UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
_____________________________________________________________________________________________
FORM 10-K
_____________________________________________________________________________________________
| ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the Fiscal Year Ended December 31 , 2023
OR
| TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |||||
For the transition period from to .
Commission File No. 001-33093

(Exact name of registrant as specified in its charter)
(State or other jurisdiction of incorporation or organization) | (IRS Employer Identification No.) | |||||||
| (Address of Principal Executive Offices) | (Zip Code) | |||||||
Registrant’s telephone number, including area code: (858 ) 550-7500
Securities registered pursuant to Section 12(b) of the Act:
| Title of Each Class | Trading Symbol | Name of Each Exchange on Which Registered | ||||||
Securities registered pursuant to Section 12(g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☒ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of 1934. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definition of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
☒ | Accelerated Filer | ☐ | Non-accelerated Filer | ☐ | Smaller reporting company | Emerging growth company | ||||||||||||||||||||||||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☒
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Exchange Act Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of the Registrant’s voting and non-voting stock held by non-affiliates was approximately $0.9 billion based on the last sales price of the Registrant’s Common Stock on the Nasdaq Global Market of the Nasdaq Stock Market LLC on June 30, 2023. For purposes of this calculation, shares of Common Stock held by directors, officers and 10% stockholders known to the Registrant have been deemed to be owned by affiliates which should not be construed to indicate that any such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the Registrant or that such person is controlled by or under common control with the Registrant.
As of February 26, 2024, the Registrant had 17,705,287 shares of Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Proxy Statement for the Registrant’s 2024 Annual Meeting of Stockholders to be filed with the Commission within 120 days of December 31, 2023 are incorporated by reference in Part III of this Annual Report on Form 10-K. With the exception of those portions that are specifically incorporated by reference in this Annual Report on Form 10-K, such Proxy Statement shall not be deemed filed as part of this Report or incorporated by reference herein.
Table of Contents
| Part I | ||||||||
| Item 1. | ||||||||
| Item 1A. | ||||||||
| Item 1B. | ||||||||
| Item 1C. | ||||||||
| Item 2. | ||||||||
| Item 3. | ||||||||
| Item 4. | ||||||||
| Part II | ||||||||
| Item 5. | ||||||||
| Item 6. | ||||||||
| Item 7. | ||||||||
| Item 7A. | ||||||||
| Item 8. | ||||||||
| Item 9. | ||||||||
| Item 9A. | ||||||||
| Item 9B. | ||||||||
| Item 9C. | ||||||||
| Part III | ||||||||
| Item 10. | ||||||||
| Item 11. | ||||||||
| Item 12. | ||||||||
| Item 13. | ||||||||
| Item 14. | ||||||||
| Part IV | ||||||||
| Item 15. | ||||||||
| Item 16. | ||||||||
| GLOSSARY OF TERMS AND ABBREVIATIONS | |||||
| Abbreviation | Definition | ||||
| 2023 Notes | $750.0 million aggregate principal amount of convertible senior unsecured notes due 2023 | ||||
| Aldeyra | Aldeyra Therapeutics, Inc. | ||||
| Amgen | Amgen, Inc. | ||||
| ASC | Accounting Standards Codification | ||||
| ASU | Accounting Standards Update | ||||
| Aziyo | Aziyo Med, LLC | ||||
| Baxter | Baxter International, Inc. | ||||
| BeiGene | BeiGene, Ltd. | ||||
| BendaRx | BendaRx Corp. | ||||
| BLA | Biologics license application | ||||
| CASI | CASI Pharmaceuticals, Inc. | ||||
| cGMP | Current Good Manufacturing Practice | ||||
| Company | Ligand Pharmaceuticals Incorporated, including subsidiaries | ||||
| Convertible Note | Senior Convertible Promissory Note | ||||
| COPD | Chronic obstructive pulmonary disease | ||||
| Cormatrix | Cormatrix Cardiovascular, Inc. | ||||
| Corvus | Corvus Pharmaceuticals, Inc. | ||||
| Credit Agreement | Credit Agreement, dated as of October 12, 2023, among Ligand Pharmaceuticals Incorporated, certain of its subsidiaries, as Guarantors (as defined therein), the Lenders (as defined therein), and Citibank, N.A., as Administrative Agent, Swingline Lender and L/C Issuer. | ||||
| CVR | Contingent value right | ||||
| CyDex | CyDex Pharmaceuticals, Inc. | ||||
| Daiichi Sankyo | Daiichi Sankyo Company, Ltd. | ||||
| Dianomi | Dianomi Therapeutics, Inc. | ||||
| DMF | Drug Master File | ||||
| ESG | Environmental, Social and Governance | ||||
| ECM | Extracellular matrix | ||||
| Eisai | Eisai Inc. | ||||
| Elutia | Elutia Inc. | ||||
| EPA | Environmental Protection Agency | ||||
| ESPP | Employee Stock Purchase Plan, as amended and restated | ||||
| EU | European Union | ||||
| Exelixis | Exelixis, Inc. | ||||
| FASB | Financial Accounting Standards Board | ||||
| FDA | U.S. Food and Drug Administration | ||||
| FSGS | Focal segmental glomerulosclerosis | ||||
| FY 2023 | The Company's fiscal year ended December 31, 2023 | ||||
| FY 2022 | The Company's fiscal year ended December 31, 2022 | ||||
| FY 2021 | The Company's fiscal year ended December 31, 2021 | ||||
| GAAP | Generally accepted accounting principles in the United States | ||||
| GCSF | Granulocyte-colony stimulating factor | ||||
| Gilead | Gilead Sciences, Inc. | ||||
| HBV | Hepatitis B Virus | ||||
| Hikma | Hikma Pharmaceuticals PLC | ||||
| Hovione | Hovione FarmCiencia, S.A. | ||||
| IM | Intramuscular | ||||
| IND | Investigational New Drug | ||||
| IRS | Internal Revenue Service | ||||
| IV | Intravenous | ||||
| Jazz | Jazz Pharmaceuticals, Inc. | ||||
| Ligand | Ligand Pharmaceuticals Incorporated, including subsidiaries | ||||
| LTP | Liver targeting prodrug | ||||
| Marinus | Marinus Pharmaceuticals, Inc. | ||||
| Melinta | Melinta Therapeutics, Inc. | ||||
| Merck | Merck & Co., Inc. | ||||
| Metabasis | Metabasis Therapeutics, Inc. | ||||
| NDA | New Drug Application | ||||
| NOLs | Net Operating Losses | ||||
| Novan | Novan, Inc. (n/k/a NVN Liquidation, Inc.) | ||||
| Novartis | Novartis AG | ||||
| Nucorion | Nucorion Pharmaceuticals, Inc. | ||||
| OmniAb | OmniAb Operations, Inc. (f/k/a OmniAb, Inc.) | ||||
| Ono | Ono Pharmaceutical Co., Ltd. | ||||
| Opthea | Opthea Limited | ||||
| Orange Book | Publication identifying drug products approved by the FDA based on safety and effectiveness | ||||
| Palvella | Palvella Therapeutics, Inc. | ||||
| PDUFA | Prescription Drug User Fee Act | ||||
| Pfenex | Pfenex Inc. | ||||
| Pfizer | Pfizer, Inc. | ||||
| Phoenix Tissue | Phoenix Tissue Repair | ||||
| PSU | Performance stock unit | ||||
| R&D | Research and Development | ||||
| Revolving Credit Facility | The revolving credit facility under the Credit Agreement | ||||
| RSU | Restricted stock unit | ||||
| Sage | Sage Therapeutics, Inc. | ||||
| Sanofi | Sanofi SA | ||||
| SARM | Selective Androgen Receptor Modulator | ||||
| SEC | Securities and Exchange Commission | ||||
| Sedor | Sedor Pharmaceuticals, Inc., or RODES, Inc. | ||||
| Seelos | Seelos Therapeutics, Inc. | ||||
| Selexis | Selexis, SA | ||||
| Sermonix | Sermonix Pharmaceuticals, LLC | ||||
| SII | Serum Institute of India | ||||
| SQ Innovation | SQ Innovation, Inc. | ||||
| Sunshine Lake Pharma | Sunshine Lake Pharma Co., Ltd. | ||||
| Takeda | Takeda Pharmaceuticals Company Limited | ||||
| Tax Act | The Tax Cuts and Jobs Act | ||||
| Teva | Teva Pharmaceuticals USA, Inc., Teva Pharmaceutical Industries Ltd. and Actavis, LLC | ||||
| Travere | Travere Inc. | ||||
| TR-Beta | Thyroid hormone receptor beta | ||||
| Vernalis | Vernalis plc | ||||
| Verona | Verona Pharma plc | ||||
| Viking | Viking Therapeutics | ||||
| Xi'an Xintong | Xi'an Xintong Medicine Research | ||||
| Zydus Cadila | Zydus Cadila Healthcare, Ltd | ||||
PART I
1
Cautionary Note Regarding Forward-Looking Statements:
You should read the following report together with the more detailed information regarding our company, our common stock and our financial statements and notes to those statements appearing elsewhere in this document.
This report contains forward-looking statements that involve a number of risks and uncertainties. Although our forward-looking statements reflect the good faith judgment of our management, these statements can only be based on facts and factors currently known by us. Consequently, these forward-looking statements are inherently subject to risks and uncertainties, and actual results and outcomes may differ materially from results and outcomes discussed in the forward-looking statements.
Forward-looking statements can be identified by the use of forward-looking words such as “believes,” “expects,” “may,” “will,” “plan,” “intends,” “estimates,” “would,” “continue,” “seeks,” “pro forma,” or “anticipates,” or other similar words (including their use in the negative), or by discussions of future matters such as those related to our future results of operations and financial position, royalties and milestones under license agreements, Captisol material sales, product development, and product regulatory filings and approvals, and the timing thereof, Ligand's status as a high-growth company, as well as other statements that are not historical. You should be aware that the occurrence of any of the events discussed under the caption “Risk Factors” could negatively affect our results of operations and financial condition and the trading price of our stock.
The cautionary statements made in this report are intended to be applicable to all related forward-looking statements wherever they may appear in this report. We urge you not to place undue reliance on these forward-looking statements, which speak only as of the date of this report. Except as required by law, we assume no obligation to update our forward-looking statements, even if new information becomes available in the future. This caution is made under the safe harbor provisions of Section 21E of the Securities Exchange Act of 1934, as amended.
References to “Ligand Pharmaceuticals Incorporated,” “Ligand,” the “Company,” “we,” “our” and “us” include Ligand Pharmaceuticals Incorporated and our wholly-owned subsidiaries.
Partner Information
Information regarding partnered products and programs comes from information publicly released by our partners and licensees.
Trademarks
This Annual Report on Form 10-K includes trademarks, trade names and service marks owned by us. Ligand®, BEPro™, Captisol®, CyDex®, LTP®, LTP Technology®, NITRICILTM, and ZELSUVMITM are protected under applicable intellectual property laws and are our property. All other trademarks, trade names and service marks including, but not limited to Pelican Expression Technology®, PeliCRM®, Pfenex Expression Technology®, OmniAb® Kyprolis®, Evomela®, Veklury®, Livogiva®, Bonteo®, Zulresso®, Rylaze®, VAXNEUVANCE™, Pneumosil®, Minnebro®, Baxdela®, Nexterone®, Noxafil®, Duavee®, FILSPARI® and are the property of their respective owners. Solely for convenience, trademarks, trade names and service marks referred to in this report may appear without the ®, ™ or SM symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent under applicable law, our rights or the right of the applicable licensor to such trademarks, trade names and service marks. Use or display by us of other parties’ trademarks, trade dress or products is not intended to and does not imply a relationship with, or endorsement or sponsorship of, us by the trademark or trade dress owners.
2
| Item 1. | Business | ||||
Overview
We are a biopharmaceutical company enabling scientific advancement through supporting the clinical development of high-value medicines. Ligand does this by providing financing, licensing our technologies or both. Our business model seeks to generate value for stockholders by creating a diversified portfolio of biopharmaceutical product revenue streams that are supported by an efficient and low corporate cost structure. Our goal is to offer investors an opportunity to participate in the promise of the biotech industry in a profitable and diversified manner. Our business model focuses on funding programs in mid- to late-stage drug development in return for economic rights, purchasing royalty rights in development stage or commercial biopharmaceutical products and licensing our technology to help partners discover and develop medicines. We partner with other pharmaceutical companies to leverage what they do best (late-stage development, regulatory management and commercialization) in order to generate our revenue. Our Captisol platform technology is a chemically modified cyclodextrin with a structure designed to optimize the solubility and stability of drugs. We have established multiple alliances, licenses and other business relationships with the world’s leading biopharmaceutical companies including Amgen, Merck, Pfizer, Jazz, Takeda, Gilead Sciences and Baxter International.
Our revenue consists of three primary elements: royalties from commercialized products, sales of our Captisol material to partners, and contract revenue from license fees and milestones payments.
Strategy and Execution
We are a biopharmaceutical royalty aggregator, focused on investing in differentiated late-stage assets and operating royalty-generating, infrastructure-light platform technologies. Since our transition to this business model in 2007, we have deployed over $1 billion of capital to build our diverse portfolio. Following the spin-offs of our OmniAb antibody discovery business in November 2022 and the Pelican Expression Technology subsidiary in September 2023, our strategy is to continue to expand our pipeline by aggregating royalty rights in mid- to late-stage development and commercial biopharma products, while maintaining a lean infrastructure and high-margin business.
Our business model is highly differentiated from a traditional biotechnology company in several key ways. First, we have limited infrastructure requirements, enabling us to maintain relative high operating margins. Second, we can enable development over a broad range of therapeutic areas and can be strategic and balanced about the size of our investments to achieve a highly diversified portfolio. Third, our business model mitigates the high volatility associated with building a business around a single or small number of assets. With this approach, we have the ability to mitigate the impact of binary clinical outcomes in the biopharmaceutical industry, thereby facilitating cash flows that are more predictable. Finally, we can target the size of our investments to achieve appropriate risk management across the portfolio.
As an organization, we bring a highly experienced team and financial strength to execute on our strategy. There is high demand for capital and low availability of structured capital in the segment of the biopharmaceutical market in which we operate, creating significant deal flow opportunity for Ligand. Unlike open-market equity investing, many of our investments take place under Confidential Disclosure Agreements (CDAs), allowing us access to in-depth, advantageous diligence materials. Our flexible investment structures are designed to mitigate risks, and also help accommodate different transaction structures based on our partners' goals. We believe our business model is highly scalable and has significant growth potential. We have assembled a talented, long-tenured team with deep industry relationships, investment experience and industry knowledge.
From a more tactical perspective, we execute our strategy using four key approaches: royalty monetization, M&A, project finance, and platform investments. With royalty monetization, we purchase rights on existing royalty contracts that are owned by inventors, academic institutions or companies. There are advantages of royalty investing as a model since royalties 1) have minimal infrastructure, 2) are non-dilutive and 3) their cash flows are often protected in bankruptcy. In M&A investments, we acquire companies with valuable assets or partnerships and realize the value of those assets by restructuring operations and/or partnering the assets. Ligand has a history of doing this successfully with deals such as:
•Pharmacopeia acquisition in 2008 which yielded Travere’s Filspari
•Metabasis acquisition in 2010 which contributed to the creation of Viking Therapeutics
•Vernalis acquisition in 2018 which yielded Verona’s ensifentrine
•Pfenex acquisition in 2020 which yielded four of our major commercial programs – Vaxneuvance, Rylaze, Pneumosil, and Teriparatide as well as our equity interest in Primrose Bio
•Novan acquisition in 2023 which yielded Zelsuvmi
3
Project finance involves the provision of development capital to fund late-stage clinical programs in return for royalty contracts that we negotiate, creating royalties on the future sales of those products. Finally, with platform technology acquisitions, we look for platforms with high operating margins and existing licensing contracts and acquire these targets. The ideal platform will provide new royalties by operating those platforms, and it will be scalable and have broad applicability. Our Captisol business is an excellent example of a successful platform technology investment.

We have a specific set of criteria we use to assess potential investments. The first criteria is time to cash flow, as we seek products that are within a few years of regulatory approval and commercialization. Typically, this means we invest in Phase 3 assets, although we also evaluate opportunities to invest in Phase 2 assets. In terms of an asset's clinical profile, we are looking for strong data supporting both efficacy and safety, and products which will ultimately deliver significant value to patients and to the healthcare system. We also look for strong market exclusivity, which can be achieved through intellectual property and/or regulatory protections. Structural alignment with our counterparty and the marketer is also a key criteria of the investments we make. Ultimately, we look for assets with favorable risk-reward profiles, which have above average probability of technical and regulatory success and can be commercialized effectively.
4

Technologies
Through a combination of research and acquisitions, we have created a partnered portfolio with a wide variety of underlying technologies. This diversification provides the added benefits of exposure to a wider breadth of scientific innovation, more licensing opportunities and lower impact of individual patent expiry.
Captisol Technology
Captisol is a patent-protected, chemically modified cyclodextrin with a structure designed to optimize the solubility and stability of drugs. This unique technology has enabled several FDA-approved products, including Gilead’s Veklury, Amgen’s Kyprolis, Baxter International’s Nexterone, Acrotech Biopharma’s and CASI Pharmaceuticals’ Evomela, Melinta Therapeutics’ Baxdela and Sage Therapeutics’ Zulresso. There are many Captisol-enabled products currently in various stages of development. We maintain a broad global patent portfolio for Captisol with the latest expiration date in 2035. Other patent applications covering methods of making Captisol, if issued, extend to 2041.
In addition to solid Captisol powder, we offer our partners access to cGMP manufactured aqueous Captisol concentrate. This product offering was established in 2017 to reduce cycle time and increase Captisol production capacity for large-volume drug products. We maintain both Type IV and Type V drug master files (DMFs) with the FDA. These DMFs contain manufacturing and safety information relating to Captisol that our licensees can reference when developing Captisol-enabled drugs. We also have active DMFs in Japan, China and Canada. In 2023, royalties on commercial products using Captisol comprised over half of our total royalty revenue.
HepDirect, LTP and BEPro Technology Platform
The HepDirect and LTP platforms are our proprietary liver-targeting prodrug technologies that can deliver many different chemical classes of drugs to the liver by using a chemical modification that renders an active pharmaceutical ingredient (API) biologically inactive until cleaved by a liver-specific enzyme. These technologies may improve the efficacy and/or safety of certain drugs and can be applied to marketed or new drug products to treat liver diseases or diseases caused by hemostasis imbalance of circulating molecules controlled by the liver.
The BEPro technology platform is a next generation prodrug technology distinct from HepDirect and LTP prodrug technologies, expanding use to non-liver related diseases. BEPro is specifically applicable to nucleotides and nucleotide analogs for the development of compounds with improved product profiles. Ligand has demonstrated benefits in cell penetration and oral, intravenous and inhaled pharmacokinetics with BEPro-enabled nucleotide analogs.
SUREtechnology Platform (owned by Selexis)
5
We acquired economic rights to various SUREtechnology Platform programs from Selexis. The SUREtechnology Platform, developed and owned by Selexis, is a novel technology that improves the way that cells are utilized in the development and manufacturing of recombinant proteins and drugs.
Pelican Expression Technology (owned by Primrose Bio, of which Ligand owns 49.9%)
The Pelican Expression Technology platform is a robust, validated, cost-effective and scalable platform for recombinant protein production, and is especially well suited for complex, large-scale proteins. Global manufacturers have demonstrated consistent success with the platform and the technology is currently outlicensed for multiple commercial and development-stage programs. The versatility of the platform has been demonstrated in the production of enzymes, peptides, antibody derivatives and engineered non-natural proteins. The platform contributes significant value to biopharmaceutical development programs by shortening timelines and reducing costs associated with research and development through commercial manufacturing of therapeutics and vaccines. Given pharmaceutical industry trends toward large molecules with increased structural complexities, the Pelican Expression Technology platform is well positioned to meet these growing needs as one of the most comprehensive and broadly available, commercially validated protein production platforms in the industry.
2023 Investment Highlights
In September 2023, we announced the sale of our Pelican business, inclusive of the Pelican Expression Technology platform, and a merger of Pelican with Primordial Genetics to form a new company, Primrose Bio. As part of the investment, Ligand retained the existing commercial royalties related to the Pelican Expression Technology platform and owns 49.9% of Primrose Bio. We also entered into a purchase and sale agreement with Primrose, whereby we invested $15 million in exchange for a portion of the economic rights from the two existing contracts of Primordial Genetics and an economic interest in potential future revenues generated from the Pelican business. Ligand retains the pre-spin royalty rights from the Pelican Expression Technology, including economic rights to Jazz’s RYLAZE, Merck’s VAXNEUVANCE and V116 vaccines, Alvogen’s Teriparatide, Serum Institute of India’s Pneumosil and MenFive vaccines, among others. We had originally acquired the Pelican business through the acquisition of Pfenex in 2020. After incubating this technology for three years, we now have five commercial royalty streams from the platform, and with the spinoff and merger, we retain a significant equity stake. We consider the acquisition of Pfenex and spin-off of Pelican Technology Holdings, Inc. to be a very successful transaction and expect these assets will continue to generate significant revenues for the Company.
In October 2023, we announced an investment of $30 million to acquire a 13% portion of the royalties and milestones owed to Ovid Therapeutics related to the potential approval and commercialization of soticlestat. Soticlestat is a Phase 3, first-in-class, novel mechanism of action molecule being studied by Takeda in two rare pediatric epilepsies: Lennox-Gastaut syndrome (LGS) and Dravet syndrome. Takeda is one of the world's leading pharmaceutical companies in neurology and rare diseases. LGS and Dravet are two very difficult treat conditions, with high unmet clinical needs despite having a few products that have been recently approved. Takeda has stated it anticipates regulatory filings for soticlestat in its fiscal year 2024. If regulatory approval is granted, commercialization and royalties to Ligand could begin a year later.
In November 2023, we closed a $20 million acquisition of Tolerance Therapeutics, a holding company owned by the inventors of TZIELD (teplizumab). TZIELD is the first disease-modifying therapy approved to treat patients with type-1 diabetes (T1D). It is a CD3-directed antibody indicated to delay the onset of stage-3 T1D in adults and in children aged eight years and older with stage-2 T1D. TZIELD was granted breakthrough therapy designation by the FDA in 2019 and was approved by the FDA in November 2022. TZIELD is marketed by Sanofi following its $2.9 billion acquisition of Provention Bio in 2023. Sanofi recently announced new data from the TZIELD PROTECT Phase 3 trial, which showed TZIELD’s potential to slow the progression of stage 3 T1D in newly diagnosed children and adolescents. These findings were published in the New England Journal of Medicine. Sanofi has a robust history in the diabetes space. Sanofi has been featuring TZIELD as one of their key launches with significant blockbuster potential. Ligand is owed a royalty of less than 1% on worldwide net sales of TZIELD.
Novan Acquisition
In September 2023, the bankruptcy court approved a $12.2 million bid from Ligand to purchase certain assets of Novan, Inc., including berdazimer gel, all assets related to the NITRICIL™ technology platform and the rights to one commercial stage asset. Prior to Novan's bankruptcy, we had a royalty interest in berdazimer topical gel, 10.3%. Berdazimer topical gel, 10.3% was approved by the FDA in January 2024, with a brand name of ZELSUVMI.
ZELSUVMI™ (berdazimer) topical gel, 10.3% is a first-in-class topical medication for the treatment of molluscum contagiosum in adults and pediatric patients one year of age or older. The FDA approved ZELSUVMI as the novel drug for the treatment of molluscum infections. ZELSUVMI is the first and only topical prescription medication that can be applied by patients, parents, or caregivers at home, outside of a physician's office, or other medical setting to treat this highly contagious viral skin infection.
6
As we incubate this newly acquired business, the Novan team is actively preparing for commercialization. Consistent with our business model, we are engaging with potential commercial partners to maximize the value for Ligand shareholders through a strategic transaction.
Commercial and Clinical Stage Partnered Portfolio
We have a large portfolio of assets currently generating royalties and future potential revenue-generating programs, including over 85 fully-funded by our partners.
Royalties on Commercial Products
The following table provides an overview of our current portfolio of royalties:
| Product | Partner | Therapeutic Area | Royalty Rate | 2023 Royalty Revenue (in millions) | Estimated 2023 Product Revenue (in millions) | ||||||||||||
| Kyprolis | Amgen/Ono/Beigene | Cancer | 1.5% - 3.0% | $35.6 | $1,503.1 | ||||||||||||
| Rylaze | Jazz | Cancer | Low single digit | $13.5 | $397.5 | ||||||||||||
| Teriparatide | Alvogen | Women's Health | 25%-40%¹ | $11.1 | $37.2 | ||||||||||||
| Evomela | Acrotech/CASI | Cancer | 20% | $10.2 | $51.0 | ||||||||||||
| Vaxneuvance | Merck | Infectious Disease | Low single digit | $4.1 | $653.9 | ||||||||||||
| Pneumosil | Serum Institute | Infectious Disease | Low single digit | $4.5 | $198.5 | ||||||||||||
| Filspari | Travere | IgA Nephropathy | 9% | $2.7 | $29.5 | ||||||||||||
| Nexterone | Baxter | Cardiovascular | Low single digit | $1.5 | $50.9 | ||||||||||||
| Other | Various | Various | Various | $0.7 | $23.6 | ||||||||||||
(1) We receive tiered profit sharing of 25% on quarterly profits less than $3.75 million, 35% on quarterly profits greater than $3.75 million but less than $7.5 million and 40% on quarterly profits greater than $7.5 million. If therapeutic equivalence is achieved, quarterly profit changes to 50% of quarterly profits.
Key Partnered Commercial Programs
The following programs represent important revenue-generating components of our current portfolio. For information about the royalties owed to us for certain of these programs, see “Royalties” later in this business section.
Kyprolis (Amgen, Ono, BeiGene)
We supply Captisol to Amgen for use with Kyprolis (carfilzomib) and granted Amgen an exclusive product-specific license under our patent rights with respect to Captisol. Kyprolis is formulated with Ligand’s Captisol technology and is approved in the United States for the following:
•In combination with dexamethasone, lenalidomide plus dexamethasone, daratumumab plus dexamethasone, or daratumumab and hyaluronidase-fihj and dexamethasone, or isatuximab and dexamethasone for the treatment of patients with relapsed or refractory multiple myeloma who have received one to three lines of therapy.
•As a single agent for the treatment of patients with relapsed or refractory multiple myeloma who have received one or more lines of therapy.
Our agreement with Amgen may be terminated by either party in the event of material breach or bankruptcy, or unilaterally by Amgen with prior written notice, subject to certain surviving obligations. Absent early termination, the agreement will terminate upon expiration of the obligation to pay royalties. Under this agreement, we are entitled to receive revenue from clinical and commercial Captisol material sales and royalties on annual net sales of Kyprolis based on our patents and applications relating to the Captisol component of Kyprolis which are not expected to expire until 2033.
Rylaze (Jazz Pharmaceuticals)
In July 2021, Jazz announced the U.S. launch of RYLAZE (asparaginase erwinia chrysanthemi (recombinant)-rywn), previously referred to as JZP458. RYLAZE, which was approved by the FDA in June 2021, is a recombinant erwinia
7
asparaginase used as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma (LBL) in adult and pediatric patients one month or older who have developed hypersensitivity to E. coli-derived asparaginase. In September 2023, Jazz announced that the European Commission (EC) had granted marketing authorization for RYLAZE, to be marketed as Enrylaze®. Jazz began a rolling launch in the second half of 2023. Additionally, Jazz is utilizing our technology for the development of PF745 (JZP341), a long-acting Erwinia asparaginase for the treatment of ALL and other hematological malignancies. Jazz has worldwide rights to develop and commercialize PF745.
Ligand is eligible to receive up to $152 million in milestone payments and tiered low-single digit royalties based on worldwide net sales of any products resulting from this collaboration, including Rylaze.
Filspari (Travere)
In early 2012, Ligand licensed the world-wide rights to sparsentan to Travere Therapeutics. Travere recently received FDA accelerated approval for FILSPARI (sparsentan) for the treatment of immunoglobulin A nephropathy (IgAN). FILSPARI is the first and only dual endothelin and angiotensin II receptor antagonist in development for rare kidney diseases and is the first non-immunosuppressive treatment indicated for IgAN. Travere anticipates a review opinion by the Committee for Medicinal Products for Human Use (CHMP) on the potential approval for sparsentan for the treatment of IgAN in Europe in the first quarter of 2024. Additionally, Travere is on track to submit a supplemental New Drug Application (sNDA) for the conversion of the existing U.S. accelerated approval in IgAN to full approval.
Travere also completed regulatory engagement on focal segmental glomerulosclerosis (FSGS) in which the FDA communicated that the Phase 3 DUPLEX study results alone are not sufficient to support an sNDA submission for an FSGS indication for sparsentan. As a result, Travere plans to conduct additional analyses of FSGS data with plans to re-engage the FDA in 2024, and implemented a strategic reorganization in the fourth quarter of 2023 to focus near-term resources on the ongoing FILSPARI launch in IgAN.
Under our license agreement with Travere, we are entitled to receive over $50 million in potential milestone payments, as well as a 9% royalty on any future worldwide sales.
Teriparatide Injection Product (PF708) (Alvogen/Adalvo)
We acquired the teriparatide injection product with the acquisition of Pfenex in October 2020. Teriparatide injection is a drug indicated for various uses including the treatment of osteoporosis in certain patients at high risk for fracture. Teriparatide injection was developed using our Pelican Expression Technology and was approved by the FDA in 2019 in accordance with the 505(b)(2) regulatory pathway, with FORTEO as the reference product. Our commercialization partner, Alvogen, launched the product in June 2020 in the United States.
Our partner, Alvogen, has exclusively licensed the rights to commercialize and manufacture the teriparatide injection product in the United States, while Adalvo has the rights to commercialize in the EU, certain countries in the Middle East and North Africa (MENA), and the rest of world (ROW) territories (the latter defined as all countries outside of the EU, U.S. and MENA, excluding Mainland China, Hong Kong, Singapore, Malaysia and Thailand). In August 2020, marketing authorization throughout the EU was received under the trade name Livogiva and in December 2020 in Saudi Arabia under the name Bonteo. In December of 2022, we terminated a license agreement with Beijing Kangchen Biological Technology Co., Ltd. (Kangchen) thereby regaining the right to commercialize PF708 in Mainland China, Hong Kong, Singapore, Malaysia and Thailand along with a non-exclusive right to conduct development activities in such countries with respect to PF708.
In accordance with our agreements with Alvogen, we are eligible to receive tiered gross profit sharing of between 25% and 40% of quarterly profits prior to an “A” therapeutic equivalence designation, which increases to a flat 50% if an “A” rating is achieved.
In accordance with our EU, MENA and ROW agreements with Adalvo, we may be eligible to receive additional upfront and milestone payments of $1.5 million and may also be eligible to receive up to 60% of gross profit derived from product sales and regional license fees, if approved, depending on geography, cost of goods sold and sublicense fees.
Evomela (Acrotech and CASI)
We supply Captisol to, and receive royalties from, Acrotech Biopharma for sales of Evomela in the U.S., and CASI Pharmaceuticals for sales in China. Evomela received marketing approval by the NMPA in August of 2019. It is the only approved and commercially available melphalan product in China. Evomela is a Captisol-enabled melphalan IV formulation which is approved by the FDA for use in two indications:
•a high-dose conditioning treatment prior to autologous stem cell transplantation (ASCT) in patients with multiple myeloma; and
•for the palliative treatment of patients with multiple myeloma for whom oral therapy is not appropriate.
8
Evomela has been granted Orphan Designation by the FDA for use as a high-dose conditioning regimen for patients with multiple myeloma undergoing ASCT. The Evomela formulation avoids the use of propylene glycol, which has been reported to cause renal and cardiac side-effects that limit the ability to deliver higher quantities of therapeutic compounds. The use of the Captisol technology to reformulate melphalan is anticipated to allow for longer administration durations and slower infusion rates, potentially enabling clinicians to safely achieve a higher dose intensity of pre-transplant chemotherapy.
Under the terms of the license agreement, Acrotech Biopharma has marketing rights worldwide excluding China and CASI Pharmaceuticals has rights to market in China. We are eligible to receive over $50 million in potential milestone payments, royalties on global net sales of the Captisol-enabled melphalan product and revenue from Captisol material sales. Acrotech and CASI’s obligation to pay royalties will expire at the end of the life of the relevant patents or when a competing product is launched, whichever is earlier, but in no event before ten years after the commercial launch. Our patents and applications relating to the Captisol component of melphalan are not expected to expire until 2033. As described herein, we have entered into a settlement agreement with Teva and Acrotech Biopharma (the holder of the NDA for Evomela) which will allow Teva to market a generic version of Evomela in the United States in 2026, or earlier under certain circumstances. Absent early termination, the agreement will terminate upon expiration of the obligation to pay royalties. The agreement may be terminated by either party for an uncured material breach or unilaterally by Acrotech and CASI by prior written notice.
VAXNEUVANCE (Merck)
VAXNEUVANCE, a 15-valent pneumococcal conjugate vaccine, also known as V114, was approved in the U.S. in July of 2021 for the prevention of invasive disease caused by Streptococcus pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F and 33F in adults 18 years of age and older, and subsequently in children 6 weeks through 17 years of age in June of 2022. VAXNEUVANCE was also approved in Europe in October 2022 for the prevention of invasive disease and pneumonia caused by Streptococcus pneumoniae in individuals 18 years and older and in infants, children and adolescents from 6 weeks to less than 18 years of age. VAXNEUVANCE utilizes CRM197 vaccine carrier protein, which is produced using the patent-protected Pelican Expression Technology™ platform. We are entitled to low single digit royalties derived from net sales of Vaxneuvance.
In December 2023, Merck announced the FDA accepted for priority review a new BLA for V116, Merck's investigational 21-valent pneumococcal conjugate vaccine specifically designed to help prevent invasive pneumococcal disease and pneumococcal pneumonia in adults. The FDA grants priority review to medicines and vaccines that, if approved, would provide a significant improvement in the safety or effectiveness of the treatment or prevention of a serious condition. The FDA has set a PDUFA date, or target action date, of June 17, 2024. If approved, Ligand is entitled to a royalty on worldwide net sales.
Pneumosil (Serum Institute of India, SII)
SII began commercialization of its 10-valent pneumococcal conjugate vaccine, Pneumosil, which is produced using CRM197 made in the Pelican Expression Technology platform, in the second quarter of 2020. Pneumosil is designed primarily to help fight against pneumococcal pneumonia among children, with an advantage of targeting the most prevalent serotypes of the bacterium causing serious illness in developing countries. Pneumosil achieved WHO Prequalification in December 2019, allowing the product to be procured by United Nations agencies and Gavi, the Vaccine Alliance, and subsequently achieved Indian Marketing Authorization in July 2020, and SII announced commercial launch of the product in India in December 2020.
TZIELD (Sanofi)
We acquired a royalty of less than 1% on net sales of TZIELD through our acquisition of Tolerance Therapeutics in the fourth quarter of 2023. TZIELD is the first disease-modifying therapy to be approved in type 1 diabetes (“T1D”). It is a CD3-directed antibody indicated to delay the onset of Stage 3 T1D in adults and children aged 8 years and older with Stage 2 T1D. TZIELD was granted Breakthrough Therapy Designation in 2019 and was approved by the FDA in November 2022. TZIELD is marketed by Sanofi, following its acquisition of Provention Bio, Inc., the developer of TZIELD, in 2023 for $2.9 billion. Sanofi recently announced new data from TZIELD’s PROTECT Phase 3 trial which showed TZIELD’s potential to slow the progression of Stage 3 T1D in newly diagnosed children and adolescents. TZIELD met the study’s primary endpoint, significantly slowing the decline of C-peptide levels, compared to placebo. Under our agreement with Tolerance, we are entitled to receive royalties through December 1, 2032.
Nexterone (Baxter)
We have a license agreement with Baxter, related to Baxter's Nexterone, a Captisol-enabled formulation of amiodarone, which is marketed in the United States and Canada. We supply Captisol to Baxter for use in accordance with the terms of the license agreement under a separate supply agreement. Under the terms of the license agreement, we will continue to earn milestone payments, royalties, and revenue from Captisol material sales. We will earn royalties on net sales of Nexterone through early 2033.
Veklury (Gilead)
9
We supply Captisol to Gilead for sales of Veklury (remdesivir). Gilead received marketing approval from the FDA in October 2020. Veklury is an antiviral treatment for COVID-19. The product has regulatory approvals for the treatment of moderate or severe COVID-19 in over 70 countries. We are supplying Captisol to Gilead under a 10-year supply agreement. We are also supplying Captisol to Gilead’s voluntary licensing generic partners who are manufacturing remdesivir for 127 low- and middle-income countries. We receive our commercial compensation for this program through the sale of Captisol.
Zulresso (Sage)
We have a license agreement with Sage, related to Sage's Zulresso, a Captisol-enabled formulation of brexanolone for the treatment of postpartum depression (PPD). Under the terms of the agreement, we receive royalties and revenue from Captisol material sales.
Noxafil-IV (Merck)
We have a supply agreement with Merck related to Merck’s NOXAFIL-IV, a Captisol-enabled formulation of posaconazole for IV use. NOXAFIL-IV is marketed in the United States, EU, Japan and Canada. We receive our commercial compensation for this program through the sale of Captisol.
Duavee or Duavive (Pfizer)
Pfizer is responsible for the marketing of bazedoxifene, a synthetic drug specifically designed to reduce the risk of osteoporotic fractures while also protecting uterine tissue. Pfizer has combined bazedoxifene with the active ingredient in Premarin to create a combination therapy for the treatment of post-menopausal symptoms in women. Pfizer is marketing the combination treatment under the brand names Duavee and Duavive in various territories. Net royalties on annual net sales of Duavee/Duavive are payable to us through the life of the relevant patent or ten years from the first commercial sale, whichever is longer, on a country-by-country basis.
Exemptia, Vivitra, Bryxta and Zybev (Zydus Cadila)
Zydus Cadila’s Exemptia (adalimumab biosimilar) is marketed in India for autoimmune diseases. Zydus Cadila uses the Selexis technology platform for Exemptia. We earn royalties on sales by Zydus Cadila for ten years following approval.
Zydus Cadila’s Vivitra (trastuzumab biosimilar) is marketed in India for breast cancer. Zydus Cadila uses the Selexis technology platform for Vivitra. We are entitled to earn royalties on sales by Zydus Cadila for ten years following approval.
Zydus Cadila’s Bryxta and Zybev (bevacizumab biosimilar) is marketed in India for various indications. Zydus Cadila uses the Selexis technology platform for Bryxta and Zybev. We earn royalties on sales by Zydus Cadila for ten years following approval.
FYCOMPA IV (Eisai)
Our partner, Eisai, is developing an intravenous Fycompa® (perampanel), formulated with Captisol, as a substitute in Japan for oral tablets as an adjunctive therapy in patients with partial onset seizures (including secondarily generalized seizures) or primary generalized tonic-clonic seizures. In January of 2023, Eisai announced that it obtained marketing authorization approval from the Japanese Ministry of Health, Labour and Welfare for the injection formulation of its in-house discovered antiepileptic drug (AED) Fycompa in Japan as an alternative therapy when oral administration is temporarily not possible. We are entitled to revenue from Captisol material sales and tiered royalties on potential future sales.
Key Partnered Pipeline Programs
We have a highly diversified partnered pipeline of development stage assets that either have or are nearing regulatory approval, or given the area of research or value of the license terms, we consider particularly noteworthy. We are eligible to receive milestone payments and royalties on these programs. This list does not include all of our partnered programs. In the case of Captisol-related programs, we are also eligible to receive revenue for the sale of Captisol material supply. The following table represents development-stage assets with disclosed royalty rates:
10
| Development stage assets with disclosed royalties | ||||||||
| Program | Licensee | Royalty Rate | ||||||
| Ciforadenant | Corvus | Mid-single digit to low-teen royalty | ||||||
| DGAT-1 | Viking | 3.0% - 7.0% | ||||||
| Ensifentrine (RPL554) | Verona | Low single digit royalty | ||||||
| FBPase Inhibitor (VK0612) | Viking | 7.5% - 9.5% | ||||||
| Lasofoxifene | Sermonix | 6.0% - 10.0% | ||||||
| MB07133 | Xi'an Xintong | 6% | ||||||
| ME-344 | MEI Pharma | Low single digit royalty | ||||||
| Oral EPO | Viking | 4.5% - 8.5% | ||||||
| Pradefovir | Xi'an Xintong | 9% | ||||||
| PTX-022 | Palvella | 8.0% - 9.8% | ||||||
| SARM (VK5211) | Viking | 7.25% - 9.25% | ||||||
| TR Beta (VK2809 and VK0214) | Viking | 3.5% - 7.5% | ||||||
| Various | Nucorion | 4.0% - 9.0% | ||||||
| Various | Seelos | 4.0% - 10.0% | ||||||
TR-Beta - VK2809 and VK0214 (Viking)
Our partner, Viking, is developing VK2809, a novel selective thyroid hormone receptor beta (TR-beta) agonist with potential in multiple indications, including hypercholesterolemia, dyslipidemia and non-alcoholic steatohepatitis (NASH). VK2809 is currently in a Phase 2b clinical trial (the VOYAGE study) in patients with biopsy-confirmed NASH. VK0214, another novel, orally available, TR-beta agonist, is in development for the potential treatment of X-linked adrenoleukodystrophy (X-ALD). VK0214 is currently being evaluated in a Phase 1b clinical trial in patients with the adrenomyeloneuropathy (AMN) form of X-ALD. Under the terms of the agreement with Viking, we may be entitled to up to $375 million of development, regulatory and commercial milestones and tiered royalties on potential future sales. Our TR Beta programs partnered with Viking are subject to CVR sharing and a portion of the cash received will be paid out to CVR holders.
In November 2023, Viking presented new results from the ongoing Phase 2b clinical trial of VK2809, a novel liver-selective thyroid hormone receptor beta agonist, in patients with biopsy-confirmed NASH. The latest findings from the VOYAGE study were featured in a late breaking poster presentation at the Liver Meeting® 2023, the annual meeting of the American Association for the Study of Liver Diseases. The newly reported findings demonstrated robust and comparable liver fat reductions in patients with or without Type 2 diabetes, as well as patients with either F2 or F3 fibrosis.
