of pre-clinical studies and clinical trials for a product candidate may be delayed by many factors, including, for example:
| ● | inability to generate sufficient pre-clinical or other in vivo or in vitro data to support the commencement of clinical trials; | |
| ● | delays in reaching a consensus with regulatory agencies on clinical trial design; and | |
| ● | the relevant regulatory authorities not allowing us to rely on previous clinical trial findings of safety and efficacy for other similar but approved products and published scientific literature. |
Moreover, as standards for pre-clinical assessment are constantly evolving and may change rapidly, even if an agreement with HSA and/or other relevant regulatory authorities on a pre-clinical trial proposal has been made, HSA and/or other relevant regulatory authorities may not accept the proposed clinical trial submission, in which case patient enrollment would be put on partial or complete hold and treatment of enrolled patients could be temporarily put on hold as the product candidate is re-assessed. Even if we commence clinical trials, we cannot guarantee the success of our clinical trials or development efforts.
The results of pre-clinical studies and early-stage clinical trials may not be indicative of future results in future clinical trials. Initial success in any clinical trials may not be indicative of future results obtained at the completion of such trials in later stage trials.
The results of our pre-clinical studies may not be indicative of future results of clinical trials, and the results of any early-stage clinical trials we commence may not be indicative of the results of the later-stage clinical trials. For example, pre-clinical models as applied to cell therapy in oncology are not sufficiently representative of the clinical setting, and thus cannot always accurately predict clinical activity nor identify all potential risks, and may not provide sufficient guidance as to appropriate dose or administration regimen of a given therapy. In addition, initial success in clinical trials may not be indicative of results obtained when such trials are completed. Interim data from clinical trials that we may conduct are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Preliminary data are required to undergo audit and verification procedures that may result in the final data being materially different from the preliminary data we previously announced. Negative differences between preliminary or interim data and final data could materially adversely affect the prospects of any product candidate that is impacted by such data updates.
Clinical trials involve a lengthy and costly process with an uncertain outcome, and we may encounter substantial delays due to a variety of reasons outside our control.
Pre-clinical and clinical trials are costly, time consuming and subject to substantial uncertainty in respect of the outcome. Due to scientific feasibility, safety, efficacy, changing standards of medical care and other variables, our product candidates may fail at any time during the trial. The results from pre-clinical testing or early clinical trials of a product candidate may not accurately indicate or ensure the outcome of later phases of clinical trials of our product candidates. HSA and/or other relevant regulatory authorities may suspend or terminate our clinical trials of any of our product candidate(s) at any time for various reasons, including, but not limited to, a perception that humans participating in such trials are exposed to unacceptable health risks or adverse side effects, or other adverse initial experiences or clinical trial findings. HSA and/or other relevant regulatory authorities may also require us to conduct additional pre-clinical studies or clinical trials due to negative or inconclusive results, or may fail to approve the raw materials, manufacturing processes or facilities of third-party manufacturers upon which we rely, and/or change their approval policies or regulations or their prior guidance to us during clinical development in a manner rendering our clinical data insufficient or unsuitable for approval. In addition, data obtained from our pre-clinical and clinical trials may not be deemed adequate to support the submission of the relevant regulatory filings in our key markets. We cannot guarantee that any pre-clinical studies or clinical trials that we may plan or initiate will be conducted as planned or completed on schedule, if at all.
We may fail at any stage in one or more of our clinical trials. We may face circumstances which prevent successful initiation, timely completion, or the desired results of our pre-clinical studies and clinical trials including, but not limited to:
| ● | delays and challenges in obtaining requisite regulatory approvals to commence pre-clinical studies and clinical trials on humans; | |
| ● | delays in reaching agreement on acceptable terms with prospective clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different trial sites; | |
| ● | our ability to recruit sufficient patients for our clinical trials in a timely manner or at all; |
| 6 |
| ● | delays in sufficiently developing, characterizing or conducting a manufacturing process suitable for advanced clinical trials; | |
| ● | delays in reaching an agreement with HSA and/or other relevant regulatory authorities on the planning, design or implementation of our pre-clinical studies and clinical trials; | |
| ● | changes in regulatory requirements or guidance that may require us to amend or submit new clinical protocols, or the introduction of unanticipated requirements; | |
| ● | changes in the standard of care on which a clinical development plan was based, which consequently may require new or additional trials; | |
| ● | lack of quality of our product candidates or other materials necessary to conduct pre-clinical studies or clinical trials of our product candidates; | |
| ● | clinical trials of our product candidates may have negative or inconclusive results, and as such, we may, or HSA and/or other relevant regulatory authorities may require us, to conduct additional clinical trials or abandon product development programs; | |
| ● | inability of enrolled patients in foreign countries to adhere to our clinical protocol and process due to differences in healthcare services or cultural customs, or additional administrative burdens associated with foreign regulatory schemes; and | |
| ● | failure of ourselves or any third-party manufacturers, contractors or suppliers to comply with regulatory requirements, maintain adequate quality controls, or be able to provide sufficient product supply to conduct and complete pre-clinical studies or clinical trials of our product candidates. |
In addition, the pandemics, epidemics, or outbreaks of infectious diseases, including future outbreaks of COVID-19 variants, Respiratory Syncytial Virus (“RSV”), or the flu, may increase the likelihood that we face such difficulties or delays in initiating, enrolling, conducting or completing our planned and ongoing pre-clinical studies and clinical trials, as applicable. If the pandemics, epidemics, or outbreaks of infectious diseases, including future outbreaks of COVID-19 variants, Respiratory Syncytial Virus (“RSV”), or the flu, could continue to prevent or otherwise obstruct regulatory authorities’ conduct of regular inspections, reviews or other regulatory activities, HSA’s (or other relevant regulatory authorities) timely review and process our regulatory filings could be severely affected. If we experience delays in the initiation, enrollment or completion of any pre-clinical study or clinical trial of our product candidates, or if any pre-clinical studies or clinical trials of our product candidates are canceled, the commercial prospects of our product candidates may be materially and adversely affected, and our ability to generate product revenues from any of these product candidates will be delayed or not realized at all. In addition, any delays in completing our clinical trials may increase our costs and hold up our product candidate development and approval process.
Our business is highly reliant on the success of our product candidates, in particular CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC, and we may fail to develop CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC successfully or to obtain regulatory approval for them.
We cannot assure that CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC will be safe and effective or will obtain the requisite approvals and licenses for commercialization, on a timely basis or at all. Although some of our employees have prior exposure to clinical trials, regulatory compliance and cGMP manufacturing, we have not previously completed any clinical trials or submitted a marketing approval application to HSA, or similar regulatory approval filings to comparable foreign authorities, for any product candidate, and we cannot be certain that CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC will be successful in clinical trials or receive the requisite regulatory approvals for the development, commercialization and marketing of CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC. HSA (or other relevant regulatory authorities) can delay, limit or refuse to grant approval of any of our product candidates for many reasons. For further details about such reasons, please refer to the risk factor entitled “Clinical trials involve a lengthy and costly process with an uncertain outcome, and we may encounter substantial delays due to a variety of reasons outside our control”. Any delay in obtaining, or inability to obtain, requisite regulatory approvals will delay or hinder our ability to successfully commercialize CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC. This may have a material and adverse impact on our business, financial condition, results of operations and growth prospects.
Furthermore, as CTM-N2D is our lead product candidate and CTM-GDT is based on the same core proprietary technology, if any of our clinical trials of CTM-N2D encounter safety, efficacy or manufacturing problems, development delays, regulatory issues or other problems, our development plans for CTM-N2D and all of our other product candidates in our pipeline could be significantly hindered, which could materially and adversely affect our business, financial condition, results of operations and growth prospects.
In the event we manage to obtain clinical proof-of-concept from our CTM-N2D Phase I trials for relapsed or refractory solid tumors of different types, we may develop CTM-N2D for additional indications. We may not be able to advance any of these clinical trial indications through the development process. Even if we receive regulatory approval to market CTM-N2D for the treatment of any of these additional clinical trial indications, any such additional clinical trial indications may not be successfully commercialized, widely accepted in the marketplace or found to be more effective than other commercially available alternatives. If we are unable to successfully develop and commercialize CTM-N2D for these additional clinical trial indications, our commercial opportunities will be limited.
| 7 |
Enrollment and retention of patients for clinical trials is costly and time-consuming and could be delayed, made more difficult or rendered impossible by multiple factors outside our control.
Identifying and qualifying suitable patients to participate in our clinical trials is vital to our development of our product candidates. We need the enrollment of a sufficient number of patients suitable to undergo such clinical trials, including patients who are suffering from the disease that the product candidate is intended to treat and who meet other eligibility criteria for any clinical trial of a new product candidate. The rate of patient enrollment, which is a pivotal factor in determining the timing of clinical trials, is dependent on many factors beyond our control, including:
| ● | our ability to locate and set up clinical trial sites; | |
| ● | the size and nature of the patient population; | |
| ● | the design and eligibility criteria of the clinical trial; | |
| ● | the physical proximity of patients to clinical sites; | |
| ● | the patient referral practices and protocols of medical practitioners; | |
| ● | changing medical practice patterns or guidelines related to the clinical trial indications we are investigating; | |
| ● | competing clinical trials or approved therapies which are seen as an attractive alternative to patients and their medical practitioners; | |
| ● | perceived risks and benefits of the product candidate under study, including as a result of adverse effects observed in similar or competing therapies; | |
| ● | our ability to obtain and maintain consent to our clinical trials from patients which may be affected as a result of various reasons, including but not limited to, patients’ unwillingness to participate in our clinical trials due to the pandemics, epidemics, or outbreaks of infectious diseases, including future outbreaks of COVID-19 variants, Respiratory Syncytial Virus (“RSV”), or the flu; | |
| ● | the risk that enrolled patients will drop out of our clinical trial or die before completion of the clinical trial; | |
| ● | our patients failing to complete a clinical trial or returning for post-treatment follow-up; and | |
| ● | our ability to manufacture or obtain the requisite materials for a patient and clinical trial. |
In addition, we will compete with many other ongoing clinical trials in sourcing for and recruiting patients for our expected clinical trials. Our clinical trials may also compete with other clinical trials for product candidates that are in a similar cellular immunotherapy area as our product candidates. This competition could reduce the number and profiles of available patients, as some patients may opt to enroll in a clinical trial conducted by one of our competitors instead of ours. Since the number of qualified clinical investigators is limited, we may conduct some of our clinical trials at the same clinical trial sites that some of our competitors use, which will reduce the number of patients who are available for our clinical trials at such clinical trial site. If we are unable to enroll a sufficient number of patients in our clinical trials in a timely manner, completion of our clinical trials may be delayed or may not be achieved, which would prevent us from commercializing our product candidates which in turn, could also cause us to fail to meet milestone deadlines required to maintain intellectual property licenses upon which we rely.
If any of our product candidates, or any competing product candidates, cause severe adverse effects (such as the development of severe or fatal cytokine release syndrome, neurotoxicity or graft-versus-host disease) we may be required to delay or discontinue further clinical development.
Our product candidates may have undesirable side effects that could result in us or HSA and/or other relevant regulatory authorities requiring us to interrupt, delay or discontinue clinical trials and could result in a more restrictive label than anticipated or the delay or denial of regulatory approvals. Our clinical trials may demonstrate a high and unacceptable level of severity and prevalence of side effects or unexpected characteristics.
As of the date of this annual report, our evaluation of our product candidates is limited to pre-clinical studies. As such, there can be no guarantee that any toxicity, and/or other adverse events, will not occur in human subjects during clinical trials.
| 8 |
While scientific data suggests that gamma delta T cell-based therapies may be better-tolerated as compared to alpha beta T cell-based therapies due to biologic differences between these cell types, there can be no assurance that patients will not experience cytokine release syndrome, neurotoxicity, GvHD or other serious adverse events. Severe adverse events associated with our product candidates, CTM-N2D, iPSC-gdNKT and CTM-GDT may also develop, since targeting NKG2DLs is not yet a well-characterized modality. NKG2D targets multiple ligands, and the landscape of ligand expression is currently not fully understood. For example, there are risks that NKG2DLs may be expressed on either known or an as-yet-underappreciated population of healthy cells. Therefore, such cells may also be targeted by CTM-N2D, iPSC-gdNKT and CTM-GDT and lead to adverse events of unknown frequency and severity. Such adverse events may cause delays in completion of our clinical programs. If unacceptable side effects arise in the development of our product candidates such that there is no longer a positive benefit risk, our clinical trials may be suspended or terminated, or the regulatory authorities may deny approval of our product candidates for any or all targeted clinical trial indications. Treatment-related side effects could also affect patient enrollment or the successful completion clinical trial by enrolled patients or result in potential product liability claims. In addition, these side effects may not be adequately identified or managed by the relevant medical personnel, and inadequate training in identifying or managing the potential side effects of our product candidates could result in patient injury or death.
Public perception of cell-based immuno-oncology therapies for treating cancer may impact public perception of our company and product candidates, or our business and operations.
Our platform utilizes a relatively new technology involving the human T cells and utilization of those modified cells in other clinical trial individuals. Public perception of our product candidates may be influenced by claims, such as that cell-based immunotherapy is unsafe, unethical, or immoral and, consequently, the public or the medical community may not be ready to accept our product candidates. Negative public reaction to and scrutiny of cell-based immunotherapy in general could result in governments implementing tighter regulations and stricter labeling requirements of cell-based immunotherapy products, including any of our product candidates, and could result a drop in the demand for any products we may develop. Adverse public attitudes may also adversely impact our ability to enroll suitable patients for our clinical trials. More restrictive government regulations or negative public opinion could have an adverse effect on our business or financial condition and may delay or impair the development and commercialization of our product candidates or demand for any products we may develop.
We may not identify or develop other product candidates and may fail to capitalize on programs or product candidates that may present a greater commercial opportunity or for which there is a greater likelihood of success.
Our business is largely based on our ability to identify, develop and commercialize successful product candidates. A key element of our strategy is to discover and develop additional product candidates based upon our core proprietary technology platform through our internal research programs. We may also explore strategic collaborations for the discovery of new product candidates. Research programs to identify product candidates require substantial technical, financial and human resources, whether or not any product candidates are ultimately identified. In addition, targets for different cancers may require changes to our core proprietary technology platform, which may delay development or prevent the manufacture our product candidates. Our research programs may initially suggest positive results in identifying potential product candidates, but fail to produce successful product candidates for clinical development for many reasons, including without limitation the following:
| ● | the research methodology(ies) and/or technology platform(s) we use may not be accurate in identifying potential product candidates; | |
| ● | competitors may develop alternatives that render our product candidates obsolete or less attractive; | |
| ● | we may choose to discontinue development if we determine that clinical results do not show promise; | |
| ● | product candidates we develop may nevertheless be covered by third parties’ patents or other exclusive rights; | |
| ● | a product candidate may be shown to have harmful side effects or other characteristics that clinical trials indicate it is unlikely to be safe, effective or otherwise does not meet applicable regulatory criteria; and | |
| ● | a product candidate may not be accepted as safe and effective by patients and/or the medical professionals. |
| 9 |
Due to our limited resources, we have to focus our time and resources on the development of specific types of treatment, or treatment for a specific type of cancer, and we may forego or delay pursuing opportunities with certain programs or product candidates or for clinical trial indications which may be shown in the future to have greater commercial potential. Our estimates regarding the potential market for our product candidates may not be accurate, and if we do not correctly identify and accurately assess the commercial potential for a particular product candidate, we may relinquish valuable rights to that product candidate through strategic collaboration, licensing or other arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate. Alternatively, we may allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a partnering arrangement.
If any of these events occur, we may be forced to delay or discontinue our development efforts with respect to a particular product candidate or we may fail to develop a potentially successful product candidate.
Although we intend to design the clinical trials for our product candidates, we plan to rely on third parties to assist us in conducting our clinical trials.
Although we manufacture our product candidates in our cGMP Facility instead of engaging and relying on an external CMO, we rely on third parties such as CROs, CMOs and specialist medical professionals, including but not limited to oncologists and hematologists, to assist us in other aspects of conducting clinical trials for our product candidates. As a result, many important aspects of our clinical development, including their conduct and timing, will not be within our direct control. Our reliance on third parties to conduct future clinical trials will also result in less direct control over the management of data developed through clinical trials than would be the case if we were relying entirely upon our own staff. Communicating with external parties can also be challenging, potentially leading to gaps in expectations and difficulties in coordinating activities. External parties may:
| ● | have staffing difficulties; | |
| ● | fail to comply with contractual obligations; | |
| ● | experience regulatory compliance issues; | |
| ● | undergo changes in priorities or become financially distressed; and/or | |
| ● | form relationships with other entities, some of which may be our competitors. |
If third parties whom we contract with do not perform our clinical trials in a satisfactory manner, breach their obligations to us and/or fail to comply with applicable legislative and regulatory requirements, we would be unable to rely on clinical data collected by these third parties and may be required to repeat, extend the duration of, or increase the size of any clinical trials we conduct, which could significantly delay the development and/or commercialization of our product candidates and significantly increase our costs.
If any of our relationships with these third parties deteriorate or terminate, we may not be able to arrange with alternative third parties on commercially reasonable terms, or at all. If third parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain are compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, any clinical trials such third parties are associated with may be extended, delayed or terminated, and we may not be able to obtain marketing approval for or successfully commercialize our product candidates. As a result, we believe that our financial results and the commercial prospects for our product candidates in the subject clinical trial would be materially and adversely affected, our costs could increase and our ability to generate revenue could be significantly delayed.
If third parties that we may rely on for our clinical trials do not successfully satisfy their contractual obligations, comply with regulatory requirements or meet targeted timelines, we may not be able to obtain requisite marketing approval for or commercialize our product candidates.
We are unable to independently conduct clinical trials though we intend to design our clinical trials for our product candidates and manufacture our product candidates in our cGMP Facility instead of engaging and relying on an external CMO. We remain dependent on medical institutions, clinical investigators, contract laboratories, and other third parties, such as CROs and CMOs to conduct or otherwise support other aspects of our clinical trials for our product candidates. In particular, in clinical trials, we are largely dependent on these parties for execution of clinical trials for our product candidates and control only certain aspects of their activities. Nevertheless, we are responsible for ensuring that our clinical trials are conducted in compliance with the applicable protocol, legal and regulatory requirements and scientific standards, and our reliance on third parties will not relieve us of our regulatory responsibilities. For any violations of laws and regulations during the conduct of our clinical trials, we could be subject to untitled letters, warning letters or enforcement action that may include civil penalties up to and including criminal prosecution.
| 10 |
We and such third parties will be required to comply with the applicable legislations, regulations and other requirements in the relevant jurisdictions in conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the trial patients are adequately informed of the potential risks of participating in clinical trials and their rights are protected. These regulations are enforced by the relevant governmental and regulatory authorities through regular inspections of clinical trial sponsors, principal investigators and trial sites. If we and/or these third parties fail to comply with applicable Good Clinical Practice and other regulations of the relevant jurisdictions, the clinical data generated in our clinical trials may be perceived as unreliable and the governmental and regulatory authorities may require us to conduct additional clinical trials before approving our marketing application filings. We cannot guarantee that, upon inspection or review, the governmental or regulatory authorities will determine that any of our future clinical trials are in compliance with applicable legislations, regulations and other requirements. In addition, our clinical trials must be conducted with product candidates produced under cGMP regulations. Our failure or the failure of these third parties to comply with these regulations may require us to conduct clinical trials again, which would delay the marketing approval process and could also subject us to enforcement action. The COVID-19 pandemic and government measures taken in response have also had a significant impact on our third-party contractors, and we expect that they will face further disruption, which may affect our commencement and execution of our pre-clinical studies and clinical trials.
As a result, we do not have direct control over several crucial aspects of our clinical development, including their conduct and timing. Our reliance on third parties to conduct future clinical trials will also result in limited direct control over the management of data developed through clinical trials than would be the case if we were relying entirely upon our own staff. Working with external parties can also be challenging, which may potentially lead to mistakes as well as difficulties in coordinating activities. External parties may:
| ● | have staffing difficulties; | |
| ● | fail to comply with contractual obligations; | |
| ● | experience legal and/or regulatory compliance issues; | |
| ● | undergo changes in priorities or become financially distressed; and/or | |
| ● | form relationships with other entities, some of which may be our competitors, which may undermine our clinical development. |
If third parties do not satisfactorily conduct our clinical trials, breach their contractual obligations to us and/or violate regulatory requirements, we cannot rely on clinical data collected by these third parties and may have to repeat, extend the duration of, or increase the size of any clinical trials we conduct, which could significantly delay commercialization and involve significantly greater expenditures.
If any of our relationships with these third parties deteriorate or terminate, we may not be able to enter into arrangements with alternative third parties on commercially reasonable terms, or at all. If third parties do not successfully carry out their contractual obligations or meet targeted timelines, if they have to be replaced with alternative third parties or if the quality or accuracy of the clinical data they obtain are compromised due to any breach of our clinical protocols, regulatory requirements or for other reasons, any clinical trials such third parties are associated with may be extended, delayed or terminated, and we may not be able to obtain the requisite marketing approval for or successfully commercialize our product candidates. As a result, we believe that our financial results and the commercial prospects for our product candidates in the clinical trials would be adversely affected, and our costs will be increased thereby affecting our ability to commercialize our products and to generate revenues.
If we fail to successfully compete with academic institutions and other biopharmaceutical companies that are developing similar product candidates or alternatives to cellular immunotherapy product candidates, our business will be materially and adversely affected.
The field of immunotherapy for treatment of cancers is characterized by intense and dynamic competition to develop new technologies and proprietary therapies. Any product candidates that we developed or are in the process of developing will have to compete with existing therapies and new therapies that may from time to time become available in the future. We face competition from various sources, including well-funded biopharmaceutical companies, established biopharmaceutical companies, as well as public and private research institutions. The areas of competition include relevant scientific and management human resources, funding for product development, establishing clinical study sites and clinical subjects participating in the trials.
| 11 |
Our known biopharmaceutical competitors working on allogeneic CAR-T therapies currently include Allogene Therapeutics, Inc. (Nasdaq:ALLO), Astellas Pharma Inc. (TSE:4503.T), Bristol-Myers Squibb (NYSE:BMY), Celyad Oncology SA (Nasdaq:CYAD), Fate Therapeutics, Inc. (Nasdaq:FATE), Gilead Sciences, Inc. (Nasdaq:GILD), NantKwest, INC. (Nasdaq:NK), Novartis International AG (NYSE:NVS), Surface Oncology, Inc. (Nasdaq:SURF), Takeda Pharmaceutical Company Limited (NYSE:TAK) and numerous other biopharmaceutical companies.
We are aware of the recent developments of our potential competitors for allogeneic CAR gamma delta T- cell therapy. Based on publicly disclosed information, in May 2023 Acepodia Biotech, Inc. announced first patient dosed in their phase 1 clinical trial of an anti-CD20 armed allogeneic gamma delta T-cell therapy to treat patients with Non-Hodgkin’s Lymphoma, as well as FDA Clearance of Investigational New Drug (IND) Application for a First-in-Class Allogeneic Anti-EGFR Cell Therapy. In first quarter 2024, Bristol Myers Squibb (NYSE: BMY) announced U.S. FDA approval for CD19-directed CAR T cell therapy as the first and only CAR T cell therapy for adults with relapsed or refractory Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL), as well as Priority Review for Follicular Lymphoma (FL) and Relapsed or Refractory Mantle Cell Lymphoma (MCL). In October 2023, Immatics N.V. (NASDAQ: IMTX) received FDA Regenerative Medicine Advanced Therapy (RMAT) designation for a TCR-T cell therapy targeting PRAME, a protein frequently expressed in a large variety of solid tumors, with current ongoing discussions with FDA to enter registration-enabling Phase 2 trial in melanoma in 2024. In December 2023, Adicet Bio, Inc. (Nasdaq: ACET) reported robust dose-dependent expansion and persistence for their gamma-delta T-cell therapy through their ongoing clinical phase I trial for advanced lymphoma, displaying a strong exposure profile and was positively associated with both PD correlates and clinical response.
Our existing competitors in the field of gamma delta T cell therapy are listed in Table 2 of the section entitled “Competition” in this annual report. We are aware of the recent development of our potential competitor for allogeneic gamma delta T- cell therapy. Based on publicly disclosed information, in November 2023, TC BioPharm (Holdings) PLC (Nasdaq: TCBP) announced FDA Clearance of a Phase 1B IND for its allogeneic unmodified gamma delta T-cell product in patients with relapsed/refractory acute myeloid leukemia. As of date of this annual report, we are not aware of any approved Phase III trial for gamma delta T cell-based cancer immunotherapy. Our gamma delta T cell product candidate may also face competition from other cell-based immunotherapy approaches derived from NK cells and T cells.
Our existing competitors in the field of iPSC-derived immune cells include Fate Therapeutics, Inc. (Nasdaq:FATE), Takeda-CiRA joint program (Takeda Pharmaceutical Company Limited (NYSE:TAK), Centre for iPS Cell Research and Application, Kyoto University) and Century Therapeutics, Inc. (Nasdaq:IPSC). In January 2024, Fate Therapeutics has announced initiation of a phase I clinical trial for an iPSC-derived CAR T-cell product targeting human epidermal growth factor receptor 2 (HER2). As of the date of this annual report, Takeda-CiRA joint program is in its pre-clinical stage.
Many of our existing or potential competitors may have greater financial and other resources, larger teams of R&D staff, and more experienced capabilities in researching, developing and testing products than we do. Many of these companies also have more experience in conducting clinical trials, obtaining relevant regulatory approvals, and manufacturing, marketing and distributing therapeutic products. Smaller or pre-clinical stage companies like us may successfully compete by establishing collaborative relationships with larger pharmaceutical companies or academic institutions. Accordingly, our competitors may be more successful than us in obtaining approval for treatments and achieving widespread market acceptance. Our competitors’ treatments may be more clinically effective, or more effectively marketed, than any treatment we may commercialize, and they may render our treatments obsolete or non-competitive before we can recover the expenses of developing and commercializing any of our treatments.
Mergers and acquisitions in the industry may result in even greater resources being concentrated in a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical study sites and subjects for clinical studies. Other small or early-stage companies may also prove to be strong competitors, particularly those with collaborative arrangements with large and established companies.
We anticipate that we will face intense and increasing competition as new therapies enter the market and advanced technologies become available from time to time. We expect that any treatments which we develop and commercialize will need to compete on, among other things, efficacy, safety, convenience of administration and delivery, price, the level of generic competition and the availability of reimbursement from government and other third-party payors.
| 12 |
Our ability to commercialize our proprietary cell products could be reduced or eliminated if our competitors develop and commercialize products that are potentially more suitable, more effective, have a better safety profile, are more convenient or are less expensive than our products. Our competitors also may obtain relevant regulatory approvals for their products more rapidly than we may be able to obtain approval for ours, which could result in our competitors obtaining a head start and establishing a frontrunner position before we are ready to commercialize. If we are not able to compete effectively against our existing and potential competitors, our business, financial condition, results of operations and growth prospects may be materially and adversely affected. Please refer to the section entitled “Competition” for further details.
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
As of the date of this annual report, we have thirty-four (34) full-time employees. We are required to continue expanding our managerial, operational, quality, manufacturing, finance, sales and other resources in order to manage our operations and clinical trials, continue our development activities and eventually commercialize our product candidates. Our existing management and personnel, systems and facilities may not be sufficient to manage and support our future growth. Our growth strategy requires that we:
| ● | discover new product candidates, develop the process and analytical methods for clinical trials and submissions and applications to the relevant regulatory authorities, complete the required clinical trial studies for each, and receive approval from the regulatory authorities to initiate clinical trials for such product candidates; | |
| ● | manage our clinical trials effectively; | |
| ● | identify, recruit, retain, incentivize and integrate additional employees; | |
| ● | continue to maintain and renew qualification of our in-house clinical cGMP Facility; and | |
| ● | continue to improve our operational, financial and management controls, reports systems and procedures. |
If we are unable to attract skilled employees, increase the size of our organization or manage our future growth effectively, it will impair our ability to execute our business strategy and our business, financial condition, results of operations and growth prospects will be materially and adversely affected.
If we fail to attract and retain senior management, clinical, and key scientific personnel, we may be unable to successfully develop our product candidates, conduct our clinical trials and commercialize our product candidates.
Our business is partially reliant on our continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel. Our senior management is crucial for our business and operations. The loss of any of our senior management could delay, hinder or prevent the successful development of our product pipeline, initiation or completion of our planned clinical trials or the commercialization of our future product candidates. We have employment and/or service agreements with all of our senior management team members.
Competition for qualified and experienced personnel in the biopharmaceuticals field is high due to the limited number of individuals with the skillset and experience required by our clinical trial industry. We will need to hire additional personnel to manage and support the expansion of our clinical development and any commercial activities we may commence. We may not be able to attract and retain suitable and qualified personnel on acceptable terms, or at all. If we cannot employ and retain the requisite qualified personnel for the operation of our business, our operation, financial condition, results of operations and growth prospects would be materially and adversely affected. In addition, to the extent we hire personnel from competitors, we may be subject to allegations that they have been improperly solicited or that they have divulged proprietary or other confidential information, or that their former employers have intellectual property rights to their research output.
| 13 |
We may incur substantial liabilities as a result of product liability lawsuits, if any, against us and this could limit commercialization of any product candidate that we may develop.
We face an inherent risk of product liability exposure related to the testing of our product candidates in clinical trials and may face an even greater risk if we commercialize any product candidate that we may develop. If we cannot successfully defend ourselves against claims that any such product candidates caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| ● | decreased demand for any product candidate that we may develop; | |
| ● | loss of revenue; | |
| ● | substantial monetary awards to trial participants or patients; | |
| ● | significant time and costs to defend the related litigation; | |
| ● | withdrawal of clinical trial participants; | |
| ● | increased insurance costs; | |
| ● | the inability to commercialize any product candidate that we may develop; and | |
| ● | loss of reputation and significant negative publicity and media attention. |
Any such outcomes could materially and adversely affect our business, financial condition, results of operations and growth prospects.
Currently, we have limited insurance coverage, and even if we obtain additional insurance policies, they may be inadequate, may not cover all of our potential liabilities and may potentially expose us to unrecoverable risks.
As of the date of this annual report, we do not carry insurance for all categories of risk that our business may encounter. We maintain clinical trial insurance coverage for the ANGELICA Trial and if we successfully commercialize any product candidate we intend to obtain a product liability insurance. Our license agreements include broad obligations to indemnify our licensors against losses, damages, costs, or expenses or claims for compensation arising of the products we are developing or licensed cell line we will use in connection with our product. Insurance availability, coverage terms and pricing continue to vary with market conditions. We will endeavor to obtain appropriate insurance coverage for insurable risks that we identify from time to time. However, we may fail to accurately predict, assess or quantify insurable risks. We may not be able to obtain appropriate insurance coverage and insurers may not respond as we intend to cover insurable events that may occur. Any significant uninsured liability may require us to pay substantial amounts, which would materially and adversely affect our business, financial condition, results of operations and growth.
In addition, although we are dependent on certain key management personnel, we do not have any key man life insurance policies in place in respect of any such individual. Therefore, if any of our key management personnel passes away or becomes disabled, we will not receive any compensation to assist with the absence of such key management personnel. The loss of such key management personnel could materially and adversely affect our business, financial condition, results of operations and growth prospects.
We do not currently carry a valid Directors’ and Officers’ insurance policy which expired on April 19, 2024. We are reviewing the requirements of the Directors’ and Officers’ insurance policy and do not intend to renew until we identify a suitable insurer. Even if we renew such insurance, any such claim that may be brought could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance, or that is in excess of the limits of the insurance coverage. We recognize the corporate litigation in general, subjecting directors and officers to expensive litigation risks. We desire to attract and retain talented and experienced individuals, to serve as directors and officers of the Company and/or one or more subsidiaries of the Company, free from undue concern for claims for damages arising out of or related to such services to the Company and/or one or more subsidiaries of the Company, we have determined and agreed to indemnify our directors and officers to the maximum extent permitted by law.
Our business involves the use of biohazardous materials and we and any third-party manufacturers and suppliers we may engage are subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these biohazardous materials.
Our R&D activities and those of any third-party manufacturers and suppliers we may engage involve the controlled storage, use and disposal of biohazardous materials. We and our manufacturers and suppliers may thus be subject to laws and regulations governing the use, manufacture, storage, handling and disposal of these biohazardous materials. In some cases, these biohazardous materials and various wastes resulting from their use are stored at our manufacturers’ facilities pending their use and disposal.
| 14 |
We cannot completely prevent the risk of contamination despite our best efforts, and any contamination could result in an obstruction of our R&D efforts and business operations, environmental damage resulting in costly clean-up and liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we believe that the safety procedures utilized by our third-party manufacturers and suppliers for handling and disposing of these materials generally comply with the standards prescribed by these laws and regulations, we cannot be absolutely certain that this is always the case and/or fully eliminate the risk of accidental contamination or injury from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources and state or federal or other relevant authorities may curtail our use of certain materials and/or interrupt our business operations. Furthermore, environmental laws and regulations are complex, subject to frequent developments and are increasingly stringent over time. We cannot anticipate the impact of such changes and cannot be certain of our future compliance with such developments. We have not obtained any biological or hazardous waste insurance coverage as of the date of this annual report. Any contamination by biohazardous materials resulting from our operations could require us to suspend and/or cease our operations and therefore materially and adversely affect our business, financial condition, results of operations and growth prospects.
We are exposed to information technology and cyber security risks and disruption of service.
Our businesses and operations rely heavily on information technology as, amongst other things, we handle, store, manage and transmit data relating to our day-to-day activities not limited to manufacturing, product analysis and data, product candidates and trials. While we have taken steps internally to back up critical data on a service provided by a reputable third-party vendor, as well as storing local copies within the Company, a risk of failure beyond our control still exists. We are therefore exposed to risks of cyber security threats, data and data privacy breaches, as well as other network security and stability risks. The scale and sophistication of cyber security threats have increased especially in recent times. We are also reliant on a number of third-party vendors to provide adequate and timely software and hardware support, that could have a material adverse effect on our systems. Disruptions to our information technology systems, whether resulting from cyber-attacks, a failure by a key vendor or otherwise, that can cause interruptions to our network and services, may result in penalties. While we have established policies and frameworks, as described in the section titled “Intellectual Property – Trade Secrets” of this annual report, we cannot assure you that such policies and frameworks are sufficient or that our business, financial condition, results of operations and prospects would not be adversely affected by such information technology and cyber security threats, data privacy breaches as well as other network security and stability risks.
RISKS RELATED TO MANUFACTURING OF CELL PRODUCTS
Our cGMP Facility may be affected by circumstances beyond our control, such as natural disasters, and we may be required to cease operations and manufacturing of our product candidates
We rely on the continued operations and maintenance of our cGMP Facility for the manufacture of our product candidates. The manufacturing of our product candidates in our cGMP Facility may be frustrated, disrupted, delayed or prevented by circumstances beyond our control, including without limitation earthquakes, fires, floods, explosion, accidents, disruptions to the electrical supply (including increased prices that may be prohibitive) leading to its shutdown, or other natural disasters. If any such event occurs, this could cause our equipment to malfunction and/or break down, and we may be forced to cease operations at our cGMP Facility notwithstanding that we have installed our own standby generator to try to mitigate such risks, until we manage to fully restore it in proper working order and condition. This will impede and delay our pre-clinical testing and progress of our proposed clinical trials and increase our costs and expenses, which will materially and adversely affect our business and operations.
In addition, as our cGMP Facility is subject to rigorous testing and standards in Malaysia, we may be subject to unannounced audit checks (or routine testing) from time to time by the relevant authorities in Malaysia, including the NPRA and others. If we fail to comply with any of the standards required of us, or if we do not comply with any of the Malaysian authorities’ directions to us within a specified time period, we may be required to suspend or cease our operations at our cGMP Facility. If this happens, we may be subject to warnings or penalties imposed by the Malaysian authorities or we may lose our license to operate the cGMP Facility. This would similarly impede and delay our pre-clinical testing and progress of our proposed clinical trials and increase our costs and expenses, which would materially and adversely affect our business, operations and prospects. As of the date of this annual statement, our Group has not been subject to any penalties issued by the Malaysian authorities for any non-compliance with the standards required for the operation and maintenance of a cGMP Facility in Malaysia.
Our current cGMP Facility may not be sufficient to handle the large-scale commercial manufacture and production of product candidates
As of the date of this annual report, we are a pre-clinical biopharmaceutical company with four product candidates: CTM-N2D, CTM-GDT, iPSC-gdNKT and CTM-MSC. Our cGMP Facility is equipped to undertake the manufacture and production of our product candidates for pre-clinical and clinical trial purposes. However, our current cGMP Facility may not have the capabilities and resources to undertake and facilitate large-scale commercial production of our product candidates. In the event that any of our product candidates reach the commercialization stage, we will need to incur additional expenses to build another facility which is equipped to handle large-scale manufacturing or outsource manufacturing and/or production.
| 15 |
Our manufacturing process is complex, and we may encounter challenges in production of our product candidates, which would delay or hinder our ability to provide a sufficient supply of our product candidates for clinical trials or our products for patients, if approved.
Our product candidates are human cells, and the process of manufacturing such product candidates, as well as engineered K562 cells, is complex, highly regulated and involves numerous risks. Manufacturing our product candidates involves harvesting blood cells from a donor, isolating the gamma delta T cells, activating and expanding the gamma delta T cells, introducing a mRNA to be expressed on the gamma delta T cells, fill and finish and eventually shipping and infusing the cell product into the patient’s body. As a result of these complexities, the cost of manufacturing our cellular product candidates is generally higher than traditional small-molecule chemical compounds or biologics, and such manufacturing process is less reliable and harder to reproduce.
Our manufacturing process is subject to product loss or failure, or product variation that may negatively impact patient outcomes, due to logistical issues associated with supply chain disruptions, the collection of starting material from the donor, shipping such material to the manufacturing site, shipping the final product back to the clinical trial recipient, preparing the product for administration, infusing the patient with the product, manufacturing issues or different product characteristics resulting from the differences in donor starting materials, variations between reagent lots, interruptions in the manufacturing process, contamination, equipment or reagent failure, improper installation or operation of equipment, vendor or operator error, inconsistency in cell growth and variability in product characteristics.
Apart from the transport of material from the donor, we may also face disruptions in the supply of other materials, especially those which have to be imported from other countries, which are essential parts of the manufacturing process. For example, we have to use a special cell culture vessel for storage of the CTM-N2D product candidate, for which we have currently only been able to identify one supplier who sends it from the United States. We also use a special cell culture medium in the manufacturing process of the CTM-N2D product candidate which has to be kept in a special freezer and therefore special arrangements have to be made for its courier. In this regard, medical courier is a lot more expensive than other third-party service providers. Therefore, in the event that there are any delays in these deliveries, or increased prices for the material and/or delivery, the manufacturing processes may be affected as the delay and/or increased costs may be prohibitive.
Slight deviations from our normal manufacturing processes could result in a decrease in production yields, product defects and other supply disruptions. If, for any reason in our CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC clinical studies, we damage and/or lose the starting material for a manufactured product for any of our patients at any point in the process, the manufacturing process for that patient would need to be repeated from the beginning and the resulting delay could require restarting the manufacturing process, or could result in such patient withdrawing from our clinical trial. If microbial, viral and/or other contaminations are discovered in any of our product candidates or in any of the manufacturing facilities in which our products or other materials are manufactured, such manufacturing facilities may need to be closed for an extended period of time to investigate and salvage the contamination. We may have to maintain a chain of identity in which materials are pegged to a specific donor as they move from the donor to the manufacturing facility, through the manufacturing process and back to the clinical trial recipient. Maintaining a chain of identity is challenging and complex and requires a high degree of accuracy and precision, and failure to do so could result in adverse patient outcomes, loss of product or regulatory action, including withdrawal of our products from the market, if licensed. Any failure in the foregoing processes could render a batch of product unusable, affect the regulatory approvals of such product candidate, cause us to incur fines or penalties or prejudice our reputation and that of our product candidates.
We may revise our manufacturing process for various reasons, such as to control costs, achieve scale, decrease processing time, and increase manufacturing success rate. As a result of changes made to our manufacturing process during the course of clinical development, we may need to demonstrate the comparability of the product used in earlier clinical phases or at earlier portions of a trial to the product used in later clinical phases or later portions of the trial. Changes to our manufacturing process made before or after commercialization could require us to show the comparability of the resulting product to the product candidate used in the clinical trials using earlier processes. We may need to collect additional non-clinical or clinical data from any modified process prior to obtaining marketing approval for the relevant product candidate produced with such modified process. If such data are not ultimately comparable to the data obtained in earlier trials or an earlier stage of the same trial in terms of safety and/or efficacy, we may be required to make further changes to our manufacturing process and/or undertake additional clinical testing, either of which could significantly delay the clinical development or commercialization of the associated product candidate, which would materially and adversely affect our business, financial condition, results of operations and growth prospects.
| 16 |
We are highly reliant on certain key suppliers for certain steps of our manufacturing processes.
Our manufacturing processes for CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC depend on the use of cell culture vessel, and other reagents, some of which might only be available from certain key suppliers. In addition, some of these reagents, at the time of procurement, typically expire after approximately four to six months. Due to the short expiration period, we are unable to store the reagents in large quantities for future needs to mitigate against the risk of shortage due to disruption of the supply chain.
Further, although many of the reagents and consumables we require for our manufacturing process are available from more than one commercial supplier, we have not yet confirmed the suitability of the use of all such reagents and consumables in our manufacturing process. Even if we are able to replace any raw materials or consumables with alternatives, such alternatives may be more expensive, result in lower yields or not be as suitable for our purposes. In addition, some of the raw materials that we use are complex materials, which is likely to be harder to obtain substitutes for. Therefore, supply disruptions could result in significant delays and additional regulatory submissions and prevent us from being able to manufacture our product candidates due to the unsuitability of the substituted reagent or consumable that we are able to obtain.
In addition, some of the reagents that we use may be restricted by limited use label licenses and may be for research use and not for commercial application of any kind. We may need to replace certain reagents in order for us to commercialize our products in the future. While there are alternatives available, such alternatives may be more expensive, may result in lower yields or may not be as suitable for our purpose. These alternative reagents may also face regulatory challenges and review and potentially result in significant delays in our clinical trials and future developments.
Any disruption in supply of these reagents could result in significant delays in our clinical trials, which would materially and adversely affect our business, financial condition, results of operations and growth prospects.
We rely on the storage of our cell bank for the engineered K562 cells and peripheral blood mononuclear cells, and any damage or loss to them would cause delays in our clinical trials and may materially and adversely affect our business.
The cell bank of the engineered K562 cells we license and our peripheral blood mononuclear cells are stored in liquid nitrogen tanks at our cGMP Facility. If these materials are damaged or lost at our cGMP Facility, including by the loss or malfunction of these liquid nitrogen tanks, or by damage from fire, power loss or other natural disasters, we would need to establish replacement master and working cell banks of the engineered K562 cells and peripheral blood mononuclear cells, which would impact clinical supply and delay our clinical trials or treatment of patients. If we are unable to find replacement storage facilities and/or re-manufacture the engineered K562 cells timeously, we may incur significant additional expenses and liability to patients whose treatment is delayed, which would materially and adversely affect our business. If we are unable to source for peripheral blood mononuclear cells which are suitable for manufacturing from healthy donors on a timely basis, we may incur significant additional expenses and liability to patients whose treatment is delayed, which would materially and adversely affect our business. Our K562 Cell License for NK cell expansion also requires us to broadly indemnify our licensor against any loss, damages, costs, expenses, or other claim for compensation in connection with the license or our licensed products under the license.
Delays in obtaining renewal of regulatory approvals for our cGMP Facility and licenses could delay our development plans and thereby limit our ability to generate revenues.
We believe that in-house cGMP manufacturing capability is important to facilitate clinical product supply, lower the risk of manufacturing disruptions and enable more cost-effective manufacturing. We have an existing cGMP Facility in Johor Bahru, Malaysia that will allow us to supply the product candidates needed for our clinical trials. In the event that our product candidates are approved for commercial use, we also plan to build an additional facility for the commercial-scale manufacture of our product candidates in the future. The design, construction, qualification and regulatory approvals for such facilities require substantial capital and technical expertise and any delay would limit our development activities and our opportunities for growth.
| 17 |
Furthermore, our cGMP Facility is subject to ongoing, periodic inspection by the NPRA to ensure compliance with cGMP and other relevant laws and regulations. Any failure to follow and document our adherence to these regulations or other regulatory requirements may lead to significant delays in the availability of products for clinical use or may result in the termination of or a hold on a clinical study. The Malaysian authorities may also make unannounced checks and audits of our cGMP Facility. Failure or omission to comply with relevant laws or regulations could also result in sanctions being imposed on us, including fines, injunctions, civil penalties, a requirement to suspend or put on hold one or more of our clinical trials, failure of regulatory authorities to grant marketing approval of our drug candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of drug candidates, operating restrictions and criminal prosecutions, any of which could materially adversely affect our business, financial condition, results of operations and growth prospects.
We also may encounter problems with regard to the following:
| ● | complying with legislation and regulations governing donor traceability, manufacturing, release of product candidates and other requirements from regulatory authorities outside Malaysia; | |
| ● | achieving adequate or clinical-grade materials that fulfill regulatory agency standards and/or specifications with consistent and acceptable production yield and costs; | |
| ● | bacterial, fungal or viral contamination in our cGMP Facility; and/or | |
| ● | shortage in qualified personnel, raw materials and/or key contractors. |
Our product candidates, if approved by the relevant regulatory authorities, may require significant commercial supply to meet market demand. In these cases, we may need to significantly increase or “scale up”, our production process. If we fail to develop sufficient manufacturing capacity and/or experience, whether internally or with a third party, in time or at all, or fail to manufacture our product candidates economically or at reasonable scale or volume or in accordance with cGMP, or if the cost of the increase or “scale-up” in our production process is not economically feasible, our development programs and commercialization of any approved products will be materially adversely affected and we may not be able to produce a sufficient quantity of our product candidates required to meet future demand and our business, financial condition, results of operations and growth prospects may be materially adversely affected.
The optimal donor and manufacturing parameters for our product candidates may have not been definitively established, which may hinder our ability to optimize our product candidates or to address any safety or efficacy issues that may arise.
If any of our clinical trials reveal issues with the safety or efficacy of any of our product candidates, modification of the donor selection criteria or the manufacturing process may be necessary to address such issues. However, we have not fully characterized or identified how donor characteristics and manufacturing process parameters affect the optimal cancer cell killing ability for our CTM-N2D and CTM-GDT product candidates for in vitro and animal efficacy studies or how such potency differences may translate into efficacy to be seen in human clinical trials, including both the proportion of patients who achieve a meaningful clinical response, and the duration of any such clinical responses. As a result, our ability to improve our manufacturing process or product potency, safety, or efficacy according to such parameters is limited and may require significant trial and error, which may cause us to incur significant costs or could result in significant delays to the clinical development and eventual commercialization of our product candidates.
We have not yet developed a validated methodology of freezing and thawing large quantities of CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC product candidates, which may be required for the storage and overseas distribution of our product candidates.
We have not yet developed a validated method of freezing and thawing CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC, which can be frozen and thawed in smaller quantities, in large quantities without damage, in a cost-efficient manner and without degradation over time. We may encounter difficulties not only in developing freezing and thawing methodologies for large scale use, but also in obtaining the necessary regulatory approvals for using such methodologies in treatment. If we cannot adequately demonstrate that our frozen product candidates are similar to our product candidates in unfrozen form to the satisfaction of the HSA and/or other regulatory authorities, we could face substantial delays in obtaining regulatory approvals from the HSA and/or other regulatory authorities. If we are unable to develop a validated method of freezing CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC for shipping purposes, our ability to promote adoption and standardization of our products candidates, as well as achieve economies of scale by centralizing our production facility, will be limited. Even if we are able to successfully freeze and CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC in large quantities, we will also need to develop a cost-effective and reliable distribution and logistics network, which we may be unable to accomplish.
| 18 |
Furthermore, we have not yet demonstrated long-term stability of cryopreserved CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC and therefore are not certain if we will be able to store the cryopreserved cells for extended periods of time. If we are unable to demonstrate such long-term stability, we will need to reduce the manufacturing batch size to ensure that the material we produce will be used before it expires. In that case, the scaling of our production processes will not deliver the efficiencies we expect, and the cost per dose of our product candidates will be substantially higher.
Due to various reasons (including the foregoing), we have not yet established the long-term stability of our cryopreserved CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC and we may not be able to commercialize CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC on a large scale or in a cost-effective manner. If any of our product candidates is found to be unstable, we will have to conduct more frequent manufacturing runs, which could cause us to incur significant additional expenses. Currently, we have identified a potential collaboration partner to develop cryopreservation method for our product candidates.
We believe we may require an updated and validated protocol for commercial-scale expansion and manufacturing of gamma delta T cells for conducting pivotal trials and for commercialization of our product candidates, if approved.
Future clinical trials that we conduct, as well as any potential commercialization of our product candidates when approved, will depend on the reliability, safety and efficacy of our processes for expanding, modifying and manufacturing CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC at scale. Our efforts to scale up production of our CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC in anticipation of future clinical trials or commercialization may reveal, an inability to overcome biology or may otherwise encounter challenges, including scrutiny from regulatory authorities. To the extent we encounter any such difficulties, our ability to conduct additional clinical trials or to scale for commercialization will be hindered or prevented, which would have an adverse effect on our business. Currently, we are in the midst of identifying potential collaboration partners to develop methods for the scaling up manufacturing of our product candidates.
RISKS RELATED TO OUR INTELLECTUAL PROPERTY
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure full protection of our proprietary rights. If our patent position does not adequately protect our product candidates, others could compete against us more directly, which would harm our business, possibly materially.
Our commercial success will depend in part on obtaining and maintaining proprietary rights including patent protection and trade secret protection of our current product candidates and future product candidates, the processes used to manufacture them and the methods for using them, as well as successfully asserting and defending these proprietary rights against third-party challenges. Our ability to stop third parties from making, using, selling, offering to sell or importing our product candidates is dependent upon the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities.
The patent positions of biotechnology and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in pharmaceutical patents has emerged to date in the U.S. or in foreign jurisdictions outside of the U.S., including Singapore. Changes in either the patent laws or interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be enforced in the patents that may be issued from the applications we currently licensed or may in the future own or license from third parties. Further, if any patents we obtain or license are deemed invalid and unenforceable, our ability to commercialize or license our product candidates or technology could be adversely affected.
Others may file patent applications covering products and technologies that are similar, identical, or competitive to ours or important to our business. We cannot be certain that any patent application owned by a third party will not have priority over patent applications filed or in-licensed by us, or that we or our licensors will not be involved in patent office proceedings such as interference, derivation, opposition, reexamination, inter partes review, reissue, post grant review or invalidity proceedings before U.S. or non-U.S. patent offices. Such proceedings are also expensive and time consuming.
| 19 |
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| ● | others may be able to make products that are similar to our product candidates, but that are not covered by the claims of our patents or our licensed patents; | |
| ● | any patents that we obtain from licensing or otherwise may not provide us with any competitive advantages; | |
| ● | any granted patents that we rely upon may be held invalid or unenforceable as a result of legal challenges by third parties; and | |
| ● | the patents of others may have an adverse effect on our business. |
We are dependent on licensed intellectual property. If we were to lose our rights to licensed intellectual property, we may not be able to continue developing or commercializing our product candidates, if approved. If we breach any of the agreements under which we license the use, development and commercialization rights to our product candidates or technology from third parties or, in certain cases, we fail to meet certain development deadlines, we could lose license rights that are important to our business.
We do not currently own any patents, and we are heavily reliant upon a number of license agreements under which we are granted rights to intellectual property that are important to our business and we may need or choose to enter into additional license agreements in the future. Our existing license agreements impose, and we expect that future license agreements will impose on us, various development, regulatory and/or commercial diligence obligations, payment of milestones and/or royalties and other obligations. If we fail to comply with our obligations under these agreements, or we are subject to a bankruptcy, the licensor may have the right to terminate the licenses, in which event we would not be able to market products covered by the licenses. Our business could suffer, for example, if any current or future licenses terminate, if the licensors fail to abide by the terms of the licenses, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms.
Licensing of intellectual property is of critical importance to our business and involves complex legal, business and scientific issues. Disputes may arise between us and our licensors regarding intellectual property subject to a license agreement, including:
| ● | the scope of rights granted under the license agreements and other interpretation-related issues; | |
| ● | whether and the extent to which our technology and processes infringe on intellectual property of the licensor that is not subject to the licensing agreement; | |
| ● | our right to sublicense patent and other rights to third parties; | |
| ● | our diligence obligations with respect to the use of the licensed technology in relation to our development and commercialization of our product candidates, and what activities satisfy those diligence obligations; | |
| ● | the ownership of inventions and know-how resulting from the joint creation or use of intellectual property by our licensors and us and our partners; | |
| ● | our right to transfer or assign the license; and | |
| ● | the effects of termination. |
If disputes over intellectual property that we have licensed prevent or impair our ability to maintain our current licensing arrangements on acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
| 20 |
We have entered into several licenses to support our various projects. Termination of any of these license agreements would have a material adverse impact on our ability to develop and commercialize derived products under each respective agreement.
We have entered into several licenses to support our various projects. We may enter into additional licenses to third-party intellectual property that are necessary or useful to our business. Our current licenses and any future licenses that we may enter into impose various royalty payment, milestone, and other obligations on us. Under some license agreements, we may not control prosecution of the licensed intellectual property, or may not have the first right to enforce the intellectual property. In those cases, we may not be able to adequately influence patent prosecution or enforcement, or prevent inadvertent lapses of coverage due to failure to pay maintenance fees. If we fail to comply with any of our obligations under a current or future license agreement, the licensor may allege that we have breached our license agreement, and may accordingly seek to terminate our license. Termination of any of our current or future licenses could result in our loss of the right to use the licensed intellectual property, which could materially adversely affect our ability to develop and commercialize a product candidate or product, if approved, as well as harm our competitive business position and our business prospects. Under some license agreements, termination may also result in the transfer of or granting in rights under certain of our intellectual property and information related to the product candidate being developed under the license, such as regulatory information.
The agreements under which we have licensed intellectual property or technology or may license intellectual property or technology in the future to or from third parties are complex, and certain provisions in such agreements may be susceptible to multiple interpretations. The resolution of any contract interpretation disagreement that may arise could narrow what we believe to be the scope of our rights to the relevant intellectual property or technology or increase what we believe to be our financial or other obligations under the relevant agreement, either of which could have a material adverse effect on our business, financial condition, results of operations and prospects. Moreover, if disputes over intellectual property that we have licensed or may license prevent or impair our ability to maintain our current licensing arrangements on commercially acceptable terms, we may be unable to successfully develop and commercialize the affected product candidates.
In addition, if our licensor fails to abide by the terms of any of the licenses, if the licensor fails to prevent infringement by third parties, if the licensed patents or other rights are found to be invalid or unenforceable, or if we are unable to enter into necessary licenses on acceptable terms, our business could suffer. Moreover, our licensor may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing, misappropriating or otherwise violating the licensor’s rights.
Similarly, if we are unable to successfully obtain rights to required third-party intellectual property rights or maintain the existing intellectual property rights we have, we may have to seek alternative options, such as developing new product candidates with design-around technologies, which may require more time and investment, or abandon development of the relevant research programs or product candidates and our business, financial condition, results of operations and prospects could suffer.
Some of the intellectual property covered by our licenses concern patent applications. We cannot assure investors that any of the currently pending or future patent applications will result in granted patents, nor can we predict how long it will take for such patents to be granted.
Some of the intellectual property covered by our current licenses from the licensor concern certain, specified patent rights (including patent applications and Patent Cooperation Treaty patent applications). While to some extent and for at least a certain period of time, the licensor has agreed to assume responsibility for the preparation, filing, prosecution and maintenance of patent applications covered by the licensed patent rights, we cannot be certain as to when or if final patents will be issued for those patent applications covered by the licensed patent rights. However, the licensor may not successfully prosecute certain patent applications, the prosecution of which the licensor controls, under which we are only a licensee and on which our business substantially depends. Even if patents issue from these applications, there is no assurance that the patents will be free from defects or survive validity or enforceability challenges, the licensors may fail to maintain these patents, may decide not to pursue litigation against third-party infringers, may fail to prove infringement or may fail to defend against counterclaims of patent invalidity or unenforceability.
Moreover, it is possible that the licensed pending patent applications will not result in granted patents, and even if such pending patent applications are granted as patents, they may not provide a basis for intellectual property protection of commercially viable products or may not provide us with any competitive advantages. Further, it is possible that, for any of the patents that may be granted in the future, others will design around the licensed patent rights or identify methods for preventing or treating diseases that do not concern the rights covered by our licenses. Further, we cannot assure investors that other parties will not challenge any patents granted to the licensor or that courts or regulatory agencies will hold licensor’s patents to be valid or enforceable. We cannot guarantee investors that, if required to defend the covered patents, we will have the funds to or be successful in defending challenges made against the licensed patents. Any successful third-party challenge to the licensed patents could result in the unenforceability or invalidity of such patents, or to such patents being interpreted narrowly or otherwise in a manner adverse to our interests. Our ability to establish or maintain a technological or competitive advantage over our competitors may be diminished because of these uncertainties.
| 21 |
Even if patents are issued based on patent applications to which we have been granted a license, because the patent positions of pharmaceutical and biotechnology products are complex and uncertain, we cannot predict the scope and extent of patent protection for our product candidates.
Any patents that may be issued based on patent applications that we have been granted licenses to will not ensure sufficient protection with respect to our activities for a number of reasons, including without limitation the following:
| ● | any issued patents may not be broad or strong enough to prevent competition from other identical or similar products; | |
| ● | if patents are not issued or if issued patents expire, there would be no protections against competitors making generic equivalents; | |
| ● | there may be prior art of which we are not aware that may affect the validity or enforceability of a patent claim; | |
| ● | there may be other patents existing, now or in the future, in the patent landscape for our product candidates that we seek to commercialize or develop, if any, that will affect our freedom to operate; | |
| ● | if patents that we have been granted licenses to are challenged, a court could determine that they are not valid or enforceable; | |
| ● | a court could determine that a competitor’s technology or product does not infringe patents that we have been granted licenses to; | |
| ● | patents to which we have been granted licenses could irretrievably lapse due to failure to pay fees or otherwise comply with regulations, or could be subject to compulsory licensing; and | |
| ● | if we encounter delays in our development or clinical trials, the period of time during which we could market our products under patent protection would be reduced. |
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and patent protection could be reduced or eliminated for noncompliance with these requirements.
Periodic maintenance fees on any issued patent are due to be paid to the United States Patent and Trademark Office (USPTO) and foreign Intellectual Property Offices in several stages over the term of the patent. Maintenance fees are also due for pending patent applications in some countries. The USPTO and various foreign intellectual property offices require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Noncompliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to office actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In such an event, our competitors might be able to enter the market, which would have a material adverse effect on our business.
The life of patent protection is limited, and third parties could develop and commercialize products and technologies similar or identical to ours and compete directly with us after a patent licensed to us expires, which could materially and adversely affect our ability to commercialize our products and technologies.
The life of a patent and the protection it affords is limited. For example, in the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional filing date. In Europe, the expiration of an invention patent is 20 years from its filing date. Even if we successfully obtain patent protection for an approved product candidate, it may face competition from biosimilar medications. Manufacturers of other drugs may challenge the scope, validity or enforceability of the patents underlying our technology in court or before a patent office, and the patent holder may not be successful in enforcing or defending those intellectual property rights and, as a result, we may not be able to develop or market the relevant product candidate exclusively, which would materially adversely affect any potential sales of that product.
| 22 |
Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such product candidates might expire before or shortly after such product candidates are commercialized. As a result, the patent pending applications licensed to us may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours. Even if we believe that the patents involved are eligible for certain (and time-limited) patent term extensions, there can be no assurance that the applicable authorities, including the FDA and the USPTO, and any equivalent regulatory authority in other countries, will agree with our assessment of whether such extensions are available, and such authorities may refuse to grant extensions to such patents, or may grant more limited extensions than requested. For example, depending upon the timing, duration and specifics of any FDA marketing approval of any product candidates we may develop, one or more of the U.S. patents licensed to us may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Action of 1984, or Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. However, we may not be granted an extension or may be granted a shorter extension because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patent, or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than requested. If we are unable to obtain patent term extension or term of any such extension is less than requested, our competitors may obtain approval of competing products following our patent expiration, and our business could be harmed. Changes in either the patent laws or interpretation of the patent laws in the United States and other countries may diminish the value of our patents or narrow the scope of our patent protection.
The patent pending applications licensed to us for our product candidates are expected to expire on various dates. Upon the expiration, we or our licensor will not be able to assert such licensed patent rights against potential competitors, which would materially adversely affect our business, financial condition, results of operations and prospects.
We may need to license intellectual property from third parties, and such licenses may not be available on commercially reasonable terms or may not be available at all.
There may be intellectual property rights existing now, or in the future, relevant to our product candidates that we seek to commercialize or develop, if any, that may affect our ability to commercialize such product candidates. Although the Company is not aware of any such intellectual property rights, a third party may hold intellectual property rights, including patent rights, which are important or necessary to the development or manufacture of our product candidates. Even if all our main product candidates are covered by patents, it may be necessary for us to use the patented or proprietary technology of third parties to commercialize our product candidates, in which case we would be required to obtain licenses from these third parties. Such licenses may not be available on commercially reasonable terms, or at all, and we could be forced to accept unfavorable contractual terms. In that event, we may be required to expend significant time and resources to redesign our technology, product candidates, or the methods for manufacturing them or to develop or license replacement technology, all of which may not be feasible on a technical or commercial basis. If we are unable to do so, our business could be harmed.
The licensing or acquisition of third-party intellectual property rights is a competitive area, and several more established companies may pursue strategies to license or acquire third party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, capital resources and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license intellectual property rights to us. We also may be unable to license or acquire third party intellectual property rights on terms that would allow us to make an appropriate return on our investment or at all. If we are unable to successfully obtain rights to required third party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of the relevant project or product candidate, which could have a material adverse effect on our business, financial condition, results of operations, and prospects.
| 23 |
We may infringe the intellectual property rights of others, which may prevent or delay our product development efforts and stop us from commercializing or increase the costs of commercializing our product candidates.
Our success will depend in part on our ability to operate without infringing the proprietary rights of third parties. We are not aware of any third-party proprietary rights that our planned products will infringe or misappropriate, but we have not conducted any freedom to operate study as we are in the earliest stages of development. We thus cannot guarantee that our product candidates, or manufacture or use of our product candidates, will not infringe third-party patents. Furthermore, a third party may claim that we are using technologies covered by the third party’s patent rights and may go to court to stop us from engaging in our normal operations and activities, including making or selling our product candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and scientific personnel. Some of these third parties may be better capitalized and have more resources than us. There is a risk that a court would decide that we are infringing the third party’s patents and would order us to stop the activities covered by the patents. In that event, we may not have a viable way around the patent and may need to halt commercialization of our product candidates. In addition, there is a risk that a court will order us to pay the other party damages for having violated the other party’s patents. In addition, we may be obligated to indemnify our licensors and collaborators against certain intellectual property infringement claims brought by third parties, which could require us to expend additional resources. The pharmaceutical and biotechnology industries have produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform.
If we are sued for patent infringement, we would need to demonstrate that our product candidates or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid, and we may not be able to do this. Proving invalidity is difficult. For example, in the U.S., proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents. Even if we are successful in these proceedings, we may incur substantial costs and diversion of management’s time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, which may not be available, defend an infringement action or challenge the validity of the patents in court. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. In addition, if we do not obtain a license, develop or obtain non-infringing technology, fail to defend an infringement action successfully or have infringed patents declared invalid, we may incur substantial monetary damages, encounter significant delays in bringing our product candidates to market and be precluded from manufacturing or selling our product candidates.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than us or the third parties from whom we license intellectual property because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
We may become involved in lawsuits to protect or enforce our intellectual property, which could be expensive, time consuming and unsuccessful.
In addition to the possibility of litigation relating to infringement claims asserted against us, we may become a party to other patent litigation and other proceedings, including inter partes review proceedings, post grant review proceedings, derivation proceedings declared by the USPTO and similar proceedings in foreign countries, regarding intellectual property rights with respect to our current or future technologies or product candidates or products. The cost to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Patent litigation and other proceedings may also absorb significant management time. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could impair our ability to compete in the marketplace.
Competitors may infringe or otherwise violate our intellectual property, including patents that may issue to or be licensed by us. As a result, we may be required to file claims in an effort to stop third-party infringement or unauthorized use. Any such claims could provoke these parties to assert counterclaims against us, including claims alleging that we infringe their patents or other intellectual property rights, and/or that any of our intellectual property, including licensed intellectual property, is invalid and/or unenforceable. This can be prohibitively expensive, particularly for a company of our size, and time-consuming, and even if we are successful, any award of monetary damages or other remedy we may receive may not be commercially valuable. In addition, in an infringement proceeding, a court may decide that our asserted intellectual property is not valid or is unenforceable, or may refuse to stop the other party from using the technology at issue on the grounds that our intellectual property does not cover the third party’s technology. An adverse determination in any litigation or defense proceedings could put our intellectual property at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing as patents.
| 24 |
If the breadth or strength of our patent or other intellectual property rights is compromised or threatened, it could allow third parties to exploit and, in particular, commercialize our technology or products or result in our inability to exploit and/or commercialize our technology and products without infringing third-party intellectual property rights. Further, third parties may be dissuaded from collaborating with us.
Interference or derivation proceedings brought by the USPTO or its foreign counterparts may be necessary to determine the priority or inventorship of inventions with respect to our licensed patents or patent applications, and we may also become involved in other proceedings, such as re-examination proceedings, before the USPTO or its foreign counterparts. Due to the substantial competition in the pharmaceutical space, the number of such proceedings may increase. This could delay the prosecution of our pending patent applications or impact the validity and enforceability of any future patents that we may obtain. In addition, any such litigation, submission or proceeding may be resolved adversely to us and, even if successful, may result in substantial costs and distraction to our management.
If we are not able to adequately prevent disclosure of trade secrets and other proprietary information, the value of our technology and product could be significantly diminished.
We also rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA, as part of its transparency initiative, is currently considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA’s disclosure policies may change in the future, if at all. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
We may be subject to claims that our employees or consultants have wrongfully used or disclosed alleged trade secrets.
As is common in the biotechnology and pharmaceutical industries, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees or consultants have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we could lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Our intellectual property may not be sufficient to protect our product candidates from competition, which may negatively affect our business as well as limit our partnership or acquisition appeal.
We may be subject to competition despite the existence of intellectual property we license or may in the future own. We can give no assurances that our intellectual property rights will be sufficient to prevent third parties from designing around patents we own or license and developing and commercializing competitive products. The existence of competitive products that avoid our intellectual property could materially adversely affect our operating results and financial condition. Furthermore, limitations, or perceived limitations, in our intellectual property may limit the interest of third parties to partner, collaborate or otherwise transact with us, if third parties perceive a higher than acceptable risk to commercialization of our product candidates or future product candidates.
| 25 |
We may elect to sue a third party, or otherwise make a claim, alleging infringement or other violation of patents, trademarks, trade dress, copyrights, trade secrets, domain names or other intellectual property rights that we either own or license from a third party. If we do not prevail in enforcing our intellectual property rights in this type of litigation, we may be subject to:
| ● | paying monetary damages related to the legal expenses of the third party; | |
| ● | facing additional competition that may have a significant adverse effect on our product pricing, market share, business operations, financial condition, and the commercial viability of our product; and | |
| ● | restructuring our company or delaying or terminating select business opportunities, including, but not limited to, R&D, clinical trial, and commercialization activities, due to a potential deterioration of our financial condition or market competitiveness. |
A third party may also challenge the validity, enforceability or scope of the intellectual property rights that we license or own and the result of these challenges may narrow the scope or claims of or invalidate patents that are integral to our product candidates in the future. There can be no assurance that we will be able to successfully defend patents we own or license in an action against third parties due to the unpredictability of litigation and the high costs associated with intellectual property litigation, amongst other factors.
Intellectual property rights may be less extensive and enforcement may be more difficult in jurisdictions outside of the U.S. Therefore, we may not be able to protect our intellectual property and third parties may be able to market competitive products that may use some or all of our intellectual property.
Changes to patent law, including the Leahy-Smith America Invests Act of 2011 and other future article of legislation, may substantially change the regulations and procedures surrounding patent applications, issuance of patents and prosecution of patents. We can give no assurances that the patents of ours or our licensor’s can be defended or will protect us against future intellectual property challenges, particularly as they pertain to changes in patent law and future patent law interpretations.
If any of our license agreements with ATPL is terminated, we could lose our rights to key components enabling our core proprietary CAR gamma delta T cell technology platform.
We are highly reliant on three licenses granted to us pursuant to the Patent License, K562 Cell License for NK cell expansion, and Know-How License by ATPL, a wholly-owned subsidiary of A*STAR and the leading R&D agency in Singapore. A*STAR is the leading body for biomedical research initiatives among the research community in Singapore. Please refer to the section entitled “Intellectual Property” for further information on these licenses. Our lead product candidates, CTM-N2D, iPSC-gdNKT and CTM-GDT, are derived from the technology we licensed from ATPL. If any of these three license agreements is terminated for any reason, we would lose the rights to use the licensed intellectual property rights that may be material or necessary to the development and/or production of our product candidates, which could impede or prevent our successful commercialization of such product candidates and materially adversely affect our business, financial condition, results of operations and growth prospects.
Furthermore, our Patent License is field-specific and has been granted to us in the field of immunotherapy, including stem cell therapy and treatment and immunotherapy drugs using any licensed patents that issue from and claim priority with and from the two Patent Cooperation Treaty applications in the Patent License.
If the licenses granted to us by ATPL are affected in any manner, it could delay our development and commercialization of our product candidates, which in turn could materially adversely affect our business, financial condition, results of operations and growth prospects.
Our development and commercialization rights to our current and future product candidates and technology are subject, in part, to the terms and conditions of patent licenses granted to us by others.
We do not have the right to control the preparation, filing, prosecution, maintenance, enforcement and defense of patents and patent applications in respect of the technology(ies) that we license from ATPL. ATPL is responsible for patent prosecution and maintenance costs in a limited number of countries and territories. Therefore, we cannot guarantee that these patent applications and patents issued therefrom will be prepared, filed, prosecuted, maintained, enforced and defended in a manner consistent with our best interests. If ATPL fails to prosecute, maintain, enforce and defend any of such patent applications or patents, or lose rights to any of those patents or patent applications, the patent rights we have licensed may be reduced or eliminated, and our right to develop and commercialize any of our products that are the subject of such licensed rights could be impaired. Additionally, we may be required to reimburse ATPL for their expenses related to the prosecution, maintenance, enforcement and defense of patents and patent applications that we license from them.
| 26 |
We may not be able to meet our commercialization milestones in our patent license agreements with ATPL, and if our license agreement is terminated as a result, our clinical trials and business will be significantly affected.
Any breach or non-compliance of any term in any of our patent license agreements with ATPL (such as our failure to meet any of the commercialization milestones relating to different phases of clinical trials or otherwise) may result in termination. Failure by us to renew or otherwise maintain the required patent licenses, or cancellation, suspension or revocation of any of our patent licenses pursuant to our patent license agreements with ATPL may result in the interruption of our clinical trials and operations if we are not able to use the patent licenses and this could potentially affect our working relationship with ATPL and/or A*STAR, which would have a materially adverse effect on our business, operations and future prospects. We may not be able to identify another licensor able and willing to license such technology(ies) to us, and we may be required to recreate our product candidates altogether which would significantly increase our costs and expenses required to operate. This would also severely delay any potential commercialization of our products and we may not be able to create a new product candidate suitable for pre-clinical or clinical trials or even commercialization.
We have entered into three addenda for our Patent License that extends the deadlines to meet the commercialization milestones. Failure to meet these extended milestone deadlines may result in termination of the license. Additionally, in the second addendum CytoMed agreed to take over responsibility for all patent prosecution maintenance costs beginning January 2022.
Pursuant to the third addendum, the four milestones have been extended from the second addendum as follows: commencement of Phase 1 clinical trial for fifteen (15) months; commencement of Phase 2 clinical trial for forty-five (45) months; commencement of Phase 3 clinical trial for fifty-seven (57) months; and commercial sale of the Licensed Product for sixty-nine (69) months.
Any patents that we have licensed from ATPL will expire, and when they expire, our business may be affected.
Our development of our product candidates is reliant on some of the technologies licensed to us pursuant to the patent licenses from ATPL as described in the section entitled “Intellectual Property”. When the patents licensed from ATPL, assuming they are issued from the pending patent applications reach the end of their lives, we may not be able to continue to preclude others from making, using or selling, any of our product candidates that are approved.
We may not be able to obtain and enforce our intellectual property rights throughout the world.
Filing, prosecuting, enforcing and defending the patents rights relating to our product candidates which have been licensed to us in every country in the world would likely be prohibitively expensive. ATPL is responsible for patent prosecution and maintenance costs in a limited number of countries and territories, and if we want to pursue protection in countries other than those for which ATPL is obligated, we will be responsible for the prosecution and maintenance costs. The extent of patent protection can vary in different countries. In addition, we have entered into three addenda for our Patent License that extends the deadlines to meet the commercialization milestones. Failure to meet these extended milestone deadlines may result in termination of the Patent License. Additionally, in the second addendum CytoMed agreed to take over responsibility for all patent prosecution and maintenance costs beginning January 2022.
The requirements for patentability may also differ in certain countries, particularly in developing countries; thus, even in countries where we do pursue patent protection, there can be no assurance that any patents will be issued by the relevant authorities in relation to our product candidates. Moreover, our ability to protect and enforce our intellectual property rights may be adversely affected by developments in international and foreign intellectual property legislation, regulations and policies.
Additionally, the laws of certain countries may not afford the same level of intellectual property protection as the laws of Singapore or other countries. Many companies have encountered significant problems in protecting and defending their intellectual property rights in certain foreign jurisdictions. It may be more difficult to enforce our patents license rights and other intellectual property rights in the legal systems of some countries, including India and China. This could make it difficult for us to obtain an injunction to stop third party infringement and/or misappropriation of our patents and/or our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. Consequently, we may not be able to prevent third parties from utilizing our technologies in certain countries outside Singapore or from selling or importing products made from our inventions in and into Singapore and/or other jurisdictions.
| 27 |
It is possible that competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop and market their own products and export such products to jurisdictions where we have patent protection, if our patent protection against infringement is not sufficiently robust. These products may compete with our product candidates, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing with us. Further, our Patent License gives ATPL the right but not the obligation to prosecute and defend any and all infringements of any licensed patent that may issue. We are obligated to obtain ATPL’s consent to pursue enforcement of our licensed patents or defend any claims asserting the invalidity of these patents (or control of such enforcement or defense) of such patent rights in all relevant jurisdictions. Proceedings to enforce our patent rights, whether or not successful, could result in substantial costs and divert our efforts and resources from other aspects of our business.
Moreover, such proceedings could put any patents at issue in such proceedings at risk of being revoked, invalidated or interpreted narrowly and our licensed patent applications at risk of not being issued and could provoke third parties to assert claims against us. We may not be successful in any lawsuits or other patent office proceedings involving the licenses granted to us and the damages or other remedies awarded, if any, may not be sufficient to compensate us for our losses and expenses and/or otherwise be commercially meaningful. Furthermore, while we intend to seek to protect the patent rights under the license agreements granted to us and if necessary, enforce rights of any patents that issue, in major markets for our products, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market our products, if approved. Accordingly, our efforts to protect our intellectual property rights in such countries may be inadequate.
If our patent protection is not sufficiently robust, our competitors could develop and commercialize products and technology similar or identical to ours.
There is intense competition in the market for cell therapy, and the market is also subject to rapid technological change. Our business depends largely on our ability to maintain a competitive edge in the development and protection of technologies and products for use in these fields.
There have been several complex legal and factual questions relating to patents of biopharmaceutical companies which has, in recent years, been the subject of much litigation. As a result, there are considerable uncertainties relating to the issuance, scope, validity, enforceability and commercial value of our licensed patent rights. Our pending and future patent applications may not result in patents being issued that protect our technology or product candidates or effectively prevent others from commercializing similar or identical technologies and product candidates.
The process to seek and obtain patent protection is costly, time-consuming and complex, and we may not be able to file, prosecute, obtain, maintain, enforce or license all required or desirable patents or patent applications at a reasonable cost or in a timely manner. We may also fail to identify patentable aspects of our R&D output before it is too late to obtain patent protection.
There may be changes in the scope of patent claims before a patent is issued, and after issuance of the patent. Even if the patent applications we license issue as patents, the patents may not be in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us or otherwise provide us with any competitive advantage. Our competitors or other third parties may be able to circumvent our patents by developing similar or alternative products in a manner which does not infringe on our patents.
