UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark one)
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the fiscal year ended |
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the transition period from to |
OR
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report
Commission file number
(Exact name of Registrant as specified in its charter)
N/A
(Translation of Registrant’s name into English)
(Jurisdiction of incorporation or organization)
+852 2121 8200
(Address of principal executive offices)
Chief Executive Officer and Chief Scientific Officer
Telephone: +
Facsimile: +
(Name, telephone, email and/or facsimile number and address of Company contact person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
Title of each class |
| Trading Symbol(s) |
| Name of each exchange on which registered |
Nasdaq Global Select Market* |
*Not for trading, but only in connection with the listing of American depositary shares on the Nasdaq Global Select Market
Securities registered or to be registered pursuant to Section 12(g) of the Act:
None
(Title of Class)
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act:
None
(Title of Class)
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the Annual Report:
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
⌧
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
◻ Yes ⌧
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
⌧
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
⌧
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See definition of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Accelerated filer ¨ | Non-accelerated filer ¨ | Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards† provided pursuant to Section 13(a) of the Exchange Act. ☐
†The term “new or revised financial accounting standard” refers to any update issued by the Financial Accounting Standards Board to its Accounting Standards Codification after April 5, 2012.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepare or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
International Financial Reporting Standards as issued | Other ◻ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
◻ Item 17 ◻ Item 18
If this is an Annual Report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
☐ Yes
(APPLICABLE ONLY TO ISSUERS INVOLVED IN BANKRUPTCY PROCEEDINGS DURING THE PAST FIVE YEARS)
Indicate by check mark whether the registrant has filed all documents and reports required to be filed by Sections 12, 13 or 15(d) of the Securities Exchange Act of 1934 subsequent to the distribution of securities under a plan confirmed by a court.
☐ Yes ☐ No
HUTCHMED (China) Limited
Table of Contents
3 | ||
5 | ||
7 | ||
7 | ||
7 | ||
7 | ||
67 | ||
158 | ||
158 | ||
181 | ||
199 | ||
203 | ||
203 | ||
204 | ||
214 | ||
214 | ||
217 | ||
217 | ||
Material Modifications to the Rights of Security Holders and Use of Proceeds | 217 | |
217 | ||
218 | ||
218 | ||
218 | ||
218 | ||
219 | ||
Purchases of Equity Securities by the Issuer and Affiliated Purchasers | 219 | |
219 | ||
219 | ||
219 | ||
Disclosure Regarding Foreign Jurisdictions that Prevent Inspection | 219 | |
219 | ||
220 | ||
221 | ||
221 | ||
221 | ||
222 | ||
224 | ||
INTRODUCTION
This annual report on Form 20-F contains our audited consolidated statements of operations data for the years ended December 31, 2023, 2022 and 2021 and our audited consolidated balance sheet data as of December 31, 2023 and 2022. Our consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles, or U.S. GAAP.
This annual report also includes audited consolidated income statement data for the years ended December 31, 2023, 2022 and 2021 and the audited consolidated statements of financial position data as of December 31, 2023 and 2022 for our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals. The financial statements of Shanghai Hutchison Pharmaceuticals have been prepared in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standard Board, or IASB.
Unless the context requires otherwise, references herein to the “company,” “HUTCHMED,” “we,” “us” and “our” refer to HUTCHMED (China) Limited, a holding company incorporated in the Cayman Islands, and its consolidated subsidiaries and joint ventures, some of which, as noted below, are incorporated and operate in the PRC. “HUTCHMED Holdings” refers to HUTCHMED Holdings Limited, a subsidiary of the Company and a holding company incorporated in the Cayman Islands. “HUTCHMED Limited” refers to “HUTCHMED Limited”, a subsidiary of HUTCHMED Holdings which is incorporated in the PRC and through which we operate our Oncology/Immunology operations in China. Our other principal operating subsidiaries for our Oncology/Immunology operations are HUTCHMED International Corporation (incorporated in Delaware), HUTCHMED Holdings (HK) Limited (incorporated in Hong Kong) and HUTCHMED (Suzhou) Limited (incorporated and operates in the PRC). “Hutchison Sinopharm” refers to Hutchison Whampoa Sinopharm Pharmaceuticals (Shanghai) Company Limited, our PRC-incorporated joint venture with Sinopharm through which we operate our principal consolidated joint venture. See Item 4. “Information on the Company—C. Organizational Structure” for a diagram illustrating our corporate structure.
Conventions Used in this Annual Report
Unless otherwise indicated, references in this annual report to:
| ● | “ADRs” are to the American depositary receipts, which evidence our ADSs; |
| ● | “ADSs” are to our American depositary shares, each of which represents five ordinary shares; |
| ● | “AstraZeneca” are to AstraZeneca AB (publ); |
| ● | “China” or “PRC” refers to the People’s Republic of China including Hong Kong and Macau and, only for the purpose of this annual report, excluding Taiwan; and only in the context of describing PRC rules, laws, regulations, regulatory authority, and any PRC entities or citizens under such rules, laws and regulations and other legal or tax matters in this annual report, excludes Taiwan, Hong Kong, and Macau; the legal and operational risks associated with operating in China also apply to our operations in Hong Kong; |
| ● | “CK Hutchison” are to CK Hutchison Holdings Limited, a company incorporated in the Cayman Islands and listed on the Hong Kong Stock Exchange, and the ultimate parent company of our largest shareholder, Hutchison Healthcare Holdings Limited; |
| ● | “Eli Lilly” are to Lilly (Shanghai) Management Company Limited; |
| ● | “E.U.” are to the European Union; |
| ● | “Guangzhou Baiyunshan” are to Guangzhou Baiyunshan Pharmaceutical Holdings Company Limited, a leading China-based pharmaceutical company listed on the Shanghai Stock Exchange and the Hong Kong Stock Exchange; |
| ● | “Hain Celestial” are to The Hain Celestial Group, Inc., a Nasdaq-listed, natural and organic food and personal care products company; |
| ● | “HK$” or “HK dollar” are to the legal currency of the Hong Kong Special Administrative Region; |
3
| ● | “Hutchison Baiyunshan” are to Hutchison Whampoa Guangzhou Baiyunshan Chinese Medicine Company Limited, which was our non-consolidated joint venture with Guangzhou Baiyunshan in which we indirectly held a 50% interest through a holding company until our disposal of such interest on September 28, 2021 (this interest was previously held through a holding company in which we have a 80% interest); |
| ● | “HUTCHMED Science Nutrition” are to HUTCHMED Science Nutrition Limited, our previous wholly owned subsidiary which we divested in December 2023; |
| ● | “Hutchison Hain Organic” are to Hutchison Hain Organic Holdings Limited, our previous joint venture with Hain Celestial in which we had a 50% interest and divested in December 2023; |
| ● | “Hutchison Healthcare” are to Hutchison Healthcare Limited, our wholly owned subsidiary; |
| ● | “HUTCHMED Limited”, our PRC-incorporated subsidiary through which we operate our Oncology/Immunology operations in China and in which we have a 99.8% interest; |
| ● | “HUTCHMED Holdings” are to HUTCHMED Holdings Limited, our subsidiary incorporated in the Cayman Islands in which we have a 99.8% interest and which is the indirect holding company of HUTCHMED Limited; |
| ● | “Hutchison Sinopharm” are to Hutchison Whampoa Sinopharm Pharmaceuticals (Shanghai) Company Limited, our PRC-incorporated joint venture with Sinopharm in which we have a 50.9% interest and through which we operate our principal consolidated joint venture; |
| ● | “Inmagene” are to Inmagene Biopharmaceuticals; |
| ● | “ordinary shares” or “shares” are to our ordinary shares, par value $0.10 per share; |
| ● | “RMB” or “renminbi” are to the legal currency of the PRC; |
| ● | “SEHK” are to The Stock Exchange of Hong Kong Limited, or the Hong Kong Stock Exchange; |
| ● | “Shanghai Hutchison Pharmaceuticals” are to Shanghai Hutchison Pharmaceuticals Limited, our non-consolidated joint venture with Shanghai Pharmaceuticals in which we have a 50% interest; |
| ● | “Shanghai Pharmaceuticals” are to Shanghai Pharmaceuticals Holding Co., Ltd., a leading pharmaceutical company in China listed on the Shanghai Stock Exchange and the Hong Kong Stock Exchange; |
| ● | “Sinopharm” are to Sinopharm Group Co. Ltd., a leading distributor of pharmaceutical and healthcare products and a leading supply chain service provider in China listed on the Hong Kong Stock Exchange; |
| ● | “Takeda” are to Takeda Pharmaceuticals International AG |
| ● | “U.S.” or “United States” are to the United States of America; |
| ● | “$” or “U.S. dollars” are to the legal currency of the United States; and |
| ● | “£” or “pound sterling” are to the legal currency of the United Kingdom. |
References in this annual report to our “Oncology/Immunology” operations are to all activities related to oncology/immunology, including sales, marketing, manufacturing and research and development with respect to our drugs and drug candidates, and references to our “Other Ventures” are to all of our other businesses.
4
Our reporting currency is the U.S. dollar. In addition, this annual report also contains translations of certain foreign currency amounts into dollars for the convenience of the reader. Unless otherwise stated, all translations of pound sterling into U.S. dollars were made at £1.00 to $1.27, all translations of RMB into U.S. dollars were made at RMB7.16 to $1.00 and all translations of HK dollars into U.S. dollars were made at HK$7.8 to $1.00, which are the exchange rates used in our audited consolidated financial statements as of December 31, 2023. We make no representation that the pound sterling, HK dollar or U.S. dollar amounts referred to in this annual report could have been or could be converted into U.S. dollars, pounds sterling or HK dollars, as the case may be, at any particular rate or at all.
Trademarks and Service Marks
We own or have been licensed rights to trademarks, service marks and trade names for use in connection with the operation of our business, including, but not limited to, the trademarks “Hutchison”, “Chi-Med”, “Hutchison China MediTech”, “HUTCHMED”, “Elunate”, “Fruzaqla”, “Sulanda”, “Orpathys”, “Tazverik” and the logos used by HUTCHMED Limited. All other trademarks, service marks or trade names appearing in this annual report that are not identified as marks owned by us are the property of their respective owners.
Solely for convenience, the trademarks, service marks and trade names referred to in this annual report are listed without the ®, ™ and (sm) symbols, but we will assert, to the fullest extent under applicable law, our applicable rights in these trademarks, service marks and trade names.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This annual report contains forward-looking statements made under the “safe harbor” provisions of the U.S. Private Securities Litigation Reform Act of 1995. These statements relate to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. The words “anticipate,” “assume,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “goal,” “intend,” “may,” “might,” “objective,” “plan,” “potential,” “predict,” “project,” “positioned,” “seek,” “should,” “target,” “will,” “would,” or the negative of these terms or other similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. These forward-looking statements are based on current expectations, estimates, forecasts and projections about our business and the industry in which we operate and management’s beliefs and assumptions, are not guarantees of future performance or development and involve known and unknown risks, uncertainties and other factors. These forward-looking statements include statements regarding:
| ● | the initiation, timing, progress and results of our or our collaboration partners’ pre-clinical and clinical studies, and our research and development programs; |
| ● | our or our collaboration partners’ ability to advance our drug candidates into, and/or successfully complete, clinical studies; |
| ● | the timing of regulatory filings and the likelihood of favorable regulatory outcomes and approvals; |
| ● | regulatory developments in China, the United States and other countries; |
| ● | the ability of our or our collaboration partners’ drug sales team to effectively develop and execute promotional and marketing activities to support the marketing and sales of our approved drug candidates; |
| ● | the timing, progress and results of our or our collaboration partners’ commercial launches, the rate and degree of market acceptance and potential market for any of our approved drug candidates; |
| ● | the pricing and reimbursement of our and our joint ventures’ products and our approved drug candidates; |
| ● | our ability to contract on commercially reasonable terms with contract research organizations, or CROs, third-party suppliers and manufacturers; |
5
| ● | the scope of protection we are able to establish and maintain for intellectual property rights covering our or our joint ventures’ products and our drug candidates; |
| ● | the ability of third parties with whom we contract to successfully conduct, supervise and monitor clinical studies for our drug candidates; |
| ● | estimates of our expenses, future revenue, capital requirements and our needs for additional financing; |
| ● | our ability to obtain additional funding for our operations; |
| ● | the potential benefits of our collaborations and our ability to enter into future collaboration arrangements; |
| ● | the ability and willingness of our collaborators to actively pursue development activities under our collaboration agreements; |
| ● | our receipt of milestone or royalty payments, service payments and manufacturing costs pursuant to our strategic alliances with AstraZeneca, Eli Lilly, Takeda and Inmagene; |
| ● | our financial performance; |
| ● | our ability to attract and retain key scientific and management personnel; |
| ● | our relationship with our joint venture and collaboration partners; |
| ● | developments relating to our competitors and our industry, including competing drug products; |
| ● | changes in our tax status or the tax laws in the jurisdictions that we operate; and |
| ● | developments in our business strategies and business plans. |
Actual results or events could differ materially from the plans, intentions and expectations disclosed in the forward-looking statements we make. As a result, any or all of our forward-looking statements in this annual report may turn out to be inaccurate. We have included important factors in the cautionary statements included in this annual report on Form 20-F, particularly in the section of this annual report on Form 20-F titled “Risk Factors,” that we believe could cause actual results or events to differ materially from the forward-looking statements that we make. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Moreover, we operate in a highly competitive and rapidly changing environment in which new risks often emerge. It is not possible for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make.
You should read this annual report and the documents that we reference herein and have filed as exhibits hereto completely and with the understanding that our actual future results may be materially different from what we expect. The forward-looking statements contained herein are made as of the date of the filing of this annual report, and we do not assume any obligation to update any forward-looking statements except as required by applicable law.
In addition, this annual report contains statistical data and estimates that we have obtained from industry publications and reports generated by third-party market research firms. Although we believe that the publications, reports and surveys are reliable, we have not independently verified the data and cannot guarantee the accuracy or completeness of such data. You are cautioned not to give undue weight to this data. Such data involves risks and uncertainties and are subject to change based on various factors, including those discussed above.
6
PART I
ITEM 1. IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS
Not applicable.
ITEM 2. OFFER STATISTICS AND EXPECTED TIMETABLE
Not applicable.
ITEM 3. KEY INFORMATION
A. Reserved.
B. Capitalization and Indebtedness.
Not applicable.
C. Reasons for the Offer and Use of Proceeds.
Not applicable.
D. Risk Factors.
HUTCHMED (China) Limited is a Cayman Islands holding company which conducts its operations in China through its PRC subsidiaries (our corporate group does not utilize any variable interest entities). We face various legal and operational risks and uncertainties as a company with substantial operations in China. The PRC government has significant authority to exert influence on the ability of a company with substantial operations in China, like us, to conduct its business, accept foreign investments or be listed on a U.S. stock exchange. For example, we face risks associated with PRC regulatory approvals of offshore offerings, anti-monopoly regulatory actions, cybersecurity, data privacy and from U.S. regulators if there is a lack of inspection from the U.S. Public Company Accounting Oversight Board, or PCAOB, on our auditors, which is further discussed below under “—Holding Foreign Companies Accountable Act” and in various risk factors in this section. The PRC government may also intervene with or influence our operations as the government deems appropriate to further regulatory, political and societal goals. The PRC government publishes from time to time new policies that can significantly affect our industry and we cannot rule out the possibility that it will in the future further release regulations or policies regarding our industry that could adversely affect our business, financial condition and results of operations. Any such action, once taken by the PRC government, could cause the value of our ADSs and ordinary shares to significantly decline or in extreme cases, become worthless.
7
Holding Foreign Companies Accountable Act
Pursuant to the Holding Foreign Companies Accountable Act, or the HFCAA, if the SEC determines that we have filed audit reports issued by a registered public accounting firm that has not been subject to inspections by the PCAOB for two consecutive years, the SEC will prohibit our shares or the ADSs from being traded on a national securities exchange or in the over-the-counter trading market in the United States. On December 16, 2021, the PCAOB issued a report to notify the SEC of its determination that the PCAOB was unable to inspect or investigate completely registered public accounting firms headquartered in mainland China and Hong Kong, including our auditor. In March 2022, the SEC conclusively listed us as a Commission-Identified Issuer under the HFCAA following the filing of our annual report on Form 20-F for the fiscal year ended December 31, 2021. On December 15, 2022, the PCAOB issued a report that vacated its December 16, 2021 determination and removed mainland China and Hong Kong from the list of jurisdictions where it is unable to inspect or investigate completely registered public accounting firms. As a result, the SEC will not provisionally or conclusively identify an issuer as a Commission-Identified Issuer if it files an annual report with an audit report issued by a registered public accounting firm headquartered in mainland China or Hong Kong on or after December 15, 2022, until such time as the PCAOB issues a new determination. Whether the PCAOB will continue to be able to satisfactorily conduct inspections of PCAOB-registered public accounting firms headquartered in mainland China and Hong Kong in the future is subject to uncertainty and depends on a number of factors out of our, and our auditor’s, control, including the uncertainties surrounding the relationship between China and the United States. If PCAOB determines in the future that it no longer has full access to inspect and investigate completely accounting firms in mainland China and Hong Kong and we continue to use an accounting firm headquartered in one of these jurisdictions to issue an audit report on our financial statements filed with the Securities and Exchange Commission, we would be identified as a Commission-Identified Issuer following the filing of the annual report on Form 20-F for the relevant fiscal year. There can be no assurance that we would not be identified as a Commission-Identified Issuer for any future fiscal year, and if we were so identified for two consecutive years, we would become subject to the prohibition on trading under the HFCAA. See Item 3.D. “Risk Factors—Risks Relating to our ADSs—The PCAOB had historically been unable to inspect our auditor in relation to their audit work performed for our financial statements and the inability of the PCAOB to conduct inspections of our auditor in the past has deprived our investors with the benefits of such inspections.” and Item 3.D. “ Risk Factors—Risks Relating to our ADSs—Our ADSs may be prohibited from trading in the United States under the HFCAA in the future if the PCAOB is unable to inspect or investigate completely auditors located in China. The delisting of the ADSs, or the threat of their being delisted, may materially and adversely affect the value of your investment.”
Permissions, Approvals, Licenses and Permits Required from the PRC Authorities for Our Operations and for the Offering of Our Securities
We conduct our business primarily through our subsidiaries and joint ventures in China. Our operations in China are governed by PRC laws and regulations. As of the date of this annual report, we and our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals, have obtained the requisite permissions, approvals, licenses and permits from the PRC government authorities that are material for the business operations of our subsidiaries and our joint ventures in China, including, among others, pharmaceutical manufacturing permits, business licenses, drug registration certificates and pharmaceutical distribution permits and no such material permission or approval has been denied. For a detailed discussion on the licenses and permits we and our non-consolidated joint venture are required to obtain as a pharmaceutical company operating in China, see Item 4.B. “Business Overview—Certificates and Permits”, “Business Overview—Regulations—Government Regulation of Pharmaceutical Product Development and Approval,” “Business Overview—Regulations—Coverage and Reimbursement” and “Business Overview—Regulations—Other Healthcare Laws.” Given the uncertainties of interpretation and implementation of relevant laws and regulations and the enforcement practice by relevant government authorities, we may be required to obtain additional requisite permissions, approvals, licenses, permits and filings for the operation of our business in the future. See also “Risks Relating to Sales of Our Internally Developed Drugs and Other Drugs—Pharmaceutical companies in China are required to comply with extensive regulations and hold a number of permits and licenses to carry on their business. Our and our joint ventures’ ability to obtain and maintain these regulatory approvals is uncertain, and future government regulation may impose additional burdens on our operations.”
8
Furthermore, the PRC government has recently indicated an intent to exert more oversight and control over offerings that are conducted overseas and/or foreign investment in China-based issuers. For example, the CSRC published the Trial Measures and Listing Guidelines (defined below) on February 17, 2023 and became effective on March 31, 2023, designed to regulate overseas securities offerings by PRC domestic companies. Given the recent nature of the introduction of the Trial Measures and Listing Guidelines, there remains significant uncertainty as to the interpretation and implementation of regulatory requirements related to overseas securities offerings and other capital markets activities. As of the date of this annual report, in connection with our historical issuance of securities to foreign investors, we are not aware of any currently effective PRC laws, regulations and regulatory rules that would require us or our non-consolidated joint venture to obtain permissions from the China Securities Regulatory Commission (the “CSRC”), and we have not received any formal notice from any PRC authority indicating that we should apply for such permission or are subject to cybersecurity review or security assessment. If (i) we mistakenly conclude that certain regulatory filings, permissions and approvals are not required or (ii) applicable laws, regulations, or interpretations change and (iii) we are required to obtain such filings, permissions or approvals in the future, but fail to receive or maintain such filings, permissions or approvals, we may face sanctions by the CSRC, the Cyberspace Administration of China (the “CAC”) or other PRC regulatory agencies. In addition, rules and regulations in China can change with little advance notice. These regulatory agencies may impose fines and penalties on our operations in China, limit our operations in China, limit our ability to pay dividends outside of China, limit our ability to list on stock exchanges outside of China or offer our securities to foreign investors or take other actions that could have a material adverse effect on our business, financial condition, results of operations and prospects, as well as the trading price of our securities. Our non-consolidated joint venture faces the same risks as well. See also “Other Risks and Risks Relating to Doing Business in China—The PRC government exerts substantial influence over the manner in which we conduct our business activities. Its oversight and discretion over our business could result in a material adverse change in our operations and the value of our ordinary shares and ADSs. Changes in laws, regulations and policies in China and uncertainties with respect to the PRC legal system could materially and adversely affect us.” and “—The PRC government has increasingly strengthened oversight in offerings conducted overseas or on foreign investment in China-based issuers, which could result in a material change in our operations and our ordinary shares and ADSs could decline in value or become worthless.”
Cash Flows Through Our Organization
HUTCHMED (China) Limited is a Cayman Islands incorporated holding company with no material operations of its own. We conduct our operations primarily in China through our PRC subsidiaries and non-consolidated joint ventures, collectively referred to as the Onshore Entities below. HUTCHMED (China) Limited has an indirect equity ownership interest in all Onshore Entities through offshore Hong Kong-incorporated holding companies, and it has received funding through various capital markets transactions. We also fund our operations through cash flows generated and dividend payments from our Oncology/Immunology and Other Ventures operations (substantially all of which have been generated in China), service and milestone and upfront payments from our collaboration partners to our PRC subsidiaries, and bank loans to our subsidiaries.
We utilize a portion of our funds outside of China to support the operations of our subsidiaries in China through capital contributions and/or shareholder loans, which are the only methods by which we can fund our subsidiaries under PRC laws and regulations. Such capital contributions and shareholder loans are subject to the satisfaction of applicable government registration and approval requirements in China and limitations on the amount of shareholder loans relative to the amount of total capital contributions. If such subsidiaries generate sufficient income, they may repay shareholder loans or distribute retained earnings through cash dividends as determined by their respective board of directors. Our PRC subsidiaries are permitted to pay dividends only out of their retained earnings, if any, as determined in accordance with PRC accounting standards and regulations. Furthermore, our PRC subsidiaries are required to make appropriations to certain statutory reserve funds or may make appropriations to certain discretionary funds, which are not distributable as cash dividends except in the event of a solvent liquidation of the companies. The amount of any repayment of shareholder loans or dividend payments can be distributed to our various offshore subsidiaries through our offshore Hong Kong-incorporated holding companies. For more information, see Item 3.D. “Risk Factors—Other Risks and Risks Relating to Doing Business in China—Restrictions on currency exchange may limit our ability to receive and use our revenue effectively.” and Item 4.B. “Business Overview—Regulations—PRC Regulation of Foreign Currency Exchange, Offshore Investment and State-Owned Assets—Regulation on Investment in Foreign invested Enterprises.” Our joint ventures in China do not require intra-group funding as they have been profitable. Service and milestone and upfront payments from our collaboration partners are received directly by our PRC subsidiaries and reinvested into their operations.
9
For the years ended December 31, 2023, 2022 and 2021, HUTCHMED provided funds to its PRC subsidiaries of $20.0 million, $310.0 million and $230.0 million, respectively, of which $20.0 million, $100.0 million and $100.0 million, respectively, were in the form of capital contributions and nil, $210.0 million and $130.0 million, respectively, were in the form of shareholder loans. Additionally, during the years ended December 31, 2023 and 2022, shareholder loans of approximately $2.6 million and $3.4 million were repaid by a PRC subsidiary, respectively. There were no transfers of assets other than transfers of cash to/from PRC subsidiaries in 2023, 2022 and 2021.
For the years ended December 31, 2023, 2022 and 2021, the Hong Kong immediate holding company of our onshore non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals, received dividends totaling approximately $42.3 million, $43.7 million and $49.9 million, respectively. These dividends were subject to a 5% withholding tax upon distribution from Shanghai Hutchison Pharmaceuticals to its Hong Kong immediate holding company.
HUTCHMED also conducts operations outside of China through subsidiaries in the U.S. and E.U. Such subsidiaries in the U.S. and E.U. have entered into service agreements with our PRC subsidiaries pursuant to which cash is transferred by our PRC subsidiaries to them to support their operations via the settlement of service invoices based on actual activities.
We have comprehensive cash management policies in place, including specific policies with respect to fund transfers through our organization. Our management regularly monitors the liquidity position and funding requirements of our subsidiaries and joint ventures. When funding is required by our operations in China, a thorough assessment is performed on the purpose of the funding (e.g., R&D investment, capital expenditures, etc.), the amount of funding and the form of injection (i.e., shareholder loans or capital contributions). Conversely, when a dividend distribution is to be made by an onshore joint venture, a similar assessment is performed on the cash flow forecast, sufficiency of funds and related factors. All necessary approvals are obtained at the chairman and chief executive officer levels and the board of directors for the relevant entities prior to any transfer. All such transfers and distributions are reviewed and approved by the relevant authorities where necessary, including the State Administration of Foreign Exchange, or SAFE, and the State Administration for Market Regulations, or SAMR. Our cash management policies and procedures also govern the management of any funds that are not yet required by our operations. Such funds are retained by our subsidiaries outside of China mainly in the form of short-term investments, such as time deposits with major banks in Hong Kong.
We have never declared or paid dividends on our ordinary shares. There have been no transfers, dividends or distributions made to U.S. investors to date. We currently expect to retain all future earnings for use in the operation and expansion of our business and do not have any present plan to pay any dividends. The declaration and payment of any dividends in the future will be determined by our board of directors in its discretion, and will depend on a number of factors, including our earnings, capital requirements, overall financial condition, and contractual restrictions. See Item 8. “Financial Information—A.8 Dividend Policy” and Item 3.D. “Risk Factors—Risks Relating to Our ADSs—We do not currently intend to pay dividends on our securities, and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of the ADSs.”
You should carefully consider all of the information in this annual report before making an investment in the ADSs. Below please find a summary of the principal risks and uncertainties we face, organized under relevant headings. In particular, as we are a China-based company incorporated in the Cayman Islands, you should pay special attention to subsections headed “Item 3. Key Information-3.D. Risk Factors-Other Risks and Risks Related to Doing Business in China.”
The following summarizes some, but not all, of the risks provided below. Please carefully consider all of the information discussed in this Item 3.D. “Risk Factors” in this annual report for a more thorough description of these and other risks.
Risks Relating to Our Financial Position and Need for Capital
| ● | Risks relating to our need for additional funding |
| ● | Risks relating to our existing and future indebtedness |
Risks Relating to Our Oncology/Immunology Operations and Development of Our Drug Candidates
| ● | Risks relating to our approach to the discovery and development of drug candidates and the lengthy, expensive and uncertain clinical development process |
10
| ● | Risks relating to expediting regulatory review, obtaining and maintaining regulatory approval and ongoing regulatory review for our drug candidates |
| ● | Risks relating to the commercialization of our drug candidates |
| ● | Risks relating to undesirable side effects of our drug candidates |
| ● | Risks relating to competition in discovering, developing and commercializing drugs |
| ● | Risks relating to our collaboration partners with respect to clinical trials, marketing and distribution |
| ● | Risks relating to our international operations |
Risks Relating to Sales of Our Internally Developed Drugs and Other Drugs
| ● | Risks relating to obtaining and maintaining permits and licenses for our and our joint ventures’ pharmaceutical operations in China |
| ● | Risks relating to leveraging our Other Ventures’ prescription drug business to commercialize our internally developed drug candidates |
| ● | Risks relating to competition in selling our approved, internally developed drugs and drugs of our Other Ventures |
| ● | Risks relating to maintaining and enhancing the brand recognition of our drugs |
| ● | Risks relating to the availability of reimbursement of our drugs, the lack of which could diminish our sales or profitability |
| ● | Risks relating to counterfeit products in China |
| ● | Risks relating to rapid changes in the pharmaceutical industry rendering our products obsolete |
| ● | Risks relating to cultivating or sourcing raw materials |
| ● | Risks relating to adverse publicity of us, our collaboration partners, our joint ventures or our products |
Risks Relating to Our Dependence on Third Parties
| ● | Risks relating to disagreements with current or future collaboration partners which we rely on for certain drug development activities including the conducting of clinical trials, manufacturing and commercialization of our medicines |
| ● | Risks relating to relying on third party suppliers for the active pharmaceutical ingredients in our drug candidate and drug products |
| ● | Risks relating to our collaboration partners or our CROs’ failure to comply with regulatory requirements pertaining to clinical trials |
| ● | Risks relating to our collaboration partners, principal investigators, CROs and other third-party contractors and consultants engaging in misconduct or other improper activities |
| ● | Risks relating to relying on distributors for logistics and distributions services |
| ● | Risks relating to the availability of benefits currently enjoyed by virtue of our association with CK Hutchison |
11
Other Risks and Risks Relating to Doing Business in China
| ● | Risks relating to compliance with privacy and cybersecurity laws, information security policies and contractual obligations related to data privacy and security and any information technology or data security failures |
| ● | Risks relating to product liability claims or lawsuits |
| ● | Risks relating to liabilities under anti-corruption laws, environmental, health and safety laws and laws relating to equity incentive plans |
| ● | Risks relating to changes in laws, regulations and policies in China and uncertainties with respect to the PRC legal system, China’s currency exchange limits and PRC government tax incentives or treatment |
Risks Relating to Intellectual Property
| ● | Risks relating to our, our joint ventures and our collaboration partners’ abilities to protect and enforce intellectual property rights and maintain confidentiality of trade secrets |
| ● | Risks relating to infringing upon third parties’ intellectual property rights |
Risks Relating to our ADSs
| ● | Risks relating to being delisted from the Nasdaq if the PCAOB is unable to inspect or investigate completely auditors located in China in the future |
| ● | Risks relating to our largest shareholder which may limit the ability of other shareholders to influence corporate matters |
You should carefully consider the following risk factors in addition to the other information set forth in this annual report. If any of the following risks were actually to occur, our company’s business, financial condition and results of operations prospects could be adversely affected and the value of our ADSs would likely suffer.
12
Risks Relating to Our Financial Position and Need for Capital
We may need substantial additional funding for our product development programs and commercialization efforts. If we are unable to raise capital on acceptable terms when needed, we could incur losses and be forced to delay, reduce or eliminate such efforts.
We expect to incur significant expenses in connection with our ongoing activities, particularly as we or our collaboration partners advance the clinical development of our clinical drug candidates which are currently in active or completed clinical studies in various countries. We will incur significant expenses as we continue research and development and initiate additional clinical trials of, and seek regulatory approval for, these and other future drug candidates. In addition, we have incurred and expect to continue to incur significant commercialization expenses related to product manufacturing, marketing, sales and distribution in China for surufatinib (marketed as Sulanda), our unpartnered drug product approved in China in December 2020, and any of our other unpartnered drug candidates that may be approved in the future. For example, the costs that may be required for the manufacture of any drug candidate that receives regulatory approval may be substantial as we may have to modify or increase the production capacity at our current manufacturing facilities or contract with third-party manufacturers. We may also incur expenses as we create additional infrastructure to support the research and development, commercialization and manufacturing of our drug products and candidates.
As a result, we have experienced negative cash flows from operations in the past. Our net cash used in operating activities was $204.2 million and $268.6 million for the years ended December 31, 2021 and 2022, respectively. Even though we generated significant amount of net cash of $219.3 million from our operating activities in 2023, this may not continue in the future as it depends on a variety of factors, including but not limited to:
| ● | the number and development requirements of the drug candidates we pursue; |
| ● | the scope, progress, timing, results and costs of researching and developing our drug candidates, and conducting pre-clinical and clinical trials; |
| ● | the cost, timing and outcome of regulatory review of our drug candidates; |
| ● | the cost and timing of commercialization activities, including product manufacturing, marketing, sales and distribution, for our drug candidates for which we have received regulatory approval; |
| ● | the amount and timing of any upfront milestone or royalty payments, service payments and reimbursement of manufacturing costs from our collaboration partners, with whom we cooperate with respect to the development and potential commercialization of certain of our drug candidates; |
| ● | the cash received from commercial sales of drug candidates for which we have received regulatory approval; |
| ● | our ability to establish and maintain strategic partnerships, collaboration, licensing or other arrangements and the financial terms of such agreements; and |
| ● | the cost, timing and outcome of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending any intellectual property-related claims. |
Accordingly, we may need to obtain substantial funding in connection with our continuing operations through public or private equity offerings, debt financings, collaborations or licensing arrangements or other sources. If we are unable to raise capital when needed or on attractive terms to supplement the cash generated from operating activities to support our operations, we could incur losses and be forced to delay, reduce or eliminate our research and development programs or any future commercialization efforts.
13
Raising capital may dilute our shareholders, restrict our operations or require us to relinquish rights to technologies or drug candidates.
We expect to finance our cash needs in part through cash flow from our operations, and we may also rely on raising capital through a combination of public or private equity offerings, debt financings and/or license and development agreements with collaboration partners. In addition, we may seek capital due to favorable market conditions or strategic considerations, even if we believe we have sufficient funds for our current or future operating plans. To the extent that we raise capital through the sale of equity or convertible debt securities (including potential further listings on other stock exchanges), the ownership interest of our shareholders may be materially diluted, and the terms of such securities could include liquidation or other preferences that adversely affect the rights of our existing shareholders. Debt financing and preferred equity financing, if available, may involve agreements that include restrictive covenants that limit our ability to take specified actions, such as incurring additional debt, making capital expenditures or declaring dividends. Additional debt financing would also result in increased fixed payment obligations.
In addition, if we raise funds through collaborations, strategic partnerships or marketing, distribution or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or drug candidates or grant licenses on terms that may not be favorable to us. We may also lose control of the development of drug candidates, such as the pace and scope of clinical trials, as a result of such third-party arrangements. If we are unable to raise funds through equity or debt financings when needed, we may be required to delay, limit, reduce or terminate our product development or future commercialization efforts or grant rights to develop and market drug candidates that we would otherwise prefer to develop and market ourselves.
Our existing and any future indebtedness could adversely affect our ability to operate our business.
Our outstanding indebtedness combined with current and future financial obligations and contractual commitments, including any additional indebtedness beyond our current loan facilities could have significant adverse consequences, including:
| ● | requiring us to dedicate a portion of our cash resources to the payment of interest and principal, and prepayment and repayment fees and penalties, thereby reducing money available to fund working capital, capital expenditures, product development and other general corporate purposes; |
| ● | increasing our vulnerability to adverse changes in general economic, industry and market conditions; |
| ● | subjecting us to restrictive covenants that may reduce our ability to take certain corporate actions or obtain further debt or equity financing; |
| ● | limiting our flexibility in planning for, or reacting to, changes in our business and the industry in which we compete; and |
| ● | placing us at a competitive disadvantage compared to our competitors that have less debt or better debt servicing options. |
We intend to satisfy our current and future debt service obligations with our existing cash and cash equivalents and short-term investments. Nevertheless, we may not have sufficient funds, and may be unable to arrange for financing, to pay the amounts due under our existing debt. Failure to make payments or comply with other covenants under our existing debt instruments could result in an event of default and acceleration of amounts due.
We have historically incurred significant net operating cash outflows, and may continue to experience net cash outflow from operating activities.
Investment in biopharmaceutical drug development is highly speculative. It entails substantial upfront expenditures and significant risk that a drug candidate might fail to gain regulatory approval or become commercially viable. Therefore, we expect to continue to incur significant expenses related to our ongoing operations, particularly research and development expenses, for the foreseeable future as we expand our development of, and seek regulatory approvals for, our drug candidates. We have historically generated net cash outflows from operations in 2021 and 2022. Although our net cash from operations turned positive in 2023, there is no guarantee that we will be able to continue to do so in the future as our operating cash flows depend on a number of variables that we may not be able to accurately predict or fully control, including the number and scope of our drug development programs and the associated cost of those programs, the cost of commercializing any approved products, our ability to generate revenue and the timing and amount of milestones and other payments we make or receive through arrangements with third parties. Our failure to generate positive cash flow from operations may adversely affect our ability to raise capital, maintain our research and development efforts, expand our business or continue our operations.
14
We face risks with our short-term investments and in collecting our accounts receivables.
Our short-term investments are bank deposits with maturities of more than three months but less than one year. Our short-term investments were $317.7 million and $602.7 million as of December 31, 2022 and 2023, respectively, and are placed with major financial institutions. These investments may earn yields substantially lower than expected. Failure to realize the benefits we expected from these investments may materially and adversely affect our business and financial results. To date, we have experienced no loss or lack of access to our invested cash or cash equivalents; however, we can provide no assurance that access to our invested cash and cash equivalents will not be impacted by adverse conditions in the financial and credit markets.
Our accounts receivable balance, net of allowance for credit losses, totaled $98.0 million and $116.9 million as of December 31, 2022 and 2023, respectively. We have policies and procedures in place to ensure that sales are made to customers with an appropriate credit history. We perform periodic credit evaluations of our customers and monitor risk factors and forward-looking information, such as country risk, when determining credit limits for customers. However, there can be no assurance such policies and procedures will effectively limit our credit risk and enable us to avoid losses, which could adversely affect our financial condition and results of operations. In addition, amounts due to us are not covered by collateral or credit insurance. If we fail to collect all or part of such accounts receivable in a timely manner, or at all, our financial condition may be materially and adversely affected.
Risks Relating to Our Oncology/Immunology Operations and Development of Our Drug Candidates
Our Oncology/Immunology operations historically operated at a net loss, and our future profitability is dependent on the performance of our Oncology/Immunology operations which rely on the successful commercialization of our drug candidates.
To date, savolitinib, fruquintinib and surufatinib (marketed as Orpathys, Elunate and Sulanda, respectively in China and in the U.S. for fruquintinib as Fruzaqla) are our only internally developed drug candidates that have been approved for sale. We do not expect our Oncology/Immunology operations to be significantly profitable unless and until we consistently generate substantial revenue from them and can successfully commercialize our other drug products.
Successful commercialization of our drug candidates is subject to many risks. Savolitinib is marketed as Orpathys in collaboration with our partner, AstraZeneca. We have partnered with Eli Lilly and Takeda on the commercialization of fruquintinib. Surufatinib is marketed by us as Sulanda without the support of a collaboration partner. Savolitinib, fruquintinib and surufatinib are the first innovative oncology drugs we, as an organization, have commercialized, and there is no guarantee that we or our collaboration partners will be able to successfully commercialize them or any of our other drug candidates for their approved indications. There are numerous examples of failures to meet expectations of market potential, including by pharmaceutical companies with more experience and resources than us. There are many factors that could cause the commercialization of savolitinib, fruquintinib and surufatinib or our other drug products to be unsuccessful, including a number of factors that are outside our control. In the case of fruquintinib, for example, the third-line metastatic colorectal cancer, or mCRC, patient population in China may be smaller than we estimate or physicians may be unwilling to prescribe, or patients may be unwilling to take, fruquintinib for a variety of reasons. Additionally, any negative development for fruquintinib, surufatinib or savolitinib in clinical development in additional indications, or in regulatory processes in other jurisdictions, may adversely impact the commercial results and potential of savolitinib, fruquintinib and surufatinib in China and globally. For example, in April 2022, the FDA issued a Complete Response Letter regarding the NDA for surufatinib for the treatment of non-pancreatic neuroendocrine tumors (NETs) and pancreatic NETs and determined that the data package submitted did not support an approval in the U.S. at the time. We subsequently withdrew our submissions to the FDA and the EMA for surufatinib. Thus, significant uncertainty remains regarding the commercial potential of savolitinib, fruquintinib and surufatinib.
Although our operations were profitable in 2023, we may not continue to achieve profitability based on the revenue to be generated from savolitinib, fruquintinib and surufatinib and/or our other drug candidates, if ever. If the commercialization of savolitinib, fruquintinib, surufatinib and/or our other drug candidates is unsuccessful or perceived as disappointing, our stock price could decline significantly and the long-term success of the product and our company could be harmed.
15
All of our drug candidates are still in development. If we are unable to obtain regulatory approval and ultimately commercialize our drug candidates, or if we experience significant delays in doing so, our business will be materially harmed.
All of our drug candidates are still in development, including those that have already received approval for the treatment of certain indications in China and United States. Although we may receive payments from our collaboration partners, including upfront payments and payments for achieving development, regulatory or commercial milestones, for certain of our drug candidates, our ability to generate significant revenue from our drug candidates is dependent on their receipt of additional regulatory approval and successful commercialization, which may never occur. Each of our drug candidates in development will require additional pre-clinical and/or clinical trials, regulatory approval in multiple jurisdictions, and substantial investment in manufacturing and significant efforts before we generate significant revenue from product sales. The success of our drug candidates will depend on several factors, including the following:
| ● | successful completion of additional pre-clinical and/or clinical trials; |
| ● | successful enrollment in, and completion of, additional clinical trials; |
| ● | receipt of additional regulatory approvals from applicable regulatory authorities for planned clinical trials, future clinical trials, drug registrations or post-approval trials; |
| ● | successful completion of all studies required to obtain regulatory approval and/or fulfillment of post-approval requirements in the United States, China, Europe, Japan and other jurisdictions for our drug candidates; |
| ● | adapting our commercial manufacturing capabilities to the specifications for our drug candidates for clinical supply and commercial manufacturing; |
| ● | obtaining and maintaining patent and trade secret protection or regulatory exclusivity for our drug candidates; |
| ● | launching commercial sales of our drug candidates, if and when approved, whether alone or in collaboration with others; |
| ● | acceptance of the drug candidates, if and when approved, by patients, the medical community and third-party payors; |
| ● | effectively competing with other therapies; |
| ● | obtaining and maintaining healthcare coverage and adequate reimbursement; |
| ● | enforcing and defending intellectual property rights and claims; and |
| ● | maintaining a continued acceptable safety profile of the drug candidates following approval. |
If we do not achieve one or more of these factors in a timely manner or at all, we could experience significant delays or an inability to successfully commercialize our drug candidates, which would materially harm our business.
Our primary approach to the discovery and development of drug candidates focuses on the inhibition of kinases, some of which are unproven.
A primary focus of our research and development efforts is on identifying kinase targets for which drug compounds previously developed by others affecting those targets have been unsuccessful due to limited selectivity, off-target toxicity and other problems. We then work to engineer drug candidates which have the potential to have superior efficacy, safety and other features as compared to such prior drug compounds. We also focus on developing drug compounds with the potential to be global best-in-class/next-generation therapies for validated kinase targets.
16
Even if we are able to develop compounds that successfully target the relevant kinases in pre-clinical studies, we may not succeed in demonstrating safety and efficacy of the drug candidates in clinical trials. Even if we are able to demonstrate safety and efficacy of compounds in certain indications in certain jurisdictions, we may not succeed in demonstrating the same in other indications or in the same indications in other jurisdictions. As a result, our efforts may not result in the discovery or development of drugs that are commercially viable or superior to existing drugs or other therapies on the market. While the results of pre-clinical studies, early-stage clinical trials as well as clinical trials in certain indications have suggested that certain of our drug candidates may successfully inhibit kinases and may have significant utility in several cancer indications, potentially in combination with other cancer drugs, chemotherapy and immunotherapies, we have not yet demonstrated efficacy and safety for many of our drug candidates in later stage clinical trials.
We may expend our limited resources to pursue a particular drug candidate or indication and fail to capitalize on drug candidates or indications that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and managerial resources, we must limit our research programs to specific drug candidates that we identify for specific indications. As a result, we may forego or delay pursuit of opportunities with other drug candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial drugs or profitable market opportunities. In addition, if we do not accurately evaluate the commercial potential or target market for a particular drug candidate, we may relinquish valuable rights to that drug candidate through collaboration, licensing or other royalty arrangements when it would have been more advantageous for us to retain sole development and commercialization rights to such drug candidate.
The regulatory approval processes of the U.S. Food and Drug Administration, or FDA, National Medical Products Administration of China, or NMPA, EMA, PDMA and comparable authorities in other countries are lengthy, time consuming and inherently unpredictable, and if we are ultimately unable to obtain regulatory approval for our drug candidates, our ability to generate revenue will be materially impaired.
Our drug candidates and the activities associated with their development and commercialization, including their design, testing, manufacture, safety, efficacy, recordkeeping, labeling, storage, approval, advertising, promotion, sale, distribution, import and export, are subject to comprehensive regulation by the FDA, NMPA, EMA, PDMA and other regulatory agencies in the United States, China, Europe, Japan and by comparable regulatory authorities in other countries. Securing regulatory approval requires the submission of extensive pre-clinical and clinical data and supporting information to the various regulatory authorities for each therapeutic indication to establish the drug candidate’s safety and efficacy. Securing regulatory approval also requires the submission of information about the drug manufacturing process to, and inspection of manufacturing facilities by, the relevant regulatory authority. Our drug candidates may not be effective, may be only moderately effective or may prove to have undesirable or unintended side effects, toxicities or other characteristics that may preclude our obtaining regulatory approval or prevent or limit commercial use.
The process of obtaining regulatory approvals in the United States, China, Europe, Japan and other countries is expensive, may take many years if additional clinical trials are required, if approval is obtained at all, and can vary substantially based upon a variety of factors, including the type, complexity and novelty of the drug candidates involved. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations, or changes in regulatory review for each submitted New Drug Application, or NDA, pre-market approval or equivalent application types, may cause delays in the approval or rejection of an application. The FDA, NMPA, EMA, PDMA and comparable regulatory authorities in other countries have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional pre-clinical, clinical or other studies. Our drug candidates could be delayed in receiving, or fail to receive, regulatory approval for many reasons, including the following:
| ● | the FDA, NMPA, EMA, PDMA or comparable regulatory authorities may disagree with the number, design, size, conduct or implementation of our clinical trials; |
| ● | we may be unable to demonstrate to the satisfaction of the FDA, NMPA, EMA, PDMA or comparable regulatory authorities that a drug candidate is safe and effective for its proposed indication; |
| ● | the results of clinical trials may not meet the level of statistical significance required by the FDA, NMPA, EMA, PDMA or comparable regulatory authorities for approval; |
| ● | we may be unable to demonstrate that a drug candidate’s clinical and other benefits outweigh its safety risks; |
17
| ● | the FDA, NMPA, EMA, PDMA or comparable regulatory authorities may disagree with our interpretation of data from pre-clinical studies or clinical trials; |
| ● | the data collected from clinical trials of our drug candidates may not be sufficient to support the submission of an NDA or other submission or to obtain regulatory approval in the United States or elsewhere; |
| ● | the FDA, NMPA, EMA, PDMA or comparable regulatory authorities may fail to approve the manufacturing processes for our clinical and commercial supplies; |
| ● | the approval policies or regulations of the FDA, NMPA, EMA, PDMA or comparable regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval; |
| ● | the FDA, NMPA, EMA, PDMA or comparable regulatory authority may prioritize treatments for emerging health crises, such as COVID-19, resulting in delays for our drug candidates; |
| ● | the FDA, NMPA, EMA, PDMA or comparable regulatory authorities may restrict the use of our products to a narrow population; and |
| ● | our collaboration partners or CROs that are retained to conduct the clinical trials of our drug candidates may take actions that materially and adversely impact the clinical trials. |
In addition, even if we were to obtain approval, regulatory authorities may approve any of our drug candidates for fewer or more limited indications than we request, may not approve the price we intend to charge for our drugs, may grant approval contingent on the performance of costly post-marketing clinical trials, or may approve a drug candidate with a label that does not include the labeling claims necessary or desirable for the successful commercialization of that drug candidate. Any of the foregoing scenarios could materially harm the commercial prospects for our drug candidates.
Furthermore, even though the NMPA has granted approval for fruquintinib and surufatinib for use in third-line mCRC and NET patients, respectively, and approval for savolitinib for lung cancer with MET exon 14 skipping alterations, we are still subject to substantial, ongoing regulatory requirements. See “—Even if we receive regulatory approval for our drug candidates, we are subject to ongoing obligations and continued regulatory review, which may result in significant additional expense.”
If the FDA, NMPA, EMA, PDMA or another regulatory agency revokes its approval of, or if safety, efficacy, manufacturing or supply issues arise with, any therapeutic that we use in combination with our drug candidates, we may be unable to market such drug candidate or may experience significant regulatory delays or supply shortages, and our business could be materially harmed.
We are currently developing combination therapies using our savolitinib, fruquintinib, surufatinib and other drug candidates with various immunotherapies, targeted therapies and/or other therapies. For example, we are currently developing savolitinib in combination with immunotherapy (Imfinzi) and targeted therapy (Tagrisso). However, we did not develop and we do not manufacture or sell Imfinzi, Tagrisso or any other therapeutic we use in combination with our drug candidates. We may also seek to develop our drug candidates in combination with other therapeutics in the future.
If the FDA, NMPA, EMA, PDMA or another regulatory agency revokes its approval, or does not grant approval, of any of these and other therapeutics we use in combination with our drug candidates, we will not be able to market our drug candidates in combination with such therapeutics. If safety or efficacy issues arise with these or other therapeutics that we seek to combine with our drug candidates in the future, we may experience significant regulatory delays, and we may be required to redesign or terminate the applicable clinical trials. In addition, if manufacturing or other issues result in a supply shortage of these or any other combination therapeutics, we may not be able to complete clinical development of savolitinib, fruquintinib, surufatinib and/or any other of our drug candidates on our current timeline or at all.
Even if one or more of our drug candidates were to receive regulatory approval for use in combination with a therapeutic, we would continue to be subject to the risk that the FDA, NMPA, EMA, PDMA or another regulatory agency could revoke its approval of the combination therapeutic, or that safety, efficacy, manufacturing or supply issues could arise with one of these combination therapeutics. This could result in savolitinib, fruquintinib, surufatinib or one of our other products being removed from the market or being less successful commercially.
18
We face substantial competition, and our competitors may discover, develop or commercialize drugs before or more successfully than we do.
The development and commercialization of new drugs is highly competitive. We face competition with respect to our current drug candidates, and will face competition with respect to any drug candidates that we may seek to develop or commercialize in the future, from major pharmaceutical companies, specialty pharmaceutical companies and biotechnology companies worldwide. There are a number of large pharmaceutical and biotechnology companies that currently market drugs or are pursuing the development of therapies in the field of kinase inhibition for cancer and other diseases. Some of these competitive drugs and therapies are based on scientific approaches that are the same as or similar to our approach, and others are based on entirely different approaches. Potential competitors also include academic institutions, government agencies and other public and private research organizations that conduct research, seek patent protection and establish collaborative arrangements for research, development, manufacturing and commercialization. Specifically, there are a large number of companies developing or marketing treatments for cancer and immunological diseases, including many major pharmaceutical and biotechnology companies.
Many of the companies against which we are competing or against which we may compete in the future have significantly greater financial resources and expertise in research and development, manufacturing, pre-clinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved drugs than we do. Mergers and acquisitions in the pharmaceutical, biotechnology and diagnostic industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize drugs that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any drugs that we or our collaborators may develop. Our competitors also may obtain FDA, NMPA, EMA, PDMA or other regulatory approval for their drugs more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we or our collaborators are able to enter the market. The key competitive factors affecting the success of all of our drug candidates, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic competition and the availability of reimbursement from government and other third-party payors.
Clinical development involves a lengthy and expensive process with an uncertain outcome.
There is a risk of failure for each of our drug candidates. It is difficult to predict when or if any of our drug candidates will prove effective and safe in humans or will receive regulatory approval. Before obtaining regulatory approval from regulatory authorities for the sale of any drug candidate, we or our collaboration partners must complete pre-clinical studies and then conduct extensive clinical trials to demonstrate the safety and efficacy of our drug candidates in humans. Clinical testing is expensive, difficult to design and implement and can take many years to complete. The outcomes of pre-clinical development testing and early clinical trials may not be predictive of the success of later clinical trials, and interim results of a clinical trial do not necessarily predict final results. Moreover, pre-clinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their drug candidates performed satisfactorily in pre-clinical studies and clinical trials have nonetheless failed to obtain regulatory approval of their drug candidates. Our current or future clinical trials may not be successful.
Commencing each of our clinical trials is subject to finalizing the trial design based on ongoing discussions with the FDA, NMPA, EMA, PDMA or other regulatory authorities. The FDA, NMPA, EMA, PDMA and other regulatory authorities could change their position on the acceptability of our trial designs or clinical endpoints, which could require us to complete additional clinical trials or impose approval conditions that we do not currently expect. Successful completion of our clinical trials is a prerequisite to submitting an NDA or analogous filing to the FDA, NMPA, EMA, PDMA or other regulatory authorities for each drug candidate and, consequently, the ultimate approval and commercial marketing of our drug candidates. We do not know whether any of our clinical trials will begin or be completed on schedule, if at all.
19
We and our collaboration partners may incur additional costs or experience delays in completing our pre-clinical or clinical trials, or ultimately be unable to complete the development and commercialization of our drug candidates.
We and our collaboration partners, including AstraZeneca, Eli Lilly, Takeda, BeiGene Ltd., or BeiGene, Inmagene, Innovent Biologics (Suzhou) Co., Inc., or Innovent, Genor Biopharma Co. Ltd., or Genor, Shanghai Junshi Biosciences Co. Ltd., or Junshi and Epizyme, Inc. (a subsidiary of Ipsen Pharma SAS), or Epizyme may experience delays in completing our pre-clinical or clinical trials, and numerous unforeseen events could arise during, or as a result of, future clinical trials, which could delay or prevent us from receiving regulatory approval, including:
| ● | regulators, institutional review boards, or IRBs, ethics committees or the China Human Genetic Resources Administration Office may not authorize us or our investigators to commence or conduct a clinical trial at a prospective trial site; |
| ● | we may experience delays in reaching, or we may fail to reach, agreement on acceptable terms with prospective trial sites and prospective CROs, who conduct clinical trials on behalf of us and our collaboration partners, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| ● | clinical trials may produce negative or inconclusive results, and we or our collaboration partners may decide, or regulators may require us or them, to conduct additional clinical trials or we may decide to abandon drug development programs; |
| ● | the number of patients required for clinical trials of our drug candidates may be larger than we anticipate, enrollment in these clinical trials may be slower than we anticipate or participants may drop out of these clinical trials or fail to return for post-treatment follow-up at a higher rate than we anticipate; |
| ● | third-party contractors used in our clinical trials may fail to comply with regulatory requirements or meet their contractual obligations in a timely manner, or at all, or may deviate from the clinical trial protocol or drop out of the trial, which may require that we or our collaboration partners add new clinical trial sites or investigators; |
| ● | we or our collaboration partners may elect to, or regulators, IRBs or ethics committees may require that we or our investigators, suspend or terminate clinical research for various reasons, including non-compliance with regulatory requirements or a finding that the participants are being exposed to unacceptable health risks; |
| ● | the cost of clinical trials of our drug candidates may be greater than we anticipate; |
| ● | the supply or quality of our drug candidates, companion diagnostics, if any, or other materials necessary to conduct clinical trials of our drug candidates may be insufficient or inadequate; and |
| ● | our drug candidates may have undesirable side effects or unexpected characteristics, causing us or our investigators, regulators, IRBs or ethics committees to suspend or terminate the trials, or reports may arise from pre-clinical or clinical testing of other cancer therapies that raise safety or efficacy concerns about our drug candidates. |
We could encounter regulatory delays if a clinical trial is suspended or terminated by us or our collaboration partners, by, as applicable, the IRBs of the institutions in which such trials are being conducted, by the Data Safety Monitoring Board, which is an independent group of experts that is formed to monitor clinical trials while ongoing, or by the FDA, NMPA, EMA, PDMA or other regulatory authorities. Such authorities may impose a suspension or termination due to a number of factors, including: a failure to conduct the clinical trial in accordance with regulatory requirements or the applicable clinical protocols, inspection of the clinical trial operations or trial site by the FDA, NMPA, EMA, PDMA or other regulatory authorities that results in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a drug, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial. Many of the factors that cause a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of our drug candidates. Further, the FDA, NMPA, EMA, PDMA or other regulatory authorities may disagree with our clinical trial design and our interpretation of data from clinical trials, or may change the requirements for approval even after it has reviewed and commented on the design for our clinical trials.
20
If we or our collaboration partners are required to conduct additional clinical trials or other testing of our drug candidates beyond those that are currently contemplated, if we or our collaboration partners are unable to successfully complete clinical trials of our drug candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, we may:
| ● | be delayed in obtaining regulatory approval for our drug candidates; |
| ● | not obtain regulatory approval at all; |
| ● | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| ● | be subject to post-marketing testing requirements; or |
| ● | have the drug removed from the market after obtaining regulatory approval. |
Our drug development costs will also increase if we experience delays in testing or regulatory approvals. We do not know whether any of our clinical trials will begin as planned, will need to be restructured or will be completed on schedule, or at all. Significant pre-clinical study or clinical trial delays also could allow our competitors to bring products to market before we do and impair our ability to successfully commercialize our drug candidates and may harm our business and results of operations. Any delays in our clinical development programs may significantly harm our business, financial condition and prospects.
If we or our collaboration partners experience delays or difficulties in the enrollment of patients in clinical trials, the progress of such clinical trials and our receipt of necessary regulatory approvals could be delayed or prevented.
We or our collaboration partners may not be able to initiate or continue clinical trials for our drug candidates if we or our collaboration partners are unable to locate and enroll a sufficient number of eligible patients to participate in these trials as required by the FDA, NMPA, EMA, PDMA or similar regulatory authorities. In particular, we and our collaboration partners have designed many of our clinical trials, and expect to design future trials, to include some patients with the applicable genomic alteration that causes the disease with a view to assessing possible early evidence of potential therapeutic effect. Genomically defined diseases, however, may have relatively low prevalence, and it may be difficult to identify patients with the applicable genomic alteration. In addition, for many of our trials, we focus on enrolling patients who have failed their first or second-line treatments, which limits the total size of the patient population available for such trials. The inability to enroll a sufficient number of patients with the applicable genomic alteration or that meet other applicable criteria for our clinical trials would result in significant delays and could require us or our collaboration partners to abandon one or more clinical trials altogether.
In addition, some of our competitors have ongoing clinical trials for drug candidates that treat the same indications as our drug candidates, and patients who would otherwise be eligible for our clinical trials may instead enroll in clinical trials of our competitors’ drug candidates.
Patient enrollment may be affected by other factors including:
| ● | the severity of the disease under investigation; |
| ● | the total size and nature of the relevant patient population; |
| ● | the design and eligibility criteria for the clinical trial in question; |
| ● | the availability of an appropriate genomic screening test/companion diagnostic; |
| ● | the perceived risks and benefits of the drug candidate under study; |
| ● | the efforts to facilitate timely enrollment in clinical trials; |
21
| ● | the patient referral practices of physicians; |
| ● | the availability of competing therapies which are undergoing clinical trials; |
| ● | the ability to monitor patients adequately during and after treatment; |
| ● | the proximity and availability of clinical trial sites for prospective patients ; and |
| ● | the impact of the spread of infectious diseases, including but not limited to the duration and scope of related government orders and restrictions. |
Enrollment delays in our clinical trials may result in increased development costs for our drug candidates, which could cause the value of our company to decline and limit our ability to obtain financing.
Our drug candidates may cause undesirable side effects that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in significant negative consequences following regulatory approval, if any.
Undesirable side effects caused by our drug candidates could cause us or our collaboration partners to interrupt, delay or halt clinical trials or could cause regulatory authorities to interrupt, delay or halt our clinical trials and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, NMPA, EMA, PDMA or other regulatory authorities. In particular, as is the case with all oncology drugs, it is likely that there may be side effects associated with the use of certain of our drug candidates. Results of our trials could reveal a high and unacceptable severity and prevalence of these or other side effects. In such an event, our trials could be suspended or terminated and the FDA, NMPA, EMA, PDMA or comparable regulatory authorities could order us to cease further development of or deny approval of our drug candidates for some or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in potential product liability claims. Any of these occurrences may harm our business, financial condition and prospects significantly.
Further, our drug candidates could cause undesirable side effects related to off-target toxicity. Many of the currently approved tyrosine kinase inhibitors or TKIs have been associated with off-target toxicities because they affect multiple kinases. While we believe that the kinase selectivity of our drug candidates has the potential to significantly improve the unfavorable adverse off-target toxicity issues, if patients were to experience off-target toxicity, we may not be able to achieve an effective dosage level, receive approval to market, or achieve the commercial success we anticipate with respect to any of our drug candidates, which could prevent us from ever generating revenue or achieving profitability. Many compounds that initially showed promise in early-stage testing for treating cancer have later been found to cause side effects that prevented further development of the compound.
Clinical trials assess a sample of the potential patient population. With a limited number of patients and duration of exposure, rare and severe side effects of our drug candidates may only be uncovered with a significantly larger number of patients exposed to the drug candidate. If our drug candidates receive regulatory approval and we or others identify undesirable side effects caused by such drug candidates (or any other similar drugs) after such approval, a number of potentially significant negative consequences could result, including:
| ● | regulatory authorities may withdraw or limit their approval of such drug candidates; |
| ● | regulatory authorities may require the addition of labeling statements, such as a “boxed” warning or a contra-indication; |
| ● | we may be required to create a medication guide outlining the risks of such side effects for distribution to patients; |
| ● | we may be required to change the way such drug candidates are distributed or administered, conduct additional clinical trials or change the labeling of the drug candidates; |
| ● | regulatory authorities may require a Risk Evaluation and Mitigation Strategy, or REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools; |
22
| ● | we may be subject to regulatory investigations and government enforcement actions; |
| ● | we may decide to remove such drug candidates from the marketplace; |
| ● | we could be sued and held liable for injury caused to individuals exposed to or taking our drug candidates; and |
| ● | our reputation may suffer. |
Any of these events could prevent us from achieving or maintaining market acceptance of the affected drug candidates and could substantially increase the cost of commercializing our drug candidates, if approved, and significantly impact our ability to successfully commercialize our drug candidates and generate revenue.
We and our collaboration partners have conducted and intend to conduct additional clinical trials for certain of our drug candidates at sites outside the United States, and the FDA may not accept data from trials conducted in such locations or may require additional U.S.-based trials.
We and our collaboration partners have conducted, currently are conducting and intend in the future to conduct, clinical trials outside the United States, particularly in China where our Oncology/Immunology operations are headquartered as well as in other jurisdictions such as Australia, Japan, South Korea and various European countries.
Although the FDA may accept data from clinical trials conducted outside the United States, acceptance of these data is subject to certain conditions imposed by the FDA. For example, the clinical trial must be well designed and conducted by qualified investigators in accordance with current good clinical practices, or GCPs, including review and approval by an independent ethics committee and receipt of informed consent from trial patients. The trial population must also adequately represent the U.S. population, and the data must be applicable to the U.S. population and U.S. medical practice in ways that the FDA deems clinically meaningful. Generally, the patient population for any clinical trial conducted outside of the United States must be representative of the population for which we intend to seek approval in the United States. In addition, while these clinical trials are subject to applicable local laws, FDA acceptance of the data will be dependent upon its determination that the trials also comply with all applicable U.S. laws and regulations. There can be no assurance that the FDA will accept data from trials conducted outside of the United States. If the FDA does not accept the data from our clinical trials conducted outside the United States, it would likely result in the need for additional clinical trials, which would be costly and time-consuming and delay or permanently halt our ability to develop and market these or other drug candidates in the United States. In April 2022, we received a Complete Response Letter from the FDA regarding the NDA for surufatinib for the treatment of pancreatic NETs and non-pancreatic NETs. The FDA determined that the data package submitted in the application, based on two successful Phase III trials in China and one bridging study in the U.S., were not sufficient to support approval in the U.S.. The Complete Response Letter indicated that a multi-regional clinical trial would be required for U.S. approval. We subsequently withdrew our submissions to the FDA and the EMA for surufatinib.
In addition, there are risks inherent in conducting clinical trials in jurisdictions outside the United States including:
| ● | regulatory and administrative requirements of the jurisdiction where the trial is conducted that could burden or limit our ability to conduct our clinical trials; |
| ● | foreign exchange fluctuations; |
| ● | manufacturing, customs, shipment and storage requirements; |
| ● | cultural differences in medical practice and clinical research; and |
| ● | the risk that patient populations in such trials are not considered representative as compared to patient populations in the United States and other markets. |
23
If we are unable to obtain and/or maintain priority review by the NMPA, fast track designation by the FDA, or another expedited registration pathway for our drug candidates, the time and cost we incur to obtain regulatory approvals may increase. Even if we receive such approvals, they may not lead to a faster development, review or approval process.
Under the Breakthrough Therapy Drug Review Procedures (For Trial Implementation), the Review and Approval Procedures for Conditional Approval of Drug Marketing Applications (For Trial Implementation), and the Priority Review and Approval Procedures for Drug Marketing Authorization (For Trial Implementation), the NMPA (or, where applicable, the National Health Commission, or the NHC) may grant priority review approval (i) to innovative drugs or new improved drugs undergoing clinical trials that are used to prevent and treat diseases that are seriously life-threatening or which seriously affect quality of life for which there is no effective prevention or treatment, or for which there is sufficient evidence to show obvious clinical advantages compared with existing treatments, (ii) to drugs undergoing clinical trials which meet the conditions for conditional approval specified in the Technical Guidelines for Conditional Approval of Drugs, (iii) to innovative drugs and new improved drugs which are in shortage, prevent and treat major infectious diseases and rare diseases, (iv) to new varieties, dosage forms and specifications that meet the physiological characteristics of children, (v) to vaccines (including innovative vaccines) urgently needed for control and prevention of diseases, and (vi) under other circumstances stipulated by the NMPA. Priority review provides a fast track process for drug registration. In the past, we received priority review status for a number of our drug candidates, including for example fruquintinib for the treatment of advanced colorectal cancer, or CRC, savolitinib for the treatment of NSCLC and surufatinib for the treatment of advanced NET. We anticipate that we may seek priority review for certain of our other drug candidates in the future.
In the United States, if a drug is intended for the treatment of a serious or life-threatening condition and the drug demonstrates the potential to address unmet medical needs for this condition, we may apply for fast track designation by the FDA. The FDA has broad discretion whether or not to grant this designation, so even if we believe a particular drug candidate is eligible for this designation, we cannot be sure that the FDA would decide to grant it. We have sought and will likely continue to seek fast track designation for some of our drug candidates. Even if we receive fast track designation for a drug candidate, we may not experience a faster development process, review or approval compared to conventional FDA procedures. The FDA may withdraw fast track designation if it believes that the designation is no longer supported by data from our clinical development program.
A failure to obtain and/or maintain priority review, fast track designation or any other form of expedited development, review or approval for our drug candidates would result in a longer time period to commercialization of such drug candidate, could increase the cost of development of such drug candidate and could harm our competitive position in the marketplace. In addition, even if we obtain priority review, there is no guarantee that we will experience a faster review or approval compared to non-accelerated registration pathways or that a drug candidate will ultimately be approved for sale.
Even if we or our collaboration partners receive regulatory approval for our drug candidates, we or our collaboration partners are subject to ongoing obligations and continued regulatory review, which may result in significant additional expense.
If the FDA, NMPA, EMA, PDMA or a comparable regulatory authority approves any of our drug candidates, we or our collaboration partners will continue to be subject to extensive and ongoing regulatory requirements. For example, even though the NMPA has granted approval of fruquintinib, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for fruquintinib continue to be subject to the NMPA’s oversight. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as continued compliance with current good manufacturing processes.
Any regulatory approvals that we or our collaboration partners receive for our drug candidates may also be subject to limitations on the approved indicated uses for which the drug may be marketed or to the conditions of approval, or contain requirements for potentially costly post-marketing testing, including post-approval testing, sometimes referred to as Phase IV clinical trials, and surveillance to monitor the safety and efficacy of the drug. In addition, regulatory policies may change or additional government regulations may be enacted that could prevent, limit or delay regulatory approval of our drug candidates. If we or our collaboration partners are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we or our collaboration partners are not able to maintain regulatory compliance, we or our collaboration partners may lose any regulatory approval that we or our collaboration partners may have obtained, which would adversely affect our business, prospects and ability to achieve or sustain profitability.
24
We may be subject to penalties if we fail to comply with regulatory requirements or experience unanticipated problems with any of our drugs that receive regulatory approval.
Once a drug is approved by the FDA, NMPA, EMA, PDMA or a comparable regulatory authority for marketing, it is possible that there could be a subsequent discovery of previously unknown problems with the drug, including problems with third-party manufacturers or manufacturing processes, or failure to comply with regulatory requirements. If any of the foregoing occurs with respect to our drug products, it may result in, among other things:
| ● | restrictions on the marketing or manufacturing of the drug, withdrawal of the drug from the market, or drug recalls; |
| ● | fines, warning letters or holds on clinical trials; |
| ● | refusal by the FDA, NMPA, EMA, PDMA or comparable regulatory authority to approve pending applications or supplements to approved applications filed by us, or suspension or revocation of drug license approvals; |
| ● | drug seizure or detention, or refusal to permit the import or export of drugs; and |
| ● | injunctions or the imposition of civil or criminal penalties. |
Any government investigation of alleged violations of law could require us to expend significant time and resources and could generate negative publicity. If we or our collaborators are not able to maintain regulatory compliance, regulatory approval that has been obtained may be lost and we may not achieve or sustain profitability, which would adversely affect our business, prospects, financial condition and results of operations.
The incidence and prevalence for target patient populations of our drug candidates are based on estimates and third-party sources. If the market opportunities for our drug candidates are smaller than we estimate or if any approval that we obtain is based on a narrower definition of the patient population, our revenue and ability to achieve profitability will be adversely affected, possibly materially.
Periodically, we make estimates regarding the incidence and prevalence of target patient populations for particular diseases based on various third-party sources and internally generated analysis and use such estimates in making decisions regarding our drug development strategy, including determining indications on which to focus in pre-clinical or clinical trials. These estimates may be inaccurate or based on imprecise data. For example, the total addressable market opportunity will depend on, among other things, their acceptance by the medical community and patient access, drug pricing and reimbursement. The number of patients in the addressable markets may turn out to be lower than expected, patients may not be otherwise amenable to treatment with our drugs, or new patients may become increasingly difficult to identify or gain access to, all of which would adversely affect our results of operations and our business.
Our future success depends on our ability to retain key executives and to attract, retain and motivate qualified personnel.
We are highly dependent on the expertise of the members of our research and development team, as well as the other principal members of our management, including Weiguo Su, Ph.D., our Chief Executive Officer, Chief Scientific Officer and director. Although we have entered into employment agreements with our executive officers, each of them may terminate their employment with us at any time with three months’ prior written notice. We do not maintain “key person” insurance for any of our executives or other employees.
Recruiting and retaining qualified management, scientific, clinical, manufacturing and sales and marketing personnel will also be critical to our success. The loss of the services of our executive officers or other key employees could impede the achievement of our research, development and commercialization objectives and seriously harm our ability to successfully implement our business strategy. Furthermore, replacing executive officers and key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to successfully develop, gain regulatory approval of and commercialize drugs. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these key personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific and clinical personnel from universities and research institutions. Failure to succeed in clinical trials may make it more challenging to recruit and retain qualified scientific personnel.
25
We have operations internationally and are subject to a variety of risks and complexities that may materially and adversely affect our business, results of operations, financial condition and growth prospects.
We have been involved in clinical and non-clinical development internationally for over a decade. Conducting our business in multiple countries subjects us to a variety of risks and complexities that may materially and adversely affect our business, results of operations, financial condition and growth prospects, including, among other things:
| ● | the increased complexity and costs inherent in managing international operations; |
| ● | diverse regulatory, financial and legal requirements, and any future changes to such requirements, in one or more countries where we are located or do business; |
| ● | country-specific tax, labor and employment laws and regulations; |
| ● | applicable trade laws, tariffs, export quotas, custom duties or other trade restrictions and any changes to them; |
| ● | challenges inherent in efficiently managing employees in diverse geographies, including the need to adapt systems, policies, benefits and compliance programs to differing labor and other regulations; |
| ● | changes in currency rates; and |
| ● | regulations relating to data security and the unauthorized use of, or access to, commercial and personal information. |
There can be no assurance that we will effectively manage the increased complexity without experiencing operating inefficiencies or control deficiencies. Such increased complexity may also lead to decisions to reposition our international operations to align them with our overall and evolving business strategy, including with our recent strategic change to focus on path to profitability. Significant management time and effort is required to effectively manage the increased complexity of our company, and our failure to successfully do so could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
We may be restricted from transferring our scientific data abroad.
On March 17, 2018, the General Office of the State Council promulgated the Measures for the Management of Scientific Data, or the Scientific Data Measures, which provides a broad definition of scientific data and relevant rules for the management of scientific data. According to the Scientific Data Measures, enterprises in China must seek governmental approval before any scientific data involving a state secret may be transferred abroad or to foreign parties. Further, any researcher conducting research funded at least in part by the Chinese government is required to submit relevant scientific data for management by the entity to which such researcher is affiliated before such data may be published in any foreign academic journal. Given that the term state secret is not clearly defined in the Scientific Data Measures, if and to the extent our research and development of drug candidates will be subject to the Scientific Data Measures and any subsequent laws as required by the relevant government authorities, we cannot assure you that we can always obtain relevant approvals for sending scientific data (such as the results of our pre-clinical studies or clinical trials conducted within China) abroad or to our foreign partners in China. The PRC Personal Information Protection Law, effective November 2021, provides that where a personal information processor needs to provide personal information outside the territory of the PRC due to business or other needs, it shall meet any of the following conditions: (i) it shall pass the security evaluation organized by the Cyberspace Administration of China (“CAC”) in accordance with the provisions of Article 40 thereof, (ii) it shall have been certified by a specialized agency for protection of personal information in accordance with the provisions of the CAC, (iii) it shall enter into a contract with the overseas recipient under the standard contract formulated by the CAC, specifying the rights and obligations of both parties, or (iv) it shall meet other conditions prescribed by laws, administrative regulations or the CAC. If we are unable to obtain necessary approvals or meet the necessary requirements in a timely manner, or at all, our research and development of drug candidates may be hindered, which may materially and adversely affect our business, results of operations, financial conditions and prospects. If the relevant government authorities consider the transmission of our scientific data to be in violation of the requirements under the Scientific Data Measures, we may be subject to fines and other administrative penalties imposed by those government authorities.
26
Any adverse developments related to the administration of our drug candidates in compassionate use programs may affect our and/or our partners’ ability to obtain regulatory approval or commercialize our drug candidates.
In many countries, physicians are permitted to administer unapproved drugs to patients who have life-threatening disease with no viable available therapy. From time to time, we and our partners participate in such programs and offer our drug candidates for patient treatment. Given that the patients receiving treatment under such programs often have very advanced diseases, there is an increased risk that they may experience more severe adverse events. If serious adverse events or other issues that call into question the potential efficacy and safety of our drug candidates occur when our drug candidates are administered through compassionate use programs, the NMPA, the FDA and other regulatory authorities may delay, limit, or deny approval of our drug candidates or require us and/or our partners to conduct additional clinical trials as a condition to marketing approval, which would increase drug development costs.
Risks Relating to Sales of Our Internally Developed Drugs and Other Drugs
Pharmaceutical companies in China are required to comply with extensive regulations and hold a number of permits and licenses to carry on their business. Our and our joint ventures’ ability to obtain and maintain these regulatory approvals is uncertain, and future government regulation may impose additional burdens on our operations.
The pharmaceutical industry in China is subject to extensive government regulation and supervision. The regulatory framework addresses all aspects of operations in the pharmaceutical industry, including approval, production, distribution, advertising, licensing and certification requirements and procedures, periodic renewal and reassessment processes, registration of new drugs and environmental protection. Violation of applicable laws and regulations may materially and adversely affect our business. In order to manufacture and distribute pharmaceutical products in China, we and our joint ventures are required to, among other things:
| ● | obtain a pharmaceutical manufacturing permit for each production facility from the NMPA; |
| ● | obtain a drug registration certificate, which includes a drug approval number, from the NMPA for each drug manufactured by us; |
| ● | obtain a pharmaceutical distribution permit from the NMPA; and |
| ● | renew the pharmaceutical manufacturing permits, the pharmaceutical distribution permits, drug registration certificates, among other requirements. |
If we or our joint ventures are unable to obtain or renew such permits or any other permits or licenses required for our or their operations, we will not be able to engage in the manufacture and distribution of our products and our business may be adversely affected.
The regulatory framework regarding the pharmaceutical industry in China is subject to change and amendment from time to time. Any such change or amendment could materially and adversely impact our business, financial condition and results of operations. The PRC government has introduced various reforms to the Chinese healthcare system in recent years and may continue to do so, with an overall objective to expand basic medical insurance coverage and improve the quality and reliability of healthcare services. Specific upcoming regulatory and policy changes remain uncertain. The implementing measures to be issued may not be sufficiently effective to achieve the stated goals and, as a result, we may not be able to benefit from such reform to the level we expect, if at all. Moreover, the reform could give rise to regulatory developments, such as more burdensome administrative procedures, which may have an adverse effect on our business and prospects.
For further information regarding government regulation in China and other jurisdictions, see Item 4.B. “Business Overview—Regulations—Government Regulation of Pharmaceutical Product Development and Approval,” “Business Overview—Regulations—Coverage and Reimbursement” and “Business Overview—Regulations—Other Healthcare Laws.”
27
As a significant portion of the operations of our Other Ventures is conducted through joint ventures, we are dependent on the success of our joint ventures, our receipt of dividends or other payments from our joint ventures for cash to fund our operations, and our investments in our joint ventures are subject to liquidity risk.
We are party to a joint venture agreement with Shanghai Pharmaceuticals, relating to our non-consolidated joint venture namely, Shanghai Hutchison Pharmaceuticals, which forms part of the operations of our Other Ventures. Our equity in earnings of such non-consolidated joint venture, net of tax, was $44.7 million, $49.7 million and $47.3 million for the years ended December 31, 2021, 2022 and 2023, respectively, as recorded in our consolidated financial statements. As such, our results of operations and financial performance have been, and will continue to be, affected by the financial performance of such joint venture as well as any other equity investees we have or may have in the future. We may also be required to recognize an impairment charge in our consolidated financial statements if there is a decline in the fair market value of our investments in such businesses below their carrying amounts for whatever reason that is determined to be other-than-temporary. Furthermore, we have consolidated joint ventures with Sinopharm which accounted for substantially all of our Other Ventures’ consolidated revenue for the years ended December 31, 2021, 2022 and 2023.
As a result, our ability to fund our operations and pay our expenses or to make future dividend payments, if any, is partly dependent on the earnings of our joint ventures and the payment of those earnings to us in the form of dividends. Payments to us by our joint ventures will be contingent upon our joint ventures’ earnings and other business considerations and may be subject to statutory or contractual restrictions. Each joint venture’s ability to distribute dividends to us is subject to approval by their respective boards of directors, which in the case of Shanghai Hutchison Pharmaceuticals is comprised of an equal number of representatives from each party. Furthermore, our ability to promptly sell one or more of our interests in our joint ventures in response to changing corporate strategy or economic, financial and investment conditions is limited. The market for such investments can be affected by various factors, such as general economic and market conditions, availability of financing, interest rates and investor demand, many of which are beyond our control. If we determine to sell any of our joint venture investments, we cannot predict if we will be successful or whether any price or other terms offered by a prospective purchaser would be acceptable to us.
Operationally, our joint venture partners have certain responsibilities and/or certain rights to exercise control or influence over operations and decision-making under the joint venture arrangements. Therefore, the success of our joint ventures depends on the efforts and abilities of our joint venture parties. For example, we appoint the general managers of Hutchison Sinopharm and Shanghai Hutchison Pharmaceuticals pursuant to the respective joint venture agreements governing these entities and therefore oversee the day-to-day management of these joint ventures. However, we still rely on our joint venture partners Sinopharm and Shanghai Pharmaceuticals to provide certain distribution and logistics services. See “—Risks Relating to Our Dependence on Third Parties—Joint ventures form an important part of our Other Ventures, and our ability to manage and develop the businesses conducted by these joint ventures depends in part on our relationship with our joint venture partners” for more information.
We may not be successful in building a commercial team to successfully manufacture, sell and market our approved drugs, and we may not be able to generate any revenue from such products.
We have leveraged our experience operating our prescription drugs business to commercialize certain of our approved, internally developed drug candidates in China. We must adapt our know-how to build a specific oncology and/or immunology focused sales and marketing team. As of December 31, 2023, we had an oncology commercial team with over 900 staff to support the commercialization of fruquintinib, surufatinib and our other drug candidates, if approved. There are risks involved in establishing an in-house oncology commercial team. For example, recruiting and/or training a sales force to detail our approved drug candidates is time consuming and could delay any drug launch. Factors that may inhibit our efforts to commercialize our drug candidates include:
| ● | our inability to recruit and retain adequate numbers of effective sales and marketing personnel; |
| ● | our inability to effectively manage the expansion of our operations and train additional qualified personnel in the relevant areas of oncology and/or immunology; |
| ● | our failure to prevent inappropriate business conducts, including behaviors that may violate anti-bribery and anti-corruption laws and regulations; |
| ● | the inability of our sales personnel to obtain access to physicians or educate adequate numbers of physicians who then prescribe any future drugs; and |
28
| ● | the lack of complementary drugs to be offered by our sales personnel, which may put us at a competitive disadvantage relative to companies with more extensive product lines. |
In such case, our business, results of operations, financial condition and prospects will be materially and adversely affected.
We face substantial competition in selling our approved, internally developed drugs and the drugs of our Other Ventures.
The marketed drugs developed and sold by our Oncology/Immunology operations and the prescription drugs business which is part of our Other Ventures’ operations face substantial competition in the pharmaceutical industry in China, which is characterized by a number of established, large pharmaceutical companies, as well as smaller emerging pharmaceutical companies, engaged in the development, production, marketing or sales of prescription drugs, in particular cardiovascular drugs. The identities of the key competitors with respect to drugs sold by our Oncology/Immunology and Other Ventures operations vary by product and, in certain cases, competitors have greater financial resources than us and may elect to focus these resources on developing, importing or in-licensing and marketing products in the PRC that are substitutes for our products and may have broader sales and marketing infrastructure with which to do so.
Such drugs may compete against products that have lower prices, superior performance, greater ease of administration or other advantages compared to our products. In some circumstances, price competition may drive our competitors to conduct illegal manufacturing processes to lower their manufacturing costs. Increased competition may result in price reductions, reduced margins and loss of market share, whether achieved by either legal or illegal means, any of which could materially and adversely affect our profit margins. We and our joint ventures may not be able to compete effectively against current and future competitors.
If we are not able to maintain and enhance brand recognition of our drugs to maintain a competitive advantage, our reputation, business and operating results may be harmed.
We believe that market awareness of our products sold through our Oncology/Immunology and Other Ventures operations, which include our joint ventures’ branded products, such as Shang Yao, and the brands of third-party products which are distributed through our joint ventures, has contributed significantly to our success. We also believe that maintaining and enhancing such brands is critical to maintaining our competitive advantage. Although the sales and marketing staff of such businesses will continue to further promote such brands to remain competitive, they may not be successful. If we or our joint ventures are unable to further enhance brand recognition and increase awareness of such products, or are compelled to incur excessive marketing and promotion expenses in order to maintain brand awareness, our business and results of operations may be materially and adversely affected. Furthermore, our results of operations could be adversely affected if the Shang Yao brand, or the brands of any other products, or our reputation, are impaired by certain actions taken by our joint venture partners, distributors, competitors or relevant regulatory authorities.
Reimbursement may not be available for the products currently sold through our Oncology/Immunology and Other Ventures operations or our drug candidates in China, the United States or other countries, which could diminish our sales or affect our profitability.
The regulations that govern pricing and reimbursement for pharmaceuticals vary widely from country to country. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after regulatory approval is granted. In some foreign markets, pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. Furthermore, once marketed and sold, government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which medications they will pay for and establish reimbursement levels. Adverse pricing reimbursement levels may hinder market acceptance of our drug candidates or other products sold by us.
29
In China, for example, the Ministry of Human Resources and Social Security of the PRC (the “MOHRSS”) or provincial or local human resources and social security authorities, together with other government authorities, review the inclusion or removal of drugs from the Medicines Catalogue for the National Basic Medical Insurance, Labor Injury Insurance and Childbirth System in China, or the National Reimbursement Drug List, or NRDL, or provincial or local medical insurance catalogues for the National Medical Insurance Program, and the category under which a drug will be classified, both of which affect the amounts reimbursable to program participants for their purchases of those medicines. These determinations are made based on a number of factors, including price and efficacy. Depending on the category under which a drug is classified in the provincial medicine catalogue, a National Medical Insurance Program participant residing in that province can be reimbursed for the full cost of Category A medicine and for the majority of the cost of a Category B medicine. In some instances, if the price range designated by the local or provincial government decreases, it may adversely affect our business and could reduce our total revenue, and if our revenue falls below production costs, we may stop manufacturing certain products. Since January 2020, January 2022 and March 2023, Elunate, Sulanda and Orpathys have been included in China’s NRDL as a Category B medicine, respectively.
In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs which may affect reimbursement rates of our drug candidates if approved. Various federal and state laws have been enacted to control drug pricing or require manufacturers to disclose information about drug pricing. For example, the Inflation Reduction Act of 2022, or IRA, was signed into law, and, among other provisions, mandates the negotiation of eligible Medicare Part B and Part D drugs; redesigns the Medicare Part D benefit; and imposes inflationary rebates for Medicare drugs that increase in price faster than the rate of inflation.
The IRA, or other federal or state laws, could affect the market conditions for, or pricing or reimbursement of, our products. There is no assurance that federal or state health care reform will not adversely affect our future business and financial results. We expect that additional U.S. state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our drug candidates or additional pricing pressures.
Moreover, eligibility for reimbursement in the United States does not imply that any drug will be paid for in all cases, or by all payors, or at a rate that covers our costs, including research, development, manufacture, sale and distribution. Interim U.S. reimbursement levels for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement rates may vary according to the use of the drug and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost drugs and may be incorporated into existing payments for other services. Net prices for drugs may be reduced by mandatory discounts or rebates required by U.S. government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the United States. Third-party payors in the United States often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement policies. Our inability to promptly obtain coverage and profitable payment rates from both government-funded and private payors for any approved drugs that we develop could have a material adverse effect on our operating results, our ability to raise capital needed to commercialize drugs and our overall financial condition.
Sales of our generic prescription drugs sold through our Other Ventures rely on the ability to win tender bids for the medicine purchases of hospitals in China.
Our prescription drugs business markets to hospitals in China that may make bulk purchases of a medicine only if that medicine is selected under a government-administered tender process that was initiated in 2018 and aimed at driving consolidation in the fragmented generic prescription drug market in China. Pursuant to this process, major cities bulk-buy certain generic drugs together, forcing companies to bid for contracts and driving down prices. The process was later expanded nationwide to cover more cities and drugs. This process, which only applies to generic prescription drugs, may reduce our Other Ventures’ product portfolio as some of our third-party generic drug partners may fail to win bids.
Periodically, a bidding process is organized on a provincial or municipal basis. Whether a drug manufacturer is invited to participate in the tender depends on the level of interest that hospitals have in purchasing this drug. The interest of a hospital in a medicine is evidenced by:
| ● | the inclusion of this medicine on the hospital’s formulary, which establishes the scope of drug physicians at this hospital may prescribe to their patients, and |
| ● | the willingness of physicians at this hospital to prescribe a particular drug to their patients. |
30
We believe that effective marketing efforts are critical in making and keeping hospitals interested in purchasing the prescription drugs sold through our Other Ventures so that we and our joint ventures are invited to submit the products to the tender. Even if we and our joint ventures are invited to do so, competitors may be able to substantially reduce the price of their products or services. If competitors are able to offer lower prices, our and our joint ventures’ ability to win tender bids during the hospital tender process will be materially affected, and could reduce our total revenue or decrease our profit.
Counterfeit products could negatively impact our revenue, brand reputation, business and results of operations.
Our products are subject to competition from counterfeit products, especially counterfeit pharmaceuticals which are manufactured without proper licenses or approvals and are fraudulently mislabeled with respect to their content and/or manufacturer. Counterfeiters may illegally manufacture and market products under our or our joint venture’s brand names, the brand names of the third-party products we or they sell, or those of our or their competitors. Counterfeit pharmaceuticals are generally sold at lower prices than the authentic products due to their low production costs, and in some cases are very similar in appearance to the authentic products. Counterfeit pharmaceuticals may or may not have the same chemical content as their authentic counterparts. If counterfeit pharmaceuticals illegally sold under our or our joint ventures’ brand names or the brand names of third-party products we or they sell result in adverse side effects to consumers, we or our joint ventures may be associated with any negative publicity resulting from such incidents. In addition, consumers may buy counterfeit pharmaceuticals that are in direct competition with products sold through our Oncology/Immunology and Other Ventures operations, which could have an adverse impact on our revenue, business and results of operations. The proliferation of counterfeit pharmaceuticals in China and globally may grow in the future. Any such increase in the sales and production of counterfeit pharmaceuticals in China, or the technological capabilities of the counterfeiters, could negatively impact our revenue, reputation, business and results of operations.
Rapid changes in the pharmaceutical industry may render our Other Ventures’ products or our internally developed drugs and drug candidates obsolete.
Future technological improvements by our competitors and continual product developments in the pharmaceutical market may render our and our joint ventures’ existing products, our or their third-party licensed products or our drug candidates obsolete or affect our viability and competitiveness. Therefore, our future success will largely depend on our and our joint ventures’ ability to:
| ● | improve existing products; |
| ● | develop innovative drug candidates; |
| ● | diversify the product and drug candidate portfolio; |
| ● | license diverse third-party products; and |
| ● | develop new and competitively priced products which meet the requirements of the constantly changing market. |
If we or our joint ventures fail to respond to this environment by improving our existing products, licensing new third-party products or developing new drug candidates in a timely fashion, or if such new or improved products do not achieve adequate market acceptance, our business and profitability may be materially and adversely affected.
31
Certain of our joint ventures’ principal products involve the cultivation or sourcing of key raw materials including botanical products, and any quality control or supply failure or price fluctuations could adversely affect our ability to manufacture our products and/or could materially and adversely affect our operating results.
The key raw materials used in the manufacturing process of certain of our joint ventures’ principal products are medicinal herbs whose properties are related to the regions and climatic conditions in which they are grown. Access to quality raw materials and products necessary for the manufacture of our products is not guaranteed. We rely on materials sourced from third-party growers and suppliers. The availability, quality and prices of these raw materials are dependent on and closely affected by weather conditions and other seasonal factors which have an impact on the yields of the harvests each year. The quality, in some instances, also depends on the operations of third-party growers or suppliers. There is a risk that such growers or suppliers sell or attempt to sell us or our joint ventures raw materials which are not authentic. If there is any supply interruption for an indeterminate period of time, our joint ventures may not be able to identify and obtain alternative supplies that comply with our quality standards in a timely manner. Any supply disruption could adversely affect our ability to satisfy demand for our products, and materially and adversely affect our product sales and operating results. Moreover, any use by us or our joint ventures of unauthentic materials illegally sold to us by third-party growers or suppliers in our or our joint ventures’ products may result in adverse side effects to the consumers, negative publicity, or product liability claims against us or our joint ventures, any of which may materially and adversely affect our operating results.
The prices of necessary raw materials and products may be subject to price fluctuations according to market conditions, and any sudden increases in demand in the case of a widespread illness such as COVID-19, SARS, MERS or avian flu may impact the cost of production. Raw material price fluctuations could increase the cost to manufacture our products and adversely affect our operating results.
Adverse publicity associated with our company or collaboration partners, our joint ventures or our or their products or third-party licensed products or similar products manufactured by our competitors could have a material adverse effect on our results of operations.
Sales of our and our joint ventures’ products are highly dependent upon market perceptions of the safety and quality of such products, including proprietary products and third-party products we and they distribute. Concerns over the safety of biopharmaceutical products manufactured in China could have an adverse effect on the reputation of our industry and the sale of such products, including products manufactured or distributed by us, our collaboration partners and our joint ventures.
We and our joint ventures could be adversely affected if any of our or our joint ventures’ products, third-party licensed products or any similar products manufactured by other companies prove to be, or are alleged to be, harmful to patients. Any negative publicity associated with severe adverse reactions or other adverse effects resulting from patients’ use or misuse of our and our joint ventures’ products or any similar products manufactured by other companies could also have a material adverse impact on our results of operations. We and our joint ventures have not, to date, experienced any significant quality control or safety problems. If in the future we or our joint ventures become involved in incidents of the type described above, such problems could severely and adversely impact our financial position and reputation.
32
We are dependent on our joint ventures’ production facilities in Shanghai, China, our manufacturing facilities in Suzhou and Shanghai, China and third-party or our collaboration partners’ manufacturing facilities for the manufacture of the principal products of our joint ventures and our own drug candidates and products.
The principal products sold by our Other Ventures are mainly produced or expected to be produced at our joint ventures’ manufacturing facilities in Shanghai, China. Our commercial supplies of finished product for fruquintinib and surufatinib sold by our Oncology/Immunology operations are manufactured at our manufacturing facility in Suzhou, China. We outsourced the manufacture of active pharmaceutical ingredients and finished product of savolitinib to a third-party manufacturer based in Shanghai, China. We have also outsourced the manufacture of the active pharmaceutical ingredients to third-party manufacturers based in China for fruquintinib and surufatinib and we have engaged a back-up supplier for fruquintinib for China only. Until our new manufacturing facility in Shanghai is fully operational and it receives all the requisite government and other approvals, we have no back-up manufacturing facility for the finished product of savolitinib and surufatinib, and our ability to produce such drugs will be negatively impacted if we experience any significant production problems at our Suzhou facility or at our third party manufacturers’ facilities. In relation to the U.S. market, finished product for fruquintinib can be supplied by either our facility in Suzhou or a third-party manufacturer in Switzerland and potentially this third-party manufacturer could supply finished product to us to be sold in China subject to relevant approvals. A significant disruption at our, our collaboration partners’, our joint ventures’ and/or our contract manufacturer’s facilities, even on a short-term basis, could impair our, our collaboration partners’ and/or our joint ventures’ ability to timely produce and ship products, which could have a material adverse effect on our business, financial position and results of operations.
Our, our collaboration partners’, our joint ventures’ and our contract manufacturer’s manufacturing operations are vulnerable to interruption and damage from natural and other types of disasters, including earthquake, fire, floods, environmental accidents, power loss, communications failures and similar events. If any disaster were to occur, our ability to operate our, our collaboration partners’, our joint ventures’ or our contract manufacturer’s business at these facilities would be materially impaired. In addition, the nature of our production and research activities could cause significant delays in our programs and make it difficult for us to recover from a disaster or switch to other contract manufacturers. We and our joint ventures maintain insurance for business interruptions to cover some of our potential losses; however, such disasters could still disrupt our operations and thereby result in substantial costs and diversion of resources.
In addition, our, our collaboration partners’, our joint ventures’ and our contract manufacturer’s production process requires a continuous supply of electricity. We and they have encountered power shortages historically due to restricted power supply to industrial users during summers when the usage of electricity is high and supply is limited or as a result of damage to the electricity supply network. Because the duration of those power shortages was brief, they had no material impact on our or their operations. Interruptions of electricity supply could result in lengthy production shutdowns, increased costs associated with restarting production and the loss of production in progress. Any major suspension or termination of electricity or other unexpected business interruptions could have a material adverse impact on our business, financial condition and results of operations.
Risks Relating to Our Dependence on Third Parties
Disagreements or disputes with our current or future collaboration partners, the amendment of any collaboration agreement or the termination of any collaboration arrangement, could cause delays in our product development and materially and adversely affect our business.
Our collaborations, including those with our oncology drug partners AstraZeneca, Eli Lilly and Takeda and our in-licensing arrangement with Epizyme, and any future collaborations that we enter into may not be successful. Disagreements or disputes between parties to a collaboration arrangement regarding issues such as clinical development and commercialization, intellectual property ownership and transfer, clinical supply of drug candidates or products, cost allocation and other matters can lead to delays in the development process or commercializing the applicable drug candidate and, in some cases, termination of the collaboration arrangement. In addition, we or our partners may seek to amend the terms of one or more our collaboration agreements to adjust, among other things, the respective roles of our company and our collaboration partners as circumstances change. Our interests may not always be aligned with those of our collaboration partners, for instance, we may be much smaller than our collaboration partners and because they or their affiliates may sell competing products. This may result in potential conflicts between our collaborators and us on matters that we may not be able to resolve on favorable terms or at all.
33
Collaborations with pharmaceutical or biotechnology companies and other third parties, including our existing agreements with AstraZeneca, Eli Lilly and Takeda, are often terminable by the other party for any reason with certain advance notice. Any such termination or expiration would adversely affect us financially and could harm our business reputation. For instance, in the event that one of the strategic alliances with a current collaborator is terminated, we may require significant time and resources to secure a new collaboration partner, if we are able to secure such an arrangement at all. As noted in the following risk factor, establishing new collaboration arrangements can be challenging and time-consuming. The loss of existing or future collaboration arrangements would not only delay or potentially terminate the possible development or commercialization of products we may derive from our technologies, but it may also delay or terminate our ability to test specific target candidates.
We rely on our collaborations with third parties for certain of our drug development activities, and, if we are unable to establish new collaborations when desired on commercially attractive terms or at all, we may have to alter our development and commercialization plans.
Certain of our drug development programs and the potential commercialization of certain drug candidates rely on collaborations, such as savolitinib with AstraZeneca and fruquintinib with Eli Lilly for China and with Takeda outside of China. In the future, we may decide to collaborate with additional pharmaceutical and biotechnology companies for the development and potential commercialization of our other drug candidates.
We face significant competition in seeking appropriate collaborators. Whether we reach a definitive agreement for collaboration will depend, among other things, upon our assessment of the collaborator’s resources and expertise, the terms and conditions of the proposed collaboration and the proposed collaborator’s evaluation of a number of factors. Those factors may include the design or results of clinical trials, the likelihood of approval by the FDA, NMPA, EMA, PDMA or similar regulatory authorities outside the United States, China, Europe, Japan and the potential market for the subject drug candidate, the costs and complexities of manufacturing and delivering such drug candidate to patients, the potential of competing drugs, the existence of uncertainty with respect to our ownership of technology, which can exist if there is a challenge to such ownership without regard to the merits of the challenge and industry and market conditions generally. The collaborator may also consider alternative drug candidates or technologies for similar indications that may be available to collaborate on and whether such collaboration could be more attractive than the one with us for our drug candidate. The terms of any additional collaboration or other arrangements that we may establish may not be favorable to us. We may also be restricted under existing collaboration agreements from entering into future agreements on certain terms with potential collaborators. Collaborations are complex and time-consuming to negotiate and document. In addition, there have been a significant number of recent business combinations among large pharmaceutical companies that have resulted in a reduced number of potential future collaborators.
We may not be able to negotiate additional collaborations on a timely basis, on acceptable terms, or eventually close the deal. If we are unable to do so, we may have to curtail the development of the drug candidate for which we are seeking to collaborate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of any sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms or at all. If we do not have sufficient funds, we may not be able to further develop our drug candidates or bring them to market and generate drug revenue.
The third-party vendors upon whom we rely for the supply of the active pharmaceutical ingredients used in some of our drug candidates and drug products are our sole source of supply, and the loss of any of these suppliers could significantly harm our business.
The active pharmaceutical ingredients used in some of our drug candidates and products are supplied to us from third-party vendors. Our ability to successfully develop our drug candidates, and to supply our commercial drugs in quantities sufficient to meet the market demand, depends in part on our ability to obtain the active pharmaceutical ingredients for these drugs in accordance with regulatory requirements and in sufficient quantities for commercialization and clinical testing. We currently obtain active pharmaceutical ingredients for each of our drug candidates from a limited number of suppliers. For example, a single supplier based in Shanghai manufactures and provides us active pharmaceutical ingredient for savolitinib. In the event any of our current suppliers of such active pharmaceutical ingredient cease operations for any reason, it may lead to an interruption in our production and supply of the product.
34
For all of our drug candidates and products, we aim to identify and qualify a manufacturer to provide such active pharmaceutical ingredient prior to submission of an NDA to the FDA and/or NMPA. We are not certain, however, that our current supply arrangements will be able to meet our demand, either because of the nature of our agreements with third party suppliers, our limited experience with third party suppliers or our relative importance as a customer to those suppliers. It may be difficult for us to assess third party vendors’ ability to timely meet our demand in the future based on past performance. While our suppliers have generally met our demand on a timely basis in the past, they may subordinate our needs in the future to their other customers.
Establishing additional or replacement suppliers for the active pharmaceutical ingredients used in our drug candidates and products, if required, may not be accomplished quickly. If we are able to find a replacement supplier, such alternative arrangements would need to be qualified and may require additional regulatory approval, which could result in further delay. While we seek to maintain adequate inventory of the active pharmaceutical ingredients used in our drug candidates and products, any interruption or delay in the supply of components or materials, or our inability to obtain such active pharmaceutical ingredient from alternate sources at acceptable prices in a timely manner could impede, delay, limit or prevent our development and commercialization efforts, which could harm our business, results of operations, financial condition and prospects.
We and our collaboration partners rely, and expect to continue to rely, on third parties to conduct certain of our clinical trials for our drug candidates. If these third parties do not successfully carry out their contractual duties, comply with regulatory requirements or meet expected deadlines, we may not be able to obtain regulatory approval for or commercialize our drug candidates and our business could be harmed.
We do not have the ability to independently conduct large-scale clinical trials. We and our collaboration partners rely, and expect to continue to rely, on medical institutions, clinical investigators, contract laboratories and other third parties, such as CROs, to conduct or otherwise support certain clinical trials for our drug candidates. Nevertheless, we and our collaboration partners (as applicable) will be responsible for ensuring that each clinical trial is conducted in accordance with the applicable protocol, legal and regulatory requirements and scientific standards, and reliance on CROs will not relieve us of our regulatory responsibilities. For any violations of laws and regulations during the conduct of clinical trials for our drug candidates, we could be subject to warning letters or enforcement action that may include civil penalties up to and including criminal prosecution.
Although we or our collaboration partners design the clinical trials for our drug candidates, CROs conduct most of the clinical trials. As a result, many important aspects of our development programs, including their conduct and timing, are outside of our direct control. Our reliance on third parties to conduct clinical trials results in less control over the management of data developed through clinical trials than would be the case if we were relying entirely upon our own staff. Communicating with outside parties can also be challenging, potentially leading to mistakes as well as difficulties in coordinating activities. Outside parties may:
| ● | have staffing difficulties; |
| ● | fail to comply with contractual obligations; |
| ● | experience regulatory compliance issues; |
| ● | undergo changes in priorities or become financially distressed; or |
| ● | form relationships with other entities, some of which may be our competitors. |
These factors may materially and adversely affect the willingness or ability of third parties to conduct our and our collaboration partners’ clinical trials and may subject us or them to unexpected cost increases that are beyond our or their control.
If any of our and our collaboration partners’ relationships with these third-party CROs terminate, we or they may not be able to enter into arrangements with alternative CROs on reasonable terms or at all. If CROs do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, any clinical trials such CROs are associated with may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for or successfully commercialize our drug candidates. As a result, we believe that our financial results and the commercial prospects for our drug candidates in the subject indication would be harmed, our costs could increase and our ability to generate revenue could be delayed.
35
We, our collaboration partners or our CROs may fail to comply with the regulatory requirements pertaining to clinical trials, which could result in fines, adverse publicity and civil or criminal sanctions.
We, our collaboration partners and our CROs are required to comply with regulations for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the trial patients are adequately informed of the potential risks of participating in clinical trials and their rights are protected. These regulations are enforced by the FDA, the NMPA and comparable foreign regulatory authorities for any drugs in clinical development. In the United States, the FDA regulates GCP through periodic inspections of clinical trial sponsors, principal investigators and trial sites. If we, our collaboration partners or our CROs fail to comply with applicable GCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or comparable foreign regulatory authorities may require additional clinical trials before approving the marketing applications for the relevant drug candidate. We cannot assure you that, upon inspection, the FDA or other applicable regulatory authority will determine that any of the future clinical trials for our drug candidates will comply with GCPs. In addition, clinical trials must be conducted with drug candidates produced under applicable manufacturing regulations. Our failure or the failure of our collaboration partners or CROs to comply with these regulations may require us or them to repeat clinical trials, which would delay the regulatory approval process and could also subject us to enforcement action. We are also required to register applicable clinical trials and post certain results of completed clinical trials on a U.S. government-sponsored database, ClinicalTrials.gov, within certain timeframes. Failure to do so can result in fines, adverse publicity and civil sanctions.
Our collaboration partners, principal investigators, CROs and other third-party contractor and consultants may engage in misconduct or other improper activities.
We are exposed to the risk that collaboration partners, principal investigators, CROs and other third-party contractor and consultants may engage in fraudulent or other illegal activity with respect to our business. Their misconduct could include intentional, reckless and/or negligent conduct or unauthorized activity that violates NMPA, FDA, EMA, PDMA or other regulations, including but not limited to those laws requiring the reporting of true, complete and accurate information. In addition, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of insurance, pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. We may not be able to identify and deter such misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us, our collaboration partners, principal investigators, CROs and other third-party contractor and consultants, and we and/or such other parties are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative penalties, damages, monetary fines, contractual damages, reputational harm, diminished profits and future earnings and disruption of our operations.
Joint ventures form an important part of our Other Ventures, and our ability to manage and develop the businesses conducted by these joint ventures depends in part on our relationship with our joint venture partners.
We are party to joint venture agreements with each of Shanghai Pharmaceuticals and Sinopharm, which together form a major portion of our Other Ventures. Under these arrangements, our joint venture partners have certain operational responsibilities and/or certain rights to exercise control or influence over operations and decision-making.
36
Our equity interests in these operating companies do not provide us with the unilateral ability to control actions which require shareholder approval. In addition, under the joint venture contracts for these entities, the consent of the directors nominated by our joint venture partners is required for the passing of resolutions in relation to certain matters concerning the operations of these companies. As a result, although we participate in the management and nominate the management and run the day-to-day operations of our joint ventures, Hutchison Sinopharm and Shanghai Hutchison Pharmaceuticals, we may not be able to secure the consent of our joint venture partners to pursue activities or strategic objectives that are beneficial to or that facilitate our overall business strategies. Furthermore, disagreements or disputes which arise between us and our joint venture partners may potentially require legal action to resolve and hinder the smooth operation of our Other Ventures or adversely affect our financial condition, results of operations and prospects.
We, our collaboration partners and our joint ventures rely on our distributors for logistics and distribution services.
We, our collaboration partners and our joint ventures rely on distributors to perform certain operational activities, including invoicing, logistics and delivery of the products we and they market to the end customers. Because we, our collaboration partners and our joint ventures rely on third-party distributors, we have less control than if we handled distribution logistics directly and can be adversely impacted by the actions of our distributors. Any disruption of our, our collaboration partners’ and our joint ventures’ distribution network, including failure to renew existing distribution agreements with desired distributors, could negatively affect product sales and materially and adversely affect our business, financial condition and results of operations.
There is no assurance that the benefits currently enjoyed by virtue of our association with CK Hutchison will continue to be available.
Historically, we have relied on the reputation and experience of, and support provided by, our founding shareholder, a wholly owned subsidiary of CK Hutchison, to advance our joint ventures and collaborations in China and elsewhere. CK Hutchison indirectly held approximately 38.2% of our total outstanding share capital as of February 15, 2024. We believe that CK Hutchison group’s reputation in China has given us an advantage in negotiating collaborations and obtaining opportunities.
We also benefit from sharing certain services with the CK Hutchison group including, among others, legal and regulatory services, company secretarial support services, tax and internal audit services, participation in the CK Hutchison group’s pension, medical and insurance plans, participation in the CK Hutchison group’s procurement projects with third-party vendors/suppliers, other staff benefits and staff training services, company functions and activities and operation advisory and support services. We pay a management fee to an affiliate of CK Hutchison for the provision of such services. In each of the years ended December 31, 2021, 2022 and 2023, we paid a management fee of approximately $1.0 million, $1.0 million and $1.0 million respectively. In addition, we benefit from the fact that two retail chains affiliated with the CK Hutchison group, PARKnSHOP and Watsons, sell certain of our Other Ventures’ products in their stores throughout Hong Kong and in other Asian countries. For the years ended December 31, 2021, 2022 and 2023, sales of our products to members of the CK Hutchison group amounted to $4.3 million, $3.6 million and $1.9 million, respectively.
Our business also depends on certain intellectual property rights licensed to us by the CK Hutchison group. See “—Risks Relating to Intellectual Property—We and our joint ventures are dependent on trademark and other intellectual property rights licensed from others. If we lose our licenses for any of our products, we or our joint ventures may not be able to continue developing such products or may be required to change the way we market such products” for more information on risks associated with such intellectual property licensed to us.
There can be no assurance the CK Hutchison group will continue to provide the same benefits or support that they have provided to our business historically. Such benefit or support may no longer be available to us, in particular, if CK Hutchison’s ownership interest in our company significantly decreases in the future.
37
Other Risks and Risks Relating to Doing Business in China
We are subject to stringent privacy and cybersecurity laws, information security policies and contractual obligations related to data privacy and security, and we may be exposed to risks related to our management of the medical data of subjects enrolled in our clinical trials and other personal or sensitive information.
We routinely receive, collect, generate, store, process, transmit and maintain medical data, treatment records and other personal details of the subjects enrolled in our clinical trials, along with other personal or sensitive information. As such, we are subject to the relevant local, state, national and international data protection and privacy laws, directives regulations, and standards that apply to the collection, use, retention, protection, disclosure, transfer and other processing of personal data in the various jurisdictions in which we operate and conduct our clinical trials. We are also subject to contractual obligations regarding the processing of personal data. Legal requirements regarding data protection and privacy continue to evolve and may result in ever-increasing public scrutiny and escalating levels of enforcement and sanctions and increased cost of compliance. Failure to comply with any of these laws could result in enforcement action against us, including investigations, civil and criminal enforcement action, fines, imprisonment of company officers and public censure, claims for damages by customers and other affected individuals, damage to our reputation and loss of goodwill, any of which could have a material adverse effect on our business, financial condition, results of operations or prospects.
