SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
OR
For the fiscal year ended
OR
For the transition period from to
Date of event requiring this shell company report . . . . . . . . . . . . . . . . . . .
Commission file number
(Exact name of Registrant as specified in its charter) |
(Jurisdiction of incorporation) |
(Address of principal executive offices) |
General Counsel Telephone: + Email: |
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person) |
Securities registered or to be registered, pursuant to Section 12(b) of the Act.
Title of each class |
| Trading Symbol |
| Name of each exchange on which registered |
evidenced by American Depositary Receipts, each American Depositary Share representing one Class B non-voting share of Grifols, S.A. | The |
Securities registered or to be registered pursuant to Section 12(g) of the Act.
None. |
(Title of Class) |
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act.
None. |
(Title of Class) |
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
⌧
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
◻ Yes ⌧
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
⌧
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
⌧
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Accelerated filer ◻ | Non-accelerated filer ◻ | Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ◻
†The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes - Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ◻
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
U.S. GAAP ◻ |
| Other ◻ |
If “Other” has been checked in response to the previous question indicate by check mark which financial statement item the registrant has elected to follow.
◻ Item 17 ◻ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
GRIFOLS, S.A.
TABLE OF CONTENTS
i | ||
iii | ||
iii | ||
iv | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
1 | ||
39 | ||
39 | ||
41 | ||
72 | ||
73 | ||
75 | ||
89 | ||
89 | ||
90 | ||
103 | ||
116 | ||
116 | ||
117 | ||
118 | ||
118 | ||
128 | ||
132 | ||
139 | ||
140 | ||
Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation. | 141 | |
142 | ||
142 | ||
144 | ||
147 | ||
147 | ||
147 | ||
150 | ||
150 | ||
150 | ||
150 | ||
150 | ||
i
155 | ||
155 | ||
155 | ||
155 | ||
155 | ||
156 | ||
166 | ||
167 | ||
167 | ||
176 | ||
176 | ||
176 | ||
176 | ||
176 | ||
177 | ||
178 | ||
178 | ||
178 | ||
178 | ||
178 | ||
180 | ||
186 | ||
186 | ||
MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS | 186 | |
186 | ||
186 | ||
Management’s Report on Internal Control over Financial Reporting | 186 | |
187 | ||
187 | ||
188 | ||
188 | ||
188 | ||
188 | ||
189 | ||
PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS | 189 | |
190 | ||
190 | ||
194 | ||
DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS | 194 | |
194 | ||
194 | ||
197 | ||
197 | ||
197 | ||
197 | ||
201 | ||
ii
GENERAL INFORMATION
As used in this annual report on Form 20-F, unless the context otherwise requires or as is otherwise indicated:
| ● | all references to “Grifols,” the “Company,” “we,” “us” and “our” refer to Grifols, S.A., a company (sociedad anónima) organized under the laws of Spain, and our consolidated subsidiaries; |
| ● | all references to the “Group” or the “Grifols Group” are to Grifols, S.A. and the group of companies owned or controlled by Grifols, S.A; and |
| ● | see “Glossary of Terms” for further explanations and/or definitions of terms referenced in this Form 20-F. |
PRESENTATION OF FINANCIAL AND OTHER INFORMATION
The basis of presentation of financial information of Grifols in this document is in conformity with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB (jointly, “IFRS-IASB”) and other legislative provisions containing the applicable legislation governing our financial information, unless indicated otherwise.
All references in this annual report on Form 20-F to (i) “euro,” “€” or “EUR” are to the common currency of the European Union and (ii) “U.S. dollar,” “$” or “USD” are to the currency of the United States.
The functional and presentation currency of Grifols is the euro. All tabular disclosures are presented in thousands of euros except share and per share amounts, percentages and as otherwise indicated. Certain monetary amounts and other figures included in this annual report on Form 20-F have been subject to rounding adjustments. Accordingly, any discrepancies in any tables between the totals and the sums of amounts listed are due to rounding.
Constant Currency
Net revenue variance in constant currency is determined by comparing adjusted current period figures, calculated using prior period monthly average exchange rates, to the prior period net revenue. The resulting percentage variance in constant currency is considered to be a non-IFRS-IASB financial measure. Net revenue variance in constant currency calculates net revenue variance without the impact of foreign exchange fluctuations. We believe that constant currency variance is an important measure of our operations because it neutralizes foreign exchange impact and illustrates the underlying change from one year to the next. We believe that this presentation provides a useful period-over-period comparison as changes due solely to exchange rate fluctuations are eliminated. Net revenue variance in constant currency, as defined and presented by us, may not be comparable to similar measures reported by other companies. Net revenue variance in constant currency has limitations, particularly because the currency effects that are eliminated constitute a significant element of our net revenue and could impact our performance significantly. We do not evaluate our results and performance without considering variances in constant currency on the one hand and changes prepared in accordance with IFRS-IASB on the other. We caution you to follow a similar approach by considering data regarding constant currency period-over-period revenue variance only in addition to, and not as a substitute for or superior to, other measures of financial performance prepared in accordance with IFRS-IASB. We present the fluctuation derived from IFRS-IASB net revenue next to the fluctuation derived from non IFRS-IASB net revenue.
See below for a reconciliation of reported net revenues to net revenues in constant currency:
Year Ended December 31, | Year Ended December 31, |
| |||||||||||
| 2023 |
| 2022 |
| % var |
| 2022 |
| 2021 |
| % var |
| |
(in millions of euros) | (in millions of euros) |
| |||||||||||
Net Revenue |
| 6,592.0 |
| 6,064.0 |
| 8.7 | % | 6,064.0 |
| 4,933.1 |
| 22.9 | % |
Variation due to exchange rate effects |
| 133.6 |
|
|
| 519.2 |
|
| |||||
Constant Currency Net Revenue |
| 6,725.6 |
| 5,544.8 |
| 10.9 | % | 5,544.8 |
| 4,933.1 |
| 12.4 | % |
iii
PRESENTATION OF MARKET INFORMATION
Market information (including market share, market position and industry data for our operating activities and those of our subsidiaries or of companies acquired by us) or other statements presented in this annual report on Form 20-F regarding our position (or that of companies acquired by us) relative to our competitors largely reflect the best estimates of our management. These estimates are based upon information obtained from customers, trade or business organizations and associations, other contacts within the industries in which we operate and, in some cases, upon published statistical data or information from independent third parties. Except as otherwise stated, our market share data, as well as our management’s assessment of our comparative competitive position, has been derived by comparing our sales figures for the relevant period to our management’s estimates of our competitors’ sales figures for such period, as well as upon published statistical data, information taken from filings with the Securities and Exchange Commission, or the SEC, and information from independent third parties, and, in particular, the reports published and the information made available by, among others, the Marketing Research Bureau, or the MRB. You should not rely on the market share and other market information presented herein as precise measures of market share or of other actual conditions.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This annual report contains statements that constitute “forward-looking statements” within the meaning of the U.S. Private Securities Litigation Reform Act of 1995. Forward-looking statements are typically identified by words such as “may,” “anticipate,” “believe,” “estimate,” “predict,” “expect,” “intend,” “forecast,” “will,” “would,” “should” or the negative of such terms or other variations on such terms or comparable or similar words or expressions.
These forward-looking statements reflect, as applicable, our management’s current beliefs, assumptions and expectations and are subject to a number of factors that may cause actual results to differ materially. These factors include, but are not limited to:
| ● | the complexity of our manufacturing processes and the susceptibility of our biological intermediates to contamination; |
| ● | our need to continually monitor our products for possible unexpected side effects; |
| ● | our ability to adhere to government regulations so that we may continue to manufacture and distribute our products; |
| ● | the impact of disruptions in our supply of plasma or in the operations of our plasma collection centers; |
| ● | the impact of competing products and pricing and the actions of competitors; |
| ● | the impact of strategies and techniques employed by short sellers or other market disruptors to drive down the market price of our shares, negatively impact our business operations and/or generate non-meritorious litigation; |
| ● | the impact of product liability claims on our business; |
| ● | our reliance on a plasma supply free of transmittable disease; |
| ● | our plans relating to our financings, including refinancing plans, interest rates and availability, and cost of financing opportunities; |
| ● | the impact of interest rate fluctuations; |
| ● | unexpected shut-downs of our manufacturing and storage facilities or delays in opening new planned facilities; |
| ● | reliance on third parties for manufacturing of products and provision of services; |
| ● | our ability to commercialize products in development; |
iv
| ● | our ability to protect our intellectual property rights. |
| ● | U.S. healthcare legislation, new legislation, regulatory action or legal proceedings affecting, among other things, the U.S. healthcare system, and pharmaceutical pricing and reimbursement, including Medicaid, Medicare and the 340B Program; |
| ● | legislation or regulations in markets outside of the United States affecting product pricing, reimbursement, access, or distribution channels; |
| ● | the magnitude of cost savings, streamlining of corporate functions, optimization of plasma costs and operations, enhancement of efficiencies and synergies, structural improvements and other measures related to our Operational Improvement Plan (as defined hereunder); and |
| ● | changes in legal requirements affecting the industries in which we operate. |
Please review a more detailed discussion of these and other risks that may impact our business set forth in this Form 20-F under “Item 3.D. Risk Factors.”
Forward-looking statements are not guarantees of future performance and involve risks, assumptions and uncertainties, including those listed above, and actual results may differ materially from those expressed or implied in the forward-looking statements.
All written and oral forward-looking statements concerning matters addressed in this annual report and attributable to us or any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this annual report. The forward-looking statements contained in this annual report speak only as of the date of this annual report. Except as required by law, we do not undertake to update any forward-looking statement to reflect events or circumstances after that date or to reflect the occurrence of unanticipated events.
v
PART I
Item 1. | IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS |
A. | Directors and Senior Management |
Not applicable.
B. | Advisers |
Not applicable.
C. | Auditors |
Not applicable.
Item 2.OFFER STATISTICS AND EXPECTED TIMETABLE
A. | Offer Statistics |
Not applicable.
B. | Method and Expected Timetable |
Not applicable.
Item 3.KEY INFORMATION
A. | [Reserved] |
B. | Capitalization and Indebtedness |
Not Applicable.
C. | Reasons for the Offer and Use of Proceeds |
Not Applicable.
D. | Risk Factors |
Summary
Our company, our business and our securities are subject to a number of risks which are described more comprehensively elsewhere in this item D. We present below a summary of our key risk factors.
Risks Relating to Our Structure:
| ● | Our substantial level of indebtedness could adversely affect our financial condition, restrict our ability to react to changes to our business, and prevent us from fulfilling our debt obligations; |
| ● | Despite our substantial indebtedness, we may still incur significantly more debt. This could exacerbate the risks associated with our substantial leverage; |
1
| ● | To service our indebtedness and other obligations, we will require a significant amount of cash. Our ability to generate cash depends on many factors, some of which may be beyond our control; |
| ● | Covenants in our debt agreements restrict our business in many ways; |
| ● | Our ability to meet our financial obligations depends in part on our ability to receive dividends and other distributions from our subsidiaries; |
| ● | We engage in transactions with related parties and such transactions may present the appearance of a possible conflict of interest that can have an adverse effect on us; |
| ● | We are a foreign private issuer under the rules and regulations of the Securities and Exchange Commission and, thus, are exempt from a number of rules under the Securities Exchange Act of 1934 and are permitted to file less information with the Securities and Exchange Commission than a company incorporated in the United States; |
| ● | Military conflicts around the world, and the global response thereto, may adversely affect our business and results of operations; and |
| ● | The ability to enforce civil liabilities under U.S. securities laws may be limited. |
Risks Relating to the Company and Our Business:
| ● | Our manufacturing processes are complex and involve biological intermediates that may be susceptible to contamination and variations in yield. Plasma and plasma derivative products are fragile, and improper handling of the plasma or plasma derivative products could adversely affect our operations; |
| ● | Once our products are approved and marketed, we must continually monitor them for signs that their use may result in serious and unexpected side effects, which could jeopardize our reputation and our ability to continue marketing our products. We may also be required to conduct post-approval clinical trials as a condition to licensing a product. Moreover, we may not be able to commercialize products in development; |
| ● | Our ability to continue manufacturing and distributing our products depends on our continued adherence to cGMP regulations at our facilities; |
| ● | A significant disruption in our supply of plasma, including as a result of macroeconomic conditions, pandemics or changes in immigration policies and enforcement could have a material adverse effect on our business and our growth plans; |
| ● | Disruption of the operations of our plasma collection centers would cause us to become supply-constrained and our financial performance would suffer; |
| ● | The COVID-19 global pandemic had, and future pandemics could have, a material adverse impact on us; |
| ● | A significant portion of our net revenue has historically been derived from sales of our immunoglobulin (“IG”) products and we expect that they will continue to comprise a significant portion of our sales. Any adverse market event with respect to these products could have a material adverse effect on us; |
| ● | We face significant competition; |
| ● | We face competition from companies with greater financial resources; |
2
| ● | Technological changes in the production of our products may require substantial investments in innovation, digital transformation and new technologies, which could render our production process uneconomical and diminish our competitiveness in the industries in which we operate; |
| ● | The discovery of new pathogens could slow our growth and adversely affect profit margins; |
| ● | Product liability claims or product recalls involving our products or products we distribute could have a material adverse effect on our business; |
| ● | Our ability to continue to produce safe and effective plasma derivative products depends on a plasma supply free of transmittable diseases; |
| ● | Our future success depends on our ability to retain members of our senior management and to attract, retain and motivate qualified personnel; |
| ● | Our business requires substantial capital to operate and grow and to achieve our strategy of realizing increased operating leverage, including the completion of several large capital projects; |
| ● | We may not be able to develop some of our international operations successfully; |
| ● | Uncertainties regarding the general regulatory and legal environment, particularly in China, could adversely affect our business; |
| ● | We are susceptible to interest rate variations; |
| ● | Our results of operations and financial condition may be affected by adverse changes in foreign currency exchange rates, especially a significant shift in the value of the euro as compared to the U.S. dollar; |
| ● | If our main facilities were to suffer a crippling accident, or if a force majeure event materially affected our ability to operate and produce saleable products, a substantial part of our manufacturing capacity could be shut down for an extended period; |
| ● | If we experience equipment difficulties or if the suppliers of our equipment or disposable goods fail to deliver key product components or supplies in a timely manner, our manufacturing ability would be impaired and our product sales could suffer; |
| ● | If our shipping or distribution channels were to become inaccessible due to a crippling accident, a pandemic, an act of terrorism, a strike, earthquake, major fire or storm, or any other force majeure event, our supply, production and distribution processes could be disrupted; |
| ● | We rely in large part on the services of third parties for the sale, distribution and delivery of our products; |
| ● | We rely on the services of third parties for the manufacture of certain products; |
| ● | We may not be able to commercialize products in development; |
| ● | Complex and evolving U.S. and international laws and regulations regarding privacy and data security and increased risk of cybersecurity incidents to our information technology systems could result in increased costs of operations and a significant disruption to our business; |
3
| ● | Cyber-attacks or other privacy and data security incidents (for example involving the personal information of our plasma or blood donors) could disrupt our business and expose us to significant losses, liability and reputational damage; |
| ● | Our success depends in large part on our ability to obtain and maintain protection in the United States and other countries of the intellectual property relating to or incorporated into our technology and products; |
| ● | In addition to patented technology, we rely on our unpatented proprietary technology, trade secrets, processes and know-how; |
| ● | We may infringe or be alleged to infringe intellectual property rights of third parties; |
| ● | We have in-licensed certain patent rights and co-own certain patent rights with third parties; |
| ● | We may not realize the expected benefits from the entry into new or amended contracts, cost-savings and business improvement initiatives; |
| ● | Climate change and increased risk of major natural disasters may adversely affect our business; and |
| ● | Changes, enactment, and/or enforcement of biometric information privacy laws of different jurisdictions, including federal and state laws in the United States, could expose us to potential liability. |
Risks Relating to the Healthcare Industry:
| ● | United States Healthcare Reform may adversely affect our business; |
| ● | Government pressures and constraints on reimbursement may adversely affect our business; |
| ● | Impact of government regulations over product development and regulatory approvals, as well as government pressures and constraints on reimbursement may adversely affect our business; |
| ● | Failure to comply with laws and regulations governing the sales and marketing of our products or an adverse decision in lawsuits may result in adverse consequences to us; |
| ● | We could be adversely affected if other government or private third-party payors decrease or otherwise limit the amount, price, scope or other eligibility requirements for reimbursement for the purchasers of our products; |
| ● | We are subject to extensive government regulatory compliance and ethics oversight; |
| ● | Failure to comply with changing regulatory requirements could materially adversely affect our business; and |
| ● | We are subject to extensive environmental, health and safety laws and regulations. |
Risks Relating to Our Shares and American Depositary Shares:
| ● | If we discover material weaknesses or significant deficiencies in our internal control over financial reporting, it may adversely affect our ability to provide timely and reliable financial information and satisfy our reporting obligations under U.S. federal securities laws, which also could affect the market price of our American Depositary Shares or our ability to remain listed on NASDAQ; |
| ● | The Grifols Family may exercise significant influence over the conduct of our business; |
4
| ● | The market price of our Class B ADSs on NASDAQ, as well as the market price of our Class A ADSs traded in over the counter markets, may be volatile; |
| ● | Techniques employed by short sellers or other market disruptors may drive down the market price of our shares, negatively impact our business operations and/or generate non-meritorious litigation; |
| ● | Fluctuations in the exchange rate between the U.S. dollar and the euro may increase the risk of holding our ADSs or shares; |
| ● | Subscription (or preemptive) rights may be unavailable to U.S. holders of our shares or ADSs; and |
| ● | ADS holders may be subject to limitations on the transfer of their ADSs. |
Risks Relating to Our Structure
Our substantial level of indebtedness could adversely affect our financial condition, restrict our ability to react to changes to our business, and prevent us from fulfilling our debt obligations.
We have a significant amount of indebtedness. As of December 31, 2023, our current and non-current financial liabilities were €11,057.2 million, of which a substantial majority (€10,033.6 million) was long-term debt.
Our high level of indebtedness could have significant adverse effects on our business, such as:
| ● | making it more difficult for us to satisfy our obligations with respect to our outstanding debt; |
| ● | making us more vulnerable to economic downturns and adverse developments in our business; |
| ● | impairing our ability to obtain additional financing for working capital, capital expenditures, acquisitions or general corporate purposes; |
| ● | reducing the funds available to us for operations and other purposes due to the substantial portion of our cash flow that we use to pay interest on our indebtedness; |
| ● | placing a prior ranking claim on the underlying assets of all of the indebtedness outstanding under our purchase money indebtedness, equipment financing and real estate mortgages; |
| ● | limiting our ability to fund a change of control offer; |
| ● | placing us at a competitive disadvantage compared to our competitors that may have proportionately less debt; |
| ● | limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate; and |
| ● | restricting us from making strategic acquisitions or exploiting other business opportunities. |
5
We expect to use cash flow from operations to pay our expenses and amounts due under our outstanding indebtedness. Our ability to make these payments depends on our future performance, which will be affected by financial, business, economic and other factors, many of which could be beyond our control. Our business may not generate sufficient cash flow from operations in the future and our anticipated growth in revenue and cash flow may not be realized, either or both of which could result in our being unable to repay indebtedness or to fund other liquidity needs. If we do not have enough liquidity, we may be required to refinance all or part of our then existing debt, sell assets or incur more debt. We may not be able to accomplish any of these alternatives on terms acceptable to us, or at all. In addition, the terms of existing or future debt agreements may restrict us from adopting any of these alternatives. The failure to generate sufficient cash flow or to achieve any of these alternatives could materially and adversely affect our business, results of operations and financial condition.
Despite our substantial indebtedness, we may still incur significantly more debt. This could exacerbate the risks associated with our substantial leverage.
We may be able to incur substantial additional indebtedness, including additional secured indebtedness, in the future. Our business is capital intensive, and we regularly seek additional capital. Although the indenture governing the 2017 Notes (as defined herein), the indenture governing the 2019 Notes (as defined herein), the indenture governing the 2021 Notes (as defined herein), the First Lien Credit Facilities (as defined herein) and the European Investment Bank Term Loans (as defined herein) contain restrictions on the incurrence of additional debt, these restrictions are subject to a number of qualifications and exceptions and, under certain circumstances, debt incurred in compliance with these restrictions, including secured debt, could be substantial. Incurring additional debt to current debt levels could exacerbate the leverage-related risks described above. For more information on our indebtedness, see Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Sources of Credit.”
To service our indebtedness and other obligations, we will require a significant amount of cash. Our ability to generate cash depends on many factors, some of which may be beyond our control.
Our ability to make payments on and to refinance our indebtedness and to fund working capital needs and planned capital expenditures will depend on our ability to generate cash in the future. A significant reduction in our operating cash flows resulting from changes in economic conditions, increased competition or other events beyond our control could increase the need for additional or alternative sources of liquidity and could have a material adverse effect on our business, financial condition, results of operations, prospects and our ability to service our debt and other obligations. If we are unable to service our indebtedness, we will be forced to adopt an alternative strategy that may include actions such as reducing capital expenditures, selling assets, restructuring or refinancing our indebtedness or seeking additional equity capital. We cannot assure you that any of these alternative strategies could be effected on satisfactory terms, if at all, or that they would yield sufficient funds to make required payments on our indebtedness.
In addition, our borrowings under the First Lien Credit Facilities are at variable rates of interest and expose us to interest rate risk. If interest rates increase, our debt service obligations on the variable rate indebtedness would increase even though the amount borrowed remained the same, and our net income would decrease.
We cannot assure you that our business will generate sufficient cash flows from operations or that future borrowings will be available to us under the First Lien Credit Facilities or otherwise in an amount sufficient to enable us to pay our indebtedness or to fund our other liquidity needs. We may need to refinance all or a portion of our indebtedness on or before the maturity of such indebtedness. We cannot assure you that we will be able to refinance any of our indebtedness, including the First Lien Credit Facilities, the 2017 Notes, the 2019 Notes, the 2021 Notes and the European Investment Bank Term Loans, on commercially reasonable terms or at all. For more information on our indebtedness, see Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Sources of Credit.”
Covenants in our debt agreements restrict our business in many ways.
The agreements governing our indebtedness and other financial obligations applicable to us contain various covenants, with customary caveats, that limit our ability and/or our restricted subsidiaries’ ability to, among other things:
| ● | incur or assume liens or additional debt or provide guarantees in respect of obligations of other persons; |
| ● | issue redeemable stock and preferred equity; |
6
| ● | pay dividends or make distributions to the shareholders of Grifols or redeem or repurchase capital stock; |
| ● | prepay, redeem or repurchase debt; |
| ● | make loans, investments and capital expenditures; |
| ● | enter into agreements that restrict distributions from our restricted subsidiaries; |
| ● | sell assets and capital stock of our subsidiaries; |
| ● | enter into certain transactions with affiliates; and |
| ● | consolidate or merge with or into, or sell substantially all of our assets to, another person. |
A breach of any of these covenants could result in a default under our debt agreements. Upon the occurrence of an event of default, the respective creditors could elect to declare all amounts outstanding under the debt agreements to be immediately due and payable and, in the case of the First Lien Credit Facilities, 2021 Notes, 2019 Notes, 2017 Notes and the EIB Term Loans, terminate all commitments to extend further credit. If we were unable to repay those amounts, the respective creditors could proceed against the collateral granted to them to secure that indebtedness. We have pledged a significant portion of our assets as collateral under the First Lien Credit Facilities, the European Investment Bank Term Loans and the 2019 Notes. If the respective creditors under our existing indebtedness accelerate the repayment of borrowings, we may not have sufficient assets to repay our indebtedness.
Our ability to meet our financial obligations depends in part on our ability to receive dividends and other distributions from our subsidiaries.
Our principal assets are the equity interests that we hold in our operating subsidiaries. As a result, we are dependent on dividends and other distributions from our subsidiaries to generate the funds necessary to meet our financial obligations, including the payment of principal and interest on our outstanding debt. Our subsidiaries may not generate sufficient cash from operations to enable us to make principal and interest payments on our indebtedness or may have preferential dividends which are required to be paid prior to any dividends to us. For example, in the case of each of Biomat USA, Inc. (“Biomat USA”) and Biomat Newco Corp (“Biomat Newco”), our U.S.-based plasma collection subsidiaries, to the extent dividends are declared by their respective shareholders, the GIC Investor (as defined Item 4 of this Part I, “Information on the Company—A. History of and Development of the Company—Important Milestones”) would be entitled to receive preferred dividends. Such dividends are equal to $4,168,421.05 per share annually payable by each of Biomat USA (in respect of its ten preferential shares) and Biomat Newco (in respect of its nine preferential shares) and carry additional rights with them as well as including redemption rights and a liquidation preference of $52,105,263.16 per share. See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Dispositions—The Biomat Transactions.”
In addition, any payment of dividends, distributions, loans or advances to us by our subsidiaries could be subject to restrictions on dividends or, in the case of foreign subsidiaries, restrictions on repatriation of earnings under applicable local law and monetary transfer restrictions in the jurisdictions in which our subsidiaries operate. In addition, payments to us by our subsidiaries will be contingent upon our subsidiaries’ earnings. Our subsidiaries are permitted under the terms of our indebtedness to incur additional indebtedness that may restrict payments from those subsidiaries to us. We cannot assure you that agreements governing current and future indebtedness of our subsidiaries will permit those subsidiaries to provide us with sufficient cash to fund payments on our indebtedness when due.
Our subsidiaries are legally distinct from us and, except for existing and future subsidiaries that guarantee certain indebtedness, have no obligation, contingent or otherwise, to pay amounts due on our debt or to make funds available to us for such payment.
7
We engage in transactions with related parties and such transactions present possible conflicts of interest that could have an adverse effect on us.
We purchase goods and services from, sell products to, and otherwise engage in other transactions with, related parties. These related party transactions create the possibility of conflicts of interest regarding to us and our management, including that:
| ● | our senior managers, executive officers and directors that hold positions of responsibility with related parties may be aware of certain business opportunities that are appropriate for presentation to us as well as to such other related parties and may present such business opportunities to such other parties; |
| ● | our senior managers, executive officers and directors that hold positions of responsibility with related parties may have significant duties with, and spend significant time serving, other entities and may have conflicts of interest in allocating time; |
| ● | such conflicts could cause an individual in our management to seek to advance his or her economic interests or the economic interests of certain related parties above ours; and |
| ● | the appearance of conflicts of interest created by related party transactions could impair the confidence of our investors. Our Audit Committee regularly reviews these transactions. Notwithstanding these reviews, it is possible that a conflict of interest could have an adverse effect on our business, financial condition and results of operations. |
For more information on our related party transactions, see Item 7 of this Part I, “Major Shareholders and Related Party Transactions—B. Related Party Transactions” and Note 31 to our audited consolidated financial statements included in this annual report on Form 20-F on page F-121.
We are a foreign private issuer under the rules and regulations of the Securities and Exchange Commission and, thus, are exempt from a number of rules under the Securities Exchange Act of 1934 and are permitted to file less information with the Securities and Exchange Commission than a company incorporated in the United States.
As a foreign private issuer under the Securities Exchange Act of 1934, as amended, or the Exchange Act, we are exempt from certain rules under the Exchange Act, including the proxy rules under Section 14 of the Exchange Act, which impose certain disclosure and procedural requirements for proxy solicitations. Moreover, we are not required to file periodic reports and financial statements with the Securities and Exchange Commission, or the SEC, as frequently or as promptly as U.S. companies with securities registered under the Exchange Act; we are not required to file financial statements prepared in accordance with United States’ generally accepted accounting principles; and we are not required to comply with SEC Regulation FD, which imposes certain restrictions on the selective disclosure of material non-public information. In addition, our officers, directors and principal shareholders are not subject to the reporting or short-swing profit recovery provisions of Section 16 of the Exchange Act or the rules under the Exchange Act with respect to their purchases and sales of our Class A shares or Class B shares. Accordingly, you may receive less information about us than you would receive about a company incorporated in the United States and may be afforded less protection under the U.S. federal securities laws than you would be afforded with respect to a company incorporated in the United States. If we lose our status as a foreign private issuer at some future time, we will no longer be exempt from such rules and, among other things, will be required to file periodic reports and financial statements as if we were a company incorporated in the United States. The costs incurred in fulfilling these additional regulatory requirements could be substantial.
Additionally, pursuant to The NASDAQ Stock Market LLC (“NASDAQ”) listing rules (the “NASDAQ Listing Rules”), as a foreign private issuer we may elect to follow our home country practice in lieu of the corporate governance requirements of the NASDAQ Listing Rule 5600 Series, with the exception of those rules that are required to be followed pursuant to the provisions of NASDAQ Listing Rule 5615(a)(3). We have elected to follow Spanish practices in lieu of the requirements of the NASDAQ Listing Rule 5600 Series to the extent permitted under NASDAQ Listing Rule 5615(a)(3). See Item 16.G. of Part II, “Corporate Governance.”
8
Military conflicts around the world, and the global response thereto, may adversely affect our business and results of operations.
The occurrence of unforeseen or catastrophic events, including geopolitical and other economic or political conditions or events such as the military conflict between Russia and Ukraine since 2022, the ongoing conflict in Gaza following the terrorist attacks that occurred in Israel in October 2023, and the more recent missile and drone attacks by Iran on Israel, depending on their scale, may cause different degrees of damage to the national and regional economies and could cause a disruption in our operations. For example, in response to the military conflict between Russia and Ukraine, the United States, United Kingdom, European Union and others have imposed significant new sanctions and export controls against Russia and certain Russian individuals and entities. This conflict has also resulted in significant volatility and disruptions to the global markets.
It is not possible to predict the long-term implications of these conflicts, which could include but are not limited to further sanctions, uncertainty about economic and political stability, increases in inflation rates and further increases in energy prices, supply chain challenges and adverse effects on currency exchange rates and financial markets. In addition, sanctions in response to these or any other future conflict have led or could lead to an increased threat of cyberattacks, which can pose risks to the security of our IT systems, our network and our service offerings, as well as the confidentiality, availability and integrity of our data. Up to the present date, the conflicts in Ukraine, Gaza and between Iran and Israel have not directly affected our business operations, but we cannot be certain that such conflicts will not affect existing and potential customers and have an adverse impact on our operations in the future.
The ability to serve process and enforce civil liabilities under U.S. securities laws may be limited.
We are a company organized under the laws of Spain, and many of our subsidiaries are also incorporated outside of the United States. A substantial portion of our assets and the assets of our subsidiaries are located outside of the United States. In addition, nearly all of our directors and officers and certain of our subsidiaries’ officers and directors are nationals or residents of countries other than the United States, and all or a substantial portion of such persons’ assets are located outside the United States. As a result, it may be difficult for investors to effect service of process within the United States upon us or certain of our subsidiaries or their directors or officers with respect to matters arising under the Securities Act of 1933 (the “Securities Act”) or to enforce against them judgments of courts of the United States predicated upon civil liability under the Securities Act. It may also be difficult to recover fully in the United States on any judgment rendered against such persons or against us or certain of our subsidiaries.
In addition, there is doubt as to the enforceability in Spain of original actions, or of actions for enforcement of judgments of U.S. courts of liabilities, predicated solely upon the securities laws of the United States. If a judgment was obtained outside Spain and efforts were made to enforce the judgment in Spain, there is some doubt that Spanish courts would agree to recognize and enforce a foreign judgment. Accordingly, even if you obtain a favorable judgment in a U.S. court, you may be required to re-litigate your claim in Spain.
We are a multinational business that operates in numerous tax jurisdictions.
We are subject to evolving and complex tax laws in the jurisdictions in which we operate, and routinely obtain advice on tax-related matters. Significant judgment is required in determining our tax liabilities, and our tax returns are periodically examined by various tax authorities. We regularly assess the likelihood of outcomes resulting from these examinations to determine the adequacy of our accrual for tax contingencies; however, due to the complexity of tax matters, the ultimate resolution of any tax matters may result in payments greater or less than the amounts accrued. In addition, we may be affected by changes in tax laws, including tax rate changes, new tax laws, and revised tax law interpretations in domestic and foreign jurisdictions and between jurisdictions, including by the E.U., which could materially adversely affect our tax expense and/or tax balances, and changes in tax policies could materially adversely impact our business. Furthermore, the integrated nature of our worldwide operations can produce conflicting claims from revenue authorities in different countries as to the profits to be taxed in the individual countries, including potential disputes relating to the prices our subsidiaries charge one another for intercompany transactions, known as transfer pricing. The occurrence of any of these risks could have a material adverse effect on our business, financial position and results of operations.
9
The Organization for Economic Co-operation and Development (“OECD”) introduced a new inclusive framework on Base Erosion and Profit Shifting (BEPS 2.0) that contains a two pillar solution to address the tax challenges arising from the digitalization of the economy. These changes are now being progressively implemented by tax authorities around the world and represent a fundamental change to the international tax framework. Pillar One provides for a new nexus standard/taxing right that allocates a portion of intangible/residual profits directly to market jurisdictions but only for the largest and most profitable companies. Pillar Two provides for a global minimum level of taxation (15%) that establishes a floor for tax competition amongst jurisdictions. Since the introduction of the OECD Inclusive Framework, over 130 countries have endorsed the framework and E.U. member states formally adopted the E.U.’s Pillar Two Directive. While the Company does not anticipate that Pillar Two will have a material impact on its tax provision or effective tax rate, it is possible that Pillar Two may result in top-up taxes in some jurisdictions in which Grifols operates and Grifols continues to monitor evolving tax legislation in the jurisdictions in which it operates.
For more information, see Note 4(q) to the audited consolidated financial statements included in this annual report on page F-47 and Note 28 to the audited consolidated financial statements included in this annual report on page F-100.
Risks Relating to the Company and Our Business
Our manufacturing processes are complex and involve biological intermediates that may be susceptible to contamination and variations in yield. Plasma and plasma derivative products are fragile, and improper handling of the plasma or plasma derivative products could adversely affect our operations.
Plasma is a raw material that is susceptible to damage and contamination and may contain human pathogens, any of which would render the plasma unsuitable for further manufacturing. For instance, contamination or improper storage of plasma by us or third-party suppliers may require us to destroy some of our raw material. If unsuitable plasma is not identified and discarded prior to its release to our manufacturing processes, it may be necessary to discard intermediate or finished product made from that plasma or to recall any finished product released to the market, resulting in a charge to cost of goods sold.
The manufacture of our plasma products is an extremely complex process of fractionation (separating the plasma into component proteins), purification, filling and finishing. Our products can become non-releasable or otherwise fail to meet our specifications through a failure of one or more of our product testing, manufacturing, process controls and quality assurance processes. We may detect instances in which an unreleased product was produced without adherence to our manufacturing procedures or plasma used in our production process was not collected or stored in a compliant manner consistent with Current Good Marketing Practice (“cGMP”) regulations enforced by the U.S. Food and Drug Administration (“FDA”) and analogous regulatory authorities of other countries, or other similar regulations, which would likely result in our determination that the impacted products should not be released and therefore should be destroyed.
Once we have manufactured our plasma-derived products, they must be handled carefully and kept at appropriate temperatures. Our failure, or the failure of third parties that supply, ship or distribute our products, to properly care for our plasma-derived products may require that such products be destroyed.
While we expect to write off small amounts of work in process inventories in the ordinary course of business due to the complex nature of plasma, our processes and our products, unanticipated events may lead to write-offs and other costs materially in excess of our expectations. Such write-offs and other costs could cause material fluctuations in our profitability. Furthermore, contamination of our products could cause investors, consumers or other third parties with whom we conduct business to lose confidence in the reliability of our manufacturing procedures, which could adversely affect our sales and profits. In addition, faulty or contaminated products that are unknowingly distributed could result in patient harm, threaten the reputation of our products and expose us to product liability damages and claims.
Due to the nature of plasma, there will be variations in the biologic properties of the plasma we collect or purchase for fractionation that may result in fluctuations in the obtainable yield of desired fractions, even if cGMP regulations are followed. Lower yields may limit production of our plasma-derived products due to capacity constraints. If such batches of plasma with lower yields impact production for extended periods, it may reduce the total volume of product that we could market and increase our cost of goods sold, thereby reducing our profitability.
10
Our manufacture of intermediate immunoassay antigens and antibodies to screen human donated blood and blood products is also a complex biologic process, subject to substantial production risks. These processes typically involve an upstream or fermentation process and a downstream or purification process. Since in the upstream process we deal with living cells, we may face a contamination by undesired cells which would eventually translate in a low yield. Yields in general can also be greatly affected by the different nutrients compositions added to the reactors in this fermentation step. Likewise during the purification step, we can face low yields due to poor resins composition, equipment failure or procedural mistakes.
Once our products are approved and marketed, we must continually monitor them for signs that their use may result in serious and unexpected side effects, which could jeopardize our reputation and our ability to continue marketing our products. We may also be required to conduct post-approval clinical trials as a condition to licensing a product.
As for all pharmaceutical products, the use of our products sometimes produces undesirable side effects or adverse reactions or events (collectively, “adverse events”). For the most part these adverse events are known, are expected to occur at some frequency and are described in the products’ labeling. Known adverse events of a number of our products include allergic or anaphylactic reactions including shock and the transmission of infective agents. Further, the use of certain products sometimes produces additional adverse events, which are detailed below.
| ● | The use of albumin sometimes produces the following adverse events: hypervolemia, circulatory overload, pulmonary edema, hyperhydration and allergic manifestations including urticaria, chills, fever and changes in respiration, pulse and blood pressure. |
| ● | The use of blood clotting Factor IX sometimes produces the following adverse events: the induction of neutralizing antibodies; thromboembolism, including myocardial infarction; disseminated intravascular coagulation; venous thrombosis and pulmonary embolism; and, in the case of treatment for immune tolerance induction, nephrotic syndrome. |
| ● | The use of the antihemophilic blood clotting Factor VIII sometimes produces the following adverse events: the induction of neutralizing antibodies, thromboembolic events and hemolytic anemia or hemolysis. |
| ● | The use of immunoglobulins sometimes produces the following adverse events: nausea, vomiting, asthenia, pyrexia, rigors, injection site reaction, allergic or anaphylactic reaction, aseptic meningitis, arthralgia, back pain, dizziness, headache, rash, pruritus, urticaria, hemolysis or hemolytic anemia, hyperproteinemia, increased serum viscosity and hyponatremia, thromboembolic reactions such as myocardial infarction, stroke, pulmonary embolism and deep vein thromboses, transfusion-related acute lung injury and renal dysfunction and acute renal failure. |
| ● | The use of anti-hepatitis B immunoglobulin sometimes produces the following adverse events: thromboembolic reactions such as myocardial infarction, stroke, pulmonary embolism and deep vein thromboses, aseptic meningitis, hemolytic anemia or hemolysis and acute renal failure. |
| ● | The use of Koate®-DVI, which we license exclusively in the United States to Kedrion S.p.A, a corporation organized under the laws of Italy, sometimes produces the following adverse events: allergic reactions, tingling in the arm, ear and face, blurred vision, headache, nausea, stomach ache and a jittery feeling. |
| ● | The use of Prolastin®, Prolastin®-C, alpha-1 proteinase inhibitor, or A1PI, sometimes produces the following adverse events: dyspnea, tachycardia, rash, chest pain, chills, influenza-like symptoms, hypersensitivity, hypotension and hypertension. |
11
In addition, the use of our products may be associated with serious and unexpected adverse events, or with less serious reactions at a greater than expected frequency. This may be especially true when our products are used in critically ill patient populations. When these unexpected events are reported to us, we must undertake a thorough investigation to determine causality and implications for product safety. These events must also be specifically reported to the applicable regulatory authorities. If our evaluation concludes, or regulatory authorities perceive, that there is an unreasonable risk associated with the product, we would be obligated to withdraw the impacted lot(s) of that product. Furthermore, an unexpected adverse event caused by a new product may be recognized only after extensive use of the product, which could expose us to product liability risks, enforcement action by regulatory authorities and damage to our reputation.
Once we produce a product, physicians are responsible for prescribing and administering the product as we have directed and for the indications described on the labeling. It is not, however, unusual for physicians to prescribe our products for unapproved, or off-label, uses or in a manner that is inconsistent with our directions or the labeling. To the extent such off-label uses and departures from our administration directions become pervasive and produce results such as reduced efficacy or other adverse effects, the reputation of our products in the marketplace may suffer.
Our ability to continue manufacturing and distributing our products depends on our continued adherence to cGMP regulations at our facilities.
The manufacturing processes for our products are governed by detailed written procedures and governmental regulations that set forth cGMP requirements for blood, blood products and other products. Our quality operations unit monitors compliance with these procedures and regulations, and the conformance of materials, manufacturing intermediates and final products to their specifications. Failure to adhere to established procedures or regulations, or to meet a specification, could require that a product or material be rejected and destroyed.
Our adherence to cGMP regulations and the effectiveness of our quality systems are periodically assessed through inspections of our facilities by the FDA, and analogous regulatory authorities of other countries. If deficiencies are noted during an inspection, we must take action to correct those deficiencies and to demonstrate to the regulatory authorities that our corrections have been effective. If serious deficiencies are noted or if we are unable to prevent recurrences, we may have to recall product or suspend operations until appropriate measures can be implemented. We are also required to report certain deviations from procedures to the FDA and analogous regulatory authorities of other countries, and even if we determine that the deviations were not material, such regulatory authorities could require us to take similar measures. Since cGMP reflects ever-evolving standards, we regularly need to update our manufacturing processes and procedures to comply with cGMP. These changes may cause us to incur costs without improving our profitability or the safety of our products. For example, more sensitive testing assays (if and when they become available) may be required or existing procedures or processes may require revalidation, all of which may be costly and time consuming and could delay or prevent the manufacturing of a product or launch of a new product.
Changes in manufacturing processes, including a change in the location where the product is manufactured or a change of a third-party manufacturer, may require prior review and approval or revalidation by the FDA or other regulatory authorities of the manufacturing processes and procedures in accordance with cGMP regulations.
To validate our manufacturing processes and procedures following completion of our upgraded facilities, we must demonstrate that the processes and procedures at the upgraded facilities are comparable to those currently in place at our other facilities. To provide such a comparative analysis, both the existing processes and the processes that we expect to be implemented at our upgraded facilities must comply with the regulatory standards prevailing at the time that our expected upgrade is completed. In addition, regulatory requirements, including cGMP regulations, continually evolve. Failure to adjust our operations to conform to new standards as established and interpreted by applicable regulatory authorities would create a compliance risk that could impair our ability to sustain normal operations.
Regulatory authorities, including the FDA and the European Medicines Office (“EMA”), routinely inspect our facilities to assess ongoing compliance with cGMP. If the FDA, the EMA or other regulatory authorities find our facilities to be out of compliance, our ongoing operations or plans to expand would be adversely affected.
12
A significant disruption in our supply of plasma, including as a result of macroeconomic conditions, pandemics or changes in immigration policies and enforcement could have a material adverse effect on our business and our growth plans.
The majority of our revenue depends on our access to U.S. source plasma (obtained through plasmapheresis), the principal raw material for our plasma derivative products. Our ability to increase revenue depends substantially on increased access to plasma. If we are unable to obtain sufficient quantities of source plasma, we may be unable to find an alternative cost-effective source of plasma and we would be limited in our ability to maintain current manufacturing levels of plasma derivative products. As a result, we could experience a substantial decrease in net revenue or profit margins, a loss of customers, a negative effect on our reputation as a reliable supplier of plasma derivative products or a substantial delay in our production growth plans.
Our current business plan envisages an increase in the production of plasma derivative products, which depends on our ability to maintain and/or increase plasma collections or improve product yield. The ability to maintain and/or increase plasma collections may be limited, our supply of plasma could be disrupted or the cost of plasma could increase substantially, as a result of numerous factors, including:
| ● | A reduction in the donor pool. |
| o | Regulators in most of the largest markets for plasma derivative products, including the United States, restrict the use of plasma collected from specific countries and regions in the manufacture of plasma derivative products. For example, the outbreak in the early 1990s of the variant Creutzfeldt-Jakob, or mad cow disease, resulted in a suspension by the United States of the use of plasma collected from certain persons based on travel, residence or transfusion in the United Kingdom and concern over the safety of blood products, which led to increased domestic and foreign regulatory control over the collection and testing of plasma and the disqualification of certain segments of the population from the donor pool, significantly reducing the potential donor pool. This suspension was only lifted in 2022. The appearance of new viral strains could further reduce the potential donor pool. More recently, the COVID-19 pandemic also adversely impacted our plasma collection volumes because of, among other things, mobility restrictions. See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Consequences of Covid-19.” |
| o | Changes in macroeconomic conditions could impact the number of donors. In the United States, the implementation of government stimulus programs issued to households during the COVID-19 pandemic had the two-pronged effect of both reducing the financial incentive for individuals to donate plasma and hindering our ability to maintain proper levels of workforce in plasma collection centers. Other macroeconomic factors such as a significant decrease in both inflation and interest rates in the United States in the future could result in increased access to cheaper debt to individuals, reducing the financial incentive for them to donate plasma; |
| o | Changes in certain immigration policies and enforcement could impair some of our supply of plasma. For example, during the COVID-19 pandemic the United States closed the U.S.-Mexico border, which prevented individuals from crossing into the United States and donating plasma in our plasma collection centers, significantly reducing the number of plasma donations. Border closures and travel bans or restrictions could limit the ability of donors to reach our plasma collection centers and reduce our plasma supply. |
| ● | Regulatory requirements. See “—Disruption of the operations of our plasma collection centers would cause us to become supply constrained and our financial performance would suffer.” |
| ● | Plasma supply sources. In recent years, there has been vertical integration in the industry as plasma derivatives manufacturers have been acquiring plasma collection centers. Any significant disruption in the supply of plasma or an increased demand for plasma may require us to obtain plasma from alternative sources, which may not be available on a timely basis. |
13
Disruption of the operations of our plasma collection centers would cause us to become supply-constrained and our financial performance would suffer.
In order for plasma to be used in the manufacturing of our products, the individual centers at which the plasma is collected must be licensed and approved by the regulatory authorities, such as the FDA and the EMA, of those countries in which we sell our products. When a new plasma collection center is opened, it must be inspected on an ongoing basis after its approval by the FDA, the EMA and other applicable regulatory authorities for compliance with cGMP and other regulatory requirements, and these regulatory requirements are subject to change. While we believe that our centers timely comply with evolving requirements, the compliance efforts necessary for evolving requirements may increase our costs. An unsatisfactory inspection could prevent a new center from being approved for operation or risk the suspension or revocation of an existing approval.
In order for a plasma collection center to maintain its governmental approval to operate, its operations must continue to conform to cGMP and other regulatory requirements. In the event that we determine a plasma collection center did not comply with cGMP and other regulatory requirements in collecting plasma, we may be unable to use and may ultimately destroy plasma collected from that center, which would be recorded as a charge to cost of goods. Additionally, if noncompliance in the plasma collection process is identified after the impacted plasma has been pooled with compliant plasma from other sources, entire plasma pools, in-process intermediate materials and final products could be impacted. Consequently, we could experience significant inventory impairment provisions and write-offs.
We plan to continue to obtain our supplies of plasma for use in our manufacturing processes through collections at our plasma collection centers and through selective acquisitions or remodeling and relocations of existing centers. This strategy is dependent upon our ability to successfully integrate new centers, to obtain FDA and other necessary approvals for any centers not yet approved by the FDA or other regulatory authorities, to maintain a cGMP compliant environment in all centers and to attract donors to our centers.
Our ability to increase and improve the efficiency of production at our plasma collection centers may be affected by: (i) changes in the economic environment and population in selected regions where we operate plasma collection centers; (ii) the entry of competitive centers into regions where we operate; (iii) our misjudging the demographic potential of individual regions where we expect to increase production and attract new donors; (iv) unexpected facility related challenges; (v) unexpected management challenges at select plasma collection centers; or (vi) unforeseen governmental changes to policies limiting the donor population or imposing regulations that make plasma donations onerous and restrictive.
The COVID-19 global pandemic had, and future pandemics could have, a material adverse impact on us.
The outbreak of the respiratory illness caused by a coronavirus named “SARS-CoV-2” (the “Coronavirus” or “COVID-19”) resulted in governments and businesses worldwide adopting emergency measures to combat the spread of the Coronavirus while seeking to maintain essential services. These measures included, without limitation, social distancing, the temporary closure of non-essential businesses, stay-at-home and work-from-home policies, self-imposed quarantine periods, border closures and travel bans or restrictions. These measures and conditions had an adverse impact on our manufacturing and supply chains, clinical trial operations and the ability of our employees to attend work or work effectively, which caused our net plasma supply to be negatively impacted from 2020 to 2022. As a result of a decrease in donors due to mobility restrictions, we increased our donor fees and, consequently, our cost per liter of plasma collected also increased during the pandemic. Although these material issues began to subside in the fourth quarter of 2021 and have since been resolved, if measures similar to those adopted in response to the COVID-19 pandemic are introduced to combat the spread of another pandemic in the future, our revenues, financial condition, profitability and cash flows may be materially adversely affected.
See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Consequences of COVID-19” for additional details.
14
A significant portion of our net revenue has historically been derived from sales of our immunoglobulin products and we expect that they will continue to comprise a significant portion of our sales. Any adverse market event with respect to these products could have a material adverse effect on us.
We have historically derived a significant portion of our net revenue from our immunoglobulin products, including our immunoglobulin products. In 2023 and 2022, our intravenous immunoglobulin (“IVIG”) products accounted for approximately 47% and 43% of our net revenue, respectively. If any of these IVIG products were to lose significant sales or were substantially or completely displaced in the market, we would lose a significant and material source of our net revenue. Similarly, if either Flebogamma®, Gamunex®-C/Gamunex® or Xembify® were to become the subject of litigation or an adverse governmental ruling requiring us to cease sales of it, our business could be adversely affected. Although we do not currently anticipate any significant decrease in the sales of any of these products, a significant decrease could result from plasma procurement and manufacturing issues resulting in lower product availability for sales and changing market conditions.
We face significant competition.
We face significant competition. Each of Takeda, CSL Behring, Kedrion Biopharma, Octapharma Plasma and Bio Products Laboratory Ltd. now has a 10% liquid IVIG product in the United States. Both Octapharma and Bio Products Laboratory also have 5% liquid IVIG products. GC Biopharma is expected to introduce its 10% IVIG product, AlygloTM, to the U.S. market sometime in 2024 and Argenx expects to obtain FDA approval in June 2024 for VYVGART Hytrulo (subcutaneous), an IVIG product that would treat patients with chronic inflammatory demyelinating polyneuropathy (“CIDP”). Such products are or will become direct competitors to some of our main products, Flebogamma®, Gamunex®-C/Gamunex® or Xembify®. As competition has increased, some of our competitors have discounted the price of immunoglobulin products as many customers have become increasingly price sensitive. If customers demand lower priced products, we may lose sales or be forced to lower our prices.
In the therapeutic area of pulmonology, specifically alpha-1-antitrypsin augmentation therapy (“AAT”), we face increased competition as a result of numerous companies such as Vertex, Kamada, Mereo Biopharma, AATec Medical and Korro Bio researching and developing novel inhaled and recombinant AAT therapies and the potential of gene therapies focusing on alpha-1. In addition, we expect increased competition will come from Sanofi due to such company’s recently announced expected acquisition from Inhibrx of all the assets and liabilities associated with INBRX-101, an optimized, recombinant AAT augmentation therapy. In the area of alpha1-proteinase inhibitor deficiency, CSL’s Respreeza® product is competing with our Prolastin product. Our current and future competitors may increase their sales, lower their prices, change their distribution model or improve their products, causing harm to our product sales and market share. Also, if the attrition rate of our A1PI patient base accelerates faster than we have forecasted, we would have fewer patients and lower sales volume.
Our competitors may also develop other new treatments, such as small molecules, recombinant products, inhaled products, gene therapies, or other novel therapies for indications for which our products are currently used. Recombinant Factor VIII and Factor IX products, which are currently available and widely used in the United States and Europe, compete with our plasma-derived product in the treatment of hemophilia A and B and are perceived by many to have lower risks of disease transmission. Additional recombinant products and new small molecules, some with extended half-lives, compete with and reduce the demand for our products. Genetech’s Hemlibra, a non-plasma product to control bleeding in patients with hemophilia A, is another competitor in the hemophilia market. The use of Hemlibra is a significant competitive risk for the use of plasma derived and recombinant Factor VIII. Additionally, numerous novel gene therapies have been approved and more are under development for the treatment of hemophilia which may further compete with our existing plasma derived therapies.
Furthermore, while we are investigating additional indications for the use of albumin, these new possibilities are countered by the fact that there are alternatives from competitors for albumin use in the main application we apply it, as a plasma volume expander.
We are only one of a number of companies that produce an alpha-1 anti-trypsin for the treatment of patients with hereditary emphysema and our competitors continually develop new products including inhaled, gene therapy, recombinant or other new various methods of administration for this therapy. Additionally, new treatments such as gene therapy, inhaled and recombinant products are in development by other companies and, regardless of the uncertainties surrounding the potential safety and efficacy of such new treatments, they increase our level of competition.
15
The introduction of products approved for alternative routes of administration, including subcutaneous, may also adversely affect sales of our products. For example, CSL Behring and Takeda introduced preparations of human immunoglobin at a 20% concentration for the treatment of people who need antibody replacement and Takeda has an immune globulin with a recombinant human hyaluronidase indicated for the treatment of primary immunodeficiency (“Pl”) in adults. Although we have FDA approval for similar concentrations and routes of administration, the level of competition remains significant and trending up as other players continue developing their operations in this expanding area of therapy.
Other companies are developing different therapies for the treatment of autoimmune diseases and other disorders that are currently treated with our immunoglobulins. If an increased use of alternative products for Factor VIII, Factor IX, albumin, alpha-1 or immunoglobulins makes it uneconomical to produce our plasma-derived products, or if further technological advances improve these products or create other competitive alternatives to our plasma derivative products, our financial condition and results of operations could be materially adversely affected. We expect in the future to face greater competition from biosimilar products which could further adversely affect our financial performance.
We do not currently sell therapeutic recombinant products. We have recombinant versions of A1PI in our pipeline, but we cannot be certain that any of these products will be approved or sold in the future. Additionally, we have recombinant versions of polyvalent and specific immune globulins under development. However, we cannot be certain that we will succeed in developing these products for licensed commercial use. As a result, our product offerings may remain plasma-derived, even if our competitors offer competing recombinant products.
We face competition from companies with greater financial resources.
We operate in highly competitive markets. Our principal competitors include Takeda, CSL Behring, Argenx and Octapharma. Some of our competitors have significantly greater financial resources than us. As a result, they may be able to devote more funds to research and development and new production technologies, as well as to the promotion of their products and business. These competitors may also be able to sustain for longer periods a deliberate substantial reduction in the price of their products or services. The development by a competitor of a similar or superior product or increased pricing competition may result in a reduction in our net revenue or a decrease in our profit margins.
Technological changes in the production of our products may require substantial investments in innovation, digital transformation and new technologies, which could render our production process uneconomical and diminish our competitiveness in the industries in which we operate.
The production of our plasma derivative, diagnostic and bio supplies products is characterized by factors such as rapid technological change, medical advances, changing consumer requirements, short device lifecycles, changing regulatory requirements and evolving industry standards. Technological advances have accelerated changes in recent years and future technological developments and medical advances could render our production processes uneconomical. Our success may depend on our ability to enhance our current technology and develop or acquire new technologies to keep pace with technological developments and evolving industry standards, while responding to changes in customer needs. Such innovation efforts may require us to invest substantial amounts of capital to upgrade our facilities and develop our products at competitive prices.
Such investments in new technologies, processes, and business models entail navigating various obstacles and risks, including discrepancies with our corporate vision and objectives, cultural barriers, skill deficiencies, resource limitations, and external disruptions. These challenges have the potential to undermine the anticipated benefits of our investments in digital transformation and diminish our competitiveness in the industries in which we operate. In addition, such investments could have a material adverse effect on our financial condition and results of operations and we may not be able to fund such investments from existing funds or raise sufficient capital to make such investments.
The discovery of new pathogens could slow our growth and adversely affect profit margins.
The possible appearance of new pathogens could trigger the need for changes in our existing inactivation and production methods, including the administration of new detection tests. Such a development could result in delays in production until the new methods are in place, as well as increased costs that may not be readily passed on to our customers. See also “—The COVID-19 global pandemic had, and future pandemics could have, a material adverse impact on us.”
16
Product liability claims or product recalls involving our products or products we distribute could have a material adverse effect on our business.
Our business exposes us to the risk of product liability claims. We face an inherent risk of product liability exposure related to the testing of our product candidates in human clinical trials and an even greater risk when we commercially sell any products. If we cannot successfully defend ourselves against claims that our product candidates or products caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in any or all of the following:
| ● | decreased demand for our products and any product candidates that we may develop; |
| ● | injury to our reputation; |
| ● | withdrawal of clinical trial participants; |
| ● | costs to defend the related litigation; |
| ● | substantial monetary awards to trial participants or patients; |
| ● | loss of revenue; and |
| ● | the inability to commercialize any products that we may develop. |
Like many plasma fractionators, we have been, and may in the future be, involved in product liability or related claims relating to our products, including claims alleging the transmission of disease through the use of such products. Plasma is a biological matter that is known to be capable of transmitting viruses and pathogens, whether known or unknown. Therefore, our plasma and plasma derivative products, if donors are not properly screened or if the plasma is not properly collected, tested, inactivated, processed, stored and transported, could cause serious disease and possibly death to the patient. Any transmission of disease through the use of one of our products or third-party products sold by us could result in claims by persons allegedly infected by such products.
Our potential product liability also extends to our Diagnostic business unit and Healthcare Solutions (as defined herein) products. In addition, we sell and distribute third-party products, and the laws of the jurisdictions where we sell or distribute such products could also expose us to product liability claims for those products. Furthermore, the presence of a defect in a product could require us to carry out a recall of such product.
A product liability claim or a product recall could result in substantial financial losses, negative reputational repercussions and an inability to retain customers. Although we have a program of insurance policies designed to protect us and our subsidiaries from product liability claims, and we self-insure a portion of this risk, claims made against our insurance policies could exceed our limits of coverage. We intend to expand our insurance coverage as our sales grow. However, as product liability insurance is expensive and can be difficult to obtain, a product liability claim could decrease our access to product liability insurance on acceptable terms. In turn, we may not be able to maintain insurance coverage at a reasonable cost and may not be able to obtain insurance coverage that will be adequate to satisfy any liability that may arise. Although we have not experienced a material liability claim, we cannot assure you that we will not experience one in the future.
17
Our ability to continue to produce safe and effective plasma derivative products depends on a plasma supply free of transmittable diseases.
Despite overlapping safeguards, including the screening of donors and other steps to remove or inactivate viruses and other infectious disease-causing agents, the risk of transmissible disease through plasma-derived products cannot be entirely eliminated. If a new infectious disease was to emerge in the human population in the future, the regulatory and public health authorities could impose precautions to limit the transmission of the disease that would impair our ability to procure plasma, manufacture our products or both. Such precautionary measures could be taken before there is conclusive medical or scientific evidence that a disease poses a risk for plasma-derived products.
In recent years, new testing and viral inactivation methods have been developed that more effectively detect and inactivate infectious viruses in collected plasma. There can be no assurance, however, that such new testing and inactivation methods will adequately screen for, and inactivate, infectious agents in the plasma used in the production of our products.
Plasma and plasma derivative products are fragile, and improper handling of our plasma or plasma derivative products could adversely affect results of operations.
Plasma is a raw material that is susceptible to damage. Almost immediately after its collection from a donor, plasma is stored and transported at temperatures that are at or below -20 degrees Celsius (-4 degrees Fahrenheit). Once we manufacture plasma derivative products, they must be handled carefully and kept at appropriate temperatures. Our failure, or the failure of third parties that supply, ship or distribute our plasma and plasma derivative products, to properly care for our plasma or plasma derivative products may require us to destroy some raw materials or products. If the volume of plasma or plasma derivative products damaged by such failures were to be significant, the loss of that plasma or those plasma derivative products could have a material adverse effect on our financial condition and results of operations.
Our future success depends on our ability to retain members of our senior management and to attract, retain and motivate qualified personnel.
We are highly dependent on the principal members of our executive and scientific teams. The loss of the services of any of these persons might impede the achievement of our research, development, operational and commercialization objectives. In particular, we believe the loss of any member of our senior management team could significantly and negatively impact our business. For details regarding the members of senior management, see Item 6 of this Part I, “Directors, Senior Management and Employees—A. Directors and Senior Management—Senior Management.” We do not maintain “key person” insurance on any of our senior management.
Recruiting and retaining qualified operations, finance and accounting, scientific, clinical and sales and marketing personnel will be critical to our success. We may not be able to attract and retain these personnel on acceptable terms, given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. If we are unable to attract, retain and motivate qualified and experienced personnel, we could lose customers and suffer reduced profitability. Even if we are successful in attracting and retaining such personnel, competition for such employees may significantly increase our compensation costs and adversely affect our financial condition and results of operations.
18
cGMP regulations also require that the personnel we employ and hold responsible for product manufacturing, including, for example, the collection, processing, testing, storage or distribution of blood or blood components, be adequate in number, educational background, training (including professional training as necessary) and experience, or a combination thereof, and have capabilities commensurate with their assigned functions, a thorough understanding of the procedures or control operations they perform, the necessary training or experience and adequate information concerning the application of relevant cGMP requirements to their individual responsibilities. Our failure to attract, retain and motivate qualified personnel may result in a regulatory violation, affect product quality, require the recall or market withdrawal of affected product or result in a suspension or termination of our license to market our products, or any combination thereof.
Our business requires substantial capital to operate and grow and to achieve our strategy of realizing increased operating leverage, including the completion of several large capital projects.
We have implemented several large capital projects to expand and improve the capacity and structure of our facilities and to improve the structure of our plasma collection centers in the United States. These projects may run over budget or be delayed. We cannot be certain that these projects will be completed in a timely manner or that we will maintain our compliance with cGMP regulations, and we may need to spend additional amounts to achieve compliance. Additionally, by the time these multi-year projects are completed, market conditions may differ significantly from our assumptions regarding the number of competitors, customer demand, alternative therapies, reimbursement and public policy, and as a result, capital returns might not be realized.
We also plan to continue to spend substantial sums on research and development, to obtain the approval of the FDA, and other regulatory authorities, for new indications for existing products, to develop new product delivery mechanisms for existing products and to develop innovative product additions. We face a number of obstacles to successfully converting these efforts into profitable products, including, but not limited to, the successful development of an experimental product for use in clinical trials, the design of clinical study protocols acceptable to the FDA and other regulatory authorities, the successful outcome of clinical trials, our ability to scale our manufacturing processes to produce commercial quantities or successfully transition technology, the approval of the FDA and other regulatory authorities of our products and our ability to successfully market an approved product or new indication.
For example, when a new product is approved, the FDA or other regulatory authorities may require post-approval clinical trials, sometimes called Phase IV clinical trials. If the results of such trials are unfavorable, this could result in the loss of the license to market the product, with a resulting loss of sales.
We continually make capital expenditures for the maintenance and enhancement of our facilities. The amount and timing of future capital spending is dependent upon a number of factors, including market conditions, regulatory requirements and the extent and timing of particular projects, among other things. Our ability to grow our business is dependent upon the timely completion of these projects and obtaining the requisite regulatory approvals.
We may not be able to develop some of our international operations successfully.
We currently conduct sales in over 100 countries. The successful operation of such geographically dispersed resources requires considerable management and financial resources. In particular, we must bridge our business culture to the business culture of each country in which we operate. In addition, international operations and the provision of services in foreign markets are subject to additional risks, such as changing market conditions, currency exchange rate fluctuations, trade wars and barriers, exchange controls, regulatory changes, changes to tax regimes (including proposed changes to U.S. tax laws by the Biden administration and the U.S. Congress), foreign investment limitations, civil disturbances, war and emerging pandemics. Furthermore, if an area in which we have significant operations or an area into which we are looking to expand suffers an economic recession or currency devaluation, our net revenues and accounts receivable collections in that region will likely decline substantially or we may not be able to successfully expand or operate in that region.
19
Uncertainties regarding the general regulatory and legal environment, particularly in China, could adversely affect our business.
Our international operations are governed by local laws and regulations applicable to foreign investments and foreign-owned enterprises. Our business could be adversely affected by the interpretation and enforcement of and changes in these laws and regulations. These laws and regulations often lack transparency, can be difficult to interpret and may be enforced inconsistently. A significant portion of our revenues is derived from our operations in China. China has not developed integrated legal systems that cover all aspects of our activities. The Chinese legal system is based on written statutes. Prior court decisions may be cited for reference but have limited precedential value. Since 1979, Chinese legislation and regulations have significantly enhanced the protections afforded to various forms of foreign investments in China. Because these laws and regulations are relatively new, and because of the limited volume of published decisions and their nonbinding nature, the interpretation and enforcement of these laws and regulations are uncertain. In addition, the Chinese legal system is based in part on government policies and internal rules that may have retroactive effect and, in some cases, are not published at all. As a result, we may not be aware of any alleged violation of these policies and rules until after the alleged violation has occurred. Any litigation in China may be protracted and result in substantial costs and diversion of resources and management attention.
We are susceptible to interest rate variations.
We use issuances of debt and bank borrowings as a source of funding. At December 31, 2023, $2.3 billion and €2.0 billion of our senior interest bearing debt, which represented 39.0% of our senior interest bearing debt, bore interest at variable rates, at a spread over the Secured Overnight Funding Rate (“SOFR”), for our U.S. dollar denominated debt and at a spread over the Euro Interbank Offered Rate (“EURIBOR”), for our euro denominated debt. Any increase in interest rates payable by us, which could be adversely affected by, among other things, our inability to meet certain financial ratios, would increase our interest expense and reduce our cash flow, which could materially adversely affect our financial condition and results of operations. See Item 11 of this Part I, “Quantitative and Qualitative Disclosures About Market Risk—Interest Rate Risk.”
Our results of operations and financial condition may be affected by adverse changes in foreign currency exchange rates, especially a significant shift in the value of the euro as compared to the U.S. dollar.
A significant portion of our business is conducted in currencies other than our reporting currency, the euro. In 2023, €5.3 billion, or 80.3%, of our net revenue of €6.6 billion was denominated in U.S. dollars. We are also exposed to currency fluctuations with respect to other currencies, such as the British pound, the Brazilian real, the Canadian dollar and the Argentine, Mexican and Chilean pesos. Currency fluctuations among the euro, the U.S. dollar and the other currencies in which we do business result in foreign currency translation gains or losses that could be significant.
We are also exposed to risk based on the payment of U.S. dollar denominated indebtedness. At December 31, 2023, we had approximately $2.8 billion of U.S. dollar denominated senior debt. See Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources” and Item 11 of this Part I, “Quantitative and Qualitative Disclosures About Market Risk—Currency Risk.”
If our main facilities were to suffer a crippling accident, or if a force majeure event materially affected our ability to operate and produce saleable products, a substantial part of our manufacturing capacity could be shut down for an extended period.
A substantial portion of our revenue is derived from plasma fractionation or products manufactured at our main facilities, including the facilities located in San Diego, Clayton, Emeryville, Los Angeles and Parets. In addition, a substantial portion of our plasma supply is stored at facilities in City of Industry, California, as well as at our Clayton and Parets facilities. If any of our main facilities, including the ones mentioned above, were to be impacted by an accident or a force majeure event such as an earthquake, major fire, storm or explosion, major equipment failure or power failure lasting beyond the capabilities of our backup generators, our revenue would be materially adversely affected. In this situation, our manufacturing capacity could be shut down for an extended period and we could experience a loss of raw materials, work-in-process or finished goods inventory. In addition, extreme weather conditions may become more severe and frequent as the temperature rises due to the effects of climate change, and such extreme weather conditions could heighten the risks and uncertainties noted above.
20
Other force majeure events such as terrorist acts, influenza pandemic or similar events could also impede our ability to operate our business. In addition, in the event of the reconstruction of our Clayton, Los Angeles or Parets facilities or our plasma storage facilities, gaining the regulatory approval for such new facilities and the replenishment of raw material plasma could be time consuming. During this period, we would be unable to manufacture all of our products at other plants due to the need for FDA and foreign regulatory authority inspection and certification of such facilities and processes.
Our property damage and business interruption insurance may be insufficient to mitigate the losses from any such accident or force majeure event. We may also be unable to recover the value of the lost plasma or work-in-process inventories, as well as the sales opportunities from the products we would be unable to produce.
If we experience equipment difficulties or if the suppliers of our equipment or disposable goods fail to deliver key product components or supplies in a timely manner, our manufacturing ability would be impaired and our product sales could suffer.
We depend on a limited number of companies that supply and maintain our equipment and provide supplies such as chromatography resins, filter media, glass and stoppers used in the manufacture of our products. If our equipment should malfunction, the repair or replacement of the machinery may require substantial time and cost, which could disrupt our production and other operations. Our plasma collection centers rely on disposable goods supplied by third parties and information technology systems hosted by third parties. Our plasma collection centers cannot operate without an uninterrupted supply of these disposable goods and the operation of these systems. Alternative sources for key component parts or disposable goods may not be immediately available. And while we have experienced periodic outages of these systems, a material outage would affect our ability to operate our collection centers.
Any new equipment or change in supplied materials may require revalidation by us or review and approval by the FDA or other regulatory authorities, including the EMA, which may be time-consuming and require additional capital and other resources. We may not be able to find an adequate alternative supplier in a reasonable time period, or on commercially acceptable terms, if at all. As a result, shipments of affected products may be limited or delayed. Our inability to obtain our key source supplies for the collection of plasma and manufacture of products may require us to delay shipments of products, harm customer relationships and force us to curtail operations.
If our shipping or distribution channels were to become inaccessible due to a crippling accident, a pandemic, an act of terrorism, a strike, earthquake, major fire or storm, or any other force majeure event, our supply, production and distribution processes could be disrupted.
Not all shipping or distribution channels are equipped to transport plasma. If any of our shipping or distribution channels becomes inaccessible due to a crippling accident, a pandemic, an act of terrorism, a strike, earthquake, major fire or storm or any other force majeure event, we may experience disruptions in our continued supply of plasma and other raw materials, delays in our production process or a reduction in our ability to distribute our products directly to our customers.
We rely in large part on third parties for the sale, distribution and delivery of our products.
We regularly enter into distribution, supply and fulfillment contracts with group purchasing organizations (“GPOs”), home care companies, alternate infusion sites, hospital groups, distributors and others. We are highly dependent on these agreements for the successful sale, distribution and delivery of our products. For example, in the United States, we rely principally on GPOs and on our distributors to sell our immunoglobulin products. If such parties breach, terminate or otherwise fail to perform under these contracts, our ability to effectively distribute our products could be impaired and our business may be materially and adversely affected. In addition, through circumstances beyond our control, such as general economic decline, market saturation or increased competition, we may be unable to successfully renegotiate our contracts or secure terms which are as favorable to us. Furthermore, we rely in certain countries on distributors for sales of our products. Disagreements or difficulties with our distributors supporting our export business could result in a loss of sales.
21
We rely on the services of third parties for the manufacture of certain products.
We have rights of sale and distribution for several different products, including Tavleese® in the European market. However, for many of these products we rely upon supply from third parties. To the extent such third parties are unable to properly and timely manufacture and deliver the necessary products and services in Europe, our business could be materially affected.
We may not be able to commercialize products in development.
Before obtaining regulatory approval for the sale of our product candidates or for the marketing of existing products for new indicated uses, we must conduct, at our own expense, extensive preclinical tests to demonstrate the safety of our product candidates in animals and clinical trials to demonstrate the safety and efficacy of our product candidates in humans. Preclinical and clinical testing is expensive, is difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, preclinical testing and the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates, including, without limitation, the following:
| ● | regulators or institutional review boards (“IRBs”) may not authorize us to commence a clinical trial or conduct a clinical trial within a country or at a prospective trial site; |
| ● | the regulatory requirements for product approvals may not be explicit, may evolve over time and may diverge by jurisdiction; |
| ● | our preclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or we may be required by regulators, to conduct additional preclinical testing or clinical trials or to abandon projects that we had expected to be promising; |
| ● | the number of patients required for our clinical trials may be larger than we anticipate, enrollment in our clinical trials may be slower than we anticipate or participants may withdraw from our clinical trials at higher rates than we anticipate, any of which would result in significant delays; |
| ● | our third-party contractors may fail to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; |
| ● | we may be forced to suspend or terminate our clinical trials if the participants are being exposed to unacceptable health risks or if any participant experiences an unexpected serious adverse event; |
| ● | regulators or IRBs may require that we hold, suspend or terminate clinical research for various reasons, including noncompliance with regulatory requirements; |
| ● | undetected or concealed fraudulent activity by a clinical researcher, if discovered, could preclude the submission of clinical data prepared by that researcher, lead to the suspension or substantive scientific review of one or more of our marketing applications by regulatory authorities and result in the recall of any approved product distributed pursuant to data determined to be fraudulent; |
| ● | the cost of our clinical trials may be greater than we anticipate; |
| ● | the supply or quality of our product candidates or other materials necessary to conduct our clinical trials may be insufficient or inadequate, as we currently do not have any agreements with third-party manufacturers for the long-term commercial supply of any of our product candidates; |
| ● | an audit of preclinical or clinical studies by the FDA or other regulatory authorities may reveal noncompliance with applicable regulations, which could lead to disqualification of the results and the need to perform additional studies; |
22
| ● | the effects of our product candidates may not achieve the desired clinical benefits or may cause undesirable side effects, or the product candidates may have other unexpected characteristics; and |
| ● | our clinical trials, or the ability of regulatory authorities to review the results of our clinical trials, may be delayed as a result of future pandemics with a similar impact to the COVID-19 pandemic. |
If we are required to conduct additional clinical trials or other testing of our product candidates beyond those that we currently contemplate, if we are unable to successfully complete our clinical trials or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may be delayed in or unable to obtain marketing approval or reimbursement for our product candidates, or be unable to obtain approval for indications that are not as broad as intended or have the product removed from the market after obtaining marketing approval.
Our product development costs will also increase if we experience delays in testing or approvals. We do not know whether any preclinical tests or clinical trials will begin as planned, will need to be restructured or will be completed on schedule, if at all. Significant preclinical or clinical trial delays could also shorten the patent protection period during which we may have the exclusive right to commercialize our product candidates or could allow our competitors to bring products to market before we do, impairing our ability to commercialize our products or product candidates.
Even if preclinical trials are successful, we still may be unable to commercialize a product due to difficulties in obtaining regulatory approval for its engineering process or problems in scaling that process to commercial production. Additionally, if produced, a product may not achieve an adequate level of market acceptance by physicians, patients, healthcare payors and others in the medical community to be profitable. The degree of market acceptance of our product candidates, if approved for commercial sale, will depend on a number of factors, some of which are beyond our control, including the following:
| ● | the prevalence and severity of any side effect; |
| ● | the efficacy and potential advantages over alternative treatments; |
| ● | the ability to offer our product candidates for sale at competitive prices; |
| ● | relative convenience and ease of administration; |
| ● | the willingness of physicians to prescribe new therapies and of the target patient population to try such therapies; |
| ● | the strength of marketing and distribution support; and |
| ● | sufficient third-party coverage or reimbursement. |
Therefore, we cannot guarantee that any products we may seek to develop will ever be successfully commercialized, and to the extent they are not successfully commercialized, such products could involve significant expense with no corresponding revenue.
Complex and evolving U.S. and international laws and regulations regarding privacy and data security and increased risk of cybersecurity incidents to our information technology systems could result in increased costs of operations and a significant disruption to our business.
Our operations are highly dependent on our information technology systems, including internet-based systems, which may be vulnerable to breakdown, cybersecurity incidents, wrongful intrusions, data breaches, malware, ransomware, and malicious attack. In addition, information security risks have generally increased in recent years, increasing our systems’ potential vulnerability, such as to data security breaches or cyber-attack, whether by employees or others, which may expose sensitive data to unauthorized persons. Such data security breaches could lead to the loss of trade secrets or other intellectual property, or could lead to the public exposure of personal information (including sensitive personal information) of our employees, customers, plasma donors and others. Data security breaches may also adversely impact the conduct of scientific research and clinical trials, including the submission of research results to support marketing authorizations.
23
Additionally, our information technology systems utilize certain third party service organizations that manage sensitive data, such as personal medical information regarding plasma donors, and our business may be adversely affected if these third party service organizations are subject to data security breaches. We may continue to incur significant expenses to comply with existing privacy and security standards and protocols imposed by law, regulation, industry standards or contractual obligations.
Federal, state and foreign governments continue to adopt new, or modify existing laws and regulations addressing data privacy and the collection, processing, storage, transfer and use of data. This includes, for example, the E.U.’s regulation, the General Data Protection Regulation (“GDPR”) and the new California Consumer Protection Act (“CCPA”), effective on January 1, 2020. In our efforts to meet the GDPR, CCPA, U.S. Health Insurance Portability and Accountability Act of 1996, as amended, and implementing regulations (“HIPAA”) and other data privacy regulations, we have made and continue to make certain operational changes to our business practices. Other governmental authorities throughout the United States and around the world are considering similar types of legislative and regulatory proposals concerning data protection. These privacy, security and data protection laws and regulations could impose increased business operational costs, require changes to our business, require notification to customers or workers of a security breach, or restrict our use or storage of personal information.
For example, health information laws and regulations, such as regulations under HIPAA and potential revisions thereto, include requirements to implement various recordkeeping, operational, notice and other practices intended to safeguard that information, limit its use to allowed purposes and notify affected individuals in the event of privacy and security breaches, establish standards regarding electronic health data transmissions and set rules for specific electronic transactions, such as transactions involving claims submissions to third party payers. Failure to comply with HIPAA and similar state laws could expose us to breach of contract claims, substantial fines, penalties and other liabilities and expenses, costs for remediation and harm to our reputation. Additionally, a U.S. federal privacy bill introduced to the U.S. House of Representatives on July 20, 2022 (and reported by the applicable committee on December 30, 2022), would establish new requirements for how companies handle personal data, including information that identifies or is reasonably linked to an individual, such as our consumers. If this bill becomes law, we may be required to implement certain security practices to protect and secure personal data against unauthorized access, and we may be subject to further requirements for complying with this requirement if any applicable agency issues related regulations.
In the United States and specifically in California, for example, the CCPA generally requires companies, such as us, to institute additional protections regarding the collection, use and disclosure of certain personal information of California residents. The California Attorney General announced the finalization of initial CCPA regulations on August 14, 2020, and two new sets of modifications to CCPA regulations have since been proposed and have completed the required public comment process, although they are still subject to internal review and finalization by the California Attorney General, the timing of which is uncertain. In addition to providing for enforcement by the California Attorney General, the CCPA also provides for a private right of action. Entities in violation of the CCPA may be liable for civil penalties. Significantly, in November 2020 California enacted the California Privacy Rights Act (“CPRA”), effective January 1, 2023, which amends the CCPA. The CPRA, among other substantive measures, expands the CCPA’s private right of action, increases consumers’ control over personal information, imposes new compliance obligations on businesses, and enacts new exceptions that may apply to our businesses. Notably, the CPRA created the California Privacy Protection Agency Board, which is responsible for enforcing the CCPA and CPRA, and which approved final CPRA regulations in February 2023. Other States in the United States either have or, presumably, will adopt similar laws.
The European Parliament and the Council of the European Union adopted the GDPR, which increased privacy rights for individuals in Europe, extended the scope of responsibilities for data controllers and data processors and imposed increased requirements and potential penalties on companies offering goods or services to individuals who are located in Europe or monitoring the behavior of such individuals (including by companies based outside of Europe). Noncompliance can result in penalties of up to the greater of €20.0 million, or 4.0% of global company revenues.
Our efforts to implement programs and controls that comply with the GDPR, CCPA, HIPAA and other data protection requirements are likely to impose additional costs on us, and we cannot predict whether the interpretations of the requirements, or changes in our practices in response to new requirements or interpretations of the requirements, could have a material adverse effect on our business.
24
Cyber-attacks or other privacy and data security incidents (for example involving the personal information of our plasma or blood donors) could disrupt our business and expose us to significant losses, liability and reputational damage.
We and our third-party service providers routinely process, store and transmit large amounts of data in our operations, including sensitive personal information as well as proprietary or confidential information relating to our business or third parties, including our plasma or blood donors. We may be subject to breaches of the information technology security systems we use both internally and externally with third-party service providers.
Cyber-attacks may penetrate our and our third-party service providers’ security controls and result in the misappropriation or compromise of sensitive personal information or proprietary or confidential information (e.g. information regarding our plasma or blood donors), including such information which is stored or transmitted on the systems used by certain of our or their products, to create system disruptions, cause shutdowns (including disruptions to our production plants), or deploy viruses, worms, ransomware, denial-of-service attacks and other malicious software programs that attack our systems. We and our third-party service providers handle the personal information of our patients and beneficiaries, patient personal data, throughout the United States and other parts of the world. We or our business associates may experience data breaches in violation of the requirements of HIPAA, GDPR and or other similar laws, for example:
| ● | data breaches involving the impermissible use, access, or disclosure of patient identifying information or unsecured personal data; |
| ● | a data breach where we or our business associates neglect to implement the required administrative, technical and physical safeguards of its electronic systems and devices, or |
| ● | a data breach that results in impermissible use, access or disclosure of personal identifying information of our employees, beneficiaries, and/or plasma or blood donors. |
When appropriate, we have filed complaints against the unknown attackers with the relevant authorities and we contacted the patients who were affected by the illegal data publication as well as other relevant regulatory authorities and stakeholders. While there has not been any material impact to our financial condition and results of operations as a result of these data breaches and attacks, future cyber-attacks against our IT systems may result in a loss of financial data or interruptions of our operations that could have a material adverse impact on our business, financial condition and results of operations in the future. The Ukraine War has increased the risk of cyber-attacks against our systems and data.
As we increase the amount of sensitive personal information or financial data that we store and share digitally, our exposure to these privacy and data breaches and cyber-attack risks increases (particularly as medical or pharmaceutical records are a high-value target), including the risk of undetected attacks, damage, loss or unauthorized disclosure or access, and the cost of attempting to protect against these risks also increases. There are no assurances that our security technologies, processes and procedures that we or our outside service providers have implemented to protect sensitive personal information and proprietary or confidential information and to build security into the design of our products will be effective.
Any failure to keep our information technology systems, financial data and our patients’ and customers’ sensitive information secure from attack, damage, loss or unauthorized disclosure or access, whether as a result of our action or inaction or that of our third-party business associates or vendors that utilize and store such personal information on our behalf, could materially adversely affect our reputation and ability to continue normal operations, expose us to mandatory public disclosure requirements, litigation and governmental enforcement proceedings, material fines, penalties and/or remediation costs, and compensatory, special, punitive and statutory damages, consent orders and other adverse actions, any of which could have a material adverse impact on our business, financial condition and results of operations. For information regarding our cybersecurity risk management and governance, see Item 16K of Part II of this annual report, “Cybersecurity.”
25
Our success depends in large part on our ability to obtain and maintain protection in the United States and other countries of the intellectual property relating to or incorporated into our technology and products.
Our success depends in large part on our ability to obtain and maintain protection in the United States and other countries for the intellectual property covering or incorporated into our technology and products, especially intellectual property related to our purification processes. The patent landscape in the field of biotechnology and pharmaceuticals generally is highly uncertain and involves complex legal and scientific questions. We may not be able to obtain additional issued patents relating to our technology or products. Even if patents are issued to us or to our licensors, they may be challenged, narrowed, invalidated, held to be unenforceable or circumvented, which could limit our ability to stop competitors from marketing similar products or limit the length of time our products have patent protection. Additionally, most of our patents relate to the processes we use to produce our products, not to the products themselves. In many cases, the plasma-derived products we produce or develop in the future will not, in and of themselves, be patentable. Since our patents relate to processes, if a competitor is able to design and utilize a process that does not rely on our protected intellectual property, such competitor could sell a plasma-derived or other product similar to one we developed or sell.
Our patents also may not afford us protection against competitors with similar technology. Because patent applications in the United States and many other jurisdictions are typically not published until 18 months after their filing, if at all, and because publications of discoveries in the scientific literature often lag behind actual discoveries, neither we nor our licensors can be certain that we or they were the first to make the inventions claimed in our or their issued patents or pending patent applications, or that we or they were the first to file for protection of the inventions set forth in such patent applications. If a third party has also filed a U.S. patent application covering our product candidates or a similar invention, we may be required to participate in an adversarial proceeding, known as an “interference proceeding,” declared by the U.S. Patent and Trademark Office to determine priority of invention in the United States. The costs of these proceedings could be substantial and our efforts in them could be unsuccessful, resulting in a loss of our anticipated U.S. patent position.
Our patents expire at various dates. Our pending and future patent applications may not issue as patents or, if issued, may not issue in a form that will provide us with any competitive advantage. Even if issued, we cannot guarantee that: any of our present or future patents or patent claims or other intellectual property rights will not lapse or be invalidated, circumvented, challenged or abandoned; our intellectual property rights will provide competitive advantages; our ability to assert our intellectual property rights against potential competitors or to settle current or future disputes will not be limited by our agreements with third parties; any of our pending or future patent applications will be issued or have the coverage originally sought; our intellectual property rights will be enforced in jurisdictions where competition may be intense or where legal protection may be weak; or we will not lose the ability to assert our intellectual property rights against, or to license our technology to, others and collect royalties or other payments. In addition, our competitors or others may design around our protected patents or technologies.
Effective protection of our intellectual property rights may be unavailable, limited or not applied for in some countries. Changes in patent laws or their interpretation in the United States and other countries could also diminish the value of our intellectual property or narrow the scope of our patent protection. In addition, the legal systems of certain countries do not favor the aggressive enforcement of patents, and the laws of foreign countries may not protect our rights to the same extent as the laws of the United States. As a result, our patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours. In order to preserve and enforce our patent and other intellectual property rights, we may need to make claims or file lawsuits against third parties. Such lawsuits could entail significant costs to us and divert our management’s attention from developing and commercializing our products.
We, like other companies in the pharmaceutical industry, may become aware of counterfeit versions of our products becoming available domestically and abroad. Counterfeit products may use different and possibly contaminated sources of plasma and other raw materials, and the purification process involved in the manufacture of counterfeit products may raise additional safety concerns, over which we have no control. Any reported adverse events involving counterfeit products that purport to be our products could harm our reputation and the sale of our products in particular and consumer willingness to use plasma-derived therapeutics in general.
26
Unauthorized use of our intellectual property may have occurred or may occur in the future. Although we have taken steps to minimize this risk, any failure to identify unauthorized use and otherwise adequately protect our intellectual property would adversely affect our business. For example, any unauthorized use of our trademarks could harm our reputation or commercial interests. Moreover, if we are required to commence litigation related to unauthorized use, whether as a plaintiff or defendant, such litigation would be time consuming, force us to incur significant costs and divert our attention and the efforts of our management and other employees, which could, in turn, result in lower revenue and higher expenses.
In addition to patented technology, we rely on our unpatented proprietary technology, trade secrets, processes and know-how.
We generally seek to protect proprietary information by entering into confidentiality agreements with our employees, consultants, scientific advisors and third parties. These agreements may not effectively prevent disclosure of confidential information, may be limited as to their term and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, our trade secrets may otherwise become known or be independently developed by our competitors or other third parties. To the extent that our employees, consultants or contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. Costly and time-consuming litigation could be necessary to determine and enforce the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position. We also rely on contractual protections with our customers, suppliers, distributors, employees and consultants and implement security measures designed to protect our trade secrets. We cannot assure you that these contractual protections and security measures will not be breached, that we will have adequate remedies for any such breach or that our suppliers, employees or consultants will not assert rights to intellectual property arising out of such contracts.
Since we rely on trade secrets and nondisclosure agreements, in addition to patents, to protect some of our intellectual property, there is a risk that third parties may obtain and improperly utilize our proprietary information to our competitive disadvantage. We may not be able to detect the unauthorized use of such information, prevent such use or take appropriate and timely steps to enforce our intellectual property rights.
We may infringe or be alleged to infringe intellectual property rights of third parties.
Our products or product candidates may infringe or be accused of infringing one or more claims of an issued patent or may fall within the scope of one or more claims in a published patent application that may be subsequently issued and to which we do not hold a license or other rights. Third parties may own or control these patents or patent applications in the United States and/or abroad. These third parties could bring claims against us or our collaborators that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us or our collaborators, we or they could be forced to stop or delay research, development, manufacturing or sales of the product or product candidate that is the subject of the suit.
If we are found to be infringing on the patent rights of a third party, or in order to avoid potential claims, we or our collaborators may choose or be required to seek a license from a third party and be required to pay license fees or royalties or both. These licenses may not be available on acceptable terms, or at all. Even if we or our collaborators were able to obtain a license, the rights may be nonexclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we or our collaborators are unable to enter into licenses on acceptable terms.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. In addition to infringement claims against us, we may become a party to other patent litigation and other proceedings, including interference proceedings declared by the U.S. Patent and Trademark Office and opposition proceedings in the European Patent Office, regarding intellectual property rights with respect to our products. The cost to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their substantially greater financial resources. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other proceedings may also absorb significant management time.
27
Many of our employees were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We take steps to ensure that our employees do not use the proprietary information or know-how of others in their work for us. We may, however, be subject to claims that we or these employees have inadvertently or otherwise used or disclosed intellectual property, trade secrets or other proprietary information of any such employee’s former employer. Litigation may be necessary to defend against these claims and, even if we are successful in defending ourselves, could result in substantial costs to us or be distracting to our management. If we fail to defend any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel.
We have in-licensed certain patent rights and co-own certain patent rights with third parties.
Our rights in certain intellectual property that we have in-licensed or co-own with third parties and the value therein may depend on our third party licensors’ or co-owners’, as applicable, performance under our intellectual property agreements with them. If one of these third parties is unable to, or does not, enforce their own rights in such intellectual property or perform under our agreements with them, it could affect our ability to effectively compete in the marketplace and operate our business.
Our in-license agreements for certain patent rights may impose payment and/or other material obligations on us as a licensee. Although we are currently in compliance with all of our material obligations under these licenses, if we were to breach any such obligations, our counterparty licensors may be entitled to terminate the licenses. Such termination may restrict, delay or eliminate our ability to develop and commercialize our products, which could adversely affect our business. We cannot guarantee that the third-party patents and technology we license will not be licensed to our competitors. In the future, we may need to obtain additional licenses, renew existing license agreements or otherwise replace existing technology. We are unable to predict whether these license agreements can be obtained or renewed or whether the technology can be replaced on acceptable terms, or at all.
We may not realize the expected benefits from the entry into new or amended contracts, cost-savings and business improvement initiatives.
We recently acquired plasma collection centers from Canadian Plasma Resources Corporation (in 2023), through the acquisition of Biotest AG (in 2022), from BPL Plasma Inc. and Kedplasma, LLC (in 2021), from South Korean firm GC Pharma (Group) (“GC Pharma”) (in 2020), and entered into plasma supply agreements with Haema Kft in Hungary and with Biotek America LLC (both in 2021). There can be no assurance we will be successful with this plasma acquisition strategy or that we will realize all of the benefits we expect from such new centers. Moreover, we have implemented a cost savings plan in to reduce headcount, become more efficient in the plasma procurement process and close certain underperforming plasma centers, among other strategies. See Item 5 of this Part I “A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Operational Improvement Plan.” Our cost savings and business improvement initiatives could result in unexpected charges and expenses that negatively impact our financial results and we could fail to achieve the desired efficiencies and estimated cost savings. In addition, if we are not able to effectively implement these initiatives, or if they fail to operate as intended, our financial results could be adversely affected. Additionally, these types of initiatives could yield unintended consequences such as distraction of management and employees, business disruption, an inability to attract or retain key personnel, which could negatively affect our business or financial condition and results of operations. If we are not able to effectively develop, implement and manage our cost savings or business improvement initiatives (including our acquisitions), we may experience operational difficulties and increased costs, which may adversely affect our results of operations.
Climate change and increased risk of major natural disasters may adversely affect our business.
Climate change is already causing extreme heat and poor air quality in some areas, which threaten to exacerbate pre-existing health conditions such as respiratory diseases. In addition, an increase in temperature and humidity may cause a proliferation of insects that carry vector-borne diseases, including dengue fever and malaria. Ultimately, climate change could undermine decades of progress in improving human health at a time when antimicrobial resistance is also rising.
28
We are exposed to climate risks such as physical risks (e.g., heat, water scarcity, sea level rise, flooding from severe weather events) and transition risks (e.g., regulatory frameworks, carbon pricing, cost of and access to capital), which could vary in magnitude and impact country by country. For example, some of our production facilities that depend on the availability of significant water supplies are located in areas where water is increasingly scarce. Other facilities are located in places that, because of increasingly violent weather events, sea level rise, or both, are increasingly at risk of substantial flooding. In regions where this risk is present, it impacts not only our own operations but also our distribution supply chain. Such events may result in increased costs, business interruptions, destruction of facilities, loss of life, and disruption to healthcare systems that patients use to access our medicines.
Climate change may trigger the adoption of new regulatory requirements across the globe. Such legislation could include increased requirements to invest in technology to reduce energy use, water use and greenhouse gas emissions, beyond what we expect to invest in our existing plans. Item 4 of this Part I, “Information on the Company—B. Business Overview—Climate Change.” In addition, legislation could include carbon pricing, climate risk disclosure mandates, and changes in zoning or building codes to increase climate resilience. The combined impact of these transition risks could increase our direct operating costs and result in the same impact across our supply chain.
Changes, enactment, and/or enforcement of biometric information privacy laws of different jurisdictions, including federal and state laws in the United States, could expose us to potential liability.
Regulatory authorities and governments around the world have implemented and are considering further legislative and regulatory proposals regarding biometric information privacy. New laws and regulations governing biometric information privacy and data protection imposing more stringent requirements may be introduced in various jurisdictions, including the United States, the European Union and the United Kingdom. Complying with laws and regulations for an increasing number of jurisdictions could require significant resources and costs, including those associated with adapting our facilities, products or processes.
In the United States, there are various laws and regulations concerning biometric data privacy and data protection, and certain U.S. state laws may be more stringent or broader in scope, or offer greater individual rights, with respect to personal information than federal, international or other state laws, and such laws may differ from each other, all of which may complicate compliance efforts. U.S. federal and state regulators, including the Federal Trade Commission (the “FTC”), have engaged in enforcement actions focused on biometric information. For example, the Biometric Information Privacy Act in Illinois (the “BIPA”), the Capture or Use of Biometric Identifier Act in Texas (the “CUBI”) and the My Health My Data Act in Washington (the “MHMD”) restrict the collection and use of biometric identifiers and biometric information. Several class action lawsuits have been brought under BIPA’s private right of action, and BIPA has generally been broadly interpreted by the courts. We were named party in, and have since settled, a class action that alleged that we failed to comply with the BIPA’s requirements when collecting fingerprint data of plasma donors. See Item 8 of this Part I, “A. Consolidated Statements and Other Financial Information—Legal Proceedings—Illinois Biometric Information Privacy Act Claim.”
Any future similar legal proceedings and any government enforcement actions we may become subject to under applicable privacy and data protection laws may cause us significant losses in addition to legal costs, which could adversely affect our business, results of operations and financial condition. In addition, any failure, or perceived failure, by us to comply with the above and other regulatory requirements or biometric information privacy and data protection-related laws, rules and regulations could result in reputational damages or proceedings or actions against us by governmental entities, consumers or other parties. Such proceedings or actions could subject us to significant penalties and negative publicity, require us to change our data and other business practices, increase our costs and severely disrupt our business or hinder our global expansion.
29
Risks Relating to the Healthcare Industry
United States Healthcare Reform may adversely affect our business.
The United States Patient Protection and Affordable Care Act and the companion Healthcare and Education Reconciliation Act, each enacted in March 2010, as amended (collectively, the “ACA”), increased federal oversight of private health insurance plans and included a number of provisions designed to reduce Medicare expenditures and the cost of health care generally, to reduce fraud and abuse, and to provide access to increased health coverage. While the ACA has materially expanded the number of individuals in the United States with health insurance, it has faced ongoing legal challenges, including federal litigation seeking to invalidate some of or all of the law or the manner in which it has been interpreted, and could have a significant impact on the United States healthcare industry.
There are also uncertainties due to federal legislative and administrative efforts to repeal (under the previous U.S. presidential administration), substantially change, replace or invalidate portions or all of the ACA. Any future legislation, guidance, rules or regulations and/or executive orders that materially alter the healthcare industry could have a significant impact on our operations. The uncertain status of the ACA affects our ability to plan, and its repeal without adequate replacement could have a material adverse effect on our United States operations.
Government pressures and constraints on reimbursement may adversely affect our business.
We engage in various manufacturing, processing, marketing and sales activities pertaining to pharmaceutical products in a number of jurisdictions around the world. These activities subject us to several governmental regulations mandating multiple types of controls over pricing and general operations in the various countries in which we operate. The growth of overall healthcare costs as a percentage of gross domestic product in many countries means that governments and payers are under intense pressure to control spending even more tightly.
In the United States, which is our main market, trends in recent years in the healthcare industry have caused significant changes including a shift towards managed or value-based care, collective purchasing agreements, consolidation in office-based healthcare providers, and other cost-saving, revenue and payment reduction measures with respect to, for example, several government healthcare programs that cover our products, including Medicaid, Medicare Parts B and D and the 340B Program. These trends could have a material adverse impact on our financial performance. Global emphasis on healthcare cost containment exerts significant pressures on the pricing of our products and on our ability to obtain and maintain reimbursement rates to cover our products, which may adversely affect our business.
In addition, the availability of federal funds to pay for our products under Medicaid and Medicare requires that we extend discounts under the 340B Program, and changes to this program could adversely affect our financial performance. The 340B Program extends discounts to a variety of eligible entities, including community health clinics and certain other entities that receive certain governmental healthcare grants, as well as hospitals that serve a disproportionate share of certain low income individuals, and certain cancer centers, children’s hospitals, critical access hospitals and rural referral centers. The 340B Program price, or ceiling price, cannot exceed the average manufacturer price (“AMP”) (as reported to the U.S. Centers for Medicare & Medicaid Services (“CMS”) under the Medicaid drug rebate program) less the Medicaid unit rebate amount. We have entered into a pharmaceutical pricing agreement (“PPA”), with the government in which we have agreed to participate in the 340B Program by charging eligible entities no more than the ceiling price for drugs intended for outpatient use. Evolving requirements with respect to this program continue to be issued by the Health Resources and Services Administration (“HRSA”) of the United States Department of Health and Human Services (“HHS”), the federal agency responsible for oversight of the 340B Program, which creates uncertainty, and certain aspects of the 340B Program, such as relating to manufacturers’ 340B pricing restrictions for prescriptions filled at contract pharmacy locations, continue to be challenged in federal courts, which adds to such uncertainty. We believe that we meet the requirements of the 340B Program, and are continuing to review and monitor these and other developments affecting the 340B Program.
Continuing efforts of certain regulatory and legislative bodies, as well as the United States Congress, are focused on pricing and reimbursement, and we expect that the healthcare industry will continue to be subject to increasing pricing and cost containment pressures in 2024 and beyond. These pricing and cost containment pressures may impact the reimbursement rates for our products and have an adverse effect on our business. For more details, see Item 4 of this Part I, “E. Regulatory Matters—Pharmaceutical Pricing and Reimbursement.”
30
Impact of government regulations over product development and regulatory approvals may adversely affect our business.
We develop and manufacture pharmaceutical products in a number of jurisdictions around the world. These activities subject us to several governmental regulations mandating specific governmental approvals that are necessary for us to develop our products in the various countries in which we operate. Obtaining market approval for our products is a lengthy, costly and complex regulatory process that requires intensive preclinical and clinical data, and the approval process can vary significantly depending on the regulatory authority of each jurisdiction. Relevant health authorities may, at the time of the filing of the application for a marketing authorization, or later during their review, impose requirements that can evolve over time, including requiring additional clinical trials, and such authorities may delay or refuse to grant approval.
Even where we have obtained marketing approval for a product in one or more major markets, we may need to invest significant time and resources in applying for approval in other markets, and there is no assurance that we will be able to obtain such approval. In recent years, health authorities have become increasingly focused on product safety and on the risk/benefit profile of pharmaceutical products, which could lead to more burdensome and costly approval processes and negatively affect our ability to obtain regulatory approval for products under development. For example, the FDA and the EMA have been implementing strict requirements for approval, particularly in terms of the volume of data needed to demonstrate a product’s efficacy and safety.
In the United States, our main market, there is an abbreviated regulatory approval pathway for biological products found to be “biosimilar” to or “interchangeable” with a biological “reference product” previously licensed under a Biologics License Applications (“BLA”). This abbreviated approval pathway is intended to permit a biosimilar product to come to market more quickly and less expensively by relying to some extent on the data generated by the reference product’s sponsor, and the FDA’s previous review and approval of the reference product.
The law provides that no biosimilar application may be accepted for FDA review until four years after the date the reference product was first licensed by the FDA, and that the FDA may not make approval of an application effective until 12 years after the reference product was first licensed. The law also includes an extensive process for the innovator biologic and biosimilar manufacturer to litigate patent infringement, validity, and enforceability, which could increase costs of protecting our reference products. Once approved, biosimilars likely would compete with, and in some circumstances may be deemed under applicable laws to be “interchangeable with,” the previously approved reference product. The extent to which a biosimilar product, once approved, will be substituted for any of our products, in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. In connection with the FDA’s authority to ensure the development of safe and effective biosimilars, in September 2022, the FDA Biosimilar User Fee Amendments of 2022 were signed into law. The FDA Biosimilar User Fee Amendments of 2022 reauthorize for an additional five years (through 2027) the Biosimilar User Fee Act of 2012, which allows the FDA to assess and collect fees for biosimilars. While the fee rates charged to biosimilar program sponsors are intended to expedite the process for reviews of biosimilar applications, it is unknown how such fees will be passed on in the future, which could also impact our financial results. We expect in the future to face greater competition from biosimilar products, including a possible increase in patent challenges, all of which could adversely affect our financial performance.
Regarding access to our products, the ACA established and provided significant funding for a Patient-Centered Outcomes Research Institute to coordinate and fund Comparative Effectiveness Research, as those terms are defined in the ACA. While the stated intent of Comparative Effectiveness Research is to develop information to guide providers to the most efficacious therapies, outcomes of Comparative Effectiveness Research could influence the reimbursement or coverage for therapies that are determined to be less cost effective than others. Should any of our products be determined to be less cost effective than alternative therapies, the levels of reimbursement for these products, or the willingness to reimburse at all, could be impacted, which could materially impact our financial results. In connection with the FDA’s authority to ensure the development of safe and effective biosimilars, in September 2022, the FDA User Fee Reauthorization Act of 2022 was signed into law, which allows the FDA to assess and collect fees for biosimilars for a five-year period through 2027. While the fee rates charged to biosimilar program sponsors are intended to expedite the process for reviews of biosimilar applications, such fees could be passed onto manufacturers and, thus, could also impact our financial results.
31
Failure to comply with laws and regulations governing the sales and marketing of our products or an adverse decision in lawsuits may result in adverse consequences to us.
We engage in various marketing, promotional and educational activities pertaining to, as well as the sale of, pharmaceutical products in a number of jurisdictions around the world. The promotion, marketing and sale of pharmaceutical products and medical devices is highly regulated and the sales and marketing practices of market participants such as us have been subject to increasing supervision by governmental authorities around the world, and we believe that this trend will continue.
For example, the laws governing our conduct in the United States are enforceable by criminal, civil and administrative penalties. Violations of laws such as the Federal Food, Drug and Cosmetic Act (“FDCA”), the Federal False Claims Act (“FCA”), the Public Health Service Act (“PHS Act”) or provisions of the U.S. Social Security Act known as the “Anti-Kickback Statute” and the “Civil Monetary Penalties Law,” or any regulations promulgated under their authority, may result in jail sentences, fines or exclusion from federal and state programs, as may be determined by Medicare, Medicaid, the Department of Defense, other regulatory authorities and the courts. There can be no assurance that our activities will not come under the scrutiny of regulators and other government authorities or that our practices will not be found to violate applicable laws, rules and regulations or prompt lawsuits by private citizen “relators” under federal or state false claims laws. For a description of fraud and abuse laws see Item 4 of this Part I, “Information on the Company—E. Regulatory Matters—Government Regulation—United States Government Regulation—Anti-fraud and Abuse Regulation.”
Failure to comply with fraud and abuse laws and regulations could also result in other significant civil and criminal penalties and costs, including the loss of licenses and the inability to participate in federal and state health care programs, and could have a material adverse effect on our business. In addition, these measures may be interpreted or applied by a prosecutorial, regulatory or judicial authority in a manner that could require us to make changes in our operations or incur substantial defense and settlement expenses. The fraud and abuse laws and regulations have been subject to heightened enforcement activity over the past few years. Since the ACA significantly strengthened provisions of the FCA, the anti-kickback provisions of Medicare and Medicaid and other healthcare antifraud provisions, there have also been a greater number of qui tam suits brought by “relator” whistleblowers, who may receive up to 30% of total government recoveries. Even unsuccessful challenges by regulatory authorities or private relators could result in reputational harm and the incurring of substantial costs. Further, many of these laws are vague or indefinite and have not been interpreted by the courts, and have been subject to frequent modification and varied interpretation by prosecutorial and regulatory authorities, increasing the risk of noncompliance. Most states have adopted similar state false claims laws, and these state laws have their own penalties which may be in addition to FCA penalties, as well as other fraud and abuse laws. While we believe that we are substantially compliant with applicable fraud and abuse laws and regulations, and have adequate compliance programs and controls in place to ensure substantial compliance, we cannot predict whether changes in applicable law, or interpretation of laws, or changes in our services or marketing practices in response to changes in applicable law or interpretation of laws, could have a material adverse effect on our business.
Failure to satisfy requirements under the FDCA can also result in penalties, as well as requirements to enter into consent decrees or orders that prescribe allowable corporate conduct. In this regard, our Los Angeles facility was previously managed pursuant to a consent decree that was entered into in February 1998 based on action by the FDA and the U.S. Department of Justice (the “DOJ”), addressing FDCA violations committed by the former owner of the facility, Alpha Therapeutic Corporation (“Alpha”). The consent decree provided for annual inspection of the plant by the FDA. On March 15, 2012, the United States District Court for the Central District of California entered an order vacating the consent decree on the Los Angeles facility.
Adverse consequences can also result from failure to comply with the requirements of the 340B Program under the PHS Act, which extends discounts to a variety of community health clinics and other entities that receive health services grants under the PHS Act (the “340B Program”). In early 2016, HRSA finalized a regulation regarding the 340B Program pricing methodology, providing. HRSA regulations prescribes when civil monetary penalties may be issued for “knowing and intentional” manufacturer overcharges of 340B Program covered entities, and provides that manufacturers who overcharge may be subject to significant monetary penalties. Such findings could also result in negative publicity that could harm the manufacturer’s reputation or cause business disruption, penalties, or CMP. Under the HRSA regulations, the CMP may be up to $5,000 for each instance of overcharging a covered entity. If we are ultimately required to change our sales or pricing practices with regard to the distribution of drugs under the 340B Program, or if we were required to pay penalties under the applicable regulations, there would be an adverse effect on our revenues and profitability.
32
In addition, companies in the United States, Canada and the European Union are generally restricted from promoting approved products for other indications that are not specifically approved by the competent regulatory authorities, nor can companies promote unapproved products. Improper promotion of unapproved drugs or devices or unapproved indications for a drug or device may subject us to warnings from, or enforcement action by, regulatory authorities, harm demand for our products, and subject us to civil and criminal sanctions. Further, sanctions under the FCA have been brought against companies accused of promoting off-label uses of drugs, because such promotion induces the use and subsequent claims for reimbursement under Medicare and other federal programs. Industry data indicates that a significant portion of IVIG volume may be used to fill physician prescriptions for indications not approved by the FDA or similar regulatory authorities. Violations or allegations of violations of the foregoing restrictions could materially and adversely affect our business.
We are required to report detailed pricing information, net of included discounts, rebates and other concessions, to CMS for the purpose of calculating national reimbursement levels, certain federal prices and certain federal and state rebate obligations. We have established systems for collecting and reporting this data accurately to CMS and have instituted a compliance program designed to assure that the information collected is complete in all respects. If we report pricing information that is not accurate to the federal government, we could be subject to fines and other sanctions (including potential FCA liability) that could adversely affect our business.
To market and sell our products outside of the United States, we must obtain and maintain regulatory approvals and comply with regulatory requirements in such jurisdictions. The approval procedures vary among countries in complexity and timing. We may not obtain approvals from regulatory authorities outside the United States on a timely basis, if at all, which would preclude us from commercializing products in those markets. In addition, some countries, particularly the countries of the European Union, regulate the pricing of prescription pharmaceuticals. In these countries, pricing discussions with governmental authorities can take considerable time after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost effectiveness of our product candidate to other available therapies. Such trials may be time consuming and expensive and may not show an advantage in efficacy for our products. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, in either the United States or the European Union, we could be adversely affected.
In the United States, under the federal Physician Payment Sunshine Act or Open Payments Program (the “PPS Act”), we are required to report and disclose payments or other transfers of value made to certain healthcare providers, including physicians, certain advanced practice providers such as physician assistants and nurse practitioners, and teaching hospitals. CMS publishes information from these reports on a publicly available website, including amounts transferred and healthcare provider identities. Under the PPS Act we are required to collect and report detailed information regarding certain financial relationships we have with covered healthcare providers. The PPS Act preempts similar state reporting laws, although we or our subsidiaries may also be required to report under certain state transparency laws that address circumstances not covered by the PPS Act, and some of these state laws are also ambiguous. We are also subject to foreign regulations requiring transparency of certain interactions between suppliers and their customers. While we believe we have substantially compliant programs and controls in place to comply with these reporting requirements, we cannot assure you that regulations will not require us to take additional compliance steps. Our compliance with these rules imposes additional costs on us.
We also are subject to certain laws and regulations concerning the conduct of our foreign operations outside the United States, including the U.S. Foreign Corrupt Practices Act (“FCPA”) and other anti-bribery laws and related laws, and laws pertaining to the accuracy of our internal books and records, which have been the focus of increasing enforcement activity in recent years. Under the FCPA, the United States has increasingly focused on regulating the conduct by U.S. businesses occurring outside of the United States, generally prohibiting remuneration to foreign officials for the purpose of obtaining or retaining business. Also, in some countries we may rely on third parties for the marketing and distribution of our products, and these parties may lack sufficient internal compliance resources, and may operate in foreign markets involving substantial corruption. If our efforts to monitor these parties fail to detect potential wrongdoing, we could be held responsible for the noncompliance of these third parties with applicable laws and regulations, which may have a material adverse effect on our business.
33
We could be adversely affected if other government or private third-party payors decrease or otherwise limit the amount, price, scope or other eligibility requirements for reimbursement for the purchasers of our products.
Certain of our products are subject to various cost-containment measures, such as government-imposed industry-wide price reductions, mandatory pricing systems, reference pricing systems, payors limiting access to treatments based on cost-benefit analyses, an increase in imports of drugs from lower-cost countries to higher-cost countries, shifting of the payment burden to patients through higher co-payments, limiting physicians’ ability to choose among competing medicines, mandatory substitution of generic drugs for the patented equivalent, and growing pressure on physicians to reduce the prescribing of patented prescription medicines. Such pressures could have a material adverse impact on our business, financial condition or results of operations, as well as on our reputation.
For example, certain pharmaceutical products, such as plasma derivative products, are subject to price controls in several of our principal markets, including Spain and countries within the European Union. In the United States, where pricing levels for our products are established by governmental payors and negotiated with private third-party payors, if the amount of reimbursement available for a product is reduced, it may cause groups or individuals dispensing the product to discontinue administration of the product, to administer lower doses, to substitute lower cost products or to seek additional price-related concessions. These actions could have a negative effect on our financial results, particularly in cases where our products command a premium price in the marketplace or where changes in reimbursement induce a shift in the location of treatment. The existence of direct and indirect price controls and pressures over our products has affected, and may continue to materially adversely affect, our ability to maintain or increase gross margins. In addition, the growth of overall healthcare costs and certain weak economic and financial environment in certain countries where we do business, as well as increased scrutiny over pharmaceutical pricing practices, such as in the United States, all enhance these pricing pressures.