Ensifentrine – RPL554 (Verona)
Ensifentrine is a first-in-class, selective, dual inhibitor of phosphodiesterase 3 and 4 enzymes combining bronchodilator and non-steroidal anti-inflammatory activities in one compound. Ligand obtained the rights to ensifentrine in 2018 in the acquisition of Vernalis. Our partner, Verona Pharma, recently completed the Phase 3 ENHANCE-21 and ENHANCE-12 trials evaluating nebulized ensifentrine for the maintenance treatment of chronic obstructive pulmonary disease (COPD) and the NDA was accepted by the U.S. FDA in September 2023. The PDUFA date for ensifentrine is June 26, 2024. Under the terms of our agreement with Verona, we are entitled to development and regulatory milestones, including a £5.0 million payment upon the first approval by any regulatory authority, and low single digit royalties on potential future sales.
Verona recently announced that it has entered into a debt financing facility providing the company with access to up to $400 million from funds managed by Oxford Finance LLC and Hercules Capital, Inc. The debt facility provides non-dilutive capital and further financial flexibility to support Verona Pharma’s continued growth, including the planned commercial launch of ensifentrine.. The debt facility replaces the existing facility of up to $150 million with an affiliate of Oxford.
Soticlestat (Takeda)
In the fourth quarter of 2023, we entered into an agreement with Ovid Therapeutics for a 13% interest in soticlestat royalties. Ovid sold its rights in soticlestat to Takeda in 2021. Under the terms of its agreement with Takeda, Ovid is eligible to receive regulatory and commercial milestone payments of up to $660 million, as well as tiered royalties on net sales of soticlestat at percentages ranging from the low-double-digits up to 20%, if soticlestat is approved and successfully commercialized. Takeda is currently studying soticlestat in two pivotal Phase 3 trials in people with Lennox-Gastaut syndrome (LGS) and Dravet
11
syndrome (DS) and announced that it anticipates regulatory filings for soticlestat in its fiscal year 2024. Ovid has no ongoing obligations or costs associated with the development of soticlestat.
Soticlestat is a potent, highly selective, first-in-class inhibitor of the enzyme cholesterol 24-hydroxylase (CH24H), with the potential to reduce seizure susceptibility and improve seizure control. CH24H is predominantly expressed in the brain, where it converts cholesterol into 24S-hydroxycholesterol (24HC) to adjust the homeostatic balance of brain cholesterol. 24HC is a positive allosteric modulator of the NMDA receptor and modulates glutamatergic signaling associated with epilepsy. Glutamate is one of the main neurotransmitters in the brain and has been shown to play a role in the initiation and spread of seizure activity. Recent literature indicates that CH24H is involved in over-activation of the glutamatergic pathway through modulation of the NMDA channel and that increased expression of CH24H can disrupt the reuptake of glutamate by astrocytes, resulting in epileptogenesis and neurotoxicity. Inhibition of CH24H by soticlestat reduces the neuronal levels of 24HC and may improve excitatory/inhibitory balance of NMDA channel activity.
Under the terms of our agreement with Ovid, we are entitled to receive regulatory and sales-based milestones of up to $85.8 million as well as 13% of royalties received by Ovid.
Ganaxalone IV (Marinus)
Our partner, Marinus, is conducting Phase 3 clinical trials with Captisol-enabled ganaxolone IV in patients with refractory status epilepticus. Marinus has exclusive worldwide rights to Captisol-enabled ganaxolone, a GABAA receptor modulator, for use in humans. We are entitled to development and regulatory milestones, revenue from Captisol material sales, and royalties on potential future sales.
Ciforadenant – CPI-444 (Corvus)
Our partner, Corvus, is conducting a Phase 1b/2 clinical trial evaluating ciforadenant as a potential first line therapy for metastatic renal cell cancer (RCC) in combination with ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1). The Phase 1b/2 study is being conducted by the Kidney Cancer Research Consortium (KCRC) and is led by The University of Texas MD Anderson Cancer Center. Under the terms of our agreement with Corvus, we are entitled to development and regulatory milestones and tiered royalties on potential future sales. The aggregate potential milestone payments from Corvus are approximately $220 million for all indications.
QTORIN (Palvella)
We acquired the economic rights to QTORIN™ 3.9% rapamycin anhydrous gel (QTORIN™ rapamycin, formerly PTX-022) from Palvella in December 2018. QTORIN™ rapamycin is a novel, topical formulation of high-strength rapamycin currently in development for the treatment of Microcystic Lymphatic Malformations (Microcystic LM). In November 2023, Palvella announced that the FDA granted Breakthrough Therapy Designation to QTORIN rapamycin for the treatment of microcystic LMs. Microcystic LMs is a chronically debilitating and lifelong genetic disease affecting an estimated more than 30,000 patients in the U.S. There are currently no FDA-approved treatments for microcystic LMs.
Lasofoxifene (Sermonix)
Lasofoxifene is a selective estrogen receptor modulator for osteoporosis treatment and other diseases, discovered through the research collaboration between Pfizer and Ligand. Our partner, Sermonix has a license for the development of oral lasofoxifene, its lead investigational drug, for the United States and additional territories. Sermonix is currently conducting the Phase 3 ELAINE-3 clinical trial to assess the efficacy of lasofoxifene in combination with Eli Lilly and Company’s CDK4/6 inhibitor abemaciclib (Verzenio®) compared to fulvestrant and abemaciclib in pre- and post-menopausal subjects with locally advanced or metastatic ER+/HER2- breast cancer with an ESR1 mutation. Under the terms of the agreement, we are entitled to receive over $45 million in potential regulatory and commercial milestone payments as well as royalties on potential future net sales.
In January 2024, Sermonix announced it entered into a strategic collaboration and exclusive license agreement with Henlius for the rights to develop, manufacture and commercialize lasofoxifene in China. Under the terms of the agreement, Henlius will receive exclusive rights and sublicenses to lasofoxifene for at least two estrogen receptor-positive (ER+)/HER2- breast cancer indications in the territory, with Sermonix retaining all other global rights. Sermonix plans to work with Henlius to accelerate the clinical development of the Phase 3 ELAINE-3 multi-regional clinical trial in China, making lasofoxifene available to Chinese patients as soon as possible. In December 2023, Sermonix activated and began enrollment for ELAINE-3 in the United States.
Pradefovir (Xi'an Xintong)
Our Chinese licensee, Xi'an Xintong Medicine Research (following its acquisition of Chiva Pharmaceuticals), is developing pradefovir, an oral liver-targeting prodrug of the HBV DNA polymerase/reverse transcriptase inhibitor adefovir, for the potential treatment of HBV infection. Pradefovir was developed using Ligand’s HepDirect technology. Xi'an Xintong
12
submitted the pradefovir NDA in May 2023, and it is under priority review by the Chinese FDA (NMPA). We are entitled to an annual licensing maintenance fee and royalties on potential future sales.
MB07133 (Xi'an Xintong)
Chinese licensee Xi'an Xintong Medicine Research is also developing MB07133, a liver specific, HepDirect prodrug of cytarabine monophosphate, for the potential treatment of hepatocellular carcinoma and intrahepatic cholangiocarcinoma. MB07133 is currently in Phase 2 in China. We are entitled to an annual licensing maintenance fee and royalties on potential future sales.
Milestone Payments
Our programs under license with our partners may generate milestone payments to us if our partners reach certain development, regulatory and commercial milestones. The following table represents the maximum value of our milestone payment pipeline by technology, development stage and partner (in millions):
| Technology* | Stage* | Partner* | |||||||||||||||||||||
| Pelican | >$195.0 | Preclinical | > $1.0 | Viking | $950.0 | ||||||||||||||||||
| Captisol | > $170.0 | Clinical | > $80.0 | Jazz | $150.0 | ||||||||||||||||||
| LTP/Hep Direct/BEPro | > $330.0 | Regulatory | > $800.0 | Travere | $50.0 | ||||||||||||||||||
| NCE/Other | > $1,200.0 | Commercial | > $900.0 | Other | >$750.0 | ||||||||||||||||||
| Total | >$1,900.0 | Total | >$1,900.0 | Total | >$1,900.0 | ||||||||||||||||||
*All tables exclude any annual access fees and collaboration revenue for development work.
Full Portfolio Details
We have assembled one of the largest portfolios of biopharmaceutical assets in the industry which provides investors the opportunity to participate in the biotech industry while mitigating the industry’s usual inherent clinical binary risks. Our portfolio consists of assets which currently generate revenue through royalties on commercial products as well as Captisol sales on commercial products. In addition to these assets, we have a substantial pipeline of development stage assets that currently generate contractual payments through milestone and license fees with future potential for royalties and Captisol material sales for those programs under our Captisol technology.
| Approved | ||||||||
| Partner Name | Program | Therapeutic Area | ||||||
| Acrotech/CASI | Evomela | Cancer | ||||||
| Alvogen/Adalvo | Teriparatide | Women's Health | ||||||
| Alvogen/Hikma/Nanjing King-Friend | Voriconazole | Infectious Disease | ||||||
| Amgen/Beigene/Ono | Kyprolis | Cancer | ||||||
| Baxter | Nexterone | Cardiovascular | ||||||
| Biocad | Teberif | Inflammatory/Metabolic | ||||||
| Eisai | FYCOMPA | Central Nervous System | ||||||
| Elutia | ECM portfolio | Medical device/Cardiology | ||||||
| Exelixis/Daiichi-Sankyo | Minnebro | Cardiovascular | ||||||
| Gilead | Veklury | Infectious Disease | ||||||
| Ingenus | ML-141 | Cancer | ||||||
| Jazz | Rylaze | Cancer | ||||||
| Melinta | Baxdela | Infectious Disease | ||||||
| Menarini | Frovatriptan | Central Nervous System | ||||||
| Fareva | Noxafil-IV | Infectious Disease | ||||||
| Merck | Vaxneuvance | Infectious Disease | ||||||
| Novan | SB206 | Infectious Disease | ||||||
13
| Novartis | Mekinist | Cancer | ||||||
| Par | Posaconazole | Infectious Disease | ||||||
| Pfizer | Duavee | Inflammatory/Metabolic | ||||||
| Pfizer | Vfend-IV | Infectious Disease | ||||||
| Sage | Zulresso | Central Nervous System | ||||||
| Sanofi | Tzield | Metabolic | ||||||
| Sedor/Lupin | Sesquient | Central Nervous System | ||||||
| Serum Institute of India | Pneumosil | Infectious Disease | ||||||
| Serum Institute of India | Meningococcal | Infectious Disease | ||||||
| Travere | Filspari | Metabolic | ||||||
| Zydus Cadila | Vivitra | Cancer | ||||||
| Zydus Cadila | Bryxta/ZyBev | Cancer | ||||||
| Zydus Cadila | Maropitant | Central Nervous System | ||||||
| Zydus Cadila | Exemptia | Inflammatory/Metabolic | ||||||
| Zydus Cadila | Vortuxi | Inflammatory/Metabolic | ||||||
| Phase 3/Pivotal or Regulatory Submission Stage | ||||||||
| Partner Name | Program | Therapeutic Area | ||||||
| Aldeyra | Reproxalap | Other/Undisclosed | ||||||
| BendaRx | Bendamustine | Oncology | ||||||
| Marinus | Ganaxalone IV | Central Nervous System | ||||||
| Merck | V116 | Pneumococcal adult | ||||||
| Ohara Pharmaceuticals | JPH203 | Cancer | ||||||
| Opthea | OPT-302 | Ophthalmology | ||||||
| Outlook Therapeutics | ONS-5010 | Other/Undisclosed | ||||||
| Palvella | PTX-022 | Other/Undisclosed | ||||||
| Sermonix | Lasofoxifene | Cancer | ||||||
| SQ Innovation | CE-Furosemide | Cardiovascular disease | ||||||
| Sunshine Lake | Vilazodone | Central Nervous System | ||||||
| Takeda | Soticlestat | Central Nervous System | ||||||
| Verona | Ensifentrine (RPL554) | Respiratory Disease | ||||||
| Xi'an Xintong | Pradefovir | Infectious Disease | ||||||
| Phase 2 | ||||||||
| Partner Name | Program | Therapeutic Area | ||||||
| Acrivon | ACR-368 | Cancer | ||||||
| Anebulo | ANEB-001 | Central Nervous System | ||||||
| Corvus | Ciforadenant | Cancer | ||||||
| CurX | CE-Topiramate | Central Nervous System | ||||||
| Phoenix Tissue | PTR-01 | Genetic Disease | ||||||
| Oncternal | Zilovertamab | Cancer | ||||||
| Sato | SB206 (Japan) | Infectious Disease | ||||||
14
| Takeda | TAK-981 | Cancer | ||||||
| Takeda | TAK-925 | Central Nervous System | ||||||
| Verona | Ensifentrine | Asthma | ||||||
| Verona | Ensifentrine | Cystic Fibrosis | ||||||
| Viking | VK5211 | Inflammatory/Metabolic | ||||||
| Viking | VK2809 | Inflammatory/Metabolic | ||||||
| Xi'an Xintong | MB07133 | Cancer | ||||||
| Phase 1 | ||||||||
| Partner Name | Program | Therapeutic Area | ||||||
| Apotex | Meloxicam | Migraine | ||||||
| Arcellx | ACLX-001 | Cancer | ||||||
| Arcellx | ACLX-002 | Cancer | ||||||
| China Resources Double Crane | CX2101A | COVID 19 | ||||||
| CSL | CSL-324 | Immunology | ||||||
| Jazz | JZP-341 | Long Acting Erwinia Asparaginase | ||||||
| Jupiter Biomedical Research | Viright | Cancer | ||||||
| MEI Pharma | ME-344 | Cancer | ||||||
| Merck | V117 | Pneumococcal | ||||||
| Novartis | MIK-665 | Cancer | ||||||
| Nucorion | NUC-1010 | Infectious disease | ||||||
| Revision Therapeutics | Rev0100 | Ophthalmology | ||||||
| Sage | SAGE-689 | Central Nervous System | ||||||
| Takeda | TAK-243 | Cancer | ||||||
| Vaxxas | Nanopatch | Infectious Disease | ||||||
| Viking | VK-0214 | Genetic Disease | ||||||
Summary of selected programs available for license
In addition to Zelsuvmi, discussed above, we have a number of unpartnered programs focused on a wide-range of potential indications or diseases with the potential for further development or licensing:
| Program | Development Stage | Targeted Indication or Disease | ||||||||||||
| CE-Iohexol | Phase 2 | Diagnostics | ||||||||||||
| Luminespib/Hsp90 Inhibitor | Phase 2 | Oncology | ||||||||||||
| CE-Sertraline, Oral Concentrate | Phase 1 | Depression | ||||||||||||
| CCR1 Antagonist | Preclinical | Oncology | ||||||||||||
| CE-Busulfan | Preclinical | Oncology | ||||||||||||
| CE-Cetirizine Injection | Preclinical | Allergy | ||||||||||||
| CE-Silymarin for Topical formulation | Preclinical | Sun damage | ||||||||||||
| FLT3 Kinase Inhibitors | Preclinical | Oncology | ||||||||||||
| GCSF Receptor Agonist | Preclinical | Blood disorders | ||||||||||||
15
Manufacturing
We contract with a third-party manufacturer, Hovione, for Captisol production. Hovione operates FDA-inspected sites in the United States, Macau, Ireland and Portugal. Manufacturing operations for Captisol are performed primarily at Hovione's Portugal and Ireland facilities. We believe we maintain adequate inventory of Captisol to meet our current partner needs and that our Captisol capacity will be sufficient to meet future partner needs.
In the event of a Captisol supply interruption, we are permitted to designate and, with Hovione’s assistance, qualify one or more alternate suppliers. If the supply interruption continues beyond a designated period, we may terminate the agreement. In addition, if Hovione cannot supply our requirements of Captisol due to an uncured force majeure event, we may also obtain Captisol from a third party and have previously identified such parties.
The current term of the agreement with Hovione is through December 2024. The agreement will automatically renew for successive two-year renewal terms unless either party gives written notice of its intention to terminate the agreement no less than two years prior to the expiration of the initial term or renewal term. In addition, either party may terminate the agreement for the uncured material breach or bankruptcy of the other party or an extended force majeure event. We may terminate the agreement for extended supply interruption, regulatory action related to Captisol or other specified events. We have ongoing minimum purchase commitments under the agreement.
Competition
Some of the drugs we and our licensees and partners are developing may compete with existing therapies or other drugs in development by other companies. Furthermore, academic institutions, government agencies and other public and private organizations conducting research may seek patent protection with respect to potentially competing products or technologies and may establish collaborative arrangements with our competitors.
Our Captisol business may face competition from other suppliers of similar cyclodextrin excipients or other technologies that are aimed to increase solubility or stability of APIs.
Our competitive position also depends upon our ability to obtain patent protection or otherwise develop proprietary products or processes. For a discussion of the risks associated with competition, see below under “Item 1A. Risk Factors.”
Corporate and Governance Highlights
We are committed to policies and practices focused on environmental sustainability, positively impacting our social community and maintaining and cultivating good corporate governance. By focusing on such ESG policies and practices, we believe we can affect a meaningful and positive change in our community and maintain our open, collaborative corporate culture. We will continue our proactive shareholder and employee engagement in 2024. See www.ligand.com for information about our ESG policies and practices. However, note that the information contained on our website is not intended to be part of this filing.
Environmental, Health and Safety (EHS)
We are committed to providing a safe and healthy workplace, promoting environmental excellence in our communities, and complying with all relevant regulations and industry standards. We establish and monitor programs to reduce pollution, prevent injuries, and maintain compliance with applicable regulations. By focusing on such practices, we believe we can affect a meaningful, positive change in our community and maintain a healthy and safe environment. During 2023, we progressed our $2.5 million solar investment at Kansas University Innovation Park; made Environmental, Social and Governance (ESG) related charitable donations; and evolved numerous programs from our ESG-focused outreach committees. We expect to continue our effort and to refine our EHS policies and practices in 2024. More information on our EHS policies and initiatives is available on our website at www.ligand.com. However, note that the information contained on our website is not intended to be part of this filing.
Government Regulation
The research and development, manufacturing and marketing of pharmaceutical products are subject to regulation by numerous governmental authorities in the United States and other countries. We and our partners, depending on specific activities performed, are subject to these regulations. In the United States, pharmaceuticals are subject to regulation by both federal and various state authorities, including the FDA. The Federal Food, Drug and Cosmetic Act and the Public Health Service Act govern the research and development, testing, manufacture, quality, safety, efficacy, labeling, storage, record keeping, approval, advertising and promotion of pharmaceutical products. These activities are subject to additional regulations that apply at the state level. There are similar regulations in other countries as well. For both currently marketed products and products in development, failure to comply with applicable regulatory requirements at any time during the product development process, approval process or after approval, can, among other things, result in delays, the suspension of regulatory approvals, regulatory enforcement
16
actions, as well as possible civil and criminal sanctions. In addition, changes in existing regulations could have a material adverse effect on us or our partners.
In particular, FDA approval is required before a drug or biological product may be marketed in the United States and they are also subject to other federal, state, and local statutes and regulations. The process required by the FDA before pharmaceutical products may be marketed in the United States generally involves the following:
•completion of extensive preclinical laboratory tests and preclinical animal studies, certain of which must performed in accordance with Good Laboratory Practice regulations and other applicable requirements ;
•submission to the FDA of an IND application, which must become effective before human clinical studies may begin;
•approval by an independent institutional review board or ethics committee at each clinical site before each clinical study may be initiated;
•performance of adequate and well-controlled human clinical studies in accordance with Good Clinical Practice (GCP) requirements to establish the safety and efficacy, or with respect to biologics, the safety, purity and potency of the product candidate for each proposed indication;
•preparation of and submission to the FDA of an NDA or BLA after completion of all pivotal clinical studies that include substantial evidence of safety, purity, and potency of the drug from analytical studies and from results of nonclinical testing and clinical trials;
•satisfactory completion of an FDA advisory committee review, where appropriate and if applicable;
•a determination by the FDA within 60 days of its receipt of an NDA or BLA to file the application for review;
•satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the proposed product is produced to assess compliance with cGMP, and potential FDA inspection of nonclinical study and clinical trial sites that generated the data in support of the NDA or BLA to ensure compliance with GCP; and
•FDA review and approval of an NDA or BLA prior to any commercial marketing or sale of the drug in the United States.
Any products manufactured or distributed pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, product sampling and distribution, and advertising and promotion of the product. There also are continuing user fee requirements, under which the FDA assesses an annual program fee for each product identified in an approved NDA or BLA. Drug and biologic manufacturers and their subcontractors are required to register their establishments with the FDA and some state agencies, and are subject to periodic unannounced inspections by the FDA and some state agencies for compliance with cGMPs, which among other things, impose certain procedural and documentation requirements upon BLA or NDA holders and any third-party manufacturers. FDA regulations also require investigation and correction of any deviations from cGMPs and impose reporting requirements upon manufacturers and their subcontractors. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMPs and other aspects of regulatory compliance.
The FDA closely regulates the marketing, labeling, advertising and promotion of drug products and biologics. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties
The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences for non-compliance include, among other things:
•restrictions on the marketing or manufacturing of a product, complete withdrawal of the product from the market or product recalls;
•fines, warning letters or holds on post-approval clinical studies;
17
•refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of existing product approvals;
•product seizure or detention, or refusal of the FDA to permit the import or export of products;
•consent decrees, corporate integrity agreements, debarment or exclusion from federal healthcare programs;
•mandated modification of promotional materials and labeling and the issuance of corrective information;
•the issuance of safety alerts, Dear Healthcare Provider letters, press releases and other communications containing warnings or other safety information about the product; or
•injunctions or the imposition of civil or criminal penalties.
For a discussion of the risks associated with government regulations, see below under “Item 1A. Risk Factors.”
Patents and Proprietary Rights
We believe that patents and other proprietary rights are important to our business. Our policy is to file patent applications to protect technology, inventions and improvements to our inventions that are considered important to the development of our business. We also rely upon trade secrets, know-how, continuing technological innovations and licensing opportunities to develop and maintain our competitive position.
Patents are issued or pending for the following key products or product families. The scope and type of patent protection provided by each patent family is defined by the claims in the various patents. Patent term may vary by jurisdiction and depend on a number of factors including potential patent term adjustments, patent term extensions, and terminal disclaimers. For each product or product family, the patents and/or applications referred to are in force in at least the United States, and for most products and product families, the patents and/or applications are also in force in European jurisdictions, Japan and other jurisdictions.
Captisol
Patents and pending patent applications covering Captisol and methods of making Captisol are owned by us. The patents covering the Captisol product with the latest expiration date is expected to be in 2033 (see, e.g., U.S. Patent No. 9,493,582 (expires Feb. 27, 2033)). Other patent applications covering methods of making Captisol, if issued, potentially have terms to 2041. We also own several patents and pending patent applications covering drug products containing Captisol as a component. Globally, we own over 400 issued patents covering all of the foregoing Captisol compositions, methods and related technology.
Ten Captisol patents in several families are listed in the Orange Book in connection with one or more prescription drugs currently on the market. These Captisol-enabled drugs include Nexterone (Baxter), Kyprolis (Amgen), Noxafil (Merck), Evomela (Acrotech/CASI), Baxdela (Melinta) and Zulresso (Sage). These patents are listed in the table below, and each patent family containing these patents has pending and/or granted counterparts in Europe, China and Japan.
Orange Book-listed Captisol Patents | |||||||||||
Country | Patent No. | Title | Expiration (nominal) ‡ | ||||||||
United States | 7635773 | Sulfoalkyl Ether Cyclodextrin Compositions | 03/13/2029 | ||||||||
United States | 8410077 | Sulfoalkyl Ether Cyclodextrin Compositions | 03/13/2029 | ||||||||
United States | 9200088 | Sulfoalkyl Ether Cyclodextrin Compositions | 03/13/2029 | ||||||||
United States | 10117951 | Sulfoalkyl Ether Cyclodextrin Compositions | 03/13/2029 | ||||||||
United States | 9750822 | Sulfoalkyl Ether Cyclodextrin Compositions | 03/13/2029 | ||||||||
United States | 9493582 | Alkylated Cyclodextrin Compositions And Processes For Preparing And Using The Same | 2/27/2033 | ||||||||
United States | 10040872 | Alkylated Cyclodextrin Compositions And Processes For Preparing And Using The Same | 10/21/2033 | ||||||||
| United States | 10864183 | Injectable Nitrogen Mustard Compositions Comprising A Cyclodextrin Derivative And Methods Of Making And Using The Same | 5/28/2030 | ||||||||
| United States | 10940128 | Injectable Melphalan Compositions Comprising A Cyclodextrin Derivative And Methods Of Making And Using The Same | 5/28/2030 | ||||||||
| United States | 11020363 | Injectable Nitrogen Mustard Compositions Comprising A Cyclodextrin Derivative And Methods Of Making And Using The Same | 5/28/2030 | ||||||||
18
‡ Expiration dates are calculated as 20 years from the earliest nonprovisional filing date to which priority is claimed, and do not take into account disclaimers or extensions that are or may be available in these jurisdictions.
Subject to compliance with the terms of the respective agreements, our rights to receive royalty payments under our licenses with our exclusive licensors typically extend for the life of the patents covering such developments. For a discussion of the risks associated with patent and proprietary rights, see below under “Item 1A. Risk Factors.”
Kyprolis
Patents protecting Kyprolis include those owned by Amgen and those owned by us. The United States patent listed in the Orange Book relating to Kyprolis owned by Amgen with the latest expiration date is not expected to expire until 2029. Patents and applications owned by Ligand relating to the Captisol component of Kyprolis are not expected to expire until 2033. Amgen filed suit against several generic drug companies over their applications to make generic versions of Kyprolis. Several generics have settled with Amgen on confidential terms. However, it has been publicly reported that the U.S. launch date for at least Breckenridge Pharmaceuticals’ generic product will be on a date that is held as confidential in 2027 or sooner, depending on certain occurrences. One generic company, Cipla Limited/Cipla USA, Inc. chose not to settle the litigation with Amgen, and proceeded to trial. The District Court upheld the validity of patent claims from three of the patents and the judgment was upheld on appeal.
Ligand UK Development Limited
Under the terms of our sale of Vernalis (R&D) Limited to HitGen in December 2020, Ligand retained a portfolio of fully-funded shots on goal, which now include S65487, a Bcl-2 inhibitor, and S64315, an Mcl-1 inhibitor for treatment of cancers, both of which are partnered with Servier in collaboration with Novartis and VER250840 (an oral, selective Chk1 inhibitor for treatment of cancer). These programs and their IP are now owned by Ligand UK Development Limited, which has a worldwide patent portfolio of over 180 granted patents in over 50 countries. This patent portfolio is mature, with expected expiry dates between 2024 and 2033.
Pelican Expression Technology Platform
In connection with the merger of Pelican and Primrose, Pfenex assigned a global patent portfolio consisting of over 200 patents and over 25 pending patent applications to Pelican, while retaining three patents and six pending patent applications directed to methods of producing Erwinia asparaginase. Additionally, as part of the merger of Pelican and Primrose, Pfenex acquired a non-exclusive, worldwide, royalty free, irrevocable, and fully sublicensable license to a portfolio of approximately 90 patents and approximately 15 pending patent applications which cover various aspects of the Pelican Expression Technology platform that are critical in helping support and retain contractual relationships including Jazz’s RYLAZE, Merck’s VAXNEUVANCE and V116 vaccines, Alvogen’s Teriparatide, and Serum Institute of India’s vaccine programs, including Pneumosil and MenFive vaccines, among others.
Novan
Through the acquisition of certain assets of Novan, we acquired a robust IP portfolio that consists of over 45 U.S. patents, 120 non-U.S. patents, and 25 pending patent applications worldwide along with substantial know-how and trade secrets. This IP portfolio provides material coverage for our platform technologies, licensed products and product candidates, in addition to ZELSUVMI, which was approved by the FDA on January 5, 2024. There are 14 issued U.S. patents covering ZELSUVMI which are expected to be listed in the Orange Book and which are expected to expire during the time period beginning in 2026 and ending in 2035.
Human Capital Management
We recognize and take care of our employees by offering a wide range of competitive pay, recognition, and benefit programs. We are proud to provide our employees the opportunity to grow and advance as we invest in their education and career development. As of December 31, 2023, we have 58 employees, of whom 24 are involved directly in scientific research and development activities.
We rely on skilled, experienced, and innovative employees to conduct the operations of our company. Our key human capital objectives include identifying, recruiting, retaining, incentivizing and integrating our existing and new employees. We frequently benchmark our compensation practices and benefits programs against those of comparable industries and in the geographic areas where our facilities are located. We believe that our compensation and employee benefits are competitive and allow us to attract and retain skilled labor throughout our organization. Our notable health, welfare and retirement benefits include:
•equity awards through our 2002 Stock Incentive Plan;
•subsidized health insurance;
19
•401(k) Plan with matching contributions;
•tuition assistance program; and
•paid time off.
We value diversity at all levels and continue to focus on extending our diversity and inclusion initiatives across our workforce. As of December 31, 2023, approximately 17% and 10% of our workforce are Asian and Hispanic, respectively. Additionally, 52% of our workforce is female and 48% is male. We believe that our business benefits from the different perspectives a diverse workforce brings.
We strive to maintain an inclusive environment free from discrimination of any kind, including sexual or other discriminatory harassment. Our employees have multiple avenues available through which inappropriate behavior can be reported, including a confidential hotline. All reports of inappropriate behavior are promptly investigated with appropriate action taken to stop such behavior.
Investor Information
Financial and other information about us is available on our website at www.ligand.com. We make available on our website, without charge, copies of our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. You may obtain copies of these documents by visiting the SEC’s website at www.sec.gov. In addition, we use X (@Ligand_LGND) and our investor relations website as a means of disclosing material non-public information and for complying with our disclosure obligations under Regulation FD. Investors should monitor our X account and our website, in addition to following our press releases, SEC filings, public conference calls and webcasts. These website addresses and the information accessible through our X account are not intended to function as hyperlinks, and the information contained in our website and in the SEC’s website is not intended to be a part of this filing.
20
| ITEM 1A. | RISK FACTORS | ||||
The following is a summary description of some of the many risks we face in our business. You should carefully review these risks in evaluating our business, including the businesses of our subsidiaries. You should also consider the other information described in this report, including the information contained in our financial statements and the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” Additional risks not presently known to us or that we currently deem immaterial also may impair our business.
Summary of Risks Related to our Business:
Our business is subject to numerous risks and uncertainties, including those described below. The principal risks and uncertainties affecting our business include, but are not limited to the following:
•Future revenue based on Kyprolis, Evomela, Teriparatide and Rylaze as well as royalties from our other partnered products, may be lower than expected;
•Future revenue from sales of Captisol material to our license partners may be lower than expected;
•We rely heavily on collaboration relationships to generate milestone and royalty payments and our collaboration partners have significant discretion when deciding whether to pursue any development program, and any failure by our partners to successfully develop a product candidate or a termination or breach of any of the related agreements, or a change in their strategy or the focus of their development and commercialization efforts with respect to our partnered programs, could reduce our milestone and license fee revenue, and potentially reduce future royalties;
•Our product candidates, and the product candidates of our partners, face significant development and regulatory hurdles prior to partnering and/or marketing which could delay or prevent licensing, sales-based royalties and/or milestone revenue;
•The royalty market may not grow at the same rate as it has in the past, or at all, and we may not be able to acquire sufficient royalties to create or sustain growth of our business;
•Information available to us about the biopharmaceutical products underlying the royalties we buy may be limited and, therefore, our ability to analyze each product and its potential future cash flow may be similarly limited;
•Third party intellectual property may prevent us or our partners from developing our potential products; our and our partners’ intellectual property may not prevent competition; and any intellectual property issues may be expensive and time consuming to resolve;
•Market acceptance and sales of any approved product will depend significantly on the availability and adequacy of coverage and reimbursement from third-party payors and may be affected by existing and future healthcare reform measures; and
•Our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations or any guidance we may provide.
Risks Related to Our Business Operations and Reliance on Third Parties:
Future revenue based on Kyprolis, Evomela, Teriparatide and Rylaze as well as royalties from our other partnered products, may be lower than expected.
A significant portion of our royalty revenue is based on sales of Kyprolis by Amgen, sales of Evomela by Acrotech Biopharma, sales of Teriparatide by Alvogen/Adalvo and sales of Rylaze by Jazz. Royalties, including payments from the foregoing partners, are expected to be a substantial portion of our ongoing revenues for the foreseeable future. Any setback that may occur with respect to any of our partners' products, and in particular Kyprolis, could significantly impair our operating results and/or reduce our revenue and the market price of our stock. Setbacks for the products could include problems with shipping, distribution, manufacturing, product safety, marketing, government regulation or reimbursement, licenses and approvals, intellectual property rights, including failure by any of the foregoing partners to enforce their respective intellectual property rights, competition with existing or new products and physician or patient acceptance of the products, as well as higher than expected total rebates, returns, discounts, or unfavorable exchange rates. These products also are or may become subject to generic competition. For example, we entered into a settlement agreement with Teva and Acrotech Biopharma (the holder of the NDA for Evomela) which will allow Teva to market a generic version of Evomela in the United States on June 1, 2026, or earlier under certain circumstances. The entry of generic competition for Evomela may materially and adversely affect the revenue we derive from Evomela sales. Also, Amgen previously settled patent litigation related to Kyprolis on confidential terms with several parties, but it was publicly reported that the U.S. launch date for at least Breckenridge Pharmaceuticals’ applicable generic product will be “on a date that is held as confidential in 2027 or sooner, depending on certain occurrences.”
21
Future revenue from sales of Captisol material to our license partners may be lower than expected.
Revenues from sales of Captisol material to our collaboration partners, including Amgen, represent a significant portion of our current revenues. Any setback that may occur with respect to Captisol could significantly impair our operating results and/or reduce the market price of our stock. Setbacks for Captisol could include problems with shipping, distribution, manufacturing, product safety, marketing, government regulation or reimbursement, licenses and approvals, intellectual property rights, competition with existing or new products and physician or patient acceptance of the products using Captisol. In addition, we may continue to generate no revenue from Captisol sales related to remdesivir due to a number of factors, including alternative treatments for COVID-19 that have been or will be developed by other companies and the decrease in COVID-19 infections, in which case the commercial opportunity could be continue to be limited.
If products or product candidates incorporating Captisol material were to cause any unexpected adverse events, the perception of Captisol safety could be seriously harmed. If this were to occur, we may not be able to sell Captisol unless and until we are able to demonstrate that the adverse event was unrelated to Captisol, which we may not be able to do. Further, the FDA could require us to submit additional information for regulatory review or approval, including data from extensive safety testing or clinical testing of products using Captisol. This would be expensive and it may delay the marketing of Captisol-enabled products and receipt of revenue related to those products, which could significantly impair our operating results and/or reduce the market price of our stock.
We obtain Captisol from Hovione, our third party manufacturer, primarily at their facilities in Ireland and Portugal. If Hovione were to cease to be able, for any reason, to supply Captisol to us in the amounts we require, or decline to supply Captisol to us, we would be required to seek an alternative source, which could potentially take a considerable length of time and impact our revenue and customer relationships. In the event of a Captisol supply interruption, we are permitted to designate and, with Hovione’s assistance, qualify one or more alternate suppliers, although there is no assurance that we could do so timely or at acceptable costs, if at all. In addition to manufacturing at Hovione’s facilities in Ireland and Portugal, we have processing capacity for Captisol in both the United States and England.
We maintain inventory of Captisol, which has a five-year shelf life, at three geographically dispersed storage locations in the United States and Europe. If we were to encounter problems maintaining our inventory, such as natural disasters, at one or more of these locations, it could lead to supply interruptions. In addition, we rely on Hovione to expand manufacturing capacity of Captisol and any failure by Hovione to timely implement such increased capacity could adversely affect our ability to supply Captisol to our partners. While we believe we maintain adequate inventory of Captisol to meet our current partner needs, and our Captisol capacity will be sufficient to meet future partner needs, our estimates and projections for Captisol demand may not be correct and any supply interruptions could materially adversely impact our operating results.
We currently depend on our arrangements with our partners and licensees to sell products using our Captisol technology. These agreements generally provide that our partners may terminate the agreements at will. If our partners discontinue sales of products using Captisol, fail to obtain regulatory approval for products using Captisol, fail to satisfy their obligations under their agreements with us, choose to utilize a competing product, or if we are unable to establish new licensing and marketing relationships, our financial results and growth prospects would be materially affected.
Further, under most of our Captisol outlicenses, the amount of royalties we receive will be reduced or will cease when the relevant patent expires. Our low-chloride patents and foreign equivalents are not expected to expire until 2033, our high purity patents and foreign equivalents, are not expected to expire until 2029 and our morphology patents and foreign equivalents are not expected to expire until 2026 in the United States, but the initially filed patents relating to Captisol expired starting in 2010 in the United States and in 2016 in most countries outside the United States. If our other intellectual property rights are not sufficient to prevent a generic form of Captisol from coming to market and if in such case our partners choose to terminate their agreements with us, our Captisol revenue may decrease significantly.
We rely heavily on collaboration relationships to generate milestone and royalty payments and our collaboration partners have significant discretion when deciding whether to pursue any development program, and any failure by our partners to successfully develop a product candidate or a termination or breach of any of the related agreements could reduce our milestone and license fee revenue, and potentially reduce future royalties.
Our strategy for developing and commercializing many of our product candidates includes entering into collaboration agreements, outlicenses, and development funding and royalty purchase agreements with corporate partners and others. These agreements give our collaboration partners significant discretion when deciding whether or not to pursue any development program. Our existing collaborations may not continue or be successful, and we may be unable to enter into future collaboration arrangements to develop and commercialize our unpartnered assets.
In addition, our collaborators may develop products, either alone or with others that compete with the types of products they are developing with us (or that we are developing on our own). This would result in increased competition for our or our partners' programs. If product candidates are approved for marketing under our collaboration programs, revenues we receive
22
will depend on the manufacturing, marketing and sales efforts of our collaboration partners, who generally retain commercialization rights under the collaboration agreements. Generally, our current collaboration partners also have the right to terminate their collaborations at will or under specified circumstances. If any of our collaboration partners breach (for example, by not making required payments when due, or at all) or terminate their agreements with us or otherwise fail to conduct their collaboration activities successfully, including due to insolvency events, ongoing product development under these agreements will be delayed or terminated. Disputes or litigation may also arise with our collaborators (with us and/or with one or more third parties), including those over ownership rights to intellectual property, know-how or technologies developed with our collaborators. Such disputes or litigation could adversely affect our rights to one or more of our product candidates. Any such dispute or litigation could delay, interrupt or terminate the collaboration research, development and commercialization of certain potential products, create uncertainty as to ownership rights of intellectual property, or could result in litigation or arbitration. The occurrence of any of these problems could be time-consuming and expensive and could adversely affect our business.
Our collaboration partners may change their strategy or the focus of their development and commercialization efforts with respect to our partnered programs, and the success of our partnered programs could be adversely affected.
If our collaboration partners terminate their collaborations with us or do not commit sufficient resources to the development, manufacture, marketing or distribution of our partnered programs, we could be required to devote additional resources to our partnered programs, seek new collaboration partners or abandon such partnered programs, all of which could reduce our revenues and otherwise have an adverse effect on our business.
In addition, biopharmaceutical development is inherently uncertain and very few therapeutic candidates ultimately progress through clinical development and receive approval for commercialization. If our partners do not receive regulatory approval for a sufficient number of therapeutic candidates originating from our partnerships, we may not be able to sustain our business model.
Our product candidates, and the product candidates of our partners, face significant development and regulatory hurdles prior to partnering and/or marketing which could delay or prevent licensing, sales-based royalties and/or milestone revenue.
Before we or our partners obtain the approvals necessary to sell any of our unpartnered assets or partnered programs, we must show through preclinical studies and human testing that each potential product is safe and effective. We and/or our partners have a number of partnered programs and unpartnered assets moving toward or currently awaiting regulatory action. Failure to show any product's safety and effectiveness could delay or prevent regulatory approval of a product and could adversely affect our business. The product development and clinical trials process is complex and uncertain. For example, the results of preclinical studies and initial clinical trials may not necessarily predict the results from later large-scale clinical trials. In addition, clinical trials may not demonstrate a product's safety and effectiveness to the satisfaction of the regulatory authorities. A number of companies have suffered significant setbacks in advanced clinical trials or in seeking regulatory approvals, despite promising results in earlier trials. The FDA may also require additional clinical trials after regulatory approvals are received. Such additional trials may be expensive and time-consuming, and failure to successfully conduct those trials could jeopardize continued commercialization of a product.
The speed at which we and our partners complete our scientific studies and clinical trials depends on many factors, including, but not limited to, the ability to obtain adequate supplies of the products to be tested and patient enrollment. Patient enrollment is a function of many factors, including the size of the patient population, the proximity of patients to clinical sites, the eligibility criteria for the trial and other potential drug candidates being studied. Delays in patient enrollment for our or our partners’ trials may result in increased costs and longer development times. In addition, our partners have rights to control product development and clinical programs for products developed under our collaborations. As a result, these partners may conduct these programs more slowly or in a different manner than expected. Moreover, even if clinical trials are completed, we or our partners still may not apply for FDA or foreign regulatory approval in a timely manner, or the FDA or foreign regulatory authority still may not grant approval.
Our product candidate discovery, early-stage development, and product reformulation programs may require substantial additional capital to complete successfully. Our partners’ development programs may require substantial additional capital to complete successfully, arising from costs to: conduct research, preclinical testing and human studies; establish pilot scale and commercial scale manufacturing processes and facilities; and establish and develop quality control, regulatory, marketing, sales and administrative capabilities to support these programs. While we expect to fund our research and development activities from cash generated from operations to the extent possible, if we are unable to do so, we may need to complete additional equity or debt financings or seek other external means of financing. These financings could depress our stock price. If additional funds are required to support our operations and we are unable to obtain them on terms favorable to us, we may be required to cease or reduce further development or commercialization of our products, to sell some or all of our technology or assets or to merge with another entity.