Data protection and privacy laws and regulations generally require clinical trial sponsors and operators and their personnel to protect the privacy of their enrolled subjects and prohibit unauthorized disclosure of personal information. We have established procedures to protect the confidentiality of medical records and personal data of subjects enrolled in our clinical trials. Access to clinical trial data has been strictly limited to authorized personnel only according to the relevant rules and regulations. External parties involved in clinical trials are also required to comply with all relevant data protection and confidentiality requirements. Data are to be used only for the intended use, as agreed by the patients and consistent with the patients’ informed consent form. While we have adopted security policies and measures to protect our proprietary data and patients’ privacy, personal patient information could be subject to leaks caused by hacking activities, human error, employee misconduct or negligence or system breakdown. We also cooperate with third parties including collaboration partners, principal investigators, hospitals, CROs and other third-party contractor and consultants for our clinical trials and operations. Any leakage or abuse of patient data by our third-party partners may be perceived by the patients as a result of our failure. Furthermore, any change in applicable laws and regulations could affect our ability to use medical data and subject us to liability for the use of such data for previously permitted purposes. For instance, we may be subject to additional regulations, laws and policies adopted by the PRC government to apply more stringent social and ethical standards in data privacy resulting from the increased global focus on this area. Any failure or perceived failure by us to prevent information security breaches or to comply with privacy policies or privacy-related legal obligations, or any compromise of information security that results in the unauthorized release or transfer of personally identifiable information or other patient data, could cause our customers to lose trust in us and could expose us to regulatory action and legal claims.
38
There are numerous U.S. federal and state laws and regulations relating to the privacy and security of personal information. In particular, regulations promulgated pursuant to the Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended, establish privacy and security standards that limit the use and disclosure of individually identifiable health information (known as “protected health information”), require the implementation of administrative, physical and technological safeguards to protect the privacy of protected health information and ensure the confidentiality, integrity and availability of electronic protected health information, and create breach reporting obligations in cases of certain unauthorized uses or disclosures. While we do not believe that we are directly subject to HIPAA as either a “covered entity” or “business associate,” U.S. sites at which we conduct clinical trials are likely to be covered entities and thus must ensure that they obtain adequate patient authorization or establish another basis under HIPAA to disclose a clinical trial subject’s individually identifiable health information to us and other entities participating in our clinical trials. In addition to federal regulation, many U.S. states have begun to focus on efforts to regulate privacy and data security. For example, in California, the California Consumer Protection Act, or CCPA, which went into effect on January 1, 2020 and was expanded by the California Consumer Privacy Rights Act, or CPRA, which went into effect on January 1, 2023, collectively establishes a privacy framework for covered businesses by creating an expanded definition of personal information, establishing new data privacy rights for consumers in the State of California, imposing special rules on the collection of consumer data from minors, and creating a new and potentially severe statutory damages framework for violations and for businesses that fail to implement reasonable security procedures and practices to prevent data breaches. A separate law, the California Confidentiality of Medical Information Act, also applies to pharmaceutical companies, including requirements for written authorization to use and disclose medical information and restrictions on the circumstances under which medical information can be used for marketing purposes. Several other states have also recently enacted or are considering comprehensive data privacy and security laws. Furthermore, all fifty states, the District of Columbia, Puerto Rico, and U.S. territories have enacted data breach notification laws that require, among other things, notifications to state governments and/or the affected individuals in the event of a data breach. These various state laws differ from one another and impose significant compliance burden. Although we take measures to protect sensitive data from unauthorized access, use or disclosure, and whenever possible contractually require third-party partners to do the same, our information technology and infrastructure and those of our third-party partners may be vulnerable to attacks by hackers or viruses or breached due to employee error, malfeasance or other malicious or inadvertent disruptions. Any such breach or interruption could compromise those networks and the information stored there could be accessed by unauthorized parties, manipulated, publicly disclosed, lost or stolen. Any such access, breach, or other loss of information relating to our information technology and infrastructure or that of our third-party partners may subject us to reputation damage, increased scrutiny and liability including legal claims or proceedings and liability under federal or state laws that protect the privacy of personal information.
39
Regulatory authorities in China have implemented a number of legislative and regulatory proposals concerning data protection. The PRC Cyber Security Law, which became effective in June 2017, created China’s first national-level data protection for “network operators,” which may include all organizations in China that provide services over the internet or another information network. The PRC Data Security Law, which took effect in September 2021, provides for a security review procedure for the data activities that may affect national security. The PRC Personal Information Protection Law, which took effect from November 2021, provides the circumstances under which a personal information processor could process personal information and the requirements for such circumstances. The PRC Personal Information Protection Law clarifies the scope of application, the definition of personal information and sensitive personal information, the legal basis of personal information processing and the basic requirements of notice and consent. The Measures for Cybersecurity Review, which took effect on February 15, 2022, provides that critical information infrastructure operators that purchase network products and services and online platform operators engaging in data processing activities that affect or may affect national security shall be subject to the cybersecurity review, and elaborates the factors to be considered when assessing the national security risks of the relevant activities. The Measures for Cybersecurity Review further stipulates that online platform operators holding personal information of over one million users shall apply with the Cybersecurity Review Office for a cybersecurity review before any public listing in a foreign country. As of the date of this annual report, we have not received any formal notice from any PRC cybersecurity regulator that we should apply for or otherwise be subject to the cybersecurity review, or subject to any investigation or received any inquiry, notice or sanction on cybersecurity review. The exact scope of “critical information infrastructure operators” under the current regulatory regime remains unclear, and the PRC government authorities may have wide discretion in the interpretation and enforcement of the applicable laws. Therefore, it is uncertain whether we would be deemed to be a critical information infrastructure operator under PRC law. If we are deemed to be a critical information infrastructure operator under the PRC cybersecurity laws and regulations, we may be subject to obligations in addition to what we have fulfilled under the PRC cybersecurity laws and regulations. In addition, on November 14, 2021, the Data Security Management Measures (Draft for Comments) was published by the CAC for public comments, which provides that data processors conducting the following activities shall apply for cybersecurity review: (i) a merger, reorganization or division of online platform operators that have acquired a large number of data resources related to national security, economic development or public interests which affect or may affect national security; (ii) a listing abroad when the data processor processes over one million users’ personal information; (iii) a listing in Hong Kong which affects or may affect national security; or (iv) other data processing activities that affect or may affect national security. It also requires data processors processing important data or listed outside China to carry out a data security assessment annually by itself or through a third party data security service provider and submit an assessment report to the local agency of the CAC. As there are still uncertainties regarding the further enactment of new laws and regulations as well as the revision, interpretation and implementation of those existing laws and regulations, we cannot assure you that we will be able to comply with such regulations in all respects.
The Measures on Security Assessment of Cross-border Data Transfer, or the Security Assessment Measures, were published on July 7, 2022, and became effective on September 1, 2022. The Security Assessment Measures specify that data controllers and/or critical information infrastructure operators will be subject to security assessment under the following circumstances: (i) data controllers exporting important data (which, under the Security Assessment Measures, is defined as data which if tampered with, damaged, leaked, or if obtained or used illegally may endanger national security, the economy, social stability, and public health and safety, etc.), (ii) critical information infrastructure operators or data controllers processing the personal information of one million people or more exporting personal information, (iii) data controllers who have exported the personal information of 100,000 people or the sensitive personal information of 10,000 people since January 1 of the previous year, or (iv) other situations provided for by the CAC that require a security assessment. As of the date of this annual report, we have not received any formal notice from any PRC cybersecurity regulator that the Company should apply for or otherwise be subject to security assessment, or subject to any investigation or received any inquiry, notice or sanction on security assessment. PRC government authorities may have wide discretion in the interpretation and enforcement of the Security Assessment Measures, including whether we have exported “important data” as defined thereunder, and thus there is uncertainty as to whether we may be subject to security assessment. Further, drafts of some of these measures have now been published, including the Measures on Security Assessment for Individual Information Cross-border Transfer (Draft for Comments) in June 2019, which may, upon enactment, require security review before transferring human health-related data out of China.
40
In addition, certain industry-specific laws and regulations affect the collection and transfer of personal data in China. For example, the Regulations of the PRC on the Administration of Human Genetic Resources, or HGR Regulations, which became effective and implemented on July 1, 2019, stipulates that use of Chinese human genetic resources, or HGR, for the purposes of carrying out collaborative international scientific research shall be approved by the administrative department of science and technology under the State Council. However, no approval is required for “international collaboration in clinical trials” that do not involve the export of HGR materials, provided that the two parties to the international collaboration shall file the type, quantity and usage of the HGR to be used with the administrative department of science and technology under the State Council before clinical trials. The PRC Biosecurity Law, which took effect on April 15, 2021, stipulates that foreign organizations and individuals, as well as institutions they establish or are the actual controllers of, must not collect or preserve HGR within the territory of China and must not provide China’s HGR to overseas. The Implementation Rules for the Administrative Regulation on Human Genetic Resources, or Implementation Rules for HGR, became effective on July 1, 2023, setting out circumstances under which the provision of or granting of access to human genetic resource information to overseas organizations, individuals or agencies controlled thereby affecting public health, national security or public interest in China would be subject to security review by the Ministry of Science and Technology, including where (i) human genetic resource information of important genetic families is involved; (ii) human genetic resource information of specific regions is involved, (iii) exome sequencing and genome sequencing information resources with of a sample exceeding 500 individuals is involved; and (iv) other circumstances that may affect the public health, national security and social public interest of China. It is possible that these laws may be interpreted and applied in a manner that is inconsistent with our practices, potentially resulting in confiscation of HGR samples and associated data and administrative fines, penalties and negative publicity.
Our clinical trial programs may implicate European data privacy laws, including the General Data Protection Regulation, or the GDPR, and local laws further implementing or supplementing the GDPR. The GDPR implements more stringent operational requirements for processors and controllers of personal data including requirements for such companies to be able to ensure and be able to demonstrate compliance with the GDPR. If our or our third-party partners’ privacy or data security measures fail to comply with the GDPR requirements, we may be subject to litigation, regulatory investigations, enforcement notices requiring us to change the way we use personal data and/or significant fines. In addition to statutory enforcement, non-compliance can lead to compensation claims by affected individuals, negative publicity and a potential loss of business. We are also subject to European laws on personal data export, as we may transfer personal data from the E.U. (or U.K.) to other jurisdictions which are not considered by the European Commission to offer “adequate” protection of personal data (such as Hong Kong or the United States). Following the Schrems II decision of the European Court of Justice in 2020, there has been intensified focus on exports of personal data which do not meet the high standards of protection expected by the E.U. Certain supervisory authorities in the E.U. have now begun to take enforcement action in this area, ordering restrictions on certain transfers of personal data to third countries such as the United States. These changes could require us to make operational changes and could increase costs and may lead to governmental enforcement actions, litigation, fines and penalties or adverse publicity that could have an adverse effect on our business.
We believe, to the best of our knowledge, our business operations do not violate any of the above laws and regulations currently in force in all material aspects. We have been taking and will continue to take reasonable measures to comply with applicable data privacy, data protection and cybersecurity laws. We cannot guarantee the effectiveness of the measures undertaken by us and business partners, and such measures may still be determined as insufficient, improper, or even as user-privacy invasive, by the relevant authorities, which may result in penalties against us. Complying with all applicable laws, regulations, standards and obligations relating to data privacy, security, and transfers may cause us to incur substantial operational costs or require us to modify our data processing practices and processes. To the extent that we need to alter our business model or practices to adapt to these announcement and provisions and future regulations, laws and policies, we could incur additional expenses. We cannot assure you we can adapt our operations to it in a timely manner. Non-compliance could result in proceedings against us by data protection authorities, governmental entities or others, including class action privacy litigation in certain jurisdictions, which would subject us to significant fines, penalties, judgments and negative publicity. In addition, if our practices are not consistent or viewed as not consistent with legal and regulatory requirements, including changes in laws, regulations and standards or new interpretations or applications of existing laws, regulations and standards, we may become subject to audits, inquiries, whistleblower complaints, adverse media coverage, investigations, loss of export privileges, severe criminal or civil sanctions and reputational damage. Any of the foregoing could have a material adverse effect on our competitive position, business, financial conditions, results of operations and prospects.
41
Product liability claims or lawsuits could cause us, our collaboration partners or our joint ventures to incur substantial liabilities.
We, our collaboration partners and our joint ventures face an inherent risk of product liability exposure related to the use of our drug candidates in clinical trials, sales of our or our joint ventures’ products or the products we or they license from third parties. If we, our collaborators and our joint ventures cannot successfully defend against claims that the use of such drug candidates in our clinical trials or any products sold by us or our joint ventures, including savolitinib, fruquintinib, surufatinib and/or any of our drug candidates which receive regulatory approval, caused injuries, we, our collaboration partners and our joint ventures could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| ● | decreased demand for our and our joint ventures’ products; |
| ● | significant negative media attention and reputational damage; |
| ● | withdrawal of clinical trial participants; |
| ● | significant costs to defend the related litigation; |
| ● | substantial monetary awards to trial participants or patients; |
| ● | loss of revenue; and |
| ● | the inability to commercialize any drug candidates that we may develop. |
Our principal insurance policies cover product liability for savolitinib, fruquintinib, surufatinib, certain prescription drugs and health supplements, property loss due to accidents or natural disasters and adverse events in clinical trials. Existing PRC laws and regulations do not require us, our collaborators or our joint ventures to have, nor do we or they, maintain liability insurance to cover product liability claims except with respect to savolitinib, fruquintinib, surufatinib, certain prescription drugs and health supplements, and liability with respect to our oncology and immunology clinical trials. Any litigation might, result in substantial costs and diversion of resources. While we maintain liability insurance for clinical trials and products, this insurance may not fully cover our potential liabilities. Inability to obtain sufficient insurance coverage at an acceptable cost or otherwise to protect against potential product liability claims could prevent or inhibit the commercialization of products that we or our collaborators develop.
An occurrence of a widespread health epidemic or other outbreaks or natural disasters could have a material adverse effect on our business, financial condition and results of operations.
Our business could be materially and adversely affected by the outbreak of a widespread health epidemic, such as COVID-19, swine flu, avian influenza, severe acute respiratory syndrome, Ebola and Zika; natural disasters, such as earthquakes, snowstorms, storm surges, floods, fires, drought and other extreme weather events and other effects of climate change; or other events, such as wars, acts of terrorism, environmental accidents, power shortages or communication interruptions. The occurrence of a disaster or a prolonged outbreak of an epidemic illness or other adverse public health developments could materially disrupt our industry and our business and operations, and have a material adverse effect on our business, financial condition and results of operations. For example, these events could cause a temporary closure of the facilities we use for our operations, significantly disrupt manufacturing and supply chain, our sales and marketing and clinical trial operations and those of our collaboration partners, and the ability to advance our research and development activities and pursue development of any of our drug candidates. Our operations could also be disrupted if any of our employees or employees of our business partners are suspected of contracting an epidemic disease, since this could require us or our business partners to quarantine some or all of these employees or disinfect the facilities used for our operations.
42
We may engage in strategic transactions, including acquisitions, investments, joint ventures or divestitures. If unsuccessful, such transaction may have an adverse effect on our business.
From time to time, we may pursue strategic transactions, including acquisitions, investments, joint ventures and divestitures. For example, we are continuing to actively evaluate non-core assets divestment opportunities as part of our strategy to focus on our core businesses, which include the potential divestment of Shanghai Hutchison Pharmaceuticals. For more information, please refer to Item 4.A. “History and Development of the Company.” Acquisitions and investments involve numerous risks such as difficulties in finding suitable partners or acquisition candidates, difficulties in obtaining financing on favorable terms, if at all, the assumption of certain known and unknown liabilities of acquired companies and difficulties in integrating operations, services, products and personnel. Joint ventures may result in issues such as conflicts in goals and corporate cultures, commercial disputes with joint venture partners as well as imbalanced contributions and benefits. Divestitures also involve numerous risks. Any divestiture could result in a dilutive impact to our future earnings and significant write-offs, including those related to goodwill and other intangible assets, which could have a material adverse effect on our results of operations and financial condition. Divestitures could also result in difficulties in the separation of operations, services, products and personnel, the diversion of management’s attention from other business concerns, the disruption of our business and the potential loss of key employees. There is also no guarantee that we can complete strategic transactions in a timely manner, on a cost-effective basis, or at all, and we may not realize the expected benefits of any transaction. We may not be successful in managing these or any other significant risks that we encounter if we engage in a strategic transaction. If we are not successful in managing the risks, uncertainties and potential disruptions, a strategic transaction could have a negative impact on our business, results of operations or financial position.
We, our collaboration partners, our joint ventures and our third party contractors may be exposed to liabilities under the U.S. Foreign Corrupt Practices Act, or FCPA, U.S. healthcare fraud and abuse laws, the Bribery Act 2010 of the United Kingdom, or U.K. Bribery Act, and Chinese anti-corruption laws, and any determination that we or they have violated these laws could have a material adverse effect on our business or our reputation.
In the day-to-day conduct of our business, we, our collaboration partners, our joint ventures and our third party contractors are in frequent contact with persons who may be considered government officials under applicable anti-corruption, anti-bribery and anti-kickback laws, which include doctors at public hospitals in China and elsewhere. Therefore, we, our collaboration partners, our joint ventures and our third party contractors are subject to risk of violations under the FCPA, the U.K. Bribery Act, and other laws in the countries where we or they do business. We, our collaboration partners, our joint ventures and our third party contractors have operations in China, agreements with third parties in China, and we and our joint ventures make most of our sales in China. The PRC laws and regulations also strictly prohibit bribery of government officials. Our and our joint ventures’ activities in China create the risk of unauthorized payments or offers of payments by the directors, employees, representatives, distributors, consultants or agents of our company, our collaboration partners, our joint ventures or our collaboration partners, even though they may not always be subject to our control. It is our policy to implement safeguards to discourage these practices by our, our collaboration partners’, our joint ventures’ and our collaboration partners’ employees and third parties. We have implemented and adopted policies designed by the R&D-based Pharmaceutical Association Committee, an industry association representing approximately 40 global biopharmaceutical companies, to ensure compliance by us and our joint ventures and our and their directors, officers, employees, representatives, distributors, consultants and agents with the anti-corruption laws and regulations. We cannot assure you, however, that our existing safeguards are sufficient or that our, our collaboration partners’, our joint ventures’ or our third party contractors’ directors, officers, employees, representatives, distributors, consultants and agents have not engaged and will not engage in conduct for which we may be held responsible, nor can we assure you that our business partners have not engaged and will not engage in conduct that could materially affect their ability to perform their contractual obligations to us or even result in our being held liable for such conduct. Violations of the FCPA, the U.K. Bribery Act or Chinese anti-corruption laws may result in severe criminal or civil sanctions, and we may be subject to other liabilities, which could have a material adverse effect on our business, reputation, financial condition, cash flows and results of operations.
When we or our collaboration partners begin to commercialize products in the United States and secure governmental reimbursement of our products, we and our collaboration partners also will be subject to the risk of violating U.S. federal and state healthcare fraud and abuse laws, including the Anti-Kickback Statute and the False Claims Act. These laws broadly prohibit providing or receiving kickbacks in connection with government-reimbursed healthcare items or services, as well submitting or causing the submission of false or fraudulent claims to government healthcare programs. Violations of these laws may result in severe criminal or civil sanctions and other administrative sanctions, which could have a material adverse effect on our business, reputation, financial condition, cash flows and results of operations.
43
Ensuring that our, our collaboration partners’, our joint ventures’ and our third party contractors’ future business arrangements with third parties comply with applicable laws could also involve substantial costs. It is possible that governmental authorities will conclude that our business practices do not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our or our joint ventures’ operations were found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, disgorgement, individual imprisonment and exclusion from government funded healthcare programs, any of which could substantially disrupt our operations. If the physicians, hospitals or other providers or entities with whom we, our collaboration partners, our joint ventures, and our third party contractors do business are found not to be in compliance with applicable laws, they may also be subject to criminal, civil or administrative sanctions, including exclusions from government funded healthcare programs.
Our employees may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements, which could have a material adverse effect on our business.
We are exposed to the risk of employee fraud or other misconduct by our employees. Misconduct by our employees could include intentional failures to comply with applicable regulations, provide accurate information to regulatory authorities or comply with healthcare fraud and abuse laws and regulations. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Such misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to our reputation. We have adopted a Code of Ethics, but it is not always possible to identify and deter employee misconduct, and the precautions we have taken to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business and results of operations, including the imposition of significant fines or other sanctions.
If we or our joint ventures fail to comply with environmental, health and safety laws and regulations, we or they could become subject to fines or penalties or incur costs that could have a material adverse effect on the success of our business.
We and our joint ventures are subject to numerous environmental, health and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment and disposal of hazardous materials and wastes. Our operations involve the use of hazardous and flammable materials, including chemical materials. Our operations also produce hazardous waste products. We and our joint ventures are therefore subject to PRC laws and regulations concerning the discharge of waste water, gaseous waste and solid waste during our manufacturing processes. We and our joint ventures are required to establish and maintain facilities to dispose of waste and report the volume of waste to the relevant government authorities, which conduct scheduled or unscheduled inspections of our facilities and treatment of such discharge. We and our joint ventures may not at all times comply fully with environmental regulations. Any violation of these regulations may result in substantial fines, criminal sanctions, revocations of operating permits, shutdown of our facilities and obligation to take corrective measures. We and our joint ventures generally contract with third parties for the disposal of these materials and waste. We and our joint ventures cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from the use of hazardous materials, we and/or our joint ventures could be held liable for any resulting damages, and any liability could exceed our resources. We and/or our joint ventures also could incur significant costs associated with civil or criminal fines and penalties.
Although we and our joint ventures maintain workers’ compensation insurance to cover costs and expenses incurred due to on-the-job injuries to our employees and third-party liability insurance for injuries caused by unexpected seepage, pollution or contamination, this insurance may not provide adequate coverage against potential liabilities. Furthermore, the PRC government may take steps towards the adoption of more stringent environmental regulations. Due to the possibility of unanticipated regulatory or other developments, the amount and timing of future environmental expenditures may vary substantially from those currently anticipated. If there is any unanticipated change in the environmental regulations, we and our joint ventures may need to incur substantial capital expenditures to install, replace, upgrade or supplement our equipment or make operational changes to limit any adverse impact or potential adverse impact on the environment in order to comply with new environmental protection laws and regulations. If such costs become prohibitively expensive, we may be forced to cease certain aspects of our or our joint ventures’ business operations.
44
We rely significantly on information technology and any failure, inadequacy, interruption or security lapse of that technology, including any cybersecurity incidents, could harm our ability to operate our business effectively.
We are heavily dependent on critical, complex and interdependent information technology systems, including internet-based systems, to support our business processes. Our information technology system security is continuously reviewed, maintained and upgraded in response to possible security breach incidents. Despite the implementation of these measures, our information technology systems and those of third parties with which we contract are vulnerable to damage from external or internal security incidents, breakdowns, malicious intrusions, cybercrimes, including State-sponsored cybercrimes, malware, misplaced or lost data, programming or human errors or other similar events. System failures, accidents or security breaches could cause interruptions in our operations and could result in inappropriately accessed, tampered with, modified or stolen scientific data or a material disruption of our clinical activities and business operations, in addition to possibly requiring substantial expenditures of resources to remedy. Such event could significantly harm our Oncology/Immunology operations, including resulting in the loss of clinical trial data which could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Such events could also lead to the loss of important information such as trade secrets or other intellectual property and could accelerate the development or manufacturing of competing products by third parties. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and our research and development programs and the development of our drug candidates could be delayed.
We have granted, and may continue to grant, options, long-term incentive scheme (“LTIP”) awards and other types of awards under our Option Schemes and our LTIP, or collectively the Schemes, which may result in increased share-based compensation expenses and give rise to potential employment related disputes.
We have adopted the Options Schemes for the purpose of granting share-based compensation awards to certain management, directors, employees and other eligible grantees as a means to retain, incentivize, reward, remunerate, compensate and/or provide benefits to eligible grantees. We recognized share-based compensation expenses of $42.0 million, $30.6 million and $36.6 million for the years ended December 31, 2021, 2022 and 2023, respectively, in our consolidated financial statements in accordance with U.S. GAAP.
We believe the granting of share-based compensation is of significant importance to our ability to attract and retain key personnel and employees, and we will continue to grant share-based compensation in the future. As a result, our expenses associated with share-based compensation may increase, which may have an adverse effect on our results of operations. We may re-evaluate the vesting schedules, exercise price or other key terms applicable to the grants under our currently effective Schemes from time to time, which may result in a substantial change in our share-based compensation expenses in the reporting periods. In addition, we could in the future become involved in disputes or legal proceedings with our employees or former employees on employment related matters (including disputes on the entitlement of options, awards and other share-based compensation or in connection with the employees’ incentive or compensation arrangements). If such disputes or legal proceedings arise, there can be no assurance that we will prevail in them, and in any event defending against these disputes or legal proceedings could cause us to incur legal and other costs. Any adverse outcome of these disputes or legal proceedings could have a material adverse effect on our reputation, business and results of operations.
For more information on the Schemes, please refer to Item 6.B. “Compensation—Equity Compensation Schemes and Other Benefit Plans.”
The PRC’s economic, political and social conditions, as well as governmental policies, could affect the business environment and financial markets in China, our ability to operate our business, our liquidity and our access to capital.
Substantially all of our and our joint ventures’ business operations are conducted in China. Accordingly, our results of operations, financial condition and prospects are subject to economic, political and legal developments in China to a significant degree. China’s economy differs from the economies of developed countries in many respects, including with respect to the amount of government involvement, level of development, growth rate, control of foreign exchange and allocation of resources. If the business environment in China deteriorates from the perspective of domestic or international investors, our or our joint ventures’ business in China may also be adversely affected.
45
Although the PRC government has implemented measures emphasizing the utilization of market forces for economic reform, the reduction of state ownership of productive assets, and the establishment of improved corporate governance in business enterprises, a substantial portion of productive assets in China is still owned by the government. In addition, the PRC government continues to play a significant role in regulating industry development by imposing industrial policies. The PRC government also exercises significant control over China’s economic growth by allocating resources, controlling payment of foreign currency-denominated obligations, setting monetary policy, regulating financial services and institutions and providing preferential treatment to particular industries or companies. See also “The PRC government exerts substantial influence over the manner in which we conduct our business activities. Its oversight and discretion over our business could result in a material adverse change in our operations and the value of our ordinary shares and ADSs. Changes in laws, regulations and policies in China and uncertainties with respect to the PRC legal system could materially and adversely affect us. In addition, rules and regulations in China can change quickly with little advance notice.”
While the PRC economy has experienced significant growth in the past 40 years, growth has been uneven across different regions and among various economic sectors of China. The PRC government has implemented various measures to encourage economic development and guide the allocation of resources. Some of these measures benefit the overall PRC economy, but may have a negative effect on us or our joint ventures. For example, our financial condition and results of operations may be adversely affected by government control over capital investments or changes in tax regulations that are applicable to us or our joint ventures.
The PRC government exerts substantial influence over the manner in which we conduct our business activities. Its oversight and discretion over our business could result in a material adverse change in our operations and the value of our ordinary shares and ADSs. Changes in laws, regulations and policies in China and uncertainties with respect to the PRC legal system could materially and adversely affect us. In addition, rules and regulations in China can change quickly with little advance notice.
We conduct a substantial portion of our business through our subsidiaries and joint ventures in China. PRC laws and regulations govern our and their operations in China. The Chinese government has exercised and continues to exercise substantial control over virtually every sector of the Chinese economy through regulation and state ownership. For example, the PRC government has recently published new policies that significantly affected certain industries such as the education and internet industries, and we cannot rule out the possibility that it will in the future release regulations or policies regarding our industry that could adversely affect our business, financial condition and results of operations. See also “The PRC’s economic, political and social conditions, as well as governmental policies, could affect the business environment and financial markets in China, our ability to operate our business, our liquidity and our access to capital” and “The PRC government has increasingly strengthened oversight in offerings conducted overseas or on foreign investment in China-based issuers, which could result in a material change in our operations and our ordinary shares and ADSs could decline in value or become worthless.”
Our ability to operate in China may be harmed by changes in its laws and regulations. The central or local governments may impose new, stricter regulations or interpretations of existing regulations that would require additional expenditures and efforts on our part to ensure our compliance with such regulations or interpretations. For instance, regulations introduced by the NMPA concerning drug inspection, investigation, evidence collection and disposal are relatively new, and because of the limited volume of published judicial decisions, which are non-binding in nature, the interpretation and enforcement of these laws and regulations are uncertain. In addition, the implementation of laws and regulations may be in part based on government policies and internal rules that are subject to the interpretation and discretion of different government agencies (some of which are not published on a timely basis or at all) that may have a retroactive effect. As a result, we may not be aware of our, our collaboration partners’ or our joint ventures’ violation of these policies and rules until sometime after the violation. The imposition of new regulations or interpretations of existing regulations can occur quickly with little advance notice. We may incur penalties for any failure to comply with PRC laws and regulations. In addition, any litigation in China, regardless of outcome, may be protracted and result in substantial costs and diversion of resources and management attention. Since PRC administrative and court authorities have significant discretion in interpreting and implementing statutory and contractual terms, it may be difficult to evaluate the outcome of administrative and court proceedings and the level of legal protection we enjoy.
46
For further information regarding government regulation in China and other jurisdictions, see Item 4.B. “Business Overview—Regulations—Government Regulation of Pharmaceutical Product Development and Approval—PRC Regulation of Pharmaceutical Product Development and Approval,” “Business Overview—Regulations—Coverage and Reimbursement—PRC Coverage and Reimbursement” and “Business Overview—Regulations—Other Healthcare Laws—Other PRC Healthcare Laws.”
The PRC government has increasingly strengthened oversight in offerings conducted overseas or on foreign investment in China-based issuers, which could result in a material change in our operations and our ordinary shares and ADSs could decline in value or become worthless.
The PRC government has indicated an intent to take actions to exert more oversight and control over offerings that are conducted overseas and/or foreign investment in China-based issuers. For example, on July 6, 2021, the relevant PRC government authorities made public the Opinions on Strictly Scrutinizing Illegal Securities Activities in Accordance with the Law, or the Opinions. These Opinions emphasized the need to strengthen the administration over illegal securities activities and the supervision of overseas listings by China-based companies and proposed to take effective measures, such as promoting the construction of relevant regulatory systems to deal with the risks and incidents faced by China-based overseas-listed companies.
On December 24, 2021, the CSRC issued the Provisions of the State Council on the Administration of Overseas Securities Offering and Listing by Domestic Companies (Draft for Comments) and the Administrative Measures for the Filing of Overseas Securities Offering and Listing by Domestic Companies (Draft for Comments), collectively the Draft Overseas Listing Regulations, for public comment until January 23, 2022.
Following issuance of the Draft Overseas Listing Regulations, on February 17, 2023, the CSRC issued the Notice on Filing Arrangements for Overseas Securities Offering and Listing by Domestic Companies (the “CSRC Filing Notice”), stating that the CSRC has published the Trial Administrative Measures of Overseas Securities Offering and Listing by Domestic Companies (the “Trial Measures”) and five supporting guidelines (the “Listing Guidelines”), collectively the Trial Measures and Listing Guidelines. Among others, the Trial Measures and Listing Guidelines provide that overseas offerings and listings by PRC domestic companies shall:
(i) | require submission of relevant materials that contain a filing report and a legal opinion, providing truthful, accurate and complete information on matters including but not limited to the shareholders of the issuer. Where the filing documents are complete and in compliance with stipulated requirements, the CSRC shall, within 20 working days after receipt of filing documents, conclude the filing procedure and publish filing results on the CSRC website. Where filing documents are incomplete or do not conform to stipulated requirements, the CSRC shall request supplementation and amendment thereto within five working days after receipt of the filing documents. The issuer should then complete supplementation and amendment within 30 working days; |
(ii) | abide by laws, administrative regulations and relevant state rules concerning foreign investment in China, state-owned asset administration, industry regulation and outbound investment, and shall not disrupt the PRC domestic market order, harm state or public interests or undermine the lawful rights and interests of PRC domestic investors; |
(iii) | abide by national secrecy laws and relevant provisions. Necessary measures shall be taken to fulfill confidentiality obligations. Divulgence of state secrets or working secrets of government agencies is strictly prohibited. Provision of personal information and important data, etc., to overseas parties in relation to overseas offering and listing of PRC domestic companies shall be in compliance with applicable laws, administrative regulations and relevant state rules; and |
(iv) | be made in strict compliance with relevant laws, administrative regulations and rules concerning national security in the spheres of foreign investment, cybersecurity, data security, etc., and issuers shall duly fulfill their obligations to protect national security. If the intended overseas offering and listing necessitates a national security review, relevant security review procedures shall be completed according to the law before the application for such offering and listing is submitted to any overseas parties such as securities regulatory agencies and trading venues; |
The Trial Measures came into effect on March 31, 2023. PRC domestic companies seeking to offer and list securities (which, for the purposes of the Trial Measures, are defined thereunder as equity shares, depository receipts, corporate bonds convertible to equity shares, and other equity securities that are offered and listed overseas, either directly or indirectly, by PRC domestic companies) in overseas markets, either via direct or indirect means, must file with the CSRC within three working days after their application for an overseas listing is submitted.
47
The Trial Measures provide that where a PRC domestic company seeks to indirectly offer and list securities in overseas markets, the issuer shall designate a major domestic operating entity, which shall, as the domestic entity responsible, file with the CSRC. The Trial Measures stipulate that an overseas listing will be determined as “indirect” if the issuer meets both of the following conditions: (1) 50% or more of any of the issuer’s operating revenue, total profit, total assets or net assets as documented in its audited consolidated financial statements for the most recent accounting year are accounted for by PRC domestic companies (“Condition I”), and (2) the main parts of the issuer’s business activities are conducted in the PRC, or its main places of business are located in the PRC, or the senior managers in charge of its business operations and management are mostly Chinese citizens or domiciled in the PRC (“Condition II”); whether Chinese citizens from Taiwan, Hong Kong, and Macau are included in the foregoing specification is not specified. The determination as to whether or not an overseas offering and listing by PRC domestic companies is indirect shall be made on a ‘substance over form’ basis. The Listing Guidelines further stipulate that if an issuer not satisfying Condition I submits an application for issuance and listing in overseas markets in accordance with relevant non-PRC issuance regulations requiring such issuer to disclose risk factors mainly related to the PRC, the securities firm(s) and the issuer’s PRC counsel should follow the principle of ‘substance over form’ in order to identify and argue whether the issuer should complete a filing under the Trial Measures.
Subsequent securities offerings of an issuer in (i) the same overseas market where it has previously offered and listed securities, and (ii) an overseas market other than one where the issuer has previously offered and listed securities shall be filed with the CSRC within three working days after offerings are completed. Additionally, the Trial Measures stipulate that after an issuer has offered and listed securities in an overseas market, the issuer shall submit a report to the CSRC within three working days after the occurrence and public disclosure of (i) a change of control thereof, (ii) investigations of or sanctions imposed on the issuer by overseas securities regulators or relevant competent authorities, (iii) changes of listing status or transfers of listing segment, and (iv) a voluntary or mandatory delisting.
The CSRC Filing Notice states that, beginning from March 31, 2023, PRC domestic enterprises which have already issued and listed securities overseas and fall within the scope of filing under the Trial Measures shall be considered “existing enterprises” (“Existing Listed Enterprises”). Existing Listed Enterprises are not required to complete filings immediately; rather, Existing Listed Enterprises should complete filings if they are subsequently involved in matters require filings, such as follow-on financing activities, in accordance with the Trial Measures.
There is a possibility that we may be deemed as an Existing Listed Enterprise as defined under the CSRC Filing Notice, and that future offerings of listed securities or listings outside China by us may be subject to CSRC filing requirements in accordance with the Trial Measures. Given that the Trial Measures and Listing Guidelines have been introduced recently, and that there remain substantial uncertainties surrounding the enforcement thereof, we cannot assure you that, if required, we would be able to complete the filings and fully comply with the relevant new rules on a timely basis, if at all.
In addition, the Measures for Cybersecurity Review, which took effect on February 15, 2022, requires, among others, prior cybersecurity review for online platform operators holding over one million users’ personal information before any public listing in a foreign country. The Measures on Security Assessment of Cross-border Data Transfer, effective on September 1, 2022, specify that data controllers and/or critical information infrastructure operators will be subject to security assessment. There remain uncertainties as to whether such measures are applicable to our business. See also “We are subject to stringent privacy and cybersecurity laws, information security policies and contractual obligations related to data privacy and security, and we may be exposed to risks related to our management of the medical data of subjects enrolled in our clinical trials and other personal or sensitive information.”
On February 24, 2023, the CSRC and other PRC governmental authorities jointly issued the Provisions on Strengthening Confidentiality and Archives Administration of Overseas Securities Offering and Listing by Domestic Companies (the “Confidentiality Provisions”), which came into effect on March 31, 2023. According to the Confidentiality Provisions, PRC domestic companies that directly or indirectly conduct overseas offerings and listings shall strictly abide by the laws and regulations on confidentiality when providing or publicly disclosing, whether directly or through their overseas listed entities, materials to securities services providers. In the event such materials contain state secrets or working secrets of government agencies, PRC domestic companies shall first obtain approval from authorities, and file with the secrecy administrative department at the same level with the approving authority; in the event that such materials, if divulged, will jeopardize national security or public interest, PRC domestic companies shall comply with procedures stipulated by national regulations. PRC domestic companies shall also provide a written statement of the specific sensitive information provided when providing materials to securities service providers, and such written statements shall be retained for inspection. As the Confidentiality Provisions were recently promulgated and are effective, their interpretation and implementation remain substantially uncertain.
48
If (i) we mistakenly conclude that certain regulatory filings, permissions and approvals are not required or (ii) applicable laws, regulations, or interpretations change and (iii) we are required to obtain such filings, permissions or approvals in the future, we may be unable to obtain them in a timely manner, or at all, and such filings, permissions or approvals may be denied or rescinded even if obtained. We may face adverse actions or sanctions by the CSRC or other PRC regulatory agencies if we are unable to comply with such requirements, which may result in fines and penalties, restrictions on our operations, having to delist from a stock exchange outside of China, the halting of securities offerings to foreign investors and other actions that could materially and adversely affect our operations and the interest of our investors and cause a significant depreciation in the price of our ordinary shares and ADSs.
Certain PRC regulations may make it more difficult for us to pursue growth through acquisitions. Any failure or perceived failure by us to comply with PRC anti-monopoly laws and regulations may result in governmental investigations or enforcement actions, litigation or claims against us and could have an adverse effect on our business, financial condition and results of operations.
We may pursue potential strategic acquisitions that are complementary to our business and operations. In doing so, we will be subject to a variety of PRC anti-monopoly laws. The Regulations on Mergers and Acquisitions of Domestic Enterprises by Foreign Investors, or the M&A Rules, adopted by six PRC regulatory agencies in 2006 and amended in 2009, established additional procedures and requirements that could make merger and acquisition activities by foreign investors more time-consuming and complex. For example, the M&A Rules require that the MOFCOM be notified in advance of any change-of-control transaction in which a foreign investor takes control of a PRC domestic enterprise if (i) any important industry is concerned, (ii) such transaction involves factors that have or may have impact on the national economic security or (iii) such transaction will lead to a change in control of a domestic enterprise which holds a famous trademark or PRC time-honored brand. The approval from the MOFCOM must be obtained in circumstances where overseas companies established or controlled by PRC enterprises or residents acquire affiliated domestic companies. Mergers, acquisitions or contractual arrangements that allow one market player to take control of or to exert decisive impact on another market player must also be notified in advance to the SAMR when the threshold under the Provisions on Thresholds for Prior Notification of Concentrations of Undertakings, or the Prior Notification Rules, issued by the State Council in 2008 and amended in 2018, is triggered. PRC national security review rules, which became effective in September 2011, require a strict review of (a) mergers and acquisitions by foreign investors that raise “national defense and security” concerns and (b) mergers and acquisitions through which foreign investors may acquire de facto control over domestic enterprises that raise “national security” concerns. The rules also prohibit any activities attempting to bypass a security review, including by structuring the transaction through a proxy or contractual control arrangement.
Further, the Measures for the Security Review of Foreign Investments promulgated by the NDRC and MOFCOM, which became effective from January 2021, require that a security review by relevant governmental authorities must be conducted for foreign investments that affect or may affect national security in accordance with the provisions thereunder.
The PRC anti-monopoly enforcement agencies have in recent years strengthened enforcement under the PRC Anti-Monopoly Law. In March 2018, the SAMR was formed as a new governmental agency to take over, among other things, the anti-monopoly enforcement functions from the relevant departments under the MOFCOM, the NDRC and SAMR. Since its inception, the SAMR has continued to strengthen anti-monopoly enforcement. In November 2021, the State Council inaugurated the National Anti-Monopoly Bureau, which aims to further implement fair competition policies and strengthen anti-monopoly supervision in the PRC, particularly to strengthen oversight and law enforcement in areas involving innovation, science and technology, information security and people’s livelihoods.
SAMR issued the Provisions on Prohibition of the Abuse of Market Dominance on March 10, 2023, which came into effect on April 15, 2023, pursuant to which an abuse of market dominance determined by the SAMR shall satisfy all the following criteria: (i) the business operator is dominating the market; (ii) the business operator has eliminated or restricted competition; (iii) the business operator has no legitimate reason to carry out such acts; and (iv) such acts by the business operator have an impact on elimination or restriction of market competition. Pursuant to the Provisions on Prohibition of Monopoly Agreements issued by SAMR and effective from April 15, 2023, entering into monopolistic agreements, which means agreements or concerted practices to eliminate or restrict competition, is prohibited, unless such agreements satisfy the specific exemptions prescribed in the Anti-Monopoly Law, such as improving technologies or increasing the efficiency and competitiveness of small and medium-sized undertakings. If business operators fail to comply with the Anti-Monopoly Law or other relevant regulations, they may be ordered to cease business activities, unwind transactions, and be subject to confiscation of unlawful profits and fines.
49
Complying with the requirements of these regulations when pursuing acquisitive transactions could be time-consuming, and any required approval processes, including obtaining approval or clearance from the MOFCOM, may delay or inhibit our ability to complete such transactions, which could affect our ability to expand our business or maintain our market share. Due to the enhanced enforcement of the Anti-Monopoly Law, we may receive greater scrutiny and attention from regulators and more frequent and rigid investigations or review by regulators, which may increase our compliance costs and subject us to heightened risks and challenges. In addition, there are significant uncertainties on the evolving legislative activities and varied local implementation practices of anti-monopoly and competition laws and regulations in China. The amended Anti-Monopoly Law, published in October 2021 in draft form for public comment, became effective in August 2022. It imposes a higher regulatory requirement to complete an acquisitive transaction. Any failure or perceived failure by us to comply with the anti-monopoly laws and regulations may result in governmental investigations or enforcement actions, lawsuits or claims against us and could have an adverse effect on our business, financial condition and results of operations. See also “Risks Relating to Sales of Our Internally Developed Drugs and Other Drugs—We may engage in strategic transactions, including acquisitions, investments, joint ventures or divestitures that may have an adverse effect on our business. If we engage in a strategic transaction, there is no assurance that the transaction will be consummated.”
Restrictions on currency exchange may limit our ability to receive and use our revenue effectively.
Substantially all of our revenue is denominated in renminbi, which currently is not a freely convertible currency. A portion of our revenue may be converted into other currencies to meet our foreign currency obligations, including, among others, payments of dividends declared, if any, in respect of our ordinary shares or ADSs. Under China’s existing foreign exchange regulations, our subsidiaries and joint ventures are able to pay dividends in foreign currencies or convert renminbi into other currencies for use in operations without prior approval from the PRC State Administration of Foreign Exchange, or the SAFE, by complying with certain procedural requirements. However, we cannot assure you that the PRC government will not take future measures to restrict access to foreign currencies for current account transactions.
Our PRC subsidiaries’ and joint ventures’ ability to obtain foreign exchange is subject to significant foreign exchange controls and, in the case of amounts under the capital account, requires the approval of and/or registration with PRC government authorities, including the SAFE. In particular, if we finance our PRC subsidiaries or joint ventures by means of foreign debt from us or other foreign lenders, the amount is not allowed to exceed either the cross-border financing risk weighted balance calculated based on a formula by the PBOC or the difference between the amount of total investment and the amount of the registered capital. Further, such loans must be filed with and registered with the SAFE or their local branches and the National Development and Reform Commission (if applicable). If we finance our PRC subsidiaries or joint ventures by means of additional capital contributions, the amount of these capital contributions must first be filed with the relevant government approval authority. These limitations could affect the ability of our PRC subsidiaries and joint ventures to obtain foreign exchange through debt or equity financing.
Our business benefits from certain PRC government tax incentives. Any changes to, or our PRC subsidiaries/joint ventures failing to continuously meet the criteria for these incentives could have a material adverse effect on our operating results by significantly increasing our tax expenses.
Certain of our PRC subsidiaries and a joint venture have been granted High and New Technology Enterprise, or HNTE, status by the relevant PRC authorities. This status allows the relevant enterprise to enjoy a reduced Enterprise Income Tax, or EIT, rate at 15% on its taxable profits. For the duration of its HNTE grant, the relevant PRC enterprise must continue to meet the relevant HNTE criteria or else the 25% standard EIT rate will be applied from the beginning of the calendar year when the enterprise fails to meet the relevant criteria. If the rules for such incentives are amended, it would be uncertain whether any criteria as amended can be met, in which case the higher EIT rate may apply resulting in increased tax burden which will impact our business, financial condition, results of operations and growth prospects.
50
We may be treated as a resident enterprise for PRC Tax purposes under China’s Enterprise Income Tax Law and Implementation Rules, or the EIT Law, and our global income may therefore be subject to PRC income tax.
China’s EIT Law defines the term “de facto management bodies” as “bodies that substantially carry out comprehensive management and control on the business operation, employees, accounts and assets of enterprises.” Under the EIT Law, an enterprise incorporated outside of China whose “de facto management bodies” are located in China is considered a “resident enterprise” and will be subject to a uniform 25% EIT rate on its global income. On April 22, 2009, China’s State Administration of Taxation, or the SAT, in the Notice Regarding the Determination of Chinese-Controlled Offshore-Incorporated Enterprises as PRC Tax Resident Enterprises on the Basis of De Facto Management Bodies, or Circular 82, further specified certain criteria for the determination of what constitutes “de facto management bodies.” If all of these criteria are met, the relevant foreign enterprise may be regarded to have its “de facto management bodies” located in China and therefore be considered a resident enterprise in China. These criteria include: (i) the enterprise’s day-to-day operational management is primarily exercised in China; decisions relating to the enterprise’s financial and human resource matters are made or subject to approval by organizations or personnel in China; (ii) the enterprise’s primary assets, accounting books and records, company seals, and board and shareholders’ meeting minutes are located or maintained in China; and (iii) 50% or more of voting board members or senior executives of the enterprise habitually reside in China. Although Circular 82 only applies to foreign enterprises that are majority-owned and controlled by PRC enterprises, not those owned and controlled by foreign enterprises or individuals, the determining criteria set forth in Circular 82 may be adopted by the PRC tax authorities as the test for determining whether the enterprises are PRC tax residents, regardless of whether they are majority-owned and controlled by PRC enterprises.
Except for our PRC subsidiaries and joint ventures incorporated in China, we believe that none of our entities incorporated outside of China is a PRC resident enterprise for PRC tax purposes. However, the tax resident status of an enterprise is subject to determination by the PRC tax authorities, and uncertainties remain with respect to the interpretation of the term “de facto management body.”
If we are treated as a PRC tax resident, dividends distributed by us to our non-PRC shareholders and ADS holders or any gains realized by non-PRC shareholders and ADS holders from the transfer of our shares or ADSs may be subject to PRC tax.
Under the EIT Law, dividends payable by a PRC enterprise to its foreign investor who is (i) a non-PRC resident enterprise with no office or premises established in China, or (ii) a non-PRC resident enterprise with an office or premises established in China but whose income (i.e. dividends received) has no de facto relationship with said office or premises, as well as gains on transfers of shares of a PRC enterprise by such a foreign investor will generally be subject to a 10% withholding tax, unless such non-PRC resident enterprise’s jurisdiction of tax residency has an applicable tax treaty with the PRC that provides for an exemption or a reduced rate of withholding tax.
If the PRC tax authorities determine that we should be considered a PRC resident enterprise for EIT purposes, any dividends payable by us to our non-PRC resident enterprise shareholders or ADS holders, as well as gains realized by such investors from the transfer of our shares or ADSs may be subject to a 10% withholding tax. Furthermore, if we are considered a PRC resident enterprise for EIT purposes, it is unclear whether our non-PRC individual shareholders (including our ADS holders) would be subject to any PRC tax on dividends or gains obtained by such non-PRC individual shareholders. If any PRC tax were to apply to dividends or gains realized by non-PRC individuals, it would generally apply at a rate of up to 20% (which in the case of dividends may be withheld at source). The foregoing rates may be reduced by an applicable tax treaty, but it is unclear if a non-PRC resident shareholder or ADS holder would be able to obtain in practice the benefits of any tax treaties between their country of tax residence and the PRC in the event that we are treated as a PRC resident enterprise. If dividends payable to our non-PRC resident shareholders, or gains from the transfer of our shares or ADSs by such shareholders are subject to PRC tax, the value of your investment in our shares or ADSs may decline significantly.
51
There is uncertainty regarding the PRC withholding tax rate that will be applied to distributions from our PRC subsidiaries and joint ventures to their respective Hong Kong immediate holding companies, which could have a negative impact on our business.
The EIT Law provides that a withholding tax at the rate of 10% is applicable to dividends payable by a PRC resident enterprise to investors who are “non-resident enterprises” (i.e., that do not have an establishment or place of business in the PRC or that have such establishment or place of business but the relevant dividend is not effectively connected with the establishment or place of business). However, pursuant to Article 10.2(1), or the Article, of the Arrangement between the Mainland of China and the Hong Kong Special Administrative Region for the Avoidance of Double Taxation and the Prevention of Fiscal Evasion with respect to Taxes on Income, or the Arrangement, withholding tax at a reduced rate of 5% may be applicable to dividends payable by PRC resident enterprises to beneficial owners of the dividends that are Hong Kong tax residents if certain requirements are met. There is uncertainty regarding whether the PRC tax authorities will consider us to be eligible to the reduced tax rate. If the Article is deemed not to apply to dividends payable by our PRC subsidiaries and joint ventures to their respective Hong Kong immediate holding companies that are ultimately owned by us, the withholding tax rate applicable to us will be the statutory rate of 10% instead of 5% which may potentially impact our business, financial condition, results of operations and growth prospects.
Any failure to comply with PRC regulations regarding our employee equity incentive plans may subject the PRC plan participants or us to fines and other legal or administrative sanctions, which could adversely affect our business, financial condition and results of operations.
In February 2012, the SAFE promulgated the Notices on Issues Concerning the Foreign Exchange Administration for Domestic Individuals Participating in Stock Incentive Plans of Overseas Publicly Listed Companies. Based on this regulation, PRC residents who are granted shares or share options by a company listed on an overseas stock market under its employee share option or share incentive plan are required to register with the SAFE or its local counterparts by following certain procedures. We and our employees who are PRC residents and individual beneficial owners who have been granted shares or share options have been subject to these rules due to our listing on the AIM market, Nasdaq and SEHK. We have registered the option schemes and the share incentive plan and will continue to assist our employees to register their share options or shares. However, any failure of our PRC individual beneficial owners and holders of share options or shares to comply with the SAFE registration requirements in the future may subject them to fines and legal sanctions and may, in rare instances, limit the ability of our PRC subsidiaries to distribute dividends to us.
In addition, the SAT has issued circulars concerning employee share options or restricted shares. Under these circulars, employees working in the PRC who exercise share options, or whose restricted shares vest, will be subject to PRC individual income tax. The PRC subsidiaries of an overseas listed company have obligations to file documents related to employee share options or restricted shares with relevant tax authorities and to withhold individual income tax of those employees related to their share options or restricted shares. Although the PRC subsidiaries currently withhold individual income tax from the PRC employees in connection with their exercise of share options, if they fail to report and pay the tax withheld according to relevant laws, rules and regulations, the PRC subsidiaries may face sanctions imposed by the tax authorities or other PRC government authorities.
We may be involved in litigation, legal disputes, claims or administrative proceedings which could be costly and time-consuming to resolve.
We may become subject, from time to time, to legal proceedings and claims that arise in the ordinary course of business or pursuant to governmental or regulatory enforcement activity. Any litigation or proceeding to which we become a party might result in substantial costs and divert management’s attention and resources. Furthermore, any litigation, legal disputes, claims or administrative proceedings which are initially not of material importance may escalate and become important to us due to a variety of factors, such as changes in the facts and circumstances of the cases, the likelihood of loss, the monetary amount at stake and the parties involved. Our insurance might not cover claims brought against us, provide sufficient payments to financially cover all of the costs to resolve such claims or continue to be available on terms acceptable to us.
52
The political relationships between China and other countries may affect our business operations.
We conduct our business primarily through our subsidiaries and joint ventures in China, but we also have clinical operations in the United States and other foreign jurisdictions. As a result, China’s political relationships with the United States and other jurisdictions may affect our business operations. There can be no assurance that our clinical trial participants or customers will not alter their perception of us or their preferences as a result of adverse changes to the state of political relationships between China and the relevant foreign jurisdictions. Any tensions and political concerns between China and the relevant foreign jurisdictions may adversely affect our business, financial condition, results of operations, cash flows and prospects.
Risks Relating to Intellectual Property
If we, our joint ventures or our collaboration partners are unable to protect our or their products and drug candidates through intellectual property rights, our competitors may compete directly against us or them.
Our success depends, in part, on our, our joint venture partners’ and our collaboration partners’ ability to protect our and our joint ventures’ and our collaboration partners’ products and drug candidates from competition by establishing, maintaining and enforcing our or their intellectual property rights. We, our joint ventures and our collaboration partners seek to protect the products and technology that we and they consider commercially important by filing PRC and international patent applications, relying on trade secrets or pharmaceutical regulatory protection or employing a combination of these methods. As of December 31, 2023, we had 274 issued patents, including 25 PRC patents, 24 U.S. patents and 13 European patents, 354 patent applications pending in major market jurisdictions, and 7 pending Patent Cooperation Treaty, or PCT, patent applications relating to the drug candidates of our Oncology/Immunology operations. For more details, see Item 4.B. “Business Overview—Patents and Other Intellectual Property.” Patents may become invalid and patent applications may not be granted for a number of reasons, including known or unknown prior art, deficiencies in the patent application or the lack of originality of the technology. In addition, the PRC and the United States have adopted the “first-to-file” system under which whoever first files an invention patent application will be awarded the patent. Under the first-to-file system, third parties may be granted a patent relating to a technology which we invented. Furthermore, the terms of patents are finite. The patents we hold and patents to be issued from our currently pending patent applications generally have a twenty-year protection period starting from the date of application.
We, our joint ventures and/or our collaboration partners may become involved in patent litigation against third parties to enforce our or their patent rights, to invalidate patents held by such third parties, or to defend against such claims. A court may refuse to stop the other party from using the technology at issue on the grounds that our or our joint ventures’ patents do not cover the third-party technology in question. Further, such third parties could counterclaim that we or our joint ventures infringe their intellectual property or that a patent we, our joint ventures or our collaboration partners have asserted against them is invalid or unenforceable. In patent litigation, defendant counterclaims challenging the validity, enforceability or scope of asserted patents are commonplace. In addition, third parties may initiate legal proceedings against us or our intellectual property to assert such challenges to our intellectual property rights.
The outcome of any such proceeding is generally unpredictable. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness or non-enablement. Patents may be unenforceable if someone connected with prosecution of the patent withheld relevant information or made a misleading statement during prosecution. It is possible that prior art of which we, our joint ventures or our collaboration partners and the patent examiner were unaware during prosecution exists, which could render our or their patents invalid. Moreover, it is also possible that prior art may exist that we, our joint ventures or our collaboration partners are aware of but do not believe is relevant to our or their current or future patents, but that could nevertheless be determined to render our patents invalid. The cost to us or our joint ventures of any patent litigation or similar proceeding could be substantial, and it may consume significant management time. We and our joint ventures do not maintain insurance to cover intellectual property infringement.
An adverse result in any litigation proceeding could put one or more of our or our joint ventures’ patents at risk of being invalidated or interpreted narrowly. If a defendant were to prevail on a legal assertion of invalidity or unenforceability of our patents covering one of our or our joint ventures’ products or our drug candidates, we could lose at least part, and perhaps all, of the patent protection covering such product or drug candidate. Competing drugs may also be sold in other countries in which our or our joint ventures’ patent coverage might not exist or be as strong. If we lose a foreign patent lawsuit, alleging our or our joint ventures’ infringement of a competitor’s patents, we could be prevented from marketing our drugs in one or more foreign countries. Any of these outcomes would have a materially adverse effect on our business.
53
Intellectual property and confidentiality legal regimes in China may not afford protection to the same extent as in the United States or other countries. Implementation and enforcement of PRC intellectual property laws may be deficient and ineffective. Policing unauthorized use of proprietary technology is difficult and expensive, and we or our joint ventures may need to resort to litigation to enforce or defend patents issued to us or them or to determine the enforceability, scope and validity of our proprietary rights or those of others. The experience and capabilities of PRC courts in handling intellectual property litigation varies, and outcomes are unpredictable. Further, such litigation may require a significant expenditure of cash and may divert management’s attention from our or our joint ventures’ operations, which could harm our business, financial condition and results of operations. An adverse determination in any such litigation could materially impair our or our joint ventures’ intellectual property rights and may harm our business, prospects and reputation.
Developments in patent law could have a negative impact on our business.
From time to time, authorities in the United States, China, Europe and Japan and other government authorities may change the standards of patentability, and any such changes could have a negative impact on our business. For example, in the United States, the Leahy-Smith America Invents Act, or the America Invents Act, which was signed into law in 2011, includes a number of significant changes to U.S. patent law. These changes include a transition from a “first-to-invent” system to a “first-to-file” system, changes to the way issued patents are challenged, and changes to the way patent applications are disputed during the examination process. As a result of these changes, patent law in the United States may favor larger and more established companies that have greater resources to devote to patent application filing and prosecution. The U.S. Patent and Trademark Office, or USPTO, has developed regulations and procedures to govern the full implementation of the America Invents Act, and many of the substantive changes to patent law associated with the America Invents Act, and, in particular, the first-to-file provisions became effective on March 16, 2013. Substantive changes to patent law associated with the America Invents Act, including continually developing case law, may affect our ability to obtain patents, and if obtained, to enforce or defend them. Accordingly, it is not clear what, if any, impact the America Invents Act will have on the cost of prosecuting our or our joint ventures’ patent applications and our or their ability to obtain patents based on our or our joint ventures’ discoveries and to enforce or defend any patents that may issue from our or their patent applications, all of which could have a material adverse effect on our business.
If we are unable to maintain the confidentiality of our, our collaboration partners’ and our joint ventures’ trade secrets, the business and competitive position of ourselves and our joint ventures may be harmed.
In addition to the protection afforded by patents and the PRC’s State Secret certification, we, our collaboration partners and our joint ventures rely upon unpatented trade secret protection, unpatented know-how and continuing technological innovation to develop and maintain our competitive position. We seek to protect our, our collaboration partners’ and our joint ventures’ proprietary technology and processes, in part, by entering into confidentiality agreements with our and their collaborators, scientific advisors, employees and consultants, and invention assignment agreements with our and their consultants and employees. We, our collaboration partners and our joint ventures may not be able to prevent the unauthorized disclosure or use of our or their technical know-how or other trade secrets by the parties to these agreements, however, despite the existence generally of confidentiality agreements and other contractual restrictions. If any of the collaborators, scientific advisors, employees and consultants who are parties to these agreements breaches or violates the terms of any of these agreements, we and our joint ventures may not have adequate remedies for any such breach or violation, and we, our collaboration partners could lose our trade secrets as a result. Enforcing a claim that a third-party illegally obtained and is using our or our joint ventures’ trade secrets, like patent litigation, is expensive and time consuming, and the outcome is unpredictable. In addition, courts in China and other jurisdictions outside the United States are sometimes less prepared or willing to protect trade secrets.
The trade secrets of our company, our collaboration partners and our joint ventures could otherwise become known or be independently discovered by our or their competitors. For example, competitors could purchase our drugs and attempt to replicate some or all of the competitive advantages we derive from our development efforts, willfully infringe our intellectual property rights, design around our protected technology or develop their own competitive technologies that fall outside of our intellectual property rights. If any of our, our collaboration partners’ or our joint ventures’ trade secrets were to be lawfully obtained or independently developed by a competitor, we and our joint ventures would have no right to prevent them, or others to whom they communicate it, from using that technology or information to compete against us or our joint ventures. If our or our joint ventures’ trade secrets are unable to adequately protect our business against competitors’ drugs, our competitive position could be adversely affected, as could our business.
54
We, our collaboration partners and our joint ventures are dependent on trademark and other intellectual property rights licensed from others. If we lose our licenses for any of our products, we, our collaboration partners or our joint ventures may not be able to continue developing such products or may be required to change the way we market such products.
We, our collaboration partners and our joint ventures are parties to licenses that give us or them rights to third-party intellectual property that are necessary or useful for our, our collaboration partners’ or our joint ventures’ businesses. In particular, the “Hutchison”, “Chi-Med”, “Hutchison China MediTech” and “HUTCHMED” brands, among others, have been licensed to us by Hutchison Whampoa Enterprises Limited, an affiliate of our largest shareholder, Hutchison Healthcare Holdings Limited. Hutchison Whampoa Enterprises Limited grants us a royalty-free, worldwide license to such brands. For more details, please see “Item 7. Major Shareholders and Related Party Transactions—Related Party Transactions—Relationship with CK Hutchison—Intellectual property licensed by the CK Hutchison group.” Under the terms of our brand license agreement, Hutchison Whampoa Enterprises Limited has the right to terminate the license if, among other things, we commit a material breach of the agreement, or within any twelve-month period the aggregate direct or indirect shareholding in our company held by CK Hutchison is reduced to less than 35%, 30% or 20%. Furthermore, the trademarks of Elunate and Orpathys are licensed to us in China by our collaboration partner Eli Lilly and AstraZeneca, respectively.
In some cases, our licensors have retained the right to prosecute and defend intellectual property rights licensed to us or our joint ventures. We depend in part on the ability of our licensors to obtain, maintain and enforce intellectual property protection for such licensed intellectual property. Such licensors may not successfully maintain their intellectual property, may determine not to pursue litigation against other companies that are infringing on such intellectual property, or may pursue litigation less aggressively than we or our joint ventures would. Without protection for the intellectual property we or our joint ventures license, other companies might be able to offer substantially identical products or branding, which could adversely affect our competitive business position and harm our business prospects.
If our, our collaboration partners’ or our joint ventures’ products or drug candidates infringe the intellectual property rights of third parties, we and they may incur substantial liabilities, and we and they may be unable to sell these products.
Our commercial success depends significantly on our, our collaboration partners and our joint ventures’ ability to operate without infringing the patents and other proprietary rights of third parties. In the PRC, invention patent applications are generally maintained in confidence until their publication 18 months from the filing date. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made and invention patent applications are filed. Even after reasonable investigation, we may not know with certainty whether any third-party may have filed a patent application without our knowledge while we or our joint ventures are still developing or producing that product. While the success of pending patent applications and applicability of any of them to our or our joint ventures’ programs are uncertain, if asserted against us or them, we could incur substantial costs and we or they may have to:
| ● | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
| ● | redesign products or processes to avoid infringement; and |
| ● | stop producing products using the patents held by others, which could cause us or them to lose the use of one or more of our or their products. |
To date, we, our collaboration partners and our joint ventures have not received any material claims of infringement by any third parties. If a third-party claims that we, our collaboration partners or our joint ventures infringe its proprietary rights, any of the following may occur:
| ● | we, our collaboration partners or our joint ventures may have to defend litigation or administrative proceedings that may be costly whether we or they win or lose, and which could result in a substantial diversion of management resources; |
| ● | we, our collaboration partners or our joint ventures may become liable for substantial damages for past infringement if a court decides that our technology infringes a third-party’s intellectual property rights; |
| ● | a court may prohibit us, our collaboration partners or our joint ventures from producing and selling our or their product(s) without a license from the holder of the intellectual property rights, which may not be available on commercially acceptable terms, if at all; and |
55
| ● | we, our collaboration partners or our joint ventures may have to reformulate product(s) so that it does not infringe the intellectual property rights of others, which may not be possible or could be very expensive and time consuming. |
Any costs incurred in connection with such events or the inability to sell our, our collaboration partners’ or our joint ventures’ products may have a material adverse effect on our business and results of operations.
We, our joint ventures and our collaboration partners may not be able to effectively enforce our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our, our collaboration partners’ or our joint venture’s products or drug candidates in all countries throughout the world would be prohibitively expensive. The requirements for patentability may differ in certain countries, particularly in developing countries. Moreover, our, our joint ventures’ or our collaboration partners’ ability to protect and enforce our or their intellectual property rights may be adversely affected by unforeseen changes in foreign intellectual property laws. Additionally, the patent laws of some foreign countries do not afford intellectual property protection to the same extent as the laws of the United States. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. The legal systems of some countries, particularly developing countries, may not favor the enforcement of patents and other intellectual property rights. This could make it difficult for us or our joint ventures to stop the infringement of our or their patents or the misappropriation of our or their other intellectual property rights. For example, many foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. Consequently, we may not be able to prevent third parties from practicing our or our joint ventures’ inventions throughout the world. Competitors may use our or our joint ventures’ technologies in jurisdictions where we or they have not obtained patent protection to develop their own drugs and, further, may export otherwise infringing drugs to territories where we or our joint ventures have patent protection, if our, our joint ventures’ or our collaboration partners’ ability to enforce our or their patents to stop infringing activities is inadequate. These drugs may compete with our drug candidates, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Proceedings to enforce our, our collaboration partners’ or our joint ventures’ patent rights in foreign jurisdictions, whether or not successful, could result in substantial costs and divert our or their efforts and resources from other aspects of our and their businesses. While we intend to protect our intellectual property rights in the major markets for our drug candidates, we cannot ensure that we will be able to initiate or maintain similar efforts in all jurisdictions in which we may wish to market our drug candidates. Furthermore, some of our collaborators are responsible for enforcing our intellectual property rights, for example, AstraZeneca is responsible for enforcing our intellectual property rights with respect to savolitinib on our behalf, we may be unable to ensure that such rights are enforced or maintained in all jurisdictions. Accordingly, our efforts to protect the intellectual property rights of our drug candidates in such countries may be inadequate.
We, our collaboration partners and our joint ventures may be subject to damages resulting from claims that we or they, or our or their employees, have wrongfully used or disclosed alleged trade secrets of competitors or are in breach of non-competition or non-solicitation agreements with competitors.
We, our collaboration partners and our joint ventures could in the future be subject to claims that we or they, or our or their employees, have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of former employers or competitors. Although we try to ensure that our and our joint ventures’ employees and consultants do not improperly use the intellectual property, proprietary information, know-how or trade secrets of others in their work for us or our joint ventures, we or our joint ventures may in the future be subject to claims that we or they caused an employee to breach the terms of his or her non-competition or non-solicitation agreement, or that we, our collaboration partners, our joint ventures, or these individuals have, inadvertently or otherwise, used or disclosed the alleged trade secrets or other proprietary information of a former employer or competitor. Litigation may be necessary to defend against these claims. Even if we, our collaboration partners and our joint ventures are successful in defending against these claims, litigation could result in substantial costs and could be a distraction to management. If our or our joint ventures’ defenses to these claims fail, in addition to requiring us and them to pay monetary damages, a court could prohibit us or our joint ventures from using technologies or features that are essential to our or their products or our drug candidates, if such technologies or features are found to incorporate or be derived from the trade secrets or other proprietary information of the former employers. An inability to incorporate such technologies or features would have a material adverse effect on our business, and may prevent us from successfully commercializing our drug candidates. In addition, we, our collaboration partners or our joint ventures may lose valuable intellectual property rights or personnel as a result of such claims. Moreover, any such litigation or the threat thereof may adversely affect our or our joint ventures’ ability to hire employees or contract with independent sales representatives. A loss of key personnel or their work product could hamper or prevent our ability to commercialize our drug candidates, which would have an adverse effect on our business, results of operations and financial condition.
56
Patent terms may be inadequate to protect the competitive position of our drug candidates for an adequate amount of time, and the absence of patent linkage, patent term extension and data and market exclusivity for NMPA-approved pharmaceutical products could increase the risk of early generic competition for our drug candidates in China.
In the United States, the Drug Price Competition and Patent Term Restoration Act of 1984, generally referred to as the Hatch-Waxman Amendments, and similar legislation in the E.U. and certain other countries, provides the opportunity for limited patent term extension. The Hatch-Waxman Amendments permit a patent-term extension of up to five years to reflect patent term lost during certain portions of product development and the FDA regulatory review process. However, a patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of drug approval; only one patent may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. The application for the extension must be submitted prior to the expiration of the patent for which extension is sought. A patent that covers multiple products for which approval is sought can only be extended in connection with one of the approvals. Depending upon the timing, duration and specifics of any FDA marketing approval process for any drug candidates we may develop, one or more of our U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Amendments. However, we may not be granted an extension because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents, or otherwise failing to satisfy applicable requirements. Moreover, the applicable period or the scope of patent protection afforded could be less than we request. In addition, to the extent we wish to pursue patent term extension based on a patent that we in-license from a third party, we would need the cooperation of that third party. If we fail to obtain patent term extensions or if the term of any such extension is less than we request, our competitors may obtain approval of competing products following our patent expiration, and thus our revenue could be reduced. Further, if this occurs, our competitors may take advantage of our investment in development and trials by referencing our clinical and pre-clinical data and launch their product earlier than might otherwise be expected, and our competitive position, business, financial condition, results of operations and prospects could be materially adversely affected.
The Hatch-Waxman Amendments also include a process for patent linkage, pursuant to which the FDA will stay approval of certain follow-on applications during the pendency of litigation between the follow-on applicant and the patent holder or licensee, generally for a period of 30 months. Moreover, the Hatch-Waxman Amendments provide for statutory exclusivities that can prevent submission or approval of certain follow-on marketing applications. For example, federal law provides a five-year period of exclusivity within the United States to the first applicant to obtain approval of a new chemical entity and three years of exclusivity protecting certain innovations to previously approved active ingredients where the applicant was required to conduct new clinical investigations to obtain approval for the modification. Similarly, the U.S. Orphan Drug Act provides seven years of market exclusivity for certain drugs to treat rare diseases, where the FDA designates the drug candidate as an orphan drug and the drug is approved for the designated orphan indication.
Chinese regulators have set forth a framework for integrating patent linkage and data exclusivity into the China regulatory regime, as well as for establishing a pilot program for patent term extension. To be implemented, this framework will require adoption of regulations. On October 17, 2020, the Standing Committee of the National People’s Congress published the Patent Law of PRC (Amended in 2020), which came into effect on June 1, 2021, or the Amended Patent Law. The Amended Patent Law provides that, among other things, the owner of the patent for an innovative new drug that has been granted the marketing authorization in China is entitled to request the Patent Administration Department under the State Council to grant a patent term extension of up to five years, in order to compensate the time required for the regulatory approval for the commercialization of such innovative new drug, provided that the patent term of such innovative new drug shall not exceed a total of 14 years. Furthermore, the PRC government entered into the Economic and Trade Agreement Between the Government of the People’s Republic of China and the Government of the United States of America with the U.S. government in January 2020 which provides that the owner of the patent for an innovative new drug that has been granted the marketing authorization in China is entitled to request a patent term extension of up to five years, provided that the patent term of such innovative new drug shall not exceed a total of 14 years from the date of marketing approval in China. If we are unable to obtain patent term extension, or the term of any such extension is less than that we request, our competitors or other third parties may obtain approval of competing products following our patent expiration. Any of the foregoing could have a material adverse effect on our competitive position, business, financial condition, results of operations and prospects.
57
Risks Relating to Our ADSs
The PCAOB had historically been unable to inspect our auditor in relation to their audit work performed for our financial statements and the inability of the PCAOB to conduct inspections of our auditor in the past has deprived our investors with the benefits of such inspections.
Our auditor, the independent registered public accounting firm that issues the audit report included elsewhere in this annual report, as an auditor of companies that are traded publicly in the United States and a firm registered with the PCAOB, is subject to laws in the United States pursuant to which the PCAOB conducts regular inspections to assess its compliance with the applicable professional standards. The auditor is located in mainland China, a jurisdiction where the PCAOB was historically unable to conduct inspections and investigations completely before 2022. As a result, we and investors in the ADSs were deprived of the benefits of such PCAOB inspections. The inability of the PCAOB to conduct inspections of auditors in China in the past has made it more difficult to evaluate the effectiveness of our independent registered public accounting firm’s audit procedures or quality control procedures as compared to auditors outside of China that are subject to the PCAOB inspections. On December 15, 2022, the PCAOB issued a report that vacated its December 16, 2021 determination and removed mainland China and Hong Kong from the list of jurisdictions where it is unable to inspect or investigate completely registered public accounting firms. However, if the PCAOB determines in the future that it no longer has full access to inspect and investigate completely accounting firms in mainland China and Hong Kong, and we use an accounting firm headquartered in one of these jurisdictions to issue an audit report on our financial statements filed with the Securities and Exchange Commission, we and investors in our ADSs would be deprived of the benefits of such PCAOB inspections again, which could cause investors and potential investors in the ADSs to lose confidence in our audit procedures and reported financial information and the quality of our financial statements.
Our ADSs may be prohibited from trading in the United States under the HFCAA in the future if the PCAOB is unable to inspect or investigate completely auditors located in China. The delisting of the ADSs, or the threat of their being delisted, may materially and adversely affect the value of your investment.
Pursuant to the HFCAA, if the SEC determines that we have filed audit reports issued by a registered public accounting firm that has not been subject to inspections by the PCAOB for two consecutive years, the SEC will prohibit our shares or ADSs from being traded on a national securities exchange or in the over-the-counter trading market in the United States.
On December 16, 2021, the PCAOB issued a report to notify the SEC of its determination that the PCAOB was unable to inspect or investigate completely registered public accounting firms headquartered in mainland China and Hong Kong and our auditor was subject to that determination. In March 2022, the SEC conclusively listed us as a Commission-Identified Issuer under the HFCAA following the filing of our annual report on Form 20-F for the fiscal year ended December 31, 2021. On December 15, 2022, the PCAOB removed mainland China and Hong Kong from the list of jurisdictions where it is unable to inspect or investigate completely registered public accounting firms. For this reason, we do not expect to be identified as a Commission-Identified Issuer under the HFCAA after we file this annual report on Form 20-F for the fiscal year ended December 31, 2023.
Each year, the PCAOB will determine whether it can inspect and investigate completely audit firms in mainland China and Hong Kong, among other jurisdictions. If the PCAOB determines in the future that it no longer has full access to inspect and investigate completely accounting firms in mainland China and Hong Kong and we use an accounting firm headquartered in one of these jurisdictions to issue an audit report on our financial statements filed with the Securities and Exchange Commission, we would be identified as a Commission-Identified Issuer following the filing of the annual report on Form 20-F for the relevant fiscal year. In accordance with the HFCAA, our securities would be prohibited from being traded on a national securities exchange or in the over-the-counter trading market in the United States if we are identified as a Commission-Identified Issuer for two consecutive years in the future. Although our ordinary shares have been listed on the SEHK and AIM and the ADSs and ordinary shares are fully fungible, we cannot assure your that an active trading market for our ordinary shares on the Hong Kong Stock Exchange or AIM of the London Stock Exchange will be sustained or that the ADSs can be converted and traded with sufficient market recognition and liquidity, if our shares and ADSs are prohibited from trading in the United States. A prohibition of being able to trade in the United States would substantially impair your ability to sell or purchase our ADSs when you wish to do so, and the risk and uncertainty associated with delisting would have a negative impact on the price of our ADSs. Also, such a prohibition would significantly affect our ability to raise capital on terms acceptable to us, or at all, which would have a material adverse impact on our business, financial condition, and prospects.
58
The listings of our shares in multiple venues may adversely affect the liquidity and value of them.
Our ADSs continue to be listed on Nasdaq, and our shares continue to be admitted to trading on the AIM. Our shares were listed on the SEHK in June 2021. The listing of the shares on the AIM and the SEHK, and the ADSs on Nasdaq, may reduce the liquidity of these securities in one or each of these markets and may adversely affect the development of an active trading market for the shares in each of these markets. The price of the shares could also be adversely affected by trading on Nasdaq. Similarly, the price of the ADSs could also be adversely affected by trading on the AIM and the SEHK. We may also seek further listings on other stock exchanges such as the Shanghai Stock Exchange, which could further affect the liquidity and value of the shares and the ADSs. Furthermore, the shares trade on the SEHK largely in electronic book-entry form. However, the ADSs are backed by physical ordinary share certificates, and the depositary for our ADS program is unable to accept book-entry interests into its custody in order to issue ADSs. As a result, if a holder of the shares wishes to deposit the shares into the ADS program and hold ADSs for trading on Nasdaq or vice versa, the issuance and cancellation process may be longer than if the depositary could accept such book-entry interests.
Our largest shareholder owns a significant percentage of our ordinary shares, which may limit the ability of other shareholders to influence corporate matters.
As of February 15, 2024, Hutchison Healthcare Holdings Limited owned approximately 38.2% of our ordinary shares. Accordingly, Hutchison Healthcare Holdings Limited can influence the outcome of any corporate transaction or other matter submitted to shareholders for approval and the interests of Hutchison Healthcare Holdings Limited may differ from the interests of our other shareholders. Under our Articles of Association, certain matters, such as amendments to our amended and restated Memorandum and Articles of Association, require the approval of not less than three-fourths of votes cast by such shareholders as, being entitled so to do, vote in person (or, in the case of such shareholders as are corporations, by their respective duly authorized representative) or by proxy. Therefore, Hutchison Healthcare Holdings Limited’s approval will be required to achieve any such threshold. In addition, Hutchison Healthcare Holdings Limited has and will continue to have a significant influence over the management and the strategic direction of our company.
Substantial future sales or perceived potential sales of our ADSs, ordinary shares or other equity or equity-linked securities in the public market could cause the price of our ADSs to decline significantly.
Sales of our ADSs, ordinary shares or other equity or equity-linked securities in the public market, or the perception that these sales could occur, could cause the market price of our ADSs to decline significantly. All of our ordinary shares represented by ADSs are freely transferable by persons other than our affiliates without restriction or additional registration under the Securities Act of 1933, or the Securities Act. The ordinary shares held by our affiliates are also available for sale, subject to volume and other restrictions as applicable under Rules 144 and 701 under the Securities Act, under sales plans adopted pursuant to Rule 10b5-1 or otherwise.
We have filed with the SEC registration statements on Form F-3, commonly referred to as a “shelf registration,” that permit us to sell any number of ADSs in a registered offering at our discretion. We have completed registered offerings raising aggregate gross proceeds of approximately $537.9 million under such shelf registration statements. Furthermore, our largest shareholder has completed registered secondary offerings raising aggregate gross proceeds of approximately $310.4 million for it as a selling shareholder under a shelf registration statement. In addition, we completed our initial public offering in Hong Kong and global offering of our ordinary shares in 2021, raising aggregate gross proceeds of approximately $614.9 million, including $80.2 million through the fulfillment of the over-allotment. We may decide to conduct future offerings from time to time, and such sales could cause the price of our ADSs to decline significantly.
In connection with the issuance of ordinary shares in private placements in 2020 and 2021, we agreed to provide certain shareholders Form F-3 registration rights. Registration of the ordinary shares held by such shareholders may result in these shares becoming freely tradable without restriction under the Securities Act immediately upon the effectiveness of the registration. Sales of these shares, or the perception that such sales could occur, could cause the price of our ADSs to decline. In addition, any changes in the investment strategies or philosophies of our major shareholders may lead to the sale of our ADSs and other securities, which could cause the price of our ADSs to decline.
59
We may be at a risk of securities litigation.
Historically, securities litigation, particularly class action lawsuits brought in the United States, have often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant share price volatility in recent years. If we were to be sued, it could result in substantial costs and a diversion of management’s attention and resources, which could harm our business.
If securities analysts do not publish research or reports about our business or if they publish negative evaluations of our business, the price of our ADSs could decline.
The trading market for our ADSs will rely in part on the research and reports that industry or financial analysts publish about us or our business. We may not be able to maintain continuous research coverage by industry or financial analysts. If one or more of the analysts covering our business downgrade their evaluations of our stock, the price of our stock could decline. If one or more of these analysts cease to cover our stock, we could lose visibility in the market for our stock, which in turn could cause our stock price to decline.
As a foreign private issuer, we are not subject to certain U.S. securities law disclosure requirements that apply to a domestic U.S. issuer, which may limit the information publicly available to our shareholders.
As a foreign private issuer we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act and therefore there may be less publicly available information about us than if we were a U.S. domestic issuer. For example, we are not required to file quarterly reports on Form 10-Q. We are also not subject to the proxy rules in the United States, and we are not required to follow the related disclosure requirements with respect to our annual general meetings, including disclosing a compensation discussion and analysis. Our disclosure with respect to our annual general meetings will be governed by the AIM Rules for Companies, or the AIM Rules, listing rules in Hong Kong and Cayman Islands requirements. In addition, our officers, directors and principal shareholders are exempt from the reporting and “short-swing” profit recovery provisions of Section 16 of the Exchange Act and the rules thereunder. Therefore, our shareholders may not know on a timely basis when our officers, directors and principal shareholders purchase or sell our ordinary shares or ADSs.
As a foreign private issuer, we are permitted to adopt certain home country practices in relation to corporate governance matters that differ significantly from Nasdaq corporate governance listing standards. These practices may afford less protection to shareholders than they would enjoy if we complied fully with corporate governance listing standards.
As a foreign private issuer, we are permitted to take advantage of certain provisions in the Nasdaq listing rules that allow us to follow Cayman Islands law for certain governance matters. Certain corporate governance practices in the Cayman Islands may differ significantly from corporate governance listing standards as, except for compliance with the obligations contained in the Companies Act and directors’ general fiduciary duties and duties of care, Cayman Islands law has no corporate governance regime which prescribes specific corporate governance standards. We intend to continue to follow Cayman Islands corporate governance practices in lieu of the corporate governance requirements of the Nasdaq Global Select Market in respect of the following: (i) the majority independent director requirement under Section 5605(b)(1) of the Nasdaq listing rules, (ii) the requirement under Section 5605(d) of the Nasdaq listing rules that a remuneration committee comprised solely of independent directors governed by a remuneration committee charter oversee executive compensation and (iii) the requirement under Section 5605(e) of the Nasdaq listing rules that director nominees be selected or recommended for selection by either a majority of the independent directors or a nominations committee comprised solely of independent directors. Cayman Islands law does not impose a requirement that our board of directors consist of a majority of independent directors, nor does Cayman Islands law impose specific requirements on the establishment of a remuneration committee or nominating committee or nominating process. Therefore, our shareholders may be afforded less protection than they otherwise would have under corporate governance listing standards applicable to U.S. domestic issuers. We have voluntarily complied with the Corporate Governance Code contained in Appendix 14 of the Rules Governing the Listing of Securities on SEHK. See Item 6.C. “Board Practice—Hong Kong Corporate Governance Code” for more details.
60
We may in the future lose our foreign private issuer status under U.S. securities laws, which could result in significant additional costs and expenses.
We are a foreign private issuer as defined in the Securities Act, and therefore, we are not required to comply with all of the periodic disclosure and current reporting requirements of the Exchange Act. The determination of foreign private issuer status is made annually on the last business day of an issuer’s most recently completed second fiscal quarter, and, accordingly, the next determination will be made with respect to us on June 30, 2024. We would lose our foreign private issuer status if, for example, more than 50% of our ordinary shares are directly or indirectly held by residents of the United States on June 30, 2024 and we fail to meet additional requirements necessary to maintain our foreign private issuer status. If we lose our foreign private issuer status on this date, we will be required to file with the SEC periodic reports and registration statements on U.S. domestic issuer forms beginning on January 1, 2025, which are more detailed and extensive than the forms available to a foreign private issuer. We will also have to mandatorily comply with U.S. federal proxy requirements, and our officers, directors and principal shareholders will become subject to the short-swing profit disclosure and recovery provisions of Section 16 of the Exchange Act. In addition, we will lose our ability to rely upon exemptions from certain corporate governance requirements under the Nasdaq listing rules. As a U.S.-listed public company, should we lose our foreign private issuer status, we will incur significant additional legal, accounting and other expenses that we would not incur as a foreign private issuer.
Fluctuations in the value of the renminbi may have a material adverse effect on your investment.
The value of the renminbi against the U.S. dollar and other currencies fluctuates and is affected by, among other things, changes in China’s and international political and economic conditions and the PRC government’s fiscal and currency policies. Since 1994, the conversion of renminbi into foreign currencies, including U.S. dollars, has been based on rates set by the PBOC, which are set daily based on the previous business day’s inter-bank foreign exchange market rates and current exchange rates on the world financial markets. It is expected that China may further reform its exchange rate system in the future.
Significant revaluation of the renminbi may have a material adverse effect on your investment. For example, to the extent that we need to convert U.S. dollars into renminbi for our operations, appreciation of the renminbi against the U.S. dollar would have an adverse effect on the renminbi amount we would receive from the conversion. Conversely, if we decide to convert our renminbi into U.S. dollars, appreciation of the U.S. dollar against the renminbi would have a negative effect on the U.S. dollar amount available to us. Appreciation or depreciation in the value of the renminbi relative to the U.S. dollar would affect our financial results reported in U.S. dollar terms regardless of any underlying change in our business or results of operations. In addition, our operating transactions and assets and liabilities in the PRC are mainly denominated in renminbi. Such amounts are translated into U.S. dollars for purpose of preparing our consolidated financial statements, with translation adjustments reflected in accumulated other comprehensive income/(loss) in shareholders’ equity. We recorded a foreign currency translation gain of $3.0 million, a foreign currency translation loss of $8.5 million and a foreign currency translation loss of $6.6 million for the years ended December 31, 2021, 2022 and 2023, respectively.
Very limited hedging options are available in China to reduce our exposure to exchange rate fluctuations. To date, we have not entered into any hedging transactions in an effort to reduce our exposure to foreign currency exchange risk. While we may decide to enter into hedging transactions in the future, the availability and effectiveness of these hedges may be limited and we may not be able to adequately hedge our exposure or at all. In addition, our currency exchange losses may be magnified by PRC exchange control regulations that restrict our ability to convert renminbi into foreign currency.
We do not currently intend to pay dividends on our securities, and, consequently, your ability to achieve a return on your investment will depend on appreciation in the price of the ADSs.
We have never declared or paid any dividends on our ordinary shares. We currently intend to invest our future earnings, if any, to fund our growth. Therefore, you are not likely to receive any dividends on your ADSs at least in the near term, and the success of an investment in ADSs will depend upon any future appreciation in its value. Consequently, investors may need to sell all or part of their holdings of ADSs after price appreciation, which may never occur, to realize any future gains on their investment. There is no guarantee that the ADSs will appreciate in value or even maintain the price at which our shareholders have purchased the ADSs.
The trading prices for our ADSs may be volatile which could result in substantial losses to you.
The market price of our ADSs has been volatile. From March 17, 2016 to February 15, 2024, the closing sale price of our ADSs ranged from a high of $42.94 to a low of $7.65 per ADS.
61
The market price for our ADSs is likely to be highly volatile and subject to wide fluctuations in response to factors, including the following:
| ● | announcements of competitive developments; |
| ● | regulatory developments affecting us, our customers or our competitors; |
| ● | announcements regarding litigation or administrative proceedings involving us; |
| ● | actual or anticipated fluctuations in our period-to-period operating results; |
| ● | changes in financial estimates by securities research analysts; |
| ● | additions or departures of our executive officers; |
| ● | release or expiry of lock-up or other transfer restrictions on our outstanding ordinary shares or ADSs; and |
| ● | sales or perceived sales of additional ordinary shares or ADSs. |
In addition, the securities markets have from time to time experienced significant price and volume fluctuations that are not related to the operating performance of particular companies. Prolonged global capital markets volatility may affect overall investor sentiment towards our ADSs, which would also negatively affect the trading prices for our ADSs.
The triple listing of our ordinary shares and the ADSs may adversely affect the liquidity and value of the ADSs.
Our ordinary shares are listed on the AIM market and on the SEHK. The triple listing of our ordinary shares and the ADSs may dilute the liquidity of these securities in one or more of these markets and may adversely affect the development of an active trading market for the ADSs in the United States or shares in Hong Kong and the United Kingdom. The price of the ADSs could also be adversely affected by trading in our ordinary shares on the AIM market and the SEHK.
Fluctuations in the exchange rate between the U.S. dollar, Hong Kong dollar and the pound sterling may increase the risk of holding the ADSs.
Our share price is quoted on the SEHK and AIM market in Hong Kong dollar and pence sterling, respectively, while the ADSs trade on Nasdaq in U.S. dollars. Fluctuations in the exchange rate between the U.S. dollar, Hong Kong dollar and the pound sterling may result in temporary differences between the value of the ADSs and the value of our ordinary shares, which may result in heavy trading by investors seeking to exploit such differences. In addition, as a result of fluctuations in the exchange rate between the U.S. dollar, Hong Kong dollar and the pound sterling, the U.S. dollar equivalent of the proceeds that a holder of the ADSs would receive upon the sale in Hong Kong of any ordinary shares or in the United Kingdom of any ordinary shares withdrawn from the depositary and the dollar equivalent of any cash dividends paid in Hong Kong dollar or pound sterling on our shares represented by the ADSs could also decline.
Securities traded on the AIM market or on the SEHK may carry or be perceived to carry a higher risk than shares traded on other exchanges and may impact the value of your investment.
Our ordinary shares are currently traded on the AIM market and on the SEHK. Investment in equities traded on AIM and the SEHK may be perceived by some to carry a higher risk than an investment in equities quoted on exchanges, such as the New York Stock Exchange or the Nasdaq. You should be aware that the value of our ordinary shares may be influenced by many factors, some of which may be specific to us and some of which may affect AIM-listed or Hong Kong-listed companies generally, including the depth and liquidity of the market, our performance, a large or small volume of trading in our ordinary shares, legislative changes and general economic, political or regulatory conditions, and that the prices may be volatile and subject to extensive fluctuations. Therefore, the market price of our ordinary shares underlying the ADSs may not reflect the underlying value of our company.
62
The depositary for our ADSs gives us a discretionary proxy to vote our ordinary shares underlying your ADSs if you do not vote at shareholders’ meetings, except in limited circumstances, which could adversely affect your interests.
Under the deposit agreement for the ADSs, the depositary gives us a discretionary proxy to vote our ordinary shares underlying your ADSs at shareholders’ meetings if you do not vote, unless:
| ● | we do not wish a discretionary proxy to be given; |
| ● | we are aware or should reasonably be aware that there is substantial opposition as to a matter to be voted on at the meeting; or |
| ● | a matter to be voted on at the meeting would materially and adversely affect the rights of shareholders. |
The effect of this discretionary proxy is that you cannot prevent our ordinary shares underlying your ADSs from being voted, absent the situations described above, and it may make it more difficult for shareholders to influence the management of our company. Holders of our ordinary shares are not subject to this discretionary proxy.
Holders of ADSs have fewer rights than shareholders and must act through the depositary to exercise their rights.
Holders of our ADSs do not have the same rights as our shareholders and may only exercise the voting rights with respect to the underlying ordinary shares in accordance with the provisions of the deposit agreement. Under our amended and restated Memorandum and Articles of Association, an annual general meeting shall be called by notice with not less than 21 clear days, and all other general meetings (including an extraordinary general meeting) shall be called by notice with not less than 14 clear days. When a general meeting is convened, you may not receive sufficient notice of a shareholders’ meeting to permit you to withdraw the ordinary shares underlying your ADSs to allow you to vote with respect to any specific matter. If we ask for your instructions, we will give the depositary notice of any such meeting and details concerning the matters to be voted upon at least 30 days in advance of the meeting date and the depositary will send a notice to you about the upcoming vote and will arrange to deliver our voting materials to you. The depositary and its agents, however, may not be able to send voting instructions to you or carry out your voting instructions in a timely manner. We will make all reasonable efforts to cause the depositary to extend voting rights to you in a timely manner, but we cannot assure you that you will receive the voting materials in time to ensure that you can instruct the depositary to vote the ordinary shares underlying your ADSs. Furthermore, the depositary will not be liable for any failure to carry out any instructions to vote, for the manner in which any vote is cast or for the effect of any such vote. As a result, you may not be able to exercise your right to vote and you may lack recourse if your ADSs are not voted as you request. In addition, in your capacity as an ADS holder, you will not be able to call a shareholders’ meeting.
You may not receive distributions on our ADSs or any value for them if such distribution is illegal or if any required government approval cannot be obtained in order to make such distribution available to you.
Although we do not have any present plan to pay any dividends, the depositary of our ADSs has agreed to pay to you the cash dividends or other distributions it or the custodian receives on ordinary shares or other deposited securities underlying our ADSs, after deducting its fees and expenses and any applicable taxes and governmental charges. You will receive these distributions in proportion to the number of ordinary shares your ADSs represent. However, the depositary is not responsible if it decides that it is unlawful or impractical to make a distribution available to any holders of ADSs. For example, it would be unlawful to make a distribution to a holder of ADSs if it consists of securities whose offering would require registration under the Securities Act but is not so properly registered or distributed under an applicable exemption from registration. The depositary may also determine that it is not reasonably practicable to distribute certain property. In these cases, the depositary may determine not to distribute such property. We have no obligation to register under the U.S. securities laws any offering of ADSs, ordinary shares, rights or other securities received through such distributions. We also have no obligation to take any other action to permit the distribution of ADSs, ordinary shares, rights or anything else to holders of ADSs. This means that you may not receive distributions we make on our ordinary shares or any value for them if it is illegal or impractical for us to make them available to you. These restrictions may cause a material decline in the value of our ADSs.
63
Your right to participate in any future rights offerings may be limited, which may cause dilution to your holdings.
We may from time to time distribute rights to our shareholders, including rights to acquire our securities. However, we cannot make rights available to you in the United States unless we register the rights and the securities to which the rights relate under the Securities Act or an exemption from the registration requirements is available. Also, under the deposit agreement, the depositary bank will not make rights available to you unless either both the rights and any related securities are registered under the Securities Act, or the distribution of them to ADS holders is exempted from registration under the Securities Act. We are under no obligation to file a registration statement with respect to any such rights or securities or to endeavor to cause such a registration statement to be declared effective. Moreover, we may not be able to establish an exemption from registration under the Securities Act. If the depositary does not distribute the rights, it may, under the deposit agreement, either sell them, if possible, or allow them to lapse. Accordingly, you may be unable to participate in our rights offerings and may experience dilution in your holdings.
If we are a passive foreign investment company for any taxable year, U.S. investors could be subject to adverse U.S. federal income tax consequences.
The rules governing passive foreign investment companies, or PFICs, can have adverse U.S. federal income tax consequences for U.S. investors of non-U.S. corporations. The PFIC status of a non-U.S. corporation for any taxable year depends upon the composition of its income and assets, the value of its assets and the classification of items of its income and assets as active or passive under the PFIC rules, as discussed further in Item 10.E. “Taxation—U.S. Taxation—Material U.S. Federal Income Tax Considerations with Respect to Ordinary Shares and ADSs.” Based on the composition of our income and assets and the estimated average value of our assets (including goodwill and other intangible assets), we believe that we were not a PFIC for our taxable year ended December 31, 2023. However, our PFIC status is a factual determination that is made on an annual basis and depends on particular facts and circumstances (such as the value of our assets, including goodwill and other intangible assets). We hold a substantial amount of cash and financial investments and while this continues to be the case, our PFIC status depends primarily on the average value of our goodwill and other intangible assets. The value of our goodwill and other intangible assets may be determined, in large part, by reference to our market capitalization, which has been, and may continue to be, volatile. Therefore, if our market capitalization declines we may be or become a PFIC. In addition, there is uncertainty as to how to apply the PFIC rules for purposes of classifying certain of our income and assets as active or passive. Furthermore, the proportionate value of our passive assets may increase over time if the value of our ownership stake in any other company in which we own less than 25% (by value) increases. In light of the foregoing, no assurance can be provided that we were not, or will not be, a PFIC for any taxable year.
If we are or become a PFIC, U.S. investors in our ordinary shares and ADSs generally will be subject to adverse U.S. federal income tax consequences, such as ineligibility for any preferential tax rates on capital gains or on actual or deemed dividends, interest charges on certain taxes treated as deferred, and additional reporting requirements under U.S. federal income tax laws and regulations. We do not expect to provide the information regarding our income that would be necessary in order for a U.S. investor to make a qualified electing fund, or QEF, election if we are a PFIC for any taxable year. U.S. investors in our ordinary shares or ADSs should consult their tax advisors regarding all aspects of the application of the PFIC rules to their ordinary shares and ADSs.
Under certain attribution rules, certain of our non-U.S. subsidiaries are expected to be treated as “controlled foreign corporations” for U.S. federal income tax purposes, and, as a result, there could be adverse U.S. federal income tax consequences to U.S. investors that own (directly or indirectly) our ordinary shares or ADSs and are treated as “Ten Percent Shareholders.”
Certain “Ten Percent Shareholders” (as defined below) in a non-U.S. corporation that is a “controlled foreign corporation” (a “CFC”) for U.S. federal income tax purposes generally are required to include in income for U.S. federal income tax purposes their pro rata share of the CFC’s “Subpart F income,” investment of earnings in U.S. property, and “global intangible low-taxed income,” even if the CFC has made no distributions to its shareholders. A non-U.S. corporation generally will be a CFC for U.S. federal income tax purposes if Ten Percent Shareholders own, directly, indirectly or constructively (through attribution), more than 50% of either the total combined voting power of all classes of stock of such corporation entitled to vote or of the total value of the stock of such corporation. A “Ten Percent Shareholder” is a United States person (as defined by the U.S. Internal Revenue Code of 1986, as amended) that owns directly or indirectly, or is considered to own constructively, 10% or more of the total combined voting power of all classes of stock entitled to vote of such corporation or 10% or more of the total value of the stock of such corporation. We are not expected to be a CFC. However, under certain “downward attribution” rules, certain of our non-U.S. subsidiaries are expected to be treated as CFCs by virtue of being constructively owned by our U.S. subsidiaries. As a non-U.S. company, we do not intend to take these U.S. tax rules into consideration in structuring its operations, nor do we intend to provide information to Ten Percent Shareholders that may be required in order for those shareholders to properly report their U.S. taxable income with respect to our operations. U.S. investors that are or may become Ten Percent Shareholders who directly or indirectly own our ordinary shares or ADSs should consult their tax advisors with respect to the application of the CFC rules to them.
64
We may be treated as a resident enterprise for U.K. corporate tax purposes, and our global income may therefore be subject to U.K. corporation tax.
U.K. resident companies are taxable in the United Kingdom on their worldwide profits. A company incorporated outside of the United Kingdom would be regarded as a resident if its central management and control resides in the United Kingdom. The place of central management and control generally means the place where the high-level strategic decisions of a company are made.
We are an investment holding company incorporated in the Cayman Islands and are admitted to trading on the AIM market of the London Stock Exchange or the AIM market. Our central management and control resides in Hong Kong, and therefore we believe that we are not a U.K. resident for corporate tax purposes. However, the tax resident status of a non-resident entity could be challenged by the U.K. tax authorities.
If the U.K. tax authorities determine that we are a U.K. tax resident, our profits will be subject to U.K. Corporation Tax rate at 19% for taxable profits below GBP 50,000 and 25% for taxable profits above GBP 250,000, subject to the potential availability of certain exemptions related to dividend income and capital gains. This may have a material adverse effect on our financial condition and results of operations.
You may have difficulty enforcing judgments obtained against us.
We are a company incorporated under the laws of the Cayman Islands, and substantially all of our assets are located outside the United States. Substantially all of our current operations are conducted in the PRC. In addition, most of our directors and officers are nationals and residents of countries other than the United States. A substantial portion of the assets of these persons are located outside the United States. As a result, it may be difficult for you to effect service of process within the United States upon these persons. It may also be difficult for you to enforce in U.S. courts judgments obtained in U.S. courts based on the civil liability provisions of the U.S. federal securities laws against us and our officers and directors, all of whom are not residents in the United States and whose assets are located outside the United States. In addition, there is uncertainty as to whether the courts of the Cayman Islands or the PRC would recognize or enforce judgments of U.S. courts against us or such persons predicated upon the civil liability provisions of the securities laws of the United States or any state.
You may be subject to limitations on transfers of your ADSs.
Your ADSs are transferable on the books of the depositary. However, the depositary may close its transfer books at any time or from time to time when it deems expedient in connection with the performance of its duties. In addition, the depositary may refuse to deliver, transfer or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary deems it advisable to do so because of any requirement of law or of any government or governmental body, or under any provision of the deposit agreement, or for any other reason.
It may be difficult for overseas regulators to conduct investigations or collect evidence within China.
Shareholder claims or regulatory investigation that are common in the United States generally are difficult to pursue as a matter of law or practicality in China. For example, in China, there are significant legal and other obstacles to providing information needed for regulatory investigations or litigation initiated outside China. Although the authorities in China may establish a regulatory cooperation mechanism with the securities regulatory authorities of another country or region to implement cross-border supervision and administration, such cooperation with the securities regulatory authorities in the Unities States may not be efficient in the absence of mutual and practical cooperation mechanisms. Furthermore, according to Article 177 of the PRC Securities Law, or Article 177, which became effective in March 2020, no overseas securities regulator is allowed to directly conduct investigations or evidence collection activities within the territory of the PRC. While detailed interpretations of or implementation rules under Article 177 have yet to be promulgated, the possible inability for an overseas securities regulator to directly conduct investigations or evidence collection activities within China may further increase difficulties you may face in protecting your interests.
65
We are a Cayman Islands company. As judicial precedent regarding the rights of shareholders under Cayman Islands law is different from U.S. law, English law or Hong Kong law, shareholders may have different shareholder rights than they would have under U.S. law, English law or Hong Kong law and may face difficulties in protecting your interests.
We are an exempted company with limited liability incorporated in the Cayman Islands. Our corporate affairs are governed by our Articles of Association (as may be further amended from time to time), the Companies Act (As Revised) of the Cayman Islands and the common law of the Cayman Islands. The rights of shareholders to take action against the directors, actions by minority shareholders and the fiduciary responsibilities of our directors are to a large extent governed by the common law of the Cayman Islands. This common law is derived in part from comparatively limited judicial precedent in the Cayman Islands as well as from English common law, which has persuasive, but not binding, authority on a court in the Cayman Islands. The laws of the Cayman Islands relating to the protection of the interests of minority shareholders differ in some aspects from those in the United States, the United Kingdom and Hong Kong. Such differences mean that the remedies available to our minority shareholders may be different from those they would have under the laws of United States, the United Kingdom, Hong Kong or other jurisdictions. In addition, some states in the United States, such as Delaware, have more fully developed and judicially interpreted bodies of corporate law than the Cayman Islands.
In addition, as a Cayman Islands exempted company, other than right to inspect and take copies of our register of members contained in our articles of association, our shareholders have no general rights under Cayman Islands law to inspect corporate records and accounts or to obtain copies of lists of shareholders of these companies with the exception that the shareholders may request a copy of the Articles of Association. Our directors have discretion under our Articles of Association to determine whether or not, and under what conditions, our corporate records may be inspected by our shareholders, but are not obliged to make them available to our shareholders. This may make it more difficult for you to obtain the information needed to establish any facts necessary for a shareholder motion or to solicit proxies from other shareholders in connection with a proxy contest. As a Cayman Islands company, we may not have standing to initiate a derivative action in U.S. federal courts, English courts or Hong Kong courts. As a result, you may be limited in your ability to protect your interests if you are harmed in a manner that would otherwise enable you to sue in U.S. federal courts, English courts or Hong Kong courts. In addition, shareholders of Cayman Islands companies may not have standing to initiate a shareholder derivative action in U.S. federal courts, English courts or Hong Kong courts.
Most of our directors and executive officers reside outside of the United States and a substantial portion of their assets are located outside of the United States. As a result, it may be difficult or impossible for you to bring an action against us or against these individuals in the United States in the event that you believe that your rights have been infringed under the securities laws of the United States or otherwise. In addition, some of our operating subsidiaries are incorporated in China. To the extent our directors and executive officers reside in China or their assets are located in China, it may not be possible for investors to effect service of process upon us or our management inside China. Even if you are successful in bringing an action, the laws of the Cayman Islands and China may render you unable to enforce a judgment against our assets or the assets of our directors and officers. Whilst there is no statutory recognition in the Cayman Islands of judgments obtained in the United States, Hong Kong or China, the courts of the Cayman Islands would recognize as a valid judgment, a final and conclusive judgment in personam obtained in such courts against the Company under which a sum of money is payable (other than a sum of money payable in respect of multiple damages, taxes or other charges of a like nature or in respect of a fine or other penalty) or, in certain circumstances, an in personam judgment for non-monetary relief, and would give a judgment based thereon provided that (a) such courts had proper jurisdiction over the parties subject to such judgment; (b) such courts did not contravene the rules of natural justice of the Cayman Islands; (c) such judgment was not obtained by fraud; (d) the enforcement of the judgment would not be contrary to the public policy of the Cayman Islands; (e) no new admissible evidence relevant to the action is submitted prior to the rendering of the judgment by the courts of the Cayman Islands; and (f) there is due compliance with the correct procedures under the laws of the Cayman Islands.
As a result of all of the above, public shareholders may have more difficulty in protecting their interests in the face of actions taken by management, members of the board of directors or controlling shareholders than they would as public shareholders of an English company, a U.S. company or a Hong Kong company.
66
We cannot assure you that our ordinary shares will remain listed on the AIM or the SEHK or our ADSs will remain listed on Nasdaq.
Although it is currently intended that our ordinary shares and ADSs will remain listed on the AIM, the SEHK and Nasdaq, as applicable, there is no guarantee of the continued listing of our securities on any of these exchanges. We may decide at some point in the future to delist voluntarily (subject to the applicable regulatory requirements) from one or more of these exchanges, or we may be delisted involuntarily if, among other factors, we do not continue to satisfy the listing requirements of the applicable exchange or comply with applicable law. For example, we could be delisted from the Nasdaq if the PCAOB continues to be unable to inspect our independent registered public accounting firm for two consecutive years. The AIM Rules for companies provide that a voluntary cancellation of admission to AIM is conditional upon the consent of not less than 75% of votes cast by its shareholders at a general meeting unless the London Stock Exchange otherwise agrees. Circumstances where the London Stock Exchange might otherwise agree that shareholder consent at a general meeting is not required would include the situation where the AIM securities are already admitted to trading on an “AIM Designated Market” (which includes Nasdaq) to enable shareholders to trade their AIM securities in the future. The SEHK rules allow an issuer whose primary listing is on SEHK and which has an alternative listing on another stock exchange to withdraw its listing with the prior approval of shareholders by ordinary resolution obtained at a duly convened meeting of the shareholders and the satisfaction of other requirements. SEHK may also cancel the listing of any securities that have been suspended from trading for a continuous period of 18 months. We cannot predict the effect a delisting of our shares on the SEHK or AIM market or our ADSs on Nasdaq would have on the market price of our shares and/or ADSs. We may also seek further listings on other stock exchanges such as the Shanghai Stock Exchange. However, there is no assurance that we would proceed with a listing and if we do proceed, that a listing would materialize.
The characteristics of the Hong Kong, U.S. and U.K. capital markets are different.
The SEHK, Nasdaq and the AIM have different trading hours, trading characteristics (including trading volume and liquidity), trading and listing rules, market regulations, and investor bases (including different levels of retail and institutional participation). As a result of these differences, the trading prices of the shares and the ADSs might not be the same, even allowing for currency differences. Circumstances peculiar to the U.S. capital markets could materially and adversely affect the price of the shares. Because of the different characteristics of the Hong Kong, U.S. and U.K. equity markets, the historical market prices of our securities may not be indicative of the performance of the shares.
We are subject to Hong Kong, Nasdaq and AIM listing and regulatory requirements concurrently.
As we are listed on the SEHK, the Nasdaq and the AIM, we are required to comply with the listing rules (where applicable) and other regulatory regimes of each stock exchange, unless otherwise agreed by the relevant regulators. We may also seek further listings on other stock exchanges such as the Shanghai Stock Exchange. Accordingly, we may incur additional costs and resources in complying with the requirements of each stock exchange.
ITEM 4. INFORMATION ON THE COMPANY
A. History and Development of the Company.
HUTCHMED (China) Limited (formerly known as Hutchison China MediTech Limited) was incorporated in the Cayman Islands on December 18, 2000 as an exempted company with limited liability under the Companies Act (As Revised) of the Cayman Islands. Our company was founded by a wholly owned subsidiary of CK Hutchison, a multinational conglomerate with operations in over 50 countries. CK Hutchison is the ultimate parent company of our largest shareholder Hutchison Healthcare Holdings Limited.
We launched our novel drug research and development operations in 2002 with the establishment of our subsidiary HUTCHMED Limited, which is focused on discovering, developing and marketing drugs for the treatment of cancer and immunological diseases. A dozen of our in-house discovered drug candidates have entered clinical trials around the world and three have so far been approved for sale. Since 2001, we have also developed drug marketing and distribution platforms in China, which primarily focus on prescription drug and consumer health products through several joint ventures and subsidiary companies and are included in our Other Ventures.
We listed our ordinary shares on the AIM market in 2006, ADSs on the Nasdaq Global Select Market in 2016 and our ordinary shares on the SEHK in 2021.
67
On March 4, 2021 we announced the consolidation of the two corporate identities that we have used since our inception. Hutchison China MediTech, or Chi-Med, which had been used as our group identity, while Hutchison MediPharma had been the identity of our novel drug research and development operations under which our oncology products had been developed and marketed. The brand HUTCHMED immediately replaced Chi-Med as our abbreviated name, and we changed our group company name at our Annual General Meeting in April 2021 from Hutchison China MediTech Limited to HUTCHMED (China) Limited.