In the United States, pricing concerns have led to heightened scrutiny and ongoing legislative efforts to increase transparency around healthcare and pharmaceutical drugs costs. For example, on November 12, 2020, CMS issued final rules imposing price transparency requirements on hospitals and group health plans, which went into effect in three stages from 2022 to 2024, and as of January 1, 2024 payers must disclose in-network provider negotiated rates (which include rates with device suppliers and manufacturers), historical out-of-network allowed amounts for all covered items and services, including all prescription drugs. States are also enacting a variety of transparency measures. The publication of our negotiated rates could impact our ability to independently negotiate sales contracts and rate agreements. In addition, uncertainty around ongoing price transparency proposals affects our ability to plan, as the proposals, if adopted, in whole or in part, could adversely affect our business.
An increasing number of states in the United States have also proposed or passed legislation that seeks to directly or indirectly regulate pharmaceutical drug pricing, such as by requiring drug manufacturers to provide advance notice of certain price increases, or to place a maximum price ceiling on pharmaceutical products purchased by state agencies. State laws regulating pharmaceutical drug pricing may cause us to experience additional pricing pressures on our affected products, and could adversely affect our business.
Also, the intended use of a drug product by a physician can affect pricing. Physicians frequently prescribe legally available therapies for uses that are not described in the product’s labeling and that differ from those tested in clinical studies and that are approved by the FDA or similar regulatory authorities in other countries. These off-label uses are common across medical specialties, and physicians may believe such off-label uses constitute the preferred treatment or treatment of last resort for many patients in varied circumstances. In the United States, many off-label uses of drug products may be reimbursed by Medicare and other third-party payors, generally based on the payors’ determination that the intended use is for a medically accepted indication, for example, based on studies published in peer-reviewed medical journals or information contained in drug compendia, such as the United States Pharmacopeia-National Formulary. However, if reimbursement for off-label uses of products, including IVG, is reduced or eliminated by Medicare or other third-party payors, including those in the United States or the European Union, we could be adversely affected.
Proposed federal and state legislation have targeted drug pricing, including direct negotiations with manufacturers over price, reimbursement and discounts. Plasma protein therapeutics have been excluded from certain aspects of the several legislations; however, there is a continuing risk that our products may be subject to new pricing restrictions.
34
We are subject to extensive government regulatory compliance and ethics oversight.
Our business is subject to extensive government regulation and oversight by the many countries in which we operate. We have enacted anticorruption, privacy, healthcare and corporate compliance policies and procedures that govern our business practices and those of our distributors and suppliers. These policies and procedures are effectuated through education, training and monitoring of our employees, distributors and suppliers. In addition, to enhance compliance with applicable healthcare laws and mitigate potential liability in the event of noncompliance, regulatory authorities, such as HHS’s Office of the Inspector General (“OIG”) of the United States, have recommended the adoption and implementation of a comprehensive healthcare compliance program that generally contains the elements of an effective compliance and ethics program described in Section 8B2.1 of the U.S. Sentencing Commission Guidelines Manual. Increasing numbers of U.S.-based pharmaceutical companies have such programs, and we have adopted U.S. healthcare compliance and ethics programs that generally incorporate the OIG’s recommendations. However, our adoption and enforcement of these various policies and procedures does not ensure that we will avoid investigation or the imposition of penalties by applicable government agencies.
Failure to comply with changing regulatory requirements could materially adversely affect our business.
We engage in various manufacturing, processing, marketing and sales activities pertaining to pharmaceutical products in a number of jurisdictions around the world. These activities subject us to several governmental regulations governing our global operations. The laws and regulations of the many jurisdictions that govern our business and operations are subject to varying and evolving interpretations that affect our ability to comply, and future changes, additions, and enforcement approaches, including in light of political changes. Changes with respect to the applicable laws and regulations may require us to update or revise our operations, services, marketing practices, and compliance programs and controls, and may impose additional and unforeseen costs on us, pose new or previously immaterial risks to us, or may otherwise have a material adverse effect on our business. There can be no assurance that current and future government regulations will not adversely affect our business, and we cannot predict new regulatory priorities, the form, content or timing of regulatory actions, and their impact on the health care industry and on our business and operations.
We are subject to extensive environmental, health and safety laws and regulations.
Our business involves the controlled use and the generation, handling, management, storage, treatment and disposal of hazardous substances, wastes and various biological compounds and chemicals. The risk of contamination or injury from these materials cannot be eliminated. If an accident, spill or release of any regulated chemicals, substances or wastes occurs, we could be held liable for resulting damages, including for investigation, remediation and monitoring of the contamination, including natural resource damages, the costs of which could be substantial. As owners and operators of real property, we could also be held liable for the presence of hazardous substances as a result of prior site uses or activities, without regard to fault or the legality of the original conduct that caused or contributed to the presence or release of such hazardous substance on, at, under or from our property. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials, chemicals and wastes.
Although we maintain workers’ compensation insurance to cover the costs and expenses that may be incurred due to injuries to our employees resulting from the use and handling of these materials, chemicals and wastes, this insurance may not provide adequate coverage against potential liabilities.
Additional or more stringent federal, state, local or foreign laws and regulations affecting our operations may be adopted in the future. We may incur substantial capital costs and operating expenses to comply with any of these laws or regulations and the terms and conditions of any permits required pursuant to such laws and regulations, including costs to install new or updated pollution control equipment, modify our operations or perform other corrective actions at our respective facilities. In addition, fines and penalties may be imposed for noncompliance with environmental and health and safety laws and regulations or for the failure to have or comply with the terms and conditions of required environmental permits.
35
Risks Relating to Our Shares and American Depositary Shares
If we discover material weaknesses or significant deficiencies in our internal control over financial reporting, it may adversely affect our ability to provide timely and reliable financial information and satisfy our reporting obligations under U.S. federal securities laws, which also could affect the market price of our American Depositary Shares or our ability to remain listed on NASDAQ.
Effective internal and disclosure controls are necessary for us to provide reliable financial reports and effectively prevent fraud and to operate successfully as a public company. If we cannot provide reliable financial reports or prevent fraud, our reputation and operating results would be harmed. A “significant deficiency” is a deficiency, or combination of deficiencies, in internal control over financial reporting that is less severe than a material weakness, yet important enough to merit attention of those responsible for oversight of our financial reporting. In addition, a “material weakness” is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis.
To the extent that any material weakness or significant deficiency exists in our internal control over financial reporting, such material weakness or significant deficiency may adversely affect our ability to provide timely and reliable financial information necessary for the conduct of our business and satisfaction of our reporting obligations under U.S. federal securities laws, which could affect our ability to remain listed on NASDAQ. Ineffective internal and disclosure controls could cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of our American Depositary Shares, or ADSs, or the rating of our debt.
See Item 15 of Part 2 “Controls and Procedures” in connection with the material weaknesses due to ineffective information technology general controls and manual recurring journal entries in 2023.
The Grifols Family may exercise significant influence over the conduct of our business.
The founders of the Company and their relatives (the “Grifols Family”) and Scranton Enterprises B.V. own, directly and indirectly, approximately 36% of our Class A shares. The Class A shares exercise 100% of the voting control of our Company. As a result, the Grifols Family and Scranton Enterprises B.V. may exercise significant influence over matters requiring shareholders’ approval, including, among other things, the election of our board of directors, or the Board, dividend policy and certain fundamental corporate action, such as the issuance of bonds, a merger or a dissolution. Conflicts may arise between the interests of the principal shareholders and those of the other shareholders, and the principal shareholders may choose to resolve the conflict in a way that does not coincide with the interests of the other shareholders. See Item 7 of this Part I, “Major Shareholders and Related Party Transactions—Related Party Transactions.”
The market price of our Class B ADSs on NASDAQ, as well as the market price of our Class A ADSs traded in over the counter markets, may be volatile.
The market price of our Class B ADSs and Class A ADSs may be volatile as a result of various factors, many of which are beyond our control. These factors include, but are not limited to, the following:
| ● | market expectations for our financial performance; |
| ● | actual or anticipated fluctuations in our results of operations and financial condition; |
| ● | changes in the estimates of our results of operations by securities analysts; |
| ● | techniques employed by short sellers or other market disruptors to drive down the market price of our shares; |
| ● | the dissemination of false or maliciously distorted information with respect to our business operations, financials or corporate disclosures; |
36
| ● | potential or actual sales of blocks of our Class B ADSs in the market by any shareholder or short selling of our Class B ADSs. Any such transaction could occur at any time or from time to time, with or without notice to us (see “—Techniques employed by short sellers may drive down the market price of our shares, negatively impact our business operations and/or generate non-meritorious litigation”); |
| ● | the entrance of new competitors or new products in the markets in which we operate; |
| ● | volatility in the market as a whole; and |
| ● | the risk factors mentioned in this section. |
The market price of our Class B ADSs may be adversely affected by any of the preceding or other factors regardless of operations and financial condition. Since December 31, 2021, our Class B ADSs have traded as high as $13.28 per ADS on May 24, 2022 and as low as $5.95 per ADS on September 29, 2022. At market close on April 16, 2024, our Class B ADSs were traded for $6.78.
Techniques employed by short sellers or other market disruptors may drive down the market price of our shares, negatively impact our business operations and/or generate non-meritorious litigation.
Short selling is the practice of selling securities that a seller does not own but rather has borrowed from a third party with the intention of buying identical securities back at a later date to return to the lender. Short sellers seek to profit from a decline in the value of the securities between the sale of the borrowed securities and the purchase of the replacement securities, as short sellers hope to pay less in that purchase than they received in the sale. As it is in short sellers’ interest for the price of the securities to decline, certain short sellers publish, or arrange for the publication of, negative research reports and allegations regarding the relevant issuer and its business prospects in order to create negative market momentum and generate profits for themselves after selling a security short. These short attacks have, in the past, created downward pressure on the price of the relevant securities.
We have been the subject of negative publicity by a short seller. For more information, see “Item 5. Operating and Financial Review and Prospects―A. Operating Results―Subsequent Events—Short Seller Reports.” It is not clear what long-term effect such negative publicity could have on us and/or whether we will continue to be subject to short seller attacks from time to time in the future. If we were to become the subject of any additional unfavorable allegations, even when such allegations are untrue, we may have to expend a significant amount of resources to investigate such allegations and/or defend ourselves. For example, in response to such negative publicity, we invested significant resources and time, including undertaking both internal and external reviews. While we would prefer to strongly defend against any such short seller attacks, we may be constrained in the manner in which we can respond to any allegations due to applicable state or federal law, or issues of commercial confidentiality. In addition, such allegations have led, and may further lead, to heightened scrutiny and investigations conducted by the SEC or the CNMV. Any future response to negative publicity from short sellers or responses to information requests from regulators could be costly and time-consuming, and could divert management’s attention from our day-to-day operations.
Even groundless or outright false allegations against us could severely impact the market price of our Class A shares, Class B shares, ADSs and our business operations. Because of the nature of our business in collecting plasma and manufacturing and selling plasma-derived therapies, we depend significantly on our brand and on customer confidence in our services. As a result, such allegations may impact our revenues by damaging our reputation, confidence in our services and relationships with our existing customers and potential new customers.
37
In the event that any lawsuit is filed against us as a result of negative publicity by a short seller, we cannot predict the timing, outcome or consequences of such actions, and we cannot assure you that our defenses will be successful or whether we will be subject to any damages, or how much. Whether or not we prevail in case any such proceedings are created against us, we may incur significant expenses defending them, which may materially and adversely affect our financial condition and results of operations.
Fluctuations in the exchange rate between the U.S. dollar and the euro may increase the risk of holding our ADSs or shares.
The Spanish securities market for equity securities consists of four stock exchanges located in Madrid, Barcelona, Bilbao and Valencia (collectively, the “Spanish Stock Exchanges”). The majority of the transactions conducted on the Spanish Stock Exchanges are done through the Spanish Automated Quotation System (Sistema de Inteconexión Bursátil Español, or SIBE).
Our Class A shares and Class B shares are listed on the Spanish Stock Exchanges and quoted on SIBE in euros. In addition, our Class B shares are traded in the United States on the NASDAQ Global Select Market in the form of ADSs, evidenced by American Depositary Receipts, or ADRs, in U.S. dollars. In addition, a number of our Class A shares are traded over the counter in the form of ADSs. Fluctuations in the exchange rate between the U.S. dollar and the euro may result in temporary differences between the value of our ADSs and the value of our shares, which may result in heavy trading by investors seeking to exploit such differences. This may increase the volatility of, and have an adverse effect on, the price of our shares or ADSs.
In addition, as a result of fluctuations in the exchange rate between the U.S. dollar and the euro, the U.S. dollar equivalent of the proceeds that a holder of our ADSs would receive upon the sale in Spain of any shares withdrawn from the ADR depositary and the U.S. dollar equivalent of any cash dividends paid in euros on our shares represented by the ADSs could also decline.
Subscription (or preemptive) rights may be unavailable to U.S. holders of our shares or ADSs.
In the case of a future increase of our registered share capital, existing shareholders will generally be entitled to subscription (or preemptive) rights pursuant to Spanish law, unless waived by a resolution of the shareholders or, if such power has been delegated to the Board pursuant to a shareholders’ resolution, by a resolution of the Board and except in certain situations, such as capital increases made for an in-kind contribution, in which subscription (or preemptive) rights are not applicable by law. Holders of the Class B shares will generally not have a right to vote on any resolution on a capital increase or on the waiver of subscription (or preemptive) rights, unless such resolution does not treat the Class B shares in the same way as the Class A shares, except in the limited circumstances set out in the Articles of Association of Grifols, S.A. as amended (the “Articles of Association”).
Holders of our ADSs representing Class A shares or, even if preemptive rights are granted, holders of our ADSs representing Class B shares, or U.S. resident shareholders may not be able to exercise voting power or subscription (or preemptive) rights, as the case may be, in which case holders of our ADSs could be substantially diluted, unless a registration statement under the Securities Act, as amended, or the Securities Act, is effective with respect to such rights and the shares for which they give such right or an exemption from the registration requirements of the Securities Act is available.
We intend to evaluate at the time of any rights offering the costs and potential liabilities associated with any such registration requirements, as well as the benefits of enabling the exercise of subscription (or preemptive) rights for the shares. In doing so, we will also evaluate any other factors that we may consider appropriate at the time.
There can be no assurance that we will decide to comply with such registration requirements. If no such registration requirements are satisfied, the depositary will sell the subscription (or preemptive) rights relating to the ADSs on deposit and will distribute the proceeds of such sale, if any, to the holders of the ADSs. If the depositary is unable to sell rights that are not exercised or not distributed or if the sale is not lawful or reasonably practicable, it will allow the rights to lapse, in which case no value will be given for these rights.
38
ADS holders may be subject to limitations on the transfer of their ADSs.
ADSs are transferable on the books of the depositary. However, the depositary may refuse to deliver, transfer or register transfers of ADSs generally when the books of the depositary are closed or if such action is deemed necessary or advisable by the depositary or by us because of any requirement of law or of any government or governmental body or commission or under any provision of the deposit agreement. Moreover, the surrender of ADSs and withdrawal of our shares may be suspended subject to the payment of fees, taxes and similar charges or if we direct the depositary at any time to cease new issuances and withdrawals of our shares during periods specified by us in connection with shareholders’ meetings, the payment of dividends or as otherwise reasonably necessary for compliance with any applicable laws or government regulations.
Item 4.INFORMATION ON THE COMPANY
A. | History of and Development of the Company |
Introduction
We were founded in 1940 in Barcelona, Spain by Dr. José Antonio Grifols i Roig, a specialist and pioneer in blood transfusions and clinical analysis and the grandfather of our current Honorary Chairperson (non-member) of the Board. We have been making and selling plasma derivative products for more than 70 years. Over the last 25 years, we have grown from a predominantly domestic Spanish company into a global company by expanding both organically and through acquisitions throughout Europe, the United States, Latin America, Africa and Asia.
We were incorporated in Spain as a limited liability company on June 22, 1987 under the name Grupo Grifols, S.A., and we changed our name to Grifols, S.A. in 2005. We conduct business under the commercial name “Grifols.” Our principal executive office is located at Avinguda de la Generalitat, 152 Parque Empresarial Can Sant Joan, 08174 Sant Cugat del Vallès, Barcelona, Spain and our telephone number is +34 93 571 0500. Our registered office is located at c/Jesús y María, 6, Barcelona, Spain.
We are a vertically integrated global producer of plasma derivatives and we believe we rank among the three largest producers in the industry in terms of total sales globally. Our activities include sourcing raw material, manufacturing various plasma derivative products and selling and distributing final products to healthcare providers. We have expanded our plasma collection network and our manufacturing capacity through a combination of organic growth and acquisitions. As of December 31, 2023 we had more than 390 operating plasma collection centers located across the United States, Germany, Austria, Hungary, Canada and Egypt; and a fractionation capacity of over 20 million liters of plasma per year.
We also research, develop, manufacture and market in vitro diagnostics products, including analytical instruments, reagents, software and associated products for use in clinical and blood bank laboratories and hospital products.
Our Class A shares have been listed on the Spanish Stock Exchanges since we completed our initial public offering on May 17, 2006 and are quoted on the SIBE under the ticker symbol “GRF.” Since January 2008, we have been part of the IBEX-35 Index, which comprises the top 35 listed Spanish companies by liquidity and market capitalization. Our Class A shares are also traded in the United States in over the counter markets in the form of ADSs, evidenced by ADRs. Each ADS traded over the counter represents two of our Class A shares. Our Class B shares were issued as part of the consideration for the acquisition of Talecris Plasma Resources, Inc. (“Talecris”), a company that has since been merged into our subsidiary Biomat USA, and are listed on the Spanish Stock Exchanges and quoted on the SIBE under the ticker symbol “GRF.P.” Our Class B shares are also traded in the United States on the NASDAQ Global Select Market in the form of ADSs, evidenced by ADRs, under the symbol “GRFS.” Each ADS traded on NASDAQ represents one of our Class B shares. Our ADSs are currently traded in U.S. dollars. In November 2011, our ADSs were added to the NASDAQ Biotechnology Index.
The SEC maintains an internet site at http://www.sec.gov that contains reports, information statements and other information regarding issuers that file electronically with the SEC.
39
Important Milestones
The following are some of our most important historical milestones:
| ● | On December 29, 2023 we entered into a Strategic Alliance and Share Purchase Agreement with Haier Group Corporation (“Haier”) for the sale of a 20% equity stake in Chinese company Shanghai RAAS Blood Products Co Ltd (“Shanghai RAAS”) in exchange for approximately $1.8 billion, while retaining an equity stake in Shanghai RAAS of 6.58%. We had acquired 26.2% of the voting and economic rights in Shanghai RAAS in exchange for 45% of the economic rights and 40% of the voting rights in our U.S. subsidiary, Grifols Diagnostic Solutions Inc. (“GDS”), on March 30, 2020. Upon successful closing of this sale, which is anticipated to occur prior to the end of the second quarter of 2024, we will maintain and extend our presence in China and use the proceeds of the sale to significantly reduce debt. The transaction is subject to customary regulatory approvals. See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting our Financial Condition and Results of Operations—Dispositions—Shanghai RAAS;” |
| ● | On April 25, 2022, we acquired all of the existing equity interest in Grifols Biotest Holdings GmbH, formerly known as Tiancheng (Germany) Pharmaceutical Holdings AG (“Biotest Holdings”), which in turn owns 89.88% of the ordinary shares and 1.08% of the preferred equity shares of German publicly traded company Biotest AG, a global company that supplies plasma protein products and biotherapeutic drugs, for a total consideration of €1,090,518,254. In addition, (1) we completed a voluntary tender offer to all remaining shareholders of Biotest AG to acquire their ordinary and preferred shares, where we acquired 1,250,298 ordinary shares (for €43.00 per share) and 8,340,577 preferred shares (for €37.00 per share) and (2) we acquired 185,359 ordinary shares in May 2022 following the exercise of a legal put right provided by applicable German corporate laws by former shareholders of Biotest AG. This has brought our interest in Biotest AG to 97.13% of the voting rights and 70.18% of the share capital. As a result of these transactions, we have acquired 28 additional plasma collection centers. See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting our Financial Condition and Results of Operations—Acquisitions—Biotest AG Acquisition;” |
| ● | On July 29, 2021, we entered into a joint venture agreement with ImmunoTek GH, LLC (“ImmunoTek”), which contract was amended in 2023, to arrange for the construction, licensing and commissioning of 28 plasma collection centers in the United States. Pursuant to the joint venture agreement, we formed a company in the United States named Biotek America LLC (“ITK JV”), through which we currently hold a 75% interest in each of the 28 plasma collection centers, while ImmunoTek holds the remaining 25%. We have the right to acquire the remaining 25% interest in all 28 plasma collection centers approximately three years after their respective openings for an aggregate amount of $634.0 million. Of the 28 centers, we expect to acquire seven in April 2024, seven in July 2024, eight in January 2025 and six in January 2026. See “—B. Business Overview—Raw Materials;” |
| ● | On December 1, 2021, we sold preferred shares representing 12.9% of Biomat Newco and 12.5% of Biomat USA, our U.S.-based plasma collection subsidiaries that are part of the Biomat group, also comprised of Biomat USA’ subsidiaries Grifols Bio Supplies, Inc., formerly known as Interstate Blood Bank, Inc., Talecris and Biomat USA South, Inc., the latter two of which have since been merged into Biomat USA and dissolved, respectively (collectively, the “Biomat Group”), to Parette Investment Pte. Ltd. (the “GIC Investor”), an affiliate of GIC Private Limited, which is a sovereign wealth fund established by the Government of Singapore. The purchase price received was $990 million. We used the net proceeds to (i) prepay $600 million of principal amount of the Revolving Loans under the First Lien Credit Facilities, (ii) prepay $142,360,501.31 of the principal amount of the Dollar Tranche B Term Loans, (iii) prepay $88,003,617.48 of the Euro Tranche B Term Loans and (iv) repurchase €97,535,000.00 of the 2019 Notes under an asset sale offer. See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Dispositions—The Biomat Transactions” and “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Sources of Credit;” |
| ● | On October 15, 2020, we acquired 100% of the equity of Alkahest, Inc. (“Alkahest”), a California biopharmaceutical company, for a total consideration of $146 million. In 2015, we had previously acquired a significant minority stake of Alkahest and, with this transaction, we gain total control of the company; |
40
| ● | On October 1, 2020, we acquired a plasma fractionation facility and two purification facilities located in Montreal, Canada, as well as 11 plasma collection centers in the United States, from South Korean firm GC Pharma, for a total consideration of $457 million. Once we finish renovations and obtain all necessary licenses and regulatory approvals for the Montreal facilities, we will become the only large-scale commercial manufacturer of plasma products in Canada, with a fractionation capacity of 1.5 million liters annually. We expect to begin manufacturing IVIG and albumin in the Canadian facilities to supply the Canadian market in 2025 and to begin plasma fractionation and gammaglobulin manufacturing by 2027; |
| ● | In June 2018, we completed the acquisition of German based pharmaceutical company Haema AG for a purchase price of €220 million; |
| ● | In August 2018, we completed the acquisition of U.S. based pharmaceutical company BPC Plasma Inc (formerly known as Biotest US Corporation), for a purchase price of $286 million; |
| ● | In December 2016, we entered into an asset purchase agreement with Hologic Inc. (“Hologic”), to acquire Hologic’s nucleic acid testing (“NAT”) Donor Screening Unit. The transaction closed in January 2017 for a purchase price of $1.9 billion; |
| ● | In January 2014, we acquired the diagnostic business of Novartis Corporation (“Novartis”), for a purchase price of $1.7 billion; |
| ● | In June 2011, we acquired U.S. based biotherapeutics company Talecris Biotherapeutics for a purchase price of $3.7 billion; and |
| ● | In July 2003, we acquired the assets of Alpha Therapeutics Corporation, including its plasma fractionation plant in Los Angeles, California, for a purchase price of $104 million. |
For further details of our principal capital expenditures and divestitures, see Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Capital Expenditures, Other Intangible Assets and Rights of Use.”
B. | Business Overview |
General
We are one of the leading global specialty plasma therapeutics companies developing, manufacturing and distributing a broad range of biological medicines based on plasma derived proteins. Plasma derivatives are proteins found in human plasma, which once isolated and purified, have therapeutic value. These protein-based therapies extend and enhance the lives of individuals who suffer from chronic and acute, often life-threatening, conditions, including primary and secondary immunological deficiencies, Chronic Inflammatory Demyelinating Polyneuropathy (“CIDP”), A1PI deficiency and related emphysema, immune-mediated ITP, Guillain Barré syndrome, Kawasaki disease, allogeneic bone marrow transplants, hemophilia A and B, von Willebrand disease, traumatic or hemorrhagic shock and severe burns. In addition, we have built a diagnostic business that focuses on researching, developing, manufacturing and marketing in vitro diagnostics products for use in clinical and blood bank laboratories. We also specialize in providing infusion solutions, nutrition products and medical devices for use in hospitals and clinics.
Our products and services are used by healthcare providers in over 100 countries to diagnose and treat patients with hemophilia, immune deficiencies, infectious diseases and a range of other medical conditions, and we have a direct presence, through the operation of commercial subsidiaries, in over 30 countries.
We are a leading producer in the industry in terms of total sales globally. We believe we have a top three market position in various segments of the plasma derivatives industry, including A1PI, IG and albumin as well as in terms of plasma collection centers and fractionation capacity. Our long-term aim is to further strengthen our leadership through the development of new and differentiated plasma-derived therapeutics, and the expansion of our global plasma collection footprint via acquisitions and greenfield projects.
41
We organize our business into five business units: Plasma Procurement, Biopharma, Diagnostic, Bio Supplies and Others.
Plasma Procurement. The Plasma Procurement business unit includes all activities relating to plasma collection, including the evaluation and screening of plasma donors and the operation of our plasma collection centers. Our Plasma Procurement business unit has no revenues, as all plasma collected in our facilities is sold to Grifols companies in the Biopharma business unit.
Biopharma (formerly Bioscience). The Biopharma business unit includes activities relating to the manufacture of plasma derivatives for therapeutic use, including the reception, analysis, quarantine, classification, fractionation and purification of plasma and the sale and distribution of end products. The main plasma products we manufacture are IG, Factor VIII, Alpha 1 (A1PI) and albumin. We also manufacture intramuscular (hyperimmune) immunoglobulins, ATIII, Factor IX and plasma thromboplastin component (“PTC”). The Biopharma business unit accounted for €5,558.3 million, or 84.3%, of our total net revenue in 2023.
Diagnostic. The Diagnostic business unit focuses on researching, developing, manufacturing and marketing in vitro diagnostics products, including analytical instruments, reagents, software and associated products for use in clinical and blood bank laboratories, covering the entire value chain from donation to transfusion. We concentrate our Diagnostic business in transfusion medicine (immunology, immunohematology) and specialty diagnostics such as hemostasis. The Diagnostic business unit’s main customers are blood donation centers, clinical analysis laboratories and hospital immunohematology services. The Nucleic Acid Testing, or NAT, Donor Screening Unit is engaged in research, development, manufacturing and commercialization of assays and instruments based on NAT technology for transfusion and transplantation screening. NAT technology makes it possible to detect the presence of infectious agents in blood and plasma donations, contributing to greater transfusion safety. The Diagnostic business unit accounted for €670.3 million, or 10.2%, of our total net revenue in 2023.
Bio Supplies. Net revenue from Bio Supplies primarily consists of revenue related to biological products for non-therapeutic use. The Bio Supplies business unit accounted for €160.0 million, or 2.4%, of our total net revenue in 2023.
Other Activities and Operations. In addition to our four business units, we have other smaller operations, activities and business lines (“Others”), which net revenue primarily originating from the provision of manufacturing services to third parties, third party plasma sales and research activities. Others also includes our Healthcare Solutions (formerly our Hospital Division). It also includes pharmaceutical products manufactured by the Group and intended for hospital pharmacies, as well as the marketing of products that complement our own products. Others accounted for €203.5 million, or 3.1%, of our total net revenue in 2023.
Geographic Markets
We believe we are a leading plasma derivatives producer globally, ranking among the three largest producers in the industry in terms of total sales, along with Takeda and CSL Group. We are the world’s largest producer of A1PI, which is used for the treatment of A1PI deficiency-related emphysema.
We currently operate in over 100 countries through distributors and subsidiaries in 30 countries. The United States is the largest sales region in the world for the plasma derivative sector. For the year ended December 31, 2023, the United States and Canada accounted for 59.1% of our total net revenue, the European Union accounted for 19.1% of our total net revenues (26.0% of which was generated in Spain) and the rest of the world accounted for 21.8% of our total net revenue.
Certain sales regions, particularly in emerging markets, have experienced continuous growth, driven by enhanced socioeconomic conditions, including a general increase in the testing of patients for immunodeficiencies, autoimmune diseases and low levels of alpha 1 protein, all conditions treatable by our products, more informed patients who are demanding better quality medical care, as well as increasing government healthcare spending on plasma derivative products. These emerging markets are expected to experience significant growth. Our presence and experience in Latin America, in countries such as Mexico, Colombia, Argentina, Chile and Brazil, where we have been marketing and selling products for over 20 years, has positioned us to benefit from this additional growth in both our Biopharma and Diagnostic business units. In the Asia-Pacific region, we have established a presence through our subsidiaries and representative offices in Malaysia, China, Thailand, Singapore, Australia, Japan, India, Hong Kong, Taiwan and Indonesia. We have also opened a Middle Eastern representative office in Dubai and Saudi Arabia.
42
We maintain a continuing focus on international expansion and acquisitions and will continue to selectively consider acquisitions that would generate operation synergies. For specific examples of acquisitions we have made to further enhance our operations, see “—A. History and Development of the Company—Important Milestones” above.
The following chart reflects a summary of net revenue by each of our geographic regions for the past three years:
| Year ended |
|
| Year ended |
|
| Year ended |
|
| ||||
December 31, | % of total | December 31, | % of total | December 31, | % of total |
| |||||||
Summary of Net Revenue by Region | 2023 | net revenue | 2022 | net revenue | 2021 | net revenue |
| ||||||
(in thousands of euros, except for percentages) |
| ||||||||||||
European Union(1) |
| 1,255,927 | 19.1 | % | 1,032,210 |
| 17.0 | % | 906,449 |
| 18.4 | % | |
United States and Canada |
| 3,898,961 | 59.1 | % | 3,855,607 |
| 63.6 | % | 3,154,549 |
| 63.9 | % | |
Rest of the World |
| 1,437,089 | 21.8 | % | 1,176,150 |
| 19.4 | % | 872,120 |
| 17.7 | % | |
Total |
| 6,591,977 | 100.0 | % | 6,063,967 |
| 100.0 | % | 4,933,118 |
| 100.0 | % | |
| (1) | Net revenue earned in the European Union includes net revenue earned in Spain. |
Principal Activities
We organize our business into five business units: Plasma Procurement, Biopharma, Diagnostic, Bio Supplies and Others. The Plasma Procurement business unit generates no revenues, as all plasma collected is sold to Grifols companies in the Biopharma business unit. As a result, our presentation of consolidated financial information excludes Plasma Procurement as an operating segment. The following chart presents our total net revenues by each of our other business units for the past three years:
| Year ended |
|
| Year ended |
|
| Year ended |
|
| ||||
December 31, | % of total | December 31, | % of total | December 31, | % of total |
| |||||||
Summary of Net Revenue by business unit | 2023 | net revenue | 2022 | net revenue | 2021 | net revenue |
| ||||||
(in thousands of euros, except for percentages) |
| ||||||||||||
Biopharma |
| 5,558,301 | 84.3 | % | 5,005,382 | 82.5 | % | 3,814,983 | 77.3 | % | |||
Diagnostic |
| 670,269 |
| 10.2 | % | 671,292 |
| 11.1 | % | 779,108 |
| 15.8 | % |
Bio Supplies |
| 159,957 |
| 2.4 | % | 146,076 |
| 2.4 | % | 115,811 |
| 2.3 | % |
Others |
| 203,450 |
| 3.1 | % | 250,165 |
| 4.1 | % | 266,461 |
| 5.4 | % |
Intersegments |
| — |
| — | (8,948) |
| (0.1) | % | (43,245) |
| (0.8) | % | |
Total |
| 6,591,977 |
| 100.0 | % | 6,063,967 |
| 100.0 | % | 4,933,118 |
| 100.0 | % |
The Biopharma Business Unit
The Biopharma business unit is responsible for the research and development, production and marketing of plasma derivative products. In 2023, the Biopharma business unit accounted for 84.3% of our total net revenue.
43
Operational Structure
The following chart illustrates its operational structure:
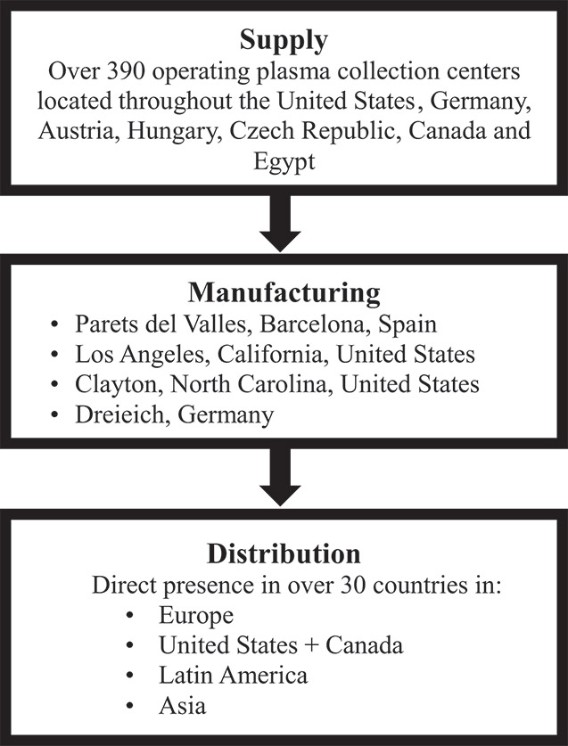
From plasma donation to therapeutic application, there are four major steps in the industry value chain process: (i) plasma collection, (ii) transport and logistics, (iii) manufacturing (fractionation and purification) and (iv) marketing and distribution. We are present at all levels of the value chain, from collection centers to distribution of the final products. This vertical integration enables us to leverage our position at each stage to control the overall process, to benefit from lower prices and to introduce complementary products, such as those offered through the Diagnostic business unit, to our customers.
Plasma Collection
Plasma is the key raw material used in the production of plasma-derived products. We have expanded our plasma collection network through a combination of organic growth by opening new plasma collection centers and acquisitions. We obtain our plasma primarily from the United States and Europe (Germany, Austria, Czech Republic, and Hungary) through more than 390 operating plasma collection centers and, to a much lesser extent, through agreements with third parties. Over the last few years, pursuant to the implementation of our business strategy, we have acquired plasma collection centers in the United States, Canada and Europe. In 2023, we increased our plasma collection by 10.0% in comparison to 2022, and the average cost per liter of plasma collected in 2023 declined by 15.2% in comparison to the average cost per liter in 2022.
44
We believe that the majority of our plasma requirements through 2024 will be met through plasma collected at our plasma collection centers and, to a lesser extent, purchased from third-party suppliers pursuant to various plasma purchase agreements. As we source the majority of our plasma internally, we are well positioned to ensure the availability of plasma for our manufacturing needs and assure the quality of the plasma throughout our manufacturing process.
We have implemented mechanisms to ensure that plasma donors meet the guidelines set forth by applicable regulations regarding, among other things, health, age and frequency of donations. Once the plasma donation is completed, as required by applicable United States and European regulations, we test every donation for pathogens such as HIV, hepatitis A, B and C, parvovirus B19 and syphilis. If we discover a unit of plasma that cannot be used in the fractionation process, we notify the donor and remove all plasma previously donated by such donor from our inventory.
Transport and Logistics
Once plasma has been collected, it is frozen at the collection center and sent to fractionation centers. One essential aspect of this process is the implementation of safety procedures to guarantee the quality and safety of the donated plasma. To ensure preservation of the proteins found in plasma, plasma must be kept at or below a temperature of -20 degrees Celsius (-4 degrees Fahrenheit). In accordance with European and United States requirements, we store our plasma at a temperature of -30 degrees Celsius (-22 degrees Fahrenheit). During transportation, plasma is kept at a temperature at or below -20 degrees Celsius. Our frozen plasma is transported by one of two transport companies, which are the same used throughout the industry.
Fractionation and Purification
Once plasma has been obtained, it may be used for plasma transfusions. It may also be frozen (as fresh frozen plasma) and manufactured into plasma derivatives through the fractionation process. The fractionation process consists of the separation of specific proteins through temperature and pH changes, as well as the use of filtration and centrifugation techniques. This process also includes a phase of introducing various viral inactivation procedures. Fractionation occurs in tanks at near freezing temperatures to maintain the integrity of the proteins. All known plasma derivative products can be fractionated from the same batch of plasma. As a result, the development of a new or higher yield plasma derivative product would likely generate incremental sales without increasing the requirement for additional plasma.
We currently operate two Biopharma manufacturing facilities in the United States (Clayton, North Carolina, and Los Angeles, Califonia), one in Spain (Parets del Vallès), one in Ireland (Dublin), one in Canada (Montreal) and one in Germany (Dreieich). Our plasma derivative products are manufactured at our Clayton, Los Angeles and Parets facilities, which have a combined fractionation capacity of over 20 liters per year. Our Clayton facility is one of the world’s largest integrated protein manufacturing sites, including fractionation, purification and aseptic filling and finishing of plasma-derived proteins.
Currently, the Clayton, Los Angeles and Parets facilities are equipped and licensed to produce certain plasma derivative products for the United States, European and other markets. For example, we produce our Flebogamma® DIF and Gamunex® IVIG products for all of our markets at the Clayton, Los Angeles and Parets facilities.
We have an agreement with Canadian Plasma Resources Corporation (“CPR”) whereby we have the right to obtain plasma donated at collection centers owned by CPR. This agreement also grants us the right to acquire plasma collection centers from CPR as early as 2026. We intend to use the plasma we acquire from CPR, as well as the plasma we collect from our own centers in Canada, to fulfill our commitments under a long-term agreement with Canadian Blood Services (“CBS”), described in the following paragraph.
45
On July 29, 2022, we executed a 15-year renewable collaboration agreement with CBS, a non-profit organization in Canada that operates on a national basis within the Canadian healthcare system. CBS provides services to patients on behalf of all provincial and territorial governments of Canada except Quebec, safeguarding Canada’s national system to provide lifesaving therapies related to blood, plasma, stem cells, organs and tissues. By means of this agreement, we committed to supply an amount of IG to CBS equivalent to 25% (2.4 million grams) of the Canadian patient consumption needs (excluding Quebec). In addition to plasma acquired from CPR as described above, we intend to fulfill this agreement with plasma collected from a Canada-based plasma collection center network owned and managed by us, manufacturing our products in our facility in Montreal, Quebec. As part of the arrangements with CBS, we are developing six wholly-owned plasma collection centers in Canada (five in the Ontario and one in the Edmonton). We have entered into a consultancy services agreement with a third-party consultant to develop such centers. The third-party consultant will collect certain fees for the services rendered, which have been disclosed in our audited consolidated financial statements and are included in our capital expenditure plan presented to the market on March 1, 2024. For each liter of Canadian plasma we use to supply IG, we will also supply to CBS all remaining manufactured paste for the processing of other plasma-derived products. We have committed to complete development of the Canadian plasma collection network and to have a fully operational plasma fractionation facility in Montreal by December 31, 2026.
In addition, on October 1, 2020, we purchased from GC Pharma a plasma fractionation facility and two purification facilities located in Montreal, Canada (as well as 11 plasma collection centers located in the United States). Once we finish renovations and obtain all necessary licenses and regulatory approvals for the Montreal facilities, we will become the only large-scale commercial manufacturer of plasma products in Canada, with a fractionation capacity of 1.5 million liters annually. We expect to begin manufacturing IVIG and albumin in the Canadian facilities to supply the Canadian market in 2025 and to begin plasma fractionation and gammaglobulin manufacturing by 2027.
We optimize utilization of our fractionation capacity by obtaining FDA and EMA licenses, and completing further requirements, that allow us to purify at any of our other facilities intermediate products that are produced at one of our facilities. We have obtained the following FDA licenses, among others:
| ● | to use at our Clayton facility the Fraction II+III made at both our Los Angeles and Parets facilities to make Gamunex®; |
| ● | to use at our Los Angeles facility the Fraction II+III made at both our Los Angeles and Clayton facilities to make Gamunex®; |
| ● | to use Fraction V obtained at our Los Angeles facility to produce albumin at our Parets facility; |
| ● | to use Fraction V obtained at both our Clayton and Parets facilities to produce Albutein® in our Los Angeles facility; |
| ● | to use Fraction IV-1 obtained at our Los Angeles facility to produce Prolastina®, an A1PI we market in Spain, at our Clayton facility; |
| ● | to use Fraction IV-1 obtained at our Los Angeles facility to produce Prolastin®-C lyophilized at our Clayton facility; |
| ● | to use Fraction IV-1 obtained at our Clayton facility to produce Prolastin®-C liquid at our Parets facility; |
| ● | to use the same method currently in place in our Parets facility to produce Alphanate® in our Los Angeles facility; |
| ● | to use paste from the fractionation facility at Clayton to produce Gamunex® and Prolastin®; |
| ● | to produce nano-filtered Gamunex® and the 40 gram vial presentation; and |
| ● | to use Cryoprecipitate obtained at our Clayton facility to produce Alphanate® at our Clayton facility, which is later sent to our Los Angeles facility to be filled. |
46
We are continuing our efforts to obtain additional FDA licenses of this nature. The flexibility provided through such licenses allows us to increase production efficiency and to better address changes in demand between the United States, the European Union and other world markets.
For more information on our manufacturing facilities, see “—D. Property, Plant and Equipment” below.
Safety
We have never experienced a mandatory recall of any batch of our finished biological products due to a safety risk. In alignment with our commitment to safety and quality, we have in the past voluntarily withdrawn some product lots not comprised of finished biological products as a precautionary measure due to a higher rate of allergic/hypersensitivity type reactions that we found to be isolated to a small subset of plasma donors. These withdrawals were conducted with the knowledge of the governmental authorities of the applicable jurisdictions. Our philosophy is that the health of the plasma donor and the patient are the paramount considerations. None of the withdrawn products involved reports of any significant impact on patients. We strongly believe that our safety philosophy is consistent with the business objective of generating profit. We also believe that we have a strong reputation for safety in our markets, thus making our products particularly attractive to customers. We further believe that our vertically integrated business model allows us to help assure the safety and quality of our plasma derivative products through the implementation of our safety standards.
The plasma collection, fractionation and purification process is long, complex and highly regulated. We have adopted and maintain rigorous safety standards that we believe exceed those required by health authorities in Europe and the United States. The Grifols Group is periodically inspected and certified for Good Manufacturing Practices (“GMP”) competent health authorities, such as European authorities, the FDA, and other relevant government authorities of other countries where our products are marketed.
We maintain standards that we believe are consistent with other industry participants with regard to plasma safety and are periodically certified by the Plasma Protein Therapeutics Association (“PPTA”), under the International Quality Plasma Program (“IQPP”), for plasma donation centers, and under the Quality Standards of Excellence, Assurance and Leadership Program (“QSEAL”), for fractionation plants. For example, source plasma inventory is held for not less than 60 days after donation, to allow for retrieval and destruction of plasma units if the donor is disqualified during this period (after seroconversion or due to high-risk behavior or international travel). We have also introduced innovative methods such as the Plasma Bottle Sampling™ system, which automatically prepares, codes and labels test samples at the time of plasma donation, and the PediGri™ On Line system, designed to provide full traceability of human plasma raw material throughout the plasma supply chain. See “—Distribution Process” below.
Our manufacturing plants have been designed to comply with the current GMP standards and applicable regulations for clean areas, and are designed to minimize clean areas as well as human intervention, with the objective of lowering the risk of contamination. The facilities are subject to a cleaning and sanitizing plan and to a corrective and preventive maintenance program. Periodically, we voluntarily shut down all of our manufacturing facilities to perform maintenance work, expansion projects and other capital investments. Our manufacturing facilities have never been subject to mandatory shut down because of regulatory noncompliance while under our operation. We believe that our voluntary shutdown procedure lowers the risk of any mandatory shutdown.
We have processes in place to ensure that all of our plasma derived products are manufactured strictly following validated and approved procedures, and in accordance with the corresponding marketing authorization. Also, each manufacturing process includes at least one validated specific virus inactivation or removal step as a precautionary measure to avoid improbable virus contamination.
Since our products are proteins that cannot be terminally sterilized, they therefore are sterilized by filtration before being aseptically filled in their final container. We have patented the Grifols Sterile Filling (“GSF”) system which minimizes the risk of microbial or particulate contamination during the aseptic filling process. During this process, sterilized containers are filled with the product under Grade A laminar air flow. The partially closed containers (vial with stopper and protector) are sterilized prior to filling. The container closure unit remains partially closed until the moment of filling, after which it is immediately sealed thus reducing the risk of contamination by reducing the product and container exposure to the controlled environment. The filling process is recorded, which enables us to identify the cause of, and rectify more easily, any related problem. These records are maintained according to our data retention policy.
47
Once aseptically filled, each unit of product is laser-marked with the objective of individually identifying each container and preventing and detecting counterfeits. This allows us to protect the integrity of our manufacturing process.
After plasma derivatives are manufactured, every unit of each lot is visually inspected in order to detect the presence of foreign particles or other imperfections in the container closure system. Each lot is also tested during production and at the end of the manufacturing process according to the licensed specifications, marketing authorization and corresponding Pharmacopoeia monographs. All processes are overseen by the quality systems in place at Grifols with the objective of ensuring that products are marketed with the appropriate quality, purity, potency and safety.
Finally, once the product is marketed, our pharmacovigilance system allows us to control all potential adverse reactions resulting from the administration of our products, thus ensuring the safety of our products globally around the world.
We continually invest in the improvement of our manufacturing facilities and plasma fractionation process, as well as in other related systems, in order to ensure the quality and safety of our products.
Distribution Process
With each batch of plasma derivatives, we deliver electronic information regarding the origin, characteristics and controls of each of the units of plasma that we use in the preparation of the batch to our customers. This feature, called the PediGri™ On Line system, allows healthcare users of our products and regulatory authorities to have immediate and easy access to this information, tangible proof of the full traceability of our products. We have had this system in place since 1996, and we believe we are the only fractionator that provides this feature to customers.
We have our own sales and distribution networks covering substantially all of our markets, staffed with highly trained personnel. A majority of our sales in 2023 were made through our own distribution network, which is experienced in the proper handling of our products. We believe that this network provides for greater safety because it allows us to track our products and react quickly in the case of a potential product recall. In countries where we do not have our own distribution network, we use carefully selected distributors who are required to follow all of our safety standards.
For further information, see “—Marketing and Distribution” below.
Biopharma Products and Services
Collected plasma, whether source or recovered, is fractionated into different component proteins. We fractionate and purify a broad range of plasma derivative products that improve patient care. Our Biopharma business unit also sells a non-plasma derivative medicinal product, Tavlesse® (fostamatinib), in Europe.
48
The chart below presents our principal products by brand name and their respective therapeutic indications:
Product Description |
| Main Therapeutic Indications |
Gamunex®/Gamunex®-C. Immune Globulin Injection (Human), 10% Caprylate/Chomatography Purified. Flebogamma® 5% and 10% DIF. Immune Globulin Intravenous (Human). | IVIG is used for the treatment of: primary and secondary immunological deficiencies and autoimmune conditions including immune-mediated ITP; Measles pre/post exposure prophylaxis in patients to whom active immunization is contraindicated or not advised; Guillain Barré syndrome; Kawasaki disease; chronic inflammatory demyelinating polyneuropathy (“CIDP”); and multifocal motor neuropathy (“MMN”). Severe acute myasthenia exacerbations is an approved indication for Gamunex®-C in Europe. | |
|
| |
Xembify®. Immune Globulin Subcutaneous (Human) - klhw 20% solution. | Used to treat Primary Humoral Immunodeficiency (“PI”). In Europe and Australia, Xembify® is also indicated to treat hypogammaglobulinaemia and recurrent bacterial infections in patients with chronic lymphocytic leukaemia (“CLL”) in whom prophylactic antibiotics have failed or are contra-indicated, hypogammaglobulinaemia and recurrent bacterial infections in multiple myeloma (“MM”) patients, and hypogammaglobulinaemia in patients pre- and post- allogeneic haematopoietic stem cell transplantation (“HSCT”). | |
|
| |
HyperRAB® Rabies Immune Globulin (Human). | Anti-rabies immunoglobulin indicated for postexposure prophylaxis, along with rabies vaccine, for all persons suspected of exposure to rabies who have not been previously vaccinated with a rabies vaccine. | |
|
| |
Prolastin®/Prolastin®-C/ Prolastin®-C Liquid/Prolasplan®/ Prolastina®/Pulmolast®/Lynspad®. Alpha 1-Proteinase Inhibitor (Human). | Used to treat adults with clinical evidence of emphysema due to severe hereditary alpha-1 antitrypsin deficiency (“A1PI deficiency”). | |
|
| |
Fanhdi™ and Alphanate®. Antihemophilic Factor/von Willebrand Factor Complex (Human). | Used for the prevention, management and control of bleeding in Factor VIII deficiency (hemophilia A) and indication for von Willebrand disease. | |
|
| |
Koate®-DVI. Antihemophilic Factor (Human). | Used for the prevention and control of bleeding in Factor VIII deficiency (hemophilia A). | |
|
| |
Albutein®/ Albutein® FlexBag® / Human Albumin Grifols®/Plasbumin®. Albumin (Human) 5%, 20% and 25%. | Used to re-establish and maintain circulation volume in the treatment of hypovolemia (i.e., traumatic or hemorrhagic shock, septic shock and severe burns) and to treat complications related to cirrhosis. | |
Vistaseal™/VeraSeal®. Human fibrinogen/human thrombin. | Used as a supplemental treatment in adults where standard surgical techniques are insufficient for improvement of haemostasis, and as suture support in vascular surgery. | |
Tavlesse®. Fostamatinib Disodium Hexahydrate Film Coated Tablets. | Used for the treatment of chronic immune thrombocytopenia (“ITP”) in adult patients who are refractory to other treatments. |
49
Gamunex-C® IVIG, a ready-to-use liquid IVIG product, is one of the leading products in the IVIG segment. We believe Gamunex-C® IVIG is one of the premium products in its category since its launch due to a comprehensive set of differentiated product characteristics. We are one of the market leaders in the production and marketing of immunoglobulin, with 23.6% market share in volume in the United States for the twelve-month period ended November 30, 2023, according to PPTA.
Xembify®, our subcutaneous immunoglobulin product for use to treat primary immunodeficiencies, was approved in Norway, Iceland and Denmark in 2023 for use to treat primary and secondary immunodeficiencies. From 2019 to 2022, Xembify® was approved for the same uses in Canada, France, Italy, Sweden, the United Kingdom, Australia, Czech Republic, Finland, Slovakia and Spain. We first launched Xembify® in the United States in 2019. In 2023, we launched Xembify® in Spain and Australia, followed by a launch in Sweden in January 2024.
We believe HyperRAB® is the world’s leading human anti-rabies immunoglobulin indicated for postexposure prophylaxis, along with rabies vaccine, for all persons suspected of exposure to rabies who have not been previously vaccinated with rabies vaccine. HyperRAB®, a 300 IU/ml formulation of which is available in the United States, is the only human rabies immunoglobulin (“HRIG”) provided as a higher-potency formulation, potentially requiring fewer injections in administration of each dose. We had an estimated 80% market share in sales of anti-rabies immunoglobulins in the United States as of December 2022.
In addition, we believe that we are the global market leader in the sales of AAT (alpha-1-antitrypsin augmentation therapy. At December 31, 2023, our AAT had 58 licenses in 27 countries worldwide, 19 of which are located in North America and Europe. Our liquid formulation of AAT (Prolastin®-C Liquid) is FDA approved as a chronic augmentation and maintenance therapy to treat emphysema related to severe hereditary A1PI deficiency. Based on our estimations we had a 72% global sales market share for AAT as of December 2022. A worldwide clinical trial is ongoing to meet post-approval regulatory commitments and obtain Prolastin®-C regulatory approval in Europe. In 2023, we sponsored over 150,000 genetic tests in connection with this clinical trial.
Between Koate®-DVI, Fanhdi and Alphanate, we had an estimated 20% market share based on volume globally in the pdFVIII hemophilia A market in 2021 (excluding Von Willebrand disease use).
We sell Grifols albumin brands globally, with an estimated 17.4% market share in volume in 2022. Our albumin products meet U.S., European and Chinese requirements, making them attractive to biotechnology companies and genetic labs, as well as hospitals and physicians. In 2021 and 2022, we launched and began to market in the United States Albutein 25% and 5% FlexBag®, a flexible container designed to add convenience, ease of use, and durability.
Tavlesse® is a novel SYK-inhibitor in-licensed from Rigel Pharmaceuticals for commercialization in Europe and additional markets in the Middle East and North Africa. Tavlesse® is indicated to treat chronic immune thrombocytopenia in adult patients who are refractory to other treatments. EMA regulatory authorization was obtained in January 2020, and commercial sales have commenced in Czech Republic, Norway, Denmark, The Netherlands, Germany, Spain, Italy, France and the United Kingdom.We plan to launch in additional countries in Europe and the Middle East. Tavlesse® is the first oral therapy commercialized by Grifols Biopharma.
We also produce antithrombin (e.g., AT, ATIII, pdAT) (Anbinex® and Thrombate® III), which is used in the prevention and treatment of thromboembolic complications in patients with antithrombin deficiency; AlphaNine®, Profilnine® and Factor IX Grifols®, which are used in the prevention and control of bleeding in patients with hemophilia B.
In addition to the products described above, we also commercialize intramuscular (hyperimmune) immunoglobulins, which are used for the prevention and treatment of tetanus, prevention and treatment of hepatitis B, and Rh factor complications during birth. Niuliva® and Igantibe® are used after liver transplants to prevent hepatitis B reinfection of the graft.
We also manufacture Vistaseal™/Veraseal®, a biological fibrin sealant composed of fibrinogen and human thrombin used in surgical operations to expedite the healing process. It is commercialized in the United States and Canada, in some European countries (i.e. Germany, United Kingdom, Switzerland, Estonia, Latvia, Lithuania, Austria, Ireland, Netherlands, Norway, Finland, Sweden, Denmark, France, Italy and Spain) as well as in Singapore and Australia by Ethicon US, LLC (a Johnson & Johnson company).
50
To sell medicinal products, we must first register the products with the relevant authorities of the jurisdictions where the products are to be marketed and sold. To comply with the regulatory requirements in a given jurisdiction, we have a core team in Spain and the United States that prepares, files and coordinates the registration process with the technical personnel at the subsidiary assigned to that jurisdiction. As of December 31, 2023, we had 1,121 Biopharma product licenses registered in 90 countries throughout Europe, the United States, Latin America, Asia and the rest of the world. As of December 31, 2023, our most significant government-issued licenses for Biopharma products are:
| ● | Gamunex®/Gamunex®-C/Flebogamma® DIF. We have 218 licenses for the marketing and sale of one or more IVIG products; |
| ● | Xembify®. We have 25 licenses for the marketing and sale of this product; |
| ● | Prolastin®/Prolastin®-C/ Prolastin®-C Liquid/Prolasplan®/Prolastina®/Pulmolast®/Lynspand®. Alpha 1-Proteinase Inhibitor (Human). We have 58 licenses for the marketing and sale of one or more of these A1PI products; |
| ● | Fanhdi™/Alphanate®/Koate®- DVI Factor VIII. We have 229 licenses for the marketing and sale of one or more of these Factor VIII products; |
| ● | Albutein®/Human Albumin Grifols®/Plasbumin®. We have 274 licenses for the marketing and sale of one or more of these albumin products in their various concentrations; |
| ● | VistaSealTM/VeraSeal®. We have 29 licenses for the marketing and sale of this product; and |
| ● | Tavlesse®. We have EMA authorization for Tavlesse® in the European Union and national authorization in the United Kingdom. Tavlesse® is currently sold in Czech Republic, Norway, Denmark, The Netherlands, Germany, Spain, Italy, France, the United Kingdom and the United Arab Emirates. |
In addition to the sale of the products described above, we have entered into a series of arrangements with many Spanish transfusion organizations to fractionate recovered plasma (plasma separated from blood obtained from a blood donation) from such organizations and manufacture plasma derivatives under our own brand name for use by hospitals. We charge the transfusion centers for the fractionation and manufacturing service. We also provide virus photo-inactivation of transfusion plasma to hospitals and clinics in Spain. The plasma is inactivated at our manufacturing facilities and then sent back to the clinic or hospital at which it was collected, where it is used for transfusions.
51
The Diagnostic Business Unit
The Diagnostic business unit, which accounted for €670.3 million, or 10.2% of total net revenue in 2023, focuses on researching, developing, manufacturing and marketing in vitro diagnostics products, including analytical instruments, reagents, software and associated products for use in diagnostic clinical and blood bank laboratories.
At December 31, 2023, more than 80.0% of our in vitro diagnostics products, including, but not limited to, Gel cards, Reagent Red Blood Cells and Ultrio Elite, had received In Vitro Diagnostic Regulation (“IVDR”) certification. IVDR is the regulatory basis for placing on the market, making available and putting into service in vitro diagnostic medical devices in the European market. We are currently implementing all remaining requirements to obtain IVDR certification in a timely manner for all of our in vitro diagnostics products.
The main areas of specialization of this business unit are transfusion medicine and clinical and specialty diagnostics.
In transfusion medicine, we believe that we have a significant market share in NAT blood screening solutions. In addition, we have increased our sales of automated immunohematology systems and reagents to hospital transfusion services and blood centers in several key global markets. We also continue to grow our portfolio of clinical and diagnostic products in select areas, including autoimmunity, and have agreements to extend the number of antigens we manufacture for use in clinical and blood bank diagnostic tests. Our principal diagnostic products are:
Product Description |
| Main Applications |
|---|---|---|
| ||
Transfusion Medicine: | ||
Procleix® Panther® systems/Procleix® Panther® with Automation Ready Technology (ART). Automated NAT blood screening systems, assays and software. | Used to detect infectious viruses and parasites in donated blood and plasma including: HIV (Types 1 & 2); Hepatitis A, Hepatitis B, Hepatitis C and Hepatitis E; parvovirus B19; West Nile Virus; Dengue Virus; Zika Virus, Plasmodium and Babesia. | |
WADiana®/Erytra® /Erytra Eflexis® analyzers. Automated immunohematology analyzers that use gel agglutination technology to enable automatic processing of DG Gel® system, including DG Gel® cards, reagent red blood cells, antisera and associated software. Manual and semiautomated equipment are also part of the DG Gel system. | Used for routine pre-transfusion compatibility testing, including blood typing, unexpected antibody screening and identification, extended phenotype and cross-match tests, among others. | |
| ||
BLOODchip ID/ IDCoreXT/ IDHPAXT/ IDRHDXT / IDCORECONTRO / BIDSXT. A multiplex blood group genotyping family of products based on Luminex technology, including kits for the main allelic variants of red blood cells, human platelet antigens, a positive control for ID COREXT, and an accompanying software. | Used to help determine blood groups in individuals who may not be able to be typed with traditional blood grouping methods due to recent transfusions, autoantibodies, or other limitations. | |
Antigens. Critical component of certain infectious disease tests. | Used in the manufacture of clinical diagnostic and blood donor screening immunoassays. | |
Clinical and Specialty Diagnostics: | ||
Promonitor®. Highly specific immunoassays for quantification of serum drug levels and anti-drug antibodies of various biological drugs. | Used to measure quantity of drug and antibodies for a number of biological drugs, commonly used in the treatment of various inflammatory diseases. | |
AlphaIDTM. Genetic test for patients for alpha-1 deficiency. | This is a free cheek swab to screen for alpha-1, the most common genetic form of Chronic Obstructive Pulmonary Disease (“COPD”). |
52
We manufacture most of our gel cards and red blood cells and assemble our immunohematology analyzers at our Parets facility in Spain. We also manufacture gel cards in Australia and red blood cells in Switzerland. We manufacture antigens at our Emeryville facility in the United States, while oligos and other critical components of the transcription-mediated amplified NAT kits for blood and plasma infectious diseases screening are manufactured at our San Diego facility in the United States. We also expect to start manufacturing gel cards and red blood cells in our San Diego facilities in 2024.
In several countries, we distribute BLOODchip® blood group genotyping tests manufactured by our subsidiary Progenika Biopharma, S.A. (“Progenika”). This product line includes ID CORE XT, which determines 37 antigens of red blood cell groups, ID RHD XT, a molecular diagnostic kit that detects the most relevant RhD variations, HPA-1 and ID HPAXT kit to determine 12 Human Platelet Antigen (“HPA”) systems. BIDSXT is the software tool that allows the analysis, interpretation and database management to transmit the results to the Laboratory information system (“LIS”).
The production, marketing and sale of many of our Diagnostic business unit products are subject to the prior registration of such products with the relevant authorities of the applicable jurisdictions. As of December 31, 2023, we had 3,335 diagnostic product licenses registered in over 80 countries in Europe, the United States, Canada, Latin America, Asia-Pacific, Middle East and Africa. In addition to the products noted above, we offer our customers solutions developed in collaboration with, or manufactured by, third-parties that we believe complement our product lines.
Our Diagnostic business unit includes a complete line of products and systems to perform blood donor screening molecular tests aimed at detecting the pathogenic agents of transfusion-related infectious diseases such as HIV (Types 1 & 2), Hepatitis A, Hepatitis B, Hepatitis C, Hepatitis E, parvovirus B19, West Nile Virus, Zika Virus, Dengue Virus, Babesia and Plasmodium. We control the research and development, manufacturing and worldwide commercialization of our Procleix® blood screening products.
Recent Efforts in Transfusion Medicine
As part of our strategy of geographic expansion, we continue to consider requests to include NAT screening for blood and plasma donations in countries as they develop their health systems. From 2020 to 2023, we entered several new countries, such as Guatemala, Paraguay and Czech Republic. In recent years we have focused on obtaining FDA and other regulatory approvals to expand our portfolio. The following table summarizes such recent efforts:
Product Description and Main Applications |
| Licensing Update |
|
Procleix Quality Controls, intended for use as an external assayed quality control material to monitor the performance of several assays performed on the Procleix® Panther® system. | FDA 510(k) clearance in 2022. | ||
Procleix® Plasmodium Assay, for use on the Procleix Panther System, in Brazil. | Approval in Brazil in 2023. |
In addition, through our U.S. subsidiaries Biomat USA and Interstate Blood Bank Inc., we have entered into an agreement with Creative Testing Solutions (“CTS”), a nonprofit blood donor testing laboratory organization in the United States. Pursuant to this agreement, as of April 1, 2022, CTS began to operate for a period of ten years (renewable for up to another ten years) our three testing laboratories located in Memphis, Tennessee and San Marcos and Austin, Texas, to perform donor screening for blood and plasma collections. Grifols Laboratory Solutions will continue to provide specialty testing, including in-process and final product testing, as well as diagnostic solutions for clinical issues related to Molecular and Serological Immunohematology. The facility in Austin, Texas, provides testing, consultation and integrated solutions to optimize patient care for specialists in Hematology, Oncology, Perinatology, Obstetrics, Pharmacy, Transplantation and Transfusion Medicine.
We operate a product line of high quality antigens, which are critical components of clinical diagnostic and blood screening immunoassay tests sold worldwide, produced through a joint business with Ortho Clinical Diagnostic. As part of this joint business, we have a contract with Abbott Laboratories for the supply of high quality antigens used in the manufacture of immunoassay diagnostics. This contract, with a total value of approximately $700.0 million, extended the supply of antigens until 2026, ensuring higher levels of recurring income in this area. We also extended our agreement with OraSure Technologies through 2022, reinforcing our position as a flexible provider of antigens.
53
The table below summarizes our recent efforts to obtain more licenses and increase the market of our immunohematology products:
Immunohematology Product Description and Main Applications |
| Licensing Update |
Blood Typing Manager, designed to aid in the interfacing and managing of data among immunohematology instruments, blood establishment computer software and laboratory information systems as part of the DG Gel® System. | CE mark in 2022; FDA 510(k) clearance in 2022; Approval in Brazil 2023. | |
Grifols sCD38, designed to counteract daratumumb-mediated pan-reactivity without significantly diluting the patients’ plasma and enables subsequent screening and identification of irregular antibodies in DG Gel® system and tube techniques, and crossmatch in DG Gel® system. Daratumumab is a CD38-directed monoclonal antibody for treating multiple myeloma. However, during treatment the drug binds to the CD38 protein on red blood cells and can alter the results of critical blood pre-transfusion tests. This can delay lifesaving transfusions for these patients. | CE mark in 2022. | |
DG Gel® 8 Direct Coombs card, used for the evaluation of the direct antiglobulin test of two different human blood samples. This product is designed to differentiate red blood cells sensitized in vivo by IgG type immunoglobulins or the complement C3d fractions. The card is intended to be used with the DG Gel® System. | BLA approval in 2022. | |
The DG Gel® DC Scan Plus, intended to be used in gel technique for the evaluation of the direct antiglobulin test. This test permits the differentiation of red blood cells sensitized in vivo by IgG, IgA and IgM type immunoglobulins and/or with the complement C3b, C3d and C4b fractions in human blood samples. The DG Gel® DC Scan Plus card is intended for the investigation of clinical situations where the presence of hemolysis has been established, or is suspected, to distinguish immune from nonimmune hemolytic anemia. The card can be used manually or with automated instruments of the DG Gel® System. | CE mark in 2022; Approval in Brazil 2023; Approval in Australia 2023. | |
Sero-Cyte Pool Dia 0.8%, designed for routine testing for the screening of irregular antibodies, including the antigen Dia, in donors. This reagent complements the existing reagent red blood cells and allows us to reinforce our presence in markets, such as Brazil, where the identification of the antigen Dia is required by local regulations. | CE mark in 2022; Approval in Brazil 2023. | |
DG Reader NET, a single card processing platform operating with the same consumables and reagents as our fully automated systems. This product is designed to be used on our DG Gel® systems. | CE mark in 2016; FDA 510(k) clearance in 2018; Approval in China 2023. |
54
Below are the main recent developments in respect of products of our Diagnostic business unit:
| ● | We obtained registrations in the Chinese market for DG Reader Net, our semi-automated blood bank system to process DG gel cards and Erytra Eflexis, our flagship fully automated blood typing platform; |
| ● | We obtained the following approvals in connection with critical Procleix Assays: |
| o | CE mark for the Procleix ArboPlex Assay 4-in-1 NAT for arbovirus screening. This assay is an in vitro nucleic acid test that detects four types of arboviruses spread through mosquito vectors: chikungunya, dengue, West Nile and Zika viruses. Arboviruses are a growing threat, with changes in climate and increasing global connectivity making the geographic spread more prevalent. This is the first CE mark under the IVDR for an acid nucleic test specifically designed for screening arboviruses; |
| o | CE mark and FDA approval for the Procleix UltrioPlex W Assay. This assay is a qualitative in vitro nucleic acid amplification test that detects five viruses in one reaction: HIV-1, HIV-2, HCV, HBV, and WNV/USUV. Due to its capabilities, we expect this assay to become the most widely used NAT blood screening assay in the United States; and |
| ● | We obtained CE mark in 2022, and are currently working to obtainin approval in the United States, for the Procleix® Plasmodium Assay, an assay to detect ribonucleic acid (“RNA”) from Plasmodium species (P. falciparum, P. knowlesi, P. malariae, P. ovale, and P. vivax) in whole blood specimens that cause Malaria disease. The assay is designed to be used for routine screening by blood banks on the Procleix® Panther® system, where we estimate that we are currently the market leader and continue our efforts to offer innovative solutions to blood banks. |
Clinical and Specialty Diagnostics
We retain the first FDA approved biological molecular test that uses the DNA of the patient for the diagnostic. This genetic test to detect alpha-1 antitrypsin deficiency (the “A1AT Genotyping Test”) can be conducted on DNA extracted from blood as well as a drop of blood collected on paper (a “Dry Blood Spot”) or human saliva samples collected as buccal swabs. This test was developed by Progenika Biopharma, a Grifols subsidiary. In 2022, A1AT Genotyping Test was also CE marked. Although highly complex, the test has been designed so any molecular biology laboratory can process it with minimal human intervention.
At the end of 2019, we also introduced AlphaID™, a new simple cheek swab that greatly simplifies the sample collection process. AlphaID™ allow physicians and healthcare providers to obtain a sufficient oral sample for alpha-1 screening, and it is completely free from ordering to results. The test is now available for distribution in the U.S. Since the approval of AlphaID™ in 2019, Progenika Biopharma has been developing a service to offer, directly to homes, a diagnostic solution to potential patients of alpha-1 antitrypsin deficiency. The service has been cleared by the FDA in 2022 as “AlphaID™ At Home Genetic Health Risk Service.” This is a complex multidisciplinary project that brings together six international companies lead by Progenika Biopharma, being the second over-the-counter clinical diagnostic service approved by FDA.
55
Through our subsidiary Progenika, we manufacture a genetic diagnosis test for Familial Hypercholesterolemia (“FH”) using next generation sequencing technology (“NGS”). The Diagnostic business unit continues its efforts to broaden the Promonitor® line, used to monitor biologic drugs as sales continue in Chile, select European Union countries, Australia and in the U.S. The Promonitor® product line includes an ELISA (enzyme-linked immunoabsorbent assay) device line also developed by Progenika to monitor patients being treated with biological medicines for rheumatoid arthritis and other chronic inflammatory diseases. We maintain CE marking of two additional tests in the Promonitor® family that enable treatment with the biological product golimumab, and several tests to permit the use of a single dilution to measure quantity of drug and antibodies for a few biological drugs, commonly used in the treatment of various inflammatory diseases, such as rheumatoid arthritis and ulcerative colitis. In 2021, we entered into a co-development agreement with DIESSE Diagnostica Senese to adapt Promonitor® ELISA kits to DIESSE’s Chorus analyzer. This adaptation to monotest and ready-to-use devices is meant to address customer and market needs for improved automation. The development of Promonitor® tests on the Chorus automated system facilitates an improved testing workflow, resulting in earlier feedback for the clinician.
Pursuant to an exclusive agreement with AESKU Diagnostics GmbH & Co. (“AESKU”), we distribute autoimmunity diagnostic products in Chile, Portugal, Spain and Mexico. We also serve key U.S. customer segments with AESKU products. One of these diagnostic products is Helios, the only fully automated platform capable of performing all immunofluorescence pipetting and reading steps in the United States, which strengthened our U.S. portfolio of products.
The Bio Supplies Business Unit
The Bio Supplies Business Unit provides human biological materials for the biotechnology industry intended for research, clinical trials and manufacturing of pharmaceutical and diagnostic products. Most materials and products come from our collection centers with vertical integration manufacturing processes (in-house production). The Bio Supplies business unit accounted for €160.0 million, or 2.4% of our total net revenue in 2023.
Products in our Bio Supplies business unit cover a broad spectrum of applications where the need for human-based biological material cannot be replaced by other types of products, such as:
Cell Therapy: providing human-based cell culture supplements, plasma proteins, and leukocyte apheresis (white blood cell concentrate obtained through apheresis and used in the development of new cell therapies).
Pharma Manufacturing: human-based plasma proteins to be used as excipients (i.e. inactive substances that serve as vehicles or mediums for a drug or other active substances in the manufacturing of therapeutic drugs). Human plasma and intermediates used to manufacture plasma derived medicines.
Diagnostic Manufacturing: human blood cells, proteins and plasma and serum for the manufacturing of quality controls and calibrators used in in vitro diagnostics tests.
Life Science Research: Human blood derived products and biospecimens used in life science basic research or by in vitro diagnostic companies to develop and validate their analytical assays.
Other Activities and Operations (“Others”)
“Others” represents operations and activities, including the provision of manufacturing services to third parties and third-party plasma sales, and pharmaceutical products manufactured by the Grifols Group intended for hospital pharmacies.
With the sale in July 2022 of substantially all assets of Goetech, LLC, which does business under the name Medkeeper (“Medkeeper”), the activities of our former “Hospital Division” were significantly reduced, which caused us to reallocate such activities and related products into a smaller business line named healthcare solutions (“Healthcare Solutions”), which is also accounted as Others. Others accounted for €203.5 million, or 3.1%, of our total net revenue in 2023. See Item 5 of this Part I, “Operating and Financial Review and Prospects—Factors Affecting Our Financial Condition and Results of Operations—Dispositions—MedKeeper.”
56
The Healthcare Solutions business line provides services and manufactures products used by hospitals, blood banks, plasma collection centers and other healthcare systems. These products include parenteral solutions, robotics and software, as well as third parties’ products that we market to supplement the products that we manufacture.
Research and Development
Research and development is a significant aspect of our business. Our principal research and development objectives are (i) to discover and develop new products, (ii) to research new applications for existing products and (iii) the improvement of our manufacturing processes to improve yields, safety and efficiency. Research and development spending was €354.9 million in 2021, €361.1 million in 2022 and €395.3 million in 2023.
At December 31, 2023, we had an amount of approximately €1.4 billion as development costs in progress (compared to €1.3 billion at December 31, 2022). This includes (1) an amount of €284.3 million in ongoing research and development projects for products for neurodegenerative disorders, neuromuscular diseases, and ophthalmological diseases acquired from Alkahest; and (2) an amount of approximately €862.0 million in ongoing research and development projects in plasma therapies acquired from Biotest (Fibrinogen and Trimodulin). See Note 7 to our audited consolidated financial statements included in this annual report.
In addition, as of December 31, 2023, we had 1,260 scientists and support staff dedicated to research and development.
We have several decades of successful innovation history. For example, we developed a unique fractionation design that reduces the risk of contamination and maintenance costs and increases the amount of product extracted per liter of plasma. We also developed the first centrifugation unit for the automated cleaning of blood cells. In addition, we were one of the first plasma fractionators to conduct double viral inactivation processes for Factor VIII and have designed and implemented a new process for the sterile filling of vials that reduces exposure to potential contaminants as compared to other existing processes. Further, we have developed a nanofiltration method of viral inactivation for our IG, alpha-1 PI, and ATIII products.
Our key therapeutic areas of study focus on immunology, hepatology and intensive care, pulmonology, hematology, neurology and infectious diseases. Our next innovations into these therapeutic areas will go beyond plasma-derived proteins, as our significant investments to increase R&D capabilities are allowing us to develop a new class of recombinant antibodies and small-molecules candidates.
Biopharma Initiatives
We have a number of research and development projects in our Biopharma business unit underway, 23 of which are in the clinical development phase. The following table reflects the total number of research and development projects in our Biopharma business unit by development phase as of the end of the last three years.
As of December 31, | ||||||
Development Phase |
| 2023 |
| 2022 |
| 2021 |
Discovery |
| 24 |
| 19 |
| 21 |
Pre-clinical |
| 23 |
| 28 |
| 30 |
Clinical |
| 22 |
| 23 |
| 22 |
Post Commercialization Studies |
| 14 |
| 39 |
| 9 |
Rest of projects |
| 16 |
| 14 |
| 14 |
Total Biopharma Research and Development Projects |
| 99 |
| 123 |
| 96 |
57
The table below presents the most important of our research and development projects:
Product Candidate |
| Therapeutic |
| Product |
| Potential Use |
| Development Phase |
Xembify® |
| Immunology |
| Plasma-derived |
| Chronic lymphocytic leukemia |
| Phase III (clinical trial program currently underway) |
Albumin | Hepatology | Plasma derived | Liver Cirrhosis | Phase III (clinical trial program currently underway) | ||||
Alpha-1 Proteinase Inhibitor | Pulmonology | Plasma-derived | Emphysema due to congenital deficiency | Phase I/II (clinical trial program in subcutaneous administration currently underway); Phase III (clinical trial program in intravenous administration currently underway) | ||||
Fibrin Sealant |
| Surgical bleeding |
| Plasma-derived |
| Vascular, organ and soft-tissue surgery |
| Launched in the U.S. in 2019 and in the E.U. in 2020. Post-marketing pediatric clinical trial is complete. E.U. approval has been granted, and U.K. and U.S approvals are pending. |
Immunoglobulin | Ophthalmology | Plasma-derived | Dry Eye Disease | Pre-clinical development | ||||
Fibrinogen | Hematology | Plasma-derived | Congenital deficiency & severe hypofibrinogen / acquired deficiency | Phase III (clinical trial program currently underway) | ||||
Trimodulin | Infectious diseases | Plasma-derived | Severe community acquired pneumonia (“sCAP”) | Phase III (clinical trial program currently underway) | ||||
Trimodulin | Infectious diseases | Plasma-derived | Community Acquired Pneumonia (“CAP”) | Phase III (clinical trial program currently underway) | ||||
CYTOTECT® | Infectious diseases | Plasma-derived | Cytomegalovirus (“CMV”) infection | The Phase III clinical trial (PreCyssion) to prevent the transmission of CMV infection from the mother to the unborn child was discontinued early at the end of 2023 with 48 patients included. |
58
Xembify® – Secondary immunodeficiency in CLL. The primary objective of this clinical trial (NCT05645107) is to obtain an indication for Xembify® in secondary immunodeficiency as result of chronic lymphocytic leukemia (CLL) as a result of secondary immunodeficiency. CLL is the proliferation and progressive accumulation of morphologically mature but immunologically less mature monoclonal B-cells in the blood, bone marrow, and lymphoid tissues. It is widely accepted that both intrinsic (genetic and epigenetic) and extrinsic (micro-environmental stimuli and B cell receptor-mediated antigenic stimulation) events can contribute to the pathogenesis of the disease. CLL is most common in adults and accounts for approximately 40% of all leukemia cases. CLL is primarily a disease of the elderly population, with less than 10% of the cases occurring in patients younger than 40 years of age. While no definitive cause of CLL has been established, targeted therapy regimens have proven to be efficacious treatments. Hypogammaglobulinemia (“HGG”) is the most predominant inherent immune defect in B-CLL patients and becomes more pronounced with longer disease duration and advanced-stage disease. Infectious complications continue to be one of the major causes of morbidity and mortality in patients with CLL. The Phase 3 clinical trial is currently enrolling patients in the United States and in European countries to support the FDA submission in the United States.
Alpha-1 Proteinase Inhibitor – Emphysema due to congenital deficiency. SPARTA (Study of Prolastin-c Randomized Therapy with Alpha-1 augmentation; NCT01983241), our phase 3 clinical trial designed to determine if alpha-1- antitrypsin deficiency (AATD) (alpha-1) patients with emphysema have a slower progression of lung tissue loss when treated weekly with two separate dose regimens of intravenous (IV) Grifols Prolastin®-C Alpha-1, is an underdiagnosed genetic disorder that can result in chronic obstructive pulmonary disease (COPD), a group of respiratory diseases that includes emphysema, which can occur when a patient has low levels of AAT, a protective protein that safeguards the lungs. The currently approved dosage is 60 mg/kg in weekly IV infusions. SPARTA, the largest randomized, double-blind, placebo-controlled study on AAT augmentation therapy to-date, is designed to evaluate the potential of Prolastin®-C to significantly reduce emphysema progression in alpha-1 patients by raising AAT protein levels through weekly administration of two active dose levels versus placebo. The clinical trial is taking place across 16 countries and more than 50 sites. It will evaluate the efficacy and safety of two separate dose regimens of Prolastin®-C (60 and 120 mg/kg/week) versus placebo for 156 weeks (i.e., three years), measuring the rate of pulmonary-tissue loss through whole lung computed tomography (CT) densitometry as the primary measure of clinical efficacy.
We are also conducting a Phase I/II study (NCT04722887) evaluating Alpha1-Proteinase Inhibitor Subcutaneous (Human) 15% (Alpha-1 15%), a subcutaneous (SC) AAT treatment being compared to Liquid Alpha1-Proteinase Inhibitor (Human) IV. This first in-human SC approach to treating alpha1-antitrypsin deficiency, if proven successful in clinical trials, could give patients the convenience and flexibility to administer their medication from home. In this multi-center, single-dose and repeat-dose study over eight weeks, Cohort 1 of Grifols Phase 1/2 study GC2008 has been completed and demonstrated no safety issues with Alpha-1 15% that would prevent the study from moving forward into Cohort 2. A SC option for AAT augmentation is not currently available and evaluating a SC dosing option in this first-in-human study for Alpha-1 patients may provide more freedom when it comes to managing their AAT deficiency by allowing patients to administer their medication from the comfort of their own home. Currently, cohort 2 is in enrolment phase and we are aiming to complete its recruitment in 2024.
Fibrin Sealant. We began clinical trials into the safety and efficacy of the use of fibrin sealant as a supportive treatment for the improvement of hemostasis in vascular, organ and soft-tissue surgery in 2008. In 2014, we completed a clinical trial in the European Union for the use of fibrin sealant in vascular surgery. Three additional clinical trials were performed: (i) a Phase III clinical trial in the United States for the use of fibrin sealant in solid organ surgery; (ii) a Phase III clinical trial in the United States for the use of fibrin sealant in soft-tissue surgery; and (iii) a Phase III clinical trial for the use of fibrin sealant in vascular surgery in the United States. All of the U.S. clinical trials for fibrin sealant were completed in 2015. Marketing authorization approvals were received from the FDA and EMA in November 2017. A distribution agreement was made with a third party, requiring an additional regulatory supplement. Vistaseal® was launched in the U.S. during 2019 and Veraseal® was launched in the E.U. in 2020. Additionally, a Phase IV study to evaluate safety and efficacy of fibrin sealant as an adjunct to haemostasis during surgery in pediatric subjects was completed in 2022 to support the FDA and EMA regulatory licenses. As of December 31, 2023, we had obtained E.U. approval, while U.K. and U.S. approvals were pending.
We incurred costs in the amount of €0.4 million and €0.1 million in connection with this project in 2022 and 2021, respectively. We hold significant granted patents on the fibrinogen and thrombin production processes.
59
Immunoglobulin. In 2023, we entered into a global collaboration and licensing agreement with Selagine, a pioneering developer of eye disease treatments, to explore the potential of an immunoglobulin eye drop to treat dry eye disease (DED), a pathology that affects more than 100 million people worldwide in 2023. In a pilot Phase I/II clinical trial, subjects treated with eye drops based on Grifols Flebogamma DIF® twice daily for eight weeks, secured a significant reduction in the signs and symptoms of DED, and with no difference in tolerability or adverse events. Several different sources of inflammation, including proteins (cytokines or chemokines), cells (neutrophils, T-cells and dendritic cells) and pathogenic antibodies, are present on the ocular surface in DED and contribute to its signs and symptoms which may benefit from application of immunoglobulin. We completed a FDA pre-IND meeting in respect of this immunoglobulin in September 2023. We are currently in pre-clinical development in the use of Flebogamma DIF® 5% in the treatment of DED and expect to move to a Phase II clinical trial in 2025.
Fibrinogen. The AdFIrst study is a prospective, active-controlled, multicenter Phase III study investigating the efficacy and safety of the fibrinogen concentrate BT524 in patients with acquired fibrinogen deficiency (“AFD”), which typically occurs during surgical procedures when there is insufficient fibrinogen to arrest bleeding. Patients who have high blood loss during planned spinal and abdominal surgery are randomized 1:1 to treatment with BT524 or FFP/Cryoprecipitate. To evaluate efficacy, further blood loss is compared between the two treatment options. The study was conducted in collaboration with ten study sites in five European countries and was concluded in September 2023. We announced in February 2024 that the AdFirst Study met its primary endpoint, demonstrating that the fibrinogen concentrate BT524 is as effective in treating AFD as the current standard of care for this condition. The concentrate reduces intraoperative blood loss in patients with AFD undergoing planned major spinal or abdominal surgery.
We expect to start regulatory authorization processes for the BT524 concentrate by the end of 2024, starting in Europe and the United States. We incurred costs in the amount of €10.3 million and €5.0 million in connection with this project in 2023 and 2022, respectively. We hold significant patents on the fibrinogen production process.
Trimodulin. Study 996: This multinational Phase III clinical trial plans to enroll about 590 adult hospitalized patients with severe sCAP (Community Acquired Pneumonia) requiring invasive mechanical ventilation. The ESsCAPE trial will be conducted worldwide and patients are treated either with trimodulin or with a placebo as add-on therapy to standard of care.
The clinical concept of this prospective, double-blind, placebo-controlled, Phase III trial was developed based on promising results from the previous Phase II clinical trial (“CIGMA”) with 160 sCAP patients requiring invasive mechanical ventilation. In the CIGMA trial, a subgroup of patients with signs of severe inflammation showed an encouraging reduction in mortality rate through rapid normalization of inflammation when treated with Trimodulin. The trial is currently being conducted in up to 20 countries, including the United States and recruitment of patients is ongoing.
Trimodulin. Study 1001: This multinational Phase III clinical trial plans to enroll about 390 adult hospitalized patients with CAP. These are patients with signs of early systemic inflammation admitted to hospitals and requiring supplemental oxygen due to the severity of their disease. We expanded the patient population to include patients with a SARS-CoV-2 infection (moderate/severe COVID-19 pneumonia) and CAP patients with any other pathogen. Patients are treated either with Trimodulin or with a placebo as add-on therapy to standard of care.
The clinical concept of this prospective, double-blind, placebo-controlled Phase III trial was developed based on positive results from the Phase II trial (“ESsCOVID”) in a subgroup of severe COVID-19 patients with early systemic inflammation and after the German authority (Paul-Ehrlich-Institute) confirmed that further development is warranted. The trial protocol with the expanded patient population has been approved in 9 countries. As of December 31, 2023, recruitment of patients is ongoing.
We incurred costs in the amount of €19.1 million and €19.0 million in connection with this project in 2023 and 2022, respectively. We received a grant from the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung, BMBF) in the amount of €8.0 million and €14.3 million for the relevant period in 2023 and 2022, respectively, which reduced expenses. We hold significant patents on the trimodulin production process.
CYTOTECT®. We terminated the Phase III clinical trial (PreCyssion; trials no. 997) of Cytotect CP Biotest to prevent the transmission of CMV infections from the mother to the unborn child was at the end of 2023. The clinical trial included 48 patients. Despite a favorable safety profile, Biotest decided to discontinue the PreCyssion study (study 997), as no statistically relevant results could be achieved with this study.
60
We incurred costs in the amount of €0.5 million and €0.4 million in connection with this project in 2023 and 2022, respectively. We hold significant granted patents on the hyperimmunoglobulin production process.
Other Biopharma research and development projects undertaken during the last two years included:
2023:
| ● | process optimization for increased IgG yields across manufacturing facilities; |
| ● | development of an improved process to provide greater product safety and yield for manufacture of Rho-D; |
| ● | development for a modified process to manufacture Plasmanate; and |
| ● | transfer of Albumin process to our subsidiary Grifols Canada Plasma, Inc. (formerly known as Prometic Plasma Resources, Inc.); |
2022:
| ● | new container closure systems for Albutein® and Xembify Prefilled syringes; |
| ● | clinical programs to evaluate new indications of Flebogamma® DIF 5%; |
| ● | A1PI. New vial sizes of Prolastin® are in development, providing important advancements in manufacturing efficiency as well as improved patient convenience; |
| ● | clinical studies to evaluate the effects of the prolonged administration of human albumin on cardiovascular, hepatic and renal function in patients with advanced cirrhosis and ascites. |
All clinical trials involve risks and uncertainties. Preclinical and clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during or as a result of preclinical testing and the clinical trial process that could delay or prevent our ability to receive regulatory approval or commercialize our product candidates. For a discussion of these unforeseen events, see Item 3 of this Part I, “Key Information—D. Risk Factors—Risks Relating to the Company and Our Business—We may not be able to commercialize products in development.” Upon the completion of each of the development stages we evaluate the results achieved as compared to the objectives pursued. Each of the key projects listed above has met our expectations with respect to results at the various development stages and we expect to move forward with the development process for each.
We believe that our current liquidity is sufficient to fund the ongoing costs of our key projects listed above through their completion as well as our other research and development initiatives.
61
Diagnostic Initiatives
Research and development in the Diagnostic business unit supports various business areas, including transfusion medicine, clinical diagnostics, and the recombinant protein business. The Diagnostic business unit focuses on the development of in vitro diagnostic reagents/assays, instrumentation, and software for donor screening, which includes pathogens detection to assure safety and blood typing tests to determine donor/recipient blood compatibility. Here, research and development focuses on opportunities to develop new assays for emerging pathogens, increase multiplex test capabilities (simultaneous detection of several analytes in a single reaction), as well as improved automation solutions in order to increase efficiency and throughput for customers. The Diagnostic business unit also develops products for clinical diagnostics, including a next generation immunoassay platform in the area of neurology and oncology, as well as a next-generation molecular testing platform covering infectious diseases.
The research and development team employs a diverse technology portfolio, including transcription mediated amplification (“TMA”), polymerase chain reaction (“PCR”), next generation sequencing (“NGS”) for molecular assays, immunologic based methods using red blood cells (“RBC”), and agglutination. We are also leveraging ultrasensitive single molecule counting detection technology to develop new immunoassays. We continue research and development for new recombinant proteins, antibodies and enzymes as critical raw materials to support internal and external diagnostics customers in various fields such as infectious diseases, neurodegenerative diseases and immunohematology. Our Diagnostics Research & Development team continues to evaluate new technologies and research tools, including artificial intelligence (AI), to assess their potential for improving our competitive advantage and for integration into our development portfolio.
In 2022, the Diagnostic business unit obtained CE mark according to new IVD Regulation 2017/746 for 17 immunohematology products in reagents, assays and software, in addition to achieving IVDD CE mark for a new kit to resolve daratumumab interference in blood typing. In Europe, our Diagnostic business unit also obtained the CE mark for Procleix Plasmodium under IVDD and CE mark for a new improved reagent equilibration system under IVDR. In the United States, we obtained FDA clearance through the 510(k) process (which means that the FDA agrees with the manufacturer that a medical device is similar to a previously approved product) for new Procleix quality controls, along with the release of a new version of pooler software. The Diagnostics business unit also achieved updated CE mark for several existing Class A products, to transition these from IVDD to IVDR regulations.
In 2023, the Diagnostics business unit continued to release innovative products for our immunohematology product line, including new software versions across the platform portfolio to increase efficiency and productivity for our customers. We launched our BTM middleware in the Unites States, while also achieving regulatory approval in China for Eflexis, our flagship blood typing platform, in order to expand our geographic footprint. We also launched DC Scan Plus, a new DG Gel test under IVDR regulation that extends specificity beyond routine analytes and improves patient care by increasing sensitivity and specificity, thereby improving clinical patient management and outcomes in the field of donor screening. We also released new versions of our pooler software, as well as software to facilitate increased automation and ease of use for testing labs. Additionally, we launched in the United States the first-ever free direct-to-consumer program (consisting of an innovative and sophisticated genotyping test and service) to screen for the genetic risk of alpha1-antitrypsin deficiency (Alpha-1).
The Diagnostic business unit filed over 30 new patents, spanning composition of matter, methods and devices, further growing our intellectual property portfolio and competitive position in the areas of infectious disease, blood typing, neurodegenerative disease and clinical diagnostics.
Other Initiatives
We are increasing our research and development activities in new fields. We conduct these activities through the creation of joint ventures participated in by Grifols Innovation and New Technologies Ltd (“GIANT”), through agreements to use patents owned by third parties and through selective acquisitions. For example, in 2021 GIANT formed a new company named Grifols Pyrenees Research Center, SL in a joint venture with the Government of Andorra (GIANT owns 80%). The new company was created to develop and manage a new research center specializing in immunology, intended to enhance the knowledge of the human immune system and develop new immunological therapies. See Notes 2 and 3 to our financial statements included in this annual report.
62
Through our subsidiary Araclon Biotech, S.L. (“Araclon”), we are dedicated to finding solutions that promote new diagnostic and therapeutic approaches to Alzheimer’s disease, to be applied in the early stages of the disease. Araclon is working on the validation of an early diagnostic test and the development of a vaccine to combat Alzheimer’s disease in the asymptomatic preclinical stage. The vaccine has passed the animal experimentation stage and a Phase I clinical trial in humans has been completed. In 2017, Araclon obtained approval by the AEMPS for a Phase II placebo-controlled trial of the AB40 vaccine in Alzheimer disease patients and completed recruitment in 2019. In 2020, a change in the design of the trial was introduced that allows cross-over of the treatment arms, so all the participants could receive the vaccine. We received the top line data of part A of the trial in mid-January 2022 and preliminary results are showing a favorable safety profile. The part B and final clinical study report became available in 2023, with results showing the vaccine’s safety, tolerability and robust immune response in early-stage Alzheimer’s patients against the Aβ40 peptide, which research suggests plays a role in cerebral amyloid angiopathy, a highly prevalent condition among such patients.
Through our subsidiary Alkahest, we focus on identifying proteins that change with age and also with the onset of disease that have a biological impact. To date, Alkahest has identified over 10,000 separate proteins using advanced molecular analysis techniques at the cellular level, some of which we have entered in our discovery and development pipeline as new targets. In 2021, Alkahest had a productive year with the completion of three Phase I and five Phase II clinical trials. In 2022, Alkahest completed one Phase II and two Phase I clinical trials. In 2023, Alkahest has pivoted to early discovery efforts and is moving forward targeting indications in Liver and CNS, as well as developing a target proteomic platform that can detect disease before it manifests phenotypically.
Through AlbaJuna Therapeutics S.L., a spin-off company from the IrsiCaixa AIDS Research Institute and our subsidiary, we promoted jointly by “la Caixa” Foundation and the Department of Health of the Government of Catalonia, and undertook the pre-clinical and clinical development of monoclonal antibodies that neutralize the effect of HIV in the body while increasing the activity of the natural killer cells that have the task of destroying infected cells. In 2020, we selected a candidate and began proof of concept studies in non-human primates (“NHP”). We also started process development and manufacturing activities by late 2020. We completed the NHP study in 2022 and the results and are currently under evaluation, along with results from other experiments, to decide how to move forward with the development.
Our subsidiary GigaGen Inc. (“GigaGen”) is developing a novel class of therapeutics, recombinant polyclonal antibodies, to address unmet needs in infectious disease. GigaGen is preparing its next recombinant polyclonal antibody therapeutic candidate, GIGA-2339, a curative treatment for chronic hepatitis B virus infection, for an IND filing and initiation of a Phase I trial in 2024. GigaGen is also developing next-generation anti-CTLA-4 monoclonal antibody therapeutic for the treatment of solid tumors, GIGA-564, which had a successful IND filing in 2023 and, as of December 31, 2023, is was initiating a Phase I trial in collaboration with the U.S. National Cancer Institute.
Finally, we signed an agreement in the first quarter of 2020 to support a consortium with the IrsiCaixa AIDS Research Institute, the Barcelona Supercomputing Center (“BSC”) and the IRTA (Institute of Agrifood Research and Technology) aimed to discover new antibodies and vaccines against COVID-19. The consortium continued in 2023 with some vaccine and antibodies candidates evaluated in in vivo studies in animals.
Seasonality
Our businesses are not significantly affected by seasonal trends.
63
Raw Materials
Plasma is the key raw material we use to make our products and therapies. We obtain our plasma primarily from our network of plasma collection centers located in the United States and Europe (Germany, Austria, Czech Republic, and Hungary), and, to a much lesser extent, from third party suppliers. See “—Principal Activities—Plasma Collection.” We are dependent on healthy individuals to donate human plasma to develop and manufacture our products. As such, our cost to acquire plasma may vary depending on the ability of donors to visit our plasma collection centers and the compensation we pay for the plasma donations. For example, during the Coronavirus pandemic, lockdowns and restricted movement, specifically the U.S. border closure with Mexico, directly affected our ability to obtain plasma donations. See Item 3 of this Part I, “D. Risk Factors— Risks Relating to the Company and Our Business—A significant disruption in our supply of plasma, including as a result of macroeconomic conditions, pandemics or changes in immigration policies and enforcement could have a material adverse effect on our business and our growth plans” and Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Consequences of Covid-19.”
Our plasma supply increased by 10.0% in 2023 in comparison to the volumes collected in 2022, mainly due to the increase in donors (the number of which surpassed 920,000), higher donation frequency and the execution of planned initiatives in our Operational Improvement Plan (as defined in Item 5 of Part I, “A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Operational Improvement Plan”) announced in February 2023. Likewise, our average cost per liter of plasma collected in 2023 declined by 15.2% in comparison to the average cost per liter in 2022. This cost per liter decrease was gradual throughout 2023, with the cost per liter at December 31, 2023 showing a decline of 22.0% in comparison to the cost per liter in July 31, 2022, which was the peak cost per liter in such year.
Our cost per liter of plasma collected decreased largely due to four factors: (1) in the wake of wider vaccination, the easing of COVID-19 constraints allowed plasma donations to resume at the U.S.-Mexico border starting in September 2022, resulting in an increase in donors, causing our plasma collections to steadily improve throughout the remainder of 2022 and through 2023; (2) the termination of government stimulus programs issued to households during the pandemic, which, while still in distribution, had the two-pronged effect of both reducing the financial incentive for individuals to donate plasma and hindering our ability to maintain proper levels of workforce in plasma collection centers; (3) the 10.0% increase in our plasma supply in 2023, as explained above; and (4) the successful execution of measures under our Operational Improvement Plan, including the reduction of our plasma donor compensation, an optimization of our plasma collection center network, with the closure or consolidation of 18 centers in 2022 and seven centers in 2023, and a streamlined organizational structure for plasma collection. See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Operational Improvement Plan.”
We closely monitor, continuously review and revise the supply sourcing strategy for our products to identify in a timely manner any risks in our supply chain, including risks arising from our dependency on outsourced manufacturing relationships with third party suppliers. Where necessary, we strategically manage our inventory levels of either key materials or finished products to address potential risks relating to operational and quality issues, production capacity and single sourcing among others. We also continue to monitor the efficiency of our plasma collection platform and have concentrated all our plasma testing into six laboratories in Austin and San Marcos, Texas, Memphis, Tennessee, Boca Raton, Florida, Leipzig, Germany and Parets, Spain.
Our long-term aim is to further strengthen our leadership through the development of new and differentiated plasma-derived therapeutics, and the expansion of our global plasma collection footprint via acquisitions and greenfield projects, as described below;
| ● | On April 25, 2022, we acquired all of the existing equity interest in Biotest Holdings from Tiancheng International Investment Limited (“TIIL”) and accepted an assignment of certain shareholder loans granted by TIIL to Biotest Holdings. Biotest Holdings in turn owns 89.88% of the ordinary shares and 1.08% of the preferred equity shares of Biotest AG, a global company that supplies plasma protein products and biotherapeutic drugs, which will give us access to 28 additional plasma collection centers. See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Acquisitions—Biotest AG Acquisition.” |
64
| ● | On July 29, 2021, we signed a joint venture agreement with ImmunoTek, which contract was amended in 2023, to arrange for the construction, licensing and commissioning of 28 plasma collection centers in the United States. Pursuant to the joint venture agreement, we formed the ITK JV, a limited liability company organized in the United States, to own all 28 centers. ITK JV has created or will create a separate series of its equity interests corresponding to each of the 28 plasma collection centers to be constructed. Through our subsidiary Grifols Bio North America, we hold or will hold 75% of each such series of ITK JV equity interests, while ImmunoTek holds or will hold the remaining 25%. Pursuant to the joint venture agreement: |
| o | we have the right to acquire ImmunoTek’s 25% stake in all 28 classes of ITK JV equity interests, each class relating to a specific plasma collection center, approximately three years following the opening of each center, for an aggregate amount of $570.7 million. Of the 28 centers, we expect to have the right to acquire seven in July 2024, eight in January 2025 and six in January 2026; |
| o | through ITK JV, we agreed to make advance payments to ImmunoTek of up to $5 million per plasma collection center ($140.0 million in the aggregate for the 28 centers), which amounts will be deducted from the purchase price of ImmunoTek’s 25% stake in the class of ITK JV equity interest corresponding to each center. As of December 31, 2023, we have made advance payments in the amount of €22.8 million; |
| o | all the plasma collected by ITK JV from the 28 plasma collection centers is sold exclusively to us. In the years ended December 31, 2023 and 2022, plasma purchases from ITK JV amounted to €233.7 million and €66.6 million, respectively; |
| o | Grifols, S.A. guarantees (1) the obligations of our subsidiary Grifols Bio North America under the joint venture agreement; and (2) the obligations of ITK JV under the real estate lease agreements for all 28 plasma collection centers; and |
| o | ImmunoTek has sole management of the plasma collection centers. |
Through our subsidiary Grifols Shared Services North America, Inc., we act as guarantor for five plasma collection center lease agreements held by ImmunoTek that are not subject to this joint venture agreement, guaranteeing up to an amount of $50 million. See Note 10 to our audited consolidated financial statements included in this annual report, on page F-62, for more details of this transaction.
| ● | On March 31, 2021, we acquired seven U.S.-based plasma donation centers from Kedplasma, LLC for a total purchase price of $55.2 million. |
| ● | On February 28, 2021, we acquired 25 U.S.-based plasma centers from BPL Plasma Inc for $385 million by means of an asset sale agreement. These plasma centers obtain, in the aggregate, a run-rate of one million liters of plasma per year. See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Acquisitions.” |
| ● | In October 2020, we purchased 11 collection centers in the United States, as well as a plasma fractionation facility and two purification facilities in Montreal, Canada, from GC Pharma for a total consideration of $457.0 million on a debt free basis. |
| ● | In April 2019, we acquired 26 plasma collection centers and ten blood donation centers in the United States as part of our acquisition of 100% of the equity stake of the Interstate Blood Bank Group (Interstate Blood Bank, Inc., Bio-Blood Components, Inc. and Plasma Biological Services, LLC), which has since been merged into our subsidiary Grifols Bio Supplies, Inc. |
65
| ● | On December 28, 2018, our subsidiary GWWO, Biotest Pharmaceuticals Corporation (“BPCorp”), which is a subsidiary of BPC Plasma Inc, Haema AG and Grifols, S.A. entered into a plasma supply agreement (the “Plasma Supply Agreement”), whereby GWWO agreed to acquire all of the plasma collected from approximately 60 plasma collection centers owned by BPCorp and Haema AG. Grifols, S.A. guarantees all GWWO obligations under the Plasma Supply Agreement, which, on January 1, 2019, was extended for a 30-year period. See Item 7 of this Part I, “Major Shareholders and Related Party Transactions—B. Related Party Transactions—Transactions involving Haema AG, BPC Plasma Inc (formerly known as Biotest US Corporation) and Scranton Enterprises B.V. and their respective subsidiaries—Plasma Supply Agreement and Advance Payments from GWWO to Scranton Plasma B.V.” |
The principal raw materials for our intravenous therapy products are plastic and glass bottles, which we purchase from various European suppliers.
Marketing and Distribution
We currently sell Biopharma, Diagnostic and Healthcare Solutions products to hospitals and clinics, GPOs, governments and other distributors in over 100 countries.
In the United States, the sales model is complex, with many intermediaries, requiring us to execute multi-faceted arrangements for the distribution of our products. Sales of finished goods are distributed through various channels such as distributors, wholesalers, specialty pharmacies, home health care companies, clinics, hospitals, government entities and directly to physician offices. Payers and purchasers also control access to products, requiring separate negotiations with payers and GPOs. GPOs are entities that act as purchasing intermediaries for their members, which are primarily hospitals. GPOs negotiate the price and volume of supplies, equipment and pharmaceutical products, including plasma derivatives, used by their members.
We market our products to healthcare providers and other decision-makers, such as those in hospitals, through focused sales presentations. Although price and volume are negotiated through contractual agreements with intermediaries, demand for our products is generated through promotional efforts by Grifols’ sales representatives. In the case of GPOs, the actual sales are made to the authorized distributor(s) of each GPO at the contract price, and the distributor then sells the products to the members of that GPO. We promote our products directly to the GPO’s members. For safety and post-sale service reasons, the distributor is required to provide us with the specifics of the ultimate delivery to the client.
The sales, marketing and distribution process is different in Europe, where the bulk of sales are generally made directly to hospitals. We have developed long-standing relationships with major hospitals in most of our European markets, and we believe that hospitals are loyal customers that recognize the high quality and safety of our products, our reliability as a supplier and the strong product expertise and service provided by our sales representatives. Due to the nature of our customer base and the prevalence of repeat sales in the industry, we market our products through focused sales presentations rather than by advertising campaigns.
Sales to Eastern Europe, the Middle East and some Asian countries are made mostly by third parties outside of our sales network. Our sales in Latin America are made mainly by our sales network.
Sales Representatives
We require our sales representatives to be able to highlight the technical differences between our products and those of our competitors. This skill requires a high degree of training, as the salesperson must be able to interact and discuss product differences with doctors, pharmacists and other medical staff. Sales representatives call on office-based healthcare providers and hospital-based healthcare providers, departmental heads, purchasing agents, senior hospital directors, lab directors and pharmacy managers. We compensate our sales representatives by means of a fixed salary and a bonus component based on sales. We divide our sales efforts along the lines of our main product categories. Our sales personnel are primarily located in Europe and the United States, but we also have sales personnel in Latin America and Asia-Pacific.
In our Biopharma business unit, we utilize mixed sales units comprised of both marketing and sales personnel. In some countries, we have product line-specific sales units for immunology & neurology, pulmonary, intensive care and coagulation factors.
66
Advertising
We participate in medical conferences, conventions and fairs and occasionally publish advertisements in medical journals and trade magazines. This promotional activity is also supported by online activities.
Distribution
We believe that having our own distribution network staffed with highly trained personnel is a critical element of a successful sales and marketing effort. Through this network, we are able to provide high-quality pre- and post-sales service, which we believe enhances brand recognition and customer loyalty. Our distribution network is experienced in the proper handling of our products and allows us to know where our products are located, enabling us to act quickly in the event of a suspected problem or product recall.
Our distribution network personnel are located in Europe, Latin America, the United States and Asia-Pacific and handle the distribution of our biological medicine, diagnostic and other medical products as well as goods manufactured by other premier healthcare companies that complement our own products.
During 2023, we distributed the majority of our products through our own distribution network. In some cases, particularly in the field of Diagnostics, we distribute products through marketing partners and third-party distributors. We have a direct presence in 30 countries and we carefully select distributors in the countries where we do not have a direct presence. We have a responsive, effective logistics organization that is able to punctually meet the needs of hospital centers and other customers throughout the world.
Our sales, marketing and distribution network included 1,550 employees as of December 31, 2023, which included 1,395 sales and distribution personnel and 155 marketing employees.
Each of our commercial subsidiaries is responsible for the requirements of its respective local market. It is our goal for each commercial subsidiary to be recognizable as one of our companies by its quality of service, ethical standards and knowledge of customer needs. Strong local knowledge enables us to build and maintain long-term relationships with customers to earn their trust and confidence.
Patents, Trademarks and Licenses
Patents and Trademarks
Through our patent ownership, co-ownership and licensing, we seek to obtain and maintain intellectual property protection for our primary products.
As of December 31, 2023, we owned 2,705 patents and patent applications in various countries throughout the world, of which 858 are in the final application process. In some countries, these patents grant a 20-year protection period. Approximately 1,283 of these patents are set to expire in the next ten years. In terms of recent expirations, the patent for the Biotest intravenously administered polyclonal immunoglobulin preparation, containing IgM and method of manufacturing (CA1341505C) expired in May 2023, the patent for the Grifols Sterile Filling system expired in January 2024, and the patent for the process of removing viruses in Fibrinogen solutions expired in March 2024.
As of December 31, 2023, we also owned 3,877 trademarks and trademark applications in various countries throughout the world, of which 137 are in the final application process. In addition, we co-own certain patents and patent applications with third parties, including patent rights co-owned with Novartis following the Novartis Acquisition.
We maintain a department with personnel in Spain, Ireland and the United States to handle the patent and trademark approval and maintenance process and to monitor possible infringements.
67
Patents for Plasma Derivative Products
As of December 31, 2023, we owned 1,980 patents and patent applications related to plasma derivatives, including 893 in Europe, 231 in the United States and Canada, and 856 in the rest of the world. The most important of these patents relate to:
| ● | a concentrated subcutaneous alpha-1 antitrypsin; |
| ● | the use of low volume plasma exchange for the treatment of Alzheimer’s disease; |
| ● | transferrin for neurodegenerative applications; |
| ● | transferrin for the treatment of Hipoxia inducible factor related conditions; |
| ● | plasmin in wound healing; |
| ● | the process for removing viruses in Fibrinogen solutions; |
| ● | a concentrated subcutaneous Immunoglobulin G injection; |
| ● | concentrated Immunoglobulin M preparations for the treatment of bacterial infections; and |
| ● | anti-thrombin to treat blunt trauma. |
Patents for Diagnostic Products and Healthcare Solutions
As of December 31, 2023, we owned 621 patents and patent applications related to our Diagnostic products and Healthcare Solutions business line, including 201 in Europe, 251 in the United States and Canada and 169 in the rest of the world. The most important of these patents relate to the:
| ● | BlisPack®, a blister handling machine; |
| ● | Erytra Eflexis®, a mid-sized instrument to perform pre-transfusion compatibility tests using DG Gel® technology; |
| ● | innovative containers for human plasma proteins; |
| ● | novel HIV antigens for blood screening; |
| ● | soluble recombinant form of CD38 receptor; |
| ● | soluble recombinant form of CD47 receptor; and |
| ● | recombinant antigens for detection of COVID-19. |
As of December 31, 2023, we owned 104 patents and patent applications related to other areas of the business, including 35 in Europe, 35 in the United States and Canada and 34 in the rest of the world.
68
Grifols’ Trademark
Marca Grifols, S.L., a company owned by certain Grifols family members, owns the rights to the “Grifols” trademark. We entered into a license agreement with Marca Grifols, S.L. on January 26, 1993, that grants us the exclusive rights to use the “Grifols” trademark for 99 years. The license agreement provides that we must pay an annual royalty fee for the license, which is based on inflation and our net sales. The fees for this license were €7.5 million, €6.6 million and €6.3 million in 2023, 2022 and 2021, respectively.
Licenses from Third Parties
We license certain intellectual property rights from third parties, including Singulex and Hologic. Singulex granted us an exclusive worldwide license under certain intellectual property rights for the use and sale of certain products and services for blood donor and plasma screening. Pursuant to an intellectual property license with Hologic, we obtained a fully paid-up license to certain of Hologic’s intellectual property for use in the NAT Donor Screening Unit.
Licenses from Government Authorities
Government authorities in the United States, at the federal, state and local level, and in other countries throughout the European Union, Latin America, Asia and elsewhere, through licenses, approvals, reviews, inspections and other requirements, extensively regulate the research, development, testing, approval, manufacturing, labeling, post-approval monitoring and reporting, packaging, promotion, storage, advertising, distribution, marketing and export and import of healthcare products such as those that we collect, manufacture, sell or are currently developing.
For example, in order to sell our plasma derivative products we must hold appropriate product licenses from applicable governmental authorities. We have 1,121 Biopharma product licenses registered in 90 countries, which include the licenses we hold from the FDA for the sale in the United States of IG, A1PI, albumin, Factor VIII, Factor IX, ATIII and PTC. The production, marketing and sale of many of our Diagnostic business unit products are subject to the prior registration of such products with the relevant authorities of the applicable jurisdictions. We have 3,335 diagnostic product licenses registered in a total of 80 countries in Europe, the United States, Canada, Latin America, Oceania and Asia.
Governmental oversight extends to the various facilities involved in our operations. For example, our Parets and Murcia facilities are subject to applicable regulations and standards of the European health authorities. With respect to oversight by the FDA, our Instituto Grifols Biopharma plant at our Parets facility has been registered with the FDA since 1995, and our other manufacturing facilities maintain FDA registration, and all are subject to FDA standards. We lease most of our plasma collection centers as well as our main laboratory facility located in Austin, Texas, and maintain licenses with the appropriate regulatory authorities, including the FDA, for all of these locations.
For more information on government licenses and regulation, see “—Principal Activities” above and “—E. Regulatory Matters” below.
Regulatory
For detailed information regarding the regulations applicable to our business, see “—E. Regulatory Matters” below.
69
Insurance Coverage
General and Product Liability
We have a program of insurance policies designed to protect us and our subsidiaries (including our United States subsidiaries) from product liability claims. Effective May 1, 2023, we have product liability insurance coverage for up to $220 million per claim and in annual aggregate for Diagnostic and Bio Supplies business units for products manufactured in all of our facilities and for third-party products we sell. That limit is of $500 million per claim and in the annual aggregate for Biopharma business unit. This policy expires on April 30, 2024, and we expect to renew it for another year prior to such expiration date. We have elected to self-insure $173.5 million per claim and in annual aggregate of our global liability program through the purchase by one of our subsidiaries of such portion of the insurance policy. See “—Self-insurance” below.