We may not identify relevant third-party patents or may incorrectly interpret the relevance, scope or expiration of third-party patents, which could materially and adversely affect our ability to develop, manufacture and market our product candidates.
There are many patents issued or applied for in the biopharmaceutical industry, and we may not be aware of patents or patent applications held by others which are relevant to our business and our product candidates.
Furthermore, after issuance of a patent, the scope of patent claims depends on an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history. Our interpretation of the relevance or the scope of a patent or a pending application may be inaccurate, and we may incorrectly determine that our product candidates are not covered by a third-party patent or may incorrectly predict whether a third party’s pending application will issue with claims of relevant scope. Our determination of the expiration date of any patent in any relevant jurisdiction that we consider relevant may also be incorrect. If we fail to correctly identify or interpret the scope of third-party patents, we may be subject to infringement claims. We cannot guarantee that we will be able to successfully settle or otherwise resolve such infringement claims and/or lawsuits. If we fail in any such dispute, in addition to being forced to pay monetary damages, we may be temporarily or permanently prohibited from commercializing our product candidates. We may also be forced to attempt to redesign our product candidates in a manner that no longer infringes third-party intellectual property rights. Any of these events, even if we were ultimately to prevail, could require us to divert substantial financial and management resources that we would otherwise be able to devote to the development and commercialization of our product candidates.
| 28 |
We may face claims for infringing, misappropriating or otherwise violating intellectual property rights of third parties or engaging in unfair competition, which would be expensive and time-consuming, and could hinder the successful development and/or commercialization our product candidates.
The success of our business is affected by our ability to develop, manufacture and market our technology and use our technology without infringing the proprietary rights of third parties. As the relevant pharmaceutical and biotechnology industries expand and more patents are issued, we face greater risks of patents being issued to third parties that relate to our product candidates and technology of which we are not aware or that we may need to circumvent or overcome to continue our business operations as currently contemplated. As a result, our technology and any future product candidates that we commercialize could be alleged to infringe patent rights and other proprietary rights of third parties, which may require costly litigation and, if we are not successful, could require us to pay substantial damages and/or limit our ability to commercialize our product candidates.
We may have to defend ourselves against litigation claims in which the scope, enforceability and validity of third-party proprietary rights are at issue, or to establish our proprietary rights. Regardless of whether any such claims that we are infringing patents or other intellectual property rights have merit, such claims can be time consuming, divert management attention and financial resources and are costly to evaluate and defend.
If a license is available from a third party who contends any product candidates for which we obtain approval infringe or misappropriate third party intellectual property rights, we may have to pay substantial license fees, annual minimum fees, royalties, upfront fees, or milestone fees, or grant cross-licenses to our intellectual property rights. We may also have to modify our product candidates so they do not infringe third-party intellectual property rights, which may not be possible or may require additional regulatory approvals, or substantial monetary expenditures and time, during which our product candidates may not be available for manufacture, use, or sale.
We may fail to identify unauthorized use of our intellectual property and enforce our intellectual property rights against infringement, and may incur substantial costs as a result of bringing litigation or other proceedings to protect our intellectual property rights.
Identifying unauthorized use of our intellectual property is challenging and expensive. From time to time, we will review products from our competitors to determine potential infringement of our rights if any. We may not be able to identify unauthorized use of, or take appropriate steps to enforce, our intellectual property rights. Any failure by us to identify any unauthorized use of our intellectual property rights by third parties could result in competitors offering products that incorporate our product features, which could in turn reduce demand for our products.
We may also, from time to time, seek to enforce our intellectual property rights against infringers when we determine that a successful outcome is probable and may result in an increase in the value of our intellectual property.
If we or our licensor choose to enforce our licensed patent rights against a party, we or our licensor may face a counterclaim that our patent is invalid and/or unenforceable. Our patents may be challenged in legal proceedings. Proceedings to challenge patents are also available internationally, including, for example, opposition proceedings and nullity actions. In general, grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Grounds for an unenforceability assertion may include an allegation that someone connected with the prosecution of the patent withheld relevant information, or made a misleading statement, during prosecution. Third parties may also raise similar claims, even outside the context of litigation. We are unable to predict the outcome of litigation suits. With respect to the validity question, for example, we cannot guarantee that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we may lose at least part, and perhaps all, of the patent protection on our product candidates.
| 29 |
In addition, such lawsuits and proceedings are costly and would take up considerable time and resources and divert the attention of managerial and scientific personnel from our business operations even if we were successful in preventing the infringement of such patents. Litigation is inherently unpredictable, and there is a risk that the court will decide that such patents are not valid and that we do not have the right to stop the other party from using the technologies. Furthermore, some of our competitors have significantly greater resources and thus may be able to sustain the costs of complex patent litigation more effectively than us. There is also the risk that, even if the validity of such patents is upheld, the court will refuse to grant an injunction against the other party on the ground that there is no infringement of our intellectual property rights.
There could also be public announcements of the results of hearings, motions or other interim proceedings or developments, and if securities analysts or investors perceive these results negatively on us, it could materially adversely affect the price of our ordinary shares. Additionally, any uncertainties resulting from the initiation and continuation of any litigation could materially and adversely affect our ability to raise the funds necessary to continue our operations.
Developments in U.S. patent law or the patent law of other jurisdictions present uncertainties in our ability to obtain patents and diminish the value of patents in general, thereby affecting our ability to protect our current and any future product candidates.
The U.S. Supreme Court and the Court of Appeals for the Federal Circuit have made, and will likely continue to make, changes in how the patent laws of the United States are interpreted. For example, in recent years, the U.S. Supreme Court has modified certain tests used by the USPTO in awarding patents, which may reduce our chances of obtaining the requisite patents and increase the likelihood of a challenge of any patents we obtain or license. Similarly, there have been international developments in how the patent laws in their respective jurisdictions are interpreted. Those changes, as well as any future changes, may materially and adversely affect our patent rights and our ability to obtain issued patents.
Developments in either the patent laws or interpretation of the patent laws in the United States could result in greater uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. Under the Leahy-Smith America Invents Act, or the America Invents Act, assuming that all other conditions for patentability are satisfied, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention.
The America Invents Act also includes a number of significant changes that affect the prosecution of patent applications and patent litigation. These include allowing third-party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO-administered post-grant proceedings, including post-grant review, inter partes review and derivation proceedings. The implementation of the America Invents Act could result in greater uncertainties and expenses relating to the prosecution of our patent applications and the enforcement or defense of patents issued therefrom, all of which could materially and adversely affect our business, financial condition, results of operations and growth prospects.
Further, the U.S. Supreme Court has made judgments on several patent cases in recent years, with a trend towards either narrowing the scope of patent protection available in certain circumstances or weakening the rights of patent owners in certain situations. This results in greater uncertainty with respect to the value of patents, if and once obtained. Depending on actions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could weaken our ability to obtain new patents or to enforce patents. Similarly, developments in patent law and regulations in other countries or jurisdictions, changes in the enforcement authorities or changes in how the relevant governmental authority enforces patent laws or regulations may weaken our ability to obtain new patents or to enforce patents, which in turn could materially adversely affect our business, financial condition, results of operations and growth prospects.
We may fail to obtain or enforce assignments of intellectual property rights from our employees and contractors.
Although we generally require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning all such intellectual property to us, we may fail to obtain an enforceable agreement with each party who in fact conceives or develops intellectual property that we regard as our own. Furthermore, our assignment agreements may be breached, and we may be forced to bring or defend claims to determine the ownership of what we regard as our intellectual property, and we may not be successful in such claims. If we fail in any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights which could materially and adversely affect our business, financial condition, results of operations and growth prospects. Even if we are successful in defending against such claims, litigation could result in substantial costs and diversion of management’s and other employees’ attention from our business operations.
| 30 |
If we fail to prevent disclosure of trade secrets and other proprietary information, the value of our technology and products could be materially diminished.
We may face difficulties in protecting our trade secrets from being disclosed or shared with third parties. We rely on trade secrets to protect our proprietary information and technologies, especially where we do not believe patent protection is appropriate or obtainable, or where the enforcement of such patents would be challenging. We rely in part on confidentiality agreements with our employees, consultants, contractors, outside scientific collaborators and other advisors to protect our trade secrets and other proprietary information. We cannot guarantee that we have entered into such agreements with each party that may have had access to our proprietary information or technologies, or that such agreements, even if in place, will not be circumvented. These agreements may not effectively prevent disclosure of proprietary information or technology and may not provide an adequate remedy in the event of unauthorized disclosure of such information or technology. In addition, others may independently discover our trade secrets and proprietary information, in which case we may have no right to prevent them from using such trade secrets or proprietary information to compete with us. We may need to engage in expensive and time-consuming litigation to enforce and determine the scope of our proprietary rights. Our failure to obtain or maintain trade secret protection could materially and adversely affect our business, financial condition, results of operations and growth prospects.
RISKS RELATED TO COMMERCIALIZATION
We have no experience as a company in obtaining regulatory approval for a drug.
As a company, we have not yet obtained regulatory approval for, or commercialized, any drug as of the date of this annual report. It is possible that the HSA and/or other relevant health and regulatory authorities may refuse to accept any or all of our planned NDAs for substantive review or may conclude after review of our data that our application is insufficient to obtain regulatory approval for any current or future product candidates. If the HSA and/or other relevant health and regulatory authorities does not approve any of our planned NDAs, we may need to conduct additional costly clinical, non-clinical or manufacturing validation studies before the HSA will reconsider our application(s). Depending on the extent of these or any other HSA and/or other relevant health and regulatory authorities-required studies, approval of any NDA or other application that we submit may be significantly delayed, possibly for several years, or may require us to expend more resources than we have available. Any failure or delay in obtaining regulatory approvals would prevent us from commercializing any of our product candidate, generating revenues and achieving and sustaining profitability. It is also possible that additional studies, if performed and completed, may not be considered sufficient by the HSA and/or other relevant health and regulatory authorities to approve any NDA or other application that we submit. If any of these outcomes occur, we may be forced to abandon the development of our product candidates, which would materially adversely affect our business and could potentially cause us to cease operations. We may face similar risks for our applications in foreign jurisdictions.
If any of our product candidates are approved for marketing and commercialization and we have not developed or secured third-party marketing, sales and distribution capabilities, we will be unable to successfully commercialize such products and may not be able to generate product revenue.
We currently do not have any sales, marketing or distribution organizational experience or capabilities. We will need to develop internal sales, marketing and distribution capabilities to commercialize any product candidate to obtain regulatory authority approval, which would be costly and time-consuming, or enter into partnerships with third parties to achieve the same. If we decide to directly market any approved products, we will have to invest substantial financial and managerial resources in a marketing and sales team with technical expertise and supporting distribution, administration and compliance capabilities. If we rely on third parties to market products or decide to co-promote products with partners, we will need to establish and maintain marketing and distribution arrangements with third parties, and there can be no assurance that we can enter into such arrangements on acceptable terms or at all. In entering into third-party marketing or distribution arrangements, any product revenue is contingent on the efforts of the third parties and we cannot guarantee that such third parties will establish adequate sales and distribution capabilities or be successful in gaining market acceptance for any approved product. If we fail to commercialize any product approved in the future, if any, either on our own or through third parties, our business, financial condition, results of operations and growth prospects could be materially adversely affected.
| 31 |
If we fail to establish biopharmaceutical collaborations on commercially reasonable terms, or at all, we may have to change our development and commercialization plans.
The advancement of our product candidates and development programs and the potential commercialization of our current and future product candidates will require substantial additional financing. For some of our programs, we may partner with pharmaceutical and biotechnology companies to develop and commercialize such product candidates. Any such collaborations may incur non-recurring and other charges, increase our near and long-term expenditures, require us to issue securities that dilute our existing shareholders, or disrupt our management and business.
We face intense competition in seeking appropriate strategic partners and the negotiation process is time-consuming and complex. Any definitive agreement for other partnerships is contingent, among other things, on our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors, including the design or results of clinical trials, the progress of our clinical trials, the likelihood of approval by the relevant regulatory authorities, the potential market for the subject product candidate, the costs and complexities of manufacturing and delivering such product candidate to patients, the potential of competing products, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and clinical trial industry and market conditions generally. The collaborator may also consider alternative product candidates or technologies for similar clinical trial indications that may be available to collaborate on and whether such a collaboration could be more attractive than the one with us for our product candidates. Further, we may fail to enter into a strategic partnership or other alternative arrangements for future product candidates because they may be deemed to be premature for collaborations and third parties may not be satisfied with the level of safety and efficacy of our product candidates. Any delays in entering into new collaborations or strategic partnership agreements related to any product candidate we develop could hinder the development and commercialization of our product candidates, which would materially and adversely affect our business prospects, financial condition, and results of operations.
We have entered into collaborations with third parties to develop or commercialize our product candidates, our prospects with respect to those product candidates will depend significantly on the success of those collaborations.
The risks of our present and future collaboration with third parties to develop and/or commercialize our product candidates include without limitation:
| ● | collaborators have significant discretion in determining the efforts and resources that they will apply to these collaborations; | |
| ● | collaborators could independently develop, or develop with third parties, products that compete directly or indirectly with our products or product candidates; | |
| ● | our agreements with collaborators include agreements that may not provide us with sole ownership of all intellectual property rights resulting from the collaborations and do not address all issues that may arise from joint ownership; | |
| ● | current and future collaboration agreements may not provide us with sufficient ownership or control of all intellectual property rights; | |
| ● | collaborators may not properly enforce, maintain or defend our intellectual property rights or may use our proprietary information in a way that gives rise to actual or threatened litigation that could jeopardize or invalidate our intellectual property or proprietary information or expose us to potential litigation, or other intellectual property proceedings; | |
| ● | disputes may arise between a collaborator and us that cause the delay or termination of the research, development or commercialization of the product candidates, or that result in costly litigation or arbitration that diverts management attention and resources; | |
| ● | if a present or future collaborator of ours were to be involved in a business combination, the continued pursuit and emphasis on our product development or commercialization program under such collaboration could be delayed, diminished or terminated; | |
| ● | collaboration agreements may restrict our right to pursue new product candidates; and | |
| ● | we may become involved in disputes over the terms of our collaboration agreements and intellectual property rights generated from our collaborations. |
| 32 |
In the event of disagreements and/or conflicts between our collaborators and us, our collaborators may act in a hostile and/or vexatious manner which could hinder the implementation of our strategies. Future collaborators may develop, either alone or with others, products in related fields that are competitive with our products or potential products that are the subject of these collaborations. Competing products, either developed by the collaborators or to which the collaborators have rights, may result in the withdrawal of support for our product candidates. Non-competition clauses in our agreements with our collaborators may prevent us from entering into collaborations with their competitors and/or being able to obtain timely regulatory approvals. Our collaborators may also terminate their agreements with us prematurely and/or fail to devote sufficient resources to the development and commercialization of products. Any of these events could hinder and severely undermine our product development efforts.
As a result, if we enter into collaboration agreements and strategic partnerships or out-license our intellectual property, products or businesses, we may not be able to realize the benefit of such transactions if we are unable to successfully integrate them with our existing operations, which could delay our timelines or otherwise adversely affect our business. We also cannot be certain that, following a strategic transaction or license, we will be able to achieve a level of revenue or specific net income that justifies such transaction.
Our product candidates could be subject to regulatory limitations following approval, if such approval is granted.
Subsequent to obtaining approval of a product candidate, if any, we must adhere to all applicable government legislation and regulations regarding the manufacture, labeling, marketing, distribution and promotion of biologic products, which are subject to change from time to time. We must adhere to labeling protocols of HSA and/or other relevant regulatory authorities, which prohibit promoting “off-label uses”. We may not be able to obtain the labeling claims necessary or desirable to successfully commercialize our products, including CTM-N2D, iPSC-gdNKT, CTM-GDT or other product candidates in development from time to time.
The HSA and/or other relevant regulatory authorities could impose substantial restrictions on the use of an approved product including restricting its use to limited clinical centers as well as through the product label, as well as on advertising, promotional and distribution activities associated with such approved product. The HSA and/or other relevant regulatory authorities could also condition their approval on the performance of post-approval clinical trials, patient monitoring or testing, which could be time-consuming and expensive. If the results of such post-marketing trials are not satisfactory, the HSA and/or other relevant regulatory authorities could withdraw marketing authorization or may impose conditions on continued marketing and/or impose or require further commitments from us or our partners that may be expensive and/or time-consuming to fulfill.
In addition, if side effects are identified after any of our products are on the market, if manufacturing problems occur subsequent to regulatory approval, or if we, our manufacturers or our partners fail to comply with regulatory requirements, including those mentioned above, we or our partners could be subject to the following:
| ● | restrictions on our ability to conduct clinical trials, including full or partial clinical holds on ongoing or planned clinical trials; | |
| ● | restrictions on such products’ manufacturing processes; | |
| ● | changes to the product label; | |
| ● | restrictions on the marketing of a product; | |
| ● | restrictions on product distribution; | |
| ● | requirements to conduct post-marketing clinical trials; | |
| ● | warnings from the regulatory authority; | |
| ● | withdrawal of the product from the market; | |
| ● | refusal to approve pending applications or supplements to approved applications that we submit; | |
| ● | recall of products; | |
| ● | fines, restitution or disgorgement of profits or revenue; | |
| ● | suspension or withdrawal of regulatory approvals; | |
| ● | refusal to permit the import or export of our products; | |
| ● | product seizure; | |
| ● | injunctions; and/or | |
| ● | imposition of civil or criminal penalties. |
Any one or a combination of the above could obstruct market acceptance of the affected product, or could substantially increase the costs and expenses of commercializing such product, which in turn could delay or prevent us from generating any revenue or profit from the sale of such product and could materially and adversely affect our business, financial condition, results of operations and growth prospects.
| 33 |
A summary of the material regulations in Singapore and Malaysia applicable to our operations to our knowledge are set out in the section titled “Regulation”. However, as the biopharmaceutical industries in Singapore and Malaysia are only being regulated in recent times, there is a possibility that the biopharmaceutical industries may become more heavily regulated and we may be subject to further legislative, regulatory and compliance requirements.
Our product candidates could be subject to more stringent limitations and procedures imposed by third-party users, including hospitals, than those imposed by regulatory authorities.
Even if we manage to obtain the necessary licenses, permits and approvals for our products, we may also be required to comply with policies, limitations and other procedures imposed by third-party users, including hospitals, of our products. Such policies, limitations and other procedures may change from time to time, depending on factors including but not limited to perceived risks and benefits of our products, public sentiment towards the biopharmaceutical industries and relevant government legislation, regulations and policies. This may restrict the distribution of our products and/or increase our compliance costs, which may materially and adversely affect our financial performance and financial condition.
There may be limited market opportunities for our product candidates, if approved, and if such market opportunities are smaller than we anticipated, our revenues and business could be materially and adversely affected.
Cancer therapies are sometimes categorized as first-line, second-line, or third-line, and the regulatory authorities often approve new therapies initially only for third-line use. When cancer is detected early enough, first-line therapy, usually chemotherapy, hormone therapy, surgery, radiation therapy or a combination of these, may be sufficient as a cure for cancer. Second and third-line therapies are administered to patients when prior therapy is not effective. Our initial planned clinical trials are expected to enroll patients who have received these standard therapies in order to first evaluate whether the product is safe and whether there is any anti-cancer activity. We currently are unable to ascertain whether CTM-N2D, iPSC-gdNKT and CTM-GDT will be safe for use in humans and whether CTM-N2D, iPSC-gdNKT and CTM-GDT will demonstrate any anti-cancer activity. Subsequently, we plan to conduct additional clinical trials depending on the anti-cancer activity we note in the initial clinical trials. If the anti-cancer activity is sufficient, we may initially seek approval of any product candidates we develop as a therapy for patients who have received one or more prior standard therapies. Subsequently, for those products that prove to be sufficiently beneficial, if any, we would expect to seek approval potentially in earlier lines of therapy, but there is no guarantee that product candidates we develop, even if approved for later lines of therapy, would be approved for earlier lines of therapy, and, prior to any such approvals, we may have to conduct additional clinical trials.
There may not be as many patients who have the type of cancers we are targeting. Additionally, the potentially addressable patient population for our current or future product candidates may be limited. Potentially addressable patient populations for our product candidates are only estimates. These estimates could prove to be inaccurate as there may not be as many potential patients in Singapore and elsewhere as estimated. It may also be that such patients may not be otherwise amenable to treatment with our product candidates, or suitable patients could become increasingly difficult to identify and access, any of which could materially and adversely affect our business, financial condition, results of operations and growth prospects.
The commercial success of any of our product candidates will depend upon such product candidate’s level of market acceptance by physicians, patients, third-party payors and others in the medical community.
Even if requisite approvals are obtained from HSA and/or other regulatory authorities, the commercial success of our product candidates will depend, in part, on the acceptance by physicians, patients and healthcare payors of cell therapy products in general, and our product candidates in particular, as medically necessary, cost-effective and safe. Physicians, patients, healthcare payors, professionals and others in the medical community may not approve of any product candidates that we commercialize. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenue and may not become profitable. The degree of market acceptance of cell therapy products and, in particular, our product candidates, if approved for commercial sale, will depend on several factors, including without limitation:
| ● | the efficacy and safety of such product candidates as demonstrated in clinical trials; | |
| ● | the potential and perceived advantages of product candidates over alternative treatments; |
| 34 |
| ● | the cost of treatment relative to alternative treatments; | |
| ● | the clinical trial indications for which the product candidate is approved by the HSA and/or other relevant regulatory authorities; | |
| ● | the willingness of physicians to prescribe new therapies; | |
| ● | the willingness of the target patient population to try new therapies; | |
| ● | the prevalence and severity of any side effects; | |
| ● | product labeling or product insert requirements imposed by HSA and/or other relevant regulatory authorities, including any limitations or warnings contained in a product’s approved labeling; | |
| ● | relative convenience and ease of administration; | |
| ● | the timing of market introduction of competitive products; | |
| ● | adverse publicity concerning our product candidates or favorable publicity about competing products and treatments; | |
| ● | sufficient third-party payor coverage, any limitations in terms of center or personnel training requirement imposed by third parties and adequate reimbursement; | |
| ● | limitations or warnings contained in the regulatory authority-approved labeling for our product candidates; | |
| ● | any regulatory authority requirement to undertake a risk evaluation and mitigation strategy; | |
| ● | the effectiveness of our sales, marketing and distribution efforts; and | |
| ● | potential product liability claims. |
Even if a product candidate displays a favorable efficacy and safety profile in pre-clinical studies and clinical trials, market acceptance of the product will not be fully known until after such product is launched. Our product candidates may not achieve broad market acceptance.
Furthermore, the policies of HSA and/or other relevant regulatory authorities may change and additional government regulations may be enacted that could prevent, limit or delay marketing approval of a product. We cannot determine the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in Singapore or abroad. If we are late or fail to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we fail to maintain regulatory compliance, we may lose any marketing approval that we may have previously obtained and as a result, we may not be able to achieve or sustain profitability.
There are several uncertainties relating to the insurance coverage and reimbursement status of newly approved products on the market. Failure to obtain or maintain adequate coverage and reimbursement for our product candidates, if approved, could hinder our ability to market such products and to generate product revenue.
The expenses incurred as a result of the administration of any of our cell therapy product candidates is anticipated to be significant, when and if regulatory approval is obtained. We expect that coverage and reimbursement by government and private payors will be essential for most patients to be able to afford these treatments. Accordingly, sales of our products, if approved, will depend substantially, both domestically and internationally, on the extent to which the costs of our product candidates will be reimbursed by government authorities, private health coverage insurers and/or other third-party payors. Coverage and reimbursement by a third-party payor could depend upon several factors, including the third-party payor’s determination that use of our product is (i) a covered benefit under its health plan, (ii) safe, effective and medically necessary, (iii) appropriate for the specific patient, (iv) cost-effective and (v) neither experimental nor investigational.
Obtaining coverage and reimbursement for a product from third-party payors is a time-consuming and expensive process that could require us to provide supporting scientific, clinical and cost-effectiveness data. We may not be able to provide adequate data to obtain acceptance with respect to coverage and reimbursement. If coverage and reimbursement are not available, or are available only at limited levels, we may fail to successfully commercialize our product candidates. Even if coverage is provided, the approved reimbursement amount may not be sufficient to generate a sufficient return on our investment.
There is significant uncertainty relating to third-party coverage and reimbursement of newly approved drug products and/or treatments.
| 35 |
Outside of Singapore, international operations may vary significantly by country and our products may be subject to extensive government price controls and other market regulations. Increasing focus on cost-containment initiatives in the European Union, Canada and other countries could result in pricing pressure. In many countries, the prices of medical products are governed by several price control mechanisms as part of national health systems. The post-approval process to secure pricing and reimbursement for a product may be time-consuming depending on the country. In general, the prices of medical and therapeutic products under such systems may be substantially lower than in Singapore. Other countries allow companies to fix their own prices for such products, but monitor and control company profits. Additional foreign price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our product candidates. Accordingly, in markets outside Singapore, the reimbursement for our products may be lower than that of Singapore and may be insufficient to generate commercially viable and sustainable product revenues and profits.
Moreover, any efforts by government and third-party payors in Singapore and abroad to limit and/or reduce healthcare costs could limit coverage and the level of reimbursement for our product candidates.
There is no certainty that the medical costs of our treatment for suitable patients will be covered by insurance. As our product candidates are novel, the treatment and medical costs may not be covered by medical and/or hospitalization insurances obtained by these patients. If so, the costs of treatment with our product candidates may be prohibitively high, and deter suitable patients from seeking help resulting in a low take-up rate for our product candidates. This will materially and adversely affect our business, growth and future prospects of our Group.
Healthcare reform initiatives and other administrative and legislative proposals may affect our business.
We cannot anticipate the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action in Singapore, Malaysia and/or any other relevant jurisdiction. If we or any third parties we may engage are late or fail to adapt to developments in existing requirements or the adoption of new requirements or policies, or if we or such third parties are not able to maintain regulatory compliance, our product candidates may lose any regulatory approval that may have been previously obtained and we may not achieve or sustain profitability. Furthermore, future price controls or other changes in pricing regulation or negative publicity related to the pricing of cell therapy products could restrict the amount that we are able to charge for our cell therapy products, which could render our product candidates, if approved, commercially unviable and materially and adversely affect our ability to raise additional capital on acceptable terms.
Obtaining and maintaining marketing approval or commercialization of our product candidates in one jurisdiction does not mean that we will be successful in obtaining marketing approval of our product candidates in other jurisdictions.
Approval procedures are different among different jurisdictions and can involve requirements and administrative review periods significantly different from, and more extensive than, those in Singapore, Malaysia and/or other relevant jurisdictions including additional pre-clinical studies or clinical trials as clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside Singapore, Malaysia and/or other relevant jurisdictions, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, we may have to obtain approval for the prices that we intend to charge for our products.
If we market products approved in jurisdictions outside Singapore, Malaysia and/or other relevant jurisdictions, we expect that we will be subject to additional risks in commercialization, including without limitation:
| ● | variations in regulatory requirements for approval of therapies in different jurisdictions; | |
| ● | decreased scope of protection of intellectual property rights; | |
| ● | unanticipated changes in tariffs, trade barriers and other regulatory requirements; | |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; | |
| ● | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; | |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country; | |
| ● | foreign reimbursement, pricing and insurance regimes; | |
| ● | workforce uncertainty in countries where labor unrest is more common; | |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and | |
| ● | business interruptions resulting from geopolitical actions, including war and terrorism, natural disasters including earthquakes, typhoons, floods and fires, and other public health crises, illnesses, epidemics or pandemics, such as the potential impact of the COVID-19 pandemic. |
We do not have any prior experience in these areas. Any of the foregoing difficulties, if encountered, could materially and adversely affect our business, financial condition, results of operations and growth prospects.
| 36 |
RISKS RELATED TO OUR ORDINARY SHARES
An active trading market for our ordinary shares or our ordinary shares may not continue and the trading price for our ordinary shares may fluctuate significantly.
Our ordinary shares are listed on the Nasdaq Capital Market. We cannot assure you that a liquid public market for our ordinary shares will continue. If an active public market for our ordinary shares does not continue, the market price and liquidity of our ordinary shares may be materially and adversely affected. As a result, investors in our securities may experience a significant decrease in the value of their ordinary shares.
In the future, our ability to raise additional capital to expand our operations and invest in our business may be limited, and our failure to raise additional capital, if required, could impair our business.
While we currently anticipate that our available funds will be sufficient to meet our cash needs for at least the next 2 years, we may need or elect to seek, additional financing at any time. Our ability to obtain financing will depend on, among other things, our development efforts, business plans, operating performance and condition of the capital markets at the time we seek financing. If we need or elect to raise additional funds, we may not be able to obtain additional debt or equity financing on favorable terms, if at all. If we raise additional equity financing, our shareholders may experience significant dilution of their ownership interests and the per-share value of our ordinary shares could decline. If we engage in additional debt financing, we may be required to accept terms that further restrict our ability to incur additional indebtedness and force us to maintain specified liquidity or other ratios and limit the operating flexibility of our business. If we need additional capital and cannot raise it on acceptable terms, we may not be able to, among other things:
| ● | Fund our operating capital requirements as we grow; | |
| ● | Continue to grow by acquiring companies; | |
| ● | Retain the leadership team and staff required; | |
| ● | Repay our liabilities as they become due; or | |
| ● | Make the necessary investments in our cell therapies. |
Our ordinary share price may in the future be volatile and, as a result, you could lose a significant portion or all of your investment.
Market prices for securities of newly-public companies have historically been particularly volatile in response to various factors, some of which are beyond our control. As a result of this volatility, you may not be able to sell your ordinary shares at or above the price you pay for your shares.
Factors affecting the trading price of our ordinary shares may include, but are not limited to:
| ● | our decision on whether or not to initiate a clinical study or to terminate an existing clinical study; | |
| ● | adverse regulatory decisions, including failure to receive regulatory approval for our products; | |
| ● | success or failure of competitive products, immunotherapy or cellular therapies more generally; | |
| ● | adverse developments concerning our manufacturers or our strategic partnerships; | |
| ● | adverse safety or other clinical results, such as those that have occurred in the past or that may occur in the future, related to cellular therapies being developed by other companies that are or may be perceived to be similar to our cellular therapies; | |
| ● | operating and share price performance of other companies that investors deem comparable to us; | |
| ● | sales of substantial amounts of ordinary shares by our Directors, executive officers or significant shareholders or the perception that such sales could occur; | |
| ● | general economic and political conditions such as recessions, interest rates, fuel prices, elections, drug pricing policies, international currency fluctuations, acts of war or terrorism, and other public health crises, illnesses, epidemics or pandemics, such as the potential impact of the COVID-19 pandemic; and | |
| ● | other factors discussed in these risk factors. |
| 37 |
Any of or a combination of the above factors could materially and adversely affect your investment in our ordinary shares, which could contribute to a loss of all or part of your investment. In such circumstances the trading price of our ordinary shares may not recover and may experience a further decline.
In addition, broad market and clinical trial industry factors could materially and adversely affect the market price of our ordinary shares, irrespective of our operating performance. The stock market in general, and Nasdaq Capital Market and the market for biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of the particular companies affected. We cannot predict the trading prices and valuations of these stocks, and of ours. For example, the trading prices for common stock of other biopharmaceutical and biotechnology companies have been highly volatile as a result of the COVID-19 pandemic. If any pandemics, epidemics, or outbreaks of infectious diseases, including future outbreaks of COVID-19 variants, Respiratory Syncytial Virus (“RSV”), or the flu, continue to rapidly evolve, the extent to which the outbreak may impact our business, pre-clinical studies and clinical trials is contingent on future developments, which are highly uncertain and cannot be accurately predicted. We may face a loss of investor confidence in the market for biopharmaceutical stocks or the stocks of other companies which investors perceive to be similar to us, the opportunities in the biopharmaceutical market or the stock market in general, which may decrease our share price regardless of our business, financial condition, results of operations or growth prospects.
We may face securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a period of volatility or decline in the market price of its securities. We are likely to face this risk as pre-clinical biopharmaceutical companies have experienced significant share price volatility in recent years. If we face such litigation, we may have to pay substantial costs (including legal costs) and this would also require a diversion of management’s attention and resources, which could materially and adversely affect our business, financial condition, results of operation and growth prospects.
If securities analysts do not publish research or reports about our business or if they publish negative reports or downgrade our shares, the price of our ordinary shares could decline.
The trading market for our ordinary shares relies in part on the research and reports that financial analysts publish about us, our business, our markets and our competitors. We have no control over the research and reports that these financial analysts publish. If we and our ordinary shares are not discussed by securities analysts, this may materially and adversely affect the market price of our ordinary shares. Furthermore, if any securities analyst chooses to downgrade our shares or issue unfavorable commentary on us or our business, our share price would likely decline. If one or more of these security analysts ceases coverage of us or fails to regularly publish reports on us, we could lose visibility in the market and interest in our ordinary shares could decrease, which in turn could cause our share price or trading volume to decline and may also impair our ability to expand our business with existing customers and attract new customers.
| 38 |
Sales of a substantial number of our ordinary shares in the public market by our existing shareholders could cause our share price to fall.
Sales of a substantial number of our ordinary shares in the public market, or the perception that these sales might occur, could depress the market price of our ordinary shares and could impair our ability to raise capital through the sale of additional equity securities. Furthermore, while our directors, officers and certain shareholders are subject to lock-up agreements with the representative of the underwriters of our Offering that restrict their ability to transfer our ordinary shares until April 2024, approximately 32.82% of outstanding ordinary shares are not subject to such lock-up and may be sold at any time. We are unable to predict the effect that sales may have on the prevailing market price of our ordinary shares.
Our principal shareholders, officers and directors beneficially own approximately 70.80% of our outstanding ordinary shares. They will therefore be able to exert significant control over matters submitted to our shareholders for approval, which could limit your ability to influence the outcome of key transactions, including a change of control, and which may result in conflicts with us or you in the future.
Our principal shareholders, officers and directors beneficially own approximately 70.80% of our ordinary shares. This significant concentration of share ownership may adversely affect the trading price for our ordinary shares because investors often perceive disadvantages in owning shares in companies with controlling shareholders. As a result, these shareholders, if they acted together, could significantly influence or even unilaterally approve matters requiring approval by our shareholders, including the election of directors and the approval of mergers or other business combination transactions. The interests of these shareholders may not always coincide with our interests or the interests of other shareholders, and which may result in conflicts with us or you in the future.
We are a “controlled company” defined under the Nasdaq Listing Rules. Although we do not intend to rely on the “controlled company” exemption under the Nasdaq Listing Rules, we could elect to rely on this exemption in the future and you will not have the same protection afforded to stockholders of companies that are subject to these corporate governance requirements.
Our principal shareholders, officers and directors beneficially hold a majority of our ordinary shares. Under the Nasdaq Listing Rules, a company of which more than 50% of the voting power is held by an individual, group or another company is a “controlled company” and is permitted to phase in its compliance with the independent committee requirements. For so long as we are a controlled company under that definition, we are permitted to elect to rely, and may rely, on certain exemptions from corporate governance rules, including:
| ● | an exemption from the rule that a majority of our Board must be independent directors; | |
| ● | an exemption from the rule that the compensation of our Chief Executive Officer must be determined or recommended solely by independent directors; and | |
| ● | an exemption from the rule that our director nominees must be selected or recommended solely by independent directors. |
As a result, you will not have the same protection afforded to stockholders of companies that are subject to these corporate governance requirements.
Although we do not intend to rely on the “controlled company” exemption under the Nasdaq Listing Rules, we could elect to rely on this exemption in the future. If we elected to rely on the “controlled company” exemption, a majority of the members of our Board might not be independent directors and our nominating and corporate governance and compensation committees might not consist entirely of independent directors upon closing of this offering. Our status as a controlled company could cause our ordinary shares to look less attractive to certain investors or otherwise harm our trading price. As a result, the investors will not have the same protection afforded to stockholders of companies that are subject to these corporate governance requirements.
We are an emerging growth company within the meaning of the Securities Act, and if we take advantage of certain exemptions from disclosure requirements available to emerging growth companies, this could make our securities less attractive to investors and may make it more difficult to compare our performance with other public companies.
We are an “emerging growth company” within the meaning of the Securities Act, as modified by the JOBS Act, and we may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act and reduced disclosure obligations regarding executive compensation in our periodic reports. As a result, our shareholders may not have access to certain information they may deem important. We could be an emerging growth company for up to five years, although circumstances could cause us to lose that status earlier, including if the market value of our ordinary shares held by non-affiliates exceeds $700 million as of any December 31 before that time, in which case we would no longer be an emerging growth company as of the following December 31. We cannot predict whether investors will find our securities less attractive because we will rely on these exemptions. If some investors find our securities less attractive as a result of our reliance on these exemptions, the trading prices of our securities may be lower than they otherwise would be, there may be a less active trading market for our securities and the trading prices of our securities may be more volatile.
Further, Section 102(b)(1) of the JOBS Act exempts emerging growth companies from being required to comply with new or revised financial accounting standards until private companies (that is, those that have not had a Securities Act registration statement declared effective or do not have a class of securities registered under the Exchange Act) are required to comply with the new or revised financial accounting standards.
The JOBS Act provides that a company can elect to opt out of the extended transition period and comply with the requirements that apply to non-emerging growth companies but any such an election to opt out is irrevocable. We have elected not to opt out of such extended transition period, which means that when a standard is issued or revised and it has different application dates for public or private companies, we, as an emerging growth company, can adopt the new or revised standard at the time private companies adopt the new or revised standard. This may make comparison of our financial statements with another public company which is neither an emerging growth company nor an emerging growth company which has opted out of using the extended transition period difficult or impossible because of the potential differences in accountant standards used.
The JOBS Act will allow us to postpone the date by which we must comply with some of the laws and regulations intended to protect investors and to reduce the amount of information we provide in our reports filed with the SEC, which could undermine investor confidence in our company and adversely affect the market price of our ordinary shares.
For so long as we remain an “emerging growth company” within the meaning of the Securities Act, as modified by the JOBS Act, we intend to take advantage of certain exemptions from various requirements that are applicable to public companies that are not “emerging growth companies” including:
| ● | the provisions of the Sarbanes-Oxley Act requiring that our independent registered public accounting firm provide an attestation report on the effectiveness of our internal control over financial reporting; | |
| ● | any rules that may be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotation or a supplement to the auditor’s report on the financial statements; and, | |
| ● | Section 107 of the JOBS Act, which provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, or the Securities Act, for complying with new or revised accounting standards. This means that an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected to delay such adoption of new or revised accounting standards. As a result of this adoption, our financial statements may not be comparable to companies that comply with the public company effective date. |
We intend to take advantage of these exemptions until we are no longer an “emerging growth company.” We will remain an emerging growth company until the earlier of (1) the last day of the fiscal year (a) following the fifth anniversary of the date of our first sale of common equity securities pursuant to an effective registration statement under the Securities Act, (b) in which we have total annual gross revenue of at least $1.235 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our ordinary shares that is held by non-affiliates exceeds $700 million as of the prior December 31, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period.
We cannot predict if investors will find our ordinary shares less attractive because we may rely on these exemptions. If some investors find our ordinary shares less attractive as a result, there may be a less active trading market for our ordinary shares, and our market prices may be more volatile and may decline.
| 39 |
We are a foreign private issuer within the meaning of the rules under the Exchange Act, and as such we are exempted from certain provisions applicable to U.S. domestic public companies.
Because we qualify as a foreign private issuer under the Exchange Act, we are exempt from certain provisions of the securities rules and regulations in the United States that are applicable to U.S. domestic issuers, including:
| ● | the rules under the Exchange Act requiring the filing with the SEC of quarterly reports on Form 10-Q or current reports on Form 8-K; | |
| ● | the sections of the Exchange Act regulating the solicitation of proxies, consents, or authorizations in respect of a security registered under the Exchange Act; | |
| ● | the sections of the Exchange Act requiring insiders to file public reports of their stock ownership and trading activities and liability for insiders who profit from trades made in a short period of time; and | |
| ● | the selective disclosure rules by issuers of material nonpublic information under Regulation FD. |
We are required to file an annual report on Form 20-F within four months of the end of each financial year. Press releases relating to financial results and material events must also be furnished to the SEC on Form 6-K. However, the information we are required to file with or furnish to the SEC is less extensive and less timely compared to that required to be filed with the SEC by U.S. domestic issuers. As a result, you may not be afforded the same protections and/or information that would be made available to you if you were investing in a U.S. domestic issuer.
As a foreign private issuer, we are subject to different U.S. securities laws and Nasdaq Capital Market governance standards than domestic U.S. issuers. This may afford less protection to holders of our ordinary shares, and you may not receive corporate and company information and disclosure that you are accustomed to receiving or in a manner in which you are accustomed to receiving it.
As a “foreign private issuer” for U.S. securities laws purposes, the rules governing the information that we will be required to disclose differ materially from those governing U.S. corporations pursuant to the Securities Exchange Act of 1934, as amended (the “Exchange Act”). The periodic disclosure required of foreign private issuers is more limited than that required of domestic U.S. issuers and there may therefore be less publicly available information about us than is regularly published by or about U.S. public companies. For example, we are not required to file quarterly reports on Form 10-Q or provide current reports on Form 8-K disclosing significant events within four days of their occurrence and our quarterly (should we provide them) or current reports may contain less or different information than required under U.S. filings. In addition, as a foreign private issuer, we are exempt from the proxy rules under Section 14 of the Exchange Act, and proxy statements that we distribute are not subject to review by the SEC. Our exemption from Section 16 rules under the Exchange Act regarding sales of ordinary shares by our insiders means that you will have less data in this regard than shareholders of U.S. companies that are subject to the Exchange Act. As a result, you may not have all the data that you are accustomed to having when making investment decisions. Also, our officers, directors and principal shareholders are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and the rules thereunder with respect to their purchases and sales of our ordinary shares.
Moreover, as a foreign private issuer, we are exempt from complying with certain corporate governance requirements of the Nasdaq Capital Market applicable to a U.S. issuer, including the requirement that a majority of our board of directors consist of independent directors. For example, we follow Singapore law with respect to the requirements for meetings of our shareholders, which are different from the requirements of the Nasdaq Capital Market. Additionally, per our Singapore legal counsel’s letter dated October 21, 2022 provided in connection with our Offering, we formally adopted home country practice and thereby opted out of the Nasdaq Capital Market rule that would otherwise require a majority of our Board of Directors to be comprised of independent directors as defined in Rule 5605(a)(2) of the Nasdaq Listing Rules, and that we must provide for a quorum of not less than 33.33% of the outstanding shares of its voting stock in relation to any meeting of its holders of its common voting stock. As the corporate governance standards applicable to us are different than those applicable to domestic U.S. issuers, you may not have the same protections afforded under U.S. securities regulation and the Nasdaq Capital Market rules as shareholders of companies that do not have such exemptions.
| 40 |
We may lose our foreign private issuer status, which would then require us to comply with the Exchange Act’s domestic reporting regime and cause us to incur additional legal, accounting and other expenses.
In order to maintain our current status as a foreign private issuer, either (1) a majority of our ordinary shares must be either directly or indirectly owned of record by non-residents of the United States or (2) (a) a majority of our executive officers or directors must not be U.S. citizens or residents, (b) more than 50 percent of our assets cannot be located in the United States and (c) our business must be administered principally outside the United States. If we lost this status, we would be required to comply with the Exchange Act reporting and other requirements applicable to U.S. domestic issuers, which are more detailed and extensive than the requirements for foreign private issuers. We may also be required to make changes in our corporate governance practices in accordance with various SEC rules and Nasdaq listing standards. The regulatory and compliance costs to us under U.S. securities laws if we are required to comply with the reporting requirements applicable to a U.S. domestic issuer may be higher than the cost we would incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs. We also expect that if we were required to comply with the rules and regulations applicable to U.S. domestic issuers, it would make it more difficult and expensive for us to obtain director and officer liability insurance. These rules and regulations could also make it more difficult for us to attract and retain qualified Board members.
If our system of internal control over financial reporting is not effective and robust, our financial results may not be accurately reported and we may not be able to prevent fraud. As a result, shareholders could deem our financial and other public reporting unreliable, which would materially and adversely affect our business and the trading price of our ordinary shares.
We need effective and robust internal controls over financial reporting in order to present reliable and accurate financial reports and prevent fraud. Any failure to implement required new or improved controls, or difficulties encountered in their implementation could result in us breaching our reporting obligations. When we lose our status both as an emerging growth company and a smaller reporting company, our independent registered public accounting firm will be required to attest to the effectiveness of our internal control over financial reporting. There are complex rules governing the standards that must be met for management to assess our internal control over financial reporting which require significant documentation, testing and possible remediation. Any testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act, or any subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed material weaknesses or that may require prospective or retroactive revisions to our financial statements or us to determine other areas for further attention or improvement. Inadequate internal controls could also cause investors to lose confidence in our reported financial information, which could materially and adversely affect the trading price of our ordinary shares.
Our disclosure controls and procedures are not risk-free and may not fully prevent and/or identify errors or acts of fraud.
We are required to satisfy periodic reporting requirements of the Exchange Act. We have planned our disclosure controls and procedures to reasonably ascertain that information required to be disclosed in reports we file or submit under the Exchange Act is accumulated and communicated to management, and recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. Any disclosure controls and procedures or internal controls and procedures, no matter how well-conceived and operated, can provide only reasonable, not absolute, certainty that the objectives of the control system are met.
We are subject to inherent limitations including the possibility that judgments in decision-making can be faulty, and that breakdowns and/or non-compliance can occur due to simple error or oversight. For example, our Directors and/or executive officers could inadvertently fail to disclose a new relationship or arrangement causing us to fail to make any related party transaction disclosures. Additionally, our disclosure controls can possibly be circumvented by the individual acts of some persons, by collusion of two or more people or by an unauthorized override of the controls as misstatements due to error or fraud may occur and not be detected in time, if at all.
We incur significantly increased costs and devote substantial management time as a result of operating as a public company.
As a public company, we incur significant legal, accounting, and other expenses that we did not incur as a private company. For example, we are subject to the reporting requirements of the Exchange Act and are required to comply with the applicable requirements of the Sarbanes-Oxley Act and the Dodd-Frank Act, as well as rules and regulations subsequently implemented by the SEC and the Nasdaq Capital Market including the establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. Compliance with these requirements increases our legal and financial compliance costs and makes some activities more time consuming and costly.
The Exchange Act requires, among other things, that we file annual and current reports with respect to our business and results of operations. We incur significant expenses and devote substantial management effort toward ensuring compliance with the auditor attestation requirements of Section 404 of the Sarbanes- Oxley Act, which will increase when we are no longer an “emerging growth company,” as defined by the JOBS Act. We may need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge. We cannot predict or estimate the amount of additional costs we may incur as a result of becoming a public company or the timing of such costs. As a result, management’s attention may be diverted from other business concerns, which could adversely affect our business and results of operations.
| 41 |
In addition, changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and as a result, their application in practice may evolve over time as regulatory and governing bodies provide new guidance. These factors could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We will continue to invest resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to their application and practice, regulatory authorities may initiate legal proceedings against us, and our business could be adversely affected.
As a result of disclosure of information as a public company, our business and financial condition have become more visible, which may result in threatened or actual litigation, including by competitors and other third parties. If the claims are successful, our business operations and financial results could be adversely affected, and even if the claims do not result in litigation or are resolved in our favor, these claims, and the time and resources necessary to resolve them, could divert the resources of our management and adversely affect our business operations and financial results. These factors could also make it more difficult for us to attract and retain qualified colleagues, executive officers and Board members.
Operating as a public company makes it more difficult and more expensive for us to obtain director and officer liability insurance on the terms that we would like. As a result, it may be more difficult for us to attract and retain qualified people to serve on our Board, our Board committees or as executive officers.
If we fail to maintain an effective system of internal control over financial reporting in the future, we may not be able to accurately report our financial condition, results of operations or cash flows, which may adversely affect investor confidence.
The Sarbanes-Oxley Act requires, among other things, that we maintain effective internal control over financial reporting and disclosure controls and procedures. We are required, under Sarbanes-Oxley Act Section404, to perform system and process evaluations and testing of internal controls over financial reporting to allow management to report annually on the effectiveness of internal control over financial reporting. This assessment requires disclosure of any material weaknesses in our internal control over financial reporting identified by management. Sarbanes-Oxley Act Section404 also generally requires an attestation from our independent registered public accounting firm on the effectiveness of internal control over financial reporting. However, for as long as we remain an emerging growth company (“EGC”), we intend to take advantage of the exemption permitting it not to comply with the independent registered public accounting firm attestation requirement.
At the time when we are no longer an Emerging Growth Company, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied with the level at which we control are documented, designed or operating. Remediation efforts may not enable us to avoid a material weakness in the future.
Compliance with Sarbanes-Oxley Act Section404 requires the incurrence of substantial accounting expense and consumes significant management efforts. We may not be able to complete evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses or significant deficiencies in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit its ability to accurately report financial condition, results of operations or cash flows. If we are unable to conclude that internal control over financial reporting is effective, or if our independent registered public accounting firm determines we have a material weakness or significant deficiency in internal control over financial reporting, it could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our ordinary shares could decline, and we could be subject to sanctions or investigations by the Nasdaq, the SEC or other regulatory authorities. Failure to remedy any material weakness in internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict future access to the capital markets.
Our results of operations may be affected by changes to, or interpretations of financial accounting standards may affect our results of operations, which may require us to change our business practices.
Our financial statements are prepared in accordance with IFRS. These accounting principles are subject to interpretation by the Financial Accounting Standards Board, the SEC and various bodies formed to interpret and create accounting rules and regulations. Changes in accounting rules can have a significant effect on our reported financial results and may affect our reporting of transactions which may be completed before the announcement of such a change. Changes to those rules or the questioning of current practices may materially adversely affect our financial results, including those contained in this filing, or the way we conduct our business.
| 42 |
Our ability to use our loss carryovers and certain other tax attributes may be limited.
As described above under the heading “We have incurred losses since our incorporation and we expect to incur significant losses for the foreseeable future”, we have incurred losses since our incorporation and anticipate that we will continue to incur significant losses for the foreseeable future. Under the Income Tax Act and subject to the agreement with the Comptroller, a company is generally allowed a deduction for trade losses, carried forward indefinitely from a prior taxable year. Under that provision, we can carry forward our trade losses to offset our future taxable income, if any, until such trade losses are fully utilized, subject to the fulfilment of certain conditions.
Furthermore, if a company undertakes substantial change in the shareholders (which is generally defined as changes of the owner of 50% or more of the company’s (or its ultimate parent company’s) total number of issued shares) as at the relevant dates (i.e. the last day of the year in which the loss was incurred and the first day of the year of assessment in which such loss is to be claimed), Section 37(12) of the Income Tax Act limits the corporation’s ability to use/carry forward the unutilized trade losses and donations to reduce its tax liability for periods after the substantial shareholder change. Our issuance of ordinary shares pursuant to our Offering may result in a limitation under Section 37(12) of the Income Tax Act, either separately or in combination with certain prior or subsequent shifts in the ownership of our ordinary shares. As a result, our ability to carry forward of our trade losses to reduce our future income tax liability may be subject to limitations. This could result in increased income tax liability for us if we generate taxable income in a future period.
The use of our tax attributes will also be limited to the extent that we do not generate positive taxable income in future tax periods. To the extent our ability to utilize our trade losses and other tax assets going forward is limited, in part or altogether, our tax liability for future periods may be greater than expected, and our business, financial condition, results of operations and growth prospects may be materially adversely affected.
Because the likelihood of paying cash dividends on our ordinary shares is remote at this time, investors must look solely to appreciation of our ordinary shares in the market to realize a gain on their investments.
We do not know when or if we will pay dividends to our shareholders, and the likelihood that we will be paying dividends on our ordinary is remote at this time. We currently intend to retain future earnings, if any, to finance the expansion of our business. Our future dividend policy is within the discretion of our board of directors and will depend upon various factors, including our business, financial condition, results of operations, capital requirements and investment opportunities. Accordingly, investors must look solely to appreciation of our ordinary shares in the market to realize a gain on their investment. This appreciation may not occur.
We currently report our financial results under IFRS, which differs in certain significant respects from U.S. GAAP.
Currently, we report our financial statements under IFRS. There have been and there may in the future be certain significant differences between IFRS and U.S. GAAP, including differences related to revenue recognition, share-based compensation expense, income tax and earnings per share. As a result, our financial information and reported earnings for historical or future periods could be significantly different if they were prepared in accordance with U.S. GAAP. In addition, we do not intend to provide a reconciliation between IFRS and U.S. GAAP unless it is required under applicable law. As a result, you may not be able to meaningfully compare our financial statements under IFRS with those companies that prepare financial statements under U.S. GAAP.
RISKS RELATED TO INVESTMENTS IN SINGAPORE COMPANIES
We are incorporated in Singapore, and our shareholders may have more difficulty in protecting their interests than they would as shareholders of a corporation incorporated in the United States.
Our corporate affairs are governed by, inter alia, our constitution and by the laws governing companies incorporated in Singapore. The rights of our shareholders and the responsibilities of the members of our Board under Singapore law may be different from those applicable to a corporation incorporated in the United States. Therefore, our public shareholders may have more difficulty protecting their interests in connection with actions taken by us, our management, members of our Board or our controlling shareholders than they would as shareholders of a corporation incorporated in the United States. For example, controlling shareholders in corporations incorporated in Delaware are subject to fiduciary duties while controlling shareholders in Singapore companies are not subject to such duties.
In addition, subject to the Singapore Companies Act and our constitution, only persons who are registered as shareholders in our register of members are recognized under Singapore law as our shareholders. Only registered shareholders have legal standing to institute shareholder actions against us or otherwise seek to enforce their rights as shareholders. Investors in our ordinary shares who are not specifically registered as shareholders in our register of members (for example, where such shareholders hold ordinary shares indirectly through the DTC) are required to be registered as shareholders in our register of members in order to institute or enforce any legal proceedings or claims against us, our Directors and/or our executive officers relating to shareholder rights. The administrative process of becoming a registered shareholder could result in delays prejudicial to any such legal proceeding or enforcement action. Please refer to the section entitled “Comparison of Shareholder Rights” for a discussion of certain differences between Singapore and Delaware corporation law.
| 43 |
Our shareholders may have more difficulty transferring their shares in the Company.
The transfer of our shares is subject to restrictions under our constitution and by the laws governing companies incorporated in Singapore. Under Singapore law, a public company shall not register a transfer of shares unless a proper instrument of transfer has been delivered to the company. In addition, our constitution provides that our Directors may decline to lodge a notice of transfer of shares with ACRA if (a) the shares are not fully paid shares, (b) the Directors do not approve of the transferee or (c) the Company has a lien on the shares. As such, it may be difficult for you to transfer your shares in the Company.
It may be difficult for you to enforce any judgment obtained in the United States against us, our Directors and officers and/or our affiliates.
A majority of our Directors and officers reside outside the United States. In addition, a majority of our assets and the assets of such persons are located outside the United States. As a result, it may be difficult to enforce in the United States any judgment obtained in the United States against us or any of such persons, including judgments based on the civil liability provisions of the U.S. securities laws. In addition, in original actions brought in courts in jurisdictions located outside the United States, it may be difficult for investors to enforce liabilities based upon U.S. securities laws.
As of the date of this annual report, there is no treaty between the United States and Singapore providing for the reciprocal recognition and enforcement of judgments in civil and commercial matters and a final judgment for the payment of money rendered by any federal or state court in the United States based on civil liability, whether or not predicated solely upon the federal securities laws, would, therefore, not be automatically enforceable in Singapore. It is not clear whether a Singapore court may impose civil liability on us, our Directors and/or officers who reside in Singapore in an action brought in the Singapore courts against us and/or such persons with respect to a violation solely of the federal securities laws of the United States.
In addition, holders of book-entry interests in the ordinary shares (for example, where such shareholders hold ordinary shares indirectly through the DTC) will be required to be registered shareholders as reflected in our register of members in order to have standing to bring a shareholder action and, if successful, to enforce a foreign judgment against us, our Directors or our executive officers in the Singapore courts. Any such enforcement action would be subject to applicable Singapore laws. The administrative process of becoming a registered shareholder could result in delays that could be prejudicial to any legal proceeding or enforcement action. In making a determination as to enforceability of a judgment of a state court or a federal court of the United States, the Singapore courts would have regard to, among others, whether the judgment was final and conclusive, given by a court of law of competent jurisdiction, expressed to be for a fixed sum of money, whether it was procured by fraud, or in breach of principles of natural justice, or whether the enforcement thereof would be contrary to public policy.
Accordingly, there can be no assurance that the Singapore courts would enforce against us, our Directors and/or our officers, judgments obtained in the United States which based on the civil liability provisions of the federal securities laws of the United States.
Subject to the general authority to allot and issue new ordinary shares as may be approved by our shareholders pursuant to the Singapore Companies Act and our constitution, our Directors may allot and issue new ordinary shares from time to time on such terms and conditions and for such purposes as may be determined by our Board of Directors in its sole discretion. Any issuance of new shares would dilute the percentage ownership of existing shareholders and could adversely impact the market price of our ordinary shares.
Under Singapore law, we may only allot and issue new ordinary shares with prior approval of our shareholders in a general meeting. Subject to the general authority to allot and issue new ordinary shares provided by our shareholders, the provisions of the Singapore Companies Act, and our constitution, we may allot and issue new ordinary shares on such terms and conditions as our Directors may think fit to impose. Such terms and conditions may be adverse to the rights of holders of our ordinary shares. Any additional issuances of new ordinary shares could dilute the percentage ownership of our existing shareholders and may adversely impact the market price of our ordinary shares.
| 44 |
Because new issuances of ordinary shares are subject to restrictions in respect of the percentage thereof under the Singapore Companies Act and the requisite shareholders’ approval, if a sufficient number of shares have not been approved for issuance in respect of one or more transactions in any given year, we may be delayed in raising capital through equity offerings or delayed or prevented from consummating an acquisition using our ordinary shares. Assuming shareholders have approved the issuance of new shares, we may seek to raise capital in the future, including to fund acquisitions, future investments and other growth opportunities. We may, for these and other purposes, issue additional ordinary shares or securities convertible into ordinary shares. Any additional issuances of new ordinary shares could dilute the percentage ownership of our existing shareholders and may also adversely impact the market price of our ordinary shares.
We are subject to the laws of Singapore, which differ in certain material respects from the laws of the United States.
As a company incorporated in Singapore, we are required to comply with the laws of Singapore, some of which are capable of extra-territorial application, as well as our constitution. In particular, we are required to comply with certain provisions of the Securities and Futures Act 2001 of Singapore, which prohibit certain forms of market conduct and information disclosures, and impose criminal and civil penalties on corporations, directors and officers in respect of any breach of such provisions. In addition, the Singapore Take-over Code specifies, among other things, the circumstances wherein a general offer shall be taken upon a change in control of a Singapore-incorporated public company and the manner and price at which voluntary and mandatory general offers are to be made.
The laws of Singapore and of the United States differ in certain significant respects. The rights of our shareholders and the obligations of our Directors and officers under Singapore law may be different from those applicable to U.S. corporations, including corporations incorporated in Delaware, in material respects, and our shareholders may have more difficulty and less clarity in protecting their interests in connection with actions taken by us, our management, members of our Board and/or our controlling shareholders than would otherwise apply to U.S. corporations, including those incorporated in Delaware. Please refer to the section titled “Comparison of Shareholder Rights” for a discussion of certain differences between Singapore and Delaware corporation law.
In addition, the application of Singapore law, in particular, the Singapore Companies Act may, in certain circumstances, impose more restrictions on us, our shareholders, Directors and officers than would otherwise be applicable to U.S. corporations, including those incorporated in Delaware. For example, the Singapore Companies Act requires a director to act with reasonable degree of diligence in the discharge of the duties of his office and, in certain circumstances, imposes criminal liability for specified contraventions of particular statutory requirements or prohibitions. In addition, pursuant to the provisions of the Singapore Companies Act, two or more shareholders holding 10% or more of the total number of issued shares as at the date of the deposit of the relevant requisition, carrying the right of voting at general meetings (disregarding paid-up shares held as treasury shares) may require our Directors to convene an extraordinary general meeting.
Singapore tax laws may differ from the tax laws of other jurisdictions.
Singapore tax laws may differ from the tax laws of other jurisdictions, including the United States. For example, Singapore does not impose tax on capital gains. However, there are no specific laws or regulations which deal with the characterization of whether a gain is income or capital in nature. Gains arising from the disposal of our ordinary shares may be construed to be of an income nature and subject to Singapore income tax, especially if they arise from or are otherwise connected with the activities which the IRAS regards as carrying on a trade or business in Singapore. In addition, holders of our ordinary shares may for the purposes of Singapore income tax be required to recognize gains or losses (not being gains or losses in the nature of capital) even though no sale or disposal of our ordinary shares is made. Prospective investors should consult their tax advisors concerning the overall tax consequences of purchasing, owning and disposing of our shares. Please also refer to the section entitled “Tax Considerations”.
| 45 |
Singapore take-over laws contain provisions that may vary from those in other jurisdictions.
The Singapore Take-over Code applies to, among others, corporations with a primary listing of their equity securities in Singapore. While the Singapore Take-over Code is drafted with, among others, listed public companies in mind, unlisted public companies with more than fifty (50) shareholders and net tangible assets of S$5.0 million or more, must also observe the letter and spirit of the general principles and rules of the Singapore Take-over Code, wherever this is possible and appropriate. Public companies with a primary listing overseas may apply to the SIC to waive the application of the Singapore Take-over Code. As of the date of this annual report, no application has been made to SIC to waive the application of the Singapore Take-over Code in relation to us.
In this regard, the Singapore Take-over Code contains certain provisions that may possibly delay, deter or prevent a future take-over or change in control of us. Under the Singapore Take-over Code, except with the consent of SIC, any person acquiring an interest, whether by a series of transactions over a period of time or not, either on his own or together with parties acting in concert with him, in 30% or more of our voting shares is required to extend a take-over offer for all remaining voting shares in accordance with the procedural and other requirements under the Singapore Take-over Code.
Except with the consent of SIC, such a take-over offer is also required to be made if a person holding between 30% and 50% (both inclusive) of our voting shares, either on his own or together with parties acting in concert with him, acquires additional voting shares representing more than 1% of our voting shares in any six (6) month period. While the Singapore Take-over Code seeks to ensure an equality of treatment among shareholders in take-over or merger situations, its provisions could substantially impede the ability of the shareholders to benefit from a change of control and, as a result, may adversely affect the market price of the ordinary shares and the ability to realize any benefit from a potential change of control.
Our operations are affected by the changes in existing Malaysian laws and regulations, and the adoption of new Malaysian laws and regulations and/or the changes in interpretation of the Malaysian laws and regulations as well as possible inconsistencies between the various Malaysian laws and regulations and/or the corresponding interpretation
Our operations in Malaysia are regulated by the laws and regulations of Malaysia, including those relating to the corporate, investment, marketing, labor, environmental protection, occupational health and safety, and taxation matters. The legal and regulatory regimes in Malaysia may be uncertain and subject to unforeseen changes. At times, the interpretation or application of laws and regulations in Malaysia may be unclear. Government policies, regulations and guidelines issued and imposed by the relevant authorities may change from time to time. We may have to incur significant capital and maintenance expenditures to comply with laws and regulations. The failure to discharge our obligations could result in the imposition of fines and penalties, damage to our reputation, delays in production or the temporary or permanent closure of our operations. In addition, existing laws, regulations or policies may become stricter or more strictly enforced. Our operations and business may face investigation, scrutiny or evaluation. The adoption of new laws and regulations or any modification to the existing laws and regulations may result in additional expenses to comply with the new laws.
While we have compliance procedures in place to ensure compliance with new legislation and every effect is taken to ensure the requirements of any new legislation are met, there is no certainty on the approach which will be taken by the relevant regulators, and we may incur additional compliance costs with the introduction of new or amended regulations. We have no control over such conditions and developments and there can be no assurance that such conditions and developments will not have a material adverse effect on our business. As a result, we may face new liabilities, reduced operating hours, additional investment requirements, or delays in the opening or expansion of operations. We may also be compelled to conduct preventive or remedial actions. These may result in increased costs. Such costs, liabilities, or disruptions in operations may materially and adversely affect our business.
We are subject to the political, economic and social conditions in the jurisdiction of Malaysia
Our cGMP Facility is located in Malaysia and our operations are substantially based in Malaysia and is therefore sensitive to the social, economic, political and legal conditions in Malaysia. Developments in Malaysia such as changes in Malaysian government policies, currency and interest rates, inflation, capital restrictions, price and wage controls, unemployment rate, taxes and duties will materially and/or adversely affect our business. We have no control over such conditions and developments and there is no assurance that such conditions and developments will not occur. These changes, if they occur, will materially and/or adversely affect our Malaysian business and operations and thus our business in general.
| 46 |
We are subject to the foreign exchange legislation and regulations in the jurisdiction of Malaysia
Local and foreign investors are subject to the FX Notices in Malaysia. The FSA and the IFSA govern the foreign exchange control framework in Malaysia. Under the FSA and the IFSA, the BNM has issued FX Notices. These FX Notices embody the BNM’s general permissions and directions. They set out the circumstances in which residents and non-residents must seek the specific approval of BNM to remit funds to and from Malaysia.
The FX Notices are reviewed regularly according to changing circumstances. As of the date of this annual report, foreign investors are allowed to repatriate from Malaysia, funds including any income earned or proceeds from divestments of Ringgit Asset (including any property in Malaysia), provided that the repatriation is made in foreign currency and the conversion of Malaysian Ringgit into foreign currency is undertaken in accordance with the FX Notices. Should the repatriation of funds be restricted in the future, this will limit our ability to extract the profits from our Malaysian business operations. In addition, our Malaysian subsidiaries may be subject to restrictions on the borrowing of foreign currency or from non-residents, which may affect our ability to raise funds in the future should the need arise. “Ringgit Asset” is defined in the FX Notices as:
| ● | Ringgit-denominated securities or Islamic securities issued in Malaysia by a resident; | |
| ● | Ringgit-denominated securities or Islamic securities issued by a non-resident as approved in writing by the Bank; | |
| ● | Ringgit-denominated Financial Instrument or Islamic Financial Instrument as approved in writing by the Bank; | |
| ● | Ringgit deposit with a Financial Institution in Malaysia including deposit-like instrument with only Ringgit delivery at the inception and maturity; or | |
| ● | any property in Malaysia. |
The relevant rules and regulations on foreign exchange control in Malaysia may also be subject to change from time to time. If there is any adverse change in the foreign exchange rules and regulations relating to the borrowing or repatriation of foreign currency, our business may be materially and adversely affected.
Regulatory risks in relation to environmental hazards, production safety and the occurrence of accidents in Malaysia
As CytoMed Malaysia’s operation in Malaysia involves handling and storing biohazardous materials or articles and sensitive equipment, CytoMed Malaysia is subject to the OSHA. The necessary facilities and measures to comply with OSHA would lead to additional costs of operation.
In the event of occurrence of accidents which causes personal injury of the staff, we may be subject to legal proceedings which may in turn result in civil or criminal liabilities if the legal proceeding taken against us turn out to be successful. Although employees who suffered injuries (including death) in the course of employment are able to claim for benefits under the SOSCO and the EIS, we as employers are not automatically immunized against any claims by virtue of contributions made towards SOSCO and EIS. In fact, courts in Malaysia have recently held that employees are allowed to claim for aggravated and exemplary damages for gross negligence on the part of employers despite having already received compensation from SOSCO.
In such circumstances, CytoMed Malaysia may also be subject to investigation by the relevant authority and if such occurrence of accidents is found to be a breach of any of the conditions of our licenses, permits and approvals, our licenses, permits and approvals for the operation of our business in Malaysia may be revoked. This could cause harm to our reputation and our financial condition may suffer. That said, claims for compensation may be mitigated by us procuring the relevant insurance or workmen’s compensation policies.
We may be subject to costs and risks associated with the monitoring, rehabilitation and compliance with environmental laws and regulations
We are subject to environmental laws and regulations, under which we are required to take steps and/or omit certain steps for the protection and preservation of the environment, including control of pollution and the conservation and management of land. Compliance with environmental laws and regulations will increase our costs and may also hinder, delay or prevent our activities, depending on what is permitted and how these requirements are interpreted and implemented by the relevant regulatory authorities. In addition, economic development and improvements in living standards may increase public awareness of environmental protection. Thus, environmental laws and regulations may become more stringent and/or more stringently enforced. If so, we may not be able to comply with these environmental laws and regulations, and thus may be subject to penalties and liabilities under environmental laws and regulations.
| 47 |
We are subject to regulations governing foreign workers in the event of employment of such foreign workers
In the event our operations in Malaysia require foreign workers, such foreign workers are regulated by the Malaysian government authorities which set a limit on the number of foreign workers which we may hire and also impose levies on each foreign worker hired by our Malaysian companies. Hence, any changes in governmental policies with respect to foreign workers in Malaysia, such as to decrease the number of foreign workers permissible to be employed by the Malaysian companies or to increase the levy payable by Malaysian companies for foreign works may materially and adversely affect our business. Any increase in demand and thus competition for foreign workers may also increase our labor costs.
Mechanism for enforcement of foreign judgments in Malaysia is limited to certain jurisdictions
In Malaysia, foreign judgments may be enforced without the need to commence a fresh action, provided that the foreign judgment in question is granted by a “reciprocating country”. The REJA provides for the mechanism to enforce foreign judgments, which is by way of ex-parte registration with the High Court of Malaya. To be eligible for registration, the foreign judgment must, amongst others, be final between parties, must provide for a sum of money to be payable (monetary judgment), the judgment must not be contrary to the public policy of Malaysia, the judgment was not obtained fraudulently or in a manner contrary to the rules of natural justice, and was pronounced by a specific court from one of the reciprocating countries set out in REJA. At the time of registration, the applicant must show that the foreign judgment was issued or pronounced within 6 years prior, has not been satisfied in full by the judgment debtor and is capable of being enforced or executed in the court of original jurisdiction. Upon registration, the judgment given by the High Court of Malaya will be treated as if it is a judgment given by the court of original jurisdiction.
The reciprocating countries under REJA are United Kingdom, Hong Kong, Singapore, New Zealand, Republic of Sri Lanka, India (excluding State of Jammu and Kashmir, State of Manipur, Tribal areas of State of Assam, Scheduled areas of the States of Madras and Andhra) and Brunei Darussalam. As mentioned, the foreign judgment eligible for registration under REJA must be one given by the specific court as prescribed. For example, in the case of Singapore, only the judgment made by the High Court of Singapore can be enforced in Malaysia through REJA.
As the mechanism provided under REJA is limited to monetary judgments in certain jurisdictions, enforcement of foreign judgments which do not fall within the ambit of REJA requires the initiation of a fresh suit and action in court.
ITEM 4. INFORMATION ON THE COMPANY
| A. | History and Development of the Company |
Our legal and commercial name is CytoMed Therapeutics Limited. We are a public company limited by shares incorporated on March 9, 2018 in Singapore. On January 19, 2023, we converted from a private company limited by shares incorporated in Singapore, known as CytoMed Therapeutics Pte. Ltd. to a public company limited by shares pursuant to the provisions of the Singapore Companies Act. As a public company limited by shares incorporated in Singapore, we are governed by the Companies Act 1967 of Singapore.
As of the date of this annual report, we are a holding company incorporated in Singapore which oversees our operations in Malaysia. We conduct our business activities primarily through our direct wholly-owned subsidiary in Malaysia, CytoMed Malaysia. We expect to conduct more research and clinical trial activities in Singapore through CytoMed moving forward. Other than CytoMed Malaysia, our subsidiaries have minimal operations.
Effective on January 17, 2023, our shareholders and directors approved a 1-for-380.83 reverse split of its ordinary shares pursuant to which the shareholders received one (1) ordinary share for every 380.83 ordinary shares held as of such date.
Our principal executive office in Singapore is located at 1 Commonwealth Lane, #08-22, Singapore 149544. Our telephone number is +65 6250 7738. Our agent for service of process in the United States is Puglisi & Associates whose address is 850 Library Avenue, Newark, Delaware 19711.
We file annual reports and other information with the SEC. You may inspect and copy any report or document we file, including this annual report and the accompanying exhibits, at the website maintained by the SEC at http://www.sec.gov. Investors should submit any inquiries to the address and telephone number of our principal executive office.
Information concerning our capital expenditures can be found in Item 5. Operating and Financial Review and Prospects.
| 48 |
| B. | Business Overview |
We are a pre-clinical biopharmaceutical company focused on harnessing our proprietary technologies into creating novel cell-based immunotherapies for treatment of human cancers and degenerative diseases. The development of our novel technologies has been inspired by the clinical success of existing CAR-T cells in treating hematological malignancies as well as the current clinical limitations and commercial challenges in extrapolating the CAR-T principle into treatment of solid tumors.
We believe that the current development of CD19-targeting CAR-T cells in treating B-cell malignancies signifies that cellular immunotherapy is becoming one of the pillars in cancer care. However, we believe that it remains challenging to apply the current CAR-T principle into treatments of other type of cancers, in particular solid tumors, due to a variety of reasons, including (i) the reliance on the limited cell quality and quantity of patients, (ii) the lack of suitable surface cancer antigens and their recognition system and (iii) the risk of cancer escape because of a single antigen-targeting strategy. To this end, we have established two novel patient blood cell-independent platform technologies to manufacture “off-the-shelf” cell-based cancer immunotherapies: one of which depends on healthy donor blood cells and the other of which depends on iPSC. Such platform technologies and the resulting product candidates exploit the multiple antigen recognition systems of NK cells and gamma delta T cells in the human body and so as to recognize and treat a broad range of cancers. Built on the proprietary platform technologies that we licensed, we are developing three cellular immunotherapy product candidates: CTM-N2D, iPSC-gdNKT and CTM-GDT. CTM-N2D is our lead product candidate and it consists of expanded gamma delta T cells grafted with NKG2DL-targeting CAR to enhance anti-cancer cytotoxicity. On March 10, 2023, we entered into the Clinical Study Agreement pursuant to which the ANGELICA Trial will be conducted by the National University Hospital Singapore to evaluate the safety of CTM-N2D in human subjects, following the HSA’s acknowledgement on January 6, 2023 that we had submitted the relevant documents to meet the approval conditions of the CTA initially granted in July 2022 relating to the use of our CTM-N2D for the aforementioned trial by HSA, and no further action was required.
Pursuant to the Clinical Study Agreement, the National University Hospital Singapore will conduct the ANGELICA Trial using our lead product candidate CTM-N2D, with Dr. Anand D Jeyasekharan, Consultant, Department of Hematology-Oncology of National University Hospital Singapore, being the principal investigator of the trial. Unless terminated earlier, the Clinical Study Agreement shall continue until completion of the trial. National University Hospital Singapore has obtained approval from the Singapore National Healthcare Group Domain Specific Review Boards in February 2023.
Pursuant to the Clinical Study Agreement, Company has agreed to (i) provide funding and supply at no cost to National University Hospital Singapore such quantities of the CTM-N2D as may be required for the ANGELICA Trial, (ii) to provide all relevant clinical information, (iii) where applicable, to provide an instruction manual/guide, product labels, storage, handling and safety instructions and advice which are required for the safe and proper use of CTM-N2D and the proper planning and conduct of the trial. In March 2023, we obtained and provided to National University Hospital Singapore a clinical trial insurance certificate required to conduct the trial in Singapore. In July 2023, we have started the clinical trial, with the donor recruitment to obtain healthy donor PBMCs which would serve as starting material for manufacturing of CTM-GDT for patients in the trial.
In all publications generated from the ANGELICA Trial, both Company and National University Hospital Singapore would be granted co-authorship. All the study results shall be jointly owned by both parties. Discoveries or inventions that relate to any enhancements or modifications to the CTM-N2D arising from the conduct of the ANGELICA Trial, whether conceived or reduced to practice either solely by or jointly with National University Hospital Singapore, will be the joint property of National University Hospital Singapore and Company.
Either party may terminate the Clinical Study Agreement at any time upon providing at least thirty (30) days’ prior written notice to the other party. National University Hospital Singapore may terminate the Clinical Study Agreement immediately in the event National University Hospital Singapore and/or the principal investigator believes on reasonable grounds that continuing the said trial poses an unacceptable risk to the rights, interests, safety or well-being of human subjects, or if a serious adverse event occurs which necessitates the discontinuance of the ANGELICA Trial.
| 49 |
We anticipate recruiting our first patient in the second quarter of 2024. Please refer to the sections entitled “Business – Our Solutions and Product Candidates: CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC” and “Business – CTM-N2D Therapy – Development Status of CTM-N2D Therapy” in this annual report for further details on the application process and progress. We expect to expand our pipeline further in Phase II trials of CTM-N2D therapy for specific cancer indications. Our second product candidate iPSC-gdNKT utilizes iPSC as a starting material to generate gdNKT, which is a synthetic hybrid of a gamma delta T cell and a NK cell. The hybrid cells express receptors of both cells which potentially allow the gdNKT cells to recognize and treat a broad range of cancers. This product has been undergoing pre-clinical process development since the fourth quarter of 2022, and is targeting to commence pre-clinical studies after 2024. Our third product candidate, CTM-GDT consists of expanded allogeneic gamma delta T cells and exploits the potential of these cells to recognize and treat a broad range of cancers. We are working with The University of Texas M.D. Anderson Cancer Center on using CTM-GDT against lymphoma and breast cancer in preclinical studies. Through a US agent, we have submitted a drug master file to US FDA and intend to pursue an investigational new drug application in the near future. CTM-GDT is considered a CGTP in Malaysia according to NPRA and we intend to manufacture the investigational product locally. Therefore, a CTX application to NPRA is required prior to commencing clinical trial. We are looking to submit a CTX application for Phase I clinical trial to NPRA, Malaysia after 2023.
As of the date of this annual report, we are a holding company incorporated in Singapore which oversees our operations in Malaysia. We conduct our business activities primarily through our direct wholly-owned subsidiary in Malaysia, CytoMed Malaysia, but expect to conduct more research and clinical trial activities in Singapore through CytoMed moving forward. Other than CytoMed Malaysia, our subsidiaries have minimal operations.
We also have an operating cGMP Facility in Johor, Malaysia (which is near Singapore) which has been built in accordance with the international PIC/S GMP standards to manufacture cell therapy products to support clinical trials. In addition, we have a well-trained operations team who conduct all essential cGMP activities including manufacture, quality control, quality assurance and documentation. We manufacture our product candidates in our cGMP Facility instead of engaging and relying on an external CMO. As of the date of this annual report, our current product pipeline and progress is summarized in Figure 1.

