UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
(Mark One)
REGISTRATION STATEMENT PURSUANT TO SECTION 12(b) OR (g) OF THE SECURITIES EXCHANGE ACT OF 1934 |
OR
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended
OR
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _________ to
OR
SHELL COMPANY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Date of event requiring this shell company report ____________________
Commission file number:
(Exact name of Registrant as specified in its charter) |
Ontario,
(Jurisdiction of incorporation or organization)
(Address of principal executive offices)
FSD Pharma Inc.
Telephone: (
Email:
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act.
Title of each class |
| Trading Symbol(s) |
| Name of each exchange on which registered |
|
|
| The |
Securities registered or to be registered pursuant to Section 12(g) of the Act. None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act. None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report.
Class A Multiple Voting Shares, no par value:
Class B Subordinate Voting Shares, no par value: 39,376,723 shares outstanding as of December 31, 2023
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.
Yes ☐
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer | ☐ | Accelerated filer | ☐ |
☒ | Emerging growth company |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C 7262(b)) by the registered public accounting firm that prepared or issued its audit report.
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
U.S. GAAP ☐ | issued by the International Accounting Standards Board ☒ | Other ☐ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
Item 17 ☐ Item 18 ☐
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).
Yes ☐ No
TABLE OF CONTENTS
6 | ||
|
| |
6 | ||
|
| |
9 | ||
|
| |
10 | ||
|
| |
| 11 | |
|
|
|
11 | ||
|
|
|
11 | ||
|
|
|
11 | ||
|
|
|
11 | ||
11 | ||
11 | ||
11 | ||
|
|
|
29 | ||
|
|
|
29 | ||
34 | ||
46 | ||
48 | ||
|
|
|
48 | ||
|
|
|
48 | ||
|
|
|
48 | ||
48 | ||
48 | ||
48 | ||
48 | ||
|
|
|
48 | ||
|
|
|
48 | ||
51 | ||
59 | ||
65 | ||
65 | ||
| 3 |
| Table of Contents |
65 | ||
|
|
|
65 | ||
67 | ||
68 | ||
|
|
|
68 | ||
|
|
|
68 | ||
71 | ||
|
|
|
71 | ||
|
|
|
| ||
71 | ||
71 | ||
71 | ||
71 | ||
71 | ||
|
|
|
72 | ||
|
|
|
72 | ||
72 | ||
72 | ||
72 | ||
73 | ||
79 | ||
79 | ||
79 | ||
79 | ||
79 | ||
|
|
|
| ||
|
| 79 |
79 | ||
|
|
|
79 | ||
79 | ||
79 | ||
79 | ||
|
|
|
|
| |
|
|
|
80 |
| 4 |
| Table of Contents |
| 5 |
| Table of Contents |
INTRODUCTION
Unless otherwise noted or the context otherwise requires, all references in this Annual Report on Form 20-F (this “Annual Report”), or this Annual Report, to “FSD,” “FSD Pharma,” “Company,” “Corporation,” “we,” “us” and “our” refer to FSD Pharma Inc., a corporation formed under the Business Corporations Act (Ontario) (the “OBCA”) and the direct or indirect subsidiary entities of FSD and any partnership interests held by FSD Pharma and its subsidiary entities, including Lucid Psycheceuticals Inc. (“Lucid”), FSD BioSciences Inc. (“FSD Biosciences”), FV Pharma Inc. (“FV Pharma”), Prismic Pharmaceuticals, Inc. (“Prismic”), FSD Strategic Investments Inc. (“FSD Strategic Investments”), FSD Pharma Australia Pty Ltd. (“FSD Australia”) and Celly Nutrition Corp. (“Celly Nu”).
Our fiscal year ends on December 31. This Annual Report includes our audited consolidated financial statements as of December 31, 2023 and 2022 and for the years ended December 31, 2023 and 2022, (the “2023 Annual Financial Statements”) which are prepared in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board. None of our financial statements were prepared in accordance with generally accepted accounting principles in the United States of America (“U.S. GAAP”).
Except where expressly indicated otherwise, our financial information is presented in U.S. dollars. All references in this Annual Report to “$” or “US$” mean United States of American (“U.S.” or “United States”) dollars, and all references in this Annual Report to “C$” mean Canadian dollars. For the convenience of the reader, in this Annual Report, unless otherwise indicated, translations from Canadian dollars into U.S. dollars were made at the rate of US$1.00 to C$1.3497, which is the average rate for the 2023 fiscal year, (2022 average rate: US$1.00=C$1.3013). Such U.S. dollar amounts are not necessarily indicative of the amounts of U.S. dollars that could actually have been purchased upon exchange of Canadian dollars at the dates indicated.
We have made rounding adjustments to some of the figures included in this Annual Report. Accordingly, numerical figures shown as totals in some tables may not be an arithmetic aggregation of the figures that preceded them.
This Annual Report includes registered and unregistered trademarks such as “Unbuzzd,” and “ALCOHOLDEATH,” which are protected under applicable intellectual property laws and are the property of the Company. Solely for convenience, our trademarks referred to in this Annual Report and in other publicly filed documents may appear without the ® or ™ symbol, but such references are not intended to indicate, in any way, that we will not assert our rights to the fullest extent under applicable law. All other trademarks used in this Annual Report are the property of their respective owners. For more information, please see “Item 4. Information on the Company. – B. Business Overview – Intellectual Property”.
We are incorporated under the laws of Ontario, Canada. Substantially all of our assets are located outside the United States. In addition, several of our directors and officers are nationals and/or residents of countries other than the United States, and all or a substantial portion of such directors’ and officers’ assets may be located outside the United States. As a result, it may be difficult for investors to effect service of process within the United States upon us or our officers or directors or to enforce against us or them judgments obtained in United States courts, including judgments predicated upon the civil liability provisions of the securities laws of the United States or any state thereof. In addition, investors should not assume that the courts of Canada (i) would enforce judgments of United States courts obtained in actions against us, our officers or directors, or other said persons, predicated upon the civil liability provisions of the U.S. federal securities laws or other laws of the United States or (ii) would enforce, in original actions, liabilities against us or such directors, officers or experts predicated upon the United States federal securities laws or any securities or other laws of any state or jurisdiction of the United States.
In addition, there is doubt as to the applicability of the civil liability provisions of the United States federal securities law to original actions instituted in Canada. It may be difficult for an investor, or any other person or entity, to assert United States securities laws claims in original actions instituted in Canada.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report contains statements that constitute forward-looking statements. All statements other than statements of historical facts contained in this Annual Report, including statements regarding our future results of operations and financial position, business strategy, product candidates, product pipeline, ongoing and planned clinical studies, including those of our collaboration partners, regulatory approvals, research and development (“R&D”) costs, timing and likelihood of success, as well as plans and objectives of management for future operations, are forward-looking statements. Many of the forward-looking statements contained in this Annual Report are often, but not always, identified by words or phrases such as “hope”, “would”, “seek”, “anticipate”, “believe”, “expect”, “plan”, “continue”, “estimate”, “will”, “predict”, “intend”, “forecast”, “future”, “target”, “project”, “capacity”, “could”, “should”, “might”, “focus”, “proposed”, “scheduled”, “outlook”, “potential”, “may” or similar expressions and includes suggestions of future outcomes, including, but not limited to statements about:
| · | discussions concerning the Company’s exploration of near-term funding strategies; |
| · | the Company’s plans to advance the R&D of its Product Candidates (as defined herein) to commercialization through studies and clinical trials, including anticipated timing and associated costs; |
| · | the application and the costs associated with such planned trials, and the Company’s ability to obtain required funding and the terms and timing thereof; |
| · | the expansion of our product offering(s); |
| · | our business objectives and the expected impacts of previously announced acquisitions and developments; |
| · | the U.S. Food and Drug Administration (“FDA”) and Health Canada, or comparable regulatory authority, application process and any review thereof and its effects on our business objectives. |
| 6 |
| Table of Contents |
Readers are cautioned not to place undue reliance on Forward-Looking Statements as the Company’s actual results may differ materially and adversely from those expressed or implied.
The Corporation has made certain assumptions with respect to the Forward-Looking Statements regarding, among other things:
| · | the Corporation’s ability to generate sufficient cash flow from operations and obtain financing, if needed, on acceptable terms or at all; |
| · | the general economic, financial market, regulatory and political conditions in which the Corporation operates; |
| · | the interest of potential purchasers in the Product Candidates; |
| · | anticipated and unanticipated costs; the government regulation of the Corporation’s activities and Product Candidates; |
| · | the timely receipt of any required regulatory approvals; |
| · | the Corporation’s ability to obtain qualified staff, equipment and services in a timely and cost efficient manner; |
| · | the Corporation’s ability to conduct operations in a safe, efficient and effective manner; and |
| · | the Corporation’s expansion plans and timeframe for completion of such plans. |
Although the Corporation believes that the expectations and assumptions on which the Forward-Looking Statements are based are reasonable, undue reliance should not be placed on the Forward-Looking Statements, because no assurance can be given that such statements will prove to be correct.
Forward-looking statements appear in a number of places in this Annual Report and include, but are not limited to, statements regarding our intent, belief, or current expectations. Forward-looking statements are based on our management’s beliefs and assumptions, and on information currently available to our management. Such statements are subject to risks and uncertainties, and actual results may differ materially from those expressed or implied in the forward-looking statements due to various factors, including, but not limited to, those identified under “Item 3. Key information - D. Risk Factors” in this Annual Report. These risks and uncertainties include multiple factors:
| · | the success of our clinical studies, our ability to obtain and maintain regulatory approval and to commercialize our Product Candidates, which include Lucid-21-302 (“Lucid-MS”), for the treatment of multiple sclerosis (“MS”) and our product for alcohol misuse in the healthcare area; |
| · | the ability of our licensing partner, Celly Nu to commercialize, sell and distribute Unbuzzd™, a functional beverage product that seeks to provide relief from inebriation and accelerate alcohol metabolism in the consumer market; |
| · | the ability of our competitors to discover, develop or commercialize competing products to Unbuzzd™, Lucid-MS or other product candidates before or more successfully than we do; |
| · | our plans to research, develop and commercialize our Product Candidates; |
| · | the identification of serious adverse, undesirable, or unacceptable side effects related to our Product Candidates; |
| · | our ability to maintain our current strategic relationships with Celly Nu, our licensing partner to commercialize Unbuzzd™; |
| · | our ability to protect and maintain our, and not infringe on third parties’, intellectual property rights throughout the world; |
| · | our ability to raise capital when needed in order to continue our product development programs and commercialization efforts; |
| · | our ability to attract and retain qualified employees and key personnel; |
| · | the acceptance by the FDA and applicable foreign regulatory authorities of data from studies for Lucid-MS that we and our collaboration partners conduct within and outside the U.S. now and in the future; |
| · | our foreign private issuer status, the loss of which would require us to comply with the Exchange Act of 1934’s, as amended (the “Exchange Act”) domestic reporting regime, and cause us to incur significant legal, accounting, and other expenses; |
| · | our incorporation in Ontario, the laws of which govern our corporate affairs and may differ from those applicable to companies incorporated in the U.S.; |
| · | the limited operating history of the Company and history of losses, and anticipated significant losses for the foreseeable future incurred to pursue commercialization of the Product Candidates; |
| · | the Company’s inability to file investigational new drug applications (“INDs”) or clinical trial application (“CTAs”) on timelines it reasonably anticipates, if at all; |
| · | the Company’s ability to identify, license or discover additional product candidates; |
| · | the Product Candidates being in the preclinical development stage; |
| · | the Company’s reliance on its Product Candidates; |
| · | the Company’s ability to successfully develop new commercialized products or find a market for their sale; |
| · | the impact of any future recall of the Company’s products; |
| · | the Company’s ability to promote and sustain its products, including any restrictions or constraints on marketing practices under the regulatory framework in which the Company operates; |
| · | failure to achieve the degree of market acceptance and demand for our products or Product Candidates by physicians, patients, healthcare payors, and others in the medical community which are necessary for commercial success, including due to the possibility that alternative, superior treatments may be available prior to the approval and commercialization of Product Candidates, should such approval be received at all; |
| 7 |
| Table of Contents |
| · | failure of clinical trials to demonstrate substantial evidence of the safety and/or effectiveness of Product Candidates, which could prevent, delay or limit the scope of regulatory approval and commercialization, including from difficulties encountered in enrolling patients in clinical trials, and reliance on third parties to conduct our clinical trials and some aspects of our research and preclinical testing, or results from future clinical testing which may demonstrate opposing evidence and draw negative conclusions regarding the effectiveness of any Product Candidate, including the effectiveness of Lucid-MS as a treatment for MS; |
| · | results of earlier studies or clinical trials not being predictive of future clinical trials and initial studies or clinical trials not establishing an adequate safety or efficacy profile for the Product Candidates to justify proceeding to advanced clinical trials or an application for regulatory approval; |
| · | potential side effects, adverse events or other properties or safety risks of the Product Candidates, which could delay or halt their clinical development, prevent their regulatory approval, cause suspension or discontinuance of clinical trials, abandonment of a Product Candidate, limit their commercial potential, if approved, or result in other negative consequences; |
| · | preliminary, interim data obtained from the Company’s clinical trials that it may announce or publish from time to time may not be indicative of future scientific observations or conclusions as more patient data becomes available, further analyses are conducted, and as the data becomes subject to subsequent audit and verification procedures; |
| · | inability to establish sales and marketing capabilities, or enter into agreements with third parties, to sell and market any Product Candidates that the Company may develop; |
| · | the ability to provide the capital required for research, product development, operations and marketing; |
| · | violations of laws and regulations resulting in repercussions; |
| · | risks inherent in an pharmaceutical business and the development and commercialization of pharmaceutical products, including the inability to accurately predict timing or amounts of expenses, requirements of regulatory authorities, and completion of clinical studies on anticipated timelines, which may encounter substantial delays or may not be able to be completed at all; |
| · | delays in clinical trials; |
| · | the Company’s inability to attain or maintain the regulatory approvals it needs in any jurisdiction to commercialize, distribute or sell any Product Candidate or other pharmaceutical products; |
| · | failure of counterparties to perform contractual obligations; |
| · | changes, whether anticipated or not, in laws, regulations and guidelines that may result in significant compliance costs for the Company, including in relation to restrictions on branding and advertising, regulation of distribution and excise taxes; |
| · | uncertainty associated with insurance coverage and reimbursement status for newly-approved pharmaceutical products, which could result in Product Candidates becoming subject to unfavorable pricing regulations, third-party coverage and reimbursement practices, or healthcare reform initiatives, including legislative measures aimed at reducing healthcare costs; |
| · | the effect that any public health crises, such as pandemics or epidemics may have on the Company’s business; |
| · | the price of our securities may be volatile due to a variety of factors, including volatility in the capital markets generally, geopolitical events, public health emergencies, macro economic pressures and natural disasters; |
| · | the inability to obtain required additional financing on terms favourable to the Corporation or at all; |
| · | the Company’s anticipated negative cash flow from operations and non-profitability for the foreseeable future; |
| · | the issuances of equity securities and the conversion of outstanding securities to Class B subordinate voting shares in the capital of the Company (the “Class B Shares”); |
| · | the Company’s dual class share structure; |
| · | the market price of the Class B Shares possibly being subject to wide price fluctuations; |
| · | whether an active trading market for the Class B Shares is sustained; |
| · | the Company’s ability to maintain compliance with Nasdaq Stock Market LLC’s (“Nasdaq”) rules for continued listing on the Nasdaq; |
| · | the Company’s ability to identify and execute future acquisitions or dispositions effectively, including the ability to successfully manage the impacts of such transactions on its operations; |
| · | lack of dividends, and reinvestment of retained earnings, if any, into the Company’s business; |
| · | the Company’s reliance on management, key persons and skilled personnel; |
| · | reliance on contract manufacturing facilities; |
| · | manufacturing problems that could result in delay of the Company’s development or commercialization programs; |
| · | the Company’s expected minimal environmental impacts; insurance and uninsured risks; |
| · | claims from suppliers; conflicts of interest between the Company and its directors and officers; |
| · | the Company’s ability to manage its growth effectively; |
| · | the Company’s ability to realize production targets; |
| · | supply chain interruptions and the ability to maintain required supplies of, equipment, parts and components; |
| · | the Company’s ability to successfully implement and maintain adequate internal controls over financial reporting or disclosure controls and procedures; |
| · | results of litigation; |
| · | the dependence of the Company’s operations, in part, on the maintenance and protection of its information technology systems, and the information technology systems of its third-party research institution collaborators, contract research organizations (“CROs”) or other contractors or consultants, which could face cyber-attacks; |
| · | failure to execute definitive agreements with entities in which the Company has entered into letters of intent or memoranda of understanding; |
| · | unfavorable publicity or consumer perception towards the Product Candidates; |
| 8 |
| Table of Contents |
| · | reputational risks to third parties with whom the Company does business; failure to comply with laws and regulations; |
| · | the Company’s reliance on its own market research and forecasts; |
| · | competition from other technologies and pharmaceutical products, including from synthetic production, new manufacturing processes and new technologies, and expected significant competition from other companies with similar businesses, and significant competition in an environment of rapid technological and scientific change; |
| · | the Company’s ability to safely, securely, efficiently and cost-effectively transport our products to consumers; |
| · | liability arising from any fraudulent or illegal activity, or other misconduct or improper activities that the Company’s directors, officers, employees, contractors, consultants, commercial partners or vendors may engage in, including noncompliance with regulatory standards and requirements; |
| · | unforeseen claims made against the Company, including product liability claims or regulatory actions; |
| · | reliance on single-source suppliers, including single-source suppliers for the acquisition of the drug substance and drug product for any of the Product Candidates; |
| · | inability to obtain or maintain sufficient intellectual property protection for the Product Candidates; |
| · | third-party claims of intellectual property infringement; |
| · | patent terms being insufficient to protect competitive position on Product Candidates; |
| · | inability to obtain patent term extensions or non-patent exclusivity; |
| · | inability to protect the confidentiality of trade secrets; |
| · | inability to protect trademarks and trade names; |
| · | filing of claims challenging the inventorship of the Company’s patents and other intellectual property; |
| · | invalidity or unenforceability of patents, including legal challenges to patents covering any of the Product Candidates; |
| · | claims regarding wrongfully used or disclosed confidential information of third parties; |
| · | risks related to the Company’s investment in Celly Nu, including the ability of Celly Nu to commercialize the exclusive rights to the recreational applications for the Company’s alcohol misuse technology for rapid alcohol detoxification; |
| · | inability to protect property rights around the world; the impact of general economic conditions on the Company’s mortgage investment activities; |
| · | risks related to the Company’s status as a foreign private issuer; |
| · | the Company taking advantage of reduced disclosure requirements applicable to emerging growth companies; |
| · | the Company’s classification as a “passive foreign investment company”; |
| · | that the Company’s international business operations, including expansion to new jurisdictions, could expose it to regulatory risks or factors beyond our control such as currency exchange rates and changes in governmental policy; |
| · | risks related to expansion of international operations; |
| · | the Company’s ability to produce and sell products in, and export products to, other jurisdictions within and outside of Canada and the United States, which is dependent on compliance with additional regulatory or other requirements; |
| · | regulatory regimes of locations for clinical trials outside of Canada and the United States; |
| · | failure to obtain approval to commercialize Product Candidates outside of Canada and the United States; |
| · | if clinical trials are conducted for Product Candidates outside of Canada and the United States, FDA, Health Canada and comparable regulatory authorities may not accept data from such trials, or the scope of such approvals from regulatory authorities may be limited; |
| · | other factors beyond the Company’s control; |
| · | the other risk factors discussed under “Item 3. Key information - D. Risk Factors”. |
These forward-looking statements are applicable only as of the date of this Annual Report, and are subject to a number of risks, uncertainties and assumptions described under the sections in this Annual Report entitled “Item 3. Key information - D. Risk Factors” and “Item 5. Operating and Financial Review and Prospects” and elsewhere in this Annual Report. Because forward-looking statements are inherently subject to risks and uncertainties, some of which cannot be predicted or quantified and some of which are beyond our control, you should not rely on these forward-looking statements as predictions of future events. The events and circumstances reflected in our forward-looking statements may not be achieved or occur and actual results could differ materially from those projected in the forward-looking statements. Moreover, we operate in an evolving environment. New risk factors and uncertainties may emerge from time to time, and it is not possible for management to predict all risk factors and uncertainties. Except as required by applicable law, we do not plan to publicly update or revise any forward-looking statements contained herein, whether as a result of any new information, future events, changed circumstances or otherwise.
MARKET AND INDUSTRY DATA
This Annual Report includes market and industry data that has been obtained from third party sources, including industry publications. The Company believes that its industry data is accurate and that its estimates and assumptions are reasonable, but there is no assurance as to the accuracy or completeness of this data. Third party sources generally state that the information contained therein has been obtained from sources believed to be reliable, but there is no assurance as to the accuracy or completeness of included information. Although the data is believed to be reliable, the Company has not independently verified any of the data from third party sources referred to in this Annual Report or ascertained the underlying economic assumptions relied upon by such sources.
| 9 |
| Table of Contents |
SUMMARY RISK FACTORS
Our business is subject to a number of risks and uncertainties, including those risks discussed at length in the section below titled “Risk Item 3. Key information - D. Risk Factors”. These risks include, among others, the following:
RISKS RELATED TO OUR PRODUCT CANDIDATES
| · | We have a limited operating history and funding, which may make it difficult to evaluate Lucid-MS, our products for alcohol misuse and their product development, product prospects and overall likelihood of success; |
| · | Our drug product candidate, Lucid-MS or our products relating to alcohol misuse, may not receive regulatory approval from Health Canada or the FDA, in a timely manner, if at all, or may receive regulatory approval on limiting terms; |
| · | We are relying on Celly Nu, our licensing partner, to develop and promote Unbuzzd™, an alcohol misuse product for the retail market; |
| · | The Company may be unable to raise the capital necessary for it to execute its strategy on favorable terms or at all; and |
| · | Drug development is a highly uncertain undertaking and involves a substantial degree of risk. |
RISKS RELATED TO THE PHARMACEUTICAL BUSINESS
| · | We rely on the UHN License (as defined herein) to use for pharmaceutical purposes certain patents and other intellectual property rights associated with Lucid-MS; |
| · | We rely on the Epitech License Agreement and the UHN License to use for pharmaceutical purposes certain patents and other intellectual property rights associated with FSD-PEA and Lucid-MS; |
| · | Even if Lucid-MS receives regulatory approval, we may nonetheless fail to achieve the degree of market acceptance of Lucid-MS by physicians, patients, healthcare payors, and others in the medical community necessary for commercial success; |
| · | We face significant competition for our Lucid-MS drug, and there is a possibility that our competitors may achieve regulatory approval for an effective treatment for MS before us or develop therapies that are safer, more advanced, or more effective than ours; |
| · | Psychedelic or psychedelic-inspired drugs may never be approved as medicines or other therapeutic applications, and violations of applicable laws and regulations, or changes in the regulatory or political discourse with respect to psychedelic or psychedelic-inspired drugs, could result in repercussions; |
| · | The loss of single-source suppliers, or their failure to supply us with the drug substance or drug product, could materially and adversely affect our business; |
| · | We currently rely on, and expect to continue to rely on, third parties to conduct drug trials and aspects of our research and preclinical testing for Lucid-MS, our products relating to alcohol misuse and other possible drug candidates; |
| · | We, our service providers, or any third-party manufacturers may fail to comply with regulatory requirements which could subject us to enforcement actions; and |
| · | The FDA, Health Canada or other comparable regulatory authorities may not accept data from trials conducted in foreign jurisdictions. |
RISKS RELATED TO OUR INTELLECTUAL PROPERTY
| · | We may be unable to obtain and maintain sufficient intellectual property protection for our Product Candidates; |
| · | Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts; and |
| · | If we are unable to adequately protect the confidentiality of our trade secrets, our trademarks or trade names, our business may be adversely affected. |
GENERAL CORPORATE RISKS
| · | Macroeconomic pressures in the markets in which we operate, including, but not limited to, the lasting effects of the COVID-19 pandemic, epidemic, or outbreak of an infectious disease, inflation, stagflation, supply chain and interest rate pressures, foreign currency exchange rate fluctuations, the ongoing conflict between Russia and Ukraine and political developments in Hong Kong and Taiwan, natural disasters and other macroeconomic and geopolitical events may materially and adversely affect our business and financial results and could cause a disruption to the development of our Product Candidates; |
| · | The Company operates in a highly regulated industry and is subject to a wide range of federal, state, and local laws, rules, and regulations, including FDA and Health Canada regulatory requirements and laws pertaining to fraud and abuse in healthcare, that affect nearly all aspects of our operations. Failure to comply with these laws, rules, and regulations, or to obtain and maintain required licenses, could subject the Company to enforcement actions, including substantial civil and criminal penalties, and might require us to recall or withdraw a product from the market or cease operations, which could materially and adversely affect our business, financial condition, and results of operations; |
| · | Any significant interruption in the supply chain for key inputs could materially impact the Company’s business; |
| · | Future sales or issuances of equity securities and the conversion of outstanding securities to Class B Shares could decrease the value of the Class B Shares and dilute investors’ voting power; |
| · | The Company’s dual class structure has the effect of concentrating voting control and the ability to influence corporate matters with a limited number of holders of Class A multiple voting shares in the capital of the Company (the “Class A Shares”); |
| · | A decline in general economic conditions may impact the viability and success of our mortgage investment activities; |
| · | We may lose our status as a foreign private issuer; |
| · | There can be no assurance that we will be able to comply with the continued listing standards of the Nasdaq and/or Canadian Securities Exchange (the “CSE”); |
| · | The Company is currently party to several legal proceedings and may become a party to potential future litigation; and |
| · | We are a passive foreign investment company for U.S. federal income tax purposes, which may result in adverse U.S. federal income tax consequences for U.S. Holders of our Class B Shares. |
| 10 |
| Table of Contents |
PART I
Item 1. Identity of Directors, Senior Management and Advisers.
A. Directors and Senior Management
Not applicable.
B. Advisers
Not applicable.
C. Auditors
Not applicable.
Item 2. Offer Statistics and Expected Timetable.
Not applicable.
Item 3. Key Information
A. [Reserved]
B. Capitalization and Indebtedness
Not applicable.
C. Reasons for the Offer and Use of Proceeds
Not applicable.
D. Risk Factors
An investment in securities of the Company should only be made by persons who can afford a significant or total loss of their investment. We are exposed to a number of risks through the pursuit of our business objectives. The following risks and uncertainties identified below are those we believe may, individually or in combination with other risks and uncertainties, have a material impact on our business, but these are not the only risks and uncertainties we face. Additional risks and uncertainties not presently known to us, or risks that we currently deem immaterial, may also impair our business operations. If any of the following risks, or any other risks and uncertainties that we have not yet identified or that we currently consider not to be material, actually occur, or become material risks, our business, financial condition, results of operations and cash flows, and consequently the price of the Class B Shares, could be materially and adversely affected.
The risks discussed below also include Forward-Looking Statements and our actual results may differ substantially from those discussed in these Forward-Looking Statements. See “Cautionary Note Regarding Forward-Looking Statements” in this Annual Report.
Risks relating to our Product Candidates
Drug development is highly uncertain undertaking and involves a substantial degree of risk. We have no product sales, which, together with our limited operating history, makes it difficult to evaluate our business and assess our future viability.
Pharmaceutical and biopharmaceutical product development is a highly speculative undertaking and involves a substantial degree of risk. We are a biotechnology corporation with a limited operating history. We have no pharmaceutical products approved for commercial sale and have not generated any revenue from pharmaceutical product sales. We are currently focused on developing Lucid-MS, a patented new chemical entity targeting the treatment of MS. The effectiveness of Lucid-MS is not yet known. We continue to incur significant research and development and other expenses related to clinical trials and other operating expenses, ongoing operations and expect to incur losses for the foreseeable future. We anticipate these losses will increase and that we will not generate any revenue from product sales of Lucid-MS unless and until after we have successfully completed clinical development and received regulatory approval, for the commercial sale of this product.
We may never be able to develop or commercialize Lucid-MS or any other drug candidate or achieve profitability. Revenue from the sale of Lucid-MS, if regulatory approval is obtained, will be dependent, in part, upon the size of the markets in the territories for which we obtain regulatory approval, the accepted price for the product, the ability to obtain reimbursement at any price and whether we own the commercial rights for that territory, as well as the efficiency and availability of any comparable products. Our growth strategy depends on our ability to generate revenue. In addition, if the number of addressable patients is less than anticipated, the indication approved by regulatory authorities is narrower than expected, or the reasonably accepted population for treatment is narrowed by competition, physician choice or treatment guidelines, we may not generate significant revenue from sales of Lucid-MS or any other drug product, even if approved. Even if we are able to generate revenue from the sale of Lucid-MS, we may not become profitable and may need to obtain additional funding to continue operations. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods. Our failure to achieve sustained profitability would depress our value and could impair our ability to raise capital, expand our business, diversify our research and development pipeline, market Lucid-MS and any other product candidates that we may identify and pursue or continue our operations.
| 11 |
| Table of Contents |
Our future success is dependent on the regulatory approval and commercialization of our Product Candidates.
We do not have any products that have gained regulatory approval. As a result, our ability to finance our operations and generate revenue, are substantially dependent on our ability to obtain regulatory approval for, and, if approved, to successfully commercialize our product candidates in a timely manner. We cannot commercialize our other product candidates in Canada or the U.S. without first obtaining regulatory approval for each product from Health Canada or the FDA; similarly, we cannot commercialize any product candidates outside of the U.S. or Canada without obtaining regulatory approval from comparable foreign regulatory authorities, including the European Medicines Agency (the “EMA”). The FDA review process typically takes years to complete and approval is never guaranteed. Before obtaining regulatory approvals for the commercial sale of Lucid-MS or any of our potential product candidates for a target indication, we must demonstrate with substantial evidence gathered in preclinical and well-controlled clinical studies, with respect to approval in Canada and in the U.S. to the satisfaction of Health Canada and the FDA, and in Europe, to the satisfaction of the EMA, that the product candidate is safe and effective for use for that target indication; and that the manufacturing facilities, processes and controls are adequate. Obtaining regulatory approval for marketing of Lucid-MS or future product candidates in one country does not ensure we will be able to obtain regulatory approval in other countries. A failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in other countries.
Even if Lucid-MS or any of our other product candidates were to successfully obtain approval from Health Canada or the FDA or comparable foreign regulatory authorities, any approval might contain significant limitations related to use restrictions for specified age groups, warnings, precautions, or contraindications, or may be subject to burdensome post-approval studies or risk management requirements. If we are unable to obtain regulatory approval for our Product Candidates in one or more jurisdictions, or any approval contains significant limitations, we may not be able to obtain sufficient funding or generate sufficient revenue to continue the development of any of our other Product Candidates that we are developing or may discover, in-license, develop or acquire in the future. Also, any regulatory approval of our Product Candidates, once obtained, may be withdrawn. Furthermore, even if we obtain regulatory approval for any of our Product Candidates, their commercial success will depend on a number of factors, including the following:
| · | development of a commercial organization within the Company or establishment of a commercial collaboration with a commercial infrastructure; |
| · | establishment of commercially viable pricing and obtaining approval for adequate reimbursement from third-party and government payers; |
| · | our ability to manufacture quantities of our Product Candidates using commercially satisfactory processes and at a scale sufficient to meet anticipated demand and enable us to reduce our cost of manufacturing; |
| · | our success in educating physicians and patients about the benefits, administration, and use of our Product Candidates; |
| · | the availability, perceived advantages, relative cost, relative safety, and relative efficacy of alternative and competing treatments; |
| · | the effectiveness of our own or our potential strategic collaborators’ marketing, sales and distribution strategy and operations; |
| · | acceptance as a safe and effective therapy by patients and the medical community; and |
| · | · a continued acceptable safety profile following approval. |
Many of these factors are beyond our control. If we are unable to successfully commercialize our Product Candidates, we may not be able to earn sufficient revenues to continue our business.
We our relying on Celly Nu, our licensing partner, to promote our Unbuzzd™ brand and if we fail to maintain a good relationship with Celly Nu our business, financial condition and results of operations could be adversely affected.
On July 31, 2023, we entered into the Celly Nu IP License Agreement (as defined herein). Pursuant to the Celly Nu IP License Agreement, we are relying on Celly Nu to promote, commercialize and distribute Unbuzzd™ to the consumer market. Although we can maintain control over Celly Nu’s products and content to a certain degree through contractual provisions in the licensing agreement, we have limited control over its marketing and commercialization strategy.
The viability of the Celly Nu IP License Agreement depends on our ability to establish and maintain good relationship with Celly Nu. The value of our Unbuzzd™ brand and the rapport that we maintain with Celly Nu is an important factor for the success of this relationship. If we are unable to maintain a good relationship with Celly Nu, it could have a material adverse effect on our results of operations. Our license agreements require us and Celly Nu to comply with operational and performance conditions that are subject to interpretation and could result in disagreements. At any given time, we could have a dispute with Celly Nu regarding the interpretation of a provision in the Celly Nu IP License Agreement. An adverse result in any such dispute could materially adversely impact our results of operations and business.
For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Overview and History”.
| 12 |
| Table of Contents |
We rely on the UHN License to use for pharmaceutical purposes certain patents and other intellectual property rights associated with Lucid-MS.
One of our principal assets is the UHN License, which provides us with an exclusive, multi-jurisdictional license to use certain patents and other intellectual property rights associated with Lucid-MS, which is owned by the University Health Network (“UHN”). We are obligated to make milestone payments and royalties to UHN under the UHN License Agreement, which may limit our future profitability and our ability to enter into marketing partnership agreements. If we materially breach any of the terms of the UHN License Agreement (and fail to cure such breach with the specified time, to the extent a cure period is available for such breach), UHN, could terminate such agreement. If we were to lose or otherwise be unable to maintain the UHN License on acceptable terms, or find that it is necessary or appropriate to secure new licenses from other third parties, we may not be able to market Lucid-MS, and our current business model and plan would be impaired, which would have a material adverse effect on our business, operating results, and financial condition.
After receiving regulatory approvals, Lucid-MS may fail to achieve a sufficient degree of market acceptance by physicians, patients, healthcare payors, and others in the medical community.
The commercial success of Lucid-MS or other drug candidates that we develop, after receiving required regulatory approvals, will depend on their degree of market acceptance by physicians, patients, third-party payors, and others in the medical community. The degree of market acceptance of Lucid-MS will depend on a number of factors, including (i) the availability of alternative, superior treatments for a MS prior to the approval and commercialization Lucid-MS for such treatment; (ii) the efficacy and safety of Lucid-MS, including side effects or unexpected characteristics; (iii) the ability to offer Lucid-MS for sale at competitive prices; (iv) the ability to manufacture Lucid-MS in sufficient quantities and to offer appropriate patient access programs, such as co-pay assistance; (v) convenience and ease of dosing and administration compared to alternative treatments; (vi) the clinical indications for which Lucid-MS is approved by the FDA or Health Canada, if it approved at all, or comparable regulatory agencies; (vii) product labeling or product insert requirements of the FDA, Health Canada or other comparable regulatory authorities, including any limitations, contraindications or warnings contained in a product’s approved labeling; (viii) restrictions on how Lucid-MS is distributed; (ix) publicity concerning Lucid-MS or competing products and treatments; (x) the strength of marketing and distribution support; (xi) favorable third-party coverage and sufficient reimbursement; and (xii) the prevalence and severity of any side effects or adverse effects.
Sales of pharmaceutical products, such as Lucid-MS if and when it is approved by regulatory authorities, will depend on the willingness of physicians to prescribe the treatment, which is likely to be based on a determination by these physicians that the products are safe, therapeutically effective and cost effective. In addition, the inclusion or exclusion of products from treatment guidelines established by various physician groups and the viewpoints of influential physicians can affect the willingness of other physicians to prescribe the treatment. We cannot predict whether physicians, physicians’ organizations, hospitals, other healthcare providers, government agencies or private insurers will determine that Lucid-MS is safe, therapeutically effective and cost effective as compared with competing treatments. If Lucid-MS does not achieve adequate levels of acceptance, we may not generate significant product revenue, and we may not become profitable.
We face significant competition for our Lucid-MS drug and there is a possibility that our competitors may develop therapies that are safer, more advanced, or more effective than ours from MS.
The development and commercialization of new drug products is highly competitive. We face competition with respect to Lucid-MS for the treatment of MS from major pharmaceutical companies, specialty pharmaceutical companies, and biotechnology companies world-wide. Potential competitors also include academic institutions, government agencies, and other public and private research organizations that conduct research, seek patent protection, and establish collaborative arrangements for research, development, manufacturing, and commercialization. Even if we are successful in achieving regulatory approval to commercialize Lucid-MS ahead of our competitors, our future pharmaceutical products may face direct competition from generic and other follow-on drug products.
More established companies may have a competitive advantage over us due to their greater size, cash flows and institutional experience. Compared to us, many of our competitors may have significantly greater financial, technical and human resources. As a result of these factors, our competitors may obtain regulatory approval of their products before we do, which will limit our ability to develop or commercialize any of our Product Candidates. In addition, many companies are developing new therapeutics to supplant or expand upon the standard of care for a number of diseases, as a result, we cannot predict what the standard of care will be as our Product Candidates progress through clinical development.
Interim, “top-line,” and preliminary data from our clinical trials that we announce or publish from time to time may change as more patient data becomes available or as additional analyses are conducted, and as the data are subject to audit and verification procedures, that could result in material changes in the final data.
From time to time, we may publish interim, “top-line,” or preliminary data from our clinical studies. Interim data from clinical trials that we may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment continues and more patient data become available. Preliminary or “top-line” data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, interim and preliminary data should be viewed with caution until the final data are available. Material adverse changes between preliminary, “top-line,” or interim data and final data could significantly harm our business prospects.
| 13 |
| Table of Contents |
We expect to rely on third parties to conduct product candidate drug trials and aspects of our research and preclinical testing.
We currently rely and expect to continue to rely on third parties, such as CROs, clinical data management organizations, medical institutions, and clinical investigators, to conduct some aspects of research and preclinical testing and clinical trials. Any of these third parties may terminate their engagements with us or be unable to fulfill their contractual obligations. If any of our relationships with these third parties terminate, we may not be able to enter into arrangements with alternative third parties on commercially reasonable terms, or at all. If we need to enter into alternative arrangements, it could delay our development activities.
Our reliance on these third parties for research and development activities reduces control over these activities but does not relieve us of our responsibilities. For example, we remain responsible for ensuring that product candidate drug trials are each conducted in accordance with the general investigational plan and protocols for each trial and applicable legal, regulatory, and scientific standards, and our reliance on third parties does not relieve us of our regulatory responsibilities. In addition, the FDA, Health Canada, and other comparable regulatory authorities require compliance with good clinical practices for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible, reproducible and accurate and that the rights, integrity, and confidentiality of trial participants are protected. Regulatory authorities enforce these good clinical practices through periodic inspections of trial sponsors, principal investigators, and trial sites. If we or any of these third parties fail to comply with applicable good clinical practice regulations, some or all of the clinical data generated in any product candidate drug trials may be deemed unreliable and the FDA, Health Canada or other comparable regulatory authorities may reject our marketing applications or require us to perform additional nonclinical or clinical trials or to enroll additional patients before approving our marketing applications. We cannot be certain that, upon inspection, such regulatory authorities will determine that any product candidate drug trial complies with the good clinical practice regulations. For any violations of laws and regulations during the conduct of clinical trials, we could be subject to untitled and warning letters or enforcement action that may include civil penalties and criminal prosecution. We also are required to register ongoing clinical trials and post the results of completed clinical trials on a government-sponsored database within certain timeframes. Failure to do so can result in fines, adverse publicity, and civil and criminal sanctions.
If these third parties do not successfully carry out their contractual duties, meet expected deadlines, or conduct clinical trials in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for a product candidate and will not be able to, or may be delayed in our efforts to, successfully commercialize product candidates. Our failure or the failure of these third parties to comply applicable regulatory requirements or our stated protocols could also subject us to enforcement action.
We also expect to rely on other third parties to store and distribute drug supplies for product candidate drug trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of any product candidates we may develop or commercialization of our medicines or other therapeutic applications, producing additional losses and depriving us of potential product revenue.
Results of earlier studies or clinical trials may not be predictive of future clinical trial results and may not justify proceeding to advanced clinical trials or an application for regulatory approval.
Success in preclinical testing and early clinical trials does not ensure that later clinical trials will generate adequate data to demonstrate the efficacy and safety of an investigational drug. A number of companies in the pharmaceutical and biotechnology industries, including those with greater resources and experience, have suffered significant setbacks in clinical trials, even after seeing promising results in earlier clinical trials. We do not know whether the clinical trials we are conducting, or may conduct, will demonstrate adequate efficacy and safety to result in regulatory approval to market any of our product candidates in any particular jurisdiction. Even if we believe that we have adequate data to support an application for regulatory approval to market our product candidates, the FDA or other comparable foreign regulatory authorities may not agree and could require us to conduct additional research studies, including late-stage clinical trials. If late-stage clinical trials do not produce favorable results, our ability to achieve regulatory approval for any of our product candidates may be adversely impacted.
Product candidates could be associated with side effects which could delay or halt clinical development, prevent regulatory approval, or result in other significant negative consequences.
As is the case with pharmaceuticals generally, it is likely that there may be side effects associated with Lucid-MS or our other drug product candidates. If Lucid-MS or our other drug product candidates are associated with undesirable side effects in preclinical studies or clinical trials or have characteristics that are unexpected, we may elect to abandon their development or limit their development to more narrow uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective, which may limit the commercial expectations for this product candidate if approved. We may also be required to modify or terminate our study plans based on findings in our preclinical studies or clinical trials.
Additionally, adverse developments in clinical trials of pharmaceutical and biopharmaceutical products conducted by others may cause the FDA, Health Canada, or other regulatory oversight bodies to suspend or terminate our clinical trials or to change the requirements for approval of Lucid-MS or our other drug product candidates.
Additionally, if we or others later identify undesirable side effects caused by Lucid-MS or our other drug product candidates once approved, several potentially significant negative consequences could result, including: (i) regulatory authorities may suspend or withdraw approvals of such product candidate; (ii) we may be required to change the way a product candidate is administered or conduct additional clinical trials; (iii) we may be required to include additional warnings on a product candidate’s labeling or the product candidate may be subject to restrictive distribution requirements; (iv) we could be sued and held liable for harm caused to patients; and (v) our reputation may suffer. Any of these occurrences may harm our business, financial condition, and prospects significantly.
In addition to side effects caused by the product candidate, the administration process or related procedures also can cause adverse side effects. If any such adverse events occur, our clinical trials could be suspended or terminated. If we are unable to demonstrate that any adverse events were caused by the administration process or related procedures, the FDA, Health Canada, or other regulatory authorities could order us to cease further development of, or deny approval of, a product candidate for any or all targeted indications. Even if we can demonstrate that all future serious adverse events are not product-related, such occurrences could affect patient recruitment or the ability of enrolled patients to complete the trial. Moreover, if we elect, or are required, to not initiate, delay, suspend or terminate any future clinical trial of Lucid-MS or any of our product candidates, the commercial prospects of Lucid-MS or such other product candidates may be harmed and our ability to generate product revenues from Lucid-MS or any of these other product candidates may be delayed or eliminated. Any of these occurrences may harm our ability to develop other product candidates, and may harm our business, financial condition, and prospects significantly.
| 14 |
| Table of Contents |
The Company may not be successful in its efforts to identify, license or discover additional product candidates.
Although a substantial amount of the Company’s effort will focus on the continued research and pre‐clinical and clinical testing, potential approval and commercialization of its Product Candidates, the success of its business also depends in part upon its ability to identify, license or discover additional product candidates. The Company’s research programs or licensing efforts may fail to yield additional product candidates for clinical development for a number of reasons, including but not limited to the following: (i) the Company’s research or business development methodology or search criteria and process may be unsuccessful in identifying potential product candidates; (ii) the Company may not be able or willing to assemble sufficient resources to acquire or discover additional product candidates; (iii) the Company’s product candidates may not succeed in pre‐clinical or clinical testing; (iv) the Company’s product candidates may be shown to have harmful side effects or may have other characteristics that may make the product candidates unmarketable or unlikely to receive marketing approval; (v) competitors may develop alternatives that render the Company’s product candidates obsolete or less attractive; (vi) product candidates the Company develops may be covered by third parties’ patents or other exclusive rights; (vii) the market for a product candidate may change during the Company’s program such that the further development of a product candidate may become undesirable; (viii) a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and (ix) a product candidate may not be accepted as safe and effective by patients, the medical community or third‐party payors.
If any of these events occurs, the Company may be forced to abandon its development efforts to identify, license or discover additional product candidates, which could have a material adverse effect on its business, prospects, results of operations and financial condition and could potentially cause the Company to cease operations. Research programs to identify new product candidates require substantial technical, financial, and human resources. The Company may focus its efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful.
The FDA, Health Canada or other comparable regulatory authorities may not accept data from trials conducted in foreign jurisdictions.
Obtaining regulatory approval in one country does not mean that regulatory approval will be obtained in any other country. We intend on submitting our initial regulatory approvals for Lucid-MS in the U.S. and Canada. Approval processes vary among countries and can involve additional product testing and validation and additional or different administrative review periods, including additional preclinical studies or clinical trials, as data from clinical trials conducted in one jurisdiction may not be accepted by regulatory authorities in other jurisdictions. In many jurisdictions outside the United States and Canada, a product candidate must be approved for reimbursement before it can be approved for sale in that jurisdiction. In some cases, the price that we intend to charge for our products is also subject to approval.
Non-U.S. and non-Canadian regulatory approval processes may include all of the risks associated with obtaining FDA or Health Canada approval, as well as additional risks. We do not have any product candidates approved for sale in any jurisdiction, including international markets, and we do not have experience in obtaining regulatory approval in international markets. If we fail to comply with regulatory requirements in international markets or to obtain and maintain required approvals, or if regulatory approval in international markets is delayed, our target market will be reduced and our ability to realize the full market potential of our Product Candidates will be harmed. In addition, if we conduct trials outside of the U.S. or Canada, the FDA or Health Canada, as applicable, may not accept the data from such trials and may require additional trials, which could be costly and time-consuming and delay aspects of our business plan.
Our suppliers could experience manufacturing problems that result in delays in our development or commercialization programs or otherwise harm our business.
Our contract manufacturing organization (“CMO”) must employ multiple steps to control the manufacturing process to assure that the process is reproducible and the product candidate is made strictly and consistently in compliance with the process. Problems with the manufacturing process, even minor deviations from the normal process, could result in product defects or manufacturing failures that result in lot failures, product recalls, product liability claims or insufficient inventory to conduct clinical trials or supply commercial markets. Furthermore, all entities involved in the preparation of product candidates for clinical trials or commercial sale, including our existing CMOs for all of our Product Candidates, are subject to extensive regulation. Components of a finished therapeutic products approved for commercial sale or used in certain clinical trials must be manufactured in accordance with good manufacturing practices (“GMP”), or similar regulatory requirements outside the United States and Canada. Our failure, or the failure of third-party manufacturers, to comply with applicable regulations could result in sanctions being imposed on us, including clinical holds, fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, license revocation, suspension of production, seizures or recalls of Product Candidates or marketed drugs, operating restrictions and criminal prosecutions, any of which could significantly and adversely affect clinical or commercial supplies of our Product Candidates and increase our costs. Consequently, there may be a material adverse effect on the business, results of operations, financial condition, and prospects of the Company.
In addition, the FDA, Health Canada, and other regulatory authorities may require us to submit samples of any lot of any approved Product Candidates together with the protocols showing the results of applicable tests at any time. Under some circumstances, the FDA, Health Canada, or other regulatory authorities may require that we not distribute a lot until the agency authorizes its release. Slight deviations in the manufacturing process, including those affecting quality attributes and stability, may result in unacceptable changes in the product that could result in lot failures or product recalls. Lot failures or product recalls could cause us to delay product launches or clinical trials, which could be costly to us and otherwise harm our business, results of operations, financial condition, and prospects.
| 15 |
| Table of Contents |
Our CMOs also may encounter problems hiring and retaining the experienced scientific, quality assurance, quality-control and manufacturing personnel needed to operate our manufacturing processes, which could result in delays in production or difficulties in maintaining compliance with applicable regulatory requirements.
Any problems in our CMOs’ manufacturing process or facilities could result in delays or cancellations of planned clinical trials, failures in satisfying ongoing regulatory obligations (before and after regulatory approval for a product candidate is obtained) and increased costs. Such problems could also make us a less attractive collaborator for potential partners, including larger biotechnology companies and academic research institutions, which could limit access to additional attractive development programs. Problems in our manufacturing process could restrict our ability to meet potential future market demand for products.
Lucid-MS, after it is approved, will be subject to extensive post-approval regulation.
After a product is approved, numerous post-approval requirements apply. Among other things, the holder of an approved NDA is subject to periodic and other FDA monitoring and reporting obligations, including obligations to monitor and report adverse events and instances of the failure of a product to meet the specifications in the NDA. Application holders must submit new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling, or manufacturing process. Application holders must also submit advertising and other promotional material to the FDA and report on ongoing clinical studies.
Depending on the circumstances, failure to meet these post-approval requirements can result in criminal prosecution, fines, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, or refusal to allow us to enter into supply contracts, including government contracts. In addition, even if we comply with FDA and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA to modify or withdraw product approval. Similar laws in other jurisdictions would also apply.
After our Product Candidates are commercialized, they may be subject to recalls for a variety of reasons, which could require the Company to expend significant management and capital resources.
Manufacturers and distributors of products are sometimes subject to the recall or return of their products for a variety of reasons, including product defects, such as contamination, unintended harmful side effects or interactions with other substances, packaging safety and inadequate or inaccurate labeling disclosure. If any of the Company’s approved and commercialized Product Candidates are recalled due to an alleged product defect or for any other reason, the Company could be required to incur the unexpected expense of the recall and any legal proceedings that might arise in connection with the recall. The Company may lose a significant amount of sales made on such products and may not be able to replace those sales at an acceptable margin or at all. In addition, a product recall may require significant management attention. Although the Company has detailed procedures in place for testing its products, there can be no assurance that any quality, potency, or contamination problems will be detected in time to avoid unforeseen product recalls, regulatory action or lawsuits. Additionally, if one of the Company’s significant brands were subject to recall, the image of that brand and the Company could be harmed. A recall for any of the foregoing reasons could lead to decreased demand for the Company’s products and could have a material adverse effect on the results of the operations and financial condition of the Company. Additionally, product recalls may lead to increased scrutiny of the Company’s operations by the FDA, Health Canada or other regulatory agencies, requiring further management attention and potential legal fees and other expenses.
If approved, Lucid-MS may face competition from generic drugs approved through an abbreviated regulatory pathway.
The Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Amendments to the Federal Food, Drug, and Cosmetic Act (the “FDC Act”), authorized the FDA to approve generic drugs that are the same as drugs previously approved for marketing under the new drug application (“NDA”) provisions of the statute pursuant to an abbreviated new drug application (“ANDA”). An ANDA relies on the preclinical and clinical testing conducted for a previously approved reference listed drug (“RLD”), and must demonstrate to the FDA that the generic drug product is identical to the RLD with respect to the active ingredients, the route of administration, the dosage form, and the strength of the drug and also that it is “bioequivalent” to the RLD. The FDA is prohibited by statute from approving an ANDA when certain marketing or data exclusivity protections apply to the RLD. If any such competitor or third party is able to demonstrate bioequivalence without infringing our patents, then this competitor or third party may then be able to introduce a competing generic product onto the market. The Hatch-Waxman Amendments also enacted the 505(b)(2) NDA pathway, which permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. A Section 505(b)(2) applicant may eliminate the need to conduct certain preclinical or clinical studies, if it can establish that reliance on studies conducted for a previously approved product is scientifically appropriate.
Market exclusivity provisions authorized under the FDC Act can delay the submission or the approval of certain marketing applications. The FDC Act provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to obtain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not approve or even accept for review an ANDA or an NDA submitted under Section 505(b)(2) by another company for another drug based on the same active moiety, regardless of whether the drug is intended for the same indication as the original innovative drug or for another indication, where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement to one of the patents listed with the FDA by the innovator NDA holder.
| 16 |
| Table of Contents |
The FDC Act also provides three years of marketing exclusivity for an NDA, or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example new indications, dosages, or strengths of an existing drug. This three-year exclusivity covers only the modification for which the drug received approval on the basis of the new clinical investigations and does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for drugs containing the active agent for the original indication or condition of use. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to any preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
If competitors are able to obtain marketing approval for generic drugs referencing our products, our products may become subject to competition from such generic drugs. The availability of competitive generic products could limit the demand, and the price we are able to charge, for any products that we may develop and commercialize.
Product liability lawsuits against us could cause us to incur substantial liabilities and could limit commercialization of any product candidates that we may develop.
We face an inherent risk of product liability exposure related to the testing of product candidates in human clinical trials and will face an even greater risk if we commercially sell any medicines or other therapeutic applications that we may develop. If we cannot successfully defend ourselves against claims that our Product Candidates, medicines, or other therapeutic applications caused injuries, we could incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in: (i) decreased demand for any product candidates, medicines or other therapeutic applications that we may develop; (ii) injury to our reputation and significant negative media attention; (iii) withdrawal of clinical trial participants; (iv) significant costs to defend the related litigation; (v) substantial monetary awards to trial participants or patients; (vi) loss of revenue; and (vii) the inability to commercialize our Product Candidates.
Although we intend to maintain product liability insurance, including coverage for clinical trials that we plan to sponsor, it may not be adequate to cover all liabilities that we may incur. We anticipate that we will need to increase our insurance coverage as we commence additional clinical trials and if we successfully commercialize any Product Candidates. The market for insurance coverage is increasingly expensive, and the costs of insurance coverage will increase as our clinical programs increase in size. We may not be able to maintain insurance coverage at a reasonable cost or in an amount adequate to satisfy any liability that may arise.
We may become liable for uninsured or uninsurable risk.
The Company may become subject to liability for risks which are uninsurable or against which the Company may opt out of insuring due to the high cost of insurance premiums or other factors. The payment of any such liabilities would reduce the funds available for usual business activities. Payment of liabilities for which insurance is not carried may have a material adverse effect on the Company’s financial position and operations.