23
The royalty market may not grow at the same rate as it has in the past, or at all, and we may not be able to acquire sufficient royalties to create or sustain growth of our business.
The growth of our business depends on our ability to acquire royalties and we may not be able to identify and acquire a sufficient number of royalties, or royalties of sufficient scale, to invest the full amount of capital that may be available to us in the future, or at our targeted amount and rate of deployment, which could prevent us from executing our growth strategy and negatively impact our results of operations. Changes in the royalty market, including its structure, participants and growth rate, changes in preferred methods of financing and capital raising in the biopharmaceutical industry, or a reduction in the growth of the biopharmaceutical industry, could lead to diminished opportunities for us to acquire royalties, fewer royalties (or fewer royalties of significant scale) being available, or increased competition for royalties. Even if we continue to acquire royalties, they may not generate a meaningful return for a period of several years, if at all, due to numerous factors including the structure of the transaction, or circumstances relating to the underlying products. As a result, we may not be able to create or sustain growth of our business as we expect or at all.
We face competition in acquiring royalties and locating suitable royalties to acquire.
There are a limited number of suitable and attractive opportunities to acquire high-quality royalties available in the market. Therefore, competition to acquire such royalties is intense and may increase. We compete with other potential acquirers for these opportunities, including companies that market the products on which royalties are paid, financial institutions and others. These competitors may be able to access lower cost capital, may be larger than us, may have relationships that provide them access to opportunities before us, or may be willing to acquire royalties for lower projected returns than we are.
Information available to us about the biopharmaceutical products underlying the royalties we buy may be limited and, therefore, our ability to analyze each product and its potential future cash flow may be similarly limited.
We may have limited information concerning the products generating the royalties we are evaluating for acquisition. Often, the information we have regarding products following our acquisition of a royalty may be limited to the information that is available in the public domain. Therefore, there may be material information that relates to such products that we would like to know but do not have and may not be able to obtain. For example, we do not always know the results of studies conducted by marketers of the products or others or the nature or amount of any complaints from doctors or users of such products. In addition, the market data that we obtain independently may also prove to be incomplete or incorrect. Due to these and other factors, the actual cash flow from a royalty may be significantly lower than our estimates.
A significant portion of our future income is dependent upon numerous royalty-specific assumptions and, if these assumptions prove not to be accurate, we may not achieve our expected rates of returns.
Our business model is based on multiple-year internal and external forecasts regarding product sales and numerous product-specific assumptions in connection with each royalty acquisition, including where we have limited information regarding the product, sales of our products and licenses to our technology. There can be no assurance that the assumptions underlying our financial models, including those regarding product sales or competition, patent expirations, exclusivity terms, license terms or license terminations for the products underlying our portfolio, products and technology, are accurate. These assumptions involve a significant element of subjective judgment and may be adversely affected by post-acquisition changes in market conditions and other factors affecting the underlying product or technology. The risks relating to these assumptions may be exacerbated for development-stage product candidates due to the uncertainties around their development, labeling, regulatory approval, commercialization timing, manufacturing and supply, competing products or related factors. With respect to our partnered programs, our assumptions regarding the financial stability or operational or marketing capabilities of the partner obligated to pay us royalties or license, milestone or other service payments, may also prove, and in the past have proven, to be incorrect. Due to these and other factors, the assets in our current portfolio or future assets, or our current or future products or technology, may not generate expected returns or returns in line with our historical financial performance or in the time periods we expect or at all, which could adversely affect our financial condition and results of operation.
The insolvency of any of our partners or third-parties who are developing or commercializing products to which we have economic rights could adversely affect our receipt of cash flows on the related milestones or royalties that we own.
If any of our partners or third-parties who are developing or commercializing products to which we have economic rights were to become insolvent and seek to reorganize under Chapter 11 of Title 11 of the U.S. Code, as amended, or the Bankruptcy Code, or liquidate under Chapter 7 of the Bankruptcy Code (or foreign equivalent), such event could delay or impede the payment of the amounts due to us under any license agreement, royalty purchase agreement or other contract under which we have acquired economic rights, pending a resolution of the insolvency proceeding. Unless we obtained a secured interest, any unpaid royalty payments under our license agreements with our partners and third-parties due for the period prior to the filing of the bankruptcy proceeding could become unsecured claims against such partner or third-party, which might not be paid in full or at all. The actual payment of such post-filing royalty payments could be delayed for a substantial period of time and might not be in the full amount due under such agreements. Given the nature of our royalty purchase agreements, royalty payments
24
due to our partners or third-parties prior to or after a bankruptcy proceeding may not be subject to the insolvency proceeding and may be considered our property, meaning there is a reduced risk of payment delay and/or non-payment. Nevertheless, a partner or third-party or another party in interest in an insolvency proceeding may attempt to recharacterize the royalty purchase agreement and claim that the royalty payments are property of the bankruptcy estate, in which case we would rely upon contractual protections related to such recharacterizations, which may not be respected in bankruptcy. In addition, certain of agreements with our partners or third-parties permit us to take a secured interest in the intellectual property underlying the licenses and royal purchase agreements, which may improve our risk profile in an insolvency proceeding.
In some cases and depending on the terms of the agreement, we are not the licensor and instead are dependent on the licensor to enforce its right to royalties under an agreement with a licensee. In any bankruptcy proceeding, the licensor would be prevented by the automatic stay from taking any action to enforce its rights without the permission of the bankruptcy court. In addition, such partner or third-party could elect to reject the license agreement. Though this would prohibit such partner or third-party from continuing to market the applicable product, it would require the licensor to undertake a new effort to market the applicable product with another distributor. Such proceedings could adversely affect the ability of a partner or other payor to make payments with respect to a royalty, and could consequently adversely affect our business, financial condition or results of operations.
If the Distribution, together with certain related transactions, failed to qualify as a reorganization under Sections 355 and 368(a)(1)(D) of the Internal Revenue Code of 1986, as amended (the “Code”), or the Merger failed to qualify as a reorganization under Section 368(a) of the Code, we could incur significant tax liabilities.
The Distribution and the Merger were conditioned upon receipt of a tax opinion from outside counsel to the effect that the Distribution qualified as a reorganization under Sections 355 and 368(a)(1)(D) of the Code, that the Merger would not cause Section 355(e) of the Code to apply to the Distribution and that the Merger would be treated as a reorganization under Section 368(a) of the Code. The opinion was delivered in connection with the closing of the Merger and was based on, among other things, certain facts, assumptions, representations and undertakings from us, OmniAb and New OmniAb, including those regarding the past and future conduct of the companies’ respective businesses and other matters. If any of these facts, assumptions, representations, or undertakings were incorrect or not satisfied, we may not be able to rely on the opinion, and we and our stockholders could be subject to significant U.S. federal income tax liabilities. In addition, the opinion is not binding on the IRS or the courts, and notwithstanding the opinion, the IRS could determine on audit that the Distribution or Merger does not qualify as a reorganization if it determines that any of the facts, assumptions, representations or undertakings on which the opinion is based are not correct or have been violated or that the Distribution or Merger should be taxable for other reasons, including as a result of a significant change in stock or asset ownership after the Distribution. If the Distribution, together with certain related transactions, is ultimately determined not to qualify as a reorganization, or the Merger is ultimately determined not to qualify as a reorganization, we and our stockholders that are subject to U.S. federal income tax could incur significant U.S. federal income tax liabilities.
The Separation and Distribution may expose Ligand to potential liabilities arising out of state and federal fraudulent conveyance laws and legal dividend requirements.
The Separation and Distribution are subject to review under various state and federal fraudulent conveyance laws. Fraudulent conveyance laws generally provide that an entity engages in a constructive fraudulent conveyance when (i) the entity transfers assets and does not receive fair consideration or reasonably equivalent value in return; and (ii) the entity: (a) is insolvent at the time of the transfer or is rendered insolvent by the transfer; (b) has unreasonably small capital with which to carry on its business; or (c) intends to incur or believes it will incur debts beyond its ability to repay its debts as they mature. An unpaid creditor or an entity acting on behalf of a creditor (including without limitation a trustee or debtor-in-possession in a bankruptcy by New OmniAb or Ligand or any of their respective subsidiaries) may bring an action alleging that the Separation or Distribution or any of the related transactions constituted a constructive fraudulent conveyance. If a court accepts these allegations, it could impose a number of remedies, including without limitation, voiding New OmniAb’s claims against Ligand, requiring New OmniAb stockholders to return to Ligand some or all of the shares of New OmniAb common stock issued via the Distribution and Merger, or providing Ligand with a claim for money damages against New OmniAb in an amount equal to the difference between the consideration received by Ligand and OmniAb’s fair market value at the time of the Distribution.
The measure of insolvency for purposes of the fraudulent conveyance laws will vary depending on which jurisdiction’s law is applied. Generally, an entity would be considered insolvent if (i) the present fair saleable value of its assets is less than the amount of its liabilities (including contingent liabilities); (ii) the present fair saleable value of its assets is less than its probable liabilities on its debts as such debts become absolute and matured; (iii) it cannot pay its debts and other liabilities (including contingent liabilities and other commitments) as they mature; or (iv) it has unreasonably small capital for the business in which it is engaged. We cannot assure you what standard a court would apply to determine insolvency or that a court would determine that New OmniAb or Ligand or any of their subsidiaries were solvent at the time of or after giving effect to the Distribution.
25
The Distribution of OmniAb common stock is also subject to review under state corporate distribution statutes. Under the DGCL, a corporation may only pay dividends to its stockholders either (i) out of its surplus (net assets minus capital) or (ii) if there is no such surplus, out of its net profits for the fiscal year in which the dividend is declared or the preceding fiscal year. Although Ligand intended to make the Distribution of OmniAb common stock entirely from surplus, we cannot assure you that a court will not later determine that some or all of the Distribution to Ligand stockholders was unlawful.
Risks Related to Intellectual Property:
Third party intellectual property may prevent us or our partners from developing our potential products; our and our partners’ intellectual property may not prevent competition; and any intellectual property issues may be expensive and time consuming to resolve.
The manufacture, use or sale of our potential products or our licensees' products or potential products may infringe the patent rights of others. If others obtain patents with conflicting claims, we may be required to obtain licenses to those patents or to develop or obtain alternative technology. We may not be able to obtain any such licenses on acceptable terms, or at all. Any failure to obtain such licenses could delay or prevent us from pursuing the development or commercialization of our potential products, platform and technology.
Generally, our success will depend on our ability and the ability of our partners to obtain and maintain patents and other intellectual property rights for our and their potential products and technologies. Our patent position is uncertain and involves complex legal and technical questions for which legal principles are unresolved. Even if we or our partners do obtain patents, such patents may not adequately protect the technology we own or have licensed.
We permit our partners to list our patents that cover their branded products in the Orange Book. If a third party submits a new drug application (NDA) or abbreviated new drug application (ANDA) for a generic drug product that relies in whole or in part on studies contained in our partner’s NDA for their branded product, the third party will have the option to certify to the FDA that, in the opinion of that third party, the patents listed in the Orange Book for our partner’s branded product are invalid, unenforceable, or will not be infringed by the manufacture, use or sale of the third party’s generic drug product. A third party certification that a new product will not infringe Orange Book-listed patents, or that such patents are invalid, is called a paragraph IV patent certification. If the third party submits a paragraph IV patent certification to the FDA, a notice of the paragraph IV patent certification must be sent to the NDA owner and the owner of the patents that are subject to the paragraph IV patent certification notice once the third-party’s NDA or ANDA is accepted for filing by the FDA. A lawsuit may then be initiated to defend the patents identified in the notice. The filing of a patent infringement lawsuit within 45 days of the receipt of notice of a paragraph IV patent certification automatically prevents the FDA from approving the generic NDA or ANDA until the earlier of the expiration of a 30-month period, the expiration of the patents, the entry of a settlement order stating that the patents are invalid or not infringed, a decision in the infringement case that is favorable to the NDA or ANDA applicant, or such shorter or longer period as the court may order. If a patent infringement lawsuit is not initiated within the required 45-day period, the third-party’s NDA or ANDA will not be subject to the 30-month stay.
Several third parties have challenged, and additional third parties may challenge, the patents covering our partner’s branded products, including Kyprolis and Evomela, which could result in the invalidation or unenforceability of some or all of the relevant patent claims. We may from time to time become party to litigation or other proceedings as a result of Paragraph IV certifications. For example, as a result of the settlement of one such matter, Teva will be permitted to market a generic version of Evomela® in the United States on June 1, 2026 or earlier under certain circumstances. The terms of the settlement agreement are otherwise confidential. Also, as noted above, Amgen previously settled patent litigation related to Kyprolis on confidential terms with several parties, but it has been publicly reported that the U.S. launch date for at least Breckenridge Pharmaceuticals’ applicable generic product will be “on a date that is held as confidential in 2027 or sooner, depending on certain occurrences.”
In addition, we cannot assure you that all of the potentially relevant prior art information that was or is deemed available to a person of skill in the relevant art prior to the priority date of the claimed invention-relating to our and our partners’ patents and patent applications has been found. If such prior art exists, it can invalidate a patent or prevent a patent from issuing from a pending patent application, and we or our partners may be subject to a third party pre-issuance submission of prior art to the USPTO. Even if our patent applications do successfully issue and even if such patents cover our or our partner’s products or potential products, third parties may initiate litigation or opposition, interference, re-examination, post-grant review, inter partes review, nullification or derivation action in court or before patent offices, or similar proceedings challenging the validity, enforceability or scope of such patents, which may result in the patent claims being narrowed or invalidated, may allow third parties to commercialize our or our partners’ products and compete directly with us and our partners, without payment to us or our partners, or limit the duration of the patent protection of our and our partners’ technology and products.
In addition, similar to what other companies in our industry have experienced, we expect our competitors and others may have patents or may in the future obtain patents and claim that making, having made, using, selling, offering to sell or importing our technologies infringes these patents. Defense of infringement and other claims, regardless of their merit, would involve
26
substantial litigation expense and would be a substantial diversion of management and employee resources from our business. Parties making claims against us may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Parties making claims against us may be able to obtain injunctive or other relief, which could block our ability to develop, commercialize and sell products or services and could result in the award of substantial damages against us, including treble damages, attorney’s fees, costs and expenses if we are found to have willfully infringed. In the event of a successful claim of infringement against us, we may be required to pay damages and ongoing royalties and obtain one or more licenses from third parties, or be prohibited from selling certain products or services. As discussed above, we may not be able to obtain these licenses on acceptable or commercially reasonable terms, if at all, or these licenses may be non-exclusive, which could result in our competitors gaining access to the same intellectual property. In addition, we could encounter delays in product or service introductions while we attempt to develop alternative products or services to avoid infringing third-party patents or proprietary rights. Defense of any lawsuit or failure to obtain any of these licenses could prevent us from commercializing products or services, and the prohibition of sale of any of our technologies could materially affect our business and our ability to gain market acceptance for our technology.
Litigation or other proceedings to enforce or defend intellectual property rights are often very complex in nature, may be very expensive and time-consuming, may divert our management’s attention from our core business, and may result in unfavorable results that could adversely impact our ability to prevent third parties from competing with our partner’s products or technologies. Any adverse outcome of such litigation or other proceedings could result in one or more or our patents being held invalid or unenforceable, which could adversely affect our ability to successfully execute our business strategy and negatively impact our financial condition and results of operations. However, given the unpredictability inherent in litigation, we cannot predict or guarantee the outcome of these matters or any other litigation. Regardless of how these matters are ultimately resolved, these matters may be costly, time-consuming and distracting to our management, which could have a material adverse effect on our business. It may be necessary for us to pursue litigation or adversarial proceedings before the patent office in order to enforce our patent and proprietary rights or to determine the scope, coverage and validity of the proprietary rights of others. The outcome of any such litigation might not be favorable to us, and even if we were to prevail, such litigation could result in substantial costs and diversion of resources and could have a material adverse effect on our business, operating results or financial condition.
In addition, periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and or applications will be due to the U.S. and various foreign patent offices at various points over the lifetime of our and our licensees’ patents and/or applications. We have systems in place to remind us to pay these fees, and we rely on our outside patent annuity service to pay these fees when due. Additionally, the U.S. and various foreign patent offices require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. We employ reputable law firms and other professionals to help us comply, and in many cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with rules applicable to the particular jurisdiction. However, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. If such an event were to occur, it could have a material adverse effect on our business.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
Any conflicts with the patent rights of others could significantly reduce the coverage of our patents or limit our ability to obtain meaningful patent protection. For example, our European patent related to Agglomerated forms of Captisol was limited during an opposition proceeding, and the rejection of our European patent application related to High Purity Captisol was upheld on appeal. In addition, any determination that our patent rights are invalid may result in early termination of our agreements with our license partners and could adversely affect our ability to enter into new license agreements. We also rely on unpatented trade secrets and know-how to protect and maintain our competitive position. We require our employees, consultants, licensees and others to sign confidentiality agreements when they begin their relationship with us. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our competitors may independently discover our trade secrets.
We may also need to initiate litigation, which could be time-consuming and expensive, to enforce our proprietary rights or to determine the scope and validity of others' rights. If this occurs, a court may find our patents or those of our licensors invalid or may find that we have infringed on a competitor's rights. In addition, if any of our competitors have filed patent applications in the United States prior to March 2013 which claim technology we also have invented, the United States Patent and Trademark Office may require us to participate in expensive interference proceedings to determine who has the right to a patent for the technology.
27
In addition, our agreements with some of our partners, suppliers or other entities with whom we do business require us to defend or indemnify these parties to the extent they become involved in infringement claims, including the types of claims described above. We could also voluntarily agree to defend or indemnify third parties in instances where we are not obligated to do so if we determine it would be important to our business relationships. If we are required or agree to defend or indemnify third parties in connection with any infringement claims, we could incur significant costs and expenses that could adversely affect our business, financial condition, results of operations and prospects. The occurrence of any of the foregoing problems could be time-consuming and expensive and could adversely affect our financial position, liquidity and results of operations.
If we are unable to obtain and maintain sufficient intellectual property protection for our products, platform and technology, or if the scope of the intellectual property protection obtained is not sufficiently broad, our competitors could develop and commercialize technologies or a platform similar or identical to ours, and our ability to successfully sell our platform and services may be impaired.
Our success depends in part on our ability to obtain and maintain adequate protection of the intellectual property we may own solely and jointly with others or otherwise have rights to, particularly patents, in the United States and in other countries with respect to our platform, our software and our technologies, without infringing the intellectual property rights of others.
We strive to protect and enhance the proprietary technologies that we believe are important to our business, including seeking patents intended to cover our platform and related technologies and uses thereof, as we deem appropriate. However, obtaining and enforcing patents in our industry is costly, time-consuming and complex, and we may fail to apply for patents on important products and technologies in a timely fashion or at all, or we may fail to apply for patents in potentially relevant jurisdictions. There can be no assurance that the claims of our patents (or any patent application that issues as a patent), will exclude others from making, using, importing, offering for sale, or selling products or services that are substantially similar to ours. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection. In countries where we have not sought and do not seek patent protection, third parties may be able to manufacture and sell our technology without our permission, and we may not be able to stop them from doing so. We may not be able to file and prosecute all necessary or desirable patent applications, or maintain, enforce and license any patents that may issue from such patent applications, at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection. We may not have the right to control the preparation, filing and prosecution of patent applications, or to maintain the rights to patents licensed to third parties. Therefore, these patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business.
It is possible that none of our pending patent applications will result in issued patents in a timely fashion or at all, and even if patents are granted, they may not provide a basis for intellectual property protection of commercially viable products or services, may not provide us with any competitive advantages, or may be challenged and invalidated by third parties or deemed unenforceable by a court. It is possible that others will design around our current or future patented technologies. As a result, our owned and licensed patents and patent applications comprising our patent portfolio may not provide us with sufficient rights to exclude others from commercializing technology and products similar to any of our products, platform and technology.
In addition, we may identify third party intellectual property and technology we may need to acquire or license in order to engage in our business, including to develop or commercialize new technologies. However, such licenses may not be available to us on acceptable terms or at all. Furthermore, geo-political actions in the United States and in foreign countries could increase the uncertainties and costs surrounding the prosecution or maintenance of our patent applications or those of any current or future license partners and the maintenance, enforcement or defense of our issued patents or those of any current or future license partners. For example, the United States and foreign government actions related to Russia’s conflict in Ukraine may limit or prevent filing, prosecution, and maintenance of patent applications in Russia. Government actions may also prevent maintenance of issued patents in Russia. These actions could result in abandonment or lapse of our or our license partners’ patents or patent applications, resulting in partial or complete loss of patent rights in Russia. If such an event were to occur, it could have a material adverse effect on our business. In addition, a decree was adopted by the Russian government in March 2022, allowing Russian companies and individuals to exploit inventions owned by patentees from the United States without consent or compensation. Consequently, we or our license partners would not be able to prevent third parties from practicing our or our inventions in Russia or from selling or importing products made using our inventions in and into Russia. Accordingly, our competitive position may be impaired, and our business, financial condition, results of operations and prospects may be adversely affected.
Issued patents directed to our platform and technology could be found invalid or unenforceable if challenged in court or before administrative bodies in the United States or abroad.
The issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability. Some of our patents or patent applications (including licensed patents) may be challenged at a future point in time in opposition, derivation, reexamination, inter partes review, post-grant review or interference. Any successful third party challenge to our patents in this
28
or any other proceeding could result in the unenforceability or invalidity of such patents or amendment to our patents in such a way that any resulting protection may lead to increased competition to our business, which could harm our business. In addition, in patent litigation in the United States, defendant counterclaims alleging invalidity or unenforceability are commonplace. The outcome following legal assertions of invalidity and unenforceability during patent litigation is unpredictable. If a defendant were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on certain aspects of our platform technologies. In addition, if the breadth or strength of protection provided by our patents and patent applications is threatened, regardless of the outcome, it could dissuade companies from collaborating with us to license, develop or commercialize current or future products, platform and technology.
We may not be aware of all third party intellectual property rights potentially relating to our products, platform and technology. Publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the United States and other jurisdictions are typically not published until approximately 18 months after filing or, in some cases, not until such patent applications issue as patents. We or our licensors might not have been the first to make the inventions included in each of our pending patent applications and we or our licensors might not have been the first to file patent applications for these inventions. There is also no assurance that all of the potentially relevant prior art relating to our patents and patent applications or licensed patents and patent applications has been found, which could be used by a third party to challenge their validity, or prevent a patent from issuing from a pending patent application.
To determine the priority of these inventions, we may have to participate in interference proceedings (with respect to patent applications filed prior to March 2013), derivation proceedings or other post-grant proceedings declared by the USPTO that could result in substantial cost to us. The outcome of such proceedings is uncertain. No assurance can be given that other patent applications will not have priority over our patent applications. In addition, changes to the patent laws of the United States allow for various post-grant opposition proceedings that have not been extensively tested, and their outcome is therefore uncertain. Furthermore, if third parties bring these proceedings against our patents, we could experience significant costs and management distraction.
The validity, scope and enforceability of any patents that cover our partners’ biologic product candidate can be challenged by third parties.
For biologics, the Biologics Price Competition and Innovation Act of 2009, BPCIA, provides a mechanism for one or more third parties to seek FDA approval to manufacture or sell biosimilar or interchangeable versions of brand name biological products. Due to the large size and complexity of biological products, as compared to small molecules, a biosimilar must be “highly similar” to the reference product with “no clinically meaningful differences between the two.” The BPCIA does not require reference product sponsors to list patents in an Orange Book and does not include an automatic 30-month stay of FDA approval upon the timely filing of a lawsuit. The BPCIA, however, does require a formal pre-litigation process which includes the exchange of information between a biosimilar applicant and a reference biologic sponsor that includes the identification of relevant patents and each parties’ basis for infringement and invalidity. After the exchange of this information, sponsors may then initiate a lawsuit within 30 days to defend the patents identified in the exchange. If the biosimilar applicant successfully challenges the asserted patent claims it could result in the invalidation of, or render unenforceable, some or all of the relevant patent claims or result in a finding of non-infringement. Such litigation or other proceedings to enforce or defend intellectual property rights are often very complex in nature, may be very expensive and time-consuming, may divert our management’s attention from our core business, and may result in unfavorable results that could limit our partners’ ability to prevent third parties from competing with their products or product candidates.
Changes in patent law in the United States and other jurisdictions could diminish the value of patents in general, thereby impairing our ability to protect our products, platform and technology.
Changes in either the patent laws or interpretation of the patent laws in the United States could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents, and may diminish our ability to protect our inventions, obtain, maintain, enforce and protect our intellectual property rights and, more generally, could affect the value of our intellectual property or narrow the scope of our future owned and licensed patents. Depending on future actions by the United States Congress, the United States courts, the USPTO and the relevant law-making bodies in other countries, the laws and regulations governing patents could change in unpredictable ways that would weaken our or our license partners’ ability to obtain new patents and patents that we or our license partners’ might obtain in the future. For example, on June 1, 2023, the European Union Patent Package (EU Patent Package) regulations were implemented with the goal of providing a single pan-European Unitary Patent and a new European Unified Patent Court (UPC) for litigation involving European patents. As a result, all European patents, including those issued prior to ratification of the EU Patent Package, now by default automatically fall under the jurisdiction of the UPC. It is uncertain how the UPC will impact granted European patents in the biotechnology and pharmaceutical industries. Our or our license partners’ European patent applications, if issued, could be challenged in the UPC. During the first seven years of the UPC’s existence, the UPC legislation allows a patent owner to opt its European patents out of the jurisdiction of the UPC. We or our license partners may decide to opt out future European patents from the UPC, but doing so may preclude us or our license partners from realizing the benefits of the
29
UPC. Moreover, if we or our license partners do not meet all of the formalities and requirements for opt-out under the UPC, our or our license partners’ future European patents could remain under the jurisdiction of the UPC. The UPC will provide our and our license partners’ competitors with a new forum to centrally revoke our European patents, and allow for the possibility of a competitor to obtain pan-European injunction. Such a loss of patent protection could have a material adverse impact on our or our license partners business and ability to commercialize our technology and product candidates and, resultantly, on our business, financial condition, prospects and results of operations.
We rely on in-licenses from third parties. If we lose these rights, our business may be materially and adversely affected, our ability to develop improvements to our technology platform may be negatively and substantially impacted, and if disputes arise, we may be subjected to future litigation, as well as the potential loss of or limitations on our ability to incorporate the technology covered by these license agreements.
We are party to royalty-bearing license agreements that grant us rights to practice certain patent rights that are related to our products, platform and technology. In spite of our efforts to comply with our obligations under our in-license agreements, our licensors might conclude that we have materially breached our obligations under our license agreements and might therefore, including in connection with any aforementioned disputes, terminate the relevant license agreement, thereby removing or limiting our ability to develop and commercialize technology covered by these license agreements. If any such in-license is terminated, or if the licensed patents fail to provide the intended exclusivity, competitors or other third parties might have the freedom to market or develop technologies similar to ours.
In addition, absent the rights granted to us under our license agreements, we may infringe the intellectual property rights that are the subject of those agreements, we may be subject to litigation by the licensor, and if such litigation by the licensor is successful we may be required to pay damages to our licensor, or we may be required to cease our development and commercialization activities that are deemed infringing, and in such event we may ultimately need to modify our activities or technologies to design around such infringement, which may be time- and resource-consuming, and which ultimately may not be successful. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
In addition, our rights to certain components of our technology platform, may be licensed to us on a non-exclusive basis. The owners of these non-exclusively licensed technologies are therefore free to license them to third parties, including our competitors, on terms that may be superior to those offered to us, which could place us at a competitive disadvantage.
Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. In addition, certain of our agreements with third parties may provide that intellectual property arising under these agreements, such as data that could be valuable to our business, will be owned by the third party, in which case, we may not have adequate rights to use such data or have exclusivity with respect to the use of such data, which could result in third parties, including our competitors, being able to use such data to compete with us.
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
We or our licensors may be subject to claims that former employees, partners or other third parties have an interest in our or our in-licensed patents, trade secrets or other intellectual property as an inventor or co-inventor. Litigation may be necessary to defend against these and other claims challenging inventorship of our or our licensors’ ownership of our owned or in-licensed patents, trade secrets or other intellectual property. If we or our licensors fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual property that is important to our systems, including our software, workflows, consumables and reagents. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees, and certain partners or partners may defer engaging with us until the particular dispute is resolved. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations and prospects.
If we are unable to protect the confidentiality of our information and our trade secrets, the value of our technology could be materially and adversely affected and our business could be harmed.
We rely on trade secrets and confidentiality agreements to protect our unpatented know-how, technology and other proprietary information, including parts of our technology platform, and to maintain our competitive position. However, trade secrets and know-how can be difficult to protect. In addition to pursuing patents on our technology, we take steps to protect our intellectual property and proprietary technology by entering into agreements, including confidentiality agreements, non-disclosure agreements and intellectual property assignment agreements, with our employees, consultants, academic institutions, corporate partners and, when needed, our advisers. However, we cannot be certain that such agreements have been entered into with all relevant parties, and we cannot be certain that our trade secrets and other confidential proprietary information will not be disclosed or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. For example, any of these parties may breach the agreements and disclose our
30
proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Such agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements, and we may not be able to prevent such unauthorized disclosure, which could adversely impact our ability to establish or maintain a competitive advantage in the market. If we are required to assert our rights against such party, it could result in significant cost and distraction.
Monitoring unauthorized disclosure and detection of unauthorized disclosure is difficult, and we do not know whether the steps we have taken to prevent such disclosure are, or will be, adequate. If we were to enforce a claim that a third party had illegally obtained and was using our trade secrets, it would be expensive and time-consuming, and the outcome would be unpredictable. In addition, some courts both within and outside the United States may be less willing, or unwilling, to protect trade secrets. Further, we may need to share our trade secrets and confidential know-how with current or future partners, collaborators, contractors and others located in countries at heightened risk of theft of trade secrets, including through direct intrusion by private parties or foreign actors, and those affiliated with or controlled by state actors.
We also seek to preserve the integrity and confidentiality of our confidential proprietary information by maintaining physical security of our premises and physical and electronic security of our information technology systems, but it is possible that these security measures could be breached. If any of our confidential proprietary information were to be lawfully obtained or independently developed by a competitor or other third party, absent patent protection, we would have no right to prevent such competitor from using that technology or information to compete with us, which could harm our competitive position. If any of our trade secrets were to be disclosed to or independently discovered by a competitor or other third party, it could harm our business, financial condition, results of operations and prospects.
Risks Related to Government Regulation and Legal Proceedings:
Market acceptance and sales of any approved product will depend significantly on the availability and adequacy of coverage and reimbursement from third-party payors and may be affected by existing and future healthcare reform measures.
Sales of the products we may market or license to our collaboration partners and the royalties we receive will depend in large part on the extent to which coverage and reimbursement is available from government and health administration authorities, private health maintenance organizations and health insurers, and other healthcare payors. Significant uncertainty exists as to the reimbursement status of healthcare products. Healthcare payors, including Medicare, are challenging the prices charged for medical products and services. Government and other healthcare payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement for medical products. Even if a product is approved by the FDA, insurance coverage may not be available, and reimbursement levels may be inadequate, to cover the costs associated with the research, development, marketing and sale of the product. If government and other healthcare payors do not provide adequate coverage and reimbursement levels for any product, market acceptance and any sales could be reduced.
From time to time, legislation is implemented to reign in rising healthcare expenditures. By way of example, the Affordable Care Act (ACA) was enacted in 2010 and included a number of provisions affecting the pharmaceutical industry, including, among other things, annual, non-deductible fees on any entity that manufactures or imports some types of branded prescription drugs and increases in Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program. Since its enactment, there have been judicial, executive and Congressional challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. Thus, the ACA will remain in effect in its current form.
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. For example, beginning April 1, 2013, Medicare payments to providers were reduced under the sequestration required by the Budget Control Act of 2011, which will remain in effect through 2032, unless additional Congressional action is taken. Additionally, on January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. On March 11, 2021, the American Rescue Plan Act of 2021 was signed into law, which eliminated the statutory Medicaid drug rebate cap, beginning January 1, 2024. Previously, the Medicaid rebate was capped at 100% of a drug’s average manufacturer price, or AMP.
Additional changes that may affect our business include the expansion of new programs such as Medicare payment for performance initiatives for physicians under the Medicare Access and CHIP Reauthorization Act of 2015, or MACRA, which was fully implemented in 2019. At this time, it is unclear how the introduction of this Medicare quality payment program will impact overall physician reimbursement. The cost of prescription pharmaceuticals in the United States has also been the subject of considerable discussion in the United States. There have been several Congressional inquiries, as well as legislative and regulatory initiatives and executive orders designed to, among other things, bring more transparency to product pricing, review
31
the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products.
Moreover, the federal government and the individual states in the United States have become increasingly active in developing proposals, passing legislation and implementing regulations designed to control drug pricing, including price or patient reimbursement constraints, discounts, formulary flexibility, marketing cost disclosure, drug price increase reporting, and other transparency measures. These types of initiatives may result in additional reductions in Medicare, Medicaid, and other healthcare funding.
Most significantly, on August 16, 2022, President Biden signed the Inflation Reduction Act of 2022 (IRA) into law. This statute marks the most significant action by Congress with respect to the pharmaceutical industry since adoption of the ACA in 2010. Among other things, the IRA requires manufacturers of certain drugs to engage in price negotiations with Medicare (beginning in 2026), with prices that can be negotiated subject to a cap; imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation (first due in 2023); and replaces the Part D coverage gap discount program with a new discounting program (beginning in 2025). The IRA permits the Secretary of the Department of Health and Human Services (HHS) to implement many of these provisions through guidance, as opposed to regulation, for the initial years. HHS has and will continue to issue and update guidance as these programs are implemented. On August 29, 2023, HHS announced the list of the first ten drugs that will be subject to price negotiations, although the Medicare drug price negotiation program is currently subject to legal challenges. The impact of the IRA on the pharmaceutical industry cannot yet be fully determined, but is likely to be significant.
We expect that these and other healthcare reform measures that may be adopted in the future may result in more rigorous coverage and payment criteria and in additional downward pressure on the prices that can be realized for any approved drug. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us or our partners from being able to generate revenue, attain profitability, or commercialize drugs. Individual states in the United States have also become increasingly active in implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for drug candidates or additional pricing pressures. We cannot predict with certainty what impact any federal or state health reforms will have on us, but such changes could impose new or more stringent regulatory requirements or result in reduced reimbursement for our products, any of which could adversely affect our business, results of operations and financial condition.
If we or our commercialization partners market products in a manner that violates healthcare laws, we may be subject to civil or criminal penalties.
We and our collaboration partners are subject to federal and state healthcare laws, including fraud and abuse, government price reporting, anti-kickback, false claims, physician payment transparency and civil monetary penalties. These laws may impact, among other things, financial arrangements with physicians, sales, marketing and education programs and the manner in which any of those activities are implemented. If our operations or those of our collaboration partners are found to be in violation of any of those laws or any other applicable governmental regulations, we or our collaboration partners may be subject to penalties, including civil and criminal penalties, damages, fines, imprisonment, exclusion from government healthcare programs or the curtailment or restructuring of operations, any of which could adversely affect our ability to operate our business and our financial condition.
Pricing and rebate calculations vary among products and programs. The calculations are complex and are often subject to interpretation by our collaboration partners, governmental or regulatory agencies, and the courts. CMS, the Department of Health & Human Services Office of Inspector General, and other governmental agencies have pursued manufacturers that were alleged to have failed to report these data to the government in a timely or accurate manner. Governmental agencies may also make changes in program interpretations, requirements or conditions of participation, some of which may have implications for amounts previously estimated or paid. We cannot assure you that any submissions by our collaboration partners to federal healthcare programs, and other governmental drug pricing programs, will not be found to be incomplete or incorrect.
Changes in and actual or perceived failures to comply with applicable data privacy, security and protection laws, regulations, standards and contractual obligations may adversely affect our business, operations and financial performance.
We and our partners may be subject to federal, state, and foreign laws and regulations that govern data privacy and security. The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing focus on privacy and data protection issues, which may affect our business and may increase our compliance costs and exposure to liability. In the United States, numerous federal and state laws and regulations govern the collection, use, disclosure, and protection of personal information, including state data breach notification laws, federal and state health
32
information privacy laws, and federal and state consumer protection laws. Each of these laws is subject to varying interpretations by courts and government agencies, creating complex compliance issues. If we fail to comply with applicable laws and regulations we could be subject to penalties or sanctions, including criminal penalties if we knowingly obtain or disclose individually identifiable health information from a covered entity in a manner that is not authorized or permitted by the Health Insurance Portability and Accountability Act, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and regulations implemented thereunder (collectively, HIPAA) or applicable state laws.
Certain states have also adopted comparable privacy and security laws and regulations, which govern the privacy, processing and protection of health-related and other personal information. Such laws and regulations will be subject to interpretation by various courts and other governmental authorities, thus creating potentially complex compliance issues for us and our future customers and strategic partners. For example, the California Consumer Privacy Act of 2018 (CCPA) went into effect on January 1, 2020. The CCPA creates individual privacy rights for California consumers and increases the privacy and security obligations of entities handling certain personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that has increased the likelihood of, and risks associated with data breach litigation. Further, the California Privacy Rights Act (CPRA) generally went into effect on January 1, 2023, and significantly amends the CCPA. It imposes additional data protection obligations on covered businesses, including additional consumer rights processes, limitations on data uses, new audit requirements for higher risk data, and opt outs for certain uses of sensitive data. It also created a new California data protection agency authorized to issue substantive regulations and could result in increased privacy and information security enforcement. Additional compliance investment and potential business process changes may be required. Similar laws have passed in other states and are continuing to be proposed at the state and federal level, reflecting a trend toward more stringent privacy legislation in the United States. The enactment of such laws could have potentially conflicting requirements that would make compliance challenging. In the event that we are subject to or affected by HIPAA, the CCPA, the CPRA or other domestic privacy and data protection laws, any liability from failure to comply with the requirements of these laws could adversely affect our financial condition.
We are also or may become subject to rapidly evolving data protection laws, rules and regulations in foreign jurisdictions. For example, the European Union General Data Protection Regulation (GDPR) governs certain collection and other processing activities involving personal data about individuals in the European Economic Area (EEA). Companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater. Among other requirements, the GDPR regulates transfers of personal data subject to the GDPR to third countries that have not been found to provide adequate protection to such personal data, including the United States, and the efficacy and longevity of current transfer mechanisms between the EEA and the United States remains uncertain. Case law from the Court of Justice of the European Union (CJEU) states that reliance on the standard contractual clauses - a standard form of contract approved by the European Commission as an adequate personal data transfer mechanism - alone may not necessarily be sufficient in all circumstances and that transfers must be assessed on a case-by-case basis. On October 7, 2022, President Biden signed an Executive Order on ‘Enhancing Safeguards for United States Intelligence Activities’ which introduced new redress mechanisms and binding safeguards to address the concerns raised by the CJEU in relation to data transfers from the EEA to the United States and which formed the basis of the new EU-US Data Privacy Framework (DPF), as released on December 13, 2022. The European Commission adopted its Adequacy Decision in relation to the DPF on July 10, 2023, rendering the DPF effective as a GDPR transfer mechanism to U.S. entities self-certified under the DPF. The DPF also introduced a new redress mechanism for EU citizens which addresses a key concern in the previous CJEU judgments and may mean transfers under standard contractual clauses are less likely to be challenged in future. With the advice of outside counsel and privacy experts, we take appropriate steps to ensure transfers of personal data outside the EEA and the UK, including to the United States, are conducted in a manner consistent with applicable law and legal requirements. We expect the existing legal complexity and uncertainty regarding international personal data transfers to continue. In particular, we expect the DPF Adequacy Decision to be challenged and international transfers to the United States and to other jurisdictions more generally to continue to be subject to enhanced scrutiny by regulators. As a result, we may have to make certain operational changes and we will have to implement revised standard contractual clauses and other relevant documentation for existing data transfers within required time frames. Since the beginning of 2021, after the end of the transition period following the United Kingdom’s departure from the European Union, we are also subject to the United Kingdom data protection regime, which imposes separate but similar obligations to those under the GDPR and comparable penalties, including fines of up to £17.5 million or 4% of a noncompliant company’s global annual revenue for the preceding financial year, whichever is greater. . On October 12, 2023, the UK Extension to the DPF came into effect (as approved by the UK Government), as a UK GDPR data transfer mechanism to U.S. entities self-certified under the UK Extension to the DPF. As we continue to expand into other foreign countries and jurisdictions, we may be subject to additional laws and regulations that may affect how we conduct business.
Furthermore, the FTC also has authority to initiate enforcement actions against entities that make deceptive statements about privacy and data sharing in privacy policies, fail to limit third-party use of personal health information, fail to implement
33
policies to protect personal health information or engage in other unfair practices that harm customers or that may violate Section 5 of the FTC Act. Failing to take appropriate steps to keep consumers’ personal information secure can constitute unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act. The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Additionally, federal and state consumer protection laws are increasingly being applied by FTC and states’ attorneys general to regulate the collection, use, storage, and disclosure of personal or personally identifiable information, through websites or otherwise, and to regulate the presentation of website content.
Compliance with applicable data privacy and security laws, rules and regulations could require us to take on more onerous obligations in our contracts, require us to engage in costly compliance exercises, restrict our ability to collect, use and disclose data, or in some cases, impact our or our partners’ ability to operate in certain jurisdictions. Each of these constantly evolving laws can be subject to varying interpretations. If we fail to comply with any such laws, rules or regulations, we may face government investigations and/or enforcement actions, fines, civil or criminal penalties, private litigation or adverse publicity that could adversely affect our business, financial condition and results of operations.
The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming and inherently unpredictable, and if we or our partners are ultimately unable to obtain regulatory approval for product candidates, our business will be substantially harmed.
