UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
(Mark One)
OR
For the fiscal year ended
OR
For the transition period from __________ to __________
OR
Date of event requiring this shell company report __________
Commission file number
(Exact name of Registrant as specified in its charter and translation of Registrant’s name into English)
(Jurisdiction of incorporation or organization)
(Address of principal executive offices)
Chief Executive Officer
Galapagos NV
Tel: +32 15 342 900 Fax: +
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
ordinary share, no nominal value per share | The | |
Ordinary shares, no nominal value per share* | The Nasdaq Stock Market LLC* |
* Not for trading, but only in connection with the registration of the American Depositary Shares.
Securities registered or to be registered pursuant to Section 12(g) of the Act. None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act. None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
Ordinary shares, no nominal value per share:
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. ☒
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934. ☐ Yes ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days ☒
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). ☒
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.:
Accelerated filer ☐ | Non-accelerated filer ☐ | Emerging growth company | |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ◻
† The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b) ◻
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
U.S. GAAP ☐ | ☒ | Other ☐ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow. ☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
TABLE OF CONTENTS
Page | ||
1 | ||
4 | ||
4 | ||
4 | ||
4 | ||
4 | ||
4 | ||
4 | ||
4 | ||
55 | ||
55 | ||
56 | ||
124 | ||
124 | ||
125 | ||
125 | ||
137 | ||
158 | ||
163 | ||
163 | ||
164 | ||
164 | ||
164 | ||
172 | ||
185 | ||
191 | ||
191 | ||
F. Disclosure of a registrant’s action to recover erroneously awarded compensation | 191 | |
192 | ||
192 | ||
195 | ||
204 | ||
204 | ||
204 | ||
205 | ||
205 | ||
205 | ||
205 | ||
205 | ||
205 | ||
205 | ||
206 | ||
206 | ||
206 | ||
206 | ||
206 | ||
206 | ||
206 | ||
218 | ||
218 | ||
218 | ||
219 | ||
219 | ||
221 | ||
221 | ||
221 | ||
221 | ||
221 | ||
224 | ||
224 | ||
Material Modifications to the Rights of Security Holders and Use of Proceeds | 224 | |
224 | ||
226 | ||
226 | ||
226 | ||
227 | ||
228 | ||
Purchases of Equity Securities by the Issuer and Affiliated Purchasers | 228 | |
228 | ||
228 | ||
231 | ||
Disclosure Regarding Foreign Jurisdictions that Prevent Inspections | 231 | |
231 | ||
231 | ||
232 | ||
232 | ||
232 | ||
232 | ||
ii
INTRODUCTION
Unless otherwise indicated or unless the context requires otherwise, “GLPG,” “the company,” “our company,” “we,” “us,” and “our” refer to Galapagos NV and its consolidated subsidiaries.
We own various trademark registrations and applications, and unregistered trademarks, including GALAPAGOS, JYSELECA (in the European countries where we commercialize filgotinib), and our corporate logo. All other trade names, trademarks and service marks referred to in this annual report on Form 20-F, or this annual report, are the property of their respective owners. Trade names, trademarks and service marks of other companies appearing in this annual report are the property of their respective holders. Solely for convenience, the trademarks and trade names in this annual report may be referred to without the ® and ™ symbols, but such references should not be construed as any indicator that their respective owners will not assert, to the fullest extent under applicable law, their rights thereto. We do not intend to use or display other companies’ trademarks and trade names to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
Our audited consolidated financial statements have been prepared in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB. Our consolidated financial statements are presented in euros. All references in this annual report to “$,” “US$,” “U.S.$,” “U.S. dollars,” “dollars,” and “USD” mean U.S. dollars and all references to “€” and “euros” mean euros, unless otherwise noted. Throughout this annual report, references to “ADSs” mean American Depositary Shares or ordinary shares represented by American Depositary Shares, as the case may be.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This annual report contains forward-looking statements within the meaning of Section 27A of the Securities Act
of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, that are based on our management’s beliefs and assumptions and on information currently available to our management. All statements other than present and historical facts and conditions contained in this annual report, including statements regarding our future results of operations and financial positions, business strategy, plans and our objectives for future operations, are forward-looking statements. When used in this annual report, the words “anticipate,” “believe,” “can,” “could,” “estimate,” “expect,” “intend,” “is designed to,” “may,” “might,” “plan,” “potential,” “predict,” “objective,” “should,” or the negative of these and similar expressions identify forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
| ● | the initiation, timing, progress and results of our preclinical studies and clinical trials, and our research and development programs; |
| ● | our ability to advance product candidates into, and successfully complete, clinical trials; |
| ● | our reliance on the success of our product candidate filgotinib and certain other product candidates; |
| ● | the timing or likelihood of regulatory filings and approvals; |
| ● | our ability to develop sales and marketing capabilities; |
| ● | the commercialization of our product candidates, if approved; |
| ● | the pricing and reimbursement of our product candidates, if approved; |
| ● | the implementation of our business model, strategic plans for our business, product candidates and technology; |
| ● | the scope of protection we are able to establish and maintain for intellectual property rights covering our product candidates and technology; |
| ● | our ability to operate our business without infringing, misappropriating or otherwise violating the intellectual property rights and proprietary technology of third parties; |
| ● | cost associated with enforcing or defending intellectual property infringement, misappropriation or violation; product liability; and other claims; |
| ● | regulatory developments in the United States, Europe, and other jurisdictions; |
| ● | estimates of our expenses, future revenues, capital requirements and our needs for additional financing; |
| ● | the potential benefits of strategic collaboration agreements and our ability to enter into strategic arrangements; |
| ● | our ability to maintain and establish collaborations or obtain additional grant funding; |
| ● | the rate and degree of market acceptance of our product candidates if approved by regulatory authorities; |
| ● | our financial performance; |
| ● | developments relating to our competitors and our industry, including competing therapies; |
| ● | our ability to effectively manage and anticipate growth; |
| ● | our ability to attract and retain qualified employees and key personnel; |
| ● | statements regarding future revenue, hiring plans, expenses, capital expenditures, capital requirements and share performance; and |
| ● | other risks and uncertainties, including those listed in the section of this annual report titled “Item 3.D.—Risk Factors.” |
You should refer to the section of this annual report titled “Item 3.D.—Risk Factors” for a discussion of important factors that may cause our actual results to differ materially from those expressed or implied by our forward-looking statements. As a result of these factors, we cannot assure you that the forward-looking statements in this annual report will prove to be accurate. Furthermore, if our forward-looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward-looking statements, you should not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time frame or at all. We undertake no obligation to publicly update any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law. Further, we cannot assess the impact of each such factor on our business or the extent to which any factor, or combination of factors, may cause actual results to be materially different from those contained in any forward-looking statement.
You should read this annual report and the documents that we reference in this annual report and have filed as exhibits to this annual report completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of our forward-looking statements by these cautionary statements. In light of the significant risks and uncertainties to which our forward-looking statements are subject, you should not place undue reliance on or regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified timeframe, or at all. We discuss many of these risks in greater detail in this annual report. For all forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995.
This annual report contains market data and industry forecasts that were obtained from third parties and industry publications. These data involve a number of assumptions and limitations, and you are cautioned not to give undue weight to such estimates. We have not independently verified any third party-information. While we believe the market position, market opportunity and market size information included in this annual report is generally reliable, such information is inherently imprecise.
Please see the Glossary of Terms at the end of Item 4 for definitions of scientific and other terms used in this annual report.
2
Summary of risk factors
We are dependent on the success of our clinical-stage product candidates, such as our oncology candidates (including GLPG5101, GLPG5201 and GLPG5301) and our immunology candidates (including GLPG3667). We cannot give any assurance that any product candidate will successfully complete clinical trials or receive regulatory approval, which is necessary before it can be commercialized.
Clinical development is a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials as well as data from any interim analysis of ongoing clinical trials may not be predictive of future trial results or approved label for clinical use. Clinical failure can occur at any stage of clinical development.
Due to our limited resources and access to capital in the past, we have decided to prioritize development of certain product candidates and may have forgone the opportunity to capitalize on product candidates or indications that may ultimately have been more profitable or for which there was a greater likelihood of success.
We may not be successful in our efforts to progress and expand our oncology and immunology portfolio and to build a pipeline of product candidates.
The regulatory approval processes of the United States Food and Drug Administration (the “FDA”), the European Medicines Agency (the “EMA”), the Ministry of Health, Labour and Welfare (the “MHLW”), the Pharmaceuticals and Medical Devices Agency (the “PMDA”), and other comparable regulatory authorities are lengthy, time consuming and inherently unpredictable, which may affect the commercial viability of our products in development. If we are unable to obtain regulatory approval for our product candidates, our business will be substantially harmed.
In connection with our global clinical trials, local regulatory authorities may have differing perspectives on clinical protocols and safety parameters, which impacts the manner in which we conduct these global clinical trials and could negatively impact our chances for obtaining regulatory approvals or marketing authorization in these jurisdictions, or for obtaining the requested label or dosage for our product candidates, if regulatory approvals or marketing authorizations are obtained.
Even if we receive regulatory approval for any of our product candidates, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. Additionally, our product candidates, if approved, could be subject to labeling and other restrictions and market withdrawal, and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products.
Our future clinical trials or those of any of our collaboration partners may reveal significant adverse events not seen in our preclinical studies or earlier clinical trials and may result in a safety profile that could inhibit regulatory approval or market acceptance of any of our product candidates.
Coverage and reimbursement decisions by third-party payers may have an adverse effect on pricing and market acceptance.
Legislative and regulatory activity may exert downward pressure on potential pricing and reimbursement for any of our product candidates, if approved, that could materially affect the opportunity to commercialize.
We face significant competition for our drug discovery and development efforts, and if we do not compete effectively, our commercial opportunities will be reduced or eliminated.
Our ability to compete may decline if we do not adequately protect our proprietary rights.
We are heavily dependent upon our global R&D collaboration with Gilead. There can be no assurance that this arrangement will deliver the benefits we expect, including but not limited to the payment of potential future milestones, opt-in and/or royalty payments by Gilead.
3
We have limited historical profit from product sales and limited historical data on product revenues, which makes it difficult to assess our future prospects and financial results.
Our business, results of operations and future growth prospects could be materially and adversely affected macroeconomic factors or global events, including the ongoing armed conflict between Russia and Ukraine.
The market price of the American Depositary Shares (the “ADSs”) could be subject to wide fluctuations.
We may be at an increased risk of securities class action litigation and of shareholder activism.
PART I
Item 1 Identity of directors, senior management and advisers
Not applicable.
Item 2 Offer statistics and expected timetable
Not applicable.
Item 3 Key information
A. Reserved
B. Capitalization and indebtedness
Not applicable.
C. Reasons for the offer and use of proceeds
Not applicable.
D. Risk factors
Our business is subject to significant risks. You should carefully consider all of the information set forth in this annual report and in our other filings with the U.S. Securities and Exchange Commission, or the SEC, including the following risk factors which we face, and which are faced by our industry. Our business, financial condition, or results of operations could be materially adversely affected by any of these risks. This report also contains forward-looking statements that involve risks and uncertainties. Our results could materially differ from those anticipated in these forward-looking statements, as a result of certain factors including the risks described below and elsewhere in this annual report and our other SEC filings. See “Special Note Regarding Forward-Looking Statements” above.
Risks related to product development and regulatory approval
We are dependent on the success of our clinical-stage product candidates, including our oncology candidates (such as GLPG5101, GLPG5201, and GLPG5301) and our immunology candidates (such as GLPG3667). We cannot give any assurance that any product candidate will complete clinical trials successfully or receive regulatory approval, which is necessary before it can be commercialized.
Our business and future success is substantially dependent on our ability to develop successfully, obtain regulatory approval for, and then successfully commercialize our clinical-stage oncology and immunology product candidates, including GLPG5101, GLPG5201, GLPG5301 and GLPG3667.
4
Our product candidates will require additional clinical development, management of clinical and manufacturing activities, regulatory approval in multiple jurisdictions (if regulatory approval can be obtained at all), securing sources of commercial manufacturing supply, building of, or partnering with, a commercial organization, substantial investment and significant marketing, sales and distribution efforts before any revenues can be generated from product sales. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA, the EMA, the Medicines and Healthcare products Regulatory Agency (the “MHRA”), or any other comparable regulatory authority such as the MHLW/PMDA, and we may never receive such regulatory approval for any of our product candidates. We cannot assure you that our clinical trials for GLPG5101, GLPG5201, GLPG5301, GLPG3667 and other product candidates will be completed in a timely manner, or at all, or that we will be able to obtain approval from the FDA, the EMA, the MHRA, the MHLW/PMDA, or any other comparable regulatory authority for any of these product candidates. We cannot be certain that we will advance any other product candidates into clinical trials. If any of GLPG5101, GLPG5201, GLPG5301, GLPG3667 or any future product candidate is not approved and commercialized, we will not be able to generate any product revenues for that product candidate. Moreover, any delay or setback in the development of any product candidate could adversely affect our business and cause the price of the ADSs, or our ordinary shares to fall.
Due to our limited resources and access to capital in the past, we have decided to prioritize development of certain product candidates and may have forgone the opportunity to capitalize on product candidates or indications that may ultimately have been more profitable or for which there was a greater likelihood of success.
Because we had limited resources in the past, we had to decide which product candidates to pursue and the resources to allocate to each. As of year-end 2022, we implemented a new innovation R&D model focusing on the therapeutic areas of oncology and immunology. Following the strategic review announced in August 2023, we transferred the commercial, medical affairs and development activities regarding filgotinib to Alfasigma in January 2024. Consequently, we are currently focused on advancing our clinical-stage pipeline, including GLPG5101, GLPG5201, GLPG5301, and GLPG3667. Our decisions concerning the allocation of research, collaboration, management, commercial and financial resources toward particular compounds, products or product candidates or therapeutic areas may not lead to the development of additional viable commercial products, we may forgo or delay the pursuit of opportunities with other product candidates or for other indications that may prove to have greater commercial potential. Similarly, our potential decisions to delay, terminate or collaborate with third parties in respect of certain product development programs may also prove not to be optimal and could cause us to miss valuable opportunities. If we make incorrect determinations regarding the market potential of our products or product candidates or misread trends in the pharmaceutical industry, our business, financial condition and results of operations could be materially adversely affected.
The regulatory approval processes of the FDA, the EMA, the MHRA, the MHLW/PMDA and other comparable regulatory authorities are lengthy, time consuming and inherently unpredictable which may affect the commercial viability of our products or product candidates in development. If we are unable ultimately to obtain regulatory approval for our products or product candidates, our business will be substantially harmed.
The time required to obtain approval by the FDA, the EMA, the MHRA, the MHLW/PMDA and other comparable regulatory authorities is unpredictable but typically takes many years following the commencement of clinical trials and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. Although we and Gilead have received regulatory approval for filgotinib in the European Union, Great Britain, and Japan for the treatment of RA and UC, it is possible that none of our other existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
5
Our product candidates could fail to receive regulatory approval for many reasons, including the following:
| ● | the FDA, the EMA, the MHRA, the MHLW/PMDA or other comparable regulatory authorities may disagree with the design or implementation of our clinical trials; |
| ● | we may be unable to demonstrate to the satisfaction of the FDA, the EMA, the MHRA, the MHLW/PMDA or other comparable regulatory authorities that a product candidate is safe and effective for its proposed indication; |
| ● | the results of clinical trials may not meet the level of statistical significance required by the FDA, the EMA, the MHRA, the MHLW/PMDA or other comparable regulatory authorities for approval; |
| ● | we may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks; |
| ● | a number of our product candidates are novel mode of action compounds, which may carry an additional risk regarding the desired level of efficacy and safety profile; |
| ● | the FDA, the EMA, the MHRA, the MHLW/PMDA or other comparable regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical trials; |
| ● | the data collected from clinical trials of our product candidates may not be sufficient to support the submission of a new drug application, or NDA, supplemental NDA, biologics license application, or BLA, or other submission or to obtain regulatory approval in the United States, Europe or elsewhere; |
| ● | the FDA, the EMA, the MHRA, the MHLW/PMDA or other comparable regulatory authorities may find deficiencies with or fail to approve the manufacturing processes or facilities of third-party manufacturers with which we contract for clinical and commercial supplies, or such processes or facilities may not pass a pre-approval inspection; and |
| ● | the approval policies or regulations of the FDA, the EMA, the MHRA, the MHLW/PMDA or other comparable regulatory authorities may change (in particular, the regulatory requirements and guidance with respect to cell/gene therapy products are still evolving) or differ from one another significantly in a manner rendering our clinical data insufficient for approval. For example, the European Commission published two legislative proposals in April (a new medicines Directive and Regulation). These proposals are subject to consideration by the European Parliament and European Council but, once the legislation has been finally approved, this will replace the current EU regulatory framework for medicines. |
This lengthy approval process as well as the unpredictability of future clinical trial results may result in our collaboration partners’ failure to obtain regulatory approval to market GLPG5101, GLPG5201, GLPG5301, GLPG3667 and/or other product candidates, which would harm our business, results of operations and prospects significantly. In addition, even if we were to obtain additional approvals, regulatory authorities may approve any of our products or product candidates for fewer or more limited indications or patient populations than we request, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a product or product candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that product candidate. In certain jurisdictions, regulatory authorities may not approve the price we intend to charge for our products. Any of the foregoing scenarios could materially harm the commercial prospects for our products or product candidates.
We cannot be certain that any of our product candidates will be successful in clinical trials or receive regulatory approval. Further, our product candidates may not receive regulatory approval even if they are successful in clinical trials. If we do not receive regulatory approvals for our product candidates, we may not be able to continue our operations. If the patient markets that we are targeting are not as significant as we estimate, we may not generate significant revenues from sales of such products or product candidates, if approved.
6
In connection with our global clinical trials, local regulatory authorities may have differing perspectives on clinical protocols and safety parameters, which impacts the manner in which we conduct these global clinical trials and could negatively impact our chances for obtaining regulatory approvals or marketing authorization in these jurisdictions, or for obtaining the requested label or dosage for our product candidates, if regulatory approvals or marketing authorizations are obtained.
In connection with our global clinical trials, we are obligated to comply with the requirements of local regulatory authorities in each jurisdiction where we execute and locate a clinical trial. Local regulatory authorities can request specific changes to the clinical protocol or specific safety measures that differ from the positions taken in other jurisdictions. For example, in November 2023, the FDA announced that it would be conducting an investigation into reports of T-cell malignancies following BCMA-directed or CD19-directed autologous chimeric antigen receptor, or CAR, -T cell immunotherapies following reports of T-cell lymphoma in patients receiving these therapies. In January 2024, the FDA determined that new safety information related to T-cell malignancies should be included in the labeling with boxed warning language on these malignancies for all BCMA- and CD-19-directed genetically modified autologous T-cell immunotherapies. Additionally, EMA’s safety committee (PRAC, Pharmacovigilance Risk Assessment Committee) started a signal procedure to review data on secondary malignancies related to T-cells, including T-cell lymphoma and leukemia, for the six approved CAR-T cell medicines. These investigations may also lead to additional scrutiny and safety follow-up measures for CAR-T cell products in development in the broader CAR-T class, which may differ between therapeutic areas or patient populations, for example, in less severe patient populations with longer life expectancies compared to heavily pretreated patients with cancer. The FDA or other regulatory authorities may approve different labels, including for whom the drug is indicated or require different warnings or precautions, or impose dosing restrictions that differ from the approved dosing regimen in other jurisdictions, and these differences could have a material adverse effect on our ability to commercialize our products in these jurisdictions. Regulatory authorities could also not approve our applications, which would adversely affect our business prospects and ability to achieve or sustain profitability.
Even if we receive regulatory approval for any of our product candidates, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expense. Additionally, our product candidates, if approved, could be subject to labeling and other restrictions and market withdrawal and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with our products.
Any regulatory approvals that we receive for our product candidates may also be subject to limitations on the approved indicated uses for which the product may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including Phase 4 clinical trials, and surveillance to monitor the safety and efficacy of the product, and we may be required to include labeling that includes significant use or distribution restrictions or significant safety warnings, including boxed warnings. For example, the FDA stated in its January 2024 final guidance document titled “Considerations for the Development of Chimeric Antigen Receptor (CAR) T Cell Products” that subjects in clinical trials treated with CAR-T cells containing an integrated transgene should be monitored for 15 years after treatment. In addition, the FDA’s November 2023 announcement regarding its investigation into T cell malignancy following BCMA-directed or CD19-directed autologous CAR-T cell immunotherapies stated that patients and clinical trial participants receiving treatment with the currently approved BCMA-directed and CD19-directed genetically modified autologous CAR-T cell immunotherapies products should be monitored life-long for new malignancies. Consequently, regulatory agencies may require extended follow-up observation periods of patients who receive treatment using CAR-T products, and we will need to adopt such observation periods for our CAR-T product candidates if required by the relevant regulatory agency, which could vary by country or region.
If the FDA, EMA, the MHRA, the MHLW/PMDA, or any other comparable regulatory authority approves any of our product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for the product will be subject to extensive and
7
ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration requirements and continued compliance with current good manufacturing practices, or cGMPs, and good clinical practices, or GCPs, for any clinical trials that we conduct post-approval. Manufacturers and other parties involved in the drug supply chain for prescription drug and biological products must also comply with product tracking and tracing requirements and for notifying the FDA of counterfeit, diverted, stolen and intentionally adulterated products or products that are otherwise unfit for distribution in the United States. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with our third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements, may result in, among other things:
| ● | restrictions on the marketing or manufacturing of the product, withdrawal of the product from the market, or voluntary or mandatory product recalls; |
| ● | fines, untitled or warning letters or holds on clinical trials; |
| ● | imposition by the FDA, the EMA, the MHLW/PMDA, or any other comparable regulatory authority of more restrictive labeling, including label updates and adjustments; |
| ● | refusal by the FDA, the EMA, the MHLW/PMDA, or any other comparable regulatory authority to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of product approvals or licenses; |
| ● | product seizure or detention, or refusal to permit the import or export of products; and |
| ● | injunctions or the imposition of civil or criminal penalties. |
The policies of the FDA, the EMA, the MHRA, the MHLW/PMDA, and other comparable regulatory authorities may change, and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our product candidates. For example, the European Commission published two legislative proposals in April (a new medicines Directive and Regulation). These proposals are subject to consideration by the European Parliament and European Council but, once the legislation has been finally approved, this will replace the current EU regulatory framework for medicines. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may lose any marketing approval that we may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
Clinical development is a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials as well as data from any interim analysis of ongoing clinical trials may not be predictive of future trial results or an approved label for clinical use. Clinical failure can occur at any stage of clinical development.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical trial process. Although product candidates may demonstrate promising results in early clinical (human) trials and preclinical (animal) studies, they may not prove to be effective in subsequent clinical trials. For example (without any limitation), testing on animals may occur under different conditions than testing in humans and therefore the results of animal studies may not accurately predict human experience. Likewise, early clinical studies may not be predictive of eventual safety or effectiveness results in larger-scale pivotal clinical trials. The results of preclinical studies and previous clinical trials as well as data from any interim analysis of ongoing clinical trials of our product candidates, as well as studies and trials of other products with similar mechanisms of action to our product candidates, may not be predictive of the results of ongoing or future clinical trials. For example (without any limitation), the results generated to date in the Phase 1/2 CAR-T trials with GLPG5101 and GLPG5201 do not ensure that later clinical trials, including any post-approval clinical trials for approved products, will continue to demonstrate similar results or observations. For our point-of -care cell therapy product candidates which are in Phase 1/2 of clinical development, dose escalation and dose expansion cohort data are required to establish the dose for pivotal trials, and durability of
8
response can only be established based on longer term follow up of patients who received therapy. Product candidates in later stages of clinical trials may fail to show the desired safety and efficacy traits despite having progressed through preclinical studies and earlier clinical trials. In addition to the safety and efficacy traits of any product candidate, clinical trial failures may result from a multitude of factors including flaws in trial design, dose selection, placebo effect and patient enrollment criteria. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier trials, and it is possible that we will as well. For example, in February 2023 we announced the topline results from our Phase 3 trial with filgotinib in Crohn’s disease. As the two induction cohorts missed the co-primary endpoints of clinical remission and endoscopic response at Week 10, we decided not to submit a Marketing Authorization Application in Europe. Based upon negative or inconclusive results, we or our collaboration partners may decide, or regulators may require us, to conduct additional clinical trials or preclinical studies. In addition, data obtained from trials and studies are susceptible to varying interpretations, and regulators may not interpret our data as favorably as we do, which may delay, limit or prevent regulatory approval.
We may experience delays in our ongoing clinical trials and we do not know whether planned clinical trials will begin on time, need to be redesigned, enroll patients on time or be completed on schedule, if at all. Clinical trials can be delayed for a variety of reasons, including delays related to:
| ● | obtaining regulatory authorization to commence a trial; |
| ● | reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| ● | obtaining Institutional Review Board, or IRB, or ethics committee approval at each site; |
| ● | obtaining regulatory concurrence on the design and parameters for the trial; |
| ● | obtaining approval for the designs of our clinical development programs for each country targeted for trial enrollment; |
| ● | recruiting suitable patients to participate in a trial, which may be impacted by the number of competing trials that are enrolling patients; |
| ● | having patients complete a trial or return for post-treatment follow-up; |
| ● | clinical sites deviating from trial protocol or dropping out of a trial; |
| ● | failing to comply with GCP or other regulatory requirements; |
| ● | adding new clinical trial sites; |
| ● | for our point-of-care cell therapy product candidates, adding and setting up new manufacturing sites; |
| ● | manufacturing sufficient quantities of product candidate or obtaining sufficient quantities of comparator drug for use in clinical trials; |
| ● | the availability of adequate financing and other resources; or |
| ● | the ongoing armed conflict between Russia and Ukraine, and sanctions against Russia. |
We could encounter delays if a clinical trial is suspended or terminated by us, our collaboration partners, by the IRBs or ethics committees of the institutions in which such trials are being conducted, or by the FDA, competent authorities of the EU member states, the MHRA, MHLW/PMDA, or other comparable regulatory authorities, or recommended for suspension or termination by the Data Monitoring Committee, or the DMC, for such trial. A suspension or termination, including in some cases a clinical hold, may be imposed due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA, competent authorities of the EU member states, the MHRA, MHLW/PMDA, or other comparable regulatory authorities, safety issues or adverse side effects, including those seen in the class to which our product candidates belong, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions, manufacturing issues or lack of adequate funding to continue the clinical trial. For example (without limitation), it is possible that safety issues or adverse side effects could be observed in trials for GLPG5101, GLPG5201 or GLPG5301 in oncology, and for GLPG3667 in immunology, which could result in a delay,
9
suspension or termination of the ongoing trials of GLPG5101, GLPG5201, GLPG5301 and GLPG3667. If we experience delays in the completion of, or experience a termination of, any clinical trial of our product candidates, the commercial prospects of our product candidates will be harmed, and our ability to generate product revenues from any of these product candidates will be delayed or prevented. In addition, any delays in completing our clinical trials will increase our costs, slow down our product candidate development and approval process and jeopardize our ability to commence product sales and generate revenues. Any of these occurrences may harm our business, financial condition and prospects significantly. In addition, many of the factors that cause or lead to a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
If GLPG5101, GLPG5201, GLPG5301, or GLPG3667 or any other product candidate is found to be unsafe or lack efficacy, we will not be able to obtain regulatory approval for it and our business would be materially harmed.
In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including changes in trial protocols, differences in composition of the patient populations, adherence to the dosing regimen and other trial protocols and the rate of dropouts among clinical trial participants. We do not know whether any Phase 2, Phase 3, or other clinical trials we or any of our collaboration partners may conduct will demonstrate consistent or adequate efficacy and safety to obtain regulatory approval to market our product candidates. If we are unable to bring any of our current or future product candidates to market, our ability to create long-term shareholder value will be limited.
The rates at which we complete our scientific studies and clinical trials depend on many factors, including, but not limited to, patient enrollment.
Patient enrollment, a significant factor in the timing of clinical trials, is affected by many factors including the size and nature of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial, the design of the clinical trial, competing clinical trials and clinicians’ and patients’ perceptions as to the potential advantages of the product candidate being studied in relation to other available therapies, including any new drugs that may be approved for the indications we are investigating.
Furthermore, our efforts to build relationships with patient communities may not succeed, which could result in delays in patient enrollment in our clinical trials. In addition, any negative results we may report in clinical trials of our drug candidate may make it difficult or impossible to recruit and retain patients in other clinical trials of that same drug candidate. Delays in the completion of any clinical trial of our product candidates will increase our costs, slow down our product candidate development and approval process and delay or potentially jeopardize our ability to commence product sales and generate revenue. In addition, some of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our product candidates.
We may not be successful in our efforts to use and expand our drug discovery efforts to build a pipeline of product candidates.
A key element of our strategy is to use and expand our drug discovery efforts to build a pipeline of product candidates and progress these product candidates through clinical development for the treatment of a variety of diseases. Although our research and development efforts to date have resulted in a pipeline of product candidates directed at various diseases, we may not be able to develop product candidates that are safe and effective. Even if we are successful in continuing to build our pipeline, the potential product candidates that we identify may not be suitable for clinical development, including as a result of being shown to have harmful side effects or other characteristics that indicate that they are unlikely to be product candidates that will receive marketing approval and achieve market acceptance. If we do not continue to develop successfully and to commercialize products, we will face difficulty in obtaining product revenues in future periods, which could result in significant harm to our financial position and adversely affect the price of the ADSs or our ordinary shares.
10
We face significant competition for our drug discovery and development efforts, and if we do not compete effectively, our commercial opportunities will be reduced or eliminated.
The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Our drug discovery and development efforts may target diseases and conditions that are already addressed by existing therapies or by product candidates that are being developed by our competitors, many of which have substantially greater resources, larger research and development staffs and facilities, more experience in completing preclinical testing and clinical trials, and more or better formulation, marketing and manufacturing capabilities than we do. As a result of these resources, our competitors may develop drug products that render our products and product candidates obsolete or noncompetitive by developing more effective drugs or by developing their products more efficiently. Our ability to develop competitive products would be limited if our competitors succeed in obtaining regulatory approvals for product candidates more rapidly than we are able to or in obtaining patent protection or other intellectual property rights that limit our drug development efforts. We depend upon our management team to develop and successfully implement strategies for us to obtain regulatory approvals for our selected product candidates more speedily than our competitors and to obtain and maintain patent protection and other intellectual property rights that protect our drug development efforts. Any drug products resulting from our research and development efforts, or from our joint efforts with collaboration partners or licensees, might not be able to compete successfully with our competitors’ existing and future products, or obtain regulatory approval in the United States, European Union or elsewhere. Further, we may be subject to additional competition from alternative forms of treatment, including generic or over-the-counter drugs.
In the field of dermatomyositis (DM), physical therapy, exercise and medication including corticosteroids, immunosuppressants or recently immunoglobulin treatment, are commonly used to treat DM. Treatment of this disease has relied for many years on off-label medication - in 2021 the FDA approved immunoglobulin treatment Octagam®, based on the Phase 3 ProDerm trial of Octapharma.
In the field of SLE, corticosteroids, antimalarials and immunosuppressants are commonly used to control lupus disease activity. Only two products are currently approved to treat SLE, both as add-on to standard therapy: Belimumab (Benlysta®) (anti-BAFF) from GSK and recently anifrolumab (Saphnelo®) (anti-IFN) from AstraZeneca. There are currently over 10 products in Phase 3 for SLE, of which the minority are oral therapies – including deucravacitinib (SotyktuTM) (TYK2) from BMS, upadacitinib (JAK) from Abbvie and cenerimod (S1P1) from Idorsia/Viatris.
In the field of hematologic malignancies, such as Non-Hodgkin’s Lymphoma (NHL), Chronic Lymphocytic Leukemia (CLL) and Multiple Myeloma (MM), there are many approved therapies or therapies in development (including but not limited to chemotherapy, BTKi, antibodies, bispecific antibodies, antibody drug conjugates, CAR-Ts, cytokines, NK and T-cell engagers, etc.) and many different types of cell therapy in development (allogeneic/autologous, CAR-T/CAR-NK, TIL, TCR-T, dendritic, etc.). As a consequence, we are operating in a highly competitive, and rapidly evolving environment. New technologies and therapies such as in vivo modification of immune cells may further disrupt this market in the mid-to-long-term. Six CAR T treatments have been approved for hematological cancers in the US and Europe: Novartis’ Kymriah® (CD19 CAR T), Gilead/Kite’s Yescarta® (CD19 CAR T), Tecartus® (CD19 CAR T), J&J’s Carvykti® (BCMA CAR T) BMS’ Breyanzi® (CD19 CAR T) and Abecma® (BCMA CAR T).
Many of our competitors have significantly greater financial, technical, and human resources than we have. Mergers and acquisitions in the pharmaceutical, medical device and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Our commercial opportunity could be reduced or eliminated if our competitors develop or market products or other novel therapies that are more effective, safer, or less costly than our current or future product candidates, or obtain regulatory approval for their products more rapidly than we may obtain approval for our product candidates. Our success will be based in part on our ability to identify, develop, and manage a portfolio of product candidates that are safer and more effective than competing products.
11
If approved, our investigational products regulated as biologics may face competition from biosimilars approved through an abbreviated regulatory pathway.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively the ACA, includes a subtitle called the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which created an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. Under the BPCIA, an application for a biosimilar product may not be submitted to the FDA until four years following the date that the reference product was first licensed by the FDA. In addition, the approval of a biosimilar product may not be made effective by the FDA until 12 years from the date on which the reference product was first licensed. During this 12-year period of exclusivity, another company may still market a competing version of the reference product if the FDA approves a BLA for the competing product containing the sponsor’s own preclinical data and data from adequate and well-controlled clinical trials to demonstrate the safety, purity, and potency of the other company’s product. The law is complex and is still being interpreted and implemented by the FDA. As a result, its ultimate impact, implementation, and meaning are subject to uncertainty.
We believe that any of our product candidates approved as a biological product under a BLA should qualify for the 12-year period of exclusivity. However, there is a risk that this exclusivity could be shortened due to congressional action or otherwise, or that the FDA will not consider our investigational medicines to be reference products for competing products, potentially creating the opportunity for generic competition sooner than anticipated. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. Moreover, the extent to which a biosimilar, once licensed, will be substituted for any one of our reference products in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
If competitors are able to obtain marketing approval for biosimilars referencing any of our product candidates, our products may become subject to competition from such biosimilars, which would impair our ability to successfully commercialize and generate revenues from sales of such products.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following marketing approval, if any.
Undesirable side effects caused by our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, the EMA, the MHRA, the MHLW/PMDA, or other comparable regulatory authorities. Results of our trials could reveal a high and unacceptable severity and prevalence of certain side effects. In such an event, our trials could be suspended or terminated and the FDA, the EMA, the MHRA, the MHLW/PMDA, or comparable regulatory authorities could order us to cease further development of or deny approval of our product candidates for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
For example (without any limitation), the ISABELA Phase 3 program in IPF was discontinued in February 2021, prior to recruitment completion. The decision was based on the recommendations of the Independent Data Monitoring Committee (IDMC) which, following a regular review of unblinded data, concluded that ziritaxestat’s benefit-risk profile no longer supported continuing these studies.
12
If one or more of our products or product candidates receives marketing approval, and we or others later identify undesirable side effects caused by such products, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may withdraw approvals of such product; |
| ● | regulatory authorities may require additional warnings on the label or impose updates to the label; |
| ● | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
| ● | we could be sued and held liable for harm caused to patients; and |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the particular product or product candidate, if approved, and could significantly harm our business, results of operations and prospects.
Our future clinical trials or those of any of our collaborators may reveal significant adverse events not seen in our preclinical studies or earlier clinical trials and result in a safety profile that could inhibit regulatory approval or market acceptance of any of our product candidates.
If significant adverse events or other side effects are observed in any of our clinical trials, we may have difficulty recruiting patients to our clinical trials, patients may drop out of our trials, we may be required to pause, delay, or abandon the trials or our development efforts of one or more product candidates altogether, we may be required to have more restrictive labeling, including updates and adjustments of product labels, or we may experience the delay or denial of regulatory approval by the FDA, EMA, MHRA, the MHLW/PMDA, or other applicable regulatory authorities. We, the FDA, or other applicable regulatory authorities, or an IRB may suspend clinical trials of a product candidate at any time for various reasons, including a belief that subjects or patients in such trials are being exposed to unacceptable health risks or adverse side effects. Some potential therapeutics developed in the biotechnology industry that initially showed therapeutic promise in early-stage trials have later been found to cause adverse events or other side effects that prevented their further development. Even if any such adverse events or other side effects do not preclude the product candidate from obtaining or maintaining marketing approval, undesirable side effects may inhibit market acceptance of the approved product due to its tolerability versus other therapies.
Cell therapies are novel, complex, and difficult to manufacture and we may not be successful in our efforts to develop and commercialize such therapies.
In June 2022, we acquired CellPoint and AboundBio with the aim to enter the space of oncology. Through the acquisitions of CellPoint and AboundBio, respectively, we gained access to an innovative, scalable, decentralized and automated point-of-care cell therapy supply model as well as fully human antibody- based therapeutics platform. CellPoint has developed, in a strategic collaboration with Lonza, a novel point -of-care supply model, which offers the potential for efficient, 7-day delivery of CAR-T therapies and avoids complex logistics, thereby addressing important limitations of current CAR-T treatments. The platform that we use consists of end-to-end xCellit® workflow management and monitoring software and Lonza’s Cocoon® platform, a functionally closed, automated manufacturing platform for cell and gene therapies.
The manufacturing processes that we use to produce our product candidates for human therapeutics are complex, novel, have not been validated for commercial use, and are subject to multiple risks. Several factors could cause production interruptions, including equipment malfunctions, facility contamination, raw material shortages or contamination, natural disasters, disruption in utility services, human error, or disruptions in the operations of our suppliers. Moreover, unlike chemical pharmaceuticals, the physical and chemical properties of a biologic therapy often cannot be fully characterized. As a result, assays of the finished product may not be sufficient to ensure that the product will perform in the intended manner. Accordingly, it is necessary to employ multiple steps to control our manufacturing process to assure that the product candidate is made strictly and consistently in compliance with the process. Problems with the manufacturing process, even minor deviations
13
from the normal process, could result in product defects or manufacturing failures that result in lot failures, product recalls, product liability claims, or insufficient inventory.
In addition, the manufacture of our point-of-care cell therapy product candidates involves complex processes, such as harvesting and transporting cells from every patient to Lonza’s Cocoon® system, engineering the cells ex vivo to express a specific biologic receptor for a specific target, and finally transporting the point-of-care cell therapy product candidates from Lonza’s Cocoon® system back to the patient for infusion into the patient. As a result of the complexities, the manufacturing process for our point-of-care cell therapy product candidates is more variable and difficult to reproduce than traditional small molecule chemical compounds or biologics. Our manufacturing process may be susceptible to product loss or failure due to logistical issues associated with the collection of patients’ cells and the infusion of the patient with our point-of-care cell therapy product candidates. Product loss or failure may also be caused by a number of factors, including manufacturing issues associated with the variability in patient material, interruptions in the manufacturing process, contamination, equipment failure, assay failures, improper installation or operation of Lonza’s Cocoon® system, vendor or operator error, inconsistency in cell growth, and variability in product characteristics.
If for any reason a patient’s starting material is lost, or if any point- of -care cell therapy product candidate does not meet the preset specifications, the manufacturing process for that patient will need to be restarted, sometimes including the re-collection of cells from the patient, and the resulting delay may adversely affect that patient’s outcome. It may even happen that failed product candidate manufacture may prevent a patient from receiving our point-of-care cell therapy product candidates. If microbial, environmental or other contaminations are discovered in our point-of-care cell therapy product candidates or in the third- party facilities in which our point- of- care cell therapy product candidates are manufactured through Lonza’s Cocoon® system, delays can occur and such facilities can be closed. If such contaminations or other product quality issues are not discovered and if as a result thereof patients are exposed to a health risk, we may be held liable. Our insurance may not cover those cases, or the financial coverage may not be sufficient. Because our point-of-care cell therapy product candidates are manufactured specifically for each individual patient, vendors and operators will be required to maintain a chain of identity with respect to the patient’s cellular material as it moves from the patient through the Cocoon® point-of-care manufacturing process, and back to the patient for infusion. Maintaining such a chain of identity is difficult and complex, and failure to do so could result in adverse patient outcomes, loss of product, or regulatory action including clinical hold or other suspension or termination of our clinical trials or withdrawal of our point-of-care cell therapy products from the market, if approved. Moreover, the patient’s starting material may lead to variable product quality or quantity which could result in an out of specification or insufficient product.
Further, as product candidates are developed through preclinical to late-stage clinical trials towards approval and commercialization, it is common that various aspects of the development program, such as point-of-care manufacturing methods, are altered along the way to optimize processes and results. Such changes carry the risk that they will not achieve their intended objectives, and any of these changes could cause our point-of-care cell therapy product candidates or our manufacturing process to perform differently and affect the results of planned clinical trials or other future clinical trials or otherwise necessitate the conduct of additional studies, which can be costly and time-consuming. We and our vendors may not successfully establish a robust point-of-care production process, including quality release and monitoring process, that fulfills the requirements of the FDA, the EMA and comparable regulatory authorities. There can be no assurance that Lonza’s Cocoon® system for our point- of-care cell therapy product candidates is viable and can be effectively scaled up or transferred to third party vendors and operators for commercialization. We are dependent on a third party for the end-to-end xCellit® workflow management and monitoring software and if there are any issues with this software, we may be unable to obtain regulatory approval for our product candidates.
Any failure to follow regulatory requirements or any delay, interruption or other issues that arise in the manufacture or storage of our product candidates as a result of a failure of our facilities or the facilities and operations of third parties to comply with regulatory requirements or pass any regulatory authority inspection could significantly impair our ability to develop and commercialize our point-of-care cell therapy product candidates. Point-of-care cell therapy product candidates that have been produced and stored for later use may degrade, become contaminated or suffer other quality defects, which may cause the affected point- of-care cell
14
therapy product candidates to no longer be suitable for their intended use. Furthermore, if our vendors or operators fail to deliver the required commercial quantities or supply of our point-of -care cell therapy product candidates on a timely basis and at reasonable costs, we would likely be unable to meet demand for our point-of-care cell therapy product candidates, and we would lose potential revenues.
In addition, the manufacturing process and facilities used to produce our point-of-care cell therapy product candidates through our Cocoon® system are subject to FDA, the EMA and comparable regulatory authority approval processes and compliance with applicable cGMPs and Good Distribution Practices, and we and our vendors and operators will need to meet all applicable regulatory authority requirements on an ongoing basis, including requirements pertaining to quality control, quality assurance, and the maintenance of records and documentation. The FDA, the EMA and comparable regulatory authorities enforce these requirements through facility inspections. In the EU, the national competent authorities are responsible for manufacturing facility inspections, but the EMA plays a coordinating role in ensuring that standards are enforced consistently throughout the EU. Facilities using our point-of-care manufacturing process, including Lonza’s Cocoon® system, must be approved by the FDA, national competent authorities in the EU or comparable regulatory authorities. Vendors and operators are also subject to continuing FDA, EMA and comparable regulatory authority compliance.
Our own or third parties’ facilities where Lonza’s Cocoon® system is in use may be unable to comply with these regulatory requirements. Poor control of production processes can lead to the introduction of contaminants, or to inadvertent changes in the properties or stability of our point-of- care cell therapy product candidates that may not be detectable in final product testing. If we or our vendors and operators are unable to reliably produce product candidates to specifications acceptable to the FDA, the EMA or comparable regulatory authorities, or in accordance with the strict regulatory requirements, we may not obtain or maintain the approvals we need to commercialize our point-of-care cell therapy product candidates. Even if we obtain regulatory approval for any of our point -of -care cell therapy product candidates, there can be no assurance that either we or our third party vendors or operators will be able to manufacture the approved product to specifications acceptable to the FDA, the EMA or comparable regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential launch of the product, or to meet potential future demand. Deviations from manufacturing requirements may further require remedial measures that may be costly and/or time-consuming for us or a third party to implement and may include the temporary or permanent closure of a facility. Any such remedial measures imposed upon us or third parties with whom we contract could materially harm our business.
Even to the extent we use vendors or operators for Lonza’s Cocoon® system, we are ultimately responsible for the manufacturing of our point-of-care cell therapy product candidates. A failure to comply with regulatory requirements may result in regulatory enforcement actions against our vendors or operators or us, including fines and civil and criminal penalties.
Patients receiving T cell-based immunotherapies may experience serious adverse events, including neurotoxicity and cytokine release syndrome. Serious adverse events or undesirable side effects associated with our CAR T product candidates may result in delays, clinical holds, or terminations of our preclinical or clinical trials, impact our ability to obtain regulatory or marketing approval, and impact the commercial potential of such product candidates, which will significantly harm our business, financial condition and prospects.
We are currently developing CAR -T product candidates, including (1) GLPG5101, a CD19 CAR-T product candidate manufactured at point-of-care, currently in Phase1/2 in rrNHL; (2) GLPG5201, CD19 CAR-T product candidates manufactured at point-of-care, currently in Phase 1/2 in rrCLL, respectively; and (3) GLPG5301, a BCMA CAR-T product candidate manufactured at point-of-care, currently in Phase 1/2 in rrMM. In previous and ongoing clinical studies, including our current studies, involving CAR-T products and product candidates, patients experienced side effects such as neurotoxicity and cytokine release syndrome. There have been life-threatening events related to severe neurotoxicity and cytokine release syndrome, requiring intense medical intervention such as intubation or vasopressor support, and in several cases, resulted in death. Severe
15
neurotoxicity is a condition that is currently defined clinically by cerebral edema, confusion, drowsiness, speech impairment, tremors, seizures, or other central nervous system side effects, when such side effects are serious enough to lead to intensive care. In some cases, severe neurotoxicity was thought to be associated with the use of certain lymphodepletion regimens used prior to the administration of the CAR- T products. Cytokine release syndrome is a condition that is currently defined clinically by certain symptoms related to the release of cytokines, which can include fever, chills, low blood pressure, when such side effects are serious enough to lead to intensive care with mechanical ventilation or significant vasopressor support. The exact cause or causes of cytokine release syndrome and severe neurotoxicity in connection with treatment of CAR-T products is not fully understood at this time. In addition, patients have experienced other adverse events in these studies, such as a reduction in the number of blood cells (in the form of neutropenia, thrombocytopenia, anemia or other cytopenias), febrile neutropenia, chemical laboratory abnormalities (including elevated liver enzymes), and renal failure.
Undesirable side effects caused by any of our CAR- T product candidates targeting rrNHL, rrCLL, or BCMA, could cause us or regulatory authorities to interrupt, delay or halt preclinical or clinical studies and could result in restrictions on the labeling, distribution, or marketing of any approved products or a requirement to conduct potentially costly post-approval studies or the delay or denial of marketing approval by the FDA or other comparable foreign regulatory authorities. Side effects and toxicities associated with any of our product candidates, if approved, as well as the warnings, precautions, and requirements listed in the prescribing information, could affect the willingness of physicians to prescribe, and patients to use, such product candidates, if approved, and negatively affect market acceptance and commercial sales. In some cases, side effects such as neurotoxicity or cytokine release syndrome have resulted in clinical holds of ongoing clinical trials and/or discontinuation of the development of the product candidate. Results of our studies could reveal a high and unacceptable severity and prevalence of side effects or unexpected characteristics. Treatment-related side effects could also affect patient recruitment or the ability of enrolled patients to complete the studies or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff, as toxicities resulting from engineered cell therapies are not normally encountered in the general patient population and by medical personnel. Medical personnel may need additional training regarding engineered cell therapies to understand their side effects. Complexity of the potential side effects of engineered cell therapies, associated with the potential presence of comorbidities, could result in deaths. Any of these occurrences may harm our business, financial condition and prospects significantly.
If we or others identify undesirable side effects caused by our product candidates, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may not approve our CAR-T product candidates, or, if such product candidates are approved, may limit or withdraw such approval; |
| ● | regulatory authorities may require the addition of labeling statements, such as a “boxed” warning or a contraindication, if our product candidates are approved; |
| ● | if our product candidates were approved, regulatory authorities may require a REMS plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries, and other risk minimization tools; |
| ● | we may be subject to regulatory investigations and government enforcement actions; |
| ● | we could be sued and held liable for injury caused to individuals exposed to or taking our product candidates; and |
| ● | our reputation may suffer. |
16
Negative public opinion and increased regulatory scrutiny of cell therapies and cellular research may damage public perception of our CAR -T product candidates, and any future products or adversely affect our ability to conduct our business or obtain and maintain marketing approvals for our CAR-T product candidates.
Public perception may be influenced by claims that cell therapy, including cell editing technologies, is unsafe, or unethical, and research activities and adverse events in the field, even if not ultimately attributable to us or our CAR-T product candidates, could result in increased governmental regulation, unfavorable public perception, challenges in recruiting patients to participate in our clinical studies, potential regulatory delays in the testing or approval of our CAR-T product candidates, labeling restrictions for any future approved CAR -T products, and a decrease in demand for any such product. For example, in November 2023, the FDA announced that it would be conducting an investigation into reports of T-cell malignancies following BCMA-directed or CD19-directed autologous CAR-T cell immunotherapies following reports of T-cell lymphoma in patients receiving these therapies. The FDA also stated that patients and clinical trial participants receiving treatment with the currently approved BCMA-directed and CD19-directed genetically modified autologous CAR-T cell immunotherapies products should be monitored life-long for new malignancies. In January 2024, the FDA determined that new safety information related to T-cell malignancies should be included in the labeling with boxed warning language on these malignancies for all BCMA- and CD-19-directed genetically modified autologous T-cell immunotherapies. Additionally, EMA’s PRAC started a signal procedure to review data on secondary malignancies related to T-cells (cancers that begin in a type of white blood cells called T-cells), including T-cell lymphoma and leukemia, for the six approved CAR-T cell medicines. More restrictive government regulations or negative public opinion would have a negative effect on our business or financial condition and may delay or impair the development and commercialization of our CAR-T product candidates or demand for any approved products.
Delays in obtaining regulatory approval of manufacturing processes and facilities or disruptions in manufacturing processes may delay or disrupt our commercialization efforts.
Before we can begin to commercially manufacture our product candidates for human therapeutics, the FDA must review for the applicable manufacturing process and facilities as part of its review of our marketing application. This will likely require the manufacturing facilities to pass a pre-approval inspection by the FDA. A manufacturing authorization must also be obtained from the appropriate EU regulatory authorities or other comparable regulatory authorities.
In order to obtain FDA approval, we will need to ensure that all of the processes, methods, and equipment are compliant with cGMP and perform extensive audits of vendors, contract laboratories, and suppliers. If any of our vendors, contract laboratories or suppliers is found to be out of compliance with cGMP, we may experience delays or disruptions in manufacturing while we work with these third parties to remedy the violation(s) or while we work to identify suitable replacement vendors. The cGMP requirements govern, among other things, quality control of the manufacturing process, raw materials, containers/closures, buildings and facilities, equipment, storage and shipment, labeling, laboratory activities, data integrity, documentation policies and procedures, and returns. In complying with cGMP, we will be obligated to expend time, resources, and efforts in production, record keeping, and quality control to assure that the product meets applicable specifications and other requirements. If we fail to comply with these requirements, we would be subject to possible regulatory action that could adversely affect our business, results of operations, financial condition, and cash flows, including the inability to sell any products that we may develop.
Risks related to commercialization of future products
The commercial success of any future products will depend upon the degree of market acceptance by physicians, healthcare payers, patients, and the medical community.
The commercial success of any future products we may develop will depend in part on acceptance by the medical community, patients, and third-party or governmental payers as medically useful, cost-effective, and safe. Any products that we and our current and future partners may bring to the market may not gain market acceptance by physicians, patients, third-party payers and others in the medical community. If these products do
17
not achieve an adequate level of acceptance, we may not generate significant product revenue and may not become profitable. The degree of market acceptance of any future products we may develop will depend on a number of factors, including:
Even if a product displays a favorable efficacy and safety profile in preclinical and clinical studies and receives regulatory approval, market acceptance of the product will not be known until after it is launched. Our efforts to educate the medical community and third-party payers on the benefits of our products may require significant resources and may never be successful. Our efforts to educate the marketplace may require more resources than are required by the conventional methods marketed by our competitors. Any of these factors may cause any future products we may develop to be unsuccessful or less successful than anticipated.
Coverage and reimbursement decisions by third-party payers may have an adverse effect on pricing and market acceptance.
There is significant uncertainty related to the third-party coverage and reimbursement of newly approved drugs. To the extent that we retain commercial rights following clinical development, we may seek approval to market our product candidates in the United States, the European Union and other selected jurisdictions. Market acceptance and sales of our product candidates, if approved, in both domestic and international markets will depend significantly on the availability of adequate coverage and reimbursement from third-party payers for any of our product candidates and may be affected by existing and future healthcare reform measures. Third-party payers, such as government authorities, private health insurers and health maintenance organizations, decide which drugs they will cover and establish payment levels. See section entitled “Information on the Company – Pharmaceutical coverage, pricing and reimbursement.”
We cannot be certain that coverage and adequate reimbursement will be available for any of our products or product candidates, if approved. Also, we cannot be certain that reimbursement policies will not reduce the demand for, or the price paid for, any of our product candidates, if approved. If reimbursement is not available or is available on a limited basis for any of our products or product candidates, if approved, we may not be able to commercialize successfully any such product candidate.
Obtaining coverage and reimbursement approval for a product from a government or other third-party payer is a time consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to the payer. We may not be able to provide data sufficient to gain acceptance with respect to coverage and reimbursement or to have pricing set at a satisfactory level. If reimbursement of our future products, if any, is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels such as may result where alternative or generic treatments are available, we may be unable to achieve or sustain profitability.
In certain countries, particularly in the European Union, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of marketing approval for a product candidate. To obtain reimbursement or
18
pricing approval in some countries, we may be required to conduct additional clinical trials that compare the cost-effectiveness of our product candidates to other available therapies. In the United States, Medicare and Medicaid are significant third party payers. Medicare is administered by the Centers for Medicare & Medicaid Services (CMS), an agency within the U.S. Department of Health and Human Services (HHS) and Medicaid is administered jointly by CMS and the individual states. Obtaining adequate coverage and reimbursement under Medicare and Medicaid is important for new drug products. Additionally, private payers may adopt coverage policies or reimbursement methodologies similar to Medicare. Reimbursement by a third-party payer may depend upon a number of factors, including the third-party payer's determination that use of a product is: a covered benefit under its health plan; safe, effective and medically necessary; appropriate for the specific patient; cost-effective; and neither experimental nor investigational. Novel and expensive cell therapies like CAR-T cell therapies have experienced and continue to experience coverage and reimbursement challenges. For example, Medicare only covers CAR-T cell therapies that meet specific criteria set forth in a national coverage decision. Other third party payers may impose coverage criteria more extensive than compliance with FDA labeling. If reimbursement of any of our products or product candidates, if approved, is unavailable or limited in scope or amount in a particular country, or if pricing is set at unsatisfactory levels, we may be unable to achieve or sustain profitability of our products in such country.
The delivery of healthcare in the European Union, including the establishment and operation of health services and the pricing and reimbursement of medicines, is almost exclusively a matter for national, rather than EU, law and policy. National governments and health service providers have different priorities and approaches to the delivery of healthcare and the pricing and reimbursement of products in that context. In general, however, the healthcare budgetary constraints in most EU member states have resulted in restrictions on the pricing and reimbursement of medicines by relevant health service providers. Coupled with ever-increasing EU and national regulatory burdens on those wishing to develop and market products, this could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities and affect our ability to commercialize any products for which we have or will obtain marketing approval.
Legislative and regulatory activity may exert downward pressure on potential pricing and reimbursement for any of our product candidates, if approved, that could materially affect the opportunity to commercialize.
The United States and several other jurisdictions are considering, or have already enacted, a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell any of our products or product candidates profitably, if approved. Among policymakers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access to healthcare. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. There have been, and likely will continue to be, legislative and regulatory proposals at the federal and state levels directed at broadening the availability of healthcare and containing or lowering the cost of healthcare. See section entitled “ Information on the Company – Patient Protection and Affordable Care Act and Healthcare Reform.” We cannot predict the initiatives that may be adopted in the future. The continuing efforts of the government, insurance companies, managed care organizations and other payers of healthcare services to contain or reduce costs of healthcare may adversely affect:
We expect that the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or, collectively, the ACA, as well as other healthcare reform measures that may be adopted in the future in any jurisdiction, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from
19
private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or successfully commercialize our products or product candidates.
Legislative and regulatory proposals have been made to expand post-approval requirements and restrict sales and promotional activities for pharmaceutical and biologic products. We cannot be sure whether additional legislative changes will be enacted, or whether FDA regulations, guidance or interpretations or regulations, guidance or interpretations of other regulatory authorities will be changed, or what the impact of such changes on the marketing approvals of our products or product candidates, if any, may be. In addition, increased scrutiny by Congress of the FDA’s approval process may significantly delay or prevent marketing approval, as well as subject us to more stringent product labeling and post-marketing testing and other requirements.
Moreover, increasing efforts by governmental and third-party payers in the United States and abroad to cap or reduce healthcare costs may cause such organizations to limit both coverage and the level of reimbursement for newly approved products and, as a result, they may not cover or provide adequate payment for our products or product candidates. There has been increasing legislative and enforcement interest in the United States with respect to specialty drug pricing practices. Specifically, there have been several recent U.S. Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs.
We expect that the healthcare reform measures that have been adopted and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product and could seriously harm our future revenues. Any reduction in reimbursement from Medicare or other government programs in the United States or in other jurisdictions may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or successfully commercialize our products.
Risks related to our financial position and need for additional capital
We have limited historical profit from product sales and limited historical data on product revenues, which makes it difficult to assess our future prospects and financial results.
Pharmaceutical product development is a highly speculative undertaking and involves a substantial degree of uncertainty. Our operations to date have been generally limited to developing our technology and undertaking preclinical studies and clinical trials of our oncology and immunology product candidates, currently including GLPG5101, GLPG5201, GLPG5301 or GLPG3667. We may not have the ability to overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the pharmaceutical area. We have transitioned from a clinical-stage to a commercial-stage company but have recently transferred the Jyseleca® business and continue as a clinical stage company in the field of oncology and immunology. We have limited historical profit from product sales and limited historical data on product revenues. Consequently, the ability to predict our future operating results or business prospects is more limited than if we had a longer operating history or approved products on the market.
With the exception of the years ended December 31, 2019 and 2023, we have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future.
With the exception of the years ended December 31, 2019 and 2023, we have incurred significant operating losses since our inception in 1999. We reported net losses of €103.2 million for the year ended December 31,
20
2021, net losses of €218.0 million for the year ended December 31, 2022, and a net profit of €211.7 million for the year ended December 31, 2023. Our losses resulted principally from costs incurred in research and development, preclinical testing, clinical development of our product and our product candidates as well as costs incurred for research programs, pre-commercial activities, commercial activities (as of 2020) and from general and administrative costs associated with our operations. In the future we intend to continue the aforementioned activities except for the commercial activities due to the discontinuation of the Jyseleca® business. Our prior losses, combined with expected future losses, have had and will continue to have an adverse effect on our shareholders’ equity and working capital. We expect to continue incurring significant research, development, and other expenses related to our ongoing operations, and to continue incurring operating losses for the foreseeable future.
If any of our product candidates fail in clinical trials or do not gain regulatory approval, or if any of our product candidates, if approved, fail to achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
Additionally, we may not achieve significant revenues from sales of products. Therefore, even if we are able to generate revenues from the sale of any approved product, we may not become profitable or sustain profitability.
We may require substantial additional funding, which may not be available to us on acceptable terms, or at all.
Our operations have consumed substantial amounts of cash since inception. We are currently conducting clinical trials for GLPG5101, GLPG5201, GLPG5301, GLPG3667 and other oncology and immunology product candidates. Developing pharmaceutical product candidates, including conducting clinical trials, is expensive. We will require substantial additional future capital in order to complete clinical development and, if we are successful, to commercialize any of our current product candidates. If the FDA, or any other comparable regulatory agency, such as the EMA, requires that we perform studies or trials in addition to those that we currently anticipate with respect to the development of our product candidates, or repeat studies or trials, our expenses would further increase beyond what we currently expect, and any delay resulting from such further or repeat studies or trials could also result in the need for additional financing and other resources.
Our existing current financial investments and cash and cash equivalents may not be sufficient for us to complete advanced clinical development of our product candidates or, if applicable, to commercialize product candidates that would be approved. Accordingly, we may continue to require substantial additional capital to continue our clinical development activities and potentially engage in commercialization activities. Because successful development of our product candidates is uncertain, we are unable to estimate the actual funds and resources we will require to complete research and development and commercialize our product candidates. The amount and timing of our future funding requirements will depend on many factors, including but not limited to:
| ● | the progress, costs, results of and timing of our ongoing and planned clinical trials; |
| ● | our ability to reach milestones under our existing collaboration arrangements and enter into additional collaborative agreements for the development and commercialization of our product candidates; |
| ● | the willingness of the FDA, EMA, the MHRA, the MHLW/PMDA, and other comparable regulatory authorities to accept our clinical trials and preclinical studies and other work as the basis for review and approval of product candidates; |
| ● | the outcome, costs and timing of seeking and obtaining regulatory approvals from the FDA, EMA, the MHRA, the MHLW/PMDA, and other comparable regulatory authorities; |
| ● | whether our collaboration partners continue to collaborate with us on the development and commercialization of our product candidates; |
| ● | the number of product candidates and indications that we pursue, whether developed internally or in-licensed; |
21
| ● | the timing and costs associated with manufacturing our product candidates for clinical trials and other studies and, if approved, for commercial sale; |
| ● | our need to expand our development activities and, potentially, our research activities; |
| ● | the timing and costs associated with establishing sales and marketing capabilities; |
| ● | the costs associated with our future product sales, marketing, commercial manufacturing, and distribution activities; |
| ● | the costs associated with adding new manufacturing sites for our point-of-care cell therapy product candidates; |
| ● | market acceptance of any approved product candidates; |
| ● | the costs of acquiring, licensing or investing in additional businesses, products, product candidates and technologies; |
| ● | the cost to maintain, expand and defend the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; |
| ● | the extent to which we may be required to pay milestone or other payments under our in-license agreements and the timing of such payments; |
| ● | our need and ability to hire additional management, development and scientific personnel; and |
| ● | our need to implement additional internal systems and infrastructure, including financial and reporting systems. |
Some of these factors are outside of our control. Based upon our current expected level of operating expenditures and our existing current financial investments and cash and cash equivalents, we believe that we will be able to fund our operating expenses and capital expenditure requirements for the coming years. This period could be shortened, but not below a period of 12 months, if there are any significant increases beyond our expectations in spending on development programs or more rapid progress of development programs than anticipated. Accordingly, we expect that we could need to raise additional funds in the future. Additional funding may not be available to us on acceptable terms, or at all. If we are unable to obtain funding from equity offerings or debt financings, including on a timely basis, we may be required to:
| ● | seek additional collaboration partners for one or more of any future proprietary product candidates at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available; |
| ● | relinquish or license on unfavorable terms our rights to technologies or any future proprietary product candidates that we otherwise would seek to develop or commercialize ourselves; |
| ● | significantly curtail one or more of our research or development programs; |
| ● | curtail our product sales, marketing, commercial manufacturing, and |
| ● | cease operations altogether. |
Raising additional capital may cause dilution to our existing shareholders, restrict our operations or require us to relinquish rights to our product candidates or technologies.
We may seek additional funding through a combination of equity offerings, debt financings, collaborations and/or licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a holder of the ADSs or our ordinary shares. The incurrence of indebtedness and/or the issuance of certain equity securities could result in increased fixed payment obligations and could also result in certain additional restrictive covenants, such as limitations on our ability to incur additional debt and/or issue additional equity, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. In addition, issuance of additional equity securities, or the possibility of such issuance, may cause the market price of the ADSs or our ordinary shares to decline. In the event that we enter into collaborations and/or licensing arrangements in order to raise capital, we may be required to accept unfavorable terms, including relinquishing or licensing to a third party on unfavorable terms our rights to technologies or product
22
candidates that we otherwise would seek to develop or commercialize ourselves or potentially reserve for future potential arrangements when we might be able to achieve more favorable terms.
Adverse developments affecting the financial services industry, such as actual events or concerns involving liquidity, defaults, or non-performance by financial institutions or transactional counterparties, could adversely affect our current and projected business operations, financial condition and results of operations.
Actual events involving limited liquidity, defaults, non-performance or other adverse developments that affect financial institutions, transactional counterparties or other companies in the financial services industry or the financial services industry generally, or concerns or rumors about any events of these kinds or other similar risks, have in the past and may in the future lead to market-wide liquidity problems. For example, the closures of Silicon Valley Bank ("SVB") and Signature Bank (“Signature”) in March 2023, and First Republic Bank (“First Republic”) in May 2023, led to disruption and volatility, including deposit outflows and increased need for liquidity, at certain banks. In addition, in March 2023, the Bank of England made the decision to sell Silicon Valley Bank UK Limited (“SVBUK”), the UK subsidiary of the SVB, to HSBC UK Bank Plc. Although depositors of these banks were largely protected, it is not certain that the Federal Reserve, FDIC or other similar agencies will treat future bank failures similarly, or would do so in a timely manner.
Although we are not a borrower or party to any such instruments with SVB, SVBUK, Signature, First Republic, or any other financial institution currently in receivership or a similar proceeding, if any financial institution with which we have banking relationships were to be placed into receivership or a similar proceeding, we may be unable to access such funds. In addition, if any of our critical vendors, suppliers, manufacturers or other third parties with whom we conduct business are unable to access funds pursuant to such instruments or lending arrangements with such a financial institution, such parties’ ability to perform their obligations under arrangements or agreements with us could be adversely affected.
Although we assess our banking relationships as we believe necessary or appropriate, our access to funding sources in amounts adequate to finance or capitalize our current and projected future business operations could be significantly impaired by factors that affect us, the financial institutions with which we have arrangements directly, or the financial services industry or economy in general. These factors could include, among others, events such as liquidity constraints or failures, the ability to perform obligations under various types of financial, credit or liquidity agreements or arrangements, disruptions or instability in the financial services industry or financial markets, or concerns or negative expectations about the prospects for companies in the financial services industry. These factors could involve financial institutions or financial services industry companies with which we have financial or business relationships, but could also include factors involving financial markets or the financial services industry generally.
In addition, investor concerns regarding the U.S. or international financial systems could result in less favorable commercial financing terms, including higher interest rates or costs and tighter financial and operating covenants, or systemic limitations on access to credit and liquidity sources, thereby making it more difficult for us to acquire financing on acceptable terms or at all. Any decline in available funding or access to our cash and liquidity resources could, among other risks, adversely impact our ability to meet our operating expenses, financial obligations or fulfill our other obligations, result in breaches of our financial and/or contractual obligations or result in violations of U.S. federal or state wage and hour laws. Any of these impacts, or any other impacts resulting from the factors described above or other related or similar factors not described above, could have material adverse impacts on our liquidity and our current and/or projected business operations, financial condition and results of operations.
23
In addition, any further deterioration in the macroeconomic economy or financial services industry could lead to losses or defaults by our critical vendors, suppliers, manufacturers or other third parties, which in turn, could have a material adverse effect on our current and/or projected business operations, financial condition and results of operations.
Risks related to our reliance on third parties
We are heavily dependent upon our collaboration arrangements with Gilead and certain other third parties for the development and commercialization of our products and there can be no assurance that these arrangements will deliver the benefits we expect.
In July 2019, we entered into a 10-year global research and development collaboration with Gilead. In connection with our entry into the option, license and collaboration agreement, we received an upfront payment of $3.95 billion and a €960 million ($1.1 billion) equity investment from Gilead. Under the option, license and collaboration agreement, we fund and lead all discovery and development autonomously until the end of the relevant Phase 2 clinical study. After the completion of a qualifying Phase 2 clinical study (or in certain circumstances, the first Phase 3 clinical study), Gilead will have the option to acquire an exclusive commercial license to that program in all countries outside of Europe. If the option is exercised, we and Gilead will co-develop the compound and share costs equally. In addition, we are heavily dependent on Gilead for the commercialization of filgotinib and the further development of filgotinib outside of Europe.
Gilead may not devote sufficient resources or give sufficient priority to the programs in respect of which it acquires a commercial license pursuant to the option, license and collaboration agreement. Furthermore, Gilead may not be successful in the commercialization of filgotinib outside of Europe and further development and commercialization of filgotinib or other programs for which it acquires a commercial license, even when they do devote resources and prioritize their efforts for such programs.
In addition, the terms of the collaboration with Gilead and any collaboration or other arrangement that we may establish may not ultimately prove to be favorable to us or may not be perceived as favorable, which may negatively impact the trading price of the ADSs or our ordinary shares. In addition, pursuant to the collaboration with Gilead, we are entitled to certain option payments and tiered royalties and milestones on certain products. There can be no assurance that such payments will be sufficient to cover the cost of development of the relevant product candidates.
We are subject to a number of additional risks associated with our dependence on our collaborations with third parties, the occurrence of which could cause our collaboration arrangements to fail. In particular, the collaboration we entered into in July 2019 is managed by a set of joint committees comprised of equal numbers of representatives from each of us and Gilead. Conflicts may arise between us and Gilead, such as conflicts concerning the interpretation of clinical data, the achievement of milestones, the interpretation of financial provisions or the ownership of intellectual property developed during the collaboration, and there can be no assurance that the joint committees will be able to resolve any such conflicts. If any such conflicts arise, Gilead could act in a manner adverse to our best interests. Any such disagreement could result in one or more of the following, each of which could delay or prevent the development or commercialization of product candidates subject to the collaboration arrangements, and in turn prevent us from generating sufficient revenues to achieve or maintain profitability:
| ● | reductions or delays in the payment of milestone payments, royalties or other payments we believe are due; |
| ● | actions taken by Gilead inside or outside our collaboration which could negatively impact our rights or benefits under our collaboration including termination of the collaboration for convenience; or |
| ● | unwillingness on the part of Gilead to keep us informed regarding the progress of its development and commercialization activities or regulatory approval or to permit public disclosure of the results of those activities. |
24
In addition to our collaboration with Gilead, we may also enter into future collaborations which will give rise to similar risks, although our ability to enter into such collaborations may be limited given the scale of our collaboration with Gilead.
If our global research and development collaboration with Gilead or other collaborations on research and development candidates do not result in the successful development and commercialization of products or if Gilead or another one of our collaboration partners terminates its agreement with us, we may not receive any future research funding or milestone or royalty payments under the collaboration. If we do not receive the funding we expect under these agreements, development of our product candidates could be delayed and we may need additional resources to develop product candidates.
We may not be successful in establishing future development and commercialization collaborations, particularly given the scale of our collaboration with Gilead, and this could adversely affect, and potentially prohibit, our ability to develop and commercialize our product candidates.
Developing pharmaceutical products, conducting clinical trials, obtaining regulatory approval, establishing manufacturing capabilities, commercializing, and marketing approved products is expensive. Accordingly, we have sought and may in the future seek to enter into collaborations with companies that have more resources and experience. In the future, however, our ability to do so may be limited given the scale of the 10-year global research and development collaboration that we entered into with Gilead in July 2019. If Gilead declines to exercise its option and we are otherwise unable to obtain a collaboration partner for our product candidates, we may be unable to advance the development of our product candidates through late-stage clinical development and seek approval in any market. In situations where we enter into a development and commercial collaboration arrangement for a product candidate, we may also seek to establish additional collaborations for development and commercialization in territories outside of those addressed by the first collaboration arrangement for such product candidate. If any of our product candidates receives marketing approval, we may enter into sales and marketing arrangements with third parties with respect to otherwise unlicensed or unaddressed territories. Furthermore, there are a limited number of potential collaboration partners, and we expect to face competition in seeking appropriate collaboration partners. If we are unable to enter into any development and commercial collaborations and/or sales and marketing arrangements on acceptable terms, or at all, we may be unable to successfully develop and seek regulatory approval for our product candidates and/or effectively market and sell approved products, if any.
We are heavily dependent upon our collaboration arrangements with Gilead and certain other third parties for the development and commercialization of our products and there can be no assurance that these arrangements will deliver the benefits we expect.
We have relied upon and plan to continue to rely upon CROs to monitor and manage data for our preclinical and clinical programs. We rely on these parties for execution of our preclinical studies and clinical trials, and we control only certain aspects of their activities. We and our CROs also rely upon clinical sites and investigators for the performance of our clinical trials in accordance with the applicable protocols and applicable legal and regulatory requirements and scientific standards. Nevertheless, we are responsible for ensuring that each of our studies and trials is conducted in accordance with the applicable protocol and applicable legal and regulatory requirements and scientific standards, and our reliance on CROs as well as clinical sites and investigators does not relieve us of our regulatory responsibilities. We are required to, and do, have mechanisms in place to adequately manage, oversee and control our clinical trials, including selection of CROs, auditing activities, strong focus on set-up (during which deliverables, timelines and roles and responsibilities are defined), and strong oversight during the conduct of clinical trials. We, our CROs, as well as the clinical sites and investigators are required to comply with current Good Clinical Practices (GCPs) and GDPs, which are regulations and guidelines enforced by the FDA, the competent authorities of the member states of the European Economic Area,
25
or EEA, and comparable regulatory authorities for all of our products in clinical development. Regulatory authorities enforce these requirements through periodic inspections of trial sponsors, investigators and clinical sites. If we, any of our CROs or any of the clinical sites or investigators fail to comply with applicable GCPs GDPs or other applicable requirements, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, EMA, the MHRA, MHLW, or comparable regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP and GDP regulations. We also cannot assure you that our CROs, as well as the clinical sites and investigators, will perform our clinical trials in accordance with the applicable protocols as well as applicable legal and regulatory requirements and scientific standards, or report the results obtained in a timely and accurate manner. In addition to GCPs, our clinical trials must be conducted with products produced under current Good Manufacturing Practice (cGMP) regulations. While we have agreements governing activities of our CROs, we have limited influence over the actual performance of our CROs as well as the performance of clinical sites and investigators. In addition, significant portions of the clinical trials for our product candidates are and will continue to be conducted outside of Belgium, which will make it more difficult for us to monitor CROs as well as clinical sites and investigators and perform visits of our clinical sites, and will force us to rely heavily on CROs to ensure the proper and timely conduct of our clinical trials in accordance with the applicable protocols and compliance with applicable regulations, including GCPs, and scientific standards. Failure to comply with applicable protocols and regulations in the conduct of the clinical trials for our product candidates may require us to repeat clinical trials, which would delay the regulatory approval process. Additionally, the performance of our CROs may also be delayed or disrupted by the ongoing armed conflict between Russia and Ukraine, and sanctions against Russia.
Some of our CROs have an ability to terminate their respective agreements with us if it can be reasonably demonstrated that the safety of the subjects participating in our clinical trials warrants such termination, or as a result of data integrity compromise, or if there is reasonable belief that good clinical practice or applicable laws or regulations will be materially violated, or if we make a general assignment for the benefit of our creditors, or if we are liquidated.
If any of our relationships with these CROs terminate, we may not be able to enter into arrangements with alternative CROs or to do so on commercially reasonable terms. In addition, our CROs are not our employees, and except for remedies available to us under our agreements with such CROs, we cannot control whether or not they devote sufficient time and resources to our preclinical and clinical programs. If CROs do not carry out their contractual duties or obligations successfully or meet expected deadlines, if they need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure (including by clinical sites or investigators) to adhere to our clinical protocols, regulatory requirements or for other reasons, our clinical trials may be extended, delayed or terminated and we may not be able to obtain regulatory approval for or successfully commercialize our product candidates. As a result, our results of operations and the commercial prospects for our product candidates would be harmed, our costs could increase substantially and our ability to generate revenues could be delayed significantly.
Switching or adding additional CROs involves additional cost and requires management time and focus. In addition, there is a natural transition period when a new CRO commences work. As a result, delays may occur, which can materially impact our ability to meet our desired clinical development timelines. Though we carefully manage our relationships with our CROs, there can be no assurance that we will not encounter challenges or delays in the future or that these delays or challenges will not have a material adverse impact on our business, financial condition and prospects.
We rely completely on third parties to manufacture our preclinical and clinical drug supplies and to produce commercial supplies of any approved product.
We do not currently have, nor do we plan to acquire, the infrastructure or capability internally to manufacture our drug supply for our approved products or preclinical and clinical drug supplies or lentiviral
26
vector. This reliance on third parties increases the risk of shortages of our drugs or drug candidates and of the availability of such drugs or drug candidates at an acceptable cost or quality. This could potentially delay, prevent or impair our development and commercialization efforts.
If, for any reason, we were to experience an unexpected loss in the supply of any of our approved products, our product candidates or placebo or comparator drug used in certain of our clinical trials, or our lentiviral vector, whether as a result of manufacturing, supply or storage issues or otherwise, we could experience delays, disruptions, suspensions or terminations of, or be required to restart or repeat, any pending or ongoing clinical trials or commercial distribution of our approved products could be negatively affected. The facilities used by our contract manufacturers or other third-party manufacturers to manufacture our approved products and our product candidates are subject to the FDA’s, EMA’s, MHRA’s, MHLW’s and other comparable regulatory authorities’ pre-approval inspections that can be conducted after we submit the required approval applications to any relevant regulatory authority, such as, for example, an NDA or BLA to the FDA. We monitor, but do not control, the implementation of the manufacturing process of, but are dependent on, our contract manufacturers or other third-party manufacturers for compliance with cGMP regulatory requirements for manufacture of any drug products. If our contract manufacturers or other third-party manufacturers do not successfully manufacture material that conforms to applicable specifications and the strict regulatory requirements of the FDA, EMA, MHRA, MHLW or others or if such authority finds deficiencies at a contract manufacturer’s facility or is unable to conduct an inspection necessary to evaluate such facility, we will not be able to secure and/or maintain regulatory approvals for our products manufactured at these facilities. This could significantly impact our ability to develop, obtain regulatory approval for or market our product candidates, if approved.
We, or our manufacturers, purchase from third-party suppliers the materials necessary to produce our product candidates for our clinical trials and our approved products. There are a limited number of suppliers for raw materials that we use to manufacture our drugs and there may be a need to assess alternate suppliers to prevent a possible disruption of the manufacture of the materials necessary to produce our product candidates for our clinical trials and our approved products. We do not have any control over the process or timing of the acquisition of raw materials by our manufacturers, and delays may result for reasons beyond our control, including the ongoing armed conflict between Russia and Ukraine.
If our manufacturers or we are unable to purchase these raw materials after regulatory approval has been obtained for our product candidates, the commercial launch of our products would be delayed or there would be a shortage in supply, which would impair our ability to generate revenues from the sale of our products. Additionally, if we receive regulatory approval for our product candidates, we may experience unforeseen difficulties or challenges in the manufacture of our products on a commercial scale compared to the manufacture for clinical purposes.
We expect to continue to depend on contract manufacturers or other third -party manufacturers for the foreseeable future.
Via the acquisition of CellPoint on June 21, 2022, we gained access to a decentralized and automated point-of-care cell therapy supply model, which CellPoint had developed in a strategic collaboration with a third party, Lonza. The platform that we license consists of the end -to-end xCellit® workflow management and monitoring software and Lonza’s Cocoon® platform, a functionally closed, automated manufacturing platform for cell and gene therapies. If, for any reason, the collaboration terminates or is otherwise materially changed and we are no longer entitled to use such technology platform, then we may be unable to secure alternatives to such technology and our research, development or other efforts may be interrupted or delayed, and our financial condition and results of operation may be materially adversely affected.
27
We rely on clinical data and results obtained by third parties that could ultimately prove to be inaccurate or unreliable.
If the third-party data and results we rely upon prove to be inaccurate, unreliable or not applicable to our product candidates, we could make inaccurate assumptions and conclusions about our product candidates and our research and development efforts could be materially adversely affected.
Risks related to our intellectual property
Our ability to compete may decline if we do not adequately protect our proprietary rights.
Future commercial success depends on obtaining and maintaining proprietary rights any future product, and our current and any future product candidates, as well as successfully defending these rights against third-party challenges. We will only be able to protect our product candidates, and their uses from unauthorized use by third parties to the extent that valid and enforceable patents, or effectively protected trade secrets, cover them. Our ability to obtain patent protection for our product candidates is uncertain due to a number of factors, including:
| ● | we may not have been the first to make the inventions covered by pending patent applications or issued patents; |
| ● | we may not have been the first to file patent applications for our product candidates or the compositions we developed or for their uses; |
| ● | others may independently develop identical, similar or alternative products or compositions and uses thereof; |
| ● | our disclosures in patent applications may not be sufficient to meet the statutory requirements for patentability; |
| ● | any or all of our pending patent applications may not result in issued patents; |
| ● | we may not seek or obtain patent protection in countries that may eventually provide us a significant business opportunity; |
| ● | any patents issued to us may not provide a basis for commercially viable products, may not provide any competitive advantages, or may be successfully challenged by third parties; |
| ● | our compositions, methods and processes may not be patentable; |
| ● | others may design around our patent claims to produce competitive products which fall outside of the scope of our patents; or |
| ● | others may identify prior art or other bases which could invalidate our patents. |
Even if we have or obtain patents covering our product candidates or compositions, we may still be barred from making, using and selling our product candidates or technologies because of the patent rights of others. Others may have filed, and in the future may file, patent applications covering compositions or products that are similar or identical to ours. If a patent owned by a third party covers one of our product candidates or its use, this could materially affect our ability to develop the product candidate or sell the resulting product if approved. Because patent applications are not published until 18 months from their priority date, there may be currently pending applications unknown to us that may later result in issued patents that our product candidates or compositions may infringe. Additionally, because the scope of claims in pending patent applications can change, there may be pending applications whose claims do not currently cover any of our product candidates but may be altered such that one or more of our product candidates are covered when the resulting patent issues. These patent applications may have priority over patent applications filed by us.
Moreover, even if we are able to obtain patent protection, such patent protection may be of insufficient scope to achieve our business objectives. For example (without any limitation), others may be able to develop a product that is similar to, or better than, ours in a way that is not covered by the claims of our patents.
Obtaining and maintaining a patent portfolio entails significant expense and resources. Part of the expense includes periodic maintenance fees, renewal fees, annuity fees, various other governmental fees on patents and/or applications due in several stages over the lifetime of patents and/or applications, as well as the cost associated with complying with numerous procedural provisions during the patent application process. We may or may not
28
choose to pursue or maintain protection for particular inventions. In addition, there are situations in which failure to make certain payments or noncompliance with certain requirements in the patent process can result in abandonment or lapse of a patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If we choose to forgo patent protection or allow a patent application or patent to lapse purposefully or inadvertently, our competitive position could suffer. Moreover, in some circumstances, we do not have the right to control the preparation, filing or prosecution of patent applications, or to maintain the patents, covering technology subject to our collaboration or license agreements with third parties. For example, under our collaboration agreement with Gilead, Gilead controls litigation on our patents for filgotinib in jurisdictions outside the European region and for any optioned programs. Therefore, these patents and patent applications may not be prosecuted or enforced in a manner consistent with the best interests of our business.
Legal actions to enforce our patent rights can be expensive and may involve the diversion of significant management time. In addition, these legal actions could be unsuccessful and could also result in the invalidation of our patents or a finding that they are unenforceable. We may or may not choose to pursue litigation or other actions against those that have infringed on our patents, or used them without authorization, due to the associated expense and time commitment of monitoring these activities. If we fail to protect or to enforce our intellectual property rights successfully, our competitive position could suffer, which could harm our results of operations.
Pharmaceutical patents and patent applications involve highly complex legal and factual questions, which, if determined adversely to us, could negatively impact our patent position.
The patent positions of biotechnology and pharmaceutical companies can be highly uncertain and involve complex legal and factual questions. The interpretation and breadth of claims allowed in some patents covering pharmaceutical compositions may be uncertain and difficult to determine and are often affected materially by the facts and circumstances that pertain to the patented compositions and the related patent claims. The standards of the United States Patent and Trademark Office, or USPTO, the European Patent Office, and other foreign counterparts are sometimes uncertain and could change in the future. Consequently, the issuance and scope of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. Certain U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to reexamination proceedings, post- grant review and/or inter partes review in the USPTO. European patents and other foreign patents may be subject also to opposition or comparable proceedings in the corresponding foreign patent office, which could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, reexamination, post-grant review, inter partes review and opposition proceedings may be costly. Accordingly, rights under any issued patents may not provide us with sufficient protection against competitive products or processes.
In addition, changes in or different interpretations of patent laws in the United States, Europe, and other jurisdictions may permit others to use our discoveries or to develop and commercialize our technology and products without providing any compensation to us, or may limit the number of patents or claims we can obtain. The laws of some countries do not protect intellectual property rights to the same extent as U.S. and European laws and those countries may lack adequate rules and procedures for defending our intellectual property rights.
If we fail to obtain and maintain patent protection and trade secret protection of our product candidates, we could lose our competitive advantage and competition we face would increase, reducing any potential revenues and adversely affecting our ability to attain or maintain profitability.
29
Developments in patent law could have a negative impact on our business.
From time to time, courts and other governmental authorities in the United States, Europe, Japan, and other jurisdictions may change the standards of patentability and any such changes could have a negative impact on our business.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to patent protection, because we operate in the highly technical field of development of therapies, we rely in part on trade secret protection in order to protect our proprietary technology and processes. However, trade secrets are difficult to protect. It is our policy to enter into confidentiality and intellectual property assignment agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors. These agreements generally require that the other party keeps confidential and does not disclose to third parties all confidential information developed by the party or made known to the party by us during the course of the party’s relationship with us. These agreements also generally provide that inventions conceived by the party in the course of rendering services to us will be our exclusive property. However, these agreements may not be honored and may not effectively assign intellectual property rights to us. Adequate remedies may not exist in the event of unauthorized use or disclosure of our confidential information. The disclosure of our trade secrets would impair our competitive position and may materially harm our business, financial condition and results of operations.
In addition to contractual measures, we try to protect the confidential nature of our proprietary information using physical and technological security measures. Such measures may not, for example (without any limitation), in the case of misappropriation of a trade secret by an employee or a third party with authorized access, provide adequate protection for our proprietary information. Our security measures may not prevent an employee or consultant from misappropriating our trade secrets and providing them to a competitor, and recourse we take against such misconduct may not provide an adequate remedy to protect our interests fully. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets, with protection varying across Europe and in other countries. Trade secrets may be independently developed by others in a manner that could prevent legal recourse by us. If any of our confidential or proprietary information, such as our trade secrets, were to be disclosed or misappropriated, or if any such information was independently developed by a competitor, our competitive position could be harmed.
We will not seek to protect our intellectual property rights in all jurisdictions throughout the world and we may not be able to adequately enforce our intellectual property rights even in the jurisdictions where we seek protection.
Filing, prosecuting and defending patents on our current and future product candidates in all countries and jurisdictions throughout the world would be prohibitively expensive, and our intellectual property rights in some countries could be less extensive than those in the United States and Europe, assuming that rights are obtained in the United States and Europe. Furthermore, even if patents are granted based on our European patent applications, we may not choose to perfect or maintain our rights in all available European countries. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as laws in the United States and Europe. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries, or from selling or importing products made using our inventions. The statutory deadlines for pursuing patent protection in individual foreign jurisdictions are based on the priority dates of each of our patent applications.
Competitors may use our technologies in jurisdictions where we do not pursue and obtain patent protection to develop their own products and further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the United States and Europe. These products may compete with our products and our patents or other intellectual property rights may not
30
be effective or sufficient to prevent them from competing. Even if we pursue and obtain issued patents in particular jurisdictions, our patent claims or other intellectual property rights may not be effective or sufficient to prevent third parties from so competing.
The laws of some foreign countries do not protect intellectual property rights to the same extent as the laws of the United States and Europe. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to pharmaceuticals or biotechnologies. This could make it difficult for us to stop the infringement of our patents, if obtained, or the misappropriation of our other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, many countries limit the enforceability of patents against third parties, including government agencies or government contractors.
In these countries, patents may provide limited or no benefit. Patent protection must ultimately be sought on a country- by - country basis, which is an expensive and time-consuming process with uncertain outcomes. Accordingly, we may choose not to seek patent protection in certain countries, and we will not have the benefit of patent protection in such countries.
Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. In addition, changes in the law and legal decisions by courts in the United States, Europe and other jurisdictions may affect our ability to obtain adequate protection for our technology and the enforcement of intellectual property. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition by potential partners or customers in our markets of interest. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively, and our business may be adversely affected.
Our inability to protect our intellectual property or failure to maintain the confidentiality and integrity of data or other sensitive company information, by cyber-attack or other event, could have a material adverse effect on our business.
Our success and competitive position are dependent in part upon our proprietary intellectual property. We rely on a combination of patents and trade secrets to protect our proprietary intellectual property, and we expect to continue to do so. Although we seek to protect our proprietary rights through a variety of means, we cannot guarantee that the protective steps we have taken are adequate to protect these rights. Patents issued to or licensed by us in the past or in the future may be challenged and held invalid. In addition, as our patents expire, we may be unsuccessful in extending their protection through patent term extensions or supplementary protection certificates. The expiration of, or the failure to maintain or extend our patents, could have a material adverse effect on us.
We also rely on confidentiality agreements with certain employees, consultants, and other third parties to protect, in part, trade secrets and other proprietary information. These agreements could be breached, and we may not have adequate remedies for such a breach. In addition, others could independently develop substantially equivalent proprietary information or gain access to our trade secrets or proprietary information.
31
Our intellectual property, other proprietary technology, and other sensitive company information is dependent on sophisticated information technology systems and is potentially vulnerable to cyber-attack, loss, damage, destruction from system malfunction, computer viruses, loss of data privacy, or misappropriation or misuse of it by those with permitted access, and other events. While we have invested to protect our intellectual property and other information, and continue to upgrade and enhance our systems to keep pace with continuing changes in information processing technology, there can be no assurance that our precautionary measures will prevent breakdowns, breaches, cyber-attacks, or other events. Such events could have a material adverse effect on our reputation, financial condition, or results of operations.
Risks related to intellectual property litigation
We may be subject to claims by third parties asserting ownership or commercial rights to inventions we develop or obligations to make compensatory payments to employees.
Third parties may in the future make claims challenging the inventorship or ownership of our intellectual property. We have written agreements with collaboration partners that provide for the ownership of intellectual property arising from our collaborations. Some of these agreements provide that we must negotiate certain commercial rights with collaboration partners with respect to joint inventions or inventions made by our collaboration partners that arise from the results of the collaboration. In some instances, there may not be adequate written provisions to address clearly the resolution of intellectual property rights that may arise from a collaboration. If we cannot successfully negotiate sufficient ownership and commercial rights to the inventions that result from the collaboration with a third-party collaboration partner, or if disputes otherwise arise with respect to the intellectual property developed in the framework of the collaboration, we may be limited in our ability to capitalize on the market potential of these inventions. In addition, we may face claims by third parties that our agreements with employees, contractors, or consultants obligating them to assign intellectual property to us are ineffective, or in conflict with prior or competing contractual obligations of assignment, which could result in ownership disputes regarding intellectual property we have developed or will develop and interfere with our ability to capture the commercial value of such inventions. Litigation may be necessary to resolve an ownership dispute, and if we are not successful, we may be precluded from using certain intellectual property, or may lose our exclusive rights in that intellectual property. Either outcome could have an adverse impact on our business.
While it is our policy to require our employees and contractors who may be involved in the development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful in executing or obtaining such an agreement with each party who, in fact, develops intellectual property that we regard as our own. In addition, such agreements may be breached or may not be self -executing, and we may be forced to bring claims against third parties, or defend claims they may bring against us, to determine the ownership of what we regard as our intellectual property. If we fail in prosecuting or defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel.
Third parties may assert that our employees or consultants have wrongfully used or disclosed confidential information or misappropriated trade secrets.
We employ individuals who were previously employed at universities, pharmaceutical companies or biopharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees and consultants do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of a former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
32
A dispute concerning the infringement or misappropriation of our proprietary rights or the proprietary rights of others could be time consuming and costly, and an unfavorable outcome could harm our business.
Our success will depend in part on our ability to operate without infringing the intellectual property and proprietary rights of third parties. We cannot assure you that our business, products, commercialization activities, and methods do not or will not infringe the patents, trademarks, or other intellectual property rights of third parties.
There is significant litigation in the pharmaceutical industry regarding patent and other intellectual property rights. While we are not currently subject to any pending intellectual property litigation, and are not aware of any such threatened litigation, we may be exposed to future litigation by third parties based on claims that our products or our product candidates, technologies or activities infringe the intellectual property rights of others. If our development activities are found to infringe any such patents, we may have to pay significant damages or seek licenses to such patents. A patentee could prevent us from using the patented drugs or compositions. We may need to resort to litigation to enforce a patent issued to us, to protect our trade secrets, or to determine the scope and validity of third-party proprietary rights. From time to time, we may hire scientific personnel or consultants formerly employed by other companies involved in one or more areas similar to the activities conducted by us. Either we or these individuals may be subject to allegations of trade secret misappropriation or other similar claims as a result of prior affiliations. Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on us. If we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court, or redesign our products. Patent litigation is costly and time consuming. We may not have sufficient resources to bring these actions to a successful conclusion. Any adverse ruling or perception of an adverse ruling in defending ourselves against these claims could have a material adverse impact on our cash position and the price of the ADSs or our ordinary shares. Any legal action against us or our collaboration partners could lead to:
| ● | payment of substantial damages for past use of the asserted intellectual property and potentially treble damages, if we are found to have willfully infringed a party’s patent rights; |
| ● | injunctive or other equitable relief that may effectively block our ability to further develop, commercialize, and sell our product candidates; or |
| ● | us or our collaboration partners having to enter into license arrangements that may not be available on commercially acceptable terms, if at all, all of which could have a material adverse impact on our cash position and business and financial condition. As a result, we could be prevented from commercializing current or future product candidates. |
Any of these risks coming to fruition could have a material adverse effect on our business, results of operations, financial condition and prospects.
33
Issued patents covering our approved product and product candidates could be found to be invalid or unenforceable if challenged in court
If we or one of our licensing partners initiated legal proceedings against a third party to enforce a patent covering our approved product or one of our product candidates, the defendant could counterclaim that the patent covering our approved product or one of our product candidates is invalid and/or unenforceable. In patent litigation, defendant counterclaims alleging invalidity and/or unenforceability are commonplace. Grounds for a validity challenge include alleged failures to meet any of several statutory requirements in most jurisdictions, including lack of novelty, obviousness or non-enablement. In the United States, grounds for unenforceability assertions include allegations that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review and equivalent proceedings in foreign jurisdictions, e.g., opposition proceedings. Such proceedings could result in revocation or amendment of our patents in such a way that they no longer cover our approved product or product candidates or competitive products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to validity, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our approved product and/or our product candidates. Such a loss of patent protection would have a material adverse impact on our business.
Risks associated to the legal uncertainty around the Unitary patent court in Europe
In Europe, the Unitary Patent Court (UPC) has opened on the 1st June 2023, and is a new court that is competent in matters of patent litigation for European patent, unless opted out, and unitary patents. Decisions from the UPC will have effect in all member states party to the UPC, and revocation in front of the UPC would potentially create a loss of rights in major jurisdictions including inter alia France, the Netherlands, Italy and Germany. Whereas we endeavor to evaluate and mitigate such a risk, new case law will emerge and at the moment remains uncertain.
Risks related to our employee matters
Our future success depends on our ability to retain the members of our Executive Committee and to attract, retain and motivate qualified scientists, development, and medical staff, consultants and advisors. If we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our industry has experienced a high rate of turnover of management and other personnel in recent years. Our ability to compete in the highly competitive biotechnology and pharmaceutical industries depends upon our ability to attract and retain highly qualified managerial, scientific and medical personnel, many of whom have been instrumental for us and have substantial experience with our therapies and related technologies. We are highly dependent on our management, scientific and medical personnel, especially our Executive Committee, which at the date of this annual report is comprised of: (i) Stoffels IMC BV (permanently represented by Dr. Paul Stoffels), our Chief Executive Officer; (ii) Thad Huston, our Chief Operating Officer, and Chief Financial Officer; (iii) Annelies Missotten, our Chief Human Resources Officer; and (iv) Valeria Cnossen, our General Counsel, each of whose services are critical to the successful implementation of our product candidates’ acquisition, development and regulatory strategies. To our best knowledge, we are not aware of any present intention of any of these individuals to leave our company. In order to induce valuable employees to continue their employment with us, we have granted subscription rights and restricted stock units (RSUs) that vest over time. The value to employees of subscription rights that vest over time is significantly affected by movements in our share price that are beyond our control, and may at any time be insufficient to counteract more lucrative offers from other companies.
Despite our efforts to retain valuable employees, members of our management, scientific, development, and medical teams may terminate their employment with us at any time, with or without notice. The loss of the
34
services of any of the members of our Executive Committee or other key employees and senior scientists could delay our research, development and other activities, and our inability to find suitable replacements could harm our business, financial condition and prospects. Our success also depends on our ability to continue to attract, retain and motivate highly skilled junior, mid-level and senior managers, as well as junior, mid-level and senior scientific and medical personnel.
We may not be able to attract or retain qualified management and scientific personnel in the future due to the intense competition for a limited number of qualified personnel among biopharmaceutical, biotechnology, pharmaceutical and other businesses. Many of the other pharmaceutical companies that we compete against for qualified personnel have greater financial and other resources, different risk profiles and a longer history in the industry than we do. They also may provide more diverse opportunities and better chances for career advancement. Some of these characteristics may be more appealing to high quality candidates than what we have to offer. Therefore, we might not be able to attract or retain these key personnel on conditions that are economically acceptable. If we are unable to continue to attract and retain high quality personnel, the rate and success at which we can develop and commercialize products or product candidates could be adversely affected.
Furthermore, we will need to recruit new managers and qualified scientific, regulatory and financial personnel to develop our business as we expand into the fields that will require additional skills and expertise, including oncology. Our inability to attract and retain these key personnel could prevent us from achieving our objectives and implementing our business strategy, which could have a material adverse effect on our business and prospects.
Risks from the improper conduct of employees, agents, contractors, CROs, consultants, vendors, or collaboration partners could adversely affect our reputation and our business, prospects, operating results, and financial condition.