Figure 1. Pipeline, milestone, and progress.
| 50 |
Strategy
Currently, there are vast unmet medical needs in cancer treatment, in particular for solid tumors. Our goal is to develop novel “off-the-shelf” cell therapies to improve the quality of life of cancer patients and to increase their overall survival. We believe that our uniquely designed platform technologies and product development strategies are expected to strengthen our capability to achieve such goal. We intend to focus on developing our two “off-the-shelf, allogeneic” platform technologies based on healthy donor blood cells and iPSCs respectively. Key elements of our strategies are as follows:
Uniquely Designed Platform Technologies to Develop Novel Cell Therapies for Cancers
Currently, many companies employ αβ T cells as main effector cells. Some companies attempt to graft NK receptors, or γδ TCRs to potentially expand antigen recognition. However, the use of αβ T cells limits such therapy to autologous use. Some companies have employed gene editing technologies to enable the use of αβ T cells allogeneically, but additional gene editing increases complexity in manufacturing and increases cost. Furthermore, genetically engineered cells may pose potential safety issue due to chances of insertional mutagenesis, leading to secondary cancers. Innate immune cells, such as γδ T cells, can be used in an allogeneic setting without requiring additional gene editing, therefore making it an ideal cell type for allogeneic therapy.
Another potential cell source is iPSC. iPSC can replicate limitlessly and can be differentiated into cell of choice. iPSC can be differentiated into innate immune cells such as NK and γδ T cells so these cells can be used for allogeneic cancer immunotherapy. As such, we employ γδ T cell derived from blood of healthy donors to generate CTM-N2D and CTM-GDT. We also use iPSC to generate iPSC-gdNKT, a hybrid between NK and γδ T cells to increase the number of surface receptors on the immune cell surface for increased antigen recognition.
We believe we have uniquely designed our platform technologies based on the following: (a) the targeted cancer antigens are ubiquitous stress-induced antigens, such as NKG2DLs and PAg; (b) the cancer antigen-recognition receptors are the built-in recognition system of innate immune cells such as γδ TCR and the grafted NKG2DL-targeting CAR; and (c) the cytotoxic effector cells are lymphocytes of innate immune system, for example, NK cells and gamma delta T cells. Such platforms allow us to use healthy donor blood cells and unlimited iPSCs as starting materials to manufacture CTM-N2D and iPSC-gdNKT product candidates, respectively. Our product candidates generated using such platform technologies possess the following desirable features: (1) “off-the-shelf”; (2) broad-spectrum; and (3) for many types of cancer patients. More importantly, both our healthy donor blood cell and iPSC-based technologies are amenable to genetic modification and thus open the possibility to further functional enhancement and indication specification.
In-house Development of the Technologies into Cell Therapies for Cancers
The cell therapy products currently in our pipeline are based on the patented, core proprietary technologies licensed from A*STAR, Singapore’s lead R&D agency in the public sector. The key inventors of these proprietary technologies licensed to us from A*STAR, Dr. ZENG Jieming and Dr. WANG Shu, have been actively involved as technology inventors in the product development of these proprietary technologies since 2018. Dr. ZENG Jieming joined us in 2019 as a full-time employee, serving as our Chief Scientific and Medical Officer to oversee product development and manufacturing. Dr. TAN Wee Kiat, a scientist who is familiar with the CTM-N2D technology, has been employed by us on a full-time basis as our Co-Chief Executive Officer and Chief Operating Officer.
The direct involvement of the inventors and key scientists during process and product development has provided valuable in-depth understanding of the technologies and experience, which are crucially important for timely and cost-efficient translation of these proprietary technologies into therapeutic products. Such benefit has been manifested by the speedy completion of our cGMP Facility and staff training. These well-trained staff members are critical to the success of quality product manufacturing and new product development.
In-house Manufacture of the Cell Therapy Products for Cancer Treatments
We believe that our in-house cGMP manufacturing capability will enable us to maintain not only the quality and availability of cell therapy products, which is crucial for our clinical success, but also allow us to manufacture cell therapy products more cost-effectively, which will be crucial for commercial viability. As such, we have constructed and fully equipped our own all-in-one cGMP Facility in Johor, Malaysia. Built in 2019, our cGMP Facility was internally validated by completion of manufacturing runs in 2020 and externally audited in 2021 by the NPRA, which is also a member of PIC/S. Our cGMP Facility also includes a quality control laboratory to support release of cell products and an R&D lab to conduct staff training and process development. We believe our current cGMP Facility is sufficient to support our planned Phase I and Phase II clinical trials.
| 51 |
Continuous Exploration of Enabling Technologies to Advance Cell Therapy Products
Both our healthy donor blood cell and iPSC-based technologies are open platforms, and we will continue to explore enabling technologies that may further enhance these platforms to generate more novel cell therapy for cancers. Such efforts may further expand and fine-tune our product pipeline for various cancer indications. Currently, Puricell is designated to explore iPSC-based enabling technologies for enhancing cell-based therapies. We are also exploring opportunities with potential partners from A*STAR to further develop proprietary manufacturing capabilities that may further increase manufacturing flexibility and scalability.
Background of Cellular Immunotherapy for Cancer and CAR-T Cell Therapy
Cellular Immunotherapy for Cancer and CAR-T Cell Therapy
Harnessing the components of the immune system to treat cancer is promising in providing targeted therapy that may reduce morbidity and mortality caused by conventional non-targeted treatments, such as chemotherapy and radiation. Immunotherapy of cancer may involve and utilize components like cytokines, antibodies, cancer vaccines and immune cells. In terms of strategy, cancer immunotherapy may be categorized into two major types: One relies largely on the patient’s own immune responses to cancers, for example, non-specific immune stimulation using cytokines, vaccination with cancer antigens, and immune checkpoint blockage to treat immunogenic cancers. The mechanism of these treatments is to stimulate or unleash the patient’s own immune cells in the body to fight cancer. In contrast, the other strategy is less reliant on the patient’s own immune response, for example, adoptive cell therapy. Typically, in adoptive cell therapy, the immune cells are derived from the cancer patient’s blood and are then genetically modified to enhance their anti-cancer activity and expanded into a clinically relevant number before reinfusion back to the patient (Figure 2). The mechanism of such cellular immunotherapy is to directly send in an army of cultured killer cells into the patient’s body to eliminate cancer. Technological progresses in antigen recognition, genetic modification, and immune cell expansion have paved the way to recent success of adoptive therapy using CAR-T for some hematologic malignancies (Figure 2).
CAR is a genetically engineered artificial receptor to recognize surface cancer antigen. It consists of three domains, namely the intracellular, transmembrane, and extracellular domain. The intracellular domain serves to activate cytotoxicity of effector cells to kill target cells. The transmembrane domain anchors the CAR molecule on the surface of effector cells. The extracellular domain is responsible for recognizing antigen of interest. Through expressing CAR, cytotoxic effector cells such as T cells may be redirected to specifically recognize a cancer-specific or cancer-associated antigen (Figure 2). With certain level of specificity, these CAR-T may recognize and destroy the cancer cells while sparing the healthy cells. The principle of CAR-T strategy has been applied to treat B-cell malignancies by targeting CD19, a B-cell lineage marker. The potency of such CD19-targeting CAR-T have been successfully demonstrated in multiple clinical trials (Kochenderfer et al. 2015; Lee et al. 2015; Locke et al. 2017; Maude et al. 2014; Neelapu et al. 2017; Schuster et al. 2017).