As our focus is the discovery and development of novel therapies in oncology and immunology, we recently disposed of our interest in some non-core operations which we believe allows us to focus resources on our primary aim of accelerating investment in our Oncology/Immunology assets. In September 2021, we disposed of our investment in Hutchison Baiyunshan, our non-core and non-consolidated over-the-counter drug joint venture business. In December 2023, we disposed of our interests in our consolidated joint venture Hutchison Hain Organic and our wholly owned subsidiary HUTCHMED Science Nutrition. We are also considering divesting other non-core businesses under our Other Ventures, including Shanghai Hutchison Pharmaceuticals. Our principal executive offices are located at 48th Floor, Cheung Kong Center, 2 Queen’s Road Central, Hong Kong. Our telephone number at that address is +852 2121 8200. The address of our registered office in the Cayman Islands is P.O. Box 309, Ugland House, Grand Cayman, KY1-1104, Cayman Islands.
See Item 5.B. “Liquidity and Capital Resources” for details on our capital expenditures for the years ended December 31, 2021, 2022 and 2023.
We are subject to the informational requirements of the Exchange Act and are required to file reports and other information with the SEC. The SEC maintains a website at www.sec.gov that contains reports, proxy and information statements, and other information regarding registrants that make electronic filings with the SEC using its EDGAR system. We also make available on our website’s investor relations page, free of charge, our annual report and the text of our reports on Form 6-K, including any amendments to these reports, as well as certain other SEC filings, as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. The address for our investor relations page is www.hutch-med.com/shareholder-information. The information contained on our website is not incorporated by reference in this annual report.
B. Business Overview.
Overview
We are a global commercial-stage biopharmaceutical company focused on the discovery, development and commercialization of targeted therapies and immunotherapies for the treatment of patients with cancer and immunological diseases. Our company started in China in 2000 and has since developed fully integrated capabilities and expanded oncology and immunology drug development operations globally. Our operational achievements and capabilities to date include:
Broad pipeline of differentiated targeted therapies and immunotherapies built for the global market. We have a pipeline of differentiated drug candidates covering both novel and validated targets, including MET, VEGFR, FGFR, CSF-1R, Syk, EZH2, PI3Kδ, IDH, ERK, BTK, CD47 and SHP2. The aim of our research is to develop drugs with high selectivity and superior safety profiles, a key benefit of which is that our drug candidates have the potential to be effectively paired with other oncology and immunology therapies at effective dosages with fewer side effects.
68
Commercially launching products while continuing to discover new assets. In China, three of our internally developed drugs, savolitinib, fruquintinib and surufatinib are commercially available to patients as Orpathys, Elunate and Sulanda, respectively. Outside of China, fruquintinib has been approved by U.S. FDA in November 2023 and marketed as Fruzaqla by our partner Takeda. To accelerate the availability of our innovative medicines for patients globally, we seek partnerships to commercialize our drugs outside of China, such as our partnership with AstraZeneca on savolitinib and with Takeda on fruquintinib. In addition, we have more than ten other drug candidates that have entered clinical development and several pre-clinical drug candidates.
Comprehensive global in-house discovery and development capabilities. We have a comprehensive drug discovery and development operation covering chemistry, biology, pharmacology, toxicology, chemistry and manufacturing controls for clinical and commercial supply, clinical and regulatory and other functions. It is led by a team of approximately 900 scientists, who have created one of the broadest global clinical pipelines among our peer oncology and immunology focused biotechnology companies. Currently, we are conducting approximately 40 different clinical studies in oncology patients globally, including over 15 Phase III registration and Phase II registration-intent studies underway.
Long-standing drug marketing and distribution experience to support the realization of in-house oncology innovations in China. We have built large-scale and profitable drug marketing and distribution capabilities through our Other Ventures operations, which primarily manufacture, market and distribute prescription drugs in China. Our more than 20 years of track record and deep institutional knowledge of the drug marketing and distribution process are being leveraged to bring our in-house oncology innovations to patients. We have built and continue to expand our in-house oncology drug sales team to approximately 930 persons at end of 2023 to support the commercialization of fruquintinib, surufatinib and our other innovative drugs, if approved, throughout China. Our oncology drug sales team covers approximately 3,500 hospitals and over 39,000 oncology physicians in China, a network that we estimate represents over 90% of oncology drug sales in China.
Our Strategies
Our vision is to be a global leader in the discovery, development and commercialization of targeted therapies and immunotherapies for the treatment of patients with cancer and immunological diseases. Key elements of our strategy are to:
Realize the global potential of our oncology drug candidates
Our first wave of innovation - namely, savolitinib (partnered globally with AstraZeneca), fruquintinib (partnered in China with Eli Lilly and outside of China with Takeda) and surufatinib (unpartnered) - are either commercialized, under review for marketing authorization or in registrational studies in multiple jurisdictions. In tandem with our ongoing progression of such drugs, we will continue to invest in the future with our deep pipeline of unpartnered next wave of oncology assets for which we own all rights globally and have significant flexibility in driving their development. We intend to accelerate our global drug development by leveraging our advanced clinical trial data from China, selectively conducting early-stage and proof-of-concept clinical trials in other jurisdictions so that the programs progress globally, then form partnerships to complete late-stage development and/or commercial launch outside China.
Continue designing and creating molecules to develop into medicines with specific and differentiated characteristics for the benefit of patients
We believe our world-class drug discovery engine is our key competitive advantage. We strive to create differentiated novel oncology and immunology treatments with global potential. Our drug discovery team has utilized our expertise in advanced medicinal chemistry to develop next-generation TKI that have both high selectivity and superior pharmacokinetic properties. Equally importantly, we will continue to design chemical and biologic drug candidates with profiles that allow them to be used in innovative combinations with other selective inhibitors, chemotherapy agents and immunotherapies. Such combination therapies enable treatment of cancer via multiple pathways and modalities simultaneously, which has the potential to significantly improve treatment outcomes.
We plan to continue to build out our global pipeline of self-discovered drug candidates by advancing a rich pipeline of early-stage drug candidates, which include small molecule drugs targeting new pathways and biologics addressing novel targets designed for use in combination with our small molecules, as well as potentially a broad range of third-party therapies.
69
Build and scale our manufacturing and commercialization capabilities
We plan to leverage our long-standing drug marketing and distribution know-how and infrastructure to support our innovative oncology product launches, focusing in particular on the Chinese market. We have a more than 20-year track record of marketing and selling products in China. We aim to steadily grow our in-house oncology drug sales team in China, currently over 900 at the end of 2023. Outside of China, we look to form collaborations with leading biopharmaceutical companies and/or contract sales organizations to fully realize the value of our assets. We will also continue to enhance our global supply chain to support the sales of our approved drugs, including through our new manufacturing plant in Shanghai and by working with third-party manufacturers.
Identify China business development opportunities to complement our internal research and development activities
We plan to explore opportunities to in-license complementary late-stage drug candidates in China to supplement our in-house research and development capabilities, with a focus on drug candidates with the potential to both complement our existing drug pipeline including through having synergistic effects and augment our oncology commercial portfolio, such as Tazverik from Ipsen. In addition, we expect to progress some of our drug candidates by pursuing business development opportunities with other biopharmaceutical companies in China such as our collaborations to evaluate combining fruquintinib with anti-PD-1 antibodies for the treatment of various solid tumor cancers. We will also continue to work with our partners, AstraZeneca, Eli Lilly and Takeda, to optimize the potential of our drug candidates savolitinib (globally with AstraZeneca) and fruquintinib (outside China with Takeda and in China with Eli Lilly).
Capitalize on regulatory reforms currently underway in China aimed at addressing existing unmet medical needs and improving the health of its people
We believe the Chinese oncology market, which comprises approximately a quarter of the global oncology patient population, represents a substantial and fast-growing market opportunity. Over the past decade, the PRC government has endeavored to foster an innovative biopharmaceutical ecosystem, and in the last few years, the pace of reforms has accelerated with a clear focus on providing Chinese patients access to world-class oncology therapies through expanded insurance reimbursement and reduced time for clinical trials and drug approvals. As a result, the oncology drug market in China is growing rapidly. Having invested in drug innovation in China for over 20 years, beginning at a time when almost no other domestic companies were involved in innovative oncology research, we believe we are well positioned to capture this market opportunity.
Oncology Commercial Operations
Fruquintinib (Elunate in China, Fruzaqla in the U.S.)
Elunate is approved for the treatment of third-line metastatic CRC for which there is an approximate incidence of 105,000 new patients per year in China. In 2023, Elunate in China achieved in-market sales of $107.5 million, up 15% (22 % at CER) versus 2022 ($93.5 million). In China, Elunate is the leading treatment for late-stage CRC with 47% of 3L treated patient share according to an IQVIA tracking study in Q2 2023.
Under the terms of our agreement with Lilly, HUTCHMED manages all on-the-ground medical detailing, promotion and local and regional marketing activities for Elunate in China. We consolidate as revenue approximately 70-80% of Elunate in-market sales from manufacturing fees, service fees and royalties paid to us by Lilly. In 2023, we consolidated $83.2 million in revenue for Elunate, equal to 77% of in-market sales.
Following negotiations with the China NHSA, Elunate continues to be included in the NRDL for a new two-year term starting in January 2024 at the same price as the 2023 NRDL price.
Fruquintinib is being marketed by our partner Takeda outside of China. Takeda launched fruquintinib (Fruzaqla) in the U.S. within 24 hours after it was approved for previously-treated metastatic CRC on November 8, 2023, with the first prescription received a day after approval. According to Takeda, uptake has been strong, with new patient starts exceeding expectations, and additional regulatory applications progressing as expected including in the EU and Japan. Since its launch until the end of 2023, Fruzaqla achieved in-market U.S. sales of $15.1 million.
70
This U.S. patient uptake was in parallel to the rapid inclusion of fruquintinib to the 2023 “NCCN Clinical Practice Guidelines for Colon Cancer” and the 2023 “NCCN Clinical Practice Guidelines for Rectal Cancer” on November 16, 2023. Fruquintinib has also been successfully recommended in six other major treatment guidelines for colorectal cancer. These will continue to drive awareness and usage of fruquintinib among doctors and patients.
In January 2024, Elunate was approved in the Hong Kong Special Administrative Region. This was the first medicine to be approved under the new mechanism for registration of new drugs (“1+” mechanism). Colorectal cancer was the second most common cancer in Hong Kong in 2021, with about 5,900 new patients diagnosed and associated with about 2,300 deaths.
Surufatinib (Sulanda in China)
Sulanda was launched in 2021 for the treatment of all advanced NETs for which there is an approximate incidence of 34,000 new patients per year in China.
Total in-market sales in 2023 increased by 36% (43% at CER) to $43.9 million (2022: $32.3 million). According to IQVIA tracking study report in the fourth quarter of 2023, Sulanda maintained its position in the market with 21% prescription share in NET treatment, ahead of Sutent and Afinitor. Following negotiations with the China NHSA, Sulanda continues to be included in the NRDL starting in January 2024. For this renewal, we kept the same price with 2023 NRDL price.
Surufatinib has been successfully recommended in 2023 Chinese medical association consensus for standardized diagnosis and treatment of pancreatic cancer neuroendocrine neoplasms and four other treatment guidelines for neuroendocrine tumors. As a result, doctors’ acceptance and patients’ access to Sulanda continues to increase.
Savolitinib (Orpathys in China)
Orpathys is the first-in-class selective MET inhibitor to be approved in China, launched and marketed by our partner, AstraZeneca for patients with MET exon 14 skipping alteration NSCLC. More than a third of the world’s lung cancer patients are in China. Among those with NSCLC globally, approximately 2-3% have tumors with MET exon 14 skipping alterations.
In 2021, 2022 and the first two months of 2023, Orpathys was sold as a self-pay drug. Following negotiations with the China NHSA in January 2023, Orpathys has been included in the updated NRDL since March 1, 2023 at a 38% discount relative to the self-pay price, broadening patient access to this medicine. Sales in 2023 were impacted by customary channel fluctuations following the announcement (in January 2023) and implementation of the NRDL listing (in March 2023), with increased volume in the latter part of 2023. In-market sales for Orpathys increased 12% (increased 19% at CER) in 2023 to $46.1 million (2022: $41.2 million) resulting in our consolidation of $28.9 million (2022: $22.3 million) in primarily revenue from manufacturing fees and royalties. Sales in the second, third and fourth quarters of 2023 were substantially higher than in the second, third and fourth quarters of 2022 before NRDL listing, increasing 104% by volume.
Market understanding of the need for MET testing has improved significantly, with approximately half of new advanced/relapsed NSCLC patients in China being tested. In the National Health Commission’s Treatment Guidelines for Primary Lung Cancer 2022 and the China Medical Association Oncology Committee Lung Cancer Group’s China Medical Association Guideline for Clinical Diagnosis and Treatment of Lung Cancer, Orpathys was identified as the only targeted therapy recommended for MET exon 14 patients, while a similar guideline from CSCO also recommended Orpathys as the standard of care for such patients. As MET testing awareness and access increases, more patients are expected to be prescribed a selective MET inhibitor.
In March 2023, Orpathys was also approved in the Macau Special Administrative Region.
71
Tazemetostat (Tazverik in Hainan and Macau, China; the U.S. and Japan)
In May 2022, Tazverik was approved by the Health Commission and Medical Products Administration of Hainan Province to be used in the Hainan Boao Lecheng International Medical Tourism Pilot Zone (Hainan Pilot Zone), under the Clinically Urgently Needed Imported Drugs scheme, for the treatment of certain patients with epithelioid sarcoma and follicular lymphoma consistent with the label as approved by the FDA. Launched in 2013 and located in China, the Hainan Pilot Zone is a destination for international medical tourism and global hub for scientific innovation, welcoming 83,900 medical tourists in 2020, according to official data. Tazemetostat was included in the 2022 CSCO guidelines for epithelioid sarcoma. 16 epithelioid sarcoma patients began treatment in 2023 (2022: 3). Tazemetostat is included in the 2023 CSCO guideline for follicular lymphoma.
In March 2023, Tazverik was approved in the Macau Special Administrative Region.
Clinical Drug Development Summary
We are the Marketing Authorization Holder in China of three internally discovered and developed innovative oncology medicines, savolitnib, fruquintinib and surufatinib, which are marketed as Orpathys, Elunate and Sulanda, respectively. Besides the three marketed drugs, we have additional drug candidates in earlier stage clinical development. Several of our oncology drug candidates are in development outside China including savolitinib, for which we are in a global partnership with AstraZeneca, and fruquintinib, for which we have licensed rights outside of China to Takeda and which has been approved by the FDA in the United States.
72
The following table summarizes the status of our clinical drug portfolio’s development as of the date of the filing of this annual report:

* Phase II registration-intent study subject to regulatory discussion.
73
** In planning.
Ù In recent discussions with China NMPA, it is clear that randomized study is now required to support registration. In view of the changing regulatory requirement, we are currently evaluating the clinical development plan and regulatory guidance before deciding the regulatory strategy for this indication.
Notes: | AITL: angioimmunoblastic T-cell lymphoma; AML: acute myeloid leukemia; BC: breast cancer; BTC: biliary tract cancer; BTK: Bruton’s tyrosine kinase; CMML: chronic myelomonocytic leukemia; CRC: colorectal cancer; CSF-1R: colony-stimulating factor 1 receptor; EGFR: epidermal growth factor receptor; EGFRm: epidermal growth factor receptor mutation-positive; EMC: endometrial cancer; ERK: extracellular signal-regulated kinase; ESCC: esophageal squamous cell carcinoma; EZH2: enhancer of zeste homolog 2; FGFR1: fibroblast growth factor receptor 1; FL: follicular lymphoma; GC: gastric cancer; GI: gastrointestinal; HCC: hepatocellular carcinoma; HL: Hodgkin lymphoma; IDH 1/2: isocitrate dehydrogenase 1/2; IHCC: intrahepatic cholangiocarcinoma; ITP: immune thrombocytopenic purpura; MAA: Marketing Authorization Application; MAPK: mitogen-activated protein kinase; mCRC: metastatic colorectal cancer; MET: mesenchymal-epithelial transition receptor; MDS: myelodysplastic syndrome; MPN: myeloproliferative neoplasm; MSS: microsatellite stable; MZL: marginal zone lymphoma; NEC: neuroendocrine carcinoma; NET: neuroendocrine tumor; NHL: non-Hodgkin lymphoma; NSCLC: non-small cell lung cancer; PD-1: programmed death-1; PD-L1: programmed death-ligand 1; PI3Kδ: phosphatidylinositol 3-kinase delta; RCC: renal cell carcinoma; SCLC: small cell lung cancer; SYK: spleen tyrosine kinase; TC: thyroid cancer; TGCT: tenosynovial giant cell tumor; TKI: tyrosine kinase inhibitor; TNBC: triple-negative breast cancer; VEGFR: vascular endothelial growth factor receptor; wAIHA = warm autoimmune hemolytic anemia |
Commercialized and Late-Stage Drug Candidates
Savolitinib – selective MET inhibitor in late-stage clinical development as a monotherapy and in combination therapies in global partnership with AstraZeneca
Savolitinib is a potent and selective small molecule inhibitor of the MET receptor tyrosine kinase, an enzyme which has been shown to function abnormally in many types of solid tumors. We designed savolitinib through chemical structure modification to specifically address kidney toxicity, the primary issue that halted development of several other selective MET inhibitors. In clinical trials to date across approximately 2,500 patients globally, savolitinib has shown promising signs of clinical efficacy in patients with multiple types of MET gene alterations in lung cancer, kidney cancer and gastric cancer with an acceptable safety profile.
In June 2021, the NMPA approved savolitinib for marketing for the treatment of NSCLC with MET exon 14 skipping alterations, making savolitinib the first-in-class selective MET inhibitor in China. This approval follows a priority review designation by the NMPA and is the first regulatory approval globally for this oral, potent and selective MET TKI. The approval by the NMPA was based on positive results from a Phase II trial conducted in China in patients with NSCLC with this mutation, including patients with the more aggressive pulmonary sarcomatoid carcinoma subtype. Savolitinib demonstrated effective anti-tumor activity based on an independent review of objective response rate (“ORR”) and disease control rate (“DCR”). The approval is conditional upon successful completion of an ongoing confirmatory study in this patient population. The results reviewed by the NMPA when it approved savolitinib were also published in The Lancet Respiratory Medicine.
We are currently developing savolitinib in global partnership with AstraZeneca, both as a monotherapy and in combination with immunotherapy and targeted therapy. Most notably, MET-aberration is a major mechanism for acquired resistance to both first-generation EGFR TKIs as well as third-generation EGFR TKIs like Tagrisso. Savolitinib has been studied extensively in these patients in the TATTON and SAVANNAH studies. Final results from the TATTON study were presented at World Conference on Lung Cancers (“WCLC”), in January 2021, and preliminary results from SAVANNAH were presented at WCLC in August 2022. Findings based on SAVANNAH and the TATTON studies supported the initiation of the SAFFRON global Phase III study in patients with EGFR-mutated, MET-driven, locally advanced or metastatic NSCLC whose disease progressed on first- or second-line treatment with Targrisso as the most recent therapy, with no prior chemotherapy in the metastatic setting allowed. China-based Phase III studies SACHI and SANOVO were also initiated. Savolitinib was granted fast track designation by the FDA for the combination treatment with Tagrisso of NSCLC patients harboring MET overexpression and/or amplification following progression on Targrisso. In comparison to potential alternative treatments, this treatment is chemotherapy-free, biomarker-specific and orally administered, aiming for a balanced efficacy, safety and quality-of-life profile for lung cancer patients.
74
Proof-of-concept studies of savolitinib in kidney cancer (as a monotherapy as well as in combination with a PD-L1 inhibitor) and gastric cancer (as a monotherapy as well as in combinations with chemotherapy) have demonstrated positive results, with subsequent clinical development ongoing or in planning. In addition, a gastric cancer patient China-registration cohort began enrolling in March 2023. For example, we initiated a global Phase III pivotal trial (SAMETA) in October 2021 for savolitinib in combination with Imfinzi, AstraZeneca’s anti-PD-L1 antibody durvalumab, in MET positive patients with papillary renal cell carcinoma or PRCC, a form of kidney cancer. Savolitinib opportunities are also continuing to be explored in multiple other MET-driven tumor settings via investigator-initiated studies.
Fruquintinib—selective VEGFR 1, 2 and 3 inhibitor with the best selectivity for its targets in global NDA submission; commercially launched as Elunate in China and as Fruzaqla in the United States in CRC in November 2018 and November 2023, respectively
Fruquintinib is a highly selective and potent oral inhibitor of vascular endothelial growth factor or VEGF receptors, known as VEGFR 1, 2 and 3. We believe that fruquintinib has the potential to become a selective small molecule VEGFR 1, 2 and 3 inhibitor for many types of solid tumors that has the highest selectivity, and we are currently studying fruquintinib in CRC, gastric cancer, endometrial cancer, kidney and other solid tumor types. Fruquintinib was designed to improve kinase selectivity to minimize off-target toxicities, improve tolerability and provide more consistent target coverage. The tolerability in patients to date, along with fruquintinib’s low potential for drug-drug interaction based on pre-clinical assessment, suggests that it may be highly suitable for combinations with other anti-cancer therapies.
Building on the data collected from our successful Phase III trial in China, known as the FRESCO study, which supported fruquintinib’s approval in China, we initiated FRESCO-2, a large randomized controlled study of fruquintinib in the United States, Europe, Japan and Australia. Based on the successful results of the multi-regional FRESCO-2 study and the FRESCO study in China, the FDA approved fruquintinib for the treatment of mCRC.
Aside from its first approved indication of third-line CRC in China, studies of fruquintinib combined with various checkpoint inhibitors (including Tyvyt and tislelizumab) are underway. An NDA is under review in China for the treatment of gastric cancer in combination with paclitaxel (FRUTIGA study), and registration-intent studies combined with checkpoint inhibitors (Tyvyt combo, in endometrial cancer and RCC) are ongoing in China.
Fruquintinib is being commercialized and developed in partnership with Eli Lilly in China, where we are responsible for development, manufacturing, on-the-ground medical detailing, promotion and local and regional marketing activities. Outside of China, we are collaborating with Takeda which holds an exclusive worldwide license from us to develop, manufacture and commercialize fruquintinib in all indications.
Surufatinib—unique angio-immuno kinase inhibitor commercially launched as Sulanda in China in advanced NETs; first VEGFR/FGFR/CSF-1R inhibitor for all advanced NETs
Surufatinib is a novel, oral angio-immuno kinase, small molecule inhibitor that selectively inhibits the tyrosine kinase activity associated with VEGFR and FGFR, which both inhibit angiogenesis, and colony stimulating factor-1 receptor, or CSF-1R, which regulates tumor-associated macrophages, promoting the body’s immune response against tumor cells. Its unique dual mechanism of action may be very suitable for possible combinations with other immunotherapies. We believe surufatinib is potentially the first VEGFR/FGFR/CSF-1R inhibitor for all advanced NETs.
Surufatinib was approved by the NMPA in December 2020 for the treatment of non-pancreatic NETs and is now being marketed by us in China under the brand name Sulanda. This NMPA approval of surufatinib was based on results from the SANET-ep study, a Phase III trial in patients with advanced non-pancreatic NETs conducted in China. The positive results of this trial were highlighted in an oral presentation at the 2019 ESMO Congress and published in The Lancet Oncology in September 2020. In June 2021, surufatinib was approved by the NMPA for the treatment of advanced pancreatic NETs. This NMPA approval of surufatinib was based on results from the SANET-p study, a Phase III trial in patients with advanced pancreatic NETs conducted in China. The positive results of this trial were highlighted in an oral presentation at the 2020 ESMO Congress and published in The Lancet Oncology in September 2020. In 2022, we presented a pooled analysis of safety data from the SANET-p and SANET-ep studies at the 2022 ASCO annual meeting.
We have various additional clinical trials of surufatinib ongoing in combination with checkpoint inhibitors.
We own all rights to surufatinib globally.
75
Sovleplenib (HMPL-523)—potentially the first selective Syk inhibitor for hematological cancer
Sovleplenib is a novel, highly selective, oral, small molecule inhibitor targeting the spleen tyrosine kinase, or Syk, for the treatment of hematological cancers and certain chronic immune diseases. Syk is a major component in B-cell receptor signaling and is an established therapeutic target in multiple subtypes of B-cell lymphomas. Because B-cell malignancies are heterogeneous and patients commonly experience relapse despite current therapies, there is a need for new therapies. Sovleplenib has been studied in clinical trials with around 600 patients to date.
We recently submitted the NDA for sovleplenib for the treatment of adult patients with primary immune thrombocytopenia (ITP), and the NDA was accepted by the NMPA in China in January 2024 after receiving priority review status in 2023. This NDA is supported by data from ESLIM-01, a randomized, double-blinded, placebo-controlled Phase III trial in China of sovleplenib in 188 adult patients with primary ITP who have received at least one prior line of standard therapy. In January 2022, sovleplenib received the Breakthrough Therapy Designation in China for treatment of primary immune thrombocytopenia.
Besides the clinical trials related to primary ITP, we also have various clinical trials of sovleplenib ongoing. In September 2022, we also initiated a randomized, double-blind, placebo-controlled Phase II/III study to evaluate the efficacy, safety, tolerability, and pharmacokinetics of sovleplenib in the treatment of warm AIHA. The enrollment of Phase II part of the study was completed in mid-2023 and primary end point has been met. We expect to initiate Phase III in the first half of 2024. In 2024, we also plan to start a Phase I/Ib dose-finding study in the United States.
We own all rights to sovleplenib globally.
Tazemetostat
In August 2021, we entered into a strategic collaboration with Epizyme, a subsidiary of Ipsen, to research, develop, manufacture and commercialize tazemetostat in Greater China, including the mainland, Hong Kong, Macau and Taiwan. Tazemetostat is an inhibitor of EZH2 developed by Ipsen that is approved by the U.S. FDA for the treatment of certain epithelioid sarcoma and follicular lymphoma patients. It received accelerated approval from the FDA based on ORR and DoR in January and June 2020 for epithelioid sarcoma and follicular lymphoma, respectively. Tazemetostat has been studied in clinical trials with around 1,300 patients to date.
We are developing and planning to seek approval for tazemetostat in various hematological and solid tumors in China. We are participating in Ipsen’s SYMPHONY-1 (EZH-302) study, leading it in China. We are generally responsible for funding all clinical trials of tazemetostat in China, including the portion of global trials conducted there. Separately, we are conducting a China bridging study in follicular lymphoma for potential conditional registration based on its U.S. approvals. The study is fully enrolled and we expect to file the NDA in mid-2024. We are responsible for the research, manufacturing and commercialization of tazemetostat in China. Tazemetostat was approved in China Hainan Pilot Zone in 2022 and the Macau Special Administrative Region in 2023.
HMPL-453—highly selective FGFR 1/2/3 inhibitor with potential in solid tumors
HMPL-453 is a novel, selective, oral inhibitor targeting FGFR 1/2/3. Aberrant FGFR signaling is associated with tumor growth, promotion of angiogenesis, as well as resistance to anti-tumor therapies. Approximately 10-15% of intrahepatic cholangiocarcinoma (IHCC) patients globally have tumors harboring FGFR2 fusion. Amongst others, we have an ongoing Phase II study in patients with advanced IHCC with FGFR2 fusion that had failed at least one line of systemic therapy where we started to enroll the registration cohort since March 2023.
We own all rights to HMPL-453 globally.
Amdizalisib (HMPL-689)—novel, highly selective PI3Kδ inhibitor with potential in hematological cancer
Amdizalisib is a novel, highly selective and potent small molecule inhibitor targeting the isoform PI3Kδ. In pre-clinical pharmacokinetic studies, amdizalisib’s pharmacokinetic properties have been found to be favorable with good oral absorption, moderate tissue distribution and low clearance. Amdizalisib received Breakthrough Therapy Designation from the CDE of the NMPA in China for the treatment of refractory follicular lymphoma in September 2021. We have multiple ongoing clinical studies of amdizalisib for various subtypes of lymphomas. Amdizalisib has been studied in clinical trials with around 500 patients to date. We own all rights to amdizalisib globally.
76
Early-Stage Drug Candidates
HMPL-306 is potentially the first dual inhibitor of IDH1 and IDH2 enzymes with applications in hematological malignancies and solid tumors. IDH1 and IDH2 mutations have been implicated as drivers of certain hematological malignancies, gliomas and solid tumors, particularly among acute myeloid leukemia patients. We have several ongoing Phase I studies in China and the United States on indications including myeloid hematological malignancies and various solid tumors. These include a Phase I trial in China was initiated in July 2020, in patients of relapsed or refractory hematological malignancies with an IDH1 and/or IDH2 mutation where we have presented the first-in-human dose-escalation phase data at EHA Annual Meeting in June 2023. Based on the pharmacodynamic, pharmacokinetic and preliminary clinical findings, a recommended Phase II dose was determined for the dose expansion phase of the study. We own all rights to HMPL-306 globally.
HMPL-760 is an investigational, highly selective, non-covalent, third-generation oral inhibitor of BTK with improved potency versus first generation BTK inhibitors against both wild type & C481S mutant enzymes. In January 2022, we initiated a Phase I trial in China in patients with previously treated chronic lymphocytic leukemia/small lymphocytic lymphoma or other types of non-Hodgkin lymphoma, including patients treated with a prior regimen containing a BTK inhibitor, whose disease carries either wild-type BTK or acquired resistance to first generation BTK inhibitors due to additional mutations to BTK. The initial dose escalation stage to determine the maximum tolerated dose and/or the RP2D is to be followed by a dose expansion phase where patients will receive HMPL‑760 to further evaluate the safety, tolerability, and clinical activity at the RP2D. We own all rights to HMPL-760 globally.
HMPL-295 is an investigative and highly selective small molecule inhibitor of ERK in the MAPK pathway with the potential to address intrinsic or acquired resistance from upstream mechanisms such as RAS-RAF-MEK. ERK is a downstream component of the RAS-RAF-MEK-ERK signaling cascade (MAPK pathway). This is our first of multiple candidates in discovery targeting the MAPK pathway. A China Phase I study of HMPL-295 as a monotherapy was initiated in July 2021. We own all rights to HMPL-295 globally.
HMPL-653 is a novel, highly selective, and potent CSF-1R inhibitor designed to target CSF-1R driven tumors as a monotherapy or in combination with other drugs. We initiated a China Phase I study in January 2022. We own all rights to HMPL-653 globally.
HMPL-A83 is an investigational IgG4-type humanized anti-CD47 monoclonal antibody that exhibits high affinity for CD47. HMPL-A83 blocks CD47 binding to Signal regulatory protein (SIRP) α and disrupts the “do not eat me” signal that cancer cells use to shield themselves from the immune system. We own all rights to HMPL-A83 globally.
HMPL-415 is an investigational SHP2 allosteric inhibitor. SHP2 is a non-receptor protein tyrosine phosphatase ubiquitously expressed mainly in the cytoplasm of several tissues. It interacts with diverse molecules in the cell, and regulates key signaling events including RAS/ERK, PI3K/AKT, JAK/STAT and PD-1 pathways. Dysregulation of SHP2 expression or activity causes many developmental diseases, and hematological and solid tumors. We initiated a China Phase I study in July 2023. We own all rights to HMPL-415 globally.
Discovery Research & Pre-clinical Development
We have built a drug discovery engine based in China, which has already produced a pipeline of around 20 differentiated clinical and late pre-clinical stage drug candidates covering both novel and validated targets of which three are now marketed. We strive to create differentiated novel oncology and immunology treatments with global potential. These include furthering both small molecule and biologic therapies which address aberrant genetic drivers and cancer cell metabolism; modulate tumor immune microenvironment; and target immune cell checkpoints. We design drug candidates with profiles that enable them to be used in innovative combinations with other therapies, such as chemotherapy, immunotherapy and other targeted therapies in order to attack disease simultaneously through multiple modalities and pathways. We believe that this approach can significantly improve treatment outcomes for patients.
Beyond these clinical and pre-clinical stage candidates, we continue to conduct research into discovering new types of drug candidates, including among others, small molecules addressing cancer-related apoptosis, cell signaling, epigenetics and protein translation; biologic drug candidates including bispecific antibodies; and novel technologies including antibody-drug conjugates and heterobifunctional small molecules.
77
Manufacturing
We use organizations in China to produce our clinical and commercial API supplies. For manufacturing drug products, we currently use a combination of contract manufacturers and our internal manufacturing facilities. We have a drug product manufacturing facility in Suzhou which manufactures both clinical and commercial supplies for fruquintinib and surufatinib. Our Suzhou facility passed a pre-approval inspection (PAI) for fruquintinib by the U.S. FDA in August 2023. We have qualified two drug product sites for supplying fruquintinib to the U.S. market: our own facility in Suzhou and a second site in Switzerland.
We have also completed construction of, qualified and obtained Drug Manufacturing Permit for a new drug product facility in Pudong, Shanghai, which will increase our novel drug product manufacturing capacity by over five times. The manufacturing and technology transfer for some of our commercial products are underway to this new facility. This is in line with our previously outlined expectations of manufacturing clinical supplies from the new facility starting in 2023 and commercial supplies around 2025, after the necessary regulatory filings and approvals.
In line with our commitment to sustainable practices and environmental stewardship, we have installed solar panels at this new facility. They contribute renewable energy directly to our operations, particularly in cooling indoor areas, significantly reducing electricity usage and greenhouse gas emissions.
We completed process validation for the API and drug product of sovleplenib at the selected commercial manufacturing facilities to support the approval of the product.
Other Ventures
In addition to our Oncology/Immunology operations, our Other Ventures include large-scale drug marketing and distribution platforms covering about 290 cities and towns in China with over 2,900 manufacturing and commercial personnel as of December 31, 2023. Built over the past 20 years, it primarily focuses on prescription drugs and consumer health products mainly through: (i) Shanghai Hutchison Pharmaceuticals, a non-consolidated joint venture with a commercial team of about 2,300 staff managing the medical detailing and marketing of its products not just in hospitals in provincial capitals and medium-sized cities, but also in the majority of county-level hospitals in China. SHPL’s Good Manufacturing Practice-certified factory holds 74 drug product manufacturing licenses and is operated by about 560 manufacturing staff and (ii) Hutchison Sinopharm, a consolidated joint venture focused on prescription drugs commercial services business, which in addition to providing certain commercial services for our own products, provides services to third-party pharmaceutical companies in China. Prior to our disposal of interest in December 2023, our Other Ventures also included Hutchison Hain Organic (Hong Kong) Limited and HUTCHMED Science Nutrition Limited, which were our joint venture and wholly-owned subsidiary principally engaged in wholesale and trading of healthcare and consumer products.
Net income attributable to our company from our Other Ventures totaled $142.9 million, $54.6 million and $50.3 million for the years ended December 31, 2021, 2022 and 2023, respectively, and are remitted to our group through dividend payments primarily from our non-consolidated joint venture mentioned above. In 2023, dividends of an aggregate amount of $42.3 million were distributed from Shanghai Hutchison Pharmaceuticals to our group, with aggregate dividends received by our group since inception of over $320 million.
Our Clinical Pipeline
The following is a summary of the clinical pipeline for our drug candidates, many of which are being investigated against multiple indications.
1. | Savolitinib (HMPL-504), MET Inhibitor |
Savolitinib is a potent and selective inhibitor of MET, an enzyme which has been shown to function abnormally in many types of solid tumors. We designed savolitinib to address human metabolite-related renal toxicity, the primary issue that halted development of several other selective MET inhibitors. In clinical studies to date, savolitinib has shown promising signs of clinical efficacy in patients with MET gene alterations in NSCLC, PRCC, CRC and gastric cancer with an acceptable safety profile. In global partnership with AstraZeneca, savolitinib has been studied in approximately 2,500 patients to date, both as a monotherapy and in combinations. For more information regarding our partnership with AstraZeneca, see “—Overview of Our Collaborations—AstraZeneca.”
78
Mechanism of Action
MET is a signaling pathway that has specific roles in normal mammalian growth and development. However, the MET pathway has also been shown to function abnormally in a range of different cancers, primarily through MET gene amplification, overexpression and gene mutations. The aberrant activation of MET has been demonstrated to be highly correlated in many cancer indications, including kidney, lung, gastric, colorectal, esophageal and brain cancer. It plays a major role in cancer pathogenesis (i.e., the development of the cancer), including tumor growth, survival, invasion, metastasis, the suppression of cell death as well as tumor angiogenesis.
MET also plays a role in drug resistance in many tumor types. For instance, MET gene aberrations has been found in NSCLC and CRC following anti-EGFR treatment, leading to drug resistance. Furthermore, MET dysregulation is considered to play a role in the immunosuppression and pathogenesis of kidney cancer.
Savolitinib Research Background
First generation selective MET inhibitors previously discovered by multinational pharmaceutical companies had positive pre-clinical data that supported their high MET selectivity and pharmacokinetic and toxicity profiles, but did not progress very far due to kidney toxicity. The issue appeared to be that certain metabolites of earlier compounds had dramatically reduced solubility and appeared to crystalize in the kidney, resulting in obstructive toxicity. With this understanding, we designed our compound, savolitinib (also known as AZD6094 and HMPL-504, formerly known as volitinib), differently while preserving high MET inhibition properties across multiple types of MET aberrations. Savolitinib has not shown any renal toxicity to date and does not appear to carry the same metabolite problems as the earlier selective MET compounds based on studies in over 2,000 patients conducted by AstraZeneca in global partnership with us.
Savolitinib Pre-clinical Evidence
In pre-clinical trials, savolitinib demonstrated strong in vitro activity against MET, affecting its downstream signaling targets and thus blocking the related cellular functions effectively, including proliferation, migration, invasion, scattering and the secretion of VEGF that plays a pivotal role in tumor angiogenesis.
One of our key areas of focus in our pre-clinical trials is to achieve superior selectivity on a number of kinases. A commonly used quantitative measure of selectivity is through comparing enzyme IC50, which represents the concentration of a drug that is required for 50% inhibition of the target kinase in vitro and the plasma concentration required for obtaining 50% of a maximum effect in vivo. High selectivity is achieved with a very low IC50 for the target cells, and a very high IC50 for the healthy cells (approximately 100 times higher than for the target cells). IC50 is measured in nM (nano-mole, a microscopic unit of measurement for the number of small molecules required to deliver the desired inhibitory effect).
In the MET enzymatic assay, savolitinib showed potent activity with IC50 of 5 nM. In a kinase selectivity screening with 274 kinases, savolitinib had potent activity against the MET Y1268T mutant (comparable to the wild-type), weaker activity against other MET mutants and almost no activity against all other kinases. Savolitinib was found to be approximately 1,000 times more potent to MET than the next non-MET kinase. Similarly, in cell-based assays measuring activity against MET phosphorylation, savolitinib demonstrated potent activity in both ligand-independent (gene amplified) and ligand-dependent (overexpressed) cells with IC50 at low nanomolar levels. In target related tumor cell function assays, savolitinib showed high potency with IC50 of less than 10 nM. Furthermore, savolitinib demonstrated cytotoxicity only on tumor cells that were MET gene amplified or MET overexpressed. In other cells, inhibition measurements demonstrated that IC50 amounts were over 30,000 nM, which is thousands of times higher than the IC50 on MET tumor cells.
The data above suggest that (i) savolitinib has potent activity against tumor cell lines with MET gene amplification in the absence of hepatocyte growth factor, or HGF, indicating that there is HGF-independent MET activation in these cells; (ii) savolitinib has potent activity in tumor cell lines with MET overexpressed, but only in the presence of HGF, indicating HGF-dependent MET activation; and (iii) savolitinib has no activity in tumor cell lines with low MET overexpression/gene amplification, suggesting that savolitinib has strong kinase selectivity.
79
Savolitinib Clinical Development
As discussed below, we have tested, and are currently testing, savolitinib in partnership with AstraZeneca in multiple indications, both as a monotherapy and in combination with other targeted therapies.
Non-small Cell Lung Cancer
The table below shows a summary of clinical trials for savolitinib in NSCLC patients.
Clinical Trials of Savolitinib in NSCLC
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Savolitinib + Tagrisso | SAVANNAH: 2L/3L EGFRm+; | Global | II | Fully enrolled | NCT03778229 | |||||
Tagrisso refractory; MET+ | Registration- | |||||||||
intent | ||||||||||
Savolitinib + Tagrisso | SAFFRON:2L/3L EGFRm+; | Global | III | Ongoing since 2022 | NCT05261399 | |||||
Tagrisso refractory; MET+ | ||||||||||
Savolitinib + Tagrisso | SACHI: 2L EGFR TKI refractory NSCLC; | China | III | Ongoing since 2021 | NCT05015608 | |||||
MET+ | ||||||||||
Savolitinib + Tagrisso |
| SANOVO: Naïve |
| China |
| III |
| Ongoing since 2021 |
| NCT05009836 |
patients with EGFRm | ||||||||||
& MET+ | ||||||||||
Savolitinib monotherapy | MET exon 14 skipping alterations | China | II Registration | Approved and launched in 2021. | NCT02897479 | |||||
Final OS analysis at ELCC | ||||||||||
2022 | ||||||||||
Savolitinib monotherapy | MET exon 14 skipping alterations | China | IIIb Confirmatory | Fully enrolled in the H1 2023. | NCT04923945 | |||||
First line cohort data at | ||||||||||
WCLC 2023 | ||||||||||
Savolitinib + Imfinzi | SOUND: MET-driven, EGFR wild type | China | II | Ongoing since 2022 | NCT05374603 |
Savolitinib Monotherapy
More than one third of the world’s lung cancer patients are in China and, among those with NSCLC, approximately 2-3% have tumors with MET exon 14 skipping alterations.
Phase II study of savolitinib monotherapy in NSCLC patients with MET exon 14 alteration (NCT02897479).
We have completed a 70-patient Phase II registration-enabling study in China of savolitinib as a monotherapy for MET exon 14 skipping NSCLC patients who have progressed following prior systemic therapy, or unable to receive chemotherapy.
At the ASCO annual meeting in June 2020, we presented interim data on 70 treated patients, of which 61 patients were efficacy evaluable at the data cut-off date of March 31, 2020. The overall data were encouraging, with efficacy in line with other selective MET inhibitors, despite the inclusion of patients with a more aggressive subtype (36% with pulmonary sarcomatoid carcinoma) and with tolerable safety. Efficacy measurements included the objective response rate, or ORR, (the percentage of patients in the study who show either partial response (tumor measurement reduction of greater than 30%) or complete response), disease control rate, median progression-free survival or PFS and median OS.
80
At subsequent data cut-off date of August 3, 2020, in the 61 evaluable patients, ORR was 49.2% and disease control rate was 93.4%. Median duration of response was 8.3 months (95% confidence interval: 5.3-16.6). Results from this study were published in The Lancet Respiratory Medicine and formed the basis for an NDA filing, which was approved by the NMPA in June 2021. Final OS and subgroup analysis was presented for this trial at European Lung Cancer Congress 2022 and published in the journal JTO Clinical and Research Reports. The updated results further confirmed the favorable benefit of savolitinib in these patients and in each subgroup and the acceptable safety profile. At final data cut off-date of June 28, 2021. In the full analysis set of 70 patients, median PFS was 6.9 months (95% confidence interval: 4.6-8.3). Median OS was 12.5 months (95% confidence interval: 10.5-21.4). A 95% confidence interval means that there is a 95% chance that the results will be within the stated range. CTC grade 3 or above TEAEs, with greater than 5% incidence related to savolitinib treatment were peripheral edema (9%), increased aspartate aminotransferase (13%) and increased alanine aminotransferase (10%). Clinical data demonstrated an acceptable safety profile with an adverse events-related discontinuations rate of 14.3%.
Phase II Study of Savolitinib Monotherapy Showing Effect in MET Exon 14 Alteration NSCLC Patients
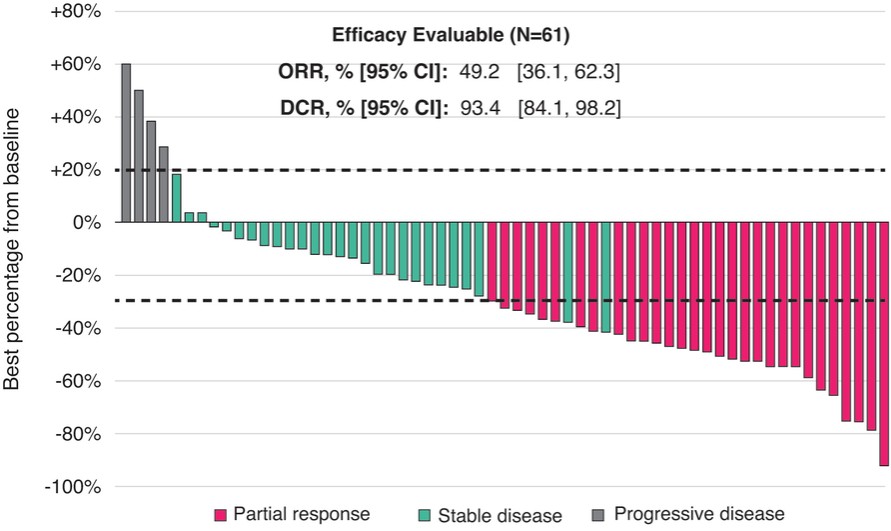
Notes: | N = number of patients; ORR = objective response rate; DCR = disease control rate; and CI = confidence interval. |
Source: | Lu S, Fang J et al. Once-daily savolitinib in Chinese patients with pulmonary sarcomatoid carcinomas and other non-small-cell lung cancers harboring MET exon 14 skipping alterations: a multicenter, single-arm, open-label, phase 2 study. Lancet Respir Med. 2021;9(10):1154-1164. doi:10.1016/S2213-2600(21)00084-9 |
A confirmatory Phase IIIb study in this patient population fully enrolled in the first half of 2023. Results from the first-line cohort of this study were disclosed at WCLC 2023. At data cut-off date of April 30, 2023, among the 84 patients in the tumor response evaluable set (TRES), ORR was 60.7% (95% CІ: 49.5% to 71.2%) and DCR was 95.2% (95% CІ: 88.3% to 98.7%), as assessed by an independent review committee. At median follow-up of 11.1 months, median PFS was 13.8 months (95% CІ: 9.7 months to not reached). Median DoR and OS have not been reached. No new safety signals were observed.
81
Savolitinib in Combination with Tagrisso
In 2015, AstraZeneca received FDA approval for Tagrisso, its drug for the treatment of T790M+ EGFRm+, TKI-resistant NSCLC. A drug with this type of activity is known as a third-generation EGFR inhibitor. In 2018, Tagrisso’s label was expanded to include previously untreated patients with EGFRm+ NSCLC. In December 2020, Tagrisso’s label was further expanded to include adjuvant therapy after tumor resection in EGFRm+ NSCLC patients. Tagrisso has been established as a new standard of care in the treatment of EGFRm+ NSCLC and has now been approved in over 80 countries. Understanding the mechanism of acquired resistance following Tagrisso treatment is a key clinical question to inform the next treatment choice. A portion of EGFRm+ TKI-resistant patients and a portion of T790M+ EGFRm+ TKI-resistant patients progress because of MET aberrations. Savolitinib was granted fast track designation by the FDA for the combination treatment with Tagrisso of NSCLC patients harboring MET overexpression and/or amplification following progression on Targrisso.
As discussed in more detail below, we and AstraZeneca are studying savolitinib in combination with Tagrisso as a treatment choice for patients who have developed a resistance to TKI (primarily Tagrisso). The acceptance and uptake of Tagrisso indicates that the market potential for savolitinib in Tagrisso-resistant, NSCLC could be material.
In January 2023, the U.S. FDA designated as a Fast Track development program the investigation of savolitinib for use in combination with Tagrisso for the treatment of patients with locally advanced or metastatic NSCLC whose tumors have MET overexpression and/or amplification, as detected by an FDA-approved test, and who have had disease progression during or following prior Tagrisso. In comparison to alternate treatments, our positioning remains chemotherapy-free, biomarker-specific and oral combinations, aiming for a balanced efficacy and safetytoxicity profile for lung cancer patients.
SAVANNAH study: Phase II study of savolitinib in combination with Tagrisso in NSCLC Tagrisso-refractory EGFRm+ patients (NCT03778229).
Based on the encouraging results of the multiple TATTON studies, we and AstraZeneca have initiated this global Phase II study in patients whose disease have progressed following Tagrisso due to MET amplification or overexpression, which has three dose cohorts of savolitinib combined with Tagrisso. In addition to continuing Tagrisso treatment, patients received savolitinib 300mg QD, 300mg BID, or 600mg QD. The study has completed an additional patient recruitment designed to further reinforce the strength of data, initially presented at WCLC 2022. We continue to evaluate the possibility of using the SAVANNAH study as the basis for U.S. accelerated approval.
The results presented at the WCLC 2022 were based on an analysis of 193 efficacy evaluable patients who received savolitinib 300mg once daily plus Tagrisso 80mg once daily at data cut-off date of August 27, 2021. Qualifying MET aberrations were FISH5+ or IHC50+. Importantly, additional analysis using a higher cut-off level of MET aberration were presented. The higher cut-off levels for MET aberration are FISH10+ and/or IHC90+. The prevalence of this higher cut-off levels of MET aberration was 34% of patients centrally tested for enrollment in this study versus 62% at the lower, qualifying cut-off level.
Results showed a trend toward improved response rates with increasing level of MET aberration. Across all patients in this analysis, ORR was 32% (95% CI: 26-39%), median DoR was 8.3 months (95% CI: 6.9-9.7 months), and median PFS was 5.3 months (95% CI: 4.2-5.8 months). These results are consistent with the TATTON and ORCHARD global studies. Among the 108 SAVANNAH patients who met the criteria for higher cut-off levels of MET aberration, ORR was 49% (95% CI: 39-59%), median DoR was 9.3 months (95% CI: 7.6-10.6 months), and median PFS was 7.1 months (95% CI: 5.3-8.0 months).
82
Importantly, among the 87 patients who did not receive prior chemotherapy, ORR was 52% (95% CI: 41-63%), median DoR was 9.6 months (95% CI: 7.6-14.9 months), and median PFS was 7.2 months (95% CI: 4.7-9.2 months). The safety profile of savolitinib plus Tagrisso was consistent with the known profiles of the combination and each treatment alone.

* | Evaluable for efficacy defined as dosed patients with measurable disease at baseline who had ≥2 on-treatment RECIST scans. |
Excludes eight patients with invalid or missing test results for IHC90+ and/or FISH10+ status, these patients were excluded from the subgroup analyses based on MET levels.
Source: | Ahn MJ, De Marinis F et al. EP08.02-140 MET Biomarker-based Preliminary Efficacy Analysis in SAVANNAH: savolitinib+osimertinib in EGFRm NSCLC Post-Osimertinib. .J Thorac Oncol. 2022 Sep;17(9):S469-S470. |
SAFFRON study: Phase III study of savolitinib in combination with Tagrisso in NSCLC Tagrisso-refractory EGFRm+ patients (NCT05261399)
Findings based on SAVANNAH and the TATTON studies supported the initiation of the SAFFRON global Phase III study in patients with EGFR-mutated, MET-driven, locally advanced or metastatic NSCLC whose disease progressed on first- or second-line treatment with Tagrisso as the most recent therapy, with no prior chemotherapy in the metastatic setting allowed. Patients are prospectively selected for the higher level of MET aberration of FISH10+ and/or IHC90+. The SAFFRON study will evaluate the efficacy and safety of savolitinib in combination with Tagrisso compared to pemetrexed plus platinum doublet-chemotherapy, the current standard-of-care treatment in this setting. The primary endpoint of the study is PFS. SAVANNAH has now activated a majority of the approximately 250 sites in over 20 countries planned for the study.
SACHI study: Phase III study of combination with Targrisso in 2L EGFR TKI refractory, MET amplified NSCLC patients (NCT05015608).
We have initiated SACHI, a China Phase III study of savolitinib in combination with Tagrisso. The Phase III trial is a multi-center, open-label, randomized, controlled study in patients with locally advanced or metastatic EGFR mutation-positive NSCLC with MET amplification after disease progression on EGFR inhibitor therapy. The study will evaluate the efficacy and safety of savolitinib in combination with Tagrisso, compared to platinum-based doublet-chemotherapy (pemetrexed plus cisplatin or carboplatin), the standard of care treatment option in this setting. The primary endpoint of the study is median PFS as assessed by investigators. Other endpoints include median PFS assessed by an independent review committee, median overall survival, ORR, duration of response, disease control rate, time to response, and safety. The first patient was dosed in November 2021. We expect to complete the enrollment of this trial in 2024.
83
SANOVO study: Phase III study of combination with Targrisso in naïve NSCLC patients with EGFR mutant and MET positive (NCT05009836).
We have initiated SANOVO, a China Phase III study of savolitinib in combination with Targrisso as a first-line treatment in certain NSCLC patients whose tumors harbor EGFR mutations and overexpress MET. The Phase III trial is a blinded, randomized, controlled study in previously untreated patients with locally advanced or metastatic NSCLC with activating EGFR mutations and MET overexpression. The study will evaluate Targrisso in combination with savolitinib comparing to Targrisso alone, a standard of care treatment option for these patients. The primary endpoint of the study is median progression free survival as assessed by investigators. Other endpoints include median progression-free survival assessed by an independent review committee, median overall survival, ORR, duration of response, disease control rate, time to response and safety. The first patient was dosed in September 2021. We expect to complete the enrollment of this trial in 2024.
Savolitinib in Combination with Imfinzi
Imfinzi is a human monoclonal antibody developed by AstraZeneca that binds to the PD-L1 protein and blocks the interaction of PD-L1 with PD-1 and CD80 proteins, countering the tumor’s immune-evading tactics and releasing the inhibition of immune responses.
SOUND study: Phase II study of combination with Imfinzi in EGFR/ALK/ROS1 wild-type, locally advanced or metastatic NSCLC patients with MET aberrations (NCT05374603).
The SOUND Phase II trial is an open-label, interventional, multicenter, exploratory Phase II study to evaluate savolitinib combined with Imfinzi in EGFR/ALK/ROS1 wild-type, locally advanced or metastatic NSCLC patients with MET aberrations. The primary endpoint is PFS.
Kidney Cancer
The table below shows a summary of the clinical trial for savolitinib in kidney cancer patients.
Clinical Trial of Savolitinib in Kidney Cancer
Treatment | Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # | |
Savolitinib + Imfinzi | SAMETA: MET-driven, unresectable and locally advanced or metastatic PRCC |
| Global |
| III |
| Ongoing since 2021 |
| NCT05043090 |
PRCC is a subtype of kidney cancer, representing about 15% of patients, with no treatments approved for patients with tumors that harbor MET-driven alterations. MET is a key genetic driver in papillary RCC, and emerging evidence suggests that combining immunotherapies with a MET inhibitor could enhance anti-tumor activity. Immune checkpoints such as PD-L1 are sometimes used by cancer cells to avoid being attacked by the immune system. As such, drugs that target these checkpoints are being developed or marketed as cancer treatments. Imfinzi is an anti-PD-L1 antibody owned by AstraZeneca. Anti-PD-L1 antibodies have been associated with clinical benefits in metastatic RCC, and MET dysregulation has been considered to play an important role in PRCC pathogenesis (including in our savolitinib Phase I and Phase II monotherapy studies) and is a mechanism of resistance against kinase inhibitors in clear cell RCC. Moreover, it is believed that the MET signaling pathway has a complex interplay with the immune system, including correlation with PD-L1 expression, immune suppression through angiogenesis and many other facets of the immune system.
During an Australian Phase I study, our investigators noted positive outcomes among PRCC patients with a strong correlation to MET gene amplification status. Out of a total of eight PRCC patients in our Australia Phase I study who were treated with various doses of savolitinib, three achieved confirmed partial responses. A further three of these eight PRCC patients achieved stable disease, which means patients without partial response but with a tumor measurement increase of less than 20%. This aggregate ORR of 38% was very encouraging for PRCC, which has no effective approved treatments. These responses were also durable as demonstrated by a patient who has been on the therapy for over 30 months and had tumor measurement reduction of greater than 85%. Importantly, the level of tumor response among these PRCC patients correlated closely with the level of MET gene amplification. The patients with consistent MET gene amplification across the whole tumor responded most to savolitinib, and with those patients with the highest level of MET gene amplification responding most to the treatment. We have conducted multiple global studies of savolitinib in PRCC patients, including the SAVOIR monotherapy and CALYPSO combination therapy global Phase II trials, that both demonstrated highly encouraging results. These results led to the initiation of a global Phase III, the SAMETA study, in 2021.
84
The CALYPSO study is an investigator-initiated open-label Phase II study of savolitinib in combination with Imfinzi. The study evaluated the safety and efficacy of the savolitinib and Imfinzi combination in PRCC patients at sites in the U.K. and Spain. 24-month follow-up of the CALYPSO study showed median PFS of 15.7 months and median OS of 27.4 months in MET-driven PRCC patients.
SAMETA study: Phase III in combination with Imfinzi PD-L1 inhibitor in MET-driven, unresectable and locally advanced or metastatic PRCC (NCT05043090)
The Phase III trial is an open-label, randomized, controlled study in treatment-naïve patients with MET-driven, unresectable and locally advanced or metastatic PRCC, to evaluate the efficacy and safety of savolitinib in combination with Imfinzi compared to single agent Imfinzi or single agent Sutent, an oral multi-kinase inhibitor considered as the standard of care treatment option in PRCC. The primary endpoint of the study is median PFS. Other endpoints include median OS, ORR, duration of response, 6-months and 12-months DCR, time to second progression, safety, pharmacokinetics and quality of life. The first patient was dosed in October 2021.
Gastric Cancer
The table below shows a summary of clinical trial for savolitinib in gastric cancer patients.
Clinical Trial of Savolitinib in Gastric Cancer
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Savolitinib monotherapy |
| 3L gastric cancer with MET amplification. Two-stage, single-arm study |
| China |
| II registration intent |
| ~64 patient registration cohort enrolling since March 2023; Breakthrough Therapy Designation |
| NCT04923932 |
MET-driven gastric cancer has a very poor prognosis. Multiple Phase II studies have been conducted in Asia to study savolitinib in MET-driven gastric cancer, which account for approximately 5% of all gastric cancer patients, demonstrated promising efficacy, including VIKTORY, which reported a 50% ORR with savolitinib monotherapy in gastric cancer patients whose tumors harbor MET amplification.
The VIKTORY study is a biomarker-based, Phase II umbrella trial in gastric cancer conducted by the Samsung Medical Center in South Korea. Patients were allocated to one of 12 biomarker-driven arms, based on a master screening protocol with tissue-based molecular analyses. Patients that tested positive for MET amplification or overexpression were treated with either savolitinib monotherapy or a combination of savolitinib and Taxotere. A total of 715 gastric cancer patients were successfully sequenced and MET amplification was observed in 3.5% of these patients (25/715). Of the 10 associated clinical trials under the VIKTORY umbrella, the highest ORR was observed in the MET amplification arm in patients treated with savolitinib monotherapy, which reported an ORR of 50% (10/20, 95% confidence interval: 28.0-71.9) and met pre-specified 6-week PFS rates. While the savolitinib and Taxotere combination was well tolerated, the VIKTORY study investigators decided to stop enrollment in the two combination cohorts in order to direct patients to the savolitinib monotherapy arm of the VIKTORY study as discussed above. The VIKTORY study investigators concluded that encouraging clinical efficacy of savolitinib in MET-amplified gastric cancer warrants further study.
Phase II study of savolitinib with potential for registration intent in 3L gastric cancer with MET amplification (NCT04923932)
This Phase II registration-intent study is a two-stage and single-arm study to evaluate the efficacy, safety and pharmacokinetics of savolitinib in locally advanced or metastatic GC or GEJ patients whose disease progressed after at least one line of standard therapy. The primary endpoint is ORR as assessed by an independent review committee. Other endpoints include 12-week and 6-month progression-free survival rates, median progression-free survival, duration of response, disease control rate, median overall survival, safety, pharmacokinetics and quality of life. The first patient was dosed in July 2021. Preliminary efficacy and safety data from an interim analysis was reported at AACR 2023, showing promising efficacy in patients with MET-amplified diseases, particularly in patients with high MET gene copy number. Confirmed ORR by independent review was 45%, or 50% in the 16 patients with high MET gene copy number. Duration of response rate at 4-months was 85.7%. The most common grade 3 or above TRAEs (more than 5%) were decreased platelet count, hypersensitivity, anemia, neutropenia and abnormal hepatic function. The BID regimen is being investigated to further evaluate the efficacy and safety of savolitinib in MET high patients. Following consultation with the NMPA with this data, a patient registration cohort began enrolling in March 2023.
85
Overview of Savolitinib Commercial Launch
Sold under the brand name Orpathys, savolitinib was granted conditional approval in China by the NMPA and launched in July 2021 by our partner, AstraZeneca. Orpathys is for the treatment of patients with non-small cell lung cancer with MET exon 14 skipping alterations who have progressed following prior systemic therapy or are unable to receive chemotherapy. The revenue we generate from Orpathys are comprised of royalty revenue and revenue from the product sales of Orpathys which we source from a third-party manufacturer and sell to AstraZeneca at cost. In 2021, we generated $11.3 million in total revenue from Orpathys sales, of which $4.8 million was royalty revenue and $6.5 million was revenue from sales of goods to AstraZeneca. In 2022, we generated $22.3 million in total revenue from Orpathys sales, of which $12.4 million was royalty revenue and $9.9 million was revenue from sales of goods to AstraZeneca. Following negotiations with the China NHSA in January 2023, starting on March 1, 2023, Orpathys was included in the updated NRDL, broadening patient access to this medicine. In 2023, we generated $28.9 million in total revenue from Orpathys sales, of which $13.8 million was royalty revenue and $15.1 million was revenue from sales of goods to AstraZeneca.
Partnership with AstraZeneca
In December 2011, we entered into a global licensing, co-development, and commercialization agreement for savolitinib with AstraZeneca. As noted above, given the complexity of many of the signal transduction pathways and resistance mechanisms in oncology, the industry is increasingly studying combinations of targeted therapies (TKI, monoclonal antibodies and immunotherapies) and chemotherapy as potentially the best approach to treating this complex and constantly mutating disease. Based on savolitinib’s clinical progress as a highly selective MET inhibitor in a number of cancers, in August 2016, December 2020 and November 2021, we and AstraZeneca amended our global licensing, co-development, and commercialization agreement for savolitinib. We believe that AstraZeneca’s portfolio of proprietary targeted therapies is well suited to be used in combinations with savolitinib, and we are studying combinations with Tagrisso (EGFRm+, T790M+) and Imfinzi (PD-L1). These combinations of multiple global first-in-class compounds are difficult to replicate, and we believe represent a significant opportunity for us and AstraZeneca. For more information regarding our partnership with AstraZeneca, see “—Overview of Our Collaborations—AstraZeneca.”
2. Fruquintinib (HMPL-013), VEGFR 1, 2 and 3 Inhibitor
Fruquintinib is a novel, selective, oral inhibitor of VEGFR 1/2/3 kinases that was designed to improve kinase selectivity with the destination of minimizing off-target toxicity and thereby improve efficacy and tolerability. Fruquintinib has been studied in clinical trials with about 5,700 patients to date, both as a monotherapy and in combination with other agents.
Aside from its first approved indication of previously-treated metastatic CRC (in China and the U.S.), several studies of fruquintinib combined with various checkpoint inhibitors (including Tyvyt and tislelizumab) are underway, some of which presented encouraging data. Registration-intent studies combined with chemotherapy (FRUTIGA study in gastric cancer) or checkpoint inhibitors (Tyvyt combo, in endometrial cancer and RCC) are completing or ongoing in China.
We are partnered with Eli Lilly in China and with Takeda outside of China.
Mechanism of Action
During the development of cancer, tumors at an advanced stage can secrete large amounts of VEGF, a protein ligand, to stimulate formation of excessive vasculature (angiogenesis) around the tumor in order to provide greater blood flow, oxygen, and nutrients to fuel the rapid growth of the tumor. Since essentially all solid tumors require angiogenesis to progress beyond a few millimeters in diameter, VEGFR drugs have demonstrated benefits in a wide variety of tumor types. VEGF and other ligands can bind to three VEGF receptors, VEGFR 1, 2 and 3, each of which has been shown to play a role in angiogenesis. Therefore, inhibition of the VEGF/VEGFR signaling pathway can act to stop the growth of the vasculature around the tumor and thereby starve the tumor of the nutrients and oxygen it needs to grow rapidly.
This therapeutic strategy has been well validated with several first-generation VEGF inhibitors having been approved globally since 2005 and 2006. These include both small molecule multi-kinase inhibitor drugs such as Nexavar and Sutent as well as monoclonal antibodies such as Avastin. The success of these drugs validated VEGFR inhibition as a new class of therapy for the treatment of cancer.
86
Fruquintinib Pre-clinical Evidence
Pre-clinical trials have demonstrated that fruquintinib is a highly selective VEGFR 1, 2 and 3 inhibitor with high potency and low cell toxicity at the enzymatic and cellular levels. In a kinase selectivity screening, fruquintinib was found to be approximately 250 times more selective to VEGFR 3 than to the next non-VEGFR kinase.
As a result of off-target side effects, existing VEGFR inhibitors are often unable to be dosed high enough to completely inhibit VEGFR, the intended target. In addition, the complex off-target toxicities resulting from inhibition of multiple signaling pathways are often difficult to manage in clinical practice. Combining such drugs with chemotherapy can lead to severe toxicities that can cause more harm than benefit to patients. To date, the first generation VEGFR TKI have been rarely used in combination with other therapies, thereby limiting their potential. Because of the potency and selectivity of fruquintinib, we believe that it has the potential to be safely combined with other oncology drugs, which could significantly expand its clinical potential.
Fruquintinib Clinical Development
Fruquintinib Monotherapy - Colorectal Cancer
The table below shows a summary of the clinical trials for fruquintinib in CRC patients. We have two additional trials in progress for fruquintinib in CRC in combination with a checkpoint inhibitor as discussed in more detail below under “— Fruquintinib Combinations with Checkpoint Inhibitors.”
Current Clinical Trials of Fruquintinib in CRC
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Fruquintinib monotherapy |
| FRESCO-2: metastatic CRC |
| U.S. / Europe / Japan / Australia |
| III |
| Approved & launched in the U.S. in Nov 2023; EMA MAA validated in Jun 2023; NDA filed in Japan in Sep 2023; Results published in The Lancet; further data presented at ASCO GI, JSMO & ASCO 2023 |
| NCT04322539 |
Fruquintinib monotherapy |
| FRESCO: ≥3L CRC; chemotherapy refractory |
| China |
| III |
| Approved and launched in 2018 |
| NCT02314819 |
Fruquintinib monotherapy |
| CRC, TN & HR+/HER2- breast cancer |
| U.S. |
| I/Ib |
| CRC data at ASCO GI 2022; results supported the initiation of the FRESCO-2 |
| NCT03251378 |
Notes: TN = triple-negative; HR+ = hormone receptor-positive; and HER2 = human epidermal growth factor receptor 2.
FRESCO study: Phase III study of fruquintinib monotherapy in third-line CRC (NCT02314819)
In 2014, we initiated the FRESCO study, which is a randomized, double-blind, placebo-controlled, multi-center, Phase III pivotal trial in China in patients with locally advanced or mCRC who had failed at least two prior systemic antineoplastic therapies, including fluoropyrimidine, Eloxatin and Camptosar. At the time, no drug was approved in third-line CRC in China with best supportive care being the general standard of care. This study followed a Phase II proof-of-concept trial in third-line CRC that met its primary endpoint of PFS in 2014.
Enrollment was completed in May 2016, and 519 patients were screened. The intent-to-treat population of 416 patients was randomized at a 2:1 ratio to receive either: 5 mg of fruquintinib orally once daily, on a three-weeks-on/one-week-off cycle, plus best supportive care (278 patients) or placebo plus best supportive care (138 patients). Randomization was stratified for prior anti-VEGF therapy and K-RAS gene status. The trial concluded in January 2017.
87
In June 2017, we presented the results of the FRESCO study in an oral presentation at the ASCO annual meeting. Results showed that FRESCO met all primary and secondary endpoints including significant improvements in OS and PFS with a manageable safety profile and lower off-target toxicities compared to other targeted therapies. The primary endpoint of median OS was 9.30 months (95% confidence interval: 8.18-10.45 months) in the fruquintinib group versus 6.57 months (95% confidence interval: 5.88-8.11 months) in the placebo group, with a hazard ratio of 0.65 (95% confidence interval: 0.51-0.83; two-sided p<0.001). The secondary endpoint of median PFS was 3.71 months (95% confidence interval: 3.65-4.63 months) in the fruquintinib group versus 1.84 months (95% confidence interval: 1.81-1.84 months) in the placebo group, with a hazard ratio of 0.26 (95% confidence interval: 0.21-0.34; two-sided p<0.001). Significant benefits were also seen in other secondary endpoints. The disease control rate in the fruquintinib group was 62% versus 12% for placebo (p<0.001), while the ORR based on confirmed responses was 5% versus 0% for placebo (p=0.012).
We have not performed a head-to-head clinical trial of fruquintinib versus Stivarga. While it is difficult to directly evaluate and compare clinical results across separate trials, data from the FRESCO study compare favorably to the data from the CONCUR study, a Phase III study of Stivarga monotherapy in CRC conducted in Asia, and the CORRECT study, a global Phase III study of Stivarga in CRC. In particular, in the Chinese patient subgroup of the CONCUR study, Stivarga had a disease control rate of 46% versus 7% in the placebo group. Median PFS was 2.0 months in the Stivarga group versus 1.7 months in the placebo group, and median OS was 8.4 months in the Stivarga group versus 6.2 months in the placebo group. In the CORRECT study, Stivarga had a disease control rate of 41% versus 15% in the placebo group. Median PFS was 1.9 months in the Stivarga group versus 1.7 months for the placebo group, and median OS was 6.4 months in the Stivarga group versus 5.0 months in the placebo group.
In terms of safety, results showed that fruquintinib had a manageable safety profile with lower off-target toxicities compared to Stivarga, the other VEGFR TKI approved for third-line CRC. Of particular interest was that the CTC grade 3 or above hepatotoxicity was similar for the fruquintinib group as compared to the placebo group, which was in contrast to Stivarga which was markedly higher and often difficult to manage in the Chinese patient population in the CONCUR study. Adverse events led to dose interruptions in 69% of patients in the Chinese patient subgroup of the CONCUR study, compared to 35% in the FRESCO study. The most frequently reported fruquintinib-related CTC grade 3 or above TEAEs included hypertension (21%), hand-foot skin reaction (11%), proteinuria (3%) and diarrhea (3%), all possibly associated with VEGFR inhibition. No other CTC grade 3 or above TEAEs exceeded 2% in the fruquintinib population, including hepatic function adverse events such as elevations in bilirubin (1%), alanine aminotransferase (<1%) or aspartate aminotransferase (<1%).
In terms of tolerability, dose interruptions or reductions occurred in only 35% and 24% of patients in the fruquintinib arm, respectively, and only 15% of patients discontinued treatment of fruquintinib due to adverse events versus 6% for placebo. The FRESCO study was published in the Journal of the American Medical Association in June 2018.
Subgroup analysis
In June 2018, a further subgroup analysis of data from the FRESCO Phase III study was presented at the ASCO annual meeting. This analysis explored possible effects of prior target therapy on the efficacy and safety of fruquintinib by analyzing the subgroups of patients with prior target therapy and those without prior target therapy.
Results showed that the benefits of fruquintinib were generally consistent across all subgroups. Among a total of 278 fruquintinib-treated patients, 111 had received prior target therapy while 55 of the 138 placebo-treated patients had received prior target therapy. In the prior target therapy subgroup, fruquintinib significantly prolonged OS and PFS. Median OS was 7.69 months for patients treated with fruquintinib versus 5.98 months for placebo (hazard ratio = 0.63; p = 0.012). Median PFS was 3.65 months for patients treated with fruquintinib versus 1.84 months for placebo (hazard ratio = 0.24; p < 0.001).
Among these 278 patients, the results showed that a subgroup of 84 patients who had received prior anti-VEGF treatment also benefited from fruquintinib. In this subgroup, the median OS was 7.20 months for fruquintinib versus 5.91 months for placebo (hazard ratio = 0.68; p = 0.066) and the median PFS was 3.48 months for fruquintinib versus 1.84 months for placebo (hazard ratio = 0.24; p < 0.001).
In the subgroup of 250 patients without prior targeted therapies, the median OS was 10.35 months for 167 patients treated with fruquintinib versus 6.93 months for 83 patients treated with placebo (hazard ratio = 0.63; p = 0.003), and the median PFS for patients treated with fruquintinib was 3.81 months versus 1.84 months for placebo (hazard ratio = 0.28; p < 0.001).
88
Additional data showed that there were no observed cumulative CTC grade 3 or above TEAEs in the subgroup of patients with prior target therapy. The CTC grade 3 or above TEAEs rates of fruquintinib were similar in the subgroups with prior target therapy (61.3%) and without prior target therapy (61.1%). This subgroup analysis is consistent with the previously reported results from the FRESCO study’s intent-to-treat population.
The results of this analysis showed that fruquintinib had clinically meaningful benefits in third-line mCRC patients regardless of prior target therapy without observed cumulative toxicity.
Quality-adjusted survival analysis
At the 2018 ASCO Annual Meeting, an analysis was presented that aimed to compare the quality-adjusted survival between the two arms of the FRESCO study using quality-adjusted time without symptoms or toxicity, or Q-TWiST, methodology and to investigate the Q-TWiST benefit of fruquintinib treatment among subgroups. Q-TWiST is a tool to evaluate relative clinical benefit-risk from a patient’s perspective and has been widely used in oncology treatment assessment. The survival time for each patient was divided into three portions: time with CTC grade 3 or above toxicity before progression, time without symptoms or CTC grade 3 or above toxicity, and time from progression or relapse until death or end of follow-up.
Patients treated with fruquintinib had longer Q-TWiST periods compared to patients treated with placebo. Q-TWiST benefits were observed regardless of prior lines of chemotherapy and prior anti-VEGF or anti-EGFR targeted therapy. The relative improvement of Q-TWiST with fruquintinib represents a clinically important quality-of-life benefit for mCRC patients.
Supported by data from the successful FRESCO study, we submitted an NDA for fruquintinib in June 2017. Fruquintinib was subsequently awarded priority review status by the NMPA in view of its clinical value in September 2017, and in September 2018, the NMPA approved fruquintinib for the treatment of patients with advanced CRC and was launched in November 2018. For more information regarding the Elunate product launch, see “—Overview of Fruquintinib Commercial Launch.”
FRESCO-2 study: Phase III study of fruquintinib monotherapy in mCRC (NCT04322539)
We initiated a global Phase III registration study, known as the FRESCO-2 study, in refractory metastatic CRC. Positive results from this double-blind, placebo-controlled, global Phase III study in 691 patients demonstrated that treatment with fruquintinib resulted in a statistically significant and clinically meaningful increase in OS and the key secondary endpoint of PFS compared to treatment with placebo. ASCO presentations showed that in subgroup analyses by prior lines of therapies up to six or more and by prior treatment with approved agents, fruquintinib improved OS and PFS for all subgroups and prior therapies, consistent with those of the overall study population. A separate study showed that during the study adverse events of special interest led to low rates of dose reduction (13.6% for patients who received fruquintinib vs 0.9% for patients who received placebo) and dose discontinuation (8.3% for patients who received fruquintinib vs 6.1% for patients who received placebo).
89
The FRESCO-2 study demonstrated that treatment with fruquintinib resulted in a statistically significant and clinically meaningful increase in the primary OS endpoint and key secondary PFS endpoint compared to treatment with placebo. The positive OS and PFS were consistent across all subgroups. Specifically, the median OS was 7.4 months for the 461 patients treated with fruquintinib compared to 4.8 months for the 230 patients in the placebo group (hazard ratio 0.66; 95% confidence interval 0.55–0.80; p<0.001). Median PFS was 3.7 months for patients treated with fruquintinib compared to 1.8 months for patients in the placebo group (HR 0.32; 95% CI 0.27–0.39; p<0.001). The DCR was 55.5% in the fruquintinib group compared to 16.1% for patients in the placebo group. Median duration of follow-up was approximately 11 months for patients in both groups.

Notes: | [1] ESMO 2022, LBA25. Dasari NA, et al. LBA25 - FRESCO-2: A global phase III multiregional clinical trial (MRCT) evaluating the efficacy and safety of fruquintinib in patients with refractory metastatic colorectal cancer. 12 Sep 2022, Proffered Paper session 2: GI, lower digestive Session. Annals of Oncology (2022) 33 (suppl_7): S808-S869. 10.1016/annonc/annonc1089. |
The safety profile of fruquintinib in FRESCO-2 was consistent with previously reported fruquintinib studies. Grade 3 or above adverse events occurred in 62.7% of patients who received fruquintinib, compared to 50.4% of patients who received placebo. Grade 3 or above adverse events that occurred in more than 5% of patients who received fruquintinib were hypertension (13.6% vs. 0.9% in the placebo group), asthenia (7.7% vs. 3.9% in the placebo group) and hand-foot syndrome (6.4% vs. 0% in the placebo group).
Phase I/Ib study of fruquintinib monotherapy in metastatic colorectal and breast cancers (NCT03251378)
We are conducting a multi-center, open-label, Phase Ib clinical study to evaluate the safety, tolerability and pharmacokinetics of fruquintinib in U.S. patients, which has established the U.S. RP2D to be 5 mg, the same as that in China. This dose was used in the FRESCO-2 study described above.
90
Fruquintinib Monotherapy - Gastric Cancer
Advanced gastric cancer is a major medical need, particularly in Asian populations, with limited treatment options for patients who have failed first-line standard chemotherapy with 5-fluorouracil and platinum doublets. The table below shows a summary of the clinical study for fruquintinib in gastric cancer patients.
Clinical Trial of Fruquintinib in Gastric Cancer
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Fruquintinib + paclitaxel |
| FRUTIGA: 2L gastric cancer |
| China |
| III |
| Supplemental NDA accepted by NMPA in Apr 2023; data at ASCO Plenary Series Feb 2024 |
| NCT03223376 |
FRUTIGA study: Phase III study of fruquintinib in combination with paclitaxel in gastric cancer (second-line) (NCT03223376)
This randomized, double-blind, Phase III study in China to evaluate fruquintinib combined with paclitaxel compared with paclitaxel monotherapy, for second-line treatment of advanced gastric cancer, enrolled 703 patients in July 2022. Its dual-primary endpoints are PFS and OS. The trial met the PFS endpoint at a statistically and clinically meaningful level. The OS endpoint was not statistically significant per the pre-specified statistical plan, although there was an improvement in median OS.
Results were presented orally at ASCO Plenary Series in February 2024. Patients on fruquintinib combined with paclitaxel achieved median PFS of 5.6 months, vs 2.7 months in the control group on paclitaxel only with HR of 0.569 and p < 0.0001. There was an improvement in OS with median OS of 9.6 months vs. 8.4 months, however this was not statistically significant. There was an imbalance of patients receiving subsequent antitumor therapies across the two groups, with 52.7% in the fruquintinib plus paclitaxel group vs. 72.2% in the paclitaxel monotherapy group. In pre-specified sensitivity analysis, when excluding patients taking subsequent antitumor therapy, OS advantage becomes nominal statistically significant for the treatment arm at 6.9 months vs 4.8 months in control arm with HR of 0.72 and p=0·0422. Fruquintinib also demonstrated a statistically significant improvement in secondary endpoints including ORR, DCR and DoR. The safety profile of fruquintinib in FRUTIGA was consistent with previously reported studies.
In April 2023, the NDA in China was accepted for review by the NMPA.
91
Fruquintinib in Combination with Checkpoint Inhibitors
The table below shows a summary of clinical trials for fruquintinib in combination with checkpoint inhibitors.
Clinical Trials of Fruquintinib with Checkpoint Inhibitors
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Fruquintinib + TYVYT (PD-1) |
| FRUSICA-1: endometrial cancer |
| China |
| II registration-intent |
| Fully enrolled; NDA filing expected in early-2024; Ib data at CSCO 2021 |
| NCT03903705 |
Fruquintinib + TYVYT (PD-1) |
| FRUSICA-2: clear cell renal cell carcinoma |
| China |
| II/III |
| Fully enrolled; topline results expected around year end 2024 |
| NCT05522231 |
Fruquintinib + TYVYT (PD-1) | Clear cell renal cell carcinoma | China | Ib/II | Fully enrolled; Updated data at ASCO 2023 | NCT03903705 | |||||
Fruquintinib + TYVYT (PD-1) | CRC | China | II | Data published in European Journal of Cancer | NCT04179084 | |||||
Fruquintinib + TYVYT (PD-1) | Gastrointestinal tumors, NSCLC, cervical cancer | China | Ib/II | Fully enrolled; Gastric cancer data at ESMO 2023; NSCLC and cervical cancer data at ESMO Asia 2023 | NCT03903705 | |||||
Fruquintinib and tislelizumab (PD-1) | MSS-CRC | U.S. | Ib/II | Ongoing since 2021; Fully enrolled; Follow-up ongoing; Conference submission pending completion of follow-up | NCT04577963 | |||||
Fruquintinib and tislelizumab (PD-1) | CRC | Korea / China | Ib/II | Fully enrolled | NCT04716634 |
In November 2018, we entered into two collaboration agreements to evaluate the safety, tolerability and efficacy of fruquintinib in combination with checkpoint inhibitors. These include a global collaboration with Innovent to evaluate the combination of fruquintinib with Innovent’s Tyvyt, a PD-1 monoclonal antibody approved in China, and a collaboration in China with Genor to evaluate the fruquintinib combination with geptanolimab, a PD-1 monoclonal antibody being developed by Genor. In May 2020, we entered into a collaboration agreement with BeiGene to evaluate the safety, tolerability and efficacy of combining two of our drug candidates, including fruquintinib, with BeiGene’s anti-PD-1 antibody tislelizumab.
Tyvyt combination for advanced endometrial cancer registration-intent cohort (NCT03903705)
Platinum-based systemic chemotherapy is the standard first-line treatment for advanced endometrial cancer. However, patients who progress following first-line chemotherapy have limited treatment options, and the prognosis remains poor. As disclosed at CSCO 2021, as of the data cutoff date of August 31, 2021, 35 patients were enrolled (NCT03903705), including 7 treatment-naïve and 28 pretreated patients. Of them, 29 were efficacy evaluable, 4 were treatment-naïve and 25 were pretreated. All 4 treatment-naïve patients experienced confirmed tumor response, for ORR of 100% (95% CI: 39.8-100.0), and median PFS was not reached. Among the 25 pretreated patients, the confirmed ORR was 32.0% (95% CI: 14.9-53.5), DCR was 92.0% (95% CI: 74.0-99.0) and the median PFS was 6.9 months (95% CI: 4.1-NR). Among the 19 proficient mismatch repair (pMMR) patients in the pretreated cohort, the confirmed ORR was 36.8% (95% CI: 16.3-61.6), DCR was 94.7% (95% CI: 74.0-99.9), median PFS was 6.9 months (95% CI: 4.1-NR), and the median OS was not reached. Among the 35 enrolled patients, treatment-related adverse events of grade 3 or above that occurred in more than 10% of patients were hypertension (4 patients, 11.4%) and proteinuria (4 patients, 11.4%). 5 (14.3%) patients reported treatment-related serious adverse events. Following encouraging data in the advanced endometrial cancer cohort, it has been expanded into a single-arm registrational Phase II study of over 130 patients.
We agreed with the NMPA to expand this cohort into a single-arm registrational Phase II study. In July 2023, the cohort fully enrolled and was granted Breakthrough Therapy Designation. If the study results are positive, we expect to file the NDA with the NMPA in this treatment setting in early-2024.
92
Tyvyt combination for advanced metastatic renal carcinoma (NCT05522231)
In first-line clear-cell renal cell carcinoma (“ccRCC”), clinical benefits have been demonstrated for the combination of antiangiogenic therapy and immunotherapy. However, there is limited evidence on the benefits of this combination in the second-line setting. Phase II (NCT03903705) data disclosed at ASCO 2023 showed encouraging anti-tumor efficacy and durability in these patients. PFS results from this exploratory study of the fruquintinib and sintilimab combination in metastatic clear-cell RCC were reported. At data cut-off on November 30, 2022, median PFS was 15.9 months in 20 previously treated patients. No new safety signals were observed.
A Phase II/III trial of fruquintinib in combination with Tyvyt as second-line treatment for locally advanced or metastatic RCC was initiated in October 2022. The study is a randomized, open-label, active-controlled study to evaluate the efficacy and safety of fruquintinib in combination with Tyvyt versus axitinib or everolimus monotherapy for the second-line treatment of advanced RCC. The primary endpoint is PFS. The enrollment was completed in December 2023. A total of 234 patients have been enrolled in the study. We expect to announce topline results around year end in 2024.
Tyvyt combination for CRC (NCT04179084)
Encouraging preliminary data presented at ASCO 2021 for fruquintinib in combination with two PD-1 inhibitors, Tyvyt and geptanolimab, in advanced CRC showed a five-fold increase in ORR and a doubling of median PFS as compared to the FRESCO study for fruquintinib as a monotherapy.
In the Tyvyt combination study (NCT04179084), at the final analysis with data cut-off date of December 30, 2021, 44 patients were enrolled into the CRC cohort (43 efficacy evaluable), 22 (21 efficacy evaluable) of whom received the RP2D. ORR was 21% for all patients and 24% for those who received the RP2D. DCR was 88% for all patients and 100% for those who received the RP2D. Median PFS was 5.6 months for all patients, and 6.9 months for those who received the RP2D. Median OS was 14.3 months for all patients and 14.8 months for those who received PRZD.
In the geptanolimab combination study (NCT03977090), for the 15 patients in the CRC cohort ORR was 26.7% (including 1 patient with unconfirmed PR) and 33% in the group that received the RP2D. DCR for all evaluable patients was 80% and median PFS was 7.3 months (95% CI: 1.9-NR). Grade 3 treatment-related adverse events occurred in 47% of patients, and no incidences of grade 4 or 5 treatment-related adverse events were observed.
Tislelizumab combinations for CRC (NCT04577963 & NCT04716634)
A MSS-CRC cohort was added to an open-label, multi-center, non-randomized Phase Ib/II study in the U.S. to assess fruquintinib in combination with tislelizumab. The Phase II study in China and Korea for fruquintinib in combination with tislelizumab was initiated and is being led by BeiGene for the treatment of advanced or metastatic, unresectable CRC.
Fruquintinib Exploratory Development
In China, we support an investigator-initiated trial program for fruquintinib, and there are about 90 of such trials ongoing in various solid tumor settings. A number of investigator-initiated trials were presented at ASCO 2023, ESMO 2023 and ASCO GI 2024, including initial results of a Phase II study of fruquintinib in combination with investigator’s choice of chemotherapy in second-line metastatic CRC with microsatellite stable (MSS) phenotype, as well as fruquintinib monotherapy for the treatment of biliary tract cancer and soft tissue sarcoma.
Overview of Fruquintinib Commercial Launch
Fruquintinib was first approved in mainland China by the NMPA in September 2018 and commercially launched in November 2018. In February 2022, it received marketing approval in Macau. In November 2023, it received marketing approval in the United States. In January 2024, it received marketing approval in Hong Kong which is the first medicine to be approved under Hong Kong’s new mechanism for registration of new drugs (“1+” mechanism) officially commenced on November 1, 2023. Fruquintinib is marketed as Elunate in China and Fruzaqla in the United States. We have partnered with Eli Lilly on fruiquintinib in China and Takeda outside of China.
93
Starting on January 1, 2020, Elunate was included on China’s NRDL at a 63% discount to its initial retail price for two years, paving the way to significantly broaden access for advanced CRC patients and rapidly build penetration in China over the coming years. The inclusion was renewed pursuant to which we agreed to a discount of 5% relative to the 2021 NRDL price, and Elunate will continue to be included in the NRDL starting January 2022 for another two years.
In 2021, we generated $53.5 million in total revenue from Elunate sales, of which $10.3 million was royalty revenue, $15.8 million was revenue from sales of goods primarily to Eli Lilly and $27.4 million was revenue from promotion and marketing services to Eli Lilly. In 2022, we generated $69.9 million in total revenue from Elunate, of which $13.9 million was royalty revenue, $14.7 million was revenue from sales of goods primarily to Eli Lilly and $41.3 million was revenue from promotion and marketing services to Eli Lilly. In 2023, we generated $83.2 million in total revenue from Elunate, of which $16.6 million was royalty revenue, $18.0 million was revenue from sales of goods primarily to Eli Lilly and $48.6 million was revenue from promotion and marketing services to Eli Lilly.
In 2023, we generated $7.2 million in total revenue from Fruzaqla sales, of which $2.1 million was royalty revenue and $5.1 million was revenue from sales of goods.
Collaboration Partnerships
Eli Lilly
In October 2013, we entered into a license and collaboration agreement with Eli Lilly in order to accelerate and broaden our fruquintinib development program in China. As a result, we were able to quickly expand the clinical development of fruquintinib into indications with unmet medical needs in China including CRC and gastric cancer, as discussed above. In December 2018, we amended our license and collaboration agreement with Eli Lilly. This amendment gives us, among other things, all planning, execution and decision making responsibilities for life cycle indication development of fruquintinib in China. Support from Eli Lilly has also helped us to establish our own manufacturing (formulation) facility in Suzhou, China, which now produces clinical and commercial supplies of fruquintinib. In July 2020, we reached an agreement with Eli Lilly to take over development and execution of all on-the-ground medical detailing, promotion and local and regional marketing activities for Elunate in China starting on October 1, 2020. Under the terms of the new agreement, we will share gross profits linked to sales target performance. Subject to meeting pre-agreed sales targets, Eli Lilly will pay us an estimated total of 70% to 80% of Elunate in-market sales in the form of royalties, manufacturing costs and service payments.
For more information regarding our partnership with Eli Lilly, see “—Overview of Our Collaborations—Eli Lilly.”
Takeda
In January 2023, we entered into an agreement with a subsidiary of Takeda whereby it received an exclusive worldwide license from us to develop, manufacture and commercialize fruquintinib in all indications and territories outside of China. Subject to the terms of the agreement, we are eligible to receive up to $1.13 billion upfront and milestone payments. We have received $435 million in payments to date, including $400 million upfront on closing of the agreement and $35 million milestone payments from Takeda pursuant to this collaboration. We are eligible to receive up to $695 million in additional potential payments relating to regulatory, development and commercial sales milestones, as well as royalties on net sales.
For more information regarding our partnership with Takeda, see “—Overview of Our Collaborations—Takeda.”
3. Surufatinib (HMPL-012), VEGFR 1, 2 and 3, FGFR1 and CSF-1R Inhibitor
Surufatinib is a novel, oral angio-immuno kinase inhibitor that selectively inhibits the tyrosine kinase activity associated with VEGFR and FGFR, both shown to be involved in tumor angiogenesis, and CSF-1R, which plays a key role in regulating tumor-associated macrophages, promoting the body’s immune response against tumor cells. Surufatinib has been studied in clinical trials with around 2,900 patients to date, both as a monotherapy and in combinations, and is approved in China. We currently retain rights to surufatinib worldwide.
94
Initial approvals for surufatinib in China are for the treatment of advanced NET patients. NETs present in the body’s organ system with fragmented epidemiology. About 58% of NETs originate in the gastrointestinal tract and pancreas, 27% in the lung or bronchus, and a further 15% in other organs or unknown origins.
Surufatinib’s ability to inhibit angiogenesis, block the accumulation of tumor associated macrophages and promote infiltration of effector T cells into tumors could help improve the anti-tumor activity of PD-1 antibodies. Several combination studies with PD-1 antibodies have shown promising data.
Mechanism of Action
Both VEGFR and FGFR signaling pathways can mediate tumor angiogenesis. CSF-1R plays an important role in the functions of macrophages. Recently, the roles in increasing tumor immune evasion of VEGFR, FGFR in regulation of T cells, tumor-associated macrophages and myeloid-derived suppressor cells have been demonstrated. Therefore, blockade of tumor angiogenesis and tumor immune evasion by simultaneously targeting VEGFR 1, 2 and 3, FGFR1 and CSF-1R kinases may represent a promising approach for oncology therapy.
Surufatinib Pre-clinical Evidence
Surufatinib inhibited VEGFR 1, 2, and 3, FGFR1 and CSF-1R kinases with IC50 in a range of 1 nM to 24 nM. It also strongly blocked VEGF-induced VEGFR2 phosphorylation in HEK293 cells and CSF-1R phosphorylation in RAW264.7 cells with an IC50 of 2 nM and 79 nM, respectively. Surufatinib also reduced VEGF- or FGF-stimulated human umbilical vein endothelial cell proliferation with an IC50 < 50 nM. In animal studies, a single oral dose of surufatinib inhibited VEGF-stimulated VEGFR2 phosphorylation in lung tissues of nude mice in an exposure-dependent manner. Furthermore, elevation of FGF23 levels in plasma 24 hours post dosing suggested suppression of FGFR signaling.
Surufatinib demonstrated potent tumor growth inhibition in multiple human xenograft models and decreased cluster of differentiation 31 expression remarkably, suggesting strong inhibition on angiogenesis through VEGFR and FGFR signaling. In a syngeneic murine colon cancer model, surufatinib demonstrated moderate tumor growth inhibition after single-agent treatment. Flow cytometry and immunohistochemistry analysis revealed an increase of certain T cells and a significant reduction in certain tumor-associated macrophages, including CSF-1R mutation positive tumor-associated macrophages in tumor tissue, indicating surufatinib has a strong effect on CSF-1R. Interestingly, a combination of surufatinib with a PD-L1 antibody resulted in enhanced anti-tumor effect. These results suggested that surufatinib has a strong effect in modulating angiogenesis and cancer immunity.
Surufatinib Clinical Development
We currently have various clinical trials of surufatinib as a monotherapy and in combination with checkpoint inhibitors ongoing or expected to begin in the near term.
Surufatinib Monotherapy - Neuroendocrine Tumors
Neuroendocrine tumors begin in the specialized cells of the body’s neuroendocrine system. Cells have traits of both hormone-producing endocrine cells and nerve cells. Neuroendocrine tumors are found throughout the body’s organ system and have complex and fragmented epidemiology, about 58% of NETs originate in the gastrointestinal tract and pancreas, 27% in the lung or bronchus, and a further 15% in other organs or unknown origins. In China, there are an estimated approximately 34,000 new patients of advanced NETs per year.
NETs can be functional, releasing hormones and peptides that cause symptoms like diarrhea and flushing, or non-functional with no symptoms. Early-stage NETs, which are often functional, can be treated with somatostatin analogue subcutaneous injections, which are approved and reimbursed in China and alleviate symptoms and slow NET growth, but have limited tumor reduction efficacy.
In late 2020, surufatinib was granted approval for drug registration by the NMPA for the treatment of non-pancreatic NET and launched in mid-January 2021 within three weeks of approval. Surufatinib was granted approval for drug registration by the NMPA for the treatment of advanced pancreatic NET and launched in June 2021. We believe that surufatinib is currently the only approved targeted therapy that can address and treat all subtypes of NETs.
95
Surufatinib received FDA Fast Track Designations in April 2020 for the treatment of pNETs and epNETs. Orphan Drug Designation for pNETs was granted in November 2019. While discussions with the FDA in 2020 suggested that two positive Phase III studies of surufatinib in patients with pNETs and epNETs in China, could form the basis to support a U.S. NDA submission, this was ultimately not accepted. A new multi-regional clinical trial (MRCT) would be required to move forward with this program in the United States, Europe and Japan. Following dialogue with the Japanese PMDA, we have decided not to file a Japanese NDA on the basis of the clinical trial data available at this time.
The table below shows a summary of clinical trials for surufatinib in neuroendocrine cancer patients.
Clinical Trials of Surufatinib in NETs
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Surufatinib monotherapy |
| SANET-ep: Non-pancreatic NET |
| China |
| III |
| Approved; Launched in 2021 |
| NCT02588170 |
Surufatinib monotherapy |
| SANET-p: Pancreatic NET |
| China |
| III |
| Approved; Launched in 2021 |
| NCT02589821 |
SANET-ep study: Phase III study of surufatinib monotherapy in non-pancreatic NETs (NCT02588170)
In 2015, we initiated the SANET-ep study, which is a Phase III study in China in patients with grade 1 and 2 advanced non-pancreatic NETs. In this study, patients were randomized at a 2:1 ratio to receive either an oral dose of 300 mg of surufatinib or a placebo once daily on a 28-day treatment cycle. The primary endpoint was PFS, with secondary endpoints including ORR, disease control rate, time to response, duration of response, OS, safety and tolerability.
A 198-patient interim analysis was conducted on SANET-ep in mid-2019, leading the independent data monitoring committee, or IDMC, to determine that it had met the pre-defined primary endpoint of PFS and should be stopped early. The positive results of this trial were highlighted in an oral presentation at the 2019 European Society for Medical Oncology Congress, and subsequently published in The Lancet Oncology in September 2020. Median PFS per investigator assessment was 9.2 months for patients treated with surufatinib, as compared to 3.8 months for patients in the placebo group (HR 0.334; 95% CI: 0.223, 0.499; p<0.0001). Efficacy was also supported by a blinded independent image review committee assessment. Surufatinib was well-tolerated in this study and the safety profile was consistent with observations in prior clinical studies. CTC grade 3 or above TEAEs in this study with greater than 5% incidence were hypertension (36%), proteinuria (19%) and anemia (7%).
96
SANET-ep Clearly Succeeded in Meeting Primary Endpoint of PFS
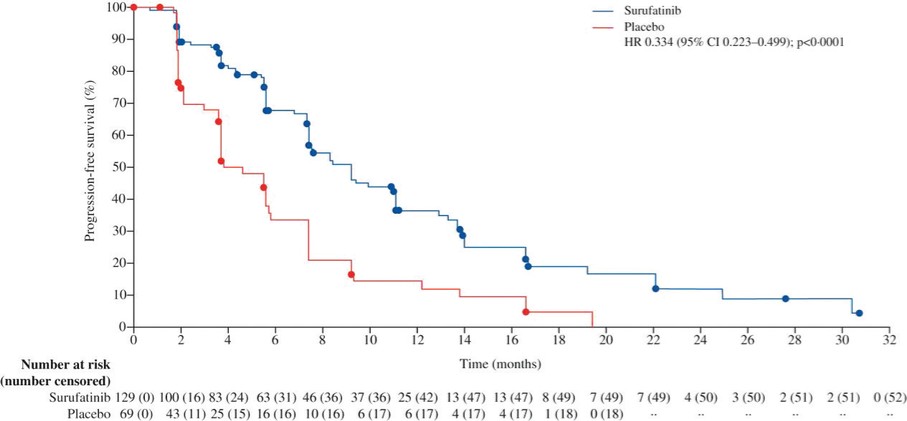
Notes: | P-value is obtained from the stratified one-sided log-rank test; Hazard ratio is obtained from stratified Cox model; CI = confidence interval; and HR = hazard ratio. |
Source: | Xu J, Shen L, Zhou Z, et al. Surufatinib in advanced extra-pancreatic neuroendocrine tumours (SANET-ep): a randomized, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21(11):1500-1512. doi:10.1016/S1470-2045(20)30496-4. |
SANET-p study: Phase III study of surufatinib monotherapy in pancreatic NETs (NCT02589821)
In 2016, we initiated the SANET-p study, which is a Phase III study in China in patients with low- or intermediate-grade, advanced pancreatic NETs. In this study, patients are randomized at a 2:1 ratio to receive either an oral dose of 300 mg of surufatinib or a placebo once daily on a 28-day treatment cycle. The primary endpoint is PFS, with secondary endpoints including ORR, disease control rate, time to response, duration of response, OS, safety and tolerability.
In early 2020, an interim analysis was conducted on SANET-p, leading the IDMC to recommend that the study stop early as the pre-defined primary endpoint of PFS had already been met. Investigator-assessed median PFS was 10.9 months for patients treated with surufatinib, as compared to 3.7 months for patients in the placebo group (HR 0.491; 95% CI: 0.319-0.755; p = 0.0011). ORRs were 19.2% for the efficacy evaluable patients in the surufatinib group versus 1.9% for the placebo group, with a DCR of 80.8% versus 66.0%, respectively. Most patients in the trial had Grade 2 disease with heavy tumor burden, including liver metastasis and multiple organ involvement. Efficacy was also supported by blinded independent image review committee assessment, with a median PFS of 13.9 months for surufatinib as compared to 4.6 months for placebo (HR 0.339; 95% CI 0.209-0.549; p<0.0001). The safety profile of surufatinib was manageable and consistent with observations in prior studies. Treatment was well tolerated for most patients, with discontinuation rates as a result of TEAEs of 10.6% in the surufatinib group as compared to 6.8% in the placebo group. CTC grade 3 or above TEAEs in this study with greater than 5% incidence were hypertension (38%), proteinuria (10%) and hypertriglyceridemia (7%).
97
SANET-p Clearly Succeeded in Meeting Primary Endpoint of PFS
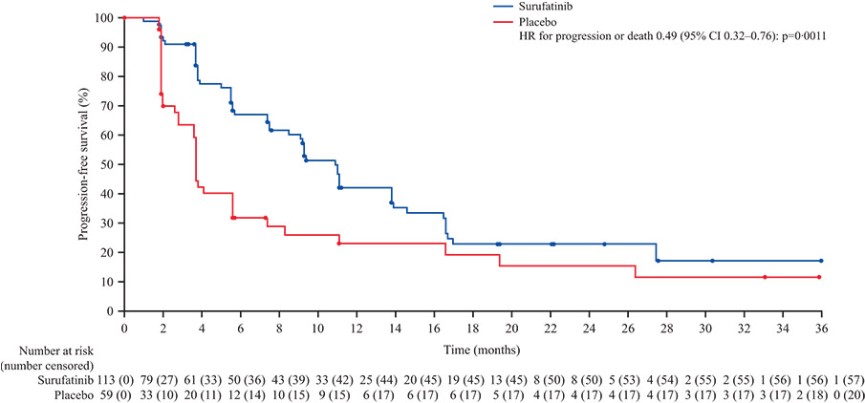
Notes: | P-value is obtained from the stratified one-sided log-rank test; Hazard ratio is obtained from stratified Cox model; CI = confidence interval; and HR = hazard ratio. |
Source: | Xu J, Shen L, Bai C, et al. Surufatinib in advanced pancreatic neuroendocrine tumours (SANET-p): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21(11):1489-1499. doi:10.1016/S1470-2045(20)30493-9. |
Surufatinib in Combination with Checkpoint Inhibitors
Surufatinib’s ability to inhibit angiogenesis, block the accumulation of tumor associated macrophages and promote infiltration of effector T cells into tumors, could help improve the anti-tumor activity of PD-1 antibodies.
The table below shows a summary of clinical trials for surufatinib in combination with checkpoint inhibitors.
Clinical Trials of Surufatinib with Checkpoint Inhibitors
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Surufatinib and Tuoyi (PD-1) |
| SURTORI-01: 2L NEC |
| China |
| III |
| Ongoing since 2021 |
| NCT05015621 |
Surufatinib and Tuoyi (PD-1) |
| NENs, GC, ESCC, SCLC, NSCLC, EMC, TC, STS, BTC |
| China |
| II |
| Fully enrolled; Data at AACR 2023 & ASCO 2023 |
| NCT04169672 |
Surufatinib and Tuoyi (PD-1) | Small cell lung cancer | China | II | Fully enrolled | NCT05509699 |
Clinical Trials with Junshi’s Tuoyi
In late 2018, we entered into a global collaboration with Junshi to evaluate the combination of surufatinib with Tuoyi. We completed a Phase I dose-finding study and presented the data at the AACR Conference in April 2020. The data showed that surufatinib plus Tuoyi were well tolerated with no unexpected safety signals observed. At the recommend Phase 2 dose, a DCR of 100% and ORR of 63.6% were reported for 11 efficacy evaluable patients, with 2 unconfirmed partial responses. Surufatinib plus Tuoyi showed encouraging antitumor activity in patients with advanced solid tumors. A Phase II China study combining surufatinib with Tuoyi enrolled patients in nine solid tumor indications. These have led to the initiation in September 2021 of the first Phase III trial combining surufatinib with a PD-1 antibody, the SURTORI-01 study in NEC and a Phase II study in SCLC in 2022.
98
NEC (subset of NENs) cohort — At the 2021 CSCO annual meeting, we presented data, with a cutoff date of July 30, 2021 for all 21 enrolled NEC patients that were efficacy evaluable. Average duration of treatment was 4.9 months (range 1-19) and median OS was 10.3 months (95% CI: 9.1-not reached). The median PFS was 4.14 months (95% CI: 1.5-5.5) and median DoR was 4.1 months (95% CI: 3.0-not reached). The confirmed ORR was 23.8% (95% CI: 8.2-47.2) and DCR was 71.4% (95% CI: 47.8-88.7). All patients experienced treatment-related adverse events, including 9 (42.9%) who experienced grade 3 or above treatment-related adverse events. 1 (4.8%) patient reported treatment-related serious adverse events. Hyperglycemia (3 patients, 14.3%), hypertension (2 patients, 9.5%) and hypertriglyceridemia (2 patients, 9.5%) were the most commonly (more than one patient) reported grade 3 or above treatment-related adverse events. No treatment-related adverse events led to treatment discontinuation or treatment-related deaths.
In September 2021, we initiated a Phase III study to evaluate the combination compared with FOLFIRI to treat patients with advanced NEC who have progression of disease or intolerable toxicity after previous first-line chemotherapy. It is a randomized, controlled, open-label, multi-center study where approximately 200 patients are expected to be enrolled. For the study group, all patients will receive study treatment in a 21-day cycle. The primary outcome measure is OS. We are the sponsor and responsible for the study’s execution. We and Junshi Biosciences are jointly funding the study.
Other cohorts —We reported the results from the advanced endometrial cancer cohorts at ASCO 2023. Amongst efficacy evaluable endometrial cancer patients, median PFS was 5.4 months and 12-month OS rate was 71.0% (median follow-up duration was 16.8 months). The combination showed a tolerable safety profile. Additionally, results from the NSCLC cohort were presented at AACR 2023 demonstrating promising anti-tumor activity in first-line setting for advanced PD-L1 positive NSCLC patients with manageable toxicity.
Surufatinib Exploratory Development
In China, we support an investigator-initiated trial program for surufatinib, with about 110 of such trials in various solid tumor settings being conducted for both combination and single agent regimens. These trials explore and answer important medical questions in addition to our own company-sponsored clinical trials. A number of investigator-initiated trials were presented at ASCO 2023, ESMO 2023 and ASCO GI 2024 for surufatinib in combination with other agents, including with chemotherapy as well as with anti-PD-1 antibodies plus different chemotherapy regimens in various solid types including pancreatic adenocarcinoma, gastric/gastroesophageal junction adenocarcinoma and biliary tract cancer. In one of these trials using surufatinib in combination with camrelizumab (an anti-PD-1) plus chemotherapy in first-line therapy for pancreatic adenocarcinoma, median PFS and OS were 9.2 months and 15.6 months, respectively, compared to 6.3 months and 8.6 months in the control group with chemotherapy only.
Overview of Surufatinib Commercial Launch
Surufatinib capsules, sold under the brand name Sulanda, were approved for marketing in China by the NMPA in December 2020 and June 2021 for the treatment of advanced non-pancreatic NETs and pancreatic NETs, respectively. In 2021, Sulanda was sold as a self-pay drug whereby patients paid for treatment out-of-pocket. We used means-test early access and patient access programs to help patients afford Sulanda. Following negotiations with the China National Healthcare Security Administration, Sulanda was included on China’s NRDL at a 52% discount on our main 50mg dosage form, relative to the 2021 self-pay price, for two years starting on January 1, 2022. The inclusion was renewed for another two years starting in January 2024 for the same discounted price.
We have also confirmed a total of around 110 investigator-initiated studies in a broad range of exploratory solid tumor indications all of which are expected to gradually expand awareness of Sulanda in China.
4. Sovleplenib (HMPL-523), Syk Inhibitor
Sovleplenib is a novel, selective, oral inhibitor targeting Syk, for the treatment of hematological malignancies and immune diseases. Syk is a component in Fc receptor and B-cell receptor signaling pathway. In December 2022, we completed recruitment of a Phase III study in China for primary ITP, for which it has received Breakthrough Therapy Designation. Positive data on both primary ITP and hematological malignancies was reported at ASH 2021 and published in Lancet Hematology in April 2023. In 2024, we plan to start a Phase I/Ib dose-finding study in the United States. We currently retain all rights to sovleplenib worldwide.
99
Mechanism of Action
Syk is a key kinase upstream to PI3Kδ and BTK within the B-cell signaling pathway and therefore thought to be an important target for modulating B-cell signaling.
Syk, a target for autoimmune diseases
The central role of Syk in signaling processes is not only in cells of immune responses but also in cell types known to be involved in the expression of tissue pathology in autoimmune, inflammatory and allergic diseases. Therefore, interfering with Syk could represent a possible therapeutic approach for treating these disorders. Indeed, several studies have highlighted Syk as a key player in the pathogenesis of a multitude of diseases, including rheumatoid arthritis, systemic lupus erythematosus and multiple sclerosis.
Syk, a target for oncology
In hematological cancer, we believe Syk is a high potential target. In hematopoietic cells, Syk is recruited to the intracellular membrane by activated membrane receptors like B-cell receptors or another receptor called Fc and then binds to the intracellular domain of the receptors. Syk is activated after being phosphorylated by certain kinases and then further induces downstream intracellular signals including B-cell linker, PI3Kδ, BTK and Phospholipase C-y2 to regulate B-cell proliferation, growth, differentiation, homing, survival, maturation, and immune responses. Syk not only involves the regulation of lymphatic cells but also signal transduction of non-lymphatic cells such as mast cells, macrophages, and basophils, resulting in different immunological functions such as degranulation to release immune active substances, leading to immunological reaction and disease. Therefore, regulating B-cell signal pathways through Syk is expected to be effective for treating lymphoma.
Syk is upstream of both BTK and PI3Kδ, and we believe it could deliver the same outcome as inhibitors of BTK and PI3Kδ, assuming no unintentional toxicities are derived from Syk inhibition.