We cannot ensure that our compliance controls, policies and procedures will in every instance protect us from acts committed by our employees, agents, contractors, CROs, consultants, vendors, or collaboration partners that would violate the laws or regulations of the jurisdictions in which we operate, including, without limitation, healthcare, employment, securities, manufacturing standards, data manipulation, scientific fraud, foreign corrupt practices, environmental, competition, and patient privacy and other privacy and data protection laws and regulations. Such improper actions could subject us to civil or criminal investigations and monetary and injunctive penalties, and could adversely impact our ability to conduct business, operating results and reputation.
In particular, our business activities may be subject to the Foreign Corrupt Practices Act, or FCPA, and similar anti-bribery or anti-corruption laws, regulations or rules of other countries in which we operate, including the U.K. Bribery Act. The FCPA generally prohibits offering, promising, giving, or authorizing others to give, anything of value, either directly or indirectly, to a non-U.S. government official in order to influence official action, or otherwise obtain or retain business. The FCPA also requires public companies to make and keep books and records that accurately and fairly reflect the transactions of the corporation and to devise, and maintain an adequate system of internal accounting controls. Our business is heavily regulated and therefore involves significant interaction with public officials, including officials of non- U.S. governments. Additionally, in many other countries, the health care providers who prescribe pharmaceuticals are employed by their government, and the purchasers of pharmaceuticals are governmental entities; therefore, our dealings with these prescribers and purchasers are subject to regulation under the FCPA. Recently the SEC and U.S. Department of Justice have increased their FCPA enforcement activities with respect to pharmaceutical companies. There is no certainty that all of our employees, agents, contractors, CROs, consultants, vendors, or collaboration partners, or those of our affiliates, will comply with all applicable laws and regulations, particularly given the high level of complexity of these laws and regulations. It is not always possible to identify and deter misconduct by employees and other third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses, or in protecting us from governmental investigations or other actions or
35
lawsuits stemming from a failure to comply with such laws or regulations. Additionally, we are subject to the risk that a person could allege such misconduct, even if none occurred. Violations of these laws and regulations could result in fines, criminal sanctions against us, our officers, or our employees, the closing down of our facilities, requirements to obtain export licenses, cessation of business activities in sanctioned countries, implementation of compliance programs, and prohibitions on the conduct of our business. Any such violations could include prohibitions on our ability to offer our products in one or more countries and could materially damage our reputation, our brand, our international expansion efforts, our ability to attract and retain employees, and our business, prospects, operating results, and financial condition.
We could be subject to liabilities under human rights, corruption, environmental, sustainability, health and safety laws or regulations, or fines, penalties or other sanctions, if we fail to comply with such laws or regulations or otherwise incur costs that could have a material adverse effect on the success of our business.
We are subject to numerous human rights, corruption, environmental, sustainability, health and safety laws, regulations, and permitting requirements, including those governing laboratory procedures, decontamination activities and the handling, transportation, use, remediation, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous materials, including chemicals, radioactive isotopes and biological materials and produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination, injury or illness from these materials or wastes either at our sites or at third- party disposal sites. In the event of such contamination, injury or illness, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs and reputational loss associated with civil or criminal fines and penalties. Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to illness or injuries to our employees resulting from the use of hazardous materials or other work-related illnesses or injuries, this insurance may not provide adequate coverage against potential liabilities.
In addition, we may incur substantial costs in order to comply with current or future environmental, sustainability, health and safety laws, regulations or permitting requirements, in which case our research, development or other efforts may be interrupted or delayed, and our financial condition and results of operation may be materially adversely affected. As of date of this annual report, we will be subject to the EU’s Corporate Sustainability Reporting Directive (CSRD) and therefore required, starting next financial year, to report in our annual report on a broad range of sustainability-related impacts, risks and opportunities on the basis of the European Sustainability Reporting Standards which require, in particular, disclosures on environmental protection, social responsibility and treatment of employees, respect for human rights, anti-corruption, bribery and diversity. In connection with these reporting obligations, we will be required to formulate long-term ESG targets, policy and strategic plans, and to conduct due diligence for our own operations and supply chain. As the CSRD requires reporting based on a double materiality assessment – we will need to be in a position to make both (i) an inside-out assessment of impact materiality (meaning a consideration of the impact of our corporate activity on sustainability matters from the perspective of citizens, consumers, employees, etc.) and (ii) an outside-in assessment of financial materiality (meaning a consideration of sustainability matters which, from the investor perspective, are material to our development, performance and financial position).These current or future laws, regulations and permitting requirements may impair our research, development or production efforts. Failure to comply with these laws, regulations and permitting requirements also may result in substantial fines, penalties or other sanctions. As an example, climate change and more specifically the related current and future regulatory requirements, as well as the accelerated transition to a low carbon economy globally, might adversely impact Galapagos’ compliance status and value chain, if not addressed adequately.
36
Risks related to our business operations and growth
We are dependent on the transfer of the Jyseleca® business, including the European and UK Marketing Authorizations, the commercial, medical affairs and development activities for Jyseleca® and approximately 400 Galapagos positions in 14 European countries, to Alfasigma.
On 30 October 2023, we announced the signing of a letter of intent contemplating a transfer of the Jyseleca® business to Alfasigma, including the European and UK marketing authorizations, the commercial, medical affairs and development activities for Jyseleca® and approximately 400 positions in 14 European countries. The completion of the intended transaction is subject to the execution of a definitive agreement and customary conditions, including regulatory approvals and consultations with works councils.
On 30 December 2023, we signed definitive agreements with Alfasigma, and on 31 January 2024, we completed the transfer of the commercial, medical affairs and development activities for Jyseleca® and approximately 400 positions in 14 European countries. Galapagos will receive a €50 million upfront payment, potential sales-based milestone payments totaling €120 million and mid-single to mid-double-digit royalties on European sales. Galapagos will contribute up to €40 million to Alfasigma by June 2025 for Jyseleca® related development activities.
Until the completion of the transfers of the European and UK marketing authorizations for filgotinib to Alfasigma, we will be subject to ongoing obligations and continued regulatory review, which may result in significant additional expenses. Additionally, filgotinib could be subject to labeling and other restrictions and market withdrawal and we may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with filgotinib. Additionally, if product liability lawsuits related to filgotinib are brought against us, we may incur substantial liabilities and may be required to limit the related commercialization.
If Alfasigma is not successful in commercializing filgotinib in Europe, we may not receive the potential milestones and royalty payments pursuant to the definitive agreement with Alfasigma.
If we fail to manage our growth effectively, our ability to develop and commercialize products could suffer.
We expect that if our drug discovery efforts continue to generate product candidates, our clinical product candidates continue to progress in development, and we continue to build our development, and medical organizations, we will require significant additional investment in personnel, management and resources. Our ability to achieve our research and development objectives depends on our ability to respond effectively to these demands to expand our internal organization, systems, controls and facilities to accommodate additional anticipated growth and upon our management developing and implementing strategies for us to realize these objectives. If we are unable to manage our growth effectively, our business could be harmed and our ability to execute our business strategy could suffer.
As a result of our limited financial, manufacturing and management recourses, we may forgo or delay pursuit of opportunities with potential product candidates that later prove to have greater market potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities.
We must maintain effective internal control over financial reporting, and if we are unable to do so, the accuracy and timeliness of our financial reporting may be adversely affected, which could have a material adverse effect on our business, investor confidence and market price.
We must maintain effective internal control over financial reporting in order to accurately and timely report our results of operations and financial condition. We often use estimates and assumptions concerning the future. We make reference to section “Critical accounting judgments and key sources of estimation uncertainty” for more information. In addition, because we are a U.S. public company, the Sarbanes- Oxley Act of 2002, or the Sarbanes-Oxley Act, requires, among other things, that we assess the effectiveness of our disclosure controls and procedures annually, and the effectiveness of our internal control over financial reporting at the end of each fiscal year.
37
Section 404 of the Sarbanes-Oxley Act, or Section 404, requires us to perform system and process evaluation and testing of our internal control over financial reporting to allow management to report on, and our independent registered public accounting firm to attest to, the effectiveness of our internal control over financial reporting. In addition, any testing by us conducted in connection with Section 404, or any subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weakness, or that may require prospective or retroactive changes to our financial statements or identify other areas for further attention or improvement. The rules governing the standards that must be met for our management to assess our internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act are complex and require significant documentation, testing and possible remediation. These stringent standards require that our Audit Committee be advised and regularly updated on management’s review of internal control over financial reporting. Our compliance with applicable provisions of Section 404 will require that we incur substantial accounting expense and expend significant management time on compliance-related issues as we implement additional corporate governance practices and comply with reporting requirements. Moreover, if we are not able to comply with the requirements of Section 404 applicable to us in a timely manner, or if we or our independent registered public accounting firm identifies deficiencies in our internal control over financial reporting that are deemed to be material weaknesses, the market price of the ADSs or our ordinary shares could decline and we could be subject to sanctions or investigations by the SEC or other regulatory authorities, which would require additional financial and management resources. Furthermore, investor perceptions of our company may suffer if deficiencies are found, and this could cause a decline in the market price of the ADSs or our ordinary shares. Irrespective of compliance with Section 404, any failure of our internal control over financial reporting could have a material adverse effect on our stated operating results and harm our reputation. If we are unable to implement these requirements effectively or efficiently, it could harm our operations, financial reporting or financial results, and could result in an adverse opinion on our internal control over financial reporting from our independent registered public accounting firm.
Our, our third party partners’ or vendors’, information technology systems and networks could face serious disruptions or suffer security breaches that could adversely affect our business.
To meet business objectives, we rely on both internal information technology (IT) systems and networks, and those of third parties and their vendors, to process and store confidential and sensitive data, including confidential research, business plans, financial information, intellectual property, patient data, customer data and personal data that may be subject to legal protection. The extensive information security and cybersecurity threats, which affect companies globally, pose a risk to the security and availability of these IT systems and networks, and the confidentiality, integrity, and availability of confidential and sensitive data.
These threats may include, but are not limited to, social-engineering attacks (including through phishing attacks), business email compromise, online and offline fraud, malicious code (such as viruses and worms), malware (including as a result of advanced persistent threat intrusions), denial-of-service attacks, access attacks (such as credential stuffing), ransomware attacks, and supply-chain attacks. These types of incidents continue to be prevalent and pervasive across industries, including in our industry, and such attacks on our systems have occurred in the past and are expected to occur in the future. In addition, we expect the amount and sophistication of the perpetrators of these attacks to continue to expand. Our systems and networks, and those of our third parties and vendors, may also be subject to software bugs, server malfunctions, software or hardware failures, loss of data or other information technology assets, adware, or telecommunications failures, earthquakes, fires, floods, and other similar threats.
We continuously assess these threats and make investments to increase internal protection, detection, and response capabilities, as well as to increase our third party providers’ capabilities and controls to address this risk.
However, because of the frequently changing attack techniques, along with the increased volume and sophistication of the attacks, there is the potential risk for us to be adversely impacted. Although Galapagos has invested time and resources in the protection of its information technology and other internal infrastructure systems, we and our vendors, like other companies in the industry, have experience attacks from time to time, and we and our vendors may experience other such attacks in the future.
38
The impact of security breaches and significant disruption in the availability of our information technology and networks could result in reputational, competitive, operational or other business harm, financial costs, litigation (including class action claims), regulatory action (for example, investigations, fines, penalties, audits and inspections), as well as interruptions in our collaborations with our partners, and delays in our research, development work, regulatory approval efforts and other work.
If product liability lawsuits are brought against us, we may incur substantial liabilities and may be required to limit commercialization of our approved product and any future approved products.
We face an inherent risk of product liability as a result of the clinical testing of our product candidates and an even greater risk in connection with our commercialization of our current and future drugs (if approved). For example (without any limitation), we may be sued if any product we develop allegedly causes injury or is found to be otherwise unsuitable during product testing, manufacturing, marketing, commercialization, use, or sale. Any such product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the product, negligence, strict liability and a breach of warranties. Physicians and patients may not comply with any warnings that identify known potential adverse effects and patients who should not use our products. Any claims against us, regardless of their merit, could be difficult and costly to defend, and could materially adversely affect the market for our products and product candidates, or any prospects for commercialization or our products and product candidates. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to stop development or, if approved, limit commercialization of our products and product candidates. Even successful defense would require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | delay or termination of clinical trials; |
| ● | injury to our reputation; |
| ● | withdrawal of clinical trial participants or difficulties in recruiting new trial participants; |
| ● | initiation of investigations by regulators; |
| ● | costs to defend or settle the related litigation; |
| ● | a diversion of management’s time and our resources; |
| ● | substantial monetary awards to trial participants or patients; |
| ● | decreased demand for our approved product, any future products, or our product candidates; |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; |
| ● | loss of revenues from product sales; and |
| ● | the inability to commercialize our approved product or any of our product candidates, if approved. |
Our inability to obtain and retain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the development or commercialization of our products and product candidates. We currently carry clinical trial liability insurance and product liability insurance at levels which we believe are appropriate for our clinical trials and our commercialization activities. Although we maintain such insurance, any claim that may be brought against us could result in a court judgment or settlement in an amount that is not covered, in whole or in part, by our insurance or that is in excess of the limits of our insurance coverage. Our insurance policies also have various exclusions, and we may be subject to a product liability claim for which we have no coverage. We will have to pay any amounts awarded by a court or negotiated in a settlement that exceed our coverage limitations or that are not covered by our insurance, and we may not have, or be able to obtain, sufficient capital or other assets to pay such amounts and our business operations could be impaired. We may not be able to maintain insurance coverage at a reasonable cost or to obtain insurance coverage that will be adequate to satisfy any liability that may arise.
39
Our relationships with customers and third-party payers may be subject, directly or indirectly, to applicable anti-kickback laws, fraud and abuse laws, false claims laws, health information privacy and security laws and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, contractual damages, reputational harm, administrative burdens and diminished profits and future earnings.
We are subject to additional healthcare statutory and regulatory requirements and enforcement by the federal government and the states and foreign governments in which we conduct our business. Healthcare providers, physicians and third-party payers play a primary role in the recommendation and prescription of any of our approved products and product candidates for which we obtain marketing approval. Our current and future arrangements with third -party payers and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships with third parties through which we market, sell and distribute any products for which we obtain marketing approval. In addition, we may be subject to privacy, data protection and security regulation of the EU, the United States and other jurisdictions in which we conduct our business. See section entitled “Information on the Company – Other healthcare laws and compliance requirements.”
The scope and enforcement of each of laws is uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Ensuring that our internal operations and future business arrangements with third parties comply with applicable healthcare laws and regulations will involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations, agency guidance or case law involving applicable fraud and abuse or other healthcare laws and regulations.
If our operations are found to be in violation of any of the laws described above or any other governmental laws and regulations that may apply to us, we may be subject to significant penalties, including administrative, civil and criminal penalties, damages, (including, but not limited to, reputational harm), fines, disgorgement, the exclusion from participation in federal and state healthcare programs, individual imprisonment, reputational harm and the curtailment or restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. Further, defending against any such actions can be costly and time consuming, and may require significant financial and personnel resources. Therefore, even if we are successful in defending against any such actions that may be brought against us, our business may be impaired. If any of the physicians or other providers or entities with whom we expect to do business are found to not be in compliance with applicable laws, they may be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs and imprisonment. If any legislative and/or regulatory initiatives and changes would lead to increased restriction on the marketing of our approved products and product candidates, or lead to limiting the funds available for healthcare in any relevant jurisdiction which may reduce reimbursement levels and is likely to affect the prices we may set, we would be negatively impacted in our ability to successfully and profitably market our approved products and product candidates. If any of the above occur, our ability to operate our business and our results of operations could be adversely affected.
We may fail to comply with evolving privacy and data protection laws and requirements in effect in the European Union and other jurisdictions.
In the European Union, or “EU”, we may face particular privacy, data security, privacy and data protection risks in connection with requirements of the Regulation (EU) 2016/679 of the European Parliament and of the Council of April 27, 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, and repealing Directive 95/46/EC, or the “GDPR”, and implementing laws and regulations. The GDPR applies inter alia to the processing of personal data in the context of the activities of an establishment of a controller in the EU. The GDPR has enhanced data protection obligations for controllers of
40
personal data, including, for example, expanded disclosures about how personal data is to be used, limitations on retention of data, enhanced requirements for securing personal data, mandatory data breach notification requirements, restrictions on transferring such personal data outside the European Economic Area, or the “EEA”, including to the United States, appointing data protection officers, conducting data protection impact assessments, and has created onerous liabilities on controllers or processors. The GDPR increases substantially the penalties to which we could be subject in the event of any non-compliance, including possible fines of up to € 20,000,000 or up to 4% of our total worldwide annual turnover of the preceding year for the most serious infringements. The GDPR may increase our responsibility and liability in relation to personal data that we process where such processing is subject to the GDPR, and we may be required to put in place additional mechanisms to ensure compliance with the GDPR. Compliance with the GDPR is a rigorous and time-intensive process that may increase our cost of doing business or require us to change our business practices, and despite those efforts, there is a risk that we may be subject to fines and penalties, litigation and reputational harm in connection with our activities. A similar legislative framework, including similar obligations and penalties, applies in Switzerland and the UK, where similar efforts are needed.
If we are investigated by a data protection authority, we may face fines and other penalties. Any such investigation or charges by data protection authorities could have a negative effect on our existing business and our ability to attract and retain new clients or pharmaceutical partners. We may also experience hesitancy, reluctance, or refusal by European or multi-national clients or pharmaceutical partners to continue to use our products due to the potential risk exposure as a result of the current (and, in particular, future) data protection obligations imposed on them by certain data protection authorities in interpretation of current law, including the GDPR. Such clients or pharmaceutical partners may also view any alternative approaches to compliance as being too costly, too burdensome, too legally uncertain, or otherwise objectionable and therefore decide not to do business with us. Any of the foregoing could materially harm our business, prospects, financial condition and results of operations.
We are also subject to evolving European privacy laws on electronic marketing and cookies. The EU is in the process of replacing the e- Privacy Directive (2002/58/EC) with a new set of rules taking the form of a regulation. While the e-Privacy Regulation was originally intended to be adopted on May 25, 2018 (alongside the GDPR), it is still going through the European legislative process. In February 2021, the EU Member States reached agreement on the European Council’s negotiating mandate for the European Parliament. While the final draft of the e- Privacy Regulation is closer to being finalized, it is unlikely that the new ePrivacy Regulation will come into effect before 2023. Preparing for and complying with the ePrivacy Regulation (if and when it becomes effective) has required, and will continue to require, us to incur substantial operational costs and may require us to change our business practices.
Despite our efforts to bring practices into compliance with the GDPR and before the effective date of the ePrivacy Regulation, we may not be successful either due to internal or external factors such as resource allocation limitations. Non-compliance could result in proceedings against us by governmental entities, customers, data subjects, consumer associations or others, as well as in penalties issued by data protection authorities as is also stated above.
We may also fail to comply with other privacy and data protection laws and regulations in other jurisdictions where we are active and/ or operating, including but not limited to the United States.
Although Galapagos has invested time and resources in the protection of its personal data and information technology and monitors its systems on an ongoing basis, some immaterial incidents have occurred in respect of which Galapagos has taken appropriate measures. To date, no material risk has been identified, and Galapagos’ business or operations have not been materially impacted by such incidents.
Business interruptions could delay us in the process of developing our product candidates.
Loss of our laboratory, warehouse or other real estate facilities through fire or other causes could have an adverse effect on our ability to continue to conduct our business. We currently have insurance coverage to compensate us for such business interruptions; however, such coverage may prove insufficient to compensate us fully for the damage to our business resulting from any significant property or casualty loss to our facilities.
41
We may undertake strategic acquisitions in the future and any difficulties from integrating such acquisitions could adversely affect our share price, and results of operations.
We may acquire companies, businesses and products that complement or augment our existing business. Because our programs may require the use of proprietary rights held by third parties, the growth of our business will likely depend in part on our ability to acquire, in-license or use these proprietary rights. We may be unable to acquire or in- license any compositions, processes or other third-party intellectual property rights from third parties that we identify as necessary for our product candidates. The acquisition and licensing of third-party intellectual property rights is a competitive area, and other companies having a competitive advantage over us due to their size, cash resources or otherwise, may pursue strategies to in-license or acquire third-party intellectual property rights that we may consider attractive. We may not be able to integrate any acquired companies, business or products successfully, or operate any acquired company, business or product profitably. Integrating any newly acquired companies, business or products could be expensive and time-consuming. Integration efforts often take a significant amount of time, place a significant strain on managerial, operational and financial resources, could result in loss of key personnel and could prove to be more difficult or expensive than we predict. The diversion of our management’s attention and any delay or difficulties encountered in connection with any future acquisitions or in-licensing we may consummate could result in the disruption of our ongoing business or inconsistencies in standards and controls that could negatively affect our ability to maintain third-party relationships. Moreover, we may need to raise additional funds through public or private debt or equity financing, or issue additional shares, to acquire any businesses or products, which may result in dilution for shareholders or the incurrence of indebtedness.
As part of our efforts to acquire companies, business or product candidates, or to enter into other significant transactions, we conduct business, legal and financial due diligence with the goal of identifying and evaluating material risks involved in the transaction. Despite our efforts, we ultimately may be unsuccessful in ascertaining or evaluating all such risks and, as a result, might not realize the intended advantages of the transaction. If we fail to realize the expected benefits from acquisitions we may consummate in the future or have consummated in the past, whether as a result of unidentified risks or liabilities, integration difficulties, regulatory setbacks, litigation with current or former employees and other events, our business, results of operations and financial condition could be adversely affected. If we acquire product candidates, we will also need to make certain assumptions about, among other things, development costs, the likelihood of receiving regulatory approval, and the market for such product candidates. Our assumptions may prove to be incorrect, which could cause us to fail to realize the anticipated benefits of these transactions.
In addition, we will likely experience significant charges to earnings in connection with our efforts, if any, to consummate acquisitions. For transactions that are ultimately not consummated, these charges may include fees and expenses for investment bankers, attorneys, accountants and other advisors in connection with our efforts. Even if our efforts are successful, we may incur, as part of a transaction, substantial charges for closure costs associated with elimination of duplicate operations and facilities and acquired in-process research and development charges. In either case, the incurrence of these charges could adversely affect our results of operations for particular periods.
Actions of activist shareholders could cause us to incur substantial costs, divert our management’s and our directors’ attention and resources, and have an adverse effect on our business and trading price.
From time to time, we may be subject to proposals by shareholders urging us to take certain corporate actions or to nominate certain individuals to our Board of Directors. If activist shareholder activities by shareholders ensue, our business could be adversely affected, as responding to actions by activist shareholders can be costly and time-consuming, disrupt our operations and divert the attention of management and Directors. For example (without any limitation), we may be required to retain the services of various professionals to advise us on activist shareholders’ matters, including legal, financial, and communications advisors, the costs of which may negatively impact our future financial results. In addition, perceived uncertainties as to our future direction, strategy or leadership created as a consequence of activist shareholders’ initiatives may result in the loss of potential business opportunities, harm our ability to attract new investors, customers, and employees, and cause the price of our ADSs or ordinary shares to experience periods of volatility or stagnation.
42
Our international operations subject us to various risks, and our failure to manage these risks could adversely affect our results of operations.
We could face significant operational risks as a result of doing business internationally, which could have a material adverse effect on our business, financial condition and results of operations, such as (without any limitation):
| ● | fluctuations in foreign currency exchange rates; |
| ● | potentially adverse and/or unexpected tax consequences, including penalties due to the failure of tax planning or due to the challenge by tax authorities on the basis of transfer pricing and liabilities imposed from inconsistent enforcement; |
| ● | potential changes to the accounting standards, which may influence our financial situation and results; |
| ● | becoming subject to the different, complex and changing laws, regulations and court systems of multiple jurisdictions and compliance with a wide variety of foreign laws, treaties and regulations; |
| ● | reduced protection of, or significant difficulties in enforcing, intellectual property rights in certain countries; |
| ● | difficulties in attracting and retaining qualified personnel; |
| ● | restrictions imposed by local labor practices and laws on our business and operations, including unilateral cancellation or modification of contracts; |
| ● | rapid changes in global government, economic and political policies and conditions, political or civil unrest or instability, terrorism or epidemics and other similar outbreaks or events, and potential failure in confidence of our suppliers or customers due to such changes or events; and |
| ● | tariffs, trade protection measures, import or export licensing requirements, trade embargoes, economic sanctions, and other trade barriers. |
Unforeseen or catastrophic events, including extreme weather events and other natural disasters, man-made disasters or the emergence of epidemics, could cause a disruption in our operations or other consequences that could have a material adverse effect on our financial condition and results of operations.
The occurrence of unforeseen or catastrophic events, including extreme weather events and other acts of god or natural disasters, man-made disasters, electricity or telecommunication interruption, geopolitical and other economic or political conditions or events (such as the armed conflict between Russia and Ukraine or the conflict in Israel and Gaza) or the emergence of epidemics or diseases, depending on their scale, may cause different degrees of damage to the national and local economies and could cause a disruption in our operations and have a material adverse effect on our financial condition and results of operations. Such events may also cause the market price and demand for our ADSs or ordinary shares to fluctuate substantially. Man-made disasters, epidemics or disease, and other events connected with the regions in which we operate could have similar effects. If a natural or man-made disaster, electricity or telecommunication interruption or other event occurred that prevented us from using all or a significant portion of our facilities, that damaged critical infrastructure, such as clinical trial sites or the manufacturing facilities of our third-party contract manufacturers, or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. The disaster recovery and business continuity plans we have in place may prove inadequate in the event of a serious disaster or similar event. We may incur substantial expenses as a result of the limited nature of our disaster recovery and business continuity plans, which, could have a material adverse effect on our business.
The increasing use of social media platforms presents risks and challenges.
We and our employees are increasingly utilizing social media tools as a means of communication both internally and externally. Despite our efforts to monitor evolving social media communication guidelines and comply with applicable rules, there is risk that the use of social media by us or our employees to communicate about our products and product candidates, operations, or business may cause us to be found in violation of applicable legal or contractual requirements. In addition, our employees may knowingly or inadvertently make use of social media in ways that may not comply with our social media policy or other legal or contractual requirements, which
43
may give rise to liability, lead to the loss of trade secrets or other intellectual property, or result in public exposure of personal information of our employees, clinical trial patients, collaboration partners, and others, and which could have an adverse effect on our business, financial conditions and results of operations. Furthermore, negative posts or comments about us or our products in social media could seriously damage our reputation, brand image and goodwill.
Risks related to tax and other financial matters
If we are unable to use tax loss carryforwards to reduce future taxable income or benefit from favorable tax legislation, our business, results of operations and financial condition may be adversely affected.
As of December 31, 2023, we had cumulative carry forward tax losses of € 776.8 million related to entities in Belgium, and €21.9 million related to the other entities of our group. These are available to carry forward and offset against possible future taxable income for an indefinite period in Belgium, but €2.2 million of these tax loss carryforwards in the United States will expire between 2028 and 2034. If we are unable to use tax loss carryforwards to reduce possible future taxable income or in case of changes in tax regulations affecting the use of tax loss carryforwards, our business, results of operations and financial condition may be adversely affected.
As a company active in research and development in Belgium and France, we have benefited from certain research and development incentives including, for example, but not limited to, the Belgian research and development tax credit and the French research tax credit (crédit d’impôt recherche). These tax credits can be offset against Belgian and French corporate income tax due, respectively. The excess portion may be refunded as from the end of a five-year fiscal period for the Belgian research and development incentive (this period of five years is reduced to four years for tax credits carried forward as from the accounting year starting 1 January 2024), and at the end of a three-year fiscal period for the French research and development incentive. As from the accounting year starting 1 January 2024, it is optional for the company in Belgium to either apply the tax credit to any corporate income tax due or to carry-over the tax credit to a subsequent taxable period (up to the four-year refund period). The research and development incentives are both calculated based on the amount of eligible research and development expenditure. The Belgian tax credit represented €20.9 million for the year ended December 31, 2021, and €17.3 million for the year ended December 31, 2022, and €26.3 million for the year ended December 31, 2023. The French tax credit amounted to €12.4 million for the year ended December 31, 2021, and €11.4 million for the year ended December 31, 2022, and €6.5 million for the year ended December 31, 2023. The Belgian and/or French tax authorities may audit each research and development program in respect of which a tax credit has been claimed and assess whether it qualifies for the tax credit regime. The tax authorities may challenge our eligibility for, or our calculation of, certain tax reductions and/or deductions in respect of our research and development activities and, should the Belgian and/or French tax authorities be successful, we may be liable for additional corporate income tax, and penalties and interest related thereto, which could have a significant impact on our results of operations and future cash flows. Furthermore, if the Belgian or French governments decide to eliminate, or reduce the scope or the rate of, the research and development incentive benefits, either of which they could decide to do at any time, our results of operations could be adversely affected.
As a company active in research and development in Belgium, we also expect to benefit from the innovation income deduction, or IID, in Belgium. The innovation income deduction regime allows net profits attributable to revenue from among others patented products (or products for which the patent application is pending) to be taxed at a lower effective tax rate than other revenues. The effective tax rate can thus be reduced down to 3.75%. At the end of 2023, we had €390.3 million of carry-forward IID in Belgium.
On December 14, 2022, the Council of the EU formally adopted the Council Directive on ensuring a global minimum level of taxation for multinational groups in the Union, laying down rules for ensuring a minimum level of effective corporate taxation of large multinational groups and large-scale purely domestic groups operating in the
44
Single Market. The Directive is largely aligned with the OECD Model Rules agreed by the Inclusive Framework and published on December 20, 2021 (the so -called “Pillar II”). The aim of the directive is to realize a 15% global minimum effective tax rate at country-per-country level. At this stage, no carve-out for patent box regimes or R&D incentives is included in the directive. This directive could have an impact on the company’s future effective tax rate and/or tax attributes. Member States will now have to transpose said directive into their national laws before December 31, 2023.
The Belgian Pillar II rules will enter into force in two steps: the income inclusion rule (or “IIR”) and the Qualified Domestic Minimum Top-up Tax (or “QDMTT”) will enter into force for taxable periods starting as of December 31, 2023, whereas the rules regarding the Undertaxed Profit Rule (or “UTPR”) will enter into force for taxable periods starting as of December 31, 2024.
Our inability to qualify for the abovementioned advantageous tax regimes, as well as the introduction of the minimum taxable base and any other future adverse changes of Belgian tax legislation, may adversely affect our business, results of operations, and financial condition.
Our shareholders residing in countries other than Belgium may be subject to double withholding taxation with respect to dividends or other distributions made by us.
Any dividends or other distributions we make to shareholders will, in principle, be subject to withholding tax in Belgium at a rate of 30%, except for shareholders which qualify for an exemption of withholding tax such as, among others, qualifying pension funds or a company qualifying as a parent company in the sense of the Council Directive (90/435/EEC) of July 23, 1990, or the Parent-Subsidiary Directive, as amended, or that qualify for a lower withholding tax rate or an exemption by virtue of a tax treaty. Various conditions may apply and shareholders residing in countries other than Belgium are advised to consult their advisers regarding the tax consequences of dividends or other distributions made by us. Our shareholders residing in countries other than Belgium may not be able to credit the amount of such withholding tax to any tax due on such dividends or other distributions in any other country than Belgium. As a result, such shareholders may be subject to double taxation in respect of such dividends or other distributions. Belgium and the United States have concluded a double tax treaty concerning the avoidance of double taxation, or the U.S.-Belgium Tax Treaty. The U.S.-Belgium Tax Treaty reduces the applicability of Belgian withholding tax to 15%, 5% or 0% for U.S. taxpayers that are the beneficial owner of the dividend income concerned, provided that the U.S. taxpayer meets the limitation on benefits conditions imposed by the U.S.-Belgium Tax Treaty. The Belgian withholding tax is generally reduced to 15% under the U.S.-Belgium Tax Treaty. The 5% withholding tax applies in cases where the U.S. shareholder, beneficial owner of the income, is a company which holds at least 10% of the voting shares in the company. A 0% Belgian withholding tax applies when the shareholder, beneficial owner of the income, is a U.S. company which has held directly at least 10% of the capital in the company for at least 12 months on the date the dividend is declared, or is, subject to certain conditions, a U.S. pension fund. The U.S. shareholders are encouraged to consult their own tax advisers to determine whether they can invoke the benefits and meet the limitation on benefits conditions as imposed by the U.S.-Belgium Tax Treaty.
Changes in tax law could adversely affect our business and financial condition.
The rules dealing with U.S. federal, state, and local and non-U.S. taxation are constantly under review by persons involved in the legislative process, the Internal Revenue Service, the U.S. Treasury Department and other taxing authorities. Changes to tax laws or tax rulings, or changes in interpretations of existing laws (which changes may have retroactive application), could adversely affect us or holders of our ADSs. These changes could subject us to additional income-based taxes and non-income taxes (such as payroll, sales, use, value-added, digital tax, net worth, property, and goods and services taxes), which in turn could materially affect our financial position and results of operations. Additionally, new, changed, modified, or newly interpreted or applied tax laws could increase our customers’ and our compliance, operating and other costs, as well as the costs of our products. In recent years, many such changes have been made, and changes are likely to continue to occur in the future. As we expand the
45
scale of our business activities, any changes in the U.S. and non-U.S. taxation of such activities may increase our effective tax rate and harm our business, financial condition, and results of operations.
We believe that we were a passive foreign investment company, or PFIC, for U.S. federal income tax purposes for the 2023 taxable year, but this conclusion is a factual determination that is made annually and thus may be subject to change. Because we believe we were a PFIC for our 2023 taxable year, this could result in adverse U.S. tax consequences to certain U.S. holders.
Generally, if, for any taxable year, at least 75% of our gross income is passive income, or at least 50% of the value of our assets is attributable to assets that produce passive income or are held for the production of passive income, including cash, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. For purposes of these tests, passive income includes dividends, interest, and gains from the sale or exchange of investment property and rents and royalties other than rents and royalties which are received from unrelated parties in connection with the active conduct of a trade or business. Our status as a PFIC depends on the composition of our income and the composition and value of our assets (for which purpose the total value of our assets may be determined in part by reference to the market value of the ADSs and our ordinary shares, which are subject to change) from time to time. Because we believe we were a PFIC for the 2023 taxable year, certain U.S. holders of the ADSs may suffer adverse tax consequences, including, but not limited to, having gains realized on the sale of the ADSs treated as ordinary income, rather than capital gain, losing the preferential rate applicable to dividends received on the ADSs by individuals who are U.S. holders, and having interest charges apply to distributions by us and the proceeds of sales of the ADSs. See “Item 10.E.—Taxation—Certain Material U.S. Federal Income Tax Considerations to U.S. Holders—Passive Foreign Investment Company Considerations.”
Based upon the value of our assets, including any goodwill, and the composition of our income and assets, we believe that we were a PFIC for our 2023 taxable year. However, our status as a PFIC is a fact-intensive determination made on an annual basis, and we cannot provide any assurances regarding our PFIC status for the current, prior or future taxable years. Because we were a PFIC for the 2022 taxable year, we will provide information necessary for our U.S. holders to make a “qualified electing fund,” or QEF, election with respect to us for the 2023 taxable year and expect to provide such information for any subsequent year if we believe we are a PFIC. We will provide such information on our website.
We believe that we were not a controlled foreign corporation, or CFC, for U.S. federal income tax purposes for the 2023 taxable year. If we were to qualify as a CFC, this could result in adverse U.S. federal income tax consequences to certain U.S. holders.
Each “Ten Percent Shareholder” (as defined below) in a non-U.S. corporation that is classified as a “controlled foreign corporation,” or a CFC, for U.S. federal income tax purposes generally is required to include in income for U.S. federal tax purposes such Ten Percent Shareholder’s pro rata share of the CFC’s “Subpart F income” and investment of earnings in U.S. property, even if the CFC has made no distributions to its shareholders. Subpart F income generally includes dividends, interest, rents and royalties, gains from the sale of securities, and income from certain transactions with related parties. For tax years beginning after December 31, 2017, each Ten Percent Shareholder of a CFC is also required to include in income such Ten Percent Shareholder’s share of “global intangible low-taxed income” with respect to such CFC. In addition, a Ten Percent Shareholder that realizes gain from the sale or exchange of shares in a CFC may be required to classify a portion of such gain as dividend income rather than capital gain. A non- U.S. corporation generally will be classified as a CFC for United States federal income tax purposes if Ten Percent Shareholders own, directly or indirectly, more than 50% of either the total combined voting power of all classes of stock of such corporation entitled to vote or of the total value of the stock of such corporation. A “Ten Percent Shareholder” is a United States person (as defined by the U.S. Internal Revenue Code of 1986, as amended (the “Code”)) who owns or is considered to own 10% or of either (1) the total combined voting power of all classes of stock entitled to vote of such corporation or (2) the total value of all classes of stock of such corporation. The determination of CFC status is complex and includes attribution rules, the application of
46
which is not entirely certain. In addition, recent changes pursuant to U.S. tax reform to the attribution rules relating to the determination of CFC status may make it difficult to determine our CFC status for any taxable year.
To our best knowledge, we do not believe that we were a CFC for the taxable year ended December 31, 2023. Furthermore, it is possible that our non-United States subsidiaries will be CFCs for the taxable year ended December 31, 2023 (or future taxable years) even if we are not a CFC for such taxable year(s). However, we cannot provide any assurances regarding our status or the status of our subsidiaries as a CFC for the 2023 taxable year or any future taxable years. U.S. holders should consult their own tax advisors with respect to the potential adverse U.S. tax consequences of becoming a Ten Percent Shareholder in a CFC. If we are classified as both a CFC and a PFIC, we generally will not be treated as a PFIC with respect to those U.S. holders that meet the definition of a Ten Percent Shareholder during the period in which we are a CFC.
We may be forced to repay the technological innovation grants if we fail to comply with our contractual obligations under the applicable grant agreements.
We have received several technological innovation grants to date, totaling €37.2 million as of December 31, 2023, from an agency of the Flemish government to support various research programs and technological innovation in Flanders. These grants carry clauses which require us to maintain a presence in the Flemish region for a number of years and invest according to pre-agreed budgets. If we fail to comply with our contractual obligations under the applicable technological innovation grant agreements, we could be forced to repay all or part of the grants received. Such repayment could adversely affect our ability to finance our research and development projects. In addition, we cannot ensure that we will then have the additional financial resources needed, the time or the ability to replace these financial resources with others.
We may be exposed to significant foreign exchange risk.
We hold portions of our cash and cash equivalents and current financial investments in currencies other than the euro, in particular, the U.S. dollar. We also incur portions of our expenses and derive revenues, in currencies other than the euro, in particular, the U.S. dollar. As a result, we are exposed to foreign currency exchange risk as our reporting currency is the euro. We currently do not engage in exchange rate hedging transactions to protect against uncertainty in future exchange rates between particular foreign currencies and the euro. Therefore, for example (without any limitation), an increase in the value of the euro against the U.S. dollar could be expected to have a negative impact on our revenue and earnings growth as U.S. dollar revenue and earnings, if any, would be translated into euros at a reduced value. We cannot predict the impact of foreign currency fluctuations, and foreign currency fluctuations in the future may adversely affect our business, financial condition, results of operations and cash flows.
The requirements of being a U.S. public company may strain our resources and divert management’s attention.
We are required to comply with various corporate governance and financial reporting requirements under the Sarbanes-Oxley Act of 2002, the Exchange Act, and the rules and regulations adopted by the SEC and the U.S. Public Corporation Accounting Oversight Board, or PCAOB, and other applicable securities rules and regulations imposing various requirements on non-U.S. public companies. Further, compliance with various regulatory reporting requires significant commitments of time from our management and our directors, which reduces the time available for the performance of their other responsibilities. Our failure to track and comply with the various rules may materially adversely affect our reputation, ability to obtain the necessary certifications to financial statements, lead to additional regulatory enforcement actions, and could adversely affect the value of the ADSs or our ordinary shares. However, these rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
47
Risks related to ownership of our ordinary shares and ADSs
The market price of the ordinary shares and ADSs could be subject to wide fluctuations.
The market price of the ordinary shares and ADSs could be subject to wide fluctuations in response to many risk factors listed in this section, and others beyond our control, including (without any limitation):
| ● | actual or anticipated fluctuations in our financial condition and operating results; |
| ● | actual or anticipated changes in our growth rate relative to our competitors; |
| ● | competition from existing products or new products that may emerge; |
| ● | announcements by us, our partners or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations, or capital commitments; |
| ● | failure to meet or exceed financial estimates and projections of the investment community or that we provide to the public; |
| ● | issuance of new or updated research or reports by securities or industry analysts; |
| ● | fluctuations in the valuation of companies perceived by investors to be comparable to us; share price and volume fluctuations attributable to inconsistent trading volume levels of our shares; |
| ● | additions or departures of key management or scientific personnel; |
| ● | disputes or other developments related to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies; |
| ● | changes to coverage policies or reimbursement levels by commercial third-party payers and government payers and any announcements relating to coverage policies or reimbursement levels; |
| ● | announcement or expectation of additional debt or equity financing efforts; |
| ● | public concern relating to the commercial value or safety of any of our products or product candidates; |
| ● | changes in government regulations; |
| ● | positive or negative results of testing and clinical trials by us, strategic partners or competitors; |
| ● | outcome of regulatory review of our product candidates; |
| ● | sales of the ordinary shares and ADSs by us, our insiders or our other shareholders; and |
| ● | general economic and market conditions. |
In addition, although the ordinary shares are listed on Euronext Brussels and Euronext Amsterdam, and the ADSs are listed on the Nasdaq Global Select Market stock exchange, we cannot assure that a trading market for those securities will be maintained.
These and other market and industry factors may cause the market price and demand for the ordinary shares and ADSs to fluctuate substantially, regardless of our actual operating performance, which may limit or prevent investors from readily selling their ordinary shares and ADSs and may otherwise negatively affect the liquidity of our capital shares. In addition, the stock market in general, and biotechnology and biopharmaceutical companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies.
We may be at an increased risk of securities class action litigation.
Historically, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant share price volatility in recent years. If we were to be sued, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
Share ownership is concentrated in the hands of our principal shareholders and management, which may have the effect of delaying or preventing a change of control of our company.
As of the date of this annual report on Form 20-F, our executive officers, Directors, current 5% or greater shareholders and their affiliated entities, including Gilead Sciences, Inc. and its affiliates, together beneficially own approximately 50.01% of our ordinary shares, including shares in the form of ADSs. This concentration of
48
ownership might have the effect of delaying or preventing a change of control of our company that other shareholders may view as beneficial, and might therefore negatively affect the market price of the ADSs.
Fluctuations in the exchange rate between the U.S. dollar and the euro may increase the risk of holding the ADSs.
Our ordinary shares currently trade on Euronext Brussels and Euronext Amsterdam in euros, while the ADSs trade on Nasdaq in U.S. dollars. Fluctuations in the exchange rate between the U.S. dollar and the euro may result in temporary differences between the value of the ADSs and the value of our ordinary shares, which may result in heavy trading by investors seeking to exploit such differences.
In addition, as a result of fluctuations in the exchange rate between the U.S. dollar and the euro, the U.S. dollar equivalent of the proceeds that a holder of the ADSs would receive upon the sale on Euronext Brussels or Euronext Amsterdam of any ordinary shares withdrawn from the depositary and the U.S. dollar equivalent of any cash dividends paid in euros on our shares represented by the ADSs could also decline.
If securities or industry analysts do not publish research or publish inaccurate research or unfavorable research about our business, the price of the ordinary shares and ADSs and trading volume could decline.
The trading market for the ordinary shares and ADSs depends in part on the research and reports that securities or industry analysts publish about us or our business. If no or few securities or industry analysts cover our company, the trading price for the ordinary shares and ADSs would be negatively impacted. If one or more of the analysts who covers us downgrades the ordinary shares and ADSs or publishes incorrect or unfavorable research about our business, the price of the ordinary shares and ADSs would likely decline. If one or more of these analysts ceases coverage of our company or fails to publish reports on us regularly, or downgrades the ordinary shares and ADSs, demand for the ordinary shares and ADSs could decrease, which could cause the price of the ordinary shares and ADSs or trading volume to decline.
We have no present intention to pay dividends on our ordinary shares in the foreseeable future and, consequently, your only opportunity to achieve a return on your investment during that time is if the price of the ordinary shares and ADSs appreciates.
We have no present intention to pay dividends in the foreseeable future. Even if future operations lead to significant levels of distributable profits, we currently intend that any and all earnings will be reinvested in our business. Any proposal by our Board of Directors to pay dividends will depend on many factors, including our financial condition (including losses carried-forward), results of operations, legal requirements, business prospects, cash requirements, new product development, and other factors. Furthermore, pursuant to Belgian law, the calculation of amounts available for distribution to shareholders, as dividends or otherwise, must be determined on the basis of our non-consolidated statutory accounts prepared in accordance with Belgian accounting rules and Belgian generally accepted accounting principles as used by us in the preparation of these accounts. In addition, in accordance with Belgian law and our Articles of Association, we must allocate each year an amount of at least 5% of our annual net profit under our non-consolidated statutory accounts to a legal reserve until such legal reserve equals 10% of our share capital. Therefore, we are unlikely to pay dividends or other distributions in the foreseeable future. If the price of the ordinary shares and ADSs declines before we pay dividends, you will incur a loss on your investment, without the likelihood that this loss will be offset in part or at all by potential future cash dividends. Accordingly, investors cannot rely on cash dividend income from ordinary shared and ADSs and any returns on an investment in the ordinary shares and ADSs will likely depend entirely upon any future appreciation in the price of the ordinary shares and ADSs.
We are a Belgian public limited liability company, and shareholders of our company may have different and in some cases more limited shareholder rights than shareholders of a U.S. listed corporation.
We are a public limited liability company incorporated under the laws of Belgium. Our corporate affairs are governed by Belgian corporate law and our Articles of Association. The rights provided to our shareholders under
49
Belgian corporate law and our articles of association may differ in certain respects from the rights that you would typically enjoy as a shareholder of a U.S. corporation under applicable U.S. federal and state laws.
Under Belgian corporate law, other than certain limited information that we must make public and except in certain limited circumstances, our shareholders may not ask for an inspection of our corporate records, while under Delaware corporate law any shareholder, irrespective of the size of its shareholdings, may do so. Shareholders of a Belgian corporation are also unable to initiate a derivative action, a remedy typically available to shareholders of U.S. companies, in order to enforce a right of our company, in case we fail to enforce such right ourselves, other than in certain cases of director liability under limited circumstances. In addition, a majority of our shareholders present or represented at our Shareholders’ Meeting may release a member of our Board of Directors from any claim of liability we may have, including if he or she has acted in bad faith or has breached his or her duty of loyalty, provided, in some cases, that the relevant acts were specifically mentioned in the convening notice to the Shareholders’ Meeting deliberating on the discharge. In contrast, most U.S. federal and state laws prohibit a company from releasing a director from liability altogether if he or she has acted in bad faith or has breached his or her duty of loyalty to the company. Finally, Belgian corporate law does not provide any form of appraisal rights in the case of a business combination. Please see the section of this annual report titled “Item 10.B.—Memorandum and Articles of Association.”
The responsibilities of members of our Board of Directors may be different from these in companies governed by U.S. laws. In the performance of its duties, our Board of Directors is required by Belgian law to consider the interests of our company, in all cases with due observation of the principles of reasonableness and fairness. It is possible that some of these parties will have interests that are different from, or in addition to, your interests as a shareholder.
As a result of these differences between Belgian corporate law and our Articles of Association, on the one hand, and the U.S. federal and state laws, on the other hand, in certain instances, you could receive less protection as an ADS holder of our company than you would as a shareholder of a listed U.S. company.
Takeover provisions in Belgian law may make a takeover difficult.
Public takeover bids on our ordinary shares and other voting securities, such as subscription rights or convertible bonds, if any, are subject to the Belgian Act of April 1, 2007, as amended, and to the supervision by the Belgian Financial Services and Markets Authority, or FSMA. Public takeover bids must be made for all of our voting securities, as well as for all other securities that entitle the holders thereof to the subscription to, the acquisition of or the conversion into voting securities. Prior to making a bid, a bidder must issue and disseminate a prospectus, which must be approved by the Belgian FSMA. The bidder must also obtain approval of the relevant competition authorities, where such approval is legally required for the acquisition of our company.
The Belgian Act of April 1, 2007, as amended, provides that a mandatory bid will be triggered if a person, as a result of its own acquisition or the acquisition by persons acting in concert with it or by persons acting on their account, directly or indirectly holds more than 30% of the voting securities in a company that has its registered office in Belgium and of which at least part of the voting securities are traded on a regulated market or on a multilateral trading facility designated by the Royal Decree of April 27, 2007, as amended, on public takeover bids. The mere fact of exceeding the relevant threshold through the acquisition of one or more shares will give rise to a mandatory bid, irrespective of whether or not the price paid in the relevant transaction exceeds the current market price.
There are several provisions of Belgian corporate law and certain other provisions of Belgian law, such as the obligation to disclose important shareholdings and merger control, that may apply to us and which may make an unfriendly tender offer, merger, change in management or other change in control, more difficult. These provisions could discourage potential takeover attempts that third parties may consider and thus deprive the shareholders of the opportunity to sell their ordinary shares at a premium (which is typically offered in the framework of a takeover bid). These provisions may also have the effect of depriving ADS holders of the opportunity to sell their ADSs at potential a premium.
50
Holders of the ADSs are not treated as shareholders of our company, do not have the same voting rights as the holders of our ordinary shares and may not receive voting materials in time to be able to exercise your right to vote.
Holders of the ADSs are not treated as shareholders of our company, unless they withdraw our ordinary shares underlying the ADSs in accordance with the deposit agreement and applicable laws and regulations. The depositary, or its nominee, is the holder of the ordinary shares underlying the ADSs. Holders of ADSs therefore do not have any rights as shareholders of our company, other than the rights that they have pursuant to the deposit agreement.
Holders of ADSs may exercise voting rights attached to the ordinary shares represented by the ADSs only in accordance with the provisions of the deposit agreement. The deposit agreement provides that, upon receipt of notice of any meeting of holders of our ordinary shares, the depositary will fix a record date for the determination of ADS holders who shall be entitled to give instructions for the exercise of voting rights. Upon timely receipt of notice from us, if we so request, the depositary shall distribute to the holders as of the record date (1) the notice of the meeting or solicitation of consent or proxy sent by us and (2) a statement as to the manner in which instructions may be given by the holders.
Holders of ADSs may instruct the depositary of their ADSs to vote the ordinary shares underlying their ADSs. Otherwise, ADS holders will not be able to exercise their right to vote, unless they withdraw the ordinary shares underlying the ADSs they hold. However, ADS holders may not know about the meeting far enough in advance to withdraw those ordinary shares. We cannot guarantee ADS holders that they will receive the voting materials in time to ensure that they can instruct the depositary to vote their ordinary shares or to withdraw their ordinary shares so that they can vote them themselves. In addition, the depositary and its agents are not responsible for failing to carry out voting instructions or for the manner of carrying out voting instructions. This means that ADS holders may not be able to exercise their right to vote, and there may be nothing they can do if the ordinary shares underlying their ADSs are not voted as they requested.
We may not be able to complete equity offerings without cancellation or limitation of the preferential subscription rights of our existing shareholders, which may as a practical matter preclude us from timely completion of offerings.
In accordance with the Belgian Companies Code, our Articles of Association provide for preferential subscription rights to be granted to our existing shareholders to subscribe on a pro rata basis and in exchange for contributions in cash, for any issue of new shares, convertible bonds or subscription rights that, unless such rights are cancelled or limited either by resolution of our Shareholders’ Meeting or by our Board of Directors in the framework of the authorized capital, as described below. The Extraordinary Shareholders’ Meeting authorized the Board of Directors to increase the share capital of Galapagos NV, in one or several times, and under certain conditions set forth in extenso in our articles of association. We refer to this authority for our Board of Directors to increase our share capital as our authorized capital. This authorization consists of two parts. A general authorization for capital increases up to 20% of the share capital at the time of convening the Shareholders’ Meeting of October 22, 2019 (i.e. €67,022,402.04) was renewed and is valid for a period of five years from the date of publication of this renewal in the Annexes to the Belgian State Gazette, i.e. November 13, 2019, so until November 12, 2024. A specific authorization for capital increases of more than 20% and up to 33% of the share capital at the time of the convening the shareholders' meeting of April 25, 2017 (i.e. € 82,561,764.93), was renewed and was valid for a period of five years from the date of publication of this renewal in the Annexes to the Belgian State Gazette, i.e. May 31, 2017, so until May 30, 2022. This specific part of the authorized capital can, however, only be used in a number of specific circumstances and upon a resolution of the Board of Directors that all its independent members within the meaning of article 7:87 of the Belgian Companies Code) approve. This specific authorization was not renewed in 2022. As of the date of this annual report, our Board of Directors may decide to issue up to 3,062,207.06 ordinary shares pursuant to the general authorization. Please see the section of this annual report titled “Item 10.B.—Memorandum and Articles of Association.” Absent renewal by our shareholders of this authorization of the Board of Directors or absent cancellation or limitation by our shareholders of the preferential subscription rights of our existing shareholders, the requirement to offer our existing shareholders the preferential right to subscribe, pro rata, for
51
new shares being offered may as a practical matter preclude us from timely raising capital on commercially acceptable terms or at all.
Shareholders may not be able to participate in equity offerings we may conduct from time to time.
If we conduct equity offerings in the future, certain shareholders, including those in the United States, may, even in the case where preferential subscription rights have not been cancelled or limited, not be entitled to exercise such rights, unless the offering is registered or the shares are qualified for sale under the relevant regulatory framework. As a result, there is the risk that investors may suffer dilution of their shareholdings should they not be permitted to participate in preference right equity or other offerings that we may conduct in the future.
Holders of ADSs may be subject to limitations on the transfer of their ADSs and the withdrawal of the underlying ordinary shares.
ADSs are transferable on the books of the depositary. However, the depositary may close its books at any time or from time to time when it deems expedient in connection with the performance of its duties. The depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary think it is advisable to do so because of any requirement of law, government or governmental body, or under any provision of the deposit agreement, or for any other reason, subject to the right of ADS holders to cancel their ADSs and withdraw the underlying ordinary shares. Temporary delays in the cancellation of your ADSs and withdrawal of the underlying ordinary shares may arise because the depositary has closed its transfer books or we have closed our transfer books, the transfer of ordinary shares is blocked to permit voting at a Shareholders’ Meeting or we are paying a dividend on our ordinary shares. In addition, ADS holders may not be able to cancel their ADSs and withdraw the underlying ordinary shares when they owe money for fees, taxes and similar charges and when it is necessary to prohibit withdrawals in order to comply with any laws or governmental regulations that apply to ADSs or to the withdrawal of ordinary shares or other deposited securities.
As a foreign private issuer, we are exempt from a number of rules under the U.S. securities laws and are permitted to file less information with the SEC than a U.S. company. This may limit the information available to holders of ADSs or our ordinary shares.
We are a “foreign private issuer,” as defined in the SEC’s rules and regulations and, consequently, we are not subject to all of the disclosure requirements applicable to public companies organized within the United States. For example (without any limitation), we are exempt from certain rules under the Securities Exchange Act 1937, as amended, (the Exchange Act) that regulate disclosure obligations and procedural requirements related to the solicitation of proxies, consents or authorizations applicable to a security registered under the Exchange Act, including the U.S. proxy rules under Section 14 of the Exchange Act. In addition, our officers and directors are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and related rules with respect to their purchases and sales of our securities. Moreover, while we currently make annual and semi-annual filings with respect to our listing on Euronext Brussels and Euronext Amsterdam and voluntarily report our results of operations on a quarterly basis, we will not be required to file periodic reports and financial statements with the SEC as frequently or as promptly as U.S. domestic issuers and will not be required to file quarterly reports on Form 10-Q or current reports on Form 8-K under the Exchange Act. Accordingly, there is and will continue to be less publicly available information concerning our company than there would be if we were not a foreign private issuer.
As a foreign private issuer, we are permitted to adopt certain home country practices in relation to corporate governance matters that differ significantly from Nasdaq corporate governance listing standards. These practices may afford less protection to shareholders than they would enjoy if we complied fully with corporate governance listing standards.
As a foreign private issuer listed on the Nasdaq Global Select Market, we are subject to corporate governance listing standards. However, rules permit a foreign private issuer like us to follow the corporate governance practices of its home country. Certain corporate governance practices in Belgium, which is our home country, may differ significantly from corporate governance listing standards applicable to U.S. domestic
52
issuers. For example (without any limitation), neither the corporate laws of Belgium nor our Articles of Association require a majority of the members of our Board of Directors to be independent, and we could include non-independent board members as members of our Nomination Committee and Remuneration Committee, and our independent board members would not necessarily hold regularly scheduled meetings at which only independent Board members are present. Currently, we intend to follow home country practice to the maximum extent possible. Therefore, our shareholders may be afforded less protection than they otherwise would have under corporate governance listing standards applicable to U.S. domestic issuers. See the sections of this annual report titled “Item 6—Directors, Senior Management and Employees” and “Item 16G —Corporate Governance.”
We may lose our foreign private issuer status in the future, which could result in significant additional cost and expense.
While we currently qualify as a foreign private issuer, and therefore we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act applicable to U.S. domestic issuers, the determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter and, accordingly, the next determination will be made with respect to us on June 30, 2024.
In the future, we would lose our foreign private issuer status if we to fail to meet the requirements necessary to maintain our foreign private issuer status as of the relevant determination date. In order to maintain our current status as a foreign private issuer, either (a) a majority of our ordinary shares must be either directly or indirectly owned of record by non-residents of the United States or (b) (i) a majority of our executive officers or Directors may not be U.S. citizens or residents, (ii) more than 50% of our assets cannot be located in the United States and (iii) our business must be administered principally outside the United States. As of March 15, 2024, a majority of our executive officers and Directors are not U.S. citizens or residents.
The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic issuer may be significantly more than costs we incur as a foreign private issuer. As a result, we expect that a loss of foreign private issuer status would increase our legal and financial compliance costs and would make some activities highly time consuming and costly. If we are not a foreign private issuer, we will be required to file periodic reports and registration statements on U.S. domestic issuer forms with the SEC, which are more detailed and extensive in certain respects than the forms available to a foreign private issuer. We would be required under current SEC rules to prepare our financial statements in accordance with U.S. GAAP, rather than IFRS, in U.S. dollars rather than euros and modify certain of our policies to comply with corporate governance practices associated with U.S. domestic issuers. Such conversion of our financial statements to U.S. GAAP will involve significant time and cost. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements on U.S. stock exchanges that are available to foreign private issuers such as the ones described above and exemptions from procedural requirements related to the solicitation of proxies.
It may be difficult for investors outside Belgium to serve process on, or enforce foreign judgments against, us or our directors and senior management.
We are a Belgian public limited liability company. Less than a majority of the members of our Board of Directors and members of our Executive Committee are residents of the United States. All or a substantial portion of the assets of such non -resident persons and most of our assets are located outside the United States. As a result, it may not be possible for investors to effect service of process within the United States upon such persons or on us or to enforce against them or us a judgment obtained in U.S. courts. Original actions or actions for the enforcement of judgments of U.S. courts relating to the civil liability provisions of the federal or state securities laws of the United States (as amended from time to time) are not directly enforceable in Belgium. The United States and Belgium do not currently have a multilateral or bilateral treaty providing for reciprocal recognition and enforcement of judgments, other than arbitral awards, in civil and commercial matters. Consequently, a final judgment for payment given by a court in the United States, whether or not predicated
53
solely upon U.S. securities laws, would not automatically be recognized or enforceable in Belgium. This will depend on the applicable Belgian national rules.
In order for a final and conclusive judgment rendered by U.S. courts based on civil liability to produce any effect on Belgian soil, it is accordingly required that this judgment be recognized or be declared enforceable by a Belgian court in accordance with Articles 22 to 25 of the 2004 Belgian Code of Private International Law, as amended. Recognition or enforcement does not imply a review of the merits of the case and is irrespective of any reciprocity requirement. A U.S. judgment will, however, not be recognized or declared enforceable in Belgium if it infringes upon one or more of the grounds for refusal that are exhaustively listed in Article 25 of the Belgian Code of Private International Law, as amended. Actions for the enforcement of judgments of U.S. courts might be successful only if the Belgian court confirms the substantive correctness of the judgment of the U.S. court and if it is satisfied that:
| ● | the effect of the enforcement judgment is not manifestly incompatible with Belgian public policy; |
| ● | the judgment did not violate the rights of the defendant; |
| ● | the judgment was not rendered in a matter where the parties transferred rights subject to transfer restrictions with the sole purpose of avoiding the application of the law applicable according to Belgian international private law; |
| ● | the judgment is not subject to further recourse under U.S. law; |
| ● | the judgment is not compatible with a judgment rendered in Belgium or with a subsequent judgment rendered abroad that might be enforced in Belgium; |
| ● | a claim was not filed outside Belgium after the same claim was filed in Belgium, while the claim filed in Belgium is still pending; |
| ● | the Belgian courts did not have exclusive jurisdiction to rule on the matter; |
| ● | the U.S. court did not accept its jurisdiction solely on the basis of either the nationality of the defendant or the location of the disputed goods; and |
| ● | the judgment submitted to the Belgian court is authentic. |
Under the Belgian Code of Private International law, in addition to recognition or enforcement and before a Belgian court, a judgment by a federal or state court in the United States against us may also serve as evidence in a similar action in a Belgian court if it meets the conditions required for the authenticity of judgments according to the law of the state where it was rendered. The findings of a federal or state court in the United States will not, however, be taken into account to the extent they appear incompatible with Belgium’s (international) public policy.
U.S. judgments ordering to pay a certain amount that are declared enforceable in Belgium are subject to the applicable registration tax in the same way as Belgian judgments. As such, a registration tax at the rate of 3% of the amount awarded is payable by the debtor(s), if the sum of money exceeds EUR 12,500. If multiple debtors were held jointly liable to pay, the debtors are also jointly liable to pay the registration tax.
In light of the above, U.S. investors may not be able to enforce against us, or members of our Board of Directors or members of our Executive Committee who are residents of Belgium or countries other than the United States any judgments obtained in U.S. courts in civil and commercial matters, including judgments under the U.S. federal securities laws (as amended from time to time).
54
Item 4 Information on the Company
A. History and development of the Company
Our legal and commercial name is Galapagos NV. We are a limited liability company incorporated in the form of a “naamloze vennootschap” / “société anonyme” under the laws of Belgium. We were incorporated in Belgium on June 30, 1999 for an unlimited duration. We are registered with the Register of Legal Entities (Antwerp, division Mechelen) under the enterprise number 0466.460.429. Our principal executive and registered offices are located at Generaal De Wittelaan L11 A3, 2800 Mechelen, Belgium and our telephone number is +32 15 342 900. Our agent for service of process in the United States is C T Corporation System, located at 28 Liberty Street, New York, New York, 10005, United States of America.
Our fiscal year ends December 31. We also maintain a corporate website at www.glpg.com. Information contained on, or that can be accessed through, our website does not constitute a part of this annual report. We have included our website address in this annual report as an inactive textual reference only.
The SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as Galapagos NV, that file electronically with the SEC.
We are currently operating as a single operating segment. Prior to the disposal of Fidelta we had two reportable segments, R&D and fee-for-service business. On January 4, 2021 however we sold our Croatian subsidiary and fee for service business Fidelta to Selvita, who acquired 100% of the outstanding shares in Fidelta. Due to the disposal of Fidelta (our fee-for-service segment), we have reported this segment as discontinued operations in our income statement of 2021.
On June 21, 2022, we acquired all outstanding shares of CellPoint B.V., a Dutch company dedicated to developing CAR-T therapies at the point-of-care, and AboundBio, Inc., a privately held U.S. biotechnology company. Each was acquired through an all-cash transaction. CellPoint was acquired for an upfront payment of €125 million, with milestone payments of up to €100 million. AboundBio was acquired for a payment of $14 million. We believe we reinforced our portfolio through the acquisitions by gaining access to a new therapeutic area, oncology as well as a decentralized and automated point-of-care cell therapy supply model and a fully human antibody-based therapeutics platform.
On October 30, 2023, we signed a letter of intent contemplating a transfer of the Jyseleca® business to Alfasigma S.p.A. (Alfasigma). The final agreement was signed on December 30, 2023 and the transaction was closed on January 31, 2024. The transfer includes the European and UK Marketing Authorizations, and the commercial, medical affairs and development activities for Jyseleca®. In connection with the completion of the transaction, approximately 400 Galapagos positions in 14 European countries transferred to Alfasigma to support business continuity and ongoing patient access for the Jyseleca® business. We received a €50 million upfront payment in connection with the transfer, and expect to receive potential milestone payments totaling €120 million and mid-single to mid-double-digit royalties on European sales. We will contribute up to €40 million by June 2025 to Alfasigma for Jyseleca® related development activities.
The transfer of our Jyseleca business has been determined to meet the criteria to be classified as held for sale and discontinued operations in our financial statements for the year ended December 31, 2023. We also presented all income statement items fully related to the Jyseleca® business to be transferred on a separate line “Net profit/loss (-) from discontinued operations, net of tax” in our consolidated income statement. The consolidated income statement for all comparative periods reported in these consolidated financial statements were restated as well to show the discontinued operations on a separate line.
Our actual capital expenditures for the years ended December 31, 2021, 2022, and 2023 amounted to €57.9 million, €36.9 million, and €19.3 million respectively. These capital expenditures primarily consisted of land, buildings, laboratory equipment, leasehold improvements, and intangible assets. We anticipate our capital expenditure in 2024 to
55
be financed from our cash reserves. For more information on our capital expenditures, see the section of this annual report titled “Item 5.B.—Liquidity and capital resources—capital expenditures.”
B. Business overview
We are a global biotechnology company with operations in Europe and the US dedicated to developing transformational medicines for more years of life and quality of life. Focusing on high unmet medical needs, we synergize compelling science, technology, and collaborative approaches to create a deep pipeline of potentially best-in-class small molecules, CAR-T therapies, and biologics in oncology and immunology. With capabilities from lab to patient, including a decentralized, point-of-care CAR-T manufacturing network, we are committed to challenging the status quo and delivering results for our patients, employees and shareholders.
Our clinical pipeline includes: 1) GLPG5101, a CD19 CAR-T product candidate manufactured near point-of-care, currently in Phase 1/2 in rrNHL; 2) GLPG5201, a CD19 CAR-T product candidate manufactured near point-of-care, currently in Phase 1/2 in rrCLL; 3) GLPG5301, a BCMA CAR-T product candidate manufactured near point-of-care, for which we initiated clinical development in rrMM, and 4) GLPG3667, a TYK2 inhibitor currently in Phase 2 clinical trial in DM and in SLE. In both our oncology and immunology portfolios, we have multiple product candidates in early research stages.
Below is an overview of our current key pipeline assets:
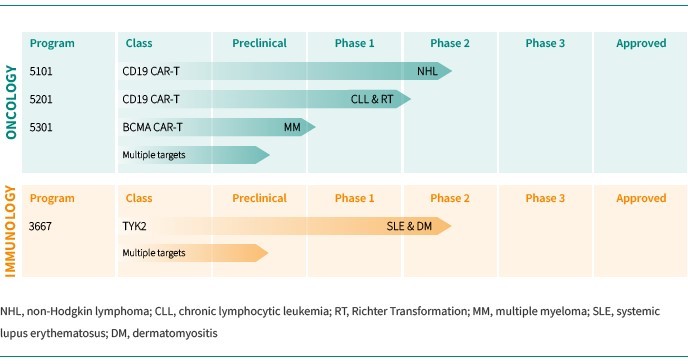
On 31 January 2024, we announced the successful completion of the transaction to transfer our Jyseleca® (filgotinib) business to Alfasigma. The transaction includes the transfer of the entire Jyseleca® business, including the European and UK Marketing Authorizations, and the commercial, medical affairs and development activities for Jyseleca®. In connection with the completion of the transaction, approximately 400 Galapagos employees in 14 European countries transferred to Alfasigma to support business continuity and ongoing patient access with respect to the Jyseleca® business.
56
Lead programs
ONCOLOGY
CAR-T pipeline programs manufactured at or near the point-of-care
| 1. | GLPG5101: CD19 CAR-T in relapsed/refractory non-Hodgkin’s lymphoma |
Non-Hodgkin’s lymphoma (NHL) is a cancer originating from lymphocytes, a type of white blood cell which is part of the body’s immune system. NHL can occur at any age although it is more common in adults over 50 years old. Initial symptoms usually are enlarged lymph nodes, fever, and weight loss. There are many different types of NHL. These types can be divided into aggressive (fast- growing) and indolent (slow-growing) types, and they can be formed from either B lymphocytes (B cells) or in lesser extent from T lymphocytes (T cells) or Natural Killer cells (NK cells). B cell lymphoma makes up about 85% of NHL cases diagnosed in the US. Prognosis and treatment of NHL depend on the stage and type of disease.
GLPG5101 is a second generation anti-CD19/4-1BB CAR-T product candidate, administered as a single fixed intravenous dose. The safety, efficacy, and feasibility of point-of-care manufactured GLPG5101 are currently being evaluated in the ATALANTA-1 Phase 1/2, open-label, multicenter study in patients with relapsed/refractory non-Hodgkin lymphoma (rrNHL).
The primary objective of the Phase 1 part of the study was to evaluate safety and to determine the recommended dose for the Phase 2 part of the study. Secondary objectives include assessment of efficacy and feasibility of near the point-of-care manufacturing of GLPG5101. The dose levels that were evaluated in Phase 1 are 50x106 (DL1), 110x106 (DL2) and 250x106 (DL3) CAR+ viable T cells. The primary objective of the Phase 2 part of the study is to evaluate the Objective Response Rate (ORR) while the secondary objectives include Complete Response Rate (CRR), duration of response, progression free survival, overall survival, safety, pharmacokinetic profile, and the feasibility of point-of-care manufacturing. Each enrolled patient will be followed for 24 months.
57

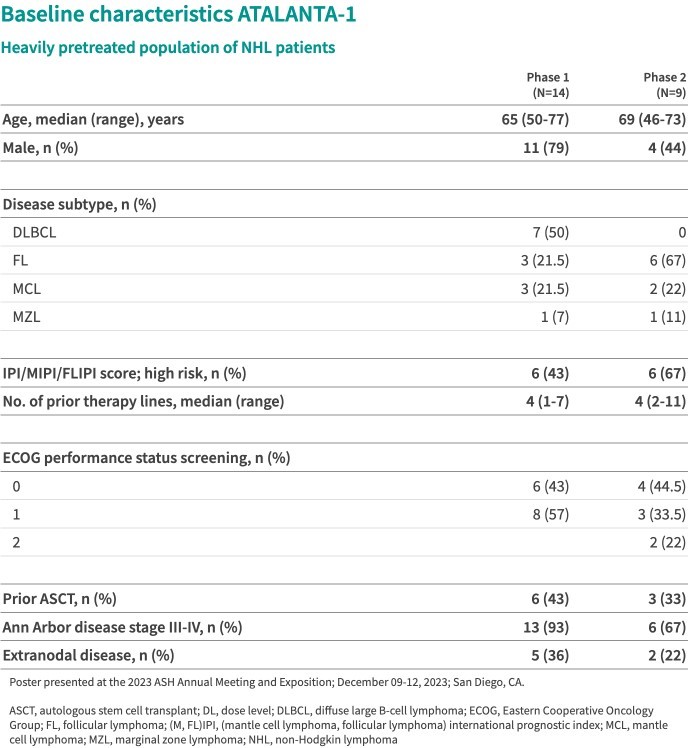
To further build a robust data package, patient recruitment of the Phase 1 dose-finding part of ATALANTA-1 is ongoing. As of 1 September 2023 (cut-off date), 14 heavily pre-treated rrNHL patients with diffuse large B cell lymphoma, mantle cell lymphoma and indolent lymphoma were enrolled (7 at DL1 and 7 at DL2). In parallel, enrollment of the Phase 2 expansion study is ongoing, and the first 9 patients have been dosed.
In December 2023, we presented promising new preliminary data from the ATALANTA-1 Phase 1 dose-finding part of the study and preliminary data of the Phase 2 expansion part during a poster session at the 65th Annual American
59
Society of Hematology (ASH) Congress San Diego (cut-off date: 1 September 2023). The detailed results are presented below.
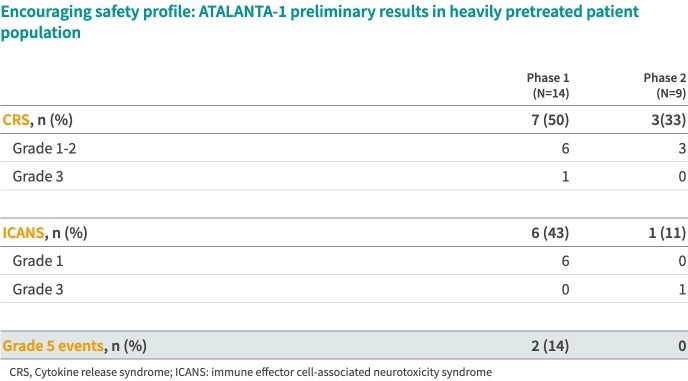
60
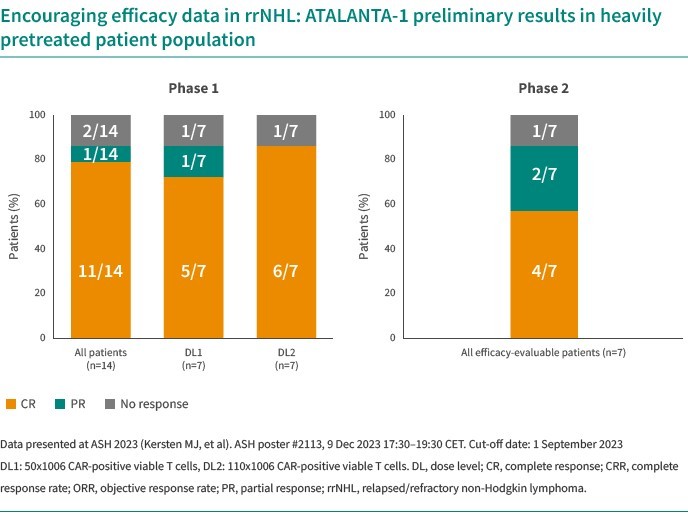
| ● | In the Phase 1 part of the study (cut-off date: 1 September 2023): |
| o | GLPG5101 showed an encouraging safety profile. Most treatment emergent adverse events (TEAEs) were Grade 1 or 2 and the majority of the few Grade ≥ 3 events were hematological. No cytokine release syndrome (CRS) Grade > 3 and no immune effector cell associated neurotoxicity syndrome (ICANS) Grade ≥ 2 were observed. |
| o | 12 of 14 evaluable patients responded to treatment (ORR of 86%), with 11 of 14 patients achieving a Complete Response (CRR of 79%). 6 of 7 patients treated with the higher dose level (DL2) responded to treatment (ORR of 86%) and achieved a Complete Response (CRR of 86%). At the time of the analysis, 8 of 12 responding patients (67%) had an ongoing response, with a duration up to 15 months (median follow-up of 8.6 months); 2 of the 4 patients who progressed after an initial response had a CD19 positive relapse and 1 had confirmed CD19-negative disease. |
61
| ● | In the Phase 2 part of the study (cut-off date: 1 September 2023): |
| o | GLPG5101 showed an encouraging safety profile with most TEAEs of Grade 1 or 2; the majority of Grade ≥ 3 events were hematological. No CRS Grade > 2 and ICANS was seen in one patient (Grade 3). |
| o | 6 of 7 evaluable patients responded to treatment (ORR of 86%) and a Complete Response was observed in 4 of 7 patients (57%). At the time of the analysis, all 6 responding patients (100%) had an ongoing response with a median follow-up of 3.2 months. |
At ASH, we also showcased encouraging preliminary translational data with regard to the status of the CAR-T cells in the GLPG5101 final product (FP).
A thorough characterization of the collected patient material in the ATALANTA-1 trial (19 patients) revealed an increased percentage of ‘early phenotype’ T cells (i.e. TN/SCM and TCM, CD4+ and CD8+) in the final product compared to the starting material (apheresed blood). This was in line with the observed decrease in more differentiated, ‘late phenotype’ T cells (i.e. TEM/EFF, CD4+ and CD8+).
This early phenotype reflects the differentiation status of the cells, which is associated with enhanced functionality and persistence of CAR-T cells after infusion in the patient.
62
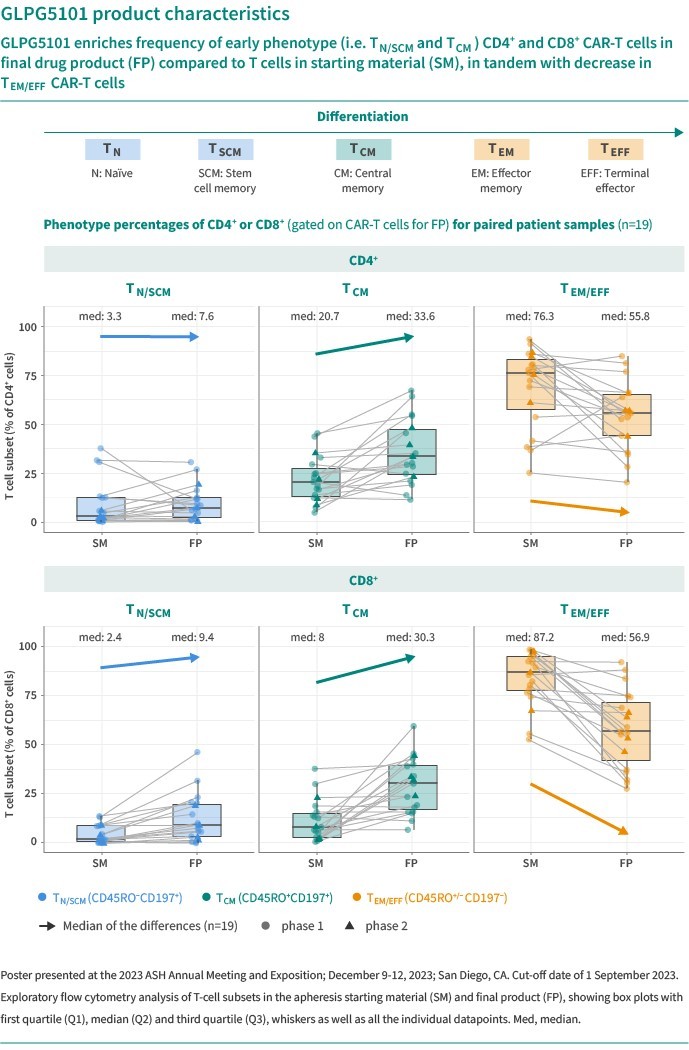
In addition, we evaluated the kinetics of expansion of the manufactured CAR-T cells in the patient by measuring the levels of CAR vector copies in blood after infusion.
Robust CAR-T cell expansion was observed in the treated patients across all dose levels with a median time to peak expansion of 14 days. In 3 out of 4 evaluable patients, we were able to detect the GLPG5101 CAR-T cells up to 9 months post-infusion (cut-off date of 1 September 2023).
These findings support the persistence of GLPG5101, which could be an early predictor of durable responses.
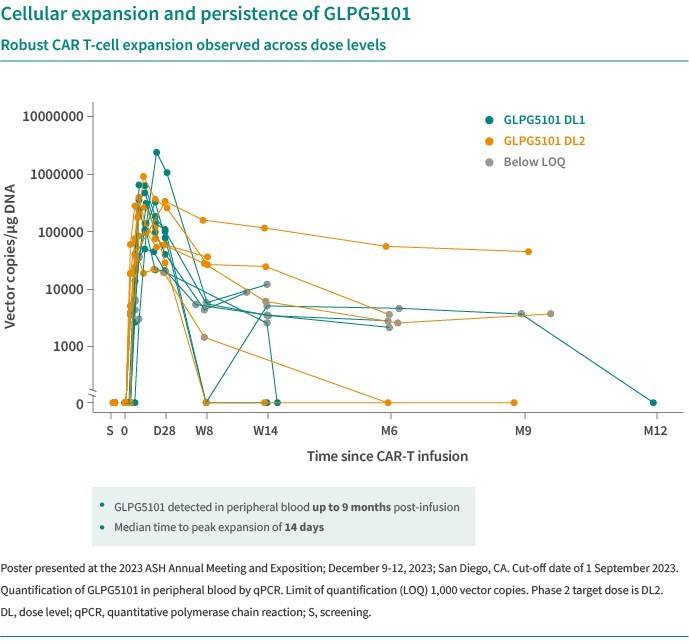
64
The ATALANTA-1 preliminary data suggest that Galapagos’ CAR-T point-of-care manufacturing platform can deliver a fit product in a median vein-to-vein time of only seven days.
| 2. | GLPG5201: CD19 CAR-T in relapsed and refractory chronic lymphocytic leukemia |
Chronic lymphocytic leukemia (CLL) is one of the chronic lymphoproliferative disorders (lymphoid neoplasms). It is characterized by a progressive accumulation of functionally incompetent lymphocytes, which are usually monoclonal in origin. CLL affects B-cells in the blood and bone marrow.1 Richter Transformation (RT) is an uncommon clinicopathological condition observed in patients with CLL. It is characterized by the sudden transformation of the CLL into a significantly more aggressive form of large cell lymphoma and occurs in approximately 2-10% of all CLL patients. CLL usually follows an indolent course and is an incurable disease. Patients who develop relapsed and refractory disease and become resistant to new agents have a dismal prognosis and a high unmet medical need for new therapeutic options such as CAR-T cells. With estimated incidence of 4.7 new cases per 100,000 individuals, CLL is the most prevalent lymphoid malignancy and is the most common adult leukemia in the US and in Europe.2 The annual incidence of patients with RT has been estimated at 1,900 new patients in the US and 2,000 in the EU5.3.
1 Wierda WG. Chronic lymphocytic leukemia/ Small lymphocytic lymphoma fact sheet. In: Foundation LR, editor: https://www.lymphoma.org/wp-content/uploads/2018/04/LRF_FACTSHEET_CLL_SLL.pdf.2018.
2 Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: A Cancer Journal for Clinicians. 2021;71(1):7-33.
3 IMARC report, 2023; 2-15% of incidence per Lightning Health literature review; Sigmund AM et al. 2022; Thompson PhA et al. 2022.IMARC report, 2023; 2-15% of incidence per Lightning Health literature review; Sigmund AM et al. 2022; Thompson PhA et al. 2022.
GLPG5201 is a second generation anti-CD19/4-1BB CAR-T product candidate, administered as a single fixed intravenous dose. The, safety, efficacy, and feasibility of point-of-care manufactured GLPG5201 are currently being evaluated in the EUPLAGIA-1 Phase 1/2, open-label, multicenter study in patients with rrCLL and rrSLL (small lymphocytic lymphoma), with or without RT.
Patients with CD19 rrCLL or rrSLL with >2 lines of therapy are eligible to participate, and patients with RT are eligible regardless of prior therapy. The primary objective of the Phase 1 part of the study is to evaluate safety and determine the recommended dose for the Phase 2 part of the study. The dose levels that are evaluated in the Phase 1 part of the study are 35x106 (DL1), 100x106 (DL2), and 300x106 (DL3) CAR+ viable T cells.
The primary objective of the Phase 2 part of the study is to assess the ORR, and the secondary objectives include the analysis of the CRR, duration of response, progression free survival, overall survival, safety pharmacokinetic profile, and feasibility of point-of- care manufacturing.
65

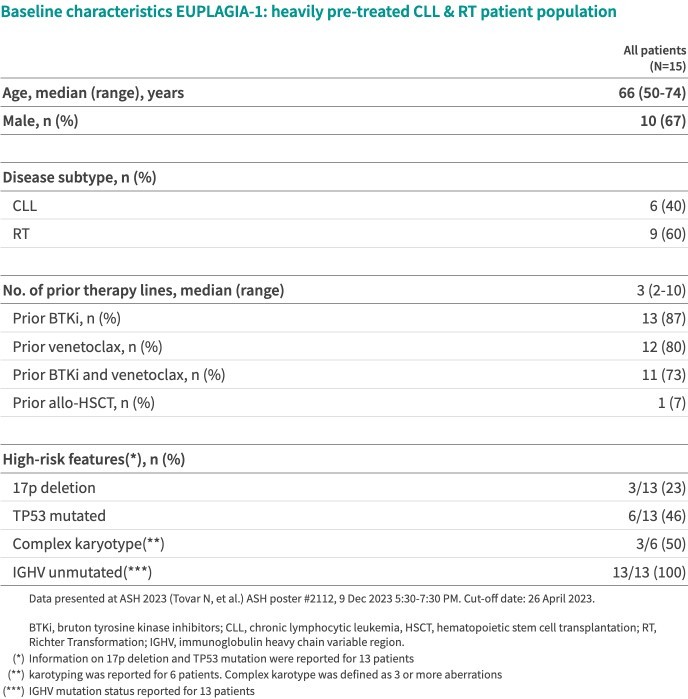
In February 2023, we presented initial encouraging safety and efficacy data (cut-off date: 9 January 2023) from the EUPLAGIA-1 Phase 1 study during a poster session at the EBMT-EHA 5th European CAR-T-cell Meeting in Rotterdam.
As of 6 September 2023, patient recruitment of the Phase 1 dose-finding part of EUPLAGIA-1 had been completed and, 15 patients (6 at dose level 1 (DL1); and 9 at dose level 2 (DL2)) were enrolled, all of whom were diagnosed with rrCLL, and 9 with additional RT. All 15 Phase 1 batches were manufactured at the point-of-care and infused as a single fresh, fit product within a median vein-to-vein time of seven days, with 80% of patients receiving the product in seven days. In December 2023, we presented promising new preliminary data from the Phase 1 dose-finding part of the study during a poster session at the 65th Annual ASH Congress in San Diego. Efficacy data as of day 28 were
67
available for 14 patients; 1 patient did not yet reach the day 28 follow-up visit at the time of the analysis. The results (cut-off date: 6 September 2023) are presented below:

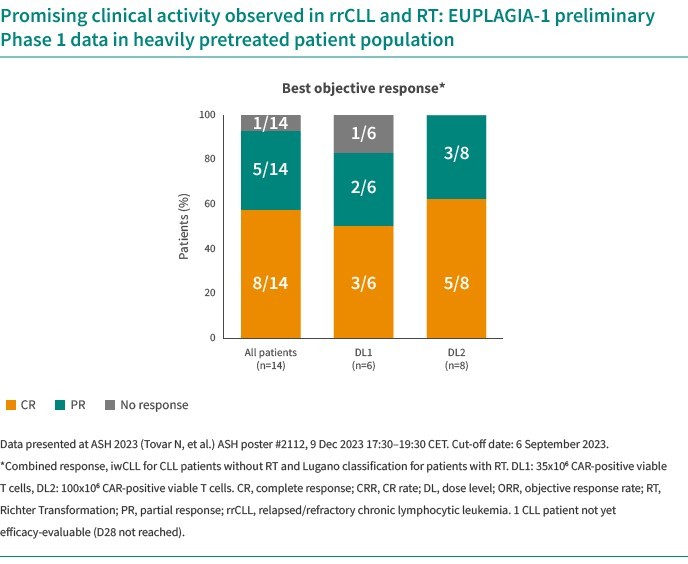
68
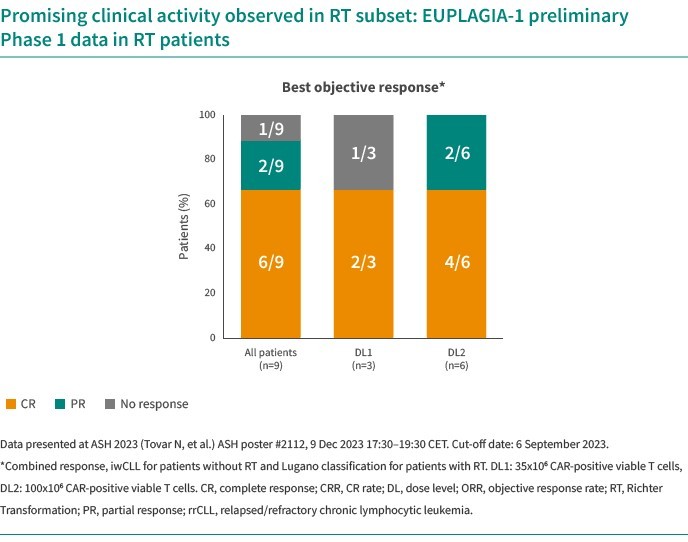
| ● | GLPG5201 showed an encouraging safety profile with most TEAEs of Grade 1 or 2, mostly hematological. CRS Grade 1 or 2 was observed in 47% of the patients, and no CRS Grade ≥ 3 or any ICANS were observed. No deaths were reported. |
| ● | Overall, 13 of 14 efficacy evaluable patients responded to treatment (Objective Response Rate (ORR) of 93%) and 8 of 14 patients achieved a Complete Response Rate (CRR of 57%). 8 of 9 patients with RT responded to treatment (ORR of 89%) and 6 of 9 RT patients achieved a Complete Response (CRR of 67 %). At time of analysis, 10 of 13 of responding patients (77%) were in ongoing response with a median follow-up of 6 months; 2 of 3 patients who progressed after an initial response had confirmed CD19-negative disease. |
69
| ● | On the higher dose level (DL2), 8 of 8 patients responded to treatment (ORR of 100%), 5 of 8 patients achieved a Complete Response (CRR of 63%), and 6 of 6 patients with RT responded to treatment (ORR of 100%). |
| ● | DL2 was selected as the recommended dose for the Phase 2 part of the study. |
At the annual EBMT-EHA congress in February 2024, we showcased encouraging preliminary translational data regarding the status of the CAR-T cells in the GLPG5201 final product (FP).
A thorough characterization of the collected patient material in the EUPLAGIA-1 trial (10 patients) revealed an increased percentage of ‘early phenotype’ T cells (i.e. TN/SCM and TCM, CD4+ and CD8+) in the final product compared to the starting material (apheresed blood). This was in line with the observed decrease in more differentiated, ‘late phenotype’ T cells (i.e. TEM/EFF, CD4+ and CD8+).
This early phenotype reflects the differentiation status of the cells, which is associated with enhanced functionality and persistence of CAR-T cells after infusion in the patient.
70
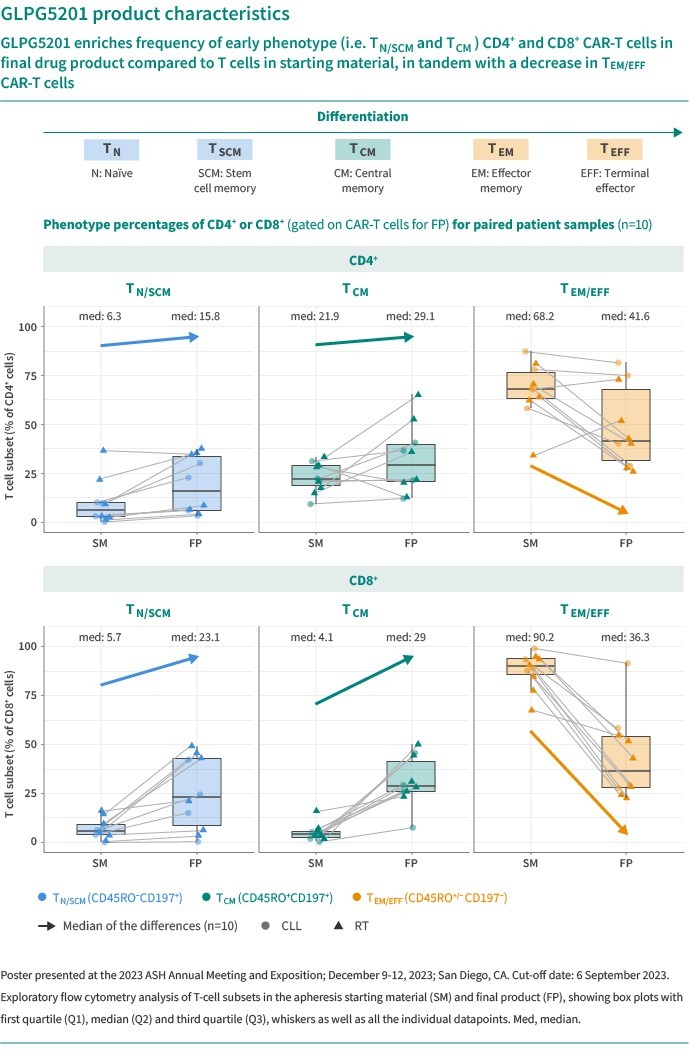
Similarly to the ATALANTA-1 study, we evaluated the kinetics of expansion of the manufactured CAR-T cells in the patient by measuring the levels of CAR vector copies in blood after infusion.
CAR-T cell expansion and persistence data was available for 13 of 15 patients. Robust expansion was observed in all patients for the dose levels tested with a median time to peak expansion of 14 days.
In 3 out of 4 evaluable patients, we were able to detect the GLPG5201 CAR-T cells up to 9 months post-infusion (cut-off date of 6 September 2023). These findings support the persistence of GLPG5201, which could be an early predictor of durable responses.
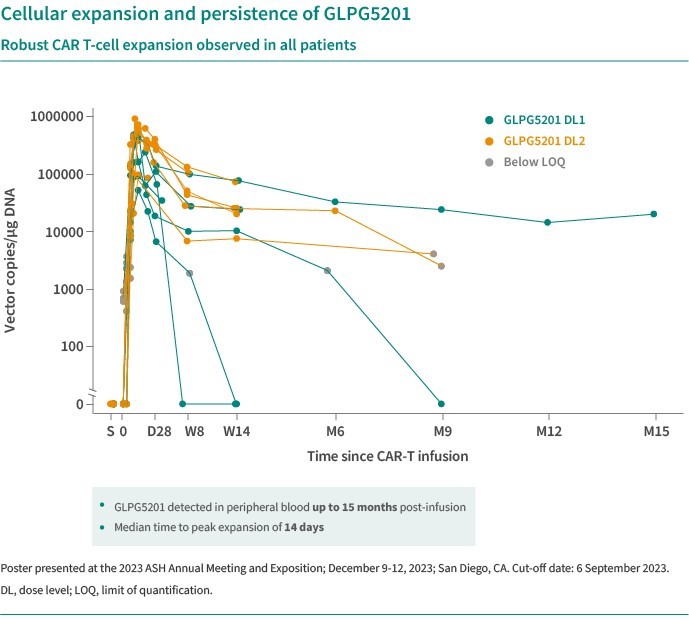
72
The EUPLAGIA-1 preliminary safety, efficacy and translational data presented above suggest that Galapagos’ CAR-T point-of-care manufacturing platform can deliver a fit product in a median vein-to-vein time of only seven days.
| 3. | GLPG5301: BCMA CAR-T in relapsed and refractory multiple myeloma |
Multiple myeloma (MM) is typically characterized by the neoplastic proliferation of plasma cells producing a monoclonal immunoglobulin. The plasma cells proliferate in the bone marrow and may result in extensive skeletal destruction with osteopenia, and osteolytic lesions with or without pathologic fractures. The diagnosis of MM is made when one (or more) of the following clinical presentations are present: bone pain with lytic lesions discovered on routine skeletal films or other imaging modalities, an increased total serum protein concentration with the presence of a monoclonal protein in the urine or serum, and anemia, hypercalcemia or renal failure. The patient may be either symptomatic or their disease may be discovered incidentally.
Despite improvements in treatment, in general, patients with MM ultimately relapse or become refractory to available regiments. Triple-refractory (refractory to CD38 monoclonal antibodies [mAbs], proteasome inhibitor [PI] and immunomodulatory imide drug [IMiD] or penta-refractory (refractory to CD38 mAbs, 2 Pls and 2 IMiDs) patients have a poor prognosis and are in urgent need of novel treatment options.
GLPG5301 is an autologous, second-generation/4-1BB B-cell maturation antigen (BCMA)-directed CAR-T product candidate, administered as an intravenous infusion of a fresh product in a single fixed dose, at the point-of-care. In December 2023, we announced that the first patient with rrMM was dosed in the Phase 1/2 PAPILIO-1 study.
PAPILIO-1 is a Phase 1/2, open-label, multicenter study to evaluate the safety, efficacy and feasibility of point-of-care manufactured GLPG5301, a BCMA CAR-T product candidate, in patients with relapsed/refractory multiple myeloma (rrMM) after ≥2 prior lines therapy. The primary objective of the Phase 1 part of the PAPILIO-1 study is to evaluate safety and determine the recommended dose for the Phase 2 part of the study. The primary objective of the Phase 2 part of the study is to evaluate the efficacy of GLPG5301, as measured by the ORR. Secondary objectives for both Phase 1 and Phase 2 include further assessment of the safety of GLPG5301, additional efficacy endpoints, including assessment of Minimal Residual Disease (MRD), as well as the feasibility of point-of-care manufacture of GLPG5301 in rrMM patients. Each enrolled patient will be followed for 24 months.
During Phase 1, up to 3 dose levels will be evaluated and at least 12 patients will be enrolled to establish the recommended Phase 2 dose. Approximately 30 additional patients will be enrolled in the Phase 2 part of the study to evaluate the safety and efficacy of GLPG5301.
73
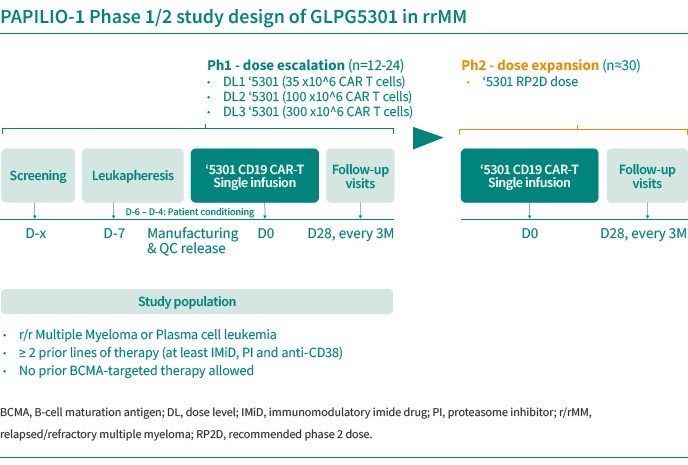
IMMUNOLOGY
Small molecules pipeline
| 1. | Jyseleca® franchise |
On 31 January 2024, we announced the successful completion of the transaction to transfer our Jyseleca® (filgotinib) business to Alfasigma S.p.A. (Alfasigma).
