UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM
For
the fiscal year ended:
or
For the transition period from _________ to ________
Commission
File Number:
(Exact name of registrant as specified in its charter)
(State or other jurisdiction of incorporation) |
(I.R.S. Employer Identification Number) |
| (Address of principal executive offices and zip code) | (Registrant’s telephone number, including area code) |
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
Securities registered pursuant to Section 12(g) of the Act: None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2)
has been subject to such filing requirements for the past 90 days.
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule
405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant
was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large accelerated filer ☐ | Accelerated filer ☐ |
| Smaller reporting company | |
| Emerging growth company |
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered
public accounting firm that prepared or issued its audit report.
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant
included in the filing reflect the correction of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No
The
registrant was
The registrant had shares of its common stock, par value $0.001, issued and outstanding as of April 12, 2024.
TABLE OF CONTENTS
| 2 |
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K (“Annual Report”) contains forward-looking statements within the meaning of the federal securities laws. All statements contained in this Annual Report, other than statements of historical fact, including statements regarding our future operating results and financial position, our business strategy and plans, potential growth or growth prospects, future research and development, sales and marketing and general and administrative expenses, and our objectives for future operations, are forward-looking statements. Words such as “believes,” “may,” “will,” “estimates,” “potential,” “continues,” “anticipates,” “intends,” “expects,” “could,” “would,” “projects,” “plans,” “targets,” and variations of such words and similar expressions are intended to identify forward-looking statements. We have based these forward-looking statements largely on our current expectations and projections about future events and trends that we believe may affect our financial condition, results of operations, business strategy, short-term and long-term business operations and objectives, and financial needs. These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described in the “Risk Factors” in this Annual Report. Readers are urged to carefully review and consider the various disclosures made in this Annual Report and in other documents we file from time to time with the Securities and Exchange Commission (the “SEC”) that disclose risks and uncertainties that may affect our business. Moreover, we operate in a very competitive and rapidly changing environment. New risks emerge from time to time. It is not possible for us to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. In light of these risks, uncertainties, and assumptions, the future events and circumstances discussed in this Annual Report may not occur and actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements.
You should not rely upon forward-looking statements as predictions of future events. The events and circumstances reflected in the forward-looking statements may not be achieved or occur. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, performance, or achievements. In addition, the forward-looking statements in this Annual Report are made as of the date of this filing, and we do not undertake, and expressly disclaim any duty, to update such statements for any reason after the date of this Annual Report or to conform statements to actual results or revised expectations, except as required by law.
You should read this Annual Report and the documents that we reference herein and have filed with the SEC as exhibits to this Annual Report with the understanding that our actual future results, performance, and events and circumstances may be materially different from what we expect.
This Annual Report also contains or may contain estimates, projections and other information concerning our industry, our business and the markets for our products, including data regarding the estimated size of those markets and their projected growth rates. Information that is based on estimates, forecasts, projections or similar methodologies is inherently subject to uncertainties and actual events or circumstances may differ materially from events and circumstances reflected in this information. Unless otherwise expressly stated, we obtained these industry, business, market and other data from reports, research surveys, studies and similar data prepared by third parties, industry and general publications, government data and similar sources. In some cases, we do not expressly refer to the sources from which these data are derived.
| 3 |
EXPLANATORY NOTE
This Annual Report on Form 10-K for the year ended December 31 2023 (“Form 10-K”) includes the restatement of our unaudited financial statements as of and for the three and nine month periods ended September 30, 2023. The Board of Directors (the “Board”) of the Company, upon recommendation of the Audit Committee of the Board and discussion with management, concluded that the Company’s previously issued unaudited financial statements as of and for the three and nine month periods ended September 30, 2023, filed with the Securities and Exchange Commission (“SEC”) on November 20, 2023, should no longer be relied upon and should be restated due to the identification of an accounting error.
As disclosed in Note 19 to the financial statements included in this Form 10-K, we restated our unaudited financial statements as of and for the three and nine month periods ended September 30, 2023 to correct an error that understated the extinguishment of debt expense and net loss in the Statement of Operations and understated additional paid in capital in the Balance Sheet. Management also concluded that the Company’s disclosure controls and procedures were not effective as of December 31, 2023 due to the existence of this material weakness.
We have not filed and do not intend to file amendments to our Quarterly Report on Form 10-Q as of and for the three and nine month periods ended September 30, 2023. Comparative amounts for 2023 presented in our 2024 Quarterly Reports on Form 10-Q will be changed retrospectively to reflect the restatement. Accordingly, investors should only rely on the financial information and other disclosures as of and for the three and nine month periods ended September 30, 2023 in this Form 10-K or in future filings with the SEC and not any previously issued or filed reports.
The impact of the restatement and the unaudited restated financial statements as of and for the three and nine month periods ended September 30, 2023 are described and included in Note 19 to our audited financial statements included in this Form 10-K.
| 4 |
PART I
ITEM 1. BUSINESS
Overview
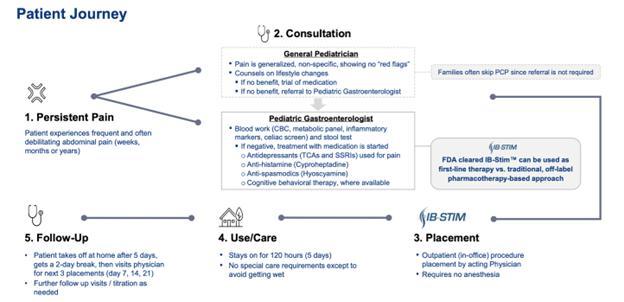
Neuraxis, Inc. (“we”, “us”, the “Company” or “Neuraxis”) is a medical technology company focused on developing neuromodulation therapies to address chronic and debilitating conditions in children and adults. We are dedicated to advancing science with our proprietary IB-Stim therapy, based on our Percutaneous Electrical Nerve Field Stimulation (PENFS) technology, which was developed internally by the Company. We believe that superior science and evidence-based research, are necessary for adoption by the medical and scientific community. With one FDA indication (functional abdominal pain associated with IBS in adolescents 11-18 years old) on the market, additional clinical trials of PENFS in multiple pediatric conditions are underway focused on unmet healthcare needs in children, see “—Our Pipeline” for more information.
Our first product, IB-Stim, is a PENFS system intended to be used in patients 11-18 years of age with functional abdominal pain associated with IBS. IB-Stim is a US FDA Class II medical device that has received one regulatory clearance: IB-Stim (DEN180057, 2019), under the regulation name of “non-implanted nerve stimulator for functional abdominal pain relief.”
Our Mission
Our mission is to provide solutions that create value and provide better and safer patient outcomes. We believe in improving lives and minimizing suffering; particularly in the pediatric population, where research and therapeutics are usually lacking. The Company already has market clearance for its IB-Stim ® that targets functional abdominal pain associated with IBS, in children, with a total addressable market of up to 6 million children. Through innovation and research, we are reimagining the future of patient care.
Our Corporate History
Neuraxis, Inc. was established in 2011 and incorporated in the state of Indiana on April 17, 2012, under the name of Innovative Health Solutions, Inc. The name was changed to Neuraxis, Inc. in March of 2022. Additionally, the Company filed a Certificate of Conversion to become a Delaware corporation on June 23, 2022. The authorized shares were increased, and a par value established. On September 7, 2021, the Company’s board of directors authorized a 4-for-1 stock split. They also increased the number of authorized common stock shares from 2,700,000 to 10,800,000. Furthermore, on September 9, 2021, the board authorized and increase of authorized shares of common stock from 10,800,000 to 13,400,000 in anticipation of a capital offering.
As part of the conversion to a Delaware corporation, the total number of shares of all classes of stock which the Corporation shall have authority to issue is 101,120,000 shares, consisting of (i) 100,000,000 shares of common stock, par value $0.001 per share, and (ii) 1,120,000 shares of Preferred Stock, par value $0.001 per share (“Preferred Stock”), 1,000,000 of which is designated as “Series A Preferred Stock” and 120,000 of which is designated as “Series Seed Preferred Stock”.
Furthermore, on January 10, 2023, the Company’s board of directors authorized a 1-for-2 reverse stock split. All per share information has been adjusted for this reverse stock split. The reverse split became effective on January 12, 2023.
We have developed three FDA cleared products, the IB-Stim (DEN180057, 2019), the NSS-2 Bridge (DEN170018, 2017), and the original 510(K) clearance (K140530, 2014), all of which were developed internally by the Company.
| ● | The IB-Stim is a PENFS device that is indicated in patients 11-18 years of age with functional abdominal pain associated with irritable bowel syndrome. The IB-Stim currently is the only product marketed and sold by the Company. | |
| ● | The NSS-2 Bridge is a percutaneous nerve field stimulator, or PNFS, device indicated for use in the reduction of the symptoms of opioid withdrawal. The NSS-2 Bridge device was licensed to Masimo in April 2020, and we received a one-time licensing fee of $250,000 from Masimo. Masimo markets and sells this product as its Masimo Bridge, and we will not receive any further licensing payments or other revenue from this product. | |
| ● | The original 510(K) device was the electroacupuncture device (“EAD”), now called NeuroStim. The EAD is no longer being manufactured, sold or distributed but reserved only for research purposes. |
| 5 |
Pediatrics Industry Overview
Pediatric providers, as a whole, had expressed concern about the lack of attention given to children with functional abdominal pain disorders (including IBS) and the limited treatment options available for a population that suffers from significant disabilities. With 20% of the United States population under age 18, our Company focus is on opportunities in pediatrics industry. The pediatrics industry has multi-billion-dollar market opportunities. The following points clearly outline the unmet need in children:
| ● | Functional abdominal pain in children is one of the most common conditions seen by pediatricians and pediatric gastroenterologists. | |
| ● | Children with functional abdominal pain report lower quality of life compared with their healthy peers and equal to those with inflammatory bowel disease. | |
| ● | Overall, 40-45% of children with functional abdominal pain disorders continue to have symptoms into adulthood, which impacts quality of life and healthcare spending. | |
| ● | A study published in 2021 demonstrates insufficient evidence for the use of medications in pediatric functional abdominal pain disorders. This lack of evidence for drugs has been supported in by the American Academy of Pediatrics and NASPGHAN. | |
| ● | IB-Stim is the only medical therapy that has shown to improve pain, global symptoms, and functional disability in children with FAP and IBS. | |
| ● | IB-Stim is the only currently used medical therapy that is better than placebo in a randomized controlled trial and received FDA clearance for pediatric IBS. |
Our Opportunity
For years, physicians and qualified healthcare professionals have resorted to the use of off-label medications without proper evidence of efficacy or safety. This is despite a technical report from the American Academy of Pediatrics and NASPGHAN which found very little evidence to endorse the use of any drugs in the treatment of FAPDs in children. Medications including tricyclic antidepressants, SSRIs and gabapentinoids continue to be used off-label despite lack of evidence to support efficacy or safety. Not only have the most commonly used medications (amitriptyline and citalopram) failed to beat placebo in clinical trials, but new studies also suggest significant risks with the potential for serious side effects with these drugs. The absence of conclusive data to support treatments based on scientific evidence, and the fact no drug therapies have been approved by the FDA for the treatment of FAPDs or IBS in children, presents a unique market opportunity for Neuraxis. Below are the current standard treatments in children with functional abdominal pain and IBS.

| 6 |
Our Solutions
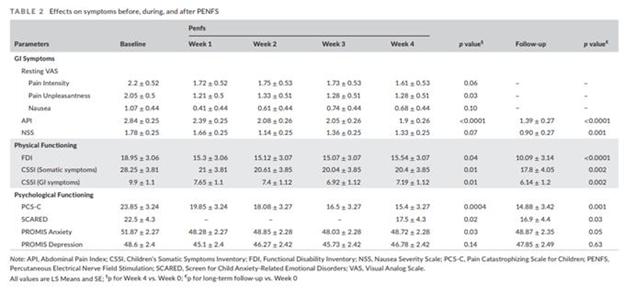
We entered the pediatric market with clinical evidence, key opinion leaders and society endorsement, including a signed letter from the American Academy of Pediatrics and NASPGHAN supporting our request for insurers to pay for our IB-Stim device. Our IB-Stim® is a non-drug alternative to reduce functional abdominal pain in patients with IBS. In June 2019, the FDA cleared IB-Stim, a non-surgical, neuromodulation device for children and adolescents who suffer from IBS, through a de novo process (DEN180057, 2019). The FDA created a new classification of PENFS for the IB-Stim device. This is based on pre-clinical and clinical studies demonstrating the mechanism of action and efficacy. Based on this new class of devices, the IB-Stim falls under 21 CFR Part 876, Subpart F – Therapeutic Devices, 876.5340, Product Code QHH. As a PENFS device, it is non-implantable and provides field stimulation to cranial nerves V, VII, IX and X in the ear to access the CNS. It stimulates remotely from the source of pain to modulate central pain regions, such as the limbic system, and relieve functional abdominal pain associated with IBS. Studies have demonstrated long-term benefits in functional disability, psychological co-morbidities, and pain. For example, the table below is from a recently published study of IB-Stim in a population of patients with chronic functional abdominal pain. The follow-up was done at 6-12 months post-treatment and shows improvements in validated questionnaires compared to baseline (API), functional disability index (FDI), pain catastrophizing scale (PCS), Screen for Childhood Anxiety Related Disorders (SCARED) and the Promis Anxiety.
Santucci NR, King C, El-Chammas KI, Wongteerasut A, Damrongmanee A, Graham K, Fei L, Sahay R, Jones C, Cunningham NR, Coghill RC. Effect of percutaneous electrical nerve field stimulation on mechanosensitivity, sleep, and psychological comorbidities in adolescents with functional abdominal pain disorders. Neurogastroenterol Motil. 2022;34:e14358.
We have only submitted one FDA De Novo request and have not submitted any additional 510(k) premarket notifications for our pipeline indications to date.
Compliance with treatment so far has been outstanding with the four weeks of therapy required to sustain long-term benefits. Compliance has been an issue with non-pharmacological treatment for children, particularly with some of the psychological approaches such as cognitive behavioral therapy or guided imagery, which sometimes requires 8-12 weeks of treatment. In fact, 95% of adolescents who used IB-Stim said that they would recommend this treatment to family and friends. Many children’s hospitals across the country are already treating children with IB-Stim successfully since it provides a better alternative for therapy in children with IBS and disability and allows them to treat them safely and effectively.
We have concentrated our marketing focus on the 260 children’s hospitals within the United States. To date, we have sold our IB-Stim product to approximately 57 children’s hospitals within our target market.

| 7 |
Competition
The competitive landscape for therapies includes off-label drugs and drugs with FDA approved only for adults with IBS while there is no FDA indicated treatments for patients 11-18 years of age with functional abdominal pain associated with IBS and prescriptions often contain FDA black box labels. Psychological treatments such as cognitive behavioral therapy (CBT) or guided imagery have been shown to be some of the most effective treatments for these conditions, however, these are limited by access to trained therapists. It also includes devices that could theoretically be used, but do not have supporting data or FDA clearance for functional bowel disorders or IBS. Digital therapeutics that offer CBT for IBS have been developed for adults with IBS with limited success in terms of reaching large numbers of patients. Virtual reality could potentially be used in the future to also deliver CBT to patients with IBS. Our method patents also limit other devices from targeting IBS through stimulation of cranial nerve branches in the ear.

Approved drugs for Adults with IBS
| 1. | Rifaximin: an intraluminal antibiotic approved for IBS-diarrhea | |
| 2. | Amitiza: a drug that stimulates fluid secretion from the intestine, approved for IBS-diarrhea | |
| 3. | Linzess: a drug that stimulates fluid secretion from the intestine, approved for IBS-constipation | |
| 4. | Plecanatide: a drug that stimulates fluid secretion from the intestine, approved IBS-constipation | |
| 5. | Eluxadoline: a schedule IV-controlled substance that is a mixed opioid receptor agonist/antagonist in the intestine approved for IBS-diarrhea |
| 8 |
Devices
| 1. | gammaCore: a transcutaneous, cervical vagal nerve stimulator cleared for cluster and migraine headaches. Recent studies using this device for adults with gastroparesis. | |
| 2. | Transcranial Magnetic Stimulation: Multiple devices cleared to treat major depressive disorder and obsessive-compulsive disorder. To date, no known gastrointestinal indications. | |
| 3. | Roo System and Sparrow therapy system: Transcutaneous auricular stimulation devices-cleared for neonatal and adult opioid withdrawal. |
The neurostimulation market is predominantly comprised of surgically implanted, invasive technologies that are not directly competitive with our technology. Several neurostimulation companies are large, publicly traded companies that have a history in the market, have significantly easier access to capital and other resources and have an established product pipeline. The combined clinical research and product development done by the industry, including by us and all our competitors, is uncovering the beneficial effects of neurostimulation which now establishes neuromodulation as a valid and scientifically supported approach to the treatment of neurological conditions, and accordingly, we expect for competition in the non-implanted space to grow in the future.
While many companies have joined the neuromodulation space, there are no companies targeting the CNS or the brain-gut axis through auricular nerves for functional bowel disorders or IBS. Currently, the Neuraxis method patents protect access to the brain, particularly the limbic systems through branches of cranial nerves in the ear.
Our Competitive Strengths
We believe that the following competitive strengths will enable us to compete effectively:
| ● | First to market | |
| ● | Strong portfolio of device and method patents | |
| ● | Large Market Opportunities | |
| ● | Strong pediatric pipeline | |
| ● | Academic Society Support | |
| ● | Lower capital expenditures in nurse, trainers, and representatives for first line therapy | |
| ● | Strong clinical data carried out in leading academic institutions in the U.S. |
Our Growth Strategies
| ● | List price of our product is $1,195 per device and $4,780 per patient | |
| ● | Strong gross margin | |
| ● | Direct sales force | |
| ● | Target customers are children’s hospitals and pediatric clinics |
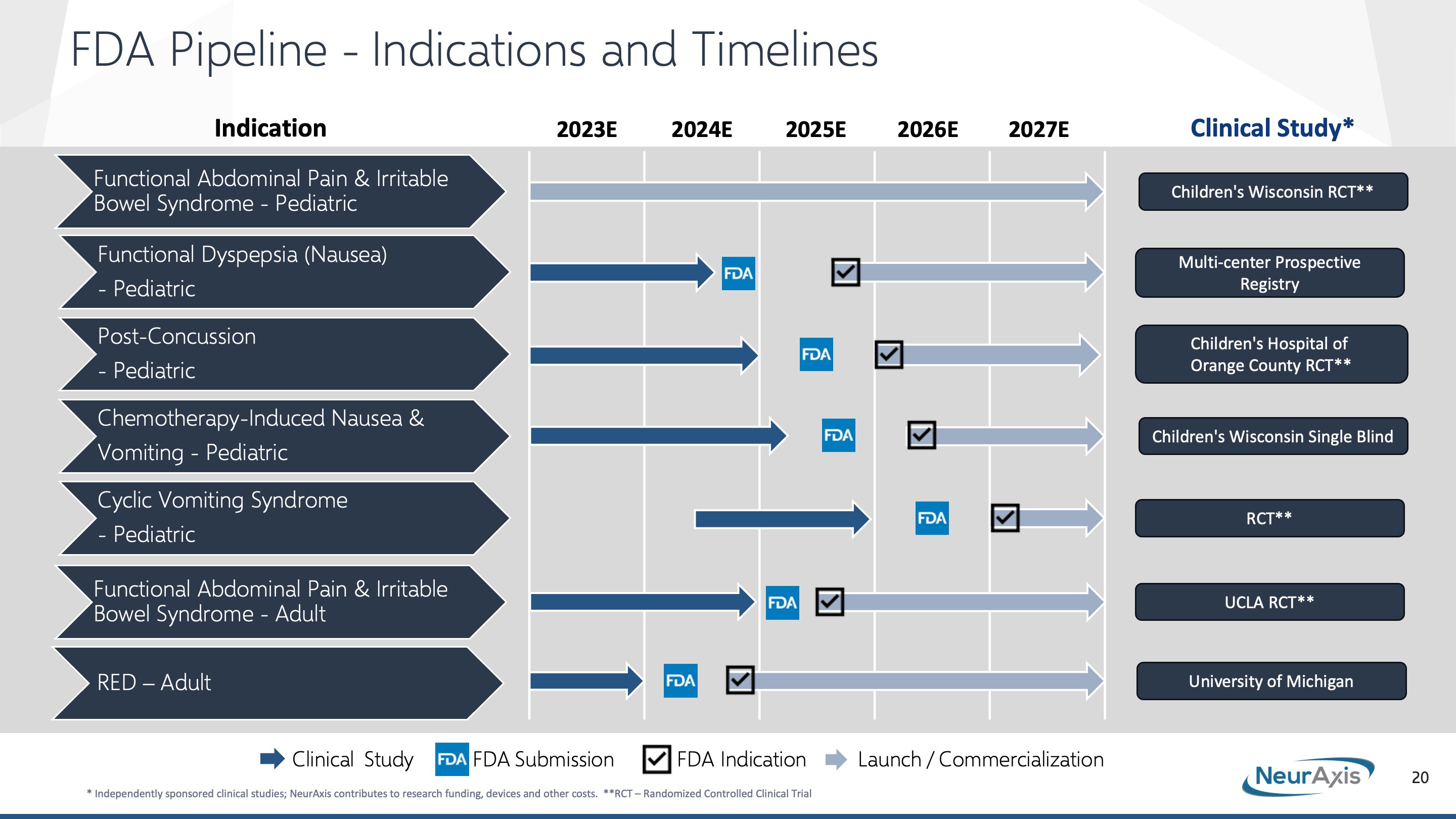
Our Pipeline
The IB-Stim device is to be used for the indication of functional abdominal pain associated with IBS and functional nausea in children. The same underlying technology will be used for the remaining pipeline indications, but we may use a name other than “IB-Stim” for marketing and commercialization purposes.
With one FDA indication—functional abdominal pain associated with IBS in adolescents 11-18 years old—on the market, additional clinical trials of PENFS in multiple pediatric conditions are underway focused on unmet healthcare needs in children. These indications consist of chronic nausea, post-concussion syndrome, chemotherapy-induced nausea and vomiting, cyclic vomiting syndrome.
The chart below shows our status in the FDA review process for IB-Stim and each of the following pediatric indications:
1. Chronic nausea: RCT completed, and data being analyzed. ClinicalTrials.gov Identifier: NCT03675321, Defining Adolescent Nausea Through Brain-Gut Physiology and Non-Invasive Neurostimulation Response. A randomized, double blind, placebo-controlled trial to evaluate the efficacy of IB-Stim in children with functional nausea. The primary endpoint was to measure improvements in nausea using the Nausea Severity Scale after IB-Stim therapy compared to a placebo device. The study enrolled 110 participants and was conducted at Children’s Wisconsin/Medical College of Wisconsin.
| 9 |
2. Post-concussion syndrome: RCT currently enrolling patients. ClinicalTrials.gov Identifier: NCT04978571, A Prospective Study on the Effect of Auricular Percutaneous Electrical Nerve Field Stimulation (PENFS) in Patients with Post-Concussion Syndrome (PCS). A randomized, double blind, placebo-controlled trial to evaluate the efficacy of IB-Stim in children with post-concussion symptoms. The primary endpoint will be to measure improvements in validated measures, including the Immediate Post-Concussion Assessment, Post-Concussion Symptom Scale, and Balance Error Scoring Symptom compared to placebo. The study will enroll 100 participants and is being conducted at Children’s Hospital of Orange County.
3. Chemotherapy-induced nausea and vomiting: RCT currently enrolling patients. ClinicalTrials.gov Identifier: NCT05143554, Efficacy of Auricular Neurostimulation for Children Adolescents and Young Adults with Chemotherapy Induced Nausea and Vomiting. Subject will be randomized to five days of active vs placebo device during administered chemotherapy known to cause moderate to severe nausea/vomiting. With the next scheduled identical chemotherapy cycle, each subject will cross over to the other device (active vs placebo). The primary endpoint will be to measure improvements in validated measures of nausea and vomiting including the Baxter Retching Faces Scale, Rhodes Index of Nausea, Vomiting and Retching, and also assessment of rescue medication. The study will enroll 50 participants and is being conducted at Children’s Wisconsin/Medical College of Wisconsin.
4. Cyclic vomiting syndrome: Pilot study completed, see ClinicalTrials.gov Identifier: NCT03434652. Auricular Neurostimulation for Children with Cyclic Vomiting Syndrome: A randomized, placebo-controlled trial. RCT anticipated to begin enrolling patients in the second half of 2023. This will be a double blind, placebo-controlled trial to evaluate efficacy of IB-Stim in pediatric patients with cyclic vomiting syndrome. The primary endpoint will be to measure decreases in the frequency and severity of cyclic vomiting episodes compared to a placebo device. The study will include a minimum of 120 patients and the site is yet to be finalized.
Each step in the FDA review process differs in duration and cannot be predicted with accuracy. Timing of FDA review and approval, if ever received, cannot be assured and the process and any approval is within the sole control and discretion of the FDA.
| 10 |
Products
The IB-Stim is a percutaneous PENFS system intended to be used in patients 11-18 years of age with functional abdominal pain associated with IBS. IB-Stim already has market clearance from FDA for functional abdominal pain associated with IBS in children. FDA has classified the non-implanted nerve stimulator for functional abdominal pain relief as Class II devices.
The IB-Stim is intended to be used for 120 hours per week for three (3) consecutive weeks, and not to exceed four (4) weeks, through application to branches of Cranial Nerves V, VII, IX and X, and the occipital nerves identified by transillumination, as an aid in the reduction of pain when combined with other therapies for IBS (DEN180057, 2019). In published studies, patients treated with IB-Stim demonstrated significant improvement in pain, disability and global symptoms with no serious adverse events, and minimal to no side effects, including localized skin irritation. See Neurostimulation for abdominal pain-related functional gastrointestinal disorders in adolescents: a randomized, double-blind, sham-controlled trial, Kovacic K, et.al., Lancet Gastroenterol Hepatol. 2017;2:727-737; Efficacy of Auricular Neurostimulation in Adolescents With Irritable Bowel Syndrome in a Randomized, Double-Blind Trial, Krasaelap A et.al., Clin Gastroenterol Hepatol. 2020;18:1987-1994; Effect of percutaneous electrical nerve field stimulation on mechanosensitivity, sleep, and psychological comorbidities in adolescents with functional abdominal pain disorders, Santucci et.al., Neurogastroenterol Motil. 2022;34:e14358.
The ability of the IB-Stim to produce systemic effects by modulating the central nervous system has been demonstrated in a pre-clinical animal model of IBS (see Business—Pre-Clinical Data). In patients with IBS, the largest effect on all pain measures, including composite pain scores, worst pain, disability and global symptoms, was seen after completing three consecutive weeks of treatment (see Business—Clinical Data). A fourth consecutive week of treatment was included in clinical testing; no safety concerns were identified with this extra consecutive week of treatment. In the trial of 115 subjects, 10 patients reported side-effects and only three discontinued the study because of side-effects. Of such 10 patients, six experienced ear discomfort (three in the PENFS group, three in the sham group), three experienced adhesive allergies (one in the PENFS group, 2 in the sham group), and one experienced syncope due to needle phobia (in the sham group). There were no serious adverse events.
Medical providers are trained to place the IB-Stim through IB-Stim Training and Certification. Once the provider is trained, the device can be placed in the outpatient clinic and can be removed by the provider in the clinic or the patient at home. IB-Stim stays on for a total of five-days to allow delivery of gentle electrical pulses to nerves below the skin that access the central nervous system. A study in adolescents showed greater improvement in functional abdominal pain and global symptom improvement with every week of treatment (up to four weeks). At the end of the four-week study, 95% of adolescents stated they would recommend the treatment to family or friends. Safety of percutaneous electrical nerve field stimulation has also been reported in a separate study of over 1200 adult patients with no serious adverse events and minimal to no side-effects.
When wearing our IB-Stim device and following an easy-to-learn and efficient procedure, patients can still attend school and extracurricular activities, exercise or play non-contact sports, shower, wear ear buds or headphones, and travel.
Our IB-Stim device costs $1,195 per device, and each patient will use four (4) devices. Potential patients with other indications are expected to use six (6) or more devices per patient.
Technology
A maladaptive central nervous system can process pain and emotions differently. This often occurs in children following a traumatic event, viral infections, inflammation or trauma. Changes in brain pathways are known to be involved in the pathophysiology of functional bowel disorders and IBS. The IB-Stim works by sending gentle electrical impulses into cranial nerve bundles located in the ear. This stimulation targets brain areas that process pain and helps reduce functional abdominal pain associated with IBS. An animal model of IBS demonstrated that the firing of neurons in the amygdala could be reduced by more than 50% in just 15 minutes of stimulation with the IB-Stim technology. A recent human study in adults with pain related to fibromyalgia suggested that the IB-Stim technology exerts its effect by modulating emotional and executive control centers related to pain processing, see Feasibility of Auricular Field Stimulation in Fibromyalgia: Evaluation by Functional Magnetic Resonance Imaging, Randomized Trial, Woodbury et.al., Pain Med. 2021;22:715-726. The field of art pertains to an electrical stimulation device, including a stimulator containing a generator to deliver electrical pulses with defined parameters, and a power supply for supplying the electrical energy through four separate needles, and at least one of which is a needle array.
| 11 |
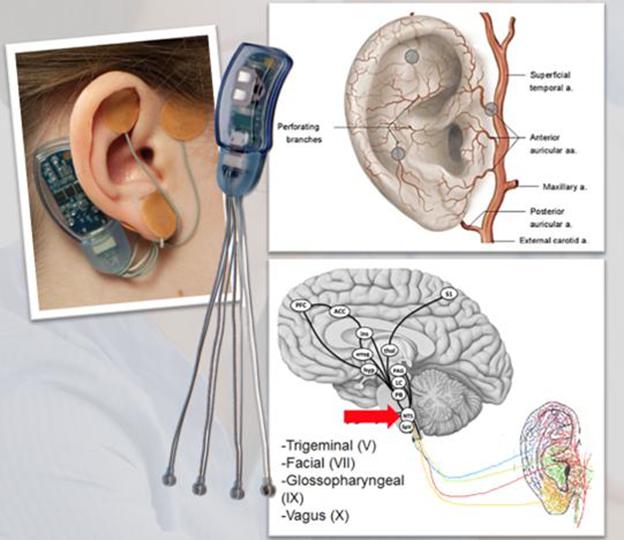
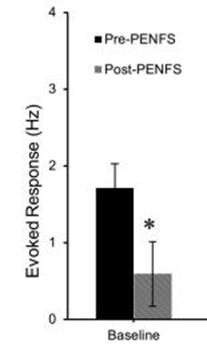
Pre-Clinical Data
In an animal model of IBS, extracellular, electrophysiologic recordings were performed from neurons in the rat amygdala before and 15 minutes after PENFS treatment. There was a 65% decrease in the spontaneous firing of these neurons after 15 minutes of PENFS.This dampening of neurons in the CNS likely accounts for the modulation of pain responses in a model of post-inflammatory visceral and somatic hyperalgesia.
| 12 |
Clinical Data
We have over 700 published patients specific to our first FDA indication which is functional abdominal pain associated with irritable bowel syndrome in patients 11-18 years of age. A published patient is defined as a patient who went through a study, the study was analyzed and now the study has been published in a peer-reviewed journal.
A randomized, controlled study in children 11-18 year of age used primary endpoint of improvements in abdominal pain. The Pain Frequency-Severity-Duration (“PFSD”) questionnaires was completed at baseline by all subjects and after each week of treatment (weeks 1–3), as well as at extended follow-up occurring in the 8–12 weeks following the end of treatment. The PFSD scale incorporates multiple aspects of the pain experience and was administered weekly during treatment and at extended follow-up appointments. The PFSD scale validated for chronic pain in children (aged 8–18 years). The PFSD was also used to rate weekly worst abdominal pain on a numerical rating scale (0 for no pain, 10 for worst pain). Patients were followed up for a median of 9.2 weeks from the last week of treatment.