Sovleplenib Research Background
The threshold of safety for a Syk inhibitor in chronic disease is extremely high, with no room for material toxicity. The failure of Tavalisse in a global Phase III registration study in rheumatoid arthritis provided important insights for us in the area of toxicity. While Tavalisse clearly showed patient benefit in rheumatoid arthritis, a critical proof-of-concept for Syk modulation, it also caused high levels of hypertension which is widely believed to be due to the high levels of off-target kinase insert domain receptor inhibition. In addition, Tavalisse has also been shown to strongly inhibit the Ret kinase, and in pre-clinical trials it was demonstrated that inhibition of the Ret kinase was associated with developmental and reproductive toxicities.
The requirement for Syk kinase activity in inflammatory responses was first evaluated with Tavalisse, which was co-developed by AstraZeneca/Rigel Pharmaceuticals, Inc. In 2013, AstraZeneca announced results from pivotal Phase III clinical trials that Tavalisse statistically significantly improved ACR20 (a 20% improvement from baseline based on the study criteria) response rates of patients inadequately responding to conventional disease-modifying anti-rheumatic drugs and a single anti-TNFα (a key pro-inflammatory cytokine involved in rheumatoid arthritis pathogenesis) antagonist at 24 weeks, but failed to demonstrate statistical significance in comparison to placebo at 24 weeks. As a result, AstraZeneca decided not to proceed. Rigel Pharmaceuticals subsequently chose to develop Tavalisse for immune thrombocytopenia instead, for which it was approved by the FDA in 2018 and the EMA in 2020.
Tavalisse was also in trials for B-cell lymphoma and T-cell lymphoma. It demonstrated some clinical efficacy in diffused large B-cell lymphoma patients with an ORR of 22%. Entospletinib has features of high potency and good selectivity toward kinases. However, entospletinib shows some inhibition of the CYP3A4, CYP2D6, and CYP1A2 enzymes involved in the metabolism of certain drugs, and therefore their inhibition could increase the risk of drug-to-drug interaction when used in combined therapy. It is no longer in development.
100
Sovleplenib Pre-clinical Evidence
The safety profile of sovleplenib was evaluated in multiple in vitro and in vivo pre-clinical trials under good laboratory practice guidelines and found to be well tolerated following single dose oral administration. Toxic findings were seen in repeat dose animal safety evaluations in rats and dogs at higher doses and found to be reversible. These findings can be readily monitored in the clinical trials and fully recoverable upon drug withdrawal. The starting dose in humans was suggested to be 5 mg. This dose level is approximately 5% of the human equivalent dose extrapolated from the pre-clinical “no observed adverse event levels,” which is below the 10% threshold recommended by FDA guidelines.
Sovleplenib Clinical Development
The table below shows a summary of the clinical trials for sovleplenib.
Current Clinical Trials of Sovleplenib
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Sovleplenib monotherapy | ESLIM-01≥2L+ Immune thrombocytopenia |
| China |
| III |
| Fully enrolled; positive topline results achieved and NDA accepted with priority review status in Jan 2024; results to be submitted at an upcoming conference in mid-2024; Breakthrough Therapy Designation |
| NCT05029635 | |
Sovleplenib monotherapy | ≥2L ITP |
| U.S. |
| Ib |
| Dose-finding study to begin in 2024 |
| Pending | |
Sovleplenib monotherapy | Warm AIHA |
| China |
| II/III |
| Phase II fully enrolled; Phase III expected in early 2024 |
| NCT05535933 |
ESLIM-01 Phase III study of sovleplenib in patients with immune thrombocytopenia (NCT05029635)
In October 2021, we initiated a randomized, double-blinded, placebo-controlled Phase III trial in China of sovleplenib in 188 adult patients with primary ITP who have received at least one prior line of standard therapy. ITP is an autoimmune disorder that can lead to increased risk of bleeding. The primary endpoint of the study is the durable response rate. In January 2022, the NMPA granted Breakthrough Therapy Designation for this indication. All endpoints were met in August 2023 and the NDA has been accepted for review and granted priority review by the NMPA in January 2024. We plan to submit the results for presentation and/or publication in mid-2024.
ESLIM-01 is supported by the results of a randomized, double-blind and placebo-controlled Phase Ib study of sovleplenib in adult patients with primary immune thrombocytopenia were presented at the 63rd American Society for Hematology’s (ASH) annual meeting. As of data cut-off date of September 30, 2021, a total of 34 patients were randomized to receive HMPL-523 and 11 patients to placebo. Among 16 patients who were randomized to receive the RP2D of 300mg once daily, 11 patients (68.8%) experienced response as defined by at least one incident of platelet count being ≥ 50×109/L in the initial 8-week double blinded phase of the study, compared to one out of 11 patients (9.1%) randomized to receive placebo. During the subsequent 16-week open-label phase of the study, one additional patient initially randomized to receive the RP2D experienced a response. Four patients randomized to placebo crossed over to receive treatment at RP2D after the initial 8-week double blinded phase of the study; all four of these patients experienced response. In total, 16 out of 20 patients (80%) experienced response during both phases of the study. Durable response, defined as platelet count being ≥ 50×109/L in at least 4 out of 6 last scheduled visits, were reported in 8 out of 20 patients (40%) who received RP2D in both phases of the study.
Safety data were presented for all 41 patients who received treatment at all doses, regardless of whether they were initially randomized to receive active treatment or crossed over during the open-label extension phase of the study. The median duration of treatment was 142 days (range: 23-170). No patients discontinued treatment due to treatment-related adverse events, and no cases of treatment-related serious adverse events were reported. There were 30 patients (73%) who experienced treatment-related adverse events, including 3 (7.3%) who experienced grade 3 or above treatment-related adverse events, one of whom received the RP2D. No treatment-related adverse events of grade 3 or above occurred in more than one patient.
101
Phase II/III Trial of sovleplenib for warm AIHA (NCT05535933)
This is a randomized, double-blind, placebo-controlled Phase II/III study to evaluate the efficacy, safety, tolerability, and pharmacokinetics of sovleplenib in the treatment of warm AIHA. AIHA is the result of destruction of red blood cells due to the production of antibodies against red blood cells which bind to antigens on the red blood cell membrane in autoimmune disorders. The first patient was enrolled in September 2022. The enrollment of Phase II part of the study was completed in mid-2023 and primary end point has been met. We expect to initiate Phase III in early 2024.
5. Tazemetostat, EZH2 Inhibitor
Tazemetostat is an inhibitor of EZH2 developed by Ipsen that is approved by the U.S. FDA for the treatment of certain epithelioid sarcoma and follicular lymphoma patients. It received accelerated approval from the FDA based on ORR and DoR in January and June 2020 for epithelioid sarcoma and follicular lymphoma, respectively.
In August 2021, we entered into a strategic collaboration with Epizyme, a subsidiary of Ipsen, to research, develop, manufacture and commercialize tazemetostat in Greater China, including the mainland, Hong Kong, Macau and Taiwan. Tazemetostat is an inhibitor of EZH2 developed by Ipsen that is approved by the U.S. FDA for the treatment of certain epithelioid sarcoma and follicular lymphoma patients. It received accelerated approval from the FDA based on ORR and DoR in January and June 2020 for epithelioid sarcoma and follicular lymphoma, respectively. Tazemetostat has been studied in clinical trials with around 1,300 patients to date.
We are developing and plan to seek approval for tazemetostat in various hematological and solid tumors in China. We are participating in Ipsen’s SYMPHONY-1 (EZH-302) study, leading it in China. We are generally responsible for funding all clinical trials of tazemetostat in China, including the portion of global trials conducted there. Separately, we are conducting a China bridging study in follicular lymphoma for potential conditional registration based on its U.S. approvals. The study is fully enrolled and, subject to the data, we plan to file the NDA in China in late-2024. We are responsible for the research, manufacturing and commercialization of tazemetostat in China. Tazemetostat was approved in China Hainan Pilot Zone in 2022 and the Macau Special Administrative Region in 2023.
Mechanism of Action
EZH2 is one member of a class of histone methyltransferases (“HMTs”). It catalyzes the methylation of histone H3 at lysine 27 (H3K27) which controls expression of various genes and in turn plays a role in the normal physiology of many cell types. Dysregulation of EZH2 has been seen in a wide range of cancers and is associated with poor clinical prognosis and outcomes. Tazemetostat inhibits EZH2 which allows transcription of genes involved in functions such as cell cycle control and terminal differentiation and thus inhibits cancer cell proliferation.
Tazemetostat Clinical Development
The table below shows a summary of our clinical trials for tazemetostat.
Clinical Trials of Tazemetostat
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Tazemetotat monotherapy | Metastatic or locally advanced | Hainan | N/A – Hainan Pilot | Approved; Launched in 2022 and 2023, respectively | N/A | |||||
Tazemetotat monotherapy |
| Relapsed/refractory 3L+ follicular lymphoma | China | II registration-intent (bridging) | Fully enrolled; NDA filing expected in late-2024 |
| NCT05467943 | |||
Tazemetostat +lenalidomide + rituximab (R2) |
| SYMPHONY-1: 2L follicular lymphoma | Global | Ib/III | Ongoing: PhIb data at ASH 2022; China portion of global Phase III trial started in the second half of 2022 |
| NCT04224493 | |||
Tazemetostat + amdizalisib |
| Relapsed/refractory lymphoma | China | II | Ongoing since February 2023 |
| NCT05713110 |
102
SYMPHONY-1 Phase Ib/III Trial of tazemetostat for follicular lymphoma (NCT04224493)
SYMPHONY-1 is a global, multicenter, randomized, double-blind, active-controlled, 3-stage, biomarker-enriched, Phase Ib/III study of tazemetostat in combination with R² (lenalidomide and rituximab) in patients with relapsed or refractory follicular lymphoma after at least one prior line of therapy. Ipsen conducted the Phase Ib portion of the study in 2021, which determined the recommended Phase III dose and also demonstrated potential efficacy in second-line follicular lymphoma. The safety profile of the combination was consistent with the previously reported safety information in the U.S. prescribing information for both tazemetostat and R², respectively.
The Phase Ib open-label portion of SYMPHONY-1 recruited 44 patients and showed ORR of 90.9%. In the 800-mg BID recommended Phase III dose cohort, 18-month PFS and DOR estimates were 94.4% and 100%. There were no dose-limiting toxicities.
In the Phase III portion of the trial, approximately 500 patients are randomly assigned to receive the recommended Phase III dose of tazemetostat of tazemetostat + R² or placebo + R². The study will also include a maintenance arm with tazemetostat or placebo following the first year of treatment with tazemetostat + R² or placebo + R². The first patient was enrolled in May 2022 and the first China patient was enrolled in September 2022.
Phase II bridging study in relapsed/refractory follicular lymphoma (NCT05467943)
In July 2022, we initiated a multicenter, open-label, Phase II study to evaluate the efficacy, safety and pharmacokinetics of tazemetostat for the treatment of patients with relapsed/refractory follicular lymphoma intended to support conditional registration in China. The primary objective is to evaluate the efficacy of tazemetostat in patients with EZH2 mutation (Cohort 1). The secondary objectives are to evaluate the efficacy of tazemetostat in patients with EZH2 wild-type (Cohort 2) and to evaluate the safety and the pharmacokinetics of tazemetostat. Enrollment of cohort 2 is complete and cohort 1 is ongoing and is expected to be completed in 2023.
6. HMPL-453, FGFR Inhibitor
HMPL-453 is a novel, selective, oral inhibitor targeting FGFR 1/2/3. Aberrant FGFR signaling is associated with tumor growth, promotion of angiogenesis, as well as resistance to anti-tumor therapies. Approximately 10-15% of IHCC patients have tumors harboring FGFR2 fusion. We currently retain all rights to HMPL-453 worldwide.
Mechanism of Action
FGFR belongs to a subfamily of receptor tyrosine kinases. Four different FGFRs (FGFR1-4) and at least 18 ligand FGFs constitute the FGF/FGFR signaling system. Activation of the FGFR pathway through the phosphorylation of various downstream molecules ultimately leads to increased cell proliferation, migration and survival. FGF/FGFR signaling regulates a wide range of basic biological processes, including tissue development, angiogenesis, and tissue regeneration. Given the inherent complexity and critical roles in physiological processes, dysfunction in the FGF/FGFR signaling leads to a number of developmental disorders and is consistently found to be a driving force in cancer. Deregulation of the FGFR can take many forms, including receptor amplification, activating mutations, gene fusions, and receptor isoform switching, and the molecular alterations are found at relatively low frequencies in most tumors. The incidence of FGFR aberrance in various cancer types is listed in the table below.
Common FGFR Alterations in Certain Tumor Types
| Gene amplification |
| Gene translocation |
| Gene mutation | |
FGFR1 |
| Lung squamous (7-15%) |
| Lung squamous (n/a) |
| Gastric (4%) |
| H&N squamous (10-17%) |
| Glioblastoma (n/a) |
| Pilocytic astrocytoma (5-8%) | |
| Esophageal squamous (9%) |
| Myeloproliferative syndrome (n/a) | |||
| Breast (10-15%) |
| Breast (n/a) | |||
FGFR2 |
| Gastric (5-10%) |
| Intra-hepatic biliary tract cancer (14%) |
| Endometrial (12-14%) |
| Breast (5-10%) |
| Breast (n/a) |
| Lung squamous (5%) | |
FGFR3 |
| Bladder (3%) |
| Bladder (3-6%) |
| Bladder (60-80% NMIBC; 15-20% MIBC) |
Salivary adenoid cystic (n/a) | Lung squamous (3%) | Cervical (5%) | ||||
Breast (1%) | Glioblastoma (3-7%) | |||||
Myeloma (15-20%) |
Notes: | H&N = head and neck; NMIBC = non-muscle invasive bladder cancer; MIBC = muscle invasive bladder cancer; and n/a = data not available. |
103
Source: | M. Touat et al., “Targeting FGFR Signaling in Cancer,” Clinical Cancer Research (2015); 21(12); 2684-94. |
HMPL-453 Research Background
We noted a growing body of evidence has demonstrated the oncogenic potential of FGFR aberrations in driving tumor growth, promoting angiogenesis, and conferring resistance mechanisms to oncology therapies. Targeting the FGF/FGFR signaling pathway has therefore attracted attention from biopharmaceutical companies and has become an important exploratory target for new anti-tumor target therapies.
The main FGFR on-target toxicities observed to date in these compounds are all mild and manageable, including hyperphosphatemia, nail and mucosal disorder, and reversible retinal pigmented epithelial detachment. However, there are still many challenges in the development of FGFR-directed therapies. Uncertainties include the screening and stratifying of patients who are most likely to benefit from FGFR targeted therapy. Intra-tumor heterogeneity observed in FGFR amplified cancer may compromise the anti-tumor activity. In addition, the low frequency of specific FGFR molecular aberrance in each cancer type may hinder clinical trial enrollment.
HMPL-453 Pre-clinical Evidence
HMPL-453 is a highly selective and potent, small molecule that targets FGFR 1/2/3 with an IC50 in the low nanomolar range. Its good selectivity was revealed in the screening against 292 kinases. HMPL-453 exhibited strong anti-tumor activity that correlated with target inhibition in tumor models with abnormal FGFR activation.
HMPL-453 has good pharmacokinetic properties characterized by rapid absorption following oral dosing, good bioavailability, moderate tissue distribution and moderate clearance in all pre-clinical animal species. HMPL-453 was found to have little inhibitory effect on major cytochrome P450 enzymes, indicating low likelihood of drug-to-drug interaction issues.
HMPL-453 Clinical Development
The table below shows a summary of our clinical trials for HMPL-453.
Clinical Trials of HMPL-453
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
HMPL-453 monotherapy | 2L Cholangiocarcinoma (IHCC with FGFR fusion) | China | II | Results presented at ASCO 2023; registration cohort enrolling since March 2023 | NCT04353375 | |||||
HMPL-453 + chemotherapies | Multiple | China | I/II | Ongoing since 2022 | NCT05173142 | |||||
HMPL-453 + Tuoyi (PD-1) | Multiple |
| China |
| I/II |
| Ongoing since 2022 |
| NCT05173142 |
Phase II HMPL-453 monotherapy in advanced IHCC (NCT04353375)
This is an open-label, single-arm Phase II study to evaluate the efficacy and safety of HMPL-453 in the treatment of patients with advanced IHCC harboring FGFR2 fusions/rearrangements after at least one line of systemic treatment failure or intolerance. Results from 25 patients treated with two different dosing regimens were presented at the ASCO 2023 annual meeting, supporting the choice of the recommended Phase II dose of 300mg oral QD (ORR of 50%). After consultation with the CDE, a monotherapy registration trial design was agreed with ORR as primary endpoint, and the first patient was enrolled in March 2023.
104
Phase Ib/II HMPL-453 in combination with chemotherapies or toripalimab in advanced solid tumors (NCT05173142)
In January 2022, we initiated a Phase Ib/II, multi-center, two-stage, open-label clinical trial of HMPL-453 in combination with chemotherapy or the anti-PD-1 therapy, toripalimab, to evaluate the safety, tolerability, pharmacokinetics and preliminary efficacy profile of HMPL-453 combination therapy in patients with specific advanced or metastatic solid tumors. The first stage of the study is a dose escalation phase to determine the dose limiting toxicity (DLT) and recommended Phase II dose of HMPL-453 in combination with chemotherapy (gemcitabine and cisplatin) or toripalimab. The second stage of the study is a dose expansion phase in solid tumor patients with either gastric cancer, intrahepatic cholangiocarcinoma, or urothelial carcinoma, harboring specific FGFR gene alterations. Each solid tumor cohort will be treated with a specific combination of HMPL-453 and a chemotherapy or anti-PD-1 therapy to further evaluate the preliminary efficacy, safety and tolerability at the recommended Phase II dose.
7. Amdizalisib (HMPL-689), PI3Kδ Inhibitor
Amdizalisib is a novel, highly selective oral inhibitor targeting the isoform PI3Kδ, a key component in the B-cell receptor signaling pathway. Amdizalisib has been studied in clinical trials with around 500 patients to date. We currently retains all rights to amdizalisib worldwide.
Mechanism of Action
Targeting the B-cell signaling pathway is emerging as a potential means to treat both hematological cancer and immunological diseases. Inhibiting different kinases found along the B-cell signaling pathway has proven to have clinical efficacy in hematological cancers, with breakthrough therapies having been recently approved by the FDA.
The high efficacy and successful approvals of Bruton’s tyrosine kinase, or BTK, inhibitors and PI3Kδ inhibitors are evidence that modulation of the B-cell signaling pathway is critical for the effective treatment of B-cell malignancies.
Class I phosphatidylinositide-3-kinases, or PI3Ks, are lipid kinases that, through a series of intermediate processes, control the activation of several important signaling proteins including the serine/threonine kinase AKT.
There are multiple sub-families of PI3K kinases, and PI3Kδ is a lipid kinase that, through a series of intermediate processes, controls the activation of several important signaling proteins, including the serine/threonine kinase B, or AKT. In most cells, AKT is a key PI3Kδ affector that regulates cell proliferation, carbohydrate metabolism, cell motility and apoptosis and other cellular processes. Upon an antigen binding to B-cell receptors, PI3Kδ can be activated through the Lyn and Syk signaling cascade.
Aberrant B-cell function has been observed in multiple immunological diseases and B-cell mediated malignancies. Therefore, PI3Kδ is considered to be a promising target for drugs that aim to prevent or treat hematologic cancer, autoimmunity and transplant organ rejection and other related inflammation diseases.
Amdizalisib Pre-clinical Evidence
Compared to other PI3Kδ inhibitors, amdizalisib shows higher potency and selectivity.
Enzyme Selectivity (IC50, in nM) of amdizalisib Versus Competing PI3Kδ Inhibitors; This Shows amdizalisib is Approximately Five- fold More Potent than Zydelig on Whole Blood Level and, unlike Copiktra, does not Inhibit PI3K-g
Enzyme IC50 (nM) |
| HMPL‑689 |
| Zydelig |
| Copiktra |
| Aliqopa |
PI3Kδ |
| 0.8 (n = 3) |
| 2 |
| 1 |
| 0.7 |
PI3Kγ (fold vs. PI3Kδ) |
| 114 (142x) |
| 104 (52x) |
| 2 (2x) |
| 6.4 (9x) |
PI3Kα (fold vs. PI3Kδ) |
| >1,000 (>1,250x) |
| 866 (433x) |
| 143 (143x) |
| 0.5 (1x) |
PI3Kδ human whole blood CD63+ |
| 3 |
| 14 |
| 15 |
| n/a |
PI3Kβ (fold vs. PI3Kδ) |
| 87 (109x) |
| 293 (147x) |
| 8 (8x) |
| 3.7 (5x) |
Source: Company.
105
Amdizalisib Clinical Development
The table below shows a summary of the clinical studies for amdizalisib.
Clinical Trials of Amdizalisib
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
Amdizalisib monotherapy | 3L Relapsed/refractory follicular lymphoma | China | II registration-intent | Met primary endpoint; Breakthrough Therapy Designation | NCT04849351 | |||||
Amdizalisib monotherapy | 2L Relapsed/refractory marginal zone lymphoma | China | II registration-intent | Ongoing since April 2021 | NCT04849351 | |||||
Amdizalisib monotherapy | Indolent non-Hodgkin’s lymphoma PTCL |
| China |
| Ib |
| Completed; Updated data presented at ICML 2023 |
| NCT03128164 |
Phase II registration-intent study of amdizalisib in patients with relapsed/refractory follicular lymphoma and relapsed/refractory marginal zone lymphoma in China (NCT04849351)
In April 2021, we commenced a registration-intent, single-arm, open-label Phase II trial in China in approximately 100 patients with relapsed/refractory follicular lymphoma and approximately 80 patients with relapsed/refractory marginal zone lymphoma, two subtypes of non-Hodgkin’s lymphoma. The trial has fully enrolled the follicular lymphoma cohort and the marginal zone lymphoma cohort enrollment is ongoing. In the follicular lymphoma cohort, the primary endpoint of ORR met its pre-specified threshold of demonstrating a clinically meaningful and a significant increase in ORR in this setting. However, in view of the changing regulatory landscape, we are currently evaluating the clinical development plan and regulatory guidance before deciding the regulatory strategy for this indication.
Phase Ib study of amdizalisib in patients with Indolent non-Hodgkin’s lymphoma in China (NCT03128164)
Our Phase I/Ib study of amdizalisib in China has successfully established a Phase II dose and has now expanded into multiple sub-categories of indolent non-Hodgkin’s lymphoma. Updated safety data as well as efficacy data were reported at ICML in June 2023. At median follow-up duration of 22.1 months, median DoR and PFS were not reached for the 26 efficacy evaluable patients in the follicular lymphoma cohort. For the marginal zone lymphoma cohort of 16 efficacy evaluable patients, at median follow-up duration of 20.3 months, median DoR was not reached and median PFS was 26.8 months. Amdizalisib showed an acceptable safety profile and promising anti-tumor activity in relapsed/refractory lymphoma.
8. HMPL-306, IDH1 and 2 Inhibitor
HMPL-306 is a novel dual-inhibitor of IDH1 and IDH2 enzymes. IDH1 and IDH2 mutations have been implicated as drivers of certain hematological malignancies, gliomas and solid tumors, particularly among acute myeloid leukemia patients. We currently retain all rights to HMPL-306 worldwide.
Mechanism of Action
IDHs are critical metabolic enzymes that help to break down nutrients and generate energy for cells. When mutated, IDH creates a molecule that alters the cell’s genetic programming and prevents cells from maturing, 2-hydroxyglutarate (“2-HG”). Reduction in 2-HG levels can be used as a marker of target engagement by an IDH inhibitor. IDH1 or IDH2 mutations are common genetic alterations in various types of blood and solid tumors, including acute myeloid leukemia, with approximately 20% of patients having mutant IDH genes, myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPNs), low-grade glioma and intrahepatic cholangiocarcinoma. Mutant IDH isoform switching, either from cytoplasmic mutant IDH1 to mitochondrial mutant IDH2, or vice versa, is a mechanism of acquired resistance to IDH inhibition in acute myeloid leukemia and cholangiocarcinoma.
Cytoplasmic mutant IDH1 and mitochondrial mutant IDH2 have been known to switch to the other form when targeted by an inhibitor of IDH1 mutant alone or IDH2 mutant alone. By targeting both IDH1 and IDH2 mutations, HMPL-306 could potentially provide therapeutic benefits in cancer patients harboring either IDH mutation and may address acquired resistance to IDH inhibition through isoform switching.
106
HMPL-306 Clinical Development
The table below shows a summary of our clinical trials for HMPL-306.
Clinical Trials of HMPL-306
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
HMPL-306 monotherapy | Myeloid hematological malignancies | China | I | Dose escalation data presented at EHA 2023; registration Phase III study planned in 2024 | NCT04272957 | |||||
HMPL-306 monotherapy | Solid tumors including but not limited to gliomas, chondrosarcomas or cholangiocarcinomas | U.S. | I | Ongoing since 2021 | NCT04762602 | |||||
HMPL-306 monotherapy | Hematological malignancies | U.S. | I | Ongoing since 2021 | NCT04764474 |
Phase I HMPL-306 monotherapy in hematological malignancies (NCT04272957)
This is a two-phase, open-label Phase I study to evaluate the safety, pharmacokinetics, pharmacodynamics and efficacy of HMPL‑306 in patients of relapsed or refractory hematological malignancies harboring IDH1 and/or IDH2 mutations. The dose escalation phase of the study is completed. The first-in-human dose-escalation phase data was presented at EHA Annual Meeting in June 2023 with ORR of 45-50%. Based on the pharmacodynamic, pharmacokinetic and preliminary clinical findings, a recommended Phase II dose was determined for the dose expansion phase of the study.
9. HMPL-760, BTK Inhibitor
HMPL-760 is an investigational, non-covalent, third generation BTK inhibitor. It is a highly potent, selective, and reversible inhibitor with long target engagement against BTK, including wild-type and C481S-mutated BTK. China phase I studies opened in early 2022 will include relapsed or refractory B-cell non-Hodgkin’s lymphoma or CLL patients with or without a prior regimen containing a BTK inhibitor. We currently retain all rights to HMPL-760 worldwide.
Mechanism of Action
BTK is a key component of the B-cell receptor signaling pathway and is an important regulator of cell proliferation and cell survival in various lymphomas. The abnormal activation of B-cell receptor signaling is closely related to the development of B-cell type hematological cancers, which represent approximately 85% of all NHL cases. BTK is considered a validated target for drugs that aim to treat certain hematological cancers, however C481S mutation of BTK is a known resistance mechanism for first and second generation BTK inhibitors.
HMPL-760 Clinical Development
The table below shows a summary of our clinical trial of HMPL-760.
Clinical Trial of HMPL-760
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # | |
HMPL-760 monotherapy | CLL, SLL, other B-NHL | China | I | Ongoing since January 2022; RP2D determined: dose expansion ongoing | NCT05190068 |
10. HMPL-295, ERK Inhibitor
HMPL-295, a novel ERK inhibitor, is our tenth in-house discovered small molecule oncology drug candidate. ERK is a downstream component of the RAS-RAF-MEK-ERK signaling cascade (MAPK pathway). This is our first of multiple candidates in discovery addressing the MAPK pathway. We currently retain all rights to HMPL-295 worldwide.
107
Mechanism of Action
RAS-MAPK pathway is dysregulated in cancer, in which mutations or nongenetic events hyper-activate the pathway in up to 50% of cancers. RAS and RAF mutations predict worse clinical prognosis in a wide variety of tumor types, mediate resistance to targeted therapies, and decrease the response to the approved standards of care, namely, targeted therapy and immunotherapy. ERK inhibition has the potential to overcome or avoid the intrinsic or acquired resistance from the inhibition of RAS, RAF and MEK upstream mechanisms. Safety and efficacy results on 22 patients with advanced solid tumors were reported during ESMO Asia 2023.
HMPL-295 Clinical Development
The table below shows a summary of our clinical trial for HMPL-295.
Clinical Trial of HMPL-295
|
|
|
|
| ||||||
Treatment | Trial Name, Patient Focus | Sites | Phase | Status/Plan | NCT # | |||||
HMPL-295 monotherapy |
| Solid tumors |
| China |
| I |
| Ongoing since 2021; data at ESMO Asia 2023 |
| NCT04908046 |
We initiated our Phase I development in China in July 2021. This is a multi-center and open-label study to evaluate the safety, tolerability, pharmacokinetics and preliminary efficacy profile of HMPL-295, and to determine the maximum tolerated dose and RP2D in patients with advanced malignant solid tumors.
11. HMPL-653, CSF-1R Inhibitor
HMPL-653 is an investigational novel, highly selective, and potent CSF-1R inhibitor designed to target CSF-1R driven tumors as a monotherapy or in combination with other drugs. We initiated a China Phase I study in Janurary 2022. We currently retain all rights to HMPL-653 worldwide.
Mechanism of Action
CSF-1R is usually expressed on the surface of macrophages and can promote growth and differentiation of macrophages. Studies have shown that blocking the CSF-1R signaling pathway could effectively modulate the tumor microenvironment, relieve tumor immunosuppression, and synergize with other anti-cancer therapies such as immune checkpoint inhibitors to achieve tumor inhibition. It has been demonstrated in several clinical studies that CSF-1R inhibitors could treat tenosynovial giant cell tumors and treat a variety of malignancies combined with immuno-oncology or other therapeutic agents. Currently no CSF-1R inhibitor has been approved in China.
HMPL-653 Clinical Development
The table below shows a summary of our clinical trial of HMPL-653.
Clinical Trial of HMPL-653
|
|
|
|
| ||||||
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
HMPL-653 monotherapy |
| Solid tumors & tenosynovial giant cell tumors |
| China |
| I |
| Ongoing since 2022, ~110 patients expected to be enrolled |
| NCT05190068 |
108
We initiated our Phase I development in China in January 2022, and the study is a multi-center, open-label and single-armed study to evaluate the safety, tolerability, pharmacokinetics and preliminary efficacy of HMPL-653 in patients with advanced or metastatic solid tumors and tenosynovial giant cell tumors. We expect to enroll around 110 patients in this study.
12. HMPL-A83, IgG4-type Humanized Anti-CD47 Monoclonal Antibody
HMPL-A83 is an investigational IgG4-type humanized anti-CD47 monoclonal antibody that exhibits high affinity for CD47. HMPL-A83 blocks CD47 binding to Signal regulatory protein (SIRP) α and disrupts the “do not eat me” signal that cancer cells use to shield themselves from the immune system. In preclinical studies, HMPL‑A83 demonstrated a high affinity for CD47 antigen on tumor cells and strong phagocytosis induction of multiple tumor cells, as well as weak affinity for red blood cells and no induction of hemagglutination, implying low risk of anemia, a potential event of special interest. HMPL-A83 has also demonstrated strong anti-tumor activity in multiple animal models. We currently retain all rights to HMPL-A83 worldwide.
HMPL-A83 Clinical Development
The table below shows a summary of our clinical trial of HMPL-A83.
Clinical Trial of HMPL-A83
Treatment |
| Trial Name, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
|
HMPL-A83 monotherapy | Advanced malignant neoplasms | China | I | Ongoing since July 2022 | NCT05429008 |
13. HMPL-415, SHP2 Allosteric Inhibitor.
HMPL-415 is a novel SHP2 allosteric inhibitor. A China Phase I study was initiated in July 2023. We currently retain all rights to HMPL-415 worldwide. SHP2 is a non-receptor protein tyrosine phosphatase ubiquitously expressed mainly in the cytoplasm of several tissues. SHP2 modulates diverse cell signaling events that control metabolism, cell growth, differentiation, cell migration, transcription and oncogenic transformation. It interacts with diverse molecules in the cell, and regulates key signaling events including RAS/ERK, PI3K/AKT, JAK/STAT and PD-1 pathways downstream of several receptor tyrosine kinases (RTKs) upon stimulation by growth factors and cytokines. This is the second of multiple candidates to have emerged from our discovery research that targets this pathway, the first being HMPL-295. Dysregulation of SHP2 expression or activity causes many developmental diseases, and hematological and solid tumors.
HMPL-415 Clinical Development
The table below shows a summary of our clinical trial of HMPL-415.
Clinical Trial of HMPL-415
Treatment |
| Name, Line, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
|
HMPL-415 monotherapy | Solid tumors | China | I | Ongoing since 2023 | NCT05886374 |
109
14. Immunology Collaboration with Inmagene
We have a strategic partnership with Inmagene, a clinical development stage company with a focus on immunological diseases, to further develop four novel preclinical drug candidates we discovered for the potential treatment of multiple immunological diseases. Funded by Inmagene, we worked together to move two drug candidates towards clinical trials. Inmagene advanced the drug candidates through global clinical development. In October 2023, Inmagene issued a notice to exercise its options to license these two drug candidates, and the parties entered into a share subscription agreement in February 2024, which, subject to customary closing conditions, entitles us to receive for common shares representing approximately 7.5% of the shares (fully diluted) in Inmagene as consideration for the exercise of the options. Following receipt of the shares, Inmagene will be granted an exclusive license to further develop, manufacture and commercialize these two drug candidates worldwide. For more details on the collaboration arrangement, please see “—Our Collaborations—Inmagene.”
The table below shows a summary of the clinical trials led by Inmagene on IMG-007 and IMG-004.
Treatment |
| Name, Line, Patient Focus |
| Sites |
| Phase |
| Status/Plan |
| NCT # |
|
IMG-007 (OX40 antibody) | Adults with alopecia areata | Global | IIa | First patient dosed | NCT06060977 | ||||||
IMG-007 (OX40 antibody) | Adults with moderate to | Global | IIa | First patient dosed | NCT05984784 | ||||||
IMG-007 (OX40 antibody) | Adult healthy volunteers | Australia | I | Single ascending | NCT05353972 | ||||||
IMG-004 (BTK inhibitor) | Adult healthy volunteers | Global | I | Single ascending | NCT05349097 |
IMG-007 in atopic dermatitis – This is a novel antagonistic monoclonal antibody targeting the OX40 receptor. OX40 is a costimulatory receptor member of the tumor necrosis factor receptor (TNFR) superfamily expressed predominantly on activated T cells. Phase I study in healthy volunteers demonstrated that up to 600 mg of IMG-007 was safe and well-tolerated, with no reports of pyrexia or chills, which were common adverse events of rocatinlimab, another OX40 antibody treatment. At projected therapeutic dose levels, IMG-007 demonstrated a mean terminal half-life of 31-37 days. The long half-life combined with a potentially improved safety profile supports IMG-007’s best-in-class potential as an OX40 targeted therapy.
Two global, proof-of-concept Phase IIa trials are ongoing. One trial evaluates the safety, pharmacokinetics and efficacy (EASI at week 12) of IMG-007 in moderate-to-severe atopic dermatitis. Patients received intravenous IMG-007 three times over four weeks. The first patient was dosed in August 2023 and Inmagene expects interim data readout in the third quarter of 2024. Another Phase II trial evaluates the safety of IMG-007 in adults with alopecia areata with SALT score ≥ 50. They will be given three doses over four weeks. First patient was dosed in October 2023 and Inmagene expects interim data readout in the third quarter of 2024.
IMG-004 in immunological diseases – This is a small molecule inhibitor that binds to BTK in a non-covalent, reversible manner. Designed specifically for inflammatory and autoimmune diseases that usually require long-term treatment, IMG-004 is potent, highly selective and brain permeable. A Phase I single ascending dose study in healthy volunteers in the U.S., initiated in August 2022, has recently completed. It showed that IMG 004 was safe and well-tolerated with a long half-life and sustained pharmacodynamic effects, supporting further clinical development. Results will be submitted to an upcoming medical conference.
Our Research and Development Approach
Our core research and development philosophy is to take a holistic approach to the treatment of cancer and immunological diseases, through multiple modalities and mechanisms, including targeted therapies, immunotherapies and other pathways. A primary objective of our research efforts has been to develop next generation drug candidates with:
| ● | unique selectivity to limit target-based toxicity; |
| ● | high potency to optimize the dose selection with the objective to lower the required dose and thereby limit compound-based toxicity; |
110
| ● | chemical structures deliberately engineered to improve drug exposure in the targeted tissue; and |
| ● | ability to be combined with other therapeutic agents, including targeted therapies, immunotherapies and chemotherapies. |
We have built a drug discovery engine, with which we strive to create differentiated novel oncology and immunology treatments with global potential. These include furthering both small molecule and biologic therapies which address aberrant genetic drivers and cancer cell metabolism; modulate tumor immune microenvironment; and target immune cell checkpoints. We design drug candidates with profiles that enable them to be used in innovative combinations with other therapies, such as chemotherapy, immunotherapy and other targeted therapy in order to attack disease simultaneously through multiple modalities and pathways. We believe that this approach can significantly improve treatment outcomes for patients.
We believe our ability to successfully develop innovative drug candidates through our Oncology/Immunology operations will be the primary factor affecting our long-term competitiveness, as well as our future growth and development. Creating high quality global first-in-class or best-in-class drug candidates requires investment of resources over a prolonged period of time, and a core part of our strategy is to continue making sustained investments in this area. As a result of this commitment, our pipeline of drug candidates has been steadily advancing and expanding, with over a dozen drug candidates put into clinical development. See “– Our Clinical Pipeline” for more details.
Beyond these clinical candidates, we continue to conduct research into discovering new types of drug candidates, including among others, small molecules addressing cancer-related apoptosis, cell signaling, epigenetics and protein translation; biologic drug candidates including bispecific antibodies; and novel technologies including antibody-drug conjugates and heterobifunctional small molecules.
Our Collaborations
Collaborations and joint ventures with corporate partners have provided us with significant funding and access to our partners’ scientific, development, regulatory and commercial capabilities. Our current oncology collaborations focus on savolitinib (collaboration with AstraZeneca) and fruquintinib (collaboration with Eli Lilly and Takeda). When we entered into these collaborations, we had already conducted the discovery research and early clinical development of each drug candidate and, following our agreements, continued to conduct the clinical development and manage or assist the engagement with regulatory authorities up to and including filing the NDAs. Our collaboration partners fund a significant portion of our research and development costs for drug candidates developed in collaboration with them. In addition, we may receive upfront payments upon our entry into these collaboration arrangements and upon the achievement of certain development milestones for the relevant drug candidate. We have received upfront payments, equity contributions and milestone payments totaling approximately $633.5 million mainly from our collaborations with AstraZeneca, Eli Lilly and Takeda as of December 31, 2023. In return, our collaboration partners are entitled to a significant proportion of any future revenue from our drug candidates developed in collaboration with them, as well as a degree of influence over the clinical development process for such drug candidates. In addition, we have entered into other clinical collaborations for combination studies with drug candidates belonging to companies such as BeiGene. We also have an immunology collaboration with Inmagene with respect to four novel pre-clinical drug candidates discovered by us and an in-licensing collaboration with Epizyme with respect to tazemetostat.
AstraZeneca
In 2008, our in-house teams started research on MET inhibitors, subsequently discovering our drug candidate, savolitinib, and conducting its pre-clinical development in-house. In 2011, we submitted applications for clinical development and initiated Phase I clinical trials. In December 2011, we entered into an agreement with AstraZeneca under which we granted to AstraZeneca co-exclusive, worldwide rights to develop, and exclusive worldwide rights to manufacture and commercialize savolitinib for all diagnostic, prophylactic and therapeutic uses. In August 2016, December 2020 and November 2021, we and AstraZeneca amended the terms of the agreement. We refer to this agreement, including the amendments thereto, as the AstraZeneca Agreement.
111
AstraZeneca paid $20.0 million upon execution of the AstraZeneca Agreement and agreed to pay royalties and additional amounts upon the achievement of development and sales milestones. Under the original terms of the AstraZeneca Agreement, we and AstraZeneca agreed to share the development costs for savolitinib in China, with AstraZeneca being responsible for the development costs for savolitinib in the rest of the world. With respect to certain clinical trials, we subsequently agreed with AstraZeneca on sharing development costs. As of December 31, 2023, we had received $64.9 million in milestone payments in addition to approximately $86.2 million in reimbursements for certain development costs. We may potentially receive future clinical development and first sales milestones payments for clinical development and initial sales of savolitinib, plus significant further milestone payments based on sales. Subject to approval of savolitinib in treating PRCC, under the amended AstraZeneca Agreement, AstraZeneca is obligated to pay us increased tiered royalties from 14% to 18% annually on all sales made of any product outside of China, which represents a five percentage point increase over the original terms, subject to a potential downward adjustment on such point increase based on the amount of any contribution by AstraZeneca to the Phase III development in patients with such indication. After total aggregate additional royalties have reached five times our contribution to the Phase III development in patients with such indication, this royalty will step down over a two-year period, to an ongoing royalty rate of 10.5% to 14.5%. AstraZeneca is also obligated to pay us a fixed royalty of 30% on all sales made of any product in China.
Development and collaboration under this agreement are overseen by a joint steering committee that is comprised of three of our senior representatives as well as three senior representatives from AstraZeneca. AstraZeneca is responsible for the development of savolitinib and all regulatory matters related to this agreement in all countries and territories other than China, and we are responsible for the development of savolitinib and all regulatory matters related to this agreement in China. Since entering the AstraZeneca Agreement, we have continued to lead the development of savolitinib in China.
Subject to earlier termination, the AstraZeneca Agreement will continue in full force and effect on a country-by-country basis as long as any collaboration product is being developed or commercialized. The AstraZeneca Agreement is terminable by either party upon a breach that is uncured, upon the occurrence of bankruptcy or insolvency of either party, or by mutual agreement of the parties. The AstraZeneca Agreement may also be terminated by AstraZeneca for convenience with 180 days’ prior written notice. Termination for cause by us or AstraZeneca or for convenience by AstraZeneca will have the effect of, among other things, terminating the applicable licenses granted by us. Termination for convenience by AstraZeneca will have the effect of obligating AstraZeneca to grant to us all of its rights to regulatory approvals and other rights necessary to commercialize savolitinib. Termination by AstraZeneca for convenience will not have the effect of terminating any license granted by AstraZeneca to us.
Eli Lilly
In 2007, our in-house research into VEGFR inhibitors led to the discovery of our drug candidate, fruquintinib. We conducted pre-clinical development in-house and initiated a Phase I clinical trial in 2010. In October 2013, we entered into an agreement with Eli Lilly whereby we granted Eli Lilly an exclusive license to develop, manufacture and commercialize fruquintinib for all uses in China and Hong Kong. In December 2018, following the commercial launch of fruquintinib in China, we and Eli Lilly amended the terms of the agreement and further amended the terms of the agreement in July 2020. We refer to this agreement, including the amendments thereto, as the Eli Lilly Agreement.
Subsequent to the entering of the Eli Lilly Agreement, we continued to lead the development of fruquintinib, including all clinical trial development. Eli Lilly reimbursed us for a majority of the development costs and provided input over the course of the development of fruquintinib. Development, collaboration and manufacture of the products under this agreement are overseen by a joint steering committee comprised of equal numbers of representatives from each party.
Eli Lilly paid a $6.5 million upfront fee following the execution of the Eli Lilly Agreement in 2013, and agreed to pay royalties and additional amounts upon the achievement of development and regulatory approval milestones. As of December 31, 2023, Eli Lilly had paid us $37.2 million in milestone payments in addition to approximately $68.5 million in reimbursements for certain development costs.
112
We could potentially receive future milestone payments for the achievement of development and regulatory approval milestones in China. Additionally, Eli Lilly is obligated to pay us tiered royalties from 15% to 20% annually on sales made of fruquintinib in China and Hong Kong, the rate to be determined based upon the dollar amount of sales made for all products in that year. Under the terms of our 2018 amendment, upon the first commercial launch of fruquintinib in China in a new life cycle indication, these tiered royalties increased to 15% to 29%. Under the terms of our 2020 amendment, we and Eli Lilly share gross profits linked to sales target performance. Subject to meeting pre-agreed sales targets, Eli Lilly will pay us an estimated total of 70% to 80% of Elunate in-market sales in the form of royalties, manufacturing costs and service payments.
Under the terms of our 2018 amendment, we are entitled to determine and conduct future life cycle indication development of fruquintinib in China beyond the three initial indications specified in the original Eli Lilly Agreement. After the 2018 amendment, we assumed responsibility for all development activities and costs for fruquintinib in China in new life cycle indications, and we have the liberty to collaborate with third-parties to explore combination therapies of fruquintinib with various immunotherapy agents. Under the terms of our 2020 amendment, we took over development and execution of all on-the-ground medical detailing, promotion and local and regional marketing activities for Elunate in China.
We are responsible in consultation with Eli Lilly for the supply of, and have the right to supply, all clinical and commercial supplies for fruquintinib pursuant to an agreed strategy for manufacturing. For the term of the Eli Lilly Agreement, such supplies will be provided by us at a transfer price that accounts for our cost of goods sold.
The Eli Lilly Agreement is terminable by either party for breach that is uncured. The Eli Lilly Agreement is also terminable by Eli Lilly for convenience with 120 days’ prior written notice or if there is a major unexpected safety issue with respect to a product. Termination by either us or Eli Lilly for any reason will have the effect of, among other things, terminating the applicable licenses granted by us, and will obligate Eli Lilly to transfer to us all regulatory materials necessary for us to continue development efforts for fruquintinib.
Takeda
In January 2023, we entered into a license agreement with a subsidiary of Takeda (the “Takeda Agreement”) to further the global development, commercialization and manufacture of fruquintinib outside of China. We have received a $400 million upfront payment and a $35 million milestone payments. We are eligible to receive up to additional $695 million potential regulatory, development and commercial sales milestone payments, plus royalties on net sales. Takeda is solely responsible for the development and commercialization of fruquintinib in all included territories.
Development and collaboration under this agreement are overseen by a joint steering committee that is comprised of an equal number of representatives from each party. Takeda is responsible for the development, manufacturing and commercialization activities with respect to fruquintinib in the included territories, other than the existing clinical trials of fruquintinib as of the effectiveness of the Takeda Agreement, which we may continue or wind-down.
Subject to earlier termination, the Takeda Agreement will continue until the expiration of the last royalty term for the last licensed product in the territory. The Takeda Agreement is terminable by Takeda after the first anniversary of the agreement effectiveness for convenience by providing a written notice in advance. Additionally, either party can terminate the Takeda Agreement for cause. Termination for convenience or for cause will have the effect of, among other things, terminating the applicable licenses granted by us. Termination will have the effect of obligating Takeda to assign us all of its rights to title, and interests in and to all clinical trial data, regulatory submissions and regulatory approvals related to fruquinitinib.
BeiGene
In May 2020, we entered into a clinical collaboration agreement with BeiGene to evaluate the safety, tolerability and efficacy of combining surufatinib and fruquintinib with BeiGene’s anti-PD-1 antibody tislelizumab, for the treatment of various solid tumor cancers, in the United States, Europe, China and Australia. Under the terms of the agreement, we and BeiGene each plan to explore development of the combination of surufatinib with tislelizumab or fruquintinib with tislelizumab in different indications and regions. We have agreed to provide mutual drug supply and other support.
113
Inmagene
In January 2021, we and Inmagene entered into a strategic partnership to further develop four novel pre-clinical drug candidates (the humanized OX40 (CD134) antagonistic monoclonal antibody (anti-OX40 mAB) (HMPL-A28), the BTK (Bruton tyrosine kinase) inhibitor (HMPL-727), a RIPK1 (receptor-interacting protein kinase 1) inhibitor (HMPL-662) and a CSF-1R (colony stimulating factor-1 receptor) inhibitor (HMPL-958)) discovered by us for the potential treatment of multiple immunological diseases. We will work together to move the drug candidates towards IND submission. If successful, Inmagene will then move the drug candidates through global clinical development.
Under the terms of the agreement, we have granted Inmagene exclusive options to four drug candidates solely for the treatment of immunological diseases. If Inmagene exercises an option, it will have the right to further develop, manufacture and commercialize that specific drug candidate worldwide, while we retain first right to co-commercialization in China. For each of the drug candidates, we will be entitled to development milestones of up to $92.5 million and up to $135 million in commercial milestones, as well as up to double-digit royalties upon commercialization. In October 2023, Inmagene issued notices to exercise the options to two of the drug candidates; namely, HMPL-A28/IMG-007 and HMPL-727/IMG-004 and as a consequence the parties entered into a Share Subscription Agreement in February 2024, which, subject to various customary closing conditions, entitles HUTCHMED to receive common shares in Inmagene representing 7.5% of the fully diluted share capital of Inmagene as consideration for the exercise of the options. Following receipt of such shares in Inmagene, Inmagene will be granted an exclusive license to further develop, manufacture and commercialize these two drug candidates worldwide. All of the rights of Inmagene under the strategic partnership in respect of the other two drug candidates, namely, HMPL-662 and HMPL-958 terminated and/or expired in March and September 2023 respectively.
Epizyme (A Subsidiary of Ipsen Pharma SAS)
In August 2021, we entered into a licensing agreement with Epizyme Inc. (a subsidiary of Ipsen Pharma SAS) pursuant to which we obtained a co-exclusive license to develop, an exclusive license to commercialize and a co-exclusive license to manufacture tazemetostat in China, Hong Kong, Taiwan and Macau for all therapeutic and palliative uses in epithelioid sarcoma, follicular lymphoma (second line and third line), diffuse large b-cell lymphoma and any other indications that are approved according to the terms of the licensing agreement.
To date, we have paid Epizyme a $25.0 million upfront payment and an aggregate of $5.0 million milestone payments. We may be required to pay an additional aggregate amount of up to $105 million in development and regulatory milestone payments and up to an additional $175 million in sales milestone payments. Epizyme is also eligible to receive, across up to eight potential indications, certain tiered royalties (from mid-teen to low-twenties-percentage) based on annual net sales of tazemetostat in the licensed territory.
We have the right to manufacture the licensed product for development and commercialization in the licensed territory and are generally responsible for funding all clinical trials of tazemetostat, including the portion of global trials conducted in the licensed territory. The agreement with Epizyme will remain in effect until, on a licensed product-by-licensed product basis, the expiration of the royalty term for each licensed product in the licensed territory.
Other Collaborations
In October and November 2018, we entered into multiple collaborations to evaluate combinations of fruquintinib and surufatinib. These include a global collaboration with Innovent to evaluate the combination of fruquintinib with Tyvyt and a global collaboration with Junshi to evaluate the combination of surufatinib with Tuoyi. In September 2019, we expanded our global collaboration agreement with Innovent to evaluate the safety and efficacy of Tyvyt in combination with surufatinib.
Other Ventures
Other Ventures is our large-scale, profitable drug marketing and distribution platform covering about 290 cities and towns in China with over 2,900 manufacturing and commercial personnel as of December 31, 2023. Built over the past 20 years, it has been focused on the sale of prescription drugs products and consumer health products conducted through the following entities:
114
Shanghai Hutchison Pharmaceuticals
Shanghai Hutchison Pharmaceuticals, our non-consolidated joint venture, primarily engages in the manufacture and sale of prescription drug products originally contributed by our joint venture partner, as well as third-party prescription drugs with a focus on cardiovascular medicine. Shanghai Hutchison Pharmaceuticals’ proprietary products are sold under the “Shang Yao” brand, literally meaning “Shanghai pharmaceuticals,” a trademark that has been used for over 50 years in the pharmaceutical retail market, primarily in Shanghai and Eastern China. The trademark is owned by the joint venture and in January 2023, the Shanghai government recognized and awarded the brand as a Shanghai heritage brand. In early 2019, Shanghai Hutchison Pharmaceuticals was awarded the 2018 State Scientific and Technological Progress Award – Second Prize, which was presented by President Xi Jinping, Premier Li Keqiang and other state leaders of the PRC at the National Science and Technology Awards Ceremony. This award was one of only two such awards given that year to studies in the botanical drug industry.
Its key product is She Xiang Bao Xin pills, a vasodilator for the long-term treatment of coronary artery and heart disease and for rapid control and prevention of acute angina pectoris, a form of chest pain. There are over one million deaths due to coronary artery disease per year in China. She Xiang Bao Xin pill is the second largest botanical prescription drug in this indication in China, with market share in January to December 2023 of 22.0% (2022: 21.0%) nationally. She Xiang Bao Xin pills’ sales represented 90% of all Shanghai Hutchison Pharmaceuticals sales in 2023.
She Xiang Bao Xin pills were first approved in 1983 and subsequently enjoyed 36 proprietary commercial protections under the prevailing regulatory system in China. In 2005, Shanghai Hutchison Pharmaceuticals was able to attain “Confidential State Secret Technology” status protection, as certified by China’s Ministry of Science and Technology and State Secrecy Bureau, which extended proprietary protection in China until late 2016. The Science and Technology Commission of Shanghai Municipality has subsequently extended such protection. Shanghai Hutchison Pharmaceuticals holds an invention patent in China covering its formulation, which extends proprietary protection through 2029. She Xiang Bao Xin pill is one of less than two dozen proprietary prescription drugs represented on China’s National Essential Medicines List, which means that all Chinese state-owned health care institutions are required to carry it. She Xiang Bao Xin pill is fully reimbursed in all of China.
Shanghai Hutchison Pharmaceuticals manufactures its products at its 78,000 square meter production facility located in Feng Pu district outside the center of Shanghai. Shanghai Hutchison Pharmaceuticals holds 74 drug product manufacturing licenses, of which 21 are included in the National Essential Medicines List, and two are in active production. The factory is operated by about 560 manufacturing staff.
As of December 31, 2023, Shanghai Hutchison Pharmaceuticals had a commercial team of about 2,300 medical sales representatives allowing for the promotion and scientific detailing of our products not just in hospitals in provincial capitals and medium-sized cities, but also in the majority of county-level hospitals in China. Shanghai Hutchison Pharmaceuticals, through its GSP-certified subsidiary, sells its products and its third-party licensed prescription drugs directly to distributors who on-sell such products to hospitals and clinics, pharmacies and other retail outlets in their respective areas, as well as to other local distributors. As of December 31, 2023, Shanghai Hutchison Pharmaceuticals engaged a group of approximately 530 primary distributors to cover China. These primary distributors in turn used over 2,400 secondary distributors to work directly with hospitals, on a local level, to manage logistics. Shanghai Hutchison Pharmaceuticals’ own prescription drugs sales representatives promote its products to doctors and purchasing managers in hospitals, clinics and pharmacies as part of its marketing efforts.
Hutchison Sinopharm
Hutchison Sinopharm is our consolidated joint venture with Sinopharm. Based in Shanghai, Hutchison Sinopharm focuses on providing logistics services to, and distributing and marketing prescription drugs in China. As of December 31, 2023, Hutchison Sinopharm had a dedicated team of over 40 commercial staff that focus on marketing over 1,000 third-party prescription drug and other products directly to about 790 public and private hospitals in the Shanghai region and through a network of approximately 125 distributors to cover all other provinces in China.
115
Starting in 2015, Hutchison Sinopharm had been the exclusive marketing agent for Seroquel tablets in China. In June 2018, AstraZeneca sold and licensed its rights to Seroquel to Luye Pharma Group, Ltd., including its rights in China. The terms of our agreement with AstraZeneca were assigned to Luye Pharma Hong Kong Ltd., or Luye Pharma HK. In May 2019, we received a notice from Luye Pharma HK purporting to terminate our agreement. We believe that Luye Pharma HK had no basis for termination and commenced confidential legal proceedings to seek damages. In December 2021, the Hong Kong International Arbitration Centre made a final award in favor of Hutchison Sinopharm against Luye Pharma Hong Kong in the amount of RMB253.2 million plus costs we incurred in the legal proceedings and interest. Luye provided a bank guarantee of up to RMB286m to cover the final award pending the outcome of the appeal process. An application was made by Luye on December 14, 2021 to set aside the final award which was heard by the High Court in Hong Kong on June 28, 2022 and dismissed by the judge on July 26, 2022. Luye obtained leave to appeal the setting aside application to the Court of Appeal in Hong Kong and a hearing in the Court of Appeal was heard on June 6, 2023 and we await the judgment. We did not have any revenue from the distribution of Seroquel for the years ended December 31, 2021, 2022 and 2023.
In 2019, we began building an in-house oncology commercial sales and marketing team at Hutchison Sinopharm to support the launch of certain of our innovative oncology drugs. By December 31, 2023, this team had grown to approximately 930 commercial sales and marketing staff in mainland China and Hong Kong.
In 2023, a significant portion of Hutchison Sinopharm’s sales were made directly to hospitals and clinics, with the remaining sales being made through distributors. As of December 31, 2023, Hutchison Sinopharm had over 920 customers of which approximately 14% were distributors, and the revenue generated from these distributors accounted for approximately 35% of the revenue of Hutchison Sinopharm for the year ended December 31, 2023.
Hutchison Healthcare
Hutchison Healthcare is our wholly owned subsidiary and is primarily engaged in the manufacture and sale of health supplements. Hutchison Healthcare’s major product is Zhi Ling Tong DHA capsules, a health supplement made from algae DHA oil for the promotion of brain and retinal development in babies and young children, which is distributed through Hutchison Sinopharm up till the end of September and from October 1, 2022 onwards, through our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals.
The majority of Hutchison Healthcare’s products are contract manufactured at a dedicated and certified manufacturing facility operated by a third party.
116
Competition
Oncology/Immunology Competition
The biotechnology and pharmaceutical industries are highly competitive. While we believe that our highly selective drug candidates, experienced development team and chemistry-focused scientific approach provide us with competitive advantages, we face potential competition from many different sources, including major pharmaceutical, specialty pharmaceutical and biotechnology companies. Any drug candidates that we successfully develop and commercialize will compete with existing drugs and/or new drugs that may become available in the future.
We compete in the segments of the pharmaceutical, biotechnology and other related markets that address inhibition of key biological pathways in cancer and immunological diseases. There are other companies working to develop kinase inhibitors and monoclonal antibodies as targeted therapies for cancer and immunological diseases. These companies include divisions of large pharmaceutical companies and biotechnology companies of various sizes.
Many of our competitors, either alone or with their strategic partners, have substantially greater financial, technical and human resources than we do and significantly greater experience in the discovery and development of drug candidates, obtaining regulatory approvals of products and the commercialization of those products. Accordingly, our competitors may be more successful than we may be in obtaining approval for drugs and achieving widespread market acceptance. Our competitors’ drugs may be more effective, or more effectively marketed and sold, than any drug we may commercialize and may render our drug candidates obsolete or non-competitive before we can recover the expenses of developing and commercializing any of our drug candidates. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available.
Below is a summary of existing therapies and therapies currently under development that may become available in the future which may compete with each of our clinical-stage drug candidates.
Savolitinib
Savolitinib is the first selective MET inhibitor approved in China, while two selective MET inhibitors are on the market in the US and Japan: Tepmetko (tepotinib) and Tabrecta (capmatinib) are approved for MET exon 14 skipping NSCLC with additional programs underway focused on lung cancer. Market Authorization Applications for Tabrecta and Tepmetko were approved in 2022 by the European Medicines Agency (EMA) for use in the treatment of MET exon 14 skipping NSCLC. Glumetinib, bozitinib and tepotinib were conditionally approved for MET exon 14 skipping NSCLC in China in 2023. Capmatinib’s NDA for MET exon 14 skipping NSCLC is currently being reviewed by NMPA. Other selective MET inhibitors in development include telisotuzumab vedotin (in Phase II for advanced solid tumors, including NSCLC), elzovantinib (TPX-0022, in Phase I/II development for advanced solid tumors), REGN-5093 and REGN5093-M114 (in Phase I for NSCLC). Sym-015 is a bi-specific antibody that binds to non-overlapping epitopes on the extracellular domain of the Met receptor tyrosine kinase (in Phase IIa development).
Fruquintinib
Approved VEGF inhibitors on the market for the treatment of CRC include Avastin (anti-VEGF monoclonal antibody), Cyramza (anti-VEGFR2 monoclonal antibody), Stivarga (VEGFR/TIE2 inhibitor) and Zaltrap (ziv-aflibercept) (VEGF inhibitor). Cyramza (ramucirumab) was approved for the treatment of second-line gastric cancer in China in 2022. TAS-102 (trifluridine/tipiracil hydrochloride) was approved for mCRC in China in 2019. Avastin is approved for NSCLC and nintedanib is approved for the treatment of lung disease associated with fibrosis (under the name Ofev) as well asadeno-NSCLC in Europe (under the name Vargatef). Other VEGFR inhibitors being developed for the treatment of NSCLC include Cabometyx, Lenvima (lenvatinib), lucitanib and Caprelsa. VEGFR inhibitors being developed for the treatment of gastric cancer include dovitinib, telatinib and Stivarga. In China, Aitan (apatinib) has been approved for the treatment of third-line gastric cancer and Focus-V (anlotinib) has been approved for the treatment of third-line NSCLC.
117
Surufatinib
Sutent (VEGFR inhibitor) and Afinitor (mTOR inhibitor) have been approved for the treatment of pancreatic NETs. Somatuline Depot (Lanreotide) is a growth hormone release inhibitor that has been approved for the treatment of gastroenteropancreatic NETs. Sandostatin (octreotide) is a growth hormone and insulin-like growth factor-1 inhibitor that has also been approved for NETs. Lutathera (Lu-dotatate), a somatostatin receptor targeting radiotherapy, has been approved by the FDA for the treatment of somatostatin receptor positive gastroenteropancreatic NETs. Furthermore, small molecules, monoclonal antibodies and radiotherapies are being developed for the treatment of NETs. Compounds undergoing development for NETs include Inlyta (axitinib, tyrosine kinase inhibitor), and Vargatef (nintedanib, a tyrosine kinase inhibitor). Cometriq (an additional brand name for cabozantinib) has been marketed for thyroid cancer and is being studied for NETs. In addition, Avastin is an anti-VEGF monoclonal antibody being studied for NETs.
Sovleplenib
There has been extensive research on oral small-molecule Syk inhibitors due to the major unmet medical need in inflammation and oncology. However, many Syk inhibitors have failed in the development stage due to their off-target toxicity as a result of lower kinase selectivity and possibly poor pharmacokinetic properties. The only small molecule drug candidate targeting Syk specifically has been approved to date is Tavalisse for the treatment of chronic immune thrombocytopenia. Lanraplenib (GS-9876) is a Syk inhibitor that has been studied for autoimmune diseases, but not currently in active development for autoimmune diseases. Syk inhibitors currently in clinical studies for hematological cancers include lanraplenib and cerdulatinib (lymphoma).
Tazemetostat
The most common treatments for follicular lymphoma are chemotherapies, usually combined with the monoclonal antibody Rituxan, or Gazyva, which is an antibody that acts against the same target as Rituxan, CD20. While Rituxan and a number of other widely used anti-cancer agents are labeled broadly for follicular lymphoma, no therapies are approved specifically for the treatment of tumors associated with EZH2 activating mutations. There are a number of companies currently evaluating investigational agents in the relapsed and refractory follicular lymphoma patient setting.
In the relapsed and refractory follicular lymphoma patient setting, both current and near-term competition exists. Current competition includes CD20 combinations along with multiple PI3K inhibitors. Near term competition includes companies currently evaluating investigational agents with varying mechanisms of action.
Other than tazemetostat, there are no therapies which have been approved specifically for the treatment of epithelioid sarcoma. Epithelioid sarcoma, an INI1-negative tumor, is typically treated with surgical resection when it presents as localized disease. When epithelioid sarcoma recurs or metastasizes, it may be treated with systemic chemotherapy or investigational agents because, other than tazemetostat, there are no approved systemic therapies specifically indicated for this disease. To the best of our knowledge there are no competitive products in development specifically for epithelioid sarcoma. However, we are aware of several clinical trials run by competitors that recruit patients with soft tissue sarcoma, which is inclusive of epithelioid sarcoma.
HMPL-453
To date, Balversa, Pemazyre and Truseltiq are the only approved therapies that specifically target the FGFR signaling pathway. Late-stage studies are underway for futibatinib and derazantinib. Additionally, a FGFR specific monoclonal antibody, bemarituzumab, is in Phase III development for gastric cancer and gastroesophageal junction (GEJ) adenocarcinoma. Several small molecule FGFR TKI are in clinical trials for solid tumors, including LOXO-435, AZD4547, rogaratinib, fisogatinib (BLU-554), famitinib, Debio 1347, E7090, ICP-192, ICP-105, ASP5878, FGF401, RLY-4008 and HH185.
118
Amdizalisib
Currently there are two PI3K inhibitors approved and on the market outside of China. Zydelig (idelalisib) is approved for the treatment of chronic lymphocytic leukemia, globally. Copiktra (duvelisib, PI3K-δ/γ dual inhibitor) is approved for CLL/SLL. In China, duvelisib and linperlisib were approved for 3L+ follicular lymphoma in 2022 and copanlisib was approved for 3L+ follicular lymphoma in 2023. TQ-B3525’s NDA for 3L+ follicular lymphoma is currently being reviewed by NMPA. Linperlisib’s NDA for peripheral T-cell lymphoma is currently being reviewed by NMPA. In addition, several drug candidates that inhibit PI3Kδ are in clinical development for hematological cancers, including tenalisib (RP6530), duvelisib (peripheral T-cell lymphoma), zandelisib (ME-401 discontinued outside of Japan), ACP 319, roginolisib (IOA-244) and BGB-10188.
HMPL-306
Tilbsovo (ivosidenib) and Rezlidnia (olutasidenib) are approved therapies that specifically inhibits IDH1 while Idhifa (enasidenib) is an approved therapy that specifically inhibits IDH2. To date, there are no approved therapies that inhibit both IDH1 and IDH2, which could be advantageous in deferring resistance to therapy. A pan-IDH inhibitor, vorasidenib, is currently in late stage development for glioma. An IDH 1/2 inhibitor, LY3410738, is in Phase 1 development for both hematological malignancies and solid tumors. Other IDH1 inhibitors in development include BAY1436032 and DS-1001b.
HMPL-760
Approved first and second generation BTK inhibitors include Imbruvica, Calquence, Tirabrutinib, Brukinsa and orelabrutinib. A third generation BTK inhibitor pirtobruntinib was approved for 3L+ in mantle cell lymphoma in January 2023. Nemtabrutinib, orelabrutinib, TG-1701 and JNJ-64264681 are in development for cancer. A number of other BTK inhibitors, such as evobrutinib, remibrutinib, tolebrutinib, rilzabrutinib, SAR444727 and fenebrutinib, are in development for immunological diseases.
HMPL-295
To date, no ERK inhibitor drug has been approved. A number of ERK inhibitors, including BVD-523, LY3214996 and LLT462, among others are being developed in clinical settings as a single agent and/or in combination with various therapeutical agents.
HMPL-653
Turalio is the only FDA approved CSF-1R inhibitor drug and currently there is no CSF-1R inhibitors approved in China. CSF-1R inhibitors in development globally include pimicotinib (ABSK021), axatilimab, BLZ945, vimseltinib, AMB-05X, NMS-03592088, ARRY-382, JNJ-40346527, emactuzumab, AMG820 and IMC-CS4.
HMPL-A83
To date, no CD47 antibody drug has been approved. A number of antibodies, including magrolimab, evorpacept, lemzoparlimab, HX009, PF-07901801, AO-176, DSP107, and IBI188, among others are being developed in clinical settings as a single agent and/or in combination with various therapeutical agents.
Other Ventures Competition
Our Other Ventures operations which focus on prescription drugs compete in the pharmaceutical industry in China, which is highly competitive and is characterized by a number of established, large pharmaceutical companies, as well as some smaller emerging pharmaceutical companies. This business faces competition from other pharmaceutical companies in China engaged in the development, production, marketing or sales of prescription drugs, in particular cardiovascular drugs.
The barrier to entry for the PRC pharmaceutical industry primarily relates to regulatory requirements in connection with the production of pharmaceutical products and new product launches. The identities of the key competitors with respect to our prescription drugs business vary by product, and, in certain cases, different competitors that have greater financial resources than us may elect to focus these resources on developing, importing or in-licensing and marketing products in the PRC that are substitutes for our products and may have broader sales and marketing infrastructure with which to do so.
119
We believe that we compete primarily on the basis of brand recognition, pricing, sales network, promotion activities, product efficacy, safety and reliability. We believe our Other Ventures’ continued success will depend on our business’s capability to: maintain profitability of its products, obtain and maintain regulatory approvals, develop drug candidates with market potential, maintain an efficient operational model, apply technologies to production lines, attract and retain talented personnel, maintain high quality standards, and effectively market and promote the products sold by our prescription drugs business.
Our Other Ventures operations which focus on consumer health products competes in a highly fragmented market in Asia, particularly in our primary market in China. We believe that this business competes primarily on the basis of brand recognition, pricing, sales network, promotion activities, product safety and reliability. We believe our continued success will depend on our business’s capability to: successfully market and distribute in-licensed products such as Earth’s Best infant formula, maintain an efficient operational model, attract and retain talented personnel, maintain high quality standards, and effectively market and promote the products sold by our business.
Patents and Other Intellectual Property
Our commercial success depends in part on our ability to obtain and maintain proprietary or intellectual property protection for our Oncology/Immunology drugs and drug candidates, our Other Ventures’ products and other know-how. Our policy is to seek to protect our proprietary and intellectual property position by, among other methods, filing patent applications in various jurisdictions related to our proprietary technology, inventions and improvements that are important to the development and implementation of our business, enforcing our patents including any patent that have been issued or may be issued that forms part of our patent portfolios, and operating without intentionally infringing valid and enforceable patent and proprietary rights of other parties. We also rely on trade secrets, know-how, continuing technological innovation, in-licensing and out-licensing opportunities to develop and strengthen our proprietary and intellectual property position.
Patents
We and our joint ventures file patent applications directed to our Oncology/Immunology drugs and drug candidates and our Other Ventures’ products in an effort to establish intellectual property positions with regard to new small molecule compounds and/or biologics, their compositions as well as their medical uses in the treatment of diseases. In relation to our Oncology/Immunology operations, we also file patent applications directed to crystalline forms, formulations, processes, key intermediates, and secondary uses as clinical trials for our drugs and drug candidates evolve. We file such patent applications and pursue additional patent protection in major market jurisdictions, including but not limited to China, the United States, Europe, Japan, Canada, South Korea, Russia, Australia, and Brazil.
Our Oncology/Immunology Patents
As of December 31, 2023, we had 274 issued patents, including 25 PRC patents, 24 U.S. patents and 13 European patents, 354 patent applications pending in the above major market jurisdictions, and 7 pending PCT patent applications relating to the drugs and drug candidates of our Oncology/Immunology operations. The intellectual property portfolios for our drugs and most advanced drug candidates are summarized below. With respect to most of the pending patent applications covering our drug candidates, prosecution has yet to commence. Prosecution is a lengthy process, during which the scope of the claims initially submitted for examination by the relevant patent office is often significantly narrowed by the time when they issue, if they issue at all. We expect this to be the case for our pending patent applications referred to below. With respect to any issued patents, we may be entitled to obtain a patent term extension of up to 5 years, subject to statutory and regulatory requirements to be met. For example, if and when a drug candidate receives approval by regulatory authority, such as FDA or NMPA, we can apply for a patent term extension on one of the issued patents covering the drug. In the U.S., the exact duration of the extension depends upon the time that we spend in clinical studies as well as getting approval from FDA. The expected expirations summarized below do not include any additional terms for patent term extensions.
120
Savolitinib—The intellectual property portfolio for savolitinib as of December 31, 2023 are summarized below:
We had a first patent family for savolitinib directed to novel small molecule compounds as well as methods of treating cancers with such compounds. In this patent family, we owned patents in various jusridictions, including patents in China, the United States, Europe and Japan, each expiring in 2030. Based on NMPA approval of savolitinib, an application has been filed with CNIPA for an extension of the Chinese patent term, which, if granted, would extend the Chinese patent term by up to five years.
We had a second patent family directed to the method for the preparation of savolitinib. In this patent family, we owned patents in various jusridictions, including patents in China and Europe, each expiring in 2039. We also had patent applications pending in this family in various jurisdictions, including patent applications in the United States and Japan, each of which, if issued, would have an expiration date in 2039. This patent family is co-owned by us and AstraZeneca.
Our collaboration partner AstraZeneca is responsible for maintaining and enforcing the intellectual property portfolio for savolitinib.
Fruquintinib—The intellectual property portfolio for fruquintinib as of December 31, 2023 are summarized below:
We had a first patent family for fruquintinib directed to novel small molecule compounds as well as methods of treating tumor angiogenesis-related disorders with such compounds. In this patent family, we owned patents in various jurisdictions, including patents in the United States and China expiring in 2028, and patents in Europe and Japan expiring in 2029.
We had a second patent family directed to crystalline forms of fruquintinib as well as methods of treating tumor angiogenesis-related disorders with such forms. In this patent family, we owned patents in various jurisdictions, including patents in the United States, China, Europe and Japan, each of which will expire in 2035.
We had a third patent family directed to the pharmaceutical composition of fruquintinib. In this patent family, we owned patent in China expiring in 2039. We also had patent applications pending in this patent family in various jurisdictions, including China, the United States, Europe and Japan, each of which, if issued, would have an expiration date in 2039.
We also had a patent in China directed to the manufacturing process of fruquintinib.
Based on FDA approval of fruquintinib, applications have been filed with USPTO for an extension of the U.S. patent term, which, if granted, would extend the US patent term by up to five years.
Our collaboration partner Takeda is responsible for maintains and enforcing the intellectual property portfolio for fruquintinib outside of China.
Surufatinib—The intellectual property portfolio for surufatinib as of December 31, 2023 are summarized below:
We had a first patent family for surufatinib directed to novel small molecule compounds as well as methods of treating tumor angiogenesis-related disorders with such compounds. In this patent family, we owned patents in various jurisdictions, including patent in China expiring in 2027, patent in the United States expiring in 2031, and patents in Europe and Japan expiring in 2028.
We had a second patent family directed to the compound and crystalline forms of surufatinib as well as methods of treating tumor angiogenesis-related disorders with such compound and forms. In this patent family, we owned patents in various jurisdictions, including two patents in China expiring in 2029 and 2030, respectively, patent in the United States expiring in 2031, and patent in Europe expiring in 2030. Based on NMPA approval of surufatinib, an application has been filed with CNIPA for an extension of the Chinese patent term, which, if granted, would extend the Chinese patent term by up to five years.
We had a third patent family directed to the formulation of a micronized active pharmaceutical ingredient used in surufatinib as well as methods of treating tumor angiogenesis-related disorders with such formulation. In this patent family, we owned patents in various jurisdictions, including patents in China, Europe and Japan, each of which will expire in 2036.
We had a fourth patent family directed to clinical indications of surufatinib. With respect to this patent family, we had a patent in Japan expiring in 2036.
121
We had a fifth patent family directed to the pharmaceutical combinations of toripalimab and surufatinib. With respect to this family, we had Chinese and Taiwan applications pending, each of which, if issued, would have an expiration date in 2041. This patent family is co-owned by us and Shanghai Junshi Biosciences Co., Ltd.
We had a sixth patent family directed to methods of using surufatinib in treating advanced pancreatic and extra-pancreatic neuroendocrine tumors. With respect to this family, we had a patent application pending in the United States, which, if issued, would have an expiration date in 2041.
We also had other patents/patent applications in China directed to the process, the formulation, and the therapeutic combinations of surufatinib.
Sovleplenib—The intellectual property portfolio for sovleplenib as of December 31, 2023 are summarized below:
We had a first patent family directed to novel small molecule compounds as well as methods of treating cancers, inflammatory diseases, allergic diseases, cell-proliferative diseases, and immunological diseases with such compounds. In this patent family, we owned patents in various jurisdictions, including the United States, China, Europe and Japan, each of which will expire in 2032.
We had a second patent family directed to the salts of sovleplenib as well as crystalline forms thereof. In this patent family, we owned patents in various jurisdictions, including the United States and Japan, each of which will expire in 2038. We also had patent applications pending in this patent family in various jurisdictions, including China, the United States and Europe, each of which, if issued, would have an expiration date in 2038.
Tazemetostat—The intellectual property portfolio for Tazemetostat is licensed from Epizyme, Inc.
We entered into a licensing agreement with Epizyme pursuant to which we obtained a co-exclusive license to develop, an exclusive license to commercialize and a co-exclusive license to manufacture tazemetostat in China, Hong Kong, Taiwan and Macau for all therapeutic and palliative uses in epithelioid sarcoma, follicular lymphoma (second line and third line), diffuse large B-cell lymphoma and any other indications that are approved according to the terms of the licensing agreement. For more details, please see “—Our Collaborations—Epizyme.”
HMPL-453—The intellectual property portfolio for HMPL-453 as of December 31, 2023 is summarized below:
We had a first patent family directed to novel small molecule compounds as well as methods of treating cancers with the compounds. In this patent family, we owned patents in various jurisdictions, including China, Europe, Japan and the United States, each of which will expire in 2034.
We had a second patent family directed to the salts of HMPL-453. In this patent family, we had patent applications pending in various jurisdictions, including China, the United States, Europe and Japan, each of which, if issued, would have an expiration date in 2040.
Amdizalisib—The intellectual property portfolio for amdizalisib as of December 31, 2023 are summarized below:
We had a first patent family directed to novel small molecule compounds as well as uses of such compounds. In this patent family, we owned patents in various jurisdictions, including the United States, Europe, China and Japan, each of which will expire in 2035.
We had a second patent family directed to crystalline forms of amdizalisib. In this patent family, we had patents in various jurisdictions, including the United States expiring in 2039. We also had patent applications pending in this family in various jurisdictions, including China, the United States, Europe and Japan, each of which, if issued, would have an expiration date in 2039.
We also had patent applications directed to the manufacturing process of amdizalisib.
HMPL-306—The intellectual property portfolio for HMPL-306 as of December 31, 2023 is summarized below:
122
We had a patent family directed to novel small molecule compounds as well as methods of treating cancers with the compounds. In this patent family, we owned patents in various jurisdictions, including China, the United States and Japan, each of which will expire in 2038. We also had patent applications pending in this patent family in various other jurisdictions, including Europe, each of which, if issued, would have an expiration date in 2038.
HMPL-760—The intellectual property portfolio for HMPL-760 as of December 31, 2023 is summarized below:
We had a patent family directed to novel small molecule compounds as well as methods of treating cancers, inflammatory diseases or auto-immune diseases with such compounds. In this family, we owned a patent in the United States, which will expire in 2041. We also had patent applications pending in this patent family in various jurisdictions, including China, the United States, Europe and Japan, each of which, if issued, would have an expiration date in 2041.
We also had patent applications directed to the method of preparing intermediates used in the manufacturing process of HMPL-760.
HMPL-295—The intellectual property portfolio for HMPL-295 as of December 31, 2023 is summarized below:
We had a patent family directed to novel small molecule compounds as well as methods of treating cancers or auto-immune diseases with such compounds. In this patent family, we had a patent in China expiring in 2040. We also had patent applications pending in various jurisdictions, including China, the United States, Europe and Japan, each of which, if issued, would have an expiration date in 2040.
HMPL-653—The intellectual property portfolio for HMPL-653 as of December 31, 2023 is summarized below:
We had a patent family directed to novel small molecule compounds as well as methods of treating cancers, inflammatory diseases or auto-immune diseases with such compounds. In this patent family, we had a patent in China expiring in 2041. We also had patent applications pending in various jurisdictions, including China, the United States, Europe and Japan, each of which, if issued, would have an expiration date in 2041.
HMPL-A83—The intellectual property portfolio for HMPL-A83 as of December 31, 2023 is summarized below:
We had a first patent family directed to novel anti-CD47 antibodies as well as methods of treating cancers with such antibodies. In this patent family, we had patent applications pending in various jurisdictions, including China, the United States, Europe and Japan, each of which, if issued, would have an expiration date in 2041.
We had a second patent family directed to the formulation of HMPL-A83. In this patent family, we had PCT, Argentina and Taiwan applications pending, each of which, if issued, would have an expiration date in 2042.
HMPL-415—The intellectual property portfolio for HMPL-415 as of December 31, 2023 is summarized below:
We had a patent family directed to novel small molecule compounds as well as methods of treating cancers, Noonan Syndrome and LEOPARD Syndrome with such compounds. In this patent family, we had patent applications pending in various jurisdictions, including China, the United States, Europe and Japan, each of which, if issued, would have an expiration date in 2042.
Other Ventures Patents
As of December 31, 2023, our joint venture Shanghai Hutchison Pharmaceuticals had (i) 87 patents in China, (ii) two patents in Canada, one patent in the U.S. and one patent in Japan granted under the Patent Cooperation Treaty, and (iii) 41 pending Chinese patent applications and ten patent applications under the Patent Cooperation Treaty, among them, two of which were filed in China, including patents for its key prescription products described below.
123
She Xiang Bao Xin Pills. As of December 31, 2023, Shanghai Hutchison Pharmaceuticals held an invention patent in China directed to the formulation of the She Xiang Bao Xin pill. Under PRC law, invention patents are granted for new technical innovations with respect to products or processes. Invention patents in China have a maximum term of 20 years. This patent will expire in 2029. The “Confidential State Secret Technology” status protection on the She Xiang Bao Xin pill technology held by Shanghai Hutchison Pharmaceuticals, as certified by China’s Ministry of Science and Technology and State Secrecy Bureau, is currently active.
Danning Tablets. As of December 31, 2023, Shanghai Hutchison Pharmaceuticals also held an invention patent in China directed to the formulation of the Danning tablet. This patent will expire in 2027.
Patent Term
The term of a patent depends upon the laws of the country in which it is issued. In most jurisdictions, a patent term is 20 years from the earliest filing date of a non-provisional patent application. In the United States, a patent’s term may be lengthened in some cases by patent term adjustment, which compensates a patentee for administrative delays by the USPTO in excess of a patent applicant’s own delays during the prosecution process, or may be shortened if a patent is terminally disclaimed over an earlier filed, commonly owned patent. In addition, the term of a patent that covers a drug or biological product may also be eligible for patent term extension when FDA approval is granted, provided statutory and regulatory requirements are met. However, the extension shall not exceed five years and the resulting total effective patent term shall not exceed 14 years from the FDA approval. In the future, if and when our drug candidates receive approval by the FDA or other regulatory authorities, we expect to apply for patent term extensions on issued patents covering those drugs, depending upon the length of the clinical trials for each drug and other factors. In China, the amended PRC Patent Law provides for both patent term adjustment and patent term extension, similar to the United States. There can be no assurance that any of our pending patent applications will be issued or that we will benefit from any patent term extension.
Similar extensions as compensation for regulatory delays are available in certain foreign jurisdictions. The actual protection afforded by a patent varies on a claim by claim and country by country basis an depends upon many factors, including the type of patent, the scope of its coverage, the availability of any patent term extensions or adjustments, the availability of legal remedies in a particular country and the validity and enforceability of the patent.
As with other pharmaceutical companies, our or our joint ventures’ ability to maintain and solidify our proprietary and intellectual property position for our drugs and drug candidates or our or their products and technologies will depend on our or our joint ventures’ success in obtaining effective patent claims and enforcing those claims if granted. However, our or our joint ventures’ pending patent applications and any patent applications that we or they may in the future file or license from third parties may not result in the issuance of patents. We also cannot predict the breadth of claims that may be allowed or enforced in our or our joint ventures’ patents. Any issued patents that we may receive in the future may be challenged, invalidated or circumvented. For example, we cannot be certain of the priority of filing covered by pending third-party patent applications. If third parties prepare and file patent applications in the United States, China, Europe, Japan or other markets that also claim technology or therapeutics to which we or our joint ventures have rights, we or our joint ventures may have to participate in interference proceedings, which could result in substantial costs to us, even if the eventual outcome is favorable to us, which is highly unpredictable. In addition, because of the extensive time required for clinical development and regulatory review of a drug candidate we may develop, it is possible that, before any of our drug candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby limiting protection such patent would afford the respective product and any competitive advantage such patent may provide.
Trade Secrets
In addition to patents, we and our joint ventures rely upon unpatented trade secrets and know-how and continuing technological innovation to develop and maintain our or their competitive position. We and our joint ventures seek to protect our proprietary information, in part, by executing confidentiality agreements with our collaborators and scientific advisors, and non-competition, non-solicitation, confidentiality, and invention assignment agreements with our employees and consultants. We and our joint ventures have also executed agreements requiring assignment of inventions with selected scientific advisors and collaborators. The confidentiality agreements we and our joint ventures enter into are designed to protect our or our joint ventures’ proprietary information and the agreements or clauses requiring assignment of inventions to us or our joint ventures, as applicable, are designed to grant us or our joint ventures, as applicable, ownership of technologies that are developed through our or their relationship with the respective counterpart. We cannot guarantee, however, that these agreements will afford us or our joint ventures adequate protection of our or their intellectual property and proprietary information rights.
124
Trademarks and Domain Names
We conduct our business using trademarks with various forms of the “Hutchison”, “Chi-Med”, “Hutchison China MediTech”, “HUTCHMED”, “Elunate”, “Fruzaqla”, “Sulanda”, “Orpathys” and “Tazverik” brands, the logos used by HUTCHMED Limited, as well as domain names incorporating some or all of these trademarks. In April 2006, we entered into a brand license agreement (as amended and restated on June 15, 2021) with Hutchison Whampoa Enterprises Limited, an indirect wholly-owned subsidiary of CK Hutchison, pursuant to which we have been granted a non-exclusive, non-transferrable, royalty-free right to use the “Hutchison”, “Hutchison China MediTech”, “Chi-Med”, “HUTCHMED” trademarks, domain names and other intellectual property rights owned by the CK Hutchison group in connection with the operation of our business worldwide. See “Connected Transactions” for further details. The “Elunate” and “Orpathys” trademarks are licensed to us in China by our collaboration partners Eli Lilly and AstraZeneca, respectively. The “Fruzaqla” trademark is owned by us and licensed exclusively outside of China to our collaboration partner, Takeda. The trademarks for the HUTCHMED Limited logo and “Sulanda” are owned by us. The “Tazverik” trademark is licensed to us in China, Hong Kong, Taiwan and Macau by our collaboration partner Epizyme.
In addition, our joint ventures seek trademark protection for their products. As of December 31, 2023, our joint venture Shanghai Hutchison Pharmaceuticals owned a total of 21 trademarks in China and one trademark in Canada related to products sold by it. For example, the name “Shang Yao” is a registered trademark of Shanghai Hutchison Pharmaceuticals in China for certain uses including pharmaceutical preparations.
Raw Materials and Supplies
Raw materials and supplies are ordered based on our or our joint ventures’ respective sales plans and reasonable order forecasts and are generally available from our or our joint ventures’ own cultivation operations and various third-party suppliers in quantities adequate to meet our needs. We typically order raw materials on short-term contract or purchase order basis and do not enter into long-term dedicated capacity or minimum supply arrangements.
For our Oncology/Immunology operations, the active pharmaceutical ingredient used in our drug candidates are supplied to us from third-party vendors. Our ability to successfully develop our drug candidates, and to ultimately supply our commercial drugs in quantities sufficient to meet the market demand, depends in part on our ability to obtain the active pharmaceutical ingredients for these drugs in accordance with regulatory requirements and in sufficient quantities for commercialization and clinical testing.
We generally aim to identify and qualify one or more manufacturers to provide such active pharmaceutical ingredients prior to submission of an NDA to the FDA and/or NMPA. We contract with two suppliers to manufacture and supply us with the active pharmaceutical ingredient for fruquintinib for commercial purposes in China and one of those suppliers is also contracted to supply us with active pharmaceutical ingredient for commercial purposes outside of China. We also contract with a single supplier to manufacture and supply us with the active pharmaceutical ingredient for surufatinib for commercial purposes. We contracted with a single supplier to provide active pharmaceutical ingredient and finished product for savolitinib and are in the process of engaging a second supplier of the active pharmaceutical ingredient for our products including savolitinib, surufatinib and sovleplenib. We manage the risk of price fluctuations and supply disruptions of active pharmaceutical ingredients by purchasing them in bulk quantities as these ingredients have a relatively long shelf life. Other than the foregoing, we do not currently have arrangements in place for a contingent or second-source supply of the active pharmaceutical ingredients for fruquintinib outside of China, surufatinib or savolitinib. In the event any of our current suppliers of such active pharmaceutical ingredients or finished product cease their operations for any reason, which may lead to an interruption in our production and operations. However, to date, while we have experienced price fluctuations associated with our raw materials, we have not experienced any material disruptions in the supply of the active pharmaceutical ingredients or the other raw materials we and our joint venture partners use. See Item 3.D. “Risk Factors—Certain of our joint venture parties principal products involve the cultivation or sourcing of key raw materials including botanical products, and any quality control or supply failure or price fluctuations could adversely affect our ability to manufacture our products and/or could materially and adversely affect our operating results.”
125
Quality Control and Assurance
We have our own independent quality control system and devote significant attention to quality control for the designing, manufacturing and testing of our products. We have established a strict quality control system in accordance with the NMPA regulations. Our laboratories fully comply with the Chinese manufacturing guidelines and are staffed with highly educated and skilled technicians to ensure quality of all batches of product release. We monitor in real time our operations throughout the entire production process, from inspection of raw and auxiliary materials, manufacture, delivery of finished products, clinical testing at hospitals, to ethical sales tactics. Our quality assurance team is also responsible for ensuring that we are in compliance with all applicable regulations, standards and internal policies. Our senior management team is actively involved in setting quality policies and managing internal and external quality performance of our company and our joint venture Shanghai Hutchison Pharmaceuticals.
Customers and Suppliers
For the years ended December 31, 2021, 2022 and 2023, we generated revenue of $188.9 million, $185.0 million and $538.0 million from our five largest customers, respectively. For the years ended December 31, 2021, 2022 and 2023, revenue from our five largest customers represented approximately 53%, 43% and 64% of our total revenue, respectively, and revenue from our largest customer in those periods represented approximately 16%, 16% and 42% of our revenue in the same periods, respectively. Save for Sinopharm, our five largest customers were independent third parties and none of our directors or their close associates or, to the knowledge of our directors, any shareholders who owned more than 5% of our issued ordinary shares had any interest in any of our five largest customers as of the date of the filing of this annual report.
In 2021, 2022 and 2023, Sinopharm, which jointly owns Hutchison Sinopharm with us, was one of our five largest customers. Sales to Sinopharm and/or its associates contributed 12%, 16% and 8% of our revenue in 2021, 2022 and 2023, respectively. Purchases from Sinopharm and/or its associates contributed approximately 1% of our total purchases in 2021, 2022 and 2023, respectively.
For the years ended December 31, 2021, 2022 and 2023, the total purchases from our five largest suppliers were $100.6 million, $90.9 million and $77.1 million, respectively. For the years ended December 31, 2021, 2022 and 2023, our purchases from our five largest suppliers represented less than 20% of our total purchases. All of our five largest suppliers were independent third parties and none of our directors or their close associates or, to the knowledge of our directors, any shareholder who owned more than 5% of our issued ordinary shares had any interest in any of our five largest suppliers as of the date of the filing of this annual report.
Contract Research Organizations
Although we or our collaboration partners design the clinical trials for our drug candidates, CROs conduct most of the clinical trials. Our agreements with CROs are usually structured as master service agreements which set out the services to be performed, payment schedule, term and confirmation that all intellectual rights arising out of or made in performance of the services are owned by us. We and our collaboration partners work with major global and Chinese CROs.
Certificates and Permits
The following sets forth the material certificates and/or permits that we have obtained for our operations in China. We have received all material certificates and permits that are, or may be, required for our operations in China. No material certificate, permission or approval for our operations has been denied by relevant authorities in China. Given the uncertainties of interpretation and implementation of relevant laws and regulations and the enforcement practice by relevant government authorities, we may be required to obtain additional licenses, permits, filings or approvals for our products and business operations in China in the future, and may not be able to maintain or renew our current licenses, permits, filings or approvals. In addition, rules and regulations in China can change quickly with little advance notice. Uncertainties due to evolving laws and regulations could impede the ability of an issuer with significant operations in China, such as us, to obtain or maintain certificates, permits or licenses required to conduct business in China. In the absence of required certificates, permits or licenses, governmental authorities could impose material sanctions or penalties on us.
HUTCHMED (Suzhou) Limited holds a pharmaceutical manufacturing permit issued by its local regulatory authority expiring on September 13, 2025. It also complies with applicable GMP standards.
126
Hutchison Sinopharm holds a pharmaceutical trading license issued by its local regulatory authority expiring on July 30, 2024. Hutchison Sinopharm also holds a good supply practice, or GSP, certificate issued by its local regulatory authority which expires on July 30, 2024. We will renew them before their expiration.
Shanghai Hutchison Pharmaceuticals holds a pharmaceutical manufacturing permit from its local regulatory authorities expiring on December 31, 2025.
Shanghai Shangyao Hutchison Whampoa GSP Company Limited, a subsidiary of Shanghai Hutchison Pharmaceuticals, holds a pharmaceutical trading license from its local regulatory authority expiring on November 17, 2024. It also holds a GSP certificate issued by its local regulatory authority expiring on November 17, 2024. We will renew them before their expiration.
Regulations
This section sets forth a summary of the most significant rules and regulations affecting our business activities in China and the United States.
Government Regulation of Pharmaceutical Product Development and Approval
PRC Regulation of Pharmaceutical Product Development and Approval
Since China’s entry to the World Trade Organization in 2001, the PRC government has made significant efforts to standardize regulations, develop its pharmaceutical regulatory system and strengthen intellectual property protection.
Regulatory Authorities
In the PRC, the NMPA is the authority that monitors and supervises the administration of pharmaceutical products and medical appliances and equipment as well as cosmetics. The NMPA’s predecessor, the State Drug Administration, or the SDA, was established on August 19, 1998 as an organization under the State Council to assume the responsibilities previously handled by the Ministry of Health of the PRC, or the MOH, the State Pharmaceutical Administration Bureau of the PRC and the State Administration of Traditional Chinese Medicine of the PRC. The SDA was replaced by the State Food and Drug Administration, or the SFDA, in March 2003 and was later reorganized into the China Food and Drug Administration, or the CFDA, in March 2013. On March 17, 2018, the First Session of the Thirteenth National People’s Congress approved the State Council Institutional Reform Proposal, according to which the duties of the CFDA were consolidated into the State Administration for Market Regulation, or the SAMR, and the NMPA was established under the management and supervision of the SAMR.
The primary responsibilities of the NMPA include:
| ● | monitoring and supervising the administration of pharmaceutical products, medical appliances and equipment as well as cosmetics in the PRC; |
| ● | formulating administrative rules and policies concerning the supervision and administration of cosmetics and the pharmaceutical industry; evaluating, registering and approving of new drugs, generic drugs, imported drugs and traditional Chinese medicine; |
| ● | undertaking the standard, registration, quality and post marketing risk management of pharmaceutical products, medical appliances and equipment as well as cosmetics; and |
| ● | examining, evaluating and supervising the safety of pharmaceutical products, medical appliances and equipment as well as that of cosmetics. |
127
The MOH is an authority at the ministerial level under the State Council and is primarily responsible for national public health. Following the establishment of the SFDA in 2003, the MOH was put in charge of the overall administration of national health in the PRC excluding the pharmaceutical industry. In March 2008, the State Council placed the SFDA under the management and supervision of the MOH. The MOH performs a variety of tasks in relation to the health industry such as establishing social medical institutes and producing professional codes of ethics for public medical personnel. The MOH is also responsible for overseas affairs, such as dealings with overseas companies and governments. In 2013, the MOH and the National Population and Family Planning Commission were integrated into the National Health and Family Planning Commission of the PRC, or the NHFPC. On March 17, 2018, the First Session of the Thirteenth National People’s Congress approved the State Council Institutional Reform Proposal, according to which the responsibilities of NHFPC and certain other governmental authorities are consolidated into the NHC, and the NHFPC shall no longer be maintained. The responsibilities of the NHC include organizing the formulation of national drug policies, the national essential medicine system and the National Essential Medicines List and drafting the administrative rules for the procurement, distribution and use of national essential medicines.
The National Healthcare Security Administration (the ‘‘NHSA’’), established in May 2018, is directly under the State Council and is responsible for the management of the healthcare security system. It is primarily responsible for drafting and implementing policies and standards on medical insurance, maternity insurance and medical assistance; supervising and administering the healthcare security funds; formulating a uniform medical insurance catalogue and payment standards on drugs, medical disposables and healthcare services; and formulating bidding procurement policies for drugs and medical consumables and supervising the implementation.
Healthcare System Reform
The PRC government has promulgated several healthcare reform policies and regulations to reform the healthcare system. On March 17, 2009, the Central Committee of the PRC Communist Party and the State Council jointly issued the Guidelines on Strengthening the Reform of Healthcare System. On March 18, 2009, the State Council issued the Implementation Plan for the Recent Priorities of the Healthcare System Reform (2009-2011). On July 22, 2009, the General Office of the State Council issued the Five Main Tasks of Healthcare System Reform in 2009.
More recently, on May 5, 2022, the General Office of the State Council issued the Key Tasks for Deepening the Reform of the Medical and Health System in 2022 (the “2022 PRC Health Care Reforms”).
Highlights of the 2022 PRC Health Care Reforms include the following:
| ● | The overall objectives of the 2022 PRC Health Care Reforms are to comprehensively promote construction of a healthy China, deeply promote the experience of Sanming’s medical reforms (which refers to certain medical reforms undertaken in Sanming, Fujian Province since 2012), promote the expansion and balanced distribution of high-quality medical resources, continue to promote the transition from centering on disease treatment to centering on people’s health, and continue to promote solutions to lack of and cost of access to medical care. |
| ● | According to the Sanming People’s Government website, the medical reforms that were undertaken in Sanming included but were not limited to (1) reforms to the personnel and salary system of public hospitals, whereby Sanming implemented target annual salaries for medical staff (being 3 times the average local salary), (2) introduction of competitive bidding processes in order to reduce the cost of medicines, and (3) integration of medical insurance management institutions to reduce coordination costs across departments. The 2022 PRC Health Care Reforms calls for promotion of Sanming’s medical reform experience, including but not limited to (1) expansion of the scope of centralized procurement, whereby state and local governments in each province should strive to have a total of more than 350 common drugs purchased; (2) reform of medical service prices, whereby all provinces shall issue documents related to the establishment of a dynamic adjustment mechanism for medical service prices before the end of June 2022, and (3) reform of the personnel and salary system of public hospitals, whereby localities should be guided to make good use of staffing resources in light of their actual circumstances, and may explore the recruitment of the best external qualified professional and technical personnel via strict and standardized procedures such as open recruitment. |
128
| ● | The 2022 PRC Health Care Reforms also promote high-quality development in medicine and healthcare, including but not limited to (1) comprehensive and steady reform of public hospitals, whereby pilot provinces shall take the lead in exploring and reviewing reform paths of public hospitals at all levels; (2) giving a greater role to government investment incentives; (3) advancement of the national medical insurance program, such as promoting the improvement of the direct settlement of expenses of inter-provincial and remote medical treatments, and unifying the scope of drugs covered by national medical insurance across the country; (4) strengthening drug supply security, for example, by accelerating the granting of market authorization to innovative drugs of clinical value; and (5) promotion of pilot projects for the revitalization of traditional Chinese medicine. The 2022 PRC Health Care Reforms also call for (i) 35,000 general practitioners and 100,000 resident doctors (including postgraduates with a master degree) to be trained through various approaches within the year, (ii) for the enrollment of professional postgraduate students to be inclined towards areas facing skills shortages, such as general practice, pediatrics, and psychiatry, and (iii) the promotion of telemecine services, which shall cover 95% of the country’s districts and counties. |
On July 21, 2023, the NHC, the NDRC, the Ministry of Finance (the “MOF”), the MOHRSS, the NHSA and the NMPA jointly issued the Key Tasks for Deepening the Reform of the Medical and Health System in the Second Half of 2023, which calls for, among others, improvement to the two-invoice system policy, strengthening and promoting the supply and use of essential medicines, additional rounds of centralized procurement of medicines and pharmaceutical consumables, and the promotion of innovation in traditional Chinese medicines and its heritage.
Drug Administration Laws and Regulations
The PRC Drug Administration Law as promulgated by the Standing Committee of the National People’s Congress in 1984 and the Implementing Measures of the PRC Drug Administration Law as promulgated by the MOH in 1989 have laid down the legal framework for the establishment of pharmaceutical manufacturing enterprises, pharmaceutical trading enterprises and for the administration of pharmaceutical products including the development and manufacturing of new drugs and medicinal preparations by medical institutions. The PRC Drug Administration Law also regulates the packaging, trademarks and the advertisements of pharmaceutical products in the PRC.
Certain revisions to the PRC Drug Administration Law took effect on December 1, 2001. They were formulated to strengthen the supervision and administration of pharmaceutical products, and to ensure the quality and the safety of pharmaceutical products for human use. The revised PRC Drug Administration Law applies to entities and individuals engaged in the development, production, trade, application, supervision and administration of pharmaceutical products. It regulates and prescribes a framework for the administration of pharmaceutical manufacturers, pharmaceutical trading companies, and medicinal preparations of medical institutions and the development, research, manufacturing, distribution, packaging, pricing and advertisements of pharmaceutical products.
The PRC Drug Administration Law was later amended on December 28, 2013 and April 24, 2015 by the Standing Committee of the National People’s Congress. It provides the basic legal framework for the administration of the production and sale of pharmaceutical products in China and covers the manufacturing, distributing, packaging, pricing and advertising of pharmaceutical products.
On August 26, 2019, the Standing Committee of the National People’s Congress promulgated the amended PRC Drug Administration Law, which took effect on December 1, 2019. The amendment brought a series of changes to the drug supervision and administration system, including but not limited to the clarification of the MAH system, pursuant to which the MAH shall assume responsibilities for non-clinical studies, clinical trials, manufacturing and marketing, post-marketing studies, monitoring, reporting and handling of adverse reactions of the drug. The amendment also stipulated that the PRC supports the innovation of drugs with clinical value and specific or special effects on human diseases, encourages the development of drugs with new therapeutic mechanisms and promotes the technological advancement of such drugs.
According to the PRC Drug Administration Law, no pharmaceutical products may be produced without a pharmaceutical production license. A manufacturer of pharmaceutical products must obtain a pharmaceutical production license from one of NMPA’s provincial level branches in order to commence production of pharmaceuticals. Prior to granting such license, the relevant government authority will inspect the manufacturer’s production facilities, and decide whether the sanitary conditions, quality assurance system, management structure and equipment within the facilities have met the required standards.
129
The PRC Drug Administration Implementation Regulations promulgated by the State Council took effect on September 15, 2002 and were later amended on February 6, 2016 and March 2, 2019 to provide detailed implementation regulations for the revised PRC Drug Administration Law. With respect to the latest revision of the PRC Drug Administration Law, promulgated on August 26, 2019 and effective on December 1, 2019, there are no corresponding revised PRC Drug Administration Implementation Regulations which have taken effect. On May 9, 2022, the draft PRC Drug Administration Implementation Regulations was published by the NMPA for comment and, as of the date of this annual report, remains in draft form, the final and effective version of which is yet to be published.
Examination and Approval of New Medicines
On January 22, 2020, the SAMR promulgated the Administrative Measures on the Registration of Pharmaceutical Products, or the Registration Measures, which became effective on July 1, 2020. According to the Registration Measures, an applicant who has obtained a drug registration certificate shall be a drug MAH. The approval process for medicines seeking marketing authorization mainly consists of the following steps:
| ● | upon the completion of pharmaceutical, pharmacological and toxicological research and related activities, an application for clinical trial will be submitted to the Center for Drug Evaluation of the NMPA, or the Center for Drug Evaluation, for review. The Center for Drug Evaluation will organize pharmacists, medical personnel and other professionals to review the application for clinical trial. A decision on approval or non-approval of the application for clinical trial of drugs will be made within 60 working days from acceptance of the application, and the applicant shall be notified of the examination and approval result through the website of the Center for Drug Evaluation. If the applicant is not notified within the stipulated period, the application shall be deemed approved. The applicant who is approved to conduct clinical trial shall act as the sponsor for the clinical trial; |
| ● | if the application for clinical trial is approved, the sponsor shall, prior to conducting subsequent phases of the clinical trial, formulate a corresponding program for the clinical trial, carry out the clinical trial after the review and approval by the Ethics Committee, and submit the corresponding program for clinical trial and supporting materials on the website of the Center for Drug Evaluation. The applicant may proceed with the relevant clinical research (which is generally conducted in three phases for a new medicine under the Registration Measures) at institutions with appropriate qualification: |
| ● | Phase I refers to the preliminary clinical trial for clinical pharmacology and body safety. It is conducted to observe the human body tolerance for new medicine and pharmacokinetics, so as to provide a basis for determining the prescription plan. |
| ● | Phase I or II refers to the stage of preliminary evaluation of clinical effectiveness. The purpose is to preliminarily evaluate the clinical effectiveness and safety of the medicine used on patients with targeted indication, as well as to provide a basis for determining the Phase III clinical trial research plan and the volume under the prescription plan. |
| ● | Phase III is a clinical trial stage to verify the clinical effectiveness. The purpose is to test and determine the clinical effectiveness and safety of the medicine used on patients with targeted indication, to evaluate the benefits and risks thereof and, eventually, to provide sufficient basis for review of the medicine registration application. |
| ● | Phase IV refers to the stage of surveillance and research after the new medicines is launched. The purpose is to observe the clinical effectiveness and adverse effects of the medicine over a much larger patient population and longer time period than in Phase I to III clinical trials, and evaluate the benefits and risks when it is administered to general or special patient population in larger prescription volume; |
| ● | the sponsor shall submit a safety update report during the research and development period on the website of the NMPA on a regular basis. The safety update report during the research and development period shall be submitted once a year, and within two months of every full year after the clinical drug trial is approved. The NMPA may require the sponsor to adjust the reporting period if deemed necessary; |
130
| ● | after (i) completing relevant pharmaceutical, pharmacological and toxicological research, clinical drug trials, and other research supporting the marketing registration of a medicine, (ii) determining medicine quality standards, (iii) completing the verification of commercial scale manufacturing process, and (iv) making preparations for drug registration inspections, the applicant shall file the application for drug marketing authorization with the Center for Drug Evaluation; |
| ● | the Center for Drug Evaluation will organize pharmaceutical, medical and other professionals to review accepted drug marketing authorization applications in accordance with relevant requirements; |
| ● | upon acceptance of an application for drug registration, the Center for Drug Evaluation will conduct a preliminary examination within 40 working days from acceptance of the application; if there is a need to conduct an examination of manufacturing premises for drug registration, the Center for Drug Evaluation will notify the Centre for Food and Drug Inspection of the NMPA to organize an examination, provide the relevant materials required, and simultaneously notify the applicant as well as the provincial drug administrative authorities where the applicant or the manufacturing enterprise is located. The Centre for Food and Drug Inspection of the NMPA shall in principle complete the examination 40 working days before expiry of the review period, and give feedback to the Center for Drug Evaluation on the status and findings etc. of the examinations; and |
| ● | if the application is approved through the comprehensive review process, the drug shall be approved for marketing and a drug registration certificate shall be issued. The drug registration certificate will state the approval number for the drug, the holder of the certificate, and information of the manufacturing enterprise. A drug registration certificate for non-prescription drugs will also state the non-prescription drug category. |
Any applicant who is not satisfied with the Center for Drug Evaluation’s decision to deny an application during the application of the drug registration period can appeal within 15 working days after it is notified by the Center for Drug Evaluation of such decision. Upon termination for examination and approval of the application for drug registration, if the applicant is dissatisfied with the administrative licensing decision, the applicant may apply for administrative review or file an administrative lawsuit.
In accordance with the Provisions on the Administration of Special Examination and Approval of Registration of New Drugs promulgated by the SFDA, issued and effective on January 7, 2009, an NDA that meets certain requirements as specified below will be handled with priority in the review and approval process, so-called “green-channel” approval. In addition, the applicant is entitled to provide additional materials during the review period besides those requested by the SFDA, and will have access to enhanced communication channels with the SFDA. As of the date of this annual report, the SFDA has been succeeded by the SAMR and NMPA.
Applicants for the registration of the following new drugs are entitled to request priority treatment in review and approval: (i) active ingredients and their preparations extracted from plants, animals and minerals, and newly discovered medical materials and their preparations that have not been sold in the China market, (ii) chemical drugs and their preparations and biological products that have not been approved for sale at its origin country or abroad, (iii) new drugs with obvious clinical treatment advantages for such diseases as AIDS, therioma, and rare diseases, and (iv) new drugs for diseases that have not been treated effectively. Under category (i) or (ii) above, the applicant for drug registration may apply for special examination and approval when applying for the clinical trial of new drugs; under category (iii) or (iv) above, the applicant may only apply for special examination and approval when applying for manufacturing.
In addition, on July 7, 2020, the NMPA released the Priority Review and Approval Procedures for Drug Marketing Authorizations (for Trial Implementation), which further clarified that a fast track process for drug registration will be available to the following drugs with distinctive clinical value: (i) (a) drugs in urgent clinical demand and in shortage and (b) innovative drugs and modified new drugs for prevention and treatment of serious infectious diseases, rare diseases and other diseases; (ii) new varieties, dosage forms and specifications of children’s drugs that conform to children’s physiological characteristics; (iii) (a) vaccines that are in urgent need for disease prevention and control and (b) innovative vaccines; (iv) drugs that have been included in the procedures for Breakthrough Therapy Designation; (v) drugs that are subject to conditional approval; and (vi) other drugs which the NMPA deems applicable. It also specified that fast track status would be given to clinical trial applications for drugs with patent expiry within three years and manufacturing authorization applications for drugs with patent expiry within one year. Concurrent applications for new drug clinical trials which are already approved in the United States or E.U. are also eligible for fast track NMPA approval.
131
Drug Technology Transfer Regulations
On August 19, 2009, the SFDA promulgated the Administrative Regulations for Technology Transfer Registration of Drugs to standardize the registration process of drug technology transfer, which includes application for, and evaluation, examination, approval and monitoring of, drug technology transfer. Drug technology transfer refers to the transfer of drug production technology by the owner to a drug manufacturer and the application for drug registration by the transferee according to the provisions in the new regulations. Drug technology transfer includes new drug technology transfer and drug production technology transfer.
Conditions for the application for new drug technology transfer
Applications for new drug technology transfer may be submitted prior to the expiration date of the monitoring period of the new drugs with respect to:
| ● | drugs with new drug certificates only; or |
| ● | drugs with new drug certificates and drug approval numbers. |
For drugs with new drug certificates only and not yet in the monitoring period, or drug substances with new drug certificates, applications for new drug technology transfer should be submitted prior to the respective expiration date of the monitoring periods for each drug registration category set forth in the new regulations and after the issue date of the new drug certificates.