Our master liability program also protects us and our subsidiaries from certain environmental liabilities arising in those countries in which our subsidiary companies have operations. This risk is covered up to a maximum of $220 million per claim and in annual aggregate, except that amount is $500 million for the Biopharma business unit.
Biomat USA, Inc., Grifols Bio Supplies, Inc., BPC Plasma Inc., Prometic Plasma Resources, Inc., Plasmavita Healthcare GmbH, Plasmavita Healthcare II GmbH, Haema AG and Haema Plasma Kft maintain a separate liability insurance policy. The policy covers their professional liability for plasmapheresis business activities and expires on April 30, 2024. The maximum amount of coverage for liability claims under the policy is $15 million per claim and in the annual aggregate. In addition, we have general liability coverage for up to $500 million per claim and in the annual aggregate for Biomat USA, Inc., Grifols Bio Supplies, Inc., BPC Plasma Inc., Prometic Plasma Resource, Inc., Plasmavita Healthcare GmbH, Plasmavita Healthcare II GmbH, Haema AG and Haema Plasma Kft.
Property Damage and Business Interruption
Our property damage and business interruption insurance program covers us and our subsidiaries (including our United States subsidiaries). This insurance program, which expires on April 30, 2024, covers damages suffered by plants and buildings, equipment and machinery. Under the current terms, the insurer will cover damages to our facilities produced by fire, smoke, lightning and explosions, among others, for up to $1.5 billion per occurrence. It also covers property damage produced by flooding, for up to $110 million per claim and in the annual aggregate.
In addition, this policy covers loss of profit for a period of 36 months with a deductible equivalent to up to five business days of lost profits. Pursuant to the loss of profit, in the event that any or all of our plants stop production due to an event not excluded under the policy, the insurer covers fixed expenses, in addition to net profits we did not earn during the term of coverage.
We also have a transit and inventory insurance program, which covers damages to raw materials, supplies, semi-finished products and finished products for up to $25 million per claim for transit and $500 million for inventory in annual aggregate.
Cyber
Our cyber insurance covers financial losses up to $50 million resulting from data breaches and ransomware attacks. This insurance program expires on April 30, 2024, and we expect to renew the policy for another year prior to its expiration date.
Self-insurance
We are self-insuring part of the risks described above through the purchase of a portion of the relevant insurance policies by Squadron Reinsurance DAC, one of our wholly owned subsidiaries. We self-insure $38.5 million per claim per year of our global liability program (except that amount is $173.5 million per claim and in the annual aggregate for the Biopharma business unit), the first $230,000 per loss for property damage and the first ten days of lost profits, the first $27,000 per claim for transit losses, the first $200,000 per claim for inventory losses and any transit or inventory losses exceeding $2 million have an additional retention of 10% of loss value with a maximum of $500,000 per loss and an annual aggregate of $3 million, and the first $5 million per loss related to cyber-security risks. These amounts are in excess of the deductibles for each of the policies that make up our insurance programs.
70
Climate Change
Aligned with the United Nations’ plan to achieve the sustainable development goals defined in its 2030 agenda, we have set commitments to help preserve the environment. Since 2020, our corporate environmental program included the reduction of emissions through the use of 68 million kWh of renewable electricity through power purchasing agreements (“PPAs”), the construction of two new photovoltaic plants (in Barcelona and Murcia) and the construction of new refrigeration plants with refrigerant gases with a global warming potential equal to zero. With the aim of replacing the most polluting technologies, we regularly analyze the technological options available on the market, with a special focus on technologies that increase the climate resilience of our facilities.
Through our corporate environmental program, which is updated every three years, we have defined short and long term science-based decarbonization targets (“SBTs”) and milestones for our business. By 2030, we are committed to reducing our greenhouse gas emissions per unit of production by 55.0% in comparison to our 2018 levels, increasing our energy efficiency per unit of production by 15.0% and ensuring that 100% of the electricity consumed in our facilities originates from renewable sources. In addition, in 2023 we committed to implementing near-term targets aligned with the goal by the Science Based Targets Initiative, a global body enabling companies and financial institutions to set emissions reductions targets in line with the latest climate science (“SBTi”), to limit global temperature rise to 1.5ºC.
Our action plan to achieve such SBTs includes (i) minimizing air travel among our different industrial and operational locations by increased use of technology such as video calls; (ii) increasing the levels of remote work by our employees with policies providing for flexible hour; (iii) optimizing logistics in our plasma transportation network, including by shipping intermediate products from our facilities in the United States to Ireland by ship, rather than air transport; (iv) minimizing the impact of employee travel and commutes with measures such as providing subsidized shared transportation service to employees in our facilities; and (vi) increasingly rely on renewable energy by purchasing energy generated from renewable sources through PPAs.
Under the SBTi, we use the GHG Protocol Corporate Accounting and Reporting Standard methodology to calculate our carbon footprint and identify the greenhouse gas emissions (“GHG”) generated by our business activity in three separate scopes:
| ● | Scope 1 are direct emissions generated by our business activity itself, including by combustion sources. In 2023, our Scope 1 emissions increased by 12.0% compared to 2022, reaching 106,450 tCO2e due to an increase in the number of operational days of our cogeneration plant; |
| ● | Scope 2 are indirect emissions from the generation of purchased energy, mainly electricity consumed by our business. In 2023, our Scope 2 emissions decreased by 8.0% (according to the market-based approach), reaching 98,106 tons of CO2e, due to an increased use of renewable energy.; and |
| ● | Scope 3 are all other indirect emissions that occur in the value chain of our business both upstream and downstream. Scope 3 includes emissions by suppliers throughout the life cycle of our products or services, business trips, employee travel and commutes, among others. In 2023, our Scope 3 emissions decreased by 33.0% compared to 2022, totaling 947,463 tons of CO2e. Goods and services remain responsible for over 50.0% of our Scope 3 emissions, followed by our contracted transportation. |
We invested €5.8 million in 2023 in environmental assets (€8.4 million in 2022 and €7.3 million in 2021), mainly intended to optimize water consumption, make improvements in wastewater treatment, eco-efficiency projects in the use of energy and the replacement of refrigerant gases with others with a lower environmental impact. Our expenses for the protection and improvement of the environment in 2023 amounted to approximately €29.6 million (€25.8 million in 2022 and €20.6 million in 2021).
71
C. | Organizational Structure |
Grifols, S.A. is the parent company of the Grifols Group, which was comprised at December 31, 2023, of 79 companies. Subsidiaries in which Grifols, S.A. directly or indirectly owned the majority of equity or voting rights have been fully consolidated, as have been companies that Grifols, S.A. controls. In addition, there were five companies that were accounted for using the equity method, because Grifols, S.A. owned between 20% and 50% of its share capital and had no power to govern its financial or operating policies.
See Notes 1 and 2(b) to our audited consolidated financial statements included in this annual report on Form 20-F for details of our consolidated and non-consolidated companies.
72
D. | Property, Plant and Equipment |
Our headquarters is in Barcelona, Spain. As of December 31, 2023, we owned or leased facilities in seven countries, with our main factory locations established in Spain, Germany and the United States. We currently own or lease manufacturing facilities in 12 sites in 11 different locations, five of which have plasma fractionation capabilities. The table below shows the geographic location and business purpose of our principal properties as of December 31, 2023.
Location |
| Facility |
| Own/Lease (2) |
| Business Purpose |
|
|---|---|---|---|---|---|---|---|
Parets del Vallès, Spain | Industrial Facility One Parets | 66% owned; 34% of the property is leased from a third party | Plasma fractionation Manufacture of plasma derivatives & business unit support activities | ||||
Industrial Facility Two Parets | 80% owned; 20% of the property is leased from a third party | Manufacture of Diagnostic and Healthcare Solutions products | |||||
Industrial Facility Three Parets | 70% owned; 30% of the property is leased from a third party | Plasma storage & other operating activities | |||||
Los Angeles, California, U.S. – Valley | Industrial Facility, U.S. | 92% owned; 8% of the property is leased from a third party | Plasma fractionation Plasma purification Manufacture of plasma derivatives | ||||
Los Angeles, California, U.S. – City of Industry | City of Industry, U.S. | 100% leased | Plasma storage | ||||
Clayton, North Carolina, U.S. | Clayton Facility | 100% owned | Plasma fractionation Plasma purification Manufacture of plasma derivatives | ||||
Durham, North Carolina, U.S. | Research Triangle Park | 22% owned, 78% of the property is leased from a third party | Research and Development Labs and Offices | ||||
Emeryville, California, U.S. | Emeryville Facility | 100% owned | Manufacture of Diagnostic products | ||||
San Carlos, California, U.S. | Alkahest site | 100% leased | Research and Development Labs and Offices | ||||
South San Francisco, California, U.S. | GigaGen site | 100% leased | Research and Development Labs and Offices | ||||
Murcia, Spain | Industrial Facility Murcia | 100% owned | Manufacture of Healthcare Solutions products | ||||
Fribourg, Switzerland | Industrial Facility Switzerland | 100% leased | Manufacture of Diagnostic products | ||||
Melbourne, Australia | Industrial Facility Australia | 100% owned | Manufacture of Diagnostic products | ||||
Vista, California, U.S. | Grifols Bio Supplies, INC | 100% leased | Manufacture of Biological products | ||||
San Marcos, Texas, U.S. | Plasma Testing Lab | 100% owned | Plasma testing | ||||
San Diego, California, U.S. | San Diego Facility | 75% owned; another 25% of the property is leased from a third party | Manufacture of components of the TMA amplified NAT kits | ||||
Dublin, Ireland | Global Operations Center | (1) | Operating activities related to the Biopharma business unit | ||||
Sant Cugat del Vallès, Barcelona, Spain | Headquarters | 100% leased | Headquarters | ||||
Derio, Bizkaia, Spain | Progenika Biopharma | 90% owned, 10% leased | Manufacture of Diagnostic products & Research and Development | ||||
Zaragoza, Spain | Araclon Biotech | 100% leased | Research and Development Labs and Offices | ||||
Boca Raton, Florida, U.S. | Grifols Bio Supplies, Inc | 100% leased | Specialty testing | ||||
Arrasate, Guipuzcoa, Spain | Kiro Grifols | 100% leased | Manufacture of Hospital equipment and Offices | ||||
Memphis, Tennessee, US | Grifols Bio Supplies, Inc | 57% owned, 43% leased | Plasma Lab | ||||
Leipzig, Germany | Haema | 100% owned | Headquarters, Lab & Warehouse | ||||
Montreal, Canada | Industrial Facility Montreal | 100% owned | Plasma fractionation Plasma purification | ||||
Dreieich, Germany | Industrial Facility Dreieich | 86% owned, 14% leased | Plasma fractionation |
(1) | We hold a 999 year leasehold interest in the property. |
(2) | Lease percentage based on property size. |
73
Plasma Fractionation Plants
Our plasma derivative products are manufactured at our Parets, Los Angeles, Clayton and Dreieich facilities. All of our fractionation facilities have FDA and EMA certification. As of December 31, 2023, our facilities had an aggregate fractionation capacity of over 20 million liters of plasma per year, and this capacity is sufficient to cover our current production needs.
The Parets facility has a fractionation capacity of 5.0 million liters per year and a unique design that separates the maintenance area from the clean areas required for the fractionation and purification procedures. This design, which we developed in house, minimizes the risk of contamination and reduces maintenance costs. In addition to licenses from the European Union and other required specific authorities for the production of various plasma derivative products, the Parets facility is also licensed by the FDA. In addition to the plasma fractionation facilities, the Parets site also has protein purification, fill and finish, packaging, storage, research and development and energy co-generation facilities for the Biopharma business unit and manufacturing for the Diagnostic business unit and Healthcare Solutions business line. The Parets facility holds GMP’s, ISO 13485 and ISO 14001 for the Biopharma, Diagnostic and Healthcare Solutions plants and ISO 9001 certifications for its diagnostic manufacturing facilities.
The Los Angeles facility has a fractionation capacity of 2.4 million liters per year. The facility contains purification and aseptic filling areas for coagulation factors, IG and albumin. The facility is licensed by the FDA and Grifols is working to certify the Los Angeles facility with ISO 14001 certification, similar to the rest of Grifols’ manufacturing plants.
We increased the capacity of our Clayton facility in North Carolina by six million liters in 2023, reaching a fractionation capacity of 11.8 million liters per year. This facility is one of the world’s largest fully integrated facilities for plasma-derived therapies, including plasma receiving, fractionation, purification, filling/freeze drying and packaging capabilities, as well as freezer storage, testing laboratories and a cGMP pilot plant for clinical supply manufacture. This facility holds the ISO 14001 certification, which recognizes excellence and continuous improvement in environmental performance. The scope of the certification includes research, development, production and quality control of pharmaceutical specialties derived from human plasma.
In October 2020, we acquired our plasma fractionation facility (together with two purification facilities) in Montreal from GC Pharma. Once we finish renovations and obtain all necessary licenses and regulatory approvals for the Montreal facilities, we will become the only large-scale commercial manufacturer of plasma products in Canada, with a fractionation capacity of 1.5 million liters annually. We expect to begin manufacturing IVIG and albumin in the Canadian facilities to supply the Canadian market in 2025 and to begin plasma fractionation and gammaglobulin manufacturing by 2027.
We have opened our new albumin purification, dosing and sterile filling plant in Dublin, Ireland. The new plant has a processing capacity of three million liters per year, quadrupling our capacity for filling albumin in flexible packaging. Aligned with our measures to combat the effects of climate change, the new Dublin plant incorporates the latest eco-efficiency technologies to save energy and water.
In April 2022, we acquired the Dreieich facility as a result of the Biotest AG acquisition. The facility has an annual manufacturing capacity of up to three million liters of plasma, as well as bulk production plants for albumin, new fibrinogen and IgM concentrate product lines and next-generation polyvalent immunoglobulins.
Global Operations Center
We have a facility located in Dublin opened a global operations center for our Biopharma business unit. The facility, located in Dublin, Ireland, occupies 22,846 square meters and centralizes decision-making with regard to commercial policy, research and development policy and supply chain global management. It houses Biopharma’s global logistics and distribution activities; warehousing of plasma, intermediate paste and finished product, labelling and secondary packaging of the product; as well as regulatory and quality activities relating to the supply of plasma and plasma derivatives. It also centralizes our treasury function and acts as our point of access to the capital markets.
74
E. | Regulatory Matters |
Government Regulation
Government authorities in the United States and in other countries extensively regulate, among other things, the research, development, testing, approval, manufacturing, labeling, post-approval monitoring and reporting, packaging, promotion, storage, advertising, distribution, marketing and export and import of healthcare products such as those we collect, manufacture, sell or are currently developing. The process of obtaining regulatory approvals and complying with all applicable laws and regulations requires the expenditure of substantial time and financial resources. The following is a summary of the overall regulatory landscape for our business.
United States Government Regulation
In the United States, the FDA regulates drugs, biologics, plasma collection and medical devices under the FDCA and, as applicable, the PHS Act, and their implementing regulations. Failure to comply with the applicable FDA requirements at any time during the product-development process, approval process or after approval may result in administrative or judicial sanctions. These sanctions could include, as applicable, the FDA’s imposition of a clinical hold on trials for drugs, devices or biologics, refusal to approve pending applications, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution or any combination of these sanctions. Any agency or judicial enforcement action could have a material adverse effect on us.
The BLA (Biologics License Application) Approval Process
Drugs that are also biological products, such as our plasma derivative products IG, A1PI, Factor VIII and albumin, and also certain in vitro diagnostic products associated with testing blood and blood components, must also satisfy the requirements of the PHS Act (Public Health Service Act) and its implementing regulations. In order for a biological drug product, or for these in vitro diagnostic tests, to be legally marketed in the United States, the product must have a BLA (Biologics License Applications) approved by the FDA. Obtaining BLA approval from the FDA is a robust process involving, among other things, completion of preclinical laboratory tests, controlled human clinical trials, submission of manufacturing and chemistry data, and multiple statistical and physical review processes by the FDA. During all phases of clinical development, regulatory authorities require extensive monitoring and auditing of all clinical activities, clinical data and clinical trial investigators, including reports regarding adverse events and safety issues.
Given the robust process, certain of our clinical trials may not be completed successfully within any specified period, if at all. Furthermore, we or the FDA may suspend or terminate clinical trials at any time on various grounds, for example, that the subjects or patients may be exposed to an unacceptable health risk, have experienced a serious and unexpected adverse event, or that continued use in an investigational setting may be unethical. Similarly, an IRB may suspend or terminate approval of research if the research is not being conducted in accordance with the IRB’s requirements or if the research has been associated with unexpected serious harm to patients.
Overall, the testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all. In most cases, the BLA must be accompanied by a substantial user fee.
75
In the United States, our main market, there is an abbreviated regulatory approval pathway for biological products found to be “biosimilar” to or “interchangeable” with a biological “reference product” previously licensed under a BLA. This abbreviated approval pathway is intended to permit a biosimilar product to come to market more quickly and less expensively by relying to some extent on the data generated by the reference product’s sponsor, and the FDA’s previous review and approval of the reference product. The law provides that no biosimilar application may be accepted for FDA review until four years after the date the reference product was first licensed by the FDA, and that the FDA may not make approval of an application effective until 12 years after the reference product was first licensed. The law also includes an extensive process for the innovator biologic and biosimilar manufacturer to litigate patent infringement, validity, and enforceability, which could increase costs of protecting our reference products. Once approved, biosimilars likely would compete with, and in some circumstances may be deemed under applicable laws to be “interchangeable with,” the previously approved reference product. The extent to which a biosimilar product, once approved, will be substituted for any of our products, in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing. In connection with the FDA’s authority to ensure the development of safe and effective biosimilars, in September 2022, the FDA Biosimilar User Fee Amendments of 2022 were signed into law. The FDA Biosimilar User Fee Amendments of 2022 reauthorize for an additional five years (through 2027) the Biosimilar User Fee Act of 2012, which allows the FDA to assess and collect fees for biosimilars.
The testing and approval processes to obtain a BLA require substantial time, effort and financial resources, and each process may take several years to complete. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The FDA may limit the indications for use or place other conditions on any approvals that could restrict the commercial application of the products.
Post-approval Requirements
After regulatory approval of a product is obtained, we are required to comply with a number of post-approval requirements. For example, as a condition of approval of a BLA, the FDA may require post-marketing testing and surveillance to monitor the product’s safety or efficacy. In addition, holders of an approved BLA are required to keep extensive records, to report certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling for their products. Also, quality control and manufacturing procedures must continue to conform to cGMP regulations and practices, as well as the manufacturing conditions of approval set forth in the BLA. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes certain procedural, substantive and recordkeeping requirements. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
Future FDA inspections may identify compliance issues at our facilities or at the facilities of our third-party suppliers that may disrupt production or distribution, or require substantial resources to correct and prevent recurrence of any deficiencies, and could result in fines or penalties by regulatory authorities. In addition, discovery of problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved BLA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing. Newly discovered or developed safety or efficacy data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications.
The ACA established and provided significant funding for a Patient-Centered Outcomes Research Institute to coordinate and fund comparative effectiveness research. While the stated intent of comparative effectiveness research is to develop information to guide providers to the most effective therapies, outcomes of comparative effectiveness research could influence the reimbursement or coverage for therapies that are determined to be less cost effective than others.
76
Orphan Drug Designation
The FDA may grant orphan drug designation to drugs intended to treat a “rare disease or condition” that affects fewer than 200,000 individuals in the United States, or that affects more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug for such a disease or condition will be recovered from sales in the United States for that drug. Orphan drug designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. Orphan drug designation can provide opportunities for grant funding towards clinical trial costs, tax advantages and FDA user fee exemptions. In addition, if a product that has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity or a meaningfully different mode of administration. Competitors may receive approval of different drugs or biologics for the indications for which the orphan product has exclusivity. However, if a company with orphan drug exclusivity is not able to supply the market, the FDA could allow another company with the same drug a license to market for said indication. The FDA granted Gamunex® IVIG orphan drug status, which provided marketing exclusivity for the CIDP indication in the United States through September 2015. Gamunex® IVIG was the first IVIG product approved for CIDP in the United States.
Fast Track Designation
The FDA’s fast track programs, one of which is fast track designation, are designed to facilitate the development and review of new drugs that are intended to treat serious or life-threatening conditions and that demonstrate the potential to address unmet medical needs for the conditions. Fast track designation applies to a combination of the product and the specific indication for which it is being studied. Thus, it is the development program for a specific drug for a specific indication that receives fast track designation.
The sponsor of a product designated as being in a fast track drug development program may engage in close early communication with the FDA, including through timely meetings and feedback on clinical trials. Products in fast track drug development programs also may receive FDA priority review or accelerated approval; in other words, the review cycle has a six-month review clock instead of a ten- or 12-month review clock. Sponsors may also be able to submit completed portions of an application before the entire application is completed; however, the review clock will not officially begin until the entire completed BLA is submitted to and filed by the FDA. The FDA may notify a sponsor that its program is no longer classified as a fast track development program if the fast track designation is no longer supported by emerging data, the designated drug development program is no longer being pursued, or another product that meets the unmet medical need for the same indication is approved first.
Plasma Collection
The FDA requires a licensing and certification process for each plasma collection center prior to opening and conducts periodic inspections of facilities and processes. Many states also regulate plasma collection, imposing similar obligations and additional inspections and audits. Collection centers are subject to periodic inspections by regulatory authorities, which if noncompliance is alleged, may result in fines, citations, the temporary closing of the centers, loss or suspension of licenses or recall of finished products.
Diagnostic Devices
Certain of our products are regulated as medical devices, which are typically subject to clearance for commercialization in the United States, often referred to as an FDA “510(k) clearance,” based on a pre-market notification to the FDA demonstrating the device to be marketed is safe and effective by proving substantial equivalence to a legally marketed device (predicate device). The manufacturers of medical devices must register their establishments with the FDA, and the production of the devices must accord with applicable current good manufacturing practices and quality system regulations. Some states also regulate the manufacture or distribution of certain medical devices, imposing additional licensing, inspection and audit requirements. With respect to the manufacture and sale of immunoassay antigens and antibodies to screen human donated blood and blood products, these products are manufactured and sold under a BLA issued by the FDA, and are subject to the heightened regulatory oversight associated with biological products.
77
Drug Supply Chain Security Act
The Federal Drug Quality and Security Act of 2013 regulates pharmaceutical supply chain requirements and pre-empts certain state laws. Title II of this act, known as the Drug Supply Chain Security Act (“DSCSA”) establishes a national electronic, interoperable system to identify and trace certain prescription drugs as they are distributed in the United States, including certain of our products. The DSCSA’s track and trace requirements (applicable to manufacturers, wholesalers, repackagers and dispensers (e.g., pharmacies) of prescription drugs) replaced the former FDA drug pedigree requirements and pre-empt state requirements that are inconsistent with, more stringent than, or in addition to, the DSCSA requirements. The DSCSA also establishes certain requirements for the licensing and operation of prescription drug wholesalers and third party logistics providers (“3PLs”), and includes the creation of national wholesaler and 3PL licenses in cases where states do not license such entities to be established in FDA regulations yet to be finalized. The DSCSA generally requires that wholesalers and 3PLs distribute drugs in accordance with certain standards regarding the recordkeeping, storage and handling of prescription drugs. Under the DSCSA, states are pre-empted from imposing any licensing requirements that are inconsistent with, less stringent than, directly related to, or covered by U.S. federal standards, however, the FDA has clarified that current state licensing requirements may remain in effect until the FDA national licensing regulations are finalized and take effect.
Anti-fraud and Abuse Regulation
Since we supply products and services that are reimbursed by U.S. federal healthcare programs such as Medicare and Medicaid, our activities are also subject to regulation by CMS (U.S. Centers for Medicare & Medicaid Services) and enforcement by the OIG (U.S. Office of the Inspector General). The federal Anti-Kickback Statute prohibits providers and others from directly or indirectly soliciting, receiving, offering or paying any remuneration with the intent of generating referrals or orders for services or items covered by a government health care program. Many states have similar laws. Courts have interpreted this law very broadly, including by holding that a violation has occurred if even one purpose of the remuneration is to generate referrals, even if there are other lawful purposes. There are statutory and regulatory exceptions, or safe harbors, that outline arrangements that are deemed lawful. However, the fact that an arrangement does not fall within a safe harbor does not necessarily render the conduct illegal under the Anti-Kickback Statute. In sum, even legitimate business arrangements between the companies and referral sources could lead to scrutiny by government enforcement agencies and require extensive company resources to respond to government investigations. Also, certain business practices, such as payment of consulting fees to healthcare providers, sponsorship of educational or research grants, charitable donations, interactions with healthcare providers that prescribe products for uses not approved by the FDA and financial support for continuing medical education programs, must be conducted within narrowly prescribed and controlled limits to avoid potential liability for wrongfully influencing healthcare providers to prescribe or purchase particular products or as a reward for past prescribing.
The FCA (Federal False Claims Act) is violated by any entity that knowingly presents or causes to be presented a false claim for payment to the U.S. federal government, or that knowingly makes or causes to be made a false record material to such a false claim, or knowingly conceals or avoids an obligation to pay to the government, including by knowingly retaining an overpayment. Payments in violation of the Anti-Kickback Statute can also form the basis for FCA liability.
The FCA has been subject to heightened enforcement activity as a result of whistleblowers (called “relators”) who file complaints in the name of the United States (and if applicable, particular states), and who may receive up to 30% of total government recoveries. FCA violators may be liable for treble damages, mandatory penalties, loss of licensing and exclusion from federal and state healthcare programs. Such penalties could have a material adverse effect on our business. Also, these measures may be interpreted or applied by a prosecutorial, regulatory or judicial authority in a manner that could require us to make changes in our operations or incur substantial defense and settlement expenses. Even unsuccessful challenges by regulatory authorities or relators could result in reputational harm and the incurring of substantial costs. Most states have adopted similar state false claims laws, which could lead to significant additional penalties.
We also are subject to certain United States and foreign laws and regulations concerning the conduct of our foreign operations, including the U.S. Foreign Corrupt Practices Act, the U.K. Bribery Act, German anti-corruption laws and other anti-bribery laws and laws pertaining to the accuracy of our internal books and records, which have been the focus of increasing enforcement activity globally in recent years.
78
The “PPS Act” (Physician Payments Sunshine Act) imposes reporting and disclosure requirements for biologic, drug and device manufacturers with regard to payments or other transfers of value made to physicians, physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists, certified nurse-midwives, teaching hospitals, manufacturers and for GPOs (group purchasing organizations), with regard to practitioner and teaching hospital ownership interests in the reporting entity. CMS publishes information from these reports on a publicly available website, including amounts transferred and practitioner and teaching hospital identities. The PPS Act preempts similar state reporting laws, although we or our subsidiaries may also be required to report under certain state transparency laws that address circumstances not covered by the PPS Act, and some of these state laws, as well as the federal law, can be ambiguous. We are also subject to foreign regulations requiring transparency of certain interactions between suppliers and their customers.
Other Health Care Regulation
In the United States, pricing concerns have led to heightened scrutiny and ongoing legislative efforts to increase transparency around healthcare and pharmaceutical drugs costs. For example, on November 12, 2020, CMS issued final rules imposing price transparency requirements on hospitals and group health plans, which went into effect in three stages from 2022 to 2024. As of January 1, 2024, hospitals and group health plans must disclose in-network provider negotiated rates (which include rates with device suppliers and manufacturers), historical out-of-network allowed amounts for all covered items and services, including all prescription drugs and drug pricing information. States have also enacted a variety of transparency rules and regulations. The publication of our negotiated rates could affect our ability to independently negotiate sales contracts and rate agreements.
Another notable Medicare healthcare reform initiative, the Medicare Access and CHIP Reauthorization Act of 2015 (“MACRA”), established a new quality payment program, which modifies certain Medicare Part B payments to “eligible clinicians,” including physicians, dentists and other practitioners. Under MACRA, certain eligible clinicians are required to participate in Medicare through the Merit-Based Incentive Payment System (“MIPS”) or the Advanced Alternative Payment Models (“APMs”), through which Medicare Part B reimbursement is adjusted up or down based on reported data related to quality, promoting interoperability, cost, and improvement activities. MIPS-eligible clinicians must report annual performance data by March 31 of the following year. Payment adjustments based on submitted data are applied to Medicare Part B claims during the performance year following data submission. MACRA provides substantial financial incentives for physicians to participate in risk contracts, while increasing physician information technology and reporting obligations. MACRA continues to evolve and its implications depend on future regulatory activity and physician activity in the marketplace. New payment and delivery system reform programs, including those modeled after such federal program, are also increasingly being rolled out at the state level through Medicaid administrators, as well as through the private sector, which may further alter the marketplace and impact our business.
Recently, a number of states in the United States have also proposed or passed legislation that seeks to directly or indirectly regulate pharmaceutical drug pricing, such as by requiring drug manufacturers to publicly report pricing information or to place a maximum price cap on pharmaceutical products purchased by state agencies. For example, in October 2017, California enacted a prescription drug price transparency law that requires prescription drug manufacturers to provide advance notice and explanation for certain drug price increases that exceed a specified threshold. Similar laws were enacted in Oregon and Washington and a number of other states are reportedly considering similar legislation. Laws of this type may cause us to experience additional pricing pressures on our affected products. For more details on these pressures, see “—Pharmaceutical Pricing and Reimbursement” below.
Antitrust and Consumer Protection
The U.S. federal government, most U.S. states and many foreign countries have antitrust laws that prohibit certain types of conduct deemed to be anti-competitive, as well as consumer protection laws that seek to protect consumers from improper business practices. At the U.S. federal level, the FTC oversees enforcement of these types of laws, and states have similar government agencies. Violations of antitrust or consumer protection laws may result in various sanctions, including criminal and civil penalties. Private plaintiffs may also bring civil lawsuits against us in the United States for alleged antitrust law violations, including claims for treble damages.
79
European Community Government Regulation
In addition to regulations in the United States, we are subject to a variety of regulations in other jurisdictions governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of countries outside the United States before we can commence marketing that product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. Also, in addition to approval of final products, plasma collection centers for manufacture into products to be distributed in the European Union must also be approved by the competent European health authority.
Medicines can be authorized in the European Union by using either the centralized authorization procedure or national authorization procedures. The EMA is responsible for the centralized authorization procedure.
Centralized Authorization Procedure
The EMA is responsible for the centralized procedure, or Community authorization procedure, for human medicines. This procedure results in Community marketing authorization, the single marketing authorization that is valid across the European Union, as well as in the European Economic Area/European Free Trade Association states Iceland, Liechtenstein and Norway.
The Community authorization procedure is compulsory for:
| ● | medicines derived from biotechnology processes, such as genetic engineering; |
| ● | advanced-therapy medicines, such as gene-therapy, somatic cell-therapy or tissue-engineered medicines; |
| ● | medicinal products for human use containing a new active substance that did not receive Community marketing authorization when the Community authorization procedure was first implemented, for which the therapeutic indication is the treatment of human immunodeficiency virus (“HIV”) or acquired immune deficiency syndrome (“AIDS”), cancer, neurodegenerative disorders, diabetes, autoimmune diseases and other immune dysfunctions or viral diseases; and |
| ● | officially designated orphan medicines (medicines for rare diseases). |
The Community authorization procedure is optional for products:
| ● | containing new active substances for indications other than those stated above; |
| ● | representing significant therapeutic, scientific or technical innovations; or |
| ● | for which the granting of a Community marketing authorization would be in the interests of European Union public health. |
Our blood derivative products are not subject to compulsory Community authorization, but it is an option for our new products. Flebogamma® DIF 50 mg/ml and 100 mg/ml, VeraSeal solutions for sealant and Tavlesse (fostamatinib) were approved through the Community authorization procedure.
Applications through the Community authorization procedure are submitted directly to the EMA. Evaluation by the EMA’s relevant scientific committee takes up to 210 days, at the end of which the committee adopts an opinion on whether the medicine should be marketed. This opinion is then transmitted to the European Commission, which has the ultimate authority for granting marketing authorizations in the European Union.
Once a Community marketing authorization has been granted, the holder of that authorization can begin to make the medicine available to patients and healthcare professionals in all European Union countries.
80
National Authorization Procedures
Each European Union member state has its own procedures for the authorization, within its own territory, of medicines that fall outside the scope of the Community authorization procedure. There are two possible routes available to companies for the authorization of such medicines in several countries simultaneously.
| ● | Decentralized procedure. Using the decentralized procedure, companies may apply for simultaneous authorization in more than one European Union country of medicines that have not yet been authorized in any European Union country and that do not fall within the mandatory scope of the centralized procedure. |
| ● | Mutual-recognition procedure. In the mutual-recognition procedure, a medicine is first authorized in one European Union member state, in accordance with the national procedures of that country. Following such authorization, further marketing authorizations can be sought from other European Union member states in a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization. |
Our products Niuliva 250 I.U./ml and Xembify 200 mg/ml were approved through the decentralized procedure. Our products Prolastina® 1000 mg/ml, and Gamunex® 10% and some of the Human Albumin Grifols / Albutein licenses were approved through the mutual-recognition procedure. All our other products were approved pursuant to individual national procedures. We expect to use the mutual-recognition procedure if we want to extend our product licenses to other European countries in the future.
In some cases, disputes arising in these procedures can be referred to the EMA for arbitration as part of a “referral procedure.”
Orphan Drug Designation
Applications for designation of orphan medicines are reviewed by the EMA through the Committee for Orphan Medicinal Products. The criteria for orphan designation are:
| ● | the medicinal product is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition affecting no more than five in 10,000 persons in the European Union at the time of submission of the designation application (prevalence criterion); |
| ● | the medicinal product is intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition, and without incentives it is unlikely that the revenue after marketing of the medicinal product would cover the investment in its development; and |
| ● | either no satisfactory method of diagnosis, prevention or treatment of the condition concerned is authorized, or, if such method exists, the medicinal product will be of significant benefit to those affected by the condition. |
Companies with an orphan designation for a medicinal product benefit from incentives such as:
| ● | protocol assistance (scientific advice for orphan medicines during the product-development phase); |
| ● | direct access to centralized marketing authorization and 10-year marketing exclusivity; |
| ● | financial incentives (fee reductions or exemptions); and |
| ● | national incentives detailed in an inventory made available by the European Commission. |
81
Since December 2011, orphan medicinal products are eligible for the following level of fee reductions:
| ● | full (100%) reduction for small- and medium-sized enterprises, or SMEs, for protocol assistance and follow-up, full reduction for non-SME sponsors for pediatric-related assistance and 75% reduction for non-SME sponsors for non-pediatric assistance; |
| ● | to determine which companies are eligible for SME incentives, the EMA applies the definition of micro-, small- and medium-sized enterprises provided in the Commission of the European Communities’ Commission Recommendation 2003/361/EC. To qualify for assistance, companies must be established in the European Economic Area, employ less than 250 employees and have an annual turnover of not more than €50 million or an annual balance sheet total of not more than €43 million; |
| ● | full reduction for pre-authorization inspections and 90% reduction for post-authorization inspections for small- and medium-sized enterprises; |
| ● | full reduction for SMEs for new applications for Community marketing authorization and 10% reduction for non-SME sponsors; and |
| ● | full reduction for post-authorization activities including annual fees only to small and medium sized enterprises in the first year after granting a marketing authorization. |
We have EMA Orphan Drug Designations for the following products:
| ● | alpha-1 proteinase inhibitor (for inhalation use) for the treatment of cystic fibrosis; and |
| ● | alpha-1 proteinase inhibitor (for inhalation use) for the treatment of congenital alpha-1 antitrypsin deficiency. |
Because each of these products is already authorized for a non-orphan indication in the EU, in order to obtain marketing authorization for any of the above-mentioned orphan indications, we would be required to apply for a separate marketing authorization through the Community authorization procedure for such indication, using a different proprietary name. It is not possible to extend the existing marketing authorization to cover the new orphan indication. Orphan and “non-orphan” indications cannot be covered by the same marketing authorization.
82
United Kingdom Regulatory Process
The United Kingdom (“U.K.”) withdrew from the E.U. on January 31, 2020, and is no longer an E.U. Member State. A transition period, during which E.U. pharmaceutical law continued to be applicable to the U.K., ended on December 31, 2020.
As of January 1, 2021, the protocol in Ireland/Northern Ireland is applicable and has an impact on marketing authorizations for medicinal products in the U.K. with respect to Northern Ireland. However, there is a new agreement between the E.U. and the U.K. called the “Windsor Framework,” which amended the Protocol of Northern Ireland and set out the long-term arrangements for the supply of medicines into Northern Ireland. The Windsor Framework will ensure that medicines can be approved and licensed on a U.K.-wide basis by the Medicines and Healthcare products Regulatory Agency (“MHRA”) and provides for the disapplication of European Union Falsified Medicines Directive (“FMD”) requirements for medicines marketed and supplied in Northern Ireland. These new arrangements will take effect on January 1, 2025.
There are several routes to obtain a marketing authorization in the U.K., England, Scotland and Wales (“Great Britain”) or Northern Ireland. The options available are determined by the intended market and the type of application. To obtain a marketing authorization, you need to use one of the following procedures:
National Routes:
| ● | National Procedure (a 150-day procedure). This national 150-day accelerated procedure is available for high-quality applications to market a medicine in the United Kingdom, Great Britain (England, Wales and Scotland) or Northern Ireland; |
| ● | Rolling review. Permits the submission of your application in module(s), to obtain a marketing authorization in the United Kingdom, Great Britain (England, Wales and Scotland) or Northern Ireland. This is a new route for marketing authorization applications, where an applicant for a new active substance in the U.K., Great Britain, or Northern Ireland submits modules of the eCTD dossier incrementally for pre-assessment by the MHRA, rather than as part of a consolidated full dossier submission. |
This rolling review is intended to streamline the development of novel medicines by offering periodic enhanced regulatory interaction and advice to reduce the risk of failure at the final phase and may be integrated with the Target Development Profile (“TDP”) to provide a clearer pathway for development of innovative medicines.
Marketing authorization applications for any new active substance based on a “full dossier,” including biological products, are eligible for the rolling review;
| ● | International recognition procedure (a 60-day to 110-day procedure). As of January 1, 2024, the EC Decision Reliance Procedure and the Mutual Recognition/Decentralized Reliance Procedure have been replaced by the new International Recognition procedure (“IRP”). This route applies to new marketing authorization applications, variations, line extensions and renewals and is open to applicants that have already received an authorization for the same product from one of MHRA’s specified Reference Regulators (“RRs”). A positive opinion from the Committee for Medicinal Products for Human Use or a Mutual Recognition/Decentralized positive end of procedure outcome is an RR authorization for the purposes of IRP. Apart from the EMA and the E.U. Member State Competent Authorities, there are other possible RRs (such as Australia, Canada and the United States). IRP will allow the MHRA to take into account the expertise and decision-making of trusted regulatory partners for the benefit of U.K. patients. The MHRA will conduct a targeted assessment of IRP applications but retain the authority to reject applications if the evidence provided is considered insufficiently robust. |
| ● | Unfettered Access from Northern Ireland (a 67-day procedure). Applicants may seek recognition in Great Britain of a marketing authorization approved in Northern Ireland under certain qualifying conditions. |
This route is available for marketing authorizations approved in Northern Ireland via European procedures (centralized, MR or DC procedures) or through the Northern Ireland national procedure.
83
For authorizations approved in E.U. procedures, applications should include the dossier as approved for marketing in Northern Ireland, accompanied by all iterations of the relevant RMS and CHMP assessment reports.
Note that national applications intended to cover marketing of a product in Northern Ireland must continue to comply with the requirements of Directive 2001/83/EC, the European Community’s code relating to medicinal products for human use, and Regulation 726/2004 on European Community procedures and supervision of medicinal products until the Windsor Framework becomes effective on January 1, 2025.
International routes (collaborative procedures):
| ● | Access consortium. The Access consortium is a medium-sized coalition of regulatory authorities that work together to promote greater regulatory collaboration and alignment of regulatory requirements for companies intending to market a medicine in the U.K., Australia, Canada, Singapore and/or Switzerland. The MHRA joined the consortium in 2020 and commenced work-sharing applications in January 2021; and |
| ● | Project Orbis. Project Orbis is a program coordinated by the FDA involving the regulatory authorities of Australia (TGA), Canada (Health Canada), the United Kingdom (MHRA), Singapore (HSA), Brazil (ANVISA), Israel (IMoH) and Switzerland (Swissmedic) to review and approve promising cancer treatments. |
In addition to the above, and until the Windsor Framework becomes effective on January 1, 2025, the following procedures can be used to obtain a marketing authorization in Northern Ireland:
| ● | Northern Ireland may be included in DC or MR procedure as a Concerned Member State (CMS). The DC and MR procedures can be used by companies intending to market a medicine in Northern Ireland and other named E.U. countries. One member state will lead the assessment of the application as the RMS. The other member states (including Northern Ireland) receiving applications are called the ‘concerned member states’ (“CMSs”). The procedure takes up to 210 days (DC procedure) or 90 days (MR procedure), excluding time taken to provide further information or data required. If the application is approved, each CMS (including Northern Ireland as a CMS) will issue a national marketing authorization for the product within 30 days of approval. |
| ● | Marketing authorizations approved in the EU’s Centralized procedure will automatically have effect in Northern Ireland. The E.U. centralized procedure, including its mandatory scope, continues to apply in Northern Ireland, and therefore, the centralized procedure results in a single marketing authorization to market a product in all E.U. member states, as well as Iceland, Liechtenstein, Norway and Northern Ireland. |
Further information can be found in the U.K.’s website for license applications (www.gov.uk).
84
Canadian Regulatory Process
Authorization to Market. Therapeutic products can be marketed in Canada after they have been subject to a review to assess their safety, efficacy and quality. A New Drug Submission must be submitted to Health Canada for review, and a Notice of Compliance (“NOC”), and/or a Drug Identification Number (“DIN”), must be received by the sponsor prior to marketing a product in Canada. Responsibility for review of pharmaceutical drug products resides with Health Canada’s Therapeutic Products Directorate (“TPD”), while responsibility for review of biological products is under the Biologics, Radiopharmaceuticals and Genetic Therapies Directorate (“BGTD”). An active DIN is required for any product being marketed in Canada. Our IG, A1PI, albumin and hyperimmune products are subject to these review and authorization processes.
Changes to Market Authorization. There are four classes of changes to existing market authorizations in Canada. Level 1 changes are considered “significantly different” and have the potential to impact safety, efficacy, quality or effectiveness of the product. These require the filing of a Supplemental New Drug Submission, and a NOC must be issued by Health Canada prior to implementation of the change. Level 2 changes are not considered “significant,” but a “Notifiable Change” submission must be filed to Health Canada for review, and approval is provided via a “No Objection” letter to the sponsor. Level 3 changes have minimal potential to impact safety, quality or effectiveness and can be made without prior approval of Health Canada; a summary of these changes is reported to Health Canada with the sponsor’s Annual Drug Notification. Level 4 changes are implemented without any notification to Health Canada, based on no expectation of risk.
Clinical Trials. A Clinical Trial Application (“CTA”), must be submitted to Health Canada prior to conducting any study protocol that proposes the use of a new product, or the use of an existing product, where the indication, target population, route of administration or dosing differs from the current market authorization. The CTA should include summaries of preclinical and clinical studies conducted and (if applicable) chemistry, manufacturing and control data, and is submitted to either TPD (for drug products) or BGTD (for biological products) for review. The TPD or BGTD are responsible for assessing protection and safety of the participants as well as quality of the product; they will issue a “No Objection” letter to sponsors for studies deemed acceptable. Research ethics board approval for each trial is also required prior to conduct of the study.
Establishment Licensing. All establishments in Canada that are involved in the fabrication, packaging/labeling, testing, import, distribution or warehousing of drug products must have a current establishment license (once an establishment license is issued, an annual report must be submitted by April 1 of each year to maintain the effectiveness of that license). As an importer/distributor, part of the licensing requirements include demonstration that any foreign (non-Canadian) facilities involved in fabrication, packaging/labeling or testing of products imported/distributed under the license comply with cGMP.
Post-Approval Requirements. The Health Products and Food Branch Inspectorate of Health Canada periodically inspects licensed establishments in Canada to verify compliance with cGMP. Manufacturers and importers are required to monitor the safety and quality of their products and must report adverse reactions to the Marketed Health Products Directorate in accordance with a prescribed timeline and format.
Regulatory Process for Markets outside the United States, Europe, United Kingdom and Canada
The majority of regulatory authorities in countries outside the United States, Canada and Europe require that a product first be approved by the FDA or European authority prior to granting the market authorization in their country. There are a limited number of countries (Bahamas, Bermuda, Guam, Oman and Qatar) that do not require further local product registration for products and they may be distributed based on the existing FDA approval.
85
In addition to requiring the submission of a license application containing documentation supporting the safety, efficacy and quality of the product, many countries require the submission of FDA Export Certificates for our products to provide assurance that such products can be legally marketed in the United States. The Certificate of Pharmaceutical Product (“CPP”), and/or the Certificate to Foreign Government (“CFG”), are issued by the FDA at the request of the manufacturer seeking licensing in the country outside the United States. The CPP conforms to the format established by the World Health Organization (“WHO”), and is intended for use by the importing country when considering whether to license the product in question for sale in that country. The CFG serves to document that the product can be legally marketed in the United States and the manufacturer is in compliance with GMP. A limited number of regulatory authorities in countries outside United States, Canada and Europe conduct onsite inspections to verify GMP compliance. Failure to maintain and document GMP compliance could result in withdrawal of marketing authorization. In addition changes to manufacturing or testing procedures for the product require approval of the change in the United States prior to the submission of the variation to the registration in the international market. These changes may require approval in each market in order to maintain product distribution. Furthermore, any changes in the distributors supporting our export business could result in a loss of sales.
Pharmaceutical Pricing and Reimbursement
In the United States and other countries, sales of our products depend, in material respects, on the availability of reimbursement from third-party payors. Third-party payors include government health programs, managed care providers, private health insurers and other organizations. These third-party payors are increasingly challenging the prices and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of healthcare products newly approved by regulatory authorities. For example, third-party payors may deny reimbursement if they do not consider the products to be cost-effective as compared to available alternative products. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
United States Pharmaceutical Pricing and Reimbursement
In the United States, our products are reimbursed or purchased under several government programs, including Medicaid, Medicare (Parts B and D) and the 340B Program. Medicaid is a joint state and federal government health plan that provides covered outpatient prescription drugs for low income individuals. Medicare is a federally run program that provides healthcare to persons aged 65 and over, as well as certain persons of any age with certain disabilities. The 340B Program is a U.S. federal government drug pricing program under the PHS Act. The availability of federal funds to pay for our products under the Medicaid and Medicare Part B programs requires that we extend discounts under the 340B Program.
Medicaid, the 340B Program and the Department of Veterans Affairs.
Medicaid includes a variety of reimbursement programs that differ state by state and imposes reimbursement requirements applicable to both fee-for-service and managed care arrangements. These requirements are applicable to certain of our products. Under Medicaid, drug manufacturers pay rebates to the states based on utilization data provided by the states and pricing information provided by manufacturers, such as AMP (average manufacturer price) and the lowest price available from the manufacturer during the rebate period to any entity in the United States in any pricing structure in the same quarter for which the AMP is computed (“Best Price”). The states share the rebates with the U.S. federal government. The rebate amount for most brand name drugs is the greater of 23.1% of the AMP per unit or the difference between the AMP and Best Price per unit as adjusted by the U.S. Consumer Price Index for All Urban Consumers (“CPI-U”), subject to certain exceptions. For example, the rebate amount for certain clotting factors such as Factor VIII and Factor IX is the greater of 17.1% of the AMP per unit or the difference between the AMP and the Best Price per unit as adjusted by the CPI-U, and the rebate amount for non-innovator multiple source (generic) drugs is equal to a minimum of 13.0% of AMP.
86
The availability of federal funds to pay for our products under Medicaid requires that we extend discounts under the 340B Program. The 340B Program extends prescription drug discounts to certain entities, including a variety of community health clinics and certain other entities that receive governmental health care grants, as well as hospitals that serve a disproportionate share of low income individuals, certain cancer centers, children’s hospitals, critical access hospitals and rural referral centers. The 340B Program prescribes a pricing methodology based on a PHS Act ceiling price that generally cannot be greater than the AMP less the unit rebate amount established under the Medicaid drug rebate program. We have entered into a PPA with the government whereby we participate in the 340B Program by charging eligible entities no more than the PHS Act ceiling price for drugs intended for outpatient use. The HRSA (Health Resources & Services Administration) of the HHS, the federal agency overseeing the 340B Program, continually issues evolving requirements with respect to the 340B Program, which creates uncertainty.
Also as a condition for Medicaid reimbursement, we make our products available for purchase by authorized government users of the Federal Supply Schedule (“FSS”) pursuant to such users’ FSS contracts with the Department of Veterans Affairs. These transactions involve establishing a FSS price in negotiation with the Department of Veterans Affairs. Companies are also required to offer discounted drug pricing to four federal agencies — the Department of Veterans Affairs, the Department of Defense, the Coast Guard and the PHS (including the Indian Health Service) — for federal funding to be made available for reimbursement of products under the Medicaid program. Sales to those four federal agencies must utilize a ceiling price methodology that generally requires drug prices to be equal to or less than 24.0% off the non-federal AMP for the prior fiscal year.
Medicare and the 340B Program.
The primary Medicare programs that may affect reimbursement for the Grifols Group are Medicare Part B, which covers physician services and outpatient care, and Medicare Part D, which provides a voluntary outpatient prescription drug benefit.
Medicare Part B reimburses providers for certain covered drugs and biologicals furnished in the outpatient setting, such as physician offices and hospital outpatient settings. The rate of reimbursement historically has generally been of average sales price (“ASP”) + 6%. Starting January 1, 2018 a CMS rule reduced Medicare Part B reimbursement to a rate of ASP - 22.5%. Such reduction was ruled unlawful by the U.S. Supreme Court on June 15, 2022, causing CMS to reinstate the general ASP + 6% rate retroactively to January 1, 2018. Following this ruling, CMS issued a new rule on November 8, 2023 determining that affected hospitals will receive a one-time lump-sum amount equivalent to the approximate payment they would have received if the 2018-2022 340B Program payment policy had never existed. The 340B Program remains subject to legal challenges involving litigation that may result in program changes, which creates uncertainty. We believe that we meet the requirements of the 340B Program and are continuing to review and monitor these and other developments affecting the 340B Program. In addition, under the Bipartisan Budget Act of 2013 and subsequent measures, Medicare is subject to a 2% reduction in federal spending, or “sequestration,” including drugs reimbursed under Medicare, for federal fiscal years 2013 through 2032. The full ramifications of this sequestration for Medicare reimbursement remain uncertain, as further Congressional action may reduce, eliminate or otherwise change this payment reduction.
Medicare Part D coverage is administered through private insurers that contract with CMS. To obtain payments under this program, which covers certain of our products, we are required to negotiate prices with private insurers operating pursuant to federal program guidance, and these prices may be lower than we might otherwise obtain. In addition, the Medicare Part D Program includes a coverage gap feature, where once beneficiaries have expended certain amounts for drugs payable under Medicare Part D, Medicare Part D benefits are decreased for a period until certain levels of out-of-pocket spending are incurred by such beneficiaries. Pursuant to this coverage gap, drug manufacturers are required to provide a 70% discount (the “Coverage Gap Discount”) regarding certain drugs falling within a beneficiary’s coverage gap. Under the federal Inflation Reduction Act of 2022, CMS is required, as part of the redesign of Medicare Part D benefit, to establish, as of January 1, 2025, a new Medicare Part D manufacturer discount program that will replace the existing Coverage Gap Discount program. Specifically, manufacturers will be required to provide a 20% price discount on brand-name drugs and a 10% discount on brand-name drugs between the deductible and the annual out-of-pocket spending cap, which replaces the 70% price discount in the coverage gap phase under the current benefit design. Because this provision is aimed at decreasing Medicare beneficiaries’ out-of-pocket costs, manufacturers’ share of those costs may increase. Consequently, our net revenue, beginning in 2025, may be adversely affected.
87
Additionally, the Inflation Reduction Act of 2022 provides Medicare, for the first time, with the ability to negotiate drug prices directly with drug manufacturers to determine the price Medicare will pay for certain drugs covered under Medicare Part D (effective as of 2026) and Medicare Part B (effective as of 2028). Certain drugs are excluded from the negotiation process, such as drugs with an orphan designation as their only FDA-approved indication and certain plasma-derived drugs. To the extent our drugs fall within any of the aforementioned excluded categories, Medicare would not have the ability to negotiate our prices for those drugs. Manufacturers that fail to comply with program requirements may be subject to civil monetary penalties and excise taxes.
Also under the Inflation Reduction Act of 2022, if a manufacturer increases the prices generally for brand name prescription drugs and biologicals covered under Medicare Part B or certain drugs and biologicals covered under Medicare Part D, in each case at a higher rate than the applicable inflation rate for such products, then such manufacturer would be required to pay rebates to Medicare. If our products are used by Medicare beneficiaries, we could be required to pay rebates to Medicare or potentially face fines representing the difference between the actual and the inflated prices. And, beginning in 2025, this law modifies a manufacturer’s liability relating to Medicare’s spending above a beneficiary’s out-of-pocket cap. In a December 13, 2023 guidance document, CMS generally outlined its calculation methodologies and processes, including certain adjustments (such as in the event of drug shortages, with, for example, certain greater reductions provided for plasma-derived products), and advised that it will begin to issue invoices to drug manufacturers for these rebates starting in 2025, including for years 2022, 2023 and 2024. Manufacturers’ failure to timely pay a rebate amount due may result in the imposition of civil monetary penalties. We are continuing to review and monitor these new Inflation Reduction Act programs under Medicare, which create uncertainty and may adversely affect our business.
Other Relevant Regulations.
The ACA imposes an annual fee on certain manufacturers and importers of branded prescription drugs and biologics in general if the entity has aggregated branded prescription drug and biologic sales of over $5.0 million to specified United States government health programs such as the Department of Veteran Affairs, or pursuant to coverage under such specified government health programs, such as Medicare Part B, Medicare Part D and Medicaid. The fee to each covered entity generally is calculated as an allocated portion of an aggregate amount of branded sales attributed to all covered entities, which aggregate amount is subject to change. From 2019 to the present date, the aggregate amount has been $2.8 billion. In addition, the Prescription Drug User Fee Act (“PDUFA”) sets forth user fees that pharmaceutical and biological companies pay to the FDA for certain applications for approvals of drugs and biologicals, licensing of certain biological products, and certain prescription drug program fees assessed annually for eligible products. The fees under PDUFA cover a substantial portion of the FDA’s operating budget, and the measure also addresses aspects of the regulatory approval process, such as timing and procedures. The PDUFA is subject to reauthorization by Congress every five years and, in September 2022, the FDA User Fee Reauthorization Act of 2022 was signed into law, reauthorizing the PDUFA for fiscal years 2023 through 2027.
Federal, state and local governments in the United States have enacted and continue to consider additional legislation to limit the growth of healthcare costs, including the costs of prescription drugs. An increasing number of states in the United States have also proposed or passed legislation that seeks to directly or indirectly regulate pharmaceutical drug pricing, such as by requiring drug manufacturers to publicly report pricing information or to place a maximum price ceiling on pharmaceutical products purchased by state agencies. For example, in October 2017, California enacted a prescription drug price transparency law that requires prescription drug manufacturers to provide advance notice and explanation for certain drug price increases that exceed a specified threshold. Laws of this type may cause us to experience additional pricing pressures on our affected products, and could adversely affect our business.
Furthermore, the marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement. Existing and future legislation could limit payments for our existing products or for drug candidates that we are developing, including possibly permitting the federal government to negotiate prices directly with manufacturers. This increasing emphasis on managed care in the United States has increased and will continue to increase the pressure on pharmaceutical pricing. For a discussion of certain risks related to reimbursement and pricing, see Item 3 of this Part I, “Key Information—D. Risk Factors—Risks Relating to the Healthcare Industry—United States Healthcare Reform may adversely affect our business.”
88
European Union Pharmaceutical Pricing and Reimbursement
Our operations in the E.U. are subject to regulations that affect the pricing and market access of our products. The governments of E.U. Member States are able to regulate the price of pharmaceutical products through cost controlling measures applied to national healthcare systems. As such, governments in the E.U. Member States have been introducing healthcare reforms to limit increases in costs, particularly with respect to prescription drugs. Some E.U. Member States have also passed legislation and health policies to impose mandatory rebates for pharmaceutical products and financial claw-backs to the pharmaceutical industry. Through health technology assessment organizations that use formal economic and clinical metrics such as cost-effectiveness, budget impact models and clinical benefits versus existing marketed (or in development) drug products being used as comparators, to determine prices, funding and reimbursement of new therapies, E.U. Member States are also seeking to limit healthcare costs. We expect that E.U. Member States will continue to pursue actions to reduce healthcare expenditures.
Pricing and Reimbursement in Other Countries
Many countries around the world have been taking steps to control healthcare costs, particularly as they relate to prescription drugs. For example, Canada is contemplating regulatory changes that seek to reduce prices for certain medicinal products, such as biologics and medicines for rare diseases. China has organized national price negotiations for certain products directly linked to national drug reimbursement. Drug prices in China may further decline due to a stated national policy of reducing healthcare costs. Furthermore, countries are utilizing tendering processes to generate competition in a bid to control prescription drugs.
Item 4.A.UNRESOLVED STAFF COMMENTS
None.
Item 5.OPERATING AND FINANCIAL REVIEW AND PROSPECTS
The following is a review of our financial condition and results of operations as of December 31, 2023 and 2022, and for the three years ended December 31, 2023, and of the key factors that have affected or are expected to be likely to affect our ongoing and future operations. You should read the following discussion and analysis in conjunction with our audited consolidated financial statements and the accompanying notes included elsewhere in this annual report on Form 20-F.
Some of the information contained in this discussion, including information with respect to our plans and strategies for our business and our expected sources of financing, contain forward-looking statements that involve risk and uncertainties. You should read “Cautionary Statement Regarding Forward-Looking Statements” in this Part I for a discussion of the risks related to those statements. You should also read Item 3 of this Part I, “Key Information—D. Risk Factors” for a discussion of certain factors that may affect our business, financial condition and results of operations.
We have prepared our audited consolidated financial statements as of December 31, 2023 and 2022, and for the three years ended December 31, 2023 in accordance with IFRS, as issued by the IASB. The financial information and related discussion and analysis contained in this item are presented in euros except as otherwise specified. Unless otherwise specified the financial information analysis in this annual report on Form 20-F is based on our actual audited consolidated financial statements as of December 31, 2023 and 2022, and for the three years ended December 31, 2023.
See “Presentation of Financial and Other Information” in this Part I for further information on our presentation of financial information.
89
A. | Operating Results |
Subsequent Events
Short Seller Reports
On January 9, February 20 and March 6, 2024, a short seller firm issued reports questioning our accounting practices, corporate disclosures and commitment to transparency in an apparent attempt to drive down the market price of our shares. These reports contained numerous false and misleading statements. Nevertheless, the price of our Class A and Class B shares declined significantly and has continued to trade at dramatically lower prices than before the reports. The market for our shares has been highly volatile since the publication of the reports. On January 8, 2024, the prices our Class A and Class B shares closed at €14.24 and €10.11, respectively. On March 7, 2024, the day following the publication of the third report by the short seller, the prices our Class A and Class B shares closed at €6.93 and €4.93, respectively.
Furthermore, following the publication of the reports, the Spanish National Securities Market Commission (Comisión Nacional del Mercado de Valores, or “CNMV”) opened an investigation in respect of the allegations made by the short seller firm and approached us with a number of inquiries, to which we have responded in a timely manner. On March 21, 2024, the CNMV issued its conclusions regarding its investigation, which confirmed that our financial statements and our indebtedness did not require a restatement. See “—CNMV Investigation’s Conclusions.” We have also been voluntarily providing information to and responding to questions posed by the SEC on an informal basis to provide clarifications. Our interactions with the SEC regarding the reports mentioned above are still ongoing. Up to the date of this annual report, the SEC has made two information requests to which we have timely responded.
We have been working to restore the confidence of markets, shareholders and other stakeholders by undertaking a number of actions, including:
| ● | Providing full cooperation and collaboration with the investigation launched by the Spanish National Securities Market Commission CNMV and the information requests from the SEC; |
| ● | Promptly communicating with all our stakeholders, sharing our clear response to the published reports by means of live conference calls and multiple official communications on our website, on the CNMV portal and on EDGAR. All press releases are publicly available on Grifols’ website (https://www.grifols.com/en/other-relevant-information); and |
| ● | We established a working group comprising the members of our Audit Committee, senior managers from the legal, communications, finance, investor relations and management teams, together with external advisors with expertise in communications. |
In addition, on January 26, 2024 we filed a complaint in the United States District Court for the Southern District of New York against the parties responsible for the false and misleading statements in the published reports to recover the financial and reputational damages caused to us and our stakeholders as a result of such statements.
CNMV’s Investigation Conclusions
As a result of the publication of certain reports by a short seller that included numerous false and misleading statements, more fully described in “—Short Seller Reports” above, the CNMV opened an investigation in respect of the allegations made by the short seller firm and approached us with a number of inquiries, to which we responded in a timely manner.
On March 21, 2024, the CNMV issued its conclusions regarding its investigation. Most importantly, the CNMV concluded that it did not identify any need to have Grifols restate its financial statements and that all analyzed related party transactions have been carried out on an arm’s length basis. The CNMV also concluded that it had found no evidence that the financial indebtedness of Grifols as reflected in its financial statements does not comport with the facts. These conclusions constitute a rejection of the claims made by the short seller firm on these points.
90
Specifically, the CNMV found reasonable and consistent with IFRS, Grifols’ accounting treatment of the following:
| ● | the consolidation of Haema AG and BPC Plasma Inc. in Grifols’ financial statements; |
| ● | the acquisition in 2021 of 25 plasma collection centers from BPL Plasma, Inc., which Grifols accounted for as a business acquisition; |
| ● | the consolidation of GDS as a controlled company as a result of the 2019 agreement with Shanghai RAAS; and |
| ● | the consolidation of Haema Plasma Kft as a controlled company since 2022. |
The CNMV did consider that the transaction with ImmunoTek (see Item 4 of this Part I, “Information on the Company—B. Business Overview—Raw Materials” and note 10 to the audited consolidated financial statements included in this annual report on page F-67) should have been accounted for as a joint arrangement pursuant to IFRS 11 instead of a financial investment. In addition, the CNMV identified the following areas for improvement in our periodic disclosures: (i) the level of detail of certain explanatory notes in our financial statements, (ii) the breakdown and level of detail of related party transactions, and (iii) the presentation of non-accounting financial performance indicators, in particular EBITDA and the debt/EBITDA ratio. At the request of the CNMV, on April 4, 2024, we published a note to the market providing additional information and explanations about these areas of improvement and other financial disclosures. Such note was furnished to the SEC as a current report on Form 6-K.
Changes in Board of Directors and Senior Management
In February 2024, the Board of Directors appointed Mr. José Ignacio (Nacho) Abia Buenache as a director, effective immediately, and the new Chief Executive Officer effective as of April 1, 2024. The Board appointed Nacho Abia as a director through co-option, which is a procedure available under Spanish corporate law under which when there is a vacancy in the board of directors, the board itself may temporarily appoint a director to fill the vacancy until the next general shareholders’ meeting. As such, Mr. Abia’s appointment as director is subject to ratification at our next general shareholders’ meeting.
Mr. Abia becoming our Chief Executive Officer, as well as the decision by Mr. Raimon Grifols Roura and Mr. Victor Grifols Deu to, following a transition period ending on May 31, 2024, step down from their current respective roles of Chief Corporate Officer and Chief Operating Officer, are part of a succession roadmap that the two resigning Grifols executives, along with the Board, set in motion back in 2022. See Item 6 of this Part I, “Directors, Senior Management and Employees—A. Directors and Senior Management.”
Acquisition of Collection Plasma Collection Centers From ImmunoTek
On April 2, 2024, through our subsidiary Grifols Bio North America, LLC, we acquired seven plasma collection centers located in the United States from ImmunoTek for $135.5 million. This transaction was carried out pursuant to the joint venture agreement entered into with ImmunotTek in 2021. See Item 4 of this Part I, “Information on the Company—B. Business Overview—Raw Materials” and Notes 10 and 32 to our audited consolidated financial statements included in this annual report on pages F-62 and F-126, respectively.
SRAAS Disposition
Pursuant to our Strategic Alliance and Share Purchase Agreement to sell our 20% equity stake in Shanghai RAAS to Haier, on February 29, 2024 the period contractually established by the parties in relation to the completion of Haier’s confirmatory due diligence has been satisfactorily concluded. Accordingly, the closing of the transaction is subject to obtaining pending ordinary regulatory approvals. In certain countries the corresponding ordinary regulatory approvals have already been received and we expect the transaction to close during the first half of 2024. See “—Factors Affecting our Financial Condition and Results of Operations—Recent Dispositions—Shanghai RAAS” and Note 12 to our audited consolidated financial statements included in this annual report on page F-70.
91
Issuance of New Senior Secured Notes by Means of a Private Placement
As of the date of this report we are actively pursuing the raising of new senior secured notes, the proceeds of which (if successful) will be used to refinance the 2017 Notes. If the transaction is successfully completed, we will promptly make further announcements to update the market.
Factors Affecting Our Financial Condition and Results of Operations
Consequences of COVID-19
Due to the COVID-19 pandemic, from March 2020 to December 2022, we experienced several impacts on our operational and financial performance. A lower level of plasma collection due to lockdowns and restricted movement, specifically the U.S. border closure with Mexico, quarantines and fear of disease, as well as the economic stimulus programs provided by the U.S. government, caused our facilities to operate with reduced capacity utilization and our plasma collection centers to offer higher donor compensation. This decrease in plasma collection levels resulted in a corresponding decline in processed volumes at our facilities, negatively affecting manufacturing cost absorption. Additionally, mainly as a result of higher compensation paid to donors, our cost per liter increased by 15% in 2020 as compared to 2019, and by 4% in 2021 as compared to 2020.
Our collection levels started to recover in the nine-month period ended December 31, 2021, and continued to improve in 2022, increasing by 25.0% in comparison to 2021 and surpassing 2019 levels. In 2023, our plasma supply increased by 10.0% in comparison to the volumes collected in 2022. This gradual recovery in plasma supply and cost per liter was largely due to our plasma collection expansion plan executed in 2021, including the acquisition of centers from BPL Plasma Inc and Kedplasma, LLC, and the easing of COVID-19 constraints, which allowed plasma donations to resume at the U.S.-Mexico border starting in September 2022. Likewise, our average cost per liter of plasma collected in 2023 declined by 15.2% in comparison to the average cost per liter in 2022.
For more details on the factors which contributed to the reduction in our cost per liter of plasma, see Item 4 of this Part I, “Information on the Company—B. Business Overview—Raw Materials. See also “—Operational Improvement Plan” and Item 3 of this Part I, “Key Information—D. Risk Factors—Risks Relating to the Company and Our Business—The COVID-19 global pandemic had, and could in the future cause, a material, adverse impact on us.”
Price Controls
Certain healthcare products, including plasma derivative products, are subject to price controls in many of the markets where they are sold, including Spain and other countries in the European Union. The existence of price controls over these products has adversely affected in the past, and may continue to adversely affect, our ability to maintain or increase our prices and gross margins.
Plasma Supply Constraints
Plasma is the key raw material used in the production of plasma-derived products. Our ability to continue to increase our revenue depends substantially on increased access to plasma. We currently obtain our plasma mainly from the United States and Europe (Germany, Austria and Hungary) primarily through our plasma collection centers and, to a much lesser extent, through agreements with third parties.
A continued increase in demand for plasma products could lead to industry supply constraints. In response, we and certain of our competitors and independent suppliers could open a number of new plasma collection centers.
As of December 31, 2023, we operated over 390 plasma collection centers located across the United States, Europe, across Germany, Austria and Hungary; Canada and Egypt. We have expanded our plasma collection network through a combination of organic growth, by opening new plasma collection centers, and acquisitions. In 2022, we acquired Biotest AG and with it, 28 new plasma collection centers. In 2021, we acquired 25 plasma collection centers from BPL Plasma Inc, seven U.S. plasma donation centers and one Canadian plasma center from Kedplasma, LLC, and entered into a joint venture agreement with ImmunoTek to develop 28 plasma collection centers in the United States. In 2020 and 2019, we acquired 37 plasma collection centers and ten blood donation centers as part of transactions involving Grifols Bio Supplies Inc (which succeeded the Interstate Bloodbank Group) and GC Pharma.
92
In addition, we are focused on optimizing our plasma collection center network by closing or consolidating underperforming centers, having closed 18 underperforming centers in 2022 and having closed or consolidated seven centers in 2023.
Recent Acquisitions
Saskatoon plasma center
On July 7, 2023, through our wholly-owned subsidiary Grifols Canada Plasma, Inc. (formerly known as Prometic Plasma Resources, Inc.), we acquired a plasma donation center in Saskatoon (Canada) from Canadian Plasma Resources Corporation for approximately €8.0 million. See Note 3(a) to our financial statements included to this annual report.
Biotest (U.K.), Ltd.
On June 1, 2023, our subsidiary Grifols U.K., Ltd., a company that focuses on the distribution and sale of therapeutic and other pharmaceutical products, especially haemoderivatives, acquired from another of our subsidiaries, Biotest AG, all shares of Biotest (U.K.) Ltd. for a total amount of €20.1 million. Effective as of November 1, 2023, Biotest (U.K., Ltd.) transferred its net assets to Grifols U.K., Ltd., resulting in an amalgamation. This transaction was meant to simplify our internal structure.
Access Biologicals, Inc.
On June 15, 2022, through our subsidiary Chiquito Acquisition Corp., we acquired 51% of the voting shares of Access Biologicals LLC, a company based in San Diego, California, for $142 million. We had acquired 49% of the voting shares of such company in January 2017 for $51 million. Access Biologicals LLC’ core business was the collection and manufacture of an extensive portfolio of biological products. Combined with a closed materials sourcing process, it provided support services for different markets such as in-vitro diagnostics, biopharmaceuticals, cell culture and diagnostic research and development. Effective as of April 1, 2023, Chiquito Acquisition Corp. and Access Biologicals LLC, as well as its subsidiaries Access Plasma, LLC and Access Cell Culture, LLC, merged with and into our subsidiary Grifols Bio Supplies Inc, which was the surviving entity of the merger.
Biotest AG Acquisition
On April 25, 2022, we acquired from TIIL 100% of the equity interests in Biotest Holdings, a German privately held stock corporation, and accepted an assignment from TIIL of certain shareholder loans granted by TIIL to Biotest Holdings. Biotest Holdings in turn owns 89.88% of the ordinary shares and 1.08% of the preferred equity shares of Biotest AG, a German stock corporation listed on the Frankfurt Stock Exchange. The purchase price for the acquisition of Biotest Holdings was €1,090,518,254, which included a loan receivable of €318 million granted by Biotest Holdings to Biotest AG. In addition, (1) we completed a voluntary tender offer to all remaining shareholders of Biotest AG to acquire their ordinary and preferred shares, where we acquired 1,250,298 ordinary shares (for €43.00 per share) and 8,340,577 preferred shares (for €37.00 per share) and (2) we acquired 185,359 ordinary shares in May 2022 following the exercise of a legal put right provided by applicable German corporate laws by former shareholders of Biotest AG. The acquisition of Biotest Holdings, the voluntary tender offer and the acquisition of shares pursuant to the put right have brought our interest in Biotest AG to 97.13% of the voting rights and 70.18% of the share capital.
In order to fund this acquisition, on October 5, 2021, our wholly-owned subsidiary Grifols Escrow Issuer, S.A.U. (the “Escrow Issuer”) issued €1,400.0 million senior notes that will mature on October 15, 2028 and bear interest at 3.875% per annum and $705.0 million senior notes that will mature on October 15, 2028 and bear interest at 4.750% per annum. Following the successful fulfilment of all applicable conditions precedent therefor, on June 27, 2023, but for accounting purposes effective as of January 1, 2023, the Escrow Issuer was merged with and into the Company, with the Company as the surviving entity and assuming all the obligations of the Escrow Issuer under the notes. See “—B. Liquidity and Capital Resources—Sources of Credit—The 2021 Notes.”
Biotest AG is a global company that specializes in innovative hematology and clinical immunology solutions. Headquartered in Dreieich (Germany), it develops, produces and markets biological medicinal products with applications in hematology, clinical immunology and intensive care. Biotest AG’s current portfolio includes 12 different products with a global commercial footprint in more than 90 countries, employing approximately 2,600 people around the world.
93
Acquisition of plasma donation centers from Kedplasma, LLC
On March 31, 2021, through our subsidiary Biomat USA, we acquired seven U.S.-based plasma donation centers from Kedplasma, LLC for a total purchase price of $55.2 million. The seven centers acquired are authorized by the FDA and European health authorities and collect approximately 240,000 liters of plasma per year.
GigaGen Acquisition
On March 8, 2021, through our subsidiary GIANT, we acquired 56% of shares of GigaGen Inc. (“GigaGen”), a pre-clinical biotherapeutics company, for $90.5 million. We had acquired 44% of GigaGen’s shares in July 2017 for $35 million. As part of this acquisition, in 2017 we entered into a research and collaboration agreement with GigaGen whereby carried out research activities to develop recombinant plyclonal immunoglobulin therapies derived from human B cells for the treatment of human diseases. Prior to the March 2021 acquisition, our stake in GigaGen was recorded using the equity method and, therefore, the difference between the fair value of the previous investment and the book value, estimated in $42 million, has been recognized as income. See Notes 3(a) and 10 to our audited consolidated financial statements included in this annual report on Form 20-F for more information regarding this transaction.
Acquisition of plasma donation centers from BPL Plasma, Inc.
On February 28, 2021, through our subsidiary Biomat USA, we acquired 25 U.S.-based plasma donation centers from BPL Plasma Inc for $385 million (such purchase price included the assumption by Biomat USA of $12 million in liabilities and $2.5 million in certain other transaction expenses). These plasma centers generate, in the aggregate, approximately 1 million liters of plasma per year. See Note 10 to our audited consolidated financial statements included in this annual report on Form 20-F for more information regarding this transaction.
Haema Plasma Kft.
On October 13, 2020, Scranton Plasma B.V., a subsidiary of one of our major shareholders Scranton Enterprises B.V., acquired 100% of the shares of Haema Plasma Kft. On February 1, 2021, we entered into a call option agreement with Scranton Plasma B.V. whereby we have the right to acquire the shares of Haema Plasma kft, exercisable until February 1, 2026. The price to exercise the call option is the amount equivalent to 13 times the EBITDA of Haema Plasma Kft, minus its net debt. Pursuant to IFRS 10, we fully consolidate Haema Plasma Kft as a Grifols subsidiary in our financial statements, as we retain control over such entity by means of the call option, potential voting rights, plasma supply agreement and management rights. Under the plasma supply agreement entered into with Haema Plasma Kft, we are entitled to acquire the vast majority of the plasma collected from the company’s seven plasma collection centers in Hungary. See Note 3(d) to our audited consolidated financial statements included in this annual report. See also Item 7 of this Part I, “Major Shareholders and Related Party Transactions—B. Related Party Transactions.”
Recent Dispositions
Shanghai RAAS
On December 29, 2023 we entered into a Strategic Alliance and Share Purchase Agreement with Haier for the sale of a 20% equity stake in Chinese company Shanghai RAAS in exchange for approximately $1.8 billion, while retaining a stake in Shanghai RAAS of 6.58%.
As part of the agreement with Haier, (1) we will retain a director on the board of directors of Shanghai RAAS; (2) we and Haier shall not transfer any of our respective shares in Shanghai RAAS for a period of three years following the closing of the transaction; and (3) we commit to (a) cause our subsidiary GDS, in which Shanghai RAAS owns 45% of the economic rights and 40% of the voting rights, to achieve an aggregate EBITDA of $850 million for the period from 2024 to 2028, provided that any deficit in such EBITDA performance would oblige us to indemnify Shanghai RAAS in an amount equivalent to a percentage of said deficit proportionate to the percentage of GDS’ capital stock held by Shanghai RAAS on such date, (b) cause GDS to distribute 50% of its distributable profit to its shareholders in the period between 2024 and 2028, and (c) assign the voting rights relating to our remaining 6.58% of Shanghai RAAS shares to Haier for a period of ten years from the payment of the purchase price by Haier. Based on the historical performance of GDS, we believe that GDS will achieve the EBITDA requirement and no indemnification will become due.
94
As part of the Strategic Alliance and Share Purchase Agreement, we will extend the term of our existing exclusive albumin distribution agreement with Shanghai RAAS through 2034, with the possibility for an additional ten-year extension, and guarantee minimum supply volumes thereunder through 2028. Upon successful closing of this transaction, which is anticipated to occur prior to the end of the second quarter of 2024, we will maintain and extend our presence in China, with our sights set on further developing the Chinese plasma industry and exploring new opportunities and synergies in the diagnostic sector, while using the proceeds of the sale to significantly reduce debt. The transaction is subject to customary regulatory approvals. See Notes 10, 12 and 29 to our audited consolidated financial statements included in this annual report on pages F-62, F-70 and F-104. See also “—Subsequent Events—Shanghai RAAS disposition.”
The Biomat Transactions
On December 1, 2021, we sold preferred shares representing 12.9% of Biomat Newco and 12.5% of Biomat USA, our U.S.-based plasma collection subsidiaries that are part of the Biomat Group, to the GIC Investor, an affiliate of GIC Private Limited, which is a sovereign wealth fund established by the Government of Singapore. The purchase price received was $990 million. We used the net proceeds from the Biomat Transactions to (i) prepay $600 million of principal amount of the Revolving Loans under the First Lien Credit Facilities, (ii) prepay $142,360,501.31 of the principal amount of the Dollar Tranche B Term Loans, (iii) prepay $88,003,617.48 of the Euro Tranche B Term Loans and (iv) repurchase €97,535,000 of the 2019 Notes under an asset sale offer. See “—B. Liquidity and Capital Resources—Sources of Credit.”
As a result of the transaction, the GIC Investor received ten class B common shares of Biomat USA and nine class B common shares of Biomat Newco. These common shares are non-voting but have the right to receive annual preferential dividends, to the extent dividends are declared, of $4,168,421.05 per share. These shares also granted the GIC Investor with redemption rights of up to one share per year beginning in 2023 at $52,105,263.16 per share. Further, the shares also carry a liquidation preference at the same share price as for redemption rights, plus all unpaid dividends. These rights would be enforceable by the GIC Investor in certain circumstances, such as in the case of a liquidation, dissolution or winding up of Biomat USA, if we cease to control or have at least a 75% voting interest in Biomat USA, or upon the exclusive licensing of all or substantially all intellectual property of Biomat USA. In addition, in the event the payment of dividends did not occur or such redemptions were not made, there would among other things be monetary penalties or holders of the shares could opt to exchange them for shares of Grifols, S.A.
As of December 31, 2023, the GIC Investor had exercised its right to redeem one class B common share of Biomat Newco, at the redemption price of $52,105,263.16, with such redemption being performed on June 30, 2023.
Subject to certain minority shareholder remedies in the charters of Biomat USA and Biomat Newco, we continue to oversee all aspects of the Biomat Group’s management and operations. All plasma collected by the Biomat Group will continue to be supplied to us for the production of plasma-derived medicines, through a long-term plasma supply agreement.
MedKeeper
In July 2022, our subsidiary Grifols Shared Services North America Inc. sold substantially all of the assets of the U.S. technology firm Geotech, LLC, doing business as Medkeeper (“MedKeeper”) to affiliates of the U.S. company Becton, Dickinson and Company for a total adjusted purchase price of $91.6 million. As a consequence of this divestment, we recognized an income of €23.1 million in our statement of profit and loss. We had acquired 51% of the equity interests of MedKeeper for $98 million in January 2018, and the remaining 49% in November 2020 for $60.2 million.
Operational Improvement Plan
In 2023, we implemented an operational improvement plan designed to reinforce our competitiveness and build a more streamlined, efficient and cost-effective global organization (the “Operational Improvement Plan”). The plan focused on three major areas: optimizing plasma costs and operations, streamlining corporate functions, and enhancing other efficiencies across the organization.
95
The first part of the plan, optimization of plasma costs and operations, improved our plasma procurement operations and enabled us to maintain desired plasma volumes while reducing the cost per liter of plasma through the following set of measures:
| ● | closure or consolidation of 18 underperforming plasma collection centers in 2022 and seven in 2023; |
| ● | reduction in compensation paid to plasma donors; |
| ● | enhanced productivity from our workforce, measured in plasma collection per full time employee; and |
| ● | installation of new and more efficient plasmapheresis equipment, which increases yield. |
The second part of the plan, streamlining corporate functions, caused a reduction in staff in 2023 of approximately 8.0% of our workforce (or approximately 2,000 employees), mainly in plasma operations in the United States, by means of initiatives such as:
| ● | centralizing and automating functions; |
| ● | more fully sharing services across business units; |
| ● | consolidating vendors; |
| ● | streamlining reporting structures; |
| ● | a labor force reduction plan (ERE) in Spain, which affected 51 employees; and |
| ● | eliminating duplicative functions and positions. |
The third part of the plan, enhancing other efficiencies across the organization, enabled us to reduce operational costs related to, among other things, global procurement, logistics, and facilities. Such reductions in operational costs required initiatives such as:
| ● | real estate rationalization affecting certain offices, but not industrial facilities; and |
| ● | establishing global organizations around commercial, industrial and supply chain functions. |
The Operational Improvement Plan increases our operating cash flow and improves our financial performance, which we believe would result in over €450 million in annualized cost savings in comparison to our overall cost base in 2022. Given the approximately nine-month inventory accounting lag applied in the plasma industry, a portion of these savings will be reflected in our income statement for the fiscal year ending on December 31, 2024. The Operational Improvement Plan resulted in a 32.0% rise in plasma collections per full-time employee, signaling improved labor productivity, and a 5.0% reduction in the manufacturing costs of our products in 2023, in each case as compared to 2022.
This is the first time we implemented such a plan. In accordance with IFRS accounting rules, the effects mentioned above are of a non-recurring nature as they relate to one-off, extraordinary measures. In 2022, we incurred costs of €36.1 million, mainly related to the closure of 18 plasma collection centers with the aim of optimizing our plasma center network. In 2023, we recorded a €159.3 million reorganization impact related to the Operational Improvement Plan. This charge relates mainly to severance payments, advisory fees, and other reorganization activities.
Other Factors
Our financial and operating prospects can also be significantly affected by a number of other internal and external factors, such as unfavorable changes in governmental regulation or interpretation, increased competition, the inability to hire or retain qualified personnel necessary to sustain planned growth, the loss of key senior managers, problems in developing some of the international operations and lack of sufficient capital, among others.
96
Operating Results
Overview
The subsequent discussion and analysis provide information that our management believes is relevant to an assessment and understanding of our consolidated results of operations. You are encouraged to read the following discussion and analysis of our financial condition and results of operations together with our audited consolidated financial statements and the related notes included elsewhere in this annual report on Form 20-F.
Year ended December 31, 2023, as compared to the year ended December 31, 2022:
| Year Ended December 31, |
| Change |
| |||||
Consolidated Statement of Profit and Loss | 2023 |
| 2022 |
| € |
| % |
| |
| (in thousands of euros, except for percentages) | ||||||||
Continuing Operations |
|
|
|
|
|
|
|
| |
Net revenue |
| 6,591,977 |
| 6,063,967 |
| 528,010 |
| 8.7 | % |
Cost of sales |
| (4,097,406) |
| (3,832,437) |
| (264,969) |
| 6.9 | % |
Gross margin |
| 2,494,571 |
| 2,231,530 |
| 263,041 |
| 11.8 | % |
Research and development |
| (395,282) |
| (361,140) |
| (34,142) |
| 9.5 | % |
Selling, general and administration expenses |
| (1,366,673) |
| (1,190,423) |
| (176,250) |
| 14.8 | % |
Operating expenses |
| (1,761,955) |
| (1,551,563) |
| (210,392) |
| 13.6 | % |
Other Income |
| 3,042 |
| 22,235 |
| (19,193) |
| (86.3) | % |
Profit/(loss) of equity accounted investees with similar activity to that of the Group |
| 63,740 |
| 103,478 |
| (39,738) |
| (38.4) | % |
Operating result |
| 799,398 |
| 805,680 |
| (6,282) |
| (0.8) | % |
Finance income |
| 62,326 |
| 33,859 |
| 28,467 |
| 84.1 | % |
Finance costs |
| (596,864) |
| (478,323) |
| (118,541) |
| 24.8 | % |
Sales of assets at amortized costs |
| (24,993) |
| (18,201) |
| (6,792) |
| 37.3 | % |
Change in fair value of financial instruments |
| 1,459 |
| 11,999 |
| (10,540) |
| (87.8) | % |
Exchange differences |
| (16,386) |
| 7,725 |
| (24,111) |
| (312.1) | % |
Finance result |
| (574,458) |
| (442,941) |
| (131,517) |
| 29.7 | % |
Profit/(loss) of equity accounted investees |
| (922) |
| (1,482) |
| 560 |
| (37.8) | % |
Profit before income tax from continuing operations |
| 224,018 |
| 361,257 |
| (137,239) |
| (38.0) | % |
Income tax expense |
| (43,349) |
| (90,111) |
| 46,762 |
| (51.9) | % |
Profit after income tax from continuing operations |
| 180,669 |
| 271,146 |
| (90,477) |
| (33.4) | % |
Consolidated profit for the year |
| 180,669 |
| 271,146 |
| (90,477) |
| (33.4) | % |
Net Revenue
Net revenue is calculated by subtracting certain chargebacks, cash discounts, volume rebates, Medicare and Medicaid discounts and other discounts from our gross revenue, which is mainly generated by the sale of goods. See Note 24 to our audited consolidated financial statements included in this annual report on Form 20-F.
Our business units reported a record performance in 2023 in terms of total consolidated net revenue. Our net revenue increased by 8.7% (10.9% at constant currency), or €528.0 million, in 2023, reaching €6.6 billion compared to €6.1 billion in 2022. Positive market dynamics, including strong demand for our key proteins, in conjunction with the recovery of our plasma collection volumes were critical to these results, with the increase in total net revenue being largely due to the performance of our Biopharma business unit, as explained below.
97
The following table reflects a summary of net revenue by each of our business units for 2023, as compared to 2022:
| Year ended |
|
| Year ended |
|
|
|
| |||||
| December 31, |
| % of total |
| December 31, |
| % of total |
|
|
| |||
Summary of Net Revenue by business unit |
| 2023 |
| net revenue |
| 2022 |
| net revenue |
| % var |
| % var CC(1) |
|
(in thousands of euros, except for percentages) |
| ||||||||||||
Biopharma | 5,558,301 | 84.3 | % | 5,005,382 | 82.5 | % | 11.1 | % | 13.3 | % | |||
Diagnostic |
| 670,269 |
| 10.2 | % | 671,292 |
| 11.1 | % | (0.2) | % | 2.3 | % |
Bio Supplies |
| 159,957 |
| 2.4 | % | 146,076 |
| 2.4 | % | 9.5 | % | 11.3 | % |
Others |
| 203,450 |
| 3.1 | % | 250,165 |
| 4.0 | % | (18.7) | % | (17.8) | % |
Intersegments |
| — |
| — |
| (8,948) |
| (0.1) | % | (100.0) | % | (100.0) | % |
Total |
| 6,591,977 |
| 100.0 | % | 6,063,967 |
| 100.0 | % | 8.7 | % | 10.9 | % |
| (1) | Net revenue variance in constant currency is determined by comparing adjusted current period net revenue, calculated using prior period monthly average exchange rates, to the prior period net revenue. See “Presentation of Financial and Other Information—Constant Currency.” |
Biopharma. Net revenue for the Biopharma business unit increased by 11.1% (13.3% at constant currency) from €5.0 billion in 2022 to €5.6 billion in 2023. This increase was mainly due to solid performance of key proteins driven by higher plasma supply, robust underlying demand, strong results from sales outside the United States and Canada and a favorable pricing environment and product mix. In 2023, there was an increase in sales of immunoglobulins (one of our main proteins representing approximately 55% of the Biopharma business units’ revenue), the net revenue of which grew by 14.9% (17.2% at constant currency) fueled by strong demand for intravenous immunoglobulin (IVIG), the launch of the subcutaneous immunoglobulin (SCIG) Xembify® in several European countries and Australia, as well as its growth in key markets such as the United States, which caused a 40.0% increase in sales of such product.
In 2023, net revenue from our global albumin sales increased by 17.6% as compared to 2022, mainly as a result of a 40.0% growth in China fueled by higher demand and price increases. Net revenue from sales of Alpha-1 decreased by 2.1% in 2023 as compared to 2022. However, a gradual recovery of alpha-1 sales in European countries led to a growth of 2.4% in the fourth quarter of 2023 on a year-on-year analysis in relation to the same period of 2022. Other commercial milestones factoring into the results of our Biopharma business unit included the commercial expansion of Tavlesse and Vistaseal.
Diagnostic. The Diagnostic business unit’s net revenue remained stable with a 0.1% decrease (a 2.3% increase at constant currency) from €671.3 million in 2022 to €670.2 million in 2023. The performance of this business unit was driven primarily by a notable growth of 6.3% (8.9% at constant currency) in net revenue derived from the sale of blood typing solutions in the main countries we sell to, including the United States, Argentina, Brazil, Spain and Saudi Arabia.
Net revenue from sales of our NAT blood and plasma screening solutions decreased by 2.6% (a 0.4% increase at constant currency). The result from sales of our NAT technology was negatively impacted by pricing concessions in the context of extending a significant contract with a key customer. This negative impact was partially offset by strong revenue from sales to the Asia-Pacific countries and progress in the U.S. tissue and organ testing markets. Successful tender wins in key regions in a partnership with the Australian Red Cross also contributed to offset the abovementioned negative impact.
Revenue from recombinant proteins increased by 1.2% (2.3% at constant currency), driven by demand in the main regions we sell to, especially the United States, and by a ten-year supply agreement with a key partner. Other important achievements fueling the growth of this business unit in 2023 were (1) the U.S. launch of AlphaID™ At Home, an innovative solution to detect alpha-1 deficiency, a genetic disease whose symptomology is similar to COPD and (2) the launch of our CD38 solution, the first soluble recombinant protein to facilitate pre-transfusion compatibility testing in patients with multiple myeloma.
Bio Supplies. Net revenue from Bio Supplies increased by 9.5% (11.3% at constant currency), from €146.1 million in 2022 to €160.0 million in 2023, mainly due to the continued integration of our subsidiary Access Biologicals (acquired in 2022), which expanded the product portfolio of this business unit, and by sales of hyperimmune plasma to third parties.
98
Others. Net revenue from Others decreased by 18.7% from €250.2 in 2022 to €203.5 million in 2023 (though at constant currency this represented a 4.0% increase). This decrease was due to the sale of certain companies and assets formerly part of our hospital division, a business unit that we shut down in 2022.
The following table reflects a summary of net revenue by each of our geographic regions for 2023 as compared to 2022:
| Year ended |
|
| Year ended |
|
|
|
| |||||
December 31, | % of total | December 31, | % of total | ||||||||||
Summary of Net Revenue by Region | 2023 | net revenue | 2022 | net revenue | % var | % var CC(1) | |||||||
(in thousands of euros, except for percentages) |
| ||||||||||||
European Union(2) | 1,255,927 | 19.1 | % | 1,032,210 | 17.0 | % | 21.7 | % | 21.7 | % | |||
United States and Canada |
| 3,898,961 |
| 59.1 | % | 3,855,607 |
| 63.6 | % | 1.1 | % | 3.4 | % |
Rest of the World |
| 1,437,089 |
| 21.8 | % | 1,176,150 |
| 19.4 | % | 22.2 | % | 25.9 | % |
Total |
| 6,591,977 |
| 100.0 | % | 6,063,967 |
| 100.0 | % | 8.7 | % | 10.9 | % |
| (1) | Net revenue variance in constant currency is determined by comparing adjusted current period net revenue, calculated using prior period monthly average exchange rates, to the prior period net revenue. See “Presentation of Financial and Other Information—Constant Currency.” |
| (2) | Net revenue earned in the European Union includes net revenue earned in Spain. |
Net revenue in the United States and Canada had a slight increase of 1.1% (3.4% at constant currency) from €3.85 billion in 2022 to €3.9 billion in 2023. This 1.1% increase represents a lower growth rate than that of the overall market for our products in the region, and is mainly due to our focus on increasing presence in other markets, combined with the loss of certain accounts in the United States in 2022 caused by temporary competitive pressures and plasma supply constraints due to the COVID-19 pandemic. With the improvement of our plasma supply and decrease in cost per liter of plasma collected, we believe we will recover such U.S. accounts in 2024. Meanwhile, net revenue in the European Union increased by 21.7% from €1.0 billion in 2022 to €1.3 billion in 2023, mainly due to a significant growth in Xembify sales and new commercial approvals that have enabled increased sales of different product mixes in the region. Net revenue in the Rest of the World increased by 22.2% (25.9% at constant currency) from €1.2 billion in 2022 to €1.4 billion in 2023, mainly due to favorable price increases globally.
Cost of sales
Cost of sales increased by 6.9% from €3.8 billion in 2022 to €4.1 billion in 2023. Cost of sales as a percentage of net revenue decreased to 62.2% in 2023 compared to 63.2% in 2022. This was mainly due to the reduction in the cost per liter of plasma collected and the reductions in our overall cost base enabled by the Operational Improvement Plan. See “—Factors Affecting our Financial Condition and Results of Operations—Operational Improvement Plan” and Item 4 of this Part I, “Information on the Company—Business Overview— Raw Materials.”
Gross Margin
The increase in gross margin from 36.8% of net revenue in 2022 to 37.8% in 2023 was mainly due to strong revenue growth and lower cost per liter of plasma as a result of (1) the easing of COVID-19 constraints, which allowed plasma donations to resume at the U.S.-Mexico border starting in September 2022, in turn resulting in an increase in donors and a 10.0% increase in our plasma supply in 2023, and (2) the cost-saving initiatives under the Operational Improvement Plan. See “—Operational Improvement Plan” and Item 4 of this Part I, “Information on the Company—Business Overview—Raw Materials.” Our 2023 income statement has started to reflect the benefits from the drop in cost per liter following its all-time high in July 2022, taking into account the approximate nine-month lag in inventory accounting in the plasma industry. The reduction in cost per liter led to a higher gross margin in the second half of 2023.
99
Research and development
Research and development spending increased by 9.5%, from to €361.1 million in 2022 to €395.3 million in 2023. These results underscore the integration of Biotest AG’s projects into our portfolio, as well as our ongoing efforts to integrate and develop cutting-edge projects such as those of Alkahest and GigaGen. See Item 4 of this Part I, “Information on the Company—B. Business Overview—Research and Development” for additional details.
Selling, general and administration expenses
Selling, general and administration expenses increased by 14.8% from €1.2 billion in 2022 to €1.4 billion in 2023. A significant part of such increases was driven by reorganization costs composed mainly of severance payments and fees paid to consultants and advisors associated with our Operational Improvement Plan. Additionally, we had some remaining cost increases in connection with the completion of the integration of Biotest AG and general inflationary pressures.
Finance result
Finance result in 2023 represented a loss of €574.5 million in 2023, compared to a loss of €442.9 million in 2022. The increase mainly results from the effect of the market increases in interest rates on our variable rate obligations under the Euro and Dollar Tranche B Term Loan under our First Lien Credit Facilities.
Income tax expense
In 2023, we had a profit before income tax of €224.0 million and income tax expense of €43.3 million, which represents a tax rate of 19.4%. Our effective tax rate decreased from 24.9% in 2022 primarily due to a change in the country mix of our taxable income.
Year ended December 31, 2022, as compared to the year ended December 31, 2021:
Year Ended December 31, | Change |
| |||||||
Consolidated Statement of Profit and Loss |
| 2022 |
| 2021 |
| € |
| % |
|
(in thousands of euros, except for percentages) |
| ||||||||
Continuing Operations |
|
|
|
|
|
|
|
| |
Net revenue |
| 6,063,967 |
| 4,933,118 |
| 1,130,849 |
| 22.9 | % |
Cost of sales |
| (3,832,437) |
| (2,970,522) |
| (861,915) |
| 29.0 | % |
Gross margin |
| 2,231,530 |
| 1,962,596 |
| 268,934 |
| 13.7 | % |
Research and development |
| (361,140) |
| (354,881) |
| (6,259) |
| 1.8 | % |
Selling, general and administration expenses |
| (1,190,423) |
| (1,061,508) |
| (128,915) |
| 12.1 | % |
Operating expenses |
| (1,551,563) |
| (1,416,389) |
| (135,174) |
| 9.5 | % |
Other Income |
| 22,235 |
| 16,302 |
| 5,933 |
| 36.4 | % |
Profit/(loss) of equity accounted investees with similar activity to that of the Group |
| 103,478 |
| 32,555 |
| 70,923 |
| 217.9 | % |
Operating result |
| 805,680 |
| 595,064 |
| 210,616 |
| 35.4 | % |
Finance income |
| 33,859 |
| 11,551 |
| 22,308 |
| 193.1 | % |
Finance costs |
| (478,323) |
| (267,702) | 210,621 | (78.7) | % | ||
Sales of assets at amortized costs | (18,201) | (10,292) | (7,909) | 76.8 | % | ||||
Change in fair value of financial instruments |
| 11,999 |
| 246 |
| 11,753 |
| 4,777.6 | % |
Exchange differences |
| 7,725 |
| (11,602) |
| 19,327 |
| 166.6 | % |
Finance result |
| (442,941) |
| (277,799) |
| (165,142) |
| 59.4 | % |
Profit/(loss) of equity accounted investees |
| (1,482) |
| 33,188 |
| (34,670) |
| (104.5) | % |
Profit before income tax from continuing operations |
| 361,257 |
| 350,453 |
| 10,804 |
| 3.1 | % |
Income tax expense |
| (90,111) |
| (85,126) |
| (4,985) |
| 5.9 | % |
Profit after income tax from continuing operations |
| 271,146 |
| 265,327 |
| 5,819 |
| 2.2 | % |
Consolidated profit for the year |
| 271,146 |
| 265,327 |
| 5,819 |
| 2.2 | % |
100
Net Revenue
Net revenue increased by 22.9% (12.4% in constant currency), or €1.1 billion, in 2022 reaching €6.1 billion compared to €4.9 billion in 2021. The increase was largely due to the performance of our Biopharma business unit, as explained below.
The following table reflects a summary of net revenue by each of our business units for 2022, as compared to 2021:
| Year ended |
|
| Year ended |
|
|
|
| |||||
December 31, | % of total | December 31, | % of total |
| |||||||||
Summary of Net Revenue by business unit | 2022 | net revenue | 2021(1) | net revenue | % var | % var CC(2) |
| ||||||
(in thousands of euros, except for percentages) |
| ||||||||||||
Biopharma | 5,005,382 | 82.5 | % | 3,814,983 | 77.3 | % | 31.2 | % | 19.6 | % | |||
Diagnostic |
| 671,292 |
| 11.1 | % | 779,108 |
| 15.8 | % | (13.8) | % | (19.7) | % |
Bio Supplies |
| 146,076 |
| 2.4 | % | 115,811 |
| 2.3 | % | 26.1 | % | 13.2 | % |
Others |
| 250,165 |
| 4.1 | % | 266,461 |
| 5.4 | % | (6.1) | % | (11.5) | % |
Intersegments |
| (8,948) |
| (0.1) | % | (43,245) |
| (0.8) | % | (79.3) | % | 80.7 | % |
Total |
| 6,063,967 |
| 100.0 | % | 4,933,118 |
| 100.0 | % | 22.9 | % | 12.4 | % |
(1) | As a consequence of the change in transactions and balances allocations by segments discussed in Item 4 of this Part I “A. Business Overview—Principal Activities—Other Activities and Operations (“Others”),” the comparative figures for the fiscal year 2021 have been adjusted accordingly. |
(2) | Net revenue variance in constant currency is determined by comparing adjusted current period net revenue, calculated using prior period monthly average exchange rates, to the prior period net revenue. See “Presentation of Financial and Other Information—Constant Currency.” |
Biopharma. Net revenue for the Biopharma business unit increased by 31.2% (19.6% at constant currency) from €3.8 billion in 2021 to €5.0 billion in 2022. This increase was mainly due to increased plasma collections, robust underlying demand for key proteins and price increases. Favorable product mix was also a notable driver with subcutaneous immunoglobulin (SCIG) Xembify® sales increasing by 33.7%, supported by higher demand and favorable customer mix, as well as Albutein® FlexBag® gaining traction after its launch in November 2021.
Diagnostic. The Diagnostic business unit’s net revenue decreased by 13.8% (19.7% at constant currency) from €779.1 million in 2021 to €671.3 million in 2022. This decrease was primarily due to non-recurring sales of TMA molecular tests, used to detect SARS-CoV-2, and the termination of mandatory Zika-virus testing, partially offset by blood typing solutions’ double-digit-growth across most geographies.
Bio Supplies. Net revenue from Bio Supplies increased by 26.1% (13.2% at constant currency), from €115.8 million in 2021 to €146.1 million in 2022, mainly driven by the acquisition of the remaining 51% capital of Access Biologicals, which positively impacted performance of Bio Supplies Biopharma’s cell culture media and plasma for diagnostics.
Others. Net revenue from Others decreased by 6.1% (11.5% at constant currency) from €266.5 million in 2021 to €250.2 in 2022. This decrease is due to the sale of substantially all of the assets of MedKeeper in July 2022 and lower third-party plasma sales.
The following table reflects a summary of net revenue by each of our geographic regions for 2022 as compared to 2021:
Year ended | Year ended |
| |||||||||||
December 31, | % of total net | December 31, | % of total |
| |||||||||
Summary of Net Revenue by Region |
| 2022 |
| revenue |
| 2021 |
| net revenue |
| % var |
| % var CC(1) |
|
(in thousands of euros, except for percentages) |
| ||||||||||||
European Union(2) |
| 1,032,210 |
| 17.0 | % | 906,449 |
| 18.4 | % | 13.9 | % | 13.5 | % |
United States and Canada |
| 3,855,607 |
| 63.6 | % | 3,154,549 |
| 63.9 | % | 22.2 | % | 8.5 | % |
Rest of the World |
| 1,176,150 |
| 19.4 | % | 872,120 |
| 17.7 | % | 34.9 | % | 25.6 | % |
Total |
| 6,063,967 |
| 100.0 | % | 4,933,118 |
| 100.0 | % | 22.9 | % | 12.4 | % |
(1) | Net revenue variance in constant currency is determined by comparing adjusted current period net revenue, calculated using prior period monthly average exchange rates, to the prior period net revenue. See “Presentation of Financial and Other Information—Constant Currency.” |
(2) | Net revenue earned in the European Union includes net revenue earned in Spain. |
101
Net revenue in the U.S. and Canada increased by 22.2% (8.5% at constant currency) from €3.1 billion in 2021 to €3.9 billion in 2022, mainly due to higher volume sales of IG as a result of higher plasma supply and strong demand, coupled with price increases. Meanwhile, sales in the European Union increased by 13.9% (13.5% at constant currency) from 906.4 million in 2021 to €1.0 billion in 2022, mainly due to higher plasma supply and strong demand, led by growth in countries like Spain, France, Italy and Germany. Sales in the Rest of the World increased by 34.9% (25.6% at constant currency) from 872.1 million in 2021 to €1.2 billion in 2022, mainly due to the strong performance of our products in China.
Cost of sales
Cost of sales increased by 29.0% from €3.0 billion in 2021 to €3.8 billion in 2022. Cost of sales as a percentage of net revenue increased to 63.2% in 2022 compared to 60.2% in 2021. This was mainly due to the high cost per liter of plasma collected in the first half of 2022 in view of increased donor compensation and labor costs. See “—Factors Affecting Our Financial Condition and Results of Operations—Consequences of COVID-19,” and Item 4 of this Part I, “Information on the Company—B. Business Overview—Raw Materials.”
Gross Margin
The decrease in gross margin from 39.8% of net revenue in 2021 to 36.8% in 2022 was mainly due to a double impact from the Biopharma and Diagnostic business units. On the one hand, we had high cost per liter of plasma collected in 2021 and the first half of 2022 due to inventory accounting (c.9-months lag). This was primarily due to elevated donor commitment compensation (“DCC”) and inflationary pressures on labor costs. On the other hand, the decrease in COVID-19 one-off testing and Zika screening adversely impacted our gross margin by 2.1% in 2022 compared to 2021.
A drop in DCC by 20.0% in the fourth fiscal quarter of 2022 in comparison to its peak in July drove a reduction of 10.0% in the cost per liter of plasma over the same reference period. Other plasma operating costs, accounting approximately for the remaining 65.0% of the full plasma cost, also declined, albeit to a lesser extent, amid the current macroeconomic backdrop.
Research and development
Research and development spending increased by 1.8%, from €354.9 million (7.2% of net revenue) in 2021 to €361.1 million (6.0% of net revenue) in 2022. These results underscore the integration of Biotest AG’s projects into our portfolio, as well as our ongoing efforts to integrate and develop cutting-edge projects such as those of Alkahest and GigaGen. See Item 4 of this Part I, “Information on the Company—B. Business Overview—Research and Development” for additional details.
Selling, general and administration expenses
Selling, general and administration expenses increased by 12.1% from €1.1 billion in 2021 to €1.2 billion in 2022, mainly as a result of the integration of Biotest AG and general inflationary pressures, partially offsetting cost savings.
Finance result
Finance result in 2022 represented a loss of €442.9 million, compared to a loss of €277.8 million in 2021. The increase mainly results from the increase of interest rates, the funds received from the GIC Investor and the issuance of the 2021 Notes (as defined herein) to finance the Biotest AG acquisition, in the amounts of €1,400,000,000 and $705,000,000, in the fourth quarter of 2021. See “—B. Liquidity and Capital Resources—Sources of Credit—The 2021 Notes.”
Income tax expense
In 2022, we had a profit before income tax of €361.3 million and income tax expense of €90.1 million, which represents a tax rate of 24.9%. Our effective tax rate increased from 24.3% in 2021 primarily due to a change in the country mix of our taxable income.
102
Regulation
For detailed information regarding the regulations applicable to our business, see Item 4 of this Part I, “Information on the Company—E. Regulatory Matters.”
Inflation and Foreign Currency Fluctuations
We historically have not been affected materially by inflation in our core geographies. However, due to the current macroeconomic context, we are having some inflation pressures on labor costs and selling, general & administrative costs. See “—Operating Results—Overview” for additional details.
For detailed information on how foreign currency fluctuations affect our business, see “—B. Liquidity and Capital Resources.” See also Item 3 of this Part I, “Key Information—Risks Relating to the Company and Our Business—Our results of operations and financial condition may be affected by adverse changes in foreign currency exchange rates, especially a significant shift in the value of the euro as compared to the U.S. dollar” and Item 11 of this Part I, “Quantitative and Qualitative Disclosures About Market Risk—Currency Risk.”
B. | Liquidity and Capital Resources |
Our principal liquidity and capital requirements consist of costs and expenses relating to:
| ● | the operation of our business (see “—Operating Results,” “—Liquidity and Capital Resources—Net Cash from Operating Activities” and “—Working Capital” for a description and quantification of costs and expenses relating to our operations); |
| ● | capital expenditures for existing and new operations (see “—Capital Expenditures” for a description and quantification of our capital expenditures, including capital expenditures on other intangible assets and rights of use additions, incurred in each of the years ended December 31, 2023, 2022 and 2021; |
| ● | the purchase price of acquisitions (see “—Recent Developments” and “—Factors Affecting Our Financial Condition and Results of Operations—Acquisitions” for a description of our most recent acquisitions); and |
| ● | debt service requirements relating to our existing and future debt (see “—Sources of Credit” for a description and quantification of our principal indebtedness). |
Historically, we have financed our liquidity and capital requirements through internally generated cash flows and debt financings. As of December 31, 2023, our cash and cash equivalents totaled €529.6 million. In addition, as of December 31, 2023, we had a liquidity position of €1,145 million, including €615.3 million in unused credit facilities available under our debt agreements that include €544.7 million available as Revolving Loans under our First Lien Credit Facilities.
We expect our cash flows from operations combined with our cash balances and availability under the Revolving Loans from the First Lien Credit Facilities to provide sufficient liquidity to fund our current obligations (primarily debt service and acquisition payments as described above), projected working capital requirements and capital expenditures for at least the next twelve months. Currently, we do not generate significant cash in any country that might have restrictions for funds repatriation, and we estimate that the existing cash located in Ireland, Spain and the United States, along with the cash generated from operations, will be sufficient to meet future cash needs in key countries.
We are committed to deleveraging in the medium term and maintaining elevated and adequate levels of liquidity through (i) internally generated cash flows, and (ii) a substantial decrease in dividend payments in the medium term. In furtherance of this commitment, we entered into an agreement to sell a 20% equity stake in Shanghai RAAS in exchange for approximately $1.8 billion, and expect to use the proceeds of this transaction to significantly reduce debt. See “—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Dispositions—Shanghai RAAS.” We also do not envision any material acquisitions in the medium term.
103
Our capital expenditures consist primarily of expanding and enhancing our production facilities, replacing fully depreciated items and promoting efficiency of our operations. In addition, we allocate cumulative industrial capital investments to expand the manufacturing capacities of the Biopharma business unit, as well as investments in the Diagnostic and Bio Supplies business units, with the goal of improving the structure of our plasma collection centers in the United States and expanding our manufacturing facilities. We are also expanding and relocating plasma donation centers and improving infrastructures related to raw materials classification, preparation and storage facilities, logistics centers and analysis laboratories.
Our principal existing contractual obligations as of December 31, 2023 are comprised of financial debt obligations, with principal and interest amortization for short- and long-term debt including, among other things, capitalized lease obligations and bilateral credit facilities bearing interest at market rate. In addition, on June 28, 2022 and on October 5, 2021, we entered into four cross-currency interest rate swaps of $705 million to hedge part of the Euro equivalent value of the 2021 Notes (see “—Sources of Credit” above and Note 30 to our audited consolidated financial statements included in this annual report on Form 20-F for further discussion regarding our debt obligations and related interest rate agreements outstanding at December 31, 2023). We have contractual obligations involving future payments for licenses and royalties based generally on volume of sales.
Historical Cash Flows
The table below presents our net cash from operating, investing and financing activities for each of the years ended December 31, 2023, 2022 and 2021.
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in thousands of euros) | ||||||
Net cash from operating activities |
| 208,283 |
| (10,867) |
| 596,975 |
Net cash (used in) investing activities |
| (397,636) |
| (1,978,823) |
| (854,149) |
Net cash from/(used in) financing activities |
| 186,045 |
| (173,493) |
| 2,297,679 |
Net Cash from Operating Activities
In the year ended December 31, 2023, our net cash from operating activities was €208.3 million, due largely to improved operational performance, a reduction in cost per liter of plasma collected and a reduced cost base throughout our business fueled by our Operational Improvement Plan. See “—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Operational Improvement Plan” and Item 4 of this Part I, “B. Business Overview—Raw Materials.” Working capital represented a loss of €369.6 million. The principal effects on working capital were as follows:
| ● | increase of €53.1 million in trade and other receivables. The average collection period remained stable at 36 days (36 days in 2022); |
| ● | increase of €427.1 million in inventory levels primarily due to increased plasma supply, partially offset by a lower cost per liter of plasma. Inventory turnover was 308 days at December 31, 2023, compared with 296 days reported at December 31, 2022; and |
| ● | increase of €103.3 million in trade and other payables. The average payment period increased from 53 days at December 31, 2022 to 59 days at December 31, 2023. |
In the year ended December 31, 2022, our net cash from operating activities represented a loss of €10.9 million due largely to an increase in inventory levels resulting from a higher cost per liter of plasma collected and upsurge in plasma donations. This context also adversely affected our working capital, which represented a loss of €609.2 million. The principal effects on working capital were as follows:
| ● | increase of €80.2 million in trade and other receivables. The average collection period increased to 36 days (32 days in 2021); |
104
| ● | increase of €600.2 million in inventory levels primarily due to a higher cost per liter of plasma collected and higher donation volumes. Inventory turnover was 296 days at December 31, 2022, compared with 278 days reported at December 31, 2021; and |
| ● | increase of €80.2 million in trade and other payables. The average payment period decreased from 64 days at December 31, 2021 to 53 days at December 31, 2022. |
In the year ended December 31, 2021, we generated net cash from operating activities of €597.0 million. The principal effects on working capital were as follows:
| ● | increase of €16.8 million in trade and other receivables. The average collection period increased to 32 days (27 days in 2020); |
| ● | increase of €157.5 million in inventory levels primarily due to an increase in period-to-period plasma volume collections through strategic acquisitions during 2021 and to the increase in the cost of plasma. Inventory turnover was 278 days at December 31, 2021, compared with 237 days reported at December 31, 2020; and |
| ● | increase of €40.4 million in trade payables. The average payment period increased from 62 days at December 31, 2020 to 64 days at December 31, 2021. |
Net Cash from/(Used) in Investing Activities
Net cash used in investing activities amounted to €397.6 million in 2023, €1,978.8 million in 2022 and €854.1 million in 2021.