The clinical development, manufacturing, labeling, storage, record-keeping, advertising, promotion, import, export, marketing and distribution of drugs and biologics are subject to extensive regulation by the FDA in the U.S. and by comparable foreign regulatory authorities in foreign markets. In the U.S., neither we nor our partners are permitted to market our product candidates in the U.S. until we receive approval of a biologics license application (BLA) or an NDA from the FDA. The process of obtaining such regulatory approval is expensive, often takes many years following the commencement of clinical trials and can vary substantially based upon the type, complexity and novelty of the product candidates involved, as well as the target indications and patient population. Approval policies or regulations may change, and the FDA and comparable regulatory authorities have substantial discretion in the approval process, including the ability to delay, limit or deny approval of a product candidate for many reasons. Despite the time and expense invested in clinical development of product candidates, regulatory approval of a product candidate is never guaranteed. Of the large number of drugs in development, only a small percentage successfully complete the FDA or foreign regulatory approval processes and are commercialized.
Prior to obtaining approval to commercialize a drug or biological product candidate in the U.S. or abroad, we or our partners must demonstrate with substantial evidence from adequate and well-controlled clinical trials, and to the satisfaction of the FDA or comparable foreign regulatory authorities, that such product candidates are safe and effective for their intended uses, and in the case of biological products in the U.S., that such product candidates are safe, pure and potent. Results from nonclinical studies and clinical trials can be interpreted in different ways. Even if we or our partners believe available nonclinical or clinical data support the safety purity, potency or efficacy of our product candidates, such data may not be sufficient to obtain approval from the FDA and comparable foreign regulatory authorities. The FDA or comparable foreign regulatory authorities, as the case may be, may also require us or our partners to conduct additional preclinical studies or clinical trials for our product candidates either prior to or post-approval, or may object to elements of clinical development programs.
The FDA or comparable foreign regulatory authorities can delay, limit or deny approval of a product candidate for many reasons, including:
•such authorities may disagree with the design or execution of clinical trials;
•negative or ambiguous results from clinical trials or results may not meet the level of statistical significance or persuasiveness required by the FDA or comparable foreign regulatory agencies for approval;
•serious and unexpected drug-related side effects may be experienced by participants in clinical trials or by individuals using drugs similar to the applicable product candidates;
•the population studied in the clinical trial may not be sufficiently broad or representative to assure safety in the full population for which we or our partners seek approval;
•such authorities may not accept clinical data from trials that are conducted at clinical facilities or in countries where the standard of care is potentially different from that of their own country;
•we or our partners may be unable to demonstrate that a product candidate’s clinical and other benefits outweigh its safety risks;
•such authorities may disagree with our or our partners’ interpretation of data from preclinical studies or clinical trials;
34
•such authorities may not agree that the data collected from clinical trials are acceptable or sufficient to support the submission of a BLA, NDA or other submission or to obtain regulatory approval in the U.S. or elsewhere, and such authorities may impose requirements for additional preclinical studies or clinical trials;
•such authorities may disagree with us or our partners regarding the formulation, labeling and/or product specifications;
•approval may be granted only for indications that are significantly more limited than those sought by us or our partners, and/or may include significant restrictions on distribution and use;
•such authorities may find deficiencies in the manufacturing processes or facilities of the third-party manufacturers utilized for clinical and commercial supplies; or
•such authorities may not accept a submission due to, among other reasons, the content or formatting of the submission.
With respect to foreign markets, approval procedures vary among countries and, in addition to the foregoing risks, may involve additional product testing, administrative review periods and agreements with pricing authorities. Even if we or our partners eventually complete clinical trials and receive approval of a BLA, NDA or comparable foreign marketing application for our product candidates, the FDA or comparable foreign regulatory authority may grant approval contingent on the performance of costly additional clinical trials and/or the implementation of burdensome monitoring requirements to address safety concerns. Any delay in obtaining, or inability to obtain, applicable regulatory approval would delay or prevent commercialization of that product candidate, which could materially and adversely impact our revenues, business and prospects.
Pharmaceutical products are subject to ongoing regulatory obligations and continued regulatory review, which may result in significant additional expense.
For any regulatory approvals that we or our partners may receive for our respective product candidates, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion, import, export and recordkeeping for our product candidates will remain subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, as well as ongoing compliance with current Good Manufacturing Practices (cGMPs) and Good Clinical Practice requirements for any clinical trials that we or they may conduct. In addition, manufacturers of drug and biological products and their facilities are subject to continual review and periodic, unannounced inspections by the FDA and other regulatory authorities for compliance with cGMP regulations and standards. In addition, regulatory approvals require the submission of periodic reports to regulatory authorities and surveillance to monitor the safety and efficacy of the product, and such approvals may contain significant limitations related to use restrictions for specified age groups, warnings, precautions or contraindications, and may include burdensome post-approval study or risk management requirements. For example, the FDA may require a Risk Evaluation and Mitigation Strategy as a condition of approval, which could include requirements for a medication guide, physician training and communication plans or additional elements to ensure safe use, such as restricted distribution methods, patient registries and other risk minimization tools.
If we, our partners or a regulatory agency discover previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facilities where the product is manufactured, a regulatory agency may impose restrictions on that product, the manufacturing facility or us, including requiring recall or withdrawal of the product from the market or suspension of manufacturing. In addition, failure to comply with FDA and other comparable foreign regulatory requirements may lead to administrative or judicially imposed sanctions, including:
•restrictions on the marketing or manufacturing of our products, withdrawal of the product from the market or voluntary or mandatory product recalls;
•restrictions on product distribution or use, or requirements to conduct post-marketing studies or clinical trials;
•fines, restitutions, disgorgement of profits or revenues, warning letters, untitled letters or holds on clinical trials;
•refusal by the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of approvals;
•product seizures or detentions, or refusal to permit the import or export of products; and
•injunctions or the imposition of civil or criminal penalties.
The occurrence of any event or penalty described above may inhibit our or our partners’ ability to commercialize and generate revenue from products and could require us or our partners to expend significant time and resources in response and could generate negative publicity. In addition, the FDA’s and other regulatory authorities’ policies may change and additional government regulations may be promulgated that could prevent, limit or delay marketing authorization of any product
35
candidates we or our partners develop. We also cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, we may be subject to enforcement action and we may not achieve or sustain profitability.
Changes in funding for the FDA and other government agencies could hinder their ability to hire and retain key leadership and other personnel, or otherwise prevent new products and services from being developed or commercialized in a timely manner, which could negatively impact our business or the business of our partners.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept the payment of user fees, and statutory, regulatory, and policy changes. Average review times at the FDA have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs and biologics to be reviewed and/or approved by necessary government agencies, which would adversely affect our business or the business of our partners. For example, over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business. If the timing of FDA’s review and approval of new products is delayed, the timing of our or our partners’ development process may be delayed which would result in delayed milestone revenues and materially harm our operations of business.
Separately, in response to the COVID-19 pandemic, the FDA postponed most inspections of domestic and foreign manufacturing facilities at various points. Even though the FDA has resumed standard inspection operations of domestic facilities where feasible, any resurgence of the virus or emergence of new variants may lead to further inspectional or administrative delays. Regulatory authorities outside the United States may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic. If a prolonged government shutdown occurs, or if global health concerns continue to hinder or prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process our or our partners’ regulatory submissions, which could have a material adverse effect on our business.
If plaintiffs bring product liability lawsuits against us or our partners, we or our partners may incur substantial liabilities and may be required to limit commercialization of our approved products and product candidates.
As is common in our industry, our partners and we face an inherent risk of product liability as a result of the clinical testing of our product candidates in clinical trials and face an even greater risk for commercialized products. Although we are not currently a party to product liability litigation, if we are sued, we may be held liable if any product or product candidate we develop causes injury or is found otherwise unsuitable during product testing, manufacturing, marketing or sale. Regardless of merit or eventual outcome, liability claims may result in decreased demand for any product candidates, partnered products or products that we may develop, injury to our reputation, discontinuation of clinical trials, costs to defend litigation, substantial monetary awards to clinical trial participants or patients, loss of revenue and product recall or withdrawal from the market and the inability to commercialize any products that we develop. We have product liability insurance that covers our clinical trials up to a $15.0 million annual limit. Our insurance coverage may not be sufficient to cover all of our product liability related expenses or losses and may not cover us for any expenses or losses we may suffer. If we are sued for any injury caused by our product candidates, partnered products or any future products, our liability could exceed our total assets.
We face risks related to handling of hazardous materials and other regulations governing environmental safety.
Our operations are subject to complex and stringent environmental, health, safety and other governmental laws and regulations that both public officials and private individuals may seek to enforce. Our activities that are subject to these regulations include, among other things, our use of hazardous materials and the generation, transportation and storage of waste. Although we have secured clearance from the EPA historically, and currently are operating in material compliance with applicable EPA rules and regulations, our business could be adversely affected if we discover that we or an acquired business is not in material compliance with these rules and regulations. In the future, we may pursue the use of other surfactant substances that will require clearance from the EPA, and we may fail to obtain such clearance. Existing laws and regulations may also be revised or reinterpreted, or new laws and regulations may become applicable to us, whether retroactively or prospectively, that may have a negative effect on our business and results of operations. It is also impossible to eliminate completely the risk of accidental environmental contamination or injury to individuals. In such an event, we could be liable for any damages that result, which could adversely affect our business.
We may also be subject to laws and regulations not specifically targeting the healthcare industry.
36
Certain regulations not specifically targeting the healthcare industry also could have material effects on our operations. For example, the California Financing Law (the “CFL”), Division 9, Sections 22000-22780.1 of the California Financial Code, could be applied to us as a result of loans or similar arrangements we enter into with partners. If a regulator were to take the position that such loans were covered by the California Financing Law, we could be subject to regulatory action that could impair our ability to continue to operate and may have a material adverse effect on our profitability and business as we currently do not hold a CFL finance lenders license. Pursuant to an exemption under the CFL, a person may make five or fewer commercial loans with a California nexus in a 12-month period without a CFL finance lenders license if such loans are “incidental” to the business of the person making the loan. This exemption, however, creates some uncertainty as to which loans could be deemed as incidental to our business. In addition, there is another exemption that would allow a person without a CFL finance lenders license to make a single commercial loan with a California nexus in a 12-month period.
Risk Related to Our Strategic Transactions:
Any difficulties from strategic acquisitions could adversely affect our stock price, operating results and results of operations.
We may acquire companies, businesses and products that complement or augment our existing business. We may not be able to integrate any acquired business successfully or operate any acquired business profitably. Integrating any newly acquired business could be expensive and time-consuming. Integration efforts often take a significant amount of time, place a significant strain on managerial, operational and financial resources and could prove to be more difficult or expensive than we predict. The diversion of our management's attention and any delay or difficulties encountered in connection with any future acquisitions we may consummate could result in the disruption of our ongoing business or inconsistencies in standards and controls that could negatively affect our ability to maintain third-party relationships. Moreover, we may need to raise additional funds through public or private debt or equity financing, or issue additional shares, to acquire any businesses or products, which may result in dilution for stockholders or the incurrence of indebtedness.
As part of our efforts to acquire companies, business or product candidates or to enter into other significant transactions, we conduct business, legal and financial due diligence with the goal of identifying and evaluating material risks involved in the transaction. Despite our efforts, we ultimately may be unsuccessful in ascertaining or evaluating all such risks and, as a result, might not realize the intended advantages of the transaction. If we fail to realize the expected benefits from acquisitions we may consummate in the future or have consummated in the past, whether as a result of unidentified risks, integration difficulties, regulatory setbacks, litigation with current or former employees and other events, our business, results of operations and financial condition could be adversely affected. If we acquire product candidates, we will also need to make certain assumptions about, among other things, development costs, the likelihood of receiving regulatory approval and the market for such product candidates. Our assumptions may prove to be incorrect, which could cause us to fail to realize the anticipated benefits of these transactions.
In addition, we will likely experience significant charges to earnings in connection with our efforts, if any, to consummate acquisitions. For transactions that are ultimately not consummated, these charges may include fees and expenses for investment bankers, attorneys, accountants and other advisors in connection with our efforts. Even if our efforts are successful, we may incur, as part of a transaction, substantial charges for closure costs associated with elimination of duplicate operations and facilities and acquired in-process research and development charges. In either case, the incurrence of these charges could adversely affect our results of operations for particular quarterly or annual periods.
Other Risks:
Our business is subject to risks arising from pandemic and epidemic diseases.
Future pandemics, including the residual effects of the COVID-19 pandemic, or other public health epidemics, pose the risk that we or our employees, contractors, including our CROs, suppliers, and other partners may be prevented from conducting business activities for an indefinite period of time, including due to spread of the disease within these groups or due to shutdowns that may be requested or mandated by governmental authorities. Although we have lifted the restrictions we previously imposed on in-person access to our facilities and currently do not believe the COVID-19 pandemic is having a material impact on our business, we cannot guarantee that pandemics, such as COVID-19 or the emergence of variants thereof, or a similar event, will not impact our operations in the future.
Several of our partners reported that their operations were impacted by the COVID-19 pandemic, with such impacts including delays in research and development programs and deprioritizing clinical trials in favor of treating patients who had contracted the virus or to prevent the spread of the virus. In addition, certain of our partners reported negative impacts on product sales which impacted our royalty revenues. Although we believe that we and our partners have adjusted our business practices to the impacts of the COVID-19 pandemic, in the future, we may experience similar pandemics or epidemic diseases that could severely impact our business, drug manufacturing and supply chain, nonclinical activities and clinical trials and our partners’ business may be impacted in similar ways, including due to delays or difficulties in enrolling patients in clinical trials, diversion of healthcare resources away from the conduct of clinical trials, interruption of, or delays in receiving, supplies of
37
Captisol or other product or product candidates from contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in delivery systems, which may result in cancellations of Captisol orders or refunds if we fail to deliver Captisol timely, interruption or delays to discovery and development pipelines and difficulties launching or commercializing products, including due to reduced access to doctors as a result of social distancing protocols.
Further, the COVID-19 pandemic impacted the trading price of shares of our common stock. The extent to which the emergence of new variants of COVID-19, or any other outbreak of a pandemic or epidemic disease, impacts our results will depend on future developments that are highly uncertain and cannot be predicted, including new information that may emerge concerning the severity of the virus and the actions to contain its impact. Further, to the extent any pandemic or epidemic disease adversely affects our business and financial results, it may also have the effect of heightening many of the other risks described in this section.
Our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations or any guidance we may provide.
Our quarterly and annual operating results may fluctuate significantly, which makes it difficult for us to predict our future operating results. These fluctuations may occur due to a variety of factors, many of which are outside of our control, including, but not limited to:
•the royalties from the sales of Kyprolis, Evomela and other products sold by our partners;
•the success of our collaboration partners’ preclinical and clinical programs;
•the timing of Captisol purchases for use in clinical trials and commercial products;
•the timing and cost of, and level of investment in, research, development, regulatory approval and commercialization activities relating to our internal development programs, which may change from time to time;
•expenditures that we may incur to acquire or develop additional product candidates and platform technologies; and
•future accounting pronouncements or changes in our accounting policies.
The cumulative effects of these factors could result in large fluctuations and unpredictability in our quarterly and annual operating results and revenues. This variability and unpredictability could result in our failing to meet the expectations of industry or financial analysts or investors for any period. If our revenue or operating results fall below the expectations of analysts or investors or below any forecasts we may provide to the market, or if the forecasts we provide to the market are below the expectations of analysts or investors, the price of our common stock could decline substantially. Such a stock price decline could occur even when we have met any previously publicly stated revenue or earnings guidance we may provide.
Changes or modifications in financial accounting standards, including those related to revenue recognition, may harm our results of operations.
From time to time, the FASB either alone or jointly with other organizations, promulgates new accounting principles that could have an adverse impact on our results of operations. For example, in May 2014, FASB issued an accounting standard for revenue recognition-Accounting Standards Codification Topic 606, Revenue from Contracts with Customers, or ASC 606-that supersedes most current revenue recognition guidance. The guidance requires a company to recognize revenue upon transfer of goods or services to a customer at an amount that reflects the expected consideration to be received in exchange for those goods or services. The guidance became effective in fiscal 2018.
Under ASC 606, Ligand estimates and books royalties in the same quarter that our partners report the sale of the underlying product. We rely on our partners’ earning releases and other information from our partners to determine the sales of our partners’ products and to estimate the related royalty revenues. If our partners report incorrect sales, or if our partners delay reporting of their earnings release, our royalty estimates may need to be revised and/or our financial reporting may be delayed.
Our ability to use our net operating loss carryforwards and certain other tax attributes to offset future taxable income may be subject to certain limitations.
As of December 31, 2023, we had U.S. federal and state net operating loss carryforwards (NOLs) of approximately $48.0 million and $165.1 million, respectively. Our federal NOLs expire through 2037 and our state NOLs begin to expire in 2028, if not utilized. Under the Tax Act, any federal NOLs arising in taxable years ending after December 31, 2017 will carry forward indefinitely. As of December 31, 2023, we had federal and California research and development tax credit carryforwards of approximately $8.5 million and $29.4 million, respectively. The federal research and development tax credit carryforwards expire in various years through 2040, if not utilized. The California research and development credit will carry forward indefinitely. Under Sections 382 and 383 of Internal Revenue Code of 1986, as amended (Code) if a corporation undergoes an “ownership change,” the corporation’s ability to use its pre-change NOLs and other pre-change tax attributes, such as research tax credits, to offset its future post-change income and taxes may be limited. In general, an “ownership change” occurs if there is a cumulative change in our ownership by “5% shareholders” that exceeds 50 percentage points over a rolling three-year period. Similar rules may apply under state tax laws. We believe we have experienced certain ownership changes in the past
38
and have reduced our deferred tax assets related to NOLs and research and development tax credit carryforwards accordingly. In the event that it is determined that we have in the past experienced additional ownership changes, or if we experience one or more ownership changes as a result future transactions in our stock, then we may be further limited in our ability to use our NOLs and other tax assets to reduce taxes owed on the net taxable income that we earn in the event that we attain profitability. Furthermore, under the Tax Act, although the treatment of tax losses generated in tax years beginning before December 31, 2017 has generally not changed, tax losses generated in tax years beginning after December 31, 2017 may only offset 80% of our taxable income. This change may require us to pay federal income taxes in future years despite having potentially generated a loss for federal income tax purposes in prior years. Any such limitations on the ability to use our NOLs and other tax assets could adversely impact our business, financial condition and operating results.
The occurrence of a catastrophic disaster could disrupt our business, damage our facilities beyond insurance limits, increase our costs and expenses, or we could lose key data which could cause us to curtail or cease operations.
We are vulnerable to damage, business disruptions and/or loss of vital data from natural or man-made disasters, such as earthquakes, tornadoes, severe weather conditions, power loss, fire, floods and similar events, as well as from accidental loss or destruction. If any disaster were to occur, our ability to operate our business could be seriously impaired. We have property, liability, and business interruption insurance which may not be adequate to cover our losses resulting from disasters or other similar significant business interruptions, and we do not plan to purchase additional insurance to cover such losses due to the cost of obtaining such coverage. Any significant losses that are not recoverable under our insurance policies could seriously impair our business, financial condition and prospects. Our ability to obtain Captisol supply from our third-party manufactures could be disrupted if the operations of these manufacturers were affected by a natural or man-made disaster or other business interruption. In addition, we rely on our partners to generate most of our revenues through royalties, Captisol sales and development activities and any disruptions to their business as a result of such disasters could negatively impact our revenues.
We rely on information technology system and any failure, inadequacy, interruption or security lapse of our information technology systems, including any cyber security incidents, could harm our ability to operate our business effectively.
Our business is increasingly dependent on critical, complex and interdependent information technology systems, including internet-based systems, to support business processes as well as internal and external communications. We operate some of these systems and networks, but we also rely on third-party providers for various products and services across our operations. Despite the implementation of security measures, our information technology systems and those of our partners and third party service providers are vulnerable to attack, damage, and interruption from cyber-attacks, computer viruses and malware (e.g. ransomware), security breaches, unauthorized access, natural disasters, terrorism, war, telecommunication and electrical failures, hacking, phishing attacks and other social engineering schemes, employee theft or misuse, human error, fraud, denial or degradation of service attacks, sophisticated nation-state and nation-state-supported actors or unauthorized access or use by persons inside our organization, or persons with access to systems inside our organization.
Attacks upon information technology systems are increasing in their frequency, levels of persistence, sophistication and intensity, and are being conducted by sophisticated and organized groups and individuals with a wide range of motives and expertise. Furthermore, because the technologies used to obtain unauthorized access to, or to sabotage or disrupt, systems change frequently and often are not recognized until launched against a target, we may be unable to anticipate these techniques or implement adequate preventative measures. We may also experience security breaches that may remain undetected for an extended period. Even if identified, we may be unable to adequately investigate or remediate incidents or breaches due to attackers increasingly using tools and techniques that are designed to circumvent controls, to avoid detection, and to remove or obfuscate forensic evidence. We may also face increased cybersecurity risks due to our reliance on internet technology and the number of our and our service providers’ employees who are (and may continue to be) working remotely, which may create additional opportunities for cybercriminals to exploit vulnerabilities. The White House, SEC and other regulators have also increased their focus on companies’ cybersecurity vulnerabilities and risks.
We and certain of our service providers are from time to time, subject to cyberattacks and security incidents. While we do not believe that we have experienced any significant system failures, accidents or security breaches, if such an event were to occur and cause interruptions in our or our critical third parties’ operations, it could lead to the loss of trade secrets or other intellectual property, as well as the public exposure of personal information of our employees and others, and could result in a material disruption of our clinical and commercialization activities and business operations, in addition to possibly requiring substantial expenditures to remedy. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and our business, reputation, and financial condition could be harmed. Any losses, costs or liabilities may not be covered by, or may exceed the coverage limits of, any or all applicable insurance policies.
The terms of our Credit Agreement may limit our flexibility in operating our business and adversely af ect our financial health and competitive position, and all of our obligations under our Credit Agreement are secured by certain of our
39
collateral and the collateral of certain of our subsidiaries, as Guarantors. If we default on these obligations, our lenders could foreclose on such assets.
In October 2023, we entered into a $75.0 million Revolving Credit Facility with Citibank, N.A. as the Administrative Agent. We, our material domestic subsidiaries, as Guarantors, and the Lenders entered into the Credit Agreement with the Administrative Agent, under which the Lenders, the Swingline Lender and the L/C Issuer agreed to make loans and other financial accommodations to us in an aggregate amount of up to $75.0 million. Borrowings under the Credit Agreement are secured by certain of our collateral and that of the Guarantors. In specified circumstances, additional guarantors are required to be added. As a result, if we default on any of our obligations under the Credit Agreement, the Lenders could foreclose on their security interest and liquidate some or all of the collateral, which would harm our business, financial condition and results of operations and could require us to reduce or cease operations.
As of the date of this report, we have been borrowed approximately $0.5 million under the Revolving Credit Facility. In order to service any indebtedness we may incur in the future, we would need to generate cash from our operating activities or other financings. Our ability to generate cash is subject, in part, to our ability to successfully execute our business strategy, as well as general economic, financial, competitive, regulatory and other factors beyond our control. Our business may not be able to generate sufficient cash flow from operations, and future borrowings or other financings may not be available to us in an amount sufficient to enable us to service our indebtedness and fund our other liquidity needs. To the extent we are required to use cash from operations or the proceeds of any future financing to service our indebtedness instead of funding working capital or other general corporate purposes, we will be less able to plan for, or react to, changes in our business, industry and in the economy generally. This could place us at a competitive disadvantage compared to our competitors that have less indebtedness.
The Credit Agreement contains customary affirmative and negative covenants that limit our ability to engage in certain transactions that may be in our long-term best interest. The affirmative covenants include, among others, covenants requiring us to maintain a leverage ratio of no greater than 2.50 to 1.00 (increasing to 3.00 to 1.00 with respect to the fiscal quarter in which a material permitted acquisition is consummated and the immediately subsequent three fiscal quarters thereafter) and maintain minimum consolidated EBITDA (as defined in the Credit Agreement) for any trailing four-quarter period of not less than $45 million. The negative covenants include, among others, limitations on our ability to incur indebtedness and certain liens, make certain investments, become liable under contingent obligations in certain circumstances, make certain restricted payments, make certain dispositions within guidelines and limits, engage in certain affiliate transactions, alter our fundamental business and make certain fundamental changes.
While we believe we are currently in compliance with the covenants contained in the Credit Agreement, we may breach these covenants in the future. Our ability to comply with these covenants may be affected by events and factors beyond our control. In the event that we breach one or more covenants, the Lenders may choose to declare an event of default and require that we immediately repay all amounts outstanding under the agreement, terminate any commitment to extend further credit and foreclose on the collateral. The occurrence of any of these events could have a material adverse effect on our business, financial condition and results of operations.
We use or draw down on our Credit Agreement or use other debt in connection with our capital deployment, which magnifies the potential for loss if the royalties acquired do not generate sufficient income to us.
We draw down on or use debt to finance a portion of our deployed capital. The use of debt creates an opportunity for an increased return but also increases the risk of loss if our assets do not generate sufficient income to us. The interest expense and other costs incurred in connection with such borrowings may not be covered by our cash flow and the level of our indebtedness could limit our ability to respond to changing business conditions. Our Credit Agreement imposes, and other debt we may incur in the future may impose, affirmative and negative covenants that could impact our operations and affect the number and size of the royalties that we may pursue. Therefore, no assurance can be given that we will be able to take advantage of favorable conditions or opportunities as a result of any restrictive covenants under our Credit Agreement or other future indebtedness. There can also be no assurance that additional debt financing, either to replace or increase existing debt financing, will be available when needed or, if available, will be obtainable on terms that are commercially reasonable.
Impairment charges pertaining to goodwill, identifiable intangible assets or other long-lived assets from our mergers and acquisitions could have an adverse impact on our results of operations and the market value of our common stock.
The total purchase price pertaining to our acquisitions in recent years have been allocated to net tangible assets, identifiable intangible assets, in-process research and development and goodwill. To the extent the value of goodwill or identifiable intangible assets or other long-lived assets become impaired, we will be required to incur material charges relating to the impairment. Any impairment charges could have a material adverse impact on our results of operations and the market value of our common stock.
40
Our investments are subject to market and credit risks that could diminish their value and these risks could be greater during periods of extreme volatility or disruption in the financial and credit markets, which could adversely impact our business, financial condition, results of operations, liquidity and cash flows.
Our investments are subject to risks of credit defaults and changes in market values. Periods of macroeconomic weakness or recession, heightened volatility or disruption in the financial and credit markets could increase these risks, potentially resulting in other than temporary impairment of assets in our investment portfolio. Any event reducing the estimated fair value of these securities, other than on a temporary basis, could have a material and adverse effect on our business, results of operations, financial condition, liquidity and cash flows. If our investment manager, fails to react appropriately to difficult market, economic and geopolitical conditions, our investment portfolio could incur material losses.
We have a risk management framework in place to identify, assess and prioritize risks, including the market and credit risks to which our investments are subject. As part of that framework, we test our investment portfolio based on various market scenarios. Under certain stressed market scenarios, unrealized losses on our investment portfolio could lead to material reductions in its carrying value.
A decline in fair value below the amortized cost of a security requires management to assess whether an impairment has occurred. The decision on whether to record an impairment is determined in part by our assessment of the financial condition and prospects of a particular issuer, projections of future cash flows and recoverability of the particular security as well as management’s assertion of whether it is more likely than not that we will sell the particular security before recovery.
Our charter documents and concentration of ownership may hinder or prevent change of control transactions.
Provisions contained in our certificate of incorporation and bylaws may discourage transactions involving an actual or potential change in our ownership. In addition, our Board of Directors may issue shares of common or preferred stock without any further action by the stockholders. Our directors, officers and certain of our institutional investors collectively beneficially own a significant portion of our outstanding common stock. Such provisions and issuances may have the effect of delaying or preventing a change in our ownership. If changes in our ownership are discouraged, delayed or prevented, it would be more difficult for our current Board of Directors to be removed and replaced, even if you or our other stockholders believe that such actions are in the best interests of us and our stockholders.
Our amended and restated bylaws provide that the Court of Chancery of the State of Delaware will be the exclusive forum for substantially all disputes between us and our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers or employees.
Our amended and restated bylaws provide that the Court of Chancery of the State of Delaware is the exclusive forum for (i) any derivative action or proceeding brought on our behalf, (ii) any action asserting a claim of breach of a fiduciary duty owed by our directors, officers or other employees to us or our stockholders, (iii) any action asserting a claim arising pursuant to any provision of the General Corporation Law of Delaware or our amended and restated certificate of incorporation or amended and restated bylaws, or (iv) any action asserting a claim governed by the internal affairs doctrine. To the extent that any such claims may be based upon federal law claims, Section 27 of the Exchange Act creates exclusive federal jurisdiction over all suits brought to enforce any duty or liability created by the Exchange Act or the rules and regulations thereunder. Furthermore, Section 22 of the Securities Act provides for concurrent jurisdiction for federal and state courts over all suits brought to enforce any duty or liability created by the Securities Act or the rules and regulations thereunder, and as such, the exclusive jurisdiction clauses set forth above would not apply to such suits. The choice of forum provisions in our amended and restated bylaws may limit a stockholder’s ability to bring a claim in a judicial forum that it finds favorable for disputes with us or our directors, officers or other employees, which may discourage such lawsuits against us and our directors, officers and other employees. By agreeing to these provisions, however, stockholders will not be deemed to have waived our compliance with the federal securities laws and the rules and regulations thereunder. Furthermore, the enforceability of similar choice of forum provisions in other companies’ certificates of incorporation and bylaws has been challenged in legal proceedings, and it is possible that a court could find these types of provisions to be inapplicable or unenforceable. If a court were to find the choice of forum provisions in our amended and restated bylaws to be inapplicable or unenforceable in an action, we may incur additional costs associated with resolving such action in other jurisdictions, which could adversely affect our business and financial condition.
Our stock price has been volatile and could experience a sudden decline in value.
The market prices for securities of biotechnology and pharmaceutical companies have historically been highly volatile, and the market has experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. Continued volatility in the overall capital markets could reduce the market price of our common stock in spite of our operating performance. Further, high stock price volatility could result in higher share-based compensation expense.
41
Our common stock has experienced significant price and volume fluctuations and may continue to experience volatility in the future. Many factors may have a significant impact on the market price of our common stock, including, but not limited to, the following factors: results of or delays in our preclinical studies and clinical trials; the success of our collaboration agreements; publicity regarding actual or potential medical results relating to products under development by us or others; announcements of technological innovations or new commercial products by us or others; developments in patent or other proprietary rights by us or others; comments or opinions by securities analysts or major stockholders or changed securities analysts’ reports or recommendations; future sales or shorting of our common stock by existing stockholders; regulatory developments or changes in regulatory guidance; litigation or threats of litigation; economic and other external factors or other disaster or crises; the departure of any of our officers, directors or key employees; period-to-period fluctuations in financial results; and price and volume fluctuations in the overall stock market.
If we are unable to remediate the identified material weakness in our internal control over financial reporting, or if we experience additional material weakness or other deficiencies or otherwise fail to maintain an effective system of internal controls, we may not be able to accurately and timely report our financial results, in which case our business may be harmed, investors may lose confidence in the accuracy and completeness of our financial reports, and the price of our common stock may decline.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting and for evaluating and reporting on the effectiveness of our system of internal control. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external reporting purposes in accordance with GAAP. We are required to furnish annually a report by management of its assessment of the effectiveness of our internal control over financial reporting as of the end of our most recent fiscal year. In addition, our independent registered public accounting firm is required to provide a related attestation report on our internal control over financial reporting.
In connection with our 2023 year-end assessment of internal control over financial reporting, we determined that the material weakness related to the ineffective process-level control activities in the business combination processes were unremediated as of December 31, 2023. For further discussion of the material weakness identified and our remedial efforts, see Item 9A. Controls and Procedures.
If we are unable to remediate successfully our existing or any future material weakness or other deficiencies in our internal control over financial reporting: the accuracy and timing of our financial reporting may be adversely affected; our liquidity, our access to capital markets, the perceptions of our creditworthiness, and our ability to complete acquisitions may be adversely affected; we may be unable to maintain compliance with applicable securities laws, Nasdaq listing requirements, and the covenants under our debt instruments regarding the timely filing of periodic reports; we may be subject to regulatory investigations and penalties; and investors may lose confidence in our financial reporting. If any such event or circumstance were to occur, our stock price could decline and our business, financial condition and results of operations could be materially adversely affected.
Our results of operations and liquidity needs could be materially negatively affected by market fluctuations and economic downturn.
Our results of operations could be materially negatively affected by economic conditions generally, both in the United States and elsewhere around the world. Concerns over inflation, energy costs, geopolitical issues, military conflicts, including the wars between Russia and Ukraine and Israel and Hamas, terrorism, public health emergencies or pandemics, the availability and cost of credit, and the U.S. financial markets have in the past contributed to, and may continue in the future to contribute to, increased volatility and diminished expectations for the economy and the markets. Sanctions imposed by the United States and other countries in response to military conflicts, including the wars between Russia and Ukraine and Israel and Hamas, may also adversely impact the financial markets and the global economy, and any economic countermeasures by the affected countries or others could exacerbate market and economic instability. Domestic and international equity markets periodically experience heightened volatility and turmoil. These events may have an adverse effect on us. In the event of a market downturn, our results of operations could be adversely affected by those factors in many ways, including making it more difficult for us to raise funds if necessary, and our stock price may further decline. We cannot provide assurance that our investments are not subject to adverse changes in market value. If our investments experience adverse changes in market value, we may have less capital to fund our operations.
| Item 1B. | Unresolved Staff Comments | ||||
None.
42
| Item 1C. | Cybersecurity | ||||
Cybersecurity Risk Management and Strategy
We have developed and implemented a cybersecurity risk management program intended to protect the confidentiality, integrity, and availability of our critical systems and information. Our cybersecurity risk management program includes a cybersecurity incident response plan.
We design and assess our program based on the National Institute of Standards and Technology (NIST), the International Organization for Standardization (ISO) and other applicable industry standards. This does not imply that we meet any particular technical standards, specifications, or requirements, only that we use the NIST, ISO and other standards as a guide to help us identify, assess, and manage cybersecurity risks relevant to our business.
Our cybersecurity risk management program is integrated into our overall enterprise risk management program, and shares common methodologies, reporting channels and governance processes that apply across the enterprise risk management program to other legal, compliance, strategic, operational, and financial risk areas.
Our cybersecurity risk management program includes:
•risk assessments designed to help identify material cybersecurity risks to our critical systems, information, products, services, and our broader enterprise information technology environment;
•a security team principally responsible for managing (i) our cybersecurity risk assessment processes, (ii) our security controls, and (iii) our response to cybersecurity incidents;
•the use of external service providers, where appropriate, to assess, test or otherwise assist with aspects of our security controls;
•cybersecurity awareness training of our employees, incident response personnel, and senior management;
•a cybersecurity incident response plan that includes procedures for responding to cybersecurity incidents; and
•a third-party risk management process for service providers, suppliers, and vendors.
We have not identified risks from known cybersecurity threats, including as a result of any prior cybersecurity incidents, that have materially affected or are reasonably likely to materially affect us, including our operations, business strategy, results of operations, or financial condition.
Cybersecurity Governance
Our Board considers cybersecurity risk as part of its risk oversight function and has delegated to the Audit Committee (the Committee) oversight of cybersecurity and other information technology risks. The Committee oversees management’s implementation of our cybersecurity risk management program.
The Committee receives regular reports from management on our cybersecurity risks. In addition, management updates the Committee, as necessary, regarding any material cybersecurity incidents, as well as any incidents with lesser impact potential.
The Committee reports to the full Board regarding its activities, including those related to cybersecurity. The full Board also receives briefings from senior management on our cyber risk management program. Board members receive presentations on cybersecurity topics from senior management, or external experts as part of the Board’s continuing education on topics that impact public companies.
Our senior management team, including the Senior Director of Information Services, is responsible for assessing and managing our material risks from cybersecurity threats. The team has primary responsibility for our overall cybersecurity risk management program and supervises both our internal cybersecurity personnel and our retained external cybersecurity consultants. The Senior Director of Information Services has over 20 years of industry experiences leading and overseeing cybersecurity programs at public and private companies.
Our senior management team supervises efforts to prevent, detect, mitigate, and remediate cybersecurity risks and incidents through various means, which may include briefings from internal security personnel; threat
43
intelligence and other information obtained from governmental, public or private sources, including external consultants engaged by us; and alerts and reports produced by security tools deployed in the information technology environment.
44
| Item 2. | Properties | ||||
The following table summarizes our principal facilities leased as of December 31, 2023, including the location and size of each facility, and their designated use. We believe our facilities are adequate for our current and near-term needs, and we will be able to locate additional facilities, as needed.
| Location | Approximate Square Feet | Operation | Lease Expiration Date | ||||||||
| Jupiter, FL | 1,650 | Corporate headquarter | October 2026 | ||||||||
| San Diego, CA | 6,850 | Office | March 2029 | ||||||||
Boston, MA(1) | 6,840 | Office | June 2029 | ||||||||
| Las Vegas, NV | 4,100 | Office | April 2028 | ||||||||
| Lawrence, KS | 3,700 | Office and laboratory | August 2032 | ||||||||
| Durham, NC | 19,300 | Office and laboratory | January 2032 | ||||||||
(1) Including lease executed in November 2023 with occupancy expected in approximately the second quarter of 2024.
| Item 3. | Legal Proceedings | ||||
See “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (11), Commitments and Contingencies—Legal Proceedings.”
| Item 4. | Mine Safety Disclosures | ||||
Not applicable.
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters, and Issuer Purchases of Equity Securities | ||||
Our common stock is traded on the Nasdaq Global Market under the symbol “LGND.” As of February 26, 2024, there were approximately 339 holders of record of the common stock.
Except for 2007, during which we declared a cash dividend on our common stock of $2.50 per share, we have not paid any dividends on our common stock in the past and currently do not expect to pay cash dividends or make any other distributions on common stock in the future. We expect to retain our future earnings, if any, for use in the operation and expansion of our business, to pay down debt and potentially for share repurchases. Any future determination to pay dividends on common stock will be at the discretion of our Board of Directors and will depend upon our financial condition, results of operations, capital requirements and such other factors as the board deems relevant.
During the fiscal year ended December 31, 2023, we did not repurchase any shares of our common stock under the stock repurchase program approved by our Board of Directors in April 2023, which allowed us to acquire up to $50 million of our common stock from time to time through April 2026.
The information required by Item 201(d) of Regulation S-K is incorporated by reference to the 2023 Annual Meeting Proxy Statement as defined in Item 10 below.
45
Performance Graph
The graph below shows the five-year cumulative total stockholder return assuming the investment of $100 and is based on the returns of the component companies weighted monthly according to their market capitalization. The graph compares total stockholder returns of our common stock, of all companies traded on the Nasdaq Stock market, as represented by the Nasdaq Composite® Index, and of the Nasdaq Biotechnology Stock Index, as prepared by The Nasdaq Stock Market Inc.
The stockholder return shown on the graph below is not necessarily indicative of future performance and we will not make or endorse any predictions as to future stockholder returns.
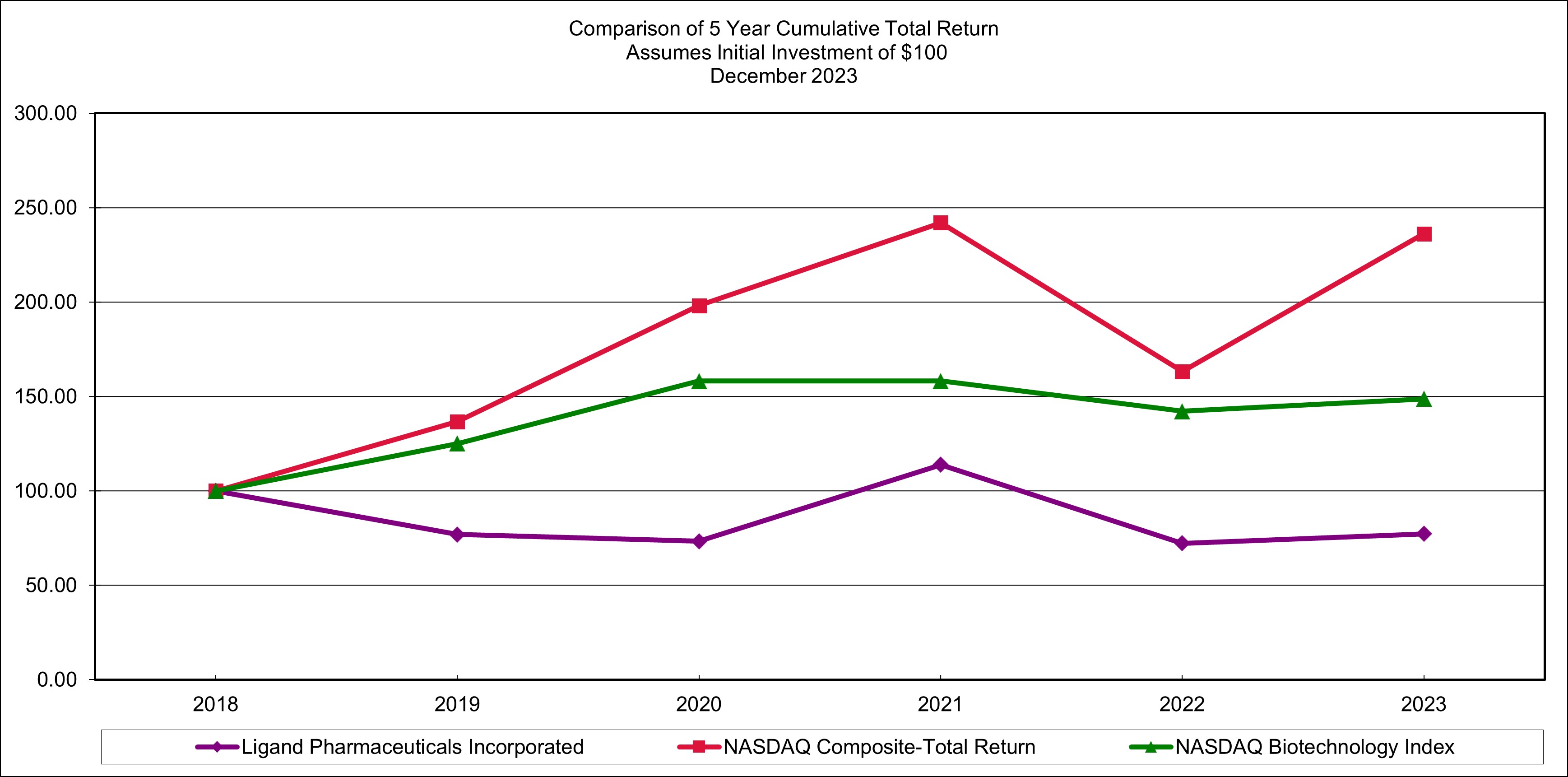
Value of $100 Invested Over Time
| 12/31/2018 | 12/31/2019 | 12/31/2020 | 12/31/2021 | 12/31/2022 | 12/31/2023 | |||||||||||||||||||||||||||||||||
| Ligand | $ | 100.00 | $ | 76.85 | $ | 73.29 | $ | 113.82 | $ | 72.27 | $ | 77.26 | ||||||||||||||||||||||||||
| NASDAQ Composite-Total Return | $ | 100.00 | $ | 136.69 | $ | 198.10 | $ | 242.03 | $ | 163.28 | $ | 236.17 | ||||||||||||||||||||||||||
| NASDAQ Biotechnology Index | $ | 100.00 | $ | 125.11 | $ | 158.17 | $ | 158.20 | $ | 142.19 | $ | 148.72 | ||||||||||||||||||||||||||
46
| Item 6. | [RESERVED] | ||||
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations | ||||
Our Management's Discussion and Analysis of Financial Condition and Results of Operations (MD&A) will help readers understand our results of operations, financial condition, and cash flows. It is provided in addition to the accompanying consolidated financial statements and notes.