Conditions for the application of drug production technology transfer
Applications for drug production technology transfer may be submitted if:
| ● | the transferor holds new drug certificates or both new drug certificates and drug approval numbers, and the monitoring period has expired or there is no monitoring period; |
| ● | with respect to drugs without new drug certificates, both the transferor and the transferee are legally qualified drug manufacturing enterprises, one of which holds over 50% of the equity interests in the other, or both of which are majority-owned subsidiaries of the same drug manufacturing enterprise; |
| ● | with respect to imported drugs with imported drug licenses, the original applicants for the imported drug registration may transfer these drugs to local drug manufacturing enterprises. |
Application for, and examination and approval of, drug technology transfer
Applications for drug technology transfer should be submitted to the provincial drug administration. If the transferor and the transferee are located in different provinces, the provincial drug administration where the transferor is located should provide examination opinions. The provincial drug administration where the transferee is located is responsible for examining application materials for technology transfer and organizing inspections on the production facilities of the transferee. Medical examination institutes are responsible for testing three batches of drug samples.
The Center for Drug Evaluation should further review the application materials, provide technical evaluation opinions and form a comprehensive evaluation opinion based on the site inspection reports and the testing results of the samples. The SFDA (which, as of the date of this annual report, has been succeeded by the SAMR and NMPA) should determine whether to approve the application according to the comprehensive evaluation opinion of the Center for Drug Evaluation. An approval letter of supplementary application and a drug approval number will be issued to qualified applications. An approval letter of clinical trials will be issued when necessary. For rejected applications, a notification letter of the examination opinions will be issued with the reasons for rejection.
132
Permits and Licenses for Manufacturing and Registration of Drugs
Production Licenses
To manufacture pharmaceutical products in the PRC, a pharmaceutical manufacturing enterprise must first obtain a Pharmaceutical Manufacturing Permit issued by the relevant pharmaceutical administrative authorities at the provincial level where the enterprise is located. Among other things, such a permit must set forth the permit number, the name, legal representative and registered address of the enterprise, the site and scope of production, issuing institution, date of issuance and effective period.
Each Pharmaceutical Manufacturing Permit issued to a pharmaceutical manufacturing enterprise is effective for a period of five years. The enterprise is required to apply for renewal of such permit within six months prior to its expiry and will be subject to reassessment by the issuing authorities in accordance with then prevailing legal and regulatory requirements for the purposes of such renewal.
Business Licenses
In addition to a Pharmaceutical Manufacturing permit, the manufacturing enterprise must also obtain a business license from the administrative bureau of industry and commerce at the local level. The name, legal representative and registered address of the enterprise specified in the business license must be identical to that set forth in the Pharmaceutical Manufacturing Permit.
Registration of Pharmaceutical Products
All pharmaceutical products that are produced in the PRC must bear a registration number issued by the NMPA, with the exception of Chinese herbs and Chinese herbal medicines in soluble form. The medicine manufacturing enterprises must obtain the medicine registration number before manufacturing any medicine.
Good Manufacturing Practices
The Guidelines on Good Manufacturing Practices, as amended in 1998 and 2010, or the Guidelines, took effect on August 1, 1999 and set the basic standards for the manufacture of pharmaceuticals. These Guidelines cover issues such as the production facilities, the qualification of the personnel at the management level, production plant and facilities, documentation, material packaging and labeling, inspection, production management, sales and return of products and customers’ complaints. On October 23, 2003, the SFDA issued the Notice on the Overall Implementation and Supervision of Accreditation of Good Manufacturing Practice Certificates for Pharmaceuticals, which required all pharmaceutical manufacturers to apply for the GMP certificates by June 30, 2004. Those enterprises that failed to obtain the GMP certificates by December 31, 2004 would have their Pharmaceutical Manufacturing Permit revoked by the drug administrative authorities at the provincial level. On October 24, 2007, the SFDA issued Evaluation Standard on Good Manufacturing Practices which became effective on January 1, 2008. On December 1, 2019, per the Announcement of the NMPA on Issues Concerning the Implementation of the PRC Drug Administration Law, GMP certificates were abolished, though manufacturers remain to be obligated to operate in accordance with the applicable requirements of the Guidelines. The Notice of the NMPA on Promulgation of the Administrative Measures for Drug Inspection (for Trial Implementation), or Trial Drug Inspection Measures, was released and effective on May 24, 2021. The Trial Drug Inspection Measures were subsequently revised on July 19, 2023. The Trial Drug Inspection Measures regulate the inspection, investigation, evidence collection and disposal and other actions carried out by medical products administrative authorities with respect to the manufacturing, distribution and use of drugs. The Trial Drug Inspection Measures stipulate that where an application for a pharmaceutical manufacturing permit is filed for the first time, on-site inspection shall be carried out in accordance with the applicable requirements of the Guidelines. Where an application for re-issuance of a pharmaceutical manufacturing permit is filed, a compliance inspection may be carried out if necessary based on the principles of risk management, taking into consideration the enterprise’s compliance with the laws and regulations on drug administration, the Guidelines, and the running of quality control systems.
Marketing Authorization Holder System
In May 2016, the State Council announced the piloting of the MAH system in ten provinces in China, where the market authorization/drug license holders are no longer required to be the actual manufacturers. The MAH system will allow for more flexibilities in contract manufacturing arrangements.
133
Under the authorization of the Standing Committee of the National People’s Congress, the State Council issued the Pilot Plan for the Drug MAH Mechanism on May 26, 2016, providing a detailed pilot plan for the MAH system in ten provinces in China. Under the MAH system, domestic drug research and development institutions and individuals in the pilot regions are eligible to be holders of drug registrations without having to become drug manufacturers. The MAHs may engage contract manufacturers for manufacturing, provided that the contract manufacturers are licensed and are also located within the pilot regions. Drugs that qualify for the MAH system include: (1) new drugs (including biological products for curative uses of Class I, Class VII and biosimilars under the Administration of Drug Registration) approved after the implementation of the MAH system; (2) generic drugs approved as Category 3 or 4 drugs under the Reform Plan for Registration Category of Chemical Medicine issued by the NMPA on March 4, 2016; (3) previously approved generics that have passed equivalence assessments against their original drugs; and (4) previously approved drugs whose licenses were held by drug manufacturers originally located within the pilot regions but have moved out of the pilot regions due to corporate mergers or other reasons.
On August 15, 2017, the CFDA issued the Circular on the Matters Relating to Promotion of the Pilot Program for the Drug MAH System, clarifying that the MAH shall be responsible for managing the whole manufacturing and marketing chain and the whole life cycle of drugs and shall assume full legal liabilities for the non-clinical drug study, clinical trials, manufacturing, marketing and distribution and adverse drug reaction monitoring. The MAH is permitted to entrust several drug manufacturers under the drug quality management system established by the MAH. The MAH shall submit a report of drug manufacturing, marketing, prescription, techniques, pharmacovigilance, quality control measures and certain other matters to the CFDA (which, as of the date of this annual report, has been succeeded by the SAMR and NMPA) within 20 working days after the end of each year.
On December 1, 2019, the latest amendment of Drug Administration Law came into effect, marking the success of the pilot work, and the MAH system has become a national system. Pursuant to the latest amendment, the legal representative and the key person-in-charge of a drug MAH shall be fully responsible for the quality of drugs.
Administrative Protection for New Drugs
The Administrative Measures Governing the Production Quality of Pharmaceutical Products, or the Administrative Measures for Production, provides detailed guidelines on practices governing the production of pharmaceutical products. A manufacturer’s factory must meet certain criteria in the Administrative Measures for Production, which include: institution and staff qualifications, production premises and facilities, equipment, hygiene conditions, production management, quality controls, product operation, maintenance of sales records and manner of handling customer complaints and adverse reaction reports.
Distribution of Pharmaceutical Products
According to the PRC Drug Administration Law and its implementing regulations, and the Measures for the Supervision and Administration of Drug Quality in Operation and Usage (issued by the SAMR on September 27, 2023 and effective on January 1, 2024), an MAH may directly distribute pharmaceutical products for which drug registration certificates have been obtained, or distribute such products via a pharmaceutical distributor. Notwithstanding, an MAH can only carry out pharmaceutical product retail activities if they have obtained a Pharmaceutical Distribution Permit.
The granting of a Pharmaceutical Distribution Permit to wholesalers shall be subject to approval of the provincial level drug regulatory authorities, while the granting of a Pharmaceutical Distribution Permit to retailers shall be subject to the approval of the drug regulatory authorities above the county level. Unless otherwise expressly approved, no pharmaceutical wholesaler may engage in the retail of pharmaceutical products, nor may pharmaceutical retailers engage in wholesaling.
A pharmaceutical distributor shall satisfy the following requirements:
| ● | personnel with pharmaceutical expertise as qualified according to law; |
| ● | business site, facilities, warehousing and sanitary environment compatible to the pharmaceutical products being distributed; |
| ● | quality management system and personnel compatible to the pharmaceutical products being distributed; and |
| ● | rules and regulations to ensure the quality of the pharmaceutical products being distributed. |
134
A pharmaceutical distributor shall establish a quality management system covering the entire process of their drug business. The purchase and sales records, storage conditions, transportation process, quality control and other records shall be completely and accurately documented, and shall not be fabricated and tampered with. MAHs and pharmaceutical distributors must keep the relevant qualifications and sale and purchase receipts and records for not less than five years and at least until one year after the expiry date of such drugs. Penalties may be imposed for any violation of record-keeping.
Penalties may be imposed on MAHs or pharmaceutical distributors that manufacture or distribute pharmaceutical products without obtaining a Pharmaceutical Manufacturing Permit or a Pharmaceutical Distribution Permit.
On December 26, 2016, the Medical Reform Office of the State Council, the National Health and Family Planning Commission, the CFDA and other five government authorities promulgated the “Two-Invoice System” Opinions, which became effective on the same date. On April 25, 2017, the General Office of the State Council further promulgated the Notice on Issuing the Key Working Tasks for Deepening the Reform of Medicine and Health System in 2017. According to these rules, a two-invoice system is encouraged to be gradually adopted for drug procurement. The two-invoice system generally requires a drug manufacturer to issue only one invoice to its distributor followed by the distributor issuing a second invoice directly to the end customer hospital. Only one distributor is permitted to distribute drug products between the manufacturer and the hospital. The system also encourages manufacturers to sell drug products directly to hospitals. Public medical institutions are required to adopt the two-invoice system, and its full implementation nationwide is targeted for 2018. As of the date of the filing of this annual report, the relevant local rules with respect to the “Two-Invoice System” have been promulgated in some provinces and municipal cities in the PRC, and the reform is still in progress. Private medical institutions are encouraged but not yet required to adopt the two-invoice system. Pharmaceutical manufacturers and distributors who fail to implement the two-invoice system may be disqualified from attending future bidding events or providing distribution for hospitals and blacklisted for drug procurement practices. These rules aim to consolidate drug distribution and reduce drug prices. The impact on our company is that Shanghai Hutchison Pharmaceuticals was required to restructure its distribution and logistics network and Hutchison Sinopharm began to shift its prior Seroquel distribution model to a fee-for-service model. For more details, please refer to Item 4.B. “Business Overview—Other Ventures.”
Foreign Investment and “State Secret” Technology Drugs
The interpretation of certain PRC laws and regulations governing foreign investment and “state secret” technology is uncertain. Under the Special Administrative Measures (Negative List) for Foreign Investment Access, or the Negative List, published by the MOFCOM and the China National Development and Reform Commission or the NDRC. Under the Catalogue, “manufacturing of modern Chinese medicines with confidential proprietary formula” has been deemed prohibited for any foreign investment. The technology and know-how of the She Xiang Bao Xin pill is classified as “state secret” technology by China’s Ministry of Science and Technology, or the MOST, and the National Administration for the Protection of State Secrets, or NAPSS.
There are currently no PRC laws or regulations or official interpretations, and therefore there can be no assurance, as to whether the use of “state secret” technology constitutes the “manufacturing of Chinese medicines with confidential proprietary formula” under the Negative List. However, under the Rules on Confidentiality of Science and Technology promulgated by the State Science and Technology Commission (the predecessor of the MOST and the NAPSS) on January 6, 1995, cooperation with foreign parties or establishing joint ventures with foreign parties in respect of state secret technology is expressly allowed, provided that such cooperation has been duly approved by the relevant science and technology authorities. The establishment of Shanghai Hutchison Pharmaceuticals as a sino-foreign joint venture, including the re-registration of licenses for She Xiang Bao Xin pills in its name, was approved by the local counterpart of the MOFCOM and the Shanghai Drug Administration in 2001. Subsequently, the “Confidential State Secret Technology” status protection for She Xiang Bao Xin pills was also granted in 2005 to Shanghai Hutchison Pharmaceuticals as a sino-foreign joint venture by the MOST and NAPSS. Consequently, we believe Shanghai Hutchison Pharmaceuticals is in compliance with all applicable PRC laws and regulations governing foreign investment and “state secret” technology. Moreover, we believe that our other joint ventures and wholly-foreign owned enterprises in the PRC are also in compliance with all applicable PRC laws and regulations governing foreign investment.
135
U.S. Regulation of Pharmaceutical Product Development and Approval
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and the Public Health Service Act, or PHSA, and their implementing regulations. The process of obtaining approvals and the subsequent compliance with appropriate federal, state and local rules and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. regulatory requirements at any time during the product development process, approval process or after approval may subject an applicant and/or sponsor to a variety of administrative or judicial sanctions, including refusal by FDA to approve pending applications, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters and other types of enforcement correspondence, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement of profits, or civil or criminal investigations and penalties brought by FDA and the U.S. Department of Justice, or DOJ, or other governmental entities. Drugs are also subject to other federal, state and local statutes and regulations.
Our drug candidates must be approved by the FDA through the NDA process before they may be legally marketed in the United States. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| ● | completion of extensive pre-clinical studies, sometimes referred to as pre-clinical laboratory tests, pre-clinical animal studies and formulation studies all performed in compliance with applicable regulations, including the FDA’s good laboratory practice regulations; |
| ● | submission to the FDA of an IND application which must become effective before human clinical trials may begin and must be updated annually; |
| ● | IRB approval before each clinical trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials in accordance with study protocols, the applicable GCPs and other clinical trial-related regulations, to establish the safety and efficacy of the proposed drug product for its proposed indication; |
| ● | preparation and submission to the FDA of an NDA; |
| ● | a determination by the FDA within 60 days of its receipt of an NDA whether the NDA is acceptable for filing; if the FDA determines that the NDA is not sufficiently complete to permit substantive review, it may request additional information and decline to accept the application for filing until the information is provided; |
| ● | in-depth review of the NDA by FDA, which may include review by a scientific advisory committee; |
| ● | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the active pharmaceutical ingredient and finished drug product are produced to assess compliance with the FDA’s cGMP; |
| ● | potential FDA audit of the pre-clinical and/or clinical trial sites that generated the data in support of the NDA; |
| ● | payment of user fees and FDA review and approval of the NDA prior to any commercial marketing or sale of the drug in the United States; and |
| ● | compliance with any post-approval requirements, such as REMS and post-approval studies required by FDA. |
136
Pre-clinical Studies
The data required to support an NDA is generated in two development stages: pre-clinical and clinical. For new chemical entities, or NCEs, the pre-clinical development stage generally involves synthesizing the active component, developing the formulation and determining the manufacturing process, evaluating purity and stability, as well as carrying out non-human toxicology, pharmacology and drug metabolism studies in the laboratory, which support subsequent clinical testing. The conduct of the pre-clinical tests must comply with federal regulations, including good laboratory practices. The sponsor must submit the results of the pre-clinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. An IND is a request for authorization from the FDA to administer an investigational drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human trials. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials and places the IND on clinical hold within that 30-day time period. In such a case, the IND sponsor must resolve with the FDA any outstanding concerns or questions before the clinical trial can begin. Some long-term pre-clinical testing, such as animal tests of reproductive adverse events and carcinogenicity, may continue after the IND is submitted. The FDA may also impose clinical holds on a drug candidate at any time before or during clinical trials due to safety concerns or non-compliance. Accordingly, submission of an IND does not guarantee the FDA will allow clinical trials to begin, or that, once begun, issues will not arise that could cause the trial to be suspended or terminated.
Clinical Studies
The clinical stage of development involves the administration of the drug product to human subjects or patients under the supervision of qualified investigators, generally physicians not employed by or under the trial sponsor’s control, in accordance with GCPs, which include the requirement that, in general, all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under written study protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Further, each clinical trial must be reviewed and approved by each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also reviews and approves the informed consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. There are also requirements governing the reporting of ongoing clinical trials and completed clinical trial results to public registries. For example, information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health for public dissemination on their ClinicalTrials.gov website.
Clinical trials are generally conducted in three sequential phases that may overlap or be combined, known as Phase I, Phase II and Phase III clinical trials.
| ● | Phase I: In a standard Phase I clinical trial, the drug is initially introduced into a small number of subjects who are initially exposed to a range of doses of the drug candidate. The primary purpose of these clinical trials is to assess the metabolism, pharmacologic action, appropriate dosing, side effect tolerability and safety of the drug. |
| ● | Phase Ib: Although Phase I clinical trials are not intended to treat disease or illness, a Phase Ib trial is conducted in patient populations who have been diagnosed with the disease for which the study drug is intended. The patient population typically demonstrates a biomarker, surrogate, or other clinical outcome that can be assessed to show “proof-of-concept.” In a Phase Ib study, proof-of-concept typically confirms a hypothesis that the current prediction of a biomarker, surrogate or other outcome benefit is compatible with the mechanism of action of the study drug. |
| ● | Phase I/II: A Phase I and Phase II trial for the same treatment is combined into a single study protocol. The drug is administered first to determine a maximum tolerable dose, and then additional patients are treated in the Phase II portion of the study to further assess safety and/or efficacy. |
| ● | Phase II: The drug is administered to a limited patient population to determine dose tolerance and optimal dosage required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, as well as identification of possible adverse effects and safety risks and preliminary evaluation of efficacy. |
137
| ● | Phase III: The drug is administered to an expanded number of patients, generally at multiple sites that are geographically dispersed, in well-controlled clinical trials to generate enough data to demonstrate the efficacy of the drug for its intended use, its safety profile, and to establish the overall benefit/risk profile of the drug and provide an adequate basis for drug approval and labeling of the drug product. Phase III clinical trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a drug during marketing. Generally, two adequate and well-controlled Phase III clinical trials are required by the FDA for approval of an NDA. A pivotal study is a clinical study that adequately meets regulatory agency requirements for the evaluation of a drug candidate’s efficacy and safety such that it can be used to justify the approval of the drug. Generally, pivotal studies are also Phase III studies but may be Phase II studies if the trial design provides a well-controlled and reliable assessment of clinical benefit, particularly in situations where there is an unmet medical need. Phase IV clinical trials are conducted after initial regulatory approval, and they are used to collect additional information from the treatment of patients in the intended therapeutic indication or to meet other regulatory requirements. In certain instances, FDA may mandate the performance of Phase IV clinical trials. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA, and more frequently if serious adverse events occur. Written IND safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events or any finding from tests in laboratory animals that suggests a significant risk to human subjects. The FDA, the IRB, or the clinical trial sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. The FDA will typically inspect one or more clinical sites to assure compliance with GCPs and the integrity of the clinical data submitted. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution, or an institution it represents, if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a trial may move forward at designated check points based on access to certain data from the trial. Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug as well as finalize a process for manufacturing the drug in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, cGMPs impose extensive procedural, substantive and recordkeeping requirements to ensure and preserve the long-term stability and quality of the final drug product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the drug candidate does not undergo unacceptable deterioration over its shelf life.
NDA Submission and FDA Review Process
Following trial completion, trial results and data are analyzed to assess safety and efficacy. The results of pre-clinical studies and clinical trials are then submitted to the FDA as part of an NDA, along with proposed labeling for the drug, information about the manufacturing process and facilities that will be used to ensure drug quality, results of analytical testing conducted on the chemistry of the drug, and other relevant information. The NDA is a request for approval to market the drug and must contain adequate evidence of safety and efficacy, which is demonstrated by extensive pre-clinical and clinical testing. The application includes both negative or ambiguous results of pre-clinical and clinical trials as well as positive findings. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a use of a drug, or from a number of alternative sources, including studies initiated by investigators. To support regulatory approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational drug product to the satisfaction of the FDA. Under federal law, the submission of most NDAs is subject to the payment of an application user fees; a waiver of such fees may be obtained under certain limited circumstances. FDA approval of an NDA must be obtained before a drug may be offered for sale in the United States.
In addition, under the Pediatric Research Equity Act of 2003, or PREA, an NDA or supplement to an NDA must contain data to assess the safety and efficacy of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers.
138
Under the Prescription Drug User Fee Act, or PDUFA, as amended, each NDA must be accompanied by an application user fee. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs for products designated as orphan drugs, unless the product also includes a non-orphan indication. The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. The FDA conducts a preliminary review of an NDA within 60 days of receipt and informs the sponsor by the 74th day after FDA’s receipt of the submission to determine whether the application is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under PDUFA, the FDA has 10 months from the filing date in which to complete its initial review of a standard NDA and respond to the applicant, and six months from the filing date for a “priority review” NDA. The FDA does not always meet its PDUFA goal dates for standard and priority review NDAs, and the review process is often significantly extended by FDA requests for additional information or clarification.
After the NDA submission is accepted for filing, the FDA reviews the NDA to determine, among other things, whether the proposed drug is safe and effective for its intended use, and whether the drug is being manufactured in accordance with cGMP to assure and preserve the drug’s identity, strength, quality and purity. The FDA may refer applications for drugs or drug candidates that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. The FDA may re-analyze the clinical trial data, which can result in extensive discussions between the FDA and us during the review process.
Before approving an NDA, the FDA will conduct a pre-approval inspection of the manufacturing facilities for the new drug to determine whether they comply with cGMPs. The FDA will not approve the drug unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the drug within required specifications. In addition, before approving an NDA, the FDA may also audit data from clinical trials to ensure compliance with GCP requirements. After the FDA evaluates the application, manufacturing process and manufacturing facilities where the drug product and/or its active pharmaceutical ingredient will be produced, it may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter usually describes all of the specific deficiencies in the NDA identified by the FDA. The Complete Response Letter may require additional clinical data and/or an additional pivotal clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, pre-clinical studies or manufacturing. If a Complete Response Letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application. Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data.
If a drug receives regulatory approval, the approval may be limited to specific diseases and dosages or the indications for use may otherwise be limited. Further, the FDA may require that certain contraindications, warnings or precautions be included in the drug labeling or may condition the approval of the NDA on other changes to the proposed labeling, development of adequate controls and specifications, or a commitment to conduct post-market testing or clinical trials and surveillance to monitor the effects of approved drugs. For example, the FDA may require Phase IV testing which involves clinical trials designed to further assess a drug’s safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved drugs that have been commercialized. The FDA may also place other conditions on approvals including the requirement for a REMS to ensure that the benefits of a drug or biological product outweigh its risks. If the FDA concludes a REMS is needed, the sponsor of the NDA must submit a proposed REMS. The FDA will not approve the NDA without an approved REMS, if required. A REMS could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of drugs. Drug approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following initial marketing.
139
Special FDA Expedited Review, Approval and Access Programs
The FDA has various programs, including fast track designation, accelerated approval, priority review and Breakthrough Therapy Designation, that are intended to expedite or simplify the process for the development and FDA review of drugs that are intended for the treatment of serious or life threatening diseases or conditions and demonstrate the potential to address unmet medical needs. The purpose of these programs is to provide important new drugs to patients earlier than under standard FDA review procedures. While these pathways can reduce the time it takes for the FDA to review an NDA, they do not guarantee that a product will receive FDA approval. In addition, expanded access programs or the Right to Try Act can provide access to unapproved, investigational treatments for patients diagnosed with life-threatening diseases or conditions who have exhausted approved treatment options and who are unable to participate in a clinical trial.
Fast Track Designation
To be eligible for a fast track designation, the FDA must determine, based on the request of a sponsor, that a drug is intended to treat a serious or life threatening disease or condition for which there is no effective treatment and demonstrates the potential to address an unmet medical need for the disease or condition. Under the fast track program, the sponsor of a drug candidate may request the FDA to designate the product for a specific indication as a fast track product concurrent with or after the filing of the IND for the drug candidate. The FDA must make a fast track designation determination within 60 days after receipt of the sponsor’s request.
In addition to other benefits, such as the ability to use surrogate endpoints and have greater interactions with the FDA, the FDA may initiate review of sections of a fast track product’s NDA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA’s time period goal for reviewing a fast track application does not begin until the last section of the NDA is submitted. A fast track drug also may be eligible for accelerated approval and priority review. In addition, the fast track designation may be withdrawn by the FDA if it believes that the designation is no longer supported by data emerging in the clinical trial process.
Priority Review
The FDA may give a priority review designation to drugs that offer major advances in treatment, or provide a treatment where no adequate therapy exists. A priority review means that the goal for the FDA to review an application is six months, rather than the standard review of ten months under current PDUFA guidelines. These six and ten month review periods are measured from the “filing” date rather than the receipt date for NDAs for new molecular entities, which typically adds approximately two months to the timeline for review and decision from the date of submission. Most products that are eligible for fast track designation are also likely to be considered appropriate to receive a priority review.
Breakthrough Therapy Designation
Under the provisions of the new Food and Drug Administration Safety and Innovation Act, or FDASIA, enacted by Congress in 2012, a sponsor can request designation of a drug candidate as a “breakthrough therapy,” typically by the end of the drug’s Phase II trials. A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are also eligible for accelerated approval. For breakthrough therapies, the FDA may take certain actions, such as intensive and early guidance on the drug development program, that are intended to expedite the development and review of an application for approval.
140
Accelerated Approval
FDASIA also codified and expanded on FDA’s accelerated approval regulations, under which FDA may approve a drug for a serious or life-threatening illness that provides meaningful therapeutic benefit over existing treatments based on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on an intermediate clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit. A surrogate endpoint is a marker that does not itself measure clinical benefit but is believed to predict clinical benefit. This determination takes into account the severity, rarity or prevalence of the disease or condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require a sponsor of a drug receiving accelerated approval to perform Phase IV or post-marketing studies to verify and describe the predicted effect on irreversible morbidity or mortality or other clinical endpoint, and the drug may be subject to accelerated withdrawal procedures. All promotional materials for drug candidates approved under accelerated regulations are subject to prior review by the FDA.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for the FDA review or approval will not be shortened. Furthermore, fast track designation, priority review, accelerated approval and Breakthrough Therapy Designation, do not change the standards for approval and may not ultimately expedite the development or approval process.
Compassionate Use and Right to Try
In the United States, investigational medical products may be made available outside of clinical trials to certain patients under expanded access or compassionate-use programs approved by FDA. These programs provide access to such investigational medical products if patients have a life-threatening disease or serious disease or condition and no comparable or satisfactory alternative therapy options are available. There is no legal obligation requiring a company to provide access to investigational medical products via expanded access pathways. Additionally, the U.S. Right to Try Act of 2018 provides a separate pathway for patients with a life-threatening disease or condition who have exhausted all other treatment options and who are unable to participate in clinical trials to access investigational drugs that have passed Phase I clinical trials. As with expanded access pathways, there is no obligation for a pharmaceutical manufacturer to make its drug products available to such eligible patients as a result of the Right to Try Act.
Pediatric Trials
Under PREA, an NDA or supplement thereto must contain data that are adequate to assess the safety and effectiveness of the drug product for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. With the enactment of FDASIA, a sponsor who is planning to submit a marketing application for a drug that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration must also submit an initial Pediatric Study Plan, or PSP, within sixty days of an end-of-Phase II meeting or as may be agreed between the sponsor and the FDA. The initial PSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including study objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach agreement on the PSP. A sponsor can submit amendments to an agreed-upon initial PSP at any time if changes to the pediatric plan need to be considered based on data collected from pre-clinical studies, early phase clinical trials, and/or other clinical development programs. The law requires the FDA to send a non-compliance letter to sponsors who do not submit their pediatric assessments as required.
Under the Best Pharmaceuticals for Children Act, or BPCA, certain therapeutic candidates may obtain an additional six months of exclusivity if the sponsor submits information requested by the FDA, relating to the use of the active moiety of the product candidate in children. Although the FDA may issue a written request for studies on either approved or unapproved indications, it may only do so where it determines that information relating to that use of a product candidate in a pediatric population, or part of the pediatric population, may produce health benefits in that population.
141
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may designate a drug product as an “orphan drug” if it is intended to treat a rare disease or condition (generally meaning that it affects fewer than 200,000 individuals in the United States, or more in cases in which there is no reasonable expectation that the cost of developing and making a drug product available in the United States for treatment of the disease or condition will be recovered from sales of the product). A company must request orphan product designation before submitting an NDA. If the request is granted, the FDA will disclose the identity of the therapeutic agent and its potential use. Orphan product designation does not convey any advantage in or shorten the duration of the regulatory review and approval process, but the product will be entitled to orphan product exclusivity, meaning that the FDA may not approve any other applications for the same product for the same indication for seven years, except in certain limited circumstances. Competitors may receive approval of different products for the indication for which the orphan product has exclusivity and may obtain approval for the same product but for a different indication. If a drug or drug product designated as an orphan product ultimately receives regulatory approval for an indication broader than what was designated in its orphan product application, it may not be entitled to exclusivity. The 21st Century Cures Act, which became law in December 2016, expanded the types of studies that qualify for orphan drug grants. Orphan drug designation also may qualify an applicant for federal and possibly state tax credits relating to research and development costs.
Post-Marketing Requirements
Following approval of a new drug, a pharmaceutical company and the approved drug are subject to continuing regulation by the FDA, including, among other things, monitoring and recordkeeping activities, reporting to the applicable regulatory authorities of adverse experiences with the drug, providing the regulatory authorities with updated safety and efficacy information, drug sampling and distribution requirements, and complying with applicable promotion and advertising requirements.
Prescription drug advertising is subject to federal, state and foreign regulations. In the United States, the FDA regulates prescription drug promotion, including standards for direct-to-consumer advertising, restrictions on promoting drugs for uses or in patient populations that are not described in the drug’s approved labeling (known as “off-label use”), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may legally prescribe drugs for off-label uses, manufacturers may not market or promote such off-label uses. Prescription drug promotional materials must be submitted to the FDA in conjunction with their first use. Modifications or enhancements to the drug or its labeling or changes of the site of manufacture are often subject to the approval of the FDA and other regulators, which may or may not be received or may result in a lengthy review process. Any distribution of prescription drugs and pharmaceutical samples also must comply with the U.S. Prescription Drug Marketing Act, a part of the FDCA.
In the United States, once a drug is approved, its manufacture is subject to comprehensive and continuing regulation by the FDA. The FDA regulations require that drugs be manufactured in specific approved facilities and in accordance with cGMP. Applicants may also rely on third parties for the production of clinical and commercial quantities of drugs, and these third parties must operate in accordance with cGMP regulations. cGMP regulations require among other things, quality control and quality assurance as well as the corresponding maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. These regulations also impose certain organizational, procedural and documentation requirements with respect to manufacturing and quality assurance activities. NDA holders using third-party contract manufacturers, laboratories or packagers are responsible for the selection and monitoring of qualified firms, and, in certain circumstances, qualified suppliers to these firms. These firms and, where applicable, their suppliers are subject to inspections by the FDA at any time, and the discovery of violative conditions, including failure to conform to cGMP, could result in enforcement actions that interrupt the operation of any such facilities or the ability to distribute drugs manufactured, processed or tested by them. Discovery of problems with a drug after approval may result in restrictions on a drug, manufacturer, or holder of an approved NDA, including, among other things, recall or withdrawal of the drug from the market, and may require substantial resources to correct.
142
The FDA also may require Phase IV testing, risk minimization action plans and post-marketing surveillance to monitor the effects of an approved drug or place conditions on an approval that could restrict the distribution or use of the drug. Discovery of previously unknown problems with a drug or the failure to comply with applicable FDA requirements can have negative consequences, including adverse publicity, judicial or administrative enforcement, warning letters from the FDA, mandated corrective advertising or communications with doctors, and civil or criminal penalties, among others. Newly discovered or developed safety or effectiveness data may require changes to a drug’s approved labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our drugs under development.
Other U.S. Regulatory Matters
Manufacturing, sales, promotion and other activities following drug approval are also subject to regulation by numerous regulatory authorities in addition to the FDA, including, in the United States, the Department of Justice, Centers for Medicare & Medicaid Services, other divisions of the Department of Health and Human Services, the Drug Enforcement Administration for Controlled Substances, the Consumer Product Safety Commission, the Federal Trade Commission, the Occupational Safety & Health Administration, the Environmental Protection Agency and state and local governments. In the United States, sales, marketing and scientific/educational programs must also comply with state and federal fraud and abuse laws. Pricing and rebate programs must comply with the Medicaid rebate requirements of the U.S. Omnibus Budget Reconciliation Act of 1990 and more recent requirements in the Affordable Care Act. If drugs are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. The handling of any controlled substances must comply with the U.S. Controlled Substances Act and Controlled Substances Import and Export Act. Drugs must meet applicable child-resistant packaging requirements under the U.S. Poison Prevention Packaging Act. Manufacturing, sales, promotion and other activities are also potentially subject to federal and state consumer protection and unfair competition laws.
The distribution of pharmaceutical drugs is subject to additional requirements and regulations, including extensive record-keeping, licensing, storage and security requirements intended to prevent the unauthorized sale of pharmaceutical drugs.
The failure to comply with regulatory requirements subjects firms to possible legal or regulatory action. Depending on the circumstances, failure to meet applicable regulatory requirements can result in criminal prosecution, fines or other penalties, injunctions, recall or seizure of drugs, total or partial suspension of production, denial or withdrawal of product approvals, or refusal to allow a firm to enter into supply contracts, including government contracts. In addition, even if a firm complies with FDA and other requirements, new information regarding the safety or efficacy of a product could lead the FDA to modify or withdraw product approval. Prohibitions or restrictions on sales or withdrawal of future products marketed by us could materially affect our business in an adverse way.
Changes in regulations, statutes or the interpretation of existing regulations could impact our business in the future by requiring, for example: (i) changes to our manufacturing arrangements; (ii) additions or modifications to product labeling; (iii) the recall or discontinuation of our products; or (iv) additional record-keeping requirements. If any such changes were to be imposed, they could adversely affect the operation of our business.
U.S. Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of the FDA approval of our drug candidates, some of our U.S. patents may be eligible for limited patent term extension under the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of an NDA plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent. The USPTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In 2018, the FDA advanced policies aimed at promoting drug competition and patient access to generic drugs, such as issuing guidance about making complex generic drugs and the circumstances in which approval of a generic product application may be delayed.
143
Marketing exclusivity provisions under the FDCA can also delay the submission or the approval of certain marketing applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a NCE. A drug is a NCE if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application, or a 505(b)(2) NDA submitted by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovator drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder.
The FDCA also provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving abbreviated new drug applications for drugs containing the active agent for the original indication or condition of use. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the pre-clinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness. Orphan drug exclusivity, as described above, may offer a seven-year period of marketing exclusivity, except in certain circumstances. Pediatric exclusivity is another type of regulatory market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms. This six-month exclusivity, which runs from the end of other exclusivity protection or patent term, may be granted based on the voluntary completion of a pediatric trial in accordance with an FDA-issued “Written Request” for such a trial.
Rest of the World Regulation of Pharmaceutical Product Development and Approval
For other countries outside of China and the United States, such as countries in Europe, Latin America or other parts of Asia, the requirements governing the conduct of clinical trials, drug licensing, pricing and reimbursement vary from country to country. In all cases the clinical trials must be conducted in accordance with GCP requirements and the applicable regulatory requirements and ethical principles.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Coverage and Reimbursement
PRC Coverage and Reimbursement
Historically, most of Chinese healthcare costs have been borne by patients out-of-pocket, which has limited the growth of more expensive pharmaceutical products. However, in recent years the number of people covered by government and private insurance has increased. According to the NHC, as of December 31, 2022, approximately 1.35 billion residents in China were enrolled in the national medical insurance program, with participation rates remaining steadily above 95%. In 2022, total income of the National Basic Medical Insurance Fund (including maternity insurance) reached RMB3,092.2 billion, an increase of 17.6% over the previous year.
Reimbursement under the National Medical Insurance Program
The National Medical Insurance Program was adopted pursuant to the Decision of the State Council on the Establishment of the Urban Employee Basic Medical Insurance Program issued by the State Council on December 14, 1998, under which all employers in urban cities are required to enroll their employees in the basic medical insurance program and the insurance premium is jointly contributed by the employers and employees. The State Council promulgated Guiding Opinions of the State Council about the Pilot Urban Resident Basic Medical Insurance on July 10, 2007, under which urban residents of the pilot district, rather than urban employees, may voluntarily join Urban Resident Basic Medical Insurance. The State Council expected the Pilot Urban Resident Basic Medical Insurance to cover the whole nation by 2010.
144
Participants of the National Medical Insurance Program and their employers, if any, are required to contribute to the payment of insurance premiums on a monthly basis. Program participants are eligible for full or partial reimbursement of the cost of medicines included in the NRDL. The Notice Regarding the Tentative Measures for the Administration of the Scope of Medical Insurance Coverage for Pharmaceutical Products for Urban Employees, jointly issued by several authorities including the Ministry of Labor and Social Security and the MOF, among others, on May 12, 1999, provides that a pharmaceutical product listed in the NRDL must be clinically needed, safe, effective, reasonably priced, easy to use, available in sufficient quantity, and must meet the following requirements:
| ● | it is set forth in the Pharmacopoeia of the PRC; |
| ● | it meets the standards promulgated by the NMPA; and |
| ● | if imported, it is approved by the NMPA for import. |
Factors that affect the inclusion of a pharmaceutical product in the NRDL include whether the product is consumed in large volumes and commonly prescribed for clinical use in the PRC and whether it is considered to be important in meeting the basic healthcare needs of the general public.
The PRC Ministry of Labor and Social Security, together with other government authorities, has the power to determine inclusion of medicines in the NRDL (also referred to as the “Drug Catalog”), which is divided into two parts, Category A and Category B. Per the Notice on the “National Basic Medical Insurance, Work Injury Insurance and Maternity Insurance Drug Catalog (2022)” issued by the National Healthcare Security Administration and the Ministry of Labor and Social Security, local authorities are required to strictly implement the Drug Catalog (2022) and must not adjust the categories of drugs, remarks and the classification of drugs in the Drug Catalog.
Patients purchasing medicines included in Category A of the NRDL are entitled to reimbursement of the entire amount of the purchase price. Patients purchasing medicines included in Category B of the NRDL are required to pay a certain percentage of the purchase price and obtain reimbursement for the remainder of the purchase price. The percentage of reimbursement for Category B medicines differs from region to region in the PRC.
The total amount of reimbursement for the cost of medicines, in addition to other medical expenses, for an individual participant under the National Medical Insurance Program in a calendar year is capped at the amounts in such participant’s individual account under such program. The amount in a participant’s account varies, depending on the amount of contributions from the participant and his or her employer.
National Essential Medicines List
On August 18, 2009, MOH and eight other ministries and commissions in the PRC issued the Provisional Measures on the Administration of the National Essential Medicines List, which was later amended in 2015, and the Guidelines on the Implementation of the Establishment of the National Essential Medicines System, which aim to promote essential medicines sold to consumers at fair prices in the PRC and ensure that the general public in the PRC has equal access to the drugs contained in the National Essential Medicines List. MOH promulgated the National Essential Medicines List (Catalog for the Basic Healthcare Institutions) on August 18, 2009, and promulgated the revised National Essential Medicines List on March 13, 2013 and September 30, 2018. According to these regulations, basic healthcare institutions funded by government, which primarily include county-level hospitals, county-level Chinese medicine hospitals, rural clinics and community clinics, shall store up and use drugs listed in the National Essential Medicines List. Per the Opinions of the General Office of the State Council on Improving the National Essential Medicines System, issued and effective on September 13, 2018, with respect to the qualifying drugs on the National Essential Medicines List, the medical insurance department shall prioritize their inclusion in the NDRL and adjust their classifications as Category A or B, respectively, in accordance with the stipulated procedures.
145
Price Controls
According to the PRC Drug Administration Law and the PRC Drug Administration Law Implementation Regulations, pharmaceutical products are subject to a directive pricing system or to be adjusted by the market. Per the Notice of the National Healthcare Security Administration on issuing the “Opinions on Doing a Good Job in the Current Drug Price Management”, or the Notice on Current Drug Price Management, effective on November 26, 2019, government guidance prices are to be implemented for narcotic drugs and Class I psychotropic drugs, while prices of other drugs are to be determined by the market. Government guidance prices refer to prices as fixed by business operators according to benchmark prices and ranges of the prices as set by the government department in charge of pricing or other related departments. According to the Pricing Catalogue Initiated by the Central Government (2020 Edition), which was promulgated by the NDRC and effective on May 1, 2020, the National Healthcare Security Administration shall be responsible for setting prices of narcotic drugs and Class I psychotropic drugs.
Further, pursuant to the Notice Regarding Further Improvement of the Order of Market Price of Pharmaceutical Products and Medical Services, or the Market Price Notice, jointly promulgated by the NDRC, the State Council Legislative Affairs Office and the State Council Office for Rectifying, the MOH, the NMPA, the MOFCOM, the MOF and Ministry of Labor and Social Security on May 19, 2006, the PRC government exercises price control over pharmaceutical products included in the NRDL and made an overall adjustment of their prices by reducing the retail price of certain overpriced pharmaceutical products and increasing the retail price of certain underpriced pharmaceutical products in demand for clinical use but that have not been produced in large quantities by manufacturers due to their low retail price level. In particular, the retail price charged by hospitals at the county level or above may not exceed 115% of the procurement cost of the relevant pharmaceutical products or 125% for Chinese herbal pieces. The Market Price Notice has been abolished per the NDRC Decision to Abolish Standardized Pricing Directories, effective May 20, 2021.
On February 9, 2015, the General Office of the State Council issued the Guiding Opinion on Enhancing Consolidated Procurement of Pharmaceutical Products by Public Hospitals, or the Opinion. The Opinion encourages public hospitals to consolidate their demands and to play a more active role in the procurement of pharmaceutical products. Hospitals are encouraged to directly settle the prices of pharmaceutical products with manufacturers. Consolidated procurement of pharmaceutical products should facilitate hospital reform, reduce patient costs, prevent corrupt conducts, promote fair competition and induce the healthy growth of the pharmaceutical industry. According to the Opinion, provincial tendering processes will continue to be used for the pricing of essential drugs and generic drugs with significant demands, and transparent multi-party price negotiation will be used for some patented drugs and exclusive drugs.
On April 26, 2014, the NDRC issued the Notice on Issues concerning Improving the Price Control of Low Price Drugs, or the Low Price Drugs Notice, together with the Low Price Drug List, or LPDL. According to the Low Price Drugs Notice, for drugs with relatively low average daily costs within the current government-guided pricing scope (low price drugs), the maximum retail prices set by the government were cancelled. Within the standards of average daily costs, the specific purchase and sale prices are fixed by the producers and operators based on the drug production costs, market supply and demand and market competition. The standards of average daily cost of low price drugs were determined by the NDRC in consideration of the drug production costs, market supply and demand and other factors and based on the current maximum retail prices set by the government (or the national average bid-winning retail prices where the government does not set the maximum retail prices) and the average daily dose calculated according to the package insert. Under the Low Price Drugs Notice, the standards for the daily cost of low price chemical pharmaceuticals and of low price traditional Chinese medicine pharmaceuticals were less than RMB3.0 ($0.46) per day and RMB5.0 ($0.76) per day respectively. The Low Price Drugs Notice has been abolished per the NDRC Decision to Abolish Standardized Pricing Directories, effective May 20, 2021.
On May 4, 2015, the NDRC, the National Health and Family Planning Commission, the NMPA, MOFCOM and three other departments issued Opinions on Promoting Drug Pricing Reform. Under these opinions, beginning on June 1, 2015, the restrictions on the prices of the drugs that were subject to government pricing were cancelled except for narcotic drugs and Class I psychotropic drugs which remained subject to maximum factory prices and maximum retail prices set by the NDRC, and following the November 2019 Notice on Current Drug Price Management, narcotic drugs and Class I psychotropic drugs prices have transitioned towards government guidance prices. The medical insurance regulatory authority now has the power to prescribe the standards, procedures, basis and methods of the payment for drugs paid by medical insurance funds. The prices of patented drugs are set through transparent and public negotiation among multiple parties. The prices for blood products not listed in the NRDL, immunity and prevention drugs that are purchased by the Chinese government in a centralized manner, and AIDS antiviral drugs and contraceptives provided by the Chinese government for free, are set through a tendering process. Except as otherwise mentioned above, the prices for other drugs may be determined by the manufacturers and the operators on their own on the basis of production or operation costs and market supply and demand.
146
Centralized Procurement and Tenders
The Guiding Opinions concerning the Urban Medical and Health System Reform, promulgated on February 21, 2000, aim to provide medical services with reasonable price and quality to the public through the establishment of an urban medical and health system. One of the measures used to realize this aim is the regulation of the purchasing process of pharmaceutical products by medical institutions. Accordingly, the MOH and other relevant government authorities have promulgated a series of regulations and releases in order to implement the tender requirements.
According to the Notice on Issuing Certain Regulations on the Trial Implementation of Centralized Tender Procurement of Drugs by Medical Institutions promulgated on July 7, 2000 and the Notice on Further Improvement on the Implementation of Centralized Tender Procurement of Drugs by Medical Institutions promulgated on August 8, 2001, medical institutions established by county or higher level government are required to implement centralized tender procurement of drugs.
The MOH promulgated the Working Regulations of Medical Institutions for Procurement of Drugs by Centralized Tender and Price Negotiations (for Trial Implementation), or the Centralized Procurement Regulations, on March 13, 2002, and promulgated Sample Document for Medical Institutions for Procurement of Drugs by Centralized Tender and Price Negotiations (for Trial Implementation), or the Centralized Tender Sample Document in November 2001, as amended in 2010, to implement the tender process requirements and ensure the requirements are followed uniformly throughout the country. The Centralized Tender Regulations and the Centralized Tender Sample Document provide rules for the tender process and negotiations of the prices of drugs, operational procedures, a code of conduct and standards or measures of evaluating bids and negotiating prices. On January 17, 2009, the MOH, the NMPA and other four national departments jointly promulgated the Opinions on Further Regulating Centralized Procurement of Drugs by Medical Institutions. According to the notice, public medical institutions owned by the government at the county level or higher or owned by state-owned enterprises (including state-controlled enterprises) shall purchase pharmaceutical products through centralized procurement. Each provincial government shall formulate its catalogue of drugs subject to centralized procurement. Specifically, the procurement could be achieved through public tendering, online bidding, centralized price negotiations and online competition platform. Except for drugs in the National Essential Medicines List (the procurement of which shall comply with the relevant rules on the National Essential Medicines List), certain pharmaceutical products which are under the national government’s special control and traditional Chinese medicines, in principle, all drugs used by public medical institutions shall be covered by the catalogue of drugs subject to centralized procurement. On July 7, 2010, the MOH and six other ministries and commissions jointly promulgated the Working Regulations of Medical Institutions for Centralized Procurement of Drugs to further regulate the centralized procurement of drugs and clarify the code of conduct of the parties in centralized drug procurement.
The centralized tender process takes the form of public tender operated and organized by provincial or municipal government agencies in principle is conducted once every year in all provinces and cities in China. Drug manufacturing enterprises, in principle, shall bid directly for the centralized tender process. Certain related parties, however, may be engaged to act as bidding agencies. Such intermediaries are not permitted to engage in the distribution of drugs and must have no conflict of interest with the organizing government agencies. The bids are assessed by a committee composed of pharmaceutical experts who will be randomly selected from a database of experts approved by the relevant government authorities. The committee members assess the bids based on a number of factors, including but not limited to, bid price, product quality, clinical effectiveness, qualifications and reputation of the manufacturer, and after-sale services. Only pharmaceuticals that have won in the centralized tender process may be purchased by public medical institutions funded by government in the relevant region.
147
4+7 Quality Consistency Evaluation
On November 15, 2018, China’s Joint Procurement Office published its Paper on Centralized Drug Procurement in “4+7 Cities,” known as the 4+7 Quality Consistency Evaluation process, or 4+7 QCE. The 4+7 QCE initiative is aimed at driving consolidation in the fragmented generic drug market in China. The 4+7 QCE initiative began as a pilot program in 11 cities: Beijing, Tianjin, Shanghai, Chongqing, Shenyang, Dalian, Xiamen, Guangzhou, Shenzhen, Chengdu and Xi’an. Under this pilot program, the public medical institutions in these 11 cities bulk-buy certain generic drugs together, forcing companies to bid for contracts and driving down prices. The 4+7 QCE initiative has expanded nationwide and now covers more varieties of drugs. On September 1, 2019, the Joint Procurement Office published its Paper on Centralized Drug Procurement in Alliance Areas (GY-YD2019-1), such areas covering 25 provinces and regions across China. On December 29, 2019, the Joint Procurement Office published its Paper on Nationwide Centralized Drug Procurement (GY-YD2019-2), promoting procurement nationwide, and on January 13, 2020, the National Healthcare Security Administration, the NHC, the NMPA, the Ministry of Industrial and Information Technology and the Logistics Support Department of the Central Military Commission promulgated the Notice on the Commencement of the Second Batch of State Organized Centralized Drug Procurement and Use, which states that the second batch of national organization of centralized procurement and use of drugs would not be carried out in selected areas but nationwide. On January 22, 2021, the General Office of the State Council issued the Opinions on Promoting the Normalization and Institutionalization of the Centralized and Quantitative Procurement of Drugs, stating that (i) the scope of procurement should focus on including drugs in the NDRL with large dosages and high purchase amounts and gradually cover all kinds of drugs that are clinically necessary and of reliable quality that are marketed in China, so as to ensure that all drugs that should be procured are exhausted, (ii) marketing authorization holders who have obtained drug registration certificates for drugs within the scope of centralized procurement can, in principle, participate in centralized drug procurement, provided they meet the requirements of centralized procurement in areas including but not limited to quality standards, production capacity and supply stability, and (iii) all public medical institutions (including military medical institutions) should participate in centralized drug procurement, and designated pharmacies shall follow the management requirements of designated agreements.
U.S. Coverage and Reimbursement
Successful sales of our products or drug candidates in the U.S. market, if approved, will depend, in part, on the extent to which our drugs will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. Patients who are provided with prescriptions as part of their medical treatment generally rely on such third-party payors to reimburse all or part of the costs associated with their prescriptions and therefore adequate coverage and reimbursement from such third-party payors are critical to new product success. These third-party payors are increasingly reducing reimbursements for medical drugs and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments, and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement, requirements for substitution of generic drugs, and pricing transparency requirements. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. Decreases in third-party reimbursement for our drug candidates, if approved, or a decision by a third-party payor to not cover our drug candidates could reduce physician usage of such drugs and have a material adverse effect on our sales, results of operations and financial condition.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, as amended by the IRA, or the MMA, established the Medicare Part D program to provide a voluntary prescription drug benefit to Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities that provide coverage of outpatient prescription drugs. Unlike Medicare Part A and B, Part D coverage is not standardized. Subject to certain limitations, Part D prescription drug plan sponsors generally are not required to pay for or cover all covered Part D drugs, and can develop their own drug formulary that identifies which drugs they will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Medicare payment for some of the cost of prescription drugs may increase demand for drugs for which we receive regulatory approval. The IRA amended the Part D benefit design to, among other items, require formulary coverage of Medicare negotiated drugs and alter manufacturer, patient, Part D plan sponsor, and government financial responsibilities in connection with the Part D benefit. Other components of the IRA affect Part D drugs by subjecting particular drugs to government negotiated prices. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
148
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. The plan for the research was published in 2012 by the U.S. Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures are made to Congress. Although the results of the comparative effectiveness studies were not intended to mandate coverage policies for public or private payors, if third-party payors do not consider a drug to be cost-effective compared to other available therapies, they may not cover such drugs as a benefit under their plans or, if they do, the level of payment may not be sufficient.
The Affordable Care Act, enacted in March 2010, has had a significant impact on the health care industry. The Affordable Care Act expanded coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceutical products, the Affordable Care Act, among other things, addressed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, established annual fees and taxes on manufacturers of certain branded prescription drugs, and created a new Medicare Part D coverage gap discount program. The IRA reformed the Medicare Part D benefit, including by sunsetting the coverage gap as of 2025, creating a new manufacturer discount agreement that goes into effect January 1, 2025, establishing a $2,000 annual cap beyond which beneficiaries will not bear any cost-sharing obligations, and restructuring manufacturer, patient, Part D plan sponsor and government financial obligations in connection with the Part D benefit.
The IRA implements other Medicare program reforms, such as mandating the negotiation of eligible Medicare Part B and Part D drugs, and imposing rebates for Medicare drugs that increase in price faster than the rate of inflation. Under the IRA’s Medicare negotiation program, the U.S. government will negotiate the Medicare prices of single-source small molecule and biologic products that have been on the market for 7 and 11 years, respectively, following FDA approval. Negotiated prices will be capped at a statutory ceiling price that is likely to represent a significant discount from average prices to wholesalers and direct purchasers. The negotiation program imposes substantial excise taxes on manufacturers that do not timely comply with the negotiation requirements and subjects manufacturers to potential civil monetary penalties for failing to offer the maximum fair price, violating the terms of the negotiation agreement, or knowingly providing false information.
Other legislative and regulatory changes have been proposed and adopted in the United States that affect reimbursement for prescription drugs. In December 2017, Congress repealed the “individual mandate,” which was an Affordable Care Act requirement that individuals obtain healthcare insurance coverage or face a penalty. This repeal could affect the total number of patients who have coverage from third-party payors that reimburse for use of our products. In July 2021, the U.S. Supreme Court dismissed a constitutional challenge to the Affordable Care Act brought by a group of Republican attorneys general seeking to invalidate the law in its entirety because of Congress’s repeal of the individual mandate.
The Budget Control Act of 2011 (“BCA”) requires automatic spending reductions to reduce the federal deficit, including Medicare spending reductions of up to 2% per fiscal year, with a uniform percentage reduction across all Medicare programs. In 2013, the Centers for Medicare & Medicaid Services (“CMS”) began imposing a 2% reduction on Medicare payments. Subsequent legislation extended sequestration for mandatory spending through FY2031 and the sequestration of Medicare benefit payments spending through FY2032. Sequestration to Medicare was suspended from May 1, 2020 through March 30, 2022, and was limited in amount from April 1, 2022 through June 30, 2022. In addition, the American Rescue Plan Act of 2021 (“ARPA”) increased the federal budget deficit in a manner that triggers an additional sequestration mandated under the Pay As You Go Act of 2010 (“PAYGO Act”). As a result, a further payment reduction of up to 4% was required to take effect in January 2022. However, Congress has delayed implementation of this payment reduction until 2023.
On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which among other things, prevented reductions in Medicare physician payment rates.
In addition, other proposed legislative and regulatory changes could affect reimbursement for prescription drugs. In November 2017, CMS announced a Final Rule that set the reimbursement rate for prescription drugs that hospitals purchased through the 340B Program at average sales price minus 22.5%, as opposed to the historical rate of average sales price plus 6%. The American Hospital Association and others successfully challenged the Final Rule. The litigation was appealed, and the U.S. Supreme Court ruled that, absent a survey of hospitals’ costs, CMS may not vary the reimbursement rates for drugs only for 340B hospitals. Congress and the U.S. administration may continue to evaluate other proposals that could affect third-party reimbursement for our drug candidates, if approved.
149
In October 2020, the U.S. Department of Health and Human Services (“HHS”) and the FDA issued a final rule and guidance concerning two new pathways for importing lower-cost drugs into the United States. The final rule allows certain prescription drugs to be imported from Canada, and the guidance describes procedures for drug manufacturers to facilitate the importation of FDA-approved drugs and biologics manufactured abroad and originally intended for sale in a foreign country into the United States. In January 2024, the FDA authorized Florida’s drug importation program, allowing the state of Florida to advance in preparing to import certain prescription drugs from Canada.
In November 2020, HHS issued a rule eliminating the safe harbor shielding Medicare Part D rebates to pharmacy benefit managers from the Anti-Kickback Statute. In response to litigation brought by a trade association on behalf of pharmacy benefit managers, the Biden administration agreed to delay the rule’s effective date until January 1, 2023. Later federal laws further delayed implementation of the final rule until 2032.
Rest of the World Coverage and Reimbursement
In some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the E.U. provides options for its member states to restrict the range of medicinal drugs for which their national health insurance systems provide reimbursement and to control the prices of medicinal drugs for human use. A member state may approve a specific price for the medicinal drug or it may instead adopt a system of direct or indirect controls on the profitability of our company placing the medicinal drug on the market. Historically, drugs launched in the E.U. do not follow price structures of the United States and generally tend to be significantly lower.
150
Other Healthcare Laws
Other PRC Healthcare Laws
Advertising of Pharmaceutical Products
In accordance with the Interim Administrative Measures for the Censorship of Advertisements for Drugs, Medical Devices, Health Food and Formula Food for Special Medical Purposes effective from March 1, 2020, the State Administration for Market Regulation is responsible for organizing and guiding the censorship of advertisements for drugs, medical devices, health foods and formula foods for special medical purposes. Any advertisement for drugs, medical devices, health food or formula food for special medical purposes shall indicate the advertisement approval number in a prominent position. The validity period of the advertisement approval number for drugs, medical devices, health food and formula food for special medical purposes shall be consistent with the shortest period of validity of the product registration certificate, record-filing certificate, or production license. Where no period of validity is prescribed in the product registration certificate, record-filing certificate or production license, the period of validity of the advertisement approval number shall be two years.
Packaging of Pharmaceutical Products
According to the Measures for The Administration of Pharmaceutical Packaging, effective on September 1, 1988, pharmaceutical packaging must comply with the provisions of the national standard and professional standard. If there are no standards, the enterprise can formulate its own standard after obtaining the approval of the provincial level drug administration or bureau of standards. The enterprise shall reapply to the relevant authorities if it needs to change the packaging standard. Drugs without packing must not be sold in PRC (except for drugs needed by the army).
Labor Protection
Under the Labor Law of the PRC, effective on January 1, 1995 and subsequently amended on August 27, 2009 and December 29, 2018, the Labor Contract Law of the PRC, effective on January 1, 2008 and subsequently amended on December 28, 2012, and the Implementing Regulations of the Labor Contract Law of the PRC, effective on September 18, 2008, employers must establish a comprehensive management system to protect the rights of their employees, including a system governing occupational health and safety to provide employees with occupational training to prevent occupational injury, and employers are required to truthfully inform prospective employees of the job description, working conditions, location, occupational hazards and status of safe production as well as remuneration and other conditions as requested by the Labor Contract Law of the PRC.
Pursuant to the Law of Manufacturing Safety of the People’s Republic of China effective on November 1, 2002 and subsequently amended on December 1, 2014 and September 1, 2021, manufacturers must establish a comprehensive management system to ensure manufacturing safety in accordance with applicable laws and regulations. Manufacturers not meeting relevant legal requirements are not permitted to commence their manufacturing activities.
Pursuant to the Administrative Measures for Production effective on March 1, 2011, manufacturers of pharmaceutical products are required to establish production safety and labor protection measures in connection with the operation of their manufacturing equipment and manufacturing process.
Pursuant to applicable PRC laws, rules and regulations, including the Social Insurance Law which became effective on July 1, 2011 and subsequently amended on December 29, 2018, the Interim Regulations on the Collection and Payment of Social Security Funds which became effective on January 22, 1999 and subsequently amended on March 24, 2019, the Interim Measures concerning the Maternity Insurance which became effective on January 1, 1995 and the Regulations on Work-related Injury Insurance which became effective on January 1, 2004 and were subsequently amended on December 20, 2010, employers are required to contribute, on behalf of their employees, to a number of social security funds, including funds for basic pension insurance, unemployment insurance, basic medical insurance, work-related injury insurance, and maternity insurance. If an employer fails to make social insurance contributions timely and in full, the social insurance collecting authority will order the employer to make up outstanding contributions within the prescribed time period and impose a late payment fee at the rate of 0.05% per day from the date on which the contribution becomes due. If such employer fails to make social insurance registration, the social insurance collecting authority will order the employer to correct within the prescribed time period. The relevant administrative department may impose a fine equivalent to three times the overdue amount and management personnel who are directly responsible can be fined RMB500 ($76.43) to RMB3,000 ($458.02) if the employer fails to correct within the prescribed time period.
151
Commercial Bribery
Medical production and operation enterprises involved in criminal, investigation or administrative procedure for commercial bribery will be listed in the Adverse Records of Commercial Briberies by provincial health and family planning administrative department. Pursuant to the Provisions on the Establishment of Adverse Records of Commercial Briberies in the Medicine Purchase and Sales Industry issued by the National Health and Family Planning Commission and effective on March 1, 2014, if medical production and operation enterprises are listed into the Adverse Records of Commercial Briberies for the first time, their production shall not be purchased by public medical institutions, and medical and health institutions receiving financial subsidies in local provincial regions for a period of two years following the publication of the Adverse Records, and public medical institutions, and medical and health institutions receiving financial subsidies in other provinces shall lower their rating in bidding or purchasing process. If medical production and operation enterprises are listed into the Adverse Records of Commercial Briberies twice or more times in five years, their production may not be purchased by public medical institutions, and medical and health institutions receiving financial subsidies nationwide in two years from public of the record.
As advised by our PRC legal advisor, from a PRC law perspective, a pharmaceutical company will not be penalized by the relevant PRC government authorities merely by virtue of having contractual relationships with distributors or third-party promoters who are engaged in bribery activities, so long as such pharmaceutical company and its employees are not utilizing the distributors or third-party promoters for the implementation of, or acting in conjunction with them in, the prohibited bribery activities. In addition, a pharmaceutical company is under no legal obligation to monitor the operating activities of its distributors and third-party promoters, and will not be subject to penalties or sanctions by relevant PRC government authorities as a result of failure to monitor their operating activities.
Product Liability
In addition to the strict new drug approval process, certain PRC laws have been promulgated to protect the rights of consumers and to strengthen the control of medical products in the PRC. Under current PRC law, manufacturers and vendors of defective products in the PRC may incur liability for loss and injury caused by such products. Pursuant to the Civil Code of the PRC, or the PRC Civil Code, promulgated on May 28, 2020 and effective on January 1, 2021, a defective product which causes property damage or physical injury to any person may subject the manufacturer or vendor of such product to civil liability for such damage or injury.
On February 22, 1993, the Product Quality Law of the PRC, or the Product Quality Law, was promulgated aiming to define responsibilities for product quality, to protect the legitimate rights and interests of the end-users and consumers and to strengthen the supervision and control of the quality of products. The Product Quality Law was amended by the Ninth National People’s Congress on July 8, 2000 and was later amended by the Eleventh National People’s Congress on August 27, 2009 and the Thirteenth National People’s Congress on December 29, 2018. Pursuant to the amended Product Quality Law, manufacturers who produce defective products may be subject to civil or criminal liability and have their business licenses revoked.
The Law of the PRC on the Protection of the Rights and Interests of Consumers was promulgated on October 13, 1993 and was amended on October 25, 2013 to protect consumers’ rights when they purchase or use goods and accept services. All business operators must comply with this law when they manufacture or sell goods and/or provide services to customers. Under the amendment on October 25, 2013, all business operators shall pay high attention to protect the customers’ privacy which they obtain during the business operation. In addition, in extreme situations, pharmaceutical product manufacturers and operators may be subject to criminal liabilities under applicable laws of the PRC if their goods or services lead to the death or injuries of customers or other third parties.
Pursuant to the PRC Civil Code, if damages to other persons are caused by defective products that are resulted from the fault of a third party such as the parties providing transportation or warehousing, the producers and the sellers of the products have the right to recover their respective losses from such third parties. If defective products are identified after they have been put into circulation, the producers or the sellers shall take remedial measures such as issuance of warning, and recall of products, etc. in a timely manner. The producers or the sellers shall be liable under tort if they cause damages due to their failure to take remedial measures in a timely manner or have not made efforts to take remedial measures, thus causing damages. If the products are produced and sold with known defects, causing deaths or severe damage to the health of others, the infringed party shall have the right to claim respective punitive damages in addition to compensatory damages.
152
Other PRC National and Provincial-Level Laws and Regulations
We are subject to changing regulations under many other laws and regulations administered by governmental authorities at the national, provincial and municipal levels, some of which are or may become applicable to our business. Our hospital customers are also subject to a wide variety of laws and regulations that could affect the nature and scope of their relationships with us.
For example, regulations control the confidentiality of patients’ medical information and the circumstances under which patient medical information may be released for inclusion in our databases, or released by us to third parties. These laws and regulations governing both the disclosure and the use of confidential patient medical information may become more restrictive in the future.
We also comply with numerous additional state and local laws relating to matters such as safe working conditions, manufacturing practices, environmental protection and fire hazard control. We believe that we are currently in compliance with these laws and regulations; however, we may be required to incur significant costs to comply with these laws and regulations in the future. Unanticipated changes in existing regulatory requirements or adoption of new requirements could therefore have a material adverse effect on our business, results of operations and financial condition.
Other U.S. Healthcare Laws
We may also be subject to healthcare regulation and enforcement by the U.S. federal government and the states where we may market our drug candidates, if approved. These laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security and physician sunshine laws and regulations.
Anti-Kickback Statute
The federal Anti-Kickback Statute prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service, or the purchase or order of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. The majority of states also have anti-kickback laws, which establish similar prohibitions and in some cases may apply to items or services reimbursed by any third-party payor, including commercial insurers. The Anti-Kickback Statute is subject to evolving interpretations. In the past, the government has enforced the Anti-Kickback Statute to reach large settlements with healthcare, pharmaceutical, and biotechnology companies based on a range of financial arrangements with physicians and other healthcare industry entities. A person or entity does not need to have actual knowledge of the Anti-Kickback Statute or specific intent to violate it in order to have committed a violation. Violations of the Anti-Kickback Statute can result in criminal, civil, or administrative liability. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for the purposes of the federal False Claims Act.
False Claims
Additionally, the civil False Claims Act prohibits knowingly presenting or causing the presentation of a false, fictitious or fraudulent claim for payment to the U.S. government. Actions under the False Claims Act may be brought by the U.S. Attorney General or as a qui tam action by a private individual in the name of the government. Analogous state law equivalents may apply and may be broader in scope than the federal requirements. Violations of the False Claims Act can result in very significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the United States, for example, in connection with the violations of the Anti-Kickback Statute, the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the False Claims Act in addition to individual criminal convictions and corporate resolutions under applicable criminal statutes. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
153
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, also created new federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Payments to Physicians
There has also been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Physician Payments Sunshine Act (“Sunshine Act”), which is a part of the Affordable Care Act, among other things, imposes annual reporting requirements on drug manufacturers for payments made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission. Certain states also mandate implementation of compliance programs, impose restrictions on drug manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians. The federal government has imposed penalties on companies that fail to appropriately report required information.
Data Privacy and Security
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conduct our business. HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and their respective implementing regulations, including the final omnibus rule published on January 25, 2013, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information that apply to most U.S. health care providers with which we interact, such as our U.S. clinical trial sites. In addition, state laws, including, notably the CCPA and other comprehensive data privacy and security laws, govern the privacy and security of personal health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts.
PRC Regulation of Foreign Currency Exchange, Offshore Investment and State-Owned Assets
PRC Foreign Currency Exchange
Foreign currency exchange regulation in China is primarily governed by the following rules:
| ● | Foreign Currency Administration Rules (1996), as last amended on August 5, 2008, or the Exchange Rules; and |
| ● | Administration Rules of the Settlement, Sale and Payment of Foreign Exchange (1996), or the Administration Rules. |
Under the Exchange Rules, the renminbi is convertible for current account items, including the distribution of dividends, interest payments, trade and service-related foreign exchange transactions. Conversion of renminbi for capital account items, such as direct investment, loan, security investment and repatriation of investment, however, is still subject to the SAFE’s scrutiny.
Under the Administration Rules, foreign-invested enterprises may only buy, sell and/or remit foreign currencies at those banks authorized to conduct foreign exchange business after providing valid commercial documents and, in the case of capital account item transactions, obtaining approval from the SAFE. Capital investments by foreign-invested enterprises outside of China are also subject to limitations, which include approvals by the MOFCOM, the SAFE and the NDRC.
154
Pursuant to the Circular on Further Improving and Adjusting the Direct Investment Foreign Exchange Administration Policies, or Circular 59, promulgated by the SAFE on November 19, 2012, effective on December 17, 2012, and amended in 2015, 2018 and 2019, approval is not required for the opening of and payment into foreign exchange accounts under direct investment, for domestic reinvestment with legal income of foreign investors in China. Circular 59 also simplified the capital verification and confirmation formalities for Chinese foreign-invested enterprises and the foreign capital and foreign exchange registration formalities required for the foreign investors to acquire the equities of Chinese party and other items. Circular 59 further improved the administration on exchange settlement of foreign exchange capital of Chinese foreign-invested enterprises.
Foreign Exchange Registration of Offshore Investment by PRC Residents
In July 2014, the SAFE issued the Notice on Relevant Issues Concerning Foreign Exchange Administration for PRC Residents to Engage in Offshore Investment and Financing and Round Trip Investment via Special Purpose Vehicles, or Circular 37, and its implementation guidelines, which abolishes and supersedes the SAFE’s Circular on Relevant Issues Concerning Foreign Exchange Administration for PRC Residents to Engage in Financing and Round Trip Investment via Overseas Special Purpose Vehicles, or Circular 75. Pursuant to Circular 37 and its implementation guidelines, PRC residents (including PRC institutions and individuals) must register with local branches of the SAFE in connection with their direct or indirect offshore investment in an overseas special purpose vehicle, or SPV, directly established or indirectly controlled by PRC residents for the purposes of offshore investment and financing with their legally owned assets or interests in domestic enterprises, or their legally owned offshore assets or interests. Such PRC residents are also required to amend their registrations with the SAFE when there is a significant change to the SPV, such as changes of the PRC individual resident’s increase or decrease of its capital contribution in the SPV, or any share transfer or exchange, merger, division of the SPV. Failure to comply with the registration procedures set forth in Circular 37 may result in restrictions being imposed on the foreign exchange activities of the relevant onshore company, including the payment of dividends and other distributions to its offshore parent or affiliate, the capital inflow from the offshore entities and settlement of foreign exchange capital, and may also subject relevant onshore company or PRC residents to penalties under PRC foreign exchange administration regulations.
In February 2012, the SAFE promulgated the Notices on Issues Concerning the Foreign Exchange Administration for Domestic Individuals Participating in Stock Incentive Plans of Overseas Publicly Listed Companies. Based on this regulation, directors, supervisors, senior management and other employees of domestic subsidiaries or branches of a company listed on an overseas stock market who are PRC citizens or who are non-PRC citizens residing in China for a continuous period of not less than one year, subject to a few exceptions, are required to register with the SAFE or its local counterparts by following certain procedures if they participate in any stock incentive plan of the company listed on an overseas stock market. Foreign exchange income received from the sale of shares or dividends distributed by the overseas listed company may be remitted into a foreign currency account of such PRC citizen or be exchanged into renminbi. Our PRC citizen employees who have been granted share options have been subject to these rules due to our admission to trading on the AIM market and the listing of our ADSs on Nasdaq.
Regulation on Investment in Foreign-invested Enterprises
Pursuant to PRC law, the registered capital of a limited liability company is the total capital contributions subscribed for by all the shareholders as registered with the company registration authority. A foreign-invested enterprise’s total investment limit was previously approved by or filed with the MOFCOM or its local counterpart by reference to both its registered capital and expected investment scale. A foreign-invested enterprise was required to obtain approval from or file with the MOFCOM or its local counterpart for any increases to its total investment limit.
During 2019 and 2020, a series of reforms concerning foreign-invested enterprises came into effect, including but not limited to the Foreign Investment Law of the PRC, effective January 1, 2020; the Implementation Rules for the Foreign Investment Law, effective January 1, 2020, and Measures on Reporting of Foreign Investment Information, effective January 1, 2020. The reformed rules do not require foreign-invested enterprises to complete the abovementioned filing or approval with the MOFCOM in relation to total investment limits; rather, pursuant to Measures on Reporting of Foreign Investment Information, during enterprise incorporation and subsequent changes in commercial registration, foreign investors and foreign-invested enterprises (as applicable) shall submit investment information to the MOFCOM or its local counterpart.
The difference between the total investment limit and the registered capital of a foreign-invested enterprise or the cross-border financing risk weighted balance calculated based on a formula by the PBOC represents the foreign debt financing quota to which the foreign-invested enterprise is entitled (i.e., the maximum amount of debt which the company may borrow from a foreign lender).
155
In accordance with these regulations, we and our joint venture partners have contributed financing to our PRC subsidiaries and joint ventures in the form of capital contributions up to the registered capital amount and/or in the form of shareholder loans up to the foreign debt quota. According to the financing needs of our PRC subsidiaries and joint ventures, we and our joint venture partners have requested and received approvals (where necessary) from the government authorities for increases to the total investment limit for certain of our PRC subsidiaries and joint ventures from time to time. As a result, these regulations have not had a material impact to date on our ability to finance such entities.
The Company Law of the PRC was amended on December 29, 2023 (such amendment, the “Revised Company Law”), and will take effect on July 1, 2024. Foreign-invested companies must comply with the Revised Company Law, unless otherwise stipulated. Among others, the Revised Company Law introduces a rule requiring the registered capital of limited liability companies to be fully paid within five years, which applies to all PRC limited liability companies. Companies incorporated before the promulgation and implementation of the Revised Company Law are required to gradually adjust to meet the deadline. In consequence, we may be required to accelerate payment of capital contributions towards the registered capital of our PRC subsidiaries and joint ventures. Specific implementation measures of the Revised Company Law shall be prescribed by the State Council, of which, as of the date of this annual report, final versions are yet to be released.
Regulation on Dividend Distribution
The principal regulations governing distribution of dividends paid by wholly foreign-owned enterprises include:
| ● | Company Law of the PRC (1993), as amended in 1999, 2004, 2005, 2013 and 2018; |
| ● | Foreign Investment Law of the PRC; and |
| ● | Implementation Rules for the Foreign Investment Law. |
| ● | Under these laws and regulations, foreign-invested enterprises in China may pay dividends only out of their accumulated profits, if any, determined in accordance with PRC accounting standards and regulations. In addition, a wholly foreign-owned enterprise in China is required to set aside at least 10.0% of its after-tax profit based on PRC accounting standards each year to its general reserves until the accumulative amount of such reserves reach 50.0% of its registered capital. These reserves are not distributable as cash dividends. |
Filings and Approvals Relating to State-Owned Assets
Pursuant to applicable PRC state-owned assets administration laws and regulations, incorporating a joint venture that will have investments of assets that are both state-owned and non-state-owned, investing in an entity that was previously owned by a state-owned enterprise and restructuring an enterprise ultimately owned by the general public require the performance of an assessment of the relevant state-owned assets and the filing of the assessment results with the competent state-owned assets administration, finance authorities or other regulatory authorities and, if applicable, the receipt of approvals from such authorities.
Our joint venture partner was required to perform a state-owned asset assessment when Shanghai Hutchison Pharmaceuticals was incorporated and our joint venture partner contributed state-owned assets, and when we invested in Hutchison Sinopharm, which was previously wholly-owned by Sinopharm, a state-owned enterprise. In addition, Hutchison Sinopharm was required to perform a state-owned asset assessment when Hutchison Sinopharm restructured from an enterprise ultimately owned by the general public into a limited liability enterprise. In all four instances, our joint venture partners have informed us that they or Hutchison Sinopharm have duly filed the relevant state-owned asset assessment results with, and obtained the requisite approvals from, the relevant governmental authorities as required by the foregoing laws and regulations. Accordingly, we believe that such joint ventures are in full compliance with all applicable laws and regulations governing the administration and restructuring of state-owned assets, although we are currently unable to obtain copies of certain filing and approval documents from our joint venture partners due to their internal confidentiality constraints. We have not received any notice of warning or been subject to any penalty or other disciplinary action from the relevant governmental authorities with respect to the applicable laws and regulations governing the administration and restructuring of state-owned assets.
156
C. Organizational Structure
The chart below shows our organizational structure, including our principal subsidiaries and joint ventures, as of February 15, 2024.
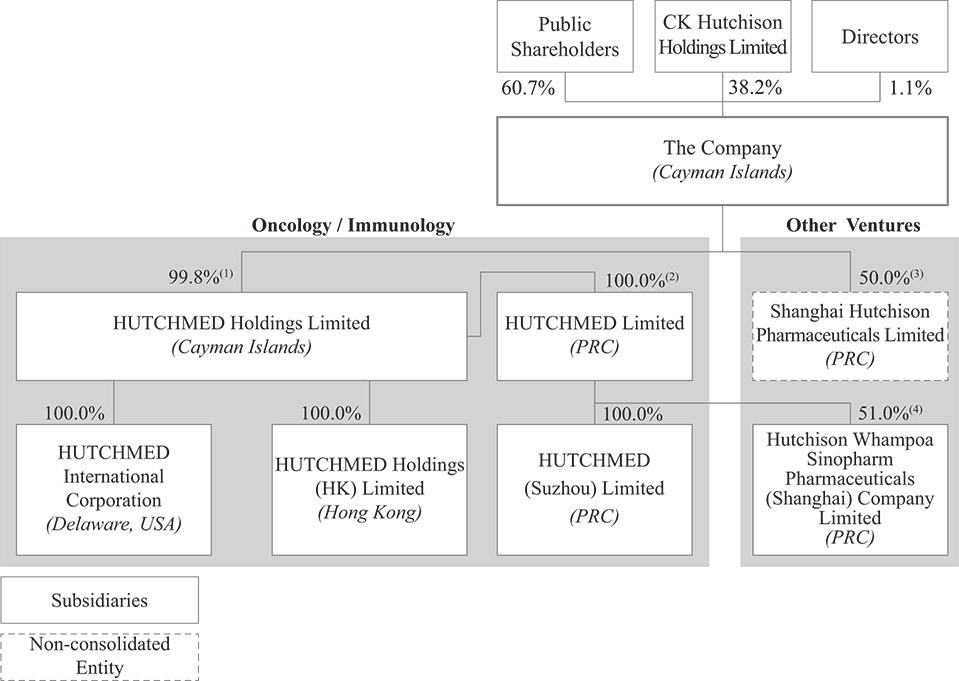
Notes:
| (1) | Employees and former employees of HUTCHMED Limited hold the remaining 0.2% shareholding in HUTCHMED Holdings Limited. |
| (2) | Held through HUTCHMED Investment (HK) Limited. HUTCHMED Limited’s revenue generated by sales of, and royalties, manufacturing costs and services fees in connection with, our current and future internally developed drug candidates are allocated to the Oncology/Immunology operations. |
| (3) | Held through our 100.0% subsidiary Shanghai HUTCHMED Investment (HK) Limited. Shanghai Pharmaceuticals Holding Co., Limited is the other 50.0% joint venture partner. |
| (4) | Sinopharm Group Co. Limited is the other 49.0% joint venture partner. |
D. Property, Plants and Equipment
We are headquartered in Hong Kong where we have our main administrative offices.
157
We rent and operate a 4,968 square meter manufacturing facility that complies with applicable GMP standards for fruquintinib and surufatinib in Suzhou, Jiangsu Province in Eastern China, and own a 5,024 square meter facility in Shanghai which houses research and development operations. We lease 9,080 square meters of office and lab space in Shanghai which houses HUTCHMED Limited’s management and staff. In 2020, we entered into a 50-year land use rights agreement for a 28,771 square meter site in Shanghai. We have recently completed the construction of an almost 55,000 square meter large-scale manufacturing facility for innovative drugs on the site. The Shanghai manufacturing facility has successfully passed an inspection by the local regulatory agency and was issued the Drug Manufacturing Permit in 2023. The clinical manufacturing and technology transfer for some of our commercial products are underway in our new facility. The Shanghai factory will be our largest manufacturing facility, with a production capacity estimated to be five times that of our facility in Suzhou.
We also lease a 12,679 square foot office in Florham Park, New Jersey to house our U.S.-based clinical and regulatory staff.
Our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals, operates a 78,000 square meter large-scale research and development and manufacturing facility in Shanghai for which it has obtained land use rights and property ownership certificates.
Our and our joint ventures’ manufacturing operations consist of bulk manufacturing and formulation, fill, and finishing activities that produce products and drug candidates for both clinical and commercial purposes. Our manufacturing capabilities have a large operation scale for our own-brand products. We and our joint ventures manufacture and sell about 2.8 billion doses of medicines a year, in the aggregate, through our well-established manufacturing base. See “—Other Ventures—Shanghai Hutchison Pharmaceuticals” for more details on our manufacturing operations.
ITEM 4A. UNRESOLVED STAFF COMMENTS
None.
ITEM 5. OPERATING AND FINANCIAL REVIEW AND PROSPECTS
You should read the following discussion and analysis of our financial condition and results of operations together with Item 3.A. “Selected Financial Data,” our consolidated financial statements and the related notes and our non-consolidated joint ventures’ consolidated financial statements and the related notes appearing elsewhere in this annual report. This report contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Exchange Act, including, without limitation, statements regarding our expectations, beliefs, intentions or future strategies that are signified by the words “expect,” “anticipate,” “intend,” “believe,” or similar language. All forward-looking statements included in this annual report are based on information available to us on the date hereof, and we assume no obligation to update any such forward-looking statements. In evaluating our business, you should carefully consider the information provided under Item 3.D. “Risk Factors.” Actual results could differ materially from those projected in the forward-looking statements.
A. Operating Results.
Overview
We are a global commercial-stage biopharmaceutical company focused on the discovery, development and commercialization of targeted therapies and immunotherapies for the treatment of patients with cancer and immunological diseases. We conduct our business through our Oncology/Immunology and Other Ventures operations.
158
Through our Oncology/Immunology operations, our team of approximately 900 scientists and staff has created, developed and in-licensed a deep portfolio of 13 drug candidates. We have advanced 13 oncology drug candidates to clinical trials in China, with four also in active clinical development in the United States and Europe. We have brought three of our internally developed drugs, savolitinib, fruquintinib and surufatinib (marketed as Orpathys, Elunate and Sulanda, respectively) to patients in China. Fruquintinib also received FDA approval in the United States in 2023 and is marketed as Fruzaqla. Moreover, tazemetostat has been approved and launched in Hainan Pilot Zone and Macau and submitted for registration in Hong Kong. We also have additional drug candidates in earlier stage clinical development (Phase I/Ib and Phase Ib/II proof-of-concept studies) and several advanced pre-clinical drug candidates. These drug candidates are being developed to treat a wide spectrum of diseases, including solid tumors, hematological malignancies and immunological diseases which we believe may address unmet medical needs and represent large commercial opportunities. Our success in research and development has led to partnerships with leading global pharmaceutical companies, including AstraZeneca, Eli Lilly and Takeda. We and our collaboration partners have invested approximately $2 billion in our Oncology/Immunology operations as of December 31, 2023, with almost all of these funds used for research and development expenses for the development of our drug candidates. Net loss attributable to our company from our Oncology/Immunology operations was $291.7 million and $385.4 million for the years ended December 31, 2021, 2022 and net income attributable to our company from our Oncology/Immunology operations was $51.2 million for the year ended December 31, 2023.
In addition, we have built large-scale and profitable drug marketing and distribution capabilities through subsidiaries and joint ventures in our Other Ventures, which primarily manufacture, market and distribute prescription drugs and healthcare products in China. Net income attributable to our company generated from our Other Ventures operations was $142.9 million, $54.6 million and $50.3 million for the years ended December 31, 2021, 2022 and 2023, respectively. In addition to helping fund our Oncology/Immunology operations, we utilize the know-how from our Other Ventures to support the launch of our internally developed Oncology/Immunology products in China.
Our consolidated revenue was $356.1 million, $426.4 million and $838.0 million for the years ended December 31, 2021, 2022 and 2023, respectively. Net loss attributable to our company was $194.6 million and $360.8 million for the years ended December 31, 2021 and 2022 and net income attributable to our company was $100.8 million for the year ended December 31, 2023.
Basis of Presentation
Our consolidated statements of operations data presented herein for the years ended December 31, 2023, 2022 and 2021 and our consolidated balance sheet data presented herein as of December 31, 2023 and 2022 have been derived from our audited consolidated financial statements, which were prepared in accordance with U.S. GAAP, and should be read in conjunction with those statements which are included elsewhere in this annual report.
We have two strategic operations, Oncology/Immunology and Other Ventures, that offer different products and services. Our Shanghai Hutchison Pharmaceuticals and Hutchison Baiyunshan (until September 28, 2021 when the disposal of our shareholding interest in Hutchison Baiyunshan was completed) joint ventures under our Other Ventures operations are accounted for under the equity accounting method as non-consolidated entities in our consolidated financial statements, and the consolidated financial statements of Shanghai Hutchison Pharmaceuticals were prepared in accordance with IFRS as issued by the IASB and audited under auditing standards generally accepted in the United States and included elsewhere in this annual report. The presentation of financial data for our business units excludes certain unallocated costs attributed to expenses incurred by our corporate head office. For more information on our corporate structure, see Item 4.A. “History and Development of the Company.”
Factors Affecting our Results of Operations
Research and Development Expenses
We believe our ability to successfully develop innovative drug candidates through our Oncology/Immunology operations will be the primary factor affecting our long-term competitiveness, as well as our future growth and development. Creating high quality global first-in-class or best-in-class drug candidates requires significant investment of resources over a prolonged period of time, and a core part of our strategy is to continue making sustained investments in this area. As a result of this commitment, our pipeline of drug candidates has been steadily advancing and expanding, with more than ten in clinical development. In addition, we are proactively making a strategic shift to focus on the most advanced assets from our internal developed pipeline, that are most likely to drive near-term value. For more information on the nature of the efforts and steps necessary to develop our drug candidates, see Item 4.B. “Business Overview—Our Clinical Pipeline” and “Business Overview—Regulations.”
159
The drug candidates of our Oncology/Immunology operations are still in development, and we have incurred and will continue to incur significant research and development costs for pre-clinical studies and clinical trials. We expect that our research and development expenses will significantly increase in future periods in line with the advancement and expansion of the development of our drug candidates.
Research and development expenses include:
| ● | employee compensation related expenses, including salaries, benefits and equity compensation expense; |
| ● | expenses incurred for payments to CROs, investigators and clinical trial sites that conduct our clinical studies; |
| ● | the cost of acquiring, developing, and manufacturing clinical study materials; |
| ● | facilities, depreciation, and other expenses, which include office leases and other overhead expenses; and |
| ● | costs associated with pre-clinical activities and regulatory operations. |
Research and development expenses incurred by our Oncology/Immunology operations totaled $299.1 million, $386.9 million and $302.0 million for the years ended December 31, 2021, 2022 and 2023, respectively, representing approximately 84.0%, 90.7% and 36.0% of our total consolidated revenue for the respective period. These research and development figures do not include payments made by our collaboration partners directly to third parties to help fund the research and development of our drug candidates.
We have been able to fund the research and development expenses for our Oncology/Immunology operations via a range of sources, including revenue generated from our commercialized drugs, payments received from our collaboration partners, cash flows generated from and dividend payments from our Other Ventures, the proceeds raised from our initial public offering on the AIM, initial public offering and follow-on offerings on Nasdaq, initial public offering on the SEHK, investments from other third parties and bank borrowings.
This diversified approach to funding allows us to not depend on any one method of funding for our research and development activities, thereby reducing the risk that sufficient financing will be unavailable as we continue to accelerate the development of our drug candidates.
For more information on the research and development expenses incurred for the development of our drug candidates, see “—Key Components of Results of Operations—Cost of Revenue and Operating Expenses—Research and Development Expenses.”
Our Ability to Commercialize Our Drug Candidates
Our ability to generate revenue from our drug candidates depends on our ability to successfully complete clinical trials for our drug candidates and obtain regulatory approvals for them in the United States, Europe, China and other major markets.
We believe that our globally-facing strategy of focusing on drug development for novel but relatively well-characterized targets and for validated targets, in combination with our development of multiple drug candidates concurrently and testing them for multiple indications and in combinations with other drugs, enhances the likelihood that our research and development efforts will yield successful drug candidates. Nonetheless, we cannot be certain if any of our drug candidates will receive regulatory approvals. Even if such approvals are granted, we will need to thereafter establish manufacturing supply and engage in extensive marketing prior to generating any revenue from such drugs. The effectiveness of our marketing will depend on the efforts of our dedicated oncology team in China and our collaboration partners in the rest of the world. The ultimate commercial success of our drugs will depend on their acceptance by patients, the medical community and third-party payors and their ability to compete effectively with other therapies on the market.
To date, surufatinib and savolitinib have been approved for sale in China and fruquintinib has been approved for sale in both China and the United States.
160
Our manufacturing site in Suzhou produces commercial supplies of fruquintinib and surufatinib. Our commercial supplies of savolitinib are outsourced and manufactured by a third-party manufacturer based in Shanghai, China. Beginning in October 2020, we assumed responsibility for the development and execution of all on-the-ground medical detailing, promotion and local and regional marketing activities in China for Elunate. Sulanda is marketed by us in China without the support of a collaboration partner. However, we have a limited history of successfully commercializing our internally developed drug candidates, which makes it difficult to evaluate our future prospects.
The competitive environment is also an important factor with the commercial success of our potential global first-in-class products, such as sovleplenib, depending on whether we are able to gain regulatory approvals and quickly bring such products to market ahead of competing drug candidates being developed by other companies.
For our drug candidates where we retain all rights worldwide, which currently include surufatinib, sovleplenib, amdizalisib, HMPL-306, HMPL-760, HMPL-453, HMPL-295, HMPL-653, HMPL-A83 and HMPL-415, we will be able to retain all the profits if any of them are successfully commercialized if they remain unpartnered, though we will need to bear all the costs associated with such drug candidates. Conversely, as discussed below, for our drug candidates which are subject to collaboration partnerships, our collaboration partners provide funding for development of the drug candidates but are entitled to retain a significant portion of any revenue generated by such drug candidates.
Our Collaboration Partnerships
Our results of operations have been, and we expect them to continue to be, affected by our collaborations with third parties for the development and commercialization of certain of our drug candidates. Currently, these include savolitinib (global collaboration with AstraZeneca) and fruquintinib (collaboration with Eli Lilly in China and with Takeda outside of China). In addition to providing us with clinical and regulatory support, the payments received from these collaborations have been critical to our ability to develop and quickly advance the pre-clinical and clinical studies of multiple drug candidates concurrently.
In particular, our partners cover a portion of our research and development costs for drug candidates developed in collaboration with them. In addition, under our collaboration agreements with AstraZeneca, Eli Lilly and Takeda, we received upfront payments upon our entry into such agreements and milestone payments upon the achievement of certain development, regulatory milestones payments for our provision of research and development services for the relevant drug candidate as well as commercial milestones and royalties. Revenue recognized in our consolidated financial statements from such agreements with AstraZeneca, Eli Lilly and Takeda totaled $107.1 million, $129.4 million and $482.0 million for the years ended December 31, 2021, 2022 and 2023, respectively.
Moreover, we have entered into and may consider entering in the future in-licensing arrangements to expand and complement our existing portfolio of novel oncology assets under which we may be obligated to make upfront, milestone and royalty payments. For example, in August 2021, we entered into an in-licensing agreement with Epizyme (a subsidiary of Ipsen Pharma SAS) to collaborate in research, development, manufacturing and commercialization of tazemetostat in Greater China, the licensed territory. In connection with this collaboration, Epizyme received a $25 million upfront payment and an aggregate of $5 million milestone payment to date and is eligible to receive up to an additional $105 million in development and regulatory milestone payments and up to an additional $175 million in sales milestone payments. Epizyme is also eligible to receive tiered royalties of mid-teen to low-twenties percent based on annual net sales of tazemetostat in the licensed territory.
The achievement of milestones for our and in-licensed drug candidates, which is dependent on the outcome of clinical studies, is subject to a high degree of uncertainty and, as a result, we cannot reasonably estimate when we can expect to receive or incur future milestone payments, revenue from related product sales, or other relevant income or expenses or at all. If we are unable to achieve development milestones for our drug candidates or if our partners were to terminate their collaborative agreements with us, payments for research and development services could also be affected.
For more information regarding our collaboration agreements, see Item 4.B. “Business Overview—Overview of Our Collaborations.”
161
China Government Insurance Reimbursement and Drug Pricing Policies
Our revenue is affected by the sales volume and pricing of our current and future internally developed drug candidates, if approved. Eligible participants in the government-sponsored medical insurance programs in China are entitled to reimbursement for varying percentages of the cost for any medicines that are included in applicable reimbursement lists. Factors that affect the inclusion of medicines in China’s NRDL and any other applicable reimbursement list may include whether the medicine is consumed in large volumes and commonly prescribed for clinical use in China and whether it is considered to be important in meeting the basic healthcare needs of the general public. For more information, see Item 4.B. “Business Overview—Coverage and Reimbursement—PRC Coverage and Reimbursement.” The inclusion of a medicine in the NRDL or other applicable reimbursement lists can substantially improve the sales volume of the medicine due to the availability of third-party reimbursements. On the other hand, such inclusion may also subject it to centralized procurement processes. The National Healthcare Security Administration has stated that centralized procurement will focus on NRDL-listed and costly-to-procure drugs. Centralized procurement may negatively affect the retail price of our drug candidates. On balance, we believe that, if priced appropriately, the benefit of the inclusion of our drug candidates in the NRDL and other applicable reimbursement lists outweighs the cost of such inclusion. Elunate was added to the NRDL in January 2020 at approximately 60% discount to its initial retail price, and such inclusion was renewed for an additional two-year term starting in January 2022 at a discount of 5% relative to the prior NRDL price. Sulanda was included in the NRDL starting in January 2022 at a 52% discount on its main dosage form, relative to its 2021 initial retail price. In January 2024, the updated NRDL will continue to include Elunate and Sulanda at the same terms as the current two year agreement. Orpathys has been included in the NRDL since March 1, 2023 at a 38% discount relative to the self-pay price.
Revenue from our Other Ventures, including the revenue of our non-consolidated joint venture Shanghai Hutchison Pharmaceuticals, is affected by the sales volume and pricing of their own-brand and third-party prescription pharmaceutical products. The sales volume of the products sold by these businesses is driven in part by the level of Chinese government spending on healthcare and the coverage of Chinese government medical insurance schemes, which is correlated with patient reimbursements for drug purchases, all of which have increased significantly in recent years as part of healthcare reforms in China. The sales volume of pharmaceutical products in China is also influenced by their representation on the NRDL, which determines eligibility for drug reimbursement, as well as their representation on the National Essential Medicines List, which mandates distribution of drugs in China. Substantially all pharmaceutical products manufactured and sold by Shanghai Hutchison Pharmaceuticals were capable of being reimbursed under the NRDL as of December 31, 2023. There were 21 of its drugs included in the National Essential Medicine List, of which two were in active production as of December 31, 2023. She Xiang Bao Xin pills, Shanghai Hutchison Pharmaceuticals’ top-selling drug, is one of the few proprietary drugs included on the National Essential Medicines List.
The NRDL and the National Essential Medicines List are subject to revision by the government from time to time, and our results could be materially and adversely affected if any of our products are removed from the NRDL or the National Essential Medicines List. For more information, see Item 3.D. “Risk Factors—Risks Relating to Sales of our Internally Developed Drugs and other Drugs—Reimbursement may not be available for the products currently sold through our Oncology/Immunology and Other Ventures operations or our drug candidates in China, the U.S. or other countries, which could diminish our sales or affect our profitability.”
In addition, the pricing of Shanghai Hutchison Pharmaceuticals’ prescription drugs is influenced by the outcomes of periodic provincial and municipal tender processes organized by the various provincial or municipal government agencies in China. For more information, see Item 4.B. “Business Overview—Coverage and Reimbursement—PRC Coverage and Reimbursement.”
Ability to Effectively Market Own-Brand and Third-Party Drugs
A key component of our Other Ventures operations is the extensive prescription drugs marketing network operated by our joint ventures Shanghai Hutchison Pharmaceuticals and Hutchison Sinopharm, which includes over 3,000 medical sales representatives covering hospitals in about 290 cities and towns in China. Our results of operations are impacted by the effectiveness of this network, including the ability of Shanghai Hutchison Pharmaceuticals to generate sales of She Xiang Bao Xin pills, which represented approximately 92%, 92% and 90% of its total revenue for the years ended December 31, 2021, 2022 and 2023, respectively. In addition, in recent years Hutchison Sinopharm has been increasingly focused on providing distribution and commercialization services for prescription drugs licensed from third parties, and we have established and continue to expand our oncology drug sales team which we utilize for our internally developed drugs for which we have commercialization rights, if approved, throughout China.
162
If the marketing efforts of these joint ventures to doctors and hospitals are not successful, our revenue and profitability may be negatively affected. Moreover, if we are unsuccessful in marketing any third party drugs, it may adversely affect our ability to enter into commercialization arrangements on acceptable terms, gain rights to market additional third-party drugs or prevent us from expanding the geographic scope of existing arrangements.
Seasonality
The results of operations of our Other Ventures operations are also affected by seasonal factors. Our Other Ventures operations typically experience higher profits in the first half of the year due to the sale cycles of our distributors, whereby they typically increase their inventories at the beginning of each year. In addition, in the second half of each year, our Other Ventures operations typically spend more on marketing activities to help reduce such inventory held by distributors. We do not experience material seasonal variations in the results of our Oncology/Immunology operations.
Critical Accounting Policies and Significant Judgments and Estimates
Our discussion and analysis of operating results and financial condition are based upon our consolidated financial statements. The preparation of consolidated financial statements requires us to estimate the effect of various matters that are inherently uncertain as of the date of the consolidated financial statements. Each of these required estimates varies with regard to the level of judgment involved and its potential impact on our reported financial results. Estimates are deemed critical when a different estimate could have reasonably been used or where changes in the estimates are reasonably likely to occur from period to period, and a different estimate would materially impact our financial position, changes in financial position or results of operations. Our significant accounting policies are discussed under note 3 to our consolidated financial statements included in this annual report. We believe the following critical accounting policies are affected by significant judgments and estimates used in the preparation of our consolidated financial statements and that the judgments and estimates are reasonable.
Revenue Recognition— Goods and Services
We generate revenue from (1) sales of goods, which are the manufacture or purchase and distribution of pharmaceutical products and other healthcare products and (2) provision of services, which are the provision of sales, distribution and marketing services to pharmaceutical manufacturers. We evaluate whether we are the principal or agent for these contracts. Where we obtain control of the goods for distribution, we are the principal (i.e. recognizes sales of goods on a gross basis). Where we do not obtain control of the goods for distribution, we are the agent (i.e. recognizes provision of services on a net basis). Control is primarily evidenced by taking physical possession and inventory risk of the goods.
Revenue from sales of goods is recognized when the customer takes possession of the goods. We have determined that this usually occurs upon completed delivery of the goods to the customer site. The amount of revenue recognized is adjusted for expected sales incentives as stipulated in the contract, which are generally issued to customers as direct discounts at the point of sale or indirectly in the form of rebates. Sales incentives are estimated using the expected value method. Additionally, sales are generally made with a limited right of return under certain conditions. Revenue are recorded net of provisions for sales discounts and returns.
Revenue from provision of services is recognized when the benefits of the services transfer to the customer over time, which is based on the proportionate value of services rendered as determined under the terms of the relevant contract. Additionally, when the amounts that can be invoiced correspond directly with the value to the customer for performance completed to date, we recognize revenue from provision of services based on amounts that can be invoiced to the customer.
Deferred revenue is recognized if consideration is received in advance of transferring control of the goods or rendering of services. Accounts receivable is recognized if the Group has an unconditional right to bill the customer, which is generally when the customer takes possession of the goods or services are rendered. Payment terms differ by subsidiary and customer, but generally range from 45 to 180 days from the invoice date.
163
Revenue Recognition— License and Collaboration Contracts
Our Oncology/Immunology reportable segment includes revenue from license and collaboration contracts, which generally contain multiple performance obligations including (1) the licenses to the development, commercialization and manufacture rights of a drug compound, (2) the research and development services for each specified treatment indication, and (3) other deliverables, which are accounted for separately if they are distinct, i.e. if a product or service is separately identifiable from other items in the arrangement and if a customer can benefit from it on its own or with other resources that are readily available to the customer.
The transaction price generally includes fixed and variable consideration in the form of upfront payment, research and development cost reimbursements, contingent milestone payments and sales-based royalties. Contingent milestone payments are not included in the transaction price until it becomes probable that a significant reversal of revenue will not occur, which is generally when the specified milestone is achieved. The allocation of the transaction price to each performance obligation is based on the relative standalone selling prices of each performance obligation determined at the inception of the contract. We estimate the standalone selling prices based on the income approach and cost plus margin approach. Control of the license to the drug compounds transfers at the inception date of the collaboration agreements and consequently, amounts allocated to this performance obligation are generally recognized at a point in time. Conversely, research and development services for each specified indication are performed over time and amounts allocated to these performance obligations are generally recognized over time using a percentage of completion method. We have determined that research and development expenses provide an appropriate depiction of measure of progress for the research and development services. Changes to estimated cost inputs may result in a cumulative catch-up adjustment. Royalty revenue is recognized as future sales occur as they meet the requirements for the sales-usage based royalty exception.
Deferred revenue is recognized if allocated consideration is received in advance of the rendering of research and development services or earning royalties on future sales. Accounts receivable is recognized based on the terms of the contract and when we have an unconditional right to bill the customer, which is generally when research and development services are rendered.
Share-based Compensation
We recognize share-based compensation expense on share options granted to employees and directors based on their estimated grant date fair value using the polynomial model. Determining the fair value of share options requires the use of subjective assumptions. This polynomial pricing model uses various inputs to measure fair value, including the market value of our underlying ordinary shares at the grant date, contractual terms, estimated volatility, risk-free interest rates and expected dividend yields. The assumptions in determining the fair value of share options are highly subjective and represent our best estimates, which involve inherent uncertainties and the application of judgment. As a result, if factors change and different assumptions are used, our level of share-based compensation could be materially different in the future.
We recognize share-based compensation expense in the consolidated statements of operations on a graded vesting basis over the requisite service period, and account for forfeitures as they occur.
Impairment of Long-lived Assets
We evaluate the recoverability of long-lived assets in accordance with authoritative guidance on accounting for the impairment or disposal of long-lived assets.
We evaluate long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying value of these assets may not be recoverable. Indicators that we consider in deciding when to perform an impairment review include significant under-performance of a business or product line in relation to expectations, significant negative industry or economic trends, and significant changes or planned changes in our use of the assets.
If indicators of impairment exist, the first step of the impairment test is performed to assess if the carrying value of the net assets exceeds the undiscounted cash flows of the assets. If yes, the second step of the impairment test is performed in order to determine if the carrying value of the net assets exceeds the fair value. If yes, impairment is recognized for the excess.
164
Allowance for Current Expected Credit Losses (“CECLs”)
We estimate our allowance for current expected credit losses (“CECLs”) based on an expected loss model, which requires the consideration of forward-looking economic variables and conditions in the reserve calculation across the portfolio.
We estimate our allowances for CECLs for accounts receivables, other receivables (except for prepayments) and amounts due from related parties by considering past events, including any historical default, current economic conditions and certain forward-looking information, including reasonable and supportable forecasts. The methodologies that the Group uses to estimate the allowance for CECLs for accounts receivables, other receivables (except for prepayments) and amounts due from related parties are as follows:
Individually evaluated—we review all accounts receivables, other receivables (except for prepayments) and amounts due from related parties considered at risk on a timely basis and perform an analysis based upon current information available about the customers and other debtors, which may include financial statements, news reports, published credit ratings as well as collateral net of repossession cost, prior collection history and current and future expected economic conditions. Using this information, we determine the expected cash flow for the accounts receivables, other receivables (except for prepayments) and amounts due from related parties and calculate an estimate of the potential loss and the probability of loss. For those accounts for which the loss is probable, we record a specific allowance.
Collectively evaluated—we determine our allowance for CECLs for collectively evaluated accounts receivables, other receivables (except for prepayments) and amounts due from related parties based on appropriate groupings.
We consider forward-looking macroeconomic variables, which may include gross domestic product, unemployment rates, equity prices and corporate profits when quantifying the impact of economic forecasts on our allowance for expected credit losses. Macroeconomic variables may vary based on historical experiences, portfolio composition and current environment. We also consider the impact of current conditions and economic forecasts relating to specific industries and client-credit ratings, in addition to performing a qualitative review of credit risk factors across the portfolio. Forward-looking estimates require the use of judgment, particularly in times of economic uncertainty.
Recent Accounting Pronouncements
See note 3 to our consolidated financial statements included in this annual report for information regarding recent accounting pronouncements.
165
Key Components of Results of Operations
The following tables set forth our selected consolidated financial data. We have derived the selected consolidated statements of operations data for the years ended December 31, 2023, 2022 and 2021 and the selected consolidated balance sheet data as of December 31, 2023 and 2022 from our audited consolidated financial statements, which were prepared in accordance with U.S. GAAP and are included elsewhere in this annual report. The following selected consolidated financial data for the years ended December 31, 2020 and 2019 and as of December 31, 2021, 2020 and 2019 have been derived from our audited consolidated financial statements for those years, which were prepared in accordance with U.S. GAAP and are not included in this annual report.
Year Ended December 31, | ||||||||||
| 2023 |
| 2022 |
| 2021 |
| 2020 |
| 2019 | |
$’000 (except share and per share data) | ||||||||||
Consolidated statement of operations data: | ||||||||||
Revenue | ||||||||||
Goods—third parties |
| 388,924 |
| 314,329 |
| 266,199 |
| 203,606 |
| 175,990 |
—related parties |
| 8,264 |
| 5,293 |
| 4,256 |
| 5,484 |
| 7,637 |
Services —commercialization—third parties |
| 48,608 |
| 41,275 |
| 27,428 |
| 3,734 |
| 2,584 |
—collaboration research and development —third parties |
| 80,397 |
| 23,741 |
| 18,995 |
| 9,771 |
| 15,532 |
—research and development—related parties |
| 481 |
| 507 |
| 525 |
| 491 |
| 494 |
Other collaboration revenue —royalties—third parties |
| 32,470 |
| 26,310 |
| 15,064 |
| 4,890 |
| 2,653 |
—licensing—third parties |
| 278,855 |
| 14,954 |
| 23,661 |
| — |
| — |
Total revenue |
| 837,999 |
| 426,409 |
| 356,128 |
| 227,976 |
| 204,890 |
Operating expenses | ||||||||||
Cost of goods—third parties |
| (331,984) |
| (268,698) |
| (229,448) |
| (178,828) |
| (152,729) |
Cost of goods—related parties |
| (4,777) |
| (3,616) |
| (3,114) |
| (3,671) |
| (5,494) |
Cost of services—commercialization —third parties |
| (47,686) |
| (38,789) |
| (25,672) |
| (6,020) |
| (1,929) |
Research and development expenses |
| (302,001) |
| (386,893) |
| (299,086) |
| (174,776) |
| (138,190) |
Selling expenses |
| (53,392) |
| (43,933) |
| (37,827) |
| (11,334) |
| (13,724) |
Administrative expenses |
| (79,784) |
| (92,173) |
| (89,298) |
| (50,015) |
| (39,210) |
Total operating expenses |
| (819,624) |
| (834,102) |
| (684,445) |
| (424,644) |
| (351,276) |
| 18,375 |
| (407,693) |
| (328,317) |
| (196,668) |
| (146,386) | |
Gain on divestment of an equity investee |
| — |
| — |
| 121,310 |
| — |
| — |
Other income/(expense) | ||||||||||
Interest income |
| 36,145 |
| 9,599 |
| 2,076 |
| 3,236 |
| 4,944 |
Other income |
| 12,949 |
| 1,833 |
| 2,426 |
| 4,600 |
| 1,855 |
Interest expense |
| (759) |
| (652) |
| (592) |
| (787) |
| (1,030) |
Other expense |
| (8,402) |
| (13,509) |
| (12,643) |
| (115) |
| (488) |
Total other income/(expense) |
| 39,933 |
| (2,729) |
| (8,733) |
| 6,934 |
| 5,281 |
Income/(loss) before income taxes and equity in earnings of equity investees |
| 58,308 |
| (410,422) |
| (215,740) |
| (189,734) |
| (141,105) |
Income tax (expense)/benefit |
| (4,509) |
| 283 |
| (11,918) |
| (4,829) |
| (3,274) |
Equity in earnings of equity investees, net of tax |
| 47,295 |
| 49,753 |
| 60,617 |
| 79,046 |
| 40,700 |
Net income/(loss) |
| 101,094 |
| (360,386) |
| (167,041) |
| (115,517) |
| (103,679) |
Less: Net income attributable to non-controlling interests |
| (314) |
| (449) |
| (27,607) |
| (10,213) |
| (2,345) |
Net income/(loss) attributable to the Company |
| 100,780 |
| (360,835) |
| (194,648) |
| (125,730) |
| (106,024) |
Earnings/(losses) per share attributable to the Company (US$ per share) | ||||||||||
—basic | 0.12 | (0.43) | (0.25) | (0.18) | (0.16) | |||||
—diluted | 0.12 | (0.43) | (0.25) | (0.18) | (0.16) | |||||
Number of shares used in per share calculation | ||||||||||
—basic | 849,654,296 | 847,143,540 | 792,684,524 | 697,931,437 | 665,683,145 | |||||
—diluted | 869,196,348 | 847,143,540 | 792,684,524 | 697,931,437 | 665,683,145 | |||||
Net income/(loss) |
| 101,094 |
| (360,386) |
| (167,041) |
| (115,517) |
| (103,679) |
Other comprehensive (loss)/income | ||||||||||
Foreign currency translation (loss)/gain |
| (6,592) |
| (8,469) |
| 2,964 |
| 9,530 |
| (4,331) |
Total comprehensive income/(loss) |
| 94,502 |
| (368,855) |
| (164,077) |
| (105,987) |
| (108,010) |
Less: Comprehensive loss/(income) attributable to non-controlling interests |
| 39 |
| 545 |
| (28,029) |
| (11,413) |
| (1,620) |
Total comprehensive income/(loss) attributable to the Company |
| 94,541 |
| (368,310) |
| (192,106) |
| (117,400) |
| (109,630) |
| 2023 |
| 2022 |
| 2021 |
| 2020 |
| 2019 | |
Consolidated balance sheet data: | ||||||||||
Cash and cash equivalents | 283,589 |
| 313,278 |
| 377,542 |
| 235,630 |
| 121,157 | |
Short-term investments | 602,747 |
| 317,718 |
| 634,158 |
| 199,546 |
| 96,011 | |
Total assets | 1,279,773 |
| 1,029,445 |
| 1,372,661 |
| 724,118 |
| 465,122 | |
Total current liabilities | 403,027 |
| 353,903 |
| 311,658 |
| 158,397 |
| 113,101 | |
Total non-current liabilities | 133,359 |
| 38,672 |
| 21,489 |
| 46,772 |
| 39,118 | |
Total shareholders’ equity | 743,387 |
| 636,870 |
| 1,039,514 |
| 518,949 |
| 312,903 | |
166
Revenue
The following table sets forth the components by contract type of our consolidated revenue for the years indicated, which does not include the revenue from our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals. In December 2023, we sold our interests in our consolidated joint venture Hutchison Hain Organic and our wholly own subsidiary HUTCHMED Science Nutrition, and their historical financial results and gain on divestment are reflected in our consolidated financial statements. In September 2021, we sold our interest in our non-consolidated joint venture, Hutchison Baiyunshan, and its historical financial results and the gain on its divestment are reflected in our consolidated financial statements.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 |
| % |
| $’000 |
| % | |
Revenue |
|
|
|
|
| |||||||
Oncology/Immunology: | ||||||||||||
Invoiced Goods—Marketed Products(1) | 83,087 | 9.9 | 57,057 | 13.4 | 33,937 | 9.5 | ||||||
Services: |
|
|
|
|
| |||||||
Commercialization—Marketed Products | 48,608 | 5.8 | 41,275 | 9.7 | 27,428 | 7.7 | ||||||
Research and Development—related parties | 481 | 0.1 | 507 | 0.1 | 525 | 0.2 | ||||||
License & Collaborations: |
|
|
|
|
|
| ||||||
Services | 80,397 | 9.6 | 23,741 | 5.5 | 18,995 | 5.3 | ||||||
Royalties—Marketed Products |
| 32,470 |
| 3.9 |
| 26,310 |
| 6.2 |
| 15,064 |
| 4.2 |
Licensing | 278,855 |
| 33.3 |
| 14,954 | 3.5 | 23,661 | 6.7 | ||||
Manufacturing Supply(1) |
| 4,718 |
| 0.5 |
| — |
| — |
| — |
| — |
Subtotal |
| 528,616 |
| 63.1 |
| 163,844 |
| 38.4 |
| 119,610 |
| 33.6 |
Other Ventures: |
|
|
|
|
|
| ||||||
Invoiced Goods(1) |
| 301,119 |
| 35.9 |
| 257,272 |
| 60.3 |
| 232,262 |
| 65.2 |
Invoiced Goods—related parties |
| 8,264 |
| 1.0 |
| 5,293 |
| 1.3 |
| 4,256 |
| 1.2 |
Subtotal |
| 309,383 |
| 36.9 |
| 262,565 |
| 61.6 |
| 236,518 |
| 66.4 |
Total | 837,999 |
| 100.0 |
| 426,409 |
| 100.0 |
| 356,128 |
| 100.0 | |
| (1) | Included in revenue from goods – third parties in our consolidated statements of operations. |
Revenue from Oncology/Immunology primarily comprises revenue from Elunate, Sulanda and Orpathys in China and revenue from Fruzaqla. The revenue we generate from Elunate is primarily comprised of revenue from the sales of Elunate to Eli Lilly which we manufacture and sell at cost, promotion and marketing services to Eli Lilly and royalty revenue. The revenue we generate from Sulanda, an unpartnered drug, is primarily comprised of revenue from sales of Sulanda to distributors. The revenue we generate from Orpathys is primarily comprised of revenue from the sales of Orpathys to AstraZeneca as well as royalty revenue. The revenue we generate from Fruzaqla is primarily comprised of revenue from manufacturing supplies to Takeda as well as royalty revenue. Additionally, Oncology/Immunology revenue includes revenue from license and collaboration agreements for upfront, milestone and research and development services payments for our drug candidates developed in collaboration with Takeda, AstraZeneca and Eli Lilly.