The transaction includes the transfer of the entire Jyseleca® business to Alfasigma, including the European and UK Marketing Authorizations, and the commercial, medical affairs and development activities for Jyseleca®. In connection with the completion of the transaction, approximately 400 Galapagos positions in 14 European countries transferred to Alfasigma to support business continuity and ongoing patient access.
| 1.1 | Jyseleca® (filgotinib) in rheumatoid arthritis (RA) |
RA is a chronic autoimmune disease that affects more than three million patients in the United States and Europe. RA is characterized by inflammation and degeneration of the joints. Patients suffer from pain, stiffness, and restricted mobility due to a persistent inflammation of multiple joints, ultimately resulting in irreversible damage of the joint cartilage and bone. The current market for RA treatments in the five major European markets (EU5) is approximately €3.3 billion. Despite progress in the treatment of RA, there remains a considerable unmet need as sustained remission remains rare1.
1 Chen Y, et al. Clin Rheumatol. 2019 Mar;38(3):727-738. doi: 10.1007/s10067-018-4340-7. Epub 2018 Oct 19.
74
Regulatory progress of Jyseleca® in RA
In 2020, Jyseleca® (filgotinib 200mg and 100mg) obtained regulatory approval in Europe, Great Britain, and Japan for the treatment of adult patients with moderate to severe active RA.
The European Summary of Product Characteristics for filgotinib, which includes contraindications and special warnings and precautions, is available at www.ema.europa.eu. The Great Britain Summary of Product Characteristics for filgotinib can be found at www.medicines.org.uk/emc and the Northern Ireland Summary of Product Characteristics for filgotinib can be found at www.emcmedicines.com/en-GB/northernireland, respectively. The interview form from the Japanese Ministry of Health, Labour and Welfare is available at www.info.pmda.go.jp.
Also in 2020, Gilead Sciences, Inc (Gilead) received a Complete Response Letter (CRL) from the U.S. Food and Drug Administration (FDA) for the New Drug Application (NDA) for filgotinib. Consequently, Gilead decided not to advance with resubmission for approval of filgotinib as a treatment for RA in the U.S.
In 2022, the Pharmacovigilance Risk Assessment Committee (PRAC) of the European Medicines Agency (EMA) concluded its Article 20 safety review of all JAK inhibitors approved in the EU for the treatment of inflammatory diseases and recommended the harmonization of all labels. PRAC concluded that JAK inhibitors should maintain their indication for the treatment of patients with RA who have responded inadequately to or who cannot tolerate disease modifying anti-rheumatic drugs (DMARDs) therapy, and for patients with UC who have responded inadequately to or who cannot tolerate conventional therapy or biologics. PRAC also recommended all JAK inhibitor product labels be updated to include a precautionary approach for use of JAK inhibitors in patients with identified risk factors only if no suitable treatment alternative is available (Section 4.4 of the product label – Warning and Precautions). On 11 November 2022, the Committee for Medicinal Products for Human Use (CHMP), the scientific committee of the EMA, adopted PRAC’s recommendation and on 10 March 2023, this decision was approved by the European Commission.
Commercialization of Jyseleca® in RA
In 2021, we took full ownership of the manufacturing and commercialization of Jyseleca® in Europe and became the Marketing Authorization Holder (MAH) in 27 countries in Europe.
Gilead is responsible for the commercialization and distribution of Jyseleca® outside of Europe, including in Japan where Jyseleca® is approved in RA and is co-marketed with Eisai.
In Central and Eastern Europe, Portugal, Greece and the Baltic countries, Swedish Orphan Biovitrum AB (Sobi) is responsible for the distribution and commercialization of Jyseleca®.
Jyseleca® reimbursement in RA in Europe
Jyseleca® in RA is currently reimbursed in Western Europe. Sobi secured reimbursement for Jyseleca® in 2023 in Poland, Slovenia, Slovakia, Estonia, Croatia, and Greece for RA.
See details on the Gilead collaboration on filgotinib in “Collaborations”.
75
Safety and efficacy in the filgotinib RA development program
Filgotinib showed favorable results in terms of onset of action, efficacy, safety, and tolerability from the FINCH Phase 3 and DARWIN Phase 2 clinical programs.
As part of the filgotinib development program, we initiated FINCH 4 in RA. The FINCH 4 study is a multi-center, open-label, long-term extension study to assess the safety and efficacy of filgotinib in patients with RA, which enrolled subjects who completed either the FINCH 1, FINCH 2, or FINCH 3 studies.
We and Gilead published integrated safety data from 7 RA studies in Annals of the Rheumatic Diseases (Winthrop et al. 2021). Data were integrated from 3 Phase 3 studies (FINCH 1 – 3), 2 Phase 2 studies (DARWIN 1, 2), and 2 long-term extension studies (DARWIN 3, FINCH 4) including up to 5.6 years of filgotinib exposure, and over a median of 1.6 years. In this pooled analysis, filgotinib was well-tolerated, and no new safety concerns were identified. Adverse events of MACE and deep venous thrombosis (DVT)/pulmonary embolism (PE) were rare and occurred in similar numbers among all treatment groups, and with a similar incidence rate across all dose groups. The data underscore the acceptable safety and tolerability profile of filgotinib as monotherapy and in conjunction with methotrexate (MTX)/csDMARDs (Conventional synthetic DMARDs) in RA.
In 2023, we presented new analyses from randomized controlled trials (RCTs) and real-world evidence (RWE) studies at the European League Against Rheumatism (EULAR) congress. These included long-term efficacy and integrated safety data, post hoc analysis identifying distinct trajectories of treatment responses in patients with RA receiving filgotinib, long-term clinical profile of filgotinib in patients with RA by cardiovascular (CV) risk factors, and the added value of filgotinib on pain relief in patients with RA achieving remission in the Phase 3 FINCH 1, 2 and 3 studies.
Furthermore, we published interim results from 500 patients on baseline characteristics as well as effectiveness and safety outcomes from the FILOSOPHY real-world evidence study.
1.2 Jyseleca® (filgotinib) in ulcerative colitis (UC)
UC is an inflammatory bowel disease (IBD) resulting in ulcerations and inflammation of the inner layer of the colon and rectum.
Regulatory progress and commercialization of Jyseleca® in UC
Filgotinib obtained regulatory approval for the treatment of adults with moderate to severe UC in the European Union in 2021, and in Great Britain and Japan in January and March 2022, respectively.
Filgotinib is marketed as Jyseleca® in Europe and Japan for the treatment of adult patients with moderate to severe active UC who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a biologic agent. Jyseleca (filgotinib) 100mg and 200mg are registered in the above-mentioned territories.
The European Summary of Product Characteristics for filgotinib, which includes contraindications and special warnings and precautions, is available at www.ema.europa.eu. The Great Britain Summary of Product Characteristics for filgotinib can be found at www.medicines.org.uk/emc and the Northern Ireland Summary of Product Characteristics for filgotinib can be found at www.emcmedicines.com/en-GB/northernireland, respectively. The interview from the Japanese Ministry of Health, Labour and Welfare is available at www.info.pmda.go.jp.
Gilead is responsible for the distribution and commercialization of Jyseleca® outside of Europe, including in Japan where Jyseleca® is approved in UC and is co-marketed with Eisai. In Central and Eastern Europe, Portugal, Greece and the Baltic countries, Swedish Orphan Biovitrum AB (Sobi) is responsible for the distribution and commercialization of Jyseleca®.
76
Jyseleca® reimbursements in UC
Jyseleca® in UC is currently reimbursed in Western Europe. Sobi secured reimbursement for Jyseleca® in 2023 in Poland, Portugal, Czech Republic, Slovakia, Estonia, and Slovenia in UC.
Financial information related to our operational segment and geographic information is contained in “Note 5—Segment information” in our consolidated financial statements appended to this annual report.
Safety and efficacy in the filgotinib UC development program
The SELECTION Phase 3 study is a multi-center, randomized, double-blind, placebo-controlled study to assess the safety and efficacy of the preferential JAK1 inhibitor filgotinib in adult patients with moderately to severely active UC. The SELECTION study comprises two induction trials and a maintenance trial. The Induction Study A enrolled biologic-naïve patients, and the Induction Study B enrolled biologic-experienced patients.
The primary objectives of SELECTION were to evaluate the efficacy of filgotinib compared with placebo in establishing clinical remission as determined by the Mayo endoscopic subscore of 0 or 1, rectal bleeding sub-score of 0, and ≥ 1-point decrease in stool frequency from baseline to achieve a sub-score of 0 or 1 at Week 10 in the induction studies and Week 58 in the maintenance study. Eligible patients who were enrolled in the SELECTION study were enrolled in the ongoing SELECTION long-term extension trial to evaluate the long-term safety of filgotinib in patients with UC. A majority of patients included in the SELECTION study (n=1348) had a Mayo Clinic Score (MCS) of 9 or higher at baseline, and 43% of biologic experienced patients (n=297/689) had insufficient response to a TNF antagonist and vedoluzimab as well. (Feagan et al., Lancet 2021; 397: 2372–84)
In 2023, we presented additional new analyses from the SELECTION program with filgotinib at the annual ECCO congress. These include new analysis from the long-term extension (LTE) study evaluating the safety and efficacy of filgotinib in UC for nearly four years; an analysis of the prolonged benefit of filgotinib in UC; an analysis exploring factors associated with the partial Mayo Clinic Score (pMCS) over time; and an analysis of the effect of filgotinib on anaemia in UC patients. Additionally, we presented pooled data from five Phase 2/3 trials, and two long-term extension trials of filgotinib designed to further understand the safety profile of filgotinib in UC and RA. Data from the SELECTIONLTE study showed that filgotinib 200mg maintained symptomatic remission and health-related quality of life (HRQoL) for up to approximately four years. Amongst subjects who completed the study, the reduction in mean pMCS in SELECTION was maintained up to LTE Week 144. In non-responders, mean pMCS decreased from LTE baseline to Week 192. The results also showed that a high proportion of completers (>80% of patients) and non-responders (>70% of patients) achieved remission according to the Inflammatory Bowel Disease Questionnaire4. The safety profile of filgotinib 200mg in the SELECTION LTE study was generally consistent with the safety profile observed in previous SELECTION studies, with no new safety signals observed.
4The Inflammatory Bowel Disease Questionnaire is a widely used questionnaire for HRQoL assessment in patients with inflammatory bowel diseases.
1.3 Filgotinib in Crohn’s disease (CD)
CD is an IBD of unknown cause, which results in chronic inflammation of the gastrointestinal (GI) tract with a relapsing and remitting course.
On 8 February 2023, we announced that both induction cohorts of the Phase 3 DIVERSITY study trial of filgotinib in CD failed to meet the co-primary endpoints of clinical remission and endoscopic response for filgotinib, 100mg and 200mg once-daily. Based on these topline data, we decided not to submit a Marketing Authorization Application in Europe for filgotinib in CD.
77
| 2. | TYK2 program: GLPG3667 |
GLPG3667 is an investigational reversible and selective TYK2 kinase domain inhibitor that was discovered by us and evaluated in a Phase 1 healthy volunteer study in 2020. The Phase 1 study was a randomized, double-blind, placebo-controlled dose escalation study evaluating safety, tolerability, pharmacokinetics (PK) and pharmacodynamics (PD) of single and multiple ascending oral doses of GLPG3667 for 13 days.
Blood was drawn at multiple time points on Day 1 and on Day 10 and stimulated ex vivo with several cytokines, including IFNα, to analyze the level of inhibition of inflammation, including the effect on phosphorylated signal transducer and activator of transcription (pSTAT) signaling as well as hematological parameters, lipids, and creatine phosphokinase (CPK) (see graphs below).
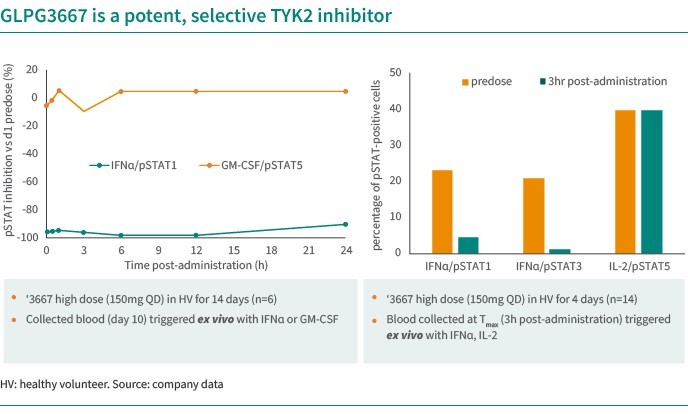
78
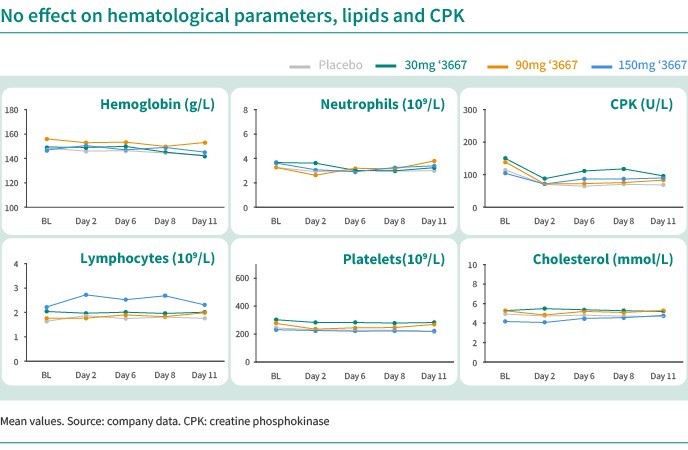
Following these results, we initiated a randomized, placebo-controlled, double-blind Phase 1b study in 31 patients with moderate to severe plaque psoriasis. Patients were randomized in a 1:1:1 ratio to a daily oral dose of GLPG3667 (low dose or high dose) or placebo, for a total of 4 weeks.
79
In July 2021, we announced positive topline results demonstrating that GLPG3667 was generally well tolerated with a positive response signal at Week 4 (see graph below):
| ● | At Week 4, 4 out of 10 patients in the high dose group had a Psoriasis Area and Severity Index (PASI)50 response, defined as at least a 50% improvement in PASI from baseline, compared to one out of 10 subjects on placebo. There were no subjects with a PASI 50 response on the low dose of GLPG3667. The 4 responders in the high dose group of GLPG3667 achieved a 52%, 65%, 74% and 81% improvement respectively in their PASI scores from baseline, while the subject randomized to placebo improved by 52%. Positive efficacy signals were also observed with the high dose for other endpoints, including affected Body Surface Area and physician and patient global assessment, versus placebo at Week 4. |
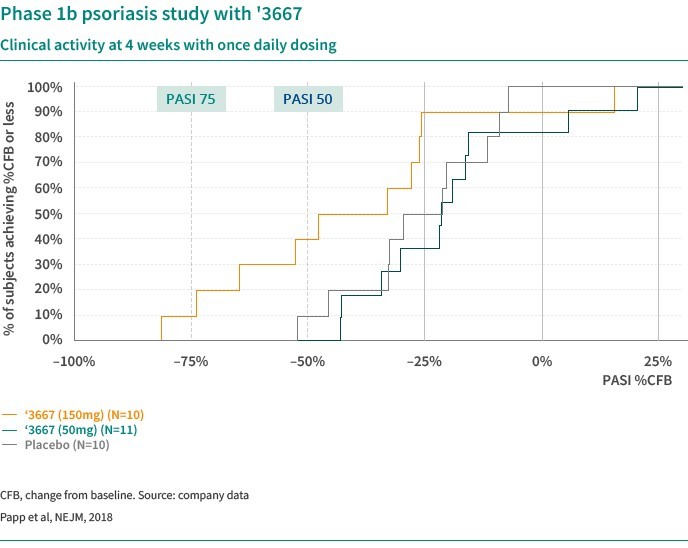
| ● | One subject in the low dose group interrupted participation in the study for one day due to exacerbation of psoriasis. The majority of treatment related adverse events (AEs) were mild in nature and transient. There were no deaths or serious adverse events (SAEs) in this 4-week study. |
80
2.1 GLPG3667 in dermatomyositis (DM)
DM is the most common form of idiopathic inflammatory myopathies (IIM) and is characterized by inflammatory and degenerative changes of the muscles and skin. Early symptoms of DM include distinct skin manifestations accompanying or preceding muscle weakness. The quality of life (QoL) of patients with DM is impaired due to muscle weakness, pain and skin disease activity5.
5 Goreshi R, et al. Quality of life in dermatomyositis. J Am Acad Dermatol. 2011 Dec;65(6):1107-16.
In April 2023, we announced that the first patient was dosed in GALARISSO, the Phase 2 study with GLPG3667 in DM patients. Topline results of the GALARISSO study are expected in 2025.
GALARISSO Phase 2 study design with GLPG3667 in DM

GALARISSO is a Phase 2 randomized, double-blind, placebo-controlled, multi-center study to evaluate the efficacy and safety of GLPG3667. A daily oral administration of GLPG3667 150mg or placebo will be investigated in approximately 62 adult patients with DM over 24 weeks. The primary endpoint is the proportion of patients with at least minimal improvement in the signs and symptoms of DM at Week 24 according to the American College of Rheumatology (ACR) and the European League Against Rheumatism (EULAR) criteria6.
6 Minimal improvement per ACR/EULAR is defined as a total improvement score (TIS) of >= 20 points. The TIS is a score derived from the evaluation of the results from 6 core set measurements of myositis disease activity.
2.2 GLPG3667 in systemic lupus erythematosus (SLE)
SLE is a chronic, inflammatory, autoimmune disease affecting nearly every organ system and thereby one of the most heterogeneous illnesses treated by physicians7. The pathogenesis of SLE is characterized by a global loss of self-tolerance with activation of autoreactive T and B cells. This leads to the production of pathogenic autoantibodies that primarily target a variety of nuclear antigens, deposit in tissues and activate complement, resulting in organ damage.
7 Rees, F. et al., (2017). The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatol. Oxf. Engl., 56(11), 1945–1961
In August 2023, we announced that the first patient was enrolled in GALACELA, the Phase 2 study with GLPG3667 in patients with SLE. Topline results of the GALACELA study are expected in 2026.
81
GALACELA Phase 2 study design with GLPG3667 in SLE
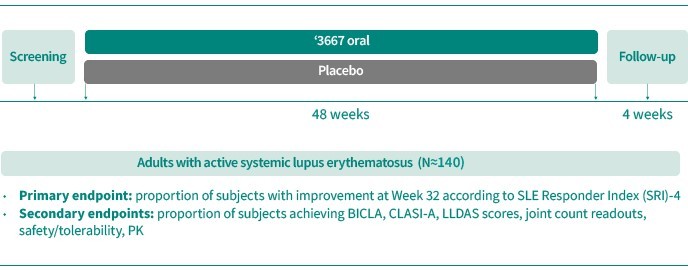
GALACELA is a Phase 2 randomized, double-blind, placebo-controlled, multi-center study to evaluate the efficacy, safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG3667 in adults with active SLE. A once-daily oral administration of GLPG3667 or placebo will be investigated in approximately 140 adult patients with SLE for 32 weeks.
The primary endpoint is the proportion of patients who achieve the SLE responder index (SRI)-4 response at Week 32.
The secondary efficacy endpoints are the proportion of patients who achieve the British Isles Lupus Assessment Group (BILAG)-based Composite Lupus Assessment (BICLA) response at Week 32, proportion of patients with >=50% reduction in Cutaneous Lupus Erythematosus Disease Area and Severity Index Activity (CLASI-A) score at Week 16, proportion of patients who achieve Lupus Low Disease Activity State (LLDAS) at Week 32 and change from baseline in the 28-joint count for tender, swollen, and tender and swollen (active) joints at Week 32.
OUR FORWARD, FASTER STRATEGY
Our goal is to bring transformational medicines to patients across the globe for more years of life and quality of life. Our focus is on conditions with high unmet medical needs.
To achieve this, we are working to synergize compelling science, technology, and approaches to develop a deep pipeline of potentially best-in-class small molecules, CAR-T therapies and biologics in oncology and immunology.
We continue to take steps to transform into an innovative pure-play biotech company by sharpening our focus on our key priority areas. Following the transfer of our entire Jyseleca® (filgotinib) business, we are moving forward with greater focus and flexibility to invest in our key technology platforms and strategic therapeutic areas.
We are committed to challenging the status quo and delivering results for patients, employees, and shareholders.
82

We have a clear path to value creation:
| ● | Pioneering for patients through our targeted R&D approach. At Galapagos, we are focusing on discovering and developing best-in-class medicines in oncology and immunology. |
| o | We are advancing our current clinical programs. |
| o | We are developing our unique decentralized CAR-T manufactured programs in hemato-oncology. |
| o | We are continuing to pursue strategic investments and partnerships to support and expand our pipeline. We are focused on validated targets and next-generation cell therapies and biologics in oncology and immunology. |
| ● | Diversifying our pipeline and making clear portfolio decisions to achieve our vision. We believe we have a greater chance of success by working with multiple drug modalities and combinations across our core therapeutic areas. By 2028, we aim to have: |
| o | A first medicine available to patients; |
| o | A robust late-stage pipeline with several programs in pivotal trials; and |
| o | A solid early-stage pipeline of small molecules, next-generation cell therapies and biologics in our core therapeutic areas. |
| ● | Accelerating and building our pipeline through strategic partnerships and M&A. To achieve our ambitious goals, we evaluate and access external innovation. We are scouting for the best science, products, and people to complement |
84
| our internal assets and capabilities, with the aim of building a balanced portfolio of best-in-class medicines across modalities and development stages in our core therapeutic areas. We are open to finding the best possible deal structure or collaboration model that benefits Galapagos and our stakeholders, with a key focus on expanding and accelerating our pipeline and bringing differentiated medicines to patients. |
| ● | Fostering a strong culture of innovation. Our success is made possible by our incredible teams. Our employees’ relentless drive for innovation, teamwork, and a quality mindset focused on efficiency is what drives our progress. We are committed to creating a purpose-driven, inclusive workplace where our people feel safe and empowered, have opportunities to learn and grow, are recognized for their contributions, and perform at their best as individuals and as a team. |
Life-changing science and innovation
We combine deep disease expertise and multiple drug modalities to accelerate time-to-patients through our internal efforts and focused business development.

CAR-T cell therapy
In 2022, we entered the field of CAR-T and antibody-therapy research and development through the acquisitions of CellPoint (in the Netherlands) and Abound Bio (in the U.S). The transactions provide us with end-to-end capabilities in CAR-T therapy development and offer the potential for a paradigm shift in the space through the implementation of a breakthrough, decentralized manufacturing model and cutting-edge fully human antibody-based capabilities to design next-generation CAR-Ts.
CAR-T cell therapy near the point-of-care
Galapagos is committed to manufacturing personalized cell therapies at or near the point-of-care. Our ambition is to reduce the manufacturing turnaround time significantly from months or weeks to days, ensuring that patients can receive their therapy in a timely manner.
85
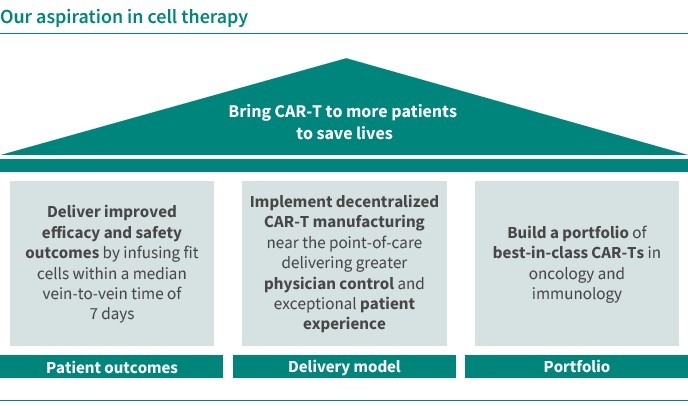
Although current CAR-T cancer therapies have made continued progress, long lead times, costly central manufacturing and complex logistics continue to be limiting factors for large-scale capacity and broad patient access globally.
86
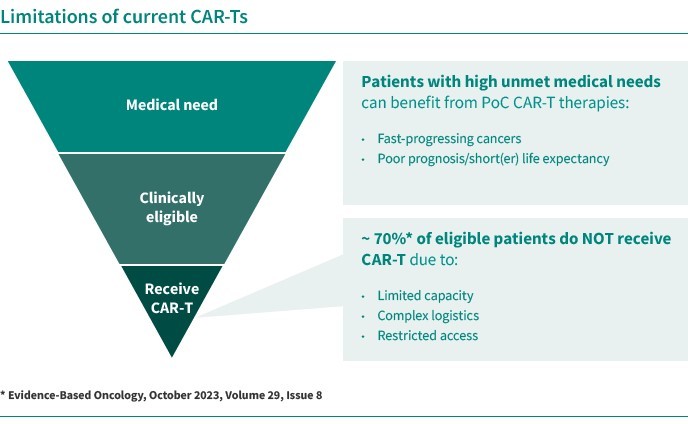
To address these challenges, we are implementing a differentiated, decentralized, point-of-care CAR-T manufacturing platform that has the potential to deliver fresh, fit cells with a seven day vein-to-vein time, enabling greater physician control and a significantly improved patient experience.
This innovative platform consists of an end-to-end xCellit® workflow management and monitoring software system, a decentralized, functionally closed, automated cell therapy manufacturing platform (using Lonza’s Cocoon®) and a proprietary quality control testing and release strategy.
87
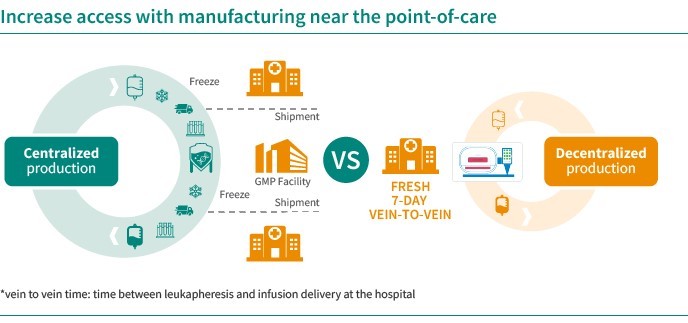
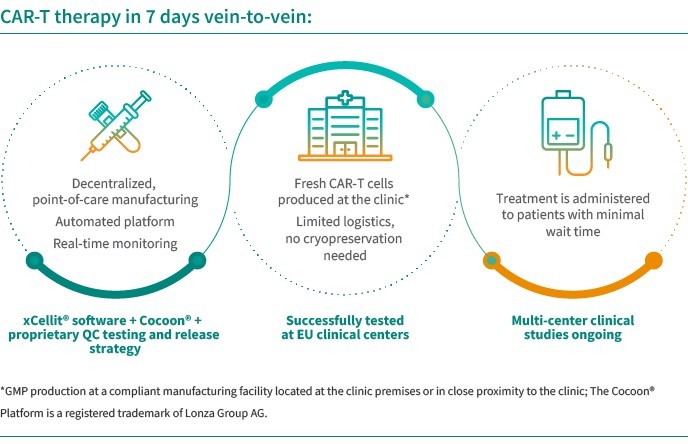
Next-generation CAR-Ts and biologics
Galapagos is developing very large, diverse human antibody libraries in standard fragments of antigen-binding fragment (Fab), single-chain variable fragments (scFv), and unique variable (VH) domain formats. These libraries enable our
88
team to discover novel high affinity binders in multiple formats rapidly (days to weeks), to optimize them for development, and to convert them for multiple applications, including multi-specific CAR-Ts, and fusion proteins.
Our proprietary methodologies have the potential to increase binder diversity, affinity and specificity, and increase the probability of identifying a lead therapeutic antibody candidate.
These unique capabilities enable us to develop next-generation CAR-T therapies that have the potential to transform patient outcomes through potentially more effective and longer-lasting treatment options, even in the event of relapse after prior CAR-T therapy. Together with the decentralized CAR-T manufacturing model, at or near the point-of-care, we aim to expand patient access and ultimately transform patient outcomes.

Small molecule research and precision medicine
In small molecule drug discovery, an assay designed to assess target activity is exposed to large collections of small chemical molecules, allowing the identification of chemical structures that interact with the target to block or activate its activity, resulting in the target's modulation in the cells and prevention of disease-causing effects.
Since our founding, we have built extensive expertise in small molecule research and development, and we are applying our small molecule approach to the discovery and development of potentially best-in-class precision medicines in our core therapeutic areas of oncology and immunology.
Our in-house capabilities include chemical library development, high throughput screening, pharmacology, and preclinical development with the goal of accelerating the time from target identification to first-in-human clinical development. In addition, we have access to the innovative research and drug discovery capabilities and expertise of NovAliX (France) through a five-year collaboration. We are actively building a deep, early-stage small molecule pipeline addressing multiple targets across a range of indications in our two therapeutic areas.
89
Intellectual property
The proprietary nature of, and protection for, our product candidates, their methods of use, and our platform technologies are an important part of our strategy to develop and commercialize novel medicines. We have obtained patents relating to certain of our product candidates, and are pursuing additional patent protection for them and for our other product candidates and technologies. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. Additionally, we have registered and unregistered trademarks, including amongst others our company name.
Our success will depend significantly on our ability to obtain and maintain patent and other proprietary protection for commercially important products, technologies, inventions and know-how related to our business and our ability to defend and enforce our patents, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen and maintain the proprietary position of our development programs.
As of March 1, 2024, patent rights held by Galapagos NV relating to our product candidates include the following:
GLPG5101 product candidate: GLPG5101 is currently being developed in our point-of-care model for the treatment of relapsed/refractory NHL. For this model, we have obtained an exclusive worldwide license from Lonza AG to use the Cocoon® for the commercial manufacture of cell therapy for the treatment of hematological malignancies at the point-of-care.
90
GLPG5201 product candidate: GLPG5201 is currently being developed in our point-of-care model for the treatment of relapsed/refractory CLL and RT For this model, we have obtained an exclusive worldwide license from Lonza AG to use the Cocoon® for the commercial manufacture of cell therapy for the treatment of hematological malignancies at the point-of-care. We also have a license and supply agreement on the materials to produce and use our GLPG5201 product candidate.
GLPG5301 product candidate: GLPG5301 is currently being developed in our point-of-care model for the treatment of relapsed/refractory MM. For this model, we have obtained an exclusive worldwide license from Lonza AG to use the Cocoon® for the commercial manufacture of cell therapy for the treatment of hematological malignancies at the point-of-care. We also have an exclusive license and supply agreement on the materials to produce and use our GLPG5301 product candidate.
GLPG3667 product candidate: We have a granted U.S. patent application, and one pending U.S. patent application. We have one patent granted via the European Patent Office (EPO) and one pending patent application at the EPO; as well as further granted patents inter alia in Japan and Australia. In addition, we have counterpart foreign patent applications that are pending in Canada, China and other foreign countries claiming GLPG3667 compositions of matter and methods of treatment using GLPG3667. Patents, if any, that issue based on this pending patent application are estimated to expire in 2038, not including any potential extensions for the marketed product that may be available via supplementary protection certificates or patent term extensions. We also have one U.S. pending patent application as well as other foreign jurisdictions claiming dosage regimen, and any patent, if granted is estimated to expire in 2042. Finally, we have four pending applications under the Patent Cooperation Treaty (PCT) disclosing solid forms, metabolites, and/or methods for treating inflammatory disorders using GLPG3667; any patents, if granted, based on these patent applications are estimated to expire in 2043.
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the date of filing the application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the United States Patent and Trademark Office, or USPTO, in granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier-filed co-owned patent. In addition, in certain instances, a patent term can be extended to recapture a portion of the term effectively lost as a result of the FDA regulatory review period. However, the restoration period cannot be longer than five years and the total patent term including the restoration period must not exceed 14 years following FDA approval. In certain foreign jurisdictions similar extensions as compensation for regulatory delays are also available. The actual protection afforded by a patent varies on a claim by claim and country to country basis for each applicable product and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory related extensions, the availability of legal remedies in a particular country, and the validity and enforceability of the patent.
Furthermore, the patent positions of biotechnology and pharmaceutical products and processes that we intend to develop and commercialize are generally uncertain and involve complex legal and factual questions. No consistent policy regarding the breadth of claims allowed in such patents has emerged to date in the United States. The patent situation outside the United States is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the United States and other countries can diminish our ability to protect our inventions and enforce our intellectual property rights; more generally, changes could affect the value of intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents.
The biotechnology and pharmaceutical industries are characterized by extensive litigation regarding patents and other intellectual property rights. Our ability to maintain and solidify our proprietary position for our product candidates and technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any of the patent applications that we may file or license from third parties will result in the issuance of any patents. The issued patents that we own or may receive in the future, may be challenged, invalidated or circumvented, and the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology. Furthermore, our competitors may be able to independently develop and commercialize similar drugs or duplicate our technology, business model, or strategy, without infringing our patents. Because of the extensive time required for clinical development and regulatory review of
91
a drug we may develop, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of any such patent.
We may rely, in some circumstances, on trade secrets and unpatented know-how to protect our technology. However, trade secrets can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our consultants, scientific advisors, and contractors and invention assignment agreements with our employees. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our consultants, contractors or collaboration partners use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Our commercial success will also depend in part on not infringing the proprietary rights of third parties. It is uncertain whether the issuance of any third-party patent would require us to alter our development or commercial strategies, or our product candidates or processes, or obtain licenses or cease certain activities. Our breach of any license agreements or failure to obtain a license to proprietary rights that we may require to develop or commercialize our product candidates may have a material adverse impact on us. If third parties have prepared and filed patent applications in the United States that also claim technology to which we have rights, we may have to participate in interference proceedings in the USPTO to determine priority of invention if the patent applications were filed before March 16, 2013, or in derivation proceedings to determine inventorship for patent applications filed after such date.
In addition, substantial scientific and commercial research has been conducted for many years in the areas in which we have focused our development efforts, which has resulted in third parties having a number of issued patents and pending patent applications relating to such areas. Patent applications in the United States and elsewhere are generally published only after 18 months from the priority date. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made. Therefore, patent applications relating to drugs similar to our current product candidates and any future drugs or discoveries and technologies we might develop may have already been filed by others without our knowledge. For more information on these and other risks related to intellectual property, see “Item 3.D.—Risk Factors—Risks Related to Our Intellectual Property.”
Collaborations
We have entered into multiple collaboration agreements with pharmaceutical partners, which have generated €4,958.3 million ($5,478.8 million converted at EUR/USD closing rate on December 31, 2023) in cash through December 31, 2023 to fund discovery and development. We expect to continue to collaborate selectively with pharmaceutical and biotechnology companies to leverage our discovery platform and accelerate product candidate development. Our current alliances include the alliances with Gilead and the restructured alliance with AbbVie.
On July 20, 2022 our exclusive Collaboration and License agreement with Molecure (formerly known as Oncoarendi Therapeutics) terminated.
Option, License and Collaboration Agreement with Gilead
In July 2019, we entered into a 10-year global research and development collaboration with Gilead. We closed the transaction on August 23, 2019.
Upon closing of the option, license and collaboration agreement, we received an upfront payment of $3.95 billion and a €960 million ($1.1 billion) equity investment from Gilead. Under the terms of its equity investment, Gilead nominated two individuals to our Board of Directors, Dr. Linda Higgins and Mr. Daniel O'Day.
92
Under the terms of the option, license and collaboration agreement, Gilead received (a) an exclusive research and development license for Gilead to conduct certain contributions contemplated by the license and collaboration agreement and (b) an option to acquire exclusive commercial licenses in all countries outside of Europe to all current and future clinical programs of Galapagos (other than filgotinib, which is already subject to an existing collaboration between the parties, and certain other programs already committed to other companies) being developed during the 10-year initial option term of the collaboration (subject to extension in certain circumstances). Under the option, license and collaboration agreement, we will continue to lead and fund all discovery and development of our programs until the end of the relevant Phase 2 clinical trials. After the completion of the relevant Phase 2 clinical study for each program, Gilead will have the option to acquire an exclusive commercial license to that program in all countries outside of Europe. If the option is exercised, Gilead and we will co-develop the compound and share costs equally.
In addition, under the option, license and collaboration agreement, Gilead was deemed to have exercised its option, and an exclusive commercial license was granted in all countries outside of Europe, to ziritaxestat, our Phase 3 candidate for idiopathic pulmonary fibrosis. US approval of ziritaxestat would have entitled us to an additional $325 million regulatory milestone fee. However, we and Gilead announced in February 2021 that all ongoing development activities with ziritaxestat would be discontinued.
For GLPG1972, a drug candidate resulting from our osteoarthritis collaboration with Servier that was subject to separate option and milestone payments under the option, license and collaboration Agreement, Gilead declined to exercise its option under the agreement in November 2020.
For all other programs included in the option, license and collaboration agreement, Gilead will make a $150 million opt-in payment per program with no subsequent milestones if Gilead decides to exercise its option. If Gilead declines to exercise its option with respect to a program, such program shall no longer be subject to the option, license and collaboration agreement and we may progress the program independently.
In addition, we will receive tiered royalties ranging from 20-24% on net sales of all products from all programs licensed by Gilead in all countries outside of Europe as part of the option, license and agreement subject to customary royalty terms and adjustments.
The collaboration is managed by a set of joint committees comprised of equal numbers of representatives from each of us and Gilead. The joint steering committee monitors and provides strategic oversight of the activities under the collaboration and facilitates communications between the parties. The joint development committee oversees and coordinates the development of the licensed products. The joint commercialization committee will oversee commercialization of licensed products. The joint communication review committee will oversee publications and other public communications related to licensed products.
Upon Gilead’s exercise of its option with respect to any of our programs, Gilead will assume responsibility for seeking regulatory approval for the optioned product and for all regulatory matters in its territory. Each party will be solely responsible for all commercialization activities and costs for the optioned product in its territory.
Upon termination of the option, license and collaboration agreement with respect to any program licensed by Gilead, all rights and licenses granted by us will terminate, and we will obtain an exclusive, perpetual and irrevocable license from Gilead under certain intellectual property rights to exploit the licensed product that is the subject of development or commercialization at the time of termination in the field in the applicable terminated region (provided that if such termination is the result of our material breach, such license will be royalty-bearing). Either we or Gilead may terminate the option, license and collaboration agreement for the other party’s uncured material breach. Either we or Gilead may terminate the option, license and collaboration agreement in the event of specified insolvency events involving the other party. Gilead may also terminate the option, license and collaboration agreement in its entirety or on a program-by-program and country-by-country basis with advance notice for convenience.
The option, license and collaboration agreement also contains customary provisions including representations and warranties of the parties, terms as to governance of the collaboration, commercialization and regulatory responsibilities of the parties, and manufacturing and supply.
93
Either party may, without the consent of the other party, assign the option, license and collaboration agreement to an affiliate or successor. If we undergo a change in control, all intellectual property of our acquirer or that becomes owned or controlled by our acquirer after such change of control shall be excluded from the scope of rights granted in the option, license and collaboration agreement.
In connection with entering into the option, license and collaboration agreement, we amended certain terms of our existing agreement with Gilead governing filgotinib, and in December 2020 and September 2021, and in October 2023, we agreed with Gilead to further amend such agreement, as further described in “Item 4 – Collaborations -- Exclusive collaboration agreement with Gilead for filgotinib.”
Exclusive collaboration agreement with Gilead for filgotinib
In December 2015, we entered into a global collaboration agreement with Gilead to develop and commercialize filgotinib for the treatment of inflammatory indications. In connection with entering into the option, license and collaboration agreement with Gilead, in August 2019 we amended and restated this agreement to increase our involvement in filgotinib’s global strategy and participate more broadly in the commercialization of filgotinib in Europe. In December 2020, we agreed to amend this agreement again, as a result of which we have assumed all development, manufacturing, commercialization and certain other rights for filgotinib in Europe through a transition largely completed at the end of 2021 and fully completed by the end of 2022. Gilead retains commercial rights and remains marketing authorization holder for filgotinib outside of Europe, including in Japan.
In connection with our entry into the collaboration agreement, we received in January 2016 an upfront payment of $725 million consisting of a onetime, non-refundable, non-creditable license fee in the amount of $300 million and a $425 million equity investment. In November 2016, Gilead initiated a Phase 3 trial in CD, for which we received a $50.0 million payment. In December 2016, Gilead initiated a Phase 2 trial in UC for which we received a $10.0 million payment. In April 2017, Galapagos initiated a Phase 2 trial in psoriatic arthritis as a new indication, for which we received a $10.0 million payment. In May 2018, Gilead initiated a phase 3 trial in UC for which we received $15.0 million. In December 2019, Gilead initiated a Phase 3 trial in psoriatic arthritis as a new indication, for which we received $10.0 million (€9.1 million). Also in December 2019, Gilead filed an NDA for filgotinib in the U.S. for which we received a $20 million payment in January 2020. In September 2020, Gilead obtained marketing authorization for filgotinib in Europe and Japan for which we received an aggregate payment of $105.0 million (€90.2 million) payment in October 2020. In connection with the agreement that we entered into with Gilead pursuant to the binding term sheet entered into in December 2020 to amend the existing arrangement for the commercialization and development of filgotinib, Gilead has agreed to irrevocably pay Galapagos €160 million, subject to certain adjustments for higher than budgeted development costs. Gilead paid €35 million in January 2021 an additional €75 million in April 2021 and €50 million in 2022. In addition, we will no longer be eligible to receive any future milestone payments relating to filgotinib in Europe. However, we will remain eligible to receive tiered royalty percentages ranging from 20% to 30% on Gilead's global net sales of filgotinib outside of Europe and future development and regulatory milestone-based payments of up to $275 million and sales-based milestone payments of up to $600 million. All payments by Gilead to us are made in U.S. dollars.
Under the terms of the collaboration, Gilead is primarily responsible for seeking regulatory approval of filgotinib in countries outside of Europe. Pursuant to the amended arrangements agreed in December 2020, we are responsible for commercializing filgotinib in Europe.
Under the amended and restated filgotinib agreement, we agreed on a 50% / 50% cost split for development costs of filgotinib, in lieu of the 20% (us) /80% (Gilead) cost split under the original filgotinib agreement. Beginning on January 1, 2021, we bore the development costs for certain studies, in lieu of the equal cost split contemplated by the 2019 agreement. These studies include the DARWIN3, FINCH4, FILOSOPHY, and Phase 4 studies and registries in RA, MANTA and MANTA-RAy, the PENGUIN1 and 2 and EQUATOR2 studies in PsA, the SEALION1 and 2 studies in AS, the HUMBOLDT study in uveitis in addition to other clinical and non-clinical expenses supporting these studies and support for any investigator sponsored trials in non-IBD conditions and non-clinical costs on all current trials. The existing 50/50 global development cost sharing arrangement will continue for the following studies: SELECTION and its long-term extension study (LTE) in UC, DIVERSITY and its LTE, DIVERGENCE 1 and 2 and their LTEs and
94
support for Phase 4 studies and registries in Crohn’s disease, pediatric studies and their LTEs in RA, UC and Crohn’s disease, and support for investigator sponsored trials in IBD. Under the 2021 amendment, the 50/50 development cost sharing arrangement ceased for DIVERSITY and its LTE starting from April 1, 2022. As from that date, Galapagos was solely responsible for all development costs. In consideration for Galapagos assuming responsibility for the DIVERSITY study, Gilead made a one-time payment to Galapagos of $15 million in 2022.
The original filgotinib agreement included a co-promotion / co-commercialization option for filgotinib, which we exercised with respect to eight European countries in December 2017. We agreed in December 2020 with Gilead to transfer the sole right to commercialize filgotinib in Europe to us after a transition period, pursuant to which most activities were transferred to us by December 31, 2021 and that we intend to complete by December 31, 2022. Until December 31, 2021, we continued to share equally with Gilead in the net profit and net losses in each of the Netherlands, Belgium, Luxembourg, France, Germany, Italy, Spain and UK. During this period, this profit and loss sharing replaces our right to receive royalties with respect to filgotinib sales by Gilead in these countries. All commercial economics on filgotinib in Europe transferred to us as of January 1, 2022, subject to payment of tiered royalties of 8 to 15 percent of net sales in Europe to Gilead, starting in 2024.
Gilead will retain sole responsibility for commercializing filgotinib outside of Europe. We will be eligible to receive tiered royalty percentages ranging from 20% to 30% on Gilead’s global net sales of filgotinib outside of Europe. The royalties payable to us under the filgotinib agreement may be reduced under certain circumstances. Our right to receive royalties under the filgotinib agreement continues, on a country-by-country basis, until the later to occur of certain specified events.
Under the amended and restated collaboration agreement, the collaboration was managed by a set of joint committees comprised of equal numbers of representatives from each of us and Gilead. The joint steering committee monitors and provides strategic oversight of the activities under the collaboration and facilitates communications between the parties. The joint development committee oversees and coordinates the development of filgotinib. The joint commercialization committee will oversee commercialization of filgotinib globally, and the shared territory joint commercialization committee will coordinate and integrate the activities of, and facilitate the communication and exchange of information between, us and Gilead with respect to the co-commercialization of filgotinib. Gilead and Galapagos will jointly prepare the global commercialization strategy. Under the transition and amendment agreement following the 2020 amendment, the joint commercialization committee and the shared territory joint commercialization committee were disbanded and the transition steering committee and joint transition team were created to oversee the transition activities. Under the second amended and restated collaboration agreement following the 2020 amendment, the transition steering committee and joint transition team were disbanded and the Jyseleca Scientific Publication Team and the Communication Points of Contact were created to facilitate the parties publication and communication activities. The filgotinib agreement, as amended, will expire on a country-by-country basis at the end of the royalty term in such country. Upon expiration of the royalty term, the licenses will become fully-paid, perpetual and irrevocable. Either we or Gilead may terminate the filgotinib agreement for the other party’s uncured material breach. Either we or Gilead may terminate the filgotinib agreement in the event of specified insolvency events involving the other party. Gilead may also terminate the filgotinib agreement in its entirety for convenience following a certain period, upon prior written notice.
If the collaboration agreement terminates in its entirety for any reason, all rights and licenses granted by either party will terminate, and we will obtain an exclusive, perpetual, irrevocable, royalty-bearing license from Gilead under certain intellectual property rights to exploit filgotinib. If the filgotinib agreement is terminated in a specific territory, all rights and licenses granted by us will be deemed to be amended not to include such territory, and we will have a corresponding license with respect to such terminated country. The filgotinib agreement also contains other termination rights specified therein.
Either party may, without the consent of the other party, assign the filgotinib agreement to an affiliate or successor. Any other assignment requires written consent of the other party. However, with respect to an assignment to an affiliate, the assigning party will remain bound by the terms of the filgotinib agreement.
In September 2021, Gilead and Galapagos agreed to further amend the collaboration. Following such amendment, Galapagos assumed sponsorship of and operational and financial responsibility for the ongoing DIVERSITY clinical
95
study, evaluating filgotinib in CD, and its long-term extension study. The transfer was intended to be completed by June 30, 2022 and completed by March 2023. Under the terms of the agreement, Gilead made a one-time payment of $15 million to Galapagos in consideration for Galapagos assuming responsibility for the DIVERSITY clinical study. From April 1, 2022, Galapagos was also solely responsible for all development costs for the DIVERSITY clinical study. In addition, if the EMA grants regulatory approval of filgotinib for the treatment of CD based on data from the DIVERSITY trial, then royalties payable by Galapagos to Gilead will be reduced by 30% across all filgotinib indications and will become 5.6 to 10.5% of net sales in Europe. On February 8, 2023, Galapagos announced that it decided not to submit a Marketing Authorization Application in Europe based on topline data from the DIVERSITY study and as a result, these adjustments to the royalties will not made. These royalties are payable as of 2024. Gilead remains responsible for commercial activities outside of Europe.
In March 2022, Gilead and Galapagos agreed to further amend the collaboration. Following such amendment, Galapagos assumed sponsorship of and operational responsibility for the MANTA study and its long-term extension. The transfer was largely completed by December 31, 2022.
On March 28, 2022 filgotinib was approved by the Japanese Ministry of Health, Labour and Welfare for UC, for which we received a $20.0 million (€18.2 million) regulatory milestone payment from Gilead in May 2022.
Also in March 2022, Gilead and Galapagos agreed to further amend the collaboration by adding the following countries to the Galapagos territory: Andorra, San Marino, Monaco, and Vatican City.
In October 2023, Gilead and Galapagos agreed to amend the collaboration to terminate the existing 50/50 global development cost sharing arrangement with Galapagos bearing the costs going forward, and to terminate Galapagos’ obligation to pay tiered royalties to Gilead on net sales of Jyseleca® in Europe, in addition to other amendments, including a simplification of the governance and certain other technical amendments.
Effective January 31, 2024, following the closing of the transaction between Galapagos and Alfasigma S.p.A. to transfer the Jyseleca® business to Alfasigma S.p.A., Galapagos assigned its rights and obligations under the filgotinib collaboration to Alfasigma S.p.A., except for Galapagos’ right to receive royalties from Gilead on net sales in the Gilead Territory under a separate agreement between Gilead and Galapagos entered into in October 2023.
Distribution agreement for Jyseleca with Sobi
In October 2021, we signed an agreement (as amended from time to time) with Swedish Orphan Biovitrum AB (‘Sobi’) regarding the distribution of Jyseleca®. Sobi acts as our distribution and commercialization partner of Jyseleca® and will distribute the medicine in Central and Eastern Europe, Greece, Portugal, and the Baltic countries. Launches or first sales of Jyseleca in the aforementioned countries trigger milestone payments. In 2022, we recorded milestones of €2.0 million triggered by the first sale of Jyseleca in the Czech Republic and Portugal by Sobi.
Effective January 31, 2024, following the closing of the transaction between Galapagos and Alfasigma S.p.A. to transfer the Jyseleca® business to Alfasigma S.p.A., Galapagos assigned its rights and obligations under the Sobi agreement to Alfasigma S.p.A.
Drug discovery collaboration transaction with NovAliX
Effective July 1, 2023, we entered into an integrated drug discovery collaboration with NovAliX, a drug-discovery focused Contract Research Organization based in Strasbourg, France. Under the terms of the agreement, Galapagos’ drug discovery and research activities conducted in Romainville, France, and Galapagos’ employees in Romainville, which were exclusively dedicated to the operation of these activities, were transferred to NovAliX who will assume all ongoing research and discovery activities in Romainville, and this for no consideration. In return, Galapagos is committed to utilizing the research capabilities and expertise of NovAliX through a five year-collaboration and within the context of the company’s R&D portfolio, during which Galapagos is committed to purchase for a total of €73.8 million services from NovAliX.
96
Seasonality
Our business is currently not materially affected by seasonality.
Manufacturing and supply
We currently do not own or operate manufacturing facilities for the production of product candidates for preclinical, clinical, or commercial use.
For non-cell therapy products, we currently outsource to a limited number of external service providers the production of all drug substances and drug products, and we expect to continue to do so to meet the preclinical and clinical requirements of our product candidates and for the production in support of our commercial sales. We have framework agreements with most of our external service providers, under which they generally provide services to us on a project-by-project basis. For our point-of-care cell therapy products, we are establishing a network of decentralised production sites at or near the hospitals, which will manufacture and test our autologous CAR-T product candidates. With each point-of-care site we need to establish the necessary agreements to manufacture and test these product candidates, and oversee the GMP activities performed at these contracted sites.
Our drug raw materials which support our clinical trials are manufactured by multiple suppliers. We have agreements for the supply of such drug materials with manufacturers or suppliers that we believe have sufficient capacity to meet our demands. In addition, we believe that adequate alternative sources for such supplies exist. However, there is a risk that, if supplies are interrupted, it would materially harm our business. We typically order raw materials and services on a purchase-order basis, and do not enter into long-term dedicated capacity or minimum supply arrangements. To date, the prices of our principal raw materials have not been volatile.
Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements, which govern record keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. The contract manufacturing organizations we use to manufacture our product candidates operate under current good manufacturing practice, or cGMP, conditions. cGMPs are regulatory requirements for the production of pharmaceuticals that will be used in humans. For most of our manufacturing processes a back-up GMP manufacturer is in place or can easily be identified.
Competition
Our industry is highly competitive and subject to rapid and significant change. While we believe that our development and commercialization experience, scientific knowledge, and industry relationships provide us with competitive advantages, we face competition from pharmaceutical, medical device, and biotechnology companies, including specialty pharmaceutical companies, generic drug companies, academic institutions, government agencies, and research institutions.
In the field of dermatomyositis (DM), physical therapy, exercise and medication including corticosteroids, immunosuppressants or recently immunoglobulin treatment, are commonly used to treat DM. Treatment of this disease has relied for many years on off-label medication - in 2021 the FDA approved immunoglobulin treatment Octagam®, based on the Phase 3 ProDerm trial of Octapharma.
In the field of SLE, corticosteroids, antimalarials and immunosuppressants are commonly used to control lupus disease activity. Only two products are currently approved to treat SLE, both as add-on to standard therapy: Belimumab (Benlysta®) (anti-BAFF) from GSK and recently anifrolumab (Saphnelo®) (anti-IFN) from AstraZeneca. There are currently over 10 products in Phase 3 for SLE, of which the minority are oral therapies – including deucravacitinib (SotyktuTM) (TYK2) from BMS, upadacitinib (JAK) from Abbvie and cenerimod (S1P1) from Idorsia/Viatris.
In the field of hematologic malignancies, such as Non-Hodgkin’s Lymphoma (NHL), Chronic Lymphocytic Leukemia (CLL) and Multiple Myeloma (MM), there are many approved therapies or therapies in
97
development (including but not limited to chemotherapy, BTKi, antibodies, bispecific antibodies, antibody drug conjugates, CAR-Ts, cytokines, NK and T-cell engagers, etc.) and many different types of cell therapy in development (allogeneic/autologous, CAR-T/CAR-NK, TIL, TCR-T, dendritic, etc.). As a consequence, we are operating in a highly competitive, and rapidly evolving environment. New technologies and therapies such as in vivo modification of immune cells may further disrupt this market in the mid-to-long-term. Six CAR T treatments have been approved for hematological cancers in the US and Europe: Novartis’ Kymriah® (CD19 CAR T), Gilead/Kite’s Yescarta® (CD19 CAR T), Tecartus® (CD19 CAR T), J&J’s Carvykti® (BCMA CAR T) BMS’ Breyanzi® (CD19 CAR T) and Abecma® (BCMA CAR T).
Many of our competitors have significantly greater financial, technical, and human resources than we have. Mergers and acquisitions in the pharmaceutical, medical device and biotechnology industries may result in even more resources being concentrated among a smaller number of our competitors. Our commercial opportunity could be reduced or eliminated if our competitors develop or market products or other novel therapies that are more effective, safer, or less costly than our current or future product candidates, or obtain regulatory approval for their products more rapidly than we may obtain approval for our product candidates. Our success will be based in part on our ability to identify, develop, and manage a portfolio of product candidates that are safer and more effective than competing products.
Government regulation
Government regulation and product approval
Government authorities in the United States at the federal, state, and local level, and in other countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, marketing, export, and import of products such as those we are developing. In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (the FDCA) and related regulations, and biological products under the FDCA and the Public Health Service Act (the PHS Act) and related regulations. Drugs and biological products also are subject to other federal, state, local and foreign statutes and regulations.
U.S. regulation
U.S. drug development and biological product process
The process of obtaining regulatory approvals and compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, or license revocation, a clinical hold, untitled or warning letters, voluntary or mandatory product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. The process required by the FDA before a drug or biological product may be marketed in the United States generally involves the following:
| ● | completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices regulations; |
| ● | submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
| ● | performance of adequate and well-controlled human clinical trials according to Good Clinical Practices, or GCP, to establish the safety and efficacy of the proposed drug or biological product for its intended use; |
| ● | preparation and submission to the FDA of a new drug application, or NDA, or biologics license application, or BLA; |
98
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product candidate, or components thereof, are produced to assess compliance with cGMP; |
| ● | potential FDA audit of the clinical trial sites that generated the data in support of the NDA or BLA; and |
| ● | FDA review and approval of the NDA or BLA. |
The testing and approval process requires substantial time, effort, and financial resources and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
Once a pharmaceutical or biological product candidate is identified for development, it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity, formulation and stability, as well as animal studies. An IND sponsor must submit the results of the preclinical tests together with manufacturing information, analytical data, and any available clinical data or literature, to the FDA as part of the IND. The sponsor must also include a protocol detailing, among other things, the objectives of the initial clinical trial, dosing procedures, subject selection, and exclusion criteria, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the initial clinical trial lends itself to an efficacy evaluation. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions related to a proposed clinical trial and places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during clinical trials due to safety concerns or non-compliance, and may be imposed on all product candidates within a certain class. The FDA also can impose partial clinical holds, for example, prohibiting the initiation of clinical trials of a certain duration or for a certain dose.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCP regulations. These regulations include the requirement that all research subjects provide informed consent in writing before their participation in any clinical trial. Further, an institutional review board, or IRB, must review and approve the plan for any clinical trial before it commences at any institution, and the IRB must conduct continuing review and reapprove the study at least annually. An IRB considers, among other things, whether the risks to individuals participating in the clinical trial are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject or his or her legal representative, and must monitor the clinical trial until completed. Each new clinical protocol and any amendments to the protocol must be submitted for FDA review, and to the IRBs for approval.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| ● | Phase 1. The product candidate is initially introduced into a small number of healthy human subjects or patients and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion and, if possible, to gain early evidence on effectiveness. In the case of some product candidates for severe or life- threatening diseases, especially when the product candidate is suspected or known to be unavoidably toxic, the initial human testing may be conducted in patients. |
| ● | Phase 2. Involves clinical trials in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product candidate for specific targeted diseases and to determine dosage tolerance and optimal dosage and schedule. |
| ● | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit relationship of the product candidate and provide an adequate basis for physician labeling. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA. Within 15 calendar days after the sponsor determines that the information qualifies for reporting, written IND safety reports
99
must be submitted to the FDA and the investigators for serious and unexpected suspected adverse events, findings from other studies or animal or in vitro testing that suggest a significant risk to humans, and any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. Phase 1, Phase 2, and Phase 3 testing may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements, or if the product candidate has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the composition and physical characteristics of the product candidate and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
A drug or biological product being studied in clinical trials may be made available to individual patients in certain circumstances. Pursuant to the 21st Century Cures Act, or Cures Act, as amended, the manufacturer of an investigational drug or biological product for a serious disease or condition is required to make available, such as by posting on its website, its policy on evaluating and responding to requests for individual patient access to such investigational product. This requirement applies on the earlier of the first initiation of a Phase 2 or Phase 3 trial of the investigational product, or, as applicable, 15 days after the drug or biological product candidate receives a designation as a breakthrough therapy, fast track product, or regenerative medicine advanced therapy. Further, the Right to Try Act of 2017, among other things, provides a federal framework for certain patients to request access to certain investigational products that have completed a Phase 1 clinical trial and that are undergoing investigation for FDA approval. There is no obligation for a pharmaceutical or biological product manufacturer to make its investigational products available to eligible patients as a result of the Right to Try Act.
U.S. review and approval processes
The results of product development, preclinical studies, and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the product candidate, proposed labeling, and other relevant information, are submitted to the FDA as part of an NDA for a new drug or BLA for biological product, requesting approval to market the product candidate. The submission of an NDA and BLA is subject to the payment of a substantial user fee; although a waiver of such fee may be obtained under certain limited circumstances. For example, the agency will waive the application fee for the first NDA or BLA that a small business or its affiliate submits for review. The sponsor of an approved NDA and BLA is also subject to an annual prescription drug product program fee.
The FDA reviews all NDAs and BLAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. The FDA may request additional information rather than accept an NDA or BLA for filing. In this event, the NDA or BLA must be re-submitted with the additional information. The re-submitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an indepth substantive review. The FDA reviews an NDA to determine, among other things, whether a drug is safe and effective for its intended use, and the FDA reviews a BLA to determine, among other things, whether a biological product is safe, pure and potent for its intended use. For both NDAs and BLAs, the FDA also reviews whether the manufacturing is cGMP-compliant to assure the product’s identity, strength, quality, and purity. Before approving an NDA or BLA, the FDA typically will inspect the facility or facilities where the product is or will be manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The FDA may refer the NDA or BLA to an advisory committee for review, evaluation, and recommendation as to whether the application should be approved and under what conditions. An advisory committee is
100
a panel of experts, including clinicians and other scientific experts, who provide advice and recommendations when requested by the FDA. The FDA is not bound by the recommendation of an advisory committee, but it considers such recommendations when making decisions.
The approval process is lengthy and difficult and the FDA may refuse to approve an NDA or BLA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information. Even if such data and information are submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA or BLA in its present form. The complete response letter usually describes all of the specific deficiencies that the FDA identified in the NDA or BLA that must be satisfactorily addressed before it can be approved. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit the NDA or BLA, addressing all of the deficiencies identified in the letter, or withdraw the application, or request an opportunity for a hearing.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages, or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings, or precautions be included in the product labeling. In addition, the FDA may require post-approval studies, including Phase 4 clinical trials, to further assess a product’s safety and effectiveness after NDA or BLA approval and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. As part of the NDA or BLA, the FDA also may require the submission of a risk evaluation and mitigation strategy, or REMS, to ensure that the benefits of the product outweigh the risks of the product. The REMS plan could include medication guides, physician communication plans, and elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools. An assessment of the REMS must be conducted at set intervals. Following product approval, a REMS also may be required by the FDA if new safety information is discovered and the FDA determines that a REMS is necessary to ensure that the benefits of the product outweigh the risks of the product.
Expedited programs
Fast track designation
The FDA has a fast track program that is intended to expedite or facilitate the process for reviewing new drugs or biological products that meet certain criteria. Specifically, new product candidates are eligible for fast track designation if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the condition. Fast track designation applies to the combination of the product candidate and the specific indication for which it is being studied. The sponsor of a new drug or biological product may request the FDA to designate the drug or biological product as a fast track product concurrently with, or at any time after, submission of an IND, and the FDA must determine if the product candidate qualifies for fast track designation within 60 days of receipt of the sponsor’s request.
In addition to other benefits, such as the ability to engage in more frequent interactions with the FDA, the FDA may initiate review of sections of a fast track drug’s NDA or BLA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of each portion of the NDA or BLA and the applicant pays applicable user fees. However, the FDA’s time period goal for reviewing an application does not begin until the last section of the NDA or BLA is submitted. Additionally, the fast track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
101
Accelerated approval
Under FDA’s accelerated approval regulations, the FDA may approve a drug or biological product for a serious or life-threatening illness that provides meaningful therapeutic benefit to patients over existing treatments based upon a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. In clinical trials, a surrogate endpoint is a marker, such as a measurement of laboratory or clinical signs of a disease or condition that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. A product candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of post-approval clinical trials sometimes referred to as Phase 4 trials to confirm the effect on the clinical endpoint. Under the Food and Drug Omnibus Reform Act of 2022, or FDORA, the FDA is now permitted to require, as appropriate, that such trials be underway prior to approval or within a specific time period after the date of approval for a product granted accelerated approval. FDORA also requires sponsors to send updates to the FDA every 180 days on the status of such studies, including progress toward enrollment targets, and the FDA must promptly post this information publicly. FDORA also gives the FDA increased authority to withdraw approval of a drug or biologic granted accelerated approval on an expedited basis if the sponsor fails to conduct such studies in a timely manner or send the necessary updates to the FDA, or if such post-approval studies fail to verify the drug’s predicted clinical benefit. Under FDORA, the FDA also is empowered to take action, such as issuing fines, against companies that fail to conduct with due diligence any post-approval confirmatory study or submit timely reports to the agency on their progress. All promotional materials for product candidates approved under accelerated regulations are subject to prior review by the FDA.
Breakthrough designation
The FDA expedites the development and review of a breakthrough therapy. A drug or biological product can be designated as a breakthrough therapy if it is intended to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that it may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. A sponsor may request that a drug or biological product be designated as a breakthrough therapy concurrently with, or at any time after, the submission of an IND, and the FDA must determine if the product candidate qualifies for breakthrough therapy designation within 60 days of receipt of the sponsor’s request. If so designated, the FDA shall act to expedite the development and review of the product candidate’s marketing application, including by meeting with the sponsor throughout the product candidate’s development, providing timely advice to the sponsor to ensure that the development program to gather preclinical and clinical data is as efficient as practicable, involving senior managers and experienced review staff in a cross-disciplinary review, assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor, and taking steps to ensure that the design of the clinical trials is as efficient as practicable.
Regenerative medicine advanced therapy designation
As part of the 21st Century Cures Act, Congress amended the FDCA to facilitate an efficient development program for, and expedite review of regenerative medicine advanced therapies, or RMATs, which include cell and gene therapies, therapeutic tissue engineering products, human cell and tissue products, and combination products using any such therapies or products. RMATs do not include those human cells, tissues, and cellular and tissue based products regulated solely under section 361 of the PHSA and 21 CFR Part 1271. This program is intended to facilitate efficient development and expedite review of regenerative medicine therapies, which are intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition and qualify for RMAT designation. A sponsor may request that the FDA designate a product candidate as a RMAT concurrently with or at any time after submission of an IND. The FDA has 60 calendar days to determine whether the product candidate meets the criteria, including whether there is preliminary clinical evidence indicating that the product candidate has the potential to address unmet medical needs for a serious or life-threatening disease or condition. A BLA for a product candidate that has received RMAT designation may be eligible for priority review or accelerated approval through use of surrogate or intermediate endpoints reasonably likely to predict long-term clinical benefit, or reliance upon data obtained from a meaningful number of sites. Benefits of
102
RMAT designation also include early interactions with FDA to discuss any potential surrogate or intermediate endpoint to be used to support accelerated approval. A product candidate with RMAT designation that is granted accelerated approval and is subject to post-approval requirements may fulfill such requirements through the submission of clinical evidence from clinical studies, patient registries, or other sources of real world evidence, such as electronic health records; the collection of larger confirmatory data sets; or post-approval monitoring of all patients treated with such therapy prior to its approval.
Priority review
Priority review is granted where there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. If a product candidate that contains a new molecular entity is granted priority review, the FDA aims to review the application six months after it accepts the application for filing. If criteria are not met for priority review, the application is subject to the standard FDA review period of ten months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Post-approval requirements
Any products which we receive FDA approval for are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, tracking and tracing requirements, complying with certain electronic records and signature requirements, and complying with FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Products may be promoted only for the approved indications and in accordance with the provisions of the approved label. Further, manufacturers must continue to comply with cGMP requirements, which are extensive and require considerable time, resources, and ongoing investment to ensure compliance. In addition, changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Manufacturers and other entities involved in the manufacturing and distribution of approved products, and those supplying products, ingredients, and components of approved products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage, and shipment of the product. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards, and test each product batch or lot prior to its release. We rely, and expect to continue to rely, on third parties for the production of clinical quantities of our product candidates. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production, or distribution, or may require substantial resources to correct.
The FDA may withdraw a product approval if compliance with regulatory requirements is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures, including a REMS, or the conduct of post-marketing studies to assess a newly discovered safety issue. FDA has authority to require post-market studies, in certain circumstances, on reduced effectiveness of a product, and FDA may require labeling changes related to new reduced effectiveness information. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, untitled or warning letters, holds on clinical trials, voluntary or mandatory product recalls, product seizures, product detention or refusal to permit the import or export of products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, injunctions, or civil or criminal penalties.
103
From time to time, legislation is drafted, introduced, and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing, and marketing of products regulated by the FDA. In addition to new legislation, FDA regulations, guidances, and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our product candidates. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.
Patent term restoration and marketing exclusivity
Depending upon the timing, duration and specifics of FDA approval of the use of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA or BLA plus the time between the submission date of an NDA or BLA and the approval of that application, less any time the applicant did not act with due diligence. Only one patent applicable to an approved drug or biological product is eligible for the extension, and the application for the extension must be submitted prior to the expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent terms extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration dates, depending on the expected length of the clinical trials and other factors involved in the filing of the relevant NDA or BLA; however, there can be no assurance that any such extension will be granted to us.
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or ANDA, or a 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
A biological product is granted twelve years of exclusivity from the time of first licensure of the biological product, which is referred to as a reference biological product. This twelve year exclusivity period was created by the Biologics Price Competition and Innovation Act of 2009, which was part of the Patient Protection and Affordable Care Act of 2010, which created an abbreviated approval pathway for biological products shown to be biosimilar to, or interchangeable with, an FDA-licensed reference biological product. This amendment to the PHS Act attempts to minimize duplicative testing. Biosimilarity, which requires that there be no clinically meaningful differences between the biological product and the reference product in terms of safety, purity, and potency, can be shown through analytical studies, animal studies, and a clinical trial or trials. Interchangeability requires that a biological product is biosimilar to the reference biological product and the product must demonstrate that it can be expected to produce the same clinical results as the reference product and, for products administered multiple times, the product and the reference product may be switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biological product. The first biological product determined to be
104
interchangeable with a branded reference product for any condition of use is also eligible for a period of exclusivity, during which time the FDA may not determine that another product is interchangeable with the same reference product for any condition of use. The FDA may approve multiple “first” interchangeable products so long as they are all approved on the same first day of marketing. This exclusivity period, which may be shared amongst multiple first interchangeable products, lasts until the earlier of: (1) one year after the first commercial marketing of the first interchangeable product; (2) 18 months after resolution of a patent infringement suit instituted under 42 U.S.C. § 262(l)(6) against the applicant that submitted the application for the first interchangeable product, based on a final court decision regarding all of the patents in the litigation or dismissal of the litigation with or without prejudice; (3) 42 months after approval of the first interchangeable product, if a patent infringement suit instituted under 42 U.S.C. § 262(l)(6) against the applicant that submitted the application for the first interchangeable product is still ongoing; or (4) 18 months after approval of the first interchangeable product if the applicant that submitted the application for the first interchangeable product has not been sued under 42 U.S.C. § 262(l)(6). Pediatric exclusivity is another type of exclusivity in the United States. A biological product or drug can obtain pediatric market exclusivity in the United States. Pediatric exclusivity, if granted, adds an additional six months to existing exclusivity periods for all formulations, dosage forms, and indications of the active moiety and, for drugs, patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection and, for drugs, patent term, may be granted based on the voluntary completion of a pediatric study in accordance with an FDA-issued “Written Request” for such a study, provided that at the time pediatric exclusivity is granted there is not less than nine months of term remaining.
Orphan drugs
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drugs or biological products intended to treat a rare disease or condition—generally a disease or condition that affects fewer than 200,000 individuals in the United States or that affects more than 200,000 individuals in the United States and for which there is no reasonable expectation that costs of research and development of the product for the indication can be recovered by sales of the product in the United States. Orphan drug designation must be requested before submitting an NDA or BLA.
After the FDA grants orphan drug designation, the generic identity of the drug or biological product and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. The first NDA or BLA applicant to receive FDA approval for a particular active ingredient to treat a particular disease or condition with FDA orphan drug designation is entitled to a seven-year exclusive marketing period in the United States for that product, for that indication. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA or BLA application user fee.
During the exclusivity period, the FDA may not approve any other applications to market the same drug or biological product for the same disease or condition, except in limited circumstances, such as if the second applicant demonstrates the clinical superiority of its product to the product with orphan drug exclusivity through a demonstration of superior safety, superior efficacy, or a major contribution to patient care. Orphan drug exclusivity does not prevent FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition.
Pediatric information
Under the Pediatric Research Equity Act of 2003, or PREA, as amended, NDAs, BLAs or supplements to NDAs or BLAs must contain data adequate to assess the safety and effectiveness of the drug or biological product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective or the biological product is safe, pure, and potent. The FDCA requires that a sponsor who is planning to submit a marketing application for a product candidate that includes a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration to submit an initial Pediatric Study Plan, or initial PSP, within sixty days of an end-of-phase 2 meeting or as may be agreed between the sponsor and the FDA. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full
105
or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of data or full or partial waivers. The FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon PSP at any time if changes to the pediatric plan need to be considered based on data collected from preclinical studies, early phase clinical trials, and/or other clinical development programs. Generally, the requirements of PREA do not apply to an application to market a drug for an orphan-designated indication.
Disclosure of clinical trial information
Sponsors of clinical trials of FDA-regulated products, including drugs and biological products, are required to register and disclose certain clinical trial information, which is publicly available at www.clinicaltrials.gov. Information related to the product, patient population, phase of investigation, study sites and investigators, and other aspects of the clinical trial is then made public as part of the registration. Sponsors are also obligated to disclose the results of their clinical trials after completion. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
Pharmaceutical coverage, pricing and reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we may obtain regulatory approval. In the United States, sales of any products for which we may receive regulatory approval for commercial sale will depend in part on the availability of coverage and reimbursement from third-party payers. Third-party payers include government programs such as Medicare and Medicaid, managed care providers, private health insurers, and other organizations. The process for determining whether a payer will provide coverage for a drug product may be separate from the process for setting the reimbursement rate that the payer will pay for the drug product. In the United States, the principal decisions about reimbursement for new medicines are typically made by the Centers for Medicare & Medicaid Services, or CMS, an agency within the U.S. Department of Health and Human Services. CMS decides whether and to what extent a new medicine will be covered and reimbursed under Medicare and private payers tend to follow CMS to a substantial degree. Third-party payers may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drugs for a particular indication. Moreover, a payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development. Third-party payers are increasingly challenging the price and examining the medical necessity and cost- effectiveness of medical products and services, in addition to their safety and efficacy. In order to obtain coverage and reimbursement for any product that might be approved for sale, we may need to conduct expensive pharmacoeconomic studies in order to demonstrate the medical necessity and cost-effectiveness of any products, in addition to the costs required to obtain regulatory approvals. Factors payers consider in determining reimbursement are based on whether the product is:
| ● | a covered benefit under its health plan; |
| ● | safe, effective and medically necessary; |
| ● | appropriate for the specific patient; |
| ● | cost-effective; and |
| ● | neither experimental nor investigational. |
Our product candidates may not be considered medically necessary or cost-effective. If third-party payers do not consider a product to be cost-effective compared to other available therapies, they may not cover the product after approval as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow a company to sell its products at a profit.
106
The U.S. government and state legislatures have shown significant interest in implementing cost containment programs to limit the growth of government-paid health care costs, including price controls, restrictions on the Affordable Care Act (ACA) contains provisions that may reduce the profitability of drug products, including, for example, increased rebates for drugs reimbursed by Medicaid programs, extension of Medicaid rebates to Medicaid managed care plans, mandatory discounts for certain Medicare Part D beneficiaries, and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. Adoption of government controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals.
Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Increasingly, third-party payors are requiring that drug companies provide them with predetermined discounts from list prices and are challenging the prices charged for medical products. We cannot be sure that reimbursement will be available for any product candidate that we commercialize and, if reimbursement is available, the level of reimbursement. In addition, many pharmaceutical manufacturers must calculate and report certain price reporting metrics to the government, such as average sales price, or ASP, and best price. Penalties may apply in some cases when such metrics are not submitted accurately and timely. Further, these prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payers fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on cost containment measures in the United States has increased and we expect will continue to increase the pressure on pharmaceutical pricing. Coverage policies and third-party reimbursement rates may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Other healthcare laws and compliance requirements
If we obtain regulatory approval of our products, we may be subject to various federal and state laws targeting fraud, waste, and abuse in the healthcare industry. These laws may impact, among other things, our proposed sales, marketing, and education programs. In addition, we may be subject to patient privacy regulation by both the federal government and the states in which we conduct our business. The U.S. laws that may affect our ability to operate include:
| ● | the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, to induce, or in return for, either the referral of an individual, or the purchase or recommendation of an item or service for which payment may be made under a federal healthcare program, such as the Medicare and Medicaid programs. A person or entity does not need to have actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it to have committed a violation. Violations are subject to civil and criminal fines and penalties for each violation, plus up to three times the remuneration involved, imprisonment, and exclusion from government healthcare programs. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act or federal civil money penalties statute (as discussed below); |
| ● | U.S. federal civil and criminal false claims laws and civil monetary penalty laws, including the federal False Claims Act, which impose criminal and civil penalties against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, including the Medicare and Medicaid programs, claims for payment that are false or fraudulent, making a false statement to avoid, decrease or |
107
| conceal an obligation to pay money to the federal government, or knowingly concealing or knowingly and improperly avoiding or decreasing such an obligation. Manufacturers can be held liable under the federal False Claims Act even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. The federal False Claims Act also permits a private individual acting as a “whistleblower” to bring actions on behalf of the federal government alleging violations of the federal False Claims Act and to share in any monetary recovery; |
| ● | the U.S. federal Health Insurance Portability and Accountability Act of 1996, or HIPAA and its implementing regulations, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and prohibit knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, and their respective implementing regulations, which impose certain obligations, including mandatory contractual terms, on covered healthcare providers, health plans, and healthcare clearinghouses, as well as their business associates, with respect to safeguarding the privacy, security and transmission of individually identifiable health information. HITECH also created new tiers of civil monetary penalties, amended HIPAA to make civil and criminal penalties directly applicable to business associates, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorneys’ fees and costs associated with pursuing federal civil actions. In addition, there may be additional federal, state and non-U.S. laws which govern the privacy and security of health and other personal information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts; |
| ● | The U.S. federal Physician Payments Sunshine Act which requires certain manufacturers of drugs, devices, biologics, and medical supplies for which payment is available under Medicare, Medicaid, or the Children’s Health Insurance Program, with specific exceptions, to report annually to the Centers for Medicare & Medicaid Services information related to payments or transfers of value made to physicians (currently defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain other licensed health care practitioners, and teaching hospitals, as well as ownership and investment interests held by the physicians described above and their immediate family members; |
| ● | federal government price reporting laws, which require us to calculate and report complex pricing metrics in an accurate and timely manner to government programs; |
| ● | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
| ● | analogous state and laws and regulations in other jurisdictions, such as state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payers, including private insurers, and state and laws in other jurisdiction governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
Many U.S. states have adopted laws similar to the federal Anti-Kickback Statute and False Claims Act, and may apply to our business practices, including, but not limited to, research, distribution, sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental payors, including private insurers. In addition, some states have passed laws that require pharmaceutical companies to comply with the April 2003 Office of Inspector General Compliance Program Guidance for Pharmaceutical Manufacturers and/or the Pharmaceutical Research and Manufacturers of America’s Code on Interactions with Healthcare Professionals. Several states also impose other
108
marketing restrictions or require pharmaceutical companies to make marketing or price disclosures to the state and require the registration of pharmaceutical sales representatives.
At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, in the event we obtain regulatory approval for any one of our products, it is possible that some of our business activities could be subject to challenge and may not comply under one or more of such laws, regulations, and guidance. Law enforcement authorities are increasingly focused on enforcing fraud and abuse laws, and it is possible that some of our practices may be challenged under these laws. Violations of these laws can subject us to administrative, civil and criminal penalties, damages, fines, disgorgement, the exclusion from participation in federal and state healthcare programs, individual imprisonment, reputational harm, and the curtailment or restructuring of our operations, as well as additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws. Efforts to ensure that our current and future business arrangements with third parties, and our business generally, will comply with applicable healthcare laws and regulations will involve substantial costs.
Patient Protection and Affordable Care Act and Healthcare Reformt
In 2010, the ACA was enacted, which included measures that significantly change the way health care is financed by both U.S. governmental and private insurers. Among other changes, the ACA:
| ● | subjected biologic products to potential competition by lower-cost biosimilars; |
| ● | increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program; |
| ● | extended the Medicaid Drug Rebate program to utilization of prescriptions of individuals enrolled in Medicaid managed care organizations; |
| ● | subjected manufacturers to new annual fees and taxes for certain branded prescription drugs; |
| ● | created a Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 70% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs to be covered under Medicare Part D; and |
| ● | provided incentives to programs that increase the federal government’s comparative effectiveness research. |
In addition, other legislative and regulatory changes have been proposed and adopted in the United States since the ACA was enacted:
| ● | The Budget Control Act of 2011 and subsequent legislation, among other things, created measures for spending reductions by Congress that include aggregate reductions of Medicare payments to providers of 2% per fiscal year, which remain in effect through 2031. Due to the Statutory Pay-As-You-Go Act of 2010, estimated budget deficit increases resulting from the American Rescue Plan Act of 2021, and subsequent legislation. Medicare payments to providers will be further reduced starting in 2025 absent further legislation. The U.S. American Taxpayer Relief Act of 2012 further reduced Medicare payments to several types of providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. |
109
| ● | On April 13, 2017, CMS published a final rule that gives states greater flexibility in setting benchmarks for insurers in the individual and small group marketplaces, which may have the effect of relaxing the essential health benefits required under the ACA for plans sold through such marketplaces. |
| ● | On May 30, 2018, the Right to Try Act, was signed into law. The law, among other things, provides a federal framework for certain patients to access certain investigational new drug products that have completed a Phase 1 clinical trial and that are undergoing investigation for FDA approval. Under certain circumstances, eligible patients can seek treatment without enrolling in clinical trials and without obtaining FDA permission under the FDA expanded access program. There is no obligation for a pharmaceutical manufacturer to make its drug products available to eligible patients as a result of the Right to Try Act. |
| ● | On May 23, 2019, CMS published a final rule to allow Medicare Advantage Plans the option of using step therapy for Part B drugs beginning January 1, 2020. |
The Inflation Reduction Act of 2022, or IRA includes several provisions that may impact our business to varying degrees, including provisions that reduce the out-of-pocket spending cap for Medicare Part D beneficiaries from $7,050 to $2,000 starting in 2025, thereby effectively eliminating the coverage gap; impose new manufacturer financial liability on certain drugs under Medicare Part D, allow the U.S. government to negotiate Medicare Part B and Part D price caps for certain high-cost drugs and biologics without generic or biosimilar competition; require companies to pay rebates to Medicare for certain drug prices that increase faster than inflation; and delay until January 1, 2032 the implementation of the HHS rebate rule that would have limited the fees that pharmacy benefit managers can charge. Further, under the IRA, orphan drugs are exempted from the Medicare drug price negotiation program, but only if they have one orphan designation and for which the only approved indication is for that disease or condition. If a product receives multiple rare disease designations or has multiple approved indications, it may not qualify for the orphan drug exemption. The implementation of the IRA is currently subject to ongoing litigation challenging the constitutionality of the IRA’s Medicare drug price negotiation program. The effects of the IRA on our business and the healthcare industry in general is not yet known.
In addition, President Biden has issued multiple executive orders that have sought to reduce prescription drug costs. In February 2023, HHS also issued a proposal in response to an October 2022 executive order from President Biden that includes a proposed prescription drug pricing model that will test whether targeted Medicare payment adjustments will sufficiently incentivize manufacturers to complete confirmatory trials for drugs approved through FDA’s accelerated approval pathway. Although a number of these and other proposed measures may require authorization through additional legislation to become effective, and the Biden administration may reverse or otherwise change these measures, both the Biden administration and Congress have indicated that they will continue to seek new legislative measures to control drug costs.
We expect that additional U.S. federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that the U.S. Federal Government will pay for healthcare drugs and services, which could result in reduced demand for our drug candidates or additional pricing pressures.
European Union regulation
Regulation in the United Kingdom
The UK formally left the EU on January 31, 2020, and the EU and the UK concluded a trade and cooperation agreement, or TCA, which was provisionally applicable since January 1, 2021 and has been formally applicable since May 1, 2021. The TCA includes specific provisions concerning pharmaceuticals, which include the mutual recognition of GMP, inspections of manufacturing facilities for medicinal products and GMP documents issued, but does not provide for wholesale mutual recognition of UK and EU pharmaceutical regulations. At present, Great Britain has implemented EU legislation on the marketing, promotion and sale of medicinal products through the Human Medicines Regulations 2012 (as amended) (under the Northern Ireland Protocol, the EU regulatory framework currently continues to apply in Northern Ireland). The regulatory regime in Great Britain therefore currently broadly aligns with EU regulations, however these regimes may diverge now that Great Britain’s regulatory system is independent from the EU
110
and the TCA does not provide for mutual recognition of UK and EU pharmaceutical legislation. For example, the new Clinical Trials Regulation which became effective in the EU on January 31, 2022 and provides for a streamlined clinical trial application and assessment procedure covering multiple EU Member States has not been implemented into UK law, and a separate application needs to be submitted for clinical trial authorization in the UK.
However, notwithstanding that there is no wholesale recognition of EU pharmaceutical legislation under the TCA, under a new international recognition procedure, which was put in place by the Medicines and Healthcare products Regulatory Agency (MHRA) on January 1, 2024, the MHRA may take account of decisions on the approval of a marketing authorization from trusted regulators in the European Union, as well as Australia, Canada, Japan, Switzerland, Singapore, and the United States (US), when considering an application for a Great Britain marketing authorization.
The Windsor Framework sets out a long-term set of arrangements for the supply of medicines into Northern Ireland. They will ensure that medicines must be approved and licensed on a UK-wide basis by the MHRA, with medicines using the same packaging and labelling across the UK. The European Medicines Agency will have no role in approving or licensing new drugs for provision in Northern Ireland. These new arrangements will take effect from 1 January 2025, with existing arrangements for supplying medicines to Northern Ireland applied in the meantime.
From 1 January 2025, joint packs, also known as common or shared packs, can no longer enter the UK supply chain (packs already released to the market may continue to be supplied until their expiry date). A joint pack is one that is shared with another EU country or countries, and which presents administrative details for both the UK and the other markets sharing the pack. From this date, medicines must be packaged in a UK-specific outer carton that is labelled ‘UK only’. Shared inner packaging components, such as multi-lingual blister foils and joint leaflets may continue to be used, provided that the UK and EU authorizations remain aligned.
Regulation in the European Union
Product development, the regulatory approval process, and safety monitoring of medicinal products and their manufacturers in the EU proceed in much the same manner as they do in the United States. Therefore, many of the issues discussed above apply similarly in the context of the EU. In addition, drugs are subject to the extensive price and reimbursement regulations of the various EU Member States.
Clinical trials
In April 2014, the EU adopted the Clinical Trials Regulation (EU) No 536/2014), which replaced the previous Clinical Trials Directive. The Clinical Trials Regulation entered into application on January 31, 2022. The transitory provisions of the Clinical Trials Regulation provide that, by January 31, 2025, all ongoing clinical trials must have transitioned to the Clinical Trials Regulation The Clinical Trials Regulation harmonises the assessment and supervision processes for clinical trials throughout the EU, via a Clinical Trials Information System (CTIS). CTIS contains the centralised EU portal and database for clinical trials provided for by the Regulation.
The goal of the Clinical Trials Regulation is to create an environment that is favourable to conducting clinical trials in the EU, with the highest standards of safety for participants and increased transparency of trial information. The Regulation requires:
| ● | consistent rules for conducting clinical trials throughout the EU; |
| ● | information on the authorisation, conduct and results of each clinical trial carried out in the EU to be publicly available. |
111
This aims to increase the efficiency of all trials in the EU with the greatest benefit for those conducted in multiple Member States. It aims to foster innovation and research, while helping avoid unnecessary duplication of clinical trials or repetition of unsuccessful trials. The key benefits of the Regulation include:
| ● | harmonised electronic submission and assessment process for clinical trials conducted in multiple Member States; |
| ● | improved collaboration, information-sharing and decision-making between and within Member States; |
| ● | increased transparency of information on clinical trials; and |
| ● | highest standards of safety for all participants in EU clinical trials |
The authorisation and oversight of clinical trials remains the responsibility of Member States, with EMA managing CTIS and supervising content publication on the public website.
Marketing approval
Marketing approvals in the EU may be obtained through a centralized, mutual recognition or decentralized procedure. The centralized procedure results in the grant of a single marketing authorization that is valid throughout the Member States, as well as the additional Member States of the European Economic Area (Norway, Iceland and Liechtenstein) (EEA).
Pursuant to Regulation (EC) No. 726/2004, as amended, the centralized procedure is mandatory for certain products, including those developed by means of specified biotechnological processes, advanced therapy medicinal products (gene therapy, somatic cell therapy, and tissue-engineered products), products for human use containing a new active substance for which the therapeutic indication is the treatment of specified diseases, including AIDS, HIV, cancer, diabetes, neurodegenerative disorders, auto-immune diseases and other immune dysfunctions and viral diseases, as well as products designated as orphan medicinal products. The Committee for Medicinal Products for Human Use, or CHMP, of the EMA also has the discretion to permit other products to use the centralized procedure if it considers them sufficiently innovative or they contain a new active substance or they may be of benefit to public health at the EU level.
In the marketing authorization application, or MAA, the applicant has to properly and sufficiently demonstrate the quality, safety, and efficacy of the drug. Under the centralized approval procedure, the CHMP, possibly in conjunction with other committees, is responsible for drawing up the opinion of the EMA on any matter concerning the admissibility of the files submitted in accordance with the centralized procedure, such as an opinion on the granting, variation, suspension or revocation of a marketing authorization, and pharmacovigilance.
The CHMP and other committees, such as the Committee for Advanced Therapies, or CAT, are also responsible for providing guidelines and have published numerous guidelines that may apply to our product candidates. These guidelines provide additional guidance on the factors that the EMA will consider in relation to the development and evaluation of drug products and may include, among other things, the preclinical studies required in specific cases; the manufacturing and control information that should be submitted in a MAA; and the post-approval measures required to monitor patients and evaluate the long-term efficacy and potential adverse reactions. Although these guidelines are not legally binding, we believe that our compliance with them is likely necessary to gain approval for any of our product candidates. Under Regulation (EC) No. 726/2004, the maximum timeframe for the evaluation of an MAA by the CHMP under the centralized procedure is 210 days after receipt of a valid application, excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP. Clock stops may extend the timeframe of evaluation of a MAA considerably beyond 210 days. When an application is submitted for a marketing authorization in respect of a drug which is of major interest from the point of view of public health and, in particular, therapeutic innovation, the applicant may request an accelerated assessment procedure. If the CHMP accepts such request, the time limit of 210 days will be reduced to 150 days (not including clock stops), but it is possible that the CHMP can revert to the standard time limit for the centralized procedure if it considers that it is no longer appropriate to conduct an accelerated assessment.
112
If the CHMP concludes that the quality, safety, and efficacy of the product are sufficiently proven, it adopts a positive opinion. This is sent to the European Commission which drafts a decision. After consulting with the Member States, the European Commission adopts a decision and grants a marketing authorization, which is valid for the whole of the EU. The marketing authorization may be subject to certain conditions, which may include, without limitation, the performance of post-authorization safety and/or efficacy studies. Pursuant to Regulation (EC) No. 726/2004, a new marketing authorization is valid for five years and may be renewed for an unlimited period on the basis of a re-evaluation of the risk-benefit balance after submission of a consolidated version of the initial marketing authorization application in addition to the pharmacovigilance data reported and all variations introduced since granting of the marketing authorization. The marketing authorization shall cease to be valid if the grant of such marketing authorization is not followed by the actual launch of the product on the market within three years or, if the product is no longer available on the market for three consecutive years.
European Union legislation also provides for a system of regulatory data and market exclusivity. According to Regulation (EC) No. 726/2004 and Directive 2001/83/EC, upon receiving marketing authorization, innovative medicinal products approved on the basis of a complete, independent data package benefit from eight years of data exclusivity and an additional two years of market exclusivity. Data exclusivity prevents generic or biosimilar applicants from referencing the innovator’s pre-clinical or clinical trials data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization, for a period of eight years from the date on which the reference product was first authorized in the EU. During the additional two-year period of market exclusivity, a generic or biosimilar marketing authorization can be submitted, and the innovator’s data may be referenced, but no generic or biosimilar medicinal product can be marketed until the expiration of the market exclusivity. The overall ten-year period will be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder, or MAH, obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. There is no guarantee that a product will be considered by the EMA to be an innovative medicinal product, and products may not qualify for data exclusivity. Even if a compound is considered to be an innovative medicine and the innovator is able to gain the period of data exclusivity, another company nevertheless could also market another version of the drug if such company obtained marketing authorization based on an MAA with a complete independent data package of pharmaceutical test, preclinical tests and clinical trials. However, products designated as orphan medicinal products enjoy, upon receiving marketing authorization, a period of 10 years of orphan market exclusivity limited to the therapeutic indication for which orphan designation has been obtained—see also “—Orphan Regulation” below. Depending upon the timing and duration of the EU marketing authorization process, products may be eligible for up to five years’ additional patent protection under a supplementary protection certificate, or SPC, pursuant to Regulation (EC) No. 469/2009. Such SPCs extend the rights under the basic patent for the drug.
Orphan regulation
In the EU, Regulation (EC) No. 141/2000, as amended, provides that a product will be designated as an orphan medicinal product if its sponsor can establish that:
| ● | (i) it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition; |
| ● | (ii) either such condition affects no more than five in ten thousand persons in the EU when the application is made, or where without incentives it is unlikely that the marketing of the product in the EU would generate sufficient return to justify the necessary investment in its development; and |
| ● | (iii)there exists no satisfactory method of diagnosis, prevention or treatment of the condition in question that has been authorized in the EU or, if such method exists, that the product will be of significant benefit to those affected by that condition. |
Regulation (EC) No. 847/2000 sets out further provisions for implementation of the criteria for designation of a products as an orphan medicinal product. An application for the designation of a product as an orphan medicinal products must be submitted before filing of a marketing authorization application.
113
If an EU-wide centralized marketing authorization in respect of an orphan product is granted or if all the EU Member States have granted marketing authorizations in accordance with the procedures for mutual recognition, the EMA and the Member States will not, for a period of 10 years, accept another application for a marketing authorization, or grant a marketing authorization or accept an application to extend an existing marketing authorization, for the same therapeutic indication, in respect of a ”similar medicinal product” to such authorized orphan product. A “similar medicinal product” is defined as a medicinal product containing a similar active substance or substances as contained in an authorized orphan medicinal product, and which is intended for the same therapeutic indication. This period may however be reduced to six years if, at the end of the fifth year, it is established, with respect to the product concerned, that the criteria for orphan medicinal product designation are no longer met, such as when it is shown on the basis of available evidence that the product is sufficiently profitable not to justify maintenance of market exclusivity (cfr. Article 8(s) of Regulation (EC) No. 141/200). Notwithstanding the foregoing, Regulation (EC) No. 141/2000 provides that a marketing authorization may be granted, for the same therapeutic indication, to a similar medicinal product if:
| ● | the holder of the marketing authorization for the original orphan medicinal product has given its consent to the second applicant; |
| ● | the holder of the marketing authorization for the original orphan medicinal product is unable to supply sufficient quantities of the product; or |
| ● | the second applicant can establish in the application that the second product, although similar to the orphan medicinal product already authorized, is safer, more effective or otherwise clinically superior. |
Other incentives available to orphan medicinal products in the EU include financial incentives such as a reduction of fees or fee waivers and protocol assistance. Orphan medicinal product designation does not convey any advantage in, or shorten the duration of the regulatory review and approval process.
Pediatric investigation plan
An application for marketing authorization of a medicinal product for human use which is not yet authorized in the EU shall be considered valid only if it includes a pediatric investigation plan, or PIP, according to Regulation (EC) No. 1901/2006, unless a waiver or deferral applies (for example if the disease or condition for which the product is intended occurs only in adult populations). The PIP or the application for waiver shall be submitted with a request for agreement, except in duly justified cases, early during the product development phase and not later than upon completion of the human pharmacokinetic studies in healthy subjects. In any case, submission of the PIP cannot be after initiation of pivotal trials or confirmatory (Phase 3) trials. The PIP sets out the timing and measures proposed to generate data to support a pediatric indication of the drug for which marketing authorization is being sought.
The Pediatric Committee, or PDCO, a scientific committee of the EMA, shall assess the content of any PIP, waivers and deferrals for a medicinal product submitted to it in accordance with the regulation on medicinal products for pediatric use and formulate an opinion thereon. If a marketing authorization is obtained and trial results in accordance with an agreed PIP are included in the product information, even when negative, the product is eligible for six months’ supplementary protection certificate extension. In the case of orphan medicinal products, a two year extension of the orphan market exclusivity may be available. This pediatric reward is subject to specific conditions and is not automatically available when data in compliance with the PIP are developed and submitted.
Manufacturing and manufacturers’ license
Pursuant to Directive 2001/83/EC and Directive (EU) 2017/1572, as transposed into the national laws of the Member States, the manufacturing of investigational medicinal products and approved products is subject to a separate manufacturer’s license and must be conducted in strict compliance with cGMP requirements, which mandate the methods, facilities, and controls used in manufacturing, processing, and packing of drugs to assure their safety and identity. Manufacturers must have at least one qualified person permanently and continuously at their disposal. The qualified person is ultimately responsible for certifying that each batch of finished product released onto the market has been manufactured in accordance with cGMP and the specifications set out in the marketing authorization or
114
investigational medicinal product dossier. cGMP requirements are enforced through mandatory registration of facilities and inspections of those facilities. Failure to comply with these requirements could interrupt supply and result in delays, unanticipated costs and lost revenues, and subject the applicant to potential legal or regulatory action, including but not limited to warning letters, suspension of manufacturing, seizure of product, injunctive action or possible civil and criminal penalties.
Wholesale distribution and license
Pursuant to Directive 2001/83/EC, the wholesale distribution of medicinal products is subject to the possession of an authorization to engage in activity as a wholesaler in medicinal products. Possession of a manufacturing authorization includes authorization to distribute by wholesale the medicinal products covered by that authorization. The distribution of medicinal products must comply with the principles and guidelines of good distribution practices, or GDP.