Our employees, directors, independent contractors, consultants, commercial partners, and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of fraud, misconduct or other illegal activity by our employees, directors, independent contractors, consultants, commercial partners, and vendors. Misconduct by these parties could include intentional, reckless and negligent conduct that fails to: (i) comply with the requirements of the FDA, Health Canada and other comparable regulatory authorities; (ii) provide true, complete and accurate information to the FDA, Health Canada and other comparable regulatory authorities; (iii) comply with manufacturing standards we have established; (iv) comply with healthcare fraud and abuse laws and similar other fraudulent misconduct laws in the United States or Canada; or (v) report financial information or data accurately or to disclose unauthorized activities appropriately. If we obtain approval of our Product Candidates from the FDA, Health Canada or other comparable regulatory authorities and begin commercializing those products in the United States, Canada or other countries, our potential exposure under such laws will increase significantly, and our costs associated with compliance with such laws are also likely to increase. In particular, research, sales, marketing, education, and other business arrangements in the healthcare industry are subject to extensive laws designed to prevent fraud, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, educating, marketing and promotion, sales and commission, certain customer incentive programs and other business arrangements generally. Activities subject to these laws and regulations also involve the improper use of information obtained in the course of patient recruitment for clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. The board of directors of the Company (the “Board”) has adopted a Code of Conduct and Ethics (the “Code”) which provides guidelines surrounding, among other items, compliance with applicable laws, conflicts of interest, certain opportunities, confidentiality and disclosure, employment practices, and use of company property and resources. However, it is not always possible to identify and deter misconduct by employees, directors and third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws and regulations. If any such actions or lawsuits are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions or lawsuits could have a significant impact on our business, including the imposition of significant fines or other sanctions.
| 17 |
| Table of Contents |
We may be unable to establish sufficient sales and marketing capabilities or enter into agreements with third parties to sell and market any product candidates in a compliant manner even if regulatory approvals are obtained.
We do not currently have a comprehensive infrastructure for the sales, marketing, and distribution of pharmaceutical drug products. The cost of establishing and maintaining such an infrastructure may exceed the cost-effectiveness of doing so. In order to market any products that may be approved by the FDA and comparable foreign regulatory authorities, we must build our sales, marketing, managerial and other nontechnical capabilities or make arrangements with third parties to perform these services for which we would incur substantial costs. If we are unable to establish adequate sales, marketing, and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenue and may not sustain profitability. We will be competing with many companies that have extensive and well-funded sales and marketing operations. Without an internal commercial organization or the support of a third party to perform sales and marketing functions, or a combination of both, we may be unable to compete successfully against more established companies.
Our Product Candidates may become subject to unfavorable pricing regulations, third-party coverage and reimbursement practices, or healthcare reform initiatives, which would harm our business.
The regulations that govern marketing approvals, pricing, coverage, and reimbursement for new drugs vary widely from country to country. Adverse pricing limitations may hinder our ability to recoup our investment in one or more Product Candidates, even if any Product Candidates we may develop obtain marketing approval.
Our ability to successfully commercialize our Product Candidates also will depend in part on the extent to which coverage and adequate reimbursement for these products and related treatments will be available from government health administration authorities, private health insurers, and other organizations. If coverage and adequate reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our Product Candidates. Even if coverage is provided, the approved reimbursement amount may not be high enough to allow us to establish or maintain pricing sufficient to realize a sufficient return on our investment.
In many countries, the prices of medical products are subject to varying price control mechanisms as part of national health systems. Other countries allow companies to fix their own prices for medicines, but monitor and control corporation profits. Additional price controls or other changes in pricing regulation could restrict the amount that we are able to charge for our Product Candidates.
Risks Related to our Intellectual Property
We may be unable to obtain and maintain sufficient intellectual property protection for our Product Candidates.
As is the case with pharmaceutical companies and other biotechnology companies, our success depends in large part on our ability to obtain and maintain protection of the intellectual property we may own solely and jointly with others, particularly patents, in the United States, Canada and other countries with respect to our Product Candidates and technology. We seek to protect our proprietary position by filing patent applications in the United States, Canada and in other countries related to the Product Candidates or other product candidates that we may identify. On April 24, 2023, the Company filed a provisional patent application with the United States Patent and Trademark Office (“USPTO”) with respect to the Company’s alcohol misuse treatment technology. We have an exclusive license from UHN to use patents and other intellectual property that is used in our Lucid-MS compound.
Obtaining and enforcing pharmaceutical and biopharmaceutical patents is costly, time consuming and complex, and we or our licensors may not be able to file and prosecute all necessary or desirable patent applications, or maintain, enforce, and license any patents that may issue from such patent applications, at a reasonable cost or in a timely manner, if at all. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. We may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the rights to patents licensed to third parties. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal, technological, and factual questions and has in recent years been the subject of much litigation. In addition, the laws of certain countries may not protect our rights to the same extent as the laws of other countries, including the United States and Canada, and vice versa. Further, we may not be aware of all third-party intellectual property rights potentially relating to our Product Candidates. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States, Canada and other jurisdictions are typically not published until 18 months after filing or, in some cases, not at all. Therefore, we cannot know with certainty whether our licensors were the first to make the inventions claimed in our licensed patents, or that our licensors were the first to file for patent protection of such inventions. Furthermore, the scope of a patent claim is determined by an interpretation of the law, the written disclosure in a patent and the patent’s prosecution history and can involve other factors such as expert opinion. Our analysis of these issues, including interpreting the relevance or the scope of claims in a patent or a pending application, determining applicability of such claims to our proprietary technologies or Product Candidates, predicting whether a third party’s pending patent application will issue with claims of relevant scope, and determining the expiration date of any patent in the United States, Canada or abroad that we consider relevant may be incorrect, which may negatively impact our ability to develop and market our Product Candidates. We do not always conduct independent reviews of pending patent applications of and patents issued to third parties. As a result, the issuance, scope, validity, enforceability, and commercial value of our patent rights, including licensed patent rights, are highly uncertain. Our future patent applications may not result in patents being issued that protect our Product Candidates, in whole or in part, or which effectively prevent others from commercializing competitive product candidates. Even if our patent applications issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors from competing with us or otherwise provide us with any competitive advantage. Our competitors may be able to circumvent our licensed patents by developing similar or alternative product candidates in a non-infringing manner.
| 18 |
| Table of Contents |
Our licensors’ ability to enforce patent rights also depends on our licensors’ ability to detect infringement. It may be difficult to detect infringers who do not advertise the components or methods that are used in connection with their products and services. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s or potential competitor’s product or service. We, along with our licensors, may not prevail in any lawsuits that we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful. If we initiate lawsuits to protect or enforce our licensed patents, or litigate against third-party claims, such proceedings would be expensive and would divert the attention of our management and technical personnel. Such proceedings could also provoke third parties to assert claims against us, including that some or all of the claims in one or more of our or our licensed patents are invalid or otherwise unenforceable.
Moreover, we may be subject to a third-party pre-issuance submission of prior art to the USPTO, or become involved in opposition, derivation, re-examination, inter partes review, post-grant review or interference proceedings challenging our or our licensors’ patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate, our licensed patents, allow third parties to commercialize our Product Candidates and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize drugs without infringing third-party patent rights. In addition, if the breadth or strength of protection provided by our licensed patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future product candidates.
In addition, the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and our licensed patents may be challenged in the courts or patent offices in the United States, Canada and abroad. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated, or held unenforceable, in whole or in part, which could limit our licensors’ abilities to stop others from using or commercializing similar or identical product candidates to ours, or limit the duration of the patent protection of our Product Candidates.
Filing, prosecuting, and defending the licensed patents on our Product Candidates in all countries throughout the world would be prohibitively expensive. Additionally, the laws of some other countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we and our licensors may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained licensed patent protection to develop their own products and may also export infringing products to territories where we have patent protection, but enforcement is not as strong as that in the U.S. or Canada. Given the amount of time required for the development, testing and regulatory review of new product candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, our patent portfolio, including licensed patents, may not provide us with sufficient rights to exclude others from commercializing drugs similar or identical to ours.
Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. However, our research, development and commercialization activities may be subject to claims that we infringe or otherwise violate patents or other intellectual property rights owned or controlled by third parties. In May 2023, GBB Drink Lab, Inc. (“GBB”) filed a lawsuit against the Company alleging a material breach of a mutual nondisclosure agreement and trade secret misappropriation in the U.S. District Court for the Southern District of Florida. This lawsuit is ongoing. For more information, please see “Item 8. Financial Information - A. Consolidated Statements and Other Financial Information – Legal Proceedings”.
There is a substantial amount of litigation, both within and outside the U.S. and Canada, involving patent, trade secret and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions and inter partes re-examination proceedings. Numerous U.S. and international issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are pursuing development candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our Product Candidates may be subject to claims of infringement of the patent rights of third parties.
Other third parties may assert that we are employing their proprietary technology without authorization. There may be other third-party patents or patent applications with claims to materials, formulations, methods of manufacture or methods for treatment related to the use or manufacture of the Product Candidates. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that the Product Candidates or other product candidates that we may identify may infringe. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of the Product Candidates or other product candidates that we may identify, any molecules formed during the manufacturing process or any final product itself, the holders of any such patents may be able to block our ability to commercialize such product candidate unless we obtained a license under the applicable patents, or until such patents expire.
Similarly, if any third-party patents were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture or methods of use, including combination therapy, the holders of any such patents may be able to block our ability to develop and commercialize the applicable product candidate unless we obtained a license or until such patent expires. In either case, such a license may not be available on commercially reasonable terms or at all, or it may be non-exclusive, which could result in our competitors gaining access to the same intellectual property.
| 19 |
| Table of Contents |
Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize the Product Candidates or other product candidates that we may identify. Defense of these claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing Product Candidates, or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
Parties making claims against us, may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation or administrative proceedings, there is a risk that some of our confidential information could be compromised by disclosure. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have material adverse effect on ability to raise additional funds or otherwise have a material adverse effect on our business, results of operations, financial condition, and prospects.
If our licensors are not able to obtain patent term extension or non-patent exclusivity in the United States under the Hatch-Waxman Act and in other countries under similar legislation, thereby potentially extending the marketing exclusivity term of our product candidates, our business may be materially harmed.
Patents have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally 20 years from its earliest U.S. non-provisional or international patent application filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited.
Depending upon the timing, duration, and specifics of FDA marketing approval of our Product Candidates, one of the U.S. patents covering each of such Product Candidates or the use thereof may be eligible for up to five years of patent term extension under the Hatch-Waxman Act. The Hatch-Waxman Act allows a maximum of one patent to be extended per FDA approved product as compensation for the patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only those claims covering such approved drug product, a method for using it or a method for manufacturing it may be extended.
Patent term extension also may be available in certain other countries upon regulatory approval of our Product Candidates. Nevertheless, our licensors may not be granted patent term extension either in the United States, Canada or in any other country because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents or otherwise failing to satisfy applicable requirements. Moreover, the term of extension, as well as the scope of patent protection during any such extension, afforded by the governmental authority could be less than requested.
If our licensors are unable to obtain patent term extension or restoration, or the term of any such extension is less than we request, the period during which we will have the right to exclusively market our product may be shortened and our competitors may obtain approval of competing products following the patent expiration sooner, and our revenue could be reduced, possibly materially.
It is possible that our licensors will not obtain patent term extension under the Hatch-Waxman Act for a U.S. patent covering a Product Candidate even where that patent is eligible for patent term extension, or if we obtain such an extension, it may be for a shorter period than we had sought. Further, for certain of our licensed patents, we do not have the right to control prosecution, including filing with the USPTO, a petition for patent term extension under the Hatch-Waxman Act. Thus, if one of our licensed patents is eligible for patent term extension under the Hatch-Waxman Act, we may not be able to control whether a petition to obtain a patent term extension is filed, or obtained, from the USPTO.
If we are unable to protect the confidentiality of our trade secrets, the value of our technology could be materially adversely affected and our business would be harmed.
We seek to protect our confidential proprietary information, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and collaborators. These agreements are designed to protect our proprietary information. However, we cannot be certain that such agreements have been entered into with all relevant parties, and we cannot be certain that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. For example, any of these parties may breach the agreements and disclose proprietary information, including trade secrets, and we may not be able to obtain adequate remedies for such breaches. We also seek to preserve the integrity and confidentiality of our confidential proprietary information by maintaining physical security of our premises and physical and electronic security of our information technology systems, but it is possible that these security measures could be breached. If any of our confidential proprietary information were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position.
| 20 |
| Table of Contents |
Unauthorized parties may also attempt to copy or reverse engineer certain aspects of our Product Candidates that we consider proprietary. We may not be able to obtain adequate remedies in the event of such unauthorized use. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret can be difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, some courts are less willing or unwilling to protect trade secrets. Trade secrets will also over time be disseminated within the industry through independent development, the publication of journal articles and the movement of personnel skilled in the art from corporation to corporation or academic to industry scientific positions. Though our agreements with third parties typically restrict the ability of our advisors, employees, collaborators, licensors, suppliers, third-party contractors, and consultants to publish data potentially relating to our trade secrets, our agreements may contain certain limited publication rights.
In addition, if any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position. Despite employing the contractual and other security precautions described above, the need to share trade secrets increases the risk that such trade secrets become known by our competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. If any of these events occurs or if we otherwise lose protection for our trade secrets, the value of this information may be greatly reduced and our competitive position, business, results of operations, financial condition and prospects would be harmed.
If our trademarks and trade names are not adequately protected, then we may not be able to build name recognition in our markets of interest and our business may be adversely affected.
Our registered or unregistered trademarks or trade names may be challenged, infringed, circumvented, or declared generic or determined to be infringing on other marks. We may not be able to protect our rights to these trademarks and trade names, which we need to build name recognition among potential collaborators or customers in our markets of interest. At times, competitors may adopt trade names or trademarks similar to ours, thereby impeding our ability to build brand identity and possibly leading to market confusion. In addition, there could be potential trade name or trademark infringement claims brought by owners of other trademarks or trademarks that incorporate variations of our registered or unregistered trademarks or trade names. Over the long term, if we are unable to establish name recognition based on our trademarks and trade names, then we may not be able to compete effectively and our business may be adversely affected. We may license our trademarks and trade names to third parties, such as distributors. Though these license agreements may provide guidelines for how our trademarks and trade names may be used, a breach of these agreements or misuse of our trademarks and tradenames by our licensees may jeopardize our rights in or diminish the goodwill associated with our trademarks and trade names. Our efforts to enforce or protect our proprietary rights related to trademarks, trade names, trade secrets, domain names, copyrights or other intellectual property may be ineffective and could result in substantial costs and diversion of resources and could adversely affect our competitive position, business, results of operations, financial condition, and prospects.
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
Our agreements with employees and our personnel policies provide that any inventions conceived by an individual in the course of rendering services to us shall be our exclusive property. Although our policy is to have all such individuals complete these agreements, we may not obtain these agreements in all circumstances, and individuals with whom we have these agreements may not comply with their terms. The assignment of intellectual property may not be automatic upon the creation of an invention and despite such agreement, such inventions may become assigned to third parties. In the event of unauthorized use or disclosure of our trade secrets or proprietary information, these agreements, even if obtained, may not provide meaningful protection, particularly for our trade secrets or other confidential information.
We or our licensors may be subject to claims that former employees, collaborators or other third parties have an interest in our owned or licensed patents, trade secrets, or other intellectual property as an inventor or co-inventor. For example, we or our licensors may have inventorship disputes arise from conflicting obligations of employees, consultants or others who are involved in developing our Product Candidates. Litigation may be necessary to defend against these and other claims challenging inventorship or our licensors’ ownership of our owned or licensed patents, trade secrets or other intellectual property. If we or our licensors fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual property that is important to our Product Candidates. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Any of the foregoing could have a material adverse effect on our competitive position, business, results of operations, financial condition, and prospects.
We may be subject to claims that our employees, consultants, or independent contractors have wrongfully used or disclosed confidential information of third parties or that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we try to ensure that our employees, consultants, and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel, which could adversely impact our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Risks relating to our Psychedelic Products
Due to funding issues, we made a decision to place all research relating to Lucid-PSYCH on hold in April 2023. If and when we resume this research, we will encounter the following risks with respect to our Lucid-PSYCH drug.
The loss of single-source suppliers, or their failure to supply us with the drug substance or drug product, could materially and adversely affect our business.
When we were actively researching Lucid-PSYCH, we relied upon a single-source supplier for the supply of drug substances and products for this compound. Although we believe that there are alternate sources of supply that could satisfy our clinical and commercial requirements, we cannot assure you that identifying alternate sources and establishing relationships with such sources would not result in significant delay in the development of the Lucid-PSYCH Product Candidate.
Our dependence on a single-source supplier exposes us to certain risks, that may materially impact our ability to progress our business, including (i) our supplier may cease or reduce production or deliveries, raise prices or renegotiate terms; (ii) delays caused by supply issues which may harm our reputation; and (iii) our single-source supplier or CMOs may experience significant business challenges, disruption or failures due to issues such as financial difficulties or bankruptcy, issues relating to regulatory or quality compliance issues, or other legal or reputational issues.
Additionally, we may not be able to enter into supply arrangements with alternative suppliers on commercially reasonable terms, or at all. A delay in the development of a Product Candidate or having to enter into a new agreement with a different third party on less favorable terms than we have with our current suppliers could have a material adverse impact upon on our business.
Psychedelic or psychedelic-inspired drugs may never be approved as medicines or other therapeutic applications and violations of applicable laws and regulations could result in repercussions.
In the United States, certain psychedelic drugs are classified as Schedule I drugs under the CSA (21 U.S.C. § 811) and the Controlled Substances Import and Export Act and as such, medical and recreational use is illegal under the U.S. federal laws.
| 21 |
| Table of Contents |
In Canada, certain substances are classified as controlled substances and are listed on Schedule III of the Controlled Drugs and Substances Act (Canada) (“CDSA”) and are also listed under the Schedule to Part J to the Food and Drug Regulations, which results in very restricted use as substances listed under Part J can generally only be used for research or clinical testing under limited circumstances. There is no guarantee that psychedelic drugs will ever be approved as medicines or other therapeutic applications in any jurisdiction in which the Company operates.
The Company's programs for Lucid-PSYCH involved controlled drugs and were conducted in strict compliance with the laws and regulations regarding the production, storage, and use of such drugs. Although the Company put a temporary hold on all research and development related to controlled drugs, if it begins research again, it will be subject to these risks. As such, all facilities engaged with such substances by or on behalf of the Company do so under current licenses and permits issued by appropriate governmental agencies. Unforeseen delays to the drug substance and drug product manufacture and supply chain may occur due to delays, errors, or other unforeseen problems with the permitting and quota process.
The failure of the Company to maintain compliance with applicable federal, state, or provincial requirements, or the loss or diversion of controlled substances, can result in significant enforcement actions. The Drug Enforcement Administration (“DEA”) and/or state authorities could seek civil penalties, refuse to renew registrations, or initiate proceedings to revoke those registrations. In certain circumstances, violations could lead to civil and criminal prosecutions, fines, penalties, and forfeitures. Overall, a violation of any laws and regulations in the jurisdictions in which the Company operates could result in significant fines, penalties, administrative sanctions, convictions, or settlements arising from civil proceedings initiated by either government entities in the jurisdictions in which the Company operates, or by private citizens, or through criminal charges. The loss of the necessary licenses, permits or exemptions, including the loss of access to licensed facilities, for use of controlled drugs could have an adverse effect on the Company's operations.
Regulatory or political change with respect to psychedelic-inspired drugs could occur.
When the Company begins actively researching psychedelic drugs, its success will depend in part, on the legality of the use of psychedelic-inspired drugs for the treatment of neuropsychiatric disorders and the acceptance of such use in the medical community. The political environment surrounding the psychedelics industry in general can be volatile and a shift in the regulatory or political realm could occur and have a drastic impact on the use of psychedelics as a whole, adversely impacting the Company's ability to successfully operate or grow its business. Furthermore, failure to follow applicable regulatory requirements will have a materially negative impact on the business of the Company.
General Corporate Risks
Macroeconomic pressures in the markets in which we operate, including, but not limited to, the lasting effects of the COVID-19 pandemic, political developments, geopolitical unrest or other conflicts or natural disasters in foreign nations, including the ongoing conflict between Russia and Ukraine, political developments in Hong Kong and Taiwan, and inflationary pressures may alter the ways in which we conduct our business operations and manage our financial capacities.
To varying degrees, the ways in which we conduct our business operations and manage our financial capacities are influenced by macroeconomic conditions that affect companies directly involved in or providing services related to the drug and biological product development. For example, real GDP growth, business and investor confidence, the lasting effects of COVID-19 pandemic, inflation, employment levels, oil prices, interest rates, tax rates, availability of consumer and business financing, housing market conditions, foreign currency exchange rate fluctuations, costs for items such as fuel and food and other macroeconomic trends can adversely affect not only our decisions and ability to engage in research and development and clinical trials, but also those of our management, employees, third-party contractors, manufacturers and suppliers, competitors, Shareholders and regulatory authorities. The ongoing military conflict between Russia and Ukraine and other geopolitical and social unrest has created extreme volatility in the global capital markets and is expected to have further global economic consequences, including disruptions of the global supply chain and energy markets. Any such volatility and disruptions may adversely affect our business or the third parties on whom we rely. If the equity and credit markets deteriorate, including as a result of political unrest, natural disasters or war, it may make any necessary debt or equity financing more difficult to obtain in a timely manner or on favorable terms, more costly or more dilutive. Increased inflation rates can adversely affect us by increasing our costs, including labor and employee benefit costs. In addition, higher inflation and macro turmoil and uncertainty could also adversely affect our customers, which could reduce demand for our products.
Economic uncertainty may adversely affect our access to capital, cost of capital and ability to execute our business plan as scheduled.
Generally, worldwide economic conditions remain uncertain. Access to capital markets is critical to our ability to operate. Traditionally, biotechnology companies have funded their research and development expenditures through raising capital in the equity markets. Declines and uncertainties in these markets in the past have severely restricted raising new capital and have affected companies’ ability to continue to expand or fund existing research and development efforts. We require significant capital for research and development for our product candidates and clinical trials. The general economic and capital market conditions, both in the U.S. and worldwide, have been volatile and at times have adversely affected our access to capital and increased the cost of capital. For example, the ongoing military conflict between Russia and Ukraine, the possibility of a wider European or global conflict, global sanctions imposed in response thereto and the possibility of a global energy crisis resulting therefrom, has created extreme volatility in the global capital markets and is expected to have further global economic consequences, including disruptions of the global supply chain and energy markets. Any such volatility and disruptions may adversely affect our business or the third parties on whom we rely. If global capital markets deteriorate, including as a result of political unrest or war, it may make any necessary financing more difficult to obtain in a timely manner or on favorable terms, more costly or more dilutive. If economic conditions become worse, our future cost of equity or debt capital and access to the capital markets could be adversely affected. If we are unable to access the capital markets on favorable terms, our ability to execute our business plan as scheduled would be compromised. Moreover, we rely and intend to rely on third parties, including clinical research organizations, contract manufacturing organizations and other important vendors and consultants. Global economic conditions may result in a disruption or delay in the performance of our third-party contractors and suppliers. If such third parties are unable to adequately satisfy their contractual commitments to us in a timely manner, our business could be adversely affected.
The Company’s limited operating history makes it difficult to evaluate its current business and future prospects and the Company may never be able to generate sufficient revenue to be profitable.
The Company’s limited operating history makes it difficult to evaluate its current business and future prospectus. The Company has never generated any material amount of revenue and has not generated any revenue from its bio-tech business. We have incurred significant losses since our inception, and we anticipate that we will continue to incur significant losses and will not be profitable or generate positive cash flow from operating activities for the foreseeable future. In addition, the Company expects to continue to increase operating expenses as it implements initiatives to continue to grow its business and pursue the commercialization of its Product Candidates. If the Company does not generate sufficient revenue to offset these expected increases in costs and operating expenses, it will not be profitable. The Company cannot predict when it will generate any revenue, or when or if it will become profitable or generate positive cash flow from operating activities, if at all.
In general, the Company is subject to many of the risks common to early-stage enterprises, including undercapitalization, cash shortages, limitations with respect to personnel, financial, and other resources and lack of revenues. There is no assurance that the Company will be successful in achieving a return on the Company’s shareholders’ (“Shareholders”) investment and the likelihood of success must be considered in light of the early stage of our operations.
Future transfers by holders of Class A Shares to arm’s length parties or other than to permitted holders will generally result in those shares converting to Class B Shares, which will have the effect, over time, of increasing the relative voting power of those holders of Class A Shares who retain their shares. Such holders could, in the future, control a significant percentage of the combined voting power of Class A Shares and Class B Shares.
Each of the Company’s directors and officers owes a fiduciary duty to the Company and must act honestly and in good faith with a view to the best interests of Company. However, any director and/or officer that is a Shareholder, even a controlling Shareholder, is entitled to vote its shares in its own interests, which may not always be in the interests of the Shareholders generally. The inability of the Class B Shares to control the matters affecting the Company, combined with the ability of holders of Class A Shares to control matters affecting the Company and to take actions that the holders of Class B Shares may not view as beneficial, may adversely affect the market price of the Class B Shares.
| 22 |
| Table of Contents |
Dilution of the percentage ownership of the Shareholders
Future sales and issuances of the Company’s Class B Shares or rights to purchase Class B Shares, including pursuant to the Company’s equity incentive plans, could result in additional dilution of the percentage ownership of the Shareholders and could cause the Company’s stock price to fall. The Company expects that significant additional capital may be needed in the future to continue its planned operations, including conducting clinical trials, commercialization efforts, expanded research and development activities, potential acquisitions, in licenses, or collaborations and costs associated with operating a public company. To raise capital, the Company may sell Class B Shares, convertible securities, or other equity securities in one or more transactions at prices and in a manner it determines from time to time. If the Company sells Class B Shares, convertible securities, or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to the Shareholders, and new investors could gain rights, preferences, and privileges senior to the holders of our Class B Shares, including Class B Shares sold in this offering upon exercise of Class B Share purchase warrants.
Failure to comply with laws and regulations could have a material adverse effect on the Company's business.
We are subject to complex laws, rules and regulations affecting our domestic and international operations in Canada, the United States and Australia relating to numerous topics, including the research and development of our pharmaceutical drugs, health care and data privacy laws, labor and employment and regulatory requirements of the CSE and Nasdaq. In addition, we are required to comply with certain U.S. Securities Exchange Commission (the “SEC”) and other legal requirements affecting public companies. Compliance with, and monitoring of, applicable laws and regulations may be difficult, time consuming and costly. Those laws and regulations and their interpretation and application may also change from time to time and those changes could have a material adverse effect on our business, investments, and results of operations. In addition, a failure to comply with applicable laws or regulations, as interpreted and applied, could have a material adverse effect on our business, including our ability to negotiate and complete our initial acquisitions, and our future results.
Although, to our knowledge, we are currently in material compliance with all applicable laws, regulations and guidelines in such jurisdictions, no assurance can be given that new laws, regulations, and guidelines will not be enacted or that existing laws, regulations, and guidelines will not be interpreted or applied in a manner which could limit or curtail our operations in such jurisdictions.
On April 17, 2023, FSD Strategic Investments entered into the CEO Mortgage Loan (as defined herein). Although the Company believes the CEO Mortgage Loan complies with the exemption contained in Section 13(k) of the Exchange Act, there can be no assurances that it complies with these regulations. There is limited regulatory guidance or legislative history as to the scope of this exemption. If the CEO Mortgage Loan does not comply, the Company could be subject to fines, penalties and/or other regulatory actions, which would have an adverse effect on our business, financial condition, and results of operations. Furthermore, amendments to current laws, regulations and guidelines, more stringent implementation, or enforcement thereof or other unanticipated events, are beyond our control and could require extensive changes to our operations, which in turn may also result in a material adverse effect on our business, financial condition, and results of operations.
For more information, see “Item 7. Major Shareholders and Related Party Transactions - B. Related Party Transactions.”
Any significant interruption in the supply chain for key inputs could materially impact the Company’s business.
Our business is dependent on a number of key inputs and their related costs including raw materials and supplies, as well as electricity, water, and other local utilities. The ability of the Company to research and develop pharmaceutical products is dependent upon, among other things, sufficient access to timely delivery of equipment, parts, and components at reasonable costs. Any significant interruption or negative change in the availability or economics of the supply chain for key inputs could materially impact our business, financial condition, and operating results. Any inability to secure required supplies and services or to do so on appropriate terms could have a material adverse impact on our business, financial condition, and operating results.
The Company may be unable to raise the capital necessary for it to execute its strategy on favorable terms or at all.
There is no guarantee that the Company will be able to execute on its strategy. Developing Lucid-MS, a biopharmaceutical products, and products for alcohol misuse, is expensive and time-consuming, and we expect to require substantial additional capital to conduct research, preclinical testing and human studies, to potentially establish pilot scale and commercial scale manufacturing processes and facilities, and to establish and develop quality control, regulatory, marketing, sales and administrative capabilities to support our existing programs and pursue potential additional programs. We are or may in the future also be responsible for the payments to third parties of expenses that may include milestone payments, license maintenance fees and royalties, including in the case of certain of our agreements with academic institutions or other companies from whom intellectual property rights underlying their respective programs have been licensed or acquired. Because the outcome of any preclinical or clinical development and regulatory approval process for Lucid-MS and other product candidates that we may develop in the future is highly uncertain, we cannot reasonably estimate the actual amounts necessary to successfully complete the development, regulatory approval process and commercialization of any product candidates we may identify.
| 23 |
| Table of Contents |
Our future funding requirements for the development of pharmaceutical products will depend on many factors, including, but not limited to: (i) the time and cost necessary to complete planned clinical trials to pursue regulatory approvals for our Lucid-MS and any other drug candidates, and to conduct post-marketing studies that could be required by regulatory authorities; (ii) the progress, timing, scope and costs of our nonclinical studies, preclinical studies, clinical trials and other related activities, including the ability to enroll patients in a timely manner for planned clinical trials described in this Annual Report and potential future clinical trials; (iii) the costs of obtaining clinical and commercial supplies of raw materials and drug products for Lucid-MS and other product candidates; (iv) our ability to successfully identify and negotiate acceptable terms for third-party supply and contract manufacturing agreements with CMO’s; (v) our ability to successfully commercialize our Product Candidates, either directly or through licensing agreements, ;(vi) the manufacturing, selling and marketing costs associated with our Product Candidates, including the cost and timing of expanding our internal sales and marketing capabilities or entering into strategic collaborations with third parties to leverage or access these capabilities; (vii) the amount and timing of sales and other revenues from our Product Candidates, if any are approved, including the sales price and the availability of adequate third-party reimbursement; (viii) the cash requirements of any future acquisitions or discovery of product candidates; (ix) the time and cost necessary to respond to technological, market, regulatory or political developments; (x) the costs of acquiring, licensing or investing in intellectual property rights (including the protection of such rights), products, product candidates and businesses; and (xi) our ability to attract, hire and retain qualified personnel.
Additional funds may not be available when we need them, on terms that are acceptable, or at all. If adequate funds are not available to us on a timely basis, we may be required to delay, limit, or terminate one or more research or development programs or the commercialization of any Product Candidates or be unable to expand operations or otherwise capitalize on business opportunities, as desired, which could materially affect our business, results of operations, financial condition, and prospects.
In addition, the continued development of the Company’s pharmaceutical operations will require significant additional financing over several years. The failure to raise such capital could result in the delay or indefinite postponement of current business strategy or the Company ceasing to carry on business. There can be no assurance that additional capital or other types of financing will be available if needed or that, if available, the terms of such financing will be favorable to the Company, at times for reasons beyond the Company’s control. For example, economic downturns or uncertain market conditions, whether affecting the economy in general or the pharmaceutical industry in particular, could adversely impact the Company’s ability to raise capital through equity or debt financing. In addition, any further issuances of equity securities could have a significant dilutive effect on the holders of Class B Shares.
In addition, from time to time, the Company may enter into transactions to acquire assets or the shares of other companies. These transactions may be financed wholly or partially with debt, which may temporarily increase the Company’s debt levels above industry standards. Any debt financing secured in the future could involve restrictive covenants relating to capital raising activities and other financial and operational matters, which may make it more difficult for the Company to obtain additional capital and to pursue business opportunities, including potential acquisitions.
Future sales or issuances of equity securities and the conversion of outstanding securities to Class B Shares could decrease the value of the Class B Shares and dilute investors’ voting power.
The Company may sell additional equity securities in future offerings, including through the sale of securities convertible into equity securities, to finance operations, acquisitions or projects, and issue additional Class B Shares, which may result in dilution.
The Board has the authority to authorize certain offers and sales of additional securities without the vote of, or prior notice to, shareholders. Based on the need for additional capital to fund expected expenditures and growth, it is likely that the Company will issue additional securities to provide such capital. Such additional issuances may involve the issuance of a significant number of Class B Shares.
Sales of substantial amounts of the Company’s securities, or the availability of such securities for sale, as well as the issuance of substantial amounts of the Class B Shares upon conversion of outstanding convertible, exercisable or exchangeable securities, could adversely affect the prevailing market prices for the Company’s securities and dilute investors’ earnings per share. A decline in the market prices of the Company’s securities could impair its ability to raise additional capital through the sale of securities should the Company desire to do so.
The success of the Company is dependent upon its senior management and key personnel and ability to hire skilled personnel.
Another risk associated with the production and sale of pharmaceutical products is the loss of important personnel. The success of the Company will be dependent upon the ability, expertise, judgment, discretion and good faith of its senior management and key personnel. While employment agreements are customarily used as a primary method of retaining the services of key employees, these agreements cannot assure the continued services of such employees. While, as of the date of this Annual Report, the Company does not anticipate any senior management turnover in the near term, there is no guarantee that the Company will be able to retain its senior management going forward. If key personnel depart, including Zeeshan Saeed, Anthony Durkacz, Nathan Coyle, Dr. Lakshmi Kotra or Donal Carroll, the Company may not be able to find appropriate replacements on a timely basis.
Furthermore, each of our executive officers may terminate their employment with us at any time. We do not maintain “key person” insurance for any of our executives or employees. Recruiting and retaining qualified scientific and clinical personnel and, if any of our Product Candidates are commercialized, sales and marketing personnel, will be critical to our success. The loss of the services of key personnel as well as the diversion of management’s and the Board’s attention to replace the services of such individuals, could have a material adverse effect on the Company’s business, operating results, or financial condition.
| 24 |
| Table of Contents |
In addition, the Company’s future success depends on its continuing ability to attract, develop, motivate, and retain highly qualified and skilled employees. Due to the specialized scientific and managerial nature of our business, the Company relies heavily on its ability to attract and retain qualified scientific, technical, and managerial personnel. In particular, specialized knowledge with respect to research and clinical development is important to the pharmaceutical industry. Qualified individuals are in high demand, and the Company may incur significant costs to attract and retain them, if it is able to hire them at all. If we are unable to identify, attract, hire, and retain qualified personnel in the future, such inability could have a material adverse effect on our business, operating results, and financial condition.
The Company’s dual class structure has the effect of concentrating voting control and the ability to influence corporate matters with a limited number of holders of Class A Shares.
The Company’s dual class structure has the effect of concentrating voting control for holders of Class A Shares and the ability to influence corporate matters with those Shareholders. Currently, there are 72 outstanding Class A Shares issued and outstanding. Class A Shares have 276,660 votes per share and Class B Shares have one vote per share. As of March 28, 2024, Shareholders who hold Class A Shares together hold approximately 33.6% of the voting power of the Company’s outstanding voting shares and therefore have significant influence over management and affairs of the Company and over all matters requiring Shareholder approval.
In addition, because of the voting ratio between Class A Shares and Class B Shares, the holders of Class A Shares collectively continue to control a majority of the combined voting power of the voting shares even where the Class A Shares represent a substantially reduced percentage of the total outstanding shares. The different voting rights could diminish the value of the Class B Shares to the extent that investors or any potential future purchasers of the Class B Shares attribute value to the superior voting or other rights of the Class A Shares. Other than as required by applicable law, holders of the Class B Shares will only have a right to vote, as a class, in limited circumstances as described in its constating documents.
The concentrated voting control of holders of Class A Shares limits the ability of holders of Class B Shares to influence corporate matters and all matters requiring Shareholder approval, including the election of directors as well as with respect to decisions regarding amendment of the Company’s share capital, creating and issuing additional classes of shares, making significant acquisitions, selling significant assets or parts of our business, merging with other companies and undertaking other significant transactions.
As a result, holders of Class A Shares have the ability to control substantially all matters affecting us and actions may be taken that our holders of Class B Shares may not view as beneficial. The market price of the Class B Shares could be adversely affected due to the significant influence and voting power of the holders of Class A Shares. Additionally, the significant voting interest of holders of Class A Shares may discourage transactions involving a change of control, including transactions in which an investor, as a holder of the Class B Shares, might otherwise receive a premium for the Class B Shares over the then-current market price, or discourage competing proposals if a going private transaction is proposed by one or more holders of Class A Shares.
The market price of the Class B Shares may be subject to wide price fluctuations.
The market price of the Class B Shares may be subject to wide fluctuations in response to many factors, including variations in the operating results of the Company and its subsidiaries, divergence in financial results from analysts’ expectations, changes in earnings estimates by stock market analysts, changes in the business prospects for the Company, general economic conditions, legislative changes, and other events and factors outside of the Company’s control. In addition, stock markets have from time to time experienced extreme price and volume fluctuations, which, as well as general economic and political conditions, could adversely affect the market price for the Class B Shares.
There is no assurance of an active or liquid market.
No assurance can be given that an active or liquid trading market for the Class B Shares will be sustained. If an active or liquid market for the Class B Shares fails to be sustained, the prices at which such securities trade may be adversely affected. Whether or not the Class B Shares will trade at lower prices depends on many factors, including the liquidity of the Class B Shares, prevailing interest rates, the markets for similar securities, general economic conditions and the Company’s financial condition, historic financial and operating performance, and future prospects.
The Company may be unable to manage its growth, including capacity constraints and pressure on its internal systems and controls.
The Company may be subject to growth-related risks including capacity constraints and pressure on its internal systems and controls. The ability of the Company to manage growth effectively will require it to continue to implement and improve its operational and financial systems and to expand, train and manage its employee base. The inability of the Company to deal with this growth may have a material adverse effect on the Company’s business, results of operations, financial condition and prospects.
Management may not be able to successfully implement and maintain adequate internal controls over financial reporting or disclosure controls and procedures.
Effective internal controls are necessary for the Company to provide reliable financial reports and to help prevent fraud. Although the Company has undertaken a number of procedures and has implemented a number of safeguards, in each case, in order to help ensure the reliability of its financial reports, including those imposed on the Company under applicable securities laws, the Company cannot be certain that such measures will ensure that the Company will maintain adequate control over financial processes and reporting. Failure to implement required new or improved controls, or difficulties encountered in their implementation, could harm the Company’s results of operations, or cause it to fail to meet its reporting obligations.
| 25 |
| Table of Contents |
Effective systems of internal control over financial reporting and disclosure are critical to the operation of a public corporation. However, we do not expect that our disclosure controls and procedures or internal control over financial reporting will prevent all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Further, the design of a control system must reflect the fact that there are resource constraints and the benefits of such controls must be considered relative to their costs. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that all control issues and instances of fraud, if any, have been detected. Due to the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and may not be detected in a timely manner or at all. If we cannot provide reliable financial reports or prevent fraud, our reputation and operating results could be materially adversely affected, which could cause investors to lose confidence in us and our reported financial information, which in turn could result in a reduction in the value of the Class B Shares.
A decline in general economic conditions may impact the viability and success of our mortgage investment activities.
FSD Strategic Investments has made and intends on continuing to make investments in loans that are secured by first or second collateral mortgages on residential real estate in the Greater Toronto Area. A decline in general economic conditions could adversely impact the ability of borrowers to service their loans and could cause default rates to increase. This could have a material adverse effect on FSD Strategic Investments’ financial condition and results of operations.
A decline in property values could adversely affect the value of the security on mortgages held by FSD Strategic Investments, thereby reducing the ability to liquidate properties held by defaulting borrowers at favorable prices.
The profits earned on mortgages depend, in part, on the spread between mortgage rates and capital market funding rates and any fee income derived therefrom. FSD Strategic Investments’ mortgage portfolios include assets whose value can fluctuate because of changing interest rates and economic and market conditions. In addition, some of these assets could be difficult to sell at any given time. Changes in interest rates and other market factors such as stock market prices and demographics could affect the preferences of its customers for different types of loan products and adversely impact our profitability. A reduction in positive spreads between mortgage rates and capital market funding rates could have a material adverse effect on FSD Strategic Investments’ financial condition and results of operations.
Investments in mortgages are relatively non-liquid assets. The nature of the assets held by FSD Strategic Investments may inhibit its ability to quickly respond to changes in broader economic or investment conditions. If the value of the properties underlying FSD Strategic Investments’ mortgages begin to deteriorate, it will be difficult for FSD Strategic Investments to liquidate certain assets in response to these changes. The liquidity profile of FSD Strategic Investments’ mortgages can create challenges for it to manage its risk exposure. Reduced asset liquidity may restrict FSD Strategic Investments’ ability to sell assets for cash without taking significant losses, which may result in a material adverse effect on FSD Strategic Investments’ financial condition and results of operations.
Risks related to our status as a foreign private issuer.
As a “foreign private issuer” under the rules and regulations of the SEC, we are permitted to, and will, file less or different information with the SEC than a company incorporated in the United States or otherwise subject to these rules, and will follow certain home country corporate governance practices in lieu of certain Nasdaq requirements applicable to U.S. issuers.
The Company is considered a “foreign private issuer” under the Exchange Act and is therefore exempt from certain rules under the Exchange Act. For example, we are not required to file current reports on Form 8-K or quarterly reports on Form 10-Q, we are exempt from the U.S. proxy rules which impose certain disclosure and procedural requirements for U.S. proxy solicitations and we will not be required to file financial statements prepared in accordance with or reconciled to U.S. GAAP so long as our financial statements are prepared in accordance with IFRS as issued by the International Accounting Standards Board. We are not required to comply with Regulation FD, which imposes restrictions on the selective disclosure of material information to shareholders, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions of Section 16 of the Exchange Act. In addition, we are not required to file periodic reports and financial statements with the SEC as frequently or within the same time frames as U.S. companies with securities registered under the Exchange Act. Accordingly, holders of the Company’s securities may receive less or different information about the Company than they may receive with respect to public companies incorporated in the United States.
In addition, as a “foreign private issuer” whose common shares are listed on Nasdaq, we are permitted to follow certain home country corporate governance practices in lieu of certain Nasdaq requirements, including those related to: shareholder approval for certain dilutive events under Nasdaq Marketplace Rule 5635, quorum requirements for shareholder meetings under Nasdaq Marketplace Rule 5620(c), certain independence requirements of certain committees of our Board under Nasdaq Marketplace Rule 5605 and proxy delivery requirements under Nasdaq Marketplace Rule 5620(b). Accordingly, the Company has opted to follow certain corporate governance practices required by its home country under the CSE, Canadian federal and provincial corporate and securities laws and the Company’s Articles, as applicable. See “Item 16G. Corporate Governance” for more details related to the differences between our home country requirements and Nasdaq requirements.
We could lose our status as a “foreign private issuer” under current SEC rules and regulations if more than 50% of our outstanding voting securities become directly or indirectly held of record by U.S. holders and one of the following is true: (i) the majority of our directors or executive officers are U.S. citizens or residents; (ii) more than 50% of our assets are located in the United States; or (iii) our business is administered principally in the United States. If we lose our status as a foreign private issuer in the future, we will no longer be exempt from the rules described above and, among other things, will be required to file periodic reports and annual and quarterly financial statements as if we were a company incorporated in the United States (including preparation of financial statements in accordance with U.S. GAAP). If this were to happen, we would likely incur substantial costs in fulfilling these additional regulatory requirements and members of our management would likely have to divert time and resources from other responsibilities to ensuring these additional regulatory requirements are fulfilled.
| 26 |
| Table of Contents |
There can be no assurance that we will be able to comply with the continued listing standards of Nasdaq and/or CSE.
Our Class B Shares are listed on Nasdaq and CSE. There can be no assurance that we will continue to meet Nasdaq and/or CSE’s listing standards. On September 27, 2022, we received a letter from the listing qualifications department staff of Nasdaq notifying us that the Company is not in compliance with the minimum bid price requirement set forth in Nasdaq’s rules for continued listing on the Nasdaq Capital Market. While we have since regained compliance with Nasdaq’s minimum bid price requirement, there can be no guarantee that we will be able to maintain such compliance in the future. If we lose our ability to maintain compliance with Nasdaq and/or the CSE’s continued listing rules, we and our Shareholders could face significant material adverse consequences, including:
| · | a limited availability of market quotations for our securities; |
| · | reduced liquidity for our securities; |
| · | a determination that our Class B Shares is a “penny stock,” in the U.S. which will require brokers trading in our Class B Shares to adhere to more stringent rules and possibly result in a reduced level of trading activity in the secondary trading market for our securities; |
| · | a limited amount of news and analyst coverage; and |
| · | decreased ability to issue additional securities or obtain additional financing in the future. |
As an “emerging growth company,” the Company cannot be certain if the reduced disclosure and governance requirements applicable to “emerging growth companies” will make its shares less attractive to investors.
As an “emerging growth company,” the Company may take advantage of certain exemptions from various reporting requirements that are applicable to other public companies, including not being required to obtain an assessment of the effectiveness of its internal controls over financial reporting from its independent registered public accounting firm pursuant to Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in its periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. In addition, the U.S. Jumpstart Our Business Startups Act (the “JOBS Act”) provides that an emerging growth company can take advantage of an extended transition period for complying with new or revised accounting standards, which the Company has elected to do.
We cannot predict if investors will find our shares less attractive because we will rely on these exemptions. If some investors find our shares less attractive as a result, there may be a less active market for our shares, our share price may be more volatile and the price at which our securities trade could be less than if we did not use these exemptions.
We expect to incur costs related to our internal control over financial reporting in the upcoming years to further improve our internal control environment. If we identify deficiencies in our internal controls over financial reporting or if we are unable to comply with the requirements applicable to us as a public company, including the requirements of Section 404 of the Sarbanes-Oxley Act, in a timely manner, we may be unable to accurately report our financial results, or report them within the timeframes required by the SEC. If this occurs, we also could become subject to sanctions or investigations by the SEC or other regulatory authorities. In addition, if we are unable to assert that our internal controls over financial reporting are effective, or if our independent registered public accounting firm is unable to express an opinion as to the effectiveness of our internal control over financial reporting, or express an adverse opinion, investors may lose confidence in the accuracy and completeness of our financial reports, we may face restricted access to the capital markets and our share price may be adversely affected.
We may not be able to successfully identify and execute future acquisitions or dispositions or to successfully manage the impacts of such transactions on our operations.
The Company has made and may continue to pursue acquisition opportunities to advance its strategic plan. The successful integration of an acquired business typically requires the management of the pre-acquisition business strategy, including the retention and addition of senior management, customers, realization of identified synergies, retention of key staff and the development of a common corporate culture. Achieving the benefits of acquisitions depends in part on successfully consolidating functions and integrating operations and procedures in a timely and efficient manner, as well as the ability to realize anticipated growth opportunities and synergies from newly formed partnerships. Any failure to integrate an acquired business or realize the anticipated benefits of new partnerships may have a material adverse effect on the Company’s business, results of operations, financial condition, and prospects, including its future prospects for acquisitions or partnerships. There is no assurance that the Company will be able to successfully integrate an acquired business in order to maximize or realize the benefits associated with an acquisition.
In addition, from time to time the Company enters into letters of intent and memoranda of understanding with respect to which definitive agreements have not yet been, but are expected to be, executed. The Company may not be able to perform under these contracts as a result of operational or other breaches or due to events beyond its control, and the Company may not be able to ultimately execute a definitive agreement in cases where one does not currently exist.
| 27 |
| Table of Contents |
Any expansion of our international operations will result in increased operational, regulatory, and other risks.
We established an Australian subsidiary in November 2022 and may in the future expand into other geographic areas, which could increase our operational, regulatory, compliance, reputational and foreign exchange rate risks. The failure of our operating infrastructure to support such expansion could result in operational failures and regulatory fines or sanctions.
The Company is currently party to several legal proceedings and may become a party to potential future litigation.
The Company is currently party to a number of proceedings; see “Item 8. Financial Information - A. Consolidated Statements and Other Financial Information – Legal Proceedings”. Such litigation could be costly and time-consuming and could divert the attention of management and other key personnel from the Company’s business and operations. The complexity of any such claims and the inherent uncertainty of commercial, employment and other litigation increases these risks. In recognition of these considerations, the Company could suffer significant litigation expenses in defending any of these claims and may enter into settlement agreements.
The Company may also become party to additional litigation in the future, including class action lawsuits, securities litigation and anti-trust and anti-competitive actions, which could adversely affect its business. Should any litigation in which the Company becomes involved be determined against the Company, such a decision could adversely affect the Company’s ability to continue operating and the market price for Company’s Class B Shares and could result in the use of significant resources. Even if the Company is involved in litigation and wins, litigation can redirect significant corporate resources and management attention.
Conflicts of interest may arise between the Company and its directors and officers as a result of other business activities undertaken by such individuals.
Certain directors and officers of the Company are, and may in the future become, directors and officers of other entities, or are otherwise engaged, and will continue to be engaged, in activities that may put them in conflict with the business strategy of the Company. In particular, certain directors and officers of the Company serve as directors or officers of entities that may compete with or have conflicting interests with the Company.
The Company’s directors and the officers are required to act honestly and in good faith with a view to its best interests. However, in conflict of interest situations, the Company’s directors and officers may owe the same duty to another corporation and will need to balance their competing interests with their duties to the Company. Circumstances (including with respect to future corporate opportunities) may arise that may be resolved in a manner that is unfavorable to the Company. These business interests could require the investment of significant time and attention by our executive officers and directors. In some cases, our executive officers and directors may have fiduciary obligations associated with business interests that interfere with their ability to devote time to our business and affairs, which could adversely affect our operations.
The Company does not anticipate paying dividends in the near future.
Effective November 29, 2023, the Corporation completed the Plan of Arrangement, which included the distribution of the Celly Nu Shares to the FSD Pharma Securityholders. For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Significant Developments in Fiscal 2023 through to March 28, 2024”.
The Company does not anticipate paying cash or stock dividends in the near future. The Company expects to retain earnings to finance the development and enhancement of its Product Candidates and to otherwise reinvest in the Company’s business. Any decision to declare and pay dividends in the future will be made at the discretion of the Board and will depend on, among other things, financial results, cash requirements, contractual restrictions, and other factors that the Board may deem relevant. As a result, investors may not receive any return on their investment in Class B Shares unless they sell them for a share price that is greater than that at which such investors purchased them.
The Company’s operations depend, in part, on the maintenance and protection of its information technology systems and the information technology systems of its third-party research institution collaborators, CROs or other contractors or consultants, which could face cyber-attacks that cause material losses to our business.
We have entered into agreements with third parties for hardware, software, telecommunications, and other information technology (“IT”) services in connection with our operations. Our operations depend, in part, on how well we, our CROs, other contractors, consultants and our suppliers protect networks, equipment, IT systems and software against damage from a number of threats, including, but not limited to, cable cuts, damage to physical plants, natural disasters, terrorism, fire, power loss, hacking, computer viruses, vandalism and theft. Our operations also depend on the timely maintenance, upgrade and replacement of networks, equipment, IT systems and software, as well as pre-emptive expenses to mitigate the risks of failures. Any of these and other events could result in information system failures, delays and/or increase in capital expenses. The failure of information systems or a component of information systems could, depending on the nature of any such failure, adversely impact our reputation and results of operations.
For example, the loss of, or damage to, clinical trial data from completed, ongoing or future preclinical or clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Likewise, we rely or expect to rely on third parties for research and development, the manufacture and supply of drug product and to conduct clinical trials, and similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or systems, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of our Product Candidates could be delayed.
| 28 |
| Table of Contents |
Certain data breaches must also be reported to affected individuals and certain regulatory bodies, and in some cases may be required to be publicly disclosed under U.S. federal and state law, federal and provincial data protection legislation in Canada and the requirements of other jurisdictions, and financial or other penalties may also apply.
Cyber incidents can result from deliberate attacks or unintentional events. Cyber-attacks could result in any person gaining unauthorized access to digital systems for purposes of misappropriating assets or sensitive information, including personally identifiable information, corrupting data, or causing operational disruption. Cyber-attacks could also result in important remediation costs, increased cyber security costs, lost revenues due to a disruption of activities, litigation and reputational harm affecting customer and investor confidence, which could materially adversely affect our business and financial results.
We have not experienced any material losses to date relating to cyber-attacks or other information security breaches, but there can be no assurance that we will not incur such losses in the future, which could be in excess of any available insurance and could materially adversely affect our business and financial results. Our risk and exposure to these matters cannot be fully mitigated because of, among other things, the evolving nature of these threats. As a result, cyber security and the continued development and enhancement of controls, processes and practices designed to protect systems, computers, software, data and networks from attack, damage or unauthorized access is a priority. As cyber threats continue to evolve, we may be required to expend additional resources to continue to modify or enhance protective measures or to investigate and remediate any security vulnerabilities.
We may be a passive foreign investment company, which may result in adverse U.S. federal income tax consequences for holders of our Class B Shares who are U.S. taxpayers.
Generally, if for any taxable year 75% or more of our gross income is passive income, or 50% or more of the average quarterly value of our assets are held for the production of, or produce, passive income, we would be characterized as a passive foreign investment company, or “passive foreign investment company” (“PFIC”), for U.S. federal income tax purposes. The determination of whether we are a PFIC is a fact-intensive determination made on an annual basis applying principles and methodologies that in some circumstances are unclear and subject to varying interpretation, and the Company’s PFIC status will depend among other things upon changes in the composition and relative value of its gross receipts and assets. We believe that we were a PFIC for the year ended December 31, 2023. In addition, although PFIC status is determined on an annual basis and generally cannot be determined until the end of the taxable year, we believe that we may be considered a PFIC for the current taxable year. Because we may continue to hold a substantial amount of cash and cash equivalents, and because the market value of the Company’s assets (including for this purpose goodwill) may be measured in large part by the market price of our shares, which is likely to fluctuate, no assurance can be given that the Company will not also be a PFIC in any future taxable year. If we are characterized as a PFIC, our shareholders who are U.S. taxpayers may suffer adverse tax consequences, including the treatment of gains realized on the sale of our Class B Shares as ordinary income, rather than as capital gain, the loss of the preferential rate applicable to dividends received on our Class B Shares by individuals who are U.S. taxpayers, and the addition of interest charges to the tax on such gains and certain distributions. For more information, please see “Item 10. Additional Information - E. Taxation - Certain Material U.S. Federal Income Tax Considerations”.
Item 4. Information on the Company.
A. History and Development of the Company
Overview and History
We were incorporated in 1998 under the OBCA under the name of Century Financial Group, Inc. On May 24, 2018, pursuant to Articles of Amendment, the Company changed its name to “FSD Pharma Inc.” From May 2018 to March 2020, the focus of the Company’s business was the cultivation, processing and sale of medical cannabis; in March 2020, however, the Company pivoted its focus to pharmaceuticals and biotechnology.
The Company is building a portfolio of innovative assets and biotech solutions for the treatment of challenging neurodegenerative and metabolic disorders and alcohol misuse disorders with drug candidates (“Product Candidates”) in different stages of development. We are currently focused on the research and development of our lead compound, Lucid-MS, a patented new chemical entity shown to prevent and reverse myelin degradation, the underlying mechanism of multiple sclerosis, in preclinical models. The Company is also focused on the research and development of novel formulations for the treatment for alcohol misuse. In addition, the Company maintains a portfolio of strategic investments through its wholly owned subsidiary, FSD Strategic Investments, which represent loans secured by residential properties.