For the active PENFS group, median worst pain at follow-up remained lower (baseline: 8.0 vs. follow-up: 6.0), whereas there was no difference at follow-up in the control group (baseline: 7.5 vs. follow-up: 7.0). The between-group differences in worst pain ratings after 3 weeks of treatment showed that the PENFS group improved to a greater extent, with the control group reporting significantly higher worst pain (median 7.0) than the PENFS group (median 5.0).
At long-term follow-up, median PFSD composite scores were 12.6 (IQR 3.6–22.5) in the PENFS group and 16.8 (4.8–33.6) in the control group. A comparison of changes in PFSD composite scores (baseline to follow-up) showed that patients in the PENFS group reported significantly greater improvement in pain (median –8.4) than those in the control group (median 0.0). This study was published in the Lancet Gastroenterology Hepatology, (Kovacic K, et.al. Lancet Gastroenterol Hepatol. 2017;2:727-737).
| 13 |

A secondary endpoint in the same study used the functional disability index (FDI) to assess functional disability in those treated with PENFS and compared to sham treatment. Those treated with PENFS changed from moderate disability to minimal at the 2–3-month follow-up while the sham device group had no change.

A separate published paper looked at 51 pediatric patients with IBS and used the symptoms response scale (SRS) to assess global symptoms improvement following PENFS treatment compared to sham. Global symptom improvement was assessed with a validated pediatric questionnaire, Symptom Response Scale (SRS). Symptoms were recorded as better, worse, or no change based on a 15-point scale across individual domains for both improvement and deterioration of overall symptoms. Findings from several studies that used the SRS have shown that using 7-point scale response options in disease-specific measures, a change score of 0.5 represents the minimal clinically important difference (Juniper et.al. J Clin Epidemiol 1994; 47: 81–87 and Guyatt GH et.al.1987; 42: 773–78). As previously noted, a minimum change in score of ≥ 2 was chosen for this study as a more stringent criterion for global improvement before and after PENFS treatment and to compare between groups. Patients and providers were blinded in terms of those who received active PENFS or sham. At the end 3 weeks of therapy using the change of ≥ 2, 81% of the PENFS group compared with 26% of the sham group (*p≤ 0.001, #p=0.002) reported overall symptom improvement. When applying an even more stringent criteria with a change ≥ 3 on the SRS, 67% of the PENFS group compared with 22% of the sham group reported symptoms improvement (p=0.002) (Krasaelap A et.al. Efficacy of Auricular Neurostimulation in Adolescents With Irritable Bowel Syndrome in a Randomized, Double-Blind Trial. Clin Gastroenterol Hepatol. 2020;18:1987-1994).
| 14 |

Recently, the largest, prospective, multicenter registry for any drug or device in pediatric patients with pain associated DGBIs was published. It evaluated outcomes of pediatric patients (8-18 years) following a 4-week course of IB-Stim in a real-world clinical setting. Overall, 292 patients met Rome IV Diagnostic criteria for any pain associated disorder of the gut-brain interaction (DGBIs). In this cohort, 92% had failed medication therapy and 61% of patients had failed 4 or more medications when they entered the study. Patients were asked to fill out several validated pediatric measures, including the abdominal pain index (API) and a validated questionnaire that assesses frequency, duration, and intensity of abdominal pain episodes. Data were collected weekly for the first 3 weeks and at 3, 6, 9 and 12 months. Compared to baseline scores, there were significant improvements in the API after 4 weeks of IB-Stim treatment at every time point, including 6 month (p<0.001) and 12 months (p<0.001). Although there were many dropouts by the end of the 12 months, the results were still significant and sustained. No serious adverse effects were recorded during the entire 12 month follow-up. (Chogle, A. et. al. A multicenter registry study on percutaneous electrical nerve field stimulation for pediatric disorders of gut-brain interaction. J Pediatr Gastroenterol Nutr. 2024 Mar 7.)
| Abdominal Pain Index (API) | ||||||
| Time point | n | Median (IQR) | p Value | |||
| Baseline | 288 | 2.68(1.84, 3.58) | N/A | |||
| 3 weeks | 209 | 1.99 (1.13, 3.27) | <0.001 | |||
| 3 months | 75 | 1.81 (0.85, 3.20) | <0.001 | |||
| 6 months | 60 | 1.70 (0.93, 2.72) | <0.001 | |||
| 9 months | 26 | 1.90 (1.33, 2.82) | 0.002 | |||
| 12 months | 22 | 220(0.41, 3.21) | <0.001 | |||
An open-label study of 20 patients treated with PENFS in a “real-world” clinical setting at Cincinnati Children’s Hospital demonstrated that after PENFS, abdominal pain (p < 0.0001), nausea (p=0.001), pain catastrophizing (p = 0.001), functional disability (p<0.0001), and anxiety (p = 0.03) exhibited significant improvements, and were sustained 6-12 months after treatment (Santucci et.al. Effect of percutaneous electrical nerve field stimulation on mechanosensitivity, sleep, and psychological comorbidities in adolescents with functional abdominal pain disorders. Neurogastroenterol Motil. 2022;34:e14358). Validated questionnairesincluded the abdominal pain index (API), nausea severity scale (NSS), functional disability index (FDI), as well as psychological measures of catastrophizing (PCS-C) and anxiety (SCARED). The table below summarizes the results pre, during and post PENFS results at long-term follow-up (Santucci et.al. Effect of percutaneous electrical nerve field stimulation on mechanosensitivity, sleep, and psychological comorbidities in adolescents with functional abdominal pain disorders. Neurogastroenterol Motil. 2022;34:e14358).

A clinically meaningful endpoint is the number needed to treat (NNT) used in treatment for abdominal pain-related functional gastrointestinal disorders in adolescents. NNT means the number of patients that need to be treated for one patient to get the targeted improvement (≥30% improvement).
| 15 |
Reimbursement
A PENFS procedure-specific Category III CPT Code (0720T) was published on December 30, 2021 and became effective for utilization on July 1,2022. Category III CPT Codes are temporary codes issued to define and track the utilization of new procedural technology. In collaboration with the American Medical Association, we withdrew our initial Category I CPT Code application submitted in 2023. We continue to work diligently with the American Academy of Pediatrics and other specialty medical societies on the pursuit of a Category I CPT Code for PENFS procedures. To expand patient access to PENFS procedures and IB-Stim technology, we launched our internal Prior Authorization team under our Guidance & Patient Support function in 2023. This continues to address the Prior Authorization process barriers for providers and children’s hospitals and streamlines a patient’s access to our Patient Advocacy and Financial Assistance offerings, if needed. Six (6) commercial health insurers, including certain Blue Cross Blue Shield licensees, have instituted formal medical policy coverage for PENFS. The total membership of these health insurers is approximately 9,000,000 covered lives; an additional health insurer, with approximately 7,000,000 members, will institute formal medical policy coverage for PENFS in the second quarter of 2024 bringing the total number of covered lives to approximately 16,000,000 over seven (7) commercial health insurers. Patients who are appropriate clinical candidates may have policy-covered access to PENFS and IB-Stim technology under their specific health plan. We continue to actively leverage clinical evidence and peer-reviewed publications to expand patient access to IB-Stim technology. In addition, we anticipate academic medical society support in the form of a position paper and an update to treatment guidelines to support the use of PENFS as a potential standard of care.
Marketing
We market our products through search engine optimization, or SEO, internet channels and to physicians via the academic society. We plan to extensively ramp-up our marketing efforts to patients and physicians as we gain additional indications.
Patients/Customers
Our current patient base is children 11-18 years of age and suffering from functional abdominal pain. Our customers are primarily children’s hospitals who serve these children.
Intellectual Property
Our intellectual property consists of patents, trademarks, and trade secrets. Our trade secrets consist of product formulas, research, and development, and unpatentable know-how, all of which we seek to protect, in part, by confidentiality agreements. To protect our intellectual property, we rely on a combination of laws and regulations, as well as contractual restrictions. Federal trademark law protects our registered trademarks. We also rely on the protection of laws regarding unregistered copyrights for certain content we create and trade secret laws to protect our proprietary technology. To further protect our intellectual property, we enter into confidentiality agreements with our executive officers and directors.
| 16 |
Trademarks
The Company has 10 registered trademarks, eight (8) of which are being used in commerce:
| Country | Trademark | Reg. No. | Reg. Date | Class/Goods | Status | |||||
| US | NEURO-STIM and Design | 5105257 | 20-Dec-2016 | 10 Int. nerve stimulator apparatus | Registered | |||||
| US | NSS THE NEUROSTIM SYSTEM and Design | 4905470 | 23-Feb-2016 | 10 Int. nerve stimulator apparatus | Registered | |||||
| US | THE NEURO-STIM SYSTEM and Design | 5105258 | 20-Dec-2016 | 10 Int. nerve stimulator apparatus | Registered | |||||
| US | NSS | 4852008 | 10-Nov-2015 | 10 Int. Medical apparatus, namely, electrical nerve stimulators; medical device, namely, a non- implantable neurological pain management generator, with percutaneously-implantable needle arrays; medical system and apparatus consisting of a non-implantable modulating frequency generator, providing neuromodulation therapy to cranial and peripheral nerves; medical system and apparatus consisting of implantable arrays for transmitting current into auricular and peri-auricular tissue; medical device for peripheral nerve and nerve field stimulation; medical system and apparatus consisting of a non-implantable modulating frequency generator and implantable needle arrays for transmitting current into auricular and peri-auricular tissue for use in pain management, namely, patient stimulators for auricular and peri-auricular peripheral nerve field neuromodulation therapy; medical apparatus, appliances and instruments for peripheral nerve field stimulation in cranial and peripheral nerves and occipital nerve branches, for pain control, headache control, control of phantom limb pain, stump pain, reflex sympathetic dystrophy (RSD), peripheral neuropathies and other types of sympathetically mediated pain | Registered | |||||
| US | IB-STIM | 5926831 | 03-Dec-2019 | 10 Int. medical apparatus, namely, electrical nerve stimulators; medical device, namely, a non- implantable modulating frequency generator, providing neuromodulation therapy to cranial and peripheral nerves; medical apparatus consisting of percutaneously implantable arrays for transmitting current into auricular and peri-auricular tissue; medical device for peripheral nerve and nerve field stimulation; medical device consisting of a non-implantable modulating frequency generator and percutaneously implantable needle arrays for transmitting current into auricular and peri-auricular tissue for use in pain management and FGID (functional gastrointestinal disorders), namely, patient stimulator for auricular and peri-auricular peripheral nerve field neuromodulation therapy; medical apparatus, for peripheral nerve field stimulation in cranial and peripheral nerves and occipital nerve branches, for pain control, FGID, irritable bowel, functional dyspepsia, functional abdominal pain, nausea, functional nausea, abdominal migraine, Crohn’s Disease, visceral hypersensitivity, chronic inflammatory bowel disease, changes in FGID co-morbidities, sleep disturbances, psychological disorders, including mood and anxiety, satiety and changes in autonomic nervous system and other types of sympathetically mediated pain | Registered |
| 17 |
| Country | Trademark | Reg. No. | Reg. Date | Class/Goods | Status | |||||
| US | IB-STIM and Design | 5926832 | 03-Dec-2019 | 10 Int. medical apparatus, namely, electrical nerve stimulators; medical device, namely, a non- implantable modulating frequency generator, providing neuromodulation therapy to cranial and peripheral nerves; medical apparatus consisting of percutaneously implantable arrays for transmitting current into auricular and peri-auricular tissue; medical device for peripheral nerve and nerve field stimulation; medical device consisting of a non-implantable modulating frequency generator and percutaneously implantable needle arrays for transmitting current into auricular and peri-auricular tissue for use in pain management and FGID (functional gastrointestinal disorders), namely, patient stimulator for auricular and peri-auricular peripheral nerve field neuromodulation therapy; Medical apparatus, for peripheral nerve field stimulation in cranial and peripheral nerves and occipital nerve branches, for pain control, FGID, irritable bowel, functional dyspepsia, functional abdominal pain, nausea, functional nausea, abdominal migraine, Crohn’s Disease, visceral hypersensitivity, chronic inflammatory bowel disease, changes in FGID co-morbidities, sleep disturbances, psychological disorders, including mood and anxiety, satiety and changes in autonomic nervous system and other types of sympathetically mediated pain | Registered | |||||
| US | IB-STIM AURICULAR STIMULATOR | 5978411 | 04-Feb-2020 | 10 Int. medical apparatus, namely, electrical nerve stimulators; Medical device, namely, a non- implantable modulating frequency generator, providing neuromodulation therapy to cranial and peripheral nerves; Medical apparatus consisting of percutaneously implantable arrays for transmitting current into auricular and peri-auricular tissue; Medical device for peripheral nerve and nerve field stimulation; Medical device consisting of a non-implantable modulating frequency generator and percutaneously implantable needle arrays for transmitting current into auricular and peri-auricular tissue for use in pain management and FGID (functional gastrointestinal disorders), namely, patient stimulator for auricular and peri-auricular peripheral nerve field neuromodulation therapy; Medical apparatus, for peripheral nerve field stimulation in cranial and peripheral nerves and occipital nerve branches, for pain control, FGID, irritable bowel, functional dyspepsia, functional abdominal pain, nausea, functional nausea, abdominal migraine, Crohn’s Disease, visceral hypersensitivity, chronic inflammatory bowel disease, changes in FGID co-morbidities, sleep disturbances, psychological disorders, including mood and anxiety, satiety and changes in autonomic nervous system and other types of sympathetically mediated pain | Registered | |||||
| US | IB-STIM AURICULAR STIMULATOR and Design | 5978412 | 04-Feb-2020 | 10 Int. medical apparatus, namely, electrical nerve stimulators; medical device, namely, a non- implantable modulating frequency generator, providing neuromodulation therapy to cranial and peripheral nerves; medical apparatus consisting of percutaneously implantable arrays for transmitting current into auricular and peri-auricular tissue; medical device for peripheral nerve and nerve field stimulation; medical device consisting of a non-implantable modulating frequency generator and percutaneously implantable needle arrays for transmitting current into auricular and peri-auricular tissue for use in pain management and FGID (functional gastrointestinal disorders), namely, patient stimulator for auricular and peri-auricular peripheral nerve field neuromodulation therapy; medical apparatus, for peripheral nerve field stimulation in cranial and peripheral nerves and occipital nerve branches, for pain control, FGID, irritable bowel, functional dyspepsia, functional abdominal pain, nausea, functional nausea, abdominal migraine, Crohn’s Disease, visceral hypersensitivity, chronic inflammatory bowel disease, changes in FGID co-morbidities, sleep disturbances, psychological disorders, including mood and anxiety, satiety and changes in autonomic nervous system and other types of sympathetically mediated pain | Registered |
| 18 |
| Country | Trademark | Reg. No. | Reg. Date | Class/Goods | Status | |||||
| US | NEURAXIS | 10 Int. Nerve stimulator apparatus; nerve stimulator apparatus for FGID, irritable bowel, functional dyspepsia, functional abdominal pain, nausea, functional nausea, abdominal migraine, Crohn’s Disease, visceral hypersensitivity, chronic inflammatory bowel disease, changes in FGID co- morbidities, sleep disturbances, psychological disorders, including mood, anxiety, and satiety, pain control, headache control, control of phantom limb pain, stump pain, reflex sympathetic dystrophy (RSD), peripheral neuropathies and other types of sympathetically mediated pain | Filed | |||||||
| US | NEURAXIS (STYLIZED) | 10 Int. Nerve stimulator apparatus; nerve stimulator apparatus for FGID, irritable bowel, functional dyspepsia, functional abdominal pain, nausea, functional nausea, abdominal migraine, Crohn’s Disease, visceral hypersensitivity, chronic inflammatory bowel disease, changes in FGID co- morbidities, sleep disturbances, psychological disorders, including mood, anxiety, and satiety, pain control, headache control, control of phantom limb pain, stump pain, reflex sympathetic dystrophy (RSD), peripheral neuropathies and other types of sympathetically mediated pain | Filed |
The Company has no unregistered trademarks.
Patents
The Company has eight (8) granted patents and nine (9) applied for patent applications in the United States and nine (9) applied for foreign patent applications.
| Country | Owner | Serial No. | Actual Filing Date | Patent No. | Issue Date | Anticipated Expiration Date | Title | Application Status | Licensing Status | |||||||||
| CA | Neuraxis, Inc. | 3096494 | 25-Apr-2019 | AURICULAR NERVE FIELD STIMULATION DEVICE | applied for | |||||||||||||
| CN | Neuraxis, Inc. | 201980027574.9 | 25-Apr-2019 | AURICULAR NERVE FIELD STIMULATION DEVICE | applied for | |||||||||||||
| EP | Neuraxis, Inc. | 19850021.7 | 25-Apr-2019 | AURICULAR NERVE FIELD STIMULATION DEVICE | applied for | |||||||||||||
| JP | Neuraxis, Inc. | 2021-509961 | 23-Oct-2020 | AURICULAR NERVE FIELD STIMULATION DEVICE | applied for | |||||||||||||
| KR | Neuraxis, Inc. | 10-2020-7034010 | 25-Apr-2019 | AURICULAR NERVE FIELD STIMULATION DEVICE | applied for | |||||||||||||
| US | Neuraxis, Inc. | 17/040766 | 23-Sep-2020 | 11369791 | 28-Jun-2022 | 21-Jun-2039 | AURICULAR NERVE FIELD STIMULATION DEVICE | granted | ||||||||||
| US | Neuraxis, Inc. | 17/715121 | 07-Apr-2022 | AURICULAR NERVE FIELD STIMULATION DEVICE | applied for | |||||||||||||
| US | Neuraxis, Inc. | 63/314028 | 25-Feb-2022 | AURICULAR NERVE FIELD STIMULATION DEVICE AND METHODS FOR USING THE SAME | applied for | |||||||||||||
| US | Neuraxis, Inc. | 63/315371 | 01-Mar-2022 | AURICULAR NERVE FIELD STIMULATION DEVICE AND METHODS FOR USING THE SAME | applied for | |||||||||||||
| US | Neuraxis, Inc. | 16/014169 | 21-Jun-2018 | 10322062 | 18-Jun-2019 | 14-May-2034 | AURICULAR PERIPHERAL NERVE FIELD STIMULATOR AND METHOD OF OPERATING SAME | granted | Out-licensed | |||||||||
| US | Neuraxis, Inc. | 16/408004 | 09-May-2019 | 11077019 | 03-Aug-2021 | 14-May-2034 | AURICULAR PERIPHERAL NERVE FIELD STIMULATOR AND METHOD OF OPERATING SAME | granted | Out-licensed |
| 19 |
| Country | Owner | Serial No. | Actual Filing Date | Patent No. | Issue Date | Anticipated Expiration Date | Title | Application Status | Licensing Status | |||||||||
| US | Neuraxis, Inc. | 17/363620 | 30-Jun-2021 | AURICULAR PERIPHERAL NERVE FIELD STIMULATOR AND METHOD OF OPERATING SAME | applied for | |||||||||||||
| US | Neuraxis, Inc. | 17/830411 | 02-Jun-2022 | DEVICE AND METHOD FOR ERADICATING PATHOGENS IN NASAL PASSAGES | applied for | |||||||||||||
| US | Neuraxis, Inc. | 17/589082 | 31-Jan-2022 | EXTERNAL AUDITORY CANAL PHOTOBIOMODULATION AND AUDIO THERAPY DEVICE | applied for | |||||||||||||
| US | Neuraxis, Inc. | 17/861646 | 11-Jul-2022 | EXTERNAL AUDITORY CANAL THERAPY DEVICE | applied for | |||||||||||||
| CA | Neuraxis, Inc. | 3143304 | 10-Dec-2021 | EXTERNAL AUDITORY CANAL PHOTOBIOMODULATION DEVICE | applied for | |||||||||||||
| CN | Neuraxis, Inc. | 202080060202.9 | 23-Jun-2020 | EXTERNAL AUDITORY CANAL PHOTOBIOMODULATION DEVICE | applied for | |||||||||||||
| EP | Neuraxis, Inc. | 20830917.9 | 10-Dec-2021 | EXTERNAL AUDITORY CANAL PHOTOBIOMODULATION DEVICE | applied for | |||||||||||||
| JP | Neuraxis, Inc. | 2021-576915 | 24-Dec-2021 | EXTERNAL AUDITORY CANAL PHOTOBIOMODULATION DEVICE | applied for | |||||||||||||
| US | Neuraxis, Inc. | 17/617364 | 08-Dec-2021 | EXTERNAL AUDITORY CANAL PHOTOBIOMODULATION DEVICE | applied for | |||||||||||||
| US | Neuraxis, Inc. | 15/488416 | 14-Apr-2017 | 10413719 | 17-Sep-2019 | 14-April-2037 | METHODS OF TREATING DISEASE USING AURICULAR PERIPHERAL NERVE FIELD STIMULATION | granted | Out-licensed | |||||||||
| US | Neuraxis, Inc. | 16/534159 | 07-Aug-2019 | 11331473 | 17-May-2022 | 14-April-2037 | METHODS OF TREATING DISEASE USING AURICULAR PERIPHERAL NERVE FIELD STIMULATION | granted | ||||||||||
| US | Neuraxis, Inc. | 15/595185 | 15-May-2017 | 9839577 | 12-Dec-2017 | 14-May-2034 | SYSTEM AND METHOD FOR AURICULAR PERIPHERAL NERVE FIELD STIMULATION | granted | Out-licensed | |||||||||
| US | Neuraxis, Inc. | 15/811278 | 13-Nov-2017 | 10010479 | 03-Jul-2018 | 14-May-2034 | SYSTEM AND METHOD FOR AURICULAR PERIPHERAL NERVE FIELD STIMULATION | granted | Out-licensed | |||||||||
| US | Neuraxis, Inc. | 14/277158 | 14-May-2014 | 9662269 | 30-May-2017 | 14-May-2034 | SYSTEMS AND METHODS FOR AURICULAR PERIPHERAL NERVE FIELD STIMULATION | granted | Out-licensed | |||||||||
| US | Neuraxis, Inc. | 17/725,761 | 21-Apr-2022 | AURICULAR NERVE FIELD STIMULATION DEVICE AND METHODS FOR USING THE SAME | Pending |
| 20 |
License Agreements
TKBMN Exclusive License Agreement
On May 7, 2020, the Company entered into an exclusive license agreement with TKBMN, LLC to obtain an exclusive license under certain patent rights (the “Patent Rights”) owned by TKBMN. Dr. Thomas Carrico, our Chief Regulatory Officer, is the manager of TKBMN. Brian Carrico, our Chief Executive Officer, and Matt Carrico, our National Sales Director, are members of TKBMN. TKBMN owns the Patent Rights set forth in the patents listed in the following table (the “TKBMN Patents”) by virtue of an assignment from Dr. Carrico, who is the sole inventor listed on the TKBMN Patents. TKBMN has assigned the auricular portion of the TKBMN Patent Rights to the Company.
Licensed TKBMN Patents
| Country | Owner | Application No. | Patent No. | Issue Date | Anticipated Expiration Date* |
Title | ||||||
| US | TKBMN, LLC | 15/981,082 | 10,792,500 | October 2, 2020 | October 18, 2037 | Systems and methods for electro-therapy treatment | ||||||
| US | TKBMN, LLC | 17/014,450 | 11,684,782 | June 7, 2023 | October 18, 2037 | Systems and methods for electro-therapy treatment |
* If all maintenance fees remain paid
Pursuant to the exclusive license agreement, TKBMN agreed to grant an exclusive, worldwide, non-transferable, royalty-free license under Patent Rights, which including three patents applications filed by TKBMN in connection with systems and methods for elector-therapy treatment, to the Company to develop, market, and sell licensed products, in the field of electro-therapy treatment by stimulation of cranial nerves, cranial nerve branches, auricular nerves, auricular nerve branches, auricular nerve bundles, and/or auricular anatomical structures in human patients (the “Field”), in consideration of a one-time license fee of $1.00. The Company has the right to grant sublicenses to the Patents Rights in the Field. The exclusive license agreement expires upon the expiration of the last to expire valid claim within the Patent Rights and may be terminated by the Company upon 60 days prior written notice. Upon expiration or termination of the exclusive license agreement, all rights in the Patent Rights will revert to TKBMN. There are no royalties or any other form of committed revenue to TKBMN or any of its members Under the agreement, the Company has agreed to cover fees and expenses associated with maintenance, prosecution, and additional associated/continuation patent filings for the TKBMN Patents.
Masimo License and Collaboration Agreement
On April 9, 2020, the Company entered into a license and collaboration agreement with Masimo. As consideration, in part, Masimo entered into a Series A Preferred Stock purchase agreement with the Company. Under the license and collaboration agreement, the Company grants an exclusive, fully paid-up, royalty-free license to specifically identified patents and trademarks in a limited Field of use. At all times, the Company remains the owner of all licensed intellectual property rights, and there is a possibility of joint ownership of collaboratively developed products and methods. The licensed patents are generally directed to a device and the treatment of opioid withdrawal symptoms. The licensed trademarks are generally directed to the NSS-2 Bridge mark. The license agreement includes a collaboration component to efficiently develop, obtain regulatory approval, and commercialize products for the limited field of use. The term of the agreement is in effect until the expiration or lapse of the last intellectual property rights. Masimo paid a one-time fee of $250,000. The license and collaboration agreement may not be terminated by the Company for any reason, and the sole remedy for any breach or default by Masimo shall be to seek monetary damages and equitable remedies. The license and collaboration agreement may be terminated by Masimo if there is material breach by the Company that remain uncured for thirty (30) days or without cause by providing thirty (30) days prior written notice. See“—Our Corporate History” for more information.
Implications of Being a Smaller Reporting Company
We are a “smaller reporting company” as defined in Rule 10(f)(1) of Regulation S-K. Smaller reporting companies may take advantage of certain reduced disclosure obligations, including, among other things, providing only two years of audited financial statements. We will remain a smaller reporting company until the last day of the fiscal year in which (1) the market value of our shares held by non-affiliates equals or exceeds $250 million as of the prior June 30th, or (2) our annual revenues equaled or exceeded $100 million during such completed fiscal year and the market value of our shares held by non-affiliates equals or exceeds $700 million as of the prior June 30th. Such reduced disclosure and corporate governance obligations may make it more challenging for investors to analyze our results of operations and financial prospects.
For additional information, see “Risk Factors – Because the Company is a ‘smaller reporting company,’ we may take advantage of certain scaled disclosures available to us, resulting in holders of our securities receiving less Company information than they would receive from a public company that is not a smaller reporting company” and “As a smaller reporting company,” we may at some time in the future choose to exempt our Company from certain corporate governance requirements that could have an adverse effect on our public stockholders.”
| 21 |
Implications of Being an Emerging Growth Company
We are an “emerging growth company” as defined in the JOBS Act. We will remain an emerging growth company until the earlier of (1) December 31, 2028, (2) the last day of the fiscal year in which we have total annual gross revenue of at least $1.235 billion, (3) the last day of the fiscal year in which we are deemed to be a “large accelerated filer” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended, or the Exchange Act, which would occur on the date on which we have issued more than $1.0 billion in non-convertible debt securities during the prior three-year period. An emerging growth company may take advantage of specified reduced reporting requirements and is relieved of certain other significant requirements that are otherwise generally applicable to public companies. As an emerging growth company, we may:
| ● | present only two years of audited financial statements, plus unaudited condensed financial statements for any interim period, and related management’s discussion and analysis of financial condition and results of operations in this prospectus; | |
| ● | avail ourselves of the exemption from the requirement to obtain an attestation and report from our auditors on the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act of 2002; | |
| ● | provide reduced disclosure about our executive compensation arrangements; and | |
| ● | not require stockholder non-binding advisory votes on executive compensation or golden parachute arrangements. |
In addition, under the JOBS Act, an emerging growth company can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. We have elected not to take advantage of the extended transition period for complying with new or revised accounting standards provided to emerging growth companies under the JOBS Act.
Government Regulation
Our products and our operations are subject to extensive regulation by the U.S. Food and Drug Administration, or FDA, and other federal, state, and local authorities in the United States, as well as comparable authorities in foreign jurisdictions. Our products are subject to regulation as medical devices in the United States under the Federal Food, Drug, and Cosmetic Act, or FDCA, and its implementing regulations.
United States Regulation
The FDA regulates, among other things, the development, design, non-clinical and clinical testing, manufacturing, safety, effectiveness, labeling, packaging, storage, installation, servicing, recordkeeping, premarket clearance or approval, adverse event reporting, advertising, promotion, marketing and distribution, and import and export and post-marketing surveillance of medical devices in the United States to ensure that medical devices distributed domestically are safe and effective for their intended uses and otherwise meet the requirements of the FDCA.
| 22 |
FDA Premarket Clearance and Approval Requirements
Unless an exemption applies, each new or significantly modified medical device commercially distributed in the United States requires FDA clearance of a 510(k) premarket notification. The 510(k) clearance can be resource intensive, expensive, and lengthy.
Under the FDCA, medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk associated with each medical device and the extent of manufacturer and regulatory control needed to ensure its safety and effectiveness. Class I devices are those for which safety and effectiveness can be assured by adherence to the FDA’s general controls for medical devices, which include compliance with the applicable portions of FDA’s current good manufacturing practices for devices, as reflected in the Quality System Regulation, or QSR, establishment registration and device listing, reporting of adverse medical events, and truthful and non-misleading labeling, advertising, and promotional materials. Some Class I devices, also called Class I reserved devices, also require premarket clearance by the FDA through the 510(k) premarket notification process described below. Most Class I devices are exempt from the premarket notification requirements.
Class II devices are subject to the FDA’s general controls, and any other special controls deemed necessary by the FDA to ensure the safety and effectiveness of the device. These special controls can include performance standards, special labeling requirements, post-market surveillance, patient registries and FDA guidance documents.
Most Class II devices are required to submit to the FDA a premarket notification under Section 510(k) of the FDCA requesting permission to commercially distribute the device. The FDA’s permission to commercially distribute a device subject to a 510(k) premarket notification is generally known as 510(k) clearance.
If a new medical device does not qualify for the 510(k) premarket notification process because no predicate device to which it is substantially equivalent can be identified, the device is automatically classified into Class III. The Food and Drug Administration Modernization Act of 1997 established a new route to market for low to moderate risk medical devices that are automatically placed into Class III due to the absence of a predicate device, called the “Request for Evaluation of Automatic Class III Designation,” or the de novo classification process. This process allows a manufacturer whose novel device is automatically classified into Class III to request down-classification of its medical device into Class I or Class II on the basis that the device presents low or moderate risk. If the manufacturer seeks reclassification into Class II, the manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and effectiveness of the medical device. The FDA may reject the reclassification petition if it identifies a legally marketed predicate device that would be appropriate for a 510(k) or that general controls would be inadequate to control the risks and special controls cannot be developed.
Obtaining FDA marketing authorization, de novo down-classification, or approval for medical devices is expensive and uncertain, and may take several years, and generally requires significant scientific and clinical data.
Some pre-amendment devices are unclassified, but are subject to FDA’s premarket notification and clearance process in order to be commercially distributed.