The following table sets forth the components of revenue of our Other Ventures by product type for the years indicated.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 |
| % |
| $’000 |
| % | |
Revenue—Other Ventures |
|
|
|
|
|
|
|
|
|
|
|
|
Prescription drug products |
| 295,396 |
| 95.5 |
| 237,293 |
| 90.4 |
| 204,091 |
| 86.3 |
Consumer health products |
| 13,987 |
| 4.5 |
| 25,272 |
| 9.6 |
| 32,427 |
| 13.7 |
Total |
| 309,383 |
| 100.0 |
| 262,565 |
| 100.0 |
| 236,518 |
| 100.0 |
Revenue from our Other Ventures primarily comprises revenue from prescription drugs including the commercial services, logistics and distribution business of our consolidated Hutchison Sinopharm joint venture with Sinopharm, a leading distributor of pharmaceutical products and a leading supply chain service provider in China.
167
Revenue from our Other Ventures also comprises revenue from sales of organic and natural products by Hutchison Hain Organic (which was divested in December 2023), Zhi Ling Tong infant nutrition and other health supplement products manufactured by Hutchison Healthcare and distributed through Hutchison Sinopharm up until the end of September and from October 1, 2022 onwards, through our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals, and certain third-party consumer products distributed and marketed by HUTCHMED Science Nutrition (which was divested in December 2023).
The revenue of our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals, the accounts of which are prepared in accordance with IFRS as issued by the IASB and whose revenue is not included in our consolidated revenue, was $332.6 million, $370.6 million and $385.5 million for the years ended December 31, 2021, 2022 and 2023, respectively. Shanghai Hutchison Pharmaceuticals is a joint venture with Shanghai Pharmaceuticals, a leading pharmaceuticals company in China, and primarily focuses on the manufacture and sale of prescription pharmaceutical products in China. We and Shanghai Pharmaceuticals each own 50% of this joint venture. We have the right to nominate the general manager and other management of this joint venture and run its day-to-day operations. The effect of Shanghai Hutchison Pharmaceuticals on our consolidated financial results is discussed below under “—Equity in Earnings of Equity Investees.”
The revenue of our former non-consolidated joint venture, Hutchison Baiyunshan, the accounts of which are prepared in accordance with IFRS as issued by the IASB and whose financial results up to September 28, 2021 are reflected in our consolidated financial statements, was $209.5 million for the period ended September 28, 2021. Hutchison Baiyunshan was a joint venture with Guangzhou Baiyunshan, a leading China-based pharmaceutical company. We sold our interest in this joint venture on September 28, 2021 and recognized a gain on divestment attributable to our Group, net of taxes, of $82.9 million from this transaction. The effect of Hutchison Baiyunshan on our consolidated financial results is discussed under “—Equity in Earnings of Equity Investees.”
Cost of Revenue and Operating Expenses
Cost of Revenue
Our cost of revenue is primarily attributable to the cost of revenue of Hutchison Sinopharm and HUTCHMED Limited. The following table sets forth the components of our cost of revenue for the years indicated.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 | % |
| $’000 |
| % | ||
Cost of Revenue | ||||||||||||
Oncology/Immunology: | ||||||||||||
Cost of Invoiced Goods | 44,040 | 11.5 | 30,403 | 9.8 | 19,133 | 7.4 | ||||||
Cost of Services | 47,686 | 12.4 | 38,789 | 12.5 | 25,672 | 9.9 | ||||||
Subtotal | 91,726 | 23.9 | 69,192 | 22.3 | 44,805 | 17.3 | ||||||
Other Ventures: | ||||||||||||
Cost of Invoiced Goods | 287,944 | 74.9 | 238,295 | 76.6 | 210,315 | 81.4 | ||||||
Cost of Invoiced Goods—related parties | 4,777 | 1.2 | 3,616 | 1.1 | 3,114 | 1.3 | ||||||
Subtotal | 292,721 | 76.1 | 241,911 | 77.7 | 213,429 | 82.7 | ||||||
Total | 384,447 | 100.0 | 311,103 | 100.0 | 258,234 | 100.0 | ||||||
The following table sets forth the components of cost of revenue of our Other Ventures by product type for the years indicated.
| Year Ended December 31, | |||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 |
| % |
| $’000 |
| % | |
Cost of Revenue—Other Ventures |
|
|
|
|
|
| ||||||
Prescription drug products | 284,927 | 97.3 | 228,968 | 94.6 | 196,375 | 92.0 | ||||||
Consumer health products | 7,794 | 2.7 | 12,943 | 5.4 | 17,053 | 8.0 | ||||||
Total |
| 292,721 |
| 100.0 |
| 241,911 |
| 100.0 |
| 213,428 |
| 100.0 |
168
Research and Development Expenses
Our research and development expenses are attributable to our Oncology/Immunology operations. These costs primarily comprise the cost of research and development for our drug candidates, including clinical trial related costs such as payments to third-party CROs, personnel compensation and related costs, and other research and development expenses. The following table sets forth the components of our research and development expenses and the clinical trial related costs incurred for the development of our main drug candidates for the years indicated.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 |
| % |
| $’000 |
| % | |
R&D Expenses | ||||||||||||
Oncology/Immunology: | ||||||||||||
Fruquintinib (targeting VEGFR1/2/3) | 40,384 | 13.4 | 52,115 | 13.5 | 57,707 | 19.3 | ||||||
Savolitinib (targeting MET) | 37,692 | 12.5 | 48,249 | 12.5 | 26,152 | 8.7 | ||||||
Surufatinib (targeting VEGFR/FGFR1/CSF-1R) | 24,746 | 8.2 | 37,635 | 9.7 | 47,971 | 16.0 | ||||||
Amdizalisib (targeting PI3Kδ) | 17,065 | 5.7 | 27,046 | 7.0 | 21,044 | 7.0 | ||||||
Sovleplenib (targeting Syk) | 14,200 | 4.7 | 23,138 | 6.0 | 8,602 | 2.9 | ||||||
HMPL-306 (targeting IDH 1/2) | 12,633 | 4.2 | 14,865 | 3.8 | 10,073 | 3.4 | ||||||
Tazemetostat (targeting EZH2) | 12,171 | 4.0 | 19,019 | 4.9 | 12,139 | 4.1 | ||||||
HMPL-453 (targeting FGFR) | 7,532 | 2.5 | 2,776 | 0.7 | 1,708 | 0.6 | ||||||
HMPL-760 (targeting BTK) | 2,467 | 0.8 | 4,954 | 1.3 | 5,288 | 1.8 | ||||||
HMPL-653 (targeting CSF-1R) | 1,981 | 0.7 | 1,778 | 0.5 | 132 | — | ||||||
HMPL-415 (targeting SHP2) | 1,121 | 0.4 | — | — | — | — | ||||||
HMPL-A83 (IgG4-type humanized anti-CD47 monoclonal antibody) | 1,096 | 0.4 | 2,840 | 0.7 | — | — | ||||||
HMPL-295 (targeting ERK) | 645 | 0.2 | 1,362 | 0.4 | 692 | 0.2 | ||||||
Others and government grant | 25,995 | 8.4 | 20,158 | 5.2 | (1,457) | (0.4) | ||||||
Total clinical trial related costs | 199,728 | 66.1 | 255,935 | 66.2 | 190,051 | 63.6 | ||||||
Personnel compensation and related costs | 93,030 | 30.8 | 119,306 | 30.8 | 91,639 | 30.6 | ||||||
Other research and development costs | 9,243 | 3.1 | 11,652 | 3.0 | 17,396 | 5.8 | ||||||
Total | 302,001 | 100.0 | 386,893 | 100.0 | 299,086 | 100.0 | ||||||
The following table summarizes our research and development expenses by location for the years indicated.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 | % |
| $’000 |
| % | ||
PRC | 195,070 | 64.6 | 215,963 | 55.8 | 159,038 | 53.2 | ||||||
U.S. and others | 106,931 | 35.4 | 170,930 | 44.2 | 140,048 | 46.8 | ||||||
Total | 302,001 | 100.0 | 386,893 | 100.0 | 299,086 | 100.0 | ||||||
We cannot determine with certainty the duration and completion costs of the current or future pre-clinical or clinical studies of our drug candidates or if, when, or to what extent we will generate revenue from the commercialization and sale of any of our drug candidates that obtain regulatory approval. We may not succeed in achieving regulatory approval for any of our drug candidates currently under development. The duration, costs, and timing of clinical studies and development of our drug candidates will depend on a variety of factors, including:
| ● | the scope, rate of progress and expense of our ongoing as well as any additional clinical studies and other research and development activities; |
| ● | future clinical study results; |
| ● | uncertainties in clinical study enrollment rate; |
169
| ● | significant and changing government regulation; and |
| ● | the timing and receipt of any regulatory approvals. |
A change in the outcome of any of these variables with respect to the development of a drug candidate could mean a significant change in the costs and timing associated with the development of that drug candidate.
For more information on the risks associated with the development of our drug candidates, see Item 3.D. “Risk Factors—Risks Relating to Our Oncology/Immunology Operations and Development of Our Drug Candidates—All of our drug candidates, other than fruquintinib for approved indications in both China and the United States, and surufatinib and savolitinib for approved indications in China, are still in development. If we are unable to obtain regulatory approval and ultimately commercialize our drug candidates, or if we experience significant delays in doing so, our business will be materially harmed.”
Selling Expenses
The following table sets forth the components of our selling expenses for the years indicated.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 |
| % |
| $’000 |
| % | |
Selling Expenses | ||||||||||||
Oncology/Immunology | 45,505 | 85.2 | 33,862 | 77.1 | 24,627 | 65.1 | ||||||
Other Ventures | 7,887 | 14.8 | 10,071 | 22.9 | 13,200 | 34.9 | ||||||
Total | 53,392 | 100.0 | 43,933 | 100.0 | 37,827 | 100.0 | ||||||
Our selling expenses primarily comprise selling expenses incurred by our Oncology/Immunology operations by HUTCHMED Limited for sales and marketing expenses and related personnel expenses for our unpartnered drug Sulanda. It also includes sales and marketing expenses and related personnel expenses incurred by our Other Ventures in their distribution and marketing of pharmaceutical and consumer health products.
Administrative Expenses
The following table sets forth the components of our administrative expenses for the years indicated.
Administrative expenses are also incurred by our corporate head office, which are not allocated to either Oncology/Immunology or Other Ventures.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 |
| % |
| $’000 |
| % | |
Administrative Expenses | ||||||||||||
Oncology/Immunology | 47,966 | 60.1 | 58,395 | 63.3 | 48,359 | 54.2 | ||||||
Other Ventures | 5,435 | 6.8 | 3,482 | 3.8 | 7,712 | 8.6 | ||||||
Corporate Head Office | 26,383 | 33.1 | 30,296 | 32.9 | 33,227 | 37.2 | ||||||
Total | 79,784 | 100.0 | 92,173 | 100.0 | 89,298 | 100.0 | ||||||
Oncology/Immunology’s administrative expenses are primarily comprised of the salaries and benefits of administrative staff, office leases and other overhead expenses incurred by HUTCHMED Limited.
Our Other Ventures’ administrative expenses are primarily comprised of the salaries and benefits of administrative staff, office leases and other overhead expenses incurred by Hutchison Sinopharm, Hutchison Healthcare and Hutchison Hain Organic (which was divested in December 2023).
Our corporate head office administrative expenses are primarily comprised of the salaries and benefits of our corporate head office employees and directors, office leases and other overhead expenses.
170
Equity in Earnings of Equity Investees
We have historically derived a significant portion of our net income from our equity in earnings of equity investees, which was primarily attributable to our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals and former non-consolidated joint venture, Hutchison Baiyunshan. Our equity in earnings of equity investees, net of tax, contributed by Shanghai Hutchison Pharmaceuticals was $44.7 million, $49.7 million and $47.3 million for the years ended December 31, 2021, 2022 and 2023 respectively. Our equity in earnings of equity investees, net of tax, contributed by Hutchison Baiyunshan was $15.9 million for the period ended September 28, 2021.
The following table shows the revenue of Shanghai Hutchison Pharmaceuticals and Hutchison Baiyunshan for the periods indicated. The consolidated financial statements of Shanghai Hutchison Pharmaceuticals are prepared in accordance with IFRS as issued by the IASB and are presented separately elsewhere in this annual report.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 |
| % |
| $’000 |
| % | |
Revenue | ||||||||||||
Other Ventures: | ||||||||||||
Shanghai Hutchison Pharmaceuticals | 385,483 | 100.0 | 370,600 | 100.0 | 332,648 | 61.4 | ||||||
Hutchison Baiyunshan(1) | — | — | — | — | 209,528 | 38.6 | ||||||
Total | 385,483 | 100.0 | 370,600 | 100.0 | 542,176 | 100.0 | ||||||
| (1) | On September 28, 2021, we completed the disposal of our equity interest in Hutchison Baiyunshan. Revenue in 2021 reflects the period from January 1, 2021 to September 28, 2021. |
The following table shows the amount of equity in earnings of equity investees, net of tax, of our non-consolidated joint ventures for the years indicated.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 |
| % |
| $’000 |
| % | |
Equity in earnings of equity investees, net of tax | ||||||||||||
Other Ventures: | ||||||||||||
Shanghai Hutchison Pharmaceuticals(1) | 47,295 | 100.0 | 49,748 | 100.0 | 44,678 | 73.7 | ||||||
Hutchison Baiyunshan(2) | — | — | — | — | 15,919 | 26.3 | ||||||
Oncology/Immunology: | ||||||||||||
Others | — | — | 5 | — | 20 | — | ||||||
Total | 47,295 | 100.0 | 49,753 | 100.0 | 60,617 | 100.0 | ||||||
| (1) | The amount for the years ended December 31, 2021, 2022 and 2023 includes elimination of unrealized profits on transactions with the Group of $36,000, $110,000 and $131,000 and GAAP difference of $1,000, $16,000 and $306,000 respectively. |
| (2) | On September 28, 2021, we completed the divestment of our shareholding interest in Hutchison Baiyunshan. Equity in earnings of Hutchison Baiyunshan reflects the period from January 1, 2021 to September 28, 2021. |
Investments in equity investees mainly consisted of our investment in Shanghai Hutchison Pharmaceuticals. The fluctuation in the investments in equity investees was primarily due to recording our equity in earnings of Shanghai Hutchison Pharmaceuticals, net of tax, offset by dividends declared.
171
The following table shows our investments in our equity investees as of the dates indicated.
| As of December 31, | |||
| 2023 |
| 2022 | |
$’000 | ||||
Shanghai Hutchison Pharmaceuticals | 48,411 |
| 73,461 | |
Other | — |
| 316 | |
Total | 48,411 |
| 73,777 | |
The following table shows the financial position of Shanghai Hutchison Pharmaceuticals as of the dates indicated.
As of December 31, | ||||
| 2023 |
| 2022 | |
$’000 | ||||
Current assets |
| 201,025 |
| 214,267 |
Non-current assets |
| 73,939 |
| 80,062 |
Current liabilities |
| (179,649) |
| (147,952) |
Non-current liabilities |
| (3,687) |
| (4,944) |
Net assets |
| 91,628 |
| 141,433 |
Results of Operations
The following table sets forth a summary of our consolidated results of operations for the years indicated, both in absolute amounts and as percentages of our revenue. This information should be read together with our consolidated financial statements and related notes included elsewhere in this annual report. Our operating results in any period are not necessarily indicative of the results that may be expected for any future period.
Year Ended December 31, | ||||||||||||
2023 | 2022 | 2021 | ||||||||||
| $’000 |
| % |
| $’000 |
| % |
| $’000 |
| % | |
Revenue |
| 837,999 |
| 100.0 |
| 426,409 |
| 100.0 |
| 356,128 |
| 100.0 |
Cost of revenue |
| (384,447) |
| (45.9) |
| (311,103) |
| (73.1) |
| (258,234) |
| (72.5) |
Research and development expenses |
| (302,001) |
| (36.0) |
| (386,893) |
| (90.7) |
| (299,086) |
| (84.0) |
Selling expenses |
| (53,392) |
| (6.4) |
| (43,933) |
| (10.3) |
| (37,827) |
| (10.6) |
Administrative expenses | (79,784) | (9.5) | (92,173) | (21.6) | (89,298) | (25.1) | ||||||
Gain on divestment of an equity investee |
| — |
| — |
| — |
| — |
| 121,310 |
| 34.1 |
Other income/(expense) |
| 39,933 |
| 4.8 |
| (2,729) |
| (0.6) |
| (8,733) |
| (2.5) |
Income tax (expense)/benefit |
| (4,509) |
| (0.5) |
| 283 |
| 0.1 |
| (11,918) |
| (3.3) |
Equity in earnings of equity investees, net of tax |
| 47,295 |
| 5.6 |
| 49,753 |
| 11.7 |
| 60,617 |
| 17.0 |
Net income/(loss) |
| 101,094 |
| 12.1 |
| (360,386) |
| (84.5) |
| (167,041) |
| (46.9) |
Net income /(loss) attributable to our company |
| 100,780 |
| 12.0 |
| (360,835) |
| (84.6) |
| (194,648) |
| (54.7) |
Taxation
Cayman Islands
HUTCHMED (China) Limited is incorporated in the Cayman Islands. The Cayman Islands currently levies no taxes on profits, income, gains or appreciation earned by individuals or corporations. In addition, our payment of dividends, if any, is not subject to withholding tax in the Cayman Islands. For more information, see Item 10.E. “Taxation—Overview of Tax Implications of Various Other Jurisdictions—Cayman Islands Taxation.”
172
People’s Republic of China
Our subsidiaries and a joint venture incorporated in the PRC are governed by the EIT Law and regulations. Under the EIT Law, the standard EIT rate is 25% on taxable profits as reduced by available tax losses. Tax losses may be carried forward to offset any taxable profits for the following five years (extended to ten years for those with HNTE status, with effective from January 1, 2018). HUTCHMED Limited and our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals, have been successful in their respective applications to renew their HNTE status for three years from January 1, 2023 to December 31, 2025. Accordingly, these entities are eligible to a preferential EIT rate of 15% for the years ended/ending December 31, 2023, 2024 and 2025. HUTCHMED (Suzhou) Limited, a wholly owned subsidiary of HUTCHMED Limited, successful renewed its HNTE status for another three years from January 1, 2021 to December 31, 2023. Accordingly, it is eligible for a preferential EIT rate of 15% for the years ended December 31, 2021, 2022 and 2023.
For more information, see Item 10.E. “Taxation—Taxation in the PRC.” Please also see Item. 3 “Key Information—Risk Factors—Other Risks and Risks Relating to Doing Business in China—Our business benefits from certain PRC government tax incentives. The expiration of, changes to, or our PRC subsidiaries/joint venture failing to continuously meet the criteria for these incentives could have a material adverse effect on our operating results by significantly increasing our tax expenses.”
According to the EIT Law and its implementation regulations, dividends declared after January 1, 2008 and paid by PRC foreign-invested enterprises to their non-PRC parent companies will be subject to PRC withholding tax at 10% unless there is a tax treaty between the PRC and the jurisdiction in which the overseas parent company is a tax resident and which specifically exempts or reduces such withholding tax, and such tax exemption or reduction is approved by the relevant PRC tax authorities. Pursuant to the tax arrangement between PRC and Hong Kong, if a shareholder of the PRC enterprise is a Hong Kong tax resident and directly holds a 25% or more equity interest in the PRC enterprise and is considered to be the beneficial owner of dividends paid by the PRC enterprise, such withholding tax rate may be lowered to 5%, subject to approval by the relevant PRC tax authorities. For more information, see Item 10.E. “Taxation—Taxation in the PRC” and “Taxation—Overview of Tax Implications of Various Other Jurisdictions— Hong Kong Taxation.”
Hong Kong
Our company and certain of its subsidiaries are subject to Hong Kong Profits Tax laws and regulations. Hong Kong has a two-tiered Profits Tax rates regime under which the first HK$2.0 million ($0.3 million) of assessable profits of qualifying corporations will be taxed at 8.25%, with the remaining assessable profits taxed at 16.5%. Hong Kong Profits Tax has been provided for at the relevant rates on the estimated assessable profits less estimated available tax losses, if any, of these entities as applicable.
Period-to-Period Comparison of Results of Operations
Year Ended December 31, 2023 Compared to Year Ended December 31, 2022
Revenue
Our revenue increased by 96.5% from $426.4 million for the year ended December 31, 2022 to $838.0 million for the year ended December 31, 2023, which resulted from increased revenue primarily in the Oncology/Immunology operations.
173
Revenue from Oncology/Immunology increased by 222.6% from $163.8 million for the year ended December 31, 2022 to $528.6 million for the year ended December 31, 2023, primarily due to revenue from Takeda of $353.1 million for the year ended December 31, 2023 (of which $280.0 million was the revenue recognized from the $400 million upfront payment received, $33.9 million was revenue related to research and development services, $32.0 million was the revenue recognized from the $35 million milestone payment received which was triggered by FDA approval of Fruzaqla in November 2023, $5.1 million was revenue from invoiced sales of goods to Takeda and $2.1 million was royalty revenue). The increase was also attributable to the sales of Elunate from $69.9 million for the year ended December 31, 2022 (of which $14.7 million was revenue from sales of goods primarily to Eli Lilly, $13.9 million was royalty revenue and $41.3 million was revenue from promotion and marketing services to Eli Lilly) to $83.2 million for the year ended December 31, 2023 (of which $18.0 million was revenue from sales of goods primarily to Eli Lilly, $16.6 million was royalty revenue and $48.6 million was revenue from promotion and marketing services to Eli Lilly). Sales of Sulanda have also contributed to the increase in revenue from $32.3 million for the year ended December 31, 2022 to $43.9 million for the year ended December 31, 2023. The increase was also attributable to the sales of Orpathys from $22.3 million for the year ended December 31, 2022 (of which $9.9 million was revenue from sales of goods and $12.4 million was royalty revenue) to $28.9 million for the year ended December 31, 2023 (of which $15.1 million was revenue from sales of goods and $13.8 million was royalty revenue). The increase has been netted off by reduction in revenue related to other (non-Takeda) research and development services which have decreased from $38.7 million for the year ended December 31, 2022 to $18.0 million for the year ended December 31, 2023, primarily attributable to receipt of a $15.0 million milestone payment upon initiating start-up activities for a Global Phase III study of Orpathys in Lung Cancer in March 2022 as well as less cost reimbursement from Eli Lilly for the year ended December 31, 2023.
Revenue from our Other Ventures increased by 17.8% from $262.6 million for the year ended December 31, 2022 to $309.4 million for the year ended December 31, 2023, primarily due to an increase in sales of prescription drugs products. Revenue from sales of prescription drugs increased by 24.5% from $237.3 million for the year ended December 31, 2022 to $295.4 million for the year ended December 31, 2023, primarily due to increased sales by our consolidated joint venture Hutchison Sinopharm. Revenue from sales of our consumer health products on the other hand has decreased by 44.7% from $25.3 million for the year ended December 31, 2022 to $14.0 million for the year ended December 31, 2023, primarily due to decreased sales by our consolidated joint venture Hutchison Hain Organic (which was divested in December 2023).
Cost of Revenue
Our cost of revenue increased by 23.6% from $311.1 million for the year ended December 31, 2022 to $384.4 million for the year ended December 31, 2023. This increase was due to increased sales by the Oncology/Immunology and Other Ventures operations.
Cost of revenue from Oncology/Immunology increased by 32.6% from $69.2 million for the year ended December 31, 2022 to $91.7 million for the year ended December 31, 2023, primarily due to an increase in sales of Elunate (including the provision of promotion and marketing services to Eli Lilly), Sulanda, Orpathys and Fruzaqla.
Cost of revenue from our Other Ventures increased by 21.0% from $241.9 million for the year ended December 31, 2022 to $292.7 million for the year ended December 31, 2023, which was primarily due to increased sales.
Cost of revenue as a percentage of our revenue decreased from 73.0% to 45.9% across these periods, primarily due to the new revenue from Takeda.
Research and Development Expenses
Our research and development expenses incurred by Oncology/Immunology decreased by 21.9% from $386.9 million for the year ended December 31, 2022 to $302.0 million for the year ended December 31, 2023, which was primarily due to a $58.6 million decrease in CROs and other clinical trial related costs and a $26.3 million decrease in personnel compensation and related costs. These decreased costs were primarily due to completion of major registration-enabling trials and strategic prioritization of our pipelines. Research and development expenses as a percentage of our revenue decreased from 90.7% to 36.0% across these periods, primarily due to the new revenue from Takeda and the aforementioned decrease in spending.
174
Selling Expenses
Our selling expenses increased by 21.5% from $43.9 million for the year ended December 31, 2022 to $53.4 million for the year ended December 31, 2023, primarily due to the expansion of our China oncology commercial team and increased marketing activities. Selling expenses as a percentage of our revenue decreased from 10.3% to 6.4% across these periods, primarily due to the new revenue from Takeda.
Administrative Expenses
Our administrative expenses decreased by 13.4% from $92.2 million for the year ended December 31, 2022 to $79.8 million for the year ended December 31, 2023. This was primarily due to a $10.4 million decrease in administrative expenses incurred by Oncology/Immunology, which was mainly due to the restructuring of our U.S. Oncology/Immunology commercial operations in 2022. Administrative expenses as a percentage of our revenue decreased from 21.6% to 9.5% across these periods, primarily due to the new revenue from Takeda and the aforementioned decrease in spending.
Other Income/(Expense)
We had net other expenses of $2.7 million for the year ended December 31, 2022 compared to net other income of $39.9 million for the year ended December 31, 2023. The change was primarily due to higher interest income of $26.5 million primarily from the interest earned on the $400 million Takeda upfront received. The change was also contributed by the net foreign exchange loss of $5.7 million for the year ended December 31, 2022 as compared to the net foreign exchange gain of $8.7 million for the year ended December 31, 2023 on USD denominated bank balances in China.
Income Tax (Expense)/Benefit
We had income tax benefit of $0.3 million for the year ended December 31, 2022 compared to income tax expense of $4.5 million for the year ended December 31, 2023, primarily due to an increase in taxable profit in Oncology/Immunology which was mainly due to the restructuring of our U.S. Oncology/Immunology commercial operations at the end of 2022.
Equity in Earnings of Equity Investees
Our equity in earnings of equity investees, net of tax, decreased by 4.9% from $49.8 million for the year ended December 31, 2022 to $47.3 million for the year ended December 31, 2023, primarily due to a decrease in gross profit margin by Shanghai Hutchison Pharmaceuticals for the year ended December 31, 2023.
Shanghai Hutchison Pharmaceuticals
The following table shows a summary of the results of operations of Shanghai Hutchison Pharmaceuticals for the years indicated. The consolidated financial statements of Shanghai Hutchison Pharmaceuticals are prepared in accordance with IFRS as issued by the IASB and are presented separately elsewhere in this annual report.
| Year Ended December 31, | |||||||
2023 | 2022 | |||||||
| ($’000) |
| % |
| ($’000) |
| % | |
Revenue |
| 385,483 |
| 100.0 |
| 370,600 |
| 100.0 |
Cost of sales |
| (101,122) |
| (26.2) |
| (89,487) |
| (24.1) |
Selling expenses |
| (150,717) |
| (39.1) |
| (144,979) |
| (39.1) |
Administrative expenses |
| (26,107) |
| (6.8) |
| (21,727) |
| (5.9) |
Other net operating income |
| 5,027 |
| 1.3 |
| 2,126 |
| 0.5 |
Taxation charge |
| (17,022) |
| (4.4) |
| (16,738) |
| (4.5) |
Profit for the year |
| 95,463 |
| 24.8 |
| 99,683 |
| 26.9 |
Equity in earnings of equity investee attributable to our company(1) |
| 47,295 |
| 12.3 |
| 49,748 |
| 13.4 |
| (1) | Equity in earnings of equity investee attributable to our company is presented under U.S. GAAP. The amount for the year ended December 31, 2023 includes elimination of unrealized profits on transactions with the Group of $131,000 and GAAP difference of $306,000. The amount for the year ended December 31, 2022 includes elimination of unrealized profits on transactions with the Group of $110,000 and GAAP difference of $16,000. |
175
Shanghai Hutchison Pharmaceuticals’ revenue increased by 4.0% from $370.6 million for the year ended December 31, 2022 to $385.5 million for the year ended December 31, 2023, primarily due to an increase in sales of She Xiang Bao Xin pills, a vasodilator used in the treatment of heart conditions. Sales of She Xiang Bao Xin pills increased by 2.1% from $341.6 million for the year ended December 31, 2022 to $348.6 million for the year ended December 31, 2023. The increase in revenue was also due to an $8.6 million increase in sales Zhi Ling Tong infant nutrition products manufactured by Hutchison Healthcare and distributed by Shanghai Hutchison Pharmaceuticals starting from October 1, 2022.
Cost of sales increased by 13.0% from $89.5 million for the year December 31, 2022 to $101.1 million for the year ended December 31, 2023, primarily due to higher sales of She Xiang Bao Xin pills. Shanghai Hutchison Pharmaceuticals’ revenue increased at a lower rate than the cost of sales mainly due to the impact of gradual price adjustment from volume-based procurement.
Selling expenses increased by 4.0% from $145.0 million for the year ended December 31, 2022 to $150.7 million for the year ended December 31, 2023, as a result of increased spending on marketing activities to support the increase in sales.
Administrative expenses increased by 20.2% from $21.7 million for the year ended December 31, 2022 to $26.1 million for the year ended December 31, 2023, primarily due to an increase in staff costs and other office expenses to support commercial activities.
Other net operating income increased by 136.5% from $2.1 million for the year ended December 31, 2022 to $5.0 million for the year ended December 31, 2023, primarily due to an increase in government grants.
Taxation charge increased by 1.7% from $16.7 million for the year ended December 31, 2022 to $17.0 million for the year ended December 31, 2023, primarily due to the reversal of certain deferred tax assets.
As a result of the foregoing, profit decreased by 4.2% from $99.7 million for the year ended December 31, 2022 to $95.5 million for the year ended December 31, 2023. Our equity in earnings of equity investees contributed by this joint venture was $49.7 million and $47.3 million for the years ended December 31, 2022 and 2023, respectively.
For more information on the financial results of our non-consolidated joint ventures, see “—Key Components of Results of Operations— Equity in Earnings of Equity Investees.”
Net Income/(Loss)
As a result of the foregoing, our net loss of 360.4 million for the year ended December 31, 2022 turned into net income of $101.1 million for the year ended December 31, 2023. Net loss attributable to our company of $360.8 million for the year ended December 31, 2022 turned into net income attributable to our Company of $100.8 million for the year ended December 31, 2023.
Year Ended December 31, 2022 Compared to Year Ended December 31, 2021
For a discussion of our results of operations for the year ended December 31, 2022 compared to the year ended December 31, 2021, see Item 5.A. “Operating Results” of our annual report on Form 20-F for the year ended December 31, 2022, filed with the SEC on February 28, 2023.
B. Liquidity and Capital Resources
To date, we have taken a multi-source approach to fund our operations, including through cash flows generated and dividend payments from our Oncology/Immunology and Other Ventures operations, service and milestone and upfront payments from our collaboration partners, bank borrowings, investments from other third parties, proceeds from our listings on various stock exchanges and follow-on offerings.
176
Primarily due to an increase in total revenue driven by Oncology/Immunology partnering, its strong commercial progress in China, and growth in third-party distribution sale, net income attributable to the Company amounted to $100.8 million for the year ended December 31, 2023, while for the years ended December 31, 2022 and 2021 the net loss attributable to the Company was $360.8 million and $194.6 million, respectively. Our Oncology/Immunology operations have historically not generated significant profits or have operated at a net loss, as creating potential global first-in-class or best-in-class drug candidates requires a significant investment of resources over a prolonged period of time. As a result, we anticipate that we may need additional financing for our Oncology/Immunology operations in future periods. See Item 3.D. “Risk Factors—Risks Relating to Our Oncology/Immunology Operations and Development of Our Drug Candidates—Our Oncology/Immunology operations have not generated significant profits and have operated at a net loss historically until recently, and our future profitability is dependent on the successful commercialization of our drug candidates.”
As of December 31, 2023, we had cash and cash equivalents of $283.6 million and short-term investments of $602.7 million and unutilized bank facilities of $68.1 million. Substantially all of our bank deposits are at major financial institutions, which we believe are of high credit quality. As of December 31, 2023, we had $79.3 million in bank loans, of which $48.2 million was related to a fixed asset loan and $31.1 million was related to a working capital loan. The total weighted average cost of bank borrowings for the year ended December 31, 2023 was 3.41% per annum. For additional information, see “—Loan Facilities.”
Certain of our subsidiaries and joint ventures, including those registered as wholly foreign-owned enterprises in China, are required to set aside at least 10.0% of their after-tax profits to their general reserves until such reserves reach 50.0% of their registered capital. In addition, certain of our joint ventures are required to allocate certain of their after-tax profits as determined in accordance with related regulations and their respective articles of association to the reserve funds upon their board approval. Profit appropriated to the reserve funds for our subsidiaries and joint ventures incorporated in the PRC was approximately $89,000, $318,000 and $168,000 for the years ended December 31, 2021, 2022 and 2023, respectively.
In addition, as a result of PRC regulations restricting dividend distributions from such reserve funds and from a company’s registered capital, our PRC subsidiaries are restricted in their ability to transfer a certain amount of their net assets to us as cash dividends, loans or advances. This restricted portion amounted to $1.0 million as of December 31, 2023. Although we do not currently require any such dividends, loans or advances from our PRC subsidiaries to fund our operations, should we require additional sources of liquidity in the future, such restrictions may have a material adverse effect on our liquidity and capital resources. For more information, see Item 4.B. “Business Overview—Regulation—PRC Regulation of Foreign Currency Exchange, Offshore Investment and State-Owned Assets—Regulation on Investment in Foreign invested Enterprises—Regulation on Dividend Distribution.”
In addition, our non-consolidated joint venture Shanghai Hutchison Pharmaceuticals held $19.1 million in cash and cash equivalents and no bank borrowings as of December 31, 2023. Such cash and cash equivalents are only accessible by us through dividend payments from the joint venture. The level of dividends declared by the joint venture is subject to agreement each year between us and our joint venture partner based on the profitability and working capital needs of the joint venture. As a result, we cannot guarantee that the joint venture will continue to pay dividends to us in the future at the same rate we have enjoyed in the past, or at all, which may have a material adverse effect on our liquidity and capital resources. For more information, see Item 3.D. “Risk Factors—Risks Relating to Sales of our Internally Developed Drugs and Other Drugs—As a significant portion of the operations of our Other Ventures is conducted through joint ventures, we are dependent on the success of our joint ventures, our receipt of dividends or other payments from our joint ventures for cash to fund our operations, and our investments in our joint ventures are subject to liquidity risk.”
177
We believe that our current levels of cash and cash equivalents, short-term investments, along with cash flows from operations, dividend payments and unutilized bank borrowings, will be sufficient to meet our anticipated cash needs for at least the next 12 months. In the long term, we believe that we can meet our need for cash through revenue generated from marketed products, public and private sales of our securities and the potential disposals of our remaining non-core businesses. However, we may require additional financing in order to fund all of the clinical development efforts that we plan to undertake to accelerate the development of our clinical-stage drug candidates. For more information, see Item 3.D. “Risk Factors—Risks Relating to Our Financial Position and Need for Capital.”
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
($’000) | ||||||
Cash Flow Data: |
|
|
| |||
Net cash generated from/(used in) operating activities |
| 219,258 |
| (268,599) |
| (204,223) |
Net cash (used in)/generated from investing activities |
| (291,136) |
| 296,588 |
| (306,320) |
Net cash generated from/(used in) financing activities |
| 48,660 |
| (82,763) |
| 650,028 |
Net (decrease)/increase in cash and cash equivalents |
| (23,218) |
| (54,774) |
| 139,485 |
Effect of exchange rate changes |
| (6,471) |
| (9,490) |
| 2,427 |
Cash and cash equivalents at beginning of the year |
| 313,278 |
| 377,542 |
| 235,630 |
Cash and cash equivalents at end of the year |
| 283,589 |
| 313,278 |
| 377,542 |
Net Cash generated from/(used in) Operating Activities
Net cash used in operating activities was $268.6 million for the year ended December 31, 2022, compared to net cash generated from operating activities of $219.3 million for the year ended December 31, 2023. The net change of $487.9 million was primarily attributable to a net loss attributable to the Company of $360.8 million for the year ended December 31, 2022 turning into a net income attributable to the Company of $100.8 million for the year ended December 31, 2023 (which included $312.0 million in upfront and milestone income recognized from Takeda).
For a discussion of our net cash used in operating activities for the years ended December 31, 2022 and 2021, see Item 5.B. “Liquidity and Capital Resources” of our annual report on Form 20-F for the year ended December 31, 2022, filed with the SEC on February 28, 2023.
Net Cash (used in)/generated from Investing Activities
Net cash generated from investing activities was $296.6 million for the year ended December 31, 2022, compared to net cash used in investing activities of $291.1 million for the year ended December 31, 2023. The net change of $587.7 million was primarily attributable to placement of more short-term investments which had net withdrawals of $316.4 million for the year ended December 31, 2022 as compared to net deposits of $285.0 million for the year ended December 31, 2023. The net change was partially offset by a $13.0 million increase in amounts received from the divestment of a former equity investee from $16.5 million during the year ended December 31, 2022 to $29.5 million during the year ended December 31, 2023.
For a discussion of our net cash generated from/(used in) investing activities for the years ended December 31, 2022 and 2021, see Item 5.B. “Liquidity and Capital Resources” of our annual report on Form 20-F for the year ended December 31, 2022, filed with the SEC on February 28, 2023.
Net Cash generated from/(used in) Financing Activities
Net cash used in financing activities was $82.8 million for the year ended December 31, 2022, compared to net cash generated from financing activities of $48.7 million for the year ended December 31, 2023. The net change of $131.5 million was mainly attributable to bank borrowings which had a net repayment of $9.2 million during the year ended December 31, 2022 as compared to net proceeds of $61.7 million during the year ended December 31, 2023. The net change was also attributable to a $39.0 million decrease in purchases of ADSs by a trustee for the settlement of equity awards of the Company from $48.1 million for the year ended December 31, 2022 to $9.1 million for the year ended December 31, 2023, as well as a $16.5 million decrease in dividends paid to non-controlling shareholders of subsidiaries from $25.6 million for the year ended December 31, 2022 to $9.1 million for the year ended December 31, 2023.
178
For a discussion of our net cash (used in)/generated from financing activities for the years ended December 31, 2022 and 2021, see Item 5.B. “Liquidity and Capital Resources” of our annual report on Form 20-F for the year ended December 31, 2022, filed with the SEC on February 28, 2023.
Contractual Obligations
The following table sets forth our contractual obligations as of December 31, 2023. For more information on bank borrowings and interest on bank borrowings, please see “—Loan Facilities.” Our purchase obligations relate to property, plant and equipment that are contracted for but not yet paid. Our lease obligations primarily comprise future aggregate minimum lease payments in respect of various factories, warehouse, offices and other assets under non-cancellable lease agreements. For more information on purchase obligations and lease obligations, please see “—Capital Expenditures.”
Payment Due by Period | ||||||||||
Less Than | More Than | |||||||||
| Total |
| 1 Year |
| 1‑2 Years |
| 2‑5 Years |
| 5 Years | |
($’000) | ||||||||||
Bank borrowings | 79,344 | 31,155 | 958 | 11,490 | 35,741 | |||||
Interest on bank borrowings |
| 11,034 |
| 2,411 |
| 1,638 |
| 4,503 |
| 2,482 |
Purchase obligations |
| 1,259 |
| 1,259 |
| — |
| — |
| — |
Lease obligations |
| 7,583 |
| 3,919 |
| 1,356 |
| 2,308 |
| — |
Total |
| 99,220 |
| 38,744 |
| 3,952 |
| 18,301 |
| 38,223 |
Shanghai Hutchison Pharmaceuticals
The following table sets forth the contractual obligations of our non-consolidated joint venture Shanghai Hutchison Pharmaceuticals as of December 31, 2023. Shanghai Hutchison Pharmaceuticals’ purchase obligations comprise capital commitments for property, plant and equipment contracted for but not yet paid. Shanghai Hutchison Pharmaceuticals’ lease obligations primarily comprise future aggregate minimum lease payments in respect of various offices under non-cancellable lease agreements.
Payment Due by Period | ||||||||||
Less Than | More Than | |||||||||
| Total |
| 1 Year |
| 1‑2 Years |
| 2‑5 Years |
| 5 Years | |
| ($’000) | |||||||||
Purchase obligations | 376 | 376 | — | — | — | |||||
Lease obligations |
| 1,459 |
| 791 |
| 651 |
| 17 |
| — |
Total |
| 1,835 |
| 1,167 |
| 651 |
| 17 |
| — |
Loan Facilities
In October 2021, HUTCHMED Limited entered into a 10-year fixed asset loan facility agreement with Bank of China Limited for the provision of a secured credit facility of RMB754.9 million ($105.5 million) with an annual interest rate at the 5-year China Loan Prime Rate less 0.80% (which was supplemented in June 2022). This credit facility is guaranteed by HUTCHMED Limited’s immediate holding company, HUTCHMED Investment (HK) Limited, and secured by the underlying leasehold land and buildings of HUTCHMED Limited, and includes certain financial covenant requirements. As of December 31, 2023, RMB344.8 million ($48.2 million) was utilized from the fixed asset loan facility.
In May 2022, HUTCHMED Group (HK) Limited entered into a 12-month revolving credit facility with HSBC in the amount of HK$390.0 million ($50.0 million) with an interest rate at HIBOR plus 0.5% per annum. This revolving facility is guaranteed by us. The revolving credit facility expired in May 2023.
In November 2023, Hutchison Whampoa Sinopharm Pharmaceuticals (Shanghai) Company entered into a short-term working capital loan facility agreement with Bank of China Limited for the provision of a credit facility of RMB300.0 million ($41.9 million) with an annual interest rate at the 1-year China Loan Prime Rate less 0.95%. As of December 31, 2023, RMB222.9 million ($31.1 million) was utilized from the loan facility.
179
Our non-consolidated joint venture Shanghai Hutchison Pharmaceuticals had no bank borrowings outstanding as of December 31, 2023.
Gearing Ratio
The gearing ratio of our group, which was calculated by dividing total interest-bearing loans by total equity, was 10.7% as of December 31, 2023, an increase from 2.8% as of December 31, 2022. The increase was primarily attributable to the increase in interest-bearing loans.
Capital Expenditures
We had capital expenditures of $16.8 million, $36.7 million and $32.6 million for the years ended December 31, 2021, 2022 and 2023, respectively. Our capital expenditures during these periods were primarily used for the purchases of leasehold land and property, plant and equipment for a new large-scale manufacturing facility for innovative drugs in Shanghai, China and to expand research facilities and our manufacturing facility in Suzhou, China. Our capital expenditures have been primarily funded by cash flows from operations, bank borrowings and proceeds from our initial public and follow-on offerings in Hong Kong and the United States and other equity offerings.
As of December 31, 2023, we had commitments for capital expenditures of approximately $1.3 million, primarily for the construction of the new manufacturing facility in Shanghai. We expect to fund these capital expenditures through cash flows from operations, bank borrowings and existing cash resources.
Our non-consolidated joint venture Shanghai Hutchison Pharmaceuticals had capital expenditures of $3.4 million, $1.9 million and $5.8 million for the years ended December 31, 2021, 2022 and 2023, respectively. These capital expenditures were primarily related to the renovation of new office and improvements to its production facilities in Shanghai. These capital expenditures were primarily funded through cash flows from operations of Shanghai Hutchison Pharmaceuticals.
C.Research and Development, Patents and Licenses, etc.
Full details of our research and development activities and expenditures are given in the “Business” and “Operating and Financial Review and Prospects” sections of this annual report above.
D.Trend Information.
Other than as described elsewhere in this annual report, we are not aware of any trends, uncertainties, demands, commitments or events that are reasonably likely to have a material adverse effect on our revenue, income, profitability, liquidity or capital resources, or that would cause our reported financial information not necessarily to be indicative of future operation results or financial condition.
E.Critical Accounting Estimates.
For information on our critical accounting estimates, please see “—Operating Results—Critical Accounting Policies and Significant Judgments and Estimates” section of this annual report above.
180
ITEM 6. DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES
A. Directors and Senior Management.
Business Experience and Qualifications of our Directors and Senior Management
Below is a list of the names and ages of our directors and officers as of February 15, 2024, and a brief account of the business experience of each of them. The business address for our directors and officers is c/o HUTCHMED (China) Limited, Level 18, The Metropolis Tower, 10 Metropolis Drive, Hunghom, Kowloon, Hong Kong.
Name |
| Age |
| Position |
TO Chi Keung, Simon |
| 72 |
| Executive Director and Chairman |
Weiguo SU |
| 66 |
| Executive Director, Chief Executive Officer and Chief Scientific Officer |
CHENG Chig Fung, Johnny |
| 57 |
| Executive Director and Chief Financial Officer |
Edith SHIH |
| 72 |
| Non-executive Director and Company Secretary |
Dan ELDAR |
| 70 |
| Non-executive Director |
Ling YANG | 44 | Non-executive Director | ||
Paul Rutherford CARTER |
| 63 |
| Senior Independent Non-executive Director |
Graeme Allan JACK |
| 73 |
| Independent Non-executive Director |
MOK Shu Kam, Tony |
| 63 |
| Independent Non-executive Director |
Michael Ming SHI |
| 58 |
| Executive Vice President, Head of R&D and Chief Medical Officer |
Karen Jane ATKIN |
| 58 |
| Executive Vice President and Chief Operating Officer |
Zhenping WU |
| 64 |
| Executive Vice President, Pharmaceutical Sciences and Manufacturing |
Hong CHEN |
| 53 |
| Executive Vice President and Chief Commercial Officer (China) |
Mark Kin Hung LEE | 46 | Senior Vice President, Corporate Management and Communications | ||
May Qingmei WANG | 60 | Senior Vice President, Business Development & Strategic Alliances | ||
Charles George Rupert NIXON |
| 54 |
| Group General Counsel |
To Chi Keung, Simon has been a director since 2000 and an executive director and chairman of our Company since 2006. He is also a member of our nomination committee, remuneration committee and technical committee. He is the managing director of Hutchison Whampoa (China) Limited (“Hutchison China”) and has been with Hutchison China for over 40 years, building its business from a small trading company to a multi-billion dollar investment group. He has negotiated major transactions with multinational corporations such as Procter & Gamble, Lockheed, Pirelli, Beiersdorf, United Airlines, and British Airways. He is currently a non-executive director of Gama Aviation Plc and formerly served as independent non-executive director on the boards of China Southern Airlines Company Limited and Air China Limited. In addition, Mr. To is a director of certain substantial shareholders (within the meaning of the Securities and Futures Ordinance) of the Company and certain companies controlled by substantial shareholders of the Company. Mr. To’s career in China spans more than 45 years. He is the original founder of the China healthcare business of Hutchison Whampoa Limited (currently a subsidiary of CK Hutchison) and has been instrumental in its acquisitions made to date. He received a Bachelor’s degree in Mechanical Engineering from Imperial College, London and a Master in Business Administration from Stanford University’s Graduate School of Business.
Weiguo Su has been an executive director since 2017 and chief executive officer of our Company since March 4, 2022. He is also our chief scientific officer since 2012. He is also a member of our technical committee. Dr. Su has headed all drug discovery and research since he joined our company, including master-minding our scientific strategy, being a key leader of our Oncology/Immunology operations, and responsible for the discovery of each and every small molecule drug candidate in our pipeline. Prior to joining our company in 2005, Dr. Su worked with the U.S. research and development department of Pfizer, Inc. In 2017, Dr. Su was granted the prestigious award by the China Pharmaceutical Innovation and Research Development Association (PhIRDA) as one of the Most Influential Drug R&D Leaders in China. Dr. Su received a bachelor of science degree in chemistry from Fudan University in Shanghai and completed a PhD and post-doctoral fellowship in chemistry at Harvard University under the guidance of Nobel Laureate Professor E. J. Corey.
181
Cheng Chig Fung, Johnny has been an executive director since 2011 and our chief financial officer since 2008. He is a member of our sustainability committee. Prior to joining our company, Mr. Cheng was vice president, finance of Bristol Myers Squibb in China and was a director of Sino-American Shanghai Squibb Pharmaceuticals Ltd. and Bristol-Myers Squibb (China) Investment Co. Ltd. in Shanghai between late 2006 and 2008. Mr. Cheng started his career as an auditor with Price Waterhouse (currently PricewaterhouseCoopers) in Australia and then KPMG in Beijing before spending eight years with Nestlé China where he was in charge of a number of finance and control functions in various operations. Mr. Cheng received a bachelor of economics, accounting major from the University of Adelaide and is a member of Chartered Accountants Australia and New Zealand.
Edith Shih has been a non-executive director since 2006, the company secretary of our company and the company secretary of group companies since 2000. She is also chairman of our sustainability committee. She has over 35 years of experience in legal, regulatory, corporate finance, compliance and corporate governance fields. She is also executive director and company secretary of CK Hutchison. She has been with the Cheung Kong (Holdings) Limited (“CKH”) group since 1989 and with Hutchison Whampoa Limited (“HWL”) since 1991. Both CKH and HWL were formerly listed on SEHK and became wholly-owned subsidiaries of CK Hutchison in 2015. She has acted in various capacities within the HWL group, including head group general counsel and company secretary of HWL as well as director and company secretary of HWL subsidiaries and associated companies. Ms. Shih is in addition a non-executive director of Hutchison Telecommunications Hong Kong Holdings Limited, Hutchison Port Holdings Management Pte. Limited as the trustee-manager of Hutchison Port Holdings Trust and a commissioner of PT Duta Intidaya Tbk. In addition, Ms. Shih is a director of certain substantial shareholders (within the meaning of the Securities and Futures Ordinance) of our company and certain companies controlled by certain substantial shareholders of our company. The aforementioned companies are either subsidiaries or associated companies of CK Hutchison of which Ms. Shih has oversight as a director of CK Hutchison. She is the past international president and current member of the council of The Chartered Governance Institute (“CGI”) as well as a past president and current honorary advisor of The Hong Kong Chartered Governance Institute (“HKCGI”). She is also a current member and past chairperson of the nomination committee of HKCGI. Further, she is also chairperson of the process review panel for the Accounting and Financial Reporting Council (formerly known as the Financial Reporting Council) and a member of the Executive Committee and Council of The Hong Kong Management Association. She was also a member of the Securities and Futures Appeal Tribunal. Ms. Shih is a solicitor qualified in England and Wales, Hong Kong and Victoria, Australia and a fellow of both the CGI and HKCGI, holding chartered secretary and chartered governance professional dual designations. She holds a bachelor of science degree and a master of arts degree from the University of the Philippines as well as a master of arts degree and a master of education degree from Columbia University, New York.
Dan Eldar has been a non-executive director of our company since 2016. He has more than 30 years of experience as a senior executive, leading global operations in telecommunications, water, biotech and healthcare. He is an executive director of Hutchison Water Israel Ltd which focuses on large scale projects including desalination, wastewater treatment and water reuse. He was formerly an independent non-executive director of Leumi Card Ltd., a subsidiary of Bank Leumi Le-Israel B.M., one of Israel’s leading credit card companies. Dr. Eldar received a doctor of philosophy degree in government from Harvard University, master of arts degree in government from Harvard University, master of arts degree in political science and public administration from the Hebrew University of Jerusalem and a bachelor of arts degree in political science from the Hebrew University of Jerusalem.
Ling Yang has been a non-executive director of our company since July 2023. She has been the managing director of Carlyle since January 2017 and co-head of Carlyle Asia Healthcare since November 2021, in charge of advising in healthcare investment and portfolio activities of Carlyle in China. She is also chairwoman and non-executive director of ADICON Holdings Limited. Prior to Carlyle Group, Ms. Yang worked in private equity at KKR Asia Limited and in investment banking at Goldman Sachs in the U.S. She was formerly a director of Shenzhen Salubris Pharmaceuticals Co., Ltd. Ms. Yang graduated summa cum laude and is a member of Phi Beta Kappa with a bachelor’s degree in economics and computer science from Smith College and she received her master of business administration degree from Harvard Business School.
Paul Rutherford Carter has been a senior independent non-executive director of our company since 2017. He is also chairman of our remuneration committee and a member of our audit committee and technical committee. He has more than 26 years of experience in the pharmaceutical industry. From 2006 to 2016, Mr. Carter served in various senior executive roles at Gilead Sciences, Inc. (“Gilead”), a research-based biopharmaceutical company, with the last position as executive vice president, commercial operations. In this role, Mr. Carter headed the worldwide commercial organization responsible for the launch and commercialization of all of the products of Gilead. He also worked as a senior executive at GlaxoSmithKline Plc (currently GSK Plc.). He is currently a director of Immatics N.V. and Kyowa Kirin International Plc. He is the chairman of Evox Therapeutics and a retained advisor to several firms active in the life sciences sector. He was formerly a director of Alder Biopharmaceuticals, Inc, Mallinckrodt plc and VectivBio Holding AG. Mr. Carter received a degree in business studies from the Ealing School of Business and Management (now merged into University of West London) and is a fellow of the Chartered Institute of Management Accountants in the United Kingdom.
182
Graeme Allan Jack has been an independent non-executive director of our company since 2017. He is also chairman of our audit committee and a member of our nomination committee and remuneration committee. He has more than 40 years of experience in finance and audit. He retired as partner of PricewaterhouseCoopers in 2006 after a distinguished career with the firm for over 33 years. He is currently an independent non-executive director of The Greenbrier Companies, Inc. (an international supplier of equipment and services to the freight rail transportation markets). He was formerly a director of COSCO SHIPPING Development Co., Ltd. (formerly known as “China Shipping Container Lines Company Limited”, an integrated financial services platform principally engaged in vessel and container leasing) and Hutchison Port Holdings Management Pte. Limited as the trustee-manager of Hutchison Port Holdings Trust (a developer and operator of deep water container terminals). Mr. Jack received a bachelor of commerce degree from University of New South Wales, Australia and is a fellow of the Hong Kong Institute of Certified Public Accountants and an associate of Chartered Accountants Australia and New Zealand.
Mok Shu Kam, Tony has been an independent non-executive director of our company since 2017. He is also chairman of our nomination committee and technical committee, a member of our audit committee and sustainability committee. Professor Mok has more than 35 years of experience in clinical oncology with his main research interest focusing on biomarker and molecular targeted therapy in lung cancer. He is currently Li Shu Fan Medical Foundation named professor and chairman of department of clinical oncology at The Chinese University of Hong Kong. Professor Mok has contributed to over 300 articles in international peer-reviewed journals, as well as multiple editorials and textbooks. In October 2018, Professor Mok was the first Chinese to be bestowed with the European Society for Medical Oncology (ESMO) Lifetime Achievement Award, one of the most prestigious international honors and recognitions given to cancer researchers, for his contribution to and leadership in lung cancer research worldwide. In September 2023, Professor Mok was awarded The Sixth Fok Ying-Tung Prize – The World Outstanding Chinese Doctor Award, for his contribution in lung cancer research. Professor Mok is a non-executive director of AstraZeneca PLC, a non-executive independent director of Lunit USA Inc. and a member of the scientific advisory board of Prenetics Global Limited (“Prenetics”). He is co-founder of Sanomics Limited (acquired by ACT Genomics Holdings Ltd. in November 2021) and Aurora Tele-Oncology Limited. He was formerly a board director of the American Society of Clinical Oncology (“ASCO”), a steering committee member of the Chinese Society of Clinical Oncology, past president of the International Association for the Study of Lung Cancer, and the chairman of the board of ACT Genomics Holdings Ltd. until it was acquired by Prenetics in December 2022. Professor Mok is also closely affiliated with the oncology community in China and has been awarded an Honorary Professorship at Guangdong Province People’s Hospital, Guest Professorship at Peking Union Medical College Hospital and Visiting Professorship at Shanghai Jiao Tong University and Distinguished Professorship at Fujian Cancer Hospital. He received his bachelor of medical science degree and a doctor of medicine from University of Alberta, Canada. He is also a fellow of the Royal College of Physicians and Surgeons of Canada, Hong Kong College of Physicians, Hong Kong Academy of Medicine, Royal College of Physicians of Edinburgh and ASCO.
Michael Ming Shi is our executive vice president, head of R&D and chief medical officer. He oversees the drug discovery and development of our Company from strategy to execution. Prior to joining our company in 2022, Dr. Shi was the global head of R&D and chief medical officer at Transcenta Holding Limited. Before that, he worked at Novartis for over 15 years, where he held various senior leadership positions including global program clinical head in clinical development. Dr. Shi is a member of American Society of Clinical Oncology, European Society of Medical Oncology, American Society of Hematology, American Association for Cancer Research, Sino-American Pharmaceutical Association and an executive committee member of the US-China Anti-cancer Association (USCACA). Dr. Shi also worked as the program director of Genetics Variation at National Institution of Health (“NIH”) and was an adjunct assistant professor at the University of Michigan Medical School. Dr. Shi holds a PhD in Molecular Pharmacology and Toxicology from the University of Southern California, and conducted postdoctoral research at the Harvard Medical School. He received his medical education from Peking Union Medical College.
Karen Jane Atkin is our executive vice president and chief operating officer. Prior to joining our company in 2021, Dr. Atkin spent 24 years at AstraZeneca in senior medical, regulatory, pharmacovigilance, R&D and commercial leadership roles, including as senior vice president of medical for biopharmaceuticals, vice president of the global infection, neuroscience and autoimmunity therapy area and the established brand business, country president of Indonesia and led China R&D for over four years. Dr. Atkin is also a registered physician with advanced level qualifications in internal medicine and pharmaceutical medicine. Dr. Atkin holds three bachelor’s degrees in physiology, medicine and surgery, respectively, from University College London. She graduated with a first class honors degree in medicine, holds a master of business administration from the Open University, and is a member of the Royal College of Physicians and a fellow of the Faculty of Pharmaceutical Medicine in the UK.
183
Zhenping Wu joined our company in 2008 and is our executive vice president of pharmaceutical sciences and manufacturing. Dr. Wu has over 29 years of experience in drug discovery and development. His past positions include senior director of pharmaceutical sciences at Phenomix Corporation, a U.S.-based biotechnology company, director of pharmaceutical development at Pfizer Global Research & Development in California (formerly Agouron Pharmaceuticals) and a group leader at Roche at its Palo Alto site. He is a past chairman and president of the board of the Sino-American Biotechnology and Pharmaceutical Association. Dr. Wu received a PhD from the University of Hong Kong and a master in business administration from the University of California at Irvine.
Hong Chen is our executive vice president and chief commercial officer (China). Prior to joining our company in 2011, Mr. Chen spent 12 years with Bristol-Myers Squibb and was last serving as its national sales & marketing director in China. Mr. Chen received a bachelor’s degree in medicine from Nanjing Medical University and an EMBA from Cheung Kong Graduate School of Business.
Mark Kin Hung Lee is our senior vice president of corporate management and communications. He began working in healthcare investment banking in the United States and Europe in 1998 and joined our company in 2009. Based in the New York and London offices of Credit Suisse, Mr. Lee was involved in the execution and origination of mergers, acquisitions, public and private financings and corporate strategy for life science companies such as AstraZeneca, Bristol-Myers Squibb and Genzyme, as well as other medical product and service companies. Mr. Lee received his bachelor’s degree in biochemical engineering with first class honors from University College London, where he was awarded a Dean’s Commendation. He also received a master of business administration from the Massachusetts Institute of Technology’s Sloan School of Management.
May Qingmei Wang is our senior vice president of business development & strategic alliances. Prior to joining our company in 2010, Dr. Wang spent 16 years with Eli Lilly where she was the head of Eli Lilly’s Asian Biology Research and responsible for establishing and managing research collaborations in China and across Asia. Dr. Wang holds numerous patents, has published more than 50 peer-reviewed articles and has given dozens of seminars and plenary lectures. Dr. Wang received a PhD in biochemistry from Purdue University.
Charles George Rupert Nixon has been our group general counsel since May 2015 and has worked with our company since 2006. Prior to joining our company, Mr. Nixon was group senior legal counsel for Hutchison Whampoa Limited (previously a listed company in Hong Kong and after a restructuring, a subsidiary of CK Hutchison) in both Hong Kong and London and prior to that senior legal counsel for Three UK, the mobile phone operator. Mr. Nixon has been with the CK Hutchison Group since 2001. Mr. Nixon received an LL.B (Hons) from Middlesex University and is a qualified solicitor in England & Wales with over 30 years of experience.
184
Board Diversity
On August 6, 2021, the SEC approved the Nasdaq Stock Market’s proposal to amend its listing standards to encourage greater board diversity and to require board diversity disclosures for Nasdaq-listed companies. Pursuant to the amended listing standards, HUTCHMED, as a foreign private issuer, is required to have at least two diverse board members or explain the reasons for not meeting this objective by 2025. Furthermore, a board diversity matrix is required to be included in the annual report on Form 20-F, containing certain demographic and other information regarding members of our board of directors. HUTCHMED currently complies with the diversity requirement, as we currently have two female and seven male members on our board of directors. The board diversity matrix is set out below.
Board Diversity Matrix (As of February 28, 2024) |
| |
Place of Principal Executive Offices | Hong Kong | |
Foreign Private Issuer | Yes | |
Disclosure Prohibited under Home Country Law | No | |
Total Number of Directors |
| 9 |
|
|
|
| Did Not Disclose | ||||
| Female |
| Male |
| Non-Binary |
| Gender | |
Part I: Gender Identity |
|
|
|
|
|
|
|
|
Directors |
| 2 |
| 7 |
| — |
| — |
Part II: Demographic Background |
|
|
|
|
|
|
|
|
Underrepresented Individual in Place of Principal Executive Offices |
| — |
| — |
| — |
| — |
LGBTQ+ |
| — | — | — | — | |||
Did Not Disclose Demographic Background |
| — | — | — | — |
B. Compensation.
Compensation Summary
Remuneration Committee organization and purpose
The Remuneration Committee comprises three members and is chaired by Mr. Paul Rutherford Carter, senior independent non-executive director, with the Chairman Mr. To Chi Keung, Simon and independent non-executive director, Mr. Graeme Allan Jack, as members. The Remuneration Committee meets towards the end of each year to determine the remuneration package of executive directors and senior management of the group and during the year to consider grants of share options and LTIP awards and other remuneration related matters. Remuneration matters are also considered and approved by way of written resolutions and where warranted, at additional meetings. The Remuneration Committee held five meetings in 2023 with 100% attendance.
The responsibilities of the Remuneration Committee are to assist the Board in achieving its objectives of attracting, retaining and motivating a broader and more diverse pool of employees of the highest caliber and experience needed to shape and execute the strategy across the group’s substantial, diverse and international business operations. It assists the group in the administration of a fair and transparent procedure for setting remuneration policies for all directors and senior management of the group. Whilst the Board retains its power to determine the remuneration of non-executive directors, the responsibility for reviewing and determining the remuneration package of individual executive directors and senior management of the group is delegated to the Remuneration Committee. The Committee is authorized to obtain, at the company’s expense, external legal or other professional advice on any matters within its Terms of Reference.
2023 Goals
In 2023, this strategy delivered significant results to our operations. As described below, a considerable number of company goals were set and achieved in 2023 on our regulatory, clinical development, business development, manufacturing, commercial, financial, organizational and sustainability operations. These included:
185
Regulatory goals. Filed regulatory submissions for the use of fruquintinib in colorectal cancer with the U.S. Food and Drug Administration, the European Medicines Agency and the Japan Pharmaceuticals and Medical Devices Agency; filed regulatory submission for the use of fruquintinib in gastric cancer with the China National Medical Products Administration; and obtained approval for the use of fruquintinib in colorectal cancer in the U.S.
Clinical Regulatory and Government Affairs goals. Listed savolitinib on the China National Reimbursement Drug List for the first time and maintained listings for fruquintinib and surufatinib; enrolled first patients in the savolitinib gastric cancer Phase II registration trial and the HMPL-453 intrahepatic cholangiocarcinoma Phase II registration trial; completed enrollment in the confirmatory savolitinib MET exon14 non-small cell lung cancer trial, the fruquintinib endometrial cancer Phase II registration trial, the fruquintinib renal cancer Phase III registration trial, the tazemetostat follicular lymphoma bridging Phase II trial and the amdizalisib follicular lymphoma Phase II trial; and filed the investigational new drug application for HMPL-415.
Business development goal. Closed an exclusive license agreement for fruquintinib outside China with Takeda.
Manufacturing goals. Established the global supply chain for fruquintinib; passed pre-approval inspections by the U.S. Food and Drug Administration; completed and certified the new Shanghai factory.
Commercial and financial goals. Reported China revenue from ELUNATE, SULANDA and TAZVERIK – the three medicines marketed by the HUTCHMED commercial team – of $128.1 million compared to $102.3 million in 2022; received $400.0 million in upfront payment from Takeda under the license agreement of fruquintinib plus regulatory milestones of $35 million; and reported net profit of $100.8 million as compared to a net loss of $360.8 million in 2022.
Organization. Maintained headcount at approximately 2,000 while improving culture and employee satisfaction, including as evidenced by the 2023 employee engagement survey that showed improvements across all industry benchmarks.
Sustainability. Improved sustainability disclosure; initiated and progressed Scope 3 and data collection programs towards meeting the new International Sustainability Standards Board disclosure frameworks; improved sustainability ratings by several key rating agencies including Hang Seng, ISS, MSCI, S&P Global, Sustainalytics and Wind; and received awards for environmental, social and governance. The awards included five from the GBA ESG Achievement Awards 2023 by Metro Finance, two from the ESG Leading Enterprise Awards by Bloomberg Businessweek, and the Top 20 Chinese Pharmaceutical Listed Companies in ESG Competitiveness by Healthcare Executive.
Remuneration components
The goal of our remuneration programs is to align remuneration delivery with performance, measured both internally against budgets and key operational achievements, and externally through share price. We believe this alignment was achieved in 2023.
In general, our compensation consists of the following components:
| ● | Base salary, to attract and retain highly skilled talent. This fixed component of pay is to provide financial stability, based on responsibilities, experience, individual contributions and peer company data; |
| ● | Annual cash bonus incentive program, to motivate, promote and reward the achievement of key short-term strategic and business goals of HUTCHMED as well as individual performance. This is a variable component of pay based on annual corporate and individual performance; and |
| ● | Equity incentives, to encourage Executive Directors, senior management and other employees to focus on out-performance and align their interests with shareholders, as well as to promote retention and to reward outstanding company and individual performance. This is in the form of grants of share options and LTIP awards, which are subject to a vesting schedule based on continued service, the value of which depends on our share price performance, to align employee interests with those of our shareholders over the longer-term. |
186
The Remuneration Committee reviewed and made recommendation to the Board on grant of share awards under the LTIP and share options under the share option scheme to incentivize talent and professional expertise to stay and grow with the Group. See “—Executive Officer Compensation” and “—Equity Compensation Schemes and Other Benefit Plans” for more details on the share awards and share options granted during 2023.
2023 review and recommendations
During the year, the Remuneration Committee reviewed background information on market data (including economic indicators, statistics and the compensation benchmarking), headcount and staff costs. It also reviewed and approved the proposed 2024 directors’ fees for executive directors and made recommendation to the board on the proposed 2024 directors’ fees for independent non-executive directors. Prior to the end of the year, the Remuneration Committee reviewed and approved the 2023 year-end bonus and 2024 remuneration package of Executive Directors and senior management of the Group. No Director or any or his/her associates is involved in deciding his/her own remuneration. The Remuneration Committee also viewed and recommended to the Board updates to its Terms of Reference based on the latest Hong Kong Corporate Governance Code which took effect on January 1, 2023.
Remuneration advisor
In addition, the Remuneration Committee has reviewed the approach to remuneration and reporting on executive remuneration in detail. Aimed at attracting and retaining top talent, the Remuneration Committee appointed an independent advisor, Aon Enterprise Solutions (Shanghai) Co., Ltd. (“Aon”) to conduct benchmarking research on the compensation of a peer group of U.S. and China biotech companies (the “Aon Benchmarking Research”). Aon has no other connection with the Company or individual Directors. The Remuneration Committee comprehensively reviewed the Group’s compensation and share-based incentives policies, the Aon Benchmarking Research and established an attractive policy to ensure the Group is able to recruit and retain top talent. Vesting of share-based awards under such policy is in line with the referenced peer group. The Committee takes seriously its responsibility to ensure that the executive remuneration practices of the Group drive strong performance, are aligned with the strategy and sustainability of the Group and are appropriate in the context of the external regulatory environment and the expectations of stakeholders.
Shareholder return comparison
The independent remuneration advisor, Aon also reviewed the total shareholder return comparator group used as a component of the company’s performance based LTIP awards for 2023, using volatility and correlation analysis to evaluate the appropriateness of this peer group. Its research from 2022 showed that about 80% of custom peer groups contained 30 peers or fewer, and encouraged the Company to use peer groups of 30 companies or more. It focused on three primary and four secondary criteria. Primary criteria for peers selection were their industry sector, centering on, commercial companies with innovative specialty biopharmaceutical medicines or drug candidates; their listing location, centering on China or the United States; and those with a three-year stock price that correlated with the Company. Secondary criteria were their therapeutic focus, prioritizing companies with marketed products and development pipelines focused on oncology therapies; their stock price volatility; their market capitalization, excluding micro-cap and large-cap companies; and their share trading history, excluding companies that have not been public for at least three years.
187
Executive Officer Compensation
Summary Compensation Table
The following table sets forth the non-equity compensation paid or accrued during the year ended December 31, 2023 to our chief executive officer and chief scientific officer, chief financial officer and other executive officers on an aggregate basis.
| Salary |
|
| Taxable |
| Non-taxable |
| Pension |
| |||
and fees | Bonus(3) | benefits | benefits | contributions | Total | |||||||
Name and Principal Position | ($) | ($) | ($) | ($) | ($) | ($) | ||||||
Weiguo SU |
| 872,520 | (1) | 1,500,000 |
| — | 7,885 |
| 70,759 |
| 2,451,164 | |
CHENG Chig Fung, Johnny |
| 413,123 | (2) | 508,241 |
| — | 10,897 |
| 29,889 |
| 962,150 | |
Other Executive Officers in the Aggregate |
| 2,524,727 |
| 3,439,341 |
| 31,977 | 155,128 |
| 154,902 |
| 6,306,075 |
Notes:
(1) | Amount includes director’s fees of $75,000. |
(2) | Amount includes director’s fees of $75,000. |
(3) | In connection with share options granted in the year ended December 31, 2016 under the 2015 Share Option Scheme, Dr. Weiguo Su and certain other executive officers were awarded retention bonuses payable when and if they exercised their options. During the year ended December 31, 2023, retention bonuses of $5,224,885 and $4,722,040 were settled respectively when Dr. Weiguo Su and certain other executive officers exercised such options, which amounts are not included in the table above. |
Employment Arrangements with our Executive Officers
Employment Agreements with Executive Officers at HUTCHMED Group (HK) Limited and HUTCHMED Holdings (HK) Limited
We have entered into employment agreements with each of our executive officers who are directly employed by HUTCHMED Group (HK) Limited or HUTCHMED Holdings (HK) Limited, namely Mr. Cheng Chig Fung, Johnny, Dr. Karen Jane Atkin, Dr. May Qingmei Wang, Mr. Mark Kin Hung Lee and Mr. Charles George Rupert Nixon. Under these employment agreements, our executives receive compensation in the form of salaries, discretionary bonuses, participation in the Hutchison Provident Fund retirement scheme, medical coverage under the CK Hutchison Group Medical Scheme, personal accident insurance and annual leave. None of the employment arrangements provide benefits to our executive officers upon termination. We may terminate employment by giving the executive officers three months’ prior written notice. The executive officer may also voluntarily terminate his/her employment with us upon not less than three months’ prior written notice to us.
Each executive officer has agreed, for the term of employment with us and thereafter, not to disclose or use for his/her own purposes any of our and our associated companies’ confidential information that the executive officer may develop or learn in the course of employment with us. Moreover, each of our executive officers has agreed, for the term of employment with us and for a period of 12 months thereafter, (i) not to undertake or be employed or interested directly or indirectly anywhere in Hong Kong in any activity which is similar to and competitive with our company or associated companies in which the executive officer had been involved in the period of 12 months prior to such termination and (ii) not to solicit for any employees of our company or our joint ventures or orders from any person, firm or company which was at any time during the 12 months prior to termination of such employment a customer or supplier of our company or associated companies.
Employment Agreements with Executive Officers at HUTCHMED Limited
We have entered into employment agreements with each of our executive officers who are employed directly by HUTCHMED Limited, namely Dr. Weiguo Su, Dr. Michael Ming Shi and Dr. Zhenping Wu. Under these employment agreements, we engage the executive officer on either an open-ended or a fixed term. Our executive officers receive compensation in the form of salaries, discretionary bonuses, annual leave, statutory maternity leave and nursing leave.
188
Under the terms of these agreements, we provide labor protection and work conditions that comply with the safety and sanitation requirements stipulated by the relevant PRC laws. The employment agreements prohibit the executive officers from engaging in any conduct and business activities which may compete with the business or interests of HUTCHMED Limited during the term of the executive officer’s employment. These executive officers also enjoy the Hutchison Provident Fund retirement scheme, medical coverage under the CK Hutchison Group Medical Scheme and personal accident insurance.
We may terminate an executive officer’s employment for cause at any time without notice. Termination for cause may include a serious breach of our internal rules and policies, serious negligence in the executive officer’s performance of his or her duties, an accusation or conviction of a criminal offence, acquisition of another job which materially affects the executive officer’s ability to perform his or her duties for our company and other circumstances stipulated by applicable PRC laws. We may terminate an executive officer’s employment with three months’ prior notice if the executive officer is unable to perform his or her duties (after the expiration of the prescribed medical treatment period) because of an illness or non-work-related injury or the executive officer is incompetent and remains incompetent after training or adjustment of his or her position.
The executive officer may voluntarily terminate his or her contract without cause with three months’ prior notice. The executive officer may also terminate the employment agreement immediately for cause, which includes a failure by us to provide labor protection and the work conditions as specified under the employment agreement. In case of termination for any reason, we agree to make any mandatory severance payments required by the relevant PRC labor laws.
Employment Agreement with Executive Officer at Hutchison Sinopharm
We have entered into an employment agreement with Mr. Hong Chen, one of our executive officers, who is directly employed by Hutchison Sinopharm. Under his employment agreement with Hutchison Sinopharm, Mr. Chen’s employment is for a fixed term, and he receives compensation in the form of salaries, discretionary bonuses, annual leave, statutory maternity leave and nursing leave.
Under the terms of this agreement, we provide labor protection and work conditions that comply with the safety and sanitation requirements stipulated by the relevant PRC laws. The employment agreement prohibits any conduct directly or indirectly which is harmful to Hutchison Sinopharm during the term of the employment.
We may terminate Mr. Chen’s employment for cause at any time without notice. We may also terminate the employment with prior notice and termination compensation if Mr. Chen is unable to perform his duties because of an illness or non-work-related injury or he is incompetent and remains incompetent after training or adjustment of his position. Mr. Chen may voluntarily terminate his employment agreement without cause with one month’s prior notice and immediately for cause.
189
Share Options
The following table sets forth information concerning the outstanding equity awards held by our chief executive officer and chief scientific officer, chief financial officer and other executive officers on an aggregate basis as of December 31, 2023.
Number of | Number of | Number of | Number of | ||||||||||||
unexercised shares | unexercised shares | shares issued | options lapsed/ | Option | |||||||||||
Date of grant of | which are | which are | upon exercise | cancelled in | expiration | ||||||||||
Name and Principal Position |
| share options(1) |
| vested |
| unvested |
| Exercise price |
| in 2023 |
| 2023 |
| date | |
Weiguo SU |
| Jun 15, 2016 | 3,000,000 |
| — | £ | 1.970 |
| 3,000,000 |
| — |
| Dec 19, 2023 | ||
Weiguo SU |
| Mar 27, 2017 | 1,000,000 |
| — | £ | 3.105 |
| — |
| — |
| Mar 26, 2027 | ||
Weiguo SU |
| Mar 19, 2018 | 1,000,000 |
| — | £ | 4.974 |
| — |
| — |
| Mar 18, 2028 | ||
Weiguo SU |
| Apr 28, 2020 | 592,275 (= 118,455 ADSs) |
| 197,425 (= 39,485 ADSs) | $ | 22.090 |
| — |
| — |
| Apr 27, 2030 | ||
Weiguo SU | Dec 14, 2020 | 14,220 (= 2,844 ADSs) | 4,740 (= 948 ADSs) | $ | 29.000 | — | — | Dec 13, 2030 | |||||||
Weiguo SU | Mar 26, 2021 | 141,200 (= 28,240 ADSs) | 141,200 (= 28,240 ADSs) | $ | 27.940 | — | — | Mar 25, 2031 | |||||||
Weiguo SU | Dec 14, 2021 | 12,460 (= 2,492 ADSs) | 12,470 (= 2,494 ADSs) | $ | 35.210 | — | — | Dec 13, 2031 | |||||||
Weiguo SU | May 23, 2022 | — | 861,220 (= 172,244 ADSs) | $ | 10.750 | — | — | May 22, 2032 | |||||||
CHENG Chig Fung, Johnny | Apr 28, 2020 | 301,425 (= 60,285 ADSs) | 100,475 (= 20,095 ADSs) | $ | 22.090 | — | — | Apr 27, 2030 | |||||||
CHENG Chig Fung, Johnny | Mar 26, 2021 | 120,250 (= 24,050 ADSs) | 120,250 (= 24,050 ADSs) | $ | 27.940 | — | — | Mar 25, 2031 | |||||||
CHENG Chig Fung, Johnny | May 23, 2022 | 111,650 (= 22,330 ADSs) | 334,950 (= 66,990 ADSs) | $ | 10.750 | — | — | May 22, 2032 | |||||||
CHENG Chig Fung, Johnny | Jun 5, 2023 | — | 61,700 (= 12,340 ADSs) | $ | 12.510 | — | — | Jun 4, 2033 | |||||||
Other Executive Officers in the Aggregate | Jun 15, 2016 | 2,736,860 | — | £ | 1.970 | 2,736,860 | — | Dec 19, 2023 | |||||||
Other Executive Officers in the Aggregate | Apr 20, 2018 | 701,100 | — | £ | 4.645 | — | — | Apr 19, 2028 | |||||||
Other Executive Officers in the Aggregate | Dec 11, 2019 | 400,000 | — | £ | 3.592 | — | — | Dec 10, 2029 | |||||||
Other Executive Officers in the Aggregate | Apr 28, 2020 | 1,112,250 (= 222,450 ADSs) | 370,750 (= 74,150 ADSs) | $ | 22.090 | — | — | Apr 27, 2030 | |||||||
Other Executive Officers in the Aggregate | Dec 14, 2020 | 44,895 (= 8,979 ADSs) | 14,995 (= 2,999 ADSs) | $ | 29.000 | — | — | Dec 13, 2030 | |||||||
Other Executive Officers in the Aggregate |
| Mar 26, 2021 | 383,250 (= 76,650 ADSs) |
| 383,250 (= 76,650 ADSs) | $ | 27.940 |
| — |
| — |
| Mar 25, 2031 | ||
Other Executive Officers in the Aggregate | Dec 14, 2021 | 172,770 (= 34,554 ADSs) | 172,800 (= 34,560 ADSs) | $ | 35.210 | — | — | Dec 13, 2031 | |||||||
Other Executive Officers in the Aggregate | May 23, 2022 | 172,350 (= 34,470 ADSs) | 517,050 (= 103,410 ADSs) | $ | 10.750 | — | — | May 22, 2032 | |||||||
Other Executive Officers in the Aggregate |
| Sep 13, 2022 | 375,000 (= 75,000 ADSs) |
| 1,125,000 (= 225,000 ADSs) | $ | 13.140 |
| — |
| — |
| Sep 12, 2032 | ||
Other Executive Officers in the Aggregate | Jun 5, 2023 | — | 263,200 (= 52,640 ADSs) | $ | 12.510 | — | — | Jun 4, 2033 | |||||||
Notes:
| (1) | The share options granted on or after April 28, 2020 were in the form of ADSs and the relevant exercise prices were stated in U.S. dollars per ADS. For purposes of this table, these share options are presented in the form of ordinary shares (with the corresponding number of ADSs where appropriate). Each ADS represents five ordinary shares. |
190
Long-Term Incentive Compensation
The following table sets forth information regarding performance based LTIP awards granted to our chief executive officer and chief scientific officer, chief financial officer and other executive officers on an aggregate basis in the year ended December 31, 2023.
| Maximum | ||
Aggregate | |||
Value of | |||
Name and Principal Position | LTIP awards(1)(2)(3) | ||
Weiguo SU, Chief Executive Officer and Chief Scientific Officer | $ | 3,289,770 | |
CHENG Chig Fung, Johnny, Chief Financial Officer | $ | 698,224 | |
Other Executive Officers in the Aggregate | $ | 3,457,643 | |
Notes:
| (1) | The amounts reflected in the table above represent the maximum aggregate value of all LTIP awards outstanding as of December 31, 2023. The LTIP awards are conditional upon the achievement of annual performance targets for the fiscal year 2023. The amounts reflected in the table above assume the maximum amount that may be paid under these contingent LTIP awards. The LTIP awards will be settled in a variable number of shares based on a fixed monetary amount awarded upon achievement of performance targets. An independent third-party trustee who administers the LTIP will purchase shares of our company on either the AIM or Nasdaq market which will be used to settle the LTIP awards. See “Outstanding Awards” for more details. |
| (2) | Vesting will occur two business days after the date of the announcement of our annual results for the financial year 2025. |
| (3) | Excluding performance based LTIP awards abovementioned, a non-performance based LTIP award granted to an Executive Officer in an amount of $1,500,000, for which 111,731 ADSs were allocated on September 13, 2022. 25% of the shares were/will be vested on September 13, 2023, September 13, 2024, September 13, 2025 and September 13, 2026 respectively. |
Director Compensation
The following table sets forth a summary of the compensation we paid to our directors other than Weiguo Su and Cheng Chig Fung, Johnny during 2023.
|
| Maximum Value of Non- | |||||
Fees Earned or | Performance Based LTIP | ||||||
Name of Director | Paid in Cash | Awards Granted | |||||
TO Chi Keung, Simon |
| $ | 85,000 | (1) | — | ||
Dan ELDAR |
| — |
| — | |||
Edith SHIH |
| — | (2) | — | |||
Ling YANG |
| — |
| — | |||
Paul Rutherford CARTER |
| $ | 117,000 |
| — | ||
Graeme Allan JACK |
| $ | 111,000 |
| — | ||
MOK Shu Kam, Tony | $ | 114,860 | — | ||||
Notes:
| (1) | Such director’s fees were paid to Hutchison Whampoa (China) Limited, a wholly owned subsidiary of CK Hutchison. Director’s fees received from our subsidiaries during the period Mr. To served as director that were paid to an intermediate holding company of our company are not included in the amounts above. |
| (2) | Director’s fees received from our subsidiaries during the period Ms. Shih served as director that were paid to a subsidiary of CK Hutchison are not included. |
| (3) | A director’s fee of $37,068 was paid to Dr. Karen Jean Ferrante, who retired as an independent non-executive director on May 12, 2023. |
191
Equity Compensation Schemes and Other Benefit Plans
We have two share option schemes. We refer to these collectively as the Option Schemes. Our shareholder adopted the first option scheme, or the 2005 Option Scheme, in June 2005, and it was subsequently approved by the shareholders of Hutchison Whampoa Limited, our then majority shareholder, in May 2006 and later amended by our board of directors in March 2007. This share option scheme expired in 2016 and no further share option can be granted. Furthermore, all outstanding share options granted under the 2005 Option Scheme have been fully exercised. In April 2015, our shareholders adopted the second option scheme, or the 2015 Option Scheme, which was later approved by the shareholders of CK Hutchison, the ultimate parent of our then majority shareholder, in May 2016. The 2015 Option Scheme was subsequently amended in April 2020.
We also have a long-term incentive scheme which was adopted by our shareholders in April 2015. We refer to this as our LTIP.
Our Option Schemes and LTIP each terminates on the tenth anniversary of their adoption. Each may also be terminated by its board of directors at any time. Any termination of a scheme is without prejudice to the awards outstanding at such time. Options are no longer being granted under the 2005 Option Scheme and all outstanding awards granted under the 2005 Option Scheme have been fully exercised.
The following describes the material terms of our Option Schemes and LTIP, or collectively the Schemes.
Awards and Eligible Grantees. The Option Schemes provide for the award of share options exercisable for ordinary shares or ADSs of our company to Eligible Employees (as defined in the Option Schemes) or non-executive directors (excluding any independent non-executive directors under the Option Schemes).
Under our LTIP, awards in the form of contingent rights to receive either shares purchased from the market by the scheme trustee or cash payments may be granted to the directors of our company, directors of our subsidiaries and employees of our company, subsidiaries, affiliates or such other companies as determined by our board of directors in its absolute discretion.
Scheme Administration. Our board of directors has delegated its authority for administering our Option Schemes and our LTIP to our remuneration committee. Each such plan administrator has the authority to, among other things, select participants and determine the amount and terms and conditions of the awards under the applicable Schemes as it deems necessary and proper, subject to the restrictions described in “—Restrictions on Grants” below.
Restrictions on Grants. Under the Option Schemes, grants may not be made to independent non-executive directors. Furthermore, those grants may not be made to any of our employees or directors if such person is also a director, chief executive or substantial shareholder of any of our direct or indirect parent companies which is listed on a stock exchange or any of its associates without approval by the independent non-executive directors of such parent company (excluding any independent non-executive director who is a proposed grantee). In addition, approval by our shareholders and the shareholders of such listed parent company is required if an option grant under our Option Schemes is to be made to a substantial shareholder or independent non-executive director of a listed parent company or any of its associates and, upon exercise of such grant and any other grants made during the prior 12-month period to that shareholder, that individual would receive an amount of our ordinary shares equal or greater than 0.1% of our total outstanding shares or with an aggregate value in excess of HK$5 million (equivalent to $0.6 million as of December 31, 2023).
In addition, options under our Option Schemes may not be granted to any individual if, upon the exercise of such options, the individual would receive an amount of shares when aggregated with all other options granted to such individual under the applicable Scheme in the 12-month period up to and including the grant date, that exceeds 1% of the total shares outstanding of the company granting the award on such date. There are no individual limits under our LTIP.
Under our LTIP, no grant to any director, chief executive or substantial shareholder of our company may be made without the prior approval of our independent non-executive directors (excluding an independent non-executive director who is a proposed grantee).
Vesting. Vesting conditions of options granted under the Schemes are determined by the respective board of directors at the time of grant.
192
Under our Option Schemes, if a participant has committed any misconduct or any conduct making such participant’s service terminable for cause, all options (whether vested or unvested) lapse unless the respective board of directors otherwise determines in its absolute discretion. Options may be exercised to the extent vested where a participant’s service ceases due to the participant’s death, serious illness, injury, disability, retirement at the applicable retirement age, or earlier if determined by the participant’s employer, or if a participant’s service ceases for any other reason other than for cause.
Under our LTIP, if a participant’s employment or service with our company or its subsidiaries is terminated for cause or if the participant breaches certain provisions in our LTIP restricting the transfer of awards by grantees and imposing non-competition obligations on grantees, all unvested awards are automatically cancelled. Where a participant’s employment or service ceases for any reason other than the reasons listed above (including due to the participant’s resignation, retirement, death or disability or upon the non-renewal of such participant’s employment or service agreement other than for cause), our board of directors may determine at its discretion whether unvested awards shall be deemed vested.
Exercise Price. The exercise price for each share pursuant to the initial options granted under the 2005 Option Scheme was a price determined by our board of directors at the date of grant, and for grants made thereafter, the exercise price was the Market Value of a share at the date of grant (as defined in our Option Schemes).
The exercise price for each share pursuant to the options granted under the 2015 Option Scheme must be the Market Value of a share at the date of grant (as defined in our Option Schemes).
Non-transferability of Awards. Awards may not be transferred except in the case of a participant’s death by the terms of each Scheme.
Takeover or Scheme of Arrangement. In the event of a general or partial offer for the shares of our company under our Option Schemes, whether by way of takeover, offer, share repurchase offer, or scheme of arrangement, the affected company is required to use all reasonable endeavors to procure that such offer is extended to all holders of options granted by such company on the same terms as those applying to shareholders. Both vested and unvested options may be exercised up until (i) the closing date of any such offer and (ii) the record date for entitlements under a scheme of arrangement, and will lapse thereafter. Certain options may also be exercised on a voluntary winding up of our company.
Under our LTIP, in the event of a general offer for all the shares of our company, whether by way of takeover or scheme of arrangement, or if our company is to be voluntarily wound up, our board of directors shall determine in its discretion whether outstanding unvested awards will vest and the period within which such awards will vest.
Amendment. Our Option Schemes require that amendments of a material nature only be made with the approval of our shareholders.
Our board of directors may alter the terms of our LTIP, but amendments which are of a material nature cannot take effect without shareholders’ approval, unless the changes take effect automatically under the terms of our LTIP.
Authorized Shares. Under our 2015 Option Scheme, our board of directors may “refresh” the scheme limit from time to time provided that the total number of shares which may be issued upon exercise of all options to be granted under our Option Schemes shall not exceed 10% of our total shares outstanding on such date. In addition, the limit on the number of shares which may be issued upon exercise of all outstanding options granted and not yet exercised under the 2015 Option Scheme and any options granted and not yet exercised under any other schemes must not exceed 10% of the outstanding shares of the company in issue from time to time. In April 2020, our shareholders approved a refresh of the 2015 Option Scheme.
Following the 2015 Option Scheme refresh discussed above, subject to certain adjustments for share splits, share consolidations and other changes in capitalization, the maximum number of shares that may be issued upon exercise of all options granted may not in the aggregate exceed 5% of our shares outstanding on April 27, 2020. Share awards under our LTIP may not exceed 5% of our shares outstanding on the adoption date of our LTIP.
193
Outstanding Awards and Grants of Awards
Share options outstanding under the 2005 Option Scheme
The 2005 Option Scheme expired in 2016, and no further share options can be granted under it. Furthermore, all outstanding share options granted thereunder have been fully exercised in 2023.
Share options outstanding and grants made in 2023 under the 2015 Option Scheme
As of December 31, 2023, options to purchase an aggregate of 29,536,655 ordinary shares, representing approximately 3.4% of our outstanding share capital, at a weighted average exercise price of £3.59 ($4.57) per ordinary share and an expiration date of 10 years from the respective date of grant remained outstanding under the 2015 Option Scheme. In the year ended December 31, 2023, we granted options to purchase an aggregate of 1,221,900 ordinary shares, representing approximately 0.1% of our outstanding share capital, at an exercise price of £1.96 ($2.50) per share under the 2015 Option Scheme. For the share options granted to Weiguo Su in 2022, the exercise of the share options is conditional upon the fulfilment of certain performance targets relating to the Group over the financial year of 2022 to 2024. Vesting will occur two business days after the date of announcement of the annual results of the Company for the financial year ending December 31, 2024. The other options vest in equal instalments of 25% over a four-year period.
Grants and vesting of LTIPs
In the year ended December 31, 2023, we granted performance based awards under our LTIP to two of our executive directors and 839 employees, giving them a conditional right to receive ordinary shares to be purchased by the third-party trustee up to an aggregate maximum cash amount of $54,935,768. These awards are related to the achievement of performance targets and will vest two business days after the date of the announcement of our annual results for the financial year 2025. For additional information on LTIP awards held by our executive officers, please see “B. Compensation—Executive Officer Compensation—Long-Term Incentive Compensation.” For additional information on LTIP awards to our directors, please see “B. Compensation—Director Compensation.”
Vesting of our LTIP awards will also depend upon the award holder’s continued employment or continued service on our board, as the case may be.
In the year ended December 31, 2023, an aggregate of 876,557 ADSs were given to award holders upon the vesting of performance based LTIP awards, and 66,284 ADSs were given to award holders upon the vesting of non-performance based LTIP awards.
C. Board Practices.
Our board of directors consists of nine directors including three executive directors, three non-executive directors and three independent non-executive directors. Pursuant to a relationship agreement dated April 21, 2006, and amended and restated on June 13, 2019, by and between our company and Hutchison Whampoa (China) Limited, a parent company of Hutchison Healthcare Holdings Limited, or the Relationship Agreement, our board of directors must consist of at least one director who is independent of the CK Hutchison group if Hutchison Whampoa (China) Limited is entitled to cast at least 50% votes eligible to be cast on a poll vote at a general meeting of our company. The Relationship Agreement will continue in effect until our ordinary shares cease to be traded on the AIM market or the CK Hutchison group individually or collectively ceases to hold at least 30% of our shares.
Under the Articles of Association, our directors are subject to retirement at an annual general meeting at least once every three years and hold office until such time as they wish to retire and not offer themselves up for re-election, are not re-elected by the shareholders, or are removed from office by ordinary resolution at a general meeting of the shareholders. Under our Articles of Association, a director will be vacated if, among other things, the director (i) becomes bankrupt or has a receiving order made against him or suspends payment or compounds with his creditors; or (ii) becomes of unsound mind. For information regarding the period during which our officers and directors have served in their respective positions, please see Item 6.A. “Directors and Senior Management.”
Board Committees
Our board of directors has established an audit committee, remuneration committee, technical committee, nomination committee and sustainability committee.
194
Audit Committee
Our audit committee consists of Graeme Allan Jack, Paul Rutherford Carter and Mok Shu Kam, Tony, with Graeme Allan Jack serving as chairman of the committee. Graeme Allan Jack, Paul Rutherford Carter and Mok Shu Kam, Tony each meets the independence requirements under the rules of the Nasdaq Stock Market and under Rule 10A-3 under the Exchange Act. We have determined that Graeme Allan Jack is an “audit committee financial expert” within the meaning of Item 407 of Regulation S-K. All members of our audit committee meet the requirements for financial literacy under the applicable rules and regulations of the SEC and the Nasdaq Stock Market.
Although we are a foreign private issuer, we are required to comply with Rule 10A-3 of the Exchange Act, relating to audit committee composition and responsibilities. Rule 10A-3 provides that the audit committee must have direct responsibility for the nomination, compensation and choice of our auditor, as well as control over the performance of their duties, management of complaints made, and selection of consultants. Under Rule 10A-3, if the governing law or documents of a listed issuer require that any such matter be approved by the board of directors or the shareholders of the company, the audit committee’s responsibilities or powers with respect to such matter may instead be advisory. Our Articles of Association provide that the appointment of our auditor must be decided by our shareholders at our annual general meeting or at a subsequent extraordinary general meeting in each year.
The audit committee formally meets at least twice a year and otherwise as required. The audit committee’s purpose is to oversee our accounting and financial reporting process and the audit of our financial statements. Our audit committee’s primary duties and responsibilities are to:
| ● | monitor the integrity of our financial statements, our annual and half-year reports and accounts and our announcements of interim or final results; |
| ● | provide advice, where requested by the board of directors, on whether the annual report and accounts, taken as a whole, are fair, balanced and understandable, and provide the information necessary for shareholders to assess our company’s position and performance, business model and strategy; |
| ● | review significant financial reporting issues and the judgments which they contain; |
| ● | review, whenever practicable without being inconsistent with any requirement for prompt reporting under applicable listing rules, other statements containing financial information such as significant financial returns to regulators and release of price sensitive information first where board of director approval is required; and |
| ● | review and challenge where necessary: |
| ● | the consistency of, and any changes to, accounting policies both on a year-on-year basis and across our company; |
| ● | the methods used to account for significant or unusual transactions where different approaches are possible; |
| ● | whether our company has followed appropriate accounting standards and made appropriate estimates and judgments, taking into account the views of the external auditor; |
| ● | the clarity of the disclosure in our financial reports and the context in which statements are made; and |
| ● | all material information presented with the financial statements, such as any operations and financial review and any corporate governance statements (insofar as it relates to the audit and risk management). |
In relation to our internal controls and risk management systems, our audit committee, among other things:
| ● | reviews the effectiveness of our internal control and risk management systems; |
195
| ● | reviews the policies and procedures for the identification, assessment and reporting of financial and non-financial risks and our management of those risks in accordance with the requirements of the Sarbanes-Oxley Act and other applicable laws, rules and regulations and the applicable requirements of any stock exchange; |
| ● | approves the appointment and removal of the head of the internal audit function; |
| ● | ensures our internal audit function has adequate standing and resources and is free from management or other restrictions; |
| ● | reviews and monitors our executive management’s responsiveness to the findings and recommendations of the internal audit function; and |
| ● | reviews with management and our independent auditors the adequacy and effectiveness of our internal control over financial reporting and disclosure controls and procedures. |
In relation to our external auditor, our audit committee, among other things:
| ● | recommends the appointment, reappointment or removal of the external auditor and considers any issues relating to their resignation, dismissal, remuneration or terms of engagement, subject to approval by the shareholders; |
| ● | considers and monitors the external auditor’s independence, objectivity and effectiveness; |
| ● | reviews and monitors the effectiveness of the audit process, considering relevant ethical or professional requirements; |
| ● | develops and implements policy on the engagement of the external auditor to provide non-audit services, taking into any relevant ethical guidance; and |
| ● | pre-approves the external auditors’ annual audit fees and the nature and scope of proposed audit coverage, subject to approval by our shareholders. |
The audit committee is authorized to obtain, at our company’s expense, reasonable outside legal or other professional advice on any matters within the scope of its responsibilities.
Remuneration Committee
Our remuneration committee consists of Paul Rutherford Carter, Graeme Allan Jack and To Chi Keung, Simon, with Paul Rutherford Carter serving as chairman of the committee. The remuneration committee is responsible for considering all material elements of remuneration policy and remuneration and incentives of our executive directors and key employees with reference to independent remuneration research and professional advice. The remuneration committee meets formally at least once each year and otherwise as required and make recommendations to our board of directors on the framework for executive remuneration and on proposals for the granting of share options and other equity incentives. Our board of directors is responsible for implementing these recommendations and agreeing the remuneration packages of individual directors. No director is permitted to participate in discussions or decisions concerning his or her own remuneration.
Technical Committee
Our technical committee consists of Mok Shu Kam, Tony, Paul Rutherford Carter, To Chi Keung, Simon and Weiguo Su, with Mok Shu Kam, Tony serving as chairman of the committee. The technical committee’s responsibility is to consider, from time to time, matters relating to the technical aspects of the research and development activities of our Oncology/Immunology operations. It invites such executives as it deems appropriate to participate in meetings from time to time.
196
Nomination Committee
Our nomination committee consists of Mok Shu Kam, Tony, Graeme Allan Jack and To Chi Keung, Simon, with Mok Shu Kam, Tony serving as chairman of the committee. Our nomination committee reviews the structure, size, diversity profile and skills set of the board against its needs and makes recommendations on the composition of the board to achieve our corporate strategy as well as promote shareholder value. It facilitates the board in the conduct of the selection and nomination of directors, makes recommendations to the board on the appointment or reappointment of directors and succession planning for directors. It also assesses director independence having regard to the criteria under the applicable corporate governance code, SEC or stock exchange rules.
Sustainability Committee
Our sustainability committee consists of Edith Shih, Cheng Chig Fung, Johnny and Mok Shu Kam, Tony, with Edith Shih serving as chairman of the committee. The sustainability committee is responsible for strengthening our corporate governance and reporting framework. It advises our board of directors and management on and oversees the development and implementation of our corporate social responsibility and sustainability initiatives, including reviewing related policies and practices as well as assessing and making recommendations on matters pertaining to our sustainability governance, strategies, planning and risk management.
Hong Kong Corporate Governance Code
Following the listing on the SEHK on June 30, 2021, our board of directors has adopted the Corporate Governance Code (“Hong Kong Corporate Governance Code”) contained in Appendix C1 of the Rules Governing the Listing of Securities on SEHK in replacement of the U.K. Corporate Governance Code 2018 and is in compliance with all code provisions of the Hong Kong Corporate Governance Code.
Code of Ethics
Our board of directors has adopted a code of ethics to set standards for our directors, officers and employees as are reasonably necessary to promote (i) honest and ethical conduct, including the ethical handling of actual or apparent conflicts of interest between personal and professional relationships; (ii) full, fair, accurate, timely and understandable disclosure in the reports and documents that we file or submit to the applicable stock exchanges, and in any other public communications; (iii) compliance with applicable governmental and regulatory laws, rules, codes and regulations; (iv) prompt internal reporting of any violations of the code of ethics; and (v) accountability for adherence to the code of ethics.
Code of Ethics for Business Partners
Our board of directors has adopted a code of ethics for our business partners, including our suppliers, vendors, customers, agents, contractors, joint venture partners and representatives. This code of ethics contains general guidelines to promote the standards outlined in our internal code of ethics as described above.
Complaints Procedures / Whistleblowing Policy
Our board of directors has adopted procedures for the confidential receipt, retention, and treatment of complaints from, or concerns raised by, employees regarding accounting, internal accounting controls and auditing matters as well as illegal or unethical matters. The complaint procedures are reviewed by the audit committee from time to time as warranted to ensure their continuing compliance with applicable laws and listing standards as well as their effectiveness.
Policy on Personal Information Governance
Our board of directors has adopted a policy on personal information governance which sets out our governance framework for the safeguard of personal information of employees, customers and other relevant personal information subjects. The senior management of each group company is accountable for the effective implementation of this policy.
197
Information Security Policy
Our board of directors has adopted an information security policy to define and help communicate the common policies for information confidentiality, integrity and availability to be applied to us and our joint ventures. The purpose of the information security policy is to ensure business continuity by preventing and minimizing the impact of security risks within our company and our joint ventures. Our information security policy applies to all of our and our joint ventures’ business entities across all countries. It applies to the creation, communication, storage, transmission and destruction of all different types of information. It applies to all forms of information, including but not limited to electronic copies, hardcopy, and verbal disclosures whether in person, over the telephone, or by other means.
Code on Dealings in Shares
Our board of directors has adopted a policy on the handling of material inside information, consisting of information which is either “inside information” under the EU Market Abuse Regulation (Regulation (EU) 596/2014), or MAR, or “material non-public information” under U.S. law. This policy, among other things, prohibits any employees, directors, other persons discharging managerial responsibilities or their connected persons dealing in our securities or their derivatives, or those of our collaborators, business partners, suppliers and customers, while in possession of material inside information. Certain members of our senior management or staff, including persons discharging managerial responsibilities, and their connected persons are subject to additional compliance requirements which are outlined in the code (including but not limited to obtaining written pre-clearance from designated members of management prior to any dealing in any such securities is allowed).
Board Diversity Policy
Our board of directors has established a board diversity policy as our board of directors recognizes the benefits of a board of directors that possesses a balance of skills, experience, expertise, independence and knowledge and diversity of perspectives appropriate to the requirements of our businesses.
We maintain that appointment to our board of directors should be based on merit that complements and expands the skills, experience, expertise, independence and knowledge of the board of directors as a whole, taking into account gender, age, professional experience and qualifications, cultural and educational background, and any other factors that our board of directors might consider relevant and applicable from time to time towards achieving a diverse board of directors. See also”—Directors and Senior Management—Board Diversity.”
D. Employees.
As of December 31, 2021, 2022 and 2023, we had 1,759, 2,025 and 1,988 full-time employees, respectively. None of our employees are represented by labor unions or covered by collective bargaining agreements. The number of employees by function as of the end of the period for our fiscal years ended December 31, 2021, 2022 and 2023 was as follows:
| 2023 |
| 2022 |
| 2021 | |
By Function: |
|
|
|
|
|
|
Oncology/Immunology |
| 960 |
| 1,022 |
| 891 |
Other Ventures |
| 987 |
| 960 |
| 820 |
Corporate Head Office |
| 41 |
| 43 |
| 48 |
Total |
| 1,988 |
| 2,025 |
| 1,759 |
As of December 31, 2023, a total of 125 employees on our Oncology/Immunology research and development team have M.D. or Ph.D. degrees. Additionally, our Other Ventures joint venture Shanghai Hutchison Pharmaceuticals employed a total of 3,005 full time employees as of December 31, 2023, and such employees are represented by labor unions and covered by collective bargaining agreements. To date, we have not experienced any strikes, labor disputes or industrial actions which had or would have a material effect on our business, and consider our relations with the union and employees to be good.
198
We recognize the importance of high-quality employees in sustaining market leadership. Salary and benefits are kept at competitive levels, while individual performance is rewarded within the general framework of the salary, bonus and incentive system of our company, which is reviewed annually. Employees are provided with a wide range of benefits that include medical coverage, provident funds and retirement plans and long service awards. We stress the importance of staff development and provides training programs on an ongoing basis. Employees are also encouraged to play an active role in community care activities.
E. Share Ownership.
See Item 6.B. “Compensation” and Item 7 “Major Shareholders and Related Party Transactions.”
F. Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation.
Not applicable.