OmniAb Separation and Spin-Off
On March 23, 2022, we entered into the Merger Agreement, by and among our company, Avista Public Acquisition Corp. II (New OmniAb) and OmniAb, Inc., a Delaware corporation and then wholly-owned subsidiary of our company (OmniAb), and Orwell Merger Sub Inc. (Merger Sub), pursuant to which New OmniAb combined with OmniAb, our then-antibody discovery business (the OmniAb Business), in a Reverse Morris Trust transaction. Pursuant to the Separation Agreement, we transferred the OmniAb Business, including certain of our related subsidiaries, to OmniAb and, in connection therewith, distributed (the Distribution) to Ligand stockholders 100% of the common stock of OmniAb. Immediately following the Distribution on November 1, 2022, in accordance with and subject to the terms and conditions of the Merger Agreement, Merger Sub merged with and into OmniAb (the Merger), with OmniAb continuing as the surviving company in the Merger and as a wholly-owned subsidiary of New OmniAb. After the Distribution, we do not beneficially own any shares of common stock in OmniAb and no longer consolidate OmniAb into our financial results for periods ending after October 31, 2022. As a result, OmniAb's historical financial results through the Separation are reflected in our consolidated financial statements as discontinued operations.
Our MD&A is organized as follows:
•Results of Operations. Detailed discussion of our revenue and expenses from continuing operations for years ended December 31, 2023, 2022 and 2021.
•Liquidity and Capital Resources. Discussion of key aspects of our consolidated statements of cash flows, changes in our financial position, and our financial commitments.
•Critical Accounting Policies and Estimates. Discussion of significant changes we believe are important to understand the assumptions and judgments underlying our consolidated financial statements.
•Recent Accounting Pronouncements. For summary of recent accounting pronouncements applicable to our consolidated financial statements, see “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (1), Basis of Presentation and Summary of Significant Accounting Policies.”
Results of Operations
Revenue
FY 2023 vs. FY 2022
| (Dollars in thousands) | 2023 | 2022 | Change | % Change | |||||||||||||||||||
| Royalties | $ | 83,910 | $ | 72,527 | $ | 11,383 | 16 | % | |||||||||||||||
| Captisol - Core | 28,372 | 16,429 | 11,943 | 73 | % | ||||||||||||||||||
| Captisol - COVID | — | 88,066 | (88,066) | (100) | % | ||||||||||||||||||
| Contract | 19,032 | 19,223 | (191) | (1) | % | ||||||||||||||||||
| Total revenue | $ | 131,314 | $ | 196,245 | $ | (64,931) | (33) | % | |||||||||||||||
Total revenue decreased by $64.9 million, or 33%, to $131.3 million in 2023 compared to $196.2 million in 2022 primarily due to the $88.1 million decrease in sales of COVID-related Captisol. The lower sales were due to reduced demand for remdesivir, a treatment for moderate or severe COVID-19. Core Captisol sales were $28.4 million in 2023 compared to $16.4 million in 2022. The higher sales were due to the timing of customer orders. Royalty revenue increased by $11.4 million, or 16%, to $83.9 million in 2023 compared to $72.5 million in 2022. The increase in royalty revenue is primarily due to the increases in sales of Kyprolis, Rylaze and Pneumosil.
47
FY 2022 vs. FY 2021
| (Dollars in thousands) | 2022 | 2021 | Change | % Change | |||||||||||||||||||
| Royalties | $ | 72,527 | $ | 48,927 | $ | 23,600 | 48 | % | |||||||||||||||
| Captisol - Core | 16,429 | 23,423 | (6,994) | (30) | % | ||||||||||||||||||
| Captisol - COVID | 88,066 | 140,827 | (52,761) | (37) | % | ||||||||||||||||||
| Contract | 19,223 | 28,367 | (9,144) | (32) | % | ||||||||||||||||||
| Total revenue | $ | 196,245 | $ | 241,544 | $ | (45,299) | (19) | % | |||||||||||||||
Total revenue from continuing operations decreased by $45.3 million, or 19%, to $196.2 million in 2022 compared to $241.5 million in 2021 primarily due to the $52.8 million decrease in sales of COVID-related Captisol. The lower sales were due to reduced demand for remdesivir, a treatment for moderate or severe COVID-19. Core Captisol sales were $16.4 million in 2022 compared to $23.4 million in 2021. The lower sales were due to the timing of customer orders. Royalty revenue increased by $23.6 million, or 48%, to $72.5 million in 2022 compared to $48.9 million in 2021. The increase in royalty revenue is driven primarily by increases in sales of drugs using the Pelican platform (Rylaze, Pneumosil and Teriparatide) along with an increase in sales of Kyprolis. Contract revenue decreased year over year in 2022 by $9.1 million primarily due to the timing of partner milestone events.
Royalty revenue is a function of our partners' product sales and the applicable royalty rate. Kyprolis royalty rate is under a tiered royalty rate structure with the highest being 3.0%. Evomela has a contractually fixed royalty rate of 20%. Teriparatide injection has a tiered gross profit share between 25% and 40% on sales that have been adjusted for certain deductible items as defined in the respective license agreement. The Rylaze royalty rate is in the low single digits. Contract revenue includes service revenue, license fees and development, regulatory and sales based milestone payments.
The following table represents royalty revenue by program:
| (in millions) | 2023 Estimated Partner Product Sales | Effective Royalty Rate | 2023 Royalty Revenue | 2022 Estimated Partner Product Sales | Effective Royalty Rate | 2022 Royalty Revenue | |||||||||||||||||
| Kyprolis | $ | 1,503.1 | 2.4% | $ | 35.6 | $ | 1,275.6 | 2.4% | $ | 30.1 | |||||||||||||
| Evomela | 51.0 | 20.0% | 10.2 | 51.0 | 20.0% | 10.2 | |||||||||||||||||
Teriparatide injection(1) | 37.2 | 29.8% | 11.1 | 47.2 | 33.5% | 15.8 | |||||||||||||||||
| Rylaze | 397.5 | 3.4% | 13.5 | 278.7 | 3.2% | 8.8 | |||||||||||||||||
| Other | 956.4 | 1.4% | 13.5 | 383.7 | 2.0% | 7.6 | |||||||||||||||||
| Total | $ | 2,945.2 | $ | 83.9 | $ | 2,036.2 | $ | 72.5 | |||||||||||||||
(1) - Teriparatide injection sales have been adjusted for certain deductible items as defined in the respective license agreement, and the royalty revenue is based on a tiered gross profit share.
Operating Costs and Expense
FY 2023 vs. FY 2022
| (Dollars in thousands) | 2023 | 2022 | Change | % Change | |||||||||||||||||||
| Cost of Captisol | $ | 10,512 | $ | 52,827 | $ | (42,315) | (80) | % | |||||||||||||||
| Amortization of intangibles | 33,654 | 34,237 | (583) | (2) | % | ||||||||||||||||||
| Research and development | 24,537 | 36,082 | (11,545) | (32) | % | ||||||||||||||||||
| General and administrative | 52,790 | 70,062 | (17,272) | (25) | % | ||||||||||||||||||
| Total operating costs and expenses | $ | 121,493 | $ | 193,208 | $ | (71,715) | (37) | % | |||||||||||||||
Total operating costs and expenses for 2023 decreased by $71.7 million or 37% compared with 2022.
Cost of Captisol decreased year over year in 2023 primarily due to lower sales of Captisol during 2023 and by the capacity ramp-up right of use asset impairment of $9.8 million recorded in the fourth quarter of 2022.
Amortization of intangibles decreased by $0.6 million in 2023 compared to 2022 with the decrease primarily due to the cessation of amortization of certain Pelican intangibles resulting from the sale of the Pelican business.
At any one time, we are working on multiple programs. As such, we generally do not track our R&D expenses on a specific program basis. Our R&D expenses decreased by $11.5 million in 2023 compared to 2022, with the decrease primarily due to lower share-based compensation and employee-related expenses, partially offset by an increase in R&D expenses due to the Novan acquisition.
48
General and administrative expenses decreased by $17.3 million in 2023 compared to 2022 primarily due to decreases in share-based compensation expense including a one-time charge associated with the retirement of our former CEO in the fourth quarter of 2022, and employee-related expenses, partially offset by an increase in G&A expenses due to the Novan acquisition.
FY 2022 vs. FY 2021
| (Dollars in thousands) | 2022 | 2021 | Change | % Change | |||||||||||||||||||
| Cost of Captisol | $ | 52,827 | $ | 62,176 | $ | (9,349) | (15) | % | |||||||||||||||
| Amortization of intangibles | 34,237 | 34,222 | 15 | — | % | ||||||||||||||||||
| Research and development | 36,082 | 32,105 | 3,977 | 12 | % | ||||||||||||||||||
| General and administrative | 70,062 | 46,790 | 23,272 | 50 | % | ||||||||||||||||||
| Other operating income | — | (37,600) | 37,600 | (100) | % | ||||||||||||||||||
| Total operating costs and expenses | $ | 193,208 | $ | 137,693 | $ | 55,515 | 40 | % | |||||||||||||||
Total operating costs and expenses from continuing operations for 2022 increased by $55.5 million, or 40% compared with 2021.
Cost of Captisol decreased year over year in 2022 primarily due to lower sales of Captisol during 2022, partially offset by the capacity ramp-up right of use asset impairment of $9.8 million recorded in the fourth quarter of 2022.
Amortization of intangibles remained steady in 2022 compared to 2021 as there have been no significant changes to the gross balance of intangible assets over these periods.
At any one time, we are working on multiple programs. As such, we generally do not track our R&D expenses on a specific program basis. Our R&D expenses increased by $4.0 million in 2022 compared to 2021 due to higher employee-related expenses and increased facility related expenses.
General and administrative expenses increased by $23.3 million in 2022 compared to 2021 primarily due to increases in stock compensation expense including a one-time charge associated with the retirement of our former CEO in the fourth quarter of 2022, headcount-related expenses and legal expenses.
Other operating income in 2021 was due to reducing the fair value of the CVR liability associated to the acquisition of Pfenex to zero, as the CVR payment expiration date passed on December 31, 2021 without achieving the triggering event. We did not have any other operating income in 2022.
We do not provide forward-looking estimates of costs and time to complete our ongoing research and development projects as such estimates would involve a high degree of uncertainty. Uncertainties include our inability to predict the outcome of research and clinical studies, regulatory requirements placed upon us by regulatory authorities such as the FDA and EMA, our inability to predict the decisions of our partners, our ability to fund research and development programs, competition from other entities of which we may become aware in future periods, predictions of market potential for products that may be derived from our work, and our ability to recruit and retain personnel or third-party contractors with the necessary knowledge and skills to perform certain research. Refer to “Item 1A. Risk Factors” for additional discussion of the uncertainties surrounding our research and development initiatives.
Gain on Sale of Pelican
The gain on sale of Pelican in the amount of $2.1 million for 2023 represents the excess of the fair value of 1) our investment in Primrose Bio and other economic rights; 2) the carrying amount of Pelican business assets and liabilities together with allocated goodwill as of September 18, 2023, the date of sale; and 3) $15.0 million consideration paid.
Other income (expense)
FY 2023 vs. FY 2022
| (Dollars in thousands) | 2023 | 2022 | Change | % Change | |||||||||||||||||||
| Gain (loss) from short-term investments | $ | 46,365 | $ | 28,540 | $ | 17,825 | (62) | % | |||||||||||||||
| Interest income | 7,711 | 2,046 | 5,665 | 277 | % | ||||||||||||||||||
| Interest expense | (656) | (1,799) | 1,143 | 64 | % | ||||||||||||||||||
| Gain on derivative instruments | 250 | — | 250 | N/A | |||||||||||||||||||
| Other income (expense), net | (1,952) | 4,187 | (6,139) | 147 | % | ||||||||||||||||||
| Total other income (expense), net | $ | 51,718 | $ | 32,974 | $ | 18,744 | (57) | % | |||||||||||||||
The fluctuation in the gain (loss) from short-term investments is primarily driven by the realized gain of $44.4 million from the sales of 5.0 million shares of Viking common stock in 2023, compared to no Viking shares sold in 2022. In addition,
49
the fluctuation is driven by the changes in the fair value of our ownership in Viking common stock (an unrealized gain of $2.6 million in 2023 as compared to an unrealized gain of $32.2 million in 2022).
Interest income consists primarily of interest earned on our short-term investments. The year over year increase in 2023 is primarily due to the significant interest rate increases by the federal reserve during 2023.
Interest expense includes the 0.75% coupon cash interest expense in addition to the non-cash accretion of discount (including the amortization of debt issuance costs) on our 2023 Notes. In May 2023, the 2023 Notes matured, and we paid the remaining $76.9 million principal amount and $0.3 million accrued interest in cash. The decrease in interest expense was primarily due to the zero debt outstanding balance after May 2023 compared to 2022. See additional information in “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (8), Debt.”
Other expense, net, increased year over year in 2023 primarily due to a current expected credit loss (CECL) adjustment over the Elutia commercial license right of $3.2 million and a Selexis commercial license right impairment loss of $0.9 million compared to no CECL adjustment over similar assets in 2022. In addition, we recorded a $4.2 million gain on our debt extinguishments in 2022 compared to no loss on debt extinguishments in 2023. See additional information in “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (8), Debt.”
FY 2022 vs. FY 2021
| (Dollars in thousands) | 2022 | 2021 | Change | % Change | |||||||||||||||||||
| Gain (loss) from short-term investments | $ | 28,540 | $ | (5,263) | $ | 33,803 | (642) | % | |||||||||||||||
| Interest income | 2,046 | 886 | 1,160 | 131 | % | ||||||||||||||||||
| Interest expense | (1,799) | (19,619) | 17,820 | 91 | % | ||||||||||||||||||
| Other income (expense), net | 4,187 | (7,650) | 11,837 | 155 | % | ||||||||||||||||||
| Total other income (expense), net | $ | 32,974 | $ | (31,646) | $ | 64,620 | 204 | % | |||||||||||||||
The fluctuation in the gain (loss) from short-term investments is primarily driven by the changes in the fair value of our ownership in Viking common stock (an unrealized gain of $32.2 million in 2022 as compared to an unrealized loss of $9.6 million in 2021).
Interest income consists primarily of interest earned on our short-term investments. The year over year increase in 2022 is primarily due to the significant interest rate increases by the federal reserve during 2022.
Interest expense includes the 0.75% coupon cash interest expense in addition to the non-cash accretion of discount (including the amortization of debt issuance costs) on our 2023 Notes. The year over year decrease was primarily due to the adoption of ASU 2020-06 which significantly reduced the debt discount balance subject to amortization. See additional information in “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (1), Basis of Presentation and Summary of Significant Accounting Policies.” In addition, we carried a lower average debt outstanding balance during 2022 as compared 2021. During 2022, we repurchased $266.4 million in principal of the 2023 Notes for $261.4 million in cash, including accrued interest of $0.5 million. See additional information in “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (8), Debt.”
Other income (expense), net, increased year over year in 2022 primarily due to a $4.2 million gain on our debt extinguishments in 2022 compared to $7.3 million loss on debt extinguishments in 2021. See additional information in “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (8), Debt.”
Income tax benefit (expense)
FY 2023 vs. FY 2022
| (Dollars in thousands) | 2023 | 2022 | Change | % Change | |||||||||||||||||||
| Income before income tax expense (benefit) from continuing operations | $ | 63,660 | $ | 36,011 | $ | 27,649 | 77 | % | |||||||||||||||
| Income tax benefit (expense) | (9,841) | (41,230) | 31,389 | (76) | % | ||||||||||||||||||
| Net income (loss) from continuing operations | $ | 53,819 | $ | (5,219) | $ | 59,038 | (1,131) | % | |||||||||||||||
| Effective Tax Rate | 15 | % | 114 | % | |||||||||||||||||||
50
Our effective tax rate for 2023 and 2022 was 15% and 114%, respectively. Our tax rate is affected by recurring items, such as the U.S. federal and state statutory tax rates and the relative amounts of income we earn in those jurisdictions, which we expect to be fairly consistent in the near term. It is also affected by discrete items that may occur in any given year, but are not consistent from year to year. In 2023, the variance from the U.S. federal statutory rate of 21% was primarily attributable to the decrease in unrecognized tax benefits. In 2022, the variance from the U.S. federal statutory rate of 21% was primarily due to limitations on the deductibility of stock-based compensation for certain officers and a discrete tax expense of $24.8 million related to the valuation allowance established during the fourth quarter of 2022 against deferred tax assets for California research and development credits and net operating losses. Beginning in 2022, the Tax Cuts and Jobs Act of 2017 requires taxpayers to capitalize and amortize R&D expenditures over five years for domestic research and fifteen years for foreign research pursuant to Section 174 of the Internal Revenue Code of 1986, as amended. We recorded an increase of $4.7 million to our current federal income tax expense and deferred tax assets for continuing operations during 2022 due to the capitalization of R&D under Section 174.
2023
•$7.2 million (11.3%) decrease from unrecognized tax benefits
•$2.2 million (3.4%) increase from the return to provision
•$1.2 million (1.9%) decrease from stock based compensaiton
•$1.0 million (1.6%) decrease from the foreign-derived intangible income deduction
•$0.8 million (1.3%) decrease from Section 162(m) limitation
2022
•$24.8 million (68.9%) increase from valuation allowance adjustments
•$5.9 million (16.3%) increase from Section 162(m) limitation
•$2.4 million (6.7%) decrease from the foreign-derived intangible income deduction
•$1.3 million (3.6%) increase due to excess tax benefits from share-based compensation which are recorded as a discrete item within the provision for income tax pursuant to ASU 2016-09
Net Loss from Discontinued Operations
Net loss from discontinued operations for the years ended December 31, 2023, 2022 and 2021 was $1.7 million, $28.1 million and $19.2 million, respectively. See additional information in “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (4), Spin-off Of OmniAb.”
Liquidity and Capital Resources
At December 31, 2023, we had approximately $170.3 million in cash, cash equivalents, and short-term investments. Cash and cash equivalents and short-term investments decreased by $41.6 million from last year, due to factors described in the “Cash Flow Summary” below. Our primary source of liquidity, other than our holdings of cash, cash equivalents, and investments, has been cash flows from operations. Our ability to generate cash from operations provides us with the financial flexibility we need to meet operating, investing, and financing needs.
Historically, we have liquidated our short-term investments and/or issued debt and equity securities to finance our business needs as a supplement to cash provided by operating activities. Our short-term investments include U.S. government debt securities, investment-grade corporate debt securities, bond funds and certificates of deposit. We have established guidelines relative to diversification and maturities of our investments in order to provide both safety and liquidity. These guidelines are periodically reviewed and modified to take advantage of trends in yields and interest rates. Additionally, we own certain securities which are classified as short-term investments that we received as a result of a milestone and an upfront license payment as well as 1.7 million shares of common stock in Viking.
On September 30, 2022, we entered into an At-The-Market Equity Offering Sales Agreement (Sales Agreement) with Stifel, Nicolaus & Company, Incorporated (Agent), under which we may, from time to time, sell shares of our common stock having an aggregate offering price of up to $100.0 million in “at the market” offerings through the Agent (ATM Offering). The shelf registration statement relating to such shares included a prospectus covering the offering, issuance and sale of up to $100.0 million of our common stock from time to time through the ATM Offering. The shares to be sold under the Sales Agreement may be issued and sold pursuant to the shelf registration statement. As of December 31, 2023 we have not issued any shares of common stock in the ATM Offering.
In May 2018, we issued the 2023 Notes with an aggregate principal amount of $750.0 million. A portion of the proceeds from such issuance totaling $49.7 million were used to repurchase 260,000 shares of our common stock. During 2021, we repurchased $152.0 million in principal of the 2023 Notes for $156.0 million in cash, including accrued interest of $0.3 million.
51
During 2022, we repurchased $266.4 million in principal of the 2023 Notes for $261.4 million in cash, including accrued interest of $0.5 million. On May 15, 2023, the 2023 Notes maturity date, we paid the remaining $76.9 million principal amount and $0.3 million accrued interest in cash.
We are obligated to make payments to operating leases, including rental commitments on leases that have not yet commenced. For information on these obligations, see detail in “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (7), Leases.”
We also have commitments under our supply agreement with Hovione for Captisol purchases. The total purchase obligation as of December 31, 2023 was $28.8 million, of which $8.6 million is expected to be paid within a year and the remaining amount is expected to be paid between 1 to 3 years.
In September 2019, our Board of Directors approved a stock repurchase program authorizing the repurchase of up to $500.0 million of our common stock from time to time over a period of up to three years. Our $500.0 million stock repurchase program expired in September 2022. In April 2023, our Board of Directors approved a stock repurchase program authorizing, but not requiring, the repurchase of up to $50.0 million of our common stock from time to time through April 2026. We expect to acquire these shares, if at all, primarily through open-market transactions in accordance with all applicable requirements of Rule 10b-18 under the Securities Exchange Act of 1934, as amended. The timing and amount of repurchase transactions will be determined by management based on our evaluation of market conditions, share price, legal requirements and other factors. See “Item 5. Market for Registrant's Common Equity, Related Stockholder Matters, and Issuer Purchase of Equity Securities.”
On October 12, 2023, we entered into a $75 million Revolving Credit Facility with Citibank, N.A. as the Administrative Agent. We, our material domestic subsidiaries, as Guarantors (as defined in the Credit Agreement), and the Lenders (each as defined in the Credit Agreement) entered into the Credit Agreement with the Administrative Agent, under which the Lenders, the Swingline Lender and the L/C Issuer (each as defined in the Credit Agreement) agreed to make loans and other financial accommodations to us in an aggregate amount of up to $75.0 million. At our option, borrowings under the Revolving Credit Facility accrue interest at a rate equal to either Term SOFR or a specified base rate plus an applicable margin linked to our leverage ratio, ranging from 1.75% to 2.50% per annum for Term SOFR loans and 0.75% to 1.50% per annum for base rate loans. The Revolving Credit Facility is subject to a commitment fee payable on the unused Revolving Credit Facility commitments ranging from 0.30% to 0.45%, depending on our leverage ratio. During the term of the Revolving Credit Facility, we may borrow, repay and re-borrow amounts available under the Revolving Credit Facility, subject to voluntary reductions of the swing line, letter of credit and revolving credit commitments.
Borrowings under the Credit Agreement are secured by certain of our collateral and that of the Guarantors. In specified circumstances, additional guarantors are required to be added. The Credit Agreement contains customary affirmative and negative covenants, including certain financial maintenance covenants, and events of default applicable to us. In the event of violation of the representations, warranties and covenants made in the Credit Agreement, we may not be able to utilize the Revolving Credit Facility or repayment of amounts owed thereunder could be accelerated.
As of December 31, 2023, we had $74.5 million in available borrowing under the Revolving Credit Facility, after utilizing $0.5 million for a letter of credit. The maturity date of the Revolving Credit Facility is October 12, 2026.
We believe that existing funds, cash generated from operations and existing sources of and access to financing are adequate to satisfy our needs for working capital; capital expenditure and debt service requirements; continued advancement of research and development efforts; potential stock repurchases; and other business initiatives we plan to strategically pursue, including acquisitions and strategic investments.
As of December 31, 2023, we had $3.2 million in fair value of contingent consideration liabilities associated with the acquisitions to be settled in future periods.
Cash Flow Summary
| (in thousands) | 2023 | 2022 | 2021 | |||||||||||||||||
| Net cash provided by (used in): | ||||||||||||||||||||
| Operating activities | $ | 49,577 | $ | 137,850 | $ | 78,798 | ||||||||||||||
| Investing activities | $ | (11,682) | $ | 163,624 | $ | 30,523 | ||||||||||||||
| Financing activities | $ | (59,947) | $ | (275,990) | $ | (137,761) | ||||||||||||||
In 2023, we generated cash from operations primarily from collections on our trade receivables. We used cash for investing activities primarily for the purchases of commercial license rights, Novan acquisition and investment in Primrose Bio, partially offset by cash from the sale and maturity of short-term investments including Viking shares. During the year, we used
52
cash for financing activities, including the repayment of the remaining $76.9 million principal amount upon maturity of the 2023 Notes and $0.3 million accrued interest in cash.
In 2022, we generated cash from operations primarily from collections on our trade receivables. We generated cash from investing activities primarily from the sale and maturity of short-term investments. During the year, we used cash for financing activities, including the payments related to the extinguishment of certain 2023 Notes.
In 2021, we generated cash from operations primarily due to the increase in net income. We generated cash from investing activities primarily from the sale and maturity of short-term investments. During the year, we used cash for financing activities, including the payments related to the extinguishment of certain 2023 Notes, partially offset by cash received from issuance of common stock under employee stock plans and bond hedge settlement.
Critical Accounting Policies and Estimates
The preparation of financial statements in conformity with GAAP requires estimates and assumptions that affect the reported amounts of assets and liabilities, revenues and expenses, and related disclosures of contingent liabilities in the consolidated financial statements and accompanying notes. The SEC has defined a company’s critical accounting policies as the ones that are most important to the portrayal of the company’s financial condition and results of operations, and which require the company to make its most difficult and subjective judgments, often as a result of the need to make estimates of matters that are inherently uncertain. Based on this definition, we have identified the critical accounting policies and judgments addressed below. We also have other key accounting policies, which involve the use of estimates, judgments, and assumptions that are significant to understanding our results. For additional information, see “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (1), Basis of Presentation and Summary of Significant Accounting Policies.” Although we believe that our estimates, assumptions, and judgments are reasonable, they are based upon information presently available. Actual results may differ significantly from these estimates under different assumptions, judgments, or conditions.
Revenue Recognition
We apply the following five-step model in accordance with ASC 606 in order to determine the revenue: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations, including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) the Company satisfies each performance obligation.
We receive royalty revenue on sales by our partners of products covered by patents that we or our partners own under contractual agreements. We do not have future performance obligations under these license arrangements. We generally satisfy our obligation to grant intellectual property rights on the effective date of the contract. However, we apply the royalty recognition constraint required under the guidance for sales-based royalties which requires a royalty to be recorded no sooner than the underlying sale occurs. Therefore, royalties on sales of products commercialized by our partners are recognized in the quarter the product is sold. Our partners generally report sales information to us on a one quarter lag. Thus, we estimate the expected royalty proceeds based on an analysis of historical experience and interim data provided by our partners including their publicly announced sales. Differences between actual and estimated royalty revenues are adjusted in the period in which they become known, typically the following quarter.
Our contracts with customers often will include variable consideration in the form of contingent milestone-based payments. We include contingent milestone based payments in the estimated transaction price when it is probable a significant reversal in the amount of cumulative revenue recognized will not occur. These estimates are based on historical experience, anticipated results and our best judgment at the time. If the contingent milestone based payment is sales-based, we apply the royalty recognition constraint and record revenue when the underlying sale has taken place. Significant judgments must be made in determining the transaction price for our sales of intellectual property. Because of the risk that products in development with our partners will not reach development based milestones or receive regulatory approval, we generally recognize any contingent payments that would be due to us upon the occurrence of the development milestone or regulatory approval.
Revenue from Captisol sales is recognized when control of Captisol material or intellectual property license rights is transferred to our customers in an amount that reflects the consideration we expect to receive from our customers in exchange for those products. A performance obligation is considered distinct from other obligations in a contract when it provides a benefit to the customer either on its own or together with other resources that are readily available to the customer and is separately identified in the contract. For Captisol material, we consider our performance obligation is satisfied at a point in time, once we have transferred control of the product, meaning the customer has the ability to use and obtain the benefit of the
53
Captisol material or intellectual property license right. We recognize revenue for satisfied performance obligations only when we determine there are no uncertainties regarding payment terms or transfer of control. Sales tax and other taxes we collect concurrent with revenue-producing activities are excluded from revenue. We have elected to recognize the cost of freight and shipping when control over Captisol material has transferred to the customer as an expense in Cost of Captisol. We expense incremental costs of obtaining a contract when incurred if the expected amortization period of the asset that we would have recognized is one year or less or the amount is immaterial. We did not incur any incremental costs of obtaining a contract during the periods reported.
We occasionally have sub-license obligations related to arrangements for which we receive license fees, milestones and royalties. We evaluate the determination of gross as a principal versus net as an agent reporting based on each individual agreement.
Goodwill and Intangible Assets — Impairment Assessments
Goodwill
Goodwill is evaluated annually for impairment using either a quantitative or qualitative analysis. Goodwill is tested for impairment at the reporting unit level, and is based on the net assets for each reporting unit, including goodwill and intangible assets. Goodwill is assigned to each reporting unit, as this represents the lowest level that constitutes a business and is the level at which management regularly reviews the operating results. The Company performs a quantitative analysis using a discounted cash flow model and other valuation techniques, but may elect to perform a qualitative analysis. In addition, goodwill is evaluated for impairment whenever an event occurs or circumstances change that would indicate that it is more likely than not that the fair value of a reporting unit is less than its carrying amount. Events or circumstances that may result in an impairment review include changes in macroeconomic conditions, industry and market considerations, cost factors, overall financial performance, other relevant entity-specific events, specific events affecting the reporting unit or sustained decrease in share price.
The annual goodwill impairment test was performed using a qualitative analysis in 2023 and 2022. A qualitative analysis is performed by assessing certain trends and factors, including projected market outlook and growth rates, forecasted and actual sales and operating profit margins, discount rates, industry data, and other relevant qualitative factors. These trends and factors are compared to, and based on, the assumptions used in the most recent quantitative analysis performed for each reporting unit. The results of the qualitative analyses did not indicate a need to perform quantitative analysis.
In 2022, during an interim period we used a quantitative assessment for goodwill and the relative fair value method to reallocate goodwill for the OmniAb business and Ligand core business due to the reorganization of the Company’s business discussed in “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (4), Spin-off of OmniAb.”
Intangible Assets
We regularly perform reviews to determine if an event occurred that may indicate the carrying values of our intangible assets are impaired. If indicators of impairment exist, we assess the recoverability of the affected long-lived assets by comparing its carrying amounts to its undiscounted cash flows. If the affected assets are not recoverable, we estimate the fair value of the assets and record an impairment loss if the carrying value exceeds the fair value. Factors that may indicate potential impairment include a significant decline in our stock price and market capitalization compared to net book value, significant changes in the ability of an asset to generate positive cash flows and the pattern of utilization of a particular asset.
In order to estimate the fair value of identifiable intangible assets, we estimate the present value of future cash flows from those assets. The key assumptions that we use in our discounted cash flow model are the amount and timing of estimated future cash flows to be generated by the asset over an extended period of time and a rate of return that considers the relative risk of achieving the cash flows, the time value of money, and other factors that a willing market participant would consider. Significant judgment is required to estimate the amount and timing of future cash flows and the relative risk of achieving those cash flows.
Assumptions and estimates about future values and remaining useful lives are complex and often subjective. They can be affected by a variety of factors, including external factors such as industry and economic trends, and internal factors such as changes in our business strategy and our internal forecasts. For example, if our future operating results do not meet current forecasts or if we experience a sustained decline in our market capitalization that is determined to be indicative of a reduction in fair value of our reporting unit, we may be required to record future impairment charges for purchased intangible assets. Impairment charges could materially decrease our future net income and result in lower asset values on our balance sheet.
54
Income Taxes
Our provision for income taxes, deferred tax assets and liabilities, and reserves for unrecognized tax benefits reflect our best assessment of estimated future taxes to be paid. Significant judgments and estimates based on interpretations of existing tax laws or regulations in the United States are required in determining our provision for income taxes. Changes in tax laws, statutory tax rates, and estimates of our future taxable income could impact the deferred tax assets and liabilities provided for in the consolidated financial statements and would require an adjustment to the provision for income taxes.
Deferred tax assets are regularly assessed to determine the likelihood they will be recovered from future taxable income. A valuation allowance is established when we believe it is more likely than not the future realization of all or some of a deferred tax asset will not be achieved. In evaluating our ability to recover deferred tax assets within the jurisdiction which they arise, we consider all available positive and negative evidence. Factors reviewed include the cumulative pre-tax book income for the past three years, scheduled reversals of deferred tax liabilities, our history of earnings and reliability of our forecasts, projections of pre-tax book income over the foreseeable future, and the impact of any feasible and prudent tax planning strategies.
We recognize the impact of a tax position in our financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Tax authorities regularly examine our returns in the jurisdictions in which we do business and we regularly assess the tax risk of our return filing positions. Due to the complexity of some of the uncertainties, the ultimate resolution may result in payments that are materially different from our current estimate of the tax liability. These differences, as well as any interest and penalties, will be reflected in the provision for income taxes in the period in which they are determined.
Share-Based Compensation
We measure and recognize compensation expense for all share-based payments, including restricted stock, ESPP and stock options, based on the estimated fair value. Restricted stock unit (RSU) and performance stock unit (PSU) are all considered restricted stock. The fair value of restricted stock is determined by the closing market price of our common stock on the date of grant. We recognize share-based compensation expense based on the fair value on a straight-line basis over the requisite service periods of the awards, taking into consideration of forfeitures as they occur. A PSU generally represents a right to receive a certain number of shares of common stock based on the achievement of corporate performance goals and continued employment during the vesting period. At each reporting period, we reassess the probability of the achievement of such corporate performance goals and any expense change resulting from an adjustment in the estimated shares to be released are treated as a cumulative catch-up in the period of adjustment. A limited amount of PSUs contain a market condition dependent upon the Company’s relative and absolute total stockholder return over a three-year period, with a range of 0% to 200% of the target amount granted to be issued under the award. Share-based compensation expense for these PSUs is measured using the Monte-Carlo simulation valuation model and is not adjusted for the achievement, or lack thereof, of the market conditions.
Conversion and Modification of Equity Awards Outstanding at Separation Date
In connection with the OmniAb Separation on November 1, 2022, under the provisions of the existing plans, we adjusted our outstanding equity awards in accordance with the Merger Agreement to preserve the intrinsic value of the awards immediately before and after the Distribution. Upon the Distribution, employees holding stock options, restricted stock units and performance restricted stock units denominated in pre-Distribution Ligand stock received a number of otherwise-similar awards either in post-Distribution Ligand stock or in a combination of post-Distribution Ligand stock and OmniAb stock based on conversion ratios outlined for each group of employees in the Merger Agreement that we entered into in connection with the Distribution. The equity awards that were granted prior to March 2, 2022 were converted under the shareholder method, wherein employees holding outstanding equity awards received equity awards in both Ligand and OmniAb. For equity awards granted after March 2, 2022, for Ligand employees, the number of awards that were outstanding at the Separation were proportionately adjusted into post-Distribution Ligand stock to maintain the aggregate intrinsic value of the awards at the date of the Separation; for OmniAb employees, the number of awards that were outstanding at the Separation were proportionately adjusted into post-Distribution OmniAb stock to maintain the aggregate intrinsic value of the awards at the date of the Separation. The conversion ratio was determined based on the relative values of Ligand common stock in the “regular way” and “ex-distribution” markets during the five-trading day period prior to the closing of the business combination.
These modified awards otherwise retained substantially the same terms and conditions, including term and vesting provisions. Additionally, we will not incur any future compensation cost related to equity awards held by OmniAb employees and directors. We will incur future compensation cost related to OmniAb equity awards held by our employees.
Recent Accounting Pronouncements
55
For the summary of recent accounting pronouncements applicable to our consolidated financial statements, see “Item 8. Financial Statements and Supplementary Data—Notes to Consolidated Financial Statements—Note (1), Basis of Presentation and Summary of Significant Accounting Policies.”
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk | ||||
We are exposed to market risk from interest rates and equity prices which could affect our results of operations, financial condition and cash flows. We manage our exposure to these market risks through our regular operating and financing activities.
Investment Portfolio Risk
At December 31, 2023, our investment portfolio included investments in available-for-sale securities of $147.4 million, including the investment in Viking common stock of $32.2 million. These securities are subject to market risk and may decline in value based on market conditions.
Foreign Currency Risk
Through our licensing and business operations, we are exposed to foreign currency risk. Foreign currency exposures arise from transactions denominated in a currency other than the functional currency and from foreign denominated revenues and profit translated into U.S. dollars. Our license partners sell our products worldwide in currencies other than the U.S. dollar. Because of this, our revenues from royalty payments are subject to risk from changes in exchange rates.
We purchase Captisol from Hovione, located in Lisbon, Portugal. Payments to Hovione are denominated and paid in U.S. dollars; however, the unit price of Captisol contains an adjustment factor which is based on the sharing of foreign currency risk between the two parties. The effect of an immediate 10% change in foreign exchange rates would not have a material impact on our financial condition, results of operations or cash flows. We do not currently hedge our exposures to foreign currency fluctuations.
Interest Rate Risk
We are exposed to changes in interest rates related primarily to our investment portfolio. Our investment policy and strategy are focused on the preservation of capital and supporting our liquidity requirements. We use a combination of internal and external management to execute our investment strategy. We typically invest in highly rated securities, with the primary objective of minimizing the risk of principal loss. Our investment policy generally requires securities to be investment grade and limits the amount of credit exposure to any one issuer. We have historically maintained a relatively short average maturity for our investment portfolio, and we believe a hypothetical 100 basis point adverse move in interest rates across all maturities would not materially impact the fair market value of the portfolio in either period.
56
| Item 8. | Consolidated Financial Statements and Supplementary Data | ||||
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Page | |||||
Report of Independent Registered Public Accounting Firm (PCAOB ID: | |||||
57
Report of Independent Registered Public Accounting Firm
To the Stockholders and the Board of Directors of Ligand Pharmaceuticals Incorporated
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Ligand Pharmaceuticals Incorporated (the Company) as of December 31, 2023 and 2022, the related consolidated statements of operations, comprehensive income (loss), stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2023, and the related notes, (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2023, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company's internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 29, 2024, expressed an adverse opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
| Impairment assessment of finite-lived intangibles | ||||||||||||||
| Description of the Matter | At December 31, 2023, the Company’s finite-lived intangible assets totaled $299.6 million. As discussed in Note 1 to the consolidated financial statements, the Company reviews finite-lived intangible assets for impairment whenever events or changes in circumstances indicate the carrying amount may not be recoverable. The Company did not identify indicators of impairment for its finite-lived intangibles at December 31, 2023. Auditing management’s assessment of impairment is challenging due to the degree of subjective auditor judgment necessary in evaluating management’s process to identify potential indicators of impairment and the related assessment of the severity of such indicators in determining whether a triggering event has occurred. A high degree of auditor judgment was required to evaluate potential triggering events which included market conditions, industry and economic trends, changes in regulations, clinical success and historical and forecasted financial results. The evaluation of triggering events could have a significant effect on the Company’s impairment assessment and the determination of whether further quantitative analysis of finite-lived intangible assets was required. | |||||||||||||
58
| How We Addressed the Matter in Our Audit | We obtained an understanding of management’s process to identify indicators of impairment, including the qualitative analysis and related inputs and assumptions used in performing the analyses. We evaluated the design and tested the operating effectiveness of the controls that address the identification of indicators of impairment. To test the Company’s evaluation of indicators of impairment for finite-lived intangibles, our audit procedures included, among others, assessing the methodologies and testing the completeness and accuracy of the Company’s analysis of events or changes in circumstances. As part of our evaluation, we considered market conditions, industry and economic trends, changes in regulations, clinical success and historical and forecasted financial results, in assessing whether an indicator of impairments exists. | |||||||||||||
| Valuation of the investment in Primrose Bio, Inc. | ||||||||||||||
| Description of the Matter | As discussed in Note 2 to the consolidated financial statements, in September 2023, the Company acquired a non-controlling interest in Primrose Bio, Inc. The Company received common shares, preferred shares and restricted shares of Primrose Bio, Inc. in consideration for the sale of 100% of its ownership in Pelican. Determining the fair value of the investment in Primrose Bio, Inc. as consideration received required management to make significant judgments about the valuation methodologies, including the unobservable inputs and other assumptions and estimates used in the valuation of the investment in Primrose Bio, Inc. Auditing management’s determination of the fair value of the consideration received involved complex judgement due to the selection of valuation methodologies and estimation used by management in determining the fair value of the Company’s investment in Primrose Bio, Inc. The Company used significant unobservable inputs which are significant to the valuation of the consideration received, such as discount rates, weight-average cost of capital, projected revenues and estimates of future cash flows. | |||||||||||||
| How We Addressed the Matter in Our Audit | We obtained an understanding, evaluated the design and tested the operating effectiveness of controls over the Company’s valuation of its investment in Primrose Bio, Inc. This included controls over management’s assessment of the significant inputs and estimates included in the determination of the fair value. Our audit procedures included, among others, evaluating the valuation methodologies used by the Company and testing significant unobservable inputs, estimates and the mathematical accuracy of the Company’s valuation calculation. We involved our valuation specialists to assist with the application of these procedures. We compared significant inputs and underlying data used in the Company’s valuation to agreements, information available from third-party sources and market data. We obtained and evaluated information that corroborates or contradicts the Company’s inputs and assumptions. | |||||||||||||
/s/ Ernst & Young LLP
We have served as the Company's auditor since 2016.