Figure 2. Manufacturing and mechanism of action of current CAR-T cell therapy. In this figure, “aAPC” refers to an artificial Antigen Presenting Cell.
| 52 |
Limitations of the Current CAR-T Cell Therapy for Cancer
Current CAR-T cell immunotherapy treatment for cancer is typically manufactured from patient’s alpha beta T cells. Using lentiviral vector, the gene of a CAR that binds to a certain surface protein on patient’s cancer cells is integrated into the genome of alpha beta T cells. Such generated CAR-T cells can then be used to treat certain cancers. The principle of CAR-T has been successfully demonstrated for treating B-cell malignancies in multiple clinical trials (Kochenderfer et al. 2015; Lee et al. 2015; Locke et al. 2017; Maude et al. 2014; Neelapu et al. 2017; Schuster et al. 2017). Such trials have led to the first two CD19-targeting CAR-T cell therapies approved by the FDA. One is tisagenlecleucel (marketed by Novartis International AG (NYSE:NVS)in the U.S. as Kymriah®) for treating acute lymphoblastic leukemia. The other is axicabtagene ciloleucel (marketed by Gilead Sciences, Inc. (Nasdaq:GILD) in the U.S. as Yescarta®)) for treating Non-Hodgkin’s lymphoma. These approved therapies target CD19, which is only expressed in normal B cells and their malignant counterparts. Such progress suggests that CD19-targeting CAR-T therapy is a breakthrough in cancer immunotherapy. However, to replicate the success of CD19-targeting CAR-T therapies in treating other types of cancers, particularly solid tumors, remains challenging. The limitations of current CAR-T strategy are manifold:
| i. | Lack of Suitable Surface Antigen for Solid Tumors |
Conceptually, the CAR-T principle is simple: grafting a CAR into T cells (typically alpha beta T cells) to redirect T cells for cancer recognition. Practically, however, the lack of suitable surface antigen limits the extrapolation of CAR-T principle into treatment of other cancers. Current CAR-T rely on the grafted CARs as the only mechanism to recognize cancer cells. The antigen targeted by CAR must be a surface antigen expressed on cancer cells but not on normal cells. The lack of surface antigens that can be safely targeted without causing clinically unmanageable side effects is a limitation in expanding CAR-T therapy beyond B-cell malignancies.
CD19, the target of the current CAR-T cell therapies is a B-cell lineage surface antigen that expresses in B-cell malignancy and normal B cells, but not other blood cell lineage. Although CD19-targeting CAR-T also kill normal B cells and cause B-cell depletion, such side effect is still clinically manageable. However, it is not easy to find a suitable surface antigen like CD19 in cancers other than for acute lymphoblastic leukemia. A limited number of other potential targets are being tested in clinical trials, such as targeting BCMA to treat multiple myeloma (Ali et al. 2016; Raje et al. 2019). In solid tumors, finding specific surface targets is even more challenging since most known tumor antigens are intracellular and thus not targetable by CAR, which targets surface antigens only.
Selecting and targeting a suitable surface antigen is of crucial importance to develop CAR-T therapy for other types of cancer. Targeting a cancer-specific antigen may provide “on-target on-cancer” therapeutic effect that outweighs its “on-target off-cancer” side effect and thus benefit the cancer patients. The delicate balance between potential therapeutic effects and unwanted side effects of the CAR-T technology is determined by the specificity of the selected cancer antigen (Figure 3).

Figure 3. Product feature of current CAR-T cell therapy.
| ii. | Targeting Single Antigen |
Currently, available CAR-T therapy can only recognize a single antigen. This is mainly due to the following technical reasons: (a) the lack of specific surface antigens in cancers; (b) the difficulties to graft multiple types of CARs in T cells using lentiviral vectors; and (c) the use of scFv of antibody as an antigen-binding domain in grafting CAR. The specificity of antibody determines that scFv-based CAR can only bind and target a single antigen. Such limitation poses a risk of cancer escape due to antigen loss, in which CAR-T cells would no longer be able recognize the cancer cells. We believe such risk has been proven in clinical studies (Jacoby et al. 2016; Yu et al. 2017).
| iii. | Complicated Manufacturing Process |
Current CAR-T manufacturing technologies pose another challenge to extend CAR-T therapy to a wide range of cancers (Levine et al. 2017). To manufacture CAR-T for B-cell malignancies, a large amount of patient’s blood cells is collected through an invasive leukapheresis procedure. Typically, such cancer patients would usually have gone through multiple lines of chemotherapy. As a result, the number and quality of blood cells especially alpha beta T cells, onto which the CARs are to be expressed, may already have been compromised. Such poor starting material makes it difficult to manufacture good quality CAR-T products. Failure in manufacturing can exclude the patients from CAR-T therapy. Moreover, lentiviral vectors are used to express the CAR in T cells and the manufacture of GMP-grade lentiviral vectors itself is a complicated process. To ensure the production of high-quality lentiviral vectors with minimal variability, several rounds of filtration and testing is required during manufacturing of a series of multiple sub-batches (Levine et al. 2017).
| 53 |
| iv. | Undesirable Product Features |
The above-mentioned technology and manufacturing limitations substantially restrict the widespread application of current CAR-T therapies and CAR-T products. There are several undesirable features of current CAR-T products:
Inaccessible: Patients may be excluded from CAR-T therapy due to the failure of blood tests and manufacturing feasibility tests. Certain patients (e.g., pediatric patients) are not eligible for CAR-T therapy due to the limited blood cells that can be collected from such patients.
Highly personalized “made-to-order” product and not “off-the-shelf”: Existing manufacturing processes of the current CAR-T products rely solely on patient’s own blood cells. Therefore, the CAR-T products are highly personalized and not “off-the-shelf”. They are not readily available if cancer patients require urgent treatment.
Safety concerns: The contamination of the starting material (patient’s blood cells) by existing cancer cells and the use of lentiviral vector to graft the CAR are not only challenging to manufacture but also pose the risk of secondary cancer, which has been described in a clinical study (Ruella et al. 2018). Due to the potential risk of insertional mutagenesis caused by lentiviral vectors, a minimum of 15-year post-treatment follow-up is required for current CAR-T products.
Expensive: It has previously been reported that Kymriah® has a one-time price of U.S.$475,000 for acute lymphoblastic leukemia (Cancer Discov, 7: OF1). The list price of Yescarta® is U.S.$373,000 (Cancer Discov, 8:5-6). These prices do not cover additional costs of hospital admission which is required in order to administer the agent and the potential treatment of side effects. Despite the clinical successes of CD19-targeting CAR-T therapies, current CAR-T therapies may be out of reach for most of the population.
Our Solutions and Product Candidates: CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC
Due to the obvious limitations of the current CAR-T technologies, we believe it is imperative to explore different cytotoxic cell sources, cancer-targeting strategies and production schemes for other types of cancers, in particular, solid tumors. To this end, we have designed and established three novel patient blood cell-independent platform technologies, in which we use healthy donor blood cells and induced pluripotent stem cells to develop and manufacture “off-the-shelf” cell-based cancer immunotherapies. Our current product candidates in the pipeline include CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC.
CTM-N2D
To produce CTM-N2D, blood cells from healthy donors are used as starting material. Through a proprietary expansion technology, high purity gamma delta T cells are obtained as effector cells. Through an mRNA electroporation technology, the expanded gamma delta T cells are further grafted with NKG2DL-targeting CAR to enhance anti-cancer cytotoxicity. Such manufacturing process is depicted in Figure 4 and this CTM-N2D technology has also been published in two peer-reviewed papers by its inventors (Cytotherapy, 2018, 20: 420; Molecular Therapy: Oncolytics, 2020, 17: 421.)
The cutting-edge features of the technology are as follows: (i) the use of healthy donor blood cells instead of patient blood cells as starting material for manufacturing to solve the product availability issue; (ii) the use of gamma delta T cells instead of alpha beta T cells as effector cells to expand the product applicability to a large population of patients; and (iii) the use of a NKG2DL-targeting CAR instead of the conventional single antigen-targeting CAR to expand the product indications to a wide range of cancer types.
Our CAR-gamma delta T cell technology is ready for Phase I clinical trial, being that we have translated the CAR-gamma delta T cell technology into a cell therapy named as CTM-N2D for further evaluation in a clinical trial, the ANGELICA Trial, with National University Hospital Singapore. We had submitted a full clinical trial dossier to HSA in April 2020. A pre-submission meeting was held with HSA in July 2020. In January 2021, HSA informed that they had reviewed our submission and, suggested that we formally submit a CTA application. CTM-N2D consists of culture-expanded cells and is considered a Class 2 CTGTP, hence, a CTA application is required for clinical trial. On March 10, 2023, we entered the Clinical Study Agreement pursuant to which the ANGELICA Trial will be conducted by the National University Hospital Singapore to evaluate the safety of CTM-N2D in human subjects, following the HSA’s confirmation on January 6, 2023 that we had satisfied the approval conditions of the CTA initially granted in July 2022, apart from continued compliance with the HPA and its subsidiary legislation, relating to the use of our CTM-N2D for the aforementioned trial by HSA. The proposed clinical trial is an open-label, single-centre, dose escalation, Phase I study. The study objectives are to determine safety, activity, and safe dosage of haploidentical or allogeneic NKG2DL-targeting CAR-gamma delta T cells, in which the proposed plan is to administer CTM-N2D in four weekly doses to patients with relapsed or refractory solid tumors of different types.
| 54 |
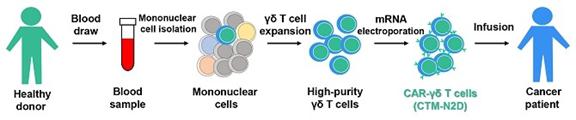
Figure 4. Manufacturing process of CAR-gamma delta T cells (CTM-N2D therapy).
iPSC-gdNKT
To exploit the cancer recognition systems of both gamma delta T cells and NK cells, we developed a proprietary induced pluripotent stem cell-derived gamma delta NKT cell platform for cancer treatment. To produce gamma delta NKT cells, iPSC lines have been derived from gamma delta T cells. The derived gamma delta T-iPSCs are used as starting material to generate gamma delta NKT cells via “NK cell-promoting” differentiation. Such a synthetic strategy to derive gamma delta NKT cells is depicted in Figure 5 and two peer-reviewed publications by the technology inventors (Stem Cell Reports, 9: 1796, 2017; PLoS ONE, 14: e0216815, 2019)

Figure 5. Synthetic strategy of iPSC-derived gamma delta NKT cells (iPSC-gdNKT therapy).
The potential novel features of the technology are as follows: (i) the use of gamma delta T-iPSCs, our proprietary cell source as starting material for manufacturing to make the production patient/donor blood cell-independent, which may ultimately solve the starting material availability issue; (ii) using a differentiation process to derive gamma delta NKT cells so that genetic modification is unnecessary, which makes manufacturing simpler and the products potentially more suitable for the patients; (iii) gamma delta NKT cells are innate lymphocytes in nature and applicable to allogeneic setting since these cells are unlikely to cause GvHD; and (iv) gamma delta NKT cells possess cancer recognition systems of gamma delta T cells and NK cells and this unique array of built-in receptors enables gamma delta NKT cells to recognize a wide spectrum of cancer types, including solid tumor and leukemia.
| 55 |
Extensive in vitro studies have demonstrated that gamma delta NKT cells are highly cytotoxic at recognizing and killing a “broad-spectrum” of cancers with great specificity (Zeng et al. 2019, a publication by the inventors of the technology). We are in the process of further evaluating gamma delta NKT cells in cancer treatment in pre-clinical studies. iPSC-gdNKT is considered a Class 2 CTGTP and hence a CTA application would be required prior to commencing clinical trial in Singapore. This product has been undergoing pre-clinical process development since fourth quarter of 2022, and is targeting to commence pre-clinical studies after 2024
CTM-GDT
Adoptive transfer of gamma delta T cells as cancer treatment has been investigated in clinical studies. Typically, a patient’s own gamma delta T cells have been first expanded in vitro using zoledronic acid and IL-2. These expanded autologous gamma delta T cells were then administrated back to the same patients in multiple doses. The clinical efficacy has been variable, which may be due to the variable quality and quantity of administrated patient-derived gamma delta T cells.
Compared with autologous gamma delta T cells expanded from patient’s own blood cells, allogeneic gamma delta T cells expanded from blood cells of healthy donors may provide a more cytotoxic cell source. Moreover, unlike autologous gamma delta T cells, the number of allogeneic gamma delta T cells that can be obtained during manufacturing is not restricted by the starting materials from the patient himself. Hence, we believe that it is valuable to further explore the potential cytotoxicity of allogeneic gamma delta T cells in clinical trials. To this end, we are also developing allogeneic gamma delta T cells into CTM-GDT therapy for cancer treatment (Figure 6). We are working with The University of Texas M.D. Anderson Cancer Center on CTM-GDT against lymphoma and breast cancer in preclinical studies. Through a US agent, we have submitted a drug master file to US FDA and intend to pursue an investigational new drug application in the near future. CTM-GDT is considered a CGTP in Malaysia according to NPRA and we intend to manufacture the investigational product locally. Therefore, a CTX application to NPRA is required prior to commencing clinical trial. We are looking to submit a CTX application for Phase I clinical trial to NPRA, Malaysia after 2023.

Figure 6. Manufacturing process of allogeneic γδ T cells (CTM-GDT therapy).
CTM-MSC
Stem cells are unspecialized cells with the capacity to regenerate and replicate as well as to develop into different types of specialized cells in the body. They are important in the human body as they are able to differentiate into specific cell types, offer the possibility of a renewable source of replacement cells and tissues to treat many diseases. Depending on the types of stem cells, different types of stem cells possess different capacity of differentiation.
We are currently researching the use of MSCs, which are multipotential adult stem cells found in the supportive framework of organs and are the building blocks for tissue renewal and repair. MSCs have the ability to differentiate into multiple cell types of mesenchymal lineage, including bone, cartilage, fat, and muscle cells as well as those of non-mesenchymal lineage depending on the environment in which they are cultured. Several studies have reported that the mechanism of MSCs in repairing tissue damage is associated with their immune-modulatory properties. One of MSCs’ vital biological functions, the immune-modulation, allows MSCs to travel to specific injury or inflammation sites in the body, and to modulate immune response to reduce inflammation. A relatable use of MSC’s tissue repair capability and immunomodulation function is demonstrated in lung tissue repair and cytokine release syndrome control in patients with severe COVID-19 disease. COVID-19 patients who have undergone MSCs treatment recovered faster and had less severe cytokine release syndrome.
It is reported that MSC therapies have been approved in several countries including Japan, South Korea and Canada. Some other specific medical conditions under global research include age-related macular degeneration, aging frailty, Alzheimer’s disease, autoimmune disease, cerebral palsy, stroke, spinal cord injury, heart disease and failure.
CytoMed is considering partnering with our associate hospital, LMC, to focus our research on umbilical cord and adipose tissue-derived MSCs as regenerative medicine. We believe that our current facility and professional research team has the capability to culture high quality MSCs for regenerative purposes.
On February 29, 2024, we entered into a research collaboration agreement with Sengkang General Hospital, a major public hospital in Singapore to advance injectable allogeneic umbilical cord derived MSC for cartilage injury. In this collaboration, we are looking to provide CTM-MSC and its conditioned media for in vivo studies and Phase I clinical trial in Singapore.
CTM-N2D Therapy
Background of CAR-gamma delta T Cell Technology
In our CAR-gamma delta T cell technology, we have used NKG2DL-targeting CAR to recognize a broad range of cancers and harnessed gamma delta T cells to carry out the anti-cancer effector functions. The rationale for using these two components is as follows:
| i. | NKG2D and NKG2DLs as Targets |
NKG2D is a naturally occurring receptor that is expressed on the cell surface of NK cells, CD8+ T cells and gamma delta T cells. NKG2D recognizes eight NKG2DLs, namely MICA/B and ULBP 1-6 (Marcus et al. 2014). These ligands are usually expressed at low levels or non-existent on the cell surface of healthy cells, but their expressions can be upregulated upon undergoing stress, infection, and malignant transformation. During tumorigenesis, DNA damage responses (such as genotoxic stress and stalled DNA replication) and poorly regulated cell proliferation may both induce NKG2DL expression on the cell surface of cancer cells. Since these biological changes are the hallmark of cancers, it is not surprising that increased expression of NKG2DLs in cancer cells has been found in patients with many different types of cancers (Table 1) (Demoulin et al. 2017). The ubiquitous expression of NKG2DLs makes them interesting targets in cancer treatment. As demonstrated by our inventors of this technology targeting NKG2DLs is achievable by exploiting the ligand-binding domain of NKG2D and integrating such domain into a NKG2DL-targeting CAR (Cytotherapy, 2018, 20: 420; Molecular Therapy: Oncolytics, 2020, 17: 421.). Using such NKG2DL-targeting CAR, it is possible to target multiple NKG2DLs and thus develop a CAR-T therapy that can be used in a wide range of cancers with lower risk of cancer escape.
| ii. | Gamma delta T Cells as Effector Cells |
As of the date of this annual report, most of the existing CAR-T therapies for cancer use alpha beta T cells as effector cells. As a result, such alpha beta T cells require the patient-specific autologous paradigm of CAR-T cell production and application (Fesnak, June, and Levine 2016; Newick et al. 2017). To manufacture CAR-T cells, patient’s own alpha beta T cells are expanded and genetically modified using lentiviral vector to express CAR. Such alpha beta T cell-based products are made strictly for autologous use only. They are not usable in an allogeneic setting for other recipients because the alpha beta T cells may recognize host’s normal cells through their TCRs and result in GvHD. Furthermore, manufacturing CAR-T is not feasible for cancer patients who suffer from profound lymphopenia due to previous chemotherapy and/or radiotherapy (Allen et al. 2017).
| 56 |

Table 1. Expression of NKG2DLs in various types of cancers (adapted from Demoulin B et al. 2017).
To make such alpha beta T cell-based CAR-T products accessible to a broad range of cancer patients and usable in an allogeneic setting, manufacturing CAR-T cells using donor alpha beta T cells while disrupting alpha beta TCR expression in alpha beta T cells using sophisticated genetic modification has been proposed as a solution to manufacture “universal” CAR-T cells (Marcus and Eshhar 2014; Torikai and Cooper 2016). However, such strategy will require additional complexities in the already complicated manufacturing process. We believe that such a strategy will be a serious challenge to the robustness, efficiency, and safety of genetic engineering technologies as well as the capability of quality control of such heavily modified products. Hence, from the viewpoint of manufacturing, we think that using other effector cells, such as gamma delta T cells, is more desirable.
Gamma delta T cells are a distinct subset of CD3+ T cells that express heterodimer of gamma delta TCR chains. In the bloodstream, approximately 1%-5% of the T cell population are gamma delta T cells which express the variable-gene segments Vγ9 and Vδ2, and thus are known as Vγ9Vδ2 T cells (Tyler et al. 2015). Biologically, gamma delta T cells differ strikingly from alpha beta T cells. Through gamma delta TCRs, gamma delta T cells recognize and interact in a non-MHC restricted fashion with various antigens, including PAgs that are produced during metabolic dysregulation in cancer cells (Tyler et al. 2015; Kabelitz 2016; Legut, Cole, and Sewell 2015). Like NK cells, gamma delta T cells also express NKG2D as a co-stimulatory receptor. Once activated, gamma delta T cells can exert cytotoxic effector functions to kill target cells by secreting cytotoxic molecules like granzyme and perforin and to activate anti-cancer immunity by secreting proinflammatory cytokines like IFN-γ and TNF (Figure 7).
| 57 |

Figure 7. Anti-cancer activities of gamma delta T cells.
Gamma delta T cells are vital in immune surveillance against cancer development. Gamma delta T cells feature an inflammatory migration profile (Brandes et al. 2003), being capable of homing to epithelial and mucosal tissues and infiltrating a broad range of human malignancies (Fournie et al. 2013). A study of approximately 18,000 human tumors across a panel of 39 tumor types has identified the presence of intra-tumoral gamma delta T cells as the most significant indicator for favorable prognosis, which is positively correlated with patient survival (Figure 8) (Gentles et al. 2015). A recent study has reported the computational identification of abundance of Vγ9Vδ2 T cells in approximately 10,000 cancer biopsies from 50 types of hematological and solid malignancies (Tosolini et al. 2017). Infiltrating Vγ9Vδ2 T cells are prominent in blood cancers (including B cell-acute lymphoblastic leukemia, acute promyelocytic leukemia (M3-AML), and chronic myeloid leukemia) and in solid tumors (including cancers of prostate, esophagus, cervix, head and neck, lung, pancreas and breast). These findings strongly indicate the therapeutic value of gamma delta T cells in cancer treatment.
| 58 |
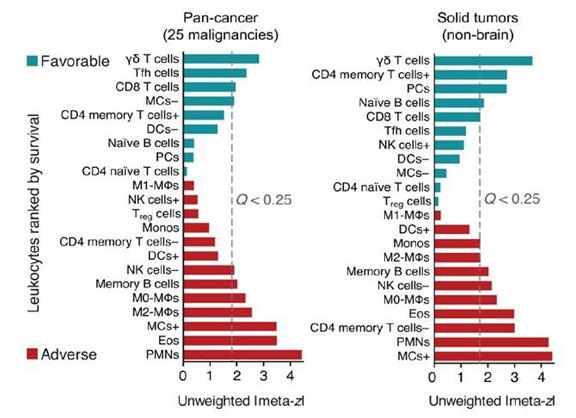
Figure 8. Intratumoral gamma delta T cell frequency is positively correlated with cancer patient survival
(adapted from Gentles AJ et al, 2015).
Adoptive transfer of Vγ9Vδ2 T cells has been used to treat cancers in several early-phase clinical trials. Such therapy was well-tolerated and linked to positive clinical outcomes (Fournie et al. 2013; Kobayashi and Tanaka 2015). GvHD was not reported in these clinical studies, suggesting that Vγ9Vδ2 T cells are a safe effector cell source for developing CAR-T therapies. In pre-clinical studies, gamma delta T cells have been redirected with CARs to target cancers. It was reported that peripheral blood Vγ9Vδ2 T cells transduced with retroviral vectors encoding either GD2 or CD19-specific CARs display antigen-specific IFN-γ secretion and cytotoxicity against cancer cells (Rischer et al. 2004). This is a demonstration that CAR-expressing gamma delta T cells may serve as potent and specific anti-cancer effector cells. Moreover, by comparing the potency of three different T-cell populations (Epstein-Barr virus-specific cytotoxic T lymphocytes (EBV-CTLs), cytokine-induced killer (CIK) cells, and Vγ9Vδ2 T cells) as effector cells after modifying them with CD33-targeting CARs, it has been found that Vγ9Vδ2 T cells are as potent as the other two population in terms of in vitro cytotoxicity (Pizzitola et al. 2011). Using polyclonal γδ T cells, it has been shown that gamma delta T cells expressing CD19-targeting CARs can reduce CD19+ leukaemia xenografts in mice (Deniger et al. 2013).
We believe the data described above strongly supports the combined use of NKG2DL-targeting CAR as cancer-recognizing receptor and gamma delta T cells (Vγ9Vδ2 T cells) as effector cells to generate CTM-N2D as a novel cancer therapy to treat a wide range of cancers. Such therapy becomes even more attractive because the built-in and grafted receptors are unlikely to recognize normal cells and cause GvHD which enables its application in allogeneic setting.
| 59 |
Product Design and Manufacturing of CTM-N2D
| i. | Product Design and Features of CTM-N2D Therapy |
The principle of CTM-N2D therapy is similar to that of the current CAR-T cell technology. However, since different components have been integrated into CTM-N2D, the key features of CTM-N2D are quite different from those of conventional CAR-T cells. In CTM-N2D, NKG2DL-targeting CAR is used to redirect the gamma delta T cells for cancer recognition (Figure 9). As such, CTM-N2D may recognize multiple stress-induced cancer antigens via the built-in receptors and the grafted NKG2DL-targeting CAR. Moreover, CTM-N2D can be produced from healthy donor blood due to the use of gamma delta T cells as effector cells. Thus, we believe CTM-N2D may be a potential solution to overcome the limitations of current CAR-T cell technology and for treating various cancers in patients.