Alcohol Misuse Disorder Product Candidates
With respect to the Product Candidates for alcohol misuse disorders, the Corporation sees two distinct routes where this segment can be developed, (i) recreational retail and (ii) healthcare; Celly Nu, through the Celly Nu IP License Agreement will be focusing on the recreational retail sector and the Corporation will be focusing on the healthcare sector as further outlined below:
(i) Alcohol Misuse: Retail Product (known as “Unbuzzd™”)
| 29 |
| Table of Contents |
A consumer recreational beverage product that will be sold via retail distribution. On July 31, 2023, the Corporation entered into a definitive exclusive intellectual property license agreement (the “Celly Nu IP License Agreement”) with Celly Nu and Lucid, which granted Celly Nu the exclusive rights to the recreational applications for the Corporation’s alcohol misuse technology for rapid alcohol detoxification, and the rights to the trademarks, Unbuzzd™ and ALCOHOLDEATH™, in exchange for securities in Celly Nu and a royalty on any product sales, so that Celly Nu could fund research and development, market and sell Unbuzzd™. As part of the Celly Nu IP License Agreement, the Corporation loaned Celly Nu C$1,000,000 on a secured basis with a term of 3 years, which bears interest at a rate of 10% per annum, payable on each anniversary, to assist Celly Nu in achieving that goal; however, the Corporation does not have an obligation to fund Celly Nu in the future (the “Celly Nu Loan Agreement”). The Celly Nu loan was secured by all of Celly Nu’s collateral (the “Celly Nu Security Agreement”).
Pursuant to the Celly Nu IP License Agreement, the Corporation will receive a 7% royalty on revenue from Celly Nu, until total royalties in the amount of C$250,000,000 have been paid to the Corporation, at which point the royalty rate is reduced to 3%. In addition, Celly Nu issued the Corporation 100,000,000 Celly Nu Shares (as defined below) as a licence fee and issued the Corporation an anti-dilution warrant, entitling the Corporation to exercise the warrant at any time, in whole or in part, for a period of five years from the date of issuance to increase their holdings in Celly Nu to 25% for nominal consideration. Upon completion of the transaction with Celly Nu, the Corporation held approximately 34.66% of the issued and outstanding Celly Nu Shares on a non-diluted basis.
The Corporation will retain all rights to medical and pharmaceutical applications under its umbrella to further develop the franchise as part of its portfolio.
Effective November 29, 2023, the Corporation completed the Plan of Arrangement (as defined herein). Upon completion of the Plan of Arrangement, the Corporation continues to hold 154,287,471 Celly Nu Shares, which represents approximately 26.15% of the issued and outstanding Celly Nu Shares on a non-diluted basis. For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Significant Developments in Fiscal 2023 through to March 28, 2024”.
The Plan of Arrangement has not had, and does not expect to have, any impact on the development of the retail product, Unbuzzd™.
The Corporation’s continued operations are not dependent on the development of Unbuzzd™.
(ii) Alcohol Misuse: Healthcare Product (the “Healthcare Product”)
The Healthcare Product has the potential to assist emergency room physicians and their medical staff with the abundance of intoxicated patients they receive as these patients are utilizing critical resources (i.e. the physicians and their medical staff) whose time can be used for more urgent and critical needs. The Corporation did not license the intellectual property with respect to the Product Candidate for the Healthcare Product to Celly Nu and will be conducting further research and development, including clinical trials, into the viability of the Healthcare Product. Although any research and development conducted by the Corporation on the Healthcare Product could be shared with, and may assist, Celly Nu in developing Unbuzzd™, the Corporation has no obligation, pursuant to the Celly Nu IP License Agreement, to share such information with Celly Nu.
The viability, development and advancement of the Healthcare Product is dependent on the Corporation obtaining requisite funding, in the amount of approximately US$10,998,811, for the Corporation to complete further research and development. The Corporation, through its initial research, has discovered that there is significant demand in the market for this type of product, an opportunity for them to capture market share and believes that if it were able to develop and sell the Healthcare Product, it would bring immense value to its shareholders. If the requisition financing is not obtained, the Corporation will be unable to develop the Healthcare Product.
The Corporation’s continued operations are not dependent on the Healthcare Product’s development.
Lucid-MS
Through Lucid, the Corporation is also currently focused on the research and development of its Lucid-MS compound. Lucid-MS is a patented new chemical entity shown to prevent and reverse myelin degradation, the underlying mechanism of MS, in preclinical models. On April 17, 2023, the Corporation announced the completion of its first-in-human dosing of Lucid-MS in the Corporation’s Phase 1 clinical trial. On May 10, 2023, the Corporation announced the completion of dosing for the first cohort of patients in the Phase 1 clinical trial of Lucid-MS.
FSD-PEA
On June 2, 2023, the Corporation terminated any further clinical development of its proprietary ultra-micronized palmitoylethanolamide (“FSD-PEA”) (also known as “FSD201”) formulation which was being developed for the treatment of inflammatory diseases. The Corporation’s team of internal medical experts conducted a profitability assessment of FSD-PEA and ultimately determined that the FSD-PEA molecule was not profitable compared against the currently available products in the market and it would not be possible to cover the Corporation’s manufacturing and research and development investments at a price that would be accepted in the market.
| 30 |
| Table of Contents |
Lucid-PSYCH
Additionally, management made the decision to put the research and development activities associated with Lucid-PSYCH (formerly Lucid-201) on hold during June 2023. This decision was made based on the cumulative cash requirements to advance the research and development of the Corporation’s portfolio of compounds. Due to cash flow prioritization strategies, management elected to prioritize the Lucid-MS compound and its alcohol misuse treatment products. The Corporation has not recognized an amount specific to Lucid-PSYCH. When the Corporation acquired Lucid, it recognized an intangible asset consisting of the world-wide exclusive license agreement with the University Health Network (the “UHN License”) for the exclusive rights to the novel Lucid-MS compound and the U.S. patent for the Lucid-MS compound covered by the UHN license. Lucid-PSYCH was not covered by the license agreement and did not have any patent protection.
Prismic
The Corporation does not operate through Prismic, however Prismic holds the right to receive certain payments based on net sales of certain products from the Corporation pursuant to an assignment agreement between Prismic and the Corporation.
FSD Strategic Investments
Through FSD Strategic Investments, the Corporation is involved in the issuance of loans secured by residential or commercial property.
The Company’s Class B Shares trades on the CSE and Nasdaq under the symbol “HUGE.”
The Company’s principal office is located at 199 Bay Street, Suite 4000, Toronto, Ontario M5L 1A9 and its telephone number is 416-854-8884. As at the date of this Annual Report, the Company is a reporting issuer in each of the provinces of Canada. The Company’s registrar and transfer agent is Marrelli Trust Company Limited. The Company’s agent for service in the United States is CT Company, 28 Liberty Street, New York, New York 10005.
For a description of our principal capital expenditures, principal acquisitions and divestitures for the three years ended December 31, 2023 and for those currently in progress, see. “Item 4. Information on the Company - A. History and Development of the Company” and “Item 4. Information on the Company - B. Business Overview” and “Item 5. - Operating and Financial Review and Prospects”.
The SEC maintains an internet site that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC, which can be found at http://www.sec.gov. Our internet address is www.fsdpharma.com. The information contained on our website is not incorporated by reference and does not form part of this Annual Report.
Significant Developments in Fiscal 2023 through to March 28, 2024
2023 Normal Course Issuer Bid
On January 12, 2023, the Board authorized a normal course issuer bid pursuant to which the Company was able to repurchase for cancellation up to 1,925,210 Class B Shares, being approximately 5% of the Company’s issued and outstanding Class B Shares as of January 12, 2023, over a 12-month period (the “2023 NCIB”). The 2023 NCIB commenced on January 18, 2023 and was terminated on January 12, 2024. Under the 2023 NCIB, the Company repurchased for cancellation 1,904,700 Class B Shares at an average price of approximately C$2.11 per Class B Shares. All Class B Shares were repurchased through the facilities of the CSE at the prevailing market price on the CSE at the time of repurchase.
Issuance of Warrants
On February 13, 2023, the Company issued warrants to purchase 500,000 Class B Shares to Jason Gold and warrants to purchase 300,000 Class B Shares to Pillow Hog Ventures Inc. in exchange for consulting services provided to the Company. The warrants vested on issuance and expire on March 30, 2024, with an exercise price ranging from US$1.50 to US$4.50.
On February 13, 2023, the Company issued warrants to purchase 500,000 Class B Shares to Zapability LLC in exchange for consulting services provided to the Company. Each tranche of warrants expires 12 months from the first day it vested, with the final tranche expiring on February 15, 2026. The warrants have an exercise price ranging from US$1.85 to US$8.00.
On February 27, 2023, the Company issued warrants to purchase 1,000,000 Class B Shares to Kevin Harrington in exchange for consulting services provided to the Company. The warrants vested on issuance and expire on February 27, 2026, with an exercise price ranging from US$1.75 USD to US$8.00.
| 31 |
| Table of Contents |
On March 24, 2023, the Company issued warrants to purchase 1,000,000 Class B Shares to Gerard David in exchange for consulting services provided to the Company. The Warrants on issuance and expire on March 24, 2026, with an exercise price ranging from US$1.75 to US$8.00.
Discontinuation of Research on Lucid-PSYCH and PEA
On June 2, 2023, the Corporation terminated any further clinical development of FSD-PEA
Interest-Bearing Mortgage Loan to CEO
On April 17, 2023, FSD Strategic Investments entered into a secured loan agreement with the CEO for C$1,200,000, with monthly payments of C$6,000 based on an annual interest rate of 6% and a blended rate of 7% (the “CEO Mortgage Loan”). The business purpose of the CEO Mortgage Loan was a treasury function to earn a rate of return on excess capital held. For more information, see “Item 7. Major Shareholders and Related Party Transactions - B. Related Party Transactions.”
Change in Board and Management
On July 4, 2023, the Company announced the appointment of Mr. Zeeshan Saeed as CEO of the Company, to succeed Mr. Anthony Durkacz, who served as interim CEO of the Company since July 2021.
At the annual general and special meeting of the Shareholders held on June 29, 2023, Messrs. Michael Zapolin and Dr. Eric Hoskins were elected as directors of the Company.
On January 24, 2024, the Company appointed Dr. Sanjiv Chopra, MD to the Board to replace Nitin Kaushal.
Celly Nu IP License Agreement
On July 31, 2023, the Company entered into the Celly Nu IP License Agreement, Celly Nu Loan Agreement and Celly Nu Licensing Agreement. For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Overview and History”.
Plan of Arrangement
On April 11, 2023, the Corporation announced its intention to complete a spin-out transaction via a statutory plan of arrangement and to hold a regarding the same at its upcoming meeting of shareholders. The Corporation ultimately made the decision to defer the spin-out transaction and did not ask its shareholders to approve the transaction at its annual general and special meeting of shareholders held on June 29, 2023.
On October 5, 2023, the Corporation announced that it had entered into a definitive arrangement agreement with Celly Nu dated October 4, 2023 (the “Arrangement Agreement”) with respect to the distribution of a portion of the Corporation’s shareholdings of Celly Nu to the FSD Pharma Securityholders (as defined herein).
Pursuant to the Arrangement Agreement, the Corporation had FSD Pharma Securityholders pass a special resolution at the special meeting of shareholders held on November 20, 2023 to approve a statutory plan of arrangement under section 182 of the OBCA (the “Plan of Arrangement”), which involved (i) an amendment to the capital structure of the Corporation (the “Share Capital Amendment”); and (ii) the distribution of a portion of the common shares in the capital of Celly Nu (“Celly Nu Shares”) to the holders of the Corporation’s Class B Shares, class A multiple voting shares (“Class A Shares”), and outstanding warrants exercisable for the purchase of Class B Shares, provided the applicable warrant certificate entitles the holder thereof to receive distributions substantially similar to those received by the holders of Class B Shares (“FSD Pharma Distribution Warrants” and together with Class A Shares and Class B Shares, the “FSD Pharma Securities”). The Shareholders and the holders of FSD Pharma Distribution Warrants (collectively, the “FSD Pharma Securityholders”) would each receive one Celly Nu Share for each Class A Share, Class B Share or FSD Pharma Distribution Warrant held.
On November 24, 2023, the Corporation received a final order from the Ontario Superior Court of Justice (Commercial List) approving the Plan of Arrangement.
The record date of the Plan of Arrangement was set at November 28, 2023 (the “Record Date”), and the ex-dividend date was set at November 27, 2023.
Effective November 29, 2023, the Corporation completed the Plan of Arrangement. Holders of FSD Pharma Securities received one Celly Nu Share for each Class A Share, Class B Share, or FSD Pharma Distribution Warrant held. FSD Pharma Securityholders also received new Class A Shares, new Class B Shares, and new FSD Pharma Distribution Warrants (“New FSD Pharma Securities”) in exchange for their Class A Shares, Class B Shares, and FSD Pharma Distribution Warrants (the “Share Exchange”). Pursuant to the Share Exchange and in accordance with the terms of the Arrangement Agreement, 24 Class A Shares were exchanged to 24 new Class B Shares. Following the closing of the Plan of Arrangement, the Corporation had 48 new Class A Shares, 39,376,723 new Class B Shares, and 6,335,758 new FSD Pharma Distribution Warrants issued and outstanding. Further details concerning the Share Exchange are set forth in the Special Meeting Circular.
| 32 |
| Table of Contents |
All Celly Nu Shares distributed to FSD Pharma Securityholders pursuant to the Plan of Arrangement are subject to restrictions on resale, and may not be transferred until May 31, 2024, provided that, Celly Nu may, in its sole discretion, waive such restrictions, in whole or in part.
The New CUSIP and ISIN numbers for Class B Shares following the completion of the Plan of Arrangement are 35954B404 and CA35954B4047, respectively, the New CUSIP and ISIN numbers for Class A Shares following the completion of the Plan of Arrangement are 35954B305 and CA35954B3056, respectively and the Celly Nu Shares distributed pursuant to the Plan of Arrangement have CUSIP and ISIN numbers of 150965200 and CA1509652006, respectively.
The Plan of Arrangement resulted in an aggregate of 45,712,529 Celly Nu Shares being distributed to the FSD Pharma Securityholders and an aggregate of 154,287,471 Celly Nu Shares retained by the Corporation, which represents approximately 26.15% of the issued and outstanding Celly Nu Shares on a non-diluted basis.
For more information regarding the Plan of Arrangement, please see the Special Meeting Circular and Arrangement Agreement, each of which is available under the Corporation’s profile on System for Electronic Document Analysis and Retrieval plus (“SEDAR+”) at www.sedarplus.ca and on EDGAR at www.sec.gov.
The Plan of Arrangement was considered a “business combination” pursuant to Multilateral Instrument 61-101 – Protection of Minority Security Holders in Special Transactions (“MI 61-101”) since (i) the FSD Pharma Securityholders’ interest in the FSD Pharma Securities may have been terminated without their consent as a result of the Share Capital Amendment; and (ii) Michael (Zappy) Zapolin (“Zapolin”), a director of the Corporation and therefore a “related party” under MI 61-101 was party to a “connected transaction” to the Plan of Arrangement. The Plan of Arrangement and subscription by Zapolin for 28,800,000 Celly Nu Shares on August 1, 2023 was a “connected transaction” (the “Related Party Purchase”). Both transactions involved Celly Nu as a common party and the Plan of Arrangement and Related Party Purchase were arguably negotiated at approximately the same time. At the time, Zapolin owned, directly or indirectly, nil Class B Shares, nil Class A Shares, nil FSD Pharma Distribution Warrants, and 500,000 warrants, each exercisable for the purchase of one Class B Share. Any FSD Pharma Securities held by Zapolin were treated in the same fashion under the Plan of Arrangement as the FSD Pharma Securities held by every other FSD Pharma Securityholder.
The Plan of Arrangement was not a “related party transaction” pursuant to MI 61-101 as a result of it being a “business combination” pursuant to MI 61-101.
The Plan of Arrangement did not have a material impact or represent a material change on the Corporation’s financial performance and condition.
Settlement of Lawsuit with Syneos Health
On August 2, 2023, the Corporation entered into a settlement agreement (the “Settlement Agreement”) with Syneos Health, LLC and Syneos Health UK Limited (collectively, the “Syneos”), whereby it was agreed that, among other things, the Corporation agreed to pay Syneos the amount of US$100,000 within five days of the execution of the Settlement Agreement and upon receipt by Syneos of such settlement payment, Syneos shall waive, release and forgive the Corporation’s payment of (i) the different between the settlement payment and the damages payment (i.e. US$1,607,831) and (ii) interest on the damages payment ordered by the award, and any other amounts that were or could have been sought in the arbitration. Pursuant to the Settlement Agreement, Syneos also agreed to withdraw its recognition application that was filed on June 30, 2023. Payment was made by the Corporation on August 4, 2023 and pursuant to the terms of the Settlement Agreement, the matter was concluded in its entirety.
Investigation of Market Activity
On July 10, 2023, the Corporation announced that it had retained Christian Attar Law, a regional litigation firm located in Houston, Texas, to co-lead, along with New York City law firm, Warshaw Burstein, LLP, an investigation of any potential naked short selling or other market manipulation of the Corporation’s securities. The Corporation has been advised that Christian Attar Law has completed their preliminary assessment of the predatory short selling and they anticipate that they will recommend to move the matter forward to the next stage but have yet to make a formal decision. The Corporation will provide updates in the event of any material progress on the investigation.
On November 22, 2023, the Corporation was provided an update from its United States counsel in connection with the possible naked short selling and market manipulation case, counsel informed the Corporation on a phone call that they plan to file a motion in the coming year. Although initially expected to be filed in February or March 2024, the Corporation is still in the information gathering process and expect to provide an update on the status of the potential naked short selling or other market manipulation in the coming months.
December Class A Share Private Placement
Effective December 4, 2023, the Corporation closed a non-brokered private placement of Class A Shares for gross proceeds of C$45.60 through the issuance of 24 Class A Shares at a price of C$1.90 per Class A Share (the “December Class A Private Placement”). All securities issued pursuant to the December Class A Private Placement were subject to a statutory hold period of four months plus a day from issuance in accordance with applicable securities laws of Canada. The Corporation intends to use the proceeds of the December Class A Private Placement for general working capital purposes. For more information, see “Item 7. Major Shareholders and Related Party Transactions - B. Related Party Transactions.”
| 33 |
| Table of Contents |
Dr. Raza Bokhari
For an update on the status of our outstanding lawsuits with Dr. Raz Bokhari in 2023, please see “Item 8. Financial Statements – Legal Proceedings.”
Shelf Prospectuses
In order to replace its prior base shelf prospectus that expired, effective December 22, 2023, the Company filed and obtained a receipt for its final short form base shelf prospectus dated December 22, 2023 (the “Canadian Prospectus”) to provide the Company with the flexibility to take advantage of financing opportunities and favourable market conditions, if and when needed, during the 25-month period that the Prospectus remains effective (the “Effective Period”). The Canadian Prospectus has been filed in each of the provinces and territories in Canada. The Canadian Prospectus enables the Company to offer, issue and sell, from time to time: Class B Shares, subscription receipts, warrants and units, or any combination thereof (collectively, the “Prospectus Securities”) for up to an aggregate offering amount of US$50,000,000, in one or more transactions during the Effective Period. Should the Company decide to offer Securities during the Effective Period, the specific terms, including the use of proceeds from any offering of Securities, will be set forth in one or more related prospectus supplements to the Canadian Prospectus.
Effective December 22, 2023, the Company also filed a registration statement on Form F-3 (File No. 333-276264) filed under the Securities Act with the SEC and declared effective on January 4, 2024 (the “Registration Statement”) containing a base shelf prospectus with the SEC (the “U.S. Base Prospectus”). The Registration Statement also qualifies the offer, issue and sale, from time to time of Securities up to an aggregate amount of US$50,000,000, subject to limitations, as applicable, under Form F-3. The Registration Statement is available for use by the Company until January 4, 2027. The terms of any Securities to be offered under the U.S. Base Prospectus will be specified in a prospectus supplement, which will be filed with the SEC in connection with any such offer.
ATM Offering
Effective February 16, 2024, the Company entered into an at-the-market offering agreement (the “ATM Agreement”) with H.C. Wainwright & Co., LLC (“Wainwright”), pursuant to which the Company, at its discretion, may offer and sell, from time to time, through Wainwright as sales agent, Class B Shares, having an aggregate offering price of up to US$11,154,232 (the “ATM Offering”). A cash commission of 3.0% on the aggregate gross proceeds raised under the ATM Offering will be paid to Wainwright in connection with its services. The ATM Offering was made in the United States pursuant to the Registration Statement and the prospectus supplement dated February 16, 2024 (“Prospectus Supplement”, together with U.S. Base Prospectus, the “U.S. Prospectus”) filed with the SEC.
Sales of the Class B Shares under the U.S. Prospectus will be made in transactions that are deemed to be "at-the-market" offering as defined in Rule 415(a)(4) promulgated under the Securities Act of 1933, as amended, (the “Securities Act”) including sales made directly on or through the Nasdaq. The Class B Shares will be distributed at the prevailing market prices at the time of each sale. As a result, prices may vary as between purchasers and during the period of distribution. No Class B Shares in the ATM Offering will be sold on the CSE or any other trading market in Canada.
The volume and timing of sales, if any, will be determined at the sole discretion of the Corporation’s management and in accordance with the terms of the ATM Agreement. If the Company chooses to sell Class B Shares under the ATM Offering, the Company intends to use the net proceeds of the ATM Offering (i) to fund our various clinical studies, trials and development programs, (ii) to fund research and development, and (iii) for general corporate purposes and working capital.
B. Business Overview
For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Overview and History”.
The Corporation currently has two (2) significant programs, which are focused on the development of treatments for challenging neurodegenerative, inflammatory, and metabolic disorders. They are:
| 1. | Lucid-MS: A potential treatment for Multiple Sclerosis with the lead candidate, Lucid-21-302; and |
| 2. | Novel Treatments for Alcohol Misuse, and related conditions. |
| 34 |
| Table of Contents |
All programs are clinical stage programs, with significant benefits to help patients, if successfully approved for clinical use. With respect to the Product Candidates for alcohol misuse disorders, the Corporation sees two distinct routes where this segment can be developed, (i) recreational retail and (ii) healthcare; Celly Nu, through the Celly Nu IP License Agreement will be focusing on the recreational retail sector and the Corporation will be focusing on the healthcare sector. Below, a brief summary on each program is provided and their corresponding status along with any partnership/licensing activities.
Lucid-MS
This program is focused on the development of novel drugs for MS. Progressive MS has no standard of care, and almost all available drugs are immunomodulatory, and do not address the neurodegeneration in the patients. The Corporation believes it has a solution that can significantly change the course of neurodegenerative decline in MS patients. The Corporation, through the acquisition of Lucid, acquired the multiple sclerosis program with the lead candidate, Lucid-MS (development code, Lucid-21-302). Lucid-21-302 exhibits moderate inhibition profile against peptidyl arginine deiminase (“PAD”) 2 and PAD 4 isozymes. There is strong evidence that hypercitrullination of myelin, mediated by increased activities of PAD 2 and potentially PAD 4, may contribute to demyelination and multiple sclerosis pathogenesis through two mechanisms: (1) destabilizing myelin integrity on neuronal axons, leading to demyelination and degeneration, and (2) generating antigenic neoepitopes, leading to immune activation. Lucid-21-302 reduced hypercitrullination, prevented demyelination and helped remyelination in various non-clinical animal models of multiple sclerosis, including functional recovery in the animals. Lucid-MS is being developed as a first-in-class, non-immunomodulatory drug for the treatment of progressive multiple sclerosis. Current data from preclinical and clinical development suggests that Lucid-21-302 could achieve the therapeutic dose to launch a proof-of-concept human study in a small number of patients (Phase 2a PoC clinical trial). This trial will pave way for a larger Phase 2b study, with appropriate biomarkers and endpoints for the treatment of progressive MS with multiple clinical sites. The Corporation has actively been planning a potential phase-2 clinical trial. The current patent on Lucid-21-302 are effective until 2036 (US10716791B2) and was licensed from University Health Network (Toronto, Canada) exclusively for development and commercialization.
Clinical Trials
On January 17, 2023, the Company submitted the CTA for a planned Phase 1 clinical trial for Lucid-MS to Health Canada. In February 2023, the Company received regulatory clearance from Health Canada to proceed with the Company’s Phase 1 clinical trial of Lucid-MS in Canada. On May 10, 2023, the Company announced the completion of dosing the first cohort of patients in the Phase 1 clinical trial of Lucid-MS.
On July 10, 2023, the Company received a “no objection letter” for a Phase 1 Lucid-MS clinical trial for the submission Clinical Trail Application, which was acknowledged on June 12, 2023. On July 19, 2023, the Company submitted a request for pre-IND meeting to the FDA, which was acknowledged August 3, 2023, and a response was received on September 21, 2023. On August 25, 2023, the Company received a “no objection letter” for the Clinical Trail Application, which was acknowledged on July 31, 2023. On September 18, 2023, the completion of study notification (after completion of five cohorts) was submitted to Health Canada. On October 2, 2023, the Company submitted a provisional patent application to the USPTO was submitted on the clinical formulation containing Lucid-MS.
The Company presented the results from its first-in-human Phase 1 study of Lucid-MS at the America’s Committee meeting for the treatment and research in multiple sclerosis in February 2024. This presentation detailed the final results including adverse events profile of Lucid-21-302 in the single-ascending dose studies. The study concluded that Lucid-21-302 is safe and well-tolerated in the dose range of 50-300 mg p.o. administered once, with no difference in pharmacokinetics between the fed and fasted states. There were no serious adverse effects, and most adverse effects (7/12) in participants receiving Lucid-21-302 were characterized as unlikely related or unrelated to study drug. In the dose range 50-300 mg, drug exposure was proportional to dose of the drug. It also demonstrated good oral absorption with ‘area under the curve’ at 300mg comparable to ‘area under the curve’ in mouse efficacy studies. Based on its internal review of the Phase 1 data, the Company believes that the positive results warrant moving to a Phase 2 clinical trial.
Phase 2 Clinical Study
Based on the positive results that the Phase-1 study yielded, the Board, on recommendation from the advisory committee, resolved to proceed with completing a Phase II MS-Study on Lucid-MS with the goal of getting the Lucid-MS to commercialization. The Company has determined that in order to get to commercialization it will cost approximately US$30,655,469.
As Lucid-MS advances from Phase-1 to Phase-2, milestone-driven investigations are planned to expedite late-stage clinical development, aligned with our chronic toxicology program. This synergy ensures enabling data for regulatory submissions for the next clinical phases, such as Phase-1b multiple ascending dose (“MAD”) cohorts, Phase-2a, and Phase-2b. To initiate Phase-2, we require data from MAD cohorts, at least three months of toxicology data, and any additional data requested by regulatory authorities. Long-term toxicology data is crucial for dosing extending up to six months or more, reflecting Lucid-MS’s potential as a chronic treatment or disease-modifying therapy for MS patients. The Lucid-MS program’s ultimate goal is to conduct regulatory clinical studies, investigating its potential as a non-immunomodulatory drug to halt disease progression and neurodegeneration in multiple sclerosis.
| 35 |
| Table of Contents |
The Company’s innovative clinical development program targets multiple sclerosis, aiming to create a groundbreaking treatment. The Company has engaged thought leaders and conducted internal discussions on regulatory guidance to design an efficient, cost-effective program spanning chemistry, product development, and data acquisition for clinical stages. The Company’s clinical trials are moving forward in Australia.
Novel Treatments for Alcohol Misuse and Related Conditions
Excess alcohol consumption (alcohol misuse or mild acute alcohol intoxication) is clinically harmful that typically follows the ingestion of excess amount of alcohol. Clinical symptoms and manifestations are heterogeneous, and can have behavioral, cardiac, gastrointestinal, pulmonary, neurological, and metabolic effects. The Company is focused on treatments to reverse inebriation and to assist accelerating alcohol metabolism in people who consumed excess alcohol, reaching blood alcohol levels around/slightly above the legal limits in various countries. Available options for the emergency response doctors and nurses are to provide a vitamin intravenous drip or let alcohol “wear off”, until medical professionals can tend to those individuals who are inebriated, occupying expensive resources in the emergency room. The Company also identified that excess alcohol consumption is a problem in the general society (consumer market), and the commonly available remedies mostly fall in the category of “hangover remedies”. Thus, there is a great need for immediate treatments that can address the challenges when one consumes excess alcohol.
Medical and R&D teams at Lucid identified several natural ingredients that are dietary supplements that can function as alcohol metabolism accelerants and enhance mental alertness; the team developed several formulations that will help enhance mental alertness, replenish cofactors, and may accelerate the rate of alcohol metabolism. This formulation may be useful in treating intoxicated individuals who wish to speed up their recovery from the effects of alcohol as well as for the treatment of intoxicated patients entering emergency departments in the hospitals. The Company will continue its R&D program and develop products for use in emergency departments and other healthcare settings. Regulatory activity in the United States, and other markets globally will be continued, aligned with the R&D and potential clinical trials (as needed) for commercialization, marketing, and distribution.
As of March 28, 2024, the Company has registered five trademarks with the Canadian Intellectual Property Office (Registrar of Trademarks) (“CIPO”) and the USPTO and 17 trademarks with the CIPO, relating to novel treatments for alcohol misuse and related conditions, including Unbuzzd™ and ALCOHOLDEATH™, which were licensed to Celly Nu pursuant to the Celly Nu IP License Agreement.
On April 24, 2023, the Company filed a provisional patent application with the USPTO with respect to the Company’s alcohol misuse treatment technology, which was licensed to Celly Nu under the Celly Nu IP License Agreement for retail use.
The Company sees two distinct routes where this segment can be developed, (i) recreational retail and (ii) healthcare. Celly Nu, through the Celly Nu IP License Agreement will be focusing on the recreational retail sector and the Company will be focusing on the healthcare sector.
Product Development on Hold or Discontinued
In June 2023, the Company terminated any further clinical development of its proprietary ultra-micronized FSD-PEA formulation for the treatment of inflammatory diseases. The Company’s team of internal medical experts conducted a profitability assessment of FSD-PEA and ultimately determined that the FSD-PEA molecule was not profitable compared against the currently available products in the market and it would not be possible to cover the Company’s manufacturing and research and development investments at a price that would be accepted in the market.
Additionally, management made the decision to put the research and development activities associated with Lucid-PSYCH (formerly Lucid-201) on hold during June 2023. This decision was made based on the cumulative cash requirements to advance the research and development of the Company’s portfolio of compounds. Due to cash flow prioritization strategies, management elected to prioritize the Lucid-MS compound and its alcohol misuse treatment products.
Milestones for Future Development
In light of the above, the Company has determined that it will prioritize the development of its viable assets, utilizing the limited in-house resources available, to maximize the chances of their successful commercialization. Therefore, the Company resolved to push forward with the development of (i) Lucid-MS and (ii) the Healthcare Product, (ii) license Unbuzzd™ to Celly and (iv), pause the development of Lucid-PSYCH and terminate the development of FSD201.
The Company’s adaptable trial planning integrates insights from clinical and non-clinical studies, regulatory guidance, and market dynamics. Our adjusted timelines, especially in 2024 and 2025, are influenced by key market regulations, ensuring efficiency and cost-effectiveness across chemistry, product development, and data acquisition.
The Company’s overarching strategy allows it to conduct studies efficiently in terms of time and cost, considering that toxicology and clinical studies can span several months to several years. It also provides flexibility to adjust or explore alternative pathways cost-effectively in case of unexpected toxicities or efficacy issues.
The table below sets forth the status of these milestones as of March 28, 2024, the estimated costs and estimated timeframe for completion thereof. The following are “forward-looking statements” and as such, there is no guarantee that such milestones will be achieved on the timelines indicated or at all. Forward-looking statements are based on management’s current expectations and are subject to a number of risks, uncertainties, and assumptions. See “Caution Regarding Forward-Looking Statements” and “Item 3. Key information - D. Risk Factors” on the Company’s ability to achieve certain of its objectives and milestones, which are contingent upon raising additional financing.
| 36 |
| Table of Contents |
Objective | Milestone(1)(2) | Estimated Cost
| Estimated Timeframe for Completion(3)(4) | Notes |
1. MAD Cohorts | ||||
|
Regulatory Agency Approval
| US$601,742 |
Q2, 2024
|
These studies are listed separately here; earlier they were a part of Clinical Studies below. Data from these studies will feed into Phase-2 clinical study designs. Timeframe is modified accordingly.
|
Site Pass Through Costs
| US$730,413 |
Q2-Q4, 2024 | ||
First Participants In
|
US$376,012
|
Q2, 2024
| ||
Last Participant In
|
US$376,012
|
Q3, 2024
| ||
Completion of Report
|
US$150,282
|
Q1, 2025
| ||
|
Sub-total
| US$2,234,461
|
|
|
2. Chronic Toxicity to initiate phase-2 (3-month study) | ||||
|
Study design for 2-species toxicity trial
|
US$37,158
|
Q2, 2024
|
These studies will be completed prior to Phase-2 initiation, and additional drug substances will be required. Proposed timeframe permits these activities.
|
First interim report
|
US$260,107
|
Q3, 2024
| ||
Second interim report
|
US$260,107
|
Q3, 2024
| ||
Final Report
|
US$185,791
|
Q4, 2024
| ||
|
Sub-total |
US$743,163 |
|
|
3. Lucid-MS Program | ||||
Non-clinical studies |
| |||
Phase 2 enabling pharmacology studies
| US$111,474 | Q3, 2024 | These non-clinical studies will be launched a few months ahead of Phase-2 studies such that continuous safety data from the non-clinical studies will advance Phase-2 dosing for chronic treatment. Reproductive toxicology and autoradiography will be required for an NDA or for Phase 3 trial application submission. | |
Chronic tox studies to complete phase-2 (2 species, up to 9 months)
|
US$1,168,582 |
Q4, 2025 | ||
Reproductive toxicology and autoradiography
|
US$1,857,907 |
Q3, 2026 | ||
Drug Substance and Product Manufacturing |
Synthesis of non-GMP drug substance for chronic toxicology studies
|
US$779,500 |
Q3, 2024 |
Proposed timeline of Q1, 2024 is required to obtain the drug substance in time for toxicology studies (3 months and 9 months). |
Development of clinical and non-clinical Formulations
|
US$334,423 |
Q4, 2024-Q1, 2025 |
This development is required for launching chronic toxicity and Phase-2 clinical studies; thus, the time frame is adjusted to fit those milestones. | |
Drug Substance for Phase 2 studies
|
US$1,114,744 |
Q1-Q2, 2025 |
Manufacturing of the drug substance for launching Phase-2 study; time frame aligns with two quarters prior to the initiation of any Phase-2 activity. | |
Drug Product for Phase 2 studies
|
US$445,898 |
Q2-Q3, 2025 |
Manufacturing of the drug substance for launching Phase-2 study; time frame aligns with one quarter prior to the initiation of any Phase-2 activity. | |
| 37 |
| Table of Contents |
Clinical Studies
|
2nd Phase 2a clinical trial site and CRO identification and deposits |
US$966,112 |
Q4, 2024-Q3, 2025 |
The time frame includes submission of regulatory files, discussions and approvals from the regulator, identification of potential clinical sites and contracts negotiations. The time frame is scheduled after completing 3-month chronic toxicology, development of clinical formulation and other Phase-2 enabling studies. |
Phase 2a proof of concept (“PoC”) clinical trial (launch, biomarkers, labs, clinical site, regulatory and other activities) |
US$4,458,977 |
Q3, 2025-Q4, 2026 |
This time frame is after the above line item, to conduct the clinical trial. | |
Phase-2b clinical trial (launch, biomarkers, biostats, labs, clinical sites, regulatory and other activities) |
US$14,863,258 |
Q3, 2025-Q4, 2026 |
Will be initiated after Phase 2a PoC, or can be in place of Phase 2a PoC, depending on market/regulatory strategy | |
Regulatory, licensing, and other support costs
|
US FDA/Health Canada/UK MHRA regulatory activities, patents maintenance/new filings, patent licensing costs. |
US$3,715,815 |
Q4, 2024-Q4, 2026 |
These are continuous activities for patents maintenance, licensing costs (to UHN), regulatory filings for early market access among others. Milestones will be based on each activity undertaken, and success of regulatory reviews. Each major milestone calls for a milestone payment to UHN. |
|
Sub-total |
US$29,816,690 |
|
|
4.Alcohol Misuse Treatments Program: Healthcare Product | ||||
Non-clinical activities |
In vitro and in vivo toxicology studies and dose range for the oral liquid formulation |
US$743,163 |
Q4, 2024-Q1, 2025 |
These non-clinical activities will be undertaken as a part of our R&D program for new formulations in late 2024, that will serve the development of hospital and consumer products. Current focus is on licensed activities for consumer market, in early 2024. |
In vitro and in vivo toxicology studies and dose range for the intravenous formulation |
US$2,229,489
|
Q4, 2024-Q2, 2025 |
These studies are aligned with the above line item for hospital product development. | |
Drug Substance and Product Manufacturing |
Oral liquid formulation development |
US$743,163 |
Q1-Q3, 2025 |
GMP R&D manufacturing of oral liquid formulation for hospital line product; aligned with completion of non-clinical activities above. |
Intravenous formulation development |
US$1,114,744 |
Q1, 2025-Q1, 2026 |
GMP R&D manufacturing of intravenous formulation for hospital line product; aligned with completion of non-clinical activities above, and after the oral formulation development, in the above line item. | |
Oral liquid formulation manufacturing for clinical study |
US$371,581
|
Q3, 2025
|
Manufacturing of clinical trial material (liquid oral), will commence after R&D during Q1-Q3, 2025
| |
GMP Sterile formulation manufacturing for clinical studies |
US$1,114,744 |
Q1-Q2, 2026 |
Manufacturing of clinical trial material (intravenous), will commence after R&D during Q1, 2025-Q1, 2026 | |
| 38 |
| Table of Contents |
Clinical Studies |
Clinical study with one oral formulation |
US$1,114,744 |
Q4, 2025-Q2, 2026 |
Clinical study using novel oral formulation is scheduled after the completion of toxicology and clinical trial materials manufacturing. |
Clinical study with one intravenous formulation for regulatory submission |
US$1,857,907 |
Q3-Q4, 2026 |
Clinical study using novel intravenous formulation is scheduled after the completion of toxicology and intravenous clinical trial materials manufacturing. | |
Regulatory, IP and other support costs |
Regulatory activities and submissions in the USA and Canada |
US$222,949 |
Q4, 2024-Q4, 2026 |
These are continuous activities for patents maintenance, licensing, regulatory filings for market access among others. Milestones will be based on each activity undertaken, and success of regulatory reviews. |
Marketing and related activities |
Medical education, pre-launch, and partnership activities |
US$1,486,326 |
Q4, 2025-Q4, 2026 |
As the clinical studies commence, marketing, outreach and partnership activities will be undertaken; the time frame is based on the clinical studies scheduling above, and the anticipated prior work for late-stage marketing and potential pre-launch for the products. |
|
Sub-total |
US$10,998,810 |
|
|
Operations |
Team members salaries, benefits, external consultants, and key opinion leaders |
US$4,087,396 |
Q4, 2024-Q4, 2026 |
These costs include additional personnel will be required for all planned clinical drug development, toxicology, project management and regulatory affairs, for all programs. |
Information technology, legal, telecommunications, facilities infrastructure, travel, shipping/logistics |
US$2,229,489 |
Q4, 2024-Q4, 2026 |
Planned programs will incur indirect costs in order to support the R&D and clinical activities. | |
Sub-total |
US$6,316,885 |
|
|
Notes:
(1) | There may be circumstances where, for sound business reasons, the Company’s reallocates the funds or determines not to proceed with a milestone. |
(2) | Subject to receipt of all necessary approvals, including any approvals required by the academic and scientific organizations with which the Company is working. |
(3) | The total expenditure may be incurred by the Company after the relevant quarter that is indicated as the target timeframe for completion. |
(4) | Based on a calendar year end. |
The materials factors or assumptions used to develop the estimated costs disclosed above are included in the “Cautionary Note Regarding Forward-Looking Statements” section above. The actual amount that the Company spends in connection with each of the intended milestones will depend on several factors, including those listed under “Item 3. Key information - D. Risk Factors” in or incorporated by reference in this Annual Report or unforeseen events. While the Company believes it has the skills and resources necessary to accomplish these business objectives, there is no guarantee that the Company will be able to do so within the timeframes indicated above, or at all. The Company will rely on third-party opinions evaluating novelty and patentability of its drug compounds, as well as data generated by tests performed by third parties indicating there is preclinical evidence of improved efficacy or safety profiles compared to currently known treatments for challenging neurodegenerative, inflammatory, and metabolic disorders based on scientifically sound preclinical studies. These tests are ongoing. While the Company believes its approach mitigates many risks associated with the challenges of obtaining regulatory approval for certain difficult to treat indications, the development of potential drugs for treatment of challenging neurodegenerative, inflammatory, and metabolic disorders involves a high degree of risk and uncertainty. The Company is committed to funding research it believes is essential for advancing the study of drugs to treat these conditions.
Research and Development
As at the date of this Annual Report, the Corporation has not generated any revenue from the sale of pharmaceutical drugs or other products. The Corporation is focused on development of pharmaceutical drugs and other products, through the research and development of novel chemical compounds and delivery mechanisms and the study of such compounds in preclinical studies. The Corporation’s preclinical studies are conducted via the various CROs and contract manufacturers it has engaged, including Ingenu CRO (a Cannvalate Pty Ltd Company) (“Ingenu”), BioPharma Services Inc. (“BioPharma”), and Vibrant Pharma Inc. (“Vibrant Pharma”). Each of Ingenu, BioPharma, and Vibrant Pharma are CROs that, in the ordinary course of the Corporation’s business, have entered into service agreements with the Corporation to provide services related to the Corporation’s preclinical studies and/or the manufacture of its various chemical compounds. The Corporation is not dependent on third party contracts. Although each of the CROs will be involved in the synthesis, or testing thereof, for the Corporation, none of these agreements allows for the various CROs to utilize any of the Corporation’s intellectual property, including its patents, formulae, trade secrets, or processes, for their own purposes. The pharmaceutical industry is a competitive and, in the event that one, or all, of these contractual relationships become unsatisfactory, the Corporation does not anticipate having difficulty retaining other services providers to perform similar services. The Corporation does not anticipate generating any revenue from any of these, or any other, service agreements.
| 39 |
| Table of Contents |
The Corporation anticipates growing its pipeline of pharmaceutical drugs and other products through its research, development, proprietary discovery programs, mergers and acquisitions, joint ventures and collaborative development agreements. The Corporation has sought protection for the intellectual property rights generated by its research and development activities through patent applications and as trade secrets. The Corporation anticipates that as these programs mature it will file additional patent applications and details about these programs will be disclosed at such time. The Corporation further anticipates that existing patent applications will result in successful patent grants by the respective intellectual property regulators of each jurisdiction in which the Corporation has submitted such applications.
The Corporation’s research and development activities (including such activities conducted by third party contractors) are conducted in strict compliance with the regulations of federal, state, local and regulatory agencies in Canada, Australia and the United States. These regulatory authorities regulate, among other things, the research, manufacture, promotion and distribution of drugs in specific jurisdictions under applicable laws and regulations.
See “Item 4. Information on the Company. – B. Business Overview - Milestones for Future Development” for further information on the Corporation’s objectives and milestones.
Intellectual Property
The following tables set forth the status for each patent applicable to the Company’s current and anticipated business for Lucid-MS and Celly Nu’s activities:
Title | Jurisdiction of Filing | Application Number | Filing Date/Patent Date/Priority Date | Status | Program |
Inhibitors of Peptidyl Arginine Deiminase (PAD) Enzymes and Uses Thereof | United States Patent and Trademark Office | Appl. No.: 15/753,208 Patent No.: US10,716,791 B2 | Filing Date: 2016-08-15 Patent Date: 2020-07-21 | Exclusive license from University Health Network (Toronto) | Lucid-MS |
Inhibitors of Peptidyl Arginine Deiminase (PAD) Enzymes and Uses Thereof | European Patent Office | Appl. No.: 22187901.8 | Filing Date: 2016-08-15 Priority Date: 2016-08-15 | Exclusive license from University Health Network (Toronto) | Lucid-MS |
Methods and Compositions Comprising a 5-HT Receptor Antagonist | United States Patent and Trademark Office | Appl. No.: 63/454,587 | Filing Date: 2023-03-24 | Provisional patent application (PAT 114860P-2) | Lucid-PSYCH(1) |
An Ingestible Formulation and Uses Thereof | United States Patent and Trademark Office | Appl. No.: 63/497,772 | Filing Date: 2023-04-24 | Provisional patent application (PAT 114119P-2) | Licensed to Celly Nu |
Note:
(1) | The Company has put any future work programs relating to Lucid-PSYCH on hold. |
| 40 |
| Table of Contents |
The Company’s wholly owned subsidiary, Lucid, has filed pending applications for the trademarks set forth in the table below with the Innovation, Science and Economic Development Canada – CIPO:
Applicant | Filing Date | Reference Number | File Number | Trademark Details | Trademark Type |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150984-1 | 2243755 | REKVRY | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150985-1 | 2243758 | DETOXIQ | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150986-1 | 2243760 | RESOBER | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150958-1 | 2243743 | ALCOHOLDEATH(1) | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150987-1 | 2243761 | Unbuzzd(1) | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150959-1 | 2243741 | DRUNQUELL | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150957-1 | 2243742 | FRESHKA | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150956-1 | 2243736 | FRESHA | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150955-1 | 224374 | ALKACLEAR | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150954-1 | 224379 | LOWBAC | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150953-1 | 2243740 | SOBRY | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150952-1 | 2243744 | BACLEAR | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 15951-1 | 2243737 | READY IN 1 | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150950-1 | 2243735 | QLARITY | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150949-1 | 2243738 | WAKEAID | Standard Characters |
Note:
(1) | Licensed to Celly Nu. |
The Company’s wholly owned subsidiary, Lucid, has filed pending trademark applications for the marks in the table below with the Innovation, Science and Economic Development Canada – CIPO:
Applicant | Filing Date | Reference Number | File Number | Trademark Details | Trademark Type |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150940-1 | 2243726 | EVERYONE MAY NEED A LITTLE IN THEIR LIFE | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150825-1 | 2243752 | THE RITUAL AFTER THE LAST CALL | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150824-1 | 2243750 | THE PROTOCOL AFTER THE LAST CALL | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150823-1 | 2243749 | HELPS REDUCE BUZZ | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150822-1 | 2243733 | A RESPONSIBLE AFTER DRINK | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150821-1 | 2243732 | THROUGH SCIENCE FIND | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150820-1 | 2243730 | THROUGH SCIENCE, FIND CLARITY | Standard Characters |
Lucid PsycheCeuticals Inc. | March 7, 2023 | TM 150819-1 | 2243729 | EVERYONE MAY NEED A LITTLE | Standard Characters |
As the Company generates new data it will continue to file or acquire additional patent applications through the Company’s development program.
Regulatory Environment
The Corporation is currently focused on obtaining regulatory approvals in the United States, Canada and Australia for the drug candidates it is developing through FSD BioSciences, Lucid and FSD Pharma Australia. In the future, the Corporation may consider seeking approvals for these drug candidates in other countries. The following is a summary of the FDA, Health Canada and the Australian Therapeutics Goods Administration (“TGA”) approval process that the Corporation and/or its related entities are undertaking with each of the Product Candidates in the United States, Canada and Australia. Assuming the Corporation is successful in obtaining the requisite approvals from the FDA, TGA or Health Canada (together the “Regulatory Approvals”) pursuant to the process set out below, it may decide to seek comparable approvals in other countries, which would be subject to different and additional regulatory requirements. Obtaining Regulatory Approval often takes several years, involves the expenditure of substantial resources, and depends on a number of factors, including the severity of the disease in question, the availability of alternative treatments, and the risks and benefits demonstrated in clinical trials.
| 41 |
| Table of Contents |
The Corporation will be subject to extensive regulations while it focuses on gaining Regulatory Approvals for treatments it is developing with each of the Product Candidates. The United States Food, Drug and Cosmetic Act of 1938, as amended, Public Health Service Act (United States), Therapeutic Goods Act 1989 (Cth) (Australia), and other federal, provincial and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labelling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling, and import and export of pharmaceutical product candidates for their respective jurisdictions. Failure to comply with applicable regulatory requirements may subject the Corporation to a variety of administrative or judicial sanctions, such as application refusals, warning or untitled letters, product candidate recalls, product candidate seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
Pharmaceutical product candidate development in the United States, Canada and Australia typically involves preclinical laboratory and animal tests, followed by a submission to commence clinical testing to, as applicable:
| (a) | the FDA for the United States (an IND); |
| (b) | Health Canada for Canada (a CTA)); or |
| (c) | in Australia, (i) where the Clinical Trial Notification (“CTN”) process is utilized, to a Human Research Ethics Committee, or (ii) where the Australian CTA process is utilized, to the TGA. |
If:
| (a) | there are no comments from the FDA within 30 days after the submission of the application in the United States; |
| (b) | a “no objection letter” is received from Health Canada; or |
| (c) | in Australia, (i) where the CTN process is utilized, the applicable Human Research Ethics Committee provides its approval and the TGA is notified by way of the due submission of a CTN, or (ii) where the Australian CTA process is utilized, the TGA provides its approval, |
then clinical trials for the drug may commence in the respective jurisdiction assuming all other requirements are met (such as institution review board approval, informed consents and any additional approvals related to the use of controlled substances). The satisfaction of pre-market approval requirements typically takes many years. The actual time required may vary substantially based upon the type, complexity and novelty of the product candidate or the diseases a product candidate targets.
Before testing any compound in human patients in the U.S., Canada or Australia, a company must generate extensive preclinical data. Preclinical testing generally includes laboratory evaluation of product chemistry and formulation, as well as toxicological and pharmacological studies in several animal species to assess the toxicity and dosing of the product candidate and its potential safety and efficacy. The conduct of the preclinical tests must comply with government regulations and requirements, including good laboratory practices. For example, in the U.S., certain animal studies must be performed in compliance with the FDA’s Good Laboratory Practice regulations and the U.S. Department of Agriculture’s Animal Welfare Act.
A Regulatory Approval must be in effect before human clinical trials may commence in the U.S., Canada or Australia, respectively. The results of preclinical testing and any previous human experience with the investigational drug are submitted to the FDA, Health Canada or TGA as part of the Regulatory Approval process in each jurisdiction, along with other information, including information about product candidate chemistry, manufacturing and controls, information about the study investigator, and a proposed clinical trial protocol.
There can be regulatory barriers to obtaining an effective Regulatory Approval based on FDA’s, Health Canada’s or TGA’s respective review of the investigative drug and, where applicable, its classification as a known controlled substance.
Clinical trials involve the administration of the product candidate that is the subject of the Regulatory Approval to healthy volunteers or study participants with the disease or condition being studied under the supervision of a qualified investigator. Clinical trials to support a NDA for marketing approval are typically conducted in three sequential phases, but the phases may overlap.
There is a process under which clinical trials may begin and involve the administration of the product candidate that is the subject of the Regulatory Approval to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted: (i) in compliance with applicable government regulations, (ii) in compliance with Good Clinical Practice, an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors, and (iii) under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol involving testing on patients and subsequent protocol amendments must be submitted to the FDA, Health Canada and/or TGA as part of the Regulatory Approval process, as applicable.
The FDA, Health Canada or TGA may order the temporary, or permanent, discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial either is not being conducted in accordance with applicable regulatory requirements or presents an unacceptable risk to the clinical trial patients. The trial protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board (“IRB”) for approval. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements or may impose other conditions.
If the trials for any of its Product Candidates are successful, the Corporation may pursue additional trials as required and may ultimately pursue a NDA, which may involve applying for additional Regulatory Approvals required to market the Corporation’s synthetic treatments in the United States or in other jurisdictions. There is no assurance that the Corporation will be successful in receiving the required approvals, and the clinical trials are subject to numerous risks.
See “Caution Regarding Forward-Looking Statements” and “Item 3. Key information - D. Risk Factors” in this Annual Report.
| 42 |
| Table of Contents |
New Drug Application and New Drug Submission Process
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a NDA requesting approval to market the product for one or more indications. The application must include the results of all preclinical, clinical, and other testing and a compilation of data relating to the product’s pharmacology and chemistry, manufacture, and controls. Under the Prescription Drug User Fee Act, a substantial application user fee is required for most NDAs, and the applicant under an approved NDA is also subject to an annual program fee for each prescription product.
After evaluating the NDA, the FDA issues either an approval letter or a complete response letter. A complete response letter generally outlines the deficiencies in the submission. Substantial additional testing or information may be required in order for the FDA to reconsider the application. If regulatory approval of a product is granted, such approval will be granted for particular indications and may entail limitations on the indicated uses for which such product may be marketed. Additionally, as a condition of approval, the FDA may impose restrictions that could affect the commercial success of a drug or require post-approval commitments, including the completion within a specified time period of additional clinical studies, which often are referred to as “Phase 4” or “post-marketing” studies. For example, as a condition of approval, the FDA may require a risk evaluation and mitigation strategy (“REMS”) to help ensure that the benefits of the drug outweigh the potential risks. REMS can include medication guides, communication plans for healthcare professionals, and elements to assure safe use, such as special training or certification for prescribing or dispensing. Moreover, product approval may require substantial post-approval testing and surveillance to monitor the drug’s safety or efficacy.
Once granted, product approvals may be withdrawn if compliance with regulatory standards is not maintained or problems are identified following initial marketing. Once an NDA is approved, a product will be subject to certain post-approval requirements, including, among other things, requirements related to record-keeping, providing the FDA with updated safety information, product sampling and distribution, and promotion and advertising. Post-approval modifications to the drug, such as changes in indications, labeling, or manufacturing processes or facilities, may require a sponsor to develop additional data or conduct additional preclinical studies or clinical trials, to be submitted in a new or supplemental NDA, which would require FDA approval.
Similarly, Health Canada (in Canada) and the TGA (in Australia) regulates, among other things, the research, development, testing, manufacture, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, post-approval monitoring, marketing and import and export of pharmaceutical products. Drug approval laws require licensing of manufacturing facilities, carefully controlled research and testing of products, and government review and approval of experimental results prior to giving approval to sell drug products.
The process required by the applicable regulatory authorities before prescription drug product candidates can be marketed in Canada or Australia requires:
| (a) | in the case of Canada, the submission of a new drug submission (“NDS”) to Health Canada; or |
|
|
|
| (b) | in the case of Australia, an application for registration in the Australian Register of Therapeutic Goods (“ARTG”), the NDS, and application for registration on the ARTG collectively referred to as “New Drug Application”. |
Health Canada and/or TGA, as the case may be, must review and approve the relevant “New Drug Application”. Health Canada must also and issue a notice of compliance and both Health Canada and TGA must issue a drug identification number prior to any commercial marketing, sale, or shipment of the drug.
Even if Health Canada approves a NDS or TGA registers the drug on the ARTG, the relevant regulatory authority may limit the approved indications for use of the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms.
The regulatory review process of a drug application in each of the U.S., Canada and Australia includes the satisfactory completion of an inspection of the manufacturing facility or facilities where the product is produced (or other evidence acceptable to the regulator) to ensure that the facilities are in compliance with current GMP (“cGMP”) requirements and are adequate to assure consistent production of the product within required specifications.