Investigational Device Process
Clinical trials are sometimes required to support a 510(k) submission. In the United States, absent certain limited exceptions, human clinical trials intended to support medical device clearance or approval or to determine safety and effectiveness of a device for an investigational use must be conducted in accordance with the FDA’s investigational device exemption, or IDE, regulations which govern investigational device labeling, prohibit promotion of the investigational device, and specify an array of recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. If the device presents a “significant risk,” to human health, as defined by the FDA, the FDA requires the device sponsor to submit an IDE application to the FDA, which must become effective prior to commencing human clinical trials. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE application must be approved in advance by the FDA for a specified number of subjects. Generally, clinical trials for a significant risk device may begin once the IDE application is approved by the FDA and the study protocol and informed consent are approved by appropriate institutional review boards at the clinical trial sites. There can be no assurance that submission of an IDE will result in the ability to commence clinical trials, and although the FDA’s approval of an IDE allows clinical testing to go forward for a specified number of subjects, it does not bind the FDA to accept the results of the trial as sufficient to prove the product’s safety and effectiveness, even if the trial meets its intended success criteria.
| 23 |
If the device under evaluation does not present a significant risk to human health, then the device sponsor is not required to submit an IDE application to the FDA before initiating human clinical trials, but must still comply with abbreviated IDE requirements when conducting such trials. A significant risk device is one that presents a potential for serious risk to the health, safety or welfare of a patient and either is implanted, used in supporting or sustaining human life, substantially important in diagnosing, curing, mitigating or treating disease or otherwise preventing impairment of human health, or otherwise presents a potential for serious risk to a subject. An IDE application must be supported by appropriate data, such as animal and laboratory test results, showing that it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE will automatically become effective thirty (30) days after receipt by the FDA unless the FDA notifies the company that the investigation may not begin. If the FDA determines that there are deficiencies or other concerns with an IDE for which it requires modification, the FDA may permit a clinical trial to proceed under a conditional approval.
Regardless of the degree of risk presented by the medical device, clinical studies must be approved by, and conducted under the oversight of, an Institutional Review Board, or IRB, for each clinical site. The IRB is responsible for the initial and continuing review of the IDE, and may pose additional requirements for the conduct of the study. If an IDE application is approved by the FDA and one or more IRBs, human clinical trials may begin at a specific number of investigational sites with a specific number of patients, as approved by the FDA. If the device presents a non-significant risk to the patient, a sponsor may begin the clinical trial after obtaining approval for the trial by one or more IRBs without separate approval from the FDA, but must still follow abbreviated IDE requirements, such as monitoring the investigation, ensuring that the investigators obtain informed consent, and labeling and record-keeping requirements. Acceptance of an IDE application for review does not guarantee that the FDA will allow the IDE to become effective and, if it does become effective, the FDA may or may not determine that the data derived from the trials support the safety and effectiveness of the device or warrant the continuation of clinical trials. An IDE supplement must be submitted to, and approved by, the FDA before a sponsor or investigator may make a change to the investigational plan that may affect its scientific soundness, study plan or the rights, safety or welfare of human subjects.
During a study, the sponsor is required to comply with the applicable FDA requirements, including, for example, trial monitoring, selecting clinical investigators and providing them with the investigational plan, ensuring IRB review, adverse event reporting, record keeping and prohibitions on the promotion of investigational devices or on making safety or effectiveness claims for them. The clinical investigators in the clinical study are also subject to FDA’s regulations and must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of the investigational device, and comply with all reporting and recordkeeping requirements. Additionally, after a trial begins, we, the FDA or the IRB could suspend or terminate a clinical trial at any time for various reasons, including the following:
| ● | The FDA or other regulatory authorities do not approve a clinical trial protocol or a clinical trial, or place a clinical trial on hold; | |
| ● | Patients do not enroll in clinical trials at the rate expected; | |
| ● | Patients do not comply with trial protocols; | |
| ● | Patient follow-up is not at the rate expected; | |
| ● | Patients experience serious adverse events; | |
| ● | Patients die during a clinical trial, even though their death may not be related to the products that are part of the trial; | |
| ● | Device malfunctions occur with unexpected frequency or potential adverse consequences; | |
| ● | Side effects or device malfunctions of similar products already in the market that change the FDA’s view toward approval of result in the imposition of new requirements or testing; |
| 24 |
| ● | Institutional review boards and third-party clinical investigators may delay or reject the trial protocol; | |
| ● | Third-party clinical investigators decline to participate in a trial or do not perform a trial on the anticipated schedule or consistent with the clinical trial protocol, investigator agreement, investigational plan, good clinical practices, the IDE regulations, or other FDA or IRB requirements; | |
| ● | Third-party investigators are disqualified by the FDA; | |
| ● | We or third-party organizations do not perform data collection, monitoring and analysis in a timely or accurate manner or consistent with the clinical trial protocol or investigational or statistical plans, or otherwise fail to comply with the IDE regulations governing responsibilities, records, and reports of sponsors of clinical investigations; | |
| ● | Third-party clinical investigators have significant financial interests related to us or our study such that the FDA deems the study results unreliable, or the company or investigators fail to disclose such interests; | |
| ● | Regulatory inspections of our clinical trials or manufacturing facilities, which may, among other things, require us to undertake corrective action or suspend or terminate our clinical trials; | |
| ● | Changes in government regulations or administrative actions; | |
| ● | The interim or final results of the clinical trial are inconclusive or unfavorable as to safety or effectiveness; or | |
| ● | The FDA concludes that our trial design is unreliable or inadequate to demonstrate safety and effectiveness. |
510(k) Clearance Process
Under the 510(k) process, the manufacturer must submit to the FDA a premarket notification submission demonstrating that the proposed device is “substantially equivalent,” as defined in the FDCA, to a legally marketed predicate device.
A predicate device is a legally marketed device that is not subject to premarket approval, i.e., a device that was legally marketed prior to May 28, 1976 (pre-amendments device) and for which a PMA is not required, a device that has been reclassified from Class III to Class II or I, or a device that was found substantially equivalent through the 510(k) process. A device is considered to be substantially equivalent if, with respect to the predicate device, it has the same intended use and has either (i) the same technological characteristics; or (ii) different technological characteristics, but the information provided in the 510(k) submission demonstrates that the device does not raise different questions of safety or effectiveness than the predicate device.
Before the FDA will accept a 510(k) premarket notification for substantive review, the FDA will first assess whether the submission satisfies a minimum threshold of acceptability. If the FDA determines that the 510(k) submission lacks necessary information for substantive review, the FDA will issue a “Refuse to Accept” letter which generally outlines the information the FDA believes is necessary to permit a substantive review and to reach a determination regarding substantial equivalence. An applicant must submit the requested information before the FDA will proceed with additional review of the submission. If a 510(k) submission is accepted for substantive review, the Medical Device User Fee Amendments sets a performance goal of 90 days for FDA review of a 510(k) submission, but the review time can be delayed if FDA raises questions or requests additional information during the review process. As a practical matter, clearance often takes longer, and clearance is never assured. Thus, as a practical matter, clearance often takes longer than 90 days. Although many 510(k) premarket notifications are cleared without clinical data, the FDA may require further information, including clinical data, to make a determination regarding substantial equivalence, which may significantly prolong the review process. If the FDA agrees that the device is substantially equivalent, it will grant clearance to commercially market the device.
| 25 |
If the FDA determines that the device is substantially equivalent to a predicate device, it will grant 510(k) clearance to commercially market the device. If the FDA determines that the device is “not substantially equivalent” to a previously cleared device, the device is automatically designated as a Class III device. The device sponsor must then fulfill more rigorous requirements of the PMA approval process, or can request a risk-based classification determination for the device in accordance with the “de novo” process, which is a route to market for certain novel medical devices that are low to moderate risk and are not substantially equivalent to a predicate device.
Medical devices can only be marketed for the indications for use for which they are cleared or approved. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change or modification in its intended use, will require a new 510(k) clearance or, depending on the modification, PMA approval or de novo reclassification. The FDA requires each manufacturer to determine whether the proposed change requires submission of a 510(k), de novo request or a PMA in the first instance, but the FDA may review this determination to evaluate the regulatory status of the modified product at any time and may require the manufacturer to cease marketing and/or request the recall of the modified device until 510(k) marketing clearance or PMA approval is obtained or a de novo request is granted. Also, in these circumstances, the manufacturer may be subject to significant regulatory fines or penalties.
Over the last several years, the FDA has proposed reforms to its 510(k) clearance process, and such proposals could include increased requirements for clinical data and a longer review period, or could make it more difficult for manufacturers to utilize the 510(k) clearance process for their products. For example, in November 2018, FDA officials announced steps that the FDA intended to take to modernize the premarket notification pathway under Section 510(k) of the FDCA. Among other things, the FDA announced that it planned to develop proposals to drive manufacturers utilizing the 510(k) pathway toward the use of newer predicates. These proposals included plans to potentially sunset certain older devices that were used as predicates under the 510(k) clearance pathway, and to potentially publish a list of devices that have been cleared on the basis of demonstrated substantial equivalence to predicate devices that are more than 10 years old. These proposals have not yet been finalized or adopted, although the FDA may work with Congress to implement such proposals through legislation.
More recently, in September 2019, the FDA issued revised final guidance describing an optional “safety and performance based” premarket review pathway for manufacturers of “certain, well-understood device types” to demonstrate substantial equivalence under the 510(k) clearance pathway by showing that such device meets objective safety and performance criteria established by the FDA, thereby obviating the need for manufacturers to compare the safety and performance of their medical devices to specific predicate devices in the clearance process. The FDA has developed and maintains a list device types appropriate for the “safety and performance based” pathway and continues to develop product-specific guidance documents that identify the performance criteria for each such device type, as well as recommended testing methods, where feasible.
PMA Approval Process
Class III devices require PMA approval before they can be marketed, although some pre-amendment Class III devices for which FDA has not yet required a PMA are cleared through the 510(k) process. The PMA process is more demanding than the 510(k) premarket notification process. In a PMA, the manufacturer must demonstrate that the device is safe and effective for its intended use, and the PMA must be supported by extensive data, including data from preclinical studies and human clinical trials. The PMA must also contain a full description of the device and its components, a full description of the methods, facilities, and controls used for manufacturing, and proposed labeling. Following receipt of a PMA, the FDA conducts an administrative review to determine whether the application is sufficiently complete to permit a substantive review. If it is not, the agency will refuse to file the PMA. If FDA accepts the application for substantive review, it has 180 days under the FDCA to complete its review of a filed PMA application, although in practice, the FDA’s review often takes significantly longer, and can take up to several years. During this review period, the FDA may request additional information or clarification of information already provided, and the FDA may issue a major deficiency letter to the applicant, requesting the applicant’s response to deficiencies communicated by the FDA. The FDA considers a PMA or PMA supplement to have been voluntarily withdrawn if an applicant fails to respond to an FDA request for information (e.g., major deficiency letter) within a total of 360 days. Before approving or denying a PMA application, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to whether the FDA should approve the submission, approve it with specific conditions, or not approve it. The FDA may or may not accept the panel’s recommendation. Prior to approval of a PMA, the FDA may conduct inspections of the clinical trial data and clinical trial sites, as well as conduct inspections of the applicant or its third-party manufacturers’ or suppliers’ manufacturing facility or facilities to, among other things, ensure compliance with the QSR. PMA applications are also subject to the payment of user fees, which for fiscal year 2021 includes a standard application fee of $365,657.
| 26 |
| ● | Overall, the FDA review of a PMA application generally takes between one and three years, but may take significantly longer. The FDA can delay, limit or deny approval of a PMA application for many reasons, including: | |
| ● | The device may not be shown safe or effective to the FDA’s satisfaction; | |
| ● | The data from pre-clinical studies and/or clinical trials may be found unreliable or insufficient to support approval; | |
| ● | The manufacturing process or facilities may not meet applicable requirements; and | |
| ● | Changes in FDA approval policies or adoption of new regulations may require additional data. |
If the FDA evaluation of a PMA is favorable, the FDA will issue either an approval letter, or an approvable letter, the latter of which usually contains a number of conditions that must be met in order to secure final approval of the PMA. When and if those conditions have been fulfilled to the satisfaction of the FDA, the agency will issue a PMA approval letter authorizing commercial marketing of the device, subject to the conditions of approval and the limitations established in the approval letter. The FDA may approve a PMA with post-approval conditions intended to ensure the safety and effectiveness of the device, including, among other things, restrictions on labeling, promotion, sale and distribution, and collection of long-term follow-up data from patients in the clinical study that supported PMA approval or requirements to conduct additional clinical studies post-approval. The FDA may condition PMA approval on some form of post-market surveillance when deemed necessary to protect the public health or to provide additional safety and effectiveness data for the device in a larger population or for a longer period of use. In such cases, the manufacturer might be required to follow certain patient groups for a number of years and to make periodic reports to the FDA on the clinical status of those patients. Failure to comply with the conditions of approval can result in material adverse enforcement action, including withdrawal of the approval. If the FDA’s evaluation of a PMA application or manufacturing facilities is not favorable, the FDA will deny approval of the PMA or issue a not approvable letter. The FDA also may determine that additional tests or clinical trials are necessary, in which case the PMA approval may be delayed for several months or years while the trials are conducted and data is submitted in an amendment to the PMA, or the PMA is withdrawn and resubmitted when the data are available. The PMA process can be expensive, uncertain and lengthy and a number of devices for which the FDA approval has been sought by other companies have never been approved by the FDA for marketing.
Certain changes to an approved medical device, such as changes in manufacturing facilities, methods, quality control procedures, sterilization (if applicable), packaging, expiration date, labeling, device specifications, materials, or design of a device, or other changes which affect the safety or effectiveness of the device that has been approved through the PMA process require submission of a new PMA or PMA supplement. PMA supplements often require submission of the same type of information as a PMA, except that the supplement is limited to information needed to support any changes from the device covered by the original, approved PMA and may not require as extensive clinical data or the convening of an advisory panel, depending on the nature of the proposed change. Certain other changes to an approved device require the submission of a new PMA, such as when the design change causes a different intended use, mode of operation, and technical basis of operation, or when the design change is so significant that a new generation of the device will be developed, and the data that were submitted with the original PMA are not applicable for the change in demonstrating a reasonable assurance of safety and effectiveness.
| 27 |
Ongoing Regulation by the FDA
Even after the FDA permits a device to be marketed, numerous and pervasive regulatory requirements continue to apply. These include:
| ● | Establishment registration and device listing with the FDA; | |
| ● | QSR requirements, which require manufacturers, including third-party manufacturers, to follow stringent design, testing, control, supplier/contractor selection, compliant handling, documentation and other quality assurance procedures during all aspects of the design and manufacturing process; | |
| ● | Labeling regulations, advertising and promotion requirements, restrictions on sale, distribution or sale of a device, each including the FDA prohibition against the promotion of products for any uses other than those authorized by the FDA, which are commonly known as “off-label” uses; | |
| ● | The Medical Device Reporting, or MDR, regulations, which require that a manufacturer report to the FDA if a device it markets may have caused or contributed to a death or serious injury, or has malfunctioned and the device or a similar device that it markets would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur; | |
| ● | Medical device correction and removal reporting regulations, which require that manufacturers report to the FDA field corrections or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the FDCA that may present a risk to health; | |
| ● | Recall requirements, including a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death; |
| ● | An order of repair, replacement, or refund; | |
| ● | Device tracking requirements; and | |
| ● | Post-market study and surveillance requirements. |
After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, will require a new 510(k). The FDA requires each manufacturer to make this determination initially, but the FDA can review any such decision and can disagree with a manufacturer’s determination. If the FDA disagrees with a manufacturer’s determination not to seek a new 510(k) clearance, the FDA may retroactively require it to seek 510(k) clearance. The FDA could also require the manufacturer to cease marketing and distribution and/or recall the modified device until 510(k) clearance is obtained. Also, in these circumstances, the manufacturer may be subject to significant regulatory fines and penalties.
FDA regulations require us to register as a medical device manufacturer with the FDA. Additionally, some states also require medical device manufacturers and/or distributors doing business within the state to register with the state or apply for a state license, which could subject our facility to state inspection as well as FDA inspection on a routine basis for compliance with the QSR and any applicable state requirements. These regulations require that we manufacture our products and maintain related documentation in a prescribed manner with respect to manufacturing, testing and control activities.
Manufacturing processes for medical devices are required to comply with the applicable portions of the QSR, which cover the methods and the facilities and controls for the design, manufacture, testing, production, processes, controls, quality assurance, labeling, packaging, distribution, installation and servicing of finished devices intended for human use. The QSR also requires, among other things, maintenance of a device master file, device history file, and complaint files. As a manufacturer, we are subject to periodic scheduled or unscheduled inspections by the FDA. Failure to maintain compliance with the QSR requirements could result in the shutdown of, or restrictions on, manufacturing operations and the recall or seizure of marketed products, which would have a material adverse effect on our business. The discovery of previously unknown problems with any of our products, including unanticipated adverse events or adverse events of increasing severity or frequency, whether resulting from the use of the device within the scope of its clearance or off-label by a physician in the practice of medicine, could result in restrictions on the device, including the removal of the product from the market or voluntary or mandatory device recalls.
| 28 |
The FDA has broad regulatory compliance and enforcement powers. If the FDA determines that a manufacturer has failed to comply with applicable regulatory requirements, it can take a variety of compliance or enforcement actions, which may result in any of the following sanctions:
| ● | Warning letters, untitled letters, fines, injunctions, consent decrees and civil penalties; | |
| ● | Recalls, withdrawals, or administrative detention or seizure of our products; | |
| ● | Operating restrictions or partial suspension or total shutdown of production; | |
| ● | Refusing or delays in processing, clearing, or approving submissions or applications for new products or modifications to existing products; | |
| ● | Suspension or withdrawal of 510(k) clearances or PMA approvals that have already been granted; | |
| ● | FDA refusal to issue certification to foreign governments needed to export our products for sale in other countries; or | |
| ● | Criminal prosecution. |
Our facilities, records and manufacturing processes are subject to periodic unscheduled inspections by the FDA. Failure to comply with the applicable United States medical device regulatory requirements could result in, among other things, warning letters, untitled letters, fines, injunctions, consent decrees, civil penalties, unanticipated expenditures, repairs, replacements, refunds, recalls or seizures of products, operating restrictions, total or partial suspension of production, the FDA’s refusal to issue certificates to foreign governments needed to export products for sale in other countries, the FDA’s refusal to grant future premarket clearances or approvals, withdrawals or suspensions of current product clearances or approvals and criminal prosecution.
Regulation of Medical Devices in the European Union
The European Union, or EU, has adopted specific directives regulating the design, manufacture, clinical investigations, conformity assessment, labeling and adverse event reporting for medical devices. EU directives must be implemented into the national laws of the EU member states and national laws may vary from one member state to another.
In the EU, there is currently no premarket government review of medical devices. However, the EU requires that all medical devices placed on the market in the EU must meet the relevant essential requirements laid down in the Council Directive 93/42/EEC, or the Medical Devices Directive, and the Council Directive 90/385/EEC, or the Active Implantable Medical Devices Directive. The most fundamental essential requirement is that a medical device must be designed and manufactured in such a way that it will not compromise the clinical condition or safety of patients, or the safety and health of users and others. In addition, the device must achieve the performances intended by the manufacturer and be designed, manufactured, and packaged in a suitable manner. The European Commission has adopted various standards applicable to medical devices. These include standards governing common requirements, such as sterilization and safety of medical electrical equipment and product standards for certain types of medical devices. There are also harmonized standards relating to design and manufacture. While not mandatory, compliance with these standards is viewed as the easiest way to satisfy the essential requirements as a practical matter. Compliance with a standard developed to implement an essential requirement also creates a rebuttable presumption that the device satisfies that essential requirement.
To demonstrate compliance with the essential requirements laid down in Annex I to the Medical Devices Directive, medical device manufacturers must undergo a conformity assessment procedure, which varies according to the type of medical device and its (risk) classification. Conformity assessment procedures require an assessment of available clinical evidence, literature data for the product, and post-market experience in respect of similar products already marketed. Except for low-risk medical devices (Class I non-sterile, non-measuring devices), where the manufacturer can self-declare the conformity of its products with the essential requirements (except for any parts which relate to sterility or metrology), a conformity assessment procedure requires the intervention of a Notified Body. Notified Bodies are independent organizations designated by EU countries to assess the conformity of devices before being placed on the market. A Notified Body would typically audit and examine a product’s technical dossiers and the manufacturers’ quality system (which must, in particular, comply with ISO 13485:2016 related to Medical Devices Quality Management Systems). If satisfied that the relevant product conforms to the relevant essential requirements, the Notified Body issues a certificate of conformity, which the manufacturer uses as a basis for its own declaration of conformity. The manufacturer may then apply the CE Mark to the device, which allows the device to be placed on the market throughout the EU.
| 29 |
Notified Body certificates of conformity are valid for a fixed duration (which shall not exceed five years). Throughout the term of the certificate, the manufacturer will be subject to periodic surveillance audits to verify continued compliance with the applicable requirements. In particular, there will be a new audit by the Notified Body before it will renew the relevant certificate(s).
As a general rule, demonstration of conformity of medical devices and their manufacturers with the essential requirements must be based, among other things, on the evaluation of clinical data supporting the safety and performance of the products during normal conditions of use. Specifically, a manufacturer must demonstrate that the device achieves its intended performance during normal conditions of use, that the known and foreseeable risks, and any adverse events, are minimized and acceptable when weighed against the benefits of its intended performance, and that any claims made about the performance and safety of the device are supported by suitable evidence. All manufacturers placing medical devices into the market in the EU must comply with the EU medical device vigilance system. Under this system, incidents must be reported to the relevant authorities of the EU member states, and manufacturers are required to take Field Safety Corrective Actions, or FSCAs, to reduce a risk of death or serious deterioration in the state of health associated with the use of a medical device that is already placed on the market. An incident is defined as any malfunction or deterioration in the characteristics and/or performance of a device, as well as any inadequacy in the labeling or the instructions for use which, directly or indirectly, might lead to or might have led to the death of a patient or user or of other persons or to a serious deterioration in their state of health. An FSCA may include the recall, modification, exchange, destruction or retrofitting of the device. FSCAs must be communicated by the manufacturer or its legal representative to its customers and/or to the end users of the device through Field Safety Notices.
The advertising and promotion of medical devices is subject to some general principles set forth by EU directives. According to the Medical Devices Directive, only devices that are CE-marked may be marketed and advertised in the EU in accordance with their intended purpose. Directive 2006/114/EC concerning misleading and comparative advertising and Directive 2005/29/EC on unfair commercial practices, while not specific to the advertising of medical devices, also apply to the advertising thereof and contain general rules, for example requiring that advertisements are evidenced, balanced and not misleading. Specific requirements are defined at national level. EU member states laws related to the advertising and promotion of medical devices, which vary between jurisdictions, may limit or restrict the advertising and promotion of products to the general public and may impose limitations on promotional activities with healthcare professionals.
Many EU member states have adopted specific anti-gift statutes that further limit commercial practices for medical devices, in particular vis-à -vis healthcare professionals and organizations. Additionally, there has been a recent trend of increased regulation of payments and transfers of value provided to healthcare professionals or entities. In addition, many EU member states have adopted national “Sunshine Acts” which impose reporting and transparency requirements (often on an annual basis), similar to the requirements in the United States, on medical device manufacturers. Certain countries also mandate implementation of commercial compliance programs.
On May 25, 2017, Regulation 2017/745, or the EU Medical Devices Regulation, entered into force, which repeals and replaces the Medical Devices Directive and the Active Implantable Medical Devices Directive. Unlike directives, which must be implemented into the national laws of the EU member states, regulations are directly applicable, without the need for adoption of EU member state laws implementing them, in all EU member states and are intended to eliminate current differences in the regulation of medical devices among EU member states. The Medical Devices Regulation, among other things, is intended to establish a uniform, transparent, predictable and sustainable regulatory framework across the EU for medical devices and ensure a high level of safety and health while supporting innovation.
The Medical Devices Regulation was originally intended to become applicable three years after publication, but in April 2020 the transition period was extended by the European Parliament and the Council of the EU by an additional year – until May 26, 2021. Devices lawfully placed on the market pursuant to the Medical Devices Directive and the Active Implantable Medical Devices Directive prior to May 26, 2021 may generally continue to be made available on the market or put into service until May 26, 2025. Once applicable, the new regulations will among other things:
| ● | Strengthen the rules on placing devices on the market and reinforce surveillance once they are available; |
| 30 |
| ● | Establish explicit provisions on manufacturers’ responsibilities for the follow-up of the quality, performance and safety of devices placed on the market; | |
| ● | Improve the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; | |
| ● | Set up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the European Union, or EU; and | |
| ● | Strengthen the rules for the assessment of certain high-risk devices, which may have to undergo an additional check by experts before they are placed on the market. |
The aforementioned EU rules are generally applicable in the European Economic Area, or EEA, which consists of the 27 EU member states plus Norway, Liechtenstein and Iceland. Other countries, such as Switzerland, have entered into Mutual Recognition Agreements and allow the marketing of medical devices that meet EU requirements.
The EU-UK Trade and Cooperation Agreement, or TCA, came into effect on January 1, 2021. The TCA does not specifically refer to medical devices. However, as a result of Brexit, the Medical Devices Regulation will not be implemented in the UK, and previous legislation that mirrored the Medical Devices Regulation in the UK law has been revoked. The regulatory regime for medical devices in the UK will continue to be based on the requirements derived from current EU legislation, and the UK may choose to retain regulatory flexibility or align with the Medical Devices Regulation going forward. CE markings will continue to be recognized in the UK, and certificates issued by EU recognized Notified Bodies will be valid in the UK, until June 30, 2023. For medical devices placed on the UK market after this period, the UK Conformity Assessment, or UKCA, marking will be mandatory. In contrast, UKCA marking and certificates issued by UK Notified Bodies will not be recognized on the EU market. The TCA does provide for cooperation and exchange of information in the area of product safety and compliance, including market surveillance, enforcement activities and measures, standardization related activities, exchanges of officials, and coordinated product recalls (or other similar actions). For medical devices that are locally manufactured but use components from other countries, the “rules of origin” criteria will need to be reviewed. Depending on which countries products will ultimately be sold in, manufacturers may start seeking alternative sources for components if this would allow them to benefit from no tariffs. The rules for placing medical devices on the Northern Ireland market will differ from those in the UK.
Healthcare Fraud and Abuse Laws
In the United States, we are subject to a number of federal and state healthcare regulatory laws that restrict business practices in the healthcare industry. These laws include, but are not limited to, federal and state anti-kickback, false claims, transparency and other healthcare fraud and abuse laws.
The U.S. federal Anti-Kickback Statute prohibits, among other things, any person or entity from knowingly and willfully offering, paying, soliciting, receiving or providing any remuneration, directly or indirectly, overtly or covertly, to induce or in return for purchasing, leasing, ordering, or arranging for or recommending the purchase, lease, or order of any good, facility, item or service reimbursable, in whole or in part, under Medicare, Medicaid or other federal healthcare programs. The term “remuneration” has been broadly interpreted to include anything of value, including cash, improper discounts, and free or reduced-price items and services. Among other things, the Anti-Kickback Statute has been interpreted to apply to arrangements between medical device manufacturers on the one hand and prescribers and purchasers on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. The government can exercise enforcement discretion in taking action against unprotected activities. Further, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation. The majority of states also have anti-kickback laws, which establish similar prohibitions, and in some cases may apply to items or services reimbursed by any third-party payor, including commercial insurers and self-pay patients.
The federal false claims, including the civil False Claims Act, prohibit, among other things, any person or entity from knowingly presenting, or causing to be presented, a false, fictitious or fraudulent claim for payment to, or approval by, the federal government, knowingly making, using, or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government, or knowingly making a false statement to avoid, decrease or conceal an obligation to pay money to the U.S. federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. Actions under the civil False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. Moreover, a claim including items or services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. In addition, various states have enacted false claim laws analogous to the federal False Claims Act, although many of these state laws apply where a claim is submitted to any third-party payor and not merely a federal healthcare program.
| 31 |
The federal Health Insurance Portability and Accountability Act of 1996 created additional federal criminal statutes that prohibit, among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Similar to the U.S. federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
The federal Physician Payments Sunshine Act requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to report annually to CMS, information related to payments or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), and teaching hospitals, and applicable manufacturers and applicable group purchasing organizations to report annually to CMS ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, such obligations will include payments and other transfers of value provided in the previous year to additional healthcare professionals, including physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, anesthesiologist assistants and certified nurse midwives.
Violations of fraud and abuse laws, including federal and state anti-kickback and false claims laws, may be punishable by criminal and civil sanctions, including fines and civil monetary penalties, the possibility of exclusion from federal healthcare programs (including Medicare and Medicaid), disgorgement and corporate integrity agreements, which impose, among other things, rigorous operational and monitoring requirements on companies. Similar sanctions and penalties, as well as imprisonment, also can be imposed upon executive officers and employees of such companies.
Coverage and Reimbursement
In the United States, our currently cleared products are not separately reimbursed by any third-party payors and if covered, are paid for as part of the procedure in which the product is used. Outside of the United States, there are many reimbursement programs through private payors as well as government programs. In some countries, government reimbursement is the predominant program available to patients and hospitals. Our commercial success depends in part on the extent to which governmental authorities, private health insurers and other third-party payors provide coverage for and establish adequate reimbursement levels for the procedures in which our products are used. Failure by physicians, hospitals, ambulatory surgery centers and other users of our products to obtain coverage and adequate reimbursement from third-party payors for procedures in which our products are used, or adverse changes in government and private third-party payors’ coverage and reimbursement policies, may adversely impact demand for our products.
Based on our experience to date, third-party payors generally reimburse for the procedures in which our products are used if medical necessity is met and a prior approval is completed with a favorable response. Some payors are moving toward a managed care system and control their healthcare costs by establishing coverage policies that categorically restrict coverage of certain procedures, or by limiting authorization for procedures, including elective procedures using our devices. No uniform policy of coverage and reimbursement among payors in the United States exists and coverage and reimbursement for procedures can differ significantly from payor to payor. Third-party payors are increasingly auditing and challenging the prices charged for medical products and services with concern for upcoding, miscoding, using inappropriate modifiers, or billing for inappropriate care settings. Some third-party payors must approve coverage for new or innovative devices or procedures before they will reimburse healthcare providers who use the products or therapies. Even though a new product may have been cleared for commercial distribution by the FDA, we may find limited demand for our product unless reimbursement approval can be obtained and/or maintained from governmental and private third-party payors.
| 32 |
In addition to uncertainties surrounding coverage policies, there are periodic changes to reimbursement levels. Third-party payors regularly update reimbursement amounts and also from time to time revise the methodologies used to determine reimbursement amounts. This includes routine updates to payments to physicians, hospitals and ambulatory surgery centers for procedures during which our products are used. These updates could directly impact the demand for our products.
We believe the overall escalating cost of medical products and services being paid for by the government and private health insurance has led to, and will continue to lead to, increased pressures on the healthcare and medical device industry to reduce the costs of products and services. Third-party payors are developing increasingly sophisticated methods of controlling healthcare costs through prospective reimbursement and capitation programs, group purchasing, redesign of benefits, and exploration of more cost-effective methods of delivering healthcare. In the United States, some insured individuals enroll in managed care programs, which monitor and often require pre-approval of the services that a member will receive. Some managed care programs pay their providers on a per capita (patient) basis, which puts the providers at financial risk for the services provided to their patients by paying these providers a predetermined payment per member per month and, consequently, may limit the willingness of these providers to use our products.
In international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted price ceilings on specific product lines and procedures. In the European Union, member states are facing increased pressure to limit public healthcare spending. There can be no assurance that procedures using our products will be covered for a specific indication, that our products will be considered cost-effective by third-party payors, that an adequate level of reimbursement will be available or that the third-party payors’ reimbursement policies will not adversely affect our ability to sell our products profitably. More and more, local, product specific reimbursement law is applied as an overlay to medical device regulation, which has provided an additional layer of clearance requirement.