Figure 9. Product design and features of CTM-N2D therapy.
| 60 |
| ii. | Manufacturing of CTM-N2D |
To produce CTM-N2D, blood cells from healthy donors (either haploidentical or allogeneic) will be used as starting material. Through a proprietary expansion technology, high purity gamma delta T cells can be obtained. Typical morphology of the expanded gamma delta T cells is shown in Figure 10. Through an mRNA electroporation technology, these gamma delta T cells are further grafted with a NKG2DL-targeting CAR to potentially enhance the potential anti-cancer activity. The manufacturing process of CTM-N2D is depicted in Figure 4.

Figure 10. Typical morphology of expanded gamma delta T cells.
Potential Advantages of CTM-N2D Therapy over current CAR-T products
We believe CTM-N2D has the potential to overcome the afore-mentioned limitations of current CAR-T products. The following are the unique features of CTM-N2D that we believe make it potentially feasible for treating solid tumors:
| i. | Targeting Stress-induced Cancer Antigens |
Stress-induced surface molecules are a category of cancer antigens that are under-utilized in current cancer therapy. For example, NKG2DLs are a group of stress-induced cancer antigens, which include ULBP1-6 and MICA/B (8 types of antigens) (Marcus et al. 2014). PAg is expressed on infected or transformed cells (Bonneville and Scotet 2006). PVR (CD155) and Nectin-2 (CD112) are expressed in cancer cells and their recognitions by DNAM-1 are essential for cell-mediated cytotoxicity (Pende et al. 2005; Carlsten et al. 2007; El-Sherbiny et al. 2007). Below are the unique features of these stress-induced cancer antigens:
Suitable targets: Unlike the lineage-specific antigens (e.g. CD19 of B-cell lineage) targeted by current CAR-T technology that are also expressed in normal cells (targeting these antigens may cause “on-target off-cancer” toxicity), the above-described stress-induced signals represent a restricted set of conserved endogenous surface molecules that are commonly observed in many types of cancer cells. These antigens are constantly being monitored by the innate immune cells of the body during cancer immunosurveillance and thus are potentially more suitable targets (Marcus et al. 2014).
Essential targets: Unlike lineage markers that are non-essential for cancer cells, this type of cancer antigen is induced by dysregulated mevalonate pathway, dysregulated proliferation and/or DNA-damage response, which are the hallmarks of cancer cells (Gober et al. 2003; Raulet, Marcus, and Coscoy 2017). Targeting such essential antigens may reduce the risk of cancer escape through immunoediting (Dunn, Old, and Schreiber 2004).
Ubiquitous targets: Since this type of cancer antigen is induced by common dysregulation biological processes of cancer cells, the antigens tend to be ubiquitously expressed by cancer cells. They appear in a wide range of cancers from leukemia to solid tumors. For example, it is well-studied that NKG2DLs are expressed in many tumor cell lines and primary tumors of diverse tissue-origins (Table 1) (Demoulin et al. 2017).
| 61 |
| ii. | Targeting Multiple Stress-induced Cancer Antigens |
To overcome cancer escape due to antigen loss, simultaneously targeting multiple antigens is critical. However, such a feature remains to be developed for CAR-T platforms due to the lack of suitable targets and the limited genetic payload (Lim and June 2017; Roybal and Lim 2017).
In contrast, CTM-N2D therapy is believed to be able to overcome these limitations by (a) targeting multiple stress-induced cancer antigens via the built-in receptors on gamma delta T cells, e.g., γδ TCR, NKG2D, DNAM-1; and (b) targeting eight known NKG2DLs expressed by cancer cells such as MICA/B, ULBP1-6 via the grafted NKG2DL-targeting CAR. We believe that simultaneously targeting these ubiquitous ligands not only reduces the risk of cancer escape but also broadens the spectrum of cancer recognition.
| iii. | Simpler Manufacturing Process |
CTM-N2D is based on gamma delta T cells instead of the traditional alpha beta T cells as cytotoxic effector cells. This significantly changes the production process and final product features. There is no using of leukapheresis and lentiviral vector during the production of CTM-N2D. The manufacturing process involves simple blood withdrawal, mononuclear cell isolation, specific gamma delta T cell expansion and mRNA electroporation.
| iv. | Unique Product Features |
The use of CTM-N2D technology may enable the following unique product features:
Targeting various types of cancers: CTM-N2D is armed with built-in receptors and grafted NKG2DL-targeting CAR to recognize ubiquitous cancer antigens. CTM-N2D may potentially recognize various types of cancers.
Applicable to a large diverse pool of patients: CTM-N2D is a donor gamma delta T cell-based product. Gamma delta T cells are unlikely to cause GvHD in recipients in an allogeneic setting and thus applicable in many patients. Hence, CTM-N2D can potentially be made and administered to a large diverse pool of patients.
Accessible: The production is based on healthy donor blood cells. By not relying on the patient’s own blood cells, the patient has a low risk of exclusion from CTM-N2D therapy due to production failure.
“Off-the-shelf”: Manufacturing of CTM-N2D starts with healthy donor’s blood cells. It has the potential to be “pre-made” and used in many patients. This “off-the-shelf” feature is favorable to meet the urgent medical need of cancer patients.
Potentially safe: Due to the use of healthy donor blood cells as starting materials and the use of mRNA electroporation to graft the CAR, there is a lower risk of secondary cancer caused by the product.
Pre-clinical Studies of CTM-N2D
We believe that the feasibility of CAR-grafted gamma delta T cells has been demonstrated in our previous publications (Du et al. 2016; Xiao et al. 2018; Ang et al. 2020). These publications suggest the feasibility of combining expansion of gamma delta T cells and mRNA electroporation to produce CAR-grafted gamma delta T cells on a large-scale basis. Depending on the type of targeted cancer antigen, different CAR constructs can be used to redirect the gamma delta T cells. To generate CTM-N2D, we use a CAR construct that targets NKG2DLs (Xiao et al. 2018; Ang et al. 2020).
| 62 |
CTM-N2D has been evaluated by in vitro cytotoxicity assay against cancer cell lines. As shown in Figure 11, gamma delta T cells grafted with NKG2Dz-γδT were significantly more cytotoxic than the controls, which are the gamma delta T cells subjected to electroporation using mGFP-γδ T. Although gamma delta T cells express natural NKG2D protein (NKG2Dp-γδT) and also showed better cytotoxicity than the controls, they were not as cytotoxic as NKG2Dz-γδT. This result suggests that NKG2DL-targeting CAR is a better construct in activating the cytotoxicity of gamma delta T cells.

Figure 11. In vitro cytotoxicity of CTM-N2D against cancer cell lines. (a) CTM-N2D against pCRC7, a colorectal cancer cell line; (b) CTM-N2D against SKOV3, an ovarian cancer cell line. mGFP-γδT; gamma delta T cells subjected to electroporation using mRNA encoding mGFP; NKG2Dp-γδT: gamma delta T cells express natural NKG2D protein; NKG2Dz-γδT: gamma delta T cells grafted with NKG2DL-targeting CAR, also known as CTM-N2D.
To exploit the cancer recognition capability of gamma delta T cells through gamma delta TCR, zoledronic acid has been used to upregulate the PAg expression in cancer cells and sensitize the cancer cells to gamma delta T cell-mediated cytotoxicity. As demonstrated in Figure 12, zoledronic acid (known in trade name as Zometa) sensitized the cancer cells to killing by gamma delta T cells and may further enhance the cytotoxicity of CTM-N2D.
To evaluate the cytotoxicity of CTM-N2D in vivo, a xenograft mouse model was established using human ovarian cancer cell line SKOV3 in non-obese diabetic scid gamma mice. Seven days later, 1x107 cells of CTM-N2D were given as treatment twice a week for a total of 3 weeks. Figure 13 shows how the injection of unmodified gamma delta T cells significantly reduces the cancer growth, while the injection of CTM-N2D significantly reduces the cancer burden and prolongs the survival of these cancer-grafted mice.
| 63 |

Figure 12. In vitro cytotoxicity of gamma delta T cells and CTM-N2D against Zometa-sensitized cancer cell lines. Cytotoxicity against (a) SW480, a colorectal cancer cell line; (b) HepG2, a hepatocellular carcinoma cell line; (c) U87, a glioblastoma cell line; and (d) SKOV3, an ovarian cancer cell line.
| 64 |

Figure 13. In vivo cytotoxicity of CTM-N2D against human cancer in xenograft mouse model. A human ovarian cancer cell line, SKOV3 was used to established cancer xenograft mouse model. Seven days later, CTM-N2D was given as treatment twice a week for total 3 weeks. Disease progress was monitored by IVIS® in vivo imaging system to show the (A) cancer growth; (B) cancer burden; and (C) survival. mGFP-γδT; gamma delta T cells subjected to electroporation using mRNA encoding mGFP; NKG2Dz-γδT: gamma delta T cells grafted with NKG2DL-targeting CAR, also known as CTM-N2D.
Clinical Studies Relevant to CTM-N2D Therapy
We believe the pre-clinical data described above provide a scientific basis for conducting clinical studies of CTM-N2D therapy. More importantly, we believe a substantial amount of relevant human data strongly supports the use of CTM-N2D therapy to treat solid tumors (Kobayashi et al. 2007; Bennouna et al. 2008; Abe et al. 2009; Nakajima et al. 2010; Noguchi et al. 2011; Sakamoto et al. 2011; Nicol et al. 2011; Kobayashi et al. 2011; Cui et al. 2014; Wada et al. 2014; Wilhelm et al. 2014; Aoki et al. 2017; Alnaggar et al. 2019; Xu et al. 2021). CTM-N2D is NKG2DL-targeting CAR-grafted gamma delta T cells. Adoptive transfer of autologous and allogeneic gamma delta T cells has been used to treat a wide range of cancers in clinical studies for more than a decade without causing severe adverse effects in patients. Regarding targeting NKG2DL, recent pre-clinical and clinical studies using NKG2DL-targeting CAR-alpha beta T cells also suggest that targeting NKG2DLs are well-tolerated. Below is a summary of these clinical findings relevant to CTM-N2D.
| i. | Adoptive Transfer of gamma delta T Cells as Cancer Treatment |
In humans, 1–10% of peripheral blood lymphocytes are gamma delta T cells, of which Vγ9Vδ2 T cells are the major subset. Such Vγ9Vδ2 T cells express Vγ9Vδ2 TCRs to recognize PAg on infected or transformed cells (Bonneville and Scotet 2006). In contrast to human leukocyte antigen (HLA)-dependent antigen recognition of alpha beta TCR, PAg recognition of Vγ9Vδ2 TCR is HLA-independent. Therefore, Vγ9Vδ2 T cells are unlikely to cause GvHD in an allogeneic setting (Lamb and Lopez 2005). Moreover, Vγ9Vδ2 T cells can kill cancer cells even without modification and are being used in clinical trials (Deniger, Moyes, and Cooper 2014; Hoeres et al. 2018).
In clinical trials, an ex vivo approach has been commonly used (Kobayashi et al. 2007; Bennouna et al. 2008; Abe et al. 2009; Nakajima et al. 2010; Noguchi et al. 2011; Sakamoto et al. 2011; Nicol et al. 2011; Kobayashi et al. 2011; Cui et al. 2014; Wada et al. 2014; Wilhelm et al. 2014; Aoki et al. 2017; Alnaggar et al. 2019; Xu et al. 2021). Patient’s own gamma delta T cells were first expanded in vitro using zoledronic acid and IL-2. These expanded autologous gamma delta T cells were then administrated back to the same patients in multiple doses. Such treatments were well-tolerated since no dose-limiting toxicities were observed at a dose level up to 4x109 cells per dose. However, the clinical efficacy has been variable, which may be due to the use of such autologous gamma delta T cells as monotherapy and/or the inherent poor quality of these patient-derived gamma delta T cells.
| 65 |
To study the potential of anti-cancer function of non-autologous gamma delta T cells, haploidentical or allogeneic gamma delta T cells have recently been used in clinical trials (Alnaggar et al. 2019; Wilhelm et al. 2014; Xu et al. 2021). There were no severe adverse events observed in these trials using non-autologous gamma delta T cells. Combining haploidentical gamma delta T cell infusion with lymphodepleting chemotherapy to facilitate graft survival, zoledronic acid to sensitize the cancer cells and IL-2 to support gamma delta T cell survival, improved clinical response has been achieved. Such combination therapy strategy can be extended to our clinical trials.
| ii. | Targeting NKG2DLs with CAR-alpha beta T Cells in Cancer Treatment |
In humans, NKG2DL are identified as MICA/B and ULBP1-6. The expression of NKG2DL is induced by various types of stress, including viral infection and malignant transformation (Raulet et al. 2013; Le Bert and Gasser 2014). Many tumor cell lines and primary tumors of various tissue-origin express NKG2DLs (Demoulin et al. 2017), which provide attractive targets for haematological cancers and solid tumors.
CAR construct has been developed to target NKG2DLs. Currently, NKG2DL-targeting CAR is a fusion of NKG2D protein and T-cell surface glycoprotein CD3 zeta. It has been used to further enhance the anti-cancer potency of alpha beta T cells. These NKG2DL-targeting CAR-alpha beta T cells have been demonstrated in a pre-clinical study to treat pancreatic cancer (Demoulin et al. 2017). A product named NKR-2, based on such technology is currently being developed by Celyad Oncology SA (Nasdaq:CYAD) in clinical trials according to Celyad Oncology SA (Nasdaq:CYAD)’s website. Preliminary data from the dose-escalation segment of this Celyad Oncology SA (Nasdaq:CYAD) clinical study demonstrated that NKR-2 cell was well-tolerated and no toxicity was observed in dose level of 1x109 cells. We believe that such data provides support for evaluating NKG2DL-targeting CAR in the context of gamma delta T cells.
Development Status of CTM-N2D Therapy
We have translated the CAR- gamma delta T cell technology into a trial-ready cell therapy CTM-N2D for further evaluation in a clinical trial. The proposed clinical trial currently being discussed is an open-label, single-center, dose escalation, Phase I study. The study objectives are to determine safety, activity, and safe dosage of haploidentical or allogeneic NKG2DL-targeting CAR-grafted gamma delta T cells, which will be given in four weekly doses to patients with relapsed or refractory solid tumors of different types. We have conducted a pre-submission meeting with HSA. A full clinical trial dossier including trial protocol, Investigator’s Brochure (IB) and Chemistry, Manufacturing, and Control (CMC) has been submitted and reviewed by HSA during the pre-submission meeting, which has concluded that they have no major issues with the documents. On March 10, 2023, we entered into the Clinical Study Agreement pursuant to which the ANGELICA Trial will be conducted by the National University Hospital Singapore to evaluate the safety of CTM-N2D in human subjects, following the HSA’s acknowledgement on January 6, 2023 that we had submitted the relevant documents to meet the approval conditions of the CTA initially granted in July 2022 relating to the use of our CTM-N2D for the aforementioned trial by HSA, and no further action was required. Pursuant to the Clinical Study Agreement, the National University Hospital Singapore will conduct the ANGELICA Trial using our lead product candidate CTM-N2D, with Dr. Anand D Jeyasekharan, Consultant, Department of Hematology-Oncology of National University Hospital Singapore, being the principal investigator of the trial. Unless terminated earlier, the Clinical Study Agreement shall continue until completion of the trial. National University Hospital Singapore has obtained approval from the Singapore National Healthcare Group Domain Specific Review Boards in February 2023. Pursuant to the Clinical Study Agreement, Company has agreed to (i) provide funding and supply at no cost to National University Hospital Singapore such quantities of the CTM-N2D as may be required for the ANGELICA Trial, (ii) to provide all relevant clinical information, (iii) where applicable, to provide an instruction manual/guide, product labels, storage, handling and safety instructions and advice which are required for the safe and proper use of CTM-N2D and the proper planning and conduct of the trial. In March 2023, we obtained and provided to National University Hospital Singapore a clinical trial insurance certificate required to conduct the trial in Singapore. In all publications generated from the ANGELICA Trial, both Company and National University Hospital Singapore would be granted co-authorship. All the study results shall be jointly owned by both parties. Discoveries or inventions that relate to any enhancements or modifications to the CTM-N2D arising from the conduct of the ANGELICA Trial, whether conceived or reduced to practice either solely by or jointly with National University Hospital Singapore, will be the joint property of National University Hospital Singapore and Company. Either party may terminate the Clinical Study Agreement at any time upon providing at least thirty (30) days’ prior written notice to the other party. National University Hospital Singapore may terminate the Clinical Study Agreement immediately in the event National University Hospital Singapore and/or the principal investigator believes on reasonable grounds that continuing the said trial poses an unacceptable risk to the rights, interests, safety or well-being of human subjects, or if a serious adverse event occurs which necessitates the discontinuance of the ANGELICA Trial. We anticipate recruiting our first patient in the second quarter of 2024. The development status of CTM-N2D therapy is depicted in Figure 1.
| 66 |
iPSC-gdNKT Therapy
Background of iPSC-derived gamma delta NKT Cell Technology
As of the date of this annual report, most CAR-T therapies for cancers are typically generated from patient’s own blood cells. Such products use alpha beta T cells as effector cells and are highly personalized and not suitable for other recipients. This is because in an allogeneic setting the TCR of alpha beta T cells may recognize host’s normal cells and cause GvHD. In contrast, immunotherapies basing on lymphocytes of innate immune system such gamma delta T cells and NK cells may be generated from donor blood cells. Gamma delta T cells and NK cells do not express alpha beta TCRs and are unlikely to cause GvHD under allogeneic setting. However, both patient blood cells and donor blood cells are limited in cell number and variable in quality. Identifying suitable blood donors and qualifying donated blood cells are necessary to ensure good quality as such cell sources form the starting material for manufacturing cell therapies. Using such limited and variable cell sources as starting material may pose significant challenges to scalability of production and standardization of products. Hence, an unlimited and standardized cell source is more desirable.
In 2007, Professor Yamanaka and his team of scientists from Kyoto University, Japan, discovered that human dermal fibroblast could be converted into pluripotent stem cells through the delivery of four genes using viral vector (Takahashi et al. 2007). Aptly named as the Yamanaka factors, these genes were integrated into the genome of the fibroblast and “reprogrammed” the mature fibroblast back to pluripotent status. Such pluripotent stem cells are “induced” from mature cell and thus named iPSCs. Functionally, iPSCs are capable of self-renewing to provide unlimited cell source and differentiating into any cell types of the body. Moreover, the scientific developments of iPSC technology have enabled the generation of iPSCs via non-viral methods that leave no “genetic footprint” in the genome, thus making iPSCs potentially more suitable for therapeutic applications.
Despite the short history of iPSCs, they are moving quickly into clinical trials. Autologous iPSCs have been used to generate retinal cells to treat blindness in a clinical trial in Japan in 2014 (Mandai, Kurimoto, and Takahashi 2017). There are ongoing trials that are investigating the use of iPSC-derived heart muscle cells for heart disease (Cyranoski 2018) and the use of iPSC-derived neurons for Parkinson’s disease (Takahashi 2020). Besides regenerative medicine, there is an ongoing trial using iPSC-derived mesenchymal stem cells to treat GvHD (Bloor et al. 2020). In late 2018, a U.S. based company gained FDA approval for using iPSC-derived NK cells to treat cancer (Shankar et al. 2020). We believe that human pluripotent stem cells, especially iPSCs, are emerging as a reliable and standardizable starting material to produce cell therapy products.
Our Group has the know-how to use pluripotent stem cells to generate therapeutic cells of various types, including cardiomyocytes, neurons, dendritic cells (Zeng et al. 2007; Du et al. 2010; Zeng et al. 2009; Zeng et al. 2012; Zeng and Wang 2014; Zeng, Wu, and Wang 2015). Recently, we have successfully reprogrammed peripheral blood cells into iPSCs using the nonviral episomal method. Typical morphology of such generated iPSCs is shown in Figure 14. We believe we have further demonstrated the use of iPSCs to generate NK cells and gamma delta NKT cells against cancers (Zeng et al. 2017; Zeng, Tang, and Wang 2019). Typical morphology of such iPSC-derived gamma delta NKT cells is shown in Figure 15. Details of iPSC-derived gamma delta NKT cells are described below.
| 67 |

Figure 14. Typical morphology of iPSCs generated from peripheral blood cells.
| 68 |

Figure 15. Typical morphology of iPSC-derived gamma delta NKT cells.
Product Design and Manufacturing Process of iPSC-gd NKT Cells
| i. | Product Design and Features of iPSC-gdNKT Cells |
To develop a clinically cytotoxic and commercially viable cell-based immunotherapy for cancers, both the product function and its manufacturing need to be taken into consideration.
Among the innate immune cells, gamma delta T cells and NK cells have an array of built-in receptors to recognize cancers, including solid tumors. Such a cancer recognition system possesses several unique features including proven suitability, pattern recognition, targeting essential antigens and targeting ubiquitous antigens (Figure 16). We believe that integrating such cancer recognition system of gamma delta T cells and NK cells into one product, namely gamma delta NKT cells would not only enhance the potential product cytotoxicity but also broaden the indication spectrum (Figure 17).
| 69 |

Figure 16. Target recognition system of gamma delta T cells and NK cells and its unique features.

Figure 17. Integrating cancer recognition capability of gamma delta T cells and NK cells into one product, gamma delta NKT cells.
| 70 |
| ii. | Manufacturing Process of iPSC-gdNKT Cells |
Ideally, such a product may be manufactured in large-scale from an unlimited cell source, which is not patient or donor blood cells, without relying on genetic modification. To this end, we are in the process of designing a proprietary manufacturing process to generate gamma delta NKT cells from iPSCs (Figure 18), which includes a one-time conversion of gamma delta T cells into iPSCs (“gamma delta T-iPSCs”) and using gamma delta T-iPSCs as starting material to manufacture gamma delta NKT cells.

Figure 18. Manufacture of gamma delta NKT cells from gamma delta T-iPSCs.
Successful Generation of iPSCs from gamma delta T Cells (gamma delta T-iPSCs)
Using our optimized reprogramming protocol, we believe we have successfully generated iPSCs from gamma delta T cells and confirmed their derivation from gamma delta T cells (Figure 19). Such generated gamma delta T-iPSCs will serve as unlimited starting material to generate iPSC-derived gamma delta NKT cells.
| 71 |

Figure 19. Generation and identification of γδ T-iPSCs. In this figure, “γδ T cell” refers to gamma delta T cell;
“GDTA/NF” means gamma delta T cell aggregate/ nucleofection
Successful Generation of gamma delta NKT Cells from gamma delta T-iPSCs
Using our “NK cell-promoting” differentiation protocol, we have successfully generated gamma delta NKT cells from gamma delta T-iPSCs. Such gamma delta NKT cells express cancer recognition receptors of both gamma delta T cells and NK cells (Figure 20).
Gamma delta NKT Cells Recognize and Kill a Broad-spectrum of Cancer Cells
We tested direct cytotoxicity of gamma delta NKT cells against cancer cells of different origins. These cancer cells were as various as follows: glioblastoma of brain (T98G, U-87); adenocarcinoma, ductal carcinoma and metastatic carcinoma of breast (MCF7, BT-474, MDA-MB-453); Burkitt’s lymphoma (Daudi, Raji); hepatocellular carcinoma (Hep G2); adenocarcinoma of ovary (SK-OV-3); metastatic melanoma of skin (FM-57, Malme-3M); squamous cell carcinoma of tongue (SCC-25); colorectal carcinoma and adenocarcinoma of colon (HCT 116, SW480); multiple myeloma (RPMI 8226); chronic myelogenous leukemia (K562); acute monocytic leukemia (THP-1). Results showed that gamma delta NKT cells efficiently killed these very different cancer cells even at low effector to target (E:T) ratios, suggesting that most surface receptors on gamma delta NKT cells may be involved in cancer recognition. See Figure 21.
| 72 |

Figure 20. Morphology and phenotype of gamma delta NKT cells.
| 73 |

Figure 21. Direct cytotoxicity of gamma delta NKT cells against a broad-spectrum of cancer cells.
| 74 |
Direct cytotoxicity of gamma delta NKT cells against SW480, a colorectal adenocarcinoma cell line, was observed under a live imaging microscope. A 48-hour time-lapse video showed that gamma delta NKT cells eliminated most SW480 cells within first 12 hours, which we believe further confirms the cytotoxicity of this novel type of killer cells. See Figure 22.

Figure 22. Seek, see, and destroy ‒ unravelling the action of gamma delta NKT cells frame by frame. In the above figure, the arrows indicate cancer cells, and the triangles indicate iPSC-derived gamma delta NKT cells.
Development Status of iPSC-derived gamma delta NKT Cell Technology
Extensive in vitro studies have demonstrated that iPSC-derived gamma delta NKT cells have the potential to recognize and kill a “broad-spectrum” of cancers with great specificity. To realize the potential of iPSC-derived gamma delta NKT cells in cancer treatment, we have been in the process of further evaluating iPSC-derived gamma delta NKT cells in pre-clinical studies since the fourth quarter of 2022, and is targeting to commence pre-clinical studies after 2024.
| 75 |
Property
Administration Office and Research Laboratory in Singapore
We own the leasehold interest of Singapore Laboratory situated at 1 Commonwealth Lane, #08-22, Singapore 149544 (Figure 23), which has an area of approximately 111 square meters housed in an industrial building within walking distance to Biopolis, a strategic district in Singapore designated for technology R&D and where A*STAR is located. The duration of the lease in respect of this property is 30 years commencing from March 1, 2008 and expiring on March 1, 2038, which is expected to be sufficient to complete our projects as stated in Figure 1. We are using the premises as our administration office and laboratory to conduct R&D and support research in our cGMP Facility in Malaysia, having fully fitted it with the necessary facilities and equipment to conduct research on GMP-grade iPSC and other cell type manufacture as well as cryopreservation of our CTM-N2D product for overseas export.

| 76 |

| 77 |


| 78 |


Figure 23. Our Administration Office and Laboratory in Singapore.
cGMP Facility in Malaysia
Our fully equipped, all-in-one cGMP Facility, consisting of office, R&D laboratory, quality control laboratory and cGMP facility is situated at 12 Jalan Permas 9/16, Bandar Baru Permas Jaya, 81750 Johor, Malaysia (Figure 24), and has the capacity to manufacture our cell therapy product candidates, CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC, for use in R&D and Phase I and Phase II clinical trials. We have also successfully completed similar trial production runs for CTM-GDT in the cGMP Facility.
For our CTM-N2D therapy product candidate, we have established standard operating procedures, BMR, in-process quality control tests and finished product quality control tests. Based on the manufacturing runs for CTM-N2D which were completed in 2020, we believe we have completed the validation of the manufacturing process of CTM-N2D and are ready to produce CTM-N2D for our Phase I clinical trial.
Our cGMP Facility is a three-story cluster facility situated on a plot of freehold land owned by us. The total gross floor area of the factory is approximately 8,600 square feet, of which 1,500 square feet is dedicated to our cGMP Facility comprising cleanrooms which support cGMP manufacturing of cell therapy products, a quality control laboratory, a cryostorage room, a control room and a material storage room.
| 79 |