Collection and use of personal data in the EU
As of May 25, 2018, the General Data Protection Regulation (GDPR) regulates the collection and use of personal data in the EU. The GDPR covers any business, regardless of its location, that provides goods or services to residents in the EU and, thus, could incorporate our activities in EU Member States. The GDPR imposes strict requirements on controllers and processors of personal data, including special protections for “sensitive information,” which includes health and genetic information of individuals residing in the EU. The GDPR grants individuals the opportunity to object to the processing of their personal information, allows them to request deletion of personal information in certain circumstances, and provides the individual with an express right to seek legal remedies in the event the individual believes his or her rights have been violated. Further, the GDPR imposes strict rules on the transfer of personal data out of the EU to regions that have not been deemed to offer “adequate” privacy protections, such as the U.S. currently. Failure to comply with the requirements of the GDPR and the related national data protection laws of the EU Member States, which may deviate slightly from the GDPR, may result in warning letters, mandatory audits, suspension of processing and financial penalties, including fines of up to 4 percent of global revenues, or €20,000,000, whichever is greater. As a result of the implementation of the GDPR, we may be required to put in place additional mechanisms ensuring compliance with the new data protection rules. In addition, once we begin to conduct business in the United Kingdom, we will be subject to stringent data protection laws that are in effect in the UK. As of January 1, 2021, the UK’s European Union (Withdrawal) Act 2018 incorporated the GDPR (as it existed on December 31, 2020 but subject to certain UK specific amendments) into UK law, referred to as the UK GDPR. The UK GDPR and the UK Data Protection Act 2018 set out the United Kingdom’s data protection regime, which is independent from but aligned to the European Union’s data protection regime. Non-compliance with the UK GDPR may result in monetary penalties of up to £17.5 million or 4% of worldwide revenue, whichever is higher. Although the UK is regarded as a third country under the EU’s GDPR, the European Commission has now issued a decision recognizing the UK as providing adequate protection under the EU GDPR and, therefore, transfers of personal data originating in the EU to the UK remain unrestricted. Like the EU GDPR, the UK GDPR restricts personal data transfers outside the UK to countries not regarded by the UK as providing adequate protection. The UK government has confirmed that personal data transfers from the UK to the EU remain free flowing.
There is significant uncertainty related to the manner in which data protection authorities will seek to enforce compliance with GDPR. For example, it is unclear whether the authorities will conduct random audits of companies doing business in the EU, or act solely after complaints are filed claiming a violation of the GDPR. The lack of compliance standards and precedent, enforcement uncertainty and the costs associated with ensuring GDPR compliance may be onerous and adversely affect our business, financial condition, results of operations and prospects.
Other regulatory requirements
A marketing authorization holder, or MAH, for a medicinal product is legally obliged to fulfill a number of obligations by virtue of its status as an MAH. The MAH can delegate the performance of related tasks to third parties, such as distributors or marketing partners, provided that this delegation is appropriately documented and the MAH maintains legal responsibility and liability.
115
The obligations of an MAH include:
Manufacturing and batch release. MAHs should ensure that all manufacturing operations comply with relevant laws and regulations (as set forth above), applicable good manufacturing practices, with the product specifications and manufacturing conditions set out in the marketing authorization and that each batch of product is subject to appropriate release formalities.
Availability and continuous supply. Pursuant to Directive 2001/83/EC, as transposed into the national laws of the Member States, the MAH for a medicinal product and the distributors of the said medicinal product actually placed on the market in a Member State shall, within the limits of their responsibilities, ensure appropriate and continued supplies of that medicinal product to pharmacies and persons authorized to supply medicinal products so that the needs of patients in the Member State in question are covered.
Pharmacovigilance. MAHs are obliged to establish and maintain a pharmacovigilance system, including a qualified person responsible for oversight, submit safety reports to the regulators and comply with the good pharmacovigilance practice guidelines adopted by the EMA.
Advertising and promotion. MAHs remain responsible for all advertising and promotion of its products, including promotional activities by other companies or individuals on their behalf and in some cases must conduct internal or regulatory pre-approval of promotional materials. Regulation in this area also covers interactions with healthcare practitioners and/or patient groups, and in some jurisdictions legal or self-regulatory obligations to disclose such interactions exist. In the EU, the promotion of prescription medicines is subject to intense regulation and control, including EU and national legislation as well as self-regulatory codes (industry codes). Advertising legislation inter alia includes a prohibition on direct-to-consumer advertising of prescription only medicinal products. All prescription medicines advertising must be consistent with the product’s approved summary of product characteristics, and must be factual, accurate, balanced and not misleading. Advertising of prescription medicines to healthcare professionals pre-approval or off-label is not allowed.
Some jurisdictions require that all promotional materials for prescription medicines be subjected to either prior internal review and approval or regulatory review and approval.
Medical affairs/scientific service. MAHs are required to disseminate scientific and medical information on its medicinal products to healthcare professionals, regulators and patients.
Preparation, filing and maintenance of the application and subsequent marketing authorization. MAHs must maintain appropriate records, comply with the marketing authorization’s terms and conditions, fulfill reporting obligations to regulators, submit renewal applications and pay all appropriate fees to the authorities. We may hold any future marketing authorizations granted for our product candidates in our own name, or appoint an affiliate or a collaboration partner to hold marketing authorizations on our behalf. Any failure by an MAH to comply with these obligations may result in regulatory action against an MAH and ultimately threaten our ability to commercialize our products.
Price and reimbursement
In the EU, the pricing and reimbursement mechanisms by private and public health insurers vary largely by country and even within countries. The public systems reimbursement for standard products is determined by guidelines established by the legislator or responsible national authority. The approach taken varies by Member State. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. Other Member States allow companies to fix their own prices for medicines, but monitor and control company profits and may limit or restrict reimbursement. The downward pressure on healthcare costs in general, particularly prescription drugs, has become very intense. As a result, increasingly high barriers are being erected to the entry of new products and some of EU Member States require the completion of studies that
116
compare the cost-effectiveness of a particular product candidate to currently available therapies in order to obtain reimbursement or pricing approval. Special pricing and reimbursement rules may apply to orphan medicinal products. Inclusion of orphan medicinal products in reimbursement systems tend to focus on the medical usefulness, need, quality and economic benefits to patients and the healthcare system as for any product. Acceptance of any medicinal product for reimbursement may come with cost, use and often volume restrictions, which again can vary by country. In addition, results based rules of reimbursement may apply.
Regulation (EU) 2021/2282 on health technology assessment (HTAR) entered into force on 11 January 2022 and will apply from 12 January 2025.The Health Technology Assessment (HTA) Regulation focuses on clinical aspects of HTA, i.e. the relative clinical effectiveness and relative clinical safety of a new health technology as compared with existing technologies. Member States' HTA bodies will conduct joint clinical assessments of new medicines and certain high-risk medical devices. They will also engage in joint scientific consultations to advise technology developers on clinical study designs that generate appropriate evidence. Moreover, “horizon scanning” exercises will identify, at an early stage, promising health technologies, to help health systems prepare for them. In addition to this mandatory scope of the HTAR, Member States may also engage in further voluntary cooperation, e.g. on health technologies other than medicines and medical devices, or on economic aspects of HTA.
Reform of general pharmaceutical legislation
On 26 April 2023, the European Commission proposed to reform the EU’s pharmaceutical legislation as part of a wider EU Pharmaceutical Strategy, including through a proposal for a new medicines Directive and Regulation, to modernize, simplify and replace the following existing legislation: Directive 2001/83/EC and Regulation (EC) No 726/2004 (referred to as the ‘general pharmaceutical legislation’), Regulation (EC) No 1901/2006 on medicines for children (‘Pediatric Regulation’), and Regulation (EC) No 141/2000 on medicines for rare diseases (‘Orphan Regulation’).
The European Commission provided the legislative proposals to the European Parliament and the European Council for their review and approval. In October 2023, the European Parliament published draft reports proposing amendments to the legislative proposals, which will be debated by the European Parliament. Once the European Commission’s legislative proposals are approved (with or without amendment), they will be adopted into EU law.
The aforementioned EU rules are generally applicable in the EEA.
Legal proceedings
From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. We are not presently a party to any legal proceedings that, if determined adversely to us, would individually or taken together have a material adverse effect on our business, results of operations, financial condition or cash flows. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Glossary of terms
Glossary of terms, to be read only in conjunction with this annual report.
Term | Description |
ADS | American Depositary Share; Galapagos has a Level 3 ADS listed on Nasdaq with ticker symbol GLPG and CUSIP number 36315X101. One ADS is equivalent to one ordinary share in Galapagos NV |
Antibody | A blood protein produced in response to and counteracting a specific antigen. Antibodies combine chemically with substances which the body recognizes as alien, such as bacteria, viruses, and foreign substances |
117
Antigen-binding fragment (Fab) | The fragment antigen-binding (Fab fragment) is a region on an antibody that binds to antigens. It is composed of one constant and one variable domain of each of the heavy and the light chain |
Assays | Laboratory tests to determine characteristics |
ATALANTA-1 | ATALANTA-1 Phase 1/2 study with point-of-care manufactured CD19 CAR-T candidate, GLPG5101, in patients with replapsed/ refractory non-Hodgkin Lymphoma (rrNHL) |
Axial spondyloarthritis (AxSpA) | Axial spondyloarthritis (axSpA) is a type of arthritis. It mostly causes pain and swelling in the spine and the joints that connect the bottom of the spine to the pelvis (sacroiliac joint). Other joints can be affected as well. It is a systemic disease, which means it may affect other body parts and organs. The disease tends to run in families |
BCMA | B cell maturation antigen (BCMA) is a member of the tumor necrosis factor receptor superfamily that plays an important role in regulating B-cell proliferation and survival. BCMA is central to the survival of multiple myeloma cells |
Biologics | Biologics, also referred to as Biologicals or Biologics, are those class of medicines which are grown and then purified from large-scale cell cultures of bacteria or yeast, or plant or animal cells. Biologicals are a diverse group of medicines which includes vaccines, growth factors, immune modulators, monoclonal antibodies, as well as products derived from human blood and plasma. What distinguishes biologicals from other medicines is that these are generally proteins purified from living culture systems or from blood, whereas other medicines are considered as small molecules and are either made synthetically or purified from plants |
Black & Scholes model | A mathematical description of financial markets and derivative investment instruments that is widely used in the pricing of European options and subscription rights |
CAR-T | Chimeric antigen receptor T cells (also known as CAR-T cells) are T cells that have been genetically engineered to produce an artificial T cell receptor for use in immunotherapy |
Cash position | Current financial investments and cash and cash equivalents |
CD19 | CD19 is a protein found on the surface of B-cells, a type of white blood cell. Since CD19 is a hallmark of B-cells, the protein has been used to diagnose cancers that arise from this type of cell - notably B-cell lymphomas |
Cell therapy | Cell therapy aims to treat diseases by restoring or altering certain sets of cells or by using cells to carry a therapy through the body. With cell therapy, cells are cultivated or modified outside the body before being injected into the patient. The cells may originate from the patient (autologous cells) or a donor (allogeneic cells) |
CHMP | Committee for Medicinal Products for Human Use is the European Medicines Agency's (EMA) committee responsible for human medicines and plays a vital role in the authorization of medicines in the European Union (EU) |
Chronic Lymphocytic Leukemia (CLL) | Chronic lymphocytic leukemia is the most common leukemia in adults. It is a type of cancer that starts in cells that become certain white blood cells (called lymphocytes) in the bone marrow. The cancer (leukemia) cells originate in the bone marrow and migrate to the bloodstream |
Complete Response Rate (CRR) | Term used for the absence of all detectable cancer after the treatment is completed |
Compound | A chemical substance, often a small molecule with drug-like properties' |
118
Contract research organization (CRO) | Organization which provides drug discovery and development services to the pharmaceutical, biotechnology and medical devices industry |
Crohn's disease (CD) | An IBD involving inflammation of the small and large intestines, leading to pain, bleeding, and ultimately in some cases surgical removal of parts of the bowel |
Cryopreservation | Process where biological material - cells, tissues, or organs - are frozen to preserve the material for an extended period of time |
Cytokine | A category of small proteins which play important roles in signaling in processes in the body |
Cytokine release syndrome (CRS) | Condition that develops when your immune system responds too aggressively to infection or after certain types of immunotherapy, such as CAR-T-cell therapy |
DARWIN | Phase 2 program for filgotinib in RA. DARWIN 1 explored three doses, in twice-daily and once-daily administration, for up to 24 weeks in RA patients with insufficient response to methotrexate (MTX) and who remained on their stable background treatment with MTX. DARWIN 2 explored three once-daily doses for up to 24 weeks in RA patients with insufficient response to methotrexate (MTX) and who washed out of their treatment with MTX. DARWIN 1 and 2 were double-blind, placebo-controlled trials which recruited approximately 900 patients globally and for which results were reported in 2015. DARWIN 3 is a long term extension trial in which all patients are on 200mg filgotinib, except for U.S. males who are on 100mg. The Week 156 results from DARWIN 3 were reported in 2019 |
Dermatomyositis (DM) | Dermatomyositis is a rare inflammatory disease. Common symptoms include distinctive skin rash, and inflammatory myopathy, or inflamed muscles, causing muscle weakness |
Development | All activities required to bring a new drug to the market. This includes preclinical and clinical development research, chemical and pharmaceutical development and regulatory filings of product candidates |
Discovery | Process by which new medicines are discovered and/or designed. At Galapagos, this is the department that oversees target and drug discovery research through to nomination of preclinical candidates |
DIVERSITY | Phase 3 program evaluating filgotinib in CD |
Dose-range finding study | Phase 2 clinical study exploring the balance between efficacy and safety among various doses of treatment in patients. Results are used to determine doses for later studies |
Double-blind | Term to characterize a clinical trial in which neither the physician nor the patient knows if the patient is taking placebo or the treatment being evaluated |
EC | European Commission |
Efficacy | Effectiveness for intended use |
EMA | European Medicines Agency, in charge of European market authorization of new medications |
End-to-end | A process that takes a system or service from beginning to end and delivers a complete functional solution, usually without strong reliance on third parties |
EUPLAGIA-1 | EUPLAGIA-1 Phase 1/2 study with point-of-care manufactured CD19 CAR-T candidate, GLPG5201, in patients with replapsed/ refractory chronic lymphocytic leukemia (rrCLL) and small lymphocytic lymphoma (rrSLL), with or without Richter transformation (RT) |
119
FDA | The U.S. Food and Drug Administration is an agency responsible for protecting and promoting public health and in charge of American market approval of new medications |
Filgotinib | Formerly known as GLPG0634, commercial name is Jyseleca®. Small molecule preferential JAK1 inhibitor, approved in RA and UC in the European Union, Great-Britain and Japan. Phase 4 studies in both RA and UC are ongoing |
FILOSOPHY | Phase 4 program evaluating filgotinib in RA |
FINCH | Phase 3 program evaluating filgotinib in RA |
FORM 20-F | Form 20-F is an SEC filing submitted to the US Securities and Exchange Commission |
FSMA | The Belgian market authority: Financial Services and Markets Authority, or Autoriteit voor Financiële Diensten en Markten |
FTE | Full-time equivalent; a way to measure an employee’s involvement in a project. For example, an FTE of 1.0 means that the equivalent work of one full-time worker was used on the project |
G&A expenses | General & administrative expenses |
GALACELA | Phase 2 study with GLPG3667 in patients with systemic lupus erythematous |
GALARISSO | Phase 2 study with GLPG3667 in patients with dermatomyositis |
GLPG0634 | Molecule number currently known as filgotinib and Jyseleca® |
GLPG3667 | A TYK2 kinase inhibitor discovered by us, topline results from the Phase 1b in psoriasis reported in July 2021 |
GLPG5101 | A second generation anti-CD19/4-1BB CAR-T product candidate currently in Phase 1/2 study in rrNHL |
GLPG5201 | A second generation anti-CD19/4-1BB CAR-T product candidate currently in Phase 1/2 study in rrCLL/SLL with or wthout RT |
GLPG5301 | A BCMA CAR-T product candidate |
IBD | Inflammatory Bowel Disease. This is a general term for an autoimmune disease affecting the bowel, including CD and UC. CD affects the small and large intestine, while UC affects the large intestine. Both diseases involve inflammation of the intestinal wall, leading to pain, bleeding, and ultimately, in some cases, surgical removal of part of the bowel |
Immune effector cell-associated neurotoxicity syndrome (ICAN) ' | Clinical and neuropsychiatric syndrome that can occur in the days to weeks following administration of certain types of immunotherapy, especially immune effector cell (IEC) and T cell engaging therapy |
Immunology | The study of the immune system and is a very important branch of the medical and biological sciences. The immune system protects humans from infection through various lines of defence. If the immune system is not functioning as it should, it can result in disease, such as autoimmunity, allergy and cancer |
In-/out-licensing | Receiving/granting permission from/to another company or institution to use a brand name, patent, or other proprietary right, in exchange for a fee and/or royalty |
Intellectual property | Creations of the mind that have commercial value and are protected or protectable, including by patents, trademarks or copyrights |
120
Investigational New Drug (IND) Application | United States Federal law requires a pharmaceutical company to obtain an exemption to ship an experimental drug across state lines, usually to clinical investigators, before a marketing application for the drug has been approved. The IND is the means by which the sponsor obtains this exemption, allowing them to perform clinical studies |
In vitro | Studies performed with cells outside their natural context, for example in a laboratory |
In vivo | Studies performed with animals in a laboratory setting |
JAK | Janus kinases (JAK) are critical components of signaling mechanisms utilized by a number of cytokines and growth factors, including those that are elevated in RA. Filgotinib is a preferential JAK1 inhibitor |
Jyseleca® | Jyseleca®; is the brand name for filgotinib |
Leukapheresis | Laboratory procedure in which white blood cells are separated from a sample of blood |
Lymphocyte | Type of white blood cell that is part of the immune system |
MACE | Major adverse cardiovascular events; a composite endpoint frequently used in cardiovascular research |
MANTA | A Phase 2 semen parameter trial with filgotinib in male patients with CD or UC |
MANTA-RAy | Phase 2 semen parameter trial with filgotinib in male patients with RA, PsA, or AS |
MHLW | Japanese Ministry of Health, Labor and Welfare (MHLW), in charge of Japanese market authorization of new medications |
MHRA | Medicines and Healthcare products Regulatory Agency in Great Britain |
Milestone | Major achievement in a project or program; in our alliances, this is usually associated with a payment |
Multiple myeloma (MM) | Multiple myeloma (MM) is typically characterized by the neoplastic proliferation of plasma cells producing a monoclonal immunoglobulin. The plasma cells proliferate in the bone marrow and can result in extensive skeletal destruction with osteolytic lesions, osteopenia, and/or pathologic fractures. |
NDA | A new drug application (NDA) is a request to the FDA for a license to market a new drug in the U.S. A NDA must show the chemical and pharmacologic description of the drug, the results of clinical trials, and the proposed drug label |
Non-Hodgkin's lymphoma (NHL) | Non-Hodgkin's lymphoma is a type of cancer that begins in the lymphatic system, which is part of the body's germ-fighting immune system. In non-Hodgkin's lymphoma, white blood cells called lymphocytes grow abnormally and form tumors throughout the body |
Objective Response Rate (ORR) | The response rate is the percentage of patients on whom a therapy has some defined effect; for example, the cancer shrinks or disappears after treatment. When used as a clinical endpoint for trials of cancer treatments, this is often called the objective response rate |
OLINGUITO | Phase 3 study with filgotinib in patients with axial spondyloarthritis |
Oncology | Field of medicine that deal with the diagnosis, treatment, prevention, and early detection of cancer |
Oral dosing | Administration of medicine by the mouth, either as a solution or solid (capsule, pill) form |
Outsourcing | Contracting work to a third party |
121
PAPILIO-1 | Phase 1/2 study with GLPG5301 in patients with relapsed/refractory multiple myeloma |
Pharmacokinetics (PK) | Study of what a body does to a drug; the fate of a substance delivered to a body. This includes absorption, distribution to the tissues, metabolism and excretion. These processes determine the blood concentration of the drug and its metabolite(s) as a function of time from dosing |
Phase 1 | First stage of clinical testing of an investigational drug designed to assess the safety and tolerability, pharmacokinetics of a drug, usually performed in a small number of healthy human volunteers |
Phase 2 | Second stage of clinical testing, usually performed in no more than several hundred patients, in order to determine efficacy, tolerability and the dose to use |
Phase 3 | Large clinical trials, usually conducted in several hundred to several thousand patients to gain a definitive understanding of the efficacy and tolerability of the candidate treatment; serves as the principal basis for regulatory approval |
Pivotal trials | Registrational clinical trials |
Placebo | A substance having no pharmacological effect but administered as a control in testing a biologically active preparation |
Point-of-care | Drug treatment is provided close to or near the patient |
PRAC | Pharmacovigilance Risk Assessment Committee of the European Medicines Agency, responsible for assessing all aspects of risk management of human medicines |
Preclinical | Stage of drug research development, undertaken prior to the administration of the drug to humans. Consists of in vitro and in vivo screening, pharmacokinetics, toxicology, and chemical upscaling |
Preclinical candidate (PCC) | A new molecule and potential drug that meets chemical and biological criteria to begin the development process |
Product candidate | Substance that has satisfied the requirements of early preclinical testing and has been selected for development, starting with formal preclinical safety evaluation followed by clinical testing for the treatment of a certain disorder in humans |
R&D operations | Research and development operations; unit responsible for discovery and developing new product candidates for internal pipeline or as part of risk/reward sharing alliances with partners |
Refractory | "Refractory" refers to a patient with cancer that is/has become resistant to, or does not respond to, treatment |
Relapsed | "Relapsed" refers to a patient with cancer that develops cancer again after a period of improvement |
Rheumatoid arthritis (RA) | A chronic, systemic inflammatory disease that causes joint inflammation, and usually leads to cartilage destruction, bone erosion and disability |
Richter transformation | Richter Transformation (RT) is an uncommon clinicopathological condition observed in patients with CLL. It is characterized by the sudden transformation of the CLL into a significantly more aggressive form of large cell lymphoma, and occurs in approximately 2-10% of all CLL patients. |
S&M expenses | Sales and marketing expenses |
SEC | Securities and Exchange Commission in the US |
122
SELECTION | Phase 3 program evaluating filgotinib in UC patients. Full results were published in The Lancet in 2021 |
SIK | Salt-inducible kinase |
Single-chain variable fragments (scFv) | Single-chain variable fragments (scFvs) are small-sized artificial constructs composed of the immunoglobulin heavy and light chain variable regions connected by a peptide linker |
Small cell lymphocyte leukemia (SLL) | Small cell lymphocyte leukemia is a type of B-cell non-Hodgkin lymphoma, where the SLL cancer is located in lymp nodes and/or the spleen |
Systemic lupus erythematosus (SLE) | An autoimmune disease, with systemic manifestations including skin rash, erosion of joints or even kidney failure |
Target | Protein that has been shown to play a role in a disease process and that forms the basis of a therapeutic intervention or discovery of a medicine |
Target discovery | Identification and validation of proteins that have been shown to play a role in a disease process |
TEAE | Treatment Emergent Adverse Event, is any event not present prior to the initiation of the treatments or any event already present that worsens in either intensity or frequency following exposure to the treatments |
TYK | Tyrosine kinase is an enzyme that can transfer a phosphate group from ATP to the tyrosine residues of specific proteins inside a cell. It functions as an "on" or "off" switch in many cellular functions. Tyrosine kinases belong to a larger class of enzymes known as protein kinases which also attach phosphates to other amino acids such as serine and threonine. GLPG3667 is a reversible and selective TYK2 kinase domain inhibitor |
Ulcerative colitis (UC) | UC is an IBD causing chronic inflammation of the lining of the colon and rectum (unlike CD with inflammation throughout the gastrointestinal tract) |
Variable heavy (VH) domain | The variable domain of an immunoglobulin heavy chain is a part of an antibody that binds to a specific antigen |
123
C. Organizational structure.
As of December 31, 2023, we had 20 subsidiaries. The following table sets out for each of our subsidiaries, the country of incorporation, and percentage ownership and voting interest held by us (directly or indirectly through our subsidiaries) (situation as per December 31, 2023).
| |||||
Company |
| Country of incorporation |
| Percentage ownership and |
|
Continuing operations | |||||
Galapagos B.V. (merged with CellPoint B.V.) | The Netherlands | 100% | |||
Galapagos GmbH | Switzerland | 100% | |||
Glpg US Holding Inc. (formerly Galapagos Inc.) | United States | 100% | |||
Galapagos SASU | France | 100% | |||
Galapagos Real Estate Belgium BV | Belgium | 100% | |||
Galapagos Real Estate Netherlands B.V. | The Netherlands | 100% | |||
Glpg US Inc. (formerly AboundBio Inc.) | United States | 100% | |||
Xenometrix, Inc. in liquidation | United States | 100% | |||
Discontinued operations | |||||
Galapagos Biopharma Belgium BV | Belgium | 100% | |||
Galapagos Biopharma Netherlands B.V. | The Netherlands | 100% | |||
Galapagos Biopharma Spain S.L.U | Spain | 100% | |||
Galapagos Biopharma Italy S.r.l. | Italy | 100% | |||
Galapagos Biopharma Germany GmbH | Germany | 100% | |||
Galapagos Biotech Ltd. | United Kingdom | 100% | |||
Galapagos Biopharma Sweden AB | Sweden | 100% | |||
Galapagos Biopharma Norway AS | Norway | 100% | |||
Galapagos Biopharma Finland Oy | Finland | 100% | |||
Galapagos Biopharma Denmark ApS | Denmark | 100% | |||
Galapagos Biopharma Austria GmbH | Austria | 100% | |||
Galapagos Biopharma Ireland Ltd | Ireland | 100% |
In 2023, CellPoint B.V. merged into Galapagos B.V.
D. Property, plants and equipment.
We have our principal executive, operational offices and laboratory space located in Mechelen, Belgium. Our main facilities owned or leased as of December 31, 2023 are set forth in the following table:
Facility location |
| Use |
| Approx. size (m2) |
| Lease expiry |
Mechelen, Belgium (leased) |
| Headquarters, R&D, Operations |
| 12,964 | December 31, 2025 | |
Oegstgeest, the Netherlands (owned) | R&D, Operations | 6,943 |
In addition to the facilities listed in the table above, we also lease office space in Basel (Switzerland), Romainville (France) and Boston (United States), as well as R&D space in Pittsburgh (United States).
In 2023 we furnished the laboratory spaces including cleanrooms and additional office space, to house our CAR-T and Cocoon-related R&D activities. This new space got occupied from early December onwards.
In Belgium, we purchased land in Mechelen at the end of 2019. In 2022, we applied and successfully received an environmental permit for the construction of a new headquarters building covering offices and laboratory space. In 2023, we have been looking for a real estate investor that could take over the construction of the new building, in a sale-and-leaseback set-up for the project. In early 2024, we decided not to construct a new building on the land in Mechelen.
124
Environmental issues
For more information on environmental issues that may affect our utilization of our facilities, please see the section of this annual report titled “Item 3.D.—Risk factors—Risks related to our organization, structure and operation—.” We could be subject to (a) liabilities under environmental, health and safety laws or regulations, and (b) if we fail to comply with such laws and regulations, fines, penalties or other sanctions, or otherwise incur costs, that could have a material adverse effect on the success or operational results of our business.
Item 4A Unresolved staff comments
Not applicable.
Item 5 Operating and financial review and prospects
Overview
We are a global biotechnology company with operations in Europe and the US dedicated to developing transformational medicines for more years of life and quality of life. Focusing on high unmet medical needs, we synergize compelling science, technology, and collaborative approaches to create a deep pipeline of potentially best-in-class small molecules, CAR-T therapies, and biologics in oncology and immunology. With capabilities from lab to patient, including a decentralized, point-of-care CAR-T manufacturing network, we are committed to challenging the status quo and delivering results for our patients, employees and shareholders.
On October 30, 2023, we signed a letter of intent contemplating a transfer of the Jyseleca® business to Alfasigma S.p.A. (Alfasigma). The final agreement was signed on December 30, 2023 and the transaction was closed on January 31, 2024. The transfer includes the European and UK Marketing Authorizations, and the commercial, medical affairs and development activities for Jyseleca®. In connection with the completion of the transaction, approximately 400 Galapagos employees in 14 European countries transferred to Alfasigma to support business continuity and ongoing patient access for the Jyseleca® business.
Our clinical pipeline includes: 1) GLPG5101, a CD19 CAR-T product candidate manufactured near point-of-care, currently in Phase 1/2 in rrNHL; 2) GLPG5201, a CD19 CAR-T product candidate manufactured near point-of-care, currently in Phase 1/2 in rrCLL; 3) GLPG5301, a BCMA CAR-T product candidate manufactured near point-of-care, for which we initiated clinical development in rrMM, and 4) GLPG3667, a TYK2 inhibitor currently in Phase 2 clinical trial in DM and in SLE. In both our immunology and oncology portfolios, we have multiple product candidates in early research stages. To date, we have funded our operations through public and private placements of equity securities, upfront payments, milestone payments, royalties received from pharmaceutical partners under our collaboration and alliance agreements, payments received from wholesalers and hospitals for Jyseleca sales, payments under our fee-for-service contracts, funding from governmental bodies, interest income as well as the net proceeds from the sale of our service division in 2014 and of our fee-for-service division in 2021. From January 1, 2021 until December 31, 2023 we received €215.9 million in payments through our collaboration and alliance agreements, €236.4 million payments from gross sales to customers for Jyseleca in Europe, and €66.6 million in net interest payments. These are items which have a significant impact upon the profitability or cash flow of our business in each year in which they are received and earned. Fee-for-service payments and payments from governmental bodies contributed €1.2 million and €101.9 million, respectively. In 2021 we realized €28.7 million of net proceeds from the disposal of Fidelta. As of December 31, 2023, we had cash and cash equivalents of €166.8 million and current financial investments of €3,517.7 million.
For the year ended December 31, 2021, we incurred a net loss of €103.2 million. For the year ended December 31, 2022, we incurred a net loss of €218.0 million. For the year ended December 31, 2023, we incurred a net income of €211.7 million. We expect to continue incurring significant research, development, and other expenses related to our ongoing operations, and to continue incurring operating losses for the foreseeable future.
125
Transfer of the Jyseleca® Business to Alfasigma
On October 30, 2023, we signed a letter of intent contemplating a transfer of the Jyseleca® business to Alfasigma S.p.A. (Alfasigma). The final agreement was signed on December 30, 2023 and the transaction was closed on January 31, 2024. The transfer includes the European and UK Marketing Authorizations, and the commercial, medical affairs and development activities for Jyseleca®. In connection with the completion of the transaction, approximately 400 Galapagos employees in 14 European countries transferred to Alfasigma to support business continuity and ongoing patient access for the Jyseleca® business. We received a €50 million upfront payment in connection with the transfer, at closing of the transaction in 2024, and are entitled to receive potential milestone payments totaling €120 million and mid-single to mid-double-digit royalties on European sales. We will contribute up to €40 million by June 2025 to Alfasigma for Jyseleca® related development activities. In addition, we plan to streamline our remaining operations and further build efficiencies, with an envisaged reduction of approximately 100 positions across the organization.
Effective 31 January 2024, following the closing of the transaction between Galapagos and Alfasigma S.p.A. to transfer the Jyseleca® business to Alfasigma S.p.A., Galapagos assigned its rights and obligations under the filgotinib collaboration to Alfasigma S.p.A., except for Galapagos’ right to receive royalties from Gilead on net sales in the Gilead Territory under a separate agreement between Gilead and Galapagos entered into in October 2023
Collaboration and alliance agreements
Our main collaborations and alliance agreements are summarized below. All U.S. dollar payment amounts which have been received in cash regarding our Gilead collaboration in this Item 5 are converted into euros as per historical exchange rates (i.e., the spot rate at the moment of the transaction).
Option, License and Collaboration Agreement with Gilead
On July 14, 2019 we entered into a 10-year global research and development collaboration with Gilead. Through this agreement, Gilead gained exclusive access to our innovative portfolio of compounds, including molecules currently in clinical trials, our preclinical programs and a proven drug discovery platform.
At inception of this collaboration in 2019 we received an upfront payment of €3,569.8 million ($3.95 billion) and a €960.1 million ($1.1 billion) equity investment from Gilead. Under the terms of its equity investment, Gilead nominated two individuals to our Board of Directors, Dr. Linda Higgins and Mr. Daniel O'Day.
On the closing date of the transaction (August 23, 2019) we concluded that the upfront payment implicitly included a premium for the future issuance of warrant A and initial and subsequent warrant B. The expected value of the warrants to be issued is treated as a contract liability ("warrant issuance liability") and reducing the transaction price until the approval date of the issuance of the underlying warrant. As from approval date, the allocation of the upfront payment to the respective warrant becomes fixed and future changes in the fair value of the respective warrant will be recognized as either a profit or loss. As such, the part of the upfront payment allocated to the warrant A and initial warrant B reflects the fair value of these financial liabilities at the warrant approval date (October 22, 2019). The value allocated to the subsequent warrant B reflects the fair value of the underlying liability as of December 31, 2021, 2022 and 2023 since subsequent warrant B is not yet approved for issuance. Subsequent warrant B is included in our deferred income in our statement of financial position.
On November 6, 2019 Gilead exercised warrant A, which resulted in an additional equity investment of €368.0 million.
At the inception of the collaboration with Gilead we identified the following three performance obligations: (i) the transfer of an extended license on ziritaxestat GLPG1690, (ii) the granting of exclusive access to our drug discovery platform (i.e. the IP, technology, expertise and capabilities) during the collaboration period and exclusive option rights on our current and future clinical programs after Phase 2 (or, in certain circumstances, the first Phase 3 study) outside Europe and (iii) an increased cost share from 20/80 to 50/50 for the global development activities of filgotinib, as a result
126
of the revised license and collaboration agreement. As part of the collaboration, Gilead also received option rights for GLPG1972, a Phase 2b candidate for osteoarthritis, in the United States.
Please refer to the note 4 “Critical accounting judgments and key sources of estimation uncertainty” - of this annual report for further explanation of critical judgments in applying accounting policies.
From the transaction proceeds received from Gilead, $738.0 million (€667.0 million) was allocated to the license on GLPG1690, $710.0 million (€641.7 million) was allocated increasing the cost share from 20/80 to 50/50 on the global development activities of filgotinib, and on December 31, 2019, $2,528.1 million (€2,284.7 million) was allocated to obtain exclusive access rights to our drug discovery platform. The amount allocated to the drug discovery platform also considered the additional effects on the transaction price from derivative financial intruments triggered by the share subscription agreement and the warrants granted to Gilead. We refer to the note 2 of this annual report tittled “Summary of significant transactions” for the allocation of the transaction price received from Gilead.
Terms of the collaboration
We will fund and lead all discovery and development autonomously until the end of Phase 2. After the completion of a qualifying Phase 2 study (or, in certain circumstances, the first Phase 3 study), Gilead will have the option to acquire a license to the compound outside Europe. If the option is exercised, we and Gilead will co-develop the compound and share costs equally. Gilead will maintain option rights to our programs through the 10-year term of the collaboration. This term can be extended for up to an additional three years thereafter for those programs, if any, that have entered clinical development prior to the end of the collaboration term. In addition, a final term extension can be granted in certain circumstances. Development of ziritaxestat GLPG1690 was discontinued in February 2021.
After the completion of the ongoing Phase 2b study of GLPG1972 concerning osteoarthritis, Gilead had the option to pay a $250 million fee to license the compound in the United States. In November 2020, Gilead declined to exercise its option to GLPG1972.
For any additional programs resulting from the collaboration, Gilead will make a $150 million opt-in payment per program and will not be subject to subsequent milestones payments. We will receive tiered royalties ranging from 20%-24% on net sales of all our products licensed by Gilead in all countries outside Europe.The collaboration is further described in “Item 4 – Collaborations – Option, License and Collaboration Agreement with Gilead”.
Filgotinib collaboration
In accordance with the collaboration agreement, as revised in 2019, we and Gilead would co-commercialize filgotinib in France, Germany, Italy, Spain and the United Kingdom and retain a 50/50 profit share in these countries pursuant to the original filgotinib license agreement. We would also share future global development costs for filgotinib equally with Gilead until a predetermined level, in lieu of the 80/20 cost split provided by the original agreement.
In December 2020, we agreed under a binding term sheet to further amend the collaboration agreement between us and Gilead, and as a result of such amendment we have assumed all development, manufacturing, commercialization and certain other rights for filgotinib in Europe. Through a phased transition during which most of the activities concerning filgotinib were transferred by Gilead to us by December 31, 2021, we became the marketing authorization holder for Jyseleca in the European economic area and Great Britain, that was completed by December 31, 2022, leaving us with the sole right to commercialize filgotinib in Europe. Until December 31, 2021, we continued to share equally with Gilead in the net profit and net losses in each of the Netherlands, Belgium, Luxembourg, France, Germany, Italy, Spain and UK. All commercial activities and economics concerning filgotinib in Europe transferred to us as of January 1, 2022, subject to payment of tiered royalties of 8% to 15% of net sales in Europe to Gilead, starting in 2024. Gilead retains commercial rights and remain marketing authorization holder for filgotinib outside of Europe, including in Japan.
Since January 1, 2021, we have been responsible for the development costs for certain studies concerning filgotinib, in lieu of the equal cost split contemplated by the previous agreement. These studies include the DARWIN3, FINCH4, FILOSOPHY, and Phase 4 studies and registries in RA, MANTA and MANTA-RAy, the PENGUIN1 and 2
127
and EQUATOR2 studies in PsA, the SEALION1 and 2 studies in AS, the HUMBOLDT study in uveitis in addition to other clinical and non-clinical expenses supporting these studies and support for any investigator sponsored trials in non-IBD conditions and non-clinical costs on all current trials. The existing 50/50 global development cost sharing arrangement between us and Gilead continues for the following studies: SELECTION and its long-term extension study (LTE) in UC, DIVERSITY and its LTE, DIVERGENCE 1 and 2 and their LTEs and support for Phase 4 studies and registries in Crohn’s disease, pediatric studies and their LTEs in RA, UC and Crohn’s disease, and support for investigator sponsored trials in IBD.
In September 2021, we and Gilead agreed to transfer the sponsorship of the DIVERSITY study and its LTE study from Gilead to us. The transfer was intended to be completed by June 30, 2022 and completed by March 2023. Since April 1, 2022, we are solely responsible for all development costs for the DIVERSITY study and the related LTE study.
In March 2022, we and Gilead agreed to transfer the sponsorship of and the operational responsibility for the MANTA study and its long-term extension to Galapagos. The transfer was substantially completed by December 31, 2022.
In connection with our entry into the initial collaboration agreement with Gilead on filgotinib, we received in January 2016 an upfront payment of $725 million consisting of a one-time, non-refundable, non-creditable license fee in the amount of $300 million and a $425 million equity investment by Gilead. In November 2016, Gilead initiated a Phase 3 trial in CD, for which we received a $50.0 million (€45.7 million) payment. In December 2016, Gilead initiated a Phase 2 trial in UC for which we received a $10.0 million (€9.4 million) payment. In April 2017, Galapagos initiated a Phase 2 trial in psoriatic arthritis as a new indication, for which we received a $10.0 million (€9.4 million) payment. In May 2018, Gilead initiated a Phase 3 trial in UC for which we received $15.0 million (€12.4 million).
In connection with the revised agreement in July 2019, $710 million (€641.7 million) of upfront consideration received from Gilead was allocated to the extended cost sharing for development costs of filgotinib.
In December 2019, Gilead initiated a Phase 3 trial in psoriatic arthritis for which we received $10.0 million (€9.1 million). In December 2019, Gilead filed an NDA for filgotinib in the U.S. for which we received a $20 million payment in January 2020. In September 2020 filgotinib was approved by both the European and the Japanese authorities, for which we received a $105.0 million (€90.2 million) payment in October 2020. In connection with the December 2020 binding term sheet that we entered into with Gilead, pursuant to which we agreed to amend the existing arrangement for the commercialization and development of filgotinib, Gilead agreed to irrevocably pay Galapagos €160 million, subject to certain adjustments for higher than budgeted development costs. Gilead paid €35 million in January 2021, €75 million in April 2021 and €50 million in March 2022. In addition, we were no longer eligible to receive any future milestone payments relating to filgotinib in Europe.
Under the terms of the agreement of September 2021, in July 2022, Gilead made a one-time payment of $15 million to us.
In March 2022 filgotinib was approved by the Japanese authorities for UC, for which we received a $20.0 million (€18.2 million) payment in May 2022.
In October 2023, Gilead and we agreed to further amend the collaboration. Gilead and we agreed to terminate the existing 50/50 global development cost sharing arrangement, with us bearing the costs going forward, and to terminate our obligation to pay tiered royalties to Gilead on net sales of Jyseleca® in Europe, in addition to other amendments. Effective 31 January 2024, following the closing of the transaction between Galapagos and Alfasigma S.p.A. to transfer the Jyseleca® business to Alfasigma S.p.A., Galapagos assigned its rights and obligations under the filgotinib collaboration to Alfasigma S.p.A., except for Galapagos’ right to receive royalties from Gilead on net sales in the Gilead Territory under a separate agreement between Gilead and Galapagos entered into in October 2023.
We remain eligible to receive tiered royalty percentages ranging from 20% to 30% on Gilead's global net sales of filgotinib outside of Europe.
128
The collaboration is further described in “Item 4 – Collaborations -- Exclusive collaboration agreement with Gilead for filgotinib”.
Terms of the equity investment
As part of the research and development collaboration Gilead also entered into a share subscription agreement with us. Gilead’s equity investment consisted of a subscription for new Galapagos shares at a price of €140.59 per share, representing at July 14, 2019 a 20% premium to Galapagos’ 30-day, volume-weighted average price. This equity subscription took place at the close of the transaction, on August 23, 2019 and increased Gilead’s stake in Galapagos from approximately 12.3% to 22.04% of the then issued and outstanding shares of Galapagos.
In addition, the extraordinary general meeting of the shareholders held on October 22, 2019 approved the issuance of warrant A and initial warrant B allowing Gilead to further increase its ownership of Galapagos to up to 29.9% of the company’s issued and outstanding shares. The initial warrant B has a term of five years and an exercise price per share equal to the greater of (i) 120% multiplied by the arithmetic mean of the 30-day daily volume weighted average trading price of Galapagos’ shares as traded on Euronext Brussels and Euronext Amsterdam, and (ii) EUR 140.59.
Subsequent warrant B is still subject to approval by an extraordinary general meeting of the shareholders. This extraordinary general meeting of shareholders shall take place between 57 and 59 months after the closing of the subscription agreement (August 23, 2019) and this warrant will have substantially similar terms, including the exercise price, to the initial warrant B. The agreement also includes a 10-year standstill restricting Gilead’s ability to propose a business combination with or acquisition of Galapagos or increase its stake in Galapagos beyond 29.9% of the company’s issued and outstanding shares, subject to limited exceptions.
On November 6, 2019 Gilead exercised warrant A and increased its ownership in Galapagos to 25.10% of the then outstanding shares of us. Gilead’s ownership at December 31, 2023 was 25.35%.
Distribution agreement for Jyseleca with Sobi
In October 2021, we signed an agreement (as amended from time to time) with Swedish Orphan Biovitrum AB (‘Sobi’) regarding the distribution of Jyseleca. Sobi acts as our distribution and commercialization partner of Jyseleca and will distribute the medicine in Central and Eastern Europe, Greece, Portugal, and the Baltic countries. Launches or first sales of Jyseleca in the aforementioned countries trigger milestone payments. We recorded milestones of €2.0 million in 2022 and milestones of €2.0 million in 2023 triggered by the first sale of Jyseleca in certain specific countries in Europe by Sobi.
Effective January 31, 2024, following the closing of the transaction between Galapagos and Alfasigma S.p.A. to transfer the Jyseleca® business to Alfasigma S.p.A., Galapagos assigned its rights and obligations under the Sobi agreement to Alfasigma S.p.A.
Drug discovery collaboration transaction with NovAliX
Effective July 1, 2023, we entered into an integrated drug discovery collaboration with NovAliX, a drug-discovery focused Contract Research Organization based in Strasbourg, France. Under the terms of the agreement, Galapagos’ drug discovery and research activities conducted in Romainville, France, and Galapagos’ employees in Romainville, which were exclusively dedicated to the operation of these activities, were transferred to NovAliX who will assume all ongoing research and discovery activities in Romainville, and this for no consideration. In return, Galapagos is committed to utilizing the research capabilities and expertise of NovAliX through a five year-collaboration and within the context of the company’s R&D portfolio, during which Galapagos is committed to purchase for a total of €73.8 million services from NovAliX.
129
Financial operations overview
Assets held for sale and discontinued operations
Where applicable and in accordance with IFRS 5, we have restated the 2021 and 2022 comparatives in the consolidated income statement and in the notes to consider the impact of classifying the Jyseleca® business as discontinued operations in 2023. (and Fidelta our fee-for-service business in 2021).
Product net sales
Revenue on the sale of Jyseleca® is recorded as “Product net sales” in our consolidated income statement (presented as dicontinued operations in this annual report).
Product net sales is the net amount of revenue recognized resulting from transferring control over our products to our customer (for example wholesalers and hospitals). Product sales revenue is recognized at a point in time when control of the goods has been transferred to the customer. This is generally when the goods are delivered to the customer depending on the specific incoterms in the contract with a customer.
The amount of revenue recognized is the amount allocated to the satisfied performance obligation taking into account variable consideration. The estimated amount of variable consideration is included in the transaction price only to the extent that it is highly probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. Variable consideration that is included in the transaction price is primarily composed of rebates, discounts, cash discounts and chargebacks granted to various customers that are part of commercial and governmental contractual arrangements or other reimbursement programs. Shelf stock adjustments are granted to some of our customers to cover the inventory held by them at the time of a price decrease becomes effective. A liability is recognized for expected rebates, cash discounts, chargebacks or other reimbursements payable directly or indirectly to customers in relation to sales made until the end of the reporting period.
The amount of variable consideration is estimated using several elements such as third-party market data, product pricing, the specific terms in the individual agreements, estimated inventory levels and the shelf life of our product. If actual results differ, these estimates will be adjusted.
Net sales are presented net of value added tax and other sales related taxes.
Collaboration Revenue
Collaboration revenues to date have consisted principally of milestones, license fees, non-refundable upfront fees and royalties received in connection with collaboration and license agreements.
The revenue recognition policy can be summarized as follows:
We recognize revenue when our customer obtains control of promised goods or services, in an amount that reflects the consideration that we expect to receive in exchange for those goods or services. To determine revenue recognition for agreements that we determine are within the scope of IFRS 15, we perform the following five steps:
(i) identify the contract
In our agreements with customers, we are mainly transferring licenses on our IP and in some cases this is combined with access rights and/or providing research and development services and/or cost sharing mechanisms. In some cases our collaborations also include an equity subscription component. If this is the case, we analyze if the criteria to combine contracts, as set out by IFRS 15, are met.
130
(ii) identify the performance obligations in the contract
Depending on the type of the agreement, there can be one or more distinct performance obligations under IFRS 15. This is based on an assessment of whether the promises in an agreement are capable of being distinct and are distinct from the other promises to transfer goods and/or services in the context of the contract. For some of our agreements we combine the transfer of the license with the performance of research and development activities because we consider that the license is not capable of being distinct and is not distinct in the context of the contract.
(iii) determine the transaction price
Collaboration and license agreements with our commercial partners for research and development activities generally include non-refundable upfront fees; milestone payments, the receipt of which is dependent upon the achievement of certain clinical, regulatory or commercial milestones; license fees, royalties on sales and sometimes reimbursement income or profits sharing arrangements.
a/ License fees or upfront payments
If the license to our intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, we recognize revenues from non-refundable upfront fees allocated to the license at the point in time the license is transferred to the customer and the customer has the right to use the license.
For licenses that are bundled with other promises, we utilize judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time. If over time, revenue is then recognized based on a pattern that best reflects the transfer of control of the service to the customer.
b/ Milestone payments other than sales based milestones
A milestone payment is only included in the transaction price to the extent that it is highly probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved (which is generally only when the milestone is achieved). We estimate the amount to be included in the transaction price using the most likely amount method, where milestone payments are included in the transaction price upon achievement of the milestone event. The transaction price is then allocated to each performance obligation on a stand-alone selling price basis, for which we recognize revenue as or when the performance obligations under the contract are satisfied. At the end of each subsequent reporting period, we re-evaluate the probability of achievement of such milestones and any related constraint, and, if necessary, adjust our estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect revenue and earnings in the period of adjustment.
c/ Reimbursement income for R&D services
Collaboration and license agreements may include reimbursement or cost sharing for research and development services: such as outsourcing costs and payment for full-time equivalents at contractual rates. R&D services are performed and satisfied over time given that the customer simultaneously receives and consumes the benefits provided by us.
Such costs reimbursements received are recognized in revenues when costs are incurred and agreed by the parties when we are acting as a principal in the scope of our stake of the R&D activities. If the later condition is not fulfilled, costs reimbursements are accounted for as a decrease of the related expenses.
d/ Sales based milestone payments and royalties
License and collaboration agreements include sales-based royalties, including commercial milestone payments based on the level of sales, and the license has been deemed to be the predominant item to which the royalties relate. Related revenue is recognized as the subsequent underlying sales occur.
131
(iv) allocate the transaction price to the performance obligations in the contract
We allocate the transaction price to each performance obligation identified in the contract based upon the stand-alone selling price. The stand-alone selling price of each performance obligation is estimated by using one of the following methods: adjusted market assessment approach, the expected cost plus a margin approach or the residual approach. If management assesses that there is only one single performance obligation, the entire transaction price would be allocated to this performance obligation.
(v) recognize revenue when (or as) the entity satisfies a performance obligation
Revenue is recognised when our customer obtains control of the goods and/or services foreseen in the contracts. The control can be transferred over time or at a point in time – which result in recognition of revenue over time and at a point in time.
In case of revenue recognition over time, we use either an input model that considers estimates of the percentage of total research and development costs that are completed each period compared to the total estimated costs (percentage of completion method) to measure the progress of the satisfaction of the underlying performance obligation (which is the method applied for the filgotinib performance obligation). In other cases, depending on specific circumstances, we recognize revenue on a straight-line basis over the estimated term of the performance obligation (which is the method applied for the performance obligation related to our drug discovery platform).
Cost of sales
Our cost of sales includes primarily the purchase cost of the goods sold and transportation costs.
R&D expenditure
Expenses on R&D activities are recognized as an expense in the period in which the expense is incurred.
Our R&D expenditure consists of costs associated with our R&D activities such as:
| ● | personnel costs associated with employing our team of R&D staff, including salaries, social security costs, and share-based compensation expenses; |
| ● | disposables and lab consumables used in the conduct of our in-house research programs; |
| ● | payments for research work conducted by sub-contractors and sponsorship of work by our network of academic collaborative research scientists; |
| ● | subcontracting costs paid to contracted research organizations, or CROs, for our preclinical studies or clinical trials, as well as costs associated with safety studies; |
| ● | Professional fees to support our R&D activities; |
| ● | costs paid to our collaboration partners and reimbursements received from our collaboration partners in the scope of the cost sharing agreements of our collaborations |
| ● | premises costs associated with our laboratory and office space to accommodate our teams; |
| ● | depreciation and impairment of fixed assets used to develop our product candidates; and |
| ● | other operating expenses, namely software and licenses, maintenance costs for equipment, travel costs, and office expenses. |
132
Our R&D expenses are expected to decrease overall, driven by the recent transfer of the filgotinib development to Alfasigma on January 31, 2024, the discontinuation of activities in fibrosis and kidney disease initiated in 2022 as a result of our new strategy, while advancing our immunology pipeline and accelerating investments in our oncology programs. Since 2021, we cumulatively have spent €1,438.3 million on R&D activities which can be split as follows between the key programs:
Year ended December 31, | |||||||||||||||
| 2023 |
| 2022 |
| 2021 |
|
| ||||||||
(Euro, in thousands) | Cumulative | ||||||||||||||
Filgotinib program | € | (190,177) | € | (245,286) | € | (171,204) | € | (606,667) | 42% | ||||||
Ziritaxestat program | — | (1,096) | (26,725) | (27,821) | 2% | ||||||||||
SIKi program | (18,900) | (47,727) | (91,957) | (158,584) | 11% | ||||||||||
TYK2 program on GLPG3667 | (31,289) | (24,467) | (27,141) | (82,897) | 6% | ||||||||||
CAR-T programs in oncology | (82,218) | (29,999) | — | (112,217) | 8% | ||||||||||
Other programs | (108,887) | (166,507) | (174,680) | (450,074) | 31% | ||||||||||
Total R&D expenses | € | (431,471) | € | (515,083) | € | (491,707) | € | (1,438,260) | 100% | ||||||
Other programs comprise expenditure for other projects in research phase and other early stage development programs in inflammation, fibrosis and other indications.
The decrease in our R&D expenditure for the year ended December 31, 2023, compared to the year before, was primarily due to reduced spend on the filgotinib development, our SIKi, and other programs. This was partly offset by cost increases for our TYK2 program in immunology and our new CAR-T programs in oncology.
The decrease in our R&D expenditure for the filgotinib development is primarily explained by the discontinuation of the DIVERSITY clinical trial in CD.
Sales and marketing expenses
Sales and marketing expenses primarily include costs associated with managing the commercial activities related to Jyseleca® in Europe.
Expenses on S&M activities are recognized as an expense in the period in which the expense is incurred.
Our sales and marketing expenses consists of costs associated with our commercial activities such as:
| ● | personnel costs associated with employing our team of commercial and supply chain staff, including salaries, social security costs, and share-based compensation expenses; |
| ● | subcontracting costs related to contracted agencies for marketing campaigns and materials, business analytics, market research and promotional expenses. |
| ● | professional fees to support our commercial activities; |
| ● | costs charged to us by Gilead or recharged by us to Gilead in the scope of our co-commercialization cost sharing agreement for filgotinib in Belgium, the Netherlands, Luxembourg, France, Italy, Spain, Germany and Great Britain until December 31, 2021 |
| ● | depreciation of fixed assets used by our commercial staff. |
| ● | other operating expenses, namely press and communication fees, operational taxes, costs of samples, travel and accommodation costs, and office expenses. |
Our sales and marketing expenses are expected to strongly decrease, as a consequence of the transfer of the Jyseleca® business to Alfasigma on January 31, 2024.
133
General and administrative expenses
General and administrative expenses consist primarily of salaries and benefits related to our executive, finance, human resources, business development, legal, intellectual property, and information technology support functions, as well as depreciation and impairment costs of fixed assets. Professional fees reported under general and administrative expenses mainly include legal fees, accounting fees, audit fees, and fees for taxation advisory and other consultancy costs. Other general and administrative operating expenses primarily encompass software and license costs, equipment maintenance and leasing costs, consultancy costs, insurance costs, Belgian tax on securities accounts, office expenses, and travel costs.
Our general and administrative expenses are expected to decrease due to a reduction of the staff as a consequence of the transfer of the Jyseleca® business to Alfasigma on January 31, 2024.
Grants and R&D incentives
We benefit from various grants and R&D incentives from certain governmental agencies. These grants and R&D incentives generally aim to partly reimburse approved expenditures incurred in our R&D efforts and are credited to the income statement, under other operating income, when the relevant expenditure has been incurred and there is reasonable assurance that the grant or R&D incentive is receivable. The main grants and R&D incentives are as follows:
| ● | Companies in Belgium are eligible to receive R&D incentives linked to R&D investments (equaling 25% of 20.5% of the investment value in 2023). This R&D tax credit results in a cash inflow to us from the tax authorities five years after the investment was made and capitalized in our standalone financial statements under Belgian GAAP for the portion that has not been used to offset the payment of corporate tax or is paid to us for the portion that remains unused. In 2021 and in 2023 we also received a grant from the National Institute for Health and Disability Insurance. This grant aims to incentivize innovative Belgian biotech companies who are performing research and development activities in order to identify new medicines. Finally, we also benefit from certain rebates on payroll withholding taxes for scientific personnel. |
| ● | In France, we benefit from R&D incentives from the French Government for R&D activities whereby 30% of qualifying R&D expenses can be recuperated. This research tax credit (crédit d’impôt recherche) results in a cash inflow to us from the tax authorities after three years, i.e., it is used to offset the payment of corporate tax or is paid to us for the portion that remains unused. Qualifying expenditures largely comprise employment costs for research staff, consumables, and certain overhead costs as well as capped outsourcing costs incurred as part of R&D projects. |
Fair value adjustments and net exchange differences
Fair value gains and losses on current financial investments consist of the interest on treasury bills which have not yet expired (until 2022, as from 2023 the interest on money market funds are included), the effect of the re-measurement at fair value of our money market funds, including the exchange differences on money market funds. These money market funds qualify for level 1 fair value measurement based upon the closing price of the investment at each reporting date.
Foreign currency exchange gain and loss comprises the realized and unrealized effects from currency exchange rate fluctuations on our balance sheet positions denominated in foreign currency. For the year ended December 31, 2023, currency exchange losses were primarily due to currency exchange rate differences on our cash held in foreign currency, and primarily in U.S. dollars. On December 31, 2023 our cash and cash equivalents and current financial investments included $865.4 million held in U.S.dollars, which could generate foreign currency exchange gain or loss in our financial results in accordance with the fluctuation of the EUR/U.S.dollar exchange rate as our functional currency is EUR.
134
Fair value re-measurement of warrants refers to initial warrant B. As this initial warrant B is not yet exercised by Gilead per December 31, 2023, we re-measured the financial liability relating to this warrant on December 31, 2021, on December 31, 2022 and on December 31, 2023 and recognized the resulting change in fair value between the year-end 2020 and year-end 2021, between year-end 2021 and year-end 2022 and between year-end 2022 and year-end 2023 in profit or loss. The recognized fair value gain of €3.0 million in 2021, €0.2 million in 2022 and of €0.02 million in 2023 are mainly the result of the change in the implied volatility of our share price and the evolution of our share price itself for these three periods. On December 31, 2023, the fair value of the financial liability related to the initial warrant B amounts to nil.
The financial liability will be re-measured at fair value at each reporting period.
Fair value losses on financial assets held at fair value through profit or loss consisted in 2021 of the effect of re-measurement of financial assets classified as equity investments held at fair value through profit or loss, which qualify for level 1 fair value measurement based upon the closing price of such securities at each reporting date, and for 2023 of an unrealized exchange loss on a participation in a non-listed company. Any gain or loss realized upon the sale of equity instruments is reported in other financial expense or in other financial income.
Other financial expense and financial income
Interest expense consists primarily of interest expense incurred on certain of our term deposits, treasury bills and leases.
Interest income consists primarily of interest earned by investing our cash reserves in interest-bearing deposit accounts, notice accounts and in current financial investments. Interest income increased due to increasing interest rates.
Other financial expenses also include the discounting component of other non-current liabilities, being the deferred consideration and milestones payable related to the acquisition of subsidiaries.
Taxation
With the exception of the year ended December 31, 2019 and 2023, we have a history of losses. We forecast to continue incurring losses as we continue to advance our immunology pipeline and accelerate investments in our oncology programs.
Consequently, we do not have any net deferred tax asset on the balance sheet as at December 31, 2023, except for subsidiaries working on a cost plus basis for which a deferred tax asset was set up for an amount of €1.1 million as of December 31, 2023. As a result of the business combination related to the acquisitions of CellPoint and AboundBio in 2022, we also recognized on acquisition date a net deferred tax liability of €23.3 million, consisting of deferred tax liabilities (€32.3 million) based upon the fair value of the acquired intangible assets less recognized deferred tax assets (€9.0 million). As a company active in research and development in Belgium, we also expect to benefit from the “innovation income deduction”, or IID in Belgium. The innovation income deduction regime allows net profits attributable to revenue from among others patented products (or products for which the patent application is pending) to be taxed at a lower effective tax rate than other revenues. The effective tax rate can thus be reduced up to 3.75%.
Operating segments
We are currently operating as a single operating segment. Prior to the disposal of Fidelta we had two reportable segments, R&D and fee-for-service business. Due to the disposal of Fidelta (our fee-for-service segment) on January 4, 2021, we reported this segment as discontinued operations in 2021.
Financial information related to our operational segment and geographic information is contained in “Note 6—Segment information” in our consolidated financial statements appended to this annual report.
135
Risks
For further information regarding governmental economic, fiscal, monetary or political policies or factors that have materially affected, or could materially affect, directly or indirectly, our operations, please see the section of this annual report titled “Item 3.D.—Risk Factors.”
Critical accounting judgments and key sources of estimation uncertainty
We refer to “Note 4-Critical accounting judgments and key sources of estimation uncertainty” in our consolidated financial statements appended to this annual report.
New standards and interpretations applicable for the annual period beginning on January 1, 2022 and for the annual period beginning on January 1, 2023
We refer to “Note 3-Material accounting policies” in our consolidated financial statements appended to this annual report.
136
| A. | Operating results |
Comparison of years ended December 31, 2023 and 2022
On January 31, 2024 we announced that we successfully completed the transfer of the Jyseleca® business to Alfasigma. The transfer of our Jyseleca® business has been determined to meet the criteria to be classified as held for sale and discontinued operations in our financial statements for the year ended December 31, 2023. We also presented all income statement items fully related to the Jyseleca® business to be transferred on a separate line “Net income/loss (-) from discontinued operations, net of tax” in our consolidated income statement. The consolidated income statement for all comparative periods reported in these consolidated financial statements were restated as well to show the discontinued operations on a separate line.
The abbreviation n.m. in the column ‘% change’ refers to ‘not meaningful’.
The following table summarizes the results of our operations for the years ended December 31, 2023 and 2022, together with the changes to those items.
Year ended December 31, | ||||||||
| 2023 |
| 2022 (*) |
| % Change | |||
(Euro, in thousands, | ||||||||
Collaboration revenues | 239,724 | 241,249 | (1%) | |||||
Total net revenues | 239,724 | 241,249 | (1%) | |||||
Research and development expenses | (241,294) | (269,797) | (11%) | |||||
Sales and marketing expenses | (5,676) | (3,480) | 63% | |||||
General and administrative expenses | (128,289) | (135,155) | (5%) | |||||
Other operating income | 47,272 | 36,127 | 31% | |||||
Operating loss | (88,263) | (131,056) | (33%) | |||||
Fair value adjustments and net exchange differences | 16,252 | 51,498 | (68%) | |||||
Other financial income | 80,249 | 18,563 | 332% | |||||
Other financial expenses | (2,613) | (9,854) | (73%) | |||||
Profit/loss (-) before tax | 5,625 | (70,849) | (108%) | |||||
Income taxes | (9,613) | (572) | 1581% | |||||
Net loss from continuing operations | (3,988) | (71,421) | (94%) | |||||
Net profit/loss (-) from discontinued operations, net of tax | 215,685 | (146,570) | (247%) | |||||
Net profit/loss (-) | € | 211,697 | € | (217,991) | ||||
Net profit/loss (-) attributable to: | ||||||||
Owners of the parent | 211,697 | (217,991) | ||||||
Basic and diluted earnings/loss (-) per share | € | 3.21 | € | (3.32) | ||||
Basic and diluted loss per share from continuing operations | € | (0.06) | € | (1.09) | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
137
Results from Discontinued Operations
On October 30, 2023 we announced that we had signed a letter of intent contemplating a transfer of the Jyseleca® business to Alfasigma, including the European and UK Marketing Authorizations, the commercial, medical and development activities for Jyseleca® and approximately 400 positions in 14 European countries.
On December 30, 2023, we signed a final share and asset purchase agreement with Alfasigma.
On December 31, 2023, the transaction was still subject to certain closing conditions such as the finalization of the consultation process with the workers councils and FDI clearance in Italy, France and Denmark. The transaction was closed on January 31, 2024, upon obtaining all necessary approvals. We received a €50.0 million upfront payment in 2024, are entitled to potential sales-based milestone payments totalling €120.0 million and mid-single to mid-double-digit royalties on European sales. We will contribute up to €40.0 million to Alfasigma by June 2025 for Jyseleca® related development activities.
The transfer of our Jyseleca® business has been determined to meet the criteria to be classified as held for sale and discontinued operations in our financial statements for the year ended December 31, 2023.
The post-tax result from discontinued operations can be disaggregated in the following items:
| i) | Financial performance |
Year ended December 31, | ||||||||
| 2023 |
| 2022 |
| % Change | |||
(Euro, in thousands, | ||||||||
Product net sales | € | 112,339 | € | 87,599 | 28% | |||
Collaboration revenues | 431,465 | 176,432 | 145% | |||||
Total net revenues | 543,804 | 264,031 | 106% | |||||
Cost of sales | (18,022) | (12,079) | 49% | |||||
Research and development expenditure | (190,177) | (245,286) | (22%) | |||||
Sales and marketing expenses | (113,356) | (144,075) | (21%) | |||||
General and administrative expenses | (17,989) | (9,776) | 84% | |||||
Other operating income | 13,003 | 10,721 | 21% | |||||
Operating profit/loss (-) | 217,262 | (136,464) | n.m. | |||||
Fair value adjustments and net currency exchange differences | (13) | (25) | (48%) | |||||
Other financial income | 679 | 15 | n.m. | |||||
Other financial expenses | (167) | (7,825) | (98%) | |||||
Profit/loss (-) before tax | 217,761 | (144,298) | n.m. | |||||
Income taxes | (2,076) | (2,272) | (9%) | |||||
Net profit/loss (-) | € | 215,685 | € | (146,570) | n.m. | |||
Basic and diluted earnings/losss (-) per share from discontinued operations | € | 3.27 | € | (2.23) | ||||
Weighted average number of shares (in thousands of shares) | 65,884 | 65,699 | ||||||
Weighted average number of shares - Diluted (in thousands of shares) | 65,933 | 65,699 | ||||||
138
Jyseleca® product net sales in Europe amounted to €112.3 million in 2023, compared to €87.6 million in 2022, of which €8.1 million realized in Belgium (€7.3 million in 2022).
Collaboration revenues in discontinued operations related to revenue recognition of the collaboration agreement with Gilead for the filgotinib development amount to €429.4 million in 2023 compared to €174.4 million for last year. Effective January 31, 2024, following the closing of the transaction between us and Alfasigma S.p.A. to transfer the Jyseleca® business to Alfasigma S.p.A., we assigned our rights and obligations under the filgotinib collaboration with Gilead to Alfasigma S.p.A., except for our right to receive royalties from Gilead on net sales in the Gilead Territory under a separate agreement between Gilead and us entered into in October 2023. As a consequence, our performance obligation towards Gilead for the development of filgotinib will come to its end, and the total estimated remaining costs to complete the filgotinib development was substantially reduced leading to a major increase in the percentage of completion of our performance obligation (applying the “cost-to-cost” input model) and a considerable positive catch-up of revenue explaining the increase in revenue recognition for the year 2023 compared to 2022. We refer to note 2 “Summary of significant transactions” of this annul report for a general description of our collaboration with Gilead.
On December 31, 2023, the remaining deferred income related to the filgotinib development amounts to €26.3 million that will mainly be released in revenue in 2024.
Additionally, we recorded in both 2023 and 2022 €2.0 million milestone payments triggered by the initial sales of Jyseleca® in specific countries by our distribution and commercialization partner Sobi.
The decrease in R&D expenditures was mainly due to the discontinuation early 2023 of the DIVERSITY clinical trials in CD. Personnel expenses decreased by €15.0 million, from €74.6 million in 2022 to €59.6 million in 2023. Subcontracting costs decreased as well by €39.0 million, from €153.7 million in 2022 to €114.7 million in 2023.
The decrease in S&M expenses is reflected in a decrease in personnel expenses by €10.8 million, from €70.2 million in 2022 to €59.3 million in 2023 due to lower bonus costs and costs of our subscription right plans, while external outsourcing costs decreased by €17.0 million, from €52.8 million in 2022 to €35.8 million in 2023 primarily explained by lower costs for marketing campaigns and promotional expenses. G&A expenses attributable to the Jyseleca® business increased primarily due to an increase in costs of our subscription right plans explained by unusually low costs in 2022 due to reversal of cost related to voluntary leavers, as well as increase in salaries. The G&A expenses for the year 2023 also include one-off legal fees related to the transaction with Alfasigma for €3.5 million.
Other operating income attributable to the Jyseleca® business increased, mainly due to higher R&D incentives income.
The movement in other financial income/expenses is primarily explained by a lower discounting effect of long-term deferred revenue for the development of filgotinib because we expect to recognize the remaining revenues in 2024. The financing component related to our filgotinib performance obligation was re-assessed on December 31, 2023 considering the reduced duration and the expected end of the performance obligation for the development of filgotinib.
139
ii) Assets and liabilities
December 31, 2023 | |||
(Euro, | |||
Property, plant and equipment | € | 4,194 | |
Deferred tax assets | 292 | ||
Other non-current assets | 598 | ||
Inventories | 737 | ||
Trade and other receivables | 15,786 | ||
Cash and cash equivalents | 7 | ||
Other current assets | 471 | ||
Total assets in disposal group classified as held for sale | 22,085 | ||
| |||
| |||
Retirement benefit liabilities | 1,160 | ||
Non-current lease liabilities | 2,327 | ||
Other non-current liabilities | 329 | ||
Current lease liabilities | 1,308 | ||
Trade and other liabilities | 25,619 | ||
Current tax payable | 1,242 | ||
Current deferred income | 59 | ||
Total liabilities directly associated with assets in disposal group classified as held for sale | 32,044 | ||
Net liability held for sale | € | (9,959) | |
Results from our continuing Operations
Collaboration revenues
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Recognition of non-refundable upfront payments and license fees | € | 230,242 | € | 230,423 | (0%) | ||||
Reimbursement income | — | 56 | n.m. | ||||||
Royalties | 9,482 | 10,770 | (12%) | ||||||
Total collaboration revenues | € | 239,724 | € | 241,249 | (1%) | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
140
We refer to note 2 “Summary of significant transactions” in our consolidated financial statements appended to this annual report for a general description of our collaboration with Gilead.
The following table summarizes details of collaboration revenues for the years ended December 31, 2023 and 2022 by collaboration and by category of revenue: upfront payments and license fees, reimbursement income and royalties.
Over time |
| Point in time | 2023 |
| 2022(*) | ||||
(Euro, in | (Euro, in | ||||||||
thousands) | thousands) | ||||||||
Recognition of non-refundable upfront payments and license fees | € | 230,242 | € | 230,423 | |||||
Gilead collaboration agreement for drug discovery platform | Ö | 230,242 | 230,423 | ||||||
Reimbursement income | - | 56 | |||||||
Novartis collaboration agreement for MOR106 | Ö | - | 56 | ||||||
Royalties | 9,482 | 10,770 | |||||||
Gilead royalties on Jyseleca | Ö | 9,466 | 10,726 | ||||||
Other royalties | Ö | 16 | 44 | ||||||
Total collaboration revenues | € | 239,724 | € | 241,249 | |||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
We recognize the consideration from Gilead allocated to the drug discovery platform on a linear basis over the 10-year period of our collaboration, of which we recognized €230.2 million in revenue in 2023. We expect to recognize the same amount in the coming years, until the end of the 10-year period.
Since signing of the letter of intent with Alfasigma in October 2023, we classified all activities that were directly related to the Jyseleca® business, including the revenue recognition related to the filgotinib performance obligation, as discontinued operations in accordance with IFRS 5. We refer to note 5 “Discontinued Operations” in our consolidated financial statements appended to this annual report for additional information.
For the year ended December 31, 2023 we also recognized in revenue €9.5 million of royalties from Gilead on filgotinib. The royalties on sales of Jyseleca® performed by Gilead in Japan were not reported as discontinued operations as we still have the right to receive those royalties on future sales made by Gilead and its commercialization partners (this right is not subject to transfer to Alfasigma as part of the transfer of the Jyseleca® business to them).
The outstanding balance of deferred income from the Gilead collaboration for the drug discovery platform at December 31, 2023 amounted to €1,299.2 million and is composed of €1,299.1 million that will be linearly recognized over the next years during the remainder of the 10 years of our collaboration and €0.1 million is related to the warrant issuance liability reserved for the subsequent warrant B.
141
R&D expenditure
The following table summarizes our R&D expenditure for the years ended December 31, 2023 and 2022, together with the changes to those items.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Personnel costs | € | (95,788) | € | (115,484) | (17%) | ||||
Subcontracting | (82,997) | (61,192) | 36% | ||||||
Disposables and lab fees and premises costs | (18,083) | (19,529) | (7%) | ||||||
Depreciation and impairment | (22,254) | (51,493) | (57%) | ||||||
Professional fees | (9,272) | (9,316) | (0%) | ||||||
Other operating expenses | (12,900) | (12,783) | 1% | ||||||
Total R&D expenses | € | (241,294) | € | (269,797) | (11%) | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The variance in our R&D expenditure in 2023 compared to 2022 was principally due to the following elements:
| ● | Depreciation and impairment decrease was primarily due to an impairment of €26.7 million of previously capitalized upfront fees related to our collaboration with Molecure on the dual chitinase inhibitor OATD-01 (GLPG4716) in fibrosis, and impairments of intangibles assets related to other discontinued projects for an amount of €8.9 million, both recorded in 2022. |
| ● | A decrease in personnel costs explained by a decrease in the R&D staff, lower bonus costs and lower accelerated non-cash cost recognition for subscription right plans related to good leavers. |
| ● | Increase in subcontracting costs following the evolution of our CAR-T program and TYK2 program. |
The table below summarizes our R&D expenditure for the years ended December 31, 2023 and 2022, broken down by program.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Ziritaxestat program | € | — | € | (1,096) | (100%) | ||||
SIKi program | (18,900) | (47,727) | (60%) | ||||||
TYK2 program on GLPG3667 | (31,289) | (24,467) | 28% | ||||||
CAR-T programs in oncology | (82,218) | (29,999) | 174% | ||||||
Other programs | (108,887) | (166,507) | (35%) | ||||||
Total R&D expenses | € | (241,294) | € | (269,797) | (11%) | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The decrease in R&D expenditure in 2023 was primarily explained by reduced spend on our SIKi and other programs primarily in kidney desease and fibrosis.
This was partly offset by higher investments in 2023 in our CAR-T programs in oncology and in our TYK2 program in immunology. Other programs comprise expenditure for other projects in research phase and other early-stage development programs.
142
Sales and marketing expenses
The following table summarizes our sales and marketing expenses for the years ended December 31, 2023 and 2022, together with the changes to those items.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Personnel costs | € | (2,997) | € | (1,693) | 77% | ||||
Depreciation | (113) | (59) | 92% | ||||||
External outsourcing costs | (1,776) | (1,267) | 40% | ||||||
Professional fees | (131) | (99) | 31% | ||||||
Other operating expenses | (659) | (363) | 82% | ||||||
Total sales and marketing expenses | € | (5,676) | € | (3,480) | 63% | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Personnel costs increased by €1.3 million in 2023 explained by an increase in staff working on oncology and other than Jyseleca® related projects.
General and administrative expenses
The following table summarizes our general and administrative expenses for the years ended December 31, 2023 and 2022, together with the changes to those items.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Personnel costs | € | (66,098) | € | (76,536) | (14%) | ||||
Depreciation and impairment | (15,978) | (8,529) | 87% | ||||||
Legal and professional fees | (23,250) | (23,715) | (2%) | ||||||
Other operating expenses | (22,963) | (26,375) | (13%) | ||||||
Total general and administrative expenses | € | (128,289) | € | (135,155) | (5%) | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The decrease in our general and administrative expenses in 2023 was mainly explained by a decrease in personnel expenses primarily due to lower accelerated non-cash cost recognition for our subscription right plans related to good leavers. This was partly offset by an impairment of €7.6 million on the construction project in Mechelen (Belgium).
Other operating income
The following table summarizes our other operating income for the years ended December 31, 2023 and 2022, together with the changes to those items.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Grant income | € | 6,618 | € | 1,873 | 253% | ||||
R&D incentives income | 32,968 | 29,104 | 13% | ||||||
Other income | 7,686 | 5,150 | 49% | ||||||
Total other operating income | € | 47,272 | € | 36,127 | 31% | ||||
143
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The grant income in 2023 mainly comprises a grant received in 2023 from the National Institute for Health and Disability Insurance amounting to €6.1 million. This grant aimed to incentivize innovative Belgian biotech companies who are performing research and development activities in order to identify new medicines.
In many cases these grant agreements carry clauses which require us to maintain a presence in the same region for a number of years and invest according to pre-agreed budgets.
R&D incentives income can be split up as follows:
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Income from innovation incentive system in France | € | 5,881 | € | 11,075 | (47%) | ||||
Income from Belgian R&D incentives | 16,535 | 10,339 | 60% | ||||||
Tax rebates on payroll withholding taxes of R&D personnel (Belgium & the Netherlands) | 10,552 | 7,689 | 37% | ||||||
Total R&D incentives income | € | 32,968 | € | 29,104 | 13% | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Other income increased mainly due to rental income and a one-off gain on the sale of fixed assets.
Fair value adjustments and net currency exchange differences
The following table summarizes our fair value adjustments and net currency exchange differences for the years ended December 31, 2023 and 2022, together with the changes to those items.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Net unrealized currency exchange gain/loss (-) | € | (20,544) | € | 41,559 | (149%) | ||||
Net realized currency exchange gain/loss (-) | (1,118) | 2,825 | (140%) | ||||||
Fair value re-measurement of warrants | 18 | 186 | (90%) | ||||||
Fair value loss on financial assets held at fair value through profit or loss | (390) | — | n.m. | ||||||
Fair value gain on current financial investments | 38,286 | 6,929 | 453% | ||||||
Total fair value adjustments and net currency exchange differences | € | 16,252 | € | 51,498 | (68%) | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The net currency unrealized exchange loss of €20.5 million in 2023 primarily related to €20.4 million of unrealized exchange loss on cash and cash equivalents and current financial investments at amortised cost held in U.S. dollars, as compared to an unrealized net exchange gain in 2022 of €41.3 million on cash and cash equivalents and current financial investments at amortized cost held in U.S. dollar. We have cash, cash equivalents and current financial investments held in U.S. dollars, which could generate foreign currency exchange gain or loss in our financial results in accordance with the fluctuation of the EUR/U.S. dollar exchange rate as our functional currency is EUR.
144
Fair value re-measurement of warrants refers to the fair value re-measurement of the Gilead initial warrant B. The fair value of the financial liability related to the Gilead initial warrant B of nil on 31 December 2023 (€0.02 million on 31 December 2022) was presented as part of trade and other liabilities in our consolidated statement of financial position and will be re-measured at each reporting period. We refer to note 2 “Summary of significant transactions” in our consolidated financial statements appended to this annual report for more information.
The fair value gain on the current financial investments reflects the exchange differences booked on the money market funds, the interest on the money market funds and the effect of the re-measurement at fair value of the money market funds on December 31, 2023. These re-measurement gains are mainly the result of the positive returns on the EUR denominated money market funds.
For more information on currency exchange fluctuations on our business, please see the section of this annual report titled “Item 11—Quantitative and qualitative disclosures about market risk—Foreign exchange risk.”
Other financial income and expense
The following table summarizes other financial income and expense for the years ended December 31, 2023 and 2022.
Year ended December 31, | |||||||||
2023 | 2022(*) |
| % Change | ||||||
(Euro, in thousands) | |||||||||
Other financial income: | |||||||||
Interest income | € | 79,290 | € | 18,094 | 338% | ||||
Discounting effect of non-current R&D incentives receivables | 617 | 93 | 563% | ||||||
Discounting effect of other non-current liabilities | 318 | — | n.m. | ||||||
Other finance income | 24 | 376 | (94%) | ||||||
Total other financial income | 80,249 | 18,563 | 332% | ||||||
Other financial expenses: | |||||||||
Interest expenses | (1,770) | (6,884) | (74%) | ||||||
Discounting effect of other non-current liabilities | — | (2,271) | (100%) | ||||||
Other finance charges | (843) | (699) | 21% | ||||||
Total other financial expense | € | (2,613) | € | (9,854) | (73%) | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Interest income was related to interests on term deposits, notice accounts and current financial investments. Interest income increased due to increased interest rates.
Interest expenses were related to interests on term deposits and treasury bills, on leases of buildings and cars. Other financial expense for 2023 also comprise the discounting effect of other non-current liabilities as deferred consideration and milestones payables related to the acquisition of subsidiaries.
145
Income Taxes
The following table summarizes our tax result for the years ended December 31, 2023 and 2022.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) | % Change | |||||
(Euro, in thousands) | |||||||||
Current tax | € | (5,928) | € | (1,738) | 241% | ||||
Deferred tax | (3,685) | 1,166 | (416%) | ||||||
Income taxes | € | (9,613) | € | (572) | 1581% | ||||
(*) The 2022 comparative has been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Current tax, consisting of corporate income taxes, and deferred tax income/loss (-) related to subsidiaries working on a cost plus basis. The increase in 2023 was primarily due to the re-assessment of net deferred tax liabilities and corporate income tax payables as a result of a one-off intercompany transaction. In addition, the deferred tax income for the year ended December 31, 2022, was largely due to the partial release of the net deferred tax liabilities related to the acquisitions of CellPoint and AboundBio.
We refer to note 11 “Income taxes” in our consolidated financial statements appended to this annual report.
Business combinations during the prior period
On June 21, 2022 we acquired, in an all-cash transaction, 100% of the shares and voting interests of CellPoint for a total agreed payment at completion of €125 million, including consideration for other liabilities associated with the transaction amounting to €10.3 million. Additional contingent consideration up to €100.0 million is due when certain development (€20.0 million), regulatory (€30.0 million) and sales-based (€50.0 million) milestones would be achieved. Total fair value at acquisition date of these milestones amounted to €20.2 million. This fair value is measured with most significant inputs being the probability of success of reaching these milestones, expected timing and the discount rate. The discounting impact is recognized in financial results.
On the same date we acquired all of the outstanding capital and voting interests of AboundBio, for a total agreed price of $14 million, including consideration for other liabilities associated with the transaction.
146
Details of the fair value of identifiable assets and liabilities acquired in both transactions, the purchase consideration, the goodwill at the acquisition date and the net cash outflow arising on acquisition are as follows:
June 21, 2022 | |||||||||||||||||||||
(Euro, in thousands) | |||||||||||||||||||||
CellPoint | AboundBio | Total | |||||||||||||||||||
| Book value |
| Adjustment |
| Fair value |
| Book value |
| Adjustment |
| Fair value |
| |||||||||
Intangible assets other than goodwill | € | 120,517 | € | 120,517 | € | 4,053 | € | 4,053 | |||||||||||||
Property, plant and equipment | € | 1,289 | 1,289 | € | 965 | 965 | |||||||||||||||
Other non-current assets | 81 | 81 | 4 | 4 | |||||||||||||||||
Trade and other receivables | 162 | 162 | - | - | |||||||||||||||||
Cash and cash equivalents | 3,179 | 3,179 | 4,279 | 4,279 | |||||||||||||||||
Other current assets | 1,254 | 1,254 | 536 | 536 | |||||||||||||||||
Deferred tax liabilities | - | (22,368) | (22,368) | - | (907) | (907) | |||||||||||||||
Trade and other liabilities | (32,789) | (32,789) | (587) | (587) | |||||||||||||||||
Current deferred income | - | - | (474) | (474) | |||||||||||||||||
Net assets acquired | (26,824) | 98,149 | 71,325 | 4,723 | 3,146 | 7,869 | |||||||||||||||
Consideration paid in cash | 107,750 | 14,976 | |||||||||||||||||||
Fair value re-measurement of previously held equity investment | - | 342 | |||||||||||||||||||
Deferred consideration | 5,808 | - | |||||||||||||||||||
Fair value of contingent consideration | 20,211 | - | |||||||||||||||||||
Fair value of total consideration | 133,769 | 15,318 | |||||||||||||||||||
Goodwill | 62,444 | 7,449 | |||||||||||||||||||
Exchange differences on goodwill | - | (80) | |||||||||||||||||||
Goodwill in the balance sheet at December 31, 2022 | € | 62,444 | € | 7,369 | € | 69,813 | |||||||||||||||
Net cash outflow arising on acquisition (2022) | |||||||||||||||||||||
Consideration paid in cash | 107,750 | 14,976 | |||||||||||||||||||
Less: cash and cash equivalents balances acquired | (3,179) | (4,279) | |||||||||||||||||||
Cash out from acquisition of subsidiaries, net of cash acquired (paid in 2022) | € | 104,571 | 10,698 | € | 115,270 | ||||||||||||||||
Cash used in operating activities for other liabilities related to the acquisition of subsidiaries (2022) | € | 28,164 | € | 28,164 | |||||||||||||||||
Cash out from acquisition of subsidiaries (payment of deferred consideration in 2023) | € | 7,000 | € | 7,000 | |||||||||||||||||
We refer to “Note 27-Business combinations during the prior period” in our consolidated financial statements appended to this annual report for more details.
Drug discovery collaboration transaction with NovAliX
We completed the integrated drug discovery collaboration transaction with NovAliX on June 30, 2023, effective as from July 1, 2023. Under the terms of the agreement, Galapagos’ drug discovery and research activities conducted in Romainville, France, and Galapagos’ employees in Romainville, which are exclusively dedicated to the operation of these activities, were transferred to NovAliX who will assume all ongoing research and discovery activities in Romainville, and this for no consideration. In return, Galapagos is committed to utilizing the research capabilities and expertise of NovAliX through a five year-collaboration and within the context of the company’s R&D portfolio, resulting in a total commitment of €73.8 million on June 30, 2023.
147
The collaboration agreement and sale and purchase agreement were negotiated as a package with one single commercial objective and with an agreed consideration for the transaction as a whole.
The impact of the transfer of activities and personnel (reference is made to the table below) was treated as an advance for future services to be obtained from NovaliX throughout the five years collaboration. This advance will gradually be released through profit or loss, in line with the purchase commitment towards NovAliX over the five year period of the collaboration between us and NovAliX. The part still to be released on December 31, 2023 has been presented in the statement of financial position as other current asset (€2.7 million) and other non-current asset (€5.6 million).
June 30, | |||
2023 | |||
(Euro, in thousands) | |||
| |||
Loss on sale of fixed assets | € | 12,506 | |
Result of transfer of retirement benefit liability | (3,022) | ||
Result of transfer of right-of-use asset | 174 | ||
Advance related to the NovAliX transaction | € | 9,658 | |
Furthermore we made an upfront payment to NovAliX of €8.3 million on closing of the transaction which is a prepayment for the future purchase commitment for the following five years. The remaining part has been presented in our statement of financial position on December 31, 2023 as other current asset (€2.4 million) and other non-current asset (€4.7 million).
148
Comparison of years ended December 31, 2022 and 2021
On January 31, 2024 we announced that we successfully completed the transfer of the Jyseleca® business to Alfasigma. The transfer of our Jyseleca® business has been determined to meet the criteria to be classified as held for sale and discontinued operations in our financial statements for the year ended December 31, 2023. We also presented all income statement items fully related to the Jyseleca® business to be transferred on a separate line “Net income/loss (-) from discontinued operations, net of tax” in our consolidated income statement. The consolidated income statement for all comparative periods reported in these consolidated financial statements were restated as well to show the discontinued operations on a separate line. The discontinued operations of the year ended December 31, 2021, also include the result from the disposal of Fidelta completed on January 1, 2021).
The following table summarizes the results of our operations for the years ended December 31, 2022 and 2021, together with the changes to those items.
Year Ended December 31, | ||||||||
| 2022 (*) |
| 2021 (*) |
| % Change | |||
(Euro, in thousands, except | ||||||||
Collaboration revenues | 241,249 | 234,384 | 3% | |||||
Total net revenues | 241,249 | 234,384 | 3% | |||||
Research and development expenses | (269,797) | (320,503) | (16%) | |||||
Sales and marketing expenses | (3,480) | (3,776) | (8%) | |||||
General and administrative expenses | (135,155) | (127,744) | 6% | |||||
Other operating income | 36,127 | 46,071 | (22%) | |||||
Operating loss | (131,056) | (171,568) | (24%) | |||||
Fair value re-measurement of share subscription agreement and warrants | 51,498 | 61,462 | (16%) | |||||
Other financial income | 18,563 | 3,057 | 507% | |||||
Other financial expenses | (9,854) | (12,353) | (20%) | |||||
Loss before tax | (70,849) | (119,402) | (41%) | |||||
Income taxes | (572) | (1,389) | (59%) | |||||
Net loss from continuing operations | (71,421) | (120,791) | (41%) | |||||
Net profit/loss (-) from discontinued operations, net of tax | (146,570) | 17,560 | (935%) | |||||
Net loss | € | (217,991) | € | (103,231) | 111% | |||
Net loss attributable to: | ||||||||
Owners of the parent | (217,991) | (103,231) | ||||||
Basic and diluted loss per share | € | (3.32) | € | (1.58) | ||||
Basic and diluted loss per share from continuing operations | € | (1.09) | € | (1.84) | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
149
Results from Discontinued Operations
We refer to the section of the results from discontinued operations in the comparison of years ended December 31, 2023 and 2022 for an introduction on the transfer of the Jyseleca® business to Alfasigma.
As a consequence of to the transfer of the Jyseleca® business to Alfasigma, we restated the consolidated income statements of the years ended December 31, 2022 and December 31, 2021, to present the Jyseleca® business as discontinued operations.
| (i) | Result from discontinued operations related to the Jyseleca® business |
Year ended December 31, | ||||||||
| 2022 |
| 2021 | % Change | ||||
(Euro, in thousands, | ||||||||
Product net sales | € | 87,599 | € | 14,753 | 494% | |||
Collaboration revenues | 176,432 | 235,709 | (25%) | |||||
Total net revenues | 264,031 | 250,462 | 5% | |||||
Cost of sales | (12,079) | (1,629) | 641% | |||||
Research and development expenditure | (245,286) | (171,205) | 43% | |||||
Sales and marketing expenses | (144,075) | (66,179) | 118% | |||||
General and administrative expenses | (9,776) | (13,155) | (26%) | |||||
Other operating income | 10,721 | 7,678 | 40% | |||||
Operating profit/loss (-) | (136,464) | 5,972 | n.m. | |||||
Fair value adjustments and net currency exchange differences | (25) | — | ||||||
Other financial income | 15 | 164 | (91%) | |||||
Other financial expenses | (7,825) | (9,733) | (20%) | |||||
Loss before tax | (144,298) | (3,597) | n.m. | |||||
Income taxes | (2,272) | (1,034) | 120% | |||||
Net loss | € | (146,570) | € | (4,631) | n.m. | |||
Basic and diluted earnings/losss (-) per share from discontinued operations | € | (2.23) | € | (0.07) | ||||
Weighted average number of shares (in thousands of shares) | 65,699 | 65,500 | ||||||
Weighted average number of shares - Diluted (in thousands of shares) | 65,699 | 65,500 | ||||||
Jyseleca® product net sales in Europe amounted to €87.6 million in 2022, compared to €14.8 million in 2021, of which €7.3 million realized in Belgium (€1.7 million in 2021).
Collaboration revenues in discontinued operations related to revenue recognition of the collaboration agreement with Gilead for the filgotinib development amount to €174.4 million in 2022 compared to €235.7 million for the same period last year. This decrease was due to a lower increase in the percentage of completion, slightly offset by higher revenue recognition of milestone payments, strongly influenced by the milestone achieved in 2022 related to the regulatory approval in Japan for UC.
150
Our R&D expenditures related to the filgotinib development in 2022 amounted to €245.3 million (€171.2 million in 2021). This was mainly driven by higher subcontracting and personnel expenses, as result of the transfer of clinical trials by Gilead to us.
Our S&M and G&A expenses related to the Jyseleca® business amounted to €153.9 million for the year 2022 compared to €79.3 million in 2021. S&M expenses increased by €77.9 million, primarily due to the termination of our 50/50 filgotinib co-commercialization cost sharing agreement with Gilead for filgotinib in 2022 and higher personnel expenses.
Other operating income attributable to the Jyseleca® business amounted to €10.7 million in 2022 compared to €7.7 million in 2021, mainly due to higher R&D tax credit income, Phd rebates income and recuperation of import taxes on delivery of API.
We reported net negative financial results of the discontinued operations of €7.8 million for the year 2022 (€9.6 million for 2021), and they mainly related to the discounting component of the filgotinib deferred income.
Disposal of FIDELTA fee-for-service business
On November 23, 2020, we signed a share purchase agreement with Selvita S.A. in relation to the disposal of Fidelta d.o.o. (our previous fee-for-service segment). The transaction was completed on January 4, 2021. The results of Fidelta were reported as discontinued operations in our consolidated income statement. Fidelta will continue performing drug discovery services for us for the next two years for which we have purchase commitments for an aggregate amount of €6.6 million on December 31, 2023.
(i) Disposal of Fidelta
a. Consideration received
| (Euro, in thousands) | ||
Cash received | € | 37,080 | |
Total cash received | € | 37,080 | |
151
b. Analysis of assets and liabilities over which control was lost
January 4, 2021 | |||
(Euro, in thousands) | |||
Intangible assets | € | 21 | |
Property, plant and equipment | 10,050 | ||
Other non-current assets | 160 | ||
Trade and other receivables | 4,428 | ||
Cash and cash equivalents | 7,884 | ||
Other current assets | 863 | ||
Total assets | 23,406 | ||
| |||
| |||
Non-current lease liabilities | 4,115 | ||
Other non-current liabilities | 70 | ||
Trade and other liabilities | 4,479 | ||
Current lease liabilities | 727 | ||
Income tax payable | 356 | ||
Total liabilities | 9,747 | ||
Net assets disposed of | € | 13,658 | |
c. Gain on disposal
(Euro, in thousands) | |||
Cash received | € | 37,080 | |
Net assets disposed of | (13,658) | ||
Effect of cumulative translation adjustment reclassified from equity on loss of control | (731) | ||
Costs associated to the sale | (500) | ||
Gain on disposal | € | 22,191 | |
d. Net cash proceedsfrom disposal of Fidelta
(Euro, in thousands) | |||
Cash received | € | 37,080 | |
Less: cash and cash equivalents balances disposed of | (7,884) | ||
Total consideration received, net of cash disposed of | 29,196 | ||
Costs associated to the sale | (500) | ||
Cash in from disposal of Fidelta, net of cash disposed of | € | 28,696 | |
152
ii) Result from discontinued operations
Year ended December 31, | ||||||
| 2022 |
| 2021 | |||
(Euro, in thousands, except share and per share data) | ||||||
Gain on disposal of subsidiaries | — | 22,191 | ||||
Operating profit | — | 22,191 | ||||
Net profit | € | — | € | 22,191 | ||
Basic and diluted earnings per share from discontinued operations | € | — | € | 0.34 | ||
Weighted average number of shares (in thousands of shares) | — | 65,500 | ||||
Weighted average number of shares - Diluted (in thousands of shares) | — | 65,831 | ||||
Results from our continuing Operations
The following table summarizes details of collaboration revenues for the years ended December 31, 2022 and 2021 by collaboration and by category of revenue: upfront payments and license fees, reimbursement income and royalties.
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Recognition of non-refundable upfront payments and license fees | € | 230,423 | € | 230,582 | (0%) | ||||
Reimbursement income | 56 | — | n.m. | ||||||
Royalties | 10,770 | 3,801 | 183% | ||||||
Total collaboration revenues | € | 241,249 | € | 234,384 | 3% | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
We recognize the consideration from Gilead allocated to the drug discovery platform on a linear basis over 10 years, of which we recognized €230.4 million in 2022. For the year ended December 31, 2022, we recognized in revenue €10.7 million of royalties from Gilead on filgotinib.
The outstanding balance of deferred income from the Gilead collaboration agreement for the exclusive access to our drug discovery platform at December 31, 2022 amounted to €1,529.4 million and was composed of €1,528.7 million that will be linearly recognized over the next years of our 10 year collaboration and €0.7 million related to the warrant issuance liability reserved for the Gilead subsequent warrant B.
We refer to note 2 “Summary of significant transactions” in our consolidated financial statements appended to this annual report for a general description of our collaboration with Gilead.
153
R&D expenditure
The following table summarizes our R&D expenditure for the years ended December 31, 2022 and 2021, together with the changes to those items.
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Personnel costs | € | (115,484) | € | (128,402) | (10%) | ||||
Subcontracting | (61,192) | (126,881) | (52%) | ||||||
Disposables and lab fees and premises costs | (19,529) | (22,933) | (15%) | ||||||
Depreciation and impairment | (51,493) | (15,709) | 228% | ||||||
Professional fees | (9,316) | (12,234) | (24%) | ||||||
Other operating expenses | (12,783) | (14,344) | (11%) | ||||||
Total R&D expenses | € | (269,797) | € | (320,503) | (16%) | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The variance in our R&D expenditure in 2022 compared to 2021 was principally due to the following elements:
| ● | Depreciation and impairment increase was primarily due to an impairment of €26.7 million of previously capitalized upfront fees related to our collaboration with Molecure on the dual chitinase inhibitor OATD-01 (GLPG4716) in fibrosis, and impairments recorded in 2022 of intangibles assets related to other discontinued projects for an amount of €8.9 million. |
| ● | A decrease in personnel costs explained by a decrease in salary expenses linked to a reduction in the staff and in non-cash cost recognition for subscription right plans.. |
| ● | Decrease in subcontracting costs following the evolution of our programs. |
The table below summarizes our R&D expenditure for the years ended December 31, 2022 and 2021, broken down by program.
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Ziritaxestat program | € | (1,096) | € | (26,725) | (96%) | ||||
SIKi program | (47,727) | (91,957) | (48%) | ||||||
TYK2 program on GLPG3667 | (24,467) | (27,141) | (10%) | ||||||
CAR-T programs in oncology | (29,999) | — | n.m. | ||||||
Other programs | (166,507) | (174,680) | (5%) | ||||||
Total R&D expenses | € | (269,797) | € | (320,503) | (16%) | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The decrease in R&D expenditure in 2022 was primarily explained by the winding down of the programs with ziritaxestat (IPF), and reduced spend on our SIKi, TYK2 and other programs. This was partly offset by new investments in CAR-T programs in oncology. Other programs comprise expenditure for other projects in research phase and other early-stage development programs focused on inflammation, fibrosis and other indications.
154
Sales and marketing expenses
The following table summarizes our sales and marketing expenses for the years ended December 31, 2022 and 2021, together with the changes to those items.
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Personnel costs | € | (1,693) | € | (1,709) | (1%) | ||||
Depreciation | (59) | (37) | 59% | ||||||
External outsourcing costs | (1,267) | (1,802) | (30%) | ||||||
Professional fees | (99) | (15) | 560% | ||||||
Other operating expenses | (363) | (214) | 70% | ||||||
Total sales and marketing expenses | € | (3,480) | € | (3,776) | (8%) | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Sales and marketing expenses for non-Jyseleca® related projects were stable for the year ended December 31, 2022 compared to the year ended December 31, 2021.
General and administrative expenses
The following table summarizes our general and administrative expenses for the years ended December 31, 2022 and 2021, together with the changes to those items.