February 29, 2024
LIGAND PHARMACEUTICALS INCORPORATED
CONSOLIDATED BALANCE SHEETS
(in thousands, except par value)
| December 31, | |||||||||||
| 2023 | 2022 | ||||||||||
| ASSETS | |||||||||||
| Current assets: | |||||||||||
Cash and cash equivalents | $ | $ | |||||||||
| Short-term investments | |||||||||||
59
| Accounts receivable, net | |||||||||||
| Inventory | |||||||||||
| Income taxes receivable | |||||||||||
| Prepaid expenses | |||||||||||
| Other current assets | |||||||||||
| Total current assets | |||||||||||
| Deferred income taxes, net | |||||||||||
| Intangible assets, net | |||||||||||
| Goodwill | |||||||||||
| Commercial license and other economic rights | |||||||||||
| Property and equipment, net | |||||||||||
| Operating lease assets | |||||||||||
| Finance lease assets | |||||||||||
| Equity method investment in Primrose Bio | |||||||||||
| Other investments | |||||||||||
| Other assets | |||||||||||
| Total assets | $ | $ | |||||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | |||||||||||
| Current liabilities: | |||||||||||
| Accounts payable | $ | $ | |||||||||
| Accrued liabilities | |||||||||||
| Current contingent liabilities | |||||||||||
| Deferred revenue | |||||||||||
| Current operating lease liabilities | |||||||||||
| Current finance lease liabilities | |||||||||||
| 2023 convertible senior notes, net | |||||||||||
| Total current liabilities | |||||||||||
| Long-term deferred revenue | |||||||||||
| Long-term contingent liabilities | |||||||||||
| Deferred income taxes, net | |||||||||||
| Long-term operating lease liabilities | |||||||||||
| Other long-term liabilities | |||||||||||
| Total liabilities | |||||||||||
| Commitments and contingencies | |||||||||||
| Stockholders’ equity: | |||||||||||
Preferred stock, $ | |||||||||||
Common stock, $ | |||||||||||
| Additional paid-in capital | |||||||||||
| Accumulated other comprehensive loss | ( | ( | |||||||||
| Retained earnings | |||||||||||
| Total stockholders’ equity | |||||||||||
| Total liabilities and stockholders’ equity | $ | $ | |||||||||
See accompanying notes to these consolidated financial statements.
LIGAND PHARMACEUTICALS INCORPORATED
CONSOLIDATED STATEMENTS OF OPERATIONS
(in thousands, except per share amounts)
| Year Ended December 31, | |||||||||||||||||
| 2023 | 2022 | 2021 | |||||||||||||||
60
| Revenues: | |||||||||||||||||
| Royalties | $ | $ | $ | ||||||||||||||
| Captisol | |||||||||||||||||
| Contract | |||||||||||||||||
| Total revenues | |||||||||||||||||
| Operating costs and expenses: | |||||||||||||||||
Cost of Captisol | |||||||||||||||||
| Amortization of intangibles | |||||||||||||||||
| Research and development | |||||||||||||||||
| General and administrative | |||||||||||||||||
| Other operating income | ( | ||||||||||||||||
| Total operating costs and expenses | |||||||||||||||||
| Gain on sale of Pelican | ( | ||||||||||||||||
| Income from continuing operations | |||||||||||||||||
| Other income (expense): | |||||||||||||||||
| Gain (loss) from short-term investments | ( | ||||||||||||||||
| Interest income | |||||||||||||||||
| Interest expense | ( | ( | ( | ||||||||||||||
| Gain on derivative instruments | |||||||||||||||||
| Other income (expense), net | ( | ( | |||||||||||||||
| Total other income (expense), net | ( | ||||||||||||||||
| Income before income tax from continuing operations | |||||||||||||||||
| Income tax benefit (expense) | ( | ( | |||||||||||||||
| Net income (loss) from continuing operations | ( | ||||||||||||||||
| Net loss from discontinued operations | ( | ( | ( | ||||||||||||||
| Net income (loss): | $ | $ | ( | $ | |||||||||||||
| Basic net income (loss) from continuing operations per share | $ | $ | ( | $ | |||||||||||||
| Basic net loss from discontinued operations per share | $ | ( | $ | ( | $ | ( | |||||||||||
| Basic net income (loss) per share | $ | $ | ( | $ | |||||||||||||
| Shares used in basic per share calculation | |||||||||||||||||
| Diluted net income (loss) from continuing operations per share | $ | $ | ( | $ | |||||||||||||
| Diluted net loss from discontinued operations per share | $ | ( | $ | ( | $ | ( | |||||||||||
| Diluted net income (loss) per share | $ | $ | ( | $ | |||||||||||||
| Shares used in diluted per share calculation | |||||||||||||||||
See accompanying notes to these consolidated financial statements.
61
LIGAND PHARMACEUTICALS INCORPORATED
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME (LOSS)
(in thousands)
| Year Ended December 31, | |||||||||||||||||
| 2023 | 2022 | 2021 | |||||||||||||||
| Net income (loss) | $ | $ | ( | $ | |||||||||||||
| Unrealized net gain (loss) on available-for-sale securities, net of tax | ( | ( | |||||||||||||||
| Comprehensive income (loss) | $ | $ | ( | $ | |||||||||||||
See accompanying notes to these consolidated financial statements.
62
LIGAND PHARMACEUTICALS INCORPORATED
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY
(in thousands)
| Common Stock | Additional paid-in capital | Accumulated other comprehensive income (loss) | Retain earnings | Total stockholders’ equity | |||||||||||||||||||||||||||||||
| Shares | Amount | ||||||||||||||||||||||||||||||||||
| Balance at December 31, 2020 | $ | $ | $ | ( | $ | $ | |||||||||||||||||||||||||||||
| Issuance of common stock under employee stock compensation plans, net of shares withheld for payroll taxes | — | — | |||||||||||||||||||||||||||||||||
| Share-based compensation | — | — | — | — | |||||||||||||||||||||||||||||||
| Unrealized net loss on available-for-sale securities, net of tax | — | — | — | ( | — | ( | |||||||||||||||||||||||||||||
| Reacquisition of equity due to 2023 debt extinguishment, net of tax | — | — | ( | — | — | ( | |||||||||||||||||||||||||||||
| Warrant and bond hedge unwind transactions | — | — | — | — | |||||||||||||||||||||||||||||||
| Net income | — | — | — | — | |||||||||||||||||||||||||||||||
| Balance at December 31, 2021 | ( | ||||||||||||||||||||||||||||||||||
| — | — | ( | — | ( | |||||||||||||||||||||||||||||||
| Issuance of common stock under employee stock compensation plans, net of shares withheld for payroll taxes | — | ( | — | — | ( | ||||||||||||||||||||||||||||||
| Share-based compensation | — | — | — | — | |||||||||||||||||||||||||||||||
| Unrealized net loss on available-for-sale securities, net of tax | — | — | ( | — | ( | ||||||||||||||||||||||||||||||
| Bond hedge transaction | — | — | — | — | |||||||||||||||||||||||||||||||
| Distribution of OmniAb | — | — | ( | — | — | ( | |||||||||||||||||||||||||||||
| Net loss | — | — | — | — | ( | ( | |||||||||||||||||||||||||||||
| Balance at December 31, 2022 | ( | ||||||||||||||||||||||||||||||||||
| Issuance of common stock under employee stock compensation plans, net of shares withheld for payroll taxes | — | — | |||||||||||||||||||||||||||||||||
| Share-based compensation | — | — | — | — | |||||||||||||||||||||||||||||||
| Unrealized net loss on available-for-sale securities, net of tax | — | — | — | ||||||||||||||||||||||||||||||||
| Final distribution of OmniAb | — | — | — | — | |||||||||||||||||||||||||||||||
| Final tax impact of OmniAb distribution | — | — | — | — | |||||||||||||||||||||||||||||||
| Net income | — | — | — | — | |||||||||||||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | $ | ( | $ | $ | |||||||||||||||||||||||||||||
See accompanying notes to these consolidated financial statements.
63
LIGAND PHARMACEUTICALS INCORPORATED
CONSOLIDATED STATEMENTS OF CASH FLOWS
(in thousands)
| Year Ended December 31, | |||||||||||||||||
| 2023 | 2022 | 2021 | |||||||||||||||
| Operating activities | |||||||||||||||||
| Net income (loss) | $ | $ | ( | $ | |||||||||||||
| Adjustments to reconcile net income (loss) to net cash provided by (used in) operating activities: | |||||||||||||||||
| Gain on sale of Pelican | ( | ||||||||||||||||
| Change in estimated fair value of contingent liabilities | ( | ( | ( | ||||||||||||||
| Depreciation of fixed assets and amortization of intangible assets | |||||||||||||||||
| Loss (gain) short-term investments | ( | ( | |||||||||||||||
Amortization/accretion of premium (discount) on investments, net | ( | ||||||||||||||||
| Amortization of debt discount and issuance fees | |||||||||||||||||
| Gain on derivative instruments | ( | ||||||||||||||||
| Loss (gain) on debt extinguishment | ( | ||||||||||||||||
| Amortization of commercial license and other economic rights | ( | ( | ( | ||||||||||||||
| Adjustment to credit loss reserves of commercial license rights | |||||||||||||||||
| Impairment loss of commercial license rights | |||||||||||||||||
| Lease amortization expense | |||||||||||||||||
| Share-based compensation | |||||||||||||||||
| Losses from equity method investment in Primrose Bio | |||||||||||||||||
| Deferred income taxes, net | ( | ||||||||||||||||
| Other | |||||||||||||||||
| Changes in operating assets and liabilities, net of acquisitions and dispositions: | |||||||||||||||||
| Accounts receivable, net | ( | ( | |||||||||||||||
| Inventory | ( | ( | |||||||||||||||
| Other economic rights | ( | ||||||||||||||||
| Accounts payable and accrued liabilities | ( | ( | |||||||||||||||
| Income taxes receivable and payable | ( | ( | |||||||||||||||
| Deferred revenue | ( | ( | |||||||||||||||
| Other assets and liabilities | ( | ( | |||||||||||||||
| Net cash provided by operating activities | |||||||||||||||||
| Investing activities | |||||||||||||||||
| Purchase of commercial license rights | ( | ||||||||||||||||
| Purchases of property and equipment | ( | ( | ( | ||||||||||||||
| Purchases of short-term investments | ( | ( | ( | ||||||||||||||
| Proceeds from commercial license rights | |||||||||||||||||
| Proceeds from sale of short-term investments | |||||||||||||||||
| Proceeds from maturity of short-term investments | |||||||||||||||||
| Cash paid for equity method investment - Nucorion | ( | ||||||||||||||||
| Cash paid for investment in Primrose Bio | ( | ||||||||||||||||
| Cash paid for Novan acquisition, net of restricted cash received | ( | ||||||||||||||||
| Other, net | ( | ( | |||||||||||||||
| Net cash (used) provided in investing activities | ( | ||||||||||||||||
| Financing activities | |||||||||||||||||
| Net cash transferred to OmniAb at separation | ( | ||||||||||||||||
| Repayment at maturity/repurchase of 2023 Notes | ( | ( | ( | ||||||||||||||
| Payments under finance lease obligations | ( | ( | ( | ||||||||||||||
| Cash paid for OmniAb transaction costs | ( | ||||||||||||||||
| Cash paid for debt issuance costs | ( | ||||||||||||||||
| Proceeds from bond hedge settlement | |||||||||||||||||
| Net proceeds from stock option exercises and ESPP | |||||||||||||||||
Taxes paid related to net share settlement of equity awards | ( | ( | ( | ||||||||||||||
64
| Repurchase of warrants | ( | ||||||||||||||||
| Payments to CVR Holders | ( | ( | |||||||||||||||
| Net cash used in financing activities | ( | ( | ( | ||||||||||||||
| Net increase (decrease) in cash, cash equivalents, and restricted cash | ( | ( | |||||||||||||||
| Cash and cash equivalents at beginning of year | |||||||||||||||||
| Cash and cash equivalents at end of year | $ | $ | $ | ||||||||||||||
| Cash paid during the year: | |||||||||||||||||
| Interest paid | $ | $ | $ | ||||||||||||||
| Taxes paid | $ | $ | $ | ||||||||||||||
| Acquisition: | |||||||||||||||||
| Fair value of tangible assets acquired, net of cash and restricted cash received | $ | ||||||||||||||||
| Deferred tax asset | |||||||||||||||||
| Goodwill | |||||||||||||||||
| Intangible assets | |||||||||||||||||
| Liabilities assumed | ( | ||||||||||||||||
| Net cash paid for Novan | $ | ||||||||||||||||
| Supplemental schedule of non-cash investing and financing activities: | |||||||||||||||||
| Accrued Primrose transaction costs | $ | $ | $ | ||||||||||||||
| Accrued royalty from commercial license rights | $ | $ | $ | ||||||||||||||
| Accrued commercial license rights purchases | $ | $ | $ | ||||||||||||||
| Accrued debt issuance costs | $ | $ | $ | ||||||||||||||
| Accrued fixed asset purchases | $ | $ | $ | ||||||||||||||
| Accrued inventory purchases | $ | $ | $ | ||||||||||||||
| Unrealized gain (loss) on available-for-sale investments | $ | $ | ( | $ | ( | ||||||||||||
See accompanying notes to these consolidated financial statements.
65
Unless the context requires otherwise, references in this report to “Ligand,” “we,” “us,” the “Company,” and “our” refer to Ligand Pharmaceuticals Incorporated and its consolidated subsidiaries.
1. Basis of Presentation and Summary of Significant Accounting Policies
Business
We are a biopharmaceutical company enabling scientific advancement through supporting the clinical development of high-value medicines. We do this by providing financing, licensing our technologies or both. We operate in one reportable segment: development and licensing of biopharmaceutical assets.
Basis of Presentation and Principles of Consolidation
Our consolidated financial statements have been prepared in accordance with U.S. GAAP and include the accounts of our parent company and its wholly-owned subsidiaries. All intercompany transactions and balances have been eliminated in consolidation.
Discontinued operations
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires the use of estimates and assumptions that affect the amounts reported in the consolidated financial statements and the accompanying notes. Actual results may differ from those estimates.
Concentrations of Business Risk
Financial instruments that potentially subject us to significant concentrations of credit risk consist primarily of cash equivalents and investments. We invest excess cash principally in United States government debt securities, investment grade corporate debt securities, mutual funds and certificates of deposit. We maintain some cash and cash equivalents balances with financial institutions that are in excess of the Federal Deposit Insurance Corporation insurance limits. We have established guidelines relative to diversification and maturities that maintain safety and liquidity. These guidelines are periodically reviewed and modified to take advantage of trends in yields and interest rates.
Revenue from significant partners, which is defined as 10% or more of our total revenue, was as follows:
| Year ended December 31, | |||||||||||||||||
| 2023 | 2022 | 2021 | |||||||||||||||
| Partner A | |||||||||||||||||
| Partner B | |||||||||||||||||
| Partner C | < | < | |||||||||||||||
We obtain Captisol primarily from two sites related to a single supplier, Hovione. If this supplier were not able to supply the requested amounts of Captisol from each site, and if our safety stocks of material were depleted, we would be unable to continue to derive revenues from the sale of Captisol until we obtained material from an alternative source, which could take a considerable length of time.
Cash Equivalents
Cash equivalents consist of highly liquid investments with maturities of three months or less from the date of acquisition.
Short-term Investments
66
Short-term investments primarily consist of investments in debt and equity securities. We classify our short-term investments as “available-for-sale”. Such investments are carried at fair value, with unrealized gains and losses on debt securities included in the statement of comprehensive income (loss), net of tax, and unrealized gains and losses on equity securities included the consolidated statement of operations. We determine the cost of investments based on the specific identification method. We determine the realized gains or losses on the sale of available-for-sale securities using the specific identification method and include net realized gains and losses as a component of other income (expense) within the consolidated statements of operations.
Debt securities consist of certificates of deposit, corporate debt securities, and securities of government-sponsored entities. Debt securities have effective maturities greater than three months and less than twenty-five months from the date of acquisition. Debt securities available-for-sale in an unrealized loss position are assessed for current expected credit losses. We start by assessing whether we intend to sell the security, or whether it is more likely than not that we will be required to sell the security before recovery of its amortized cost basis. If either of the criteria regarding intent or requirement to sell is met, the security’s amortized cost basis is written down to fair value through earnings. For debt securities available-for-sale that do not meet the aforementioned criteria, we evaluate whether the decline in fair value has resulted from credit losses or other factors. In making this assessment, we consider the extent to which fair value is less than amortized cost, any changes in interest rates, and any changes to the rating of the security by a rating agency, among other factors. If this assessment indicates that a credit loss exists, the present value of cash flows expected to be collected from the security is compared to the amortized cost basis of the security. If the present value of cash flows expected to be collected is less than the amortized cost basis, a credit loss exists and an allowance for credit losses is recorded, limited by the amount that the fair value is less than the amortized cost basis. Any impairment that has not been recorded through an allowance for credit losses is recognized in other comprehensive income or loss, as applicable.
Accounts Receivable and Allowance for Credit Losses
Inventory
Property and Equipment
Property and equipment are stated at cost, subject to review for impairment, and depreciated over the estimated useful lives of the assets, which generally range from to nine years , using the straight-line method. Amortization of leasehold improvements is recorded over the shorter of the lease term or estimated useful life of the related asset. Maintenance and repairs are charged to operations as incurred. When assets are sold, or otherwise disposed of, the cost and related accumulated depreciation are removed from the accounts and any gain or loss is included in operating income or expense.
Acquisitions
We first determine whether a set of assets acquired constitute a business and should be accounted for as a business combination. If the assets acquired are not a business, we account for the transaction as an asset acquisition. Business combinations are
67
accounted for by using the acquisition method of accounting which requires us to use significant estimates and assumptions, especially with respect to intangible assets. We record the excess consideration over the aggregate fair value of tangible and intangible assets, net of liabilities assumed, as goodwill.
Under the acquisition method of accounting, we recognize separately from goodwill the identifiable assets acquired, the liabilities assumed, including contingent consideration and all contractual contingencies, generally at the acquisition date fair value. Contingent purchase consideration to be settled in cash are remeasured to estimated fair value at each reporting period with the change in fair value recorded in statement of operations. Costs that we incur to complete the business combination such as investment banking, legal and other professional fees are not considered part of consideration and we charge them to general and administrative expense as they are incurred.
Should the initial accounting for a business combination be incomplete by the end of a reporting period that falls within the measurement period, we report provisional amounts in our financial statements. During the measurement period, we adjust the provisional amounts recognized at the acquisition date to reflect new information obtained about facts and circumstances that existed as of the acquisition date that, if known, would have affected the measurement of the amounts recognized as of that date and we record those adjustments to our financial statements in the period of change, if any.
Contingent Liabilities
In connection with the acquisition of CyDex in January 2011, we recorded a contingent liability for amounts potentially due to holders of the CyDex CVRs and former license holders. The liability is periodically assessed based on events and circumstances related to the underlying milestones, royalties and material sales.
In connection with the acquisition of Metabasis in January 2010, we issued Metabasis stockholders four tradable CVRs for each Metabasis share. The fair values of the CVRs are remeasured at each reporting date through the term of the related agreement.
Any change in fair value is recorded in our consolidated statement of operations. For additional information, see “Note (6), Fair Value Measurement and Note (9), Balance Sheet Account Details.”
Goodwill, Intangible Assets and Other Long-Lived Assets
Goodwill, which has an indefinite useful life, represents the excess of cost over fair value of net assets acquired. Goodwill is reviewed for impairment at the reporting unit level at least annually during the fourth quarter, or more frequently if an event occurs indicating the potential for impairment. During the goodwill impairment review, we assess qualitative factors to determine whether it is more likely than not that the fair value of a reporting unit is less than the carrying amount, including goodwill. The qualitative factors include, but are not limited to, macroeconomic conditions, industry and market considerations, and the overall financial performance. If, after assessing the totality of these qualitative factors, we determine that it is not more likely than not that the fair value of our reporting unit is less than the carrying amount, then no additional assessment is deemed necessary. Otherwise, we proceed to perform the quantitative assessment. We will then evaluate goodwill for impairment by comparing the estimated fair value of the reporting unit to its carrying value, including the associated goodwill. To determine the fair value, we generally use a combination of market approach based on Ligand and comparable publicly traded companies in similar lines of businesses and the income approach based on estimated discounted future cash flows. Our cash flow assumptions consider historical and forecasted revenue, operating costs and other relevant factors. We may also elect to bypass the qualitative assessment in a period and elect to proceed to perform the quantitative assessment for the goodwill impairment test. We performed the annual assessment for goodwill impairment at the reporting unit level during the fourth quarter of 2023, noting no impairment.
68
Commercial license and other economic rights
Commercial license and other economic rights represent a portfolio of future milestone and royalty payment rights acquired that are passive in nature (i.e., we do not own the intellectual property or have the right to commercialize the underlying products).
For commercial license rights, we account them in accordance with ASC 310, Receivables, and are measured at amortized cost using the prospective effective interest method described in ASC 835-30, Imputation of Interest. If management determines it can reliably estimate future cash flows of the asset, we amortize the asset using the effective interest method whereby we forecast expected cash flows over the term of the arrangement to arrive at an annualized effective interest. If cash flows are not reliably estimable, the asset is accounted for using the non-accrual method.
We evaluate commercial license rights for impairment on an individual basis. If the effective interest rate is lower for the current period than the prior period, and if the gross cash flows have declined (expected and collected), we record provision expense for the change in expected cash flows.
We recognize an allowance for current expected credit losses under ASC 326 – Financial Instruments – Credit Losses on our commercial license rights. The credit rating, which is primarily based on publicly available data and updated quarterly, is the primary credit quality indicator used to determine the credit loss provision.
Provisions for changes in projected cash flows and credit losses are recorded as part of general and administrative expenses on the consolidated statements of operations.
For other economic rights, which are characterized as a funded research and development arrangement, thus we account for them in accordance with ASC 730-20, Research and Development Arrangement, and reduce the asset as the funds are expended.
Equity Method Investment
Investments that we do not consolidate but in which we have significant influence over the operating and financial policies of the investee are classified as equity method investments and are accounted for using the equity method of accounting.
In applying the equity method of accounting, investments are initially recorded at cost and are subsequently adjusted based on our proportionate share of net income or loss of the investee, net of any distributions received from the investee.
Other Investments
Other investments present equity securities that primarily include common stocks, preferred stocks we invested. Equity securities without readily determinable or estimable fair values are measured using the measurement alternative, which is cost less impairment, if any, and adjustments resulting from observable price changes in orderly transactions for the identical or similar investment of the same issuer.
Revenue Recognition
Our revenue is generated primarily from royalties on sales of products commercialized by our partners, Captisol material sales, and contract revenue for services, license fees and development, regulatory and sales based milestone payments.
We apply the following five-step model in accordance with ASC 606, Revenue from Contracts with Customers, in order to determine the revenue: (i) identification of the promised goods or services in the contract; (ii) determination of whether the promised goods or services are performance obligations, including whether they are distinct in the context of the contract; (iii) measurement of the transaction price, including the constraint on variable consideration; (iv) allocation of the transaction price to the performance obligations; and (v) recognition of revenue when (or as) the Company satisfies each performance obligation.
Royalties
We receive royalty revenue on sales by our partners of products covered by patents that we or our partners own under contractual agreements. We do not have future performance obligations under these license arrangements. We generally satisfy our obligation to grant intellectual property rights on the effective date of the contract. However, we apply the royalty recognition constraint required under the guidance for sales-based royalties which requires a royalty to be recorded no sooner than the underlying sale occurs. Therefore, royalties on sales of products commercialized by our partners are recognized in the quarter the product is sold. Our partners generally report sales information to us on a one quarter lag. Thus, we estimate the expected royalty proceeds based on an analysis of historical experience and interim data provided by our partners including their publicly announced sales. Differences between actual and estimated royalty revenues are adjusted in the period in which they become known, typically the following quarter.
Captisol Sales
69
Revenue from Captisol sales is recognized when control of Captisol material or intellectual property license rights is transferred to our customers in an amount that reflects the consideration we expect to receive from our customers in exchange for those products. A performance obligation is considered distinct from other obligations in a contract when it provides a benefit to the customer either on its own or together with other resources that are readily available to the customer and is separately identified in the contract. For Captisol material, we consider our performance obligation is satisfied at a point in time, once we have transferred control of the product, meaning the customer has the ability to use and obtain the benefit of the Captisol material or intellectual property license right. We recognize revenue for satisfied performance obligations only when we determine there are no uncertainties regarding payment terms or transfer of control. Sales tax and other taxes we collect concurrent with revenue-producing activities are excluded from revenue. We have elected to recognize the cost of freight and shipping when control over Captisol material has transferred to the customer as an expense in Cost of Captisol. We expense incremental costs of obtaining a contract when incurred if the expected amortization period of the asset that we would have recognized is one year or less or the amount is immaterial. We did not incur any incremental costs of obtaining a contract during the periods reported.
Contract Revenue
Our contracts with customers often will include variable consideration in the form of contingent milestone-based payments. We include contingent milestone based payments in the estimated transaction price when it is probable a significant reversal in the amount of cumulative revenue recognized will not occur. These estimates are based on historical experience, anticipated results and our best judgment at the time. If the contingent milestone based payment is sales-based, we apply the royalty recognition constraint and record revenue when the underlying sale has taken place. Significant judgments must be made in determining the transaction price for our sales of intellectual property. Because of the risk that products in development with our partners will not reach development based milestones or receive regulatory approval, we generally recognize any contingent payments that would be due to us upon the development milestone or regulatory approval. Depending on the terms of the arrangement, we may also defer a portion of the consideration received if we have to satisfy a future obligation, which typically occurs with our contracts for R&D services.
For R&D services we recognize revenue over time and we measure our progress using an input method. The input methods we use are based on the effort we expend or costs we incur toward the satisfaction of our performance obligation. We estimate the amount of effort we expend, including the time it will take us to complete the activities, or the costs we may incur in a given period, relative to the estimated total effort or costs to satisfy the performance obligation. This results in a percentage that we multiply by the transaction price to determine the amount of revenue we recognize each period. This approach requires us to make numerous estimates and use significant judgement. If our estimates or judgements change over the course of the collaboration, they may affect the timing and amount of revenue that we recognize in the current and future periods.
We occasionally have sub-license obligations related to arrangements for which we receive license fees, milestones and royalties. We evaluate the determination of gross as a principal versus net as an agent reporting based on each individual agreement.
Deferred Revenue
Disaggregation of Revenue
Royalty revenue for 2023, 2022 and 2021 for continuing operations are reported as below (in thousands):
| Year ended December 31, | ||||||||||||||||||||
| 2023 | 2022 | 2021 | ||||||||||||||||||
| Kyprolis | $ | $ | $ | |||||||||||||||||
| Evomela | ||||||||||||||||||||
| Teriparatide injection | ||||||||||||||||||||
| Rylaze | ||||||||||||||||||||
| Other | ||||||||||||||||||||
| $ | $ | $ | ||||||||||||||||||
70
The following table represents disaggregation of Captisol and contract revenue for continuing operations (in thousands):
| Year ended December 31, | ||||||||||||||||||||
| 2023 | 2022 | 2021 | ||||||||||||||||||
| Captisol | ||||||||||||||||||||
| Captisol - Core | $ | $ | $ | |||||||||||||||||
Captisol - COVID(a) | ||||||||||||||||||||
| $ | $ | $ | ||||||||||||||||||
| Contract | ||||||||||||||||||||
| Service Revenue | $ | $ | $ | |||||||||||||||||
| License Fees | ||||||||||||||||||||
| Milestone | ||||||||||||||||||||
| Other | ||||||||||||||||||||
| $ | $ | $ | ||||||||||||||||||
(a) Captisol - COVID represents revenue on Captisol supplied for use in formulation with remdesivir, an antiviral treatment for COVID-19.
Research and Development Expenses
Share-Based Compensation
We incur share-based compensation expense related to restricted stock, ESPP, and stock options.
Restricted stock unit (RSU) and performance stock unit (PSU) are all considered restricted stock. The fair value of restricted stock is determined by the closing market price of our common stock on the date of grant. We recognize share-based compensation expense based on the fair value on a straight-line basis over the requisite service periods of the awards, taking into consideration of forfeitures as they occur. PSU generally represents a right to receive a certain number of shares of common stock based on the achievement of corporate performance goals and continued employment during the vesting period. At each reporting period, we reassess the probability of the achievement of such corporate performance goals and any expense change resulting from an adjustment in the estimated shares to be released are treated as a cumulative catch-up in the period of adjustment. A limited amount of PSUs contain a market condition dependent upon the Company’s relative and absolute total stockholder return over a three-year period, with a range of 0 % to 200 % of the target amount granted to be issued under the award. Share-based compensation expense for these PSUs is measured using the Monte-Carlo simulation valuation model and is not adjusted for the achievement, or lack thereof, of the market conditions.
The Black-Scholes-Merton option-pricing model is used to estimate the fair value of stock purchases under our ESPP and stock options granted. The model assumptions include expected volatility, term, dividends, and the risk-free interest rate. We look to historical and implied volatility of our stock to determine the expected volatility. The expected term of an award is based on historical forfeiture experience, exercise activity, and on the terms and conditions of the stock awards. The expected dividend yield is determined to be 0 % given that except for 2007, during which we declared a cash dividend on our common stock of $2.50 per share, we have not paid any dividends on our common stock in the past and currently do not expect to pay cash dividends or make any other distributions on common stock in the future. The risk-free interest rate is based upon U.S. Treasury securities with remaining terms similar to the expected term of the share-based awards.
We grant options, RSUs and PSUs to employees and non-employee directors. Non-employee directors are accounted for as employees. Options and RSUs granted to certain non-employee directors typically vest one year from the date of grant. Options granted to employees typically vest 1/8 on the six month anniversary of the date of grant, and 1/48 each month thereafter for forty-two months . RSUs and PSUs granted to employees vest over three years . All option awards generally expire ten years from the date of grant.
71
Income Taxes
The provision for income taxes is computed using the asset and liability method, under which deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the financial reporting and tax bases of assets and liabilities, and for the expected future tax benefit to be derived from tax loss and credit carryforwards. Deferred tax assets and liabilities are determined using the enacted tax rates in effect for the years in which those tax assets are expected to be realized. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in the provision for income taxes in the period that includes the enactment date.
Deferred tax assets are regularly assessed to determine the likelihood they will be recovered from future taxable income. A valuation allowance is established when we believe it is more likely than not the future realization of all or some of a deferred tax asset will not be achieved. In evaluating the ability to recover deferred tax assets within the jurisdiction which they arise we consider all available positive and negative evidence. Factors reviewed include the cumulative pre-tax book income for the past three years, scheduled reversals of deferred tax liabilities, history of earnings and reliable forecasting, projections of pre-tax book income over the foreseeable future, and the impact of any feasible and prudent tax planning strategies.
We recognize the impact of a tax position in our financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Tax authorities regularly examine our returns in the jurisdictions in which we do business and we regularly assess the tax risk of our return filing positions. Due to the complexity of some of the uncertainties, the ultimate resolution may result in payments that are materially different from our current estimate of the tax liability. These differences, as well as any interest and penalties, will be reflected in the provision for income taxes in the period in which they are determined.
Income (Loss) Per Share
Basic income (loss) per share is calculated by dividing net income (loss) by the weighted-average number of common shares outstanding during the period. Diluted income per share is computed based on the sum of the weighted average number of common shares and potentially dilutive common shares outstanding during the period. Diluted loss per share is computed based on the sum of the weighted average number of common shares outstanding during the period.
Potentially dilutive common shares consist of shares issuable under the 2023 Notes, stock options and restricted stock. The 2023 Notes have a dilutive impact when the average market price of the Company’s common stock exceeds the applicable conversion price of the respective notes. It is our intent and policy to settle conversions through combination settlement, which essentially involves payment in cash equal to the principal portion and delivery of shares of common stock for the excess of the conversion value over the principal portion. Potentially dilutive common shares from stock options and restricted stock are determined using the average share price for each period under the treasury stock method. In addition, the following amounts are assumed to be used to repurchase shares: proceeds from exercise of stock options and the average amount of unrecognized compensation expense for stock options and restricted stock. In loss periods, basic net loss per share and diluted net loss per share are identical since the effect of otherwise dilutive potential common shares is anti-dilutive and therefore excluded.
| Year Ended December 31, | |||||||||||||||||
| 2023 | 2022 | 2021 | |||||||||||||||
| Weighted average shares outstanding: | |||||||||||||||||
| Dilutive potential common shares: | |||||||||||||||||
| Restricted stock | |||||||||||||||||
| Stock options | |||||||||||||||||
| 2023 Convertible Senior Notes | |||||||||||||||||
| Shares used to compute diluted income per share | |||||||||||||||||
| Potentially dilutive shares excluded from calculation due to anti-dilutive effect | |||||||||||||||||
Comprehensive Income (Loss)
Comprehensive income (loss) represents net income (loss) adjusted for the change during the periods presented in unrealized gains and losses on available-for-sale debt securities, foreign currency translation adjustments, and reclassification adjustments for realized gains or losses included in net income (loss). The unrealized gains or losses are reported on the Consolidated Statements of Comprehensive Income (Loss).
72
Accounting Standards Updates, Recently Adopted
Effective January 1, 2023, we adopted ASU 2021-08, Business Combinations (Topic 805): Accounting for Contract Assets and Contract Liabilities from Contracts with Customers (“ASU 2021-08”). The amendments in ASU 2021-08 require that an acquiring entity recognize and measure contract assets and contract liabilities acquired in a business combination in accordance with Accounting Standards Codification (“ASC”) Topic 606, Revenue from Contracts with Customers (“ASC Topic 606”). At the acquisition date, an acquirer should account for the related revenue contracts in accordance with ASC Topic 606 as if it had originated the contracts. The adoption of this standard did not have a material impact on our consolidated financial statements and disclosures.
Accounting Standards Not Yet Adopted
In November 2023, the Financial Accounting Standards Board (“FASB”) issued updated accounting guidance related to annual and interim segment disclosures. The updated accounting guidance, among other things, requires disclosure of certain significant segment expenses. We will adopt the updated accounting guidance in our Annual Report on Form 10-K for the year ended December 31, 2024. We do not expect impact from the adoption of the new accounting guidance will have material impact to our segment disclosures.
In December 2023, the FASB issued ASU 2023-09, Income Taxes (Topic 740): Improvements to Income Tax Disclosures. The update requires a public business entity to disclose, on an annual basis, a tabular rate reconciliation using both percentages and currency amounts, broken out into specified categories with certain reconciling items further broken out by nature and jurisdiction to the extent those items exceed a specified threshold. In addition, all entities are required to disclose income taxes paid, net of refunds received disaggregated by federal, state/local, and foreign and by jurisdiction if the amount is at least 5% of total income tax payments, net of refunds received. Adoption of the ASU allows for either the prospective or retrospective application of the amendment and is effective for annual periods beginning after December 15, 2024, with early adoption permitted. The Company has not yet completed its assessment of the impact of ASU 2023-09 on the Company’s Consolidated Financial Statements.
We do not believe that any other recently issued, but not yet effective accounting pronouncements, if adopted, would have a material impact on our consolidated financial statements or disclosures
73
2. Sale of Pelican Business and Investment in Primrose Bio
On September 18, 2023, we entered into a merger agreement, pursuant to which our subsidiary, Pelican Technology Holdings, Inc. (“Pelican”) became a wholly owned subsidiary of Primrose Bio. Primrose Bio is a private company focused on synthetic biology. Pelican has developed technology related to PET (protein expression technology) and PelicCRM197 (vaccine material), and has property and equipment, as well as leased property in San Diego, CA. As part of the transaction, we received 2,146,957 common shares, 4,278,293 preferred shares and 474,746 restricted shares of Primrose Bio. Simultaneous with the merger, we entered into a Purchase and Sale Agreement with Primrose Bio and contributed $15.0 million in exchange for 50 % of potential development milestones and certain commercial milestones from two contracts previously entered into by Primordial Genetics. In addition, starting January 1, 2025, we will receive 25 % of sales revenue of PeliCRM197 above $3.0 million and 35 % of all PeliCRM197 licensing revenue in perpetuity.
We retained contractual relationships utilizing the Pelican Expression Technology, including the commercial royalty rights to Jazz’s RYLAZE, Merck’s VAXNEUVANCE and V116 vaccines, Alvogen’s Teriparatide, Serum Institute of India’s vaccine programs, including Pneumosil and MenFive vaccines, among others.
We determined that the sale of Pelican meets the definition of a deconsolidation of a business. Net assets sold together with allocated goodwill and cash consideration paid were as follows (in thousands):
| Property and equipment, net | $ | ||||
| Intangible assets | |||||
| Other assets | |||||
| Operating lease right-of-use assets | |||||
| Financing lease right-of-use assets | |||||
| Accrued liabilities | ( | ||||
| Deferred revenue | ( | ||||
| Long-term operating lease liabilities | ( | ||||
| Other liabilities | ( | ||||
| Net assets sold | |||||
| Allocated goodwill | |||||
| Cash consideration paid | |||||
| $ | |||||
Fair value of the consideration received includes the following (in thousands):
| Equity method investment | $ | ||||
| Equity securities | |||||
| Derivative assets | |||||
| $ | |||||
Goodwill allocated to the selling business based on the relative fair value of the Pelican business and Ligand that was written off was $4.1 million, resulting in a $2.1 million gain on sale of Pelican recorded to income (loss) from operations for the year ended December 31, 2023.
Transaction costs of $1.2 million were allocated to the equity method investment and equity securities based on the relative fair value.
As described above, we will receive 25 % of sales revenue of PeliCRM197 above $3.0 million and 35 % of all PeliCRM197 licensing revenue in perpetuity. The considerations were recognized as contingent consideration under the loss recovery model and they will be measured based on the gain contingency model under ASC 450, Contingencies, and thus, will be recognized as the underlying contingencies are resolved.
In addition, we will receive 50 % of potential development milestones and certain commercial milestones from two contracts previously entered into by Primordial Genetics. The considerations were recognized as derivative assets with a fair value of $3.2 million, at the disposition date, which was included under other long-term asset in our consolidated balance sheet . They
74
are recognized as derivative assets under ASC 815, Derivatives and Hedging, as they have two underlyings (development and commercial milestones) and (i) the commercial milestones are dependent on the development milestones and (ii) the commercial milestone underlying is not determined to be predominate. The derivative assets are recorded at fair value as of September 18, 2023, and has been marked to fair value as of December 31, 2023 with an adjustment of $0.3 million. Any change in fair value is recorded to Other income (expense) in our consolidated statement of operations. For additional information, see “Note (6), Fair Value Measurement”.
Investment in Primrose Bio
We apply the equity method to investments in common stock and to other investments in entities that have risk and reward characteristics that are substantially similar to an investment in the investee’s common stock. Since the preferred stock and restricted share investment in Primrose Bio has a substantive liquidation preference, it is not substantially similar to the common stock investment and is therefore recorded as an equity security under ASC 321.
Investments - Equity Securities
We account for our common stock investment in Primrose Bio under the equity method as we have the ability to exercise significant influence over its operating and financial results. In applying the equity method, we record the investment at fair value. Ligand's proportionate share of net loss of Primrose Bio is recorded in our consolidated statements of operations for the year ended December 31, 2023. Our equity method investments are reviewed for indicators of impairment at each reporting period and are written down to fair value if there is evidence of a loss in value that is other-than-temporary. Our share of the net loss of Primrose Bio since the divestiture date for year ended December 31, 2023 was $1.8 million, which reduced Ligand's equity method investment accordingly. Any income of loss from our equity method investments are recorded to Other income (expense) in our consolidated statement of operations.
We determined that the Series A preferred stock investment in Primrose Bio did not have a readily determinable fair value and therefore elected the measurement alternative in ASC 321 to subsequently record the investment at cost, less any impairments, plus or minus changes resulting from observable price changes in orderly transactions for identical or similar investments of the same issuer. When fair value becomes determinable, from observable price changes in orderly transactions, our investment will be marked to fair value. There have been no observable price changes or impairments identified since September 18, 2023.
During the fourth quarter of 2023, our President and Chief Operating Officer, Matt Korenberg, became a board member of Primrose Bio.
75
3. Acquisition
Novan
On September 27, 2023, we closed the transaction to acquire certain assets of Novan, Inc. (“Novan”) pursuant to the agreement we entered into with Novan on July 17, 2023 for $15.0 million in cash (which agreement contemplated Novan filing for bankruptcy relief) and provided up to $15.0 million in debtor-in-possession (“DIP”) financing inclusive of a $3.0 million bridge loan funded on the same day. Novan filed for Chapter 11 reorganization on July 17, 2023. On September 27, 2023, the bankruptcy court approved our $12.2 million bid to purchase from Novan its lead product candidate berdazimer topical gel, 10.3 %, all other assets related to the NITRICIL technology platform and the rights to one commercial stage asset. The remaining commercial assets of Novan will be sold to other parties. The approved $12.2 million bid was credited to the $15.0 million DIP financing, with the balance of $2.8 million and accrued interest repaid to us.
The acquisition was accounted for as business combination. We recorded $3.1 million of acquisition-related costs for legal, due diligence and other costs in connection with the acquisition within operating expenses in our consolidated statement of operations for the year ended December 31, 2023.