Figure 24. Our cGMP Facility.
| 80 |
Intellectual Property
Our intellectual property rights and proprietary technology are protected through a combination of intellectual property rights protection regimes, including trade secrets and patents, confidentiality and non-disclosure obligations and contracts and contractual provisions in respect of innovations and invention assignments which clarify and address intellectual property ownership. We seek not only to protect our intellectual property rights and proprietary technology in key markets, but also to supplement our intellectual property portfolio to enhance such protection for our technological innovations and branding efforts by filing new patent and trademark applications when and where appropriate. We subscribe to a continuing intellectual property rights protection framework which includes guidelines on R&D, trade secrets, know-how, acquisition, exploitation, monitoring and enforcement of intellectual property rights.
We currently do not own any patents. As of the date of this annual report, the Company and/or its subsidiary, Puricell, have entered into three license agreements with ATPL, relating to developing, using, making, having made, manufacturing, distribute, marketing, importing, exporting, and selling our CTM-N2D, iPSC-gdNKT, CTM-GDT or future pipeline product candidates, upon obtaining regulatory approval.
The licenses include the Patent License, Know-How License, and K562 License for NK cell expansion. We and Puricell will rely on these licenses in developing products which we plan to seek and to obtain regulatory approval for and, upon receiving approval, plan to market and sell them worldwide.
Patent License
In June 2018, we entered into an exclusive, worldwide, non-sublicensable, non-transferable and revocable-for-cause license with ATPL under two pending patent applications filed under the Patent Cooperation Treaty (PCT) related to NKG2DL-targeting CAR-gamma delta T cell and iPSC-gdNKT cell technology, and any granted patent(s) and application(s) claiming common priority with and from such applications in the field of immunotherapy, including stem cell therapy, which was subsequently amended by an addendum entered into in December 2020, a second addendum entered into in September 2021, and a third addendum entered into in October 2022 (“Patent License”). Both Dr. ZENG Jieming and Dr. WANG Shu are the inventors of the CTM-N2D, CTM-GDT and iPSC-gdNKT cell technologies described in the Patent License, as well as co-founders and shareholders of the Company. The patent for the iPSC-gdNKT cell technology has been granted protection in Japan and China to ATPL until February 7, 2038, being 20 years from the filing date of February 8, 2018. The patent for NKG2DL-targeting CAR-gamma delta T cell has been granted protection in the United States, Malaysia and China to ATPL until February 7, 2038, being 20 years from the filing date of February 8, 2018.
ATPL is not obligated under the Patent License to render any technical assistance, maintenance or support services with respect to the Company. ATPL and its affiliates retained the right to use and develop the licensed patents solely for internal R&D purposes. ATPL made no warranties as to the licensed patents or that use of them would be free from infringement of third party rights.
The Company owns any new versions of, modifications, additions, improvements, upgrades, and development to the licensed technology developed by the Company (“Enhancements”)
Payments to ATPL pursuant to the Patent License include an upfront licensing fee (in the mid-five figure Singapore dollar) and royalties, subject to an annual minimum payment (in the low-five figure Singapore dollar). Royalties which are calculated based on tiered low single-digit percentage of net sales, and an annual minimum royalty, payable until the end of the term of the Patent License. To date, we have paid APTL an aggregate of S$117,900, including an upfront license fee, annual minimum royalty and the milestone payment. Under this Patent License, we are also required to pay an aggregate of S$650,000 for various milestones related to the clinical development and to meet commercialization milestones of our product candidates.
Unless terminated earlier, the term of the Patent License extends until expiration of the last of the patent rights licensed to us by ATPL. Patents issuing from these patent applications, if any, are not expected to expire before 2038. We may terminate the Patent License at any time upon providing at least thirty (30) days’ prior written notice to ATPL. ATPL may terminate the Patent License in circumstances including a breach by us remaining unremedied by us after thirty (30) days’ notice, including failure to meet development and commercialization milestones, an encumbrance taking possession or a receiver being appointed of any of our property or assets, any voluntary arrangement between us and our creditors, and/or cessation of our business, liquidation or receivership.
| 81 |
The aforementioned addendum in relation to the Patent License was entered into in respect of, amongst others, the NKG2DL-targeting CAR-gamma delta T cell and iPSC-gdNKT cell technology, which granted us the right to sublicense our license rights to our affiliates subject to certain conditions, such as ATPL being informed of any such sublicense within thirty (30) days of the execution of the same and such sublicense being on a non-exclusive basis only. Royalty payments charged under the sublicenses shall not be lower than the royalty payable by us to ATPL under the Patent License.
The aforementioned second addendum extends the deadlines to meet the commercialization milestones in exchange for CytoMed agreeing to take over responsibility for all patent prosecution and maintenance costs beginning January 2022 and includes the correct application number for one of the two licensed PCT patent application in relation to the NKG2DL-targeting CAR-gamma delta T cell.
Pursuant to the aforementioned third addendum, the four milestones have been extended from the second addendum as follows: commencement of Phase 1 clinical trial for fifteen (15) months; commencement of Phase 2 clinical trial for forty-five (45) months; commencement of Phase 3 clinical trial for fifty-seven (57) months; and commercial sale of the Licensed Product for sixty-nine (69) months.
Two Patent Families
CAR-gdT
Four patent grants and 4 pending patent applications made by A*STAR are related to our CTM-N2D and CTM-GDT cells technologies, and include manufacturing process, treatment and compositions of matter claims. The issued patent grants are the European, the United States, China and Malaysia patents and are licensed from ATPL. The pending patent applications include applications in Singapore, United Kingdom, Germany and France. Of these issued and pending patent applications, none are owned or co-owned by us. The expiration date of the issued patent and pending patent applications are approximately 2038.
iPSC-gdNKT
Two patent grants and 6 pending patent applications made by A*STAR are related to our iPSC-gdNKT cell technology, and include manufacturing process, treatment and compositions of matter claims. The issued patent grants are the Japan and China patents and are licensed from ATPL. The pending patent applications include applications in the United States, Malaysia, Singapore, United Kingdom, Germany and France. Of these issued and pending patent applications, none are owned or co-owned by us. The expiration date of the issued patent and pending patent applications are approximately 2038.
Know-How License
In October 2020, Puricell entered into a license agreement with ATPL, under which ATPL granted Puricell and its affiliates a worldwide, fee bearing, royalty bearing, non-exclusive, non-sublicensable, non-transferable and revocable-for-cause license to use ATPL’s GMP-compatible reprogramming of human blood cells know-how for research, development, and manufacturing of GMP-grade human iPSC derived from the licensed technology and to use, make, have made, manufacture, distribute, market, import, export, sell and have sold GMP-grade human iPSC which incorporate the licensed know-how or which cannot be developed, manufactured, used, sold, performed or provided without infringing ATPL’s rights in the licensed know-how (“Know-How License”). Payments to ATPL pursuant to the Know-How License include a nominal upfront license fee and royalties which are calculated based on a percentage ranging from the low-single digit to mid-single digit of net revenue up to an agreed maximum amount and an annual minimum royalty (in the low-four figure Singapore dollar). To date, we have paid ATPL an aggregate of S$8,570, including an upfront license fee and annual minimum royalty. The last royalty payment will be on December 15, 2030. The term of the Know-How License is ten (10) years, unless terminated earlier.
ATPL made no warranties concerning the licensed know-how, including that use of the licensed know-how would be free from third party infringement claims.
The Know-How License does not prejudice ATPL and its affiliates from further developing, using, commercializing, and licensing the licensed technology. Except as provided in the Know-How License, Puricell shall not adapt, translate, alter, copy reproduce, deal in, reverse engineer, decompile, disassemble, or create any derivative works based in whole or part on the licensed know-how.
| 82 |
Puricell is required to indemnify ATPL and its affiliates, officers, employees, and scientists against any loss, damages, costs, expenses or other claim for compensation which arises out or in connection with the Know-How License or in any way related to the licensed know-how, except to the extent caused by ATPL’s negligence or willful misconduct.
Puricell is entitled to terminate on provision of at least thirty (30) days’ written notice. ATPL may terminate for a breach by Puricell that remains uncured after 30 days’ notice, an encumbrance takes possession of Puricell or a receiver is appointed of any Puricell property or assets, Puricell makes a voluntary arrangement with its creditors, Puricell goes into liquidation, except for purposes of amalgamation or reconstruction and the resulting licensee agrees to be bound by or assume the license, or Puricell ceases or threatens to cease to carry on its business.
The Know-How License is not to be assigned without ATPL’s prior written consent, but there is a novation provision that permits a new entity that takes over the whole or substantially the whole of ATPL’s operations to take on ATPL’s rights and obligations.
K562 Cell License for NK cell expansion
In December 2020, ATPL granted us and our affiliates, a non-exclusive, royalty-free, fee bearing, non-transferable, non-sublicensable, and revocable-for-cause license to use modified human K562 cells in the territories of Singapore and Malaysia for internal academic, research or investigation only and include the results of such uses in marketing materials, such uses which are important for the development of our future product candidates. Unless terminated earlier, the license has a five (5) year term which may be renewed by mutual agreement on similar terms. To date, we have paid a nominal upfront license fee of S$3,210 to ATPL pursuant to the K562 Cell License for NK cell expansion.
ATPL retained the right to use all products and/or services the Company creates which contain or incorporate the modified human K562 cells. Any new intellectual property generated by ATPL is owned by ATPL and its affiliates. ATPL also retained the rights of ATPL and its affiliates to use, license, or otherwise commercialize modified human K562 cells.
The K562 Cell License for NK cell expansion includes use restrictions. The licensed product, or any new intellectual property ATPL generates, is owned by ATPL and the Company does not acquire any rights in it. Except for the Company’s affiliates, the cell line shall not be transferred or sublicensed without the express written permission of ATPL.
The K562 Cell License for NK cell expansion does not preclude the Company from commercializing products or services developed by the Company and its affiliates, but only to the extent that any such commercial products shall not incorporate any part of the licensed product or otherwise infringe ATPL intellectual property rights. ATPL acknowledged that the Company and its affiliates’ customers may use commercial products without a sublicense and without making additional payments to ATPL. The Company also agreed not to file any patent or other application claiming rights over the licensed product or the subject of the licensed products, but ATPL agreed that the results of research that the Company solely conducts using the licensed product will be owned by the Company. The Company also must comply with all applicable laws, rules, and regulations that may be required to accept delivery of the K562 cells.
ATPL made no warranties concerning the Licensed Products or K562 cells, including that their use would be free from third party infringement claims. The K562 Cell License for NK cell expansion required the Company to acknowledge that the K562 cells or modifications licensed under the K562 Cell License for NK cell expansion are or may be the subject of a patent application not licensed under the K562 Cell License for NK cell expansion, and that nothing in the K562 Cell License for NK cell expansion grants any implied or express license or right under any patents, know-how, trade secrets, or proprietary rights to the licensed products or any related process for profit-making or commercial purposes.
ATPL shall have the right to prosecute and defend any and all infringements and all benefits obtained therefrom belong to ATPL.
The Company is required to indemnify ATPL and its affiliates, officers, employees, and scientists against any loss, damages, costs, expenses or other claim for compensation which arises out or in connection with the K562 Cell License for NK cell expansion or in any way related to the licensed products or modified human K562 cells, except to the extent caused by ATPL’s negligence or willful misconduct.
| 83 |
Unless earlier terminated, the K562 Cell License for NK cell expansion has a five (5) year term extendable on request.
The Company is entitled to terminate the K562 Cell License for NK cell expansion on provision of at least two (2) months’ written notice. ATPL may terminate for a Company breach that remains uncured after thirty (30) days’ notice, an encumbrance takes possession, or a receiver is appointed to the property or assets of the Company, the Company goes into liquidation, except for purposes of amalgamation or reconstruction and the resulting Licensee agrees to be bound by or assume the K562 Cell License for NK cell expansion, or the Company ceases to carry on its business.
The K562 Cell License for NK cell expansion is not to be assigned without ATPL’s prior written consent, but there is a novation provision that permits a new entity that takes over the whole or substantially the whole of ATPL’s operations to take on ATPL’s rights and obligations.
Kyoto University Technology
We are dedicated to developing cutting-edge induced pluripotent stem cell technology as a source material for allogeneic cancer immunotherapy, and understand we may need a license to intellectual proper rights of Kyoto University. We have notified Kyoto University of our research progress, understand that a license is available, but understand that we do not need to enter into a licensing agreement with Kyoto University until we have begun clinical trials. A license is not yet required for our current stage of R&D.
Other intellectual property protection
We also rely on trade secrets, know-how, continuing technological innovation and our confidential information to develop and maintain our proprietary position and protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and others who may have access to proprietary information, under which they are bound to assign to us inventions made during the term of their employment or term of service.
Trade Secrets
Trade secrets may be afforded some measure of protection by a number of agreements amongst contracting member states and legislation in certain countries, of which remedies available consequent of a breach varies from destruction of products manufactured as a result of illegally obtained proprietary information, compulsory and punitive damages, injunctive relief, provisional relief to prevent infringement and preserve evidence to criminal sanctions. As we rely on trade secrets to protect our proprietary information, know- how and technologies and appreciate that the value of our trade secrets lies in its secrecy, we have and will continuously and consistently explore and take proactive steps to safeguard our trade secrets, relying in part on confidentiality agreements with our employees, and third parties and/or refrain from conducting our business where such protection is wanting.
However, these agreements may be breached, and we may not have adequate remedies for such breaches. In addition, our trade secrets may otherwise be acquired or independently discovered by competitors. To the extent that our contractors, commercial partners, collaborators, employees, and consultants use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. For more information, please refer to the section titled “Risk Factors—Risks Related to Our Intellectual Property”.
We also seek to preserve the integrity and confidentiality of our data and trade secrets by ensuring constant physical security for our premises, physical and electronic security of our information technology systems and maintaining back-ups of our information.
| 84 |
Trademarks
We also seek to protect our brand through trademark rights. As of the date of this annual report, “CytoMed Therapeutics” and our logo has been registered in Singapore on June 21, 2021 and will be due for renewal on June 21, 2031. Our trademark registration application in Malaysia has been registered on June 24, 2021 and will be due for renewal on June 24, 2031. On March 25, 2024, we filed a trademark registration application in People’s Republic of China which the examination process is currently underway.
Domain Names
As of the date of this annual report, the Company owns the domain name “Cytomed.sg”, which was registered in 2018 and is due for renewal in 2026.
Competition
The field of immunotherapy for treatment of cancers is characterized by intense and dynamic competition to develop new technologies and proprietary therapies. Any product candidates that we are in the process of developing will have to compete with existing therapies and new therapies that may from time to time become available in the future. We face competition from various sources, including well-funded biopharmaceutical companies, established biopharmaceutical companies, as well as public and private research institutions. The areas of competition include relevant scientific and management human resources, funding for product development, establishing clinical study sites and clinical subjects participating in the trials.
Our known biopharmaceutical competitors working on allogeneic CAR-T therapies currently include Allogene Therapeutics, Inc. (Nasdaq:ALLO), Astellas Pharma Inc. (TSE:4503.T), Bristol-Myers Squibb (NYSE:BMY), Celyad Oncology SA (Nasdaq:CYAD), Fate Therapeutics, Inc. (Nasdaq:FATE), Gilead Sciences, Inc. (Nasdaq:GILD), NantKwest, INC. (Nasdaq:NK), Novartis International AG (NYSE:NVS), Surface Oncology, Inc. (Nasdaq:SURF), Takeda Pharmaceutical Company Limited (NYSE:TAK) and numerous other biopharmaceutical companies.
We are aware of the recent developments of our potential competitors for allogeneic CAR gamma delta T- cell therapy. Based on publicly disclosed information, in May 2023 Acepodia Biotech, Inc. announced first patient dosed in their phase 1 clinical trial of an anti-CD20 armed allogeneic gamma delta T-cell therapy to treat patients with Non-Hodgkin’s Lymphoma, as well as FDA Clearance of Investigational New Drug (IND) Application for a First-in-Class Allogeneic Anti-EGFR Cell Therapy. In first quarter 2024, Bristol Myers Squibb (NYSE: BMY) announced U.S. FDA approval for CD19-directed CAR T cell therapy as the first and only CAR T cell therapy for adults with relapsed or refractory Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL), as well as Priority Review for Follicular Lymphoma (FL) and Relapsed or Refractory Mantle Cell Lymphoma (MCL). In October 2023, Immatics N.V. (NASDAQ: IMTX) received FDA Regenerative Medicine Advanced Therapy (RMAT) designation for a TCR-T cell therapy targeting PRAME, a protein frequently expressed in a large variety of solid tumors, with current ongoing discussions with FDA to enter registration-enabling Phase 2 trial in melanoma in 2024. In December 2023, Adicet Bio, Inc. (Nasdaq: ACET) reported robust dose-dependent expansion and persistence for their gamma-delta T-cell therapy through their ongoing clinical phase I trial for advanced lymphoma, displaying a strong exposure profile and was positively associated with both PD correlates and clinical response.
Our existing competitors in the field of gamma delta T cell therapy are listed in Table 2 of the section entitled “Competition” in this annual report. We are aware of the recent development of our potential competitor for allogeneic gamma delta T- cell therapy. Based on publicly disclosed information, in November 2023, TC BioPharm (Holdings) PLC (Nasdaq: TCBP) announced FDA Clearance of a Phase 1B IND for its allogeneic unmodified gamma delta T-cell product in patients with relapsed/refractory acute myeloid leukemia. As of date of this annual report, we are not aware of any approved Phase III trial for gamma delta T cell-based cancer immunotherapy. Our gamma delta T cell product candidate may also face competition from other cell-based immunotherapy approaches derived from NK cells and T cells.
Our existing competitors in the field of iPSC-derived immune cells include Fate Therapeutics, Inc. (Nasdaq:FATE), Takeda-CiRA joint program (Takeda Pharmaceutical Company Limited (NYSE:TAK), Centre for iPS Cell Research and Application, Kyoto University) and Century Therapeutics, Inc. (Nasdaq:IPSC). In January 2024, Fate Therapeutics has announced initiation of a phase I clinical trial for an iPSC-derived CAR T-cell product targeting human epidermal growth factor receptor 2 (HER2). As of the date of this annual report, Takeda-CiRA joint program is in its pre-clinical stage.
| 85 |
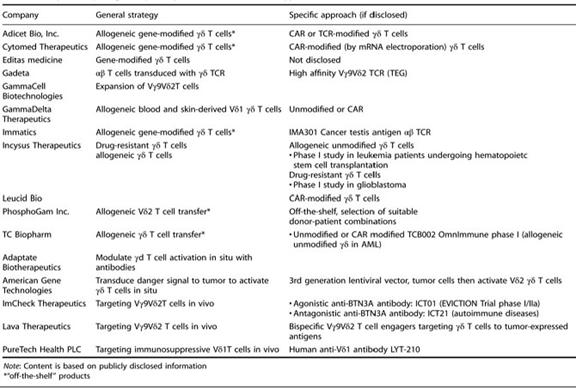
Table 2. Companies exploring γδ T cell-based immunotherapy (adapted from Kabeliz D et al, 2020).
Many of our existing or potential competitors may have greater financial and other resources, larger teams of R&D staff, and more experienced capabilities in researching, developing and testing products than we do. Many of these companies also have more experience in conducting clinical trials, obtaining relevant regulatory approvals, and manufacturing, marketing and distributing therapeutic products. Smaller or pre-clinical stage companies like us may successfully compete by establishing collaborative relationships with larger pharmaceutical companies or academic institutions. Accordingly, our competitors may be more successful than us in obtaining approval for treatments and achieving widespread market acceptance. Our competitors’ treatments may be more clinically effective, or more effectively marketed, than any treatment we may commercialize, and they may render our treatments obsolete or non-competitive before we can recover the expenses of developing and commercializing any of our treatments.
Mergers and acquisitions in the industry may result in even greater resources being concentrated in a smaller number of our competitors. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical study sites and subjects for clinical studies. Other small or early-stage companies may also prove to be strong competitors, particularly those with collaborative arrangements with large and established companies.
We anticipate that we will face intense and increasing competition as new therapies enter the market and advanced technologies become available from time to time. We expect that any treatments which we develop and commercialize will need to compete on, among other things, efficacy, safety, convenience of administration and delivery, price, the level of generic competition and the availability of reimbursement from government and other third-party payors.
Our ability to commercialize our proprietary cell products could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have a better safety profile, are more convenient or are less expensive than our products. Our competitors also may obtain relevant regulatory approvals for their products more rapidly than we may be able to obtain approval for ours, which could result in our competitors obtaining a head start and establishing a frontrunner position before we are ready to commercialize. If we are not able to compete effectively against our existing and potential competitors, our business, financial condition, results of operations and growth prospects may be materially and adversely affected.
| 86 |
Prospects, Business Strategies and Future Plans
Industry Overview and Prospects
Immunotherapy is gaining recognition and acceptance as an alternative therapy to treatment of cancer, in particular, in cancer patients who do not respond well to conventional cancer treatment such as surgery, chemotherapy or radiation.
CAR-T cell therapy as a form of immunotherapy
Immunotherapy harnesses the immune system’s ability to fight cancer. CAR-T cell therapy is a form of immunotherapy which uses specially altered T-cells to fight cancer. Cancer patients have naturally occurring T-cells which are often capable of targeting cancer cells. These T-cells are a powerful form of immune cells in our human body, and come in several types. The NKT-cells, in particular, are capable of recognizing and eliminating cancer cells in a precise manner. However, the existence of these T-cells alone is not sufficient to eliminate all cancer cells. A potential roadblock is that these T-cells must first become activated before they can effectively eliminate cancer cells, and they must be able to maintain that activity for a sufficiently long period of time in order to sustain an effective anti-cancer response. Another limitation is that these T-cells might not exist in sufficient quantity.
CAR-T cell therapy market
Since the approval of the first CAR-T cell therapeutic treatment in 2017 by the FDA, there has been widespread research and an exponential increase in clinical trial activity in the field of immunotherapy. Between 2017 and 2022, there have been at least 6 CAR-T products that have reached the market, in which the earliest approvals, Kymriah® and Yescarta®, have been commercially available since 2018, and have been infused into many patients worldwide.
However, all approved CAR-T cell therapies thus far take an autologous treatment approach. Autologous CAR-T cells are expensive to produce because they are manufactured on a patient-by-patient basis, and can be hampered by a shortage of CAR-T cells or viral vectors. Hence, current CAR-T cell therapies are usually recommended for end-stage cancer patients who have exhausted all conventional treatment options.
CAR-T cell therapy has been a game-changer in the cancer treatment industry, creating hope that it could usher in a new era of cancer treatment. However, the success stories have typically come from targeting CD19, which is now considered an antigen that holds the key to a limited range of blood cancers. CAR-T cell therapy treatment for other types of cancers, in particular, solid tumors, has been severely limited so far.
Cell Therapy in Singapore and Malaysia
The cell therapy sector in Singapore and Malaysia is at a nascent stage, with only a handful of biopharmaceutical companies within the cellular immunotherapy space in the treatment of cancer. Singapore approved the first CAR-T therapy for cancer treatment in March 2021, Kymriah® by Novartis (Singapore) Pte Ltd, which is the first commercially approved CAR-T therapy in Singapore currently in use for certain specific forms of B-cell malignancies. Yescarta is the second commercially approved CAR-T therapy for a similar indication. SpheChon is the third approved cell therapy, for degenerative disease, particularly for joints. Malaysia has not yet approved any CAR-T therapy for treatment of cancer for commercial use.
Singapore established ACTRIS in April 2020 to meet the potential demand of cell therapy process development and product manufacturing to enable clinical utility. ACTRIS is the national center for facilitating discovery, process development and manufacturing of cellular-based therapeutics in immunotherapy and regenerative medicine. ACTRIS works with other government agencies such as A*STAR and industry players such as hospitals, universities, research organizations and clinicians. ACTRIS is a business unit under CRIS, a wholly-owned subsidiary of the Ministry of Health Singapore.
We intend to capitalize on our close connections with A*STAR to tap into the latest developments and advances in cell-based therapeutics in the field of cancer treatment.
| 87 |
Business Strategies and Future Plans
CytoMed is a pre-clinical biopharmaceutical company and aims to establish itself as one of the leading biopharmaceutical companies in Asia engaged in the immunotherapy and degenerative diseases R&D, with the goal of providing affordable cell-based cancer immunotherapy for unmet needs and MSC for treatment of degenerative diseases. We are headquartered in Singapore with our production facilities in Malaysia for cost efficiency.
We intend to focus on developing our two (2) “off-the-shelf, allogeneic” technology platforms centered on gamma delta T cells and iPSCs respectively for cell based cancer immunotherapy, and MSC for degenerative diseases. Please refer to the “Business” section for further details.
Our aim is to be amongst the pioneers of cellular immunotherapy treatment for cancer to serve the emerging economies within South-east Asia. We aim to strategically position ourselves as a bridge between the cutting-edge of cellular therapy in the U.S. and our origin amongst emerging economies in South-east Asia, with our headquarter located in Singapore. We believe that the U.S. has the depth and breadth of expertise and experience to accelerate our R&D efforts in cellular therapy. We aim to establish our profile with like-minded biopharmaceutical companies, researchers, scientists and sources of investment capital within the U.S and Asia.
We have embarked on our China strategy via a collaborative effort on using our CTM-GDT for cancer immunotherapy. We intend to conduct an investigator initiated clinical trial through local partners in Chongqing, one of the most populous regions in China.
In addition, we anticipate that cellular science will advance and gain more mainstream acceptance in the future. We are considering conducting research in regenerative medicine and in particular, stem cell application in an aging population in Asia. We believe we are well-placed with the necessary expertise, experience and resources to take advantage of such prospects.
Close connections with scientific and medical community
As described in the section entitled “Management”, we have industry advisors on our Scientific Advisory Board and our key executive officers have the relevant specialized technical and medical expertise in cellular therapy.
Our core technologies are licensed from A*STAR, Singapore’s lead public sector R&D agency. By working closely with research organizations and government bodies, we are continually kept abreast of the latest developments in the field of cellular therapy. We believe that our strategic positioning, experience and expertise will give us an edge to take advantage of the growing and developing cellular therapy market.
In addition, we also seek collaboration, joint-ventures and out-licensing rights to jointly develop our two novel “off-the-shelf” technology platforms using our infrastructure in Singapore and Malaysia with industry players so as to speed up commercialization of our product candidates. In 2021, through our wholly-owned subsidiary, Advance Cancer Centre Pte Ltd, we acquired a 20% equity stake in LMC which operates a licensed private hospital in Malaysia with an Oncology & Hematology Department. LMC is a licensed private hospital offering obstetrics and gynecological services, minimally invasive surgery, cancer treatment and care, plastic and reconstructive surgery, orthopedics and clinical research. In 2019, LMC received approval from the National Committee of Clinical Research of Malaysia to set up an independent ethics committee for the purpose of facilitating ethical review and approval of clinical trials on-site. We intend to work with LMC to conduct clinical trials.
In addition, we have acquired the premises at 1 Commonwealth Lane, #08-22, Singapore 149544. Please refer to the section titled “Administration Office and Research Laboratory in Singapore” below for more information. We use the premises, which is fitted with relevant equipment to perform research on GMP-grade iPSC and other cell type manufacture as well as cryopreservation of our CTM-N2D product for overseas delivery, as our administrative office and research laboratory.
We may also explore setting up a joint laboratory in the U.S. depending on the extent of our collaboration with suitable partners there.
Lower business and operating costs in South-east Asia
The relatively lower business and operating costs in South-east Asia should position us well to achieve our vision of developing our donor blood cell-based and iPSC-based platform technologies into off-the-shelf cancer immunotherapies with the ultimate goal to offer lower cost cell therapies as a form of cancer treatment to patients.
| 88 |
We have invested in and completed construction of our own cGMP Facility which is fully equipped with state-of-the-art equipment, which we believe is capable of handling the manufacturing for Phase I and Phase II clinical trials for our clinically-ready CAR T therapy, which is trade-named “CTM-N2D”. Our cGMP Facility has been audited by the NPRA of the Ministry of Health Malaysia, which has determined that our cGMP Facility has a satisfactory level of compliance. We believe our proprietary product candidate, CTM-N2D, is clinically ready to commence clinical trials while our iPSC-gdNKT platform is at the final development stage for clinical trials as of the date of this annual report. On March 10, 2023, we entered into the Clinical Study Agreement pursuant to which the ANGELICA Trial will be conducted by the National University Hospital Singapore to evaluate the safety of CTM-N2D in human subjects, following the HSA’s acknowledgement on January 6, 2023 that we had submitted the relevant documents to meet the approval conditions of the CTA initially granted in July 2022 relating to the use of our CTM-N2D for the aforementioned trial by HSA, and no further action was required. We have already recruited a competent team of scientists and technicians and have provided them with the necessary training to manufacture the investigational product for clinical trial. We anticipate recruiting our first patient in the second quarter of 2024. Our iPSC-gdNKT platform is in the pre-clinical development stage as of the date of this annual report.
REGULATION
Cell-based therapies are still considered novel and a new medical science in South-east Asia. The CTGTP Regulations in Singapore became effective on March 1, 2021. Malaysia’s CGTP regulations became effective on January 1, 2021. We expect that such regulations will be amended as more experience and expertise are gained by both regulators, clinicians and scientific community. Although future regulations may hinder or be detrimental to cell-based therapies, the medical community generally considers that such cellular “living” medicine has the potential to enhance patient treatment modalities for many diseases. Reflecting this, Singapore set up a national agency in April 20, 2020 known as ACTRIS comprising, inter alia, all the national universities, hospitals, research institutions and funding agencies in Singapore.
| 89 |
Singapore
It is intended that the Singapore Laboratory will be used, amongst others, to conduct research in Singapore. On March 10, 2023, we entered into the Clinical Study Agreement pursuant to which the ANGELICA Trial will be conducted by the National University Hospital Singapore to evaluate the safety of CTM-N2D in human subjects, following the HSA’s acknowledgement on January 6, 2023 that we had submitted the relevant documents to meet the approval conditions of the CTA initially granted in July 2022 relating to the use of our CTM-N2D for the aforementioned trial by HSA, and no further action was required. In July 2023, we have started the clinical trial, with the donor recruitment to obtain healthy donor PBMCs which would serve as starting material for manufacturing of CTM-GDT for patients in the trial.
Being a company incorporated in Singapore, we are subject to all relevant laws and regulations of Singapore and may be affected by new laws, regulations and policies which are introduced by the Singapore government from time to time. We have identified the main laws and regulations (apart from those pertaining to general business requirements) that we anticipate may materially affect our operations, the relevant regulatory bodies and the licenses, permits and approvals typically required for the conduct of our business in Singapore.
The following description is a summary of material laws and regulations applicable to our operations. The laws and regulations set out below are not exhaustive and are only intended to provide general information to investors and are neither designed nor intended to be a substitute for professional advice. Prospective investors should consult their own advisers regarding the implication of the relevant laws and regulations on us.
Overview
In Singapore, the health products industry is regulated by the HSA and operates under the oversight of the MOH Singapore. The HSA administers health-related laws and regulations and regulates the health products sector, ensuring that drugs, innovative therapeutics and other health-related products are regulated and meet safety, quality and efficacy standards. Our registered medical professionals are also regulated by the SMC under legislation and regulations including the Medical Registration Act 1997, the Medical Registration Regulations 2010 and the relevant guidelines in Singapore.
Apart from the laws and regulations mentioned below for which HSA or MOH Singapore is the regulatory authority, compliance with the applicable circulars, directives and guidance released by HSA or MOH Singapore from time to time must be ensured as well.
Our business generally falls under the regulatory framework consisting of the HPA, HBRA, and the partially in-force HCSA, with subsidiary legislation and guidelines as promulgated by HSA, MOH Singapore and the SMC.
In particular, human biomedical research is regulated by the HBRA and its sub-legislation, namely, the Human Biomedical Research Regulations 2017 and the Human Biomedical Research (Restricted Research) Regulations 2017. Legislation is supplemented by the Ethics Guidelines for Human Biomedical Research released by the Bioethics Advisory Committee of Singapore.
Apart from the above, it is also important to note privacy issues that may arise out of research or clinical trials are regulated under the PDPA Singapore, if not already covered by subsidiary legislation of the main legislation set out in this section.
HSA
The HSA, established by the HSA Act, operates under the oversight of MOH Singapore. The general functions, objects and duties of the HSA as stated in the HSA Act, include in particular:
| (i) | to regulate the manufacture, import, export, sale, supply, advertisement and use of health products, tobacco products, radioactive materials and irradiating apparatuses in accordance with the applicable written laws; |
| (ii) | to conduct technological assessments of health products for the purpose of determining their quality, safety, efficacy and suitability for consumption and use in Singapore and to advise the Singapore government thereon; |
| (iii) | to provide professional, investigative, analytical and other services in health sciences and chemical metrology (relating to human health) to the Singapore government and to any other person or body (whether in Singapore or elsewhere); and |
| (iv) | to conduct or engage any other person to conduct research in health sciences, and generally to promote the development of health sciences. |
| 90 |
MA
Pharmaceutical products (also known as chemical or biological drugs) were previously regulated under the MA and Poisons Act 1938 of Singapore. They are now regulated as “therapeutic products” under the HPA and the Health Products (Therapeutic Products) Regulations 2016, along with other health products (medical devices and cosmetic products). This is part of the HSA’s initiative to transfer regulatory control of all health products from the MA to the HPA. We will also be required to ensure compliance with circulars, directives and guidance relating to “therapeutic products”, including the Guidance on Therapeutic Product Registration in Singapore released by the HSA in August 2021, where applicable.
Cell, tissue and gene therapy products, in which our business is involved, are classified as a new category of health product under the HPA. This is further elaborated on below.
Import, supply, presentation and advertisement of cell, tissue or gene therapy products
HPA
The HPA, amongst others, regulates the manufacture, import, supply, presentation and advertisement of health products. It was amended by the HPA Order 2021 on March 1, 2021 to introduce a new category of health products, namely, CTGTP in the First Schedule to the HPA. The HPA therefore provides the legislative basis for regulating the manufacture, import, supply, presentation, and advertisement of CTGTP.
Prior to this, regulatory controls for CTGTPs were provided for in the MA.
Our research has been conducted in our laboratories in Malaysia and we are preparing to conduct trials in Singapore. While we intend to manufacture any approved product candidates in Malaysia, we may potentially conduct research, trials, presentations, advertisements, and treatments in Singapore. As our CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC cell technologies may be considered as CTGTP, we are therefore mindful of the restrictions under the HPA and its regulations that could require us to obtain approvals and/or licensing in Singapore.
CTGTP Regulations
The CTGTP Regulations are given effect under the HPA, and came into operation on March 1, 2021. They provide for the regulations relating to the manufacture, import, supply, presentation, registration, duties, and obligations of manufacturers, importers and other persons carrying out such activities.
In summary, CTGTPs are health products intended for use in humans for therapeutic, preventive, palliative, or diagnostic purposes. CTGTP can contain any of the following and achieves its primary intended action by pharmacological, immunological, physiological, metabolic or physical means:
| (i) | viable or non-viable human cells or tissues; | |
| (ii) | viable animal cells or tissues; or | |
| (iii) | recombinant nucleic acids. |
Regulation 2 of the CTGTP Regulations states that CTGTPs are risk-stratified into two classes:
| (i) | Class 1 CTGTPs are deemed lower risk and satisfy all of the following criteria: |
| (a) | minimally manipulated; | |
| (b) | intended for homologous use (performing same function and administered at the same anatomical site or histological environment in the recipient as in the donor); and | |
| (c) | not combined or used in conjunction with therapeutic products or medical devices as defined in the First Schedule to the HPA. |
| 91 |
Examples of Class 1 CTGTPs include bone grafts, amniotic membrane, and skin.
| (ii) | Class 2 CTGTPs are deemed higher risk, and are CTGTPs other than Class 1 CTGTPs. Examples include gene modified cells, cells grown on scaffold, culture expanded cells, vectors with therapeutic gene, and xeno-based products. |
The level of control over companies dealing in the activities of manufacture, import and supply (“dealers”) is calibrated to the degree of manipulation of the CTGTP.
| (i) | The CTGTP Regulations provide that CTGTPs that are minimally manipulated are subject to lower controls. |
Dealers dealing in only minimally manipulated CTGTPs are not subject to licensing requirements and need only provide a written notification to HSA prior to the start of the manufacturing, importing or wholesaling activity (“dealer’s notice”). Upon successful submission of a notice, the dealer will be considered a known importer, known wholesaler, or known manufacturer (“known dealer”). Failure to submit a notice is an offence under the CTGTP Regulations. Renewal of a notice is not required, but the dealer must submit an update of the dealer’s notice whenever there are any changes to the information which was provided earlier.
However, a dealer which already has a license for (ii) below does not need to submit a notice for import, wholesale, or manufacture of minimally manipulated CTGTP.
| (ii) | CTGTPs that are more than minimally manipulated require a license. |
Known dealers are required to comply with the respective duties and obligations under the CTGTP Regulations, including to:
| a. | maintain records of manufacture; | |
| b. | maintain records of receipt and supply; | |
| c. | maintain systems of traceability of the CTGTP and its starting and raw materials, including any substances that come into contact with the cells and tissue; | |
| d. | maintain records of defects and adverse effects; | |
| e. | report defects and adverse effects; | |
| f. | notify HSA concerning recall; and | |
| g. | notify HSA before supply of Class 2 CTGTPs. |
In addition, dealers are required to comply with the relevant regulatory guidance where appropriate, which include:
| (i) | For manufacturers – MOH Singapore’s Guidelines for Healthcare Institutions Providing Tissue Banking and HSA’s Guidelines on Good Manufacturing Practice for CTGTPs; and | |
| (ii) | For importers and wholesalers – HSA’s Good Distribution Practice Standard for Medical Devices, ISO 13485, and HSA’s Guidance Notes on Good Distribution Practice. |
Additional guidance can also be sought from other regulatory guidance released by the relevant regulatory authorities, including HSA’s Guidance on Cell, Tissue and Gene Therapy Products Registration in Singapore.
Advertisement Regulations
Advertisements of CTGTPs do not require prior approval from the HSA. However, the advertisements must comply with the principles and requirements as stated in the HPA and Advertisement Regulations. The Advertisement Regulations are given effect under the HPA and came into effect on November 1, 2016. Some of such key principles and requirements are highlighted below.
Advertisements of CTGTPs refer to any information that can promote the sale or use of the CTGTP, and can be in any forms or media including but not limited to publication in a newspaper, magazine, journal or other periodical, display of posters or notices, circulars, brochures, pamphlets, books, letters addressed to individuals or organizational bodies, and/or any other activity intended to introduce, publicize or raise the profile or public awareness or visibility of any CTGTP for the purpose of promoting the sale or use of it.
| 92 |
Advertisements of CTGTPs must be aligned with the intended uses per its registration or notification made to HSA. Importantly, they cannot be directed at the public (i.e. cannot be made available in any public places) and, amongst others, must not:
| (i) | advertize an unregistered or unnotified product; | |
| (ii) | advertize an unregistered indication of a registered or notified product; | |
| (iii) | make any false or misleading claims or representations; | |
| (iv) | make unsubstantiated claims; | |
| (v) | make claims that mislead by emphasis, contrast or omission with regard to the safety, quality or efficacy of the product; | |
| (vi) | make claims that give rise to any unrealistic expectations with regard to the effectiveness of the product; | |
| (vii) | encourage inappropriate or excessive use; | |
| (viii) | suggest guaranteed results without side effects; | |
| (ix) | falsely claim any endorsement by public authority; | |
| (x) | include endorsements or recommendations by any healthcare professional or a person of celebrity, social or professional status; | |
| (xi) | use the names or logos of HSA and any of professional groups; and/or | |
| (xii) | offer refunds, in full or partial amounts, to users of the product. |
Clinical trials and research
In the event that clinical trials, research and/or treatments are undertaken in Singapore, we will be required to obtain and maintain the necessary licenses and approvals, and compliance with the following legislation and regulations (where applicable) must be ensured. Depending on the requirements of the specific clinical trials, research and/or treatments and industry standards from time to time, we will also be required to adhere to international guidelines such as the Guidelines for Good Clinical Practice of the International Conference on Harmonization.
| 93 |
PHMCA
ECEG 2016 and HME 2016
The ECEG 2016 provides guidance for inculcating good medical practice in Singapore based on the fundamental tenets of medical ethics.
In particular, Guideline B6 of the ECEG 2016 stipulates that in general, untested treatments that are not generally accepted by the medical profession can only be offered to patients in two forms: (i) in the context of formal and approved clinical trials (which would be subject to the ethics of research), or (ii) as innovative therapy (which may only be offered when conventional therapy is unhelpful and it is a desperate or dire situation).
Guideline B6.1 of the HME 2016 defines “innovative therapy” as “a completely novel or significantly modified standard therapy with little or nothing in the way of studies or scanty evidence of efficacy, effects or side effects”. Under the HME 2016 and ECEG 2016, doctors may offer such innovative therapy to patients in desperate or dire situations, and where conventional therapy is unhelpful. In such instances, patients’ informed consent must be obtained, failing which the doctor may be struck off the Singapore Registrar of Medical Practitioners.
Guideline B8 of the ECEG 2016 also stipulates that any medical research on human subjects or trials of any treatment on patients must be approved by the Institutional Review Board or Ethics Committee (as referred to in the ECEG 2016) and conform to the Singapore’s Good Clinical Practice guidelines and other existing guidelines on human biomedical research.
CRM Regulations
The CRM Regulations governs the import, manufacture and supply of clinical research materials for use in clinical research studies (including clinical trials). The CRM Regulations is given effect under the HPA and came into effect on November 1, 2016.
CRM refers to any registered or unregistered therapeutic product, medicinal product, medicinal device, applicable CTGTP or placebo, that is manufactured, imported or supplied for the purpose of being used in clinical research, by way of administration to a trial participant in accordance with the research protocol or for a clinical purpose.
The manufacture, import and supply of CRM in Singapore for (i) Class 2 CTGTPs or (ii) Class 1 CTGTPs for which a manufacturer / importer / wholesaler notification has not been made to HSA, must comply with the CRM Regulations.
All local dealers of CRM must only supply the CRM for use in a clinical research, unless the CRM is locally registered and is manufactured, imported or supplied with a valid dealer’s license. The local sponsor of the clinical research must also ensure that the CRM is only used in a research study approved by an institutional review board, and in accordance with the research protocol, and must ensure that any unused CRM is disposed of or exported to another country within 6 months of study conclusion.
A CRM notification is not needed under certain scenarios listed in the CRM Regulations. For products imported solely for export to overseas trial sites, a CRM notification is not applicable and separate approval to import them must be applied for separately.
Additional requirements such as prior approval from the HSA must be complied with if a CRM comprises controlled drugs and psychotropic substances, poisons and radiopharmaceuticals.
| 94 |
Clinical Trials Regulations
Following the HPA Order 2021 coming into effect, which specified and defined CTGTPs as a distinct category of products under the HPA, all Class 2 CTGTPs used in research are subject to the Clinical Trials Regulations with effect from March 1, 2021. Prior to this, the Medicines (Clinical Trials) Regulations 2016 under the MA applied to in-house manufactured Class 2 CTGTPs used in research.
The Clinical Trials Regulations is given effect under the HPA, and came into effect on November 1, 2016. It provides a risk-based approval to the regulation of clinical trials, whereby the requirements and extent of pre-trial regulatory review are risk-stratified according to the local registration status of the investigational product used in the clinical trial. The risk stratification of the clinical trials is intended to improve the overall resource efficiency while ensuring trial participants’ safeties.
A CTA is required for a “higher risk” clinical trial of a locally unregistered therapeutic product or Class 2 CTGTP, or involving an unapproved use of a locally registered therapeutic product or Class 2 CTGTP. On the other hand, a “lower risk” clinical trial of a locally registered product or Class 2 CTGTP that is used in accordance with its approved label will only be required to be notified to HSA through a Clinical Trial Notification (“CTN”). As locally registered products would already have been reviewed by HSA for product registration, CTN submissions will be subjected to a simplified regulatory screening and verification process that leverages the review by the Institutional Review Board (as referred to in the Clinical Trials Regulations). In most instances, this is expected to shorten clinical trial start-up timelines as compared to clinical trials that require authorization.
Further guidance can be sought from HSA’s regulatory guidance, in particular from its Clinical Trials Guidance dated March 1, 2021 on determination of whether a clinical trial requires a CTA, CTN or Clinical Trial Certificate.
HBRA
Generally, the HBRA regulates the conduct of human biomedical research (as defined under Section 3 of the HBRA) conducted under the supervision and control of a research institution and the handling of human tissue for use in research and the research has been reviewed by an institutional review board appointed by that research institution. It first came into force on July 1, 2016 and was implemented in phases, and includes provisions on consent, institutional review boards, savings and transition of research conduct, regulation of human biomedical research, prohibited and restricted human biomedical research, human tissue activities and tissue banks, enforcement powers, etcetera.
Read with the HBRA, subsidiary legislation under the HBRA, namely the Human Biomedical Research Regulations 2017 and Human Biomedical Research (Restricted Research) Regulations 2017, cumulatively this legislation regulates the conduct of human biomedical research and, in particular, subject certain types of research to stricter controls. This includes the introduction of human stem cells (pluripotent or not) into animals. The Ethics Guidelines for Human Biomedical Research, released by the Bioethics Advisory Committee of Singapore, does not have statutory force but operates alongside these subsidiary legislations to provide guidance and emphasize the fundamental principles of solidarity, respect for persons, justice, proportionality, sustainability, beneficence and research integrity.
In particular, the HBRA provides clarity with regard to the roles and responsibilities of parties involved in human biomedical research and the handling of human tissue for use in research. Clinical trials using Class 1 CTGTPs have to ensure compliance with the HBRA, which is regulated by MOH Singapore.
No human biomedical research is permitted to be conducted except under the supervision and control of a research institution with (a) a place of business in Singapore; and (b) at least two (2) individuals ordinarily resident in Singapore who are responsible on behalf of the research institution for the supervision and control of the biomedical research. Any person who contravenes this shall be guilty of an offence and shall be liable on conviction to a fine not exceeding S$50,000 or to imprisonment for a term not exceeding five (5) years or to both.
A research institution is required to submit a notification in the prescribed form and manner before the commencement of the first human biomedical research conducted under that research institution’s supervision and control. It must also submit in the prescribed form a declaration of compliance in respect of all human biomedical research conducted under its supervision and control in the preceding 12 months, or such other period of time as the Director of Medical Services (as referred to in the HBRA) may require. Any person who fails to comply with either of the above shall be guilty of an offence and shall be liable on conviction to a fine not exceeding S$10,000 or to imprisonment for a term not exceeding 12 months or to both.
| 95 |
The HBRA requires that every research institution must, in respect of any human biomedical research which is carried out under its supervision and control, among others:
| (a) | supervise, review and proactively monitor the conduct of the research; |
| (b) | designate a principal person in charge to be responsible for ensuring that the research institution complies with the provisions of the HBRA; |
| (c) | formula and implement appropriate standards, policies and procedures to supervise, review and monitor the conduct of the research; |
| (d) | establish a data and safety monitoring board if its institutional review board considers that it is necessary for the purposes of any particular research proposal; and |
| (e) | investigate any areas of concern and take such remedial measures as appropriate. |
The Second Schedule of the HBRA sets out the research, studies and matters excluded from the definition of human biomedical research, which includes clinical trials of health products and medicinal products conducted in accordance with the HPA or MA, respectively. The applicability of the HBRA therefore depends on the matters that are ultimately the subject of the clinical trials conducted by us.
Conduct in laboratory
Any research facility that uses animals for scientific purposes must obtain a license from the Agri-Food and Veterinary Authority of Singapore. The research facility must also comply with the Guiding Principles for the Care and Use of Animals for Scientific Purposes released by the National Advisory Committee for Laboratory Animal Research, and establish an Institutional Animal Care and Use Committee to oversee and evaluate the animal care and use programs of an institution as required under the Animals and Birds (Care and Use of Animals for Scientific Purposes) Rules given effect under the Animals and Birds Act 1965 of Singapore.
Singapore also adheres to the OECD’s Mutual Acceptance of Data scheme. This is therefore an endorsement that Singapore’s generated research data complies with the OECD’s Principles of Good Laboratory Practice and such data can then be accepted automatically by other OECD countries and facilities the sharing of research.
Third parties with research facilities that use animals for scientific purposes that we may collaborate with for the R&D of our product candidates will be required to obtain the requisite licenses.
HCSA
The phased implementation of the HCSA commenced from January 3, 2022, with 3 phases implemented as thus far. As at December 18, 2023, the PHMCA is repealed.
MOH Singapore acknowledged the need to replace the PHMCA with the HCSA primarily due to significant changes to the healthcare landscape in Singapore – in particular, from emerging new healthcare services and models (e.g. home and community-based care and telemedicine services) and new technological advancements (e.g. proton beam therapy, cell, tissue and gene therapy, and clinical genetic testing services). The HCSA therefore introduces a services-based licensing regime, replacing the premises-based licensing regime under the PHMCA, and generally groups the licensable healthcare services into 6 broad service categories – hospital services, ambulatory care services, long-term residential care services, non-premises based services, health support services and special services. A provider may be required to have multiple licenses depending on the services provided. License conditions will depend on the type of service provided and some services will require other licenses as a pre-requisite.
| 96 |
Section 3 of the HCSA defines “healthcare service” to mean any of the following services, whether or not provided for reward:
| (a) | assessment, diagnosis, treatment, prevention or alleviation of an ailment, a condition, disability, disease, disorder or an injury affecting any part of the human body or mind; | |
| (b) | nursing or rehabilitative care of an individual suffering from an ailment, a condition, disability, disease, disorder or an injury mentioned in paragraph (a); | |
| (c) | conduct of any clinical procedure to change, or that is intended to change, the appearance or anatomy of an individual; | |
| (d) | assessment of the health of an individual; | |
| (e) | any other service of a medical or healthcare nature that is prescribed; |
Licensable healthcare services mean any of the healthcare services listed in the First Schedule of the HCSA.
CTGT is a newly regulated service covered under the HCSA. As of the date of this annual report, specific regulations in relation to CTGT under the HCSA have not been released.
Notwithstanding that specific regulations in relation to CTGT have yet to be released, the HCSA generally states the following likely to be generally relevant to CTGT:
| (i) | for selected services, mandates the appointments of key personnel in addition to the licensee – namely the key appointment holder, Principal Officer, and Clinical Governance Officer (as defined in the HCSA) – to strengthen governance and oversight of healthcare services; |
| (ii) | mandates the institution of quality assurance committees and/or clinical ethics committees for select licensees, and/or service review committees for selected services or programs that are deemed higher risk, more complex or of greater public interest; |
| (iii) | enhances MOH Singapore’s powers to gather data for the purposes of patient safety, care, welfare, and public health interest, and to publish information about non-compliant licensees and unlicensed providers; |
| (iv) | places restrictions on licensees’ names to minimize public misperception; |
| (v) | places restrictions on the provision of licensable healthcare services together with other unrelated or unlicensed services at a premise or a conveyance; and |
| (vi) | only a licensee or a person acting on the authority of a HCSA licensee may advertise licensable healthcare services. |
Regulations such as the Healthcare Services (General) Regulations, Healthcare Services (Advertisement) Regulations, and other regulations in relation to the specific services, will have to be complied with under the HCSA. Given the legislation and regulations relating to CTGTs in Singapore, we and/or the hospitals and/or third parties that we may collaborate with will be required to, inter alia, ensure compliance with all applicable legislation and regulations in force, implement controls, obtain the necessary licenses and approvals, depending on factors including the location(s) of our clinical trials and research and healthcare services using CTGTs.
Malaysia
CytoMed Malaysia is a pre-clinical biopharmaceutical company which does not have any clinical operations and has not commenced any clinical trials in Malaysia.
The following description is a summary of material laws and regulations applicable to our operations in Malaysia. The regulations and policies set out below are not exhaustive and are only intended to provide general information to investors and are neither designed nor intended to be a substitute for professional advice. Prospective investors should consult their own advisers regarding the implication of the laws and regulations on us.
| 97 |
Operation of business
The following laws and regulations are generally applicable to the operation of CytoMed Malaysia’s business in Malaysia:
ICA
Section 3(1) of the ICA provides that, no person shall engage in any manufacturing activity unless he is issued a license in respect of such manufacturing activity. Manufacturing activity is defined under the ICA as the making, altering, blending, ornamenting, finishing or otherwise treating or adapting any article or substance with a view to its use, sale, transport, delivery or disposal and includes the assembly of parts and ship repairing but shall not include any activity normally associated with retail or wholesale trade.
Pursuant to section 3(2) of the ICA, any person who fails to comply with the provision of subsection (1) is guilty of an offence and is liable on conviction to a fine not exceeding RM2,000 or to a term of imprisonment not exceeding 6 months and to a further fine not exceeding RM1,000 for every day during which such default continues.
Local Government Act 1976 and Johor Bahru By-Laws
Pursuant to section 102(s) of the Local Government Act 1976, every local authority may from time to time make, amend and revoke by-laws in respect of all such matters as are necessary or desirable for the maintenance of the health, safety and well-being of the inhabitants or for the good order and government of the local authority area and in particular, amongst others, to control and supervise, by registration, licensing or otherwise, including in proper cases by prohibition, a trade, business or industry which is of an obnoxious nature or which could be a source of nuisance to the public or a class of the public.
Pursuant to by-law 3(1) of the Johor Bahru By-Laws, no person shall use a premise for any trade, business or industrial activity without a business premise license issued pertaining to the premise by the Mayor of Johor Bahru. Under by-law 3(3) of the Johor Bahru By-Laws, any person who contravenes by-law 3(1) shall be guilty of an offence and shall, upon conviction, be liable to a fine of not exceeding RM2,000 or to imprisonment for a term of not exceeding 1 year or to both.
| 98 |
OSHA
There are no licenses or certificates issued pursuant to the OSHA. However, there are obligations imposed under the OSHA, including but not limited to:
| (a) | ensure, so far as is practicable, the safety, health and welfare at work of all employees; |
| (b) | without prejudice to the above, the matters to which the duty extends include in particular: |
| (i) | the provision and maintenance of plant and systems of work that are, so far as is practicable, safe and without risks to health; | |
| (ii) | the making of arrangements for ensuring, so far as is practicable, safety and absence of risks to health in connection with the use or operation, handling, storage and transport of plant and substances; | |
| (iii) | the provision of such information, instruction, training and supervision as is necessary to ensure, so far as is practicable, the safety and health at work of employees; | |
| (iv) | so far as is practicable, as regards any place of work under the control of the employer or self-employed person, the maintenance of it in a condition that is safe and without risks to health and the provision and maintenance of the means of access to and egress from it that are safe and without such risks; and | |
| (v) | the provision and maintenance of a working environment for employees that is, so far as is practicable, safe, without risks to health, and adequate as regards facilities for their welfare at work; and |
| (c) | except in such cases as may be prescribed, to prepare and as often as may be appropriate revise a written statement of general policy with respect to the safety and health at work of employees and the organization and arrangements for the time being in force for carrying out that policy, and to bring the statement and any revision of it to the notice of all employees. |
Contravention of any of the above provisions shall, on conviction, be liable to a fine not exceeding RM50,000 or to imprisonment for a term not exceeding 2 years or to both.
| 99 |
Notwithstanding the above, the Occupational Safety and Health (Amendment) Act 2022 has been enacted but has not come into force as of the date of this annual report. Amendments to the OSHA include but are not limited to the following:
| (a) | imposition of new duties on principal, employer and self-employed person to ensure the safety of contractors and sub-contractors, to conduct risk assessment, and to develop and implement procedures to deal with emergencies; |
| (b) | requirement to appoint an occupational safety and health coordinator; |
| (c) | grant of right to employees to remove themselves from imminent danger at the workplace; |
| (d) | increased penalties and punishments for employers, self-employed persons, principals and manufacturers who breach their duties under the OSHA; and |
| (e) | joint and several liability of directors and specified office bearers for offences committed by the company or other relevant body. |
EQA
Licensing requirements
Pursuant to section 21 of the EQA, there are acceptable conditions specified for the emissions, discharge or deposit of environmentally hazardous substances, pollutants or wastes or the emission of noise into any area, segment or element of the environment. The EQA provides that no person shall, unless licensed:
| (a) | emit or discharge of any environmentally hazardous substances, pollutants or wastes into the atmosphere; |
| (b) | pollute any soil or surface of any land; |
| (c) | emit, discharge or deposit of any environmentally hazardous substances, pollutants or wastes into any inland waters; or |
| (d) | emit of any noise greater in volume, intensity or quality, |
in contravention of the acceptable conditions specified.
Contravention of the above shall be guilty of an offence and shall not be liable to a fine not exceeding RM100,000 or to imprisonment for a period not exceeding 5 years or to both and to a further fine not exceeding RM1,000 a day (RM500 a day for noise pollution) for every day that the offence is continued after a notice by the Director General, requiring him to cease the act specified therein has been served upon the offender.
Pursuant to section 41 of the EQA, every omission or neglect to comply with, and every act done or attempted to be done contrary to, the provisions of the EQA or any regulations made thereunder or any breach of the conditions and restrictions subject to, or upon which, any license is issued under the EQA or any regulations made thereunder shall be an offence against the EQA and in respect of any such offence for which no penalty is expressly provided the offender shall be liable to a fine not exceeding RM10,000 or to imprisonment for a period not exceeding 2 years or to both.
CytoMed Malaysia has taken steps to manage environmentally hazardous wastes in compliance with the EQA. Notably, CytoMed Malaysia has engaged the services of a biohazard waste collector and has designated a disposal area for the collection of biohazard wastes.
Written notification to the Director General
Generally, an owner of occupier of a premise must not, without giving prior written notification to the Director General under the Clean Air Regulations:
| (a) | carry out any change in operation of his premises; |
| (b) | carry out any work on any premises that may result in a source of emission; |
| (c) | construct on any land, any building or premises designed or used for a purpose that may result in a new source of emission; |
| (d) | make, cause or permit to be made any change of, to, or in any plant, machine or equipment used or installed at the premises that causes a material change in the quantity or quality of emission from an existing source; or |
| (e) | carry out any changes or modifications to an existing air pollution control system. |
| 100 |
The written notification must be submitted to the Director General not less than 30 days before the commencement of such work in such form as determined by the Director General. Regulation 29 of the Clean Air Regulations states that, any person who contravenes or fails to comply with any provisions of the Clean Air Regulations shall be guilty of an offence and shall be liable to a fine not exceeding RM100,000 or to imprisonment for a term not exceeding 2 years or to both.
Laboratory work
The following laws and regulations are applicable to the laboratory work undertaken by CytoMed Malaysia:
PA 1952
The PA 1952 governs the licenses issued to use poisons. Licenses are categorized into Type A, B, C, D and E licenses. The licenses will be subject to terms and conditions as the licensing officer may in his discretion impose, which are not inconsistent with PA 1952 or of any regulations made thereunder, subject to appeal to the Minister of Health of Malaysia charged with the responsibility for medical and health services.
Pursuant to section 32 of the PA 1952, any person guilty of an offence against the PA 1952, which includes failure to obtain license to use poisons, shall be punishable by a fine not exceeding RM50,000 or by imprisonment for a term not exceeding 5 years or both. If the act or omission is in the opinion of the court of such a nature as to amount to willful default or culpable negligence, which endangered or was likely to endanger human life, such person shall be liable, on conviction, to a fine not exceeding RM200,000 or to imprisonment for a term not exceeding 10 years or both.
CytoMed Malaysia does not hold any license to use poison, as the use of poison is for the sole purpose of research only.
Prevention and Control of Infectious Diseases Act 1988 and PCID Regulations
Pursuant to the PCID Regulations issued by the Minister of Health of Malaysia under section 31 of the Prevention and Control of Infectious Diseases Act 1988, no person shall import or export human tissues or any part thereof and any pathogenic organisms or substances or any part thereof, unless with the approval of the authorized officer. Upon approval, the authorized officer shall issue a permit.
Pursuant to regulation 9 of the PCID Regulations, any person who fails to comply with any provisions under these PCID Regulations commits an offence and shall on conviction be liable:
| (a) | in respect of a first offence, to imprisonment for a term not exceeding 1 year or to a fine not exceeding RM3,000 or to both; |
| (b) | in respect of a second offence or subsequent offence, to imprisonment for a term not exceeding 3 years or to a fine not exceeding RM5,000 or to both; |
| (c) | in respect of a continuing offence, to a further fine not exceeding RM100 for every day during which such offence continues. |
CytoMed Malaysia currently holds an import and an export license which allow it to import or export human tissues or any part thereof and any pathogenic organisms or substances or any part thereof.
Development and manufacturing of CGTPs
Subject to any further regulatory interpretation, amendments, guidelines, policies or orders to be issued or taken by the relevant governmental authorities (including the Ministry of Health Malaysia), the following laws and regulations may be applicable to CytoMed Malaysia once CytoMed Malaysia passes the clinical trials and commences operations to develop and manufacture CGTPs:
CDCR
The CDCR is a regulation provided under section 26 of the Sale of Drugs Act 1952 of Malaysia. Under CDCR, a product means a drug in a dosage unit or otherwise, for use wholly or mainly by being administered to one or more human beings or animals for a medicinal purpose or a drug to be used as an ingredient of a preparation for a medicinal purpose.
| 101 |
The Guidelines on CGTPs for CGTPs were issued under regulation 29 of the CDCR. It has come into effect on January 1, 2021 and provides that CGTPs fit within the meaning of medicinal products under the CDCR. In accordance to the CDCR, CGTPs would be classified as biological products, as defined above. As such, the Guidelines on CGTPs should be read in conjunction with CDCR.
The Guidelines on CGTPs addresses the development, manufacturing and quality control as well as non-clinical and clinical development of CGTPs which include somatic cell therapy, tissue engineering and gene therapy products. These guidelines are intended for products entering the registration process at the NPCB. In order to investigate the use of CGTPs in a local clinical trial, an application that reports data from pre-clinical studies on the likely safety and efficacy of the investigational product must be filed by NPCB. An approval for a CTIL or a CTX is mandatory before an unregistered CGTP is administered to human trial subjects in Malaysia. Following January 1, 2021, only CGTPs that are registered with the NPCB can market its products in Malaysia.
The registration of CGTPs under the Guidelines on CGTPs are in line with regulation 7 of the CDCR, which provides that no person shall manufacture, sell, supply, import or possess or administrator any product unless:
| (a) | the product is a registered product; and |
| (b) | the person holds the appropriate license required and issued under CDCR. |
Further, pursuant to regulation 8 of the CDCR, the Drug Control Authority of Malaysia may, on application made in such manner or form as it may require, register any product subject to such conditions as it may impose. Other than the registration of products, the CDCR provides that the following circumstances below may require licenses:
| (a) | manufacturer’s license authorizing the licensee to manufacture the registered products in the premises specified in the license and to sell by wholesale or supply the products; |
| (b) | a wholesaler’s license authorizing the licensee to sell by wholesale or supply the registered products from the address of the business premises specified in the license; |
| (c) | a clinical trial import license authorizing the licensee to import any product for purposes of clinical trials, notwithstanding that the product is not a registered product; and |
| (d) | an import license authorizing the licensee to import and sell by wholesale or supply the registered products from the address of the premises specified in the license. |
Any person who contravenes any of the provisions of the CDCR or any condition of any license issued under the CDCR or any condition subject to which a product is registered under the CDCR commits an offence.
According to the CDCR, all registered products and notified cosmetics are to be manufactured within GMP compliant premises. ‘Manufacturing’ is defined as (a) the making of assembling of the product, (b) the enclosing or packing of the product in any container in a form suitable for administration or application, and the labelling of the container, and (c) the carrying out of any process in the course of any of the foregoing activities.
GMP is a standard imposed by the Ministry of Health Malaysia where manufacturers of registered pharmaceutical/veterinary/health supplements/traditional products and/or notified cosmetics to ensure that the product manufactured is safe, efficacious and of quality. GMP compliance is one of the requirements for product registration and/or notification of cosmetics, as well as to apply for a Manufacturer’s License with the Drug Control Authority. GMP compliant premises should be licensed by the local town council, Department of Environment and Fire and Rescue Department. A GMP Certificate will be issued for the purposes of exporting locally manufactured registered products. It endorses that the local manufacturer complies with the current GMP requirements. These certificates are required by the overseas regulatory agencies for the purpose of product registration in the respective countries.
| 102 |
Any person who contravenes any of the provisions of the CDCR or any condition of any license issued under the CDCR or any condition subject to which a product is registered under the CDCR commits an offence. Additionally, the manufacturer’s license may be revoked by the Director of Pharmaceutical Services (as defined in the CDCR), according to regulation 17(1) of the CDCR if the manufacturer does not comply with GMP standards and requirements.
The Guidance Note for CTGPs Manufacturing Facility in Malaysia issued by NPRA, which only applicable to Class II CGTPs (which are higher risk cell therapy products) is established to facilitate and provide guidance in setting up a manufacturing facility for CGTPs which are regulated under the NPRA.
Among others, the guidance note has set out the types of inspection to be conducted on CGTPs manufacturing facility, which depends on the inspection objective and product category as follows:
| (a) | Pre-certification inspection: an inspection conducted on CGTPs manufacturing facility during the voluntary registration period (before year 2023). |
| (b) | Pre-licensing inspection: an inspection conducted on CGTPs manufacturing facility for the purpose of licensing and product registration. |
| (c) | Pre-approval inspection: an inspection conducted on additional manufacturing line/new sources of cell in an existing inspected CGTPs manufacturing facility. |
| (d) | Routine inspection: a subsequent inspection conducted on existing inspected CGTPs manufacturing facility according to a planned schedule by NPRA. |
| (e) | Verification inspection: an inspection conducted following a punitive action. Depending on the condition, verification inspection can be combined with routine inspection. |
| (f) | Investigation inspection: investigation inspection is an inspection conducted on premises based on complaint received and product recall activity. |
PHFSA
Pursuant to section 3 of the PHFSA, any person intending to establish or maintain private healthcare facilities or services including a private blood bank shall obtain a license from the Ministry of Health Malaysia. Private blood bank is defined to mean any premises, other than a government blood bank, used or intended to be used for collecting, screening, processing, storing or distributing natural human blood or blood product.
Pursuant to section 5 of the PHFSA, failure to obtain a license is an offence and any individual person found guilty shall be liable, on conviction to a fine not exceeding RM300,000 or to an imprisonment for a term not exceeding 6 years or to both and for a continuing offence, to a fine not exceeding RM1,000 for every day or part of a day during which the offence continues after conviction. Where the offence is committed by a body corporate, the penalty is a fine not exceeding RM500,000 and for a continuing offence, a fine not exceeding RM5,000 for every day or part of a day during which the offence continues after conviction. The person responsible for the body corporate shall also be guilty of the offence and shall be liable, on conviction to a fine not exceeding RM300,000 or to imprisonment for a term not exceeding 6 years or to both, and for a continuing offence, to a fine not exceeding RM1,000 for every day or part of a day during which the offence continues after conviction.
Employment matters
The following laws and regulations are generally applicable to employment matters:
EPFA
Pursuant to section 43(1) of the EPFA, it is compulsory for employees and their employers to make monthly contributions on the amount of wages at the rate respectively set out in the Third Schedule of the EPFA to the Employment Provident Fund, which is a statutory retirement fund. The contributions are made to the account of the individual employee.
| 103 |
ESSA
Monthly contributions will also have to be made to SOCSO both by the employer and employees irrespective of the amount of wages as per the rates set out in the Third Schedule of the ESSA. The SOCSO administers:
| (a) | the EIS which provides employees with coverage by way of cash benefits and medical care in the event of any disablement or death due to employment injury; and |
| (b) | the Invalidity Pension Scheme which provides 24-hours coverage to employees against invalidity and death due to any cause before attaining the age of 60 years. |
EISA
The EISA implemented the EIS, effective since January 2018 by the SOCSO. The EISA sets out provisions to provide certain benefits and a re-employment placement program for insured persons in the event of loss of employment which will promote active labor market policies. All private sector employers need to pay monthly contributions for each employee. Civil servants, domestic servants and those who are self-employed are exempted. All employees from 18 to 59 years of age have to contribute. Employees aged between 57 and 60 who have never contributed to the SOCSO are exempted from this protection plan.
The employer is liable to pay interest on such amount at 6% per annum for each day of late contributions in respect of any period during which such amount remains unpaid provided that:
| (a) | if the amount of interest so calculated is less than RM5.00, the interest payable shall be RM5.00 in respect of each month or part of a month; and |
| (b) | if the amount of interest exceeds RM5.00, the interest payable shall be calculated to the next highest multiple of RM5.00 in respect of each such month or part of a month. |
Immigration Regulations 1963
Pursuant to regulation 9 of the Immigration Regulations 1963, an employment pass is required for the entry of any foreign person into Malaysia to take up employment under a contract of service. Such contract must be for a minimum period of 2 years employment with a company or firm approved and under which such person is entitled to a salary of not less than RM1,200 per month. Any person who without reasonable cause fails to comply with any condition imposed in respect of any pass shall be guilty of an offence under regulation 39 and shall be liable on conviction to a term of imprisonment not exceeding 6 months or to a fine not exceeding RM1,000 or to both.
Others
PDPA Malaysia
The PDPA Malaysia applies to any person who processes and has control over the processing of personal data in respect of commercial transactions. Personal data refers to any information that relates directly or indirectly to an individual data subject, who is identified or identifiable from that information or from that and other information in the possession of a data user, including any sensitive personal data and expression of opinion about the data subject; but does not include any information that is processed for the purpose of a credit reporting business carried on by a credit reporting agency under the Credit Reporting Agencies Act 2010 of Malaysia.
Data user refers to a person who either alone or jointly or in common with other persons processes any personal data or has control over or authorizes the processing of any personal data, but does not include a data processor. A data user is required to comply with all the personal data protection principles under the PDPA Malaysia, including the notice and choice principle under section 7 of the PDPA Malaysia, which requires the issuance of written notices in the Malay and English languages informing data subjects that, amongst others, the personal data of the data subject is being processed by or on behalf of the data user and the purposes for which the personal data is being or is to be collected and further processed.
| 104 |
Pursuant to section 5(2) of the PDPA Malaysia, a data user who contravenes the personal data protection principles under the PDPA Malaysia commits an offence and shall, on conviction, be liable to a fine not exceeding RM300,000 or to imprisonment for a term not exceeding 2 years or to both.
Permits, licenses and approvals
As of the date of this annual report, we have the necessary licenses, permits and approvals in Malaysia required for the operations of CytoMed Malaysia.
| C. | Organization structure |
As of the date of this annual report, we are a holding company incorporated in Singapore which oversees our operations in Malaysia. We conduct our business activities primarily through our direct wholly-owned subsidiary in Malaysia, CytoMed Malaysia. We expect to conduct more research and clinical trial activities in Singapore through CytoMed moving forward. Other than CytoMed Malaysia, our subsidiaries have minimal operations.
| D. | Property, plants and equipment |
For a description of our property, plants and equipment, see Item 4.B. Business Overview.
ITEM 4.A. UNRESOLVED STAFF COMMENTS
None.
ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
| A. | Operating Results |
The following discussion of the results of our operations and our financial condition should be read in conjunction with the consolidated financial statements and the related notes to those statements included in this annual report. The following discussion includes forward-looking statements. These forward-looking statements are subject to risks, uncertainties and other factors that could cause our actual results to differ materially from those expressed or implied by the forward-looking statements. Factors that could cause or contribute to these differences include, but are not limited to, those discussed elsewhere in this annual report. Please refer to the sections titled “Cautionary Statement Regarding Forward-Looking Statements” and “Item 3. Key Information–D. Risk Factors” for further details.
All translations from Singapore dollars to U.S. dollars and from U.S. dollars to Singapore dollars in this annual report are made at a rate of S$1.3193 to U.S.$1.00, the exchange rate in effect as of December 31, 2023 as set forth in the H.10 statistical release of the U.S. Board of Governors of the Federal Reserve System.
Overview
We are a pre-clinical biopharmaceutical company focused on the R&D of allogeneic, “off-the-shelf” cell-based immunotherapies for treatment of human cancers. The development of our novel technologies has been inspired by the clinical success of existing CAR-T cells in treating hematological malignancies as well as the current clinical limitations and commercial challenges in extrapolating the CAR-T principle into treatment of solid tumors. All of our product candidates are designed to be allogeneic, meaning they are produced using cells from a different person than the patient treated, as well as on an “off-the-shelf” basis, unlike existing autologous cell therapies. Built on our proprietary platform technologies, we are developing four product candidates: CTM-N2D, iPSC-gdNKT, CTM-GDT and CTM-MSC.
On April 18, 2023, we completed our Offering, whereby we issued and sold 2,412,369 ordinary shares at a price to the public of U.S.$4.00 per share for aggregate gross proceeds of S$12.94 million (U.S.$9.81 million). We received S$10.31 million (U.S.$7.81 million) in net proceeds after deducting underwriting discounts and commissions and offering expenses.
| 105 |
The following table presents our consolidated statements of profit or loss and comprehensive loss data for the financial years ended December 31, 2021, 2022 and 2023:
| For the year ended December 31, | ||||||||||||||||
| 2021 | 2022 | 2023 | ||||||||||||||
| S$ | S$ | S$ | U.S.$ | |||||||||||||
| Revenue | - | - | - | - | ||||||||||||
| Other operating income | 154,610 | 485,374 | 803,904 | 609,341 | ||||||||||||
| Other gains/(losses) including fair value changes on financial instruments - net | 3,185 | (1,116,622 | ) | (492,910 | ) | (373,615 | ) | |||||||||
| Research expenses | (1,090,623 | ) | (1,522,765 | ) | (1,589,693 | ) | (1,204,952 | ) | ||||||||
| Depreciation of property, plant and equipment | (98,721 | ) | (109,246 | ) | (101,866 | ) | (77,212 | ) | ||||||||
| Amortization of intangible assets | (2,512 | ) | (2,500 | ) | (2,187 | ) | (1,658 | ) | ||||||||
| Employee benefits expenses | (168,802 | ) | (210,657 | ) | (454,143 | ) | (344,230 | ) | ||||||||
| Finance expenses | (117,696 | ) | (125,175 | ) | (51,282 | ) | (38,871 | ) | ||||||||
| Other expenses | (518,766 | ) | (494,675 | ) | (2,213,142 | ) | (1,677,512 | ) | ||||||||
| Share of results of associate | (212,578 | ) | (33,546 | ) | (31,198 | ) | (23,647 | ) | ||||||||
| Loss before income tax | (2,051,903 | ) | (3,129,812 | ) | (4,132,517 | ) | (3,132,356 | ) | ||||||||
| Income tax expense | - | (1,640 | ) | (653 | ) | (495 | ) | |||||||||
| Loss for the year | (2,051,903 | ) | (3,131,452 | ) | (4,133,170 | ) | (3,132,851 | ) | ||||||||
| Other comprehensive loss: | ||||||||||||||||
| Items that may be reclassified subsequently to profit or loss: | ||||||||||||||||
| Foreign currency translation loss | (22,628 | ) | (96,504 | ) | (89,918 | ) | (68,155 | ) | ||||||||
| Total comprehensive loss for the year | (2,074,531 | ) | (3,227,956 | ) | (4,223,088 | ) | (3,201,006 | ) | ||||||||
| Loss attributable to: | ||||||||||||||||
| Equity holders of the Company | (2,051,650 | ) | (3,131,209 | ) | (4,132,896 | ) | (3,132,643 | ) | ||||||||
| Non-controlling interests | (253 | ) | (243 | ) | (274 | ) | (208 | ) | ||||||||
| Total | (2,051,903 | ) | (3,131,452 | ) | (4,133,170 | ) | (3,132,851 | ) | ||||||||
| Total comprehensive loss attributable to: | ||||||||||||||||
| Equity holders of the Company | (2,074,278 | ) | (3,227,713 | ) | (4,222,814 | ) | (3,200,798 | ) | ||||||||
| Non-controlling interests | (253 | ) | (243 | ) | (274 | ) | (208 | ) | ||||||||
| Total | (2,074,531 | ) | (3,227,956 | ) | (4,223,088 | ) | (3,201,006 | ) | ||||||||
| Loss per share for loss attributable to equity holders of the Company | ||||||||||||||||
| Basic and diluted | (0.30 | ) | (0.40 | ) | (0.39 | ) | (0.29 | ) | ||||||||
| Weighted average number of ordinary shares used in computing loss per share | ||||||||||||||||
| Basic and diluted | 6,934,824 | 7,915,101 | 10,630,344 | 10,630,344 | ||||||||||||
Key Factor Affecting Our Results of Operations
Our financial condition and results of operation have been and will continue to be affected by a number of factors, many of which may be beyond our control, including those factors set out in the section headed ‘‘Risk Factors’’ in this annual report and those set out below:
No product candidates approved for commercial sale
We are a pre-clinical biopharmaceutical development stage company and we do not have any products approved for commercial sale as of the date of this annual report. We are focused on developing human cells as therapeutics and our technologies are new and unproven. Since our incorporation in 2018, we have devoted most of our resources to support our product candidate development efforts, building our intellectual property portfolio, developing our supply chain, conducting business planning, constructing our centralized cGMP Facility, hiring and training staff, raising capital and providing general and administrative support for these operations. We have not generated any revenue. Given that we will continue to invest most of our resources in developing our product candidates, we expect to continue to incur increasing losses for the foreseeable future.
| 106 |
Key Components of Our Results of Operations
Revenue
Since our incorporation, we have not generated any revenue and do not expect to generate any revenue from the commercial sale of products in the foreseeable future. As of the date of this annual report, we have no therapeutic products approved for sale commercially. If our development efforts for one or more of our product candidates are successful and result in regulatory approval, or if we enter into collaboration with third parties, we may generate revenue from a combination of product sales or payments from collaboration in the future.
Other Operating Income
Other operating income amounted to S$154,610, S$485,374 and S$803,904 for the financial years ended December 31, 2021, 2022 and 2023, respectively. The income was primarily consisted government grants received in view of the COVID-19 pandemic and support in relation to the technology innovation, research income and interest income. The following table sets forth the breakdown of our other operating income for these periods:
| Year ended December 31, | ||||||||||||
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Government grants: | ||||||||||||
| Jobs Support Scheme | 4,880 | - | - | |||||||||
| Wage Subsidy Program | 7,385 | 3,099 | - | |||||||||
| Enterprise Development Grant | 10,400 | 8,640 | - | |||||||||
| Jobs Growth Incentive | 3,750 | 5,550 | - | |||||||||
| Hiring Incentive and Training Programme | 7,709 | 9,077 | 3,459 | |||||||||
| Startup SG Tech - Proof-of-concept grant | - | 84,091 | - | |||||||||
| Others | 3,080 | 2,880 | 4,112 | |||||||||
| Research income | 113,841 | 363,912 | 507,736 | |||||||||
| Interest income | 3,369 | 2,097 | 285,719 | |||||||||
| Others | 196 | 6,028 | 2,878 | |||||||||
| Total | 154,610 | 485,374 | 803,904 | |||||||||
The Hiring Incentive and Training Programme (“Hiring Incentive”) was an economic recovery incentive introduced in Malaysia to promote job creation among employers while increasing employment prospects. For the financial years ended December 31, 2021, 2022 and 2023, the Hiring Incentive amounted to S$7,709, S$9,077 and S$3,459, respectively.
The Startup SG Tech - Proof-of-concept (“SSG Tech POC”) grant is a scheme to drive the growth of startups based on proprietary technology and to foster the spirit of deep-tech innovation among startups. Under the scheme, qualifying companies may receive early-stage funding for developing and commercializing the innovations. For the financial year ended December 31, 2022, SSG Tech POC grant amounted to S$84,091 and the development project was terminated during the financial year ended December 31, 2023.
Research income was derived from the manufacturing of limited quantities of cells for researchers and institutions on a non-profit, cost recovery basis for the purpose of research. Research income was S$113,841 for the financial year ended December 31, 2021. For the financial years ended December 31, 2022 and 2023, research income increased by 219.7% and 39.5% as compared to the respective previous year, mainly because of the increase in demand from the researchers and institutions.
Interest income from fixed deposits was S$3,369 and S$2,097 for the financial years ended December 31, 2021 and 2022, respectively. For the financial years ended December 31, 2023, interest income rose to S$285,719 mainly from the placement of Offering proceeds in fixed deposits.
Other gains/(losses) including fair value changes on financial instruments – net
Other gains/(losses) including fair value changes on financial instruments – net, mainly consist of the net currency exchange losses and the fair value changes on convertible loans and warrants. We recorded a net other gain of S$3,185 for the financial years ended December 31, 2021. The net other loss for the financial year ended December 31, 2022 increased by S$1.12 million as compared to previous year, primarily pertaining to an increase of S$1.10 million in fair value of the convertible loans. The net other loss dropped to S$492,910 mainly due to the convertible loan were converted into ordinary shares and warrants granted to underwriters during the financial year ended December 31, 2023.
| 107 |
Research Expenses
Our research expenses consist primarily of costs incurred for our research and pre-clinical activities. We expense research costs as incurred. Non-refundable advance payments for goods or services to be received in the future for use in R&D activities are recorded as prepaid expenses. The prepaid amounts are expensed as the related goods are delivered or the services are performed. The following table sets forth the breakdown of our research expenses for these periods:
| Year ended December 31, | ||||||||||||
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Employee benefits expenses | 400,250 | 500,038 | 642,533 | |||||||||
| Depreciation of property, plant and equipment | 217,945 | 270,421 | 272,886 | |||||||||
| Amortization of intangible assets | 10,700 | 10,700 | 4,209 | |||||||||
| Consumables expenses | 304,120 | 310,938 | 489,112 | |||||||||
| Royalty expenses | 10,700 | 9,592 | 30,382 | |||||||||
| Professional fees | 63,387 | 317,613 | 20,779 | |||||||||
| Electricity expenses | 60,366 | 64,968 | 62,515 | |||||||||
| Others | 23,155 | 38,495 | 67,277 | |||||||||
| Total | 1,090,623 | 1,522,765 | 1,589,693 | |||||||||
Research expenses were S$1.09 million for the financial year ended December 31, 2021, compared to S$1.52 million for the financial year ended December 31, 2022. For the financial year ended December 31, 2023, research expenses increased by S$66,928, primarily attributable to the increase in consumables expenses by S$178,174, higher employee benefits by S$142,495 resulting from increase in headcount and employee salary rates, higher royalty expenses by S$20,790. This was partially offset by the decrease in professional fee of S$296,834.
Our R&D activities are central to our business. We expect that our research expenses will continue to increase in absolute amounts in the foreseeable future as we continue pre-clinical development for our product candidates and continue our advances in scientific research and product development.
| 108 |
Employee benefits expenses
Employee benefits expenses were S$168,802 for the financial years ended December 31, 2021. Employee benefits expenses increased by S$243,486 from S$210,657 for the financial year ended December 31, 2022 to S$454,143 for the financial year ended December 31, 2023, mainly driven by the increase in headcount and employee salary rates.
Finance Expenses
Finance expenses consisted interest expenses on bank borrowing, convertible loans, third party loans and lease liabilities.
Finance expenses were S$117,696 for the financial years ended December 31, 2021. Finance expenses decreased by S$73,893 from S$125,175 for the financial year ended December 31, 2022 to S$51,282 for the financial year ended December 31, 2023. This is mainly resulted from the decrease in convertible loans interest as the convertible loans were converted into ordinary shares during the financial year ended December 31, 2023.
Other Expenses
Other expenses consist mainly of Offering expenses and costs associated with being a public listed company, including annual listing fee, directors’ and officers’ insurance and investor relationship expenses. Other expenses also include, inter alia, professional fees for legal, auditing, tax and consulting services, and other expenses that are not attributable to R&D activities. The following table sets forth the breakdown of our other expenses for these periods:
| For the year ended December 31, | ||||||||||||
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Advertising | 2,481 | - | 33,307 | |||||||||
| Annual listing fee | - | - | 64,370 | |||||||||
| Company insurance | - | - | 282,797 | |||||||||
| Entertainment | 310 | 3,260 | 30,977 | |||||||||
| Investor relationship expenses | - | - | 204,808 | |||||||||
| Legal fees | - | - | 115,272 | |||||||||
| Low-value assets rental | 1,997 | 2,201 | 1,703 | |||||||||
| Offering expenses | 316,805 | 199,625 | 758,563 | |||||||||
| Professional fees | 118,036 | 184,253 | 396,915 | |||||||||
| Property tax | 4,081 | 7,045 | 6,661 | |||||||||
| Printing and stationery | 5,747 | 6,176 | 11,941 | |||||||||
| IT expenses | 9,482 | 13,051 | 18,255 | |||||||||
| Repair and maintenance | 3,504 | 8,720 | 3,728 | |||||||||
| Reversal of grant | - | - | 84,091 | |||||||||
| Service fees | 11,660 | 11,268 | 13,209 | |||||||||
| Subscription fee | 833 | 2,165 | 1,083 | |||||||||
| Transportation and travelling expenses | 867 | 3,364 | 144,998 | |||||||||
| Tools and supplies | 1,086 | 3,516 | 2,333 | |||||||||
| Utilities | 15,766 | 21,201 | 23,941 | |||||||||
| Others | 26,111 | 28,830 | 14,190 | |||||||||
| Total | 518,766 | 494,675 | 2,213,142 | |||||||||
Other expenses were S$518,766 and S$494,675 for the financial years ended December 31, 2021 and 2022, respectively. The decrease of S$24,091 was due mainly to the decrease in Offering expenses and partially offset by the increase in professional fees and utilities. For the financial year ended December 31, 2023, other expenses increased by S$1,718,467 to S$2,213,142. The increase was mainly due to the increase in Offering expenses by S$558,938, costs associated with being a public listed of S$550,746, higher professional fees by S$212,662, higher transportation and travelling expenses by S$141,634, higher legal fees by S$115,272 and the reversal of SSG Tech POC grant of S$84,091.
Share of results of associate
Share of results of associate consisted of the share of post-acquisition results of an associate incorporated in Malaysia during the financial year and its impairment loss.
Share of loss of associate recognized by S$33,546 for the financial year ended December 31, 2022, while for the financial year ended December 31, 2023, share of loss of associate was recorded at S$31,198.
| 109 |
Loss for the year
As a result of the foregoing, we had loss of S$2.05 million, S$3.13 million and S$4.13 million for the financial years ended December 31, 2021, 2022 and 2023, respectively. If the non-recurring Offering expenses (S$758,563) and the costs associated with being a public listed company (S$550,746) are excluded, the loss for the year will be significantly reduced to S$2.82 million (approximately to US$2.14 million). We expect to continue to incur losses for the foreseeable future in connection with our ongoing activities. However, our cash position remains strong with S$9.00 million cash and cash equivalent as at December 31, 2023 which enables us to support the R&D activities, including the ANGELICA Trial and the research collaboration with Sengkang General Hospital considering our low-cost structure. Our ability to generate product revenue sufficient to achieve profitability will depend on the successful development and commercialization of one or more of our product candidates.
Quantitative and Qualitative Disclosures about Market Risks
We are exposed to market risks in the ordinary course of our business. These risks primarily include currency risk and interest rate risk.
Currency risk
We operate in Asia with dominant operations in Singapore and Malaysia. We regularly transact in currencies other than our respective functional currencies (“foreign currencies”). Currency risk arises when transactions are denominated in foreign currencies other than functional currency. In addition, we are exposed to currency translation risk on the net assets in foreign operations.
Interest rate risk
As at December 31, 2021, 2022 and 2023, we had cash and cash equivalents of S$2.51 million, S$1.58 million and S$9.00 million. Our exposure to interest rate sensitivity is impacted by changes in the underlying U.S. bank interest rates. We have not entered into investments for trading or speculative purposes.
| B. | Liquidity and Capital Resources |
Overview
Since our incorporation, we have not generated any revenue from commercial sales of product and have incurred losses and negative cash flows from our operations and expect these conditions to continue for the foreseeable future. We have not yet commercialized any of our product candidates, which are in various phases of pre-clinical development, and we do not expect to generate revenue from commercial sales of any products for the foreseeable future.
As at December 31, 2021, 2022 and 2023, we had accumulated losses of S$5.07 million, S$8.20 million and S$12.33 million, respectively and net cash used in operating activities of S$1.71 million, S$1.40 million and S$3.43 million, respectively. We expect that our expenses and capital requirements will increase significantly in connection with our ongoing activities as we continue to develop and seek regulatory approvals for our product candidates, engage in other R&D activities to expand our pipeline of product candidates, maintain and expand our intellectual property portfolio, and ultimately establish a sales organization and operate as a public company.
As at December 31, 2021, 2022 and 2023, we had S$2.51 million, S$1.58 million and S$9.00 million, respectively, in cash and cash equivalents. Our liquidity and working capital requirements primarily related to our operating expenses. Historically, we have funded our operations primarily through private equity financing and issuance of convertible loans. Going forward, we expect to fund our working capital and other liquidity requirements from various sources, including but not limited to the net proceeds from our Offering and other equity and debt financings as and when appropriate.
Cash Flows
The following table summarizes our cash flows for the years presented:
| For the year ended December 31, | ||||||||||||||||
| 2021 | 2022 | 2023 | ||||||||||||||
| S$ | S$ | S$ | U.S.$ | |||||||||||||
| Net cash used in operating activities | (1,712,985 | ) | (1,398,409 | ) | (3,432,830 | ) | (2,602,010 | ) | ||||||||
| Net cash used in investing activities | (823,666 | ) | (473,305 | ) | (2,829,175 | ) | (2,144,451 | ) | ||||||||
| Net cash generated from financing activities | 4,173,396 | 968,716 | 10,919,885 | 8,277,030 | ||||||||||||
| Net changes in cash and cash equivalents | 1,636,745 | (902,998 | ) | 4,657,880 | 3,530,569 | |||||||||||
| Cash and cash equivalents at beginning of year | 885,272 | 2,512,768 | 1,579,718 | 1,197,391 | ||||||||||||
| Effect of foreign currency translation on cash and cash equivalents | (9,249 | ) | (30,052 | ) | (13,411 | ) | (10,165 | ) | ||||||||
| Cash and cash equivalents at end of year | 2,512,768 | 1,579,718 | 6,224,187 | 4,717,795 | ||||||||||||
Operating Activities
During the financial year ended December 31, 2021, net cash used in operating activities amounted to S$1.71 million. This was primarily attributable to the loss for the year of S$2.05 million, adjusted for non-cash charges of S$377,892 for depreciation and amortization, fair value loss on convertible loans and unrealized currency translation losses, S$117,696 in interest expenses, S$3,369 in interest income, S$212,578 in share of results of associate and a S$369,248 net change in working capital.
| 110 |
During the financial year ended December 31, 2022, net cash used in operating activities amounted to S$1.40 million. This was primarily attributable to the loss for the year of S$3.13 million, adjusted for non-cash charges of S$1.46 million for depreciation and amortization, fair value gain on convertible loans and unrealized currency translation gains, S$125,175 in interest expenses, S$2,097 in interest income, S$33,546 in share of results of associate and a S$109,902 net change in working capital.
During the financial year ended December 31, 2023, net cash used in operating activities amounted to S$3.43 million. This was primarily attributable to the loss for the year of S$4.13 million, adjusted for non-cash charges of S$477,368 for depreciation and amortization, fair value gain on convertible loans and unrealized currency translation losses, S$223,845 in fair value changes on warrant liabilities, S$51,282 in interest expenses, S$285,719 in interest income, S$31,198 in share of results of associate and a S$104,918 net change in working capital.
Investing Activities
Net cash used in investing activities during the financial year ended December 31, 2021 was S$823,666, mainly due to the purchase of property, plant and equipment of S$582,354 and the balance payment for acquisition of an associate for S$241,312.
Net cash used in investing activities during the financial year ended December 31, 2022 was S$473,305, mainly due to the purchase of property, plant and equipment of S$473,795.
Net cash used in investing activities during the financial year ended December 31, 2023 was S$2.83 million, mainly due to the placement of fixed deposits of S$2.77 million.
Financing Activities
During the financial year ended December 31, 2021, net cash generated from financing activities was S$4.17 million mainly due to the proceeds from issuance of ordinary shares of S$2 million, proceeds from share application monies of S$2 million, and proceeds from a third-party loan of S$350,000. This was partially offset by the interest payment of S$117,696 and repayment of bank borrowings of S$51,452.
During the financial year ended December 31, 2022, net cash generated from financing activities was S$968,716 mainly due to the proceeds from issuance of ordinary shares of S$1.22 million, proceeds from a third party loan of S$300,000. This was partially offset by the repayment of a third party loan of S$350,000, interest payment of S$125,175 and repayment of bank borrowings of S$71,204.
During the financial year ended December 31, 2023, net cash generated from financing activities was S$10.92 million mainly due to the net proceeds from issuance of ordinary shares of S$11.31 million, partially offset by the repayment of a third party loan of S$300,000, interest payment of S$46,713 and repayment of bank borrowings of S$32,984.
Funding Requirements
Our plan of operation is to continue implementing our business strategy, continue R&D of CTM-N2D and our other product candidates and continue to expand our research pipeline and our internal R&D capabilities. We expect our expenses to increase substantially in connection with our ongoing activities, particularly as we conduct the ANGELICA Trial with National University Hospital Singapore, research collaboration with Sengkang General Hospital and advance the pre-clinical activities of our product candidates. In addition, we expect to incur additional costs associated with operating as a public company. Our future capital requirements will depend on many factors, including:
| ● | the scope, timing, progress, costs, and results of discovery, pre-clinical development, and clinical trials for our current and future product candidates; | |
| ● | the number of clinical trials required for regulatory approval of our current and future product candidates; | |
| ● | the costs, timing, and outcome of regulatory review of any of our current and future product candidates; | |
| ● | the cost of manufacturing clinical and commercial supplies of our current and future product candidates; |
| 111 |
| ● | the costs and timing of future commercialization activities, including manufacturing, marketing, sales, and distribution, for any of our product candidates for which we receive marketing approval; | |
| ● | the costs and timing of preparing, filing, and prosecuting patent applications, maintaining and enforcing our intellectual property rights, and defending any intellectual property-related claims, including any claims by third parties that we are infringing on their intellectual property rights; | |
| ● | the cost of maintaining our own R&D and centralized cGMP Facility and future facility expansion plans; | |
| ● | our ability to maintain existing, and establish new, strategic collaborations, licensing, or other arrangements and the financial terms of any such agreements, including the timing and amount of any future milestone, royalty, or other payments due under any such agreement; | |
| ● | the revenue, if any, received from commercial sales of our product candidates for which we receive marketing approval; | |
| ● | the expenses to attract, hire and retain, skilled personnel; | |