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Personnel costs | € | (76,536) | € | (60,364) | 27% | ||||
Depreciation and impairment | (8,529) | (16,338) | (48%) | ||||||
Legal and professional fees | (23,715) | (25,345) | (6%) | ||||||
Other operating expenses | (26,375) | (25,697) | 3% | ||||||
Total general and administrative expenses | € | (135,155) | € | (127,744) | 6% | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The increase in our general and administrative expenses in 2022 was mainly explained by an increase in personnel expenses primarily due to accelerated non- cash cost recognition for our subscription right plans related to good leavers. This was partly offset by a decrease in depreciation and impairment costs due to an impairment cost of €9.3 million in 2021 on other tangible fixed assets following our decision to reassess the construction project in Mechelen (Belgium).
Other operating income
The following table summarizes our other operating income for the years ended December 31, 2022 and 2021, together with the changes to those items.
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Grant income | € | 1,873 | € | 7,334 | (74%) | ||||
R&D incentives income | 29,104 | 37,416 | (22%) | ||||||
Other income | 5,150 | 1,325 | 289% | ||||||
Total other operating income | € | 36,127 | € | 46,075 | (22%) | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
155
The grant income in 2021 mainly comprises a grant received in 2021 from the National Institute for Health and Disability Insurance amounting to €5.4 million. This grant aimed to incentivize innovative Belgian biotech companies who are performing research and development activities in order to identify new medicines.
R&D incentives income was primarily composed of:
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Income from innovation incentive system in France | € | 11,075 | € | 12,245 | (10%) | ||||
Income from Belgian R&D incentives | 10,339 | 14,835 | (30%) | ||||||
Tax rebates on payroll withholding taxes of R&D personnel (Belgium & the Netherlands) | 7,689 | 10,336 | (26%) | ||||||
Total R&D incentives income | € | 29,104 | € | 37,416 | (22%) | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Other income increased mainly due to rental income and a one-off sale of side products from our R&D activities.
Fair value adjustments and net currency exchange differences
The following table summarizes our fair value adjustments and net currency exchange differences for the years ended December 31, 2022 and 2021, together with the changes to those items.
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) |
| % Change | ||||
(Euro, in thousands) | |||||||||
Fair value adjustments and net currency exchange differences: | |||||||||
Net unrealized currency exchange gain | € | 41,559 | € | 56,966 | (27%) | ||||
Net realized currency exchange gain/loss (-) | 2,825 | (308) | (1,017%) | ||||||
Fair value re-measurement of warrants | 186 | 2,960 | (94%) | ||||||
Fair value loss on financial assets held at fair value through profit or loss | — | (4,919) | (100%) | ||||||
Fair value gain on current financial investments | 6,929 | 6,763 | 2% | ||||||
Total fair value adjustments and net currency exchange differences | € | 51,498 | € | 61,462 | (16%) | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The net unrealized currency exchange gain of €41.6 million in 2022 primarily related to € 41.3 million of unrealized exchange gains on cash and cash equivalents and current financial investments at amortised cost held in U.S. dollars, as compared to an unrealized net exchange gain in 2021 of € 56.6 million on cash and cash equivalents and current financial investments at amortized cost held in U.S. dollar. We have cash, cash equivalents and current financial investments held in U.S. dollars, which could generate foreign currency exchange gain or loss in our financial results in accordance with the fluctuation of the EUR/U.S. dollar exchange rate as our functional currency is EUR.
Fair value re-measurement of warrants refers to the fair value re- measurement of the Gilead initial warrant B. The fair value of the financial liability related to the Gilead initial warrant B of €0.02 million on December 31, 2022 (€0.2 million on December 31, 2021) is presented as part of trade and other liabilities in our consolidated statement of financial position and will be re-measured at each reporting period. We refer to note 2 “Summary of significant transactions” in our consolidated financial statements appended to this annual report for more information.
156
For the year ended December 31, 2021, fair value loss on financial assets held at fair value through profit or loss consisted of negative effects from the fair value re-measurement of financial assets classified as equity investments which qualify for level 1 fair value measurement based upon the closing price of such securities at each reporting date, and of an impairment loss on a participation in a non-listed company. This resulted in a net book value of zero of the financial assets held at fair value through profit or loss on December 31, 2021. The fair value gain on the current financial investments reflects the positive exchange differences booked on the money market funds, compensated by the interest on treasury bills which have not yet expired and the effect of the re-measurement at fair value of the money market funds on December 31, 2022. These re-measurement losses are mainly the result of the negative returns on the EUR denominated money market funds.
For more information on currency exchange fluctuations on our business, please see the section of this annual report titled “Item 11—Quantitative and qualitative disclosures about market risk—Foreign exchange risk.”
Other financial income and expense
The following table summarizes other financial income and expense for the years ended December 31, 2022 and 2021.
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) |
|
| % Change | |||
(Euro, in thousands) | |||||||||
Other financial income: | |||||||||
Interest income | € | 18,094 | € | 2,865 | 532% | ||||
Discounting effect of non-current R&D incentives receivables | 93 | 93 | — | ||||||
Discounting effect of other non-current liabilities | — | — | |||||||
Other finance income | 376 | 99 | 280% | ||||||
Total other financial income | 18,563 | 3,057 | 507% | ||||||
Other financial expenses: | |||||||||
Interest expenses | (6,884) | (11,592) | (41%) | ||||||
Discounting effect of other non-current liabilities | (2,271) | — | n.m. | ||||||
Other finance charges | (699) | (761) | (8%) | ||||||
Total other financial expense | (9,854) | (12,353) | (20%) | ||||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Interest expenses were related to interests on term deposits, treasury bills that came to maturity and on leases of buildings and cars. Other financial expense for 2022 also comprise the discounting effect of other non-current liabilities as deferred consideration and milestones payables related to the acquisition of subsidiaries.
Interest income was related to interests on term deposits, notice accounts and current financial investments. Interest income increased due to increased interest rates.
Income Taxes
The following table summarizes our tax result for the years ended December 31, 2022 and 2021.
Year ended December 31, | |||||||||
| 2022(*) |
| 2021(*) | % Change | |||||
(Euro, in thousands) | |||||||||
Current tax | € | (1,738) | € | (884) | 97% | ||||
Deferred tax | 1,166 | (505) | (331%) | ||||||
Income taxes | € | (572) | € | (1,389) | (59%) | ||||
157
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Current tax, consisting of corporate income taxes, and deferred tax income/loss (-) related to subsidiaries working on a cost plus basis. In addition, the deferred tax income for the year ended December 31, 2022, was largely due to the partial release of the net deferred tax liabilities related to the acquisitions of CellPoint and AboundBio.
We refer to note 11 of our consolidated financial statements ‘Income taxes’.
Business combinations
We refer to the comparison of years ended December 31, 2023 and 2022 for more information on the business combinations with CellPoint and AboundBio, acquired in June 2022.
B. Liquidity and capital resources
With the exception of the years ended December 31, 2019 and 2023, we have incurred significant operating losses. We have funded our operations through public and private placements of equity securities, upfront and milestone payments and royalties received from pharmaceutical partners under our collaboration agreements, payments under our fee-for-service contracts, funding from governmental bodies, interest income as well as the net proceeds from the sale of our service division and our fee-for-service division. As from the year ended December 31, 2021, net product sales also started to contribute funding our operations. Our cash flows may fluctuate and are difficult to forecast and will depend on many factors. As at December 31, 2023, our current financial investments and cash and cash equivalents amounted to €3,684.5 million. For more information on our policies regarding financial instruments, please see “Note 3—Material accounting policies—Financial instruments” included in our consolidated financial statements appended to this annual report.
Cash flows
The working capital is sufficient for our present requirements.
Comparison for the years ended December 31, 2023 and 2022
The following table summarizes the results of our audited consolidated statement of cash flows for the years ended December 31, 2023 and 2022.
| 2023 |
| 2022 |
| Variance | ||||
(Euro, in thousands) | |||||||||
Cash and cash equivalents at beginning of the period | € | 508,117 | € | 2,233,368 | € | (1,725,252) | |||
Net cash flows used in operating activities | (405,970) | (500,544) | 94,574 | ||||||
Net cash flows generated from/used in (-) investing activities | 71,186 | (1,245,514) | 1,316,700 | ||||||
Net cash flows used in financing activities | (5,001) | (1,487) | (3,514) | ||||||
Effect of exchange rate differences on cash and cash equivalents | (1,522) | 22,293 | (23,815) | ||||||
Cash and cash equivalents at end of the period | € | 166,810 | € | 508,117 | € | (341,306) | |||
| 2023 |
| 2022 |
| Variance | ||||
(Euro, in thousands) | |||||||||
Current financial investments at end of the period | € | 3,517,698 | € | 3,585,945 | € | (68,247) | |||
Cash and cash equivalents at end of the period | 166,803 | 508,117 | (341,314) | ||||||
Cash and cash equivalents classified as assets held for sale | 7 | — | 7 | ||||||
Current financial investments and cash and cash equivalents at end of the period | € | 3,684,508 | € | 4,094,062 | € | (409,554) | |||
158
The net decrease of €341.3 million in cash and cash equivalents for the year ended December 31, 2023, consisted of negative unrealized exchange differences of €1.5 million and decrease in cash and cash equivalents of €339.8 million. This latter was composed of (i) €414.8 million of operational cash burn, (ii) €1.8 million of cash proceeds from capital and share premium increase from exercise of subscription rights in 2023, (iii) the net sale of current financial investments of €94.2 million, (iv) €7.0 million cash out from the acquisitions of CellPoint, and (v) €14.0 million acquisition of financial assets held at fair value through profit or loss.
The operational cash burn/cash flow is defined as the decrease or increase in our cash and cash equivalents (excluding the effect of exchange rate differences on cash and cash equivalents), minus:
i. the net proceeds, if any, from share capital and share premium increases included in the net cash flows generated from/used in (-) financing activities
ii.the net proceeds or cash used, if any, in acquisitions or disposals of businesses; the acquisition of financial assets held at fair value through profit or loss, the movement in restricted cash and movement in current financial investments, if any, the loan and advances given to third parties, if any, included in the net cash flows generated from/used in (-) investing activities
iii. the cash used for other liabilities related to the acquisition of businesses, if any, included in the net cash flow generated from/used in (-) operating activities.
This alternative liquidity measure is in our view an important metric for a biotech company in the development stage.
The following table presents a reconciliation of the operational cash burn, to the closest IFRS measures, for each of the periods indicated:
| 2023 |
| 2022 | |||
(Euro, in thousands) | ||||||
Decrease in cash and cash equivalents (excluding effect of exchange differences) | € | (339,785) | € | (1,747,545) | ||
Less: | ||||||
Net proceeds from capital and share premium increases | (1,770) | (6,695) | ||||
Net purchase/sale (-) of current financial investments | (94,233) | 1,087,032 | ||||
Acquisition of financial assets held at fair value through profit or loss | 13,965 | — | ||||
Cash out from acquisition of subsidiaries, net of cash acquired | 7,000 | 115,270 | ||||
Cash advances and loans to third parties | — | 10,000 | ||||
Cash used for other liabilities related to the acquisition of subsidiaries | — | 28,164 | ||||
Total operational cash burn | € | (414,824) | € | (513,774) | ||
The net cash flow used in operating activities for the year ended December 31, 2023 decreased by €94.6 million as compared to the year ended December 31, 2022, mainly caused by higher interest income received and higher product net sales in 2023.
The variance in net cash flow generated from/used in (-) investing activities for the year ended December 31, 2023, can be primarily explained by the net sale of current financial investments of €94.2 million for the year ended December 31, 2023 as compared to the net purchase of current financial investments of €1,087.0 million for the year ended December 31, 2022. For the year ended December 31, 2023 cash out from acquisition of subsidiaries and cash advances amounted to €7.0 million as opposed to €125.3 million for the year ended December 31, 2022. We also reported lower investments in (in)tangible fixed assets, which decreased from €36.9 million for the year ended December 31, 2022 to €19.3 million for the year ended December 31, 2023.
159
The decrease in net cash flow used in financing activities for the year ended December 31, 2023, can primarily be attributed to lower proceeds received on exercises of subscription rights, which were for the years ended December 31, 2023 and 2022 respectively €1.8 million and €6.7 million.
The consolidated cash flow table above included both continuing and discontinued operations. The table below summarizes our statement of cash flows from discontinued operations included in the consolidated cash flow table for the years ended December 31, 2023 and 2022.
| 2023 |
| 2022 |
| Variance | ||||
(Euro, | |||||||||
Net cash flow used in operating activities | € | (175,627) | € | (191,095) | € | 15,468 | |||
Net cash flow used in investing activities | (105) | (136) | 31 | ||||||
Net cash flow used in financing activities | (1,928) | (1,841) | (87) | ||||||
Net cash flow used in discontinued operations | € | (177,660) | € | (193,072) | € | 15,412 | |||
Comparison for the years ended December 31, 2022 and 2021
We refer to the “item 5 – Operating and financial review and prospects – Financial operations overview” of our year ended December 31, 2022 Form 20-F for the comparison of the years ended December 31, 2022 and 2021.
The consolidated cash flow table in item 5 of the Form 20-F of the year ended December 31, 2022, included both continuing and discontinued operations.
The table below summarizes our statement of cash flows from discontinued operations included in the consolidated cash flow table for the years ended December 31, 2022 and 2021. (for the year ended December 31, 2021, it includes cash flows related to the Jyseleca business as well as the proceeds from the sale of Fidelta on January 4, 2021).
| 2022 |
| 2021 |
| Variance | ||||
(Euro, | |||||||||
Net cash flow used in operating activities | € | (191,095) | € | (119,524) | € | (71,571) | |||
Net cash flow generated from/used (-) in investing activities | (136) | 28,539 | (28,675) | ||||||
Net cash flow used in financing activities | (1,841) | (872) | (969) | ||||||
Net cash flow from discontinued operations | € | (193,072) | € | (91,857) | € | (101,215) | |||
Cash and funding sources
For the years ended December 31, 2023, 2022 and 2021, excluding cash proceeds from subscription right exercises, we did not make use of other equity financing.
As of December 31, 2023, we had no financial debt.
Our ongoing financial commitments are listed in the section of this annual report titled “Item 5.F.—Tabular disclosure of contractual obligations” and mainly consist of purchase commitments.
160
Payment of dividends by subsidiaries
The amount of dividends payable by our subsidiaries to us is subject to, among other restrictions, general limitations imposed by the corporate laws, capital transfer restrictions and exchange control restrictions of the respective jurisdictions where those subsidiaries are organized and operate.
Of our current financial investments and cash and cash equivalents held outside of our Belgian entities as of December 31, 2023 and 2022, the amount of cash that would have been subject to withholding taxes if transferred to us by way of dividends and the amount of cash that could not have been transferred by law was in each case immaterial.
Funding requirements
Based on conservative assumptions, that may prove to be wrong, we believe that our existing current financial investments and cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements at least for a period of 12 months.
Our present and future funding requirements will depend on many factors, including, among other things:
| ● | the terms and timing of milestones, in-licensing payments and expense reimbursement payments, if any, from our collaboration and alliance agreements; |
| ● | the progress, timing, scope and costs of preclinical testing and clinical trials for any current or future compounds; |
| ● | the number and characteristics of potential new compounds we identify and decide to develop; |
| ● | our need to expand our development activities and, potentially, our research activities; |
| ● | the costs involved in filing patent applications and maintaining and enforcing patents; |
| ● | the cost, timing and outcomes of regulatory approvals; |
| ● | selling and marketing activities undertaken in connection with the commercialization of our products or aniticpated commercialization of any of our current or future compounds; and |
| ● | the amount of revenues, if any, we may derive either directly or in the form of royalty payments from future sales of our products. |
We may raise additional capital through the sale of equity or convertible debt securities. In such an event, your ownership interest may be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a holder of the ADSs or our ordinary shares.
For more information as to the risks associated with our future funding needs, see the section of this annual report titled “Item 3.D.—Risk Factors—Risks Related to Our Financial Position and Need for Additional Capital.”
Capital expenditures
Our commitments for capital expenditures as of December 31, 2023 amounted to €3.9 million.
Our capital expenditures amounted to €19.3 million, €36.9 million and €57.9 million for the years ended December 31, 2023, 2022 and 2021 respectively.
In 2023, our capital expenditures consisted of €6.8 million for land and building additions, laboratory and computer and other equipment for €11.9 million, €0.6 million of intangible assets related to software development.
161
In 2022, our capital expenditures consisted of €19.2 million for land and building additions, laboratory and computer and other equipment for €8.1 million, €9.6 million of intangible assets related to license fees (€7.5 million), milestone payments (€0.9 million) and software development (€1.1 million).
In 2021, our capital expenditures consisted of €46.2 million for land and building additions, laboratory and computer and other equipment for €8.0 million, €3.7 million of intangible assets related to license fees (€1.3 million) and software development (€2.4 million).
Off-balance sheet arrangements
During the periods presented, we did not and do not currently have any off-balance sheet arrangements as defined under SEC rules, such as relationships with unconsolidated entities or financial partnerships, which are often referred to as structured finance or special purpose entities, established for the purpose of facilitating financing transactions that are not required to be reflected on our balance sheets.
Contractual obligations and commitments
We have certain purchase commitments with contract research organization subcontractors and with Gilead principally. Future events could cause actual payments to differ from these estimates. On December 31, 2023, we had outstanding obligations for purchase commitments, which become due as follows:
| Total |
| Less than |
| 1 - 3 |
| 3 - 5 |
| More than 5 | ||||||
(Euro, in thousands) | |||||||||||||||
Purchase commitments | € | 408,521 | € | 237,495 | € | 143,532 | € | 25,768 | € | 1,727 | |||||
On December 31, 2022, we had outstanding obligations for purchase commitments, which become due as follows:
| Total |
| Less than |
| 1 - 3 |
| 3 - 5 |
| More than 5 | ||||||
(Euro, in thousands) | |||||||||||||||
Purchase commitments | € | 398,627 | € | 240,237 | € | 136,560 | € | 20,797 | € | 1,032 | |||||
Our purchase commitments at the end of the year 2023 included €239.6 million related to projects in development phase (2022: €243.6 million), €79.0 million for projects in discovery research phase (2022: €20.9 million), €45.9 million for shared services (2022: €49.4 million), €29.9 million for commercial and medical affairs (2022: €36.0 million), and €14.2 million related to Jyseleca® product supply chain (2022: €48.8 million).
Effective July 1, 2023, under the terms of the agreement, our drug discovery and research activities conducted in Romainville, France, andour employees in Romainville, which were exclusively dedicated to the operation of these activities, were transferred to NovAliX who will assume all ongoing research and discovery activities in Romainville. In return, we are committed to utilizing the research capabilities and expertise of NovAliX through a five year-collaboration and within the context of the company’s R&D portfolio, during which we are committed to purchase for a total of €73.8 million services from Novalix. At year end 2023, our purchase commitments towards Novalix amounts to €63.9 million and are included in the €79.0 million related to discovery research.
At the end of the year 2023, €139.1 million of our purchase commitments related to the Jyseleca® discontinued operations transferred to Alfasigma on January 31, 2024, of which €110.8 million related to the filgotinib clinical development and €21.2 million related to commercial and medical affairs activities.
162
In addition to the table above, we have a contractual cost sharing obligation related to our collaboration agreement with Gilead for filgotinib, which is disclosed under the sections of this annual report titled “Item 5–Operating and Financial Review and Prospects.–Collaboration and Alliance Agreements–Option, License and Collaboration Agreement with Gilead”, and “Item 7.B.–Related Party Transactions.–Transaction with Major Shareholder”. The contractual cost sharing obligation amounted to €12.3 million at December 31, 2023 (€281.6 million at December 31, 2022).
We entered into a license agreement with another pharmaceutical company. Under the terms of this agreement we have the obligation to pay potential milestones, which are dependent on successful completion of certain development and commercial milestones, as detailed in the agreement. At December 31, 2023 this commitment amounts to €243.5 million on an undiscounted and non-risk adjusted basis. This amount represents the maximum amount that would be paid if all milestones would be achieved but excludes variable royalty payments based on unit sales.
The table above does not include retirement benefit liabilities, non-current lease liabilities, non-current deferred income and other non-current liabilities.
We provide retirement benefit plans for all of our qualifying employees. We classify these benefits on the basis of the type of benefit provided and in particular as defined contribution plans, defined benefit obligations and other provisions for employees. At December 31, 2023 the net liability for such obligations amounted to €2.3 million (€5.5 million at December 31, 2022).
Non-current lease liabilities amounted to €4.9 million at December 31, 2023 (€14.7 million at December 31, 2022) and related to the non-current portion of liabilities linked to right-of-use under IFRS 16 primarily for leased buildings and leased cars.
Non-current deferred income was €1,071.2 million at December 31, 2023 (€1,623.6 million at December 31, 2022) and mainly related to the non-current portion of the transaction price from Gilead still to be recognized as collaboration revenue in the future for our collaboration on filgotinib and the exclusive access rights to our drug discovery platform. See “Note 26 – Deferred income” in our consolidated financial statements appended to this annual report.
Other non-current liabilities amounted to €31.6 million at December 31, 2023 (€21.8 million at December 31, 2022) and primarily consisted of the fair value of the long-term portion of the deferred considerations and milestones payable in the scope of the acquisition of CellPoint and AboundBio. They also included payables related to RSU plans granted in 2021, 2022 and 2023. See “Note 3 – Material accounting policies” and “Note 31 – Share based payments” in our consolidated financial statements appended to this annual report. Members of the Executive Committee and other employees were granted RSU’s in 2019, 2020, 2021, 2022, and 2023. An RSU is a grant that takes the form of a promise that employees will receive Galapagos stock in the future and it will be payable, at the company’s discretion in cash or in shares, upon completion of a certain vesting period. Each RSU reflects the value of one Galapagos share. The RSU’s are measured based on the average share price over the 30-calendar day period preceding the measurement date. We recognize the corresponding expense and liability over the vesting period. The fair value of the liability is re-measured at each reporting date because currently it is management’s intention to settle the RSU’s in cash.
C. Research and development, patents and licenses, etc
For a discussion of our R&D activities, see “Item 4.B.—Business Overview” and “Item 5.A.—Operating Results.”
D. Trend information
Other than as disclosed elsewhere in this annual report, we are not aware of any trends, uncertainties, demands, commitments or events for the period from January 1, 2023 to December 31, 2023 that are reasonably likely to have a material adverse effect on our net revenues, income, profitability, liquidity or capital resources, or that caused the disclosed financial information to be not necessarily indicative of future operating results or financial conditions. For a discussion of trends, see “Item 4.B.—Business overview,” “Item 5.A.—Operating results,” and “Item 5.B.—Liquidity and capital resources.”
163
E. Critical Accounting Estimates
We refer to “Note 4 - Critical accounting judgments and key sources of estimation uncertainty” in our consolidated financial statements appended to this annual report.
Item 6 Directors, senior management and employees
A. Directors and senior management
Board of Directors
As a listed company with its registered office in Mechelen (Belgium), Galapagos NV (“Galapagos” or the “Company”) is required to apply the Belgian Code of Companies and Associations (the “Belgian Companies Code”) and 2020 Belgian Corporate Governance Code (the “2020 Code”), both of which entered into force on January 1, 2020 and as amended from time to time.
For the reporting year beginning on January 1, 2023, the 2020 Code was our reference code. On March 21, 2023, as a consequence of the establishment of the Management Committee, i.e., an informal Committee providing advice and assistance to the Executive Committee, the Board of Directors approved an amendment to the Company’s Corporate Governance Charter. On September 19, 2023, the Board of Directors approved another amendment that refers to the establishment of the Science and Development Committee as a specialized Board Committee to provide advice on certain matters to the Board of Directors, and that provides that non-executive Directors may only be natural persons. On December 11, 2023, the Board of Directors approved a further amendment to describe the responsibilities of the Audit Committee and management for overseeing and managing cybersecurity risks. Galapagos NV’s Corporate Governance Charter is available on its website: www.glpg.com (this website does not form part of this annual report). This Corporate Governance Charter applies in addition to the applicable laws and regulations (including without limitation, the Belgian Companies Code and the 2020 Code) and Galapagos’ Articles of Association The Company’s Corporate Governance Charter describes the main aspects of corporate governance at Galapagos, including its governance structure, the terms and functioning of the Board of Directors (including its Board Committees), the Executive Committee and the rules of conduct.
The 2020 Code requires companies to make an explicit choice for one of the governance structures provided for in the Belgian Companies Code.
Since April 26, 2022, Galapagos has adopted a one-tier governance structure as provided by the Belgian Companies Code, with the Board of Directors as the ultimate decision-making body, who has delegated certain powers to manage the Company to the Executive Committee..
Our one-tier governance structure

164

The role of the Board of Directors is to pursue sustainable value creation by the Company, by setting the Company’s strategy, putting in place effective, responsible and ethical leadership and monitoring the Company’s performance. The Board of Directors is the ultimate decision-making body, with the overall responsibility for the management and control of the Company, as well as the general policy and strategy of the Company. The Board is authorized to carry out all actions that are necessary or useful for the realization of the Company’s object, with the exception of those reserved to the Shareholders’ Meeting by applicable law. The Board acts as a collegiate body.
The Board of Directors has delegated certain powers to manage the Company to the Executive Committee, led by the Chief Executive Officer (“CEO”). The Executive Committee is responsible and accountable to the Board of Directors for the discharge of its responsibilities, including, but not limited to, the management of the Galapagos group, and the research, identification and development of strategic possibilities and proposals. Furthermore, the Board of Directors has delegated the day-to-day management of the Company to one Executive Committee member, i.e. our CEO.
In order to efficiently fulfill its tasks and in view of the size and activities of the Company, the Board of Directors has established an Audit Committee, a Nomination Committee, a Remuneration Committee, and a Scientific and Development Committee. These Board Committees serve in an advisory capacity to the Board of Directors on the matters delegated to them respectively as set forth in the applicable laws and the Galapagos’ Corporate Governance Charter.
We currently have nine Board members, less than a majority of whom are citizens or residents of the United States.
Under Galapagos’ Articles of Association, our Board of Directors consists of at least five and no more than nine members, of which at least three members are independent Directors as defined by the Belgian Companies Code. Except for Stoffels IMC BV (permanently represented by Dr. Paul Stoffels), all members of our Board of Directors are non-executive Directors. Within the aforementioned limits, the number of members of the Board of Directors is determined by our Shareholders’ Meeting. The members of our Board of Directors are elected, re-elected and may be removed at a Shareholders’ General Meeting with a simple majority vote of our shareholders. Pursuant to the Company’s Articles of Association, our Board members serve terms of up to four years. Members of the Board of Directors whose mandate has come to an end, may be re-appointed.
When a position on the Board of Directors becomes vacant, the remaining Board members may temporarily fill the mandate by cooptation and until appointment of a new Director at the next Shareholders’ Meeting. Each member of the Board of Directors appointed as such by the Shareholders’ Meeting shall complete the tenure of the Board member he / she replaces, unless the Shareholders’ Meeting decides otherwise. The Nomination Committee nominates, for approval by the Board of Directors, candidates to fill vacancies as they arise, and advises on proposals for appointment originating from shareholders, in each case taking into account Galapagos’ needs and the selection criteria determined by the Board of Directors. In proposing candidates, particular consideration will be given to gender diversity and diversity in general, as well as complementary skills, knowledge and experience.
165
Subject to the approval of Galapagos’ Shareholders’ Meeting and certain other conditions, Gilead has, under the terms of the share subscription agreement dated 14 July 2019, as amended, the right to have two designees appointed to our Board of Directors. Our Special Shareholders’ Meeting of October 22, 2019 approved the appointment of Daniel O’Day and Linda Higgins as directors of Galapagos for a period of fours years, effective immediately. Our Annual Shareholders’ Meeting held on April 25, 2023 approved the re-appointment of Daniel O’Day and Linda Higgins as directors of Galapagos for a period of four years, effective immediately.
The tenure of Dr. Rajesh Parekh and Dr. Mary Kerr as members of the Board of Directors came to an end during the financial year ending on December 31, 2023. We thank Dr. Rajesh Parekh and Dr. Mary Kerr for their contributions and commitment to the Company over the years.
During its meeting of June 12, 2023, the Board of Directors appointed Dr. Susanne Schaffert by cooptation as a non-executive independent Director, replacing Dr. Rajesh Parekh who stepped down on June 10, 2023.
During its meeting of September 19, 2023, the Board of Directors appointed Mr. Simon Sturge by cooptation as a non-executive independent Director, replacing Dr. Mary Kerr who stepped down on September 18, 2023. Dr. Susanne Schaffert’s and Mr. Simon Sturge’s appointments will be submitted to the confirmation of the Company’s Annual Shareholders’ Meeting which will be held on April 30, 2024.
The following table sets forth certain information with respect to the current members of our Board of Directors, including their ages, as of December 31, 2023:
Name |
| Age |
| Date service began in current term |
| Date of expiration of current term (1) |
| Position(s) |
Stoffels IMC BV (permanently represented by Dr. Paul Stoffels) (2) | n/a | 2022 | 2026 | Chair of the Board | ||||
Jérôme Contamine (3) | 66 | 2022 | 2026 | Board member | ||||
Dan Baker (4) | 73 | 2022 | 2026 | Board member | ||||
Peter Guenter (5) | 61 | 2019 | 2027 | Board member | ||||
Daniel O'Day | 59 | 2019 | 2027 | Board member | ||||
Linda Higgins | 61 | 2019 | 2027 | Board member | ||||
Elisabeth Svanberg (6) | 61 | 2020 | 2024 | Board member | ||||
Dr. Susanne Schaffert (7) | 56 | 2023 | 2028 (9) | Board member | ||||
Mr. Simon Sturge (8) | 64 | 2023 | 2028 (9) | Board member | ||||
(1) The term of the mandates of the Board member will expire immediately after the annual shareholders' meeting to be held in the year set forth next to the member's name. | ||||||||
(2) Management company incorporated in 2022, age of Dr. Paul Stoffels: 61. Member of the Nomination Committee and the Science and Development Committee. | ||||||||
(3) Chair of the Audit Committee, member of the Nomination Committee and the Remuneration Committee. | ||||||||
(4) Chair of the Science and Development Committee, member of the Remuneration Committee. | ||||||||
(5) Member of the Audit Committee. | ||||||||
(6) Chair of the Nomination Committee and the Remuneration Committee, member of the Science and Development Committee. | ||||||||
(7) Member of the Science and Development Committee. | ||||||||
(8) Member of the Audit Committee. | ||||||||
(9) Appointed by the Board by cooptation in 2023, subject to confirmation by the Shareholders' Meeting to be held on April 30, 2024. | ||||||||
The address for the members of our Board of Directors is Generaal De Wittelaan L11 A3, 2800 Mechelen, Belgium.
Six out of nine of the members of the Board of Directors are independent under the Nasdaq Stock Market listing and US Securities Exchange Act requirements, and under Belgian law: Peter Guenter, Jérôme Contamine, Dan Baker,
166
Elisabeth Svanberg, Susanne Schaffert, and Simon Sturge. In 2023, the Board of Directors was therefore composed of a majority of independent Directors.
The following is the biographical information of the members of our Board of Directors (members per December 31, 2023):
Paul Stoffels1 joined Galapagos as Chief Executive Officer in April 2022, and is an executive member and the Chairman of our Board of Directors since April 26, 2022. He also is a member of the Executive Committee at Galapagos. Prior to that, he was Vice Chairman of the Executive Committee and Chief Scientific Officer of Johnson & Johnson where he set the company's wide innovation agenda and led its pharmaceutical R&D-pipeline, as well as other external initiatives. Before that, he was worldwide Chairman of Pharmaceuticals of Johnson & Johnson which, under his leadership, significantly rejuvenated its product pipeline and adopted a transformational R&D-operating model, which resulted in the launch of 25 innovative medicines across the globe. Dr. Stoffels joined Johnson & Johnson in 2002, following the acquisition of Virco and Tibotec, where he was Chief Executive Officer and Chairman respectively, and where he led the development of several breakthrough products for the treatment of HIV. Dr. Stoffels also is a member of the Supervisory Board of Philips Healthcare in the Netherlands.
Peter Guenter is a non-executive independent member of our Board of Directors since April 30, 2019. Mr. Guenter is a member of the Executive Board of Merck and Chief Executive Officer of Merck Healthcare since January 2021. Before joining Merck, he served as Chief Executive Officer at Almirall from 2017 to 2020. Prior to joining Almirall, he worked at Sanofi for 22 years, most recently as Executive Vice President Diabetes and Cardiovascular Global Business Unit. During his tenure at Sanofi, he held many senior positions including Vice President Eastern Europe and Northern Europe, Vice President Business Management and Support, General Manager Germany, Senior Vice President Europe, Executive Vice President Global Commercial Operations, and Executive Vice President General Medicine and Emerging Markets. He was a member of Sanofi’s Executive Committee from 2013 until August 2017. Before joining Sanofi, he held different positions in sales and marketing at Smith Kline and Ciba Geigy. Mr. Guenter also is a member of the Board of the European Federation of Pharmaceutical Industries and Associations (EFPIA). He holds a Master’s Degree in Physical Education from the Faculty of Medicine and Health Sciences, University of Ghent.
Daniel O’Day is a non-executive member of our Board of Directors since October 22, 2019. Mr. O’Day is the Chairman of the Board of Directors and Chief Executive Officer of Gilead Sciences, which employs more than 17,000 people worldwide. Prior to joining Gilead in 2019, Mr. O’Day served as the Chief Executive Officer of Roche Pharmaceuticals. His career at Roche spanned more than three decades, during which he held several executive positions in the company’s pharmaceutical and diagnostics divisions in North America, Europe and Asia. He served as a member of Roche’s Corporate Executive Committee, as well as on a number of public and private Boards, including Genentech, Flatiron Health and Foundation Medicine. Mr. O’Day also serves on the Board of Directors for the Pharmaceutical Research and Manufacturers of America Organization and Georgetown University. Mr. O’Day holds a Bachelor’s Degree in Biology from Georgetown University and a MBA from Columbia University in New York. On March 26, 2024, Mr. O’Day resigned as member of Galapagos’ Board of Directors. He was replaced by Gilead’s CFO, Mr. Andrew Dickinson, effective March 27, 2024.
Linda Higgins is a non-executive member of our Board of Directors since October 22, 2019. Linda Slanec Higgins, PhD, joined Gilead Sciences, Inc. in 2010 and is currently Sr. Vice President Research Strategy, Innovation & Portfolio. In her first ten years at Gilead, she led the Biology division, significantly expanding the therapeutic area scope and capabilities of the department. She founded External Innovation as integral component for Research. She previously served as President & Chief Executive Officer of InteKrin Therapeutics, and as Head of Research at Scios, a Johnson & Johnson company, where she provided leadership for drug discovery, preclinical development and translational medicine. Dr. Higgins is passionate about biopharmaceutical discovery and development, and has been dedicated to excellence in applied scientific research since 1991. She has led projects and departments in multiple therapeutic areas
1 Stoffels IMC BV, permanently represented by Dr. Paul Stoffels
167
including central nervous system, fibrosis, inflammation, cardiovascular, virology and oncology. Dr. Higgins built many of these as new areas at Scios and Gilead. Dr. Higgins earned an A.B. in Behavioral Physiology from Kenyon College, a Ph.D. in Neurosciences from the University of California, San Diego School of Medicine, and completed Post-Doctoral training in Molecular Genetics at the Howard Hughes Medical Institute of the University of California, Berkeley. She has authored over 50 original peer reviewed scientific papers and invited articles, and is an inventor of over a dozen patents. Dr. Higgins also serves as a non-executive director on the Board of Arcus Bioscience.
Elisabeth Svanberg is a non-executive independent member of our Board of Directors since April 28, 2020. Dr. Svanberg received her MD and PhD from the University of Gothenburg (Sweden), and is a Board-Certified General Surgeon and Associate Professor of Surgery. Dr. Svanberg joined Serono International in 2000, initially in the field of metabolism, and subsequently held roles of increasing responsibilities before joining Bristol Myers Squibb in the United States in 2007. At BMS, Dr. Svanberg served as Development Leader for a first-in-class novel diabetes medicine, and subsequently as Head of Medical Affairs for the Intercontinental region. In 2014, Dr. Svanberg joined Janssen Pharmaceuticals (a Johnson & Johnson company) as Vice President, Head of the Established Products group where she was managing a portfolio of 90 products, used by an estimated 150 million patients globally. Dr. Svanberg subsequently served as Chief Development Officer at Ixaltis, and as Chief Medical Officer at Kuste Biopharma, specialty pharmaceutical companies developing proprietary therapeutics to treat genitourinary (GU) disorders with unmet medical need. Dr. Svanberg is a partner at Ventac Partners (since 2023) and also serves as a non-executive director on the Boards of Egetis (formerly PledPharma) (since 2017), Amolyt Pharma (since 2021), LEO Pharma (since 2022), and EPICS Therapeutics (since 2022).
Jérôme Contamine is a non-executive independent member of our Board of Directors since April 26, 2022. Mr. Contamine served as Chief Financial Officer of Sanofi for more than nine years from 2009 until 2018. Prior to joining Sanofi, he was Chief Financial Officer of Veolia from 2000 to 2009. He previously held various operating functions at Total, and served four years as an auditor at the Cour des Comptes (the supreme body responsible for auditing the use of public funds in France). Mr. Contamine is a graduate of France’s École Polytechnique, ENSAE (École Nationale de la Statistique et de l’Administration Économique) and École Nationale d’Administration. He held the position of non-executive director at Valeo from 2006 to 2017 and Total Energies from 2020 to 2023. Mr. Contamine also serves as a non-executive director on the Board of Société Générale. Effective as of March 21, 2023, Jérôme Contamine is appointed as the new Lead Non-Executive Director of Galapagos, replacing Dr. Rajesh Parekh.
Daniel G. Baker is a non-executive independent member of our Board of Directors since April 26, 2022. Dr. Baker joined Janssen/Centocor in 2000 and as Vice President Immunology R&D his responsibilities included the clinical development of Remicade, Simponi and Stelara, as well as other programs in rheumatology and dermatology. He supervised many Phase I-III trials in multiple disease areas, and oversaw more than 15 regulatory approvals in the US, Europe and Japan. Throughout his time at Janssen, he was responsible for evaluating business development opportunities in the immunology space. In 2015 he took on a new role as Disease Area Stronghold Leader at Janssen where he was responsible for Phase II & III clinical development plans for rheumatology products and the overall portfolio strategy in rheumatology and immunology. This included the early research strategy for immunology discovery, managing the early portfolio development and approving all late-stage efforts. Since his retirement from Janssen in 2019, he has continued to be involved in bringing therapies to patients. He raised capital (>$20MM) to fund and start an immunology company, KiRA Biotech, where he now acts as Chief Executive Officer and as Executive Director. Dr. Baker received his B.A. in Biology from Gettysburg College and his Medical Degree from the University of Pennsylvania. He completed his Medical Residency at Hershey Medical Center and Fellowship in Rheumatology and Immunology at the University of Pennsylvania, followed by a Research Fellowship in Rheumatology at Mass General Hospital. He continued on as part of the faculty of the University of Pennsylvania for 18 years before taking on industry roles.
Susanne Schaffert is a non-executive independent member of our Board of Directors since June 12, 2023, and is the former Global President, Novartis Oncology, and a member of the Executive Committee of Novartis. For more than 25 years, Dr. Schaffert has dedicated her career at Novartis to helping patients live longer, better lives. Before assuming her role as President of Novartis Oncology, Dr. Schaffert served as Chairperson and President of Advanced Accelerator Applications since its acquisition by Novartis in January 2018. Prior to this, Dr. Schaffert was the Head of Region Europe at Novartis Oncology, where she was responsible for leading Novartis’ Oncology Business Unit in the European Region, marketing key products in lung, breast and renal cancer, as well as hematology and coordinating the entire
168
Oncology operations for EU countries. From 2010 to 2012, Dr. Schaffert served as the Head of Investor Relations for Novartis Group and prior thereto, she served as the Novartis Global Franchise Head for Immunology and Infectious Diseases. Dr. Schaffert first joined Novartis Germany in 1995 as a sales representative, and she has held a series of positions in Sales & Marketing with increasing responsibilities in both national and global functions. Dr. Schaffert has experience from various Boards and Committees, and beyond serving Galapagos NV as non-executive independent Board member, she is also an independent non-executive Director on the Board of Incyte Corporation, a Board member and member of the Advisory Group at Novo Holdings in Denmark and serves as independent Board Director on the boards of ARTBio, US and Vetter Pharma, Germany. She is also a member of the Board of Partners of E. Merck KG. Dr. Schaffert holds an M.Sc. in Chemistry and a Ph.D. with honors in Organic Chemistry from University of Erlangen (Germany).
Simon Sturge is a non-executive independent member of our Board of Directors since September 19, 2023, and was the former CEO of Kymab, a biotech company focused on immune-mediated diseases and immuno-oncology therapeutics, until its acquisition by Sanofi in 2021. Mr. Sturge brings over 40 years of global experience in the pharmaceutical industry, including manufacturing expertise from decades of leadership roles at Celltech Biologics (now Lonza), Boehringer Ingelheim and Merck KGgA. He is currently chairing three biotechnology companies in Switzerland, Belgium, and the United States. He also runs his family investment fund and consultancy company and is a Trustee of Weizmann UK. Mr. Sturge joined Kymab as CEO in 2019 before selling it to Sanofi two years later. Before Kymab, he spent six years at Merck Group, based at their corporate headquarters in Darmstadt, Germany, as Executive Vice President Global Strategy, Business Development & Global Operations and previously as Chief Operating Officer of Merck Healthcare, responsible for the company’s global commercial and manufacturing operations. In this capacity, he was responsible for the continued growth in global sales at Merck KGaA, as well as the commercial launches of Bavencio® (anti-PD-L1 antibody, avelumab) in solid tumors and Mavenclad® (cladribine) for relapsing multiple sclerosis. Prior to that, Mr. Sturge served as Corporate Senior Vice President, Biopharmaceuticals at Boehringer Ingelheim, where he was responsible for the company’s global biopharmaceuticals manufacturing business as well as its biosimilars portfolio. Mr. Sturge was also founder and CEO of Ribotargets (now Vernalis), which was acquired by British Biotech. Mr. Sturge holds a BSc degree in Biology from Sussex University.
Pursuant to the new Nasdaq Stock Market Listing Rule and US Securities Exchange Act requirements, we disclose aggregated statistical information about the Board’s voluntary self-identified gender and racial characteristics and LGBTQ+ status for 2023. Diverse means an individual who self-identifies as one or more of the following: Female, LGBTQ+, or an underrepresented individual based on national, racial, ethnic, indigenous, cultural, religious or linguistic identity in the country of the Company’s principal executive offices.
Board of Directors Diversity Matrix (As of March 15, 2024) |
|
| ||||
Country of Principal Executive Offices |
| Belgium | ||||
Foreign Private Issuer |
| Yes | ||||
Disclosure prohibited under Home Country Law | No | |||||
Total number of Board members | 9 | |||||
Female | Male | Non-binary | Did not disclose gender | |||
Part I: Gender Identity | ||||||
Directors (members of the Board of Directors) | 3 | 6 | 0 | 0 | ||
Part II: Demographic Background | ||||||
Underrepresented individual in Home Country Jurisdiction | 0 | |||||
LGBTQ+ | 0 | |||||
Did not disclose demographic background | 0 |
As a foreign private issuer, under the listing requirements and rules of Nasdaq, we satisfy the Board diversity requirement with two female Directors. In 2023, we have three women (except between June 12, 2023 and September 18, 2023 when the Board consisted of four women) and six men (except between June 12, 2023 and September 18, 2023
169
when the Board consisted of five men) in our Board of Directors. During 2023, Galapagos also complied with its obligations with respect to gender diversification in the Board of Directors as set forth in article 7:86 of the Belgian Companies Code, and our Board will continue to monitor further compliance. In proposing candidates, particular consideration is given to diversity in gender, age, nationality, educational and professional background, as well as complementary skills, knowledge and experience.
Executive Committee
As from April 26, 2022, Galapagos has adopted a one-tier governance structure as provided for by the Belgian Companies Code, with the Board of Directors replacing the (former) Supervisory Board and the Executive Committee replacing the (former) Management Board.
The following table sets forth certain information with respect to the members of our Executive Committee as of December 31, 2023:
| ||||
Name |
| Age |
| Position(s) |
Stoffels IMC BV (permanently represented by Dr. Paul Stoffels) (1) |
| Chief Executive Officer | ||
Michele Manto (2) |
| 50 | Chief Commercial Officer | |
Valeria Cnossen |
| 50 | General Counsel, Executive Vice President | |
Annelies Missotten |
| 51 | Chief Human Resources Officer, Executive Vice President | |
Thad Huston |
| 53 | Chief Financial Officer & Chief Operating Officer, Executive Vice President | |
| ||||
(1) Management company incorporated in 2022, age of Dr. Paul Stoffels: 61. | ||||
(2) Executive Committee member until December 31, 2023. |
| |||
The address for the Executive Committee members is Generaal De Wittelaan L11 A3, 2800 Mechelen, Belgium.
In 2023, there was no conflict of interests between the private interests or other duties of the members of the Executive Committee listed above and their duties to us, except for the conflicts of interests between us and a Board / Executive Committee member as further described in “Item 7.B – Related Party Transaction”.
Bart Filius’ mandate as President, Chief Financial Officer & Chief Operating Officer and Executive Committee member ended per June 30, 2023.
Thad Huston was appointed as our Chief Financial Officer & Chief Operating Officer and Executive Member as from July 1, 2023.
Michele Manto’s mandate as Chief Commercial Officer and Executive Committee member ended per December 31, 2023.
Below are the biographies of the members of our Executive Committee (members per December 31, 2023):
Paul Stoffels 2 joined Galapagos as Chief Executive Officer in April 2022, and is an executive member and the Chairman of our Board of Directors since April 26, 2022. He also is a member of the Executive Committee at Galapagos. Prior to that, he was Vice Chairman of the Executive Committee and Chief Scientific Officer of Johnson & Johnson where he set the company's wide innovation agenda and led its pharmaceutical R&D-pipeline, as well as other external initiatives. Before that, he was worldwide Chairman of Pharmaceuticals of Johnson & Johnson which, under his leadership, significantly rejuvenated its product pipeline and adopted a transformational R&D-operating model, which resulted in the launch of 25 innovative medicines across the globe. Dr. Stoffels joined Johnson & Johnson in 2002,
2 Stoffels IMC BV, permanently represented by Dr. Paul Stoffels
170
following the acquisition of Virco and Tibotec, where he was Chief Executive Officer and Chairman respectively, and where he led the development of several breakthrough products for the treatment of HIV. Dr. Stoffels also is a member of the Supervisory Board of Philips Healthcare in the Netherlands.
Thad Huston joined Galapagos in June 2023 as Chief Financial Officer, Chief Operating Officer, and member of the Executive Committee. He previously served a Senior Vice President, Finance and Corporate Operations of Kite Pharma, a Gilead Company, where he was responsible for all financial aspects of the market leading cell therapy business worldwide. He was also a member of the Kite Leadership Team, the Gilead CFO Leadership Teams and the Fosun-Kite Board. Before joining Kite in 2021, Thad served as Chief Financial Officer at LivaNova PLC, a medical device company specializing in cardiovascular and neuromodulation products, where he played a key role in external R&D innovation and M&A and led the global, cross-functional teams across the group. Prior to LivaNova, he spent over 25 years in leadership positions at Johnson & Johnson (J&J), which included roles as Chief Financial Officer and Chief Operating Officer of J&J Pharmaceutical Research and Development, Chief Financial Officer of J&J’s Global Surgery and Medical Devices groups managing up to $21 billion in annual revenue, and President of Xian-Janssen, leading J&J’s pharmaceutical division in China. Before that, he held senior financial roles at various J&J locations in the U.S., Belgium, Russia, and Hungary.
Michele Manto is appointed as Chief Commercial Officer in January 2020, and member of the Executive Committee at Galapagos. He joined Galapagos in September 2017 as Senior Vice President Commercial Operations to build and lead Galapagos’ commercial organization and capabilities. Previously, Mr. Manto held various commercial leadership roles at AbbVie, most recently as General Manager, Global Marketing Rheumatology and General Manager in the Netherlands. Prior to this, he led AbbVie’s commercial activities and launches in rheumatology, gastroenterology and dermatology in Germany and other European countries. He started his professional career as a management and strategy consultant at McKinsey & Company. Mr. Manto holds an MBA from INSEAD and a Degree in Engineering from the Politecnico of Milan. The mandate of Michele Manto as Chief Commercial Offier and member of the Executive Committee at Galapagos ended on December 31, 2023.
Annelies Missotten is appointed as Chief Human Resources Officer and member of the Executive Committee at Galapagos. She joined Galapagos as Vice President Human Resources in February 2018 to transform and build an expert HR team to enable business growth, and leading the transformation of Galapagos into an integrated biopharmaceutical company with an international set-up. In 2020, she was appointed Senior Vice President Human Resources and strategic advisor to the CEO and Executive Committee. Before joining Galapagos, she held various senior global HR positions at GSK. She started her career at Proximus, and acquired deep expertise over time in key HR Centres of Expertise, including Training & Development, Talent Acquisition and Reward, and HR Business partnership roles. Ms. Missotten holds a Master's Degree in Roman Philology from KU Leuven, a DEA in Italian Culture and Linguistics from the Paris IV Sorbonne (France) and L’Università Cattolica di Milano. Over the years, she completed her education with several systemic psychology and coaching certifications and business courses, amongst others, from INSEAD, Fontainebleau (France).
Valeria Cnossen is appointed as General Counsel, responsible for Compliance & Ethics, the Corporate Secretary Office and Intellectual Property, and member of the Executive Committee at Galapagos. Ms. Cnossen joined Galapagos on 1 August 2022. She previously was General Counsel of the Consumer Health Group at Johnson & Johnson where she was a strategic partner and key advisor on laws and regulations, transactions and emerging areas, impacting the business such as digital, transparency, sustainability and public policy. Prior to that, she held leadership roles within the Medical Devices and Pharmaceutical Sectors of Johnson & Johnson. Ms. Cnossen joined Johnson & Johnson in 2011 through the acquisition of Crucell, where she was Head of Legal and Compliance. Prior to joining Crucell, Ms. Cnossen was in private legal practice at De Brauw Blackstone Westbroek in the Netherlands, and Cravath, Swaine & Moore in New York City. Ms. Cnossen is a purpose-driven leader, known for her ability to develop high-performing teams and the careers of others, especially as a mentor for women.
The Executive Committee has been entrusted by the Board of Directors with the executive management and running of the Company. Without prejudice to the overall responsibility and tasks of the Board of Directors regarding
171
the management and control of the Company, the tasks of the Executive Committee include the following matters (without limitation): the research, identification and development of strategic possibilities and proposals which may contribute to our development in general, the management of the Company and the Galapagos group, the supervision of the performance of the business in comparison with the strategic goals, plans and budgets, and the support of the Chief Executive Officer with the day-to-day management of the Company and the Galapagos group.
The Executive Committee meets as often as necessary to ensure to its effective operation, and in principle once per month.
Family relationships
There are no family relationships among any of the members of our Executive Committee or Board of Directors.
B. Compensation
The aggregate compensation paid and benefits in kind granted by us to our (former) members of the Executive Committee and Board of Directors, excluding equity-related compensation, for the year ended December 31, 2023, was €4,559,519. This includes remuneration paid in 2023 to Directors that stepped down in 2023, and to Executive Committee members whose mandate came to an end in 2023.
For the year ended December 31, 2023, the total amounts set aside or accrued to provide pension, retirement or similar benefits to our Executive Committee amounted to €208,804.00.
For a discussion of our management agreements with the Executive Committee members, see the section of this annual report titled “Item 7.B.—Related Party Transactions.— Agreements with Our Board Members and Executive Committee Members.” For more information regarding subscription right grants, see “—Subscription Right Plans” below and regarding RSU grants, see “—RSU Plans” below.
Remuneration policy
In 2020, pursuant to the expected implementation in Belgium of the Directive (EU) 2017/828 of the European Parliament and of the Council of 17 May 2017 amending Directive 2007/36/EC as regards the encouragement of long-term shareholder engagement, or SRD II, a revised version of the Remuneration Policy was submitted to a binding vote by our 2020 Shareholders’ Meeting and was approved. On May 6, 2020, the Belgian Act of April 28, 2020 transposing the SRD II into Belgian law was published in the Belgian Official Journal. The policy was applicable during the reporting years 2020 and 2021.
In light of the proposed introduction of a one-tier governance structure at the occasion of the Extraordinary Shareholders’ Meeting held on April 26, 2022, a revised Remuneration Policy was submitted to a binding vote by our 2022 Shareholders’ Meeting and approved during this Shareholders’ Meeting. Our Remuneration Policy was prepared in accordance with the Belgian Companies Code. The amended remuneration policy applied during the reporting years beginning on January 1, 2022.
A further revised Remuneration Policy will be submitted to a binding vote by our 2024 Shareholders’ Meeting. Subject to approval, this revised Remuneration Policy will apply as from January 1, 2024.
The objective of our Remuneration Policy is intended to attract, engage, and retain the diverse qualified and expert individuals that we need to pursue our strategic and operational objectives whilst reinforcing our culture and sustainability ambitions for the benefit of patients, our people and planet. Our specific goals for remuneration are to offer competitive opportunities for talented employees by benchmarking against appropriate peer groups, to incentivize exceptional and sustainable performance, aligned with corporate achievements, to provide differential rewards based on individual performance, to avoid differentiation on any grounds except for performance and other proper factors, and to reinforce an open and equitable culture.
172
Compensation of our Board of Directors
The remuneration of our members of the Board of Directors is submitted by our Board of Directors for approval to the Shareholders’ Meeting and is only implemented after such approval. The procedure for establishing the remuneration policy and setting remuneration for members of our Board of Directors is determined by our Board of Directors on the basis of proposals from the Remuneration Committee, taking into account relevant benchmarks from the biotechnology industry.
The Annual Shareholders’ Meeting of April 28, 2020 determined, upon recommendation of the Nomination and Remuneration committee, that the compensation (excluding expenses) of the members of the Board of Directors, other than the Board members representing a shareholder, for the exercise of their mandate during the financial year ending December 31, 2023 is as follows:
(a) fixed cash remuneration: (i) Chair of the Board of Directors (i.e. Stoffels IMC BV, permanently represented by Dr. Paul Stoffels: N/A ; (ii) other non-executive members of the Board of Directors (i.e. Raj Parekh (until June 10, 2023), Mary Kerr (until September 18, 2023), Peter Guenter, Elisabeth Svanberg, Dan Baker, Jérôme Contamine, Susanne Schaffert (as of June 12, 2023), and Simon Sturge (as of September 19, 2023): €50,000 each; (iii) annual additional compensation for membership of a Board committee: €15,000; (iv) annual additional compensation for the Chairmanship of a Board committee: €20,000;
(b) equity-based remuneration: (i) Chair of the Board of Directors (i.e. Stoffels IMC BV, permanently represented by Dr. Paul Stoffels: N/A; (ii) other non-executive members of the Board of Directors (i.e. Raj Parekh (until June 10, 2023), Mary Kerr (until September 18, 2023), Peter Guenter, Elisabeth Svanberg, Dan Baker, Jérôme Contamine, Susanne Schaffert (as of June 12, 2023), and Simon Sturge (as of September 19, 2023): €50,000 each; in each case (i) and (ii) subject to the requirement to use the net amount (after taxes) to acquire Galapagos shares. These latter payments make up the equivalent of an equity component of the Board members’ remuneration and the resulting shares are to be held until at least one year after the member of the Board of Directors leaves the Board of Directors and at least three years after the time of acquisition.
The same 2020 Annual Shareholders’ Meeting resolved that the mandate of a member of the Board of Directors representing a shareholder on the Board of Directors will not be remunerated, being Daniel O’Day and Linda Higgins.
Effective from April 26, 2022, our new Chief Executive Officer, Stoffels IMC BV (permanently represented by Dr. Paul Stoffels) has been appointed as the Chair of our Board of Directors. The Chief Executive Officer will only be remunerated for the performance of its executive functions as CEO and will not be entitled to any additional remuneration for its mandates of member and Chairman of the Board of Directors or member of any Committee.
The remuneration of the members of the Board of Directors does not contain a variable part; hence no performance criteria apply to their remuneration.
173
The following table sets forth the fees received by our members of the Board of Directors for the performance of their mandate as a Board member during the year ended December 31, 2023.
Board of Directors | Audit committee | Nomination Committee | Remuneration Committee | Science and Development Committee (2) | ||||||||||||||||||||||||||||||||||
Cash remuneration | Equity-based remuneration | Cash remuneration | Cash remuneration | Cash remuneration | Cash remuneration | |||||||||||||||||||||||||||||||||
Chair | Member | Cash granted | Acquired | Chair | Member | Chair | Member | Chair | Member | Chair | Member | |||||||||||||||||||||||||||
Fees earned | Fees earned | to acquire | GLPG | Fees earned | Fees earned | Fees earned | Fees earned | Fees earned | Fees earned | Fees earned | Fees earned | |||||||||||||||||||||||||||
Name |
| (Euro) |
| (Euro) |
| GLPG shares (1) |
| shares (1) |
| (Euro) |
| (Euro) |
| (Euro) |
| (Euro) |
| (Euro) |
| (Euro) |
| (Euro) |
| (Euro) | ||||||||||||||
Stoffels IMC BV, permanently represented by Dr. Paul Stoffels (3) | ||||||||||||||||||||||||||||||||||||||
Dr. Rajesh Parekh (4) | € | 22,253 | € | 22,000 | 264 | € | 4,389 | € | 4,389 | |||||||||||||||||||||||||||||
Dr. Mary Kerr (5) |
| 35,870 | 36,000 | 441 | € | 10,761 | ||||||||||||||||||||||||||||||||
Mr. Peter Guenter |
| 50,000 | 50,000 | 644 | 15,000 | |||||||||||||||||||||||||||||||||
Dr. Elisabeth Svanberg (6) |
| 50,000 | 50,000 | 635 | 15,611 | 15,611 | € | 3,292 | € | 4,239 | ||||||||||||||||||||||||||||
Mr. Jérôme Contamine | 50,000 | 50,000 | 644 | € | 20,000 | € | 15,000 | 15,000 | ||||||||||||||||||||||||||||||
Dr. Dan Baker (7) | 50,000 | 50,000 | 635 | 11,708 | € | 5,652 | ||||||||||||||||||||||||||||||||
Mr. Daniel O’Day (8) | ||||||||||||||||||||||||||||||||||||||
Dr. Linda Higgins (8) | ||||||||||||||||||||||||||||||||||||||
Dr. Susanne Schaffert (9) | 27,610 | 28,000 | 360 | 4,239 | ||||||||||||||||||||||||||||||||||
Mr. Simon Sturge (10) | 14,130 | 14,000 | 180 | 4,239 | ||||||||||||||||||||||||||||||||||
Total | € | - | € | 299,863 | € | 300,000 | 3,803 | € | 20,000 | € | 30,000 | € | 20,000 | € | 15,000 | € | 20,000 | € | 30,000 | € | 5,652 | € | 8,478 | |||||||||||||||
(1) The company grants a gross amount equal to the respective Board member’s annual cash remuneration, to use the net portion (after taxes) to acquire shares of Galapagos in the open market. | ||||||||||||||||||||||||||||||||||||||
(2) Established on September 19, 2023. | ||||||||||||||||||||||||||||||||||||||
(3) Chair of the Board of Directors as of April 26, 2022. Stoffels IMC BV does not receive any remuneration for its mandate as Chair of the Board of Directors or Committee member. | ||||||||||||||||||||||||||||||||||||||
(4) Director until June 10, 2023, Chair of the Nomination Committee and the Renumeration Committee until March 20, 2023. | ||||||||||||||||||||||||||||||||||||||
(5) Director and Audit Committee member until September 18, 2023. | ||||||||||||||||||||||||||||||||||||||
(6) Chair of the Nomination Committee and the Renumeration Committee as of March 21, 2023, Science and Development Committee member as of September 19, 2023. | ||||||||||||||||||||||||||||||||||||||
(7) Chair of the Science and Development Committee as of September 19, 2023. | ||||||||||||||||||||||||||||||||||||||
(8) Mr. O’Day and Dr. Higgins, both Gilead representatives, do not receive any remuneration for their mandate as members of the Board of Directors. | ||||||||||||||||||||||||||||||||||||||
(9) Director as of June 12, 2023, Science and Development Committee member as of September 19, 2023. | ||||||||||||||||||||||||||||||||||||||
(10) Director and Audit Committee member as of September 19, 2023. | ||||||||||||||||||||||||||||||||||||||
The table below provides an overview as of December 31, 2023 of the subscription rights held by the (former) members of the Board of Directors. Upon recommendation of our Nomination and Remuneration Committee, the Board of Directors decided in February 2020 to discontinue the grant of subscription rights to members of the Board of Directors going forward, taking into account the stricter rules of the Belgian Companies Code and provision 7.6 of the 2020 Code.
Subscription right award (1) | ||||||||||||||||
Subscription | Number of ordinary | Number of | Number of | |||||||||||||
Subscription right | right |
| shares underlying | subscription rights | subscription rights | |||||||||||
Grant | Vesting | exercise |
| expiration |
| subscription rights | exercisable | exercised | ||||||||
Name |
| Plan |
| date |
| period |
| price (Euro) |
| date |
| per Dec. 31, 2023 |
| per Dec. 31, 2023 |
| during 2023 |
Raj Parekh | WP 2017 | 8/30/2017 | 36 months | 80.57 | 5/16/2025 | 15,000 | 15,000 | |||||||||
WP 2018 | 6/18/2018 | 1/36 per | 79.88 | 4/18/2026 | 15,000 | 15,000 | ||||||||||
WP 2019 | 07/12/2019 | month | 95.11 | 04/10/2027 | 15,000 | 15,000 | ||||||||||
Total |
|
|
|
| 45,000 | 45,000 | — | |||||||||
| ||||||||||||||||
Mary Kerr | WP 2017 | 8/30/2017 | 36 months | 80.57 | 5/16/2025 | 7,500 | 7,500 | |||||||||
WP 2018 | 6/18/2018 | 1/36 per | 79.88 | 4/18/2026 | 7,500 | 7,500 | ||||||||||
WP 2019 | 07/12/2019 | month | 95.11 | 04/10/2027 | 7,500 | 7,500 | ||||||||||
Total | 22,500 | 22,500 | — | |||||||||||||
Peter Guenter | WP 2019 | 07/12/2019 | 36 months | 95.11 | 04/10/2027 | 7,500 | 7,500 | |||||||||
1/36 per | ||||||||||||||||
month | ||||||||||||||||
Total | 7,500 | 7,500 | — | |||||||||||||
(1) The Subscription Rights under Warrant Plan 2015.B became nul and void on their expiration date December 21, 2023. | ||||||||||||||||
Dr. Elisabeth Svanberg, Dr. Dan Baker, Mr. Jérôme Contamine, Mr. Daniel O’ Day, Dr. Linda Higgins, Dr. Susanne Schaffert and Mr. Simon Sturge don’t possess any subscription rights.
No loans, quasi-loans or other guarantees were given to the members of the Board of Directors during the year ended December 31, 2023.
174
Compensation of members of the Executive Committee
The compensation of the members of our Executive Committee is determined by our Board of Directors based on the recommendations by our Remuneration Committee.
The remuneration of the members of our Executive Committee consists of different components:
| ● | Fixed cash remuneration: a basic fixed fee (cash base salary) designed to fit responsibilities, relevant experience and competences, in line with market rates for equivalent positions. The amount of fixed remuneration is evaluated and determined by the Board of Directors every year for the Chief Executive Officer, upon recommendation of the Remuneration Committee and for the other members of the Executive Committee upon recommendation of the Chief Executive Officer. |
| ● | Variable remuneration (short-term): members of the Executive Committee may be entitled to a short-term cash component, being a cash bonus. The award of a bonus is merit-driven and based on the group’s performance management system that is based on annual individual performance (including exceptional deliverables) in combination with our overall performance, compared to the level of achievement of individual and corporate objectives that are established annually. As from the year that ended December 31, 2019 and under our Remuneration Policy, the maximum short-term cash bonus of the Chief Executive Officer is set at 75% of his yearly fixed cash salary. The actual bonus of the Chief Executive Officer is determined by our Board of Directors, upon recommendation of the Remuneration Committee, and is based on the achievement of corporate and individual objectives. The maximum aggregate cash bonus pot for the other members of the Executive Committee is set at maximum 50% of their combined fixed cash salaries for the short-term cash bonus. The actual bonuses of these other Executive Committee members are determined by our Board of Directors, upon recommendation of the Chief Executive Officer, and are based on the achievement of corporate and individual objectives. In addition, exceptional special bonuses, outside the scope of the regular bonus schemes, can be considered by the Board of Directors, upon recommendation of the Remuneration Committee, in the event of and for exceptional achievements. |
| ● | Variable remuneration (long-term) – Incentive plans: as from the year that ended December 31, 2019 and under our Remuneration Policy, the Chief Executive Officer is eligible to receive the equivalent number of restricted stock units, or RSUs, to 75% of his cash base salary, and the other members of the Executive Committee are eligible to receive the equivalent number of RSUs to 50% of the total amount of their aggregate cash base salaries as an annual long term incentive. The number of granted RSUs will be equivalent to the respective cash bonus of the Chief Executive Officer or the other Executive Committee members for the respective financial year. They may receive additional RSUs under other RSU plans that were put in place. For a description of the main characteristics of our RSU plans for Executive Committee members, see “RSU Plans” below. |
In addition, subscription rights have been granted and may be granted in the future to the members of the Executive Committee. For a description of the main characteristics of our subscription right plans, see “Subscription Right Plans” below.
| ● | Other: pursuant to our remuneration policy, the Executive Committee members are eligible to benefits such as a pension, company car, insurances, tax advisory services and payments for invalidity and healthcare cover and other fringe benefits of non-material value. |
No loans, quasi-loans or other guarantees were given to members of our Executive Committee during the year ended December 31, 2023.
175
The following table sets forth information concerning the compensation earned by the Executive Committee members during the year ended December 31, 2023:
| (Euro) | ||||||||||||||||||||
Fixed remuneration | Variable remuneration | Total | |||||||||||||||||||
Executive |
| Base salary |
| Other components (1) |
| Pension |
| Cash bonus (2) |
| Multi-year variable | remuneration | ||||||||||
Committee member |
|
|
|
|
| Vested RSUs (3) |
| Granted SRs (4) |
| ||||||||||||
Stoffels IMC BV | € | 750,000 | € | — | € | — | € | 506,250 | € | 647,536 | € | 67,500 | € | 1,971,286 | |||||||
Other Executive Committee members (5) | € | 1,605,839 | € | 304,633 | € | 208,804 | € | 735,000 | € | 2,127,645 | € | 101,250 | € | 5,083,171 | |||||||
(1) Other components are the value of the benefits and perquisites awarded, such as a company car, tax advisory services, and health and disability insurance. | |||||||||||||||||||||
(2) The one-year variable is the short-term cash bonus awarded to each Executive Committee member in respect of 2023 and paid in April 2024. | |||||||||||||||||||||
(3) During financial year 2023 RSUs vested under RSU plans 2019.II, 2020.I, 2020.II, 2021.I, 2021.II, 2021.IV, 2022.I and 2022.II and pay-outs occurred accordingly. | |||||||||||||||||||||
(4) The value of the subscription rights ("SRs") granted during the financial year 2023 is calculated by comparing the exercise price with the average share price of the share as quoted on Euronext Brussels and Amsterdam during the financial year 2023. | |||||||||||||||||||||
(5) Pursuant to the applicable Belgian legislation for the one-tier governance system, we hereby disclose the remuneration of the other Executive Committee members on an aggregated basis. This includes remuneration paid to Bart Filius until June 30, 2023 and to Thad Huston as of July 1, 2023. | |||||||||||||||||||||
The total remuneration table above sets forth the value of the number of RSUs vested and paid out in 2023 for each Executive Committee member. Each RSU represents the right to receive, at Galapagos’ discretion, one Galapagos share or a payment in cash of an amount equivalent to the volume-weighted average price of the Galapagos share on Euronext Brussels over the 30-calendar day period preceding the relevant vesting date. However, for members of the Executive Committee, any vesting prior to the third anniversary of the offer date will always give rise to a payment in cash rather than a delivery of shares as an incentive.
During 2023, there were RSU vestings under eight different RSU Plans: Plan 2019.II, Plan 2020.I, Plan 2020.II, Plan 2021.I, Plan 2021.II, Plan 2021.IV, Plan 2022.I, and Plan 2022.II. The pay-outs to the Executive Committee members occurred accordingly and the aggregate amounts are set forth in the total remuneration table above.
Subscription rights
In addition, the members of the Executive Committee during the year ended December 31, 2023, were granted and accepted subscription rights.
In 2023, our Chief Executive Officer, Stoffels IMC BV (permanently represented by Dr. Paul Stoffels) was offered 50,000 subscription rights under Subscription Right Plan 2023 BE. He accepted all the granted subscription rights. The exercise price of these subscription rights is €35.11. The subscription rights under Subscription Right Plan 2023 BE vest only and fully on the first day of the fourth calendar year following the calendar year in which the grant was made. These subscription rights are exercisable as from January 1, 2027.
The following members of the Executive Committee were offered new subscription rights under Subscription Right Plan 2023 BE and each accepted all subscription rights granted as per the following: Mr. Michele Manto (25,000 subscription rights), Annelies Missotten (25,000 subscription rights), Valeria Cnossen (25,000 subscription rights) and Thad Huston (200,000 subscription rights). The exercise price of these subscription rights is €35.11 except for the subscription rights of Thad Huston of which the exercise price is €38.58. For all the beneficiaries under Subscription Right Plan 2023 BE, the subscription rights vest only and fully on the first day of the fourth calendar year following the calendar year in which the grant was made. These subscription rights are exercisable as from January 1, 2027.
176
The table below provides an overview as of December 31, 2023 of the subscription rights held by, awarded to and exercised by the (former) members of our Executive Committee during the year ended December 31, 2023.
Subscription rights awarded | ||||||||||||||||||
Number of | Subscription rights exercised | |||||||||||||||||
Subscription | ordinary shares | Number of | Number of | Number of | ||||||||||||||
rights | Subscription | underlying | subscription | subscription | subscription | |||||||||||||
| exercise | rights | subscription rights |
| rights exercisable | rights awarded | rights exercised | |||||||||||
Grant | Vesting | price | expiration | Per Dec 31, | per Dec 31, | during | per Dec 31, | |||||||||||
Name |
| Plan |
| date |
| period |
| (Euro) |
| date |
| 2023 |
| 2023 |
| 2023 |
| 2023 |
Stoffels IMC BV, permanently represented by Dr. Paul Stoffels | SR Plan 2022 (B) | 03/25/2022 | 100% 3rd year | 50.00 | 01/25/2030 | 1,000,000 | ||||||||||||
SR Plan 2023 BE | 8/5/2023 | after year of grant | 35.11 | 5/5/2031 | 50,000 | — | 50,000 | — | ||||||||||
Total |
|
|
| 1,050,000 |
| — |
| 50,000 | — | |||||||||
Bart Filius | WP 2017 | 8/30/2017 | 100% 3rd year | 80.57 | 5/16/2025 | 60,000 | 60,000 | |||||||||||
WP 2018 | 6/18/2018 | after year of |
| 79.88 | 4/18/2026 | 80,000 |
| 80,000 | ||||||||||
WP 2019 | 12/7/2019 | grant | 95.11 | 10/4/2027 | 65,000 | 65,000 | ||||||||||||
SR Plan 2020 | 6/16/2020 | 168.42 | 4/17/2028 | 50,000 | ||||||||||||||
SR Plan 2021 BE | 2/7/2021 | 64.76 | 4/30/2029 | 50,000 | ||||||||||||||
SR Plan 2022 BE | 7/7/2022 | 57.46 | 6/5/2030 | 68,000 | ||||||||||||||
Total |
|
|
| 373,000 |
| 205,000 |
| — | — | |||||||||
Michele Manto | WP 2017 | 8/30/2017 | 100% 3rd year | 80.57 | 5/16/2025 | 60,000 | 60,000 | |||||||||||
WP 2018 | 6/18/2018 | after year of | 79.88 | 4/18/2026 | 30,000 |
| 30,000 | |||||||||||
WP 2019 | 12/7/2019 | grant | 95.11 | 10/4/2027 | 40,000 | 40,000 | ||||||||||||
SR Plan 2020 | 6/16/2020 | 168.42 | 4/17/2028 | 30,000 | ||||||||||||||
SR Plan 2021 BE | 2/7/2021 | 64.76 | 4/30/2029 | 30,000 | ||||||||||||||
SR Plan 2022 BE | 7/7/2022 | 57.46 | 6/5/2030 | 24,000 | ||||||||||||||
SR Plan 2023 BE | 08/28/2023 | 35.11 | 5/5/2031 | 25,000 | 25,000 | |||||||||||||
Total |
|
| 239,000 |
| 130,000 |
| 25,000 | — | ||||||||||
Annelies Missotten | WP 2018 | 06/18/2018 | 100% 3rd year | 79.88 | 04/18/2026 | 26,000 |
| 26,000 | ||||||||||
WP 2019 | 12/7/2019 | after year of | 95.11 | 10/4/2027 | 20,000 | 20,000 | ||||||||||||
SR Plan 2020 | 06/16/2020 | grant | 168.42 | 04/17/2028 | 15,000 | |||||||||||||
SR Plan 2021 BE | 2/7/2021 | 64.76 | 04/30/2029 | 22,500 | ||||||||||||||
SR Plan 2022 BE | 7/7/2022 | 57.46 | 5/6/2030 | 18,000 | ||||||||||||||
SR Plan 2023 BE | 7/7/2023 | 35.11 | 5/5/2031 | 25,000 | 25,000 | |||||||||||||
Total |
|
| 126,500 |
| 46,000 |
| 25,000 | — | ||||||||||
Valeria Cnossen | SR Plan 2022 BE | 9/11/2022 | 100% 3rd year | 51.58 | 6/5/2030 | 30,000 | ||||||||||||
SR Plan 2023 BE | 08/28/2023 | after year of | 35.11 | 5/5/2031 | 25,000 | 25,000 | ||||||||||||
grant | ||||||||||||||||||
Total |
|
| 55,000 |
| — |
| 25,000 | — | ||||||||||
Thad Huston | SR Plan 2023 BE | 08/28/2023 | 100% 3rd year | 38.58 | 5/5/2031 | 200,000 | — | 200,000 | ||||||||||
after year of | ||||||||||||||||||
Total | grant |
|
| 200,000 |
| — |
| 200,000 | — | |||||||||
177
RSU plans
Upon recommendation of the Nomination and Remuneration committee, the Board of Directors has updated the Remuneration Policy in 2020 to also include the grant of RSUs as a long-term incentive for the members of the Executive Committee, starting from the year ended on December 31, 2019. The revised 2022 Remuneration Policy still includes the grant of RSUs.
We currently have the following restricted stock unit (‘RSU’) programs:
| ● | Plan 2020.I, Plan 2021.I, Plan 2022.I, and Plan 2023.I: these plans are intended to provide a long-term incentive to certain of our employees and Executive Committee members and, as of 2020, replaces the deferred portion of the bonus under the old Senior Management Bonus Scheme; |
| ● | Plan 2019.II, 2020.II, Plan 2021.II, Plan 2021 IV, Plan 2022.II, and Plan 2023.II: these plans are designed with the aim to retain a specific group of our key employees and Executive Committee members whose retention is considered so important for the future performance of Galapagos that an additional incentive is desireable. The beneficiaries are nominated by the Remuneration Committee and the Board of Directors approves the list of beneficiaries. The four-year vesting period is designed to be aligned with long-term shareholder interests; and |
| ● | Plan 2021.III and Plan 2022.III: these plans are intended to compensate employees who transferred from Gilead to Galapagos in the framework of the transfer of European commercialization rights for the long-term incentive plans within Gilead under which unvested RSU awards lapsed upon transfer out of the Gilead group. These employees received an RSU grant from Galapagos. |
The RSU plans are intended to provide certain members of the Executive Committee and certain employees of Galapagos the opportunity to receive RSUs as an incentive. Their purpose is to retain and encourage participants to contribute to the performance of Galapagos and its affiliates by aligning their financial interests with those of the shareholders.
The main characteristics of these plans are as follows:
| ● | The RSUs are offered for no consideration; |
| ● | Generally, three or four year vesting periods apply, as set forth per plan in the table below; |
| ● | Each RSU reflects the value of one Galapagos share and payout will be in cash or shares, at Galapagos’ discretion, it being understood that in respect of members of the Executive Committee, any vesting prior to the third anniversary of the offer date will always give rise to a payment in cash rather than a delivery of shares as an incentive; and |
| ● | In case of termination of service before the vesting date, bad and good leaver rules apply. Any unvested RSUs are forfeited upon termination of service before the vesting date. |
In 2023, the Executive Committee members were offered new RSUs under the 2023 RSU Annual Long-Term Incentive Plan and the 2023 RSU Retention Plan, subject to acceptance. The members of the Executive Committee accepted all RSUs offered to them. The first RSU grant will vest in full three years after the offer date. The second RSU grant has a four-year vesting period, with 25% vesting each year and a first vesting date on May 1, 2024. Except for the RSUs granted to and accepted by Stoffels IMC BV (permanently represented by Dr. Paul Stoffels), the RSUs are not transferable. The table below sets forth the number of RSUs offered to each Executive Committee member in 2023: Stoffels IMC BV (permanently represented by Dr. Paul Stoffels): 138,971 RSUs, , Mr. Michele Manto: 47,812 RSUs, Ms Annelies Missotten: 46,338 RSUs, Ms Valeria Cnossen: 47,401 RSUs and Mr Thad Huston: 50,544 RSUs.
178
No RSUs expired during the year ended December 31, 2023. The table below sets out the main characteristics of RSU plans issued to the (former) Executive Committee members during 2019, 20220, 20221, 2022, and 2023, the RSUs awarded to and accepted by each (former) Executive Committee member under the respective RSU Plan and vested for and paid out to each (former) Executive Committee member during 2023:
Name |
| Plan |
| Grant |
| Vesting period |
| Vesting |
| Number |
| Number |
Stoffels IMC BV, permanently represented by Dr. Stoffels | Plan 2022.II | 5/5/2022 | 25%/year, | 1/5/2023 | 74,408 | 18,602 | ||||||
Four-year vesting period | 1/5/2024 | |||||||||||
1/5/2025 | ||||||||||||
1/5/2026 | ||||||||||||
Plan 2023.I | 5/8/2023 | 100% three years after offer date | 5/8/2026 | 9,695 | ||||||||
Plan 2023.II | 5/9/2023 | 25%/year | 1/5/2024 | 129,276 | ||||||||
Four-year vesting period | 1/5/2025 | |||||||||||
1/5/2026 | ||||||||||||
1/5/2027 | ||||||||||||
Total | 213,379 | 18,602 | ||||||||||
Bart Filius (1) | Plan 2019.I | 10/16/2019 | 100% three years after offer date | 10/16/2022 | 5,000 | |||||||
Plan 2019.II | 10/16/2019 | 25%/year | 1/5/2020 | 17,924 | ||||||||
Four-year vesting period | 1/5/2021 | |||||||||||
1/5/2022 | ||||||||||||
1/5/2023 | 4,481 | |||||||||||
Plan 2019.III | 10/16/2019 | 50% two year after offer date | 10/16/2021 | 16,922 | ||||||||
50% three years after offer date | 10/16/2022 | |||||||||||
Plan 2020.I | 5/6/2020 | 100% three years after offer date | 5/6/2023 | 1,452 | 1,452 | |||||||
Plan 2020.II | 5/6/2020 | 25%/year | 1/5/2021 | 11,148 | ||||||||
Four-year vesting period | 1/5/2022 | |||||||||||
1/5/2023 | 2,787 | |||||||||||
1/5/2024 | ||||||||||||
Plan 2021.I | 5/5/2021 | 100% three years after offer date | 5/5/2024 | 1,011 | ||||||||
Plan 2021.IV | 5/5/2021 | 25%/year | 1/5/2022 | 61,719 | ||||||||
Four-year vesting period | 1/5/2023 | 15,429 | ||||||||||
1/5/2024 | ||||||||||||
1/5/2025 | ||||||||||||
Plan 2022.I | 5/3/2022 | 100% three years after offer date | 5/3/2025 | 3,570 | ||||||||
Plan 2022.II | 5/5/2022 | 25%/year | 1/5/2023 | 57,872 | 14,468 | |||||||
Four-year vesting period | 1/5/2024 | |||||||||||
1/5/2025 | ||||||||||||
1/5/2026 | ||||||||||||
Total | 176,618 | 38,617 | ||||||||||
Michele Manto | Plan 2019.II | 10/16/2019 | 25%/year | 1/5/2020 | 5,121 |
179
Four-year vesting period | 1/5/2021 | |||||||||||
1/5/2022 | ||||||||||||
1/5/2023 | 1,280 | |||||||||||
Plan 2020.I | 5/6/2020 | 100% three years after offer date | 5/6/2023 | 612 | 612 | |||||||
Plan 2020.II | 5/6/2020 | 25%/year | 1/5/2021 | 5,308 | ||||||||
Four-year vesting period | 1/5/2022 | |||||||||||
1/5/2023 | 1,327 | |||||||||||
1/5/2024 | ||||||||||||
Plan 2021.I | 5/5/2021 | 100% three years after offer date | 5/5/2024 | 835 | ||||||||
Plan 2021.IV | 9/24/2021 | 25%/year | 1/5/2022 | 30,859 | ||||||||
Four-year vesting period | 1/5/2023 | 7,714 | ||||||||||
1/5/2024 | ||||||||||||
1/5/2025 | ||||||||||||
Plan 2022.I | 5/3/2022 | 100% three years after offer date | 5/3/2025 | 2,550 | ||||||||
Plan 2022.II | 5/5/2022 | 25%/year | 1/5/2023 | 24,804 | 6,201 | |||||||
Four-year vesting period | 1/5/2024 | |||||||||||
1/5/2025 | ||||||||||||
1/5/2026 | ||||||||||||
Plan 2023.I | 5/8/2023 | 100% three years after offer date | 5/8/2026 | 4,720 | ||||||||
Plan 2023.II | 5/9/2023 | 25%/year | 1/5/2024 | 43,092 | ||||||||
Four-year vesting period | 1/5/2025 | |||||||||||
1/5/2026 | ||||||||||||
1/5/2027 | ||||||||||||
Total | 117,901 | 17,134 | ||||||||||
Annelies Missotten | Plan 2019.II | 10/16/2019 | 25%/year | 1/5/2020 | 1,536 | |||||||
Four-year vesting period | 1/5/2021 | |||||||||||
1/5/2022 | ||||||||||||
1/5/2023 | 384 | |||||||||||
Plan 2020.I | 5/6/2020 | 25%/year | 1/5/2021 | 332 | ||||||||
Four-year vesting period | 1/5/2022 | |||||||||||
1/5/2023 | 83 | |||||||||||
1/5/2024 | ||||||||||||
Plan 2020.II | 5/6/2020 | 25%/year | 1/5/2021 | 956 | ||||||||
Four-year vesting period | 1/5/2022 | |||||||||||
1/5/2023 | 239 | |||||||||||
1/5/2024 | ||||||||||||
Plan 2021.I | 5/5/2021 | 25%/year | 1/5/2022 | 1,488 | ||||||||
Four-year vesting period | 1/5/2023 | 372 | ||||||||||
1/5/2024 | ||||||||||||
1/5/2025 | ||||||||||||
Plan 2021.II | 5/6/2021 | 25%/year | 1/5/2022 | 2,708 | ||||||||
Four-year vesting period | 1/5/2023 | 677 | ||||||||||
1/5/2024 | ||||||||||||
1/5/2025 | ||||||||||||
Plan 2022.I | 5/3/2022 | 25%/year | 1/5/2023 | 1,766 | 444 |
180
Four-year vesting period | 1/5/2024 | |||||||||||
1/5/2025 | ||||||||||||
1/5/2026 | ||||||||||||
Plan 2022.II | 5/5/2022 | 25%/year | 1/5/2023 | 2,980 | 745 | |||||||
Four-year vesting period | 1/5/2024 | |||||||||||
1/5/2025 | ||||||||||||
1/5/2026 | ||||||||||||
Plan 2023.I | 5/8/2023 | 100% three years after offer date | 5/8/2026 | 3,246 | ||||||||
Plan 2023.II | 5/9/2023 | 25%/year | 1/5/2024 | 43,092 | ||||||||
Four-year vesting period | 1/5/2025 | |||||||||||
1/5/2026 | ||||||||||||
1/5/2027 | ||||||||||||
Total | 58,104 | 2,944 | ||||||||||
Valeria Cnossen | Plan 2022.II | 5/8/2022 | 25%/year | 1/5/2023 | 9,512 | 2,378 | ||||||
Four-year vesting period | 1/5/2024 | |||||||||||
1/5/2025 | ||||||||||||
1/5/2026 | ||||||||||||
Plan 2023.I | 5/8/2023 | 100% three years after offer date | 5/8/2026 | 4,309 | ||||||||
Plan 2023.II | 5/9/2023 | 25%/year | 1/5/2024 | 43,092 | ||||||||
Four-year vesting period | 1/5/2025 | |||||||||||
1/5/2026 | ||||||||||||
1/5/2027 | ||||||||||||
Total | 56,913 | 2,378 | ||||||||||
Thad Huston | Plan 2023.II | 6/15/2023 | 25%/year | 1/5/2024 | 50,544 | |||||||
Four-year vesting period | 1/5/2025 | |||||||||||
1/5/2026 | ||||||||||||
1/5/2027 | ||||||||||||
Total | 50,544 | — | ||||||||||
(1) On the leaver date of Mr. Bart Filius his outstanding RSUs became null and void, being 81,632 RSUs. | ||||||||||||
Limitations on liability and indemnification matters
Under Belgian law, the members of the Board of Directors and Executive Committee members of a company may be liable for damages to the company in case of improper performance of their duties. Our members of the Board of Directors and members of the Executive Committee may be liable to our company and to third parties for infringement of our Articles of Association or Belgian company law. Under certain circumstances, members of the Board of Directors and members of the Executive Committee may be criminally liable.
We maintain liability insurance for our members of the Board of Directors and members of the Executive Committee, including insurance against liability under the Securities Act.
The Belgian Companies Code includes a cap on liability for directors (i.e. members of the Board of Directors and persons in charge of daily management, such as our Chief Executive Officer) for any damages they cause due to mismanagement, including breaches of the articles of association and the Belgian Companies Code. This liability cap applies towards the company and third parties. For Galapagos NV, the cap amounts to €12,000,000. The cap applies irrespective of the number of claimants or defendants for the same (set of) facts. However, the cap does not apply to
181
repetitive minor misconduct, serious error or cases of fraud. Furthermore, the cap does not apply to directors’ liability under the special liability regimes relating to payment of withholding tax, VAT and social security contributions.
Certain of our members of the Board of Directors may through their relationships with their employers or partnerships be insured and/or indemnified against certain liabilities in their capacity as members of our Board of Directors.
In the underwriting agreements we entered into in connection with our May 2015 global offering and subsequent follow-on U.S. public offerings, the underwriters agreed to indemnify, under certain conditions, us, the members of our Board of Directors and persons who control our company within the meaning of the Securities Act against certain liabilities, but only to the extent that such liabilities are caused by information relating to the underwriters furnished to us in writing expressly for use in the applicable registration statements and certain other disclosure documents.
Subscription right plans
Various subscription right plans were approved for the benefit of our employees, and for members of the Board of Directors, members of the Executive Committee and independent consultants of Galapagos NV (“subscription rights” is the new term for instruments formerly referred to as “warrants” under the new Belgian Companies Code). For subscription right plans issued prior to 2011, the subscription rights offered to the employees and independent consultants vest according to the following schedule: 10% of the warrants vest on the date of the grant; an additional 10% vest at the first anniversary of the grant; an additional 20% vest at the second anniversary of the grant; an additional 20% vest at the third anniversary of the grant; and an additional 40% vest at the end of the third calendar year following the grant.
The subscription rights granted under subscription right plans created from 2011 onwards vest and become exercisable at the end of the third calendar year following the year of the grant, with no intermediate vesting, with the exception of the subscription rights granted (i) under Warrant Plan 2015 (B), Warrant Plan 2015 RMV, and Warrant Plan 2016 (B), which vest on the third anniversary of the notary deed enacting the acceptance and issuance of the subscription rights and (ii) under Subscription Right Plan 2021 RMV, Subscription Right Plan 2021 ROW, Subscription Right Plan 2022 (A), Subscription Right Plan 2022 RMV, Subscription Right Plan 2022 ROW, Subscription Right Plan 2023 RMV, and Subscription Right Plan 2023 ROW vest and become exercisable in instalments, with 25% of each grant vesting on the first day of the second calendar year following the calendar year in which the grant was made, 25% vesting on the first day of the third calendar year following the calendar year in which the grant was made and 50% vesting on the first day of the fourth calendar year following the calendar year in which the grant was made. In 2023, Subscription Right Plans 2015, 2015 (B) and 2015 RMV expired.
The subscription rights offered to members of the Board of Directors vest over a period of 36 months at a rate of 1/36th per month. As from January 1, 2020, Galapagos no longer grants any subscription rights to members of the Board of Directors, taking into account the stricter rules of the Belgian Companies Code and provision 7.6 of the 2020 Corporate Governance Code.
Pursuant to a resolution of our extraordinary shareholders’ meeting of May 23, 2011, in the event of a change of control over Galapagos NV, all outstanding subscription rights vest immediately and will be immediately exercisable.
After the reverse 4:1 share split approved by the Extraordinary Shareholders’ Meeting of March 29, 2005, four warrants under Warrant Plan 2002 Belgium entitle the subscription right holder to subscribe for one ordinary share. For the subscription right plans created from 2005 onwards, one subscription right entitles the subscription right holder to subscribe for one ordinary share. In the summaries and tables below, the numbers of subscription rights issued under Warrant Plan 2002 Belgium are divided by four to avoid confusion in entitlements and rights.
182
Generally, unless our Board of Directors at the time of the grant of the subscription right determines a higher exercise price, the exercise price of a subscription right will at least be equal to:
| ● | the last closing price of our ordinary shares on Euronext Amsterdam prior to the date on which the subscription right is offered; or |
| ● | the average closing price of our ordinary shares on Euronext Amsterdam over the thirty-day period preceding the date on which the subscription right is offered. |
However, for the subscription rights offered under Warrant Plan 2002 Belgium, since the ordinary shares of our company were not yet traded or listed on a stock exchange at the time of the relevant offers, the exercise price was to be determined by our Board of Directors at the time of the offer and had to be at least equal to the market value of the former Class D shares, as determined by the Board of Directors at that time and as certified by the auditor of our Company. In addition, the exercise price could not be lower than (i) the book value of the existing shares as appearing from the last approved annual accounts of the company at the date of the offer and (ii) €1.
From 2002 until December 31, 2023, an aggregate of 21,654,809 subscription rights were granted. Of these 21,654,809 subscription rights:
| ● | 3,549,430 subscription rights lapsed because they were not timely exercised by their beneficiaries, due to their beneficiaries no longer being employed by the Company or because another condition for vesting was not met; and |
| ● | 6,632,859 subscription rights were exercised. |
As a result, as of December 31, 2023, there were 11,472,520 subscription rights outstanding, representing approximately 17.41% of the total number of all our issued and outstanding voting financial instruments.
The table below sets forth the details of all subscription rights granted under the subscription right plans for employees, members of the Board of Directors, members of the Executive Committee and independent consultants in force as per December 31, 2023, including the plan under which the subscription rights were granted, the offer date, exercise price, expiry date, number of subscription rights exercised, number of subscription rights voided and number of subscription rights outstanding. Aside from the subscription rights set forth in the below table, there are currently no other stock options, options to purchase securities, convertible securities or other rights to subscribe for or purchase outstanding securities.