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. Current and future legislative proposals to further reform healthcare or reduce healthcare costs may limit coverage of or lower reimbursement for the procedures associated with the use of our products. The cost containment measures that payors and providers are instituting and the effect of any healthcare reform initiative implemented in the future could impact our revenue from the sale of our products.
The implementation of the Affordable Care Act (the “ACA”) in the United States, for example, has changed healthcare financing and delivery by both governmental and private insurers substantially, and affected medical device manufacturers significantly. The ACA, among other things, provided incentives to programs that increase the federal government’s comparative effectiveness research, and implemented payment system reforms including a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models. Additionally, the ACA expanded eligibility criteria for Medicaid programs and created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. Since its enactment, there have been judicial, executive and political challenges to certain aspects of the ACA. On June 17, 2021, the U.S. Supreme Court dismissed the most recent judicial challenge to the ACA brought by several states without specifically ruling on the constitutionality of the ACA. It is unclear how healthcare reform measures of the Biden administration or other efforts, if any, to challenge, repeal or replace the ACA will impact the law or our business.
| 33 |
In addition, other legislative changes have been proposed and adopted since the ACA was enacted. For example, the Budget Control Act of 2011, among other things, reduced Medicare payments to providers by 2% per fiscal year, effective on April 1, 2013 and, due to subsequent legislative amendments to the statute, will remain in effect through 2030, with the exception of a temporary suspension from May 1, 2020 through March 31, 2021, unless additional Congressional action is taken. Additionally, the American Taxpayer Relief Act of 2012, among other things, further reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. The Medicare Access and CHIP Reauthorization Act of 2015 repealed the formula by which Medicare made annual payment adjustments to physicians and replaced the former formula with fixed annual updates and a new system of incentive payments that began in 2019 that are based on various performance measures and physicians’ participation in alternative payment models, such as accountable care organizations.
We expect additional state and federal healthcare reform measures to be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our products or additional pricing pressure.
Data Privacy and Security Laws
Numerous state, federal and foreign laws, including consumer protection laws and regulations, govern the collection, dissemination, use, access to, confidentiality and security of personal information, including health-related information. In the United States, numerous federal and state laws and regulations, including data breach notification laws, health information privacy and security laws, including HIPAA, and federal and state consumer protection laws and regulations (e.g., Section 5 of the FTC Act), that govern the collection, use, disclosure, and protection of health-related and other personal information could apply to our operations or the operations of our partners. In addition, certain state and non-U.S. laws, such as the CCPA, the CPRA and the GDPR, govern the privacy and security of personal information, including health-related information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties and private litigation. Privacy and security laws, regulations, and other obligations are constantly evolving, may conflict with each other to complicate compliance efforts, and can result in investigations, proceedings, or actions that lead to significant civil and/or criminal penalties and restrictions on data processing.
In Europe, the GDPR went into effect on May 25, 2018 and introduces strict requirements for processing the personal data of European Union data subjects. Companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global revenues of the preceding financial year of the noncompliant company, whichever is greater.
Among other requirements, the GDPR regulates transfers of personal data subject to the GDPR to third countries that have not been found to provide adequate protection to such personal data, including the United States, and the efficacy and longevity of current transfer mechanisms between the EU and the United States remains uncertain. For example, in 2016, the EU and United States agreed to a transfer framework for data transferred from the EU to the United States, called the Privacy Shield, but the Privacy Shield was invalidated in July 2020 by the Court of Justice of the European Union.
Further, from January 1, 2021, companies have to comply with the GDPR and also the United Kingdom General Data Protection Regulation, or the UK GDPR, which, together with the amended UK Data Protection Act 2018, retains the GDPR in UK national law. The UK GDPR mirrors the fines under the GDPR, i.e., fines up to the greater of €20 million (£17.5 million) or 4% of global turnover. The relationship between the United Kingdom and the European Union in relation to certain aspects of data protection law remains unclear, and it is also unclear how United Kingdom data protection laws and regulations will develop in the medium to longer term, and how data transfers to and from the United Kingdom will be regulated in the long term. Currently there is a four- to six-month grace period agreed in the EU and United Kingdom Trade and Cooperation Agreement, ending June 30, 2021 at the latest, while the parties discuss an adequacy decision. The European Commission published a draft adequacy decision on February 19, 2021. If adopted, the decision will enable data transfers from EU member states to the United Kingdom for a four-year period, subject to subsequent extensions.
| 34 |
Environmental Matters
Based on our current operations, environmental protection requirements do not have a significant financial and operational effect on the capital expenditures, earnings and competitive position of our Company in the current financial year and are not expected to have a significant effect in the reasonably foreseeable future.
Manufacturing Services Agreement
On August 21, 2020, the Company entered into a Manufacturing Services Agreement with GMI Corporation (“GMI”), dated as of August 21, 2020 (“MSA”), for the manufacture and supply of the Company’s IB-STIM device based upon the Company’s product specifications as set forth in the MSA.
The Company provides the necessary equipment to GMI and retains ownership. GMI bears the risk of loss of and damage to the equipment and consigned materials. Performance under the MSA is initiated by orders issued by the Company and accepted by GMI.
The initial term of the MSA was 24 months and automatically renews for terms of twelve months unless either party provides a written termination notice to the other party within 180 days prior to the end of the then-current term.
GMI was established in 1990 and manufactures our IB-Stim device in its 69,000 square foot facility that including offices, factory, environmentally controlled room, warehouse, parts processing, assembly, quality control, of which approximately 1,000 square feet is dedicated to producing our product, located in Indiana.
With controlled, repeatable, monitored production process, GMI has kit production capacity that is sufficient for all our projected needs. There is a new, dedicated, environmentally controlled build room built in 2022 for our equipment and production. All of our material are now maintained in this room.
GMI has a quality management system that is ISO 13485:2016 certified, FDA registered, and ITAR registered.
In connection with the MSA, the Company entered into a quality agreement with GMI, dated as of August 24, 2020, for GMI to test product provided by the Company and perform quality assurance services.
Employees
As of December 31, 2023, we had 19 full-time employees.
ITEM 1A. RISK FACTORS
Risks Relating to Our Business and Our Product
Our business and prospects depend entirely on our current product, IB-Stim. Even though we have received FDA clearance for our product, it will remain subject to ongoing regulatory review. If we are unable to maintain regulatory clearance and commercialize our product or are significantly delayed or limited in our commercialization efforts, our business and prospects will be materially harmed.
Almost all of our revenues have been derived from sales and royalties from sales of IB-Stim, and we expect to develop, market, and sell other neuromodulation therapy devices for the treatment of chronic and debilitating conditions in children. The commercial success of our products and our ability to generate and maintain revenues from the sale of our products will depend on a number of factors, including:
| ● | our ability to develop and obtain additional regulatory clearances and further commercialize our products for additional indications; | |
| ● | our ability to expand into new markets and future indications; | |
| ● | the acceptance of our products by patients and the healthcare community, including physicians and third-party payers (both private and governmental), as therapeutically effective and safe; | |
| ● | the accomplishment of various scientific, engineering, clinical, regulatory and other goals, which we sometimes refer to as milestones, on our anticipated timeline; |
| 35 |
| ● | the relative cost, safety and efficacy of alternative therapies; | |
| ● | our ability to obtain and maintain sufficient coverage or reimbursement by private and governmental third-party payers and to comply with applicable health care laws and regulations; | |
| ● | the ability of our third-party manufacturers to manufacture our products in sufficient quantities with acceptable quality; | |
| ● | our ability to provide marketing, distribution and customer support for our products; | |
| ● | the potential presence of competitive products in our active indications; | |
| ● | results of future clinical studies relating to our products or other competitor products for similar indications; | |
| ● | compliance with applicable laws and regulatory requirements; | |
| ● | the maintenance of our existing regulatory clearance; and | |
| ● | the consequences of any reportable adverse events involving our products. |
In addition, the promotion of our products is limited to approved indications, which vary by geography. The labelling for our device in the U.S. is limited in certain respects, which may limit the number of patients to whom it is prescribed.
Our ability to generate future revenues will also depend on achieving regulatory approval of, and eventual commercialization of, our products for additional indications and in additional geographies, which is not guaranteed. Our near-term prospects are substantially dependent on our ability to obtain regulatory approvals on the timetable we have anticipated, and thereafter to further successfully commercialize our products for additional indications. Regulatory changes or actions in areas in which we operate or propose to operate may further affect our ability to obtain regulatory clearances on our anticipated timetable. If we are not able to receive such approvals, meet other anticipated milestones, or further commercialize our products, or are significantly delayed or limited in doing so, our business and prospects will be materially harmed and we may need to reduce expenses by delaying, reducing or curtailing the development of our products and we may need to raise additional capital to fund our operations, which we may not be able to obtain on favorable terms, if at all.
To date, we have not generated any operating profits, and due to our long-term research and development efforts, we have a history of incurring substantial operating losses.
We were founded in 2011 and have a history of incurring substantial operating losses. We anticipate continuing to incur significant costs associated with developing and commercializing our products for approved indications including signal development, device hardware and software development, product sales, marketing, manufacturing, and distribution expenses. We expect our research, development, and clinical study expenses to increase in connection with our ongoing activities and as additional indications enter clinical development and as we advance our product development. Our expenses could increase beyond expectations if, for example, we are required by the FDA, or other regulatory agencies or similar governing bodies, to change manufacturing processes for our products or to perform clinical, nonclinical or other types of studies in addition to those that we currently anticipate. Our revenues are dependent, in part, upon the size of the markets in the jurisdictions in which we receive regulatory approval, the accepted price for our products and the ability to obtain reimbursement at the accepted applicable price. If the number of addressable patients is not as significant as we or our strategic partners and licensees estimate, the indications approved by regulatory authorities are narrower than we expect or the eligible population for treatment is narrowed by competition, regulatory approvals, physician choice or treatment guidelines, we may not generate significant revenues. If we are not able to generate significant revenues, we may never be sustainably profitable.
Our clinical studies could be delayed or otherwise adversely affected by many factors, including difficulties in enrolling patients.
Clinical testing can be costly and take many years, and the outcome is uncertain and susceptible to varying interpretations. Moreover, success in pre-clinical and early clinical studies does not ensure that large-scale studies will be successful or predict final results. Acceptable results in early studies may not be replicable in later studies. A number of companies in therapeutics industries have suffered significant setbacks in advanced clinical studies, even after promising results in earlier studies. Negative or inconclusive results or adverse events or incidents during a clinical study could cause the clinical study to be redone or terminated. In addition, failure to appropriately construct clinical studies could result in high rates of adverse events or incidents, which could cause a clinical study to be suspended, redone or terminated. Our failure or the failure of third-party participants in our studies to comply with their obligations to follow protocols and/or legal requirements may also result in our inability to use the affected data in our submissions to regulatory authorities.
| 36 |
The timely completion of clinical studies depends, among other things, on our ability to enroll a sufficient number of patients who remain in the study until its conclusion. We may experience difficulties in patient enrollment in our clinical studies for a variety of reasons, including:
| ● | the severity of the disease under investigation; | |
| ● | the limited size and nature of the patient population; | |
| ● | the patient eligibility criteria defined in our protocol and other clinical study protocols; | |
| ● | the nature of the study protocol, including the attractiveness of, or the discomforts and risks associated with, the treatments received by enrolled subjects; | |
| ● | difficulties and delays in clinical studies that may occur as a result of the COVID-19 pandemic; | |
| ● | the ability to obtain IRB approval at clinical study locations; | |
| ● | clinicians’ and patients’ perceptions as to the potential advantages, disadvantages and side effects of our products in relation to other available therapies, including any new drugs or treatments that may be approved for the indications we are pursuing; | |
| ● | availability of other clinical studies that exclude use of our products; | |
| ● | the possibility or perception that enrolling in a product’s clinical study may limit the patient’s ability to enroll in future clinical studies for other therapies due to protocol restrictions; | |
| ● | the possibility or perception that our software is not secure enough to maintain patient privacy; | |
| ● | patient referral practices of physicians; | |
| ● | the ability to monitor patients adequately during and after treatment; | |
| ● | the availability of appropriate clinical study investigators, support staff, drugs and other therapeutic supplies and proximity of patients to clinical sites; | |
| ● | physicians’ or our ability to obtain and maintain patient consents; and | |
| ● | the risk that when we collaborate with a third-party for research of a product in a particular institution, we can expect to relinquish some or all of the control over the future success of that study to the third-party. |
If we have difficulty enrolling and retaining a sufficient number or diversity of patients to conduct our clinical studies as planned, or encounter other difficulties, we may need to delay, terminate or modify ongoing or planned clinical studies, any of which would have an adverse effect on our business.
If we are unable to develop an adequate sales and marketing organization or contract with third parties to assist us, we may not be able to successfully commercialize our products for current and future indications.
To achieve commercial success for our products, we must compliantly develop and grow our sales and marketing organization and, as necessary, enter into sales and distribution relationships with third parties to market and sell our products. Developing and managing a sales and marketing organization is a difficult, expensive and time consuming process. We may not be able to successfully develop adequate sales and marketing capabilities to achieve our growth objectives. We compete with other medical device, pharmaceutical and life sciences companies to recruit, hire, train and retain the sales and marketing personnel that we anticipate we will need, and the nature of our products may make it more difficult to compete for sales and marketing personnel. In addition, because our current products require, and we anticipate our future products will require, physician training and education, our sales and marketing organization may need to grow substantially as we expand our approved indications and markets. As a consequence, our expenses associated with building up and maintaining our sales force and marketing capabilities may be disproportionate to the revenues we may be able to generate on sales of our products.
If we are unable to establish adequate sales and marketing capabilities or successful sales and distribution relationships, we may fail to realize the full revenue potential of our products for current and future indications, and we may not be able to achieve the necessary growth in a cost-effective manner or realize a positive return on our investment. In our future sales and distribution agreements with other companies, we generally may not have control over the resources or degree of effort that any of these third parties may devote to our products, and if they fail to devote sufficient time and resources to the marketing of our products, or if their performance is substandard, our revenues may be adversely affected.
| 37 |
The success of our business may be dependent on the actions of our collaborative partners.
Our business strategy includes, in part, the consummation of collaborative arrangements with companies who will support the development and commercialization of our products and technology. We may also enter into clinical collaborations with third parties to test our products and technology together with other products and technologies.
When we collaborate with a third party for commercialization of a product in a particular territory, we can expect to relinquish some or all of the control over the future success of that product to the third party in that territory. In addition, our collaborative partners may have the right to terminate applicable agreements, including payment obligations, prior to or upon the expiration of the agreed-upon terms. We may not be successful in establishing or maintaining collaborative arrangements on acceptable terms or at all, collaborative partners may terminate funding before completion of projects, our products may not achieve the criteria for milestone payments, our collaborative arrangements may not result in successful product commercialization, our products may not receive acceptable pricing and we may not derive any revenue from such arrangements. Additionally, our collaborators may not perform their obligations as expected or in compliance with study protocols or applicable laws. Acts or omissions by collaborators may disqualify study data for use in regulatory submissions and/or create liability for us in the jurisdictions in which we operate. Any disagreements with collaborators, including disagreements over proprietary rights, contract interpretation or the preferred course of commercialization, might cause delays or termination of the commercialization of products, might lead to additional responsibilities for us with respect to commercializing products, or might result in litigation or arbitration, any of which would be time-consuming and expensive. To the extent that we are not able to develop and maintain collaborative arrangements, we would need to devote substantial capital to undertake commercialization activities on our own in order to further expand our reach, and we may be forced to limit the territories in which we commercialize our products.
We may not be successful in achieving market acceptance of our products by healthcare professionals, patients and/or third-party payers in the timeframes we anticipate, or at all, which could have a material adverse effect on our business, prospects, financial condition and results of operations.
We may not achieve market acceptance of our products for current or future indications within the timeframes we have anticipated, or at all, for a number of different reasons, including the following factors:
| ● | it may be difficult to gain broad acceptance of our products because they are new technologies and involve a novel or derivative mechanism of action and, as such, physicians may be reluctant to prescribe our products without prior experience or additional data or training; | |
| ● | physicians may be reluctant to prescribe our products due to their perception that the supporting clinical study designs have limitations, as they are, for example, unblinded; | |
| ● | physicians at large academic universities and medical centers may prefer to enroll patients into clinical studies instead of prescribing our products; | |
| ● | it may be difficult to gain broad acceptance at community hospitals where the number of patients seeking treatment may be more limited than at larger medical centers, and such community hospitals may not be willing to invest in the resources necessary for their physicians to become trained to use our products, which could lead to reluctance to prescribe our products; | |
| ● | patients may be reluctant to use our products for various reasons, including a perception that the treatment is untested or difficult to use or a perception that our software is not secure; | |
| ● | our products may have side effects and our products cannot be worn in all circumstances; and | |
| ● | each patient will use more than one device and therefore, as the duration of the treatment course increases, the overall price will increase correspondingly and, when used in combination with other treatments, the overall cost of treatment will be greater than using a single type of treatment. |
In particular, our products may not achieve market acceptance for current or future indications because of the following additional factors:
| ● | achieving patient acceptance could be difficult because not all patients are willing to comply with requirements of treatment with our products, and other patients may forego our products for financial, privacy, cosmetic, visibility or mobility reasons; |
| 38 |
| ● | achieving patient compliance may be difficult because the recommended use of our products is 120 hours per week for three (3) consecutive weeks, and not to exceed four (4) weeks, which to some extent restricts physical mobility because our products cannot be worn in all circumstances, and the patient or a caregiver must ensure that it remains continuously operable and this may also impact the pool of patients to whom physicians may be willing to prescribe our products; | |
| ● | there may be certain perceived limitations to our study designs or data obtained from our clinical studies; | |
| ● | efficacy may also be limited in instances where patients take a break from the device when experiencing skin rashes, or while bathing or swimming (because our products should not be immersed in water); and | |
| ● | patients may decline therapy or prescribers may be unwilling to prescribe our products due to certain adverse events attributable to the device reported in clinical studies by patients treated with our products. |
In addition, even if we are successful in achieving market acceptance of our products for IBS or other indications, we may be unsuccessful in achieving market acceptance of our products for other indications.
There may be other factors that are presently unknown to us that also may negatively impact our ability to achieve market acceptance of our products. If we do not achieve market acceptance of our products in the timeframes we anticipate, or are unable to achieve market acceptance at all, our business, prospects, financial condition and results of operations could be materially adversely affected.
Failure to secure and maintain adequate coverage and reimbursement from third-party payers could adversely affect acceptance of our products and reduce our revenues.
We expect that the majority of our revenues will come from third-party payers, primarily children’s hospitals, either directly to us in markets where we provide our products or plan to provide our device candidates to patients or indirectly via payments made to hospitals or other entities providing our products or which may in the future provide our device candidates to patients.
In the U.S., private payers cover the largest segment of the population, with the remainder either uninsured or covered by governmental payers. The majority of the third-party payers outside the U.S. are government agencies, government sponsored entities or other payers operating under significant regulatory requirements from national or regional governments.
Third-party payers may decline to cover and reimburse certain procedures, supplies or services. Additionally, some third-party payers may decline to cover and reimburse our products for a particular patient even if the payer has a favorable coverage policy addressing our products or previously approved reimbursement for our products. Additionally, private and government payers may consider the cost of a treatment in approving coverage or in setting reimbursement for the treatment.
Private and government payers are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of governments. Adoption of additional price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our revenues and operating results. If third-party payers do not consider our products or the combination of our products with additional treatments to be cost-justified under a required cost-testing model, they may not cover our products for their populations or, if they do, the level of reimbursement may not be sufficient to allow us to sell our products on a profitable basis.
Reimbursement for the treatment of patients with medical devices is governed by complex mechanisms. These mechanisms vary widely among countries, can be informal, somewhat unpredictable, and evolve constantly, reflecting the efforts of these countries to reduce public spending on healthcare. As a result, obtaining and maintaining reimbursement for the treatment of patients with medical devices has become more challenging. We cannot guarantee that the use of our products will receive reimbursement approvals and cannot guarantee that our existing reimbursement approvals will be maintained in any country.
Our failure to secure or maintain adequate coverage or reimbursement for our products by third-party payers in the U.S. or in the other jurisdictions in which we market our products could have a material adverse effect on our business, revenues and results of operations and cause our stock price to decline.
| 39 |
We may not be successful in maintaining reimbursement codes necessary to facilitate accurate and timely billing for our products or physician services attendant to our products.
Third-party payers, healthcare systems, government agencies or other groups often issue reimbursement codes to facilitate billing for products and physician services used in the delivery of healthcare. Our technology specific CAT III CPT Code (0720T) was published on December 30, 2021 and effective on July 1, 2022. We may not be able to maintain the CPT code for physician services related to our products. Our future revenues and results may be affected by the absence of CPT codes, as physicians may be less likely to prescribe the therapy when there is no certainty that adequate reimbursement will be available for the time, effort, skill, practice expense and malpractice costs required to provide the therapy to patients.
Outside the U.S., we have not secured codes to describe our products or to document physician services related to the delivery of therapy using our products. The failure to obtain and maintain these codes could affect the future growth of our business.
We may depend on single-source suppliers for some of our components. The loss of these suppliers could prevent or delay shipments of our products, delay our clinical studies or otherwise adversely affect our business.
In certain jurisdictions, we may source some of the components of our products from only a single vendor. If any one of these single-source suppliers were to fail to continue to provide components to us on a timely basis, or at all, our business and reputation could be harmed. We will seek and maintain second-source suppliers, but we can provide no assurance that we will secure or maintain such suppliers. We have developed or are in the process of developing and obtaining regulatory approval for second sources for components in all jurisdictions. Various steps must be taken before securing these suppliers, including qualifying these suppliers in accordance with regulatory requirements, but we may never receive such approvals. The risks associated with the failure of our suppliers to comply with strictly enforced regulatory requirements as described below are exacerbated by our dependence on single-source suppliers.
If we experience any deficiency in the quality of, delay in or loss of availability of any components supplied to us by third-party suppliers, or if we switch suppliers or components, we may face additional regulatory delays and the manufacture and delivery of our products would be interrupted for an extended period of time, which could materially adversely affect our business, prospects, financial condition and results of operations. If we are required to obtain prior regulatory approval from the FDA or regulatory authorities or similar governing bodies in other jurisdictions or to conduct a new conformity assessment procedure for our products, regulatory approval for our products may not be received on a timely basis, or at all, which would have a material adverse effect on our business, prospects, financial condition and results of operations.
Quality control problems with respect to devices and components supplied by third-party suppliers could have a material adverse effect on our reputation, our clinical studies or the commercialization of our products and, as a result, a material adverse effect on our business, prospects, financial condition and results of operations.
Our products, which are manufactured by third parties, are highly technical and are required to meet exacting specifications. Any quality control problems that we experience with respect to the devices and components supplied by third-party suppliers could have a material adverse effect on our reputation, our attempts to complete our clinical studies, our operating expenses or the commercialization of our products. The failure of our suppliers to comply with strictly enforced regulatory requirements could expose us to regulatory action, including warning letters, product recalls, suspension or termination of distribution, product seizures or civil penalties. If we experience any delay in the receipt or deficiency in the quality of products supplied to us by third-party suppliers, or if we have to switch to replacement suppliers, we may face additional regulatory delays and the manufacture and delivery of our products would be interrupted for an extended period of time, which would materially adversely affect our business, prospects, financial condition and results of operations.
| 40 |
We currently do not own a manufacturing facility and rely on a sole manufacturer for the production of our product. Any significant disruption to the sole manufacturer’s operations or facilities could have a material adverse effect on our business, financial condition and results of operations.
We rely on a sole manufacturer for the production of our products. We do not have control over the operations of the facilities of the third-party manufacturer that we use. A significant disruption to our manufacturer could have a material adverse effect on our business, financial condition and results of operations. Our reliance on our manufacturer poses a number of risks, including lack of control over the manufacturing process and ultimately over the quality and timing of delivery of our product. A change in our relationship with our manufacturer could result in a material adverse effect on our business, financial condition and results of operations. A decision to change manufacturers would result in longer times for design and production as we secure any necessary licenses or clearances, develop quality control measures, and implement manufacturing processes.
Continued testing of our products may not yield successful results and could reveal currently unknown aspects or safety hazards associated with our products.
Our research and development programs are designed to test the safety and efficacy of our products through extensive pre-clinical and clinical testing. Even if our ongoing and future pre-clinical and clinical studies are completed as planned, we cannot be certain that their results will support our claims or that the FDA and other regulatory authorities will agree with our conclusions. Success in pre-clinical studies and early clinical studies does not ensure that later clinical studies will be successful, and we cannot be sure that the later studies will replicate the results of prior studies and pre-clinical studies. The clinical study process may fail to demonstrate that our device candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a device candidate and may delay development of others. It is also possible that patients enrolled in clinical studies will experience adverse side effects that have not been previously observed. In addition, our pre-clinical and clinical studies for our device candidates involve a relatively small patient population and, as a result, these studies may not be indicative of future results.
We may experience numerous unforeseen events during, or as a result of, the testing process that could delay or prevent further commercialization of our products, including the following:
| ● | pre-clinical and clinical testing for our products may not produce the desired effect, may be inconclusive or may not be predictive of safety or efficacy results obtained in future clinical studies, following long-term use or in much larger populations; | |
| ● | unanticipated adverse events or other side effects that are not currently known may occur during our clinical studies that may preclude additional regulatory approval or result in additional limitations to commercial use if approved; and | |
| ● | the data collected from our clinical studies may not reach statistical significance or otherwise not be sufficient to support FDA or other regulatory approval. |
If unacceptable side effects arise in the development of our products for future indications, we could suspend or terminate our clinical studies or the FDA or other regulatory authorities could order us to cease clinical studies or deny approval of our device candidates for any or all targeted indications, narrow the approved indications for use or otherwise require restrictive product labeling or marketing or require further clinical studies, which may be time-consuming and expensive and may not produce results supporting FDA or other regulatory approval of our products in a specific indication. Treatment-related side effects could also affect patient recruitment or the ability of enrolled patients to complete the study or result in potential product liability claims. In addition, these side effects may not be appropriately recognized or managed by the treating medical staff. We expect to have a need to train medical personnel using our devices for clinical studies and upon any commercialization of our products for future indications. Inadequate training in recognizing or managing the potential side effects of our products could result in patient injury or death. Any of these occurrences may harm our business, prospects and financial condition significantly.
Any delay or termination of our clinical studies will delay the filing of submissions for regulatory approvals of our products and ultimately our ability to commercialize our products and generate revenues. Furthermore, we may abandon our products for indications that we previously believed to be promising. Any of these events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
| 41 |
As we expand, we may experience difficulties managing our growth.
Our anticipated growth will place a significant strain on our management and on our operational and financial resources and systems. We could face challenges inherent in efficiently managing a more complex business with an increased number of employees over large geographic distances, including the need to implement appropriate systems, policies, benefits and compliance programs. Failure to manage our growth effectively could materially adversely affect our business. Additionally, our anticipated growth will increase the demands placed on our third-party suppliers, resulting in an increased need to carefully monitor the available supply of components and services and to scale up our quality assurance programs. There is no guarantee that our suppliers will be able to support our anticipated growth. Any failure by us to manage our growth effectively could have an adverse effect on our ability to achieve our development and commercialization goals.
The size and expected growth of our available market has not been established with precision and may be smaller than we estimate.
Our data on the available market for our current products and future products is based on a number of internal and third-party research reports, estimates and assumptions. While we believe that such research, our assumptions and the data underlying our estimates are reasonable, these assumptions and estimates may not be correct. In addition, the statements in this prospectus relating to, among other things, the expected growth in the market for our IB-Stim are based on a number of internal and third-party data, estimates and assumptions, and may prove to be inaccurate. If the actual number of consumers who would benefit from our products, the price at which we can sell future products or the available market for our products is smaller than we estimate, it could have a material adverse effect on our business, financial condition and results of operations.
If physicians and patients do not accept our current and future products or if the market for indications for which any product candidate is approved is smaller than expected, we may be unable to generate significant revenue, if any.
Even when any of our product candidates obtain regulatory approval, they may not gain market acceptance among physicians, patients, and third-party payers. Physicians may decide not to recommend our treatments for a variety of reasons including:
| ● | timing of market introduction of competitive products; | |
| ● | demonstration of clinical safety and efficacy compared to other products; | |
| ● | cost-effectiveness; | |
| ● | limited or no coverage by third-party payers; | |
| ● | convenience and ease of administration; | |
| ● | prevalence and severity of adverse side effects; | |
| ● | restrictions in the label of the device; | |
| ● | other potential advantages of alternative treatment methods; and | |
| ● | ineffective marketing and distribution support of its products. |
If any of our product candidates is approved but fails to achieve market acceptance or such market is smaller than anticipated, we may not be able to generate significant revenue and our business would suffer.
Because of the specialized nature of our business, the termination of relationships with our key employees, consultants and advisors may prevent us from successfully operating our business, including developing our products, conducting clinical studies, commercializing our products and obtaining any necessary financing.
We are highly dependent on the members of our executive team, the loss of whose services may adversely impact the achievement of our objectives. While we have entered into employment agreements with each of our key executives, any of them could leave our employment at any time. We do not have “key person” insurance on any of our employees. The loss of the services of one or more of our current employees might impede the achievement of our business objectives.
The competition for qualified personnel in the medical device fields is intense, and we rely heavily on our ability to attract and retain qualified scientific, technical and managerial personnel. Our future success depends upon our ability to attract, retain and motivate highly skilled employees. In order to commercialize our products successfully, we will be required to expand our workforce, particularly in the areas of research and development and clinical studies, sales and marketing and supply chain management. These activities will require the addition of new personnel and the development of additional expertise by existing management personnel. We face intense competition for qualified individuals from numerous pharmaceutical, biopharmaceutical and biotechnology companies, as well as academic and other research institutions. We may not be able to attract and retain these individuals on acceptable terms or at all. Failure to do so could materially harm our business.
| 42 |
Customer or third-party complaints or negative reviews or publicity about our company or our products could harm our reputation and brand.
We are heavily dependent on customers who use our IB-Stim device to provide good reviews and word-of-mouth recommendations to contribute to our growth. Customers who are dissatisfied with their experiences with our products or services may post negative reviews. We may also be the subject of blog, forum or other media postings that include inaccurate statements and/or create negative publicity. In addition, any negative news regarding similar products may adversely impact our business. Any negative reviews or publicity, whether real or perceived, disseminated by word-of-mouth, by the general media, by electronic or social networking means or by other methods, could harm our reputation and brand and could severely diminish consumer confidence in our products.
Adverse global economic conditions could have a negative effect on our business, results of operations and financial condition and liquidity.
A general slowdown in the global economy, including a recession, or in a particular region or industry, an increase in trade tensions with U.S. trading partners, inflation or a tightening of the credit markets could negatively impact our business, financial condition and liquidity. Adverse global economic conditions have from time to time caused or exacerbated significant slowdowns in the industries and markets in which we operate, which have adversely affected our business and results of operations. Macroeconomic weakness and uncertainty also make it more difficult for us to accurately forecast revenue, gross profit and expenses, and may make it more difficult to raise or refinance debt. Sustained uncertainty about, or worsening of, current global economic conditions and further escalation of trade tensions between the U.S. and its trading partners, could result in a global economic slowdown and long-term changes to global trade. Such events may also (i) cause our customers and consumers to reduce, delay or forgo spending, (ii) result in customers sourcing products from other suppliers not subject to such restrictions or tariffs, (iii) lead to the insolvency or consolidation of key suppliers and customers, and (iv) intensify pricing pressures. Any or all of these factors could negatively affect demand for our products and our business, financial condition and results of operations.
Additionally, economic conditions in certain regions may also be affected by natural disasters and public health emergencies, such as extreme weather events, and could have a significant adverse effect on our business, including interruption of our commercial and clinical operations, supply chain disruption, endangerment of our personnel, fewer patient visits, increased patient drop-out rates, delays in recruitment of new patients, and other delays or losses of materials and results.