Investments made in 2023 focused primarily on capital expenditures, particularly on the Biopharma business unit’s new production facilities, including investments in plasma fractionation, immunoglobulin purification and albumin plants in Montreal (Canada), a new sterile albumin purification dosing and filling plant in Dublin, and various IT and digitalization-related projects. See”—Capital Expenditures.”
Investments made in 2022 included the Biotest AG acquisition for a total of €1.4 billion, as well as execution of the call option to acquire the remaining 51% of Access Biologicals’ share capital for a total of $142 million, and €375.5 million allocated to property, plant and equipment and intangible assets. Capital expenditures focused mainly on new Biopharma manufacturing facilities, including a new albumin plant in Dublin, the upgrade of the Montreal plasma fractionation, immunoglobulin purification and albumin plants, as well as several IT and digitalization-related projects. See “—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Acquisitions;” and “—Capital Expenditures, Other Intangible Assets and Rights of Use.”
Investments made in 2021 included the acquisition of 25 plasma collection centers from BPL for $370 million, seven plasma collection centers from Kedplasma, LLC for $55.2 million, the first payment related to the acquisition of GigaGen for an amount of €38.2 million and the remaining payment for the Alkahest acquisition of $126 million. In addition to the aforementioned acquisitions, we invested €315.1 million in property, plant and equipment and other intangibles in 2021. See “—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Acquisitions.”
105
Net Cash from/(Used in) Financing Activities
Net cash from financing activities was €186.0 million in 2023, primarily due to a drawdown of €360.2 million from our Revolving Credit Facility (see Note 21(b) to our audited consolidated financial statements included in this annual report on Form 20-F), partially offset primarily by lease payments of €105.9 million (see Note 8(b) to our audited consolidated financial statements included in this annual report on Form 20-F), the redemption by the GIC Investor of one preferred share of Biomat Newco for €47.9 million and the repayment of €31.9 million of the EIB Term Loans.
Net cash used in financing activities was €173.5 million in 2022, primarily as a result of net debt repayments.
Net cash from financing activities was €2,297.7 million in 2021, primarily as a result of the issuance of the 2021 Notes to finance the Biotest AG acquisition, in the amounts of €1,400,000,000 and $705,000,000 (see “—B. Liquidity and Capital Resources—Sources of Credit—The 2021 Notes.”), and dividend payouts of €259 million. On the other hand, sale of investments includes the Biomat Transactions, under which we received a $990 million investment in our subsidiary Biomat USA. See “—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Dispositions.”
Working Capital
Our working capital, which is driven primarily by our trade receivables turnover and inventory aging, can vary significantly from period to period depending on the activity. Our capital requirements will depend on many factors, including our rate of sales growth, acceptance of our products, continued access to adequate manufacturing capacities, maintaining cGMP compliant facilities, the timing and extent of research and development activities, and changes in operating expenses, including costs of production and sourcing of plasma, all of which are subject to uncertainty. We anticipate that our cash needs will be significant and that we may need to increase our borrowings under current or future debt agreements in order to fund our operations and strategic initiatives. We anticipate that our working capital will increase in absolute terms in order to grow our business.
Inventory Aging
Inventory aging average increased from 2022 to 2023, primarily as a result of the progressive impact of the improved cost per liter of plasma in a context of increased supply. Inventory aging increased to 308 days at December 31, 2023, compared to 296 days at December 31, 2022. Inventory aging increased from 2021 to 2022, primarily as a result of the higher cost per liter of plasma collected and higher plasma donation volumes. Inventory aging increased to 296 days at December 31, 2022, compared to 278 days at December 31, 2021.
See Item 4 of this Part I, “Information on the Company—B. Business Overview—Raw Materials” for additional details.
Trade Receivables
We routinely sell receivables with maturity dates no shorter than 30 days (“Eligible Receivables”) to financial institutions (factors) in varied contractual arrangements with or without recourse. In sales of Eligible Receivables without recourse, all material risks and benefits inherent to the ownership of the assigned receivables, including the right to unilaterally transfer the assigned receivables to unrelated third parties, are transferred to the factor. These sales are considered as factoring without recourse and therefore the consideration paid to us by factors for such assigned receivables is not accounted as debt in our balance sheet.
In the fiscal years ended December 31, 2023, 2022 and 2021, we sold without recourse €2,858.1 million, €3,174.3 million and €2,975.3 million, respectively, of receivables to third parties. We estimate the volume of invoices we sold without recourse to financial institutions which, based on their due date, would not have been collected at December 31, 2023, to be €391.9 million (€445.2 million and €317.1 million at December 31, 2022 and 2021, respectively).
We also sell Eligible Receivables to financial institutions while retaining the risks and benefits inherent to the ownership thereof. These sales are considered as factoring with recourse and the amount of such assigned receivables remains on our balance sheet, while the amount advanced to us by the factors is recognized on our balance sheet as short-term debt. At December 31, 2023, 2022 and 2021, we had €17.0 million, €16.5 million and €23.5 million, respectively, recorded in our balance sheet as short-term debt in respect of factoring transactions with recourse.
106
For the fiscal year ended December 31, 2023, the finance cost we recorded in our statement of profit and loss in respect of receivables sold totaled €25.0 million (€18.2 million and €10.3 million in the fiscal years ended December 31, 2022 and 2021, respectively). Our receivables had an aging average of 36, 36 and 32 days at December 31, 2023, 2022 and 2021, respectively. See notes 14 and 27 of our consolidated audited financial statements included in this annual report on pages F-72 and F-99 respectively.
Capital Expenditures, Other Intangible Assets and Rights of Use
In 2023, we advanced our capital investment plan to expand and improve the production facilities of our business units. In line with our Operational Improvement Plan, we optimized capital expenditure resource allocations considering the significant investments already made from 2018 to 2022. In 2023, our capital expenditures totaled €281.5 million (€291.7 million in 2022). The following table presents our capital expenditure, other intangible assets and rights of use additions in the years ended December 31, 2023, 2022 and 2021, by business unit.
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021(1) | |
(in thousands of euros) | ||||||
Biopharma |
| 458,216 |
| 402,672 |
| 349,890 |
Diagnostic |
| 29,107 |
| 49,890 |
| 19,991 |
Bio Supplies |
| 9,066 |
| 98 |
| 13,836 |
Others |
| 3,884 |
| 30,192 |
| 28,597 |
Unallocated |
| 48,618 |
| 59,866 |
| 55,380 |
Total |
| 548,891 |
| 542,718 |
| 467,694 |
(1) | As a consequence of the change in transactions and balances allocations by segments discussed in Item 4 of this Part I “A. Business Overview—Principal Activities—Other Activities and Operations (“Others”),” the comparative figures for the fiscal year 2021 have been adjusted accordingly. |
January 2021 through December 2023
Facilities. The most important capital projects relating to the expansion and improvement of our manufacturing facilities during 2023, 2022 and 2021 were:
Parets site (Barcelona, Spain):
| ● | investments to increase purification capacity of fibrin sealant and topic thrombin of €3.7 million in 2022 and €1.9 million in 2021; |
| ● | investments to increase Factor VIII manufacturing capacity of €0.1 million in 2022 and €1.0 million in 2021; |
| ● | investments to increase the production of intravenous solutions bags of €0.5 million in 2023, €1.4 million in 2022 and €3.4 million in 2021; |
| ● | investments to improve the manufacturing lines of the production of intravenous solutions in glass bottles in the Parets facility of €0.1 million in 2022 and €1.3 million in 2021; and |
| ● | investments of €0.5 million in 2022 and €4.2 million in 2021, to build the extension of the existing Grifols International building and 2-8ºC chamber for 2,000 pallets. |
Clayton site (North Carolina, United States):
| ● | investments for the construction of a new immunoglobulins’ purification and filling plant of €11.4 million in 2023, €20.7 million in 2022 and €23.3 million in 2021; |
| ● | investments for the construction of a new 6-million-liter fractionation plant of €1.0 million in 2023, €32,176 in 2022 and €2.4 million in 2021,; |
107
| ● | investments to expand packaging incubators of €0.1 million in 2022 and €2.0 million in 2021; |
| ● | investments for the construction of a plasma logistic center of €1.7 million in 2021 to increase the overall plasma storage capacity; and |
| ● | expansion of Grifols’ current waste water pretreatment plant in Clayton to meet Town of Clayton permit limits, with investments of €4.3 million in 2023, €12.4 million in 2022 and €11.1 million in 2021. |
Los Angeles (California, United States):
| ● | investments to increase our albumin purification capacity and including a new presentation in ready-to-use flexible bags of €0.5 million in 2022 and €1.5 million in 2021; and |
| ● | investments to increase Factor VIII manufacturing capacity of €0.3 million in 2021. |
Dublin (Ireland):
| ● | investments to build a new headquarters, global operations and logistics center to serve as part of the new global operations center of the Biopharma business unit of €0.8 million in 2023, €2.0 million in 2022 and €0.8 million in 2021; and |
| ● | investment in a new albumin purification and filling plant for bags of €20.5 million in 2023, €17.6 million in 2022 and €33.0 million in 2021. |
Quebec (Canada):
| ● | investments of €44.8 million in 2023, €56.9 million in 2022 and €11.3 million in 2021 to remodel Canada facility for fractionation increase, Albumin manufacturing and Gamunex addition. |
San Diego (California, United States):
| ● | investments of €0.2 million in 2023, €0.3 million in 2022, and €0.5 million in 2021 to expand manufacturing capacity for our NAT Diagnostic business, including quality control, research and development labs and an R&D pilot plant; and |
| ● | investments of €4.7 million in 2023, €6.8 million in 2022 and €3.0 million in 2021 to build a new immunohematology manufacturing facility in building 10895. |
Emeryville (California, United States):
| ● | investments of €0.1 million in 2023, €0.3 million in 2022 and €4.4 million in 2021, for the new protein manufacturing process and scale up labs based on mammalian cell cultures. |
Other Investments. Other relevant capital projects relating to the expansion and improvement of our manufacturing facilities during 2023, 2022 and 2021 were:
| ● | investments in serialization to enhance manufacturing and packaging identification of €0.5 million in 2023, €1.3 million in 2022 and €1.2 million in 2021; |
| ● | investments in new donor centers and donor center expansions in the United States of €4.6 million in 2023, €23.8 million in 2022 and €38.6 million in 2021; |
108
| ● | investments of €0.4 million in 2023, €3.8 million in 2022 and €8.1 million in 2021 to expand our overall lab testing capacity; |
| ● | investments for a new data center building in Los Angeles to support all IT services and to address current risks with the existing data center of €0.2 million in 2023, €0.9 million in 2022 and €1.7 million in 2021; |
| ● | investments of €1.7 million in 2021 to increase our plastic manufacturing capacity and create vertical integration for the group with synergies between the Biopharma business unit and Healthcare Solutions line; |
| ● | investments of €3.8 million in 2022 and €3.0 million in 2021 for the acquisition of a new property of 79,180 square meters adjacent to our Barcelona manufacturing facilities to expand our industrial and research capabilities, adding to our current workforce in the region by more than 3,500 employees; |
| ● | investments to remodel our commercial offices worldwide of €1.9 million in 2021, including new offices in Beijing, Singapore, Chile, Marseille, Mexico, Tokyo, Czech Republic, Shanghai and a new warehouse in the U.K.; |
| ● | investments to update manufacturing facilities to EMA regulation related to the manufacturing of sterile medicinal products of €2.3 million in 2023, €5.1 million in 2022 and €4.6 million in 2021; and |
| ● | investments to increase our IVIG purification capacity of €1.0 million in 2023, €0.8 million in 2022 and €1.2 million in 2021. |
January 2024 through December 2025
In 2023, we completed a €1.4 billion investment plan initiated in 2018 that involved, among other investments, cumulative industrial capital investments to expand the manufacturing capacities of the Biopharma business unit, as well as investments in the Diagnostic business unit and Healthcare Solutions business line. We are currently focusing our capital expenditures on the maintenance of our facilities while we formulate a large-scale strategy for 2024 and 2025.
The majority of our capital investments benefit our Biopharma business unit, systemically enhancing our manufacturing facilities. We aim to optimize utilization of our fractionation capacity by obtaining FDA and EMA licenses and completing other requirements to purify any of our intermediate products at any of our plants. We are also relocating and renovating plasma donation centers and improving infrastructures related to raw materials classification, preparation and storage facilities, logistics centers and analysis laboratories. See “—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Operational Improvement Plan.”
The most important planned capital projects relating to the improvement of our manufacturing facilities are:
| ● | Montreal: continue upgrading facilities for plasma fractionation and purification; |
| ● | Bilbao: construction of a new building for single molecule counting near Progenika; |
| ● | Clayton: continue the investment to upgrade the waste treatment plant, investment in the initial Phase II of the purification and filling facility and expansion of the IV Fibrinogen capacity; |
| ● | San Diego: rebuilding of a laboratory, offices and warehouse, and continue the expansion of the blood testing and immunohematology systems; |
| ● | Dreieich: investments in several sectors to be made by Biotest AG, as well as in plasma collection centers; and |
| ● | Egypt: construction of a new manufacturing plant (phase I Plasma Logistic Center), including a new plasma laboratory and a finished product and raw material warehouse. |
109
Sources of Credit
European Investment Bank Term Loans
On October 28, 2015, GWWO entered into a loan agreement with the European Investment Bank for a term loan of €100 million under the European Fund for Strategic Investments (the “2015 EIB Term Loan”), which was amended on December 5, 2017, April 15, 2019, and on November 15, 2019. The financial terms of the loan agreement include a fixed interest rate of 2.40% for a tenor of ten years from October 28, 2015, and a repayment schedule with amortization in years three through ten. The proceeds of this loan are being used to support our research and development, primarily focusing on the search for new indications for plasmatic proteins, including the treatment of Alzheimer’s disease, vascular disease, cardiovascular surgery and arterial thrombosis, amongst others.
On December 5, 2017, Grifols obtained a new long-term loan with the European Investment Bank totaling €85 million (the “2017 EIB Term Loan”), which was amended on April 15, 2019 and on November 15, 2019. The financial terms of the loan include a fixed interest rate of 2.019% for a tenor of ten years and a two-year grace period before any payment of principal becomes due and payable. The proceeds of this loan are being used for research and development initiatives, notably the discovery and development of new products (plasma proteins), the finding of new therapeutic indications for existing plasma proteins and the improvement of manufacturing processes to increase yields, safety and efficiency.
On September 7, 2018, Grifols obtained a new long-term loan with the European Investment Bank totaling €85 million (the “2018 EIB Term Loan” and, together with the 2015 EIB Term Loan and the 2017 EIB Term Loan, the “EIB Term Loans”), which was amended on April 15, 2019 and on November 15, 2019. The financial terms of the loan agreement include a fixed interest rate of 2.145% for a tenor of 10 years and a two-year grace period before any payment of principal becomes due and payable. The proceeds of this loan are being used for research and development initiatives, notably the discovery of new therapeutic indications for plasma-derived protein therapies.
The EIB Term Loans are guaranteed by the same entities that guarantee the First Lien Credit Facilities described below and are secured by a perfected first priority security interest (subject to permitted liens, as defined in the documentation governing the EIB Term Loans) on the same collateral securing the First Lien Credit Facilities and the 2019 Notes, each as described below (noting that the blood plasma inventory of GWWO located in Spain is not charged to secure the 2019 Notes), subject to a customary pari passu intercreditor agreement entered into by and among Grifols, GWWO, certain subsidiaries of Grifols party thereto, the European Investment Bank, Bank of America, N.A., as collateral agent under the First Lien Credit Facilities and The Bank of New York Mellon, London branch, as collateral agent under the 2019 Notes.
We entered into an amendment to the EIB Term Loans on August 6, 2021 to permit (i) the consummation of the Biomat Transactions; and (ii) upon the consummation of the Biomat Transactions, the release of Biomat USA and Talecris from their respective guarantees provided under the corresponding guarantee agreement for the EIB Term Loans and that release the liens granted over the assets of Biomat USA and Talecris. The Biomat Transactions were consummated on December 1, 2021.
On September 28, 2022, we entered into Amended and Restated Accession Agreements in connection with the EIB Term Loans to add Biotest Holdings as guarantor thereunder.
As of December 31, 2023, we had €116.9 million in aggregate principal amount outstanding of EIB Term Loans.
110
First Lien Credit Facilities
On November 15, 2019, we entered into a Credit and Guaranty Agreement (the “First Lien Credit Facilities”) with a syndicate led by Bank of America Europe Designated Activity Company (formerly known as Bank of America Merrill Lynch International Limited Designated Activity Company), Bank of America, N.A., BNP Paribas S.A., Sucursal en España, HSBC France, Banco Bilbao Vizcaya Argentaria S.A., and JP Morgan Securities PLC, as the arrangers (the “Arranging Banks”), which consist of the “Term Loans” and the “Revolving Loans.” The initial Term Loans (consisting of a Dollar Tranche B Term Loan and a Euro Tranche B Term Loan) were fully drawn down on November 15, 2019. Both the Dollar Tranche B Term Loan (in original principal amount equal to $2,500,000,000) and the Euro Tranche B Term Loan (in original principal amount equal to €1,360,000,000) mature eight years from November 15, 2019 and have a repayment schedule with quarterly amortization starting on the last business day of the fiscal quarter ending on March 31, 2020, equal to 0.25% of the aggregate principal amount of the initial Dollar Tranche B Term Loan (or Euro Tranche B Term Loan, as the case may be) outstanding on November 15, 2019, with the remainder payable at maturity.
The Revolving Loans, which initially provided for a commitment of $500,000,000, are available during the period commencing from November 15, 2019 and ending on the sixth anniversary of November 15, 2019. On May 7, 2020, we signed an upsize to the Revolving Loans to increase the lender commitments thereunder from $500,000,000 to $1,000,000,000 with the existing and new revolving lenders. The terms and conditions of which are similar to those entered into on November 15, 2019. As part of the upsize, the applicable margin for Revolving Loans was increased from 0.50% to 1.50% in the case of Base Rate Loans and from 1.50% to 2.50% in the case of Eurocurrency Rate Loans. Additionally, the commitment fee payable in respect of the unused Revolving Commitments was increased from 0.50% to 0.875%. The purpose of the upsize of the Revolving Loans was to reinforce our liquidity position.
The borrower under the revolving facility is GWWO, an Irish entity and our wholly owned direct subsidiary. The borrower under the Euro denominated tranche B facility is Grifols. The borrower under the Dollar-denominated tranche B facility is Grifols Worldwide Operations USA, Inc. (“Grifols Worldwide Operations USA”), a Delaware corporation and a direct wholly owned subsidiary of GWWO. The First Lien Credit Facilities are governed by New York law; however, certain collateral documents are governed under the local law of other jurisdictions.
The interest rates on the Revolving Loans are either (a) the base rate (i.e., the greatest among (i) the prime rate, (ii) the federal funds rate plus 0.50% and (iii) the Term Secured Overnight Funding Rate (“Term SOFR”), with a one-month interest period, plus 1.00%) plus 1.50% or (b) Term SOFR (if denominated in dollars) or EURIBOR (if denominated in Euros) plus 2.50%. The interest rate on the Dollar Tranche B Term Loan is either (a) the base rate plus 1.00% or (b) Term SOFR plus 2.00%. The interest rate on the Euro Tranche B Term Loan is EURIBOR plus 2.25%.
Borrowings under the First Lien Credit Facilities are subject to mandatory prepayment upon the occurrence of certain events, including the incurrence of certain debt and the sale or other disposition of certain assets. In addition, a portion of the borrowings under the First Lien Credit Facilities are subject to mandatory prepayment in the event we have excess cash flow, as defined therein. Both the Term Loans and the Revolving Loans are guaranteed by Grifols (solely in respect of the obligations of Grifols Worldwide Operations USA and GWWO) and certain subsidiaries of Grifols that together with Grifols represented, as of December 31, 2022, in aggregate, at least 60% of the earnings before interest, tax, depreciation and amortization of Grifols and its subsidiaries (calculated in accordance with the formula set forth in the First Lien Credit Facilities, the “Guarantor Coverage Test”), and are secured by a perfected first priority security interest (subject to permitted liens, as described in the First Lien Credit Facilities) in all of the tangible and intangible assets of the U.S. credit parties and plasma inventory of GWWO and pledges of equity of certain subsidiaries of Grifols (subject to certain exclusions and limitations). As of the date of this annual report on Form 20-F, the First Lien Credit Facilities are guaranteed by our subsidiaries Grifols Biologicals LLC, Grifols Shared Services North America, Inc., Grifols Therapeutics LLC, Instituto Grifols, S.A., Grifols International S.A., Grifols USA, LLC, and Grifols Biotest Holdings GmbH and the co-borrowers (and guarantors) GWWO and Grifols Worldwide Operations USA.
The First Lien Credit Facilities include customary affirmative and negative covenants and events of default. Negative covenants include, among other limitations, limitations on additional debt, liens, asset sales and affiliate transactions. Events of defaults include, among other events, violation of covenants, material breaches of representations, cross default to other material debt, bankruptcy and insolvency and material judgments.
111
The terms of the First Lien Credit Facilities contain limitations on our ability to pay ordinary dividends. We may pay dividends (a) in the ordinary course of business consistent with our dividend policy in an amount not to exceed in respect of any fiscal year, 40% of the consolidated net income of Grifols and its subsidiaries for such fiscal year, which may be paid in installments, the first, no earlier than December of such fiscal year and the last, no later than the following fiscal year or (b) whether or not in the ordinary course of business so long as after giving effect thereto, the leverage ratio is not greater than 3.75x. We may make regularly scheduled payments of interest in respect of the 2017 Notes and the Senior Refinancing Notes (as defined in the First Lien Credit Facilities) to the extent required by the terms of the indenture governing the 2017 Notes or the Senior Refinancing Notes Documents (as defined in the First Lien Credit Facilities), as the case may be.
The First Lien Credit Facilities and related security documents were amended on August 13, 2021 to (i) permit the consummation of the Biomat Transactions, (ii) reduce the Guarantor Coverage Test to 60%, and (iii) upon the consummation of the Biomat Transactions, release Biomat USA and Talecris from their respective guarantees provided under the First Lien Credit Facilities and release the liens granted over the assets of Biomat USA and Talecris.The Biomat Transactions were consummated on December 1, 2021, and we used part of the net proceeds therefrom to (i) prepay $600 million of principal amount of the Revolving Loans under the First Lien Credit Facilities, (ii) prepay $142,360,501.31 of the Dollar Tranche B Term Loans and (iii) prepay the Euro equivalent of $88,003,617.48 of the Euro Tranche B Term Loans.
On April 21, 2022 and April 25, 2022, we entered into Counterpart Agreements in connection with the First Lien Credit Facilities to add, respectively, the Escrow Issuer (which has since been merged with and into the Company) and Biotest Holdings as guarantors thereunder. On September 28, 2022, we amended and restated the Counterpart Agreement entered into on April 25, 2022 by Biotest Holdings to amend and restate the provisions relating to the guaranty limitations for German guarantors to account for Biotest Holdings conversion from a stock corporation (Aktiengesellschaft) to a limited liability company (Gesellschaft mit beschränkter Haftung).
The First Lien Credit Facilities were amended on May 3, 2023, to provide for the replacement of LIBOR as a reference interest rate by SOFR.
As of December 31, 2023, we had €1,255.3 million and $2,307.9 million in aggregate principal amount outstanding of Term Loans, and no amounts outstanding under our Revolving Loans.
The 2017 Notes
On April 26, 2017, Grifols issued €1.0 billion aggregate principal amount of senior unsecured notes that will mature on May 1, 2025 and bear interest at 3.20% per annum (the “2017 Notes”). On May 2, 2017, the 2017 Notes were listed on the Global Exchange Market of the Irish Stock Exchange.
The 2017 Notes pay interest semi-annually in arrears on May 1 and November 1, commencing on November 1, 2017. The 2017 Notes are currently guaranteed on a senior unsecured basis by Grifols and the subsidiaries of Grifols that are guarantors and co-borrowers under the First Lien Credit Facilities.
We may redeem the 2017 Notes, in whole or in part, at a redemption price of 100.000% of the principal amount plus accrued and unpaid interest, if any, on the 2017 Notes redeemed, to the applicable redemption date. We are not required to make mandatory redemption or sinking fund payments with respect to the 2017 Notes.
If we experience a change of control, we must give holders of the 2017 Notes the opportunity to sell to us their 2017 Notes at 101% of their principal amount, plus accrued and unpaid interest.
Grifols and the guarantors of the 2017 Notes may incur additional indebtedness if the fixed charge coverage ratio (as defined in the indenture governing the 2017 Notes) for Grifols and the restricted subsidiaries (as defined in the indenture governing the 2017 Notes) on a consolidated basis for the most recently ended four full fiscal quarters immediately preceding the date on which such additional indebtedness is incurred would have been at least 2.00 to 1.00, determined on a pro forma basis.
112
The indenture governing the 2017 Notes contains certain covenants limiting, subject to exceptions, carve-outs and qualifications, Grifols’ ability and its restricted subsidiaries’ ability to: (i) pay dividends or make certain other restricted payments or investments; (ii) incur additional indebtedness or provide guarantees of indebtedness and issue disqualified stock; (iii) create liens on assets; (iv) merge, consolidate, or sell all or substantially all of our and our restricted subsidiaries’ assets; (v) enter into certain transactions with affiliates; (vi) create restrictions on dividends or other payments by our restricted subsidiaries; and (vii) create guarantees of indebtedness by restricted subsidiaries. The indenture also contains certain customary events of default.
On August 6, 2021, we entered into an indenture supplement amending the indenture governing the 2017 Notes to (i) permit the consummation of the Biomat Transactions, and (ii) upon the consummation of the Biomat Transactions, release Biomat USA and Talecris from their guarantees provided under the indenture governing the 2017 Notes. The Biomat Transactions were consummated on December 1, 2021. On April 21 and April 25, 2022, we entered into indenture supplements amending the indenture governing the 2017 Notes to add, respectively, the Escrow Issuer (which has since merged with and into the Company) and Biotest Holdings as guarantors for the 2017 Notes. On July 21, 2023, we entered into an indenture supplement amending the indenture governing the 2017 Notes to acknowledge that the Escrow Issuer has been merged with and into the Company, thereby releasing the guarantee granted by the Escrow Issuer.
The 2019 Notes
On November 15, 2019 Grifols issued €905.0 million senior secured notes that will mature on February 15, 2025 and bear interest at 1.625% per annum (the “1.625% Notes”) and €770.0 million senior secured notes that will mature on November 15, 2027 and bear interest at 2.250% per annum (the “2.250% Notes” and together with the 1.625% Notes, the “2019 Notes”).
The 2019 Notes are currently guaranteed on a senior secured basis by the wholly-owned subsidiaries of Grifols that are guarantors and co-borrowers under the First Lien Credit Facilities. Subject to permitted liens, all obligations under the 2019 Notes, and the guarantees of those obligations, are secured on a first-priority basis by the tangible and intangible assets of the domestic guarantors, the blood plasma inventory of GWWO (with the exception of blood plasma inventory located in Spain) and pledges of equity of certain subsidiaries of Grifols (subject to certain exclusions and limitations). The collateral securing the 2019 Notes also secures the First Lien Credit Facilities and the EIB Term Loans, subject to the Intercreditor Agreement.
We are not required to make mandatory redemption or sinking fund payments with respect to the 2019 Notes.
If we experience a change of control, we must give holders of the 2019 Notes the opportunity to sell to us their 2019 Notes at 101% of their principal amount, plus accrued and unpaid interest.
Grifols and the guarantors of the 2019 Notes may incur additional indebtedness if the fixed charge coverage ratio (as defined in the indenture governing the 2019 Notes) for Grifols and the restricted subsidiaries (as defined in the indenture governing the 2019 Notes) on a consolidated basis for the most recently ended four full fiscal quarters immediately preceding the date on which such additional indebtedness is incurred would have been at least 2.00 to 1.00, determined on a pro forma basis. In addition, Grifols and the guarantors of the 2019 Notes may incur additional secured indebtedness if the secured leverage ratio (as defined in the indenture governing the 2019 Notes) for Grifols and the restricted subsidiaries (as defined in the indenture governing the 2019 Notes) on a consolidated basis for the most recently ended four full fiscal quarters immediately preceding the date on which such additional indebtedness is incurred would not exceed 4.50 to 1.00, determined on a pro forma basis.
The indenture governing the 2019 Notes contains certain covenants limiting, subject to exceptions, carve-outs and qualifications, Grifols’ ability and its restricted subsidiaries’ ability to: (i) pay dividends or make certain other restricted payments or investments; (ii) incur additional indebtedness or provide guarantees of indebtedness and issue disqualified stock; (iii) create liens on assets; (iv) merge, consolidate, or sell all or substantially all of our and our restricted subsidiaries’ assets; (v) enter into certain transactions with affiliates; (vi) create restrictions on dividends or other payments by our restricted subsidiaries; and (vii) create guarantees of indebtedness by restricted subsidiaries. The indenture also contains certain customary events of default.
On November 15, 2019 the 2019 Notes were listed on the Global Exchange Market of the Irish Stock Exchange.
113
On August 6, 2021, we entered into an indenture supplement amending the indenture governing the 2019 Notes to (i) permit the consummation of the Biomat Transactions, and (ii) upon the consummation of the Biomat Transactions, release Biomat USA and Talecris from their guarantees and collateral provided under the indenture governing the 2019 Notes. The Biomat Transactions were consummated on December 1, 2021, and, on January 11, 2022, we repurchased an aggregate principal amount of €97,535,000 (€67,144,000 of 1.625% Notes and €30,391,000 of 2.250% Notes) of the 2019 Notes under an asset sale offer to bondholders.
On April 21 and April 25, 2022, we entered into indenture supplements amending the indenture governing the 2019 Notes to add, respectively, the Escrow Issuer (which has since merged with and into the Company) and Biotest Holdings as guarantors for the 2019 Notes. On July 21, 2023, we entered into an indenture supplement amending the indenture governing the 2019 Notes to acknowledge that the Escrow Issuer has been merged with and into the Company, thereby releasing the guarantee granted by the Escrow Issuer.
A. The 1.625% Notes
The 1.625% Notes pay interest semi-annually in arrears on February 15 and August 15, commencing on February 15, 2020. Grifols may redeem the 1.625% Notes, in whole or in part, at the redemption prices (expressed as percentages of principal amount) set forth below plus accrued and unpaid interest, if any, on the 1.625% Notes redeemed, to the applicable redemption date (subject to the right of the holders of the 1.625% Notes on the relevant record date to receive interest due on the relevant interest payment date), if redeemed during the twelve-month period beginning on February 15 of the years indicated below:
Year |
| Percentage |
|
2024 and thereafter |
| 100.000 | % |
B. The 2.250% Notes
The 2.250% Notes pay interest semi-annually in arrears on May 15 and November 15, commencing on May 15, 2020. Grifols may redeem the 2.250% Notes, in whole or in part, at the redemption prices (expressed as percentages of principal amount) set forth below plus accrued and unpaid interest, if any, on the 2.250% Notes redeemed, to the applicable redemption date (subject to the right of the holders of the 2.250% Notes on the relevant record date to receive interest due on the relevant interest payment date), if redeemed during the twelve-month period beginning on November 15 of the years indicated below:
Year |
| Percentage |
|
2023 and thereafter |
| 100.5625 | % |
2024 and thereafter |
| 100.000 | % |
The 2021 Notes
On October 5, 2021, the Escrow Issuer, a newly formed wholly owned subsidiary of Grifols that did not hold or otherwise have any interest in any material assets, issued €1,400,000,000 senior notes that will mature on October 15, 2028 and bear interest at 3.875% per annum (the “Euro notes”) and $705,000,000 senior notes that will mature on October 15, 2028 and will bear interest at 4.750% per annum (the “Dollar notes” and together with the Euro notes, the “2021 Notes”). The 2021 Notes were issued to fund the acquisition of Biotest Holdings (and indirectly Biotest AG) and a voluntary tender offer for the shares in Biotest AG not owned by Biotest Holdings. See “—A. Factors Affecting Our Financial Condition and Results of Operations—Acquisitions—Biotest AG Acquisition.”
On the date of issuance of the 2021 Notes, the gross proceeds from the offering were deposited into segregated escrow accounts for the benefit of the holders of the 2021 Notes, to be released upon the fulfillment of the conditions precedent to the Biotest AG acquisition. Such conditions were fulfilled on April 21, 2022, and the funds were released to the Escrow Issuer Following the successful fulfilment of all conditions precedent therefor. Effective as of June 27, 2023, the Escrow Issuer was merged with and into the Company, the results of such merger being that the Company is the surviving entity, assuming (by operation of law) all assets and obligations of the Escrow Issuer, and the Escrow Issuer ceased to exist (the “Escrow Issuer Merger”).
114
Prior to the Escrow Issuer Merger, the 2021 Notes remained general unsecured obligations of the Escrow Issuer unconditionally guaranteed on a senior unsecured basis by the Company and each of our wholly-owned subsidiaries that that are guarantors and co-borrowers under the First Lien Credit Facilities. From and after the Escrow Issuer Merger, the 2021 Notes became general unsecured obligations of the Company and are unconditionally guaranteed on a senior unsecured basis by our wholly-owned subsidiaries that are guarantors and co-borrowers under the First Lien Credit Facilities.
We are not required to make mandatory redemption or sinking fund payments with respect to the 2021 Notes.
If we experience a change of control, we must give holders of the 2021 Notes the opportunity to sell to us their 2021 Notes at 101% of their principal amount, plus accrued and unpaid interest.
Grifols and the guarantors of the 2021 Notes may incur additional indebtedness if the fixed charge coverage ratio (as defined in the indenture governing the 2021 Notes) for Grifols and the restricted subsidiaries (as defined in the indenture governing the 2021 Notes) on a consolidated basis for the most recently ended four full fiscal quarters immediately preceding the date on which such additional indebtedness is incurred would have been at least 2.00 to 1.00, determined on a pro forma basis.
The indenture governing the 2021 Notes contains certain covenants limiting, subject to exceptions, carve-outs and qualifications, Grifols’ ability and its restricted subsidiaries’ ability to: (i) pay dividends or make certain other restricted payments or investments; (ii) incur additional indebtedness or provide guarantees of indebtedness and issue disqualified stock; (iii) create liens on assets; (iv) merge, consolidate, or sell all or substantially all of our and our restricted subsidiaries’ assets; (v) enter into certain transactions with affiliates; (vi) create restrictions on dividends or other payments by our restricted subsidiaries; and (vii) create guarantees of indebtedness by restricted subsidiaries. The indenture also contains certain customary events of default.
On October 11, 2021 the 2021 Notes were listed on the Global Exchange Market of the Irish Stock Exchange.
On September 28, 2022, we entered into an indenture supplement amending the indenture governing the 2021 Notes to add Biotest Holdings as guarantor for the 2021 Notes. On July 21, 2023, we entered into an indenture supplement amending the indenture governing the 2021 Notes to acknowledge that the Escrow Issuer has been merged with and into the Company, and that the company became the issuer of the 2021 Notes, assuming all obligations previously held by the Escrow Issuer.
A. The Dollar notes
The Dollar notes accrue interest at the rate of 4.750% per annum pay interest semi-annually in arrears on April 15 and October 15, commencing on April 15, 2022. We may redeem the Dollar notes, in whole or in part, at any time on and after October 15, 2024 at the redemption prices (expressed as percentages of principal amount) set forth below plus accrued and unpaid interest, if any, on the Dollar notes redeemed, to the applicable redemption date (subject to the right of holders of the Dollar notes on the relevant record date to receive interest due on the relevant interest payment date), if redeemed during the twelve-month period beginning on October 15 of the years indicated below:
Fiscal Year |
| Percentage |
|
2024 |
| 102.375 | % |
2025 |
| 101.188 | % |
2026 and thereafter |
| 100.000 | % |
Prior to October 15, 2024, we may redeem up to 40% of the aggregate principal amount of the Dollar notes at a redemption price equal to 104.750% of the principal amount thereof with an amount equal to or less than the net cash proceeds that we raise in one or more equity offerings, plus accrued and unpaid interest on such notes, if any, to, but not including, the redemption date.
115
B. The Euro notes.
The Euro notes accrue interest at the rate of 3.875% per annum pay interest semi-annually in arrears on April 15 and October 15, commencing on April 15, 2022. We may redeem the Euro notes, in whole or in part, at any time on and after October 15, 2024 at the redemption prices (expressed as percentages of principal amount) set forth below plus accrued and unpaid interest, if any, on the Euro notes redeemed, to the applicable redemption date (subject to the right of holders of the Euro notes on the relevant record date to receive interest due on the relevant interest payment date), if redeemed during the twelve-month period beginning on October 15 of the years indicated below:
Fiscal Year |
| Percentage |
|
2024 |
| 101.938 | % |
2025 |
| 100.969 | % |
2026 and thereafter |
| 100.000 | % |
Prior to October 15, 2024, we may redeem up to 40% of the aggregate principal amount of the Euro notes at a redemption price equal to 103.875% of the principal amount thereof with an amount equal to or less than the net cash proceeds that we raise in one or more equity offerings, plus accrued and unpaid interest on such notes, if any, to, but not including, the redemption date.
The Biomat Transactions
On December 1, 2021, we sold preferred shares representing 12.9% of Biomat Newco and 12.5% of Biomat USA, our U.S.-based plasma collection subsidiaries that are part of the Biomat Group, to the GIC Investor. Specifically, the GIC Investor acquired preferred shares. The purchase price received was $990 million.
As a result of the transaction, the GIC Investor received ten class B common shares of Biomat USA and nine class B common shares of Biomat Newco. While named common shares, such shares are non-voting and have annual preferential dividends of $4,168,421.05 per share of Biomat USA and Biomat Newco. These preferred shares also granted the GIC Investor with redemption rights of up to one share per year beginning in 2023 at $52,105,263.16 per share. This investment was originally recorded as equity, but was later restated as debt in our consolidated financial statements for the year ended December 31, 2021, prepared under pursuant to IFRS-EU and filed with the CNMV in Spain. The investment by the GIC Investor is recorded as debt in our financial statements, and the reclassification did not affect compliance with the covenants under our debt instruments.
Other Debt
Certain other credit facilities and lease obligations are in place with various lenders and consist of long-term and short-term indebtedness of both us and Grifols subsidiaries. As of December 31, 2023, we had €276.0 million of aggregate short-term credit under these facilities. The short-term credit facilities have maturity dates occurring in the next 12 months. See Note 21 to our consolidated audited financial statements included in this annual report on page F-90.
C. | Research and Development, Patents and Licenses |
For detailed information regarding our research and development initiatives, see Item 4 of this Part I, “Information on the Company—B. Business Overview—Research and Development.”
D. | Trend Information |
Plasma-derived protein therapies are essential to extend and improve the lives of individuals suffering from chronic, acute and life-threatening conditions including infectious diseases, such as hepatitis, immunological diseases, such as multiple sclerosis, hemophilia, von Willebrand disease, liver dialysis and acute conditions such as burns and severe blood loss. For this reason, the administration of these products cannot be interrupted or postponed without putting patients’ lives at risk. This ensures a stable demand for such products. In addition, because of the nature of the diseases treated, the reimbursement rates for plasma derivative products in the United States are high. Any changes to such rates would likely elicit a strong lobbying response in the United States.
116
Based on MRB reports, in 2021, the worldwide plasma proteins market (without recombinant products) reached $29.2 billion, a 23.5% increase from 2018, or 7.3% per year of such three-year period. We believe that many plasma derivative products are underutilized and will continue to benefit from strong demand. Additionally, new indications are being explored for a number of plasma-derived therapies, such as the treatment of Alzheimer’s disease. We believe that the volume of global sales of plasma derivative products will continue to grow driven primarily by the same factors that have contributed to its historical growth, including:
| ● | population growth; |
| ● | the discovery and approval of new applications and indications for plasma-based products; |
| ● | an increase in the number of diagnosed patients and diagnosed but previously-untreated patients; |
| ● | geographic expansion; and |
| ● | physicians’ greater awareness of conditions and treatments. |
Approximately 19.1% of our sales were generated in the European Union in 2023, as compared to 17.0% in 2022 and 18.4% in 2021. We anticipate that the percentage of our sales generated in the European Union will not significantly increase in 2024.
There are significant barriers to entry into the plasma derivative products industry, as the industry is highly regulated and requires significant expertise and capital investments. We do not expect these barriers to decrease in the near term.
Regulatory Environment. In order to operate in the plasma derivatives industry, manufacturers and distributors must comply with extensive regulation by the FDA, the EMA and comparable authorities worldwide. As a result, significant investments are required to develop, equip and maintain the necessary storage, fractionation and purification facilities and to develop appropriate sale, marketing and distribution infrastructures. Additionally, only proteins derived from plasma collected at FDA-approved centers can be marketed in the United States, so securing an adequate supply of U.S. source plasma is required to operate in the United States. We expect these regulatory restrictions to continue.
Product Pipeline.
We have an expanded portfolio of key products as a result of our recent acquisitions and will continue to invest in research and development with respect to new product and new indications for existing products. Some key research and development projects underway include clinical studies of the use of albumin, diagnostic and vaccine therapies to treat Alzheimer’s disease, of albumin to treat advance cirrhosis and ascites, and of antithrombin in heart surgery.
Our product pipeline offers a strategic balance between risk and value and includes over 30 clinical trials across diverse phases. In 2023, our subsidiary Biotest AG achieved positive results in its phase 3 clinical trial for fibrinogen concentrate, marking a significant step in treating acquired fibrinogen deficiency. Other innovations reinforce our position in plasma-derived medicine, such as IG Yimmugo®, a newly developed immunoglobulin for the treatment of immunodeficiencies and autoimmune diseases, and Trimodulin, an antibody composition purified from human plasma in clinical development to treat severe community-acquired pneumonia (sCAP) and severe COVID-19.
E. | Critical Accounting Estimates |
The preparation of consolidated financial statements in accordance with IFRS requires us to make estimates and judgments in certain circumstances that affect the reported amounts of assets, liabilities, revenue, expenses and the related disclosures of contingent assets and liabilities. A detailed description of our significant accounting policies is included in the notes to our audited consolidated financial statements included elsewhere in this annual report on Form 20-F.
117
Certain of our significant accounting policies require subjective and complex judgments, often requiring the use of estimates about the effects of matters that are inherently uncertain. We apply estimation methodologies consistently from year to year. Other than changes required due to the issuance of new accounting guidance, there have been no significant changes in our application of critical accounting estimates during the periods presented. We periodically review our critical accounting estimates and estimates with the Audit Committee of our Board.
Critical accounting estimates include depreciation, subsequent recognition, impairment, goodwill and amortization, among others. See Notes 2, 4 and 6 to our audited consolidated financial statements included in this annual report on Form 20-F for more information regarding our critical accounting estimates and Goodwill, respectively.
More information on recently issued accounting standards is included in Note 2 to our audited consolidated financial statements included in this annual report on Form 20-F.
Item 6.DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
A. | Directors and Senior Management |
Directors
From January 2023 until the date of this annual report, the following changes were made to our Board of Directors:
| ● | In February 2023, Mr. Steven F. Mayer resigned from his position of Executive Chairperson of the Board of the Directors and as a member of the Board of Directors due to personal reasons. Mr. Thomas Glanzmann was then unanimously appointed by our Board as the new Executive Chairperson, replacing Mr. Steven F. Mayer; |
| ● | In February 2023, as a consequence of the above, Mr. Thomas Glanzmann resigned from his position as Vice-Chairperson of our Board and Mr. Raimon Grifols Roura was unanimously appointed by our Board as its new Vice-Chairperson, replacing Mr. Thomas Glanzmann; |
| ● | In December 2023, Mr. Victor Grifols Roura resigned from our Board of Directors due to his retirement. His vacancy was filled by Mr. Albert Grifols Coma-Cros, who was unanimously appointed as a director by the Board through the co-option procedure. His appointment is subject to ratification at our next general shareholders’ meeting; |
| ● | In February 2024, Mr. José Ignacio (Nacho) Abia Buenache was appointed as a member of our Board of Directors, filling the vacancy that had been created when Mr. Steven F. Mayer resigned. Mr. Abia was also appointed as a director by the Board through a co-option procedure, and therefore his appointment is subject to ratification at our next general shareholders’ meeting. |
118
Set forth below are the names and current positions of the members of the Board:
Name |
| Age |
| Title |
| Type |
| Director |
| Term |
|
José Ignacio (Nacho) Abia Buenache | 55 | Director and Chief Executive Officer | Executive | Feb 2024 | next GSM(2) | ||||||
Thomas H. Glanzmann | 65 | Executive Chairperson | Executive | Apr 2006 | Oct 2024 | ||||||
Raimon Grifols Roura | 60 | Director, Vice-Chairperson of the Board | Executive | May 2015 | June 2027 | ||||||
Víctor Grifols Deu | 47 | Director | Executive | May 2016 | Oct 2024 | ||||||
Tomás Dagá Gelabert | 68 | Director | Other External | Apr 2000 | June 2027 | ||||||
Enriqueta Felip Font | 60 | Director | Independent | May 2019 | June 2027 | ||||||
Montserrat Muñoz Abellana | 56 | Director | Independent | June 2022 | June 2026 | ||||||
Susana González Rodríguez | 48 | Director | Independent | June 2022 | June 2026 | ||||||
Carina Szpilka Lázaro | 55 | Director and Lead Independent Director(1) | Independent | May 2015 | June 2027 | ||||||
James Costos | 61 | Director | Independent | Oct 2020 | Oct 2024 | ||||||
Iñigo Sánchez-Asiaín Mardones | 60 | Director | Independent | May 2015 | June 2027 | ||||||
Albert Grifols Coma-Cros | 46 | Director | Proprietary | Dec 2023 | next GSM(2) | ||||||
Nuria Martín Barnés | 65 | Secretary, non-member, of the Board of Directors | n/a | May 2015 | n/a | ||||||
Laura de la Cruz Galán | 35 | Vice-Secretary, non-member, of the Board of Directors | n/a | Dec 2023 | n/a |
| (1) | The lead independent director is a figure introduced by Law 31/2014, adopted on December 3, 2014, that amended the Spanish Companies Act in matters of corporate governance, or Law 31/2014. It is mandatory to appoint a lead independent director when the office of Chairperson of the Board and that of chief executive officer is held by the same person. The lead independent director must, among other responsibilities, (i) be an independent director and be authorized to request the calling of a board meeting or the inclusion of new points on the agenda of a board meeting already convened, (ii) coordinate and gather the non-executive directors, (iii) direct, when applicable, the Chairperson’s periodic evaluation by the Board, (iv) maintain contacts with investors and shareholders in order to find out their points of view for the purpose of forming an opinion on their concerns, in particular, in relation to the corporate governance of the Company; and (v) coordinate the plan for succession of the Chairperson. The Board, in its meeting held on June 16, 2023, agreed to re-elect Carina Szpilka Lázaro as the Company’s Lead Independent Director. |
| (2) | The term expires on our next general shareholders’ meeting, expected to take place as legally required prior to June 30, 2024. Appointed to the Board by means of the co-option procedure under Spanish corporate law. See Item 5 of this Part I, “Operating Results—Subsequent Events—Changes in Board of Directors and Senior Management.” |
119
Director Biographies
José Ignacio (Nacho) Abia Buenache
Mr. Jose Ignacio (Nacho) Abia Buenache has served as board member of Grifols, S.A. since February 2024, having assumed the office of Chief Executive Officer of the Company on April 1, 2024. Previously, he was President and Chief Executive Officer of Olympus Corporation of the Americas based in Pennsylvania. He also served as Executive Officer and COO and CSO at Olympus Corporation, a publicly traded company in the Tokyo stock exchange. Mr. Abia has over two decades of experience in the medical technology and life sciences industries, with half of that time spent in the United States and the other half in Europe, while also spending significant time in the Asia-Pacific region to develop businesses. Prior to joining Olympus, he held several positions in the information technologies and consumer electronics industries working for a variety of technology companies, including Sony and Techdata.
In the academic field, Nacho holds a Bachelor’s Degree in Telecommunications and Electronics Engineering from the Universitat Politécnica de Catalunya (UPC), a Master of Business Administration (MBA) degree from the business school (EAE) of Barcelona, and has completed the Senior Management Program (PADE) at IESE (University of Navarra). Mr. Abia also serves on the board of directors of AdvaMed (Advanced Medical Technology Association) in the United States, the U.S.-Spain Chamber of Commerce and is a also member of the board of trustees of the Lehigh Valley Health Network, a hospital system composed of 13 hospitals and multiple care centers in Pennsylvania. In the past, he was a member of the board of directors of Evident Corporation, a company in the field of life sciences and industrial solutions.
Thomas H. Glanzmann
Mr. Thomas H. Glanzmann has served as a director of Grifols since April 2006 and since February 21, 2023, he is the Executive Chairperson of the Company. He serves as a director on the board and as a member of several committees at Alcon, Inc. (among others, the sustainability, compensation and innovation committees). He is also a founder and General Partner in Medical Technology Venture Partners in California. From 2006 until 2011 he was the CEO and Chairman of Gambro AB. Prior to this Mr. Glanzmann was the CEO and Managing Director of HemoCue AB. Between 1988 and 2004 he held various positions at Baxter Healthcare Corporation: Senior Vice President and Senior Corporate Officer of Baxter Healthcare Corporation; President of Baxter Bioscience; Chief Executive Officer of Immuno International; and President of the European Biotech Group, among others. Between 1984 and 1988 he worked at Philip Morris where he was the country manager for Norway, Denmark and Iceland. Mr. Glanzmann holds an MBA from IMD in Lausanne-Switzerland, a B.A. in Political Science from Dartmouth College, U.S., and a Board of Directors Certification from the UCLA Anderson School of Management, U.S.
Raimon Grifols Roura
Mr. Raimon Grifols Roura is the non-executive Vice-Chairperson of the Board and our Chief Corporate Officer. He was Grifols’ joint and several Chief Executive Officer together with Mr. Víctor Grifols Deu from 2017 to 2023. He succeeded his brother, Mr. Víctor Grifols Roura in the position. He is a member of the administration bodies of several companies within the Grifols Group. From 2001 to 2015 he held the role of secretary, non-member, of the Board of Directors of Grifols, and in 2015 began serving as director and Vice Secretary of the Board of Directors. In May 2016, the Board accepted his resignation as Vice Secretary. Until his appointment as executive director in July 2016, Mr. Grifols Roura was a partner at the law firm Osborne Clarke in Spain. Mr. Grifols Roura earned his law degree from the University of Barcelona (Universidad de Barcelona).
Mr. Raimon Grifols Roura is a shareholder of Deria S.A. (a non-controlling shareholder, pursuant to the Spanish Securities Market Act). He is also a shareholder of Scranton Enterprises, B.V. (a non-controlling shareholder, pursuant to the Spanish Securities Market Act).
120
Víctor Grifols Deu
Mr. Víctor Grifols Deu is a member of our Board and our Chief Operating Officer. He was Grifols’ joint and several Chief Executive Officer together with Mr. Raimon Grifols Roura from 2017 to 2023. He succeeded his father, Mr. Víctor Grifols Roura in the position. He also served as a member of the administration bodies of several companies within the Grifols Group and was appointed executive director in May 2016. He joined the Company in 2001 as an analyst in the Planning and Control Department of the Company. In 2008 he became the director of the Planning and Control Department and was also appointed a member of the Executive Committee. He has been part of the team that analyzed and was responsible for the integration of operations after the acquisition of Alpha Therapeutics, Talecris Biotherapeutics and Novartis’ Transfusion Diagnostic Unit. He graduated in Business Administration and Management from the Ramon Llull University — Sarrià Chemical Institute and holds a postgraduate degree in Business Administration and Management from Michael Smurfit Business School in Dublin.
Mr. Víctor Grifols Deu is a shareholder of Ralledor Holding Spain S.L. (a non-controlling shareholder, pursuant to the Spanish Securities Market Act).
Tomás Dagá Gelabert
Mr. Tomás Dagá Gelabert has served as director of Grifols since April 2000 and was also our Vice Secretary of the Board from May 2016 to December 2023. He is a partner and founder of the law firm Osborne Clarke in Spain. He was the managing partner of the law firm Osborne Clarke in Spain until June 30, 2017. Prior to joining Osborne Clarke, he worked in the corporate and tax department of Peat Marwick Mitchell & Co. in Barcelona. His professional career has provided him with a wide range of accounting, financial and audit skills. He is currently a member of the administrative bodies of several companies within the Grifols Group, as well as a board member of Shanghai RAAS. Mr. Dagá earned his law degree from the University of Barcelona (Universidad de Barcelona).
Mr. Tomás Dagá Gelabert is a shareholder of Scranton Enterprises, B.V. (a non-controlling shareholder, pursuant to the Spanish Securities Market Act).
Enriqueta Felip Font
Dr. Enriqueta Felip Font has served as a director of Grifols since May 2019. She received her degree in Medicine and Surgery from the Autonomous University of Barcelona (UAB), where she also completed her studies for a PhD in Medical Oncology. She was an Associate Professor at the UAB from 2010 to May 2019. She is a Professor of Medicine at the Universitat de Vic (UVicc-UCC). Dr. Felip Font has an extensive professional career and accredited experience in the oncology sector, as well as knowledge in the scientific and research field. She is currently the Section Chief of the Medical Oncology Service at Vall d’Hebron University Hospital and the Principal Investigator of the Vall d’Hebron Institute of Oncology’s Thoracic Tumors and Head and Neck Cancer Group. Dr. Enriqueta Felip Font has made a significant contribution to cancer research, especially in the field of thoracic tumors, and has collaborated in the development of lung cancer approaches that define the current standard of care for the disease. Dr. Enriqueta Felip Font has been involved in several initiatives with scientific organizations, among them, as member of the Board of Directors of the International Association for the Study of Lung Cancer (IASLC, 2017-2021). She is currently the President of the Oncology Comission at Vall d’Hebron University Hospital and a member of the Scientific Committee of the Institut d’Investigació i Innovació Parc Taulí. Throughout her career, she has obtained several recognitions for her work in the oncology field. In 2015, she was awarded with the first Women for Oncology Award from the European Society of Medical Oncology (ESMO). In May 2022, she was awarded with the Prize “La Vanguardia de la Ciencia,” which awards cutting-edge scientific works aiming to give visibility to frontier scientific research in Spain.
Most recently, she featured on Clarivate Analytics’ annual Global Highly Cited Researchers List 2018, 2019, 2020, 2021, 2022 and 2023. Dr. Enriqueta Felip Font has authored more than 350 peer-reviewed articles. Her professional background has provided her with expertise and knowledge on scientific and innovation matters.
Montserrat Muñoz Abellana
Ms. Muñoz Abellana earned a degree in Chemical Engineering from the Institut Químic de Sarrià in Barcelona (Universitat Ramon Lull) and several Executive Development Programs at IESE, INSEAD and the London Business School.
121
She began her professional career in the Consumer Goods sector at Procter & Gamble where she held different roles in Operations across Europe. For the last 17 years and until end of 2022, she has been a Senior Executive at Danone where she has served as Global Medical Nutrition Operations Vice President, Iberia Medical Nutrition General Manager and Value Chain Digital Transformation Vice President.
Ms. Muñoz Abellana is also an independent director and the Chairperson of the Audit and Compliance Committee at Uriach and independent director at Comexi and has served from 2015 to early 2022 as Board member in related National Industry Groups (Asociación Española de Nutrición Enteral and Federación Española de la Industria de Alimentación y Bebidas). Her professional background has provided her with expertise and knowledge on digital transformation matters, as well as accounting, financial and audit skills.
Susana González Rodríguez
Ms. González Rodríguez earned a degree in Business Administration from the Asturias Business School and an MBA from the San Francisco State University. She began her professional career in the electronics industry sector at TE Connectivity where she held different roles. She is currently a Senior Executive at Rockwell Automation where, since January 2019, she has served as President of the Europe, Middle East, and Africa regions. Likewise, she is a managing director at Rockwell Automation B.V. and is a member of the Global Business Women Leaders Council by The Conference Board and a member of the Spanish Board Directors (Instituto de Consejeros-Administradores). Her professional background has provided her with expertise and knowledge on strategy, sales and digital transformation matters.
Carina Szpilka Lázaro
Ms. Carina Szpilka Lázaro has served as a director of Grifols since May 2015 and is the Lead Independent director of Grifols’ Board since February 2022. She earned a degree in Business Administration from the Universidad Pontificia de Comillas in Madrid (ICADE) and an Executive MBA from the Instituto de Empresa de Madrid. She began her professional career in the financial sector working at Banco Santander and Argentaria (now known as BBVA). In 1998 she was part of the team that founded ING Direct in Spain, where she held the position of CEO from 2010 to 2013, having previously held that position in ING Direct France from 2008 to 2010. She is currently an independent director at Abanca and Meliá Hotels International, as well as a partner at KFund Venture Capital. She has received numerous awards. Among others, in 2011 she was given the “Female Executive of the Year” award by the Spanish Federation of Female Directors, Executives, Professionals and Entrepreneurs (Federación Española de Mujeres Directivas - FEDEPE). For four years, she was also a member of the UNICEF Foundation. Her academic and professional background has provided her with financial, accounting and audit skills as well as in the field of digitalization.
James Costos
Mr. James Costos has served as a director of Grifols since October 2020. He is an American diplomat who holds a degree in Political Science from the University of Massachusetts. He has an extensive professional career and accredited experience in different sectors including international relations and the digital and communications sectors. From 2013 to 2017, he was the U.S. Ambassador to the Kingdom of Spain and to the Principality of Andorra. He is currently the President of Secuoya Studios in Madrid. He is a member of the Board of Directors of PJT Partners, a firm providing financial advisory services in investment banking, and Senior Managing Director in the Venture Technology Group at Dentons. He is member of various cultural and humanitarian organizations, among others the Hispanic Society of America, as well as he is a member of the board of the Human Rights Campaign. Likewise, he has been appointed as member of the J. William Fullbright Foreign Scholarship Board. His professional background has provided him with international expertise and knowledge on matters such as the development of diversity and equality policies.
122
Iñigo Sánchez-Asiaín Mardones
Mr. Iñigo Sánchez-Asiaín Mardones has been the Lead Independent director of the Board since May 2015 until 2022 and he has served as director of Grifols, S.A. since May 2015. He earned a degree in Business Administration from the Universidad Pontificia de Comillas in Madrid (ICADE) and an MBA from Harvard Business School. In 2010 he founded Portobello Capital, where he remains a partner and a member of the Executive Committee and Investment Committee, leading the investments in companies such as Angulas Aguinaga, a company where he is Vice-Chairman and member of the Executive Committee, and Hotels & Resorts Blue Sea, S.L., where he is a member and Chairperson of the Board of Directors. He is also a member of the Executive Committee at the Harvard Club of Spain, which he has previously chaired. Previously, from 1993 to 2005, he was Deputy General Director at Banco Santander and from 2005-2010 was a partner and member of the board of directors of Ibersuizas Gestión SGECR, S.A, through which, together with his academic training, he has gained experience and knowledge on matters such as accounting, audit and risk management, both financial and non-financial.
Albert Grifols Coma-Cros
Mr. Albert Grifols Coma-Cros joined the Grifols Group in 2004 as an analyst in the Planning and Control Department. In 2007, he moved to the Finance Department as a financial analyst. In 2013, Mr. Grifols Coma-Cros held the position of Corporate Treasury Director and moved to Ireland to develop and implement the Grifols Group’s internal banking transactions. From 2018 to 2020, he was appointed Managing Director of Grifols in Ireland, ensuring the correct collusion between Irish culture and Grifols’ own values in the gradual growth of our subsidiary in the country. From 2021 to 2023, Mr. Grifols Coma-Cros served as Chief Scientific Innovation Officer, being responsible for consolidating all of the Grifols Group’s scientific knowledge previously dispersed under a single scientific organization. He holds a degree in Business Administration from the Universitat Autònoma de Barcelona and has completed several Management Development Programs at ESADE, Georgetown University or the Institut Estudis Financers (IEF).
Mr. Grifols Coma-Cros has also been a director of the board of directors of Fisa 14, S.A., a real estate company, since 2015, and president of Bansabadell 18, FP, a pension fund, since 2018.
Biography of the Secretary and Vice-Secretary, Non-Members, of the Board
Nuria Martín Barnés
Ms. Nuria Martín Barnés served as Vice-Secretary, non-member, of the Board of Directors from 2001 to 2015, and has served as Secretary, non-member, of the Board of Directors since 2015. Ms. Martín has been the managing Partner at Osborne Clarke Spain since July 1, 2017 until December 31, 2022. Prior to joining Osborne Clarke she worked in the Corporate and Tax Department of KPMG Peat Marwick from 1982 to 1986. Ms. Martín is a trustee and Chairperson of the Probitas Fundación Privada foundation. She is secretary and member of the board of directors of Compañía General de Inversiones, SICAV, S.A., and Gesiuris CAT Patrimonis, as well as Secretary, non-member, of the Board of Gesiuris Asset Management, S.G.I.I.C., S.A. Ms. Martín earned her law degree from the University of Barcelona.
Laura de la Cruz Galán
Ms. Laura de la Cruz Galán has been the Vice-Secretary of our Board since December 2023 and Secretary, non-member, of our Audit Committee since April 2024. She is a Counsel of the Spanish law firm Osborne Clarke, where she has been since 2012. Ms. De la Cruz specializes in corporate governance with respect to listed companies, as well as international M&A transactions. She holds a Law Degree from ESADE Law School in Barcelona, where she graduated in 2011. In 2023, she was selected to join the 8th edition of the “Women’s Talent Pool Leadership Programme” organized by the European Network for Women in Leadership. Furthermore, she is a trustee and Secretary of the Board of Trustees of Fundació Víctor Grifols i Lucas.
123
Senior Management
We have continued to effect changes in our senior management aligned with our strategic plan of streamlining corporate functions, separating ownership from our senior management and enhancing other efficiencies across the organization. See Item 5 of this Part I “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Operational Improvement Plan.”
In February 2023, the Board of Directors appointed Mr. Thomas Glanzmann as Grifols, S.A.’ new Executive Chairperson, following the resignation of Mr. Steven F. Mayer for personal reasons. Thomas has been a member of our Board of Directors for more than 16 years and its non-Executive Vice-Chairperson since 2017, in addition to chairing Grifols’ Sustainability Committee since 2020. Most of Thomas’ career has been associated with the plasma industry, as he has held key roles in several prominent companies in the past, such as serving as president of Baxter Bioscience, CEO of Immuno International and president of the European Baxter Biotech Group.
In May 2023, to further optimize and accelerate our performance and increase shareholder value, our Board of Directors realigned our senior management team and executive governance bodies to streamline our corporate governance and ensure continued focus on results. In particular, our Board condensed the functions of Executive Chairperson and Chief Executive Officer (CEO), both assumed by Mr. Thomas Glanzmann, while the positions of Co-Chief Executive Officers evolved into the Senior Executive Leadership Team (“SELT”), led by Mr. Glanzmann and comprising Raimon Grifols Roura, now serving as Chief Corporate Officer (CCO) and focusing on optimizing the value of our Corporate affiliates and partnerships as well as leading key corporate initiatives; Víctor Grifols Deu, now serving as Chief Operating Officer (COO) and focusing on managing the day to day business with all operating functions reporting to him; and Mr. Alfredo Arroyo, maintaining his position as Chief Financial Officer (CFO). Mr. Grifols Deu and Mr. Grifols Roura each kept his respective position as member of our Board of Directors.
As of January 1, 2024, Mr. Albert Grifols Coma-Cros stepped down from his executive position of Chief Scientific Innovation Officer, while remaining a member of the Board of Directors as a proprietary director, representing one of our major shareholders, Ponder Trade, S.L.
In February 2024, the Board appointed Mr. José Ignacio (Nacho) Abia Buenache as the new Chief Executive Officer of Grifols, a position Mr. Abia assumed on April 1, 2024. His appointment as director is subject to the ratification of our next general shareholders’ meeting. Following a transition period working alongside our Chief Operating Officer and Chief Corporate Officer, Mr. Abia will assume the responsibilities previously held by such positions, while Mr. Víctor Grifols Deu and Mr. Raimon Grifols Roura will step down from their executive positions and remain as members of our Board.
Other notable changes in our senior management were:
| ● | Roland Wandeler was appointed as the new president of our Biopharma business unit, replacing Maria Pia D’Urbano; |
| ● | Camille Alpi was appointed as our Chief Human Resources and Talent Officer, replacing Montserrat Gaja Llamas; |
| ● | Dr. Jörg Schüttrumpf was appointed Chief Scientific Innovation Officer (CSIO) and will focus on accelerating the development of differentiated plasma and non-plasma medicines in key therapeutic areas, building on Grifols’ strong innovation portfolio; and |
| ● | Mr. Miguel Louzan was named as our Chief Digital Information Officer (CDIO) to lead digital and data transformation, focusing his work on accelerating the company’s use of digital platforms and new technologies to transform and reinforce critical business activities such as plasma donor and customer relationships, manufacturing operations, new therapy development and cybersecurity. |
| ● | Laura Carratalà was appointed as the Vice President of our Bio Supplies business unit, replacing Mr. Albert Grifols Roura; the former President of the Bio Supplies business unit. |
124
Our senior management currently consists of the following persons:
Name |
| Age |
| Title |
| Since |
|
José Ignacio (Nacho) Abia | 55 | Chief Executive Officer | 2024 | ||||
Alfredo Arroyo Guerra | 67 | Chief Financial Officer | 2013 | ||||
Víctor Grifols Deu | 47 | Chief Operating Officer | 2023 | ||||
Raimon Grifols Roura | 59 | Chief Corporate Officer | 2023 | ||||
Thomas H. Glanzmann | 65 | Executive Chairperson of the Board | 2023 | ||||
Camille Alpi | 43 | Chief Human Resources and Talent Officer | 2024 | ||||
David Ian Bell | 69 | Chief Corporate Development, Legal & Data Protection Officer | 2022 | ||||
Daniel Fleta Coit | 53 | Chief Industrial Services Officer | 2022 | ||||
Laura Carratalà Peña | 36 | VP, Bio Supplies | 2024 | ||||
Maria Teresa Rioné Llano | 59 | Chief Communications Officer | 2018 | ||||
Jörg Schüttrumpf | 49 | Chief Scientific Innovation Officer | 2024 | ||||
Antonio Martinez Martinez | 57 | President of the Diagnostic business unit | 2022 | ||||
Francisco Javier Guix Huguet | 62 | Vice President, Healthcare Solutions | 2022 | ||||
Lluis Pons Gomez | 41 | Senior Vice President, Strategy& COO Office | 2022 | ||||
Ignacio Ramal Subira | 56 | Chief Internal Audit & Enterprise Risk Manager | 2022 | ||||
Miguel Louzan | 50 | Chief Digital Information Officer | 2023 | ||||
Jordi Balsells Valls | 51 | President, Plasma Procurement | 2022 | ||||
Roland Wandeler | 51 | President, Biopharma | 2024 |
Senior Management Biographies
The following are the biographies of our senior management who are not also directors:
Alfredo Arroyo Guerra
Mr. Arroyo has served as our Corporate Vice President and Chief Financial Officer since January 2007. Previously, Mr. Arroyo served as a CFO and in various Senior Finance positions in companies including KPMG, Carrefour, Chupa Chups, Reckitt Benckiser and Winterthur. Mr. Arroyo received a degree in Economics and is a Certified Public Accountant in Spain.
Camille Alpi
Mr. Alpi joined Grifols in February 2024 as our new Chief Human Resources and Talent Office. Mr. Alpi is a human resources executive with 20 years of experience across Europe, the U.S., South America and Asia. Prior to joining Grifols, he was Vice President Human Resources for Danone Specialized Nutrition, the company’s healthcare division. Mr. Alpi has a master’s degree in business and economics from Uppsala University, Sweden. He is based in the company’s global headquarters, in Sant Cugat (Barcelona), and takes over from Montserrat Gaja, who has retired.
125
David Ian Bell
Mr. Bell has been our General Counsel since 2003 and Chief Corporate Development Officer since 2021. He currently serves as Chief Corporate Development, Legal and Data Protection Officer. He previously held the position of Chief Innovation Officer from 2016 to 2020. Mr. Bell joined us as a Corporate Vice President of Grifols Shared Services North America, Inc. (previously Grifols, Inc.) in July 2003. He also served as a member of our Executive Committee in Spain. He additionally serves on the boards of numerous companies affiliated to Grifols. Mr. Bell is responsible for all legal activities of our U.S. operations, including litigation, mergers and acquisitions, real estate transactions, intellectual property and contracts. Prior to joining us, Mr. Bell was Vice President and General Counsel for Alpha. He also spent time as a partner at the U.S. law firm of Knapp, Petersen & Clarke where he specialized in complex litigation involving healthcare, pharmaceutical and biotechnology regulation and liability. Mr. Bell attended the University of California, Irvine, Southwestern University School of Law and a postgraduate program at Harvard Law School. He is a member of the California State Bar and is admitted to practice before the United States Supreme Court as well as numerous federal appellate and district courts.
Daniel Fleta Coit
Mr. Fleta joined us in 2001 and, since June 2022, he has been our Chief Industrial Services Officer. From 2018 to 2022, he served as our Chief Industrial Officer. Previously, Mr. Fleta served as Managing Director of Grifols Engineering S.A. from 2011 to 2018. Beginning in 2005, he has served as Director Pharmaceutical Projects. Mr. Fleta received a degree in Industrial Engineering from the Institut Químic de Sarrià in 1995.
Laura Carratalà Peña
Ms. Carratalà joined Grifols in 2015 as part of the Plasma Corporate Financial Controlling Team. She has held various positions within the company in the Plasma and Bio Supplies business units, as well as in our finance corporate department. She has been VP of Bio Supplies since September 2023, taking over the leadership of such business unit from Albert Grifols following his retirement in February 2024.
Maria Teresa Rioné Llano
Ms. Rioné joined Grifols in 2018 and currently serves as the Chief Communications Officer. Prior to joining Grifols, Ms. Rioné was Senior Director of Communications Western Europe at Nike Corporation. Ms. Rioné is a graduate in Law with honors in Commercial Law from Universitat de Barcelona and a Master’s in Marketing and Sales Management from IE Business School.
Jörg Schüttrumpf
Dr. Jörg Schüttrumpf joined Grifols in September 2024 as the new Chief Scientific Innovation Officer (CSIO). As the head of innovation for the entire Grifols Group, Dr. Schüttrumpf concentrates on accelerating the development of differentiated plasma and non-plasma medicines in key therapeutic areas. Since early 2022, Dr. Schüttrumpf has been Chief Scientific Officer of our subsidiary Biotest AG, where he had been Head of Global Research since 2012 and Head of Research and Development since 2015. Dr. Schüttrumpf is a physician scientist specializing in Transfusion Medicine and Hemostaseology with degrees from Goethe University Frankfurt, Albert-Ludwigs-University Freiburg, and IMD Business School. During his career he worked in leading research institutions both in Germany and in the United States.
Antonio Martinez Martinez
Dr. Martinez joined Grifols in 2020 and, since 2022, serves as President of the Diagnostic business unit. Previously, he was the President of Diagnostic Scientific & R&D. Prior to Grifols, he served as Chief Executive Officer of Progenika Biopharma S.A., a leading molecular diagnostic company dedicated to personalized medicine he co-founded in 2000 and that was acquired by Grifols in 2013. Mr. Martinez has received the Ernst & Young Most Innovative Entrepreneur Award (2010) and the Ruban d´Honneur in the European Business Awards, HSBC Bank (2011). Before receiving a MOD from the Instituto de Empresa, Dr. Martinez obtained his PhD from the University of Navarra with a project aimed at the development of a diagnostic method for cystic fibrosis, an aim that he accomplished in 1992. The output of Dr. Martinez’s research and development work includes more than 70 publications in scientific journals and 20 patent applications on diagnostic methods for genotyping or gene expression.
126
Francisco Javier Guix Huguet
Mr. Guix joined Grifols in 1989 and, since June 2022, he has been our Vice-President, Healthcare Solutions. From 2018 to 2022, Mr. Guix served as President, Hospital Sales & Commercial Operations IBAM&ROW. Since he started in Grifols as a Medical Devices Product Manager he has occupied several commercial positions in Marketing and Sales Management. He has been a member of the board of directors of our subsidiary Kiro Grifols, S.L. since 2014. He occupies also a place in the Constant Group board. Mr. Guix received a degree in Biochemistry by the Universitat Autonoma de Barcelona in 1987.
Lluis Pons Gomez
Mr. Pons joined Grifols in 2017. He currently serves as Senior Vice-President, Strategy & COO Office. Previously, Mr. Pons served in Bain&Co. as a management consultant. Mr. Pons holds an MBA from Hong Kong UST and London Business School and received a degree in Industrial Engineering from the Universitat Politecnica de Catalunya in 2000.
Ignacio Ramal Subira
Mr. Ramal joined Grifols in 2008. He currently serves as Chief Internal Audit & Enterprise Risk Manager. Previously, Mr. Ramal served as external auditor in Ernst & Young. Mr. Ramal received a degree in Economics and is a Certified Public Accountant in Spain.
Jordi Balsells Valls
Mr. Balsells joined Grifols in 2022 as President of Plasma Procurement. Previously, Mr. Balsells served in various executive roles at FC Barcelona and Desigual. Mr. Balsells received a degree in Economics and Business Administration from Universitat Pompeu Fabra (Barcelona) and an Executive MBA degree from Esade Business School.
Roland Wandeler
Roland Wandeler, Ph.D., is the new President of our Biopharma Business Unit, having joined us in February 2024. Dr. Wandeler brings more than two decades of global biopharmaceutical experience to Grifols. He has a track record of driving commercial, operational and organizational excellence throughout his career in both Europe and the United States. This includes being Chief Commercial Officer at MorphoSys, where he helped build a fully integrated biopharmaceutical company. Prior to that, Mr. Wandeler worked for 14 years at Amgen, where he held executive positions of increasing responsibility, including Corporate Vice President and General Manager Bone Health and Cardiology in the U.S., a multibillion growth business. He replaces Joel Abelson, who has retired. Dr. Wandeler is a graduate of the Swiss Federal Institute of Technology (ETH), in Zurich. He holds a doctorate in technical sciences as well as Master of Science in chemical engineering. He is located in Grifols’ corporate offices in North Carolina’s Research Triangle Park, not far from the company’s flagship plasma-medicines manufacturing site in Clayton, N.C.
Family Relationships
Mr. Raimon Grifols Roura, director and Chief Corporate Officer, Mr. Alberto Grifols Roura, former President of the Bio Supplies business unit and Mr. Víctor Grifols Roura, a former director and Honorary Chairperson (non-member) of the Board, are brothers.
Mr. Raimon Grifols Roura, our Chief Corporate Officer is the uncle of Mr. Víctor Grifols Deu, our Chief Operating Officer, both being directors and former Co-Chief Executive Officers.
Mr. Alberto Grifols Roura, former President of the Bio Supplies business unit, is the uncle of Mr. Victor Grifols Deu, a director, our Chief Operating Officer and former Co-Chief Executive Officer.
Mr. Víctor Grifols Deu, our Chief Operating Officer, director and former Co-Chief Executive Officer, is the son of Mr. Víctor Grifols Roura, a former director and the Honorary Chairperson (non-member) of the Board.
127
Messrs. Víctor Grifols Roura, Alberto Grifols Roura and Raimon Grifols Roura are the grandchildren of Mr. José Antonio Grifols i Roig, our founder.
Mr. Raimon Grifols Roura, our Chief Corporate Officer, director and former Co-Chief Executive Officer, Mr. Alberto Grifols Roura, former President of the Bio Supplies business unit and Mr. Victor Grifols Roura, a former director and Honorary Chairperson (non-member) of the Board, are uncles of Mr. Albert Grifols Coma-Cros, a director.
Arrangements Pursuant to Which Certain Directors or Senior Management Were Selected
We have no arrangements.
B. | Compensation |
The compensation of our directors is determined pursuant to our director remuneration policy. Pursuant to Article 15 of the Regulations of the Internal Functioning of the Board of Directors of Grifols, S.A. (Reglamento de funcionamiento interno del consejo de administración, or the “Board Regulations”), the appointments and remuneration committee of the Board (Comisión de Nombramientos y Retribuciones, or the “Appointments and Remuneration Committee”) must, as a core responsibility, propose to the Board of Directors (for approval and further proposal to the shareholders) the remuneration policy applicable to our directors. Further, the Appointments and Remuneration Committee also proposes to the Board of Directors, for approval, the remuneration of our senior managers and employees performing top-level management duties under the direct supervision of the Board.
As part of this core responsibility, in 2023, the Appointments and Remuneration Committee performed a thorough review of our previous director remuneration policy, obtained comments from shareholders, investors and other stakeholders, and obtained advise from independent consultant Mercer LLC. As a result of this process, our shareholders approved our current director remuneration policy at the general shareholders’ meeting held on June, 16, 2023 (the “Director Remuneration Policy”). The purpose of the new policy is to reinforce the alignment of our remuneration systems with our long-term strategic plan, shareholders’ interests and sustainability, all with prudent risk management and avoiding conflicts of interest.
The main changes in relation to the previous director remuneration policy are as follows: (i) the metrics and weight allocation of the short-term variable cash remuneration paid to our executive directors were modified, becoming directly linked to the results of the Grifols Group, and no longer including partial payment in Company Class B shares (Restricted Stock Units), now being fully payable in cash; (ii) a new long-term incentive plan for our Chief Operating Officer and Chief Corporate Officer, consisting of the award of stock options in respect to Class A shares and, under separate terms and conditions, the award of Class A shares to the Executive Chairperson; (iii) the main terms of the agreement entered into with our Executive Chairperson were set forth and (iv) the remuneration of the Company’s Honorary Chairperson was determined.
The new Director Remuneration Policy was in force and applicable during 2023 and will remain in force for fiscal years 2024 and 2025, unless our shareholders modify it at a general shareholders’ meeting.
128
Compensation of Members of the Board
Our Articles of Association generally set forth the processes for the determination of the compensation paid to the members of the Board. Article 20.bis of the Articles of Association provides that the directors’ remuneration shall be approved by the general shareholders’ meeting and shall apply for a maximum of three fiscal years. New directors’ remuneration policies must be approved by the general shareholders’ meeting prior to the last year of applicability of the previous policy, and any new policies approved may apply from the date of approval up to the three following years if so determined by the general shareholders’ meeting.
Pursuant to Article 26 of the Board Regulations, the Director Remuneration Policy (i) with respect to directors in their role as such, must necessarily determine the maximum amount of aggregate annual remuneration to be paid to all directors, as well as the criteria for its distribution taking into account the duties and responsibilities attributed to each of them and (ii) with respect to directors performing executive duties, must include the amount of annual fixed remuneration, the different parameters to set the variable components and the main terms and conditions of their contracts, including, in particular, duration, severance payments or compensations for the termination of the employment relationship, exclusivity, post-contractual non-competition, and retention or loyalty agreements. The Board then determines how much of the shareholder-approved aggregate compensation amount will be allocated to each director as compensation, taking into account a prior report of our Appointments and Remuneration Committee, in each case pursuant to the framework of the Articles of Association, the Director Remuneration Policy and the relevant agreements, as applicable.
Our directors are entitled to receive compensation in a fixed amount for serving as directors on our Board, pursuant to our Articles of Association and the Director Remuneration Policy. Director compensation for the performance of executive duties may consist of (i) a fixed amount, (ii) a variable amount based on financial and non-financial metrics, (iii) if applicable, compensations in certain cases of termination or dismissal, and (iv) may include the delivery of shares, or share options or amounts referenced to the value of the shares, subject to the requirements established by the legislation from time to time, in each case pursuant to the Articles of Association and the Director Remuneration Policy, as well as with the agreements approved in accordance with the provisions of the Spanish Companies Act.
The total compensation accrued by directors in the year ended December 31, 2023, in the aggregate, amounted to €12.2 million. This amount includes the remuneration of both executive and non-executive directors.
Non-executive Directors
Our independent directors, proprietary directors and those categorized as “Other External Directors” are non-executive directors (consejeros no ejecutivos). Our compensation philosophy, as set forth in Article 27 of the Board Regulations, provides that the remuneration of non-executive directors shall provide incentives for our directors to be dedicated and involved, while not creating an obstacle to their independence. To that end, Article 27 further establishes that the Board, following the advice of the Appointments and Remuneration Committee, shall take the necessary measures to ensure that non-executive directors’ remuneration adheres to the following guidelines: (a) their remuneration should be relative to their dedication, qualification and responsibility; and (b) they are excluded from any plans (x) consisting of the delivery of equity awards or options or other instruments linked to the value of our shares, (y) linked to our performance or (z) including retirement benefits. However, non-executive directors may be remunerated with our shares provided that they agree to hold such shares for the duration of the term as members of the Board.
Pursuant to the Director Remuneration Policy, our shareholders set the maximum gross annual amount at €100,000 payable per non-executive director, other than those non-executive directors that render paid professional services to us. Any director that is a member of one of the Board committees (Audit Committee, Appointments and Remuneration Committee and Sustainability Committee) receives an additional gross annual remuneration of €25,000 (thus, their total annual remuneration would amount to €125,000). Similarly, each committee chairperson receives an additional €25,000 (thus, their total annual remuneration would amount to €150,000). The lead independent director receives an additional remuneration amounting to €50,000 (thus, lead independent director’s total annual remuneration would amount to €150,000). Under no circumstances the remuneration of a non-executive director may exceed €150,000 per year.
129
Our executive directors and directors who render paid professional services to the Grifols Group do not receive any of remuneration solely for their capacity as director. Directors categorized as “other external directors” did not receive any remuneration in 2023. None of our directors received attendance fees for meetings of the Board or committees of the Board. Finally, pursuant to Article 20.bis of the Articles of Association, our directors are reimbursed for all expenses incurred in connection with their service as directors.
As of the date of this annual report, Ms. Enriqueta Felip Font, Mr. James Costos, Ms. Montserrat Muñoz Abellana, Ms. Susana González Rodríguez, Ms. Carina Szpilka Lázaro and Mr. Iñigo Sánchez-Asiaín Mardones are our independent directors in conformity with Exchange Act requirements and NASDAQ Listing Rules. Mr. Dagá serves as other external director (and not independent) and Mr. Albert Grifols Coma-Cros serves as proprietary director (and not independent) in conformity with Spanish corporate governance rules.
Honorary Chairperson
On September 30, 2022, Mr. Victor Grifols Roura stepped down from his position as chairperson of the Board, while remaining as a non-executive director, and was appointed as Honorary Chairperson of the Board. As the Honorary Chairperson, in addition to his attributions as director, Mr. Victor Grifols Roura assumed additional duties, including a role as a public ambassador of the Company.
We determined the compensation of Mr. Victor Grifols Roura for the years ended December 31, 2023 and 2022 taking into account his proven experience as director and former chairperson of the Board, in addition to his knowledge in the sector where we operate. When deciding his remuneration, which is the same fixed amount he had when he held an executive position (excluding variable amounts), we considered the additional duties that he began carrying out, as well as those set out in the Spanish Companies’ Act, the Articles of Association and the Remuneration Policy for the position of Honorary Chairperson of the Board. In the years ended December 31, 2023 and 2022, we paid Mr. Victor Grifols Roura for his roles as director and Honorary Chairperson a fixed annual amount of €965,000.
On December 18, 2023, Mr. Victor Grifols Roura resigned from his position as a member of the Board of Directors, due to his retirement. He now serves only as our Honorary Chairperson, non-member, of the Board, and will not receive any remuneration in 2024.
Executive Directors
Certain members of our Board of Directors also perform executive functions for the Company (our current Executive Chairperson, Chief Executive Officer, Chief Operating Officer and Chief Corporate Officer).
The compensation for executive directors relating to their executive functions may consist of (i) a fixed amount, (ii) a variable amount based on financial and non-financial metrics including, among others, environmental, social and governance (“ESG”) objectives (in 2023, 10.0% of variable remuneration was linked to ESG factors, 25.0% of which are environmental, 40.0% social and 35% good corporate governance), (iii) if applicable, compensations in certain cases of termination or dismissal, and (iv) the delivery of shares, or share options or amounts referenced to the value of the shares, subject to the requirements established by the legislation from time to time.
Our Chief Corporate Officer and Chief Operating Officer accrued, in the aggregate, €3.4 million in cash remuneration in 2023 (€1.9 million in fixed compensation and €1.5 million in variable compensation, which has been paid).
130
We determined the individual compensation of our executive directors accrued during 2023 in accordance with the Director Remuneration Policy applicable for the year ended December 31, 2023. However, part of the variable compensation we paid to our Chief Operating Officer and Chief Corporate Officer in 2023 corresponds to amounts computed pursuant to the variable remuneration plan established in our previous directors’ remuneration policy, which became effective on June 10, 2022 and is no longer in force. In February 2023, the Board awarded to each of our Chief Corporate Officer and Chief Operating Officer variable remuneration for the year ended December 31, 2022, in an amount equal to 60.66% of their respective annual fixed remunerations. After voluntarily waiving the right to receive 50% of such variable remuneration, our Chief Corporate Officer and Chief Operating Officer each received the amount of €271,444.55 in March 2023. We did not allocate any restricted stock units (RSUs, with a two-year vesting period) in the year ended December 31, 2021. Hence, we awarded no Class B shares to our executive directors in 2023.
For the year ended December 31, 2023, Mr. Thomas Glanzmann, our Executive Chairperson of the Board accrued €1.1 million as fixed remuneration and €0.8 million in variable compensation, both payable in cash. In addition, we awarded Mr. Thomas Glanzmann on a one-time basis stock options to acquire 700,000 of our Class A shares, with a vesting period of two years, pursuant to our Executive Chairman share-based payment plan. See “—E. Share Ownership—Executive Chairman share-based payment plan.”
Mr. Steven F. Mayer, who served as the Executive Chairperson of the Board from September 30, 2022, to February 21, 2023, received the aggregate amount of €751,354 in 2023. He received this amount from our U.S. subsidiary Grifols Shared Services North America, Inc. for the exercise of executive functions inherent to his position as Executive Chairperson of said subsidiary. Further to this amount, Mr. Mayer received €5.0 million due to the termination of his contractual relationship with Grifols Shared Services North America, Inc. Mr. Mayer did not receive any remuneration from Grifols, S.A. for his former position as Executive Chairperson of Grifols, S.A.
Other Members of Senior Management
In 2023, members of our senior management (excluding those who also served as members of the Board) accrued compensation amounting to €23.7 million, in the aggregate. This figure includes accruals for contingent or deferred compensation earned in respect of 2023 service. In 2022, members of our senior management (excluding those who also served as members of the Board) were paid compensation amounting to €13.9 million in the aggregate. This figure includes accruals for contingent or deferred compensation earned in respect of 2022 service. In 2021, members of our senior management (excluding those who also served as members of the Board) were paid compensation amounting to €15.1 million in the aggregate. This figure includes accruals for contingent or deferred compensation earned in respect of 2021 service.
The breakdown of the aggregate amount paid to our senior management (excluding those who also served as members of the Board) for discharging their duties in the years ended December 31, 2023, 2022 and 2021 is set forth in the table below.
Amount paid in the year | ||||||
ended December 31, | ||||||
Component |
| 2023 |
| 2022 |
| 2021 |
(in euros) | ||||||
Salaries |
| 14,567,449 |
| 8,489,991 |
| 11,074,903 |
Variable Compensation |
| 9,131,372 |
| 5,400,772 |
| 4,061,044 |
Stock options or other securities |
| — |
| — |
| — |
Other — e.g., life and health insurance |
| 34,864 |
| 68,301 |
| 119,510 |
Other — e.g., pensions/savings |
| 30,776 |
| 124,677 |
| 125,327 |
131
Executive Compensation Clawback Policy
On October 26, 2022, the SEC amended Rule 10D-1 and other rules under Section 10D of the Exchange Act to implement the incentive-based compensation recovery provision, referred to as the “clawback” provision, of the Dodd-Frank Wall Street Reform and Consumer Protection Act (“Clawback Rules”). The Clawback Rules required national securities exchanges (such as NASDAQ) to adopt listing standards under which issuers must implement and enforce policies that require the clawback of incentive-based compensation received by any current or former executive officer during the three fiscal years immediately preceding the date of a required restatement of an issuer’s filed financial statements due to the issuer’s material noncompliance with any financial reporting requirement under the securities laws.
In June 2023, the SEC approved the proposed clawback listing standards issued by NASDAQ. On October 19, 2023, our Board of Directors approved our “Clawback Policy for the Recovery of Erroneously Awarded Compensation for the Senior Management,” to comply with the Clawback Rules. The policy is attached to this annual report as Exhibit 97.1.
Employment and Severance Arrangements
We have entered into employment contracts with eight members of our senior management that entitle them to unilaterally rescind their employment contracts and receive termination benefits of two to five years’ salary in the event that we undergo a change of control. In addition to this, five members of our senior management are contractually entitled to termination benefits of one to two years’ salary under certain circumstances other than a change of control.
See Notes 29(c) and 31(a) to our audited consolidated financial statements included in this annual report for further details of the payments received by employees.
Equity and Other Incentive Programs
See “—E. Share Ownership” for a description of our equity-based compensation plans available to eligible employees, including members of our senior management.
Pension and Retirement Compensation Programs
Our directors and senior management employed by our U.S. subsidiaries participate in a tax-qualified 401(k) plan on the same terms as our other employees. The aggregate amount of employer contributions to the 401(k) plans for our directors and senior management during 2023 was €30.8 million. In 2023, neither we nor our subsidiaries set aside or accrued any other amounts to provide pension, retirement or similar benefits for our directors or senior management.
C. | Board Practices |
Board of Directors
Pursuant to the Articles of Association, we are managed by a board of directors (the “Board”), which may be composed of not less than three and not more than 15 directors. Our current Board has 12 directors. Directors must be individuals. Under Spanish law, the Board is responsible for management, administration and representation in all matters concerning the business, subject to the provisions of the Articles of Association and the powers conferred at the general shareholders’ meeting.
Appointment and Dismissal
Pursuant to Spanish law and our Articles of Association, directors are elected by our shareholders to serve for a term of four years and may be reelected to serve for an unlimited number of terms, except in the case of independent directors, who pursuant to Spanish law and the regulations of our Board originally approved at the Board meeting held on April 5, 2006, as amended from time to time (the “Board Regulations”), shall not serve as such for more than 12 years. We do not provide for the reelection of directors at staggered intervals or cumulative voting for such directors or otherwise.
132
A director must be an individual. If a director ceases to hold office prior to the expiration of his or her term, the Board may fill the vacancy by appointing a new director to replace the outgoing director. Any director so appointed will hold office until the next general shareholders’ meeting when the appointment may be confirmed or revoked by our shareholders. If such appointment takes place between the time that a general shareholders’ meeting is called and the time the meeting takes place, then the director so appointed will hold office until the next general shareholders’ meeting, when this appointment is to be confirmed or revoked. Any such appointment will be only for the remainder of the term of the outgoing director, without prejudice to such director’s eventual election. A director may resign, or be removed, from office by a resolution of our general shareholders’ meeting at any time. A director who is also a shareholder may vote freely on any of our shareholders’ resolutions relating to the appointment and dismissal of directors (including the appointment or dismissal of that director).
In addition, pursuant to the Board Regulations, a director must tender a resignation to the Board and the Board may accept such resignation, in its discretion, under the following circumstances: (i) when the director ceases to hold the executive position to which such director’s appointment to the Board was related; (ii) when circumstances arise that might harm the Company’s name or reputation, related or not to their actions within the Company; (iii) when the director becomes unable to hold the office due to a legal cause of ineligibility or incompatibility; (iv) when any criminal charges are brought against or a formal inquiry is opened against him or her by a regulator; (v) when the director has been severely admonished by our Audit Committee for having breached his or her duties as director; (vi) when the director’s participation on the Board may jeopardize our interests or when the reasons for his or her appointment cease to exist; and (vii) in the case of a proprietary director, when the relevant shareholder ceases to hold its stake in us, or reduces its stake below the level that reasonably justified the appointment of such director. When a director leaves his/her position, whether by resignation or resolution of the general shareholders’ meeting before his/her tenure expires, he/she shall explain, in sufficient detail, the reasons behind this decision or, in the case of non-executive directors, his/her opinion of the reasons for the general shareholders’ meeting resolution, in a letter that must be sent to the members of the board via the chairperson or the secretary.
In addition, under Spanish corporate law, a holder of voting shares (or group of shareholders of voting shares acting together) may, subject to availability of seats on the Board, appoint a number of directors proportionate to that shareholder’s (or group of shareholders’) interest in our voting capital. If the voting capital stock represented by the shares held by such shareholder (or group of shareholders) is equal to or greater than the result of dividing our total voting capital stock by the number of directors, such shareholder (or group of shareholders) shall have the right to appoint a proportionate number of directors. For example, a shareholder holding 20 voting shares out of a total of 100 voting shares in a company with five directors will be entitled to appoint one director. Should this power be exercised, shares so pooled shall not participate in the voting for the other members of the Board. However, they may exercise their voting rights with respect to the removal of existing directors. Since such rights apply only to voting shares or Class B shares that have recovered their voting rights, our Class B shares and the Class B ADSs that represent them in the United States do not count towards the proportional representation right.
The Board must appoint a Chairperson of the Board from among its members. Mr. Thomas Glanzmann is the current Executive Chairperson. The Board may also designate one or more Vice Chairperson, who shall be numbered consecutively, and who shall replace the Chairperson in the event of impossibility to act or absence. Mr. Raimon Grifols Roura is the current Vice Chairperson. The Board may appoint an Honorary Chairperson that is not required to be a member of the Board. The Honorary Chairperson shall have duties of honorary representation and will provide advice to the Board to the Chairperson and to the Vice Chairperson of the Board. Currently, Mr. Victor Grifols Roura is the Honorary Chairperson, and a non-member of the Board.
The Board must also appoint a Secretary and may also designate one or more Vice-Secretaries. Neither the Secretary nor the Vice-Secretary is required to be a member of the Board; however, the Secretary or the Vice-Secretary will not be entitled to vote on matters before the Board unless he or she is a member of the Board. Ms. Nuria Martín Barnés is the current Secretary non-member of the Board, while Ms. Laura de la Cruz Galán is the current Vice-Secretary, non-member, of the Board.
133
Meetings of the Board
Pursuant to the Articles of Association, a meeting of the Board may be called by the Chairperson whenever he considers such a meeting necessary or suitable. The Chairperson is also required to call a meeting at the request of one-third of the directors. Meetings of the Board are called using any means of notice at least ten days before the date of the meeting, unless exigent circumstances require a shorter term. Such notice of a meeting of the Board must state the place, date and time as well as the issues to be discussed. The Board is required by Spanish law to hold a meeting at least every three months. Our Articles of Association provide that a majority of the directors (half plus one of the directors present at a meeting) of the Board (represented in person or by proxy by another director on the Board; non-executive directors may only appoint another non-executive director to represent them) constitutes a quorum. Except as otherwise provided by law or specified in the Articles of Association, resolutions of the Board must be passed by an absolute majority of the directors present or represented at a meeting, with the Chairperson having the right to cast a deciding vote in the event of a tie.
Pursuant to the Articles of Association the Board may hold meetings by videoconference, conference call or by any other distance communication systems as long as said communications take place in real time and therefore, in one sole act, and both the identity of the participating or voting individual and the security of the electronic communications, are properly guaranteed.
Delegation of Powers
Pursuant to Spanish law and our Articles of Association, the Board may delegate its powers either to an executive committee (Comisión Ejecutiva) or to one or more chief executive officers. Spanish corporate law provides that resolutions appointing an executive committee, any chief executive officer or authorizing the permanent delegation of all, or part of, such board of directors’ powers, requires a two-thirds majority of the members of such board of directors and the registration of such resolution in the Spanish Commercial Registry (Registro Mercantil). The Board may also revoke such powers at any time. In addition, when a member of the Board is appointed chief executive officer or vested with executive functions, he/she will need to enter into an agreement with the Company, which shall be approved by a two-thirds majority of the Board. The director in question will have to refrain from participating in the deliberation and voting process of such agreement.
Under Spanish corporate law, a board of directors may also grant general or specific powers of attorney to any person whether or not that person is a director or a shareholder. General powers of attorney must be registered in the Commercial Registry. However, Spanish law provides that the following powers, among others, may not be delegated by the Board: (i) the formulation and submission for approval of the yearly financial statements at the general shareholders’ meeting; and (ii) those powers granted to the board of directors by a general shareholders’ meeting (unless otherwise provided in the relevant shareholders’ resolution).
Mr. José Ignacio (Nacho) Abia Buenache is the current Chief Executive Officer of the Company, with delegation of all powers legally delegable from the Board. Further, Mr. Thomas Glanzmann currently serves as Executive Chairperson of the Board, with delegation of all powers legally delegable from the Board.
Expiration of Current Terms
The periods during which our directors and senior management have served in their offices, as well as the date of expiration of each director’s term, are shown in the tables under “—A. Directors and Senior Management” above.
Termination Benefits
We have entered into employment contracts with all members of our senior management that entitle them to unilaterally rescind their employment contracts and receive termination benefits of two to five years’ salary in the event that we undergo a change of control. In addition to this, six members of our senior management are contractually entitled to termination benefits of one to four years’ salary under certain circumstances other than a change of control.
See Notes 28(c) and 30(a) to our audited consolidated financial statements included in this annual report on Form 20-F for further details of the payments received by employees.
134
Diversity
On August 6, 2021, the U.S. Securities and Exchange Commission approved NASDAQ’s proposal to amend its listing standards to encourage greater board diversity and to require board diversity disclosures for NASDAQ-listed companies. Pursuant to the amended listing standards, Grifols, as a foreign private issuer, is presently required to have at least one diverse member of the Board and two diverse members of the Board by August 6, 2025, or explain the reasons for not meeting this objective. Furthermore, a Board diversity matrix is required to be included in the Annual Report on Form 20-F, containing certain demographic and other information regarding members of the Board.
Grifols currently complies with the diversity requirement, as we currently have four female members on our Board of Directors. The Board diversity matrix is set out below.
Board Diversity Matrix (status as of the date of this annual report)
Country of Principal Executive Offices | Spain | |||
Foreign Private Issuer | Yes | |||
Disclosure Prohibited under Home Country Law | Yes(1) | |||
Total Number of Directors | 12 | |||
Female | Male | Non-Binary | Did not Disclose Gender | |
Part I: Gender Identity | ||||
Directors | 4 | 8 | n.a. | n.a. |
Part II: Demographic Background | ||||
Underrepresented Individual in Home Country Jurisdiction | n.a. | |||
LGBTQ+ | n.a. | |||
(1) | In accordance with Article 9.1 of the EU General Data Protection Regulation (GDPR) (Regulation (EU) 2016/679 of 27 April 2016), the categories of data contained in Part I and Part II of the above table are considered “special categories of personal data” (which includes, among others, personal data revealing racial or ethnic origin, or a natural person’s sex life or sexual orientation). The same article prohibits the processing of these special categories of personal data, unless any of the derogations regulated in Article 9.2 of GDPR apply. |
One of the derogations to this prohibition is contained in Article 9.2.a) of GDPR, which provides that the data subject has given explicit consent to the processing of those personal data for one or more specified purposes, except where Union or Member State law provide that this prohibition may not be lifted by the data subject. Precisely on this point, Article 9.1 of the Spanish Organic Act 3/2018 (Spanish Data Protection Act) provides that, in order to avoid discriminatory situations, the sole consent of natural persons is not enough to lift the general prohibition of processing said special categories of personal data in Spain. Given this general prohibition in Spain, we consider that the lawful manner by which Grifols could process such categories of personal data from its Directors would either be based: (a) on the existence of a EU or EU Member State legal instrument with the status of law applicable to Grifols that enables said processing for reasons of substantial public interest (based on the derogation contained in Article 9.2.g) of GDPR, jointly with Article 9.2 of the Spanish Data Protection Act), or; (b) on special categories of personal data which are manifestly made public by the corresponding Director (based on the derogation contained in Article 9.2.e) of GDPR).
In conclusion, Grifols cannot collect nor share such personal data because (i) there is no legal instrument with the status of law applicable to Grifols that enables such processing and, further, and (ii) Grifols would not be in a position to ascertain whether or not the corresponding Director has manifestly made public such sensitive personal data.
The Policy on Director Diversity in the composition of the Board of Directors was initially approved by the Board of Directors of Grifols on February 22, 2019 and amended on December 11, 2020, with the aim of formalizing the procedures and guidelines followed during the selection process of candidates to form part of the Board of Directors of Grifols to favor an appropriate composition of the same.
135
In accordance with the recommendations set out in the Code of Good Governance of Listed Companies (Código de Buen Gobierno de las Sociedades Cotizadas) published by the CNMV, the policy has always had two aims:
| ● | to guarantee that any proposal for the appointment or re-election of members of the Board of Directors be based on a prior analysis of the required skills by the Board of Directors; and |
| ● | to support knowledge, experience, age and gender diversity. |
The Board of Directors of Grifols must ensure that the selection process promotes balance and diversity in age, gender, experience and knowledge, as well as that it be free from any bias that may infer any kind of discrimination, in particular, on grounds of gender, disability or any other personal condition. In this respect, any discriminatory circumstance by reason of gender obstructing or hindering the appointment of a female candidate to the Board of Directors must be avoided. The policy provides that efforts must be made to ensure that the Board of Directors has a diverse and balanced composition as a whole, so that its composition enriches the analysis and debate and contributes plural points of view and positions in matters within its competence. In this respect, Grifols must continue to promote measures that encourage Grifols to have a significant number of women in senior management roles.
Committees of the Board
The Board has an Audit Committee, an Appointments and Remuneration Committee and a Sustainability Committee. The following is a brief description of such committees.
Audit Committee
The Board established an Audit Committee in compliance with Articles 24.bis and 24.ter of the Articles of Association and Article 14 of the Board Regulations.
The regulations applicable to the Audit Committee are set forth in the provisions referred to above, as well as the bylaws of the Audit Committee, which were approved by the Board and the Audit Committee on December 9, 2008, and modified in February 2023 in order to adapt its content to the current recommendations of the Good Governance Code of Listed Companies. In connection with the Talecris Biotherapeutics acquisition, at a Board meeting held on May 24, 2011, the Articles of Association and Board Regulations were amended to conform to NASDAQ Listing Rules and to facilitate the listing of our Class B ADSs on NASDAQ. Furthermore, the bylaws of the Audit Committee were modified at a Committee meeting held on March 31, 2015, to adapt them to the requirements imposed by Law 31/2014. In 2017, article 24.ter of the Articles of Association and Article 14 of the Board Regulations concerning the composition and functions of the Audit Committee were amended in order to adequate their content to the latest amendments of the Spanish Companies Act introduced by the currently in force Spanish Audit Act.
Pursuant to our Spanish corporate governance requirements and our Articles of Association and the Board Regulations, the Audit Committee consists of a minimum of three directors and a maximum of five directors who are appointed by the Board based on such directors’ knowledge, competence and experience in accounting, audit and risk management matters (both financial and non-financial). All of the members of the Audit Committee must be non-executive directors, and the majority must be independent directors. As a group, the members of the Committee must have the pertinent technical knowledge in relation to the sector of activity of the Company. In addition, all members of the Audit Committee, including the chairperson, must meet the independence, experience and other requirements set forth in the Exchange Act and NASDAQ Listing Rules.
The responsibilities of the Audit Committee include:
| ● | reporting to the shareholders at general shareholders’ meetings regarding matters for which the Audit Committee is responsible; |
| ● | recommending to the Board the selection, appointment, re-election, hiring and replacement of the external auditor regardless of the faculties vested in the general shareholders’ meeting and the Board with regard to the approval of such resolutions under Spanish law; |
136
| ● | oversight of our internal audit department, including selecting, appointing and dismissing its manager, monitoring its budget, receiving periodic information on the department’s activities and ensuring that management takes the conclusions and recommendations of the department’s reports into account; |
| ● | setting up and supervising procedures for the receipt, retention and treatment of complaints regarding accounting, internal controls or auditing matters, as well as the confidential and anonymous submission by employees and other persons related to Grifols of concerns regarding questionable accounting or auditing matters; |
| ● | exercising oversight of the process for gathering financial and non-financial information and the related internal control system; reviewing the financial statements and the periodic financial statements that should be submitted to the securities regulatory authorities and ensuring that the appropriate accounting standards are followed; reporting to the Board on any change in the accounting standards and on balance sheet and off balance sheet risks; |
| ● | supervising and evaluating the efficiency of our internal control, internal audit and risk control and management systems, financial and non-financial, including any operative, technological, cybersecurity, legal, social, environmental, political, reputational or corruption related risks; |
| ● | receiving information from the auditors including relating to auditor independence and conduct of audits of the financial statements, and issuing on an annual basis a written opinion on the independence of the auditor; |
| ● | ensuring that the external auditor holds an annual meeting with the full Board of Directors to report on the work carried out and on the evolution of our accounting and risk situation; |
| ● | ensuring that the remuneration paid to the external auditor for its work does not compromise its quality nor its independence; |
| ● | reporting on the related-party transactions to be approved by the general meeting or the board of directors and supervise the internal procedure for those whose approval has been delegated; |
| ● | supervising any transactions entered into with significant shareholders as set forth in the Board Regulations; and |
| ● | (i) ensuring compliance with the Internal Code of Conduct of Grifols in Matters Relating to the Stock Market, or Stock Market Code of Conduct, the Code of Conduct for Grifols’ Employees, the Board Regulations (each available on our website, at www.grifols.com) and, in general, any other corporate regulations and (ii) making any necessary proposals to improve such regulations. |
The Audit Committee currently consists of Mr. Iñigo Sánchez-Asiaín Mardones, Ms. Montserrat Muñoz Abellana and Ms. Carina Szpilka Lázaro. Each of Mr. Sánchez-Asiaín, Ms. Muñoz and Ms. Szpilka is independent in conformity with Exchange Act requirements and NASDAQ Listing Rules, as well as in conformity with the Spanish Companies Act. Ms. Laura de la Cruz Galán serves as Secretary, non-member, of the Audit Committee, having replaced Mr. Tomás Dagá in such role on April 17, 2024.
On April 13, 2023, we received a deficiency letter from the Listing Qualifications Staff of NASDAQ. The notice was prompted by our inadvertent falling out of compliance with NASDAQ Listing Rule 5605(c)(2)(A), which requires that issuers have an audit committee of at least three members who each meet the criteria for independence set forth in Rule 10A-3(b)(1) under the Securities Exchange Act of 1934. The letter does not affect our listing on NASDAQ or our operations.
We fell out of compliance due to the appointment of Mr. Tomás Dagá to our audit committee, on September 30, 2022. Although Mr. Dagá’s appointment was compliant with the audit committee composition requirements of our home Spanish regulator, it did not meet the independence requirements of NASDAQ Listing Rule 5605(c)(2)(A) and Rule 10A-3(b)(1) under the Securities Exchange Act of 1934 as Mr. Dagá is an external non-independent director.
As acknowledged by NASDAQ in its deficiency letter, the appointment on April 13, 2023, of Ms. Montserrat Muñoz to the audit committee to replace Mr. Dagá and cured this deficiency.
137
Appointments and Remuneration Committee
The Board established an Appointments and Remuneration Committee in compliance with Article 24.bis and 24. quater of the Articles of Association and Article 15 of the Board Regulations.
Pursuant to Spanish corporate governance requirements and Article 15 of the Board Regulations, the Appointments and Remuneration Committee is required to consist of between three and five members, all of which must be non-executive directors, which includes at least two independent directors.
The responsibilities of the Appointments and Remuneration Committee include:
| ● | assisting in the nomination of directors, including evaluating potential nominees in light of the level of knowledge, competence and experience necessary to serve on the Board; |
| ● | establishing a representation target for the gender that is least represented on the Board and prepare guidelines to achieve said target; |
| ● | reporting and making proposals to the Board on the appointment of members to the various committees of the Board and on the persons who should hold the office of Secretary and Vice-Secretary of the Board; |
| ● | examining and organizing the orderly and planned succession of the Chairperson of the Board and the Chief Executive Officer; |
| ● | reporting on proposals for the appointment and removal of any members of senior management made by the Chief Executive Officer; |
| ● | making proposals on the remuneration plans for the Board and senior management; |
| ● | periodically reviewing the remuneration plans of senior management, including considering their suitability and performance; |
| ● | reporting on transactions in which directors may have a conflict of interest and ensuring that potential conflicts of interest do not impair the independence of any external advice provided to the committee; and |
| ● | periodically reviewing the remuneration policy applied to directors and senior management and ensuring that their individual remuneration is proportionate to that paid to other directors and senior management. |
Consistent with NASDAQ Listing Rules for foreign private issuers, our Appointments and Remuneration Committee currently consists of Ms. Carina Szpilka Lázaro, Mr. Tomás Dagá Gelabert and Ms. Susana González Rodríguez as directors. Each of Ms. Szpilka and Ms. González is independent, in conformity with Exchange Act requirements and NASDAQ Listing Rules and Mr. Dagá is considered an “Other External” director under the Spanish Companies Act. Ms. Nuria Martín Barnés serves as Secretary, non-member, of the Appointments and Remuneration Committee.
Sustainability Committee
In its meeting held on December 11, 2020, the Board resolved to amend certain articles of the Board Regulations, in order to adapt its content to certain recommendations of the reform of the Good Governance Code of Listed Companies published in June 2020 by the CNMV, and created a Sustainability Committee.
138
The regulations applicable to the Sustainability Committee are set forth in article 15 bis. of the Board Regulations, as well as in the Regulations of the Sustainability Committee, which were approved by the Board on February 19, 2021 and amended in February 2023. Pursuant to Article 15 bis of the Board Regulations and the Regulations of the Sustainability Committee, the Sustainability Committee is required to consist of between three and five members, all of which must be non-executive directors, the majority of them being independent.
The responsibilities of the Sustainability Committee include:
| ● | monitoring compliance with the Company’s internal codes of conduct and corporate governance rules, and ensuring that the corporate culture is aligned with its purpose and values; |
| ● | monitoring the implementation of the general policy regarding the disclosure of economic-financial, non-financial and corporate information, as well as communication with shareholders and investors, proxy advisors and other stakeholders. Similarly, the way in which the Company communicates and relates with small and medium-sized shareholders should be monitored; |
| ● | periodically evaluating the effectiveness of the Company’s corporate governance system and environmental, climate change and social policy to confirm that it is fulfilling its mission to promote the corporate interest and catering, as appropriate, to the legitimate interests of remaining stakeholders; |
| ● | ensuring the Company’s environmental, climate change and social practices are in accordance with the established strategy and policy; and |
| ● | monitoring and evaluating the Company’s interaction with its stakeholder groups. |
The Sustainability Committee currently consists of Mr. James Costos, Ms. Montserrat Muñoz Abellana and Ms. Enriqueta Felip Font. Each of the members is independent, in conformity with Exchange Act requirements and NASDAQ Listing Rules. Ms. Nuria Martín Barnés serves as Secretary, non-member, of the Sustainability Committee.
The Sustainability Steering Committee is a multidisciplinary and international team created in 2021 coordinated by the Investor Relations and Sustainability Department, which reports to the Sustainability Committee. Among its functions, the committee fosters ongoing dialogue to identify, establish, implement and confirm compliance with Grifols Master Plan objectives, and generates and coordinates the reporting of nonfinancial and corporate sustainability information.