| ● | the costs of operating as a public company; | |
| ● | our ability to establish a commercially viable pricing structure and obtain approval for coverage and adequate reimbursement from third-party and government payors; | |
| ● | the effect of competing technological and market developments; and | |
| ● | the extent to which we acquire or invest in businesses, products and technologies. |
A change in the outcome of any of these variables with respect to the development of a product candidate could mean a significant change in the costs and timing associated with the development of that product candidate.
Until such time as we can generate significant revenue from sales of our product candidates, if ever, we expect to finance our operations from the sale of additional equity or debt financings, or other capital which comes in the form of strategic collaborations, licensing, or other arrangements. In the event that additional financing is required, we may not be able to raise it on terms favorable to us, or at all. If we raise additional funds through the issuance of equity or convertible debt securities, it may result in dilution to our existing shareholders. Debt financing or preferred equity financing, if available, may result in increased fixed payment obligations, and the existence of securities with rights that may be senior to those of our ordinary shares. If we incur indebtedness, we could become subject to covenants that would restrict our operations.
If we raise funds through strategic collaboration, licensing or other arrangements, we may relinquish significant rights or grant licenses on terms that are not favorable to us. Our ability to raise additional funds may be adversely impacted by potential worsening global economic conditions and the recent disruptions to, and volatility in, the credit and financial markets in the United States and worldwide. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market products or product candidates that we would otherwise prefer to develop and market ourselves.
Contractual Obligations and Commitments
The following table summarizes our contractual obligations and commitments as of December 31, 2023:
| Payment Due by Period | ||||||||||||||||||||
| Total | Less
than 1 year | Between
1 and 2 years | Between
2 and 5 years | Over
5 years | ||||||||||||||||
| S$ | S$ | S$ | S$ | S$ | ||||||||||||||||
| Contractual Obligations: | ||||||||||||||||||||
| Bank borrowings (1)(2) | 554,799 | 53,261 | 53,261 | 159,782 | 288,495 | |||||||||||||||
| Lease liabilities (2) | 6,462 | 6,462 | - | - | - | |||||||||||||||
| Commitment: | ||||||||||||||||||||
| Minimum royalty commitments | 160,302 | 11,880 | 11,990 | 35,970 | 100,462 | |||||||||||||||
Capital commitments (3) | 387,720 | 387,720 | - | - | - | |||||||||||||||
Loan commitments (4) | 1,000,000 | 1,000,000 | - | - | - | |||||||||||||||
| (1) | Includes scheduled principal payments on bank borrowings outstanding as of December 31, 2023. | |
| (2) | Includes interest payments. | |
| (3) | Relating to the purchase of a property in Malaysia for business expansion purposes. | |
| (4) | Loan to a third party at 5.0% interest per annum to set up our presence in China (refer to the sections titled “Business Strategies and Future Plans” for further details). |
Other than as shown above, we did not have any significant capital and other commitments, long term obligations or guarantees as of December 31, 2023.
| 112 |
| C. | R&D Expenses |
We record as expenses all costs incurred in conducting research and pre-clinical activities in the periods in which such costs are incurred. R&D expenses include employees’ benefits, laboratory supplies, fees paid to entities that conduct certain quality testing activities as well as facility-related expenses.
As part of the process of preparing our financial statements, we are required to estimate our accrued R&D expenses. We make estimates of our accrued expenses as of each balance sheet date in the financial statements based on facts and circumstances known to us at that time. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the expense. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from the estimate, we adjust the accrual or the amount of prepaid expenses accordingly. Although we do not expect our estimates to be materially different from amounts actually incurred, our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in reporting amounts that are too high or too low in any particular period. To date, there have not been any material adjustments to our prior estimates of accrued R&D expenses.
| D. | Trend Information |
See “Item 5A. Operating Results” within this annual report.
| E. | Critical Accounting Estimates |
Our management’s discussion and analysis of financial condition and results of operations is based on our financial statements, which have been prepared in accordance with IFRS. The preparation of our financial statements and related disclosures requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities, costs and expenses and the disclosure of contingent assets and liabilities in our financial statements. We base our estimates on historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate our estimates and assumptions on an ongoing basis. Our actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are described in greater detail in Note 2 to our financial statements in this annual report, we believe that the following accounting policies are those most critical to the judgments and estimates used in the preparation of our financial statements.
Recently Issued Accounting Pronouncements
A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 2 to our financial statements in this annual report.
| 113 |
Emerging Growth Company Status
We are an “emerging growth company” under the JOBS Act. The JOBS Act, permits that an “emerging growth company” may take advantage of the extended transition period for complying with new or revised accounting standards applicable to public companies until those standards would otherwise apply to private companies. We have elected to avail ourselves of delayed adoption of certain accounting standards. Accordingly, our financial statements may not be comparable to the financial statements of public companies that comply with such new or revised accounting standards. We intend to rely on other exemptions provided by the JOBS Act, including without limitation, not being required to comply with the auditor attestation requirements of Section 404(b) of Sarbanes-Oxley Act.
We will remain an emerging growth company until the earliest of (i) the last day of the financial year in which we have more than U.S.$1.235 billion in annual revenue, (ii) the date we qualify as a “large accelerated filer” as defined in Rule 12b-2 under Exchange Act, which would occur if the market value of our ordinary shares held by non-affiliates exceeded U.S.$700 million, (iii) the issuance, in any three-year period, by us of more than U.S.$1 billion in non-convertible debt securities, and (iv) the last day of the financial year ending after the fifth anniversary of our initial public offering.
Recent Developments
The Company has assessed all events occurred from December 31, 2023, up through April 22, 2024, which is the date that these consolidated financial statements are available to be issued. Other than the events disclosed below, there are not any material subsequent events that would require disclosure in these consolidated financial statements.
On February 1, 2024, we completed the purchase of a property in Johor, Malaysia (“Purchase”) for business expansion by receiving the notice of transfer of ownership for the property from the Malaysian land office in which the sale and purchase agreement of such Purchase was entered with a third party on September 29, 2023 for a total purchase consideration of approximately S$431,000 (equivalent to approximately RM1.50 million).
On February 29, 2024, we entered into a research collaboration agreement with Sengkang General Hospital, a major public hospital in Singapore to advance injectable allogeneic umbilical cord derived MSC for cartilage injury. In this collaboration, we are looking to provide CTM-MSC and its conditioned media for in vivo studies and Phase I clinical trial in Singapore.
On March 25, 2024, we received the letter of acceptance for the acquisition of certain assets relating to the business of providing cord blood banking for a total offer price of approximately S$661,000 (equivalent to RM2.30 million). As at the date of this report, earnest deposit of approximately S$13,000 (equivalent to RM46,000) is paid and the management is in the midst of reviewing the master agreement relating to this acquisition.
ITEM 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
| A. | Directors and Senior Management |
Board of Directors
The following table sets forth information regarding the current members of our Board of Directors as of the date hereof.
| Name | Age | Position/Title | ||
Executive Directors CHOO Chee Kong |
66 |
Director and Chairman | ||
| Dr. Lucas LUK Tien Wee | 40 | Director and Chief Clinical Officer | ||
| Dr. ZENG Jieming | 51 | Director and Chief Scientific and Medical Officer | ||
| Dr. TAN Wee Kiat | 37 | Director and Co-Chief Executive Officer and Chief Operating Officer | ||
| Independent Directors | ||||
| Prof. LOH Yuin Han (1) | 47 | Independent Director | ||
| Dr. YEW Chak Hua (2) | 43 | Independent Director | ||
| Mark LEONG Kei Wei (3) | 48 | Independent Director | ||
| WU Tao Thomas (4) | 59 | Independent Director | ||
| Dr. TOH Keng Kiat (5) | 83 | Independent Director |
| (1) | Member of the Nominating and Corporate Governance Committee. |
| (2) | Member of the Compensation Committee. |
| (3) | Audit Committee financial expert, Chair of the Audit Committee and Chair of the Compensation Committee. |
| (4) | Member of the Audit Committee. |
| (5) | Chair of the Nominating and Corporate Governance Committee and Member of the Audit Committee. |
| 114 |
Executive Officers
The following table sets forth information regarding our current executive officers as of the date thereof.
| Name | Age | Position/Title | ||
| CHOO Chee Kong | 66 | Chairman and Director | ||
| Dr. Lucas LUK Tien Wee | 40 | Chief Clinical Officer and Director | ||
| Dr. ZENG Jieming | 51 | Chief Scientific and Medical Officer and Director | ||
| Dr. TAN Wee Kiat | 37 | Co-Chief Executive Officer and Chief Operating Officer and Director | ||
| GOH Yvonne | 35 | Chief Financial Officer | ||
| TAN Yoong Ying | 36 | Chief Corporate Officer |
The following sets forth certain biographical information with respect to our Directors and executive officers. Unless otherwise stated, the business address of our Directors and executive officers is 1 Commonwealth Lane, #08-22 Singapore 149544.
Mr. CHOO Chee Kong has been our Chairman and a Director since the incorporation of our Company in March 2018. Mr. Choo has over 20 years of experience in corporate finance. He is presently the executive vice chairman of CNMC Goldmine Holdings Limited (CNMC Group) which is listed on the SGX-ST (SGX-ST stock code: 5TP) since 2008 where he is responsible for the formulation of the strategic direction and expansion plans as well as the corporate governance of CNMC Group. He previously worked at DBS Bank Ltd of Singapore from 1986 to 2000. In the year 2000, Mr. Choo started his own corporate advisory and stockbroking boutique, Westcomb Financial Group Limited, which he listed on SGX-ST in 2004 (SGX-ST stock code: 5EC) and served as its Chief Executive Officer from 2000 to 2008. He served as an independent director at SGX-ST listed Second Chance Properties Ltd (SGX-ST stock code: 528) from February 2009 to November 2015 and SGX-ST listed FDS Networks Group Ltd (SGX-ST stock code: F07) from June 2008 to March 2015. He also served as a non-executive director at SGX-ST listed Advance SCT Limited (SGX-ST stock code: 5FH) from May 2012 to June 2015. He started his career in October 1981 as a Project Engineer with Esso Singapore Pte Ltd. Mr. Choo received a Bachelor of Engineering in Mechanical Engineering (First Class Honours) from University of Liverpool (United Kingdom) in July 1981 and a Master in Business Administration from the University of Bradford (United Kingdom) in December 1985. We believe that Mr. Choo’s extensive knowledge of our Company as founder and his experience in executive roles across multiple industries qualifies him to serve on our Board.
In October 2015, Mr. Choo, was publicly reprimanded by the Singapore Exchange for failing to disclose and seek shareholder approval for an interested party transaction which occurred in May 2012 where he was the non-executive director to Advance SCT Limited, the company in question, and also the 50% owner of CNCM Capital Pte Ltd, the other company party to the transaction. There were no fines imposed on him and as of the date of this annual report, he remains as an executive director of a SGX-ST listed company, CNMC Goldmine Holdings Limited (CNMC Group) (SGX-ST stock code: 5TP).
On October 5, 2009, Mr. Choo was appointed as a non-executive director and chairman of Falmac Limited (“Falmac”), a company listed on the SGX-ST’s Catalist Board when Falmac was being explored as a potential target for a reverse takeover. However, the reverse takeover exercise did not materialize and Falmac was delisted by the SGX-ST on August 22, 2011. Following the delisting of Falmac, Mr. Choo resigned as a non-executive director and chairman of Falmac on August 29, 2011. Subsequently in or around August 2013, a creditor petitioned to the Singapore High Court to wind up Falmac and the winding up order was granted on May 15, 2014.
Dr. Lucas LUK Tien Wee, M.D., has served as a member of our Board since January 2021 and as our Chief Clinical Officer since April 18, 2023. He has a keen interest in cellular therapy and has attended numerous certificate courses pertaining to clinical cell therapy. He is our designated Principal Investigator for our registered Phase I Clinical Trials in Malaysia, pertaining to Mesenchymal Stem Cell Therapy and CAR-T Cell therapy. Dr. Lucas also serves as a member of the Science Industry Advisory Committee for PSB Academy’s School of Life Sciences (Singapore), since 2019. He is also the Medical Director and Consultant Obstetrician & Gynaecologist (O&G), and founding Chairman of the Independent Ethics Committee, at LMC, a private licensed hospital in Malaysia since 2019. As an advocate for women’s health, Dr. Lucas has held the position of Secretary for the Royal College of Obstetricians & Gynaecologists (RCOG) International Representative Committee (IRC) – Malaysia, from 2018 until October 2021. He is also actively involved in medical education and has been on board the central planning and training committee for the Malaysian National MRCOG Parallel Pathway Programme for Malaysian doctors training in the specialty of Obstetrics & Gynaecology, since November 2017. He is an invited medical lecturer at Monash University (Johor Campus) since March 2021. Dr. Lucas received his M.D. MRCOG (UK) from Royal College of Obstetricians & Gynaecologists (United Kingdom) in 2016 and MB BCh BAO from Royal College of Surgeons (Ireland) in 2008, and has a Good Clinical Practice qualification. We believe that Dr. Luk’s extensive background in the practice of medicine and academic tenure qualifies him to serve on our Board.
| 115 |
Dr. ZENG Jieming, M.D., Ph.D., is one of our scientific founders. He was appointed to the Board on April 18, 2023. He has served as our Chief Scientific and Medical Officer overseeing the product development and manufacturing as well as the clinical trial application since August 2019. Equipped with over 20 years of research experience and in-depth knowledge in the field, he has dedicated himself to translating the Company’s platform technologies into clinical applications and therapeutics. Prior to joining CytoMed, he has been working as a research scientist in the A*STAR, Singapore from June 2004 until July 2019. He has been the key scientist of several research projects and the lead authors of 11 original research articles, for which he both carried out and led the research as well as wrote and published the papers. His research has been focused on developing efficient non-viral and viral vectors for gene therapy (Zeng, et al. J Gene Med 6: 1247, 2004; Zeng, et al. Biomaterials 26: 679, 2005; Zeng, et al. Biomaterials 28: 1443, 2007; Zeng, et al. Stem Cells 25: 1055, 2007; Zeng, et al. Molecular Therapy 17: 1585, 2009; Du, Zeng et al. J Biosci Bioeng 109: 1, 2010) as well as developing novel cancer immunotherapies from human pluripotent stem cells (“hPSCs”), including human embryonic stem cells (“hESCs”) and induced pluripotent stem cells (“iPSCs”) (Zeng, et al. The Journal of Immunology 188: 4297, 2012; Zeng, et al. Stem Cells Transl Med 3: 69, 2014; Zeng, et al. Scientific Reports 5: 15262, 2015; Zeng, et al. Stem Cell Reports 9: 1796, 2017; Zeng, et al. PLoS One 14: e02168152019). During his 15-year stint in A*STAR, he successfully generated dendritic cells for cancer vaccination and NK cells for adoptive cell transfer from hPSCs (Zeng, et al. The Journal of Immunology 188: 4297, 2012; Zeng, et al. Stem Cells Transl Med 3: 69, 2014; Zeng, et al. Scientific Reports 5: 15262, 2015; Zeng, et al. Stem Cell Reports 9: 1796, 2017). These hPSC-based immune cell technologies open a new frontier in the development of cell-based cancer immunotherapy. In 2017, he has invented an iPSC-based technology to produce a novel type of synthetic immune cells, γδ NKT cells to recognize and kill a wide range of cancers (Zeng, et al. PloS One 14: e02168152019) and the technology has been licensed to us in 2018. Dr. Zeng received his Ph.D. from National University of Singapore (Singapore) in 2004 and M.D. from Sun Yat-Sen University (China) in 1996. We believe that Dr. Zeng’s professional achievements in translational medicine qualifies him to serve on our Board.
Dr. TAN Wee Kiat, Ph.D., has served as our Chief Operating Officer since February 2019 and was appointed to the Board on April 18, 2023. He has also been serving as the Company’s Co-Chief Executive Officer since January 1, 2024. He oversees operations of the Company, including facility management, human resource, manpower allocation, manufacturing, quality control and assurance of cell therapy products, clinical trial operations, R&D, budgeting of operation, compliance and support business development function. Prior to that, he served as the Company’s Chief Production Officer and Chief Technology Officer from July 2018 to January 2019 where he was tasked to develop the Company’s technology into a clinically ready platform for clinical trial application. Dr. Tan also serves as a director of Puricell Lab Pte Ltd since its incorporation in October 2020. Dr Tan has also been appointed as a director of CytoMed Therapeutics (Malaysia) Sdn Bhd starting from November 11, 2022. Prior to joining CytoMed, he served in Tessa Therapeutics Pte Ltd, a biotechnology company engaged in the use of virus specific T cells to treat cancer, as a research scientist from August 2017 to July 2018. Dr. Tan completed his Ph.D. in 2016 following cancer research at the Institute of Bioengineering and Nanotechnology, A*STAR. He graduated with a Bachelor of Science in 2012, both degrees from the National University of Singapore (Singapore). Dr. Tan has a GCP qualification. We believe that Dr. Tan’s background in biopharmaceutical industry operations and academic research qualifies him to serve on our Board.
Ms. GOH Yvonne has served as our Chief Financial Officer since March 2020. She oversees the functions relating to finance, accounting, reporting and procurement. Prior to joining CytoMed, Ms. Goh served as Finance Manager at SBI Offshore Limited) (SGX-ST stock code: 5PL), a public company listed in Singapore engaged in the oil and gas industry, from September 2017 to February 2020. Prior to assuming the position of Finance Manager at SBI Offshore Limited, Ms. Goh was the Assistant Finance Manager from December 2016 to September 2017, Accountant from May 2016 to November 2016 and Senior Accounts Executive from April 2015 to April 2016. From July 2010 until April 2015, Ms. Goh was the Senior Executive – Finance at Leeden National Oxygen Limited, a welding, gas and safety integration specialist based in Singapore. Prior to that, Ms. Goh held accounting roles in various private companies. Ms. Goh is a Chartered Accountant (CA) of the Institute of Singapore Chartered Accountants (ISCA) since 2016. We believe that Ms. Goh’s broad operational management experience in listed companies and experience in accounting qualifies her to serve as our Chief Financial Officer.
| 116 |
Ms. TAN Yoong Ying has served as our Chief Corporate Officer since June 2018 where she oversees legal compliance, business administration, public and government relation, facility management, human resource and corporate secretarial. She has been a director of all our subsidiaries in Malaysia and Singapore since their incorporation, including CytoMed Malaysia, starting from June 2018, IPSCBank Pte Ltd, IPSC Depository Sdn. Bhd. Starting from June 2019, Puricell, starting from October 2020 and Advance Cancer Centre Pte Ltd, starting from February 2020. Prior to these roles with the Company’s subsidiaries, Ms. Tan was a director of Advantage Mining Sdn Bhd from March 2016 until June 2018. From November 2014 to March 2017, she served as the Department Manager of Pulai Mining Sdn Bhd, a private gold mining company in Malaysia, where she participated actively in daily operation management, corporate restructuring and acquisition. Ms. Tan holds a Postgraduate Degree in Legal Practice from BPP Law School (United Kingdom) graduated in 2014, LLM from University of Aberdeen (United Kingdom) in 2012, and a Bachelor Degree in Laws from Jinan University (China) in 2011.
Prof. LOH Yuin Han, Ph.D. was appointed to our Board and its Nominating and Corporate Governance Committee on April 18, 2023. He has served as a Research Director at A*STAR’s IMCB since April 2021, where he also serves as the Division Director overseeing research in Cell Therapies. Concurrently, he is an Associate Professor (Adjunct) at the NUS Yong Loo Lin School of Medicine, National University of Singapore (NUS) Faculty of Science and NUS Graduate School of Integrative Sciences and Engineering starting from July 2018. Prior to joining IMCB, Prof. Loh was a Postdoctoral fellow at the Harvard Medical School, Boston Children’s Hospital, Division of Pediatric Hematology and Oncology from July 2018 to July 2011. There he was trained under the tutelage of Dr. George Q. Daley, one of the first scientists to successfully convert human skin cells to iPSCs. Prof. Loh’s research focuses on dissecting the mechanisms regulating cell fate changes and developing novel tools for the use of stem cells in clinical cell-based therapies. He was the first in the world to describe the regulatory factors which play essential roles in maintaining the self-renewal and pluripotency of embryonic stem cells (Loh et al. Nature Genetics. 2006), and uncovered the method to reverse the cell fate of human blood cells into reprogrammed stem cells (Loh et al. Cell Stem Cells. 2010), which facilitated the modelling of congenital and somatic hematologic diseases using iPSCs (Agarwal S and Loh YH et al. Nature 2010). He was part of the team which demonstrated the use of synthetic mRNA to reprogramming human fibroblast cells (Warren et al. Cell Stem Cells 2010). The work was selected as one of the Top 10 breakthroughs of the year by Science journal, and the technology was spun off as Moderna Therapeutics. In recent years, his group pioneered the discoveries of endogenous retro-element (Yang et al. Cell 2015) and histone chaperones (Fang et al. Nature Communications 2018) as guardians of embryonic stem cell fates. Prof. Loh’s publications are highly regarded by the research community, and have been cited close to 13,000 times by peers worldwide. His research work has earned him several prestigious national and international accolades including the National Research Foundation Investigatorship Award 2018, Stem Cell Society Singapore Young Investigator Award 2015, World Technology Network Fellowship (Biotechnology) 2012, MIT TR35 Asia Pacific Award 2012, A*STAR Investigatorship Research Award 2011, Singapore Youth Award for Science and Technology 2010 (The highest accolade for youth under 35 year old), Singapore National Academy of Science Young Scientist Award 2009, and Quest Technology Award 1997. He serves on both the International and the Education Committee of the International Society for Stem Cell Research (ISSCR) since January 2018, the Executive Council for the Singapore Association for the Advancement of Science (SAAS) since 2013, and the Executive Committee of the Stem Cell Society Singapore (SCSS) since 2012. From 2003 to 2008, Prof. Loh did his Ph.D. study as a pioneer batch of A*STAR Graduate Scholar at the NUS Graduate School of Integrative Sciences and Engineering, and was awarded the Philip Yeo Award (prize money contributed by Nobel Laureate Sydney Brenner) for the best publication. Prof. Loh studied Biology and graduated with B.Sc. 1st Class Honours in 2003 from the National University of Singapore (Singapore), where he was awarded the HSBC Scholarship. We believe that Prof Loh’s strong academic background in cancer research and his recognition in such fields qualifies him to serve on our Board.
Dr. YEW Chak Hua, M.D. was appointed to our Board and its Compensation Committee on April 18, 2023. He has served as a medical consultant in the specialty of regenerative medicine in various clinics and centers in Malaysia since March 2020. With more than 15 years medical practice experience, Dr. Yew has a deep interest in cellular therapy and stem cells-derived cytokines to treat various chronic degenerative disease such as glaucoma, diabetes, osteoarthritis, sexual dysfunction, sports injury, renal impairment, Alzheimer’s disease, and stroke. Dr. Yew obtained his Letter of Credentialing and Privileging in Aesthetic Medicine Practice in 2017, and passed the membership from International Council of Ophthalmology (UK) in the same year. Prior to that, Dr. Yew served as Medical Officer in various hospitals, healthcare group and clinics across Malaysia. Dr. Yew received his M.SC in Regenerative Medicine from UCSI University (Malaysia) in 2014, and M.B.B.S from University of Malaya (Malaysia) in 2007. We believe that Dr. Yew’s record in practice of medical practice and his broad understanding of regenerative medicine qualifies him to serve on our Board.
| 117 |
Mr. Mark LEONG Kei Wei was appointed to our Board on April 18, 2023. Also on April 18, 2023, he was appointed as the Audit Committee financial expert, Chair of the Audit Committee and Chair of the Compensation Committee. He presently serves as an independent director and audit committee chairman of four SGX-ST listed companies – LMIRT Management Ltd (SGX-ST stock code: D5IU), the manager of Lippo Malls Indonesia Retail Trust, HS Optimus Holdings Limited (SGX-ST stock code: 504), mDR Limited (SGX-ST stock code: Y3D) and 9R Limited (SGX-ST stock code: 1Y1). He also serves on an ASX listed company – as an executive chairman of Osteopore Limited (ASX stock code: OSX). From April 2023 to December 2023, he was the executive chairman of Lifebrandz Ltd (SGX-ST stock code: 1D3). From March 2021 to April 2023, he was a director of a financial advisory services firm Auspac Financial Advisory Pty Ltd in Australia, and a director of its Singapore subsidiary Auspac Management Services Pte Ltd since September 2021. Mr. Leong was the Chief Operating Officer of SBI Offshore Limited (SGX-ST stock code: 5PL), a company in the Oil & Gas industry from November 2017 to August 2020. Prior to this, he was a director of Pulai Mining Sdn Bhd, an exploration and mining company, from January 2016 to February 2017. From May 2012 to March 2013, he held the role of Vice President (Finance and Investment) of a family office, Colossus Holdings Pte Ltd. From January 2010 to April 2012, Mr. Leong held the dual role of Chief Development Officer and Deputy Chief Executive Officer of Atos Wellness Ltd (ASX stock code: ATW). Between 2002 and 2009, he undertook Chief Financial Officer roles in two SGX-ST listed companies, Westcomb Financial Group Limited (SGX-ST stock code: 5EC) and a subsidiary within Swiber Holdings Ltd (SGX-ST stock code: BGK). Prior to that, he was a senior auditor with KPMG from August 1999 to October 2001. Mr. Leong is a Fellow of the Association of Chartered Certified Accountants (“ACCA”), a Chartered Accountant of the Institute of Singapore Chartered Accountants (“ISCA”) and a Member of the Singapore Institute of Directors (“SID”). We believe that Mr. Leong’s history of serving as executive officer at a publicly traded companies and his broad accounting experience qualifies him to serve on our Board.
Mr. WU Tao Thomas was appointed to our Board and the Audit Committee on April 18, 2023. He has nearly three decades of experience in finance services. He has served as Chief Executive Officer at Noah Holdings Singapore Pte Ltd (wholly owned subsidiary of Noah Holdings Limited) since June 2019. From September 2017 until August 2018, he was Managing Director, Head of Asia Pacific Corporate Strategy at Julius Baer. Prior to that, Mr. Wu was Chief Financial Officer at Xunlei Ltd (Nasdaq stock code: XNET) from November 2013 to September 2017, and Noah Holdings Limited (NYSE stock code: NOAH) from March 2010 to November 2013. He also served as an Independent Director and Audit Committee member of Seven Days Inn (NYSE stock code: SVN) from October 2011 to October 2013. From March 2007 to February 2010, Mr. Wu was a senior portfolio manager at Alliance Bernstein L.P. based in San Francisco and New York, where he served many Asian institutional clients and retail partners. Prior to that, Mr. Wu was a senior high yield analyst at Moody’s Investors Services based in New York from February 2005 to March 2007, a senior vice president at the investment banking division of Development Bank of Singapore based in Hong Kong from 2001 to 2005 and a Vice President at the mergers and acquisitions division at J.P. Morgan & Company from 1994 to 2001. Mr. Wu received his Master’s Degree in Public Administration from Syracuse University (United States) in 1992 and his Bachelor’s Degree in Mathematics from Grinnell College (United States) in 1987. We believe that Mr. Wu’s strong background in financial analysis and proven record in management positions in investment management qualifies him to serve on our Board.
Dr. TOH Keng Kiat, M.D. was appointed to our Board on April 18, 2023. Also on April 18, 2023, he was appointed as the Chair of the Nominating and Corporate Governance Committee and as a member of the Audit Committee. He is a medical doctor by training and practicing haematologist. He is currently practicing as a Consultant Haemato-Oncologist, beginning from November 2020, at the International Cancer Specialists Pte Ltd in Singapore and also as the Medical Director, starting from August 2017, of Cryoviva Singapore, a AABB accredited cord blood and cord tissue bank with operations in India, Bangkok, Singapore, the Middle East and Vietnam. Dr. Toh was an Associate Professor of Medicine (Practice) of Monash University Malaysia campus since 2007 and retired in January 2023. He was Medical Director of Singapore’s first private cord blood bank, Cordlife Group Limited (now a SGX-listed company. SGX: P8A), from 2005 to 2008. In 1975, Dr. Toh’s early career was with the Singapore Blood Bank and the Ministry of Health Singapore Department of Haematology at the Singapore General Hospital. He remained a visiting consultant there until 2007 before going into private practice. Dr. Toh was a nominated member of Parliament in the Singapore Government from 1992 to 1994, a member of Rotary International and was President from 1998 to 1999 during the Rotary International Convention in Singapore. Dr. Toh received his FRCP© in 1980, M.R.C.P (United Kingdom) in 1970, both from University of Edinburgh (United Kingdom), and M.B.B.S. from University of Singapore in 1965. He is a Fellow of the Royal Colleges of Physicians of London, Edinburgh, Glasgow and the International Society of Haematologists. We believe that Dr. Toh’s decades of experience in haematology and managerial experience as a medical director qualifies him to serve on our Board.
| 118 |
Scientific Advisory Board
Our Scientific Advisory Board (“SAB”) is comprised of the following individuals:
| Name | Age | Position | ||
| Dr. Harry ZEMON | 58 | Scientific Advisory Board | ||
| Dr. TOO Heng Phon | 65 | Scientific Advisory Board | ||
| Dr. Ahmad Radzi Bin AHMAD BADRUDDIN | 66 | Scientific Advisory Board | ||
| Dr. WANG Shu | 68 | Scientific Advisory Board |
The following sets forth certain biographical information with respect to the members of our SAB:
Dr. Harry ZEMON, M.D., FACS was appointed to our SAB on April 18, 2023, He is a practicing surgical oncologist with a focus on solid foregut tumors and has been in practice for over 20 years. He is board certified by the American Board of Surgery since 2006 and is affiliated with White Plains Hospital (United States) and Greenwich Hospital (United States). Dr. Zemon has been the Chair of Surgery at Westmed Medical Group in Westchester, New York since 2014. Prior to that, he served as attending surgeon in North Shore University Hospital, Lake Success (United States) and Long Island Jewish Medical Center (United States) from 2007 to 2013, and Hudson Valley Surgical Group, Sleepy Hollow (United States) from 2006 to 2008. Dr. Zemon has been a member of American College of Surgeons since 2005. He has authored several peer-reviewed publications and book chapters with the topics focusing on laparoscopic surgery and organs transplantation. Dr. Zemon completed fellowships at the Johns Hopkins University (United States) in surgical oncology in 2006 and cancer immunobiology in 2003. His fellowship in immunobiology focused on adoptive immunotherapy under the guidance of Dr. Jonathan Schneck of the Johns Hopkins’ Schneck’s Immunology and Cancer Immunotherapy Laboratory (United States). He completed his medical school and surgical training at George Washington University School of Medicine (United States) in 1998.
Dr. TOO Heng Phon Ph.D. was appointed to our SAB on April 18, 2023. He has been a faculty member in the Department of Biochemistry, National University of Singapore since 1994, and was a Scientific Advisor to the Biotransformation Innovation Platform and a Scientist in the Bioprocess Technological Institute, A*STAR since September 2014. His laboratory was funded by Roche Diagnostics (United States & Asia Pacific) and National Institute of Health (United States) to develop qPCR assays for infectious diseases. Recently, his team developed a non-viral gene delivery technology to modify mesenchymal stem cells. There are intellectual property protections of his specific diagnostic and biotechnology platforms with various agencies and with Massachusetts Institute of Technology, United States. He is also a co-founder and Chairman of MiRXES Pte Ltd, a Singapore molecular diagnostic company since 2014. Prior to that, from June 1986 to March 1993, he was a Research & Teaching Fellow at Department of Anesthesiology and Department of Biological Chemistry & Molecular Pharmacology Harvard Medical School where he was a recipient of the Merck Sharpe Dohme Academic Development Fellowship. He also received training in the Medical Research Council, Cambridge (United Kingdom), where he was a Procter & Gamble Fellow from January 1985 to December 1986, Dr. Too was a Fellow of the Singapore Massachusetts Institute of Technology Alliance in the Molecular Engineering of Biological & Chemical Systems program in 2000 and co-chaired the Chemical & Pharmaceutical Engineering program in 2018. Dr. Too received his PhD from Imperial College of Science & Technology (United Kingdom) and Institute of Ophthalmology (United Kingdom) in 1985 and BSC (Hon), ARCS from Imperial College of Science & Technology (United Kingdom) in 1982.
Dr. Ahmad Radzi Bin AHMAD BADRUDDIN was appointed to our SAB on April 18, 2023. He has been the Chief Executive Officer and Consultant Clinical Oncologist at the Integrated Oncology Centre in Johor Bahru, Malaysia since April 2012. Dr. Radzi currently serves as the Sessional Consultant Clinical Oncologist both at the Gleneagles Medini Hospital, starting from November 2019, in Nusajaya Johor, Malaysia and at Hospital Pantai Kuala Lumpur, beginning from March 2014, in Kuala Lumpur, Malaysia. He is also concurrently serving as Visiting Consultant Clinical Oncologist both at the Sunway Medical Center, starting from August 2016 in Selangor, Malaysia and at the Oncolife Centre, beginning from July 2017, in Kuala Lumpur, Malaysia. Dr. Radzi was Advisory Board Member for Roche in Malaysia, from 2010 to 2017, for Roche’s chemotherapy treatments, where he was the Chairman for the Tarceva Subcommittee and the Co-Chairman for the Avastin Subcommittee. He was also an Advisory Board Member for AstraZeneca in Malaysia from 2010 until 2017. Dr. Radzi received his Medical Doctor degree from the Universiti Kebangsaan Malaysia (Malaysia) in 1984, his Diploma in Medical Radiotherapy specializing in clinical oncology from the Royal College of Radiologists at the University of London (United Kingdom) in 1989, and his Fellowship of the Faculty of the Royal College of Surgeons, also specializing in clinical oncology, at the Royal College of Surgeons Ireland (Ireland) in 1991.
| 119 |
Dr. WANG Shu, M.D., Ph.D. was appointed to our SAB on April 18, 2023. He is one of our scientific founders. Dr. Wang is awarded with the prestigious title of Emeritus Professorship on July 1, 2022 for his service as a Professor at the Department of Biological Sciences, National University of Singapore (Singapore) since 2002, specializing in immunotherapy, stem cell therapy, and gene therapy with research emphasis on developing platform technologies applicable to cancer treatment. Prior to that, from May 2003 to June 2019, he served as a Lab Principal Investigator, Principal Research Scientist at the Institute of Bioengineering and Nanotechnology (Singapore), focusing on gene therapy. Dr. Wang has published more than 160 peer-reviewed scientific articles in the gene therapy areas a 58 Google Scholar. His publications have over 12,000 citations worldwide. In recent years, he has been working towards building platform technologies that support the development of novel immune cell-based therapeutics. Powerful cell generation, expansion, and modification technologies used in the production process for cancer immunotherapy have been established. His research focuses in several new cellular therapeutics in the area of chimeric antigen receptor (CAR) adoptive immunotherapy and with potential for use against a broad range of cancers are also under testing currently. Dr. Wang received his M.D. and Ph.D. in Cell Biology and Neurobiology from Medical College, University of Göteborg (Sweden) in 1993.
In December 2017, Dr. WANG Shu was the subject of a formal investigation by A*STAR, Institute of Bioengineering and Nanotechnology, Singapore for not adequately supervising a doctoral student that fabricated data in his research. As a consequence of the findings, it was deemed necessary that five (5) publications which incorporated the doctoral student’s fabricated data and which he co-authored be retracted. Dr. WANG is a professor at the National University of Singapore as of the date of this annual report.
Family Relationships
There are no family relationships or other arrangements among our Directors and executive officers. We are not aware of any conflicts of interests related to our Directors and executive officers arising from the management and operations of our business.
| B. | Compensation |
The table below sets forth information regarding the total compensation awarded to and earned by the Company’s NEOs for services rendered during the financial year ended December 31, 2023:
| Name and Principal Position | Year | Salary (U.S.$) | Bonus (U.S.$) | All Other Compensation (U.S.$) | Total (U.S.$) | |||||||||||||||
| CHOO Chee Kong, | 2023 | 23,043 | - | 4,273 | 27,316 | |||||||||||||||
| Chairman (1)(2)(3) | ||||||||||||||||||||
| Dr. ZENG Jieming, | 2023 | 88,226 | 1,516 | - | 89,742 | |||||||||||||||
| Chief Scientific and Medical Officer (3) | ||||||||||||||||||||
| Dr. TAN Wee Kiat, | 2023 | 86,614 | 1,516 | - | 88,130 | |||||||||||||||
| Co-Chief Executive Officer and Chief Operating Officer (3) | ||||||||||||||||||||
(1) During the financial year ended December 31, 2023, Mr. Choo was party to a service agreement with CytoMed Malaysia, dated September 8, 2021, for which he received a monthly service fee.
(2) The monthly service fee for Mr. Choo reported above was paid in Malaysian Ringgit and is reported above based on a rate of MYR4.5903 to U.S.$1.00, the exchange rate in effect as of December 31, 2023, as set forth in the H.10 statistical release of the U.S. Board of Governors of the Federal Reserve System.
(3) The salary and bonus for Mr. Choo, Drs. Zeng and Tan reported above was paid in Singapore dollars and is reported above based on a rate of S$1.3193 to U.S.$1.00, the exchange rate in effect as of December 31, 2023, as set forth in the H.10 statistical release of the U.S. Board of Governors of the Federal Reserve System.
Service Agreements with NEOs
Effective April 18, 2023, we entered into service agreements with each of our NEOs. Such service agreements do not have a specified term and provide that such services agreements are terminable for cause at any time. The terms of these service agreements are substantially similar to each other. An NEO may terminate his employment at any time upon three-months prior written notice. We may immediately terminate in the event of default, misconduct or any breach or non-observance by the employee.
Each NEO has agreed to hold in strict confidence and not to use, except for the benefit of our Company, any proprietary information, technical data, trade secrets and know-how of our Company or the confidential or proprietary information of any third party, including our subsidiaries and our clients, received by our Company. Each of our NEOs has agreed to be bound by restrictive covenants governing confidentiality, a non-solicitation and non-competition restriction that applies to employees, client, customers and executives for one year following termination, and confirmation that all intellectual property developed during the term of his employment belongs exclusively to the Company.
Director Compensation
Our Board has executed an Independent Director Appointment Letter with each of our non-employee Directors, effective from April 2023. Each appointment letter includes reimbursement for out-of-pocket direct expenses incurred in connection with meeting attendance and provides that we will pay each of our non-employee Directors annual cash retainers for service on the Board and for service on each committee on which the Director is a member. The chairperson of each committee will receive a higher annual retainer for such service. These retainers are payable in arrears in four equal installments on the last day of each quarter, provided that the amount of such payment will be pro-rated for any portion of such quarter that the Director is not serving on our Board or the applicable committee. The retainers to be paid to non-employee Directors for service on the Board and for service on each committee of the Board on which the Director is a member are as follows:
| Position | Member Annual Retainer | Chairperson Annual Retainer | ||||||
| Board of Directors | U.S.$ | 12,000 | U.S.$ | 3,600 | ||||
| Audit Committee | U.S.$ | 4,200 | U.S.$ | 2,100 | ||||
| Compensation Committee | U.S.$ | 3,000 | U.S.$ | 1,500 | ||||
| Nominating and Corporate Governance Committee | U.S.$ | 3,600 | U.S.$ | 1,800 | ||||
Subject to shareholders’ approval at a general meeting, the Compensation Committee and the Directors shall have the discretion to increase the non-employee Directors’ compensation. In addition, non-employee Directors will be eligible to participate in the Incentive Plan and may be granted share options and/or restricted shares under the Incentive Plan from time to time.
Executive Clawback Compensation Policy
On November 16, 2023, the Board adopted an Executive Compensation Recovery Policy (the “Clawback Policy”) providing for the recovery of certain incentive-based compensation from current and former executive officers of the Company in the event the Company is required to restate any of its financial statements filed with the SEC under the Exchange Act in order to correct an error that is material to the previously-issued financial statements, or that would result in a material misstatement if the error were corrected in the current period or left uncorrected in the current period. Adoption of the Clawback Policy was mandated by new Nasdaq listing standards introduced pursuant to Exchange Act Rule 10D-1. The Clawback Policy is in addition to Section 304 of the Sarbanes-Oxley Act of 2002 which permits the SEC to order the disgorgement of bonuses and incentive-based compensation earned by a registrant issuer’s chief executive officer and chief financial officer in the year following the filing of any financial statement that the issuer is required to restate because of misconduct, and the reimbursement of those funds to the issuer. A copy of the Clawback Policy has been filed herewith as Exhibit 97.1.
| C. | Board Practices |
Board of Directors
Our Board consists of 9 directors, including 4 executive (non-independent) directors and 5 independent directors. We established an Audit Committee, a Nominating and Corporate Governance Committee and a Compensation Committee on April 18, 2023. We adopted a charter for each of the three committees. Each of the committees of our Board has the composition and responsibilities described below.
Board Diversity
The table below sets forth the board diversity matrix of our board of directors as of the date of this annual report pursuant to NASDAQ’s Board Diversity Rule:
Board Diversity Matrix (As of April 22, 2024)
| ||||
Country of Principal Executive Offices:
|
Singapore | |||
Foreign Private Issuer:
|
Yes | |||
Disclosure Prohibited under Home Country Law:
|
No | |||
Total Number of Directors:
|
9 | |||
| Female | Male | Non-Binary | Did Not Disclose Gender
| |
Part I: Gender Identity
|
||||
Directors
|
0 | 9 | 0 | 0 |
Part II: Demographic Background
|
||||
Underrepresented in Home Country Jurisdiction
|
0 | |||
LGBTQ+
|
0 | |||
Did Not Disclose Demographic Background
|
0 | |||
Audit Committee
The Board has established an audit committee of the Board. Our audit committee is comprised of three (3) independent Directors, namely Mark LEONG Kei Wei, Dr. TOH Keng Kiat and WU Tao Thomas, with Mark LEONG Kei Wei serving as chairperson. Our Board has determined that Mark LEONG Kei Wei is an “audit committee financial expert” as defined by applicable SEC rules and the Nasdaq Listing Rules and regulations.
| 120 |
The Board has adopted an audit committee charter, which details the principal functions of the audit committee, including the following:
| ● | general oversight of the integrity of our financial statements, qualifications and independence of our independent auditors and internal financial and accounting controls; | |
| ● | appointing, approving compensation arrangements, retaining, evaluating, terminating and overseeing our independent registered public accounting firm; | |
| ● | reviewing and discussing with our independent registered public accounting firm its independence from us; | |
| ● | reviewing with our independent registered public accounting firm the matters required to be reviewed by applicable auditing requirements; | |
| ● | approving all audit and permissible non-audit services to be performed by our independent registered public accounting firm; | |
| ● | overseeing the financial reporting process and discussing with management and our independent registered public accounting firm the interim and annual financial statements that we file with the SEC and ACRA; | |
| ● | reviewing and approving related person transactions; | |
| ● | reviewing and monitoring our internal controls, disclosure controls and procedures and compliance with legal and regulatory requirements; and | |
| ● | establishing procedures for the confidential, anonymous submission of concerns regarding questionable accounting, internal accounting controls, and auditing matters. |
Compensation Committee
The Board has established a compensation committee of the Board comprise of two (2) independent Directors, namely Mark LEONG Kei Wei and Dr. YEW Chak Hua, with Mark LEONG Kei Wei serving as chairperson.
The Board has adopted a compensation committee charter, which details the principal functions of the compensation committee, including the following:
| ● | reviewing compensation goals, policies, plans and programs for our executive officers; | |
| ● | reviewing and approving the compensation of our executive officers; | |
| ● | reviewing and approving employment agreements and other similar arrangements between us and our executive officers; and | |
| ● | reviewing and recommending to our Board the compensation to be provided to our Directors. |
Nominating and Corporate Governance Committee
The Board has established a nominating and corporate governance committee of the Board. Our nominating and corporate governance committee consists of two (2) independent Directors namely Dr. TOH Keng Kiat and Prof. LOH Yuin Han, with Dr. TOH Keng Kiat serving as chairperson.
The Board has adopted a nominating and corporate governance committee charter, which detailed the principal functions of the nominating and corporate governance committee, including the following:
| ● | identifying individuals qualified to become members of our Board, consistent with criteria approved by our Board; | |
| ● | overseeing the organization of our Board to discharge the Board’s duties and responsibilities properly and efficiently; and | |
| ● | reviewing and recommending to our Board appropriate changes to our corporate governance guidelines. |
Compensation Committee Interlocks and Insider Participation
None of the members of the compensation committee is or has been at any time, one of our executive officers or employees. None of our executive officers serve as a member of the Board or compensation committee of any entity that has one or more executive officers serving as a member of our Board or on our compensation committee.
| 121 |
Duties of Directors
Under Singapore law, directors of a Singapore company owe certain fiduciary duties towards the company, including a duty to act in good faith in the best interests of the company, a duty to act honestly and to use reasonable diligence in the discharge of the duties of their office. Directors generally owe fiduciary duties to the Company, and not to the Company’s shareholders. Our shareholders may not have a direct cause of action against our Directors for any breaches of fiduciary duties. The Company has a right to seek damages if a duty owed by Directors is breached, subject to the applicable laws on quantification of damages.
Terms of Directors and Officers
Each of our Executive Directors and Executive Officers (each an “Executive” for the purposes of this section) have a service agreement that may be terminated by either party with the provision of at least three (3) months’ prior written notice, or payment of three (3) months’ salary in-lieu of notice, to the other party. We may also terminate the employment of any Executive at any time without notice or payment in-lieu of notice under certain circumstances, such as if the Executive becomes bankrupt, is or may be suffering from a mental disorder, is convicted of any criminal offence, save for an offence under road traffic legislation for which he is not sentenced to any term of imprisonment, and sentenced to any term of imprisonment, becomes prohibited by our Constitution, any law or regulatory or governmental authority, from being or ceases to be, an employee or executive or director of the Company, for any reason, commits any act that is likely to discredit the Executive or the Company through association, has acted in willful misconduct in the performance of his duties, or breaches any material provision of the service agreement. The service agreements will automatically terminate if the Executive is found guilty of any willful misconduct in the discharge of his duties or responsibilities, or series or repeated breach or non-observance by the Executive of any term in the service agreement.
Our officers, such as our Chairman and our Chief Financial Officer, are appointed by and serve at the discretion of our board of directors.
Subject to the provisions of any relevant laws, pursuant to our Constitution, one-third of the Directors for the time being or, if the number is not a multiple of three, the number nearest to but not less than one-third with a minimum of one, shall retire from office at each annual general meeting of our Company.
| D. | Employees |
As of the date of this Report, we have thirty-four (34) full-time employees. Of these employees, twenty-eight (28) employees are engaged in our R&D and manufacturing activities. The rest of our employees carry out office administration and finance functions.
Our operations are currently managed by Chief Scientific and Medical Officer, Dr. ZENG Jieming, and Co-Chief Executive Officer and Chief Operating Officer, Dr. TAN Wee Kiat. Pursuant to our employment agreements with Dr. ZENG Jieming and Dr. TAN Wee Kiat, both Dr. ZENG Jieming and Dr. TAN Wee Kiat are required to keep all proprietary and confidential information entrusted to them confidential during and after their employment and all intellectual properties developed by Dr. ZENG Jieming and Dr. TAN Wee Kiat belong exclusively to the Company.
We rely heavily on non-disclosure agreements with our employees to protect our confidential and proprietary information. Both Dr. ZENG Jieming and Dr. WANG Shu are the inventors of our CTM-N2D, CTM-GDT and iPSC-gdNKT cell technologies and the co-founders and shareholders of the Company.
We currently rely on substantial growth activities on our existing R&D team including key aspects of clinical development and manufacturing and only outsource certain quality services and tests and BAS software to the qualified independent organizations, advisors and consultants.
We have generally been able to retain valuable employees, members of our management, scientific and development team. We maintain a good working relationship with our employees and we have not experienced any labor disputes.
Incentive Plan
For future equity awards, our Board has adopted the Incentive Plan to provide an additional means through which to the grant of awards to attract, motivate, retain and reward selected key employees and other eligible persons, including our NEOs. We have also obtained approval for the Incentive Plan from our shareholders. A summary of our Incentive Plan is set out below.
Shares Subject to the Incentive Plan
A total of 1,279,117 ordinary shares will be available for issuance under the Incentive Plan. If an award granted under the Incentive Plan is forfeited, canceled, settled, or otherwise terminated without a distribution of shares, the shares underlying that award will again become available for issuance under the Incentive Plan. If shares delivered under the Incentive Plan are tendered or withheld to pay the exercise price of a share option or to satisfy withholding taxes, such shares will again become available for issuance under the Incentive Plan.
| 122 |
Administration of the Incentive Plan
Our Board or a committee appointed by the Board will administer the Incentive Plan as the plan administrator and will have broad authority to:
| ● | select participants and determine the types of awards that they are to receive; |
| ● | determine the number of shares that are to be subject to awards and the terms and conditions of awards, including the price (if any) to be paid for the shares or the award and establish the vesting conditions (if applicable) of such shares or awards; |
| ● | cancel, modify or waive our rights with respect to, or modify, discontinue, suspend or terminate any or all outstanding awards, subject to any required consents; |
| ● | construe and interpret the terms of the Incentive Plan and any agreements relating to the Incentive Plan; |
| ● | determine whether awards will be settled in cash, ordinary shares, other securities, other property, or in any combination thereof; |
| ● | prescribe, amend, and rescind rules and regulations relating to the Incentive Plan; and |
| ● | make all other determinations deemed necessary or advisable for administering the Incentive Plan. |
Participation
Employees, officers, directors, members of our SAB and consultants that provide services to us may be selected to receive awards under the Incentive Plan.
Types of Awards
The Incentive Plan permits the granting of awards in the form of share options and restricted shares.
Share Options
A share option entitles the participant to purchase ordinary shares at a fixed exercise price. The exercise price per share will be determined by the plan administrator in the applicable award agreement in its sole discretion at the time of the grant, but the exercise price cannot be less than the closing sales price for our ordinary shares on the grant date. The exercise price may be paid in cash, check, by surrender of ordinary shares already held by the participant, or by cashless or net exercise. The maximum term of each share option shall be fixed by the plan administrator, but in no event shall an option be exercisable more than ten (10) years after the date such option is granted.
Restricted Shares
A restricted share award is an award of ordinary shares that vests in accordance with the terms and conditions established by the plan administrator.
Equitable Adjustments
In the event of a merger, consolidation, recapitalization, share split, reverse share split, reorganization, split-up, spin-off, combination, repurchase, or other change in corporate structure affecting the ordinary shares, the maximum number and kind of shares reserved for issuance or with respect to which awards may be granted under the Incentive Plan will be adjusted to reflect such event, and the plan administrator will make such adjustments as it deems appropriate and equitable in the number, kind and exercise price of ordinary shares covered by outstanding awards made under the Incentive Plan.
Change in Control
In the event of any change in control (as defined in the Incentive Plan), the plan administrator will take any action it deems appropriate, which action may include, without limitation, the following: (i) the continuation of the awards, if the Company is the surviving corporation; (ii) the assumption of the awards by the surviving corporation or its parent or subsidiary; (iii) the substitution by the surviving corporation or its parent or subsidiary of equivalent awards; (iv) accelerated vesting of the award, with all performance objectives and other vesting criteria deemed achieved at targeted levels, and a limited period during which to exercise the awards prior to closing of the change in control, or (v) settlement of any award for the change in control price (less, to the extent applicable, the per share exercise price).
Term
The Incentive Plan, unless terminated, will continue in effect for a term of ten (10) years from January 18, 2023, the date the Incentive Plan was adopted by the Board.
| 123 |
Amendment and Termination
The Board may at any time amend, alter, suspend or terminate the Incentive Plan, although no such action may, without the written consent of the participant, impair the rights of any participant with respect to outstanding awards.
No options were issued to date.
| E. | Share Ownership |
Please see Item 7. Major Shareholders and Related Party Transactions of this annual report for information relating to ownership of our securities by our directors, officers and certain major shareholders.
| F. | DISCLOSURE OF A REGISTRANT’S ACTION TO RECOVER ERRONEOUSLY AWARDED COMPENSATION. |
Not Applicable.
ITEM 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
| A. | Major Shareholders |
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as of the date of this annual report for:
| ● | each beneficial owner of 5% or more of our outstanding ordinary shares; | |
| ● | each of our directors and NEOs; and | |
| ● | all of our directors and NEOs as a group. |
The following section and tables set forth certain information with respect to the beneficial ownership of our ordinary shares and as adjusted to reflect the sale of the ordinary shares offered by us in the Offering and the 1-for-380.83 reverse split of our ordinary shares effective January 17, 2023.
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include ordinary shares issuable upon the exercise of options that are immediately exercisable or exercisable within sixty (60) days of the date of this annual report. Percentage ownership calculations are based on 11,540,000 ordinary shares, outstanding as of the date of this annual report.
Except as otherwise indicated, all of the shares reflected in the table are ordinary shares and all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
| 124 |
Except as otherwise indicated in the table below, addresses of our directors, executive officers and 5% or greater beneficial owners are in care of CytoMed Therapeutics Limited, 1 Commonwealth Lane, #08-22, Singapore 149544.
Number of shares | %(3) | |||||||
| Shareholders with 5% or more ordinary shares in the Company: | ||||||||
| Glorious Finance Limited | 4,653,604 | 40.33 | % | |||||
| WANG Shu | 883,858 | 7.66 | % | |||||
| mDR Limited (2) | 1,336,923 | 11.59 | % | |||||
| ZENG Jieming | 589,239 | 5.11 | % | |||||
| Directors and NEOs: | ||||||||
| CHOO Chee Kong (1) | 3,157,497 | 27.36 | % | |||||
| ZENG Jieming | 589,239 | 5.11 | % | |||||
| Lucas LUK Tien Wee | 92,563 | * | ||||||
| TAN Wee Kiat | 5,000 | * | ||||||
| Dr. TOH Keng Kiat | - | - | ||||||
| YEW Chak Hua | - | - | ||||||
| Mark LEONG Kei Wei | - | - | ||||||
| WU Tao Thomas | - | - | ||||||
| LOH Yuin Han | ||||||||
| All directors and NEOs as a group (9 persons) | 3,844,299 | 33.31 | % | |||||
* Less than 1%.
| (1) | Consists of: (a) 2,559,482 ordinary shares held by Glorious Finance Limited (“Glorious Finance”). CHOO Chee Kong (“Mr. Choo”) owns 55.0% of shares in Glorious Finance. Mr. Choo disclaims beneficial ownership of 2,094,122 ordinary shares held by Glorious Finance to the extent that he does not have an economic interest therein; (b) 410,515 ordinary shares held by EP Capital Inc. (“EP Capital”). Mr. Choo is deemed to beneficially own the 410,515 ordinary shares held by EP Capital, and (c) 187,500 ordinary shares held directly by Mr. Choo. |
| (2) | mDR Limited is a public listed company on the Mainboard of the SGX-ST. |
| (3) | Rounded to the nearest two decimal places. |
| B. | Related Party Transactions |
Other than compensation arrangements for our Directors and executive officers, which are described elsewhere in this annual report, the following includes a summary of transactions since our incorporation on March 9, 2018 and any currently proposed transactions, to which we were or are to be a participant, in which:
| ● | the amount involved exceeded or will exceed the lesser of (1) U.S.$120,000 or (2) 1% of the average of our total assets; and |
| ● | any of our Directors, executive officers or holders of more than 5% of our ordinary shares, or member of the immediate family of, or person sharing the household with, the foregoing persons, had or will have a direct or indirect material interest. |
Personal Guarantee by CHOO Chee Kong
On December 28, 2018, CytoMed Malaysia obtained credit facilities of RM2 million (approximately U.S.$435,701) from Hong Leong Bank Berhad to purchase the property at 12, Jalan Permas 9/16, Bandar Baru Permas Jaya, 81750 Masai, Johor Darul Ta’zim in Malaysia for the construction of the cGMP Facility, which is secured by a personal guarantee from CHOO Chee Kong, our Director and Chairman. This personal guarantee is expected to be discharged in 2024.
| 125 |
Guarantee by CHOO Chee Kong pursuant to the Convertible Loan Agreement with mDR Limited
On December 10, 2019, the Company entered into the mDR Convertible Loan of up to S$1.50 million to fund the abovementioned purchase of the shares in LMC, expenses for the proposed listing, and working capital purposes. As security for this loan, CHOO Chee Kong and Messiah Limited executed personal and corporate guarantees, respectively, in favor of mDR Limited guaranteeing the obligations of the Company under the agreement. CHOO Chee Kong is also the sole director and a shareholder of Messiah Limited.
On April 21, 2023, the Company converted the mDR Convertible Loan of S$1,500,000 into 589,509 ordinary shares. The personal and corporate guarantees have been thereby discharged after the conversion.
Subscription of Shares
On June 26, 2019, and September 28, 2020, the then-shareholders of the Company agreed to a waiver of their pre-emption rights to allot and issue certain numbers of shares to Glorious Finance Limited, at the consideration sum of S$1 million and S$500,000 respectively.
On February 19, 2021, the then-shareholders of the Company agreed to a waiver of their pre-emption rights to allot and issue certain numbers of shares to Dr Lucas LUK Tien Wee, a director of our Company, at the consideration sum of S$190,355.
On April 15, 2021, we entered into a subscription agreement with Glorious Finance Limited in relation to its subscription of 237,274 ordinary shares in our Company, at consideration sum of S$500,000. The subscription of shares was completed on April 22, 2021.
On April 15, 2021, we entered into a subscription agreement with mDR Limited in relation to its subscription of 711,822 ordinary shares in our Company at consideration sum of S$1.50 million. The subscription of shares was completed on April 22, 2021. In relation to this agreement, CHOO Chee Kong, our Director and Chairman, and mDR Limited also executed a put option agreement on April 15, 2021, allowing mDR Limited to have the right and option to require CHOO Chee Kong to purchase and acquire a certain number of shares in our Company from mDR Limited.
Landmark Investment Agreement
On January 20, 2020, we entered into an investment agreement with, inter alia, LMC, as varied by a supplemental agreement entered into on January 20, 2021 and a further supplemental letter dated October 28, 2021, to acquire new ordinary shares representing 20.0% of the total number of shares in LMC at a subscription consideration of RM1.50 million. The subscription of shares in LMC was completed on September 6, 2021. Dr Lucas LUK Tien Wee, one of the shareholders of LMC and parties of the investment agreement, was appointed a Director of our Company on January 1, 2021.
As our NEO and Chief Clinical Officer, Dr Lucas LUK Tien Wee is deeply involved in our preparation for clinical trial application in Singapore and Malaysia. As Chief Executive Officer and major shareholder of LMC, Dr Lucas LUK Tien Wee brings valuable expertise, experience and connections to facilitate clinical trial preparation including protocol documentation. With deep interest in research, LMC has been purchasing small quantities of R&D products from us since beginning of 2020. The aggregate sum of such transactions for the financial year ended December 31, 2023 is amounted to S$183,518. After we become publicly listed on Nasdaq, such transactions will be subject to the Audit Committee’s scrutiny to ensure they are conducted on arm’s length basis.
Joint Venture Share Award
On October 7, 2020, our Group incorporated Puricell as part of our plans to develop regenerative medicine and awarded 5.0% shareholding amounting to a nominal S$50 in Puricell to LOH Yuin Han, one of our independent Directors. LOH Yuin Han is a senior staff at the IMCB, a research institute under A*STAR. Depending on milestone achievement, LOH Yuin Han’s stake can increase to 20.0%. Such awards are free of cost to LOH Yuin Han.
Policies and Procedures for Related Party Transactions
As of the dates of the related party transactions set forth above, we had not yet adopted a formal policy for the review, approval or ratification of related party transactions. Accordingly, the transactions discussed above were not reviewed, approved or ratified in accordance with any such policy.
However, effective on April 18, 2023, we adopted a code of business conduct and ethics requiring us to avoid, wherever possible, all conflicts of interests, except under guidelines or resolutions approved by our Board (or the appropriate committee of our Board) or as disclosed in our public filings with the SEC. Under our code of business conduct and ethics, conflict of interest situations include any financial transaction, arrangement or relationship (including any indebtedness or guarantee of indebtedness) involving the Company. Our code of business conduct and ethics as adopted is filed as an exhibit to this annual report.
| 126 |
In addition, our audit committee, pursuant to its written charter is responsible for reviewing and approving related party transactions to the extent that we enter into such transactions. An affirmative vote of a majority of the members of the audit committee present at a meeting at which a quorum is present is required in order to approve a related party transaction. A majority of the members of the entire audit committee constitute a quorum. Without a meeting, the unanimous written consent of all of the members of the audit committee is required to approve a related party transaction. The audit committee charter is filed as an exhibit this annual report is a part. We also require each of our Directors and executive officers to complete a Directors’ and officers’ questionnaire that elicits information about related party transactions.
These procedures are intended to determine whether any such related party transaction impairs the independence of a Director or presents a conflict of interest on the part of a Director, employee or officer.
| C. | Interests of Experts and Counsel |
Not applicable for annual reports on Form 20-F.
ITEM 8. FINANCIAL INFORMATION
The financial statements required by this item can be found at the end of this report beginning on page F-1.
Legal or Arbitration Proceedings
As of the date of the annual report, the Company has not been subject to any legal or arbitration proceedings, including those relating to bankruptcy, receivership or similar proceedings and those involving any third party, which may have, or have had in the recent past, significant effects on the company’s financial position or profitability. However, from time to time, we may be subject to legal proceedings arising in the ordinary course of business.
Dividend Policy
Subject to the provisions of the Companies Act and any rights for the time being attaching to any class or classes of shares: (i) our directors may declare dividends or distributions out of our funds which are lawfully available for that purpose and (ii) our shareholders may, by ordinary resolution, declare dividends but no such dividend shall exceed the amount recommended by the directors.
Subject to the requirements of the Companies Act regarding the application of a company’s share premium account and with the sanction of an ordinary resolution, dividends may also be declared and paid out of any share premium account. The directors when paying dividends to shareholders may make such payment either in cash or in specie.
Unless provided by the rights attached to a share, no dividend shall bear interest.
We do not know when or if we will pay dividends to our shareholders (including our public shareholders), and the likelihood that we will be paying dividends on our ordinary is remote at this time. We currently intend to retain future earnings, if any, to finance the expansion of our business. Our future dividend policy is within the discretion of our board of directors and will depend upon various factors, including our business, financial condition, results of operations, capital requirements and investment opportunities.
Significant Changes
There have been no significant changes since the date of the consolidated financial statements included in this annual report.
ITEM 9. THE OFFER AND LISTING
Our ordinary shares are listed on the Nasdaq Capital Market under the symbol “GDTC.”
| A. | Offering and Listing Details |
Not applicable for annual reports on Form 20-F.
| B. | Plan of Distribution |
Not applicable for annual reports on Form 20-F.
| C. | Markets |
Not applicable for annual reports on Form 20-F.
| D. | Dilution |
Not applicable for annual reports on Form 20-F.
| E. | Expense of the Issue |
Not applicable for annual reports on Form 20-F.
| 127 |
ITEM 10. ADDITIONAL INFORMATION
| A. | Share Capital |
Not applicable for annual reports on Form 20-F.
| B. | Constitution of the Company |
A copy of our Constitution is attached as Exhibit 1.1 to this annual report. The information called for by this Item is incorporated by reference to the sections titled “Description of Share Capital” and “Comparison of Shareholder Rights” included in our Rule 424(b)(4) prospectus filed with the SEC on April 17, 2023.
| C. | Material Contracts |
Attached as exhibits to this annual report or incorporated by reference herein are the contracts we consider to be both material and outside the ordinary course of business immediately preceding the date of this annual report. We refer you to “Item 4. Information on the Company”, “Employment Agreements” under “Item 6. Directors, Senior Management and Employees”, and under “Item 7. Major Shareholders and Related Party Transactions” of this annual report for a discussion of these contracts. Other than as discussed in this annual report, we have no material contracts, other than contracts entered into in the ordinary course of business, to which we are a party.
| D. | Exchange Controls |
There are no exchange control regulations or currency restrictions in the Republic of Singapore.
| E. | Taxation |
The following are material Singaporean and U.S. federal income tax considerations relevant to an investment in our ordinary shares. This discussion does not address all of the tax consequences that may be relevant in light of the investor’s particular circumstances. Potential investors should consult their tax advisers regarding the Singaporean, U.S. federal, state and local, and non-U.S. tax consequences of owning and disposing of our ordinary shares in their particular circumstances.
Material Singaporean Tax Considerations
The statements made herein regarding taxation are general in nature and based on certain aspects of current tax laws of Singapore and administrative guidelines issued by the relevant authorities in force as of the date of this annual report and are subject to any changes in such laws or administrative guidelines, or in the interpretation of these laws or guidelines, occurring after such date, which changes could be made on a retrospective basis. These laws and guidelines are also subject to various interpretations and the relevant tax authorities or the courts could later disagree with the explanations or conclusions set out below. The statements below are not to be regarded as advice on the tax position of any holder of our ordinary shares or of any person acquiring, selling or otherwise dealing with our ordinary shares or on any tax implications arising from the acquisition, sale or other dealings in respect of our ordinary shares. The statements made herein do not purport to be a comprehensive or exhaustive description of all of the tax considerations that may be relevant to a decision to acquire, own or dispose of our ordinary shares and do not purport to deal with the tax consequences applicable to all categories of investors, some of which (such as dealers in securities) may be subject to special rules. Prospective holders of our ordinary shares are advised to consult their own tax advisers as to the Singapore or other tax consequences of the acquisition, ownership of or disposal of our ordinary shares. The statements below regarding the Singapore tax treatment of dividends received in respect of our ordinary shares are based on the assumption that the Company is tax resident in Singapore for Singapore income tax purposes. It is emphasized that neither the Company nor any other persons involved in this annual report accepts responsibility for any tax effects or liabilities resulting from the subscription for, acquisition, holding or disposal of our ordinary shares.
Individual Income Tax
An individual is a tax resident in Singapore in a year of assessment (“YA”) if, in the year preceding the YA, he was physically present in Singapore or exercised an employment in Singapore (other than as a director of a company) for 183 days or more, or if he resides in Singapore except for such temporary absences therefrom as may be reasonable and not inconsistent with a claim by such individual to be resident in Singapore.
| 128 |
Individual taxpayers who are Singapore tax residents are subject to Singapore income tax on income accruing in or derived from Singapore. All foreign-sourced income received in Singapore on or after January 1, 2004 by a Singapore tax resident individual (except for income received through a partnership in Singapore) is exempt from Singapore income tax if the Comptroller is satisfied that the tax exemption would be beneficial to the individual. Currently, a Singapore tax resident individual is taxed at progressive rates ranging from 0% to 22%. The maximum tax rate will be increased to 24% with effect from the year of assessment 2024.
Non-resident individuals, subject to certain exceptions and conditions, are subject to Singapore income tax on income accruing in or derived from Singapore at the rate of 22%, and 24% with effect from the year of assessment 2024.
Corporate Income Tax
A corporate taxpayer is regarded as resident in Singapore for Singapore tax purposes if the control and management of its business is exercised in Singapore.
Corporate taxpayers who are Singapore tax residents are subject to Singapore income tax on income accruing in or derived from Singapore and, subject to certain exceptions, on foreign-sourced income received or deemed received in Singapore. Foreign-sourced income in the form of dividends, branch profits and service income received or deemed received in Singapore by Singapore tax resident companies on or after June 1, 2003 are exempt from tax if certain prescribed conditions are met, including the following:
| (i) | such income is subject to tax of a similar character to income tax under the law of the jurisdiction from which such income is received; | |
| (ii) | at the time the income is received in Singapore, the highest rate of tax of a similar character to income tax (by whatever name called) levied under the law of the territory from which the income is received on any gains or profits from any trade or business carried on by any company in that territory at that time is not less than 15%; and | |
| (iii) | the Comptroller is satisfied that the tax exemption would be beneficial to the person resident in Singapore. |
Certain concessions and clarifications have also been announced by IRAS with respect to such conditions.
A non-resident corporate taxpayer is subject to income tax on income that is accrued in or derived from Singapore, and on foreign-sourced income received or deemed received in Singapore, subject to certain exceptions.
The corporate tax rate in Singapore is currently 17%. Preferential tax rate(s) may be available under certain tax incentives / schemes. In addition, three-quarters of up to the first S$10,000 of a company’s annual normal chargeable income, and one-half of up to the next S$190,000, is exempt from corporate tax from the YA 2020 onwards. The remaining chargeable income (after the tax exemption) will be fully taxable at the prevailing corporate tax rate.
New start-up companies will also, subject to certain conditions and exceptions, be eligible for tax exemption on three-quarters of up to the first S$100,000 of a company’s annual normal chargeable income, and one-half of up to the next S$100,000, a year for each of the company’s first three YAs from YA 2020 onwards. The remaining chargeable income (after the tax exemption) will be taxed at the applicable corporate tax rate.
Dividend Distributions
All Singapore-resident companies are currently under the one-tier system.
Dividends received in respect of our ordinary shares by either a resident or non-resident of Singapore are not subject to Singapore withholding tax, on the basis that we are a tax resident of Singapore and under the one-tier system.
| 129 |
Under the one-tier system, the tax on corporate profits is final and dividends paid by a Singapore resident company are tax exempt in the hands of a shareholder, regardless of whether the shareholder is a company or an individual and whether or not the shareholder is a Singapore tax resident.
Gains on Disposal of our Ordinary Shares
Singapore does not impose tax on capital gains. There are no specific laws or regulations which deal with the characterization of whether a gain is income or capital in nature. Gains arising from the disposal of our ordinary shares may be construed to be of an income nature and subject to Singapore income tax, especially if they arise from or are otherwise connected with the activities which the IRAS regards as carrying on a trade or business in Singapore.
Holders of our ordinary shares who apply, or who are required to apply, the FRS 39, FRS 109 or SFRS(I) 9 (as the case may be) may for the purposes of Singapore income tax be required to recognize gains or losses (not being gains or losses in the nature of capital) in accordance with the provisions of FRS 39, FRS 109 or SFRS(I) 9 (as modified by the applicable provisions of Singapore income tax law) even though no sale or disposal of our ordinary shares is made.
Holders of our ordinary shares who may be subject to this tax treatment should consult their accounting and tax advisers regarding the Singapore income tax consequences of their acquisition, holding and disposal of our ordinary shares.
Stamp Duty
There is no stamp duty payable on the subscription and issuance for our ordinary shares.
Where our ordinary shares evidenced in certificated form are acquired in Singapore, stamp duty is payable on the instrument of their transfer at the rate of 0.2% of the consideration for, or market value of, our ordinary shares, whichever is higher, and is rounded down to the nearest dollar, subject to a minimum duty of S$1.
Stamp duty is borne by the purchaser unless there is an agreement to the contrary. Where an instrument of transfer is executed outside Singapore or no instrument of transfer is executed, no stamp duty is generally payable on the acquisition of our ordinary shares. However, stamp duty may be payable if the instrument of transfer is executed outside Singapore and is received in Singapore.
If the instrument of transfer has been executed, it has to be stamped within (a) 14 days after signing the document if it is executed in Singapore; or (b) within 30 days after receiving the document in Singapore if the document is executed outside Singapore.
Pursuant to recent amendments to the Stamp Duties Act, stamp duty is payable on certain electronic instruments that effect a transfer of interest in our ordinary shares, where such instruments are regarded or deemed to be executed in Singapore, or executed outside Singapore and received in Singapore. In this regard, an electronic instrument that is executed outside Singapore is received in Singapore if (a) it is retrieved or accessed by a person in Singapore; (b) an electronic copy of it is stored on a device (including a computer) and brought into Singapore; or (c) an electronic copy of it is stored on a computer in Singapore.
On the basis that any transfer instruments in respect of any interests in our ordinary shares (whether traded on Nasdaq or JSE) are executed outside Singapore through the transfer agent(s), share registrar(s) and/or administrative depositary agent(s) in the United States and/or South Africa for registration in our share register(s) and/or administrative depositary register(s) (including branch register(s) of members) maintained in the United States and/or South Africa respectively, no stamp duty should be payable in Singapore on such transfers to the extent that the instruments of transfer (including electronic instruments) are not received in Singapore and all electronic records and any information relating to such transfers are not electronically received by persons in Singapore, stored on any server or device in Singapore or made accessible to any person in Singapore.
Estate Duty
Singapore estate duty was abolished with respect to all deaths occurring on or after February 15, 2008.
| 130 |
GST
The sale of our ordinary shares by a GST-registered investor belonging in Singapore for GST purposes to another person belonging in Singapore is an exempt supply not subject to GST. Any input GST incurred by the GST-registered investor in making this exempt supply is generally not recoverable from the Singapore Comptroller of GST.
Where our ordinary shares are sold by a GST-registered investor in the course of or furtherance of a business carried on by such investor contractually to and for the direct benefit of a person belonging outside Singapore, the sale should generally, subject to satisfaction of certain conditions, be considered a taxable supply subject to GST at 0%. Any input GST incurred by the GST-registered investor in making such a supply in the course of or furtherance of a business may be fully recoverable from the Singapore Comptroller of GST. Investors should seek their own tax advice on the recoverability of GST incurred on expenses in connection with the purchase and sale of our ordinary shares.
Services consisting of arranging, brokering, underwriting or advising on the issue, allotment or transfer of ownership of our ordinary shares rendered by a GST-registered person to an investor belonging in Singapore for GST purposes in connection with the investor’s purchase, sale or holding of our ordinary shares will be subject to GST at the standard rate of 9.0%. Similar services rendered by a GST registered person contractually to and for the direct benefit of an investor belonging outside Singapore should generally, subject to the satisfaction of certain conditions, be subject to GST at 0%.
Material U.S. Federal Income Tax Considerations
The following are material U.S. federal income tax consequences to the “U.S. Holders” described below of owning and disposing of ordinary shares, but this discussion does not purport to be a comprehensive description of all of the tax considerations that may be relevant to a particular person’s decision to acquire ordinary shares.
This discussion applies only to a U.S. Holder that holds the ordinary shares as capital assets for U.S. federal income tax purposes. In addition, it does not describe all of the tax consequences that may be relevant in light of a U.S. Holder’s particular circumstances, including any alternative minimum tax or Medicare contribution tax considerations, or consequences applicable to U.S. Holders subject to special rules, such as:
| ● | certain financial institutions; |
| ● | insurance companies, regulated investment companies, real estate investment trusts and broker-dealers; |
| ● | dealers or traders in securities that use a mark-to-market method of tax accounting; |
| ● | entities subject to the “applicable financial statement” accounting rules under Section 451(b) of the Code; |
| ● | persons holding ordinary shares as part of a straddle, integrated or similar transaction; |
| ● | persons whose functional currency for U.S. federal income tax purposes is not the U.S. dollar; |
| ● | entities classified as partnerships for U.S. federal income tax purposes and their partners; |
| ● | tax-exempt entities, “Individual Retirement Accounts” or “Roth IRAs”; |
| ● | persons that own or are deemed to own 10% or more of our stock by voting power or value; |
| ● | persons whose “tax residence” is outside the United States; or |
| ● | persons holding ordinary shares in connection with a trade or business outside the United States. |
If a partnership (or other entity that is classified as a partnership for U.S. federal income tax purposes) owns ordinary shares, the U.S. federal income tax treatment of a partner will generally depend on the status of the partner and the activities of the partnership. Partnerships that intend to own ordinary shares and their partners should consult their tax advisers as to their particular U.S. federal income tax consequences of owning and disposing of ordinary shares.
| 131 |
This discussion is based on the Code, administrative pronouncements, judicial decisions, and final, temporary and proposed United States Treasury regulations, all as of the date hereof, any of which is subject to change, possibly with retroactive effect.
This discussion does not address the effects of any state, local or non-U.S. tax laws, or any U.S. federal tax laws other than income tax laws (such as U.S. federal estate or gift tax laws). U.S. Holders should consult their tax advisers concerning the U.S. federal, state, local and non-U.S. tax consequences of owning and disposing of ordinary shares in their particular circumstances.
Except as described below under the section titled “Passive Foreign Investment Company Rules”, this discussion assumes that we are not, and will not be, a PFIC for any taxable year.
Taxation of Distributions
Subject to the PFIC discussion below, distributions paid on our ordinary shares, other than certain pro rata distributions of ordinary shares, will be treated as dividends to the extent paid out of our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. Because we do not maintain calculations of our earnings and profits under U.S. federal income tax principles, U.S. Holders generally should expect that all distributions will be treated as dividends. Dividends will not be eligible for the dividends-received deduction generally available to U.S. corporations under the Code. Subject to applicable limitations, dividends paid by “qualified foreign corporations” to certain non-corporate U.S. investors are taxable at a preferential rate applicable to long-term capital gains. For example, one of the applicable limitations is that in order for a dividend paid by a “qualified foreign corporation” to be eligible for the preferential rate, it must be paid with respect to shares that the non-corporate U.S. investor has owned for more than 60 days during the 121-day period beginning 60 days before the date on which the shares become ex-dividend. A non-U.S. corporation is treated as a qualified foreign corporation with respect to dividends paid on stock that is readily tradable on certain U.S. securities markets, such as Nasdaq, where the Company’s ordinary shares are listed. The preferential rate does not apply if the non-U.S. corporation is a PFIC for the year the dividend is paid or the preceding year. Non-corporate U.S. Holders should consult their tax advisers regarding the availability of the preferential rate and any limitations that may apply in their particular circumstances.
Dividends will be included in a U.S. Holder’s income on the date of receipt. The amount of any dividend income paid in a currency other than the U.S. dollar will be the U.S. dollar amount calculated by reference to the spot rate in effect on the date of receipt, regardless of whether the payment is in fact converted into U.S. dollars on such date. If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder generally should not be required to recognize foreign currency gain or loss in respect of the amount received. A U.S. Holder may have foreign currency gain or loss if the dividend is converted into U.S. dollars after the date of receipt. Dividends will be treated as foreign-source income for foreign tax credit purposes, which may be relevant to U.S. Holders in calculating their foreign tax credit limitation. Foreign currency gain or loss generally will be treated as U.S.-source income or loss for foreign tax credit purposes.
Sale or Other Taxable Disposition of Ordinary Shares
Subject to the PFIC discussion below, a U.S. Holder will generally recognize capital gain or loss on a sale or other taxable disposition of ordinary shares, which will be long-term capital gain or loss if, at the time of the sale or disposition, the U.S. Holder has owned the ordinary shares for more than one year. The amount of gain or loss will equal the difference between the amount realized on the sale or disposition and the U.S. Holder’s tax basis in the ordinary shares disposed of, in each case as determined in U.S. dollars. A U.S. Holder’s gain or loss will generally be treated as U.S.-source income or loss for foreign tax credit purposes. U.S. Holders that sell ordinary shares for an amount denominated in a non-U.S. currency should consult their tax advisers regarding the exchange rate at which the amount received should be translated to U.S. dollars, and whether any U.S.-source foreign currency gain or loss may be required to be recognized as a result of the sale. Long-term capital gains recognized by non-corporate U.S. Holders are taxed at a rate that is lower than the rate applicable to ordinary income. The deductibility of capital losses is subject to limitations.
| 132 |
Passive Foreign Investment Company Rules
In general, a non-U.S. corporation is a PFIC for U.S. federal income tax purposes for any taxable year in which (i) 50% or more of the value of its assets (generally determined based on the average of the quarterly values of its gross assets) consists of assets that produce, or are held for the production of, passive income, or (ii) 75% or more of its gross income consists of passive income. For purposes of the above calculations, a non-U.S. corporation that owns, directly or indirectly, at least 25% by value of the shares of another corporation is treated as if it held its proportionate share of the assets of the other corporation and received directly its proportionate share of the income of the other corporation. Passive income generally includes dividends, interest, rents, royalties and gains from the sale or exchange of investment property. Cash is a passive asset for these purposes. Goodwill is generally characterized as an active asset if it is associated with business activities that produce active income.
Based on the current and expected composition of our income and assets and the value of our assets, including the estimated value of our goodwill, we do not expect to be a PFIC for our current taxable year or in the foreseeable future. However, our PFIC status for any taxable year is an annual determination that can be made only after the end of that year, and will depend on the composition of our income and assets and the value of our assets from time to time (including the value of our goodwill, which may be determined in part by reference to the market price of the ordinary shares, which could be volatile). We may be viewed as holding a substantial amount of cash for tax purposes as a result of the Offering and our PFIC status for any taxable year may also depend on how, and how quickly, we use our liquid assets and cash (including cash raised in the Offering). Because the value of our goodwill may be determined by reference to our market capitalization, we could become a PFIC for any taxable year if the price of our ordinary shares declines significantly while we hold a substantial amount of cash and financial investments. In addition, the application of the PFIC rules is subject to a number of uncertainties and the proper characterization of some of our income and assets is not entirely clear. Accordingly, there can be no assurance that we will not be a PFIC for our current or any future taxable year.
If we were a PFIC for any taxable year and any entity in which we own equity interests were also a PFIC (a “Lower-tier PFIC”), U.S. Holders would be deemed to own a proportionate amount (by value) of the shares of each Lower-tier PFIC and would be subject to U.S. federal income tax according to the rules described in the next paragraph on (i) certain distributions by the Lower-tier PFIC and (ii) dispositions of shares of the Lower-tier PFIC, in each case as if the U.S. Holders held such shares directly, even though the U.S. Holder would not receive any proceeds of those distributions or dispositions.
In general, if we were a PFIC for any taxable year during which a U.S. Holder held ordinary shares, gain recognized by such U.S. Holder on a sale or other disposition (including certain pledges) of its ordinary shares would be allocated ratably over its holding period. The amounts allocated to the taxable year of the sale or disposition and to any year before we became a PFIC with respect to such U.S. Holder would be taxed as ordinary income. The amount allocated to each other taxable year would be subject to tax at the highest rate in effect for individuals or corporations, as appropriate, for that taxable year, and an interest charge would be imposed on the resulting tax liability for each such year. Furthermore, to the extent that distributions received by a U.S. Holder in any year on its ordinary shares exceeded 125% of the average of the annual distributions on the ordinary shares received during the preceding three years or the U.S. Holder’s holding period, whichever is shorter, such distributions would be subject to taxation in the same manner. If we were a PFIC for any taxable year during which a U.S. Holder owned ordinary shares, we would generally continue to be treated as a PFIC with respect to the U.S. Holder for all succeeding years during which the U.S. Holder owned the ordinary shares, even if we ceased to meet the threshold requirements for PFIC status. Certain elections may be available that would result in alternative treatments (such as mark-to-market treatment) of the ordinary shares. U.S. Holders should consult their tax advisers to determine whether any of these elections would be available, and, if so, what the consequences of the alternative treatments would be in their particular circumstances.
If we were a PFIC (or with respect to a particular U.S. Holder were treated as a PFIC) for a taxable year in which we paid a dividend or for the prior taxable year, the preferential tax rate described above with respect to dividends paid to certain non-corporate U.S. Holders would not apply.
We do not intend to provide information necessary for U.S. Holders to make qualified electing fund elections which, if available, would result in tax treatment different from the general tax treatment for PFICs described above.
If we were a PFIC for any taxable year during which a U.S. Holders owned any ordinary shares, the U.S. Holder would generally be required to file annual reports on United States Internal Revenue Service Form 8621. Substantial penalties and other adverse tax consequences may apply for failure to timely file such reports. U.S. Holders should consult their tax advisers regarding the determination of whether we are a PFIC for any taxable year and the potential application of the PFIC rules to their ownership of ordinary shares.
| 133 |
Information Reporting and Backup Withholding
Payments of distributions and sales proceeds that are made within the United States or through certain U.S. related financial intermediaries may be subject to information reporting and backup withholding, unless (i) the U.S. Holder is a corporation or other “exempt recipient” and (ii) in the case of backup withholding, the U.S. Holder provides a correct taxpayer identification number and certifies that it is not subject to backup withholding. The amount of any backup withholding from a payment to a U.S. Holder will be allowed as a credit against its U.S. federal income tax liability and may entitle it to a refund, provided that the required information is timely furnished to the Internal Revenue Service.
Certain U.S. Holders who are individuals (or certain specified entities) may be required to report information relating to their ownership of ordinary shares or non-U.S. accounts through which ordinary shares are held on Form 8938 with the Internal Revenue Service. Substantial penalties and other tax consequences may apply for failure to timely file such reports. U.S. Holders should consult their tax advisers regarding their reporting obligations with respect to our ordinary shares.
| F. | Dividends and paying agents |
Not applicable for annual reports on Form 20-F.
| G. | Statement by experts |
Not applicable for annual reports on Form 20-F.
| H. | Documents on display |
We file annual reports and other information with the SEC. You may inspect and copy any report or document we file, including this annual report and the accompanying exhibits, at the website maintained by the SEC at http://www.sec.gov.
We will also provide without charge to each person, including any beneficial owner of our ordinary shares, upon written or oral request of that person, a copy of any and all of the information that has been incorporated by reference in this annual report. Please direct such requests to Choo Chee Kong, Chairman and Director c/o CytoMed Therapeutics Limited at 1 Commonwealth Lane, #08-22, Singapore 149544. Our telephone number is +65 6250 7738.
| I. | Subsidiary information |
Not applicable.
| J. | Annual Report to Security Holders |
Not applicable.
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Quantitative and Qualitative Disclosures about Market Risks
We are exposed to market risks in the ordinary course of our business. These risks primarily include currency risk and interest rate risk.
Currency risk
We operate in Asia with dominant operations in Singapore and Malaysia. We regularly transact in currencies other than our respective functional currencies (“foreign currencies”). Currency risk arises when transactions are denominated in foreign currencies other than functional currency. In addition, we are exposed to currency translation risk on the net assets in foreign operations.
Interest rate risk
As of December 31, 2021, 2022 and 2023, we had cash and cash equivalents of S$2.51 million, S$1.58 million and S$9.00 million. Our exposure to interest rate sensitivity is impacted by changes in the underlying U.S. bank interest rates but is minimal. We have not entered into investments for trading or speculative purposes.
| 134 |
ITEM 12. DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
Not applicable.
PART II
ITEM 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
None.
ITEM 14. MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OR PROCEEDS
| A.—D. | Material Modifications to the Rights of Security Holders |
See “Item 10. Additional Information—B. Constitution” for a description of the rights of shareholders, which remain unchanged.
| E. | Use of Proceeds |
The following “Use of Proceeds” information relates to the registration statement on Form F-1, as amended, initially filed on November 18, 2022 (File No. 333-268456) (the “Registration Statement”) in relation to our Offering of 2,412,369 ordinary shares at an initial offering price of U.S.$4.00 per share. The Registration Statement was declared effective by the SEC on March 31, 2023. The Benchmark Company LLC acted as the representatives of the underwriters. We received gross proceeds in the amount of S$12.94 million (U.S.$9.81 million) and approximately S$10.31 million (U.S.$7.81 million) in net proceeds after deducting underwriting discounts and commissions and offering expenses.
As of December 31, 2023, there is no material change in the use of proceeds as described in our registration statement on Form F-1.
ITEM 15. CONTROLS AND PROCEDURES
2. Disclosure Controls and Procedures
An evaluation was performed under the supervision and with the participation of our management, including our Chairman and Chief Financial Officer, regarding the effectiveness of our design and operation of our disclosure controls and procedures as of December 31, 2023. Based on that evaluation, our management, including our Chairman and Chief Financial Officer, has concluded that our disclosure controls and procedures as of December 31, 2023 were ineffective due to the lack of sufficient financial reporting and accounting personnel with appropriate knowledge of U.S. GAAP and SEC reporting requirements to properly address complex U.S. GAAP accounting issues and to prepare and review our consolidated financial statements and related disclosures to fulfil U.S. GAAP and SEC financial reporting requirements.
Disclosure controls and procedures are designed to enable us to record, process, summarize and report information required to be included in the reports that we file or submit under the Exchange Act within the time period required and also effective to ensure that information required to be disclosed in the reports that it files or submits under the Exchange Act is accumulated and communicated to our management, including its principal executive and principal financial officer, to allow timely decisions regarding required disclosure.
It should be noted that while our disclosure controls and procedures as of December 31, 2023 were designed at the reasonable assurance level, our management does not expect that our disclosure controls and procedures or internal financial controls will prevent all errors or fraud. A control system, no matter how well conceived or operated, can provide only reasonable, not absolute, assurance that the objectives of the control system are met.
| B. | Management’s Annual Report on Internal Control over Financial Reporting |
This annual report does not include a report of management’s assessment regarding internal control reporting due to a transition period established by rules of the SEC for newly public companies.
| C. | Attestation Report of the Registered Public Accounting Firm |
This annual report does not include an attestation report of the Company’s registered public accounting firm due to a transition period established by the rules of the SEC for newly public companies.
| D. | Changes in Internal Controls Over Financial Reporting |
There have been no changes in our internal controls over financial reporting that occurred during the period covered by this annual report that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| 135 |
ITEM 16. RESERVED
ITEM 16A. AUDIT COMMITTEE FINANCIAL EXPERT
Our board of directors has determined that Mark LEONG Kei Wei, an independent director and a member and chairperson of our audit committee, is an “Audit Committee Financial Expert” under Section 407(d)(5) of Regulation S-K promulgated under the Securities and Exchange Act of 1934, as amended, and the corporate governance rules of the Nasdaq Capital Market.
ITEM 16B. CODE OF ETHICS
Our Code of Business Conduct and Ethics (“Code of Ethics”) applies to all of our employees, including our chairman, chief financial officer and principal accounting officer. Our Code of Ethics will be available on our corporate website https://www.cytomed.sg/ and is filed as an exhibit to this annual report. Information contained on, or that can be accessed through, our website is not a part of, and shall not be incorporated by reference into, this annual report We will disclose on our website any amendment to, or waiver from, a provision of our Code of Ethics that applies to our directors or executive officers to the extent required under the rules of the SEC.
ITEM 16C. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Our Audit Committee has enacted the policies and procedures as set forth within and pursuant to our Audit Committee Charter, which can be found within our website at https://investor.cytomed.sg/governance/governance-documents/default.aspx.
The following table sets forth the aggregate fees for audit and other services provided by our independent registered public accounting firm, WWC, P.C. for the years ended December 31, 2022 and 2023:
| $’000 | 2022 | 2023 | ||||||
| Audit Fees1 | U.S.$ | 70,000 | U.S.$ | 85,000 | ||||
| Audit-Related Fees2 | U.S.$ | 18,000 | U.S.$ | 20,000 | ||||
| Tax Fees | - | - | ||||||
| All Other Fees | - | - | ||||||
(1) “Audit Fees” represent the aggregate fees billed or to be billed for each of the financial years listed for professional services rendered by our principal auditor for the audit of our annual financial statements.
(2) “Other audit service fees” are the aggregate fees billed for assurance and related services that are reasonably related to the performance of the audit and are not reported under “Audit Fees”. These fees primarily include the review of documents filed with the SEC.
We have not engaged WWC, P.C. to provide tax compliance, tax advice, tax planning or any other services.
ITEM 16D. EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES
Not applicable.
ITEM 16E. PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS
The Company did not repurchase its ordinary shares on our behalf, or by or on behalf of an “affiliated purchaser” (as defined under Rule 10b-18(a)(3) under the Exchange Act), for the year ended December 31, 2023.
| 136 |
ITEM 16F. CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT
Not applicable.
ITEM 16G. CORPORATE GOVERNANCE
Home Country Practice Exemption as a Foreign Private Issuer
As a foreign private issuer, as defined in Rule 3b-4 under the Exchange Act, we are permitted to follow certain corporate governance rules of our home country, the Republic of Singapore) in lieu of Nasdaq Capital Market’s corporate governance rules. Our corporate governance practices do not deviate from Nasdaq Capital Market corporate governance rules and we are in full compliance with all other applicable Nasdaq Capital Market corporate governance standards; provided, however, that per our Singapore legal counsel’s letter dated October 21, 2022 provided in connection with our Offering, we formally adopted home country practice and thereby opted out of the Nasdaq Capital Market rule that would otherwise require a majority of our Board of Directors to be comprised of independent directors as defined in Rule 5605(a)(2) of the Nasdaq Listing Rules, and that we must provide for a quorum of not less than 33.33% of the outstanding shares of its voting stock in relation to any meeting of its holders of its common voting stock. Currently, we do not plan to rely on the home country practice exemption with respect to its corporate governance other than the quorum requirements and the rule that would otherwise require a majority of our Board of Directors to be comprised of independent directors. Our constitution provides that two members present in person or by proxy shall form a quorum, as contrasted with the Nasdaq requirement of one-third of a company’s outstanding voting securities. Further, the Singapore Companies Act does not require that a majority of our Board of Directors consists of Independent Directors, as contrasted with the Nasdaq requirement of a majority of independent directors.
Implications of being a Controlled Company
Our principal shareholders, officers and directors beneficially hold a majority of our ordinary shares and will continue to be a controlled company pursuant to “controlled company” defined under the Nasdaq Listing Rules. Accordingly, we are a controlled company under the applicable Nasdaq listing standards. For so long as we are a controlled company under that definition, we are permitted to elect to rely, and may rely, on certain exemptions from corporate governance rules, including:
| ● | an exemption from the rule that a majority of our board of directors must be independent directors; | |
| ● | an exemption from the rule that the compensation of our chief executive officer must be determined or recommended solely by independent directors; and | |
| ● | an exemption from the rule that our director nominees must be selected or recommended solely by independent directors. |
As a result, shareholders not have the same protection afforded to stockholders of companies that are subject to these corporate governance requirements. Please see “Risk Factors — We are a “controlled company” defined under the Nasdaq Listing Rules. Although we do not intend to rely on the “controlled company” exemption under the Nasdaq Listing Rules, we could elect to rely on this exemption in the future and you will not have the same protection afforded to stockholders of companies that are subject to these corporate governance requirements.”
ITEM 16H. MINE SAFETY DISCLOSURE
Not applicable.
ITEM 16I. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
ITEM 16J. INSIDER TRADING POLICIES
We have adopted insider trading policies governing the purchase, sale, offer to purchase, or offer to sell of any securities of the Company by directors, officers and employees, as well as advisors, consultants or contractors. A copy of the insider policies is attached as an exhibit to this annual report.
ITEM 16K. CYBERSECURITY
As a biopharmaceutical company focused on harnessing our proprietary technologies into creating novel cell-based immunotherapies for treatment of human cancers and degenerative diseases, we fully recognize the critical importance of establishing robust administrative and technical measures to safeguard our information management security systems and protect the confidentiality, integrity, and availability of data, in compliance with the necessary legislation and regulations in the jurisdictions in which we operate.
In order to ensure our adherence to the necessary legislation and regulations in the jurisdictions in which we operate, we have implemented protocols to protect against cybersecurity threats and prevent unauthorized access to sensitive data, and conduct regular assessment of the Company’s cybersecurity risks and vulnerabilities, by identifying potential threats, assessing the likelihood and potential impact of cyberattacks. We also conduct ongoing evaluation of the industry trends and regulatory environments to ensure we are in full compliance with applicable cybersecurity laws and regulations in all jurisdictions where we operate. We have set in place an efficient risk mitigation and control and incident response protocols to identify potential risks, detect, effectively respond to, and recover from cybersecurity breaches.
Overall, we believe that we have established a robust framework to protect against cybersecurity threats, mitigate risks, preserve customer trust and reputation, and support the sustainable growth of our Company.
Our cybersecurity program is managed by our Chief Corporate Officer, Ms. TAN Yoong Ying, who is responsible for the implementation of any company-wide protocols and measures required for compliance with the necessary legislation and regulations, and reports to our board of directors and our Chairman.
| 137 |
PART III
ITEM 17. FINANCIAL STATEMENTS
See Item 18 “Financial Statements.”
ITEM 18. FINANCIAL STATEMENTS
The financial information required by this item, together with the report of WWC, P.C. Certified Public Accountants, is set forth on pages F-1 through F-44 and are filed as part of this annual report.
ITEM 19. EXHIBITS
| 138 |
| + | Indicates a management contract or compensatory plan. |
| # | Pursuant to Item 601(b)(10)(iv) of Regulation S-K, certain identified information marked with [*****] has been excluded from the exhibit because it is both (i) not material and (ii) the type that the registrant treats as private or confidential. |
| 139 |
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| CYTOMED THERAPEUTICS LIMITED | ||
| By: | Choo Chee Kong | |
| Name: | Choo Chee Kong | |
| Title: | Chairman & Director | |
Date: April 22, 2024
| 140 |
CYTOMED THERAPEUTICS LIMITED
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| F-1 |