The FDA, Health Canada and TGA also conduct regular, periodic visits to re-inspect equipment, facilities, and processes following the initial approval of a product. Failure to comply with applicable cGMP requirements and other conditions of product approval may lead the regulatory authority to take enforcement action or seek sanctions, including fines, issuance of warning letters, civil penalties, injunctions, suspension of manufacturing operations, operating restrictions, withdrawal of approval, seizure or recall of products, and criminal prosecution.
Controlled Substances - United States
In June 2023, the Company decided to stop any development efforts for Lucid-PSYCH, which is considered a controlled-substance in the United States, Canada, and Australia. If and when the Company continues with the development of Lucid- PSYCH for psycho functions, it will need to comply with the controlled substances laws in various jurisdictions.
Drugs and other substances that are determined to have a potential for abuse are also regulated under the United States Comprehensive Drug Abuse Prevention and Control Act of 1970, as amended, also known as the Controlled Substances Act (the "CSA") and its implementing regulations, as "controlled substances." The CSA establishes a closed chain of distribution for entities handling controlled substances, which include researchers, manufacturers, distributors, pharmacies and physicians, importers, and exporters. The CSA and regulations enforced by the DEA impose registration, security, quotas inventory, recordkeeping, reporting, storage, manufacturing, distribution, importation, exportation, and other requirements on entities handling controlled substances. Practitioners such as pharmacies and physicians, as well as other types of entities that handle controlled substances, such as researchers and analytical laboratories, are also subject to DEA registration and other requirements related to controlled substances.
| 43 |
| Table of Contents |
The CSA categorizes controlled substances into one of five schedules - Schedule I, II, III, IV, or V - depending on the potential for abuse and physical or psychological dependence. Schedule I substances by definition have a high potential for abuse, have no currently accepted medical use in treatment in the United States, and lack accepted safety for use under medical supervision. They may not be marketed or sold for dispensing to patients in the United States. Certain "hallucinogens" or psychedelic drugs are currently regulated as Schedule I controlled substances, as is any substance that includes any of a Schedule I substance's salts, isomers (e.g., optical, position, and geometric isomers), or salts of isomers, whenever the existence of such salts, isomers, and salts of isomers is possible within the specific chemical. Pharmaceutical products having a currently accepted medical use and that are otherwise approved for marketing may be listed as Schedule II, III, IV, or V substances, with Schedule II substances presenting the highest potential for abuse and physical or psychological dependence, and Schedule V substances presenting the lowest relative potential for abuse and dependence.
Whether a new drug or substance is ultimately controlled or not is a fact specific determination that the DEA makes based on the input of the Department of Health and Human Services (including the FDA), which provides scientific and medical findings and recommendations to the DEA. During the FDA approval process, the FDA will generally conduct an abuse potential evaluation of any substance that could have an effect on the central nervous system. If FDA finds that a new drug or substance may have an abuse potential that would require the drug to be controlled, FDA notifies the DEA and provides information/recommendation to the DEA on its scheduling. The DEA must conduct notice and comment rulemaking to propose scheduling of a new substance. If a drug being approved contains a substance already controlled under the CSA, that drug will generally be controlled in the same schedule absent findings or recommendations that it should be placed in another schedule.
Lucid-PSYCH is a Schedule I listed substance under the CSA. Its use in the United States is highly restricted under Federal law, even though there have been a few state and local laws seeking to loosen restrictions. A facility that seeks to manufacture, distribute, import, or export any Schedule I controlled substance must register with the DEA. The DEA registration is specific to the particular location, activity, and controlled substance. A DEA registered facility must maintain records documenting all activities, including the manufacture, receipt, and distribution, of controlled substances. The import or export of a Schedule I substance requires a permit and may need to comply with international drug control treaties as well as DEA requirements.
Any Schedule I drug or substance approved by the FDA must be rescheduled (or descheduled) to another schedule before it can be commercially marketed in the United States. Rescheduling or descheduling a Schedule I substance to another schedule is dependent on FDA approval and FDA recommendation as to the appropriate schedule. Any rescheduling or descheduling action requires the DEA to conduct notice and comment rulemaking. Such action will be subject to public comment and requests for hearing which could affect the scheduling of these substances.
Controlled Substances – Canada
A controlled substance is a type of drug that the Government of Canada has categorized as having a higher-than-average potential for abuse or addiction and is listed in one of the schedules (I to V) of the CDSA. Lucid-PSYCH is a controlled substance in Canada. The possession, sale or distribution of controlled substances is prohibited unless specifically permitted by the government.
Under section 56 of the CDSA the Minister of Health may exempt a person or a class of persons or any controlled substance or class thereof from the application of all or any provision of the CDSA or regulations if necessary for a medical or a scientific purpose or is otherwise in the public interest. Researchers requiring a controlled substance for research, including clinical trials, must receive an exemption under the CDSA, which can permit the importation, possession and/or use of a specified quantity of the controlled substance for a specified purpose. The Minister of Health can impose any terms and conditions that the Minister considers necessary in respect of the exemption. Through agreements with third parties, the Company has access to facilities that have experience and licenses required to handle Controlled Substances listed under the CDSA. Similar to the United States, in Canada certain scheduled substances would require reclassification to a different schedule in order to permit commercial marketing.
Controlled Substances – Australia
Like in the United States and Canada, a controlled drug is a type of drug that the Australian Government has categorized as having a higher-than-average potential for abuse or addiction and is listed in one of the schedules (1 to 10) of the Poisons Standard.
Substances with therapeutic uses are generally contained in Schedules 2, 3, 4 and 8 with progression through these Schedules signifying increasingly restrictive regulatory controls. Schedule 9 details substances that should be available only for teaching, training, medical or scientific research including clinical trials conducted with the approval of Commonwealth and/or State and Territory health authorities.
Lucid-PSYCH is currently a prohibited drug in Australia, meaning its supply is largely limited to clinical trials.
FSD Strategic Investments
The Company’s Strategic Investment segment generates interest income earned on a portfolio of finance receivables, which represent loans secured by residential or commercial property, with FSD Strategic Investments having a first collateral mortgage on the secured property for a sum equal to the interest payments plus the principal amount. FSD Strategic Investments earns interest through fixed rate lending arrangements, which includes faith-based loans, that have an average term to maturity of two years from the date of issuance.
| 44 |
| Table of Contents |
Strategic Investments has historically not incurred any significant operating expenditures as the loans are arranged through a third-party financing intermediary, with the borrower being responsible for covering all administrative related costs.
The Board has developed criteria for making investments decisions, as follows: i) the maximum loan-to-value ratio is 55%; ii) the maximum dollar value for any given secured loan is not to exceed C$1,200,000; and iii) the residential property must be located in the Greater Toronto Area. Before issuing a secured loan, the Company undertakes extensive due diligence to ensure that adequate care is exercised in the funding of mortgage or loan transactions, including checking personal identification, verifying title documents, attending the property, or conducting an on-site appraisal to satisfy as to the value of the property, and reviewing application and supporting documentation with legal counsel.
As at December 31, 2023, the Company has a finance receivable balance of $8,095,354 and minimum contractual payments receivable at the end of the loan terms totaling $8,527,569. The loans will begin to mature in the second quarter of fiscal 2024.
Other Significant Operations and Principal Activities – Fiscal 2021 and 2022
2021 and 2022 At-The-Market Financings
Between July 2020 and February 2021, the Company issued and sold Class B Shares for gross proceeds of approximately US$20,000,000 under an equity distribution agreement dated July 10, 2020 entered into with A.G.P./Alliance Global Partners (the “Sales Agent”), reaching the maximum amount allowed under such agreement. On February 11, 2021, the Company entered into a new equity distribution agreement (the “2021 Equity Distribution Agreement”) with the Sales Agent and issued and sold Class B Shares for gross proceeds of US$18,167,511 between February 11, 2021 and March 12, 2021. Sales of Class B Shares under the equity distribution agreements were made through "at-the-market" offerings as defined in Rule 415(a)(4) promulgated under the Securities Act including sales made directly on or through the Nasdaq. As a result, prices may varied as between purchasers and during the period of distribution. No offers or sales of Class B Shares were made in Canada on the CSE or other trading markets in Canada.
Licensing Agreement with University Health Network
Prior to its acquisition by the Company, Lucid entered into a licensing agreement dated May 19, 2021 (the “UHN License Agreement”) with the UHN. Under the terms of the UHN License Agreement, the Company pays an annual license maintenance fee of C$100,000 to UHN until the first commercial sale of a product utilizing the intellectual property licensed to the Company under the UHN License Agreement, including Lucid-MS, is made. In addition, the Company is committed to making milestone payments totaling up to C$12,500,000 to UHN if all product development and regulatory milestones are met. Furthermore, the Company will also pay revenue milestone payments and royalties if revenue milestones from commercial sales are achieved as well as a percentage of sublicensing revenue received by the Company under any sublicense. Milestones can be extended by mutual agreement. Unless otherwise terminated in accordance with its terms, the UHN License Agreement will remain in force until the expiration of the last valid claim under the last licensed patent covering the products licensed from UHN.
Lucid Acquisition
On August 25, 2021, the Company entered into a definitive agreement (the “Master Agreement”) to acquire 100% of the issued and outstanding shares of Lucid, an early-stage Canadian-based specialty biotechnology company focused on the development of therapies to treat critical neurodegenerative diseases, for total consideration of 4,502,392 Class B Shares, 161,091 Options and 112,162 warrants to purchase Class B Shares (the “Lucid Acquisition”). 304,880 Class B Shares and all of the warrants issued as part of the consideration for the Lucid Acquisition were issued to First Republic Capital Corporation, a company controlled by Anthony Durkacz, (“First Republic”) the CEO of the Company, in exchange for securities of Lucid held by First Republic prior to the completion of the Lucid Acquisition.
On September 13, 2021, shareholder approval for the Lucid Acquisition was obtained at a special meeting of Lucid shareholders. On September 21, 2021, the transaction was completed by way of a three-cornered amalgamation between Lucid, the Company and a wholly owned subsidiary of the Company pursuant to an amalgamation agreement dated September 20, 2021, entered into among the Company, Lucid and a wholly owned subsidiary of the Company in respect of the Lucid Acquisition. The Lucid Acquisition involved the issuance of approximately 4.5 million Class B Shares as the acquisition consideration, at a deemed price of approximately US$1.56 per Class B Share. Additionally, all of the outstanding Lucid stock options and warrants became exercisable into Class B Shares, with the number and exercise price of such securities adjusted in accordance with the transaction’s exchange ratio. In connection with the closing of the Lucid Acquisition, Dr. Lakshmi Kotra, maintained his position as Lucid’s CEO.
Covar Agreement
On October 1, 2021, the Company entered into an agreement with Covar Pharmaceuticals Inc. (“Covar”), a contract development and manufacturing services organization, to commence work on providing research quantities of the Company’s drug candidate, Lucid-PSYCH, on an exclusive basis for further clinical evaluation (the “Covar Agreement”). Lucid-PSYCH is a psychoactive compound that is being researched by the Company through Lucid in connection with the treatment of major depressive disorder. Covar’s research and development facility is licensed to handle psychoactive compounds such as Lucid-PSYCH, which are “controlled substances” listed under the Controlled Drugs and Substances Act (Canada).
| 45 |
| Table of Contents |
2022 Normal Course Issuer Bid
On December 21, 2021, the Board authorized a normal course issuer bid pursuant to which the Company was able to repurchase for cancellation up to 2,000,000 Class B Shares, being approximately 5% of the Company’s issued and outstanding Class B Shares as of December 21, 2021, over a 12-month period (the “2022 NCIB”). The 2022 NCIB commenced on January 4, 2022, and was terminated on December 21, 2022. Under the 2022 NCIB, the Company repurchased for cancellation 1,999,800 Class B Shares at an average price of approximately C$1.20 per Class B Shares. All Class B Shares were repurchased through the facilities of the CSE at the prevailing market price on the CSE at the time of repurchase.
Sale of FV Pharma’s Facility
On May 6, 2022, the Company decided to focus its efforts and resources on the pharmaceutical business and initiated the process to exit the medical cannabis industry. In connection with that decision, the Company sold FV Pharma’s facility located at 520 William Street, Cobourg, Ontario, K9A 3A5 (and the 64-acre property on which the facility was located for total consideration of $12,730,942 (C$16,400,000).
Specialized Knowledge and Personnel
The Board and executive officers of the Company, led by Zeeshan Saeed, as CEO and Co-Chairman, Anthony Durkacz, Co-Chairman, and Dr. Lakshmi P. Kotra, as CEO of Lucid, have a wide combination of the skills, knowledge and experience that are necessary for the successful advancement of the Company’s business plan. Our future growth and success depend on our ability to recruit, retain, manage, and motivate our qualified employees. The inability to hire or retain experienced personnel in the pharmaceutical field could adversely affect our ability to execute our business plan and harm our operating results. Due to the specialized scientific and managerial nature of our business, we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. The competition for qualified personnel in the pharmaceutical field is intense. Due to this intense competition, we may be unable to continue to attract and retain qualified personnel necessary for the development of our business or to recruit suitable replacement personnel.
Competitive Conditions
The pharmaceutical industry market for MS drugs is highly competitive and subject to rapid change. The industry continues to expand and evolve as an increasing number of competitors and potential competitors enter the market. There are few approved therapies for progressive multiple sclerosis such as ocrelizumab by Roche and siponimod by Novartis), all of which are immunomodulatory drugs. Bruton's Tyrosine Kinase Inhibitors are also being investigated as therapies for progressive MS in Phase 3 clinical trials; for example fenebrutinib by Roche (NCT04544449) and tolebrutinib by Sanofi (NCT04458051. Kyverna Therapeutics recently announced their Phase-2 clinical trial on the cell-based therapy candidate KYV-101, in people with treatment-resistant progressive multiple sclerosis (MS). Immunic Therapeutics is developing vidofludimus as a potential treatment for all MS types; the Phase 2 CALLIPER clinical trial (NCT05054140) is testing it against a placebo specifically in people with progressive forms of the disease. Tiziana Life Sciences is developing an antibody, foralumab that is designed to reduce inflammation in the brain and spinal cord by blocking CD3, a protein found on the surface of T-cells. This type of immune cells is involved in MS progression. All known drugs and those under development are immunomodulatory, several are biologics and do not directly address demyelination, which is the hallmark feature in MS, to the best of our knowledge. Even if Lucid-MS is approved, it will compete with product offerings from large and well-established companies that have greater marketing and sales experience and capabilities than we have or our third-party research collaborators. Other companies with greater resources than us may announce similar plans in the future.
However, we believe that Lucid-MS will be superior to the other MS products for individuals with progressive because based on our current clinical work, it appears to restore myeline growth and prevents degration.
Environmental Matters
The Company expects the financial and operational effects of environmental protection requirements on its capital expenditures, profit, and competitive position in the current and future financial years to be minimal.
Employees
As at December 31, 2023, the Company directly employed eight full-time employees and one part-time employee. All of these employees are based in the Toronto, Ontario area. The Company believes its relationship with its employees, consultants and contractors is good. None of the Company’s employees are represented by a labour union or subject to a collective bargaining agreement.
Reorganizations
The Company has not completed any material reorganization within the three most recently completed financial years, except for the reorganization that occurred pursuant to the Plan of Arrangement.
C. Organizational Structure
As at the date of this Annual Report, the Company has seven subsidiaries, Lucid, FSD BioSciences, Prismic, FSD Strategic Investments, FSD Australia, FV Pharma and Celly Nu. The corporate chart of the Company including the Company’s subsidiaries, together with the jurisdiction of incorporation of the Company and its subsidiary and the percentage of voting securities beneficially owned, controlled, or directed, directly or indirectly, by the Company is as follows:
| 46 |
| Table of Contents |
The Company has 4 active subsidiaries, which are:
Lucid
Lucid is a focused on the development of therapies to treat critical neurodegenerative diseases. Lucid is currently focused on research and development of Lucid-MS, which is a molecular compound identified for potential treatment of MS. Lucid conducted research on LUCID-Psych, which was being considered as a treatment for major depressive orders, which research is currently on hold due to funding concerns.
Celly Nu
Celly Nu. Is focused on the commercialization of Unbuzzd™. The Company has entered into the Celly Nu IP License with Celly Nu in June 2023.
FSD Strategic Investments
FSD Strategic Investments was incorporated in Ontario on May 13, 2022. During Fiscal 2022, the Company invested approximately C$7 million dollars in loans secured by real property primarily located in the Greater Toronto Area. These loans are considered to be highly collateralized as they are issued up to 55% of the appraised value of the secured property. FSD Strategic Investments is focused on generating returns and cashflow through the issuance of loans secured by residential or commercial property.
FSD Australia
On November 24, 2022, the Company incorporated a new Australian subsidiary, FSD Australia, to facilitate its development of Lucid-PSYCH and other assets. The registered and head office of FSD Australia is Level 7 330 Collins Street, Melbourne VIC, 3000. FSD Australia was established to facilitate the Company’s development of Lucid-PSYCH by running Australian clinical trials in respect of Lucid-PSYCH, and potentially other assets. Subject to satisfaction of relevant eligibility criteria, FSD Australia may be entitled to claim the Australian research and development tax incentive for eligible expenditure it incurs on eligible research and development activities.
Inactive Subsidiaries
FV Pharma
The Company suspended all activities by FV Pharma Inc, which had engaged in the cannabis business, as of September 2020 and in May 2022, substantially all of the assets of FV Pharma were sold. FV Pharma has accumulated historic tax losses and continues to exist as an entity wholly owned by the Company. Upon termination of FSD-PEA, Prismic did not have any assets or remaining liabilities other than outstanding notes payable which were assumed on the acquisition of Prismic and are classified as current liabilities of the Company. Prismic has accumulated historic tax losses and continues to exist as an entity wholly owned by the Company. For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Other Significant Operations and Principal Activities – Fiscal 2021 and 2022”.
FSD BioSciences
FSD BioSciences was focused on the research and development of FSD-PEA (also known as FSD-201), an ultra-micronized PEA. On June 2, 2023, the Company terminated any further clinical development of FSD-PEA formulation for the treatment of inflammatory diseases.
Prismic
The Company does not operate through Prismic; however, Prismic holds the right to receive certain payments based on net sales of certain products under an amended and restated licensing agreement between Epitech Group SpA dated dated January 8, 2020, as amended, (the “Epitech License Agreement”) and the Company pursuant to an assignment agreement between Prismic and the Company (the “Prismic Assignment Agreement”). On June 2, 2023, the Company terminated any further clinical development of FSD-PEA formulation for the treatment of inflammatory diseases.
| 47 |
| Table of Contents |
Property, Plants and Equipment
The Company’s current operating plan does not include building infrastructure. The Company operates from its head office located in Toronto, Ontario, Canada. The Toronto office space costs approximately C$163,093 per annum (excluding operating costs and taxes) and is rented on a fixed term, ending on August 31, 2024. The Company believes that its current facilities are adequate to meet its ongoing needs and that, if the Company requires additional space, it will be able to obtain additional facilities on commercially reasonable terms.
Item 4A. Unresolved Staff Comments.
Not applicable.
Item 5. Operating and Financial Review and Prospects
A. Operating Results
See the Company’s Management’s Discussion and Analysis of Financial Condition and Results of Operations for the three months ended and fiscal years ended December 31, 2023 and 2022 (the “2023 Annual MD&A”) attached hereto as Exhibit 15.1.
B. Liquidity and Capital Resources
See the 2023 Annual MD&A attached hereto as Exhibit 15.1.
C. Research and Development, Patents and Licenses, etc.
For a discussion of our research and development activities, see “Item 4. Information on the Company. – B, Business Overview - Products and Sales” and the 2023 Annual MD&A attached hereto as Exhibit 15.1.
D. Trend Information
Other than as disclosed elsewhere in this Annual Report, we are not aware of any trends, uncertainties, demands, commitments, or events for the period from January 1, 2023 to December 31, 2023 that are reasonably likely to have a material adverse effect on our net revenues, income from continuing operations, profitability, liquidity or capital resources, or that would cause our reported financial information not necessarily to be indicative of future operating results or financial condition. For a discussion of trends, see “Item 4.B.-Business Overview” and the 20223 Annual MD&A attached hereto as Exhibit 15.1.
E. Critical Accounting Estimates
See Notes 2 and 3 to our 2023 Annual Financial Statements in Item 18.
Item 6. Directors, Senior Management and Employees
A. Directors and Senior Management.
The following table sets forth certain information with respect to our executive officers and directors as of March 28, 2024:
Name |
| Age |
| Position(s) with the Company |
| Other Directorships |
| Date of Initial Appointment |
Anthony Durkacz |
| 48 |
| Co-Executive Chairman and Director |
| Stock Trend Capital Inc. |
| May 18, 2018 |
Zeeshan Saeed |
| 54 |
| CEO, President, Co-Executive Chairman and Director |
| Celly Nu |
| May 24, 2018(1) |
Nathan Coyle |
| 43 |
| Chief Financial Officer (“CFO”) |
| N/A |
| May 5, 2021 |
Donal Carroll |
| 48 |
| Chief Operating Officer (“COO”) |
| Bird River Resources Inc.; The Hast Corporation (formerly, Senternet Phi Gamma Inc.) |
| May 18, 2018(3) |
Dr. Lakshmi P. Kotra |
| 53 |
| Director, CEO of Lucid, President of FSD Biosciences and CEO of FSD Australia |
| Celly Nu |
| November 25, 2023 |
Adnan Bashir |
| 54 |
| Director |
| N/A |
| June 1, 2021 |
Dr. Eric Hoskins |
| 63 |
| Director |
| Celly Nu; Cybin Inc.; Think Research Corporation (formerly, AIM4 Ventures Inc.) |
| June 29, 2023 |
Dr. Sanjiv Chopra (2) |
| 74 |
| Director |
| N/A |
| January 29, 2024 |
Michael (Zappy) Zapolin |
| 57 |
| Director |
| N/A |
| June 29, 2023 |
| 48 |
| Table of Contents |
Notes:
(1) | Mr. Saeed departed from his position as President and director of the Company effective January 25, 2021 but was re-elected as a director of the Company on May 14, 2021 and re-appointed as President of the Company on July 27, 2021. |
(2) | Dr. Sanjiv Chopra replaced Nitin Kaushal on the Board effective January 29, 2024. |
(3) | Mr. Carroll was initially appointed to the Board on May 18, 2018 until August 2018. On July 27, 2018, he was appointed interim CFO and on January 2, 2020, he became the permanent CFO until May 4, 2021. On May 14, 2021, he once again was a Board member until January 29, 2024. In August 2021, he became COO. |
Term of Office
Each director is to serve until his or her successor is elected and qualified or until his death, resignation, or removal. Our Board appoints our officers and each officer is to serve until his successor is appointed and qualified or until his or her death, resignation, or removal.
Executive Officers
Anthony Durkacz
Mr. Durkacz served as the Company’s interim CEO from July 2021 to July 2023, and has served as its Executive Co-Chairman since May 2021 and has served as a member of the Board since June 2018. Mr. Durkacz is also a director and the Executive Vice-President of First Republic and has served in those roles since 2014. In addition, Mr. Durkacz is the Chairman of World Class Extractions Inc. (CSE: PUMP; OTCQB: WCEXF) and has served in that role since 2018. Prior to co-founding the Company, from January 2013 to December 2013, Mr. Durkacz was President of Capital Ideas Investor Relations. He previously served as the CFO and a director of Snipp Interactive Inc. (TSXV: SPN.V), a global marketing solutions company that provides a modular software-as-a-service technology suite from January 2011 to January 2013. Mr. Durkacz was instrumental in the financing and public listing of Snipp Interactive Inc. with operations in Canada, the United States, Mexico, and India. From 2006 to 2009, he served as COO and CFO of MKU Canada Inc. and engaged in mergers and acquisitions of companies around the world. From 2002 to 2006, Mr. Durkacz served as the CFO and a director of Astris Energi Inc., a dual-listed public company in the United States and Canada which was acquired by an international conglomerate. Mr. Durkacz began his career at TD Securities on the capital markets trading floor. He holds an Honours Bachelor of Business Administration degree from Brock University with a major in both Accounting and Finance.
Zeeshan Saeed
Mr. Saeed, a co-founder of the Company, has served as the Company’s President and Executive Co-Chairman since May 2021. He became the Company’s CEO on July 4, 2023. Previously, he served as President of the Company from May 2019 to January 2021 and as a director from May 2018 to January 2021. From December 2017 to May 2019, Mr. Saeed served as Executive Vice President of FV Pharma, a subsidiary of the Company and a former licensed producer of cannabis in Canada under the Cannabis Act (Canada). From October 2013 to December 2017, he provided consulting services to FV Pharma from April 2003 to December 2017, Mr. Saeed served as President of ZZ Telecommunications Inc., a long-distance telecommunications common carrier. Mr. Saeed was the founder and CEO of Platinum Telecommunications Inc. from 2011 to 2013. He has a Bachelor of Science in Mechanical Engineering from the University of Engineering and Technology Lahore.
Nathan Coyle
Nathan Coyle has served as the Company’s CFO since May 2021. He previously served as the Company’s Corporate Controller from January 2020 to May 2021 and as Controller of Chem-Ecol Ltd. from July 2013 to January 2020. From July 2013 to January 2020, Mr. Coyle worked with Turtle Holdings Limited, a family investment company, implementing corporate strategies to maximize growth. From 2005 to 2013, Mr. Coyle was with Illinois Tool Works, where he was a key player in restructuring the organization, shaping the growth, and streamlining businesses within his industrial packaging segment. Mr. Coyle’s involvement in multiple mergers and acquisitions and integrating those organizations was key to company growth. Mr. Coyle holds a Bachelor of Business Administration with honours from Brock University and is a Chartered Professional Accountant.
Dr. Lakshmi P. Kotra
Dr. Lakshmi P. Kotra has served as CEO of Lucid since September 2020, which he co-founded in 2020 and has served as a director on the Board since November 2022. Dr. Kotra received his Ph.D. in Pharmacy (Medicinal Chemistry) from the University of Georgia under Prof. David Chu’s supervision, and completed postdoctoral training at Wayne State University under Prof. Shahriar Mobashery’s supervision. He joined the Faculty of Pharmacy, University of Toronto in 2000, and University Health Network in 2006, where he led a research group and drug discovery program with multiple portfolios. An academic entrepreneur, Dr. Kotra has contributed to a number of important drug discovery and development projects, including metabolic disorders, neurodegenerative and immunological disorders, anti-HIV drugs, antibacterials, and antimalarials. He has authored/co-authored over 130 publications and delivered over 140 scientific talks internationally. Dr. Kotra is the recipient of several awards for his accomplishments, including the Julia Levy Award in 2021 from the Society of Chemical Industry (SCI) Canada in recognition of his substantial contribution to the successful commercialization of innovation in Canada in the field of biomedical science and engineering. In addition to Lucid, he co-founded WinSanTor Biosciences, a San Diego, CA-based company developing treatments for peripheral neuropathies, and CannScience Innovations focused on medical cannabis and cannabinoids. Dr. Kotra has been serving the Company as Chief Executive Officer of its wholly owned subsidiary, Lucid, since the completion of the Company’s acquisition of Lucid in September 2021.
| 49 |
| Table of Contents |
Donal Carroll
Mr. Carroll joined the Company as interim CFO in 2018 and was appointed to the position on a permanent basis in December 2019, where he served until May 2021. Mr. Carroll was appointed as COO of the Company on August 15, 2021. Mr. Carroll has also served as a director on the Company’s Board from May 2018 to July 2018 and from May 2021 to January 2024. Mr. Carroll has 20 years of corporate finance leadership and public company experience, as well as experience in syndicate investing both in equity and debt securities. From June 2005 to January 2008, he served as an Accounting Supervisor with Alberto Culver (now Unilever (NYSE:UL)), from February 2008 to October 2013, Mr. Carroll has served as Controller with Videojet Technologies, and from October 2013 to July 2017, he served as a Corporate Controller with Cardinal Meats, where he was instrumental in major restructuring activities, mergers and acquisitions and the implementations of new internal controls and ERP systems. Mr. Carroll has been a Director of Bird River Resources Inc. since August 2019 and was a Director of Climb Credit Inc. He holds a CPA-CMA designation as well as a Bachelor of Commerce degree from University College Dublin.
Non-Employee Directors
Adnan Bashir
Mr. Bashir has over 14 years of experience in strategic management and operations. He is the founder and President of 58Northwest Inc., a management consulting and marketing services company, and has held the role since 2018. From 2005-2018, Mr. Bashir was General Manager for Al Batha Group, a diversified business conglomerate based in Dubai, UAE. Mr. Bashir was responsible for overseeing the management and operations of 4 companies within the group and was instrumental in acquiring and developing new businesses and partners from Europe, the US and China. During his tenure at Al Batha Group, Mr. Bashir gathered extensive experience in executing turnaround strategies, transforming weak businesses into sustainable and profitable ones and implementing new technologies. Mr. Bashir holds a Bachelor of Science Degree in Mechanical Engineering from University of Engineering and Technology Lahore and has completed extensive executive education, including in strategic management, audit, sales management, and technical management.
Dr. Eric Hoskins
Dr. Eric Hoskins is a medical doctor and public health expert with more than 30 years’ experience in healthcare, public policy, economic development, and international trade. Dr. Hoskins recently served as the Chair of the Federal Advisory Council on the Implementation of National Pharmacare.
He previously served as president of War Child Canada and was awarded the Order of Canada in 2007 for his humanitarian work. During Dr. Hoskins’ nearly 10 years as a member of provincial parliament in Ontario, he held several cabinet positions including Minister of Health and Long-Term Care; Economic Development, Trade and Employment; Children and Youth Services; as well as Citizenship and Immigration. As a tireless health advocate, Dr. Hoskins has many years of experience creating and delivering health programs in Africa and the Middle East.
Dr. Sanjiv Chopra
Sanjiv Chopra, MD, is Professor of Medicine and served as Faculty Dean for Continuing Medical Education at Harvard Medical School for 12 years. He serves as a Marshall Wolf Distinguished Clinician Educator Brigham and Women’s Hospital.
Dr. Chopra has more than 170 publications and ten books to his credit. Dr. Chopra is Editor-in-Chief of the Hepatology Section of UpToDate, the most widely used electronic textbook in the world subscribed to by more than 1.5 million physicians in 195 countries.
He is a sought after inspirational speaker across the United States and abroad, addressing diverse audiences on topics related to medicine, leadership, happiness, and living with purpose.
Michael (Zappy) Zapolin
Zappy Zapolin is a well-known futurist, psychedelic concierge to the stars, and award-winning filmmaker who is dedicated to expanding human consciousness.
As the youngest Vice President in the history of investment bank Bear Stearns, Zappy is a frequent commentator on investment opportunities in the biotech and emerging psychedelic industry.
| 50 |
| Table of Contents |
Certain Proceedings involving Directors
Mr. Durkacz has been serving as director of FSD since June 18, 2018. On March 5, 2021, FSD was subject to a court order with respect to the 2021 Annual and Special Meeting which, among other things, prohibited the Company’s then CEO and directors, other than Mr. Durkacz, from voting certain of their shares at the 2021 Annual and Special Meeting. On April 9, 2021, the Court ordered an injunction restraining the Company’s then CEO and former directors, other than Mr. Durkacz, from authorizing or undertaking any transaction by FSD other than in the ordinary course of business, issuing any Class B Shares or authorizing the payment of any form of compensation to such former CEO and directors prior to the 2021 Annual and Special Meeting.
Family Relationships
There are no family relationships among any of our executive officers or directors.
Special Arrangements
Not applicable.
B. Compensation.
Executive Compensation
The purpose of this Compensation Discussion and Analysis is to provide information about the Company’s philosophy, objectives, and processes regarding executive compensation. This disclosure is intended to communicate compensation provided to: (i) the CEO; (ii) the CFO; (iii) each of the three most highly compensated executive officers of the Company, including any of its subsidiaries, or the three most highly compensated individuals acting in a similar capacity, other than the CEO and CFO, as at the end of the most recently completed financial year whose total compensation was, individually, more than C$150,000; and (iv) each individual who would be a NEO under paragraph (c) but for the fact that the individual was neither an executive officer of the Company or its subsidiaries, nor acting in a similar capacity, as at December 31, 2023, (collectively, the “NEOs”) and (v) the directors of the Company.
During the year ended December 31, 2023, the NEOs of the Company were as follows:
| 1) | Zeeshan Saeed, CEO and Co-Executive Chairman, and Director of the Company; |
| 2) | Anthony Durkacz, Co-Executive Chairman, and Director of the Company; |
| 3) | Nathan Coyle, CFO of the Company; |
| 4) | Donal Carroll, COO of the Company; and |
| 5) | Dr. Lakshmi Kotra, Director of the Company, CEO of Lucid, President of FSD Biosiences, and CEO FSD Australia. |
The description of the Company’s compensation philosophy and objectives and the elements of such compensation for the year ended December 31, 2023 is set forth below:
Compensation Philosophy and Objectives
The executive compensation program adopted by the Company and applied to its executive officers is designed to attract and retain qualified and experienced executives who will contribute to the success of the Company. The executive compensation program attempts to ensure that the compensation of the senior executive officers provides a competitive base compensation package and a strong link between corporate performance and compensation. Senior executive officers are motivated through the program to enhance long-term shareholder value and rewarded for their yearly individual contribution in the context of overall annual corporate performance.
Elements of Compensation
The executive compensation program during the year, ended December 31, 2023 consisted of three principal components: (i) base compensation; (ii) potential annual incentive award; (iii) Options to Class B Shares (“Options”); (iv) restricted share units (“RSUs”); and (v) performance share units (“PSUs”). Options, RSUs, and PSUs are granted pursuant to the Company’s equity incentive plan (the “Equity Incentive Plan”) which replaced the Company’s rolling stock option plan (the “Stock Option Plan”). For the year ended December 31, 2023, all executive compensation was determined and administered by the Board based on recommendations by the compensation, nominating and governance committee of the Company (“Compensation, Nominating and Governance Committee”).
Compensation Governance
The Compensation, Nominating and Governance Committee is currently comprised of three directors, Zeeshan Saeed (Chair), Michael (Zappy) Zapolin, and Adnan Bashir. Zeeshan Saeed is not an independent director, and Michael (Zappy) Zapolin, and Adnan Bashir are independent directors.
The primary goal of the Company’s executive compensation program is to attract and retain the key executives necessary for the Company’s long term success, to encourage executives to further the development of the Company and its operations, and to motivate top quality and experienced executives.
| 51 |
| Table of Contents |
The Compensation, Nominating and Governance Committee has been tasked with establishing an executive compensation program, which, prior to May 16, 2022, included equity compensation by way of share awards and Options granted under the Stock Option Plan. As of May 16, 2022, the executive compensation program includes equity compensation by way of share awards, RSUs, PSUs and Options granted under the Equity Incentive Plan.
The Compensation, Nominating and Governance Committee reviews the adequacy of remuneration for the executive officers by evaluating their performance in light of the Company’s goals and objectives, the bonus opportunities contained in their employment agreements, and by comparing the performance of the Company with other reporting issuers of similar size in the same industry.
The Board is of the view that all elements of the total program should be considered, rather than any single element. As such, the Company does not use fixed criteria in determining the mix of compensation and instead determines compensation based on a contextual analysis of the Company. While the Company does not have a formally established peer group in determining compensation, the Compensation, Nominating and Governance Committee will on occasion reference other comparable publicly traded Canadian companies to align its compensation practices with market practice.
The terms of any proposed compensation for the directors of the Company who are not also officers of the Company (including any Options, RSUs and PSUs to be granted) will be determined by the Compensation, Nominating and Governance Committee. The compensation program is designed to provide income certainty, to attract and retain executives and to provide incentives for the achievement of both short-term and long-term objectives of the Company.
Compensation Process
The Compensation, Nominating and Governance Committee, through discussion without any formal objectives, criteria, or analysis, determines the compensation of the Company’s executive officers. The Compensation, Nominating and Governance Committee has no formal criteria or goals tied to total compensation or any significant element of total compensation. The Board, through the Compensation, Nominating and Governance Committee, is responsible for determining all forms of compensation, including share-based compensation and long-term incentives in the form of Options, RSUs and PSUs to be granted to the Company’s executive officers and directors, and for reviewing the recommendations respecting compensation of other officers of the Company from time-to-time, to ensure such arrangements reflect the responsibilities and risks associated with each position. The Compensation, Nominating and Governance Committee determines compensation by considering: (i) recruiting and retaining executives critical to the Company’s success and the enhancement of shareholder value; (ii) providing fair and competitive compensation; (iii) balancing the interests of management and the Shareholders; and (iv) rewarding performance, both on an individual basis and with respect to the Company’s operations in general.
Annual Incentive Awards
Annual incentive awards are designed to motivate NEOs to achieve the Company’s short-term corporate goals, and rewards individual and overall performance. Annual incentives are based on objective, identifiable measures set at the beginning of each financial year at the discretion of the Compensation, Nominating and Governance Committee, which may vary from year to year and incentive payments are expected to be determined by the Board on the recommendation of the Compensation, Nominating and Governance Committee.
Option Awards
Long-term incentives in the form of Options are intended to align the interests of the Company’s directors and its executive officers with those of its Shareholders, to provide a long-term incentive that rewards these individuals for their contribution to the creation of Shareholder value, and to reduce the cash compensation the Company would otherwise pay. The Equity Incentive Plan is administered by the Board. While the Company does not have a formally established peer group in determining compensation, in considering the number of Options to be granted to the NEOs, reference is made to the number of Options granted to officers of other comparable publicly traded Canadian companies. The Compensation, Nominating and Governance Committee also considers previous grants of equity incentives and the overall number of equity incentives that are outstanding relative to the number of outstanding Shares in determining whether to make any new grants of Options and the size and terms of any such grants, as well as the level of effort, time, responsibility, ability, experience, and level of commitment of the executive officer in determining the level of Option compensation.
Share Unit Awards
The Equity Incentive Plan provides that Eligible Persons (as defined in the Equity Incentive Plan) may be allocated share units in the form of RSUs or PSUs (collectively, “Share Units”), which represent the right to receive an equivalent number of Class B Shares or the Market Price (as defined in the Equity Incentive Plan) in cash on the vesting date. The issuance of Class B Shares may be subject to vesting requirements similar to those described above with respect to the exercise of Options, including such time- or performance-based conditions as may be determined from time to time by the Board in its discretion.
The Equity Incentive Plan provides for the express designation of Share Units as either RSUs, which have time-based vesting conditions, or PSUs, which have performance-based vesting conditions over a specified period. The Equity Incentive Plan provides that if Share Units are scheduled to settle during a blackout period, such settlement shall be postponed until the trading day following the date on which the blackout period ends (or as soon as practicable thereafter, and in any event, within 10 business days following the end of the blackout period), and the Market Price of any RSUs or PSUs settled in cash will be determined as of the trading day immediately prior to the settlement date.
| 52 |
| Table of Contents |
The Compensation, Nominating and Governance Committee also considers previous grants of equity incentives and the overall number of equity incentives that are outstanding relative to the number of outstanding Shares in determining whether to make any new grants of Share Units and the size and terms of any such grants, as well as the level of effort, time, responsibility, ability, experience, and level of commitment of the executive officer in determining the level of Share Unit compensation.
Insider Trading and Blackout Period Policy
All of the Company’s executives, other employees and directors are subject to the Company’s Insider Trading and Blackout Period Policy, which prohibits trading in the Company’s securities while in possession of material undisclosed information about the Company. Under this policy, such individuals are also prohibited from entering into hedging transactions involving securities of the Company, such as short sales, puts and calls. Furthermore, subject to certain limited exceptions, the Company permits executives, including the NEOs, to trade in the Company’s securities only during prescribed trading windows.
Risk Analysis
The Board and Compensation, Nominating and Governance Committee considered risks associated with executive compensation and do not believe that the Company’s executive compensation policies and practices encourage its executive officers to take inappropriate or excessive risks. Aside from a fixed base salary, NEOs are compensated through the granting of awards, which are compensation that is both “at risk” and associated with long-term value creation. The value of such compensation is dependent upon Shareholder return over award vesting periods which reduces the incentive for executives to take inappropriate or excessive risks as their long-term compensation is at risk.
Performance graph
The following performance graph compares the total cumulative return to a Shareholder who invested C$100 in Shares on January 1, 2018, assuming reinvestment of dividends, with the cumulative total return on the S&P/TSX Composite Total Return Index and SPDR S&P Biotech ETF for each year following January 1, 2018. The performance of the Shares as set out in the graph below does not necessarily indicate future performance.
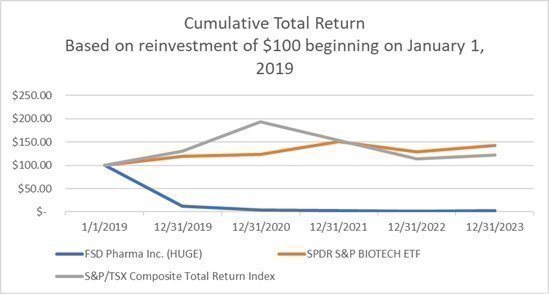
| 53 |
| Table of Contents |
|
| December 31, 2019 (C$) |
|
| December 31, 2020 (C$) |
|
| December 31, 2021 (C$) |
|
| December 31, 2022 (C$) |
|
| December 31, 2023 (C$) |
| |||||
FSD Pharma Inc. |
|
| 11.85 |
|
|
| 3.40 |
|
|
| 2.23 |
|
|
| 1.72 |
|
|
| 2.01 |
|
SPDR S&P Biotech ETF (NYSEARCA: XBI) |
|
| 118.93 |
|
|
| 123.96 |
|
|
| 151.55 |
|
|
| 129.57 |
|
|
| 143.46 |
|
S&P/TSX Composite Total Return Index |
|
| 130.54 |
|
|
| 193.22 |
|
|
| 153.66 |
|
|
| 113.92 |
|
|
| 122.55 |
|
The Company is of the view that compensation levels for the directors and executive officers cannot and should not be directly compared to year-over-year relative market performance. Market performance is impacted by a number of external factors beyond the control of management and an increase or decrease in the market price and thus should not be a determining factor in establishing the annual compensation of the Company’s directors and executive officers. The stock price directly impacts the benefits enjoyed by the directors and executive officers from the Stock Option Plan and Equity Incentive Plan. As a result, the trend shown in the above graph does not necessarily correspond to the Company’s compensation to its directors and executive officers for the same period.
The Company’s compensation package is based on competitive compensation trends and the value of the services provided and is designed to attract and retain top quality personnel for the longer term in order to manage and grow the business through both adverse and favorable economic cycles. These factors may not yield immediate results in stock price.
Summary Compensation Table
Name and Principal Position | Year | Salary (US$) | Share-Based Awards(1) (US$) | Option Based Awards(2) (US$) | Non-Equity Incentive Plan Compensation (US$) | Pension Value | All Other Compensation(3) (US$) | Total Compensation (US$) | |
Annual incentive plans |
Long-term incentive plans | ||||||||
Anthony Durkacz(4) Co-Executive Chairman and Director
| 2023 | 222,625 | Nil | 377,248 | Nil | Nil | Nil | 599,873 | |
2022 | 239,269 | 316,834(5) | Nil | Nil | Nil | 136,710(6) | 692,813 | ||
2021 | 103,703 | Nil | 777,798 | Nil | Nil | Nil | 881,501 | ||
Nathan Coyle(7) CFO | 2023 | 178,208 | Nil | Nil | Nil | Nil | Nil | 178,208 | |
2022 | 191,891 | Nil | Nil | Nil | Nil | Nil | 191,891 | ||
2021 | 189,186 | Nil | 46,586 | 15,734 | Nil | Nil | 251,506 | ||
Zeeshan Saeed(8) CEO& Co-Executive Chairman | 2023 | 222625 | Nil | 377,248 | Nil | Nil | Nil | 599,873 | |
2022 | 239,269 | 398,010 (5) | Nil | Nil | Nil | 136,710 (6) | 773,989 | ||
2021 | 225,271 | Nil | 777,798 | Nil | Nil | Nil | 1,003,069 | ||
Donal Carroll(9) COO | 2023 | 225,625 | Nil | 377,248 | Nil | Nil | Nil | 602,873 | |
2022 | 239,269 | 321,543(5) | 34,519 | Nil | Nil | 136,710 (6) | 732,041 | ||
2021 | 351,406 | Nil. | 777,798 | 78,670 | Nil | 4,779(6) | 1,212,653 | ||
Dr. Lakshmi Kotra Director, CEO of Lucid, President of FSD Biosiences, and CEO FSD Australia | 2023 | 144,724 | Nil. | 377,248 | Nil | Nil | Nil | 521,972 | |
2022 | 331,510(10) | 80,212(5)(10) | Nil | Nil | Nil | Nil | 411,722 | ||
2021 | 44,430(10) | Nil | 172,799 | Nil | Nil | Nil | 217,228.96 | ||
| 54 |
| Table of Contents |
Notes:
(1) | “Share-based Award” means an award of Class B Shares and includes PSUs and RSUs. The dollar amount disclosed is based on the closing price per Class B Share at the date of each grant. |
(2) | “Option-based Award” means an award of Options under the Stock Option Plan or the Equity Incentive Plan. This does not represent cash paid to the NEO. This figure is based on the grant date fair value of such Options. The grant date fair value was determined in accordance with International Financial Reporting Standards. This methodology was chosen in order to be consistent with the accounting fair value used by the Company in its financial statements, and the Black-Scholes option pricing model is a commonly used methodology for valuing options which provides an objective and reasonable estimate of fair value. Calculating the value of stock options using the Black-Scholes option pricing model is very different from a simple “in-the-money” value calculation. Accordingly, caution must be exercised in comparing grant date fair value amounts with cash compensation or an in-the-money option value calculation. |
(3) | Includes Company-paid health and life insurance benefits and car allowances for all NEOs. |
(4) | Mr. Durkacz has been a director of the Company since June 18, 2018 and was appointed as interim CEO of the Company on July 27, 2021. |
(5) | This figure represents PSUs granted under the Equity Incentive Plan. This does not represent cash paid to the NEO. This figure is based on the incremental grant date fair value of such PSUs. The grant date fair value was determined in accordance with International Financial Reporting Standards. The incremental fair value is the difference between the fair value of the PSUs based on the share price on the grant date and the fair value of the share options cancelled as measured on date of modification. This methodology was chosen in order to be consistent with the accounting fair value used by the Company in its financial statements. For further details on the valuation of PSUs, see Note 16 to the 2023 Annual Financial Statements, starting at page F-1. |
(6) | These amounts represent cash bonuses paid to the NEO. |
(7) | Mr. Coyle was appointed as the CFO of the Company on May 4, 2021, initially on interim and then on a permanent basis. |
(8) | Mr. Saeed departed from his position as President and director of the Company effective January 25, 2021 but was re- elected as a director of the Company on May 14, 2021 and re-appointed as President of the Company on July 27, 2021. |
(9) | Mr. Carroll resigned as CFO of the Company on May 4, 2021 and was elected as a director of the Company at the 2021 Meeting on May 14, 2021. Mr. Carroll was appointed as COO of the Company on August 15, 2021. |
(10) | Dr. Kotra is not paid a salary by the Company, but was paid a consulting fee of $331,510.00 for the year ended December 31, 2022 and $44,430.00 for the year ended December 31, 2022. Dr. Kotra’s PSU awards for the year ended December 31, 2022 includes 51,500 PSUs issued to ILace Therapeutics International Inc., a corporation controlled by Dr. Kotra. |
Outstanding Option-Based and Share-Based Awards
The following table sets forth information with respect to Options, RSUs and PSUs held by the NEOs which were outstanding as of December 31, 2023:
|
| Option-based Awards |
| Share-based Awards |
| |||||||||||||||||
Name |
| Number of securities underlying unexercised options (#) (b) |
|
| Option exercise price ($) (c) |
|
| Option expiration date (d) |
| Value of unexercised in-the-money options ($) (e) |
| Number of shares or units of shares that have not vested (#) (f) |
| Market or payout value of share- based awards that have not vested ($) (g) |
|
| Market or payout value of vested share-based awards not paid out or distributed ($) (h) |
| ||||
Zeeshan Saeed CEO, Co-Executive Chairman, and Director |
|
| 500,000 |
|
|
| 1.30 |
|
| 2028-01-25 |
| Nil |
| Nil |
| Nil |
|
|
| 596,080 |
| |
Nathan Coyle |
|
| 5,000 |
|
|
| 2.88 |
|
| 2024-03-18 |
| Nil |
| Nil |
| N/A |
|
|
| N/A |
| |
CFO |
|
| 30,000 |
|
|
| 1.73 |
|
| 2024-06-01 |
| Nil |
| Nil |
| N/A |
|
|
| N/A |
| |
Anthony Durkacz Co-Executive Chairman & Director |
|
| 500,000 |
|
|
| 1.30 |
|
| 2028-01-25 |
| Nil |
| Nil |
| Nil |
|
|
| 596,080 |
| |
Lakshmi Kotra Director, CEO of Lucid, President of FSD Biosiences, and CEO FSD Australia |
|
| 500,000 |
|
|
| 1.30 |
|
| 2028-01-25 |
| Nil |
| Nil |
| Nil |
|
|
| 184,744 |
| |
Donal Carroll COO |
|
| 500,000 |
|
|
| 1.30 |
|
| 2028-01-25 |
| Nil |
| Nil |
| Nil |
|
|
| 600,596 |
| |
| 55 |
| Table of Contents |
Incentive plan awards – value vested or earned during the year
Name (a) |
| Option-based awards – Value vested during the year ($) (b) |
|
| Share-based awards – Value vested during the year ($) (c) |
| Non-equity incentive plan compensation – Value earned during the year ($) (d) | ||
Zeeshan Saeed CEO, Co-Executive Chairman, and Director |
| 377,248 |
|
| Nil |
| Nil | ||
Nathan Coyle CFO |
| Nil |
|
| Nil |
| Nil | ||
Anthony Durkacz Co-Executive Chairman & Director |
| 377,248 |
|
| Nil |
| Nil | ||
Lakshmi Kotra Director, CEO of Lucid, President of FSD Biosiences, and CEO FSD Australia |
| 377,248 |
|
| Nil |
| Nil | ||
Donal Carroll COO |
| 377,248 |
|
| Nil |
| Nil | ||
Stock option plans and other incentive plans
Equity Incentive Plan
On May 16, 2022, the Board adopted the Equity Incentive Plan, which was effective on June 29, 2023, upon the Company receiving shareholder approval at the annual general and special meeting. The Equity Incentive Plan replaced the Stock Option Plan.
Purpose of the Equity Incentive Plan
The principal purposes of the Equity Incentive Plan is to: (i) promote a further alignment of interests between officers, employees and other eligible service providers and the Shareholders, (ii) associate a portion of the compensation payable to officers, employees and other eligible service providers with the returns achieved by the Shareholders, and (iii) attract and retain officers, employees and other eligible service providers with the knowledge, experience and expertise required by the Company.
The Equity Incentive Plan contains provisions applicable to all grants of Options, RSUs, and PSUs (collectively, “Awards”) in the capital of the Company, to Eligible Persons (as defined in the Equity Incentive Plan).
Equity Incentive Plan Maximum, Limits and Vesting Restrictions
| 56 |
| Table of Contents |
The Equity Incentive Plan provides that: (i) the aggregate number of Class B Shares that may be issued pursuant to the Equity Incentive Plan, together with all other security-based compensation arrangements of the Company, shall be equal to 10% of the issued and outstanding Class B, from time to time; (ii) the aggregate number of Class B Shares reserved for issuance pursuant to the Equity Incentive Plan to any one participant, together with all other security-based compensation arrangements of the Company, must not exceed 5% of the aggregate issued and outstanding Class B Shares; (iii) the maximum number of Class B Shares (a) issued to Insiders (as defined in the Equity Incentive Plan) within any one year period, and (b) issuable to Insiders, at any time, under the Equity Incentive Plan or when combined with all other security-based compensation arrangements of the Company, shall not exceed 10% of the number of the aggregate issued and outstanding Class B Shares; and (iv) the maximum number of Class B Shares issued within any 12-month period to an individual Employed (as defined in the Equity Incentive Plan) by the Company or any of its subsidiaries, or a service provider engaged in investor relations activities must not exceed 1% of the issued and outstanding Class B Shares. Unless otherwise designated by the Board in the applicable grant agreement, 25% of the Options granted under the Equity Incentive Plan shall vest and become exercisable on the first anniversary of the grant date and the remaining 75% of the Options shall vest and become exercisable in equal quarterly installments beginning on the 15-month anniversary of the grant date and ending on the four-year anniversary of the grant date.
As at December 31, 2023 the Company had issued an aggregate total of 2,460,615 Awards outstanding under the Equity Incentive Plan, comprised of 2,460,615 Options, Nil RSUs, Nil PSUs and a total of 1,477,057 Class B Shares remained authorized for issuance under the Equity Incentive Plan.
The Equity Incentive Plan is administered by the Board, which has full and complete discretionary authority with respect to the granting of all awards thereunder. The Board may, subject to applicable law and certain restrictions, delegate its powers, rights, and duties under the Equity Incentive Plan to a committee of the Board, a person or persons, as it may determine, from time to time, on terms and conditions as it may determine. Awards may be granted under the Equity Incentive Plan to such service providers of the Company and its subsidiaries, if any, as the Board may from time to time designate. With respect to Options, exercise prices will be determined by the Board but will, in no event, be less than the market price of the Class B Shares on the grant date or the lowest price permitted by the policies of any stock exchange on which the Class B Shares may be listed. Generally, all Options granted under the Equity Incentive Plan will expire not later than the date that is ten years from the date that such Options are granted.
With respect to RSUs and PSUs, the number of such awards granted shall be determined by dividing the grant value for such grant, as determined by the Board, by the market price of a subordinate voting share as at the grant date (or otherwise determined by the Board). Settlement of RSUs and PSUs shall be made by the issuance of one Class B Share for each RSU or PSU then being settled, a cash payment equal to the market price of one subordinate voting share on the vesting date of the RSUs or PSUs being settled in cash or a combination of Class B Shares and cash, all as determined by the Board in its discretion, or as specified in applicable grant agreement. Subject to certain exceptions, any awards granted under the Equity Incentive Plan are not transferable or assignable other than by testamentary disposition or pursuant to the laws of intestate succession.
Stock Option Plan
The Stock Option Plan was replaced by the Equity Incentive Plan. A summary of the material terms of the Stock Option Plan can be found in the management information circular dated May 19, 2023. There are no predecessor Option outstanding under the Stock Option Plan.
Pension Disclosure
The Company established a 401(k) plan on January 31, 2021, but the plan was terminated on December 31, 2021. The Company currently does not have any pension plans that provide for payments or benefits at, following, or in connection with retirement.
Termination and Change of Control
The Company has entered into executive employment agreements with each of the NEOs (the “Executive Agreements”) other than Nathan Coyle. Each Executive Agreement provides for the NEO’s annual base salary, vacation entitlement and benefits.
The following is a description of material provisions of the Executive Agreements as they relate to termination and change of control.