D. | Employees |
The table below indicates the number of employees by department as of December 31, 2023, 2022 and 2021:
As of December 31, | ||||||
Department | 2023 | 2022 | 2021 | |||
Manufacturing |
| 18,979 |
| 21,235 |
| 18,735 |
Research & development — technical area |
| 1,255 |
| 1,271 |
| 1,088 |
Administration and others |
| 1,686 |
| 1,870 |
| 1,609 |
General management |
| 267 |
| 302 |
| 313 |
Marketing |
| 155 |
| 167 |
| 203 |
Sales and distribution |
| 1,395 |
| 1,469 |
| 1,285 |
Total |
| 23,737 |
| 26,314 |
| 23,233 |
139
The table below indicates the number of employees by geographic region as of December 31, 2023, 2022 and 2021:
As of December 31, | ||||||
Geographic Region |
| 2023 |
| 2022 |
| 2021 |
Spain |
| 4,181 |
| 4,224 |
| 4,163 |
North America |
| 14,076 |
| 16,862 |
| 16,393 |
Rest of the World |
| 5,480 |
| 5,228 |
| 2,677 |
Total |
| 23,737 |
| 26,314 |
| 23,233 |
The decrease in the number of employees is related to the Operational Improvement Plan. See Item 5 of this Part I, “A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Operational Improvement Plan.”
We actively train our employees. The Grifols Academy opened in Spain during the second quarter of 2011. It is a meeting point for advanced training on all processes related to the preparation and production of plasma-derived medicines. The Grifols Academy acts as a center of technical, scientific and management training for the Grifols Group’s personnel, fostering a continued exchange among experts and external bodies, such as professional healthcare associations, hospitals, schools and universities. Through the Grifols Academy, we offer to our employees technical training and professional development opportunities, including an educational expenses reimbursement program, a number of long-term leadership development initiatives and onboarding processes conducted with virtual reality technology. In the last three years, an annual average of 12,100 of our employees participated in the Grifols Academy’s professional development and plasmepherisis training programs.
In 2023, we launched our Skillsoft Percipio platform, accessible to all Grifols employees. Using this platform, our employees throughout the various countries we operate in can enhance their skillsets from any device by accessing 8,400 virtual courses available in 18 languages and covering a wide array of topics, including digital transformation, leadership, diversity, equality and inclusion, collaboration, personal wellbeing, productivity and process improvements.
Our Spanish employees are mainly represented by three labor unions, the Workers’ Commissions (Comisiones Obreras - CCOO), the Workers General Union (Unión General de Trabajadores - UGT) and the General Labor Confederation (Confederación General del Trabajo - CGT). The employees of some of our subsidiaries in Spain, Germany, Italy, France, Argentina and Brazil are covered by collective bargaining agreements. The remainder of our employees are not represented by labor unions. We have not experienced any significant work stoppages in the last 15 years. We generally consider our employee relations to be good.
We subscribe to an insurance policy that covers death or permanent disability of employees caused by work accidents. All of our employees are covered under this policy. We implemented a defined contribution pension plan for all our Spanish entities beginning on January 1, 2002, which excludes top management and which requires us to make matching payments to these employees. Our contribution to this pension plan was €1.2 million in the year ended December 31, 2023, compared to €1.0 million and €0.9 million in the years ended December 31, 2022 and 2021, respectively. We also sponsor a savings plan for the benefit of U.S. employees, which qualifies as a defined contribution plan under Section 401(a) of the Internal Revenue Code of 1986, as amended. We make fully vested matching contributions to the savings plan, which totaled $33.4 million in 2023, compared to $34.1 million and $31.8 million for the years ended December 31, 2022 and 2021 respectively. For certain employees in Germany, we have a defined benefit pension plan, as required by statutory law. The pension cost relating to this plan is not material.
E. | Share Ownership |
For information on the direct, indirect and represented holdings of our current directors and executive officers with respect to our Class A shares as of December 31, 2023 see Item 7 of this Part I, “Major Shareholders and Related Party Transactions—A. Major Shareholders.”
In March 2022, we established a Restricted Share Plan (“RSU”) under which eligible employees are entitled to receive up to 50% of their annual bonus in Class B Shares or Class B ADSs and we match this with an additional 50% contribution in RSUs. The Class B shares or ADSs delivered under this plan were valued at the date of grant of the employee bonus. The RSUs have a vesting period of two years and one day, after which period the RSUs may be exchanged for Class B Shares or ADSs. If an eligible employee leaves the company or is terminated prior to the vesting period, he/she will not be entitled to the additional RSUs. As of December 31, 2023, the share-based payments cost of employees under the RSU Plan was to €8.3 million.
140
In 2023, our Board of Directors made modifications to our Remunerations Policy, including the discontinuation of this partial payment of variable compensation in RSUs. Instead, our Board created two new variable compensation plans, which are described below. For more details regarding our Remunerations Policy, see “—B. Compensation.” For more information regarding our variable compensation plans, see Note 29(c) to our audited consolidated financial statements included in this annual report.
Equity-settled share-based payment plan
In 2023, we approved a four-year long-term incentive plan for certain executive directors, members of the senior management of Grifols, S.A. and our subsidiaries. Under this plan, these individuals, including each of our Chief Operating Officer and our Chief Corporate Officer, have the right to receive a certain number of options representing the right to acquire certain Class A shares for an exercise price of €8.96 per share. Given our transition to a new Chief Executive Officer, these stock options will not vest for the Chief Operating Officer and Chief Corporate Officer, as such positions will no longer exist following the transition. Of the options granted, 40% will vest on the second anniversary of the plan and the remaining 60% will vest upon the fourth anniversary of the plan, in each case subject to certain vesting conditions. A maximum of 4.0 million stock options, representing the right to acquire 4.0 million Class A Shares, will be granted under the equity-settled share-based payment plan. As of December 31, 2023, the total accumulated amount recognized in our Consolidated Statement of Profit and Loss in connection with this plan was €2.6 million.
Cash-settled share-based payment plan
In May 2023, our Board of Directors approved a four-year long-term incentive plan for certain members of our management team. The plan is based on the award of restricted stock units (RSUs), of which 50% will vest upon the second anniversary of the plan and the remaining 50% will vest at the end of the fourth year of the plan. The RSUs granted under this plan will be settled in cash for the amount equivalent to the average price of the Class A Shares during the five business days preceding the settlement date. As of December 31, 2023, the total accumulated amount recognized in our Consolidated Statement of Profit and Loss in connection with this plan was €1.7 million.
Executive Chairman share-based payment plan
In June 2023, our shareholders approved a long-term variable remuneration for our Executive Chairperson, Mr. Thomas Glanzmann. Under such plan, he was awarded 700,000 stock options representing the right to acquire 700,000 Class A Shares at an exercise price established by the shareholders at a general shareholders’ meeting. The stock options will vest on the second anniversary of the award date, subject to Mr. Thomas Glanzmann passing a performance evaluation to be conducted by the Board of Directors and the Appointments and Remuneration Committee.
This plan was created to reward the efforts required to execute our Operational Improvement Plan. It grants a one-time basis award made in 2023 of 700,000 Class A shares.
F.Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation.
There was no erroneously awarded compensation paid to our executive officers in 2023.
141
Item 7.MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A. | Major Shareholders |
The following table sets forth certain information, including information regarding beneficial ownership of our Class A (voting) shares as of the date of this annual report, for (i) our major shareholders, including, in accordance with applicable Spanish regulations, each person or entity that is known to us to be the beneficial owner of more than 3% of our Class A shares or 1% of our Class A shares in the event of a person or entity domiciled in a tax haven, (ii) each of our directors and (iii) each member of our senior management.
Since our Class A shares are represented through book entries, their exact ownership structure cannot be known, except through the information that the shareholders provide voluntarily or in compliance with applicable regulations, and information provided by the Sociedad de Gestión de los Sistemas de Registro, Compensación y Liquidación de Valores, S.A., or Iberclear, on which the shares are settled and cleared, and its participant entities (entidades participantes).
Beneficial ownership is determined in accordance with applicable Spanish regulations.
| Number of |
| Percentage of |
| |
Voting | Voting |
| |||
Name of Beneficial Owner | Shares | rights |
| ||
Major Shareholders |
|
|
|
| |
Deria S.A.(1) | 39,183,692 |
| 9.195 | % | |
Scranton Enterprises B.V.(2) | 35,812,622 |
| 8.404 | % | |
Ponder Trade, S.L. | 30,209,093 |
| 7.089 | % | |
The Goldman Sachs Group, Inc.(3) | 26,475,668 | 6.214 | % | ||
Ralledor Holding Spain S.L. (4) | 26,224,374 |
| 6.154 | % | |
Capital Research and Management Company(5) | 19,412,817 |
| 4.555 | % | |
Blackrock, Inc.(6) | 18,341,400 |
| 4.304 | % | |
JP Morgan Chase & Co. (7) | 16,410,073 | 3.851 | % | ||
Europacific Growth Fund(8) | 13,831,807 | 3.227 | % | ||
Jeffries Financial Group Inc.(9) | 13,115,305 | 3.078 | % | ||
BNP Paribas Asset Management Europe (10) | 13,001,544 | 3.051 | % | ||
Rokos Global Macro Master Fund LP(11) | 4,843,786 | 1.137 | % | ||
Melqart Opportunities Master Fund Ltd(12) | 4,790,811 | 1.124 | % | ||
Directors |
|
|
|
| |
Thomas H. Glanzmann (13) | 221,322 | * | |||
José Ignacio (Nacho) Buenache | 92,807 |
| * | ||
Tomás Dagá Gelabert (14) | 303,661 |
| * | ||
Víctor Grifols Deu(15) | 107,834 |
| * | ||
Raimon Grifols Roura | 41,118 |
| * | ||
Carina Szpilka Lázaro | 1,490 |
| * | ||
Albert Grifols Coma-Cros | 66,000 |
| * | ||
Senior Management |
|
|
|
| |
David Ian Bell | 20,000 |
| * | ||
Victor Grifols Deu | 107,834 |
| * | ||
Raimon Grifols Roura | 41,118 | * | |||
José Ignacio (Nacho) Buenache | 92,807 |
| * | ||
Maria Teresa Rioné Llano | 5,289 |
| * | ||
Francisco Javier Guix Huguet | 1,367 |
| * | ||
Jordi Balsells Valls | 806 | * |
* | Less than 1%. |
(1) | Members of the Grifols Roura family hold their respective shares indirectly through Deria S.A. |
142
(2) | Scranton Enterprises B.V. in which certain of our directors own shares. Some members of the Grifols Family who are directors or executive officers hold part of their shares indirectly through Scranton Enterprises B.V. See “—B. Related Party Transactions.” |
(3) | Of the total number of 26,475,668 voting rights, 3,271,331 voting rights are held indirectly by The Goldman Sachs Group, Inc. through rights over Class A shares, 12,007,015 through rights to recall loaned Class A shares and 10,988,207 through equity swap, 162,977 through call options and 46,138 through call warrants. Further, of the total voting rights, Goldman Sachs International (London) holds indirectly 0.502% of the total voting rights and 5.422% of the total voting rights through financial instruments. |
(4) | On February 21, 2022, Ms. Nuria Roura Carreras informed the shareholders of Ralledor Holding Spain S.L., that due to her advanced age, she no longer holds the indirect voting rights of Grifols, S.A. and, therefore, from that date forward the voting rights of Grifols, S.A. have been held directly by Ralledor Holding Spain S.L. |
(5) | Of the total number of 19,412,817 voting rights, 8,092,817 voting rights are held indirectly by Capital Research and Management Company through rights over Class A shares and 11,320,000 are rights to recall loaned Class A shares. |
(6) | Of the total number of 18,341,400 voting rights, 8,465,470 voting rights are held indirectly by Blackrock Inc. through rights over Class A shares; 9,797,513 are rights to recall loaned Class A shares and 78,417 voting rights are held through contracts for differences (CFD). |
(7) | Of the total number of 16,410,073 voting rights, 798,850 voting rights are held indirectly by JP Morgan Chase & Co through rights over Class A shares, 3,522 through third party depositary receipts (right-of-use held), and 15,607,701 through equity swaps. |
(8) | Of the total number of 13,831,807 voting rights, 2,431,807 voting rights are held directly by Europacific Growth Fund and 11,320,000 are rights to recall loaned Class A shares. |
(9) | Of the total number of 13,115,305 voting rights, 3,533,349 are rights to recall loaned Class A shares and 9,581,956 through equity swaps. |
(10) | Of the total number of 13,001,544 voting rights, 8,378,309 voting rights are held directly by BNP Paribas Asset Management Europe and 4,623,235 voting rights are held indirectly, in each case, through rights over Class A shares. |
(11) | The 4,843,786 voting rights are held through equity swaps. |
(12) | The 4,790,811 voting rights are held through contracts for differences (CFD). |
(13) | 24,000 Class A shares are held indirectly through Glanzmann Enterprises AG. |
(14) | Of the total number of 303,661voting shares attributed to Mr. Tomás Dagá Gelabert, 35,000 are held indirectly through Prismiberica, S.A. |
(15) | Of the total number of 107,834 voting shares attributed to Mr. Victor Grifols Deu, 93,214 are held indirectly through New Fiction 2012, S.L. |
To our knowledge, we are not controlled, directly or indirectly, by any other corporation, government or any other natural or legal person. We do not know of any arrangements which would result in a change in our control.
Significant Changes in Ownership
Beginning on January 1, 2023 until the date of this filing, in accordance with Spanish reporting requirements, the following transfers of shares were reported to the CNMV:
| ● | The Goldman Sachs Group, Inc. acquired a 6.214% stake. |
| ● | Capital Research and Management Company decreased its stake from 4.600% to 4.555%. |
| ● | Blackrock, Inc. decreased its stake from 4.309% to 4.304%. |
| ● | JP Morgan Chase & Co. acquired a 3.851% stake. |
| ● | Europacific Growth Fund increased its stake from 3.033% to 3.227%. |
| ● | Jefferies Financial Group Inc. acquired a 3.078% stake. |
| ● | BNP Paribas Asset Management Europe acquired a 3.051% stake. |
| ● | Rokos Global Macro Master Fund LP acquired a 1.137% stake. |
| ● | Melqart Opportunities Master Fund Ltd acquired a 1.124% stake. |
143
Voting Rights
Each of our Class A shares is entitled to one vote, except that the voting rights of Class A shares held in treasury by us or by any of our direct subsidiaries are suspended. Class A shares held by our major shareholders, directors or senior management do not entitle such shareholders to different voting rights.
Holders of our Class B shares generally do not have voting rights, except with respect to certain extraordinary matters that require approval by a majority of our outstanding Class B shares. However, each of our Class B shares entitles its holder to receive a minimum annual preferred dividend out of the distributable profits at the end of each fiscal year the share is outstanding equal to €0.01 per Class B share. In any given fiscal year, we will pay a preferred dividend to the holders of our Class B shares before any dividend out of the distributable profits for such fiscal year is paid to the holders of our Class A shares.
See in Item 10 of this Part I, “Additional Information—B. Memorandum and Articles of Association—Shareholder Rights—Class B Shares—Separate Vote at General Shareholder Meetings on Extraordinary Matters” and “Additional Information — B. Memorandum and Articles of Association — Shareholder Rights” for further details regarding our Class A shares and Class B shares.
B. | Related Party Transactions |
From time to time we have entered into transactions with related parties, including with entities involving certain members of our Board of Directors or senior management As required by our corporate governance guidelines described below, these transactions have been agreed on an arm’s length basis.
We have an Internal Code of Conduct on Matters Related to the Securities Market, which sets forth rules and principles to ensure transparency and arm’s-length terms in our transactions with related parties and other situations of potential conflicts of interest. This policy establishes that the Audit Committee of our Board of Directors is responsible for monitoring and assessing related party transactions, producing a report that requires approval by our shareholders at our general shareholders’ meeting or our Board, as the case may be. Our Audit Committee is also responsible for, among other things, reviewing and supervising the fulfillment of our internal procedures for related party transactions. For further details regarding our related party transactions, see Note 31 to our audited consolidated financial statements included in this annual report. on pages F-121 and following.
Transactions involving Haema AG, BPC Plasma Inc (formerly known as Biotest US Corporation) and Scranton Enterprises B.V. and their respective subsidiaries
Sale of the entities to Scranton Plasma B.V., Vendor Loan to Scranton Plasma B.V. and Call Option Agreement
On December 28, 2018, we sold our 100% stake in each of Haema AG and BPC Plasma Inc to Scranton Plasma B.V., a subsidiary of Scranton Enterprises B.V., one of our major shareholders and a related party, for a total of $538 million. Scranton Plasma B.V. financed the purchase through a loan in the principal amount of $360 million (the “Acquisition Financing”). The lender of the transaction required GWWO to extend a vendor loan to Scranton Plasma B.V. with a maximum amount of $150 million. The initial principal amount was equivalent to $95 million, with a maturity date of December 28, 2025, and an interest rate of EURIBOR plus 200 basis points. In 2023, GWWO loaned an additional €15.0 million to Scranton Plasma B.V. under the same terms and conditions of the vendor loan. As of the date of this annual report on Form 20-F, the euro equivalent of $115.9 million was outstanding on the vendor loan, which was also the largest amount outstanding since January 1, 2022. See Note 11(b) to the audited consolidated financial statements included in this annual report on pages F-70.
Also on December 28, 2018, we entered into a call option agreement with Scranton Plasma B.V. whereby we may repurchase the shares of Haema AG and BPC Plasma Inc at any time. The exercise price of the option as set forth in the agreement would be equal to the greater of: (i) the same price for which the shares were sold to Scranton Plasma B.V., plus the expenses related to the transaction and the increase in net working capital from the time of the sale (December 28, 2018) to the exercise of the option, and (ii) the amount necessary to repay the Acquisition Financing, i.e. $360 million plus accrued interest and any other amounts necessary to cancel such debt.
144
Although we currently do not hold any equity interests in either Haema AG or BPC Plasma Inc, we retain control over such entities due to, among other factors, (1) the call option agreement to reacquire their shares and the financial capability to exercise such call option, (2) potential voting rights derived from the call option, (3) the fact that we acquire all of the plasma collected in the plasma collection centers owned by Haema AG and BPC Plasma Inc under the Plasma Supply Agreement described below and (4) the ability to manage such entities under management agreements. As a result of these factors, pursuant to IFRS 10 we fully consolidate such entities as Grifols subsidiaries in our financial statements. For more details, see Note 19 and to the audited consolidated financial statements included in this annual report on pages F-79.
Plasma Supply Agreement and Advance Payments from GWWO to Scranton Plasma B.V.
On December 28, 2018, our subsidiary GWWO, Biotest Pharmaceuticals Corporation (“BPCorp”), which is a subsidiary of BPC Plasma Inc, Haema AG and Grifols, S.A. entered into the Plasma Supply Agreement, whereby GWWO agreed to acquire all of the plasma collected from approximately 60 plasma collection centers owned by BPCorp and Haema AG. Grifols, S.A. guarantees all GWWO obligations under the Plasma Supply Agreement, which, on January 1, 2019, was extended for a 30-year period.
The price GWWO pays for the plasma acquired under the Plasma Supply Agreement is established based on the full cost of production, plus a fixed margin, and there is exclusivity of sale. Subject to certain conditions and procedures, the agreement also grants to BPCorp and Haema AG the right to receive payments from GWWO in advance for plasma to be delivered in the future. As of the date of this annual report, GWWO had a balance of €46.0 million in advance payments for future delivery of plasma under the Plasma Supply Agreement. During 2023, the highest balance of advance payments amounted to €88.4 million.
Cash-Pooling Financing Agreement between BPC Plasma Inc, Haema AG and Scranton Plasma B.V.
In February 2019, Haema AG and BPC Plasma Inc entered into a Cash-Pooling Financing Agreement with Scranton Plasma B.V. with a maturity date in 2024. Under this agreement, Haema AG and BPC Plasma Inc transfer funds from time to time to their parent company, Scranton Plasma B.V. which advances may be set off by Scranton Plasma B.V. against upstream dividends distributed from time to time by Haema AG and BPC Plasma Inc. As of December 31, 2023, the balance of the advances under this cash-pooling agreement was €101.2 million. In 2023, BPC Plasma Inc. distributed dividends without cash outflow to Scranton Plasma B.V. in an amount of €266.4 million, corresponding to the BPC Plasma Inc results of the four immediately preceding fiscal years. During 2023, the highest balance under this cash-pooling agreement amounted to €361.8 million.
Transactions involving Haema Plasma Kft.
Call Option to Acquire the shares of Haema Plasma Kft; Management Agreement; Plasma Supply Agreement
On February 1, 2021 Scranton Plasma B.V. acquired 100% of the shares of Haema Plasma Kft. Scranton is a shareholder of Grifols. On February 1, 2021 the Grifols Group entered into a call option agreement with Scranton Plasma B.V. whereby we have the right to acquire the shares of Haema Plasma kft, exercisable by the Grifols Group only 12 months after signing and with an expiry of 48 months from the date on which the option becomes exercisable. The option price was set at thirteen times EBITDA minus net debt. Grifols did not make any monetary consideration for the purchase option agreement when signing the agreement. The Group has potential voting rights arising from the option to purchase the shareholding and these are substantive, based on:
| • | A call option for Grifols which gives it the irrevocable and exclusive right (not an obligation) to acquire the Haema Plasma Kft shareholding at any time after February 1, 2022. |
| • | The Company is committed to providing support services in the business of collecting, processing and distributing plasma from the donation centers. There is also a Plasma Supply Agreement whereby the plasma produced by these entities will be used almost entirely to cover Grifols' needs. There is no sales exclusivity. |
| • | There are no shareholder agreements that provide for relevant decisions to be approved in a manner other than by majority vote. |
145
The above are indicators of the power that Grifols acquired over this entity, considering that the call option is likely to be exercised and Grifols will have the financial capacity to carry it out. Similarly to Haema AG and BPC Plasma Inc, we fully consolidate Haema Plasma Kft as a Grifols subsidiary in our financial statements pursuant to IFRS 10, as we retain control over such entity by means of the call option, potential voting rights, plasma supply agreement and management rights. See Note 3(d) to the audited consolidated financial statements included in this annual report on page F-24.
Cash Pooling Financing with GWWO
In 2021, our subsidiary GWWO entered into a cash pooling financing agreement with Haema Plasma Kft. Under such agreement, GWWO advances resources from time to time to a centralized treasury mechanism for purposes of providing Haema Plasma Kft with cash availability. Since 2022, financial information on Haema Plasma Kft is part of our consolidated financial statements and this transaction has been reported under our consolidated balance sheets. The outstanding balance of this cash pooling agreement at December 31, 2023 was €3.6 million, while the highest balance of such arrangement during 2023 was €6.5 million.
Promissory Notes issued by our subsidiary Instituto Grifols S.A.
Deria S.A., one of our major shareholders, Messrs. Raimon Grifols Roura and Albert Grifols Coma-Cros, and Padolç, S.L., a company controlled by Mr. Victor Grifols Roura, own promissory notes issued by our subsidiary Instituto Grifols S.A.
Instituto Grifols S.A. has been issuing bearer form promissory notes on an annual basis since 1987 to provide additional working capital and liquidity to the Grifols Group. The promissory notes are non-negotiable securities and may not be transferred to third parties (except by Instituto Grifols, S.A. itself for subsequent redemption) until the date of the notes’ redemption. The issuance of the promissory notes is not considered a public offering for the subscription of securities under Spanish law and does not require verification by the CNMV. As of December 31, 2023, and 2022, the outstanding balances of the promissory notes owned by our related parties was €16 million and €15 million, respectively. These amounts were also the highest the outstanding balances were throughout 2023 and 2022, as applicable.
These bearer promissory notes have one year maturity and a nominal value of €3,000 each. Instituto Grifols, S.A. redeems all outstanding promissory notes at their maturity date of May 4 every year, regardless of the date of acquisition of such notes. The interest rate for the promissory notes is set based on the weighted average cost of the Grifols Group’s debt. The notes maturing on May 4, 2024, accrue interest at an annual rate of 4.0%. See Note 21 to our consolidated audited financial statements included in this annual report on page F-90.
Receivables owed by Mr. Victor Grifols Roura
In December 2023, on behalf of the Honorary Chairperson of our Board of Directors, Mr. Victor Grifols Roura, we entered into a deferred income insurance policy with an insurance company, handling the administrative mechanics of the insurance policy’s procurement and bearing the related costs. At December 31, 2023, we had an account receivable against Mr. Victor Grifols Roura in the amount of €5.6 million, which was also the highest outstanding balance in respect of such transaction in 2023. In January 2024, Mr. Victor Grifols Roura reimbursed such amount to the Company. Due to the nature of the transaction, there was no fund disposition by Mr. Victor Grifols Roura.
Transactions with Centurión Real Estate S.A.U.
We have entered into a number of lease agreements with Centurión Real Estate S.A.U. whereby we pay for the rights-of-use of certain real estate properties located at Sant Cugat del Vallès, Barcelona, Spain, which we use as office buildings, including for our Spanish headquarters. The sole shareholder of Centurión Real Estate S.A.U. is Scranton Enterprises B.V., one of our major shareholders. These lease agreements were entered into on an arm’s-length basis. In the years ended December 31, 2023, 2022 and 2021, we paid to Centurión Real Estate S.A.U. the amounts of €7.2 million, €6.3 million and €5.3 million, respectively. These lease agreements will expire on March 1, 2045.
146
Charitable Contributions
In 2023, we contributed to three charitable foundations, the Víctor Grifols i Lucas Foundation, the J.A. Grifols Foundation and the Probitas Private Foundation, which were formed by us, and certain of our current officers and directors serve as patrons of the Probitas Private Foundation.
The Víctor Grifols i Lucas Foundation provides grants to further the study of bioethics. It was created in 1998 with the mission of promoting bioethics through dialogue between specialists in a range of areas. The Víctor Grifols i Lucas Foundation seeks to foster ethical attitudes in organizations, companies and individuals active in the field of human health, offering a discussion platform that provides a forum for the exchange of different perspectives. Mr. Víctor Grifols i Lucas is our former Chief Executive Officer and is the father of both Mr. Raimon Grifols Roura, an executive director, and Mr. Victor Grifols Roura, our Honorary Chairperson (non-member) of the Board. We contributed €0.4 million, €0.5 million and €0.4 million to the Víctor Grifols i Lucas Foundation in the years ended December 31, 2023, 2022 and 2021, respectively.
The J.A. Grifols Foundation provides support intended for civic, social, environmental or educational programs that address the needs of the communities where our plasma collection centers are located as a means to strengthen community bonds. It was established in 2008 with the mission of contributing to the health and well-being of plasma donors and the communities where they live. In the years ended December 31, 2023 and 2022, we contributed €0.4 million and €0.4 million, respectively, to the J.A. Grifols Foundation.
The Probitas Private Foundation provides medical and sanitary assistance to international communities that lack medical and sanitary resources or that have an urgent and essential need for such services due to catastrophes. The Probitas Private Foundation was founded by us in 2008. Mr. Tomás Dagá Gelabert, one of our directors, was a patron of the Probitas Private Foundation until May 27, 2021. We contributed €1.3 million, €3.4 million and €3.6 million to the Probitas Private Foundation in the years ended 2023, 2022 and 2021, respectively.
Loans
Except as otherwise described above in this section, we have not extended any advances or loans to members of the Board or key management personnel nor have we assumed any guarantee commitments on their behalf. We also have not assumed any pension or life insurance obligations on behalf of former or current members of the Board or key management personnel.
C. | Interests of Experts and Counsel |
Not Applicable.
Item 8.FINANCIAL INFORMATION
A. | Consolidated Statements and Other Financial Information |
Financial Statements
See our audited consolidated financial statements and the related notes starting on page F-1 of this annual report on Form 20-F.
Legal Proceedings
We are involved in various legal proceedings in the ordinary course of our business. In the event of adverse outcomes of these proceedings, we believe that resulting liabilities will either be covered by insurance or not have a material adverse effect on our financial condition or results of operations.
147
OFAC Request for Interpretative Guidance
Through our subsidiary Biotest AG, we have entered into product-supply agreements with five different Iranian entities, whereby the Iranian entities send plasma collected in Iran to Biotest AG for processing. Biotest AG then uses the plasma to manufacture pharmaceutical products for the Iranian entities, including IG products, Albumin, Factor VIII and Factor IX. These product-supply agreements were entered into by Biotest AG prior to our acquisition of the company in 2022. See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting our Financial Condition and Results of Operations—Acquisitions—Biotest AG Acquisition.”
On February 21, 2023, Grifols filed a request with the Licensing Division of the U.S. Office of Foreign Assets Control (“OFAC”) for interpretative guidance or, in the alternative, for license authorization, under the Iranian Transactions and Sanctions Regulations (“ITSR”), which restrict commercial interaction with Iran and its governmental entities and provide for sanctions in the case of failure to comply. In its filing, Grifols asserts that the ITSR should not apply in this case, but that if OFAC determines otherwise, that OFAC should authorize the issuance of a license permitting Biotest AG’s current activity involving Iran on humanitarian grounds. As of the date of this annual report on Form 20-F, we have not received a response from the OFAC’s Licensing Division.
We cannot assure you that OFAC will agree with our assertion that the ITSR should not apply or that it will issue the requested license. In such instance, we would have to terminate Biotest AG’s activities in Iran and may be subject to penalties. The termination of Biotest AG’s activities in Iran would not have a material adverse effect on our operations or financial condition.
Illinois Biometric Information Privacy Act Claim
The plaintiffs allege that we committed certain administrative violations of the Illinois Biometric Information Privacy Act (BIPA) in a class action. Such plaintiffs donated plasma at one of our Illinois-based plasma donation centers. As part of the FDA approved donation process, donors were required to scan at least one fingerprint to verify their identity. The Plaintiffs allege that we failed to comply with the BIPA’s requirements when collecting their fingerprint data. Specifically, they allege that we violated the BIPA by (a) failing to develop a publicly available data-retention policy and guidelines for permanently destroying biometric data, and (b) collecting, using, and storing their donors’ biometric data without obtaining informed written consent.
Although we denied all of the plaintiff’s allegations, we have settled this class action, without any admission or determination of liability or the strength of the parties’ claims and defenses, for the amount of $16,750,000. On March 9, 2023, the court preliminarily approved the settlement and signed an order granting final approval of the parties’ class action settlement on September 19, 2023.
Third Arbitration involving GDS and Ortho-Clinical Diagnostics against Siemens
In 2022, GDS and Ortho-Clinical Diagnostics (“Ortho”) initiated a third arbitration against Siemens alleging the failure to pay royalties under certain agency and license agreements. Siemens counterclaimed against GDS and Ortho alleging that those royalties were not owed, among other claims. We settled this dispute, without any admission or determination of liability or the strength of the parties’ claims and defenses, in April 2023.
148
Unpaid Royalties Dispute
GDS, GWWO, Abbott Laboratories, or Abbott, and Novartis Vaccines and Diagnostics, Inc. are in dispute over unpaid royalties payable by Abbot to GDS and Ortho, under an HIV License and Option agreement dated August 16, 2019 (the “HIV License”). On September 12, 2019, GDS and Ortho filed a Notice of Arbitration in the U.S. District Court for the Northern District of Illinois. On October 3, 2019, Abbott terminated the HIV License and filed for declaratory relief seeking to invalidate the licensed patent. GDS filed motions to dismiss and to compel arbitration, but the Court continued all pending motions and referred the parties to a Magistrate for a mandatory settlement conference. On the February 5, 2020, the parties attended a mandatory settlement conference ordered by the District Judge, with the magistrate judge presiding. No satisfactory settlement was reached. On March 16, 2020, GDS and Ortho filed an answer and counterclaim to the litigation, while simultaneously pursuing arbitration for the pre-termination amount owed by Abbot. The arbitration hearing was on June 15-16, 2020, and the arbitrator awarded $4 million to GDS/Ortho. The court litigation is continuing. Abbot’s motion to dismiss was denied December 1, 2020. Fact discovery concluded on October 25, 2021. Expert Discovery was concluded on October 14, 2022, and the parties filed dispositive motions, including a motion for summary judgement by Abbott and an opposition filed by GDS. The Court in pertinent part denied Abbott’s motion for summary judgement. GDS and Ortho contend that the patent is valid and they believe that Abbott will be unsuccessful in its Declaratory Relief action. A mediation meeting took place on January 31, 2024, without success. We have scheduled a status conference to discuss the matter again and set further trial and pre-trial hearings. We expect trial is expected to take place in late 2024 or early 2025.
See Note 29(e) to our annual consolidated financial statements included in this annual report for additional information regarding the legal proceedings in which we are involved.
Antitrust Approval of Biotest Pharmaceuticals Corporation Acquisition
In August 2018, the FTC issued a consent order which allowed the acquisition of 24 donor centers and required the divestiture of three centers to Kedrion. The consent order requires annual reports to be made to the FTC for a period of 10 years. We have delivered each annual compliance report since we completed the acquisition and there has been no further action by the FTC.
CFIUS Approval on certain transactions
In September 2019, as a consequence of the share exchange agreement we entered with Shanghai RAAS, Grifols and the Committee on Foreign Investments in the United States (“CFIUS”) entered into a National Security Agreement to ensure the protection of certain data obtained as required from donors of human source plasma collected in the United States and maintained in donor management systems (“DMS”), and pursuant to this agreement, we are obligated to make bi-annual compliance reports to CFIUS. The most recent report was filed and accepted in February 1, 2024 and the next report is due to be filed in August 2024.
Dividend Policy
Class A Shares
Our dividend policy is to pay out approximately 40% of our net consolidated profits. However, the First Lien Credit Facilities and some other documents governing our financial indebtedness contain limitations on our ability to pay cash dividends in the ordinary course of business in accordance with our dividend policy depending on our debt levels and the availability of certain restricted payments baskets. For a further discussion of the terms of the First Lien Credit Facilities and our other financing arrangements, see Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Sources of Credit” In any event, as a result of our commitment to reduce our debt level, we do not expect to pursue any distribution of cash dividends until debt leverage is below 4.00:1.00.
The declaration and payment of dividends is reviewed annually by the Board based upon a review of our balance sheet and cash flow, the ratio of current assets to current liabilities, our expected capital and liquidity requirements, the provisions of our governing documents and the provisions in our financing arrangements governing cash dividends. The payment of future dividend will be determined by the Board, based upon the factors described above and other factors that it deems relevant at the time that declaration of a dividend is considered. There can be no assurance as to whether or in what amounts any future dividend might be paid.
149
In addition, the availability of the reserves for distribution is subject to limitations under Spanish law. The distributable reserves of us and our Spanish subsidiaries are limited by the amount of mandatory reserves, which include, for us and each of our Spanish subsidiaries, the legal reserves and the amount of capitalized research and developments pending to be amortized by us and each of our Spanish subsidiaries. This limitation on distributable reserves due to capitalized research and developments expenditure amounted, on a consolidated basis, to €23.9 million at December 31, 2023.
During 2023, no dividend was paid. As mentioned above, the Board has not proposed the distribution of dividends to shareholders at the upcoming annual general meeting of shareholders.
Class B Shares
Each Class B share entitles its holder to receive a minimum annual preferred dividend out of the distributable profits at the end of each fiscal year the share is outstanding equal to €0.01 per Class B share, if the aggregate preferred dividend does not exceed the distributable profits for that year and provided that the distribution of dividends has been approved by our shareholders. In any given fiscal year, we will pay a preferred dividend to the holders of the Class B shares before any dividend out of the distributable profits for such fiscal year is paid to the holders of Class A shares. The preferred dividend on all issued Class B shares will be paid by us within the nine months following the end of that fiscal year, in an amount not to exceed the distributable profits obtained during that fiscal year. During 2023, no dividend was paid.
B. | Significant Changes |
See Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Subsequent Events.”
Item 9.THE OFFER AND LISTING
A. | Offer and Listing Details |
Our Class A shares have been listed on the Spanish Stock Exchanges since we completed our initial public offering on May 17, 2006 and are quoted on the Spanish Automated Quotation System under the ticker symbol “GRF.”
Our Class B shares have been listed on the Spanish Stock Exchanges since June 2, 2011 and quoted on the Spanish Automated Quotation System under the ticker symbol “GRF.P.”
Our Class A ADSs are not listed on a national exchange and have traded on the Over the Counter Bulletin Board, an electronic stock listing service provided by NASDAQ, since July 2009.
Our Class B ADSs have been listed and traded on the NASDAQ Global Select Market under the symbol “GRFS” since June 2, 2011. Each Class B ADS represents one Class B share.
B. | Plan of Distribution |
Not Applicable
C. | Market |
Our Class A shares have been listed on the Spanish Stock Exchanges since May 17, 2006 and are quoted on the Spanish Automated Quotation System under the ticker symbol “GRF.” Our Class B shares were issued as part of the consideration for the Talecris acquisition and were listed on the Spanish Stock Exchanges on June 2, 2011 and quoted on the Spanish Automated Quotation System under the ticker symbol “GRF.P.”
Our Class B ADSs have been listed and traded on the NASDAQ Global Select Market under the symbol “GFRS” since June 2, 2011.
150
Spanish Securities Market
The Spanish Stock Exchanges consist of four stock exchanges located in Madrid, Barcelona, Bilbao and Valencia. The majority of the transactions conducted on them are done through the Spanish Automated Quotation System. During 2023, the Spanish Automated Quotation System accounted for the majority of the total trading volume of equity securities on the Spanish Stock Exchanges.
Spanish Automated Quotation System
The Spanish Automated Quotation System was introduced in 1989 and links the Spanish Stock Exchanges, providing those securities listed on it with a uniform continuous market that eliminates most of the differences among the Spanish Stock Exchanges.
The principal feature of the system is the computerized matching of buy and sell orders at the time of entry of the order. Each order is executed as soon as a matching order is entered, but can be modified or canceled until executed. The activity of the market can be continuously monitored by investors and brokers. The Spanish Automated Quotation System is operated and regulated by the Sociedad de Bolsas, a corporation owned by the companies that manage the Spanish Stock Exchanges. All trades on the Spanish Automated Quotation System must be placed through a bank, brokerage firm, an official stock broker or a dealer firm member of a local exchange directly.
There is a pre-opening session held from 8:30 a.m. to 9:00 a.m. CET (UTC+1) each trading day, during which orders are placed. The computerized trading hours are from 9:00 a.m. to 5:30 p.m. CET (UTC+1). Each session ends with a five-minute auction, between 5:30 and 5:35 p.m. CET (UTC+1), with a random closedown of 30 seconds. The price resulting from each auction is the closing price of the session.
On March 29, 2021, new rules came into effect regarding the functioning of the Spanish Automated Quotation System, which, among other things, regulates the maximum price fluctuations in the price of stocks. Under the new rules, each stock in the continuous market is assigned a static and a dynamic range within which the price can fluctuate. The price of a stock may rise or fall by its static range (which is public and calculated according to the stock’s average historic price volatility) above or below its opening price (which is the closing price of the previous session). When the stock trades outside of this range, the trading of the stock is suspended for five minutes, during which an auction takes place. After this auction, the price of the stock can once again rise or fall by its static range above or below its last auction price (which will be considered as the new static price before triggering another auction). Furthermore, the price of a stock cannot rise or fall by more than its dynamic price range (which is public and calculated according to the stock’s average intra-day volatility), from the last price at which it has traded. If the price variation exceeds the stock’s dynamic range, a five-minute auction is triggered.
Moreover, there is a block market (el mercado de bloques) allowing for block trades between buyers and sellers from 9:00 a.m. to 5:30 p.m. CET (UTC+1) during the trading session. Under certain conditions, this market allows cross-transactions of trades at prices different from prevailing market prices. Trading in the block market is subject to certain limits with regard to price deviations and volumes.
Between 5:40 p.m. and 8:00 p.m. CET (UTC+1), certain trades may occur benefiting from an exemption to the pre-trade transparency requirements.
Clearance and Settlement System
Until April 1, 2003, transactions carried out on the Spanish Stock Exchanges and the continuous market were cleared and settled through the Servicio de Compensación y Liquidación de Valores, S.A. (whose commercial name is Iberclear). Since April 1, 2003, the settlement and clearance of all trades on the Spanish Stock Exchanges, the Public Debt Market (Mercado de Deuda Pública), the AIAF Fixed Income Market (Mercado AIAF de Renta Fija) and the Market for Latin-American Stocks in Euros (Mercado de Valores Latinoamericanos en Euros) as well as any securities trading on other official regulated markets and multilateral trading systems that have appointed Iberclear for such purposes, have been made through Iberclear, which was formed as a result of a merger between the Servicio de Compensación y Liquidación de Valores, S.A and Central de Anotaciones del Mercado de Deuda Pública, which was managed by the Bank of Spain.
151
Book-entry System
Ownership of shares listed on any Spanish Stock Exchange is required to be represented by entries in a register maintained by Iberclear, and transfers or changes in ownership are effected by entries in such register. The securities register system is structured in two levels: the central registry managed by Iberclear, which keeps the securities balances of the participants, and a detailed registry managed by the participants where securities are listed by holder’s name.
Securities Market Legislation
The Spanish Securities Market Act (today known as Ley 6/2023, de 17 de marzo, de los Mercados de Valores y de los Servicios de Inversión), or Securities Market Act, which first came into effect in 1989, among other things:
| ● | established an independent regulatory authority, the CNMV, to supervise the securities markets; |
| ● | established a framework for the regulation of trading practices, tender offers and insider trading; |
| ● | required stock exchange members to be corporate entities; |
| ● | required companies listed on a Spanish Stock Exchange to file annual audited financial statements and to make public semi-annual financial information; |
| ● | established a framework for integrating quotations on the Spanish Stock Exchanges by computer; |
| ● | exempted the sale of securities from transfer and value added taxes; |
| ● | deregulated brokerage commissions as of 1992; and |
| ● | provided for transfer of shares by book-entry or by delivery of evidence of title. The Securities Market Act was amended by, among others, Law 37/1998, which implemented two European Union directives that innovated the Securities Market Act. The first was the recognition that both Spanish and other European Union member state companies authorized to provide investment services have full access to the official secondary securities markets, with full capacity to operate, thereby enabling the direct admission of banking entities into the stock exchange area. The second innovation was that the scope of the Securities Market Act was enlarged to include a list of financial instruments, such as financial exchange contracts, or installment financial contracts, which expanded the categories of securities included. |
The Securities Market Act was further amended by Law 44/2002 (November 22, 2002) on reform measures of the financial system, which introduced certain modifications to the laws governing financial markets and corporations generally, including:
| ● | provisions requiring listed companies to establish an audit committee, redefining the reporting requirements for relevant events, establishing rules relating to the treatment of confidential and insider information and related party transactions, preventing manipulative and fraudulent practices with respect to market prices and otherwise regarding market transparency; |
| ● | the establishment of Iberclear; and |
| ● | the authorization of the Ministry of Economy and Finance (Ministerio de Economía y Hacienda) to regulate financial services electronic contracts. |
152
On July 17, 2003, the Securities Market Act was amended by Law 26/2003 in order to reinforce the transparency of listed companies. It introduced:
| ● | information and transparency obligations including detailed requirements of the contents of the corporate website of listed companies and the obligation to file with the CNMV an annual corporate governance report; and |
| ● | the obligation to implement a series of corporate governance rules including, among others, regulations regarding the boards of directors and the general shareholders’ meeting. |
On March 11, 2005, Royal Decree Law 5/2005 was approved, modifying the Securities Market Act in order to implement Directive 2003/71/EC of the European Parliament and of the Council of the European Union, or Council, on the prospectus to be published when securities are offered to the public or admitted to trading. The Directive (i) harmonizes the requirements for the process of approval of prospectuses, which enables a prospectus to be valid throughout the European Union and (ii) incorporates the application of the country-of-origin principle later set forth in Spanish Royal Decree, or Royal Decree, 1362/2007.
Law 6/2007, of April 12, 2007, amended the Securities Market Act to modify the rules for takeover bids and for issuer transparency. This Law came into effect on August 13, 2007, and partially integrates into the Spanish legal system Directive 2004/25/EC of the European Parliament and of the Council, of April 21, 2004, on takeover bids and Directive 2004/109/EC of the European Parliament and of the Council, of December 15, 2004, on the harmonization of transparency requirements in relation to information about issuers whose securities are admitted to trading on a regulated market and amending Directive 2001/34/EC. This Law was further developed by Royal Decree 1066/2007, of July 27, 2007, on rules applicable to takeover bids for securities; by Royal Decree 1362/2007, of October 19, 2007, on transparency requirements for issuers of listed securities; and by Royal Decree 1698/2012, of December 21, 2012, to implement Directive 2010/73/EC of the European Parliament and of the Council, of December 24, 2010 (amending Directive 2003/71/CE and Directive 2004/109/EC).
Law 6/2007 (i) introduced several changes to the periodic financial information (annual, biannual and quarterly) to be published by issuers of listed securities and (ii) introduced new developments to the system that establishes the duty to provide notice of significant stakes in an enterprise. These duties include notification requirements such as:
| ● | anyone with a right to acquire, transfer or exercise voting rights granted by the shares, regardless of the actual ownership of the shares, and anyone owning, acquiring or transferring other securities or financial instruments that grant a right to acquire shares with voting rights must provide notice of the holding of a significant stake in accordance with the regulations; |
| ● | directors of listed companies, in addition to providing notice of any transaction concerning the shares or other securities or financial instruments of the issuer that are linked to these shares, must inform the CNMV of their stake upon appointment or resignation; and |
| ● | listed companies must provide notice of transactions concerning their treasury shares in certain cases, which will be established in the developing regulations. |
Law 12/2006, of May 16, 2006, amended the Securities Market Act by (i) introducing a new article relating to notifications to the CNMV of transactions that might constitute insider dealing or market manipulation, (ii) completing the regulation of Bolsas y Mercados Españoles, which operates the Spanish Stock Exchanges and financial markets and (iii) clarifying the regulation of significant participations in the entities that manage the clearing and settlement of securities and the Spanish secondary securities markets.
Law 12/2010, of June 30, 2010, amended the Securities Market Act to require listed companies to create electronic shareholders forums on their websites to facilitate communication prior to the holding of general meetings. It also established that shareholders of listed companies may create associations to exercise their rights and coordinate the defense of their common interests. Such associations must enroll in a special CNMV registry. Finally, Law 12/2010 also amended the Securities Market Act to change the regulations regarding the composition and functions of audit committees.
153
Royal Legislative Decree 1/2010, of July 2, 2010, approved the Spanish Companies Act in order to consolidate and clarify the laws applicable to public limited companies, limited share partnerships and limited liability companies.
Law 2/2011, of March 4, 2011, on Sustainable Economy (Ley de Economía Sostenible) amended the Securities Market Act’s provisions related to the requirements for annual reports on corporate governance and management reports. The Law also made certain corporate governance and shareholder disclosure recommendations in the Spanish Unified Good Governance Code for Listed Companies (Código Unificado de Buen Gobierno de las Sociedades Cotizadas, the “CNMV Governance Code), regarding the composition of boards of directors and its committees and the qualification of directors as executive, proprietary or independent mandatory. The CNMV Governance Code for Listed Companies was approved in 2015 and further revised in June 2020. It unified the recommendations and principles that are applicable to Spanish listed companies; removed some principles and recommendations of the CNMV Governance Code that were written into Spanish legislation and introduced some recommendations on the corporate social responsibility of listed companies.
Law 25/2011, of August 1, 2011, amended the Securities Market Act to implement Directive 2007/36/CE of the European Parliament and of the Council, regarding the exercise of certain rights of the shareholders of listed companies, to simplify and promote the right to information and shareholder voting rights.
Law 1/2012, of June 22, 2012, amended the Spanish Companies Act by making corporate websites mandatory for listed companies and introducing other new requirements regarding the creation, amendment, transfer and removal of corporate websites, as well as the obligations of directors arising in connection with the contents of such websites.
Law 31/2014, of December 3, 2014, amended the Spanish Companies Act to improve the corporate governance practices, increase management efficiency and increase the transparency of companies listed on a Spanish Stock Exchange.
Regulation (EU) No. 596/2014, on market abuse, which was directly applicable in all European Union member states, came into force in 2016 with the aim to ensure that European Union regulation keeps pace with market developments in order to combat market abuse on financial markets as well as across commodity and related derivative markets.
Regulation (EU) No. 2017/1129, of June 14, 2017, which was directly applicable in all European Union member states, came into full force in 2019 to regulate the prospectus to be published when securities are offered to the public or admitted to trading on a regulated market, and repealing Directive 2003/71/EC.
Royal Legislative Decree 19/2018, of November 23, 2018, on payment services and other urgent financial measures amends among others, the Securities Market Act in order to integrate into the Spanish legal system, and Regulation (EU) No. 596/2014, on market abuse. The main novelties introduced to the Securities Market Act are (i) the distinction between the concepts of inside information and relevant information, (ii) the removal of the obligation to have an internal code of conduct for securities markets and (iii) the reduction of the notification threshold of people with management responsibilities.
On December 28, 2018, the Spanish Commercial Code, the Companies Act and the Audit Act were amended by Law 11/2018 in order to reinforce the disclosure of non-financial and diversity information, among others, of listed companies. It introduced information and diversity obligations including (i) the obligation to prepare a non-financial information statement on environmental matters, social and employee-related matters, respect for human rights, anti-corruption and bribery matters and society matters and (ii) the obligation to ensure that the selection procedures for Company directors facilitate diversity in relation to age, disability and training as well as gender, experience and knowledge.
On June 26, 2020, the CNMV approved the partial review of the CNMV Governance Code. The review updated and adapted various recommendations of such code to various intervening legal amendments approved since its publication and clarified the scope of others that had raised certain doubts. The four key elements of the reform were: (i) promoting the presence of women in boards of directors; (ii) greater importance of non-financial information and sustainability; (iii) more attention to reputational risk and, in general, non-financial risks; and (iv) clarification of aspects related to the remuneration of the board members.
154
As a result of the amendment of the CNMV Governance Code referred to above, Circular 1/2020 of October 6, 2020, amended (i) Circular 5/2013 of June 12, 2020, by establishing a new template for the annual corporate governance report for listed public companies, savings banks and other entities that issue securities admitted for trading on official securities markets and (ii) Circular 4/2013 of June 12, 2020, by establishing a new template for the annual report on remuneration of directors of listed public companies and members of boards of directors and control committees of savings banks that issue securities admitted to trading on official securities markets.
Law 5/2021, of April 12, 2021, among other regulations, amended the Spanish Companies Act, as well as the Spanish Securities Market Act. The purpose of this law is to transpose into Spanish Law Directive (EU) 2017/828 of the European Parliament and of the Council of 17 May 2017 amending Directive 2007/36/EC with respect to the encouragement of long-term shareholder engagement in listed companies.
As a result of the amendment of the Spanish Companies Act and the Spanish Securities Market Act referred to above, Circular 3/2021 of September 28, 2021, amended (i) Circular 4/2013 of June 12, 2020, by establishing a new template for the annual report on remuneration of directors of listed public companies and members of boards of directors and control committees of savings banks that issue securities admitted to trading on official securities markets and (ii) Circular 5/2013 of June 12, 2020, by establishing a new template for the annual corporate governance report for listed public companies, savings banks and other entities that issue securities admitted for trading on official securities markets.
On May 26, 2022, Circular 2/2022 of the CNMV was approved. This circular establishes the new forms to be used to report significant shareholding in entities whose securities are admitted to trading on a regulated market and to report any transaction that an issuer makes with its treasury shares and includes certain new provisions applicable to market makers.
Law 6/2023, of March 17, 2023, approved the Securities Market Act, repealing the former securities market act from 2015. The new Securities Market contains certain adjustments in relation to the 2015 legislation meant to improve its organization and eliminate a number of inconsistencies. In order to develop the Securities Market Act, the following regulations were approved: (i) Royal Decree 813/2023, on the legal regime of investment services companies and other entities that provide investment services (effective on November 29, 2023); (ii) Royal Decree 814/2023, on financial instruments, admission to trading, registration of negotiable securities and market infrastructures (effective on November 29, 2023, except for the provision contained in article 111, which entered into force on November 10, 2023); and (iii) Royal Decree 815/2023, in relation to the official registries of the National Securities Market Commission, cooperation with other authorities and supervision of investment services companies (effective on November 10, 2023).
D. | Selling Shareholders |
Not Applicable.
E. | Dilution |
Not Applicable.
F. | Expense of the Issue |
Not Applicable.
Item 10.ADDITIONAL INFORMATION
A. | Share Capital |
Not Applicable.
155
B. | Memorandum and Articles of Association |
The following is a summary of the material terms of our Articles of Association and Board Regulations, as amended and currently in effect. This summary is not meant to be complete and is qualified in its entirety by reference to each of the Articles of Association and Board Regulations. Because this is a summary, it does not contain all the information that may be important to you. You should read the Articles of Association and Board Regulations carefully. The current Articles of Association are included as Exhibit 1.1 and Exhibit 1.2 (English translation) to this annual report on Form 20-F. The Articles of Association and the Board Regulations are also available on our website, which does not form part of this annual report on Form 20-F, at www.grifols.com under the headings “Investors—Corporate Governance—Articles of Association” and “Investors—Corporate Governance—Board of Directors, Regulations.”
The Articles of Association were originally approved and incorporated with the Commercial Registry on June 22, 1987. The Board Regulations were initially approved by the Board on April 5, 2006.
At the general shareholders’ meeting held on May 29, 2015, the shareholders voted to amend our Articles of Association on matters pertaining to corporate governance in order to ensure compliance with the amended Spanish Companies Act. The shareholders renewed the delegation of authority to the Board to effect a two-to-one split of the Class A and Class B shares, within one year following the date of the meeting, by reducing the nominal value and increasing the number of such shares, without changing the total nominal value of the share capital. Finally, the shareholders provided the Board authorization for the derivative acquisition of treasury stock thereby revoking and leaving without effect the authorization granted to the Board during the shareholder meeting on extraordinary matters held on January 25, 2011.
At the general shareholders’ meeting held on May 27, 2016, the shareholders voted to delegate to the Board, with full power of substitution in any of its members, the authority to increase the Company’s share capital at once or in several times and at any given moment, within a maximum term of five (5) years as from the date of the May 27, 2016, general meeting, and in an amount that in no case may exceed half of the Company’s share capital at the time of this authorization. Pursuant to this authorization, the share capital increases will be carried out, if appropriate, by issuing and placing in circulation the new shares (whether of Class A and Class B, exclusively Class A or exclusively Class B), with or without share premium, with a consideration consisting in cash contributions. As long as there are non-voting Class B shares in circulation, the capital increases will observe, when applicable, the provisions of the Company’s Articles of Association as regards the pre-emptive right of acquisition that may correspond in said capital increases. Likewise, as long as Class B shares hold the redemption rights foreseen in paragraph 4 of article 6.bis of the Articles of Association, the nominal value of the Class B shares that may be issued in the execution of this delegation of authorities cannot exceed one fourth of the total amount of the share capital resulting from the capital increase resolution.
At the general shareholders’ meeting held on May 26, 2017, the shareholders voted to amend our Articles of Association concerning the composition and functions of the Audit Committee, in order to conform its content to the latest amendments of the Companies Act introduced by the Audit Act currently in force. The shareholders also voted to amend the regulation of the general shareholders’ meeting, concerning the competences of the general shareholders’ meeting, in order to adapt its content to the latest amendments of the Companies Act, introduced by Law 5/2015 of promotion of business financing (Ley 5/2005 de fomento de la financiación empresarial), on matters of issuance of bonds and other securities. The amendment consists of eliminating the issuance of numbered series of bonds or other securities, whether convertible or not, that may recognize or create a debt expressly as a competence of the general shareholders’ meeting. The shareholders also renewed the delegation of authority to apply for the listing of the Class A shares on NASDAQ, via Class A ADSs, within three years following the date of the meeting.
At the general shareholders’ meeting held on May 24, 2019, the shareholders voted to amend our Articles of Association and the regulations of the general shareholders’ meeting with respect to the valid casting of votes through distance voting systems of the general shareholders’ meeting in order to extend the deadline for receipt of votes until immediately before midnight on the day prior to the date that the general shareholders’ meeting is scheduled at its first call or second call.
156
At the general shareholders’ meeting held on October 9, 2020, the shareholders voted to amend our Articles of Association and the regulations of the general shareholders’ meeting with respect to the right to attend, proxy granting and representation at the general shareholders’ meeting, with the purpose of expressly establishing the possibility of attending the general shareholders’ meeting by remote, simultaneous and bidirectional connection via telematics means. The shareholders also renewed the delegation of authority to the Board for the derivative acquisition of treasury stock, thereby revoking and voiding the authorization granted to the Board during the general shareholders’ meeting held on May 29, 2015. Such authorization is granted for a maximum term of five years. Further, the shareholders renewed the delegation of authority to apply for the listing of the Class A shares on NASDAQ, via Class A ADSs, within three years following the date of the meeting.
At the general shareholders’ meeting held on May 21, 2021, the shareholders voted to renew the delegation of authority to the Board to increase the company’s share capital, thereby revoking and voiding the authorization granted to the Board during the general shareholders’ meeting held on May 27, 2016. Such authorization is granted for a maximum term of five years as from the date of the general shareholders’ meeting.
At the general shareholders’ meeting held on June 10, 2022, the shareholders voted to amend our Articles of Association and regulations of the general shareholders’ meeting in order to conform their wording to the latest amendments of the Spanish corporate law rules and regulations, with amendments regarding (1) the right to attend, proxy granting and representation at the general shareholders’ meeting and the casting of votes through distance voting systems; (2) the remuneration of the Board of Directors; (3) the Audit Committee and annual accounts; (4) the information rights available for shareholders prior to the holding of the general meeting; and (5) the virtual attendance, distance voting and the minutes of the general shareholders’ meeting.
At the general shareholders’ meeting held on June 16, 2023, the shareholders voted to amend our Articles of Association in order to include the delivery of shares or shares options or amounts referred to the value of the shares, as remuneration to directors for the performance of executive duties.
The Board, with full power of substitution in any of its members, has the authority to set the terms and conditions of the capital increases and the characteristics of the shares in all aspects not foreseen by the general shareholders’ meeting, as well as to freely offer the new unsubscribed shares within the term(s) of exercise of the pre-emptive right of subscription; establish that, in the event of an incomplete subscription, the share capital will be increased only in the amount of the subscriptions effectively carried out; redraft the articles of the Articles of Association related to share capital and number of shares; exclude, pursuant to the provisions of article 506 of the Companies Act, the pre-emptive right in the terms and conditions set forth therein and up to a maximum of 20% of the Company’s share capital; apply for, when appropriate, the listing of the shares issued pursuant to this authorization, as well as to carry out all the necessary actions and procedures and to file the documents that might be required before the competent bodies of the above-mentioned stock exchange markets, for admission to listing of the new shares issued as a consequence of the agreed capital increase; it is expressly put on record that Grifols agrees to be bound by already existing and future rules related to the Stock Exchange matters and, specially, as regards contracting, permanence and exclusion from official listing; request the inclusion of the new shares in the accounting registries of the company Sociedad de Gestión de los Sistemas de Registro, Compensación y Liquidación de Valores, S.A.U. (Iberclear).
The full text of the amendments to the Articles of Association detailed above is available on our website, which does not form part of this annual report on Form 20-F, at www.grifols.com under the heading “Investors—Corporate Governance.”
General
As of December 31, 2023, our share capital was €119,603,705 and comprised:
| ● | Class A shares: 426,129,798 ordinary shares with a par value of €0.25 each. All of the Class A shares belong to the same class and series. |
| ● | Class B shares: 261,425,110 non-voting preference shares with a par value of €0.05 each. All of the Class B shares belong to the same class and series and have the preferential rights set forth in the Articles of Association. |
157
All of our shares are fully paid and non-assessable. Both share classes are issued in book-entry form, governed by the Securities Market Act, as amended, and such other provisions as may be applicable. The book-entry registry is maintained by Iberclear and its participant entities.
Register
We are a public limited trading company registered with the Commercial Registry of Barcelona. Our fiscal identification number is A-58389123.
Our principal executive office is located at Avinguda de la Generalitat, 152 Parque Empresarial Can Sant Joan, 08174 Sant Cugat del Vallès, Barcelona, Spain. Our registered office is located at c/Jesús y María, 6, Barcelona (08022). We were incorporated on June 22, 1987. Our fiscal year runs from January 1 to December 31.
Corporate Purpose
Article 2 of the Articles of Association states that our corporate purpose is to provide administration, management and supervision services of companies and businesses as well as investments in personal and real estate assets.
Board of Directors
Under Article 31 of the Board Regulations, a director shall abstain from attending or intervening in deliberations that affect matters in which he/she (or any person related to him/her) is personally involved, directly or indirectly. A director cannot carry out professional or commercial transactions with us, directly or indirectly, unless he/she previously informs the Board about the conflict of interest, and the Board, following a report from our Appointments and Remuneration Committee, approves the transaction.
Under Article 15 of the Board Regulations, the Appointments and Remuneration Committee will in all cases be fully composed of non-executive directors, two of which shall be independent directors, and the chairperson must be an independent director.
The Board, with the advice of the Appointments and Remuneration Committee, sets director compensation. As set forth in Article 20.bis of our Articles of Association the directors’ remuneration shall be approved by the general shareholders’ meeting and shall apply for a maximum of three fiscal years. New directors’ remuneration policies must be approved by the general shareholders’ meeting prior to the last year of applicability of the previous policy, and any new policies approved may apply from the date of approval up to the three following years if so determined by the general shareholders’ meeting. As set forth in Article 27 of the Board Regulations, non-executive directors should be excluded from receiving remuneration linked to our profits or welfare systems, other than shares in Grifols, that they must hold until their resignation as directors. Further, the establishment of equity compensation plans in which members of the Board participate must be authorized in the Articles of Association and requires the shareholders’ prior approval at a shareholders’ meeting. Additionally, the amount of non-executive directors’ remuneration should be calculated in order to incentivize dedication but not become an obstacle to independence.
For more information regarding related party transactions, see Item 7 of this Part I, “Major Shareholders and Related Party Transactions—B. Related Party Transactions.”
We do not impose an age limit requirement for the retirement or non-retirement of directors. We also do not impose a shareholding requirement for director qualification. Article 6 of the Board Regulations does provide, however, that a director cannot qualify as an independent external director if he or she has a significant shareholding in us.
For information regarding the provisions of the Articles of Association as applied to the Board, see Item 6 of this Part I, “Directors, Senior Management and Employees—A. Directors and Senior Management—Directors” and “Directors, Senior Management and Employees—C. Board Practices.”
158
Shareholder Rights
The following summary of material considerations concerning our share capital briefly describes certain material provisions of the Articles of Association and Spanish law relating to our share capital. Because it is a summary, it is not meant to be complete, is qualified by reference to the applicable Spanish laws and our Articles of Association and does not contain all the information that may be important to you.
Neither Spanish law nor our Articles of Association limit the right to own our securities, including the rights of non-resident or foreign shareholders to hold or exercise voting rights on the securities.
Under Spanish law, the rights of shareholders may be changed only by an amendment to the articles of association of a company that complies with the requirements explained below under “—Class A Shares—shareholders’ meetings and Voting Rights.” Our Articles of Association do not further specify what actions or quorums are required to change the rights of our shareholders, other than that they classify an amendment thereto as an extraordinary matter, as described below in “—Class B Shares—Separate Vote at General Shareholder Meetings on Extraordinary Matters.”
Class A Shares
Shareholders’ Meetings and Voting Rights
Pursuant to Article 13 of our Articles of Association and the Spanish Companies Act, the annual general shareholders’ ordinary meeting shall be held during the first six months of each fiscal year on a date fixed by the Board. Resolutions presented at duly constituted general shareholders’ meetings are, except as indicated herein, passed by a simple majority vote of the voting capital present or represented at the meeting.
Extraordinary meetings may be called by the Board whenever it deems it appropriate or at the request of one or more shareholders representing at least 3% of our share capital. The requesting shareholders must state in their request the matters to be addressed at the meeting. Per Spanish Law and the Articles of Association, we are required to publish a “calling of the meeting”, which sets forth the matters to be voted on at each general shareholders’ meeting, at least one month prior to the date set for the meeting in at least: (i) the Official Gazette of the Commercial Registry (Boletín Oficial del Registro Mercantil) or one of the local newspapers of wide circulation in the province where we are domiciled (currently Barcelona, Spain); (ii) CNMV’s website; and (iii) our website.
Holders of ordinary and Class B shares duly registered in the book-entry records maintained by Iberclear and its participant entities at least five days prior to the day on which a shareholders’ meeting is scheduled, in the manner provided in the notice for such meeting, may attend such meeting (in person or represented by proxy) and, where so entitled, may vote. Holders of our Class B shares generally do not have voting rights, except with respect to certain extraordinary matters that require approval by a majority of our outstanding Class B shares, as set forth below in “—Class B Shares—Separate Vote at General Shareholder Meetings on Extraordinary Matters.”
For an ordinary or extraordinary general meeting of shareholders to be duly constituted on the first call, the presence in person or by proxy of shareholders representing 25% of our issued voting share capital is required to constitute a quorum and proceed. If a quorum is not obtained on the first call, a meeting is validly convened on the second call regardless of the share capital in attendance.
Under Spanish law, the following shareholder actions require approval by the affirmative vote of the holders of a majority of our Class A shares present in person or represented by proxy at a duly constituted meeting of holders of our Class A shares at which meeting, if (i) on first call, a quorum of at least 50% of the issued voting share capital is present or represented by proxy or (ii) on second call, a quorum of at least 25% of the issued voting share capital is present or represented by proxy (unless on such second call less than 50% of the issued voting share capital is present or represented by proxy, in which case those matters require the affirmative vote of at least two-thirds of the share capital present or represented at such meeting):
| ● | the issuance of bonds; |
159
| ● | an increase or reduction of the share capital, or the suppression/limitation of pre-emptive rights in issuances of new shares; |
| ● | the transformation of Grifols (change in corporate nature); |
| ● | a merger, de-merger, split, spin-off or other structural change subject to Law 3/2009; |
| ● | any other amendment of the Articles of Association; and |
| ● | a dissolution. |
For purposes of determining the quorum, those shareholders who vote by mail or through the internet are counted as being present at the meeting, as provided by the regulations of the general shareholders’ meeting of Grifols, S.A (Reglamento de la Junta General de Accionistas). Such regulations are available on our website, which does not form part of this annual report on Form 20-F, at www.grifols.com under the heading “Investors—Corporate Governance—Shareholders’ General Meeting—Regulations of the general shareholders’ meeting.”
In general, resolutions passed at a general shareholders’ meeting are binding upon all shareholders. In very limited circumstances, Spanish law gives dissenting or absent shareholders, including those holding Class B shares, the right to have their Grifols’ shares redeemed by us at prices determined in accordance with established formulas or criteria.
Dividends
Payment of dividends must be proposed by the Board and authorized by our shareholders at a general shareholders’ meeting. Interim dividends may be declared by the Board on account of profits for the then current fiscal year, subject to certain limitations.
Spanish law requires each company to apply at least 10% of its net income each year to a legal reserve until the balance of such reserve is equivalent to at least 20% of such company’s issued share capital. A company’s legal reserve is not available for distribution to its shareholders except upon such company’s liquidation. According to Spanish law, dividends may only be paid out of profits (after deduction of any amounts required to be applied to the legal reserve) or distributable reserves and only if the value of a company’s net worth is not, and as a result of distribution would not be, less than such company’s share capital.
In addition, no profits may be distributed unless the amount of the distributable reserves is at least equal to the amount of research and development expenses recorded as an asset on a company’s consolidated balance sheet.
Spanish law also requires the creation of a non-distributable reserve equal to the amount of goodwill recorded as an asset on a company’s consolidated balance sheet and that an amount at least equal to 5% of such goodwill be transferred from the profit from each financial year to such non-distributable reserve until such time as the non-distributable reserve is of an amount at least equal to the goodwill recorded on such company’s consolidated balance sheet. If, in any given financial year, there are no or insufficient profits to transfer an amount equal to 5% of the goodwill recorded as an asset on a company’s consolidated financial statement, Spanish law requires that the shortfall be transferred from freely distributable reserves to the non-distributable legal reserve.
In the event of a reduction in share capital to offset losses, dividends may not be distributed until the legal reserve reaches 10% of the new share capital.
Distributions of dividends to our Class A shareholders will be made in proportion to the capital that they have paid up. The shareholders at the general shareholders’ meeting shall decide the amount, time and form of payment of the dividends. If these details are not so determined, the dividend will be payable at our registered office on the day following the date of the resolution.
The right to a dividend lapses and reverts to us if it is not claimed within five years after it becomes payable. Dividends payable by us to non-residents of Spain may be subject to a Spanish withholding tax of 19%, effective January 1, 2016. However, residents of certain countries are entitled to the benefits of the Convention Between the United States of America and the Kingdom of Spain for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income, as described below in “—E. Taxation—Spanish Tax Considerations.”
160
As set forth below under “—Class B Shares—Preferred Dividend,” since the issuance of our Class B shares, the dividend rights of our Class A shareholders have been subordinated to the €0.01 per share preferred dividend of our Class B shares.
Liquidation Rights
Upon our winding-up and liquidation, holders of our Class A shares and Class B shares will be entitled to receive a pro rata portion of any assets remaining after the payment of our debts, taxes and the expenses of the liquidation as follows: (i) before any amount is distributed to the holders of Class A shares, the holders of Class B shares will receive the nominal value and share premium paid up for such Class B shares at the time of issuance and (ii) once such liquidation preference is received, the holders of the Class A shares and Class B shares will share pari passu in the amounts distributed.
Subscription (or Preemptive) Rights and Increases of Share Capital
Pursuant to the Spanish Companies Act, shareholders and holders of convertible bonds have subscription (or preemptive) rights to subscribe for any new shares (or other securities convertible into, or exchangeable for, shares) issued by a company in a capital increase via monetary contributions.
In accordance with the Spanish Companies Act, such subscription (or preemptive) rights may be waived under special circumstances by a resolution passed at a meeting of shareholders or the Board (such as when we listed on the Spanish Stock Exchanges), and the general shareholders’ meeting delegates to the Board the right to increase the share capital or to issue securities convertible into, or exchangeable for, shares and to waive subscription (or preemptive) rights). See Item 3 of this Part I, “Key Information—D. Risk Factors—Risks Relating to Our Shares and American Depositary Shares—Subscription (or preemptive) rights may be unavailable to U.S. holders of our shares or ADSs.”
Further, subscription (or preemptive) rights, in any event, will not be available in the event of certain capital increases, such as those in which we receive an in-kind contribution, those effected to meet the requirements of a convertible bond issue or those for a merger in which shares are issued as consideration. Subscription (or preemptive) rights are transferable, may be traded on the Spanish Automated Quotation System and may be of value to existing shareholders because new shares may be offered for subscription at prices lower than prevailing market prices. In the case of a share capital increase against reserves, the same rule applies to the free allotment (derecho de asignación gratuita) rights.
Finally, as described below in “—Class B Shares—Subscription Rights,” in connection with an issuance of securities where subscription (or preemptive) rights apply, our Class B shares may only be granted preemptive rights with respect to additional Class B shares if our Class A shares are granted preemptive rights with respect to additional Class A shares. The preemptive rights of each class must be otherwise equal.
Registration and Transfers
Our Class A shares are in book-entry form on Iberclear and are indivisible. Joint holders of one share must designate a single person to exercise their shareholders’ rights, but they are jointly and severally liable to us for all the obligations flowing from their status as shareholders, such as the payment of any pending capital calls.
Iberclear maintains the central registry reflecting the number of shares held by each of its participant entities. Each participant entity, in turn, maintains a registry of the owners of such shares.
Transfers of shares quoted on the Spanish Stock Exchanges are normally made through credit entities or investment companies that are members of the Spanish Stock Exchanges.
161
Reporting Requirements
Pursuant to Royal Decree 1362/2007, any individual or legal entity that, by whatever means, acquires or transfers shares with voting rights in a company for which Spain is listed as the Country of Origin (Estado Miembro) (as defined therein) and which is listed on an official secondary securities market or other regulated market in the European Union must notify the issuer and the CNMV, if, as a result of such transaction, the proportion of voting rights held by that individual or legal entity reaches, exceeds or thereafter falls below a 3% threshold of that company’s total voting rights. The notification obligations are also triggered at thresholds of 5% and multiples thereof (excluding 55%, 65%, 85%, 95% and 100%). The applicable threshold is 1% (or its successive multiples thereof) for persons or entities located in designated “tax havens” (as defined in Royal Decree 1080/1991) or other jurisdictions lacking adequate supervision.
The individual or legal entity obliged to provide the notification must serve the notification by means of the form approved by the CNMV from time to time for such purpose, within four business days from the date on which the transaction is acknowledged. Royal Decree 1362/2007 deems that a transaction is “acknowledged” within two business days from the date on which such transaction is entered into.
The reporting requirements apply not only to the purchase or transfer of voting shares, but also to those transactions in which, without a purchase or transfer, the proportion of voting rights of an individual or legal entity reaches, exceeds or thereafter falls below the threshold that triggers the obligation to report as a consequence of a change in the total number of voting rights of a company on the basis of the information reported to the CNMV and disclosed by such individual or legal entity.
Regardless of the actual ownership of the voting shares, any individual or legal entity with a right to acquire, transfer or exercise voting rights of the shares, and any individual or legal entity that owns, acquires or transfers, whether directly or indirectly, other securities or financial instruments that grant a right to acquire shares with voting rights, will also have an obligation to notify us and the CNMV of the holding of a significant stake in accordance with the regulations.
Furthermore, all members of the Board must report to both us and the CNMV the percentage and number of voting rights in Grifols held by them at the time of becoming or ceasing to be a member of the Board. All members of the Board must also report any change in the percentage of voting rights they hold, regardless of the amount, as a result of any acquisition or disposition of our shares or voting rights, or financial instruments that carry a right to acquire or dispose of shares that have voting rights attached, including any stock-based compensation that they may receive pursuant to any of our compensation plans.
In addition, pursuant to the Securities Market Act, any member of the Board and any parties closely related to any member of the Board must similarly report any acquisition or disposal of our shares (in this case, either Class A or Class B shares), derivatives or other financial instruments relating to our shares regardless of the size, including information on the percentage of voting rights which they hold as a result of the relevant transaction within five business days of such transaction. In this respect, Regulation (EU) No. 596/2014, on market abuse, introduces certain changes as regards notifications from directors. From a practical viewpoint, the transactions that may be notified are broadened, the notification period is reduced from 5 to 3 business days and the prohibition against directors and executives to trade during 30 calendar days before the publication of an interim or annual financial report (restricted periods or “blackouts”) is regulated. Royal Decree-Law 19/2018, which amends the Securities Market Act and implements Regulation (EU) No. 594/2014, on market abuse, establishes that persons discharging managerial responsibilities, as well as persons closely associated with them, must report to Grifols and the CNMV any acquisition or disposal of our shares (in this case, either Class A or Class B shares), derivatives or other financial instruments relating to our shares, once the sum of the amounts of all transactions made within a calendar year reaches the amount of €20,000.
Additional disclosure obligations apply in respect of voting agreements. In this respect, the Spanish Companies Act requires parties to disclose certain types of shareholders’ agreements that affect the exercise of voting rights at a general shareholders’ meeting or contain restrictions or conditions on the transferability of shares or bonds that are convertible or exchangeable into shares.
Moreover, persons holding a net aggregate short position in our shares must report the short position to the CNMV on a confidential basis whenever it reaches 0.2% and notify the CNMV of any subsequent decrease or increase by 0.1% (and successive multiples thereof) within the day immediately following the relevant trade. The CNMV publishes individual net short positions of 0.5% or more and aggregate information on net short positions between 0.2% and 0.5%.
162
The Articles of Association do not contain additional provisions governing the ownership threshold above which shareholder ownership must be disclosed.
Class B Shares
Our Class B shares have substantially similar dividend and other economic rights as our Class A shares, summarized above in “—Class A Shares,” but differ from the Class A shares in some important respects that are outlined below.
Voting Rights
Holders of our Class B shares generally do not have voting rights, except with respect to certain extraordinary matters, with respect to which approval by a majority of our outstanding Class B shares is required.
Separate Vote at General Shareholder Meetings on Extraordinary Matters
Notwithstanding the lack of voting rights of our Class B shares generally, resolutions on the matters detailed below (each, an “extraordinary matter”) require the approval of a majority of our outstanding Class B shares.
| ● | Any resolution (i) authorizing us or any of our subsidiaries to repurchase or acquire any of our Class A shares, except for pro rata repurchases available equally to holders of our Class B shares on the same terms and at the same price as offered to holders of our Class A shares or (ii) approving the redemption of any of our shares and any share capital reductions (through repurchases, cancellation of shares or otherwise), other than (a) those redemptions required by law and (b) those redemptions which affect equally our Class A shares and Class B shares and in which each Class B share is treated the same as a Class A share in such transaction. |
| ● | Any resolution approving the issuance, granting or sale (or authorizing the Board to issue, grant or sell) (i) any of our shares, (ii) any rights or other securities exercisable for or exchangeable or convertible into our shares or (iii) any options, warrants or other instruments giving the right to the holder thereof to purchase, convert, subscribe or otherwise receive any of our securities, except if (a) each Class B share is treated the same as a Class A share in the relevant issuance, grant or sale and, therefore, has a preferential subscription right (derecho de suscripción preferente) or a free allotment right in the relevant issuance, grant or sale to the same extent, if any, as a Class A share or (b) if the issuance is made in accordance with the subscription rights described in “— Subscription Rights” below. |
| ● | Any resolution approving unconditionally or not (i) a transaction subject to Law 3/2009 (including, without limitation, a merger, split-off, cross-border redomiciliation or global assignment of assets and liabilities), except if in such transaction each Class B share is treated the same as a Class A share or (ii) our dissolution or winding-up, except where such resolution is required by law. |
| ● | Any resolution for the delisting of any Grifols shares from any stock exchange. |
| ● | Generally, any resolution and any amendment of the Articles of Association that directly or indirectly adversely affects the rights, preferences or privileges of our Class B shares (including any resolution that adversely affects our Class B shares relative to our Class A shares or that positively affects our Class A shares relative to our Class B shares, or that affects the provisions in the Articles of Association relating to our Class B shares). |
The general shareholders’ meeting has the power to decide on all matters assigned to it by law or by the Articles of Association and, in particular, without limitation to the foregoing, shall be the only corporate body or office entitled to decide on these extraordinary matters.
163
Preferred Dividend
Each of our Class B shares entitles its holder to receive a minimum annual preferred dividend out of the distributable profits at the end of each fiscal year the share is outstanding equal to €0.01 per Class B share. In any given fiscal year, we will pay a preferred dividend to the holders of our Class B shares before any dividend out of the distributable profits for such fiscal year is paid to the holders of our Class A shares. The preferred dividend on all issued Class B shares will be paid by us within the nine months following the end of that fiscal year, in an amount not to exceed the distributable profits obtained by us during that fiscal year.
If, during a fiscal year, we have not obtained sufficient distributable profits to pay in full, out of those profits, the preferred dividend on all the Class B shares outstanding, the preferred dividend amount exceeding the distributable profits obtained by us will not be paid and will not be accumulated as a dividend payable in the future.
Lack of payment, total or partial, of the preferred dividend during a fiscal year due to insufficient distributable profits to pay in full the preferred dividend for that fiscal year will not cause our Class B shares to recover any voting rights.
As set forth above in “—Class A Shares—Dividends,” the dividend rights of our Class A shareholders are subordinated to the preferred dividend described in this section.
Other Dividends
Each Class B share is entitled to receive, in addition to the preferred dividend referred to above, the same dividends and other distributions (in each case, whether in cash, securities of Grifols or any of our subsidiaries, or any other securities, assets or rights) as one Class A share. Each Class B share is treated as one Class A share for the purpose of any dividends or other distributions made on our Class A shares, including as to the timing of the declaration and payment of any such dividend or distribution.
Redemption Rights
Each holder of our Class B shares is entitled to redeem those shares as set forth in this section if a tender offer for all or part of our share capital is made and settled (in whole or in part), except if holders of our Class B shares were entitled to (i) participate in such offer and (ii) have their shares acquired in such offer equally and on the same terms as holders of our Class A shares (including, without limitation, for the same consideration).
Upon the closing and settlement (in whole or in part) of a tender offer for our shares in which holders of our Class B shares were not entitled to (i) participate and (ii) have their shares acquired in such offer equally and on the same terms as holders of our Class A shares (including, without limitation, for the same consideration), the redemption process will follow the process detailed below.
| ● | We will, within ten days of the date on which the redemption event occurred (i.e., the date on which the triggering tender offer settled), publish in the Commercial Registry Gazette, the Spanish Stock Exchanges’ Gazettes and in at least two of the newspapers with widest circulation in Barcelona an announcement informing the holders of our Class B shares of the redemption event and the process for the exercise of redemption rights in connection with such redemption event. |
| ● | Each holder of our Class B shares will be entitled to exercise its redemption right for two months from the first date of settlement of the tender offer triggering the redemption right by notifying us of its decision. We will ensure that mechanisms are in place so that the notification of the exercise of the redemption right may be made through Iberclear. |
164
| ● | The redemption price to be paid by us for each Class B share for which the redemption right has been exercised will be the sum of (i) the amount in euro of the highest consideration paid in the tender offer triggering the redemption right plus (ii) interest on the amount referred to in (i), from the date such tender offer is first settled until the date of full payment of the redemption price, at a rate equal to the one-year EURIBOR plus 300 basis points. For the purposes of this calculation, the amount in euro corresponding to any non-cash consideration paid in the tender offer will be the market value of such non-cash consideration as of the date the tender offer is first settled. The calculation of such market value shall be supported by at least two independent experts designated by us from auditing firms of international repute. |
| ● | We will, within 40 days of the date on which the period for notification of the exercise of redemption rights following a tender offer lapses, take all the necessary actions to (i) effectively pay the redemption price for our Class B shares for which the redemption right has been exercised and complete the capital reduction required for the redemption and (ii) reflect the amendment to Article 6 of the Articles of Association (related to share capital) deriving from the redemption. |
The number of our Class B shares redeemed shall not represent a percentage over our total Class B shares issued and outstanding at the time the tender offer is made in excess of the percentage that the sum of our Class A shares (i) to which the tender offer is addressed, (ii) held by the offerors in that offer and (iii) held by persons acting in concert with the offerors or by persons having reached an agreement relating to the offer with the offerors represent over the total Class A shares issued and outstanding at the time the tender offer causing the redemption of our Class B shares is made.
Payment of the redemption price will be subject to us having sufficient distributable reserves but, after a tender offer occurs and until the redemption price for our Class B shares is paid in full, we will not be able to declare or pay any dividends nor any other distributions to our shareholders (in each case, whether in cash, securities of Grifols or any of our subsidiaries, or any other securities, assets or rights).
Liquidation Rights
Each Class B share entitles its holder to receive, upon our winding-up and liquidation, an amount equal to the sum of (i) the nominal value of such Class B share and (ii) the share premium paid up for such Class B share when it was subscribed for.
We will pay the liquidation amount to the holders of our Class B shares before any amount on account of liquidation is paid to the holders of our Class A shares.
Each of our Class B shares entitles its holder to receive, in addition to the liquidation preference amount, the same liquidation amount paid for a Class A share.
Subscription Rights
Each Class B share entitles its holder to the same rights (including preferential subscription rights and free allotment rights) as one Class A share in connection with any issuance, granting or sale of (i) any shares in Grifols, (ii) any rights or other securities exercisable for, exchangeable or convertible into shares in Grifols or (iii) any options, warrants or other instruments giving the right to the holder thereof to purchase, convert, subscribe or otherwise receive any securities in Grifols.
As an exception, the preferential subscription rights and the free allotment rights of the Class B shares will only be for new Class B shares or for instruments giving the right to purchase, convert, subscribe for or otherwise receive Class B shares, and the preferential subscription right and the free allotment right of an Class A share will only be for new Class A shares or for instruments giving the right to purchase, convert, subscribe or otherwise receive Class A shares, for each capital increase or issuance that meets the following three requirements: (i) the issuance of Class A shares and Class B shares is in the same proportion of our share capital as they represent at the time the resolution on the capital increase is passed; (ii) grants of preferential subscription rights or free allotment rights, as applicable, to the Class B shares for the Class B shares are under the same terms as the preferential subscription rights or free allotment rights, as applicable, granted to the Class A shares for the Class A shares; and (iii) no other shares or securities are issued.
165
Registration and Transfers
Class B shares are in book-entry form on Iberclear and are indivisible, as indicated with respect to Class A shares above in “—Class A Shares—Registration and Transfers.”
Change in Control
The Articles of Association do not contain any provisions that would have the effect of delaying, deferring or preventing a change in control of Grifols.
Changes in Share Capital
Changes in share capital are considered extraordinary matters and must be approved by our shareholders in accordance with the procedures explained above in “—Class A Shares—shareholders’ meetings and Voting Rights” and “—Class B Shares—Separate Vote at General Shareholder Meetings on Extraordinary Matters.”
A capital increase may be effected by issuing new shares or by increasing the par value of existing shares. A capital reduction may be effected by reducing the par value of existing shares or by redeeming or repurchasing existing shares.
426,129,798 Class A Shares are currently issued and outstanding with a par value of €0.25 per share and 261,425,110 Class B Shares are currently issued and outstanding with a par value of €0.05 per share. As of December 31, 2023, our total share capital stands at €119,603,705.
Sinking Fund
The Articles of Association do not contain any sinking fund provisions.
C. | Material Contracts |
The following contracts have been entered into by us within the two years immediately preceding the date of this annual report on Form 20-F or contain provisions under which we or another member of the Grifols Group has an obligation or entitlement that is material to us:
2017 Notes
For a summary of the material terms of the 2017 Notes, see Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Sources of Credit—The 2017 Notes.”
2019 Notes
For a summary of the material terms of the 2019 Notes, see Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Sources of Credit—The 2019 Notes.”
2021 Notes
For a summary of the material terms of the 2021 Notes, see Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Sources of Credit—The 2021 Notes.”
First Lien Credit Facilities
For a summary of the material terms of the First Lien Credit Facilities, see Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Sources of Credit—First Lien Credit Facilities.”
166
European Investment Bank Term Loans
For a summary of the material terms of the European Investment Bank Term Loans, see Item 5 of this Part I, “Operating and Financial Review and Prospects—B. Liquidity and Capital Resources—Sources of Credit—European Investment Bank Term Loans.”
Acquisitions
For a summary of the material terms of our acquisitions substantially completed in 2023, 2022 and 2021, see Item 5 of this Part I, “Operating and Financial Review and Prospects—A. Operating Results—Factors Affecting Our Financial Condition and Results of Operations—Acquisitions.”
D. | Exchange Controls |
Restrictions on Foreign Investment
Under present regulations, foreign investors may transfer invested capital, capital gains and dividends out of Spain without limitation on the amount other than applicable taxes. Law 19/2003, of July 4, 2003, as amended, updated Spanish exchange control and money laundering prevention provisions, by recognizing the principle of freedom of the movement of capital between Spanish residents and nonresidents unless they fall within the scope of article 7 bis of Law 19/2003, enacted in December 2022 through Royal Decree-Law 20/2022, or - solely with respect to investments in the defense sector - article 11 of Royal Decree 664/1999, of April 23, 1999.
The law establishes procedures for the declaration of capital movements for purposes of administrative or statistical information and authorizes the Spanish government to take measures which are justified on grounds of public policy or public security and public health. Article 7 bis of Law 19/2003 establishes a screening mechanism for certain investments made by non-E.U. and non-EFTA residents, based on public order, public health and public security reasons. It also provides the mechanism to take exceptional measures with regard to third countries if such measures have been approved by the European Union or by an international organization to which Spain is a party.
Violating the screening mechanism can invalidate transactions, with fines up to the value of the investment. Non-Spanish foreign investors, excluding those in tax havens, must notify the Spanish Registry of Foreign Investments post-investment for statistical purposes (as established in Royal Decree 571/2023, of July 4). Residents of tax havens require prior and subsequent notifications, except in specific cases. Additional regulations apply to certain industries such as air transportation, mining, and defense-related activities. E.U. residents are generally exempt from these restrictions, except in defense-related sectors and the manufacturing of weapons and explosives for non-military purposes.
Exchange Controls
Law 10/2010, on the prevention of money laundering and funding of terrorism, was adopted on April 28, 2010 and entered into force on April 30, 2010. This Law requires a person moving (i) paper money and coins in any currency, (ii) bearer checks in any currency or (iii) any other physical medium, including electronic media, designed for use as payment to the bearer to declare such payment to the Spanish exchange control authorities if it exceeds €10,000 (or the foreign currency equivalent).
E. | Taxation |
In General
Treatment of Holders of ADSs
This section describes the material United States federal income and Spanish tax consequences of owning shares or ADSs. It applies to you only if you hold your shares or ADSs as capital assets for tax purposes. This section does not apply to you if you are a member of a special class of holders subject to special rules, including:
| ● | a dealer in securities; |
167
| ● | a trader in securities that elects to use a mark-to-market method of accounting for securities holdings; |
| ● | a tax-exempt organization; |
| ● | a life insurance company; |
| ● | a person liable for alternative minimum tax under the Code (as defined below); |
| ● | a person that actually or constructively owns 10% or more of our voting stock; |
| ● | a person that holds shares or ADSs as part of a straddle or a hedging or conversion transaction; or |
| ● | a U.S. Holder (as defined below) whose functional currency is not the U.S. dollar. |
This section is based on the Internal Revenue Code of 1986, as amended, or the Code, its legislative history, existing and proposed regulations, published rulings and court decisions, in each case as in effect as of the date hereof and all of which are subject to change, possibly on a retroactive basis, as well as the tax laws of Spain and regulations thereunder and the Convention Between the United States of America and the Kingdom of Spain for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with Respect to Taxes on Income, or the Treaty, in each case as in effect as of the date hereof and subject to change.
You are a “U.S. Holder” if you are a beneficial owner of shares or ADSs and you are:
| ● | a citizen or resident of the United States; |
| ● | a corporation or other entity taxable as a corporation, created or organized in or under the laws of the United States, any state thereof or the District of Columbia; |
| ● | an estate whose income is subject to United States federal income tax regardless of its source; or |
| ● | a trust if a United States court can exercise primary supervision over the trust’s administration and one or more United States persons are authorized to control all substantial decisions of the trust. |
An “eligible U.S. Holder” is a U.S. Holder that:
| ● | is a resident of the United States for purposes of the Treaty; |
| ● | does not maintain a permanent establishment or fixed base in Spain to which shares or ADSs are attributable and through which the U.S. Holder carries on or has carried on business (or, in the case of an individual, performs or has performed independent personal services); and |
| ● | is otherwise eligible for benefits under the Treaty with respect to income and gain from the shares or ADSs. |
A “non-U.S. Holder” is a beneficial owner of shares or ADSs that is not a U.S. Holder.
In addition, if a partnership (including any entity treated as a partnership for United States federal income tax purposes) is a beneficial owner of shares or ADSs, the tax treatment of a partner in the partnership will generally depend upon the status of the partner and the activities of the partnership. A beneficial owner of shares or ADSs that is a partnership, and partners in such partnership, should consult their own tax advisors regarding the tax consequences of owning and disposing of shares or ADSs.
You should consult your own tax advisor regarding the United States federal, state and local and the Spanish and other tax consequences of owning and disposing of shares and ADSs in your particular circumstances. In particular, you should confirm your status as an eligible U.S. Holder with your advisor and should discuss any possible consequences of failing to qualify as an eligible U.S. Holder.
168
This discussion addresses only United States federal income taxation and Spanish income taxation, gift and inheritance taxation, wealth taxation and transfer taxation.
Treatment of Holders of ADRs
In general, and taking into account the earlier assumptions, for United States federal income and Spanish tax purposes, if you hold ADRs evidencing ADSs, you will be treated as the owner of the shares represented by those ADRs. Exchanges of shares for ADRs, and ADRs for shares, generally will not be subject to United States federal income or to Spanish tax.
Spanish Tax Considerations
This discussion of Spanish tax consequences applies only to owners of ADSs or shares who are eligible U.S. Holders. The following is a summary of material Spanish tax matters and is not exhaustive of all the possible tax consequences to individuals or entities of the acquisition, ownership and disposition of ADSs or shares.
Taxation of Dividends
Under Spanish law, including Royal Legislative Decree 5/2004, of March 5, 2004, as amended by Law 26/2014 (which is effective from January 1, 2015), on the Non-Resident Income Tax Law, dividends paid by a Spanish resident company to a holder of ordinary shares or ADSs not residing in Spain for tax purposes and not operating through a permanent establishment in Spain are subject to Spanish Non-Resident Income Tax of 19%.
We will levy an initial withholding tax on the gross amount of dividends at a 19% tax rate, following the procedures set forth by the Spanish Ministerial Order, or Order, of April 13, 2000. However, under the Treaty and subject to the fulfillment of certain requirements, individuals and entities may be entitled to a reduced rate of 15%.
To benefit from the Treaty’s reduced rate of 15%, an individual or entity must provide the depositary with a certificate from the U.S. Internal Revenue Service, or IRS, stating that, to the knowledge of the IRS, it is a resident of the United States within the meaning of the Treaty. The IRS certificate may be obtained by filing an IRS Form 8802 and is valid for a period of one year from the date of issue, unless it includes a specific year for which a tax resident is considered, in which case the certificate will be deemed applicable during that year.
According to the Order of April 13, 2000, to get a direct application of the Treaty’s reduced rate of 15%, the certificate referred to above must be provided to the depositary before the tenth day following the end of the month in which the dividends were distributable by us. If an individual or entity fails to timely provide the depositary with the required documentation, it may obtain a refund of the 4% in excess withholding that would result from the Spanish tax authorities in accordance with the procedures below.
Spanish Refund Procedure
According to Royal Decree 1776/2004, of July 30, 2004, as amended, which further develops the Royal Legislative Decree 5/2004 on the Non-Resident Income Tax Law, a refund of the amount withheld in excess of the rate provided by the Treaty can be obtained from the relevant Spanish tax authorities. An eligible U.S. Holder may pursue the refund claim by filing all of the following:
| ● | a Spanish 210 Form; |
| ● | the certificate from the IRS referred to above in “—Taxation of Dividends;” and |
| ● | evidence that non-resident income tax was withheld with respect to it. |
The Spanish Revenue Office must make the refund within six months after the refund claim is filed. If such period lapses without receipt of the refund, the holder is entitled to receive interest for late payment on the amount of the refund claimed.
169
The refund claim must be filed within four years of the date on which the withheld tax was collected by the Spanish tax authorities. According to Order EHA/3316, of December 17, 2010, for dividends paid as of January 2011, the 210 Form must be filed as from February 1st of the calendar year following the year in which the dividend was paid.
Individuals and entities are urged to consult their own tax advisers regarding refund procedures and any U.S. tax implications of refund procedures.
Taxation of Capital Gains
Under Spanish law, any capital gains derived from securities issued by persons residing in Spain for tax purposes are considered to be Spanish source income and, therefore, are taxable in Spain. For U.S. residents, income from the sale of ADSs or shares will be treated as capital gains for Spanish tax purposes. Spanish Non-Resident Income Tax is levied at a 19% rate on capital gains realized by persons not residing in Spain for tax purposes who are not entitled to the benefit of any applicable treaty for the avoidance of double taxation. Capital gains and losses will be calculated separately for each transaction and losses may not be offset against capital gains.
Notwithstanding the above, capital gains derived from the transfer of shares on an official Spanish secondary securities market by any holder who is a resident of a country that has entered into a treaty for the avoidance of double taxation with Spain containing a clause of “exchange of information” (as defined in Law 36/2006, of November 30, 2006, related to measures to prevent tax fraud) will be exempt from taxation in Spain. In addition, under the Treaty, capital gains realized by an individual or entity upon the disposition of ADSs or shares will not be taxed in Spain. An individual or entity is required to establish that it is entitled to this exemption by providing to the relevant Spanish tax authorities an IRS certificate of residence in the United States, together with the appropriate Spanish 210 tax form, between January 1st and January 20th of the calendar year following the year in which the transfer of shares took place.
Spanish Wealth Tax
Individuals not resident in Spain for tax purposes who hold shares or ADSs located in Spain are subject to the Spanish wealth tax (Law 19/1991), which imposes a tax on property and rights located in Spain or that can be exercised within the Spanish territory on the last day of any year. The Spanish tax authorities may take the view that all shares of Spanish corporations and all ADSs representing such shares are located in Spain for Spanish tax purposes. If the tax authorities take this view, non-residents of Spain who held shares or ADSs on the last day of any year would be subject to the Spanish wealth tax for such year on the average market value of such shares or ADSs during the last quarter of such year (this average price of listed shares is published in the official State Gazette every year). Law 4/2008 amended the Spanish wealth tax law, introducing a 100% tax rebate and eliminating the obligation to file any form for tax periods starting as of 1 January 2008. However, this 100% tax rebate was temporarily abolished with effect as of the 2011 fiscal year, and since then this situation has been extended every year. From 2021, the abolition of the rebate has been established as indefinite, thus eliminating the need to stipulate extensions. Notwithstanding the above, there is a tax-free allowance of €700,000.
As a result of the above legislation, non-residents of Spain who hold or held shares, ADSs, or other assets or rights located in Spain according to Spanish wealth tax law, on the last day of the year, the combined value of which exceeds €700,000 might be subject to the Spanish wealth tax on that excess amount at marginal rates varying between 0.2% and 3.5% (the highest bracket increased by 1% since 2021), and would be obliged to file the corresponding wealth tax return.
Law 11/2021 amends the Wealth Tax Law to clarify that all non-residents taxpayers (and not only those who are resident in the EU and EEA jurisdictions) are entitled to apply the tax reliefs approved by the Spanish Regions.
170
Solidarity Tax on Large Fortunes
On December 29, 2022, a new solidarity tax on large fortunes came into force, meant to be supplementary to the wealth tax described above. The new solidarity tax is a direct and personal tax levied on individuals with a net wealth above €3.0 million as of December 31 of each year. Non-Spanish residents would be subject to this tax only taking into account their Spanish net wealth. This new tax is consistent with the provisions of the Spanish wealth tax in most essential aspects (such as exemptions, taxable and net taxable bases, tax rates and limit on amount of tax payable). The solidarity tax had been designed to be temporary and remain in force for two calendar years (i.e. 2022 and 2023), although the law included a review clause allowing the legislator to assess whether to extend it at the end of the initial period. Following publication of the recent Royal Decree-Law 8/2023, the solidarity tax has been extended indefinitely (with effects starting in 2024), until the wealth tax is revised in the context of the autonomous community finance system. In order to avoid double taxation, the rules applicable to the new solidarity tax allows individuals to deduct amounts already paid as Spanish wealth tax from their total payable solidarity tax
Spanish Inheritance and Gift Taxes
Under Law 29/1987, transfers of shares or ADSs upon death or by gift are subject to Spanish inheritance and gift taxes if the transferee is a resident of Spain for tax purposes, or if the shares or ADSs are located in Spain at the time of gift or death, or the rights attached thereto could be exercised or have to be fulfilled in the Spanish territory, regardless of the residence of the beneficiary. In this regard, the Spanish tax authorities may determine that all shares of Spanish corporations and all ADSs representing such shares are located in Spain for Spanish tax purposes. The applicable tax rate, after applying all relevant factors, ranges between 0% and 81.6% for individuals. Law 11/2021 amends the Gift and Inheritance Tax Law to clarify that all non-residents taxpayers (and not only those who are resident in the EU and EEA jurisdictions) are entitled to apply the tax reliefs approved by the Spanish Regions, following the case-law of the Spanish Supreme Court.
Gifts granted to corporations not resident in Spain are subject to Spanish Non-Resident Income Tax of 19% of the fair market value of the shares as a capital gain. If the donee is a United States corporation, the exclusions available under the Treaty described above in “—Taxation of Capital Gains” will be applicable.
Expenses of Transfer
Transfers of ADSs or shares will be exempt from any Spanish transfer tax or value-added tax. Additionally, no Spanish stamp tax will be levied on such transfers.The transfer of shares or ADSs may be subject to the Spanish Financial Transaction tax (the “Spanish FTT”). The Spanish law which implements the Spanish FTT (the “FTT Law”) was approved on October 7, 2020, and published in the Spanish Official Gazette (Boletín Oficial del Estado) on October 16, 2020. The Spanish FTT came into force three months after the publication of the FTT Law (i.e., on January 16, 2021) and will charge a 0.2% rate on specific acquisitions of listed shares issued by Spanish companies (including ADSs) whose market capitalization exceeds 1 billion euros (€1 billion) (this may be the case of Grifols), regardless of the jurisdiction of residence of the parties involved in the transaction. Transactions in the primary market (such as a capital increase) are exempt from the Spanish FTT. However, the Spanish FTT will subject other transactions involving the acquisition of shares or ADSs depending on the market capitalization of Grifols. Prospective investors are advised to seek their own professional advice in relation to the Spanish FTT.
171
United States Federal Income Tax Considerations
Taxation of Dividends
U.S. Holders
Under the United States federal income tax laws, and subject to the passive foreign investment company (“PFIC”) rules discussed below, if you are a U.S. Holder, the gross amount of any dividend (including any preferred dividends on our Class B shares) we pay out of our current or accumulated earnings and profits (as determined for United States federal income tax purposes) is subject to United States federal income taxation. If you are a noncorporate U.S. Holder, dividends (including any preferred dividends on our Class B shares) paid to you that constitute qualified dividend income will be taxable to you at a maximum tax rate of 20%, provided that you hold the shares or ADSs for more than 60 days during the 121-day period beginning 60 days before the ex-dividend date and meet other holding period requirements. Dividends we pay (including any preferred dividends on our Class B shares) with respect to the shares or ADSs generally will be qualified dividend income.
With respect to any dividends we pay (including any preferred dividends on our Class B shares) you must include any Spanish tax withheld from the dividend payment in the gross amount of such dividend even though you do not in fact receive it. Dividends are taxable to you when you, in the case of shares, or the Depositary, in the case of ADSs, receive such dividends, actually or constructively. Such dividends will not be eligible for the dividends-received deduction generally allowed to United States corporations in respect of dividends received from other United States corporations. The amount of a dividend distribution that you must include in your income as a U.S. Holder will be the U.S. dollar value of the euro payments made, determined at the spot euro/U.S. dollar rate on the date the dividend distribution is includible in your income, regardless of whether the payment is in fact converted into U.S. dollars. Generally, any gain or loss resulting from currency exchange fluctuations during the period from the date you include a dividend payment in income to the date you convert the payment into U.S. dollars will be treated as ordinary income or loss and will not be eligible for the special tax rate applicable to qualified dividend income. The gain or loss generally will be income or loss from sources within the United States for foreign tax credit limitation purposes. Distributions in excess of current and accumulated earnings and profits, as determined for United States federal income tax purposes, will be treated as a non-taxable return of capital to the extent of your adjusted tax basis in the shares or ADSs and, thereafter, as capital gain.
Subject to certain limitations, the Spanish tax withheld in accordance with the Treaty and paid over to Spain will be creditable or deductible against your United States federal income tax liability. Special rules apply in determining the foreign tax credit limitation with respect to dividends that are subject to the maximum 20% tax rate. To the extent a refund of the tax withheld is available to you under Spanish law or under the Treaty, the amount of tax withheld that is refundable will not be eligible for credit against your United States federal income tax liability. See “—Spanish Tax Considerations—Spanish Refund Procedure” above for the procedures for obtaining a tax refund.
Dividends will be income from sources outside the United States, and dividends paid will, depending on your circumstances, be “passive” or “general” income which, in either case, is treated separately from other types of income for purposes of computing the foreign tax credit allowable to you.
A U.S. Holder may make an election to treat all foreign taxes paid as deductible expenses in computing taxable income, rather than as a credit against tax, subject to generally applicable limitations. Such an election, once made, applies to all foreign taxes paid for the taxable year subject to the election. The rules governing foreign tax credits are complex and, therefore, U.S. Holders are strongly encouraged to consult their own tax advisors to determine whether they are subject to any special rules that may limit their ability to make effective use of foreign tax credits and whether or not an election would be appropriate based on their particular circumstances.
172
Non-U.S. Holders
If you are a non-U.S. Holder, dividends (including any preferred dividends on our Class B shares) paid to you in respect of shares or ADSs will not be subject to United States federal income tax unless such dividends are “effectively connected” with your conduct of a trade or business within the United States, and such dividends are attributable to a permanent establishment that you maintain in the United States if that is required by an applicable income tax treaty as a condition for subjecting you to United States taxation on a net income basis. In such cases you generally will be taxed in the same manner as a U.S. Holder. If you are a corporate non-U.S. Holder, “effectively connected” dividends may, under certain circumstances, be subject to an additional “branch profits tax” at a 30% rate or at a lower rate if you are eligible for the benefits of an income tax treaty that provides for a lower rate.
Taxation of Capital Gains
U.S. Holders
Subject to the PFIC rules discussed below, if you are a U.S. Holder and you sell or otherwise dispose of your shares or ADSs, you will recognize capital gain or loss for United States federal income tax purposes equal to the difference between the U.S. dollar value of the amount that you realize and your adjusted tax basis, determined in U.S. dollars, in your shares or ADSs. Capital gain of a noncorporate U.S. Holder is generally taxed at a maximum rate of 20% where such noncorporate U.S. Holder has a holding period greater than one year. Such gain or loss will generally be income or loss from sources within the United States for foreign tax credit limitation purposes.
Non-U.S. Holders
If you are a non-U.S. Holder, you will not be subject to United States federal income tax on gain recognized on the sale or other disposition of your shares or ADSs unless:
| ● | the gain is “effectively connected” with your conduct of a trade or business in the United States, and the gain is attributable to a permanent establishment that you maintain in the United States if that is required by an applicable income tax treaty as a condition for subjecting you to United States taxation on a net income basis; or |
| ● | you are an individual, you are present in the United States for 183 or more days in the taxable year of the sale, and certain other conditions exist. |
If you are a corporate non-U.S. Holder, “effectively connected” gains that you recognize may also, under certain circumstances, be subject to an additional “branch profits tax” at a 30% rate or at a lower rate if you are eligible for the benefits of an income tax treaty that provides for a lower rate.
173
Passive Foreign Investment Company Considerations
We believe that our shares and ADSs should not be treated as stock of a PFIC for United States federal income tax purposes, but this conclusion is a factual determination that is made annually and thus may be subject to change. If we were to be treated as a PFIC, gain realized on the sale or other disposition of your shares or ADSs would in general not be treated as capital gain. Instead, if you are a U.S. Holder, you would be treated as if you had realized such gain and certain “excess distributions” ratably over your holding period for the shares or ADSs and would be taxed at the highest tax rate in effect for each such year to which the gain was allocated, together with an interest charge in respect of the tax attributable to each such year. With certain exceptions, your shares or ADSs will be treated as stock in a PFIC if we were a PFIC at any time during your holding period in your shares or ADSs. Certain elections (such as the mark-to-market election or the qualified electing fund (“QEF”) election) may be available that would result in alternative treatments of the ADSs or shares. Dividends that you receive from us will not be eligible for the special tax rates applicable to qualified dividend income if we are treated as a PFIC with respect to you either in the taxable year of the distribution or the preceding taxable year, but instead will be taxable at rates applicable to ordinary income.
Medicare Contribution Tax on Unearned Income
A U.S. Holder that is an individual is subject to a 3.8% tax on the lesser of (1) such U.S. Holder’s “net investment income” for the relevant taxable year and (2) the excess of such U.S. Holder’s modified adjusted gross income for the taxable year over a certain threshold (between $125,000 and $250,000, depending on the individual’s circumstances). A U.S. Holder that is an estate, or a trust that does not fall into a special class of trusts that is exempt from such tax, is subject to a 3.8% tax on the lesser of (1) such U.S. Holder’s undistributed “net investment income” for the relevant taxable year and (2) the excess of such U.S. Holder’s adjusted gross income for the taxable year over the amount at which the highest tax bracket begins for that taxable year ($14,450 for 2023). A U.S. Holder’s net investment income will generally include, among other items, the amount of gross dividend income and the amount of any net gains from such U.S. Holder’s disposition of your shares or ADSs, unless such dividends or net gains are derived in the ordinary course of the conduct of a trade or business (other than a trade or business that consists of certain passive or trading activities). U.S. Holders that are individuals, estates or trusts should consult their own tax advisors regarding the applicability of this tax to income and gains in respect of their investment in the shares or ADSs.
Backup Withholding and Information Reporting
If you are a noncorporate U.S. Holder, information reporting requirements, on Internal Revenue Service Form 1099, generally will apply to:
| ● | dividend payments or other taxable distributions made to you within the United States; and |
| ● | the payment of proceeds to you from the sale of shares or ADSs effected at a United States office of a broker. |
Additionally, backup withholding may apply to such payments if you are a noncorporate U.S. Holder that:
| ● | fails to provide an accurate taxpayer identification number; |
| ● | is notified by the Internal Revenue Service that you have failed to report all interest and dividends required to be shown on your federal income tax returns; or |
| ● | in certain circumstances, fails to comply with applicable certification requirements. |
If you are a non-U.S. Holder, you are generally exempt from backup withholding and information reporting requirements with respect to:
| ● | dividend payments made to you outside the United States by us or another non-United States payor; and |
174
| ● | other dividend payments and the payment of the proceeds from the sale of shares or ADSs effected at a United States office of a broker, if the income associated with such payments is otherwise exempt from United States federal income tax; and: |
| ● | the payor or broker does not have actual knowledge or reason to know that you are a United States person and you have furnished the payor or broker one of the following: |
| ● | an Internal Revenue Service Form W-8BEN, Form W-8BEN-E or an acceptable substitute form upon which you certify, under penalties of perjury, that you are a non-United States person, or |
| ● | other documentation upon which it may rely to treat the payments as made to a non-United States person in accordance with U.S. Treasury regulations, or |
| ● | you otherwise establish an exemption. |
Payment of the proceeds from the sale of shares or ADSs effected at a foreign office of a broker generally will not be subject to information reporting or backup withholding. However, a sale of shares or ADSs that is effected at a foreign office of a broker will be subject to information reporting and backup withholding if:
| ● | the proceeds are transferred to an account maintained by you in the United States; |
| ● | the payment of proceeds or the confirmation of the sale is mailed to you at a United States address; or |
| ● | the sale has some other specified connection with the United States as provided in U.S. Treasury regulations, unless the broker does not have actual knowledge or reason to know that you are a United States person and the documentation requirements described above are met or you otherwise establish an exemption. |
In addition, a sale of shares or ADSs effected at a foreign office of a broker will be subject to information reporting if the broker is:
| ● | a United States person; |
| ● | a controlled foreign corporation for United States federal income tax purposes; |
| ● | a foreign person 50% or more of whose gross income is effectively connected with the conduct of a United States trade or business for a specified three-year period; or |
| ● | a foreign partnership, if at any time during its tax year: |
| ● | one or more of its partners are “U.S. persons,” as defined in U.S. Treasury regulations, which in the aggregate hold more than 50% of the income or capital interest in the partnership, or |
| ● | such foreign partnership is engaged in the conduct of a United States trade or business, unless the broker does not have actual knowledge or reason to know that you are a United States person and the documentation requirements described above are met or you otherwise establish an exemption. Backup withholding will apply if the sale is subject to information reporting and the broker has actual knowledge that you are a United States person. |
Backup withholding is not an additional tax. You generally may obtain a refund of any amounts withheld under the backup withholding rules that exceed your income tax liability by filing a refund claim with the United States Internal Revenue Service.
175
Disclosure of Information with Respect to Foreign Financial Assets
Certain U.S. individuals who hold any interest in “specified foreign financial assets,” including our shares or ADSs, during such holder’s taxable year must attach to their U.S. tax return for such year certain information with respect to each such asset if the aggregate value of all of such assets exceeds $50,000 (or a higher dollar amount prescribed by the Internal Revenue Service), unless such shares or ADSs are held in an account maintained by a U.S. payor, such as a U.S. financial institution or the U.S. branch of a foreign bank or insurer. For this purpose, a “specified foreign financial asset” includes any depositary, custodial or other financial account maintained by a foreign financial institution, and certain assets that are not held in an account maintained by a financial institution, including any stock or security issued by a person other than a U.S. person. A taxpayer subject to these rules who fails to furnish the required information may be subject to a penalty of $10,000, and an additional penalty may apply if the failure continues for more than 90 days after the taxpayer is notified of such failure by the Internal Revenue Service, unless the taxpayer demonstrates a reasonable cause for such failure to comply. An accuracy-related penalty of 40% is imposed for an underpayment of tax that is attributable to an “undisclosed foreign financial asset understatement,” which for this purpose is the portion of the understatement of gross income for any taxable year that is attributable to any transaction involving an “undisclosed foreign financial asset,” including any asset that is subject to information reporting requirements under these rules, which would include our shares or ADSs if the dollar threshold described above were satisfied.The applicable statute of limitations for assessment of U.S. federal income taxes is extended to six years if a taxpayer omits from gross income more than $5,000 and such omission is attributable to a foreign financial asset as to which reporting is required under the rules described in the preceding paragraph or would be so required if such rules were applied without regard to the dollar threshold or any other exceptions specified by the Internal Revenue Service. In addition, the statute of limitations will be suspended if a taxpayer fails to provide in a timely manner either information with respect to specified foreign financial assets required to be reported or the annual information reports required for holders of PFIC stock, including PFIC stock for which a QEF election is made. You should consult your own tax advisor concerning any obligation you may have to furnish information to the Internal Revenue Service as a result of holding our shares or ADSs.