Number of | ||||||||||||||||
Number of | Number of | Number of | subscription rights | |||||||||||||
Subscription | Exercise | subscription rights | subscription rights | subscription rights | still | Exercisable | ||||||||||
right plan |
| Offer date |
| price (€) |
| granted |
| exercised |
| voided |
| outstanding |
| from |
| Expiry date |
2002 Belgium |
| 03/06/2002 |
| 4.00 |
| 553,705 |
| 423,698 |
| 130,007 |
| — |
| 01/01/2006 |
| 03/06/2010 |
| 09/02/2002 |
| 4.00 |
| 27,125 |
| 14,150 |
| 12,975 |
| — |
| 01/01/2006 |
| 09/02/2010 | |
| 03/06/2003 |
| 4.00 |
| 5,250 |
| 1,287 |
| 3,963 |
| — |
| 01/01/2007 |
| 03/31/2007 | |
| 04/01/2003 |
| 4.00 |
| 7,500 |
| 7,500 |
| — |
| — |
| 01/01/2007 |
| 04/01/2011 | |
| 06/15/2004 |
| 4.00 |
| 2,000 |
| 2,000 |
| — |
| — |
| 01/01/2008 |
| 06/15/2012 | |
| 07/09/2004 |
| 4.00 |
| 31,250 |
| 31,250 |
| — |
| — |
| 01/01/2008 |
| 02/01/2017 | |
| 07/22/2004 |
| 4.00 |
| 7,500 |
| — |
| 7,500 |
| — |
| 01/01/2008 |
| 03/31/2008 | |
| 01/31/2005 |
| 6.76 |
| 159,375 |
| 115,000 |
| 44,375 |
| — |
| 01/01/2009 |
| 02/01/2017 | |
Total |
|
|
|
|
| 793,705 |
| 594,885 |
| 198,820 |
| — |
|
|
|
|
2005 |
| 07/04/2005 |
| 6.91 |
| 145,000 |
| 145,000 |
| — |
| — |
| 01/01/2009 |
| 07/03/2018 |
| 11/23/2005 |
| 8.35 |
| 125,000 |
| 75,000 |
| 50,000 |
| — |
| 01/01/2009 |
| 11/22/2018 | |
| 12/15/2005 |
| 8.60 |
| 12,500 |
| 12,500 |
| — |
| — |
| 01/01/2009 |
| 12/14/2018 | |
| 02/13/2006 |
| 8.61 |
| 40,000 |
| 8,000 |
| 32,000 |
| — |
| 01/01/2010 |
| 03/31/2010 | |
| 02/13/2006 |
| 8.73 |
| 53,500 |
| 50,972 |
| 2,528 |
| — |
| 01/01/2010 |
| 03/31/2010 | |
| 11/22/2006 |
| 8.65 |
| 82,600 |
| 61,285 |
| 21,315 |
| — |
| 01/01/2010 |
| 11/21/2019 | |
Total |
|
|
|
|
| 458,600 |
| 352,757 |
| 105,843 |
| — |
|
|
|
|
2006 BNL |
| 02/13/2006 |
| 8.61 |
| 112,953 |
| 100,662 |
| 12,291 |
| — |
| 01/01/2010 |
| 02/12/2019 |
| 11/22/2006 |
| 8.65 |
| 87,090 |
| 16,450 |
| 70,640 |
| — |
| 01/01/2010 |
| 11/21/2019 | |
| 02/14/2007 |
| 9.57 |
| 102,900 |
| 9,170 |
| 93,730 |
| — |
| 01/01/2011 |
| 08/31/2011 | |
| 05/04/2007 |
| 9.22 |
| 17,500 |
| 17,500 |
| — |
| — |
| 01/01/2011 |
| 05/03/2020 | |
| 06/28/2007 |
| 8.65 |
| 735 |
| 735 |
| — |
| — |
| 01/01/2011 |
| 06/27/2020 | |
| 12/21/2007 |
| 7.12 |
| 25,110 |
| 13,171 |
| 11,939 |
| — |
| 01/01/2011 |
| 12/20/2020 | |
Total |
|
|
|
|
| 346,288 |
| 157,688 |
| 188,600 |
| — |
|
|
|
|
2006 UK |
| 06/01/2006 |
| 8.70 |
| 302,191 |
| 230,963 |
| 71,228 |
| — |
| 01/01/2010 |
| 09/30/2014 |
| 11/22/2006 |
| 8.65 |
| 13,965 |
| 11,907 |
| 2,058 |
| — |
| 01/01/2010 |
| 11/21/2014 |
183
| 12/19/2006 |
| 9.18 |
| 77,700 |
| 31,885 |
| 45,815 |
| — |
| 01/01/2010 |
| 12/18/2014 | |
| 06/28/2007 |
| 8.43 |
| 30,585 |
| 20,085 |
| 10,500 |
| — |
| 01/01/2011 |
| 06/27/2015 | |
| 12/21/2007 |
| 7.25 |
| 945 |
| 945 |
| — |
| — |
| 01/01/2011 |
| 12/20/2015 | |
Total |
|
|
|
|
| 425,386 |
| 295,785 |
| 129,601 |
| — |
|
|
|
|
2007 |
| 06/28/2007 |
| 8.65 |
| 108,126 |
| 108,126 |
| — |
| — |
| 01/01/2011 |
| 06/27/2015 |
| 06/28/2007 |
| 8.65 |
| 256,314 |
| 203,141 |
| 53,173 |
| — |
| 01/01/2011 |
| 06/27/2020 | |
Total |
|
|
|
|
| 364,440 |
| 311,267 |
| 53,173 |
| — |
|
|
|
|
2007 RMV |
| 10/25/2007 |
| 8.65 |
| 108,850 |
| 103,950 |
| 4,900 |
| — |
| 01/01/2011 |
| 10/24/2020 |
Total |
|
|
|
|
| 108,850 |
| 103,950 |
| 4,900 |
| — |
|
|
|
|
2008 |
| 06/26/2008 |
| 5.60 |
| 201,445 |
| 194,119 |
| 7,326 |
| — |
| 01/01/2012 |
| 06/25/2021 |
Total |
|
|
|
|
| 201,445 |
| 194,119 |
| 7,326 |
| — |
|
|
|
|
2008 (B) |
| 06/26/2008 |
| 5.60 |
| 57,500 |
| 50,000 |
| 7,500 |
| — |
| 01/01/2012 |
| 06/25/2013 |
Total |
|
|
|
|
| 57,500 |
| 50,000 |
| 7,500 |
| — |
|
|
|
|
2009 |
| 04/01/2009 |
| 5.87 |
| 555,000 |
| 490,000 |
| 65,000 |
| — |
| 01/01/2013 |
| 03/31/2017 |
Total |
|
|
|
|
| 555,000 |
| 490,000 |
| 65,000 |
| — |
|
|
|
|
2009 (B) |
| 02/06/2009 |
| 7.09 |
| 135,100 |
| 131,670 |
| 3,430 |
| — |
| 01/01/2013 |
| 06/01/2014 |
Total |
|
|
|
|
| 135,100 |
| 131,670 |
| 3,430 |
| — |
|
|
|
|
2010 |
| 04/27/2010 |
| 11.55 |
| 466,500 |
| 416,750 |
| 49,750 |
| — |
| 01/01/2014 |
| 04/26/2018 |
| 04/27/2010 |
| 11.55 |
| 40,000 |
| 40,000 |
| — |
| — |
| 04/27/2014 |
| 04/26/2018 | |
Total |
|
|
|
|
| 506,500 |
| 456,750 |
| 49,750 |
| — |
|
|
|
|
2010 (B) |
| 04/27/2010 |
| 11.55 |
| 195,040 |
| 190,108 |
| 4,932 |
| — |
| 01/01/2014 |
| 04/26/2015 |
Total |
|
|
|
|
| 195,040 |
| 190,108 |
| 4,932 |
| — |
|
|
|
|
2010 (C) |
| 12/23/2010 |
| 11.74 |
| 75,000 |
| 75,000 |
| — |
| — |
| 01/01/2014 |
| 12/22/2018 |
Total |
|
|
|
|
| 75,000 |
| 75,000 |
| — |
| — |
|
|
|
|
2011 |
| 05/23/2011 |
| 9.95 |
| 561,500 |
| 432,500 |
| 129,000 |
| — |
| 01/01/2015 |
| 05/22/2019 |
| 05/23/2011 |
| 9.95 |
| 57,500 |
| 50,000 |
| 7,500 |
| — |
| 05/23/2015 |
| 05/22/2019 | |
Total |
|
|
|
|
| 619,000 |
| 482,500 |
| 136,500 |
| — |
|
|
|
|
2011 (B) |
| 05/23/2011 |
| 9.95 |
| 129,220 |
| 127,750 |
| 1,470 |
| — |
| 01/01/2015 |
| 05/22/2016 |
Total |
|
|
|
|
| 129,220 |
| 127,750 |
| 1,470 |
| — |
|
|
|
|
2012 |
| 09/03/2012 |
| 14.19 |
| 448,640 |
| 345,490 |
| 103,150 |
| — |
| 01/01/2016 |
| 09/02/2020 |
| 09/03/2012 |
| 14.19 |
| 32,500 |
| 22,500 |
| 10,000 |
| — |
| 09/03/2016 |
| 09/02/2020 | |
Total |
|
|
|
|
| 481,140 |
| 367,990 |
| 113,150 |
| — |
|
|
|
|
2013 |
| 05/16/2013 |
| 19.38 |
| 602,790 |
| 431,840 |
| 170,950 |
| — |
| 01/01/2017 |
| 05/15/2021 |
Total |
|
|
|
|
| 602,790 |
| 431,840 |
| 170,950 |
| — |
|
|
|
|
2013 (B) |
| 09/18/2013 |
| 15.18 |
| 75,000 |
| 30,000 |
| 45,000 |
| — |
| 01/01/2017 |
| 06/30/2017 |
Total |
|
|
|
|
| 75,000 |
| 30,000 |
| 45,000 |
| — |
|
|
|
|
2014 |
| 07/25/2014 |
| 14.54 |
| 571,660 |
| 536,660 |
| 35,000 |
| — |
| 01/01/2018 |
| 07/24/2022 |
Total |
|
|
|
|
| 571,660 |
| 536,660 |
| 35,000 |
| — |
|
|
|
|
2014 (B) |
| 10/14/2014 |
| 11.93 |
| 150,000 |
| 150,000 |
| — |
| — |
| 01/01/2018 |
| 10/13/2022 |
Total |
|
|
|
|
| 150,000 |
| 150,000 |
| — |
| — |
|
|
|
|
2015 |
| 04/30/2015 |
| 28.75 |
| 532,053 |
| 513,390 |
| 18,663 |
| — |
| 01/01/2019 |
| 04/29/2023 |
Total |
|
|
|
|
| 532,053 |
| 513,390 |
| 18,663 |
| — |
|
|
|
|
2015 (B) |
| 12/22/2015 |
| 49.00 |
| 399,000 |
| 157,500 |
| 241,500 |
| — |
| 03/02/2019 |
| 12/21/2023 |
Total |
|
|
|
|
| 399,000 |
| 157,500 |
| 241,500 |
| — |
|
|
|
|
2015 RMV |
| 12/22/2015 |
| 49.00 |
| 97,500 |
| 62,500 |
| 35,000 |
| — |
| 03/02/2019 |
| 12/21/2023 |
Total |
|
|
|
|
| 97,500 |
| 62,500 |
| 35,000 |
| — |
|
|
|
|
2016 |
| 06/01/2016 |
| 46.10 |
| 514,250 |
| 177,750 |
| 11,000 |
| 325,500 |
| 01/01/2020 |
| 05/31/2024 |
Total |
|
|
|
|
| 514,250 |
| 177,750 |
| 11,000 |
| 325,500 |
|
|
|
|
2016 RMV |
| 06/01/2016 |
| 46.10 |
| 120,000 |
| 51,000 |
| — |
| 69,000 |
| 01/01/2020 |
| 05/31/2024 |
Total |
|
|
|
|
| 120,000 |
| 51,000 |
| — |
| 69,000 |
|
|
|
|
2016 (B) | 01/20/2017 | 62.50 | 150,000 | 140,000 | — | 10,000 | 04/06/2020 | 01/19/2025 | ||||||||
Total | 150,000 | 140,000 | — | 10,000 | ||||||||||||
2017 | 05/17/2017 | 80.57 | 595,500 | — | 10,500 | 585,000 | 01/01/2021 | 05/16/2025 | ||||||||
Total | 595,500 | — | 10,500 | 585,000 | ||||||||||||
2017 RMV | 05/17/2017 | 80.57 | 127,500 | — | 5,000 | 122,500 | 01/01/2021 | 05/16/2025 | ||||||||
Total | 127,500 | — | 5,000 | 122,500 | ||||||||||||
2018 | 04/19/2018 | 79.88 | 1,097,745 | — | 132,750 | 964,995 | 01/01/2022 | 04/18/2026 | ||||||||
Total | 1,097,745 | — | 132,750 | 964,995 | ||||||||||||
2018 RMV | 04/19/2018 | 79.88 | 137,500 | — | 5,000 | 132,500 | 01/01/2022 | 04/18/2026 | ||||||||
Total | 137,500 | — | 5,000 | 132,500 | ||||||||||||
2019 | 04/10/2019 | 95.11 | 1,504,940 | — | 296,700 | 1,208,240 | 01/01/2023 | 04/10/2027 | ||||||||
Total | 1,504,940 | — | 296,700 | 1,208,240 | ||||||||||||
2019 RMV | 04/10/2019 | 95.11 | 194,750 | — | 17,500 | 177,250 | 01/01/2023 | 04/10/2027 | ||||||||
Total | 194,750 | — | 17,500 | 177,250 | ||||||||||||
2020 | 04/17/2020 | 168.42 | 1,925,185 | — | 555,568 | 1,369,617 | 01/01/2024 | 04/17/2028 | ||||||||
Total | 1,925,185 | — | 555,568 | 1,369,617 | ||||||||||||
2020 RMV | 04/17/2020 | 168.42 | 248,150 | — | 54,850 | 193,300 | 01/01/2024 | 04/17/2028 | ||||||||
Total | 248,150 | — | 54,850 | 193,300 | ||||||||||||
2021 BE | 04/30/2021 | 64.76 | 1,117,603 | — | 84,997 | 1,032,606 | 01/01/2025 | 04/30/2029 | ||||||||
Total | 1,117,603 | — | 84,997 | 1,032,606 | ||||||||||||
2021 RMV | 04/30/2021 | 64.76 | 291,725 | — | 65,429 | 226,296 | 01/01/2023 | 04/30/2029 | ||||||||
Total | 291,725 | — | 65,429 | 226,296 | ||||||||||||
2021 ROW | 04/30/2021 | 64.76 | 1,084,105 | — | 416,609 | 667,496 | 01/01/2023 | 04/30/2029 | ||||||||
Total | 1,084,105 | — | 416,609 | 667,496 | ||||||||||||
2022 (A) | 01/13/2022 | 46.18 | 30,000 | — | — | 30,000 | 01/01/2023 | 01/13/2030 | ||||||||
Total | 30,000 | — | — | 30,000 | ||||||||||||
2022 (B) | 01/26/2022 | 50.00 | 1,000,000 | — | — | 1,000,000 | 01/01/2024 | 01/25/2030 | ||||||||
Total | 1,000,000 | — | — | 1,000,000 | ||||||||||||
2022 BE | 05/06/2022 | 57.46 | 839,400 | — | 21,572 | 817,828 | 01/01/2026 | 05/60/2030 | ||||||||
08/05/2022 | 51.58 | 78,000 | — | — | 78,000 | 01/01/2026 | 05/60/2030 | |||||||||
Total | 917,400 | — | 21,572 | 895,828 | ||||||||||||
2022 RMV | 05/06/2022 | 57.46 | 244,389 | — | 40,925 | 203,464 | 01/01/2024 | 05/60/2030 | ||||||||
Total | 244,389 | — | 40,925 | 203,464 |
184
2022 ROW | 05/06/2022 | 57.46 | 875,450 | — | 169,950 | 705,500 | 01/01/2024 | 05/60/2030 | ||||||||
08/05/2022 | 51.58 | 60,000 | — | — | 60,000 | |||||||||||
Total | 935,450 | — | 169,950 | 765,500 | ||||||||||||
2023 BE | 05/05/2023 | 35.11 | 611,000 | — | 1,972 | 609,028 | 01/01/2027 | 05/05/2031 | ||||||||
06/15/2023 | 38.58 | 200,000 | — | — | 200,000 | 01/01/2027 | 05/05/2031 | |||||||||
Total | 811,000 | — | 1,972 | 809,028 | ||||||||||||
2023 RMV | 05/05/2023 | 35.11 | 110,000 | — | 7,500 | 102,500 | 01/01/2025 | 05/60/2031 | ||||||||
Total | 110,000 | — | 7,500 | 0 | 102,500 | |||||||||||
2023 ROW | 05/05/2023 | 35.11 | 597,400 | — | 35,500 | 561,900 | 01/01/2025 | 05/60/2031 | ||||||||
11/17/2023 | 32.99 | 20,000 | — | — | 20,000 | |||||||||||
Total | 617,400 | — | 35,500 | 581,900 | ||||||||||||
Grand Total |
|
|
|
|
| 21,654,809 | 6,632,859 | 3,549,430 | 11,472,520 |
|
|
|
|
In addition to the subscription right plans for our employees, Board members, Executive Comittee members, and independent consultants described above, on October 22, 2019, our Extraordinary Shareholders’ Meeting approved the issuance of two subscription rights for the benefit of Gilead Therapeutics A1 Unlimited Company, called the initial Warrant A and the initial Warrant B. These subscription rights entitle the holder thereof to subscribe, during the entire term of the respective subscription right, upon each exercise of a subscription right, for a maximum number of shares that is sufficient to bring the shareholding of Gilead and its affiliates to 25.1% and 29.9%, respectively, of the actually issued and outstanding shares after the exercise of the relevant subscription right (rounded down to the nearest whole share). The initial Warrant A has a term of one year and an exercise price of €140.59 per share and expired during 2020. The initial Warrant B has a term of five years and an exercise price per share equal to the greater of (i) 120% multiplied by the arithmetic mean of the 30-day daily volume weighted average trading price of Galapagos’ shares as traded on Euronext Brussels and Euronext Amsterdam, and (ii) €140.59. The initial Warrant B will expire on 22 August 2024. In accordance with the subscription agreement between Galapagos and Gilead, the issuance of subsequent Warrant B is on the agenda of the Extraordinary Shareholders’ Meeting to be held on April 30, 2024. The subsequent Warrant B has a term of five years and an exercise price per share equal to the greater of (i) 120% multiplied by the arithmetic mean of the 30-day daily volume weighted average trading price of Galapagos’ shares as traded on Euronext Brussels and Euronext Amsterdam, and (ii) €140.59.
C. Board practices
As from April 26, 2022, Galapagos has adopted a one-tier governance structure as provided for by the Belgian Companies Code, with the Board of Directors replacing the (former) Supervisory Board and the Executive Committee replacing the (former) Management Board. For further information on our governance structure, see the section of this annual report titled “Item 6.A – Directors and Senior Management – Board of Directors”.
Our Board of Directors can set up specialized Board Committees to analyze specific issues and advise the Board on those issues. The Board Committees are advisory bodies only, and the final decision-making power remains within the collegial responsibility of the Board of Directors. The Board determines the terms of reference of each Board Committee with respect to its organization, procedures, policies and activities.
In order to efficiently fulfil its tasks and in view of the size and activities of the Company, our Board of Directors has set up and appointed an Audit Committee, a Nomination Committee, a Remuneration Committee and a Science and Development Committee (established on September 19, 2023) . Until May 2, 2022, there was a combined Nomination and Remuneration Committee. As from May 2, 2022, our Board of Directors has set up a separate Nomination Committee and Remuneration Committee. The composition, role and functioning of all Board Committees complies with all applicable requirements of the Belgian Companies Code, the 2020 Code, the Exchange Act, the exchanges on which our ordinary shares and ADSs are listed and SEC rules and regulations, however taking into account the diffirences as set out below and our status as a foreign private issuer. For information regarding the (temporary) deviation from the provisions with respect to the composition of our Nomination Committee as set forth in the 2020 Code, see “item 16G – Corporate Governance”.
185
The members of each Board Committee are appointed by the Board of Directors, and can be dismissed by the Board of Directors at any time. The duration of the mandate of a Board Committee member cannot exceed that such Director’s membership. In deciding on the specific composition of each Board Committee, consideration is given to the needs and qualifications required for the optimal functioning of that Board Committee.
Except the arrangements described in the section of this annual report titled “Item 7.B.—Related-Party Transactions— Agreements with Our Board Members and Executive Committee,” there are no arrangements or understanding between Galapagos and any Executive Committee member or Board member providing for benefits upon termination of their employment, other than as required by applicable law. For information regarding the expiration of our Board members’ current terms of office and the period each member of our Board of Directors has served in that office, see “Item 6.A.— Directors and Senior Management.— Our Board of Directors.”
Lead Non-Executive Director
Pursuant to the Company’s Corporate Governance Charter and as a counter balancing governance structure for the combined CEO & Chair role within the Board, the Board of Directors appointed a Lead Non-Executive Director. The Lead Non-Executive Director is also automatically the Vice-Chair of the Board of Directors. The Lead Non-Executive Director is entrusted with the responsibilities and powers as set out in Galapagos’ Corporate Governance Charter, including, but not limited to, serving as principal liaison between the non-executive directors and the Chair of the Board. Dr. Rajesh Paresh was appointed as the Lead Non-Executive Director of Galapagos, effective as of May 2, 2022. Effective as of March 21, 2023, Jérôme Contamine is appointed as the new Lead Non-Executive Director of Galapagos, replacing Dr. Rajesh Parekh.
Director independence
As a foreign private issuer, under the listing requirements and rules of Nasdaq, we are not required to have independent directors on our Board of Directors, except that our Audit Committee is required to consist fully of independent directors, subject to certain phase-in schedules. However, our Board of Directors has determined that, under current listing requirements and rules of Nasdaq and taking into account any applicable committee independence standards, Rajesh Parekh (until June 10, 2023), Jérôme Contamine, Peter Guenter, Mary Kerr (until September 18, 2023), Elisabeth Svanberg, Dan Baker, Susanne Schaffert, and Simon Sturge are “independent Directors.” In making such determination, our Board of Directors considered the relationships that each non-executive Board member has with us and all other facts and circumstances our Board of Directors deemed relevant in determining the relevant Board member's independence, including the number of ordinary shares beneficially owned by the relevant Board member and his or her affiliated entities (if any).
The independence criteria under the applicable Nasdaq Stock Market Listing Rules and US Securities Exchange Act requirements differ from the independence criteria set forth in article 7:87 of the Belgian Companies Code and provision 3.5 of the 2020 Code. Under article 7:87 of the Belgian Companies Code and provision 2020, Jérôme Contamine, Peter Guenter, Mary Kerr (until September 18, 2023), Elisabeth Svanberg, Dan Baker, Susanne Schaffert, and Simon Sturge are “independent members of our Board of Directors”. Pursuant to Belgian law, our Board of Directors is composed of a majority of independent non-executive Directors.
Role of the Board of Directors in risk oversight
Our Board of Directors is responsible for the oversight of our risk management activities, and has delegated to the Audit Committee the responsibility to assist our Board in this task. While our Board of Directors oversees our risk management, our Executive Committee is responsible for day-to-day risk management processes. Our Board of Directors expects our Executive Commmittee members to consider risk and risk management in each business decision, to proactively develop and monitor risk management strategies and processes for day-to-day activities and to effectively implement risk management strategies adopted by the Board. We believe this division of responsibilities is the most effective approach for addressing the risks we face.
186
Corporate governance practices
Along with our Articles of Association, we initially adopted a Corporate Governance Charter in accordance with the rules and recommendations set out in the Belgian Corporate Governance Code issued on March 12, 2009 by the Belgian Corporate Governance Committee. In light of the new Belgian Companies Code, the Belgian Corporate Governance Committee adopted a new 2020 Belgian Corporate Governance Code (the “2020 Code”), published on May 9, 2019. The 2020 Code applies compulsorily to reporting years beginning on or after January 1, 2020.
Our Board of Directors has adopted the 2020 Code for the reporting period beginning on January 1, 2020. Our Corporate Governance Charter is available on our website: www.glpg.com (this website does not form part of this annual report), and applies in addition to the applicable laws and regulations, our Articles of Association, and the corporate governance provisions included in the Belgian Companies Code and the 2020 Code. For the reporting year beginning on January 1, 2022, the 2020 Code was our reference code.
The 2020 Code is based on a “comply or explain” system: Belgian listed companies are expected to follow the 2020 Code, but can deviate from specific provisions and guidelines (though not the principles) provided they disclose the justification for such deviations.
For the reporting year beginning on January 1, 2023, our Board of Directors strove to comply with the rules and recommendations of the 2020 Code as much as possible. At the same time, our Board believes that certain deviations from its provisions were justified in view of our particular situation. Provision 3.12 of the 2020 Code recommends that, in case of a one-tier governance structure, (a) there should be a clear division of the responsibilities between the person presiding over the Board of Directors (the Chair) and the person assuming executive responsibility for running the company’s business (the CEO), and (b) the Chair of the Board of Directors and CEO should not be the same individual. In deviation from this provision, Stoffels IMC BV (permanently represented by Dr. Paul Stoffels) who is our CEO since April 1, 2022, was also appointed as Chair of the Board of Directors as from April 26, 2022. In light of the prevailing circumstances, the Board of Directors considered that the one-tier governance structure and the combined role as CEO/Chair allows the Company to fully leverage the leadership of Dr. Paul Stoffels, and to efficiently set and implement the Company's direction and strategy (including in the field of business development). Furthermore, the Board of Directors is of the opinion that such combined role had a positive impact on the functioning and efficiency of the Board, as well as on the provision of information to the Board of Directors, allowing the Board of Directors to monitor the Company’s (and group’s) performance more effectively. In order to ensure sufficient balance, the Board adopted a counter balancing governance structure that includes the election of a Lead Non-Executive Director acting as the principal liaison between the Chair and the non-executive members of the Board of Directors. Dr. Rajesh Parekh was appointed as the Lead Non-Executive Director of Galapagos, effective as of May 2, 2022. Effective as of March 21, 2023, Jérôme Contamine is appointed as the new Lead Non-Executive Director of Galapagos, replacing Dr. Rajesh Parekh. The Lead Non-Executive Director is entrusted with the responsibilities and powers as set out in the Corporate Governance Charter of Galapagos.
For information regarding the (temporary) deviation from the provisions with respect to the composition of our Nomination Committee as set forth in the 2020 Code, see "Item 16G – Corporate Governance”
Our Board of Directors regularly reviews its Corporate Governance Charter and makes such changes as it deems necessary and appropriate. Additionally, our Board of Directors adopted written terms of reference for the Executive Committee, the Audit Committee, the Nomination Committee and the Remuneration Committee, which are part of our Corporate Governance Charter. On April 26, 2022, as a consequence of the introduction of one-tier governance structure at the Company through the amendment of our Articles of Association, Galapagos’ Board of Directors approved an updated Corporate Governance Charter. On March 21, 2023, our Board of Directors approved an amendment to the Corporate Governance Charter. The amended Corporate Governance Charter refers to the establishment of the Management Committee supporting the Executive Committee, allows the same person to be Lead Non-Executive Director and Chair of the Audit Committee, provides that the Lead Non-Executive Director is member or Chair of the Nomination Committee, and clarifies that the Lead Non-Executive Director supports the Chair in ensuring the prevention and managing of conflicts of interest involving potentially a Director. On September 19, 2023, the Board of Directors approved another amendment that refers to the establishment of the Science and Development Committee as a
187
specialized Board Committee to provide advice on certain matters to the Board of Directors, and that provides that non-executive Directors may only be natural persons. On December 11, 2023, the Board of Directors approved a further amendment to describe the responsibilities of the Audit Committee and management for overseeing and managing cybersecurity risks. Galapagos’ Corporate Governance Charter is available on our website: www.glpg.com (information contained on, or that can be accessed through, our website does not constitute a part of this annual report). The Corporate Governance Charter applies in addition to the applicable laws and regulations, Galapagos’ Articles of Association and the corporate governance provisions included in the Belgian Companies Code and the 2020 Code. The Corporate Governance Charter describes the main aspects of corporate governance at Galapagos, including its governance structure, the terms and functioning of the Board of Directors (including its Board Committees), the Executive Committee and the rules of conduct.
Board Committees
The Board of Directors has established an Audit Committee, a Nomination Committee, a Remuneration Committee and a Science and Development Committee, which operate pursuant to the written terms of reference for each of the Board Committees that are part of Galapagos’ Corporate Governance Charter adopted by our Board. The composition and functioning of all Board Committees complies with all applicable requirements of the Belgian Companies Code and the 2020 Code, the Exchange Act, the exchanges on which our ordinary shares and ADSs are listed and the SEC rules and regulations, however taking into account the differences set out below and our status as a foreign private issuer. For information regarding the (temporary) deviation from the provisions with respect to the composition of our Nomination Committee as set forth in the 2020 Code, see "Item 16G – Corporate Governance”.
The Listing Rules of the Nasdaq Stock Market include certain accommodations in the corporate governance requirements that allow foreign private issuers to follow “home country” corporate governance practices in lieu of the otherwise applicable corporate governance standards of the Nasdaq Stock Market. We intend to rely on certain exceptions for foreign private issures. The application of such exceptions requires that we disclose each of the Nasdaq Stock Market Listing Rules that we do not follow and describe the Belgian corporate governance practices we do follow in lieu of the relevant Nasdaq Stock Market corporate governance standard.
We follow Belgian corporate governance practices in lieu of the otherwise applicable corporate governance requirements of the Nasdaq Stock Market in respect of the following rules applicable to our Board Committees:
| ● | Compensation committee. We have opted out of (a) Nasdaq Stock Market Listing Rule 5605(d)(2) which requires that compensation of officers must be determined by, or recommended to the Board of Directors for determination, either by a majority of the independent directors, or a compensation committee comprised solely of independent directors, and (b) Nasdaq Stock Market Listing Rule 5605(e) which requires that director nominees be selected, or recommended for selection, either by a majority of the independent directors or a nominations committee comprised solely of independent directors. Under Belgian law, we are not subject to such composition requirements. Pursuant to article 7:100 of the Belgian Companies Code and the rules and recommendations of the 2020 Code, we are required to set up a Remuneration Committee within our Board of Directors, consisting of a majority of independent Board members. In addition, the 2020 Code provides that the Board of Directors should set up a Nomination Committee, which can be combined with the Remuneration Committee. Until May 2, 2022, there was a combined Nomination and Remuneration Committee. As from May 2, 2022, our Board of Directors has set up a separate Nomination Committee and Remuneration Committee. Although we have chosen not to comply with Nasdaq Listing Rule 5605(d) regarding the independence of our Remuneration Committee, all members of our Remuneration Committee meet the heightened independence requirements under these rules. |
188
| ● | Charters. Nasdaq Stock Market Listing Rules 5605(c)(1), (d)(1) and (e)(2) require that each committee of the Board of Directors must have a formal written charter. Pursuant to the 2020 Code, our Board of Directors has drawn up a Corporate Governance Charter including, amongst others, the internal rules with respect to the composition, role and functionnning of our Board Committees. |
Audit Committee
Our Audit Committee consists of three members (situation as of December 31, 2023): Jérôme Contamine (Chair), Mary Kerr (until September 18, 2023), Peter Guenter, and Simon Sturge (as of September 19, 2023).
Our Board of Directors has determined that all members of our Audit Committee are non-executive members of our Board of Directors, and all of the Audit Committee members are also independent Board members under Rule 10A-3 of the Exchange Act and the applicable rules of the Nasdaq Stock Market, as amended, and that, as per December 31, 2022, Jérôme Contamine qualifies as an “Audit Committee financial expert” as defined under the Exchange Act. Collectively, the members of the Audit Committee shall have sufficient relevant expertise to fulfill their roles effectively, notably in financial matters and the life sciences industry.
Our Audit Committee assists our Board of Directors in fulfilling its monitoring responsibilities with respect to financial reporting and control and risk management in the broadest sense.
Our Audit Committee’s duties and responsibilities to carry out its purposes include, among others, without any limitation:
| ● | monitoring the integrity of the Company’s financial statements, and the Company’s accounting and financial reporting process and financial statement audits; |
| ● | monitoring the effectiveness of the Company’s internal control and risk management systems, incl. the cybersecurity risk magment program and related controls; |
| ● | monitoring the internal audit function and its effectiveness; |
| ● | monitoring the performance of the external auditor and the audit of the statutory and consolidated accounts, including any follow-up on questions and recommendations made by the external auditor; |
| ● | reviewing and monitoring the independence of the external auditor; and |
| ● | informing the Board on the Company’s ESG activities. |
The Audit Committee regularly reports to our Board of Directors on the discharge of its functions. It informs our Board about all areas in which action or improvement is necessary in its opinion and produces recommendations concerning the necessary steps that need to be taken in that regard. The audit review and the reporting on that review cover us and our subsidiaries as a whole. The members of the Audit Committee are entitled to receive all information which they need for the performance of their function, from our Board of Directors, Executive Committee and employees. Every member of the Audit Committee shall exercise this right in consultation with the Chairman of the Audit Committee.
Since 2019, the Audit Committee also reviews Environmental, Social and Governance (ESG) initiatives, as included in the annual Sustainability report, ensuring that we implement our planned initiatives and communicate them effectively and accurately to our employees and shareholders. The Sustainability report 2023 provides the non-financial information required by article 3:6 §4 and article 3:32 §2 of the Belgian Companies Code; a copy of our Sustainability report 2023 is available on our company website at http://www.glpg.com/financial-reports (this website does not form part of this annual report on Form 20-F).
189
Nomination Committee
Our Nomination Committee consists of three members (situation as of December 31, 2023): Dr. Rajesh Parekh (Chair until March 20, 2023), , Elisabeth Svanberg (Chair as of March 21, 2023), Jérôme Contamine, and Stoffels IMC BV (permanently represented by Dr. Paul Stoffels).
Our Board of Directors has determined that a majority of all members of our Nomination Committee are non-executive Board members.
Concerning our Company’s nomination policy, the Nomination Committee’s duties and responsibilities to carry out its purposes include, among others, without any limitation:
| ● | providing recommendations to the Board of Directors on the appointment of our Board members, CEO and Executive Committee members, as well as the size and composition of our Board and Board Committees; |
| ● | preparing selection criteria and procedures for our Board members, CEO and Executive Committee members; |
| ● | selecting and proposing suitable candidates for vacant Board of Directors mandates, reviewing candidates as proposed by a shareholder in accordance with our Articles of Association and advising on proposals to re-appoint current members of the Board of Directors; |
| ● | recommending individuals for appointment as CEO or Executive Committee member (in consultation with the CEO as regards the appointment of other members of the Executive Committee); and |
| ● | assessing the performance of the CEO and, in cooperation with the CEO, of other Executive Committee members and senior management. |
Remuneration Committee
Our Remuneration Committee consists of three members (situation as of December 31, 2023): Dr. Rajesh Parekh (Chair until March 20, 2023), Elisabeth Svanberg (Chair as of March 21, 2023), Jérôme Contamine, and Dan Baker (member as of March 21, 2023).
Our Board of Directors has determined that all members of our Remuneration Committee are non-executive Board members, a majority of which is also an independent member of our Board of Directors.
Concerning our company’s Remuneration Policy, the Remuneration Committee’s duties and responsibilities to carry out its purposes include, among others, without any limitation:
| ● | making recommendations to the Board of Directors on the remuneration of our Board members, CEO and Executive Committee members, including, but not limited to, variable remuneration and long-term incentives, whether or not stock-related, in the form of stock options or other financial instruments; |
| ● | establishing the Remuneration Policy and strategy for our Board members, Executive Committee members and senior management; and |
| ● | preparing and submitting a remuneration report to the Board of Directors for inclusion in the annual report. |
190
Science and Development Committee
Our Science and Development Committee, established on September 19, 2023, consists of five members (situation as of December 31, 2023): Dan Baker (Chair), Stoffels IMC BV (permanently represented by Dr. Paul Stoffels), Elisabeth Svanberg, Linda Higgins, and Susanne Schaffert.
The Science and Development Committee provides input and advice to the Board of Directors on matters relating to the Company’s Research and Development (“R&D”) strategy, and serves as a resource, as needed, regarding scientific, medical and product safety matters.
The majority of members of the Science and Development Committee are non-executive independent Directors. The Chair of the Science and Development Committee is a non-executive independent Director. Collectively, the Science and Development Committee members have sufficient relevant experience to fulfill their roles effectively.
D. Employees
As of December 31, 2023, we had 1,123 employees. Our employees in Austria, Belgium, France, Germany and the Netherlands have established employee representation through works councils. We have never experienced any employment-related work stoppages, and we consider our relations with our employees to be generally good. We have also engaged and may continue to engage independent contractors to assist us with our clinical activities. At each date shown, we had the following number of employees, broken out by department and geography:
December 31, | ||||||
|
| 2023 |
| 2022 |
| 2021 |
Function: |
|
|
|
| ||
Executive officers |
| 5 | 4 | 5 | ||
Research |
| 143 | 267 | 299 | ||
Development |
| 305 | 342 | 407 | ||
Commercial and medical affairs | 373 | 420 | 292 | |||
Corporate and support |
| 297 | 305 | 306 | ||
Total |
| 1,123 | 1,338 | 1,309 | ||
Geography: |
| |||||
Leiden, the Netherlands |
| 203 | 147 | 141 | ||
Mechelen, Belgium |
| 457 | 547 | 544 | ||
Romainville, France |
| 95 | 254 | 257 | ||
Boston & Pittsburgh, United States | 36 | 20 | 6 | |||
Basel, Switzerland | 52 | 53 | 57 | |||
Cambridge, United Kingdom | 58 | 71 | 78 | |||
Milan, Italy | 59 | 64 | 60 | |||
Madrid, Spain | 48 | 63 | 58 | |||
Munich, Germany | 83 | 88 | 85 | |||
Vienna, Austria | 12 | 12 | 9 | |||
Copenhagen, Denmark | 8 | 6 | 5 | |||
Stockholm, Sweden | 8 | 10 | 6 | |||
Helsinki, Finland | 1 | 1 | 2 | |||
Oslo, Norway | 1 | 1 | — | |||
Dublin, Ireland | 2 | 1 | 1 | |||
Total |
| 1,123 | 1,338 | 1,309 | ||
E. Share Ownership
For information regarding the share ownership of our members of the Board of Directors and members of the Executive Committee, see “Item 6.B.— Compensation” and “Item 7.A.—Major shareholders.”
F. . Disclosure of a registrant’s action to recover erroneously awarded compensation
[Not Applicable]
191
Item 7 Major shareholders and related party transactions
A. Major shareholders
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as of March 15, 2024 for:
| ● | each person who is known by us to own beneficially more than 5% of our outstanding ordinary shares; |
| ● | each member of our Board of Directors; |
| ● | each member of our Executive Committee; and |
| ● | all members of our Board of Directors and Executive Committee as a group. |
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include ordinary shares that can be acquired within 60 days of March 15, 2024. The percentage ownership information shown in the table is based upon 65,897,071 ordinary shares outstanding as of March 15, 2024.
Except as otherwise indicated, all of the shares reflected in the table are ordinary shares or ADSs, and all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
In computing the number of ordinary shares beneficially owned by a person and the percentage ownership of that person, we deemed outstanding ordinary shares subject to subscription rights held by that person that are immediately exercisable or exercisable within 60 days of March 15, 2024. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. Beneficial ownership representing less than 1% is denoted with an asterisk (*). The information in the table below is based on information known to us or ascertained by us from public filings made by the shareholders.
192
Except as otherwise indicated in the table below, addresses of the members of the Board of Directors, members of our Executive Committee and named beneficial owners are in care of Galapagos NV, Generaal De Wittelaan L11 A3, 2800 Mechelen, Belgium.
Shares beneficially owned |
| ||||
Name of beneficial owner |
| Number |
| Percentage |
|
5% shareholders: |
|
|
|
|
|
Gilead Sciences, Inc. |
| 16,707,477 | (1)(2) | 25.35 | % |
Van Herk Investments B.V. |
| 4,635,672 | (1)(3) | 7.03 | % |
EcoR1 Capital LLC | 6,505,890 | (1)(4) | 9.87 | % | |
FMR LLC | 5,095,393 | (1)(5) | 7.73 | % | |
Members of the Board of Directors and Executive Committee: |
|
| |||
Stoffels IMC BV | — | — |
| ||
Peter Guenter | 9,492 | (6) | * |
| |
Jérôme Contamine | 1,010 | (7) | * | ||
Dan Baker | 998 | (8) | * | ||
Daniel O'Day | — | — | |||
Linda Higgins | — | — | |||
Elisabeth Svanberg | 1,883 | (9) | * | ||
Susanne Schaffert | 360 | (10) | — | ||
Simon Sturge | 180 | (11) | — | ||
Executive Committee members excluding Stoffels IMC BV |
| 63,600 | (12) | * | % |
All members of our Board of Directors and Executive Committee as a group (12 persons) |
| 77,523 | (13) | * | % |
| (1) | At the time of the most recent transparency notification or filing of a statement of beneficial ownership with the SEC. |
| (2) | Consists of 16,707,477 shares held by Gilead Therapeutics A1 Unlimited Company, which is a subsidiary of Gilead Sciences Ireland Unlimited Company, which is in turn a subsidiary of Gilead Biopharmaceutics US, LLC which is in turn a subsidiary of Gilead Sciences, Inc., which has the sole voting and investment power with respect to these shares. The address of Gilead Sciences, Inc. is 333 Lakeside Drive, Foster City, CA 94404, United States of America. |
| (3) | Consists of 4,635,672 shares held by Van Herk Investments B.V., as reported in a Schedule 13D filed on November 2, 2021 by (i) Van Herk Investments B.V., a private company with limited liability incorporated under the laws of the Netherlands (“VHI”), with respect to Common Stock (as defined below) beneficially owned by it, (ii) Van Herk Investments THI B.V., a private company with limited liability incorporated under the laws of the Netherlands (“VHIT”), with respect to Common Stock beneficially owned by VHI, (iii) Van Herk Private Equity Investments B.V., a private company with limited liability incorporated under the laws of the Netherlands (“VHPI”), with respect to Common Stock beneficially owned by VHI and VHIT, (iv) Stichting Administratiekantoor Penulata, a foundation organized under the laws of the Netherlands (“Penulata”), with respect to Common Stock beneficially owned by VHI, VHIT and VHPI, (v) Van Herk Management Services B.V., a private company with limited liability incorporated under the laws of the Netherlands (“VHMS”), with respect to Common Stock beneficially owned by VHI, VHIT and VHPI, (vi) Onroerend Goed Beheer- en Beleggingsmaatschappij A. van Herk B.V., a private company with limited liability incorporated under the laws of the Netherlands (“OGBBA”), with respect to Common Stock beneficially owned by VHI, VHIT, VHPI and VHMS, (vii) A. van Herk Holding B.V., a private company with limited liability incorporated under the laws of the Netherlands (“Holdings”), with respect to Common Stock beneficially owned by VHI, VHIT, VHPI, VHMS and OGBBA, (viii) Stichting Administratiekantoor Abchrys, a foundation organized under the laws of the Netherlands (“Abchrys”), with respect to Common Stock beneficially owned by VHI, VHIT, VHPI, VHMS, OGBBA and Holdings, and (ix) Adrianus van Herk (“Mr. van Herk”) with respect to Common Stock beneficially owned by VHI, VHIT, VHPI, VHMS, OGBBA, Holdings, Penulata and Abchrys. Mr. van Herk is (i) an investor, (ii) the holder of all of the depositary receipts issued by Penulata and Abchrys, (iii) the sole board member of Penulata and Abchrys, and (iv) the sole managing director of VHMS, OGBBA and Holdings. Penulata holds substantially all of the issued and outstanding shares of VHPI. VHPI is the sole shareholder of VHIT. VHIT is the sole shareholder of VHI. VHI is principally engaged in |
193
| making investments. Abchrys holds substantially all of the issued and outstanding shares of Holdings. Holdings is the sole shareholder of OGBBA. OGBBA is the sole shareholder of VHMS and is principally engaged in making investments. VHMS is the sole managing director of VHI, VHIT and VHPI. Each of Mr. van Herk, VHIT, VHPI, Penulata, VHMS, OGBBA, Holdings and Abchrys disclaims beneficial ownership of the securities covered by such Schedule 13D statement. The address of each of VHI, VHIT, VHPI, Penulata, VHMS, OGBBA, Holdings, Abchrys and Mr. van Herk, is Lichtenauerlaan 30, 3062 ME Rotterdam, the Netherlands. |
| (4) | Consists of ordinary shares and American Depository Receipts. EcoR1 Capital LLC, controlled by Mr. Nodelman, controls investment fund EcoR1 Capital Fund Qualified LP, which all together hold 6,505,890 of Galapagos’ voting rights. The address of EcoR1 Capital LLC is 357 Tehama Street #3, San Francisco, CA 94103, United States of America. |
| (5) | Consists of 5,095,393 shares. FMR LLC, controls investment funds Fidelity Management & Research Company LLC, FIAM Holdings LLC, FIAM LLC, Fidelity Management Trust Company, Fidelity Advisory Holdings LLC, Strategic Advisers LLC, and Fidelity Institutional Asset Management Trust Company, which all together hold 5,095,393 Galapagos’ voting rights. The address of FMR LLC is 245 Summer Street, Boston, Massachusetts 02210. |
| (6) | Consists of (i) 1,992 shares and (ii) 7,500 shares issuable upon the exercise of subscription rights that are immediately exercisable or exercisable within 60 days of March 15, 2024. |
| (7) | Consists of (i) 1,010 shares. |
| (8) | Consists of (i) 998 shares. |
(9) | Consists of 1,883 shares. |
(10) | Consists of 360 shares. |
(11) | Consists of 180 shares. |
(12) | Consists of (i) 2,600 shares and (ii) 61,000 shares issuable upon the exercise of subscription rights that are immediately exercisable or exercisable within 60 days of March 15, 2024. |
(13) | Consists of (i) 9,023 shares and (ii) 68,500 shares issuable upon the exercise of subscription rights that are immediately exercisable or exercisable within 60 days of March 15, 2024. |
Each of our shareholders is entitled to one vote per ordinary share. All shareholders have identical voting rights per share. We are not aware of any arrangement that may result in a change of control of our company.
As of March 15, 2024, reviewing identified ownership in depository received reports and independent shareholder identification of 93% of outstanding shares (ordinary shares and ADSs), approximately 50% of those are held by institutional investors domiciled in the United States, excluding Gilead Sciences, Inc., or Gilead. We estimate that shares were held in the United States by approximately 144 institutional holders of record, excluding Gilead Sciences, Inc., or Gilead. As of March 15, 2024, there were outstanding 20,315,443 ADSs, each representing one ordinary share, and in the aggregate representing approximately 30.8% of our outstanding ordinary shares. The actual number of holders is greater than these numbers of record holders, and includes beneficial owners whose ADSs are held in street name by brokers and other nominees. This number of holders of record also does not include holders whose shares may be held in trust by other entities.On February 14, 2024, EcoR1 Capital, LLC filed a Schedule 13G/A with the SEC to report that as of February 14, 2024, it held 9.9% of the outstanding shares of Galapagos as of February 14, 2024.
On February 9, 2024, FMR LLC filed a Schedule 13G/A with the SEC to report that as of February 8, 2024, it held 7.73% of the outstanding shares of Galapagos as of February 8, 2024.
On July 6, 2023, EcoR1 Capital LLC filed a Schedule 13G with the SEC to report that as of June 7, 2023, it held 9.87% of the outstanding shares of Galapagos as of June 7, 2023.
On January 9, 2023, we received a transparency notification from FMR LLC, to report that as of January 5, 2023, it together with its affiliates held 5.93% of the outstanding shares of Galapagos as of January 5, 2023. FMR LLC thus crossed above the 5% threshold of Galapagos voting rights by purchase of voting securities on January 5, 2023.
During 2022, FMR LLC filed multiple transparency notifications. On May 31, 2022 we received a transparency notification from FMR LLC, to report that as of May 27, 2022, it together with its affiliates held 5.04% of the outstanding shares of Galapagos as of May 27, 2022. FMR LLC thus crossed above the 5% threshold of Galapagos voting rights by purchase of voting securities on May 27, 2022. On August 3, 2022 we received a transparency
194
notification from FMR LLC, to report that as of July 29, 2022, it together with its affiliates held 5.69% of the outstanding shares of Galapagos as of July 29, 2022. On August 15, 2022 we received a transparency notification from FMR LLC, to report that as of August 9, 2022, it held together with its affiliates 5.69% of the outstanding shares of Galapagos as of August 9, 2022. On December 22, 2022 we received a transparency notification from FMR LLC, to report that as of August 23, 2022, it together with its affiliates held 5.65% of the outstanding shares of Galapagos as of August 23, 2022. On August 25, 2022 we received a transparency notification from FMR LLC, to report that as of December 20, 2022, it together with its affiliates held 5.90% of the outstanding shares of Galapagos as of December 20, 2022.
On February 3, 2022, EcoR1 Capital LLC filed a Schedule 13G with the SEC to report that as of January 27, 2022, it held 5.2% of the outstanding shares of Galapagos as of January 27, 2022. EcoR1 Capital LLC thus crossed above the 5% threshold of Galapagos voting rights by purchase of voting securities on January 27, 2022.
On November 2, 2021, Van Herk Investments BV filed a Schedule 13D with the SEC to report that, as of November 2, 2021, it held 7.1% of the outstanding shares of Galapagos as of September 20, 2021.
On May 10, 2021, The Capital Group Companies, Inc. filed a Schedule 13G with the SEC to report that, as of April 30, 2021, it held 4.5% of the outstanding shares of Galapagos as of April 30, 2021. The Capital Group Companies, Inc. thus crossed below the 5% threshold of Galapagos voting rights by disposing of voting securities on April 30, 2021.
On February 26, 2021, we received a transparency notification from The Capital Group Companies, Inc., who notified that it holds 9.91% of the outstanding Galapagos shares. The Capital Group Companies, Inc. thus crossed below the 10% threshold of Galapagos’ voting rights by disposing of voting securities on February 22, 2021. On February 5, 2021, we received a transparency notice from The Capital Group Companies, Inc., that it held 10.03% of Galapagos shares. The Capital Group Companies, Inc. thus crossed above the 10% threshold of Galapagos’ voting rights by purchase of voting securities on 22 January 2021. Capital Group controls Capital Bank & Trust Company and Capital Research & Management Company through its direct subsidiary Capital Group International, Inc. (“CGII”), controls four CGII investment management companies (Capital International, Inc.; Capital International Limited, Capital International Sàrl; and Capital International K.K.), which all together hold 6,563,320 of Galapagos’ voting rights, consisting of ordinary shares (6,559,874) and equivalent financial instruments (right to recall lent American Depository Shares) (3,446), which represents 10.03% of Galapagos’ 65,411,767 outstanding shares as of March 15, 2021. On January 6, 2021, we received a transparency notice from Gilead Sciences, Inc., who notified that certain changes occurred in the chain of intermediary companies through which Gilead holds its shares in Galapagos, as a result of which Gilead holds its shares in Galapagos as of December 31, 2020 through its direct subsidiary Gilead Biopharmaceutics US, LLC, which through Gilead Sciences Ireland UC controls Gilead Therapeutics A1 Unlimited Company, which in turn holds 16,707,477 of Galapagos' voting rights, consisting of 16,707,477 shares (unchanged). Those 16,707,477 shares represent 25.54% of Galapagos' 65,411,767 outstanding shares as of March 15, 2021.
B. Related party transactions
Since January 1, 2019, we have engaged in the following major transactions with Gilead Sciences, Inc., and/or any of its affiliates.
On July 14, 2019, we and Gilead announced that we entered into a 10-year global research and development collaboration. In the context of the transaction, Gilead also made an equity investment in Galapagos. Finally, we amended and restated the license agreement for filgotinib that we originally entered into with Gilead on December 16, 2015.
On August 23, 2019, the closing of the transaction took place and we received an upfront payment of $3.95 billion (or €3,569.8 million) and a $1.1 billion (or €960.1 million) equity investment from Gilead.
195
On December 15, 2020, we and Gilead amended the existing arrangement for the commercialization and development of Jyseleca® (filgotinib).
On September 6, 2021, we and Gilead agreed to transfer the sponsorship of and operational and financial responsibility for the ongoing DIVERSITY clinical study, evaluating filgotinib in CD, and its long-term extension study from Gilead to Galapagos. Under the terms of the agreement, Gilead will make a one-time payment of $15 million to Galapagos in consideration for Galapagos assuming responsibility for the DIVERSITY clinical study.
In March 2022, Gilead and we agreed to transfer the sponsorship of and the operational responsibility for the MANTA study, a safety study in men with moderately to severely active UC, CD to assess semen parameter while taking filgotinib, and its long-term extension to Galapagos. The transfer was largely completed by December 31, 2022. The (former) Supervisory Board (currently Board of Directors) resolved and confirmed, as far as needed, that the related party transaction approval mechanism as set forth in article 7:116 of the Belgian Companies Code did not have to applied, since (i) the value of the aforementioned related party transaction is less than 1% of Galapagos’ consolidated net equity (based on the consolidated financial statements of Galapagos NV for the year ended December 31, 2021), and (ii) Galapagos is therefore able to rely on the materiality exemption as set out in article 7:116, §2 of the Belgian Companies Code. Furthermore, we entered into some mainly technical and non-material amendments to the existing transactions between us and Gilead (or its affiliates) during 2022.
In October 2023, Gilead and Galapagos agreed to amend the collaboration to terminate the existing 50/50 global development cost sharing arrangement with Galapagos bearing the costs going forward, and to terminate Galapagos’ obligation to pay tiered royalties to Gilead on net sales of Jyseleca® in Europe, in addition to other amendments, including a simplification of the governance and certain other technical amendments.
Effective January 31, 2024, following the closing of the transaction between Galapagos and Alfasigma S.p.A. to transfer the Jyseleca® business to Alfasigma S.p.A., Galapagos assigned its rights and obligations under the filgotinib collaboration to Alfasigma S.p.A., except for Galapagos’ right to receive royalties from Gilead on net sales in the Gilead Territory under a separate agreement between Gilead and Galapagos entered into in October 2023.
Share subscription agreement
As part of the research and development collaboration, Gilead entered into a share subscription agreement with us. On August 23, 2019, Gilead Therapeutics A1 Unlimited Company subscribed to 6,828,985 new Galapagos shares at a price of €140.59 per share, including issuance premium.
On October 22, 2019, our extraordinary shareholders’ meeting further issued a warrant to Gilead Therapeutics A1 Unlimited Company, known as warrant A, that confers the right to subscribe for a number of new shares sufficient to bring the number of shares owned by Gilead and its affiliates to 25.1% of the issued and outstanding shares. Warrant A expires one year after the issue date and the exercise price per share is EUR 140.59. On November 6, 2019, Gilead exercised Warrant A and increased its ownership in Galapagos to 25.10% of the then issued and outstanding shares. Warrant A expired in October 2020.
On October 22, 2019, Gilead Therapeutics A1 Unlimited Company was also issued another warrant, known as the initial warrant B, that confers the right to subscribe for a number of new shares sufficient to bring the number of shares owned by Gilead and its affiliates to 29.9% of the issued and outstanding shares. Such warrant will expire on August 23, 2024. The exercise price per share will be the greater of (i) 120% multiplied by the arithmetic mean of the 30-day daily volume weighted average trading price of the Galapagos shares preceding the date of the exercise notice with respect to such exercise, and (ii) €140.59. Between 57 and 59 months of August 23, 2019, subject to and upon approval by our Shareholders’ Meeting, Gilead Therapeutics A1 Unlimited Company will be issued a subsequent warrant with substantially similar terms, including as to exercise price, to the initial Warrant B. This subsequent Warrant B will expire on the earlier of the date that is five years after the fifth anniversary of the closing and the date that the warrant is issued. The issuance of this warrant is on the agenda of the Extraordinary Shareholders’ Meeting of April 30, 2024.
196
Gilead and Gilead Therapeutics A1 Unlimited Company are subject to certain standstill restrictions until the date that is 10 years following the closing. Among other things, during this time Gilead and its affiliates and any party acting in concert with them may not, without our priorconsent, acquire voting securities of Galapagos exceeding more than 29.9% of the then issued and outstanding voting securities, and Gilead and Gilead Therapeutics A1 Unlimited Company may not propose a business combination with or acquisition of Galapagos. The standstill restrictions are subject to certain exceptions as provided in the share subscription agreement.
Pursuant to the terms of the share subscription agreement, Gilead and Gilead Therapeutics A1 Unlimited Company also agreed to certain lock-up provisions. They shall not, and shall cause their affiliates not to, without our prior consent, dispose of any equity securities of Galapagos prior to the second anniversary of the closing. During the period running from the date that is two years following the closing until the date that is five years following the closing, Gilead and its affiliates shall not, without our prior consent, dispose of any equity securities of Galapagos if after such disposal they would own less than 20.1% of the then issued and outstanding voting securities of Galapagos. The lock-up restrictions are subject to certain exceptions as provided in the share subscription agreement and may terminate upon certain events. In April 2021, Gilead and Galapagos agreed to amend the share subscription agreement to extend the full lock-up of all of Gilead’s securities of Galapagos to a period of five years until August 22, 2024. We and Gilead agreed to amend the share subscription agreement for conformity with our change from a two-tier to a one-tier governance system.
Global research and development collaboration
We will fund and lead all discovery and development autonomously until the end of Phase 2. After the completion of a qualifying Phase 2 study (or, in certain circumstances, the first Phase 3 study), Gilead will have the option to acquire a license to the compound outside Europe. If the option is exercised, we and Gilead will co-develop the compound and share costs equally. If GLPG1690 had been approved in the United States, Gilead would have paid us an additional $325 million regulatory milestone fee. Development of GLPG1690 was discontinued in February 2021.
For GLPG1972, after the completion of the ongoing Phase 2b study in osteoarthritis, Gilead had the option to pay a $250 million fee to license the compound in the United States. If certain secondary efficacy endpoints for GLPG1972 had been met, Gilead would have paid us up to an additional $200 million. Following opt-in on GLPG1972, we would have been eligible to receive up to $550 million in regulatory and sales based milestones. In November 2020, Gilead declined to exercise its option to GLPG1972. For all other programs resulting from the collaboration, Gilead will make a $150 million opt-in payment per program and will owe no subsequent milestones. We will receive tiered royalties ranging from 20-24% on net sales of all our products licensed by Gilead in all countries outside Europe as part of the agreement. With respect to GLPG1690, reimbursement of development costs under the cost split mechanism by Gilead to us amounted to €0.3 million for the year ended December 31, 2023 (€0.4 million for the year ended December 31, 2022 and €18.1 million for the year ended December 31, 2021).
For further information on our exclusive option, license and collaboration agreement with Gilead, see the section of this annual report titled “Item 4.B.—Business overview.—Collaborations— Option, License and Collaboration Agreement with Gilead.”.
Filgotinib collaboration
Under the agreement as revised in 2019, we and Gilead would co-commercialize filgotinib in France, Germany, Italy, Spain and the United Kingdom and retain the 50/50 profit share in these countries that was part of the original filgotinib license agreement. The parties would also share future global development costs for filgotinib equally until a predetermined level, in lieu of the 80/20 cost split provided by the original agreement.
In December 2020, we and Gilead entered into a binding term sheet pursuant to which we agreed to amend this agreement again. Under the terms of the new arrangement, we assumed all development, manufacturing, commercialization and certain other rights for filgotinib in Europe. Most of the activities transferred to Galapagos by December 31, 2021, and the transition was completed by December 31, 2022. Gilead retains commercial rights and remains marketing authorization holder for filgotinib outside of Europe, including in Japan. All commercial economics
197
on filgotinib in Europe transferred to us as of January 1, 2022, subject to payment of tiered royalties of 8 to 15 percent of net sales in Europe to Gilead, starting in 2024.
Since January 1, 2021, we bear the future development costs for certain studies, in lieu of the equal cost split contemplated by the previous agreement. These studies included the DARWIN3, FINCH4, FILOSOPHY, and Phase 4 studies and registries in RA, MANTA and MANTA-RAy, the PENGUIN1 and 2 and EQUATOR2 studies in PsA, the SEALION1 and 2 studies in AS, the HUMBOLDT study in uveitis in addition to other clinical and non-clinical expenses supporting these studies and support for any investigator sponsored trials in non-IBD conditions and non-clinical costs on all current trials. The existing 50/50 global development cost sharing arrangement continued for the following studies: SELECTION and its long-term extension study (LTE) in UC, DIVERSITY and its LTE, DIVERGENCE 1 and 2 and their LTEs and support for Phase 4 studies and registries in Crohn’s disease, pediatric studies and their LTEs in RA, UC and Crohn’s disease, and support for investigator sponsored trials in IBD. In September 2021, we and Gilead agreed to transfer the sponsorship of the DIVERSITY study and its LTE study from Gilead to Galapagos. This transfer was intended to be completed by June 30, 2022 and completed by March 2023. From April 1, 2022, Galapagos will also be solely responsible for all development costs for the DIVERSITY study and its LTE study. In March 2022, Gilead and we agreed to transfer the sponsorship of and the operational responsibility for the MANTA study and its long-term extension to Galapagos. This transfer was largely completed by December 31, 2022.
Under the original exclusive license and collaboration agreement, we received from Gilead $60.0 million (or €55.1 million) in milestone payments in the year ended December 31, 2016, $10.0 million (or €9.4 million) in milestone payments in the year ended December 31, 2017, and $15.0 million (or €12.4 million) in milestone payments in the year ended December 31, 2018. In December 2019, Gilead initiated a Phase 3 trial in psoriatic arthritis for which we received $10.0 million (€9.1 million). In December 2019, Gilead filed an NDA for filgotinib in the U.S. for which we received a $20.0 million (€ 18.2 million) payment in January 2020. In September 2020 filgotinib was approved by both the European and the Japanese authorities, for which we received a $105.0 million (€90.2 million) payment in October 2020.
In connection with the December 2020 amendments to the existing arrangement for the commercialization and development of filgotinib, Gilead has agreed to irrevocably pay Galapagos €160 million, subject to certain adjustments for higher than budgeted development costs. Gilead paid €35 million in January 2021, an additional €75 million in April 2021, and €50 million in March 2022.
Under the terms of the agreement of September 2021, Gilead made a one-time payment of $15 million to us in July 2022 in consideration for Galapagos assuming responsibility for the DIVERSITY Study.
On March 28, 2022 filgotinib was approved by the Japanese Ministry of Health, Labour and Welfare for UC, for which we received a $20.0 million (€18.2 million) regulatory milestone payment from Gilead in May 2022.
In addition, from January 1, 2021 until December 31, 2023, we received €22.0 million of royalties on Jyseleca® from Gilead.
We are no longer eligible to receive any future milestone payments relating to filgotinib in Europe. However, we remain eligible to receive tiered royalty percentages ranging from 20% to 30% on Gilead's global net sales of filgotinib outside of Europe.
We incurred €190.2 million in development costs for the year ended December 31, 2023 for the development of filgotinib in collaboration with Gilead. The reimbursement of research and development costs under the cost split mechanism by us to Gilead amounted to nil for the year ended December 31, 2023 (€2.4 million for the year ended December 31, 2022 and €81.3 million for the year ended December 31, 2021). The reimbursement of research and development costs under the cost split mechanism by Gilead to us amounted to €3.6 million for the year ended December 31, 2023 (nil for the year ended December 31, 2022 and nil for the year ended December 31, 2021).
The reimbursement of commercialization costs under the cost split mechanism by us to Gilead amounted to nil for the year ended December 31, 2023 (nil for the year ended December 31, 2022 and nil million for the year ended December 31, 2021).
198
The reimbursement of commercialization costs under the cost split mechanism by Gilead to us amounted to nil million for the year ended December 31, 2023 (€0.1 million for the year ended December 31, 2022 and €66.7 million for the year ended December 31, 2021).
We purchased raw materials, semi-finished products and finished products of Jyseleca® from Gilead for an amount of nil million for the year ended December 31, 2023 (€13.5 million for the year ended December 31, 2022 and €24.9 million for the year ended December 31, 2021).
For further information on our exclusive license and collaboration agreement with Gilead, see the section of this annual report titled “Item 4.B.— Business overview.—Collaborations—Exclusive collaboration agreement with Gilead for filgotinib.”
Transactions with related companies
From time to time, in the ordinary course of our business, we may contract for services from companies in which certain of the members of our Board of Directors or Executive Committee may serve as director or advisor. The cost of these services is negotiated on an arm’s length basis, and none of these arrangements are material to us.
Agreements with our Board members and Executive Committee members
Management arrangements
As from January 1, 2020, all members of the Executive Committee (the former Management Board) provide their services under a management agreement with Galapagos NV, subject to Belgian law, that contains a notice period of six months and no other severance payments. These management agreements were replaced in April 2020 to take into account the implementation of the new Belgian Companies Code by our Company. During the period as from January 1, 2022 until April 26, 2022, Galapagos had a two-tier governance structure as provided by the Belgian Companies Code, with two governance bodies: the Supervisory Board and the Management Board. As from April 26, 2022, Galapagos has adopted a one-tier governance structure as provided by the Belgian Companies Code, with the Board of Directors replacing the (former) Supervisory Board, and the Executive Committee replacing the (former) Management Board. The paragraphs below set forth the main terms of the agreements.
Stoffels IMC BV, permanently represented by Dr. Paul Stoffels
On January 26, 2022, we entered into a management agreement, subject to Belgian law, with Stoffels IMC BV (permanently represented by Dr. Paul Stoffels) for the position of Chief Executive Officer for an indefinite period. Effective April 1, 2022, Stoffels IMC BV (permanently represented by Dr. Paul Stoffels) is our new Chief Executive Officer. For the year ended December 31, 2023, Stoffels IMC BV, permanently represented by Dr. Paul Stoffels, received a base remuneration from Galapagos NV of €750,000.
Bart Filius
On September 15, 2014, Galapagos B.V. entered into an employment agreement, subject to Dutch law, with Bart Filius for the position of Chief Financial Officer, starting December 1, 2014 for an indefinite period. Effective December 1, 2014, Mr. Filius’ employment agreement with Galapagos B.V. was reduced from a full-time basis to a part-time basis, for approximately 60% of his time, and he entered into a management agreement, subject to Belgian law, with Galapagos NV for approximately 40% of his time. In addition to his role as Chief Financial Officer, Mr. Filius has served as Chief Operating Officer since September 2017. On May 6, 2020, we entered into a new management agreement, subject to Belgian law, with Bart Filius for the position of Chief Operating Officer and Chief Financial Officer for an indefinite period. The management agreement with Mr. Bart Filius was terminated on June 30, 2023. For the six-month period from January 1, 2023 until June 30, 2023, Mr. Filius received a base remuneration from Galapagos NV of €266,250.
199
Thad Huston
On June 15, 2023, Galapagos NV entered into a management agreement with Thad Huston, subject to Belgian law, for the position of Chief Financial Officer and Chief Operating Officer, for an indefinite period. For the year ended December 31, 2023, Mr. Huston received a base remuneration from Galapagos NV of €254,939.
Michele Manto
On May 6, 2020, we entered into a new management agreement, subject to Belgian law, with Michele Manto for the position of Chief Commercial Officer for an indefinite period. For the year ended on December 31, 2023, Mr. Manto received a base remuneration from Galapagos NV of €377,150. The position of Mr. Manto as Chief Commercial Officer and member of Galapagos’ Executive Committee was terminated on December 31, 2023.
Valeria Cnossen
On July 22, 2022 we entered into a management agreement, subject to Belgian law, with Valeria Cnossen for the position of General Counsel for an indefinite period. On February 15, 2023 we amended the management agreement to reflect her appointment as Executive Committee member as of January 1, 2023. For the year ended on December 31, 2023, Ms. Cnossen received a base remuneration from Galapagos NV of €382,500.
Annelies Missotten
On December 16, 2022 we entered into a management agreement, subject to Belgian law, with Annelies Missotten for the position of Chief Human Resources Officer for an indefinite period. She became an Executive Committee member as of January 1, 2023. Prior to the entry into force of the management agreement, Annelies Missotten was bound by an employment agreement for indefinite duration. For the year ended on December 31, 2023, Ms. Missotten received a base remuneration from Galapagos NV of €325,000.
Severance payments upon change of control
The abovementioned agreements with the members of our Executive Committee do not provide for severance compensation. They do not contain notice periods that exceed nine months for the CEO and six months for the other Executive Committee members. However, we entered into undertakings with the Executive Committee members providing that, in case their contract with us is terminated as a result of a change of control of our Company, they would be entitled to a severance compensation of twelve months’ base salary for our CEO and nine months’ base salary for the other Executive Committee members.
Severance payments for departing Executive Committee or (former) Management Board members
On May 2, 2023, Galapagos announced the departure of Mr. Bart Filius, President, COO and CFO and Executive Committee member per 30 June 2023. Upon substantiated recommendation of the Remuneration Committee, the Board approved a termination compensation of €1,650,000, consisting of compensation for a non-compete obligation for 12 months in an amount of €545,000, and a termination amount of €1,105,000 taking into account loss of 2023 bonus and loss of unvested RSUs. Effective July 1, 2023, Mr. Filius was no longer a member of the Executive Committee. He exercised an advisory role until December 31, 2023, for a total consultancy fee of €330,000. The Board determined this arrangement would best serve the interests of Galapagos, in particular given the critical role played by the outgoing President, COO and CFO in onboarding the then new CEO as well as remaining with Galapagos to support business continuity as a successor was found. Mr Bart Filius was not eligible for any equity grants (RSUs and subscription rights) in 2023. He qualifies as a good leaver under the terms and conditions of the relevant subscription right plans and this is not part of his termination package.
200
On January 2, 2024, Galapagos announced the departure of Mr. Michele Manto, CCO and Executive Committee member per December 31, 2023. No termination compensation was awarded. From January 1, 2024 until May 31, 2024, Mr. Manto will execute an advisory role to support the Jyseleca® transition to Alfasigma, for which he wil receive a total fee of €191,900 and remain entitled to RSU pay-outs during this period. Mr. Manto will be entitled to his cash bonus for 2023, but will not be eligible for any equity grants (RSUs and subscription rights) in 2024. He qualifies as a good leaver under the terms and conditions of the relevant subscription right plans.
Board of Directors and Executive Committee compensation
See the sections of this annual report in “Item 6.B.—Compensation.” titled “—Compensation of Our Board or see the sections of this annual report in “Item 6.B.—Compensation.” titled “—Compensation of Our Board of Directors” and “— Compensation of Members of the Executive Committee” and the section titled “Item 7.A.—Major Shareholders.” for information regarding compensation of the members of our Board of Directors and Executive Committee.
Equity awards
Since January 1, 2020, we have granted subscription rights and RSUs to the members of our Executive Committee. We do not grant RSUs to the members of our Board of Directors and have discontinued the grant of subscription rights to members of our Board of Directors as of 2020, taking into account the stricter rules under the Belgian Companies Code and 2020 Code.
See the sections of this annual report in “Item 6.B.—Compensation.” titled “—Compensation of Our Board of Directors” and “— Compensation of Members of our Executive Committee” and the section titled “Item 7.A.—Major Shareholders.” for information regarding equity awards to the members of our Executive Committee.
Bonus plans
See the section of this annual report titled “Item 6.B.—Compensation.—Compensation of Members of the Executive Committee” for information regarding bonus plans for members of our Executive Committee.
Related-party transactions policy
Article 7:97 of the Belgian Companies Code provides for a special procedure that applies to intra-group or related party transactions. The procedure applies to decisions or transactions between us and our related parties that are not one of our subsidiaries. Prior to any such decision or transaction, our Board of Directors must appoint a special committee consisting of three independent Board members, who can opt to be assisted by one or more independent experts. This special committee must assess the business advantages and disadvantages of the relevant decision or transaction, quantify its financial consequences, and determine whether the relevant decision or transaction causes a disadvantage to us that is manifestly illegitimate in view of our policy. If the special committee determines that the relevant decision or transaction is not illegitimate but will prejudice us, it must analyze the advantages and disadvantages of such decision or transaction and set out such considerations as part of its advice. Our Board of Directors must then make a decision, taking into account the opinion of the special committee. Any deviation from the special committee’s advice must be justified. Members of the Board of Directors who have a conflict of interest are not entitled to participate in the deliberation and vote. The special committee’s advice and the decision of the Board of Directors must be notified to our external auditor, who must render a separate opinion to assess that there is no material inconsistency between the accounting and financial information included in the minutes of the Board of Directors and in the advice of the special committee of the independent Board members compared to the information that the external auditor has within the framework of its mandate. The conclusion of the special committee and the opinion by the external auditor must publicly disclosed at the time the transaction is entered into. This procedure does not apply to any decisions or transactions in the ordinary course of business under customary market conditions and security documents, or to transactions or decisions with a value of less than 1% of our net assets as shown in our consolidated annual accounts.
201
On 30 October 2023, we entered into a related party transaction with Gilead within the meaning of article 7:97 of the Belgian Companies Code, by agreeing to further amend the collaboration agreement. Gilead and Galapagos agreed to terminate the existing 50/50 global development cost sharing arrangement, with Galapagos bearing the costs going forward, and to terminate Galapagos’ obligation to pay tiered royalties to Gilead on net sales of Jyseleca® in Europe, in addition to other amendments.
The Board of Directors applied the related party transaction approval procedure as set forth in article 7:97 of the Belgian Companies Code. Within the context of this procedure, a committee of three independent members of the Board of Directors of Galapagos (the “Committee”) issued an advice to the Board of Directors in which the Committee assessed the amended terms of the collaboration agreement. In its advice to the Board of Directors, the Committee concluded the following: “The Committee believes that, under the circumstances, the proposed amendments to the filgotinib collaboration between Gilead and Galapagos are reasonable and fair from the point of view of Galapagos and its shareholders, and in line with the strategy of the Company. The proposed amendments offer an important opportunity to have autonomy on development and commercial activities in Europe in its ongoing collaboration with Gilead. The proposed amendments also come with a number of challenges and risks, but these are not unreasonable and can be managed going forward. The Committee therefore believes that the proposed amendments to the collaboration with Gilead in relation to filgotinib are in the interest of Galapagos, and in any event not manifestly abusive. In view hereof, the Committee issues a favourable and unqualified opinion to the Board of Directors of Galapagos.” The Board of Directors did not deviate from the Committee’s advice.
The assessment by the Statutory Auditor of Galapagos of the advice of the Committee and the minutes of the Board of Directors is as follows: “Based on our assessment, nothing has come to our attention that causes us to believe that the financial and accounting data reported in the advice of the Ad hoc committee of the independent members of the Board of Directors dated 30 October 2023 and in the minutes of the Board of Directors dated 30 October 2023, which justify the proposed transaction, are not consistent, in all material respects, compared to the information we possess in the context of our assignment.”
In addition to this, our Corporate Governance Charter provides for guidelines for transactions between Galapagos and members of our Board of Directors or Executive Committee.
According to such guidelines:
| ● | it is expected from all members of our Board of Directors and Executive Committee that they avoid all acts, standpoints or interests which are conflicting with, or which give the impression that they are conflicting with, the interests of our Company; |
| ● | all transactions between Galapagos and the members of our Board of Directors or Executive Committee need the approval of our Board of Directors, as relevant in accordance with article 7:97 of the Belgian Companies Code and the procedure described in the Related Person Transaction Policy. The members of our Board of Directors and Executive Committee are, by way of non-exhaustive example, not allowed, directly or indirectly, to enter into agreements with Galapagos which relate to supply of materials or delivery of services (other than in the framework of their mandate for the Company), except with the explicit approval of our Board of Directors; |
| ● | any transaction between the Company and members of the Board of Directors or the Executive Committee can only be entered into at arm’s length (i.e. normal market conditions); |
| ● | in the event any member of our Board of Directors, or, in case pertaining, its permanent representative is confronted with a direct or indirect interest of a monetary nature that conflicts with the interests of the Company in respect of a decision or an act falling within the scope of the responsibilities of the Board of Directors, the provisions of article 7:96 of the Belgian Companies Code shall apply, and the relevant Board member shall inform the other Board members prior to the deliberation on the subject matter. Therefore in the event article 7:96 of the Belgian Companies Code applies and a conflict of interests exists between us and a |
202
| member of our Board of Directors, or, in case pertaining, its permanent representative, the relevant Board member shall not participate in the deliberation and vote concerning such point on the agenda; |
| ● | in the event article 7:96 of the Belgian Companies Code does not apply, the existence of a conflict of interest shall be reported by the relevant Board member, its existence shall be included in the minutes (but shall not be published) and the relevant Board member shall not vote on the matter. |
In addition, Galapagos’ Corporate Governance Charter stipulates that, if a member of the Executive Committee has a direct or indirect interest of monetary nature that conflicts with the interests of the Company in respect of a decision or an act falling within the scope of the responsibilities of the Executive Committee, the Executive Committee shall refrain from making any decision. The Executive Committee shall instead escalate the matter to the Board of Directors. The Board of Directors shall decide whether or not to approve such decision or act, and shall apply the conflict of interest procedure as set out in article 7:96 of the Belgian Companies Code.
In the event a conflict of interest exists within the Executive Committee that falls outside the scope of article 7:96 of the Belgian Companies Code, the existence of such conflict shall be reported by the relevant Executive Committee member, its existence shall be included in the minutes (but shall not be published) and the relevant Executive Committee member shall not vote on the matter.
We have adopted a Related-Person Transaction policy that sets forth our procedures for the identification, review, consideration and approval or ratification of related-party transactions. For purposes of this policy only, a related-party transaction is a transaction in which we are a participant and a related party has a direct or indirect material interest. For purposes of this policy, a related person is any member of the Executive Committee, any member of the Board of Directors (or nominee) or beneficial owner of more than 5% of any class of our voting securities, including any of their immediate family members and any entity owned or controlled by such persons. Under this policy, if a transaction has been identified as a related-party transaction, our Audit Committee will review and consider information regarding the related-party transaction. In reviewing any related-party transaction, the Audit Committee will take into account, among other factors it deems appropriate, (i) whether the transaction is on terms no less favorable to us than terms generally available in a transaction with an unaffiliated third party under the same or similar circumstances; and (ii) the extent of the related party’s interest in the related-party transaction. Additionally, we will provide the Audit Committee with all material information regarding the related-party transaction, the interest of the related party, and any potential disclosure obligations in connection therewith. In addition, under our Code of Conduct, our employees, Board members and Executive Committee members have an affirmative responsibility to disclose any transaction or relationship that reasonably could be expected to give rise to a conflict of interest.
In 2023, the following conflicts of interest between Galapagos NV and a Director within the meaning of article 7:96 of the Belgian Companies Code were noted:
| ● | In a meeting of the Board of Directors held on February 20, 2023, the following was reported in connection with the proposed compensation of the CEO (cash bonus and RSUs): the Chair informed the Board of Directors of a conflict of interest, concerning the proposed compensation of the CEO. The Board considered that said compensation was a justified reward for the results achieved by the CEO in 2022. The Board shared the opinion of the Remuneration Committee that the proposed compensation is justified and reasonable. The Chair did not take part in the deliberation and vote concerning this decision. |
| ● | In a meeting of the Board of Directors held on May 2, 2023, the following was reported in accordance with article 7:96 of the Belgian Companies Code in connection with the proposed grants of subscription rights and RSUs to the CEO under the 2023 plans: the Chair informed the Board of Directors of a conflict of interest, concerning the proposed grants of subscription rights and RSUs to the CEO under the 2023 plans. The Board considered that said compensation was a justified reward for the results achieved by the CEO in 2022, in line with the contractual arrangement with the CEO executed in 2022 and with the Company’s Remuneration Policy. Furthermore, the Board deemed the proposed grants to be an important tool in the retention of Stoffels IMC BV as CEO of the Company and considered that these grants have no material impact on the financial position of the Company. The Board shared the opinion of the Remuneration Committee that the |
203
| proposed compensation is justified and reasonable. The Chair did not take part in the deliberation and the vote concerning this decision. |
| ● | In a meeting of the Board of Directors held on May 5, 2023, the following was reported in accordance with article 7:96 of the Belgian Companies Code in connection with the proposed issuance of the 2023 subscription right plans: The CEO and also Chair of the Board of Directors, Stoffels IMC BV, reported prior to this meeting that he had a conflict of interest within the meaning of article 7:96 of the Belgian Companies Code in connection with the issuance of the number of subscription rights under the Subscription Right Plan 2023 BE, Subscription Right Plan 2023 RMV, and Subscription Right Plan 2023 ROW, for the benefit of employees of the Company and its subsidiaries, with cancellation of the preferential subscription right of the existing shareholders in the framework of the issuance of these subscription rights and the related possible future capital increase, as the CEO will be a beneficiary under Subscription Right Plan 2023 BE. The Board of Directors, upon the recommendation of the Remuneration Committee, is of the opinion that the proposed agenda items and the proposed grant of subscription rights to the CEO are consistent with the Company’s Remuneration Policy and are justified and reasonable. The nature of the proposed decision and the financial impact on the Company are described in more detail in the above-mentioned special report of the Board of Directors. In accordance with the procedure provided for in article 7:96 of the Belgian Companies Code, the CEO and also Chair of the Board of Directors, Stoffels IMC BV, does not attend this meeting and will not take part in the deliberation and the vote. |
| ● | In a meeting of the Board of Directors held on 30 October 2023, the following was reported in accordance with article 7:96 of the Belgian Companies Code in connection with the proposed amendment to the Galapagos-Gilead filgotinib agreement: The Chair reported that, prior to this meeting, Daniel O’Day and Dr. Linda Higgins had informed him that, since they are representatives of Gilead, they might have a conflict of interest in relation to the resolutions to be passed by the Board of Directors in relation to this agenda topic. Accordingly, Daniel and Linda had recused themselves for this part of the meeting, and did not take part in the deliberation and resolutions in relation to this agenda topic. |
C. Interests of experts and counsel
Not applicable.
Item 8 Financial information
A. Consolidated statements and other financial information
Consolidated financial statements
Our consolidated financial statements are appended at the end of this annual report, starting at page F-1, and incorporated herein by reference.
Legal proceedings
From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. We are not presently a party to any legal proceedings that, if determined adversely to us, would individually or taken together have a material adverse effect on our business, results of operations, financial condition or cash flows. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
204
Dividend distribution policy
We have never declared or paid any cash dividends on our ordinary shares. We do not anticipate paying cash dividends on our equity securities in the foreseeable future and intend for the foreseeable future to retain all available funds and any future earnings for use in the operation and expansion of our business. Additionally, under the Option, License and Collaboration Agreement, dated as of July 14, 2019, by and between the Company and Gilead Sciences, Inc. (the “OLCA”), we are generally prohibited from using any Upfront Consideration (as defined in the OLCA), plus all net proceeds paid to us by Gilead pursuant to the 2019 share subscription agreement, to pay dividends on our ordinary shares. In general, distributions of dividends proposed by our Board of Directors require the approval of our shareholders at a shareholders’ meeting with a simple majority vote, although our Board of Directors may declare interim dividends without shareholder approval, subject to the terms and conditions of the Belgian Companies Code.
Pursuant to Belgian law, the calculation of amounts available for distribution to shareholders, as dividends or otherwise, must be determined on the basis of our non-consolidated statutory financial accounts. In addition, under the Belgian Companies Code, we may declare or pay dividends only if, following the declaration and issuance of the dividends, the amount of our net assets on the date of the closing of the last financial year according to our statutory annual accounts (i.e., the amount of the assets as shown in the balance sheet, decreased with provisions and liabilities, all as prepared in accordance with Belgian accounting rules), decreased with the non-amortized costs of incorporation and expansion and the non-amortized costs for research and development, does not fall below the amount of the paid-up capital (or, if higher, the called capital), increased with the amount of nondistributable reserves. Finally, prior to distributing dividends, we must allocate at least 5% of our annual net profits (under our non-consolidated statutory accounts prepared in accordance with Belgian accounting rules) to a legal reserve, until such legal reserve amounts to 10% of our share capital.
B. Significant changes
None.
Item 9 The offer and listing
A. Offer and listing details
The ADSs have been listed on the Nasdaq Global Select Market, or Nasdaq, under the symbol “GLPG” since May 14, 2015. Prior to that date, there was no public trading market for the ADSs. Our ordinary shares have been trading on Euronext Amsterdam and Euronext Brussels under the symbol “GLPG” since May 6, 2005. Prior to that date, there was no public trading market for the ADSs or our ordinary shares. Our global offering in May 2015 was priced at $42.05 per ADS and €37.00 per ordinary share based on an exchange rate of $1.1365 per euro.
B. Plan of distribution
Not applicable.
C. Markets
The ADSs have been listed on Nasdaq under the symbol “GLPG” since May 14, 2015, and our ordinary shares have been listed on Euronext Amsterdam and Euronext Brussels under the symbol “GLPG” since May 6, 2005.
D. Selling shareholders
Not applicable.
E. Dilution
Not applicable.
205
F. Expenses of the issue
Not applicable.
Item 10 Additional information
A. Share capital
Not applicable.
B. Memorandum and Articles of Association
The information set forth in our Registration Statement on Form F-3ASR (File No. 333-230639), automatically effective upon filing with the SEC on March 29, 2019, under the heading “Description of Share Capital”, as further supplemented by Exhibit 2.3 to this Annual Report (“Description of Securities”), is deemed incorporated by simple reference.
C. Material contracts
For information on our material contracts, please see the sections of this annual report titled “Item 4—Information on the Company” and “Item 7— Major shareholders and related party transactions.”
D. Exchange controls
There are no Belgian exchange control regulations that impose limitations on our ability to make, or the amount of, cash payments to residents of the United States.
We are in principle under an obligation to report to the National Bank of Belgium certain cross-border payments, transfers of funds, investments and other transactions in accordance with applicable balance-of-payments statistical reporting obligations. Where a cross-border transaction is carried out by a Belgian credit institution on our behalf, the credit institution will in certain circumstances be responsible for the reporting obligations.
E. Taxation
Certain material U.S. federal income tax considerations to U.S. holders
The following is a summary of certain material U.S. federal income tax considerations relating to ownership and disposition of ADSs by a U.S. holder (as defined below). This summary addresses only the U.S. federal income tax considerations for U.S. holders of the ADSs and that will hold such ADSs as capital assets for U.S. federal income tax purposes. This summary does not address all U.S. federal income tax matters that may be relevant to a particular U.S. holder. This summary does not address all tax considerations that may be applicable to a holder of ADSs that may be subject to special tax rules including, without limitation, the following:
| ● | banks, financial institutions or insurance companies; |
| ● | brokers, dealers or traders in securities, currencies, commodities, or notional principal contracts; |
| ● | tax-exempt entities or organizations, including an “individual retirement account” or “Roth IRA” as defined in Section 408 or 408A of the Code (as defined below), respectively; |
| ● | real estate investment trusts, regulated investment companies or grantor trusts; |
206
| ● | persons that hold the ADSs as part of a “hedging,” “integrated” or “conversion” transaction or as a position in a “straddle” for U.S. federal income tax purposes; |
| ● | partnerships (including entities classified as partnerships for U.S. federal income tax purposes) or other pass-through entities (including S Corportions), or persons that will hold the ADSs through such an entity; |
| ● | persons that received the ADSs as compensation for the performance of services; |
| ● | certain former citizens or long-term residents of the United States; |
| ● | holders that own directly, indirectly, or through attribution 10% or more of the voting power or value of the ADSs and shares; and |
| ● | holders that have a “functional currency” for U.S. federal income tax purposes other than the U.S. dollar. |
Further, this summary does not address the U.S. federal estate, gift, or alternative minimum tax considerations, or any U.S. state, local, or non-U.S. tax considerations of the ownership and disposition of the ADSs.
This description is based on the U.S. Internal Revenue Code of 1986, as amended, or the Code; existing, proposed and temporary U.S. Treasury Regulations promulgated thereunder, administrative and judicial interpretations thereof; and the income tax treaty between Belgium and the United States in each case as of and available on the date hereof. All the foregoing is subject to change, which change could apply retroactively, and to differing interpretations, all of which could affect the tax considerations described below. There can be no assurances that the U.S. Internal Revenue Service, or the IRS, will not take a contrary or different position concerning the tax consequences of ownership and disposition of the ADSs or that such a position would not be sustained. Holders should consult their own tax advisers concerning the U.S. federal, state, local and non-U.S. tax consequences of owning, and disposing of the ADSs in their particular circumstances.
For the purposes of this summary, a “U.S. holder” is a beneficial owner of ADSs that is (or is treated as), for U.S. federal income tax purposes:
| ● | an individual who is a citizen or resident of the United States; |
| ● | a corporation, or other entity that is treated as a corporation for U.S. federal income tax purposes, created or organized in or under the laws of the United States, any state thereof, or the District of Columbia; |
| ● | an estate, the income of which is subject to U.S. federal income taxation regardless of its source; or |
| ● | a trust, if a court within the United States is able to exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of the substantial decisions of such trust or has a valid election in effect under applicable U.S. Treasury Regulations to be treated as a United States person. |
If a partnership (or any other entity treated as a partnership for U.S. federal income tax purposes) holds ADSs, the U.S. federal income tax consequences relating to an investment in the ADSs will depend in part upon the status of the partner and the activities of the partnership. Such a partner or partnership should consult its tax advisor regarding the U.S. federal income tax considerations of owning and disposing of the ADSs in its particular circumstances.
In general, a U.S. holder who owns ADSs will be treated as the beneficial owner of the underlying shares represented by those ADSs for U.S. federal income tax purposes. Accordingly, no gain or loss will generally be recognized if a U.S. holder exchanges ADSs for the underlying shares represented by those ADSs.
The U.S. Treasury has expressed concern that parties to whom ADSs are released before shares are delivered to the depositary (“pre-release”), or intermediaries in the chain of ownership between holders and the issuer of the security
207
underlying the ADSs, may be taking actions that are inconsistent with the claiming of foreign tax credits by holders of ADSs. These actions would also be inconsistent with the claiming of the reduced rate of tax, described below, applicable to dividends received by certain non-corporate holders. Accordingly, the creditability of Belgian taxes, and the availability of the reduced tax rate for dividends received by certain non-corporate U.S. holders, each described below, could be affected by actions taken by such parties or intermediaries.
As indicated below, this discussion is subject to U.S. federal income tax rules applicable to a “passive foreign investment company,” or a PFIC.
Persons considering an investment in the ADSs should consult their own tax advisors as to the particular tax consequences applicable to them relating to ownership and disposition of the ADSs, including the applicability of U.S. federal, state and local tax laws and non-U.S. tax laws.
Distributions. Although we do not currently plan to pay dividends, and subject to the discussion under “—Passive Foreign Investment Company Considerations” below, the gross amount of any distribution (before reduction for any amounts withheld in respect of Belgian withholding tax) actually or constructively received by a U.S. holder with respect to ADSs will be taxable to the U.S. holder as a dividend to the extent of the U.S. holder’s pro rata share of our current and accumulated earnings and profits as determined under U.S. federal income tax principles. Distributions in excess of earnings and profits will be non-taxable to the U.S. holder to the extent of, and will be applied against and reduce, the U.S. holder’s adjusted tax basis in the ADSs. Distributions in excess of earnings and profits and such adjusted tax basis will generally be taxable to the U.S. holder as either long-term or short-term capital gain depending upon whether the U.S. holder has held the ADSs for more than one year as of the time such distribution is received. However, since we do not calculate our earnings and profits under U.S. federal income tax principles, it is expected that any distribution will be reported as a dividend, even if that distribution would otherwise be treated as a non-taxable return of capital or as capital gain under the rules described above. Noncorporate U.S. holders may qualify for the preferential rates of taxation with respect to dividends on ADSs applicable to long-term capital gains (i.e., gains from the sale of capital assets held for more than one year) applicable to qualified dividend income (as discussed below) if we are a “qualified foreign corporation” and certain other requirements (discussed below) are met. A non-U.S. corporation (other than a corporation that is classified as a PFIC for the taxable year in which the dividend is paid or the preceding taxable year) generally will be considered to be a qualified foreign corporation (a) if it is eligible for the benefits of a comprehensive tax treaty with the United States which the Secretary of Treasury of the United States determines is satisfactory for purposes of this provision and which includes an exchange of information provision, or (b) with respect to any dividend it pays on ADSs which are readily tradable on an established securities market in the United States. The ADSs are listed on the Nasdaq Global Select Market, or Nasdaq, which is an established securities market in the United States, and we expect the ADSs to be readily tradable on Nasdaq. However, there can be no assurance that the ADSs will be considered readily tradable on an established securities market in the United States in later years. We are incorporated under the laws of Belgium, and we believe that we qualify as a resident of Belgium for purposes of, and are eligible for the benefits of, The Convention between the Government of the United States of America and the Government of the Kingdom of Belgium for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income, signed on November 27, 2006, or the U.S.-Belgium Tax Treaty, although there can be no assurance in this regard. Further, the IRS has determined that the U.S.-Belgium Tax Treaty is satisfactory for purposes of the qualified dividend rules and that it includes an exchange-of-information program. Therefore, subject to the discussion under “—Passive Foreign Investment Company Considerations” below, such dividends will generally be “qualified dividend income” in the hands of individual U.S. holders, provided that a holding period requirement (more than 60 days of ownership, without protection from the risk of loss, during the 121-day period beginning 60 days before the ex-dividend date) and certain other requirements are met. The dividends will not be eligible for the dividends-received deduction generally allowed to corporate U.S. holders.
A U.S. holder generally may claim the amount of any Belgian withholding tax as either a deduction from gross income or a credit against U.S. federal income tax liability. However, the foreign tax credit is subject to numerous complex limitations that must be determined and applied on an individual basis. Generally, the credit cannot exceed the same proportion of a U.S. holder’s U.S. federal income tax liability which such U.S. holder’s “foreign source” taxable income bears to such U.S. holder’s worldwide taxable income. In applying this limitation, a U.S. holder’s various items of income and deduction must be classified, under complex rules, as either “foreign source” or “U.S. source.” In
208
addition, this limitation is calculated separately with respect to specific categories of income. The amount of a distribution with respect to the ADSs that is treated as a “dividend” may be lower for U.S. federal income tax purposes than it is for Belgian income tax purposes, potentially resulting in a reduced foreign tax credit for the U.S. holder. Furthermore, Belgian income taxes that are withheld in excess of the rate applicable under the U.S.-Belgium Tax Treaty or that are refundable under Belgian law will not be eligible for credit against a U.S. holder’s federal income tax liability. Each U.S. holder should consult its own tax advisors regarding the foreign tax credit rules.
In general, the amount of a distribution paid to a U.S. holder in a foreign currency will be the dollar value of the foreign currency calculated by reference to the spot exchange rate on the day the U.S. holder receives the distribution, regardless of whether the foreign currency is converted into U.S. dollars at that time. Any foreign currency gain or loss a U.S. holder realizes on a subsequent conversion of foreign currency into U.S. dollars will be U.S. source ordinary income or loss. If dividends received in a foreign currency are converted into U.S. dollars on the day they are received, a U.S. holder should not be required to recognize foreign currency gain or loss in respect of the dividend.
Sale, Exchange or Other Taxable Disposition of the ADSs. A U.S. holder will generally recognize gain or loss for U.S. federal income tax purposes upon the sale, exchange or other taxable disposition of ADSs in an amount equal to the difference between the U.S. dollar value of the amount realized from such sale or exchange and the U.S. holder’s tax basis for those ADSs. Subject to the discussion under “—Passive Foreign Investment Company Considerations” below, this gain or loss will generally be a capital gain or loss. The adjusted tax basis in the ADSs generally will be equal to the cost of such ADSs. Capital gain from the sale, exchange or other taxable disposition of ADSs of a non-corporate U.S. holder is generally eligible for a preferential rate of taxation applicable to capital gains, if the non-corporate U.S. holder’s holding period determined at the time of such sale, exchange or other taxable disposition for such ADSs exceeds one year (i.e., such gain is a long-term taxable gain). The deductibility of capital losses for U.S. federal income tax purposes is subject to limitations. Any such gain or loss that a U.S. holder recognizes generally will be treated as U.S. source income or loss for foreign tax credit limitation purposes.
For a cash basis taxpayer, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the settlement date of the purchase or sale. In that case, no foreign currency exchange gain or loss will result from currency fluctuations between the trade date and the settlement date of such a purchase or sale. An accrual basis taxpayer, however, may elect the same treatment required of cash basis taxpayers with respect to purchases and sales of the ADSs that are traded on an established securities market, provided the election is applied consistently from year to year. Such election may not be changed without the consent of the IRS. For an accrual basis taxpayer that does not make such an election, units of foreign currency paid or received are translated into U.S. dollars at the spot rate on the trade date of the purchase or sale. Such an accrual basis taxpayer may recognize exchange gain or loss based on currency fluctuations between the trade date and the settlement date. Any foreign currency gain or loss a U.S. holder realizes will be U.S. source ordinary income or loss.
Net Investment Income Tax. Certain U.S. holders that are individuals, estates or trusts are subject to a 3.8% tax on all or a portion of their “net investment income,” which may include all or a portion of their dividend income and net gains from the disposition of ADSs. Each U.S. holder that is an individual, estate or trust is urged to consult its tax advisors regarding the applicability of the Net Investment Income tax to its income and gains in respect of its investment in the ADSs.
Passive Foreign Investment Company Considerations. If we are a PFIC for any taxable year, a U.S. holder would be subject to special rules generally intended to reduce or eliminate any benefits from the deferral of U.S. federal income tax that a U.S. holder could derive from investing in a non-U.S. company that does not distribute all of its earnings on a current basis.
A corporation organized outside the United States generally will be classified as a PFIC for U.S. federal income tax purposes in any taxable year in which, after applying certain look-through rules with respect to the income and assets of its subsidiaries, either: (i) at least 75% of its gross income is “passive income” or (ii) at least 50% of the average quarterly value of its total gross assets, for which purpose, assuming we are treated as a publicly traded company pursuant to Section 1297(e)(3) of the Code, the total value of our assets may be determined in part by reference to the
209
market value of its ADSs and ordinary shares, which is subject to change) is attributable to assets that produce “passive income” or are held for the production of “passive income.”
Passive income for this purpose generally includes dividends, interest, royalties, rents, gains from commodities and securities transactions, the excess of gains over losses from the disposition of assets which produce passive income, and includes amounts derived by reason of the temporary investment of cash, including the funds raised in offerings of the ADSs. If a non-U.S. corporation owns directly or indirectly at least 25% by value of the stock of another corporation, the non-U.S. corporation is treated for purposes of the PFIC tests as owning its proportionate share of the assets of the other corporation and as receiving directly its proportionate share of the other corporation’s income for purposes of the PFIC tests. If we are classified as a PFIC for any year with respect to which a U.S. holder owns ADSs, we will continue to be treated as a PFIC with respect to such U.S. holder in all succeeding years during which the U.S. holder owns ADSs, regardless of whether we continue to meet the tests described above.
Whether we are a PFIC for any taxable year will depend on the composition of our income and the projected composition and estimated fair market values of our assets in each year, and because this is a factual determination made annually after the end of each taxable year, there can be no assurance that we will not be considered a PFIC for any taxable year. The market value of our assets may be determined in large part by reference to the market price of the ADSs and our ordinary shares, which is likely to fluctuate. Based on the foregoing, with respect to our 2022 taxable year, we anticipate that we will be a PFIC based upon the expected value of our assets, including any goodwill, and the expected composition of our income and assets, however, as previously mentioned, we cannot provide any assurances regarding our PFIC status for the current, prior or future taxable years.
Because we believe we were a PFIC for the 2022 taxable year, unless you make one of the elections described below, a special tax regime will apply to both (a) any “excess distribution” by us to you (generally, your ratable portion of distributions in any year which are greater than 125% of the average annual distribution received by you in the shorter of the three preceding years or your holding period for the ADSs) and (b) any gain realized on the sale or other disposition of the ADSs. Under this regime, any excess distribution and realized gain will be treated as ordinary income and will be subject to tax as if (a) the excess distribution or gain had been realized ratably over your holding period, (b) the amount deemed realized in each year had been subject to tax in each year of that holding period at the highest marginal rate for such year (other than income allocated to the current period or any taxable period before we became a PFIC, which would be subject to tax at the U.S. holder’s regular ordinary income rate for the current year and would not be subject to the interest charge discussed below), and (c) the interest charge generally applicable to underpayments of tax had been imposed on the taxes deemed to have been payable in those years. In addition, dividend distributions made to you will not qualify for the lower rates of taxation applicable to long-term capital gains discussed above under “—Distributions.”
Certain elections exist that would result in an alternative treatment (such as mark-to-market treatment) of the ADSs. If a U.S. holder makes the mark-to-market election, the U.S. holder generally will recognize as ordinary income any excess of the fair market value of the ADSs at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ADSs over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). If a U.S. holder makes the election, the U.S. holder’s tax basis in the ADSs will be adjusted to reflect these income or loss amounts. Any gain recognized on the sale or other disposition of ADSs in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). The mark-to-market election is available only if we are a PFIC and the ADSs are “regularly traded” on a “qualified exchange.” The ADSs will be treated as “regularly traded” in any calendar year in which more than a de minimis quantity of the ADSs are traded on a qualified exchange on at least 15 days during each calendar quarter (subject to the rule that trades that have as one of their principal purposes the meeting of the trading requirement as disregarded). Nasdaq is a qualified exchange for this purpose and, consequently, if the ADSs are regularly traded, the mark-to-market election will be available to a U.S. holder.
Because we believe we were a PFIC for the 2022 taxable year, we must generally continue to be treated as a PFIC by that U.S. holder for all succeeding years during which the U.S. holder holds the ADSs, unless we cease to meet the
210
requirements for PFIC status and the U.S. holder makes a “deemed sale” election with respect to the ADSs. If such election is made, the U.S. holder will be deemed to have sold the ADSs it holds at their fair market value on the last day of the last taxable year in which we qualified as a PFIC, and any gain from such deemed sale would be subject to the consequences applicable to sales of PFIC shares described above. After the deemed sale election, the U.S. holder’s ADSs with respect to which the deemed sale election was made will not be treated as shares in a PFIC unless we subsequently become a PFIC.
The tax consequences that apply if we are a PFIC would also be different from those described above if a U.S. holder were able to make a valid “qualified electing fund,” or QEF, election. Because we were a PFIC for the 2022 taxable year, we will provide information necessary for our shareholders to make a QEF election with respect to us for the 2022 taxable year and expect to provide such information for any subsequent year if we believe we are a PFIC. We will provide such information on our website. A U.S. holder that makes a QEF election with respect to our shares is required to include a pro rata share of our income on a current basis, whether or not we make distributions. U.S. holders should consult their tax advisors to determine whether any of these above elections would be available and if so, what the consequences of the alternative treatments would be in their particular circumstances.
Because we believe we were a PFIC for the 2022 taxable year, the general tax treatment for U.S. holders described in this section would apply to indirect distributions and gains deemed to be realized by U.S. holders in respect of any of our subsidiaries that also may be determined to be PFICs (“lower-tier PFICs”).
If a U.S. holder owns ADSs during any taxable year in which we are a PFIC, the U.S. holder generally will be required to file an IRS Form 8621 (Information Return by a Shareholder of a Passive Foreign Investment Company or Qualified Electing Fund) with respect to the company and any lower-tier PFICs, generally with the U.S. holder’s federal income tax return for that year.
The U.S. federal income tax rules relating to PFICs are complex. Prospective U.S. investors are urged to consult their own tax advisers with respect to ownership and disposition of the ADSs, the consequences to them of an investment in a PFIC, any elections available with respect to the ADSs and the IRS information reporting obligations with respect to ownership and disposition of the ADSs.
Backup Withholding and Information Reporting. U.S. holders generally will be subject to information reporting requirements with respect to dividends on ADSs and on the proceeds from the sale, exchange or disposition of ADSs that are paid within the United States or through U.S.-related financial intermediaries, unless the U.S. holder is an “exempt recipient.” In addition, U.S. holders may be subject to backup withholding on such payments, unless the U.S. holder provides a correct taxpayer identification number and a duly executed IRS Form W-9 or otherwise establishes an exemption. Backup withholding is not an additional tax, and the amount of any backup withholding will be allowed as a credit against a U.S. holder’s U.S. federal income tax liability and may entitle such holder to a refund, provided that the required information is timely furnished to the IRS.