The COVID-19 pandemic could materially adversely impact our business.
As the COVID-19 pandemic continues around the globe, we have experienced and will likely continue to experience disruptions that could severely impact our business and clinical studies, which could include:
| ● | delays and/or difficulties in onboarding active patients and enrolling patients in our clinical studies; | |
| ● | delays and/or difficulties in clinical site initiation, including difficulties in recruiting clinical site investigators and clinical site staff; | |
| ● | declines in prescriptions written due to a perception that our products are difficult to administer remotely or if patients are unwilling to travel to treatment sites or receive in-home treatment assistance from us or other caregivers; | |
| ● | reductions in third-party reimbursements, which could materially affect our revenue, as most of our patients rely on third-party payers to cover the cost of our products and a material number of our patients could lose access to their private health insurance plan if they or someone in their family lose their job; | |
| ● | diversion of healthcare resources away from conducting clinical studies, including the diversion of hospitals serving as our clinical study sites and hospital staff supporting the conduct of our clinical studies; |
| 43 |
| ● | interruption of key clinical study activities, such as clinical study site monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others; | |
| ● | staff disruptions and turnover internally and at treatment sites and third-party providers who provide support, either directly as a result of illness or indirectly as a result of vaccine mandates and other changes in terms of employment; | |
| ● | delays in receiving approval from local regulatory authorities or IRBs to initiate our planned clinical studies; | |
| ● | delays in clinical sites receiving the supplies and materials needed to conduct our clinical studies; | |
| ● | interruption in shipping that may affect the transport of active patient and clinical study materials; | |
| ● | changes in local regulations as part of a response to the COVID-19 outbreak that may require us to change the ways in which our clinical studies are conducted, which may result in unexpected costs, or to discontinue the clinical studies altogether; | |
| ● | delays in necessary interactions with local regulators, ethics committees and other important agencies and contractors due to limitations in employee resources or forced furlough of government employees; | |
| ● | disruption of our supply chain as our suppliers and common carriers are unable to meet our requirements to provide us the materials we need for clinical study and active patient care needs; | |
| ● | indirect consequences of the COVID-19 pandemic on the economy in general, such as an increase in bankruptcies of our key suppliers, or the inability of our third-party payers to meet their obligations reimburse us in a timely fashion or at all; | |
| ● | postponements and cancellations of key conferences and meetings and travel restrictions could interfere with our ability to interact with key thought leaders in the field, leading to a disruption in the rate of adoption of our technology; | |
| ● | access restrictions at offices, hospitals, and treatment centers, and stakeholder illness could interfere with the ability of our sales force to engage in face-to-face visits with providers, leading to a disruption in the rate of adoption of our technology; | |
| ● | increases in expenditures for technology and other tools necessary to provide patient care in an environment where both patient and care-giver travel is restricted and access to in-person interaction is limited; | |
| ● | refusal of the FDA to accept data from clinical studies in affected geographies outside the United States; and | |
| ● | patient delays in seeking or receiving treatment, either due to fear of infection or lack of access to treatment and study sites, leading to fewer diagnoses of the indications our products are approved to treat or more advanced procession of the disease, which may contraindicate the use of our products or disqualify the patient from participating in a given study. |
The global status of the COVID-19 pandemic continues to rapidly evolve. The extent to which the pandemic may impact our business and clinical studies will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the disease, the duration of the pandemic, travel restrictions and social distancing guidelines, business closures or business disruptions and the effectiveness of actions taken to contain and treat the disease. The response to the pandemic may result in permanent changes to the environment in which we operate as described above in ways we are unable to predict. The COVID-19 pandemic may also have the effect of heightening many of the other risks described herein.
Developing medical technology entails significant technical, regulatory and business risks.
We may fail to adapt our technology to user requirements or emerging treatment standards. Neuromodulation therapies are not currently considered standard of care for IBS and may not ever be considered standard of care. Treatment standards may not evolve to incorporate our product. New industry standards for the development, manufacture and marketing of medical devices may evolve and we may not be able to conform to the changes, meet new standards in a timely fashion or maintain a competitive position in the market. In particular, regulatory standards for electrical treatments of medical conditions are evolving. If we face material delays in introducing our products and new technology, we may fail to attract new customers.
Our Company has an evolving business strategy and investors must be willing to accept a substantial degree of uncertainty.
The Company’s strategic focus is on the development of developing neuromodulation therapies to address chronic and debilitating conditions in children. The Company may engage in ongoing discussions with potential licensees, other strategic partners and institutional or private financing sources, the result of which could add to or alter its current strategic focus, cash needs or ownership structure. Investors must be willing to accept a substantial degree of uncertainty and must be willing to rely upon the Company’s board of directors and management to complete an appropriate business strategy to commercially exploit targeted business opportunities.
| 44 |
We may not be able to compete with treatments now being marketed and developed, or which may be developed and marketed in the future by other companies.
Our products will compete with existing and new therapies and treatments for chronic and debilitating conditions in children. We are aware of a number of companies currently seeking to develop alternative therapies or treatment for such diseases and conditions at least in part. Numerous pharmaceutical, biotechnology, drug delivery and medical device companies, hospitals, research organizations, individual scientists, and nonprofit organizations are engaged in the development of alternatives to our technology. Some of these companies have greater research and development capabilities, experience, manufacturing, marketing, financial, and managerial resources than we do. Collaborations or mergers between large pharmaceutical or biotechnology companies with competing treatment technologies could enhance our competitors’ financial, marketing, and other resources. Developments by other medical device companies could make our products or technologies uncompetitive or obsolete. Accordingly, our competitors may succeed in developing competing technologies, obtaining FDA clearances and/or approval for products or gaining market acceptance more rapidly than we can.
Due in part to our limited financial resources, we may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable indications or therapeutic areas for our product candidates, and/or we may be unable to pursue the clinical trials that we would like to pursue.
We have limited technical, managerial, and financial resources to determine the indications on which we should focus the development efforts related to our product candidates. Due to our limited available financial resources, we may have curtailed clinical development programs and activities that might otherwise have led to more rapid progress of our product candidates through the regulatory and development processes.
We may make incorrect determinations with regard to the indications and clinical trials on which to focus the available resources that we do have. Furthermore, we cannot assure you that we will be able to retain adequate staffing levels to run our operations and/or to accomplish all of the objectives that we otherwise would seek to accomplish. Our decisions to allocate our research, management, and financial resources toward particular indications or therapeutic areas for our product candidates may not lead to the development of viable commercial products and may divert resources from better opportunities. Similarly, our decisions to delay or terminate product development programs may also cause us to miss valuable opportunities.
We have material weaknesses in our internal control over financing reporting. If we fail to establish and maintain proper and effective internal control over financial reporting, our operating results and our ability to operate our business could be harmed.
Ensuring that we have adequate internal financial and accounting controls and procedures in place so that we can produce accurate financial statements on a timely basis is a costly and time-consuming effort that needs to be re-evaluated frequently. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with generally accepted accounting principles. Due to accounting resource constraints, we have had limited review controls. These constraints have resulted in (1) a lack of segregation of duties, since we have a limited administrative staff, (2) lack of internal controls structure review and (3) misapplication of U.S. GAAP. As a result of these constraints and the restatement our unaudited financial statements as of and for the three and nine month periods ended September 30, 2023, management concluded material weaknesses in our internal control over financial reporting existed as of December 31 2023 and, accordingly, our internal control over financial reporting and disclosure controls and procedures were not effective as of such date. Specifically, management identified a material accounting error that understated extinguishment of debt expense and net loss in the Statements of Operations and understated additional paid in capital in the Balance Sheet as of and for the three and nine month periods ended September 30, 2023 as described and restated in Note 19 to the audited financial statements included in this Form 10-K.
Our management is composed of a small number of individuals resulting in a situation where limitations on segregation of duties exist. All responsibility for accounting entries and the creation of financial statements is held by a single person, though the Company engages multiple accounting consultants for accounting, tax and audit support. To remedy this situation, we would need to hire additional staff or financial consultant support.
| 45 |
We document, review and improve our internal controls and procedures for compliance with Section 404 of the Sarbanes-Oxley Act, which require annual management assessment of the effectiveness of our internal control over financial reporting. To comply with the requirements of being a public company, the Company has undertaken various actions, and will take additional actions, such as remediating the material weaknesses described above, implementing additional internal controls and procedures and hiring internal audit staff or financial consultants. Testing and maintaining internal controls can divert our management’s attention from other matters that are important to the operation of our business. Additionally, when evaluating internal controls over financial reporting, the Company may identify additional material weaknesses that it may not be able to remediate in time to meet the applicable deadline imposed upon us for compliance with the requirements of Section 404 of the Sarbanes-Oxley Act. If the Company identifies any additional material weaknesses in its internal control over financial reporting or is unable to remediate the material weakness described above or comply with the requirements of Section 404 of the Sarbanes-Oxley Act in a timely manner or if the Company’s independent registered public accounting firm is unable to express an unqualified opinion as to the effectiveness of our internal control over financial reporting once it is no longer an emerging growth company, or if the Company is unable to conclude in our quarterly and annual reports that our disclosure controls and procedures are effective, investors may lose confidence in the accuracy and completeness of the Company’s financial reports and the market price of our common stock could be negatively affected, and the Company could become subject to investigations by the stock exchange on which our securities are listed, the SEC or other regulatory authorities, which could require additional financial and management resources.
In addition, if the Company fails to remediate any material weakness, including the material weaknesses described above, our financial statements could be inaccurate and the Company could face restricted access to capital markets. Our small size and internal control deficiencies may adversely affect our financial condition, results of operation and access to capital. Moreover, our internal control over financial reporting will not prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud will be detected. If we cannot provide reliable financial reports or prevent fraud, we may not be able to manage our business as effectively as we would if an effective control environment existed, and our business and reputation with investors may be harmed.
Risks Related to Legal and Regulatory Matters
Product liability suits, whether or not meritorious, could be brought against us due to alleged defective devices or for the misuse of our products, which could result in expensive and time-consuming litigation, payment of substantial damages and/or expenses and an increase in our insurance rates.
If our current or future devices are defectively designed or manufactured, contain defective components or are misused, or if someone claims any of the foregoing, whether or not meritorious, we may become subject to substantial and costly litigation. For example, we may be sued if our products cause or are perceived to cause injury or are found to be otherwise unsuitable during clinical testing, manufacturing, marketing or sale. This may occur if our products are misused or damaged, have a sudden failure or malfunction (including with respect to safety features) or are otherwise impaired due to wear and tear. Even absent a product liability suit, malfunctions of our products or misuse by physicians or patients would need to be remedied swiftly in order to maintain continuous use and ensure efficacy of our products.
Any product liability claims may include allegations of defects in manufacturing, defects in design, a failure to warn of dangers inherent in the device, negligence, strict liability or a breach of warranties. Claims could also be asserted under state consumer protection acts. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products. Even successful defense may require significant financial and management resources. Regardless of the merits or eventual outcome, liability claims may result in:
| ● | decreased demand for our products; | |
| ● | injury to our reputation; | |
| ● | withdrawal of clinical study participants and inability to continue clinical studies; | |
| ● | initiation of investigations by regulators; |
| 46 |
| ● | costs to prepare for and defend the related litigation; | |
| ● | a diversion of management’s time and our resources; | |
| ● | substantial monetary awards to study participants or patients; | |
| ● | product recalls, withdrawals or labeling, marketing or promotional restrictions; | |
| ● | loss of revenues; | |
| ● | exhaustion of any available insurance and our capital resources; | |
| ● | the inability to commercialize any device candidate; and | |
| ● | a decline in our share price. |
Product liability claims could divert management’s attention from our core business, be expensive to defend and result in sizable damage awards against us. We may not have sufficient insurance coverage for all claims. Any product liability claims brought against us, with or without merit, could increase our product liability insurance rates or prevent us from securing continuing coverage, could harm our reputation in the industry and could reduce revenues. Product liability claims in excess of our insurance coverage would be paid out of cash reserves, if any, which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Even if our agreements with our third-party manufacturers and suppliers entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
Other future litigation and regulatory actions could have a material adverse impact on the Company.
From time to time, we may be subject to litigation and other legal and regulatory proceedings relating to our business or investigations or other actions by governmental agencies. No assurances can be given that the results of these or new matters will be favorable to us. An adverse resolution of lawsuits, arbitrations, investigations or other proceedings or actions could have a material adverse effect on our financial condition and results of operations, including as a result of non-monetary remedies. Defending ourselves in these matters may be time-consuming, expensive and disruptive to normal business operations and may result in significant expense and a diversion of management’s time and attention from the operation of our business, which could impede our ability to achieve our business objectives. Additionally, any amount that we may be required to pay to satisfy a judgment, settlement, fine or penalty may not be covered by insurance. Subject to the Delaware General Corporation Law, our certificate of incorporation permit us to indemnify any director against any liability, to purchase and maintain insurance against any liability for any director and to provide any director with funds (whether by loan or otherwise) to meet expenditures incurred or to be incurred by such director in defending any criminal, regulatory or civil proceedings or in connection with an application for relief (or to enable any such director to avoid incurring such expenditure). In addition, under our Articles of Incorporation and bylaws (the “Bylaws”) we are obligated to indemnify each of our directors and officers against certain liabilities and expenses arising from their being a director or officer to the maximum extent permitted by Delaware law. In the event we are required to make such payments to our directors and officers, there can be no assurance that any of these payments will not be material.
We are subject to consumer protection laws that regulate our marketing practices and prohibit unfair or deceptive acts or practices. Our actual or perceived failure to comply with such obligations could harm our business, and changes in such regulations or laws could require us to modify our products or marketing or advertising efforts.
In connection with the marketing or advertisement of our products, we could be the target of claims relating to false, misleading, deceptive or otherwise noncompliant advertising or marketing practices, including under the auspices of the FTC and state consumer protection statutes. If we rely on third parties to provide any marketing and advertising of our products, we could be liable for, or face reputational harm as a result of, their marketing practices if, for example, they fail to comply with applicable statutory and regulatory requirements.
If we are found to have breached any consumer protection, advertising, unfair competition or other laws or regulations, we may be subject to enforcement actions that require us to change our marketing and business practices in a manner that may negatively impact us. This could also result in litigation, fines, penalties and adverse publicity that could cause reputational harm and loss of customer trust, which could have a material adverse effect on our business, financial condition and results of operations.
| 47 |
We are increasingly dependent on information technology systems and are subject to privacy and security laws. Our products and our systems and infrastructure face certain risks, including from cyber security breaches and data leakage.
We increasingly rely upon technology systems and infrastructure. Our technology systems, including our products, are potentially vulnerable to breakdown or other interruption by fire, power loss, system malfunction, unauthorized access and other events. Likewise, data privacy breaches by employees and others with both permitted and unauthorized access to our products and our systems may pose a risk that protected patient information (“PI”) may be exposed to unauthorized persons or to the public, or may be permanently lost. The increasing use and evolution of technology, including cloud-based computing, creates additional opportunities for the unintentional dissemination of information, intentional destruction of confidential information stored in our systems or in non-encrypted portable media or storage devices. We could also experience a business interruption, information theft of confidential information, or reputational damage from industrial espionage attacks, malware or other cyber incidents, which may compromise our system infrastructure or lead to data leakage, either internally or at our third-party service providers or other business partners.
The size and complexity of our computer systems, and scope of our geographic reach, make us potentially vulnerable to information technology system breakdowns, internal and external malicious intrusion, cyberattacks and computer viruses. Because the techniques used to obtain unauthorized access, or to sabotage systems, change frequently and generally are not recognized until launched against a target, we may be unable to anticipate these techniques or to implement adequate preventative measures. If we do not allocate and effectively manage the resources necessary to build and sustain the proper technology infrastructure or properly manage third-party contractors who perform data management services on our behalf, then a security breach could subject us to, among other things, transaction errors, business process inefficiencies, the loss of customers, damage to our reputation, business disruptions or the loss of or damage to intellectual property. Such security breaches could expose us to a risk of loss of information, litigation, penalties, remediation costs and potentially significant liability to customers, employees, business partners and regulatory authorities, including, for example, under the Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) in the United States and Regulation 2016/679 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data under GDPR in the EU. If our data management systems (including third party data management systems) do not effectively collect, secure, store, process and report relevant data for the operation of our business, whether due to equipment malfunction or constraints, software deficiencies, or human error, our ability to effectively plan, forecast and execute our business plan and comply with applicable laws and regulations will be impaired. Any such impairment could materially and adversely affect our financial condition and results of operations.
While we have invested heavily in the protection of data and information technology and in related training, there can be no assurance that our efforts will prevent significant breakdowns, breaches in our systems or other cyber incidents or ensure compliance with all applicable security and privacy laws, regulations, and standards, including with respect to third-party service providers that utilize sensitive personal information, including PI, on our behalf.
A security breach, whether of our products, systems or third-party hosting services we utilize, could disrupt treatments being provided by our products, disrupt access to our customers’ stored information, such as patient treatment data and health information, and could lead to the loss of, damage to or public disclosure of such data and information, including patient health information. Such an event could have serious negative consequences, including possible patient injury, regulatory action, fines, penalties and damages, reduced demand for our products, an unwillingness of customers to use our products, harm to our reputation and brand and time-consuming and expensive litigation, any of which could have a material adverse effect on our financial results. We currently carry cyber and privacy liability insurance with an aggregate limit of $1,000,000, but the amount of insurance coverage that we purchased and may purchase in the future may be inadequate. In the future, our insurance coverage may be expensive or not be available on acceptable terms or in sufficient amounts, if at all.
| 48 |
We may choose to, or may be required to, suspend, repeat or terminate our clinical studies if they are not conducted in accordance with regulatory requirements, the results are negative or inconclusive or the studies are not well designed.
Clinical studies must be conducted in accordance with the FDA’s cGCPs and the equivalent laws and regulations applicable in other jurisdictions in which the clinical studies are conducted. The clinical studies are subject to oversight by the FDA, regulatory agencies in other jurisdictions, ethics committees and institutional review boards at the medical institutions where the clinical studies are conducted. In addition, clinical studies must be conducted with device candidates produced under the FDA’s QSR and in accordance with the applicable regulatory requirements in the other jurisdictions in which the clinical studies are conducted. The conduct of clinical studies may require large numbers of test patients.
The FDA or regulatory agencies in other jurisdictions might delay or terminate our clinical studies of a device candidate for various reasons, including:
| ● | the device candidate may have unforeseen adverse side effects or may not appear to be more effective than current therapies; | |
| ● | we may not agree with the FDA, a regulatory authority in another jurisdiction or an ethics committee regarding the protocol for the conduct of a clinical study; | |
| ● | new therapies may become the standard of care while we are conducting our clinical studies, which may require us to revise or amend our clinical study protocols or terminate a clinical study; or | |
| ● | fatalities may occur during a clinical study due to medical problems that may or may not be related to clinical study treatments. |
Furthermore, the process of obtaining and maintaining regulatory approvals in the U.S. and other jurisdictions is lengthy, expensive and uncertain. It can vary substantially, based on the type, complexity and novelty of the product involved. Accordingly, any of our device candidates could take a significantly longer time than we expect to, or may never, gain regulatory approval, which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
Legislative and regulatory changes in the U.S. and in other countries regarding healthcare insurance and government-sponsored reimbursement programs (such as Medicare in the United States) may adversely affect our business and financial results.
We rely to a material degree on highly regulated private and government-run health insurance programs for our revenue in most of the countries in which we operate. The laws and regulations regarding health care programs, both public and private, are driven by public policy considerations that may be unrelated to the direct provision of patient care, such as lowering costs or requiring or limiting access to healthcare options. These laws and regulations are very complicated and there are many requirements we must satisfy in order for our products to become and remain eligible for reimbursement under these programs. In many cases we may have limited negotiating power when negotiating reimbursement rates for our products.
In the future, lawmakers and regulators could also pass additional healthcare laws and implement other regulatory changes at both the national and local levels. These laws and regulations could potentially affect coverage and reimbursement for our products. However, we cannot predict the ultimate content, timing or effect of any future healthcare initiatives or the impact any future legislation or regulation will have on us.
With respect to countries outside the U.S., the national competent authorities in the EU member states, the UK, Switzerland, Israel, Japan, and other jurisdictions are also increasingly active in their goal of reducing public spending on healthcare. We cannot, therefore, guarantee that the treatment of patients with our products would be reimbursed in any particular country or, if successfully included on reimbursement lists, whether we will remain on such lists.
We are subject to extensive post-marketing regulation by the FDA and comparable authorities in other jurisdictions, which could impact the sales and marketing of our products and could cause us to incur significant costs to maintain compliance. In addition, we may become subject to additional regulation in other jurisdictions if we market and sell our products outside of the U.S.
We market and sell our products subject to extensive regulation by the FDA and numerous other federal, state and governmental authorities in other jurisdictions. These regulations are broad and relate to, among other things, the conduct of pre-clinical and clinical studies, product design, development, manufacturing, labeling, testing, product storage and shipping, premarket clearance and approval, conformity assessment procedures, premarket clearance and approval of modifications introduced in marketed products, post-market surveillance and monitoring, reporting of adverse events and incidents, pricing and reimbursement, interactions with healthcare professionals, interactions with patients, information security, advertising and promotion and product sales and distribution. Although IB-Stim already has market clearance from FDA for functional abdominal pain associated with IBS in children, we will require additional FDA clearances to market our products for treating other indications.
| 49 |
In addition, before our products can be marketed in the EU, our products must obtain a CE Certificate from a notified body. New intended uses of CE marked medical devices falling outside the scope of the current CE Certificate require a completely new conformity assessment before the device can be CE marked and marketed in the EU for the new intended use. The process required to gather necessary information and draw up documentation in order to obtain CE Certification of a medical device in the EU can be expensive and lengthy and its outcome can be uncertain. We may make modifications to our products in the future that we believe do not or will not require notifications to our notified body or new conformity assessments to permit the maintenance of our current CE Certificate. If the competent authorities of the EU member states or our notified body disagree and require the conduct of a new conformity assessment, the modification of the existing CE Certificate or the issuance of a new CE Certificate, we may be required to recall or suspend the marketing of the modified versions of our products.
In Japan, new medical devices or new therapeutic uses of medical devices falling outside the scope of the existing approval by the MHLW require a new assessment and approval for each such new device or use. Accordingly, we may be required to obtain a new approval from MHLW before we launch a modified version of our products or the use of our products for additional indications. Approval time frames from the MHLW vary from simple notifications to review periods of one or more years, depending on the complexity and risk level of the device. In addition, importation into Japan of medical devices is subject to “Quality Management System (QMS) Ordinance,” which includes the equivalent of “Good Import” regulations in the U.S. As with any highly regulated market, significant changes in the regulatory environment could adversely affect our ability to commercialize our products in Japan.
In the U.S. and other jurisdictions, we also are subject to numerous post-marketing regulatory requirements, which include regulations under the QSR related to the manufacturing of our products, labeling regulations and medical device reporting regulations, which require us to report to the FDA or comparable regulatory authorities in other jurisdictions and our notified body if our products cause or contributes to a death or serious injury, or malfunction in a way that would likely cause or contribute to a death or serious injury. In addition, these regulatory requirements may in the future change in a way that adversely affects us. If we fail to comply with present or future regulatory requirements that are applicable to us, we may be subject to enforcement action by the FDA or comparable regulatory authorities in other jurisdictions and notified bodies, which may include any of the following sanctions:
| ● | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; | |
| ● | unanticipated expenditures to address or defend such actions; | |
| ● | patient notification, or orders for repair, replacement or refunds; | |
| ● | voluntary or mandatory recall, withdrawal or seizure of our current or future devices; | |
| ● | administrative detention by the FDA or other regulatory authority in another jurisdiction of medical devices believed to be adulterated or misbranded; | |
| ● | operating restrictions, suspension or shutdown of production; | |
| ● | refusal or delay of our requests for approval for new intended uses for or modifications to our products or for approval of new devices; | |
| ● | refusal or delay in obtaining CE Certificates for new intended uses for or modifications to our products; | |
| ● | suspension, variation or withdrawal of the CE Certificates granted by our notified body in the EU; | |
| ● | prohibition or restriction of products being placed on the market; | |
| ● | operating restrictions; | |
| ● | suspension or withdrawal of approvals that have already been granted; | |
| ● | refusal to grant export approval for our products or any device candidates; or | |
| ● | criminal prosecution. |
The occurrence of any of these events could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
| 50 |
Over time, we expect to make modifications to our products that are designed to improve efficacy, reduce side effects, enhance the user experience or for other purposes. Modifications to our products may require approvals, modified or new CE Certificates and analogous regulatory approvals in other jurisdictions or even require us to cease promoting or to recall the modified versions of our products until such clearances, approvals or modified or new CE Certificates are obtained, and the FDA, comparable regulatory authorities in other jurisdictions or our notified body may not agree with our conclusions regarding whether new approvals are required.
In addition, any substantial change introduced to a medical device or to the quality system certified by our notified body requires a new conformity assessment of the device and can lead to changes to the CE Certificates or the preparation of a new CE Certificate of Conformity. Substantial changes may include, among others, the introduction of a new intended use of the device, a change in its design or a change in the Company’s suppliers. Responsibility for determination that a modification constitutes a substantial change lies with the manufacturer of the medical device. We must inform the notified body that conducted the conformity assessment of the products we market or sell in the EU of any planned substantial changes to our quality system or changes to our products that could, among other things, affect compliance with the MDR or the devices’ intended use. The notified body will then assess the changes and verify whether they affect the product’s conformity with the Essential Requirements laid down in Annex I to the MDD or the conditions for the use of the device. If the assessment is favorable, the notified body will issue a new CE Certificate or an addendum to the existing CE Certificate attesting compliance with the Essential Requirements laid down in Annex I to the MDD. There is a risk that the competent authorities of the EU member states or our notified body may disagree with our assessment of the changes introduced to our products. The competent authorities of the EU member states or our notified body also may come to a different conclusion than the FDA on any given product modification.
In addition, medical devices that have obtained a CE Certification under the MDD may in principle continue to be marketed under such CE Certificate until the CE Certificate expires and at the latest until May 27, 2024, provided that the manufacturer complies with the MDR’s additional requirements related to post-marketing surveillance, market surveillance, vigilance, and registration of economic operators and of devices. However, if such medical devices undergo a significant change in their design or intended use, we would need to obtain a new CE Certificate under the MDR for these devices.
If the FDA disagrees with us and requires us to submit a new application for then-existing modifications and/or the competent authorities of the EU member states or our notified body disagree with our assessment of the change introduced in a product, its design or its intended use, we may be required to cease promoting or to recall the modified product until we obtain approval and/or until a new conformity assessment has been conducted in relation to the product, as applicable. In addition, we could be subject to significant regulatory fines or other penalties. Furthermore, our products could be subject to recall if the FDA, comparable regulatory authorities in other jurisdictions, or our notified body determine, for any reason, that our products are not safe or effective or that appropriate regulatory submissions were not made. Any recall or requirement that we seek additional approvals or clearances could result in significant delays, fines, increased costs associated with modification of a product, loss of revenues and potential operating restrictions imposed by the FDA, comparable foreign regulatory authorities in other jurisdictions, or our notified body. Delays in receipt or failure to receive approvals/certification, or the failure to comply with any other existing or future regulatory requirements, could reduce our sales, profitability and future growth prospects.
| 51 |
In addition to FDA requirements, we will spend considerable time and money complying with other federal, state, local and foreign rules, regulations and guidance and, if we are unable to fully comply with such rules, regulations and guidance, we could face substantial penalties.
We are subject to extensive regulation by the U.S. federal government and the states and other countries in which we conduct our business. U.S. federal government healthcare laws apply when we submit a claim on behalf of a U.S. federal healthcare program beneficiary, or when a customer submits a claim for an item or service that is reimbursed under a U.S. federal government-funded healthcare program, such as Medicare or Medicaid. The laws that affect our ability to operate our business in addition to the Federal Food, Drug, and Cosmetic Act and FDA regulations include, but are not limited to, the following:
| ● | the U.S. federal Anti-Kickback Statute, an intent-based federal criminal statute which prohibits knowingly and willfully offering, providing, soliciting or receiving remuneration of any kind to induce or reward, or in return for, referrals or the purchase, lease, order or recommendation or arranging of any items or services reimbursable by a federal healthcare program; | |
| ● | the Federal Civil False Claims Act, which imposes civil penalties, including through civil whistleblower or “qui tam” actions, for knowingly submitting or causing the submission of false or fraudulent claims of payment to the federal government, knowingly making, using or causing to be made or used a false statement or record material to payment of a false claim or avoiding, decreasing or concealing an obligation to pay money to the federal government; | |
| ● | the Federal Criminal False Claims Act, which is similar to the Federal Civil False Claims Act and imposes criminal liability on those that make or present a false, fictitious or fraudulent claim to the federal government; | |
| ● | Medicare laws and regulations that prescribe requirements for coverage and reimbursement, and laws prohibiting false claims or unduly influencing selection of products for reimbursement under Medicare and Medicaid; | |
| ● | healthcare fraud statutes that prohibit false statements and improper claims to any third-party payer; | |
| ● | the Federal Physician Self-Referral Law, commonly known as the Stark law, which, absent an applicable exception, prohibits physicians from referring Medicare and Medicaid patients to an entity for the provision of certain designated health services (“DHS”), if the physician (or a member of the physician’s immediate family) has an impermissible financial relationship with that entity and prohibits the DHS entity from billing for such improperly referred services; | |
| ● | the Federal Beneficiary Anti-Inducement Statute, which prohibits the offering of any remuneration to a beneficiary of Medicare or Medicaid that is likely to influence that beneficiary’s choice of provider or supplier. This can include, but is not limited to, inappropriate provision of patient services including financial assistance. Recent government investigations have focused on this particular prohibition. There are established exceptions from liability, but we cannot guarantee that all of our practices will fall squarely within those exceptions; | |
| ● | the U.S. Foreign Corrupt Practices Act, which can be used to prosecute companies in the U.S. for arrangements with physicians or other parties outside the U.S. if the physician or party is a government official of another country and the arrangement violates the law of that country; | |
| ● | the Federal Trade Commission Act, the Lanham Act and similar federal and state laws regulating truthfulness in advertising and consumer protection; and | |
| ● | the Federal Physician Payments Sunshine Act, the French Sunshine Act and similar state and foreign laws, which require periodic reporting of payments and other transfers of value made to U.S. and French-licensed physicians, teaching hospitals, and in the U.S., physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists, and certified nurse-midwives. |
Similar laws exist in the EU, individual EU member states and other countries. These laws are complemented by EU or national professional codes of practices.
HIPAA provides data privacy and security provisions for safeguarding medical information. Additionally, states in the U.S. are enacting local privacy laws (e.g., California). In the EU, the GDPR harmonizes data privacy laws and rules on the processing of personal data, including patient and employee data, across the EU. The GDPR has a number of strict data protection and security requirements for companies processing data of EU residents, including when such data is transferred outside of the EU. Additionally, we need to comply with analogous privacy laws in other jurisdictions in which we operate, such as the Israeli Privacy Protection Law, the Asia Pacific Economic Cooperation Privacy Framework, and Japan’s Act on the Protection of Personal Information.