F. | Dividends and Paying Agents |
Not Applicable.
G. | Statement by Experts |
Not Applicable.
H. | Documents on Display |
We are subject to the information requirements of the Exchange Act, except that, as a foreign private issuer, we are not subject to the proxy rules or the short-swing profit disclosure rules of the Exchange Act. In accordance with these information requirements, we file or furnish reports and other information with the SEC. Reports and other information filed or furnished by us with the SEC may be inspected and copied at the public reference facilities maintained by the SEC at Room 1024, 100 F Street, N.E., Washington, D.C. 20549, and at the SEC’s regional offices at 233 Broadway, New York, New York 10279 and Northwestern Atrium Center, 500 West Madison Street, Suite 1400, Chicago, Illinois 60661-2511.
Copies of such material may also be inspected at the offices of NASDAQ, 4 Times Square, New York, New York 10036, on which our ADSs are listed. In addition, information filed electronically with the SEC is publicly available on the SEC’s website, which does not form part of this annual report on Form 20-F, at http://www.sec.gov.
I. | Subsidiary Information |
Not Applicable.
J. | Annual Report to Security Holders |
If we are required to provide an annual report to security holders in response to the requirements of Form 6-K, we will submit the annual report to security holders in electronic format in accordance with the EDGAR Filer Manual.
176
Item 11.QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
The risks inherent in our market-sensitive instruments are potential losses that may arise from adverse changes to interest rates, foreign exchange rates and market prices. We are subject to market risk resulting from changes in interest rates because such changes may affect the cost at which we obtain financing. We are subject to exchange rate risk with respect to our debt denominated in foreign currencies.
See Note 30 to our audited consolidated financial statements included in this annual report on Form 20-F on page F-110.
Currency Risk
We operate internationally and are exposed to currency risks when operating in foreign currencies, in particular with respect to the U.S. dollar. Currency risk is associated with future commercial transactions, recognized assets and liabilities and net investments in foreign operations.
We hold several investments in foreign operations, the net assets of which are exposed to currency risk. Currency risk affecting net assets of our foreign operations in U.S. dollars are mitigated primarily through borrowings in the relevant foreign currency. Our main exposure to currency risk is to the U.S. dollar, which is used in a significant percentage of our transactions in foreign currencies.
If the U.S. dollar had strengthened by 10% against the euro at December 31, 2023 and 2022, equity would have increased by €820.6 million and €892.8 million, respectively, and profit would have decreased by €20.6 million and €17.4 million, respectively. This analysis assumes that all other variables are held constant, especially that interest rates remain constant. A 10% weakening of the U.S. dollar against the euro at December 31, 2023 and December 31, 2022 would have had the opposite effect for the amounts shown above, all other variables being held constant.
We use hedge accounting to partially hedge our currency risk exposure. See Note 30(d) to our audited consolidated financial statements included in this annual report on Form 20-F on page F-119.
Interest Rate Risk
Our interest rate risks arise from current and non-current borrowings. Borrowings at variable interest rates expose us to cash flow interest rate risks. The purpose of managing interest rate risk is to balance the debt structure, maintaining part of borrowings at fixed rates and hedging part of variable rate debt.
As of December 31, 2023, $2.3 billion and €2.0 billion of our indebtedness had a variable rate (SOFR or EURIBOR, respectively). This variable-rate debt represented 39.0% of our total debt at December 31, 2023 (37.0% at December 31, 2022) and includes mainly senior secured debt. See Item 3 of this Part I, “Key Information—D. Risk Factors—Risks Relating to the Company and Our Business—We are susceptible to interest rate variations.”
177
As of the date of this annual report, we are not participating in interest rate hedges of Euros or U.S. dollars. In previous years, the fair value of interest rate swaps, contracted to reduce the impact of increases in variable interest rates (SOFR and EURIBOR), were accounted for on a monthly basis. These derivative financial instruments comply with hedge accounting requirements.
If the interest rate had been 100 basis points higher at December 31, 2023, the interest expense would have increased by €34.1 million. A 100 basis points decrease in interest rates at December 31, 2023 would have had the opposite effect for the amounts shown above. As we do not have any hedging derivatives in place, the net effect on cash interest payments would have increased by the same amount.
Market Price Risk
We are subject to price risk with respect to raw materials, which is mitigated by the vertical integration of the hemoderivatives business in a sector that is highly concentrated.
Item 12.DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
A. | Debt Securities |
Not Applicable.
B. | Warrants and Rights |
Not Applicable.
C. | Other Securities |
Not Applicable.
D. | American Depositary Shares |
Deutsche Bank Trust Company Americas serves as the depositary for both our Class A ADSs and our Class B ADSs, and its principal executive office is located at 60 Wall Street, New York, NY 10005, USA. The custodian is Deutsche Bank Sociedad Anónima Española, and its principal office in Spain is located at Ronda General Mitre 72-74, 08017 Barcelona, Spain.
Each Class A ADS represents the right to receive one half of one Class A ordinary share of Grifols. Each Class B ADS represents the right to receive one Class B non-voting preference share of Grifols.
178
The following is a summary of the fee provisions of the deposit agreements for each of the Class A ADSs and Class B ADSs. For more complete information, you should read each deposit agreement in its entirety.
Associated Fee |
| Depositary Action |
$5.00 (or less) per 100 ADSs (or portion of 100 ADSs) | Issuance of ADSs, including issuance resulting from a distribution of shares or rights or other property. Cancellation of ADSs for the purpose of withdrawal, including if the deposit agreement terminates. | |
$2.00 (or less) per 100 ADSs (or portion of 100 ADSs) | Distribution of cash proceeds, including cash dividends or sale of rights and other entitlements. | |
$2.00 (or less) per 100 ADSs (or portion of 100 ADSs) per calendar year, provided that this fee, when combined with the fee for distribution of cash proceeds, including cash dividends or sale of rights and other entitlements, shall not exceed $2.00 (or less) per 100 ADSs (or portion of 100 ADSs) in any calendar year | Depositary operation and maintenance costs. | |
Annual fee of $1.00 per 100 ADSs | Inspections of the relevant share register. | |
Registration or transfer fees | Transfer and registration of our shares on its share register to or from the name of the depositary or its agent when you deposit or withdraw our shares. | |
Expenses of the depositary | Cable, telex and facsimile transmissions (when expressly provided in the deposit agreement). Converting foreign currency to U.S. dollars. | |
Taxes and other governmental charges the depositary or the custodian has to pay on any ADS or share underlying an ADS, including any applicable interest and penalties thereon and any share transfer or other taxes or governmental charges, for example, stock transfer taxes, stamp duty or withholding taxes | As necessary. | |
Any fees and expenses incurred by the depositary in connection with the conversion of a foreign currency in compliance with the applicable exchange control and other regulations, and the delivery of deposited securities, including any fees of a central depository, and any additional fees, charges, costs, or expenses, that may be incurred by the depositary from time to time | As necessary. | |
Any additional fees, charges, costs or expenses that may be incurred by the depositary from time to time. | As necessary. |
The depositary collects its fees for issuance and cancellation of our ADSs directly from investors depositing shares or surrendering our ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by deduction from cash distributions, by directly billing investors or by charging the book-entry system accounts of participants acting for such investors. The depositary may generally refuse to provide fee-attracting services until its fees for those services are paid.
179
The fees and charges holders of our ADSs may be required to pay may vary over time and may be changed by us and by the depositary. Our ADS holders will receive prior notice of such changes.
Fees Paid by the Depositary to Grifols
Deutsche Bank Trust Company Americas, as depositary, has agreed to reimburse or pay on behalf of Grifols certain reasonable expenses related to our ADR programs and incurred by us in connection with the programs, such as investor relations activities and ongoing maintenance expenses and listing fees. It has covered all such expenses incurred by us during 2023 for an amount of $2.2 million. The amounts the depositary reimbursed or paid are not perforce related to the fees it collected from ADS holders.
GLOSSARY OF TERMS
“340B Program” means the federal health care program established by Section 340B of the PHS Act, which requires manufacturers participating in Medicaid to agree to provide outpatient drugs to covered entities, including a variety of community health clinics and certain other entities that receive certain governmental health care grants, as well as hospitals that serve a disproportionate share of certain low income individuals, certain cancer centers, children’s hospitals, critical access hospitals and rural referral centers, at significantly reduced prices.
“AAT” means alpha1-antitrypsin, a protein that protects the lungs.
“ACA” refers to the U.S. Affordable Care Act and the companion Healthcare and Education Reconciliation Act, each enacted in March 2010, as amended.
“AlphaID” is a free cheek swab to test for alpha-1 deficiency in patients.
“AEMPS” refers to the Spanish Agency of Medicines and Medical Products.
“AMP” means generally the average manufacturer price, as defined based on methodologies set forth in federal regulations, that wholesalers and other large purchasers pay manufacturers for certain outpatient prescription drugs covered by Medicaid, and is used, among other things, to help calculate the rebates paid by certain drug manufacturers that are shared by the U.S. and state governments for Medicaid-covered outpatient drugs.
“Alzheimer’s disease” is the most common form of dementia. This incurable, degenerative, and terminal disease was first described by German psychiatrist and neuropathologist Alois Alzheimer in 1906 and was named after him.
“Albumin” is the most abundant blood plasma protein and is produced in the liver and forms a large proportion of all plasma. Albumin normally constitutes about 60% of human plasma. It is important in regulating blood volume by maintaining the oncotic pressure of the blood compartment.
“ASP” means the average sales price of certain outpatient drugs covered by Medicare Part B, and is used to help calculate reimbursement of such drugs.
“Assays” are systems designed to detect antibodies, antigens or the nucleic acid of an infectious agent. For instance, the WNV assay detects the presence of the West Nile virus in blood donations. The main types of assay used for blood screening are Immunoassays and Nucleic acid technology, or NAT assays.
“ATIII” means intramuscular (hyperimmune) immunoglobulins.
“A1PI” means alpha-1 proteinase inhibitor.
“BIDSXT” means a software tool that allows the analysis, interpretation and database management to transmit the results to the LIS.
180
“BLA” (Biologics License Application) is a biological license application issued by the FDA, and serves as a U.S. marketing authorization for certain biological drug products.
“BlisPack” a blister handling machine.
“BLOODchip” blood group genotyping tests manufactured by Progenika, a company in which Grifols has a majority stake.
“Best Price” means generally the lowest price, as defined based on methodologies set forth in federal regulations, available from a manufacturer for certain outpatient drugs reimbursed by Medicaid, with respect to specified purchasers, such as wholesalers, retailers, health care providers and other designated entities. Best Price is used, among other things, among other things, to help calculate the rebates paid by certain drug manufacturers that are shared by the U.S. and state governments for Medicaid-covered outpatient drugs.
“CCPR” refers to the California Consumer Protection act, a regulation passed by the U.S. state of California.
“CFIUS” refers to the Committee on Foreign Investment in the United States.
“cGMP” means current Good Manufacturing Practice.
“CIDP” means chronic inflammatory demyelinating polyneuropathy, a neurological disease resulting in weakness, numbness, pain and difficulty in walking.
“Cirrhosis” is a medical condition which is a result of advanced liver disease. It is characterized by the replacement of liver tissue by fibrosis (scar tissue) and regenerative nodules (lumps that occur due to attempted repair of damaged tissue).
“Congenital Alpha-1 Antitrypsin Deficiency” is an inherited disease characterized by reduced levels in the blood of the substance Alpha-1 Antitrypsin, or AAT. This substance is a protein that is normally made by the liver and reaches other organs (such as the lungs) after being released into the blood circulation.
“CLL” means chronic lymphocytic leukaemia.
“CMS” refers to the U.S. Centers for Medicare & Medicaid Services.
“CMV” means Cytomegalovirus, a common virus that infects people of all ages.
“CNMV” means the Comisión Nacional del Mercado de Valores.
“CPI-U” means the Consumer Price Index For All Urban Consumers, which measures the changes in the price of a basket of goods and services purchased by urban consumers.
“CPP” is the certificate of pharmaceutical product, a certificate issued in the format recommended by the WHO, which establishes the status of a pharmaceutical product and of the applicant for a certificate in the relevant exporting country.
“CSRC” refers to the Chinese Securities Regulatory Commission.
“DHPR” means dihydropyridine receptors.
“Diabetes” is a metabolic disease in which a person has high blood sugar, either because the pancreas does not produce enough insulin, or because cells do not respond to the insulin that is produced.
“DOJ” refers to the United States Department of Justice.
“ELISA” means enzyme-linked immunosorbent assay.
181
“EMA” refers to the European Medicines Agency.
“Erytra Eflexis” a fully automated, mid-size analyzer that performs pretransfusion compatibility testing using DG Gel technology.
“Factor VIII” or “FVIII” is an essential blood clotting factor also known as anti-haemophilic factor, or AHF. In humans, Factor VIII is encoded by the F8 gene. Defects in this gene results in hemophilia A, which is a sex-linked disease and occurs predominantly in males. FVIII concentrated from donated blood plasma, or alternatively recombinant FVIII, or rFVIII, can be given to hemophiliacs to restore hemostasis.
“Factor IX” is an important blood clotting factor also known as Christmas factor or plasma thromboplastin component, or PTC. It is one of the serine proteases of the coagulation system and belongs to the peptidase family S1. In humans, a deficiency of this protein causes haemophilia B, which is a sex-linked disease and occurs predominantly in males.
“FDA” is the U.S. Food and Drug Administration.
“Fibrin Sealant” is surgical adhesive material that is utilized in a variety of surgical situations.
“Fractionation” is the process of fractionating plasma, or separating it into its different components or plasma derivatives.
“FSS” refers to the Federal Supply Schedule, a schedule managed by the U.S. Department of Veterans Affairs, which makes available discounted drug pricing for authorized government users.
“GMP” means good manufacturing practices.
“GPO” means group purchasing organization.
“GDPR” refers to the General Data Protection Regulation, an EU regulation.
“Gri-fill System” is a process for the sterile filling of flexible material bags.
“HBV” means hepatitis B virus.
“HBC” means hepatitis C virus.
“Hematology” is the study of blood, blood-forming organs, and blood diseases.
“Hemoderivative” is a substance obtained by fractionation of human blood plasma.
“Hemophilia A” is a genetic deficiency in clotting Factor VIII, which causes increased bleeding (usually affects males).
“Hemostasis” is a complex process which causes the bleeding process to stop. It refers to the process of keeping blood within a damaged blood vessel (the opposite of hemostasis is hemorrhage). Most of the time this includes the changing of blood from a fluid to a solid state. Intact blood vessels are central to moderating blood’s tendency to clot. Hemostasis has three major steps: 1) vasoconstriction, 2) temporary blockage of a break by a platelet plug, and 3) blood coagulation, or formation of a clot that seals the hole until tissue are repaired.
“HHS” refers to the U.S. Department of Health and Human Services.
“HIPAA” refers to the U.S. Health Insurance Portability and Accountability Act of 1996, as amended, and the regulations promulgated thereunder, a U.S. health information privacy, security and breach notification.
“HIV” refers to the human immunodeficiency virus.
182
“HRSA” means the Health Resources & Services Administration, the subagency of HHS responsible for, among other things, oversight of the 340B Program. HRSA is primarily responsible for ensuring access to health care services for people who are uninsured, including through grant funding programs.
“IFX” means infliximab, a medication used to treat Crohn’s Disease and Ulcerative Colitis.
“IG” means immunoglobulin, which contains the pooled IgG (immunoglobulin (antibody) G) extracted from plasma.
“Immunohematology” is a branch of hematology relating to the study of antigens and antibodies and their effects on blood and the relationships between disorders of the blood and the immune system.
“Immunology” is a broad branch of biomedical science that covers the study of all aspects of the immune system in organisms. It deals with the physiological functioning of the immune system in states of both health and disease; malfunctions of the immune system in immunological disorders (autoimmune diseases, hypersensitivities, immune deficiency, transplant rejection); the physical, chemical and physiological characteristics of the components of the immune system in vitro, in situ, and in vivo.
“IND” means investigational new drug application, which is an application that must be accepted by the FDA and in effect prior to certain drug sponsors commencing clinical trials involving human subjects.
“IRB” refers to institutional review boards, oversight committees that approve and monitor clinical trials to protect the rights and welfare of human subjects.
“ITP” means idiopathic thrombocytopenic purpura.
“IVIG” means intravenous immunoglobulin, which is a blood product administered intravenously. It contains the pooled IgG (immunoglobulin (antibody) G) extracted from plasma. It is mainly used as treatment in four major categories: (i) immune deficiencies, (ii) inflammatory and autoimmune diseases, (iii) neurological diseases and (iv) acute infections.
“Kawasaki disease” is a rare autoimmune disease that mostly affects children and causes inflammation of vessels, fever and rashes. This disease can be treated with IVIG.
“Koate-DVI” is a medication is used to control and prevent bleeding episodes in people with low levels of factor VIII (hemophilia A).
“LIS” means Laboratory information system.
“Medicaid” is a social healthcare program in the United States for individuals with low income and resources.
“Medicare” is a U.S. federal health insurance program for individuals 65 years old and over and certain younger individuals with disabilities.
“Medicare Part B” is a portion of the Medicare program generally which provides fee-for-service reimbursement for medical services such as by physicians and other health care practitioners, and certain outpatient services, equipment, supplies, and certain drugs, including physician-administered drugs and drugs provided in the hospital outpatient setting.
“Medicare Part D” is a portion of the Medicare program generally which includes optional coverage for brand name and generic prescription drugs dispensed to Medicare beneficiaries, generally by retail pharmacies. Like, Medicare Part C, Medicare Part D is offered by private health insurance companies that are contracted with Medicare. Individuals enrolled in Medicare Part D generally can choose to receive benefits through stand-alone prescription drug plans, or through Medicare Part C prescription drug plans that provide integrated medical coverage, including drugs.
“MM” means multiple myeloma.
183
“MRB” refers to the Market Research Bureau, Inc., an independent market research firm which supplies blood and plasma products industry data on a global level.
“NAT” means nucleic acid testing.
“NVD” means the share and asset agreement, executed with Novartis Vaccines and Diagnostics, Inc.
“OIG” is the HHS Office of the Inspector General, which is charged with protecting the integrity of HSS programs, including the Medicare and Medicaid programs.
“Orphan drug” is a pharmaceutical agent that has been developed specifically to treat a rare medical condition, the condition itself being referred to as an orphan disease. The assignment of orphan status to a disease and to any drugs developed to treat it is a matter of public policy in many countries, and has resulted in medical breakthroughs that may not have otherwise been achieved due to the economics of drug research and development The Orphan Drug Act (ODA) of January 1983, passed in the United States, with lobbying from the National Organization for Rare Disorders, is meant to encourage pharmaceutical companies to develop drugs for diseases that have a small market. Under the law, companies that develop such a drug (a drug for a disorder affecting fewer than 200,000 people in the United States) may be granted seven years of market exclusivity, and may get clinical trial tax incentives.
“Open Payments Program” imposes new reporting and disclosure requirements for pharmaceutical and medical device manufacturers with regard to payments or other transfers of value made to certain U.S. covered healthcare practitioners, such as physicians, and to academic medical centers, and with regard to certain ownership interests held such practitioners in reporting entities.
“PDUFA” is the Prescription Drug User Fee Act, which levies a user fee on certain human drug applications.
“Plasma” is the liquid part of the blood. The majority of plasma is composed of water. The remainder is essential proteins and antibodies that help sustain our body’s vital functions. A shortage of any one of these plasma proteins, such as albumin or immunoglobulins, can give rise to one of many life-threatening illnesses.
“Plasmapheresis” is a technique which separates plasma from other blood components, such as red blood cells, platelets, and other cells. These unused blood components are suspended in saline solution and immediately re-injected back into the donor while the plasma collection process is taking place. Because the donor is only providing plasma and not whole blood, the recovery process is faster and better tolerated, and the donor is therefore able to make donations more frequently. Plasmapheresis was developed by Jose Antonio Grifols Lucas in the year 1951. It is the only procedure that is capable of obtaining sufficient quantities of plasma to cover the needs of manufacturing our many different plasma protein therapies.
“Plasma derivatives” are proteins found in human plasma, which once isolated and purified, have therapeutic value.
“PTC” means plasma thromboplastin component.
“Prolastin” is a concentrated form of alpha1-antitrypsin, or AAT, produced by Grifols and derived from human plasma and approved only for chronic, or ongoing, replacement therapy in people with emphysema caused by genetic AAT deficiency. Given as prescribed, Prolastin raises the levels of AAT in the blood and lungs. Raising the AAT level may help reduce the damage to the lungs caused by destructive enzymes.
“Promonitor” Highly specific ELISA kits for quantification of serum drug levels and anti-drug antibodies of various biological drugs
“Q-Coagulometer, Q-Smart Q-Next and Q-Expert analyzers” Fully automated hemostasis analyzers that use reagents to measure blood coagulation levels.
“RFID” means Radio-Frequency Identification.
“RNA” means ribonucleic acid.
184
“sCAP” means severe community acquired pneumonia.
“SCIG” means subcutaneous immune globulin, which is a blood product administered subcutaneously. It contains the pooled IgG (immunoglobulin (antibody) G) extracted from plasma and is mainly used as treatment in four major categories: (i) immune deficiencies, (ii) inflammatory and autoimmune diseases, (iii) neurological diseases and (iv) acute infections.
“SME” means small and medium-sized enterprises.
“SYK-inhibitor” a new group of small molecule inhibitors which have been proposed as a therapy for both lymphoma and chronic lymphocytic leukemia.
“TMA” transcription mediated amplification, a technology that allows a clinical laboratory to perform assays for blood screening with fewer steps, less processing time, and faster results. It is used in molecular biology, forensics, and medicine for the rapid identification and diagnosis of pathogenic organisms.
“Triturus analyzers” Open and fully automated analyzer for ELISA (enzyme-linked immunoabsorbent assay), tests with multi-test/multi-batch capability.
“Von Willebrand Disease” is the most common hereditary coagulation abnormality described in humans, although it can also be acquired as a result of other medical conditions. It arises from a qualitative or quantitative deficiency of von Willebrand factor, a multimeric protein that is required for platelet adhesion.
“WADiana/Erytra analyzers” Automated immunohematology analyzers that use gel agglutination technology to enable automatic processing of DG Gel® blood determination cards.
“WHO” refers to the World Health Organization.
185
PART II
Item 13.DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
None.
Item 14.MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS
None.
Item 15.CONTROLS AND PROCEDURES
A. | Evaluation of Disclosure Controls and Procedures |
Our Chief Executive Officers and our Chief Financial Officer, after evaluating the effectiveness of our disclosure controls and procedures (as defined in Rule 13a-15(e) under the Exchange Act ) as of the end of the period covered by this annual report on Form 20-F, have concluded that, as of such date, our disclosure controls and procedures were not effective due to the material weaknesses described below.
B. | Management’s Report on Internal Control over Financial Reporting |
Our management, under the supervision of our Chief Executive Officer and our Chief Financial Officer, is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control system is designed to provide reasonable assurance as to the reliability of financial reporting and the preparation of the published financial statements under generally accepted accounting principles. For Grifols, S.A., “generally accepted accounting principles” means IFRS as issued by IASB.
Our internal control over the financial reporting system includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of our company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of our company are being made only in accordance with authorizations of management and directors of our company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our company assets that could have a material effect on the financial statements.
Any internal control system, no matter how well designed, has inherent limitations, including the possibility of human error and the circumvention or overriding of the controls and procedures, which may not prevent or detect misstatements.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2023. In making this assessment, they used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control—Integrated Framework. Based on their assessment under these criteria, our management believes that, as of December 31, 2023, our internal control over financial reporting was not effective because of the material weaknesses described below.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of the registrant’s annual or interim financial statements will not be prevented or detected on a timely basis.
186
Management has identified a material weakness related to information technology general controls (“ITGCs”), specifically the identification of risks, ineffective design and implementation and insufficient training of individuals to operate controls in the areas of user access and program change-management over certain information technology (IT) systems that support the Company’s financial reporting processes. As a result of these deficiencies, the related process-level IT dependent manual and automated application controls were deemed ineffective because they could not be relied upon.
Management has also identified a material weakness related to the identification of risks related to certain routine or recurring manual journal entries, resulting in a lack of controls over the review and approval of those journal entries.
The Company’s independent registered public accounting firm, KPMG Auditores, S.L. has issued an adverse audit report on the effectiveness of the Company’s internal control over financial reporting as of December 31, 2023, which appears in Item15.C of this annual report on Form 20-F. KPMG Auditores, S.L. has issued an unqualified opinion on our financial statements, which is included in page F-3 of this annual report on Form 20-F.
The material weaknesses did not result in any misstatements to the financial statements, and there were no changes to previously reported financial results. However, a reasonable possibility exists that material misstatements to the consolidated financial statements will not be prevented or detected on a timely basis.
C.Attestation Report of the Registered Public Accounting Firm
KPMG Auditores, S.L., an independent registered public accounting firm, who also audits the Group’s consolidated financial statements for 2023, has audited the effectiveness of Grifols S. A.’s internal control over financial reporting, and has issued an adverse audit report thereon, which is included on page F-6 of this annual report on Form 20-F.
D.Changes in Internal Control over Financial Reporting
Except for the material weaknesses in ITGCs and manual recurring journal entries described above, there were no changes in our internal control over financial reporting that occurred during the period covered by this annual report that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting. After December 31, 2023, we began the remediation efforts described below.
Remediation Plan
Management is committed to the remediation of the material weaknesses described above, as well as the continued improvement of our internal control over financial reporting. In order to remediate the material weaknesses, our management, supported by IT governance external specialists and under the oversight of the Audit Committee, has initiated measures intended to remediate the material weaknesses and strengthen our internal controls over financial reporting including, but not limited to: (1) developing and deploying a training program regarding the operation and importance of ITGCs and policies, including educating control owners on the principles and requirements of each control, with a focus on controls involving user access to IT systems and change management of IT systems that support financial reporting processes; (2) developing and maintaining documentation of ITGCs to facilitate knowledge transfer in the event of personnel and function changes; (3) developing enhanced risk assessment procedures and controls related to changes in IT systems to improve the identification of financially relevant applications and the selection, development, and monitoring of control activities and procedures; (4) redefining roles and responsibilities to improve the operation, assurance, and oversight of ITGCs, enhancing communication and coordination with business areas, and creating a dynamic, responsive environment; (5) enhancing quarterly reporting on the remediation measures to the Audit Committee of the Board of Directors; and (6) developing and deploying an automated approval workflow for park and post management review step for manual journal entries that will strengthen controls over the aforementioned procedures.
While we are taking steps to implement our remediation plan, the material weaknesses will not be considered remediated until the enhanced controls operate for a sufficient period of time and management has concluded, through testing, that the related controls are operating effectively. We will monitor the effectiveness of the remediation plan and refine the remediation efforts as appropriate. As we continue to validate and test our internal control over financial reporting, we may determine that additional measures or modifications to the remediation plan are necessary or appropriate.
187
Item 16.[RESERVED]
Item 16.AAUDIT COMMITTEE FINANCIAL EXPERT
The Board has determined that Mr. Íñigo Sánchez-Asiaín Mardones is an “audit committee financial expert,” as defined in Item 16A of Form 20-F, and is an independent director under Rule 10A-3 under the Exchange Act.
Item 16.BCODE OF ETHICS
We have adopted the Employee Code of Conduct, which applies to all of our employees, directors and officers, including our principal executive officer, principal financial officer and principal accounting officer. This Code is intended to meet the definition of “code of ethics” under Item 16B of Form 20-F.
If the Code of Conduct for Grifols’ Employees is amended, or if a waiver is granted, we will disclose such amendment or waiver on our website.
Item 16.CPRINCIPAL ACCOUNTANT FEES AND SERVICES
The table below sets forth the total fees paid to KPMG Auditores, S.L., our principal accountants, for services performed in the years 2023 and 2022:
| 2023 |
| 2022 | |
(in thousands of euros) | ||||
Audit services | 1,832 |
| 1,778 | |
Other assurance services(1) | 571 |
| 560 | |
Total | 2,403 |
| 2,338 | |
| (1) | These are fees paid for assurance services or other work traditionally provided to us by external audit firms in their role as statutory auditors. Other assurance services include limited reviews of the interim financial statements and the audit of the financial statements under applicable accounting standards. |
The table below sets forth the total fees paid to other member firms of the KPMG international organization, for services performed in the years 2023 and 2022, and breaks down these amounts by category of service:
| 2023 |
| 2022 | |
(in thousands of euros) | ||||
Audit services |
| 3,779 |
| 4,115 |
Other assurance services(1) |
| 1,380 |
| 1,013 |
Tax advisory services |
| 4 |
| 3 |
Other fees(2) |
| 127 |
| 206 |
Total |
| 5,290 |
| 5,337 |
| (1) | These are fees paid for assurance services or other work traditionally provided to us by external audit firms in their role as statutory auditors. Other assurance services include limited reviews of the interim financial statements and the audit of the financial statements under applicable accounting standards. |
| (2) | All other fees primarily relate to the review of non-financial information. |
188
The table below sets forth the total fees paid to other auditors for services performed in the years 2023 and 2022, and breaks down these amounts by category of service:
| 2023 |
| 2022 | |
(in thousands of euros) | ||||
Audit fees | 229 |
| 84 | |
Audit-related fees | — |
| — | |
Tax fees | — |
| — | |
All other fees | — |
| — | |
Total | 229 |
| 84 | |
Pre-approval Policies and Procedures
Subject to shareholder approval of the independent auditor in accordance with Spanish law, the Audit Committee makes recommendations to the Board regarding the appointment, retainer and replacement of the independent auditor. The Audit Committee is also directly responsible for the compensation and oversight of the work of the independent auditor. We have developed a policy regarding the engagement of professional services by our external auditor, in accordance with the Spanish Audit Law and the Sarbanes-Oxley Act of 2002. This policy generally provides that we will not engage our independent auditors to render audit or non-audit services unless the service is specifically approved in advance by the Audit Committee.
In accordance with the pre-approval policy, all audit and permitted non-audit services performed for us by our principal accountants, or any of its affiliates, were approved by the Audit Committee, which concluded that the provision of such services by the independent accountants was compatible with the maintenance of that firm’s independence in the conduct of its auditing functions.
Item 16.DEXEMPTION FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES
Not applicable.
Item 16.EPURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS
We did not repurchase any of our equity securities during the year ended December 31, 2023.
As of December 31, 2023, we held 3,944,430 Class A shares and 4,518,199 Class B shares in treasury. During 2023, we delivered 681,585 Class B treasury shares to eligible employees as compensation for the Restricted Share Unit Retention Plan, of which 50,784 were given to Grifols, S.A. employees. See Note 29 to our audited consolidated financial statements included in this annual report on page F-104.
The Buy-back Program
On its meeting held on March 11, 2021, our Board of Directors resolved to implement a buy-back program of Grifols’ own shares (the “Buy-back Program”), in accordance with the authorization granted by Grifols’ ordinary general shareholders’ meeting held on October 9, 2020, under item 12 of its agenda.
The Buy-back Program was carried out pursuant to the provisions of the Regulation (EU) No. 596/2014 of the European Parliament and of the Council of April 16, 2014, on market abuse regulation (the “MAR”) and Commission Delegated Regulation (EU) 2016/1052 of March 8, 2016, supplementing Regulation (EU) No 596/2014 of the European Parliament and of the Council (the “Delegated Regulation”) with regard to regulatory technical standards for the conditions applicable to buy-back programs and stabilization measures (with the exception of article 2 para. 1(a) of the Delegated Regulation) (the Delegated Regulation, together with the MAR, the “Buy-back Programme Rules”).
The maximum number of shares allowed to be acquired under the Buy-back Program was 6,875,549, in the aggregate. Specifically, 4,261,298 Class A shares and 2,614,251 Class B shares, representing approximately 1% of our share capital were allowed to be bought back. The maximum net investment allowed was €125 million. The purchase of Class A and Class B shares was to be made on a pro-rata basis, in accordance with the Articles of Association.
189
Grifols entrusted the execution of the Buy-back Program to an independent bank. This bank made its decisions regarding the number of shares, share price and time at which any share purchase was carried out without any influence of Grifols, in accordance with the Buy-back Program Rules. We did not exercise control over the bank’s decisions in this respect.
The Buy-back Program started on March 12, 2021, and remained in force until June 14, 2021, and the total of 3,944,430 Class A Shares and 2,419,896 Class B Shares were bought back.
Item 16.FCHANGES IN REGISTRANT’S CERTIFYING ACCOUNTANT
The term of office of KPMG Auditores, S.L., statutory auditor of the Company for the fiscal year ended December 31, 2023, cannot be extended because it reached the maximum legal duration. During our annual shareholders’ meeting held on June 16, 2023, we appointed Deloitte, S.L. as statutory auditor of the Company’s consolidated annual accounts for a term of three years, starting on January 1, 2024. Such appointment will therefore comprise the audit of the annual accounts for the fiscal years ending on December 31, 2024, 2025 and 2026.
The selection procedure of the auditors to be appointed by the general shareholders’ meeting in 2023 was overseen by the Audit Committee, following which a recommendation to the Board of Directors was issued. The Board of Directors approved our Audit Committee’s recommendation and decided to propose the appointment of Deloitte, S.L. as statutory auditor. Consequently, the Board of Directors proposed to the general shareholders’ meeting held on June 16, 2023 to appoint Deloitte, S.L. as new statutory auditor for a three-year term.
The report of KPMG Auditores, S.L. on the consolidated financial statements for each of the years ended December 31, 2023 and 2022 did not contain an adverse opinion or a disclaimer of opinion and was not qualified or modified as to uncertainty, audit scope or accounting principles other than the material weaknesses in our internal control over financial reporting disclosed in Item 15 of this annual report due to ineffective information technology general controls in the areas of user access and program change-management over certain information technology systems that support our financial reporting processes. In addition, during each of the years ended December 31, 2023 and 2022:
| ● | there were no “disagreements” (as that term is described in Item 16.F (a)(1)(iv) of the Instructions to Form 20-F and the instructions to Item 16.F) between Grifols and KPMG Auditores, S.L. on any matters of accounting principles or practices, financial statement disclosure, or auditing scope or procedures, which disagreement(s), if not resolved to KPMG Auditores, S.L.’ satisfaction, would have caused KPMG Auditores, S.L. to make reference to the subject matter of the disagreement(s) in connection with its report; and |
| ● | there were no “reportable events” (as that term is defined in Item 16.F (a)(1)(v) of the Instructions to Form 20-F). |
We have provided KPMG Auditores, S.L. with a copy of the foregoing disclosure and have requested that they furnish us with a letter addressed to the SEC stating whether they agree with such disclosure and, if not, stating the respects in which they do not agree. A copy of KPMG Auditores S.L.’s letter, dated April 19, 2024, in which KPMG Auditores, S.L. states that they agree with such disclosure, is filed herewith as Exhibit 15.1.
Item 16.GCORPORATE GOVERNANCE
Pursuant to NASDAQ Listing Rules, as a foreign private issuer, we may elect to follow our home country practice in lieu of the corporate governance requirements of the NASDAQ Listing Rule 5600 Series, with the exception of those rules that are required to be followed pursuant to the provisions of NASDAQ Listing Rule 5615(a)(3). We have elected to follow Spanish practices in lieu of the requirements of the NASDAQ Listing Rule 5600 Series to the extent permitted under NASDAQ Listing Rule 5615(a)(3). Set forth below is a summary of the significant differences between the corporate governance practices we follow under Spanish law (as in effect as of December 31, 2023) and those followed by NASDAQ-listed U.S. domestic issuers.
190
Corporate Governance
Under NASDAQ Listing Rules, a U.S. domestic issuer is required to establish a quorum as specified in its bylaws for any meeting of the holders of common stock, provided, however, that such quorum is not permitted to be less than 33% of the outstanding shares of voting stock. The Articles of Association provide that, on the first call of our general shareholders’ meetings, a duly constituted meeting requires a quorum of at least 25% of our subscribed share capital with voting rights, and, if a quorum is not obtained on the first call, a meeting is validly convened on the second call regardless of the share capital in attendance. However, certain major corporate actions (such as issuing additional ordinary shares, increasing or decreasing our share capital, issuing debt securities, amending the Articles of Association or approving merger transactions) require shareholder approval at a meeting at which at least 50% of our subscribed share capital with voting rights is present or represented on the first call or at least 25% of the share capital with voting rights present or represented on second call. However, when the number of shareholders attending our meeting represents less than 50% of our subscribed share capital with voting rights, resolutions on any of these major corporate actions must be adopted by the affirmative vote of at least two-thirds of the share capital present or represented at such meeting.
In addition, all actions described in Article 6.bis of the Articles of Association, which are considered to affect the economic rights of our Class B shares, must be approved at a shareholders’ meeting by the holders of at least a majority of Class B shares.
Under NASDAQ Listing Rules, U.S. domestic issuers are required to solicit proxies, provide proxy statements for all shareholders’ meetings and provide copies of such proxy materials to NASDAQ. As a foreign private issuer, we are generally exempt from the SEC rules governing the solicitation of shareholder proxies. However, under Spanish law and per the Articles of Association, we are required to publish a calling of the meeting at least one month prior to the date set for each general shareholders’ meeting in at least: (i) the Official Gazette of the Commercial Registry or one of the local newspapers of wide circulation in the province where we are domiciled (currently Barcelona, Spain); (ii) CNMV’s website; and (iii) our website. We distribute a copy of the notice of the meeting and a form of proxy to our U.S. shareholders and also make these materials available through our website in advance of such meeting.
Under NASDAQ Listing Rules, shareholders of U.S. domestic issuers must be given the opportunity to vote on equity compensation plans and material revisions thereto, with limited exceptions set forth in NASDAQ Listing Rules, including an exception for foreign private issuers who follow the laws of their home country. Under Spanish law, equity compensation plans involving the issuance of our securities require prior shareholder approval. Additionally, equity compensation plans in which our officers and employees participate can be approved by the Board without shareholder approval. However, the establishment of equity compensation plans in which members of the Board participate must be authorized in the Articles of Association and requires the shareholders’ prior approval at a shareholders’ meeting.
Under NASDAQ Listing Rules, shareholders of U.S. domestic issuers must approve the issuance of securities when such issuance would result in a change in control of such issuer. Under Spanish law, any issuance of our securities, regardless of whether such issuance would result in a change of control, requires prior shareholder approval.
In Spain, companies with securities listed on a Spanish Stock Exchange are:
(i)recommended to follow the provisions of the CNMV Governance Code;
(ii)required by law to publish an Annual Report on Corporate Governance as well as corporate governance information on their websites;
(iii)required by law to publish an Annual Report on Remuneration of the members of the Board; and
(iv)required by law to comply with the regulations with respect to audit committees and appointment and remuneration committees set forth in the Spanish Companies Act, as amended.
191
Board Practices
Independence of Directors
Pursuant to NASDAQ Listing Rules, a majority of the directors of a listed U.S. company are required to be “independent,” as such term is defined by NASDAQ Listing Rules. As a foreign private issuer, we are exempt from such requirement, and Spanish law does not contain any such requirements.
Spanish law establishes the category of directors and the indispensable requirements to determine their independence. The Board Regulations, consistent with Spanish law, recognize two main categories of directors: (i) executive directors; and (ii) external directors, who can be divided into (a) proprietary directors, (b) independent directors and (c) other directors who cannot be considered proprietary or independent.
The definition of “independent director,” as set forth by Spanish law, provides that the persons listed below may not be nominated or designated as independent directors.
(i)Employees or executive directors of any Group companies, unless three or five years have elapsed, respectively, since the termination of the relationship.
(ii)Persons that have received some payment from us or from the Group in addition to their directors’ remuneration, unless the amount involved is not significant to the director. Dividends or pension supplements received by a director for prior employment or professional services are excluded, provided that such payments are non-contingent (i.e., the paying company has no discretionary power to suspend, modify or revoke the payment).
(iii)Persons that have been, during the last three years, partners of the external auditors or the firm responsible for the audit report, whether with respect to the audit of us or any other company in the Group for those years.
(iv)Executive directors or senior officers of other companies in which any of our executive directors or senior officers is an external director.
(v)Persons that have or had, during the last year, material business relationships with us or with any other company in the Group, whether in their own name or as a significant shareholder, director or senior officer of a company that has or had such a relationship. For purposes of this paragraph (v), “business relationships” means any relationship with suppliers of goods or services, including financial, advisory and consultancy services.
(vi)Significant shareholders, executive directors or senior officers of an entity which receives or has received, during the last three years, significant donations from us or the Group. This provision does not apply to those who are merely trustees of a foundation receiving donations.
(vii)Spouses or related persons maintaining an analogous relationship or close relatives of one of our executive directors or senior officers.
(viii)Any person not proposed for appointment or renewal by the Appointments and Remuneration Committee.
(ix)Persons in any of the situations set out in (i), (v), (vi) or (vii) above with regard to a significant shareholder or a shareholder with Board representation. In the case of the family relations set out in (vii) above, the limitation applies not only in connection with the shareholder but also with our proprietary directors.
(x)Persons that have been directors for 12 consecutive years.
The proprietary directors who lose this status as a consequence of the sale of the shareholding by the shareholder they represent, can be reelected as independent directors only when such shareholder has sold the total amount of its shares.
192
Finally, any member of the Board that owns our shares can be considered independent, as long as the shareholding is not significant and satisfies all the above-mentioned conditions.
We have not determined whether our directors would be considered independent under NASDAQ Listing Rules, except for the three directors who are members of the Audit Committee and as such must meet NASDAQ independence criteria. As of the date of this report, six members of the Board are independent directors in accordance with the Board Regulations and the CNMV Governance Code.
Furthermore, we follow the Spanish Companies Act, which does not, unlike NASDAQ Listing Rules, require independent directors to hold meetings where only such independent directors are present.
For a detailed discussion of the composition, responsibilities and terms of our Audit Committee, see Item 6 of Part I, “Directors, Senior Management and Employees—C. Board Practices—Committees of the Board—Audit Committee.”
Audit Committee
Responsibilities and Terms. In accordance with NASDAQ Listing Rules, our Audit Committee is in charge of the appointment, compensation, retention and oversight of the services of any registered public accounting firm engaged for the purpose of preparing and issuing any audit report, or for performing other audit reviews or related services. Notwithstanding the above, Spanish laws provide our shareholders with the authority to appoint and replace the independent auditor at a general shareholders’ meeting.
Independence of the Audit Committee. All of the members of our Audit Committee meet the independence criteria set out in NASDAQ Listing Rules. Subsequent to the entry into force of Law 31/2014 and Law 22/2015, Spanish law requires that (a) the Audit Committee be composed of external directors (the majority of them being independent and one of them being appointed due to his knowledge and experience in accounting or auditing matters) and (b) the chairperson of the Audit Committee is an independent director. For a further discussion regarding the composition of our Audit Committee, see Item 6 of Part I, “Directors, Senior Management and Employees—C. Board Practices—Committees of the Board—Audit Committee.”
Internal Audit Department. We have an internal audit department responsible for internal audit matters and ensuring the efficiency of the internal audit control process of our different business units. Our internal audit department reports directly to the Audit Committee, supporting the adequate performance of all its functions.
Appointments and Remuneration Committee
Pursuant to NASDAQ Listing Rules, foreign private issuers are exempt from the requirements regarding independent nominating and compensation committees. Foreign private issuers are permitted to follow their home country corporate governance practice in this respect.
Spanish law requires that all Spanish listed companies have an appointments and remuneration committee comprised of external directors, at least two of whom must be independent, and that the chairperson of the appointments and remuneration committee be an independent director.
Our Appointments and Remuneration Committee is comprised exclusively of external directors and is chaired by an independent director. For a detailed discussion of our Appointments and Remuneration Committee, see Item 6 of Part I, “Directors, Senior Management and Employees—C. Board Practices—Committees of the Board—Appointments and Remuneration Committee.”
Internal Code of Conduct on Matters Related to the Securities Market and Business Ethics
Under NASDAQ Listing Rules, we are required to adopt a code of business conduct and ethics applicable to all directors, officers and employees, which must be publicly available. Under Spanish law, listed companies were previously required to have an internal code of conduct on matters related to the securities markets. However, with the entry into force of Royal Legislative Decree 19/2018, of November 23, 2018, on payment services and other urgent financial measures, this obligation has been removed.
193
Notwithstanding the above, Grifols will continue to apply the internal code of conduct for securities markets that was approved by the Board in its meeting held on October 28, 2016, in order to prevent insider trading, misconduct, and to control possible conflicts of interest. The Internal Code of Conduct on Matters Related to the Securities Market is included in this annual report as Exhibit 11.1.
Additionally, the Board Regulations set out in detail the directors’ main obligations relating to conflicts of interest concerning business opportunities, use of Grifols’ assets, confidentiality and non-competition. Although not mandatory under Spanish laws, the Board of Directors also approved the Code of Conduct for Grifols Employees. Both the Both the Board Regulations and the Code of Conduct for Grifols Employees, which do not form part of this annual report on Form 20 F, are publicly available at www.grifols.com.
Item 16.HMINE SAFETY DISCLOSURE
Not applicable.
Item 16.IDISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
Item 16.JINSIDER TRADING POLICIES
We have policies and procedures in place governing the purchase, sale and other dispositions of our securities by directors, senior management and employees. Our insider trading policy and procedures is included in our Internal Code of Conduct in Matters Relating to The Securities Market that is attached to this annual report as Exhibit 11.1.
Item 16.KCYBERSECURITY
Risk Management and Strategy
Cybersecurity risk management is an integral part of our overall enterprise risk management program. Our cybersecurity risk management program provides a framework for handling cybersecurity threats and incidents, including threats and incidents associated with the use of services provided by third-party service providers, and is designed to facilitate coordination across different departments of the Grifols Group in the handling of such cybersecurity threats and incidents. This framework includes steps for assessing the severity of a cybersecurity threat, identifying the source of a cybersecurity threat, including whether the cybersecurity threat is associated with a third-party service provider, implementing cybersecurity countermeasures and mitigation strategies and, as later explained in greater detail, informing management and our Board of Directors of material cybersecurity threats and incidents. Our cybersecurity risk management program is regularly updated to align with industry best practices established by internationally accepted security standards and its effectiveness in mitigating the risks that the we are exposed to is periodically assessed.
As cyberattacks evolve and become more sophisticated, we have strengthened our prevention and monitorization efforts. During the past few years, we ramped up cybersecurity and information security measures with the aim to ensure an adequate protection of our information and the assets supporting business processes. In September 2023, we appointed Miguel Louzan as our first Chief Digital Information Officer (“CDIO”). In this new role for our business, our CDIO is tasked with accelerating our companies’ use of digital platforms, data science and new technologies to transform and strengthen critical business activities such as relationships with plasma donors and customers as well as manufacturing operations, the development of new therapeutics and cybersecurity. Accordingly, our CDIO focuses on greater application of artificial intelligence, point-of-contact technologies and other automation tools that will help protect us against cybersecurity risks effectively and efficiently.
On November 16, 2023, we adopted our Cybersecurity Policy, which sets forth the principles of our cybersecurity strategy:
| ● | Maintain robust, updated, and resilient systems for processing of personal information, supported by encryption, anonymization, and other relevant measures; |
194
| ● | Define a systematic approach for the continuous identification and assessment of cybersecurity risks, including third party risks, as well as for the response to any cybersecurity incident; |
| ● | Implement security measures to protect the confidentiality, integrity and availability of information systems and associated processes (including information systems managed by third parties), and continuously monitor their effectiveness to ensure ongoing improvement; |
| ● | Implement procedures and invest in tools to facilitate agile adaptation to changing conditions in the technological environment; |
| ● | Ensure that effective response and recovery programs are in place, encompassing people, processes, information systems and technology to: detect, assess, respond within a reasonable time frame to, remedy and, if necessary pursuant to applicable legislation, disclose to investors actual or potential cybersecurity incidents and threats; effectively recover from cybersecurity incidents; escalate cybersecurity incidents to management and the cybersecurity team; and notify authorities of any incidents as required by applicable laws and regulations; |
| ● | Maintain a highly qualified cybersecurity team, comprised of management, information technology and legal personnel, by defining adequate hiring criteria and establishing rigorous training plans; |
| ● | Ensure training is provided to employees, executives and directors regarding cybersecurity risks, and protection of sensitive and personal data. Training shall include protecting against phishing attacks, guidance on the use of email, internet, and social media to ensure sensitive information is appropriately handled and protected and the escalation process for employees to follow in the event of an identified cybersecurity incident or threat; and |
| ● | Collaborate with peer companies, industry associations and government agencies to share best practices and effective solutions against cybersecurity threats. |
We have adopted security measures in the past few years intended to: (i) ensure end-to-end protection of business processes, considering logical and physical security, privacy and fraud management concerns, (ii) ensure compliance with the security and privacy by design principles; and (iii) improve client access control and authentication services related to online services, from a security and user experience perspective, including by enhancing the use of facial biometrics, behavioral biometrics, advanced analytics models and the implementation of dynamic card verification values.
Further, system monitoring capabilities, as well as incident prevention, detection and response capabilities have also been strengthened through the use of integrated information sources, improved analytical capabilities and automated platforms, improving information security management from a preventive and proactive approach. We routinely review, reinforce and tests our security processes and procedures through simulation exercises in the areas of physical security and digital security. Specialized teams periodically perform security technical tests in order to detect and correct possible security vulnerabilities. These tests include technical tests of technological platforms as well as malicious users’ simulated attacks. The outcome of such exercises is a fundamental part of a feedback process designed to improve the our cybersecurity strategies. We are further evolving our cybersecurity tools and platforms to be able to respond to new threats generated by artificial intelligence and other new technologies. We are also expanding our active cybersecurity monitoring to additional industrials and IOT areas.
In addition, we continuously carry out training and awareness initiatives related to security and privacy, promoting training and awareness campaigns for our employees, clients and society. Some of the topics covered include protection of personal information, secure password management, device protection (laptops, smartphones, etc.), social engineering (phishing, smishing, vishing), malware and other technical attacks detection, detection of scams, security on online purchases and how to react if there is a security incident.
195
In 2023, we did not identify any cybersecurity threats that materially affected or are reasonably likely to materially affect our business strategy, results of operations or financial condition. However, despite our efforts, we cannot eliminate all risks from cybersecurity threats, or provide assurances that we have not experienced an undetected cybersecurity incident. For more information about these risks, please see Item 3 of Part I, “D. Risk Factors—Risks Relating to the Company and Our Business—Cyber-attacks or other privacy and data security incidents could disrupt our business and expose us to significant losses, liability and reputational damage.”
Governance
Our Board of Directors, through the Audit Committee, is responsible for supervising and evaluating the efficiency of the control and management on cybersecurity. Our Internal Audit and Enterprise Risk Management Department supports the Audit Committee in the fulfilment of this responsibility, which includes oversight of our threat landscape, posture, performance, and strategy related to cybersecurity. The Audit Committee is also charged with overseeing the cybersecurity incident trends and potentially significant incidents that have been handled.
To support the deployment of the principles of our cybersecurity strategy and processes, in addition to appointing our CDIO, we have created and implemented the Information Security Process and Management System (the “ISMS”). The ISMS is based on the appropriate definition of objectives, roles and responsibilities, policies and procedures, and technology to: (i) identify cybersecurity threats and related risks; (ii) protect critical assets; (iii) detect and respond to cybersecurity threats and cybersecurity incidents; and (iv) recover business services due to a cybersecurity incident.
Our Head of the Information Security Office (“ISEC”) reports to the CDIO and has the authority to develop and implement the our cybersecurity policies, standards, procedures, and oversee the implementation and effectiveness of the ISMS. To that end, we are in. the process of selecting and appointing the members for the new Global Cybersecurity Committee. This committee will facilitate the alignment of cybersecurity initiatives with business objectives; ensure global coverage of the ISMS; collaborate in the prioritization and execution of security initiatives and projects; and promote a culture of protection against cybersecurity threats throughout the Grifols Group entities. This committee will be comprised of representatives of business units, information technology and legal personnel, as well as operations and services areas.
The Head of Internal Audit shall update the Audit Committee, at least twice per year, regarding the control and management on cybersecurity. For these updates the Audit Committee may require the assistance of the CDIO and/or the Head of the ISEC.
196
PART III
Item 17.FINANCIAL STATEMENTS
We have elected to provide financial statements pursuant to Item 18 of this Part III.
Item 18.FINANCIAL STATEMENTS
The audited consolidated financial statements as required under Item 18 of this Part III are attached hereto starting on page F-1 of this annual report on Form 20-F. The audit report of KPMG, our independent registered public accounting firm, is included herein preceding the audited consolidated financial statements.
Item 19.EXHIBITS
Exhibit |
| Description |
|---|---|---|
1.1 | Articles of Association (Estatutos) of Grifols, S.A. (English translation)* | |
2.1 | ||
2.2 | ||
2.3 | ||
2.4 | ||
2.5 | ||
2.6 | ||
2.7 | ||
197
Exhibit |
| Description |
|---|---|---|
2.8 | ||
2.9 | ||
2.10 | ||
2.11 | ||
2.12 | ||
2.13 | ||
2.14 | ||
2.14 | ||
2.15 | ||
198
Exhibit |
| Description |
|---|---|---|
2.16 | ||
2.17 | ||
4.1 | ||
4.2 | ||
4.3 | ||
4.4 | ||
4.5 | ||
4.6 | ||
4.7 | ||
8.1 | ||
10.1 | ||
11.1 | Internal Code of Conduct of Grifols, S.A. in Matters Relating to the Securities Market* | |
12.1 | Principal Executive Officer Certification pursuant to Section 302 of the Sarbanes-Oxley Act of 2002* | |
12.2 | Principal Financial Officer Certification pursuant to Section 302 of the Sarbanes-Oxley Act of 2002* | |
199
Exhibit |
| Description |
|---|---|---|
13.1 | ||
15.1 | ||
97.1 | ||
101.INS | Inline XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document | |
101.SCH | Inline XBRL Taxonomy Extension Schema | |
101.CAL | Inline XBRL Taxonomy Extension Schema Calculation Linkbase | |
101.DEF | Inline XBRL Taxonomy Extension Schema Definition Linkbase | |
101.LAB | Inline XBRL Taxonomy Extension Schema Label Linkbase | |
101.PRE | Inline XBRL Taxonomy Extension Schema Presentation Linkbase | |
104 | Cover Page Interactive Date File (formatted as Inline XBRL and contained in Exhibit 101) |
* | Filed herewith. |
200
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
GRIFOLS, S.A. | ||
By: | /s/ Thomas Glanzmann | |
Name: Thomas Glanzmann | ||
Title: Executive Chairperson of the Board of Directors | ||
GRIFOLS, S.A. | ||
Date: April 19, 2024. | ||
201
GRIFOLS, S.A. AND SUBSIDIARIES
Consolidated Financial Statements
31 December 2023 and 2022
SUMMARY
● | ||
Report of Independent Registered Public Accounting Firm ( | F - 3 | |
F - 8 | ||
F - 10 | ||
F - 11 | ||
F - 12 | ||
F - 13 | ||
● | ||
F - 14 | ||
F - 14 | ||
F - 21 | ||
F - 33 | ||
F - 49 | ||
F - 51 | ||
F - 56 | ||
F - 59 | ||
F - 60 | ||
F - 62 | ||
F - 69 | ||
F - 70 | ||
F - 71 | ||
F - 72 | ||
F - 72 | ||
F - 73 | ||
F - 74 | ||
F - 77 | ||
F - 79 | ||
F - 83 | ||
F - 88 | ||
F - 94 | ||
F - 95 | ||
F - 95 | ||
F - 97 | ||
F - 98 | ||
F - 99 | ||
F - 100 | ||
F - 104 | ||
F - 110 | ||
F - 121 | ||
F - 125 |
F-1
GRIFOLS, S.A. AND SUBSIDIARIES
Consolidated Financial Statements
31 December 2023 and 2022
SUMMARY
● | ||||
| F - 127 | |||
F - 148 | ||||
F - 150 | ||||
F - 152 | ||||
F - 154 | ||||
F-2
Report of Independent Registered Public Accounting Firm
To the Shareholders and Board of Directors of Grifols, S.A.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of Grifols, S.A. and subsidiaries (the Company) as of 31 December 2023 and 2022, the related consolidated statements of profit and loss, comprehensive income, changes in consolidated equity, and cash flows for each of the years in the three-year period ended 31 December 2023, and the related notes and Appendix I to V (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of 31 December 2023 and 2022, and the results of its operations and its cash flows for each of the years in the three year period ended 31 December 2023, in conformity with International Financial Reporting Standard as issued by the International Accounting Standard Board and International Financial Reporting Standards as adopted by the European Union.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of 31 December 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated 19 April 2024 expressed an adverse opinion on the effectiveness of the Company’s internal control over financial reporting.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Evaluation of the Diagnostic goodwill impairment analysis
As discussed in Notes 4 and 6 to the consolidated financial statements, the goodwill balance as of 31 December 2023 was Euros 6,802,127 thousand, of which Euros 2,679,357 thousand related to the Diagnostic cash generating unit (CGU). The Company calculates the recoverable amount of goodwill on an annual basis and whenever there is an indication that goodwill may be impaired.
F-3
We identified the evaluation of the goodwill impairment analysis for the Diagnostic CGU as a critical audit matter. Significant auditor judgment was required to evaluate the Company’s impairment test which was performed using a discounted cash flow model. The discounted cash flow model included assumptions related to sales projections for the Nucleic Acid Testing (NAT), the Blood Typing Solutions (BTS) and Clinical Diagnostics (CDx) lines of business, perpetual growth rate and the discount rate. Minor changes to these assumptions, could have a significant effect on the Company’s assessment of the carrying value of the goodwill.
The primary procedures we performed to address this critical audit matter included the following:
We evaluated the design and tested the operating effectiveness of certain internal controls related to the Company’s goodwill impairment assessment process, including controls related to the determination of the fair value less costs of disposals/recoverable amount of the Diagnostic CGU, and the development of the sales projections of the NAT, BTS and CDx lines of business, perpetual growth rate and discount rate assumptions.
We involved a valuation professional with specialized skills and knowledge, who assisted in:
‒ | Evaluating the Company’s perpetual growth rate for the Diagnostic CGU, by comparing it with publicly available market data for comparable entities. |
‒ | Evaluating the discount rate by comparing it against a discount rate range that was independently developed using publicly available market data for comparable entities. |
We performed sensitivity analyses over the significant assumptions to assess their impact on the recoverable amount of the Diagnostic CGU.
We challenged the sales projections for the NAT, BTS and CDx business lines, by examining publicly available data of past experiences of the evolution of similar technologies and industry reports.
We evaluated the Company’s ability to forecast sales projections for the NAT, BTS and CDx business lines by comparing the historical projections to actual results and the business plans approved by the Company’s governing bodies.
Classification of assets held for sale of a 20% stake in Shanghai RAAS
As discussed in notes 2, 4 and 12 to the consolidated financial statements, on 29 December 2023, the Company reached an agreement with Haier Group Corporation (“Haier”) for the sale of a 20% stake in the associate Shanghai RAAS (SRAAS) for RMB 12.5 billion (approximately USD 1,800 million), which includes certain future commitments with the buyer. The closing of the transaction is subject to certain normal conditions such as the relevant regulatory approvals. The Company has classified the related interest held in SRAAS of Euros 1,433,867 as non-current assets held for sale.
We identified the assessment of the classification of the interest held in SRAAS as non-current assets held for sale as a critical audit matter. Specifically, a high degree of auditor judgment and specialized skills were required to evaluate management’s assessment of the probability of achieving the relevant regulatory approvals necessary for the sale to be completed.
F-4
The following are the primary procedures we performed to address this critical audit matter:
‒ | we evaluated the design and tested the operating effectiveness of an internal control related to the classification of non-current assets held for sale; and |
‒ | we involved legal professionals with specialized skills and knowledge, who assisted in evaluating the probability of achieving the necessary regulatory approvals for the sale to be completed by assessing the legal confirmation received from the Company’s external legal counsel and performing further inquiries of the Company’s external legal counsel to evaluate their basis for conclusion. |
KPMG Auditores, S.L.
We have served as the Company’s auditor since 1990
Barcelona, Spain
19 April 2024
F-5
Report of Independent Registered Public Accounting Firm
To the Shareholders and Board of Directors of Grifols, S.A.
Opinion on Internal Control Over Financial Reporting
We have audited Grifols, S.A. and subsidiaries (the Company) internal control over financial reporting as of 31 December, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. In our opinion, because of the effect of the material weakness, described below, on the achievement of the objectives of the control criteria, the Company has not maintained effective internal control over financial reporting as of 31 December, 2023, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of 31 December, 2023 and 2022, the related consolidated statements of profit and loss, comprehensive income, changes in consolidated equity, and cash flows for each of the years in the three-year period ended 31 December, 2023 and the related notes and Appendix I to V (collectively, the consolidated financial statements), and our report dated 19 April, 2024 expressed an unqualified opinion on those consolidated financial statements.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the Company’s annual or interim financial statements will not be prevented or detected on a timely basis. Material weaknesses have been identified and included in management’s assessment related to (i) identification of risks and ineffective design and implementation and insufficient training of individuals to operate controls related to information technology general controls (ITGCs); and (ii) identification of risks related to the review and approval of certain manual recurring journal entries. These material weaknesses did not result in any identified misstatements in the financial statements. The material weaknesses were considered in determining the nature, timing, and extent of audit tests applied in our audit of the 31 December 2023 consolidated financial statements, and this report does not affect our report on those consolidated financial statements.
Basis for Opinions
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
F-6
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
KPMG Auditores, S.L.
Barcelona, Spain
19 April 2024
F-7
GRIFOLS, S.A. AND SUBSIDIARIES
Consolidated Balance Sheet
at 31 December 2023 and 2022
(Expressed in thousands of Euros)
Assets |
| Reference |
| 31/12/23 |
| 31/12/22 |
Goodwill | Note 6 | | | |||
Other intangible assets | Note 7 | | | |||
Rights of use | Note 8 | | | |||
Property, plant and equipment | Note 9 | | | |||
Investment in equity-accounted investees | Note 10 | | | |||
Non-current financial assets | ||||||
Non-current financial assets measured at fair value | | | ||||
Non-current financial assets at amortized cost | | | ||||
Total non-current financial assets | Note 11 | | | |||
Other non-current assets | Note 10 | | — | |||
Deferred tax assets | Note 28 | | | |||
Total non-current assets | | | ||||
Non-current assets held for sale | Note 12 | | | |||
Inventories | Note 13 | | | |||
Current contract assets | Note 14 | | | |||
Trade and other receivables | ||||||
Trade receivables | | | ||||
Other receivables | | | ||||
Current income tax assets | | | ||||
Trade and other receivables | Note 15 | | | |||
Other current financial assets | Note 11 | |||||
Current financial assets measured at fair value | | | ||||
Current financial assets at amortized cost | | | ||||
Total current financial assets | Note 11 | | | |||
Other current assets | | | ||||
Cash and cash equivalents | Note 16 | | | |||
Total current assets | | | ||||
Total assets | | |
The accompanying notes form an integral part of the consolidated financial statements.
F-8
GRIFOLS, S.A. AND SUBSIDIARIES
Consolidated Balance Sheet
at 31 December 2023 and 2022
(Expressed in thousands of Euros)
Equity and liabilities |
| Reference |
| 31/12/23 |
| 31/12/22 |
Share capital | | | ||||
Share premium | | | ||||
Reserves | | | ||||
Treasury stock | ( | ( | ||||
Profit for the year attributable to the Parent | | | ||||
Total equity | | | ||||
Cash Flow hedges | | ( | ||||
Other comprehensive Income | ( | ( | ||||
Other comprehensive income from non-current assets held for sale | | — | ||||
Translation differences | | | ||||
Other comprehensive expenses | | | ||||
Equity attributable to the Parent | Note 17 | | | |||
Non-controlling interests | Note 19 | | | |||
Total equity | | | ||||
Liabilities |
| |||||
Grants | | | ||||
Provisions | Note 20 | | | |||
Non-current financial liabilities | Note 21 | | | |||
Other non-current liabilities | — | | ||||
Deferred tax liabilities | Note 28 | | | |||
Total non-current liabilities | | | ||||
Provisions | Note 20 | | | |||
Current financial liabilities | Note 21 | | | |||
Trade and other payables |
| |||||
Suppliers | | | ||||
Other payables | | | ||||
Current income tax liabilities | | | ||||
Total trade and other payables | Note 22 | | | |||
Other current liabilities | Note 23 | | | |||
Total current liabilities | | | ||||
Total liabilities | | | ||||
Total equity and liabilities | | |
The accompanying notes form an integral part of the consolidated financial statements.
F-9
GRIFOLS, S.A. AND SUBSIDIARIES
Consolidated Statements of Profit and Loss
at 31 December 2023, 2022 and 2021
(Expressed in thousands of Euros)
| Reference |
| 31/12/23 |
| 31/12/22 |
| 31/12/21 | |
Continuing Operations |
|
|
|
|
|
| ||
Net revenue | Note 5 and 24 |
| |
| |
| | |
Cost of sales |
| ( |
| ( |
| ( | ||
Gross Margin |
| |
| |
| | ||
Research and development |
| ( |
| ( |
| ( | ||
Selling, general and administration expenses |
| ( |
| ( |
| ( | ||
Operating Expenses |
| ( |
| ( |
| ( | ||
Other Income | | |||||||
Profit of equity accounted investees with similar activity to that of the Group | Note 10 | | ||||||
Operating Result |
|
|
| | ||||
Finance income |
|
|
| | ||||
Finance costs |
| ( |
| ( |
| ( | ||
Sale of assets at amortized cost | Note 15 | ( | ( | ( | ||||
Change in fair value of financial instruments |
|
|
| | ||||
Exchange differences |
| ( |
| |
| ( | ||
Finance result | Note 27 |
| ( |
| ( |
| ( | |
Profit/(loss) of equity accounted investees | Note 10 |
| ( |
| ( |
| | |
Profit before income tax from continuing operations |
| |
| |
| | ||
Income tax expense | Note 28 |
| ( |
| ( |
| ( | |
Profit after income tax from continuing operations |
|
|
| | ||||
Consolidated profit for the year |
|
|
| | ||||
Profit attributable to the Parent |
|
|
| | ||||
Profit attributable to non-controlling interest | Note 19 |
|
|
| | |||
Basic earnings per share (Euros) | Note 18 |
|
|
| ||||
Diluted earnings per share (Euros) | Note 18 |
|
|
|
The accompanying notes form an integral part of the consolidated financial statements.
F-10
GRIFOLS, S.A. AND SUBSIDIARIES
Consolidated Statements of Comprehensive Income
for the years ended 31 December 2023, 2022 and 2021
(Expressed in thousands of Euros)
| Reference |
| 31/12/23 |
| 31/12/22 |
| 31/12/21 | |
Consolidated profit for the year | | | | |||||
Items that will not be reclassified to profit or loss | ||||||||
Items for reclassification to profit or loss | ||||||||
Translation differences | ( | | | |||||
Equity accounted investees / Translation differences | Note 10 | | | ( | ||||
Other comprehensive income from non-current assets held for sale | | | | |||||
Cash flow hedges - effective portion of changes in fair value | ( | | | |||||
Cash flow hedges - amounts taken to profit or loss | | ( | | |||||
Tax effect | ( | | ( | |||||
Other | ( | ( | | |||||
Other comprehensive income for the year, after tax | ( | | | |||||
Total comprehensive income for the year | ( | | | |||||
Total comprehensive income attributable to the Parent | ( | | | |||||
Total comprehensive income attributable to non-controlling interests | | | |
The accompanying notes form an integral part of the consolidated financial statements.
F-11
GRIFOLS, S.A. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
at 31 December 2023, 2022 and 2021
(Expressed in thousands of Euros)
| Reference |
| 31/12/23 |
| 31/12/22 |
| 31/12/21 | |
Cash flows from operating activities |
|
| ||||||
Profit before tax | | | | |||||
Adjustments for: | | | | |||||
Amortization and depreciation | Note 26 | | | | ||||
Other adjustments: | | | | |||||
(Profit) / losses on equity accounted investments | Note 10 | ( | ( | ( | ||||
Impairment of assets and net provision charges | | | | |||||
(Profit) / losses on disposal of fixed assets | Notes 7, 8 and 9 | | ( | | ||||
Government grants taken to income | ( | ( | ( | |||||
Finance cost / (income) | | | | |||||
Other adjustments | ( | ( | ( | |||||
Change in operating assets and liabilities | ( | ( | ( | |||||
Change in inventories | ( | ( | ( | |||||
Change in trade and other receivables | ( | ( | ( | |||||
Change in current financial assets and other current assets | | ( | ( | |||||
Change in current trade and other payables | | | | |||||
Other cash flows used in operating activities | ( | ( | ( | |||||
Interest paid | Note 21d | ( | ( | ( | ||||
Interest received | | | | |||||
Income tax paid | ( | ( | ( | |||||
Other paid | | ( | ( | |||||
Net cash from/used in operating activities | | ( | | |||||
Cash flows from investing activities | ||||||||
Payments for investments | ( | ( | ( | |||||
Group companies, associates and business units | Notes 3 and 10 | ( | ( | ( | ||||
Property, plant and equipment and intangible assets | ( | ( | ( | |||||
Property, plant and equipment | Note 7 | ( | ( | ( | ||||
Intangible assets | Note 9 | ( | ( | ( | ||||
Other financial assets | ( | ( | ( | |||||
Proceeds from the sale of investments | | | | |||||
Group companies, associates and business units | Notes 3 and 10 | | | | ||||
Property, plant and equipment | | | | |||||
Other financial assets | | | | |||||
Net cash used in investing activities | ( | ( | ( | |||||
Cash flows from financing activities | ||||||||
Proceeds from and payments for equity instruments | | ( | ( | |||||
Payments for treasury stock | | ( | ( | |||||
Proceeds from and payments for financial liability instruments | | ( | | |||||
Issue | | | | |||||
Redemption and repayment | ( | ( | ( | |||||
Lease payments | Note 8 and 21d | ( | ( | ( | ||||
Dividends and interest on other equity instruments | | | ( | |||||
Dividends paid | | ( | ( | |||||
Dividends received | Note 10 | | | | ||||
Other cash flows used in financing activities | | ( | ( | |||||
Financing costs included in the amortized cost of the debt | | | ( | |||||
Other amounts from / (used in) financing activities | | ( | | |||||
Net cash from/(used in) financing activities | | ( | | |||||
Effect of exchange rate fluctuations on cash | ( | | | |||||
Net increase / (decrease) in cash and cash equivalents | ( | ( | | |||||
Cash and cash equivalents at beginning of the year | | | | |||||
Cash and cash equivalents at year end | Note 16 | | | |
The accompanying notes form an integral part of the consolidated financial statements.
F-12
GRIFOLS, S.A. AND SUBSIDIARIES
Statement of Changes in Consolidated Equity
for the years ended 31 December 2023, 2022 and 2021
(Expressed in thousands of Euros)
Attributable to shareholders of the Parent | ||||||||||||||||||||||||||||
|
|
|
|
|
| Accumulated other comprehensive income | Equity |
|
| |||||||||||||||||||
Profit attributable | Other | Other comprehensive | attributable | |||||||||||||||||||||||||
Share | Share | to | Interim | Treasury | Translation | comprehensive | income from non-current | Cash flow | to | Non-controlling | ||||||||||||||||||
| Reference |
| Capital |
| Premium |
| Reserves |
| Parent |
| dividend |
| Stock |
| differences |
| income |
| assets held for sale |
| hedges |
| Parent |
| interests |
| Equity | |
Balance at 31 December 2020 | | | | | — | ( | ( | ( | — | — | — | | | |||||||||||||||
Translation differences | — | — | — | — | — | — | | — | — | — | | | | |||||||||||||||
Cash flow hedges | Note 30 | — | — | — | — | — | — | — | — | — | | | — | | ||||||||||||||
Other comprehensive income | — | — | — | — | — | — | — | | — | — | | — | | |||||||||||||||
Other comprehensive income / (expense) for the year | — | — | — | — | — | — | | | — | | | | | |||||||||||||||
Profit/(loss) for the year | — | — | — | | — | — | — | — | — | — | | | | |||||||||||||||
Total comprehensive income / (expense) for the year | — | — | — | | — | — | | | — | | | | | |||||||||||||||
Net change in treasury stock | Note 17 (d) | — | — | — | — | — | ( | — | — | — | — | ( | — | ( | ||||||||||||||
Acquisition / Divestment of non-controlling interests | Note 17 (c) | — | — | ( | — | — | — | — | — | — | — | ( | | ( | ||||||||||||||
Other changes | — | — | ( | — | — | — | — | — | — | — | ( | | ( | |||||||||||||||
Distribution of 2020 profit: | ||||||||||||||||||||||||||||
Reserves | — | — | | ( | — | — | — | — | — | — | — | — | — | |||||||||||||||
Dividends | — | — | ( | — | — | — | — | — | — | — | ( | ( | ( | |||||||||||||||
Interim dividend | — | — | — | - | — | — | — | — | — | — | — | — | — | |||||||||||||||
Operations with shareholders or owners | — | — | | ( | — | ( | — | — | — | — | ( | ( | ( | |||||||||||||||
Balance at 31 December 2021 | | | | | — | ( | | ( | — | | | | | |||||||||||||||
Translation differences | — | — | — | — | — | — | | — | — | — | | | | |||||||||||||||
Cash flow hedges | Note 30 | — | — | — | — | — | — | — | — | — | ( | ( | — | ( | ||||||||||||||
Other comprehensive income | — | — | — | — | — | — | — | ( | — | — | ( | — | ( | |||||||||||||||
Other comprehensive income / (expense) for the year | — | — | — | — | — | — | | ( | — | ( | | | | |||||||||||||||
Profit/(loss) for the year | — | — | — | | — | — | — | — | — | — | | | | |||||||||||||||
Total comprehensive income / (expense) for the year | — | — | — | | — | — | | ( | — | ( | | | | |||||||||||||||
Net change in treasury stock | Note 17 (d) | — | — | — | — | — | | — | — | — | — | | — | | ||||||||||||||
Acquisition / Divestment of non-controlling interests | Note 17 (c) | — | — | — | — | — | — | — | — | — | — | — | | | ||||||||||||||
Other changes | — | — | | — | — | — | — | — | — | — | | | | |||||||||||||||
Distribution of 2021 profit: | ||||||||||||||||||||||||||||
Reserves | — | — | | ( | — | — | — | — | — | — | — | — | — | |||||||||||||||
Dividends | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||
Interim dividend | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||
Operations with shareholders or owners | — | — | | ( | — | | — | — | — | — | | | | |||||||||||||||
Balance at 31 December 2022 | | | | | — | ( | | ( | — | ( | | | | |||||||||||||||
Translation differences | — | — | — | — | — | — | ( | — | — | — | ( | ( | ( | |||||||||||||||
Cash flow hedges | Note 30 | — | — | — | — | — | — | — | — | — | | | — | | ||||||||||||||
Other comprehensive income | — | — | — | — | — | — | — | ( | — | — | ( | — | ( | |||||||||||||||
Other comprehensive income from non-current assets held for sale | — | — | — | — | — | — | — | — | | — | | — | | |||||||||||||||
Other comprehensive income / (expense) for the year | — | — | — | — | — | — | ( | ( | | | ( | ( | ( | |||||||||||||||
Profit/(loss) for the year | — | — | — | | — | — | — | — | — | — | | | | |||||||||||||||
Total comprehensive income / (expense) for the year | — | — | — | | — | — | ( | ( | | | ( | | ( | |||||||||||||||
Net change in treasury stock | Note 17 (d) | — | — | — | — | — | | — | — | — | — | | — | | ||||||||||||||
Acquisition / Divestment of non-controlling interests | Note 17 (c) | — | — | ( | — | — | — | — | — | — | — | ( | | ( | ||||||||||||||
Other changes | Note 10 | — | — | ( | — | — | — | — | — | — | — | ( | ( | ( | ||||||||||||||
Distribution of 2022 profit: | ||||||||||||||||||||||||||||
Reserves | — | — | | ( | — | — | — | — | — | — | — | — | — | |||||||||||||||
Dividends | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||
Interim dividend | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||
Operations with shareholders or owners | — | — | | ( | — | | — | — | — | — | ( | ( | ( | |||||||||||||||
Balance at 31 December 2023 | | | | | — | ( | | ( | | | | | | |||||||||||||||
The accompanying notes form an integral part of the consolidated financial statements.
F-13
(1) Nature, Principal Activities and Subsidiaries
Grifols, S.A. (hereinafter the Company) was incorporated with limited liability under Spanish law on 22 June 1987. Its registered and tax offices are in Jesús i Maria, 6, 08022, Barcelona. The Company’s statutory activity consists of providing corporate and business administrative, management and control services, as well as investing in assets and property. Its principal activity involves rendering administrative, management and control services to its subsidiaries.
On 17 May 2006 the Company completed its flotation on the Spanish securities market, which was conducted through the public offering of
The Company’s shares were floated on the Spanish stock exchange IBEX-35 index on 2 January 2008.
All of the Company’s shares are listed on the Barcelona, Madrid, Valencia and Bilbao securities markets and on the Spanish Automated Quotation System (SIBE/Continuous Market). On 2 June 2011, Class B non-voting shares (ADRs) were listed on the NASDAQ (USA) and on the Spanish Automated Quotation System (SIBE/Continuous Market).
Grifols, S.A. is the Parent of the subsidiaries listed in Appendix I of this note to the consolidated financial statements.
Grifols, S.A. and subsidiaries (hereinafter the Group) act on an integrated basis and under common management and their principal activity is the procurement, manufacture, preparation and sale of therapeutic products, especially hemoderivatives.
The main factory locations of the Group’s Spanish companies are in Parets del Vallés (Barcelona) and Torres de Cotilla (Murcia), while the US companies are located in Los Angeles (California), Clayton (North Carolina), Emeryville (California), and San Diego (California).
(2) Basis of Presentation
The consolidated financial statements have been prepared on the basis of the accounting records of Grifols, S.A. and of the Group companies. The consolidated financial statements for 2023 have been prepared under International Financial Reporting Standards as issued by the International Accounting Standard Board (IFRS-IASB) which for Grifols Group purposes, are identical to the International Financial Reporting Standards as adopted by the European Union (IFRS-EU) to present fairly the consolidated equity and consolidated financial position of Grifols, S.A. and subsidiaries at 31 December 2023, as well as the consolidated results from their operations, consolidated cash flows and consolidated changes in equity for the year then ended.
At their meeting held on 17 April 2024 the Board of Directors of Grifols, S.A. authorized for issue the 2023 consolidated financial statements.
The consolidated financial statements are presented in thousands of Euros, which is the functional and presentation currency of the Parent.
These consolidated financial statements for 2023 show comparative figures for 2022 and voluntarily show figures for 2021 from the consolidated statement of profit and loss, consolidated statement of comprehensive income, consolidated statement of changes in equity and consolidated statement of cash flows and their corresponding notes thereto. For the purposes of comparing the consolidated statement of profit and loss for 2023, 2022 and 2021 and the consolidated balance sheet for 2023 and 2022, the effects of the application new standards described in note 2 must be taken into account.
The Group adopted IFRS-EU for the first time on 1 January 2004 and has been preparing its financial statements under International Financial Reporting Standards, as adopted by the European Union (IFRS-EU) as required by Spanish capital market regulations governing the presentation of financial statements by companies whose debt or own equity instruments are listed on a regulated market.
F-14
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
In accordance with the provision of section 357 of the Irish Companies Act 2014, the Company has irrevocably guaranteed all liabilities of an Irish subsidiary undertaking, Grifols Worldwide Operations Limited (Ireland) (see Appendix I), for the financial year ended 31 December 2023 as referred to in subsection 1(b) of that Act, for the purposes of enabling Grifols Worldwide Operations Limited to claim exemption from the requirement to file their own financial statements in Ireland.
(a) Relevant accounting estimates, assumptions and judgments used when applying accounting principles
The preparation of the consolidated financial statements in conformity with IFRS-IASB requires management to make judgments, estimates and assumptions that affect the application of Group accounting policies. The following notes include a summary of the relevant accounting estimates and judgments used to apply accounting policies which have the most significant effect on the amounts recognized in the consolidated financial statements.
| ● | Determination of the fair value of assets, liabilities and contingent liabilities in relation to business combinations. The fair value methods used by the Group are detailed in note 3. During fiscal year 2023, there were no significant business combinations. |
| ● | Assumptions used to test non-current assets and goodwill for impairment. Relevant cash generating units are tested annually for impairment. These are based on risk-adjusted future cash flows discounted using appropriate interest rates. The key assumptions used are specified in note 6. Assumptions relating to risk-adjusted future cash flows and discount rates are based on business forecasts and are therefore inherently subjective. Future events could cause a change in business forecasts, with a consequent adverse effect on the future results of the Group. To the extent considered a reasonably possible change in key assumptions could result in an impairment of goodwill, a sensitivity analysis has been disclosed to show the effect of changes to these assumptions and the effect of the cash generating unit (CGU) on the recoverable amount. |
| ● | Evaluation of the capitalization of development costs (see note 4(d)). The key assumption is related to the estimation of sufficient future economic benefits of the projects. |
| ● | The calculation of the income tax expense requires tax legislation interpretations in the jurisdictions where Grifols operates. The decision as to whether the tax authority will accept a given uncertain tax treatment and the expected outcome of outstanding litigation requires significant estimates and judgements. Likewise, Grifols recognizes deferred tax assets, mainly from tax credits and rights to deduct to the extent that it is probable that sufficient taxable income will be available against which temporary differences can be utilized, based on management assumptions regarding amount and payments of future taxable profits (see notes 4(q) and 28). |
| ● | Determination of chargebacks made to certain customers in the United States (see note 4 (p)). |
| ● | The assumptions used for the calculation of the fair value of financial instruments (see notes 3, 29 and 31). |
| ● | Evaluation of whether Grifols controls a subsidiary or not, analyzing factors such as rights derived from contractual agreements, as well as actual and potential voting rights, considering for these purposes the potential voting rights held by Grifols exercisable at the closing date (see note 10 and 19). |
In addition, the Group has considered as a relevant judgment the classification as a non-current asset held for sale of the
No changes have been made to prior year judgments relating to existing uncertainties.
The Group is also exposed to interest rate and currency risks. Refer to sensitivity analysis in note 30.
F-15
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(b) Basis of consolidation
Appendix I shows details of the percentages of direct or indirect ownership of subsidiaries by the Company at 31 December 2023, 2022 and 2021, as well as the consolidation method used in each case for preparation of the accompanying consolidated financial statements.
Subsidiaries in which the Company directly or indirectly owns the majority of equity or voting rights have been fully consolidated. Associates in which the Company owns between 20% and 50% of share capital and over which it has no control but does have significant influence, have been accounted for under the equity method.
Although the Group holds
On the other hand, the Group holds the
The entities Haema AG, BPC Plasma, Inc. and Haema Plasma Kft., of which Grifols does not hold shares, but there exists control over them (see notes 3(d) and 19), have been fully consolidated.
Grifols (Thailand) Ltd. has
Changes in associates and jointly controlled entities are detailed in note 10.
Changes in subsidiaries
In 2023:
| ● | Grifols Escrow Issuer, S.A. and Gripdan Invest, S.L. |
With effect as of 1 January 2023, Grifols Escrow Issuer, S.A., Gripdan Invest, S.L and Grifols, S.A. entered into a merger agreement, with Grifols, S.A. being the surviving company.
‘This operation has had no impact on the Consolidated Financial Statements.
| ● | Access Biologicals LLC. and Chiquito Acquisition Corp. |
With effect as of 1 April 2023, Access Biologicals, L.L.C, Chiquito Acquisition Corp. and Grifols Bio Supplies, Inc. (formerly Interstate Blood Bank, Inc. (IBBI)) entered into a merger agreement, with Grifols Bio Supplies, Inc. being the surviving company.
‘This operation has had no impact on the Consolidated Financial Statements.
| • | Goetech LLC |
On 30 June 2023, the company Geotech LLC (D/B/A Medkeeper) has been dissolved.
This operation has had no impact on the Consolidated Financial Statements.
F-16
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
| • | Kiro Grifols, S.L. |
On 27 July 2023, Grifols reached an agreement to acquire the remaining
| • | AlbaJuna Therapeutics, S.L. |
On 9 October 2023, Grifols, through its wholly owned subsidiary Grifols Innovation and New Technologies Limited, Inc., reached an agreement to acquire the remaining
| • | Biotest (U.K.), Ltd. |
On 1st June 2023, Grifols U.K., Ltd. reached an agreement with Biotest AG to acquire the total shares of Biotest (U.K. Ltd.) for a total amount of Euros
The following companies were formed during 2023 and became part of the Grifols Group consolidated:
| ● | Biomat Holdings, LLC |
| ● | Canada, Inc. (subsequently changed its name to Grifols Plasma Canada - Ontario Inc.) |
In 2022:
| ● | Albimmune, S.L. |
On 13 January 2022, Grifols, through its wholly owned subsidiary Grifols Innovation and New Technologies Limited, Inc., reached an agreement to acquire
| ● | VCN Biosciences, S.L. |
On 10 March 2022, Grifols, together with the other shareholders, reached an agreement to sell percent of the issued and outstanding shares of VCN Bioscience, S.L. for US Dollars
As a result of this divestment, the Group has recognized income of Euros
| ● | Biomat USA, Inc. |
Effective 1 April 2022, Biomat USA Inc. and Talecris Plasma Resources, Inc. entered into a merger agreement, and the resulting company was Biomat USA, Inc.
| ● | Biotest AG and Grifols Biotest Holdings GmbH |
On 25 April 2022, and once all regulatory approvals had been obtained, Grifols completed the acquisition of
F-17
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
| ● | Access Biologicals Inc. |
On 15 June 2022, Grifols, through its wholly owned subsidiary Chiquito Acquisition Corp., reached an agreement to acquire all the shares of Access Biologicals LLC, exercising the call option for the remaining
| ● | Grifols México, S.A. de C.V. |
Effective 15 December 2022, Grifols México, S.A. de C.V. and Logística Grifols, S.A. de C.V. entered into a merger agreement, and the resulting company was Grifols México, S.A. de C.V.
In 2021:
| ● | Grifols Pyrenees Research Center, SL |
Grifols, through its wholly-owned subsidiary Grifols Innovation and New Technologies Limited (“GIANT”), owns
The remaining
| ● | Gigagen, Inc. |
On 8 March 2021, Grifols, through its wholly owned subsidiary Grifols Innovation and New Technologies Limited (“GIANT”), reached an agreement to acquire all of the shares of Gigagen, Inc. for a total consideration of US Dollars
With the acquisition of
| • | Grifols Canada Plasma, Inc. (formerly Prometic Plasma Resources, Inc.) |
On 31 December 2021, Grifols, through its wholly owned subsidiary Grifols Canada Therapeutics Inc., reached an agreement to acquire all of the shares of Prometic Plasma Resources Inc. for a total consideration of US Dollars
| ● | Grifols Escrow Issuer, S.A. |
On August 26, 2021, Grifols, S.A. acquired all of the shares of Grifols Escrow Issuer, S.A. for a total consideration of US Dollars
| ● | Araclon Biotech, SL |
On October 2021 Araclon Biotech, S.L carried out a share capital increases of Euros
F-18
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
| ● | Haema Plasma Kft. |
On 1 February 2021, Scranton Plasma B.V. acquired
The following companies were incorporated during 2021 and were included in the consolidated Grifols Group.
| ● | Grifols Middle East&Africa, LLC |
| ● | Grifols Bio North America, LLC |
| ● | Biomat Holdco, LLC |
| ● | Biomat Newco, Corp |
(c) Amendments to IFRS in 2023, 2022 and 2021
In accordance with IFRS, the following should be noted in connection with the scope of application of IFRS and the preparation of these consolidated financial statements of the Group.
Effective in 2023
The following standards published by the IASB and the IFRS Interpretations Committee and adopted by the European Union for application in Europe came into force in 2023 and, therefore, have been taken into account in the preparation of these consolidated financial statements:
|
|
| Mandatory application for annual periods | |||
beginning on or after: | ||||||
Normas | EU effective date |
| IASB effective date | |||
IAS 12 |
| Amendments to IAS 12 Income Taxes: Deferred Tax related to Assets and Liabilities arising from a Single Transaction (issued on 7 May 2021) | 1 January 2023 | 1 January 2023 | ||
| ||||||
IFRS 17 |
| Insurance Contracts (issued on 18 May 2017); including Amendments to IFRS 17 (issued on 25 June 2020) | 1 January 2023 | 1 January 2023 | ||
IAS 8 |
| Amendments to IAS 8 Accounting policies, Changes in Accounting Estimates and Errors: Definition of Accounting Estitmates (issued on 12 February 2021) | 1 January 2023 | 1 January 2023 | ||
|
| |||||
IAS 1 | Amendments to IAS 1 Presentation of Financial Statements and IFRS Practice Statement 2: Disclosure of Accounting policies (issued on 12 February 2021) | 1 January 2023 | 1 January 2023 | |||
IAS 12 |
| Amendments to IAS 12 Income taxes: International Tax Reform – Pillar Two Model Rules (issued 23 May 2023) | 1 January 2023 | 1 January 2023 | ||
|
| |||||
IFRS 17 |
| Amendments to IFRS 17 Isurance contracts: Initial Application of IFRS 17 and IFRS 9 - Comparative Information (issued on 9 December 2021) | 1 January 2023 | 1 January 2023 | ||
The application of these standards and interpretations has had no significant impact on these consolidated financial statements.
F-19
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Standards issued but not effective in 2023
At the date these consolidated financial statements were authorized for issue, the following IFRS and amendments have been published by the IASB but their application is not mandatory until the future periods indicated below:
|
|
| Mandatory application for annual periods | |||
beginning on or after: | ||||||
Standards | EU effective date | IASB effective date | ||||
IAS 1 |
| Amendments to IAS 1 Presentation of Financial Statements: - Classification of Liabilities as Current or Non-current Date (issued on 23 January 2020); - Classification of Liabilities as Current or Non-current - Deferral of Effective Date (issued on 15 July 2020); and - Non-current Liabilities with Covenants (issued on 31 October 2022) |
| 1 January 2024 |
| 1 January 2024 |
IFRS 16 |
| Amendments to IFRS 16 Leases: Lease Liability in a Sale and Leaseback (issued on 22 September 2022) | 1 January 2024 | 1 January 2024 | ||
IAS 7 |
| Amendments to IAS 7 Cash flow statement and IFRS 16 Financial instruments: information to disclose: Financial agreements with suppliers (issued on 25 May 2023). |
| pending |
| 1 January 2024 |
IAS 21 | Amendments to IAS 21 The Effects of Changes in Foreign Exchange Rates: Lack of Exchangeability (issued on 15 August 2023) | pending | 1 January 2025 | |||
The Group has not applied any of these standards or interpretations in advance of their effective date.
The application of these standards and interpretations would not have significant impact on these consolidated financial statements.
Effective in 2022
Mandatory application for annual periods | ||||||
beginning on or after: | ||||||
Standards |
|
| EU effective date |
| IASB effective date | |
Amendments issued 14 May 2020 to: |
|
| ||||
– IFRS 3 Business Combinations: references to the Conceptual Framework; | ||||||
Various | – IAS 16 Property, Plant and Equipment: Proceeds before Intended Use; | 1 January 2022 | 1 January 2022 | |||
– IAS 37 Provisions, Contingent Liabilities and Contingent Assets: Onerous Contracts — Cost of Fulfilling a Contract ; and | ||||||
– Annual Improvements to IFRSs 2018-2020: IFRS 1, IFRS 9, IFRS 16 and IAS 41. | ||||||
F-20
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Effective in 2021
| Mandatory application for annual periods | |||||||
beginning on or after: | ||||||||
Standards | EU effective date | IASB effective date | ||||||
IFRS 4 |
| Amendments to IFRS 4 Insurance Contracts – deferral of IFRS 9 (issued on 25 June 2020) |
| 1 January 2021 |
| 1 January 2021 | ||
Various |
| Amendments to IFRS 9, IAS 39, IFRS 7, IFRS 4 and IFRS 16 Interest Rate Benchmark Reform – Phase 2 (issued on 27 August 2020) | 1 January 2021 | 1 January 2021 | ||||
IFRS 16 |
| Amendment to IFRS 16 Leases Covid 19-Related Rent Concessions beyond 30 June 2021 (issued on 31 March 2021) | 1 April 2021 | 1 April 2021 | ||||
(3) Business Combinations and Divestments
2023
a)Saskatoon plasma center
On 7 July, 2023, Grifols, through its
Aggregate details of the cost of the business combination, provisional the fair value of the net assets acquired and the provisional goodwill at the acquisition date are shown below:
|
|
|
|
| Thousands of | |
Reference | Thousands of Euros | Canadian Dollars | ||||
Cost of the business combination |
|
|
|
|
|
|
Consideration paid |
|
|
| |
| |
Total consideration paid |
|
|
| |
| |
Fair value of net assets acquired |
|
|
| |
| |
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
| Note 6 |
| |
| |
The amounts determined at the acquisition date of the assets acquired are as follows:
| Fair Value | |||
Thousands of | ||||
Thousands of Euros | Canadian Dollars | |||
Property, plant and equipment |
| |
| |
Inventories |
| |
| |
Total Assets |
| |
| |
Total net assets acquired |
| |
| |
F-21
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The resulting goodwill was allocated to the Biopharma segment and includes the donor database, licenses and workforce. The entire goodwill is considered tax deductible.
b)Albajuna Therapeutics, S.L.
On 9 October, 2023, Grifols, through its
In 2016, Grifols made a capital investment of
Aggregate details of the cost of the business combination, the provisional fair value of the net assets acquired and the provisional goodwill at the acquisition date are shown below:
| Reference |
| Thousands of Euros | |
Cost of the business combination |
|
|
|
|
Consideration paid |
|
|
| |
Total consideration paid |
|
|
| — |
Fair value of net assets acquired |
|
|
| ( |
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
| Note 6 |
| |
The amounts determined at the acquisition date of the assets, liabilities and contingent liabilities acquired are as follows:
| Fair Value | |
Thousands of Euros | ||
Non-current financial assets |
| |
Deferred tax assets |
| |
Trade and other receivables |
| |
Cash and cash equivalents |
| |
Total assets |
| |
Non-current financial liabilities |
| ( |
Current financial liabilities |
| ( |
Trade and other payables |
| ( |
Total Liabilities and contingent liabilities |
| ( |
Total net assets acquired |
| ( |
As future economic benefits cannot be estimated at the acquisition date, the total amount allocated to goodwill has been totally impaired at the moment of its posting (See note 6).
F-22
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
2022
c)Grifols Canada Plasma, Inc. (formerly Prometic Plasma Resources, Inc.)
On 31 December 2021, Grifols, through its wholly owned subsidiary Grifols Canada Therapeutics, Inc., acquired all the shares of Prometic Plasma Resources Inc. for a total of Canadian Dollars
Aggregate details of the cost of the business combination, the fair value of the net assets acquired and the goodwill at the acquisition date are shown below:
Thousands of | ||||||
| Reference |
| Thousands of Euros |
| Canadian Dollars | |
Cost of the business combination |
|
|
|
|
|
|
Consideration paid |
|
|
| |
| |
Total consideration paid |
|
|
| |
| |
Fair value of net assets acquired |
|
|
| |
| |
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
| Note 6 |
| |
| |
At transaction date, total consideration paid was allocated to goodwill, and the amount was restated based on the fair value of the net assets acquired during the following year. Consequently, the amount reflected in note 6 is the movement between both effects, while the amount in the previous table shows the final balance.
The amounts determined at the acquisition date of the assets, liabilities and contingent liabilities acquired are as follows:
Fair Value | ||||
|
| Thousands of Canadian | ||
Thousands of Euros | Dollars | |||
Other Intangible Assets |
| |
| |
Rights of Use |
| |
| |
Property, plant and equipment |
| |
| |
Inventories |
| |
| |
Trade and other reeceivables |
| |
| |
Other current assets |
| |
| |
Cash and cash equivalents |
| |
| |
Total Assets |
| |
| |
Non-current financial liabilities |
| ( |
| ( |
Current financial liabilities |
| ( |
| ( |
Trade and other payables |
| ( |
| ( |
Total Liabilities |
| ( |
| ( |
Total net assets acquired |
| |
| |
The resulting goodwill was allocated to the Biopharma segment and includes the donor database, licenses and workforce.
F-23
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Grifols Canada Plasma, Inc. (formerly Prometic Plasma Resources, Inc.) acquisition had an impact of Euros
d)Haema Plasma Kft.
On 1 February 2021, Scranton Plasma B.V. acquired
On 1 February 2021 the Group signed a call option on the shares of Haema Plasma kft, exercisable by the Group only
The Group has potential voting rights arising from the option to purchase the shareholding and these are substantive, based on:
| ● | A call option for Grifols which gives it the irrevocable and exclusive right (not an obligation) to acquire the Haema Plasma Kft shareholding at any time after 1 February 2022. |
| ● | Grifols is committed to providing support services in the business of collecting, processing and distributing plasma from the donation centers. There is also a Plasma Supply Agreement whereby the plasma produced by these entities will be used almost entirely to cover Grifols’ needs. There is no sales exclusivity. |
| ● | There are no shareholder agreements that provide for relevant decisions to be approved in a manner other than by majority vote. |
The above are indicators of the power that Grifols acquires over this entity, considering that the call option is likely to be exercised and Grifols will have the financial capacity to carry it out.
Consequently, at the time the option becomes exercisable, the option empowers Grifols, even though it has not yet been exercised, and Haema Plasma Kft. is therefore consolidated in Grifols’ consolidated financial statements from 2022.
Aggregate details of the cost of the business combination, the fair value of the net assets acquired and the goodwill at the acquisition date are shown below:
|
| Thousands of |
| Thousands of | ||
Reference | Euros | Hungarian Forint | ||||
Call option price |
|
|
| |
| |
Total call option price |
|
|
| |
| |
Fair value of net assets acquired |
|
|
| |
| |
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
| Note 6 |
| |
| |
F-24
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Grifols did not give any monetary consideration for this purchase option.
The amounts determined at the date of consolidation of the assets, liabilities and contingent liabilities acquired are as follows:
Fair Value | ||||
|
| Thousands of Hungarian | ||
Thousands of Euros | Forint | |||
Other Intangible assets |
| |
| |
Rights of Use |
| |
| |
Property, plant and equipment |
| |
| |
Other non-current assets |
| |
| |
Deferred tax assets |
| |
| |
Inventories |
| |
| |
Trade and other receivables |
| |
| |
Other current assets |
| |
| |
Cash and cash equivalents |
| |
| |
Total Assets |
| |
| |
Provisions |
| ( |
| ( |
Non-current financial liabilities |
| ( |
| ( |
Current financial liabilities |
| ( |
| ( |
Trade and other payables |
| ( |
| ( |
Other current liabilities |
| ( |
| ( |
Total Liabilities and contingent liabilities |
| ( |
| ( |
Total net assets acquired |
| |
| |
The resulting goodwill was allocated to the Biopharma segment and includes the donor database, licences and workforce.
The entire goodwill is not considered tax deductible.
As of 31 December 2023, the option is in the money since the exercise price is approximately equal to the price of Haema Plasma, Kft shares. On the other hand, since the valuation of the option is based on non-observable market variables, it corresponds to Level 3 of the fair value hierarchy. Taking into account the uncertainties underlying the valuation of the option as it involves non-observable variables, and the value of the option not being significant, said value has not been recognized as of 31 December 2023 and 2022.
e)VCN Biosciences, S.L.
On 10 March 2022, Grifols, together with the other shareholders, reached an agreement to sell one hundred percent of the issued and outstanding shares of VCN Bioscience, S.L. for US Dollars
As a result of this divestment, the Group recognized an income of Euros
F-25
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
f)Biotest AG
On 25 April 2022, and once all regulatory approvals were obtained, Grifols completed the acquisition of
| ● | Grifols acquired the entire share capital of Tiancheng (Germany) Pharmaceutical Holdings AG for Euros |
| ● | At the same time as the transaction, Grifols closed the voluntary takeover bid to all shareholders, which involved the payment of |
The investment in Biotest will significantly strengthen Grifols’ capabilities, including its scientific and technical capabilities, helping to strengthen the availability of plasma medicines, its commercial presence and its R&D pipeline. With the opening of
Aggregate details of the cost of the business combination, the fair value of the net assets acquired and the goodwill at the acquisition date are shown below:
| Reference |
| Thousands of Euros | |
Cost of the business combination |
|
|
|
|
Consideration paid |
|
|
| |
Total consideration paid |
|
|
| |
Fair value of net assets acquired |
|
|
| |
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
| Note 6 |
| |
The resulting goodwill was allocated to the Biopharma segment.
F-26
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The amounts determined at the date of consolidation of the assets, liabilities and contingent liabilities acquired are as follows:
Fair Value | ||
| Thousand of Euros | |
Other Intangible Assets |
| |
Rights of Use |
| |
Property, plant and equipment |
| |
Other non-current assets |
| |
Deferred Tax Assets |
| |
Inventories |
| |
Contract Assets |
| |
Trade and other receivables |
| |
Other current assets |
| |
Cash and cash equivalents |
| |
Total assets |
| |
Non-controlling interests |
| ( |
Non-current provisions |
| ( |
Non-current financial liabilities |
| ( |
Other non-current liabilities |
| ( |
Deferred tax liabilities |
| ( |
Current Provisions |
| ( |
Current financial liabilities |
| ( |
Trade and other payables |
| ( |
Other current liabilities |
| ( |
Total Liabilities and contingent liabilities |
| ( |
Total net assets acquired |
| |
As part of the purchase price allocation, the company determined that identifiable intangible assets are the research and development projects in progress, the current product portfolio as well as certain distribution agreements.
The fair value of intangible assets was estimated using an income approach and the projected cash flows were discounted using rates between
The fair value of research and development projects in progress involving plasma therapies (Fibrinogen, IgM and IgG) were estimated in accordance with an income approach based on the Multiple-Period Excess Earnings Method for the application of which the results of such projects were adjusted for the probability of success according to the clinical phase of the project at the date of the transaction.
The current product portfolio comprises regulatory approvals, trademarks, patient relationships and physician relationships related to products currently marketed by Biotest. The distribution agreements identified as intangible assets relate to the distribution of certain products in different geographic regions. In both cases, the fair value was determined using the Multiple-Period Excess Earnings Method.
Research and development projects in progress, the current product portfolio and distribution agreements are amortized on a straight-line basis over an average period of
F-27
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
If the acquisition had taken place as of January 1, 2022, the revenue would have changed by Euros
Biotest Group’s acquisition had an impact of Euros
The Group recognized under operating expenses in the consolidated statement of profit and loss an amount of Euros
g)Access Biologicals Inc.
On 15 June 2022, Grifols, through its wholly owned subsidiary Chiquito Acquisition Corp., reached an agreement to acquire all the shares of Access Biologicals LLC, exercising the call option for the remaining
Access Biologicals’ core business is the collection and manufacture of an extensive portfolio of biological products. Combined with a closed materials sourcing process, it provides support services for different markets such as in-vitro diagnostics, biopharmaceuticals, cell culture and diagnostic research and development.
Aggregate details of the cost of the business combination, the fair value of the net assets acquired and the goodwill at the acquisition date are shown below:
|
|
| Thousands of US | |||
Reference | Thousands of Euros | Dollars | ||||
Cost of the business combination |
|
|
|
|
|
|
First share purchase |
|
|
| |
| |
Second share purchase (present value) |
| |
| | ||
Total consideration paid |
|
|
| |
| |
Gain on the previously held investment |
| |
| | ||
Accumulated gain for equity method before acquisition date |
| |
| | ||
Step-up of the previously held investment |
| |
| | ||
Fair value of net assets acquired |
|
|
| ( |
| ( |
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
| Note 6 |
| |
| |
F-28
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The amounts determined at the date of consolidation of the assets, liabilities and contingent liabilities acquired are as follows:
Fair Value | ||||
| Thousands of |
| Thousands of US | |
Euros | Dollars | |||
Other Intangible Assets |
| |
| |
Property, plant and equipment |
| |
| |
Other non-current assets |
| |
| |
Inventories |
| |
| |
Trade and other receivables |
| |
| |
Other current assets |
| |
| |
Cash and cash equivalents |
| |
| |
Total Assets |
| |
| |
Trade and other payables |
| ( |
| ( |
Deferred tax liabilities |
| ( |
| ( |
Other non-current liabilities |
| ( |
| ( |
Total Liabilities and contingent liabilities |
| ( |
| ( |
Total net assets acquired |
| |
| |
The resulting provisional goodwill was allocated to the Bio-Supplies segment.
As part of the purchase price allocation, the Company determined that identifiable intangible assets are customer relationships.
Customer relationships were valued using the Multiple-Period Excess Earnings Method, for the application of which a discount rate of
If the acquisition had taken place as of January 1, 2022, the revenue would have changed by Euros
Access Biologicals, Inc acquisition had an impact of Euros
The Group recognized under operating expenses in the consolidated statement of profit and loss an amount of Euros
h)Goetech, LLC
In July 2022, Grifols closed an agreement to sell in cash substantially all of the assets of its subsidiary Goetech LLC, whose trade name is MedKeeper, for a US Dollars
F-29
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
As a consequence of this divestment, the Group recognized an income of Euros
2021
i)Gigagen, Inc.
On 8 March 2021, Grifols, through its wholly owned subsidiary Grifols Innovation and New Technologies Limited (“GIANT”), reached an agreement to acquire all of the shares of Gigagen, Inc. for a total consideration of US Dollars
GigaGen is a U.S. biotechnology company specializing in the discovery and early development of recombinant biotherapeutic drugs. GigaGen’s research focuses on the discovery of new biological treatments based on antibodies derived from millions of donor-derived immune system cells.
With the acquisition of
From the total amount agreed, as of 31 December 2021, an amount of Euros
The Group recognized an amount of Euros
Aggregate details of the cost of the business combination, the fair value of the net assets acquired and the goodwill at the acquisition date are shown below:
| Thousands of |
| Thousands of | |
Euros | US Dollars | |||
Consideration paid |
|
|
| |
First share purchase |
| |
| |
Second share purchase (present value) |
| |
| |
Total consideration paid |
| |
| |
Fair value of the previous investment in the company |
| |
| |
Fair value of net assets acquired |
| |
| |
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
| |
| |
F-30
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The amounts determined at the acquisition date of the assets, liabilities and contingent liabilities acquired are as follows:
Fair value | ||||
Thousands of | Thousands of | |||
| Euros |
| US Dollars | |
Development costs in progress | |
| | |
Property, plant and equipment | |
| | |
Non-current financial assets | |
| | |
Trade and other receivables | |
| | |
Other current assets | |
| | |
Cash and cash equivalents | |
| | |
Total assets | |
| | |
Non-current liabilities | ( |
| ( | |
Current liabilities | ( |
| ( | |
Total liabilities and contingent liabilities | ( |
| ( | |
Total net assets identified | |
| | |
The fair value of the R&D projects in progress was estimated based on market approach of comparable transactions.
The resulting goodwill was allocated to the others segment and includes the specialized R&D workforce and the portfolio of future early-stage products.
The acquired business generated consolidated results for the Group during the period from the acquisition date to year-end in the amount of Euros
If the acquisition had occurred as of 1 January 2021, the Group’s net revenues and results would not have changed significantly.
Gigagen acquisition had an impact of Euros
j)BPL Plasma, Inc.
On 28 February 2021, Biomat USA, Inc. the Group’s American subsidiary, acquired
The transaction received the necessary regulatory approvals and was financed with its own resources, without issuing debt.
Grifols will obtain approximately
The Group recognized transaction costs of Euros
F-31
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Aggregate details of the cost of the business combination, the definitive fair value of the net assets acquired and the definitive goodwill at the acquisition date are shown below:
| Thousands of Euros |
| Thousands of US Dollars | |
Consideration paid | ||||
First payment made |
| |
| |
Cash paid at the transaction closing date |
| |
| |
Total consideration paid |
| |
| |
Fair value of net assets acquired |
| |
| |
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
| |
| |
The amounts determined at the acquisition date of the assets, liabilities and contingent liabilities acquired are as follows:
| Fair value | |||
Thousands of Euros |
| Thousands of US Dollars | ||
Property, plant and equipment |
| |
| |
Non-current financial assets |
| |
| |
Inventories |
| |
| |
Total assets |
| |
| |
Current liabilities |
| ( |
| ( |
Total liabilities and contingent liabilities |
| ( |
| ( |
Total net assets identified |
| |
| |
The resulting goodwill was allocated to the Biopharma segment and included the donor database, licenses and workforce.
The entire goodwill is considered tax deductible.
k)Acquisition of plasma centers from Kedplasma, LLC.
On 31 March 2021, Biomat USA, Inc., the Group’s American subsidiary, acquired
Grifols will have immediate access to the plasma obtained at these centers, which obtain approximately
The transaction received the necessary regulatory approvals and was financed with equity without issuing debt.
The Group recognized transaction costs of Euros
F-32
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Aggregate details of the cost of the business combination, the definitive fair value of the net assets acquired and the definitive goodwill at the acquisition date are shown below:
| Thousands of Euros |
| Thousands of US Dollars | |
Consideration paid |
| |||
Cash paid | |
| | |
Total consideration paid |
| |
| |
Fair value of net assets acquired |
| |
| |
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
| |
| |
The amounts determined at the acquisition date of the assets, liabilities and contingent liabilities acquired are as follows:
| Fair value | |||
Thousands of Euros |
| Thousands of US Dollars | ||
Property, plant and equipment |
| |
| |
Inventories |
| |
| |
Total assets |
| |
| |
Total net assets identified |
| |
| |
The resulting goodwill was allocated to the Biopharma segment and included the donor database, licenses and workforce. The entire goodwill is considered tax deductible.
On 31 December 2021, Grifols, through its wholly owned subsidiary Grifols Canada Therapeutics Inc., acquired all of the shares of Prometic Plasma Resources Inc. for a total consideration of US Dollars
(4)Significant Accounting Policies
(a) | Consolidation |
Dependents
Subsidiaries are considered to be those over which the Group exercises control. A subsidiary is controlled when, due to its involvement in it, it is exposed, or has the right, to variable returns and has the capacity to influence such returns through the power it exercises over it.
The income, expenses and cash flows of subsidiaries are included in the consolidated financial statements from the date of acquisition, which is the date on which the Group effectively obtains control of the subsidiaries. Subsidiaries are excluded from consolidation from the date on which control is lost.
Transactions and balances with Group companies and unrealized gains or losses have been eliminated in consolidation.
The accounting policies of the subsidiaries have been adapted to the Group’s accounting policies for transactions and other events that, being similar, have occurred in similar circumstances.
The financial statements of the subsidiaries used in the consolidation process are as of the same reporting date and the same period as those of the Parent Company.
F-33
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Appendix I includes information on the subsidiaries included in the Group’s consolidation.
Business combinations
The acquisition method is used to account for the acquisition of subsidiaries in a business combination. The acquisition date is the date on which the Group obtains control of the acquired business.
The acquisition cost of a subsidiary is determined at the acquisition date and comprises (i) the fair values of assets delivered, (ii) liabilities incurred or assumed, (iii) equity instruments issued, (iv) the fair value of any asset or liability resulting from a contingent consideration arrangement and (v) the fair value of any previous interest in the subsidiary. Any disbursement that is not part of the exchange for the acquired business is excluded.
Acquisition-related costs are expensed as incurred.
The Group recognizes identifiable assets acquired and liabilities and contingent liabilities assumed at fair value at the acquisition date. Non-current assets held for sale, liabilities for employee compensation, transactions with payments based on equity instruments, deferred tax assets and liabilities and right-of-use assets and liabilities and lease liabilities are excluded from the application of this criterion.
The excess of the consideration transferred the amount of any non-controlling interest in the acquired subsidiary and the acquisition-date fair value of any previous interest in the acquired subsidiary over the fair value of the identifiable net assets is recorded as goodwill. If these amounts are less than the fair value of the identifiable net assets of the acquired subsidiary, the difference is recognized in profit or loss as a bargain purchase.
When settlement of any part of the cash consideration is deferred, amounts payable in the future are discounted to their present value at the date of exchange.
Contingent consideration is classified as equity or a financial liability. Amounts classified as a financial liability are subsequently remeasured at fair value with changes in fair value recognized in profit or loss.