Foreign Asset Reporting. Certain U.S. holders who are individuals and certain entities controlled by individuals may be required to report information relating to an interest in the ADSs, subject to certain exceptions (including an exception for shares held in accounts maintained by U.S. financial institutions) by filing IRS Form 8938 (Statement of Specified Foreign Financial Assets) with their federal income tax return. U.S. holders are urged to consult their tax advisors regarding their information reporting obligations, if any, with respect to their ownership and disposition of the ADSs.
THE DISCUSSION ABOVE IS A GENERAL SUMMARY. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A PROSPECTIVE INVESTOR. EACH PROSPECTIVE INVESTOR IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT OF AN INVESTMENT IN ADSS IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
211
Belgian tax consequences
The following paragraphs are a summary of material Belgian tax consequences of the ownership and disposal by an investor of ADSs representing our shares. The summary is based on laws, treaties and regulatory interpretations in effect in Belgium on the date of this Form 20-F, all of which are subject to change, including changes that could have retroactive effect.
The summary only discusses Belgian tax aspects which are relevant to U.S. holders of ADSs representing our shares, or “Holders”. This summary does not address Belgian tax aspects which are relevant to persons who are fiscally resident in Belgium or who are engaged in a business in Belgium through a permanent establishment or a fixed base in Belgium to which the ADSs are effectively connected.
This summary does not purport to be a description of all of the tax consequences of the ownership and disposal of ADSs representing our shares, and does not take into account the specific circumstances of any particular investor, some of which may be subject to special rules, or the tax laws of any country other than Belgium. This summary does not describe all tax consequences of investors that are subject to special rules, such as banks, insurance companies, collective investment undertakings, dealers in securities or currencies, persons that hold, or will hold, ADSs representing our shares as a position in a straddle, share-repurchase transaction, conversion transactions, synthetic security or other integrated financial transactions. Investors should consult their own advisers regarding the tax consequences of an investment in ADSs representing our shares in the light of their particular circumstances, including the effect of any state, local or other national laws.
In addition to the assumptions mentioned above, it is also assumed in this discussion that for purposes of the domestic Belgian tax legislation, the owners of ADSs will be treated as the owners of the ordinary shares represented by such ADSs. However, the assumption has not been confirmed by or verified with the Belgian Tax Authorities.
Dividend withholding tax
For Belgian income tax purposes, the gross amount of all benefits paid on or attributed to the ordinary shares represented by the ADSs is generally treated as a dividend distribution. By way of exception, the repayment of fiscal capital carried out in accordance with the Belgian Companies and Associations Code is not treated as a dividend distribution to the extent that such repayment is imputed to the fiscal capital (subject to certain conditions and the pro rata rule, see below). This fiscal capital includes, in principle, the actual paid-up statutory share capital and, subject to certain conditions, the paid-up issuance premiums and the cash amounts subscribed to at the time of the issue of profit sharing certificates. However, for any decision of capital reduction, in accordance with the Belgian Companies and Associations Code, the amount of the capital reduction will be deemed to be derived proportionally (a) from the fiscal capital of our company, on the one hand and (b) on the other hand, from certain reserves (i.e., and in the following order: (i) certain taxed reserves incorporated in the capital of our company, (ii) certain taxed reserves not incorporated into the capital of our company and (iii) certain tax-exempt reserves incorporated into the capital of our company). Only the part of the capital reduction that is deemed to be paid out of the fiscal capital may, subject to certain conditions, not be considered as a dividend distribution for Belgian tax purposes. The part of the capital reduction that is deemed to be derived from the abovementioned taxed (irrespective of whether they are incorporated into the capital) and/or tax-exempt reserves incorporated into the capital will be treated as a dividend distribution from a tax perspective and be subject to Belgian withholding tax, if applicable. Such portion is determined on the basis of the ratio of the taxed reserves (except for the legal reserve up to the legal minimum and certain unavailable retained earnings) and the tax-exempt reserves incorporated into the capital (with a few exceptions) over the aggregate of such reserves and the fiscal capital.
As a general rule, a withholding tax of 30% is levied on the gross amount of dividends paid on or attributed to the ordinary shares represented by the ADSs, subject to such relief as may be available under applicable domestic or tax treaty provisions. In case of a redemption by us of our own shares represented by ADSs, the redemption distribution (after deduction of the portion of fiscal capital represented by the redeemed shares) will be treated as a dividend which in principle is subject to the withholding tax of 30%, subject to such relief as may be available under applicable domestic or tax treaty provisions. No Belgian withholding tax will be triggered if this redemption is carried out on a stock exchange
212
and meets certain conditions. In case of a liquidation of our company, any amounts distributed in excess of the fiscal capital will also be treated as a dividend, and will in principle be subject to a 30% withholding tax, subject to such relief as may be available under applicable domestic or tax treaty provisions. For non-residents the dividend withholding tax, if any, will be the only tax on dividends in Belgium, unless the non-resident is engaged in a business in Belgium through a fixed base in Belgium or a Belgian permanent establishment to which the ADSs are effectively connected. Prospective Holders should consult their own advisors regarding the tax consequences in case the ADSs are effectively connected to a fixed base or a permanent establishment in Belgium.
Relief of Belgian Dividend Withholding Tax
Under the U.S.-Belgium Tax Treaty, under which we are entitled to benefits accorded to residents of Belgium, there is a reduced Belgian withholding tax rate of 15% on dividends paid by us to a U.S. resident which beneficially owns the dividends and is entitled to claim the benefits on the U.S.-Belgium Tax Treaty under the limitation on benefits article included in the U.S.-Belgium Tax Treaty, or “Qualifying Holders”.
If such Qualifying Holder is a company that owns directly at least 10% of our voting stock, the Belgian withholding tax rate is further reduced to 5%. No withholding tax is however applicable, if the Qualifying Holder does not carry on a business in Belgium through a permanent establishment situated therein, with which our shares, represented by the ADSs, are effectively connected and is either of the following:
| ● | a company that is a resident of the United States that has directly owned our shares, represented by the ADSs, representing at least 10% of our capital for a twelve-month period ending on the date the dividend is declared, or |
| ● | a pension fund in the meaning of Article 3, (1), (k) of the U.S.-Belgium Tax Treaty, that is a resident of the United States, provided that such dividends are not derived from the carrying on of a business by the pension fund or through an associated enterprise. |
Under the normal procedure, we or our paying agent must withhold the full Belgian withholding tax, without taking into account the reduced U.S.- Belgium Tax Treaty rate. Qualifying Holders may then make a claim for reimbursement for amounts withheld in excess of the rate defined by the U.S.- Belgium Tax Treaty. The reimbursement form (Form 276 Div-Aut.) can be obtained as follows:
| ● | by letter from KMO Centrum Specifieke Materies - Team 6 - Kruidtuinlaan 50, mailbox 3429, B-1000 Brussels, Belgium; |
| ● | by telephone at +32 (0)2 572 57 57 + call code 17486; |
| ● | via e-mail at foreigners.team6@minfin.fed.be; or at |
| ● | https://finance.belgium.be/en/private-individuals/international/reimbursement-withholding-tax-movable-property. |
The reimbursement form is to be sent to KMO Centrum Specifieke Materies - Team 6 - Kruidtuinlaan 50, mailbox 3429, B-1000 Brussels, Belgium as soon as possible and in each case within a term of five years starting from the first of January of the year the withholding tax was paid to the Belgian Treasury.
Qualifying Holders may also, subject to certain conditions, obtain the reduced U.S.-Belgium Tax Treaty rate at source. Qualifying Holders should deliver a duly completed Form 276 Div-Aut., accompanied by a duly stamped and signed form 6166 to us no later than ten days after the date on which the dividend has been paid or attributed (whichever comes first).
213
Additionally, pursuant to Belgian domestic tax law, dividends paid or attributed to non-resident individuals who do not use our shares represented by ADSs in the exercise of a professional activity may be exempt from non-resident individual income tax up to the amount of 800 EUR (for income year 2023). Consequently, if Belgian withholding tax has been levied on dividends paid or attributed to our shares represented by ADSs, such Belgian non-resident may request in his or her non-resident income tax return that any Belgian withholding tax levied on dividends up to the amount of EUR 800 (for income year 2023) be credited and, as the case may be, reimbursed. However, if no Belgian non-resident income tax return has to be filed by the non-resident individual, any Belgian withholding tax levied on dividends up to such an amount could in principle be reclaimed by filing a request thereto addressed to the designated tax official. Such a request has to be made at the latest on December 31 of the calendar year following the calendar year in which the relevant dividend(s) have been received, together with an affidavit confirming the non-resident individual status and certain other formalities which are determined by Royal Decree. For the avoidance of doubt, all dividends paid or attributed to the non-resident individual are taken into account to assess whether the maximum amount of EUR 800 (for income year 2023) is reached (and hence not only the amount of dividends paid or attributed on our shares represented by ADSs).
Additionally, pursuant to Belgian domestic tax law, dividends distributed to corporate Holders beneficially owning the dividends that qualify as a parent company will be exempt from Belgian withholding tax, provided that the shares which are represented by ADSs held by the Holder amount to at least 10% of our share capital upon payment or attribution of the dividends and such minimum participation is held or will be held during an uninterrupted period of at least one year, and provided the general and specific anti-abuse provisions do not apply. A Holder-beneficial owner qualifies as a parent company (i) if it has a legal form similar to the ones listed in the annex to the Parent-Subsidiary Directive as amended from time to time, (ii) if it is considered to be a tax resident according to the laws of the United States of America and the U.S.-Belgium Tax Treaty, and (iii) if it is subject to a tax similar to the Belgian corporate income tax without benefiting from a tax regime that derogates from the ordinary tax regime. Please note that this withholding tax exemption will not be applicable to dividends which are connected to an arrangement or a series of arrangements ("rechtshandeling of geheel van rechtshandelingen"/"acte juridique ou un ensemble d'actes juridiques") for which the Belgian Tax Administration, taking into account all relevant facts and circumstances, has proven, unless evidence to the contrary, that this arrangement or this series of arrangements is not genuine ("kunstmatig"/"non authentique") and has been put in place for the main purpose or one of the main purposes of obtaining the dividend received deduction, the above dividend withholding tax exemption or one of the advantages of the Parent-Subsidiary Directive in another EU Member State. An arrangement or a series of arrangements is regarded as not genuine to the extent that they are not put into place for valid commercial reasons which reflect economic reality.
In order to benefit from this exemption, the Holder must provide us or our paying agent with a certificate confirming its qualifying status and the fact that it satisfies the abovementioned conditions.
If the Holder holds the above-mentioned minimum participation for less than one year, at the time the dividends are paid on or attributed to the shares represented by the ADSs, we must levy the withholding tax but we do not need to transfer it to the Belgian Treasury provided that the Holder provides us or our paying agent, at the latest upon the attribution of the dividends, its qualifying status, with a certificate confirming – in addition to its qualifying status and the fulfilment of the relevant conditions – , the date as of which the Holder has held the minimum participation, and the Holder’s commitment to hold it for an uninterrupted period of at least one year. The Holder must also inform us or our paying agent when the one-year period has expired or if its shareholding drops below 10% of our share capital before the end of the one-year holding period. Upon satisfying the one-year shareholding requirement, the dividend withholding tax which was temporarily withheld will be paid to the Holder.
Dividends paid or attributed to a corporate Holder beneficially owning the dividend income will also be exempt from withholding tax, provided that (i) the Holder is subject to corporate income tax or a similar tax without benefiting from a tax regime that derogates from the ordinary tax regime, (ii) upon the date of payment or attribution of the dividends, the Holder holds a participation in us with an acquisition value of at least € 2,500,000, but representing less than 10% of our capital, (iii) the dividends relate to shares represented by the ADSs which are or will be held in full ownership for at least one year without interruption, (iv) the Holder has a legal form similar to the ones listed in the annex to the Parent-Subsidiary Directive, as amended from time to time and (v) the general anti-abuse provision is not be applicable. The exemption from withholding tax is only applicable to the extent that the ordinary Belgian withholding
214
tax, which would be due in the absence of said exemption, is, in principle, neither creditable nor reimbursable in the hands of the Holder. The latter condition is arguably contrary to EU law based on a recent judgement of the Court of Justice of the EU dated June 16, 2022 (C-572/20). Shareholders are encouraged to consult their own tax advisor to determine whether they can invoke this argument if need be. In order to benefit from the above exemption of withholding tax, the corporate Holder must provide us or our paying agent with a certificate confirming (i) that it has a legal form as described above, (ii) that it is subject to corporate income tax or a similar tax without benefiting from a tax regime that deviates from the ordinary domestic tax regime, (iii) that it holds a participation of less than 10% in our capital, but with an acquisition value of at least € 2,500,000 upon the date of payment or attribution of the dividend, (iv) that the dividends relate to shares in us represented by the ADSs which it has held or will hold in full legal ownership for an uninterrupted period of at least one year, (v) to which extent it could in principle, in case this exemption would not exist, credit the levied Belgian withholding tax or obtain a reimbursement thereof according to the legal provisions applicable on December 31st of the year preceding the year of the payment or attribution of the dividends, and (vi) its full name, legal form, address and fiscal identification number, if applicable. Furthermore, we or our paying agent may also request confirmation from the Holder that the Holder commits to keep the participation with an acquisition value of at least € 2,500,000 until the completion of the minimum holding period of one year and that the Holder immediately notifies us or our paying agent of the completion of said one year holding period.
Withholding tax is also not applicable, pursuant to Belgian domestic tax law, on dividends paid to a U.S. pension fund which satisfies the following conditions:
| (i) | to be a legal entity with separate legal personality and fiscal residence in the United States, |
| (ii) | whose corporate purpose consists solely in managing and investing funds collected in order to pay legal or complementary pensions, |
| (iii) | whose activity is limited to the investment of funds collected in the exercise of its statutory mission, without any profit making aim, |
| (iv) | which is exempt from income tax in the United States, and |
| (v) | provided that it (save in certain particular cases as described in Belgian law) is not contractually obligated to redistribute the dividends to any ultimate beneficiary of such dividends for whom it would manage our shares or ADSs, nor obligated to pay a manufactured dividend with respect to our shares or ADSs under a securities borrowing transaction. |
The exemption will only apply if the U.S. pension fund provides an affidavit confirming that it is the full legal owner or usufruct holder of our shares or ADSs and that the above conditions are satisfied. The organization must then forward that affidavit to us or our paying agent.
Please note that the above withholding tax exemption will not be applicable to dividends which are connected to an arrangement or a series of arrangements ("rechtshandeling of geheel van rechtshandelingen"/"acte juridique ou un ensemble d'actes juridiques") for which the Belgian Tax Administration, taking into account all relevant facts and circumstances, has proven, unless evidence to the contrary, that this arrangement or this series of arrangements is not genuine ("kunstmatig"/"non authentique") and has been put in place for the main purpose or one of the main purposes of obtaining the dividend received deduction, the above dividend withholding tax exemption or one of the advantages of the Parent-Subsidiary Directive in another EU Member State. An arrangement or a series of arrangements is regarded as not genuine to the extent that it is not put into place for valid commercial reasons which reflect economic reality. There is a rebuttable presumption that dividends are deemed to be connected to an artificial transaction if the shares have not been held by the pension fund in full legal ownership for an uninterrupted period of at least 60 days within 15 days from the date of the attribution or payment of the income.
Prospective Holders are encouraged to consult their own tax advisers to determine whether they qualify for an exemption or a reduction of the withholding tax rate upon payment of dividends and, if so, the procedural requirements for obtaining such an exemption or a reduction upon the payment of dividends or making claims for reimbursement.
215
Capital gains and losses
Pursuant to the U.S.-Belgium Tax Treaty, capital gains and/or losses realized by a Qualifying Holder entitled to claim the benefits of the U.S.- Belgium Tax Treaty under the limitation of benefits article in the U.S.-Belgium Tax Treaty from the sale, exchange or other disposition of our shares represented by ADSs are exempt from tax in Belgium.
Capital gains realized on our shares represented by ADSs by a corporate Holder who is not such a Qualifying Holder are generally not subject to taxation in Belgium unless these ADSs are held in connection with a business conducted in Belgium through a Belgian permanent establishment or a fixed place in Belgium to which the ADSs are effectively connected (in which case a 25% or 0% tax on the capital gain may apply, depending on the particular circumstances). Capital losses are generally not tax deductible in Belgium.
Private individual Holders who are not such Qualifying Holders and who are holding our shares represented by ADSs as a private investment and within the bounds of the normal management of one’s private estate will, as a rule, not be subject to tax in Belgium on any capital gains arising out of a disposal of our shares represented by ADSs.
Capital losses will, as a rule, not be tax deductible in Belgium. Capital gains realized by a Holder upon the redemption of shares represented by ADSs or upon our liquidation will generally be taxable as a dividend. See “— Dividend Withholding Tax” above.
Estate and gift tax
There is no Belgian estate tax on the transfer of our shares represented by ADSs on the death of a Belgian non-resident. Donations of our shares represented by ADSs made in Belgium may or may not be subject to gift tax depending on the modalities under which the donation is carried out.
Belgian Tax on Securities Accounts
Parliament adopted on February 11, 2021 a bill submitted by the Belgian federal government introducing an annual tax on securities accounts. This bill was published in the Belgian State Gazette on February 25, 2021 and entered into force on February 26, 2021.
An annual tax of 0.15% is levied on securities accounts of which the average value of the taxable financial instruments (covering, amongst others, financial instruments such as our shares represented by ADSs) held thereon during a reference period of twelve consecutive months (in principle) starting on October 1 and ending on September 30 of the subsequent year, would exceed EUR 1 million. The first reference period begins on February 26, 2021 and ends on September 30, 2021.
The amount of the tax due is limited to 10% of the difference between said average value of the taxable financial instruments, and the threshold of EUR 1 million.
The tax targets, among others, securities accounts held by non-resident individuals, companies and legal entities with a financial intermediary established or located in Belgium.
A financial intermediary is defined as (i) the National Bank of Belgium, the European Central Bank and foreign central banks performing similar functions, (ii) a central securities depository included in article 198/1, §6, 12° of the Belgian Income Tax Code, (iii) a credit institution or a stockbroking firm as defined by Article 1, §3 of the Law of April 25, 2014 on the status and supervision of credit institutions and investment companies and (vi) the investment companies as defined by Article 3, §1 of the Law of October 25, 2016 on access to the activity of investment services and on the legal status and supervision of portfolio management and investment advice companies, which are, pursuant to national law, admitted to hold financial instruments for the account of customers.
216
There are various exemptions, such as securities accounts (in)directly held by non-residents for their own account at central securities depositories or at a depositary bank accredited by the National Bank of Belgium. This exemption is subject to the condition that the securities accounts are not attributable to a Belgian branch of the non-residents.
Belgian tax on stock exchange transactions
The purchase and the sale and any other acquisition or transfer for consideration by a Holder of existing shares represented by ADSs (secondary market transactions) is subject to the Belgian tax on stock exchange transactions (“taks op de beursverrichtingen” / “taxe sur les opérations de bourse”) if it is entered into or carried out in Belgium through a professional intermediary. The tax on stock exchange transactions is not due upon the issuance of new shares represented by ADSs (primary market transactions). The tax on stock exchange transactions is levied at a rate of 0.35% of the purchase/sales price, capped at € 1,600 per transaction and per party. A separate tax is due by each party to any such transaction, and both taxes are in principle collected by the professional intermediary.
Belgian non-residents who purchase or otherwise acquire or transfer, for consideration, existing shares represented by ADSs in Belgium for their own account through a professional intermediary may be exempt from the stock exchange tax if they deliver a certificate to the financial intermediary in Belgium confirming their non-resident status.
In addition to the above, no tax on stock exchange transactions is due on transactions entered into by the following parties, provided they are acting for their own account: (i) professional intermediaries described in Article 2, 9° and 10° of the Law of August 2, 2002, (ii) insurance companies described in Article 2, §1 of the Law of July 9, 1975 (as replaced by Article 5 of the Law of March 13, 2016 on the status and supervision of insurance and reinsurance undertakings), (iii) professional retirement institutions referred to in Article 2, §1 of the Law of October 27, 2006 relating to the control of professional retirement institutions, (iv) collective investment institutions, or (v) regulated real estate companies, (vi) the aforementioned non-residents (upon delivery of a certificate of non-residency in Belgium).
No stock exchange tax will thus be due by Holders on the subscription, purchase or sale of existing shares represented by ADSs, if the Holders are acting for their own account. In order to benefit from this exemption, the Holders must deliver a certificate to their financial intermediary in Belgium confirming their non-resident status for Belgian tax purposes.
The European Commission has published a proposal for a Directive for a common financial transactions tax (the “FTT”). The proposal currently stipulates that once the FTT enters into force, the participating Member States shall not maintain or introduce taxes on financial transactions other than the FTT (or VAT as provided in the Council Directive 2006/112/EC of November 28, 2006 on the common system of value added tax). For Belgium, the tax on stock exchange transactions should thus be abolished once the FTT enters into force. The proposal is still subject to negotiation between the participating Member States and therefore may be changed at any time.
Common Reporting Standard
Following recent international developments, the exchange of information is governed by the Common Reporting Standard (“CRS”). On May 16, 2023, the total of jurisdictions that have signed the multilateral competent authority agreement (“MCAA”) amounts to 120. The MCAA is a multilateral framework agreement to automatically exchange financial and personal information, with the subsequent bilateral exchanges coming into effect between those signatories that file the subsequent notifications.
Under CRS, financial institutions resident in a CRS country are required to report, according to a due diligence standard, financial information with respect to reportable accounts, which includes interest, dividends, account balance or value, income from certain insurance products, sales proceeds from financial assets and other income generated with respect to assets held in the account or payments made with respect to the account. Reportable accounts include accounts held by individuals and entities (which include trusts and foundations) with fiscal residence in another CRS country. The standard includes a requirement to look through passive entities to report on the relevant controlling persons.
217
On December 9, 2014, EU Member States adopted Directive 2014/107/EU on administrative cooperation in direct taxation (“DAC2”), which provides for mandatory automatic exchange of financial information as foreseen in CRS. DAC2 amends the previous Directive on administrative cooperation in direct taxation, Directive 2011/16/EU and replaces the EC Council Directive 2003/48EC on the taxation of savings income (commonly referred to as the “Savings Directive”) as from January 1, 2016. Austria has been nonetheless allowed to exchange information under DAC2 as from January 1, 2017.
On May 27, 2015, Switzerland signed an agreement with the European Union in order to implement, as from January 1, 2017, an automatic exchange of information based on the CRS. This new agreement will replace the agreement on the taxation of savings that entered into force in 2005. As from January 1, 2017, financial institutions in the EU and Switzerland apply the due diligence procedures envisaged under the new agreement to identify customers who are reportable persons, i.e., for Switzerland residents of any EU Member State. This data was exchanged for the first time in autumn 2018.
As a result of the Law of December 16, 2015, the mandatory automatic exchange of information applies in Belgium (i) as of income year 2016 (first information exchange in 2017) towards the EU Member States (including Austria, irrespective of the fact that the automatic exchange of information by Austria towards other EU Member States is only foreseen as of income year 2017), (ii) as of income year 2014 (first information exchange in 2016) towards the US and (iii), with respect to any other non-EU States that have signed the MCAA, as of income year 2016 (first information exchange in 2017) for a first list of 18 countries, as of income year 2017 (first information exchange in 2018) for a second list of 44 countries, as of income year 2018 (first information exchange in 2019) for a third list of 1 country, as of income year 2019 (first information exchange in 2020) for a fourth list of 6 countries, as of income year 2020 (first information exchange in 2021) for a list of two countries and as of income year 2021 (first information exchange in 2022) for a list of 3 countries.
Investors who are in any doubt as to their position should consult their professional advisers.
F. Dividends and paying agents
Not applicable.
G. Statement by experts
Not applicable.
H. Documents on display
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and under those requirements will file reports with the SEC. Those reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an annual report containing financial statements that have been examined and reported on, with and opinion expressed by an independent registered public accounting firm.
We maintain a corporate website at www.glpg.com. We intend to post a link to our annual report on Form 20-F as filed with the SEC on our website promptly following it being filed with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this annual report. We have included our website address in this annual report solely as an inactive textual reference.
You may also review a copy of this annual report, including exhibits and any schedule filed herewith, and obtain copies of such materials at prescribed rates, at the SEC’s Public Reference Room in Room 1580, 100 F Street, NE,
218
Washington, D.C. 20549-0102. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as Galapagos NV, that file electronically with the SEC.
With respect to references made in this annual report to any contract or other document of Galapagos NV, such references are not necessarily complete and you should refer to the exhibits attached or incorporated by reference to this annual report for copies of the actual contract or document..
I. Subsidiary information
Not applicable.
Item 11 Quantitative and qualitative disclosures about market risk
Our financial risks are managed centrally. Our finance department coordinates the access to national and international financial markets and considers and manages continuously the financial risks concerning our activities. These relate to the following financial markets risks: credit risk, liquidity risk, currency and interest rate risk. Our interest rate risk is limited because we have no financial debt and have a strong cash position. In case of decreasing interest rates we will face a reinvestment risk on our strong cash position. We do not buy or trade financial instruments for speculative purposes. For additional information on general risk factors, please see the section of this annual report titled “Item 3.D.—Risk Factors.”
Liquidity risk
Our cash and cash equivalents and current financial investments amounted to respectively €166.8 million and €3,517.7 million on December 31, 2023. Cash used in operating activities amounted to €406.0 million for the year ended December 31, 2023. Management forecasts our liquidity requirements to ensure that there is sufficient cash to meet operational needs. Based upon our current expected level of operating expenditures and our existing cash and cash equivalents, we believe that we will be able to fund our operating expenses and capital expenditure requirements at least for a period of 12 months. We have no credit lines. Such forecasting is based on realistic assumptions with regards to product sales, royalties, milestone and upfront payments to be received, taking into account our past track record, including the assumption that not all new projects that are being planned will be realized.
All our cash and cash equivalents have only an insignificant liquidity risk as they are all convertible upon a maximum three month notice period and without incurring a significant penalty.
Credit risk
The term “credit risk” refers to the risk that a counterparty will default on its contractual obligations resulting in financial loss.
We grant credit to our clients in the framework of our normal business activities. Usually, we require no pledge or other collateral to cover the amounts due. Management continuously evaluates the client portfolio for creditworthiness. All receivables are considered collectable.
We did not account for a provision for expected credit losses relating to our trade and other receivables given that there is no history of material credit losses, nor does forward looking information reveals any potential risk and due to the high quality nature of our customers.
219
Aging balance of receivables that are due, but are still considered collectable:
December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
(Euro, in thousands) | |||||||||
60 - 90 days | € | 3 | € | 424 | € | 141 | |||
90 - 120 days | 3 | 208 | 92 | ||||||
more than 120 days | € | 117 | € | 473 | € | 113 | |||
Our cash and cash equivalents are invested primarily in current, notice and term accounts. For banks and financial institutions, only independently rated parties with a minimum rating of ‘A’ are accepted at the beginning of the term. Our current financial investments are also kept within different financial institutions and include term deposits, money market funds and treasury bills with an AAA rating. The money market funds are invested in a well-diversified portfolio of highly rated assets.
Interest rate risk
The only variable interest-bearing financial instruments are cash and cash equivalents and current financial investments.
Changes in interest rates may cause variations in interest income and expenses resulting from short term interest-bearing assets. Management does not expect the short term interest rates to decrease significantly in the immediate foreseeable future, which limits the interest exposure on our cash and cash equivalents and current financial investments.
Effect of interest rate fluctuation
A 100 basis point increase in interest rates at balance sheet date would have increased profit or loss, and equity, by approximately €36.8 million (2022: €40.9 million, 2021: €47.0 million); a 100 basis point decrease in interest rates would have decreased profit or loss, and equity, by approximately €36.8 million (2022: €40.9 million, 2021: €47.0 million).
Foreign exchange risk
We are exposed to foreign exchange risk arising from various currency exposures. Our principal functional currency is euro, but we receive payments from our business partners Gilead in U.S. dollars and acquire some consumables and materials in U.S. dollars, Swiss Francs, and GB Pounds.
To limit this risk, we attempt to align incoming and outgoing cash flows in currencies other than the euro. In addition, contracts closed by our different entities are mainly in the functional currencies of that entity, except for the collaboration agreements signed with Gilead for which payments are denominated in U.S. dollars.
The exchange rate risk in case of a 10% change in the exchange rate amounts to:
December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
Net book value | (Euro, in thousands) | ||||||||
Increase in Euros - U.S. Dollars | € | (78,013) | € | (85,140) | € | (83,996) | |||
Increase in Euros - GB Pounds | 666 | 960 | 1,093 | ||||||
Increase in Euros - CH Francs | 385 | 557 | 233 | ||||||
The exchange rate risk on the U.S. dollar is primarily related to our cash and cash equivalents and current financial investments held in U.S dollars.
220
Capital risk factors
We manage our capital to safeguard that we will be able to continue as a going concern. At the same time, we want to ensure the return to our shareholders through the results from our research and development activities.
Our capital structure consists of current financial investments, cash and cash equivalents, and equity attributed to the holders of our equity instruments, such as capital, reserves and results carried forward, as mentioned in the consolidated statement of changes in equity.
We manage our capital structure and make the necessary adjustments in the light of changes of economic circumstances, the risk characteristics of underlying assets and the projected cash needs of the current research and development activities.
The adequacy of the capital structure will depend on many factors, including scientific progress in the research and development programs, the magnitude of those programs, the commitments to existing and new clinical contract research organizations, the ability to establish new alliance or collaboration agreements, the capital expenditures, market developments and any future acquisition.
Neither we nor any of our subsidiaries are subject to any externally imposed capital requirements, other than those imposed by generally applicable company law requirements.
Item 12 Description of securities other than equity securities
A. Debt securities
Not applicable.
B. Warrants and rights
Not applicable.
C. Other securities
Not applicable.
D. American Depositary Shares
Citibank, N.A., as depositary, registers and delivers ADSs. Each ADS represents one ordinary share (or a right to receive one ordinary share) deposited with Citibank International Limited (located at EGSP 186, 1 North Wall Quay, Dublin 1, Ireland) or any successor, as custodian for the depositary. Each ADS will also represent any other securities, cash or other property which may be held by the depositary in respect of the depositary facility. The depositary’s corporate trust office at which the ADSs are administered is located at 388 Greenwich Street, New York, New York 10013.
A deposit agreement among us, the depositary and the ADS holders sets out the ADS holder rights as well as the rights and obligations of the depositary. New York law governs the deposit agreement and the ADSs. A copy of the deposit agreement is incorporated by reference as an exhibit to this annual report.
221
Fees and charges
Pursuant to the terms of the deposit agreement, the holders of ADSs will be required to pay the following fees:
Service |
| Fees |
● Issuance of ADSs | Up to U.S. $0.05 per ADS issued | |
● Cancellation of ADSs | Up to U.S. $0.05 per ADS canceled | |
● Distribution of cash dividends or other cash distributions | Up to U.S. $0.05 per ADS held | |
● Distribution of ADSs pursuant to stock dividends, free stock distributions or exercise of rights. | Up to U.S. $0.05 per ADS held | |
● Distribution of securities other than ADSs or rights to purchase additional ADSs | Up to U.S. $0.05 per ADS held | |
● ADS Services | Up to U.S. $0.05 per ADS held on the applicable record date(s) established by the depositary |
The holders of ADSs will also be responsible to pay certain fees and expenses incurred by the depositary and certain taxes and governmental charges such as:
| ● | taxes (including applicable interest and penalties) and other governmental charges; |
| ● | the registration fees as may from time to time be in effect for the registration of ordinary shares on the share register and applicable to transfers of ordinary shares to or from the name of the custodian, the depositary or any nominees upon the making of deposits and withdrawals, respectively; |
| ● | certain cable, telex and facsimile transmission and delivery expenses; |
| ● | the expenses and charges incurred by the depositary in the conversion of foreign currency; |
| ● | the fees and expenses incurred by the depositary in connection with compliance with exchange control regulations and other regulatory requirements applicable to ordinary shares, ADSs and ADRs; and |
| ● | the fees and expenses incurred by the depositary, the custodian, or any nominee in connection with the servicing or delivery of deposited property. |
ADS fees and charges payable upon (i) deposit of ordinary shares against issuance of ADSs and (ii) surrender of ADSs for cancellation and withdrawal of ordinary shares are charged to the person to whom the ADSs are delivered (in the case of ADS issuances) and to the person who delivers the ADSs for cancellation (in the case of ADS cancellations). In the case of ADSs issued by the depositary into the Depositary Trust Company, or DTC, or presented to the depositary via DTC, the ADS issuance and cancellation fees and charges may be deducted from distributions made through DTC, and may be charged to the DTC participant(s) receiving the ADSs or the DTC participant(s) surrendering the ADSs for cancellation, as the case may be, on behalf of the beneficial owner(s) and will be charged by the DTC participant(s) to the account(s) of the applicable beneficial owner(s) in accordance with the procedures and practices of the DTC participant(s) as in effect at the time. ADS fees and charges in respect of distributions and the ADS service fee are charged to the holders as of the applicable ADS record date. In the case of distributions of cash, the amount of the applicable ADS fees and charges is deducted from the funds being distributed. In the case of (i) distributions other than cash and (ii) the ADS service fee, holders as of the ADS record date will be invoiced for the amount of the ADS fees and charges and such ADS fees and charges may be deducted from distributions made to holders of ADSs. For ADSs held
222
through DTC, the ADS fees and charges for distributions other than cash and the ADS service fee may be deducted from distributions made through DTC, and may be charged to the DTC participants in accordance with the procedures and practices prescribed by DTC and the DTC participants in turn charge the amount of such ADS fees and charges to the beneficial owners for whom they hold ADSs.
In the event of refusal to pay the depositary fees, the depositary may, under the terms of the deposit agreement, refuse the requested service until payment is received or may set off the amount of the depositary fees from any distribution to be made to the ADS holder. Note that the fees and charges you may be required to pay may vary over time and may be changed by us and by the depositary. You will receive prior notice of such changes. The depositary may reimburse us for certain expenses incurred by us in respect of the ADR program, by making available a portion of the ADS fees charged in respect of the ADR program or otherwise, upon such terms and conditions as we and the depositary agree from time to time.
223
PART II
Item 13 Defaults, dividend arrearages and delinquencies
Not applicable.
Item 14 Material modifications to the rights of security holders and use of proceeds
Not applicable.
Item 15 Controls and procedures
| A. | Disclosure controls and procedures |
Our management, with the participation of our Chief Executive Officer and Chief Financial Officer, has evaluated the effectiveness of the design and operation of our disclosure controls and procedures pursuant to Exchange Act Rule 13a-15(b) as of December 31, 2023. While there are inherent limitations to the effectiveness of any system of disclosure controls and procedures, including the possibility of human error and the circumvention or overriding of the controls and procedures, our disclosure controls and procedures are designed to provide reasonable assurance of achieving their objectives. Based upon our evaluation, as of December 31, 2023, our Chief Executive Officer and Chief Financial Officer have concluded that the disclosure controls and procedures, in accordance with Exchange Act Rule 13a-15(e), (i) are effective at that level of reasonable assurance in ensuring that information required to be disclosed in the reports that are filed or submitted under the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the Commission’s rules and forms, and (ii) are effective at that level of reasonable assurance in ensuring that information to be disclosed in the reports that are filed or submitted under the Exchange Act is accumulated and communicated to the management of our company, including our Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosure.
| B. | Management’s Annual Report on internal control over financial reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting (as such term is defined in Rule 13a-15(f) and Rule 15d-15(f) of the Exchange Act, as amended). Our internal control over financial reporting is a process designed, under the supervision of our Chief Executive Officer and Chief Financial Officer, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of our financial statements for external reporting purposes in accordance with generally accepted accounting principles.
Our internal control over financial reporting includes policies and procedures that pertain to the maintenance of records that, in reasonable detail, accurately and fairly, reflect transactions and dispositions of assets, provide reasonable assurance that transactions are recorded in the manner necessary to permit the preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures are only carried out in accordance with the authorization of our management and directors, and provide reasonable assurance regarding the prevention or timely detection of any unauthorized acquisition, use or disposition of our assets that could have a material effect on our financial statements. Our internal control over financial reporting also includes controls over relevant IT systems that have an impact on financial reporting including accuracy and completeness of our account balances.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect all misstatements. Moreover, projections of any evaluation of the effectiveness of internal control to future periods are subject to a risk that controls may become inadequate because of changes in conditions and that the degree of compliance with the policies or procedures may deteriorate.
Our management has assessed the effectiveness of internal control over financial reporting based on the Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in 2013. Based on this assessment, our management has concluded that our internal control over financial reporting as of December 31, 2023, was effective.
224
The effectiveness of internal control over financial reporting as of December 31, 2023 has also been audited by BDO Bedrijfsrevisoren BV, our independent registered public accounting firm. Their audit report on our internal control over financial reporting, is included below.
| C. | Report of Independent Registered Public Accounting Firm |
Shareholders and Board of Directors
Galapagos NV
Mechelen, Belgium
Opinion on Internal Control over Financial Reporting
We have audited Galapagos’ (the “Company’s”) internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (the “COSO criteria”). In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2023, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the consolidated statement of financial position of the Company as of December 31, 2023, the related consolidated income statement, statement of comprehensive income/loss, changes in equity, and cash flows for the year then ended, and the related notes, and our report dated March 28, 2024 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying “Item 15 , Management’s Annual Report on Internal Control over Financial Reporting”. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit of internal control over financial reporting in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely
225
detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ BDO Bedrijfsrevisoren BV Zaventem, Belgium, March 28, 2024 |
| D. | Changes in internal control over financial reporting |
There were no changes in our internal control over financial reporting identified in connection with the evaluation required by Rule 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the year ended December 31, 2023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 15T. Controls and procedures
Not applicable.
Item 16 Reserved
Not applicable.
Item 16A Audit Committee financial expert
Our Board of Directors has determined that Jérôme Contamine is an Audit Committee financial expert as defined by SEC rules, and has the requisite financial sophistication under the applicable rules and regulations of the Nasdaq Stock Market. Furthermore, Jérôme Contamine is an independent director as such term is defined in Rule 10A-3 under the Exchange Act and under the listing standards of the Nasdaq Stock Market.
Item 16B Code of Conduct
We have established a Code of Conduct to ensure that our members of the Board of Directors and Executive Committee and employees are making ethical and compliant decisions and acting with integrity, ethics and respect for human rights when conducting Galapagos’ business and performing their day-to-day duties. We expect any conflicts of interest to be addressed appropriately, and corruption and fraud prevented. To this end, we give various trainings, including on our Code of Conduct to all our employees and consultants. This year, 94% of our employees completed the Code of Conduct training and we measure against all employees, including those that may be on long term leave or ill. Our Code of Conduct is available on our website at www.glpg.com/governance-information.
At the beginning of 2023, we made some updates to our Code of Conduct to ensure that it continues to reflect who we are as an organization, including an explicit applicability of our Code of Conduct to our suppliers and business partners and more ESG related provisions.
One breach of our Code of Conduct was escalated to the Audit Committee in 2023. Appropriate measures were taken to address this breach.
.
226
Item 16C Principal Accountant fees and services
BDO Bedrijfsrevisoren BV (BDO) was appointed as statutory auditor by the Shareholders’ Meeting held on April 25, 2023, for a term of three years expiring immediately after the Annual Shareholders’ meeting to be held in 2026 which will have decided upon the annual accounts for the financial year to be ended on December 31, 2025.
Deloitte Bedrijfsrevisoren BV (Deloitte) ceased to be our statutory auditor as of the date of the Annual Shareholders’ Meeting held on April 25, 2023.
Our principal accountants billed the following fees to us for professional services rendered in 2023 (BDO) and 2022 (Deloitte):
Year ended December 31, | ||||||
| 2023 |
| 2022 | |||
(Euro, in thousands) | ||||||
Audit Fees | € | 1,124.0 | € | 1,127.1 | ||
Audit-Related Fees | 20.2 | 26.9 | ||||
Tax fees | 68.0 | — | ||||
All Other Fees | 6.6 | 429.5 | ||||
Total | € | 1,218.8 | € | 1,583.5 | ||
“Audit Fees” are the aggregate fees billed for the audit of our annual financial statements. This category also includes services that generally the independent accountant provides, such as consents and assistance with and review of documents filed with the SEC.
“Audit-Related Fees” are the aggregate fees billed for assurance and related services that are reasonably related to the performance of the audit and are not reported under Audit Fees.
“Tax Fees” are the aggregate fees billed for tax assistance relating to personal payroll taxes related to prior year filings.
“All Other Fees” are any additional amounts billed for products and services provided by the principal accountant. For the year ended December 31, 2023, they relate to non-audit fees, in particular related to assurance services in relation to ESG reporting. For the year ended December 31, 2022, they relate to non-audit fees, in particular related to advisory services in relation to IT and quality management.
Audit and non-audit services pre-approval policy
The Audit Committee has responsibility for, among other things, appointing, setting compensation of and overseeing the work of our independent registered public accounting firm, or external auditor. In recognition of these responsibilities, the Audit Committee has adopted a policy governing the pre-approval of all audit and permitted non-audit services performed by our external auditor to ensure that the provision of such services does not impair the external auditor’s independence from us and our management. Unless a type of service to be provided by our external auditor has received general pre-approval from the Audit Committee, it requires specific pre-approval by the Audit Committee. The payment for any proposed services in excess of pre-approved cost levels requires specific pre-approval by the Audit Committee.
Pursuant to its pre-approval policy, the Audit Committee may delegate its authority to pre-approve services to the Chairman of the Audit Committee. The decisions of the Chairman to grant pre-approvals must be presented to the full Audit Committee at its next scheduled meeting. The Audit Committee may not delegate its responsibilities to pre-approve services to the management.
The Audit Committee has considered the non-audit services provided by BDO Bedrijfsrevisoren BV in 2023 and Deloitte Bedrijfsrevisoren BV in 2022 as described above and believes that they are compatible with their independence as our external auditors in those years. In accordance with Regulation S-X, Rule 2-01, paragraph (c)(7)(i), no fees for professional services were approved pursuant to any waivers of the pre-approval requirement.
227
Item 16D Exemptions from the listing standards for Audit Committees
Not applicable.
Item 16E Purchases of equity securities by the issuer and affiliated purchasers
Not applicable.
Item 16F Change in registrant’s certifying accountant
Under the EU Audit Regulation 537/2014 we are required to rotate our external audit firm, which required us to change our external auditor for the financial year ended December 31, 2023. In accordance with Belgian law, the independent auditor of our statutory financial statements is appointed by the shareholders on the proposal of the Board of Directors.
Accordingly, the Board of Directors, upon recommendation of the Audit Committee, proposed that the 2023 Annual Shareholders’ Meeting of the Company appoint BDO Bedrijfsrevisoren BV (BDO), a limited liability company, with its registered office at Da Vincilaan 9/E.6, 1930 Zaventem, Belgium, permanently represented by Ellen Lombaerts, as independent auditor of the Company for the fiscal years 2023, 2024 and 2025. At the Annual Shareholders’ Meeting held on April 25, 2023, our shareholders appointed BDO Bedrijfsrevisoren BV, as proposed by the Board of Directors. Following the appointment of BDO as described above, from the date of the 2023 Annual Shareholders’ Meeting, Deloitte Bedrijfsrevisoren BV (Deloitte) ceased to be the independent auditor of the Company.
The reports of Deloitte on our financial statements for the past two years did not contain an adverse opinion or a disclaimer of opinion and were not qualified or modified as to uncertainty, audit scope or accounting principles.
In connection with the audits of our financial statements for the two fiscal years ended December 31, 2021 and December 31, 2022, and in the subsequent interim period through the date of this Annual Report, (i) there were no disagreements with Deloitte on any matters of accounting principles or practices, financial statement disclosure, or auditing scope and procedures which, if not resolved to the satisfaction of Deloitte would have caused Deloitte to make reference to the matter in their report; and (ii) there were no “reportable events” as defined in Item 16F(a)(1)(v) of Form 20-F.
We have requested Deloitte to furnish a letter addressed to the SEC stating whether it agrees with the foregoing disclosure and, if not, stating the respects in which it does not agree. A copy of that letter dated March 28, 2024 is filed as Exhibit 15.2 to this Form 20-F.
During the two fiscal years ended December 31, 2021 and 2022, and in the subsequent interim period through the date of the appointment of BDO on April 25, 2023, neither the Company nor anyone on its behalf consulted with BDO with respect to either (i) the application of accounting principles to a specified transaction, either completed or proposed, or the type of audit opinion that might be rendered on the Company’s financial statements, and no written report or oral advice was provided by BDO to the Company that BDO concluded was an important factor considered by the Company in reaching a decision as to the accounting, auditing or financial reporting issue, or (ii) any matter that was either the subject of a disagreement (as defined in 16F(a)(1)(iv) of Form 20-F and the related instructions to that Item) or a reportable event (as described in 16F(a)(1)(v) of Form 20-F).
Item 16G Corporate governance
As a Belgian naamloze vennootschap / société anonyme, we are subject to various corporate governance requirements under Belgian law, including the Belgian Code of Companies and Associations (the “Belgian Companies Code”), and the 2020 Belgian Corporate Governance Code (the “2020 Code”). In addition, as we qualify a foreign private issuer listed on the Nasdaq Global Select Market, we will be subject to Nasdaq corporate governance listing standards. However, the Nasdaq Global Select Market’s listing standards include certain accommodations in corporate governance requirements that allow foreign private issuers to follow home country corporate governance practices in lieu of the otherwise applicable corporate governance standards of the Nasdaq Global Select Market, with certain
228
exceptions. We intend to rely on certain exemptions for foreign private issuers, and follow Belgian corporate governance practices in lieu of the Nasdaq corporate governance rules.
In 2019, the Belgian Companies Code was approved by the Belgian Parliament. For existing companies, like us, there was in principle a transition regime providing for a staggered applicability of the new provisions. However, certain parts of the (new) Belgian Companies Code already applied to Galapagos as of January 1, 2020, and the full transition was completed at our Extraordinary Shareholders’ Meeting of April 28, 2020, which resolved to amend our Articles of Association as a consequence of the newly applicable Belgian Companies Code.
The full text of our Articles of Association (coordinated as per March 20, 2023) is made available as an exhibit to this annual report.
From January 1, 2022 until April 26, 2022, Galapagos had a two-tier governance structure as provided by the Belgian Companies Code, with two governance bodies: the Supervisory Board and the Management Board. As from April 26, 2022, Galapagos has adopted a one-tier governance structure as provided by the Belgian Companies Code, with the Board of Directors replacing the (former) Supervisory Board and the Executive Committee replacing the (former) Management Board.
The role of the Board of Directors is to pursue sustainable value creation by the Company, by setting the Company’s strategy, putting in place effective, responsible and ethical leadership and monitoring the Company’s performance. The Board of Directors is the ultimate decision-making body, with the overall responsibility for the management and control of the Company, and is authorized to carry out all actions that are necessary or useful for the realization of the Company’s object, with the exception of those reserved to the Shareholders’ Meeting by applicable law. The Board of Directors also supervises the Executive Committee. The Board of Directors acts as a collegiate body.
The Board of Directors has delegated certain powers to manage the Company to the Executive Committee, led by our Chief Executive Officer. The Executive Committee is responsible and accountable to the Board of Directors for the discharge of its responsibilities. Furthermore, the Board of Directors has delegated the day-to-day management of the Company to one Executive Committee member, i.e. our Chief Executive Officer.
For further information on our governance structure, see the section of this annual report titled “Item 6.A – Directors and Senior Management”
On April 26, 2022, as a consequence of the introduction of a one-tier governance structure at the Company through the amendment of our Articles of Association, Galapagos’ Board of Directors approved an updated Corporate Governance Charter. On March 21, 2023, our Board of Directors approved an amendment to the Corporate Governance Charter. The amended Corporate Governance Charter refers to the establishment of the Management Committee supporting the Executive Committee, allows the same person to be Lead Non-Executive Director and Chairman of the Audit Committee, provides that the Lead Non-Executive Director is member or Chairman of the Nomination Committee, and clarifies that the Lead Non-Executive Director supports the Chairman in ensuring the prevention and managing of conflicts of interest involving potentially a director. On September 19, 2023, the Board of Directors approved another amendment that refers to the establishment of the Science and Development Committee as a specialized Board Committee to provide advice on certain matters to the Board of Directors, and that provides that non-executive Directors may only be natural persons. On December 11, 2023, the Board of Directors approved a further amendment to describe the responsibilities of the Audit Committee and management for overseeing and managing cybersecurity risks. Galapagos’ Corporate Governance Charter is available on our website: www.glpg.com (this website does not form part of this annual report). The Corporate Governance Charter applies in addition to the applicable laws and regulations, Galapagos’ Articles of Association and the corporate governance provisions included in the Belgian Companies Code and the 2020 Code. The Corporate Governance Charter describes the main aspects of corporate governance at Galapagos, including its governance structure, the terms and functioning of the Board of Directors (including its Board Committees), the Executive Committee and the rules of conduct.
The 2020 Code is based on a “comply or explain” principle: Belgian listed companies are expected to follow the 2020 Code, but can deviate from specific provisions and guidelines (though not the principles) provided they disclose the justification for such deviations.
229
For the reporting year beginning on January 1, 2023, our Board of Directors strove to comply with the rules and recommendations of the 2020 Code as much as possible. At the same time, our Board of Directors believes that certain deviations from its provisions were justified in view of our particular situation. Provision 4.19 of the 2020 Code recommends that the Board of Directors should set up a Nomination Committee with the majority of its members comprising independent non-executive directors. In deviation from this provision, the Nomination Committee consisted between January 1, 2023 and March 20, 2023 of one executive Director, one independent non-executive Director and one non-executive Director. The latter (Dr. Rajesh Parekh) no longer qualifies as independent pursuant to article 7:87 of the Belgian Companies Code and provision 3.5 of the 2020 Code given his long tenure at Galapagos. In 2022, the Board felt it was appropriate to appoint him as a member and Chair of the Nomination Committee in view of his experience as former Chairman of the Board, and to ensure a smooth transition to the new Chair, after the implementation of the one-tier governance model. Effective as of March, 21 2023, Dr. Elisabeth Svanberg was appointed as member and Chair of the Nomination Committee, replacing Dr. Rajesh Parekh.
Differences between our corporate governance practices and the listing rules of the Nasdaq stock market
The following is a summary of the significant ways in which our corporate governance practices differ from those required by the Nasdaq Listing Rules with which we are not required to comply:
| ● | Quorum At Shareholders’ Meetings. Nasdaq Stock Market Listing Rule 5620(c) requires that for any shareholders’ meeting, the quorum must be no less than 33% (1/3) of the outstanding ordinary shares. There is no quorum requirement generally applicable to our Shareholders’ Meetings pursuant to the Belgian Companies Code and/or our Articles of Association, except as provided for by Belgian law in relation to decisions regarding certain matters, e.g. amendment of the Articles of Association. |
| ● | Committees. We have opted out of (a) Nasdaq Stock Market Listing Rule 5605(d)(2) which requires that compensation of officers must be determined by, or recommended to, the Board of Directors for determination, either by a majority of the independent directors, or a compensation committee comprised solely of independent directors, and (b) Nasdaq Stock Market Listing Rule 5605(e) which requires that director nominees be selected, or recommended for selection, either by a majority of the independent directors or a nominations committee comprised solely of independent Directors. Under Belgian law, we are not subject to such composition requirements. Pursuant to Article 7:100 of the Belgian Companies Code and the rules and recommendations of the 2020 Code, we are required to set up a Remuneration Committee within our Board of Directors, consisting of a majority of independent members of our Board of Directors. In addition, the 2020 Code provides that the Board of Directors should set up a Nomination Committee, which can be combined with the Remuneration Committee. Until May 2, 2022, there was a combined Nomination and Remuneration Committee. As from May 2, 2022, our Board of Directors has set up a separate Nomination Committee and Remuneration Committee. Although we have chosen not to comply with Nasdaq Listing Rule 5605(d) regarding the indepence of our Remuneration Committee, all Remuneration Committee members meet the heightened independence requirements under these rules. |
| ● | Executive Session. Nasdaq Stock Market Listing Rule 5605(b)(2) requires that independent directors must have regularly scheduled meetings at which only independent directors are present. There is no corresponding requirement under Belgian law. We do not intend to require the independent Board members to meet separately from the full Board on a regular basis, although the Board of Directors is supportive of its independent members voluntarily arranging to meet separately from the other Board members when and if they wish to do so, and although our Corporate Governance Charter states that as long as the CEO serves as the Chair, the Lead Non-Executive Director shall chair sessions of the non-executive Directors to be held at least one time per year without the presence of the Chair. |
| ● | Committee Charters. Nasdaq Stock Market Listing Rules 5605(c)(1), (d)(1) and (e)(2) require that each Board Committee must have a formal written charter. Pursuant to the 2020 Code, our Board of Directors has drawn up a Corporate Governance Charter including, amongst others, the internal rules with respect to the composition, role and functionning of our Board Committees. |
| ● | Shareholder Approval for Certain Issuances of Securities. Nasdaq Stock Market Listing Rule 5635 requires that a company obtains shareholder approval prior to making certain issuances of securities. Pursuant |
230
| to the Belgian Companies Code and subject to the conditions set forth therein and in our Articles of Association, our Board of Directors is allowed to increase the share capital of Galapagos in one or several times, and to issue new Galapagos’ shares through the use of authorized capital limited to the maximum amount of our share capital. The authorized capital may however not be used for (i) capital increases by contribution in kind exclusively reserved for one of our shareholders holding shares to which more than 10% of the voting rights are attached, (ii) the issuance of shares with multiple voting rights, (iii) the issuance of a new class of securities, or (iv) the issuance of subscription rights intended mainly for one or more specified persons other than our or our subsidiaries’ staff. Restrictions on the use of the authorized capital also exist in case a public take-over bid on us has been announced. |
Item 16H Mine safety disclosure
Not applicable.
Item 16I Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
Item 16J Insider Trading Policies
Not applicable.
Item 16K Cybersecurity
Cybersecurity Risk Management and Strategy
Galapagos has made significant efforts and investments to increase our cybersecurity posture. We currently maintain an information security risk management framework designed to protect the Company from risks from cybersecurity threats. This framework is informed by a security controls matrix that incorporates elements of an industry-standard cybersecurity framework. Our risk management framework operationalizes risk management principles designed to identify and assess upcoming risks, and assign the risks to risk owners with appropriate mitigation steps. We also conduct internal and external penetration testing and vulnerability assessments, and engage third-party cybersecurity experts to conduct periodic cybersecurity risk assessments.
We are in the process of implementing a new enterprise risk management framework, tailored to the size and structure of the Company, to help us identify risks that should be reported/escalated to senior management. This new framework includes a risk forum that discusses changes in the risk environment, including key cybersecurity risks identified and managed by the IT Security function. The Head of IT Security is a standing member of the forum.
We also maintain written information security policies and procedures designed to ensure confidentiality, integrity and availability of the Galapagos data and IT systems. Our incident response plans and playbooks address clearly identified control objectives, which are based on industry standards, and have been operationalized in policies, procedures and work instructions. We have also engaged an external security partner to support us on a broad set of cybersecurity activities the daily management of our security operations and on-demand services.
Cybersecurity threats associated with the use of third-party vendors are managed and overseen through specific policies and procedures. Each vendor is evaluated on their cybersecurity risk, and remediation programs for identified risks are set up and periodically reviewed based on the initial assessment.
231
Governance Related to Cybersecurity Risks
The Chief Information Security Officer (“CISO”) is responsible for our information security risk management process, and reports periodically on the Galapagos information security risk environment to various Company stakeholders (internal control, data privacy, quality and compliance teams). Currently, the CISO position is held by an individual with over 20 years of work experience in life sciences IT management positions, and who previously had responsibility over information security and risk management.
We have also established a cross-functional Cybersecurity Disclosure team (“CSD team”) and Cybersecurity Disclosure board (“CSD board”) to oversee and manage the prioritization of risks from cybersecurity threats. The CSD team is comprised of the CISO and members of Galapagos’ legal and internal control teams. The CSD team members have experience in cybersecurity, legal and financial reporting. The CSD board is comprised of the CISO, the Senior Director, Head of Internal Control & Finance Operations, the General Counsel, and the Chief Financial Officer and Chief Operating Officer. The CSD board members have experience in the areas of cybersecurity, legal, internal control and finance.
The Company’s Board of Directors is responsible for the oversight of our risk management activities, and has delegated to the Audit Committee the responsibility to assist our Board in this task. While our Board of Directors oversees our risk management, our Executive Committee is responsible for day-to-day risk management processes.
Specifically, Galapagos’ Audit Committee is responsible for evaluating Galapagos’ system of internal control and risk management, including the cybersecurity risk management program and related controls. The CISO and the Head of IT review risks from cybersecurity threats and incidents with the Audit Committee during an annual review, and report incidents that require escalation per set procedures. The Audit Committee is responsible for the final approval of any disclosure of a material cybersecurity incident.
The Executive Committee is responsible for setting up and maintaining policies related to the risk profile of the Company and its systems to identify, assess, manage and monitor cybersecurity risks.
PART III
Item 17 Financial statements
Not applicable.
Item 18 Financial statements
See pages F-1 through F-69 of this annual report.
Item 19 Exhibits
The Exhibits listed in the Exhibit Index at the end of this annual report are filed as Exhibits to this annual report.
232
Index to the Consolidated Financial Statements
FINANCIAL SECTION
Audited consolidated financial statements as of December 31, 2023 and 2022 and for the years ended December 31, 2023, 2022 and 2021.
Contents
Report of Independent Registered Public Accounting Firm – BDO Bedrijfsrevisoren BV (Firm ID: | F-2 |
Report of Independent Registered Public Accounting Firm – Deloitte Bedrijfsrevisoren/Réviseurs d’Entreprises BV/SRL (Firm ID:1133) | F-5 |
F-6 | |
F-7 | |
F-8 | |
F-9 | |
F-10 | |
F-11 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Shareholders and Board of Directors
Galapagos NV
Mechelen, Belgium
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated statement of financial position of Galapagos NV (the “Company”) as of December 31, 2023, the related consolidated income statement, statement of comprehensive income/loss, changes in equity, and cash flows for the year ended December 31, 2023, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2023, and the results of its operations and its cash flows for the year then ended, in conformity the International Financial Reporting Standards as issued by the International Accounting Standards Boards.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (“PCAOB”), the Company's internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (“COSO”) and our report dated March 28, 2024 expressed an unqualified opinion thereon.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Determination of the percentage of completion used for revenue recognition related to the filgotinib performance obligation under the license and collaboration agreement with Gilead, reported within the results from discontinued operations
As described in notes 2, 4, 5 and 26 to the consolidated financial statements, the Company recognized collaboration revenues of 429,4 million EUR in 2023 from upfront payments and milestone payments related to the filgotinib performance obligation under the license and collaboration agreement with Gilead (the “agreement”). The Company
F-2
recognized revenue using the cost-to-cost input method, which management believes best depicts the transfer of control to the customer. The extent of progress towards completion is measured based on the ratio of actual costs incurred to date, to the total estimated costs expected upon satisfying the filgotinib performance obligation.
Significant management judgment is required in determining the total estimated costs still to be incurred and the period over which the Company is expected to complete its performance obligation, impacting the revenue recognition. This significant estimate is the principal consideration for our conclusion that procedures relating to the determination of the estimated costs to complete the performance obligation, impacting the revenue recognition for the filgotinib performance obligation, is a critical audit matter. This increased level of judgment by management led to a high degree of auditor judgment, complexity, and effort in performing procedures and in evaluating audit evidence related to management’s assumptions of the estimation of total costs to complete.
The primary procedures we performed to address this critical audit matter included:
| ● | Testing the design and operating effectiveness of controls over management’s assessment on determining the estimate of total costs to complete the performance obligation, which included evaluating the reasonableness of significant assumptions related to the estimate. |
| ● | Testing the accuracy and completeness of actual costs incurred to date based on a sample. |
| ● | Evaluating management’s ability to reasonably estimate the costs to complete the performance obligation, including: |
| o | Evaluating the appropriateness of changes made during the period to management’s estimates of total costs to complete. |
| o | Performing a comparison of management’s prior period cost estimates to actual costs incurred and approved. |
| o | Evaluating the period over which management is expecting the Company to complete its performance obligation. |
| o | Comparing certain costs to third-party supporting evidence. |
| o | Considering the impact of any subsequent events on management’s assumptions. |
Presentation of discontinued operations and assets and liabilities held for sale related to the transfer of the Jyseleca® business to Alfasigma
As described in note 2 and 5 to the consolidated financial statements, the Company announced on October 30, 2023, the signing of a letter of intent contemplating the transfer of the Jyseleca® business to Alfasigma. On December 30, 2023 the Share and Asset Purchase Agreement (‘SAPA’) was signed with the final closing of the transaction being subject to closing conditions. On January 31, 2024 these conditions, including the signing of the Transition agreement, were fulfilled and the transaction was closed.
Significant management judgement was used in the determination and classification of the expenses incurred directly in relation to the Jyseleca® business as discontinued operations, which involved certain judgments in allocating expenses and in the identification of the assets and liabilities that form part of the disposal group relating to the transfer of the Jyseleca® business, considering the transition period and conditions agreed with Alfasigma. This significant judgment is the principal consideration for our conclusion that procedures relating to auditing management’s judgements applied to the determination and classification of the expenses and in the identification of the assets and liabilities is a critical audit matter. Significant audit effort was involved in performing these procedures.
The primary procedures we performed to address this critical audit matter included:
| ● | Testing the design and operating effectiveness of controls over management’s accounting treatment for discontinued operations, including those over the determination of the assets and liabilities allocated to the disposal group. |
| ● | Evaluating management’s judgements over the identification of all assets and liabilities belonging to the disposal group by reading relevant agreements and assessing the Company’s ongoing involvement during the transition period agreed with Alfasigma. |
| ● | Assessing the reasonableness of the judgements applied by management in allocating expenses to the discontinued operations by inspecting supporting documentation, and inquiring management regarding specific assumptions made. |
F-3
/s/ We have served as the Company's auditor since 2023. March 28, 2024 | |
| |
F-4
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the Board of Directors of Galapagos NV
Opinion on the Financial Statements
We have audited the accompanying consolidated statement of financial position of Galapagos NV and subsidiaries (the "Company") as of December 31, 2022, the related consolidated statements of operations, comprehensive income/loss(-), changes in equity and cash flows, for the years ended December 31, 2022 and 2021, and the related notes (collectively referred to as the "financial statements"). In our opinion, the 2022 and 2021 financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2022, and the results of its operations and its cash flows for the years ended December 31, 2022 and 2021, in conformity with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Deloitte Bedrijfsrevisoren/Réviseurs d’Entreprises BV/SRL
Zaventem, Belgium
March 23, 2023 (March 28, 2024 as to the effects of the discontinued operations described in Note 5)
We began serving as the Company’s auditor in 2000. In 2023 we became the predecessor auditor.
F-5
Consolidated Statements of Financial Position
December 31, | |||||||||
| 2023 |
| 2022 |
| Notes | ||||
(Euro, in thousands) | |||||||||
Assets | |||||||||
Goodwill | € | | € | | 13 | ||||
Intangible assets other than goodwill | | | 14 | ||||||
Property, plant and equipment | | | 15 | ||||||
Deferred tax assets | | | 23 | ||||||
Non-current R&D incentives receivables | | | 17 | ||||||
Other non-current assets | | | 16 | ||||||
Non-current assets | | | |||||||
Inventories | | | 18 | ||||||
Trade and other receivables | | | 19 | ||||||
Current R&D incentives receivables | | | 17 | ||||||
Current financial investments | | | 20 | ||||||
Cash and cash equivalents | | | 21 | ||||||
Other current assets | | | 19 | ||||||
Current assets from continuing operations | | | |||||||
Assets in disposal group classified as held for sale | | — | 5 | ||||||
Total current assets | | | |||||||
Total assets | € | | € | | |||||
Equity and liabilities | |||||||||
Share capital | € | | € | | 22 | ||||
Share premium account | | | 22 | ||||||
Other reserves | ( | ( | |||||||
Translation differences | ( | ( | |||||||
Accumulated losses | ( | ( | |||||||
Total equity | | | |||||||
Retirement benefit liabilities | | | |||||||
Deferred tax liabilities | | | 23 | ||||||
Non-current lease liabilities | | | 24 | ||||||
Other non-current liabilities | | | 25 | ||||||
Non-current deferred income | | | 26 | ||||||
Non-current liabilities | | | |||||||
Current lease liabilities | | | 24 | ||||||
Trade and other liabilities | | | 25 | ||||||
Current tax payable | | | |||||||
Current deferred income | | | 26 | ||||||
Current liabilities from continuing operations | | | |||||||
Liabilities directly associated with assets in disposal group classified as held for sale | | — | 5 | ||||||
Total current liabilities | | | |||||||
Total liabilities | | | |||||||
Total equity and liabilities | € | | € | | |||||
The accompanying notes form an integral part of these financial statements.
F-6
Consolidated Income Statements
Year ended December 31, | ||||||||||
2023 |
| 2022 (*) |
| 2021 (*) |
| Notes | ||||
(Euro, in thousands, except per share data) | ||||||||||
Collaboration revenues | | | | 7 | ||||||
Total net revenues | | | | |||||||
Research and development expenses | ( | ( | ( | 8 | ||||||
Sales and marketing expenses | ( | ( | ( | 8 | ||||||
General and administrative expenses | ( | ( | ( | 8 | ||||||
Other operating income | | | | 8 | ||||||
Operating loss | ( | ( | ( | |||||||
Fair value adjustments and net exchange differences | | | | 10 | ||||||
Other financial income | | | | 10 | ||||||
Other financial expenses | ( | ( | ( | 10 | ||||||
Profit/loss (-) before tax | | ( | ( | |||||||
Income taxes | ( | ( | ( | 11 | ||||||
Net loss from continuing operations | ( | ( | ( | |||||||
Net profit/loss (-) from discontinued operations, net of tax | | ( | | 5 | ||||||
Net profit/loss (-) | € | | € | ( | € | ( | ||||
Net profit/loss (-) attributable to: |
|
|
| |||||||
Owners of the parent | | ( | ( | |||||||
Basic and diluted earnings/loss (-) per share | € | | € | ( | € | ( | 12 | |||
Basic and diluted loss (-) per share from continuing operations | € | ( | € | ( | € | ( | ||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The accompanying notes form an integral part of these financial statements.
F-7
Consolidated Statements of Comprehensive Income/Loss (-)
Year ended December 31, | ||||||||
2023 |
| 2022 (*) |
| 2021 (*) | ||||
(Euro, in thousands) | ||||||||
Net profit/loss (-) | € | | € | ( | € | ( | ||
Items that will not be reclassified subsequently to profit or loss: | ||||||||
Re-measurement of defined benefit obligation | ( | | | |||||
Items that may be reclassified subsequently to profit or loss: | ||||||||
Translation differences, arisen from translating foreign activities | | | | |||||
Realization of translation differences upon sale/liquidation of foreign operations | — | — | | |||||
Other comprehensive income/loss (-), net of income tax | ( | | | |||||
Total comprehensive income/loss (-) attributable to: | ||||||||
Owners of the parent | | ( | ( | |||||
Total comprehensive income/loss (-) attributable to owners of the parent arises from: | ||||||||
Continuing operations | ( | ( | ( | |||||
Discontinued operations | | ( | | |||||
Total comprehensive income/loss (-), net of income tax | € | | € | ( | € | ( | ||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The accompanying notes form an integral part of these financial statements.
F-8
Consolidated Statements of Changes in Equity
| Share |
| Share |
| Translation |
| Other |
| Accumul. |
| Total | |||||||
(Euro, in thousands) | ||||||||||||||||||
On December 31, 2020 | € | | € | | € | ( | € | ( | € | ( | € | | ||||||
Net loss | ( | ( | ||||||||||||||||
Other comprehensive income | | | | |||||||||||||||
Total comprehensive income/loss (-) | | | ( | ( | ||||||||||||||
Share-based compensation | | | ||||||||||||||||
Exercise of subscription rights | | | | |||||||||||||||
On December 31, 2021 | € | | € | | € | ( | € | ( | € | ( | € | | ||||||
Net loss | ( | ( | ||||||||||||||||
Other comprehensive income | | | | |||||||||||||||
Total comprehensive income/loss (-) | | | ( | ( | ||||||||||||||
Share-based compensation | | | ||||||||||||||||
Exercise of subscription rights | | | | |||||||||||||||
On December 31, 2022 | € | | € | | € | ( | € | ( | € | ( | € | | ||||||
Net profit | | | ||||||||||||||||
Other comprehensive income/loss (-) | | ( | ( | |||||||||||||||
Total comprehensive income/loss (-) | | ( | | | ||||||||||||||
Share-based compensation | | | ||||||||||||||||
Exercise of subscription rights | | | | |||||||||||||||
On December 31, 2023 | € | | € | | € | ( | € | ( | € | ( | € | | ||||||
The accompanying notes form an integral part of these financial statements.
F-9
Consolidated Statements of Cash Flows
| 2023 |
| 2022 |
| 2021 |
| Notes | ||||
(Euro, in thousands) | |||||||||||
Net profit/loss (-) | € | | € | ( | € | ( | |||||
Adjustment for non-cash transactions | | | | 29 | |||||||
Adjustment for items to disclose separately under operating cash flow | ( | ( | | 29 | |||||||
Adjustment for items to disclose under investing and financing cash flows | ( | ( | ( | 29 | |||||||
Change in working capital other than deferred income | ( | | | 29 | |||||||
Cash used for other liabilities related to the acquisition of subsidiaries | — | ( | — | 27 | |||||||
Decrease in deferred income | ( | ( | ( | 26 | |||||||
Cash used in operations | ( | ( | ( | ||||||||
Interest paid | ( | ( | ( | ||||||||
Interest received | | | | ||||||||
Corporate taxes paid | ( | ( | ( | ||||||||
Net cash flows used in operating activities | ( | ( | ( | ||||||||
Purchase of property, plant and equipment | ( | ( | ( | 15 | |||||||
Purchase of and expenditure in intangible fixed assets | ( | ( | ( | 14 | |||||||
Proceeds from disposal of property, plant and equipment | | | — | 15 | |||||||
Purchase of current financial investments | ( | ( | ( | 20 | |||||||
Investment income received related to current financial investments | | | | 20 | |||||||
Sale of current financial investments | | | | 20 | |||||||
Disposals of subsidiaries, net of cash disposed | — | — | | 5 | |||||||
Cash out from acquisition of subsidiaries, net of cash acquired | ( | ( | — | 27 | |||||||
Cash advances and loans to third parties | — | ( | — | 27 | |||||||
Acquisition of financial assets held at fair value through profit or loss | ( | — | — | 16 | |||||||
Proceeds from sale of financial assets held at fair value through profit or loss | — | — | | ||||||||
Net cash flows generated from/used in (-) investing activities | | ( | | ||||||||
Payment of lease liabilities | ( | ( | ( | 24 | |||||||
Proceeds from capital and share premium increases from exercise of subscription rights | | | | 22 | |||||||
Net cash flows used in financing activities | ( | ( | ( | ||||||||
Increase/decrease (-) in cash and cash equivalents | € | ( | € | ( | € | | |||||
Cash and cash equivalents at beginning of year | € | | € | | € | | 21 | ||||
Increase/decrease (-) in cash and cash equivalents | ( | ( | | ||||||||
Effect of exchange rate differences on cash and cash equivalents | ( | | | ||||||||
Cash and cash equivalents at end of year | € | | € | | € | | 21 | ||||
The accompanying notes form an integral part of these financial statements.
F-10
Notes to Consolidated Financial Statements
1. General information
Galapagos NV is a limited liability company incorporated in Belgium and has its registered office at Generaal De Wittelaan L11 A3, 2800 Mechelen, Belgium. In the notes to the consolidated financial statements, references to “we,” “us,” “the group” or “Galapagos” include Galapagos NV together with its subsidiaries. We refer to note 33 for a list of consolidated companies.
We are a global biotechnology company with operations in Europe and the US dedicated to developing medicines focusing on oncology and immunology.
The components of the result presented in the financial statements include the results of the companies mentioned in note 33 Consolidated companies as of December 31, 2023.
Our operations had
Effective as from July 1, 2023 we transferred our drug discovery and research activities in Romainville, France, and
On January 31, 2024 we announced that we successfully completed the transfer of the Jyseleca® business to Alfasigma, including the European and UK Marketing Authorizations, the commercial, medical and development activities for Jyseleca® and approximately
Our continuing operations had
We refer to note 33 for a list of companies included in the discontinued operations and to note 5 for more details on the discontinued operations.
F-11
2. Summary of significant transactions
TRANSFER OF JYSELECA® BUSINESS TO ALFASIGMA
On October 30, 2023, we signed a letter of intent contemplating a transfer of the Jyseleca® business to Alfasigma. The final share and asset purchase agreement was signed on December 30, 2023 and the transaction was closed on January 31, 2024. The transfer includes the European and UK Marketing Authorizations, and the commercial, medical affairs and development activities for Jyseleca®. In connection with the completion of the transaction, approximately
Effective January 31, 2024, following the closing of the transaction between us and Alfasigma for the transfer the Jyseleca® business, we assigned our rights and obligations under the Gilead filgotinib collaboration to Alfasigma, except for our right to receive royalties from Gilead on net sales in the Gilead Territory under a separate agreement between Gilead and us entered into in October 2023.
On January 31, 2024, we also signed a transition agreement with Alfasigma enacting the responsibilities and services that will be provided by the parties during a transition period for the transfer of the business. The gradual transfer of our remaining inventories to Alfasigma is also governed by this agreement.
We refer to note 5 for more details on the discontinued operations.
GILEAD COLLABORATION AGREEMENT
On July 14, 2019 we and Gilead announced that we entered into a
We identified the following three performance obligations as part of this collaboration: (i) the transfer of an extended license on ziritaxestat GLPG1690 (this performance obligation was satisfied completely in 2019), (ii) the granting of exclusive access to our drug discovery platform (i.e. the IP, technology, expertise and capabilities) during the collaboration period and exclusive option rights on our current and future clinical programs after Phase 2 (or, in certain circumstances, the first Phase 3 study) outside Europe and (iii) an increased cost share from /80 to
In the years thereafter (2020-2023), the collaboration agreement relating to filgotinib was restated several times (see further in this chapter).
We however retain the following performance obligations, (i) the granting of exclusive access to our drug discovery platform (i.e. the IP, technology, expertise and capabilities) during the collaboration period and exclusive option rights on our current and future clinical programs after Phase 2 (or, in certain circumstances, the first Phase 3 study) outside Europe and (ii) an increased cost share from /80 to
F-12
This second performance obligation was transferred to Alfasigma on 31 January 2024, when we closed the transaction for the transfer of the Jyseleca business to Alfasigma and the (amended and restated) collaboration agreement relating to filgotinib was assigned to Alfasigma as a consequence thereof.
Terms of the collaboration relating to our drug discovery platform
We will fund and lead all discovery and development autonomously until the end of Phase 2. After the completion of a qualifying Phase 2 study (or, in certain circumstances, the first Phase 3 study), Gilead will have the option to acquire a license to the compound outside Europe. If the option is exercised, we and Gilead will co-develop the compound and share costs equally. Gilead will maintain option rights to our programs through the
Gilead will make a $
Revised filgotinib collaboration
Since the revised agreement of December 2020, we assumed all development, manufacturing, commercialization and certain other rights for filgotinib in Europe. Since January 1, 2021, we bear the full future development costs for certain studies (defined as “Group A activities”), in lieu of the equal cost split contemplated by the previous agreement. The
Since the amendment of December 2020, we are also no longer eligible to receive any future milestone payments relating to filgotinib in Europe. Other terms of the original license agreement remained in effect.
On October 30, 2023, we and Gilead agreed to amend the Filgotinib Agreement by terminating the existing 50/50 global development cost sharing arrangement with us bearing the costs going forward, and to terminate our obligation to pay tiered royalties to Gilead on net sales of Jyseleca® in Europe, in addition to other amendments.
Effective January 31, 2024, following the closing of the transaction between Galapagos and Alfasigma for the transfer of the Jyseleca® business, Galapagos assigned its rights and obligations under the filgotinib collaboration to Alfasigma, except for Galapagos’ right to receive royalties from Gilead on net sales in the Gilead Territory under a separate agreement between Gilead and Galapagos entered into in October 2023.
Gilead remains responsible for commercial activities outside of Europe.
Terms of the equity investment
As part of the research and development collaboration of 2019 Gilead also entered into a share subscription agreement with us. Gilead’s equity investment consisted of a subscription for new Galapagos shares. This equity subscription took place at closing of the transaction, on August 23, 2019 and increased Gilead’s stake in Galapagos from approximately
In addition, the extraordinary general meeting of shareholders of October 22, 2019 approved the issuance of Warrant A and initial Warrant B allowing Gilead to further increase its ownership of Galapagos to up to
F-13
subscription agreement (August 23, 2019) and this warrant will have substantially similar terms, including as to exercise price, to the initial Warrant B. On 31 December 2023 the value of the subsequent Warrant B decreased to €
The agreement also includes a
Gilead’s ownership amounted to
Since October 22, 2019, Gilead has
Evolution of the total transaction price
The transaction price is currently composed of a fixed part, being non-refundable upfront and license fees and a variable part, being milestone payments, sales based milestones and sales based royalties, and cost reimbursements for R&D activities delivered. Milestone payments are included in the transaction price of the arrangement to the extent that it is highly probable that a significant reversal of revenue will not occur. Milestone payments received from Gilead are recognized in revenue over time till the end of the development plan. Sales based milestones and sales based royalties are also part of the arrangement and are recognized as revenues at a point in time at the moment they occur.
The €
The below table summarizes the changes in the transaction price during 2022 and 2023 of our collaboration with Gilead:
| December 31, |
| Other |
| December 31, |
| Other |
| December 31, | ||||||
(Euro, in thousands) | |||||||||||||||
Allocation of transaction price | |||||||||||||||
Upfront consideration | € | | € | | € | | |||||||||
Milestones achieved | | € | | | |||||||||||
Royalties | | | | € | | | |||||||||
Impact initial valuation of share subscription | | | | ||||||||||||
| | | | | |||||||||||
Less: | |||||||||||||||
Warrants issuance liabilities | |||||||||||||||
Warrant A | ( | ( | ( | ||||||||||||
Initial warrant B | ( | ( | ( | ||||||||||||
Subsequent warrant B | ( | ( | ( | ||||||||||||
| | | | | |||||||||||
Allocation to performance obligations | |||||||||||||||
Ziritaxestat | | | | ||||||||||||
Filgotinib (1) | | | | | | ||||||||||
Drug discovery platform (10 years) | € | | € | | € | | € | | € | | |||||
(1) With regard to the additional consideration received as a result of the Option, License and Collaboration agreement (July 14, 2019) allocated to the filgotinib performance obligation, we assumed the existence of a significant financing component estimated to €
F-14
TRANSFER OF THE DRUG DISCOVERY AND RESEARCH ACTIVITES IN ROMAINVILLE (FRANCE) TO NOVALIX
Effective as from July 1, 2023 we transferred our drug discovery and research activities in Romainville and employees exclusively dedicated to the operation of these activities to NovAliX, who assumes all ongoing research and discovery activities in Romainville. In return, we are committed to utilizing the research capabilities and expertise of NovAliX through a five year-collaboration and within the context of our R&D portfolio during which we are committed to purchase for a total of €
We refer to note 28 of these consolidated financial statements for more details on this transaction.
3. Material accounting policies
Our material accounting policies are summarized below.
BASIS OF PREPARATION AND GOING CONCERN ASSUMPTION
The consolidated financial statements are prepared in accordance with the International Financial Reporting Standards (IFRS), issued by the International Accounting Standards Board (IASB) and the interpretations issued by the IASB’s International Financial Reporting Interpretation Committee. The consolidated financial statements provide a general overview of our activities and the results achieved. They give a true and fair view of our financial position, our financial performance and cash flows, on a going concern basis.
The consolidated financial statements are presented in Euros, which is also our functional currency. Amounts are rounded to the nearest thousand, unless otherwise stated.
RECLASSIFICATION OF ACCRUED INTERESTS ON FINANCIAL INVESTMENTS – COMPARATIVE PERIODS NOT RESTATED
Accrued interests on financial assets were in the past recorded on the “current financial investments” line for treasury bills while reported on a separate current assets/current liabilities line for the other financial investments measured at amortized cost (mainly term deposits).
During the third quarter of 2023 we decided to align the presentation of all our financial investments at amortized cost and present all accrued interests as part of the amortized cost measurement on the “current financial investments” / “cash and cash equivalents” line.
As the impact of this classification misstatement on the prior period comparative numbers was deemed immaterial for all comparative periods presented, we did not restate the prior period financial statements. This is in accordance with IAS 8.42 that requires to only adjust the comparatives for material prior period errors.
The impact of this error on December 31, 2022 amounted to €
F-15
DISCONTINUED OPERATIONS PRESENTATION – COMPARATIVE PERIODS RESTATED
Our income statement and statement of comprehensive income of the comparative periods presented in these financial statements have been restated to present the activities related to the Jyseleca® business on a separate line “Net income/loss (-) from discontinued operations, net of tax”.
We refer to note 5 for more detailed information on these discontinued operations.
NEW STANDARDS AND INTERPRETATIONS APPLICABLE FOR THE ANNUAL PERIOD BEGINNING ON JANUARY 1, 2023
New standards and interpretations applicable for the annual period beginning on January 1, 2023 did not have a material impact on our consolidated financial statements except for the application of the amendments made to IAS 1 Disclosure of Accounting Policies. As a result of this amendment, we reassessed our accounting policies in order to remain with only the material accounting policies.
STANDARDS AND INTERPRETATIONS PUBLISHED, BUT NOT YET APPLICABLE FOR THE ANNUAL PERIOD BEGINNING ON JANUARY 1, 2023
A number of new standards are effective for annual periods beginning on or after January 1, 2024 with earlier adoption permitted. However we have not early adopted new or amended standards in preparing our consolidated financial statements. We are currently still assessing the impact of these new accounting standards and amendments that are not yet effective but we expect no standard to have a material impact on our financial statements in the period of initial application.
The following amendments are effective for the period beginning January 1, 2024:
The following amendments are effective for the period beginning January 1, 2025:
BUSINESS COMBINATIONS
Business combinations are accounted for using the acquisition method. In the statement of financial position, all identifiable assets, liabilities and contingent liabilities are initially recognized at their fair value at the acquisition date. The results of acquired operations are included in our consolidated income statement from the date on which control is obtained. Any contingent consideration to be transferred by us will be recognized at fair value at the acquisition date. Subsequent changes to the fair value of the contingent consideration, which is deemed to be an asset or liability, will be recognized in our consolidated income statement. The excess of the fair value of the total purchase consideration transferred over the fair value of the acquired assets and assumed liabilities is recognized as goodwill. The valuations in support of fair value determinations are based on information available at the acquisition date. Acquisition related costs are expensed as incurred.
Any contingent consideration to be transferred by us in relation to businesses acquired that are linked to milestone payments and are initially recognized at fair value as a financial liability. They are adjusted for the probability of their likelihood of payment and are appropriately discounted to reflect the impact of time.
Changes in the fair value of these contingent consideration liabilities in subsequent periods are recognized in our consolidated income statement on the line “other operating income/expense”. The effect of unwinding the discount over time is recognized on the line “other financial expenses”.
F-16
Contingent amounts payable or paid by us to former shareholders of acquired companies, who continue to be employed by us, but which would be automatically forfeited (or become repayable) upon termination of employment before a specific date, are classified as remuneration for post-combination services in our consolidated income statement. These cash-settled contingent amounts are recognized in accordance with IAS 19 and are recorded in the statement of financial position on the lines “other (non-) current assets” and “other non-current/trade and other liabilities” depending on the timing of the payment by us.
GOODWILL
Goodwill is initially measured as the excess of the total purchase consideration transferred and the fair value of the acquired assets and assumed liabilities. Subsequently, goodwill is stated at cost less impairments.
As goodwill is considered to have an indefinite life, it is tested for impairment at least once a year (at each year-end), and whenever there is an indication that it may be impaired, by comparing its carrying amount with its recoverable amount.
Any impairment costs are recorded in our consolidated income statement on the line “Other operating income/expenses”.
INTANGIBLE ASSETS OTHER THAN GOODWILL
Expenditure on research activities is recognized as an expense in the period in which it is incurred.
An internally generated intangible asset arising from our development activities is recognized only if all of the following conditions are met:
| ● | Technically feasible to complete the intangible asset so that it will be available for use or sale |
| ● | We have the intention to complete the intangible assets and use or sell it |
| ● | We have the ability to use or sell the intangible assets |
| ● | The intangible asset will generate probable future economic benefits, or indicate the existence of a market |
| ● | Adequate technical, financial and other resources to complete the development are available |
| ● | We are able to measure reliably the expenditure attributable to the intangible asset during its development |
| (i) | Internally generated intangible assets |
The amount capitalized as internally generated intangible assets is the sum of the development costs incurred as of the date that the asset meets the conditions described above. Because of risks and uncertainties inherent to the regulatory authorizations and to the development process itself, management estimates that the conditions for capitalization are not met until we obtain regulatory approval from the competent authorities.
Currently we recognize all development costs as an expense in the period in which they are incurred, even for approved products because they do not generate separately identifiable incremental future economic benefits that can be reliably measured.
| (ii) | Licences, rights and in-process research and development |
Acquired in-process research and development obtained through in-licensing agreements, business combinations, collaboration agreements or separate acquisitions are capitalized as an intangible asset provided that they are separately identifiable, controlled by us and expected to provide economic benefits. As the probability criterion in IAS 38 is always considered to be satisfied for separately acquired research and
F-17
development assets, upfront and milestone payments to third parties for products or compounds for which regulatory approval has not yet been obtained are recognized as intangible assets. We consider such intangible assets as not yet available for use until the moment that the underlying asset is approved and commercially launched. Amortization will commence when the underlying asset is approved for commercialization and the asset will be amortized over its useful life.
Intangible assets may also consist of upfront fees paid to third party institutions in exchange for an option to negotiate a license to any of the third party’s rights in technology resulting from the collaboration. The upfront fee paid in exchange for this option is capitalized as intangible asset and amortized over the expected duration of the option.
Exclusivity contracts and technology acquired through business combinations are valued independently as part of the fair value of the businesses acquired and are amortized over their estimated useful lives. The estimated useful life is based on the lower of the contract life or the economic useful life.
In the event an asset has an indefinite life, this fact is disclosed along with the reasons for being deemed to have an indefinite life. Intangible assets with an indefinite useful life and intangible assets which are not yet available for use are tested for impairment annually, and whenever there is an indication that the asset might be impaired.
| (iii) | Software and databases |
Acquired software is recognized at cost less accumulated amortization and any impairment loss. Amortization is recognized so as to write off the cost of assets over their useful lives (generally between
| (iv) | Contract costs |
Contract costs only include success fees that were capitalized in relation to the Gilead agreement of 2019. These costs are currently amortized on a straight-line basis over a period of 10 years, reflecting the term of our collaboration with Gilead.
We review at each balance sheet date the carrying amount of our intangible assets to determine whether there is any indication that those assets have suffered an impairment loss. If any such indication exists, the recoverable amount of the asset is estimated in order to determine the extent of the impairment loss (if any). Where the asset does not generate cash flows that are independent from other assets, we estimate the recoverable amount of the cash-generating unit to which the asset belongs. If the recoverable amount of an asset or cash generating unit is estimated to be less than the carrying amount, the carrying amount of the asset is reduced to its recoverable amount. An impairment loss is recognized as an expense immediately.
PROPERTY, PLANT AND EQUIPMENT
Property, plant and equipment are recognized at cost less accumulated depreciation and any impairment loss.
Depreciation of an asset begins when it is available for use, ie when it is in the location and condition necessary for it to be capable of operating in the manner intended by management.
Depreciation is recognized so as to write off the cost of assets over their useful lives, using the straight-line method, on the following bases:
| ● | Buildings: |
| ● | Installation & machinery: |
| ● | Furniture, fixtures & vehicles: |
F-18
Leasehold improvements are depreciated
The other tangible assets category mainly consists of assets under construction. Assets under construction are not depreciated.
Any gain or loss incurred at the disposal of an asset is determined as the difference between the sale proceeds and the carrying amount of the asset and is recognized in profit or loss.
We review at each balance sheet date the carrying amount of our property, plant and equipment to determine whether there is any indication that those assets have suffered an impairment loss. If any such indication exists, the recoverable amount of the asset is estimated in order to determine the extent of the impairment loss (if any).
LEASES
All leases are accounted for by recognizing a right-of-use asset and a corresponding lease liability except for:
| ● | Leases of low value assets; and |
| ● | Leases with a duration of 12 months or less |
Liabilities arising from a lease are initially measured on a present value basis. Lease liabilities include the net present value of the lease payments that are not paid at the commencement date, discounted using the incremental borrowing rate. Our lease payments generally only include fixed payments and extension option payments if we are reasonably certain to exercise this option.
After initial recognition, the lease liability will be measured at amortized cost using the discount rate determined at commencement and will be re-measured (with a corresponding adjustment to the related right-of-use asset) when there is a change in future lease payments, generally in case of reassessment of options.
At the commencement date, the right-of-use assets are measured at cost, comprising the amount of the initial lease liability, less any lease incentives received from the lessors.
After initial recognition, the right-of-use assets are measured at cost and generally depreciated over the the lease term on a straight-line basis. The right-of-use assets will be adjusted for any re-measurements of the lease liability as a result of lease modifications. The right-of-use assets are subject to impairment testing if there is an indicator for impairment, as for property, plant and equipment. The right-of-use assets are presented in the statement of financial position under the caption “property, plant and equipment” and the lease liabilities are presented as current and non-current lease liabilities.
We only include extension options (or periods after termination options) in the lease term if the lease is reasonably certain to be extended (or not terminated). The assessment is reviewed if a significant event or a significant change in circumstances occurs which affects this assessment and that is within our control.
INVENTORIES
Inventories consist of raw materials, semi-finished products and finished products. These inventories are initially recognized at cost, and subsequently at the lower of cost and net realizable value. Cost comprises all costs of purchase, conversion costs and transportation costs, and is determined using the FIFO-method.
F-19
FINANCIAL INSTRUMENTS
Financial assets and financial liabilities are recognized in our statement of financial position when we become a party to the contractual provisions of the instrument.
(i) Financial assets
Financial assets are initially recognized either at fair value or at their transaction price. All recognized financial assets will subsequently be measured at either amortized cost or fair value under IFRS 9 on the basis of both our business model for managing the financial assets and the contractual cash flow characteristics of the financial asset.
| ● | a financial asset that (i) is held within a business model whose objective is to collect the contractual cash flows and (ii) has contractual cash flows that are solely payments of principal and interest on the principal amount outstanding is measured at amortized cost (net of any write down for impairment), unless the asset is designated at fair value through profit or loss (FVTPL) under the fair value option; |
| ● | all other financial assets are measured at FVTPL. |
A financial asset is classified as current when the cash flows expected to flow from the instrument mature within one year.
We derecognize a financial asset when the contractual rights to the cash flows from the asset expire, or we transfer the rights to receive the contractual cash flows on the financial asset in a transaction in which substantially all the risks and rewards of ownership of the financial asset are transferred.
Financial assets at fair value through profit or loss
Financial assets are designated at fair value through profit or loss if we manage such investments and make purchase and sale decisions based on their fair value in accordance with the investment strategy. Attributable transaction costs are recognized in profit or loss as incurred. Financial assets at fair value through profit or loss are measured at fair value, and changes therein, which take into account any dividend income, are recognized in profit or loss.
Equity instruments
We hold investments in equity instruments, which based on IFRS 9, are designated as financial assets at fair value through profit or loss. The fair value of listed investments is based upon the closing price of such securities on Euronext at each reporting date. If there is no active market for an equity instrument, we establish the fair value by using valuation techniques.
Current financial investments measured at fair value through profit or loss
Current financial investments include financial assets measured at fair value through profit or loss and may comprise short term bond funds that have a maturity equal or less than 12 months, and money market funds.
Cash equivalents measured at fair value through profit or loss
Cash equivalents measured at fair value through profit or loss may comprise bonds and money market funds that are readily convertible to cash and are subject to an insignificant risk of changes in value.
Financial assets at amortized cost
Receivables
Receivables are designated as financial assets measured at amortized cost. They are initially measured either at fair value or at transaction price, in the absence of a significant financing component.
F-20
All receivables are subsequently measured in the balance sheet at amortized cost, which generally corresponds to nominal value less expected credit loss provision.
Receivables mainly comprise trade and other receivables and current/non-current R&D incentives receivables.
The R&D incentives receivables relate to refunds resulting from R&D incentives on research and development expenses in France and Belgium. This is a grant receivable that is based on annual declarations and is only refunded in case it cannot be offset by a tax payable. Research and development incentives receivables are discounted over the period until maturity date according to the appropriate discount rates. We refer to the accounting policy on grants and R&D incentives.
Current financial investments measured at amortized cost
Current financial investments measured at amortized cost include treasury bills that have a maturity equal to or less than 12 months. We apply settlement date accounting for the recognition and de-recognition of current financial investments measured at amortized cost. Current financial investments measured at amortized cost also include term deposits with maturities exceeding three months from the acquisition date.
Cash and cash equivalents measured at amortized cost
Cash and cash equivalents measured at amortized cost mainly comprise of notice accounts and term deposits that are readily convertible to cash within three months or less, that are subject to an insignificant risk of changes in their value and that are held for the purpose of meeting short-term cash commitments.
Cash and cash equivalents exclude restricted cash, which is presented in the line other non-current assets in the statement of financial position.
Impairment
The impairment loss of a financial asset measured at amortized cost is calculated based on the expected loss model.
For trade receivables, in the absence of a significant financing component, the loss allowance is measured at an amount equal to lifetime expected credit losses. Those are the expected credit losses that result from all possible default events over the expected life of those trade receivables.
Impairment losses are recognized in the consolidated income statement.
(ii) Financial liabilities
Financial liabilities are initially measured either at fair value or at their transaction price. Subsequent to initial recognition, financial liabilities are measured at amortized cost or at fair value.
Financial liabilities measured at amortized cost mainly comprise trade and other liabilities.
Trade and other liabilities are comprised of liabilities that are due less than one year from the balance sheet date and are in general not interest bearing and settled on an ongoing basis during the financial year. They also include accrued expenses related to our research and development project costs.
We derecognize a financial liability when its contractual obligations are discharged, cancelled or expire.
TAXATION
Income tax in the profit or loss accounts represents the sum of the current tax and deferred tax.
Current tax is the expected tax payable on the taxable profit of the year. The taxable profit of the year differs from the profit as reported in the financial statements as it excludes items of income or expense that are taxable or deductible
F-21
in other years and it further excludes items that are never taxable or deductible. Our liability for current tax is calculated using tax rates that have been enacted or substantively enacted by the balance sheet date.
Deferred income tax is provided in full, using the liability-method, on temporary differences arising between the tax bases of assets and liabilities and their carrying amounts in the financial statements. However, the deferred income tax is not accounted for if it arises from the initial recognition of an asset or liability in a transaction other than a business combination that at the time of the transaction affects neither accounting nor taxable profit nor loss.
Deferred income tax is determined using tax rates (and laws) that have been enacted or substantively enacted by the balance sheet date and are expected to apply when the related deferred income tax asset is realized or the deferred income tax liability is settled. Deferred tax assets are recognized to the extent that it is probable that future taxable profit will be available against which the temporary differences can be utilized. As such, a deferred tax asset for the carry forward of unused tax losses will be recognized to the extent that is probable that future taxable profits will be available.
REVENUE RECOGNITION
Revenues to date have consisted principally of collaboration revenues, which consist of milestones, license fees, non-refundable upfront fees and royalties received in connection with collaboration and license agreements. Starting in 2021 we also have commercial revenues from the sales of Jyseleca, which are reported as “Product net sales” on the discontinued operations line in our consolidated income statement.
The revenue recognition policies can be summarized as follows:
We recognize revenue when our customer obtains control of promised goods or services, in an amount that reflects the consideration that we expect to receive in exchange for those goods or services. To determine revenue recognition for agreements that we determine are within the scope of IFRS 15, we perform the following five steps:
COLLABORATION REVENUES
(i) identify the contract
In our agreements with customers we are mainly transferring licenses on our IP and in some cases this is combined with access rights and/or providing research and development services and/or cost sharing mechanisms. In some cases our collaborations also include an equity subscription component. If this is the case, we analyze if the criteria to combine contracts, as set out by IFRS 15, are met.
(ii) identify the performance obligations in the contract
Depending on the type of the agreement, there can be one or more distinct performance obligations under IFRS 15. This is based on an assessment of whether the promises in an agreement are capable of being distinct and are distinct from the other promises to transfer goods and/or services in the context of the contract. For some of our agreements we combine the transfer of the license with the performance of research and development activities because we consider that the license is not capable of being distinct and is not distinct in the context of the contract.
(iii) determine the transaction price
Collaboration and license agreements with our commercial partners for research and development activities generally include non-refundable upfront fees; milestone payments, the receipt of which is dependent upon the achievement of certain clinical, regulatory or commercial milestones; license fees, royalties on sales and sometimes reimbursement income or profits sharing arrangements.
a/ License fees or upfront payments
If the license to our intellectual property is determined to be distinct from the other performance obligations identified in the arrangement, we recognize revenues from non-refundable upfront fees allocated to the license at the point in time the license is transferred to the customer and the customer has the right to use the license.
F-22
For licenses that are bundled with other promises, we utilize judgment to assess the nature of the combined performance obligation to determine whether the combined performance obligation is satisfied over time or at a point in time. If the performance obligation is satisfied over time, revenue is then recognized based on a pattern that best reflects the transfer of control of the service to the customer.
b/ Milestone Payments other than sales based milestones
A milestone payment is only included in the transaction price to the extent that it is highly probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. Where milestone payments are included in the transaction price we estimate the amount to be included in the transaction price using the most likely amount method. The transaction price is allocated to each performance obligation on a stand-alone selling price basis. We recognize revenue as or when the performance obligations under the contract are satisfied. At the end of each subsequent reporting period, we re-evaluate the probability of achievement of such milestones and any related constraint. If necessary we adjust our estimate of the overall transaction price. Any such adjustments are recorded on a cumulative catch-up basis, which would affect revenue and earnings in the period of adjustment.
c/ Reimbursement Income for R&D Services
Collaboration and license agreements may include reimbursement or cost sharing for research and development services: such as outsourcing costs and payment for FTEs at contractual rates. R&D services are performed and satisfied over time given that the customer simultaneously receives and consumes the benefits provided by us.