Anthony Durkacz (Co-Executive Chairman and Director)
Mr. Durkacz has an executive employment agreement with the Company. In the event of both a change of control transaction and Mr. Durkacz ceasing to be employed by the Company for any reason, all outstanding unvested Options held as of the date Mr. Durkacz ceases to be employed by the Company shall immediately vest and remain outstanding and exercisable for a period of five years from that date. All vested Options held by Mr. Durkacz that are outstanding on that date shall remain outstanding and be exercisable for five years following the applicable vesting date of the Options. Additionally, in the event of any change of control transaction, the expiry date of each Option issued to Mr. Durkacz prior to the date, and during the term, of his Executive Agreement shall be the date that is five years from the applicable vesting date of such Option. In the event Mr. Durkacz’s employment is terminated as a result of a change of control transaction, he would be entitled to receive C$50,000.
Zeeshan Saeed (CEO, Co-Executive Chairman and Director)
Mr. Saeed has an executive employment agreement with the Company. In the event the Company terminates Mr. Saeed’s employment without cause or in the event of any termination of the agreement by either party following a change of control, the Company will provide Mr. Saeed with a cash payment in an amount equal to 24-months (2 years) compensation, being the sum of: (i) base salary, (ii) the applicable target bonus, and (iii) the cash value of any stock grants provided in the last 12-months. In the event of a change of control transaction and Mr. Saeed ceasing to be employed by the Company for any reason, all outstanding unvested Options held by Mr. Saeed as of the date that he ceases being employed by the Company shall immediately vest and shall remain outstanding and be exercisable for five years following that date. All vested Options held by Mr. Saeed that are outstanding on that date will remain outstanding and be exercisable for five years following the applicable vesting date of the Options. Additionally, in the event of any change of control transaction, the expiry date of each Option issued to Mr. Saeed prior to the date, and during the term, of his Executive Agreement shall be the date that is five years from the applicable vesting date of such Option. In the event Mr. Saeed’s employment is terminated without cause or as a result of a change of control transaction, he would be entitled to receive C$600,000.
| 57 |
| Table of Contents |
Donal Carroll (COO)
Mr. Carroll has an executive employment agreement with the Company. In the event the Company terminates Mr. Carroll’s employment without cause or in the event of any termination of the agreement by either party following a change of control, the Company shall pay Mr. Carroll a cash payment in an amount equal to 24 months (two years) compensation, being the sum of: (i) base salary, (ii) the applicable target bonus, and (iii) the cash value of any stock grants provided in the last 12-months. In the event of a change of control transaction and Mr. Carroll ceasing to be employed by the Company for any reason, all outstanding unvested Options, as of the date Mr. Carroll ceases to be employed by the Company shall immediately vest and shall remain outstanding and be exercisable for a period of five years following that date. All vested Options held by Mr. Carroll that are outstanding as of that date shall remain outstanding and be exercisable for five years following the applicable vesting date of the Options. Additionally, in the event of any change of control transaction, the expiry date of each Option issued to Mr. Carroll prior to the date, and during the term, of his Executive Agreement shall be the date that is five years from the applicable vesting date of such Option. In the event Mr. Carroll’s employment is terminated without cause or as a result of a change of control transaction, he would be entitled to receive C$600,000.
Nathan Coyle (CFO)
Mr. Coyle has an executive employment agreement with the Company. In the event the Company terminates Mr. Coyle’s employment without cause or in the event of any termination of the agreement by either party following a change of control, the Company shall pay Mr. Coyle a cash payment in an amount equal to one month’s salary per year of employment up to a maximum of 12 months and no less than four months compensation. In the event of a change of control transaction and Mr. Coyle ceasing to be employed by the Company for any reason, the expiry date of each Option issued to Mr. Coyle prior to the date, and during the term, of his Executive Agreement shall be the date that is five years from the applicable vesting date of such Option.
Liability Insurance of Directors and Officers
The Company has directors’ and officers’ liability insurance coverage for losses to the Company if the Company is required to reimburse directors and officers, where permitted, and for direct indemnity of directors and officers where corporate reimbursement is not permitted by law. This insurance protects the Company against liability (including costs), subject to standard policy exclusions, which may be incurred by directors and/or officers acting in such capacity for the Company. All directors and officers are covered by the policy and the amount of insurance applies collectively to all. The annual cost for this insurance during the year ended December 31, 2023 was C$108,000 per annum.
Indemnification
Amended and Restated By-Law No. 1 provides for indemnification of each of the Company’s directors, former directors, officers, former officers, respective heirs and legal representatives of directors and officers and each individual who acts or acted at the Company’s request as a director or officer of a body corporate or an individual acting in a similar capacity of another entity (each, an “Indemnified Person”) against all costs, charges and expenses reasonably incurred in respect of any civil, criminal or administrative, investigative or other proceeding to which that Indemnified Person is made a party by reason of being or having been at the relevant time a director or officer of the Company or of a body corporate of which the Company is or was a shareholder or creditor, or by reason of having acted in a similar capacity for any other entity that is or was at the relevant time directly or indirectly owned or controlled by the Company, as required by the OBCA.
The Company has entered into indemnity agreements with each director and officer providing that if such director or officer is or was involved in any threatened, pending or completed proceeding by reason of the fact that such director or officer is or was a director or officer of the Company or is or was serving at the Company’s request as a director or officer of another entity, such director or officer will be indemnified and held harmless by the Company to the fullest extent authorized by and in the manner set forth in the Amended and Restated By-Law No.1 and OBCA against all expense, liability and loss reasonably incurred or suffered by such director or officer in connection therewith. Under such indemnity agreements, to the fullest extent allowable under applicable law, the Company shall also indemnify against any costs actually and reasonably paid or incurred by a director or officer in connection with any action or proceeding by such director or officer for (i) indemnification or reimbursement of any costs, or payment of any cost advance, by the Company under any provision of the agreements, or under any other agreement or provision of our constating documents and (ii) recovery under any directors’ and officers’ liability insurance policies maintained by the Company, regardless of whether the director or officer ultimately is determined to be entitled to such indemnification or insurance recovery, as the case may be.
A new Board was elected on June 29, 2023 at the annual general and special meeting of shareholders. The following describes the director compensation program that was in effect for the portion of the year ended after the 2023 Meeting. A cash retainer of C$50,000 per year is paid in monthly installments in arrears. In addition, C$10,000 per year is paid to the Chair of the audit committee of the Company (the “Audit Committee”) and C$20,000 per year is paid to the Chairs of each of the Compensation, Nominating and Governance Committee and the Disclosure Committee, paid in monthly installments in arrears in each case. No additional fees are paid to the members for attending the meetings of our Board and meetings of our standing committees. Under the director compensation program for the year ended December 31, 2023, newly appointed directors were granted stock options that vested immediately.
| 58 |
| Table of Contents |
Director Compensation Table
The following table sets forth information concerning compensation accrued or paid to the Company’s non-employee directors, other than NEOs whose compensation is reported in the Summary Compensation Table above, during the year ended December 31, 2023:
Name(1) |
| Fees earned ($) (b) |
|
| Share-based awards ($) (c) |
| Option-based awards ($) (d) |
| Non-equity incentive plan compensation ($) (e) |
|
| Pension value ($) (f) |
| All other compensation ($) (g) |
|
| Total ($) (h) |
| ||||
Michael (Zappy) Zapolin |
|
| 26,000 |
|
| Nil |
| Nil |
| Nil |
|
| Nil |
| 100,000 |
|
|
| 126,000 |
| ||
Adnan Bashir |
|
| 44,525 |
|
| Nil |
| Nil |
|
| 94,848 |
|
| Nil |
| Nil |
|
|
| 139,373 |
| |
Nitin Kaushal |
|
| 56,599 |
|
| Nil |
| Nil |
|
| 94,848 |
|
| Nil |
| Nil |
|
|
| 151,447 |
| |
Dr. Eric Hoskins |
|
| - |
|
| Nil |
| Nil |
| Nil |
|
| Nil |
| Nil |
|
|
| - |
| ||
Lawrence (Larry) Latowsky(2) |
|
| 25,992 |
|
| Nil |
| Nil |
|
| 94,848 |
|
| Nil |
| Nil |
|
|
| 120,840 |
| |
Joseph L. Romano(3) |
|
| 22,023 |
|
| Nil |
| Nil |
| Nil |
|
| Nil |
| Nil |
|
|
| 22,023 |
| ||
Notes:
(1) | The relevant disclosure pertaining to directors who are NEOs (meaning, Zeeshan Saeed and Anthony Durkacz) are reflected in the summary compensation table set forth above. |
(2) | Lawrence (Larry) Latowsky resigned from the Board effective June 29, 2023. |
(3) | Joseph L. Romano resigned from the Board effective June 29, 2023. |
Director Compensation - Share-based Awards, Option-Based Awards and Non-Equity Incentive Plan Compensation
Name |
| Option-based awards – Value vested during the year ($) |
| Share-based awards – Value vested during the year ($) |
| Non-equity incentive plan compensation – Value earned during the year ($) |
| |
Michael (Zappy) Zapolin |
| Nil |
| Nil |
| Nil |
| |
Adnan Bashir |
| Nil |
| Nil |
|
| 94,848 |
|
Nitin Kaushal |
| Nil |
| Nil |
|
| 94,848 |
|
Dr. Eric Hoskins |
| Nil |
| Nil |
| Nil |
| |
Lawrence (Larry) Latowsky |
| Nil |
| Nil |
|
| 94,848 |
|
Joseph L. Romano |
| Nil |
| Nil |
| Nil |
| |
C. Board Practices
Our Board is responsible for our stewardship and strategic direction. It does not actively manage but rather supervises the management of our business and affairs to ensure a consistent focus on increasing shareholder value. In exercising their powers and discharging their duties, our directors shall (a) act honestly and in good faith with a view to the best interests of the Company; and (b) exercise the care, diligence, and skill that a reasonably prudent person would exercise in comparable circumstances.
Board
Our Board currently consists of seven members. The Board is responsible for the stewardship of the Company and for the supervision of management to protect Shareholder interests. The Board oversees the development of the Company’s strategic plan and the ability of management to continue to deliver on the corporate objectives.
The independent judgment of the Board in carrying out its responsibilities is the responsibility of all directors. The Board facilitates independent supervision of management through meetings of the Board and through frequent informal discussions among independent members of the Board and management. In addition, the Board has free access to the Company’s external auditors, external legal counsel and to the Company’s officers.
The Board is responsible for assessing the effectiveness of the Board as a whole, the committees of the Board and the contribution of individual directors. Each Board member has considerable experience in the guidance and management of public companies and the Board has found this has been sufficient to meet the needs of the Company to date.
Composition and Independence of the Board
The Board is currently comprised of seven directors: Anthony Durkacz, Zeeshan Saeed, Dr. Sanjiv Chopra, Dr. Eric Hoskins, Adnan Bashir, Dr. Lakshmi P. Kotra and Michael (Zappy) Zapolin.
| 59 |
| Table of Contents |
The Board has determined that four of the seven directors, namely Dr. Sanjiv Chopra, Dr. Eric Hoskins, Adnan Bashir, and Michael (Zappy) Zapolin, have no material relationship with the Company, either directly or indirectly, which could, in the view of the Board, be reasonably expected to interfere with the exercise of such individual’s independent judgment, and are “independent” (as determined under Rule 5605(a)(2) of the Nasdaq Stock Market Rules and as such term is defined in National Instrument 52-110 - Audit Committees (“NI 52-110”)).
Anthony Durkacz, Zeeshan Saeed and Dr. Lakshmi Kotra are not “independent” (as determined under Rule 5605(a)(2) of the Nasdaq Stock Market Rules and as such term is defined in NI 52-110), as Anthony Durkacz is the Co-Executive Chairman of the Company and a holder, through a corporation he owns and controls, of Class A Shares, Zeeshan Saeed is the CEO and Co-Executive Chairman of the Company and has an interest in Class A Shares held in a trust for his economic benefit and Dr. Lakshmi Kotra is the CEO of Lucid, President of FSD Biosciences and CEO of FSD Australia.
Board Meetings
Although the independent directors do not hold regularly scheduled meetings at which non-independent directors and members of management are not in attendance, the Board has adopted the practice of following each meeting with an independent directors’ discussion. The Board ensures open and candid discussion among its independent directors by continuously monitoring situations where a conflict of interest or perceived conflict of interest with respect to a director may exist. In cases where such a conflict of interest or perceived conflict of interest is identified, it is addressed in accordance with the Business Corporations Act (Ontario) and the Board Mandate. The Board may determine that it is appropriate to hold an in-camera session excluding a director with a conflict of interest or perceived conflict of interest, or such director may consider that it is appropriate to recuse him or herself from considering and voting with respect to the matter under consideration.
The Co-Executive Chairmen of the Board, Anthony Durkacz and Zeeshan Saeed, are non-independent directors. The independent chair of the Audit Committee, Dr. Eric Hoskins, provides leadership for its independent directors. Messrs. Durkacz, Saeed, and Hoskins are responsible for encouraging open and candid discussion among the independent directors, as discussed above, as well as facilitating Board meetings.
Messrs. Durkacz, Saeed, and Hoskins collaborate when setting the agenda of Board meetings and both work with the broader Board to promote good governance and ethics in the decision-making process of the Board.
Considering the guidelines contained in National Policy 58-201 – Corporate Governance Guidelines, the Board convenes meetings, as deemed necessary, of the independent directors at which non-independent directors and members of the management are not in attendance. The Board is of the opinion that no formal leadership of independent directors is required given the size of the Board and the ability of the independent directors to convene meetings of independent directors.
The attendance record of each of the Company’s directors for Board meetings and committee meetings held during the year ended December 31, 2023 was as follows:
Name | Board Meetings Attended/Held | Audit Committee Meetings Attended/Held | Compensation, Nominating and Governance Committee Meetings Attended/Held | Disclosure Committee Meetings Attended/Held |
Anthony Durkacz | 7/7 | - | - | 1/1 |
Zeeshan Saeed | 7/7 | - | 1/1 | - |
Donal Carroll(1) | 4/4 | - | - | - |
Adnan Bashir | 7/7 | 4/4 | 1/1 | 1/1 |
Nitin Kaushal | 7/7 | 4/4 | - | - |
Lawrence (Larry) Latowsky(1) | 4/4 | 2/2 | 1/1 | 1/1 |
Joseph Romano(1) | 4/4 | - | - | - |
Dr. Lakshmi P. Kotra | 7/7 | - | - | - |
Dr. Eric Hoskins(1) | 3/3 | - | - | - |
Michael (Zappy) Zapolin(1) | 3/3 | 2/2 | - | - |
| 60 |
| Table of Contents |
Notes:
(1) | On May 25, 2023, Donal Carroll resigned from the Board and Dr. Eric Hoskins was appointed. At the annual general and special meeting of the Company’s shareholders held on June 29, 2023, Michael (Zappy) Zapolin and Dr. Eric Hoskins were elected as directors of the Company to replace Joseph Romano and Lawrence (Larry) Latowsky. The numbers in these rows reflect attendance with respect to Board and committee meetings held in the period in 2023 during which such individual was a member of the Board. |
Board Mandate
The duties and responsibilities of the directors of the Board are to supervise the management of the business and affairs of the Company; and to act in the best interests of the Company. In discharging its mandate, the directors of the Company are responsible for the oversight and review of the development of, among other things, the following matters:
| · | the strategic direction of the Company; |
| · | identifying the principal business risks of the Company and ensuring that procedures and people are in place to appropriately manage these risks; |
| · | succession planning, including appointing, training, and monitoring senior management; |
| · | a communications policy for the Company to facilitate communications with investors and other interested parties; and |
| · | the integrity of the internal controls and procedures (including adequate management information systems and the oversight of the testing of internal controls) within the Company. |
The Board also has the mandate to assess the effectiveness of the Board as a whole, its committees and the contributions of individual directors. The Board discharges its responsibilities and obligations either directly or through its committees, currently consisting of the Audit Committee, Compensation, Nominating and Governance Committee and the Disclosure Committee.
Position Descriptions
There are currently no position descriptions for the Co-Chairmen of the Corporation and the Chair of each committee. The persons acting as chairs of Board committees have the experience and expertise necessary to assess the role they must play in the context of a public company. The Chairmen and the Chair of each committee presides over all meetings (of the Board or committee, as applicable), participates in the development of meeting calendars and agenda, and ensures the orderly and efficient use of time in the meetings. Each committee chair reports to the Board on a regular basis. The primary role for each is to lead the Board and its committees in fulfilling the duties set out in their charters and/or mandates.
There are also no position descriptions in place for each of the CEO, CFO or COO, respectively. Their roles and responsibilities are delineated through the involvement of the Board in the Corporation’s affairs and the ongoing formal and informal communication between the Board and the CEO, CFO and COO, respectively.
Feedback Policy
There are no formal measures in place for receiving feedback from Shareholders.
Orientation and Continuing Education
New directors are provided orientations which include meetings with management on business directions, operational issues, and financial aspects of the Company.
The Compensation, Nominating and Governance Committee ensures that new directors receive orientation materials describing the Company’s business and its corporate governance policies and procedures. New directors will have meetings with the Co-Executive Chairmen of the Board and CEO, and with the CFO, and are expected to visit the Company’s principal offices. The Compensation, Nominating and Governance Committee is responsible for confirming that procedures are in place and resources are made available to provide directors with appropriate continuing education opportunities.
Management updates the Board on a regular basis regarding the business and activities of the Company to ensure that the directors have the necessary knowledge to meet their obligations as directors. Directors are encouraged to communicate with management, the auditors and the Company’s legal counsel to keep themselves current with the Company’s business. Directors are also provided with full access to the Company’s records.
Ethical Business Conduct
All Board members and employees are committed to maintaining the highest standards of integrity and ethical business conduct in the management of the Company and their interaction with all key Shareholders. These standards can only be achieved by the Company by adhering to the values and principles of conduct.
The Company expects all Board members and employees to conduct themselves in an ethical and law-abiding manner, in all areas, including but not limited to conflicts of interest and the protection and proper use of corporate assets, information and opportunities.
The Board has adopted the Code, which provides guidelines surrounding, among other items, compliance with applicable laws, conflicts of interest, certain opportunities, confidentiality and disclosure, employment practices, and use of company property and resources. The Code is available on the Company’s SEDAR+ profile accessible at www.sedarplus.ca and the Company’s website, https://fsdpharma.com/for-investors/.
| 61 |
| Table of Contents |
The Board has delegated responsibility for monitoring compliance with the Code and for investigating and enforcing matters related to the Code to management, who will report breaches of the Code to the Company’s general counsel or human resources.
Directors are required by applicable law and the Code to promptly disclose any potential conflict of interest that may arise. If a director has a material interest in an agreement or transaction, applicable law, the Code, and principles of sound corporate governance require them to declare the interest in writing or request to have such interest entered in the minutes of meetings of directors and, where required by applicable law, abstain from voting with respect to the agreement or transaction.
Conflicts of Interest
When faced with a conflict, it is required that business judgment of responsible persons, uninfluenced by considerations other than the best interests of the Company, will be exercised in compliance with the guidelines set out in the Code. Pursuant to the OBCA, any officer or director of the Company with a conflict of interest must disclose the nature and extent of such conflict to the Board and recuse themselves from a matter that materially conflicts with that individual’s duty as a director or senior officer of the Company.
Protection and Proper Use of Corporate Assets, Information and Opportunities
Confidential information is not to be used for any purposes other than those of the Company. This requirement of confidentiality extends beyond the duty not to discuss private information, whether about the Company and/or its management and also applies to any asset of the Company, including trade secrets, patient, supplier or customer lists, business plans, computer software, company records and other proprietary information. The Code adopted by the Board provides for certain specific guidelines around the duty of confidentiality of employees, officers, and directors of the Company.
In the situation of contracts with third parties such as suppliers and service providers, management is to share only that information which is needed to satisfy the conditions of the contract and only to those individuals who need to know.
The duty of confidentiality applies to all Board members and employees even after leaving the Company regardless of the reason for departing.
Compliance with Laws, Rules, and Regulations
It is required that the Company is in compliance with all legislation applicable to the Company’s business operations, including but not restricted to the laws of the Province of Ontario, all Canadian provincial laws and legislation, and any other similar legislation in jurisdictions where the Company operates.
All Board members and employees have a duty to know, understand and comply with any specific legislation pertaining to the business of the Company and any legislation applicable to their duties and responsibilities.
The Board has adopted the Code which provides guidelines surrounding, among other items, compliance with applicable laws, conflicts of interest, certain opportunities, confidentiality and disclosure, employment practices, and use of company property and resources.
Board Committees
Audit Committee
The Board is responsible for reviewing and approving the unaudited interim financial statements, and annual audited financial statements, together with other financial information of the Company and for ensuring that Management fulfills its financial reporting responsibilities. The Audit Committee assists the Board in fulfilling this responsibility. The Audit Committee meets with management to review the financial reporting process, unaudited interim financial statements, and annual audited financial statements, together with other financial information of the Company. The Audit Committee reports its findings to the Board for its consideration in approving the unaudited interim financial statements, and annual audited financial statements, together with other financial information of the Company for issuance to the Shareholders.
The Audit Committee meets regularly on at least a quarterly basis. The members of the Audit Committee do not have fixed terms and are appointed and replaced from time to time by resolution of the Board.
Pursuant to NI 52-110, the Audit Committee is required to have a charter, which is incorporated by reference hereto as Exhibit 99.1.
Composition of the Audit Committee
| 62 |
| Table of Contents |
As of the date of this Annual Report, the following are the members of the Audit Committee:
Name |
| Independent(1) |
| Financially Literate(2) |
Dr. Eric Hoskins |
| Yes |
| Yes |
Michael (Zappy) Zapolin |
| Yes |
| Yes |
Adnan Bashir |
| Yes |
| Yes |
Notes:
| 1. | Within the meaning of subsection 1.4 of NI 52-110 and as determined under Exchange Act Rule 10A-3 and Rule 5605(a)(2) of the Nasdaq Stock Market Rules. |
| 2. | Within the meaning of subsection 1.6 of NI 52-110. |
Relevant Education and Experience
The Board believes that the composition of the Audit Committee reflects financial literacy and expertise. Currently, all members of the Audit Committee have been determined by the Board to be “independent” and “financially literate” (as determined under Rule 5605(a)(2) of the Nasdaq Stock Market Rules and as such terms are defined under NI 52-110). The Board has made these determinations based on the education as well as breadth and depth of experience of each member of the Audit Committee.
All the members of the Audit Committee have the education and/or practical experience required to understand and evaluate financial statements that present a breadth and level of complexity of accounting issues that are generally comparable to the breadth and complexity of issues that can reasonably be expected to be raised by the Company’s financial statements. For more information related to the experience of each of the members of the Audit Committee, see “Item 6.A. Directors and Senior Management”.
Audit Committee Oversight
At no time since the commencement of the Company’s most recently completed fiscal year was a recommendation of the Audit Committee to nominate or compensate an external auditor not adopted by the Board.
Reliance on Certain Exemptions
At no time since the commencement of the Company’s most recently completed fiscal year has the Company relied on an exemption from the provisions of NI 52-110.
Pre-Approval Policies and Procedures
The Audit Committee pre-approves all audit services to be provided to the Company by its independent auditors. Non-audit services that are prohibited to be provided to the Company by its independent auditors may not be pre-approved. In addition, prior to the granting of any pre-approval, the Audit Committee must be satisfied that the performance of the services in question will not compromise the independence of the independent auditors. All non-audit services performed by the Company’s auditor for the fiscal year ended December 31, 2023 were pre-approved by the Audit Committee. No non-audit services were approved pursuant to the de minimis exemption to the pre-approval requirement set forth in Rule 2-01(c)(7)(i)(C) of Regulation S-X.
External Auditor Service Fees
Please see, “Item 16.C. Principal Accountant Fees and Services”.
Compensation, Nominating and Governance Committee
Nomination of Directors
The Compensation, Nominating and Governance Committee is currently comprised of three directors of the Company: Dr. Eric Hoskins (Chair), Zeeshan Saeed and Adnan Bashir and. Dr. Eric Hoskins and Adnan Bashir are considered to be “independent” (as determined under Rule 5605(a)(2) of the Nasdaq Stock Market Rules and as such term is defined under NI 52-110).
The Compensation, Nominating and Governance Committee is responsible for recommending to the Board a list of candidates for nomination for election to the Board at each annual meeting of Shareholders. In addition, as the need arises, it will identify and recommend to the Board new candidates for Board membership. The Compensation, Nominating and Governance Committee selects potential directors with the desired background and qualifications, taking into account the needs of the Board at the time. In making its recommendations, the Compensation, Nominating and Governance Committee considers whether each candidate is or would be “independent” and “financially literate” (as determined under Rule 5605(a)(2) of the Nasdaq Stock Market Rules and as such terms are defined under NI 52-110) and possesses the competencies and skills that: (i) are considered to be necessary for the Board, as a whole, to possess; (ii) are considered to be necessary for each existing director to possess; and (iii) each new nominee will bring to the boardroom. The Compensation, Nominating and Governance Committee also considers whether or not each new nominee can devote sufficient time and resources to his or her duties as a Board member.
The Compensation, Nominating and Governance Committee is also responsible for examining the size of the Board and recommending to the Board a size that facilitates effective decision making, reviewing the overall composition of the Board, taking into consideration factors such as business experience, areas of expertise and competency of each director and evaluating whether the necessary and appropriate committees exist to support the work of the Board.
| 63 |
| Table of Contents |
Compensation
The Compensation, Nominating and Governance Committee is also responsible for determining the compensation for the directors and the executive officers. The Compensation, Nominating and Governance Committee reviews the adequacy of remuneration for the executive officers by evaluating their performance in light of the Company’s goals and objectives, and by comparing with other reporting issuers of similar size in the same industry.
The Compensation, Nominating and Governance Committee also periodically reviews the adequacy and form of directors’ compensation and recommends to the Board a compensation model that appropriately compensates directors for the responsibilities and risks involved in being a director and a member of one or more committees, as applicable. The Compensation, Nominating and Governance Committee is also responsible for reviewing the executive compensation disclosure before the Company discloses this information publicly.
The Compensation, Nominating and Governance Committee is also responsible for: (i) ensuring that the mission and strategic direction of the Company is reviewed annually; (ii) ensuring that the Board and each of its committees carry out its functions in accordance with due process; (iii) assessing the effectiveness of the Board as a whole, each committee of the Board, and the contribution of each individual director; (iv) identifying, recruiting, endorsing, appointing, and orienting new directors; (v) reviewing and making compensation related recommendations and determinations regarding senior executives and directors; and (vi) the Company’s human resources and compensation policies and processes.
Governance
The Compensation, Nominating and Governance Committee is also responsible for, among other things: (i) assisting the Company and the Board in fulfilling their respective corporate governance responsibilities under applicable securities laws, instruments, rules and mandatory policies and regulatory requirements; (ii) promoting a culture of integrity throughout the Company; and (iii) developing the Company’s approach to governance issues and establishing sound corporate governance practices that are in the interests of shareholders and that contribute to effective and efficient decision-making.
Disclosure Committee
The Disclosure Committee is currently comprised of three directors of the Company: Michael (Zappy) Zapolin (Chair), Anthony Durkacz and Adnan Bashir. Michael (Zappy) Zapolin and Adnan Bashir are considered to be “independent” (as determined under Rule 5605(a)(2) of the Nasdaq Stock Market Rules and as such term is defined under NI 52-110).
The Disclosure Committee is responsible for, among other things, ensuring that the Company complies with its timely continuous disclosure obligations, overseeing and monitoring compliance with the Company’s disclosure policies, guidelines, and procedures, promoting effective communication and preserving confidentiality of material information.
Assessments
The Board and its individual directors are assessed on an informal basis continually as to their effectiveness and contribution. The Board encourages discussion among members as to evaluation of its effectiveness as a whole, of each individual director and each of its committees. The Board does not consider that formal assessments would be useful at this stage of the Company’s development. To assist in its review, the Board may conduct informal surveys of its directors. In addition, the Board works closely with management and, accordingly, are in a position to assess individual director’s performance on an ongoing basis.
Director Term Limits and Other Mechanisms of Board Renewal
The Board has not adopted a term limit for directors and, as part of the Board’s assessment process, the Board considers the benefit of renewal among directors in the context of the needs of the Board from time to time. In light of the nature of the industry in which the Company operates, the Board does not believe that adopting a term limit for directors is necessary or appropriate at this time.
Policies Regarding the Representation of Women on the Board
The Company does not have a written policy relating to the identification and nomination of women directors. When considering and recommending qualified director nominees, the Compensation, Nominating and Governance Committee evaluates all candidates on their skills and experience in the context of what the Board as a whole requires to be effective, taking the background and diversity, including gender, of all directors and nominees into consideration.
| 64 |
| Table of Contents |
Consideration of the Representation of Women in the Director Identification and Selection Process
The Compensation, Nominating and Governance Committee goes through a rigorous process when considering a director nominee, including an evaluation of the skills and experience of the current directors, determining the gaps in skills and experience that exist and finding potential candidates to fill those gaps and round out the skills and experience of the Board as a whole. Diversity (including the representation of women on the Board and in executive officer positions) is a factor considered in determining the optimal composition of the Board. The final recommendation for nomination or appointment to the Board has been based on the best combination of skills and experience for the position, with due regard for the benefits of diversity on the Board.
Consideration Given to the Representation of Women in Executive Officer Appointments
The Board encourages the consideration of women who have the necessary skills, knowledge, experience, and character when considering new potential candidates for executive officer positions.
Issuer’s Targets Regarding the Representation of Women on the Board and in Executive Officer Positions
The Board does not have specific targets in respect of appointing women to the Board and in respect of executive officer appointments.
Number of Women on the Board and in Executive Officer Positions
As of the date of this Annual Report there are no women on the Board or in executive officer positions (0%).
D. Employees
As of December 31, 2023, we had 8 full-time employees. None of our employees are represented by collective bargaining agreements. We believe that we maintain good relations with our employees. At each date shown, we had the following number of full-time employees, broken out by function.
|
| December 31, |
| |||||||||
|
| 2023 |
|
| 2022 |
|
| 2021 |
| |||
Function: |
|
|
|
|
|
| ||||||
Research and development |
|
| 3 |
|
|
| 12 |
|
|
| 4 |
|
General and administrative |
|
| 5 |
|
|
| 5 |
|
|
| 6 |
|
Total |
|
| 8 |
|
|
| 17 |
|
|
| 10 |
|
E. Share Ownership.
For information regarding the share ownership of our directors and executive officers, see “Item 6.B.-Compensation” and “Item 7.A.-Major Shareholders.”
F. Disclosure of a Registrant’s Action to Recover Erroneously Awarded Compensation.
Not applicable.
Item 7. Major Shareholders and Related Party Transactions.
A. Major Shareholders
The following table provides information with respect to the beneficial ownership of our Class A Shares and Class B Shares as of March 28, 2024:
| · | each person, or group of affiliated persons, known by us to beneficially own five percent (5%) or more of any class of our shares; |
| · | each of our NEOs; |
| · | each of our directors; and |
| · | all of our directors and executive officers as a group. |
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities and include shares that can be acquired within 60 days of March 28, 2024. The percentage ownership information shown in the table is based upon 72 Class A Shares and 40,429,569 Class B Shares outstanding as of March 28, 2024. Each Class A Share is convertible into one Class B Share at the option of the Class A Share holder. Class A Shares have 276,660 votes per share and Class B Shares have one vote per share. For further information regarding the voting rights of the Class A Shares and the Class B Shares, see Exhibit 2, “Description of Securities”.
Except as otherwise indicated, all persons listed below have sole voting and investment power with respect to the shares beneficially owned by them, subject to applicable community property laws. The information is not necessarily indicative of beneficial ownership for any other purpose.
| 65 |
| Table of Contents |
In computing the number of shares beneficially owned by a person and the percentage ownership of that person, we deemed outstanding shares subject to Options and Warrants held by that person that are immediately exercisable or exercisable within 60 days of March 28, 2024. We did not deem these shares outstanding, however, for the purpose of computing the percentage ownership of any other person. The information in the table below is based on information known to us or ascertained by us from public filings made by the shareholders. Except as otherwise indicated, addresses of the directors, executive officers and named beneficial owners are in the care of FSD at 199 Bay Street, Suite 4000, Toronto, Ontario M5L 1A9.
For additional information regarding the Options reported in the following table, see “Item 6. Directors, Senior Management and Employees - B. Compensation”.
|
| Class A Shares(1) |
| Class B Shares |
| Total Voting Power | |||||||||
Name of Beneficial Owner |
| Number |
|
| Percent |
| Number |
|
| Percent |
| Percent | |||
|
|
|
|
|
|
|
| ||||||||
Five Percent or Greater Holders: |
|
|
|
|
|
|
| ||||||||
Rehan Saeed(3) |
|
| 36 |
|
| 50% |
|
| 731,086 |
|
| 1.8% |
| 17.7% | |
|
|
|
|
|
|
|
|
|
|
|
|
| |||
Current Directors and Named Executive Officers: |
|
|
|
|
|
|
|
|
|
|
|
| |||
Anthony Durkacz Co-Executive Chairman and Director(4) |
|
| 36 |
|
| 50% |
|
| 1,623,165 |
|
| 4.0% |
| 19.2% | |
Zeeshan Saeed CEO, President, Co-Executive Chairman and Director(5) |
|
| 36 |
|
| 50% |
|
| 2,741,146 |
|
| 6.8% |
| 21.0% | |
Nathan Coyle CFO(6) |
|
|
|
| 0% |
|
| 32,500 |
|
| 0.1% |
| 0.1% | ||
Donal Carroll COO(7) |
|
|
|
| 0% |
|
| 973,268 |
|
| 2.4% |
| 1.6% | ||
Adnan Bashir Director(8) |
|
|
|
| 0% |
|
| 9,393 |
|
| 0.0% |
| 0.0% | ||
Dr. Eric Hoskins Director |
|
|
|
| 0% |
|
| - |
|
| 0.0% |
| 0.0% | ||
Dr. Sanjiv Chopra Director |
|
|
|
| 0% |
|
| - |
|
| 0.0% |
| 0.0% | ||
Michael (Zappy) Zapolin Director |
|
|
|
| 0% |
|
| - |
|
| 0.0% |
| 0.0% | ||
Dr. Lakshmi P. Kotra Director, CEO of Lucid, President of FSD Biosciences and CEO of FSD Australia (9) |
|
|
|
| 0% |
|
| 1,922,197 |
|
| 4.8% |
| 3.2% | ||
All current directors and executive officers (9 individuals) |
|
| 72 |
|
| 100% |
|
| 7,301,669 |
|
| 18.1% |
| 45.1% | |
Notes:
(1) | Class A Shares have 276,660 votes per share. |
(2) | The reported number of Class A Shares consists of shares held by Mr. Bokhari. The reported number of Class B Shares consists of 24 Class B Shares issuable upon conversion of Class A Shares. |
(3) | The reported number of Class A Shares consists of shares held by the Xorax Family Trust (“Xorax”), as to which Mr. Rehan Saeed, the trustee of Xorax, has shared voting and dispositive power (and which such Class A Shares are held for the benefit of Mr. Zeeshan Saeed). The reported number of Class B Shares consists of (a) 40,055 outstanding Class B Shares held by Mr. Rehan Saeed, and (b) the following shares as to which Mr. Rehan Saeed has shared voting and dispositive power: (i) 441,031 outstanding Class B Shares held by Xorax, (ii) 24 Class B Shares issuable upon conversion of Class A Shares held by Xorax, (iii) 100,000 Class B Shares issuable upon exercise of outstanding Warrants held by Legacy Family Trust (“Legacy”), of which Mr. Rehan Saeed is the trustee, exercisable within 60 days of March 28, 2024 and (iv) 150,000 outstanding Class B Shares held by Legacy. |
(4) | The reported number of Class A Shares consists of shares held by Fortius Research and Trading Corporation (“Fortius”), as to which Mr. Durkacz, who controls Fortius, has shared voting and dispositive power. The reported number of Class B Shares consists of (a) the following shares as to which Mr. Durkacz has sole voting and dispositive power: (i) 975,122 outstanding Class B Shares and (ii) 500,000 Class B Shares issuable upon exercise of outstanding Options exercisable within 60 days of March 28, 2024 and (b) the following shares as to which Mr. Durkacz has shared voting and dispositive power: (i) 106,043 outstanding Class B Shares held by Fortius, (ii) 24 Class B Shares issuable upon conversion of Class A Shares held by Fortius and (iii) 42,000 outstanding Class B Shares held by Jacqueline Burns, the spouse of Mr. Durkacz. The reported number of Class B Shares does not include 373,671 outstanding Class B Shares held by First Republic, of which Mr. Durkacz is a director, Executive Vice President and majority shareholder. Mr. Durkacz does not have or share voting or investment power over the Class B Shares held by First Republic. |
| 66 |
| Table of Contents |
(5) | The reported number of Class A Shares consists of 36 Class A Shares held by Xorax, of which Mr. Zeeshan Saeed has shared voting and dispositive power. The reported number of Class B Shares consists of the following shares as to which Mr. Zeeshan Saeed (a) has sole voting and dispositive power: (i) 1,800,115 outstanding Class B Shares and (ii) 500,000 Class B Shares issuable upon exercise of outstanding Options exercisable within 60 days of March 28, 2024 and (b) has shared voting and dispositive power: (i) 36 Class B Shares issuable upon conversion of Class A Shares held by Xorax and (ii) 441,031 outstanding Class B Shares held by Xorax. |
(6) | The reported number of Class B Shares consists of the following shares as to which Mr. Nathan Coyle has sole voting and dispositive power: (a) 2,500 outstanding Class B Shares and (b) 35,000 Class B Shares issuable upon exercise of outstanding Options exercisable within 60 days of March 28, 2024 as to which Mr. Nathan Coyle has sole voting and dispositive power. |
(7) | The reported number of Class B Shares consists of (a) 971,268 outstanding Class B Shares and (b) 500,000 Class B Shares issuable upon exercise of outstanding Options exercisable within 60 days of March 28, 2024. |
(8) | The reported number of Class B Shares consists of (a) 9,200 outstanding Class B Shares as to which Mr. Adnan Bashir has sole voting and dispositive power, (b) 98 outstanding Class B Shares held by 58 Northwest, a corporation controlled by Mr. Bashir, and (c) 95 outstanding Class B Shares held by TFSA, a corporation controlled by Mr. Bashir. |
(9) | The reported number of Class B Shares consists of the following shares as to which Dr. Lakshmi Kotra has sole voting and dispositive power: (a) 515,797 outstanding Class B Shares, (b) 865,200 outstanding Class B Shares held by ILace Therapeutics International Inc., a corporation controlled by Dr. Kotra, (c) 41,200 outstanding Class B Shares held by Kotra Trust, a trust of which Dr. Kotra is the trustee, and (d) 500,000 Class B Shares issuable upon exercise of outstanding Options exercisable within 60 days of March 28, 2024. |
(12)
| As noted above in Notes 3 and 5 to this chart, the 36 Class A Shares and 441,031 of the Class B Shares reported under Mr. Rehan Saeed and Mr. Zeeshan Saeed’s shareholdings represent the same Class A Shares and Class B Shares, which are held by Xorax, of which Mr. Rehan Saeed and Mr. Zeeshan Saeed have shared voting and dispositive power. |
As of March 28, 2024, we estimate that approximately 53.7% of our outstanding Class B Shares, which equates to 21,720,976 Class B Shares, were held in the United States by 34 holders of record. This represents 36% of the voting control of the Company. The number of holders of record does not include beneficial owners whose Class B Shares are held in street name by brokers and other nominees. The number of holders of record also does not include holders whose shares may be held in trust by other entities.
B. Related Party Transactions
Since January 1, 2023, we have engaged in the following transactions with our related parties. For this purpose, our related parties include (a) enterprises that directly or indirectly control or are controlled by, or are under common control with, us; (b) our associates; (c) shareholders beneficially owning 10% or more of our voting power and other individuals with significant influence over us, and close members of any such individual’s family; (d) our directors and executive officers, and close members of their families; and (e) enterprises in which a substantial interest in the voting power is owned, directly or indirectly, by any person described in (c) or (d) or over which such a person is able to exercise significant influence. Our related parties include enterprises owned by directors or major shareholders and enterprises that have a member of key management in common with us. All of the transactions have been reviewed and approved by our Board or another independent committee of the Board.
Transactions with Celly Nu
For more information on the Celly Nu IP License Agreement, Celly Nu Loan Agreement, Celly Nu Security Agreement and Plan of Arrangement, please see “Item 4. Information on the Company. A. History and Development of the Company Overview and History”.
For accounting purposes, the Company determined that it obtained control of Celly Nu on July 31, 2023, and control was maintained at all times from July 31, 2023, through December 31, 2023. Celly Nu is significantly dependent on the Company as a result of the IP License Agreement and Celly Nu Loan Agreement. The Company concluded it has control of Celly Nu, as the Company, together with persons or entities considered to be de facto agents of the Company, hold a combined 52.05% of the voting rights of Celly Nu as of December 31, 2023. In addition, key management personnel of the Company hold three of the four board of director positions of Celly Nu. The assessment of control is performed on a continuous basis.
Mortgage Loan to CEO
On April 17, 2023, FSD Strategic Investments entered into the CEO Mortgage Loan with the CEO. The CEO Mortgage Loan matures on April 26, 2025, and is part of FSD Strategic Investments’ portfolio of loans. As of December 31, 2023, the amount due on the CEO Mortgage Loan was C$1,200,000. The business purpose of the CEO Mortgage Loan was a treasury function to earn a rate of return on excess capital held.
FSD Strategic Investments secures each mortgage loan with collateral in the residential property at least equal to 55% of the value of the loan. In the case of the CEO Mortgage Loan, although all the other loans under FSD Strategic Investments’ portfolio of loans prior to this one were a first charge mortgage on the underlying residential property, the CEO Mortgage Loan was a second charge mortgage. FSD Strategic Investments determined that because the CEO Mortgage Loan met its strict underwriting requirements and had an undertaking requirement (as described herein), it would take a second charge on the residential property. The CEO’s residential property had a Home Equity Line of Credit (“HELOC”), of which no funds have been withdrawn against the property. FSD Strategic Investments viewed this as appropriate security as the HELOC had a zero balance and the CEO undertook to FSD Strategic Investment that the HELOC would only be used by the CEO to pay the CEO Mortgage Loan. FSD Strategic Investments determined that this undertaking by the CEO to use the HELOC funds for payment of the CO Mortgage Loan provided sufficient security under the CEO Mortgage Loan. In addition, the CEO met the underwriting requirements of FSD Strategic Investments.
| 67 |
| Table of Contents |
The CEO Mortgage Loan was made to the CEO on the same terms and conditions that a member of the general public would receive, based on the underwriting requirements of FSD Strategic Investments. In determining the interest rate for each borrower under its portfolio of loans, FSD Strategic Investments took the following criteria into consideration: (i) their credit score; (ii) employment status; (iii) salary; (iv) property location; (v) equity on the property; and (vi) the Bank of Canada rate at the time of the mortgage. In the case of the CEO Mortgage Loan, although all the other loans under FSD Strategic Investments’ portfolio of loans, are a first charge mortgage on the underlying residential property, his was a second charge. The Company determined that because the CEO Mortgage Loan was made to an individual with a salary that is more than double that of the other borrowers, with a very clear picture of longevity, the exception was made as the first charge on his residential property was for a HELOC which no funds have been withdrawn against. The Company viewed this as appropriate security as the HELOC had a zero balance and the borrower undertook to the Company that the HELOC will not be used unless for payment towards the Company’s second mortgage.
Before the Board approved the Mortgage Loan, FSD Strategic Investments sought the advice of counsel to guide the Board with its decision in the CEO Mortgage Loan transaction. Under Section 13(k) of the Exchange Act, an issuer, who is in the consumer credit business, can provide loans to its executive officers and directors that (i) are made in the ordinary course of the issuer’s consumer credit business, (ii) are of a type that is generally made available by the issuer to the public and (iii) made on market terms that are no more favorable than those offered by the issuer to the general public. “Consumer credit” generally includes credit extended to an individual primarily for personal, family or household purposes. A standard loan made to an individual to purchase his or her primary residence should normally fall within the definition of consumer credit.
The Company and FSD Strategic Investments believe that the CEO Mortgage Loan complies with this exemption in that FSD Strategic Investments is in the business or providing mortgage loans and the CEO Mortgage Loan was made in the ordinary course of business, is of a type generally made available by it to the public and is made on market terms to the CEO that are no more favorable than those offered by it to the general public. However, there is limited regulatory and legislative guidance regarding the scope of this exemption and FSD Strategic Investments cannot provide assurances that it in full compliance with United States laws. Any failure to comply with any laws and regulations, may adversely affect its business, and results of operations” See “Item 3. Key information - D. Risk Factors Failure to comply with laws and regulations could have a material adverse effect on the Company's business”.
With respect to Canadian law, in completing the CEO Mortgage Loan, the Company relied on exemptions from the formal valuation and minority approval requirements of MI 61-101 on the basis that at the time the CEO Mortgage Loan was agreed to, neither the fair market value of the CEO Mortgage Loan, nor the fair market value of the consideration for the CEO Mortgage Loan, insofar as it involved the interested party, exceeded 25% of the Company’s market capitalization, as determined in accordance with MI 61-101.
December Class A Share Private Placement
Effective December 4, 2023, the Corporation closed the December Class A Private Placement.
Xorax Family Trust, a trust of which Zeeshan Saeed, the CEO and Co-Chairman of FSD Pharma is a beneficiary, and Fortius Research and Trading Corp., a corporation of which Anthony Durkacz, a director of FSD is a director, purchased all of the Class A Shares issued pursuant to the December Class A Private Placement. The participation by such insiders is considered a “related-party transaction” within the meaning of MI 61-101. The Corporation relied on exemptions from the formal valuation and minority shareholder approval requirements of MI 61-101 contained in respectively, sections 5.5(a) and 5.7(1)(a) of MI 61-101 in respect of related party participation in the December Class A Private Placement as neither the fair market value (as determined under MI 61-101) of the subject matter of, nor the fair market value of the consideration for, the transaction, insofar as it involved the related parties, exceeded 25% of the Corporation’s market capitalization (as determined under MI 61-101). The Corporation did not file a material change report more than 21 days before the expected closing of the December Class A Private Placement because the details of the participation therein by related parties to the Corporation were not settled until shortly prior to the closing, and the Corporation wished to close on an expedited basis for business reasons.
Directors and Officers Liability Insurance
We maintain directors’ and officers’ liability insurance policies for the liability of our directors and officers arising out of the performance of their duties and for our liability arising out of securities claims. The policies provide coverage in respect of a maximum total liability of C$3,000,000, and includes specific exclusions described in the policy.
D&O Indemnification Agreements
See “Item 6.B. Compensation - Indemnification” for details.
Lucid Acquisition
See “Item 4.A. History and Development of the Company-General Development of the Business - Three Year History-Lucid Acquisition” for further details.
C. Interests of Experts and Counsel
Not applicable.
Item 8. Financial Information
A. Consolidated Statements and Other Financial Information
Consolidated Financial Statements
Our 2023 Annual Financial Statements are appended at the end of this Annual Report, starting at page F-1, and incorporated herein by reference.
Dividend Distribution Policy
In connection with the Plan of Distribution, the Company distributed an aggregate of approximately 45,712,529 Celly Nu Shares to its shareholders. As of December 31, 2023, the Company owns 26.15% Celly Nu Shares on a non-diluted basis.
| 68 |
| Table of Contents |
Except for the Celly Nu shares distributed in the Plan of Arrangement, the Company has not paid any dividends or distributions on its outstanding Class B Shares, and we have no current intention to declare dividends on our Class B Shares in the foreseeable future. Any decision to pay dividends on our Class B Shares in the future will be at the discretion of our Board and will depend on, among other things, our results of operations, current and anticipated cash requirements and surplus, financial condition, any future contractual restrictions and financing agreement covenants, solvency tests imposed by corporate law and other factors that our Board may deem relevant.
Legal Proceedings
The Company is engaged in certain legal proceedings, as further described below. Litigation has been, and is expected to be, costly and time-consuming and could divert the attention of management and key personnel from our business operations. Although we intend to vigorously defend ourselves against any pending claims, and future claims that may occur, we cannot assure that we will succeed in defending any of these claims and that the judgments will not be upheld against us. If we are unsuccessful in our defense of these claims or unable to settle the claims in a manner satisfactory to us, we may be faced with outcomes that could have a material adverse effect on the Company and its financial condition. Except as otherwise disclosed below, there are no material outstanding legal proceedings or regulatory actions to which the Company is party, nor, to Company’s knowledge, are there any such proceedings or actions contemplated.
GBB Drink Lab Litigation
On May 12, 2023, the Corporation announced receipt of a lawsuit filed in the United States District Court for the Southern District of Florida (the “U.S. District Court”) by GBB against the Corporation, alleging breach of a mutual non-disclosure agreement and misappropriation of trade secrets, valued, as of August 30, 2022 (prior to the misappropriation and material breach) at US$53,047,000. The Corporation believes the allegations are without merit and intends to defend itself in the lawsuit. On June 23, 2023, the Corporation filed a motion to dismiss the complaint. On July 3, 2023, GBB responded in opposition to the Corporation’s motion to dismiss the complaint. The Motion to Dismiss the Amended Complaint filed on June 23, 2023 has been fully briefed and is awaiting adjudication by the U.S. District Court. In the meantime, on August 24, 2023, the parties filed a proposed joint scheduling report with the U.S. District Court, which set forth various deadlines that would govern this action. Under the proposed joint schedule, which still needs to be approved by the U.S. District Court, the case would be trial-ready by November 30, 2024.
Dr. Raza Bokhari
Following the contested meeting held on, the former CEO, Dr. Raza Bokhari commenced five actions against the Company and management, which resulted in counterclaims and additional unexpected legal and operating expenses. As at March 28, 2024, the status of the matters is as follows:
Wrongful Dismissal Arbitration
On July 15, 2021, the Corporation’s former CEO, Dr. Raza Bokhari, filed an arbitration notice seeking C$30.2 million for breach of contract, severance, and damages, along with C$500,000 for punitive damages and legal fees. Dr. Bokhari had been placed on administrative leave after the May 14, 2021 shareholder meeting and was terminated for cause on July 27, 2021, following an investigation by a special committee of the Board. The Company defended the arbitration and counterclaimed against Dr. Bokhari for reimbursement of expenses he directed the Company to pay to himself, as well as losses the Company sustained as a result of Dr. Bokhari’s decision to authorize a series of dilutive share issuances.
The arbitration concluded in August 2022. In its 174 page arbitral award, the arbitrator dismissed Dr. Bokhari’s claims in their entirety. The arbitrator also ordered Dr. Bokhari to repay certain monies to the Company in respect of the Company’s counterclaim, while also awarding the Company its costs of the arbitration which he subsequently fixed at approximately C$2.8 million, plus interest.
On December 9, 2022, Dr. Bokhari sought to set aside the award, citing unfair treatment and inadequate reasoning. On October 4, 2023, the Company announced that the Ontario Superior Court of Justice had dismissed Dr. Bokhari’s motion to set aside the arbitration award. Dr. Bokhari was required to put up C$150,000 as security for costs before the motion was heard, which he has forfeited. In addition, Dr. Bokhari was ordered to pay C$175,000 to cover the Company’s legal costs for his failed set aside motion.
On October 13, 2023, Dr. Bokhari served notices of motion on the Company for leave to appeal the set aside and enforcement orders issued by the Ontario Superior Court of Justice’s on October 4, 2023. On December 1, 2023, the Company filed a petition to confirm the arbitration award in the United States District Court for Eastern District of Pennsylvania. On December 15, 2023, the Company submitted a responding party’s factum to the Court of Appeal for Ontario. On February 6, 2024, the Company announced that the Ontario Superior Court of Justice affirmed judgment and awarded an additional C$5,000 in costs in light of Dr. Bokhari’s failed motion for leave to appeal. As of the date hereof, the litigation is ongoing.
| 69 |
| Table of Contents |
Restraining Order and Class B Share Cancellation Application
On January 21, 2021 and February 10, 2021, the Board authorized the issuance of an aggregate of 1,349,765 Class B Shares as share based awards to certain directors and officers of the Corporation, including Dr. Bokhari. Upon determining that 1,198,146 of these Class B Shares (the “Contested Shares”) had been inappropriately issued contrary to applicable laws, the Board resolved to cancel the Contested Shares on June 1, 2021, and later directed the Corporation’s transfer agent to cancel and return the Contested Shares to treasury. On July 2, 2021, Dr. Bokhari, filed an action against the Corporation seeking to prevent the Corporation from cancelling his portion of the Contested States. The motion was heard and denied on July 27, 2021. On July 21, 2021, the Corporation commenced a legal proceeding against Dr. Bokhari, former members of the Board, including James Datin, Robert Ciaruffoli, Stephen Buyer and Gerald Goldberg, Dr. Bokhari’s brokerages’ Haywood Securities Inc. and Haywood Securities (US) Inc., and the Corporation’s transfer agent. The Corporation made an application before the Superior Court stating that the Contested Shares were issued contrary to section 23(2) of the OBCA and validly cancelled by resolution of the Board passed on June 1, 2021. The Corporation was able to reach an agreement with all of the former directors other than Dr. Bokhari under which they did not oppose the Corporation’s application and agreed to be bound by the decision in the application, and the Corporation agreed not to seek costs against them. Neither the Corporation’s transfer agent nor any of Dr. Bokhari’s brokerages took any position on the application. On March 8, 2022, the court issued a mixed decision in the application, permitting the Contested Share grant to Dr. Bokhari until the date of his termination but cancelling 504,888 Contested Shares relating to services that were to be provided after the date of termination.
Bokhari v. FSD Pharma Inc. Et al.
On July 2, 2021, Dr Bokhari filed an action against the Corporation, FSD BioSciences, Anthony Durkacz and Zeeshan Saeed. The case was placed in civil suspense pending resolution of arbitration. Therefore, no further activity will occur in this case unless and until the aforementioned arbitration concludes. As of the date hereof, the litigation is ongoing.
Bokhari Wrongful Means Action
In June 2023, Dr. Bokhari commenced an action against the Company and FSD Biosciences by way of notice of action issued out of the Ontario Superior Court of Justice in Toronto, Canada. He subsequently filed a statement of claim, of July 7, 2023 and served the notice of action and statement of claim on a former director of the Company on December 19, 2023. The action seeks USD $1.5 million in damages for intentional interference with economic relations, misrepresentation, negligence, and other causes of action to be specified in a statement of claim. We delivered a notice of intent to defend in the action on January 5, 2024, but thus far not been required to provide a statement of defense. We believe these claims are without merit.
Bokhari Employment Claim
By way of notice of action issued on May 11, 2023, Dr. Bokhari commenced an action for damages for breach of contract against the Company in the Ontario Court of Justice in Toronto Canada. He subsequently filed a statement of claim in which he specified the claim as a claim for USD $30.2 million in damages on the basis that the Company breached his employment agreement by not providing him notice of default before terminating his employment. On November 10, 2023, the last day on which he could do so, Dr. Bokhari served the notice of action and statement of claim on an FSD director. We served a notice of intent to defend in this action on November 22, 2023 but have not been required to serve a statement of defense. We note that to the extent he wished to advance this claim, it is a claim that Dr. Bokhari should have advanced in the employment arbitration, but did not do so As such, and bearing in mind the decision the arbitrator reached in that proceeding in our view, we believe that this claim has no merit.