The laws and codes of practices applicable to us are subject to evolving interpretations. Moreover, certain U.S. federal and state laws regarding healthcare fraud and abuse and certain laws in other jurisdictions regarding interactions with healthcare professionals and patients are broad and we may be required to restrict certain of our practices to be in compliance with these laws. Healthcare fraud and abuse laws also are complex and even minor, inadvertent irregularities, or even the perception of impropriety, can potentially give rise to claims that a statute has been violated.
| 52 |
Any violation of these laws could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Similarly, if there is a change in law, regulation or administrative or judicial interpretations, we may have to change our business practices or our existing business practices could be challenged as unlawful, which likewise could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline. Fines and penalties for violations of these laws and regulations could include severe criminal and civil penalties, including, for example, significant monetary damages, exclusion from participation in the federal healthcare programs and permanent disbarment of key employees. Any penalties, damages, fines, curtailment or restructuring of our operations would adversely affect our ability to operate our business, our prospects and our financial results. In addition, any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses, divert our management’s attention from the operation of our business and damage our reputation.
In addition, although we believe that we have the required licenses, permits and accreditation to dispense our products in the future, a regulator could find that we need to obtain additional licenses or permits. We also may be subject to mandatory reaccreditation and other requirements in order to maintain our billing privileges. Failure to satisfy those requirements could cause us to lose our privileges to bill governmental and private payers. If we are required to obtain permits or licenses that we do not already possess, we also may become subject to substantial additional regulation or incur significant expense.
To ensure compliance with Medicare, Medicaid and other regulations, federal and state governmental agencies and their agents, including MACs, may conduct audits of our operations to support our claims submitted for reimbursement of items furnished to beneficiaries and health care providers. Depending on the nature of the conduct found in such audits and whether the underlying conduct could be considered systemic, the resolution of these audits could adversely impact our revenue, financial condition and results of operations.
If we, our collaborative partners, our contract manufacturers or our component suppliers fail to comply with the FDA’s QSR or equivalent regulations established in other countries, the manufacturing and distribution of our products could be interrupted, and our product sales and results of operations could suffer.
We, our collaborative partners, our contract manufacturers and our component suppliers are required to comply with the FDA’s QSR and the equivalent quality system requirements imposed by the laws and regulations in other jurisdictions, which are a complex regulatory framework that covers the procedures and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our products. We cannot assure you that our facilities or our contract manufacturers’ or component suppliers’ facilities would pass any future quality system inspection. If our or any of our contract manufacturers’ or component suppliers’ facilities fails a quality system inspection, the manufacturing or distribution of our products could be interrupted and our operations disrupted. Failure to take adequate and timely corrective action in response to an adverse quality system inspection could force a suspension or shutdown of our packaging and labeling operations or the manufacturing operations of our contract manufacturers, and lead to suspension, variation or withdrawal of our regulatory approvals or a recall of our products. If any of these events occurs, we may not be able to provide our customers with our products on a timely basis, our reputation could be harmed and we could lose customers, any or all of which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
Our products may in the future be subject to recalls that could harm our reputation, business and financial results.
The FDA and similar governmental authorities in other jurisdictions have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture. In the case of the FDA, the authority to require a recall must be based on an FDA finding that there is a reasonable probability that the device would cause serious injury or death. In addition, governmental bodies in other jurisdictions have the authority to require the recall of our products in the event of material deficiencies or defects in design or manufacture. Distributors and manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. A government-mandated or voluntary recall by us or one of our manufacturers could occur as a result of component failures, manufacturing errors, design or labeling defects or other deficiencies and issues. The FDA requires that certain classifications of recalls be reported to the FDA within ten working days after the recall is initiated. Requirements for the reporting of product recalls to the competent authorities are imposed in other jurisdictions in which our products are or would be marketed in the future. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA or to the competent authorities of other countries. In the future, we may initiate voluntary recalls involving our products that we determine do not require notification of the FDA or to other equivalent non-U.S. authorities. If the FDA or the equivalent non-U.S. authorities disagree with our determinations, they could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA and the equivalent non-U.S. authorities could take enforcement action if we fail to report the recalls when they were conducted. Recalls of our products would divert managerial and financial resources and could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
| 53 |
If our products cause or contribute to a death or a serious injury, or malfunction in certain ways, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA Medical Device Reporting regulations and the equivalent regulations applicable in other jurisdictions in which our products are or may be marketed in the future, medical device manufacturers are required to report to the FDA and to the equivalent non-U.S. authorities information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. If we fail to report these events to the FDA or to the equivalent authorities in other jurisdictions within the required time frames, or at all, the FDA or the equivalent authorities in other jurisdictions could take enforcement action against us. Any such adverse event involving our products also could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results.
We may be subject to fines, penalties or injunctions if we are determined to be promoting the use of our products for unapproved or off-label uses.
Medical devices may be marketed only for the indications for which they are approved. Our promotional materials and training materials must comply with FDA regulations and other applicable laws and regulations governing the promotion of our products in the U.S. and other jurisdictions.
If the FDA or the competent authorities in other jurisdictions determine that our promotional materials or training constitutes promotion of an unapproved use, they could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions, including the issuance of an untitled or warning letter, an injunction, seizure, civil fines and criminal penalties. It is also possible that authorities in other federal, state or national enforcement in other jurisdictions might take action if they consider our promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. In that event, our reputation could be damaged and the commercialization of our products could be impaired.
We are affected by and subject to environmental laws and regulations that could be costly to comply with or that may result in costly liabilities.
We are subject to environmental laws and regulations, including those that impose various environmental controls on the manufacturing, transportation, storage, use and disposal of hazardous chemicals and other materials used in, and hazardous waste produced by, the manufacturing of our products. We incur and expect to continue to incur costs to comply with these environmental laws and regulations. Additional or modified environmental laws and regulations, including those relating to the manufacture, transportation, storage, use and disposal of materials used to manufacture our products or restricting disposal or transportation of batteries, may be imposed that may result in higher costs.
In addition, we cannot predict the effect that additional or modified environmental laws and regulations may have on us, our third-party suppliers of equipment and our products or our customers.
The pediatrics and medical device industries are characterized by patent and other intellectual property litigation and disputes, and any litigation, dispute or claim against us may cause us to incur substantial costs, could place a significant strain on our financial resources, divert the attention of management from our business, harm our reputation and require us to remove certain devices from the market.
Whether a product infringes a patent or violates other intellectual property rights involves complex legal and factual issues, the determination of which is often uncertain. Any intellectual property dispute, even a meritless or unsuccessful one, would be time consuming and expensive to defend and could result in the diversion of our management’s attention from our business and result in adverse publicity, the disruption of research and development and marketing efforts, injury to our reputation and loss of revenues. Any of these events could negatively affect our business, prospects, financial condition and results of operations.
| 54 |
Third parties may assert that our products, the methods employed in the use of our products or other activities infringe on their patents. Such claims may be made by competitors seeking to obtain a competitive advantage or by other parties, many of whom have significantly larger intellectual property portfolios than we have. Additionally, in recent years, individuals and groups have begun purchasing intellectual property assets for the purpose of making claims of infringement and attempting to extract settlements from companies like ours. With respect to our current products, the risk of infringement claims is exacerbated by the fact that there are numerous issued and pending patents relating to the treatment of cancer. Because patent applications can take many years to issue, and in many cases remain unpublished for many months after filing, there may be applications now pending of which we are unaware that may later result in issued patents that our products may infringe.
There could also be existing patents that one or more components of our products or other device candidates may inadvertently infringe. As the number of competitors in the market or other device candidates grows, the possibility of inadvertent patent infringement by us or a patent infringement claim against us increases. To the extent we gain greater market visibility, our risk of being subject to such claims is also likely to increase. If a third party’s patent was upheld as valid and enforceable and we were found to be infringing, we could be prevented from making, using, selling, offering to sell or importing our products or other device candidates, unless we were able to obtain a license under that patent or to redesign our systems to avoid infringement. A license may not be available at all or on terms acceptable to us, and we may not be able to redesign our products to avoid any infringement. Modification of our products or development of device candidates to avoid infringement could require us to conduct additional clinical studies and to revise our filings with the FDA and other regulatory bodies, which would be time-consuming and expensive. If we are not successful in obtaining a license or redesigning our devices, we may be unable to make, use, sell, offer to sell or import our devices and our business could suffer. We may also be required to pay substantial damages and undertake remedial activities, which could cause our business to suffer.
We may also be subject to claims alleging that we infringe or violate other intellectual property rights, such as copyrights or trademarks, may have to defend against allegations that we misappropriated trade secrets, and may face claims based on competing claims of ownership of our intellectual property. The confidentiality and assignment of inventions agreements that our employees, consultants and other third parties sign may not in all cases be enforceable or sufficient to protect our intellectual property rights. In addition, we may face claims from third parties based on competing claims to ownership of our intellectual property.
We may employ individuals who were previously employed at other medical device companies, and as such we may be subject to claims that such employees have inadvertently or otherwise used or disclosed the alleged trade secrets or other proprietary information of their former employers. Any such litigation, dispute or claim could be costly to defend and could subject us to substantial damages, injunctions or other remedies, which could have a material adverse effect on our business, prospects, financial condition and results of operations and cause our stock price to decline.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our devices.
As is the case with other medical device companies, our success is heavily dependent on our intellectual property rights, and particularly on our patent rights. Obtaining and enforcing patents in the medical device industry involves both technological and legal complexity, and is therefore costly, time consuming and inherently uncertain. In addition, the U.S. has recently enacted and is currently implementing wide-ranging patent reform legislation. Certain U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents once obtained. Depending on decisions by the U.S. Congress, the federal courts and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could further negatively impact the value of our patents, narrow the scope of available patent protection or weaken the rights of patent owners.
| 55 |
Future regulatory action remains uncertain.
We operate in a highly regulated and evolving environment with rigorous regulatory enforcement. Any legal or regulatory action could be time-consuming and costly. If we or the manufacturers or distributors that supply our products fail to comply with all applicable laws, standards, and regulations, action by the FDA or other regulatory agencies could result in significant restrictions, including restrictions on the marketing or use of the products we sell or the withdrawal of the products we sell from the market. Any such restrictions or withdrawals could materially affect our reputation, business and operations.
Our product candidates will remain subject to ongoing regulatory review even after they receive marketing clearances, and if we fail to comply with continuing regulations, we could lose these clearances and the sale of any of our approved commercial products could be suspended.
Even as we received regulatory clearance to market the IB-Stim, the manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion, and record keeping related to IB-Stim will remain subject to extensive regulatory requirements. If we fail to comply with the regulatory requirements of the FDA and other applicable domestic and foreign regulatory authorities or discover any previously unknown problems with any approved product, manufacturer, or manufacturing process, we could be subject to administrative or judicially imposed sanctions, including:
| ● | restrictions on the products, manufacturers, or manufacturing processes; | |
| ● | warning letters; | |
| ● | civil or criminal penalties; | |
| ● | fines; | |
| ● | injunctions; | |
| ● | product seizures or detentions; | |
| ● | pressure to initiate voluntary product recalls; | |
| ● | suspension or withdrawal of regulatory clearances and/or approvals; and | |
| ● | refusal to approve pending applications for marketing clearances and/or approval of new products or supplements to approved applications. |
Intellectual property litigation and infringement claims could cause us to incur significant expenses or prevent us from selling certain of our products.
The therapeutic medical device and pharmaceutical industries are characterized by extensive intellectual property litigation and, from time to time, we may become the subject of claims of infringement or misappropriation. Regardless of outcome, such claims are expensive to defend and divert management and operating personnel from other business issues. A successful claim or claims of patent or other intellectual property infringement against us could result in payment of significant monetary damages and/or royalty payments or negatively impact our ability to sell current or future products in the affected category.
We depend extensively on our patents and proprietary technology and the patents, and we must protect those assets in order to preserve our business.
Although we expect to seek patent protection for any devices, in silico products (if any), systems, and processes we discover and/or for any specific use we discover for new or previously known compounds, devices, biologics, products, systems, or processes, any or all of these may not be subject to effective patent protection. In addition, our issued patents may be declared invalid or our competitors may find ways to avoid the claims in the patents.
Our success will depend, in part, on our ability to obtain patents, protect our trade secrets and proprietary knowledge and operate without infringing on the proprietary rights of others. We are the sole assignee of numerous granted United States patents, pending United States patent applications and international patents. The patent position of pharmaceutical and biotechnology firms like us are generally highly uncertain and involves complex legal and factual questions, resulting in both an apparent inconsistency regarding the breadth of claims allowed in United States patents and general uncertainty as to their legal interpretation and enforceability. Accordingly, patent applications assigned to us may not result in patents being issued, any issued patents assigned to us may not provide us with competitive protection or may be challenged by others, and the current or future granted patents of others may have an adverse effect on our ability to do business and achieve profitability.
| 56 |
Moreover, others may independently develop similar products, may duplicate our products, or may design around our patent rights. In addition, as a result of the assertion of rights by a third-party or otherwise, we may be required to obtain licenses to patents or other proprietary rights of others in or outside of the United States. Any licenses required under any such patents or proprietary rights may not be made available on terms acceptable to us, if at all. If we do not obtain such licenses, we could encounter delays in product market introductions during our attempts to design around such patents or could find that the development, manufacture or sale of products requiring such licenses is foreclosed. In addition, we could incur substantial costs in defending suits brought against us or in connection with patents to which we hold licenses or in bringing suit to protect our own patents against infringement.
Due to legal and factual uncertainties regarding the scope and protection afforded by patents and other proprietary rights, we may not have meaningful protection from competition.
Our long-term success will substantially depend upon our ability to protect our proprietary technologies from infringement, misappropriation, discovery and duplication, and avoid infringing the proprietary rights of others. Our patent rights and the patent rights of biotechnology and pharmaceutical companies in general, are highly uncertain and include complex legal and factual issues. Because of this, our pending patent applications may not be granted. These uncertainties also mean that any patents that we own or will obtain in the future could be subject to challenge, and even if not challenged, may not provide us with meaningful protection from competition. Due to our financial uncertainties, we may not possess the financial resources necessary to enforce our patents. Patents already issued to us or our pending applications may become subject to dispute, and any dispute could be resolved against us. Because a substantial number of patents have been issued in the field of neuromodulation therapy and because patent positions can be highly uncertain and frequently involve complex legal and factual questions, the breadth of claims obtained in any application or the enforceability of our patents cannot be predicted. Consequently, we do not know whether any of our pending or future patent applications will result in the issuance of patents or, to the extent patents have been issued or will be issued, whether these patents will be subject to further proceedings limiting their scope, will provide significant proprietary protection or competitive advantage, or will be circumvented or invalidated.
Also, because of these legal and factual uncertainties, and because pending patent applications are held in secrecy for varying periods in the United States and other countries, even after reasonable investigation, we may not know with certainty whether any products that we (or a licensee) may develop will infringe upon any patent or other intellectual property right of a third party. We believe that the patents that we own or have applied for do not infringe any third-party patents; however, we cannot know for certain whether we could successfully defend our position, if challenged. We may incur substantial costs if we are required to defend our intellectual property in patent suits brought by third parties. These legal actions could seek damages and seek to enjoin testing, manufacturing and marketing of the accused product or process. In addition to potential liability for significant damages, we could be required to obtain a license to continue to manufacture or market the accused product or process.
If the third parties on which we rely for the conduct of our clinical trials and results do not perform our clinical trial activities in accordance with good clinical practices and related regulatory requirements, we may be unable to obtain regulatory approval for or commercialize our product candidates.
We may use independent clinical investigators and other third-party service providers to conduct and/or oversee the clinical trials of our product candidates for the foreseeable future.
The FDA requires us and our clinical investigators to comply with regulations and standards, commonly referred to as good clinical practices, for conducting, recording, and reporting the results of clinical trials to assure that data and reported results are credible and accurate, and that the trial participants are adequately protected. Our reliance on third parties that we do not control does not relieve us of these responsibilities and requirements. Third parties may not complete activities on schedule or may not conduct our clinical trials in accordance with regulatory requirements or the respective trial plans and protocols. The failure of these third parties to carry out their obligations could delay or prevent the development, approval, and commercialization of our product candidates or result in enforcement action against us.
| 57 |
Risks Related to Our Common Stock
We may not be able to maintain a listing of our common stock on NYSE American.
Our common stock is listed on NYSE American. We must meet certain financial and liquidity criteria to maintain such listing. If we violate NYSE American’s listing requirements, or if we fail to meet any of NYSE American’s listing standards, our common stock may be delisted. In addition, our board of directors may determine that the cost of maintaining our listing on a national securities exchange outweighs the benefits of such listing. A delisting of our common stock from NYSE American may materially impair our stockholders’ ability to buy and sell our common stock and could have an adverse effect on the market price of, and the efficiency of the trading market for, our common stock. The delisting of our common stock could significantly impair our ability to raise capital and the value of your investment.
We do not expect to declare or pay dividends in the foreseeable future.
We do not expect to declare or pay dividends in the foreseeable future, as we anticipate that we will invest future earnings in the development and growth of our business. Therefore, holders of our common stock will not receive any return on their investment unless they sell their securities, and holders may be unable to sell their securities on favorable terms or at all.
If securities industry analysts do not publish research reports on us, or publish unfavorable reports on us, then the market price and market trading volume of our common stock could be negatively affected.
Any trading market for our common stock may be influenced in part by any research reports that securities industry analysts publish about us. We do not currently have and may never obtain research coverage by securities industry analysts. If no securities industry analysts commence coverage of us, the market price and market trading volume of our common stock could be negatively affected. In the event we are covered by analysts, and one or more of such analysts downgrade our securities, or otherwise reports on us unfavorably, or discontinues coverage of us, the market price and market trading volume of our common stock could be negatively affected.
Future issuances of our common stock or securities convertible into, or exercisable or exchangeable for, our common stock, or the expiration of lock-up agreements that restrict the issuance of new common stock or the trading of outstanding common stock, could cause the market price of our common stock to decline and would result in the dilution of your holdings.
Future issuances of our common stock or securities convertible into, or exercisable or exchangeable for, our common stock, or the expiration of lock-up agreements that restrict the issuance of new common stock or the trading of outstanding common stock, could cause the market price of our common stock to decline. We cannot predict the effect, if any, of future issuances of our securities, or the future expirations of lock-up agreements, on the price of our common stock. In all events, future issuances of our common stock would result in the dilution of your holdings. In addition, the perception that new issuances of our securities could occur, or the perception that locked-up parties will sell their securities when the lock-ups expire, could adversely affect the market price of our common stock. In addition to any adverse effects that may arise upon the expiration of these lock-up agreements, the lock-up provisions in these agreements may be waived, at any time and without notice. If the restrictions under the lock-up agreements are waived, our common stock may become available for resale, subject to applicable law, including without notice, which could reduce the market price for our common stock.
| 58 |
Future issuances of debt securities, which would rank senior to our common stock upon our bankruptcy or liquidation, and future issuances of preferred stock, which could rank senior to our common stock for the purposes of dividends and liquidating distributions, may adversely affect the level of return you may be able to achieve from an investment in our common stock.
In the future, we may attempt to increase our capital resources by offering debt securities. Upon bankruptcy or liquidation, holders of our debt securities, and lenders with respect to other borrowings we may make, would receive distributions of our available assets prior to any distributions being made to holders of our common stock. Moreover, if we issue preferred stock, the holders of such preferred stock could be entitled to preferences over holders of common stock in respect of the payment of dividends and the payment of liquidating distributions. Because our decision to issue debt or preferred stock in any future offering, or borrow money from lenders, will depend in part on market conditions and other factors beyond our control, we cannot predict or estimate the amount, timing or nature of any such future offerings or borrowings. Holders of our common stock must bear the risk that any future offerings we conduct or borrowings we make may adversely affect the level of return, if any, they may be able to achieve from an investment in our common stock.
If our shares of common stock become subject to the penny stock rules, it would become more difficult to trade our shares.
The SEC has adopted rules that regulate broker-dealer practices in connection with transactions in penny stocks. Penny stocks are generally equity securities with a price of less than $5.00, other than securities registered on certain national securities exchanges or authorized for quotation on certain automated quotation systems, provided that current price and volume information with respect to transactions in such securities is provided by the exchange or system. If we do not retain a listing on NYSE American or another national securities exchange and if the price of our common stock is less than $5.00, our common stock could be deemed a penny stock. The penny stock rules require a broker-dealer, before a transaction in a penny stock not otherwise exempt from those rules, to deliver a standardized risk disclosure document containing specified information. In addition, the penny stock rules require that before effecting any transaction in a penny stock not otherwise exempt from those rules, a broker-dealer must make a special written determination that the penny stock is a suitable investment for the purchaser and receive (i) the purchaser’s written acknowledgment of the receipt of a risk disclosure statement; (ii) a written agreement to transactions involving penny stocks; and (iii) a signed and dated copy of a written suitability statement. These disclosure requirements may have the effect of reducing the trading activity in the secondary market for our common stock, and therefore stockholders may have difficulty selling their shares.
We are subject to ongoing public reporting requirements that are less rigorous than Exchange Act rules for companies that are not emerging growth companies, and our stockholders could receive less information than they might expect to receive from more mature public companies.
We are required to publicly report on an ongoing basis as an “emerging growth company” (as defined in the JOBS Act) under the reporting rules set forth under the Exchange Act. For so long as we remain an emerging growth company, we may take advantage of certain exemptions from various reporting requirements that are applicable to other Exchange Act reporting companies that are not emerging growth companies, including but not limited to:
| ● | not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act; | |
| ● | being permitted to comply with reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements; and | |
| ● | being exempt from the requirement to hold a non-binding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. |
We expect to take advantage of these reporting exemptions until we are no longer an emerging growth company. We would remain an emerging growth company for up to five years, although if the market value of our common stock that is held by non-affiliates exceeds $700 million as of any June 30 before that time, we would cease to be an emerging growth company as of the following December 31.
Because we are subject to ongoing public reporting requirements that are less rigorous than Exchange Act rules for companies that are not emerging growth companies, our stockholders could receive less information than they might expect to receive from more mature public companies. We cannot predict if investors will find our common stock less attractive if we elect to rely on these exemptions, or if taking advantage of these exemptions would result in less active trading or more volatility in the price of our common stock.
| 59 |
Because the Company is a “smaller reporting company,” we may take advantage of certain scaled disclosures available to us, resulting in holders of our securities receiving less Company information than they would receive from a public company that is not a smaller reporting company.
We are a “smaller reporting company” as defined in the Exchange Act. As a smaller reporting company, we may take advantage of certain of the scaled disclosures available to smaller reporting companies and will be able to take advantage of these scaled disclosures for so long as (i) our common stock held by non-affiliates is less than $250 million measured on the last business day of our second fiscal quarter, or (ii) our annual revenue is less than $100 million during the most recently completed fiscal year and our common stock held by non-affiliates is less than $700 million measured on the last business day of our second fiscal quarter. To the extent we take advantage of any reduced disclosure obligations, it may make it harder for investors to analyze the Company’s results of operations and financial prospectus in comparison with other public companies.
As a smaller reporting company, we are permitted to comply with scaled-back disclosure obligations in our SEC filings compared to other issuers, including with respect to disclosure obligations regarding executive compensation in our periodic reports and proxy statements. We have elected to adopt the accommodations available to smaller reporting companies. Until we cease to be a smaller reporting company, the scaled-back disclosure in our SEC filings will result in less information about our company being available than for other public companies.
If investors consider our common stock less attractive as a result of our election to use the scaled-back disclosure permitted for smaller reporting companies, there may be a less active trading market for our common stock and our share price may be more volatile.
As a “smaller reporting company,” we may at some time in the future choose to exempt our Company from certain corporate governance requirements that could have an adverse effect on our public shareholders.
Under NYSE American rules, a “smaller reporting company,” as defined in Rule 12b-2 under the Exchange Act, is not subject to certain corporate governance requirements otherwise applicable to companies listed on NYSE American. For example, a smaller reporting company is exempt from the requirement of having a compensation committee composed solely of directors meeting certain enhanced independence standards, as long as the compensation committee has at least two members who do meet such standards. Although we have determined not to avail ourselves of this or other exemptions from NYSE American requirements that are or may be afforded to smaller reporting companies while we will seek to maintain our shares on NYSE American, in the future we may elect to rely on any or all of these exemptions. By electing to utilize any such exemptions, our Company may be subject to greater risks of poor corporate governance, poorer management decision-making processes, and reduced results of operations from problems in our corporate organization. Consequently, if we were to avail ourselves of these exemptions, our stock price might suffer, and there is no assurance that we would be able to continue to meet all continuing listing requirements of NYSE American from which we would not be exempt, including minimum stock price requirements.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 1C. CYBERSECURITY
CYBERSECURITY RISK MANAGEMENT AND STRATEGY. NeurAxis has developed and implemented a cybersecurity framework intended to assess, identify and manage risks from threats to the security of our information, systems, products and network using a risk-based approach. The framework is informed in part by the National Institute of Standards and Technology (NIST) Cybersecurity Framework and International Organization for Standardization 27001 (ISO 27001) Framework, although this does not imply that we meet all technical standards, specifications, or requirements under the NIST or ISO 27001.
Our key cybersecurity processes include the following:
| ● | Risk-based controls for information systems and information on NeurAxis’ networks: We seek to maintain an information technology infrastructure that implements physical, administrative and technical controls that are calibrated based on risk and designed to protect the confidentiality, integrity and availability of our information systems and information stored on NeurAxis’ networks, including customer information, personal information, PHI/PII, intellectual property and proprietary information. |
| ● | Cybersecurity incident policies: We have cybersecurity incident policies, an incident response plan and a dedicated team to respond to cybersecurity incidents, including experienced counsel. When a cybersecurity incident occurs or we identify a vulnerability, the dedicated team is responsible for leading the initial assessment of priority and severity, including external experts that may also be engaged as appropriate. NeurAxis’ response to incidents depends on the severity level and seeks to improve its cybersecurity incident response plan. NeurAxis, through experienced cybersecurity and HIPAA/HITECH counsel, has developed a security manual and a privacy policy. |
| 60 |
| ● | Training: We provide security awareness training to help our employees understand their information protection and cybersecurity responsibilities at NeurAxis. We also provide additional role-based training to some employees based on customer requirements, regulatory obligations, and industry risks. |
| ● | Supplier risk assessments: We have participated in several third-party risk assessment processes that include expectations regarding information and cybersecurity. NeurAxis also seeks contractual commitments from key suppliers to appropriately secure and maintain their information technology systems and protect NeurAxis information that is processed or stored on their systems. This may or may not include business associate agreements, downstream vendor agreements and vendor auditing in some cases. |
| ● | Third-party assessments of NeurAxis: We have third-party cybersecurity companies engaged to assess NeurAxis’ cybersecurity readiness and to assist in identifying and remediating risks from cybersecurity threats. NeurAxis has a “real time” cybersecurity partner that monitors our servers 24/7/365 for any attempted intrusions. |
We have not identified risks from known cybersecurity threats, that have materially affected us, including our operations, business strategy, results of operations, cash flows or financial condition. We face certain ongoing risks from cybersecurity threats, including active interactions with children’s hospitals while assisting with insurance prior approvals, that, if realized, are reasonably likely to materially affect us, including our operations, business strategy, results of operations, cash flows or financial condition.
CYBERSECURITY GOVERNANCE. NeurAxis’ Board of Directors is responsible for oversight of cybersecurity risk. The Board receives reporting about NeurAxis’ practices, programs, notable threats or incidents and other developments related to cybersecurity throughout the year, including through periodic updates from NeurAxis’ Chief Regulatory Officer/Privacy Officer and VP of IT/Security Officer.
NeurAxis’ Security Officer reports to NeurAxis’ Chief Regulatory Officer and together, they lead the Company’s overall cybersecurity function. The Security Officer has over 12 years of experience in managing and leading IT or cybersecurity teams and participates in various cyber security trainings frequently. The Security Officer collaborates with NeurAxis personnel and our outside IT vendors to identify and analyze cybersecurity risks to NeurAxis, considers industry trends, implement controls, as appropriate and feasible, to mitigate these risks and enables business leaders to make risk-based business decisions that impact cybersecurity considerations. The Security Officer meets with senior leadership to review and discuss NeurAxis’ cybersecurity program, including emerging cyber risks, threats, and industry trends. The Security Officer also supervises efforts to prevent, detect, mitigate, and remediate cybersecurity risks and incidents through various means, including by collaborating with external security personnel and business stakeholders, and incorporating threat intelligence and other information obtained from governmental, public, or private sources to strengthen our cybersecurity technologies and processes.
ITEM 2. PROPERTIES
Our corporate headquarters are located in Carmel, Indiana, where we lease office space for employees, pursuant to a lease agreement with SePRO Development Company II, LLC. The term of this lease commenced on March 1, 2021 and ended on February, 28, 2024. Over the term of the lease, monthly base rent was $1,676 through March 1, 2023 and $1,717 thereafter. Prior to the end of the lease term, the Company moved its corporate headquarters to another location in Carmel, Indiana, pursuant to a lease agreement with Zeller-Carmel Property, LLC. The term of this lease commenced on January 1, 2024 and is scheduled to end on May 31, 2029. Over the term of the lease, the monthly base rent is $6,721 with an annual 2.5% escalator. We received a 50% reduction in our monthly rent for the first 10 months of the lease.
We also lease office space in Versailles, Indiana, pursuant to two lease agreements with Hashbo Properties, LLC. The original lease term of each agreement commenced January 1, 2022 for a period of one year with automatic one year renewals unless termination notice is provided 60 days in advance. The monthly base rent is $2,308 with an annual 4% escalator upon renewal.
| 61 |
ITEM 3. LEGAL PROCEEDINGS
From time to time, the Company may be involved in litigation relating to claims arising out of operations in the normal course of business. As of April 16, 2024, other than those described below, there were no pending or threatened legal proceedings that could reasonably be expected to have a material effect on the results of the Company’s operations. There are also no proceedings in which any of the Company’s directors, officers or affiliates is an adverse party to the Company or has a material interest adverse to the Company’s interest.
On February 6, 2019, plaintiff Ritu Bhambhani, M.D., initiated a lawsuit against Innovative Health Solutions, Inc. and others in the United States District Court for the District of Maryland. Plaintiffs Bhambhani and Sudhir Rao subsequently amended the complaint, with the Third Amended Complaint (“Complaint”) containing the most recent set of allegations. The Complaint asserted claims under the RICO Act, as well as of fraudulent misrepresentation, intentional misrepresentation by concealment, and civil conspiracy and sought compensatory damages in excess of $5 million, pre-judgment interest, punitive damages, attorney’s fees, court costs and designation of the case as a class action. The Complaint states that the Company, distributors of the Company’s product, and medical billing and coding consultants allegedly made misrepresentations to the plaintiffs that the Company’s NeuroStim device and related procedures could be billed to, and reimbursed by, Medicare and other insurance payors as a surgically implantable neurostimulator. Plaintiffs claim to have suffered damages when Medicare administrative contractors declined to pay plaintiffs for their use of the device.
On February 11, 2022, the Company filed a motion for summary judgment based upon the plaintiffs not being proper parties to assert claims against the Company. On June 14, 2022, the Court granted the Company’s motion for summary judgment and dismissed the Complaint.
On July 14, 2022, plaintiffs Ritu Bhambhani and Sudhir Rao filed a notice of appeal with the Fourth Circuit Court of Appeals. The Company filed a motion to dismiss. On January 4, 2023, the Court issued an order that stated it was deferring a ruling on the motion to dismiss the appeal and that it would address those arguments at the same time that it addressed the substantive merits of the case. While it is too early to predict the ultimate outcome of this matter, we continue to believe we have meritorious defenses, that the dismissal of the Complaint should be upheld, and intend to continue to defend this matter vigorously.