Such costs reimbursements received are recognized in revenues when costs are incurred and agreed by the parties when we are acting as a principal in the scope of our stake of the R&D activities. If the later condition is not fulfilled, costs reimbursements are accounted for as a decrease of the related expenses.
d/ Sales based milestone payment and Royalties
License and collaboration agreements include sales-based royalties, including commercial milestone payments based on the level of sales, and the license has been deemed to be the predominant item to which the royalties relate. Related revenue is recognized as the subsequent underlying sales occur.
(iv) allocate the transaction price to the performance obligations in the contract
We allocate the transaction price to each performance obligation identified in the contract based upon the stand-alone selling price. The stand-alone selling price of each performance obligation is estimated by using one of the following methods: adjusted market assessment approach, the expected cost plus a margin approach or the residual approach. If management assesses that there is only one single performance obligation, the entire transaction price would be allocated to this performance obligation.
(v) recognize revenue when (or as) the entity satisfies a performance obligation
Revenue is recognized when our customer obtains control of the goods and/or services foreseen in the contracts. The control can be transferred over time or at a point in time – which results in recognition of revenue over time or at a point in time.
In case of revenue recognition over time, we use an input model that considers estimates of the percentage of total research and development costs that are completed each period compared to the total estimated costs (percentage of completion method) to measure the progress of the satisfaction of the underlying performance obligation (which is the applied method for the filgotinib performance obligation). In other cases, depending on specific circumstances, we recognize revenue on a straight-line basis over the estimated term of the performance obligation (which is the applied method for the performance obligation related to our drug discovery platform).
F-23
PRODUCT NET SALES
Revenue on the sale of Jyseleca is recorded as “Product net sales” on the discontinued operations line in our consolidated income statement.
Product net sales is the net amount of revenue recognized resulting from transferring control over our products to our customer (for example wholesalers and hospitals). Product sales revenue is recognized at a point in time when control of the goods has transferred to the customer. This is generally when the goods are delivered to the customer depending on the specific incoterms in the contract with a customer.
The amount of revenue recognized is the amount allocated to the satisfied performance obligation taking into account variable consideration. The estimated amount of variable consideration is included in the transaction price only to the extent that it is highly probable that a significant reversal in the amount of cumulative revenue recognized will not occur when the uncertainty associated with the variable consideration is subsequently resolved. Variable consideration that is included in the transaction price is primarily composed of rebates, discounts, cash discounts and chargebacks granted to various customers that are part of commercial and governmental contractual arrangements or other reimbursement programs. Shelf stock adjustments are granted to some of our customers to cover the inventory held by them at the time of a price decrease becomes effective. A liability is recognized for expected rebates, cash discounts, chargebacks or other reimbursements payable directly or indirectly to customers in relation to sales made until the end of the reporting period.
The amount of variable consideration is estimated using several elements such third-party market data, product pricing, the specific terms in the individual agreements, estimated inventory levels and the shelf life of our product. If actual results differ, these estimates will be adjusted.
Net sales are presented net of value added tax and other sales related taxes.
COST OF SALES
Cost of sales includes primarily the purchase cost of the goods sold and transportation costs.
OTHER OPERATING INCOME
Grants and R&D incentives
As we carry out extensive research and development activities, we benefit from various grants and R&D incentives from certain governmental agencies. These grants and R&D incentives generally aim to partly reimburse (approved) expenditures incurred in our research and development efforts and are credited to the income statement, under other income, when the relevant expenditure has been incurred and there is reasonable assurance that the grants or R&D incentives are receivable.
SHARE-BASED PAYMENTS
a/ Equity-settled share based payments
We grant equity-settled incentives to certain employees, members of the Executive Committee and consultants in the form of subscription rights. Equity-settled subscription rights are measured at fair value at the date of acceptance. The fair value determined at the acceptance date of the subscription rights is expensed over time until the end of the vesting period, based on our estimate of subscription right warrants that are expected to be exercised. Fair value is measured by use of the Black & Scholes model. The expected life used in the model has been adjusted, based on management’s best estimate, for the effects of non-transferability, exercise restrictions, and behavioral considerations.
b/ Long-term incentive plans in RSUs (Restricted Stock Units)
Executive Committee members and other employees are granted RSUs. An RSU is a grant that takes the form of a promise that employees will receive Galapagos stock in the future and it will be payable, at the company’s discretion in cash or in shares, upon completion of a certain vesting period. Each RSU reflects the value of one Galapagos share.
F-24
The RSUs are measured based on the average share price over the 30-calendar day period preceding the measurement date. We recognize the corresponding expense and liability over the vesting period. The fair value of the liability is re-measured at each reporting date because currently it is management’s intention to settle the RSUs in cash.
SEGMENT REPORTING
We currently have
ASSETS HELD FOR SALE AND DISCONTINUED OPERATIONS
A discontinued operation is a component of an entity that either has been disposed of, or that is classified as held for sale. It must either: represent a major separate line of business or geographical area of operations; be part of a single coordinated disposal plan; or be a subsidiary acquired exclusively with a view to resale.
Intercompany transactions between continuing and discontinued operations are eliminated against discontinuing operations.
Non-current assets and disposal groups are classified as assets held for sale if their carrying amount is to be recovered principally through a sale transaction rather than through continuing use. This condition is regarded as met only when the sale is highly probable and the asset (or disposal group) is available for immediate sale in its present condition. A transaction is assumed to be highly probable if there are no significant risks of completion of the transaction, which depends on the specific circumstances but usually required at least an agreed binding term sheet.
They are stated at the lower of carrying amount and fair value less costs to sell with any resulting impairment recognized. Assets related to discontinued operations and assets of disposal group held for sale are not depreciated.
On November 23, 2020 we signed a share purchase agreement with Selvita S.A. in relation to the disposal of Fidelta d.o.o. (our previous fee-for-service segment). The transaction was completed on January 4, 2021 for a total consideration of €
On October 30, 2023, we signed a letter of intent to transfer our Jyseleca® business to Alfasigma and the final agreement was signed on December 30, 2023. We classified the assets and the associated liabilities of the Jyseleca® business as held for sale in our financial statements for the year ended December 31, 2023. The transaction was closed on January 31, 2024.
Where applicable and in accordance with IFRS 5, we have restated the 2022 and 2021 comparatives in the consolidated income statement and in the notes to consider the impact of classifying the Jyseleca® business as discontinued operations in 2023.
We refer to note 5 of our consolidated financial statements.
4. Critical accounting judgments and key sources of estimation uncertainty
In the application of the accounting policies, we are required to make judgments, estimates and assumptions about the carrying amounts of assets and liabilities that are not readily apparent from other sources. The estimates and associated assumptions are based on historical experience and other factors that are considered to be relevant. Actual results may differ from these estimates.
Our estimates and assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimate is revised if the revision affects only that period or in the period of the revisions and future periods if the revision affects both current and future periods.
The following are the critical judgments that we have made in the process of applying the accounting policies and the key sources of estimation that have the most significant effect on the amounts recognized in the consolidated financial statements presented elsewhere in this annual report.
F-25
Critical judgments in applying accounting policies
IFRS 15 – Revenue recognition of the collaboration with Gilead for the development of filgotinib (reported within the results from discontinued operations)
Our critical judgments were as follows:
Identification of the contract
| ● | Despite the recent additional amendment to the collaboration with Gilead for the development of filgotinib (reference is made to note 2), management judged that all activities are still beneficial for the further development of filgotinib, for which Gilead still owns the ex-Europe rights. All contract modifications have thus been analyzed following the requirements of IFRS 15 as we concluded that Gilead is still to be considered as a customer. This is also supported by the fact that we concluded that there continues to be only one performance obligation with respect to filgotinib. |
Identification of the performance obligation
| ● | The recent modifications to the collaboration with Gilead (reference is made to note 2) did not give rise to new performance obligations. There was only a change in scope and price of the existing filgotinib performance obligation, which was only partly satisfied at the time of the modification. Based on this, the contract modification has been treated on a cumulative catch-up basis under IFRS 15. |
Allocation of the total transaction price
| ● | We assessed that the contract modification only changes the scope of the filgotinib performance obligation and the change in both fixed and variable consideration is reflective of the updated stand-alone selling price for the remaining activities of this performance obligation. If we would have concluded that the increased consideration was not, or only partially, related to the filgotinib performance obligation, the consideration would have been potentially allocated to other performance obligations in the contract, which would alter the timing of revenue recognition. |
| ● | The denominator used in the calculation of the percentage of completion reflects our best estimate of our total costs to complete the filgotinib performance obligation. These costs were assessed considering management’s best estimate of the design and duration of ongoing and planned clinical trials and the expected closing of the transaction with Alfasigma. As a result of this transaction, the contract with Gilead relating to filgotinib will be transferred to Alfasigma and we will be released from our performance obligation. The remaining costs per December 31, 2023 mainly reflect the costs that we still estimate to incur before the transfer to Alfasigma. |
IFRS 5 – Classification of group of assets/liabilities held for sale (disposal group) and discontinued operations
| ● | Management determined that selling the Jyseleca® business represents a “discontinued operation” in accordance with IFRS 5. We assessed that the Jyseleca® business represents a component of the group for which the related operations and cashflows could be distinguished from the rest of the entity. Jyseleca® is our only commercialized product and represents a major line of business. |
| ● | Management assessed that, at the reporting date, the sale of the Jyseleca® business to Alfasigma was highly probable. A letter of intent was signed on October 30, 2023 and included a customary break-up fee in the event that the parties would not proceed with definitive agreements (share and asset purchase agreement and transition agreement). These definitive agreements were signed on December 30, 2023 and only included usual and customary closing conditions. Based on this, we assessed that the sale was highly probable and classified the disposal group as held for sale per December 31, 2023. |
| ● | Our inventories were not considered to be part of the disposal group held for sale. The inventories will not transfer to Alfasigma on closing of the sale transaction but will gradually be transferred to Alfasigma over the coming years. In the meantime, we will bear all risks related to these inventories. |
We refer to note 5 for more information about the discontinued operations and disposal group held for sale.
F-26
Key sources of estimation uncertainty
The following are the key sources of estimation uncertainty that have the most significant effect on the amounts recognized in our consolidated financial statements for the year ended December 31, 2023.
Costs to complete the filgotinib performance obligation
The denominator used in the calculation of the percentage of completion reflects our best estimate of the total costs to complete the filgotinib performance obligation (which is composed of the actual costs already incurred at reporting date and our best estimate of the remaining costs to complete the performance obligation). As our estimate of the costs is depending on the evolution of the development activities and the expected closing date of the transfer of the Jyseleca® business to Alfasigma, it may be subject to change in the future. If the outcome of certain activities would be different from the assumptions that we made, it could lead to a material adjustment to the total estimated costs, resulting in a reallocation of revenue between current and future periods. Our total deferred income balance related to this filgotinib performance obligation amounts to €
At reporting date, had our best estimate of the remaining cost to complete the filgotinib performance obligation been increased by
We refer to note 5 for more information on the results from discontinued operations.
Goodwill impairment
Determining whether goodwill is impaired requires an estimation of the recoverable amount of the cash-generating unit to which the goodwill has been allocated. The calculation of this recoverable amount includes forecasts of future cash flows of the cash-generating unit (highly dependent upon the probability of success linked to the progress of our clinical programs) that cover a period of 17 years and an appropriate discount rate is required to calculate present values, a process which involves estimates. Given that the calculation contains cashflows that go beyond the 5-years horizon it becomes less verifiable and more assumptions are used. Unexpected events, inherent in the business, can cause that results are completely different than the ones predicted. These estimates are constantly monitored and an impairment test will be executed as soon as there is an impairment indicator and at least annually. The carrying value of goodwill at December 31, 2023 is €
We refer to note 13 for more information about the goodwill and impairment of goodwill.
Contingent consideration
The contingent consideration included in the consideration payable for the acquisition of CellPoint was recorded at fair value at the date of acquisition and is updated at each reporting date. The carrying amount at 31 December 2023 amounts to €
We refer to note 25 for more information about the contingent consideration payable for the acquisition of CellPoint.
5. Discontinued operations and assets held for sale
TRANSFER OF JYSELECA® BUSINESS TO ALFASIGMA
On October 30, 2023 we announced that we had signed a letter of intent contemplating a transfer of the Jyseleca® business to Alfasigma, including the European and UK Marketing Authorizations, the commercial, medical and
F-27
development activities for Jyseleca and approximately
On December 31, 2023, the transaction was still subject to certain closing conditions such as the finalization of the consultation process with the workers councils and FDI clearance in Italy and France and Denmark. The transaction was closed on January 31, 2024, upon obtaining all necessary approvals. We received a €
On January 31, 2024, we also signed a transition agreement with Alfasigma enacting the responsibilities and services that will be provided by the parties during a transition period for the transfer of the business. The gradual transfer of our remaining inventories to Alfasigma is also governed by this contract.
The transfer of our Jyseleca® business has been determined to meet the criteria to be classified as held for sale and discontinued operations in our financial statements for the year ended December 31, 2023.
The post-tax result from discontinued operations can be disaggregated in the following items:
Year ended December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
(Euro, in thousands, except share and per share data) | |||||||||
Product net sales | € | | € | | € | | |||
Collaboration revenues | | | | ||||||
Total net revenues | | | | ||||||
Cost of sales | ( | ( | ( | ||||||
Research and development expenditure | ( | ( | ( | ||||||
Sales and marketing expenses | ( | ( | ( | ||||||
General and administrative expenses | ( | ( | ( | ||||||
Other operating income | | | | ||||||
Operating profit/loss (-) | | ( | | ||||||
Fair value adjustments and net currency exchange differences | ( | ( | — | ||||||
Other financial income | | | | ||||||
Other financial expenses | ( | ( | ( | ||||||
Profit/loss (-) before tax | | ( | ( | ||||||
Income taxes | ( | ( | ( | ||||||
Net profit/loss (-) | € | | € | ( | € | ( | |||
Basic and diluted earnings/loss (-) per share from discontinued operations | € | | € | ( | € | ( | |||
Weighted average number of shares (in thousands of shares) | | | | ||||||
Weighted average number of shares - Diluted (in thousands of shares) | | | | ||||||
Jyseleca® product net sales in Europe amounted to €
F-28
Collaboration revenues in discontinued operations related to revenue recognition of the collaboration agreement with Gilead for the filgotinib development amount to €
Effective January 31, 2024, following the closing of the transaction between us and Alfasigma S.p.A. to transfer the Jyseleca® business to Alfasigma, we assigned our rights and obligations under the filgotinib collaboration with Gilead to Alfasigma, except for our right to receive royalties from Gilead on net sales in the Gilead Territory under a separate agreement between Gilead and us entered into in October 2023. As a consequence, our performance obligation towards Gilead for the development of filgotinib will come to its end, and the total estimated remaining costs to complete the filgotinib development was substantially reduced leading to a major increase in the percentage of completion of our performance obligation (applying the “cost-to-cost” input model) and a considerable positive catch-up of revenue explaining the increase in revenue recognition for the year 2023 compared to 2022.
We refer to note 2 “Summary of significant transactions” for a general description of our collaboration with Gilead.
On December 31, 2023, the remaining deferred income related to the filgotinib development amounts to €
The following major classes of assets and liabilities relating to these operations have been classified as held for sale in the consolidated statement of financial position on December 31, 2023:
Assets and liabilities held for sale
December 31, 2023 | |||
(Euro, | |||
Property, plant and equipment | € | ||
Deferred tax assets | |||
Other non-current assets | |||
Inventories | |||
Trade and other receivables | |||
Cash and cash equivalents | |||
Other current assets | |||
Total assets in disposal group classified as held for sale | | ||
| |||
| |||
Retirement benefit liabilities | |||
Non-current lease liabilities | |||
Other non-current liabilities | |||
Current lease liabilities | |||
Trade and other liabilities | |||
Current tax payable | |||
Current deferred income | |||
Total liabilities directly associated with assets in disposal group classified as held for sale | | ||
Net liability held for sale | € | ( | |
This disposal group contains all assets and liabilities of the Galapagos subsidiaries that were fully dedicated to the Jyseleca® business and that will be transferred to Alfasigma in the current transaction. The divestiture includes
F-29
Biopharma Sweden AB, Galapagos Biopharma Finland Oy, Galapagos Biopharma Ireland Ltd., Galapagos Biopharma Norway AS, Galapagos Biopharma Austria GmbH. In addition, and as part of the same transaction, we will transfer all assets, liabilities and employees directly related to the Jyseleca® business but belonging to Galapagos NV or other Galapagos subsidiaries, of which the main asset is the worldwide IP relating to Jyseleca®. Our inventories were not considered as part of the disposal group as these did not transfer to Alfasigma on closing of the transaction on January 31, 2024 but will gradually transfer to Alfasigma during the coming years and we will bear the risks associated with it as long as it is not transferred.
Held for sale assets are stated at their carrying amount, which is lower than the fair value less costs to sell. We concluded that the expected present value of the purchase price to be obtained from Alfasigma for the sale of the Jyseleca® business approximates the fair value less costs to sell of the disposal group.
Cash flow
| 2023 |
| 2022 |
| 2021 | ||||
(Euro, | |||||||||
Net cash flow used in operating activities | € | ( | € | ( | € | ( | |||
Net cash flow used in investing activities | ( | ( | ( | ||||||
Net cash flow used in financing activities | ( | ( | ( | ||||||
Net cash flow used in discontinued operations | € | ( | € | ( | € | ( | |||
DISPOSAL OF FIDELTA
On November 23, 2020 we signed a share purchase agreement with Selvita S.A. in relation to the disposal of Fidelta d.o.o. (our previous fee-for-service segment).
The transaction was completed on January 4, 2021 for a total consideration of €
Consideration received
| (Euro, in thousands) | ||
Cash received | € | | |
Total cash received | € | | |
F-30
Analysis of assets and liabilities over which control was lost
January 4, 2021 | |||
(Euro, in thousands) | |||
Intangible assets | € | ||
Property, plant and equipment | |||
Other non-current assets | |||
Trade and other receivables | |||
Cash and cash equivalents | |||
Other current assets | |||
Total assets | | ||
| |||
| |||
Non-current lease liabilities | |||
Other non-current liabilities | |||
Trade and other liabilities | |||
Current lease liabilities | |||
Income tax payable | |||
Total liabilities | | ||
Net assets disposed of | € | | |
Gain on disposal of Fidelta
(Euro, in thousands) | |||
Cash received | € | ||
Net assets disposed of | ( | ||
Effect of cumulative translation adjustment reclassified from equity on loss of control | ( | ||
Costs associated to the sale | ( | ||
Gain on disposal | € | | |
Net cash proceeds from the disposal of Fidelta
(Euro, in thousands) | |||
Cash received | € | ||
Less: cash and cash equivalents balances disposed of | ( | ||
Total consideration received, net of cash disposed of | | ||
Costs associated to the sale | ( | ||
Cash in from disposal of Fidelta, net of cash disposed of | € | | |
F-31
RESULT FROM DISCONTINUED OPERATIONS
Year ended December 31, | ||||||||||
| 2023 |
| 2022 | 2021 | ||||||
(Euro, in thousands, except share and per share data) | ||||||||||
Gain on disposal of subsidiaries | € | — | € | — | € | | ||||
Operating profit | — | — | | |||||||
Net profit | € | — | € | — | € | | ||||
Basic and diluted earnings per share from discontinued operations | € | — | € | — | € | | ||||
Weighted average number of shares (in thousands of shares) | — | — | | |||||||
Weighted average number of shares - Diluted (in thousands of shares) | — | — | | |||||||
CASH FLOW FROM DISCONTINUED OPERATIONS
| 2023 |
| 2022 |
| 2021 | ||||
(Euro, in thousands) | |||||||||
Net cash flows generated from investing activities | € | — | € | — | € | | |||
Net cash flow from discontinued operations | € | — | € | — | € | | |||
6. Segment information
We currently operate as a single operating segment.
GEOGRAPHICAL INFORMATION
In 2021, 2022 and 2023, our continuing operations were mainly located in Belgium, France, the Netherlands, Switzerland and United States. The revenues from our collaboration partner Gilead represented nearly
Following table summarizes the net revenues by destination of customer:
Year ended December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
(Euro, in thousands) | |||||||||
United States of America | € | | € | | € | | |||
Europe | | | | ||||||
Total net revenues | € | | € | | € | | |||
minus: | |||||||||
United States of America | € | | € | | € | | |||
Europe | | | | ||||||
Total net revenues from discontinued operations | € | | € | | € | | |||
United States of America | € | | € | | € | | |||
Europe | | | | ||||||
Total net revenues from continuing operations | € | | € | | € | | |||
F-32
As of December 31, 2023, we held €
December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
(Euro, in thousands) | |||||||||
Belgium | € | | € | | € | | |||
France | | | | ||||||
The Netherlands | | | | ||||||
Switzerland | | | | ||||||
Spain | — | | | ||||||
United States of America | | | | ||||||
Other | — | | | ||||||
Total non-current assets | € | | € | | € | | |||
F-33
7. Total revenues
COLLABORATION REVENUES
The following table summarizes details of collaboration revenues for the years ended December 31, 2023, 2022 and 2021 by collaboration and by category of revenue: upfront payments and license fees, reimbursement income, and royalties.
Over time |
| Point in time | 2023 |
| 2022(*) |
| 2021(*) | |||||
(Euro, in | (Euro, in | (Euro, in | ||||||||||
thousands) | thousands) | thousands) | ||||||||||
Recognition of non-refundable upfront payments and license fees | € | € | € | |||||||||
Gilead collaboration agreement for drug discovery platform | Ö | |||||||||||
Reimbursement income | - | - | ||||||||||
Novartis collaboration agreement for MOR106 | Ö | - | - | |||||||||
Royalties | ||||||||||||
Gilead royalties on Jyseleca | Ö | |||||||||||
Other royalties | Ö | |||||||||||
Total collaboration revenues | € | | € | | € | |||||||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
We recognize the consideration from Gilead allocated to the drug discovery platform on a linear basis over the 10-year period of our collaboration, of which we recognized €
Since signing of the letter of intent with Alfasigma in October 2023, we classified all activities that were directly related to the Jyseleca® business, including the revenue recognition related to the filgotinib performance obligation, as discontinued operations in accordance with IFRS 5. We refer to note 5 “Discontinued Operations” for additional information.
For the year ended 31 December 2023 we also recognized in revenue €
Collaboration with Gilead
We refer to note 2 of these consolidated financial statements for a general description of our collaboration with Gilead.
In addition, we concluded as follows for the remaining performance obligations:
Access rights to the drug discovery platform, option rights and R&D activities
F-34
8. Operating costs and other operating income
RESEARCH AND DEVELOPMENT EXPENDITURE
The following table summarizes research and development expenditure for the years ended December 31, 2023, 2022 and 2021.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| 2021(*) | ||||
(Euro, in thousands) | |||||||||
Personnel costs | € | ( | € | ( | € | ( | |||
Subcontracting | ( | ( | ( | ||||||
Disposables and lab fees and premises costs | ( | ( | ( | ||||||
Depreciation | ( | ( | ( | ||||||
Professional fees | ( | ( | ( | ||||||
Other operating expenses | ( | ( | ( | ||||||
Total R&D expenses | € | ( | € | ( | € | ( | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The table below summarizes our research and development expenditure for the years ended December 31, 2023, 2022 and 2021, broken down by program.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| 2021(*) | ||||
(Euro, in thousands) | |||||||||
Ziritaxestat program | € | — | € | ( | € | ( | |||
SIKi program | ( | ( | ( | ||||||
TYK2 program on GLPG3667 | ( | ( | ( | ||||||
CAR-T programs in oncology | ( | ( | — | ||||||
Other programs | ( | ( | ( | ||||||
Total R&D expenses | € | ( | € | ( | € | ( | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
SALES AND MARKETING EXPENSES
The following table summarizes the sales and marketing expenses for the years ended December 31, 2023, 2022 and 2021.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| 2021(*) | ||||
(Euro, in thousands) | |||||||||
Personnel costs | € | ( | € | ( | € | ( | |||
Depreciation | ( | ( | ( | ||||||
External outsourcing costs | ( | ( | ( | ||||||
Professional fees | ( | ( | ( | ||||||
Other operating expenses | ( | ( | ( | ||||||
Total sales and marketing expenses | € | ( | € | ( | € | ( | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
F-35
GENERAL AND ADMINISTRATIVE EXPENSES
The following table summarizes the general and administrative expenses for the years ended December 31, 2023, 2022 and 2021.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| 2021(*) | ||||
(Euro, in thousands) | |||||||||
Personnel costs | € | ( | € | ( | € | ( | |||
Depreciation and impairment | ( | ( | ( | ||||||
Legal and professional fees | ( | ( | ( | ||||||
Other operating expenses | ( | ( | ( | ||||||
Total general and administrative expenses | € | ( | € | ( | € | ( | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
OTHER OPERATING INCOME
The following table summarizes other operating income for the years ended December 31, 2023, 2022 and 2021.
Year ended December 31, | |||||||||
2023 | 2022(*) | 2021(*) | |||||||
(Euro, in thousands) | |||||||||
Grant income | € | | € | | € | | |||
R&D incentives income | | | | ||||||
Other income | | | | ||||||
Total other operating income | € | | € | | € | | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The grant income in 2023, 2022 and 2021was fully related to grants from a Flemish agency and the Belgian government. In many cases these grant agreements carry clauses which require us to maintain a presence in the same region for a number of years and invest according to pre-agreed budgets. Grant income in 2023 also included a grant of €
R&D incentives income was primarily composed of:
Year ended December 31, | |||||||||
2023 | 2022(*) | 2021(*) | |||||||
(Euro, in thousands) | |||||||||
Income from innovation incentive system in France | € | | € | | € | | |||
Income from Belgian R&D incentives | | | | ||||||
Tax rebates on payroll withholding taxes of R&D personnel (Belgium & the Netherlands) | | | | ||||||
Total R&D incentives income | € | | € | | € | | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
F-36
9. Staff costs
The table below summarizes the number of employees of our continuing operations on December 31, 2023, 2022 and 2021:
| 2023 |
| 2022 |
| 2021 | ||||
Number of employees on December 31 | | ||||||||
The average number of FTE’s of our continuing operations during the years 2023, 2022 and 2021 was:
Year ended December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
Members of the Executive Committee | | | | ||||||
Research and development | | | | ||||||
Commercial and medical affairs | | | | ||||||
Corporate and support | | | | ||||||
Total | | | | ||||||
Their aggregate remuneration comprised:
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| 2021(*) | ||||
(Euro, in thousands) | |||||||||
Wages and salaries | € | ( | € | ( | € | ( | |||
Social security costs | ( | ( | ( | ||||||
Pension costs | ( | ( | ( | ||||||
Costs related to subscription right plans | ( | ( | ( | ||||||
Other personnel costs | ( | ( | ( | ||||||
Total personnel costs | € | ( | € | ( | € | ( | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Reference is made to note 31 “Share-based payments” for more information on our subscription right plans.
F-37
10. Financial result
The following table summarizes the financial result, consisting of fair value adjustments and net currency exchange differences, other financial income and other financial expenses for the years ended December 31, 2023, 2022 and 2021.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| 2021(*) | ||||
(Euro, in thousands) | |||||||||
Fair value adjustments and net currency exchange differences: | |||||||||
Net unrealized currency exchange gain/loss (-) | € | ( | € | | € | | |||
Net realized currency exchange gain/loss (-) | ( | | ( | ||||||
Fair value re-measurement of warrants | | | | ||||||
Fair value loss on financial assets held at fair value through profit or loss | ( | — | ( | ||||||
Fair value gain on current financial investments | | | | ||||||
Total fair value adjustments and net currency exchange differences | | | | ||||||
Other financial income: | |||||||||
Interest income | | | | ||||||
Discounting effect of non-current R&D incentives receivables | | | | ||||||
Discounting effect of other non-current liabilities | | — | — | ||||||
Other finance income | | | | ||||||
Total other financial income | | | | ||||||
Other financial expenses: | |||||||||
Interest expenses | ( | ( | ( | ||||||
Discounting effect of other non-current liabilities | — | ( | — | ||||||
Other finance charges | ( | ( | ( | ||||||
Total other financial expense | ( | ( | ( | ||||||
Total net other financial income | € | | € | | € | | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
The net currency unrealized exchange loss of million in 2023 primarily related to €
The fair value gain on the current financial investments reflects the exchange differences booked on the money market funds, the interest on the money market funds and the effect of the re-measurement at fair value of the money market funds on December 31, 2023. These re-measurement gains are mainly the result of the positive returns on the EUR denominated money market funds.
Interest income was related to interests on term deposits, notice accounts and current financial investments. Interest income increased due to increased interest rates.
Interest expenses were related to interests on term deposits and treasury bills, on leases of buildings and cars. Other financial expense for 2022 also comprise the discounting effect of other non-current liabilities as deferred consideration and milestones payables related to the acquisition of subsidiaries.
F-38
11. Income taxes
The following table summarizes the income taxes recognized in profit or loss for the years ended December 31, 2023, 2022 and 2021.
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| 2021(*) | ||||
(Euro, in thousands) | |||||||||
Continuing operations | |||||||||
Current tax | € | ( | € | ( | € | ( | |||
Deferred tax | ( | | ( | ||||||
Income taxes | € | ( | € | ( | € | ( | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
Current tax, consisting of corporate income taxes, and deferred tax income/cost (–) related to subsidiaries of our continuing operations working on a cost plus basis. The increase in 2023 as compared to 2022 was primarily due to the re-assessment of net deferred tax liabilities and corporate income tax payables as a result of a one-off intercompany transaction.
TAXES RECOGNIZED IN INCOME STATEMENT
For the purpose of the disclosure below corporation tax was calculated at
Year ended December 31, | |||||||||
| 2023 |
| 2022(*) |
| 2021(*) | ||||
(Euro, in thousands) | |||||||||
Profit/loss (-) before tax | € | | € | ( | € | ( | |||
Income tax debit/credit (-), calculated using the Belgian statutory tax rate on the accounting income/loss (-) before tax (theoretical) | | ( | ( | ||||||
Tax expenses in income statement (effective) | | | | ||||||
Difference in tax expense/income to explain | € | | € | | € | | |||
Effect of tax rates in other jurisdictions | € | ( | € | ( | € | ( | |||
Effect of non-taxable income | ( | ( | ( | ||||||
Effect of share based payment expenses without tax impact | | | | ||||||
Effect of expenses/income (-) not subject to tax | ( | ( | ( | ||||||
Effect of non tax-deductible expenses | | | | ||||||
Effect of recognition of previously non-recognized deferred tax assets | ( | ( | ( | ||||||
Effect of tax losses (utilized) reversed | ( | — | — | ||||||
Effect of under or over provision in prior periods | ( | | ( | ||||||
Effect of non-recognition of deferred tax assets | | | | ||||||
Effect of derecognition of previously recognized deferred tax assets | | | | ||||||
Effect of use of investment deduction | — | — | ( | ||||||
Effect of use of innovation income deduction | ( | — | — | ||||||
Total explanations | € | | € | | € | | |||
(*) The 2022 and 2021 comparatives have been restated to reflect the impact of classifying the Jyseleca® business as discontinued operations in 2023.
F-39
Non-taxable income for the years ended December 31, 2023, 2022 and 2021 related to non-taxable grants and tax credits.
12. Earnings/loss (-) per share
Year ended December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
Earnings/loss (-) per share: | |||||||||
Net profit/loss (-) attributable to owners of the parent (Euro, in thousands) | € | | € | ( | € | ( | |||
Number of shares (thousands) | |||||||||
Weighted average number of shares for the purpose of basic earnings/loss (-) per share | | | | ||||||
Basic earnings/loss (-) per share (Euros) | € | | € | ( | € | ( | |||
Net profit/loss (-) attributable to owners of the parent (Euro, in thousands) | € | | € | ( | € | ( | |||
Number of shares (thousands) | |||||||||
Weighted average number of shares for the purpose of diluted earnings/loss (-) per share | | | | ||||||
Number of dilutive potential ordinary shares | | — | — | ||||||
Diluted earnings/loss (-) per share (Euros) | € | | € | ( | € | ( | |||
As our operations reported a net loss in 2022 and 2021, the outstanding subscription rights (specified in note 31) have an anti-dilutive effect rather than a dilutive effect. Consequently, basic and diluted loss per share were the same for 2022 and 2021.
Reference is also made to note 2 where an explanation is provided about the terms and conditions of the outstanding Gilead Warrant B that can, potentially, be exercised by Gilead and lead to a dilutive effect. Due to the exercise price mechanism of the Gilead Warrant B, this warrant was out-of-the-money for all years presented.
13. Goodwill and impairment of goodwill
The following table illustrates the goodwill at December 31, 2023:
Cost | (Euro, in thousands) | ||
On January 1, 2022 | € | — | |
Recognized on acquisition of subsidiaries | |||
Exchange differences on goodwill | ( | ||
On December 31, 2022 | € | | |
Exchange differences on goodwill | ( | ||
On December 31, 2023 | € | | |
The goodwill resulting from both the acquisition of CellPoint (€
The valuation technique that was applied to determine the fair value less costs of disposal of the cash-generating unit is a discounted cash flow method (“DCF”) with projected cash flows that cover a period of years (in accordance with management's assumptions on patent protection of the underlying assets). The period considered exceeds five years because the main sales are expected for the period beyond 2029. The key assumptions used in this valuation (level 3 in the fair value hierarchy) of the recoverable amount of the underlying cash-generating unit were:
| ● | Probability of success of our clinical programs that is based on benchmarks in combination with management estimate. Probabilities of success are continuously evaluated in light of the progress of our portfolio. |
| ● | Terminal growth rate of - |
| ● | Discount rate of |
F-40
| ● | Future revenue and investment assumptions are based on management estimates of the overall cell therapy market, consistent with the assumptions that a market participant would make. Estimates about sales volumes and prices were verified against several external databases. |
Reference is made to note 27 “Business combinations during the prior period” for a detailed description of both business combinations.
14. Intangible assets other than goodwill
| Software & |
| Licenses, technology and in-process R&D | Exclusive rights | Contract costs | Total | |||||||||
(Euro, in thousands) | |||||||||||||||
Acquisition value | |||||||||||||||
On January 1, 2022 | € | | € | | € | — | € | | € | | |||||
Impact of acquisitions of businesses | | | | | |||||||||||
Additions | | | | ||||||||||||
Sales and disposals | ( | ( | ( | ||||||||||||
Translation differences | ( | ( | |||||||||||||
On December 31, 2022 | | | | | € | | |||||||||
Additions | | — | — | | |||||||||||
Sales and disposals | ( | ( | ( | ||||||||||||
Translation differences | ( | ( | |||||||||||||
On December 31, 2023 | € | | € | | € | | € | | € | | |||||
Amortization and impairment | |||||||||||||||
On January 1, 2022 | € | | € | | € | — | € | | € | | |||||
Amortization | | | | | | ||||||||||
Impairment | | | |||||||||||||
Sales and disposals | ( | ( | ( | ||||||||||||
Translation differences | ( | ( | |||||||||||||
On December 31, 2022 | | | | | | ||||||||||
Amortization | | | | | | ||||||||||
Sales and disposals | ( | ( | ( | ||||||||||||
Translation differences | ( | ( | |||||||||||||
On December 31, 2023 | € | | € | | € | | € | | € | | |||||
Carrying amount | |||||||||||||||
On December 31, 2022 | € | | € | | € | | € | | € | | |||||
On December 31, 2023 | € | | € | | € | | € | | € | | |||||
Impact of acquisition of businessess in 2022 refers to the acquisition of CellPoint and AboundBio. We refer to note 27 ‘Business combinations during the prior period’.
The exclusive rights refer to our exclusivity contract with Lonza and are depreciated until the beginning of March 2030, in accordance with the contract.
In 2022 we recorded an impairment of €
F-41
On December 31, 2023, our statement of financial position did not hold any internally generated assets capitalized as intangible asset.
15. Property, plant and equipment
FULLY OWNED |
| Land, buildings & |
| Installation & |
| Furniture, |
| Other |
| Total | |||||
(Euro, in thousands) | |||||||||||||||
Acquisition value | |||||||||||||||
On January 1, 2022 | € | | € | | € | | € | | € | | |||||
Impact of acquisitions of businesses | | | | | |||||||||||
Additions | | | | | | ||||||||||
Sales and disposals | ( | ( | ( | ( | |||||||||||
Reclassifications | | | | ( | — | ||||||||||
Translation differences | | ( | | | |||||||||||
On December 31, 2022 | | | | | | ||||||||||
Additions | | | | | | ||||||||||
Sales and disposals | ( | ( | ( | ( | ( | ||||||||||
Reclassifications | | | | ( | — | ||||||||||
Reclassifications to assets in disposal group classified as held for sale | ( | ( | ( | ||||||||||||
Translation differences | | ( | | | |||||||||||
On December 31, 2023 | € | | € | | € | | € | | € | | |||||
Depreciations and impairment | |||||||||||||||
On January 1, 2022 | € | | € | | € | | € | — | € | | |||||
Depreciation | | | | | |||||||||||
Sales and disposals | ( | ( | ( | ( | |||||||||||
Translation differences | | ( | | | |||||||||||
On December 31, 2022 | | | | — | | ||||||||||
Depreciation | | | | | |||||||||||
Impairment | | | |||||||||||||
Sales and disposals | ( | ( | ( | ( | ( | ||||||||||
Reclassifications to assets in disposal group classified as held for sale | ( | ( | ( | ||||||||||||
Translation differences | | ( | | | |||||||||||
On December 31, 2023 | € | | € | | € | | € | — | € | | |||||
Carrying amount | |||||||||||||||
On December 31, 2022 | € | | € | | € | | € | | € | | |||||
On December 31, 2023 | € | | € | | € | | € | | € | | |||||
The sales and disposals of 2023 mainly relate to the transaction with NovAlix. We refer to note 28 “Details of the NovAlix transaction” for more information.
The other tangible assets primarily consist of assets under construction, which are not yet available for use and therefore not yet depreciated as per December 31, 2023. In 2023 we recorded an impairment of €
During 2022, the construction of our new building in Oegstgeest (the Netherlands) was completed which explains the reclassification from “other tangible assets” to “land, building and building improvements” for €
F-42
RIGHT-OF-USE |
| Land & |
| Installation & |
| Furniture, |
| Total | ||||
(Euro, in thousands) | ||||||||||||
Acquisition value | ||||||||||||
On January 1, 2022 | € | | € | | € | | € | | ||||
Additions | | | | |||||||||
Sales and disposals | ( | ( | ( | ( | ||||||||
Translation differences | | ( | | |||||||||
On December 31, 2022 | | | | | ||||||||
Additions | | | | |||||||||
Sales and disposals | ( | ( | ( | ( | ||||||||
Reclassifications to assets in disposal group classified as held for sale | ( | ( | ( | |||||||||
Translation differences | | | | |||||||||
On December 31, 2023 | € | | € | | € | | € | | ||||
Depreciations and impairment | ||||||||||||
On January 1, 2022 | € | | € | | € | | € | | ||||
Depreciation | | | | | ||||||||
Sales and disposals | ( | ( | ( | ( | ||||||||
Translation differences | | ( | | |||||||||
On December 31, 2022 | | | | | ||||||||
Depreciation | | | | | ||||||||
Sales and disposals | ( | ( | ( | ( | ||||||||
Reclassifications to assets in disposal group classified as held for sale | ( | ( | ( | |||||||||
Translation differences | | | | |||||||||
On December 31, 2023 | € | | € | | € | | € | | ||||
Carrying amount | ||||||||||||
On December 31, 2022 | € | | € | | € | | € | | ||||
On December 31, 2023 | € | | € | | € | | € | | ||||
December 31, | ||||||||||||
2023 | 2022 | |||||||||||
(Euro, in thousands) | ||||||||||||
Carrying amount | ||||||||||||
Property, plant and equipment fully owned | € | | € | | ||||||||
Right-of-use | | | ||||||||||
Total property, plant and equipment | € | | € | | ||||||||
The sales and disposals of 2023 mainly relate to the transaction with NovAlix. We refer to note 28 “Details of the NovAlix transaction” for more information.
We refer to note 24 Lease liabilities for a detail of the lease liabilities related to these right-of-use assets.
There are no pledged items of property, plant and equipment. There are also no restrictions in use on any items of property, plant and equipment.
F-43
16. Other non-current assets
Other non-current assets consisted of following items:
December 31, | ||||||
| 2023 |
| 2022 | |||
(Euro, in thousands) | ||||||
Non-current restricted cash | € | | € | | ||
Financial assets held at fair value through profit or loss | | — | ||||
Non-current portion of upfront payment to NovAlix | | — | ||||
Non-current portion of advance related to the NovAliX transaction | | — | ||||
Other non-current assets | | | ||||
Total other non-current assets | € | | € | | ||
Financial assets held at fair value through profit or loss on December 31, 2023 consisted of an equity instrument of a non-listed company. We have no restrictions on the sale of this equity instrument and the asset is not pledged under any of our liabilities. The fair value of this equity instrument was determined by reference to the initial transaction price (classified as level 3 in the fair value hierarchy).
We refer to note 28 “Details of the NovAlix transaction” for more information related to the upfront payment and the advance.
17. Research and Development incentives receivables
The table below illustrates the R&D incentives receivables related captions in the balance sheet at December 31, 2023 and 2022:
December 31, | ||||||
| 2023 |
| 2022 | |||
(Euro, in thousands) | ||||||
Non-current R&D incentives receivables | € | | € | | ||
Current R&D incentives receivables | | | ||||
Total R&D incentives receivables | € | | € | | ||
The table below provides detailed information on the maturity of the non-current R&D incentives receivables reported in our statement of financial position at December 31, 2023.
December 31, 2023 | ||||||||||||||||||
Maturity date | ||||||||||||||||||
| 2025 |
| 2026 |
| 2027 |
| 2028 |
| 2029-2033 |
| Total | |||||||
(Euro, in thousands) | ||||||||||||||||||
French non-current R&D incentives receivables - discounted value | € | | | | — | — | € | | ||||||||||
Belgian non-current R&D incentives receivables - discounted value | | | | | | | ||||||||||||
Total non-current R&D incentives receivables - discounted value | € | | € | | | | | € | | |||||||||
F-44
18. Inventories
December 31, | ||||||
| 2023 |
| 2022 | |||
(Euro, in thousands) | ||||||
Raw materials | € | | € | | ||
Semi-finished products | | | ||||
Finished products | | | ||||
Total inventories | € | | € | | ||
Finished goods consisted in full out of Jyseleca® finished products.
19. Trade and other receivables and other current assets
December 31, | ||||||
| 2023 |
| 2022 | |||
(Euro, in thousands) | ||||||
Trade receivables | € | | € | | ||
Prepayments | | | ||||
Other receivables | | | ||||
Trade and other receivables | | | ||||
Accrued income | | | ||||
Deferred charges | | | ||||
Other current assets | | | ||||
Total trade and other receivables & other current assets | € | | € | | ||
Trade and other receivables decreased primarily due to the classification to assets held for sale for an amount of €
On December 31, 2023, we did not have any provision for expected credit losses since we don’t have a history of credit losses and we are not aware of any forward-looking information that could materially influence the credit risk.
20. Current financial investments
December 31, | ||||||
| 2023 |
| 2022 | |||
(Euro, in thousands) | ||||||
Money market funds | € | | € | | ||
Treasury bills | | | ||||
Term deposits | | | ||||
Total current financial investments | € | | € | | ||
Term deposits refer to non-cancellable term deposits with a maturity exceeding three months from the acquisition date. Our portfolio of treasury bills contains only AAA rated paper, issued by Belgium, Germany, France and Europe. Our money market funds portfolio consists of AAA short-term money market funds with a diversified and highly rated underlying portfolio managed by established fund management companies with a proven track record leading to an insignificant risk of changes in value. The funds have an important daily liquidity and can be easily converted to cash.
On December 31, 2023, our current financial investments included $
F-45
We refer to note 34 for more information on these current financial investments and to note 10 for more details about the fair value re-meaurements and currency exchange gains or losses in our consolidated income statement.
21. Cash and cash equivalents
December 31, | ||||||
2023 | 2022 | |||||
(Euro, in thousands) | ||||||
Cash at banks | € | | € | | ||
Term deposits | | | ||||
Cash and cash equivalents from continuing operations | € | | € | | ||
Cash and cash equivalents included in assets held for sale | | — | ||||
Total cash and cash equivalents | € | | € | | ||
Cash and cash equivalents may comprise cash at banks, bank deposits and money market funds that are readily convertible to cash and are subject to an insignificant risk of changes in value. Cash and cash equivalents at December 31, 2023 comprised a term deposit of €
On December 31, 2023 our cash and cash equivalents included $
22. Share capital
| 2023 |
| 2022 |
| 2021 | ||||
(Euro, in thousands) | |||||||||
On January 1 | € | | € | | € | | |||
Share capital increase | | | | ||||||
Costs of capital increase | — | — | — | ||||||
Share capital on December 31, | € | | € | | € | | |||
Aggregate share capital | € | | € | | € | | |||
Costs of capital increase (accumulated) | ( | ( | ( | ||||||
Share capital on December 31, | € | | € | | € | | |||
F-46
HISTORY OF SHARE CAPITAL
The history of the share capital of Galapagos NV between January 1, 2021 and December 31, 2023 is as follows:
Date |
|
|
| Share capital |
| Number of |
| Aggregate |
| Aggregate | ||||||||
January 1, 2021 | | € | | |||||||||||||||
March 19, 2021 | | | ||||||||||||||||
June 7, 2021 | | | ||||||||||||||||
September 20, 2021 | | | ||||||||||||||||
December 3, 2021 | | | ||||||||||||||||
December 31, 2021 |
| | | |||||||||||||||
March 18, 2022 | | | ||||||||||||||||
June 20, 2022 | | | ||||||||||||||||
September 27, 2022 | | | ||||||||||||||||
December 31, 2022 | | | ||||||||||||||||
March 20, 2023 | | | ||||||||||||||||
December 31, 2023 |
| | € | | ||||||||||||||
On December 31, 2023, Galapagos NV’s share capital amounted to €
All of the share issuances listed above were for cash consideration.
F-47
The below table summarizes the capital increases for the years 2021, 2022 and 2023.
(Euro, in thousands, except share data) |
| Number of shares |
| Share |
| Share |
| Share capital |
| Average exercise price subscription right |
| Closing share price on date of capital increase | ||||||
( in Euro/ subscription right) | ( in Euro/ share) | |||||||||||||||||
On January 1, 2021 | | € | | € | | € | | |||||||||||
March 19, 2021 : exercise of subscription rights | | | | | | | ||||||||||||
June 7, 2021 : exercise of subscription rights | | | | | | | ||||||||||||
September 20, 2021 : exercise of subscription rights | | | | | | | ||||||||||||
December 3, 2021 : exercise of subscription rights | | | | | | | ||||||||||||
On December 31, 2021 | | | | | ||||||||||||||
March 18, 2022 : exercise of subscription rights | | | | | | | ||||||||||||
June 20, 2022 : exercise of subscription rights | | | | | | | ||||||||||||
September 27, 2022 : exercise of subscription rights | | | | | | | ||||||||||||
On December 31, 2022 | | | | | ||||||||||||||
March 20, 2023 : exercise of subscription rights | | | | | | | ||||||||||||
On December 31, 2023 | | € | | € | | € | | |||||||||||
Other information
|
| Ordinary shares |
| Total | |||||
Par value of shares (€) | | | |||||||
The Board of Directors is authorized for a period of
| ◾ | A general authorization for capital increases up to |
| ◾ | A specific authorization for capital increases of more than |
As of December 31, 2023, an amount of €
F-48
23. Deferred tax
The following table shows the movements in deferred tax assets and deferred tax liabilities:
DEFERRRED TAX ASSETS | |||||||||||||||
| Retirement |
| Tax loss |
| Property, plant |
| Other |
| Total | ||||||
(Euro, in thousands) | |||||||||||||||
On January 1, 2022 | € | | € | | € | | € | | |||||||
Credited/charged (-) to profit or loss | | ( | | ( | |||||||||||
Reclassification | ( | ( | |||||||||||||
Charged to other comprehensive income | ( | ( | |||||||||||||
Exchange differences | | ( | | ||||||||||||
On December 31, 2022 | | | € | — | | | |||||||||
Credited/charged (-) to profit or loss | ( | | | ( | |||||||||||
Reclassification to assets/liabilities held for sale | ( | ( | |||||||||||||
Charged to other comprehensive income | | | |||||||||||||
Exchange differences | | ( | ( | ( | |||||||||||
On December 31, 2023 | € | | € | — | € | | € | | € | | |||||
DEFERRED TAX LIABILITIES | |||||||||||||||
| Intangible |
| Other |
| Total | ||||||||||
(Euro, in thousands) | |||||||||||||||
On January 1, 2022 | € | — | € | — | |||||||||||
Impact of acquisition of businesses | ( | ( | |||||||||||||
Credited/charged (-) to profit or loss | | | |||||||||||||
Reclassification | | | |||||||||||||
On December 31, 2022 | ( | € | — | ( | |||||||||||
Credited/charged (-) to profit or loss | ( | ( | ( | ||||||||||||
Exchange differences | | | |||||||||||||
On December 31, 2023 | € | ( | € | ( | € | ( | |||||||||
The unrecognized deferred tax assets on December 31, 2023 amount to €
The total amount of tax attributes and deductible temporary differences at December 31, 2023 amounted to €
F-49
The available tax losses carried forward that can be offset against possible future taxable profits amounted to €
With the exception of 2019 and 2023, we have a history of losses. We forecast to continue incurring taxable losses in the foreseeable future as we continue to invest in clinical and preclinical development programs and discovery platforms. Consequently, no net deferred tax asset was recognized as at December 31, 2023, except for our subsidiaries operating on a cost plus basis for which deferred tax assets were recognized for €
Net deferred tax liabilities were initially calculated based on the fair value of the intangible assets identified from the acquisition of CellPoint and AboundBio, adjusted by considering the related recognizable deferred tax assets. We refer to note 27 for more information on the purchase price allocation of the business combinations.
24. Lease liabilities
December 31, | December 31, | |||||||||||
2023 |
| 2022 | 2023 |
| 2022 | |||||||
(Euro, in thousands) | (Euro, in thousands) | |||||||||||
| Lease payments |
| Present value of lease payments | |||||||||
Lease liabilities | ||||||||||||
Within one year | € | | € | € | | € | ||||||
In the second to fifth years inclusive | | | ||||||||||
After five years | — | | — | | ||||||||
€ | | € | | € | | € | | |||||
Less future finance charges | ||||||||||||
Present value of lease liabilities | € | | € | | ||||||||
Less amount due for settlement within 12 months | ||||||||||||
Amount due for settlement after 12 months | € | | € | | € | | € | | ||||
We refer to note 15 Property, plant and equipment, for details on the right-of-use assets.
F-50
25. Trade and other liabilities and other non-current liabilities
December 31, | ||||||
| 2023 |
| 2022 | |||
(Euro, in thousands) | ||||||
Trade and other liabilities | € | | € | | ||
Current contingent consideration related to milestones CellPoint | — | | ||||
Current deferred consideration payable CellPoint | — | | ||||
Current financial instruments | — | | ||||
Accrued charges | | | ||||
Total trade and other liabilities | € | | € | | ||
Non-current contingent consideration related to milestones CellPoint | € | | € | | ||
Other non-current liabilities | | | ||||
Total other non-current liabilities | € | | € | | ||
The decrease in total trade and other liabilities can be largely explained by the payment of deferred consideration related to the acquisition of CellPoint.
The contingent consideration arrangement relating to the acquisition of CellPoint requires us to pay the former owners of CellPoint additional considerations up to €
The fair value measurement is based on significant inputs that are not observable in the market, which are classified as Level 3 inputs. Key assumptions in the valuation at December 31, 2022 included a discount rate of
As per December 31, 2023 changes were made to the discount rate (
F-51
26. Deferred income
The movement in the non-current and current deferred income is detailed in the table below.
| Gilead collaboration |
| Gilead collaboration |
| Other |
| Total | |||||
(Euro, in thousands) | ||||||||||||
| ||||||||||||
On January 1, 2022 | € | | € | | € | — | € | | ||||
Of which current portion : | | | — | | ||||||||
Upfront received | | | ||||||||||
Significant financing component (2) | | | ||||||||||
Revenue recognition of upfront | ( | ( | ( | |||||||||
Revenue recognition of milestones | ( | ( | ||||||||||
Other movements | | | ||||||||||
On December 31, 2022 | € | | € | | € | | | |||||
Of which current portion : | | | | | ||||||||
Reclassification to liabilities directly associated with assets in disposal group classified as held for sale | ( | ( | ||||||||||
Significant financing component (2) | ( | ( | ||||||||||
Revenue recognition of upfront | ( | ( | ( | |||||||||
Revenue recognition of milestones | ( | ( | ||||||||||
Other movements | ( | ( | ||||||||||
On December 31, 2023 | € | | € | | € | | € | | ||||
Of which current portion : | | | | | ||||||||
| (1) | The upfront received and the outstanding balance comprise the issuance liabilities for the warrants and the upfront payment allocated to the drug discovery platform. |
| (2) | With regard to the additional consideration received for the extended cost sharing for filgotinib, we assume the existence of a sigificant financing component reflecting the time value of money on the estimated recognition period |
We refer to note 2 for a detail of the allocation of the transaction price received from Gilead and to note 5 and note 7 for a description of our revenue recognition.
27. Business combinations during the prior period
On June 21, 2022 we acquired, in an all-cash transaction,
On the same date we acquired all of the outstanding capital of AboundBio, for a total agreed price of $
The main reason for these acquisitions was to position ourselves in next-generation cancer therapy market and to significantly broaden our portfolio and capabilities. The goal is to expand the current market for CAR-T therapies and have an important impact on patients in need of additional and improved treatment options.
F-52
Details of the fair value of identifiable assets and liabilities acquired in both transactions, the purchase consideration, the goodwill at the acquisition date and the net cash outflow arising on acquisition are as follows:
June 21, 2022 | |||||||||||||||||||||
(Euro, in thousands) | |||||||||||||||||||||
CellPoint | AboundBio | Total | |||||||||||||||||||
| Book value |
| Adjustment |
| Fair value |
| Book value |
| Adjustment |
| Fair value |
| |||||||||
Intangible assets other than goodwill | € | € | € | € | |||||||||||||||||
Property, plant and equipment | € | € | |||||||||||||||||||
Other non-current assets | |||||||||||||||||||||
Trade and other receivables | - | - | |||||||||||||||||||
Cash and cash equivalents | |||||||||||||||||||||
Other current assets | |||||||||||||||||||||
Deferred tax liabilities | - | ( | ( | - | ( | ( | |||||||||||||||
Trade and other liabilities | ( | ( | ( | ( | |||||||||||||||||
Current deferred income | - | - | ( | ( | |||||||||||||||||
Net assets acquired | ( | | | | | | |||||||||||||||
Consideration paid in cash | |||||||||||||||||||||
Fair value re-measurement of previously held equity investment | - | ||||||||||||||||||||
Deferred consideration | - | ||||||||||||||||||||
Fair value of contingent consideration | - | ||||||||||||||||||||
Fair value of total consideration | | | |||||||||||||||||||
Goodwill | | | |||||||||||||||||||
Exchange differences on goodwill | - | ( | |||||||||||||||||||
Goodwill in the balance sheet at December 31, 2022 | € | | € | | € | | |||||||||||||||
Net cash outflow arising on acquisition (2022) | |||||||||||||||||||||
Consideration paid in cash | |||||||||||||||||||||
Less: cash and cash equivalents balances acquired | ( | ( | |||||||||||||||||||
Cash out from acquisition of subsidiaries, net of cash acquired (paid in 2022) | € | | | € | | ||||||||||||||||
Cash used in operating activities for other liabilities related to the acquisition of subsidiaries (2022) | € | | € | | |||||||||||||||||
Cash out from acquisition of subsidiaries (payment of deferred consideration in 2023) | € | | € | | |||||||||||||||||
As part of the acquisitions, we identified the following acquired intangible assets:
| ● | IPR&D: in-process research and development related to |
| ● | Exclusive rights: through the acquisition of Cellpoint we acquired on the one hand a collaboration agreement between Cellpoint and Lonza providing the exclusive right to use the automated Lonza Cocoon® Platform in the development and commercialization of CAR-T cell products, and secondly, a collaboration agreement between Cellpoint and Hypertrust providing exclusivity to use the jointly developed XCellit® software for workflow management and monitoring for the manufacturing of the CAR-T cells using the Lonza Cocoons® Platform. The fair values at acquisition date amounted to € |
| ● | Technology: through the acquisition of AboundBio, we acquired a fully human antibody-based therapeutics platform which was valued at € |
F-53
We assessed that the carrying value of all other acquired assets and assumed liabilities approximate their fair value at acquisition date.
The goodwill arising from both transactions totaling €
The acquisition costs related to both transactions were considered not to be material and were recognized in our consolidated income statement on the line "general & administrative expenses”.
28. Details of the NovAlix transaction
We completed the integrated drug discovery collaboration transaction with NovAliX on June 30, 2023, effective as from July 1, 2023. Under the terms of the agreement, Galapagos’ drug discovery and research activities conducted in Romainville, France, and Galapagos’ employees in Romainville, which are exclusively dedicated to the operation of these activities, were transferred to NovAliX who will assume all ongoing research and discovery activities in Romainville, and this for
The collaboration agreement and sale and purchase agreement were negotiated as a package with one single commercial objective and with an agreed consideration for the transaction as a whole.
The impact of the transfer of activities and personnel (reference is made to the table below) was treated as an advance for future services to be obtained from NovaliX throughout the
| June 30, | ||
2023 | |||
(Euro, in thousands) | |||
Loss on sale of fixed assets | € | ||
Result of transfer of retirement benefit liability | ( | ||
Result of transfer of right-of-use asset | |||
Advance related to the NovAliX transaction | € | | |
Furthermore we made an upfront payment to NovAliX of €
F-54
29. Note to the cash flow statement
The following table details the adjustments related to the operating cash flow:
December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
(Euro, in thousands) | |||||||||
Adjustment for non-cash transactions | |||||||||
Depreciation and impairment | € | | € | | € | | |||
Share-based compensation expenses | | | | ||||||
Increase/decrease (-) in retirement benefit obligations and provisions | | | ( | ||||||
Unrealized exchange losses/gains (-) and non-cash other financial result | | ( | ( | ||||||
Discounting effect of non-current deferred income | ( | | | ||||||
Discounting effect of other non-current liabilities | ( | | — | ||||||
Fair value re-measurement of warrants | ( | ( | ( | ||||||
Net change in fair value of current financial investments | ( | ( | ( | ||||||
Fair value adjustment financial assets held at fair value through profit or loss | | — | | ||||||
Other non-cash expenses | | | | ||||||
Total adjustment for non-cash transactions | € | | € | | € | | |||
Adjustment for items to disclose separately under operating cash flow | |||||||||
Interest expense | € | | € | | € | | |||
Interest income | ( | ( | ( | ||||||
Tax expense | | | | ||||||
Total adjustment for items to disclose separately under operating cash flow | € | ( | € | ( | € | | |||
Adjustment for items to disclose under investing and financing cash flows | |||||||||
Gain on disposal of subsidiaries | € | — | € | — | € | ( | |||
Gain on sale of fixed assets | ( | ( | — | ||||||
Realized exchange gain on sale of current financial investments | — | — | ( | ||||||
Investment income related to current financial investments | ( | ( | ( | ||||||
Total adjustment for items to disclose separately under investing and financing cash flow | € | ( | € | ( | € | ( | |||
Change in working capital other than deferred income | |||||||||
Increase in inventories | € | ( | € | ( | € | ( | |||
Increase (-)/ decrease in receivables | ( | | | ||||||
Increase/decrease (-) in liabilities | | ( | ( | ||||||
Total change in working capital other than deferred income | € | ( | € | | € | | |||
F-55
30. Off-balance sheet arrangements
CONTRACTUAL OBLIGATIONS AND COMMITMENTS
On December 31, 2023, we had outstanding obligations for future purchase commitments, which become due as follows:
| Total |
| Less than |
| 1 - 3 |
| 3 - 5 |
| More than 5 | ||||||
(Euro, in thousands) | |||||||||||||||
Purchase commitments | € | | € | | | | | ||||||||
On December 31, 2022, we had outstanding obligations for future purchase commitments, which become due as follows:
| Total |
| Less than |
| 1 - 3 |
| 3 - 5 |
| More than 5 | ||||||
(Euro, in thousands) | |||||||||||||||
Purchase commitments | € | | € | | € | | € | | € | | |||||
Our purchase commitments at the end of the year 2023 included €
At year end 2023, our purchase commitments towards NovAliX amounted to €
At the end of the year 2023, €
At the end of the year 2023, we have remaining short-term contractual cost sharing obligations related to the termination of our collaboration agreement with Gilead for filgotinib, before its transfer to Alfasigma, amounting to €
We entered into a license agreement with another pharmaceutical company. Under the terms of this agreement we have the obligation to pay potential milestones, which are dependent on successful completion of certain development and commercial milestones, as detailed in the agreement. At December 31, 2023 this commitment amounts to €
31. Share based payments
SUBSCRIPTION RIGHT PLANS
Presented below is a summary of subscription right activities for the reported periods. Various subscription right plans were approved for the benefit of our employees, and for members of the Executive Committee and of the Board of Directors and independent consultants of Galapagos NV.
The subscription rights offered to members of the Board of Directors vest over a period of
F-56
On May 5, 2023 the Board of Directors issued “Subscription Right Plan 2023 BE”, “Subscription Right Plan 2023 RMV” and “Subscription Right Plan 2032 ROW”, for a total of
Following table shows when a subscription right becomes exercisable, per issued plan:
Subscription right exercisable as from: | Cliff vesting | Graded vesting | ||||||
| First tranche of | Second tranche of | Third tranche of | |||||
Subscription right plans before 2021 | First day after end of third calenderyear following the grant | — | — | — | ||||
Subscription right plan 2021BE | First day after end of third calenderyear following the grant | — | — | — | ||||
Subscription right plan 2021RMV and ROW | — | January 1, 2023 | January 1, 2024 | January 1, 2025 | ||||
Subscription right plan 2022 (A) | — | January 1, 2023 | January 1, 2024 | January 1, 2025 | ||||
Subscription right plan 2022 (B) | January 1, 2026 | — | — | — | ||||
Subscription right plan 2022BE | January 1, 2026 | — | — | — | ||||
Subscription right plan 2022RMV and ROW | — | January 1, 2024 | January 1, 2025 | January 1, 2026 | ||||
Subscription right plan 2023BE | January 1, 2027 | — | — | — | ||||
Subscription right plan 2023RMV and ROW | — | January 1, 2025 | January 1, 2026 | January 1, 2027 | ||||
In the event of a change of control over Galapagos NV, all outstanding subscription rights vest immediately and will be immediately exercisable.
The table below sets forth a summary of subscription rights outstanding and exercisable at December 31, 2023, per subscription right plan:
|
|
|
| Outstanding |
|
|
|
|
| Outstanding |
| Exercisable | ||||||||
per | Granted | Exercised | Forfeited | Expired | per | per | ||||||||||||||
Allocation | Expiry | Exercise | January 1, | during | during | during | during | December 31, | December 31, | |||||||||||
Subscription right plan | date | date | price (€) | 2023 | year | year | year | year | 2023 | 2023 | ||||||||||
2015 | 04/30/2015 | 04/29/2023 | | | ( | ( | — | — | ||||||||||||
2015 (B) | 12/22/2015 | 12/21/2023 | | | ( | — | — | |||||||||||||
2015 RMV | 12/22/2015 | 12/21/2023 | | | ( | — | — | |||||||||||||
2016 | 1/6/2016 | 05/31/2024 | | | | | ||||||||||||||
2016 RMV | 1/6/2016 | 05/31/2024 | | | | | ||||||||||||||
2016 (B) | 01/20/2017 | 01/19/2025 | | | | | ||||||||||||||
2017 | 05/17/2017 | 05/16/2025 | | | ( | | | |||||||||||||
2017 RMV | 05/17/2017 | 05/16/2025 | | | ( | | | |||||||||||||
2018 | 04/19/2018 | 04/18/2026 | | | ( | | | |||||||||||||
2018 RMV | 04/19/2018 | 04/18/2026 | | | ( | | | |||||||||||||
2019 | 10/4/2019 | 9/4/2027 | | | ( | | | |||||||||||||
2019 RMV | 10/4/2019 | 9/4/2027 | | | ( | | | |||||||||||||
2020 | 04/17/2020 | 04/16/2028 | | | ( | | | |||||||||||||
2020RMV | 04/17/2020 | 04/16/2028 | | | ( | | | |||||||||||||
2021BE | 04/30/2021 | 04/29/2029 | | | ( | | ||||||||||||||
2021RMV | 04/30/2021 | 04/29/2029 | | | ( | | | |||||||||||||
2021ROW | 04/30/2021 | 04/29/2029 | | | ( | | | |||||||||||||
2022 (A) | 01/13/2022 | 12/1/2030 | | | | | ||||||||||||||
2022 (B) | 01/26/2022 | 01/25/2030 | | | | |||||||||||||||
2022BE | 6/5/2022 | 5/5/2030 | | | ( | | ||||||||||||||
2022BE | 5/8/2022 | 5/5/2030 | | | | |||||||||||||||
2022RMV | 6/5/2022 | 5/5/2030 | | | ( | | | |||||||||||||
2022ROW | 6/5/2022 | 5/5/2030 | | | ( | | | |||||||||||||
2022ROW | 5/8/2022 | 4/8/2030 | | | | | ||||||||||||||
2023BE | 5/5/2023 | 4/5/2031 | | — | | ( | | |||||||||||||
2023RMV | 5/5/2023 | 4/5/2031 | | — | | ( | | |||||||||||||
2023ROW | 5/5/2023 | 4/5/2031 | | — | | ( | | |||||||||||||
2023BE | 06/15/2023 | 06/14/2031 | | — | | | ||||||||||||||
2023ROW | 11/17/2023 | 4/5/2031 | | — | | | ||||||||||||||
Total | | | ( | ( | ( | | |
F-57
|
| Weighted | ||||
average | ||||||
exercise | ||||||
Subscription rights | price (Euro) | |||||
Outstanding on January 1, 2021 | | € | | |||
Exercisable on December 31, 2020 | | | ||||
Granted during the period | | | ||||
Forfeited during the year | ( | | ||||
Exercised during the period | ( | | ||||
Expired during the year | — | — | ||||
Outstanding on December 31, 2021 | | € | | |||
Exercisable on December 31, 2021 | | | ||||
Granted during the period | | | ||||
Forfeited during the year | ( | | ||||
Exercised during the period | ( | | ||||
Expired during the year | — | — | ||||
Outstanding on December 31, 2022 | | € | | |||
Exercisable on December 31, 2022 | | | ||||
Granted during the period | | | ||||
Forfeited during the year | ( | | ||||
Exercised during the period | ( | | ||||
Expired during the year | ( | | ||||
Outstanding on December 31, 2023 | | € | | |||
Exercisable on December 31, 2023 | | | ||||
F-58
The table below sets forth the inputs into the valuation of the subscription rights.
2023BE |
| 2023RMV/ROW |
| 2022 (A) |
| 2022 (B) |
| ||||||||||||||
May 5 & June 15 | May 5 & November 17 | January 13 | January 26 | ||||||||||||||||||
Weighted average exercise price (€) | € | | € | | € | | € | | |||||||||||||
Weighted average share price at acceptance date (€) | € | | € | | € | | € | | |||||||||||||
Weighted average fair value at the acceptance date (€) | € | | € | | € | | € | | |||||||||||||
Weighted average estimated volatility (%) | | | | | |||||||||||||||||
Weighted average expected life of the subscription rights (years) | |||||||||||||||||||||
Weighted average risk free rate (%) | | | ( | | |||||||||||||||||
Expected dividends | None | None | None | None | |||||||||||||||||
2022BE | 2022 RMV/ROW | 2022BE/ROW | 2021 | 2021 RMV/ROW | |||||||||||||||||
May 6 | May 6 | August 6 |
| April 30 | April 30 | ||||||||||||||||
Exercise Price (€) | € | | € | | € | | € | | € | | |||||||||||
Weighted average share price at acceptance date (€) | € | | € | | € | | € | | € | | |||||||||||
Weighted average fair value at the acceptance date (€) | € | | € | | € | | € | | € | | |||||||||||
Weighted average estimated volatility (%) | | | | | | ||||||||||||||||
Weighted average expected life of the subscription rights (years) | |||||||||||||||||||||
Weighted average risk free rate (%) | | | | ( | ( | ||||||||||||||||
Expected dividends | None | None | None | None | None | ||||||||||||||||
Subscription right Plans
The exercise price of the subscription rights is determined pursuant to the applicable provisions of the Belgian Law of March 26, 1999.
The weighted average estimated volatility is calculated on the basis of the implied volatility of the share price over the weighted average expected life of the subscription rights.
The weighted average expected life of the subscription right is calculated as the estimated duration until exercise, taking into account the specific features of the plans.
Our share based compensation expense in 2023 in relation to subscription right plans amounted to €
The following table provides an overview of the outstanding subscription rights per category of subscription right holders at December 31, 2023, 2022 and 2021.
F-59
Category
December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 (1) | ||||
(in number of subscription rights) | |||||||||
Members of the Board of Directors | | | | ||||||
Members of the Executive Committee | | | | ||||||
Personnel | | | | ||||||
Total subscription rights outstanding | | | | ||||||
The outstanding subscription rights at the end of the accounting period have an average exercise price of €
RESTRICTED STOCK UNITS RSUs
Each RSU represents the right to receive, at Galapagos’ discretion,
We currently have the following restricted stock unit (RSU) programs:
| ● | Plan 2020.I, Plan 2021.I and Plan 2022.I and Plan 2023.1: these plans are intended to provide a long-term incentive to certain of our employees and members of the Executive Committee; |
| ● | Plan 2020.II, Plan 2021.II, Plan 2021.IV, Plan 2022.II and Plan 2023.II: these plans are designed with the aim to retain a specific group of our key employees and members of the Executive Committee whose retention is considered so important for our future performance that an additional incentive is desired. The beneficiaries are nominated by the Remuneration Committee and the Board of Directors approves the list of beneficiaries. The four-year vesting period is designed to be aligned with long-term shareholder interests; |
| ● | Plan 2021.III and Plan 2022.III: these plans are intended to compensate employees who transferred from Gilead to us in the framework of the transfer of European commercialization rights for the long-term incentive plans within Gilead under which unvested RSU awards lapsed upon transfer out of the Gilead group. These employees received a one-time Restricted Stock Units grant from us. |
The main characteristics of all these plans are as follows:
| ● | the RSUs are offered for no consideration; |
| ● | generally |
| ● | payout will be in cash or shares, at Galapagos’ discretion, it being understood that in respect of members of the Executive Committee, any vesting prior to the third anniversary of the offer date will always give rise to a payment in cash rather than a delivery of shares as an incentive; |
| ● | any unvested RSUs are forfeited upon termination of service before the vesting date. |
F-60
The table below sets forth a summary of RSUs outstanding at December 31, 2023, per RSU plan:
|
| Outstanding |
|
|
|
| Outstanding | |||||
per | Granted | Forfeited | Paid in cash | per | ||||||||
Offer | January 1, | during | during | during | December 31, | |||||||
RSU plan | date | 2023 | year | year | year | 2023 | ||||||
Plan 2019.II. | 10/16/2019 | | ( | ( | — | |||||||
Plan 2020.I. | 06/05/2020 | | ( | ( | | |||||||
Plan 2020.II. | 07/05/2020 | | ( | ( | | |||||||
Plan 2021.I. | 05/05/2021 | | ( | ( | | |||||||
Plan 2021.II. | 05/06/2021 | | ( | ( | | |||||||
Plan 2021.III. | 06/03/2021 | | ( | ( | | |||||||
Plan 2021.IV. | 09/24/2021 | | ( | ( | | |||||||
Plan 2022.I. | 03/05/2022 | | ( | ( | | |||||||
Plan 2022.II. | 5/05/2022 - 5/08/2022 | | ( | ( | | |||||||
Plan 2022.III. | 07/06/2022 | | ( | ( | | |||||||
Plan 2023.I. | 08/05/2023 | | ( | | ||||||||
Plan 2023.II. | 9/05/2023 - 15/06/2023 - 17/11/2023 | | ( | | ||||||||
Total | | | ( | ( | |
2023 |
| 2022 | 2021 | ||||||
(in number of RSUs) | |||||||||
Outstanding on January 1, | | | | ||||||
Granted during the year | | | | ||||||
Forfeited during the year | ( | ( | ( | ||||||
Paid in cash during the year | ( | ( | ( | ||||||
Outstanding on December 31, | | | | ||||||
The RSUs are measured based on the volume-weighted average price of the Galapagos share on Euronext Brussels over the
The following table provides an overview of the outstanding RSUs per category of RSU holders on December 31, 2023, 2022 and 2021.
December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
(in number of RSUs) | |||||||||
Members of the Executive Committee | | | | ||||||
Personnel | | | | ||||||
Total outstanding RSUs | | | | ||||||
32. Related parties
Relationship and transactions with entities with control of, or significant influence over, Galapagos
Gilead
Gilead exercises significant influence over Galapagos as from the equity subscription on August 23, 2019. As a result of the equity subscription we received a transparency notification from Gilead on August 28, 2019 confirming they held
By exercising Warrant A on November 6, 2019 Gilead increased its ownership in Galapagos to
F-61
The presumption of significant influence is also confirmed by the fact that Gilead has the right, for as long as it holds more than
The following table details our relation with Gilead:
December 31, | |||||||||
2023 |
| 2022 | 2021 | ||||||
(Euro, in thousands) | |||||||||
Trade and other receivables (1) | € | | € | | € | | |||
Trade and other payables | | € | | ||||||
Year ended December 31, | |||||||||
2023 |
| 2022 | 2021 | ||||||
(Euro, in thousands) | |||||||||
Revenues recognized related to the performance obligation for the drug discovery platform | € | | € | | € | | |||
Revenues recognized related to the filgotinib performance obligation (2) | | | | ||||||
Royalty income related to the commercialization of filgotinib | | | | ||||||
Cost reimbursements related to the development of GLPG1690 (3) | | | | ||||||
Cross charges from and to Gilead relating to filgotinib (4) | | ( | ( | ||||||
Costs (-)/deduction of costs relating to our 50/50 profit/(cost) share mechanism (5) | |||||||||
included in sales and marketing expenses | — | | | ||||||
included in research and development expenditure | — | ( | | ||||||
Purchase of raw materials, semi-finished products and finished products of Jyseleca | € | — | € | | € | | |||
(1) Consisting on December 31, 2023 of filgotinib development cost sharing receivables of €
(2) Upfront and milestone payments recognized in accordance with the percentage of completion of the underlying obligation.
(3) Shown as decrease of research and development expenditure.
(4) Net amount shown as an (increase)/decrease of research and development expenditure.
(5) Profit/cost share mechanism came to an end beginning of 2022.
As at December 31, 2023 we have
A detailed explanation of our transactions with Gilead in 2021, 2022 and 2023 can be found in the section titled Agreements with major Galapagos NV shareholders. There are no other shareholders or other entities who, solely or jointly, control Galapagos or exercise significant influence over Galapagos.
Relationship and transactions with subsidiaries
Please see note 33 for an overview of the consolidated companies of the group, which are all wholly-owned subsidiaries of Galapagos NV.
F-62
Relationship and transactions with key management personnel
Our key management personnel consists of the members of the Executive Committee and members of the Board of Directors. All amounts mentioned in this section are based on expenses recognized in the financial statements for the relevant financial year.
Remuneration of key management personnel
On December 31, 2023, our Board of Directors consisted of nine members: Stoffels IMC BV (permanently represented by Dr. Paul Stoffels), Mr. Simon Sturge, Dr. Susanne Schaffert, , Mr. Peter Guenter, Mr. Daniel O’Day, Dr. Linda Higgins, Dr. Elisabeth Svanberg, Mr. Jérôme Contamine and Dr. Dan Baker.
During its meeting of June 12, 2023, the Board of Directors appointed Dr. Susanne Schaffert by cooptation as a non-executive independent Director, replacing Dr. Rajesh Parekh who stepped down on June 10, 2023.
During its meeting of September 19, 2023, the Board of Directors appointed Mr. Simon Sturge by cooptation as a non-executive independent Director, replacing Dr. Mary Kerr who stepped down on September 18, 2023.
Dr. Susanne Schaffert’s and Mr. Simon Sturge’s appointments will be submitted for confirmation to the Company’s Annual Shareholders’ Meeting which will be held on April 30, 2024.
Effective from January 1, 2020, Galapagos no longer grants any subscription rights to members of the Board of Directors, taking into account the stricter rules of the Belgian Companies Code. Prior to 2020, Board members were granted subscription rights.
Effective from April 26, 2022, our new CEO, Stoffels IMC BV, permanently represented by Dr. Paul Stoffels, has been appointed as the Chair of the Board of Directors of Galapagos. The CEO will only be remunerated for the performance of its executive functions as CEO and is not entitled to any additional remuneration for its mandates of Chair of the Board of Directors or of any Committee.
F-63
The remuneration package of the members of key management personnel comprises:
Year ended December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
Remuneration of key management personnel: | |||||||||
Euro, in thousands (except for the number of subscription rights and RSUs) | |||||||||
Short-term benefits to executive Committee members as a group (1) | € | | € | | € | | |||
Board fees for members of the Board of Directors | | | | ||||||
Post-employment benefits (2) | | | | ||||||
Severance package (3) | | — | | ||||||
Subscription rights granted in the year | |||||||||
Number of subscription rights granted in the year to Executive Committee members as a group | | | | ||||||
Total cost of subscription rights granted in the year under IFRS 2 | € | | € | | € | | |||
Number of RSUs granted in the year | |||||||||
Total number of RSUs granted in the year to Executive Committee members as a group (1)(4) | | | | ||||||
(1) Dr. Wigerinck was a member of the Executive Committee (formerly Management Board) until November 30, 2021. His remuneration and benefits are included in the overview for the financial year 2021. Mr. Onno Van de Stolpe was our CEO and Executive Committee member until March 31, 2022, Dr. Andre Hoekema was our CBO and Executive Committee member until October 31, 2022 and Dr. Walid Abi-Saab was our CMO and Executive Committee member until December 31, 2022. Their (prorated) remuneration and benefits are included in the overview for the financial years 2021 and 2022. Effective as of April 1, 2022, Stoffels IMC BV, permanently represented by Dr. Paul Stoffels, is our CEO and Chair of the Executive Committee. His (prorated) remuneration is included in the overview for the financial years 2022 and 2023. Mr. Bart Filius was a member of the Executive Committee until June 30, 2023 and Mr. Michele Manto was a member of the Executive Committee until December 31, 2023. Their (prorated) remuneration and benefits are included in the overview for the financial years 2021, 2022 and 2023. Ms. Valeria Cnossen and Ms. Annelies Missotten were members of the Executive Committee as of January 1, 2023. Mr. Thad Huston was a member of the Executive Committee as of July 1, 2023. Their (prorated) remuneration and benefits are included in the overview for the financial year 2023.
(2) Only Executive Committee members receive post-employment benefits.
(3) In 2021, we disclosed Dr. Wigerinck's and Mr. Van de Stolpe's severance packages. In 2021, an amount of €
(4) This is the sum of the RSUs awarded during the respective financial year, excluding the RSUs representing the deferred portion of the bonus for 2021 in FY2021, for 2022 in FY2022, and for 2023 in FY2023 (each time to be granted in the following financial year). Only Executive Committee members were awarded RSUs.
OTHER
No loans, quasi-loans or other guarantees were given by Galapagos NV or any of its subsidiaries to members of the Board of Directors and of the Executive Committee. We have not entered into transactions with our key management personnel, other than as described above with respect to remuneration arrangements relating to the exercise or termination of their mandates as members of the Executive Committee and the Board of Directors.
F-64
33. Consolidated companies as of December 31, 2023
Year ended December 31, | ||||||||||
2023 | 2022 | 2021 | ||||||||
Name of the subsidiary |
| Country |
| % voting right |
| Change in % |
| % voting right |
| % voting right |
CONTINUING OPERATIONS | ||||||||||
Glpg US Inc. (formerly AboundBio Inc.) | United States | |||||||||
CellPoint B.V. (till 30/11/2023) | The Netherlands | |||||||||
Galapagos B.V. (merged with CellPoint B.V.) | The Netherlands | |||||||||
Galapagos GmbH | Switzerland | |||||||||
Glpg US Holding Inc. (formerly Galapagos, Inc.) | United States | |||||||||
Galapagos NV | Belgium | Parent company | Parent company | Parent company | ||||||
Galapagos Real Estate Belgium BV (former Galapagos Real Estate 1 BV) | Belgium | |||||||||
Galapagos Real Estate Netherlands B.V. | The Netherlands | |||||||||
Galapagos SASU | France | |||||||||
Xenometrix, Inc. in liquidation | United States | |||||||||
DISCONTINUED OPERATIONS | ||||||||||
Galapagos Biopharma Belgium BV | Belgium | |||||||||
Galapagos Biopharma Netherlands B.V. | The Netherlands | |||||||||
Galapagos Biopharma Spain S.L.U. | Spain | |||||||||
Galapagos Biopharma Italy S.r.l. | Italy | |||||||||
Galapagos Biopharma Germany GmbH | Germany | |||||||||
Galapagos Biopharma Sweden AB | Sweden | |||||||||
Galapagos Biopharma Norway AS | Norway | |||||||||
Galapagos Biopharma Finland Oy | Finland | |||||||||
Galapagos Biopharma Denmark ApS | Denmark | |||||||||
Galapagos Biopharma Austria GmbH | Austria | |||||||||
Galapagos Biopharma Ireland Ltd | Ireland | |||||||||
Galapagos Biotech Ltd (formerly Inpharmatica Ltd.) | United Kingdom | |||||||||
There are no significant restrictions on the group’s ability to access or use assets and settle liabilities of one of the group’s subsidiaries.
34. Financial risk management
Financial risk factors
Our financial risks are managed centrally. Our finance department coordinates the access to national and international financial markets and considers and manages continuously the financial risks concerning our activities. These relate to the following financial markets risks: credit risk, liquidity risk, currency risk and interest rate risk. Our interest rate risk is limited because we have no financial debt. In case of decreasing interest rates we will face a reinvestment risk on our strong cash and cash equivalents and current financial investments balance. We do not buy or trade financial instruments for speculative purposes.
F-65
Categories of financial assets and liabilities (the below table does not contain the financial assets and liabilities included in the disposal group held for sale – reference is made to note 5 for more information):
December 31, | ||||||||||
| 2023 |
| 2022 |
| 2021 | Notes | ||||
(Euro, in thousands) | ||||||||||
Financial assets held at fair value through profit or loss | ||||||||||
Equity instruments | € | | — | — | 16 | |||||
Current financial investments | | | | 20 | ||||||
Financial assets at amortized cost | ||||||||||
Current financial investments | | | | 20 | ||||||
Cash and cash equivalents | | | | 21 | ||||||
Restricted cash (current and non-current) | | | | 16 | ||||||
Other non-current assets | | | | 16 | ||||||
Trade receivables | | | | 19 | ||||||
Total financial assets | € | | € | | € | | ||||
Financial liabilities held at fair value through profit or loss | ||||||||||
Current financial instruments | € | — | € | | € | | 25 | |||
Current contingent consideration related to milestones CellPoint | — | | — | 25 | ||||||
Non-current contingent consideration related to milestones CellPoint | | | — | 25 | ||||||
Financial liabilities at amortized cost | ||||||||||
Trade liabilities | | | | 25 | ||||||
Lease liabilities | | | | 24 | ||||||
Current deferred consideration payable CellPoint | — | | — | 25 | ||||||
Total financial liabilities | € | | € | | € | | ||||
The carrying amounts of trade and other payables and trade and other receivables are considered to be the same as their fair values, due to their short-term nature.
Financial assets held at fair value through profit or loss
Financial assets held at fair value through profit or loss consisted of an equity instrument of a non-listed companies and current financial investments.
We have no restrictions on the sale of this equity instrument and the asset is not pledged under any of our liabilities.
The fair value of the equity instrument in the non-listed company has been determined mainly by reference to the initial transaction price (classified as level 3 in the fair value hierarchy).
Current financial investments include money market funds in EUR and USD, which all classify for level 1 fair value measurement.
Liquidity risk
Current financial investments and cash and cash equivalents amounted to €
All our and cash and cash equivalents have only an insignificant liquidity risk as they are all convertible upon a maximum three month notice period and without incurring a significant penalty in normal market circumstances.
F-66
Credit risk
The term “credit risk” refers to the risk that counterparty will default on its contractual obligations resulting in financial loss for us.
We grant credit to our clients in the framework of our normal business activities. Usually, we require no pledge or other collateral to cover the amounts due. All our receivables are considered collectable.
We did not account for a provision for expected credit losses relating to our trade and other receivables given that there is no history of material credit losses, nor does forward looking information reveals any potential risks and due to the high quality nature of our customers.
Aging balance of receivables that are due, but that are still considered collectable:
December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
(Euro, in thousands) | |||||||||
60 - 90 days | € | | € | | € | | |||
90 - 120 days | | | | ||||||
more than 120 days | € | | € | | € | | |||
Our cash and cash equivalents are invested primarily in current, notice and term accounts. For banks and financial institutions, only independently rated parties with a minimum rating of ‘A’ are accepted at the beginning of the term. Our current financial investments are also kept within different financial institutions and include term deposits, money market funds and treasury bills with an AAA rating. The money market funds are invested in a well-diversified portfolio of highly rated assets.
Interest rate risk
The only variable interest-bearing financial instruments are cash and cash equivalents and current financial investments.
Changes in interest rates may cause variations in interest income and expenses resulting from short term interest-bearing assets. Management does not expect the short term interest rates to decrease significantly in the immediate foreseeable future, which limits the interest exposure on our cash and cash equivalents and current financial investments.
Effect of interest rate fluctuation
A
Foreign exchange risk
We are exposed to foreign exchange risk arising from various currency exposures. Our principal functional currency is euro, but we receive payments from our main collaboration partner Gilead in U.S. dollars and acquire some consumables and materials in U.S. dollars, Swiss Francs and GB Pounds.
To limit this risk, we attempt to align incoming and outgoing cash flows in currencies other than EUR. In addition, contracts closed by our different entities are mainly in the functional currencies of that entity, except for the collaboration agreement signed with Gilead for which payments are denominated in U.S. dollars.
F-67
The exchange rate risk in case of a
December 31, | |||||||||
| 2023 |
| 2022 |
| 2021 | ||||
Net book value | (Euro, in thousands) | ||||||||
Increase in Euros - U.S. Dollars | € | ( | € | ( | € | ( | |||
Increase in Euros - GB Pounds | | | | ||||||
Increase in Euros - CH Francs | | | | ||||||
The exchange rate risk on the U.S. dollar is primarily related to our cash and cash equivalents and current financial investments held in U.S dollars.
Capital risk factors
We manage our capital to safeguard that we will be able to continue as a going concern. At the same time, we want to ensure the return to our shareholders through the results from our research and development activities.
Our capital structure consists of current financial investments, cash and cash equivalents, and equity attributed to the holders of our equity instruments, such as capital, reserves and results carried forward, as mentioned in the consolidated statement of changes in equity.
We manage our capital structure and make the necessary adjustments in the light of changes of economic circumstances, the risk characteristics of underlying assets and the projected cash needs of the current research and development activities.
The adequacy of the capital structure will depend on many factors, including scientific progress in the research and development programs, the magnitude of those programs, the commitments to existing and new clinical CROs, the ability to establish new alliance or collaboration agreements, the capital expenditures, the new commercial activities, market developments and any future acquisition.
Neither Galapagos NV nor any of its subsidiaries are subject to any externally imposed capital requirements, other than those imposed by generally applicable company law requirements.
35. Auditor’s remuneration
BDO Bedrijfsrevisoren BV (BDO) was appointed as statutory auditor by the Shareholders’ Meeting held on April 25, 2023, for a term of three years expiring immediately after the Annual Shareholders’ meeting to be held in 2026 which will have decided upon the annual accounts for the financial year to be ended on December 31, 2025.
Deloitte Bedrijfsrevisoren BV (Deloitte) ceased to be our statutory auditor as of the date of the Annual Shareholders’ Meeting held on April 25, 2023.
Our principal accountants billed the following fees to us for professional services rendered in 2023 (BDO) and 2022 (Deloitte).
The statutory auditor’s fees for carrying out its mandate at group level amounted to €
F-68
36. Events after balance sheet date
On January 3, 2024, we signed a strategic collaboration and license agreement with BridGene Biosciences to further strengthen our growing early-stage oncology precision medicine pipeline. Under the terms of the agreement, BridGene will receive from us up to $
On January 4, 2024, we announced that we entered into a strategic collaboration agreement with Thermo Fisher Scientific for CAR-T manufacturing and kitting services for our point-of-care CAR-T product candidate in the San Francisco area. Under the terms of the agreement, Thermo Fisher will provide GMP manufacturing as well as BioServices and Specialty Logistics for our CAR-T hemato-oncology clinical program in the San Francisco area, effective January 2024. We will initiate the technology transfer to enable Thermo Fisher’s manufacturing activities.
On January 31, 2024, we successfully completed the transaction with Alfasigma for the transfer of the Jyseleca® business after having met all closing conditions. As part of the transaction, the amended Filgotinib Agreement between us and Gilead has been assigned by us to Alfasigma. Alfasigma paid us an upfront payment of €
On January 31, 2024 we participated for $
F-69
EXHIBIT INDEX
Incorporated by Reference | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Schedule/ | File Date | |||||||||
Exhibit |
| Description |
| Form |
| File Number |
| Exhibit |
| (mm/dd/yyyy) |
1.1# | Articles of Association March 20, 2023 (English translation) | Form 20-F | 001-37384 | 1.1 | 3/23/2023 | |||||
2.1 | Form F-1/A | 333-203435 | 4.1 | 04/30/2015 | ||||||
2.2 | 424(b)3 | 333-203584 | A | 10/15/2018 | ||||||
2.3# | ||||||||||
4.1 | Lease dated June 30, 1999 between the registrant and Innotech N.V., as amended (English translation) | Form F-1 | 333-203435 | 10.1 | 04/15/2015 | |||||
4.2† | Form F-1/A | 333-203435 | 10.3 | 05/11/2015 | ||||||
4.6## | Form F-1 | 333-203435 | 10.7 | 04/15/2015 | ||||||
4.7† | Form S-8 | 333-208697 | 99.1 | 12/22/2015 | ||||||
4.8** | Form 6-K | 001-37384 | 10.1 | 01/19/2016 | ||||||
4.10† | Form S-8 | 333-211834 | 99.1 | 06/03/2016 | ||||||
4.11† | Form S-8 | 333-215783 | 99.1 | 01/27/2017 | ||||||
4.12† | Form 20-F | 001-37384 | 4.12 | 03/23/2017 | ||||||
4.13 | Form 20-F | 001-37384 | 4.13 | 03/23/2017 | ||||||
4.14† | Form S-8 | 333-218160 | 99.1 | 05/22/2017 | ||||||
4.15† | Form 20-F | 001-37384 | 4.15 | 03/23/2018 | ||||||
4.16 | Form 20-F | 001-37384 | 4.16 | 03/23/2018 | ||||||
4.17 | Form 20-F | 001-37384 | 4.17 | 03/23/2018 | ||||||
4.18 | Form 20-F | 001-37384 | 4.18 | 03/29/2019 | ||||||
4.19 | Form 20-F | 001-37384 | 4.19 | 03/29/2019 | ||||||
4.20† | Form S-8 | 333-225263 | 99.1 | 05/29/2018 | ||||||
4.21† | Form 20-F | 001-37384 | 4.21 | 03/29/2019 | ||||||
4.22† | Form S-8 | 333-231765 | 99.1 | 05/24/2019 | ||||||
4.23† | Form S-8 | 333-231765 | 99.2 | 05/24/2019 | ||||||
177
Incorporated by Reference | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Schedule/ | File Date | |||||||||
Exhibit |
| Description |
| Form |
| File Number |
| Exhibit |
| (mm/dd/yyyy) |
4.24## | Form 6-K | 001-37384 | 99.2 | 08/29/2019 | ||||||
4.25## | Form 6-K | 001-37384 | 99.3 | 08/29/2019 | ||||||
4.26 | Form 6-K | 001-37384 | 99.4 | 08/29/2019 | ||||||
4.27† | Form 20-F | 001-37384 | 4.27 | 03/27/2020 | ||||||
4.28† | Form 20-F | 001-37384 | 4.28 | 03/27/2020 | ||||||
4.29† | Form 20-F | 001-37384 | 4.29 | 03/27/2020 | ||||||
4.30 | Form 20-F | 001-37384 | 4.30 | 03/27/2020 | ||||||
4.31 | Form 20-F | 001-37384 | 4.31 | 03/27/2020 | ||||||
4.32 | Deed of purchase between the registrant and NMBS (English translation) | Form 20-F | 001-37384 | 4.32 | 03/27/2020 | |||||
4.33† | Form S-8 | 333-249416 | 99.1 | 10/09/2020 | ||||||
4.34† | Form 20-F | 001-37384 | 4.34 | 03/25/2021 | ||||||
4.35† | Form 20-F | 001-37384 | 4.35 | 03/25/2021 | ||||||
4.36† | Form 20-F | 001-37384 | 4.36 | 03/25/2021 | ||||||
4.37 | Form 20-F | 001-37384 | 4.37 | 03/25/2021 | ||||||
4.38 | Form 20-F | 001-37384 | 4.38 | 03/25/2021 | ||||||
4.39 | Form 20-F | 001-37384 | 4.39 | 03/25/2021 | ||||||
4.40† | Form S-8 | 333-260500 | 99.2 | 10/26/2021 | ||||||
4.41† | Form S-8 | 333-260500 | 99.3 | 10/26/2021 | ||||||
4.42† | Form S-8 | 333-260500 | 99.1 | 10/26/2021 | ||||||
4.43† | Form 20-F | 001-37384 | 4.43 | 03/24/2022 | ||||||
178
Incorporated by Reference | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Schedule/ | File Date | |||||||||
Exhibit |
| Description |
| Form |
| File Number |
| Exhibit |
| (mm/dd/yyyy) |
4.44† | Form 20-F | 001-37384 | 4.44 | 03/24/2022 | ||||||
4.45† | Form 20-F | 001-37384 | 4.45 | 03/24/2022 | ||||||
4.46† | Form 20-F | 001-37384 | 4.46 | 03/24/2022 | ||||||
4.47#† | ||||||||||
4.48#† | Form 20-F | 001-37384 | 4.48 | 3/23/23023 | ||||||
4.49† | Form S-8 | 333-268756 | 99.2 | 12/12/2022 | ||||||
4.50† | Form S-8 | 333-268756 | 99.3 | 12/12/2022 | ||||||
4.51† | Form S-8 | 333-268756 | 99.1 | 12/12/2022 | ||||||
4.52#† | Plan 2022.I – RSU Long-Term Incentive Plan 2022 (English translation) | Form 20-F | 001-37384 | 4.52 | 3/23/2023 | |||||
4.53#† | Plan 2022.II – RSU Retention Plan 2022 (English translation) | Form 20-F | 001-37384 | 4.53 | 3/23/2023 | |||||
4.54#† | Plan 2022.III – RSU GSI Replacement Plan 2022 (English translation) | Form 20-F | 001-37384 | 4.54 | 3/23/2023 | |||||
4.55# | Form 20-F | 001-37384 | 4.55 | 3/23/2023 | ||||||
4.56# | Form 20-F | 001-37384 | 4.56 | 3/23/2023 | ||||||
4.57#† | Plan 2023.I – RSU Long-Term Incentive Plan 2023 (English translation) | |||||||||
4.58#† | Plan 2023.II – RSU Retention Plan 2023 (English translation) | |||||||||
4.59#† | ||||||||||
4.60#† | ||||||||||
4.61#† | ||||||||||
4.62##** | Form 6-K | 001-37384 | 99.3 | 5/11/2021 | ||||||
8.1# | ||||||||||
12.1# | ||||||||||
179
Incorporated by Reference | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Schedule/ | File Date | |||||||||
Exhibit |
| Description |
| Form |
| File Number |
| Exhibit |
| (mm/dd/yyyy) |
12.2# | ||||||||||
13.1* | ||||||||||
13.2* | ||||||||||
15.1# | ||||||||||
15.2# | ||||||||||
15.3# | ||||||||||
97.1 | ||||||||||
101.INS# | Inline XBRL Instance Document | |||||||||
101.SCH# | Inline XBRL Taxonomy Extension Schema Document | |||||||||
101. CAL# | Inline XBRL Taxonomy Extension Calculation Linkbase Document | |||||||||
101. DEF# | Inline XBRL Taxonomy Extension Definition Linkbase Document | |||||||||
101.LAB# | Inline XBRL Taxonomy Extension Label Linkbase Document | |||||||||
101.PRE# | Inline XBRL Taxonomy Extension Presentation Linkbase Document | |||||||||
104 | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101) | |||||||||
# Filed herewith.
* Furnished herewith.
† Indicates a management contract or any compensatory plan, contract or arrangement.
## Certain exhibits and schedules to these agreements were omitted from the registration statement pursuant to Item 601(b)(2) of Regulation S-K. The registrant will furnish copies of any of the exhibits and schedules to the U.S. Securities and Exchange Commission upon request.
** Confidential treatment status has been granted as to certain portions thereto, which portions are omitted and filed separately with the U.S. Securities and Exchange Commission.
180
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
GALAPAGOS NV | |||
/s/ Stoffels IMC BV, permanently represented by Dr. Paul Stoffels | |||
By: | Stoffels IMC BV, permanently represented by Dr. Paul Stoffels | ||
Title: | Chief Executive Officer (Principal Executive Officer) | ||
Date: March 28, 2024 | |||
181