When the business combination could only be determined on a provisional basis, the identifiable net assets are initially recorded at their provisional values, recognizing the adjustments made during the measurement period as if they had been known at the acquisition date, restating comparative figures for the previous year, if applicable. The adjustments to the provisional values only incorporate information relating to facts and circumstances that existed at the acquisition date and which, had they been known, would have affected the amounts recognized at that date. The measurement period should not exceed twelve months from the date of acquisition.
If the business combination is carried out in stages, the acquisition-date carrying amount of the previously held equity interest of the acquiree is remeasured at its acquisition-date fair value, with any resulting gain or loss recognized in profit or loss.
Non-controlling interests
Non-controlling interests in subsidiaries are recorded at the acquisition date at their percentage of interest in the fair value of the identifiable net assets, without considering potential voting rights. In addition, the profit or loss for the year and each component of other comprehensive income allocated to the non-controlling interest is allocated in proportion to its percentage of ownership. Non-controlling interests in the results and equity of subsidiaries are shown separately in the consolidated statement of profit and loss, the consolidated statement of comprehensive income, the consolidated statement of changes in equity and the consolidated balance sheet, respectively.
F-34
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The increase and reduction of non-controlling interests in a subsidiary while maintaining control is recognized as an equity transaction in reserves.
Associated
Associated entities are those over which the Group exercises significant influence, understood as the capacity to intervene in financial and operating decisions, without the existence of control or joint control.
Investments in associates are initially recognized at acquisition cost, including costs directly attributable to the acquisition and any active or passive contingent consideration that depends on future events or the fulfillment of certain conditions.
Subsequently, investments in associates are accounted for by the equity method from the date on which significant influence is exercised until the date on which the Company can no longer justify the existence of significant influence.
The excess between the cost of the investment and the Group’s share of the fair values of the identifiable net assets is recorded as goodwill, which is included in the carrying amount of the investment. The shortfall, once the amounts of the cost of the investment and the identification and valuation of the net assets of the associate have been evaluated, is recorded as income in the determination of the investor’s share in the results of the associate for the year in which it was acquired.
The accounting policies of the associated companies have been subject to time and valuation homogenization in the same terms as those referred to in the subsidiaries.
The Group’s share in the profits or losses of associates obtained from the date of acquisition is recorded as an increase or decrease in the value of the investments with a credit or debit to “Profit of equity accounted investees with similar activity to that of the Group” when the investee companies carry out the same activity as the corporate purpose of the Group described in note 1 and, otherwise, in “Profit /(loss) of equity accounted investees”. Likewise, the Group’s share in the other comprehensive income of associates obtained since the acquisition date is recorded as an increase or decrease in the value of the investments in associates, with the balancing entry by nature being recognized in other comprehensive income. Dividend distributions are recorded as decreases in the value of investments. To determine the Group’s share of profits or losses, including impairment losses recognized by associates, income or expenses arising from the acquisition method are considered.
When the Group’s share of losses on an equity accounted investment equals or exceeds its interest in the entity, the Group does not recognize additional losses unless it has incurred obligations or made payments on behalf of the other entity.
The Group’s share in the profits or losses of associates and changes in equity is determined on the basis of the ownership interest at year-end, without considering the possible exercise or conversion of potential voting rights. However, the Group’s share is determined considering the possible exercise of potential voting rights and other derivative financial instruments that, in substance, grant current access to the economic benefits associated with ownership interests, i.e. the right to participate in future dividends and changes in the value of associates.
After applying the equity method, the Group assesses whether there is objective evidence of impairment of the net investment in the associate. Some of the main evidence include significant cumulative losses, contractual default, financial difficulties and adverse changes in technology, industry or economy affecting the associate. The impairment calculation is determined by comparing the carrying amount of the net investment in the associate with its recoverable amount, where recoverable amount is the higher of value in use or fair value less costs of disposal. In this regard, the value in use is calculated based on the Group’s share of the present value of the estimated cash flows from ordinary activities and the amounts that could result from the final disposal of the associate. The recoverable amount of the investment in an associate is assessed in relation to each associate (see note 10), unless it does not constitute a cash-generating unit (CGU). Impairment losses are not allocated to goodwill or other assets implicit in the investment in associates arising from the application of the acquisition method. In subsequent years, reversals of the value of investments are recognized against income, to the extent that there is an increase in the recoverable value. Impairment losses are presented separately from the Group’s share in the results of associates.
F-35
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Appendix I includes information on subsidiaries and associates included in the Group’s consolidation.
Joint agreements
Joint arrangements are those in which there is a contractual agreement to share control over an economic activity, so that decisions on the relevant activities require the unanimous consent of the Group and the other operators. Investments in joint arrangements are classified as joint operations or joint ventures, depending on the contractual rights and obligations of each investor, rather than the legal structure of the joint arrangement.
Joint transactions are considered when the participants in the joint arrangement are entitled to the assets and obligations in respect of the liabilities. This type of arrangement is consolidated proportionally integrating the assets and liabilities related to the transaction as described in note 10.
Joint ventures are those when the participants in the agreement have a right to the net assets. This type of arrangement is included in the consolidated financial statements using the equity method, as described in note 10.
(b)Transactions and balances in foreign currencies
Transactions in foreign currencies are translated to the functional currency using the average exchange rate of the previous month provided that it does not differ significantly from the exchange rate at the date of the transaction. Foreign currency gains and losses resulting from the settlement of these transactions and from the translation of monetary assets and liabilities denominated in foreign currencies at closing exchange rates are recognized in profit or loss except when there are qualified cash flow hedges and qualified net investment hedges that are deferred to equity.
The effect of exchange rate changes on cash and cash equivalents denominated in foreign currencies is presented separately in the statement of cash flows as “Effect of exchange rate changes on cash”.
The translation of foreign operations whose functional currency is not that of a hyperinflationary country has been made by applying the following criteria:
| m) | Assets and liabilities, including goodwill and adjustments to net assets arising from the acquisition of businesses, are translated at the closing exchange rate at each balance sheet date; |
| n) | Revenues and expenses are translated at the average exchange rate of the previous month, as an approximation of the exchange rate at the date of the transaction; |
| o) | Translation differences resulting from the application of the above criteria are recognized in other comprehensive income. |
(c)Goodwill
After initial recognition, goodwill is recorded at cost, less any accumulated impairment loss, which is not reversible.
Goodwill is not amortized, but is tested for impairment on an annual basis or more frequently in the event that events indicative of a potential loss in the value of the asset have been identified. For these purposes, goodwill resulting from business combinations is allocated to each of the cash generating units (CGUs) or groups of CGUs that are expected to benefit from the synergies of the combination and the criteria referred to in note 6 are applied. CGUs or groups of CGUs are identified at the lowest level that goodwill is controlled for the purpose of internal management (Note 6).
F-36
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(d)Intangible assets
Intangible assets are recorded at cost (acquisition or development) or at fair value when acquired in a business combination, less accumulated amortization and any accumulated impairment losses.
Any costs incurred during the research phase of projects are recognized as an expense when incurred.
Costs related to development activities for internally generated intangible assets are capitalized to the extent that:
| ● | The Group has technical studies that justify the viability of the production process; |
| ● | There is a commitment by the Group to complete production of the asset so that it is in a condition for sale or internal use; |
| ● | The asset will generate sufficient economic benefits; |
| ● | The Group has the technical and financial resources to complete the development of the asset and has developed budget control and analytical accounting systems that make it possible to monitor the budgeted costs, the modifications introduced and the costs actually charged to the various projects. |
In relation to the development costs of new products or drugs, they are capitalized as long as their economic profitability is reasonably assured and when they are either in a pivotal phase or correspond to projects related to products that are currently being marketed in various markets, in both cases with expected technical feasibility. Development costs previously recognized as an expense are not recognized as an asset in a subsequent period.
The separate acquisition or through a business combination of an research and development project in progress is capitalized in any case, in accordance with the provisions of IAS 38, since the price paid for the acquisition reflects expectations about the probability that the future economic benefits of the asset are used by the Group. Subsequent costs are recorded following the provisions for internally generated intangible assets.
The Group amortizes its intangible assets with finite useful lives by distributing the cost of the assets on a straight-line basis according to the following criteria:
| Amortisation method |
| Rates | |
Development expenses |
| Straight line |
| |
Concessions, patents, licenses, trademarks and similar |
| Straight line |
| |
Computer software |
| Straight line |
| |
Currently marketed products |
| Straight line |
|
Intangible assets with indefinite useful lives are not subject to amortization but are tested for impairment at least once a year.
The Group reviews the useful lives of intangible assets at the end of each year. Changes in the initially established criteria are recognized as a change in estimate.
(e)Property, plant and equipment
Property, plant and equipment are stated at cost, less accumulated depreciation and, if applicable, accumulated impairment losses.
Cost includes, among other items, direct labor costs used in the construction of the asset and a portion of the costs indirectly attributable to the asset.
F-37
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Finance costs incurred that are directly attributable to the acquisition or construction of the asset until the asset is ready for use also form part of the cost.
Likewise, expansion or improvement costs are included as an increase in the value of the asset when they represent an increase in its capacity or an extension of its useful life. However, maintenance costs are recognized in income when incurred.
Depreciation of property, plant and equipment is provided on a straight-line basis over the estimated useful lives of the assets, less their residual value.
Depreciation of property, plant and equipment is determined by applying the following criteria:
| Depreciation method |
| Rates | |
Buildings |
| Straight line |
| |
Other property, technical equipment and machinery |
| Straight line |
| |
Other property, plant and equipment |
| Straight line |
|
The Group reviews the residual value, useful life and depreciation method of property, plant and equipment at the end of each reporting period. Changes in the initially established criteria are recognized as a change in estimate.
(f)Leases
Lessee
The determination of whether a contract is or contains a lease is based on an analysis of the contractual arrangement and requires an assessment of whether the lessee has the right to control the use of the identified asset and to obtain all of the economic benefits from the use of the asset throughout the lease term.
The lease term is the non-cancelable period considering the initial term of each contract unless the Group has a unilateral extension or termination option and there is reasonable certainty that such option will be exercised in which case the corresponding extension or early termination term will be considered.
In lease contracts where the Group acts as lessee, it is recognized at the lease commencement date (i.e. the date on which the underlying asset is available for use):
| ● | A liability for the present value of the installments to be paid over the lease term, using the incremental borrowing or implicit interest rate as the discount rate when expressly indicated in the contract and, |
| ● | A right-of-use asset representing the right to use the underlying leased asset during the term of the lease. |
Lease liabilities include fixed lease payments less any incentives, as well as variable payments that depend on an index or interest rate known at the date of inception of the lease. Also included is the exercise price of the purchase option when the lessee is reasonably certain of exercising it. After initial recognition, the liability is increased by the interest on the lease liability and reduced by the payments made. The liability is also remeasured if there are changes in the amounts payable and the lease terms. Payments included in the lease payments corresponding to maintenance, electricity, water, gas, security, cleaning, among others, are not part of the lease liability and are recognized as an expense.
The incremental borrowing rate is determined taking into account: (i) geographic areas, (ii) financial term, (iii) lease term, (iv) risk-free rate as reference rate and (v) financial spread.
F-38
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Rights-of-use assets are measured at cost, less accumulated amortization and impairment losses (if any) and adjusted as a result of the remeasurement of the lease liability. Cost includes the amount of the initial valuation of the lease liability, as well as any amounts previously paid to the lessor prior to or at the commencement date of the lease less any incentives received by the lessor and estimated costs to decommission the leased asset. Amortization of rights of use is provided on a straight-line basis over the shorter of the estimated useful life of the asset or the lease term.
The Group applies the exception to recognition for those contracts where the lease term is 12 months or less or where the value of the leased asset (individually) when new, is less than US Dollars 5,000 or its equivalent in another currency. Consequently, in these cases, the amounts accrued will be recognized as an expense during the lease term.
Lessor
When the Group acts as lessor, it classifies contracts between operating and finance leases. Leases in which the Group acts as lessor while retaining a significant portion of the risks and rewards incidental to ownership of the leased asset are treated as operating leases. Otherwise, the lease is treated as a finance lease.
(g)Impairment of non-financial assets
Goodwill and intangible assets that have an indefinite useful life are not subject to amortization and are tested for impairment annually, or more frequently in the event of events or changes in circumstances that indicate that they may be impaired.
Other assets are tested for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable.
When the recoverable amount is less than the carrying amount of the asset, an impairment loss is recognized in the consolidated statement of profit and loss for the difference between both amounts.
The recoverable amount is the higher of an asset’s fair value less costs of disposal and the estimated value in use based on discounted future cash flows expected to arise from the use of the asset. The estimate of value in use considers expectations about possible variations in the amount or timing of cash flows, the time value of money, the price to be paid for bearing the uncertainty related to the asset and other factors that affect the valuation of future cash flows related to the asset.
For the purpose of assessing impairment losses, assets are grouped at the lowest levels for which there are separately identifiable cash inflows that are largely independent of the cash inflows of other assets or groups of assets (cash-generating units). Impairment losses on non-financial assets (other than goodwill) are reviewed for possible reversal at the end of each reporting period.
Losses related to the impairment of CGUs are initially allocated to reduce, if applicable, the value of goodwill attributed to the CGU and then to the other assets of the CGU, pro rata based on the carrying amount of each asset, with the limit for each asset being the higher of its fair value less costs of disposal, its value in use and zero.
Impairment losses related to goodwill are not reversible.
F-39
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(h)Financial instruments
Financial assets
Ranking
The classification of financial assets is determined based on the characteristics of the contractual cash flows of those assets and the business model that represents how the financial assets are managed to achieve a particular business objective. In determining whether the cash flows are obtained through the receipt of contractual cash flows from the assets, consideration is given to the frequency, value and timing of sales in prior periods, the reasons for those sales and expectations regarding future sales activity. This information provides indicative data on how the Group’s stated objective regarding the management of financial assets is achieved and, more specifically, how cash flows are obtained.
Therefore, financial assets are classified according to the following valuation categories based on the business model and are only reclassified when, and only when their business model for managing them changes:
| a. | Financial assets at amortized cost: includes financial assets, including those admitted to trading on an organized market, for which the Group holds the investment under a business model whose objective is to hold financial assets to receive cash flows from the execution of the contract, and the contractual terms of the asset give rise, at specified dates, to cash flows that are solely collections of principal and interest on the principal amount outstanding. |
In general, the following are included in this category:
i.Trade receivables: arising from the sale of goods or the rendering of services for trade transactions with deferred payment, and
ii.Receivables from non-trade operations: these arise from loans or credits granted by the Group whose collections are of a determined or determinable amount.
b. | Financial assets at fair value through other comprehensive income: this category includes financial assets whose contractual conditions give rise, at specified dates, to cash flows that are solely collections of principal and interest on the principal amount outstanding, and are held within the framework of a business model whose objective is achieved by obtaining contractual cash flows and selling financial assets. Investments in equity instruments irrevocably designated by the Group at the time of their initial recognition are also included in this category, provided that they are not held for trading and are not to be valued at cost. |
c. | Financial assets at fair value through profit or loss: includes financial assets held for trading and those financial assets that have not been classified in any of the above categories. Also included in this category are financial assets that are optionally designated by the Group at the time of initial recognition, which otherwise would have been included in another category, because such designation eliminates or significantly reduces a valuation inconsistency or accounting missmatch that would otherwise arise. |
Initial measurement
Financial assets are recorded, in general terms, initially at the fair value of the consideration given plus directly attributable transaction costs. However, transaction costs directly attributable to assets recorded at fair value through profit or loss are recognized in the statement of profit and loss for the year.
F-40
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Subsequent measurement
Financial assets at amortized cost are recorded by applying this valuation criterion, charging to the statement of profit and loss the interest accrued by applying the effective interest rate method.
Financial assets included in the fair value category through other comprehensive income are recorded at fair value, without deducting any transaction costs that may be incurred in their disposal. Changes in fair value are recorded directly in equity until the financial asset is derecognized or impaired, at which time the amount so recognized is taken to the statement of profit and loss.
Financial assets at fair value through profit or loss are measured at fair value and the result of changes in fair value is recorded in the statement of profit and loss.
Disposals of financial assets
Financial assets are derecognized when the rights to receive cash flows related to them have expired or have been transferred and the Group has substantially transferred the risks and rewards of ownership. Similarly, they are disposed from the balance sheet when there are transfers of collection rights, whose certain risks are shared with the factor, such as the risk of default, but exists a transfer of control to the factor, understood as the unilateral capacity to sell those assets to a non-related third party without the necessity of enforcing additional restrictions to the sale.
Impairment
The Group assesses, on a prospective basis, the expected credit losses associated with its debt instruments carried at amortized cost and at fair value through other comprehensive income The methodology applied for impairment depends on whether there has been a significant increase in credit risk.
For trade receivables, the Group applies the simplified approach permitted by IFRS 9 which requires expected losses to be recorded from the initial recognition of the receivables, so that the Group determines expected credit losses as a probability-weighted estimate of such losses over the expected life of the financial instrument.
The practical solution used is the use of a provisioning matrix based on segmentation into homogeneous asset groups, applying historical information on default rates for these groups and applying reasonable information on future economic conditions.
Default rates are calculated based on current default experience over the past year, as it is a very dynamic market, and are adjusted for differences between current and historical economic conditions and considering projected information, which is reasonably available.
Financial liabilities
Financial liabilities assumed or incurred by the Group are classified in the following measurement categories:
a. | Financial liabilities at amortized cost: are those debits and payables of the Group that have arisen from the purchase of goods and services for trading operations, or those which, without having a commercial origin, not being derivative instruments, arise from loan or credit operations received by the Group. |
These liabilities are initially measured at the fair value of the consideration received, adjusted for directly attributable transaction costs. Any difference between the amount received and its repayment value is recognized in the consolidated statement of profit and loss during the repayment period of the debt, applying the effective interest rate method.
F-41
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
b.Financial liabilities at fair value through profit or loss.
Liability derivative financial instruments are measured at fair value, following the same criteria as those corresponding to financial assets at fair value through profit or loss described in the preceding section.
The Group derecognizes financial liabilities when the obligations that generated them are extinguished.
Assets and liabilities are presented separately in the balance sheet and are only presented at their net amount when the Group has the enforceable right to offset the recognized amounts and, in addition, intends to settle the amounts on a net basis or to realize the asset and settle the liability simultaneously.
Equity instruments
The Group holds financial assets, mainly equity instruments, which are measured at fair value. When Group management has opted to present gains and losses in the fair value of equity investments in other comprehensive income, after initial recognition, the equity instruments are measured at fair value, recognizing the gain or loss in other comprehensive income. Amounts recognized in other comprehensive income are not reclassified to profit or loss, but are reclassified to reserves when the instruments are derecognized. Dividends from such investments continue to be recognized in profit or loss as other income when the Group’s right to receive payments is established.
(i)Derivative financial instruments and hedging activities
Financial derivatives are recognized at fair value at the date of the contract and at each year-end. The method for recognizing the gain or loss depends on whether the derivative is classified as a hedging instrument, and if so, the nature of the hedged asset.
For accounting purposes, they are classified as follows:
(i) Derivatives qualifying for cash flow hedge accounting
Hedging effectiveness
Hedge effectiveness is determined at the inception of the hedging relationship, and through periodic prospective effectiveness assessments to ensure that there is an economic relationship between the hedged item and the hedging instrument.
In derivatives such as the euro/Dollar cross-currency swap, the Group uses the hypothetical derivative method to assess effectiveness. This hypothetical derivative is constructed without the inclusion of credit risk and currency spread. Under the hypothetical derivative method, the cumulative change in the fair value of the actual currency swap, excluding the effect of the currency spread, will be compared to the cumulative change in the fair value of the hypothetical swap. Therefore, the hypothetical derivative is constructed as a cross-currency swap with fixed euro payment, fixed U.S. Dollar receipt without the inclusion of credit risk and foreign currency spread and with a fair value of zero at the date of designation.
Recognition
At the inception of the hedging relationship, the Group documents the economic relationship between the hedging instruments and the hedged items, including whether changes in cash flows of the hedging instruments are expected to offset changes in cash flows of the hedged items. The Group documents its risk management objective and strategy for undertaking its hedging transactions.
The effective portion of changes in the fair value of derivatives designated and classified as cash flow hedges is recognized in equity under “Cash flow hedge reserve”. In the case of cross-currency swaps, the currency spread of the hedging relationship is excluded and treated as hedging costs in equity. The gain or loss corresponding to the ineffective portion is recognized immediately in profit or loss for the year under the heading “Change in fair value of financial instruments”.
F-42
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Amounts accumulated in the hedging reserve included in shareholders’ equity are transferred to profit or loss when the hedged item affects profit or loss or when ineffectiveness is identified.
The fair value of derivatives designated as hedges is detailed in note 30. Movements in the hedging reserve included in shareholders’ equity are shown in note 17 (c).
(ii) Derivatives that do not qualify for hedge accounting
When derivatives do not meet the criteria for hedge accounting, they are classified as “held for trading”. Changes in fair value are recognized immediately in the consolidated statement of profit and loss.
(j)Own equity instruments
The acquisition of treasury stock is recorded at acquisition cost, reducing equity until the time of disposal. Gains or losses on the disposal of treasury stock are recorded under “Reserves” in the consolidated balance sheet. Transaction costs related to own equity instruments, net of taxes, are recorded as a reduction of equity.
(k)Inventories
Inventories are stated at the lower of weighted average cost or net realizable value. Net realizable value is the estimated selling price in the normal course of business, less the estimated costs to complete production and those necessary to make the sale. For raw materials and other supplies it is the replacement cost.
The cost includes direct materials, direct labor and an appropriate proportion of indirect variable and fixed costs, the latter being allocated on the basis of the normal working capacity of the means of production. The cost of plasma stocks includes the amount delivered to donors, or the amount invoiced by the seller when purchased from third parties, as well as the cost of products and devices used in the collection process, and rental and storage costs. The costs of purchased inventories are determined after deducting discounts and rebates when it is probable that the conditions determining their concession will be met. Indirect costs such as management and administrative overheads are recognized as expenses in the period in which they are incurred.
Any previously recognized inventory impairment adjustment is reversed against income under “Cost of sales” when the circumstances that caused the impairment no longer exist or when there is clear evidence of an increase in the net realizable value as a result of a change in economic circumstances. The reversal of the write-down is limited to the lower of cost and the new net realizable value of inventories.
(l)Cash and cash equivalents
Cash and cash equivalents include cash on hand, demand deposits with banks, other short-term highly liquid investments with an original maturity of three months or less that are readily convertible to known amounts of cash and which are subject to an insignificant risk of changes in value.
(m)Government grants
Government grants are recognized when there is reasonable assurance that the conditions attached to the grant will be met and that the grant will be collected.
Non-refundable capital grants are recorded on the liability side of the consolidated balance sheet at the original amount granted and are recognized in the consolidated statement of profit and loss as the related assets financed are depreciated.
F-43
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Grants received as compensation for expenses or losses already incurred or for the purpose of providing immediate financial support not related to future expenses are credited to the consolidated statement of profit and loss.
Financial liabilities that incorporate implicit aid in the form of the application of below-market interest rates are recognized initially at fair value. The difference between this value, adjusted where appropriate for the costs of issuing the financial liability and the amount received, is recorded as a government grant based on the nature of the grant.
(n)Employee benefits
(i) Defined contribution plans
The Group records the contributions to be made to defined contribution plans as they accrue. The amount of accrued contributions is recorded under “Personnel expenses” in the consolidated statement of profit and loss in the year to which the contribution relates.
(ii) Defined benefit plans
The liability recognized corresponds to the present value of the obligation at the consolidated balance sheet date less the fair value of plan assets. The defined benefit obligation is calculated annually by independent actuaries using the projected unit credit method. The present value of the obligation is determined by discounting the estimated future cash flows at interest rates of bonds denominated in the currency in which the benefits will be paid and with maturities similar to those of the related obligations. Actuarial gains and losses arising from changes in actuarial assumptions or differences between assumptions and reality are recognized in equity under “Other comprehensive income”. Past service costs are recognized in the consolidated statement of profit and loss under “Personnel expenses”.
(iii) Termination benefits
Termination benefits are recognized on the earlier of the following dates: (a) when the Group can no longer withdraw the offer or (b) when the Group recognizes costs of a restructuring within the scope of IAS 37 and this results in the payment of termination benefits.
(iv) Short-term employee benefits
The Group recognizes the expected cost of short-term compensation in the form of paid leave whose rights accrue as employees render the services that entitle them to receive it. If the leave is not accrued, the expense is recognized as the leave is taken.
The Group recognizes the expected cost of profit sharing or employee incentive plans when there is a present legal or constructive obligation as a result of past events and a reliable estimate can be made of the value of the obligation.
(v) Share-based payments
The Group has granted different remuneration plans based on equity instruments to certain members of the management team who are rendering service to the company, which will be settled with equity instruments or cash, depending on the plan.
The equity instruments granted become vested when the employees complete a certain period of service and meet the objectives established in the incentive plan. Grifols recognizes the services received from its employees as such services are rendered during the vesting period as a personnel expense in the consolidated income statement and a corresponding increase in equity if the transaction is equity-settled or a corresponding liability if the transaction is cash-settled, at an amount based on the value of the equity instruments.
F-44
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
In transactions with employees that are equity-settled, the amount recognized corresponds to the amount that will be settled once the agreed conditions are met and will not be reviewed or revalued during the vesting period, as the commitment is equity-settled. If an employee resigns from his or her position before the end of the vesting period, he or she will only receive the agreed share-based incentive. The fair value of services received is estimated by estimating the fair value of the shares granted at the grant date, net of estimated dividends to which the employee is not entitled, during the performance period.
For plans that are settled in cash, the services received and the corresponding liability are recognized at the fair value of the liability, referring to the date on which the requirements for recognition are met. Subsequently, and until settlement, the corresponding liability is measured at its fair value at the closing date of each year, with any changes in valuation occurring during the year being recognized in the consolidated income statement. The fair value is determined by reference to the market value of the shares at the date of the estimate, net of estimated dividends to which the employee is not entitled, during the performance period.
(o)Provisions
Provisions are recognized when the Group has a present legal or constructive obligation as a result of a past event, it is probable that an outflow of resources embodying economic benefits will be required to settle the obligation and a reliable estimate can be made of the amount of the obligation. Provisions are not recognized for future operating losses.
The amount of the provision corresponds to the best estimate at the closing date of the disbursements required to settle the present obligation, after taking into account the risks and uncertainties related to the provision and, when significant, the financial effect of discounting, provided that the disbursements to be made in each period can be reliably determined.
(p)Revenue recognition
Revenue from the sale of goods or services is recognized at an amount that reflects the consideration the Group expects to be entitled to receive in exchange for transferring goods or services to a customer, at the time the customer obtains control of the goods or services rendered, i.e. when the customer has the ability to direct the use of the goods or services. The consideration committed in a contract with a customer may include fixed amounts, variable amounts, or both. The amount of consideration may vary due to discounts, rebates, incentives, performance bonuses, penalties or other similar items. Contingent consideration is only included in the transaction price when it is highly probable that the amount of revenue recognized will not be subject to significant future reversals. Revenue is presented net of value added tax and any other amounts or taxes, which in substance correspond to amounts received on behalf of third parties.
(i)Sales of goods
Revenue from the sale of goods is recognized when the Group satisfies the performance obligation by transferring the committed goods to the customer. An asset is transferred when the customer obtains control of that asset. In assessing the satisfaction of the performance obligation, the Group considers the following indicators of the transfer of control, which include, but are not limited to, the following:
| ● | The Group has a present right to payment for the asset. |
| ● | The customer has the legal right to the asset |
| ● | The Group has transferred the physical possession of the asset |
| ● | Customer has the significant risks and rewards of asset ownership |
| ● | The customer has accepted the asset |
F-45
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The nature of the assets that the Group undertakes to transfer are mainly: sale of goods, sale of equipment, toll contracts, maintenance and technical service contracts, training, licenses, royalties and know-how and engineering contracts, among others.
In determining the transaction price, it is assumed that the goods and/or services are transferred in accordance with the terms of the contract. The consideration committed to a customer may include fixed amounts, variable amounts, or both. The price should be estimated taking into account the effect of variable consideration (as applicable) for returns, chargebacks/volume discounts or other incentives, provided that the same is highly probable.
The Group participates in state Medicaid programs in the United States. Provision for Medicaid rebates is recorded at the time the sale is recorded in an amount equal to the estimated Medicaid rebate claims attributable to such sale. The Group determines the estimate of the accrual for Medicaid rebates primarily based on historical Medicaid rebate experience, legal interpretations of applicable laws related to the Medicaid program and any new information regarding changes in Medicaid program guidelines and regulations that could affect the amount of the rebates. The Group considers pending Medicaid claims, Medicaid payments, and inventory levels in the distribution channel and adjusts the provision periodically to reflect actual experience. Although rebate payments typically occur with a lag of one to two quarters, adjustments for actual experience have not been material.
As is standard industry practice, certain customers have entered into contracts with the Group for purchases that are eligible for a price discount based on a minimum purchase quantity, volume discounts or cash discounts. These discounts are accounted for as a reduction in sales and accounts receivable in the same month in which the sales are invoiced based on a combination of the customer’s actual purchase data and historical experience when the customer’s actual purchase data is later known.
In the United States, the Group enters into agreements with certain customers to establish contractual prices for products, which these entities purchase from the authorized wholesaler or distributor (collectively, “wholesalers”) of their choice. Accordingly, when these entities purchase the products from the wholesalers at the contractual price which is lower than the price charged by the Group to the wholesaler, the Group provides the wholesaler with a credit known as a chargeback. The Group accounts for the accrual of chargebacks at the time of sale. The allowance account for chargebacks is based on the Group’s estimate of the wholesaler’s inventory levels and the expected direct sale of the products by the wholesalers at the contract price based on past chargeback history and other factors. The Group periodically monitors factors influencing the estimation for rebates and applies adjustments when it believes that actual rebates may differ from the established allowance accounts. These adjustments occur over a relatively short period of time. As these refunds are typically settled within
The amount at closing for the remaining discounts is settled in the following year within
(ii) Provision of services
Revenue from the rendering of services is recognized over time provided that the following criteria are met (i) the client simultaneously receives and consumes the benefits provided by Grifols’ activity as it is carried out, (ii) Grifols produces or improves an asset that the client controls as the asset is produced and (iii) Grifols produces a specific asset for the client, to which cannot give an alternative use, and has an enforceable right of collection of the activity carried out so far. If the performance obligation is fulfilled over time, income is recognized as it is satisfied considering the percentage of completion. If the performance obligation does not meet the above conditions, the following indicators are evaluated to determine that control of the asset has been transferred to the client: (i) through physical possession of the asset where Grifols has the right to demand payment for it and (ii) the client has accepted the asset, the significant risks and rewards inherent in ownership of the asset and has legal title. If the performance obligation is met on a specific date, the corresponding revenue is recognized on that date.
F-46
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(q)Income tax
The income tax expense or tax credit for the year comprises both current tax and deferred tax.
Current tax is the amount payable on the taxable income for the current year based on the applicable tax rate for each jurisdiction. It is calculated on the basis of the laws enacted or about to be enacted at the balance sheet date in the countries where subsidiaries and associates operate and generate taxable income. The Group periodically evaluates the positions taken in tax returns with respect to situations where the applicable tax regulations are subject to interpretation and considers such uncertainty in uncertain tax treatments when determining the corresponding tax gain or loss, tax bases, unused tax credits or tax rates.
Deferred taxes are recognized on temporary differences arising between the tax bases of assets and liabilities and their carrying amounts in the consolidated financial statements. It is determined using tax rates (and laws) enacted or about to be enacted at the balance sheet date that are expected to apply when the related deferred tax asset is realized or the deferred tax liability is settled.
Deferred tax liabilities and assets are recognized:
| ● | Recognition of deferred tax liabilities: |
The Group recognizes deferred tax liabilities in all cases except those which:
| o | arise from the initial recognition of goodwill or an asset or liability in a transaction that is not a business combination, on the date of the transaction it does not affect either the accounting result or the taxable base and on the date of the transaction do not give raise to taxable and deductible temporary differences for the same amount. |
| o | correspond to differences related to investments in subsidiaries, associates and joint ventures over which the Group has the ability to control the timing of their reversal and it is not probable that their reversal will occur in the foreseeable future. |
| ● | Recognition of deferred tax assets: |
The Group recognizes deferred tax assets whenever:
| o | it is probable that there will be sufficient future tax profits to offset them or when tax legislation contemplates the possibility of future conversion of deferred tax assets into a claim payable against the Public Administration. However, assets that arise from the initial recognition of assets or liabilities in a transaction that is not a business combination, on the date of the transaction do not affect either the accounting result or the taxable base and on the date of the transaction do not give raise to taxable and deductible temporary differences for the same amount, are not recognized. |
| o | they correspond to temporary differences related to investments in subsidiaries, associates and joint ventures to the extent that the temporary differences will reverse in the foreseeable future and positive future tax profits are expected to be generated to offset the differences. |
Deferred tax assets and liabilities are not recognized for temporary differences between the carrying amount and tax base of investments in foreign operations when the company is able to control the date on which the temporary differences will reverse and it is probable that the temporary differences will not reverse in the foreseeable future. Likewise, deferred tax liabilities are not recognized if they arise from the initial recognition of goodwill. Lastly, deferred tax assets are only recognized if it is probable that sufficient future taxable profit will be available against which they can be utilized.
F-47
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Deferred tax assets and liabilities are offset when there is a legally enforceable right to offset current tax assets and liabilities and when the deferred tax balances relate to the same taxation authority. Current tax assets and liabilities are offset when the entity has a legally enforceable right to offset and intends either to settle on a net basis or to realize the asset and settle the liability simultaneously.
Current or deferred income tax is recognized in profit or loss, unless it arises from a transaction or economic event that has been recognized in other comprehensive income or directly in equity. In such cases, the tax is also recognized in other comprehensive income or directly in equity, respectively.
(r)Segment reporting
An operating segment is a component of the Group that engages in business activities from which it may earn revenues and incur expenses, whose operating results are regularly reviewed by the Group’s chief operating decision maker in order to decide on the resources to be allocated to the segment, evaluate its performance and for which discrete financial information is available.
(s)Environment
The Group carries out operations whose main purpose is to prevent, reduce or repair damage to the environment as a result of its activities.
Items of property, plant and equipment acquired for the purpose of being used on a lasting basis in its activity and whose main purpose is the minimization of environmental impact and the protection and improvement of the environment, including the reduction or elimination of future pollution from the Group’s operations, are recognized as assets through the application of measurement, presentation and disclosure criteria consistent with those mentioned in note 4(e).
(t)Non-current assets held for sale
The criteria for held for sale classification is regarded as met only when the Group determines the sale to be highly probable, management is committed to a decision to sell and all actions required to complete the sale indicate that it is unlikely that significant changes to the sale will be made or that the decision will be withdrawn. These assets are measured at the lower of their carrying value and fair value less costs for its alienation. Once classified as held for sale they are no longer depreciated or amortized.
In addition, the asset or disposal group is available for immediate sale in its present condition (subject only to terms that are usual and customary for such transactions) and the sale is expected to be completed within one year from the date of the classification. In case of having some delays caused by events or circumstances outside Grifols control and there is sufficient evidence of this commitment to sell, the Group will present those assets as “Non-current assets held for sale”.
The non-current assets held for sale are presented separately in the statement of financial position as “Non-current assets and disposal groups held for sale” and “Liabilities associated with non-current assets and disposal groups held for sale” for the liabilities, if exist.
Additionally, the Group considers as discontinued operations the components (cash-generating units) which represent a separate major line of business or geographic area, that is significant and can be considered separately from the rest, which are sold or disposed in an alternative way or meet the requirements to be presented as held for sale. Likewise, it is considered as discontinued operations those entities acquired exclusively with the finality to be resold. The result after taxes of these discontinued operations are presented in a unique line in the consolidated statement of profit and loss, as “Result from discontinued operations after tax”.
F-48
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(5) Segment Reporting
In accordance with IFRS 8 “Operating Segments”, financial information for operating segments is reported in the accompanying Appendix II, which forms an integral part of this note to the consolidated financial statements.
Group companies are divided into
Assets, liabilities, income and expenses for segments include directly and reliably attributable items. Items which are not attributed to segments by the Group are:
| ● | Balance sheet: equity, cash and cash equivalents and loans and borrowings. |
| ● | Statement of profit and loss: finance result and income tax. |
(a) | Operating segments |
The operating segments defined by the Steering Committee are as follows:
| ● | Biopharma (formerly Bioscience): concentrates all activities related to products derived from human plasma for therapeutic use. |
| ● | Diagnostic: including the marketing of diagnostic testing equipment, reagents and other equipment, manufactured by Group or other companies. |
| ● | Bio Supplies: this groups together transactions related to biological products for non-therapeutic use. The part relating to sales of plasma to third parties has been reclassified from Bio Supplies to Other. |
| ● | Others: includes the provision of manufacturing services to third parties, plasma sales to third parties and research activities. It also includes pharmaceutical products manufactured by the Group and intended for hospital pharmacies, as well as the marketing of products that complement the Group’s own products. |
Details of sales by groups of products for 2023, 2022 and 2021 are as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 (*) | |
Biopharma |
|
|
| |||
Haemoderivatives |
| |
| |
| |
Diagnostic |
| |||||
Transfusional medicine |
| |
| |
| |
Other diagnostic |
| |
| |
| |
Bio supplies | | | | |||
Others |
| |
| | | |
Total |
| |
| | | |
* As a consequence of the review of transactions and balances allocations by segments made in the year 2022, the comparative figures for the fiscal year 2021 was adjusted accordingly.
F-49
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
At 31 December 2023,
As of 31 December 2023,
The Group has concluded that hemoderivative products are sufficiently alike to be considered as a whole for the following reasons:
| ● | All these products are human plasma derivatives and are manufactured in a similar way. |
| ● | The customers and methods used to distribute these products are similar. |
| ● | All these products are subject to the same regulations regarding production and the same regulatory environment. |
(b)Geographical information
Geographical information is grouped into
| ● | United States of America and Canada |
| ● | Spain |
| ● | Rest of the European Union |
| ● | Rest of the world |
The definition of these
The financial information reported for geographical areas is based on sales to third parties in these markets as well as the location of assets.
(c)Main customers
In 2023, the revenue from a customer in the Biopharma segment represented approximately
F-50
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(6) Goodwill
Details of and movement in this caption of the consolidated balance sheet at 31 December 2023 are as follows:
Thousands of Euros | ||||||||||||||||
|
|
| Balance at |
| Business |
|
|
| Translation |
| Balance at | |||||
Segment | Reference | 31/12/2022 | Combination | Impairment | Transfers | differences | 31/12/2023 | |||||||||
Net value |
|
|
|
| ||||||||||||
Grifols UK, Ltd. (UK) |
| Biopharma |
| | — | — | — | | | |||||||
Grifols Italia.S.p.A. (Italy) |
| Biopharma |
| | — | — | — | — | | |||||||
Biomat USA, Inc.(USA) |
| Biopharma |
| | — | — | — | ( | | |||||||
Grifols Australia Pty Ltd. (Australia) / Medion Diagnostics AG (Switzerland) |
| Diagnostic |
| | — | — | — | ( | | |||||||
Grifols Therapeutics, Inc. (USA) |
| Biopharma |
| | — | — | — | ( | | |||||||
Progenika Biopharma, S.A. (Spain) |
| Diagnostic |
| | — | — | — | — | | |||||||
Grifols Diagnostic (Novartis & Hologic) (USA, Spain and Hong Kong) |
| Diagnostic |
| | — | — | — | ( | | |||||||
Kiro Grifols, S.L. (Spain) | Others | | — | — | — | — | | |||||||||
Haema, AG. (Germany) | Biopharma | | — | — | — | — | | |||||||||
BPC Plasma, Inc. (USA) | Biopharma | | — | — | — | ( | | |||||||||
Plasmavita Healthcare GmbH (Germany) | Biopharma | | — | — | — | — | | |||||||||
Alkahest, Inc (USA) | Others | | — | — | — | ( | | |||||||||
Grifols Canada Therapeutics, Inc (Canada) | Biopharma | | — | — | — | ( | | |||||||||
GigaGen, Inc (USA) | Others | | — | — | — | ( | | |||||||||
Haema Plasma Kft. (Hungary) | Biopharma | Note 3 | | — | — | — | | | ||||||||
Grifols Canada Plasma, Inc. (formerly Prometic Plasma Resources, Inc.) | Biopharma | Note 3 | | | — | — | ( | | ||||||||
Grifols Biotest Holdings GmbH / Biotest AG (Germany) | Biopharma | Note 3 | | — | — | — | — | | ||||||||
Access Biologicals, LLC (USA) | Bio Supplies | Note 3 | | — | — | ( | ( | — | ||||||||
Grifols Bio Supplies Inc (USA) | Bio Supplies | — | — | — | | ( | | |||||||||
AlbaJuna Therapeutics, S.L (Spain) | Others | Note 3 | — | | ( | — | — | — | ||||||||
|
|
| | | ( | | ( | | ||||||||
(See note 3) | ||||||||||||||||
F-51
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Details of and movement in this caption of the consolidated balance sheet at 31 December 2022 were as follows:
Thousands of Euros | ||||||||||||||||
|
|
| Balance at |
| Business |
|
|
| Translation |
| Balance at | |||||
Segment | Reference | 31/12/2021 | Combination | Disposals | Transfers | differences | 31/12/2022 | |||||||||
Net value |
|
|
|
| ||||||||||||
Grifols UK.Ltd. (UK) |
| Biopharma |
| | — | — | — | ( | | |||||||
Grifols Italia.S.p.A. (Italy) |
| Biopharma |
| | — | — | — | — | | |||||||
Biomat USA, Inc.(USA) |
| Biopharma |
| | — | — | | | | |||||||
Grifols Australia Pty Ltd. (Australia) / Medion Diagnostics AG (Switzerland) |
| Diagnostic |
| | — | — | — | | | |||||||
Grifols Therapeutics, Inc. (USA) |
| Biopharma |
| | — | — | — | | | |||||||
Progenika Biopharma, S.A. (Spain) |
| Diagnostic |
| | — | — | — | — | | |||||||
Grifols Diagnostic (Novartis & Hologic) (USA, Spain and Hong Kong) |
| Diagnostic |
| | — | — | — | | | |||||||
Kiro Grifols S.L. (Spain) | Others | | — | — | — | — | | |||||||||
Goetech LLC (USA) | Others | Note 3 | | — | ( | — | | — | ||||||||
Haema AG (Germany) | Biopharma | | — | — | — | — | | |||||||||
BPC Plasma, Inc. (USA) | Biopharma | | — | — | — | | | |||||||||
Interstate Blood Bank, Inc. (USA) | Biopharma | | — | — | ( | | — | |||||||||
Plasmavita Healthcare GmbH (Germany) |
| Biopharma |
| | — | — | — | — | | |||||||
Alkahest, Inc (USA) | Others | | — | — | — | | | |||||||||
Grifols Canada Therapeutics, Inc (Canada) | Biopharma | | — | — | — | ( | | |||||||||
GigaGen, Inc (USA) | Others | | — | — | — | | | |||||||||
Grifols Canada Plasma, Inc. (formerly Prometic Plasma Resources, Inc.) | Biopharma | Note 3 | | ( | — | — | ( | | ||||||||
Haema Plasma Kft. (Hungary) | Biopharma | Note 3 | — | | — | — | ( | | ||||||||
Grifols Biotest Holdings GmbH / Biotest AG (Germany) | Biopharma | Note 3 | — | | — | — | — | | ||||||||
Access Biologicals, LLC (USA) | Bio Supplies | Note 3 | — | | — | — | ( | | ||||||||
| | ( | — | | | |||||||||||
(See note 3) | ||||||||||||||||
Impairment testing:
CGUs correspond to the reporting segments except for the Others segment which corresponds to Kiro Grifols, Alkahest and GigaGen as separated CGUs.
As a result of the acquisition of Talecris in 2011, and for impairment testing purposes, the Group combines the CGUs allocated to the Biopharma segment, grouping them together at segment level, because substantial synergies were expected to arise on the acquisition of Talecris, and due to the vertical integration of the business and the lack of an independent organized market for the products. Because the synergies benefit the Biopharma segment globally they cannot be allocated to individual CGUs. The Biopharma segment represents the lowest level to which goodwill is allocated and is subject to control by Group management for internal control purposes.
As a result of the acquisition of Novartis’ Diagnostic business unit in 2014, the Group decided to combine Araclon, Progenika, Australia and Hologic’s share of NAT donor screening unit acquisition into a single CGU for the Diagnostic business as the acquisition is supporting not only the vertically integration business but also cross-selling opportunities. In addition, for management purposes, the Group’s management is focused on the business more than geographical areas or individual companies.
F-52
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The Hospital division is no longer a reportable segment since it does not meet any of the quantitative thresholds described in IFRS 8 Operating Segments. The segment information included in the Hospital CGU in previous years is currently grouped into an Others segment.
In addition, due to the acquisition of the remaining
The CGUs established by Grifols management are:
| ● | Biopharma |
| ● | Diagnostic |
| ● | Bio Supplies |
| ● | Kiro Grifols |
| ● | GigaGen |
| ● | Alkahest |
Alkahest’s goodwill was generated as a counterpart to the deferred tax liability corresponding to the intangible assets recognized as a result of the allocation of the excess purchase price over the acquired net assets.
The recoverable amount of the Biopharma CGU and Bio Supplies CGU has been calculated based on its value in use calculated as the present value of the five-year future cash flows discounted at a discount rate considering the related inherent risk.
The recoverable amount of the Diagnostic CGU has been calculated based on its fair value less costs to sell calculated as the present value of future cash flows approved by Management discounted at a discount rate considering the inherent risk. Due to the reorganization to boost the business units, a long-term strategic plan was approved in order to transform the Diagnostic business unit by investments which will lead to a beyond five-year growth. Consequently, management has estimated future cash flows for the period 2024-2034.
The recoverable amount of the Kiro Grifols CGU has been calculated based on its fair value less costs to sell calculated as the present value of the five-year cash flows discounted at a discount rate considering the related inherent risk.
For the calculation of the recoverable amount, management has considered:
| ● | Gross margin based on historical performance and actual situation |
| ● | Development prospects in the international market |
| ● | Current investments |
| ● | Investments which will imply a significant growth of the production capacity for those cases whose fair value has been considered |
Cash flows estimated as of the year in which stable growth in the CGU has been reached are extrapolated using the estimated growth rates indicated below. Perpetual growth rates are consistent with the forecasts included in industry reports.
The recoverable amount of the GigaGen CGU has been determined based on the fair value less costs to sell, calculated as the present value of the future cash flows mainly of a research and development project that have been approved by management, adjusted by the probability of success and discounted at a discount rate that includes their inherent risk. Cash flows have been estimated taking into consideration a useful life of 20 years from the product launch and their reduction as of the sixth year.
F-53
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The key assumptions used in calculating impairment testing of the CGUs for 2023 have been as follows:
| Perpetual Growth rate |
| Pre-tax discount rate |
| |
Biopharma |
| | % | | % |
Diagnostic |
| | % | | % |
Bio Supplies | | % | | % | |
Kiro Grifols | | % | | % | |
GigaGen | N/A | | % |
Additionally, the following key assumptions have been used for the GigaGen CGU impairment testing:
| Sink rate |
| Success rate |
| |
GigaGen |
| | % | | % |
Likewise, for the impairment test of the Diagnostic CGU, the sales of Nucleic Acid Test (NAT), Blood Typing Solution (BTS) and those of the Clinical Diagnostic have been considered as key assumptions.
The discount rate used reflects specific risks relating to the CGUs and the countries in which they operate. The main assumptions used for determining the discount rate are as follows:
| ● | Risk free rate: normalized government bonds at |
| ● | Market risk premium: premium based on market research |
| ● | Unlevered beta: average market beta |
| ● | Debt to equity ratio: average market ratio |
The key assumptions used in calculating impairment testing of the CGUs for 2022 were as follows:
| Perpetual Growth rate |
| Pre-tax discount rate | |
Biopharma |
|
| ||
Diagnostic |
|
| ||
Bio Supplies |
|
| ||
Kiro Grifols |
|
| ||
GigaGen |
| N/A |
|
Likewise, for the impairment test of the Diagnostic CGU, the sales of Blood Typing Solution (BTS) and those of the Clinical Diagnostic were considered as key assumptions.
Additionally, the following key assumptions were used for the GigaGen CGU impairment testing:
| Sink rate |
| Success rate |
| |
GigaGen |
| | % | | % |
F-54
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
In 2023, and according to the current economic context, the reasonably possible changes considered for the CGUs impairment testing are a variation in the discount rate, as well as in the estimated perpetual growth rate, with independent movements of each other, as follows:
| Perpetual Growth rate |
| Pre-tax discount rate | |
Biopharma |
| +/- |
| +/- |
Diagnostic | +/- | +/- | ||
Bio Supplies | +/- | +/- | ||
Kiro Grifols | +/- | +/- | ||
GigaGen |
| N/A |
| +/- |
Additionally, for the impairment test of the Diagnostic CGU, the following sensitivity scenarios to variations in sales of the NAT, BTS and CDx business lines have also been considered:
| ● | NAT sales sensitivity scenario: a lower sales projection than initially projected has been estimated by approximately |
| ● | BTS sales sensitivity scenario: a lower sales projection than initially projected has been estimated by approximately |
| ● | CDx sales sensitivity scenario: a projection has been estimated so that CDx sales from 2030 onwards represent on average approximately |
| ● | Aggregate sensitivity scenario to NAT, BTS and CDx sales: a scenario has been estimated as a result of the previous sensitivity scenarios. |
In addition, the following reasonably possible change has been considered for the GigaGen CGU impairment testing:
| Sink rate | |
GigaGen |
| +/- |
The reasonably possible changes in key assumptions considered by management in the calculation of the recoverable amount of the Biopharma, Bio Supplies, Kiro Grifols and GigaGen CGU’s would not cause the carrying amount to exceed its recoverable amount.
The reasonably possible changes in key assumptions considered by management in the calculation of the Diagnostic CGU recoverable amount would cause the carrying amount to exceed its recoverable amount as follows:
| % Asset Value |
| |
Aggregate sensitivity scenario to NAT, BTS and CDx sales |
| ( | % |
Detail of the assets by segment value is shown in Annex II.
In 2022, the reasonably possible changes considered for the CGUs impairment testing were a variation in the discount rate, as well as in the estimated perpetual growth rate, with independent movements of each other, as follows:
| Perpetual Growth rate |
| Pre-tax discount rate | |
Biopharma |
| +/- |
| +/- |
Diagnostic |
| +/- |
| +/- |
Bio Supplies |
| +/- |
| +/- |
Kiro Grifols |
| +/- |
| +/- |
GigaGen |
| N/A |
| +/- |
F-55
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Additionally, for the impairment test of the Diagnostic CGU, two scenarios of sensitivity to variations in the sales of the Blood Typing Solutions (BTS) business line and the Clinical Diagnostics (CDx) business line were also considered. In the first case, sales projections were estimated to be approximately
In addition, the following reasonably possible change was considered for the GigaGen CGU impairment testing:
| Sink rate | |
GigaGen |
| +/- |
At 31 December 2023 Grifols’ stock market capitalization totals Euros
(7) Other Intangible Assets
Details of other intangible assets and movement during the years ended 31 December 2023 and 2022 are included in Appendix III, which forms an integral part of these notes to the consolidated financial statements.
Intangible assets acquired from Talecris mainly include currently marketed products. Identifiable intangible assets correspond to Gamunex and have been recognized at fair value at the acquisition date of Talecris and classified as currently marketed products. Intangible assets recognized comprise the rights on the Gamunex product, its commercialization and distribution license, trademark, as well as relations with hospitals. Each of these components is closely linked and fully complementary, are subject to similar risks and have a similar regulatory approval process.
Intangible assets acquired from Progenika mainly include currently marketed products. Identifiable intangible assets correspond to blood, immunology and cardiovascular genotyping. These assets have been recognized at fair value at the acquisition date of Progenika and classified as currently marketed products.
The intangible assets acquired from Biotest mainly include the acquired product portfolio. The identifiable intangible assets correspond to the plasma therapies segment and have been recorded at fair value at the date of acquisition of Biotest and classified as an acquired product portfolio.
The intangible assets acquired from Access Biologicals mainly include customer relationships. This asset has been recorded at fair value at the date of acquisition of Access Biologicals and classified as acquired customer relationships.
F-56
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The cost and accumulated amortization of currently marketed products and customer relationships acquired from Talecris,Progenika, Biotest and Access at 31 December 2023 was as follows:
Thousands of Euros | ||||||||
| Balance at |
|
| Translation |
| Balance at | ||
31/12/2022 | Additions | differences | 31/12/2023 | |||||
Cost of currently marketed products - Gamunex |
| | — |
| ( | | ||
Cost of currently marketed products - Progenika |
| |
| — |
| — |
| |
Cost of currently marketed products - Biotest | | — | — | | ||||
Cost of customer relationships - Access | | — | ( | | ||||
Accumulated amortisation of currently marketed products - Gamunex |
| ( |
| ( |
| |
| ( |
Accumulated amortisation of currently marketed products - Progenika |
| ( |
| ( |
| — |
| ( |
Accumulated amortisation of currently marketed products - Biotest | ( | ( | — | ( | ||||
Accumulated amortisation of customer relationships - Access | ( | ( | | ( | ||||
Carrying amount of currently marketed products and customer relationships |
| |
| ( |
| ( |
| |
The cost and accumulated amortization of currently marketed products and customer relationships acquired from Talecris, Progenika, Biotest and Acces at 31 December 2022 was as follows:
Thousands of Euros | ||||||||||
| Balance at |
| Business |
|
| Translation |
| Balance at | ||
31/12/2021 | Combination | Additions | differences | 31/12/2022 | ||||||
Cost of currently marketed products - Gamunex |
| | — |
| — | | | |||
Cost of currently marketed products - Progenika |
| |
| — |
| — |
| — |
| |
Cost of currently marketed products - Biotest | — | | — | ( | | |||||
Cost of customer relationships - Access | — | | — | — | | |||||
Accumulated amortisation of currently marketed products - Gamunex | ( | — | ( | ( | ( | |||||
Accumulated amortisation of currently marketed products - Progenika |
| ( |
| — | ( |
| — |
| ( | |
Accumulated amortisation of currently marketed products - Biotest |
| — |
| — |
| ( |
| — |
| ( |
Accumulated amortisation of customer relationships - Access | — | — | ( | | ( | |||||
Carrying amount of currently marketed products and customer relationships |
| |
| |
| ( |
| |
| |
The estimated useful life of the currently marketed products acquired from Talecris is considered limited, has been estimated at
At 31 December 2023 the residual useful life of currently marketed products is
F-57
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The estimated useful life of the currently marketed products acquired from Progenika is considered limited, has been estimated at
The estimated useful life of the product portfolio acquired from Biotest is considered limited and has been estimated at
The estimated useful life of the customer relationships acquired from Access Biologicals is considered limited and has been estimated at
(a) | Self — constructed intangible assets |
At 31 December 2023 the Group has recognized Euros
(b) | Purchase commitments |
At 31 December 2023 the Group has
(c) | Other intangibles in progress |
At 31 December 2023 the Group has an amount of Euros
(d) | Results on disposal of intangible assets |
The total losses on disposals and sale of intangible assets amounts to Euros
(e) | Impairment testing |
Indefinite-lived intangible assets have been allocated to the corresponding cash-generating unit (CGU). These assets have been tested for impairment together with goodwill (see note 6).
Impairment testing has been analyzed for each of the intangible assets in progress by calculating its recoverable amount based on their fair value based on the discount of free cash flows adjusted by the probability of success according to the clinical phase of the project.
F-58
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(8) Leases
Details of leases in the consolidated balance sheet at 31 December 2023 and 2022 are as follows:
Right-of-use assets |
| Thousands of Euros | ||
| 31/12/2023 |
| 31/12/2022 | |
Land and buildings |
| | | |
Machinery |
| | | |
Computer equipment |
| | | |
Vehicles |
| | | |
| | | ||
Lease liabilities |
| Thousands of Euros | ||||
| Reference | 31/12/2023 |
| 31/12/2022 | ||
Non-current |
| Note 21 | | | ||
Current |
| Note 21 | | | ||
| | | ||||
The composition of lease liabilities as of 31 December 2023 and 2023 is shown below. Undiscounted future payments classified on a maturity basis are presented together with the effect of the financial discount:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Maturity: |
|
|
|
|
Within one year |
| |
| |
In the second year |
| |
| |
In the third to fifth years |
| |
| |
After the fifth year |
| |
| |
| |
| | |
Discounting effect |
| |
| |
Total lease liabilities |
| |
| |
Details by maturity of lease liabilities are shown under “Liquidity risk” in note 30.
At 31 December 2023, the Group has recognized an amount of Euros
F-59
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
At 31 December 2023 and 2022, the amounts recognized in the consolidated statement of profit and loss related to lease agreements are:
Right-of-use depreciation |
| Thousands of Euros | ||
| 31/12/2023 |
| 31/12/2022 | |
Buildings |
| | | |
Machinery |
| | | |
Computer equipment |
| | | |
Vehicles |
| | | |
| | | ||
| Thousands of Euros | |||||
| Reference |
| 31/12/2023 |
| 31/12/2022 | |
Finance lease expenses |
| Note 27 | | | ||
| | | ||||
| Thousands of Euros | |||
| 31/12/2023 |
| 31/12/2022 | |
Expenses related to short-term contracts |
| | | |
Expenses related to low-value contracts | | | ||
Other operating lease expenses |
| | | |
| | | ||
At 31 December 2023, the Group has paid a total of Euros
The total amount recognized in the balance sheet corresponds to lease contracts in which the Group is the lessee.
(9) Property, Plant and Equipment
Details of property, plant and equipment and movement in the consolidated balance sheet at 31 December 2023 and 2023 are included in Appendix V, which forms an integral part of this note to the consolidated financial statements.
Property, plant and development under construction at 31 December 2023 and 2022 mainly comprise investments made to extend the companies’ equipment and to increase their productive capacity.
In 2023, the Group has capitalized interests for a total amount of Euros
a) | Insurance |
Group policy is to contract sufficient insurance coverage for the risk of damage to property, plant and equipment. At 31 December 2023 the Group has a combined insurance policy for all Group companies, which more than adequately covers the carrying amount of all the Group’s assets.
b) | Losses on disposal of property, plant and equipment |
Total losses incurred on disposals of property, plant and equipment for 2023 amount to Euros
F-60
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
c) | Self - constructed property, plant and equipment |
At 31 December 2023 the Group has recognized Euros
d) | Purchase commitments |
At 31 December 2023 the Group has property, plant and equipment purchase commitments amounting to Euros
e) | Fixed assets under construction |
The fixed assets under construction as of 31 December 2023 amount to Euros
f) | Impairment testing |
During 2023, the Group disposed of property, plant and equipment as part of the reorganization of the USA donor center network. In this regard, the impairment corresponding to these assets which belong to the Biopharma segment have been written off for a total amount of Euros
As a result of the reorganization of the USA donor center network, an impairment for some property, plant and equipment allocated to the relocated donor centers was recognized for a total amount of Euros
Tangible assets have been assigned to the corresponding cash-generating unit (CGU) and their impairment has been analyzed jointly with the impairment of goodwill (see note 6).
g) | Transfers |
At 31 December 2022, transfers included the reclassification of Euros
F-61
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(10) Equity-Accounted Investees and Joint Business
Details of this caption in the consolidated balance sheet at 31 December 2023 and 2022 are as follows:
|
| Thousands of Euros |
|
| Thousands of Euros | |||
% ownership | 31/12/2023 | % ownership | 31/12/2022 | |||||
Shanghai RAAS Blood Products Co., Ltd. |
| | % | |
| | % | |
Grifols Egypt Plasma Derivatives |
| | % | |
| | % | |
BioDarou P.J.S. Co. | | % | | | % | | ||
Total equity accounted investees with similar activity to that of the Group |
| |
| | ||||
Albajuna Therapeutics, S.L |
| | % | — |
| | % | |
Mecwins, S.A. | | % | | | % | | ||
Total of the rest of equity accounted investees | | | ||||||
Total equity-accounted investees | | |
Movement in the investments in equity-accounted investees for the year ended 31 December 2023 is as follows:
Thousands of Euros | ||||||||||||||||
2023 | ||||||||||||||||
Equity accounted investees with similar activity to that of the Group | Rest of equity accounted investees | |||||||||||||||
Shanghai | ||||||||||||||||
RAAS Blood | Grifols Egypt | Albajuna | ||||||||||||||
Products Co., | Plasma | BioDarou P.J.S. | Therapeutics, | Mecwins, | ||||||||||||
| Ltd. |
| Derivatives | Co. | Total |
| S.L |
| S.A. |
| Total |
| Total | |||
Balance at 1 January |
| |
| | | |
| |
| |
| |
| | ||
Acquisitions |
| — |
| | — | |
| — |
| — |
| — |
| | ||
Transfers |
| — |
| — | — | — |
| — |
| — |
| — |
| — | ||
Share of profit / (losses) |
| |
| ( | | |
| ( |
| ( |
| ( |
| | ||
Share of other comprehensive income / translation differences |
| ( |
| ( | | ( |
| |
| — |
| |
| ( | ||
Collected dividends |
| ( |
| — | — | ( |
| — |
| — |
| — |
| ( | ||
Uncollected dividends | — | — | ( | ( | — | — | — | ( | ||||||||
Transfers | ( | — | — | ( | — | — | — | ( | ||||||||
Balance at 31 December |
| |
| | | |
| — |
| |
| |
| | ||
F-62
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Movement in the investments in equity-accounted investees for the year ended 31 December 2022 is as follows:
| Thousands of Euros | |||||||||||||||||
2022 | ||||||||||||||||||
Equity accounted investees with similar activity to that of the Group | Rest of equity accounted investees | |||||||||||||||||
Shanghai RAAS | Grifols Egypt | Albajuna | ||||||||||||||||
Access | Blood Products | Plasma | BioDarou | Therapeutics, | ||||||||||||||
| Biologicals LLC |
| Co., Ltd. |
| Derivatives |
| P.J.S. Co. |
| Total |
| S.L |
| Mecwins, S.A. |
| Total |
| Total | |
Balance at 1 January |
| |
| |
| |
| — | |
| |
| |
| |
| | |
Acquisitions |
| — |
| — |
| — |
| | |
| — |
| — |
| — |
| | |
Transfers |
| ( |
| — |
| — |
| — | ( |
| — |
| — |
| — |
| ( | |
Share of profit / (losses) |
| |
| |
| |
| ( | |
| ( |
| ( |
| ( |
| | |
Share of other comprehensive income / translation differences |
| |
| ( |
| ( |
| | ( |
| — |
| — |
| — |
| ( | |
Collected dividends |
| ( |
| ( |
| — |
| — | ( |
| — |
| — |
| — |
| ( | |
Others | — | — | | — | | — | — | — | | |||||||||
Balance at 31 December |
| — |
| |
| |
| | |
| |
| |
| |
| | |
Movement in the investments in equity-accounted investees for the year ended 31 December 2021 is as follows:
Thousands of Euros | ||||||||||||||||||
2021 | ||||||||||||||||||
Equity accounted investees with similar activity to that of the Group | Rest of equity accounted investees | |||||||||||||||||
Access | Shanghai RAAS | Grifols Egypt | Albajuna | |||||||||||||||
Biologicals | Blood Products | Plasma | Therapeutics, | |||||||||||||||
| LLC |
| Co., Ltd. |
| Derivatives |
| Total |
| S.L |
| GigaGen, Inc. |
| Mecwins, S.A. |
| Total |
| Total | |
Balance at 1 January | | | — | | | | | | | |||||||||
Acquisitions |
| — |
| — |
| |
| |
| — |
| — |
| |
| |
| |
Transfers |
| — |
| — |
| — |
| — |
| — |
| ( |
| — |
| ( |
| ( |
Share of profit / (losses) |
| |
| |
| ( |
| |
| ( |
| |
| ( |
| |
| |
Share of other comprehensive income / translation differences |
| |
| |
| |
| |
| ( |
| |
| — |
| |
| |
Collected dividends |
| ( |
| ( |
| — |
| ( |
| — |
| — |
| — |
| — |
| ( |
Balance at 31 December |
| |
| |
| |
| |
| |
| — |
| |
| |
| |
The main movements of the equity-accounted investees with similar activity to that of the Group are explained below:
Grifols Egypt for Plasma Derivatives (S.A.E.)
On 29 July 2021, a cooperation agreement was signed with the National Service Projects Organization (NSPO) to help build a platform to bring self-sufficiency in plasma-derived medicines to Egypt. The Company made a first contribution of US Dollars
Shanghai RAAS Blood Products Co. Ltd.
In March 2019, Grifols entered into a share exchange agreement with Shanghai RAAS Blood Products Co. Ltd. (hereinafter SRAAS), through which Grifols would deliver
After receiving all relevant authorizations, at 31 December 2019, Grifols delivered
F-63
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
On 30 March 2020, the share exchange agreement was closed and Grifols received SRAAS shares corresponding to
Consequently, the consolidated balance sheet at 31 December 2020, did not longer show any financial asset related to the contractual right, but the interest in SRAAS was recorded as an investment in an associate company because the Group exercises significant influence in accordance with the criteria established in IAS 28 – Investment in Associates and Joint Ventures. SRAAS’ equity-accounted investment was recognized at the value of the shares at the closing date of the transaction. The difference between the contractual right value recognized at 31 December 2019 and SRAAS quoted value at 30 March 2020 was Euros
The impact on the consolidated statement of profit and loss related to the equity method result was included in the Operating Result under “Profit/(loss) of equity accounted investees with similar activity to that of the Group”, since SRAAS is a company dedicated to the plasma product sector.
The transaction costs were recognized as part of the investment value and totaled Euros
On 29 December 2023, Grifols announced a Strategic Alliance and Share Purchase Agreement with Haier Group Corporation (Haier) for the sale of approximately a
According to the fair value implicit in the transaction with Haier, there is no impairment indication in SRAAS investment as of December 31, 2023. At 31 December 2023 Shanghai RAAS Blood Products Co. Ltd. stock market capitalization totals RMB
| Agreed price in transaction with Haier |
| 31/12/2023 |
| Date of acquisition | |
SRAAS shares price |
| CNY |
| CNY |
| CNY |
As of 31 December 2022, the recoverable value of the investment in SRAAS was determined in accordance with its value in use, calculated as the present value of future cash flows discounted at a discount rate that reflects the inherent risk thereof.
The key assumptions used to perform the impairment test of the investment in SRAAS for 2022 were as follows:
| Perpetual Growth rate |
| Pre-tax discount rate |
| |
SRAAS |
| | % | | % |
The reasonably possible changes considered for SRAAS were a variation in the discount rate, as well as in the estimated perpetual growth rate, according to the following detail:
| Perpetual Growth rate |
| Pre-tax discount rate |
| |
SRAAS |
| +/- |
| +/- |
Due to the aforementioned Share Purchase Agreement with Haier Group Corporation, as of December 31, 2023, the amount equivalent to
Access Biologicals LLC.
On 12 January 2017, the group announced the acquisition of
F-64
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The principal business activity of Access Biologicals is the collection and manufacturing of an extensive portfolio of biological products. Combined with closed-loop material sourcing, it provides critical support for various markets such as in-vitro diagnostic manufacturing, biopharmaceutical, cell culture and diagnostic research & development.
On 15 June 2022, Grifols, through its wholly-owned subsidiary Chiquito Acquisition Corp., reached an agreement to acquire all the shares of Access Biologicals LLC, exercising the call option for the remaining
BioDarou P.J.S. Co.
On 25 April 2022, and after obtaining all regulatory approvals, Grifols closed the acquisition of
The company’s goal is to collect plasma, process it into immunoglobulins, factors and human albumin through Biotest AG and then sell the finished products in Iran.
The main movements for the rest of the equity-accounted investees are explained below:
Albajuna Therapeutics, S.L.
In 2016, Grifols made a capital investment of
On 9 October, 2023, Grifols, through its
Medcom Advance, S.A.
In February 2019, the Group completed the acquisition of
Mecwins, S.A.
On 22 October 2018 Grifols allocated Euros
Mecwins is a spin-off of the Institute of Micro and Nanotechnology of the Center for Scientific Research (CSIC), specialized in the development of innovative nanotechnological analysis tools for the diagnosis and prognosis of diseases.
F-65
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Mecwins has developed ultrasensitive optical reading immunoassay technology from nanosensors for the detection of protein biomarkers in blood. This technology has potential applications in fields such as oncology, cardiovascular and infectious diseases.
The injection of capital, in which CRB Inverbio also participated with an additional Euros
In 2021, Mecwins, S.A. acquired own shares from Progenika Biopharma, S.A. to generate treasury stock. This acquisition caused the percentage of ownership in Mecwins, S.A. to decrease to
GigaGen Inc.
On 5 July 2017, Grifols through its
GIANT and GigaGen entered into a Research and Collaboration Agreement whereby in exchange of a collaboration fee of US Dollars
On 8 March 2021, Grifols, through its wholly owned subsidiary Grifols Innovation and New Technologies Limited (“GIANT”), reached an agreement to acquire all of the shares of Gigagen, Inc. for a total amount of US Dollars
The most recent financial statements available of the main equity-accounted investments of Grifols are as follows:
Balance sheet:
Thousands of Euros | ||||
31/12/2023 | 31/12/2022 | |||
| SRAAS |
| SRAAS | |
Non-current assets | ||||
Current assets |
| |
| |
Cash and cash equivalents |
| |
| |
Non-current liabilities |
| ( |
| ( |
Non-current financial liabilities |
| ( |
| ( |
Current liabilities |
| ( |
| ( |
Net assets |
| |
| |
P&L:
Thousands of Euros | ||||
31/12/2023 | 31/12/2022 | |||
| SRAAS |
| SRAAS | |
Net revenue | ||||
Profit for the year |
|
| ||
F-66
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Joint arrangement
Biotek America LLC
In July 2021, Grifols signed a collaboration agreement with ImmunoTek GH, LLC (ImmunoTek), an operator with recognized experience in the market, for the opening and management of 21 plasma collection centers. This agreement was subsequently amended to increase the number of centres to 28 plasma centres. The transaction was instrumentalized through the creation of a transparent company for tax purposes in the United States, Biotek America LLC (“ITK JV”), which created a series of shares for each center. Grifols holds
During the 2023 financial year, the Group and Immunotek signed an amendment to the initial agreement. As of 31 December 2023 and 2022, this collaboration agreement has involved:
| ● | The construction, licensing, and commissioning by ImmunoTek of a total of |
| ● | The sale of each center to Grifols approximately 3 years after its opening, for an approximate amount of US Dollars |
| ● | Grifols made advances of up to US Dollars |
| ● | All of the plasma collected by ITK JV through the |
| ● | ImmunoTek exclusively holds the management of the centers in exchange for a fee for its management that amounted to Euros |
| ● | As a manager, you can carry out all the acts you deem necessary under your sole and sole responsibility, but always within the activities agreed by the parties. It can only be terminated with the unanimous consent of the parties. However, the manager does not act with a delegated power, insofar as it has exposure for management fees and the achievement of objectives to maximise the selling price of each of the series. |
| ● | In the event of liquidation of ITK JV, once the creditors of ITK JV or each of the series have been paid, the advances contributed by the unitholders must then be returned, in this case, the advances contributed by the Grifols Group and the remainder, if any, will be distributed to each of the shareholders in proportion to their participation in the share capital (Immunotek |
| ● | None of the series should be responsible for expenses incurred or attributed to the other series. All profit, loss, income and expense items will be allocated to ImmunoTek, including any tax benefits derived therefrom. However, all assets and liabilities correspond to each of the series. Therefore, each of the series has a separate legal personality, with assets and liabilities isolated from the rest, i.e. each series is a SILO. |
| ● | Grifols, through Grifols Shared Services North America, Inc. acts as guarantor of |
F-67
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
As of 31 December 2023, the Group has made advance payments for the acquisition of the
| Thousand | |||
Year | US Dollar |
| Euros | |
2024 |
| |
| |
2025 |
| |
| |
2026 |
| |
| |
Total |
| |
| |
Regardless of whether Grifols holds a
Therefore, to the extent that there is joint control and each series is representative of a SILO and has been designed and created to sell all the plasma collected to Grifols and advances the necessary funds for the development of the series and guarantees the obligations, they should be considered joint agreements.
However, there is a disproportion between Grifols’ percentage stake in the series, which amounts to
In accordance with the above, up to 2022, Grifols has recognized the participation in the series applying the participation method and, to the extent that the result attributed to Grifols is zero, such participation has been valued at a zero amount. On the other hand, it has recognized as an asset the advances granted to Immunotek for the development of the centers. The advances granted will be deducted from the purchase price agreed by the centres, so they will be cancelled with the acquisition of each of the series.
As indicated, the transaction has been accounted for as a forward contract to acquire a business and, therefore, there is a derivative financial instrument that does not meet the requirement for exclusion under IFRS 9.
Since the strike price of the call options as well as the forward contract have been established considering a price per liter of market, the value of the options is not relevant. In addition, although a forward contract implies a term obligation, that fact does not imply recognition of the contractual obligation to acquire an asset or business, to the extent that it is not controlled in accordance with IAS 32.
Notwithstanding the above, in 2023 the series has been integrated in accordance with IFRS 11 Joint Agreements, with the aim of improving transparency. The integration was carried out prospectively from 1 January 2023, recognising an adjustment in reserves amounting to Euros
F-68
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Below is a breakdown of the aggregate balances of the
Thousands of | ||||||||
US Dollars | Euros | |||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2023 |
| 31/12/2022 | |
Non-current assets |
| |
| |
| |
| |
Current assets |
| |
| |
| |
| |
Total assets |
| |
| |
| |
| |
Non-current liabilities |
| |
| |
| |
| |
Current liabilities |
| |
| |
| |
| |
Total liabilities |
| |
| |
| |
| |
Total Equity |
| ( |
| ( |
| ( |
| ( |
Thousands of | ||||||||
US Dollars | Euros | |||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2023 |
| 31/12/2022 | |
Net revenue |
| |
| |
| |
| |
Profit for the year |
| ( |
| ( |
| ( |
| ( |
(11) Financial Assets
Details of non-current financial assets on the consolidated balance sheet at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||||
| Reference |
| 31/12/2023 |
| 31/12/2022 | |
Other non-current investments |
| |
| | ||
Non-current derivatives | Note 30 | | | |||
Total Non-current financial assets measured at fair value | | | ||||
Non-current guarantee deposits |
| |
| | ||
Other non-current financial assets |
| (a) | |
| | |
Non-current loans to third parties | (b) | | | |||
Total Non-current financial assets measured at amortized cost |
| |
| | ||
In Non-current guarantee deposits, there are long-term deposits with related parties that amount
F-69
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Details of current financial assets on the consolidated balance sheet at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||||
| Reference |
| 31/12/2023 |
| 31/12/2022 | |
Current derivatives | Note 31 | | | |||
Total Non-current financial assets measured at fair value |
| |
| | ||
Thousands of Euros | ||||||
| Reference |
| 31/12/2023 |
| 31/12/2022 | |
Deposits and guarantees |
| |
| | ||
Other current financial assets | (a) | | | |||
Current loans to third parties |
| (b) | |
| | |
Total other current financial assets measured at amortized cost |
| |
| | ||
(a) | Other non-current and current financial assets |
Details of other non-current and current financial assets are as follows:
Thousands of Euros | ||||||
| Reference |
| 31/12/2023 | 31/12/2022 | ||
Other financial assets with related parties |
| Note 31 | |
| | |
Other financial assets with associated parties | Note 31 | | — | |||
Other financial assets with third parties |
| |
| | ||
Total other non-current and current financial assets |
| |
| | ||
Other financial assets with related parties includes the open balance of the cash pooling that Haema AG and BPC Plasma, Inc. have with Scranton Plasma B.V. (see note 31). Those balances have been reclassified from non-current to curent based on their maturity. In 2023, the balance was significantly reduced because BPC Plasma Inc. distributed a dividend without cash outflow compensating “other non-current financial assets”. The dividend corresponds to the result of the previous 4 years for a value of Euros
| (b) | Non-current and current loans |
Details of non-current and current loans are as follows:
Thousands of Euros | ||||||
Reference | 31/12/2023 | 31/12/2022 | ||||
Loans to related parties |
| Nota 31 |
| |
| |
Loans to third parties |
|
|
| |
| |
Total current and non-current loans |
|
|
| |
| |
(12) Non-current assets held for sale
On 29 December 2023, Grifols reached an agreement with Haier Group Corporation (“Haier”) for the sale of a
The closing of the transaction is subject to the relevant regulatory approvals and confirmatory due diligence by the buyer. Both parties estimate that the closing of the transaction will occur in June 2024, although it could be postponed if any regulatory approvals are pending at that date.
F-70
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
As part of the agreement with Haier, the parties have agreed that Grifols will retain a director on the Board of Directors of SRAAS. Grifols and SRAAS will amend the Exclusive Distribution Agreement with SRAAS to supply increased quantities of human serum Albumin in the Chinese market, to extend its current term for an initial period of
| • | achieve an aggregate EBITDA in Grifols Diagnostic Solutions of US Dollar |
| • | distribute |
| • | Under the voting proxy agreement, the Group will cede the exercise of voting rights relating to the |
With this transaction, Grifols will mainatin its presence in China, will continue with its commercial agreements with SRAAS, and at the same time, will fulfill its commitment to deleverage.
At December 31, 2023, the amount equivalent to
(13) Inventories
Details of inventories at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Goods for resale |
| |
| |
Raw materials and supplies |
| |
| |
Work in progress and semi-finished goods |
| |
| |
Finished goods |
| |
| |
| |
| | |
Movement in the inventory provision was as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Balance at 1 January |
| |
| |
| |
Net charge for the year |
| |
| ( |
| |
Cancellations for the year |
| ( |
| ( |
| ( |
Translation differences |
| ( |
| |
| |
Balance at 31 December |
| |
| |
| |
F-71
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
As a result of the discontinuation of the Blood Collection Systems activity, an impairment of some inventory was recognized for a total amount of Euros
The cost of inventory amounts to Euros
(14) Contract assets
Contract assets from contract fractionation relate to contractual obligations from contract fractionation agreements entered into by Biotest AG. The resulting performance obligations are generally fulfilled by Biotest over a period of up to
Details of contract assets at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2023 | |
Contract assets (gross) |
| |
| |
Allowances for expected credit losses |
| ( |
| ( |
Contract assets (net) |
| |
| |
Default risks are accounted for by making value adjustments to the contract assets. The allowance for expected credit losses is calculated as the difference between the nominal amount of the contract assets and the estimated recoverable amount.
Movement in allowance for expected credit losses corresponding to contract assets is included in note 30.
(15) Trade and Other Receivables
Details at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||||
| Reference |
| 31/12/2023 |
| 31/12/2022 | |
Trade receivables |
| |
| | ||
Receivables from associates |
| Note 31 | |
| | |
Impairment losses |
| Note 30 (i) | ( |
| ( | |
Trade receivables |
| |
| | ||
Other receivables |
| Note 30 (i) | |
| | |
Personnel |
| |
| | ||
Advance payments |
| Note 30 (i) | |
| | |
Taxation authorities, VAT recoverable |
| |
| | ||
Other public entities |
| |
| | ||
Other receivables |
| |
| | ||
Current income tax assets |
| |
| | ||
Total trade and other receivables |
| |
| | ||
F-72
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
At 31 December 2022, Advance payments included prepayments to Biotek America, LLC (ImmunoTek) for a total amount of
‘Assignment of credit rights
During 2023, 2022 and 2021 the Grifols Group has sold receivables without recourse to some financial institutions (factors), to which the risks and benefits inherent to the ownership of the assigned credits are substantially transferred. Also, the control over the assigned credits, understood as the factor’s ability to sell them to an unrelated third party, unilaterally and without restrictions, has been transferred to the factor.
The main conditions of these contracts include the advanced collection of the assigned credits that vary between
These contracts have been considered as without recourse factoring and the amount advanced by the factors has been derecognized from the balance sheet.
Likewise, in financial years 2023, 2022, and 2021, some receivables assignment contracts were signed with a financial institution, in which the Group retains the risks and benefits inherent to the ownership of the assigned credits. These contracts have been considered as factoring with recource and the assigned amount remains in the consolidated balance sheet at 31 December 2023 and a short-term debt has been recognized for an amount equal to the consideration received from the factor for the assignment. The amount recognized in Euros
Total receivables without recourse sold to financial institutions through the aforementioned contracts in 2023 amount to Euros
At 31 December 2023 the finance cost of credit rights sold for the Group totals Euros
Details of balances with related parties are shown in note 31.
The volume of invoices sold without recourse to various financial institutions which, based on their due date would not have been collected at 31 December 2023, totals Euros
(16) Cash and Cash Equivalents
Details of this caption of the consolidated balance sheet at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Current deposits |
| |
| |
Cash in hand and at banks |
| |
| |
Total cash and cash equivalents |
| |
| |
| (1) |
F-73
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(17) Equity
Details of consolidated equity and movement are shown in the consolidated statement of changes in equity.
(a) | Share capital |
At 31 December 2023 and 2022, the Company’s share capital amounts to Euros
| ● | Class A shares: |
| ● | Class B shares: |
The main characteristics of the Class B shares are as follows:
| ● | Each Class B share entitles its holder to receive a minimum annual preferred dividend out of the distributable profits at the end of each year equal to Euros |
| ● | Each Class B share is entitled to receive, in addition to the above-mentioned preferred dividend, the same dividends and other distributions as for one Grifols ordinary share. |
| ● | Each Class B share entitles the holder to its redemption under certain circumstances, if a takeover bid for all or part of the shares in the Company has been made, except if holders of Class B shares have been entitled to participate in the bid on the same terms as holders of Class A shares. The redemption terms and conditions reflected in the Company’s by-laws limit the amount that may be redeemed, requiring that sufficient distributable reserves be available, and limit the percentage of shares to be redeemed in line with the ordinary shares to which the bid is addressed. |
| ● | In the event the Company were to be wound up and liquidated, each Class B share entitles the holder to receive, before any amounts are paid to holders of ordinary shares, an amount equal to the sum of (i) the par value of the Class B share, and (ii) the share premium paid for the Class B share when it was subscribed. In addition to the Class B liquidation preference amount, each holder is entitled to receive the same liquidation amount that is paid for each ordinary share. |
These shares are freely transferable.
Since 23 July 2012 the ADSs (American Depositary Shares) representing Grifols’ Class B shares (non-voting shares) have had an exchange ratio of 1:1 in relation to Class B shares, ie.1 ADS represents
The Company’s knowledge of its shareholders is based on information provided voluntarily or in compliance with applicable legislation. According to the information available to the Company, there are
At 31 December 2023 and 2022, the number of outstanding shares is equal to the total number of Company shares, less treasury stock.
F-74
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Movement in outstanding shares during 2023 is as follows:
| Reference |
| Class A shares |
| Class B shares | |
Balance at 1 January 2023 |
| |
| | ||
(Acquisition) / disposal of treasury stock |
| Note 17 (d) | — |
| | |
Balance at 31 December 2023 |
| |
| |
Movement in outstanding shares during 2022 is as follows:
| Reference |
| Class A shares |
| Class B shares | |
Balance at 1 January 2022 |
| |
| | ||
(Acquisition) / disposal of treasury stock |
| Note 17 (d) | — |
| ( | |
Balance at 31 December 2022 |
| |
| |
(b) | Share premium |
Movement in the share premium is described in the consolidated statement of changes in equity, which forms an integral part of this note to the consolidated financial statements.
(c) | Reserves |
The drawdown of accumulated gains is subject to legislation applicable to each of the Group companies.
The movement in this caption of the consolidated balance sheet during the years ended at 31 December 2023, 2022 and 2021 is reflected in the consolidated statement of changes in equity, the most significant movements being detailed below:
Legal reserve
Companies in Spain are obliged to transfer
At 31 December 2023 and 2022 the legal reserve of the Parent amounts to Euros
Distribution of the legal reserves of Spanish companies is subject to the same restrictions as those of the Company and at 31 December 2023 the balance of the legal reserve of other Spanish companies amounts to Euros
Other foreign Group companies have a legal reserve amounting to Euros
Unavailable reserve
At 31 December 2023, Euros
F-75
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Hedging reserve
The hedging reserve includes the cash flow hedge reserve and the costs of hedging reserve, see note 4(i) for details. The cash flow hedge reserve is used to recognise the effective portion of gains or losses on derivatives that are designated and qualify as cash flow hedges, as described in note 30.
The group defers the changes in the forward element of forward contracts and the time value of option contracts in the costs of hedging reserve.
(d) | Treasury stock |
The Parent held Class A and B treasury stock equivalent to
Treasury stock Class A
During the years ended at 31 December 2023 and 2022, there have been no movements in Class A treasury shares, with a total of
Treasury stock Class B
Movement in Class B treasury stock during 2023 was as follows:
| No. of Class B |
| ||
shares | Thousands of Euros | |||
Balance at 1 January 2023 |
| |
| |
Disposal Class B shares |
| ( |
| ( |
Balance at 31 December 2023 |
| |
| |
In March, May and October 2023, the Group delivered
Movement in Class B treasury stock during 2022 is as follows:
| No. of Class B |
| ||
shares | Thousands of Euros | |||
Balance at 1 January 2022 |
| |
| |
Disposal Class B shares |
| ( |
| ( |
Acquisition Class B shares | | | ||
Balance at 31 December 2022 |
| |
| |
In March 2022, the Group delivered
(e) | Distribution of profit and dividends |
The profits of Grifols, S.A. and subsidiaries will be distributed as agreed by respective shareholders at their general meetings.
F-76
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The proposed distribution of profit of the Parent Grifols, S.A. for the years ended 31 December 2023, and the distribution of profit approved for 2022, presented at the general meeting held on 16 June 2023, is as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Voluntary reserve |
| ( |
| ( |
Lossest of the Parent |
| ( |
| ( |
The distribution of profit corresponding to the year ended 31 December 2023 and 2022 are presented in the statement of changes in consolidated equity.
During 2023 and 2022
(f) Restricted Share Unit Retention Plan
The Group has set up a Restricted Share Unit Retention Plan (hereinafter RSU Plan) and a long-term incentive plan for certain employees (see note 29). This commitment will be settled using equity instruments and the cumulative accrual amounts to Euros
The incentive plan has been granted to certain employees as part of their compensation package, subject to the achievement of various metrics, both financial and non-financial. The plan has been assessed by calculating the unit value of the options at the valuation date and multiplying it by the total number of options to be granted. Subsequently, this unit value will be adjusted based on the likelihood of achieving the specified objectives.
(18) Earnings Per Share
(a) | Basic Earnings per share |
The calculation of basic earnings per share is based on the profit for the year attributable to the shareholders of the Parent divided by the weighted average number of ordinary shares in circulation throughout the year, excluding treasury stock.
Details of the calculation of basic earnings per share are as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Profit for the year attributable to shareholders of the Parent (Thousands of Euros) |
| |
| |
| |
Weighted average number of ordinary shares outstanding |
| |
| |
| |
Basic earnings per share (Euros per share) |
| |
| |
| |
The weighted average number of ordinary shares outstanding (basic) is as follows:
Number of shares | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Issued shares outstanding at 1 January |
| |
| |
| |
Effect of shares issued |
| |
| |
| ( |
Weighted average number of ordinary shares outstanding (basic) at 31 December |
| |
| |
| |
F-77
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(b) Diluted Earnings per share
Diluted earnings per share are calculated by dividing profit for the year attributable to shareholders of the Parent by the weighted average number of ordinary shares in circulation considering the diluting effects of potential ordinary shares.
The RSU Plan granted by the Group and payable in shares, assumes the existence of dilutive potential shares. Diluted earnings per share have been calculated as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Profit for the year attributable to shareholders of the Parent (Thousands of Euros) |
| | | | ||
Weighted average number of ordinary shares outstanding (diluted) |
| |
| |
| |
Diluted earnings per share (Euros per share) |
| |
| |
| |
The weighted average number of ordinary shares outstanding diluted has been calculated as follows:
Number of shares | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Ordinary shares outstanding at 1 January |
| |
| |
| |
Shares committed under RSU plan |
| ( |
| ( |
| ( |
Effect of treasury stock |
| |
| |
| ( |
Weighted average number of ordinary shares outstanding (diluted) at 31 December |
| |
| |
| |
| (2) |
F-78
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(19) Non-Controlling Interests
Details of non-controlling interests and movement at 31 December 2023 are as follows:
Thousands of Euros | ||||||||||||||||
|
|
| Business |
|
|
|
| |||||||||
combinations / | ||||||||||||||||
|
| Balance at |
|
| Perimeter |
| Other |
| Translation |
| Balance at | |||||
Reference | 31/12/2022 | Additions | additions |
| Dividends |
| movements | differences | 31/12/2023 | |||||||
Grifols (Thailand) Pte Ltd |
| | | — | ( | — | ( | | ||||||||
Grifols Malaysia Sdn Bhd |
| | | — | — | — | ( | | ||||||||
Araclon Biotech, S.A. |
| ( | ( | — | — | — | — | ( | ||||||||
Kiro Grifols, S.L. |
| ( | ( | | — | — | — | | ||||||||
Haema AG |
| | | — | — | — | | |||||||||
BPC Plasma, Inc | | | — | ( | | ( | | |||||||||
Grifols Diagnostics Solutions Inc. | | | — | — | | ( | | |||||||||
Plasmavita Healthcare | | | — | — | — | — | | |||||||||
Haema Plasma Kft | | | — | — | — | | | |||||||||
G Pyrenees Research Cntr | ( | ( | — | — | | — | | |||||||||
Albimmune SL | ( | ( | — | — | — | — | ( | |||||||||
Biotest AG | Note 3 | | ( | | — | ( | | | ||||||||
| | | | ( | | ( | | |||||||||
During the 2023 financial year, BPC Plasma, Inc. distributed a dividend without cash outflow compensating Other non-current financial assets. This dividend corresponds to the result of the previous 4 financial years, valued at Euros
F-79
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Details of non-controlling interests and movement at 31 December 2022 are as follows:
Thousands of Euros | ||||||||||||||
Business | ||||||||||||||
combinations / | ||||||||||||||
| Balance at |
|
| Perimeter | Other |
| Translation |
| Balance at | |||||
| Reference |
| 31/12/2021 |
| Additions |
| additions |
| movements |
| differences |
| 31/12/2022 | |
Grifols (Thailand) Pte Ltd |
| | | — | ( | | | |||||||
Grifols Malaysia Sdn Bhd |
| | | — | — | | | |||||||
Araclon Biotech, S.A. |
| | ( | — | — | — | ( | |||||||
VCN Bioscience, S.L |
| | — | — | ( | — | — | |||||||
Kiro Grifols, S.L. |
| | ( | — | | — | ( | |||||||
Haema AG | | ( | — | — | — | | ||||||||
BPC Plasma, Inc | | | — | — | | | ||||||||
Diagnostics Solutions Inc. | | | — | | | | ||||||||
Plasmavita Healthcare | | ( | — | — | — | | ||||||||
Haema Plasma Kft | — | ( | | — | ( | | ||||||||
G Pyrenees Research Cntr | — | ( | | — | — | ( | ||||||||
Albimmune SL | — | ( | | — | — | ( | ||||||||
Biotest AG |
| Note 3 | — | ( | | | | | ||||||
| | | | | | |||||||||
On 25 April 2022, the Group acquired
At 31 December 2023 and 2022, the main items of the statement of financial positions of the most significant non-controlling interests are as follows:
Thousands of Euros | ||||||||||||||
31/12/2023 | ||||||||||||||
Total Consolidated | ||||||||||||||
Non- | Equity (except for | % Non- | Non- | |||||||||||
current | Current | Non-current | Current | intercompany | controlling | controlling | ||||||||
assets | assets | liabilities | liabilities | eliminations) | Interest | interests | ||||||||
Grupo Biotest |
| |
| |
| ( |
| ( |
| |
| | % | |
Grupo GDS |
| |
| |
| ( |
| ( |
| |
| | % | |
Haema AG |
| |
| |
| ( |
| ( |
| |
| | % | |
BPC Plasma, Inc |
| |
| |
| ( |
| ( |
| |
| | % | |
| |
| |
| ( |
| ( |
| |
| | |||
Thousands of Euros | ||||||||||||||
31/12/2022 | ||||||||||||||
Total Consolidated | ||||||||||||||
Non- | Equity (except for | % Non- | Non- | |||||||||||
current | Current | Non-current | Current | intercompany | controlling | controlling | ||||||||
assets | assets | liabilities | liabilities | eliminations) | Interest | interests | ||||||||
Grupo Biotest |
| |
| |
| ( |
| ( |
| |
| | % | |
Grupo GDS |
| |
| |
| ( |
| ( |
| |
| | % | |
Haema AG |
| |
| |
| ( |
| ( |
| |
| | % | |
BPC Plasma, Inc |
| |
| |
| ( |
| ( |
| |
| | % | |
| |
| |
| ( |
| ( |
| |
| | |||
F-80
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Thousands of Euros | Thousands of Euros | |||||||||||||||
31/12/2023 | 31/12/2022 | |||||||||||||||
Consolidated | % Non- | Non- | Consolidated | % Non- | Non- | |||||||||||
Ordinary | Net | controlling | controlling | Ordinary | Net | controlling | controlling | |||||||||
Income | Income | Interest | interests | Income | Income | Interest | interests | |||||||||
Grupo Biotest |
| |
| ( |
| | % | ( |
| |
| ( |
| | % | ( |
Grupo GDS |
| |
| |
| | % | |
| |
| |
| | % | |
Haema AG |
| |
| |
| | % | |
| |
| ( |
| | % | ( |
BPC Plasma, Inc |
| |
| |
| | % | |
| |
| |
| | % | |
| |
| |
| |
| |
| |
| | |||||
Detail of cash flows of the most significant non-controlling interests is as follows:
| Thousands of Euros | |||||||||||||||
| 31/12/2023 | 31/12/2022 | ||||||||||||||
Haema |
| BPC Plasma |
| Biotest, AG |
| Grupo GDS |
| Haema |
| BPC Plasma |
| Biotest, AG |
| Grupo GDS | ||
Net cash flows from operating activities |
| | |
| ( | | ( | | ( | | ||||||
Net cash flows from investing activities |
| ( | ( |
| | ( | ( | ( | ( | ( | ||||||
Net cash flows from financing activities |
| — | — |
| ( | ( | — | — | | | ||||||
( | ( | ( | | ( | ( | | ( | |||||||||
Haema AG and BPC Plasma, Inc.
In mid-2018, Grifols acquired
The following indicators support the power that Grifols maintains over these companies, even after their sale to Scranton and that, therefore, it retains control over Haema and BPC in accordance with IFRS 10:
- | Grifols has an option to repurchase |
- | There are no shareholder agreements that establish that relevant decisions are approved in a manner different from by majority vote. |
- | Grifols has the financial capacity to exercise the purchase option; |
- | Although Grifols does not have voting rights, it maintains power in both companies, through its ability to exercise the repurchase option which grants it potential voting rights; |
- | Furthermore, Grifols is the manager of both companies through the management contract in the plasma collection business of the donation centers, which includes general management and joint approval of the business plan, granting the intellectual property license and know-how. |
F-81
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
- | Additionally, there is a plasma supply agreement for |
Therefore, although Scranton owns all of the voting rights, Grifols manages the businesses and acquires
As a result of all of the above, Grifols has the power to direct the relevant activities of these companies, since it manages them and jointly determines their business plan, having the unilateral right to repurchase
In relation to the purchase option and given that it is based on a variable number of shares and a variable acquisition price, said instrument is a derivative financial instrument that must be valued at fair value with changes in the profit and loss account.
Based on the contractual conditions, Grifols has estimated the price of the option as (i) the price for which the shares have been sold to Scranton (US Dollars
Likewise, both the shares of Haema AG and the shares of BPC Plasma Inc. are currently pledged as collateral for the loan from Scranton Plasma BV with Bank of America. If a default occurs under the loan agreement, as long as the financing banks have not executed the corresponding pledge, Grifols may exercise the purchase option. Grifols will pay the bank in preference to Scranton Plasma BV until the amount of the debt at the time of acquisition is settled. There is no time limitation in the loan agreement for Grifols to exercise the repurchase option.
GDS Group
There is an indirect participation through SRAAS:
- | Grifols owns a |
- | SRAAS owns a |
Since IAS 28 does not address how to account for cross-participations, Grifols has opted to: in the equity method of integration of the result of SRAAS, the result that SRAAS recognizes when integrating the result of GDS by its percentage of participation (
F-82
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
When determining the allocation of the GDS result attributed to the non-controlling interest (SRAAS), SRAAS’s percentage of participation in GDS is adjusted by
Grifols, S.A. has control over Grifols Diagnostic Solutions, Inc (hereinafter GDS) through Grifols Shared Services North America, Inc (hereinafter GSSNA), following the entry of the new shareholder Shanghais RAAS Blood Products Co Ltd (hereinafter SRAAS).
Grifols, S.A., through GSSNA, owns
Both shareholders have the right of first refusal in the event of a sale of the stake by each of the parties. In addition, SRAAS has certain veto rights, although Grifols has control over GDS for the following reasons:
| ● | Grifols holds |
| ● | It has been expressly endorsed by the parties in their agreements; |
| ● | In the meetings of the Board there is no reference or formal approval of the business and investment plan by SRAAS, and only very generic presentations of results are made and at no time do they mention or compare with the budget, but comparisons are made with respect to the previous comparative period; |
| ● | Grifols only requires approval for investments or divestments in relevant assets, understood as such amounts greater than |
| ● | The absence of control or joint control implies a risk to the performance of SRAAS and to mitigate this, the minimum EBITDA guarantee mentioned in note 19 was signed; |
| ● | GDS is directed, operated and managed directly by Grifols, without SRAAS having any relevant involvement; |
(20) Provisions
Details of provisions at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Provisions for pensions and similar obligations (a) |
| |
| |
Other provisions |
| |
| |
Non-current provisions | | | ||
Trade provisions | | | ||
Other provisions | | | ||
Current provisions |
| |
| |
F-83
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The movement in non-current and current provisions is as follows:
Thousands of Euros | ||||||||
| Reference |
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Opening balance |
| | | | ||||
Business combinations | Note 3 |
| | | | |||
Net charges | | | | |||||
Net cancellations | ( | ( | ( | |||||
Transfers | ( | ( | ( | |||||
Translation differences | ( | | | |||||
Closing balance |
| | | | ||||
| (a) | Pension plan |
At 31 December 2023, 2022 and 2021, the balance of provisions for pensions and similar mainly includes provisions made by the Biotest Group in relation to retirement benefit obligations and foreign personal commitments with employment.
Benefits are based on the employee’s length of service and salary. Retirement benefit obligations relate mainly to employees of the Group’s German companies. Similar obligations are foreign obligations payable in a lump sum on retirement and obligations of the pension savings plan. These plans are voluntary pension plans not subject to statutory or legal obligations. The amount of the pension obligations is mainly dependent on interest rate movements and the life expectancy of the participants.
In financial year 2023, assets of Euros
At 31 December 2023 and 2022, the net defined benefit liability of the Group comprises the following:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
From pension plans | |
| | |
From similar obligations | |
| | |
Net present value of defined benefit obligations | | | ||
For pension plans | | | ||
For similar obligations | | | ||
Fair value of plan assets | | | ||
From pension plans | | | ||
From similar obligations | | | ||
Net defined benefit liability | |
| | |
F-84
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The costs for the defined benefit plans consist of the following components:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Current service cost |
| | | |
Net interest expenses |
| | | |
Total expenses recognised in profit and loss | | | ||
Actuarial losses due to experience adjustments | ( | | ||
Actuarial gains due to changes in financial assumptions | | ( | ||
Actuarial gains from changes in demographic assumptions | | ( | ||
Return on plan assets (excluding amounts included in net interest expense) | ( | | ||
Revaluation recognised directly in other comprehensive income | | ( | ||
Defined benefit costs |
| | ( | |
In financial year 2023, actuarial losses of Euros (
F-85
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The following table shows the reconciliation of the net present value of the defined benefit obligation (DBO):
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Net present value of defined benefit obligation |
| | | |
Current service cost |
| | | |
Interest expense | | | ||
Expenses recognised in the statement of profit and loss | | | ||
Actuarial losses due to experience adjustments | ( | | ||
Actuarial gains due to changes in financial assumptions | | ( | ||
Actuarial gains from changes in demographic assumptions | | ( | ||
Revaluation recognised directly in other comprehensive income | | ( | ||
Pension benefits paid | ( | ( | ||
Net present value of defined benefit obligations at 31 December |
| | | |
The following table shows the reconciliation of the fair value of plan assets:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Fair value of plan assets |
| | | |
Interest income |
| | | |
Income recognised in the consolidated statement of income | | | ||
Return on plan assets (excluding amounts included in net interest expenses) | ( | ( | ||
Revaluations recognised directly in the statement of comprehensive income | ( | ( | ||
Contribution by the employer | | | ||
Payments from plan assets | ( | | ||
Fair value of plan assets as of 31 December |
| | | |
The following payments are expected to be made in subsequent years based on the current pension obligations of the Group:
Thousands of Euros | ||||
| 31/12/2023 | 31/12/2022 | ||
In the next 12 months |
| | | |
Between 2 and 5 years |
| | | |
Between 5 and 10 years | | | ||
After 10 years | | | ||
Total expected payments | | | ||
The weighted average term of the defined benefit plans is
F-86
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Plan assets of the Group were invested in the following asset classes as of the reporting date:
| Thousands of Euros | |||
| 31/12/2023 | 31/12/2022 | ||
Cash and cash equivalents |
| | | |
Financial investment |
| | | |
Fund shares | | | ||
Total expected payments | | | ||
The plan assets transferred are invested in accordance with defined investment principles, whereby the maturity or termination option of the financial instruments must always be selected in such a way that the association can meet its payment obligations. In accordance with the investment principles, the assets can be invested in Euro time deposits as well as domestic government bonds, mortgage bonds or fund units in money market funds or corporate bonds, all in Euro. Loans can also be issued to the Group companies against the corresponding guarantees. A minimum rating of A- is required for all financial instruments.
The calculation of the pension plans is based on the following actuarial assumptions:
| 31/12/2023 | 31/12/2022 | |||
Discount rate |
| | % | | % |
Expected return on plan assets |
| | % | | % |
Rate of increase for wages and salaries | | % | | % | |
Rate of interest for pensions | | % | | % | |
Employee turnover rate | | % | | % |
Actuarial assumptions are mainly based on historical empirical values with the exception of the discount rate. The calculation was based on the published Heubeck 2018 G mortality tables.
Under IAS 19.145, the effect of any possible changes to parameters for the underlying assumptions used to calculate the pension obligations must be disclosed in the sensitivity analysis. Only changes that are realistically expected to occur in the following financial year are to be considered.
The actuarial rate of interest, salary trend, pension trend and life expectancy are regarded as material assumptions. These parameters are shown in the following overview together with information on the parameter changes and their impact on the net present value calculation as of 31 December 2023.
Thousands of Euros | ||||
Impact on the | ||||
Parameter change | pension obligation | |||
Rate of interest |
| Increase by |
| ( |
Rate of interest |
| Decrease by |
| |
Salary trend |
| Increase by |
| |
Salary trend |
| Decrease by |
| ( |
Pension trend |
| Increase by |
| |
Pension trend |
| Decrease by |
| ( |
Life expectancy |
| Increase by one year |
| |
F-87
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The impact on the net present value calculation as of 31 December 2022 is as follows:
| Thousands of Euros | |||
Impact on the | ||||
| Parameter change |
| pension obligation | |
Rate of interest |
| Increase by |
| ( |
Rate of interest |
| Decrease by |
| |
Salary trend | Increase by | | ||
Salary trend | Decrease by | ( | ||
Pension trend | Increase by | | ||
Pension trend | Decrease by | ( | ||
Life expectancy |
| Increase by one year |
| |
An amount of Euros
Thousands of Euros | ||||
| 31/12/2023 | 31/12/2022 | ||
Defined contribution plans of the Company |
| | | |
Employer contributions to statutory pension scheme |
| | | |
| | |||
| (3) |
(21) Financial Liabilities
This note provides information on the contractual conditions of the Group’s financial liabilities, which are measured at amortized cost, except for the financial derivatives that are valued at fair value. For further information on exposure to interest rate risk, currency risk and liquidity risk and the fair values of financial liabilities, please refer to note 30.
Details at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||||
Financial liabilities |
| Reference |
| 31/12/2023 |
| 31/12/2022 |
Non-current bonds |
| (a) | |
| | |
Senior secured debt |
| (b) | |
| | |
Other loans |
| (b) | |
| | |
Other non-current financial liabilities |
| (c) | |
| | |
Non-current financial derivatives | Note 30 | | | |||
Non-current lease liabilities | Note 8 | | | |||
Loan transaction costs | ( | ( | ||||
Total non-current financial liabilities |
| |
| | ||
Current bonds |
| (a) | |
| | |
Senior secured debt |
| (b) | |
| | |
Other loans |
| (b) | |
| | |
Other current financial liabilities |
| (c) | |
| | |
Current financial derivatives |
| Note 30 | |
| | |
Current lease liabilities | Note 8 | | | |||
Loan transaction costs | ( | ( | ||||
Total current financial liabilities |
| |
| | ||
F-88
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(a) | Senior Notes |
Detail of Senior Notes at 31 December 2023 are as follows:
| Thousands of Euros | |||||||||||
| Issue date |
| Company |
| Nominal value |
| Currency |
| Annual coupon |
| Maturity | |
18/04/2017 |
| Grifols, S.A. |
| |
| Euros |
| | % | 2025 | ||
Unsecured senior notes | 05/10/2021 |
| Grifols, S.A. | (*) | |
| Euros |
| | % | 2028 | |
05/10/2021 |
| Grifols, S.A. | (*) | |
| US Dollar |
| | % | 2028 | ||
Secured senior notes | 15/11/2019 |
| Grifols, S.A. |
| |
| Euros |
| | % | 2027 | |
15/11/2019 |
| Grifols, S.A. |
| |
| Euros |
| | % | 2025 | ||
(*)As a consecuence of the merge between Grifols Escrow Issuer, S.A. and Grifols, S.A. in 2023 (see note 2)
The bonds issued by Grifols, S.A. in 2017 and 2019 were admitted to listing on the Irish Stock Exchange on the same issue date.
On 5 October 2021, Grifols Escrow Issuer, S.A. closed the issuance of a senior unsecured corporate bond (Senior Unsecured Notes) in
The proceeds from the bonds were used to finance the Euros
Details of movement in the Senior Notes at 31 December 2023 are as follows:
Thousands of Euros | ||||||
| Opening |
|
| |||
outstanding balance | Exchange | Closing outstanding | ||||
01/01/23 | differences | balance 31/12/23 | ||||
Senior unsecured corporate notes 2017 |
| |
| — |
| |
Senior secured corporate notes 2019 |
| |
| — |
| |
Senior unsecured corporate notes Euros 2021 |
| |
| — |
| |
Senior unsecured corporate notes US Dollars 2021 |
| |
| ( |
| |
| |
| ( |
| | |
Details of movement in the Senior Notes at 31 December 2022 are as follows:
Thousands of Euros | ||||||||
Opening outstanding | Exchange | Closing outstanding | ||||||
| balance 01/01/22 |
| Repurchase |
| differences |
| balance 31/12/22 | |
Senior unsecured corporate notes 2017 |
| |
| | | | ||
Senior secured corporate notes 2019 | | ( | | | ||||
Senior unsecured corporate notes Euros 2021 | | | | | ||||
Senior unsecured corporate notes US Dollars 2021 | | | | | ||||
| |
| ( | | | |||
F-89
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
On 2 December 2021, Grifols, S.A. announced a repurchase offer for the same price plus unpaid accrued interests of the mentioned bonds, up to the equivalent in Euros of US Dollars
At 31 December 2023 and 2022 the current obligations caption includes the issue of bearer promissory notes to Group employees, as follows:
Thousands of Euros |
| ||||
| 31/12/2023 |
| 31/12/2022 |
| |
Issue date |
| 05/05/2023 |
| 04/05/2022 | |
Maturity date |
| 04/05/2024 |
| 04/05/2023 | |
Nominal amount of promissory notes (Euros) |
| |
| | |
Interest rate |
| | % | | % |
Promissory Notes subscribed |
| |
| | |
Buy-backs or redemptions |
| ( |
| ( | |
Interest pending accrual |
| ( |
| ( | |
(b)Loans and borrowings
Details of loans and borrowings at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||||||||||||||
31/12/2023 | 31/12/2022 | |||||||||||||||
Amount | Carrying | Amount | Carrying | |||||||||||||
Credit |
| Currency |
| Interest rate |
| Date awarded |
| Maturity date |
| extended |
| amount |
| extended |
| amount |
Senior debt - Tranche B | Euros | Euribor + | 15/11/2019 | 15/11/2027 | | | | | ||||||||
Senior debt - Tranche B |
| US Dollars | SOFR + | 15/11/2019 | 15/11/2027 | | | | | |||||||
Total senior debt |
| | | | | |||||||||||
EIB Loan |
| Euros | 20/11/2015 | 20/11/2025 | | | | | ||||||||
EIB Loan | Euros | 22/12/2017 | 22/12/2027 | | | | | |||||||||
EIB Loan | Euros | 25/09/2018 | 25/09/2028 | | | | | |||||||||
Total EIB Loan | | | | | ||||||||||||
Revolving Credit |
| US Dollars | SOFR + | 15/11/2019 | 15/11/2025 | | | | — | |||||||
Total Revolving Credit |
| | | | — | |||||||||||
Other non-current loans |
| Euros | — | — | | | ||||||||||
Loan transaction costs |
| — | ( | — | ( | |||||||||||
Non-current loans and borrowings |
|
|
|
|
|
|
| | | | | |||||
Thousands of Euros | ||||||||||||||||
31/12/2023 | 31/12/2022 | |||||||||||||||
Amount | Carrying | Amount | Carrying | |||||||||||||
Credit |
| Currency |
| Interest rate |
| Date awarded |
| Maturity date |
| extended |
| amount |
| extended |
| amount |
Senior debt - Tranche B |
| Euros | Euribor + | 15/11/2019 | 15/11/2027 | (*) | | (*) | | |||||||
Senior debt - Tranche B | US Dollars | SOFR + | 15/11/2019 | 15/11/2027 | (*) | | (*) | | ||||||||
Total senior debt |
| — | | — | | |||||||||||
EIB Loan |
| Euros | 20/11/2015 | 20/11/2025 | (*) | | (*) | | ||||||||
EIB Loan | Euros | 22/12/2017 | 22/12/2027 | (*) | | (*) | | |||||||||
EIB Loan | Euros | 25/09/2018 | 25/09/2028 | (*) | | (*) | | |||||||||
Total EIB Loan | — | | — | | ||||||||||||
Other current loans |
| | | | | |||||||||||
Loan transaction costs |
| — | ( | — | ( | |||||||||||
Current loans and borrowings |
|
|
|
|
|
| | | | | ||||||
(*) See amount granted under non-current debt
Current loans and borrowings include accrued interest amounting to Euros
F-90
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Between 2015 and 2018, the Group arranged
“Other current loans” includes a secured loan from the group company Biotest, AG with an original term of
Senior Secured debt
The Senior Secured debt consists of an
| ◾ | US Dollar Tranche B: |
| ● | Original principal amount of US Dollars |
| ● | Applicable margin of |
| ● | Quasi-bullet repayment structure. |
| ● | Maturity in 2027. |
| ◾ | Tranche B in Euros: |
| ● | Original principal amount of Euros |
| ● | Applicable margin of |
| ● | Quasi-bullet repayment structure. |
| ● | Maturity in 2027. |
Details of Tranche B by maturity at 31 December 2023 are as follows:
US Tranche B | Tranche B in Euros | |||||||||
Principal in Thousands | Principal in | Principal in Thousands of | ||||||||
| Currency |
| of US Dollars |
| Thousands of Euros |
| Currency |
| Euros | |
Maturity | ||||||||||
2024 | US Dollars | | | Euros | | |||||
2025 | US Dollars | | | Euros | | |||||
2026 | US Dollars | | | Euros | | |||||
2027 | US Dollars | | | Euros | | |||||
Total |
| US Dollars |
| |
| | Euros | | ||
The borrowers of the total Senior secured debt are Grifols, S.A. and Grifols Worldwide Operations USA, Inc.