Cunningham Assessment Application
By notice of application dated September 26, 2023, Dr. Bokhari applied to the Ontario Superior Court of Justice for an order directing an assessment of the accounts/billing rendered by former Justice Douglas Cunningham as arbitrator in the Wrongful Dismissal Arbitration noted above. In late January 2024, Dr. Bokhari served Arbitrator Cunningham with the notice of application and a supporting affidavit sworn January 24, 2024. Under the terms of the retainer agreement between FSD, Dr. Bokhari and Arbitrator Cunningham, FSD is jointly and severally liable for any costs Arbitrator Cunningham might incur as a result of this proceeding. The liability exposure that FSD could have in this matter is approximately C$182,777.50, which represents half of the arbitration fees, plus any costs in defending the arbitrator To protect its interest, FSD has instructed its legal counsel to move to have FSD joined to the proceeding. We believe this claim is without merit.
Bokhari Indemnification Application
On November 12, 2021, Dr. Bokhari commenced an application in the Ontario Superior Court of Justice, Commercial List, seeking an order appointing an arbitrator to arbitrate his claim to be entitled to indemnification of his legal expenses associated with the litigation he commenced against FSD or in which he was named as a party by FSD. FSD denied the validity of the underlying indemnification agreement and therefore opposed the application. In April 2022, the parties agreed to allow Dr. Bokhari to adjourn the application indefinitely. Last year, Dr. Bokhari retained new counsel who indicated that it intended to pursue the application. To date that new counsel has taken no steps to do so. We believe this claim is without merit.
On April 6, 2022, Dr. Bokhari commenced an application in the Superior Court seeking an order appointing an arbitrator to arbitrate his claim to be entitled to indemnification of his legal expenses associated with the litigation he has commenced against the Corporation or in which he has been named as a defendant against the Corporation. The Corporation denied the validity of the underlying indemnification agreement and opposed the application. In April 2022, the parties agreed to adjourn the application without setting a new hearing date. As of the date hereof, the litigation is ongoing.
| 70 |
| Table of Contents |
FSD Petition against Raza Bokhari to Confirm Arbitration Award
On December 1, 2023, the Company filed a Petition to Confirm Arbitration (the “Petition”), in the Eastern District of Pennsylvania, which seeks to (a) confirm the four awards entered in an arbitration in Ontario, Canada, in favor of the Company and against former CEO Raza Bokhari and (b) enter final judgment against Bokhari in an amount in excess of C$3,000,000. The petition was filed in the U.S. District Court for the Eastern District of Pennsylvania. Bokhari filed a response on February 9, 2024. The Company filed a response thereafter and the litigation is ongoing.
Parkway Clinical Laboratories
On July 8, 2021, Parkway Clinical Laboratories, a company wholly owned by Dr. Bokhari, filed an action against the Company, which was subsequently settled following a conference between the parties on October 20, 2021.
On July 20, 2021, a shareholder of the Corporation filed a claim in the Delaware Chancery Court against the Corporation and its directors and officers seeking to remedy harm they believe the directors and officers of the Corporation have caused by their actions. The shareholder has filed the claim on count of breach of fiduciary duties and corporate waste against the directors and officers with no dollar amount being claimed. On September 13, 2021, the Corporation filed a motion to dismiss in its entirety and the motion was heard on February 8, 2022. The claim was dismissed by the court May 6, 2022.
Employment Litigation
From time to time, the Company is involved with employment litigation, in the normal course of its business.
Subsequent to the resignation of two former FSD BioSciences employees (the “Former Employees”), on July 9, 2021, the Former Employees filed a joint claim against the Corporation. On September 17, 2021, the Corporation filed a motion to dismiss the claim in its entirety. On December 13, 2021, the Court granted the Corporation’s motion and dismissed the case. One of the Former Employee’s claims was dismissed with prejudice while the other was not.
On July 18, 2023, Kevin Cassidy, a former employee sued the Company and Lucid and the Company in the Ontario Superior Court of Justice. The claim is for wrongful dismissal and the amounts claimed are C$497,000, plus additional damages, which have not been quantified, plus interest and costs. Pleadings have been exchanged and, on January 26, 2024, Cassidy’s counsel served a motion for summary judgment. The hearing date for the summary judgment motion has not been scheduled as the parties are currently in the process of discussing a timetable for all necessary steps which must be completed prior to the hearing. Once all such steps are completed, the parties will attend at court to schedule the hearing date. It is premature to estimate the likely outcome of this claim.
On October 12, 2023, Dr. Sima Salahshor, a former employee sued the Company and Lucid in the Ontario Superior Court of Justice. The claim is for wrongful dismissal and the amounts claimed are C$97,500, plus other damages, which have not been quantified, plus interest and costs. The claim was served. This matter is at the pleadings stage and we expect that the defense will be served shortly. It is premature to estimate the likely outcome of this claim.
B. Significant Changes
A discussion of the significant changes in our business can be found under “Item 4. Information on the Company—A. History and Development of the Company” and “Item 4. Information on the Company—B. Business Overview.”
Item 9. The Offer and Listing
A. Offer and Listing Details
No offering is made by this Annual Report. The Company’s Class B Shares commenced trading on the CSE on May 29, 2018 under the symbol “HUGE”. Prior to the CSE listing there was no public trading in any securities of the Company. The Class B Shares commenced trading on the Nasdaq in the United States on January 9, 2020 under the symbol “HUGE”. The Company’s Class B Shares are listed on the Frankfurt Stock Exchange under the symbol “0K9A”. Trading on the FSE market is minimal.
B. Plan of Distribution
Not applicable.
C. Markets
See Item 9.A “Offer and listing details.”
D. Selling Shareholders
Not applicable.
E. Dilution
Not applicable.
F. Expenses of the Issue
Not applicable.
| 71 |
| Table of Contents |
Item 10. Additional Information
Not applicable.
B. Memorandum and Articles of Association
See Exhibit 2.1 to this Annual Report on Form 20-F for a summary of certain material provisions of our articles of incorporation, as amended; bylaws, as amended; and certain related sections of the OBCA. See Exhibit 1.1 to this Annual Report on Form 20-F for our articles of incorporation, as amended, and Exhibit 1.2 for our bylaws, as amended.
C. Material Contracts.
Except as set forth below, the material terms of our material contracts are described elsewhere in this Annual Report. Below is a a summary of certain material contracts, together with references to the relevant sections of this Annual Report where the material terms of such contracts are described. Please also see Item 19. Exhibits for a complete summary of all material contracts.
The summaries provided below and elsewhere in this Annual Report are not meant to be exhaustive and are qualified in their entirety by the full text of the relevant agreements, copies of which are filed as exhibits to this Annual Report.
Celly Nu IP License Agreement, Celly Nu Loan Agreement, Celly Nu Security Agreement and Plan of Arrangement
For more information on the Celly Nu IP License Agreement, Celly Nu Loan Agreement, Celly Nu Security Agreement and Plan of Arrangement, please see “Item 4. Information on the Company. - A. History and Development of the Company Overview and History”.
Coattail Agreement
In accordance with the rules of the CSE designed to ensure that, in the event of a take-over bid, the holders of Class B Shares will be entitled to participate on an equal footing with holders of Class A Shares, the holders of not less than 80% of the outstanding Class A Shares have entered into the coattail agreement dated May 24, 2018 among the Company, Computershare Investor Services Inc., the Company’s previous registrar and transfer agent of the Company, and certain of the shareholders holding at least 80% of the Class A Shares. The Coattail Agreement contains provisions customary for dual class, publicly traded Ontario corporations designed to prevent transactions that otherwise would deprive the holders of Class B Shares of rights under the take-over bid provisions of applicable Canadian securities legislation to which they would have been entitled if the Class A Shares had been Class B Shares.
See Exhibit 2.1, “Description of Securities,” for details.
UHN License Agreement
For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Other Significant Operations and Principal Activities – Fiscal 2021 and 2022”.
Epitech License Agreement and Prismic Assignment Agreement
For more information, please see “Item 4. Information on the Company. – C. Organizational Structure” and Exhibits 4.9, 4.10 and 4.11..
ATM Agreement
For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Significant Developments in Fiscal 2023 through to March 28, 2024”.
Lucid Amalgamation Agreement and Master Agreement
The Lucid Amalgamation Agreement is the amalgamation agreement dated September 20, 2021, entered into among the Company, Lucid and a wholly owned subsidiary of the Company (“Subco”) in connection with the Lucid Acquisition. Pursuant to the Lucid Amalgamation Agreement, Lucid Psycheceuticals Inc. and Subco agreed to amalgamate into Lucid.
For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Other Significant Operations and Principal Activities – Fiscal 2021 and 2022”.
D. Exchange Controls
The Company was formed under and subject to the laws of the Province of Ontario, Canada. Subject to the next paragraph and the disclosure under “Exhibit 2.1 - Description of Securities-Competition Act” and “Exhibit 2.1 - Description of Securities - Investment Canada Act” below, there is no law or governmental decree or regulation in Canada that restricts the export or import of capital, or affects the payment of dividends or interest or other amounts to a non-resident holder of Class B Shares, other than withholding tax requirements.
| 72 |
| Table of Contents |
There is no limitation imposed by Canadian law or by the charter or other constituent documents of the Company on the right of a non-resident to hold or vote Class B Shares of the Company. However, the Competition Act (Canada) and the Investment Canada Act (Canada) have rules regarding certain acquisitions of shares by certain persons, including non-residents, along with other requirements under that legislation.
See “Item 10. Additional Information - E. Taxation” for additional information regarding the material U.S. and Canadian federal income tax consequences relating to the ownership and disposition of our Class B Shares by U.S. Holders (as defined therein).
E. Taxation
Certain Material U.S. Federal Income Tax Considerations
The following is a general summary of certain material U.S. federal income tax considerations applicable to a U.S. Holder (as defined below) arising from and relating to the acquisition, ownership, and disposition of Class B Shares acquired pursuant to the offering.
This summary is for general information purposes only and does not purport to be a complete analysis or listing of all potential U.S. federal income tax considerations that may apply to a U.S. Holder arising from and relating to the acquisition, ownership, and disposition of Class B Shares. In addition, this summary does not take into account the individual facts and circumstances of any particular U.S. Holder that may affect the U.S. federal income tax consequences to such U.S. Holder, including, without limitation, specific tax consequences to a U.S. Holder under an applicable income tax treaty. Accordingly, this summary is not intended to be, and should not be construed as, legal or U.S. federal income tax advice with respect to any U.S. Holder. This summary does not address the U.S. federal alternative minimum, U.S. federal estate and gift, U.S. state and local, and non-U.S. tax consequences to U.S. Holders of the acquisition, ownership, and disposition of Class B Shares. In addition, except as specifically set forth below, this summary does not discuss applicable tax reporting requirements. Each prospective U.S. Holder is urged to consult its own tax advisor regarding the U.S. federal income, U.S. federal alternative minimum, U.S. federal estate and gift, U.S. state and local, and non-U.S. tax consequences relating to the acquisition, ownership, and disposition of Class B Shares.
No ruling from the Internal Revenue Service (the “IRS”) has been requested, or will be obtained, regarding the U.S. federal income tax consequences of the acquisition, ownership, and disposition of Class B Shares. This summary is not binding on the IRS, and the IRS is not precluded from taking a position that is different from, and contrary to, the positions taken in this summary. In addition, because the authorities on which this summary are based are subject to various interpretations, the IRS and the U.S. courts could disagree with one or more of the conclusions described in this summary.
Scope of this Summary
This summary is based on the Internal Revenue Code of 1986, as amended (the “Code”), Treasury Regulations (whether final, temporary, or proposed), published rulings of the IRS, published administrative positions of the IRS, the Convention Between Canada and the United States of America with Respect to Taxes on Income and on Capital, signed September 26, 1980, as amended (the “Canada-U.S. Tax Convention”), and U.S. court decisions that are applicable, and, in each case, as in effect, as of the date of this document. Any of the authorities on which this summary is based could be changed in a material and adverse manner at any time, and any such change could be applied retroactively. This summary does not discuss the potential effects, whether adverse or beneficial, of any proposed legislation.
For purposes of this summary, the term “U.S. Holder” means a beneficial owner of Class B Shares acquired pursuant to the offering that is for U.S. federal income tax purposes:
| · | an individual who is a citizen or resident of the United States; |
| · | a corporation (or other entity treated as a corporation for U.S. federal income tax purposes) organized under the laws of the United States, any state thereof or the District of Columbia; |
| · | an estate whose income is subject to U.S. federal income taxation regardless of its source; or |
| · | a trust that (i) is subject to the primary supervision of a court within the United States and the control of one or more U.S. persons for all substantial decisions or (ii) has a valid election in effect under applicable Treasury Regulations to be treated as a U.S. person. |
For purposes of this summary, the term “U.S. Holder” means a beneficial owner of Class B Shares acquired pursuant to the offering that is for U.S. federal income tax purposes:
| 73 |
| Table of Contents |
This summary does not address the U.S. federal income tax considerations applicable to U.S. Holders that are subject to special provisions under the Code, including, but not limited to, U.S. Holders that: (a) are tax-exempt organizations, qualified retirement plans, individual retirement accounts, or other tax-deferred accounts; (b) are financial institutions, underwriters, insurance companies, real estate investment trusts, or regulated investment companies; (c) are broker-dealers, dealers, or traders in securities or currencies that elect to apply a mark-to-market accounting method; (d) have a “functional currency” other than the U.S. dollar; (e) own Class B Shares as part of a straddle, hedging transaction, conversion transaction, constructive sale, or other arrangement involving more than one position; (f) acquire Class B Shares in connection with the exercise of employee stock options or otherwise as compensation for services; (g) hold Class B Shares other than as a capital asset within the meaning of Section 1221 of the Code (generally, property held for investment purposes); (h) are required to accelerate the recognition of any item of gross income with respect to Class B Shares as a result of such income being recognized on an applicable financial statement; or (i) own, have owned or will own (directly, indirectly, or by attribution) 10% or more of the total combined voting power or value of the outstanding shares of the Company. This summary also does not address the U.S. federal income tax considerations applicable to U.S. Holders who are: (a) U.S. expatriates or former long-term residents of the U.S.; (b) persons that have been, are, or will be a resident or deemed to be a resident in Canada for purposes of the Tax Act; (c) persons that use or hold, will use or hold, or that are or will be deemed to use or hold Class B Shares in connection with carrying on a business in Canada; (d) persons whose Class B Shares constitute “taxable Canadian property” under the Tax Act; or (e) persons that have a permanent establishment in Canada for the purposes of the Canada-U.S. Tax Convention. U.S. Holders that are subject to special provisions under the Code, including, but not limited to, U.S. Holders described immediately above, should consult their own tax advisor regarding the U.S. federal income, U.S. federal alternative minimum, U.S. federal estate and gift, U.S. state and local, and non-U.S. tax consequences relating to the acquisition, ownership, and disposition of Class B Shares.
If an entity or arrangement that is classified as a partnership (or other “pass-through” entity) for U.S. federal income tax purposes holds Class B Shares, the U.S. federal income tax consequences to such entity and the partners (or other owners) of such entity generally will depend on the activities of the entity and the status of such partners (or owners). This summary does not address the tax consequences to any such partner (or owner). Partners (or other owners) of entities or arrangements that are classified as partnerships or as “pass-through” entities for U.S. federal income tax purposes should consult their own tax advisors regarding the U.S. federal income tax consequences arising from and relating to the acquisition, ownership, and disposition of Class B Shares.
Ownership and Disposition of Class B Shares
The following discussion is subject in its entirety to the rules described below under the heading “Passive Foreign Investment Company Rules”.
Taxation of Distributions
A U.S. Holder that receives a distribution, including a constructive distribution, with respect to a Class B Share will be required to include the amount of such distribution in gross income as a dividend (without reduction for any foreign income tax withheld from such distribution) to the extent of the current or accumulated “earnings and profits” of the Company, as computed for U.S. federal income tax purposes. To the extent that a distribution exceeds the current and accumulated “earnings and profits” of the Company, such distribution will be treated first as a tax-free return of capital to the extent of a U.S. Holder’s tax basis in the Class B Shares and thereafter capital gain to the extent of the excess over the U.S. Holder’s tax basis, Capital gain will be taxed in the manner described below at “Sale or Other Taxable Disposition of Class B Shares”. The Company may not maintain the calculations of its earnings and profits in accordance with U.S. federal income tax principles, and each U.S. Holder may have to assume that any distribution by the Company with respect to the Class B Shares will constitute dividend income. Dividends received on Class B Shares by corporate U.S. Holders generally will not be eligible for the “dividends received deduction”. Subject to applicable limitations and provided the Company is eligible for the benefits of the Canada-U.S. Tax Convention or the Class B Shares are readily tradable on a United States securities market, dividends paid by the Company to non-corporate U.S. Holders, including individuals, generally will be eligible for the preferential tax rates applicable to long-term capital gains for dividends, provided certain holding period and other conditions are satisfied, including that the Company not be classified as a PFIC in the tax year of distribution or in the preceding tax year. The dividend rules are complex, and each U.S. Holder should consult its own tax advisor regarding the application of such rules.
Sale or Other Taxable Disposition of Class B Shares
A U.S. Holder will recognize gain or loss on the sale or other taxable disposition of Class B Shares in an amount equal to the difference, if any, between (a) the amount of cash plus the fair market value of any property received and (b) such U.S. Holder’s tax basis in such Class B Shares sold or otherwise disposed of. Any such gain or loss generally will be capital gain or loss, which will be long-term capital gain or loss if, at the time of the sale or other disposition, such Class B Shares are held for more than one year.
Preferential tax rates apply to long-term capital gains of a U.S. Holder that is an individual, estate, or trust. Deductions for capital losses are subject to significant limitations under the Code.
Passive Foreign Investment Company Rules
If the Company were to constitute a PFIC for any year during a U.S. Holder’s holding period, then certain potentially adverse rules would affect the U.S. federal income tax consequences to a U.S. Holder resulting from the acquisition, ownership, and disposition of Class B Shares. The Company believes that it was a PFIC for the prior tax year ended December 31, 2023, and based on current business plans and financial expectations, the Company expects to be a PFIC for the current tax year. If the Company is a PFIC in the taxable year in which a U.S. Holder first invests in the Company, the adverse rules described below will apply indefinitely unless the Company no longer is a PFIC in a subsequent taxable year and the U.S. Holder makes a timely “purging election” as described below. No opinion of legal counsel or ruling from the Internal Revenue Service (“IRS”) concerning the status of the Company as a PFIC has been obtained or is currently planned to be requested. However, PFIC classification is fundamentally factual in nature, generally cannot be determined until the close of the tax year in question, and is determined annually. In addition, the analysis depends, in part, on the application of complex U.S. federal income tax rules, which are subject to differing interpretations.
| 74 |
| Table of Contents |
General PFIC Rules
In any year in which the Company is classified as a PFIC, a U.S. Holder will be required to file an annual report with the IRS containing such information as Treasury Regulations and/or other IRS guidance may require. In addition to penalties, a failure to satisfy such reporting requirements may result in an extension of the time period during which the IRS can assess a tax. U.S. Holders should consult their own tax advisors regarding the requirements of filing such information returns under these rules, including the requirement to file an IRS Form 8621 annually.
The Company generally will be a PFIC if, after the application of certain “look-through” rules with respect to subsidiaries in which the Company holds at least 25% of the value of such subsidiary, for a tax year, (a) 75% or more of the gross income of the Company for such tax year is passive income (the “income test”) or (b) 50% or more of the value of the Company’s assets either produce passive income or are held for the production of passive income (the “asset test”), based on the quarterly average of the fair market value of such assets. “Gross income” generally includes all sales revenues less the cost of goods sold, plus income from investments and from incidental or outside operations or sources, and “passive income” generally includes, for example, dividends, interest, certain rents and royalties, certain gains from the sale of stock and securities, and certain gains from commodities transactions. Active business gains arising from the sale of commodities generally are excluded from passive income if substantially all of a foreign corporation’s commodities are stock in trade or inventory, depreciable property used in a trade or business or supplies regularly used or consumed in the ordinary course of its trade or business, and certain other requirements are satisfied.
If the Company were a PFIC in any tax year during which a U.S. Holder held Class B Shares, and subject to a U.S. Holder making a “QEF Election” or “Mark-to-Market Election” as described below, such holder generally would be subject to special rules with respect to “excess distributions” made by the Company on the Class B Shares and with respect to gain from the disposition of Class B Shares. An “excess distribution” generally is defined as the excess of distributions with respect to the Class B Shares received by a U.S Holder in any tax year over 125% of the average annual distributions such U.S. Holder has received from the Company during the shorter of the three preceding tax years, or such U.S. Holder’s holding period for the Class B Shares. Generally, a U.S. Holder would be required to allocate any excess distribution or gain from the disposition of the Class B Shares ratably over its holding period for the Class B Shares. Such amounts allocated to the year of the disposition or excess distribution would be taxed as ordinary income, and the preferential tax rates applicable to capital gains or dividends received on our Class B shares would not be available. In addition, amounts allocated to prior tax years would be taxed as ordinary income at the highest tax rate in effect for each such year and an interest charge would apply at a rate applicable to underpayments. These adverse tax consequences would not apply to a pension or profit-sharing trust or other tax-exempt organization that did not borrow funds or otherwise utilize leverage in connection with its acquisition of Class B Shares. In addition, if a non-electing U.S. Holder who is an individual dies while owning our Class B Shares, such U.S. Holder’s successor generally would not receive a step-up in tax basis with respect to such Class B Shares, but instead would have a tax basis equal to the lower of the fair market value of such Class B Shares or the decedent’s tax basis in such Class B Shares.
QEF Election
The tax consequences described above upon a PFIC determination may be mitigated if a U.S. Holder makes a timely “qualified electing fund” election (a “QEF election”) with respect to its interest in the PFIC, provided the Company provides the U.S Holder with the necessary information regarding its ordinary earnings and net capital gain. Consequently, if the Company is classified as a PFIC, it would likely be advantageous for a U.S. Holder to elect to treat the investment as a “qualified electing fund” (a “QEF”) with respect to such U.S. Holder in the first year in which it holds Class B Shares. If a U.S. Holder makes a timely QEF election with respect to the Company, the electing U.S. Holder would be required in each taxable year that the Company is considered to be a PFIC to include in gross income (i) as ordinary income, the U.S. Holder’s pro rata share of the ordinary earnings of the Company and (ii) as capital gain, the U.S. Holder’s pro rata share of the net capital gain (if any) of the Company, whether or not the ordinary earnings or net capital gain are distributed. An electing U.S. Holder’s basis in its Class B Shares will be increased to reflect the amount of any taxed but undistributed income. Distributions of income that had previously been taxed will result in a corresponding reduction of basis in the Class B Shares and will not be taxed again as distributions to the U.S. Holder. Gain realized from the sale of our Class B Shares covered by a QEF election would be taxed as a capital gain and the denial of the basis step-up at death described above would not apply. Generally, a QEF election must be made by the U.S. Holder in a timely filed tax return for the first taxable year in which the U.S. Holder held our Class B Shares that includes the close of our taxable year for which we met the PFIC gross income test or asset test. A separate QEF election would need to be made for any of our subsidiaries that are classified as a PFIC. A QEF election is made on IRS Form 8621.
The U.S. federal income tax on any gain from the disposition of Class B Shares or from the receipt of Excess Distributions may be greater than the tax that would apply if a timely QEF election is made. If the Company does not provide the required information with regard to the QEF election, U.S. Holders will not be able to make a QEF election and will, subject to the discussion of the mark-to-market election below, continue to be subject to the general PFIC rules as described above. U.S. Holders are urged to consult their own tax advisors regarding the advisability and availability of making a QEF election with respect to the Company.
Mark-to-Market Election
Alternatively, if the Company were to be classified as a PFIC, a U.S. Holder could also avoid certain of the general PFIC rules described above by making a timely mark-to-market election on Form 8621 (instead of a QEF election), provided the Class B Shares are treated as regularly traded on a qualified exchange or other market within the meaning of the applicable Treasury regulations. U.S. Holders are urged to consult their own tax advisers regarding the potential availability and consequences of a mark-to-market election. A U.S. Holder who makes the mark-to-market election generally must include as ordinary income each year the increase in the fair market value of the Class B Shares and deduct from gross income the decrease in the value of such shares during each of its taxable years, but with losses limited to the amount of previously recognized net gains. The U.S. Holder’s tax basis in the Class B Shares would be adjusted to reflect any income or loss recognized as a result of the mark-to-market election. If a mark-to-market election with respect to our Class B Shares is in effect on the date of a U.S. Holder’s death, the tax basis of the Class B Shares in the hands of a U.S. Holder who acquired them from a decedent will be the lesser of the decedent’s tax basis or the fair market value of the Class B Shares. Any gain from a sale, exchange or other disposition of the Class B Shares in any taxable year in which we are a PFIC (i.e., when we meet the gross income test or asset test described above) would be treated as ordinary income and any loss from a sale, exchange or other disposition would be treated first as an ordinary loss (to the extent of any net mark-to-market gains previously included in income) and thereafter as a capital loss. If we cease to be a PFIC, any gain or loss recognized by a U.S. Holder on the sale or exchange of the Class B Shares would be classified as a capital gain or loss. The Class B Shares should be marketable stock as long as they are listed on the Nasdaq Capital Market and are regularly traded. A mark-to-market election will not apply to the Class B Shares for any taxable year during which we are not a PFIC but will remain in effect with respect to any subsequent taxable year in which we again become a PFIC. Such election will not apply to any subsidiary that we own. Accordingly, a U.S. Holder may continue to be subject to the PFIC rules with respect to any lower-tier PFICs notwithstanding the U.S. Holder’s mark-to-market election.
| 75 |
| Table of Contents |
Purging Election
If we are a PFIC at any time when a U.S. Holder holds our Class B Shares, we will generally continue to be treated as a PFIC with respect to the U.S. Holder for all succeeding years during which the U.S. Holder holds our Class B Shares even if we cease to meet the PFIC gross income test or asset test in a subsequent year. However, if we cease to meet these tests, a U.S. Holder can avoid the continuing impact of the PFIC rules by making a special election (a Purging Election) to recognize gain by making a “deemed sale” election with respect to all of the U.S. Holder’s Class B Shares and have such Class B Shares deemed to be sold at their fair market value on the last day of the last taxable year during which we were a PFIC. Under another type of purging election, the Company will be deemed to have made a distribution to the U.S. Holder of such U.S. Holder’s pro rata share of the Company’s earnings and profits as determined for U.S. federal income tax purposes. In order for the U.S. Holder to make this second election, the Company must also be determined to be a “controlled foreign corporation” as defined by the U.S. Tax Code (which may not be the case, but please see the Controlled Foreign Corporation section below). The shareholder makes a purging election under Code section 1298(b)(1) and regulations section 1.1298–3 on IRS Form 8621 attached to the shareholder’s tax return (including an amended return), or requests the consent of the IRS Commissioner to make a late election under Code section 1298(b)(1) and regulations section 1.1298–3(e) (late purging election) on Form 8621-A. In addition, for a U.S. Holder making such an election, a new holding period would be deemed to begin for our Class B Shares for purposes of the PFIC rules. After the Purging Election, the Class B Shares with respect to which the Purging Election was made will not be treated as shares in a PFIC unless we subsequently again become a PFIC.
Each U.S. person who is a shareholder of a PFIC generally must file an annual report (on IRS Form 8621) with the IRS containing certain information, and the failure to file such report could result in the imposition of penalties on such U.S. person and in the extension of the statute of limitations with respect to federal income tax returns filed by such U.S. person.
U.S. Holders should be aware that, for each tax year, if any, that the Company is a PFIC, the Company can provide no assurances that it will satisfy the record keeping requirements or make available to U.S. Holders the information such U.S. Holders require to make a QEF Election with respect to the Company or any subsidiary that also is classified as a PFIC. U.S. Holders should consult their own tax advisors regarding the potential application of the PFIC rules to the ownership and disposition of Class B Shares, and the availability of certain U.S. tax elections under the PFIC rules.
Additional Considerations
Controlled Foreign Corporation
As a result of the enactment of the Tax Cuts and JOBS Act and the repeal of Code section 958(b)(4), it is possible to accidentally create a controlled foreign corporation (“CFC”) without having a direct or indirect United States shareholder. Historically, Code section 958(b)(4) prevented stock owned by a foreign person from being attributed downward to a U.S. person (e.g., a partnership, corporation, trust, or estate) owned by such foreign person. Effective as of the last taxable year of a foreign corporation beginning before January 1, 2018, stock may be attributed downward from a person to a corporation if “50 percent or more in value of the stock in a corporation is owned, directly or indirectly by or such person.” As a result of the repeal of Code section 958(b)(4), it is thus possible to accidentally create CFCs without a direct or indirect United States shareholder. This is because, since the repeal, a U.S. corporation owned by a foreign parent corporation may be treated as constructively owning the stock of the foreign parent or even the foreign parent’s foreign subsidiaries (i.e., foreign brother-sister companies). The Company believes that certain case law along with the legislative intent of the repeal may substantiate that it is not a CFC, although it is possible that the IRS may disagree. In discussing the repeal of Code section 958(b)(4), the Senate amendment that was followed by the Conference Report provides: “[f]urthermore, the Senate Finance Committee explanation states that the provision is not intended to cause a foreign corporation to be treated as a controlled foreign corporation regarding a U.S. shareholder as a result of attribution of ownership under section 318(a)(3) to a U.S. person that is not a related person (within the meaning of section 954(d)(3)) to such U.S. shareholder as a result of the repeal of section 958(b)(4).” The Tax Court case of Nettie Miller v. Commissioner stands for the premise that reading the current rules strictly would result in an absurd result that the Company’s U.S. subsidiary (a non-Code section 958(a) shareholder) is treated as constructively owning the Company. Furthermore, in Rev. Rul. 74-605, the IRS concluded that a subsidiary could not be attributed ownership of its direct or indirect parent corporations for purposes of applying Code section 304. The IRS’s rationale is that if a subsidiary were treated as constructively owning its parent (or grandparent) under the constructive ownership rules (that is, Code section 318(a)(3)(C)), it would further be treated as owning its own stock. The IRS concluded that treating a subsidiary as owning its own stock would violate regulation section 1.318-1(b)(1), which provides that “a corporation shall not be considered to own its own stock by reason of section 318(a)(3)(C).” Thus, the Company may not be a CFC as a result of Rev. Rul. 74-605, Nettie Miller, and the legislative intent of the repeal of Code section 958(b)(4). It should be noted, however, that the rationale for the Company not being classified as a CFC may not extend to the Company’s non-U.S. subsidiaries. As such, any U.S. Holders that own 10 percent or more of the total combined voting power of all classes of stock entitled to vote of the Company, or 10 percent or more of the total value of shares of all classes of stock of the Company should consider whether the overlap rules for CFCs and PFICs apply to them pursuant to Code section 1297(d) when determining their U.S. tax obligations for the Company or any of its non-U.S. subsidiaries. U.S. Holders should consult their own tax advisors regarding the application of this rule since the attribution rules related to ownership are very complex.
| 76 |
| Table of Contents |
Additional Tax on Passive Income
Certain U.S. Holders that are individuals, estates and trusts whose income exceeds certain thresholds will be required to pay a 3.8% surtax on “net investment income” including, among other things, dividends, and net gain from disposition of property (other than property held in certain trades or businesses). U.S. Holders should consult their own tax advisors regarding the application, if any, of this tax on their ownership and disposition of Class B Shares.
Receipt of Foreign Currency
The amount of any distribution paid to a U.S. Holder in foreign currency, or on the sale, exchange, or other taxable disposition of Class B Shares, generally will be equal to the U.S. dollar value of such foreign currency based on the exchange rate applicable on the date of receipt (regardless of whether such foreign currency is converted into U.S. dollars at that time). A U.S. Holder will have a basis in the foreign currency equal to its U.S. dollar value on the date of receipt. Any U.S. Holder who converts or otherwise disposes of the foreign currency after the date of receipt may have a foreign currency exchange gain or loss that would be treated as ordinary income or loss, and generally will be U.S. source income or loss for foreign tax credit purposes. Different rules apply to U.S. Holders who use the accrual method of tax accounting. Each U.S. Holder should consult its own U.S. tax advisor regarding the U.S. federal income tax consequences of receiving, owning, and disposing of foreign currency.
Foreign Tax Credit
Subject to the PFIC rules discussed above, a U.S. Holder that pays (whether directly or through withholding) Canadian income tax with respect to dividends paid on the Class B Shares generally will be entitled, at the election of such U.S. Holder, to receive either a deduction or a credit for such Canadian income tax. Generally, a credit will reduce a U.S. Holder’s U.S. federal income tax liability on a dollar-for-dollar basis, whereas a deduction will reduce a U.S. Holder’s income that is subject to U.S. federal income tax. This election is made on a year-by-year basis and applies to all foreign taxes paid (whether directly or through withholding) by a U.S. Holder during a year.
Complex limitations apply to the foreign tax credit, including the general limitation that the credit cannot exceed the proportionate share of a U.S. Holder’s U.S. federal income tax liability that such U.S. Holder’s “foreign source” taxable income bears to such U.S. Holder’s worldwide taxable income. In applying this limitation, a U.S. Holder’s various items of income and deduction must be classified, under complex rules, as either “foreign source” or “U.S. source.” Generally, dividends paid by a foreign corporation should be treated as foreign source for this purpose, and gains recognized on the sale of stock of a foreign corporation by a U.S. Holder should be treated as U.S. source for this purpose, except as otherwise provided in an applicable income tax treaty, and if an election is properly made under the Code. However, the amount of a distribution with respect to the Class B Shares that is treated as a “dividend” may be lower for U.S. federal income tax purposes than it is for Canadian federal income tax purposes, resulting in a reduced foreign tax credit allowance to a U.S. Holder. In addition, this foreign tax credit limitation is calculated and applied separately with respect to specific categories of income. The foreign tax credit rules are complex, and each U.S. Holder should consult its own U.S. tax advisor regarding the foreign tax credit rules.
Backup Withholding and Information Reporting
Under U.S. federal income tax law and Treasury Regulations, certain categories of U.S. Holders must file information returns with respect to their investment in, or involvement in, a foreign corporation. For example, U.S. return disclosure obligations (and related penalties) are imposed on individuals who are U.S. Holders that hold certain specified foreign financial assets in excess of certain threshold amounts. The definition of specified foreign financial assets includes not only financial accounts maintained in foreign financial institutions, but also, unless held in accounts maintained by a financial institution, any stock or security issued by a non-U.S. person, any financial instrument or contract held for investment that has an issuer or counterparty other than a U.S. person and any interest in a foreign entity. U. S. Holders may be subject to these reporting requirements unless their Class B Shares are held in an account at certain financial institutions. Penalties for failure to file certain of these information returns are substantial. U.S. Holders should consult their own tax advisors regarding the requirements of filing information returns, including the requirement to file an IRS Form 8938.
Payments made within the United States or by a U.S. payor or U.S. middleman, of dividends on, and proceeds arising from the sale or other taxable disposition of, Class B Shares will generally be subject to information reporting and backup withholding tax, at the rate of 24%, if a U.S. Holder (a) fails to furnish such U.S. Holder’s correct U.S. taxpayer identification number (generally on Form W-9), (b) furnishes an incorrect U.S. taxpayer identification number, (c) is notified by the IRS that such U.S. Holder has previously failed to properly report items subject to backup withholding tax, or (d) fails to certify, under penalty of perjury, that such U.S. Holder has furnished its correct U.S. taxpayer identification number and that the IRS has not notified such U.S. Holder that it is subject to backup withholding tax. However, certain exempt persons generally are excluded from these information reporting and backup withholding rules. Backup withholding is not an additional tax. Any amounts withheld under the U.S. backup withholding tax rules generally will be allowed as a credit against a U.S. Holder’s U.S. federal income tax liability, if any, or will be refunded, if such U.S. Holder furnishes required information to the IRS in a timely manner.
The discussion of reporting requirements set forth above is not intended to constitute a complete description of all reporting requirements that may apply to a U.S. Holder. A failure to satisfy certain reporting requirements may result in an extension of the time period during which the IRS can assess a tax, and under certain circumstances, such an extension may apply to assessments of amounts unrelated to any unsatisfied reporting requirement. Each U.S. Holder should consult its own tax advisor regarding the information reporting and backup withholding rules.
| 77 |
| Table of Contents |
THE ABOVE SUMMARY IS NOT INTENDED TO CONSTITUTE A COMPLETE ANALYSIS OF ALL TAX CONSIDERATIONS APPLICABLE TO U.S. HOLDERS WITH RESPECT TO THE ACQUISITION, OWNERSHIP, AND DISPOSITION OF CLASS B SHARES. U.S. HOLDERS SHOULD CONSULT THEIR OWN TAX ADVISORS AS TO THE TAX CONSIDERATIONS APPLICABLE TO THEM IN THEIR OWN PARTICULAR CIRCUMSTANCES.
Certain Canadian Federal Income Tax Considerations
The following summary describes, as of the date hereof, the material Canadian federal income tax considerations generally applicable to a Shareholder who is a beneficial owner of our Class B Shares and who, at all relevant times, for the purposes of the application of the Income Tax Act (Canada) and the Income Tax Regulations (collectively, the “Canadian Tax Act”), (1) is not, and is not deemed to be, resident in Canada for purposes of the Canadian Tax Act and any applicable income tax treaty or convention; (2) deals at arm’s length with us; (3) is not affiliated with us; (4) does not use or hold, and is not deemed to use or hold, Class B Shares in a business or part of a business carried on in Canada; (5) has not entered into, with respect to the Class B Shares, a “derivative forward agreement”, as that term is defined in the Canadian Tax Act and (6) holds the Class B Shares as capital property (a “Non-Canadian Holder”). This summary does not apply to a Non-Canadian Holder that is an insurer carrying on an insurance business in Canada and elsewhere or an “authorized foreign bank”, as that term is defined in the Canadian Tax Act. Such Non-Canadian Holders should consult their own tax advisors for advice having regards to their particular circumstances.
This summary is based on the current provisions of the Canadian Tax Act and the Canada-United States Tax Convention (1980), as amended (the “Canada-U.S. Tax Treaty”), and an understanding of the current administrative policies and assessing practices of the Canada Revenue Agency published in writing prior to the date hereof. It takes into account all specific proposals to amend the Canadian Tax Act and the Canada-U.S. Tax Treaty, publicly announced by or on behalf of the Minister of Finance (Canada) prior to the date hereof (the “Proposed Amendments”) and assumes that all Proposed Amendments will be enacted in the form proposed. However, no assurances can be given that the Proposed Amendments will be enacted as proposed, or at all. This summary does not otherwise take into account or anticipate any changes in law or administrative policy or assessing practice whether by legislative, regulatory, administrative or judicial decision or action nor does it take into account tax legislation or considerations of any province, territory or foreign jurisdiction, which may differ from those discussed herein.
This summary is of a general nature only and is not, and is not intended to be, legal or tax advice to any particular shareholder, and no representations with respect to the income tax consequences to any particular shareholder are made. This summary is not exhaustive of all Canadian federal income tax considerations. Accordingly, you should consult your own tax advisor with respect to your particular circumstances.
Generally, for purposes of the Canadian Tax Act, all amounts relating to the acquisition, holding or disposition of the Class B Shares must be converted into Canadian dollars based on the exchange rate quoted by the Bank of Canada on the date such amount arose or such other rate of exchange as is acceptable to the Minister of National Revenue (Canada).
Dividends
Dividends paid or credited on the Class B Shares or deemed to be paid or credited on the Class B Shares to a Non-Canadian Holder will be subject to Canadian withholding tax at the rate of 25% on the gross amount of the dividend, subject to any reduction in the rate of withholding to which the Non-Canadian Holder is entitled under any applicable income tax treaty or convention between Canada and the country in which the Non-Canadian Holder is resident. For example, under the Canada-U.S. Tax Treaty, where dividends on the Class B Shares are considered to be paid to or derived by a Non-Canadian Holder that is a beneficial owner of the dividends and is a U.S. resident for the purposes of, and is entitled to the full benefits of, the Canada-U.S. Tax Treaty, the applicable rate of Canadian withholding tax is generally reduced to 15%. We will be required to withhold the applicable withholding tax from any dividend and remit it to the Canadian government for the Non-Canadian Holder’s account. Non-Canadian Holders are urged to consult their own advisors to determine their entitlement to relief under an applicable income tax treaty or convention.
Dispositions
A Non-Canadian Holder will not be subject to tax under the Canadian Tax Act on any capital gain realized on a disposition or deemed disposition of a Class B Share, unless the Class B Share is “taxable Canadian property” to the Non-Canadian Holder for purposes of the Canadian Tax Act at the time of disposition and the Non-Canadian Holder is not entitled to relief under an applicable income tax treaty or convention between Canada and the country in which the Non-Canadian Holder is resident.
Generally, the Class B Shares will not constitute “taxable Canadian property” to a Non-Canadian Holder at a particular time provided that the Class B Shares are listed at that time on a “designated stock exchange” (as defined in the Canadian Tax Act), which currently includes the CSE and the Nasdaq, unless at any particular time during the 60-month period that ends at that time the following two conditions are met concurrently:
| · | at least 25% of the issued shares of any class or series of our capital stock was owned by or belonged to any combination of (a) the Non-Canadian Holder, (b) persons with whom the Non-Canadian Holder does not deal at arm’s length for purposes of the Canadian Tax Act, and (c) partnerships in which the Non-Canadian Holder or a person described in (b) holds a membership interest directly or indirectly through one or more partnerships, and |
| · | more than 50% of the fair market value of the Class B Shares was derived, directly or indirectly, from one or any combination of: (i) real or immoveable property situated in Canada, (ii) “Canadian resource properties” (as that term is defined in the Canadian Tax Act), (iii) “timber resource properties” (as that term is defined in the Canadian Tax Act) or (iv) options in respect of, or interests in, or for civil law rights in, a property described in any of the foregoing whether or not the property exists. |
| 78 |
| Table of Contents |
Notwithstanding the foregoing, in certain circumstances, Class B Shares could be deemed to be “taxable Canadian property” to a Non-Canadian Holder. Non-Canadian Holders whose Class B Shares are, or may constitute, “taxable Canadian property” should consult their own tax advisors.
F. Dividends and Paying Agents
Not applicable.
G. Statement by Experts
Not applicable.
H. Documents on Display
We are subject to the information reporting requirements of the Exchange Act applicable to foreign private issuers and under those requirements will file reports with the SEC. Those reports may be inspected without charge at the locations described below. As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders are exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file periodic reports and financial statements with the SEC as frequently or as promptly as United States companies whose securities are registered under the Exchange Act. Nevertheless, we will file with the SEC an Annual Report on Form 20-F containing financial statements that have been examined and reported on, with and opinion expressed by an independent registered public accounting firm.
We maintain a corporate website at www.fsdpharma.com. We intend to post our Annual Report on Form 20-F on our website promptly following its filing with the SEC. Information contained on, or that can be accessed through, our website does not constitute a part of this Annual Report. We have included our website address in this Annual Report solely as an inactive textual reference.
The SEC maintains a website (www.sec.gov) that contains reports, proxy and information statements and other information regarding registrants, such as us, that file electronically with the SEC.
This Annual Report, copies of our financial statements and other continuous disclosure documents required under the Securities Act (Ontario) are available for viewing on the Company’s SEDAR+ profile accessible at www.sedarplus.ca. All of the documents referred to are in English.
With respect to references made in this Annual Report to any contract or other document of our company, such references are not necessarily complete, and you should refer to the exhibits attached or incorporated by reference to this Annual Report for copies of the actual contract or document.
I. Subsidiary Information
Not applicable.
J. Annual Report to Security Holders
Not applicable.
Item 11. Quantitative and Qualitative Disclosures About Market Risk.
Not applicable.
Item 12. Description of Securities Other than Equity Securities
A. Debt Securities
Not applicable.
B. Warrants and Rights
Not applicable.
C. Other Securities
Not applicable.
D. American Depositary Shares
Not applicable.
| 79 |
| Table of Contents |
PART II
Item 13. Defaults, Dividend Arrearages and Delinquencies
Not applicable.
Item 14. Material Modifications to the Rights of Security Holders and Use of Proceeds.
A.-D.
Not applicable.
E. Use of Proceeds
Use of Proceeds Reconciliation from 2020 Prospectuses
2020 Prospectuses(1)(2)(3) | |||
| Allocated | Spent to Date | Difference |
Acquisitions(4)(5) | US$5,000,000 |
| US$5,000,000 |
Investments(6) Strategic Investments | US$6,000,000
|
US$7,720,190 | US$6,000,000 (US$7,720,190) |
Capital Expenditures (Research and Development)(5) FSD201 Lucid-MS Lucid-Psych Alcohol Misuse Segment (UNBUZZED and the Healthcare Product) | US$35,000,000
|
US$10,578,381 US$4,957,100 US$2,383,733 US$1,544,210 | US$35,000,000 (US$10,578,381) (US$4,957,100) (US$2,383,733) (US$1,544,210) |
Working Capital and General Corporate((7)(8)9)(10)(11) Legal Expenses, Litigation and Contested Meeting Operating Expenses | US$10,000,000
|
US$11,681,072 US$10,416,756 | US$10,000,000 (US$11,681,072) (US$10,416,756) |
Strategic Initiatives(12) Share Buyback 2022 Share Buyback 2023 |
|
US$1,730,255 US$2,895,487 |
(US$1,730,255) (US$2,895,487) |
Total: | US$56,000,000 | US$53,907,184 | US$2,092,816 |
Notes:
1. | Pursuant to the Company’s previous shelf registration statement dated on February 8, 2020 filed on Form F-10, SEC File No. 333- 236780, which became effective on June 17, 2020 (as amended, the "2020 Registration Statement") and the Company’s short form base shelf prospectus dated June 16, 2020 (the “2020 Base Shelf Prospectus” and together with the 2020 Registration Statement, the “2020 Prospectus”), the Corporation planned to distribute up to an aggregate initial offering price of US$100,000,000 (or the equivalent in other currencies or currency units based on the applicable exchange rate at the time of the offering) of its securities. | ||||||||
2. | Pursuant to the 2020 Prospectuses, the Corporation allocated anticipated proceeds for the development of FSD201, future acquisitions and investments, to financial capital expenditures and for working capital and general corporate purposes. | ||||||||
3. | Under the 2020 Prospectuses, the Corporation, completed four raises totaling US$59,453,014. | ||||||||
i. | An at-the-market offering of up to US$19,976,512 pursuant to a prospectus supplement dated July 10, 2020 (the “First ATM”). Under the First ATM, the Corporation raised US$19,976,512 through the issuance of 7,412,574 Class B Shares at an average price of US$2.69 per Class B Share. For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Other Significant Operations and Principal Activities – Fiscal 2021 and 2022”. | ||||||||
ii. | An offering of up to US$9,999,996 pursuant to a prospectus supplement dated July 31, 2020 (the “July 2020 Offering”). Under the July 2020 Offering, the Corporation raised US$9,999,996 by issuing a total of 2,762,430 Class B Shares at a price of US$3.62 per Class B Share and warrants to purchase 1,381,215 Class B Shares. Each warrant had an exercise price of US$4.26 per Class B Share and expires five years from the date of issuance. | ||||||||
iii. | An offering of up to US$9,499,994 pursuant to a prospectus supplement dated October 16, 2020 (the “October 2020 Offering”). Under the October 2020 Offering, the Corporation raised US$9,499,994 by issuing a total of 4,318,179 Class B Shares at a price of US$2.20 per Class B Share and warrants to purchase 3,454,543 Class B Shares. Each warrant had an exercise price of US$2.60 per Class B Share and expires five years from the date of issuance. | ||||||||
iv. | An at-the-market offering of up to US$19,994,712 pursuant to a prospectus supplement dated February 11, 2021 (the “Second ATM”). Under the Second ATM, the Corporation raised US$19,994,713 through the issuance of 8,124,136 Class B Shares at an average price of US$2.46. For more information, please see “Item 4. Information on the Company. - A. History and Development of the Company - Other Significant Operations and Principal Activities – Fiscal 2021 and 2022”. | ||||||||
4. | On September 21, 2021, the Corporation acquired Lucid, which was satisfied in Class B Shares. This transaction did not require a cash outlay and thus the Corporation repurposed the funds towards two share repurchase programs, as further outlined in Note 12 below. | ||||||||
5. | Includes funds allocated to complete one or more strategic corporate transactions (such as acquisitions) intended to accelerate the growth and development of the Corporation’s businesses, which funds remain subject to reallocation by the Corporation towards budgets and expenditures which are identified and/or otherwise unidentified or unforeseen, as at the relevant date. | ||||||||
6. | During the effectiveness of the 2020 Prospectuses, the Corporation explored multiple investment opportunities, but none of them met the investment criteria of the Corporation and the Corporation repurposed the funds towards secured loans with an average maturity of two years from the date of issuance that earn fees at fixed rates. | ||||||||
7. | Includes (i) funds allocated for the payment of (a) general corporate and administrative expenses, (b) ongoing legal fees and other professional and consulting fees, and salaries, and (c) ongoing costs associated with being a reporting issuer, and (ii) funds generally reserved for (x) allocation towards future expenditures which are otherwise unidentified or unforeseen as of the relevant date, and (y) re-allocation towards the budgets allocated for identified purposes as of the relevant date. | ||||||||
| 80 |
| Table of Contents |
8. | On January 4, 2021, certain shareholders of the Corporation (the “Requisitioning Shareholders”) requisitioned a meeting of shareholders of the Corporation in compliance with section 105 of the OBCA (the “Contested Meeting”). On January 21, 2021, the Board called the Contested Meeting to be held on June 29, 2021; however, the Requisitioning Shareholders filed an application with the Commercial List of the Superior Court of Justice (the “Superior Court”) to, among other things, have the date of the Contested Meeting accelerated. This led to a series of appearances before the Superior Cout and subsequently the divisional court. On March 30, 2021, the Corporation commenced an action against the Requisition Shareholders alleging that their dissident circular was mislead and deficient, however, this claim was dismissed in its entirety by the Superior Court on May 10, 2021. In addition, the Requisitioning Shareholders filed an additional statement of claim on April 6, 2021, against the Corporation and Board stating that, among other things, their conduct against shareholders, including the Requisition Shareholders, was oppressive (the “Oppression Claim”). The Oppression Claim has been dormant since the conclusion of the Contested Meeting. In April 2021, Mr. Durkacz brought a motion before the Superior Court seeking various relief in relation to the conduct of the Contested Meeting, which was partially granted by the Superior Court on May 10, 2021. At the Contest Meeting, held on May 14, 2021, the previous Board members were relieved of there duties. The Contested Meeting and associated events resulted in additional unexpected legal and operating expenses of C$3,113,170 (approximately US$2,338,795, converted at a price of C$1.00:US$0.7513 based on the Bank of Canada exchange rate as of December 21, 2023). | |
9. | Following the Contested Meeting, the former CEO, Dr. Raza Bokhari commenced five actions against the Corporation and management, which resulted in counterclaims and additional unexpected legal and operating expenses of C$7,402,079 (approximately US$5,560,874, converted at a price of C$1.00:US$0.7513 based on the Bank of Canada exchange rate as of December 21, 2023): |
i. | Dr. Raza Bokhari: On July 15, 2021, the Corporation’s former CEO, Dr. Raza Bokhari, filed an arbitration notice seeking C$30.2 million for breach of contract, severance, and damages, along with C$500,000 for punitive damages and legal fees. Dr. Bokhari had been placed on administrative leave after the May 14, 2021 shareholder meeting and was terminated for cause on July 27, 2021, following an investigation by a special committee. The arbitration concluded in August 2022 with Dr. Raza Bokhari’s claims dismissed. Dr. Bokhari was also ordered to repay certain funds to the Corporation and cover arbitration costs in the amount of approximately C$2.8M plus interest. On December 9, 2022, Dr. Bokhari sought to set aside the award, citing unfair treatment and inadequate reasoning. On October 4, 2023, the Corporation announced that the Ontario Superior Court of Justice had dismissed Dr. Bokhari’s motion to set aside the arbitration award. Dr. Bokhari was required to put up C$150,000 as security for costs before the motion was heard, which he has forfeited. In addition, Dr. Bokhari was ordered to pay C$175,000 to cover the Corporation’s legals costs for his failed set aside motion. On December 1, 2023, the Corporation filed a petition to confirm the arbitration award in the United States District Court for Eastern District of Pennsylvania. On October 13, 2023, Dr. Bokhari served notices of motion on the Corporation for leave to appeal the set aside and enforcement orders issued by the Ontario Superior Court of Justice’s on October 4, 2023. On December 15, 2023, the Corporation submitted a responding party’s factum to the Court of Appeal for Ontario. As of the date hereof, the litigation is ongoing. | ||||||||
ii. | Parkway Clinical Laboratories: On July 8, 2021, Parkway Clinical Laboratories, a company wholly owned by Dr. Bokhari, filed an action against the Corporation, which was subsequently settled following a conference between the parties on October 20, 2021. | ||||||||
iii. | Bokhari v. FSD Pharma Inc. Et al.: On July 2, 2021, Dr Bokhari filed an action against the Corporation, FSD BioSciences, Anthony Durkacz and Zeeshan Saeed. The case was placed in civil suspense pending resolution of arbitration. Therefore, no further activity will occur in this case unless and until the aforementioned arbitration concludes. As of the date hereof, the litigation is ongoing. | ||||||||
iv. | Restraining Order and Class B Share Cancellation Application: On January 21, 2021 and February 10, 2021, the Board authorized the issuance of an aggregate of 1,349,765 Class B Shares as share based awards to certain directors and officers of the Corporation, including Dr. Bokhari. Upon determining that the Contested Shares had been inappropriately issued contrary to applicable laws, the Board resolved to cancel the Contested Shares on June 1, 2021, and later directed the Corporation’s transfer agent to cancel and return the Contested Shares to treasury. On July 2, 2021, Dr. Bokhari, filed an action against the Corporation seeking to prevent the Corporation from cancelling his portion of the Contested States. The motion was heard and denied on July 27, 2021. On July 21, 2021, the Corporation commenced a legal proceeding against Dr. Bokhari, former members of the Board, including James Datin, Robert Ciaruffoli, Stephen Buyer and Gerald Goldberg, Dr. Bokhari’s brokerages’ Haywood Securities Inc. and Haywood Securities (US) Inc., and the Corporation’s transfer agent. The Corporation made an application before the Superior Court stating that the Contested Shares were issued contrary to section 23(2) of the OBCA and validly cancelled by resolution of the Board passed on June 1, 2021. The Corporation was able to reach an agreement with all of the former directors other than Dr. Bokhari under which they did not oppose the Corporation’s application and agreed to be bound by the decision in the application, and the Corporation agreed not to seek costs against them. Neither the Corporation’s transfer agent nor any of Dr. Bokhari’s brokerages took any position on the application. On March 8, 2022, the court issued a mixed decision in the application, permitting the Contested Share grant to Dr. Bokhari until the date of his termination but cancelling 504,888 Contested Shares relating to services that were to be provided after the date of termination. | ||||||||
v. | Indemnity Application: On April 6, 2022, Dr. Bokhari commenced an application in the Superior Court seeking an order appointing an arbitrator to arbitrate his claim to be entitled to indemnification of his legal expenses associated with the litigation he has commenced against the Corporation or in which he has been named as a defendant against the Corporation. The Corporation denied the validity of the underlying indemnification agreement and opposed the application. In April 2022, the parties agreed to adjourn the application without setting a new hearing date. As of the date hereof, the litigation is ongoing. | ||||||||
| 10. | On July 20, 2021, a shareholder of the Corporation filed a claim in the Delaware Chancery Court against the Corporation and its directors and officers seeking to remedy harm they believe the directors and officers of the Corporation have caused by their actions. The shareholder filed the claim on count of breach of fiduciary duties and corporate waste against the directors and officers with no dollar amount being claimed. On September 13, 2021, the Corporation filed a motion to dismiss in its entirety and the motion was heard on February 8, 2022. The claim was dismissed by the court May 6, 2022. |
| 11. | FSD BioSciences Employees: Subsequent to the resignation of the Former Employees, on July 9, 2021, the Former Employees filed a joint claim against the Corporation. On September 17, 2021, the Corporation filed a motion to dismiss the claim in its entirety. On December 13, 2021, the Court granted the Corporation’s motion and dismissed the case. One of the Former Employee’s claims was dismissed with prejudice while the other was not. |
| 12. | As the Corporation had excess proceeds from the 2020 Prospectuses and did not identify any strategic acquisitions and/or investment opportunities, the Corporation conducted two share repurchase programs, one beginning in December 2021 and the other in January 2023, as the Board determined that the market price of the Class B Shares was undervalued, and these repurchases would strategically return value to shareholders. |
In all of these offerings, the Company used the proceeds in a manner consistent with that set forth in the applicable prospectus supplements. None of the net proceeds was paid to any of our directors or officers or their associates, or any 10% stockholder or any affiliate of ours.
| 81 |
| Table of Contents |
For more information on the legal proceedings, please see “Item 8. Financial Information - A. Consolidated Statements and Other Financial Information – Legal Proceedings”.