On July 14, 2022, plaintiffs Ritu Bhambhani, LLC; Box Hill Surgery Center, LLC; Pain and Spine Specialists of Maryland, LLC; and SimCare ASC, LLC initiated a lawsuit against the Company and others in the United States District Court for the District of Maryland. The plaintiffs in this lawsuit are business entities owned or partially owned by the plaintiffs that initiated the litigation described above. The Complaint asserted claims under the RICO Act, as well as fraudulent misrepresentation, intentional misrepresentation by concealment, and civil conspiracy and seeks compensatory damages in excess of $75,000, pre-judgment interest, punitive damages, attorney’s fees, and court costs. The Complaint states that the Company, distributors of the Company’s product, and medical billing and coding consultants allegedly made misrepresentations to the plaintiffs that the Company’s NeuroStim device and related procedures could be billed to, and reimbursed by, Medicare and other insurance payors as a surgically implantable neurostimulator. Plaintiffs claim to have suffered damages when Medicare administrative contractors declined to pay plaintiffs for their use of the device.
On September 28, 2022, the Company filed a motion to dismiss all claims. On May 25, 2023, the Court issued an Order and a Memorandum Opinion which dismissed the plaintiffs’claims related to the RICO Act. The remaining claims are still pending, and no trial date has been set for the case.
The Court has vacated its Scheduling Order at the parties’ request so that the parties could try to resolve the disputes in both cases through an independent third-party mediator. No mediation date has been set. While it is too early to predict the ultimate outcome of this matter, we believe the Company has meritorious defenses and intends to defend this matter vigorously.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
| 62 |
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information and Holders of Common Stock
Our common stock is listed on the NYSE American, under the symbol “NRXS”.
As of April 12, 2024, there were approximately 425 record holders of our common stock. The number of record holders does not include beneficial owners of common stock whose shares are held in the names of banks, brokers, nominees or other fiduciaries and holders of unissued shares common stock.
The last reported sales price for our Common Stock as reported on the NYSE American on April 12, 2024 was $3.10.
Dividends
We have not declared or paid any cash dividends on our common stock, and we do not anticipate declaring or paying cash dividends for the foreseeable future. We are not subject to any legal restrictions respecting the payment of dividends, except that we may not pay dividends if the payment would render us insolvent. Any future determination as to the payment of cash dividends on our common stock will be at the discretion of our board of directors and will depend on our financial condition, operating results, capital requirements and other factors that the board of directors considers to be relevant.
Recent Sales of Unregistered Securities
On November 9, 2023, the Company entered into a securities purchase agreement (the “SPA”) with Flagstaff International, LLC (“Flagstaff International”) for the issuance and purchase of the Company’s Series B Convertible Preferred Stock, par value $0.001 per share (the “Series B Preferred Stock Financing”), at price per share of $2.38 (the “Series B Preferred Financing”). The aggregate purchase price is $3,000,000, to be paid in 15 monthly installments of $200,000 each, commencing on the later of January 10, 2024 or a date after stockholders approve of an amendment to the Company’s Certificate of Incorporation to authorize the creation of the Series B Preferred Stock (the “Stockholder Approval”). The Series B Preferred Stock is convertible at any time into shares of common stock of the Company without any further consideration. The closing of this transaction is subject to the Stockholder Approval following which the Company will file the Series B Preferred Stock Certificate of Designation with the Secretary of State of the State of Delaware. Following the issuance of the Series B Preferred Stock, it will rank senior to the common stock with respect to payments upon the liquidation, dissolution and winding up of the Company. As of December 31, 2023, Flagstaff International, LLC has not yet paid for or been issued any Series B Preferred Stock shares.
The Company previously issued (i) warrants exercisable for an aggregate of 793,655 shares of common stock with an exercise price of $5.25 per share to Leonite Fund I, LP, Emmis Capital II, LLC, Bigger Capital Fund, LP, District 2 Capital Fund, LP, and Exchange Listing, from June through November 2022 (the “2022 Warrants”), (ii) warrants exercisable for an aggregate of 505,570 shares of common stock with a strike price at $5.25 per share to certain investors from March through July 2023 (the “the 2023 Warrants”), and (iii) 120,235 warrants to the placement agent for the 2023 financing with the same terms as the 2023 Warrants (the “PA Warrants”, and together with the 2022 Warrants and the 2023 Warrants, the “Warrants”). In connection with the Series B Preferred Stock Financing, pursuant to Section 2(c) of the 2022 Warrants and pursuant to Section 3(b) of the 2023 Warrants and the PA Warrants, as a result of the execution of the SPA, the Company adjusted the exercise price of each Warrant to $2.38 per share and issued (i) new 2023 Warrants to purchase 609,760 shares of common stock and (ii) new PA Warrants to purchase 144,990 shares of common stock pursuant to Section 3(b) of such warrants. The number of shares of the Company’s common stock issuable upon exercise of the 2022 Warrants remain unchanged.
ITEM 6. RESERVED
| 63 |
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
We are a growth stage company focused on developing neuromodulation therapies to address chronic and debilitating conditions in children. Our mission is to provide solutions that create value and provide better and safer patient outcomes. Our IB-Stim device is a PENFS system intended to be used in patients 11-18 years of age with functional abdominal pain associated with IBS. Our device already has market clearance from FDA for functional abdominal pain associated with IBS in children. Other indications in our pipeline are comprised of functional nausea in children, post-concussion syndrome in children, and cyclic vomiting syndrome in children. For more information, see “Business—Our Pipeline” and “—Products.”
Since our inception, we have incurred significant operating losses. Our net loss was $14,626,683 and $4,780,061 million for the years ended December 31, 2023 and 2022, respectively. Our auditors have expressed substantial doubt about our ability to continue as a going concern in their audit opinion. We expect to incur significant expenses and operating losses for the foreseeable future as we continue to pursue widespread insurance coverage of our IB-Stim device and seek FDA clearance of our device for other indications. There are a number of milestones and conditions that we must satisfy before we will be able to generate sufficient revenue to fund our operations, including FDA clearance of our IB-Stim device to treat future indications.
Factors Affecting our Business and Results of Operations
Revenue
Our revenue is derived from the sale of our IB-Stim device to healthcare companies, primarily hospitals and clinics. Sales generally are not seasonal and only mildly correlated with economic cycles. Our IB-Stim device sells for $1,195 per device, and each child being treated for functional abdominal pain associated with IBS will use three to four devices. Potential patients with future indications are expected to use six or more devices per patient.
Our sales typically are made on a purchase order basis rather than through long-term purchase commitments. We enter into sales agreements with customers for IB-Stim devices based on purchase orders and standard terms, which vary slightly based on the customer’s form, and conditions of sale. Standard payment terms generally are that payment is due within 30 days. Our largest sales were to three hospitals representing approximately 45% and 52% of total sales for the years ended December 31, 2023 and 2022, respectively.
Inflation did not have a material impact on our operations for any applicable period, and we do not expect inflation to have a material impact on our operations for the foreseeable future.
Expenses
We have four categories of expenses: cost of goods sold, selling expenses, research and development (“R&D”), and general and administrative (“G&A”).
Costs of goods sold consist of costs paid for the IB-Stim device to our contract manufacturer along with periodic inventory adjustments and expired inventory write-offs. We ramped up production in 2018 and 2019 to meet expected demand and avoid any inventory shortages. This resulted in some excess inventory that did expire. The costs of expired inventory were $25,008 and $10,026 for the years ended December 31, 2023 and 2022, respectively, representing 8.2% and 3.4% of our costs of goods sold, respectively. Expired inventory expense is related to our FDA clearance for our device in the treatment of functional abdominal pain associated with IBS in children. Specifically, a certain component of our IB-Stim device is cleared for a two-year period after the date the device is manufactured, and if the device is not sold in such period, we must take the device out of inventory and write it off. Expired inventory has not been material to our results, averaging 1% or less of revenue over the past five years. We have a fixed-price contract with the manufacturer of our IB-Stim device to produce the device. We expect production costs to remain relatively constant and only nominal inventory expirations in the foreseeable future.
| 64 |
Our core selling expenses primarily consist of commissions and shipping costs. These expense items are generally correlated with sales.
Research and development expense is attributable to our clinical trials and related efforts to have our IB-Stim device cleared by the FDA for other indications.
General and administrative expense primarily consists of wages and benefits, professional fees including legal and audit, insurance, investor relations, advertising, facility costs, utilities and travel.
Gross Profit and Gross Margin
Our management uses gross profit and gross margin to evaluate the efficiency of operations and as a key component to determining the effectiveness and allocation of resources. We calculate gross profit as revenue less cost of goods sold, and gross margin as gross profit divided by revenue. Our gross margin has been and will continue to be affected by a variety of factors, primarily the average selling price of our IB-Stim device, production volume, order flows, change in mix of customers, third-party manufacturing costs related to components of our IB-Stim device, and cost-reduction strategies. We expect our gross profit to increase for the foreseeable future as our revenue grows, both through broader insurer acceptance of our IB-Stim device in the near term and approval of our technology for the treatment of other indications over the longer term. Our gross margin may fluctuate from quarter to quarter due to changes in average selling prices, particularly as we introduce enhancements to our IB-Stim device and new products to address other indications, and as we adopt new manufacturing processes and technologies.
Results of Operations
Comparison of Year Ended December 31, 2023 and Year Ended December 31, 2022
The following table presents our statements of operations for the years ended December 31, 2023 and 2022:
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Net sales | $ | 2,460,049 | $ | 2,684,735 | ||||
| Cost of goods sold | 303,345 | 297,060 | ||||||
| Gross profit | 2,156,704 | 2,387,675 | ||||||
| Selling expenses | 323,569 | 410,883 | ||||||
| Research and development | 169,315 | 225,610 | ||||||
| General and administrative | 8,328,315 | 5,123,420 | ||||||
| Operating loss | (6,664,495 | ) | (3,372,238 | ) | ||||
| Other (expense) income, net: | ||||||||
| Financing charges | (2,772 | ) | (2,322,216 | ) | ||||
| Interest expense | (476,416 | ) | (318,666 | ) | ||||
| Change in fair value of warrant liability | 844,854 | 606,049 | ||||||
| Change in fair value of derivative financial instruments | 198,551 | 713,989 | ||||||
| Amortization of debt discount and issuance cost | (4,881,622 | ) | (98,935 | ) | ||||
| Extinguishment of debt liabilities | (3,649,561 | ) | - | |||||
| Other income | 4,778 | 11,956 | ||||||
| Total other (expense) income, net | (7,962,188 | ) | (1,407,823 | ) | ||||
| Net loss | $ | (14,626,683 | ) | $ | (4,780,061 | ) | ||
| 65 |
Net Sales
Net sales declined $224,686, or 8.4%, to $2,460,049 in the year ended December 31, 2023 due to (i) management’s focus on gaining further insurance coverage for our products, funding and the initial public offering and (ii) fewer shipments to certain customers as they manage through the insurance reimbursement process.
Gross Profit and Gross Margin
Gross profit declined $230,971, or 9.7%, to $2,156,704 in the year ended December 31, 2023 due to (i) lower sales volume and (ii) gross margin declining 120 bps from 88.9% to 87.7% in the years ended December 31, 2023 and 2022, respectively, due to growth in our patient assistance program that provides discounts to patients without insurance coverage.
Selling Expenses
Selling expenses declined $87,314, or 21.3%, to $323,569 in the year ended December 31, 2023 due to (i) lower sales volume and (ii) a reduction in the commission rate paid to the sales team in the middle of the year ended December 31, 2022.
Research and Development
Research and development expense declined $56,295, or 25.0%, to $169,315 in the year ended December 31, 2023 due to more patient trials conducted in the year ended December 31, 2023 as the Company prepared for more FDA submissions.
General and Administrative
General and administrative expense increased $3,204,895, or 62.6%, to $8,328,315 in the year ended December 31, 2023 due to (i) incremental headcount to build out market access and patient assistance teams including recruiting costs, (ii) higher insurance, investor relations and board of director costs post-IPO, (iii) one-time IPO advisory costs and (iv) higher advertising costs in order to expand market awareness.
Operating Loss
Our operating loss increased $3,292,257, or 97.6%, to $6,664,495 in the year ended December 31, 2023 primarily due to lower sales volume and higher general and administrative expenses.
| 66 |
Other (Expense) Income
Other expense increased $6,554,365, or 465.6%, to $7,962,188 in the year ended December 31, 2023 due to the full amortization of the unamortized debt discount upon the IPO, a loss on the extinguishment of debt upon the IPO, higher interest expense and a lower year-over-year change in the fair value of the derivative financial instrument liability partially offset by the absence of convertible note financing fees in the year ended December 31, 2023 and a larger year-over-year change in the fair value of the warrant liability.
Net Loss
Our net loss increased $9,846,622, or 206.0%, to $14,626,683 in the year ended December 31, 2023 primarily due to lower sales volume, higher general and administrative expenses, the full amortization of the debt discount upon the IPO and the loss on the extinguishment of debt upon the IPO.
Liquidity and Capital Resources
We had cash on hand of $78,560 and $253,699 as of December 31, 2023 and 2022, respectively. We maintained negative working capital of $1,643,058 and $6,501,101 as of December 31, 2023 and 2022, respectively. The increase in working capital was primarily due to the conversion of notes payable upon the IPO along with the corresponding elimination or reduction of the fair value liability of warrants and derivatives and payments of employee bonuses and to vendors with proceeds from the IPO.
We have incurred losses since inception and have funded our operations primarily with a combination of sales, debt, and the sale of capital stock. As of December 31, 2023, we had an stockholders’ deficit of $1,403,241 with no outstanding long-term debt.
Our future capital requirements will depend upon many factors, including progress with developing, manufacturing, and marketing our technologies, the time and costs involved in preparing, filing, prosecuting, maintaining, and enforcing patent claims and other proprietary rights, our ability to establish collaborative arrangements, marketing activities and competing technological and market developments, including regulatory changes and overall economic conditions in our target markets. Our ability to generate revenue and achieve profitability requires us to successfully market and secure purchase orders for our products from customers currently identified in our sales pipeline and to new customers as well. The primary activity that will drive all customers and revenues is the adoption of insurance coverage by commercial insurance carriers nationally, so this is a top priority of the Company. These activities, including our planned research and development efforts, will require significant uses of working capital through the rest of 2024 and beyond. Based on our current operating plans, we believe that our existing cash at the time of this filing will only be sufficient to meet our anticipated operating needs through the end of 2024.
Additionally, we will have to meet all the financial disclosure and reporting requirements associated with being a publicly reporting company. Our management will have to spend additional time on policies and procedures to make sure it is compliant with various regulatory requirements, especially that of Section 404 of the Sarbanes-Oxley Act. This additional corporate governance time required of management could limit the amount of time our management has to implement our business plan and may delay our anticipated growth plans.
| 67 |
The following table summarizes our cash flows from operating, investing, and financing activities for the years ended December 31, 2023 and 2022:
| Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Net cash used in operating activites | $ | (6,693,978 | ) | $ | (2,298,004 | ) | ||
| Net cash used in investing activities | (71,781 | ) | (61,205 | ) | ||||
| Net cash provided by financing activities | 6,590,620 | 2,292,050 | ||||||
| Net decrease in cash and cash equivalents | (175,139 | ) | (67,159 | ) | ||||
| Cash and cash equivalents at beginning of period | 253,699 | 320,858 | ||||||
| Cash and cash equivalents at end of period | $ | 78,560 | $ | 253,699 | ||||
Operating Activities – Net cash used in operating activities increased $4,395,974, or 191.3%, in the year ended December 31, 2023 primarily due to higher general and administrative expenses and the payment of vendors and employee bonuses, partially offset by the issuance of common stock for non-cash consideration of IPO services.
Investing Activities – Net cash used in investing activities increased $10,576, or 17.3%, in the year ended December 31, 2023 due to purchases of equipment, trademarks and licenses.
Financing Activities – Net cash provided by financing activities increased $4,298,570, or 187.5%, in the year ended December 31, 2023 due to proceeds from the issuance of common stock via an IPO and convertible notes partially offset by repayments of notes payable and the payment of IPO costs.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
See Index to Financial Statements and Financial Statement Schedules from page F-1 of this annual report on Form 10-K, which are incorporated herein by reference.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES
Application of U.S. GAAP
The Company’s financial statements are maintained in accordance with U.S. GAAP. Our application and compliance with U.S. GAAP is based on guidance and interpretations issued by the Financial Accounting Standards Board through the Accounting Standards Codification and Accounting Standards Updates. Our conclusions are communicated to our management, including our principal executive officer and principal financial officer, to allow timely and accurate decisions and reporting. Our management, with the participation of our principal executive and principal financial officer, evaluated our Company’s controls over the application of U.S. GAAP as of the end of the period covered by this Form 10-K. Based on this evaluation, our principal executive officer and principal financial officer concluded that as of December 31, 2023, our controls over the application of U.S. GAAP were not effective as evidenced by the restatement of our unaudited financial statements as of and for the three and nine month periods ended September 30, 2023 included in Note 19 to this Form 10-K that constituted a material weakness.
Disclosure Controls and Procedures
We maintain “disclosure controls and procedures,” as that term is defined in Rule 13a-15(e), promulgated by the SEC pursuant to the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Disclosure controls and procedures include controls and procedures designed to ensure that information required to be disclosed in our Company’s reports filed under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our principal executive officer and principal financial officer to allow timely decisions regarding required disclosure. Our management, with the participation of our principal executive officer and principal financial officer, evaluated our Company’s disclosure controls and procedures as of the end of the period covered by this Form 10-K. Based on this evaluation, our principal executive officer and principal financial officer concluded that as of December 31, 2023, our disclosure controls and procedures were not effective. The ineffectiveness of our disclosure controls and procedures was due to material weaknesses as a result of the (i) lack of segregation of duties, (ii) lack of internal controls structure review and (iii) misapplication of GAAP.
| 68 |
Management’s Annual Report on Internal Control Over Financial Reporting
This Annual Report on Form 10-K does not include a report of management’s assessment regarding internal control over financial reporting due to a transition period established by the rules of the SEC for newly public companies.
Changes in Internal Control Over Financial Reporting
There were no changes in the Company’s internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) of the Exchange Act) during the year ended December 31, 2023, that has materially affected, or is reasonably likely to materially affect, the Company’s internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
Not applicable.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
| 69 |
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The following table and biographical summaries set forth information, including principal occupation and business experience, about our directors and executive officers as of April 12, 2024:
| Name | Age | Position | ||
| Brian Carrico | 42 | President, Chief Executive Officer, and Director | ||
| Timothy Henrichs (1) | 51 | Chief Financial Officer | ||
| Dr. Adrian Miranda | 54 | Chief Medical Officer, Senior Vice President of Science and Technology | ||
| Dr. Thomas Carrico | 67 | Chief Regulatory Officer, Compliance Officer and Privacy Officer | ||
| Dr. Christopher Robin Brown | 70 | Director | ||
| Bradley Mitch Watkins | 49 | Director | ||
| Beth Keyser | 55 | Director | ||
| Kristin Ferge | 50 | Director |
| (1) | On January 30, 2024, our former Chief Financial Officer John Seale resigned from his position, effective as of the close of business on January 30th. On the same day, Timothy R. Henrichs resigned as a member of the board of directors, effective February 2, 2024. On January 26, 2024, the board of directors appointed Mr. Henrichs to serve as the CFO, effective on February 5, 2024. |
Brian Carrico, President, Chief Executive Officer and Director
Brian Carrico joined the Company in 2012. During his tenure, Mr. Carrico has held multiple leadership positions of increasing responsibility, including Vice President of Sales and President before becoming CEO on January 1, 2018. As an early employee in the Company’s life cycle, Mr. Carrico was instrumental in setting the strategic agenda for the Company, raising start-up capital, championing new product development, and bringing the Company’s technology to market. Prior to joining Neuraxis, Mr. Carrico worked selling in the operating room at Bard Medical and in the Cath lab at St. Jude Medical. He attended Indiana State University and holds a Bachelor of Science in Business Marketing.
Timothy Henrichs, Chief Financial Officer
Timothy Henrichs currently serves as the Company’s Chief Financial Officer since February 5, 2024 and was a director of the Company from August 9, 2023 to February 2, 2024. Mr. Henrichs’ global leadership experience spans over 20 years and across several industries, including healthcare, home improvement, retail, software and education. Previously Mr. Henrichs served as the Chief Financial Officer of Renovo Home Partners since 2022. He also served as the Executive Vice President and Chief Financial Officer of Follett Corporation from 2008 to 2022 and Global Controller of General Electric Company’s Healthcare Clinical Systems division responsible for the manufacture and distribution of medical devices to the ultrasound, patient monitoring and anesthesiology markets form 2005 to 2008 in addition to leadership positions at Federal Signal Corporation and Ernst & Young LLP. Mr. Henrichs earned his bachelor’s degree in accounting from the University of Notre Dame and is a Certified Public Accountant with an inactive license in the State of Illinois.
| 70 |
Dr. Adrian Miranda, Chief Medical Officer, Senior Vice President of Science and Technology
Dr. Adrian Miranda has served as our Chief Medical Officer since 2018 and brings a unique background of research and clinical expertise to his role. Prior to joining Neuraxis, Dr. Miranda was an Assistant professor at the Medical College of Wisconsin. He is a board-certified pediatric gastroenterologist. He obtained his undergraduate degree in Biology from San Diego State University and obtained his medical degree from the Medical College of Wisconsin. He completed his residency and subspecialty training in pediatric gastroenterology at Children’s Hospital of Wisconsin.
As a physician scientist, he has spent the past 20 years of his career investigating the pathophysiology of visceral and somatic pain, as well as exploring new therapeutic options. His focus has been on studying the effects of adverse early life events, neuroplasticity and the development of chronic pain. He has an extensive publication record and has lectured nationally and internationally.
Dr. Thomas Carrico, Chief Regulatory Officer, Compliance Officer and Privacy Officer
Dr. Thomas Carrico has served as our Chief Regulatory Officer since November 2017. He joined the Company in February 2012 as Director of Regulatory Affairs. Prior to and during his early years with Neuraxis, he was President & Clinic Director at Spine and Neuromuscular Associates in Lawrenceburg, Indiana from January 2002 to December 2018. He has over 40 years of experience in the healthcare field and has been involved in the study and application of techniques and treatments that directly affect the autonomic nervous system, especially regarding homeostasis and balance of the parasympathetic and the sympathetic nervous system. Dr. Carrico has a history of working with attorneys while serving on state and national boards, which has positioned him to integrate into regulatory responsibilities at the Company. Dr. Carrico received his undergraduate education from Indiana University and his Doctorate from Palmer College of Chiropractic.
Dr. Christopher Robin Brown, Director
Dr. Chirstopher Robin Brown is a co-founder of the Company. He developed clinical protocol, initial practice guidelines, designed and implemented the practitioner certification program and personally financed the first two years of the Company. After developing the technique of transillumination to isolate auricular neurovascular bundles, he authored and designed the initial studies establishing neurovascular and tissue energy transfer theories upon which the devices’ use are based. Dr. Brown is listed as the sole or principal inventor on all Neuraxis patents and is currently active in further device development working closely with compliance, product design and engineering.
Upon graduation from the Indiana University School of Dentistry in 1982, while serving as clinic chief in the United States Army Reserve (USAR) dental corps at Fort Benjamin Harrison in Indianapolis, Indiana, Dr. Brown started a private practice (current) concentrating in head, neck, and facial pain developing the first hospital based facial pain clinic in Indiana. He received his master’s degree in Biomechanical Trauma in 1996 from Lynn University, one of only 12 dentists in the United States to hold the combination of DDS and MPS degrees. Dr. Brown has authored several textbook chapters, published peer reviewed articles on the physics of soft tissue trauma, pain, financial management, was regional editor for a national facial pain management Journal, and has lectured extensively nationally and internationally. He served on the Board of Directors of the American Academy of Pain Management for 15 years, helping grow the organization from 800 members to over 5,000. Throughout his tenure, he developed educational tracks, served as Industry liaison, one term as treasurer and one term as President. He served on the national board of The Alliance of TMD practitioners, serving one term as president.
Throughout his career, Dr. Brown has been active in the purchasing and management of several destressed clinics and re-structuring them into profitable enterprises. He has performed extensive volunteer work overseas providing surgical care in the Dominican Republic, local dental clinics serving the underprivileged, and recently provided dental screenings for the deployment of soldiers in the USAR and National Guard.
| 71 |
Bradley Mitch Watkins, Director
Bradley Mitch Watkins has overseen four companies through their early commercialization periods within the medical device sector over the last 20 years. He has directly reported to the CEO or board of directors and operated as the lead for all field operations. Over his 20 years in a multitude of medical device markets, Mr. Watkins has overseen $410 million in company acquisitions in an array of leadership roles. He has thrived in early commercialization, recruitment, and strategic company direction. These duties have groomed Mr. Watkins with a wide array of responsibilities beyond sales, including marketing, clinical study design, manufacturing, R&D, FDA submissions, and fiscal oversight. Mr. Watkins has been the National Sales Manager of Terumo Interventional Systems since 2015, where he has led multiple new technology sales teams within the peripheral IV and Electrophysiology markets. Mr. Watkins received his bachelor’s degree in behavioral science from the University of Maryland.
Beth Keyser, Director
With more than 20 years of experience in executive roles in population health, Beth Keyser is skilled at understanding the unique, complex needs of multiple market segments and devises solutions that meet their specific goals. Ms.Keyser is the President, BCBS of Indiana at Anthem, Inc. since 2020. From 2018 to 2020, Ms. Keyser served as the President, Create at Brighton Health Plan Solutions. From 2015 to 2020, Ms. Keyser served as the Senior Vice President, International and Hawaii Markets at Sharecare, Inc. Ms. Keyser received her master’s degree in Executive Master of Science, Health Administration, from University of Alabama at Birmingham.
Kristin Ferge, Director
Kristin Ferge has been President and Chief Financial Officer of Capri Communities and Bridges Home Healthcare, a Wisconsin-based privately held senior living corporation, since 2016. Prior to joining Capri, Ms. Ferge was an executive for 18 years with Brookdale Senior Living Inc. or one of its predecessors. Ms. Ferge ended her tenure at Brookdale, a publicly traded senior living company, as Executive Vice President, Treasurer, and Chief Accounting Officer. Prior to Brookdale, Ms. Ferge was an auditor with KPMG. Ms. Ferge is a certified public accountant.
Family Relationships
Brian Carrico, our Chief Executive Officer and Director, is the son of Dr. Carrico, our Chief Regulatory Officer. There are no other family relationships between or among any of our executive officers or other directors.
Role of the Board
It is the paramount duty of the board to oversee our management in the competent and ethical operation of the Company on a day-to-day basis and to assure that the long-term interests of the stockholders are being served. To satisfy this duty, the directors take a proactive, focused approach to their positions, and set standards to ensure that we are committed to business success through maintenance of ambitious standards of responsibility and ethics.
Director Terms; Qualifications
Our directors are elected for a term of one year and until their successors qualified, nominated, and appointed or elected.
When considering whether directors and nominees have the experience, qualifications, attributes and skills to enable the board of directors to satisfy its oversight responsibilities effectively in light of the Company’s business and structure, the board of directors focuses primarily on the industry and transactional experience, and other background, in addition to any unique skills or attributes associated with a director.
Director or Officer Involvement in Certain Legal Proceedings
There are no material proceedings to which any director or officer, or any associate of any such director or officer, is a party that is adverse to our Company or any of our subsidiaries or has a material interest adverse to our Company or any of our subsidiaries. No director or executive officer has been a director or executive officer of any business which has filed a bankruptcy petition or had a bankruptcy petition filed against it during the past ten years. No director or executive officer has been convicted of a criminal offense or is the subject of a pending criminal proceeding during the past ten years. No director or executive officer has been the subject of any order, judgment or decree of any court permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities or banking activities during the past ten years. No director or officer has been found by a court to have violated a federal or state securities or commodities law during the past ten years.
| 72 |
Directors and Officers Liability Insurance
The Company maintains directors’ and officers’ liability insurance insuring its directors and officers against liability for acts or omissions in their capacities as directors or officers, subject to certain exclusions. Such insurance may also insure the Company against losses, which it may incur in indemnifying its officers and directors. In addition, officers and directors also have indemnification rights under applicable laws, and the Company’s Certificate of Incorporation and Bylaws.
Director Independence
The listing rules of NYSE American require that independent directors must comprise a majority of a listed company’s board of directors. In addition, the rules of NYSE American require that, subject to specified exceptions, each member of a listed company’s audit, compensation, and nominating and governance committees be independent. Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Exchange Act. Under the rules of NYSE American, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
Our board of directors has undertaken a review of the independence of our directors and considered whether any director has a material relationship with it that could compromise his or her ability to exercise independent judgment in carrying out his or her responsibilities. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, the board of directors has determined that three are “independent” as that term is defined under the applicable rules and regulations of the SEC and the listing standards of NYSE American. In making these determinations, our board of directors considered the current and prior relationships that each non-employee director has with the Company and all other facts and circumstances our board of directors deemed relevant in determining their independence.
Board Committees
As of December 31, 2023, the following three standing committees have been established: Audit Committee; Compensation Committee; and Nominating and Corporate Governance Committee. Each of our independent directors, Bradley Mitch Watkins, Beth Keyser and Kristin Ferge, serve on each committee. Our board has adopted written charters for each of these committees. Copies of the charters are available on our website. Our board may establish other committees as it deems necessary or appropriate from time to time.
Audit Committee
The Audit Committee, among other things, is responsible for:
| ● | appointing; approving the compensation of; overseeing the work of; and assessing the independence, qualifications, and performance of the independent auditor; | |
| ● | reviewing the internal audit function, including its independence, plans, and budget; |
| ● | approving, in advance, audit and any permissible non-audit services performed by our independent auditor; | |
| ● | reviewing our internal controls with the independent auditor, the internal auditor, and management; | |
| ● | reviewing the adequacy of our accounting and financial controls as reported by the independent auditor, the internal auditor, and management; | |
| ● | overseeing our financial compliance system; and | |
| ● | overseeing our major risk exposures regarding the Company’s accounting and financial reporting policies, the activities of our internal audit function, and information technology. |
| 73 |
The board of directors has affirmatively determined that each member of the Audit Committee meets the additional independence criteria applicable to audit committee members under SEC rules and listing standards of NYSE American All members of the Audit Committee are able to read and understand fundamental financial statements, are familiar with finance and accounting practices and principles and are financially literate. The board of directors has adopted a written charter setting forth the authority and responsibilities of the Audit Committee. The board of directors has affirmatively determined that each member of the Audit Committee is financially literate, and that Kristin Ferge meets the qualifications of an Audit Committee financial expert.
The Audit Committee consists of Kristin Ferge, Bradley Mitch Watkins, and Beth Keyser and Ms. Ferge serves as chair of the Audit Committee. The functioning of the Audit Committee complies with the applicable requirements of the rules and listing standards of NYSE American and the SEC.
Compensation Committee
The Compensation Committee is responsible for:
| ● | reviewing and making recommendations to the Board with respect to the compensation of our officers and directors, including the CEO; | |
| ● | overseeing and administering the Company’s executive compensation plans, including equity-based awards; | |
| ● | negotiating and overseeing employment agreements with officers and directors; and | |
| ● | overseeing how the Company’s compensation policies and practices may affect the Company’s risk management practices and/or risk-taking incentives. |
The Compensation Committee consists of Bradley Mitch Watkins, Beth Keyser, and Kristin Ferge, and Mr. Watkins serves as chair of the Compensation Committee. The board of directors has affirmatively determined that each member of the Compensation Committee meets the independence criteria applicable to compensation committee members under SEC rules and listing standards of NYSE American. The Company believes that the composition of the Compensation Committee meets the requirements for independence under, and the functioning of such Compensation Committee comply with, any applicable requirements of the rules and regulations of listing standards of NYSE American and the SEC.