The following table sets forth an allocation of the preliminary purchase price to the identifiable tangible and intangible assets acquired and liabilities assumed, with the excess recorded to goodwill (in thousands):
| Restricted Cash | $ | ||||
| Property and equipment, net | |||||
| Right-of-use asset | |||||
| Other assets | |||||
| Deferred tax asset | |||||
| Intangible assets acquired | |||||
| Goodwill | |||||
| Deferred revenue | ( | ||||
| Lease liabilities | ( | ||||
| Other liabilities | ( | ||||
| Cash paid for Novan, including restricted cash received | |||||
| DIP loan fees and interest | |||||
| Total consideration | $ | ||||
None of the goodwill is deductible for tax purposes. Acquired intangible assets of $10.7 million related to core technology. The fair value of the core technology was based on the discounted cash flow method that estimated the present value of the potential royalties, milestones, and collaboration revenue streams derived from the licensing of the related technologies. These projected cash flows were discounted to present value using a discount rate of 29 %. The fair value of the core technology is being amortized on a straight-line basis over the estimated useful life of 15 years.
Acquired other liabilities of $13.7 million related to a royalty and milestone payments purchase agreement, entered by Novan in 2019 and assumed as part of the acquisition, which previously provided Novan $25.0 million of funding used primarily in the clinical development of berdazimer topical gel, 10.3 %. Pursuant to the purchase agreement, Novan will pay ongoing quarterly payments, calculated based on an applicable percentage per product of any upfront fees, milestone payments, royalty payments or equivalent payments received by Novan pursuant to any out-license agreement, net of any upfront fees, milestone payments, royalty payments or equivalent payments paid by Novan to third parties pursuant to any agreements under which Novan has in-licensed intellectual property with respect to such products. If Novan decides to commercialize any product on its own following regulatory approval, as opposed to commercializing through an out-license agreement or other third-party arrangement, Novan will be obligated to pay a low single digits royalty on net sales of such products. This contract liability was fair valued based on the discounted cash flow method that estimated the present value of the potential royalties, milestones, and collaboration revenue streams derived from the related programs mentioned above, by applying a discount rate of 14.0 % (revenue risk-adjusted discount rate).
The estimated fair values of assets acquired, liabilities assumed and purchased intangibles are provisional. Specifically, the provisional amounts include estimated projections on the completion of the clinical development process and projected revenue related to commercializing products based on the underlying technology as well as the assumed underlying contracts. The accounting for these amounts falls within the measurement period and, therefore, we may adjust these provisional amounts to reflect new information obtained about facts and circumstances that existed as of the acquisition date.
76
4. Spin-off of OmniAb
On March 23, 2022, we entered into the Separation Agreement to separate our OmniAb Business and the Merger Agreement, pursuant to which APAC would combine with OmniAb, and acquire Ligand's OmniAb Business, in a Reverse Morris Trust transaction (collectively, the “Transactions”).
After the closing date of the Transactions on November 1, 2022, the historical financial results of OmniAb have been reflected in our consolidated financial statements as discontinued operations under GAAP for all periods presented through the date of the Distribution. Pursuant to the Transaction Agreements, Ligand contributed to OmniAb cash and certain specific assets and liabilities constituting the OmniAb Business. Pursuant to the Distribution, Ligand distributed on a pro rata basis to its shareholders as of October 26, 2022 shares of the common stock of OmniAb representing 100 % of Ligand’s interest in OmniAb. Immediately following the Distribution, Merger Sub merged with and into OmniAb, with OmniAb continuing as the surviving company in the Merger and as a wholly owned subsidiary of New OmniAb. The entire transaction was completed on November 1, 2022, and following the Merger, New OmniAb is an independent, publicly traded company whose common stock trades on NASDAQ under the symbol “OABI.” After the Distribution, we do not beneficially own any shares of common stock in OmniAb and no longer consolidate OmniAb into our financial results for periods ending after November 1, 2022.
Discontinued operations
In connection with the Merger, the Company determined its antibody discovery business qualified for discontinued operations accounting treatment in accordance with ASC 205-20. We recognized a $1.7 million tax provision adjustment related to deferred taxes during the year ended December 31, 2023 that was attributable to the discontinued operations. The following table summarizes revenue and expenses of the discontinued operations for the years ended December 31, 2022 and 2021 (in thousands):
| Year Ended December 31, | |||||||||||
| 2022 | 2021 | ||||||||||
| Revenues: | |||||||||||
| Royalties | $ | $ | |||||||||
| Contract revenue | |||||||||||
| Total revenues | |||||||||||
| Operating costs and expenses: | |||||||||||
| Amortization of intangibles | |||||||||||
| Research and development | |||||||||||
| General and administrative | |||||||||||
| Total operating costs and expenses | |||||||||||
| Loss from operations | ( | ( | |||||||||
| Other income (expense): | |||||||||||
| Gain from short-term investments | |||||||||||
| Interest expense | ( | ||||||||||
| Other income (expense), net | ( | ||||||||||
| Total other expense, net | |||||||||||
| Loss before income tax | ( | ( | |||||||||
| Income tax benefit | |||||||||||
| Net loss | $ | ( | $ | ( | |||||||
The following table summarizes the significant non-cash items, capital expenditures of the discontinued operations, and financing activities that are included in the consolidated statements of cash flows for the years ended December 31, 2022 and 2021 (in thousands):
77
| Year Ended December 31, | |||||||||||
| 2022 | 2021 | ||||||||||
| Operating activities: | |||||||||||
| Change in fair value of contingent consideration | $ | ( | $ | ||||||||
| Depreciation and amortization | |||||||||||
| Stock-based compensation expense | |||||||||||
| Investing activities: | |||||||||||
Cash paid for acquisition, net of cash acquired | $ | $ | |||||||||
| Purchase of property, plant and equipment | ( | ( | |||||||||
| Payments to CVR Holders | ( | ( | |||||||||
| Financing activities: | |||||||||||
| Payments to CVR Holders | $ | ( | $ | ( | |||||||
| Supplemental cash flow disclosures: | |||||||||||
| Purchases of property, plant and equipment included in accounts payable and accrued expenses | $ | $ | |||||||||
5. Commercial License and Other Economic Rights
As of December 31, 2023 and 2022, commercial license and other economic rights consist of the following (in thousands):
| December 31, 2023 | December 31, 2022 | |||||||||||||||||||||||||||||||||||||
| Gross | Adjustments(1) | Net | Gross | Adjustments(2) | Net | |||||||||||||||||||||||||||||||||
| Elutia and CorMatrix | $ | $ | ( | $ | $ | $ | ( | $ | ||||||||||||||||||||||||||||||
| Selexis and Dianomi | ( | ( | ||||||||||||||||||||||||||||||||||||
Ovid | ( | |||||||||||||||||||||||||||||||||||||
| Tolerance | ( | |||||||||||||||||||||||||||||||||||||
| Palvella | ||||||||||||||||||||||||||||||||||||||
| Total | $ | $ | ( | $ | $ | $ | ( | $ | ||||||||||||||||||||||||||||||
(1) Amounts represent accumulated amortization to principal of $11.2 million, credit loss adjustments of $8.1 million, and impairment of $0.9 million as of December 31, 2023.
Commercial license and other economic rights represent a portfolio of future milestone and royalty payment rights acquired from Selexis, S.A. (Selexis) in April 2013 and April 2015, CorMatrix Cardiovascular, Inc. (CorMatrix) in May 2016, which was later acquired by Aziyo (Aziyo changed its corporate name to Elutia Inc. ("Elutia") in September 2023) in 2017, Dianomi Therapeutics, Inc. in January 2019, Ovid Therapeutics Inc. in October 2023, Tolerance Therapeutics, Inc. in November 2023, and Palvella Therapeutics, Inc. in December 2023. Commercial license and other economic rights acquired are accounted for as financial assets as further discussed below.
For commercial license rights, we have elected a prospective approach to account for changes in estimated cash flows and selected a method for determining when an impairment would be recognized and how to measure that impairment. In circumstances where our new estimate of expected cash flows is greater than previously expected, we will update our yield prospectively. In circumstances where our new estimate of expected cash flows is less than previously expected and below our original estimated yield we record an impairment. Impairment is recognized by reducing the financial asset to an amount that represents the present value of our most recent estimate of expected cash flows discounted by the original effective interest rate. In circumstances where our new estimate of expected cash flows is less than previously expected, but not below our original estimated yield, we update our yield prospectively. We recorded a $0.9 million impairment loss for Selexis commercial license rights during the year ended December 31, 2023 as a result of recently reduced programs.
In May 2017, we entered into a royalty agreement with Elutia Inc. pursuant to which we will receive royalties from certain marketed products that Elutia acquired from CorMatrix. Pursuant to the agreement, we received $10.0 million in 2017 from Elutia to buydown the royalty rates on the products CorMatrix sold to Elutia. The agreement closed on May 31, 2017, in connection with the closing of the asset sale from CorMatrix to Elutia (the “CorMatrix Asset Sale”). Per the agreement, we will receive a 5 % royalty on the products Elutia acquired in the CorMatrix Asset Sale, reduced from the original 20 % royalty from CorMatrix pursuant to the previously disclosed interest purchase agreement, dated May 3, 2016 (the “Original Interest Purchase
78
Agreement”) between CorMatrix and us. In addition, Elutia has agreed to pay us up to $10.0 million of additional milestones tied to cumulative net sales of the products Elutia acquired in the CorMatrix Asset Sale and to extend the term on these royalties by one year. The royalty agreement will terminate on May 31, 2027. In addition, in May 2017, we entered into an amended and restated interest purchase agreement (the “Amended Interest Purchase Agreement”) with CorMatrix, which supersedes in its entirety the Original Interest Purchase Agreement. Other than removing the commercial products sold to Elutia in the CorMatrix Sale, the terms of the Amended Interest Purchase Agreement remain unchanged with respect to the CorMatrix developmental pipeline products, including the royalty rate of 5 % on such pipeline products. The Amended Interest Purchase Agreement will terminate 10 years from the date of the first commercial sale of such products.
We account for the Elutia commercial license right as a financial asset in accordance with ASC 310. During the year ended December 31, 2023, we further considered the current and expected future economic and market conditions and recorded a $3.2 million credit loss adjustment to Elutia commercial license rights based on the assessment of current company performance and nonpayment by Elutia in recent quarters. This credit loss adjustment was recorded to General and administrative expense in our consolidated statement of operations. As of December 31, 2023, management is in process of modifying the payment terms with Elutia and has placed the loan on the non-accrual method during the year ended December 31, 2023 until we are able to reliably estimate future cash flows.
We account for the Selexis commercial license right as a financial asset in accordance with ASC 310, and amortize the commercial license right using the effective interest method. The annual effective interest associated with the forecasted cash flows from the royalty agreement with Selexis as of December 31, 2023 is 2.5 %. Revenue is calculated by multiplying the carrying value of the commercial license right by the effective interest. The payments received in the year ended December 31, 2023 and 2022 were allocated accordingly between revenue and the amortization of the commercial license rights.
We had accounted for commercial license rights related to Dianomi on a non-accrual basis and had fully reserved the credit loss as of December 31, 2022. Dianomi Therapeutics, Inc. was dissolved in July 2023; therefore, we removed the related commercial license rights as of December 31, 2023.
In October 2023, we made an investment of $30 million to acquire a 13 % portion of the royalties and milestones owed to Ovid Therapeutics related to the potential approval and commercialization of soticlestat. We performed predominant analysis and determined that the predominant characteristics of the underlyings are the sales-based milestones and royalties since the value of the projected cash flows tied to the underlyings are highly correlated with changes in the projected cash flows from royalties and commercial milestones and not the regulatory milestones. Therefore, the Ovid contract qualifies for the royalty scope exception under ASC815, Derivatives and Hedging, and is accounted for as a financial asset in accordance with ASC 310. As soticlestat is in Phase 3 clinical trials, management has placed the investment on the non-accrual method during the year ended December 31, 2023 until we are able to reliably estimate future cash flows.
In November 2023, we acquired Tolerance Therapeutics, Inc. (“Tolerance Therapeutics”) for $20 million in cash. Tolerance Therapeutics is a holding company, owned by the inventors of TZIELD (teplizumab), that is owed a royalty of less than 1 % on worldwide net sales. TZIELD is marketed by Sanofi in 2023. We account for the Tolerance commercial license right as a financial asset in accordance with ASC 310. Due to the early stages of TZIELD's commercialization, management has placed the investment on the non-accrual method during the year ended December 31, 2023 until we are able to reliably estimate future cash flows.
In December 2023, we announced the expansion of its strategic partnership with Palvella to accelerate Phase 3 development of QTORIN rapamycin for the treatment of Microcystic Lymphatic Malformations ("Microcystic LMs"). According to the terms of the second amendment to the development funding and royalties agreement, Palvella received an upfront payment of $5.0 million from Ligand. In return for the upfront payment, among other contractual changes, the tiered royalty payable by Palvella to Ligand was increased to between 8.0 % and 9.8 % based on annual aggregate worldwide net sales of QTORIN rapamycin. We are not obligated to provide additional funding to Palvella for development or commercialization of OTORIN.
We determined the economic rights related to Palvella should be characterized as a funded research and development arrangement, thus we account for them in accordance with ASC 730-20, Research and Development Arrangement, and reduce our asset as the funds are expended by Palvella. As of December 31, 2023, of the $5.0 million upfront funding related to the second amendment with Palvella, none of the funding to Palvella was expended, and we will reduce our asset as the funds are expended by Palvella in the future. Our CEO and director, Todd Davis, is a director of Palvella, who beneficially owns less than 2 % of Palvella's outstanding equity. Mr. Davis recused himself from all of the board's consideration of the agreement between us and Palvella, including any financial analysis, the terms of the amendment and the vote to approve the purchase agreement and the related transactions.
79
6. Fair Value Measurement
We measure certain financial assets and liabilities at fair value on a recurring basis. Fair value is a market-based measurement that should be determined using assumptions that market participants would use in pricing an asset or liability. We establish a three-level hierarchy to prioritize the inputs used in measuring fair value. The levels are described in the below with level 1 having the highest priority and level 3 having the lowest:
Level 1 - Observable inputs such as quoted prices in active markets
Level 2 - Inputs other than the quoted prices in active markets that are observable either directly or indirectly
Level 3 - Unobservable inputs in which there is little or no market data, which require the Company to develop its own assumptions
The following table provides a summary of the assets and liabilities that are measured at fair value on a recurring basis as of December 31, 2023 and 2022 (in thousands):
| Fair Value Measurements at Reporting Date Using | |||||||||||||||||||||||
December 31, 2023 | Quoted Prices in Active Markets for Identical Assets | Significant Other Observable Inputs | Significant Unobservable Inputs | ||||||||||||||||||||
| Total | (Level 1) | (Level 2) | (Level 3) | ||||||||||||||||||||
| Assets: | |||||||||||||||||||||||
Short-term investments, excluding Viking(1) | $ | $ | $ | $ | |||||||||||||||||||
| Investment in Viking common stock | |||||||||||||||||||||||
| Total assets | $ | $ | $ | $ | |||||||||||||||||||
| Liabilities: | |||||||||||||||||||||||
| Contingent liabilities - CyDex | $ | $ | $ | $ | |||||||||||||||||||
Contingent liabilities - Metabasis(3) | |||||||||||||||||||||||
| Total liabilities | $ | $ | $ | $ | |||||||||||||||||||
| Fair Value Measurements at Reporting Date Using | |||||||||||||||||||||||
December 31, 2022 | Quoted Prices in Active Markets for Identical Assets | Significant Other Observable Inputs | Significant Unobservable Inputs | ||||||||||||||||||||
| Total | (Level 1) | (Level 2) | (Level 3) | ||||||||||||||||||||
| Assets: | |||||||||||||||||||||||
Short-term investments, excluding Viking (1) | $ | $ | $ | $ | |||||||||||||||||||
| Investment in Viking common stock | |||||||||||||||||||||||
| Total assets | $ | $ | $ | $ | |||||||||||||||||||
| Liabilities: | |||||||||||||||||||||||
| Contingent liabilities - CyDex | $ | $ | $ | $ | |||||||||||||||||||
Contingent liabilities - Metabasis(3) | |||||||||||||||||||||||
| Liability for amounts owed to a former licensor | |||||||||||||||||||||||
| Total liabilities | $ | $ | $ | $ | |||||||||||||||||||
(1) Excluding our investment in Viking, our short-term investments in marketable debt and equity securities are classified as available-for-sale securities based on management's intentions and are at level 2 of the fair value hierarchy, as these investment securities are valued based upon quoted prices for identical or similar instruments in markets that are not active, and model-based valuation techniques for which all significant assumptions are observable in the market. Short-term investments in bond funds are valued at their net asset value (NAV) on the last day of the period. We have classified marketable securities with original maturities of greater than one year as short-term investments based upon our ability and intent to use any and all of those marketable securities to satisfy the liquidity needs of our current operations. In addition, we have investment in warrants resulting from Seelos Therapeutics Inc. milestone payments that were settled in shares during the first quarter of 2019 and are at level 3 of the fair value hierarchy, based on Black-Scholes value estimated by management on the last day of the period.
(2) In connection with the Purchase and Sale Agreement with Primrose Bio, we received 50 % of potential development milestones and certain commercial milestones from two contracts previously entered into by Primordial Genetics. The considerations were recognized as derivative assets included under other
80
long-term asset in our consolidated balance sheet. They are recognized as derivative assets under ASC 815, Derivatives and Hedging, as they have two underlyings (development and commercial milestones) and (i) the commercial milestones are dependent on the development milestones and (ii) the commercial milestone underlying is not determined to be predominate. The fair value of the derivative assets was determined using a discounted cash flow approach using a discount rate in line with the stages of the underlying contracts.
A reconciliation of the level 3 financial instruments as of December 31, 2023 is as follows (in thousands):
| Assets | |||||
Fair value of level 3 financial instruments as of December 31, 2022 | $ | ||||
| Fair value adjustments to equity security warrants | ( | ||||
| Fair value of derivative assets | |||||
Fair value of level 3 financial instruments as of December 31, 2023 | $ | ||||
| Liabilities | |||||
Fair value of level 3 financial instruments as of December 31, 2022 | $ | ||||
| Payments to CVR holders and other contingent payments | ( | ||||
Fair value of level 3 financial instruments as of December 31, 2023 | $ | ||||
A reconciliation of the level 3 financial instruments as of December 31, 2022 is as follows (in thousands):
| Assets | |||||
Fair value of level 3 financial instruments as of December 31, 2021 | $ | ||||
| ( | |||||
Fair value of level 3 financial instruments as of December 31, 2022 | $ | ||||
| Liabilities | |||||
Fair value of level 3 financial instruments as of December 31, 2021 | $ | ||||
| Fair value adjustments to contingent liabilities | ( | ||||
Fair value of level 3 financial instruments as of December 31, 2022 | $ | ||||
Assets Measured on a Non-Recurring Basis
We apply fair value techniques on a non-recurring basis associated with valuing potential impairment losses related to our goodwill, indefinite-lived intangible assets and long-lived assets.
We evaluate goodwill and indefinite-lived intangible assets annually for impairment and whenever circumstances occur indicating that goodwill might be impaired. We determine the fair value of our reporting unit based on a combination of inputs, including the market capitalization of Ligand, as well as Level 3 inputs such as discounted cash flows, which are not observable from the market, directly or indirectly. We determine the fair value of our indefinite-lived intangible assets using the income approach based on Level 3 inputs.
There was no no no
Fair Value of Financial Instruments
81
Our cash and cash equivalents, accounts receivable, other current assets, accounts payable, accrued liabilities, deferred revenue, current operating lease liabilities, current financing lease liabilities are financial instruments and are recorded at cost in the consolidated balance sheets. The estimated fair value of these financial instruments approximates their carrying value due to their short-term nature.
Financial Assets Not Measured at Fair Value
Commercial license rights are measured and carried on the balance sheet at amortized cost using the effective interest method or on a non-accrual basis. Management calculates the fair value of commercial license rights using a forecasted royalty receipts. The projected future cash flows derive from royalty payments and milestones, then discounted using appropriate individual discount rates. The fair value of commercial license and other economic rights assets is classified as Level 3 within the fair value hierarchy since it is determined based upon inputs that are both significant and unobservable. The estimated fair value and related carrying values of commercial license rights as of December 31, 2023 were $75.9 million and $62.3 million, respectively. The estimated fair value and related carrying value of the commercial license rights as of December 31, 2022 were $13.1 million and $10.2 million, respectively.
7. Leases
Finance lease
In May 2020 and January 2021, we entered into an agreement and the first amendment with Hovione, our third-party manufacturer, to increase our manufacturing of Captisol, respectively. The agreements are considered to include an embedded finance lease under ASC 842, Leases, as it provides the Company the right to use the underlying equipment to exclusively manufacture Captisol. As of December 31, 2021, we have fully paid consideration of $69.1 million for prepaid inventory and capacity ramp-up fee. We allocated consideration in the agreements between lease and non-lease components using relative standalone prices. Since the inception of the agreements, we have allocated $50.2 million of the consideration paid to the non-lease component which is accounted for as prepaid inventory and being amortized to cost of Captisol based on the usage. The remaining balance of $18.9 million was recognized as a right of use asset.
Given the current COVID status, our forecast for COVID-related Captisol has been significantly reduced, which triggered an indicator of impairment of the right of use asset as of December 31, 2022. We performed a recoverability test at the asset group level by comparing the sum of the estimated undiscounted future cash flows attributable to the asset group to its carrying value and identified the asset was impaired. We recorded a $9.8 million of impairment charge based on the fair value of the right of use asset which has been recognized in cost of Captisol in our consolidated statement of operations for the year ended December 31, 2022. As of December 31, 2022 the remaining right of use asset balance is $4.0 million which will be amortized straight-line over the remaining 6 years lease term. In the year-ended December 31, 2023, we recorded no impairment to this asset group as there have been no indicators of impairment, and as of December 31, 2023, the remaining right of use asset balance is $3.4 million.
Operating lease
We lease certain office facilities and equipment primarily under various operating leases. Our operating leases have remaining contractual terms up to nine years , some of which include options to extend the leases for up to five years . Our lease agreements do not contain any material residual value guarantees, material restrictive covenants, or material termination options. Our operating lease costs are primarily related to facility leases for administration offices and research and development facilities.
Lease assets and lease liabilities are recognized at the commencement of an arrangement where it is determined at inception that a lease exists. Lease assets represent the right to use an underlying asset for the lease term, and lease liabilities represent the obligation to make lease payments arising from the lease. These assets and liabilities are initially recognized based on the present value of lease payments over the lease term calculated using our incremental borrowing rate generally applicable to the location of the lease asset, unless the implicit rate is readily determinable. Lease assets also include any upfront lease payments made and lease incentives. Lease terms include options to extend or terminate the lease when it is reasonably certain that those options will be exercised.
In addition to base rent, certain of our operating leases require variable payments, such as insurance and common area maintenance. These variable lease costs, other than those dependent upon an index or rate, are expensed when the obligation for those payments is incurred. Leases with an initial term of 12 months or less are not recorded on the balance sheet, and the expense for these short-term leases and for operating leases is recognized on a straight-line basis over the lease term.
The depreciable life of lease assets and leasehold improvements is limited by the expected lease term, unless there is a transfer of title or purchase option reasonably certain of exercise.
82
During the year ended December 31, 2023, we entered into an amendment to the lease agreement for our office located in San Diego, California, which resulted in a $1.1
Operating and Finance Lease Assets and Liabilities (in thousands):
| December 31, 2023 | December 31, 2022 | ||||||||||
| Assets | |||||||||||
| Operating lease assets | $ | $ | |||||||||
| Finance lease assets | |||||||||||
| Total lease assets | $ | $ | |||||||||
| Liabilities | |||||||||||
| Current operating lease liabilities | $ | $ | |||||||||
| Current finance lease liabilities | |||||||||||
| Long-term operating lease liabilities | |||||||||||
| Total lease liabilities | $ | $ | |||||||||
Maturity of Operating and Finance Lease Liabilities as of December 31, 2023 (in thousands):
| Maturity Dates | Operating Leases | Finance Leases | ||||||||||||
| 2024 | $ | $ | ||||||||||||
| 2025 | ||||||||||||||
| 2026 | ||||||||||||||
| 2027 | ||||||||||||||
| 2028 | ||||||||||||||
| Thereafter | ||||||||||||||
| Total lease payments | ||||||||||||||
| Less tenant improvement allowance | ( | |||||||||||||
| Less imputed interest | ( | ( | ||||||||||||
| Present value of lease liabilities | $ | $ | ||||||||||||
As of December 31, 2023, our operating leases have a weighted-average remaining lease term of 7.4 years and a weighted-average discount rate of 7.7 %. As of December 31, 2022, our operating leases have a weighted-average remaining lease term of 9.3 years and a weighted-average discount rate of 7.1 %. Cash paid for amounts included in the measurement of operating lease liabilities was $1.4 million and $1.7 million for the years ended December 31, 2023 and 2022, respectively. Operating lease expense was $1.4 million (net of sublease income of $0.3 million) and $0.7 million (net of sublease income of $0.7 million) for the years ended December 31, 2023 and 2022, respectively.
As of December 31, 2023, our finance leases have a weighted-average remaining lease term of 3.4 years and a weighted-average discount rate of 6.8 %. As of December 31, 2022, our finance leases have a weighted-average remaining lease term of 1.1 years and a weighted-average discount rate of 4.1 %. We excluded the Hovione equipment lease in the calculation of weighted average remaining lease term and weighted average discount rate because the Hovione lease was fully paid off as of December 31, 2021. Cash paid for amounts included in the measurement of these finance lease liabilities were both $0.05 million for the years ended December 31, 2023 and 2022. Finance lease expense was $0.7 million and $2.3 million for the years ended December 31, 2023 and 2022, respectively.
8. Debt
In May 2018, we issued $750 million aggregate principal amount of 2023 Notes, bearing cash interest at a rate of 0.75 % per year, payable semi-annually. The net proceeds from the offering, after deducting the initial purchasers' discount and offering expenses, were approximately $733.1 million.
83
In connection with the issuance of the 2023 Notes, we incurred $16.9 million of issuance costs, which primarily consisted of underwriting, legal and other professional fees and is being amortized to interest expense using the effective interest method over the five years expected life of the 2023 Notes. The effective interest rate for the year ended December 31, 2023 is 0.5 %. During the year ended December 31, 2023 we recognized a total of $0.6 million in interest expense which includes $0.4 million in contractual interest expense and $0.2 million in amortized issuance costs.
During 2021, we repurchased $152.0 million in principal of the 2023 Notes for $156.0 million in cash, including accrued interest of $0.3 million. We accounted for the repurchase as a debt extinguishment, which resulted in (1) a loss of $7.3 million reflected in other income (expense), net, in our consolidated statement of operations for the year ended December 31, 2021, (2) a $13.7 million reduction in debt discount, and (3) a $10.2 million reduction to additional paid in capital, related to the reacquisition of the equity component in our consolidated balance sheet as of December 31, 2021.
During 2022, we repurchased $266.4 million in principal amount of the 2023 Notes for $261.4 million in cash, including accrued interest of $0.5 million We accounted for the repurchase as a debt extinguishment, which resulted in a gain of $4.2 million reflected in other income (expense), net, in our consolidated statement of operations for the year ended December 31, 2022, and a $1.3 million reduction in debt discount.
On May 15, 2023, the 2023 Notes maturity date, we paid the remaining $76.9 million principal amount and $0.3 million accrued interest in cash.
Convertible Bond Hedge and Warrant Transactions
In conjunction with the 2023 Notes, in May 2018, we entered into convertible bond hedges and sold warrants covering 3,018,327 shares of our common stock to minimize the impact of potential dilution to our common stock and/or offset the cash payments we are required to make in excess of the principal amount upon conversion of the 2023 Notes. The convertible bond hedges have an exercise price of $206.65 per share and are exercisable when and if the 2023 Notes are converted. We paid $140.3 million for these convertible bond hedges. If upon conversion of the 2023 Notes, the price of our common stock is above the exercise price of the convertible bond hedges, the counterparties will deliver shares of common stock and/or cash with an aggregate value approximately equal to the difference between the price of common stock at the conversion date and the exercise price, multiplied by the number of shares of common stock related to the convertible bond hedge transaction being exercised. The convertible bond hedges and warrants described below are separate transactions entered into by us and are not part of the terms of the 2023 Notes. Holders of the 2023 Notes and warrants did not have any rights with respect to the convertible bond hedges.
Concurrently with the convertible bond hedge transactions, we entered into warrant transactions whereby we sold warrants covering 3,018,327 shares of common stock with an exercise price of $315.38 per share, subject to certain adjustments. We received $90.0 million for these warrants. The warrants have various expiration dates ranging from August 15, 2023 to February 6, 2024. The warrants will have a dilutive effect to the extent the market price per share of common stock exceeds the applicable exercise price of the warrants, as measured under the terms of the warrant transactions. The common stock issuable upon exercise of the warrants will be in unregistered shares, and we do not have the obligation and do not intend to file any registration statement with the SEC registering the issuance of the shares under the warrants.
In January 2021, in connection with the repurchases of approximately $20.3 million in principal of the 2023 Notes for approximately $19.1 million in cash, including accrued interest of $0.1 million, during the quarter ended December 31, 2020, we entered into amendments with Barclays Bank PLC, Deutsche Bank AG, London Branch, and Goldman Sachs & Co. LLC to the convertible note hedges transactions we initially entered into in connection with the issuance of the 2023 Notes. The amendments provide that the options under the convertible note hedges corresponding to such repurchased 2023 Notes will remain outstanding notwithstanding such repurchase.
During the year ended December 31, 2021, in connection with the repurchases of $152.0 million in principal of the 2023 Notes for $156.0 million in cash, including accrued interest of $0.3 million, we entered into Warrant Early Unwind Agreements and Bond Hedge Unwind Agreements with Barclays Bank PLC, Deutsche Bank AG, and Goldman Sachs & Co. LLC to unwind a portion of the convertible note hedges transactions we initially entered into in connection with the issuance of the 2023 Notes. We paid $18.4 million as part of the Warrant Early Unwind Agreements reducing the number of shares covered by the warrants from 3,018,327 to 2,559,254 . As of December 31, 2023, the number of warrants that remain outstanding is 545,000 .
In August 2022, in connection with the repurchases of $227.8 million in principal of the 2023 Notes for $223.7 million in cash, including accrued interest of $0.4 million made during the six months ended June 30, 2022, we entered into Bond Hedge Unwind Agreements with Barclays Bank PLC, Deutsche Bank AG, and Goldman Sachs & Co. LLC to unwind a portion of the convertible note hedges transactions we initially entered into in connection with the issuance of the 2023 Notes.
84
The following table summarizes information about the 2023 Notes (in thousands) as of December 31, 2022.
| December 31, 2022 | |||||
| Principal amount of 2023 Notes outstanding | $ | ||||
| Unamortized discount (including unamortized debt issuance cost) | ( | ||||
| Total long-term portion of notes payable | $ | ||||
| Fair value of convertible senior notes outstanding (Level 2) | $ | ||||
Revolving Credit Facility
On October 12, 2023, we entered into a $75.0 million revolving credit facility (the “Revolving Credit Facility”) with Citibank, N.A. as the Administrative Agent. We, our material domestic subsidiaries, as Guarantors (as defined in the Credit Agreement), and the Lenders (as defined in the Credit Agreement) entered into a credit agreement (the “Credit Agreement”) with the Administrative Agent, under which the Lenders, the Swingline Lender and the L/C Issuer (each as defined in the Credit Agreement) agreed to make loans and other financial accommodations to us in an aggregate amount of up to $75.0 million. At our option, borrowings under the Revolving Credit Facility accrue interest at a rate equal to either Term Secured Overnight Financing Rate ("Term SOFR") or a specified base rate plus an applicable margin linked to our leverage ratio, ranging from 1.75 % to 2.50 % per annum for Term SOFR loans and 0.75 % to 1.50 % per annum for base rate loans. The Revolving Credit Facility is subject to a commitment fee payable on the unused Revolving Credit Facility commitments ranging from 0.30 % to 0.45 %, depending on our leverage ratio. During the term of the Revolving Credit Facility, we may borrow, repay and re-borrow amounts available under the Revolving Credit Facility, subject to voluntary reductions of the swing line, letter of credit and revolving credit commitments.
Borrowings under the Credit Agreement are secured by certain of our collateral and that of the Guarantors. In specified circumstances, additional guarantors are required to be added. The Credit Agreement contains customary affirmative and negative covenants, including certain financial maintenance covenants, and events of default applicable to us. In the event of violation of the representations, warranties and covenants made in the Credit Agreement, we may not be able to utilize the Revolving Credit Facility or repayment of amounts owed thereunder could be accelerated.
As of December 31, 2023, we had $74.5 million in available borrowing under the Revolving Credit Facility, after utilizing $0.5 million for letter of credit. The maturity date of the Revolving Credit Facility is October 12, 2026.
As of December 31, 2023, there were no events of default or violation of any covenants under our financing obligations.
9. Balance Sheet Account Details
Short-term Investments
85
Excluding our investments in Viking, the following table summarizes the various investment categories at December 31, 2023 and 2022 (in thousands):
| Cost | Gross unrealized gains | Gross unrealized losses | Estimated fair value | ||||||||||||||||||||
December 31, 2023 | |||||||||||||||||||||||
| Short-term investments | |||||||||||||||||||||||
| Bond Fund | $ | $ | $ | ( | $ | ||||||||||||||||||
| Bank deposits | ( | ||||||||||||||||||||||
| Corporate bonds | ( | ||||||||||||||||||||||
| Commercial paper | ( | ||||||||||||||||||||||
| U.S. government securities | ( | ||||||||||||||||||||||
| Municipal bonds | ( | ||||||||||||||||||||||
| Corporate equity securities | ( | ||||||||||||||||||||||
| $ | $ | $ | ( | $ | |||||||||||||||||||
December 31, 2022 | |||||||||||||||||||||||
| Short-term investments | |||||||||||||||||||||||
| Bond fund | $ | $ | $ | ( | $ | ||||||||||||||||||
| Commercial paper | |||||||||||||||||||||||
| Corporate bonds | ( | ||||||||||||||||||||||
| Bank deposits | ( | ||||||||||||||||||||||
| U.S. government securities | ( | ||||||||||||||||||||||
| Corporate equity securities | ( | ||||||||||||||||||||||
| Warrants | |||||||||||||||||||||||
| $ | $ | $ | ( | $ | |||||||||||||||||||
Gain (loss) from short-term investments on our consolidated statements of operations includes both realized and unrealized gain (loss) from our short-term investments in public equity and warrant securities, and realized gain (loss) from available-for-sale debt securities.
The following table summarizes our available-for-sale debt securities by contractual maturity (in thousands):
| December 31, 2023 | |||||||||||
| Amortized Cost | Fair Value | ||||||||||
| Within one year | $ | $ | |||||||||
| After one year through five years | |||||||||||
| Total | $ | $ | |||||||||
The following table summarizes our available-for-sale debt securities in an unrealized loss position (in thousands):
86
| Less than 12 months | 12 months or greater | Total | |||||||||||||||||||||||||||||||||
| Gross Unrealized Losses | Estimated Fair Value | Gross Unrealized Losses | Estimated Fair Value | Gross Unrealized Losses | Estimated Fair Value | ||||||||||||||||||||||||||||||
| December 31, 2023 | |||||||||||||||||||||||||||||||||||
| Bank deposits | $ | ( | $ | $ | $ | $ | ( | $ | |||||||||||||||||||||||||||
| Corporate bonds | ( | ( | ( | ||||||||||||||||||||||||||||||||
| Commercial paper | ( | ( | |||||||||||||||||||||||||||||||||
| Municipal bonds | ( | ( | |||||||||||||||||||||||||||||||||
| U.S. Government Securities | ( | ( | |||||||||||||||||||||||||||||||||
| Total | $ | ( | $ | $ | ( | $ | $ | ( | $ | ||||||||||||||||||||||||||
| December 31, 2022 | |||||||||||||||||||||||||||||||||||
| Bank deposits | $ | ( | $ | $ | $ | $ | ( | $ | |||||||||||||||||||||||||||
| Corporate bonds | ( | ( | ( | ||||||||||||||||||||||||||||||||
| Commercial paper | |||||||||||||||||||||||||||||||||||
| U.S. Government Securities | ( | ( | |||||||||||||||||||||||||||||||||
| Total | $ | ( | $ | $ | ( | $ | $ | ( | $ | ||||||||||||||||||||||||||
Our investment policy is capital preservation and we only invested in U.S.-dollar denominated investments. We held a total of 31 positions which were in an unrealized loss position with a total of $0.01 million unrealized losses as of December 31, 2023. We believe that we will collect the principal and interest due on our debt securities that have an amortized cost in excess of fair value. The unrealized losses are largely due to changes in interest rates and not to unfavorable changes in the credit quality associated with these securities that impacted our assessment on collectability of principal and interest. We do not intend to sell these securities nor do we believe that we will be required to sell these securities before the recovery of the amortized cost basis. Accordingly, no credit losses were recognized for the year ended December 31, 2023.
Short-term Investments: Investment in Viking:
Our ownership in Viking was approximately 1.7 % as of December 31, 2023, and we account for it as an investment in available-for-sale equity securities, which is measured at fair value, with changes in fair value recognized in net income.
As of December 31, 2023 and December 31, 2022, our investment in Viking is $32.2 million and $63.1 million, respectively, and included in short-term investments on the balance sheet. During the year ended December 31, 2023, we sold 5.0 million shares of Viking common stock and recognized a realized gain of $44.4 million. During the year ended December 31, 2021, we sold 0.6 million shares of Viking common stock and recognized a realized gain of $3.6 million. There were no sales of Viking common stock during the year ended December 31, 2022.
Property and equipment are stated at cost and consists of the following (in thousands):
| December 31, | |||||||||||
| 2023 | 2022 | ||||||||||
| Lab and office equipment | $ | $ | |||||||||
| Leasehold improvements | |||||||||||
| Computer equipment and software | |||||||||||
| Less accumulated depreciation and amortization | ( | ( | |||||||||
| $ | $ | ||||||||||
Depreciation of equipment is computed using the straight-line method over the estimated useful lives of the assets which ranges from to nine years . Leasehold improvements are amortized using the straight-line method over their estimated useful lives or their related lease term, whichever is shorter. Depreciation expense of $2.9 million, $3.8 million, and $2.4 million was recognized for the years ended December 31, 2023, 2022, and 2021, respectively, and was included in operating expenses.
87
Goodwill and identifiable intangible assets consist of the following (in thousands):
| December 31, | |||||||||||
| 2023 | 2022 | ||||||||||
| Indefinite-lived intangible assets | |||||||||||
| Goodwill | $ | $ | |||||||||
| Definite-lived intangible assets | |||||||||||
| Completed technology | |||||||||||
| Less: Accumulated amortization | ( | ( | |||||||||
| Trade name | |||||||||||
| Less: Accumulated amortization | ( | ( | |||||||||
| Customer relationships | |||||||||||
| Less: Accumulated amortization | ( | ( | |||||||||
| Contractual relationships | |||||||||||
| Less: Accumulated amortization | ( | ( | |||||||||
| Total goodwill and other identifiable intangible assets, net | $ | $ | |||||||||
Amortization of finite-lived intangible assets is computed using the straight-line method over the estimated useful life of the asset of up to 20 years. Amortization expense of $33.7 million, $34.2 million, and $34.2 million was recognized for the years ended December 31, 2023, 2022, and 2021, respectively. Estimated amortization expense for the years ending December 31, 2024 through 2028 is $32.8 no
Accrued liabilities consist of the following (in thousands):
| December 31, | |||||||||||
| 2023 | 2022 | ||||||||||
| Compensation | $ | $ | |||||||||
| Professional fees | |||||||||||
| Customer deposit | |||||||||||
| Amounts owed to former licensees | |||||||||||
| Royalties owed to third parties | |||||||||||
| Subcontractor | |||||||||||
| Supplier | |||||||||||
| Other | |||||||||||
| $ | $ | ||||||||||
Other long-term liabilities consist of the following (in thousands):
| December 31, | |||||||||||
| 2023 | 2022 | ||||||||||
| Unrecognized tax benefits | $ | $ | |||||||||
| Novan contract liability | |||||||||||
| Other long-term liabilities | |||||||||||
| $ | $ | ||||||||||
Contingent liabilities:
In connection with the acquisition of CyDex in January 2011, we issued a series of CVRs and also assumed certain contingent liabilities. We may be required to make additional payments upon achievement of certain clinical and regulatory milestones to the CyDex shareholders and former license holders.
In connection with the acquisition of Metabasis in January 2010, we entered into four CVR agreements with Metabasis shareholders. The CVRs entitle the holders to cash payments as frequently as every six months as proceeds are received by us upon the sale or licensing of any of the Metabasis drug development programs and upon the achievement of specified milestones.
The following table summarizes roll-forward of contingent liabilities as of December 31, 2023 and 2022 (in thousands):
88
December 31, 2021 | Payments | Fair Value Adjustment | December 31, 2022 | Payments | Fair Value Adjustment | Repurchases | December 31, 2023 | |||||||||||||||||||
| Cydex | $ | $ | $ | ( | $ | $ | ( | $ | $ | $ | ||||||||||||||||
| Metabasis | ( | |||||||||||||||||||||||||
| Total | $ | $ | $ | ( | $ | $ | ( | $ | ( | $ | $ | |||||||||||||||
10. Stockholders’ Equity
Share-based Compensation Expense
The following table summarizes share-based compensation expense from continuing operations (in thousands):
| December 31, | |||||||||||||||||
| 2023 | 2022 | 2021 | |||||||||||||||
| Share-based compensation expense as a component of: | |||||||||||||||||
| Research and development expenses | $ | $ | $ | ||||||||||||||
| General and administrative expenses | |||||||||||||||||
| $ | $ | $ | |||||||||||||||
Conversion and Modification of Equity Awards Outstanding at Separation Date
In connection with the OmniAb Separation on November 1, 2022, under the provisions of the existing plans, we adjusted our outstanding equity awards in accordance with the Merger Agreement to preserve the intrinsic value of the awards immediately before and after the Distribution. Upon the Distribution, employees holding stock options, restricted stock units and performance restricted stock units denominated in pre-Distribution Ligand stock received a number of otherwise-similar awards either in post-Distribution Ligand stock or in a combination of post-Distribution Ligand stock and OmniAb stock based on conversion ratios outlined for each group of employees in the Merger Agreement that we entered into in connection with the Distribution. The equity awards that were granted prior to March 2, 2022 were converted under the shareholder method, wherein employees holding outstanding equity awards received equity awards in both Ligand and OmniAb. For equity awards granted after March 2, 2022, for Ligand employees, the number of awards that were outstanding at the Separation were proportionately adjusted into post-Distribution Ligand stock to maintain the aggregate intrinsic value of the awards at the date of the Separation; for OmniAb employees, the number of awards that were outstanding at the Separation were proportionately adjusted into post-Distribution OmniAb stock to maintain the aggregate intrinsic value of the awards at the date of the Separation. The conversion ratio was determined based on the relative values of Ligand common stock in the “regular way” and “ex-distribution” markets during the five-trading day period prior to the closing of the business combination.