Item 15. Controls and Procedures
A. Disclosure Controls and Procedures
We maintain disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which are designed to ensure that information required to be disclosed in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms and that such information is accumulated and communicated to our management, including our CEO and CFO, as appropriate to allow timely decisions regarding required disclosure.
Under the supervision and with the participation of our CEO and CFO, our management has evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2023, the end of the period covered by this annual report. Based on that evaluation, our CEO and CFO concluded that our disclosure controls and procedures were effective as of December 31, 2023.
The effectiveness of our disclosure controls and procedures and our internal control over financial reporting is subject to various inherent limitations, including cost limitations, judgments used in decision making, assumptions about the likelihood of future events, the soundness of our systems, the possibility of human error, and the risk of fraud. Moreover, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions and the risk that the degree of compliance with policies or procedures may deteriorate over time. Because of these limitations, there can be no assurance that any system of disclosure controls and procedures or internal control over financial reporting will be successful in preventing all errors or fraud or in making all material information known in a timely manner to the appropriate levels of management.
B. Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control over financial reporting is the process designed by and under the supervision of our CEO and CFO to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of our financial statements for external reporting in accordance with accounting principles generally accepted in the United States of America. Management has evaluated the effectiveness of our internal control over financial reporting using the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission in Internal Control-Integrated Framework (2013).
Under the supervision and with the participation of our CEO and CFO, our management has assessed the effectiveness of our internal control over financial reporting as of December 31, 2023 and concluded that it was effective.
C. Attestation Report of the Registered Public Accounting Firm
Not applicable.
D. Changes in Internal Control Over Financial Reporting
Under the supervision and with the participation of our CEO and CFO, our management has evaluated changes in our internal control over financial reporting that occurred during the period covered by this Annual Report. Based on that evaluation, our CEO and CFO did not identify any change in our internal control over financial reporting that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Item 16. [Reserved.]
Item 16A. Audit Committee Financial Expert.
Our Board has determined that Nitin Kaushal, the Chair of the Audit Committee of our Board, is an “audit committee financial expert” as defined by SEC rules and has the requisite financial sophistication under the listing standards of the Nasdaq Stock Market. Mr. Kaushal meets the standards of independence applicable to audit committees under Rule 10A-3 under the Exchange Act and under the listing standards of the Nasdaq Stock Market.
| 82 |
| Table of Contents |
Item 16B. Code of Ethics.
We have adopted the Code that is applicable to all of our directors, executive officers, and employees, including our principal executive officer, principal financial officer, principal accounting officer and controller. A copy of the Code is available on our website at www.fsdpharma.com.
During 2023, except for the CEO Mortgage Loan, no provision of the Code applicable to our principal executive officer, principal financial officer, principal accounting officer or controller was amended (other than technical, administrative, or other non-substantive amendments), nor did we grant any waiver (including an implicit waiver) of any provision of the Code to any such officer.
We intend to disclose any amendments to the Code applicable to our principal executive officer, principal financial officer, principal accounting officer, controller or persons performing similar functions (other than technical, administrative, or other non-substantive amendments) and any waiver of the Code for any such persons on our website to the extent required by the rules and regulations of the SEC, including the instructions to Item 16B of Form 20-F. Any waiver of the Code for these covered persons may be made only by the Board or the Chief Executive Officer and will be disclosed promptly to stockholders and others, as required by applicable law. The Company must disclose changes to and wavers of the Code in accordance with applicable law.
Item 16C. Principal Accountant Fees and Services.
MNP LLP, auditors of the Company since November 29, 2019, served as our independent registered public accounting firm for the years ended December 31, 2023, 2022 and 2021. The following table provides a summary of the fees for professional services rendered by MNP for the years ended December 31, 2023 and 2022:
Auditors’ Fees
The following table sets forth the fees billed by the Company’s auditor during the years ended December 31, 2023 and December 31, 2022:
Fee |
| 2023 |
|
| 2022 | |||
Audit Fees(1) |
| C$ | 380,138 |
|
| C$ | 386,599 | |
Audit-Related Fees(2) |
| C$ | 57,432 |
|
|
| Nil | |
Tax Fees(3) |
| C$ | 63,990.28 |
|
| C$ | 45,176 | |
All Other Fees(4) |
| $ | 57,432 |
|
| Nil | ||
Total |
| $ | 558,503 |
|
| C$ | 431,775 | |
Notes:
(1) | “Audit Fees” include fees necessary to perform the annual audit and quarterly reviews of the Company’s consolidated financial statements. Audit Fees include fees for review of tax provisions and for accounting consultations on matters reflected in the financial statements. Audit Fees also include audit or other attest services required by legislation or regulation, such as comfort letters, consents, reviews of securities filings and statutory audits. |
(2) | “Audit-Related Fees” include services that are traditionally performed by the auditor. These audit-related services include employee benefit audits, due diligence assistance, accounting consultations on proposed transactions, internal control reviews and audit or attest services not required by legislation or regulation. |
(3) | “Tax Fees” include fees for all tax services other than those included in “Audit Fees” and “Audit-Related Fees”. This category includes fees for tax compliance, tax planning and tax advice. Tax planning and tax advice includes assistance with tax audits and appeals, tax advice related to mergers and acquisitions, and requests for rulings or technical advice from tax authorities. |
(4) | “All Other Fees” include all other non-audit services. |
All permissible categories of non-audit services require pre-approval by the Audit Committee, subject to certain statutory exemptions.
Item 16D. Exemptions from the Listing Standards for Audit Committees.
Not applicable.
Item 16E. Purchases of Equity Securities by the Issuer and Affiliated Purchasers.
On January 12, 2023, the Board authorized the 2023 NCIB, pursuant to which the Company was able to repurchase for cancellation up to 1,925,210 Class B Shares, being approximately 5% of the Company’s issued and outstanding Class B Shares as of January 12, 2023, over a 12-month period. The 2023 NCIB commenced on January 18, 2023 and was terminated on January 12, 2024. Under the 2023 NCIB, the Company repurchased for cancellation 1,904,700 Class B Shares at an average price of approximately C$2.11 per Class B Shares. All Class B Shares were repurchased through the facilities of the CSE at the prevailing market price on the CSE at the time of repurchase.
| 83 |
| Table of Contents |
The following table specifies the number of shares purchased by the Company under the 2023 NCIB:
Period |
| Total Number of Shares Purchased |
|
| Average Price Paid Per Share (C$) |
|
| Total Number of Shares Purchased as Part of Publicly Announced Plans or Programs |
|
| Maximum Number of Shares that May Yet Be Purchased Under the Plans or Programs |
| ||||
|
|
|
|
|
|
|
|
| ||||||||
Month #1 (January 1, 2023 - January 31, 2023) |
|
| 289,600 |
|
|
| 1.39 |
|
|
| 289,600 |
|
|
| 1,635,610 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #2 (February 1, 2023 - February 28, 2023) |
|
| 1,328,700 |
|
|
| 2.27 |
|
|
| 1,618,300 |
|
|
| 306,910 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #3 (March 1, 2023 - March 31, 2023) |
|
| 286,400 |
|
|
| 2.10 |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #4 (April 1, 2023 - April 30, 2023) |
|
| - |
|
|
| - |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #5 (May 1, 2023 - May 31, 2023) |
|
| - |
|
|
| - |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #6 (June 1, 2023 - June 30, 2023) |
|
| - |
|
|
| - |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #7 (July 1, 2023 - July 31, 2023) |
|
| - |
|
|
| - |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #8 (August 1, 2023 - August 31, 2023) |
|
| - |
|
|
| - |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #9 (September 1, 2023 - September 30, 2023) |
|
| - |
|
|
| - |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #10 (October 1, 2023 - October 31, 2023) |
|
| - |
|
|
| - |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #11 November 1, 2023 - November 30, 2023 |
|
| - |
|
|
| - |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Month #12 (December 1, 2023 - December 31, 2023) |
|
| - |
|
|
| - |
|
|
| 1,904,700 |
|
|
| 20,510 |
|
| 84 |
| Table of Contents |
Item 16F. Change in Registrant’s Certifying Accountant.
Not applicable.
Item 16G. Corporate Governance.
The Company is a foreign private issuer and its Class B Shares are listed on Nasdaq. Nasdaq Marketplace Rule 5615(a)(3) permits a foreign private issuer to follow its home country practices in lieu of most of the requirements of the 5600 Series of the Nasdaq Marketplace Rules. In order to claim such an exemption, the Company must disclose the significant differences between its corporate governance practices and those required to be followed by U.S. domestic issuers under Nasdaq’s corporate governance requirements. Set forth below is a brief summary of such differences:
Independent Director Requirements
Nasdaq Marketplace Rule 5605(b)(1) requires a majority of the board of directors of each issuer to be comprised of independent directors, as that term is defined under Rule 5605(a)(2). The Company has a majority of independent directors and follows the Nasdaq Marketplace Rule and complies with the applicable CSE rules and applicable Canadian and Ontario corporate and securities regulatory requirements.
Shareholder Approval Requirements
Nasdaq Marketplace Rule 5635 requires each issuer to obtain shareholder approval prior to certain dilutive events, including a transaction other than a public offering involving the sale of 20% or more of the issuer’s outstanding shares of common stock prior to the transaction for less than the greater of book or market value of the stock. The Company does not follow this Nasdaq Marketplace Rule. Instead, and in accordance with the Nasdaq exemption, the Company complies with Ontario corporate and securities laws, which do not require shareholder approval for dilutive events unless the Company were to dispose of all or substantially all of its undertaking. In addition, the Company follows the CSE policies which require shareholder approval on the occurrence of a “fundamental change,” defined by the policies of the CSE to be a “major acquisition” (whereby for the next 12-month period at least 50% of the issuer’s assets will be comprised of, or anticipated revenues are expected to be derived from, the assets, properties, businesses or other interests that are the subject of the major acquisition) accompanied or preceded by a “change of control.” In such context, a “change of control” would include the distribution of a number of equity securities of the issuer equal to or greater than 100% of the number outstanding prior to the transaction, as well as a substantial change of management or the board of directors of the issuer.
In addition, Nasdaq Marketplace Rule 5635 requires shareholder approval of most equity compensation plans and material revisions to such plans. We do not follow this Nasdaq Marketplace Rule. Instead, and in accordance with the Nasdaq exemption, we comply with Ontario corporate and securities laws, which do not require shareholder approval of equity compensation plans. In addition, the Company intends to follow the CSE policies and certain provisions of Canadian securities laws which require limitations on the number of equity compensation securities that can be distributed to persons performing investor relations services to 1% of the issued and outstanding amount of listed securities in a 12-month period, and further limit the number of equity compensation securities that can be distributed to a director, officer or a related entity of the issuer, or an associate thereof (each a “related person”), on a fully diluted basis to not exceed 5% of the outstanding securities of the issuer, or collectively to related persons exceeds 10% of the outstanding securities of the issuer.
Quorum Requirement
Nasdaq Marketplace Rule 5620(c) requires that each company that is not a limited partnership shall provide for a quorum as specified in its by-laws for any meeting of holders of common stock; provided, however, that in no case shall such quorum be less than 33-1/3% of the outstanding shares of the Company’s common voting stock. The Company does not presently follow this Nasdaq Marketplace Rule. Instead, the Company complies with Ontario corporate and securities laws and its by-laws which do not require a quorum of no less than 33-1/3% of the outstanding shares of the Company’s common voting stock and provides that the quorum for the transaction of business at a meeting of shareholders is at least two voting persons holding or representing, in the aggregate, not less than 10% of the issued and outstanding shares of the applicable class.
| 85 |
| Table of Contents |
Independent Director Oversight of Executive Compensation and Board Nominations
Nasdaq’s Marketplace Rule 5605(d) requires independent director oversight of executive officer compensation arrangements by approval of such compensation by a committee comprised solely of independent directors, and Marketplace Rule 5605(e) requires similar oversight with respect to the process of selecting nominees to the board or oversight by a majority of the independent directors. Under the exemption available to foreign private issuers under Rule 5615(a)(3), the Company is not required to comply with Nasdaq Marketplace Rules 5605(d) or 5605(e). Instead, and in accordance with the Nasdaq exemption, the Company complies with the applicable CSE rules and applicable Canadian corporate and securities regulatory requirements.
Proxy Delivery Requirements
Nasdaq Marketplace Rule 5620(b) requires that a listed company that is not a limited partnership to solicit proxies and provide proxy statements for all meetings of shareholders, and also provide copies of such proxy solicitation materials to Nasdaq. The Company is a “foreign private issuer” as defined in Rule 3b-4 under the Exchange Act, and the equity securities of the Company are accordingly exempt from the proxy rules set forth in Sections 14(a), 14(b), 14(c) and 14(f) of the Exchange Act. The Company solicits proxies in accordance with applicable rules and regulations in Canada.
Item 16H. Mine Safety Disclosure
Not applicable.
Item 16I. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
Item 16J. Insider Trading Policies
This disclosure item does not yet apply to the Company.
Item 16K. Cybersecurity.
We have established policies and processes for assessing, identifying, and managing risks from cybersecurity threats, and have integrated these processes into our overall risk management systems and processes. We routinely assess risks from cybersecurity threats, including any potential unauthorized occurrence on or conducted through our information systems that may result in adverse effects on the confidentiality, integrity, or availability of our information systems or any information residing therein.
We conduct periodic risk assessments to identify cybersecurity threats, as well as assessments in the event of a material change in our business practices that may affect information systems that are vulnerable to such cybersecurity threats. These risk assessments include identification of reasonably foreseeable internal and external risks, the likelihood and potential damage that could result from such risks, and the sufficiency of existing policies, procedures, systems, and safeguards in place to manage such risks.
Following these risk assessments, we evaluate whether and how to re-design, implement, and maintain reasonable safeguards to minimize identified risks; reasonably address any identified gaps in existing safeguards; and regularly monitor the effectiveness of our safeguards. We devote significant resources and designate high-level personnel, including our CFO, who reports to our Board, to manage the risk assessment and mitigation process.
As part of our overall risk management system, we monitor our safeguards and train our employees on these safeguards. Personnel at all levels and departments are made aware of our cybersecurity policies through trainings integrated into new employee onboarding processes and annual employee re-training.
We engage consultants, experts, or other third parties in connection with our risk assessment processes. These third parties assist us in designing and implementing our cybersecurity policies and procedures, as well as in monitoring and testing our safeguards.
We require each third-party service provider who may have access to our systems and/or our sensitive data to confirm that it has the ability to implement and maintain appropriate security measures, consistent with all applicable laws, to implement and maintain reasonable security measures in connection with their work with us, and to promptly report any suspected breach of its security measures that may affect our company.
We have not experienced any cybersecurity incidents that have been determined to be material in the past, however, like other life sciences technology companies, we have experienced cybersecurity incidents and may continue to experience them in the future. For additional information regarding whether any risks from cybersecurity threats, including as a result of any previous cybersecurity incidents, materially affected or are reasonably likely to materially affect our company, including our business strategy, results of operations, or financial condition, please refer to “Item 3. Key information - D. Risk Factors” in this Annual Report on Form 20-F.
| 86 |
| Table of Contents |
Governance
One of the key functions of our Board is informed oversight of our risk management process, including risks from cybersecurity threats. Our Board is responsible for monitoring and assessing strategic risk exposure, and our executive officers are responsible for the day-to-day management of the material risks we face. Our Board administers its cybersecurity risk oversight function directly as a whole, as well as through the audit committee.
Our CFO and our management committee on cybersecurity, and outside consultants, who collectively possess significant experience in evaluating, managing, and mitigating security and other risks, including cybersecurity risks, are primarily responsible to assess and manage our material risks from cybersecurity threats.
Our CFO and our management committee on cybersecurity oversee our cybersecurity policies and processes, including those described in “Risk Management and Strategy” above. The processes by which our CFO and representatives from our management committee on cybersecurity are informed about and monitor the prevention, detection, mitigation, and remediation of cybersecurity incidents includes the following:
| · | monitoring of Company computer and information systems for potential malware, ransomware and other malicious activity, and remediation of identified issues, including mitigation of identified risks and containment and elimination of any malicious software; |
|
|
|
| · | mandatory cybersecurity training as part of new employee onboarding along with required annual employee cybersecurity re-training; |
|
|
|
| · | monitoring of systems and network infrastructure by security information and event management application; |
|
|
|
| · | prompt incident reporting directly to the Board; and |
|
|
|
| · | escalation to the Company’s Audit Committee and Board as warranted based upon the nature of the identified issue. |
Our CFO and/or representatives from our management committee on cybersecurity provide periodic briefings to the audit committee regarding our company’s cybersecurity risks and activities, including any recent cybersecurity incidents and related responses, cybersecurity systems testing, activities of third parties, and the like. Our Audit Committee provides regular updates to the Board on such reports.
| 87 |
| Table of Contents |
PART III
Item 17. Financial Statements
Not applicable.
Item 18. Financial Statements
See pages F-1 through F-44 appearing at the end of this Annual Report on Form 20-F following the signature page.
Item 19. Exhibits.
2.1* | Description of Securities |
| 88 |
| Table of Contents |
101.1* | Interactive Data File. |
101.INS* | Inline XBRL Instance Document–the instance document does not appear in the Interactive Data File as its XBRL tags are embedded within the Inline XBRL document. |
101.SCH* | Inline XBRL Taxonomy Extension Schema Document. |
101.CAL* | Inline XBRL Taxonomy Extension Calculation Linkbase Document. |
101.DEF* | Inline XBRL Taxonomy Extension Definition Linkbase Document. |
101 LAB* | Inline XBRL Taxonomy Extension Label Linkbase Document |
101 PRE* | Inline XBRL Taxonomy Extension Presentation Linkbase Document |
101 | LAB* Inline XBRL Taxonomy Extension Label Linkebase Document |
101 | PRE Inline XBRL Taxonomy Exension Presentation Linkbase Document |
104 | Cover Page Interactive Data File (embedded within the Inline XBRL and contained in Exhibit 101) |
* Filed herewith.
** Furnished herewith.
† Indicates a management contract or any compensatory plan, contract, or arrangement.
| 89 |
| Table of Contents |
SIGNATURES
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| FSD Pharma Inc. |
| |
|
|
|
|
| By: | /s/ Zeeshan Saeed |
|
| Name: | Zeeshan Saeed |
|
| Title: | Chief Executive Officer |
|
|
|
|
|
| Date: | April 1, 2024 |
|
87 |

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM |
To the Board of Directors and Shareholders of FSD Pharma Inc. Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated statements of financial position of FSD Pharma Inc. (the “Company”) as of December 31, 2023 and 2022 and the related consolidated statements of loss and comprehensive loss, changes in shareholders’ equity, and cash flows for each of the years in the three-year period ended December 31, 2023, and the related notes (collectively referred to as the “consolidated financial statements”).
In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as of December 31, 2023 and 2022, and the results of its consolidated operations and its consolidated cash flows for each of the years in the three-year period ended December 31, 2023, in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Material Uncertainty Related to Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has an accumulated deficit and has suffered a net loss that raise substantial doubt about its ability to continue as a going concern. Management's plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Chartered Professional Accountants Licensed Public Accountants
We have served as the Company’s auditor since 2019
PCAOB ID:
March 28, 2024
|
| |
Suite 900, 50 Burnhamthorpe Road W, Mississauga ON, L5B 3C2 |
| T: 416.626.6000 F: 416.626.8650 |
|
| MNP.ca |

| F-1 |
FSD Pharma Inc.
Consolidated financial statements
For the years ended December 31, 2023, 2022 and 2021
(expressed in United States dollars, except per share amounts)
| F-2 |
FSD PHARMA INC. |
|
|
|
|
|
| |||||||||||||
|
|
|
|
|
|
| |||||||||||||
CONSOLIDATED STATEMENTS OF FINANCIAL POSITION | |||||||||||||||||||
[expressed in United States dollars] | |||||||||||||||||||
|
|
|
|
|
|
| |||||||||||||
As at |
|
|
| December 31, |
|
| December 31, |
| |||||||||||
|
|
|
| 2023 |
|
| 2022 |
| |||||||||||
|
| Notes |
|
| $ |
|
| $ |
| ||||||||||
|
|
|
|
|
|
|
|
|
| ||||||||||
ASSETS |
|
|
|
|
|
|
|
|
| ||||||||||
Current assets |
|
|
|
|
|
|
|
|
| ||||||||||
Cash and cash equivalents |
|
|
|
|
|
|
|
|
| ||||||||||
Other receivables |
|
| 6 |
|
|
|
|
|
|
| |||||||||
Prepaid expenses and deposits |
|
| 7 |
|
|
|
|
|
|
| |||||||||
Investments |
|
| 9 |
|
|
|
|
|
|
| |||||||||
Finance receivables, net |
|
| 8 |
|
|
|
|
|
|
| |||||||||
Net investment in lease |
|
|
|
|
|
|
|
|
|
| |||||||||
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Non-current assets |
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Equipment, net |
|
|
|
|
|
|
|
|
|
| |||||||||
Investments |
|
| 9 |
|
|
|
|
|
|
| |||||||||
Right-of-use asset, net |
|
|
|
|
|
|
|
|
|
| |||||||||
Finance receivables, net |
|
| 8 |
|
|
|
|
|
|
| |||||||||
Intangible assets, net |
|
| 10 |
|
|
|
|
|
|
| |||||||||
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
LIABILITIES |
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Current liabilities |
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Trade and other payables |
|
| 11 |
|
|
|
|
|
|
| |||||||||
Lease obligations |
|
|
|
|
|
|
|
|
|
| |||||||||
Warrants liability |
|
| 12 |
|
|
|
|
|
|
| |||||||||
Notes payable |
|
|
|
|
|
|
|
|
|
| |||||||||
|
|
|
|
|
|
|
|
|
|
| |||||||||
Non-current liabilities |
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Lease obligations |
|
|
|
|
|
|
|
|
|
| |||||||||
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
SHAREHOLDERS' EQUITY |
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Class A share capital |
|
| 13 |
|
|
|
|
|
|
| |||||||||
Class B share capital |
|
| 13 |
|
|
|
|
|
|
| |||||||||
Warrants |
|
| 13 |
|
|
|
|
|
|
| |||||||||
Contributed surplus |
|
|
|
|
|
|
|
|
|
| |||||||||
Foreign exchange translation reserve |
|
|
|
|
|
|
|
|
|
| |||||||||
Accumulated deficit |
|
|
|
|
|
| ( | ) |
|
| ( | ) | |||||||
Equity attributable to shareholders of the Company |
|
|
|
|
|
|
|
|
|
| |||||||||
Non-controlling interests |
|
| 15 |
|
|
| ( | ) |
|
|
| ||||||||
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
|
|
|
|
|
|
|
|
|
| |||||||||
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Going concern uncertainty |
|
| 1 |
|
|
|
|
|
|
|
|
| |||||||
Commitments and contingencies |
|
| 20 |
|
|
|
|
|
|
|
|
| |||||||
Subsequent events |
|
| 24 |
|
|
|
|
|
|
|
|
| |||||||
The accompanying notes are an integral part of these consolidated financial statements. |
| |
|
|
|
On behalf of the Board: |
|
|
|
|
|
|
|
"Signed" | "Signed" |
|
|
Director - Zeeshan Saeed | Director - Eric Hoskins |
|
|
| F-3 |
FSD PHARMA INC. |
|
|
|
|
|
|
|
|
|
|
|
CONSOLIDATED STATEMENTS OF LOSS AND COMPREHENSIVE LOSS | |||||
[expressed in United States dollars, except number of shares] | |||||
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
For the years ended December 31, |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
|
|
| 2023 |
|
| 2022 |
|
| 2021 |
| |||||
|
| Notes |
|
| $ |
|
| $ |
|
| $ |
| ||||
Expenses |
|
|
|
|
|
|
|
|
|
|
|
| ||||
General and administrative |
|
| 17 |
|
|
|
|
|
|
|
|
|
| |||
External research and development fees |
|
|
|
|
|
|
|
|
|
|
|
|
| |||
Share-based payments |
|
| 14 |
|
|
|
|
|
|
|
|
|
| |||
Depreciation and amortization |
|
| 10 |
|
|
|
|
|
|
|
|
|
| |||
Impairment loss |
|
| 10 |
|
|
|
|
|
|
|
|
|
| |||
Total operating expenses |
|
|
|
|
|
|
|
|
|
|
|
|
| |||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from continuing operations |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income |
|
| 18 |
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Finance expense, net |
|
|
|
|
|
|
|
|
|
|
|
|
| |||
Gain on remeasurement of financial liability |
|
| 20 |
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Gain on change in fair value of derivative liability |
|
| 12 |
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Loss on changes in fair value of investments |
|
| 9 |
|
|
|
|
|
|
|
|
|
| |||
Net loss from continuing operations |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net income (loss) from discontinued operations |
|
| 5 |
|
|
|
|
|
|
|
|
| ( | ) | ||
Net loss |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other comprehensive loss |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Items that may be subsequently reclassified to loss: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Exchange (loss) gain on translation of foreign operations |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
| ||
Comprehensive loss |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net loss attributable to: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Equity owners of the Company |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Non-controlling interests |
|
| 15 |
|
|
| ( | ) |
|
|
|
|
|
| ||
|
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net (loss) income per share |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic and diluted - continuing operations |
|
| 16 |
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Basic and diluted - discontinued operations |
|
| 16 |
|
|
|
|
|
|
|
|
| ( | ) | ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Weighted average number of shares outstanding – basic and diluted |
|
| 16 |
|
|
|
|
|
|
|
|
|
| |||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these consolidated financial statements. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| F-4 |
FSD PHARMA INC. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||
CONSOLIDATED STATEMENTS OF CHANGES IN SHAREHOLDERS' EQUITY | ||||||||||||||||||||||||||||||||||||||||||||
For the years ended December 31, 2023, 2022 and 2021 | ||||||||||||||||||||||||||||||||||||||||||||
[expressed in United States dollars, except number of shares] | ||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||
|
| Class A shares |
|
| Class B shares |
|
| Warrants |
|
| Contributed surplus |
|
| Non-controlling interests |
|
| Foreign exchange translation reserve |
|
| Accumulated deficit |
|
| Total |
| ||||||||||||||||||||
|
| # |
|
| $ |
|
| # |
|
| $ |
|
| # |
|
| $ |
|
| $ |
|
| $ |
|
| $ |
|
| $ |
|
| $ |
| |||||||||||
Balance, December 31, 2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
| ||||||||||
Shares issued [note 13] |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
Share-based payments [note 14] |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||
Share cancellation [note 13] |
|
| — |
|
|
|
|
|
| ( | ) |
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Lucid acquisition [note 4] |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||||||
Warrants expired |
|
| — |
|
|
|
|
|
| — |
|
|
| — |
|
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Comprehensive loss for the year |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | ||||||
Balance, December 31, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
| ||||||||||
Share repurchase [note 13] |
|
| — |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) | ||||||
Share-based payments [note 14] |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
Share cancellation [note 13] |
|
| — |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Warrants expired [note 13] |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
PSUs converted to shares [note 14] |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Comprehensive loss for the year |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) | ||||||
Balance, December 31, 2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
| ||||||||||
Initial recognition of non-controlling interests |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
| ( | ) |
|
| ( | ) | |||||
Plan of arrangement [note 13] |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
| ||||||||
Share repurchase [note 13] |
|
| — |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) | ||||||
Share-based payments [note 14] |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
Share options exercised [note 13] |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
| ||||||||
PSUs converted to shares [note 14] |
|
| — |
|
|
|
|
|
|
|
|
|
|
|
| — |
|
|
|
|
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
| ||||||||
Warrants issued [note 13] |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||||
Warrants expired [note 13] |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||||
Comprehensive loss for the year |
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
| — |
|
|
|
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) | ||||
Balance, December 31, 2023 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
| ( | ) |
|
|
| |||||||||
The accompanying notes are an integral part of these consolidated financial statements.
| F-5 |
FSD PHARMA INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
For the years ended December 31, 2023, 2022 and 2021
[expressed in United States dollars]
|
| December 31, 2023 |
|
| December 31, 2022 |
|
| December 31, 2021 |
| |||
|
| $ |
|
| $ |
|
| $ |
| |||
|
|
|
|
|
|
|
|
|
| |||
Operating activities |
|
|
|
|
|
|
|
|
| |||
Net loss from continuing operations |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Add (deduct) items not affecting cash |
|
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
|
|
|
|
|
|
| |||
Interest expense |
|
|
|
|
|
|
|
|
| |||
Share-based payments |
|
|
|
|
|
|
|
|
| |||
Change in fair value of investments |
|
|
|
|
|
|
|
|
| |||
Change in fair value of derivative liability |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Unrealized foreign exchange (gain) loss |
|
| ( | ) |
|
|
|
|
|
| ||
Gain on remeasurement of financial liability |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Impairment loss |
|
|
|
|
|
|
|
|
| |||
Gain on net investment in lease |
|
|
|
|
| ( | ) |
|
|
| ||
Changes in non-cash working capital balances |
|
|
|
|
|
|
|
|
|
|
|
|
Finance receivables |
|
| ( | ) |
|
| ( | ) |
|
|
| |
Other receivables |
|
|
|
|
|
|
|
| ( | ) | ||
Prepaid expenses and deposits |
|
|
|
|
|
|
|
| ( | ) | ||
Note receivable |
|
| ( | ) |
|
|
|
|
|
| ||
Trade and other payables |
|
|
|
|
| ( | ) |
|
|
| ||
Cash used in continuing operating activities |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Cash used in discontinued operating activities |
|
|
|
|
| ( | ) |
|
| ( | ) | |
Cash used in operating activities |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Investing activities |
|
|
|
|
|
|
|
|
|
|
|
|
Cash acquired from acquisition of Lucid Psycheceuticals Inc. |
|
|
|
|
|
|
|
|
| |||
Purchase of investments |
|
| ( | ) |
|
| ( | ) |
|
|
| |
Purchase of equipment |
|
|
|
|
| ( | ) |
|
|
| ||
Additions to intangible assets |
|
|
|
|
| ( | ) |
|
| ( | ) | |
Net cash upon control of subsidiary |
|
|
|
|
|
|
|
|
| |||
Proceeds from sale of investments |
|
|
|
|
|
|
|
|
| |||
Cash (used in) provided by continuing investing activities |
|
| ( | ) |
|
| ( | ) |
|
|
| |
Cash provided by discontinued investing activities |
|
|
|
|
|
|
|
|
| |||
Cash (used in) provided by investing activities |
|
| ( | ) |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
Financing activities |
|
|
|
|
|
|
|
|
|
|
|
|
Share repurchase |
|
| ( | ) |
|
| ( | ) |
|
|
| |
Proceeds from issuance of shares, net |
|
|
|
|
|
|
|
|
| |||
Repayment of notes payable |
|
|
|
|
|
|
|
| ( | ) | ||
Payment of lease obligation |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Share options exercised |
|
|
|
|
|
|
|
|
| |||
Cash (used in) provided by continuing financing activities |
|
| ( | ) |
|
| ( | ) |
|
|
| |
Cash (used in) provided by financing activities |
|
| ( | ) |
|
| ( | ) |
|
|
| |
|
|
|
|
|
|
|
|
|
|
|
|
|
Net decrease |
|
| ( | ) |
|
| ( | ) |
|
|
| |
Cash and cash equivalents, beginning of the year |
|
|
|
|
|
|
|
|
| |||
Cash and cash equivalents, end of the year |
|
|
|
|
|
|
|
|
| |||
|
|
|
|
|
|
|
|
|
|
|
|
|
The accompanying notes are an integral part of these consolidated financial statements. |
|
|
|
|
|
|
|
|
|
|
|
|
| F-6 |
FSD PHARMA INC.
Notes to the consolidated financial statements
(expressed in United States dollars)
December 31, 2023 and 2022
1. Nature of business
FSD Pharma Inc. (“FSD” or the “Company”) is a biopharmaceutical company dedicated to building a portfolio of innovative assets and biotech solutions for the treatment of challenging neurodegenerative, inflammatory and metabolic disorders and alcohol misuse disorders with drug candidates in different stages of development. Through its wholly-owned subsidiary, Lucid Psycheceuticals Inc. (“Lucid”), FSD is focused on the research and development of its lead compound, Lucid-MS (formerly Lucid-21-302) (“Lucid-MS”). Lucid-MS is a patented new chemical entity shown to prevent and reverse myelin degradation, the underlying mechanism of multiple sclerosis, in preclinical models. FSD is also focused on the research and development of a treatment for alcohol misuse for application in hospitals and other medical practices. FSD maintains a portfolio of strategic investments through its wholly-owned subsidiary, FSD Strategic Investments Inc., which represent loans secured by residential property.
The Company’s registered office is located at 199 Bay Street, Suite 4000, Toronto, Ontario, M5L 1A9. The Company’s shares are listed on the Nasdaq Capital Market and on the Canadian Securities Exchange under the symbol “HUGE”.
On July 31, 2023, the Company entered into an exclusive intellectual property license agreement (the “License Agreement”) with Celly Nutrition Corp. (“Celly”). The License Agreement provides Celly access to proprietary information for the purposes of consumer product development and marketing. The License Agreement grants Celly the rights to a proprietary formulation of natural ingredients, vitamins, and minerals to help with liver and brain function for the purposes of potentially quickly relieving from the effects of alcohol consumption, such as inebriation, and restoring normal lifestyle. The License Agreement also grants Celly rights to certain trademarks. In exchange, FSD received
Going concern uncertainty
The consolidated financial statements (“financial statements”) of the Company for the years ended December 31, 2023, 2022 and 2021, have been prepared on the basis of accounting principles applicable to a going concern, which assumes that the Company will continue in operation for the foreseeable future and will be able to realize its assets and discharge its liabilities and commitments in the normal course of operations. These financial statements do not include any adjustments to the amounts and classification of assets and liabilities that would be necessary should the Company be unable to continue as a going concern. Such adjustments could be material.
The Company is in the preliminary stages of its planned operations and has not yet determined whether its processes and business plans are economically viable. The continued operations of the Company and the recoverability of amounts shown for intangible assets are dependent upon the ability of the Company to obtain sufficient financing to complete the research and development program of Lucid-MS. As well as fund the research and development of a treatment for alcohol misuse for application in hospitals and other medical practices.
| F-7 |
FSD PHARMA INC.
Notes to the consolidated financial statements
(expressed in United States dollars)
December 31, 2023 and 2022
As at December 31, 2023, the Company has an accumulated deficit of $
Subsidiaries
The Company has the following subsidiaries:
|
|
| Ownership percentage as at |
| ||||||||||
Entity Name |
| Country |
| December 31, 2023 |
|
| December 31, 2022 |
|
| December 31, 2021 |
| |||
|
|
|
| % |
|
| % |
|
| % |
| |||
FSD Biosciences Inc. |
| USA |
|
|
|
|
|
|
|
|
| |||
Prismic Pharmaceuticals Inc. |
| USA |
|
|
|
|
|
|
|
|
| |||
FV Pharma Inc. |
| Canada |
|
|
|
|
|
|
|
|
| |||
Lucid Psycheceuticals Inc. |
| Canada |
|
|
|
|
|
|
|
|
| |||
FSD Strategic Investments Inc. |
| Canada |
|
|
|
|
|
|
|
| — |
| ||
FSD Pharma Australia Pty Ltd |
| Australia |
|
|
|
|
|
|
|
| — |
| ||
Celly Nutrition Corp. |
| Canada |
|
|
|
|
| — |
|
|
| — |
| |
2. Basis of presentation
[a] Statement of compliance
These financial statements have been prepared by management in accordance with International Financial Reporting Standards (“IFRS”) as issued by the International Accounting Standards Board (“IASB”). The policies set out below have been consistently applied to all periods presented, unless otherwise noted.
These financial statements were approved and authorized for issuance by the Board of Directors (the “Board”) of the Company on March 26, 2024.
[b] Basis of measurement
These financial statements have been prepared on a historical cost basis, except for certain financial instruments which are measured at fair value. Historical costs are generally based upon the fair value of the consideration given in exchange for goods and services received.
Fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date, regardless of whether that price is directly observable or estimated using another valuation technique. In estimating the fair value of an asset or a liability, the Company takes into account the characteristics of the asset or liability if market participants would take those characteristics into account when pricing the asset or liability at the measurement date. Fair value for measurement and/or disclosure purposes in these financial statements is determined on such a basis, except for share-based payment transactions that are within the scope of IFRS 2, Share-based Payment (“IFRS 2”) and measurements that have some similarities to fair value, but are not fair value, such as value in use in IAS 36, Impairment of Assets (“IAS 36”).
| F-8 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
[c] Basis of presentation
The accompanying financial statements include the accounts of FSD and its subsidiaries. The financial statements incorporate the assets and liabilities of the Company and its subsidiaries as at December 31, 2023 and 2022 and the results for the Company and its subsidiaries for the years ended December 31, 2023, 2022 and 2021.
Subsidiaries are those entities over which the Company has control. The Company controls an entity when it is exposed to, or has rights to, variable returns from its involvement with the entity and has the ability to affect those returns through its power over the entity. Subsidiaries are fully consolidated from the date on which control is transferred to the Company. All intra-entity assets and liabilities, revenues, expenses and cash flows relating to transactions between subsidiaries of the Company are eliminated in full on consolidation.
Non-controlling interests (“NCI”) represent ownership interests in consolidated subsidiaries by parties that are not shareholders of the Company. They are shown as a component of total equity in the consolidated statements of financial position, and the share of income (loss) attributable to non-controlling interests is shown as a component of net income (loss) in the consolidated statements of loss and comprehensive loss. Changes in the parent company’s ownership that do not result in a loss of control are accounted for as equity transactions.
[d] Functional currency and presentation currency
The financial statements of each company within the consolidated group are measured using their functional currency, which is the currency of the primary economic environment in which an entity operates. The Company’s functional currency is the United States dollar and the functional currencies of its subsidiaries are as follows:
| FSD Biosciences Inc. | United States Dollar |
| Prismic Pharmaceuticals Inc. | United States Dollar |
| FV Pharma Inc. | Canadian Dollar |
| Lucid Psycheceuticals Inc. | Canadian Dollar |
| FSD Strategic Investments Inc. | Canadian Dollar |
| FSD Pharma Australia Pty Ltd | Australian Dollar |
| Celly Nutrition Corp. | Canadian Dollar |
[e] Use of estimates and judgments
The preparation of these financial statements in conformity with IFRS requires management to make estimates, judgments and assumptions that affect the application of accounting policies and the reported amounts of assets and liabilities as at the date of the financial statements and the reported amounts of revenue and expenses during the reporting periods. Actual results could differ from these estimates.
Estimates are based on management’s best knowledge of current events and actions that the Company may undertake in the future. Estimates and underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimate is revised if the revision affects only that period, or in the period of the revision and future periods if the revision affects both current and future periods.
| F-9 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The following are the critical judgments, apart from those involving estimations, that management has made in the process of applying the Company’s accounting policies and that have the most significant effect on the amounts recognized in the financial statements:
[i] Going concern
At each reporting period, management assesses the basis of preparation of the financial statements. These financial statements have been prepared on a going concern basis in accordance with IFRS. The going concern basis of presentation assumes that the Company will continue its operations for the foreseeable future and be able to realize its assets and discharge its liabilities and commitments in the normal course of business.
[ii] Contingencies
From time to time, the Company is named as a party to claims or involved in proceedings, including legal, regulatory and tax related, in the ordinary course of its business. While the outcome of these matters may not be estimable at the reporting date, the Company makes provisions, where possible, for the estimated outcome of such claims or proceedings. Should a loss result from the resolution of any claims or proceedings that differs from these estimates, the difference will be accounted for as a charge to profit or loss in that period. The actual results may vary and may cause significant adjustments.
[iii] Intangible assets
The Company employs significant estimates to determine the estimated useful lives of intangible assets, considering the nature of the asset, contractual rights, expected use and review of asset useful lives. The Company reviews amortization methods and useful lives annually or when circumstances change and adjusts its amortization methods and assumptions prospectively.
The Company reviews intangible assets for impairment annually or when impairment indicators exist. If the recoverable amount of the respective intangible asset is less than its carrying amount, it is considered to be impaired. In the process of measuring the recoverable amount, management makes assumptions about future events and circumstances. The actual results may vary and may cause significant adjustments.
[iv] Valuation of share-based payments and warrants
Management measures the costs for share-based payments and warrants, including certain warrant liabilities, using market-based option valuation techniques. Assumptions are made and estimates are used in applying the valuation techniques. These include estimating the future volatility of the share price, expected dividend yield, expected term, expected risk-free interest rate and the rate of forfeiture. For performance share units (“PSUs”), management is required to estimate when the vesting conditions will be met. Such estimates and assumptions are inherently uncertain. Changes in these assumptions affect the fair value estimates of share-based payments, warrants and warrant liabilities.
[v] Allowance for credit losses
Judgment is required as to the timing of establishing an allowance for credit losses and to estimate the amount of expected credit losses taking into consideration counterparty creditworthiness, the fair value of underlying collateral, current and future economic trends, the expected residual value of the underlying assets and past experience.
[vi] Valuation of investments
The Company holds investments that have do not have quoted prices in active markets. In determining the fair value of investments, management is required to make certain estimates and assumptions regarding the fair value as of the reporting date. Assumptions are made and estimates are used in applying the valuation techniques to determine fair value. These include observable inputs other than quoted prices in active markets. Such investments are classified as Level 2 within the fair value hierarchy. The value at which the Company could ultimately realize upon disposition of these investments may differ from their carrying value and such differences could be material.
| F-10 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The financial information of private companies may not always be available, or such information may be insufficient or unreliable for valuation purposes. In determining the fair value of shares held in private company investments, management is required to make certain estimates and assumptions regarding the fair value as of the reporting date. Assumptions are made and estimates are used in applying the valuation techniques to determine fair value. These include the most recently available financial statements of the investee, price for most recently completed financing, as well as closely comparable public companies and general market and economic conditions. Such investments are classified as Level 3 within the fair value hierarchy. The value at which the Company could ultimately realize upon disposition of these investments may differ from their carrying value and such differences could be material.
[vii] Functional currency
The Company and its subsidiaries are required to determine their functional currencies based on the primary economic environment in which each entity operates. In order to do that, management has to analyze several factors, including which currency mainly influences the cost of undertaking the business activities, in which currency the entity has received financing, and in which currency it keeps its receipts from operating activities. Management uses its judgment to determine which factors are most important when the above indicators are mixed and the functional currency is not obvious.
[viii] Disclosure of interests in other entities
To assess the investment in Celly, judgment was required to determine if the Company has significant influence or control of Celly. The Company considered the relevant guidance in IFRS 10 – Consolidated Financial Statements, IAS 24 – Related Party Disclosures and IAS – 28 Investments in Associates and Joint Ventures.
Judgment is applied in determining when the Company controls an investment even if the Company holds less than a majority of the investee’s voting rights (the existence of de facto control). The Company concluded it has control of Celly even though the Company only holds 26.15% of the voting rights as of December 31, 2023. The Company concluded it has control of Celly as the Company, together with persons or entities considered to be de facto agents of the Company, holds a combined 52.05% of the voting rights of Celly. In addition, key management personnel of the Company hold three of the four board of director positions of Celly. The assessment of control is performed on a continuous basis. The Company determined that it obtained control of Celly on July 31, 2023, and control was maintained at all times from July 31, 2023, through December 31, 2023. Celly is significantly dependent on the Company as a result of the License Agreement and the loan. The NCI component of Celly is included as a separate component in equity (Note 15).
3. Material accounting policies
[a] Equipment
Equipment is measured at cost less accumulated depreciation and impairment losses. The cost of an item of equipment includes expenditures that are directly attributable to the acquisition or construction of the asset.
Depreciation is based on the estimated useful lives of the assets provided as follows:
| Computer equipment | |
| Furniture and fixtures Lease improvements |
An item of equipment and any significant part initially recognized are derecognized upon disposal or when no future economic benefits are expected from their use or disposal. Any gain or loss arising on derecognition of the asset (calculated as the difference between the net disposal proceeds and the carrying amount of the asset) is included in the consolidated statements of loss and comprehensive loss when the asset is derecognized. The assets’ residual values, useful lives and methods of depreciation and the depreciation charge are adjusted prospectively, if appropriate.
| F-11 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
[b] Intangible Assets
Intangible assets are recorded at cost less accumulated amortization and accumulated impairment losses. Amortization is recognized in the consolidated statements of loss and comprehensive loss on a straight-line basis over the useful life, as follows:
| Intellectual Property |
Expenditures on internally generated intangible assets during the research phase are expensed as incurred. Expenditures on internally generated intangible assets during the development phase, which comprise deferred development costs, are initially capitalized and recognized in the consolidated balance sheet if they meet the recognition criteria. Subsequent to initial recognition, deferred development costs are accounted for at cost less accumulated amortization and are amortized on a straight-line basis over an estimated useful life beginning once the deferred development costs are used in commercial production.
[c] Foreign Currency Transactions
Foreign currency transactions are translated into functional currencies at exchange rates in effect on the date of the transactions. At the end of each reporting period, monetary assets and liabilities denominated in foreign currencies are translated into functional currencies at the foreign exchange rate applicable at that period-end date. Non-monetary assets and liabilities that are measured in terms of historical cost in a foreign currency are translated using the exchange rate at the date of the transaction. Realized and unrealized exchange gains and losses are recognized in the consolidated statements of loss and comprehensive loss.
On consolidation, assets and liabilities of operations with a functional currency other than United States dollar are translated into United States dollar at period end foreign currency rates. Expenses of such operations are translated into the United States dollar at average rates for the period. Foreign currency translation gains and losses are recognized in other comprehensive income. The relevant amount in cumulative foreign currency translation adjustment is reclassified into earnings upon disposition of a foreign operation.
[d] Financial Instruments
Financial assets and financial liabilities are recognized when the Company becomes a party to the contractual provisions of the instruments.
Financial assets and financial liabilities are initially measured at fair value. Transaction costs that are directly attributable to the acquisition or issue of financial assets and financial liabilities (other than financial assets and financial liabilities at fair value through profit or loss) are added to or deducted from the fair value of the financial assets or financial liabilities, as appropriate, on initial recognition. Transaction costs directly attributable to the acquisition of financial assets or financial liabilities at fair value through profit or loss are recognized immediately in the consolidated statements of loss and comprehensive loss.
· | Financial assets
On initial recognition, a financial asset is classified as measured at amortized cost, fair value through other comprehensive income (‘‘FVOCI’’), or fair value through profit and loss (‘‘FVTPL’’). The classification of financial assets is based on the business model in which a financial asset is managed and its contractual cash flow characteristics. |
| F-12 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
| A financial asset is measured at amortized cost if it meets both of the following conditions and is not designated as at FVTPL: | |
|
|
|
| · | It is held within a business model whose objective is to hold assets to collect contractual cash flows; and |
|
|
|
| · | Its contractual terms give rise on specified dates to cash flows that are solely payments of principal and interest on the principal amount outstanding. |
|
|
|
| A financial asset (unless it is a trade receivable without a significant financing component that is initially measured at the transaction price) is initially measured at fair value plus, for an item not at FVTPL, transaction costs that are directly attributable to its acquisition.
The following accounting policies apply to the subsequent measurement of financial assets. | |
Financial assets at FVTPL | Subsequently measured at fair value. Net gains and losses, including any interest or dividend income, are recognized in the consolidated statements of loss and comprehensive loss. |
Financial assets at amortized cost | Subsequently measured at amortized cost using the effective interest method, less any impairment losses. Interest income, foreign exchange gains and losses and impairment losses are recognized in profit or loss. Any gain or loss on derecognition is recognized in consolidated statements of loss and comprehensive loss. |
· | Financial liabilities |
|
The Company initially recognizes financial liabilities at fair value on the date at which the Company becomes a party to the contractual provisions of the instrument.
The Company classifies its financial liabilities as either financial liabilities at FVTPL or amortized cost.
Subsequent to initial recognition, other liabilities are measured at amortized cost using the effective interest method. Financial liabilities at FVTPL are stated at fair value with changes being recognized in consolidated statements of loss and comprehensive loss.
The Company derecognizes a financial liability when its contractual obligations are discharged or cancelled or expire. |
· | Financial liabilities and equity instruments |
| · | Classification as debt or equity |
|
|
Debt and equity instruments issued by the Company are classified as either financial liabilities or as equity in accordance with the substance of the contractual arrangements and the definitions of a financial liability and an equity instrument. The Company does not reclassify financial liabilities or equity after initial recognition due to a change in circumstance. |
|
|
|
| · | Equity instruments |
|
|
|
|
| An equity instrument is any contract that evidences a residual interest in the assets of an entity after deducting all of its liabilities. Equity instruments issued by a group entity are recognized at the proceeds received, net of direct issue costs.
Repurchase of the Company’s own equity instruments is recognized and deducted directly in equity. No gain or loss is recognized in the consolidated statements of loss and comprehensive loss on the purchase, sale, issue or cancellation of the Company’s own equity instruments. |
| F-13 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
· | Classification of financial instruments |
|
The Company classifies its financial assets and liabilities depending on the purpose for which the financial instruments were acquired, their characteristics and management intent as outlined below: |
| Cash and cash equivalent | Amortized cost |
| Other receivables | Amortized cost |
| Investments | Fair value through profit or loss |
| Finance receivables | Amortized cost |
| Trade and other payables | Amortized cost |
| Warrants liability | Fair value through profit or loss |
| Notes payable | Amortized cost |
· | Impairment of financial assets |
| · | Other receivables |
|
|
An expected credit loss (“ECL”) model applies to financial assets measured at amortized cost. The Company’s financial assets measured at amortized cost and subject to the ECL model consist primarily of finance and other receivables. The Company applies the simplified approach to impairment for finance and other receivables by recognizing a loss allowance based on lifetime expected losses at each reporting date taking into considerations historical credit loss experience and financial factors specific to the debtors and general economic conditions. The Company has assessed the impairment of its finance and other receivables using the expected credit loss model. |
| · | Finance receivables |
|
|
Finance receivables are a financial asset initially recognized at fair value and are subsequently carried at amortized cost using the effective interest method. The Company’s business model is to hold these receivables to collect contractual cash flows that represent solely payments of principal and interest. Finance receivables are assessed for impairment at the end of each reporting period in accordance with IFRS 9 as outlined below.
The ECL model is based on the credit losses expected to arise over the life of the assets, unless there has been no significant increase in credit risk since origination, in which case the allowance is based on the 12 months’ expected credit loss. The ECL model uses a three-stage impairment approach based on changes in the credit risk of the finance receivable since initial recognition. The three stages are as follows:
Stage 1– Finance receivables that have not experienced a significant increase in credit risk since initial recognition.
Stage 2– Finance receivables that have experienced a significant increase in credit risk since initial recognition.
Stage 3 – Finance receivables for which there is objective evidence of impairment. |
| F-14 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The Company considers a number of factors when assessing if there has been a significant increase in credit risk, including the number of days past due, changes in the financial condition of the borrower, responsiveness of the borrower and other borrower specific information that may be available, without consideration of collateral.
In determining its estimation of the ECL allowances, the Company also considers past events, current market conditions including interest rates, real estate market statistics, and supportable forward-looking information, including macro-economic factors, such as housing price and interest rate forecasts.
The ECL model requires the recognition of credit losses equal to 12-month ECLs for Stage 1 and recognition of lifetime expected credit losses for Stage 2 and 3. The 12-month ECLS are lifetime ECLS that are expected to occur within 12 months after the reporting date. The lifetime ECLs represent the expected loss in value due to possible default events over the life of a mortgage receivable weighted by the likelihood of a loss. Three factors are primarily used to measure ECLs: probability of default (PD), loss given default (LGD) and exposure at default (EAD).
[e] Impairment of long-lived assets
Long-lived assets, including equipment and intangible assets are tested for impairment at each reporting date when there are indicators of impairment or whenever events or changes in circumstances indicate that the carrying amount of an asset exceeds its recoverable amount. Intangible assets with an indefinite useful life are tested for impairment at least annually in the fourth quarter and whenever there is an indication that the asset may be impaired. The Company has no indefinite life intangible assets.
For the purpose of impairment testing, assets that cannot be tested individually are grouped together into the smallest group of assets that generates cash inflows from continuing use that are largely independent of the cash inflows of other assets or groups of assets (the cash-generating unit, or “CGU”). The recoverable amount of an asset or a CGU is the higher of its fair value less costs to sell and its value in use. If the carrying amount of an asset exceeds its recoverable amount, an impairment charge is recognized immediately in net loss equal to the amount by which the carrying amount exceeds the recoverable amount. Where an impairment loss subsequently reverses, the carrying amount of the asset is increased to the lesser of the revised estimate of the recoverable amount, and the carrying amount that would have been recorded had no impairment loss been recognized previously.
[f] Income Taxes
Income tax expense comprises current and deferred tax. Current tax and deferred tax are recognized in net profit or loss except to the extent that it relates to a business combination or items recognized directly in equity or in other comprehensive loss.
Current income taxes are recognized for the estimated income taxes payable or receivable on taxable income or loss for the current year and any adjustment to income taxes payable in respect of previous years. Current income taxes are determined using tax rates and tax laws that have been enacted or substantively enacted by the year-end date.
Deferred tax assets and liabilities are recognized where the carrying amount of an asset or liability differs from its tax base, except for taxable temporary differences arising on the initial recognition of goodwill and temporary differences arising on the initial recognition of an asset or liability in a transaction which is not a business combination and at the time of the transaction affects neither accounting nor taxable profit or loss.