ITEM 7. MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS
A. Major Shareholders.
We had 871,256,270 ordinary shares outstanding as of February 15, 2024. The following table and accompanying footnotes set forth information relating to the beneficial ownership of our ordinary shares as of February 15, 2024 by:
| ● | each person, or group of affiliated persons, known by us to beneficially own more than 5% of our outstanding ordinary shares; |
| ● | each of our directors; and |
| ● | each of our named executive officers. |
199
Our major shareholders do not have voting rights that are different from our shareholders in general. Beneficial ownership is determined in accordance with the rules and regulations of the SEC. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, we have included shares that the person has the right to acquire within 60 days of February 15, 2024, including through the exercise of any option, warrant, or other right or the conversion of any other security. These shares, however, are not included in the computation of the percentage ownership of any other person.
|
| Number of |
| ||||
Number of | American | ||||||
Ordinary | Depositary | Percent of Issued | |||||
Name of beneficial owner | Share held | Share held | Share Capital** | ||||
Executive Officers and Directors: |
|
|
|
|
|
| |
Weiguo SU |
| * |
| * | * | ||
CHENG Chig Fung, Johnny |
| * |
| * | * | ||
TO Chi Keung, Simon |
| * |
| * |
| * | |
Edith SHIH |
| * |
| * |
| * | |
Dan ELDAR |
| * | * | * | |||
Ling YANG |
| — |
| — |
| — | |
MOK Shu Kam, Tony |
| — |
| * |
| * | |
Paul Rutherford CARTER |
| * |
| * |
| * | |
Graeme Allan JACK |
| — |
| * |
| * | |
Michael Ming SHI |
| — |
| * |
| * | |
Karen Jane ATKIN |
| — |
| * |
| * | |
Zhenping WU | * | * | * | ||||
Mark Kin Hung LEE | * | * | * | ||||
May Qingmei WANG |
| * | * | * | |||
Hong CHEN | * | * | * | ||||
Charles George Rupert NIXON | * | * | * | ||||
All Executive Officers and Directors as a Group | 12,288,882 | 1,525,217 | 2.3 | % | |||
Principal Shareholders: | |||||||
Hutchison Healthcare Holdings Limited(1) | 332,478,770 | — | 38.2 | % |
* | Less than 1% of our total outstanding ordinary shares. |
** | For each person and group included in this table, percentage ownership is calculated by dividing the number of shares beneficially owned by such person or group by the sum of (i) 871,256,270 ordinary shares outstanding as of February 15, 2024, and (ii) the number of ordinary shares or ADSs underlying share options held by such person or group that are exercisable within 60 days of February 15, 2024. |
| (1) | Hutchison Healthcare Holdings Limited, a British Virgin Islands company, is an indirect wholly owned subsidiary of CK Hutchison, a company incorporated in the Cayman Islands and listed on The Hong Kong Stock Exchange. The registered address of Hutchison Healthcare Holdings Limited is Vistra Corporate Services Centre, Wickhams Cay II, Road Town, Tortola VG1110, British Virgin Islands. |
As of February 15, 2024, based on public filings with the SEC, AIM and SEHK, there are no other major shareholders holding 5% or more of our ordinary shares or ADSs representing ordinary shares except as described above. As of February 15, 2024, there were three ordinary shareholders of record with an address in the United States. Deutsche Bank Trust Company America, as depositary of our ADS program, held 107,984,025 ordinary shares as of that date in the name of DB London (Investors Services) Nominees Limited.
To our knowledge, except as disclosed above, we are not owned or controlled, directly or indirectly, by another corporation, by any foreign government or by any other natural or legal person or persons, severally or jointly. To our knowledge, there are no arrangements or operations of which may at a subsequent date result in us undergoing a change in control. Our major shareholders do not have different voting rights than any of our other shareholders.
200
B. Related Party Transactions.
Relationship with CK Hutchison
Letters of awareness with respect to loans
CK Hutchison has provided letters of awareness to certain of our lenders stating that it is aware that loan facilities have been provided to us and that its current intention is that for so long as amounts are outstanding under such loan facilities, it will not reduce its direct or indirect shareholding as to result in it ceasing to be the single largest indirect shareholder of our company.
Relationship Agreement with the CK Hutchison group
We entered into a relationship agreement dated April 21, 2006, which was amended and restated on June 13, 2019 with effect from June 3, 2015, with Hutchison Whampoa (China) Limited, which is an indirect wholly owned subsidiary of CK Hutchison, with a view to ensuring that our company is capable of carrying on its business independent of the CK Hutchison group. We refer to this agreement as the Relationship Agreement. The Relationship Agreement provides, among other things, that all transactions between any of us or our joint ventures, on the one hand, and the CK Hutchison group, on the other, will be on an arm’s length basis, on normal commercial terms and in a manner consistent with the AIM Rules. The Relationship Agreement further provides that the approval of our board of directors shall be required for any transaction between any of us or our joint ventures, on one hand, and the CK Hutchison group, on the other hand and that in approving any such transaction, our board of directors must consist of at least one director who is independent of CK Hutchison. Our board of directors must consist of at least one director who is independent of the CK Hutchison group if Hutchison Whampoa (China) Limited is entitled to cast at least 50% votes eligible to be cast on a poll vote at a general meeting of our company, see Item 6.C. “Directors, Senior Management and Employees—Board Practices.” Hutchison Whampoa (China) Limited has also agreed to procure that each member of the Hutchison Whampoa (China) Limited group will not exercise its voting rights and powers so as to amend our Memorandum or Articles of Association in a manner which is inconsistent with the Relationship Agreement. The Relationship Agreement will continue to be effective until the first to occur of: (i) our shares ceasing to be traded on the AIM market or; (ii) the CK Hutchison group individually or collectively cease to hold or control the exercise of at least 30% or more of the rights to vote at our general meetings.
Products sold to group companies of CK Hutchison
We have entered into agreements with members of the CK Hutchison group, including the retail grocery and pharmacy chains PARKnSHOP and Watsons which are owned and operated by the A.S. Watson Group, an indirect subsidiary of CK Hutchison, in respect of the distribution of certain of our consumer health products. For the year ended December 31, 2023, sales of our products to members of the CK Hutchison group amounted to $1.9 million (amounts covered from January until divestment of Hutchison Hain Organic on December 7, 2023). In addition, for the year ended December 31, 2023, we paid approximately $0.2 million (amounts covered from January until divestment of Hutchison Hain Organic on December 7, 2023) to members of the CK Hutchison group for the provision of marketing services associated with these products. Our sales to CK Hutchison group companies are made pursuant to purchase orders issued by each purchaser periodically, the terms of which are on an arm’s length basis on normal commercial terms. Following the disposal of the Group’s 100% interest in Hutchison Hain Organic and 100% interest in HUTCHMED Science Nutrition, the aforementioned agreements were terminated in December 2023.
See Item 3.D. “Risk Factors—Risks Relating to Our Dependence on Third Parties—There is no assurance that the benefits currently enjoyed by virtue of our association with CK Hutchison will continue to be available” for more information on the risks associated with our relationship with CK Hutchison’s group companies.
Intellectual property licensed by the CK Hutchison group
We conduct our business using trademarks with various forms of the “Hutchison”, “Chi-Med”, “Hutchison China MediTech”, “HUTCHMED”, “Elunate” and “Sulanda” brands, the logos used by HUTCHMED Limited, as well as domain names incorporating some or all of these trademarks. We have entered into a brand license agreement dated April 21, 2006 (as amended and restated on June 13, 2019 with effect from June 3, 2015 and as further amended and restated on June 15, 2021 with effect from March 4, 2021) with Hutchison Whampoa Enterprises Limited, which is an indirect wholly owned subsidiary of CK Hutchison, pursuant to which we have been granted a non-exclusive, non-transferrable, royalty-free right to use the “Hutchison,” “Hutchison China MediTech”, “Chi-Med”, “HUTCHMED” trademarks, domain names and other intellectual property rights owned by the CK Hutchison group in connection with the operation of our business worldwide. We refer to this amended and restated agreement as the Brand License Agreement. We are also permitted to sub-license such intellectual property rights to our affiliates.
201
The Brand License Agreement contains provisions on quality control pursuant to which we are obliged to use the brands and related materials in compliance with the brand guidelines, industry best practice and other quality directives issued by Hutchison Whampoa Enterprises Limited from time to time. Under this agreement, we assign all intellectual property rights, including future copyrights in any works incorporating brand-related material or translations thereof, to Hutchison Whampoa Enterprises Limited (subject to any third-party rights).
Hutchison Whampoa Enterprises Limited may terminate the Brand License Agreement (or any sub-license) if, among other things, we commit a material breach of the agreement, or within any twelve-month period aggregate direct or indirect shareholding in our company held by CK Hutchison, our indirect shareholder, is reduced to less than 35%, 30% or 20%. On termination of the Brand License Agreement, we (and any sub-licensees) must immediately cease using the brands and are obliged to withdraw from the sale of any products bearing the brands; provided that if the agreement is terminated following a change in CK Hutchison’s aggregate direct or indirect shareholding in our company, we will have a six-month transitional period during which we can continue to use the licensed rights.
On December 21, 2023, the brand license royalty agreement with Hutchison Whampoa Enterprises Limited was renewed with effect from January 1, 2024 for a period of three years up to and including December 31, 2026, pursuant to which we will pay an annual fee of HK$12 million (up to an aggregate royalty payable of no more than HK$120 million) in consideration of the grant of the royalty-free right to use the trademarks owned by Hutchison Whampoa Enterprises Limited to Hutchison Baiyunshan and HBYS JV companies upon the completion of the disposal of shareholding interest in Hutchison Baiyunshan.
Sharing of services with the CK Hutchison group
Pursuant to an amended and restated services agreement dated January 1, 2016 between us and Hutchison Whampoa (China) Limited, an indirect wholly owned subsidiary of CK Hutchison, we share certain services with and receive operational support from the CK Hutchison group including, among others, legal and regulatory services, company secretarial support services, tax and internal audit services, shared use of accounting software system and related services, participation in the CK Hutchison group’s pension, medical and insurance plans, participation in the CK Hutchison group’s procurement projects with third-party vendors/suppliers, other staff benefits and staff training services, company functions and activities and operation advisory and support services. We refer to this amended and restated agreement as the Services Agreement. The Services Agreement replaces our prior services agreement with Hutchison Whampoa (China) Limited, dated April 21, 2006, which had substantially similar terms. We pay a management fee to Hutchison Whampoa (China) Limited for the provision of such services. In addition, we make payments under the Services Agreement to Hutchison Whampoa (China) Limited for our executive offices in Hong Kong. Furthermore, pursuant to the terms of the Services Agreement, Hutchison Whampoa (China) Limited charges us management fees and other costs through Hutchison Healthcare Holdings Limited, its wholly owned subsidiary.
The Services Agreement may be terminated by either party by giving three months’ written notice. Hutchison Whampoa (China) Limited may also immediately terminate if its shareholding in our company falls below 30%. The services provided under the Services Agreement are provided on an arm’s length basis, on normal commercial terms.
Any amount unpaid after 30 days accrues interest at the rate of 1.5% per annum. In the year ended December 31, 2023, we paid a management fee of approximately $1.0 million under the Services Agreement. As of December 31, 2023, we had $0.5 million in unpaid fees outstanding to Hutchison Whampoa (China) Limited.
Agreements with Our Directors and Executive Officers
Director and Executive Officer Compensation
See Item 6.B. “Compensation—Executive Officer Compensation” and “Compensation—Director Compensation” for a discussion of our compensation of directors and executive officers.
Equity Compensation
See Item 6.B. “Compensation—Equity Compensation Schemes and Other Benefit Plans.”
202
Employment Agreements
We have entered into employment agreements with our executive officers. For more information regarding these agreements, see Item 6.B. “Compensation—Executive Officer Compensation—Employment Arrangements with our Executive Officers.” No director has a service contract with us not terminable by us within one year without payment of compensation (other than statutory compensation).
Indemnification Agreements
We have entered into indemnification agreements with each of our directors and executive officers. We also maintain a general liability insurance policy which covers certain liabilities of our directors and executive officers arising out of claims based on acts or omissions in their capabilities as directors or officers.
C. Interests of Experts and Counsel.
Not applicable.
ITEM 8. FINANCIAL INFORMATION
A. Consolidated Financial Statements and Other Financial Information.
See Item 18 “Financial Statements.”
A.7 Legal Proceedings.
There are no material legal proceedings pending or, to our knowledge, threatened against us. We are also not aware of any incidents of non-compliance with laws and regulations that may have a significant impact on us which would have a material adverse effect on our financial condition or results of operations. From time to time we become subject to legal proceedings and claims in the ordinary course of our business, including claims of alleged infringement of patents and other intellectual property rights. Such legal proceedings or claims, even if not meritorious, could result in the expenditure of significant financial and management resources.
A.8 Dividend Policy.
We have never declared or paid dividends on our ordinary shares. We currently expect to retain all future earnings for use in the operation and expansion of our business and do not have any present plan to pay any dividends. The declaration and payment of any dividends in the future will be determined by our board of directors in its discretion, and will depend on a number of factors, including our earnings, capital requirements, overall financial condition, and contractual restrictions.
B. Significant Changes.
We have not experienced any significant changes since the date of our audited consolidated financial statements included in this annual report.
ITEM 9. THE OFFER AND LISTING
Not applicable except for Item 9.A.4 and Item 9.C.
Our ADSs are listed on the Nasdaq Global Select and our ordinary shares are admitted to trading on the AIM market under the symbol “HCM.” In addition, our ordinary shares are listed on the SEHK under stock code “0013.”
203
ITEM 10. ADDITIONAL INFORMATION
A. Share Capital.
Not applicable.
B. Memorandum and Articles of Association.
On May 29, 2019, we conditionally adopted an amended and restated memorandum and articles of association by special resolution and effective on the date on which our shares are listed on the SEHK (the “Amended and Restated Articles”). On June 30, 2021, the listing date of our shares on the SEHK, the Amended and Restated Articles replaced the then existing articles of association of our company adopted by at the annual general meeting held on April 27, 2020.
C. Material Contracts.
Except as otherwise disclosed in this annual report (including the exhibits hereto), we are not currently, and have not been in the last two years, party to any material contract, other than contracts entered into in the ordinary course of our business.
D. Exchange Controls.
Foreign currency exchange in the PRC is primarily governed by the Foreign Exchange Administration Rules issued by the State Council on January 29, 1996 and effective as of April 1, 1996 (and amended on January 14, 1997 and August 5, 2008) and the Regulations of Settlement, Sale and Payment of Foreign Exchange which came into effect on July 1, 1996.
Under the Foreign Exchange Administration Rules, renminbi is freely convertible for current account items, including the distribution of dividends payments, interest payments, and trade and service-related foreign exchange transactions. Conversion of renminbi for capital account items, such as direct investment, loans, securities investment and repatriation of investment, however, is still generally subject to the approval or verification of the SAFE.
Under the Regulations of Settlement, Sale and Payment of Foreign Exchange, foreign invested enterprises including wholly foreign owned enterprises, may buy, sell or remit foreign currencies only at those banks that are authorized to conduct foreign exchange business after providing such banks with valid commercial supporting documents and, in the case of capital account item transactions, after obtaining approvals from the SAFE. Capital investments by foreign invested enterprises outside the PRC are also subject to limitations, which include approvals by the MOFCOM, the SAFE and the NDRC.
In March 2015, the SAFE released the Circular on Reforming the Management Approach regarding the Foreign Exchange Capital Settlement of Foreign-invested Enterprises, or FIEs, or the Foreign Exchange Capital Settlement Circular, which became effective from June 1, 2015. This circular replaced the SAFE’s previous related circulars, including the Circular on Issues Relating to the Improvement of Business Operation with Respect to the Administration of Foreign Exchange Capital Payment and Settlement of Foreign Invested Enterprises. The Foreign Exchange Capital Settlement Circular clarifies that FIEs may settle a specified proportion of their foreign exchange capital in banks at their discretion, and may choose the timing for such settlement. The proportion of foreign exchange capital to be settled at FIEs’ discretion for the time being is 100% and the SAFE may adjust the proportion in due time based on the situation of international balance of payments. The circular also stipulates that FIEs’ usage of capital and settled foreign exchange capital shall comply with relevant provisions concerning foreign exchange control and be subject to the management of a negative list. The Notice of the SAFE on Policies for Reforming and Regulating Control over Foreign Exchange Settlement under the Capital Account, which became effective from June 9, 2016 and supplements the Foreign Exchange Capital Settlement Circular, stipulates that the FIEs’ capital and Renminbi capital gained from the settlement of foreign exchange capital may not be directly or indirectly used for expenditure beyond the business scope of the FIEs or as prohibited by laws and regulations of the PRC. Such capital also may not be directly or indirectly used for granting loans to non-affiliated enterprises except as permitted by the business scope of the FIE or for construction or purchase of real estate other than self-use (exceptions only apply for real estate enterprises).
204
In addition, the payment of dividends by entities established in the PRC is subject to limitations. Regulations in the PRC currently permit payment of dividends only out of accumulated profits as determined in accordance with accounting standards and regulations in the PRC. Each of our PRC subsidiaries that is a domestic company is also required to set aside at least 10.0% of its after-tax profit based on PRC accounting standards each year to its general reserves or statutory capital reserve fund until the accumulative amount of such reserves reach 50.0% of its respective registered capital. These restricted reserves are not distributable as cash dividends. In addition, if any of our PRC subsidiaries or joint ventures incurs debt on its own behalf in the future, the instruments governing the debt may restrict its ability to pay dividends or make other distributions to us.
For more information about foreign exchange control, see Item 3.D. “Risk Factors—Other Risks and Risks Relating to Doing Business in China—Restrictions on currency exchange may limit our ability to receive and use our revenue effectively.”
E. Taxation.
The following is a general summary of certain PRC, Hong Kong, Cayman Islands and U.S. federal income tax consequences relevant to the acquisition, ownership and disposition of our ADSs. The discussion is not intended to be, nor should it be construed as, legal or tax advice to any particular individual. The discussion is based on laws and relevant interpretations thereof in effect as of February 27, 2024, all of which are subject to change or different interpretations, possibly with retroactive effect. The discussion does not address U.S. state or local tax laws, or tax laws of jurisdictions other than the PRC, Hong Kong, the Cayman Islands and the United States. You should consult your own tax advisors with respect to the consequences of acquisition, ownership and disposition of our ADSs and ordinary shares.
Taxation in the PRC
PRC Enterprise Income Tax
Under the EIT Law, which was promulgated on March 16, 2007 and subsequently amended on February 24, 2017 and December 29, 2018, and its implementation rules which became effective on January 1, 2008 and subsequently amended on April 23, 2019, the standard tax rate of 25% applies to all enterprises (including FIEs) with exceptions in special situations if relevant criteria are met and subject to the approval of the PRC tax authorities.
An enterprise incorporated outside of the PRC whose “de facto management bodies” are located in the PRC is considered a “resident enterprise” and will be subject to a uniform EIT rate of 25% on its global income. In April 2009, the SAT, in Circular 82, specified certain criteria for the determination of what constitutes “de facto management bodies.” If all of these criteria are met, the relevant foreign enterprise will be deemed to have its “de facto management bodies” located in the PRC and therefore be considered a resident enterprise in the PRC. These criteria include: (a) the enterprise’s day-to-day operational management is primarily exercised in the PRC; (b) decisions relating to the enterprise’s financial and human resource matters are made or subject to approval by organizations or personnel in the PRC; (c) the enterprise’s primary assets, accounting books and records, company seals, and board and shareholders’ meeting minutes are located or maintained in the PRC; and (d) 50% or more of voting board members or senior executives of the enterprise habitually reside in the PRC. Although Circular 82 only applies to foreign enterprises that are majority-owned and controlled by PRC enterprises, not those owned and controlled by foreign enterprises or individuals, the determining criteria set forth in Circular 82 may be adopted by the PRC tax authorities as the test for determining whether the enterprises are PRC tax residents, regardless of whether they are majority-owned and controlled by PRC enterprises. However, it is not entirely clear how the PRC tax authorities will determine whether a non-PRC entity (that has not already been notified of its status for EIT purposes) will be classified as a “resident enterprise” in practice.
Except for our PRC subsidiaries and joint ventures incorporated in China, we believe that none of our entities incorporated outside of China is a PRC resident enterprise for PRC tax purposes. However, the tax resident status of an enterprise is subject to determination by the PRC tax authorities, and uncertainties remain with respect to the interpretation of the term “de facto management body.”
205
If a non-PRC enterprise is classified as a “resident enterprise” for EIT purposes, any dividends to be distributed by that enterprise to non-PRC resident shareholders or ADS holders or any gains realized by such investors from the transfer of shares or ADSs may be subject to PRC tax. If the PRC tax authorities determine that we should be considered a PRC resident enterprise for EIT purposes, any dividends payable by us to our non-PRC resident enterprise shareholders or ADS holders with no office or premises established in China, or with an office or premises established in China but whose income (i.e. dividends received) has no de facto relationship with said office or premises, as well as gains realized by such investors from the transfer of our shares or ADSs may be subject to a 10% withholding tax. Furthermore, if we are considered a PRC resident enterprise for EIT purposes, it is unclear whether our non-PRC individual shareholders (including our ADS holders) would be subject to any PRC tax on dividends or gains obtained by such non-PRC individual shareholders. If any PRC tax were to apply to dividends realized by non-PRC individuals, it would generally apply at a rate of up to 20% (which in the case of dividends may be withheld at source). The foregoing rates may be reduced by an applicable tax treaty, but it is unclear if a non-PRC resident shareholder or ADS holder would be able to obtain in practice the benefits of any tax treaties between their country of tax residence and the PRC in the event that we are treated as a PRC resident enterprise.
According to the EIT Law, dividends declared after January 1, 2008 and paid by PRC FIEs to their non-PRC parent companies will be subject to PRC withholding tax at 10% unless there is a tax treaty between the PRC and the jurisdiction in which the overseas parent company is a tax resident and which specifically exempts or reduces such withholding tax, and such tax exemption or reduction is approved by the relevant PRC tax authorities. Pursuant to the Arrangement, if the non-PRC immediate holding company is a Hong Kong tax resident and directly holds a 25% or more equity interest in the PRC enterprise and is considered to be the beneficial owner of dividends paid by the PRC enterprise, such withholding tax rate may be lowered to 5%, subject to approval by the relevant PRC tax authorities in accordance with relevant tax regulations upon the assessment of beneficial ownership.
Overview of Tax Implications of Various Other Jurisdictions
Cayman Islands Taxation
According to our Cayman Islands counsel, Conyers Dill & Pearman, the Cayman Islands currently levies no taxes on individuals or corporations based upon profits, income, gains or appreciation and there is no taxation in the nature of inheritance tax or estate duty. There are no other taxes likely to be material to us levied by the government of the Cayman Islands except for stamp duties which may be applicable on instruments executed in, or brought within the jurisdiction of the Cayman Islands. The Cayman Islands is a party to a double tax treaty entered into with the United Kingdom in 2010 but it is otherwise not a party to any double tax treaties that are applicable to any payments made to or by our company. There are no exchange control regulations or currency restrictions in the Cayman Islands.
Pursuant to the Tax Concessions Act of the Cayman Islands, HUTCHMED (China) Limited has obtained an undertaking: (a) that no law which is enacted in the Cayman Islands imposing any tax to be levied on profits or income or gains or appreciations shall apply to us or our operations; and (b) that the aforesaid tax or any tax in the nature of estate duty or inheritance tax shall not be payable (i) on its shares, debentures or other obligations or (ii) by way of the withholding in whole or in part of any relevant payment as defined in the Tax Concessions Act.
The undertaking is for a period of twenty years from December 31, 2020.
Hong Kong Taxation
Profits Tax
HUTCHMED (China) Limited is a Hong Kong tax resident. Hong Kong tax residents are subject to Hong Kong Profits Tax in respect of profits arising in or derived from Hong Kong at the current rate of 16.5% (except portions eligible for the two-tiered profits tax as discussed above). Dividend income earned by a Hong Kong tax resident is generally not subject to Hong Kong Profits Tax. To keep in line with reform of foreign-sourced income regime, the Inland Revenue (Amendment) (Taxation on Foreign-sourced Disposal Gains) Ordinance 2023 (the 2023 Amendment Ordinance) was enacted on 8 December 2023 with a view to bringing the regime into force from 1 January 2024. The scope of assets covered for foreign sourced disposal gains is expanded to cover all types of property.
Hong Kong tax on shareholders and ADS holders
No tax is payable in Hong Kong in respect of dividends paid by a Hong Kong tax resident to their shareholders, including our ADS holders.
206
Hong Kong Profits Tax will not be payable by our shareholders, including our ADS holders (other than shareholders / ADS holders carrying on a trade, profession or business in Hong Kong and holding the shares / ADSs for trading purposes), on any capital gains made on the sale or other disposal of the shares or ADSs. Shareholders, including our ADS holders, should take advice from their own professional advisors as to their particular tax position.
U.S. Taxation
Corporate Tax
Our subsidiaries in the United States, HUTCHMED International Corporation and HUTCHMED US Corporation, are subject to a federal corporate tax of 21%.
Material U.S. Federal Income Tax Considerations with Respect to Ordinary Shares and ADSs
The following summary, subject to the limitations set forth below, describes the material U.S. federal income tax consequences for a U.S. Holder (as defined below) of the ownership and disposition of ordinary shares and ADSs. It is not a comprehensive description of all tax considerations that may be relevant to a particular person’s ownership of our securities. This discussion is limited to U.S. Holders that hold such ordinary shares or ADSs as capital assets within the meaning of Section 1221 of the Internal Revenue Code of 1986, as amended, or the Code (generally, property held for investment). For the purposes of this summary, a “U.S. Holder” is a person that is, for U.S. federal income tax purposes, a beneficial owner of an ordinary share or ADS and:
| ● | a citizen or individual resident of the United States; |
| ● | a corporation (or any other entity treated as a corporation for U.S. federal income tax purposes) organized in or under the laws of the United States or any state thereof, or the District of Columbia; |
| ● | an estate the income of which is subject to U.S. federal income taxation regardless of its source; or |
| ● | a trust if (i) it has a valid election in effect to be treated as a U.S. person for U.S. federal income tax purposes or (ii) a U.S. court can exercise primary supervision over its administration and one or more U.S. persons have the authority to control all of its substantial decisions. |
This summary does not purport to consider all aspects of U.S. federal income taxation that may be relevant to U.S. Holders in light of their particular circumstances, including the possible effect of the special tax accounting rules under Section 451 of the Code, or alternative minimum or Medicare contribution tax consequences. In addition, it does not address aspects of U.S. federal income taxation that may be applicable to U.S. Holders subject to special rules, including:
| ● | banks or other financial institutions; |
| ● | insurance companies; |
| ● | real estate investment trusts; |
| ● | regulated investment companies; |
| ● | grantor trusts; |
| ● | tax-exempt organizations, “individual retirement accounts or “Roth IRAs”; |
| ● | partnerships (or other entities or arrangements treated as partnerships for U.S. federal income tax purposes) or S corporations holding our ordinary shares or ADSs, and their partners or shareholders; |
| ● | dealers or electing traders in securities that use a mark-to-market method of tax accounting; |
207
| ● | persons whose functional currency is not the U.S. dollar; |
| ● | persons that acquired ordinary shares or ADSs as compensation; |
| ● | persons holding ordinary shares or ADSs in connection with a trade or business conducted outside of the United States. |
| ● | persons holding our ordinary shares or ADSs as part of a straddle, integrated or similar transaction for U.S. federal income tax purposes; or |
| ● | direct, indirect or constructive owners of 10% or more of our equity (by vote or value). |
If an entity or arrangement treated as a partnership for U.S. federal income tax purposes owns our ordinary shares or ADSs, the tax treatment of the partnership and a partner in such partnership generally will depend on the status of the partner and the activities of the partnership. Such partnerships and partners should consult their tax advisors as to the U.S. federal income tax consequences of acquiring, owning and disposing of our ordinary shares or ADSs.
This discussion does not address the effects of any state, local or non-U.S. tax law or any U.S. federal taxes other than income taxes (such as U.S. federal estate or gift tax consequences). We have not received nor do we expect to seek a ruling from the U.S. Internal Revenue Service, or the IRS, regarding any matter discussed herein. No assurance can be given that the IRS would not assert, or that a court would not sustain, a position contrary to any of those set forth below. Each investor should consult its tax advisors with respect to the U.S. federal, state, local and non-U.S. tax consequences of acquiring, owning and disposing of our ordinary shares and ADSs.
This discussion is based on the Code, final and proposed U.S. Treasury Regulations promulgated thereunder and administrative and judicial interpretations thereof, and the income tax treaty between the PRC and the United States, or the U.S.- PRC Tax Treaty, each as of the date hereof, all of which are subject to change or differing interpretations, possibly with retroactive effect, which could affect the tax consequences described herein. In addition, this summary assumes that the deposit agreement, and all other related agreements, will be performed in accordance with their terms.
INVESTORS SHOULD CONSULT THEIR TAX ADVISORS WITH REGARD TO THE PARTICULAR TAX CONSEQUENCES APPLICABLE TO THEIR SITUATIONS AS WELL AS THE APPLICATION OF ANY U.S. FEDERAL, STATE, LOCAL, NON-U.S. OR OTHER TAX LAWS, INCLUDING GIFT AND ESTATE TAX LAWS.
ADSs
A U.S. Holder of ADSs will generally be treated, for U.S. federal income tax purposes, as the owner of the underlying ordinary shares that such ADSs represent. Accordingly, no gain or loss will be recognized if a U.S. Holder exchanges ADSs for the underlying shares represented by those ADSs.
208
Taxation of Dividends
The following is subject to the discussion under “—Passive Foreign Investment Company Considerations” below.
As described in Item 8. “Financial Information—A.8 Dividend Policy” above, we do not currently anticipate paying any distributions on our ordinary shares or ADSs in the foreseeable future. However, to the extent there are any distributions made with respect to our ordinary shares or ADSs, the gross amount of any such distribution (including withheld taxes, if any) made out of our current or accumulated earnings and profits (as determined for U.S. federal income tax purposes) will generally be taxable to a U.S. Holder as ordinary dividend income on the date such distribution is actually or constructively received. Distributions in excess of our current and accumulated earnings and profits will be treated as a non-taxable return of capital to the extent of the U.S. Holder’s adjusted tax basis in the ordinary shares or ADSs, as applicable, and thereafter as capital gain. However, because we do not maintain calculations of our earnings and profits in accordance with U.S. federal income tax accounting principles, U.S. Holders should expect that distributions paid with respect to our ordinary shares or ADSs will be reported as dividends. Dividends paid to corporate U.S. Holders will not qualify for the dividends received deduction that may otherwise be allowed under the Code.
The amount of income from dividends paid in a non-U.S. currency will be the U.S. dollar amount of the dividend calculated by reference to the exchange rate in effect on the date of receipt, regardless of whether the payment is in fact converted into U.S. dollars. If the dividend is converted into U.S. dollars on the date of receipt, a U.S. Holder generally should not be required to recognize foreign currency gain or loss in respect of the dividend income. A U.S. Holder may have foreign currency gain or loss, taxable as ordinary income or loss, if the dividend is converted into U.S. dollars after the date of receipt. Foreign currency gain or loss generally will be treated as U.S.-source gain or loss.
Dividends paid to a non-corporate U.S. Holder by a “qualified foreign corporation” may be subject to reduced rates of U.S. federal income taxation if certain holding period and other requirements are met. A qualified foreign corporation generally includes a foreign corporation (other than a PFIC) if (1) its ordinary shares (or ADSs backed by ordinary shares) are readily tradable on an established securities market in the United States or (2) it is eligible for benefits under a comprehensive U.S. income tax treaty that includes an exchange of information program and which the U.S. Treasury Department has determined is satisfactory for these purposes. We are not eligible for the benefits of any U.S. income tax treaty. However, because our ADSs are listed on the Nasdaq, a non-corporate U.S. Holder of ADSs may be eligible for the preferential tax rates on dividends, subject to applicable limitations (including a minimum holding period and other requirements) and provided that we are not a PFIC (and are not treated as a PFIC with respect to the U.S. Holder) for the taxable year of distribution of the preceding taxable year.
For purposes of the foreign tax credit rules, dividends will be treated as foreign-source income. As described in “—Taxation in the PRC” above, if we are deemed to be a “resident enterprise” under PRC tax law, U.S. Holders may be subject to PRC withholding taxes on dividends paid by us. In that case, subject to certain conditions and limitations and the discussion below regarding the impact of certain Treasury regulations, such PRC taxes withheld from dividend payments (at a rate not exceeding the applicable rate provided in the U.S.-PRC Tax Treaty for U.S. Holders eligible for the benefits of the U.S.-PRC Tax Treaty) generally will be eligible for credit against a U.S. Holder’s U.S. federal income tax liability under the U.S. foreign tax credit rules. The U.S. foreign tax credit rules are complex. For example, under Treasury regulations, in the absence of an election to apply the benefits of an applicable income tax treaty, in order to be creditable, non-U.S. income tax rules must be consistent with certain U.S. federal income tax principles, and we have not determined whether the PRC income tax system meets these requirements. The IRS released notices that provide relief from certain of the provisions of the Treasury regulations described above for taxable years ending before the date that a notice or other guidance withdrawing or modifying the temporary relief is issued (or any later date specified in such notice or other guidance). A U.S. Holder that is not entitled, or does not elect, to claim a foreign tax credit for PRC tax withheld may instead be eligible to claim a deduction in respect of such withholding, but only for a year in which such U.S. Holder elects to do so for all creditable foreign income taxes and subject to other applicable limitations. U.S. Holders should consult their tax advisors regarding the foreign tax credit and deduction rules in light of their particular circumstances.
209
Taxation of Capital Gains
The following is subject to the discussion under “—Passive Foreign Investment Company Considerations” below.
Upon the sale, exchange, or other taxable disposition of our ordinary shares or ADSs, a U.S. Holder generally will recognize gain or loss in an amount equal to the difference between the amount realized on such sale or exchange and the U.S. Holder’s adjusted tax basis in such ordinary shares or ADSs, in each case determined in U.S. dollars. A U.S. Holder’s initial tax basis will be the U.S. Holder’s U.S. dollar purchase price for such ordinary shares or ADSs.
Such gain or loss generally will be capital gain or loss, and will be long-term capital gain or loss if a U.S. Holder held the ordinary share or ADS for more than one year. Long-term capital gains of non-corporate U.S. Holders are taxed at a preferential tax rate. The deductibility of capital losses is subject to limitations.
As described in “—Taxation in the PRC” above, if we are deemed to be a “resident enterprise” under PRC tax law, any gain on the sale of ordinary shares or ADSs may be subject to PRC taxes. Under the Code, capital gains of U.S. persons generally are treated as U.S.-source income. However, if a U.S. Holder is eligible for the benefits of the U.S.-PRC Tax Treaty, the holder may be able to elect to treat such disposition gain as PRC-source gain under the U.S.-PRC Tax Treaty for U.S. foreign tax credit purposes and claim a foreign tax credit in respect of PRC taxes on such gains. A U.S. Holder will be eligible for U.S.-PRC Tax Treaty benefits if (for the purposes of the treaty) such holder is a resident of the United States and satisfies the “limitations of benefits” requirements specified in the U.S.-PRC Tax Treaty. Because the determination of treaty benefit eligibility is fact-intensive and depends upon a U.S. Holder’s particular circumstances, U.S. Holders should consult their tax advisors regarding their eligibility for the U.S.-PRC Tax Treaty benefits. Treasury regulations generally preclude a U.S. Holder from claiming a foreign tax credit with respect to PRC income taxes on gains from dispositions of ordinary shares or ADSs if a U.S. Holder is not eligible for, or does not elect to apply the benefits of, the U.S.-PRC Tax Treaty. As discussed above under “—Taxation of Dividends,” the IRS released notices that provide relief from certain of these Treasury regulations’ provisions (including the limitation described in the preceding sentence) for taxable years ending before the date that a notice or other guidance withdrawing or modifying the temporary relief is issued (or any later date specified in such notice or other guidance). However, even if these Treasury regulations do not prohibit a U.S. Holder from claiming a foreign tax credit with respect to PRC taxes on disposition gains, other limitations under the foreign tax credit rules may preclude a U.S. Holder from claiming a foreign tax credit. If PRC taxes (if any) on disposition gains are not creditable, they may be deductible or reduce the amount realized on the disposition. An election to deduct creditable non-U.S. taxes instead of claiming foreign tax credits applies to all creditable non-U.S. taxes paid or accrued in the taxable year. The rules governing foreign tax credits and the deductibility of non-U.S. taxes are complex. U.S. Holders are also encouraged to consult their tax advisors regarding the tax consequences in the event PRC tax is imposed on a disposition of ordinary shares or ADSs, including the U.S.-PRC Tax Treaty’s resourcing rule, any reporting requirements with respect to a treaty-based return position and the creditability or deductibility of any non-U.S. tax on disposition gains in their particular circumstances (including any applicable limitations).
Passive Foreign Investment Company Considerations
Status as a PFIC. The rules governing PFICs can result in adverse U.S. federal income tax consequences to U.S. Holders. We generally will be a PFIC for U.S. federal income tax purposes if, for any taxable year, either: (1) 75% or more of our gross income consists of certain types of passive income, or (2) 50% or more of the average value of our assets (generally determined on a quarterly basis) consists of our assets that produce, or are held for the production of, passive income. Passive income generally includes dividends, interest, rents and royalties (other than certain rents and royalties treated under the PFIC rules as derived in the active conduct of a trade or business), annuities and gains from assets that produce passive income. If a non-U.S. corporation owns at least 25% (by value) of the stock of another corporation, the non-U.S. corporation is treated for the purposes of the PFIC tests as owning its proportionate share of the assets of the other corporation and as receiving directly its proportionate share of the other corporation’s income. Ownership stakes of less-than-25% (by value) in other corporations are treated as passive assets. Cash and cash equivalents are generally treated as passive assets. Goodwill is generally treated as an active asset to the extent associated with activities that generate non-passive income.
210
Based on the composition of our income and assets and the estimated average value of our assets (including goodwill and other intangible assets), we believe that we were not a PFIC for our taxable year ended December 31, 2023. However, our PFIC status is a factual determination that is made on an annual basis and depends on particular facts and circumstances (such as the value of our assets, including goodwill and other intangible assets). We hold a substantial amount of cash and financial investments and while this continues to be the case, our PFIC status depends primarily on the average value of our goodwill and other intangible assets. The value of our goodwill and other intangible assets may be determined, in large part, by reference to our market capitalization, which has been, and may continue to be, volatile. Therefore, if our market capitalization declines we may be or become a PFIC. In addition, there is uncertainty as to how to apply the PFIC rules for purposes of classifying certain of our income and assets as active or passive. Furthermore, the proportionate value of our passive assets may increase over time if the value of our ownership stake in any other company in which we own less than 25% (by value) increase. In light of the foregoing, no assurance can be provided that we were not, or will not be, a PFIC for any taxable year.
U.S. federal income tax treatment of a shareholder of a PFIC generally. If we are a PFIC for any taxable year during which a U.S. Holder owns ordinary shares or ADSs, the U.S. Holder, absent certain elections, generally will be subject to adverse rules (regardless of whether we continue to be a PFIC) with respect to (1) any “excess distributions” (generally, the extent that any distributions received by the U.S. Holder on its ordinary shares or ADSs in a taxable year exceed 125% of the average annual distributions received by the U.S. Holder in the three preceding taxable years or, if shorter, the U.S. Holder’s holding period) and (2) any gain realized on the sale or other disposition, including, in certain cases, a pledge of such ordinary shares or ADSs.
Under these rules (a) any gain or excess distribution will be allocated ratably over the U.S. Holder’s holding period, (b) the amount allocated to the current taxable year and any taxable year prior to the first taxable year in which we became a PFIC will be taxed as ordinary income and (c) the amount allocated to each other taxable year during the U.S. Holder’s holding period (i) will be subject to tax at the highest rate of tax in effect for the applicable category of taxpayer for that year and (ii) will be subject to an interest charge at a statutory rate with respect to the resulting tax attributable to each such other taxable year. In addition,a non-corporate U.S. Holder will not be eligible for reduced rates of taxation on any dividends received from us if we are a PFIC (or are treated as a PFIC with respect to the U.S. Holder) in the taxable year in which such dividends are paid or in the preceding taxable year.
If we are a PFIC for any taxable year during which a U.S. Holder owns ordinary shares or ADSs, we generally will continue to be treated as a PFIC with respect to that U.S. Holder in all succeeding taxable years, even if we cease to meet the threshold requirements for PFIC status described above, unless the U.S. Holder makes a timely “deemed sale election.” If we are a PFIC for any taxable year and then cease to be a PFIC, a U.S. Holder may make a “deemed sale election” to be treated for U.S. federal income tax purposes as having sold such U.S. Holder’s ordinary shares or ADSs on the last day of our taxable year during which we were a PFIC. A U.S. Holder that makes a deemed sale election would then cease to be treated as owning stock in a PFIC if we cease to be a PFIC for subsequent taxable years. However, gain recognized as a result of making the deemed sale election would be subject to the adverse rules described above and loss would not be recognized.
If we are a PFIC for any taxable year, a U.S. Holder will be treated as owning a proportionate amount (by value) of stock or shares owned by us in any direct or indirect subsidiaries that are also PFICs (any such entity, a “Lower-tier PFIC”) and will be subject to similar adverse rules with respect to any distributions we receive from, and dispositions we make of, the stock or shares of such subsidiaries, in each case as if the U.S. Holder owned its proportionate share of the Lower-tier PFIC directly, even though the U.S. Holder will not receive the proceeds of those distributions or dispositions directly. U.S. Holders are urged to consult their tax advisors about the application of the PFIC rules to any of our subsidiaries.
PFIC “mark-to-market” election. In certain circumstances, a holder of “marketable stock” of a PFIC will be subject to tax consequences different that those described above by making a timely mark-to-market election with respect to such stock. For the purposes of these rules “marketable stock” is generally stock which is “regularly traded” (traded in greater than de minimis quantities on at least 15 days during each calendar quarter) on a “qualified exchange.” Nasdaq, on which the ADSs are listed, is a “qualified exchange” for this purpose. A non-U.S. exchange is a “qualified exchange” if it is regulated by a governmental authority in the jurisdiction in which the exchange is located and with respect to which certain other requirements are met. The IRS has not identified specific non-U.S. exchanges that are “qualified” for this purpose.
211
A U.S. Holder that makes a timely mark-to-market election must include in gross income, as ordinary income, for each taxable year that we are a PFIC an amount equal to the excess, if any, of the fair market value of the U.S. Holder’s ordinary shares or ADSs that are “marketable stock” at the close of the taxable year over the U.S. Holder’s adjusted tax basis in such ordinary shares or ADSs. An electing U.S. Holder may also claim an ordinary loss deduction for the excess, if any, of the U.S. Holder’s adjusted tax basis in such ordinary shares or ADSs over their fair market value at the close of the taxable year, but this deduction is allowable only to the extent of any net mark-to-market gains previously included in income pursuant to the timely mark-to-market election. The adjusted tax basis of a U.S. Holder’s ordinary shares or ADSs with respect to which the timely mark-to-market election applies would be adjusted to reflect amounts included in gross income or allowed as a deduction because of such election. If a U.S. Holder makes an effective mark-to-market election with respect to our ordinary shares or ADSs, gains from an actual sale or other disposition of such ordinary shares or ADSs in a year in which we are a PFIC will be treated as ordinary income, and any losses incurred on such sale or other disposition will be treated as ordinary losses to the extent of any net mark-to-market gains previously included in income (with any excess loss treated as a capital loss).
If we are a PFIC for any taxable year during a U.S. Holder’s holding period prior to the first taxable year with respect to which the U.S. Holder made a mark-to-market election, the general PFIC rules described above under “—U.S. federal income tax treatment of a shareholder of a PFIC generally” will apply with respect to the excess of the fair market value of the ADSs or ordinary shares at the end of that first taxable year over the U.S. Holder’s tax basis in the ADSs or ordinary shares. Otherwise, a timely mark-to-market election will be effective for the taxable year for which the election is made and all subsequent taxable years unless the ordinary shares or ADSs are no longer regularly traded on a qualified exchange or the IRS consents to the revocation of the election.
There is no law, regulation or administrative guidance that provides for a right to make a mark-to-market election for equity interests in any Lower-tier PFIC the shares of which are not regularly traded on a qualified exchange. As a result, even if a U.S. Holder makes a mark-to-market election with respect to our ordinary shares or ADSs, such U.S. Holder could nevertheless be subject to the PFIC rules described under “—U.S. federal income tax treatment of a shareholder of a PFIC generally” with respect to such U.S. Holder’s indirect interest in any Lower-tier PFIC. U.S. Holders should consult their tax advisors regarding the availability of, and the procedure for, and the effect of making, a mark-to-market election, and whether making the election would be advisable, including in light of their particular circumstances.
No QEF election. We do not expect to provide the information regarding our income that would be necessary in order for a U.S. Holder to make a timely “qualifying electing fund” election, or QEF election, if we were a PFIC, which, if available, could materially affect the tax consequences of the ownership and disposition of the ordinary shares and ADSs if we are a PFIC for any taxable year. Therefore, U.S. Holders will not be able to make this election.
PFIC information reporting requirements. If we are (or are treated with respect to a particular U.S. Holder as) a PFIC for any year in which a U.S. Holder owns ordinary shares or ADSs, such U.S. Holder generally will be required to file an annual information return on IRS Form 8621 with respect to us and any Lower-tier PFIC.
NO ASSURANCE CAN BE GIVEN THAT WE ARE NOT CURRENTLY A PFIC OR THAT WE WILL NOT BECOME A PFIC IN THE FUTURE. U.S. HOLDERS SHOULD CONSULT THEIR TAX ADVISORS WITH RESPECT TO THE OPERATION OF THE PFIC RULES AND RELATED REPORTING REQUIREMENTS IN LIGHT OF THEIR PARTICULAR CIRCUMSTANCES, INCLUDING THE ADVISABILITY AND EFFECTS OF MAKING ANY ELECTION THAT MAY BE AVAILABLE.
Backup Withholding and Information Reporting and Filing Requirements
Backup withholding and information reporting requirements may apply to distributions on, and proceeds from the sale or disposition of, ordinary shares and ADSs that are held by U.S. Holders. The payor will be required to withhold tax (currently at a rate of 24%) on such payments made within the United States, or by a U.S. payor or a U.S. intermediary (and certain subsidiaries thereof) to a U.S. Holder, other than an exempt recipient, if the U.S. Holder is not otherwise exempt and:
| ● | the holder fails to furnish the holder’s taxpayer identification number, which for an individual is ordinarily his or her social security number; |
| ● | the holder furnishes an incorrect taxpayer identification number; |
212
| ● | the applicable withholding agent is notified by the IRS that the holder previously failed to properly report payments of interest or dividends; or |
| ● | the holder fails to certify under penalties of perjury that the holder has furnished a correct taxpayer identification number and that the IRS has not notified the holder that the holder is subject to backup withholding. |
Backup withholding is not an additional tax. Amounts withheld as backup withholding may be credited against a U.S. Holder’s U.S. federal income tax liability (if any) or refunded provided the required information is furnished to the IRS in a timely manner. U.S. Holders should consult their tax advisors regarding their qualification for an exemption from backup withholding and the procedures for obtaining such an exemption.
Certain U.S. Holders of specified foreign financial assets with an aggregate value in excess of the applicable dollar threshold may be required to report information relating to their holding of ordinary shares or ADSs, subject to certain exceptions (including an exception for securities held in accounts maintained by certain financial institutions) with their tax returns for each year in which they hold such interests. U.S. Holders should consult their own tax advisors regarding the information reporting obligations that may arise from their acquisition, ownership or disposition of our ordinary shares or ADSs.
THE ABOVE DISCUSSION DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A PARTICULAR INVESTOR. INVESTORS ARE STRONGLY URGED TO CONSULT THEIR TAX ADVISORS ABOUT THE TAX CONSEQUENCES OF AN INVESTMENT IN OUR ORDINARY SHARES OR ADSs.
F. Dividends and Payment Agents.
Not applicable.
G. Statement by Experts.
Not applicable.
H. Documents on Display.
We are subject to the informational requirements of the Exchange Act and are required to file reports and other information with the SEC. Shareholders may access our reports and other information filed with the SEC by viewing them on the SEC’s website, at www.sec.gov. We also make available on our website’s investor relations page, free of charge, our annual report and the text of our reports on Form 6-K, including any amendments to these reports, as well as certain other SEC filings, as soon as reasonably practicable after they are electronically filed with or furnished to the SEC. The address for our investor relations page is www.hutch-med.com/ shareholder-information. The information contained on our website is not incorporated by reference in this annual report.
We are a “foreign private issuer” as such term is defined in Rule 405 under the Securities Act, and are not subject to the same requirements that are imposed upon U.S. domestic issuers by the SEC. Under the Exchange Act, we are subject to reporting obligations that, in certain respects, are less detailed and less frequent than those of U.S. domestic reporting companies. As a result, we do not file the same reports that a U.S. domestic issuer would file with the SEC, although we are required to file or furnish to the SEC the continuous disclosure documents that we are required to file on the AIM market.
We will furnish Deutsche Bank Trust Company Americas, the depositary of our ADSs, with our annual reports, which will include a review of operation and annual audited consolidated financial statements prepared in conformity with U.S. GAAP, and all notices of shareholders’ meetings and other reports and communications that are made generally available to our shareholders. The depositary will make such notices, reports and communications available to holders of ADSs and, upon our requests, will mail to all record holders of ADSs the information contained in any notice of a shareholders’ meeting received by the depositary from us.
I. Subsidiary information.
Not applicable.
213
J. Annual Report to Security Holders.
The Company intends to submit annual report provided to security holders in electronic format as an exhibit to a current report on Form 6-K.
ITEM 11. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Foreign Exchange Risk
A substantial portion of our revenue and expenses are denominated in renminbi, and our consolidated financial statements are presented in U.S. dollars. We do not believe that we currently have any significant direct foreign exchange risk and have not used any derivative financial instruments to hedge our exposure to such risk. Although, in general, our exposure to foreign exchange risks should be limited, the value of your investment in our ADSs will be affected by the exchange rate between the U.S. dollar and the renminbi because the value of our business is effectively denominated in renminbi, while the ADSs will be traded in U.S. dollars.
The value of the renminbi against the U.S. dollar and other currencies may fluctuate and is affected by, among other things, changes in China’s political and economic conditions. The conversion of renminbi into foreign currencies, including U.S. dollars, has been based on rates set by the PBOC. On July 21, 2005, the PRC government changed its decade-old policy of pegging the value of the renminbi to the U.S. dollar. Under the revised policy, the renminbi is permitted to fluctuate within a narrow and managed band against a basket of certain foreign currencies. This change in policy resulted in a more than 20% appreciation of the renminbi against the U.S. dollar in the following three years. Between July 2008 and June 2010, this appreciation halted, and the exchange rate between the renminbi and U.S. dollar remained within a narrow band. In June 2010, the PBOC announced that the PRC government would increase the flexibility of the exchange rate, and thereafter allowed the renminbi to appreciate slowly against the U.S. dollar within the narrow band fixed by the PBOC. At various times since then, the PBOC has significantly devalued the renminbi against the U.S. dollar. If we decide to convert renminbi into U.S. dollars for the purpose of making payments for dividends on our ordinary shares or ADSs or for other business purposes, appreciation of the U.S. dollar against the renminbi would have a negative effect on the U.S. dollar amounts available to us.
Credit Risk
Substantially all of our bank deposits are in major financial institutions, which we believe are of high credit quality. We limit the amount of credit exposure to any single financial institution. We make periodic assessments of the recoverability of trade and other receivables and amounts due from related parties. Our historical experience in collection of receivables falls within the recorded allowances, and we believe that we have made adequate provision for uncollectible receivables.
Interest Rate Risk
We have no significant interest-bearing assets except for bank deposits. Our exposure to changes in interest rates is mainly attributable to our bank borrowings, which bear interest at floating interest rates and expose us to cash flow interest rate risk. We have not used any interest rate swaps to hedge our exposure to interest rate risk. We have performed sensitivity analysis for the effects on our results for the year from changes in interest rates on floating rate borrowings. The sensitivity to interest rates used is based on the market forecasts available at the end of the reporting period and under the economic environments in which we operate, with other variables held constant. According to the analysis, the impact on our results of a 1.0% interest rate shift would be a maximum increase/decrease of $0.1 million for the year ended December 31, 2023.
ITEM 12. DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES
| A. | Debt Securities. |
Not applicable.
| B. | Warrants and Rights. |
Not applicable.
214
| C. | Other Securities. |
Not applicable.
| D. | American Depositary Shares. |
Our ADSs representing our ordinary shares are currently traded on Nasdaq. Dealings in our ADSs on Nasdaq are conducted in U.S. dollars.
ADSs may be held either:
(a)directly: (i) by having an American Depositary Receipt, also referred to as an ADR, which is a certificate evidencing a specific number of ADSs registered in the holder’s name; or (ii) by having uncertificated ADSs registered in the holder’s name; or
(b)indirectly, by holding a security entitlement in ADSs through a broker or other financial institution that is a direct or indirect participant in The Depository Trust Company, also called DTC.
The depositary for our ADSs is Deutsche Bank Trust Company Americas, whose office is located at 1 Columbus Circle, New York, NY 10019, United States.
Fees and charges our ADS holders may have to pay
ADS holders will be required to pay the following service fees to Deutsche Bank Trust Company America, the depositary of our ADS program, and certain taxes and governmental charges (in addition to any applicable fees, expenses, taxes and other governmental charges payable on the deposited securities represented by ADSs):
Service |
| Fees |
● To any person to which ADSs are issued or to any person to which a distribution is made in respect of ADS distributions pursuant to stock dividends or other free distributions of stock, bonus distributions, stock splits or other distributions (except where converted to cash) | Up to $0.05 per ADS issued | |
● Cancellation or withdrawal of ADSs, including the case of termination of the deposit agreement | Up to $0.05 per ADS cancelled | |
● Distribution of cash dividends | Up to $0.05 per ADS held | |
● Distribution of cash entitlements (other than cash dividends) and/or cash proceeds from the sale of rights, securities and other entitlements | Up to $0.05 per ADS held | |
● Distribution of ADSs pursuant to exercise of rights | Up to $0.05 per ADS held | |
● Depositary services | Up to $0.05 per ADS held on the applicable record date(s) established by the depositary bank (an annual fee) |
215
ADS holders will also be responsible for paying certain fees and expenses incurred by the depositary bank and certain taxes and governmental charges (in addition to any applicable fees, expenses, taxes and other governmental charges payable on the deposited securities represented by any of your ADSs) such as:
| ● | Fees for the transfer and registration of ordinary shares charged by the registrar and transfer agent for the ordinary shares in the Cayman Islands (i.e., upon deposit and withdrawal of ordinary shares). |
| ● | Expenses incurred for converting foreign currency into U.S. dollars. |
| ● | Expenses for cable, telex and fax transmissions and for delivery of securities. |
| ● | Taxes and duties upon the transfer of securities, including any applicable stamp duties, any stock transfer charges or withholding taxes (i.e., when ordinary shares are deposited or withdrawn from deposit). |
| ● | Fees and expenses incurred in connection with the delivery or servicing of ordinary shares on deposit. |
| ● | Fees and expenses incurred in connection with complying with exchange control regulations and other regulatory requirements applicable to ordinary shares, ordinary shares deposited securities, ADSs and ADRs. |
| ● | Any applicable fees and penalties thereon. |
The depositary fees payable upon the issuance and cancellation of ADSs are typically paid to the depositary bank by the brokers (on behalf of their clients) receiving the newly issued ADSs from the depositary bank and by the brokers (on behalf of their clients) delivering the ADSs to the depositary bank for cancellation. The brokers in turn charge these fees to their clients. Depositary fees payable in connection with distributions of cash or securities to ADS holders and the depositary services fee are charged by the depositary bank to the holders of record of ADSs as of the applicable ADS record date.
The depositary fees payable for cash distributions are generally deducted from the cash being distributed or by selling a portion of distributable property to pay the fees. In the case of distributions other than cash (i.e., share dividends, rights), the depositary bank charges the applicable fee to the ADS record date holders concurrent with the distribution. In the case of ADSs registered in the name of the investor (whether certificated or uncertificated in direct registration), the depositary bank sends invoices to the applicable record date ADS holders. In the case of ADSs held in brokerage and custodian accounts (via DTC), the depositary bank generally collects its fees through the systems provided by DTC (whose nominee is the registered holder of the ADSs held in DTC) from the brokers and custodians holding ADSs in their DTC accounts. The brokers and custodians who hold their clients’ ADSs in DTC accounts in turn charge their clients’ accounts the amount of the fees paid to the depositary banks.
In the event of refusal to pay the depositary fees, the depositary bank may, under the terms of the deposit agreement, refuse the requested service until payment is received or may set off the amount of the depositary fees from any distribution to be made to the ADS holder.
Fees and other payments made by the depositary to us
The depositary has agreed to pay certain amounts to us in exchange for its appointment as depositary. We may use these funds towards our expenses relating to the establishment and maintenance of the ADR program, including investor relations expenses, or otherwise as we see fit.
Ordinary Shares and Conversions
Our ordinary shares are admitted to trading on AIM and trade on the SEHK. Dealings in our ordinary shares on the AIM and SEHK are conducted in pound sterlings and H.K. dollars, respectively.
In connection with the initial public offering of our ordinary shares in Hong Kong in June 2021, we established a branch register of members in Hong Kong, or the Hong Kong share register, which will be maintained by our Hong Kong Share Registrar, Computershare Hong Kong Investor Services Limited. Our principal register of members, or the Cayman share register, will continue to be maintained by our Principal Share Registrar, Computershare Investor Services (Jersey) Limited. All ordinary shares offered in our initial public offering in Hong Kong were registered on the Hong Kong share register in order to be listed and traded on the SEHK.
Details on the conversion process between SEHK, Nasdaq and AIM are available at https://www.hutch-med.com/shareholder-information/investor-faqs/.
216
PART II
ITEM 13. DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES
None.
ITEM 14. MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS
A-D. Material Modifications to the Rights of Security Holders; Assets Securing Securities; Trustees; Paying Agents.
None.
E. Use of Proceeds.
Not applicable.
ITEM 15. CONTROLS AND PROCEDURES
A. Evaluation of Disclosure Controls and Procedures.
As required by Rule 13a-15 under the Exchange Act, management, including our chief executive officer and our chief financial officer, has evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this report. Disclosure controls and procedures refer to controls and other procedures designed to ensure that information required to be disclosed in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in our reports that we file or submit under the Exchange Act is accumulated and communicated to management, including our principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding our required disclosure. Based on such evaluation, our management has concluded that, as of December 31, 2023, our disclosure controls and procedures were effective.
B. Management’s Annual Report on Internal Control over Financial Reporting.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rule 13a-15(f) and 15d-15(f) promulgated under the Securities Exchange Act of 1934. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of consolidated financial statements in accordance with U.S. GAAP and includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of a company’s assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of consolidated financial statements in accordance with generally accepted accounting principles, and that a company’s receipts and expenditures are being made only in accordance with authorizations of a company’s management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of a company’s assets that could have a material effect on the consolidated financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness of our internal control over financial reporting to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management, with the participation of our chief executive officer and chief financial officer, has assessed the effectiveness of our internal control over financial reporting as of December 31, 2023. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013 Framework). Based on this assessment, management concluded that our internal control over financial reporting was effective as of December 31, 2023.
217
C. Attestation Report of the Independent Registered Public Accounting Firm.
Our independent registered public accounting firm, PricewaterhouseCoopers Zhong Tian LLP (“PricewaterhouseCoopers Zhong Tian”), has audited the effectiveness of our internal control over financial reporting as of December 31, 2023, as stated in its report, which appears in this annual report.
D. Changes in Internal Control over Financial Reporting.
There were no changes in our internal controls over financial reporting during the fiscal year ended December 31, 2023 that have materially and adversely affected, or are reasonably likely to materially and adversely affect, our internal control over financial reporting.
ITEM 16. RESERVED
ITEM 16A. AUDIT COMMITTEE FINANCIAL EXPERTS
Our audit committee consists of Graeme Allan Jack, Paul Rutherford Carter and Mok Shu Kam, Tony, with Graeme Allan Jack serving as chairman of the committee. Graeme Allan Jack, Paul Rutherford Carter and Mok Shu Kam, Tony each meet the independence requirements under the rules of the Nasdaq Stock Market and under Rule 10A-3 under the Exchange Act. We have determined that Graeme Allan Jack is an “audit committee financial expert” within the meaning of Item 407 of Regulation S-K. All members of our audit committee meet the requirements for financial literacy under the applicable rules and regulations of the SEC and the Nasdaq Stock Market. For information relating to qualifications and experience of each audit committee member, see Item 6. “Directors, Senior Management and Employees.”
ITEM 16B. CODE OF ETHICS
Our board of directors has adopted a code of ethics applicable to all of our employees, officers and directors, including our principal executive officer, principal financial officer, principal accounting officer or controller, and persons performing similar functions. This code is intended to qualify as a “code of ethics” within the meaning of the applicable rules of the SEC. Our code of ethics is available on our website at https://www.hutch-med.com/shareholder-information/corporate-governance/code-of-ethics/. Information contained on, or that can be accessed through, our website is not incorporated by reference into this annual report. See Item 6.C. “Board Practices—Code of Ethics” for more information.
ITEM 16C. PRINCIPAL ACCOUNTANT FEES AND SERVICES
Principal Accountant Fees and Services
The following table summarizes the fees charged by PricewaterhouseCoopers Zhong Tian, our principal external auditors, for certain services rendered to our company, including some of our subsidiaries and joint ventures, during 2023 and 2022.
Year ended | ||||
December 31, | ||||
2023 | 2022 | |||
($’000) | ||||
Audit fees(1) |
| 2,682 |
| 2,200 |
Tax fees(2) |
| 189 |
| 337 |
Total(3) |
| 2,871 |
| 2,537 |
Notes:
| (1) | “Audit fees” means the aggregate fees billed in each of the fiscal years for professional services rendered by our principal external auditors for the audit of our annual financial statements and review of our interim financial statements. |
| (2) | “Tax fees” means the aggregate fees billed in each of the fiscal years for professional services rendered by our principal external auditors for tax compliance and tax advice. |
218
| (3) | The fees disclosed are exclusive of out-of-pocket expenses and taxes on the amounts paid, which totaled approximately $52,000 and $68,000 in 2022 and 2023, respectively. |
Audit Committee Pre-approval Policies and Procedures
Our audit committee reviews and pre-approves the scope and the cost of audit services related to us and permissible non-audit services performed by the independent auditors, other than those for de minimis services which are approved by the audit committee prior to the completion of the audit. All of the services related to our company provided by PricewaterhouseCoopers Zhong Tian, our principal external auditors, listed above have been approved by the audit committee.
ITEM 16D. EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES
Not applicable.
ITEM 16E. PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS
None.
ITEM 16F. CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT
Not applicable.
ITEM 16G. CORPORATE GOVERNANCE
As permitted by Nasdaq, in lieu of the Nasdaq corporate governance rules, but subject to certain exceptions, we may follow the practices of our home country which for the purpose of such rules is the Cayman Islands. Certain corporate governance practices in the Cayman Islands may differ significantly from corporate governance listing standards as, except for general fiduciary duties and duties of care, Cayman Islands law has no corporate governance regime which prescribes specific corporate governance standards. For example, we follow Cayman Islands corporate governance practices in lieu of the corporate governance requirements of the Nasdaq Global Select Market in respect of the following:
(i)the majority independent director requirement under Section 5605(b)(1) of the Nasdaq listing rules,
(ii)the requirement under Section 5605(d) of the Nasdaq listing rules that a remuneration committee comprised solely of independent directors governed by a remuneration committee charter oversee executive compensation, and
(iii)the requirement under Section 5605(e) of the Nasdaq listing rules that director nominees be selected or recommended for selection by either a majority of the independent directors or a nominations committee comprised solely of independent directors.
Cayman Islands law does not impose a requirement that our board of directors consist of a majority of independent directors, nor does Cayman Islands law impose specific requirements on the establishment of a remuneration committee or nominating committee or nominating process. We voluntarily comply with Hong Kong Corporate Governance Code. See Item 6.C. “Board Practice—Hong Kong Corporate Governance Code” for more details.
ITEM 16H. MINE SAFETY DISCLOSURE
Not applicable.
ITEM 16I. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTION
Not applicable.
ITEM 16J. INSIDER TRADING POLICIES
Not Applicable.
219
ITEM 16K. CYBERSECURITY
Cybersecurity risk management is an integral part of our overall enterprise risk management program. Our cybersecurity risk management program is designed based on N.I.S.T cybersecurity framework. This framework includes steps for (a) identifying cybersecurity threats, assessing the severity, identifying the source and whether the threat is associated with a third-party service provider; (b) reporting material cybersecurity incidents to management and our board of directors; (c) implementing safeguards, countermeasures and mitigation strategies; and (d) remediation and restoration of the affected systems. Our cybersecurity team also engages third-party security experts for defense protection capability assessment and system enhancements. In addition, our cybersecurity team provides training to all employees annually.
Our board of directors has overall oversight responsibility for our risk management, and delegates cybersecurity risk management oversight to the audit committee of the board of directors. The audit committee is responsible for ensuring that management has processes in place designed to identify and evaluate cybersecurity risks to which the company is exposed and implement processes and programs to manage cybersecurity risks and mitigate cybersecurity incidents. The audit committee also reports material cybersecurity risks to our full board of directors.
Management is responsible for identifying, considering and assessing material cybersecurity risks on an ongoing basis, establishing processes to ensure that such potential cybersecurity risk exposures are monitored, putting in place appropriate mitigation measures and maintaining cybersecurity programs. Our cybersecurity programs are under the direction of an IT Working Group established by the audit committee consisting currently of Dr. Dan Eldar, a Non- Executive Director, Mr. James Lai, Head of Corporate Internal Audit for CKHH, and Mr. Cheng Chig Fung, Johnny, Chief Financial Officer and Executive Director, who receive reports from our cybersecurity team led by the Head of IT and Security and monitors the prevention, detection, mitigation, and remediation of cybersecurity incidents.
Our Head of IT and Security and dedicated IT personnel are experienced information systems security professionals and information security managers with more than 15 years of relevant experience. The IT Working Group regularly updates the audit committee on the company’s cybersecurity programs, material cybersecurity risks and mitigation strategies and provides cybersecurity reports quarterly that cover, among other topics, third-party assessments of the company’s cybersecurity programs, developments in cybersecurity and updates to the company’s cybersecurity programs and mitigation strategies.
In 2023, we did not identify any cybersecurity threats that have materially affected or are reasonably likely to materially affect our business strategy, results of operations, or financial condition. However, despite our efforts, we cannot eliminate all risks from cybersecurity threats, or provide assurances that we have not experienced an undetected cybersecurity incident. For more information about these risks, please see “Risk Factors – We rely significantly on information technology and any failure, inadequacy, interruption or security lapse of that technology, including any cybersecurity incidents, could harm our ability to operate our business effectively” in this annual report.
220
PART III
ITEM 17. FINANCIAL STATEMENTS
See Item 18 “Financial Statements.”
ITEM 18. FINANCIAL STATEMENTS
Our consolidated financial statements and the consolidated financial statements of our non-consolidated joint venture, Shanghai Hutchison Pharmaceuticals, are included at the end of this annual report.
221
4.11 | ||
4.12 | ||
4.13 | ||
4.14 | ||
4.15+ | ||
4.16+ | ||
8.1* | ||
12.1* | Certification of Chief Executive Officer Required by Rule 13a-14(a) | |
12.2* | Certification of Chief Financial Officer Required by Rule 13a-14(a) | |
13.1† | ||
13.2† | ||
15.1* | ||
15.2* | ||
15.3* | ||
15.4* | ||
101.INS* | XBRL Instance Document | |
101.SCH* | XBRL Taxonomy Extension Schema Document | |
101.CAL* | XBRL Taxonomy Extension Calculation Linkbase Document | |
101.LAB* | XBRL Taxonomy Extension Label Linkbase Document | |
101.PRE* | XBRL Taxonomy Extension Presentation Linkbase Document | |
101.DEF* | XBRL Taxonomy Extension Definitions Linkbase Document | |
104* | Cover Page Interactive Data File (embedded within the Inline XBRL document) |
* Filed herewith.
† Furnished herewith.
+ Portions of the exhibit have been omitted because they are both (i) not material and (ii) would likely cause competitive harm to the company if publicly disclosed.
223
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on annual report on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
HUTCHMED (China) Limited | |||
By: | /s/ Weiguo Su | ||
Name: | Weiguo Su | ||
Title: | Chief Executive Officer | ||
Date: February 28, 2024 | |||
224
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Audited Consolidated Financial Statements of HUTCHMED (China) Limited | |
Report of Independent Registered Public Accounting Firm (PCAOB ID | F-2 |
As at December 31, 2023 and December 31, 2022: | |
F-5 | |
For the Years Ended December 31, 2023, 2022 and 2021: | |
F-6 | |
F-7 | |
F-8 | |
F-9 | |
F-10 | |
Audited Consolidated Financial Statements of Shanghai Hutchison Pharmaceuticals Limited | |
F-51 | |
For the Years Ended December 31, 2023, 2022 and 2021: | |
F-53 | |
F-54 | |
As at December 31, 2023 and December 31, 2022: | |
F-55 | |
For the Years Ended December 31, 2023, 2022 and 2021: | |
F-56 | |
F-57 | |
F-58 |
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders of HUTCHMED (China) Limited
Opinions on the Financial Statements and Internal Control over Financial Reporting
We have audited the accompanying consolidated balance sheets of HUTCHMED (China) Limited and its subsidiaries (the “Company”) as of December 31, 2023 and 2022, and the related consolidated statements of operations, of comprehensive income/(loss), of changes in shareholders’ equity and of cash flows for each of the three years in the period ended December 31, 2023, including the related notes (collectively referred to as the “consolidated financial statements”). We also have audited the Company’s internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2023 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control - Integrated Framework (2013) issued by the COSO.
Basis for Opinions
The Company’s management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Annual Report on Internal Control over Financial Reporting appearing under Item 15 of Form 20-F. Our responsibility is to express opinions on the Company’s consolidated financial statements and on the Company’s internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
F-2
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that (i) relate to accounts or disclosures that are material to the consolidated financial statements and (ii) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Allocation of transaction price in relation to the license and collaboration agreement with Takeda Pharmaceuticals International AG
As described in Note 18 to the consolidated financial statements, the Company and Takeda Pharmaceuticals International AG entered into an exclusive out-licensing agreement (the “Takeda Agreement”) to further the global development, commercialization and manufacturing of Fruquintinib, under the brand name of Fruzaqla, in territories outside of Mainland China, Hong Kong and Macau (the “Territory”), which resulted in the recognition of US$278.9 million of licensing revenue, US$9.8 million of revenue from manufacturing supply, US$62.4 million of research and development services revenue and US$2.1 million of royalties revenue for the year ended December 31, 2023. The Company evaluated the Takeda Agreement under ASC 606, Revenue from Contracts with Customers (“ASC 606”) and identified three material performance obligations within the arrangement: 1) the licenses for the development and commercialization of Fruquintinib in the Territory and the manufacture of Fruquintinib for use in the Territory (“License obligation”); 2) manufacturing supply of Fruquintinib to support the development and commercialization activities in the Territory (“Manufacturing supply obligation”); and 3) services for research and development of ongoing clinical trials, regulatory submissions and manufacturing technology transfer (“Services obligation”). The standalone selling price of the License obligation and Manufacturing supply obligation are determined using a discounted cash flow method based on the probability-weighted present value of forecasted cash flows associated with out-licensing Fruquintinib in the Territory. The standalone selling price of the Services obligation is determined using a cost plus margin approach based on the present value of estimated future services costs plus a reasonable margin. Management exercises significant estimates and judgments in determining the standalone selling prices for each performance obligation identified and allocates the transaction price to each performance obligation based on the relative standalone selling prices.
The principal considerations for our determination that performing procedures relating to the allocation of transaction price in relation to the Takeda Agreement is a critical audit matter are the significant estimates and judgments by management involved in determining the standalone selling prices for each performance obligation identified, which will affect the timing and amount of revenue recognized for each performance obligation. The estimates of standalone selling price involved management’s key assumptions such as forecasted revenue, probabilities of regulatory approvals, estimated future service costs, margin rates and discount rates. These significant assumptions are subject to a high degree of estimation uncertainty and could be affected by future economic, regulatory and market conditions, which in turn led to a high degree of auditor judgment and significant audit effort in evaluating the audit evidence related to management’s significant assumptions.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included testing the effectiveness of internal controls relating to how management formulated the accounting estimates and assumptions involved in determining the standalone selling prices for each performance obligations identified, and the resulting allocation of transaction price to each performance obligation in the Takeda Agreement. The procedures also included, among others, testing management’s process of allocation of transaction price in relation to the Takeda Agreement, evaluating the valuation methodologies and the discount rates used by management to determine the standalone selling prices of the identified performance obligations with the assistance of our internal valuation specialists, assessing the capability of management’s expert, evaluating the reasonableness of the significant assumptions including forecasted revenue, probabilities of regulatory approvals, estimated future service costs and margin rates, by comparing these significant assumptions to industry, business and market data and information available from third-party sources, evaluating the relevance and reasonableness of the underlying data used by management, and evaluating the sensitivity of the significant assumptions by assessing the changes to revenue recognition amounts for the year ended December 31, 2023 from changes in these assumptions.
F-3
Allowances for credit losses on accounts receivable, other receivables (except for prepayments) and amounts due from related parties
As described in Note 6 to the consolidated financial statements, as of December 31, 2023, the gross balance of accounts receivable was US$117.1 million and an allowance for credit losses of US$0.2 million was made. As described in Note 7 to the consolidated financial statements, as of December 31, 2023, the gross balance of other receivables was US$14.9 million which consisted of the balance of prepayments of US$7.1 million, and no allowance for credit losses was made. As described in Note 24 to the consolidated financial statements, as of December 31, 2023, the gross balance of amounts due from related parties was US$28.5 million, and no allowance for credit losses was made. The allowances for credit losses were made based on estimate of the current expected credit losses to be incurred over the expected life of these receivables.
The principal considerations for our determination that performing procedures relating to the allowances for credit losses on accounts receivable, other receivables (except for prepayments) and amounts due from related parties is a critical audit matter are the significant estimates and judgments by management when developing the current expected credit losses to be incurred over the expected life of these receivables, which in turn led to a high degree of auditor judgment and significant audit effort in evaluating the audit evidence related to management’s significant estimates and judgments, including portfolio groups of accounts receivable, other receivables (except for prepayments) and amounts due from related parties and estimated loss rates.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included testing the effectiveness of internal controls relating to management’s estimate of allowances for credit losses on accounts receivable, other receivables (except for prepayments) and amounts due from related parties. The procedures also included, among others, testing management’s process of developing the allowances for credit losses, testing the accuracy and completeness of the underlying data and the mathematical accuracy of the allowances for credit losses, evaluating the appropriateness of the model and methodology used by management, assessing the reasonableness of portfolio groups of the receivables used by management involved evaluating their credit risk characteristics, assessing the reasonableness of estimated loss rates used by management involved evaluating the historical default rates and application of forward-looking information, with the assistance of our internal valuation specialists.
/s/
February 28, 2024
We have served as the Company’s auditor since 2021.
F-4
CONSOLIDATED FINANCIAL STATEMENTS
HUTCHMED (CHINA) LIMITED
CONSOLIDATED BALANCE SHEETS
(IN US$’000, EXCEPT SHARE DATA)
December 31, | ||||||
| Note |
| 2023 |
| 2022 | |
Assets | ||||||
Current assets | ||||||
Cash and cash equivalents | 5 | |
| | ||
Short-term investments | 5 | |
| | ||
Accounts receivable | 6 | |
| | ||
Other receivables, prepayments and deposits | 7 | |
| | ||
24 | |
| | |||
Inventories | 8 | |
| |||
Total current assets |
| |
| | ||
Property, plant and equipment | 9 |
| ||||
Right-of-use assets | 10 | |||||
Deferred tax assets | 25(ii) |
| ||||
Investments in equity investees | 11 |
| ||||
Other non-current assets | ||||||
Total assets |
|
| ||||
Liabilities and shareholders’ equity | ||||||
Current liabilities | ||||||
Accounts payable | 12 |
| ||||
Other payables, accruals and advance receipts | 13 |
| ||||
Short-term bank borrowings | 14 | — | ||||
Deferred revenue | 18 | |||||
Income tax payable | 25(iii) | |||||
Lease liabilities | 10 | |||||
Total current liabilities |
|
| ||||
Lease liabilities, non-current portion | 10 | |||||
Deferred tax liabilities | 25(ii) |
| ||||
Long-term bank borrowings | 14 |
| ||||
Deferred revenue, non-current portion | 18 |
| ||||
Other non-current liabilities |
|
| ||||
Total liabilities |
| |
| | ||
Commitments and contingencies | 15 | |||||
Company’s shareholders’ equity | ||||||
Ordinary shares; $ | 16 |
| ||||
Additional paid-in capital |
|
| ||||
Accumulated losses |
| ( |
| ( | ||
Accumulated other comprehensive loss |
| ( |
| ( | ||
Total Company’s shareholders’ equity |
|
| ||||
Non-controlling interests |
|
| ||||
Total shareholders’ equity |
|
| ||||
Total liabilities and shareholders’ equity |
|
| ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
HUTCHMED (CHINA) LIMITED
CONSOLIDATED STATEMENTS OF OPERATIONS
(IN US$’000, EXCEPT SHARE AND PER SHARE DATA)
Year Ended December 31, | ||||||||
| Note |
| 2023 |
| 2022 |
| 2021 | |
Revenue | ||||||||
Goods —third parties | | |
| | ||||
—related parties | 24(i) | | |
| | |||
Services —commercialization—third parties | | | | |||||
—research and development—related parties | 24(i) | | |
| | |||
—collaboration research and development—third parties | | |
| | ||||
Other collaboration revenue | ||||||||
—royalties—third parties | | | | |||||
—licensing—third parties | | | | |||||
Total revenue | 18 | |
| |
| | ||
Operating expenses | ||||||||
Costs of good—third parties |
| ( | ( |
| ( | |||
Costs of good—related parties |
| ( | ( |
| ( | |||
Costs of service—commercialization—third parties | ( | ( | ( | |||||
Research and development expenses | 20 | ( | ( |
| ( | |||
Selling expenses |
| ( | ( |
| ( | |||
Administrative expenses |
| ( | ( |
| ( | |||
Total operating expenses |
| ( |
| ( |
| ( | ||
| |
| ( |
| ( | |||
Gain on divestment of an equity investee | 22 | — | — | | ||||
Other income/(expense) | ||||||||
Interest income | 27 | | |
| | |||
Other income | 23 | | |
| | |||
Interest expense | 27 | ( | ( |
| ( | |||
Other expense | 23 | ( | ( |
| ( | |||
Total other income/(expense) |
| |
| ( |
| ( | ||
Income/(loss) before income taxes and equity in earnings of equity investees |
| |
| ( |
| ( | ||
Income tax (expense)/benefit | 25(i) | ( |
| |
| ( | ||
Equity in earnings of equity investees, net of tax | 11 | |
| |
| | ||
Net income/(loss) |
| |
| ( |
| ( | ||
Less: Net income attributable to non-controlling interests |
| ( |
| ( |
| ( | ||
Net income/(loss) attributable to the Company |
| |
| ( |
| ( | ||
Earnings/(losses) per share attributable to the Company (US$ per share) | ||||||||
—basic | 26 | | ( | ( | ||||
—diluted | 26 | | ( | ( | ||||
Number of shares used in per share calculation |
|
| ||||||
—basic | 26 | | | |||||
—diluted | 26 | |||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-6
HUTCHMED (CHINA) LIMITED
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME/(LOSS)
(IN US$’000)
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
Net income/(loss) |
| | ( |
| ( | |
Other comprehensive (loss)/income | ||||||
Foreign currency translation (loss)/gain |
| ( | ( |
| | |
Total comprehensive income/(loss) |
| |
| ( |
| ( |
Less: Comprehensive loss/(income) attributable to non-controlling interests |
| | |
| ( | |
Total comprehensive income/(loss) attributable to the Company |
| |
| ( |
| ( |
The accompanying notes are an integral part of these consolidated financial statements.
F-7
HUTCHMED (CHINA) LIMITED
CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS’ EQUITY
(IN US$’000, EXCEPT SHARE DATA IN ’000)
Accumulated | Total | |||||||||||||||
Ordinary | Ordinary | Additional | Other | Company’s | Non- | Total | ||||||||||
Shares | Shares | Paid-in | Accumulated | Comprehensive | Shareholders’ | controlling | Shareholders’ | |||||||||
| Number |
| Value |
| Capital |
| Losses |
| Income/(Loss) |
| Equity |
| Interests |
| Equity | |
As at January 1, 2021 | |
| |
| |
| ( |
| |
| |
| | | ||
Net (loss)/income |
| — |
| — |
| — |
| ( |
| — | ( |
| |
| ( | |
Issuance in relation to public offering | | | | — | — | | — | | ||||||||
Issuance in relation to private investment in public equity | | | | — | — | | — | | ||||||||
Issuance costs | — | — | ( | — | — | ( | — | ( | ||||||||
Issuances in relation to share option exercises |
| |
| |
| |
| — |
| — |
| |
| — |
| |
Share-based compensation | ||||||||||||||||
Share options |
| — |
| — |
| |
| — |
| — |
| |
| |
| |
Long-term incentive plan (“LTIP”) |
| — |
| — |
| |
| — |
| — |
| |
| |
| |
| — |
| — |
| |
| — |
| — |
| |
| |
| | |
LTIP—treasury shares acquired and held by Trustee |
| — |
| — |
| ( |
| — |
| — |
| ( |
| — |
| ( |
Dividends declared to non-controlling shareholders of subsidiaries (Note 24 (iii)) |
| — |
| — |
| — |
| — |
| — |
| — |
| ( | ( | |
Transfer between reserves | — | — | | ( | — | — | — | — | ||||||||
Divestment of an equity investee (Note 22) | — | — | ( | — | ( | ( | ( | ( | ||||||||
Foreign currency translation adjustments |
| — |
| — |
| — |
| — |
| |
| |
| |
| |
As at December 31, 2021 |
| |
| |
| |
| ( |
| |
| |
| |
| |
Net (loss)/income | — | — | — | ( | — | ( | | ( | ||||||||
Issuances in relation to share option exercises | | | | — | — | | — | | ||||||||
Share-based compensation | ||||||||||||||||
Share options | — | — | | — | — | | | | ||||||||
LTIP | — | — | | — | — | | | | ||||||||
— | — | | — | — | | | | |||||||||
LTIP—treasury shares acquired and held by Trustee (Note 17(ii)) | — | — | ( | — | — | ( | — | ( | ||||||||
Dividends declared to non-controlling shareholders of subsidiaries (Note 24 (iii)) | — | — | — | — | — | — | ( | ( | ||||||||
Transfer between reserves | — | — | | ( | — | — | — | — | ||||||||
Foreign currency translation adjustments | — | — | — | — | ( | ( | ( | ( | ||||||||
As at December 31, 2022 | | | | ( | ( | | | | ||||||||
Net income | — | — | — | | — | | | | ||||||||
Issuances in relation to share option exercises | | | | — | — | | — | | ||||||||
Share-based compensation | ||||||||||||||||
Share options | — | — | | — | — | | | | ||||||||
LTIP | — | — | | — | — | | ( | | ||||||||
— | — | | — | — | | | | |||||||||
LTIP—treasury shares acquired and held by Trustee (Note 17(ii)) | — | — | ( | — | — | ( | — | ( | ||||||||
Dividends declared to non-controlling shareholders of subsidiaries (Note 24 (iii)) | — | — | — | — | — | — | ( | ( | ||||||||
Transfer between reserves | — | — | | ( | — | — | — | — | ||||||||
Divestment of subsidiaries | — | — | ( | — | ( | ( | ( | ( | ||||||||
Divestment of other equity investee | — | — | ( | — | | ( | — | ( | ||||||||
Foreign currency translation adjustments | — | — | — | — | ( | ( | ( | ( | ||||||||
As at December 31, 2023 | | | | ( | ( | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-8
HUTCHMED (CHINA) LIMITED
CONSOLIDATED STATEMENTS OF CASH FLOWS
(IN US$’000)
Year Ended December 31, | ||||||||
| Note |
| 2023 |
| 2022 |
| 2021 | |
Net cash generated from/(used in) operating activities |
| 28 |
| | ( |
| ( | |
Investing activities | ||||||||
Purchases of property, plant and equipment |
|
| ( | ( |
| ( | ||
Purchase of leasehold land | — | — | ( | |||||
Refund of leasehold land deposit | — | — | | |||||
Deposits in short-term investments |
|
| ( | ( |
| ( | ||
Proceeds from short-term investments | | | | |||||
Purchase of a warrant | 19 | — | — | ( | ||||
Dividend and proceeds received from divestment of Hutchison Whampoa Guangzhou Baiyunshan Chinese Medicine Company Limited (“HBYS”) | 22 | | | | ||||
Proceeds from divestment of other equity investee | — | | — | |||||
Proceeds from divestment of subsidiaries | 24(i) | | — | — | ||||
Cash disposed from divestment of subsidiaries | ( | — | — | |||||
Net cash (used in)/generated from investing activities |
|
| ( | |
| ( | ||
Financing activities | ||||||||
Proceeds from issuances of ordinary shares |
|
| | |
| | ||
Purchases of treasury shares |
| 17(ii) |
| ( | ( |
| ( | |
Dividends paid to non-controlling shareholders of subsidiaries |
| 24(iii) |
| ( | ( |
| ( | |
Repayment of loan to a non-controlling shareholder of a subsidiary |
|
| — | — |
| ( | ||
Proceeds from bank borrowings |
|
| | |
| — | ||
Repayment of bank borrowings |
|
| — | ( |
| — | ||
Payment of issuance costs |
|
| — | ( |
| ( | ||
Net cash generated from/(used in)financing activities |
|
| | ( |
| | ||
Net (decrease)/increase in cash and cash equivalents |
|
| ( | ( |
| | ||
Effect of exchange rate changes on cash and cash equivalents |
|
| ( | ( |
| | ||
|
| ( | ( |
| | |||
Cash and cash equivalents | ||||||||
Cash and cash equivalents at beginning of year |
|
| | |
| | ||
Cash and cash equivalents at end of year |
|
| | |
| | ||
Supplemental disclosure for cash flow information | ||||||||
Cash paid for interest |
|
| | |
| | ||
Cash paid for tax, net of refunds |
| 25(iii) |
| | |
| | |
Supplemental disclosure for non-cash activities | ||||||||
Increase in accrued capital expenditures | | | | |||||
Vesting of treasury shares for LTIP | 17(ii) | | | | ||||
The accompanying notes are an integral part of these consolidated financial statements.
F-9
HUTCHMED (CHINA) LIMITED
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
1. Organization and Nature of Business
HUTCHMED (China) Limited (the “Company”) and its subsidiaries (together the “Group”) are principally engaged in researching, developing, manufacturing and marketing pharmaceutical products. The Group and its equity investee have research and development facilities and manufacturing plants in the People’s Republic of China (the “PRC”) and sell their products mainly in the PRC, including Hong Kong and Macau. In addition, the Group has established international operations in the United States of America (the “U.S.”) and Europe.
The Company’s ordinary shares are listed on the Main Board of The Stock Exchange of Hong Kong Limited (“HKEX”) and the AIM market of the London Stock Exchange, and its American depositary shares (“ADS”) are traded on the Nasdaq Global Select Market.
Liquidity
As at December 31, 2023, the Group had accumulated losses of US$
Based on the Group’s operating plan, the existing cash and cash equivalents, short-term investments and unutilized bank borrowing facilities are considered to be sufficient to meet the cash requirements to fund planned operations and other commitments for at least the next twelve months from the issuance date of the consolidated financial statements.
F-10
2. Particulars of Principal Subsidiaries and Equity Investee
Equity interest | ||||||||
Place of | attributable to | |||||||
establishment | the Group | |||||||
and | December 31, | |||||||
Name |
| operations |
| 2023 |
| 2022 |
| Principal activities |
Subsidiaries | ||||||||
HUTCHMED Limited |
| PRC |
| | % | | % | Research, development, manufacture and commercialization of pharmaceutical products |
HUTCHMED International Corporation |
| U.S. |
| | % | | % | Provision of professional, scientific and technical support services |
Hutchison Whampoa Sinopharm Pharmaceuticals (Shanghai) Company Limited (“HSPL”) |
| PRC |
| | % | | % | Provision of sales, distribution and marketing services to pharmaceutical manufacturers |
Hutchison Healthcare Limited |
| PRC |
| | % | | % | Manufacture and distribution of healthcare products |
Hutchison Hain Organic (Hong Kong) Limited (“HHOHK”) (note) |
| Hong Kong |
| — | % | | % | Wholesale and trading of healthcare and consumer products |
HUTCHMED Science Nutrition Limited (“HSN”) (note) |
| Hong Kong |
| — | % | | % | Wholesale and trading of healthcare and consumer products |
Equity investee | ||||||||
SHPL | PRC | | % | | % | Manufacture and distribution of prescription drug products | ||
Note: On December 7, 2023, the Group completed a transaction to divest its entire investment in HHOHK and HSN to Hutchison Whampoa (China) Limited, an indirect subsidiary of CK Hutchison Holdings Limited (“CK Hutchison”) (Note 24(i)).
3. Summary of Significant Accounting Policies
Principles of Consolidation and Basis of Presentation
The accompanying consolidated financial statements reflect the accounts of the Company and all of its subsidiaries in which a controlling interest is maintained. When a subsidiary is deconsolidated from the date that control ceases, any gain or loss on the divestment of the interest sold is recognized in profit or loss. Amounts previously recognized in other comprehensive income/(loss) for the subsidiary are transferred to the consolidated statements of operations as part of the gain or loss on the divestment. All inter-company balances and transactions have been eliminated in consolidation. The consolidated financial statements have been prepared in conformity with generally accepted accounting principles in the U.S. (“U.S. GAAP”).
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting period.
Foreign Currency Translation
The Company’s presentation currency and functional currency is the U.S. dollar (“US$”). The financial statements of its subsidiaries with a functional currency other than the US$ have been translated into the Company’s presentation currency. All assets and liabilities of the subsidiaries are translated using year-end exchange rates and revenue and expenses are translated at average exchange rates for the year. Translation adjustments are reflected in accumulated other comprehensive income /(loss) in shareholders’ equity.
Net foreign currency exchange gains/(losses) of US$
F-11
Foreign Currency Risk
The Group’s operating transactions and its assets and liabilities in the PRC are mainly denominated in Renminbi (“RMB”), which is not freely convertible into foreign currencies. The Group’s cash and cash equivalents denominated in RMB are subject to government controls. The value of the RMB is subject to fluctuations from central government policy changes and international economic and political developments that affect the supply and demand of RMB in the foreign exchange market. In the PRC, certain foreign exchange transactions are required by law to be transacted only by authorized financial institutions at exchange rates set by the People’s Bank of China (the “PBOC”). Remittances in currencies other than RMB by the Group in the PRC must be processed through the PBOC or other PRC foreign exchange regulatory bodies which require certain supporting documentation in order to complete the remittance.
Allowance for Current Expected Credit Losses and Concentration of Credit Risk
Financial instruments that potentially expose the Group to credit risk consist primarily of cash and cash equivalents, short-term investments, and financial assets not carried at fair value including accounts receivable and other receivables.
The Group recognizes an allowance for current expected credit losses (“CECLs”) on financial assets not carried at fair value. CECLs are calculated over the expected life of the financial assets on an individual or a portfolio basis considering information available about the counterparties’ credit situation and collectability of the specific cash flows, including information about past events, current conditions and future forecasts.
The Group places substantially all of its cash and cash equivalents and short-term investments in major financial institutions, which management believes are of high credit quality. The Group has a practice to limit the amount of credit exposure to any particular financial institution. Additionally, the Group has policies in place to ensure that sales are made to customers with an appropriate credit history and the Group performs periodic credit evaluations of its customers. Normally the Group does not require collateral from trade debtors. The Group has not had any material credit losses.
Cash and Cash Equivalents
The Group considers all highly liquid investments purchased with original maturities of three months or less to be cash equivalents. Cash and cash equivalents consist primarily of cash on hand and bank deposits and are stated at cost, which approximates fair value.
Short-term Investments
Short-term investments include deposits placed with banks with original maturities of more than three months but less than one year.
Accounts Receivable
Accounts receivable are stated at the amount management expects to collect from customers based on their outstanding invoices. The allowance for CECLs reflects the Group’s current estimate of credit losses expected to be incurred over the life of the receivables. The Group considers various factors in establishing, monitoring, and adjusting its allowance for CECLs including the aging of the accounts and aging trends, the historical level of charge-offs, and specific exposures related to particular customers. The Group also monitors other risk factors and forward-looking information, such as country risk, when determining credit limits for customers and establishing adequate allowances for CECLs. Accounts receivable are written off after all reasonable means to collect the full amount (including litigation, where appropriate) have been exhausted.
Inventories
Inventories are stated at the lower of cost or net realizable value. Cost is determined using the weighted average cost method. The cost of finished goods comprises raw materials, direct labor, other direct costs and related production overheads based on normal operating capacity. Net realizable value is the estimated selling price in the ordinary course of business, less applicable variable selling expenses. A provision for excess and obsolete inventory will be made based primarily on forecasts of product demand and production requirements. The excess balance determined by this analysis becomes the basis for excess inventory charge and the written-down value of the inventory becomes its cost. Written-down inventory is not written up if market conditions improve.
F-12
Property, Plant and Equipment
Property, plant and equipment consist of buildings, leasehold improvements, plant and equipment, furniture and fixtures, other equipment and motor vehicles. Property, plant and equipment are stated at cost, net of accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the depreciable assets.
Buildings |
| |
Plant and equipment |
| |
Furniture and fixtures, other equipment and motor vehicles |
| |
Leasehold improvements |
| Shorter of (a) |
Additions and improvements that extend the useful life of an asset are capitalized. Repairs and maintenance costs are expensed as incurred.
Impairment of Long-Lived Assets
The Group evaluates the recoverability of long-lived assets in accordance with authoritative guidance on accounting for the impairment or disposal of long-lived assets. The Group evaluates long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying value of these assets may not be recoverable. If indicators of impairment exist, the first step of the impairment test is performed to assess if the carrying value of the net assets exceeds the undiscounted cash flows of the assets. If yes, the second step of the impairment test is performed in order to determine if the carrying value of the net assets exceeds the fair value. If yes, impairment is recognized for the excess.
Investments in Equity Investees
Investments in equity investees over which the Group has significant influence are accounted for using the equity method. The Group evaluates equity method investments for impairment when events or circumstances suggest that their carrying amounts may not be recoverable. An impairment charge would be recognized in earnings for a decline in value that is determined to be other-than-temporary after assessing the severity and duration of the impairment and the likelihood of recovery before disposal. The investments are recorded at fair value only if impairment is recognized.
Leasehold Land
Leasehold land represents fees paid to acquire the right to use the land on which various plants and buildings are situated for a specified period of time from the date the respective right was granted and are stated at cost less accumulated amortization and impairment loss, if any. Amortization is computed using the straight-line basis over the lease period of
Goodwill
Goodwill represents the excess of the purchase price plus fair value of non-controlling interests over the fair value of identifiable assets and liabilities acquired. Goodwill is not amortized, but is tested for impairment at the reporting unit level on at least an annual basis or when an event occurs or circumstances change that would more likely than not reduce the fair value of a reporting unit below its carrying amount. When performing an evaluation of goodwill impairment, the Group has the option to first assess qualitative factors, such as significant events and changes to expectations and activities that may have occurred since the last impairment evaluation, to determine if it is more likely than not that goodwill might be impaired. If as a result of the qualitative assessment, that it is more likely than not that the fair value of the reporting unit is less than its carrying amount, the quantitative fair value test is performed to determine if the fair value of the reporting unit exceeds its carrying value.
Other Intangible Assets
Other intangible assets with finite useful lives are carried at cost less accumulated amortization and impairment loss, if any. Amortization is computed using the straight-line basis over the estimated useful lives of the assets.
F-13
Borrowings
Borrowings are recognized initially at fair value, net of debt issuance costs incurred. Borrowings are subsequently stated at amortized cost; any difference between the proceeds (net of debt issuance costs) and the redemption value is recognized in the consolidated statements of operations over the period of the borrowings using the effective interest method.
Ordinary Shares
The Company’s ordinary shares are stated at par value of US$
The Company’s ordinary shares are traded in the form of ordinary shares and ADS. Each ADS represents
Treasury Shares
The Group accounts for treasury shares under the cost method. The treasury shares are purchased for the purpose of the LTIP and held by a trustee appointed by the Group (the “Trustee”) prior to vesting.
Share-Based Compensation
Share options
The Group recognizes share-based compensation expense on share options granted to employees and directors based on their estimated grant date fair value using the Polynomial model. This Polynomial pricing model uses various inputs to measure fair value, including the market value of the Company’s underlying ordinary shares at the grant date, contractual terms, estimated volatility, risk-free interest rates and expected dividend yields. The Group recognizes share-based compensation expense in the consolidated statements of operations on a graded vesting basis over the requisite service period, and accounts for forfeitures as they occur.
Share options are classified as equity-settled awards. Share-based compensation expense, when recognized, is charged to the consolidated statements of operations with the corresponding entry to additional paid-in capital.
LTIP
The Group recognizes the share-based compensation expense on the LTIP awards based on a fixed or determinable monetary amount on a straight-line basis for each annual tranche awarded over the requisite period. For LTIP awards with performance targets, prior to their determination date, the amount of LTIP awards that is expected to vest takes into consideration the achievement of the performance conditions and the extent to which the performance conditions are likely to be met. Performance conditions vary by awards, and may include targets for shareholder returns, financings, revenue, net income after taxes and the achievement of clinical, regulatory, business development and manufacturing milestones.
These LTIP awards are classified as liability-settled awards before the determination date (i.e. the date when the achievement of any performance conditions are known), as they settle in a variable number of shares based on a determinable monetary amount, which is determined upon the actual achievement of performance targets. As the extent of achievement of the performance targets is uncertain prior to the determination date, a probability based on management’s assessment of the achievement of the performance targets has been assigned to calculate the amount to be recognized as an expense over the requisite period.
After the determination date or if the LTIP awards have no performance conditions, the LTIP awards are classified as equity-settled awards. If the performance target is achieved, the Group will pay the determined monetary amount to the Trustee to purchase ordinary shares of the Company or the equivalent ADS. Any cumulative compensation expense previously recognized as a liability will be transferred to additional paid-in capital. If the performance target is not achieved, no ordinary shares or ADS of the Company will be purchased and the amount previously recorded in the liability will be reversed and included in the consolidated statements of operations.
F-14
Defined Contribution Plans
The Group’s subsidiaries in the PRC participate in a government-mandated multi-employer defined contribution plan pursuant to which certain retirement, medical and other welfare benefits are provided to employees. The relevant labor regulations require the Group’s subsidiaries in the PRC to pay the local labor and social welfare authority’s monthly contributions at a stated contribution rate based on the monthly basic compensation of qualified employees. The relevant local labor and social welfare authorities are responsible for meeting all retirement benefits obligations and the Group’s subsidiaries in the PRC have no further commitments beyond their monthly contributions. The contributions to the plan are expensed as incurred.
The Group also makes payments to other defined contribution plans for the benefit of employees employed by subsidiaries outside the PRC. The defined contribution plans are generally funded by the relevant companies and by payments from employees.
The Group’s contributions to defined contribution plans for the years ended December 31, 2023, 2022 and 2021 were US$
Revenue Recognition
Revenue is measured based on consideration specified in a contract with a customer, and excludes any sales incentives and amounts collected on behalf of third parties. Taxes assessed by a governmental authority that are both imposed on and concurrent with a specific revenue-producing transaction, that are collected by the Group from a customer, are also excluded from revenue. The Group recognizes revenue when it satisfies a performance obligation by transferring control over a good, service or license to a customer.
| (i) | Goods and services |
The Group principally generates revenue from (1) sales of goods, which are the manufacture or purchase and distribution of pharmaceutical products and other consumer health products, and (2) provision of services, which are the provision of sales, distribution and marketing services to pharmaceutical manufacturers. The Group evaluates whether it is the principal or agent for these contracts. Where the Group obtains control of the goods for distribution, it is the principal (i.e. recognizes sales of goods on a gross basis). Where the Group does not obtain control of the goods for distribution, it is the agent (i.e. recognizes provision of services on a net basis). Control is primarily evidenced by taking physical possession and inventory risk of the goods.
Revenue from sales of goods is recognized when the customer takes possession of the goods. This usually occurs upon completed delivery of the goods to the customer site. The amount of revenue recognized is adjusted for expected sales incentives as stipulated in the contract, which are generally issued to customers as direct discounts at the point-of-sale or indirectly in the form of rebates. Sales incentives are estimated using the expected value method. Additionally, sales are generally made with a limited right of return under certain conditions. Revenue is recorded net of provisions for sales discounts and returns.
Revenue from provision of services is recognized when the benefits of the services transfer to the customer over time, which is based on the proportionate value of services rendered as determined under the terms of the relevant contract. Additionally, when the amounts that can be invoiced correspond directly with the value to the customer for performance completed to date, the Group recognizes revenue from provision of services based on amounts that can be invoiced to the customer.
Deferred revenue is recognized if consideration is received in advance of transferring control of the goods or rendering of services. Accounts receivable is recognized if the Group has an unconditional right to bill the customer, which is generally when the customer takes possession of the goods or services are rendered. Payment terms differ by subsidiary and customer, but generally range from
| (ii) | License and collaboration contracts |
The Group’s Oncology/Immunology reportable segment includes revenue generated from license and collaboration contracts, which generally contain multiple performance obligations including (1) the licenses to the development, commercialization and manufacture rights of a drug compound, (2) the research and development services for each specified treatment indication, and (3) other deliverables, which are accounted for separately if they are distinct, i.e. if a product or service is separately identifiable from other items in the arrangement and if a customer can benefit from it on its own or with other resources that are readily available to the customer.
F-15
The transaction price generally includes fixed and variable consideration in the form of upfront payment, research and development cost reimbursements, contingent milestone payments and sales-based royalties. Contingent milestone payments are not included in the transaction price until it becomes probable that a significant reversal of revenue will not occur, which is generally when the specified milestone is achieved. The allocation of the transaction price to each performance obligation is based on the relative standalone selling prices of each performance obligation determined at the inception of the contract. The Group estimates the standalone selling prices based on the income approach and cost plus margin approach. Control of the license to the drug compounds transfers at the inception date of the collaboration agreements and consequently, amounts allocated to this performance obligation are generally recognized at a point in time. Conversely, research and development services for each specified indication are performed over time and amounts allocated to these performance obligations are generally recognized over time using a percentage-of-completion method. The Group has determined that research and development expenses provide an appropriate depiction of measure of progress for the research and development services. Changes to estimated cost inputs may result in a cumulative catch-up adjustment. Royalty revenue is recognized as future sales occur as they meet the requirements for the sales-usage based royalty exception.
Deferred revenue is recognized if allocated consideration is received in advance of the Group rendering research and development services or earning royalties on future sales. Accounts receivable is recognized based on the terms of the contract and when the Group has an unconditional right to bill the customer, which is generally when research and development services are rendered.
Research and Development Expenses
Research and development expenses include the following: (i) research and development costs, which are expensed as incurred; (ii) acquired in-process research and development (“IPR&D”) expenses, which include the initial costs of externally developed IPR&D projects, acquired directly in a transaction other than a business combination, that do not have an alternative future use; and (iii) milestone payment obligations for externally developed IPR&D projects incurred prior to regulatory approval of the product in the in-licensed territory, which are accrued when the event requiring payment of the milestone occurs (milestone payment obligations incurred upon regulatory approval are recorded as other intangible assets).
Collaborative Arrangements
The Group enters into collaborative arrangements with collaboration partners that fall under the scope of Accounting Standards Codification (“ASC”) 808, Collaborative Arrangements (“ASC 808”). The Group records all expenditures for such collaborative arrangements in research and development expenses as incurred, including payments to third party vendors and reimbursements to collaboration partners, if any. Reimbursements from collaboration partners are recorded as reductions to research and development expenses and accrued when they can be contractually claimed.
Government Grants
Grants from governments are recognized at their fair values. Government grants that are received in advance are deferred and recognized in the consolidated statements of operations over the period necessary to match them with the costs that they are intended to compensate. Government grants in relation to the achievement of stages of research and development projects are recognized in the consolidated statements of operations when amounts have been received and all attached conditions have been met. Non-refundable grants received without any further obligations or conditions attached are recognized immediately in the consolidated statements of operations.
Leases
In an operating lease, a lessee obtains control of only the use of the underlying asset, but not the underlying asset itself. An operating lease is recognized as a right-of-use asset with a corresponding liability at the date which the leased asset is available for use by the Group. The Group recognizes an obligation to make lease payments equal to the present value of the lease payments over the lease term. The lease terms may include options to extend or terminate the lease when it is reasonably certain that the Group will exercise that option.
F-16
Lease liabilities include the net present value of the following lease payments: (i) fixed payments; (ii) variable lease payments that depend on an index or a rate; and (iii) payments of penalties for terminating the lease if the lease term reflects the lessee exercising that option, if any. Lease liabilities exclude the following payments that are generally accounted for separately: (i) non-lease components, such as maintenance and security service fees and value added tax, and (ii) any payments that a lessee makes before the lease commencement date. The lease payments are discounted using the interest rate implicit in the lease or if that rate cannot be determined, the lessee’s incremental borrowing rate being the rate that the lessee would have to pay to borrow the funds in its currency and jurisdiction necessary to obtain an asset of similar value, economic environment and terms and conditions.
An asset representing the right to use the underlying asset during the lease term is recognized that consists of the initial measurement of the operating lease liability, any lease payments made to the lessor at or before the commencement date less any lease incentives received, any initial direct cost incurred by the Group and any restoration costs.
After commencement of the operating lease, the Group recognizes lease expenses on a straight-line basis over the lease term. The right-of-use asset is subsequently measured at cost less accumulated amortization and any impairment provision. The amortization of the right-of-use asset represents the difference between the straight-line lease expense and the accretion of interest on the lease liability each period. The interest amount is used to accrete the lease liability and to amortize the right-of-use asset. There is no amount recorded as interest expense.
Payments associated with short-term leases are recognized as lease expenses on a straight-line basis over the period of the leases.
Subleases of right-of-use assets are accounted for similar to other leases. As an intermediate lessor, the Group separately accounts for the head-lease and sublease unless it is relieved of its primary obligation under the head-lease. Sublease income is recorded on a gross basis separate from the head-lease expenses. If the total remaining lease cost on the head-lease is more than the anticipated sublease income for the lease term, this is an indicator that the carrying amount of the right-of-use asset associated with the head-lease may not be recoverable, and the right-of-use asset will be assessed for impairment.
Income Taxes
The Group accounts for income taxes under the liability method. Under the liability method, deferred income tax assets and liabilities are determined based on the differences between the financial reporting and income tax bases of assets and liabilities and are measured using the income tax rates that will be in effect when the differences are expected to reverse. A valuation allowance is recorded when it is more likely than not that some of the net deferred income tax asset will not be realized.
The Group accounts for an uncertain tax position in the consolidated financial statements only if it is more likely than not that the position is sustainable based on its technical merits and consideration of the relevant tax authority’s widely understood administrative practices and precedents. If the recognition threshold is met, the Group records the largest amount of tax benefit that is greater than 50 percent likely to be realized upon ultimate settlement.
The Group recognizes interest and penalties for income taxes, if any, under income tax payable on its consolidated balance sheets and under other expense in its consolidated statements of operations.
Earnings/(losses) per Share
Basic earnings/(losses) per share is computed by dividing net income/(loss) attributable to the Company by the weighted average number of outstanding ordinary shares in issue during the year. Weighted average number of outstanding ordinary shares in issue excludes treasury shares.
Diluted earnings/(losses) per share is computed by dividing net income/(loss) attributable to the Company by the weighted average number of outstanding ordinary shares in issue and dilutive ordinary share equivalents outstanding during the year. Dilutive ordinary share equivalents include ordinary shares and treasury shares issuable upon the exercise or settlement of share-based awards or warrants issued by the Company using the treasury stock method. The computation of diluted earnings/(losses) per share does not assume conversion, exercise, or contingent issuance of securities that would have an anti-dilutive effect.
F-17
Segment Reporting
Operating segments are reported in a manner consistent with the internal reporting provided to the chief executive officer who is the Group’s chief operating decision maker. The chief operating decision maker reviews the Group’s internal reporting in order to assess performance and allocate resources.
Profit Appropriation and Statutory Reserves
The Group’s subsidiaries and equity investee established in the PRC are required to make appropriations to certain non-distributable reserve funds.
In accordance with the relevant laws and regulations established in the PRC, the Company’s subsidiaries registered as wholly-owned foreign enterprise have to make appropriations from their after-tax profits (as determined under generally accepted accounting principles in the PRC (“PRC GAAP”)) to reserve funds including general reserve fund, enterprise expansion fund and staff bonus and welfare fund. The appropriation to the general reserve fund must be at least
In addition, Chinese domestic companies must make appropriations from their after-tax profits as determined under PRC GAAP to non-distributable reserve funds including statutory surplus fund and discretionary surplus fund. The appropriation to the statutory surplus fund must be
The use of the general reserve fund, enterprise expansion fund, statutory surplus fund and discretionary surplus fund is restricted to the offsetting of losses or increases to the registered capital of the respective company. The staff bonus and welfare fund is a liability in nature and is restricted to fund payments of special bonus to employees and for the collective welfare of employees. All these reserves are not permitted to be transferred to the company as cash dividends, loans or advances, nor can they be distributed except under liquidation.
4. Fair Value Disclosures
Cash equivalents, short-term investments, accounts receivable, other receivables, accounts payable and other payables are carried at cost, which approximates fair value due to the short-term nature of these financial instruments. Bank borrowings are floating rate instruments and carried at amortized cost, which approximates fair values.
5. Cash and Cash Equivalents and Short-term Investments
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Cash and Cash Equivalents | ||||
Cash at bank and on hand |
| |
| |
Bank deposits maturing in three months or less |
| |
| |
|
| | ||
Short-term Investments | ||||
Bank deposits maturing over three months (note) | | | ||
| | | ||
Note: The maturities for short-term investments ranged from
F-18
Certain cash and bank balances denominated in RMB, US$ and UK Pound Sterling (“£”) were deposited with banks in the PRC. The conversion of these balances into foreign currencies is subject to the rules and regulations of foreign exchange control promulgated by the PRC government. Cash and cash equivalents and short-term investments were denominated in the following currencies:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
US$ | | | ||
RMB | | | ||
Hong Kong dollar (“HK$”) | | | ||
£ | | | ||
Others | | | ||
| |
| | |
6. Accounts Receivable
Accounts receivable from contracts with customers consisted of the following:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Accounts receivable—third parties |
| |
| |
Accounts receivable—related parties (Note 24(ii)) |
| |
| |
Allowance for credit losses | ( | ( | ||
Accounts receivable, net |
| |
| |
Substantially all accounts receivable are denominated in RMB, US$ and HK$ and are due within one year from the end of the reporting periods. The carrying values of accounts receivable approximate their fair values due to their short-term maturities.