These modified awards otherwise retained substantially the same terms and conditions, including term and vesting provisions. Additionally, we will not incur any future compensation cost related to equity awards held by OmniAb employees and directors. We will incur future compensation cost related to OmniAb equity awards held by our employees.
Stock Plans
In June 2022, our stockholders approved the amendment and restatement of the Ligand Pharmaceuticals Incorporated 2002 Stock Incentive Plan (the “2002 Plan”). The amended and restated 2002 Plan, which is referred to herein as the “Restated Plan” was amended to increase the shares available for issuance by 1.0 million.
On July 29, 2022, our board of directors (the “Board”) approved the Ligand Pharmaceuticals Incorporated 2022 Employment Inducement Plan (the “2022 Inducement Plan”). The terms of the 2022 Inducement Plan are substantially similar to the terms of the Restated Plan with the exception that incentive stock options may not be issued under the 2022 Inducement Plan and awards under the 2022 Inducement Plan may only be issued to eligible recipients under the applicable Nasdaq Listing Rules. The 2022 Inducement Plan was adopted by the Board without stockholder approval pursuant to Rule 5635(c)(4) of the Nasdaq Listing Rules. The Board has initially reserved 300,000 shares of the Company’s common stock for issuance pursuant to awards granted under the 2022 Inducement Plan.
As of December 31, 2023, there were 1.0 million shares available for future option grants or direct issuance under the Restated Plan and the 2022 Inducement Plan.
89
Following is a summary of our stock option plan activity and related information:
| Shares | Weighted Average Exercise Price | Weighted Average Remaining Contractual Term in Years | Aggregate Intrinsic Value (In thousands) | ||||||||||||||||||||
| Balance at January 1, 2021 | $ | $ | |||||||||||||||||||||
| Granted | $ | ||||||||||||||||||||||
| Exercised | ( | $ | |||||||||||||||||||||
| Forfeited | ( | $ | |||||||||||||||||||||
| Balance at December 31, 2021 | $ | $ | |||||||||||||||||||||
| Exercisable at December 31, 2021 | $ | $ | |||||||||||||||||||||
| Options vested and expected to vest as of December 31, 2021 | $ | $ | |||||||||||||||||||||
| Granted | $ | ||||||||||||||||||||||
| Exercised | ( | $ | |||||||||||||||||||||
| Forfeited | ( | $ | |||||||||||||||||||||
| Balance at October 31, 2022 | $ | $ | |||||||||||||||||||||
| Exercisable at October 31, 2022 | $ | $ | |||||||||||||||||||||
| Options vested and expected to vest as of October 31, 2022, before Separation and Regrant | $ | $ | |||||||||||||||||||||
| Cancellation due to Separation, Before Regrant | ( | ||||||||||||||||||||||
| Balance at November 1, 2022, Before Regrant | |||||||||||||||||||||||
Granted (1) | $ | ||||||||||||||||||||||
| Exercised | ( | $ | |||||||||||||||||||||
| Forfeited | ( | $ | |||||||||||||||||||||
| Balance at December 31, 2022 | $ | $ | |||||||||||||||||||||
| Exercisable at December 31, 2022 | $ | $ | |||||||||||||||||||||
| Options vested and expected to vest as of December 31, 2022 | $ | $ | |||||||||||||||||||||
| Granted | $ | ||||||||||||||||||||||
| Exercised | ( | $ | |||||||||||||||||||||
| Forfeited | ( | $ | |||||||||||||||||||||
| Balance at December 31, 2023 | $ | $ | |||||||||||||||||||||
| Exercisable at December 31, 2023 | $ | $ | |||||||||||||||||||||
| Options vested and expected to vest as of December 31, 2023 | $ | $ | |||||||||||||||||||||
The weighted-average grant-date fair value of all stock options granted during 2023, 2022 and 2021 was $36.65 , $28.90 , and $80.08 per share, respectively. The total intrinsic value of all options exercised during 2023, 2022 and 2021 was approximately $12.0 million, $4.6 million, and $77.3 million, respectively.
Cash received from options exercised, net of fees paid, in 2023, 2022 and 2021 was $22.2 million, $2.6 million and $33.0 million, respectively.
90
Following is a further breakdown of the options outstanding as of December 31, 2023:
| Range of exercise prices | Options outstanding | Weighted average remaining life in years | Weighted average exercise price | Options exercisable | Weighted average exercise price | ||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
$ | $ | $ | |||||||||||||||||||||||||||
| $ | $ | ||||||||||||||||||||||||||||
The assumptions used for the specified reporting periods and the resulting estimates of weighted-average grant date fair value per share of options granted:
| Year Ended December 31, | |||||||||||||||||
2023 | 2022 | 2021 | |||||||||||||||
| Risk-free interest rate | |||||||||||||||||
| Expected volatility | |||||||||||||||||
| Expected term | |||||||||||||||||
As of December 31, 2023, there was $26.2 million of total unrecognized compensation cost related to non-vested stock options under the 2002 Plan. That cost is expected to be recognized over a weighted average period of 2.5 years.
As of December 31, 2023, there was $1.1 million of total unrecognized compensation cost related to non-vested OmniAb stock options received upon aforementioned spin-off conversion. That cost is expected to be recognized over a weighted average period of 0.9 years.
Restricted Stock Activity
The following is a summary of our restricted stock activity and related information:
| Shares | Weighted-Average Grant Date Fair Value | ||||||||||
| Outstanding at December 31, 2021 | $ | ||||||||||
| Granted | $ | ||||||||||
| Vested | ( | $ | |||||||||
| Forfeited | ( | $ | |||||||||
| Outstanding at October 31, 2022, before Separation and Regrant | $ | ||||||||||
| Forfeited due to Separation, Before Regrant | ( | ||||||||||
| Balance at November 1, 2022, Before Regrant | |||||||||||
| Granted | $ | ||||||||||
| Vested | ( | $ | |||||||||
| Forfeited | ( | $ | |||||||||
| Outstanding at December 31, 2022 | $ | ||||||||||
| Granted | $ | ||||||||||
| Vested | ( | $ | |||||||||
| Forfeited | ( | $ | |||||||||
| Outstanding at December 31, 2023 | $ | ||||||||||
91
As of December 31, 2023, unrecognized compensation cost related to non-vested stock awards under the 2002 Plan amounted to $15.2 million. That cost is expected to be recognized over a weighted average period of 1.6 years.
As of December 31, 2023, there was $0.1 million of total unrecognized compensation cost related to non-vested OmniAb stock awards received upon aforementioned spin-off conversion. That cost is expected to be recognized over a weighted average period of 0.1 years.
Employee Stock Purchase Plan
As of December 31, 2023, 30,801 shares of our common stock are available for future issuance under the Amended Employee Stock Purchase Plan, or ESPP. The ESPP permits eligible employees to purchase up to 1,250 shares of Ligand common stock per calendar year at a discount through payroll deductions. The price at which stock is purchased under the ESPP is equal to 85 % of the fair market value of the common stock on the first of a month offering period or purchase date, whichever is lower. There were 5,080 , 8,479 and 8,448 shares issued under the ESPP in 2023, 2022 and 2021, respectively.
Share Repurchases
In April 2023, our Board of Directors has approved a stock repurchase program authorizing, but not requiring, the repurchase of up to $50.0 million of our common stock from time to time through April 2026. We expect to acquire shares, if at all, primarily through open-market transactions in accordance with all applicable requirements of Rule 10b-18 under the Securities Exchange Act of 1934, as amended. The timing and amount of repurchase transactions will be determined by management based on our evaluation of market conditions, share price, legal requirements and other factors. During the years ended December 31, 2023, 2022 and 2021, we did no
At-the Market Equity Offering Program
On September 30, 2022, we filed a registration statement on Form S-3 (the “Shelf Registration Statement”), which became automatically effective upon filing, covering the offering of common stock, preferred stock, debt securities, warrants and units.
On September 30, 2022, we also entered into an At-The-Market Equity Offering Sales Agreement (the “Sales Agreement”) with Stifel, Nicolaus & Company, Incorporated (the “Agent”), under which we may, from time to time, sell shares of our common stock having an aggregate offering price of up to $100.0 million in “at the market” offerings through the Agent (the “ATM Offering”). The Shelf Registration Statement included a prospectus covering the offering, issuance and sale of up to $100.0 million of our common stock from time to time through the ATM Offering. The shares to be sold under the Sales Agreement may be issued and sold pursuant to the Shelf Registration Statement. To date, we have not issued any shares of common stock in the ATM Offering.
11. Commitment and Contingencies: Legal Proceedings
Legal Proceedings
We record an estimate of a loss when the loss is considered probable and estimable. Where a liability is probable and there is a range of estimated loss and no amount in the range is more likely than any other number in the range, we record the minimum estimated liability related to the claim in accordance with ASC 450, Contingencies. As additional information becomes available, we assess the potential liability related to our pending litigation and revises our estimates. Revisions in our estimates of potential liability could materially impact our results of operations.
On October 31, 2019, we received three civil complaints filed in the U.S. District Court for the Northern District of Ohio on behalf of several Indian tribes. The Northern District of Ohio is the Court that the Judicial Panel on Multi-District Litigation (“JPML”) has assigned more than one thousand civil cases which have been designated as a Multi-District Litigation (“MDL”) and captioned In Re: National Prescription Opiate Litigation. The allegations in these complaints focus on the activities of defendants other than the Company and no individualized factual allegations have been advanced against us in any of the three complaints. We reject all claims raised in the complaints and intend to vigorously defend these matters.
From time to time, we may also become subject to other legal proceedings or claims arising in the ordinary course of our business. We currently believe that none of the claims or actions pending against us is likely to have, individually or in the aggregate, a material adverse effect on our business, financial condition or results of operations. Given the unpredictability inherent in litigation, however, we cannot predict the outcome of these matters.
12. Income Taxes
92
The components of the income tax expense (benefit) for continuing operations are as follows (in thousands):
| Year Ended December 31, | |||||||||||||||||
| 2023 | 2022 | 2021 | |||||||||||||||
| Current expense (benefit): | |||||||||||||||||
| Federal | $ | ( | $ | $ | |||||||||||||
| State | ( | ||||||||||||||||
| Foreign | |||||||||||||||||
| ( | |||||||||||||||||
| Deferred expense (benefit): | |||||||||||||||||
| Federal | ( | ( | |||||||||||||||
| State | ( | ||||||||||||||||
| ( | |||||||||||||||||
| Total income tax expense (benefit) | $ | $ | $ | ( | |||||||||||||
A reconciliation of income tax expense (benefit) from continuing operations to the amount computed by applying the statutory federal income tax rate to the net income (loss) from continuing operations is summarized as follows (in thousands):
| Year Ended December 31, | |||||||||||||||||
| 2023 | 2022 | 2021 | |||||||||||||||
| Tax at federal statutory rate | $ | $ | $ | ||||||||||||||
| State, net of federal benefit | ( | ||||||||||||||||
| FDII | ( | ( | ( | ||||||||||||||
| Rate change for changes in federal, foreign or state law | ( | ( | |||||||||||||||
| Change in uncertain tax positions | ( | ( | |||||||||||||||
| Contingent liabilities | ( | ( | |||||||||||||||
| Foreign tax differential on income/loss of foreign subsidiaries | ( | ( | |||||||||||||||
| Research and development credits | ( | ( | |||||||||||||||
| Debt repurchases | |||||||||||||||||
| Subpart F income | |||||||||||||||||
| Share-based compensation | ( | ||||||||||||||||
| Provision to return adjustments | ( | ||||||||||||||||
| Officer compensation | |||||||||||||||||
| Change in valuation allowance | ( | ||||||||||||||||
| Other | ( | ||||||||||||||||
| $ | $ | $ | ( | ||||||||||||||
We remeasured certain deferred tax assets and liabilities based on the rates at which they are expected to reverse in the future, which is generally 21%. Significant components of our deferred tax assets and liabilities as of December 31, 2023 and 2022 are shown below. We assess the positive and negative evidence to determine if sufficient future taxable income will be generated to use the existing deferred tax assets. Our evaluation of evidence resulted in management concluding that the majority of our deferred tax assets will be realized. However, we maintain a valuation allowance to offset certain net deferred tax assets as management believes realization of such assets are uncertain as of December 31, 2023, 2022 and 2021. The valuation allowance amount in connection with the deferred taxes decreased $1.2 million in 2023, increased $24.8 million in 2022 and increased $11.2 million in 2021.
We offset all deferred tax assets and liabilities by jurisdiction, as well as any related valuation allowance, and present them on our consolidated balance sheet as a non-current deferred income tax asset or liability (as applicable). Deferred tax assets
93
(liabilities) are comprised of the following:
| December 31, | |||||||||||
| 2023 | 2022 | ||||||||||
| (in thousands) | |||||||||||
| Deferred tax assets: | |||||||||||
| Net operating loss carryforwards | $ | $ | |||||||||
| Research credit carryforwards | |||||||||||
| Capitalized R&D | |||||||||||
| Stock compensation | |||||||||||
| Other | |||||||||||
| Valuation allowance for deferred tax assets | ( | ( | |||||||||
| Net deferred tax assets | $ | $ | |||||||||
| Deferred tax liabilities: | |||||||||||
| Identified intangibles | ( | ( | |||||||||
| Other | ( | ( | |||||||||
| Net deferred tax liabilities | $ | ( | $ | ( | |||||||
| Deferred income taxes, net | $ | ( | $ | ( | |||||||
As of December 31, 2023, we had federal net operating loss carryforwards set to expire through 2037 of $48.0 million and $165.1 million of state net operating loss carryforwards that begin to expire in 2028. We also have $8.5 million of federal research and development credit carryforwards, which expire through 2040. We have $29.4 million of California research and development credit carryforwards that have no expiration date. In addition, we have approximately $95.5 million of non-U.S. net operating loss carryovers and approximately $16.5 million of non-U.S. capital loss carryovers that have no expiration date.
As of December 31, 2022, we had federal net operating loss carryforwards set to expire through 2037 of $81.1 million and $168.3 million of state net operating loss carryforwards that begin to expire in 2028. We also had $8.5 million of federal research and development credit carryforwards, which expire through 2040. We had $29.0 million of California research and development credit carryforwards that have no expiration date. In addition, we had approximately $96.1 million of non-U.S. net operating loss carryovers and approximately $15.6 million of non-U.S. capital loss carryovers that have no expiration date.
Pursuant to Section 382 and 383 of the Internal Revenue Code of 1986, as amended, utilization of our net operating losses and credits may be subject to annual limitations in the event of any significant future changes in its ownership structure. These annual limitations may result in the expiration of net operating losses and credits prior to utilization. The deferred tax assets as of December 31, 2023 are net of any previous limitations due to Section 382 and 383.
We account for income taxes by evaluating a probability threshold that a tax position must meet before a financial statement benefit is recognized. The minimum threshold is a tax position that is more likely than not to be sustained upon examination by the applicable taxing authority, including resolution of any related appeals or litigation processes, based on the technical merits of the position. Our remaining liabilities for uncertain tax positions are presented net of the deferred tax asset balances on the accompanying consolidated balance sheet.
A reconciliation of the amount of unrecognized tax benefits at December 31, 2023, 2022 and 2021 is as follows (in thousands):
| December 31, | |||||||||||||||||
| 2023 | 2022 | 2021 | |||||||||||||||
| Balance at beginning of year | $ | $ | $ | ||||||||||||||
| Additions based on tax positions related to the current year | |||||||||||||||||
| Additions for tax positions of prior years | |||||||||||||||||
| Reductions for tax positions of prior years | ( | ( | ( | ||||||||||||||
| Balance at end of year | $ | $ | $ | ||||||||||||||
Included in the balance of unrecognized tax benefits at December 31, 2023 is $20.6 million of tax benefits that, if recognized would impact the effective rate. There are no positions for which it is reasonably possible that the uncertain tax benefit will significantly increase or decrease within twelve months.
94
We recognize interest and penalties related to uncertain tax positions in income tax expense. As of December 31, 2023 and December 31, 2022, we recognized an immaterial amount of interest and penalties. We file income tax returns in the United States, various state jurisdictions, and United Kingdom with varying statutes of limitations. The federal statute of limitation remains open for the 2020 tax year to the present. The state income tax returns generally remain open for the 2019 tax year through the present. Net operating loss and research credit carryforwards arising prior to these years are also open to examination if and when utilized. The Company's 2019 and 2020 California tax returns are under examination by the California Franchise Tax Board. The Company does not anticipate that the examination will result in a material adjustment to its financial statements. No other income tax returns are currently under examination. We believe our reserve for unrecognized tax benefits and contingent tax issues is adequate with respect to all open years.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | ||||
None.
| Item 9A. | Controls and Procedures | ||||
(a) Evaluation of Disclosure Controls and Procedures
We are responsible for maintaining disclosure controls and procedures designed to provide reasonable assurance that information required to be disclosed in reports we file under the Exchange Act is recorded, processed, summarized and reported within the specified time periods and accumulated and communicated to management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. The design of any system of controls is based in part upon certain assumptions about the likelihood of future events, and there can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions; over time, controls may become inadequate because of changes in conditions, or the degree of compliance with policies or procedures may deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected. As of the end of the period covered by this Annual Report on Form 10-K, we have carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of our disclosure controls and procedures, as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, and have concluded our disclosure controls and procedures were not effective at a reasonable assurance level as of December 31, 2023 due to the material weakness described below.
In light of the material weakness described below, management performed additional analyses and other procedures to ensure that our consolidated financial statements were prepared in accordance with GAAP. Accordingly, management believes that the consolidated financial statements included in this Annual Report fairly present, in all material respects, our financial position, results of operations, and cash flows as of and for the periods presented, in accordance with GAAP.
There have been no changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2023 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
(b) Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting for the Company. Internal control over financial reporting is a process to provide reasonable assurance regarding the reliability of our financial reporting for external purposes in accordance with accounting principles generally accepted in the United States of America. Internal control over financial reporting includes maintaining records that in reasonable detail accurately and fairly reflect our transactions; providing reasonable assurance that transactions are recorded as necessary for preparation of our financial statements in accordance with generally accepted accounting principles; providing reasonable assurance that receipts and expenditures are made in accordance with our management and directors; and providing reasonable assurance that
unauthorized acquisition, use or disposition of company assets that could have a material effect on our financial statements would be prevented or detected on a timely basis. Because of its inherent limitations, internal control over financial reporting is not intended to provide absolute assurance that a misstatement of our financial statements would be prevented or detected.
Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework established by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) as set forth in the 2013 Internal Control-Integrated Framework. Based on our evaluation under the 2013 framework in Internal Control -
95
Integrated Framework, management concluded that our internal controls over financial reporting were not effective as of December 31, 2023 as a result of the material weakness discussed in the paragraphs that follow below.
A material weakness is a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected on a timely basis.
The Company's management concluded that the Company did not implement and maintain effective controls related to the valuation of the acquired intangible and contract liability, specifically controls over the review of cash flow forecasts used in the valuation of the acquired intangible asset and the discount rate used in the valuation of the contract liability. The deficiencies in our internal control over financial reporting due to the material weakness described above did not result in any misstatement in our consolidated financial statements or other disclosures. These deficiencies created, however, a reasonable possibility that a material misstatement in our consolidated financial statements would not be prevented or detected on a timely basis.
Management’s Remediation Plan
In an effort to address the identified material weakness and enhance our internal controls related to our business combination purchase price allocation process, we continue to maintain our financial reporting process we followed to prepare consolidated financial statements in accordance with GAAP for audit committee meetings on a quarterly and annual basis. We have hired additional accounting personnel and third party consultants with appropriate knowledge, experience, and/or training commensurate with our technical accounting and financial reporting requirements to enhance the process going forward. Our ongoing remediation efforts are focused on continued employee training related to internal control over financial reporting. Management will continue to work to improve the respective process and controls over our business combination purchase price allocation process. However, the material weakness will not be considered remediated until the applicable remedial controls operate for a sufficient period of time and management has concluded, through testing, that these controls are operating effectively.
Ernst & Young LLP, an independent registered public accounting firm, has audited the Company’s consolidated financial statements included in this Annual Report on Form 10-K and has issued an attestation report, included herein, on the effectiveness of our internal control over financial reporting as of December 31, 2023.
96
Report of Independent Registered Public Accounting Firm
The Stockholders and Board of Directors of Ligand Pharmaceuticals Incorporated
Opinion on Internal Control Over Financial Reporting
We have audited Ligand Pharmaceuticals Incorporated’s internal control over financial reporting as of December 31, 2023, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, because of the effect of the material weakness described below on the achievement of the objectives of the control criteria, Ligand Pharmaceuticals Incorporated (the Company) has not maintained effective internal control over financial reporting as of December 31, 2023, based on the COSO criteria.
A material weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected on a timely basis. The following material weakness has been identified and included in management’s assessment. Management has identified a material weakness in the design and operation of controls related to the Company’s valuation of the acquired intangible asset and contract liability in a business combination, specifically controls over the review of cash flow forecasts used in the valuation of the acquired intangible asset and the discount rate used in the valuation of the contract liability.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of December 31, 2023 and 2022, the related consolidated statements of operations, comprehensive income (loss), stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2023, and the related notes. This material weakness was considered in determining the nature, timing and extent of audit tests applied in our audit of the 2023 consolidated financial statements, and this report does not affect our report dated February 29, 2024 which expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
San Diego, California
97
February 29, 2024
| Item 9B. | Other Information | ||||
Rule 10b5-1 Trading Arrangements
From time to time, our officers (as defined in Rule 16a-1(f) of the Exchange Act) and directors may enter into Rule 10b5-1 or non-Rule 10b5-1 trading arrangements (as each such term is defined in Item 408 of Regulation S-K). During the three months ended December 31, 2023, none of our officers or directors adopted terminated
98
| Item 9C. | Disclosure Regarding Foreign Jurisdictions that Prevent Inspections | ||||
None
99
Part III
| Item 10. | Directors, Executive Officers and Corporate Governance | ||||
Code of Conduct
The Board of Directors has adopted a Code of Conduct and Ethics Policy (“Code of Conduct”) that applies to all officers, directors and employees. The Company will promptly disclose (1) the nature of any amendment to the Code of Conduct that applies to our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions and (2) the nature of any waiver, including an implicit waiver, from a provision of our Code of Conduct that is granted to one of these specified officers, the name of such person who is granted the waiver and the date of the waiver on our website in the future. The Code of Conduct can be accessed via our website (http://www.ligand.com), Corporate Overview page. You may also request a free copy by writing to: Investor Relations, Ligand Pharmaceuticals Incorporated, 3911 Sorrento Valley Boulevard, Suite 110, San Diego, CA 92121.
The other information under Item 10 is hereby incorporated by reference to Ligand’s Definitive Proxy Statement to be filed with the SEC within 120 days of December 31, 2023.
| Item 11. | Executive Compensation | ||||
Item 11 is hereby incorporated by reference to Ligand’s Definitive Proxy Statement to be filed with the SEC within 120 days of December 31, 2023.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | ||||
Item 12 is hereby incorporated by reference to Ligand’s Definitive Proxy Statement to be filed with the SEC within 120 days of December 31, 2023.
| Item 13. | Certain Relationships and Related Transactions, and Director Independence | ||||
Item 13 is hereby incorporated by reference to Ligand’s Definitive Proxy Statement to be filed with the SEC within 120 days of December 31, 2023.
| Item 14. | Principal Accountant Fees and Services | ||||
Item 14 is hereby incorporated by reference to Ligand’s Definitive Proxy Statement to be filed with the SEC within 120 days of December 31, 2023.
100
PART IV
| Item 15. | Exhibits and Financial Statement Schedule | ||||
(a) The following documents are included as part of this Annual Report on Form 10-K.
(1) Financial statements
(2) Schedules not included herein have been omitted because they are not applicable or the required information is in the consolidated financial statements or notes thereto.
(3) The following exhibits are filed as part of this Form 10-K and this list includes the Exhibit Index.
| Incorporated by Reference | ||||||||||||||||||||
Exhibit Number | Description of Exhibit | Form | File Number | Date of Filing | Exhibit Number | Filed Herewith | ||||||||||||||
| Agreement and Plan of Merger, dated as of March, 23, 2022, by and among Avista Public Acquisition Corp. II, Ligand Pharmaceuticals Incorporated, OmniAb, Inc. and Orwell Merger Sub Inc. | 8-K | 001-33093 | March 24, 2022 | 2.1 | ||||||||||||||||
| Separation and Distribution Agreement, dated as of March 23, 2022, by and among Avista Public Acquisition Corp. II, Ligand Pharmaceuticals Incorporated and OmniAb, Inc. | 8-K | 001-33093 | March 24, 2022 | 2.2 | ||||||||||||||||
| Amended and Restated Certificate of Incorporation of the Company. | S-4 | 333-58823 | July 9, 1998 | 3.1 | ||||||||||||||||
| Certificate of Amendment of the Amended and Restated Certificate of Incorporation of the Company, dated June 14, 2000 | 10-K | 0-20720 | March 29, 2001 | 3.5 | ||||||||||||||||
| Certificate of Amendment of the Amended and Restated Certificate of Incorporation of the Company, dated June 30, 2004 | 10-Q | 0-20720 | August 5, 2004 | 3.6 | ||||||||||||||||
| Certificate of Amendment of the Amended and Restated Certificate of Incorporation of the Company, dated November 17, 2010 | 8-K | 001-33093 | November 19, 2010 | 3.1 | ||||||||||||||||
| Certificate of Amendment of the Amended and Restated Certification of Incorporation of the Company, dated June 19, 2018 | S-8 | 333-233130 | August 8, 2019 | 3.6 | ||||||||||||||||
| Fourth Amended and Restated Bylaws of the Company | 8-K | 001-33093 | October 30, 2020 | 3.1 | ||||||||||||||||
| Specimen stock certificate for shares of the common stock of the Company | 10-K | 001-33093 | March 1, 2018 | 4.1 | ||||||||||||||||
| Description of Registered Securities | 10-K | 001-33093 | February 24, 2021 | 4.3 | ||||||||||||||||
| 2002 Stock Incentive Plan (as amended and restated effective June 10, 2022) | DEF 14A | 001-33093 | April 22, 2022 | Appendix A | ||||||||||||||||
| 2002 Employee Stock Purchase Plan (as amended and restated effective June 6, 2019) | DEF | 001-33093 | April 24, 2019 | Appendix B | ||||||||||||||||
| Form of Stock Option Grant Notice and Stock Option Agreement under the Company’s 2002 Stock Incentive Plan | 10-K | 001-33093 | February 24, 2014 | 10.5 | ||||||||||||||||
101
| Form of Stock Issuance Agreement for non-employee directors under the Company’s 2002 Stock Incentive Plan | S-1 | 333-131029 | January 13, 2006 | 10.289 | ||||||||||||||||
| Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Agreement under the Company’s 2002 Stock Incentive Plan | 10-K | 001-33093 | March 1, 2018 | 10.6 | ||||||||||||||||
| Form of Restricted Stock Unit Grant Notice and Restricted Stock Unit Agreement under the Company’s 2002 Stock Incentive Plan - Performance-Based RSU Form | 10-K | 001-33093 | March 1, 2018 | 10.7 | ||||||||||||||||
| Form of Executive Officer Change in Control Severance Agreement | 8-K | 001-33093 | August 22, 2007 | 10.1 | ||||||||||||||||
| Form of Change in Control Severance Agreement | 10-Q | 001-33093 | May 8, 2023 | 10.3 | ||||||||||||||||
| Amended and Restated Severance Plan, effective November 1, 2022 | 10-K | 001-33093 | February 28, 2023 | 10.8 | ||||||||||||||||
| Director Compensation and Stock Ownership Policy, as amended and restated, effective August 4, 2023 | 10-Q | 001-33093 | August 9, 2023 | 10.1 | ||||||||||||||||
| 2022 Employment Inducement Plan | 10-Q | 001-33093 | August 9, 2022 | 10.2 | ||||||||||||||||
| Amendment to 2022 Employee Inducement Plan | X | |||||||||||||||||||
| Form of Stock Option Agreement under the Company’s 2022 Employment Inducement Plan | 10-Q | 001-33093 | August 9, 2022 | 10.3 | ||||||||||||||||
| Form of Restricted Stock Unit Award Agreement under the Company’s 2022 Employment Inducement Plan | 10-Q | 001-33093 | August 9, 2022 | 10.4 | ||||||||||||||||
| Form of Performance-Based Restricted Stock Unit Award Agreement under the Company’s 2022 Employment Inducement Plan | 10-Q | 001-33093 | August 9, 2022 | 10.5 | ||||||||||||||||
| Separation Agreement , effective December 12, 2022, by and between Ligand Pharmaceuticals Incorporated and John Higgins | 10-K | 001-33093 | February 28, 2023 | 10.14 | ||||||||||||||||
| Severance Agreement, effective December 5, 2022, by and between Ligand Pharmaceuticals Incorporated and Todd C. Davis | 10-K | 001-33093 | February 28, 2023 | 10.15 | ||||||||||||||||
| Tax Matters Agreement, dated as of November 1, 2022, by and among OmniAb, Inc.(f/k/a Avista Public Acquisition Corp. II) Ligand Pharmaceuticals Incorporated and OmniAb Operations, Inc. (f/k/a OmniAb, Inc.) | 8-K | 001-33093 | November 4, 2022 | 10.1 | ||||||||||||||||
| Amended and Restated Employee Matters Agreement, dated as of August 18, 2022, by and among Ligand Pharmaceuticals Incorporated, OmniAb Operations, Inc. (f/k/a OmniAb, Inc.), OmniAb, Inc. (f/k/a Avista Public Acquisition Corp. II) and Orwell Merger Sub Inc. | 10-Q | 001-33093 | November 8, 2022 | 10.1 | ||||||||||||||||
| TR Beta Contingent Value Rights Agreement, dated January 27, 2010, among the Company, Metabasis Therapeutics, Inc., David F. Hale and Mellon Investor Services LLC | 8-K | 001-33093 | January 28, 2010 | 10.2 | ||||||||||||||||
| General Contingent Value Rights Agreement, dated January 27, 2010, among the Company, Metabasis Therapeutics, Inc., David F. Hale and Mellon Investor Services LLC | 8-K | 001-33093 | January 28, 2010 | 10.4 | ||||||||||||||||
102
| Amendment of General Contingent Value Rights Agreement, dated January 26, 2011, among the Company, Metabasis Therapeutics, Inc., David F. Hale and Mellon Investor Services LLC | 8-K | 001-33093 | January 31, 2011 | 10.1 | ||||||||||||||||
| Amendment of General Contingent Value Rights Agreement dated May 20, 2014 among the Company, Metabasis Therapeutics, Inc., David F. Hale and Computershare Inc. | 8-K | 001-33093 | May 22, 2014 | 10.1 | ||||||||||||||||
| Amendment of TR Beta Contingent Value Rights Agreement dated May 20, 2014 among the Company, Metabasis Therapeutics, Inc., David F. Hale and Computershare, Inc. | 8-K | 001-33093 | May 22, 2014 | 10.2 | ||||||||||||||||
| Captisol® Supply Agreement, dated December 20, 2002, among CyDex, Inc., Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited and Hovione International Limited | 10-K | 001-33093 | March 3, 2011 | 10.1 | ||||||||||||||||
| 1st Amendment to Captisol® Supply Agreement, dated July 29, 2005, among CyDex, Inc., Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited and Hovione International Limited | 10-K | 001-33093 | March 3, 2011 | 10.101 | ||||||||||||||||
| 2nd Amendment to Captisol® Supply Agreement, dated March 1, 2007, among CyDex, Inc., Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | 10-K | 001-33093 | March 3, 2011 | 10.102 | ||||||||||||||||
| 3rd Amendment to Captisol® Supply Agreement, dated January 25, 2008, among CyDex, Inc., Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited, and Hovione International Limited | 10-K | 001-33093 | March 3, 2011 | 10.103 | ||||||||||||||||
| 4th Amendment to Captisol® Supply Agreement, dated September 28, 2009, among CyDex Pharmaceuticals, Inc., Hovione LLC, Hovione FarmaCiencia S.A., Hovione Pharmascience Limited and Hovione International Limited | 10-K | 001-33093 | March 3, 2011 | 10.104 | ||||||||||||||||
| License Agreement, dated September 3, 1993, between CyDex L.C. and The University of Kansas | 10-K | 001-33093 | March 3, 2011 | 10.105 | ||||||||||||||||
| First Amendment to License Agreement, dated February 24, 1998, between CyDex, Inc. and The University of Kansas | 10-K | 001-33093 | March 3, 2011 | 10.106 | ||||||||||||||||
| Second Amendment to License Agreement, dated August 4, 2004, between CyDex, Inc. and The University of Kansas | 10-K | 001-33093 | March 3, 2011 | 10.107 | ||||||||||||||||
| Acknowledgement Agreement, dated February 22, 2008, between CyDex, Inc. and The University of Kansas | 10-K | 001-33093 | March 3, 2011 | 10.111 | ||||||||||||||||
| Exclusive License Agreement, dated June 4, 1996, between Pfizer, Inc. and The University of Kansas | 10-K | 001-33093 | March 3, 2011 | 10.108 | ||||||||||||||||
| Addendum to Nonexclusive License Agreement, dated December 11, 2001, between CyDex, Inc. and Pfizer, Inc. | 10-K | 001-33093 | March 3, 2011 | 10.11 | ||||||||||||||||
| License Agreement, by and between CyDex Pharmaceuticals, Inc. and Spectrum Pharmaceuticals, Inc., dated as of March 8, 2013 | 10-Q | 001-33093 | May 8, 2013 | 10.2 | ||||||||||||||||
| Supply Agreement, by and between CyDex Pharmaceuticals, Inc. and Spectrum Pharmaceuticals, Inc., dated as of March 8, 2013 | 10-Q | 001-33093 | May 8, 2013 | 10.3 | ||||||||||||||||
| Addendum, dated May 22, 2019, by and among Ligand Pharmaceuticals Incorporated, CyDex Pharmaceuticals, Inc., and Acrotech Biopharma LLC (as successor-in-interest to Spectrum Pharmaceuticals, Inc.), to that certain License Agreement between Ligand Pharmaceuticals Incorporated and Spectrum Pharmaceuticals, Inc., dated March 8, 2013 | 10-Q | 001-33093 | August 8, 2019 | 10.1 | ||||||||||||||||
103
| Royalty Stream and Milestone Payments Purchase Agreement, dated April 29, 2013, between the Company and Selexis S.A. | 10-Q | 001-33093 | August 1, 2013 | 10.2 | ||||||||||||||||
| Master License Agreement dated May 21, 2014 among the Company, Metabasis Therapeutics, Inc. and Viking Therapeutics, Inc. | 10-Q | 001-33093 | August 5, 2014 | 10.2 | ||||||||||||||||
| First Amendment to Master License Agreement dated September 6, 2014 among the Company, Metabasis Therapeutics, Inc. and Viking Therapeutics, Inc. | 10-Q | 001-33093 | October 31, 2014 | 10.9 | ||||||||||||||||
| Second Amendment to Master License Agreement, dated April 8, 2015, among the Company, Metabasis Therapeutics, Inc. and Viking Therapeutics, Inc. | 10-Q | 001-33093 | August 5, 2015 | 10.1 | ||||||||||||||||
| Development Funding and Royalties Agreement, dated December 13, 2018, by and between Ligand Pharmaceuticals Incorporated and Palvella Therapeutics, Inc. | 10-K | 001-33093 | February 28, 2019 | 10.48 | ||||||||||||||||
| Amendment No. 1 to Development Funding and Royalties Agreement, dated as of May 22, 2020, by and between the Company and Palvella Therapeutics, Inc. | X | |||||||||||||||||||
| Amendment No. 2 to Development Funding and Royalties Agreement, dated as of November 29, 2023, by and between the Company and Palvella Therapeutics, Inc. | X | |||||||||||||||||||
| Sublicense Agreement between the Company, Pharmacopeia, Inc. and Retrophin LLC dated as of February 16, 2012, as amended through Amendment No. 5 to Sublicense Agreement, dated March 20, 2018. | 10-K | 001-33093 | February 28, 2022 | 10.37 | ||||||||||||||||
| Interest Purchase Agreement, dated May 3, 2016, between the Company and CorMatrix Cardiovascular, Inc. | 8-K/A | 001-33093 | May 9, 2016 | 10.1 | ||||||||||||||||
| Amended and Restated Interest Purchase Agreement, dated May 31, 2017, between the Company and CorMatrix Cardiovascular, Inc. | 10-Q | 001-033093 | August 9, 2017 | 10.2 | ||||||||||||||||
| Form of Indemnification Agreement between the Company and each of its directors | 10-K | 001-33093 | March 1, 2018 | 10.60 | ||||||||||||||||
| Form of Indemnification Agreement between the Company and each of its officers | 10-K | 001-33093 | March 1, 2018 | 10.61 | ||||||||||||||||
| Addendum, dated May 22, 2019, by and among Ligand Pharmaceuticals Incorporated, CyDex Pharmaceuticals, Inc., and Acrotech Biopharma LLC (as successor-in-interest to Spectrum Pharmaceuticals, Inc.), to that certain License Agreement between Ligand Pharmaceuticals Incorporated and Spectrum Pharmaceuticals, Inc., dated March 8, 2013 | 10-Q | 001-33093 | August 8, 2019 | 10.1 | ||||||||||||||||
| At-the-Market Equity Offering Sales Agreement, dated September 30, 2022, by and between the Registrant and Stifel, Nicolaus & Company, Incorporated | S-3ASR | 333-267678 | September 30, 2022 | 1.2 | ||||||||||||||||
| Credit Agreement, dated as of October 12, 2023, by and among the Registrant, certain of its subsidiaries, as Guarantors (as defined therein), the Lenders (as defined therein), and Citibank, N.A., as Administrative Agent, Swingline Lender and L/C Issuer | 8-K | 001-33093 | October 18, 2023 | 10.1 | ||||||||||||||||
| Subsidiaries of the Company | X | |||||||||||||||||||
| Consent of Independent Registered Public Accounting Firm | X | |||||||||||||||||||
104
| Certification by Principal Executive Officer, Pursuant to Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | X | |||||||||||||||||||
| Certification by Principal Financial Officer, Pursuant to Rules 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | X | |||||||||||||||||||
| Certifications by Principal Executive Officer and Principal Financial Officer, Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | X | |||||||||||||||||||
| Policy for Recovery of Erroneously Awarded Compensation | X | |||||||||||||||||||
| 101 | The following financial information from our Annual Report on Form 10-K for the fiscal year ended December 31, 2023, formatted in iXBRL (inline eXtensible Business Reporting Language): (i) Consolidated Balance Sheets, (ii) Consolidated Statements of Operations, (iii) Consolidated Statement of Comprehensive Income, (iv) Consolidated Statements of Stockholders' Equity, (v) Consolidated Statements of Cash Flows, and (vi) the Notes to Consolidated Financial Statements. | X | ||||||||||||||||||
| 104 | The cover page from the Company's Annual Report on Form 10-K for the fiscal year ended December 31, 2023, formatted in Inline XBRL and contained in Exhibit 101. | X | ||||||||||||||||||
† Confidential treatment has been requested for portions of this exhibit. These portions have been omitted and submitted separately to the Securities and Exchange Commission.
# Indicates management contract or compensatory plan.
* Certain schedules and annexes have been omitted in accordance with Item 601(a)(5) of Regulation S-K. A copy of any omitted schedule and/or annex will be furnished as a supplement to the U.S. Securities and Exchange Commission upon request.
** Pursuant to Item 601(b)(10) of Regulation S-K, certain confidential portions of this exhibit were omitted by means of marking such portions with an asterisk because the identified confidential portions (i) are not material and (ii) would be competitively harmful if publicly disclosed.
| Item 16. | Form 10-K Summary | ||||
None
105
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| LIGAND PHARMACEUTICALS INCORPORATED | |||||
| By: | /S/ TODD C. DAVIS | ||||
| Todd C. Davis, | |||||
| Chief Executive Officer | |||||
Date: February 29, 2024
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||||||||||||
| /s/ TODD C. DAVIS | Chief Executive Officer and Director (Principal Executive Officer) | February 29, 2024 | ||||||||||||
| Todd C. Davis | ||||||||||||||
| /s/ OCTAVIO ESPINOZA | Chief Financial Officer (Principal Financial and Accounting Officer) | February 29, 2024 | ||||||||||||
| Octavio Espinoza | ||||||||||||||
| /s/ JOHN W. KOZARICH | Director and Chairman of the Board | February 29, 2024 | ||||||||||||
| John W. Kozarich | ||||||||||||||
| /s/ JASON M. ARYEH | Director | February 29, 2024 | ||||||||||||
| Jason M. Aryeh | ||||||||||||||
| /s/ NANCY R. GRAY | Director | February 29, 2024 | ||||||||||||
| Nancy R. Gray | ||||||||||||||
| /s/ JASON HAAS | Director | February 29, 2024 | ||||||||||||
| Jason Haas | ||||||||||||||
| /s/ JOHN L. LAMATTINA | Director | February 29, 2024 | ||||||||||||
| John L. LaMattina | ||||||||||||||
| /s/ STEPHEN L. SABBA | Director | February 29, 2024 | ||||||||||||
| Stephen L. Sabba | ||||||||||||||
| /s/ MARTINE ZIMMERMANN | Director | February 29, 2024 | ||||||||||||
| Martine Zimmermann | ||||||||||||||
106