Recognition of deferred tax assets for unused tax losses, tax credits and deductible temporary differences is restricted to those instances where it is probable that future taxable profit will be available against which the deferred tax asset can be utilized. At the end of each reporting period, the Company reassesses unrecognized deferred tax assets. The Company recognizes a previously unrecognized deferred tax asset to the extent that it has become probable that future taxable profit will allow the deferred tax asset to be recovered.
| F-15 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
[g] Share-based Compensation
Share options and warrants awarded to non-employees are accounted for using the fair value of the instrument awarded or service provided, whichever is considered more reliable. Share options, PSUs and warrants awarded to employees are accounted for using the fair value method. The fair value of the share options, PSUs and warrants granted are recognized as an expense on a proportionate basis consistent with the vesting features of each tranche of the grant. The fair value of share options and warrants are calculated using the Black-Scholes option pricing model with assumptions applicable at the date of grant. The fair value of PSUs is calculated using market share prices at the date of grant.
[h] Net Loss per Share
Net loss per share is calculated based on the loss for the financial year and the weighted average number of common shares outstanding during the year. Diluted net loss per share is calculated using the loss for the financial year adjusted for the effect of any dilutive instruments and the weighted average diluted number of shares (ignoring any potential issue of common shares that would be anti-dilutive) during the year.
[i] External research and development
External research and development costs are expensed in the periods in which they are incurred, with the exception of development costs for new products with proven technical feasibility and for which a defined future market exists. Such development costs are capitalized in accordance with the Company’s policy for intangible assets. The Company’s external research and development costs relate primarily to third-party contract research organizations.
[j] Discontinued operations
Discontinued operations are reported when a component of the Company, representing a separate major line of business or area of operations with clearly distinguishable cash flows, has been disposed of or is held for sale. Classification as a discontinued operation occurs upon disposal or when the operation meets the criteria to be classified as held for sale, if earlier. Discontinued operations are reported as a separate element of net income or loss on the consolidated statements of loss and comprehensive loss for both the current and comparative periods. When a disposal group is classified as held for sale, assets and liabilities are aggregated and presented as separate line items, respectively, on the consolidated statements of financial position. Comparative periods are not restated on the consolidated statements of financial position. Assets held for sale are not depreciated and are measured at the lower of carrying value and fair value less costs to sell.
New standards, amendments and interpretations recently adopted by the Company
IAS 1, Presentation of financial statements (“IAS 1”)
In January 2020, the IASB issued Classification of Liabilities as Current or Non-current (Amendments to IAS 1). The amendments aim to promote consistency in applying the requirements by helping companies determine whether, in the consolidated statements of financial position, debt and other liabilities with an uncertain settlement date should be classified as current (due or potentially due to be settled within one year) or non-current. The amendments include clarifying the classification requirements for debt a company might settle by converting it into equity.
The amendments are effective for annual reporting periods beginning on or after January 1, 2024, with earlier application permitted. The Company early adopted these amendments effective January 1, 2023. The impact of adopting these amendments on the Company’s financial statements was not material.
| F-16 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
IAS 8, Accounting Policies, Changes in Accounting Estimates and Errors (“IAS 8”)
In February 2021, the IASB issued Definition of Accounting Estimates, which amends IAS 8. The amendment clarifies how to distinguish changes in accounting policies from changes in accounting estimates. Under the new definition, accounting estimates are “monetary amounts in financial statements that are subject to measurement uncertainty”. The amendment provides clarification to help entities to distinguish between accounting policies and accounting estimates.
The amendments are effective for annual periods beginning on or after January 1, 2023. The impact of adopting these amendments on the Company’s financial statements was not material.
IAS 12, Income Taxes (“IAS 12”)
In May 2021, the IASB issued Deferred Tax related to Assets and Liabilities arising from a single transaction (Amendments to IAS 12). The amendment narrows the scope of the initial recognition exemption so that it does not apply to transactions that give rise to equal taxable and deductible temporary differences. As a result, companies will need to recognize a deferred tax asset and deferred tax liability for temporary differences arising on initial recognition of transactions such as leases and decommissioning obligations.
The amendments are effective for annual reporting periods beginning on or after January 1, 2023 and are to be applied retrospectively. The impact of adopting these amendments on the Company’s financial statements was not material.
New standards, amendments and interpretations not yet adopted by the Company
IFRS 16 – Leases (“IFRS 16”)
In September 2022, the IASB issued amendments to IFRS 16, Leases, which add to requirements explaining how a company accounts for a sale and leaseback after the date of the transaction.
The amendments are effective for annual reporting periods beginning on or after January 1, 2024. Earlier application is permitted.
All other IFRSs and amendments issued but not yet effective have been assessed by the Company and are not expected to have a material impact on the financial statements.
4. Acquisition of Lucid
On September 21, 2021, the Company acquired all of the issued and outstanding common shares of Lucid, an early-stage Canadian-based specialty pharmaceutical company focused on the development of therapies to treat critical neurodegenerative diseases, for total consideration of $
Prior to the acquisition, the Company’s Executive Co-Chairman of the Board beneficially held approximately
It was determined that the acquisition of Lucid did not qualify as a business combination in accordance with IFRS 3, and therefore it was accounted for as an asset acquisition. The individual identifiable assets acquired and liabilities assumed were identified. The purchase consideration was first allocated to the fair values of the acquired cash and cash equivalents, other receivables, prepaid expenses and deposits and trade and other payables, as their carrying values were determined to equal their fair values. The remaining purchase price was allocated to the acquired intangible assets.
| F-17 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The total consideration for the purchase of Lucid was $7,290,731. The purchase consideration consisted of $
|
| Warrants |
|
| Share Options |
| ||
Grant date share price |
| $ |
|
| $ |
| ||
Exercise Price |
| $ |
|
| $ |
| ||
Expected dividend yield |
|
| — |
|
|
| — |
|
Risk free interest rate |
|
| % |
|
| |||
Expected life (years) |
|
|
|
| ||||
Annualized volatility |
|
| % |
|
| % | ||
The allocation of the total consideration to the fair value of the identifiable assets acquired and liabilities assumed as at the date of the acquisition was as follows:
|
| Fair value recognized on acquisition |
| |
|
| $ |
| |
Cash and cash equivalents |
|
|
| |
Other receivables |
|
|
| |
Prepaid expenses and deposits |
|
|
| |
Intangible assets |
|
|
| |
Trade and other payables |
|
| ( | ) |
|
|
|
| |
The Company also capitalized $
5. Discontinued operations
In March 2020, the Company decided to focus its efforts and resources on the pharmaceutical business and initiated the process to exit the medical cannabis industry and sell FV Pharma’s facility located at 520 William Street, Cobourg, Ontario, K9A 3A5 (the “Facility”) and the 64-acre property on which the Facility is located (the “Facility Property”). On May 6, 2022, the Company closed the sale of the Facility and the Facility Property for total consideration of $
Results of operations related to the Disposal Group are reported as discontinued operations for the years ended December 31, 2022 and 2021.
| F-18 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
Net income (loss) from discontinued operations for the years ended December 31, 2022 and 2021 is comprised of the following:
|
|
|
|
|
|
|
|
|
| |||
|
|
|
|
| For the years ended December 31, |
| ||||||
|
| Notes |
|
| 2022 $ |
|
| 2021 $ |
| |||
Expenses |
|
|
|
|
|
|
|
|
| |||
General and administrative |
|
| 17 |
|
|
|
|
|
|
| ||
Total operating expenses |
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
Loss from discontinued operations |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Other income |
|
|
|
|
|
| ( | ) |
|
| ( | ) |
Gain on sale of property and plant |
|
|
|
|
|
| ( | ) |
|
|
| |
Net income (loss) from discontinued operations |
|
|
|
|
|
|
|
|
| ( | ) | |
Cash flows from discontinued operations for the year ended December 31, 2022 and 2021 is comprised of the following:
|
| For the years ended December 31, |
| |||||
|
| 2022 |
|
| 2021 |
| ||
|
| $ |
|
| $ |
| ||
Operating activities |
|
|
|
|
|
| ||
Net income (loss) from discontinued operations |
|
|
|
|
| ( | ) | |
Add (deduct) items not affecting cash |
|
|
|
|
|
|
|
|
Changes in non-cash working capital balances |
|
|
|
|
|
|
|
|
Gain on sale of property and plant |
|
| ( | ) |
|
|
| |
Other receivables |
|
| ( | ) |
|
|
| |
Prepaid expenses and deposits |
|
|
|
|
| ( | ) | |
Trade and other payables |
|
|
|
|
| ( | ) | |
Cash used in operating activities |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
Proceeds from sale of property and plant, net |
|
|
|
|
|
| ||
Cash provided by investing activities |
|
|
|
|
|
| ||
There were no discontinued operations for the year ended December 31, 2023.
6. Other receivables
The Company’s other receivables are comprised of the following:
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
|
| $ |
|
| $ |
| ||
Sales tax recoverable |
|
|
|
|
|
| ||
Interest receivable |
|
|
|
|
|
| ||
Other receivables |
|
|
|
|
|
| ||
|
|
|
|
|
|
| ||
| F-19 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
7. Prepaid expenses and deposits
The Company’s prepaid expenses and deposits include the following:
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
|
| $ |
|
| $ |
| ||
Research and development |
|
|
|
|
|
| ||
Insurance |
|
|
|
|
|
| ||
Other prepaids and deposits |
|
|
|
|
|
| ||
|
|
|
|
|
|
| ||
8. Finance receivables
Finance receivables consist of secured loan receivables measured at amortized cost, net of allowance for expected credit losses.
Finance receivables as at December 31, 2023 are as follows:
|
|
| $ |
|
Balance – January 1, 2022 |
|
| — |
|
Additions |
|
|
| |
Add: Interest income |
|
|
| |
Less: Interest payments |
|
| ( | ) |
Less: Principal payments |
|
| ( | ) |
Effects of foreign exchange |
|
| ( | ) |
Balance – December 31, 2022 |
|
|
| |
Additions |
|
|
| |
Add: Interest income |
|
|
| |
Less: Interest payments |
|
| ( | ) |
Less: Principal payments |
|
| ( | ) |
Effects of foreign exchange |
|
|
| |
Balance – December 31, 2023 |
|
|
| |
Current |
|
|
| |
Non-current |
|
|
| |
Balance – December 31, 2023 |
|
| 8,095,354 |
|
Allowances for expected credit losses as at December 31, 2023, were $nil. Finance receivables earn fees at fixed rates and have an average term to maturity of two years from the date of issuance. The loans are secured by residential property with a first or second collateral mortgage on the secured property, except for the loan issued to a related party (Note 21). Loans are issued up to 55% of the initial appraised value of the secured property at the time of issuance.
| F-20 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
Finance receivables include the following:
|
| December 31, 2023 |
| |
|
| $ |
| |
Minimum payments receivable |
|
|
| |
Unearned income |
|
| ( | ) |
Net investment |
|
|
| |
Allowance for credit losses |
|
|
| |
Finance receivables, net |
|
|
| |
As at December 31, 2023, all loans were classified as stage 1 and there were no changes between stages during the year.
9. Investments
The following tables outline changes in investments during the periods:
Entity |
| Instrument |
| Note |
| Balance at December 31, 2022 |
|
| Proceeds from sale |
|
| Additions |
|
| Change in fair value through profit or loss |
|
| Effects of foreign exchange |
|
| Balance at December 31, 2023 |
| ||||||
|
|
| $ |
|
| $ |
|
| $ |
|
| $ |
|
| $ |
|
| $ |
| |||||||||
Solarvest BioEnergy Inc. |
| Shares |
| (i) |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
| |||||
Solarvest BioEnergy Inc. |
| Convertible debenture |
| (i) |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
| |||||
A2ZCryptoCap Inc. |
| Shares |
| (ii) |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
| |||||
Lions Bay Fund |
| Shares |
| (iii) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Royal Bank of Canada |
| Guaranteed Investment Certificate |
| (iv) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
|
|
|
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Current |
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Non-Current |
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
Entity |
| Instrument |
| Note |
| Balance at December 31, 2021 |
|
| Proceeds from sale |
|
| Additions |
|
| Change in fair value through profit or loss |
|
| Balance at December 31, 2022 |
| |||||
|
|
| $ |
|
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||||||
True Pharma Strip Inc. |
| Shares |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
HUGE Shops |
| Shares |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
SciCann Therapeutics |
| Shares |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Solarvest BioEnergy Inc. |
| Shares |
| (i) |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
| ||||
Solarvest BioEnergy Inc. |
| Convertible debenture |
| (i) |
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
| ||||
A2ZCryptoCap Inc. |
| Shares |
| (ii) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
Lions Bay Fund |
| Shares |
| (iii) |
|
|
|
|
| — |
|
|
|
|
|
|
|
|
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ( | ) |
|
|
| |||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Current |
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Non-Current |
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
(i) Solarvest BioEnergy Inc. (“Solarvest”)
The Company holds
As at December 31, 2023, the fair value of the shares was determined to be $nil given the halt in trading of Solarvest’s shares as a result of the entity failing to maintain a transfer agent and due to the significant financial and operational challenges being faced by the entity. The fair value of the convertible debenture was determined to be $nil as well. The shares have been classified as level 1 within the fair value hierarchy – quoted market price, and the convertible debenture has been classified as level 2 – valuation technique with observable market inputs.
| F-21 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
As at December 31, 2022, the fair value of the shares was determined based on the quoted market price of the shares of C$
(ii) A2ZCryptoCap Inc. (“A2Z”)
On June 23, 2022, the Company acquired
(iii) Lions Bay Fund (“Fund”)
During the year ended December 31, 2022, the Company invested $
(iv) On August 9, 2023, the Company purchased a Guaranteed Investment Certificate (“GIC”) in the amount of $
10. Intangible assets
Intangible assets as at December 31, 2023 are as follows:
|
| Innovet |
|
| Prismic |
|
| Lucid |
|
| Total |
| ||||
Cost |
| $ |
|
| $ |
|
| $ |
|
| $ |
| ||||
As at December 31, 2021 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Additions |
|
|
|
|
|
|
|
|
|
|
|
| ||||
As at December 31, 2022 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Impairment |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
| ( | ) | |
As at December 31, 2023 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated amortization |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As at December 31, 2021 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Amortization |
|
|
|
|
|
|
|
|
|
|
|
| ||||
As at December 31, 2022 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Amortization |
|
|
|
|
|
|
|
|
|
|
|
| ||||
Impairment |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
| ( | ) | |
As at December 31, 2023 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Net book value |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As at December 31, 2022 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
As at December 31, 2023 |
|
|
|
|
|
|
|
|
|
|
|
| ||||
During the year ended December 31, 2023, the Company recognized an impairment loss of $
During the year ended December 31, 2023, the Company recognized an impairment loss of $
| F-22 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The Company’s intangible asset for Lucid represents the license agreement with the University Health Network giving the Company world-wide exclusive rights to the Lucid-MS compound and related patents.
11. Trade and other payables
Trade and other payables consist of the following:
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
|
| $ |
|
| $ |
| ||
Trade payables |
|
|
|
|
|
| ||
Accrued liabilities (i) |
|
|
|
|
|
| ||
|
|
|
|
|
|
| ||
(i) Accrued liabilities consist of the following:
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
|
| $ |
|
| $ |
| ||
External research and development fees |
|
|
|
|
|
| ||
Operational expenses |
|
|
|
|
|
| ||
Professional and other fees |
|
|
|
|
|
| ||
Accrued interest |
|
|
|
|
|
| ||
|
|
|
|
|
|
| ||
12. Warrants Liability
In August 2020, the Company issued
On initial recognition the Company determined that these warrants did not meet the IFRS definition of equity due to the exercise price being denominated in United States dollar, which was not the functional currency of the Company at the time resulting in variability in exercise price. The change in functional currency on October 1, 2020, was determined to be a change in circumstance and, as such, the Company has made an accounting policy choice to continue to recognize the warrants as a financial liability classified at fair value through profit or loss.
The fair value of the warrants liability as at December 31, 2023, was $
The fair value was determined using the Black-Scholes option pricing model and the following assumptions:
|
| December 31, 2023 |
|
| December 31, 2022 |
| ||
Share price |
| $ |
|
| $ |
| ||
Exercise price |
| $ |
|
| $ |
| ||
Expected dividend yield |
|
| — |
|
|
| — |
|
Risk free interest rate |
|
| % |
|
| % | ||
Expected life |
|
|
|
|
|
| ||
Expected volatility |
|
| % |
|
| % | ||
| F-23 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
13. Share capital
[a] Authorized
The Company is authorized to issue an unlimited number of Class A multiple voting shares (“Class A shares”) and an unlimited number of Class B subordinate voting shares (“Class B shares”), all without par value. All shares are ranked equally with regard to the Company’s residual assets.
The holders of Class A shares are entitled to 276,660 votes per Class A share held. Class A shares are held by the Chief Executive Officer (“CEO”), President, Executive Co-Chairman of the Board and the Director and Executive Co-Chairman of the Board. The holders of Class B shares are entitled to one (1) vote per share held.
[b] Issued and outstanding
Reconciliation of the Company’s share capital is as follows:
|
| Class A shares |
|
| Class B shares |
|
| Warrants |
| |||||||||||||||
|
| # |
|
| $ |
|
| # |
|
| $ |
|
| # |
|
| $ |
| ||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Balance, December 31, 2020 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| 4,968,958 |
| |||||
Shares issued [a] |
|
| — |
|
|
| — |
|
|
|
|
|
| 38,341,407 |
|
|
| — |
|
|
| — |
| |
Share-based payments [b] |
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Share cancellation [b] |
|
| — |
|
|
| — |
|
|
| ( | ) |
|
| — |
|
|
| — |
|
|
| — |
|
Lucid acquisition [c] |
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
|
|
|
|
| ||||
Warrants expired |
|
| — |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
| ( | ) |
|
| ( | ) |
Balance, December 31, 2021 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Share repurchase [d] |
|
| — |
|
|
| — |
|
|
| ( | ) |
|
| ( | ) |
|
| — |
|
|
| — |
|
Share-based payments [e] |
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
| — |
|
|
| — |
| ||
Share cancellation [f] |
|
| — |
|
|
| — |
|
|
| ( | ) |
|
| ( | ) |
|
| — |
|
|
| — |
|
PSU converted to shares |
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
| — |
|
|
| — |
| ||
Warrants expired |
|
| — |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
| ( | ) |
|
| ( | ) |
Balance, December 31, 2022 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Plan of arrangement [k] |
|
| — |
|
|
|
|
|
|
|
|
| — |
|
|
| — |
|
|
| — |
| ||
Share repurchase [g] |
|
| — |
|
|
| — |
|
|
| ( | ) |
|
| ( | ) |
|
| — |
|
|
| — |
|
Share-based payments [j] |
|
| — |
|
|
| — |
|
|
|
|
|
| 36,000 |
|
|
| — |
|
|
| — |
| |
Share options exercised [i] |
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
| — |
|
|
| — |
| ||
PSU converted to shares |
|
| — |
|
|
| — |
|
|
|
|
|
|
|
|
| — |
|
|
| — |
| ||
Warrants issued [h] |
|
| — |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
|
|
|
|
| ||
Warrants expired |
|
| — |
|
|
| — |
|
|
| — |
|
|
| — |
|
|
| ( | ) |
|
| ( | ) |
Balance, December 31, 2023 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Activity during the period from December 31, 2020 to December 31, 2021:
| [a] | During the year ended December 31, 2021, the Company issued |
|
|
|
| [b] | On February 17, 2021, the Company issued |
|
|
On July 26, 2021, the Company issued |
| F-24 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
|
| During the year ended December 31, 2021, the Company issued |
|
|
|
| [c] | On September 21, 2021, the Company issued |
|
|
|
Activity during the period from December 31, 2021 to December 31, 2022: | ||
|
|
|
| [d] | During the year ended December 31, 2022, the Company repurchased and cancelled |
|
|
|
| [e] | During the year ended December 31, 2022, the Company issued |
|
|
|
| [f] | On March 29, 2022, the Company cancelled |
|
|
|
Activity during the period from December 31, 2022 to December 31, 2023: | ||
|
|
|
| [g] | During the year ended December 31, 2023, the Company repurchased and canceled |
|
|
|
| [h] | During the year ended December 31, 2023, the Company issued |
|
|
|
| [i] | During the year ended December 31, 2023, the Company issued |
|
|
|
| [j] | During the year ended December 31, 2023, the Company issued |
|
|
|
| [k] | In November 2023, the Company completed the Plan of Arrangement reorganization. The Company cancelled all 72 Class A Shares of the Company and reissued 24 new Class B shares and 48 new Class A Shares. The Company cancelled all 39,376,699 Class B shares outstanding and reissued 39,376,698 new Class B shares. There was 1 previously issued Class B share that was removed due to an administrative adjustment. The Company also cancelled and reissued 6,335,758 FSD Pharma New Distribution Warrants. Each holder of the Company’s Class A shares, Class B shares and the FSD Pharma New Distribution Warrants was distributed a share of Celly from the Company for each Class A share, Class B share and New Distribution Warrant held. As a result, the Company issued |
|
|
The Company issued |
| F-25 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The changes in the number of warrants outstanding during the year ended December 31, 2023, 2022 and 2021 were as follows:
|
| Number of warrants |
|
| Weighted average exercise price |
| ||
|
| # |
|
| C$ |
| ||
Outstanding as at December 31, 2020 |
|
|
|
|
|
| ||
Issued |
|
|
|
|
|
| ||
Expired |
|
| ( | ) |
|
|
| |
Outstanding as at December 31, 2021 |
|
|
|
|
|
| ||
Expired |
|
| ( | ) |
|
|
| |
Outstanding as at December 31, 2022 |
|
|
|
|
|
| ||
Issued |
|
|
|
|
|
| ||
Expired |
|
| ( | ) |
|
|
| |
Outstanding as at December 31, 2023 |
|
|
|
|
|
| ||
Measurement of fair values
The fair value of the warrants issued during the year ended December 31, 2023 and 2021, were estimated at the date of grant using the Black-Scholes option pricing model with the following inputs:
|
| 2023 |
|
| 2021 |
| ||
Grant date share price |
| C$ |
|
| C$ |
| ||
Exercise price |
| C$ |
|
| C$ |
| ||
Expected dividend yield |
|
| — |
|
|
| — |
|
Risk free interest rate |
|
|
|
| ||||
Expected life |
|
|
|
| ||||
Expected volatility |
|
|
|
| ||||
There were no warrants granted during the year ended December 31, 2022.
| F-26 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The following table is a summary of the Company’s warrants outstanding as at December 31, 2023:
|
|
|
| Exercise price |
|
| Number outstanding |
| ||||
Expiry Date |
|
|
| C$ |
|
| # |
| ||||
March 14, 2024 |
|
| (i) |
|
|
|
|
|
| |||
March 14, 2024 |
|
| (i) |
|
|
|
|
|
| |||
March 14, 2024 |
|
| (i) |
|
|
|
|
|
| |||
March 30, 2024 |
|
| (i) |
|
|
|
|
| 300,000 |
| ||
March 30, 2024 |
|
| (i) |
|
|
|
|
|
| |||
March 30, 2024 |
|
| (i) |
|
| 5.95 |
|
|
|
| ||
May 24, 2024 |
|
| (i) |
|
| 1.98 |
|
|
|
| ||
February 27, 2025 |
|
| (i) |
|
|
|
|
|
| |||
February 27, 2025 |
|
| (i) |
|
|
|
|
|
| |||
February 27, 2025 |
|
| (i) |
|
|
|
|
|
| |||
March 15, 2025 |
|
|
|
|
|
|
|
|
|
| ||
March 15, 2025 |
|
|
|
|
|
|
|
|
|
| ||
March 23, 2025 |
|
|
|
|
|
|
|
|
|
| ||
March 24, 2025 |
|
| (i) |
|
|
|
|
|
| |||
March 24, 2025 |
|
| (i) |
|
|
|
|
|
| |||
March 24, 2025 |
|
| (i) |
|
|
|
|
|
| |||
May 4, 2025 |
|
|
|
|
|
|
|
|
|
| ||
May 10, 2025 |
|
|
|
|
|
|
|
|
|
| ||
May 17, 2025 |
|
|
|
|
|
|
|
|
|
| ||
May 31, 2025 |
|
|
|
|
|
|
|
|
|
| ||
June 8, 2025 |
|
|
|
|
|
|
|
|
|
| ||
August 6, 2025 |
|
| (i) |
|
|
|
|
|
| |||
October 20, 2025 |
|
| (i) |
|
|
|
|
|
| |||
January 16, 2026 |
|
|
|
|
|
|
|
|
|
| ||
January 20, 2026 |
|
|
|
|
|
|
|
|
|
| ||
May 15, 2028 |
|
|
|
|
|
| 1.50 |
|
|
|
| |
|
|
|
|
|
|
|
|
|
|
| ||
(i) Warrants were issued in US$
| F-27 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The following table is a summary of the Company’s warrants outstanding as at December 31, 2022:
|
|
|
| Exercise price |
|
| Number outstanding |
| ||||
Expiry Date |
|
|
| C$ |
|
| # |
| ||||
May 20, 2023 |
|
|
|
|
|
|
|
|
| |||
June 23, 2023 |
|
|
|
|
| 2.50 |
|
|
|
| ||
July 24, 2023 |
|
|
|
|
|
|
|
|
| |||
September 11, 2023 |
|
|
|
|
|
|
|
|
| |||
May 4, 2025 |
|
|
|
|
|
|
|
|
| |||
May 10, 2025 |
|
|
|
|
|
|
|
|
| |||
May 17, 2025 |
|
|
|
|
|
|
|
|
| |||
May 31, 2025 |
|
|
|
|
|
|
|
|
| |||
June 8, 2025 |
|
|
|
|
|
|
|
|
| |||
August 6, 2025 |
|
| (i) |
|
|
|
|
|
| |||
October 20, 2025 |
|
| (i) |
|
|
|
|
|
| |||
January 16, 2026 |
|
|
|
|
|
|
|
|
|
| ||
January 20, 2026 |
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
| ||
(i) Warrants were issued in US$
The following table is a summary of the Company’s warrants outstanding as at December 31, 2021:
|
|
|
| Exercise price |
|
| Number outstanding |
| ||||
Expiry Date |
|
|
| C$ |
|
| # |
| ||||
May 24, 2022 |
|
|
|
|
|
|
|
|
| |||
September 15, 2022 |
|
|
|
|
|
|
|
|
| |||
November 30, 2022 |
|
|
|
|
|
|
|
|
| |||
December 31, 2022 |
|
|
|
|
|
|
|
|
| |||
May 20, 2023 |
|
|
|
|
|
|
|
|
| |||
June 23, 2023 |
|
|
|
|
| 2.50 |
|
|
|
| ||
July 24, 2023 |
|
|
|
|
|
|
|
|
| |||
September 11, 2023 |
|
|
|
|
|
|
|
|
| |||
May 4, 2025 |
|
|
|
|
|
|
|
|
| |||
May 10, 2025 |
|
|
|
|
|
|
|
|
| |||
May 17, 2025 |
|
|
|
|
|
|
|
|
| |||
May 31, 2025 |
|
|
|
|
|
|
|
|
| |||
June 8, 2025 |
|
|
|
|
|
|
|
|
| |||
August 6, 2025 |
|
| (i) |
|
|
|
|
|
| |||
October 20, 2025 |
|
| (i) |
|
|
|
|
|
| |||
January 16, 2026 |
|
|
|
|
|
|
|
|
|
| ||
January 30, 2026 |
|
|
|
|
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
|
| ||
(i) Warrants were issued in US$
| F-28 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
14. Share-based compensation
The Company has established a share option plan (the “Option Plan”) for directors, officers, employees and consultants of the Company. The Company’s Board determines, among other things, the eligibility of individuals to participate in the Option Plan, the term and vesting periods, and the exercise price of options granted to individuals under the Option Plan.
Each share option converts into one common share of the Company on exercise. No amounts are paid or payable by the individual on receipt of the option. The options carry neither rights to dividends nor voting rights. Options may be exercised at any time from the date of vesting to the date of their expiry.
[i] Share-based payment arrangements
During the year ended December 31, 2023, the Company granted
The change in the number of share options outstanding during the year ended December 31, 2023, were as follows:
|
| Number of options |
|
| Weighted average exercise price |
| ||
|
| # |
|
| C$ |
| ||
Outstanding as at December 31, 2022 |
|
|
|
|
| 3.71 |
| |
Granted |
|
|
|
|
| 1.52 |
| |
Forfeited |
|
| ( | ) |
|
| 2.09 |
|
Exercised |
|
| ( | ) |
|
| 1.30 |
|
Expired |
|
| ( | ) |
|
| 4.03 |
|
Outstanding as at December 31, 2023 |
|
|
|
|
| 1.56 |
| |
Exercisable as at December 31, 2023 |
|
|
|
|
| 1.54 |
| |
The change in the number of share options outstanding during the year ended December 31, 2022, were as follows:
|
| Number of options |
|
| Weighted average exercise price |
| ||
|
| # |
|
| C$ |
| ||
Outstanding as at December 31, 2021 |
|
|
|
|
| 2.75 |
| |
Granted |
|
|
|
|
| 1.30 |
| |
Forfeited |
|
| ( | ) |
|
| 3.75 |
|
Expired |
|
| ( | ) |
|
| 3.71 |
|
Cancelled |
|
| ( | ) |
|
| 2.56 |
|
Outstanding as at December 31, 2022 |
|
|
|
|
| 3.71 |
| |
Exercisable as at December 31, 2022 |
|
|
|
|
| 3.71 |
| |
The change in the number of share options outstanding during the year ended December 31, 2021, were as follows:
|
| Number of options |
|
| Weighted average exercise price |
| ||
|
| # |
|
| C$ |
| ||
Outstanding as at December 31, 2020 |
|
|
|
|
| 6.11 |
| |
Granted |
|
|
|
|
| 2.26 |
| |
Forfeited |
|
| ( | ) |
|
| 4.83 |
|
Expired |
|
| ( | ) |
|
| 4.87 |
|
Cancelled |
|
| ( | ) |
|
| 9.85 |
|
Outstanding as at December 31, 2021 |
|
|
|
|
| 2.75 |
| |
Exercisable as at December 31, 2021 |
|
|
|
|
| 2.72 |
| |
| F-29 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
During the year ended December 31, 2023,
During the year ended December 31, 2022, the Company cancelled
Measurement of fair values
The fair value of share options granted during the year ended December 31, 2023, 2022 and 2021, were estimated at the date of grant using the Black-Scholes option pricing model with the following inputs:
|
| 2023 |
|
| 2022 |
|
| 2021 |
| |||
Grant date share price |
| C$ |
|
| C$ |
|
| C$ |
| |||
Exercise price |
| C$ |
|
| C$ |
|
| C$ |
| |||
Expected dividend yield |
|
| — |
|
|
| — |
|
|
| — |
|
Risk free interest rate |
|
|
|
| % |
|
| |||||
Expected life |
| |
|
| |
|
| |
| |||
Expected volatility |
|
|
|
| % |
|
| |||||
Expected volatility was estimated by using the annualized historical volatility of the Company. The expected option life represents the period of time that options granted are expected to be outstanding. The risk-free interest rate is based on Canadian government bonds with a remaining term equal to the expected life of the options.
The following table is a summary of the Company’s share options outstanding as at December 31, 2023:
Options outstanding |
|
| Options exercisable |
| ||||||||||||||
Exercise price |
|
| Number outstanding |
|
| Weighted average remaining contractual life [years] |
|
| Exercise price |
|
| Number exercisable |
| |||||
C$ |
|
| # |
|
| # |
|
| C$ |
|
| # |
| |||||
|
|
|
|
|
|
| 4.07 |
|
|
| 1.30 |
|
|
|
| |||
|
|
|
|
|
|
| 1.87 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.42 |
|
|
|
|
|
|
| ||||
| 2.31 |
|
|
|
|
|
| 2.16 |
|
|
| 2.31 |
|
|
|
| ||
|
|
|
|
|
|
| 2.23 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 2.15 |
|
|
| 2.45 |
|
|
|
| |||
|
|
|
|
|
|
| 2.00 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.21 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 2.86 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.28 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 3.66 |
|
|
| 1.54 |
|
|
|
| |||
| F-30 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The following table is a summary of the Company’s share options outstanding as at December 31, 2022:
Options outstanding |
|
| Options exercisable |
| ||||||||||||||
Exercise price |
|
| Number outstanding |
|
| Weighted average remaining contractual life [years] |
|
| Exercise price |
|
| Number exercisable |
| |||||
C$ |
|
| # |
|
| # |
|
| C$ |
|
| # |
| |||||
|
|
|
|
|
|
| 2.58 |
|
|
| 1.30 |
|
|
|
| |||
|
|
|
|
|
|
| 2.21 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.04 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 3.00 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.21 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
|
|
|
|
|
|
|
| |||||
|
|
|
|
|
|
| 0.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.28 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.52 |
|
|
| 3.71 |
|
|
|
| |||
The following table is a summary of the Company’s share options outstanding as at December 31, 2021:
Options outstanding |
|
| Options exercisable |
| ||||||||||||||
Exercise price |
|
| Number outstanding |
|
| Weighted average remaining contractual life [years] |
|
| Exercise price |
|
| Number exercisable |
| |||||
C$ |
|
| # |
|
| # |
|
| C$ |
|
| # |
| |||||
|
|
|
|
|
|
| 3.46 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 2.42 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 4.00 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 3.92 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 3.21 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 0.71 |
|
|
| 4.42 |
|
|
|
| |||
|
|
|
|
|
|
| 3.29 |
|
|
| 4.75 |
|
|
|
| |||
|
|
|
|
|
|
| 1.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 4.00 |
|
|
| 7.63 |
|
|
|
| |||
|
|
|
|
|
|
| 1.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.49 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 1.24 |
|
|
| 18.09 |
|
|
|
| |||
|
|
|
|
|
|
| 2.28 |
|
|
|
|
|
|
| ||||
|
|
|
|
|
|
| 2.50 |
|
|
| 2.72 |
|
|
|
| |||
[ii] Performance Share Units (“PSUs”)
In May 2022, the Company established a performance share unit plan (“PSU Plan”), for directors, offers, employees and consultants of the Company. The Company’s Board determines the eligibility of individuals to participate in the PSU Plan in order to align their interests with those of the Company’s shareholders.
| F-31 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
No amounts are paid or payable by the individual on receipt of the PSUs. Each PSU converts into one common share of the Company at $nil exercise price. The Company’s PSU Plan provides that the number of common shares reserved for issuance may not exceed 10% of the aggregate number of common shares that are outstanding unless the Board has increased such limit by a Board resolution.
The change in the number of PSUs during the years ended December 31, 2023 and 2022, is as follows:
|
| Number of PSUs |
| |
|
| # |
| |
Outstanding as at December 31, 2021 |
|
|
| |
Granted |
|
|
| |
Converted to Class B Common shares |
|
| ( | ) |
Outstanding as at December 31, 2022 |
|
|
| |
Granted |
|
|
| |
Forfeited |
|
| ( | ) |
Converted to Class B Common shares |
|
| ( | ) |
Outstanding as at December 31, 2023 |
|
|
| |
During the year ended December 31, 2023, the Company converted
The Company recognized share-based compensation for the years ended December 31, 2023, 2022 and 2021 as follows:
|
| For the years ended December 31, |
| |||||||||
|
| 2023 |
|
| 2022 |
|
| 2021 |
| |||
|
| $ |
|
| $ |
|
| $ |
| |||
Share options |
|
|
|
|
|
|
|
|
| |||
PSUs |
|
|
|
|
|
|
|
|
| |||
Class B Common Shares issued for services |
|
|
|
|
|
|
|
|
| |||
Class B Common Shares issued for compensation |
|
|
|
|
|
|
|
|
| |||
Warrants issued for services |
|
|
|
|
|
|
|
|
| |||
Other (i) |
|
|
|
|
|
|
|
|
| |||
|
|
|
|
|
|
|
|
|
| |||
| (i) | Share-based compensation related to share options and restricted share units issued by Celly and convertible into common shares of Celly. |
15. Non-controlling interests
Through the License Agreement, FSD acquired
Reconciliation of non-controlling interest is as follows:
|
|
| $ |
|
Balance, December 31, 2022 |
|
|
| |
Initial recognition of non-controlling interests |
|
| ( | ) |
Share-based payments |
|
|
| |
Deemed dividend (Note 13) |
|
|
| |
Comprehensive loss for the period |
|
| ( | ) |
Balance, December 31, 2023 |
|
| ( | ) |
| F-32 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
16. Loss per share
Net loss per common share represents net loss attributable to common shareholders divided by the weighted average number of common shares outstanding during the year.
For all the periods presented, diluted loss per share equals basic loss per share due to the anti-dilutive effect of warrants, share options and PSUs. The outstanding number and type of securities that could potentially dilute basic net loss per share in the future but would have decreased the loss per share (anti-dilutive) for the years ended December 31, 2023, 2022 and 2021 are as follows:
|
| December 31, 2023 |
|
| December 31, 2022 |
|
| December 31, 2021 |
| |||
|
| # |
|
| # |
|
| # |
| |||
Warrants |
|
|
|
|
|
|
|
|
| |||
Share Options |
|
|
|
|
|
|
|
|
| |||
PSUs |
|
|
|
|
|
|
|
|
| |||
|
|
|
|
|
|
|
|
|
| |||
17. General and administrative
Components of general and administrative expenses for the years ended December 31, 2023, 2022 and 2021 were as follows:
|
| For thee years ended December 31, |
| |||||||||
|
| 2023 |
|
| 2022 |
|
| 2021 |
| |||
|
| $ |
|
| $ |
|
| $ |
| |||
Professional fees |
|
|
|
|
|
|
|
|
| |||
General office, insurance and administration expenditures |
|
|
|
|
|
|
|
|
| |||
Consulting fees |
|
|
|
|
|
|
|
|
| |||
Salaries, wages and benefits |
|
|
|
|
|
|
|
|
| |||
Investor relations |
|
|
|
|
|
|
|
|
| |||
Building and facility costs |
|
|
|
|
|
|
|
|
| |||
Foreign exchange (gain) loss |
|
| ( | ) |
|
|
|
|
|
| ||
|
|
|
|
|
|
|
|
|
| |||
Allocated to: |
|
|
|
|
|
|
|
|
|
|
|
|
Continuing operations |
|
|
|
|
|
|
|
|
| |||
Discontinued operations |
|
|
|
|
|
|
|
|
| |||
18. Segment information
Reportable segments are reported in a manner consistent with the internal reporting provided to the chief operating decision maker, with appropriate aggregation. The chief operating decision maker is the CEO who is responsible for allocating resources, assessing the performance of the reportable segment and making key strategic decisions. The Company operates in two segments: Biopharmaceutical and Strategic Investments.
The Company’s Biopharmaceutical segment is focused on furthering the research and development of the Company’s drug candidates and the development of a treatment for alcohol misuse for application in hospitals and other medical practices. The Biopharmaceutical segment primarily earns interest income on excess cash on hand invested in short-term guaranteed investment certificates.
| F-33 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The Company’s Strategic Investments segment is focused on generating returns and cashflow through the issuance of loans secured by residential property, with FSD Strategic Investments having a first or second collateral mortgage on the secured property.
The following tables summarize the Company’s total current and non-current assets and current and non-current liabilities as of December 31, 2023 and 2022, on a segmented basis:
|
| As at December 31, 2023 |
| |||||||||
|
| Biopharmaceutical |
|
| Strategic Investments |
|
| Consolidated |
| |||
|
| $ |
|
| $ |
|
| $ |
| |||
Current assets |
|
|
|
|
|
|
|
|
| |||
Non-current assets |
|
|
|
|
|
|
|
|
| |||
Current liabilities |
|
|
|
|
|
|
|
|
| |||
Non-current liabilities |
|
|
|
|
|
|
|
|
| |||
|
| As at December 31, 2022 |
| |||||||||
|
| Biopharmaceutical |
|
| Strategic Investments |
|
| Consolidated |
| |||
|
| $ |
|
| $ |
|
| $ |
| |||
Current assets |
|
|
|
|
|
|
|
|
| |||
Non-current assets |
|
|
|
|
|
|
|
|
| |||
Current liabilities |
|
|
|
|
|
|
|
|
| |||
Non-current liabilities |
|
|
|
|
|
|
|
|
| |||
The following tables summarize the Company’s interest income, total operating expenses, and net loss for the years ended December 31, 2023 and 2022 on a segmented basis:
|
| For the year ended December 31, 2023 |
| |||||||||
|
| Biopharmaceutical |
|
| Strategic Investments |
|
| Consolidated |
| |||
|
| $ |
|
| $ |
|
| $ |
| |||
Interest income |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Total operating expenses |
|
|
|
|
|
|
|
|
| |||
Net loss |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| For the year ended December 31, 2022 |
| |||||||||
|
| Biopharmaceutical |
|
| Strategic Investments |
|
| Consolidated |
| |||
|
|
| $ |
|
| $ |
|
| $ |
| ||
Interest income |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Total operating expenses |
|
|
|
|
|
|
|
|
| |||
Net loss |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
| F-34 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
19. Income taxes
The reconciliation of income tax expense for the years ended December 31, 2023, 2022 and 2021 consists of the following:
|
| 2023 |
|
| 2022 |
|
| 2021 |
| |||
|
| $ |
|
| $ |
|
| $ |
| |||
Loss from continuing operations before income taxes |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Statutory federal and provincial tax rate |
|
| % |
|
| % |
|
| % | |||
Income tax recovery at the statutory tax rate |
|
| ( | ) |
|
| ( | ) |
|
| ( | ) |
Permanent differences |
|
|
|
|
|
|
|
|
| |||
Book to filing adjustments |
|
| ( | ) |
|
|
|
|
|
| ||
Share issuance cost booked directly to equity |
|
|
|
|
|
|
|
| ( | ) | ||
Impact of tax rate changes |
|
| ( | ) |
|
|
|
|
|
| ||
Foreign exchange |
|
| ( | ) |
|
|
|
|
| ( | ) | |
Change in tax benefits not recognized |
|
|
|
|
|
|
|
|
| |||
|
|
|
|
|
|
|
|
|
| |||
Deferred tax assets have not been recognized in respect of the following temporary differences as at December 31, 2023 and 2022:
|
| 2023 |
|
| 2022 |
|
| 2021 |
| |||
|
|
| $ |
|
| $ |
|
| $ |
| ||
Non-capital losses - Canada |
|
|
|
|
|
|
|
|
| |||
Net-operating loss - US |
|
|
|
|
|
|
|
|
| |||
Unrealized foreign exchange loss |
|
|
|
|
|
|
|
|
| |||
Share-issuance costs |
|
|
|
|
|
|
|
|
| |||
Capital losses carried forward |
|
|
|
|
|
|
|
|
| |||
Other investments |
|
|
|
|
|
|
|
|
| |||
IFRS 16 |
|
|
|
|
|
|
|
|
| |||
Property, plant and equipment |
|
|
|
|
|
|
|
|
| |||
Total |
|
|
|
|
|
|
|
|
| |||
The Company’s Canadian non-capital income tax losses expire as follows:
|
|
| $ |
|
2038 |
|
|
| |
2039 |
|
|
| |
2040 |
|
|
| |
2041 |
|
|
| |
Thereafter |
|
|
| |
|
|
|
|
The company has cumulative US federal net operating loss carryforwards of approximately $
| F-35 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
20. Commitments and contingencies
Commitments
Lucid-MS Agreement
The Company has entered into a license agreement that governs the Lucid-MS compound. Under the terms of the agreement, the Company shall pay a yearly license maintenance fee of C$
Under the agreement the Company is committed to minimum milestone payments of $nil and maximum milestone payments of C$
Contingencies
Legal Matters
From time to time, the Company is named as a party to claims or involved in proceedings, including legal, regulatory and tax related, in the ordinary course of its business. While the outcome of these matters may not be estimable at the reporting date, the Company makes provisions, where possible, for the estimated outcome of such claims or proceedings. Should a loss result from the resolution of any claims or proceedings that differs from these estimates, the difference will be accounted for as a charge to the consolidated statements of loss and comprehensive loss in that period.
Contract Research Organization (“CRO”) Dispute
The Company was involved in arbitration proceedings with a CRO regarding amounts claimed to be owed to the CRO by the Company. The CRO was claiming it is owed amounts outstanding for work on clinical trials in the United States.
In November 2022, evidentiary hearings were held in New York. The parties submitted post-hearing briefs in December 2022. On May 19, 2023, an arbitrator arrived at a non-binding decision that both parties breached the agreements and awarded the CRO $1.7 million plus interest on past due amounts. On June 30, 2023, the CRO filed a motion to make the May 19, 2023 award recognized and enforceable in Ontario, Canada.
On August 2, 2023, the Company entered into a settlement agreement with the CRO for $
As at December 31, 2023, all matters have been resolved.
Raza Bokhari
On July 15, 2021, the Company’s former CEO, Raza Bokhari, filed a notice of arbitration seeking relief and support for breach of contract and severance and damages in the amount of $
Raza Bokhari was placed on administrative leave from his role as the Company’s Chief Executive Officer following the Company’s annual general and special meeting of shareholders on May 14, 2021, pending the outcome of an investigation of various concerns by a Special Committee comprised of independent directors using independent legal counsel. Upon the recommendation of the Special Committee, Raza Bokhari’s employment was terminated for cause by the Company’s board on July 27, 2021.
| F-36 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The Company disputed the allegations and counterclaimed against Raza Bokhari for losses sustained as a result of his alleged breaches of his duties to the Corporation. The arbitration hearing concluded in August 2022 and the arbitrator issued his decision in November 2022. Raza Bokhari’s claim for USD $
On December 9, 2022, Raza Bokhari filed an application in the Ontario Superior Court seeking to set aside the arbitral award of the court on the grounds that he was not treated equally and fairly and the arbitrator’s written award provided inadequate reasons for his decision.
On December 20, 2022, the Company’s legal counsel wrote to the Commercial List of the Ontario Superior Court of Justice seeking to transfer the application from the Civil List to the Commercial List. The request was granted on January 12, 2023.
On April 28, 2023, the court ordered the case to be heard at the Commercial List on September 27, 2023.
On September 27 and 28, 2023, the application to set aside the award and cost of ground of unfairness was dismissed. As Raza Bokhari lost the set aside application, the court ordered Raza Bokhari to pay the Company C$
On October 13, 2023, Raza Bokhari filed a “Notice of Motion for Leave to Appeal” with the Court of Appeal for Ontario.
On December 15, 2023, the Company submitted a responding party’s factum to the Court of Appeal for Ontario.
On February 6, 2024, the Ontario Superior Court of Justice affirmed judgment and awarded an additional C$
GBB Drink Lab, Inc.
GBB Drink Lab, Inc. (“GBB”) has filed a complaint with the United States District Court of Southern District of Florida, Fort Lauderdale Division against FSD Biosciences, Inc. and FSD Pharma, Inc. claiming a material breach of a mutual non-disclosure agreement and misappropriation of trade secrets, which GBB claims has and continues to cause irreparable harm, valued, as of August 30, 2022 (prior to the misappropriation and material breach) at $53,047,000. On June 23, 2023, the Company filed a motion to dismiss the complaint. On July 3, 2023, GBB responded in opposition to the Company’s motion to dismiss the complaint. On August 24, 2023, the parties filed a proposed joint scheduling report with the U.S. District Court, which set forth various deadlines that would govern this action. Under the proposed joint schedule, which still needs to be approved by the U.S. District Court, the case would be trial-ready by November 30, 2024.
The ultimate outcome of the matter cannot be determined at this time.
21. Related party transactions
Key management personnel are those persons having the authority and responsibility for planning, directing and controlling activities of the entity, directly or indirectly.
Transactions with key management and directors comprised the following:
| a) | In fiscal 2023, the Company paid independent directors’ compensation of C$ |
| F-37 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
| b) | During the year ended December 31, 2023, the Company granted |
|
|
|
| c) | During the year ended December 31, 2023, the Company granted the previous interim CEO, the current CEO, the Chief Operating Officer (“COO”) and the CEO of Lucid, |
|
|
|
| d) | During the year ended December 31, 2023, the Company entered into a secured loan agreement with the CEO, President, and Executive Co-Chairman of the Board in the amount of C$ |
|
|
|
| e) | During the year ended December 31, 2023, the Company issued |
|
|
|
| f) | In November 2023, the Company issued |
|
|
|
| g) | In February 2021, as compensation, the Company issued |
|
|
|
| h) | For the year ended December 31, 2023, the Company paid expenses of $nil (2022 – $nil and 2021 – $ |
|
|
|
| i) | For the year ended December 31, 2023, the Company reimbursed $ |
|
|
|
| j) | During the year ended December 31, 2021, the Company reimbursed certain directors C$ |
| F-38 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
Key management personnel compensation during the years ended December 31, 2023, 2022 and 2021 is comprised of:
|
| 2023 |
|
| 2022 |
|
| 2021 |
| |||
|
| $ |
|
| $ |
|
| $ |
| |||
Salaries, benefits, bonuses and consulting fees |
|
|
|
|
|
|
|
|
| |||
Share-based payments |
|
|
|
|
|
|
|
|
| |||
Total |
|
|
|
|
|
|
|
|
| |||
As at December 31, 2023, the Company owes an executive officer $
22. Capital Management
The Company’s capital management objectives are to maintain financial flexibility in order to complete the research and development of a proprietary formulation of natural ingredients, vitamins, and minerals to help with liver and brain function for the purposes of quickly relieving individuals from the effects of alcohol consumption.
The Company defines capital as the aggregate of its capital stock and borrowings.
As at December 31, 2023, the Company’s Share Capital was $
The Company manages its capital structure in accordance with changes in economic conditions. In order to maintain or adjust its capital structure, the Company may elect to issue or repay financial liabilities, issue shares, repurchase shares or undertake any other activities as deemed appropriate under the specific circumstances. The Company is not subject to any externally imposed capital requirements.
23. Financial Instruments and Risk Management
Credit risk
Credit risk is the risk of financial loss to the Company if a customer or counterparty to a financial instrument fails to meet its contractual obligations and arises principally from deposits with banks and outstanding other receivables and finance receivables. The Company trades only with recognized, creditworthy third parties.
The Company does not hold any collateral as security for its outstanding finance receivables but mitigates this risk by dealing only with, what management believes to be, financially sound counterparties and, accordingly, does not anticipate significant loss for non-performance. The loans are secured by real estate properties and the Company is granted a first or second collateral charge mortgage on the properties for a sum equal to the interest payments plus the principal amount. The Company performs assessments on factors such as: timing of payments, loan to value, communications with the borrower and external macro factors such as interest rates and economic conditions to mitigate risks.
Liquidity risk
Liquidity risk is the risk the Company will not be able to meet its financial obligations as they come due. The Company’s exposure to liquidity risk is dependent on the Company’s ability to raise additional financing to meet its commitments and sustain operations. The Company mitigates liquidity risk by management of working capital, cash flows, the issuance of share capital and if desired, the issuance of debt. The Company’s trade and other payables and notes payables are all due within twelve months from the date of these financial statements.
| F-39 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
If unanticipated events occur that impact the Company’s ability to carry out the planned clinical trials, the Company may need to take additional measures to increase its liquidity and capital resources, including issuing debt or additional equity financing or strategically altering the business forecast and plan. In this case, there is no guarantee that the Company will obtain satisfactory financing terms or adequate financing. Failure to obtain adequate financing on satisfactory terms could have a material adverse effect on the Company’s results of operations or financial condition.
Market risk
Market risk is the risk the fair value or future cash flows of a financial instrument will fluctuate because of changes in market prices. Market risk comprises three types of risk: foreign currency risk, interest rate risk and other price risk.
· | Foreign currency risk |
|
|
| Foreign currency risk arises on financial instruments that are denominated in a currency other than the functional currency in which they are measured. The Company’s primary exposure with respect to foreign currencies is from Canadian dollar denominated cash, investments and trade and other payables. A 1% change in the foreign exchange rates would not result in any significant impact to the financial statements. |
|
|
· | Interest rate risk |
|
|
| Interest rate risk is the risk the fair value or future cash flows of a financial instrument will fluctuate because of changes in market interest rates. The Company does not have any material long-term borrowings outstanding subject to variable interest rates. Therefore, the Company is not exposed to interest rate risk as at December 31, 2023. |
|
|
· | Other price risk |
|
|
| Other price risk is the risk the fair value or future cash flows of a financial instrument will fluctuate because of changes in market prices (other than those arising from interest rate risk or currency risk), whether those changes are caused by factors specific to the individual financial instrument or its issuer, or factors affecting all similar financial instruments traded in the market. The Company is not exposed to other price risk as at December 31, 2023. |
Fair values
The carrying values of cash, other receivables, trade and other payables and notes payable approximate fair values due to the short-term nature of these items or they are being carried at fair value or, for notes payable, interest payables are close to the current market rates. The risk of material change in fair value is not considered to be significant. The Company does not use derivative financial instruments to manage this risk.
Financial instruments recorded at fair value on the consolidated statement of financial position are classified using a fair value hierarchy that reflects the significance of the inputs used in making the measurements. The Company categorizes its fair value measurements according to a three-level hierarchy. The hierarchy prioritizes the inputs used by the Company’s valuation techniques. A level is assigned to each fair value measurement based on the lowest-level input significant to the fair value measurement in its entirety. The three levels of the fair value hierarchy are defined as follows:
| · | Level 1 – Unadjusted quoted prices as at the measurement date for identical assets or liabilities in active markets. |
|
|
|
| · | Level 2 – Observable inputs other than quoted prices included in Level 1, such as quoted prices for similar assets and liabilities in active markets; quoted prices for identical or similar assets and liabilities in markets that are not active; or other inputs that are observable or can be corroborated by observable market data. |
|
|
|
| · | Level 3 – Significant unobservable inputs that are supported by little or no market activity. The fair value hierarchy also requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. |
| F-40 |
FSD PHARMA INC. Notes to the consolidated financial statements (expressed in United States dollars) December 31, 2023 and 2022 |
The fair value hierarchy requires the use of observable market inputs whenever such inputs exist. A financial instrument is classified to the lowest level of the hierarchy for which a significant input has been considered in measuring fair value. During the year, there were no transfers of amounts between levels.
24. Subsequent events
On January 24, 2024, the Company entered into an agreement with SBS Intl Group LLC. (“SBS”) to assist the Company in enhancing its market awareness and foster productive, continuing dialogues with shareholders and other market participants. The agreement grants SBS
On January 2024, the Company entered into an agreement with Draper, Inc. (“Draper”) and Carriage House Capital, Corp. (“Carriage House”) to assist the Company in enhancing its market awareness and foster productive, continuing dialogues with shareholders and other market participants. The agreement grants Draper and Carriage
On February 23, 2024, the Company entered into a settlement agreement to issue
On February 23, 2024, the Company entered into a settlement agreement to issue
On February 23, 2024, the Company granted
On March 26, 2024,
Subsequent to December 31, 2023, the Company entered into an at-the-market offering agreement (the “ATM Agreement”) with H.C Wainwright & Co., LLC to sell Class B shares, having an aggregate offering price up to $
| F-41 |