Revolving credit facility
On 7 May 2020, the Group concluded the upsize of the multi-currency revolving credit facility from US Dollars
F-91
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Movement in the Revolving Credit Facility is as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Drawn opening balance |
| |
| |
Drawdowns |
| |
| |
Repayments |
| ( |
| ( |
Translation differences |
| ( |
| ( |
Drawn closing balance |
| |
| |
Guarantors
The Notes, the Senior Term Loans and the Revolving Loans are secured by Grifols, S.A. and certain significant subsidiaries of Grifols, S.A., which together with Grifols, S.A., represent, in the aggregate, at least
The Notes are guaranteed on a senior secured basis by subsidiaries of Grifols, S.A. that are guarantors and co-borrower under the New Credit Facilities. The guarantors are Grifols Worldwide Operations Limited, Grifols Biologicals Inc., Grifols Shared Services North America, Inc., Grifols Therapeutics, Inc., Instituto Grifols, S.A., Grifols Worldwide Operations USA, Inc., Grifols USA, Llc. and Grifols International, S.A.
(c)Other financial liabilities
Details of other financial liabilities at 31 December 2023 and 2022 are as follows:
Thousands of Euros | ||||||
Other financial liabilities |
| Reference |
| 31/12/2023 |
| 31/12/2022 |
Non-current debt with GIC (sovereign wealth fund in Singapore) |
| (i) |
| |
| |
Non-current preferential loans |
| |
| | ||
Other non-current financial liabilities |
| (ii) |
| |
| |
Total other non-current financial liabilities |
| |
| | ||
Current debt with GIC (sovereign wealth fund in Singapore) |
| (i) |
| |
| |
Current preferential loans |
| |
| | ||
Other current financial liabilities |
| (ii) |
| |
| |
Total other current financial liabilities |
| |
| | ||
(i)Debt with GIC – Singapore sovereign wealth fund
In November 2021 approval was received from the pertinent authorities to close the agreement with GIC (Sovereign Fund of Singapore), announced in June 2021, whereby the Group received an amount of US Dollars
The main terms and conditions of the agreement with GIC were:
| ● | The distribution of annual preferential dividends to GIC equivalent to US Dollar |
| ● | The redemption right with respect to Class B stock for US Dollars |
F-92
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
| ● | From 1 December 2036, holders of Class B shares of Biomat USA will have the right to request Biomat USA to redeem up to the total of the Class B shares they hold at a value of US Dollars |
| ● | In the event that the dividends or the annual redemption at Biomat USA or Biomat NewCo, where applicable, is not approved, is partially paid, or is otherwise not paid, GIC holds the right to obtain in exchange thereof an undetermined number of shares among the following alternatives (i) an additional number of shares in Biomat USA, in lieu of the non-payment occurred at Biomat USA, (ii) an additional number of shares in Biomat NewCo, in lieu of the non-payment occurred at Biomat NewCo; or (iii) a number of ADRs of Grifols, S.A. in lieu of either (i) or (ii). |
| ● | Grifols holds the right to redeem all of the Class B stock from the fifth year onwards; |
| ● | In the event of liquidation of Biomat USA and Biomat Newco, GIC shall have the right to the preferential liquidation of US Dollars |
At 31 December 2023, Current debt with GIC includes Euros
Grifols did not have the discretional right to avoid payment in cash and therefore, the instrument is recorded as a financial liability.
The Group does not lose control of Biomat USA and continues overseeing all aspects of the Biomat Group’s administration and operations.
(ii)Other non-current and current financial liabilities
At 31 December 2023, “other non-current financial liabilities” include mainly an unsecured long-term loan in the amount of Euros
At 31 December 2023, “other current financial liabilities” include mainly distributor commission liabilities of Euros
Details of the maturity of other financial liabilities are as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Maturity at: |
|
|
|
|
Up to one year |
| |
| |
Two years |
| |
| |
Three years |
| |
| |
Four years |
| |
| |
Five years |
| |
| |
Over five years |
| |
| |
| |
| | |
F-93
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(d)Changes in liabilities derived from financing activities
Thousands of Euros | ||||||||||||
Senior Secured | ||||||||||||
debt & Other | Finance lease | Other financial | ||||||||||
| Reference |
| Bonds |
| loans |
| liabilities |
| liabilities |
| Total | |
Carrying amount at 1 January 2021 |
| |
| |
| |
| |
| | ||
New financing |
| |
| |
| — |
| |
| | ||
Refunds |
| ( |
| ( |
| ( |
| ( |
| ( | ||
Interest accrued |
| |
| |
| |
| |
| | ||
Other movements |
| ( |
| |
| |
| |
| | ||
Interest paid/received |
| ( |
| ( |
| — |
| — |
| ( | ||
Business combinations |
| Note 3 | — |
| — |
| — |
| ( |
| ( | |
Foreign exchange differences |
| |
| |
| |
| |
| | ||
Balance at 31 December 2021 |
| |
| |
| |
| |
| | ||
New financing | | | — | | | |||||||
Refunds | ( | ( | ( | ( | ( | |||||||
Interest accrued | | | | | | |||||||
Other movements | | ( | | — | | |||||||
Interest paid/received | ( | ( | — | ( | ( | |||||||
Business combinations | Note 3 | ( | | | | | ||||||
Foreign exchange differences | | | | | | |||||||
Balance at December 31 2022 | | | | | | |||||||
New financing | | | — | | | |||||||
Refunds | ( | ( | ( | ( | ( | |||||||
Interest accrued | | | | | | |||||||
Other movements | — | — | | | | |||||||
Interest paid/received | ( | ( | — | ( | ( | |||||||
Business combinations | Note 3 | — | — | — | | | ||||||
Foreign exchange differences | ( | ( | ( | ( | ( | |||||||
Balance at 31 December 2023 | | | | | | |||||||
(22) Trade and Other Payables
Details are as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Suppliers |
| |
| |
VAT payable |
| |
| |
Taxation authorities, withholdings payable |
| |
| |
Social security payable |
| |
| |
Other public entities |
| |
| |
Other payables |
| |
| |
Current income tax liabilities |
| |
| |
| |
| | |
F-94
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Suppliers
Details of balances with related parties are shown in note 31.
The Group’s exposure to currency risk and liquidity risk associated with trade and other payables is described in note 30.
(23) Other Current Liabilities
Details at 31 December are as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Salaries payable |
| |
| |
Other payables |
| |
| |
Deferred income |
| |
| |
Advances received |
| |
| |
Other current liabilities |
| |
| |
At 31 December 2023, and 31 December 2022, the advances received are contract liabilities relate to unperformed performance obligations for which Grifols has received a consideration from the customer.
(24) Net Revenues
Net revenues are mainly generated from the sale of goods.
The distribution of net consolidated revenues for 2023, 2022 and 2021 by segment is as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 (*) | |
Biopharma |
| |
| |
| |
Diagnostic |
| |
| |
| |
Bio supplies | | | | |||
Others |
| |
| |
| |
Intersegments | | ( | ( | |||
| |
| |
| | |
*As a consequence of the review of transactions and balances allocations by segments made in the year 2022, the comparative figures for the fiscal year 2021 have been adjusted accordingly.
The geographical distribution of net consolidated revenues is as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 (*) | |
USA and Canada |
| |
| |
| |
Spain |
| |
| |
| |
European Union |
| |
| |
| |
Rest of the world |
| |
| |
| |
Consolidated |
| |
| |
| |
F-95
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Details of discounts and other reductions in gross income are as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 (*) | |
Gross sales |
| |
| |
| |
Chargebacks |
| ( |
| ( |
| ( |
Cash discounts |
| ( |
| ( |
| ( |
Volume rebates |
| ( |
| ( |
| ( |
Medicare and Medicaid |
| ( |
| ( |
| ( |
Other discounts |
| ( |
| ( |
| ( |
Net sales |
| |
| |
| |
Movement in discounts and other reductions in gross income during 2023 is as follows:
Thousands of Euros |
| ||||||||||||
|
|
| Cash |
| Volume |
| Medicare / |
| Other |
|
|
| |
Chargebacks | discounts | rebates | Medicaid | discounts | Total |
| |||||||
Balance at 31 December 2022 |
| |
| |
| |
| |
| |
| | |
Current estimate related to sales made in current and previous periods (1) |
| |
| |
| |
| |
| |
| | |
(Actual returns or credits in current period related to sales made in current period) (2) |
| ( |
| ( |
| ( |
| ( |
| ( |
| ( | |
(Actual returns or credits in current period related to sales made in prior periods) (3) |
| ( |
| ( |
| ( |
| ( |
| ( |
| ( | |
Translation differences |
| ( |
| |
| ( |
| ( |
| ( |
| ( | |
Balance at 31 December 2023 |
| |
| |
| |
| |
| |
| | |
| (1) | Net impact in income statement: estimate for the current year plus prior years’ adjustments. Adjustments made during the year corresponding to prior years’ estimates have not been significant. |
| (2) | Amounts credited and posted against provisions for current period |
| (3) | Amounts credited and posted against provisions for prior period |
F-96
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Movement in discounts and other reductions to gross income during 2022 was as follows:
Thousands of Euros |
| ||||||||||||
|
|
| Cash |
| Volume |
| Medicare / |
| Other |
|
|
| |
Chargebacks | discounts | rebates | Medicaid | discounts | Total |
| |||||||
Balance at 31 December 2021 |
| |
| |
| |
| |
| |
| | |
Current estimate related to sales made in current and previous periods (1) |
| |
| |
| |
| |
| |
| | |
(Actual returns or credits in current period related to sales made in current period) (2) |
| ( |
| ( |
| ( |
| ( |
| ( |
| ( | |
(Actual returns or credits in current period related to sales made in prior periods) (3) |
| ( |
| ( |
| ( |
| ( |
| ( |
| ( | |
Translation differences |
| |
| |
| |
| |
| |
| | |
Balance at 31 December 2022 |
| |
| |
| |
| |
| |
| | |
Movement in discounts and other reductions to gross income during 2021 was as follows:
Thousands of Euros |
| ||||||||||||
|
|
| Cash |
| Volume |
| Medicare / |
| Other |
|
|
| |
Chargebacks | discounts | rebates | Medicaid | discounts | Total |
| |||||||
Balance at 31 December 2020 |
| |
| |
| |
| |
| |
| | |
Current estimate related to sales made in current and previous periods (1) |
| |
| |
| |
| |
| |
| | |
(Actual returns or credits in current period related to sales made in current period) (2) |
| ( |
| ( |
| ( |
| ( |
| ( |
| ( | |
(Actual returns or credits in current period related to sales made in prior periods) (3) |
| ( |
| ( |
| ( |
| ( |
| ( |
| ( | |
Translation differences |
| |
| |
| |
| |
| |
| | |
Balance at 31 December 2021 |
| |
| |
| |
| |
| |
| | |
(25) Personnel Expenses
Details of personnel expenses by function are as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Cost of sales |
| |
| |
| |
Research and development |
| |
| |
| |
Selling, general & administration expenses |
| |
| |
| |
| |
| |
| | |
F-97
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Details by nature are as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Wages and salaries | |
| |
| | |
Contributions to pension plans | |
| |
| | |
Other social charges | |
| |
| | |
Social Security | |
| |
| | |
|
| |
| | ||
On February 15, 2023, the Group announced the implementation of a comprehensive operational improvement plan with significant savings. The plan included the optimization of plasma costs and operations, the streamlining of corporate functions, and other initiatives to improve efficiency in the organization. It also included a reduction in staff in 2023 that affected approximately
(26) Expenses by Nature
(a) Amortization and depreciation
Expenses for the amortization and depreciation of intangible assets, right of use assets and property, plant and equipment, incurred during 2023, 2022 and 2021 classified by functions are as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Cost of sales |
| |
| |
| |
Research and development |
| |
| |
| |
Selling, general & administration expenses |
| |
| |
| |
| |
| |
| | |
(b) Other operating income and expenses
Other operating income and expenses incurred during 2023, 2022 and 2021 by function are as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Cost of sales |
| |
| |
| |
Research and development |
| |
| |
| |
Selling, general & administration expenses |
| |
| |
| |
| |
| |
| | |
F-98
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Details by nature are as follows:
Thousands of Euros | ||||||||
| Reference |
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Changes in trade provisions |
| |
| |
| | ||
Professional services |
| |
| |
| | ||
Commissions |
| |
| |
| | ||
Supplies and auxiliary materials |
| |
| |
| | ||
Operating leases |
| Note 8 | |
| |
| | |
Freight |
| |
| |
| | ||
Repair and maintenance expenses |
| |
| |
| | ||
Advertising |
| |
| |
| | ||
Insurance |
| |
| |
| | ||
Royalties |
| |
| |
| | ||
Travel expenses |
| |
| |
| | ||
External services |
| |
| |
| | ||
R&D Expenses |
| |
| |
| | ||
Gains on disposal of assets | ( | ( | — | |||||
Other |
| |
| |
| | ||
Other operating income&expenses |
| |
| |
| | ||
On February 15, 2023, the Group announced the implementation of a comprehensive operational improvement plan with significant savings. The plan included the optimization of plasma costs and operations, the streamlining of corporate functions, and other initiatives to improve efficiency in the organization. As of 31 December 2023, the Group recognized an expense of approximately Euros
(27) Finance Result
Details are as follows:
Thousands of Euros | ||||||||
| Reference |
| 31/12/2023 |
| 31/12/2023 |
| 31/12/2023 | |
Finance income |
| |
| |
| | ||
Finance costs from Senior Unsecured Notes |
| ( |
| ( |
| ( | ||
Finance costs from senior debt |
| Note 21 (b) | ( |
| ( |
| ( | |
Finance costs from other financial liabilities | ( | ( | — | |||||
Capitalized interest |
| Note 9 | |
| |
| | |
Finance lease expenses | ( | ( | ( | |||||
Other finance costs |
| Note 8 | ( |
| ( |
| ( | |
Finance costs |
| ( |
| ( |
| ( | ||
Finance costs from sale of receivables | Note 15 | ( | ( | ( | ||||
Change in fair value of financial instruments |
| |
| |
| | ||
Exchange differences |
| ( |
| |
| ( | ||
Finance result |
| ( |
| ( |
| ( | ||
The finance costs from other financial liabilities heading for 2023 includes finance costs related to the interest on the funds received by GIC amounting
During 2023 the Group has capitalized interest at a rate of between
F-99
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(28) Taxation
Grifols, S.A. is authorized to file consolidated tax returns in Spain with Grifols Movaco, S.A., Laboratorios Grifols, S.A., Instituto Grifols, S.A., Biomat, S.A., Grifols Viajes, S.A., Grifols International, S.A., Grifols Engineering, S.A., Gripdan Invest, S.L., Araclon Biotech, Aigües Minerals de Vilajuiga, S.A. and VCN Biosciences, S.L. Grifols, S.A., in its capacity as Parent, is responsible for the filing and settlement of the consolidated tax return. Under prevailing tax law, Spanish companies pay
The North American company Grifols Shared Services North America, Inc. is also authorized to file consolidated tax returns in the USA with Grifols Biologicals Inc., Grifols USA, LLC., Biomat USA, Inc., Grifols Therapeutics Inc., Talecris Plasma Resources, Inc, Interstate Blood Bank, Inc. and Goetech, LLC.. The profits of the companies domiciled in the USA, determined in accordance with prevailing tax legislation, are subject to tax of approximately
Grifols assesses the effect of uncertain tax treatments and recognizes the effect of the uncertainty on taxable earnings. At 31 of December 2023 and 2022, the potential obligations deriving from tax claims are properly covered. There are no lawsuits or uncertain tax treatments that are individually material.
In 2021, the OECD released the Model Rules for Pillar 2 to address tax challenges arising from the digitization of the economy. This international tax system reform focuses on the geographic allocation of profits for tax purposes and is designed to ensure that multinational enterprises are subject to a minimum effective tax rate of 15%.
On 15 December 2022, the Council of the European Union formally adopted the European Directive on Pillar 2. As of 31 December 2023, Spain has approved the Draft Law transposing the European Directive to ensure a global minimum taxation of 15% for multinational corporations. This legislation will apply prospectively to accounting periods beginning on January 1, 2024.
On 23 May 2023, the International Accounting Standards Board (IASB) published the International Tax Reform - Second Pillar Model Rules. Proposed amendments to IAS 12, which will be applicable for periods beginning on 1 January 2023. The amendments to IAS 12 provide for a mandatory temporary exemption in recognizing deferred tax balances arising from the implementation of Pillar 2 legislation.
The Group has developed an accounting policy consistent with the amendments to IAS 12, whereby the Group does not record adjustments to deferred tax assets and liabilities resulting from the introduction of the minimum effective tax rate of 15%. In developing this accounting policy, the Group has also adopted the exemption provided in paragraph 98M of the amendments to IAS 12 to avoid providing detailed information on the amendments for transitional periods beginning on 1 January 2023.
As of 31 December 2023, the Group continues to assess the implications of Pillar 2 reforms, including quantifying the impact on current tax resulting from the approval of the regulations. The assessment of potential exposure to Pillar 2 income taxes is based on the most recent tax returns, country-by-country reports, and financial statements of the Group’s constituent entities. According to the assessment, effective tax rates of Pillar 2 in most jurisdictions where the Group operates are above 15%. However, there are a limited number of jurisdictions where the safe harbor transitional exemption does not apply, and the effective tax rate of Pillar 2 is close to 15%. The Group does not anticipate significant exposure to Pillar 2 income taxes in those jurisdictions.
On 18 January 2024, the Constitutional Court declared unconstitutional various tax precepts contained in Royal Decree-Law 3/2016. The company has assessed the impact that these provisions had in 2017 and subsequent years, and considers that, as they did not have a significant impact, it will not challenge the tax assessments for these years.
F-100
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(a)Reconciliation of accounting and taxable income
Details of the income tax expense and income tax related to profit for the year are as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Profit before income tax from continuing operations |
| |
| |
| |
Tax at |
| |
| |
| |
Permanent differences |
| ( |
| ( |
| |
Effect of different tax rates |
| |
| |
| ( |
Tax credits (deductions) |
| ( |
| |
| ( |
Prior year income tax expense |
| |
| |
| |
Other income tax expenses/(income) |
| |
| |
| ( |
Total income tax expense |
| |
| |
| |
Deferred tax |
| ( |
| ( |
| |
Current tax |
| |
| |
| |
Total income tax expense |
| |
| |
| |
The effect of the different tax rates is basically due to a change of country mix in profits
F-101
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(b)Deferred tax assets and liabilities
Details of deferred tax assets and liabilities are as follows:
Thousands of Euros | ||||||
Tax effect | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Assets |
|
|
|
|
|
|
Provisions |
| |
| |
| |
Inventories |
| |
| |
| |
Tax credits (deductions) |
| |
| |
| |
Tax loss carryforwards |
| |
| |
| |
Fixed assets, amortisation and depreciation | | — | — | |||
Other |
| |
| |
| |
Subtotal, assets |
| |
| |
| |
Goodwill |
| ( |
| ( |
| ( |
Fixed assets, amortisation and depreciation |
| ( |
| ( |
| |
Intangible assets |
| — |
| ( |
| ( |
Other | ( | ( | — | |||
Subtotal, net liabilities |
| ( |
| ( |
| ( |
Deferred assets, net |
| |
| |
| |
Liabilities |
|
|
| |||
Goodwill |
| ( |
| ( |
| ( |
Intangible assets |
| ( |
| ( |
| ( |
Fixed assets |
| ( |
| ( |
| ( |
Debt cancellation costs |
| ( |
| ( |
| ( |
Others | ( | — | — | |||
Subtotal, liabilities |
| ( |
| ( |
| ( |
Tax loss carryforwards |
| |
| |
| |
Tax credits (deductions) | | | — | |||
Inventories |
| |
| |
| |
Provisions | | | | |||
Other |
| |
| |
| |
Subtotal, net assets |
| |
| |
| |
Net deferred Liabilities |
| ( |
| ( |
| ( |
Movement in deferred tax assets and liabilities is as follows:
Thousands of Euros | ||||||
Deferred tax assets and liabilities |
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 |
Balance at 1 January |
| ( |
| ( |
| ( |
Movements during the year |
| |
| |
| ( |
Business combination (note 3) |
| |
| ( |
| ( |
Translation differences |
| |
| ( |
| ( |
Balance at 31 December |
| ( |
| ( |
| ( |
F-102
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The Spanish companies have opted to apply accelerated depreciation to certain additions to property, plant and equipment, which has resulted in the corresponding deferred tax liability.
The remaining assets and liabilities recognized in 2023, 2022 and 2021 were recognized in the statement of profit and loss.
Estimated net deferred tax assets to be reversed in a period of less than 12 months amount to Euros
The majority of the tax deductions pending application from Spanish companies related mainly to research and development, mature in
Tax loss carryforwards pending to be offset derived from the US companies are available for
The Group has not recognized as deferred tax assets the tax effect of the unused tax loss carryforwards of Group companies, which amount to Euros
The amount of unrecognized deferred tax liabilities associated with investments in subsidiaries amounted to Euros
The commitments from Spanish companies from the reversal of deferred tax related to provisions of investments in subsidiaries are not significant.
(c)Years open to inspection
Under prevailing legislation, taxes cannot be considered to be definitively settled until the returns filed have been inspected by the taxation authorities, or the prescription period has elapsed.
The main tax audits currently open in the Group are as follows:
| ● | Certain companies of the Group domiciled in Spain were subject to an inspection by the Spanish State Tax Administration Agency in relation to Corporate Income Tax for the years 2014, 2015 and 2016 and Value Added Tax for the years 2015 and 2016. |
As a result of said procedure, the State Tax Administration Agency issued assessments containing the results of the inspection, where it is indicated that the treatment of certain transactions and computations mainly related to Transfer Pricing should be adjusted, taking into consideration different interpretations related to the allocation of taxable bases between different jurisdictions. With respect to Corporate Income Tax, the deductibility of certain expenses for the computation of the tax payable has been questioned. These assessments were signed in conformity by the Group on 8 November 2021. It should be noted that no penalties were imposed on any of the Group companies for any of the taxes subject to verification.
The results of the inspection did not have a significant impact on the Group’s consolidated financial statements, and the differences determined by the State Tax Administration Agency were recorded as part of the current tax included under the heading “Current tax liabilities” in the Consolidated Balance Sheet as of 31 December 2021.
F-103
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
If the result of the procedure is considered to be replicable to years not reviewed and open to inspection, the Group estimated that it was not necessary to record provisions in the consolidated financial statements mainly because the number of transactions that gave rise to the aforementioned assessments has significantly decreased since the years in which they were inspected.
Likewise, having adjusted the allocation of taxable income in accordance with the aforementioned assessments for the purposes of their consideration for the determination of Transfer Pricing, the Group now has a legal right to recover certain amounts from the corresponding Administration, in accordance with the provisions of the European Convention on International Commercial Arbitration with respect to international double taxation. The minimum amount to be recovered, upon which its realization is virtually certain, was recorded as a non-current receivable included in the caption “other payable” as of 31 December 2021.
| ● | Grifols Shared Services North America, Inc. and subsidiaries: In 2020 notification of an inspection was received relating to the State Income Tax for the fiscal years 2017 and 2018. |
| ● | Certain Group companies domiciled in Spain were notified in July 2022 of the inspection by the Spanish State Tax Administration Agency in relation to Corporation Tax for the years 2017 to 2019 and Value Added Tax, personal income tax, non-resident income and capital income for the years 2018 and 2019. |
Group management does not expect any significant liability to derive from these inspections.
Based on its experience of the different tax inspections in the different jurisdictions in which Grifols operates, the Group considers it unlikely that there will be a scenario of discrepancy with the taxation authorities that will require significant adjustments to be made to the tax result or to the asset and/or liability balances relating to corporate income tax.
(29) Other Commitments with Third Parties and Other Contingent Liabilities
(a) | Guarantees |
The Group has no significant guarantees extended to third parties.
(b) | Guarantees committed with third parties |
Since 30 June 2023, Grifols, through Grifols Shared Services North America, Inc, acts as a guarantor for
In March 2019, Grifols entered into a share exchange agreement with Shanghai RAAS Blood Products Co. Ltd. The sales contract establishes a consideration of Shanghai RAAS shares for Grifols Diagnostic Solutions Inc. shares and a contingent consideration in the form of a minimum guarantee for the EBITDA (Earnings before interests, tax, amortization and depreciation) differential to be generated by Grifols Diagnostic Solutions Inc. at the end of
The contingent consideration is part of the acquisition price of SRAAS shares and is subsequently valued at fair value with changes in profit and loss. Both at the initial moment and in each year, the fair value of the financial liability has been
F-104
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Additionally, under the framework of the Strategic Alliance and Share Purchase Agreement with Haier Group Corporation announced on 29 December 2023, for the sale of a
Additionally, the Group has significant guarantees extended to third parties described in note 21.
(c)Obligations with personnel
The Group’s annual contribution to defined contribution pension plans of Spanish Group companies for 2023 has amounted to Euros
In successive years this contribution will be defined through labor negotiations.
In the event that control is taken of the Company, the Group has agreements with
The Group has contracts with
Restricted Share Unit Retention Plan
In March 2022, the Group established a Restricted Stock Share Plan (hereinafter RSU) for certain employees. Under this plan, an employee may elect to receive up to
Class B Grifols shares and Grifols ADSs are valued at the date of grant of the bonus.
These RSUs will have a vesting period of and will subsequently be exchanged for Grifols Class B Shares or Grifols ADSs (American Depositary Shares representing
If an eligible employee leaves the company or is terminated prior to the vesting period, he/she will not be entitled to the additional RSUs.
At 31 December 2023 the Group has settled the 2020 RSU plan for an amount of Euros
This commitment is treated as equity-settled and the accumulated amount recognized at 31 December 2023 as share-based payments cost of employees is Euros
F-105
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Equity-settled share-based payment plan
In May 2023, the Board of Directors approved a proposal to the Ordinary General Meeting on 16 June, 2023, which approved it, a long term incentive plan. based on the granting of stock options for certain executive directors, members of the senior management of Grifols and its subsidiaries. The plan has a term of
Settlement date |
| Number of shares assigned |
| Unit fair value (Euros) |
2025 |
| |
| |
2027 |
| |
| |
Additionally, there is a special remuneration plan referenced to the value of the share settled in equity instruments for certain executives with an exercise price of Euros
Settlement date |
| Number of RSUs assigned |
| Unit fair value (Euros) |
28/02/2024 |
| |
| |
22/02/2025 |
| |
| |
28/02/2025 |
| |
| |
The recognized amount in Equity as of 31 December 2023 amounts to Euros
Cash-settled share-based payment plan
In May 2023, the Board of Directors of Grifols, S.A. approved a new long-term incentive plan based on restricted stock units (RSUs) aimed at certain members of the management team of the Company and its subsidiaries. The plan has a total duration of
Settlement date |
| Number of RSUs assigned |
| Unit fair value (Euros) |
2025 |
| |
| |
2027 |
| |
| |
Savings plan and profit-sharing plan
The Group has a defined contribution plan (savings plan), which qualifies as a deferred salary arrangement under Section 401 (k) of the Internal Revenue Code (IRC). Once eligible, employees may elect to contribute a portion of their salaries to the savings plan, subject to certain limitations. The Group matches
F-106
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Other plans
The Group has a defined benefit pension plan for certain former Talecris Biotherapeutics, GmbH employees in Germany as required by statutory law. The pension cost relating to this plan is not material for the periods presented.
(d)Purchase commitments
Details of the Group’s raw material purchase commitments at 31 December 2023 are as follows:
| Thousands of Euros | |
2024 |
| |
2025 |
| |
2026 |
| |
2027 |
| |
2028 |
| |
More than 5 years | |
Purchase option on BPC Plasma Inc. and Haema AG
On 28 December 2018, the Group sold BPC Plasma Inc. and Haema AG to Scranton Enterprises B.V. The sales contract included a purchase option for Grifols that grants it the irrevocable and exclusive right (not an obligation) to acquire the shares sold to Scranton Enterprises B.V. (both at the same time) at any time from the effective date of sale.
The exercise price of the option will be equal to the greater of: (i) the same price at which the shares were sold to Scranton, adding the expenses related to the transaction and the increase in net working capital from the time of exercise of the option and the time at which the sale occurred, and (ii) the amount necessary to cancel the debt contracted by Scranton with the financing entity of the transaction for an amount of US Dollars
National Service Projects Organization (NSPO)
On July 29, 2021, Grifols signed an agreement with the Egyptian company National Service Projects Organization (“NSPO”) through which Grifols and NSPO has incorporated a new entity in Egypt for the construction and operation of
F-107
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Canadian Blood Services
In September 2022, Grifols signed a collaboration agreement with Canadian Blood Services (CBS) to supply them with
Euros | ||||||
2024 |
| 2025 |
| 2026 |
| 2027 |
| |
| |
| | |
(e)Judicial procedures and arbitration
Details of legal proceedings in which the Company or Group companies are involved are as follows:
| ● | ABBOTT LABORATORIES v. GRIFOLS DIAGNOSTIC SOLUTIONS INC., GRIFOLS WORLDWIDE OPERATIONS LIMITED AND NOVARTIS VACCINES AND DIAGNOSTICS, INC. |
Served: 8 October 2019
US District Court, Northern District of Illinois
Patent Infringement, Civil Action No. 1:19-cv-6587
Abbott Laboratories (“Abbott”), GDS, GWWO and Novartis Vaccines and Diagnostics, Inc. are in dispute over unpaid royalties payable by Abbott to GDS and Ortho-Clinical Diagnostics (“Ortho”) under an HIV License and Option agreement dated 16 August 2019 (the “HIV License”). On 12 September 2019, GDS and Ortho filed Notice of Arbitration. On 3 October 2019, Abbott terminated the HIV License and filed for Declaratory Relief seeking to invalidate the licensed patent. On March 16, 2020, Grifols and Ortho filed an answer and counterclaim to the litigation, while simultaneously pursuing arbitration for the pre-termination amount owed by Abbott. The arbitration hearing was 15-16 June 2020. Grifols/Ortho were awarded $
NEXT ACTION: Expert Discovery was concluded on October 14th 2022 and the parties filed dispositive motions, including a motion for summary judgement by Abbott, which was unsuccessful to dispose of the litigation. GDS and Ortho contend that the patent is valid and they believe that Abbott will be unsuccessful in its Declaratory Relief action. A mediation took place on 31 January 2024 without success. A status conference is scheduled to discuss the matter again and set further dates for trial and pre-trial hearings. Trial is expected to be in late 2024 or early 2025.
| ● | RAMIREZ-VIVAR, ALFONSO v. GRIFOLS DIAGNOSTIC SOLUTIONS, INC. |
Served: 11 March 2021
Superior Court, CA County of Alameda
Case No.: RG21089519
Wage & Hour Class Action
Plaintiff claiming violation of CA wage & hour statutes, including a claim under the Private Attorney’s General Act.
F-108
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
NEXT STEP: The Hearing on the class certification motion was heard on 28 October 2022. Court granted class certification encompassing all persons employed in California by GDS as hourly non-exempt employees during period of February 22, 2017 through November 4, 2022, relating to only
CLASS POTENTIAL: Approx.
| ● | CERUS CORPORATION v. LABORATORIOS GRIFOLS, S.A. |
Cerus Corporation (“Cerus”) and Laboratorios Grifols, S.A. (“Grifols”) entered into a Manufacturing and Supply Agreement executed in 2016, pursuant to which Grifols was to manufacture and supply to Cerus processing and filters sets to be used by Cerus in its own product (the “Agreement”). As a result of Grifols’ decision to discontinue the manufacturing, sale and support of its blood bag product business worldwide, Grifols was unable to comply with the Agreement.
In December 2021, Cerus filed a notice of arbitration in the UK pursuant to the terms of the Agreement alleging wrongful termination of the Agreement by Grifols. Furthermore, in January 2022, Cerus filed injunctive measures with the Courts of Rubí (Barcelona) requiring the suspension of the closure of Grifols’ blood bags production facility until the arbitration proceedings is finalized.
NEXT ACTION: In March 2024, the Parties agreed to further suspend the proceedings, which was granted by the Tribunal until 7 June 2024. However, the Tribunal has called the parties to a short remote hearing on 30 May 2024 in order to avoid that this matter is drifted indefinitely. In the meantime, the companies are working on activating the manufacturing and supply activities within the terms of the Agreement.
| ● | THE STATE CO. FOR MARKETING DRUGS AND MEDICAL APPLIANCES IN IRAQ (KIMADIA) v. LABORATORIOS GRIFOLS, S.A. |
The State Co. for Marketing Drugs and Medical Appliances in Iraq (“KIMADIA”) awarded a tender for the supply of blood bags to Laboratorios Grifols, S.A. (“Grifols”). Grifols, through Hali/Tiba (its agent in Iraq), informed KIMADIA on Grifols’ inability to supply the blood bags pursuant to the tender awarded, due to its decision to discontinue the manufacturing, sale and support of its blood bag product business.
The tender documents set forth a list of penalties and compensations in case the awardee is unable to supply the products to KIMADIA. Further, Hali/Tiba also claims Grifols a compensation for the services performed in relation to the tender.
NEXT ACTION: Grifols has received verbal information that KIMADIA has been able to sourced alternative product for an agreeable pricing and that discussions among Hali/Tiba and KIMADIA had not continue on the topic of possible sanctions. However, given the absence of any written confirmation on the latter, Grifols prefers to let some time go by to assure that the possible claim will not occur.
F-109
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(30) Financial Instruments
(a)Classification
Below is a breakdown of the financial instruments by nature, category and fair value. The Group does not provide details of the fair value of certain financial instruments as their carrying amount is very similar to their fair value because of its short term.
Thousands of Euros | ||||||||||||||||||||||
31/12/2023 | ||||||||||||||||||||||
Carrying amount | Fair Value | |||||||||||||||||||||
Financial assets | Financial | Financial | Other | |||||||||||||||||||
at amortised | Financial assets | assets at FV | liabilities at | financial | ||||||||||||||||||
| costs |
| at FVTPL |
| through OCI |
| Hedges |
| amortised costs |
| liabilities |
| Total |
| Level 1 |
| Level 2 |
| Level 3 |
| Total | |
Non-current financial assets |
| — | |
| | — |
| — |
| — |
| |
| |
| — |
| |
| | ||
Derivative instruments | — | — | — | | — | — | | — | | — | | |||||||||||
Trade receivables | — | — | | — | — | — | | — | | — | | |||||||||||
Financial assets measured at fair value | — | |
| | |
| — |
| — |
| |
|
|
|
|
|
|
| ||||
Non-current financial assets | | — |
| — | — |
| — |
| — |
| | |||||||||||
Other current financial assets |
| | — |
| — | — |
| — |
| — |
| |
|
|
|
|
|
|
|
| ||
Trade and other receivables |
| | — |
| — | — | — |
| — |
| |
|
|
|
|
|
|
|
| |||
Cash and cash equivalents |
| | — |
| — | — |
| — |
| — |
| |
|
|
|
|
|
|
|
| ||
Financial assets measured at amortized cost |
| | — |
| — | — |
| — |
| — |
| |
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
| |||||||||||||||
Derivatives instruments | — | ( | — | — | — | ( | — | ( | — | — | ||||||||||||
Financial liabilities measured at fair value | — | ( | — | — | — | — | ( | |||||||||||||||
Senior Unsecured & Secured Notes |
| — | — |
| — | — |
| ( |
| — |
| ( | ( |
| — |
| — |
| ( | |||
Promissory Notes |
| — | — |
| — | — |
| ( |
| — |
| ( |
|
|
|
|
|
|
| |||
Senior secured debt |
| — | — |
| — | — |
| ( |
| — |
| ( |
| — |
| ( |
| — |
| ( | ||
Other bank loans |
| — | — |
| — | — |
| ( |
| — |
| ( |
|
|
|
|
|
|
|
| ||
Lease liabilities |
| — | — |
| — | — |
| ( |
| — |
| ( |
|
|
|
|
|
|
|
| ||
Other financial liabilities |
| — | — |
| — | — |
| ( |
| — |
| ( |
|
|
|
|
|
|
|
| ||
Trade and other payables |
| — | — |
| — | — |
| ( |
| — |
| ( |
|
|
|
|
|
|
| |||
Other current liabilities |
| — | — |
| — | — |
| — |
| ( |
| ( |
|
|
|
|
|
|
|
| ||
Financial liabilities measured at amortized cost |
| — | — |
| — | — |
| ( |
| ( |
| ( |
|
|
|
|
|
|
|
| ||
| | ( |
| | |
| ( |
| ( |
| ( |
|
|
|
|
|
|
|
| |||
F-110
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Thousands of Euros | ||||||||||||||||||||||
31/12/2022 | ||||||||||||||||||||||
Carrying amount | Fair Value | |||||||||||||||||||||
Financial assets | Financial | Financial | Financial | Other | ||||||||||||||||||
at amortised | assests at | assets at FV | liabilities at | financial | ||||||||||||||||||
| costs |
| FVTPL |
| through OCI |
| Hedges |
| amortised costs |
| liabilities |
| Total |
| Level 1 |
| Level 2 |
| Level 3 |
| Total | |
Non-current financial assets |
| — | |
| | — | — |
| — |
| |
| |
| — |
| |
| | |||
Derivative instruments | — | — | — | | — | — | | — | | — | | |||||||||||
Trade receivables |
| — | — |
| | — | — |
| — |
| |
| — |
| |
| — |
| | |||
Financial assets measured at fair value |
| — | |
| | | — |
| — |
| |
|
|
|
|
|
|
| ||||
— | ||||||||||||||||||||||
Non-current financial assets |
| | — |
| — | — | — |
| — |
| |
|
|
|
|
|
|
|
| |||
Other current financial assets |
| | — |
| — | — | — |
| — |
| |
|
|
|
|
|
|
|
| |||
Trade and other receivables |
| | — |
| — | — | — | — |
| |
|
|
|
|
|
|
|
| ||||
Cash and cash equivalents |
| | — |
| — | — | — |
| — |
| |
|
|
|
|
|
|
|
| |||
Financial assets measured at amortized cost |
| | — |
| — | — | — |
| — |
| |
|
|
|
|
|
|
|
| |||
Derivatives instruments | — | ( | — | — | — | ( | — | ( | — | ( | ||||||||||||
Financial liabilities measured at fair value | — | ( | — | — | — | — | ( | |||||||||||||||
Senior Unsecured & Secured Notes |
| — | — |
| — | — | ( |
| — |
| ( |
| ( |
| — |
| — |
| ( | |||
Promissory Notes |
| — | — |
| — | — | ( |
| — |
| ( |
|
|
|
|
|
|
| ||||
Senior secured debt |
| — | — |
| — | — | ( |
| — |
| ( |
| — |
| ( |
| — |
| ( | |||
Other bank loans |
| — | — |
| — | — | ( |
| — |
| ( |
|
|
|
|
|
|
| ||||
Lease liabilities |
| — | — |
| — | — | ( |
| — |
| ( |
|
|
|
|
|
|
| ||||
Other financial liabilities |
| — | — |
| — | — | ( |
| — |
| ( |
|
|
|
|
|
|
| ||||
Other non-current debts | — | — | — | — | — | ( | ( | |||||||||||||||
Trade and other payables |
| — | — |
| — | — | ( |
| — |
| ( |
|
|
|
|
|
|
| ||||
Other current liabilities |
| — | — |
| — | — | — |
| ( |
| ( |
|
|
|
|
|
|
| ||||
Financial liabilities measured at amortized cost |
| — | — |
| — | — | ( |
| ( |
| ( |
|
|
|
|
|
|
| ||||
| | ( |
| | | ( |
| ( |
| ( |
|
|
|
|
|
|
| |||||
(b)Measurement of fair value
In order to determine the fair value of financial assets or liabilities, the Group uses the following hierarchy based on the relevance of the variables used:
| ● | Level 1: estimations based on quoted prices of the instrument. |
| ● | Level 2: estimations based on significant observable variables coming directly from the market. |
| ● | Level 3: estimations based on valuation techniques other than observable variables in the market, mainly discounted cash flows. |
(c)Financial risk management
This item provides information on the Group’s exposure to risk associated with the use of financial instruments, the Group’s objectives and procedures to measure and mitigate this risk, and the Group’s capital management strategy.
The Group is exposed to the following risks:
| ● | Credit risk |
| ● | Liquidity risk |
| ● | Market risk: includes interest rate risk, currency risk and other price risks. |
F-111
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The Group’s risk management policies are established to identify and analyze the risks faced by the Group, define appropriate risk limits and controls and to control risks and comply with limits. Risk management policies and procedures are reviewed regularly so that they reflect changes in market conditions and the Group’s activities. The Group’s management procedures and rules are designed to create a strict and constructive control environment in which all employees understand their duties and obligations.
The Group’s Audit Committee supervises how management controls compliance with the Group’s risk management procedures and policies and reviews whether the risk management policy is suitable considering the risks to which the Group is exposed. This committee is assisted by Internal Audit which acts as supervisor. Internal Audit performs regular and ad hoc reviews of the risk management controls and procedures and reports its findings to the Audit Committee.
(i)Credit risk
Credit risk is the risk to which the Group is exposed in the event that a customer or counterparty to a financial instrument fails to discharge a contractual obligation, and mainly results from trade receivables and the Group’s investments in financial assets.
Trade receivables
The main risk is that of late payments, which is mitigated through the possibility of claiming interest as foreseen by prevailing legislation. No significant bad debt or late payment issues have been detected for sales to private entities.
The Group recognizes impairment based on its best estimate of the expected losses on trade and other receivables. The main impairment losses recognized are due to specific losses relating to individually identified risks. At year end, these impairment losses are immaterial.
Concentration of credit risk
For trade receivables the Group uses the simplified approach, estimating lifetime expected credit losses, while for all other financial assets the Group uses the general approach for calculating expected credit losses. In both cases, due to the customers’ credit rating, as well as the internal classification systems currently in place for new customers and considering that collection periods are mostly under
Exposure to credit risk
The carrying amount of financial assets represents the maximum exposure to credit risk. At 31 December 2023 and 2022 the maximum level of exposure to credit risk is as follows:
Thousands of Euros | ||||||
Carrying amount |
| Reference |
| 31/12/2023 |
| 31/12/2022 |
Non-current financial assets |
| Note 11 |
| |
| |
Other current financial assets |
| Note 11 |
| |
| |
Contractual assets | Note 14 | | | |||
Trade receivables |
| Note 15 |
| |
| |
Other receivables |
| Note 15 |
| |
| |
Cash and cash equivalents |
| Note 16 |
| |
| |
|
|
| |
| | |
F-112
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The maximum level of exposure to risk associated with receivables and contractual assets at 31 December 2023 and 2022, by geographical area, is as follows.
Thousands of Euros | ||||
Carrying amount |
| 31/12/2023 |
| 31/12/2022 |
Spain |
| |
| |
EU countries |
| |
| |
United States of America |
| |
| |
Other European countries |
| |
| |
Other regions |
| |
| |
| |
| | |
Impairment losses
A breakdown of the trade and other receivables and contractual assets net of the impairment losses by ageing at 31 December 2023 is as follows:
Thousands of Euros | ||||||||
Total net third | ||||||||
Total gross carrying | party trade | |||||||
| ECL Rate |
| amount |
| Provision |
| receivables | |
Not matured |
| | % | |
| ( |
| |
Past due 0-30 days |
| | % | |
| ( |
| |
Past due 31-60 days |
| | % | |
| ( |
| |
Past due 61-90 days |
| | % | |
| ( |
| |
Past due 91-180 days |
| | % | |
| ( |
| |
Past due 181-365 days |
| | % | |
| ( |
| |
More than one year |
| | % | | ( | | ||
Customers with objective evidence of impairment |
|
|
| | ( | — | ||
| | ( | | |||||
An impairment matrix based on the length of time overdue was used to monitor receivables portfolios that do not show any specific indications of impairment in individual cases. For trade receivables related to customers from the Middle East which are overdue by more than one year, the flat-rate percentages from the impairment matrix were adjusted due to special default patterns.
F-113
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
A breakdown of the trade and other receivables and contractual assets net of the impairment losses by ageing as of 31 December 2022 is as follows:
Thousands of Euros | ||||||||
Total net third | ||||||||
Total gross carrying | party trade | |||||||
| ECL Rate |
| amount |
| Provision |
| receivables | |
Not matured |
| | % | |
| ( |
| |
Past due 0-30 days |
| | % | |
| ( |
| |
Past due 31-60 days |
| | % | |
| ( |
| |
Past due 61-90 days |
| | % | |
| ( |
| |
Past due 91-180 days |
| | % | |
| ( |
| |
Past due 181-365 days |
| | % | |
| ( |
| |
More than one year |
| | % | |
| ( |
| — |
Customers with objective evidence of impairment |
|
|
| |
| ( |
| — |
| |
| ( |
| | |||
Movement in the bad debt provision was as follows:
Thousands of Euros | ||||||
| 31/12/2023 |
| 31/12/2022 |
| 31/12/2021 | |
Opening balance |
| |
| |
| |
Net charges for the year |
| |
| |
| |
Net cancellations for the year |
| ( |
| ( |
| ( |
Transfers | | | — | |||
Translation differences |
| ( |
| |
| |
Closing balance |
| |
| |
| |
The Group does not have significant credit risk, with both treasury placements and the contracting of derivatives being carried out with highly solvent financial institutions.
(ii)Liquidity risk
Liquidity risk is the risk that the Group cannot meet its financial obligations as they fall due. The Group’s approach to managing liquidity is to ensure where possible, that it always has sufficient liquidity to settle its obligations at the maturity date, both in normal conditions and in times of tension, to avoid incurring unacceptable losses or tarnishing the Group’s reputation.
The Group manages liquidity risk on a prudent basis, based on availability of cash and sufficient committed unused long-term credit facilities, enabling the Group to implement its business plans and carry out operations using stable and secure sources of financing.
At 31 December 2023 the Group has total cash and cash equivalents of Euros
F-114
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The Group is able to provide sufficient liquidity to fund its current obligations based on cash flows from operations combined with cash balances and availability of unused credit lines, and it is committed to maintaining elevated and adequate levels of liquidity through internally generated cash flows, and a decrease in dividend payments in the medium term. Additionally, currently the Group does not generate significant cash in any country that might have restrictions on the repatriation of funds.
As in previous years, the Group continues with its quarterly program for optimization of working capital, which is mainly based on contracts to sell receivables without recourse.
The main contractual obligations existing at the end of the fiscal year comprise mainly long-term financial debt obligations with capital repayments and interest payments (see note 21).
The Group’s treasury budget plans to pay all its commitments in the next 12 months. Additionally, the cash received from the divestment in Shanghai RAAS (see Notes 10 and 12) and the improvement in operating cash flow will be used to continue reducing the level of indebtedness initiated in previous years. On the other hand, the Group has various additional financing alternatives such as negotiating with debt holders, accessing the debt market or possible divestments in non-strategic assets, to optimize the debt structure and its financial cost.
Details of the contractual maturity dates of financial liabilities including committed interest calculated using interest rate forward curves are as follows:
Thousands of Euros | ||||||||||||||||
|
| Carrying |
|
|
|
|
|
|
|
|
|
| ||||
amount at | Contractual | 6 months | 6 - 12 | 1-2 | 2 - 5 | More than | ||||||||||
Carrying amount |
| Reference |
| 31/12/23 |
| flows |
| or less |
| months |
| years |
| years |
| 5 years |
Financial liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bank loans |
| Note 21 |
| |
| |
| |
| |
| |
| |
| — |
Other financial liabilities |
| Note 21 |
| |
| |
| |
| |
| |
| |
| |
Bonds and other marketable securities |
| Note 21 |
| |
| |
| |
| |
| |
| |
| — |
Lease liabilities |
| Note 21 |
| |
| |
| |
| |
| |
| |
| |
Payable to suppliers |
| Note 22 |
| |
| |
| |
| |
| — |
| — |
| — |
Other current liabilities |
| Note 23 |
| |
| |
| |
| |
| — |
| — |
| — |
Financial derivatives | Note 30 (d) | | | | — | | — | — | ||||||||
Other commitments | Note 10 | — | | | | | | — | ||||||||
Total |
|
|
| |
| |
| |
| |
| |
| |
| |
F-115
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Thousands of Euros | ||||||||||||||||
|
|
| Carrying |
|
|
|
|
|
|
|
|
|
| |||
amount at | Contractual | 6 months | 6 - 12 | 1-2 | 2 - 5 | More than | ||||||||||
Carrying amount | Reference | 31/12/22 | flows | or less | months | years | years | 5 years | ||||||||
Financial liabilities |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bank loans |
| Note 21 |
| |
| |
| |
| |
| |
| |
| — |
Other financial liabilities |
| Note 21 |
| |
| |
| |
| |
| |
| |
| |
Bonds and other marketable securities |
| Note 21 |
| |
| |
| |
| |
| |
| |
| — |
Lease liabilities |
| Note 21 |
| |
| |
| |
| |
| |
| |
| |
Payable to suppliers |
| Note 22 |
| |
| |
| |
| |
| — |
| — |
| — |
Other current liabilities |
| Note 23 |
| |
| |
| |
| |
| — |
| — |
| — |
Financial derivatives | Note 30 (d) | | | | — | | | — | ||||||||
Total |
|
|
| |
| |
| |
| |
| |
| |
| |
In addition, on 31 December 2023 and 2022, the Group has a call option that grants it the irrevocable and exclusive right (not an obligation) to acquire the companies Haema AG and BPC Plasma Inc. (see note 29).
(iii)Currency risk
The Group operates internationally and is therefore exposed to currency risk when operating with foreign currencies, especially with regard to the US Dollar. Currency risk is associated with future commercial transactions, recognized assets and liabilities, and net investments in foreign operations.
The Group holds significant investments in foreign operations, the net assets of which are exposed to currency risk. The conversion risk affecting net assets of the Group’s foreign operations in US Dollars is mitigated primarily through borrowings in this foreign currency.
The Group’s main exposure to currency risk is with regard to the US Dollar, which is used in a significant percentage of transactions in foreign functional currencies.
The financing obtained in Euros represents
As mentioned in note 21, part of the US Dollar debt of the Group is covered by a currency swap to hedge the exposure to the associated currency risk.
The Group applies the cost of hedging method. This method enables the Group to exclude the currency basis spread from the designated hedging instrument and, subject to certain requirements, changes in their fair value attributable to this component are recognized in other comprehensive income.
F-116
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Details of the Group’s exposure to currency risk is as follows:
Thousands of Euros | ||||
31/12/2023 | ||||
| Euros (*) |
| US Dollars (**) | |
Trade receivables |
| |
| |
Receivables from Group companies |
| |
| |
Loans to Group companies |
| |
| |
Cash and cash equivalents |
| |
| |
Trade payables |
| ( |
| ( |
Payables to Group companies |
| ( |
| ( |
Loans from Group companies |
| ( |
| — |
Bank loans |
| ( |
| — |
|
|
|
| |
Balance sheet exposure |
| ( |
| ( |
(*) Balances in Euros in subsidiaries with US Dollars functional currency
(**) Balances in US Dollars in subsidiaries with Euros functional currency
Thousands of Euros | ||||
31/12/2022 | ||||
| Euros (*) |
| US Dollars (**) | |
Trade receivables |
| |
| |
Receivables from Group companies |
| |
| |
Loans to Group companies |
| |
| |
Cash and cash equivalents |
| |
| |
Trade payables |
| ( |
| ( |
Payables to Group companies |
| ( |
| ( |
Loans from Group companies |
| ( |
| — |
Bank loans |
| ( |
| — |
|
|
|
| |
Balance sheet exposure |
| |
| |
(*) Balances in Euros in subsidiaries with US Dollar functional currency
(**) Balances in US Dollar in subsidiaries with Euros functional currency
The most significant exchange rates applied at 2023 and 2022 year ends are as follows:
Closing exchange rate | ||||
Euros |
| 31/12/2023 |
| 31/12/2022 |
US Dollars |
|
| | |
A sensitivity analysis for foreign exchange fluctuations is as follows:
Had the US Dollar strengthened by
F-117
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
A
The Group uses hedge accounting to partially hedge the currency risk exposure (See note 30 (d)).
(iv)Interest rate risk
The Group’s interest rate risks arise from current and non-current borrowings. Borrowings at variable interest rates expose the Group to cash flow interest rate risks. Fixed-rate borrowings expose the Group to fair value interest rate risk.
The objective of the management of interest rate risk is to achieve a balance in the structure of the debt, keeping part of the external resources issued at a fixed rate and covering part of the variable rate debt through hedges.
A significant part of the financing obtained accrues interest at fixed rates, representing
Variable-rate debt represents
To date, the profile of interest on interest-bearing financial instruments is as follows:
Thousands of Euros | ||||
| 31/12/2023 |
| 31/12/2022 | |
Fixed-interest financial instruments |
|
|
|
|
Financial liabilities |
| ( |
| ( |
| ( |
| ( | |
Variable-interest financial instruments | ||||
Financial liabilities |
| ( |
| ( |
| ( |
| ( | |
( | ( | |||
Had the interest rate been
(v) Market price risk
Price risk affecting raw materials is mitigated by the vertical integration of the hemoderivatives business in a highly concentrated sector.
F-118
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(d)Financial derivatives
At 31 December 2023 and 2022 the Group has recognized the following derivatives:
Thousands of Euros | ||||||||||||
Notional | Notional | |||||||||||
amount at | amount at | Value at | Value at | |||||||||
Financial derivatives |
| Currency |
| 31/12/2023 |
| 31/12/2022 |
| 31/12/23 |
| 31/12/22 |
| Maturity |
Cross currency interest rate swap |
| US Dollar |
| |
| |
| |
| |
| 15/10/2024 |
Cross currency interest rate swap |
| US Dollar |
| |
| |
| ( |
| |
| 15/10/2024 |
Foreign exchange rate forward |
| Swiss Franc |
| |
| |
| |
| |
| 05/02/2024 |
Foreign exchange rate forward |
| Canadian dollar |
| |
| |
| |
| |
| 07/02/2024 |
Foreign exchange rate forward |
| Pound Sterling |
| — |
| |
| — |
| |
| 29/11/2023 |
Foreign exchange rate forward | Czech crown | | — | | — | 12/02/2024 | ||||||
Foreign exchange rate forward | Mexican Peso | | — | | — | 12/02/2024 | ||||||
Foreign exchange rate forward | Turkish lira | | — | | — | 31/01/2024 | ||||||
Foreign exchange rate forward |
| US Dollar |
| |
| |
| |
| |
| 29/02/2024 |
Foreign exchange rate forward | Euro | | | | | 22/01/2024 | ||||||
Energy PPA | Euro / KwH | — | — | | — | 31/12/2032 | ||||||
Total assets (note 11) |
|
|
| |
| |
|
| ||||
Cross currency interest rate swap |
| US Dollar |
| |
| |
| ( |
| ( |
| 15/10/2024 |
Foreign exchange rate forward |
| Canadian dollar |
| |
| |
| ( |
| ( |
| 05/01/2024 |
Foreign exchange rate forward |
| US Dollar |
| |
| |
| ( |
| ( |
| 30/01/2023 |
Foreign exchange rate forward |
| Czech crown |
| |
| — |
| ( |
| — |
| 12/02/2024 |
Foreign exchange rate forward |
| Pound Sterling |
| |
| — |
| ( |
| — |
| 12/02/2024 |
Foreign exchange rate forward | Japanese Yen | | — | ( | — | 07/02/2024 | ||||||
Total liabilities (note 20) |
|
|
| ( |
| ( | ||||||
(i)Hedging derivative financial instruments
On 5 October 2021, the Group subscribed
- The Group receives a loan of Euros
- The Group grants a US Dollars
On 28 June 2022, the Group subscribed one cross currency interest-rate swap of US Dollars
- The Group receives a Euros
- The Group grants a US Dollars
The derivative complies with the criteria required for hedge accounting. See further details in notes 4 (i).
F-119
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(ii)Derivative financial instruments at fair value through profit and loss
The Group has subscribed various foreign exchange forwards to partially hedge the foreign currency value of intercompany loan. Since the Group chooses not to apply hedge accounting criteria, gains or losses resulting from changes in the fair value of derivatives are taken directly to “Change in fair value of financial instruments” in the consolidated statement of profit and loss. At 31 December 2023, the Group has recognized a net finance cost of Euros
(iii)Electricity derivative
At the beginning of 2023, the Company contracted a hedge on the variation of the price of electricity. This contract has served in its entirety to cover the purchase price of electricity against potential market price increases. The energy price hedging derivatives meet the requirements to apply hedge accounting, so the variations in the value of this financial instrument are recorded (by the net amount of taxes) in equity.
The movement in derivative financial instruments is as follows:
| Thousands of Euros | |||
| 31/12/2023 | 31/12/2022 | ||
Opening balance | | | ||
Business combination |
| — |
| ( |
Changes in fair value recognized in equity |
| |
| ( |
Transfer to profit or loss |
| |
| |
Transfer to profit or loss - translation differences |
| ( |
| |
Tax effect |
| ( |
| |
Collections / Payments |
| ( |
| ( |
Closing balance |
| |
| |
(e)Capital management
The directors’ policy is to maintain a solid capital base in order to ensure investor, creditor and market confidence and sustain future business development. The board of directors defines and proposes the level of dividends paid to shareholders.
The capital structure is periodically reviewed through the preparation of strategic plans focused mainly on a sequential improvement of EBITDA (Earnings before interest, tax, amortization and depreciation), generation of operating cash and discipline in the allocation of capital; with the objective and commitment to reduce the leverage ratio.
In accordance with the senior secured debt contract, the Group is subject to compliance with some covenants. At 31 December 2023 and 2022, the Group complies with the covenants in the contract.
F-120
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
The credit rating of the Group is as follows:
|
| January 2024 |
| September 2023 |
| March 2023 | September 2022 | |||
Moody's Investors |
| Corporate rating |
| B2 | B1 | |||||
| Senior secured debt |
| Ba3 | Ba3 | ||||||
| Senior Unsecured debt |
| Caa1 | B3 | ||||||
| Perspective |
| Negative | Negative | ||||||
Standard & Poor's |
| Corporate rating | B+ |
|
| B+ | ||||
| Senior secured debt | BB- |
|
| BB- | |||||
| Senior Unsecured debt | B- |
|
| B- | |||||
| Perspective | Stable |
|
| Stable | |||||
Fitch Ratings |
| Corporate rating |
| BB- |
| BB- | ||||
| Senior secured debt |
| BB+ |
| BB+ | |||||
| Senior Unsecured debt |
| B+ |
| B+ | |||||
| Perspective |
| Stable |
| Stable |
The Parent held Class A and B treasury stock equivalent to
(31) Balances and Transactions with Related Parties
(a)Group balances with related parties
Details of balances with related parties at 31 December 2023 are as follows:
Thousands of Euros | ||||||||||
Carrying amount | Reference | Associates | Key management personeel | Other related parties |
| Board of directors | ||||
Receivables |
| 15 |
| |
| — |
| |
| — |
Other financial assets |
| 11 |
| |
| — |
| |
| — |
Loans |
| 11 |
| — |
| — |
| |
| — |
Guarantee deposits |
| 11 |
| — |
| — |
| |
| — |
Total debtors |
|
|
| |
| — |
| |
| — |
Debts |
|
|
| — |
| ( |
| ( |
| ( |
Total creditors |
|
|
| — |
| ( |
| ( |
| ( |
Total |
|
|
| |
| ( |
| |
| ( |
F-121
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Details of balances with related parties at 31 December 2022, restated to be comparative with details of balances with related parties for 2023, are as follows:
Thousands of Euros | ||||||||||||
Carrying amount | Reference | Associates | Joint ventures | Key management personeel | Other related parties | Board of directors | ||||||
Receivables |
| 15 |
| |
| — |
| — |
| — |
| — |
Current contract assets |
| 15 |
| |
| — |
| — |
| — |
| — |
Other financial assets |
| 11 |
| — |
| |
| — |
| |
| — |
Advanced payments |
| 15 |
| — |
| |
| — |
| — |
| — |
Loans |
| 11 |
| — |
| — |
| — |
| |
| — |
Guarantee deposits |
| 11 |
| — |
| — |
| — |
| |
| — |
Total debtors |
|
|
| |
| |
| — |
| |
| — |
Trade payables |
|
|
| ( |
| ( |
| — |
| — |
| — |
Debts |
|
|
| — |
| — |
| ( |
| ( |
| ( |
Total creditors |
|
|
| ( |
| ( |
| ( |
| ( |
| ( |
Total |
|
|
| |
| |
| ( |
| |
| ( |
The heading “Receivables” corresponding to associates includes outstanding balances from sales to associated companies, mainly corresponding to Anhui Tonrol Pharmaceutical Co. (company of the Shanghai RAAS Blood Products, Co. Ltd. Group) (Euros
The heading “Loans” mainly includes a loan signed by Scranton Plasma, BV. with the group on 28 December 2018 for an initial amount of US Dollars
The heading “Other financial assets” balance corresponding to other related parties corresponds to a cash-pooling financing agreement that BPC Plasma, Inc and Haema, AG have with Scranton Plasma, BV with maturity in 2024 (see note 11).
The heading of “debts” includes an amount of Euros
F-122
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(b) | Group transactions with related parties |
Group transactions with related parties during 2023 are as follows:
Thousands of Euros | ||||||||
|
| Key management |
| Other related |
| Board of directors | ||
Associates | personnel | parties | of the Company | |||||
Net sales |
| | — |
| |
| — | |
Purchases | ( | — |
| ( |
| — | ||
Rendering of services | ( | — | ( | — | ||||
Remuneration |
| — | ( |
| — |
| ( | |
Payments for rights of use |
| — | — |
| ( |
| — | |
Finance income | — | — | | — | ||||
Dividends paid/received | | — | ( | — | ||||
Loans | — | — | | — | ||||
| | ( |
| ( |
| ( | ||
Group transactions with related parties during 2022 were as follows:
Thousands of Euros | ||||||||||
|
| Joint |
| Key management |
| Other related |
| Board of directors | ||
Associates | ventures | personnel | parties | of the Company | ||||||
Net sales |
| | — |
| — |
| — |
| — | |
Purchases |
| ( | ( | — | — | — | ||||
Rendering of services | ( | — | — | ( | — | |||||
Remuneration |
| — | — |
| ( |
| — |
| ( | |
Payments for rights of use | — | — | — | ( | — | |||||
Purchase of property, plant and equipment | — | — | — | | — | |||||
Finance income | — | — | — | | — | |||||
Dividends paid/received | | — | — | — | — | |||||
Loans | — | — | — | | — | |||||
| | ( |
| ( |
| |
| ( | ||
Group transactions with related parties during 2021 were as follows:
Thousands of Euros | ||||||||
|
| Key management |
| Other related |
| Board of directors | ||
Associates | personnel | parties | of the Company | |||||
Net sales |
| | — |
| — |
| — | |
Purchases |
| ( | — | — | — | |||
Rendering of services | ( | — | ( | — | ||||
Remuneration |
| — | ( |
| — |
| ( | |
Payments for rights of use | — | — | ( | — | ||||
Purchase of property, plant and equipment | — | — | | — | ||||
Finance income | | — |
| |
| — | ||
Dividends paid/received |
| | — | — | — | |||
Loans | — | — | | — | ||||
| | ( |
| |
| ( | ||
F-123
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Every year the Group contributes
“Net sales” includes sales to associated companies mainly corresponding to Anhui Tonrol Pharmaceutical Co. (company of the Shanghai RAAS Blood Products, Co. Ltd. Group) (Euros
“Purchases” mainly included in 2022 purchases of plasma from the centers related to the collaboration agreement with Biotek America, LLC (see note 10).
“Other service expenses” includes an amount of Euros
“Payments for right-of-use assets” corresponds to the office buildings of Grifols in Sant Cugat del Vallès. All lease contracts have a maturity date of 1 March 2045.
“Finance income” mainly includes accrued interest (Euros
The dividends received correspond to the associated companies Shanghai RAAS Blood Products Co. Ltd., Bio Darou P.J.S. Co. and Access Biologicals LLC. Additionally, the dividends distributed correspond to BPC Plasma Inc. (see note 11).
“Loans” mainly includes the net amounts disbursed under the cash-pooling financing agreement that BPC Plasma, Inc and Haema, AG have with Scranton Plasma, BV mentioned above.
Directors representing shareholders´ interests have received remuneration of Euros
The Group has not extended any advances or loans to the members of the board of directors or key management personnel nor has it assumed any guarantee commitments on their behalf. It has also not assumed any pension or life insurance obligations on behalf of former or current members of the board of directors or key management personnel. In addition, certain Company directors and key management personnel have termination benefit commitments (see note 29).
(c) | Conflicts of interest concerning the directors |
The Company’s directors and their related parties have not entered into any conflict of interest that should have been reported in accordance with article 229 of the revised Spanish Companies Act.
F-124
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
(32) Subsequent events
Gotham City Research Report
On 9 January 2024, a short seller investor issued a report based on speculation and false information regarding Grifols’ accounting and financial information. Although the company’s fundamentals remain sound and unchanged and all financial information was duly reported in the audited financial statements, this action had a significant impact on Grifols’ share price and corporate reputation.
The company is currently working to restore the confidence of markets, shareholders and other stakeholders in three key areas:
| o | Communication and collaboration with the Spanish regulator (CNMV). |
| o | Transparent communication with all our stakeholders: sharing our clear response to the published report through live conference calls and multiple official communications on the company’s website and on the CNMV portal. All press releases are publicly available on Grifols’ website |
| o | Clear and transparent communication with our teams and employee representatives, including major unions. |
| o | Reinforced communication with investors, official communications, direct phone calls, video calls and e-mails. |
| o | The company filed a complaint in the United States District Court for the Southern District of New York against Daniel Yu, Gotham City Research LLC, General Industrial Partners LLP, Cyrus de Weck, and their affiliates to claim for the financial and reputational damages caused to Grifols and their stakeholders as a result of the defendants’ actions. |
| o | The company established a dedicated working group comprising senior managers from the legal, communications, finance, investor relations and management teams, together with external advisors with expertise in communications. |
As a result of the information published by Gotham City Research LLC, in relation to the accounting and financial information of Grifols, S.A. and subsidiaries, the National Securities Market Commission (CNMV), in the exercise of its supervisory powers, has made various requests for information to the Group. The Parent Company has responded to the requirements received and the National Securities Market Commission has shared its analysis of the Group financial information concluding that no significant errors were identified.
SRAAS Share Purchase Agreement
As indicated in note 12, Grifols and Haier Group Corporation (“Haier”) entered into a Strategic Alliance and Share Purchase Agreement to transfer the
Senior debt issuance
On 10 April 2024 the Group informed the market through the National Securities Market Commission that it is actively working to issue senior secured notes, the funds of which, if successful, will be used to refinance senior unsecured notes maturing in 2025.
F-125
GRIFOLS, S.A. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(in thousand Euros)
Acquisition of
On 2 April 2024, the Group, through its
Aggregate details of the cost of the business combination, the provisional fair value of the net assets acquired and the provisional goodwill (or the amount by which the business combination cost exceeds the fair value of the net assets acquired) at the acquisition date are shown below. The values shown in the table below should be considered provisional.
For practical purposes, for the present transaction, the Euro / Dollar exchange rate of
| Thousands of Euros |
| Thousands of US Dollars | |
Cost of the business combination | ||||
Consideration paid | ||||
Total consideration paid | ||||
Fair value of net assets acquired | ||||
Goodwill (excess of the cost of the business combination over the fair value of net assets acquired) |
The amounts determined at the acquisition date of the assets, liabilities and contingent liabilities acquired are as follows:
| Fair value | |||
Thousands of Euros |
| Thousands of US Dollars | ||
Assets |
|
|
|
|
Rights of Use |
| |
| |
Property, plant and equipment |
| |
| |
Inventories |
| |
| |
Total Assets |
| |
| |
Liabilities |
|
|
|
|
Non-current financial liabilities |
| ( |
| ( |
Current financial liabilities |
| ( |
| ( |
Total Liabilities |
| ( |
| ( |
Total net assets acquired |
| |
| |
The resulting goodwill will be allocated to the Biopharma segment.
F-126
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
Acquisition / | 12/31/2023 | 12/31/2022 | 12/31/2021 |
| |||||||||||||||||||
Registered | Incorporation | % shares | % shares | % shares |
| ||||||||||||||||||
Name |
| Offices |
| Office |
| date |
| Activity |
| Statutory Activity |
| Direct |
| Indirect |
| Direct |
| Indirect |
| Direct |
| Indirect |
|
Fully Consolidated Companies |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||
Diagnostic Grifols, S.A. |
| Spain | Polígono Levante |
| 1987 |
| Industrial |
| Development and manufacture of diagnostic equipment, instruments and reagents. |
| — |
| | % | — |
| | % | — |
| | % | |
Instituto Grifols, S.A. |
| Spain | Polígono Levante |
| 1987 |
| Industrial |
| Plasma fractioning and the manufacture of haemoderivative pharmaceutical products. |
| | % | | % | | % | | % | | % | | % | |
Laboratorios Grifols, S.A. |
| Spain | Polígono Levante |
| 1989 |
| Industrial |
| Production of glass- and plastic-packaged parenteral solutions, parenteral and enteral nutrition products |
| | % | — | | % | — | | % | | % | |||
Biomat, S.A. |
| Spain | Polígono Levante |
| 1991 |
| Industrial |
| Analysis and certification of the quality of plasma used by Instituto Grifols, S.A. It also provides transfusion centres with plasma virus inactivation services (I.P.T.H). |
| | % | | % | | % | | % | | % | | % | |
F-127
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
Grifols Engineering, S.A. |
| Spain | Polígono Levante |
| 2000 |
| Industrial |
| Design and development of the Group’s manufacturing installations and part of the equipment and machinery used at these premises. The company also renders engineering services to external companies. |
| | % | | % | | % | | % | | % | | % | |
Biomat USA, Inc. |
| United States | 2410 Lillyvale Avenue |
| 2002 |
| Industrial |
| Procuring human plasma. |
| — |
| | % | — |
| | % | — |
| | % | |
Grifols Biologicals, LLC. |
| United States | 5555 Valley Boulevard |
| 2003 |
| Industrial |
| Plasma fractioning and the production of haemoderivatives. |
| — |
| | % | — |
| | % | — |
| | % | |
Grifols Australia Pty Ltd. |
| Australia | Unit 5/80 Fairbank |
| 2009 |
| Industrial |
| Distribution of pharmaceutical products and the development and manufacture of reagents for |
| | % | — |
| | % | — |
| | % | — |
| |
Medion Grifols Diagnostic AG |
| Switzerland | Bonnstrasse,9 |
| 2009 |
| Industrial |
| Development and manufacturing activities in the area of biotechnology and diagnostics. |
| — |
| | % | — |
| | % | — |
| | % | |
Grifols Therapeutics, LLC. |
| United States | 4101 Research Commons |
| 2011 |
| Industrial |
| Plasma fractioning and the production of haemoderivatives. |
| — |
| | % | — |
| | % | — |
| | % | |
F-128
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
Talecris Plasma Resources, Inc. (merged with Biomat USA, Inc.) |
| United States | 4101 Research Commons |
| 2011 |
| Industrial |
| Procurement of human plasma. |
| — |
| — | — |
| — | — |
| | % | |||
Grifols Worldwide Operations Limited |
| Grange Castle Business Park, |
| 2012 |
| Industrial |
| Packaging, labelling, storage, distribution, manufacture and development of pharmaceutical products |
| | % | — |
| | % | — |
| | % | — |
| ||
Progenika Biopharma, S.A. |
| Parque Tecnológico de Vizcaya, |
| 2013 |
| Industrial |
| Development, production and commercialisation of biotechnological solutions. |
| | % | | % | | % | | % | | % | | % | ||
Grifols Diagnostics Solutions, Inc. |
| 4560 Horton Street |
| 2013 |
| Industrial |
| Manufacture and sale of blood testing products |
| — | | % | — | | % | — | | % | |||||
Grifols Worldwide Operations USA Inc. |
| 13111 Temple Avenue, City of |
| 2014 |
| Industrial |
| Manufacture, warehousing, and logistical support for biological products. |
| — |
| | % | — |
| | % | — |
| | % | ||
Grifols Asia Pacific Pte, Ltd |
| Singapore | 501 Orchard Road nº20-01 |
| 2003 |
| Commercial |
| Distribution and sale of medical and pharmaceutical products. |
| | % | — |
| | % | — |
| | % | — |
| |
Grifols Movaco, S.A. |
| Polígono Levante |
| 1987 |
| Commercial |
| Distribution and sale of reagents, chemical products and other pharmaceutical specialities, and |
| | % | | % | | % | | % | | % | | % |
F-129
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
of medical and surgical materials, equipment and instruments for use by laboratories and health centres. | |||||||||||||||||||||||
Grifols Portugal Productos Farmacéuticos e Hospitalares, Lda. |
| Portugal | Rua de Sao Sebastiao,2 |
| 1988 |
| Commercial |
| Import, export and commercialisation of pharmaceutical and hospital equipment and products, particularly Grifols products. |
| | % | | % | | % | | % | | % | | % | |
Grifols Chile, S.A. |
| Chile | Avda. Americo Vespucio, 2242 |
| 1990 |
| Commercial |
| Development of pharmaceutical businesses, which can involve the import, production, commercialisation and export of related products. |
| | % | — |
| | % | — |
| | % | — |
| |
Grifols USA, LLC. |
| United States | 2410 Lillyvale Avenue |
| 1990 |
| Commercial |
| Distribution and marketing of company products. |
| — |
| | % | — |
| | % | — |
| | % | |
Grifols Argentina, S.A. |
| Argentina | Bartolomé Mitre 3690/3790, |
| 1991 |
| Commercial |
| Clinical and biological research. Preparation of reagents and therapeutic and diet products. Manufacture and commercialisation of other pharmaceutical specialities. |
| | % | | % | | % | | % | | % | | % | |
Grifols s.r.o. |
| Czech Republic | Calle Zitna,2 |
| 1992 |
| Commercial |
| Purchase, sale and distribution of chemical- |
| | % | — |
| | % | — |
| | % | — |
|
F-130
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
pharmaceutical products, including human plasma. | |||||||||||||||||||||||
Grifols (Thailand) Ltd |
| Thailand | 191 Silom Complex Building, |
| 2003 |
| Commercial |
| Import, export and distribution of pharmaceutical products. |
| — |
| | % | — |
| | % | — |
| | % | |
Grifols Malaysia Sdn Bhd |
| Malaysia | Level 18, The Gardens North |
| 2003 |
| Commercial |
| Distribution and sale of pharmaceutical products. |
| — |
| | % | — |
| | % | — |
| | % | |
Grifols International, S.A. |
| Spain | Polígono Levante |
| 1997 |
| Commercial |
| Coordination of the marketing, sales and logistics for all the Group’s subsidiaries operating in other |
| | % | | % | | % | | % | | % | | % | |
Grifols Italia S.p.A |
| Italy | Via Carducci, 62d |
| 1997 |
| Commercial |
| Purchase, sale and distribution of chemical-pharmaceutical products. |
| | % | — |
| | % | — |
| | % | — |
| |
Grifols UK Ltd. |
| United Kingdom | Gregory Rowcliffe & Milners, 1 |
| 1997 |
| Commercial |
| Distribution and sale of therapeutic and other pharmaceutical products, especially haemoderivatives. |
| | % | — | | % | — |
| | % | — |
| ||
Grifols Brasil, Lda. |
| Brazil | Rua Umuarama, 263 |
| 1998 |
| Commercial |
| Import and export, preparation, distribution and sale of pharmaceutical and chemical products for |
| | % | — | | % | — | | % | — |
F-131
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
equipment and instruments. | |||||||||||||||||||||||
Grifols France, S.A.R.L. |
| France | Arteparc, Rue de la Belle du Canet, |
| 1999 |
| Commercial |
| Commercialisation of chemical and healthcare products. |
| | % | | % | | % | | % | | % | | % | |
Grifols Polska Sp.z.o.o. |
| Poland | Grzybowska 87 street00-844 |
| 2003 |
| Commercial |
| Distribution and sale of pharmaceutical, cosmetic and other products. |
| | % | — |
| | % | — |
| | % | — |
| |
Logística Grifols, S.A. de C.V. (merged with Grifols México, S.A. de C.V.) |
| Mexico | Calle Eugenio Cuzin, nº 909-913 |
| 2008 |
| Commercial |
| Manufacture and commercialisation of pharmaceutical products for human and veterinary use. |
| — | — | — | — | | % | | % | |||||
Grifols México, S.A. de C.V. |
| Mexico | Calle Eugenio Cuzin, nº 909-913 |
| 1993 |
| Commercial |
| Production, manufacture, adaptation, conditioning, sale and purchase, commissioning, representation |
| | % | — | | % | — | | % | | % | |||
Grifols Nordic, AB |
| Sweden | Sveavägen 166 |
| 2010 |
| Commercial |
| Research and development, production and marketing of pharmaceutical products, medical devices and |
| | % | — |
| | % | — |
| | % | — |
|
F-132
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
deriving from the aforementioned activities. | |||||||||||||||||||||||
Grifols Colombia, Ltda |
| Colombia | Carrera 7 No. 71 52 Torre B piso |
| 2010 |
| Commercial |
| Sale, commercialisation and distribution of medicines, pharmaceutical (including but not limited to |
| | % | | % | | % | | % | | % | | % | |
Grifols Deutschland GmbH |
| Germany | Lyoner Strasse 15, D- |
| 2011 |
| Commercial |
| Procurement of the official permits and necessary approval for the production, commercialisation and |
| | % | — |
| | % | — |
| | % | — |
|
F-133
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
Grifols Canada, Ltd. |
| Canada | 5060 Spectrum Way, Suite 405 |
| 2011 |
| Commercial |
| Distribution and sale of biotechnological products. |
| | % | — | | % | — | — |
| | % | |||
Grifols Pharmaceutical Technology (Shanghai) Co., Ltd. |
| Unit 901-902, Tower 2, No. |
| 2013 |
| Commercial |
| Pharmaceutical consultancy services (except for diagnosis), technical and logistical consultancy |
| | % | — |
| | % | — |
| | % | — |
| ||
Grifols (H.K.), Limited |
| Units 1505-7 BerKshire House, |
| 2014 |
| Commercial |
| Distribution and sale of diagnostic products. |
| — |
| | % | — |
| | % | — |
| | % | ||
Grifols Japan K.K. |
| Hilton Plaza West Office Tower, 19th floor. 2-2, Umeda 2-chome, Kita-ku Osaka-shi Japón | Hilton Plaza West Office Tower, |
| 2014 |
| Commercial |
| Research, development, import and export and commercialisation of pharmaceutical products, devices |
| | % | — |
| | % | — |
| | % | — |
| |
Grifols India Healthcare Private Ltd |
| Regus Business Centre |
| 2014 |
| Commercial |
| Distribution and sale of pharmaceutical products. |
| | % | | % | | % | | % | | % | | % | ||
Grifols Diagnostics Equipment Taiwan Limited |
| 8F., No.367, Fuxing N. RD., |
| 2016 |
| Commercial |
| Distribution and sale of diagnostic products. |
| | % | — |
| | % | — |
| | % | — |
| ||
Grifols Viajes, S.A. |
| Spain | Can Guasch, 2 |
| 1995 |
| Services |
| Travel agency exclusively serving Group companies. |
| | % | | % | | % | | % | | % | | % |
F-134
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
Squadron Reinsurance Designated Activity Company |
| Ireland | The Metropolitan Building, 3rd |
| 2003 |
| Services |
| Reinsurance of Group companies’ insurance policies. |
| — |
| | % | — |
| | % | — |
| | % | |
Grifols Shared Services North America, Inc. |
| United States | 2410 Lillivale Avenue |
| 2011 |
| Services |
| Support services for the collection, manufacture, sale and distribution of plasma derivatives and related |
| | % | — |
| | % | — |
| | % | — |
| |
Gripdan Invest, S.L (merged with Grifols S.A.) |
| Avenida Diagonal 477 Barcelona, |
| 2015 |
| Services |
| Rental of industrial buildings |
| — | — |
| | % | — |
| | % | — |
| |||
Araclon Biotech, S.L. |
| Spain | Paseo de Sagasta, 17 2º izqda. |
| 2012 |
| Research |
| Creation and commercialisation of a blood diagnosis kit for the detection of Alzheimer's and |
| — |
| | % | — |
| | % | — |
| | % | |
VCN Bioscience, S.L. |
| Avenida de la Generalitat 152 |
| 2012 |
| Research |
| Research and development of therapeutic approaches for tumours for which there is currently no |
| — |
| — | — |
| — | — |
| | % | ||||
Grifols Innovation and New Technologies Limited |
| Grange Castle Business Park, |
| 2016 |
| Research |
| Biotechnology research and development |
| — |
| | % | — |
| | % | — |
| | % | ||
Kiro Grifols S.L | Spain | Polígono Bainuetxe, 5, 2º planta, Aretxabaleta, Guipúzcoa | 2014 | Research | Development of machines and equipment to automate and | | % | — | | % | — | | % | — |
F-135
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
control key points of hospital processes, and hospital pharmacy processes. | |||||||||||||||||||||||
Chiquito Acquisition Corp. (merged with Grifols Bio Supplis Inc.) | 2711 Centerville Road Suite 400, Wilmington, Delaware, New Castle County, United States | 2017 | Corporate | Engage in any lawful act or activity for which a corporation may be organized under the General Corporation Law of the State of Delaware, as amended from time to time (the "DGCL"). | — | — | — | | % | — | | % | |||||||||||
Aigües Minerals de Vilajuiga, S.A. | Carrer Sant Sebastià, 2, 17493 Vilajuïga, Girona | Carrer Sant Sebastià, 2, 17493 Vilajuïga, Girona, Spain | 2017 | Industrial | Collection and use of mineral-medicinal waters and obtaining of all necessary administrative concessions for the optimum and widest use of these. | | % | | % | | % | | % | | % | | % | ||||||
Goetech LLC (D/B/A Medkeeper) | 7600 Grandview Avenue, Suite 210, Arvada, CO 80002, United States | 2018 | Industrial | Development and distribution of web and mobile-based platforms for hospital pharmacies | — | — | — | | % | — | | % | |||||||||||
Grifols Bio Supplies Inc. (before Interstate Blood Bank, Inc.) | 5700 Pleasantville Road | 2016 | Industrial | Procurement of human plasma. | — | | % | — | | % | — | | % | ||||||||||
Haema, AG | LandsteinerstraBe 1, 04103 Leipzig - Germany | 2018 | Industrial | Procurement of human plasma. | — | — | — | — | — | — | |||||||||||||
BPC Plasma, Inc (formerly Biotest Pharma Corp) | 901 Yamato Rd., Suite 101, Boca Raton FL 33431 - United States | 2018 | Industrial | Procurement of human plasma. | — | — | — | — | — | — | |||||||||||||
F-136
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
Haema Plasma Kft. | Bajcsy-Zsilinszky út 12., 1051 Budapest (Hungría) | 2021 | Industrial | Procurement of human plasma. | — | — | — | — | — | — | |||||||||||||
Alkahest, Inc. | 3500 South DuPont Hwy, Dover, County of Kent Estados Unidos | 3500 South DuPont Hwy, | 2015 | Research | Development of novel plasma-based products for the treatment of cognitive decline in aging and disorders of the central nervous system (CNS). | — | | % | — | | % | — | | % | |||||||||
Plasmavita Healthcare GmbH | Colmarer Strasse 22, 60528 Frankfurt am Main - Germany | Colmarer Strasse 22, 60528 Frankfurt am Main - Germany | 2018 | Industrial | Procurement of human plasma. | — | | % | — | | % | — | | % | |||||||||
Plasmavita Healthcare II GmbH | Garnisongasse 4/12, 1090 Vienna, Austria | Garnisongasse 4/12, 1090 Vienna, Austria | 2019 | Industrial | Procurement of human plasma. | — | | % | — | | % | — | | % | |||||||||
Grifols Canada Therapeutics Inc. (formerly Green Cross Biotherapeutics; Inc) | 2911 Avenue Marie Curie, Arrondissement de Saint-Laurent, Quebec | 2020 | Industrial | Conducting business in Pharmceuticals and Medicines Industry | | % | | % | | % | | % | | % | — | ||||||||
Grifols Laboratory Solutions, Inc | Corporation Trust Center, 1209, Orange Street, Wilmington, New Castle Country, Delaware, 19801 | 2020 | Services | Engage in any lawful act or activity for which corporations may be organized under the General Corporation Law of Delaware | — | | % | — | | % | — | | % | ||||||||||
Grifols Korea Co., Ltd. | 302 Teheran-ro, Gangnam-gu, Seoul (Yeoksam-dong) | 2020 | Commercial | Import, export of diagnostic in vitro products and solutions. | | % | — | | % | — | | % | — | ||||||||||
Grifols Middle East & Africa LLC | Office No. 534, 5th floor, NamaaBuilding No.155, Ramses Extension Street, Al Hay Al Sades, Nasr City, Cairo | 2021 | Services | Providing consultation (except for those stipulated in Article 27 of the Capital Market Law and its executive regulations) and carry out those | | % | | % | | % | | % | | % | | % |
F-137
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
commercial activities that are permitted by the law. | |||||||||||||||||||||||
GigaGen Inc. | 407 Cabot Road | 2017 | Industrial | Engage in any lawful act or activity for which corporations may be organized under General Corporation Law. | — | | % | — | | % | — | | % | ||||||||||
Grifols Pyrenees Research Center, S.L. | C/ Prat de la Creu, 68-76, Planta 3ª, Edifici Administratiu del Comú d'Andorra la Vella | 2021 | Industrial | Constitution, development and management of operations of a research and development center in all areas of immnology, dedicated to find possible solutions for therapeutic applications. | — | | % | — | | % | — | | % | ||||||||||
Grifols Bio North America LLC | 251 Little Falls Drive, Wilmington, New Castle County, 19808, Delaware | 2021 | Industrial | Engage in any lawful business permitted by the Act or the laws of any jurisdiction in which the Company may do business. | — | | % | — | | % | — | | % | ||||||||||
Biomat Holdings LLC | FALTA | 2410 Grifols Way, Los Angeles, California, 90032, United States. | 2023 | Services | Administration and financing services to Immunotek donor centers. | — | | % | — | — | — | — | |||||||||||
Biomat Holdco, LLC. | 251 Little Falls Drive, Wilmington, New Castle County, Delaware, 19808 | 2021 | Services | Engage in any lawful act or activity for which corporations may be organized under | — | | % | — | | % | — | | % |
F-138
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
General Corporation Law of Delaware. | |||||||||||||||||||||||
Biomat Newco, Corp. | 251 Little Falls Drive, Wilmington, New Castle County, Delaware, 19808 | 2021 | Services | Engage in any lawful act or activity for which corporations may be organized under General Corporation Law of Delaware. | — | | % | — | | % | — | | % | ||||||||||
Grifols Escrow Issuer, S.A. (merged with Grifols, S.A.) | Parque Empresarial Can Sant Joan, Avda de la Generalitat, 152-156, Sant Cugat del Vallès, 08174, Barcelona | 2021 | Services | Administration, management and control services for companies and businesses, as well as investment in property, as well as providing advisory services of any investee entities or group companies. | — | — | | % | — | | % | — | |||||||||||
Grifols Canada Plasma, Inc. (formerly Prometic Plasma Resources, Inc.) | 531 Boul. Des Prairies, Building 15 | 2021 | Industrial | Procurement of human plasma. | — | | % | — | | % | | % | — | ||||||||||
Grifols Canada Plasma – Ontario Inc. (formerly Canada Inc.) | 2911 av. Marie-Curie, Montreal, Quebec, H4S0B7, Canada | 2023 | Services | Administration, operating management and control services of plasma recollecting centers, directly or indirectly, through its affiliates. | — | | % | — | — | — | — | ||||||||||||
Access Biologicals, LLC (merged with Grifols Bio Supplies, Inc.) | 955, Park Center Drive, Vista, CA 92801, United States | 2017 | Industrial | Manufacture of biological products such as specific serum and | — | — | — | | % | — | | % |
F-139
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
plasma reagents that are used by biotechnological and biopharmaceutical companies for in-vitro diagnosis, cell culture and research and development in the field of diagnostics. | |||||||||||||||||||||||
Access Biologicals IC-DISC, Inc. (merged with Grifols Bio Supplies, Inc.) | 995 Park Center Dr, Vista, CA 92081, United States | 2017 | Industrial | Manufacture of biological products, including specific sera and plasma-derived reagents, which are used by biotechnology and biopharmaceutical companies for in-vitro diagnostics, cell culture, and research and development in the diagnostic field. | — | — | — | | % | — | | % | |||||||||||
Access Cell Culture, LLC. (merged with Grifols Bio Supplies, Inc.) | 995 Park Center Dr, Vista, CA 92081, United States | 2017 | Industrial | Manufacture of biological products, including specific sera and plasma-derived reagents, which are used by biotechnology and biopharmaceutical companies for in-vitro diagnostics, cell culture, and research and development in the diagnostic field. | — | — | — | | % | — | | % |
F-140
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
Access Plasma, LLC. (merged with Grifols Bio Supplies, Inc.) | 995 Park Center Dr, Vista, CA 92081, United States | 2017 | Industrial | Manufacture of biological products, including specific sera and plasma-derived reagents, which are used by biotechnology and biopharmaceutical companies for in-vitro diagnostics, cell culture, and research and development in the diagnostic field. | — | — | — | | % | — | | % | |||||||||||
Albimmune, S.L. | Parque Empresarial Can Sant Joan, Avda de la Generalitat, 152-156, Sant Cugat del Vallès, 08174, Barcelona España | 2022 | Research | The purpose of the company is the research, development and exploitation of a project on the application of the use of albumin as a medicine | — | | % | — | | % | — | — | |||||||||||
Biotest, AG | Landsteinerstr. 5, D-63303 Dreieich, Germany | 2022 | Industrial | Development, manufacture and distribution of biological, chemical, pharmaceutical, human and veterinary medical, cosmetic and dietary products as well as containers, devices, machines and accessories for medical, pharmaceutical and analytical purposes, as well as research in these fields. | | % | | % | | % | | % | — | — |
F-141
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
Furthermore the activity (especially research development, production and distribution) in the field of plant protection and plant breeding, the field of testing and purification of soil, water and air and in the field of products, materials and techniques used in space. | |||||||||||||||||||||||
Biotest Austria, GmbH | Einsiedlergasse 58, A-1050, Vienna, Austria | 2022 | Industrial | Distribution of pharmaceutical products. | — | | % | — | | % | — | — | |||||||||||
Biotest Italia, S.R.L. | Via Leonardo da Vinci 43, I-20090 Trezzano sul Naviglio MI, Italy | 2022 | Industrial | Distribution of pharmaceutical products. | | % | — | — | | % | — | — | |||||||||||
Biotest (UK) Ltd. (merged with Grifols UK, Ltd.) | 17 High Street, B31 2UQ Longbridge Birmingham, United Kingdom | 2022 | Industrial | Distribution of pharmaceutical products. | — | — | — | | % | — | — | ||||||||||||
Biotest (Schweiz) AG | Schützenstrasse 17, CH-5102 Rupperswil, Switzerland | 2022 | Industrial | Distribution of pharmaceutical products. | — | | % | — | | % | — | — | |||||||||||
Biotest Hungaria Kft | Torbágy utca 15/ A, Törökbálint 2045, Hungary | 2022 | Industrial | Procurement of human plasma. | — | | % | — | | % | — | — | |||||||||||
Biotest Farmacêutica LTDA | Rua José Ramos Guimarães, 49 A Centro, 12955-000, Bom Jesus dos Perdões – SP, Brasil | 2022 | Industrial | Distribution of pharmaceutical products. | | % | — | — | | % | — | — | |||||||||||
Biotest Hellas M.E.P.E. | 45 Michalakopoulou Str., 11528 Athens, Greece | 2022 | Research | Research and development of solutions in the Biopharma area. | — | | % | — | | % | — | — | |||||||||||
Biotest France SAS | 45/47 rue d'Hauteville, 75010 Paris, France | 2022 | Services | The purpose of the company is to act as an agent and support the | | % | — | — | | % | — | — |
F-142
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
group companies. | |||||||||||||||||||||||
Biotest Pharmaceuticals Ilaç Pazarlama Anonim Sirketi | Nishstanbul, Cobançesme Mahallesi, 34197 Bahçeliever, Istanbul, Turkey | 2022 | Research | Research and development of solutions in the Biopharma area. | — | | % | — | | % | — | — | |||||||||||
Biotest Medical, S.L.U. | C/ Frederic Mompou, nº 5, 6º 3ª A, 08960 Sant Just Desvern, Barcelona, Spain | 2022 | Industrial | Distribution of pharmaceutical products. | | % | — | — | | % | — | — | |||||||||||
Biotest Pharma, GmbH | Landsteinerstr. 5, D-63303 Dreieich, Germany | 2022 | Industrial | Carry out the development and production activities in the Biopharma area. | — | | % | — | | % | — | — | |||||||||||
Biotest Lux S.à.r.l. | 17, Boulevard F.W. Raiffeisen L-2411 Luxembourg | 2023 | Services | Providing financing and centralisation of services for Biotest companies. | — | | % | — | — | — | — | ||||||||||||
BioDarou PLC | Sarparast St., Italia St. Felestin Ave, 1416653163 Tehran, Iran | 2022 | Industrial | Procurement of human plasma. | — | | % | — | | % | — | — | |||||||||||
Biotest Grundstücksverwaltungs GmbH | Landsteinerstr. 5, D-63303 Dreieich, Germany | 2022 | Services | Management of own assets. | — | | % | — | | % | — | — | |||||||||||
Plasma Service Europe GmbH | Landsteinerstr. 5, D-63303 Dreieich, Germany | 2022 | Industrial | Procurement of human plasma. | — | | % | — | | % | — | — | |||||||||||
Cara Plasma s.r.o. | Jungmannova 745/24 - Nové Město, 110 00 Praha 1 , Czech Republic | 2022 | Industrial | Procurement of human plasma. | — | | % | — | | % | — | — | |||||||||||
Plazmaszolgálat Kft | Torbágy utca 15/ A, Törökbálint 2045, Hungary | 2022 | Industrial | Procurement of human plasma. | — | | % | — | | % | — | — | |||||||||||
Grifols Biotest Holdings GmbH | Colmarer Str. 22, 60528 Frankfurt am Main, Germany | 2022 | Services | Management of own assets as well as the acquisition, sale, holding and management of shares in other companies in Germany and abroad in the company's own name and on its | | % | — | | % | — | — | — |
F-143
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
own account (not third parties), in particular in Biotest AG with registered offices in Dreiech. | |||||||||||||||||||||||
AlbaJuna Therapeutics, S.L | Hospital Germans Trias i Pujol, carretera de Canyet, s/n, Badalona, Spain | 2016 | Research | Development and manufacture of therapeutic antibodies against HIV. | | % | — | — | | % | — | | % | ||||||||||
Equity-accounted investees and others | |||||||||||||||||||||||
Aradigm Corporation | 3929 Point Eden Way | 2013 | Research | Development and commercialisation of drugs delivered by inhalation for the prevention and treatment of severe respiratory diseases. | — | — | — | — | — | | % | ||||||||||||
Mecwins, S.L. | Avenida Fernandos Casas Novoa, 37 | 2013 | Research | Research and production of nanotechnological, biotechnological and chemical solutions. | — | | % | — | | % | — | | % | ||||||||||
Albajuna Therapeutics, S.L (becomes part of the group) | Hospital Germans Trias i Pujol, carretera de Canyet, s/n, Badalona | 2016 | Research | Development and manufacture of therapeutic antibodies against HIV. | — | — | — | | % | — | | % | |||||||||||
Singulex, Inc. | 4041 Forest Park Avenue St. Louis, Missouri | 2016 | Research | Development of the Single Molecule Counting (SMC™) technology for clinical diagnostic and scientific discovery. | — | — | — | — | — | | % |
F-144
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
Access Biologicals, LLC. (becomes part of the group) | 995 Park Center Dr, Vista, CA 92081, United States | 2017 | Industrial | Manufacture of biological products, including specific sera and plasma-derived reagents, which are used by biotechnology and biopharmaceutical companies for in-vitro diagnostics, cell culture, and research and development in the diagnostic field. | — | — | — | — | — | | % | ||||||||||||
Access Biologicals IC-DISC, Inc. (becomes part of the group) | 995 Park Center Dr, Vista, CA 92081, United States | 2017 | Industrial | Manufacture of biological products, including specific sera and plasma-derived reagents, which are used by biotechnology and biopharmaceutical companies for in-vitro diagnostics, cell culture, and research and development in the diagnostic field. | — | — | — | — | — | | % | ||||||||||||
Access Cell Culture, LLC. (becomes part of the group) | 995 Park Center Dr, Vista, CA 92081, United States | 2017 | Industrial | Manufacture of biological products, including specific sera and plasma-derived reagents, which are used by biotechnology and biopharmaceuti | — | — | — | — | — | | % |
F-145
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
cal companies for in-vitro diagnostics, cell culture, and research and development in the diagnostic field. | |||||||||||||||||||||||
Access Plasma, LLC. (becomes part of the group) | 995 Park Center Dr, Vista, CA 92081, United States | 2017 | Industrial | Manufacture of biological products, including specific sera and plasma-derived reagents, which are used by biotechnology and biopharmaceutical companies for in-vitro diagnostics, cell culture, and research and development in the diagnostic field. | — | — | — | — | — | | % | ||||||||||||
Medcom Advance, S.A | Av. Roma, 35 Entresuelo 1, 08018 Barcelona; Spain | 2019 | Research | Research and development of nanotechnological solutions. | — | | % | — | | % | — | | % | ||||||||||
Shanghai RAAS Blood Products Co. Ltd. | 2009 Wangyuan Road, Fengxian District, Shanghai | 2020 | Industrial | Introducing advanced and applicable technologies, instruments and scientific management systems for manufacturing and diagnosis of blood products, in order to raise the production capacity and enhance quality standards of blood products to the | | % | — | | % | — | | % | — |
F-146
APPENDIX I
GRIFOLS, S.A. AND SUBSIDIARIES
Information on Group Companies, Associates and others for the years ended 31 December 2023, 2022 and 2021
international level. | |||||||||||||||||||||||
Grifols Egypt for Plasma Derivatives (S.A.E.) | Tolip El Narges Hotel, Teseen Streett, Fifth Settlement, Cairo | 2021 | Industrial | Establish and operate a plasma fractionation plant, regardless of whether the plasma is collected locally or imported, as well as its filling and packaging. | | % | — | | % | — | | % | — | ||||||||||
Biotek America LLC ("ITK JV") | 1430 East Southlake Blvd | 2021 | Industrial | Build and manage until the opening of donor plasma centers in the United States. | | % | — | | % | — | | % | — |
This appendix is part of note 2 from the consolidated financial statements.
F-147
APPENDIX II
GRIFOLS, S.A. AND SUBSIDIARIES
Operating Segments for the years ended 31 December 2023, 2022 and 2021
(Expressed in thousands of Euros)
Biopharma | Diagnostic | Bio Supplies | Others | Intersegments | Consolidated | |||||||||||||||||||||||||||||||
| 2023 |
| 2022 |
| 2021 (*) | 2023 |
| 2022 |
| 2021 (*) |
| 2023 |
| 2022 |
| 2021 (*) |
| 2023 |
| 2022 |
| 2021 (*) |
| 2023 |
| 2022 |
| 2021 (*) | 2023 |
| 2022 |
| 2021 (*) | |||
Revenues from external customers |
| |
| |
| | |
| |
| |
| |
| |
| |
| |
| |
| |
| — |
| ( |
| ( | |
| |
| | ||
| ||||||||||||||||||||||||||||||||||||
Total operating income |
| |
| |
| | |
| |
| |
| | | | | | | — | ( | ( | |
| |
| | ||||||||||
Profit/(Loss) for the segment |
| |
| |
| | |
| |
| |
| | | | | ( | ( | | | ( | |
| |
| | ||||||||||
Unallocated expenses |
|
|
|
|
|
|
|
|
|
|
|
| ( |
| ( |
| ( | |||||||||||||||||||
Operating profit/(loss) |
|
|
|
|
|
|
|
|
|
|
|
| |
| |
| | |||||||||||||||||||
Finance result |
|
|
|
|
|
|
|
|
|
|
|
| ( |
| ( |
| ( | |||||||||||||||||||
|
| |||||||||||||||||||||||||||||||||||
Share of profit/(loss) of equity- accounted investee | — | — | — | — | — | — | — | — | — | ( | ( | | — | — | — | ( | ( | | ||||||||||||||||||
Income tax expense |
|
|
|
|
|
|
|
|
|
|
|
| ( |
| ( |
| ( | |||||||||||||||||||
Profit for the year after tax |
|
|
|
|
|
|
|
|
|
|
|
| |
| |
| | |||||||||||||||||||
Segment assets |
| |
| |
| | |
| |
| |
| | | | | | | — | ( | ( | |
| |
| | ||||||||||
Equity-accounted investments |
| |
| |
| | — |
| — |
| — |
| — | — | | | | | — | — | — | |
| |
| | ||||||||||
Unallocated assets |
| — |
| — |
| — | — |
| — |
| — |
| — | — | — | — | — | — | — | — | — | |
| |
| | ||||||||||
Total assets |
|
|
|
|
|
|
|
|
|
|
|
| |
| |
| | |||||||||||||||||||
Segment liabilities |
| | | | | | |
| | | | | | | — | — | — | |
| |
| | ||||||||||||||
Unallocated liabilities |
| — | — | — | — | — | — |
| — | — | — | — | — | — | — | — | — | |
| |
| | ||||||||||||||
Total liabilities |
|
|
|
|
|
|
|
|
|
|
|
| |
| |
| | |||||||||||||||||||
Other information: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||||||||||||
Allocated amortisation and depreciation |
| |
| |
| | |
| |
| |
| | | | | | | — | — | — | |
| |
| | ||||||||||
Unallocated amortisation and depreciation |
| — |
| — |
| — | — |
| — |
| — |
| — | — | — | — | — | — | — | — | — | |
| |
| | ||||||||||
Allocated expenses that do not require cash payments |
| |
| ( |
| | |
| |
| |
| | | | ( | ( | | — | — | — | |
| ( |
| | ||||||||||
Unallocated expenses that do not require cash |
| — |
| — |
| — | — |
| — |
| — |
| — | — | — | — | — | — | — | — | — | |
| ( |
| | ||||||||||
Allocated additions for the year of property, plant & |
| |
| |
| | |
| |
| |
| | | | | | | — | — | — | |
| |
| | ||||||||||
Unallocated additions for the year of property, plant |
| — |
| — |
| — | — |
| — |
| — |
| — | — | — | — | — | — | — | — | — | |
| |
| | ||||||||||
* As a consequence of the review of transactions and balances allocations by segments done in 2022, the comparative figures for the fiscal year 2021 have been adjusted accordingly.
This appendix forms an integral part of note 5 to the consolidated financial statements.
F-148
APPENDIX II
GRIFOLS, S.A. AND SUBSIDIARIES
Reporting by geographical area
for the years ended 31 December 2023, 2022 and 2021
(Expressed in thousands of Euros)
Spain | Rest of European Union | USA + Canada | Rest of World | Consolidated | ||||||||||||||||||||||||||
| 2023 |
| 2022 |
| 2021 |
| 2023 |
| 2022 |
| 2021 |
| 2023 |
| 2022 |
| 2021 |
| 2023 |
| 2022 |
| 2021 |
| 2023 |
| 2022 |
| 2021 | |
Net Revenue |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
Assets by geographical area |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
Other information: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Additions for the year of property, plant & equipment, intangible assets and rights of use |
| |
| |
| |
| | |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| | |
This appendix forms an integral part of note 5 to the consolidated financial statements.
F-149
APPENDIX III
GRIFOLS, S.A. AND SUBSIDIARIES
Changes in Other Intangible Assets
for the year ended
31 December 2023
(Expressed in thousands of Euros)
Balance at |
|
|
| Translation | Balance at | |||||||
| 12/31/2022 |
| Additions |
| Transfers |
| Disposals |
| differences |
| 12/31/2023 | |
Development costs |
| |
| |
| — |
| — |
| ( |
| |
Concessions, patents, licenses brands & similar |
| |
| |
| ( |
| ( |
| ( |
| |
Computer software |
| |
| |
| |
| ( |
| ( |
| |
Currently marketed products |
| |
| — |
| — |
| — |
| ( |
| |
Other intangible assets |
| |
| |
| ( |
| ( |
| ( |
| |
Total cost of intangible assets |
| |
| |
| |
| ( |
| ( |
| |
Accum. amort. of development costs |
| ( |
| ( |
| — |
| — |
| |
| ( |
Accum. amort of concessions, patents, licenses, brands & similar |
| ( |
| ( |
| |
| |
| |
| ( |
Accum. amort. of computer software |
| ( |
| ( |
| ( |
| |
| |
| ( |
Accum. amort. of currently marketed products |
| ( |
| ( |
| — |
| — |
| |
| ( |
Accum. amort. of other intangible assets |
| ( |
| ( |
| — |
| |
| |
| ( |
Total accum. amort intangible assets |
| ( |
| ( |
| ( |
| |
| |
| ( |
Impairment of other intangible assets |
| ( |
| ( |
| — |
| |
| |
| ( |
Carrying amount of intangible assets |
| |
| ( |
| |
| |
| ( |
| |
This appendix forms an integral part of note 7 to the consolidated financial statements.
F-150
APPENDIX III
GRIFOLS, S.A. AND SUBSIDIARIES
Changes in Other Intangible Assets
for the year ended
31 December 2022
(Expressed in thousands of Euros)
Balance at |
| Business |
| Translation | Balance at | |||||||||
| 31/12/2021 |
| Additions | combinations | Transfers | Disposals | differences | 12/31/2022 | ||||||
Development costs |
| |
| | |
| — |
| ( |
| |
| | |
Concessions, patents, licenses brands & similar |
| |
| | |
| |
| ( |
| |
| | |
Computer software |
| |
| | |
| |
| ( |
| |
| | |
Currently marketed products |
| |
| — | — |
| — |
| — |
| |
| | |
Other intangible assets |
| |
| | |
| ( |
| ( |
| |
| | |
Total cost of intangible assets |
| |
| | |
| |
| ( |
| |
| | |
Accum. amort. of development costs |
| ( |
| ( | — |
| — |
| |
| ( |
| ( | |
Accum. amort of concessions, patents, licenses, brands & similar |
| ( |
| ( | ( |
| — |
| |
| ( |
| ( | |
Accum. amort. of computer software |
| ( |
| ( | ( |
| |
| |
| ( |
| ( | |
Accum. amort. of currently marketed products |
| ( |
| ( | — |
| — |
| — |
| ( |
| ( | |
Accum. amort. of other intangible assets |
| ( |
| ( | — |
| — |
| |
| ( |
| ( | |
Total accum. amort intangible assets |
| ( |
| ( | ( |
| |
| |
| ( |
| ( | |
Impairment of other intangible assets |
| ( |
| ( | — |
| |
| |
| ( |
| ( | |
Carrying amount of intangible assets |
| |
| ( | |
| |
| ( |
| |
| |
(See note 3)
This appendix forms an integral part of note 7 to the consolidated financial statements.
F-151
APPENDIX IV
GRIFOLS, S.A. AND SUBSIDIARIES
Movement in Rights of Use
for the year ended
31 December 2023
(Expressed in thousands of Euros)
Balance at |
|
|
| Translation | Balance at | |||||||
| 12/31/2022 |
| Additions |
| Transfers |
| Disposals |
| differences |
| 12/31/2023 | |
Land and buildings |
| |
| | — |
| ( |
| ( |
| | |
Machinery |
| |
| | ( |
| ( |
| ( |
| | |
Computer equipment |
| |
| | ( |
| ( |
| ( |
| | |
Vehicles |
| |
| | ( |
| ( |
| ( |
| | |
|
|
|
|
| ||||||||
Total cost of rights of use | | | ( | ( | ( | | ||||||
Accum. depr. of land and buildings |
| ( |
| ( | — |
| |
| |
| ( | |
Accum. depr. of machinery |
| ( |
| ( | |
| |
| |
| ( | |
Accum. depr. of computer equipment | ( | ( | | | | ( | ||||||
Accum. depr. of vehicles |
| ( |
| ( | |
| |
| |
| ( | |
|
|
|
|
| ||||||||
Total accum. Depr. of rights of use |
| ( |
| ( | |
| |
| |
| ( | |
Carrying amount of rights of use |
| |
| | ( |
| ( |
| ( |
| |
This appendix forms an integral part of note 8 to the consolidated financial statements.
F-152
APPENDIX IV
GRIFOLS, S.A. AND SUBSIDIARIES
Movement in Rights of Use
for the year ended
31 December 2022
(Expressed in thousands of Euros)
Balance at |
| Business |
| Translation | Balance at | |||||||||
| 12/31/2021 |
| Additions | combinations |
| Transfers |
| Disposals |
| differences |
| 12/31/2022 | ||
Land and buildings |
| |
| | | ( |
| ( |
| |
| | ||
Machinery |
| |
| | | ( |
| ( |
| |
| | ||
Computer equipment |
| |
| | | ( |
| ( |
| |
| | ||
Vehicles |
| |
| | | ( |
| ( |
| |
| | ||
|
|
|
|
| ||||||||||
Total cost of rights of use | | | | ( | ( | | | |||||||
Accum. depr. of land and buildings |
| ( |
| ( | ( | |
| |
| ( |
| ( | ||
Accum. depr. of machinery |
| ( |
| ( | ( | |
| |
| |
| ( | ||
Accum. depr. of computer equipment | ( | ( | — | | | ( | ( | |||||||
Accum. depr. of vehicles |
| ( |
| ( | — | |
| |
| ( |
| ( | ||
|
|
|
|
| ||||||||||
Total accum. depr. of rights of use |
| ( |
| ( | ( | |
| |
| ( |
| ( | ||
Carrying amount of rights of use |
| |
| | | ( |
| ( |
| |
| |
This appendix forms an integral part of note 8 to the consolidated financial statements.
F-153
APPENDIX V
GRIFOLS, S.A. AND SUBSIDIARIES
Movement in Property, Plant and Equipment
for the year ended
31 December 2023
(Expressed in thousands of Euros)
Balances at | Business | Translation | Balances at | |||||||||||
31/12/2022 | Additions | combination | Transfers | Disposals | differences | 31/12/2023 | ||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
Cost: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Land and buildings |
| |
| |
| — |
| |
| ( |
| ( |
| |
Plant and machinery |
| |
| |
| |
| |
| ( |
| ( |
| |
Fixed Assets under construction |
| |
| |
| — |
| ( |
| — |
| ( |
| |
| |
| |
| |
| |
| ( |
| ( |
| | |
Accumulated depreciation: |
|
|
|
|
|
|
|
|
|
|
|
|
| |
Buildings |
| ( |
| ( |
| — |
| |
| |
| |
| ( |
Plant and machinery |
| ( |
| ( |
| ( |
| ( |
| |
| |
| ( |
| ( |
| ( |
| ( |
| ( |
| |
| |
| ( | |
Impairment of other property, plant and equipment | ( |
| ( |
| — |
| — |
| |
| |
| ( | |
Carrying amount |
| |
| |
|
| ( |
| ( |
| ( |
| | |
(See note 3) |
This appendix forms an integral part of note 9 to the consolidated financial statements.
F-154
APPENDIX V
GRIFOLS, S.A. AND SUBSIDIARIES
Movement in Property, Plant and Equipment
for the year ended
31 December 2022
(Expressed in thousands of Euros)
Balances at | Business | Translation | Balances at | |||||||||||
| 31/12/2021 |
| Additions |
| combination |
| Transfers |
| Disposals |
| differences |
| 31/12/2022 | |
Cost: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Land and buildings |
| |
| |
| |
| |
| ( |
| |
| |
Plant and machinery |
| |
| |
| |
| |
| ( |
| |
| |
Fixed Assets under construction |
| |
| |
| — |
| ( |
| — |
| |
| |
| |
| |
| |
| ( |
| ( |
| |
| | |
Accumulated depreciation: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Buildings |
| ( |
| ( |
| — |
| |
| |
| ( |
| ( |
Plant and machinery |
| ( |
| ( |
| ( |
| |
| |
| ( |
| ( |
| ( |
| ( |
| ( |
| |
| |
| ( |
| ( | |
Impairment of other property, plant and equipment | ( |
| ( |
| — |
| |
| |
| ( |
| ( | |
Carrying amount |
| |
| |
| |
| ( |
| ( |
| |
| |
|
|
| (See note 3) |
This appendix forms an integral part of note 9 to the consolidated financial statements.
F-155