An aging analysis for accounts receivable—third parties based on the relevant invoice dates is as follows:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Not later than 3 months |
| |
| |
Between 3 months to 6 months |
| |
| |
Between 6 months to 1 year |
| |
| |
Later than 1 year |
| |
| |
Accounts receivable—third parties |
| |
| |
Movements on the allowance for credit losses:
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
As at January 1 |
| | |
| | |
Increase in allowance for credit losses |
| | |
| | |
Decrease in allowance due to subsequent collection | ( | ( | ( | |||
Exchange difference |
| ( | ( |
| | |
Divestment of subsidiaries | ( | — | — | |||
As at December 31 |
| |
| |
| |
F-19
7. Other receivables, prepayments and deposits
Other receivables, prepayments and deposits consisted of the following:
| December 31, | |||
| 2023 |
| 2022 | |
| (in US$’000) | |||
Prepayments | | | ||
Interest receivables | | | ||
Value-added tax receivables |
| |
| |
Deposits | | | ||
Dividend receivables (Note 22) | — | | ||
Others |
| |
| |
| |
| | |
8. Inventories
Inventories, net of provision for excess and obsolete inventories, consisted of the following:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Raw materials |
| |
| |
Finished goods |
| |
| |
| |
| | |
F-20
9. Property, Plant and Equipment
Property, plant and equipment consisted of the following:
Furniture | ||||||||||||
and | ||||||||||||
fixtures, | ||||||||||||
other | ||||||||||||
Plant | equipment | |||||||||||
Leasehold | and | and motor | Construction | |||||||||
| Buildings |
| improvements |
| equipment |
| vehicles |
| in progress |
| Total | |
(in US$’000) | ||||||||||||
Cost |
|
|
|
|
|
|
|
|
|
|
|
|
As at January 1, 2023 |
| |
| |
| |
| |
| |
| |
Additions |
| — |
| |
| |
| |
| |
| |
Disposals |
| — |
| — |
| ( |
| ( |
| — |
| ( |
Divestment of subsidiaries | — | ( | — | ( | — | ( | ||||||
Transfers |
| |
| |
| |
| |
| ( |
| — |
Exchange differences |
| ( |
| ( |
| ( |
| ( |
| ( |
| ( |
As at December 31, 2023 |
| |
| |
| |
| |
| |
| |
Accumulated depreciation and impairment |
|
|
|
|
|
|
|
|
|
|
| |
As at January 1, 2023 |
| |
| |
| |
| |
| — |
| |
Depreciation |
| |
| |
| |
| |
| — |
| |
Impairment | — | | | | — | | ||||||
Disposals |
| — |
| — |
| ( |
| ( |
| — |
| ( |
Divestment of subsidiaries | — | ( | — | ( | — | ( | ||||||
Exchange differences |
| ( |
| ( |
| ( |
| ( |
| — |
| ( |
As at December 31, 2023 |
| |
| |
| |
| |
| — |
| |
Net book value |
|
|
|
|
|
| ||||||
As at December 31, 2023 |
| |
| |
| |
| |
| |
| |
F-21
Furniture | ||||||||||||
and | ||||||||||||
fixtures, | ||||||||||||
other | ||||||||||||
Plant | equipment | |||||||||||
Leasehold | and | and motor | Construction | |||||||||
| Buildings |
| improvements |
| equipment |
| vehicles |
| in progress |
| Total | |
(in US$’000) | ||||||||||||
Cost |
|
|
|
|
|
|
|
|
|
|
|
|
As at January 1, 2022 |
| |
| |
| |
| |
| |
| |
Additions |
| — |
| |
| |
| |
| |
| |
Disposals |
| — |
| ( |
| ( |
| ( |
| — |
| ( |
Transfers |
| — |
| |
| |
| |
| ( |
| — |
Exchange differences |
| ( |
| ( |
| ( |
| ( |
| ( |
| ( |
As at December 31, 2022 |
| |
| |
| |
| |
| |
| |
Accumulated depreciation |
|
|
|
|
|
|
|
|
|
| ||
As at January 1, 2022 |
| |
| |
| |
| |
| — |
| |
Depreciation |
| |
| |
| |
| |
| — |
| |
Disposals |
| — |
| ( |
| ( |
| ( |
| — |
| ( |
Transfers | — | — | ( | | — | — | ||||||
Exchange differences |
| ( |
| ( |
| ( |
| ( |
| — |
| ( |
As at December 31, 2022 |
| |
| |
| |
| |
| — |
| |
Net book value |
|
|
|
|
|
|
|
|
|
|
| |
As at December 31, 2022 |
| |
| |
| |
| |
| |
| |
10. Leases
Leases consisted of the following:
| December 31, | |||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Right‑of‑use assets |
|
|
|
|
Offices |
| |
| |
Factories | | | ||
Warehouse (note) |
| |
| |
Others |
| |
| |
Total right‑of‑use assets |
| |
| |
Lease liabilities, current portion |
| |
| |
Lease liabilities, non‑current portion |
| |
| |
Total lease liabilities |
| |
| |
Note: Comprised of a warehouse in Suzhou that is leased through June 2026 in which the contract has a termination option with
F-22
Lease activities are summarized as follows:
| Year Ended December 31, | |||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Lease expenses: |
|
|
|
|
Short‑term leases with lease terms equal or less than 12 months |
| |
| |
Leases with lease terms greater than 12 months |
| |
| |
Impairment |
| |
| — |
| | |||
Cash paid on lease liabilities |
| |
| |
Non-cash: Lease liabilities recognized from obtaining right-of-use assets | | | ||
Non-cash: Lease liabilities changed in relation to modifications and terminations |
| — |
| ( |
Lease contracts are typically within a period of
Future lease payments are as follows:
| December 31, | |
| 2023 | |
(in US$’000) | ||
Lease payments: |
|
|
Not later than 1 year |
| |
Between 1 to 2 years |
| |
Between 2 to 3 years |
| |
Between 3 to 4 years |
| |
Between 4 to 5 years |
| |
Total lease payments |
| |
Less: Discount factor |
| ( |
Total lease liabilities |
| |
11. Investments in Equity Investees
Investments in equity investees consisted of the following:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
SHPL |
| |
| |
Other (note) |
| — |
| |
| |
| | |
Note: On April 13, 2023, the Group completed a transaction to divest its entire investment in a former equity investee to a third party.
The equity investees are private companies and there are no quoted market prices available for their shares.
F-23
Summarized financial information for the significant equity investees, SHPL and HBYS (divested in 2021), is as follows:
| (i) | Summarized balance sheets |
SHPL | ||||
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Current assets |
| |
| |
Non-current assets |
| |
| |
Current liabilities |
| ( |
| ( |
Non-current liabilities |
| ( | ( | |
Net assets |
| | | |
| (ii) | Summarized statements of operations |
SHPL | HBYS | |||||||
Period Ended | ||||||||
Year Ended December 31, | September 28, | |||||||
| 2023 |
| 2022 |
| 2021 |
| 2021(note (a)) | |
(in US$’000) | ||||||||
Revenue |
| | | |
| | ||
Gross profit |
| | | |
| | ||
Interest income |
| | | |
| | ||
Profit before taxation |
| | | |
| | ||
Income tax expense (note (b)) |
| ( | ( | ( |
| ( | ||
Net income (note (c)) | | | | | ||||
Non-controlling interests | — | — | — | ( | ||||
Net income attributable to the shareholders of equity investee |
| | | | | |||
Notes:
| (a) | The summarized statement of operations for HBYS for the year ended December 31, 2021 includes the period when HBYS was the Group’s equity investee from January 1, 2021 to September 28, 2021, the completion date of the divestment. The Group has accounted for the investment in HBYS under the equity method up to September 28, 2021. |
| (b) | The main entity within the SHPL group has been granted the High and New Technology Enterprise (“HNTE”) status. Accordingly, the entity was eligible to use a preferential income tax rate of |
| (c) | Net income is before elimination of unrealized profits on transactions with the Group. The amounts eliminated were approximately US$ |
F-24
| (iii) | Reconciliation of summarized financial information |
Reconciliation of the summarized financial information presented to the carrying amount of investments in equity investees is as follows:
SHPL | HBYS | |||||||
| 2023 |
| 2022 |
| 2021 |
| 2021(note) | |
(in US$’000) | ||||||||
Opening net assets after non-controlling interests as at January 1 |
| | |
| | | ||
Net income attributable to the shareholders of equity investee |
| | |
| | | ||
Dividends declared |
| ( | ( |
| ( | ( | ||
Other comprehensive income/(loss) | | ( | | | ||||
Closing net assets after non-controlling interests as at December 31/September 28 |
| | |
| | | ||
Group’s share of net assets |
| | |
| | | ||
Goodwill | | | | — | ||||
Elimination of unrealized profits on downstream sales | ( | ( | — | — | ||||
Divestment (Note 22) | — | — | — | ( | ||||
Carrying amount of investments as at December 31 |
| | |
| | — | ||
Note: The summarized financial information for HBYS for the year ended December 31, 2021 includes the period when HBYS was the Group’s equity investee from January 1, 2021 to September 28, 2021, the completion date of the divestment. The Group has accounted for the investment in HBYS under the equity method up to September 28, 2021.
SHPL had the following capital commitments:
December 31, | ||
| 2023 | |
(in US$’000) | ||
Property, plant and equipment | ||
Contracted but not provided for |
| |
12. Accounts Payable
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Accounts payable |
| |
| |
Substantially all accounts payable are denominated in RMB, EUR and US$ and due within one year from the end of the reporting period. The carrying values of accounts payable approximate their fair values due to their short-term maturities.
An aging analysis based on the relevant invoice dates is as follows:
December 31, | ||||
2023 | 2022 | |||
(in US$’000) | ||||
Not later than 3 months | |
| | |
Between 3 months to 6 months | |
| | |
Between 6 months to 1 year | |
| | |
Later than 1 year | |
| | |
|
| | ||
F-25
13. Other Payables, Accruals and Advance Receipts
Other payables, accruals and advance receipts consisted of the following:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Accrued research and development expenses |
| |
| |
Accrued salaries and benefits |
| |
| |
Accrued capital expenditures |
| |
| |
Accrued selling and marketing expenses | | | ||
Accrued administrative and other general expenses | | | ||
| | |||
Deposits | | | ||
Deferred government grants |
| |
| |
Others |
| |
| |
| |
| | |
14. Bank Borrowings
Bank borrowings consisted of the following:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Current |
| |
| — |
Non-current |
| |
| |
The weighted average interest rate for outstanding bank borrowings for the years ended December 31, 2023 and 2022 was
| (i) | Short-term working capital loan facility |
In November 2023, a subsidiary entered into a short-term working capital loan facility with a bank in the amount of RMB
(ii) | 10-year fixed asset loan facility |
In October 2021, a subsidiary entered into a
(iii) | 1-year revolving loan facility |
In May 2022, the Group through its subsidiary, entered into a
F-26
The Group’s bank borrowings are repayable as from the dates indicated as follows:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Not later than 1 year | | — | ||
Between 1 to 3 years |
| |
| |
Between 3 to 4 years | | | ||
Between 4 to 5 years | | | ||
Later than 5 years |
| |
| |
| |
| | |
As at December 31, 2023 and 2022, the Group had unutilized bank borrowing facilities of US$
15. Commitments and Contingencies
The Group had the following capital commitments:
December 31, | ||
| 2023 | |
(in US$’000) | ||
Property, plant and equipment | ||
Contracted but not provided for |
| |
The Group does not have any other significant commitments or contingencies.
16. Ordinary Shares
As at December 31, 2023, the Company is authorized to issue
Each ordinary share is entitled to
F-27
17. Share-based Compensation
(i) | Share-based Compensation of the Company |
The Company conditionally adopted a share option scheme on April 24, 2015 (as amended on April 27, 2020) (the “Hutchmed Share Option Scheme”). Pursuant to the Hutchmed Share Option Scheme, the Board of Directors of the Company may, at its discretion, offer any employees and directors (including Executive and Non-executive Directors but excluding Independent Non-executive Directors) of the Company, holding companies of the Company and any of their subsidiaries or affiliates, and subsidiaries or affiliates of the Company share options to subscribe for shares of the Company.
As at December 31, 2023, the aggregate number of shares issuable under the Hutchmed Share Option Scheme was
Share options granted are generally subject to a
A summary of the Company’s share option activity and related information is as follows:
|
|
| Weighted average |
| ||||
Weighted average | remaining | Aggregate | ||||||
Number of | exercise price in | contractual life | intrinsic value | |||||
share options | US$ per share | (years) | (in US$’000) | |||||
Outstanding at January 1, 2022 |
| |
| |
| | ||
Granted (note) |
| |
| | ||||
Exercised |
| ( |
| | ||||
Cancelled |
| ( |
| | ||||
Expired | ( | | ||||||
Outstanding at December 31, 2022 |
| |
| |
|
| | |
Granted |
| |
| | ||||
Exercised |
| ( |
| | ||||
Cancelled |
| ( |
| | ||||
Expired | ( | | ||||||
Outstanding at December 31, 2023 |
| |
| | | |||
Vested and exercisable at December 31, 2022 |
| |
| |
|
| | |
Vested and exercisable at December 31, 2023 |
| | | |
Note: Includes
F-28
In estimating the fair value of share options granted, the following assumptions were used in the Polynomial model for awards granted in the periods indicated:
Year Ended December 31, | |||||
2023 | 2022 | ||||
Weighted average grant date fair value of share options (in US$ per share) | | | |||
Significant inputs into the valuation model (weighted average): | |||||
Exercise price (in US$ per share) | | | |||
Share price at effective date of grant (in US$ per share) | | | |||
Expected volatility (note (a)) | | % | | % | |
Risk-free interest rate (note (b)) | | % | | % | |
Contractual life of share options (in years) | |||||
Expected dividend yield (note (c)) | | % | | % | |
Notes:
| (a) | The Company calculated its expected volatility with reference to the historical volatility prior to the issuances of share options. |
| (b) | The risk-free interest rates reference the U.S. Treasury yield curves because the Company’s ADS are currently listed on the NASDAQ and denominated in US$. |
| (c) | The Company has not declared or paid any dividends and does not currently expect to do so prior to the exercise of the granted share options, and therefore uses an expected dividend yield of |
The Company will issue new shares to satisfy share option exercises. The following table summarizes the Company’s share option exercises:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Cash received from share option exercises |
| | | | ||
Total intrinsic value of share option exercises |
| | | | ||
The Group recognizes compensation expense on a graded vesting approach over the requisite service period. The following table presents share-based compensation expense included in the Group’s consolidated statements of operations:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Research and development expenses |
| | | | ||
Selling and administrative expenses |
| | | | ||
Cost of revenue | | | | |||
| | | | |||
As at December 31, 2023, the total unrecognized compensation cost was US$
| (ii) | LTIP |
The Company grants awards under the LTIP to participating directors and employees, giving them a conditional right to receive ordinary shares of the Company or the equivalent ADS (collectively the “Awarded Shares”) to be purchased by the Trustee up to a cash amount. Vesting will depend upon continued employment of the award holder with the Group and will otherwise be at the discretion of the Board of Directors of the Company. Additionally, some awards are subject to change based on annual performance targets prior to their determination date.
F-29
LTIP awards prior to the determination date
Performance targets vary by award, and may include targets for shareholder returns, financings, revenue, net income/(loss) after taxes and the achievement of clinical, regulatory, business development and manufacturing milestones. As the extent of achievement of the performance targets is uncertain prior to the determination date, a probability based on management’s assessment on the achievement of the performance target has been assigned to calculate the amount to be recognized as an expense over the requisite period with a corresponding entry to liability.
LTIP awards after the determination date
Upon the determination date, the Company will pay a determined monetary amount, up to the maximum cash amount based on the actual achievement of the performance target specified in the award, to the Trustee to purchase the Awarded Shares. Any cumulative compensation expense previously recognized as a liability will be transferred to additional paid-in capital. Based on the actual achievement of performance target, the amount previously recorded in the liability will be adjusted through share-based compensation expense.
Granted awards under the LTIP are as follows:
| Maximum cash amount |
| Covered |
| Performance target | |
Grant date | (in US$ millions) | financial years | determination date | |||
May 23, 2022 | 2022 | note (a) | ||||
September 13, 2022 | 2022 | note (a) | ||||
September 13, 2022 | note (b) | note (b) | ||||
June 5, 2023 | 2023 | note (a) |
Notes:
| (a) | The annual performance target determination date is the date of the announcement of the Group’s annual results for the covered financial year and vesting occurs two business days after the the s results for the financial year |
| (b) | This award does not stipulate performance targets and is subject to a vesting schedule of |
The Trustee has been set up solely for the purpose of purchasing and holding the Awarded Shares during the vesting period on behalf of the Company using funds provided by the Company. On the determination date, if any, the Company will determine the cash amount, based on the actual achievement of each annual performance target, for the Trustee to purchase the Awarded Shares. The Awarded Shares will then be held by the Trustee until they are vested.
The Trustee’s assets include treasury shares and funds for additional treasury shares, trustee fees and expenses. The number of treasury shares (in ordinary shares equivalent) held by the Trustee were as follows:
Number of | Cost | |||
| treasury shares |
| (in US$’000) | |
As at January 1, 2022 | | | ||
Purchased | | | ||
Vested | ( | ( | ||
As at December 31, 2022 | | | ||
Purchased | | | ||
Vested | ( | ( | ||
As at December 31, 2023 | | |
Based on the estimated achievement of performance conditions for 2023 financial year LTIP awards, the determined monetary amount was US$
F-30
For the years ended December 31, 2023 and 2022, US$
The following table presents the share-based compensation expenses recognized under the LTIP awards:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Research and development expenses |
| |
| |
| |
Selling and administrative expenses |
| |
| |
| |
Cost of revenue | | | | |||
| |
| |
| | |
Recorded with a corresponding credit to: |
|
|
| |||
Liability |
| |
| |
| |
Additional paid-in capital |
| |
| |
| |
| |
| |
| | |
For the years ended December 31, 2023, 2022 and 2021, US$
As at December 31, 2023, the total unrecognized compensation cost was approximately US$
18. Revenue
The following table presents revenue disaggregated by contract type:
Year Ended December 31, 2023 | ||||||
Oncology/ | Other | |||||
| Immunology |
| Ventures |
| Total | |
(in US$’000) | ||||||
Invoiced Goods—Marketed Products | | — | | |||
—Distribution |
| — |
| |
| |
Services —Commercialization of Marketed Products | | — | | |||
—Research and Development | | — | | |||
License & Collaborations—Services | | — | | |||
—Royalties |
| |
| — |
| |
—Licensing | | — | | |||
—Manufacturing supply | | — | | |||
| |
| |
| | |
Third parties |
| |
| |
| |
Related parties (Note 24(i)) |
| |
| |
| |
| |
| |
| | |
F-31
Year Ended December 31, 2022 | ||||||
Oncology/ | Other | |||||
| Immunology |
| Ventures |
| Total | |
(in US$’000) | ||||||
Invoiced Goods—Marketed Products | | — | | |||
—Distribution | — |
| |
| | |
Services —Commercialization of Marketed Products | | — | | |||
—Research and Development | | — | | |||
License & Collaborations—Services | | — | | |||
—Royalties |
| |
| — |
| |
—Licensing | | — | | |||
| |
| |
| | |
Third parties |
| |
| |
| |
Related parties (Note 24(i)) |
| |
| |
| |
| |
| |
| | |
Year Ended December 31, 2021 | ||||||
Oncology/ | Other | |||||
| Immunology |
| Ventures |
| Total | |
(in US$’000) | ||||||
Invoiced Goods—Marketed Products | | — | | |||
—Distribution | — | | | |||
Services —Commercialization of Marketed Products |
| | — | | ||
—Research and Development | | — | | |||
License & Collaborations—Services | | — | | |||
—Royalties | |
| — |
| | |
—Licensing | | — | | |||
| |
| |
| | |
Third parties |
| |
| |
| |
Related parties (Note 24(i)) |
| |
| |
| |
| |
| |
| | |
The following table presents liability balances from contracts with customers:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Deferred revenue | ||||
Current—Oncology/Immunology segment (note (a)) |
| |
| |
Current—Other Ventures segment (note (b)) |
| |
| |
| |
| | |
Non-current—Oncology/Immunology segment (note (a)) |
| |
| |
Total deferred revenue (note (c) and (d)) | | | ||
Notes:
| (a) | Oncology/Immunology segment deferred revenue relates to unamortized upfront and milestone payments, invoiced amounts for royalties where the customer has not yet completed the in-market sale and advance consideration received for cost reimbursements which are attributed to research and development services that have not yet been rendered as at the reporting date. |
| (b) | Other Ventures segment deferred revenue relates to payments in advance from customers for goods that have not been transferred and services that have not been rendered to the customer as at the reporting date. |
F-32
| (c) | Estimated deferred revenue to be recognized over time as from the date indicated is as follows: |
| December 31, | |||
2023 |
| 2022 | ||
(in US$’000) | ||||
Not later than 1 year |
| | | |
Between 1 to 2 years |
| | | |
Between 2 to 3 years | | | ||
Between 3 to 4 years |
| | — | |
Later than 4 years | | — | ||
| | | ||
(d) | As at January 1, 2023, deferred revenue was US$ |
License and collaboration agreement with Takeda Pharmaceuticals
On January 23, 2023, the Group and Takeda Pharmaceuticals International AG (“Takeda”) entered into an exclusive out-licensing agreement (the “Takeda Agreement”) in territories outside of Mainland China, Hong Kong and Macau (the “Territory”) to further the global development, commercialization and manufacturing of Fruzaqla, also known as fruquintinib, a targeted oncology therapy for the treatment of various types of solid tumors. Under the terms of the Takeda Agreement, the Group is entitled to receive a series of payments up to US$
Upfront and milestone payments according to the Takeda Agreement received up to December 31, 2023 are summarized as follows:
| (in US$’000) | |
Upfront payment |
| |
Regulatory approval milestone payment achieved |
| |
As of December 31, 2023, the total revenue recognized under the Takeda Agreement is US$
The Takeda Agreement has the following material performance obligations: (1) the licenses for the development and commercialization of Fruzaqla in the Territory and the manufacture of Fruzaqla for use in the Territory, (2) manufacturing supply and (3) services for research and development including ongoing clinical trials and regulatory submissions and manufacturing technology transfer.
The transaction price for these performance obligations includes the upfront payment, service cost reimbursements, milestone payments and sales-based royalties. Milestone payments are not included in the transaction price until they become probable that a significant reversal of revenue would not occur, which is generally when the criteria to receive the specified milestone are achieved.
F-33
The allocation of the transaction price to each relevant performance obligation was based on the relative standalone selling price of each performance obligation determined at the inception of the contract. Variable consideration is allocated entirely to a performance obligation or to a distinct good or service that forms part of a single performance obligation if the terms of the variable consideration relate to the satisfaction of the respective performance obligation and the amount allocated is consistent with the amount expected to be received for the satisfaction of the respective performance obligation. The standalone selling price of the licenses for the development and commercialization of Fruzaqla in the Territory and the manufacture of Fruzaqla for use in the Territory and manufacturing supply was determined using a discounted cash flow method based on the probability-weighted present value of forecasted cash flows associated with out-licensing Fruzaqla in the Territory, and the standalone selling price of the services for research and development of ongoing clinical trials, regulatory submissions and manufacturing technology transfer was determined using a cost plus margin approach based on the present value of estimated future service costs plus a reasonable margin. Significant assumptions included in the determination of the standalone selling prices for each performance obligation identified including forecasted revenue, probabilities of regulatory approvals, estimated future service costs, margin rates and discount rates. Based on these estimations, proportionate amounts of transaction price to be allocated to the licenses, and other performance obligations were
Revenue recognized under the Takeda Agreement is as follows:
Year Ended | ||
| December 31, 2023 | |
(in US$’000) | ||
Manufacturing supply—Invoiced Marketed Products sales | | |
—Allocated from upfront payment | | |
Services—Research and Development | | |
—Allocated from upfront and milestone payments |
| |
Royalties—Marketed Products |
| |
Licensing—Allocated from upfront and milestone payments | | |
| |
License and collaboration agreement with Eli Lilly
On October 8, 2013, the Group entered into a licensing, co-development and commercialization agreement in China with Eli Lilly and Company (“Lilly”) relating to Elunate (“Lilly Agreement”), as the China brand name for fruquintinib. Under the terms of the Lilly Agreement, the Group is entitled to receive a series of payments up to US$
In December 2018, the Group entered into various amendments to the Lilly Agreement (the “2018 Amendment”). Under the terms of the 2018 Amendment, the Group is entitled to determine and conduct future life cycle indications (“LCI”) development of Elunate in China beyond the
F-34
In July 2020, the Group entered into an amendment to the Lilly Agreement (the “2020 Amendment”) relating to the expansion of the Group’s role in the commercialization of Elunate across all of China. Under the terms of the 2020 Amendment, the Group is responsible for providing promotion and marketing services, including the development and execution of all on-the-ground medical detailing, promotion and local and regional marketing activities, in return for service fees on sales of Elunate made by Lilly. In October 2020, the Group commenced such promotion and marketing services. In addition, development and regulatory approval milestones for an initial indication under the Lilly Agreement were increased by US$
Upfront and cumulative milestone payments according to the Lilly Agreement received up to December 31, 2023 are summarized as follows:
| (in US$’000) | |
Upfront payment |
| |
Development milestone payments achieved |
| |
The Lilly Agreement has the following performance obligations: (1) the license for the commercialization rights to Elunate and (2) the research and development services for the specified indications. The transaction price includes the upfront payment, research and development cost reimbursements, milestone payments and sales-based royalties. Milestone payments were not included in the transaction price until it became probable that a significant reversal of revenue would not occur, which is generally when the specified milestone is achieved. The allocation of the transaction price to each performance obligation was based on the relative standalone selling prices of each performance obligation determined at the inception of the contract. Based on this estimation, proportionate amounts of transaction price to be allocated to the license to Elunate and the research and development services were
The 2018 Amendment is a separate contract as it added distinct research and development services for the LCIs to the Lilly Agreement. The 2020 Amendment related to the promotion and marketing services is a separate contract as it added distinct services to the Lilly Agreement. Such promotion and marketing services are recognized over time based on amounts that can be invoiced to Lilly. The 2020 Amendment related to the additional development and regulatory approval milestone amounts is a modification as it only affected the transaction price of research and development services for a specific indication under the Lilly Agreement, and therefore, such additional milestone amounts will be included in the transaction price accounted under the Lilly Agreement once the specified milestones are achieved.
Revenue recognized under the Lilly Agreement and subsequent amendments is as follows:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Goods—Invoiced Marketed Products sales |
| | |
| | |
Services—Commercialization of Marketed Products |
| | |
| | |
—Research and Development | | | | |||
—Allocated from upfront and milestone payments | | | — | |||
Royalties—Marketed Products | | | | |||
| |
| |
| | |
F-35
License and collaboration agreement with AstraZeneca
On December 21, 2011, the Group and AstraZeneca AB (publ) (“AZ”) entered into a global licensing, co-development, and commercialization agreement for Orpathys (“AZ Agreement”), also known as savolitinib, a novel targeted therapy and a highly selective inhibitor of the c-Met receptor tyrosine kinase for the treatment of cancer. Under the terms of the AZ Agreement, the Group is entitled to receive a series of payments up to US$
In August 2016 (as amended in December 2020), the Group entered into an amendment to the AZ Agreement whereby the Group shall pay the first approximately US$
Upfront and cumulative milestone payments according to the AZ Agreement received up to December 31, 2023 are summarized as follows:
| (in US$’000) | |
Upfront payment |
| |
Development milestone payments achieved |
| |
First-sale milestone payment achieved | |
The AZ Agreement has the following performance obligations: (1) the license for the commercialization rights to Orpathys and (2) the research and development services for the specified indications. The transaction price includes the upfront payment, research and development cost reimbursements, milestone payments and sales-based royalties. Milestone payments were not included in the transaction price until it became probable that a significant reversal of revenue would not occur, which is generally when the specified milestone is achieved. The allocation of the transaction price to each performance obligation was based on the relative standalone selling prices of each performance obligation determined at the inception of the contract. Based on this estimation, proportionate amounts of transaction price to be allocated to the license to Orpathys and the research and development services were
Revenue recognized under the AZ Agreement and subsequent amendments is as follows:
Year Ended December 31, | ||||||
2023 | 2022 | 2021 | ||||
(in US$’000) | ||||||
Goods—Invoiced Marketed Products sales |
| |
| |
| |
Services—Research and Development |
| |
| |
| |
—Allocated from upfront and milestone payments | | | | |||
Royalties—Marketed Products |
| |
| |
| |
Licensing—Allocated from upfront and milestone payments |
| — |
| |
| |
| |
| |
| | |
F-36
19. In-Licensing arrangement
On August 7, 2021, the Group and Epizyme, Inc. (“Epizyme”) entered into a license agreement (the “In-license Agreement”) for tazemetostat, a novel inhibitor of EZH2 that is approved by the U.S. Food and Drug Administration for the treatment of certain patients with epithelioid sarcoma and follicular lymphoma. The Group is responsible for the development and commercialization of tazemetostat in the PRC, Hong Kong, Macau and Taiwan (the “Territory”) and also holds rights to manufacture tazemetostat for the Territory. The Group also received a
Under the terms of the In-license Agreement and warrant, the Group paid Epizyme a US$
The US$
The warrant was recorded as a financial asset at fair value with changes to fair value recognized to the consolidated statements of operations. During the year ended December 31, 2022, an affiliate of Ipsen S.A. acquired all outstanding shares of Epizyme and the warrant expired under the terms of the In-license Agreement and warrant. For the years ended December 31, 2022 and 2021, fair value losses of US$
20. Research and Development Expenses
Research and development expenses are summarized as follows:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Clinical trial related costs |
| |
| |
| |
Personnel compensation and related costs |
| |
| |
| |
Other research and development expenses |
| |
| |
| |
| |
| |
| | |
The Group has entered into multiple collaborative arrangements under ASC 808 to evaluate the combination of the Group’s drug compounds with the collaboration partners’ drug compounds. For the years ended December 31, 2023, 2022 and 2021, the Group has incurred research and development expenses of US$
21. Government Grants
Government grants in the Oncology/Immunology segment are primarily given in support of the construction of a manufacturing plant in Shanghai and R&D activities which are conditional upon i) the Group spending a predetermined amount, regardless of success or failure of the research and development projects and/or ii) the achievement of certain stages of research and development projects being approved by the relevant PRC government authority. They are refundable to the government if the conditions, if any, are not met. Government grants in the Other Ventures segment are primarily given to promote local initiatives. These government grants may be subject to ongoing reporting and monitoring by the government over the period of the grant.
Government grants, which are deferred and recognized in the consolidated statements of operations over the period necessary to match them with the costs that they are intended to compensate, are recognized in other payables, accruals and advance receipts (Note 13) and other non-current liabilities. For the years ended December 31, 2023, 2022 and 2021, the Group received government grants of US$
F-37
Government grants were recognized in the consolidated statements of operations as follows:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Research and development expenses |
| |
| |
| |
Other income |
| |
| |
| |
| |
| |
| | |
22. Gain on Divestment of An Equity Investee
In March 2021, the Group entered into a sale and purchase agreement (the “SPA”) with a third party to sell its entire investment in HBYS with closing subject to regulatory approval in the PRC. On September 28, 2021, the Group completed the divestment for cash consideration of US$
On May 13, 2021 and September 23, 2021, HBYS had declared dividends to shareholders of US$
In addition, the Group and Hutchison Whampoa Enterprises Limited, an affiliate of CK Hutchison, entered into a license agreement on June 15, 2021, conditional upon the completion of the divestment, to grant a continuing right to use the “Hutchison Whampoa” brand by HBYS for
The gain on divestment of an equity investee was recognized in the consolidated statements of operations as follows:
| Year Ended December 31, | |
2021 | ||
(in US$’000) | ||
Proceeds |
| |
Dividend receivables–third party |
| |
| | |
Less: Group’s share of net assets of HBYS (Note 11(iii)) |
| ( |
Dividend receivables–HBYS |
| ( |
Withholding tax liability on dividend receivables–HBYS |
| |
Branding liability |
| ( |
Accumulated other comprehensive income and reserves |
| |
Transaction costs and others |
| |
Gain on divestment of an equity investee |
| |
Less: Capital gain tax |
| ( |
Less: Gain on divestment of an equity investee attributable to non-controlling interests |
| ( |
Gain on divestment of an equity investee attributable to the Group |
| |
F-38
23. Other income/(expense)
Year Ended December 31, | ||||||
2023 | 2022 | 2021 | ||||
(in US$’000) | ||||||
Other income: |
|
|
|
|
|
|
Foreign exchange gains |
| |
| — |
| |
Government grants |
| |
| |
| |
Others |
| |
| |
| |
| |
| |
| | |
Other expense: |
|
|
|
|
|
|
Impairment of property, plant and equipment |
| ( |
| — |
| — |
Impairment of right-of-use assets |
| ( |
| — |
| — |
Foreign exchange losses |
| — |
| ( |
| — |
Fair value losses on warrant |
| — |
| ( |
| ( |
Others |
| ( |
| ( |
| ( |
| ( |
| ( |
| ( | |
24. Significant Transactions with Related Parties and Non-Controlling Shareholders of Subsidiaries
The Group has the following significant transactions with related parties and non-controlling shareholders of subsidiaries, which were carried out in the normal course of business at terms determined and agreed by the relevant parties:
| (i) | Transactions with related parties: |
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Sales to: | ||||||
Indirect subsidiaries of CK Hutchison |
| | |
| | |
An equity investee | | | — | |||
| | | ||||
Revenue from research and development services from: | ||||||
An equity investee |
| | |
| | |
Purchases from: | ||||||
Equity investees |
| | |
| | |
Rendering of marketing services from: | ||||||
Indirect subsidiaries of CK Hutchison |
| | |
| | |
An equity investee |
| — | |
| — | |
| | |
| | ||
Rendering of management services from: | ||||||
An indirect subsidiary of CK Hutchison | | | | |||
Entered brand license agreement with: | ||||||
An indirect subsidiary of CK Hutchison (note (a)) |
| — | — |
| | |
Divestment of subsidiaries to: | ||||||
An indirect subsidiary of CK Hutchison (note (b)) | | — | — | |||
F-39
| (ii) | Balances with related parties included in: |
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Accounts receivable—related parties | ||||
Indirect subsidiaries of CK Hutchison (note (c)) |
| — |
| |
An equity investee (note (c)) |
| |
| |
| | |||
Amounts due from related parties | ||||
An indirect subsidiary of CK Hutchison (note (c)) | | — | ||
An equity investee (note (c) and (d)) | | | ||
| | |||
Other payables, accruals and advance receipts | ||||
Indirect subsidiaries of CK Hutchison (note (e) and (g)) | | | ||
An equity investee (note (c) and (f)) | | | ||
| | |||
Other non-current liabilities | ||||
An equity investee (note (f)) | | | ||
An indirect subsidiary of CK Hutchison (note (g)) |
| |
| |
| | |||
Notes:
| (a) | The branding rights for HBYS from an indirect subsidiary of CK Hutchison were recognized in the consolidated statements of operations through the gain on divestment of an equity investee (Note 22). For each of the years ended December 31, 2023, 2022 and 2021, the Group paid US$ |
| (b) | On December 7, 2023, the Group completed a transaction to divest HHOHK and HSN to an indirect subsidiary of CK Hutchison for proceeds of US$ |
| (c) | Balances with related parties are unsecured, repayable on demand and interest-free. The carrying values of balances with related parties approximate their fair values due to their short-term maturities. |
| (d) | As at December 31, 2023, dividends receivable of US$ |
| (e) | Amounts due to indirect subsidiaries of CK Hutchison are unsecured, repayable on demand and interest-bearing if not settled within |
| (f) | Includes other deferred income representing amounts recognized from granting of commercial, promotion and marketing rights. |
| (g) | As at December 31, 2023 and 2022, a branding liability payable of US$ |
F-40
| (iii) | Transactions with non-controlling shareholders of subsidiaries: |
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Sales |
| |
| |
| |
Purchases |
| |
| |
| |
Dividends declared |
| |
| |
| |
Distribution service fee | | — | — | |||
| (iv) | Balances with non-controlling shareholders of subsidiaries included in: |
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Accounts receivable |
| |
| |
Accounts payable |
| |
| |
Other payables, accruals and advance receipts | | — | ||
25. Income Taxes
(i) | Income tax expense/(benefit) |
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Current tax | ||||||
HK (note (a)) |
| | |
| | |
PRC (note (b) and (c)) |
| | |
| | |
U.S. and others (note (d)) | | | | |||
Total current tax | | | | |||
Deferred income tax expense/(benefit) |
| | ( |
| ( | |
Income tax expense/(benefit) |
| | ( |
| | |
Notes:
| (a) | The Company, |
| (b) | Taxation in the PRC has been provided for at the applicable rate on the estimated assessable profits less estimated available tax losses, if any, in each entity. Under the PRC Enterprise Income Tax Law (the “EIT Law”), the standard enterprise income tax rate is |
F-41
Pursuant to the EIT law, a
Pursuant to PRC Bulletin on Issues of Enterprise Income Tax and Indirect Transfers of Assets by Non-PRC Resident Enterprises, an indirect transfer of a PRC resident enterprise by a non-PRC resident enterprise, via the transfer of an offshore intermediate holding company, shall be subject to PRC withholding tax under certain conditions.
(c) | Current tax in the PRC for the year ended December 31, 2021 includes US$ |
(d) | The Company’s subsidiary in the U.S. with operations primarily in New Jersey is subject to U.S. taxes, primarily federal and state taxes, which have been provided for at approximately |
The reconciliation of the Group’s reported income tax expense to the theoretical tax amount that would arise using the tax rates of the Company against the Group’s income/(loss) before income taxes and equity in earnings of equity investees is as follows:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Income/(loss) before income taxes and equity in earnings of equity investees | | ( | ( | |||
Tax calculated at the statutory tax rate of the Company | | ( | ( | |||
Tax effects of: | ||||||
Different tax rates applicable in different jurisdictions | | | | |||
Tax valuation allowance | | | | |||
Preferential tax rate difference | ( | ( | ( | |||
Preferential tax deduction and credits | ( | ( | ( | |||
Expenses not deductible for tax purposes | | | | |||
Withholding tax on undistributed earnings of PRC entities | | | | |||
Income not subject to tax | ( | ( | ( | |||
Temporary difference | ( | ( | | |||
Others | | | | |||
Income tax expense/(benefit) | | ( | | |||
F-42
(ii) | Deferred tax assets and liabilities |
The significant components of deferred tax assets and liabilities are as follows:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Deferred tax assets | ||||
Cumulative tax losses | | | ||
Others | | | ||
Total deferred tax assets | | | ||
Less: Valuation allowance | ( | ( | ||
Deferred tax assets | | | ||
Deferred tax liabilities | ||||
Undistributed earnings from a PRC entity | | | ||
Others | | | ||
Deferred tax liabilities | | | ||
The movements in deferred tax assets and liabilities are as follows:
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
As at January 1 |
| | |
| ( | |
Movement of previously recognized withholding tax on undistributed earnings |
| | |
| | |
(Charged)/Credited to the consolidated statements of operations | ||||||
Withholding tax on undistributed earnings of PRC entities |
| ( | ( |
| ( | |
Deferred tax on amortization of intangible assets |
| | |
| | |
Deferred tax on temporary differences, tax loss carried forward and research tax credits |
| | |
| | |
Reclassification from current tax | | — | — | |||
Divestment of subsidiaries | ( | — | — | |||
Divestment of an equity investee | — | — | | |||
Exchange differences |
| ( | |
| ( | |
As at December 31 |
| | |
| | |
The deferred tax assets and liabilities are offset when there is a legally enforceable right to set off and when the deferred income taxes relate to the same fiscal authority.
The cumulative tax losses can be carried forward against future taxable income and will expire in the following years:
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
No expiry date |
| | | |
2024 | | | ||
2025 | | | ||
2026 | | | ||
2027 | | | ||
2028 | | | ||
2029 | | | ||
2030 | | | ||
2031 | | | ||
2032 | | | ||
2033 | | — | ||
| | | ||
F-43
The Company believes that it is more likely than not that future operations outside the U.S. will not generate sufficient taxable income to realize the benefit of the deferred tax assets. Certain of the Company’s subsidiaries have had sustained tax losses, which will expire within
A U.S. subsidiary of the Company has approximately US$
The table below summarizes changes in the deferred tax valuation allowance:
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
As at January 1 |
| | |
| | |
Charged to consolidated statements of operations |
| | |
| | |
Utilization of previously unrecognized tax losses |
| ( | ( |
| ( | |
Write-off of tax losses |
| ( | ( |
| — | |
Divestment of subsidiaries | ( | — | — | |||
Others |
| — | — |
| ( | |
Exchange differences |
| ( | ( |
| | |
As at December 31 |
| | |
| | |
As at December 31, 2023, 2022 and 2021, the Group did not have any material unrecognized uncertain tax positions.
| (iii) | Income tax payable |
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
As at January 1 | | | | |||
Current tax | | | | |||
Withholding tax upon dividend declaration from PRC entities | | | | |||
Tax paid (note) | ( | ( | ( | |||
Reclassification (from)/to prepaid tax | ( | ( | | |||
Reclassification to deferred tax | | — | — | |||
Divestment of subsidiaries | ( | — | — | |||
Divestment of an equity investee (Note 22) | — | — | ( | |||
Exchange difference | ( | ( | | |||
As at December 31 | | | | |||
Note: The amount for 2022 includes US$
26. Earnings/(Losses) Per Share
(i) | Basic earnings/(losses) per share |
Basic earnings/(losses) per share is calculated by dividing the net income/(loss) attributable to the Company by the weighted average number of outstanding ordinary shares in issue during the year. Treasury shares held by the Trustee are excluded from the weighted average number of outstanding ordinary shares in issue for purposes of calculating basic earnings/(losses) per share.
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
Weighted average number of outstanding ordinary shares in issue |
| | |
| | |
Net income/(loss) attributable to the Company (US$’000) | | ( |
| ( | ||
Basic earnings/(losses) per share attributable to the Company (US$ per share) | | ( |
| ( | ||
F-44
(ii) | Diluted earnings/(losses) per share |
Diluted earnings/(losses) per share is calculated by dividing net income/(loss) attributable to the Company by the weighted average number of outstanding ordinary shares in issue and dilutive ordinary share equivalents outstanding during the year. Dilutive ordinary share equivalents include shares issuable upon the exercise or settlement of share options, LTIP awards and warrants issued by the Company using the treasury stock method.
Year Ended December 31, | ||||||
2023 | 2022 | 2021 | ||||
Weighted average number of outstanding ordinary shares in issue |
| |
| |
| |
Effect of share options and LTIP awards |
| |
| — |
| — |
Weighted average number of outstanding ordinary shares in issue and dilutive ordinary share equivalents outstanding |
| |
| |
| |
Net income/(loss) attributable to the Company (US$’000) |
| |
| ( |
| ( |
Diluted earnings/(losses) per share attributable to the Company (US$ per share) |
| |
| ( |
| ( |
For the years ended December 31, 2022 and 2021, the share options, LTIP awards and warrants issued by the Company were not included in the calculation of diluted losses per share because of their anti-dilutive effect.
27. Segment Reporting
The Group’s operating segments are as follows:
| (i) | Oncology/Immunology: focuses on discovering, developing, and commercializing targeted therapies and immunotherapies for the treatment of cancer and immunological diseases. Oncology/Immunology is further segregated into |
| (a) | R&D: comprises research and development activities covering drug discovery, development, manufacturing and regulatory functions, out-licensing of in-house developed drugs, as well as administrative activities to support research and development operations; and |
| (b) | Marketed Products: comprises the invoiced sales, marketing, manufacture and distribution of drugs developed from research and development activities including out-licensed marketed products. |
| (ii) | Other Ventures: comprises other commercial businesses which include the sales, marketing, manufacture and distribution of other prescription drugs and healthcare products. |
The performance of the reportable segments is assessed based on segment net income/(loss) attributable to the Company.
F-45
The segment information is as follows:
Year Ended December 31, 2023 | ||||||||||||||||||||
Oncology/Immunology | ||||||||||||||||||||
Other | ||||||||||||||||||||
R&D | Marketed Products | Ventures | ||||||||||||||||||
U.S. and | U.S. and | |||||||||||||||||||
| PRC |
| Others |
| Subtotal |
| PRC | Others | Subtotal | Subtotal |
| PRC |
| Unallocated |
| Total | ||||
(in US$’000) | ||||||||||||||||||||
Revenue from external customers |
| | | | | | | | | — | | |||||||||
Interest income |
| | | | — | — | — | | | | | |||||||||
Interest expense | ( | — | ( | — | — | — | ( | ( | ( | ( | ||||||||||
Equity in earnings of equity investees, net of tax |
| — | — | — | — | — | — | — | | — | | |||||||||
Income tax (expense)/benefit |
| ( | ( | ( | ( | — | ( | ( | ( | ( | ( | |||||||||
Net (loss)/income attributable to the Company |
| ( | | | | | | | | ( | | |||||||||
Depreciation/amortization |
| ( | ( | ( | — | — | — | ( | ( | ( | ( | |||||||||
Additions to non-current assets (other than financial instruments and deferred tax assets) | | | | — | — | — | | | | | ||||||||||
December 31, 2023 | ||||||||||||||||||||
Oncology/Immunology | ||||||||||||||||||||
Other | ||||||||||||||||||||
R&D | Marketed Products | Ventures | ||||||||||||||||||
U.S. and | U.S. and | |||||||||||||||||||
| PRC |
| Others |
| Subtotal |
| PRC |
| Others |
| Subtotal |
| Subtotal |
| PRC |
| Unallocated |
| Total | |
(in US$’000) | ||||||||||||||||||||
Total assets |
| | | | | | | | | | | |||||||||
Property, plant and equipment |
| | | | — | — | — | | | | | |||||||||
Right-of-use assets | | | | — | — | — | | | | | ||||||||||
Leasehold land |
| | — | | — | — | — | | — | — | | |||||||||
Goodwill |
| — | — | — | — | — | — | — | | — | | |||||||||
Other intangible asset |
| — | — | — | — | — | — | — | | — | | |||||||||
Investments in equity investees |
| — | — | — | — | — | — | — | | — | | |||||||||
Year Ended December 31, 2022 | ||||||||||||||||||||
Oncology/Immunology | ||||||||||||||||||||
Other | ||||||||||||||||||||
R&D | Marketed Products | Ventures | ||||||||||||||||||
U.S. and | U.S. and | |||||||||||||||||||
| PRC |
| Others |
| Subtotal |
| PRC | Others | Subtotal | Subtotal |
| PRC |
| Unallocated |
| Total | ||||
(in US$’000) | ||||||||||||||||||||
Revenue from external customers |
| | — | | | — | | | | — | | |||||||||
Interest income |
| | | | — | — | — | | | | | |||||||||
Interest expense | — | — | — | — | — | — | — | — | ( | ( | ||||||||||
Equity in earnings of equity investees, net of tax |
| | — | | — | — | — | | | — | | |||||||||
Income tax (expense)/ benefit |
| ( | | | ( | — | ( | | ( | ( | | |||||||||
Net (loss)/income attributable to the Company |
| ( | ( | ( | | — | | ( | | ( | ( | |||||||||
Depreciation/amortization |
| ( | ( | ( | — | — | — | ( | ( | ( | ( | |||||||||
Additions to non-current assets (other than financial instruments and deferred tax assets) | | | | — | — | — | | | | | ||||||||||
F-46
December 31, 2022 | ||||||||||||||||||||
Oncology/Immunology | ||||||||||||||||||||
Other | ||||||||||||||||||||
R&D | Marketed Products | Ventures | ||||||||||||||||||
U.S. and | U.S. and | |||||||||||||||||||
| PRC |
| Others |
| Subtotal |
| PRC |
| Others | Subtotal | Subtotal |
| PRC |
| Unallocated |
| Total | |||
(in US$’000) | ||||||||||||||||||||
Total assets |
| | | | | — | | | | | | |||||||||
Property, plant and equipment |
| | | | — | — | — | | | | | |||||||||
Right-of-use assets | | | | — | — | — | | | | | ||||||||||
Leasehold land |
| | — | | — | — | — | | — | — | | |||||||||
Goodwill |
| — | — | — | — | — | — | — | | — | | |||||||||
Other intangible asset |
| — | — | — | — | — | — | — | | — | | |||||||||
Investments in equity investees |
| | — | | — | — | — | | | — | | |||||||||
Year Ended December 31, 2021 | ||||||||||||||||||||
Oncology/Immunology | ||||||||||||||||||||
Other | ||||||||||||||||||||
R&D | Marketed Products | Ventures | ||||||||||||||||||
U.S. and | U.S. and | |||||||||||||||||||
| PRC |
| Others |
| Subtotal |
| PRC | Others | Subtotal | Subtotal |
| PRC |
| Unallocated |
| Total | ||||
(in US$’000) | ||||||||||||||||||||
Revenue from external customers |
| | — | | | — | | | | — | | |||||||||
Interest income |
| | | | — | — | — | | | | | |||||||||
Interest expense | — | — | — | — | — | — | — | — | ( | ( | ||||||||||
Equity in earnings of equity investees, net of tax |
| | — | | — | — | — | | | — | | |||||||||
Income tax benefit /(expense) |
| | | | ( | — | ( | | ( | ( | ( | |||||||||
Net (loss)/income attributable to the Company |
| ( | ( | ( | | — | | ( | | ( | ( | |||||||||
Depreciation/amortization | ( | ( | ( | — | — | — | ( | ( | ( | ( | ||||||||||
Additions to non-current assets (other than financial instruments and deferred tax assets) |
| | | | — | — | — | | | | | |||||||||
Revenue from external customers is after elimination of inter-segment sales. Sales between segments are carried out at mutually agreed terms. The amounts eliminated attributable to sales between PRC and U.S. and others under R&D in Oncology/Immunology segment were US$
A summary of customers which accounted for over 10% of the Group’s revenue for the years ended December 31, 2023, 2022 and 2021 is as follows:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Customer A | | — | — | |||
Customer B |
| | |
| | |
Customer C |
| (note) | |
| | |
Customer D |
| (note) |
| |
| |
Note: Customer did not account for over 10% of the Group’s revenue during the year.
Customer A, B and C are included in Oncology/Immunology and Customer D is primarily included in Other Ventures.
Unallocated expenses mainly represent corporate expenses which include corporate administrative costs, corporate employee benefit expenses and the relevant share-based compensation expenses, net of interest income. Unallocated assets mainly comprise cash and cash equivalents and short-term investments.
F-47
28. Note to Consolidated Statements of Cash Flows
Reconciliation of net income/(loss) for the year to net cash generated from/(used in) operating activities:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
| (in US$’000) | |||||
Net income/(loss) |
| |
| ( |
| ( |
Adjustments to reconcile net income/(loss) to net cash generated from/(used in) operating activities | ||||||
Depreciation and amortization |
| |
| |
| |
Amortization of finance costs |
| — |
| |
| |
Loss on disposals of property, plant and equipment | | | | |||
Impairment of property, plant and equipment | | — | — | |||
Impairment of right-of-use assets | | — | — | |||
Provision for excess and obsolete inventories, net |
| |
| |
| ( |
Provision for credit losses, net | | | ( | |||
Share-based compensation expense—share options |
| |
| |
| |
Share-based compensation expense—LTIP |
| |
| |
| |
Equity in earnings of equity investees, net of tax |
| ( |
| ( |
| ( |
Dividends received from SHPL |
| |
| |
| |
Impairment of investment in other equity investee | — | | — | |||
Changes in right-of-use assets | | | ( | |||
Fair value losses on warrant | — | | | |||
Gain from divestment of HBYS | — | — | ( | |||
Gain from divestment of subsidiaries | ( | — | — | |||
Gain from divestment of other equity investee | ( | — | — | |||
Unrealized currency translation (gain)/loss |
| ( |
| |
| ( |
Changes in income tax balances |
| |
| ( |
| |
Changes in operating assets and liabilities | ||||||
Accounts receivable |
| ( |
| ( |
| ( |
Other receivables, prepayments and deposits |
| |
| |
| ( |
Amounts due from related parties |
| ( |
| |
| ( |
Inventories |
| |
| ( |
| ( |
Accounts payable |
| ( |
| |
| |
Other payables, accruals and advance receipts |
| ( |
| |
| |
Lease liabilities | ( | ( | | |||
Deferred revenue |
| |
| |
| |
Others | ( | | ( | |||
Total changes in operating assets and liabilities | | | | |||
Net cash generated from/(used in) operating activities |
| |
| ( |
| ( |
29. Litigation
From time to time, the Group may become involved in litigation relating to claims arising from the ordinary course of business. The Group believes that there are currently
F-48
On May 17, 2019, Luye Pharma Hong Kong Ltd. (“Luye”) issued a notice to the Group purporting to terminate a distribution agreement that granted the Group exclusive commercial rights to Seroquel in the PRC for failure to meet a pre-specified target. The Group disagrees with this assertion and believes that Luye have no basis for termination. As a result, the Group commenced legal proceedings in 2019 in order to seek damages. On October 21, 2021 (and a decision on costs and interest in December 2021), the Group was awarded an amount of RMB
30. Restricted Net Assets
Relevant PRC laws and regulations permit payments of dividends by the Company’s subsidiaries in the PRC only out of their retained earnings, if any, as determined in accordance with PRC accounting standards and regulations. In addition, the Company’s subsidiaries in the PRC are required to make certain appropriations of net after-tax profits or increases in net assets to the statutory surplus fund prior to payment of any dividends. In addition, registered share capital and capital reserve accounts are restricted from withdrawal in the PRC, up to the amount of net assets held in each subsidiary. As a result of these and other restrictions under PRC laws and regulations, the Company’s subsidiaries in the PRC are restricted in their ability to transfer their net assets to the Group in terms of cash dividends, loans or advances, with restricted portions amounting to US$
In addition, the Group has an equity investee in the PRC, where the Group’s equity in undistributed earnings amounted to US$
31. Subsequent Events
The Group evaluated subsequent events through February 28, 2024, which is the date when the consolidated financial statements were issued.
In February 2024, pursuant to the strategic partnership with Inmagene Biopharmaceuticals (“Inmagene”), Inmagene has exercised its options to license two drug candidates discovered by HUTCHMED, IMG-007 and IMG-004, for approximately
F-49
Report of Independent Auditors
To the Board of Directors of Shanghai Hutchison Pharmaceuticals Limited
Opinion
We have audited the accompanying consolidated financial statements of Shanghai Hutchison Pharmaceuticals Limited and its subsidiaries (the “Company”), which comprise the consolidated statements of financial position as of December 31, 2023 and 2022, and the related consolidated income statements, consolidated statements of comprehensive income, changes in equity and cash flows for each of the three years in the period ended December 31, 2023, including the related notes (collectively referred to as the “consolidated financial statements”).
In our opinion, the accompanying consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2023 in accordance with IFRS Accounting Standards as issued by the International Accounting Standards Board.
Basis for Opinion
We conducted our audit in accordance with auditing standards generally accepted in the United States of America (US GAAS). Our responsibilities under those standards are further described in the Auditors’ Responsibilities for the Audit of the Consolidated Financial Statements section of our report. We are required to be independent of the Company and to meet our other ethical responsibilities, in accordance with the relevant ethical requirements relating to our audit. We believe that the audit evidence we have obtained is sufficient and appropriate to provide a basis for our audit opinion.
Responsibilities of Management for the Consolidated Financial Statements
Management is responsible for the preparation and fair presentation of the consolidated financial statements in accordance with IFRS Accounting Standards as issued by the International Accounting Standards Board, and for the design, implementation, and maintenance of internal control relevant to the preparation and fair presentation of consolidated financial statements that are free from material misstatement, whether due to fraud or error.
In preparing the consolidated financial statements, management is responsible for assessing the Company’s ability to continue as a going concern for at least, but not limited to, twelve months from the end of the reporting period, disclosing, as applicable, matters related to going concern and using the going concern basis of accounting unless management either intends to liquidate the Company or to cease operations, or has no realistic alternative but to do so.
Auditors’ Responsibilities for the Audit of the Consolidated Financial Statements
Our objectives are to obtain reasonable assurance about whether the consolidated financial statements as a whole are free from material misstatement, whether due to fraud or error, and to issue an auditors’ report that includes our opinion. Reasonable assurance is a high level of assurance but is not absolute assurance and therefore is not a guarantee that an audit conducted in accordance with US GAAS will always detect a material misstatement when it exists. The risk of not detecting a material misstatement resulting from fraud is higher than for one resulting from error, as fraud may involve collusion, forgery, intentional omissions, misrepresentations, or the override of internal control. Misstatements are considered material if there is a substantial likelihood that, individually or in the aggregate, they would influence the judgment made by a reasonable user based on the consolidated financial statements.
In performing an audit in accordance with US GAAS, we:
| ● | Exercise professional judgment and maintain professional skepticism throughout the audit. |
| ● | Identify and assess the risks of material misstatement of the consolidated financial statements, whether due to fraud or error, and design and perform audit procedures responsive to those risks. Such procedures include examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. |
F-51
Auditors’ Responsibilities for the Audit of the Consolidated Financial Statements (Continued)
| ● | Obtain an understanding of internal control relevant to the audit in order to design audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control. Accordingly, no such opinion is expressed. |
| ● | Evaluate the appropriateness of accounting policies used and the reasonableness of significant accounting estimates made by management, as well as evaluate the overall presentation of the consolidated financial statements. |
| ● | Conclude whether, in our judgment, there are conditions or events, considered in the aggregate, that raise substantial doubt about the Company’s ability to continue as a going concern for a reasonable period of time. |
We are required to communicate with those charged with governance regarding, among other matters, the planned scope and timing of the audit, significant audit findings, and certain internal control-related matters that we identified during the audit.
/S/ PricewaterhouseCoopers Zhong Tian LLP
Shanghai, the People’s Republic of China
February 28, 2024
F-52
Shanghai Hutchison Pharmaceuticals Limited
Consolidated Income Statements
(in US$’000)
Year Ended December 31, | ||||||||
| Note |
| 2023 |
| 2022 |
| 2021 | |
Revenue |
| 5 | 385,483 |
| 370,600 |
| 332,648 | |
Cost of sales | (101,122) |
| (89,487) |
| (77,559) | |||
Gross profit | 284,361 |
| 281,113 |
| 255,089 | |||
Selling expenses | (150,717) |
| (144,979) |
| (131,821) | |||
Administrative expenses | (26,107) |
| (21,727) |
| (22,627) | |||
Other net operating income |
| 6 | 5,027 |
| 2,126 |
| 4,759 | |
Operating profit |
| 7 | 112,564 |
| 116,533 |
| 105,400 | |
Finance costs | 15 | (79) | (112) | (116) | ||||
Profit before taxation |
| 112,485 |
| 116,421 |
| 105,284 | ||
Taxation charge |
| 8 | (17,022) |
| (16,738) |
| (15,896) | |
Profit for the year | 95,463 |
| 99,683 |
| 89,388 | |||
The accompanying notes are an integral part of these consolidated financial statements.
F-53
Shanghai Hutchison Pharmaceuticals Limited
Consolidated Statements of Comprehensive Income
(in US$’000)
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
Profit for the year |
| 95,463 | 99,683 |
| 89,388 | |
Other comprehensive income/(loss) that has been or may be reclassified subsequently to profit or loss: |
|
| ||||
Exchange translation differences |
| 2,320 | (16,581) |
| 3,341 | |
Total comprehensive income |
| 97,783 | 83,102 |
| 92,729 | |
The accompanying notes are an integral part of these consolidated financial statements.
F-54
Shanghai Hutchison Pharmaceuticals Limited
Consolidated Statements of Financial Position
(in US$’000)
December 31, | ||||||
| Note |
| 2023 |
| 2022 | |
Assets |
|
|
|
| ||
Current assets |
|
|
|
| ||
Cash and cash equivalents |
| 10 | 19,129 |
| 33,923 | |
Trade and bills receivables |
| 11 | 15,601 |
| 21,856 | |
Other receivables, prepayments and deposits |
| 12 | 2,269 |
| 3,672 | |
Inventories |
| 13 | 164,026 |
| 154,816 | |
Total current assets |
| 201,025 |
| 214,267 | ||
Property, plant and equipment |
| 14 | 57,930 |
| 62,831 | |
Right-of-use assets | 15 | 1,092 | 1,717 | |||
Leasehold land |
| 5,967 |
| 6,291 | ||
Other intangible assets |
| 519 |
| 823 | ||
Deferred tax assets |
| 16 | 8,330 |
| 8,327 | |
Other non-current assets | 641 | — | ||||
Total assets |
| 275,504 |
| 294,256 | ||
Liabilities and shareholders’ equity |
|
| ||||
Current liabilities |
|
| ||||
Trade payables |
| 17 | 23,836 |
| 23,095 | |
Other payables, accruals and advance receipts |
| 18 | 153,937 |
| 121,354 | |
Current tax liabilities |
| 19 | 1,163 |
| 2,791 | |
Lease liabilities | 15 | 713 | 712 | |||
Total current liabilities |
| 179,649 |
| 147,952 | ||
Deferred income |
| 3,030 |
| 3,585 | ||
Lease liabilities | 15 | 657 | 1,360 | |||
Total liabilities |
| 183,336 |
| 152,897 | ||
Shareholders’ equity |
|
| ||||
Share capital |
| 33,382 |
| 33,382 | ||
Reserves |
| 58,786 |
| 107,977 | ||
Total shareholders’ equity |
| 92,168 |
| 141,359 | ||
Total liabilities and shareholders’ equity |
| 275,504 |
| 294,256 | ||
The accompanying notes are an integral part of these consolidated financial statements.
F-55
Shanghai Hutchison Pharmaceuticals Limited
Consolidated Statements of Changes in Equity
(in US$’000)
| Share |
| Exchange |
| General |
| Retained |
| Total | |
capital | reserve | reserves | earnings | equity | ||||||
As at January 1, 2021 | 33,382 | 2,605 | 998 | 115,723 | 152,708 | |||||
Profit for the year |
| — |
| — |
| — |
| 89,388 |
| 89,388 |
Other comprehensive income |
|
|
|
|
| |||||
Exchange translation differences |
| — |
| 3,341 |
| — |
| — |
| 3,341 |
Total comprehensive income |
| — |
| 3,341 |
| — |
| 89,388 |
| 92,729 |
Transfer between reserves | — | — | 31 | (31) | — | |||||
Dividends declared to shareholders |
| — |
| — |
| — |
| (99,744) |
| (99,744) |
As at December 31, 2021 |
| 33,382 |
| 5,946 |
| 1,029 |
| 105,336 |
| 145,693 |
Profit for the year | — | — | — | 99,683 | 99,683 | |||||
Other comprehensive income | ||||||||||
Exchange translation differences | — | (16,581) | — | — | (16,581) | |||||
Total comprehensive (loss)/income | — | (16,581) | — | 99,683 | 83,102 | |||||
Transfer between reserves | — | — | 14 | (14) | — | |||||
Dividends declared to shareholders | — | — | — | (87,436) | (87,436) | |||||
As at December 31, 2022 | 33,382 | (10,635) | 1,043 | 117,569 | 141,359 | |||||
Profit for the year |
| — |
| — |
| — |
| 95,463 |
| 95,463 |
Other comprehensive income |
|
|
|
|
| |||||
Exchange translation differences |
| — |
| 2,320 |
| — |
| — |
| 2,320 |
Total comprehensive income |
| — |
| 2,320 |
| — |
| 95,463 |
| 97,783 |
Transfer between reserves |
| — |
| — |
| 30 |
| (30) |
| — |
Dividends declared to shareholders (Note 18) |
| — |
| — |
| — |
| (146,974) |
| (146,974) |
As at December 31, 2023 |
| 33,382 |
| (8,315) |
| 1,073 |
| 66,028 |
| 92,168 |
The accompanying notes are an integral part of these consolidated financial statements.
F-56
Shanghai Hutchison Pharmaceuticals Limited
Consolidated Statements of Cash Flows
(in US$’000)
Year Ended December 31, | ||||||||
| Note |
| 2023 |
| 2022 |
| 2021 | |
Operating activities |
|
|
|
|
|
|
|
|
Net cash generated from operations |
| 20 |
| 96,080 |
| 96,270 |
| 93,970 |
Interest received |
|
|
| 645 |
| 1,219 |
| 1,116 |
Income tax paid |
| 19 |
| (18,709) |
| (19,003) |
| (15,976) |
Net cash generated from operating activities |
|
|
| 78,016 |
| 78,486 |
| 79,110 |
Investing activities |
|
|
|
|
| |||
Purchase of property, plant and equipment |
|
| (6,488) |
| (1,865) |
| (3,362) | |
Purchase of intangible asset | — | (410) | — | |||||
Proceeds from disposal of property, plant and equipment |
|
|
| 12 |
| 20 |
| 32 |
Net cash used in investing activities |
|
|
| (6,476) |
| (2,255) |
| (3,330) |
Financing activities |
|
|
|
|
|
|
|
|
Dividends paid to shareholders |
| 18 |
| (84,615) |
| (87,436) |
| (99,744) |
Lease payments |
| 15 |
| (810) |
| (809) |
| (303) |
Net cash used in financing activities |
|
|
| (85,425) |
| (88,245) |
| (100,047) |
Net decrease in cash and cash equivalents |
|
|
| (13,885) |
| (12,014) |
| (24,267) |
Effect of exchange rate changes on cash and cash equivalents |
|
|
| (909) |
| (4,101) |
| 1,827 |
| (14,794) |
| (16,115) |
| (22,440) | |||
Cash and cash equivalents |
|
|
|
|
| |||
Cash and cash equivalents at beginning of year |
|
|
| 33,923 |
| 50,038 |
| 72,478 |
Cash and cash equivalents at end of year |
|
|
| 19,129 |
| 33,923 |
| 50,038 |
The accompanying notes are an integral part of these consolidated financial statements.
F-57
Shanghai Hutchison Pharmaceuticals Limited
Notes to the Consolidated Financial Statements
1. General Information
Shanghai Hutchison Pharmaceuticals Limited (the “Company”) and its subsidiaries (together the “Group”) are principally engaged in manufacturing, selling and distribution of prescription drug products. The Group has manufacturing plants in the People’s Republic of China (the “PRC”) and sells mainly in the PRC.
The Company was incorporated in the PRC on April 30, 2001 as a Chinese-Foreign Equity joint venture. The Company is jointly controlled by Shanghai HUTCHMED Investment (HK) Limited (“SHHCMI(HK)L”) and Shanghai Traditional Chinese Medicine Co., Ltd (“SHTCML”).
These consolidated financial statements are presented in United States dollars (“US$”), unless otherwise stated and have been approved for issue by the Company’s Board of Directors on February 28, 2024.
2. Summary of Accounting Policies
The consolidated financial statements of the Company have been prepared in accordance with International Financial Reporting Standards (“IFRS Accounting Standards”) as issued by the International Accounting Standards Board (“IASB”) and interpretations issued by the IFRS Interpretations Committee applicable to companies reporting under IFRS Accounting Standards. These consolidated financial statements have been prepared under the historical cost convention.
During the year, the Group has adopted all of the new and revised standards, amendments and interpretations issued by the IASB that are relevant to the Group’s operations and mandatory for annual periods beginning January 1, 2023. The adoption of these new and revised standards, amendments and interpretations did not have any material effects on the Group’s results of operations or financial position.
The following standards, amendments and interpretations were issued but not yet effective for the financial year ended December 31, 2023 and have not been early adopted by the Group:
IAS 1 (Amendments)(1) | Classification of Liabilities as Current or Non-current |
IAS 1 (Amendments)(1) | Non-current Liabilities with Covenants |
IFRS 16 (Amendments)(1) | Lease Liability in a Sale and Leaseback |
IAS 7 and IFRS 7 (Amendments)(1) | Supplier Finance Arrangements |
IAS 21 (Amendments)(2) | Lack of Exchangeability |
IFRS 10 and IAS 28 (Amendments)(3) | Sale or Contribution of Assets between an Investor and its Associate or Joint Venture |
(1)Effective for the Group for annual periods beginning on or after January 1, 2024.
(2)Effective for the Group for annual periods beginning on or after January 1, 2025.
(3)Effective date to be determined by the IASB.
The adoption of standards, amendments and interpretations listed above in future periods is not expected to have any material effects on the Group’s results of operations or financial position.
(a) Material Accounting Policies
(i) Property, Plant and Equipment
Property, plant and equipment other than construction in progress are stated at historical cost less accumulated depreciation and any accumulated impairment losses. Historical cost includes the purchase price of the asset and any directly attributable costs of bringing the asset to its working condition and location for its intended use.
F-58
Subsequent costs are included in the asset’s carrying amount or recognized as a separate asset, as appropriate, only when it is probable that future economic benefits associated with the item will flow to the Group and the cost of the item can be measured reliably. All other repairs and maintenance are charged to the consolidated income statements during the financial period in which they are incurred.
Depreciation is calculated using the straight-line method to allocate asset costs less accumulated impairment losses over their estimated useful lives. The principal estimated useful lives are as follows:
Buildings |
| 20 years |
Leasehold improvements | Over the unexpired period of the lease or 5 years, whichever is shorter | |
Plant and equipment | 10 years | |
Furniture and fixtures, other equipment and motor vehicles | 5 years |
The assets’ useful lives are reviewed and adjusted, if appropriate, at the end of each reporting period. An asset’s carrying amount is written down immediately to its recoverable amount if the asset’s carrying amount is greater than its estimated recoverable amount.
Gains and losses on disposals are determined by comparing net sales proceeds with the carrying amount of the relevant assets and are recognized in the consolidated income statements.
Construction in progress represents buildings, plant and machinery under construction and pending installation and is stated at cost less accumulated impairment losses, if any. Cost includes the costs of construction of buildings and the costs of plant and machinery. No provision for depreciation is made on construction in progress until such time as the relevant assets are completed and ready for its intended use. When the assets concerned are brought into use, the costs are transferred to property, plant and equipment and depreciated.
(ii) Research and Development
Research expenditure is recognized as an expense as incurred. Costs incurred on development projects (relating to the design and testing of new or improved products) are recognized as intangible assets when it is probable that the project will generate future economic benefits by considering its commercial and technological feasibility, and costs can be measured reliably. Other development expenditures are recognized as an expense as incurred. Development costs previously recognized as an expense are not recognized as an asset in a subsequent period. Development costs with a finite useful life that have been capitalized, if any, are amortized on a straight line basis over the period of expected benefit not exceeding five years. The capitalized development costs are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset exceeds its recoverable amount.
Where the research phase and the development phase of an internal project cannot be clearly distinguished, all expenditure incurred on the project is charged to the consolidated income statements.
(iii) Financial Liabilities and Equity Instruments
Financial liabilities and equity instruments issued by the Group are classified according to the substance of the contractual arrangements entered into and the definitions of a financial liability and an equity instrument. Financial liabilities (including trade and other payables) are initially measured at fair value, and are subsequently measured at amortized cost, using the effective interest method. An equity instrument is any contract that does not meet the definition of a financial liability and evidences a residual interest in the assets of the Group after deducting all of its liabilities.
Ordinary shares are classified as equity. Incremental costs, net of tax, directly attributable to the issue of new shares are shown in equity as a deduction from the proceeds.
F-59
(iv) Current and Deferred Income Tax
(1) Current income tax
The current income tax charge is calculated on the basis of the tax laws enacted or substantively enacted at the balance sheet date in the country where the Group operates and generates taxable income. Management periodically evaluates positions taken in tax returns with respect to situations in which applicable tax regulation is subject to interpretation. It establishes provisions where appropriate on the basis of amounts expected to be paid to the tax authorities.
(2) Deferred income tax
Inside basis differences
Deferred income tax is provided in full, using the liability method, on temporary differences arising between the tax bases of assets and liabilities and their carrying amounts in the consolidated financial statements. However, deferred tax liabilities are not recognised if they arise from the initial recognition of goodwill. Deferred income tax is also not accounted for if it arises from initial recognition of an asset or liability in a transaction other than a business combination that at the time of the transaction affects neither accounting nor taxable profit or loss and does not give rise to equal taxable and deductible temporary differences. Deferred income tax is determined using tax rates (and laws) that have been enacted or substantively enacted by the end of the reporting period and are expected to apply when the related deferred income tax asset is realised or the deferred income tax liability is settled.
Deferred income tax assets are recognized only to the extent that it is probable that future taxable profit will be available against which the temporary differences can be utilized. Deferred income tax assets and deferred income tax liabilities are offset when there is a legally enforceable right to set off and when the deferred income taxes related to the same fiscal authority.
Outside basis differences
Deferred income tax liabilities are provided on taxable temporary differences arising from investments in subsidiaries, except for deferred income tax liabilities where the timing of the reversal of the temporary difference is controlled by the Group and it is probable that the temporary difference will not reverse in the foreseeable future.
Deferred income tax assets are recognized on deductible temporary differences arising from investments in subsidiaries, only to the extent that it is probable the temporary difference will reverse in the future and there is sufficient taxable profit available against which the temporary difference can be utilized.
(v) Revenue and Income Recognition
Revenue is measured based on consideration specified in a contract with a customer, and excludes any sales incentives and amounts collected on behalf of third parties. Taxes assessed by a governmental authority that are both imposed on and concurrent with a specific revenue-producing transaction, that are collected by the Group from a customer, are also excluded from revenue. The Group recognizes revenue when it satisfies a performance obligation by transferring control over a good to a customer.
The Group principally generates revenue from sales of goods. Revenue from sales of goods is recognized when the customer takes possession of the goods. This usually occurs upon completed delivery of the goods to the customer site. The amount of revenue recognized is adjusted for expected sales incentives as stipulated in the contract, which are generally issued to customers as direct discounts at the point-of-sale or indirectly in the form of rebates. Sales incentives are estimated using the expected value method. Additionally, sales are generally made with a limited right of return under certain conditions. Revenues are recorded net of provisions for sales discounts and returns.
Revenue from provision of services is recognized when the benefits of the services transfer to the customer over time, which is based on the proportionate value of services rendered as determined under the terms of the relevant contract. Additionally, when the amounts that can be invoiced correspond directly with the value to the customer for performance completed to date, the Group recognizes revenue from provision of services based on amounts that can be invoiced to the customer.
F-60
Payments in advance from customers are deferred if consideration is received in advance of transferring control of the goods or rendering of services. Accounts receivable is recognized if the Group has an unconditional right to bill the customer, which is generally when the customer takes possession of the goods or services are rendered. Payment terms differ by subsidiary and customer, but generally range from 45 to 180 days from the invoice date.
(b) Other Accounting Policies
(i)Basis of Consolidation
The consolidated financial statements of the Group include the financial statements of the Company and its subsidiaries.
The accounting policies of subsidiaries have been changed where necessary to ensure consistency with the policies adopted by the Group.
Intercompany transactions, balances and unrealized gains on transactions between group companies are eliminated. Unrealized losses are also eliminated unless the transaction provides evidence of an impairment of the transferred asset.
(ii) Subsidiaries
Subsidiaries are all entities over which the Group has control. The Group controls an entity when the Group is exposed, or has rights, to variable returns from its involvement with the entity and has the ability to affect those returns through its power to direct the activities of the entity. In the consolidated financial statements, subsidiaries are accounted for as described in Note 2(b)(i) above.
Subsidiaries are fully consolidated from the date on which control is transferred to the Group. They are de-consolidated from the date that control ceases.
(iii) Foreign Currency Translation
Items included in the financial statements of each of the Group’s companies are measured using the currency of the primary economic environment in which the entity operates (the “functional currency”). The functional currency of the Company and its subsidiaries is Renminbi (“RMB”) whereas the consolidated financial statements are presented in US$, which is the Company’s presentation currency.
Foreign currency transactions are translated into the functional currency using the exchange rates at the dates of the transactions. Foreign currency gains and losses resulting from the settlement of such transactions and from the translation of monetary assets and liabilities denominated in foreign currencies at year end exchange rates are generally recognized in the consolidated income statements.
The financial statements of the Company and its subsidiaries are translated into the Company’s presentation currency using the year end rates of exchange for the statements of financial position items and the average rates of exchange for the year for the income statement items. Exchange translation differences are recognized directly in other comprehensive income.
F-61
(iv) Impairment of Non-Financial Assets
Assets are reviewed for impairment to determine whether there is any indication that the carrying value of these assets may not be recoverable and have suffered an impairment loss. If any such indication exists, the recoverable amount of the asset is estimated in order to determine the extent of the impairment loss, if any. The recoverable amount is the higher of an asset’s fair value less costs to sell and value in use. Such impairment loss is recognized in the consolidated income statements. Assets that have an indefinite useful life such as goodwill or intangible assets not ready to use are not subject to amortization and are tested for impairment annually and when there are indications that the carrying value may not be recoverable.
(v) Inventories
Inventories are stated at the lower of cost or net realizable value. Cost is determined using the weighted average cost method. The cost of finished goods comprises raw materials, direct labor, other direct costs and related production overheads (based on normal operating capacity). Net realizable value is the estimated selling price in the ordinary course of business, less applicable variable selling expenses.
(vi) Trade and Other Receivables
Trade and other receivables are recognized initially at the amount of consideration, which is unconditional. Trade and other receivables solely represent payments of principal and interest, if any, and the Group holds such financial assets with the objective to collect its contractual cash flows. Therefore, the Group measures them subsequently at amortized cost using the effective interest method, less any loss allowance. The Group applies the IFRS 9 simplified approach to measuring expected credit losses which uses a lifetime expected loss allowance for all trade receivables. To measure the expected credit losses, trade receivables have been grouped based on shared credit risk characteristics and the days past due. All other receivables at amortized cost are considered to have low credit risk, and the loss allowance recognized during the period was therefore limited to 12 months expected losses. The amount of the provision is recognized in the consolidated income statements.
(vii) Cash and Cash Equivalents
In the consolidated statements of cash flows, cash and cash equivalents include cash on hand, bank deposits and other short-term highly liquid investments with original maturities of three months or less that are readily convertible to known amounts of cash and which are subject to an insignificant risk of changes in value, if any.
(viii) Employee Benefits
The employees of the Group participate in defined contribution retirement benefit plans managed by the relevant municipal and provincial governments in the PRC. The assets of these plans are held separately from the Group. The Group is required to make monthly contributions to the plans calculated as a percentage of the employees’ salaries. The municipal and provincial governments undertake to assume the retirement benefit obligations to all existing and future retired employees under the plans described above. Other than the monthly contributions, the Group has no further obligations for the payment of the retirement and other post-retirement benefits of its employees.
(ix) Leases
A lease is recognized as a right-of-use asset with a corresponding liability at the date which the leased asset is available for use by the Group. The Group recognizes an obligation to make lease payments equal to the present value of the lease payments over the lease term. The lease terms may include options to extend or terminate the lease when it is reasonably certain that the Group will exercise that option.
F-62
Lease liabilities include the net present value of the following lease payments: (i) fixed payments; (ii) variable lease payments that depend on an index or a rate; and (iii) payments of penalties for terminating the lease if the lease term reflects the lessee exercising that option, if any. Lease liabilities exclude the following payments that are generally accounted for separately: (i) non-lease components, such as maintenance and security service fees and value added tax, and (ii) any payments that a lessee makes before the lease commencement date. The lease payments are discounted using the interest rate implicit in the lease or if that rate cannot be determined, the lessee’s incremental borrowing rate being the rate that the lessee would have to pay to borrow the funds in its currency and jurisdiction necessary to obtain an asset of similar value, economic environment and terms and conditions.
An asset representing the right to use the underlying asset during the lease term is recognized that consists of the initial measurement of the lease liability, any lease payments made to the lessor at or before the commencement date less any lease incentives received, any initial direct cost incurred by the Group and any restoration costs.
After commencement of the lease, each lease payment is allocated between lease liability and finance costs. The finance costs are recognized over the lease term so as to produce a constant periodic rate of interest on the remaining balance of the lease liability for each period. The right-of-use asset is depreciated on a straight-line basis over the period of the lease.
Payments associated with short-term leases are recognized as lease expenses on a straight-line basis over the period of the leases.
Leasehold land is accounted under IFRS 16.
(x) Government Incentives
Incentives from government are recognized at their fair values where there is a reasonable assurance that the incentives will be received and all attached conditions will be complied with.
Government incentives relating to costs are deferred and recognized in the consolidated income statements over the period necessary to match them with the costs that they are intended to compensate.
Government grants relating to property, plant and equipment are included in other payables, accruals and advance receipts and non-current liabilities as deferred income and credited to the consolidated income statements on a straight-line basis over the expected lives of the related assets.
(xi) Segment Reporting
Operating segments are reported in a manner consistent with the internal reporting provided to the chief operating decision makers. The Company’s Board of Directors, which is responsible for allocating resources and assessing performance of the operating segments, has been identified as the steering committee that makes strategic decisions.
(xii) General Reserves
In accordance with the laws applicable to Foreign Investment Enterprises established in the PRC, the Company makes appropriations to certain non-distributable reserve funds including the general reserve fund, the enterprise expansion fund and the staff bonus and welfare fund. The amount of appropriations to these funds are made at the discretion of the Company’s Board of Directors.
3. Financial Risk Management
(a) Financial risk factors
The Group’s activities expose it to a variety of financial risks, including credit risk and liquidity risk. The Group does not use any derivative financial instruments for speculative purposes.
F-63
(i) Credit risk
The carrying amounts of cash and cash equivalents, trade and bills receivables and other receivables included in the consolidated statements of financial position represent the Group’s maximum exposure to credit risk of the counterparty in relation to its financial assets.
Substantially all of the Group’s cash and cash equivalents are deposited in major financial institutions, which management believes are of high credit quality. The Group has a practice to limit the amount of credit exposure to any financial institution.
Bills receivables are mostly settled by state-owned banks or other reputable banks and therefore the management considers that they will not expose the Group to any significant credit risk.
The Group has no significant concentrations of credit risk. The Group has policies in place to ensure that the sales of products are made to customers with appropriate credit history and the Group performs periodic credit evaluations of its customers.
Management periodically assesses the recoverability of trade and bills receivables and other receivables. The Group’s historical loss rates reflect current and forward-looking information on specific factors affecting the ability of the customers to settle the receivables, and historical experience collecting receivables falls within the recorded allowances. The Group has not had any material credit losses.
(ii) Liquidity risk
Prudent liquidity management implies maintaining sufficient cash and cash equivalents and the availability of funding when necessary. The Group’s policy is to regularly monitor current and expected liquidity requirements to ensure that it maintains sufficient cash balances and adequate credit facilities to meet its liquidity requirements in the short and long term.
As at December 31, 2023 and 2022, in addition to future lease payments due based on the lease term (Note 15) , the Group’s all other current financial liabilities are mainly due for settlement within twelve months and the Group expects to meet all liquidity requirements.
(b) Capital risk management
The Group’s objectives when managing capital are to safeguard the Group’s ability to provide returns for shareholders and benefits for other stakeholders and to maintain an optimal capital structure to reduce the cost of capital.
The Group regularly reviews and manages its capital structure to ensure an optimal balance between higher shareholders’ return that might be possible with higher levels of borrowings and the advantages and security afforded by a sound capital position, and makes adjustments to the capital structure in light of changes in economic conditions.
The Group monitors capital on the basis of the liabilities to assets ratio. This ratio is calculated as total liabilities divided by total assets as shown on the consolidated statements of financial position.
The liabilities to assets ratio as at December 31, 2023 and 2022 was as follows:
December 31, | |||||
| 2023 |
| 2022 | ||
| (in US$’000) | ||||
Total liabilities | 183,336 |
| 152,897 | ||
Total assets | 275,504 |
| 294,256 | ||
Liabilities to assets ratio | 66.5 | % | 52.0 | % | |
F-64
(c) Fair value estimation
The Group does not have any financial assets or liabilities which are carried at fair value. The carrying amounts of the Group’s current financial assets, including cash and cash equivalents, trade and bills receivables and other receivables, and current financial liabilities, including trade payables and other payables and accruals, approximate their fair values due to their short-term maturities. The carrying amounts of the Group’s financial instruments carried at cost or amortized cost are not materially different from their fair values.
The face values less any estimated credit adjustments for financial assets and liabilities with a maturity of less than one year are assumed to approximate their fair values. The fair value of financial liabilities for disclosure purposes is estimated by discounting the future contractual cash flows at the current market interest rate that is available to the Group for similar financial instruments.
4. Critical Accounting Estimates and Judgements
Note 2(a) includes a summary of the material accounting policies used in the preparation of the consolidated financial statements. The preparation of consolidated financial statements often requires the use of judgements to select specific accounting methods and policies from several acceptable alternatives. Furthermore, significant estimates and assumptions concerning the future may be required in selecting and applying those methods and policies in the consolidated financial statements. The Group bases its estimates and judgements on historical experience and various other assumptions that it believes are reasonable under the circumstances. Actual results may differ from these estimates and judgements under different assumptions or conditions.
The following is a review of the more significant assumptions and estimates, as well as the accounting policies and methods used in the preparation of the consolidated financial statements.
(a) Sales rebates
Certain sales rebates are provided to customers when their business performance for an agreed period within the year and the whole year meets certain criteria as stipulated in the contracts. Sales rebates are considered variable consideration and the estimate of sales rebates during the year is based on estimated sales transactions for the entire period stipulated and is subject to change based on actual performance and collection status.
(b) Useful lives of property, plant and equipment
The Group has made substantial investments in property, plant and equipment. Changes in technology or changes in the intended use of these assets may cause the estimated period of use or value of these assets to change.
(c) Deferred income tax
Deferred tax is recognized using the liability method on temporary differences arising between the tax bases of assets and liabilities against which the deductible temporary differences and the carry forward of unused tax losses and tax credits can be utilized. Deferred income tax assets are recognized only to the extent that it is probable that future taxable profit will be available against which the temporary differences can be utilized. Where the final outcomes are different from the estimations, such differences will impact the carrying amount of deferred tax in the period in which such determination is made.
5. Revenue and Segment Information
Management has reviewed the Group’s internal reporting in order to assess performance and allocate resources, and has determined that the Group has two reportable operating segments as follows:
—Manufacturing business—manufacture and distribution of drug products
—Distribution business—provision of sales, distribution and marketing services to pharmaceutical manufacturers and healthcare products
F-65
The operating segments are strategic business units that offer different products and services. They are managed separately because each business requires different technology and marketing approaches. The performance of each of the reportable segments is assessed based on a measure of operating profit/(loss).
The segment information is as follows:
Year Ended December 31, 2023 | ||||||
| Manufacturing |
| Distribution |
| ||
business | business | Total | ||||
| PRC | |||||
(in US$’000) | ||||||
Revenue from external customers | 373,376 |
| 12,107 |
| 385,483 | |
Interest income | 427 |
| 327 |
| 754 | |
Operating profit/(loss) | 113,468 |
| (904) |
| 112,564 | |
Finance costs | 76 |
| 3 |
| 79 | |
Depreciation/amortization | 8,365 |
| 163 |
| 8,528 | |
Additions to non-current assets (other than financial instruments and deferred tax assets) | 4,859 |
| 54 |
| 4,913 | |
December 31, 2023 | ||||||
| Manufacturing |
| Distribution |
| ||
business | business | Total | ||||
| PRC | |||||
| (in US$’000) | |||||
Total segment assets | 272,104 |
| 3,400 |
| 275,504 | |
Year Ended December 31, 2022 | ||||||
Manufacturing | Distribution | |||||
business |
| business |
| Total | ||
| PRC | |||||
(in US$’000) | ||||||
Revenue from external customers |
| 367,512 |
| 3,088 |
| 370,600 |
Interest income |
| 501 |
| 479 |
| 980 |
Operating profit/(loss) |
| 117,210 |
| (677) |
| 116,533 |
Finance costs |
| 110 |
| 2 |
| 112 |
Depreciation/amortization |
| 9,151 |
| 89 |
| 9,240 |
Additions to non-current assets (other than financial instruments and deferred tax assets) |
| 3,636 |
| 532 |
| 4,168 |
December 31, 2022 | ||||||
| Manufacturing |
| Distribution |
| ||
business | business | Total | ||||
| PRC | |||||
(in US$’000) | ||||||
Total segment assets |
| 291,877 |
| 2,379 |
| 294,256 |
F-66
Year Ended December 31, 2021 | ||||||
| Manufacturing |
| Distribution |
| ||
business | business | Total | ||||
PRC | ||||||
| (in US$’000) | |||||
Revenue from external customers |
| 331,097 |
| 1,551 |
| 332,648 |
Interest income |
| 629 |
| 587 |
| 1,216 |
Operating profit/(loss) |
| 107,361 |
| (1,961) |
| 105,400 |
Finance costs | 114 | 2 | 116 | |||
Depreciation/amortization |
| 9,118 |
| 50 |
| 9,168 |
Additions to non-current assets (other than financial instruments and deferred tax assets) |
| 5,867 |
| 82 |
| 5,949 |
Revenue from external customers is after elimination of inter-segment sales. The amount eliminated was US$80.5 million for 2023 (2022: US$87.3 million; 2021: US$77.8 million). Sales between segments are carried out at mutually agreed terms. Revenue from external customers from the manufacturing business is for sales of goods which are recognized at a point in time. Revenue from external customers from the distribution business is for provision of services which are recognized over time.
6. Other Net Operating Income
Year Ended December 31, | ||||||
2023 |
| 2022 |
| 2021 | ||
| (in US$’000) | |||||
Interest income |
| 754 |
| 980 |
| 1,216 |
Net foreign exchange (loss)/gain |
| (78) |
| (83) |
| 25 |
Government incentives |
| 4,414 |
| 2,198 |
| 2,999 |
Other operating (loss)/income |
| (63) |
| (969) |
| 519 |
| 5,027 |
| 2,126 |
| 4,759 | |
7. Operating Profit
Year Ended December 31, | ||||||
2023 |
| 2022 |
| 2021 | ||
| (in US$’000) | |||||
Operating profit |
| 112,564 |
| 116,533 |
| 105,400 |
Operating profit is stated after charging/(crediting) the following:
Year Ended December 31, | ||||||
2023 |
| 2022 |
| 2021 | ||
| (in US$’000) | |||||
Cost of inventories recognized as expense |
| 70,397 |
| 63,079 |
| 50,637 |
Research and development expense | 8,621 | 7,169 | 9,350 | |||
Depreciation of property, plant and equipment |
| 7,417 |
| 8,148 |
| 8,100 |
Loss on disposal of property, plant and equipment |
| 32 |
| 449 |
| 60 |
Amortization of leasehold land |
| 158 |
| 166 |
| 172 |
Amortization of other intangible assets |
| 288 |
| 245 |
| 233 |
Depreciation charge of right-of-use assets and lease expenses |
| 872 |
| 917 |
| 1,171 |
Movement on the provision for excess and obsolete inventories | 2,121 | (65) | (141) | |||
Auditor’s remuneration |
| 221 |
| 227 |
| 223 |
Employee benefit expenses (Note 9) |
| 117,126 |
| 111,200 |
| 100,311 |
F-67
8. Taxation Charge
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
| (in US$’000) | |||||
Current tax (Note 19) | 17,197 |
| 18,082 |
| 15,082 | |
Deferred income tax (Note 16) | (175) |
| (1,344) |
| 814 | |
Taxation charge | 17,022 |
| 16,738 |
| 15,896 | |
The taxation charge on the Group’s profit before taxation differs from the theoretical amount that would arise using the Group’s weighted average tax rate as follows:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
| (in US$’000) | |||||
Profit before taxation | 112,485 |
| 116,421 |
| 105,284 | |
Tax calculated at the statutory tax rates of respective companies | 28,121 |
| 29,105 |
| 26,321 | |
Tax effects of: |
|
| ||||
Expenses not deductible for tax purposes | 1,628 |
| 1,397 |
| 1,946 | |
Utilization of unrecognized temporary differences | (518) |
| (898) |
| (55) | |
Tax concession (note) | (12,540) |
| (13,000) |
| (12,420) | |
Under provision in prior years | 331 |
| 134 |
| 104 | |
Taxation charge | 17,022 |
| 16,738 |
| 15,896 | |
Note: The Company has been granted the High and New Technology Enterprise (“HNTE”) status. Accordingly, the Company is subject to a preferential income tax rate of 15% in 2023 and successfully renew the HNTE status in 2023 (2022: 15%; 2021: 15%). Certain research and development expenses are also eligible for super-deduction such that 200% of qualified expenses incurred are deductible against taxable profits for tax purposes (2022: 200%; 2021: 200%).
The weighted average tax rate calculated at the statutory tax rates of respective companies was 25%. The effective tax rate for the year ended December 31, 2023 was 15.1% (2022: 14.4%; 2021: 15.1%).
9. Employee Benefit Expenses
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
(in US$’000) | ||||||
Wages, salaries and bonuses | 90,372 |
| 86,330 |
| 77,335 | |
Pension costs—defined contribution plans | 10,444 |
| 9,701 |
| 8,713 | |
Staff welfare | 16,310 |
| 15,169 |
| 14,263 | |
117,126 |
| 111,200 |
| 100,311 | ||
Employee benefit expenses of approximately US$22.8 million for the year ended December 31, 2023 (2022: US$19.8 million; 2021: US$20.1 million) are included in cost of sales.
10. Cash and Cash Equivalents
December 31, | ||||
| 2023 |
| 2022 | |
(in US$’000) | ||||
Cash and cash equivalents | 19,129 |
| 33,923 | |
F-68
The cash and cash equivalents denominated in RMB were deposited with banks in the PRC. The conversion of these RMB denominated balances into foreign currencies is subject to the rules and regulations of foreign exchange control promulgated by the PRC government.
11. Trade and Bills Receivables
December 31, | |||||
| 2023 |
| 2022 | ||
| (in US$’000) | ||||
Trade receivables—third parties | 11,461 |
| 12,845 | ||
Trade receivables—related parties (Note 22(b)) | 1,303 |
| 3,695 | ||
Bills receivables | 2,837 |
| 5,316 | ||
15,601 |
| 21,856 | |||
All trade and bills receivables are denominated in RMB and are due within one year from the end of the reporting period. The carrying values of trade and bills receivables approximate their fair values due to their short-term maturities.
No allowance for credit losses has been made for trade and bills receivables for the years ended December 31, 2023, 2022 and 2021.
12. Other Receivables, Prepayments and Deposits
December 31, | ||||
| 2023 |
| 2022 | |
| (in US$’000) | |||
Prepayments to suppliers | 1,179 |
| 2,624 | |
Interest receivables | 132 |
| 25 | |
Deposits | 676 |
| 778 | |
Others | 282 |
| 245 | |
2,269 |
| 3,672 | ||
13. Inventories
December 31, | ||||
| 2023 |
| 2022 | |
| (in US$’000) | |||
Raw materials |
| 40,808 |
| 22,804 |
Work in progress |
| 91,351 |
| 108,168 |
Finished goods |
| 31,867 |
| 23,844 |
| 164,026 |
| 154,816 | |
F-69
14. Property, Plant and Equipment
Furniture | ||||||||||||
and | ||||||||||||
fixtures, | ||||||||||||
other | ||||||||||||
Plant | equipment | |||||||||||
Leasehold | and | and motor | Construction | |||||||||
| Buildings | improvements | equipment | vehicles | in progress | Total | ||||||
(in US$’000) | ||||||||||||
Cost | ||||||||||||
As at January 1, 2023 |
| 69,582 |
| 815 |
| 25,098 |
| 14,582 |
| 521 |
| 110,598 |
Additions |
| 76 |
| — |
| 1,339 |
| 1,163 |
| 1,605 |
| 4,183 |
Disposals |
| (23) |
| — |
| (106) |
| (222) |
| — |
| (351) |
Transfers |
| 15 |
| 97 |
| 102 |
| 1,475 |
| (1,689) |
| — |
Exchange differences |
| (1,865) |
| (24) |
| (696) |
| (434) |
| (12) |
| (3,031) |
As at December 31, 2023 |
| 67,785 |
| 888 |
| 25,737 |
| 16,564 |
| 425 |
| 111,399 |
Accumulated depreciation |
|
|
|
|
|
| ||||||
As at January 1, 2023 |
| 21,376 |
| 314 |
| 16,116 |
| 9,961 |
| — |
| 47,767 |
Depreciation |
| 3,494 |
| 307 |
| 2,079 |
| 1,537 |
| — |
| 7,417 |
Disposals |
| (7) |
| — |
| (87) |
| (213) |
| — |
| (307) |
Exchange differences |
| (636) |
| (14) |
| (467) |
| (291) |
| — |
| (1,408) |
As at December 31, 2023 |
| 24,227 |
| 607 |
| 17,641 |
| 10,994 |
| — |
| 53,469 |
Net book value |
|
|
|
|
|
| ||||||
As at December 31, 2023 |
| 43,558 |
| 281 |
| 8,096 |
| 5,570 |
| 425 |
| 57,930 |
Furniture | ||||||||||||
and | ||||||||||||
fixtures, | ||||||||||||
other | ||||||||||||
Plant | equipment | |||||||||||
Leasehold | and | and motor | Construction | |||||||||
| Buildings |
| improvements |
| equipment |
| vehicles |
| in progress |
| Total | |
(in US$’000) | ||||||||||||
Cost |
|
|
|
|
|
|
|
|
|
|
|
|
As at January 1, 2022 |
| 75,587 |
| 848 |
| 26,438 |
| 15,033 |
| 136 |
| 118,042 |
Additions |
| 27 |
| 38 |
| 117 |
| 516 |
| 2,924 |
| 3,622 |
Disposals |
| (886) |
| — |
| (227) |
| (178) |
| — |
| (1,291) |
Transfers |
| 1,058 |
| — |
| 974 |
| 478 |
| (2,510) |
| — |
Exchange differences |
| (6,204) |
| (71) |
| (2,204) |
| (1,267) |
| (29) |
| (9,775) |
As at December 31, 2022 |
| 69,582 |
| 815 |
| 25,098 |
| 14,582 |
| 521 |
| 110,598 |
Accumulated depreciation |
|
|
|
|
|
| ||||||
As at January 1, 2022 |
| 19,983 |
| 94 |
| 14,817 |
| 9,498 |
| — |
| 44,392 |
Depreciation |
| 3,606 |
| 238 |
| 2,830 |
| 1,474 |
| — |
| 8,148 |
Disposals |
| (439) |
| — |
| (205) |
| (178) |
| — |
| (822) |
Exchange differences |
| (1,774) |
| (18) |
| (1,326) |
| (833) |
| — |
| (3,951) |
As at December 31, 2022 |
| 21,376 |
| 314 |
| 16,116 |
| 9,961 |
| — |
| 47,767 |
Net book value |
|
|
|
|
|
| ||||||
As at December 31, 2022 |
| 48,206 |
| 501 |
| 8,982 |
| 4,621 |
| 521 |
| 62,831 |
F-70
15. Leases
Leases consisted of the following:
| December 31, | |||
2023 |
| 2022 | ||
(in US$’000) | ||||
Right-of-use assets: | ||||
Offices |
| 1,092 | 1,717 | |
Lease liabilities—current |
| 713 | 712 | |
Lease liabilities—non-current |
| 657 | 1,360 | |
| 1,370 | 2,072 | ||
Lease activities are summarized as follows:
| Year Ended December 31, | |||
2023 |
| 2022 | ||
| (in US$’000) | |||
Lease expenses: Short-term leases with lease terms equal or less than 12 months |
| 207 | 236 | |
Depreciation charge of right-of-use assets |
| 665 | 681 | |
Interest expense (included in finance costs) |
| 79 | 112 | |
Cash paid on lease liabilities |
| 810 | 809 | |
Cash paid on short-term leases | 175 | 186 | ||
Non-cash: Lease liabilities recognized from obtaining right-of-use assets |
| 78 | 135 | |
Lease contracts are typically within a period of 1 to 5 years. The weighted average remaining lease term and weighted average discount rate as at December 31, 2023 was 1.8 years (2022: 2.7 years) and 4.69% (2022: 4.70%) respectively.
Future lease payments are as follows:
| December 31, | |||
2023 |
| 2022 | ||
(in US$’000) | ||||
Lease payments: |
|
| ||
Not later than 1 year |
| 758 | 791 | |
Between 1 to 2 years |
| 651 | 755 | |
Between 2 to 3 years |
| 17 | 660 | |
Total lease payments |
| 1,426 | 2,206 | |
Less: Discount factor |
| (56) | (134) | |
Total lease liabilities |
| 1,370 | 2,072 | |
F-71
16. Deferred Tax Assets
The significant components of deferred tax assets and liabilities are as follows:
| December 31, | |||
2023 | 2022 | |||
(in US$’000) | ||||
Deferred tax assets |
|
|
|
|
Accrued expenses |
| 8,124 |
| 8,919 |
Others |
| 2,774 |
| 1,582 |
Deferred tax assets |
| 10,898 |
| 10,501 |
Deferred tax liabilities |
|
|
|
|
Accelerated depreciation allowances and others |
| 2,568 |
| 2,174 |
Deferred tax liabilities |
| 2,568 |
| 2,174 |
Net deferred tax assets |
| 8,330 |
| 8,327 |
The movements in deferred tax assets and liabilities are as follows:
| 2023 |
| 2022 |
| 2021 | |
| (in US$’000) | |||||
As at January 1 | 8,327 |
| 7,715 |
| 8,315 | |
Credited/(debited) to the consolidated income statements |
|
|
|
| ||
—Accrued expenses, provisions, deferred income, accelerated depreciation and other temporary differences | 175 |
| 1,344 |
| (814) | |
Exchange differences | (172) |
| (732) |
| 214 | |
As at December 31 | 8,330 |
| 8,327 |
| 7,715 | |
The Group’s deferred tax assets are mainly temporary differences including accrued expenses, provisions, deferred income, accelerated depreciation and other temporary differences. There is no deferred tax assets in respect of tax losses which have not been recognized in the consolidated financial statements as at December 31, 2023 (2022: US$24,000).
These unrecognized tax losses can be carried forward against future taxable income and will expire in the following years:
December 31, | ||||
| 2023 |
| 2022 | |
| (in US$’000) | |||
2024 | — |
| 76 | |
2025 | — |
| 7 | |
2026 | — |
| 6 | |
2027 | — |
| 5 | |
— |
| 94 | ||
17. Trade Payables
December 31, | ||||
| 2023 |
| 2022 | |
| (in US$’000) | |||
Trade payables—third parties | 18,268 |
| 19,737 | |
Trade payables—related parties (Note 22(b)) | 5,568 |
| 3,358 | |
23,836 |
| 23,095 | ||
All trade payables are denominated in RMB and due within one year from the end of the reporting period. The carrying value of trade payables approximates their fair values due to their short-term maturities.
F-72
18. Other Payables, Accruals and Advance Receipts
December 31, | ||||
| 2023 |
| 2022 | |
| (in US$’000) | |||
Accrued salaries and benefits | 21,899 |
| 21,100 | |
Accrued sales rebates and marketing expenses | 61,996 |
| 73,721 | |
Value‑added tax and tax surcharge payables | 3,139 |
| 5,204 | |
Payments in advance from customers (note a) | 7,093 |
| 14,004 | |
Dividend payable (note b) | 54,260 | — | ||
Others | 5,550 |
| 7,325 | |
153,937 |
| 121,354 | ||
Note a: Substantially all customer balances as at December 31, 2022 were recognized to revenue during the year ended December 31, 2023. Additionally, substantially all customer balances as at December 31, 2023 are expected to be recognized to revenue within one year upon transfer of goods or services as the contracts have an expected duration of one year or less.
Note b: On January 31, 2023, the Company declared a dividend of RMB988.3 million (US$147.0 million), of which RMB600.0 million (US$84.6 million) has been distributed during the year ended December 31, 2023 and RMB388.3 million (US$54.3 million) is recorded as dividend payable under other payables, accruals and advance receipts as at December 31, 2023.
19. Current Tax Liabilities
| 2023 |
| 2022 |
| 2021 | |
| (in US$’000) | |||||
As at January 1 | 2,791 |
| 4,089 |
| 5,032 | |
Current tax (Note 8) | 17,197 |
| 18,082 |
| 15,082 | |
Tax paid | (18,709) |
| (19,003) |
| (15,976) | |
Exchange difference | (116) |
| (377) |
| 108 | |
Transfer from other receivables | — | — | (157) | |||
As at December 31 | 1,163 |
| 2,791 |
| 4,089 | |
F-73
20. Notes to the Consolidated Statements of Cash Flows
(a) Reconciliation of profit for the year to net cash generated from operations:
| 2023 |
| 2022 |
| 2021 | |
| (in US$’000) | |||||
Profit for the year | 95,463 |
| 99,683 |
| 89,388 | |
Adjustments to reconcile profit for the year to net cash generated from operations |
|
| ||||
Taxation charge | 17,022 |
| 16,738 |
| 15,896 | |
Finance costs | 79 |
| 112 |
| 116 | |
Interest income | (754) |
| (980) |
| (1,216) | |
Depreciation on property, plant and equipment | 7,417 |
| 8,148 |
| 8,100 | |
Loss on disposal of property, plant and equipment | 32 |
| 449 |
| 60 | |
Amortization of leasehold land | 158 |
| 166 |
| 172 | |
Amortization of other intangible assets | 288 |
| 245 |
| 233 | |
Depreciation charge of right-of-use assets | 665 |
| 681 |
| 663 | |
Provision for excess and obsolete inventories | 2,121 |
| (65) |
| (141) | |
Exchange differences | (3,019) |
| (5,682) |
| (693) | |
Changes in operating assets and liabilities: |
|
| ||||
Trade and bills receivables | 6,255 |
| (4,374) |
| 939 | |
Other receivables, prepayments and deposits | 1,510 |
| (580) |
| (80) | |
Inventories | (11,331) |
| (35,361) |
| (37,575) | |
Trade payables | 741 |
| 10,684 |
| 1,237 | |
Other payables, accruals and advance receipts | (20,012) |
| 7,804 |
| 18,608 | |
Deferred income | (555) |
| (1,398) |
| (1,737) | |
Total changes in operating assets and liabilities | (23,392) |
| (23,225) |
| (18,608) | |
Net cash generated from operations | 96,080 |
| 96,270 |
| 93,970 | |
(b) Supplemental disclosure for non-cash activities
During the years ended December 31, 2023, in addition to non-cash activities of lease liabilities as disclosed in Note 15, there was a decrease in accruals made for purchases of property, plant and equipment of US$1.7 million (2022 and 2021: an increase of US$1.8 million and a decrease of US$0.3 million respectively).
21. Capital Commitments
The Group had the following capital commitments:
| December 31, | |
2023 | ||
| (in US$’000) | |
Property, plant and equipment | ||
Contracted but not provided for |
| 376 |
Capital commitments for property, plant and equipment are mainly for improvements to the Group’s plant.
F-74
22. Significant Related Party Transactions
The Group has the following significant transactions with related parties which were carried out in the normal course of business at terms determined and agreed by the relevant parties:
(a) Transactions with related parties:
Year Ended December 31, | ||||||
| 2023 |
| 2022 |
| 2021 | |
| (in US$’000) | |||||
Sales of goods to: | ||||||
—A fellow subsidiary of SHTCML | 9,329 |
| 13,861 |
| 12,181 | |
—A fellow subsidiary of SHHCMI(HK)L | 3,651 |
| 4,231 |
| 3,492 | |
12,980 |
| 18,092 |
| 15,673 | ||
Purchase of goods from: |
|
| ||||
—SHTCML | 12,173 |
| 11,072 |
| 10,002 | |
—Fellow subsidiaries of SHTCML | 1,130 |
| 683 |
| 1,311 | |
—A fellow subsidiary of SHHCMI(HK)L | 6,350 | 1,683 | — | |||
19,653 | 13,438 | 11,313 | ||||
Rendering of research and development services from: |
|
| ||||
—A fellow subsidiary of SHHCMI(HK)L | 481 |
| 507 |
| 525 | |
Provision of marketing services to: |
|
| ||||
—A fellow subsidiary of SHTCML | 1,241 |
| 952 |
| 1,146 | |
—A fellow subsidiary of SHHCMI(HK)L | — |
| 127 |
| — | |
1,241 |
| 1,079 |
| 1,146 | ||
Purchase of intangible asset from: | ||||||
—A fellow subsidiary of SHHCMI(HK)L | — | 410 | — | |||
Leasing office from: | ||||||
—SHTCML | — |
| — |
| 247 | |
No transactions have been entered into with the directors of the Company (being the key management personnel) during the year ended December 31, 2023 (2022 and 2021: nil).
F-75
(b) Balances with related parties included in:
December 31, | ||||
| 2023 |
| 2022 | |
| (in US$’000) | |||
Trade and bills receivables | ||||
—A fellow subsidiary of SHTCML | 1,303 |
| 3,622 | |
—A fellow subsidiary of SHHCMI(HK)L | — |
| 73 | |
1,303 |
| 3,695 | ||
Other receivables, prepayments and deposits |
|
|
| |
—A fellow subsidiary of SHTCML | 391 |
| 402 | |
—A fellow subsidiary of SHHCMI(HK)L | 72 | — | ||
463 | 402 | |||
Trade payables |
| |||
—SHTCML | 3,630 | 1,266 | ||
— Fellow subsidiaries of SHTCML | 294 |
| 152 | |
—A fellow subsidiary of SHHCMI(HK)L | 1,644 | 1,940 | ||
5,568 | 3,358 | |||
Other payables, accruals and advance receipts | ||||
—SHTCML (Note 18) | 27,130 | — | ||
—SHHCMI(HK)L (Note 18) | 27,130 | — | ||
—Fellow subsidiaries of SHHCMI(HK)L | 1,356 | 1,256 | ||
55,616 | 1,256 | |||
Balances with related parties are unsecured, interest-free and repayable on demand. The carrying values of balances with related parties approximate their fair values due to their short-term maturities.
23. Particulars of Principal Subsidiaries
Equity | ||||||||||||||
Nominal value | interest | |||||||||||||
Place of | of registered | attributable |
| |||||||||||
establishment | capital | to the Group | ||||||||||||
| and | December 31, | ||||||||||||
Name |
| operation |
| 2023 |
| 2022 |
| 2023 |
| 2022 |
| Type of legal entity |
| Principal activity |
| (in RMB’000) | |||||||||||||
Shanghai Shangyao Hutchison Whampoa GSP Company Limited |
| PRC |
| 20,000 |
| 20,000 |
| 100% | 100% | Limited liability company |
| Distribution of drug products | ||
Hutchison Heze Bio Resources & Technology Co., Limited (note) |
| PRC |
| — |
| 1,500 |
| — | 100% | Limited liability company |
| Agriculture and sales of Chinese herbs | ||
Note: Hutchison Heze Bio Resources & Technology Co., Limited was liquidated in June 2023.
24. Subsequent Events
The Group evaluated subsequent events through February 28, 2024, which is the date when the consolidated financial statements were issued.
F-76