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
| To: | The Board of Directors and Shareholders of |
| CytoMed Therapeutics Limited |
Opinion on the Financial Statements
We have audited the accompanying consolidated statements of financial positions of CytoMed Therapeutics Limited and its subsidiaries (collectively the “Company”) as of December 31, 2022 and 2023, and the related consolidated statements of profit or loss and other comprehensive loss, changes in equity, and cash flows in each of the years for the three-year period ended December 31, 2023 and the related notes (collectively referred to as the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2022 and 2023, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2023, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ WWC, P.C.
Certified Public Accountants
PCAOB
ID No.
We have served as the Company’s auditor since 2022.
April 22, 2024

| F-2 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF FINANCIAL POSITIONS
As of December 31, 2022 and 2023
| Note | 2022 | 2023 | 2023 | |||||||||||||
| S$ | S$ | U.S.$ | ||||||||||||||
| ASSETS | ||||||||||||||||
| Current assets | ||||||||||||||||
| Other receivables | 5 | |||||||||||||||
| Cash and cash equivalents | 4 | |||||||||||||||
| Total current assets | ||||||||||||||||
| Non-current assets | ||||||||||||||||
| Investment in associate | 6 | |||||||||||||||
| Property, plant and equipment | 7 | |||||||||||||||
| Intangible assets | 8 | |||||||||||||||
| Total non-current assets | ||||||||||||||||
| Total assets | ||||||||||||||||
| LIABILITIES AND EQUITY | ||||||||||||||||
| Current liabilities | ||||||||||||||||
| Trade and other payables | 9 | |||||||||||||||
| Contract Liabilities | 10 | |||||||||||||||
| Borrowings | 11 | |||||||||||||||
| Convertible loans | 12 | |||||||||||||||
| Lease liabilities | 13 | |||||||||||||||
| Warrant liabilities | 14 | |||||||||||||||
| Current Tax Liability | ||||||||||||||||
| Total current liabilities | ||||||||||||||||
| Non-current liabilities | ||||||||||||||||
| Borrowings | 11 | |||||||||||||||
| Lease liabilities | 13 | |||||||||||||||
| Total non-current liabilities | ||||||||||||||||
| Total liabilities | ||||||||||||||||
| Capital and reserves | ||||||||||||||||
| Share capital* | 15 | |||||||||||||||
| Warrant reserve | 14 | |||||||||||||||
| Translation reserves | ( | ) | ( | ) | ( | ) | ||||||||||
| Accumulated losses | ( | ) | ( | ) | ( | ) | ||||||||||
| Attributable to equity holders of the Company | ||||||||||||||||
| Non-controlling interests | ( | ) | ( | ) | ( | ) | ||||||||||
| Total equity | ||||||||||||||||
| Total liabilities and equity | ||||||||||||||||
| * | Giving retroactive effect to the reverse share split effected which are detailed in Note 15 to the consolidated financial statements |
The accompanying notes are an integral part of these consolidated financial statements.
| F-3 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF PROFIT OR LOSS AND OTHER COMPREHENSIVE LOSS
For the years ended December 31, 2021, 2022 and 2023
| Note | 2021 | 2022 | 2023 | 2023 | ||||||||||||||||
| S$ | S$ | S$ | U.S.$ | |||||||||||||||||
| Revenue | ||||||||||||||||||||
| Other operating income | 16 | |||||||||||||||||||
| Other gains/(losses) including fair value changes on financial instruments - net | 17 | ( | ) | ( | ) | ( | ) | |||||||||||||
| Research expenses | 18 | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||
| Depreciation of property, plant and equipment | 7 | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||
| Amortization of intangible assets | 8 | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||
| Employee benefits expenses | 19 | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||
| Finance expenses | 20 | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||
| Other expenses | 21 | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||
| Share of results of associate | 6 | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||
| Loss before income tax | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Income tax expense | 22 | ( | ) | ( | ) | ( | ) | |||||||||||||
| Loss for the year | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Other comprehensive loss: | ||||||||||||||||||||
| Items that may be reclassified subsequently to profit or loss: | ||||||||||||||||||||
| Foreign currency translation loss | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Total comprehensive loss for the year | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Loss attributable to: | ||||||||||||||||||||
| Equity holders of the Company | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Non-controlling interests | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Total | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Total comprehensive loss attributable to: | ||||||||||||||||||||
| Equity holders of the Company | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Non-controlling interests | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Total | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Loss per share attributable to equity holders of the Company: | ||||||||||||||||||||
| Basic and diluted | ) | ) | ) | ) | ||||||||||||||||
| Weighted average number of ordinary shares used in computing loss per share | ||||||||||||||||||||
| Basic and diluted* | ||||||||||||||||||||
| * | Giving retroactive effect to the reverse share split effected which are detailed in Note 15 to the consolidated financial statements |
The accompanying notes are an integral part of these consolidated financial statements.
| F-4 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CHANGES IN EQUITY
For the years ended December 31, 2021, 2022 and 2023
| Attributable to equity holders of the Company | ||||||||||||||||||||||||||||||||
| Share capital* | Share application monies | Warrant reserves | Translation reserves | Accumulated losses | Total | Non-controlling interests | Total equity | |||||||||||||||||||||||||
| S$ | S$ | S$ | S$ | S$ | S$ | S$ | S$ | |||||||||||||||||||||||||
| Balance at January 1, 2021 | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||
| Total comprehensive loss for the year | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||
| Issuance of shares | ||||||||||||||||||||||||||||||||
| Issuance of shares – transferred from share application monies | ( | ) | ||||||||||||||||||||||||||||||
| Share issued for service | ||||||||||||||||||||||||||||||||
| Share application monies received | ||||||||||||||||||||||||||||||||
| Total transactions with owners of the Company | ||||||||||||||||||||||||||||||||
| Balance at December 31, 2021 | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||
| Total comprehensive loss for the year | | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||||||||||||
| Issuance of shares | ( | ) | ||||||||||||||||||||||||||||||
| Balance at December 31, 2022 | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||
| Total comprehensive loss for the year | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||
| Warrant exercised before year end | ||||||||||||||||||||||||||||||||
| Issuance of shares | ||||||||||||||||||||||||||||||||
| Total transactions with owners of the Company | | |||||||||||||||||||||||||||||||
| Balance at December 31, 2023 | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||
| * | Giving retroactive effect to the reverse share split effected which are detailed in Note 15 to the consolidated financial statements |
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
For the years ended December 31, 2021, 2022 and 2023
| 2021 | 2022 | 2023 | 2023 | |||||||||||||
| S$ | S$ | S$ | U.S.$ | |||||||||||||
| Operating activities | ||||||||||||||||
| Loss before income tax | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Adjustments for: | ||||||||||||||||
| Amortization of intangible assets | ||||||||||||||||
| Depreciation of property, plant and equipment | ||||||||||||||||
| Fair value loss/(gain) on convertible loans | ( | ) | ||||||||||||||
| Share of results of associate | ||||||||||||||||
| Fair value changes on warrant liabilities | ||||||||||||||||
| Interest expense | ||||||||||||||||
| Interest income | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Gain from disposal of property, plant and equipment | ( | ) | ||||||||||||||
| Write off of property, plant and equipment | ||||||||||||||||
| Unrealized currency translation losses/(gains) | ( | ) | ( | ) | ( | ) | ||||||||||
| Operating cash flows before movements in working capital | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Other receivables | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Trade and other payables | ||||||||||||||||
| Cash used in operations | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Income tax paid | ( | ) | ( | ) | ||||||||||||
| Interest received | ||||||||||||||||
| Net cash used in operating activities | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Investing activities | ||||||||||||||||
Fixed deposits with maturities over 3 months | ( | ) | ( | ) | ||||||||||||
| Purchase of property, plant and equipment | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Proceeds from disposal of property, plant and equipment | ||||||||||||||||
| Acquisition of an associate | ( | ) | ||||||||||||||
| Net cash used in investing activities | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Financing activities | ||||||||||||||||
| Proceeds from issuance of ordinary shares | ||||||||||||||||
| Proceeds from a third party loan | ||||||||||||||||
| Receipts from share application monies | ||||||||||||||||
| Principal payment of third party loan | ( | ) | ( | ) | ( | ) | ||||||||||
| Principal payment of bank borrowings | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Principal payment of lease liabilities | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Interest paid | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Net cash from financing activities | ||||||||||||||||
| Net increase/(decrease) in cash and cash equivalents | ( | ) | ||||||||||||||
| Cash and cash equivalents at beginning of year | ||||||||||||||||
| Effects of foreign currency translation on cash and cash equivalents | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||
| Cash and cash equivalents at end of year | ||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-6 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
1 GENERAL
On March 8, 2018, CytoMed Therapeutics Limited (the former name: CytoMed Therapeutics Pte. Ltd.) (the “Company”) was incorporated in the Republic of Singapore. The Company is a private limited company incorporated and domiciled in Singapore. The address of its registered office is at 1 Commonwealth Lane, #08-22, Singapore 149544. The Company is headquartered in Singapore and conducts its operations domestically and in Malaysia. On January 19, 2023, the Company had converted to a public limited company and the name of the Company has changed from “CytoMed Therapeutics Pte. Ltd.” to “CytoMed Therapeutics Limited” and that the name “CytoMed Therapeutics Limited” has substituted for “CytoMed Therapeutics Pte. Ltd.” wherever “CytoMed Therapeutics Pte. Ltd.” appears in the Constitution of the Company.
The Company’s immediate and ultimate holding corporation is Glorious Finance Limited, incorporated in the British Virgin Islands.
The principal activities of the Company are to carry on the business of innate immune cell-based immunotherapy, pluripotent stem cell-based therapy and undertaking the research and development of immune cell and stem cell-based therapy. The Company conducts its primary operations through its directly held wholly owned subsidiary that is incorporated and domiciled in Malaysia, namely CytoMed Therapeutics (Malaysia) Sdn. Bhd., which is principally engaged in manufacturing innate immune cell-based immunotherapy and pluripotent stem cell-based therapy and consultancy services and undertaking the research and development of immune cell and stem cell-based therapy for advancing cellular immunotherapy to treat cancer.
The principal activities of the subsidiaries of the Company (the “Company” or collectively known as the “Group”) are as follows:
| Name of entity | Principal activities | Country of business / incorporation | Group’s effective equity interest held | |||||||||
| 2022 | 2023 | |||||||||||
| % | % | |||||||||||
| Held by IPSCBank Pte Ltd | ||||||||||||
The principal activities of the associate are disclosed in Note 6 to the consolidated financial statements.
| F-7 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
2 MATERIAL ACCOUNTING POLICY INFORMATION
BASIS OF PREPARATION – These financial statements have been prepared in accordance with International Financial Reporting Standards (“IFRS”) under the historical cost convention, except as disclosed in the accounting policies below.
The preparation of these financial statements in conformity with IFRS requires management to exercise its judgement in the process of applying the Group’s accounting policies. It also requires the use of certain critical accounting estimates and assumptions. The areas involving a higher degree of judgement or complexity, or areas where estimates and assumptions are significant to the financial statements are disclosed in Note 3.
Adoption of IFRS
On January 1, 2022, the Group adopted the new or amended IFRS and Interpretations of IFRS (“INT IFRS”) that are mandatory for application for the financial year. Changes to the Group’s accounting policies have been made as required, in accordance with the transitional provisions in the respective IFRS and INT IFRS.
BASIS OF ACCOUNTING – The financial statements have been prepared in accordance with the historical cost basis, except as disclosed in the accounting policies below, and are drawn up in accordance with the provisions of the IFRS.
Historical cost is generally based on the fair value of the consideration given in exchange for goods and services.
Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date, regardless of whether that price is directly observable or estimated using another valuation technique. In estimating the fair value of an asset or a liability, the Company takes into account the characteristics of the asset or liability which market participants would take into account when pricing the asset or liability at the measurement date. Fair value for measurement and/or disclosure purposes in these financial statements is determined on such a basis.
In addition, for financial reporting purposes, fair value measurements are categorized into Level 1, 2 or 3 based on the degree to which the inputs to the fair value measurements are observable and the significance of the inputs to the fair value measurement in its entirety, which are described as follows:
| ● | Level 1 inputs are quoted prices (unadjusted) in active markets for identical assets or liabilities that the entity can access at the measurement date; | |
| ● | Level 2 inputs are inputs, other than quoted prices included within Level 1, that are observable for the asset or liability, either directly or indirectly; and | |
| ● | Level 3 inputs are unobservable inputs for the asset or liability. |
The Company’s policy is to recognize transfers into and transfers out of any of the three levels as of the date of the event or change in circumstances that caused the transfer.
| F-8 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
ADOPTION OF NEW AND REVISED STANDARDS – At the date of authorization of these financial statements, the management anticipates that the adoption of the above/other IFRS and amendments to IFRS in future periods will not have a material impact on the financial statements of the Group in the period of their initial adoption.
NEW AND REVISED IFRS STANDARDS IN ISSUE BUT NOT YET EFFECTIVE – At the date of authorization of these financial statements, the Group and the Company have not adopted the new and revised IFRS, IFRS INT and amendments to IFRS that have been issued but are not yet effective to them. The Group does not anticipate that the adoption of these new and revised IFRS pronouncements in future periods will have a material impact on the Group’s and the Company’s financial statements in the period of their initial adoption.
BASIS OF CONSOLIDATION
| (a) | Consolidation |
Subsidiaries are all entities (including structured entities) over which the Group has control. The Group controls an entity when the Group is exposed to, or has rights to, variable returns from its involvement with the entity and has the ability to affect those returns through its power over the entity. Subsidiaries are fully consolidated from the date on which control is transferred to the Group. They are deconsolidated from the date on that control ceases.
In preparing the consolidated financial statements, transactions, balances and unrealized gains on transactions between group entities are eliminated. Unrealized losses are also eliminated unless the transaction provides evidence of an impairment indicator of the transferred asset. Accounting policies of subsidiaries have been changed where necessary to ensure consistency with the policies adopted by the Group.
Non-controlling interests comprise the portion of a subsidiary’s net results of operations and its net assets, which is attributable to the interests that are not owned directly or indirectly by the equity holders of the Company. They are shown separately in the consolidated statements of profit or loss and other comprehensive loss, consolidated statements of changes in equity, and consolidated statements of financial positions. Total comprehensive loss is attributed to the non-controlling interests based on their respective interests in a subsidiary, even if this results in the non-controlling interests having a deficit balance.
| (b) | Acquisitions |
The acquisition method of accounting is used to account for business combinations entered into by the Group.
The consideration transferred for the acquisition of a subsidiary or business comprises the fair value of the assets transferred, the liabilities incurred and the equity interests issued by the Group. The consideration transferred also includes any contingent consideration arrangement and any pre-existing equity interest in the subsidiary measured at their fair values at the acquisition date.
Acquisition-related costs are expensed as incurred.
| F-9 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
Identifiable assets acquired and liabilities and contingent liabilities assumed in a business combination are, with limited exceptions, measured initially at their fair values at the acquisition date.
On an acquisition-by-acquisition basis, the Group recognizes any non-controlling interest in the acquiree at the date of acquisition either at fair value or at the non-controlling interest’s proportionate share of the acquiree’s identifiable net assets.
The excess of (a) the consideration transferred, the amount of any non-controlling interest in the acquiree and the acquisition-date fair value of any previous equity interest in the acquiree over the (b) fair value of the identifiable net assets acquired is recorded as goodwill. Please refer to the paragraph “Intangible assets – Goodwill” for the subsequent accounting policy on goodwill.
CONVENIENCE TRANSLATION
Translations
of amounts in the consolidated statements of financial positions, consolidated statements of profit or loss and other comprehensive loss,
and consolidated statement of cash flows from SGD into USD as of and for the year ended December 31, 2023 are solely for the convenience
of the reader and were calculated at the noon buying rate of USD
FINANCIAL ASSETS
| (a) | Classification and measurement |
The Group classifies its financial assets at amortized cost.
The classification depends on the Group’s business model for managing the financial assets as well as the contractual terms of the cash flows of the financial assets.
The Group reclassifies debt instruments when and only when its business model for managing those assets changes.
| F-10 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
At initial recognition
At initial recognition, the Group measures a financial asset at its fair value plus, in the case of the financial assets not a fair value through profit or loss, transaction cost that are directly attributable to the acquisition of the financial asset. Transaction costs of financial assets carried at fair value through profit or loss are expensed in profit or loss.
At subsequent measurement
Debt instrument – Debt instruments mainly comprised of cash and cash equivalents and other receivables (excluding prepayments).
Debt instruments that are held for collection of contractual cash flows where those cash flows represent solely payments of principal and interest are measured at amortized cost. A gain or loss on a debt instrument that is subsequently measured at amortized cost and is not part of a hedging relationship is recognized in profit or loss when the asset is derecognized or impaired. Interest income from these financial assets is included in interest income using the effective interest rate method.
| (b) | Impairment |
The Group assesses on a forward-looking basis the expected credit losses associated with its debt financial assets carried at amortized cost. The impairment methodology applied depends on whether there has been a significant increase in credit risk. The note to the consolidated financial statements on credit risk details how the Group determines whether there has been a significant increase in credit risk.
| (c) | Recognition and derecognition |
Purchases and sales of financial assets are recognized on trade date – the date on which the Group commits to purchase or sell the asset.
Financial assets are derecognized when the rights to receive cash flows from the financial assets have expired or have been transferred and the Group has transferred substantially all risks and rewards of ownership.
On disposal of a debt instrument, the difference between the carrying amount and the sale proceeds is recognized in profit or loss.
| F-11 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
FINANCIAL LIABILITIES AND EQUITY INSTRUMENTS
| (a) | Classification as debt or equity |
Debt and equity instruments issued by a Group entity are classified as either financial liabilities or as equity in accordance with substance of the contractual arrangements and the definitions of a financial liability and an equity instrument.
| (b) | Equity instruments |
An equity instrument is any contract that evidences a residual interest in the assets of an entity after deducting all of its liabilities. Equity instruments issued by a Group are recognized at the proceeds received, net of direct issue costs.
| (c) | Financial liabilities |
Except for convertible loans and warrant liabilities which are stated at fair value through profit or loss, all other financial liabilities are subsequently measured at amortized cost using the effective interest method.
The effective interest method is a method of calculating the amortized cost of a financial liability and of allocating interest expense over the relevant period. The effective interest rate is the rate that exactly discounts estimated future cash payments (including all fees and points paid or received that form an integral part of the effective interest rate, transaction costs and other premiums or discounts) through the expected life of the financial liability, or (where appropriate) a shorter period, to the amortized cost of a financial liability.
The warrants issued by the Company are accounted for as financial liabilities. The warrants are initially recognized at fair value, and in subsequent periods measured at fair value through profit or loss with any changes in fair value recognized in profit or loss until the warrants are exercised or expire.
| (d) | Derecognition of financial liabilities |
The Group derecognizes financial liabilities when, and only when, the Group’s obligations are discharged, cancelled or expired. The difference between the carrying amount of the financial liability derecognized and the consideration paid and payable, including any non-cash assets transferred or liabilities assumed, is recognized in profit or loss.
| (e) | Offsetting financial instruments |
Financial assets and liabilities are offset and the net amount reported in the consolidated statements of financial positions when there is a legally enforceable right to offset and there is an intention to settle on a net basis or realize the asset and settle the liability simultaneously.
INVESTMENT
IN AN ASSOCIATE – An associate is an entity over which the Group has significant influence, but not control or joint control,
over the financial and operating policies of the entity. Significant influence is presumed to exist when the Group holds
The Group accounts for its investment in the associate using the equity method.
On acquisition of the investment, any excess of the cost of the investment over the Group’s share of the net fair value of the investee’s identifiable assets and liabilities represents goodwill and is included in the carrying amount of the investment. Any excess of the Group’s share of the net fair value of the investee’s identifiable assets and liabilities over the cost of the investment is included as income in the determination of the Group’s share of the associate’s profit or loss in the period in which the investment is acquired.
Under the equity method, the investment in an associate is carried at cost plus post-acquisition changes in the Group’s share of net assets of the associate. Goodwill relating to the associate is included in the carrying amount of the investment and is not tested for impairment separately.
After application of the equity method, the Group determines whether it is necessary to recognize an impairment loss on its investment in its associate. At each reporting date, the Group determines whether there is objective evidence that the investment in the associate is impaired. If there is such evidence, the Group calculates the amount of impairment as the difference between the recoverable amount of the associate and its carrying value, and then recognizes the loss within “Share of results of associate” in the profit or loss.
| F-12 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
PROPERTY, PLANT AND EQUIPMENT
| (a) | Measurement |
| (i) | Property, plant and equipment |
Property, plant and equipment are initially recognized at cost and subsequently carried at cost less accumulated depreciation and accumulated impairment losses.
| (ii) | Components of costs |
The cost of an item of property, plant and equipment initially recognized includes its purchase price and any cost that is directly attributable to bringing the asset to the location and condition necessary for it to be capable of operating in the manner intended by management.
| (b) | Depreciation |
Freehold land is not depreciated. Depreciation on other items of property, plant and equipment is calculated using the straight-line method to allocate their depreciable amounts over their estimated useful lives as followed;
| Building | - | |||||||
| Laboratory equipment | - | |||||||
| Motor vehicle | - | |||||||
| Furniture, fittings and equipment | - | |||||||
| Renovation | - |
The residual values, estimated useful lives and depreciation method of property, plant and equipment are reviewed, and adjusted as appropriate, at each balance sheet date. The effects of any revision are recognized in profit or loss when the changes arise.
| (c) | Subsequent expenditure |
Subsequent expenditure relating to property, plant and equipment that has already been recognized is added to the carrying amount of the asset only when it is probable that future economic benefits associated with the item will flow to the entity and the cost of the item can be measured reliably. All other repair and maintenance expenses are recognized in profit or loss when incurred.
| F-13 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
INTANGIBLE ASSETS
| (a) | Goodwill |
Goodwill on acquisitions of subsidiaries and businesses, represents the excess of (i) the sum of the consideration transferred, the amount of any non-controlling interest in the acquiree and the acquisition-date fair value of any previous equity interest in the acquiree over (ii) the fair value of the identifiable net assets acquired. Goodwill on subsidiaries is recognized separately as intangible assets are carried at cost less accumulated impairment losses.
| (b) | Acquired intellectual properties licenses |
Intellectual
properties licenses acquired are initially recognized at cost and are subsequently carried at cost less accumulated amortization and
accumulated impairment losses. These costs are amortized to profit or loss using the straight-line method over
| (c) | Acquired computer software licenses |
Acquired computer software licenses are initially recognized at cost which includes the purchase prices and other directly attributable costs of preparing the asset for its intended use. Costs associated with maintaining the computer software are expensed off when incurred.
Computer
software licenses are subsequently carried at cost less accumulated amortization and accumulated impairment losses. These costs are amortized
to profit or loss using the straight-line method over their estimated useful lives of
The amortization period and amortization method of intangible assets other than goodwill are reviewed at least at each balance sheet date. The effects of any revision are recognized in profit or loss when the changes arise.
IMPAIRMENT OF NON-FINANCIAL ASSETS
| (a) | Goodwill |
Goodwill recognized separately as an intangible asset is tested for impairment annually and whenever there is indication that the goodwill may be impaired.
For the purpose of impairment testing of goodwill, goodwill is allocated to each of the Group’s cash-generating-units (“CGU”) expected to benefit from synergies arising from the business combination.
| F-14 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
An impairment loss is recognized when the carrying amount of a CGU, including the goodwill, exceeds the recoverable amount of the CGU. The recoverable amount of a CGU is the higher of the CGU’s fair value less cost to sell and value-in-use.
The total impairment loss of a CGU is allocated first to reduce the carrying amount of goodwill allocated to the CGU and then to the other assets of the CGU pro-rata on the basis of the carrying amount of each asset in the CGU.
An impairment loss on goodwill is recognized as an expense and is not reversed in a subsequent period.
| (b) | Intangible assets (other than Goodwill) and Property, plant and equipment |
Intangible assets and property, plant and equipment are tested for impairment whenever there is any objective evidence or indication that these assets may be impaired.
For the purpose of impairment testing, the recoverable amount (i.e. the higher of the fair value less cost to sell and the value-in-use) is determined on an individual asset basis unless the asset does not generate cash inflows that are largely independent of those from other assets. If this is the case, the recoverable amount is determined for the CGU to which the asset belongs.
If the recoverable amount of the asset (or CGU) is estimated to be less than its carrying amount, the carrying amount of the asset (or CGU) is reduced to its recoverable amount. The difference between the carrying amount and recoverable amount is recognized as an impairment loss in profit or loss.
An impairment loss for an asset is reversed if, and only if, there has been a change in the estimates used to determine the asset’s recoverable amount since the last impairment loss was recognized. The carrying amount of this asset is increased to its revised recoverable amount, provided that this amount does not exceed the carrying amount that would have been determined (net of any accumulated amortization or depreciation) had no impairment loss been recognized for the asset in prior years.
A reversal of impairment loss for an asset other than goodwill is recognized in profit or loss.
| (c) | Investment in an associate |
The Group determines whether it is necessary to recognize an impairment loss on its investment in its associate. At each reporting date, the Group determines whether there is objective evidence that the investment in the associate is impaired. If there is such evidence, the Group calculates the amount of impairment as the difference between the recoverable amount of the associate and its carrying value, and then recognizes the loss within ‘Share of results of associate’ in the statements of profit or loss.
The annual impairment test of investment in an associate involved significant management assessment and judgement required in determining the assumptions to be used to estimate the recoverable amount. Such recoverable amount is based on the higher of the value in use or fair value less costs of disposal, has been derived from discounted forecast cash flow models, including estimates of futures sales volumes and prices, operating costs, revenue growth rates and the weighted-average cost of capital (discount rate).
TRADE AND OTHER PAYABLES – Trade and other payables represent liabilities for goods and services provided to the Group prior to the end of financial year which are unpaid. They are classified as current liabilities if payment is due within one year or less (or in the normal operating cycle of the business if longer). Otherwise, they are presented as non-current liabilities.
Trade and other payables are initially recognized at fair value, and subsequently carried at amortized cost using the effective interest method.
| F-15 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
BORROWINGS – Borrowings are presented as current liabilities unless the Group has an unconditional right to defer settlement for at least 12 months after the balance sheet date, in which case they are presented as non-current liabilities.
| (a) | Borrowings |
Borrowings are initially recognized at fair value (net of transaction costs) and subsequently carried at amortized cost. Any difference between the proceeds (net of transaction costs) and the redemption value is recognized in profit or loss over the period of the borrowings using the effective interest method.
| (b) | Convertible loans |
The component parts of the convertible loans are classified separately as financial liability and equity in accordance with the substance of the contractual arrangements and the definitions of a financial liability and an equity instrument. A conversion option that will be settled by the exchange of a fixed amount of cash or another financial asset for a fixed number of the Company’s own equity instruments is an equity instrument. A conversion option that will be settled other than by the exchange of a fixed amount of cash or another financial asset for a fixed number of the Group’s own equity instruments is a conversion option derivatives. Convertible loans issued by the Company are designated at fair value through profit or loss (“FVTPL”) on initial recognition. Convertible loans are measured at fair value with changes in fair value recognized in profit or loss.
Transaction costs that relate to the issue of convertible loans are charged to profit or loss immediately.
| (c) | Borrowing costs |
All borrowing costs are recognized in profit or loss in the period in which they are incurred.
LEASES
When the Group is the lessee
At the inception of the contract, the Group assesses if the contract contains a lease. A contract contains a lease if the contract conveys the right to control the use of an identified asset for a period of time in exchange for consideration. Reassessment is only required when the terms and conditions of the contract are changed.
| F-16 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
| ● | Lease liabilities |
The initial measurement of a lease liability is measured at the present value of the lease payments discounted using the implicit rate in the lease, if the rate can be readily determined. If that rate cannot be readily determined, the Group shall use its incremental borrowing rate.
Lease payments include the following:
| - | Fixed payment (including in-substance fixed payments), less any lease incentives receivables; | |
| - | Variable lease payment that are based on an index or rate, initially measured using the index or rate as at the commencement date; | |
| - | Amount expected to be payable under residual value guarantees; | |
| - | The exercise price of a purchase option if is reasonably certain to exercise the option; and | |
| - | Payment of penalties for terminating the lease, if the lease term reflects the Group exercising that option. |
For contracts that contain both lease and non-lease components, the Group allocates the consideration to each lease component on the basis of the relative stand-alone price of the lease and non-lease component. The Group has elected to not separate lease and non-lease component for property leases and account these as one single lease component.
Lease liability is measured at amortized cost using the effective interest method. Lease liability shall be remeasured when:
| - | There is a change in future lease payments arising from changes in an index or rate; | |
| - | There is a change in the Group’s assessment of whether it will exercise an extension option; or | |
| - | There is modification in the scope or the consideration of the lease that was not part of the original term. |
| F-17 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
| ● | Short-term and low-value leases |
The Group has elected to not recognized right-of-use assets and lease liabilities for short-term leases that have lease terms of 12 months or less and leases of low value leases. Lease payments relating to these leases are expensed to profit or loss on a straight-line basis over the lease term.
EMPLOYEE BENEFITS – Employee benefits are recognized as an expense, unless the cost qualifies to be capitalized as an asset.
| (a) | Defined contribution plans |
Defined contribution plans are post-employment benefit plans under which the Group pays fixed contributions into separate entities such as the Central Provident Fund on a mandatory, contractual or voluntary basis. The Group has no further payment obligations once the contributions have been paid.
| (b) | Employee leave entitlement |
Employee entitlements to annual leave are recognized when they accrue to employees. A provision is made for the estimated liability for annual leave as a result of services rendered by employees up to the balance sheet date.
PROVISIONS – Provisions are recognized when the Group has a present obligation (legal or constructive) as a result of a past event, it is probable that the Group will be required to settle the obligation, and a reliable estimate can be made of the amount of the obligation.
The amount recognized as a provision is the best estimate of the consideration required to settle the present obligation at the end of the reporting period, taking into account the risks and uncertainties surrounding the obligation. Where a provision is measured using the cash flows estimated to settle the present obligation, its carrying amount is the present value of those cash flows.
When some or all of the economic benefits required to settle a provision are expected to be recovered from a third party, the receivable is recognized as an asset if it is virtually certain that reimbursement will be received and the amount of the receivable can be measured reliably.
| F-18 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
REVENUE RECOGNITION – Revenue is measured based on the consideration to which the Group expects to be entitled in exchange for transferring promised goods or services to the customer, excluding amounts collected on behalf of third parties.
Revenue is recognized when the Group satisfies a performance obligation by transferring promised goods or services to the customer, which is when the customer obtains control of the goods or services. A performance obligation may be satisfied at a point in time or over time. The amount of revenue recognized is the amount allocated to the satisfied performance obligation.
Advance payments received from customers are recognized as contract liabilities as the Group has not yet satisfied its performance obligation. Contract liabilities are recognized as other income when the Group satisfied its performance obligation.
Research income – Research income is recognized at point in time when the Group satisfied its performance obligation by transferring control of promised goods to the customers. The transaction price is the amount of the consideration in the contract (mainly on costs recovery basis) to which the Group expects to be entitled in exchange for transferring the promised goods.
Interest Income – Interest income from a financial asset is accrued on a time basis using the effective interest method, by reference to the principal outstanding and at the effective interest rate applicable, which is the rate that exactly discounts the estimated future cash receipts through the expected life of the financial asset to that asset’s net carrying amount on initial recognition.
RESEARCH EXPENSES – Research expenses are charged to profit or loss in the period in which it occurred. Non-refundable advance payments for goods or services to be received in the future for use in R&D activities are recorded as prepaid expenses. The prepaid amounts are expensed as the related goods are delivered or the services are performed. We allocate our direct R&D expenses across each product candidate.
R&D expenses consist primarily of costs incurred for our research and pre-clinical activities, which include:
| ● | salaries, welfare benefits and other related expenses for the employees engaged in R&D functions; | |
| ● | costs of manufacturing product candidates and consultants engaged in research, pre-clinical studies and potential future clinical trials; | |
| ● | expenses related to the upkeep of the facilities we use, which include expenses for maintenance of facilities and laboratory equipment | |
| ● | costs related to regulatory compliance; and | |
| ● | royalty payments for our intellectual properties used in research activities. |
GOVERNMENT GRANTS AND SUBSIDIES – Grants from the government are recognized as a receivable at their fair value when there is reasonable assurance that the grant will be received and the Group will comply with all the attached conditions.
Government grants receivable are recognized as income over the periods necessary to match them with the related costs which they are intended to compensate, on a systematic basis. Government grants relating to expenses are shown separately as other income.
Grants related to assets are presented as deferred income under trade and other payables.
| F-19 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
SEGMENT REPORTING – An operating segment is a component of an entity:
| ● | that engages in business activities from which it may earn revenues and incur expenses (including revenues and expenses relating to transactions with other components of the same entity); | |
| ● | whose operating results are regularly reviewed by the entity’s chief operating decision maker to make decisions about resources to be allocated to the segment and assess its performance; and | |
| ● | for which discrete financial information is available. |
The Group has identified one operating segment i.e. the business of innate immune cell-based immunotherapy, pluripotent stem cell-based therapy and undertaking the research and development of immune cell and stem cell-based therapy.
The assessment of reportable segments is based upon having similar economic characteristics and if the operating segments are similar in the following respects:
| ● | the nature of the products and services; | |
| ● | the nature of the production processes; | |
| ● | the type or class of customer for their products and services; | |
| ● | the methods used to distribute their products or provide their services; and | |
| ● | if applicable, the nature of the regulatory environment, for example, banking, insurance, or public utilities. |
Reportable segments are distinguished due to their differences in their operations and economics. They are managed separately because they require different business, technological, and marketing strategies. The Group’s Chairman is considered to be the Group’s Chief Operating Decision Maker (“CODM”). The CODM reviews non-financial information, for purposes of allocating resources. Based on the internal financial information provided to the CODM, the Group has determined that the identified operating segment as one reportable segment.
The CODM evaluates the assets and liabilities despite disaggregated financial information being available, the accounting policies used in the determination of the segment amounts are the same as those used in the preparation of the Group’s financial statements.
SHARE CAPITAL – Ordinary shares are classified as equity. Incremental costs directly attributable to the issuance of new ordinary shares are deducted against the share capital account.
| F-20 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
INCOME TAX – Current income tax for current and prior periods is recognized at the amount expected to be paid to or recovered from the tax authorities, using the tax rates and tax laws that have been enacted or substantively enacted by the balance sheet date. Management periodically evaluates positions taken in tax returns with respect to situations in which applicable tax regulation is subject to interpretation and considers whether it is probable that a tax authority will accept an uncertain tax treatment. The Group measures its tax balances either based on the most likely amount or the expected value, depending on which method provides a better prediction of the resolution of the uncertainty.
Deferred income tax is recognized for all temporary differences arising between the tax bases of assets and liabilities and their carrying amounts in the financial statements except when the deferred income tax arises from the initial recognition of goodwill or an asset or liability in a transaction that is not a business combination and affects neither accounting nor taxable profit or loss at the time of the transaction.
A deferred income tax liability is recognized on temporary differences arising on investments in subsidiaries, associates, except where the Group is able to control the timing of the reversal of the temporary difference and it is probable that the temporary difference will not reverse in the foreseeable future.
A deferred income tax asset is recognized to the extent that it is probable that future taxable profit will be available against which the deductible temporary differences and tax losses can be utilized.
Deferred income tax is measured:
| (i) | at the tax rates that are expected to apply when the related deferred income tax asset is realized or the deferred income tax liability is settled, based on tax rates and tax laws that have been enacted or substantively enacted by the balance sheet date; and | |
| (ii) | based on the tax consequence that will follow from the manner in which the Group expects, at the balance sheet date, to recover or settle the carrying amounts of its assets and liabilities. |
Current and deferred income taxes are recognized as income or expense in profit or loss, except to the extent that the tax arises from a business combination or a transaction which is recognized directly in equity. Deferred tax arising from a business combination is adjusted against goodwill on acquisition.
The Group accounts for investment tax credits (for example, productivity and innovation credit) similar to accounting for other tax credits where a deferred tax asset is recognized for unused tax credits to the extent that it is probable that future taxable profit will be available against which the unused tax credits can be utilized.
| F-21 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
FOREIGN CURRENCY TRANSACTIONS
| (a) | Functional and presentation currency |
Items included in the financial statements of each entity in the Group are measured using the currency of the primary economic environment in which the entity operates (“functional currency”). The financial statements are presented in Singapore Dollars, which is the functional currency of the Company.
| (b) | Transactions and balances |
Transactions in a currency other than the functional currency (“foreign currency”) are translated into the functional currency using the exchange rates at the dates of the transactions. Currency exchange differences resulting from the settlement of such transactions and from the translation of monetary assets and liabilities denominated in foreign currencies at the closing rates at the balance sheet date are recognized in profit or loss. Monetary items include primarily financial assets (other than equity investments) and financial liabilities. However, in the consolidated financial statements, currency translation differences arising from borrowings in foreign currencies and net investment in foreign operations, are recognized in other comprehensive loss and accumulated in the currency translation reserve.
Foreign exchange gains and losses that relate to borrowings are presented in the profit or loss within “Finance expenses”. All other foreign exchange gains and losses impacting profit or loss are presented in the profit or loss within “Other (losses)/gains – net”.
Non-monetary items measured at fair values in foreign currencies are translated using the exchange rates at the date when the fair values are determined.
| (c) | Translation of Group entities’ financial statements |
The results and financial position of all the Group entities (none of which has the currency of a hyperinflationary economy) that have a functional currency different from the presentation currency are translated into the presentation currency as follows:
| (i) | assets and liabilities are translated at the closing exchange rates at the reporting date; | |
| (ii) | income and expenses are translated at average exchange rates (unless the average is not a reasonable approximation of the cumulative effect of the rates prevailing on the transaction dates, in which case income and expenses are translated using the exchange rates at the dates of the transactions); and |
| F-22 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
| (iii) | all resulting currency translation differences are recognized in other comprehensive loss and accumulated in the currency translation reserve. These currency translation differences are reclassified to profit or loss on disposal or partial disposal with loss of control of the foreign operation. |
Goodwill and fair value adjustments arising on the acquisition of foreign operations are treated as assets and liabilities of the foreign operations and translated at the closing rates at the reporting date.
CASH AND CASH EQUIVALENTS – For the purpose of presentation in the consolidated statement of cash flows, cash and cash equivalents include cash on hand, deposits with financial institutions which are subject to an insignificant risk of change in value.
RELATED PARTIES
A relate party is a person or entity that is related to the Group.
(A) A person or a close member of that person’s family is related to the Group if that person:
(i) has control or joint control over the Group;
(ii) has significant influence over the Group; or
(iii) is a member of the key management personnel of the Company or of a parent of the Company.
(B) An entity is related to the Group if any of the following conditions applies:
(i) The entity and the Company are members of the same group (which means that each parent, subsidiary and fellow subsidiary is related to the others);
(ii) One entity is an associate or joint venture of the other entity (or an associate or joint venture of a member of a group of which the other entity is a member);
(iii) Both entities are joint ventures of the same third party;
(iv) One entity is a joint venture of a third entity and the other entity is an associate of the third entity;
(v) The entity is a post-employment benefit plan for the benefit of employees of either the Group or an entity related to the Group. If the Group is itself such a plan, the sponsoring employers are also related to the Group;
(vi) The entity is controlled or jointly controlled by a person identified in (A);
(vii) A person identified in (A)(i) has significant influence over the entity or is a member of the key management personnel of the entity (or of a parent of the entity); or
(viii) The entity, or any member of a group of which it is a part, provides key management personnel services to the Company or to a parent of the Company.
| F-23 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
RESERVES
(i) Translation reserves
Translation reserves represent the foreign currency translation difference arising from the translation of the financial statements of companies within the Group from their functional currency to the Group’s presentation currency.
(ii) Accumulated losses
Accumulated losses comprise the cumulative net losses recognized in the Group’s consolidated statements of profit or loss.
3 CRITICAL ACCOUNTING JUDGEMENTS AND KEY SOURCES OF ESTIMATION UNCERTAINTY
In the application of the Company’s accounting policies, which are described in Note 2 to the financial statements, management is required to make judgements, estimates and assumptions about the carrying amounts of assets and liabilities that are not readily apparent from other sources. The estimates and associated assumptions are based on historical experience and other factors that are considered to be relevant. Actual results may differ from these estimates.
The estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimate is revised if the revision affects only that period, or in the period of the revision and future periods.
Key sources of estimation uncertainty
The key assumptions concerning the future and other key sources of estimation uncertainty at the end of the reporting period, that have a significant risk of causing a material adjustment to the carrying amounts of assets and liabilities within the next financial year, are disclosed below:
| (a) | Impairment assessment for property, plant, and equipment (Note 7) and investment in an associate (Note 6) |
Property, plant and equipment and investment in associate, are tested for impairment when there is any objective evidence or indication that these assets may be impaired. Impairment exists when the carrying value of an asset or CGU exceeds its recoverable amount.
The recoverable amounts of property, plant and equipment and investment in associate have been determined based on higher of the fair value less costs to sell or value-in use (“VIU”) calculations. If the carrying amounts exceed the recoverable amounts, an impairment is recognized to profit or loss for the differences.
| F-24 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
Property, plant and equipment mainly consist of freehold land, building, and laboratory equipment. Management has assessed that there were no objective evidence or indication that the carrying amounts of the Group’s property, plant and equipment may not be recoverable as at the end of reporting date. Accordingly, impairment assessment is not required.
| (b) | Valuation of warrants (Note 14) |
A warrant that provides the holder with the right to buy a fixed number of equity instruments of the Company of the warrant for an exercise price that will be fixed at a future date. At initial recognition, because of the variability in the exercise price, the Company in applying paragraph 16 of IAS 32 Financial Instruments: Presentation classifies the warrants as financial liabilities at FVTPL as this derivative financial instrument does not meet the criteria of settlement by the Company in exchanging a fixed amount of cash or another financial asset for a fixed number of its own equity instruments so called “fixed-for-fixed condition”.
At the date of inception, no quoted prices in an active market are available for these financial liabilities. These financial liabilities were valued by the directors of the Company with reference to valuations carried out by an internal specialist (“Valuer”). The fair value of these financial liabilities is established by using valuation techniques as set out in the Note to the consolidated financial statements. Valuation modals established by the Valuer maximise the use of market inputs to the extent possible. Management estimates and assumptions are reviewed periodically and are adjusted if necessary. Should any of the estimates and assumptions changed, it may lead to a change in the fair value to be recognized in profit or loss. As at the end of the reporting period, key assumptions assessed by the management were disclosed in the Note 14 to the consolidated financial statements.
| (c) | Valuation of convertible loans (Note 12) |
In
the previous financial year, the convertible loans issued by the Company are classified as financial liabilities at FVTPL. No quoted
prices in an active market are available for these financial liabilities. These financial liabilities were valued by the directors of
the Company with reference to valuations carried out by an independent qualified professional valuer. The fair value of these financial
liabilities is established by using valuation techniques as set out in the Note to the consolidated financial statements. Valuation modals
established by the valuer maximise the use of market inputs to the extent possible. Management estimates and assumptions are reviewed
periodically and are adjusted if necessary. Should any of the estimates and assumptions changed, it may lead to a change in the fair
value to be recognized in profit or loss. As at the end of the reporting periods, management has assessed that the probability of conversion
of the convertible loans was anticipated to be
4 CASH AND CASH EQUIVALENTS
For the purpose of the consolidated statements of cash flows, cash and cash equivalents comprise the following:
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Cash at banks | ||||||||
| Cash on hand | ||||||||
| Short-term fixed deposits | ||||||||
| Less: Fixed deposits with maturities over 3 months | ( | ) | ||||||
| Cash and cash equivalents on consolidated statements of cash flows | ||||||||
Fixed
deposits are placed for a periods ranging between of
5 OTHER RECEIVABLES
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Deposit paid for purchase of property | ||||||||
| Prepayments | ||||||||
| Prepaid initial public offering expenses | ||||||||
| Prepaid consumables | ||||||||
| Sundry deposits | ||||||||
| Sundry receivables | ||||||||
| Total | ||||||||
Amounts due from key management personnel are generally advances extended to them during the course of business. The balance is unsecured, interest-free, and repayable on demand.
| F-25 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
6 INVESTMENT IN AN ASSOCIATE
The Group’s investment in the associate is summarized below:
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Beginning of financial year | ||||||||
| Share of post-acquisition losses | ( | ) | ( | ) | ||||
| Currency realignment | ( | ) | ||||||
| End of financial year | ||||||||
Management
has assessed the recoverable amount of the investment in associate calculation based on its VIU, using discounted cash flow forecasts
covering a five-year period in which the management made judgements over certain key inputs in relation to cash flows, revenue growth
rates and discount rate. It was concluded that the fair value less costs of disposal did not exceed the VIU. As a result of this analysis
Key assumptions used to determine the value in use of the investment in associate are as follows:
| 2022 | 2023 | |||||||
| % | % | |||||||
| Revenue growth rate | ||||||||
| Discount rate | ||||||||
| Name of entity | Principal activities | Country of business / incorporation | Group’s effective equity interest held | |||||||||
| 2022 | 2023 | |||||||||||
| % | % | |||||||||||
The following table illustrates the summarized financial information of the Group’s material associate (and not the Group’s share of those amounts), adjusted for difference in accounting policies between the Group and the associate, if any.
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Current assets | ||||||||
| Non-current assets and fair value of hospital license | ||||||||
| Current liabilities | ( | ) | ( | ) | ||||
| Non-current liabilities | ( | ) | ( | ) | ||||
| Net assets of the associate | ||||||||
| Revenue | ||||||||
| Loss for the year | ( | ) | ( | ) | ||||
Reconciliation of the summarized financial information presented to the carrying amount of the Group’s investment in the associate is as follows:
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Net assets with fair value of hospital license | ||||||||
| Group’s equity interest | % | % | ||||||
| Group share of net assets | ||||||||
| Carrying value | ||||||||
| F-26 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
7 PROPERTY, PLANT AND EQUIPMENT
| Building | Freehold land | Laboratory equipment | Motor vehicle | Furniture, fittings and equipment | Renovation | Total | ||||||||||||||||||||||
| S$ | S$ | S$ | S$ | S$ | S$ | S$ | ||||||||||||||||||||||
| Cost: | ||||||||||||||||||||||||||||
| At January 1, 2022 | ||||||||||||||||||||||||||||
| Addition | ||||||||||||||||||||||||||||
| Disposal and write off | ( | ) | ( | ) | ||||||||||||||||||||||||
| Currency realignment | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||
| At December 31, 2022 | ||||||||||||||||||||||||||||
| Addition | ||||||||||||||||||||||||||||
| Currency realignment | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||
| At December 31, 2023 | ||||||||||||||||||||||||||||
| Accumulated depreciation: | ||||||||||||||||||||||||||||
| At January 1, 2022 | ||||||||||||||||||||||||||||
| Addition | ||||||||||||||||||||||||||||
| Disposal and write off | ( | ) | ( | ) | ||||||||||||||||||||||||
| Currency realignment | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||
| At December 31, 2022 | ||||||||||||||||||||||||||||
| Addition | ||||||||||||||||||||||||||||
| Currency realignment | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||
| At December 31, 2023 | ||||||||||||||||||||||||||||
| Carrying amount: | ||||||||||||||||||||||||||||
| At December 31, 2022 | ||||||||||||||||||||||||||||
| At December 31, 2023 | ||||||||||||||||||||||||||||
| F-27 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
Motor vehicle is acquired under finance leasing arrangement. Details of the lease liability is disclosed in Note 13.
As
of December 31, 2023, bank borrowing is secured by the freehold land and building of the Group with carrying amount of S$
Depreciation expense included in the consolidated statements of profit or loss and other comprehensive loss is analyzed as follows:
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Depreciation of property, plant and equipment (per above) | ||||||||
| Less: Accounted under | ||||||||
| “Research expenses” (Note 18) | ( | ) | ( | ) | ||||
8 INTANGIBLE ASSETS
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Goodwill | ||||||||
| Intellectual properties licenses (Note (a)) | ||||||||
| Computer software licenses (Note (b)) | ||||||||
| Total | ||||||||
| (a) | Intellectual properties licenses |
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Cost: | ||||||||
| At beginning and end of financial year | ||||||||
| Accumulated amortization: | ||||||||
| At beginning of financial year | ||||||||
| Amortization charge | ||||||||
| At end of financial year | ||||||||
| Carrying value | ||||||||
| F-28 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
| (b) | Computer software licenses |
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Cost: | ||||||||
| At beginning of financial year | ||||||||
| Currency realignment | ( | ) | ( | ) | ||||
| At end of financial year | ||||||||
| Accumulated amortization: | ||||||||
| At beginning of financial year | ||||||||
| Amortization charge | ||||||||
| Currency realignment | ( | ) | ( | ) | ||||
| At end of financial year | ||||||||
| Carrying value | ||||||||
| (c) | Amortization expense is analyzed as followed: |
| Intellectual properties licenses | ||||||||
| Computer software licenses | ||||||||
| Total | ||||||||
| Less: Accounted under | ||||||||
| “Research expenses” (Note 18) | ( | ) | ( | ) | ||||
Management has evaluated, assessed and concluded that there is no indication of impairment for the intangible assets and accordingly no impairment allowance is necessary.
9 TRADE AND OTHER PAYABLES
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Trade payable | ||||||||
| Amount due to a director | ||||||||
| Other payables | ||||||||
| Accrued operating expenses | ||||||||
| Deferred grant income | ||||||||
| Total | ||||||||
The amount due to a director related to reimbursement of expenses.
Trade and other payables including amount due to a director were unsecured, interest-free, and repayable on demand.
| F-29 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
10 CONTRACT LIABILITIES
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Contract liabilities | ||||||||
Contract liabilities are fees which the Group billed and received consideration ahead of the transfer of goods and services. Management expects that all the unsatisfied performance obligations as of the end of the reporting periods may be recognized as other income within the next twelve months.
The
changes in contract liabilities of S$
11 BORROWINGS
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Current | ||||||||
| Bank loan | ||||||||
| Third party loan | ||||||||
| Total – current | ||||||||
| Non-current | ||||||||
| Bank loan | ||||||||
| Total | ||||||||
Note:
| Bank loan: | Principal
amount of RM |
The exposure of the borrowing of the Group to interest rate changes and the contractual repayment dates at the balance sheet date are as follows:
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| 6 months or less | ||||||||
| 6 – 12 months | ||||||||
| 1 – 5 years | ||||||||
| Over 5 years | ||||||||
| Total | ||||||||
The loan is secured over building and freehold land (Note 7) and personal guarantees by a director.
| Third party loan: | Principal
amount of S$ |
| F-30 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
12 CONVERTIBLE LOANS
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Convertible loans | ||||||||
| Loan 1 | Loan 1 | |||||||||||||||
| (Tranche 1) | (Tranche 2) | Loan 2 | Total | |||||||||||||
| S$ | S$ | S$ | S$ | |||||||||||||
| At January 1, 2022 | ||||||||||||||||
| Fair value adjustment | ||||||||||||||||
| At December 31, 2022 | ||||||||||||||||
| Fair value adjustment | ||||||||||||||||
| Converted to ordinary shares during the year | ( |
) | ( |
) | ( |
) | ( |
) | ||||||||
| At December 31, 2023 | ||||||||||||||||
Loan 1
The
convertible loan of S$
As at the conversion date, the convertible loan
was valued at S$
Loan 2
The
convertible loan of S$
As
at the conversion date, the convertible loan was valued at S$
The convertible loans were stated at fair value which was valued by the directors of the Company with reference to an independent qualified professional valuer.
As at year end, key valuation assumptions used to determine the fair value of the convertible loans are as follows:
| As at December 31, 2022 | % | |||
| Risk-free interest rate | ||||
| Discount rate for redemption scenario | ||||
| Discount rate for conversion scenario | ||||
| Conversion discount rate | ||||
| As at December 31, 2023 | % | |||
| Risk-free interest rate | ||||
| Discount rate for redemption scenario | ||||
| Discount rate for conversion scenario | ||||
| Conversion discount rate | ||||
The Group measures the convertible loans at fair value through profit or loss. The fair value measurement of the convertible loans is categorized as Level 3 under the Fair Value Hierarchy with the following details:
| Valuation technique input(s) | Significant unobservable input(s) | Inter-relationship between significant unobservable inputs and fair value measurements | |||
| Debt component : Discount cash flow method | |||||
| Conversion option component: | |||||
| Number of Conversion Shares to be issued, Y = A/B * C | |||||
| A - | Loan conversion amount / (1-discount) | ||||
| B - | Conversion valuation | ||||
| C - | total number of issued Shares as at the date of the Conversion Notice + Y | ||||
During the year, there were no transfers into or out of Level 3.
| F-31 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
13 LEASE LIABILITIES
Set out below are the carrying amounts of lease liabilities and the movements during the years:
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| At beginning of year | ||||||||
| Currency realignment | ( | ) | ( | ) | ||||
| Interest charged (Note 20) | ||||||||
| Payment made | ( | ) | ( | ) | ||||
| At end of year | ||||||||
Presentation on
Consolidated Statements of Financial Positions:
| Current | ||||||||
| Non-current | ||||||||
| Total |
The
effective interest rate applied to the lease liabilities recognized in the statements of financial positions is
Lease liabilities of the Group are secured over a motor vehicle (Note 7).
Total
cash outflow for leases amounts to S$
14 WARRANT LIABILITIES AND WARRANT RESERVE
WARRANT LIABILITIES
| Number of warrants | Amount S$ | Weighted Average Exercise Price US$ | ||||||||||
| 2022 | ||||||||||||
| Beginning of financial year | ||||||||||||
| Fair value changes to profit or loss | ||||||||||||
| Cashless exercise of warrants | ||||||||||||
| Currency realignment | - | |||||||||||
| End of financial year | ||||||||||||
| Number of warrants | Amount S$ | Weighted Average Exercise Price US$ | ||||||||||
| 2023 | ||||||||||||
| Beginning of financial year | ||||||||||||
| Fair value at inception date | ||||||||||||
| Fair value changes to profit or loss | ||||||||||||
| Cashless exercise of warrants classified under warrant reserve | ( | ) | ( | ) | ||||||||
| Currency realignment | ( | ) | - | |||||||||
| End of financial year | ||||||||||||
On April 13, 2023, the Company entered into underwriting agreements (the “Underwriting Agreements”) with various third parties as representative of the several underwriters (the “Representative”), relating to the Initial Public Offering (“Offering”) of shares of the Company’s ordinary shares, with par value, at an Offering price of US$ per share. Pursuant to the Underwriting Agreements, the Company agreed to issue the underwriters warrant (the “Representative’s Warrants”) to purchase an aggregate of of the Company’s ordinary shares, which is equal to five percent (%) of the shares sold in the Offering, excluding the over-allotment option, at an exercise price of US$, which is equal to 100% of the Offering price. The Representative’s Warrants can be exercised on a cashless basis by the holder into a variable number of shares based on the volume weighted average observable price of the Company’s ordinary shares at the time of exercise. The Representative’s Warrants may be exercised beginning on October 11, 2023 until April 14, 2028 and will expire in five () years from the date of the issuance.
The outstanding Representative’s Warrants are recognized as warrant liabilities as of December 31, 2023 and are measured at fair value at their inception date and subsequently remeasured at each reporting period with changes being recorded in the statement of profit or loss.
| F-32 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
The Representative’s Warrants are considered at Level 2 fair value hierarchy. The fair value of the warrants was determined by using Black-Scholes option pricing model using the key assumptions as follows:
| As at December 31, 2022 | ||||
| Expected volatility | ||||
| Risk-free interest rate | ||||
| Expected term (years) | ||||
| Exercise price | ||||
| Spot price | ||||
| Fair value of warrant | ||||
| As at December 31, 2023 | ||||
| Expected volatility | % | |||
| Risk-free interest rate | % | |||
| Expected term (years) | ||||
| Exercise price | US$ | |||
| Spot price | US$ | |||
| Fair value of warrant | US$ | |||
WARRANT RESERVE
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| At beginning of year | ||||||||
| Cashless exercise of warrants | ||||||||
| At end of year | ||||||||
Warrant
reserve amounting to S$
| 2022 | 2023 | |||||||
| Number of shares: | ||||||||
| At beginning of financial year | ||||||||
| Conversion from convertible loans | ||||||||
| Issuance of ordinary shares | ||||||||
| At end of financial year | ||||||||
| Paid-up capital (S$): | ||||||||
| At beginning of financial year | ||||||||
| Conversion from convertible loans | ||||||||
| Issuance of ordinary shares | ||||||||
| Transfer from share application monies | ||||||||
| At end of financial year | ||||||||
| Share application monies (S$): | ||||||||
| At beginning of financial year | ||||||||
| Transferred upon issuance of ordinary shares to share capital | ( | ) | ||||||
| At end of financial year | ||||||||
The paid-up ordinary shares have no par value and carry one vote per share and carry a right to dividends as and when declared by the Company.
Share application monies
On
April 18, 2022, the Company has issued for ordinary shares for S$
The newly issued shares rank pari passu in all aspects with the previously issued shares.
| F-33 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
Paid-up capital
For the financial year ended December 31, 2022:
The Company issued:
| (i) | ordinary shares for a total consideration of S$ by offsetting the share application monies to provide funds for the expansion of the Group’s operations; and |
| (ii) | ordinary shares for a total consideration of S$ of cash to provide funds for the expansion of the Group’s operations. |
For the financial year ended December 31, 2023:
The newly issued shares rank pari passu in all aspects with the previously issued shares.
On
January 17, 2023, the Company effected a
On January 26, 2023, the Company converted
the convertible loan of S$
On
April 18, 2023, the Company completed the Offering on the Nasdaq Capital Market, whereby issued and sold ordinary shares at
a price to the public of U.S.$ per share for aggregate gross proceeds of S$. The offering expenses of S$
On
April 21, 2023, the Company converted the convertible loan of S$
16 OTHER OPERATING INCOME
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Government grants: | ||||||||||||
| Jobs Support Scheme | ||||||||||||
| Wage Subsidy Program | ||||||||||||
| Enterprise Development Grant | ||||||||||||
| Jobs Growth Incentive | ||||||||||||
| Hiring Incentive and Training Programme | ||||||||||||
| Startup SG Tech - Proof-of-concept grant | ||||||||||||
| Others | ||||||||||||
| Research income | ||||||||||||
| Interest income | ||||||||||||
| Others | ||||||||||||
| Total | ||||||||||||
The Jobs Support Scheme of the Singapore Government provides wage support and helps employers retain their local employees during a period of economic uncertainty.
The Wage Subsidy Program is financial assistance introduced in Malaysia paid to employers and helps employers affected economically by the COVID-19 pandemic to continue operations and avoid the loss of jobs and income streams for all enterprises.
| F-34 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
The Enterprise Development Grant (Industry) supports projects that aim to help Small to Medium Enterprises in different stages of their technology innovation journey, to overcome their challenges in terms of resources and research and development capabilities.
The Jobs Growth Incentive of the Singapore Government supports employers to accelerate their hiring of local workforce, so as to create good and long-term jobs for locals.
The Hiring Incentive and Training Programme is an economic recovery incentive introduced in Malaysia to promote job creation among employers while increasing employment prospects.
The Startup SG Tech - Proof-of-concept grant is a scheme to drive the growth of startups based on proprietary technology and to foster the spirit of deep-tech innovation among startups. Under the scheme, qualifying companies may receive early stage funding to help them develop and commercialize their innovations.
17 OTHER GAINS/(LOSSES) INCLUDING FAIR VALUE CHANGES ON FINANCIAL INSTRUMENTS – NET
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Fair value gain/(loss) on convertible loans (Note 12) | ( | ) | ( | ) | ||||||||
| Foreign currency exchange loss - net | ( | ) | ( | ) | ( | ) | ||||||
| Gain on disposal of property, plant and equipment | ||||||||||||
| Fair value on warrants liabilities at inception date (Note 14) | ( | ) | ||||||||||
| Fair value changes on warrant liabilities (Note 14) | ( | ) | ||||||||||
| Total | ( | ) | ( | ) | ||||||||
18 RESEARCH EXPENSES
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Employee benefits expenses (Note 19) | ||||||||||||
| Depreciation of property, plant and equipment (Note 7) | ||||||||||||
| Amortization of intangible assets (Note 8) | ||||||||||||
| Consumables expense | ||||||||||||
| Royalty expenses | ||||||||||||
| Professional fees | ||||||||||||
| Electricity expenses | ||||||||||||
| Others | ||||||||||||
| Total | ||||||||||||
19 EMPLOYEE BENEFITS EXPENSES
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Wages and salaries | ||||||||||||
| Director’s fee | ||||||||||||
| Defined contribution plans | ||||||||||||
| Other short-term benefits | ||||||||||||
| Total | ||||||||||||
| Less: Accounted under | ||||||||||||
| “Research expenses” (Note 18) | ( | ) | ( | ) | ( | ) | ||||||
| Total | ||||||||||||
| F-35 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
20 FINANCE EXPENSES
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Interest expenses on: | ||||||||||||
| Bank loan | ||||||||||||
| Third party loan | ||||||||||||
| Convertible loans | ||||||||||||
| Lease liabilities (Note 13) | ||||||||||||
| Total | ||||||||||||
21 OTHER EXPENSES
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Advertising | ||||||||||||
| Annual listing fee | ||||||||||||
| Company insurance | ||||||||||||
| Entertainment | ||||||||||||
| Investor relationship expenses | ||||||||||||
| Legal fees | ||||||||||||
| Low-value assets rental | ||||||||||||
| Offering expenses | ||||||||||||
| Professional fees | ||||||||||||
| Property tax | ||||||||||||
| Printing and stationery | ||||||||||||
| IT expenses | ||||||||||||
| Repair and maintenance | ||||||||||||
| Reversal of grant | ||||||||||||
| Service fees | ||||||||||||
| Subscription fee | ||||||||||||
| Transportation and travelling expenses | ||||||||||||
| Tools and supplies | ||||||||||||
| Utilities | ||||||||||||
| Others | ||||||||||||
| Total | ||||||||||||
22 INCOME TAX EXPENSE
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Current tax liability | ||||||||||||
| F-36 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
The tax on the Group’s loss before tax differs from the theoretical amount that would arise using the Singapore standard rate of income tax as follows:
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Loss before income tax | ( | ) | ( | ) | ( | ) | ||||||
| Tax calculated at tax rate of 17% | ( | ) | ( | ) | ( | ) | ||||||
| Effects of: | ||||||||||||
| - different tax rates in other countries | ( | ) | ( | ) | ||||||||
| - deferred tax assets not recognized | ||||||||||||
| - expenses not deductible for tax purposes | ||||||||||||
| - income not subject to tax | ( | ) | ( | ) | ( | ) | ||||||
| - Under provision of prior year tax | ||||||||||||
| Income tax expenses | ||||||||||||
The
Group has unused tax losses of approximately S$
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Unutilized tax losses | ||||||||||||
| At beginning of year | ||||||||||||
| Addition | ||||||||||||
| Over-provision in respect of prior years | ( | ) | ||||||||||
| At end of year | ||||||||||||
| Unabsorbed capital allowance | ||||||||||||
| At beginning of year | ||||||||||||
| (Utilization)/Addition | ( | ) | ||||||||||
| Over-provision in respect of prior years | ( | ) | ||||||||||
| At end of year | ||||||||||||
| Total | ||||||||||||
| Unrecorded approximate deferred tax benefits @ 17% | ||||||||||||
| F-37 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
23 COMMITMENTS
| (a) | Operating lease commitments |
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Low value leases | ||||||||||||
The Group has elected not to recognize right-of-use assets and lease liabilities for short-term leases that have lease terms of 12 months or less and leases of low value leases. Lease payments relating to these leases are charged to profit or loss on a straight-line basis over the lease term.
| (b) | Capital commitments |
Capital expenditures contracted for at the balance sheet date but not recognized in the financial statements are as follows:
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Purchase of a property in Malaysia | ||||||||||||
| Loan to a third party* | ||||||||||||
| * |
| (c) | Other commitments |
The Group entered into an exclusive, worldwide, non-sublicensable, non-transferable and revocable-for-cause license with an independent company for the NKG2DL-targeting CAR-gamma delta T cell and iPSC-gdNKT cell technology. As of December 31, 2023, the total future minimum payments under non-cancellable licencing agreements are as follows:
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Within 1 year | ||||||||||||
| After 1 year but within 5 years | ||||||||||||
| Over 5 years | ||||||||||||
| Total | ||||||||||||
| F-38 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
24 FINANCIAL INSTRUMENTS, FINANCIAL RISKS AND CAPITAL RISKS MANAGEMENT
| a) | Categories of financial instruments |
The following table sets out the financial instruments as at the end of the reporting period:
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Financial asset | ||||||||||||
| Financial assets at amortized cost: | ||||||||||||
| Other receivables | ||||||||||||
| Cash and cash equivalents | ||||||||||||
| Financial liabilities | ||||||||||||
| Financial liabilities at amortized cost: | ||||||||||||
| Trade and other payables | ||||||||||||
| Lease liabilities | ||||||||||||
| Borrowings other than convertible loans | ||||||||||||
| Financial liabilities at FVTPL: | ||||||||||||
| Convertible loans | ||||||||||||
| Warrant liabilities | ||||||||||||
| b) | Financial instruments subject to offsetting, enforceable master netting arrangements and similar agreements |
The Company does not have any financial instruments which are subject to enforceable master netting arrangements or similar netting agreements.
| c) | Financial risk management policies and objectives |
The management of the Company monitors and manages the financial risks relating to the operations of the Company to ensure appropriate measures are implemented in a timely and effective manner. These risks include market risk (including currency risk and interest rate risk), credit risk and liquidity risk.
| F-39 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
| (i) | Market risk management |
The Group activities are exposed primarily to the financial risks of changes in foreign currency exchange rates and interest rates. Management monitors risks associated with changes in foreign currency exchanges rates and interest rates and will consider appropriate measures should the need arise.
There has been no significant change to the Group’s exposure to market risk or the manner in which it manages and measures the risk.
| (ii) | Foreign currency risk management |
The Group operates in Singapore and Malaysia. Entities in the Group regularly transact in currencies other than their respective functional currencies (“foreign currencies”).
Currency risk arises when transactions are denominated in foreign currencies other than the Group entities’ respective functional currencies.
In addition, the Group is exposed to currency translation risk on the net assets in foreign operations.
The Group’s currency exposure based on the information provided to key management is as follows:
| SGD | MYR | USD | GBP | Total | ||||||||||||||||
| S$ | S$ | S$ | S$ | S$ | ||||||||||||||||
| December 31, 2023 | ||||||||||||||||||||
| Financial assets | ||||||||||||||||||||
| Cash and bank balances | ||||||||||||||||||||
| Trade and other receivables | ||||||||||||||||||||
| Intra-group receivables | ||||||||||||||||||||
| Financial assets | ||||||||||||||||||||
| Financial liabilities | ||||||||||||||||||||
| Trade and other payables | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Borrowings | ( | ) | ( | ) | ||||||||||||||||
| Warrant liabilities | ( | ) | ( | ) | ||||||||||||||||
| Intra-group payables | ||||||||||||||||||||
| Financial liabilities | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Net financial (liabilities)/assets | ( | ) | ||||||||||||||||||
| Add: Net financial assets/(liabilities) denominated respective entities’ functional currencies | ( | ) | ||||||||||||||||||
| Currency exposure of financial assets, net of those denominated in the Company’s functional currency | ||||||||||||||||||||
| SGD | MYR | USD | GBP | Total | ||||||||||||||||
| S$ | S$ | S$ | S$ | S$ | ||||||||||||||||
| December 31, 2022 | ||||||||||||||||||||
| Financial assets | ||||||||||||||||||||
| Cash and bank balances | ||||||||||||||||||||
| Trade and other receivables | ||||||||||||||||||||
| Intra-group receivables | ||||||||||||||||||||
| Financial liabilities | ||||||||||||||||||||
| Trade and other payables | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||
| Borrowings | ( | ) | ( | ) | ( | ) | ||||||||||||||
| Intra-group payables | ( | ) | ( | ) | ( | ) | ||||||||||||||
| ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | |||||||||||
| Net financial (liabilities)/assets | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Add: Net financial assets/(liabilities) denominated respective entities’ functional currencies | ||||||||||||||||||||
| Currency exposure of financial assets, net of those denominated in the Company’s functional currency | ( | ) | ( | ) | ||||||||||||||||
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
If
the MYR and USD change against the SGD by
| Approximate increase/(decrease) | ||||||||
| 2023 | 2022 | |||||||
| Loss after tax | Loss after tax | |||||||
| S$ | S$ | |||||||
| MYR against SGD | ||||||||
| - strengthened | ||||||||
| - weakened | ||||||||
| USD against SGD | ||||||||
| - strengthened | ( | ) | ||||||
| - weakened | ( | ) | ||||||
| GBP against SGD | ||||||||
| - strengthened | ||||||||
| - weakened | ( | ) | ||||||
| F-40 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
| (iii) | Interest rate risk management |
The
Group is exposed to interest rate risk as the Group has bank loan amounting to S$
The sensitivity analysis below has been determined based on the exposure to interest rate for non-derivative instruments at the end of the reporting period. A 50 basis point increase or decrease is used when reporting interest rate risk internally to key management personnel and represents management’s assessment of the reasonably possible change in interest rates.
If
interest rates on loans had been 50 basis points higher/lower and all other variables were held constant, the Group’s loss for
the year would increase/decrease by approximately S$
| (iv) | Credit risk management |
Credit risk refers to the risk that a counterparty will default on its contractual obligations resulting in a financial loss to us. Our credit risk is primarily attributable to our cash and cash equivalents.
Our credit risk arising from cash and cash equivalents is limited because the counterparties are banks and financial institutions with good credit ratings which we consider to have low credit risk.
| (v) | Liquidity risk management |
Prudent liquidity risk management implies sufficient cash to finance the Group’s and the Company’s operations and development activities. The Group manages the liquidity risk by maintaining a level of cash and cash equivalents deemed adequate to finance the Group’s business operations and development activities. The Group’s objective is to maintain a balance between continuing of funding and flexibility through the use of borrowings.
Cash
flow from operations and capital contributions and loans from shareholders have been utilized to finance the working capital requirements
of the Group. As of December 31, 2023, the Group has negative cash flow from operating activities of S$
The table below analyses non-derivative financial liabilities of the Group and the Company into relevant maturity groupings based on the remaining period from the balance sheet date to the contractual maturity date.
| Less than 1 year or on demand | Between 1 and 2 years | Between 2 and 5 years | Over 5 years | Total undiscounted cash flow | ||||||||||||||||
| S$ | S$ | S$ | S$ | S$ | ||||||||||||||||
| December 31, 2023 | ||||||||||||||||||||
| Trade and other payables | ||||||||||||||||||||
| Lease liabilities | ||||||||||||||||||||
| Borrowings (excluding lease liabilities) | ||||||||||||||||||||
| December 31, 2022 | ||||||||||||||||||||
| Trade and other payables | ||||||||||||||||||||
| Lease liabilities | ||||||||||||||||||||
| Borrowings (excluding lease liabilities) | ||||||||||||||||||||
| F-41 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
| (vi) | Fair value of financial assets and financial liabilities |
The management considers that the carrying amounts of the Company’s financial assets and financial liabilities approximate their respective fair values due to the relatively short-term maturity of these financial instruments.
The fair values of the Group’s borrowings (other than convertible loans and warrant liabilities which are disclosed in Notes 12 and 14 to the consolidated financial statements) are determined by using the discounted cash flows method using discount rate that reflects the issuer’s borrowing rate as at the end of the reporting period. The Group’s non-performance risk as at December 31, 2022 and 2023 was assessed to be insignificant.
The following table shows an analysis of each class of financial instruments measured at fair value at the reporting date:
| Fair value measurement at the reporting date using | ||||||||||||||||
Quoted prices in active markets for identical instruments | Significant observant | Significant | ||||||||||||||
| (Level 1) | (Level 2) | (Level 3) | Total | |||||||||||||
| S$ | S$ | S$ | S$ | |||||||||||||
| 2023 | ||||||||||||||||
| Financial liabilities: | ||||||||||||||||
| Warrant liabilities | ||||||||||||||||
| 2022 | ||||||||||||||||
| Financial liabilities: | ||||||||||||||||
| Convertible loans | ||||||||||||||||
The fair values of other classes of financial assets and liabilities are disclosed in the respective notes to consolidated financial statements.
| (d) | Capital risk management policies and objectives |
The management manages its capital to ensure that the Group will be able to continue as a going concern in order to provide returns for shareholders and benefits for other stakeholders and to maintain an optimal capital structure to reduce cost of capital.
The capital structure of the Company consists of equity attributable to owners of the Company, comprising issued capital and accumulated losses as disclosed in the notes to financial statements.
Management monitors capital based on debt-to-equity ratio. The debt-to-equity ratio is calculated as total debt divided by total equity.
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Total borrowings (Notes 11-13) | ||||||||
| Total equity | ||||||||
| Debt-to-equity % | % | % | ||||||
The Group is not subject to externally imposed capital requirements for the financial years ended December 31, 2022 and 2023.
The Group’s overall strategy remains unchanged from prior year.
25 SEGMENT INFORMATION
Operating segments are identified on the basis of internal reports about components of the Group that are regularly reviewed by the COO (“Chief Operating Officer”) for the purpose of resource allocation and performance assessment. Segment results, assets and liabilities include items directly attributable to a segment as well as those that can be allocated on a reasonable basis. The Group operates in a single business segment which is the business of innate immune cell-based immunotherapy, pluripotent stem cell-based therapy and undertaking the research and development of immune cell and stem cell-based therapy. No operating segments have been aggregated to form the following reportable operating segment.
Geographical information:
Non-current assets (excluding investment in the associate) information based on the location of assets are as follows:
| 2022 | 2023 | |||||||
| S$ | S$ | |||||||
| Malaysia | ||||||||
| Singapore | ||||||||
| Total | ||||||||
Non-current assets information presented above consist of property, plant and equipment and intangible assets as presented in the consolidated statements of financial positions.
| F-42 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
26 RECONCILIATIONS OF LIABILITIES ARISING FROM FINANCING ACTIVITIES
| Cash Flows | Non-cash changes | |||||||||||||||||||||||||||
At beginning of year | Proceeds from borrowings | Payments | Interest expenses | Conversion of ordinary shares | Fair value changes | At end of year | ||||||||||||||||||||||
| S$ | S$ | S$ | S$ | S$ | S$ | |||||||||||||||||||||||
| 2023 | ||||||||||||||||||||||||||||
| Bank loan | ( | )# | ||||||||||||||||||||||||||
| Warrant liabilities | ( |
) | ^ | |||||||||||||||||||||||||
| Third party loan | ( | ) | ||||||||||||||||||||||||||
| Convertible loans | ( | ) | ( |
) | ||||||||||||||||||||||||
| Lease liabilities | ( | )* | ||||||||||||||||||||||||||
| 2022 | ||||||||||||||||||||||||||||
| Bank loan | ( | ) | ||||||||||||||||||||||||||
| Third party loan | ( | ) | ||||||||||||||||||||||||||
| Convertible loans | ( | ) | ||||||||||||||||||||||||||
| Lease liabilities | ( | )* | ||||||||||||||||||||||||||
| 2021 | ||||||||||||||||||||||||||||
| Bank loan | ( | ) | ||||||||||||||||||||||||||
| Third party loan | ( | ) | ||||||||||||||||||||||||||
| Convertible loans | ( | ) | ( | ) | ||||||||||||||||||||||||
| Lease liabilities | ( | )* | ||||||||||||||||||||||||||
| ^ | |
| # | |
| * |
Other significant non-cash transactions arising from the financing activities in the Consolidated Statements of Cash Flows were as follows:
During the financial year ended December 31, 2021, the Group had the following significant non-cash transactions:
Issuance of ordinary shares for a total consideration of S$ for settlement of a professional fees of the equivalent amount.
During the financial year ended December 31, 2023, the Group had the following significant non-cash transactions:
Issuance of warrants for S$
27 RELATED PARTY TRANSACTIONS
Some of the Company’s transactions and arrangements are with related parties and the effect of these on the basis determined between the parties is reflected in these financial statements. The balances are unsecured, interest-free and repayable on demand unless otherwise stated.
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Transactions with related parties: | ||||||||||||
| Research income from associate | ||||||||||||
| Purchase from associate | ||||||||||||
| Service fee paid to directors | ||||||||||||
| Amount due to a director | ||||||||||||
| Balances with related parties: | ||||||||||||
| Trade payables to associate | ||||||||||||
| Trade receivable from associate | ||||||||||||
Compensation of directors and key management personnel
The remuneration of directors and key management personnel during the year were as follows:
| 2021 | 2022 | 2023 | ||||||||||
| S$ | S$ | S$ | ||||||||||
| Wages and salaries | ||||||||||||
| Defined contribution plans | ||||||||||||
| Other short-term benefits | ||||||||||||
| Total | ||||||||||||
| Analyzed by: | ||||||||||||
| Directors of the Company | ||||||||||||
| Other key management personnel | ||||||||||||
| Total | ||||||||||||
| F-43 |
CYTOMED THERAPEUTICS LIMITED AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Years ended December 31, 2021, 2022 and 2023
28 HOLDING COMPANY AND RELATED COMPANY TRANSACTIONS
The Company is a subsidiary of Glorious Finance Limited, incorporated in the British Virgin Islands, which is also the Company’s ultimate holding company. Related companies in these financial statements refer to members of the ultimate holding company’s group of companies.
Some of the Company’s transactions and arrangements are between members of the group and the effect of these on the basis determined between the parties is reflected in these financial statements. The intercompany balances are unsecured, interest-free and repayable on demand, unless otherwise stated.
29 EVENTS OCCURRING AFTER BALANCE SHEET DATE
The Company has assessed all events occurred from December 31, 2023, up through April 22, 2024, which is the date that these consolidated financial statements are available to be issued. Other than the events disclosed below, there are not any material subsequent events that would require disclosure in these consolidated financial statements.
On
February 1, 2024, the Group completed the purchase of a property in Johor, Malaysia (“Purchase”) for business expansion by
receiving the notice of transfer of ownership for the property from the Malaysian land office in which the sale and purchase agreement
of such Purchase was entered into between the Company and a third party on September 29, 2023 for a
total purchase consideration of approximately
S$
On February 29, 2024, the Company entered into a research collaboration agreement with Sengkang General Hospital, a major public hospital in Singapore to advance injectable allogeneic umbilical cord derived MSC for cartilage injury. In this collaboration, the Company looking to provide CTM-MSC and its conditioned media for in vivo studies and Phase I clinical trial in Singapore.
On
March 25, 2024, the Group received the letter of acceptance for the acquisition of certain assets (“Acquisition”) of a
third party (“Acquiree”) relating to the business of providing cord blood banking for a total offer price of
approximately S$
| F-44 |