Nominating and Corporate Governance Committee
The Nominating and Corporate Governance Committee, among other things, is responsible for:
| ● | reviewing and assessing the development of the executive officers and considering and making recommendations to the Board regarding promotion and succession issues; | |
| ● | evaluating and reporting to the Board on the performance and effectiveness of the directors, committees and the board of directors as a whole; |
| ● | working with the Board to determine the appropriate and desirable mix of characteristics, skills, expertise and experience, including diversity considerations, for the full Board and each committee; | |
| ● | annually presenting to the Board a list of individuals recommended to be nominated for election to the board; | |
| ● | reviewing, evaluating, and recommending changes to the Company’s committee charters; | |
| ● | recommending to the Board individuals to be elected to fill vacancies and newly created directorships; | |
| ● | overseeing the Company’s compliance program, including the Code of Conduct; and | |
| ● | overseeing and evaluating how the Company’s corporate governance and legal and regulatory compliance policies and practices, including leadership, structure, and succession planning, that may affect the Company’s major risk exposures. |
The Nominating and Corporate Governance Committee consists of Bradley Mitch Watkins, Beth Keyser, and Kristin Ferge and Ms. Keyser serves as chair of the Nominating and Corporate Governance Committee. The Company’s board of directors has determined that each member of the Nominating and Corporate Governance Committee is independent within the meaning of the independent director requirements for independence under the NYSE American listing standards and SEC rules and regulations.
| 74 |
Compensation Committee Interlocks and Insider Participation
None of the Company’s executive officers serves, or in the past has served, as a member of the board of directors or compensation committee, or other committee serving an equivalent function, of any entity that has one or more executive officers who serve as members of the Company’s board of directors or its compensation committee. None of the members of the Company’s compensation committee is, or has ever been, an officer or employee of the Company.
Code of Business Conduct and Ethics
The Company’s board of directors has adopted a code of business conduct and ethics (“Code of Conduct”) applicable to its employees, directors and officers, in accordance with applicable U.S. federal securities laws and the corporate governance rules of NYSE American. The Code of Conduct is effective and is publicly available on the Company’s website. Any substantive amendments or waivers of the Code of Conduct may be made only by the Company’s board of directors and will be promptly disclosed as required by applicable U.S. federal securities laws and the corporate governance rules of NYSE American.
Item 11. Executive Compensation
Summary Compensation Table
The following table sets forth information concerning all compensation earned by our Chief Executive Officer and two other persons who served as executive officers as, at, or during the year ended December 31, 2023, and who earned compensation exceeding $100,000 during 2023 (the “Named Executive Officers”), for services as executive officers for the last two years.
| Name and Principal Position | Year | Salary ($) | Bonus ($) | Insurance ($) | Total ($) | |||||||||||||||
| Brian Carrico | 2023 | 375,000 | 441,346 | 23,052 | 839,398 | |||||||||||||||
| Chief Executive Officer | 2022 | 373,578 | — | 18,601 | 392,179 | |||||||||||||||
| Dr. Thomas Carrico | 2023 | 278,837 | 116,716 | 15,544 | 411,097 | |||||||||||||||
| Chief Regulatory Officer | 2022 | 271,962 | — | 15,234 | 287,196 | |||||||||||||||
| Dr. Adrian Miranda | 2023 | 300,000 | — | 23,052 | 323,052 | |||||||||||||||
| Chief Medical Officer | 2022 | 266,346 | — | 18,601 | 284,947 | |||||||||||||||
| (1) | Represents payments to our executive officers pursuant to their respective employment agreements. For more information, see “—Employment Agreements.” |
| 75 |
Outstanding Equity Awards at Fiscal Year-End
The following table sets forth information regarding equity awards held by the Named Executive Officers as of December 31, 2023.
| Name | Number of Securities Underlying Unexercised Options, Exercisable (#) | Number of Securities Underlying Unexercised Options, Not Exercisable (#) | Option Exercise Price ($) | Option Expiration Date | ||||||||||
| Brian Carrico | 320,000 | — | 6.94 | 09/13/29 | ||||||||||
| Dr. Thomas Carrico | 306,236 | — | 6.94 | 09/13/29 | ||||||||||
| Dr. Adrian Miranda | 337,204 | — | 6.94 | 09/13/29 | ||||||||||
| (1) | All option awards were granted under the Innovative Health Solutions, Inc. 2017 Stock Compensation Plan and vested fully upon grant. |
Innovative Health Solutions, Inc. 2017 Stock Compensation Plan, As Amended
On October 12, 2017, the Company adopted the Innovative Health Solutions, Inc. 2017 Stock Compensation Plan, as amended on September 13, 2019, September 9, 2021, and November 1, 2022 (collectively, the “2017 Plan”). The purpose of the 2017 Plan is to grant incentive stock options, nonqualified stock options, or restricted stock awards to our officers, employees, directors, advisors, and consultants. The maximum numbers of shares of common stock that may be issued pursuant to awards granted were 1,319,394. Cancelled and forfeited stock options and stock awards may again become available for grant under the 2017 Plan. As of the date of December 31, 2023, options to purchase all 1,319,394 shares of common stock have been granted under the 2017 Plan and remain outstanding, and no shares remain available for issuance under the 2017 Plan. The following summary briefly describes the principal features of the 2017 Plan and is qualified in its entirety by reference to the full text of the 2017 Plan, which is filed as an exhibit to the registration statement of which this prospectus forms a part.
Purpose of the 2017 Plan: The purposes of the 2017 Plan are to encourage ownership of shares by eligible employees and key non-employees in order to attract and retain such eligible employees in the employ of the Company or an affiliated entity, or to attract such key non-employees to provide services to the Company or an affiliated entity, and to provide additional incentive for such persons to promote the long-term success of the Company or an affiliated entity.
Administration of the Plan: The 2017 Plan is administered by the board of directors, or the committee to which the board of directors delegates the power to act. Among other things, the administrator has the authority to select persons who will receive awards, determine the types of awards and the number of shares to be covered by awards, and to establish the terms, conditions, restrictions and other provisions of awards. The administrator has authority to establish, amend and rescind rules and regulations relating to the 2017 Plan.
Eligible Recipients: Persons eligible to receive awards under the 2017 Plan are those officers, employees, directors, advisors, and consultants of the Company or an affiliated entity who are selected by the administrator.
Shares Available under the 2017 Plan: The maximum number of shares of our common stock that may be delivered to participants under the 2017 Plan is 1,319,394 shares, subject to adjustment for certain corporate changes affecting the shares, such as stock splits. No new grants will be made under the 2017 Plan, and shares subject to an award under the 2017 Plan for which the award is canceled, forfeited or expires will become available for grants under the 2022 Plan described below. Shares subject to an award that is settled in cash will not again be made available for grants under the 2017 Plan.
| 76 |
Stock Options
General. Subject to the provisions of the 2017 Plan, the administrator has the authority to determine all grants of stock options, although there are currently no shares of common stock remaining reserved for grants under the 2017 Plan.
Option Price. The exercise price for stock options is determined at the time of grant. The exercise price may not be less than the fair market value on the date of grant. Additionally, incentive stock option grants to any person owning more than 10% of our voting stock must have an exercise price of not less than 110% of the fair market value on the grant date.
Exercise of Options. An option may be exercised only in accordance with the terms and conditions for the option agreement as established by the administrator at the time of the grant. The option must be exercised by notice to us, accompanied by payment of the exercise price. Payments may be made in cash or, at the option of the administrator, by actual or constructive delivery of shares of common stock to the holder of the option based upon the fair market value of the shares on the date of exercise.
Expiration or Termination. Options, if not previously exercised, will expire on the expiration date established by the administrator at the time of grant. In the case of incentive stock options, such term cannot exceed ten years provided that in the case of holders of more than 10% of our voting stock, such term cannot exceed five years. Options will terminate before their expiration date only if the holder’s service with our Company or an affiliate terminates before the expiration date and the holder is terminated for cause. The option may remain exercisable until the expiration date of the option after terminations of employment for any reason other than for cause, including terminations as a result of death, disability or retirement.
Incentive and Non-Qualified Options. An incentive stock option is an option that is intended to qualify under certain provisions of the Internal Revenue Code, for more favorable tax treatment than applies to non-qualified stock options. Any option that does not qualify as an incentive stock option will be a non-qualified stock option. Under the Code, certain restrictions apply to incentive stock options. For example, the exercise price for incentive stock options may not be less than the fair market value of the shares on the grant date and the term of the option may not exceed ten years. In addition, an incentive stock option may not be transferred, other than by will or the laws of descent and distribution and is exercisable during the holder’s lifetime only by the holder. In addition, no incentive stock options may be granted to a holder that is first exercisable in a single year if that option, together with all incentive stock options previously granted to the holder that also first become exercisable in that year, relate to shares having an aggregate market value in excess of $100,000, measured at the grant date.
Restricted Stock Awards: Restricted stock awards could have also been granted under the 2017 Plan, although there are currently no shares of common stock remaining reserved for grants under the 2017 Plan. A restricted stock award is a grant of shares of common stock or of a right to receive shares in the future.
Other Material Provisions: Awards are evidenced by a written agreement, in such form as may be approved by the administrator. In the event of various changes to the capitalization of our Company, such as stock splits, stock dividends and similar re-capitalizations, an appropriate adjustment will be made by the administrator to the number of shares covered by outstanding awards or to the exercise price of such awards. The administrator is also permitted to include in the written agreement provisions that provide for certain changes in the award in the event of a change of control of our Company, including acceleration of vesting. Except as otherwise determined by the administrator at the date of grant, awards will not be transferable, other than by will or the laws of descent and distribution. Prior to any award distribution, we are permitted to deduct or withhold amounts sufficient to satisfy any employee withholding tax requirements. Our board of directors also has the authority, at any time, to discontinue the granting of awards. The Plan may be amended by the board of directors and such amendment shall become effective upon adoption by the board of directors; provided, however, that any amendment shall be subject to the approval of the stockholders of the Company at or before the next annual meeting of the stockholders of the Company if such stockholder approval is required by applicable laws. No amendment that would adversely affect any outstanding award made under the Plan can be made without the consent of the holder of such award.
No new grants can be made under the 2017 Plan. The terms and conditions of awards granted under the 2017 Plan prior to the effective date of the 2022 Plan will not be affected by the adoption or approval of the 2022 Plan, and the 2017 Plan will remain effective with respect to such awards.
| 77 |
Neuraxis, Inc. 2022 Omnibus Securities and Incentive Plan, As Amended
On November 1, 2022, the Company adopted the Neuraxis, Inc. 2022 Omnibus Securities and Incentive Plan (as amended January 18, 2023, the “2022 Plan”). The purpose of the 2022 Plan is to attract, retain and provide incentives to key management employees and non-employee directors of, and non-employee consultants to, the Company and its affiliates, and to align the interests of such employees, non-employee directors and non-employee consultants with those of the Company’s stockholders. The maximum numbers of shares of common stock that may be issued pursuant to awards granted are 300,000. Cancelled and forfeited stock options and stock awards may again become available for grant under the 2022 Plan. As of the date of this prospectus, no awards have been granted under the 2022 Plan, and 300,000 shares of common stock remain available for issuance under the 2022 Plan. The following summary briefly describes the principal features of the 2022 Plan and is qualified in its entirety by reference to the full text of the 2022 Plan.
Purpose of the 2022 Plan: The purposes of the 2022 Plan is to benefit the stockholders of the Company, by assisting the Company to attract, retain and provide incentives to key management employees and non-employee directors of, and non-employee consultants to, the Company and its affiliates, and to align the interests of such employees, non-employee directors and non-employee consultants with those of the Company’s stockholders.
Administration of the 2022 Plan: The 2022 Plan shall be administered by the board of directors or the committee designated by the board of directors. Among other things, the administrator has the authority to select persons who will receive awards, determine the time or times when an award shall be made, what type of award shall be granted, the term of an award, the date or dates on which an award vests (including acceleration of vesting), the form of any payment to be made pursuant to an award, the terms and conditions of an award (including the forfeiture of the award (and/or any financial gain) if the holder of the award violates any applicable restrictive covenant thereof), the restrictions under a restricted stock award and the number of common stock which may be issued under an award, all as applicable. In addition, subject to the express provisions of the 2022 Plan, the administrator is authorized to construe the 2022 Plan and the respective award agreements executed thereunder, to prescribe such rules and regulations relating to the 2022 Plan as it may deem advisable to carry out the intent of the 2022 Plan, to determine the terms, restrictions and provisions of each award, and to make all other determinations necessary or advisable for administering the 2022 Plan.
Eligible Recipients: Persons eligible to receive awards under the 2022 Plan will be those officers, employees, directors, advisors, and consultants of the Company or an affiliated entity who are selected by the administrator.
Shares Available under the Plan: The maximum number of shares of our common stock that may be delivered to participants under the 2022 Plan is 300,000 shares, subject to adjustment for certain corporate changes affecting the shares, such as stock splits. Shares subject to an award under the 2022 Plan for which the award is lapses, expires, is canceled, terminated unexercised or ceases to be exercisable again become available for grants under the 2022 Plan.
Stock Options
General. Subject to the provisions of the 2022 Plan, the administrator has the authority to determine all grants of stock options.
Option Price. The exercise price for stock options is determined at the time of grant. The exercise price may not be less than the fair market value on the date of grant. Additionally, incentive stock option grants to any person owning more than 10% of our voting stock must have an exercise price of not less than 110% of the fair market value on the grant date.
Exercise of Options. An option may be exercised only in accordance with the terms and conditions for the option agreement as established by the administrator at the time of the grant. The option must be exercised by notice to us, accompanied by payment of the exercise price. Payments may be made in cash or, at the option of the administrator, by actual or constructive delivery of shares of common stock to the holder of the option based upon the fair market value of the shares on the date of exercise.
| 78 |
Expiration or Termination. Options, if not previously exercised, will expire on the expiration date established by the administrator at the time of grant. In the case of incentive stock options, such term cannot exceed ten years provided that in the case of holders of more than 10% of our voting stock, such term cannot exceed five years. Options will terminate before their expiration date if the holder’s service with our Company or a subsidiary terminates before the expiration date. The option may remain exercisable for specified periods after certain terminations of employment, including terminations as a result of death, disability or retirement, with the precise period during which the option may be exercised to be established by the administrator and reflected in the grant evidencing the award.
Incentive and Non-Qualified Options. As described elsewhere in this summary, an incentive stock option is an option that is intended to qualify under certain provisions of the Code, for more favorable tax treatment than applies to non-qualified stock options. Any option that does not qualify as an incentive stock option will be a non-qualified stock option. Under the Code, certain restrictions apply to incentive stock options. For example, generally, the exercise price for incentive stock options may not be less than the fair market value of the shares on the grant date and the term of the option may not exceed ten years. In addition, an incentive stock option may not be transferred, other than by will or the laws of descent and distribution, and is exercisable during the holder’s lifetime only by the holder. In addition, to the extent that the aggregate fair market value of common stock with respect to which incentive stock options are exercisable for the first time by an individual during any calendar year under all plans of the Company and any parent corporation or subsidiary corporation thereof which provide for the grant of incentive stock options exceeds $100,000, the portion of such incentive stock options that exceeds such threshold shall be treated as non-qualified stock options. Incentive stock options shall be granted to employees only.
Restricted Stock Awards: Restricted stock awards can be granted under the 2022 Plan. A restricted stock award is a grant of shares of common stock or of a right to receive shares in the future. These awards will be subject to such conditions, restrictions and contingencies as the administrator shall determine at the date of grant. Those may include requirements for continuous service and/or the achievement of specified performance goals.
Unrestricted Stock Awards: Unrestricted stock awards can also be granted under the 2022 Plan. An unrestricted stock award is a grant of shares of common stock which is not subject to restrictions, in consideration for past services rendered thereby to the Company or an affiliate or for other valid consideration.
Restricted Stock Unit Awards: Restricted stock unit awards (“RSUs”) can be granted under the 2022 Plan upon the satisfaction of predetermined individual service related vesting requirements. The holder of a restricted stock unit shall be entitled to receive a cash payment equal to the fair market value of shares of common stock, for each unit awarded to the holder.
Performance Stock Unit Awards: Performance stock unit awards can be granted under the 2022 Plan. A holder of performance stock units shall be entitled to receive a cash payment equal to the dollar value or number of shares of common stock assigned to such units if the holder and/or the Company satisfy the predetermined performance goals and objectives.
Distribution Equivalent Rights: Distribution equivalent right awards can be granted under the 2022 Plan. A distribution equivalent right award entitles the holder to receive bookkeeping credits, cash payments and/or common stock distributions equal in amount to the distributions that would have been made to the holder had the holder held a specified number of common stock during the period the holder held the distribution equivalent right.
Stock Appreciation Rights: Stock appreciation rights can also be granted under the 2022 Plan, which is a right, granted alone or in connection with a related Option, to receive a payment on the date of exercise. The base value of the stock appreciation right shall be set forth by the administrator and shall not be less than the fair market value of the common stock at the date of grant for the stock appreciation right which is not a tandem stock appreciation right. No stock appreciation right shall be exercisable after the expiration of ten (10) years from the date of its grant. Upon the exercise of some or all of the portion of a stock appreciation right, the holder shall receive a payment from the Company, in cash or in the form of common stock having an equivalent fair market value or in a combination of both. If the administrator grants a stock appreciation right which is intended to be a tandem stock appreciation right, the tandem stock appreciation right shall be granted at the same time as the related option.
| 79 |
Other Material Provisions: Awards are evidenced by a written agreement, in such form as may be approved by the administrator. In the event of various changes to the capitalization of our Company, such as stock splits, stock dividends and similar re-capitalizations, an appropriate adjustment will be made by the administrator to the number of shares covered by outstanding awards or to the exercise price of such awards. The administrator is also permitted to include in the written agreement provisions that provide for certain changes in the award in the event of a change of control of our Company, including acceleration of vesting. Except as otherwise determined by the administrator at the date of grant, awards will not be transferable, other than by will or the laws of descent and distribution. Prior to any award distribution, we are permitted to deduct or withhold amounts sufficient to satisfy any employee withholding tax requirements. Our board of directors also has the authority, at any time, to discontinue the granting of awards. The 2022 Plan may be amended by the board of directors and such amendment shall become effective upon adoption by the board of directors; provided, however, that any amendment shall be subject to the approval of the stockholders of the Company at or before the next annual meeting of the stockholders of the Company if such stockholder approval is required by applicable laws. No amendment that would adversely affect any outstanding award made under the 2022 Plan can be made without the consent of the holder of such award. The 2022 Plan shall continue in effect, unless sooner terminated, until the tenth (10th) anniversary of the date on which it is adopted by the board of directors.
Employment Agreements
Brian Carrico, our Chief Executive Officer, entered into an employment agreement with the Company, dated August 9, 2022 and amended on May 4, 2023, which has a five-year initial term and provides for a base salary of $330,000, which shall be increased each year by not less than 3% per annum. Mr. Carrico also will receive a one-time incentive payment in the amount of $435,577, which amount consists of accrued and unpaid salary and a bonus to incentivize Mr. Carrico to remain with the Company for future service. Neither the accrued and unpaid salary nor bonus was the subject of any contract or agreement between Mr. Carrico and the Company prior to the execution of the employment agreement. The agreement contemplated that the incentive payment would be paid by December 15, 2022, but due to administrative impracticability, payment was made with a portion of the proceeds from the IPO in August 2023. In addition, Mr. Carrico is entitled to payment of a deferred bonus in an amount equal to (i) the aggregate of the strike price or exercise price of all 320,000 unexercised options to purchase stock or shares of the Company held by Mr. Carrico (the “Aggregate Strike Price”) plus (ii) a tax gross-up payment on the Aggregate Strike Price reasonably calculated by the Company at the highest marginal rates so that after payment of all ordinary income taxes on such Aggregate Strike Price, there remains an amount sufficient to pay such ordinary income taxes. The special deferred bonus will be paid in substantially equal 20% installments (the “Annual Deferred Bonus Payment”) on January 2 on each of 2024, 2025, 2026, 2027, and 2028 (the “Scheduled Payment Dates”), with a condition that on or before each Scheduled Payment Date, Mr. Carrico shall exercise at least 64,000 of the stock option. None of stock options has been exercised, therefore, no special deferred bonus has been paid as of the date of this prospectus. Mr. Carrico may exercise any portion of the stock options after a Scheduled Payment Date; provided, however, an exercise after the applicable Scheduled Payment Date shall result in forfeiture of the related Annual Deferred Bonus Payment.
If the employment agreement is terminated by the Company without cause (as defined in the employment agreement), Mr. Carrico will receive any accrued compensation (as defined in the employment agreement) and is entitled to severance payments as follows:
| ● | If termination occurs during the initial term, the severance payment shall be the amount equal to the greater of (a) three times Mr. Carrico’s base salary as of the termination date; and (b) three times the total amount of Mr. Carrico’s bonus payments the Company paid Mr. Carrico over the one year prior to the termination date, to be paid in substantially equal monthly installments over the course of the three years. | |
| ● | If termination occurs after the initial term, the severance payment shall be the amount equal to the greater of (a) one and one half (1.5) times Mr. Carrico’s base salary as of the termination date; and (b) one and one half (1.5) times the total amount of Mr. Carrico’s bonus payments the Company paid Mr. Carrico over the one (1) year prior to the termination date, to be paid in substantially equal monthly installments over the course of 18 months following the termination date. | |
| ● | In addition, as part of the severance payment, we agreed to pay Mr. Carrico monthly COBRA premiums for continuation of health coverage for 18 months post termination. |
| 80 |
If the employment agreement is terminated by the Company for cause, Mr. Carrico will forfeit any unpaid Annual Deferred Bonus Payment, but will receive any unpaid base salary that has been earned at the time of such termination, reimbursement of any expenses properly incurred prior to the Mr. Carrico’s termination date; and accrued and unused paid time off (“PTO”), if any, in accordance with the Company’s PTO policy in effect on Mr. Carrico’s termination date.
Mr. Carrico may terminate the employment agreement without good reason upon more than thirty (30) days’ prior written notice or for good reason without prior written consent, and will receive accrued compensation (as defined in the employment agreement) and the unpaid balance of the deferred bonus.
In the event that Mr. Carrico is terminated without cause, non-forfeited portions of the deferred bonus will be paid in equal installments over the remaining scheduled payment dates, if vested options as scheduled are exercised before the scheduled payment dates. A deferred bonus tranche will be forfeited if its corresponding options have expired.
On a change in control (as defined in the employment agreement), full vesting of the Annual Deferred Bonus Payment will occur and be paid in a single lump sum within 30 days after the change in control. The offering shall not be considered a change in control.
Pursuant to the employment agreement, Mr. Carrico also agreed to (i) not disclose to any unauthorized person or use for his own account any confidential information without the prior written consent of the Company or the board of directors, (ii) will not, directly or indirectly encourage, solicit, induce (or attempt to encourage, solicit or induce) any employee or agent of the Company that was employed (or otherwise engaged) at the time of his separation during his employment and for 24 months after his separation from that employment for any reason; (iii) will not, directly or indirectly, have any ownership interest in, work for, advise, manage, act as an agent or consultant for, or have any business connection or business or employment relationship with any entity or person which competes with Company; (iv) will not, directly or indirectly, have any ownership interest in, work for, advise, manage, act as an agent or consultant for, or have any business connection or business or employment relationship with any entity or person which competes with the Company during his employment and, (v) will not, directly or indirectly and in a competitive capacity own, manage, finance, operate, control or participate in ownership, management, or operation of, act as an agent, consultant, or be employed with, any business engaged in the design, manufacture, marketing, sale or servicing of any service or product with which Mr. Carrico was involved during his last year of employment with the Company; or which the Company is developing, producing, marketing, selling or servicing (or plans to develop, produce, market, sale or service) and about which Mr. Carrico gained any confidential information in the course of his employment with the Company for a period of 24 months after his separation from the Company.
Dr. Adrian Miranda, our Chief Medical Officer and Senior Vice President of Science and Technology, entered into an employment agreement with the Company, dated August 17, 2022, which has a two-year initial term and provides for a base salary of $300,000 with annual compensation increase. In addition, Dr. Miranda shall be entitled to payment of a special deferred bonus in an amount equal to (i) the aggregate of the strike price or exercise price of all 337,204 unexercised options to purchase stock or shares of the Company held by Dr. Miranda (the “Aggregate Strike Price”) plus (ii) a tax gross-up payment on the Aggregate Strike Price reasonably calculated by the Company at the highest marginal rates so that after payment of all ordinary income taxes on such Aggregate Strike Price, there remains an amount sufficient to pay such ordinary income taxes. The deferred bonus will be paid in substantially equal 20% installments (the “Annual Deferred Bonus Payment”) on January 2 on each of 2024, 2025, 2026, 2027, and 2028, with a condition that on or before each scheduled payment date, Dr. Miranda shall exercise at least 67,441 of the stock options.
| 81 |
If the employment agreement is terminated by the Company without cause (as defined in the employment agreement) occurs during the term of the agreement, Dr. Miranda will receive any accrued compensation (as defined in the employment agreement) and is entitled to certain amount of severance payments as follows:
| ● | If termination occurs during the initial term, the Company shall provide Dr. Miranda with severance compensation in the form of salary continuation at his Base Salary as of the termination date and ending the later of (i) 6 months or (ii) on the expiration date of the initial term. | |
| ● | If termination occurs after the initial term, the severance payment shall be the amount equal to one half (1/2) of Dr. Miranda’s Base Salary as of the termination date. | |
| ● | In addition, if termination occurs during the initial term, as part of the severance payment, we agreed to pay Dr. Miranda reimbursement of his monthly COBRA premiums for continuation of health coverage for 18 months post termination. |
If the employment agreement is terminated by the Company for cause, Dr. Miranda will forfeit any unpaid Annual Deferred Bonus Payment, but will receive any unpaid base salary that has been earned at the time of such termination, reimbursement of any expenses properly incurred prior to the Dr. Miranda’s termination date; and (iii) accrued and unused PTO, if any, in accordance with the Company’s PTO policy in effect on Dr. Miranda’s termination date.
Dr. Miranda may terminate the employment agreement without good reason upon more than thirty (30) days’ prior written notice or for good reason without prior written consent, and will receive accrued compensation (as defined in the employment agreement) and the unpaid balance of the special deferred bonus.
In the event that Dr. Miranda is terminated without cause, non-forfeited portions of the deferred bonus will be paid in equal installments over the remaining scheduled payment dates, if vested options as scheduled are exercised before the scheduled payment dates. A deferred bonus tranche will be forfeited if its corresponding options have expired.
On a change in control (as defined in the employment agreement), full vesting of the Annual Deferred Bonus Payment will occur and be paid in a single lump sum within 30 days after the change in control. The offering shall not be considered a change in control.
Pursuant to the employment agreement, Dr. Miranda also agreed to (i) not disclose to any unauthorized person or use for his own account any confidential information without the prior written consent of the Company or the board of directors, (ii) will not, directly or indirectly encourage, solicit, induce (or attempt to encourage, solicit or induce) any employee or agent of the Company that was employed (or otherwise engaged) at the time of his separation during his employment and for 24 months after his separation from that employment for any reason; (iii) will not, directly or indirectly, have any ownership interest in, work for, advise, manage, act as an agent or consultant for, or have any business connection or business or employment relationship with any entity or person which competes with Company; (iv) not, directly or indirectly, have any ownership interest in, work for, advise, manage, act as an agent or consultant for, or have any business connection or business or employment relationship with any entity or person which competes with the Company during his employment and (v) will not, directly or indirectly and in a competitive capacity own, manage, finance, operate, control or participate in ownership, management, or operation of, act as an agent, consultant, or be employed with, any business engaged in the design, manufacture, marketing, sale or servicing of any service or product with which Dr. Miranda was involved during his last year of employment with the Company; or which the Company is developing, producing, marketing, selling or servicing (or plans to develop, produce, market, sale or service) and about which Dr. Miranda gained any confidential information in the course of his employment with the Company for a period of 24 months after his separation from the Company.
| 82 |
Dr. Thomas Carrico, Chief Regulatory Officer, entered into an employment agreement with the Company, dated August 9, 2022 and amended on May 4, 2023, which has a two-year initial term and provides for a base salary of $275,000 with annual compensation increase. Dr. Carrico will also receive a one-time incentive payment in the amount of $135,655, which amount includes accrued and unpaid salary and a bonus to incentivize Dr. Carrico to remain with the Company for future service. Neither the accrued and unpaid salary nor bonus was the subject of any contract or agreement between Dr. Carrico and the Company prior to the execution of the employment agreement. The agreement contemplated that the incentive payment would be paid by December 15, 2022, but due to administrative impracticability, payment was made with a portion of the proceeds from the IPO in August 2023. In addition, Dr. Carrico shall be entitled to payment of a deferred bonus in an amount equal to (i) the aggregate of the strike price or exercise price of all 306,236 unexercised options to purchase stock or shares of the Company held by Dr. Carrico (the “Aggregate Strike Price”) plus (ii) a tax gross-up payment on the Aggregate Strike Price reasonably calculated by the Company at the highest marginal rates so that after payment of all ordinary income taxes on such Aggregate Strike Price, there remains an amount sufficient to pay such ordinary income taxes. The special deferred bonus will be paid in substantially equal 20% installments (the “Annual Deferred Bonus Payment”) on January 2 on each of 2024, 2025, 2026, 2027, and 2028 (the “Scheduled Payment Dates”), with a condition that on or before each Scheduled Payment Date, Dr. Carrico shall exercise at least 61,274 of the stock option. None of stock options has been exercised, therefore, no special deferred bonus has been paid as of the date of this prospectus. Dr. Carrico may exercise any portion of the stock options after a Scheduled Payment Date; provided, however, an exercise after the applicable Scheduled Payment Date shall result in forfeiture of the related Annual Deferred Bonus Payment.
If the employment agreement is terminated by the Company without cause (as defined in the employment agreement), Dr. Carrico will receive any accrued compensation (as defined in the employment agreement) and is entitled to certain amount of severance payments as follows:
| ● | If termination occurs during the initial term, the Company shall provide Dr. Carrico with severance compensation in the form of salary continuation at his Base Salary as of the termination date and ending the later of (i) 6 months or (ii) on the expiration date of the initial term. | |
| ● | If termination occurs after the initial term, the severance payment shall be the amount equal to one half (1/2) of Dr. Carrico’s Base Salary as of the termination date. | |
| ● | In addition, if termination occurs during the initial term, as part of the severance payment, we agreed to pay Dr. Carrico reimbursement of his Medicare, Medicare Supplement and prescription drug coverage insurance premiums for continuation of health coverage for 18 months post termination. |
If the employment agreement is terminated by the Company for cause, Dr. Carrico will forfeit any unpaid Annual Deferred Bonus Payment, but will receive any unpaid base salary that has been earned at the time of such termination, reimbursement of any expenses properly incurred prior to the Dr. Carrico’s termination date; and (iii) accrued and unused PTO, if any, in accordance with the Company’s PTO policy in effect on Dr. Carrico’s termination date.
Dr. Carrico may terminate the employment agreement without good reason upon more than thirty (30) days’ prior written notice or for good reason without prior written consent, and will receive accrued compensation (as defined in the employment agreement) and the unpaid balance of the special deferred bonus.
In the event that Dr. Carrico is terminated without cause after this offering, non-forfeited portions of the deferred bonus will be paid in equal installments over the remaining scheduled payment dates, if vested options as scheduled are exercised before the scheduled payment dates. A deferred bonus tranche will be forfeited if its corresponding options have expired.
On a change in control (as defined in the employment agreement), full vesting of the Annual Deferred Bonus Payment will occur and be paid in a single lump sum within 30 days after the change in control. The offering shall not be considered a change in control.
Pursuant to the employment agreement, Dr. Carrico also agreed to (i) not disclose to any unauthorized person or use for his own account any confidential information without the prior written consent of the Company or the board of directors, (ii) will not, directly or indirectly encourage, solicit, induce (or attempt to encourage, solicit or induce) any employee or agent of the Company that was employed (or otherwise engaged) at the time of his separation during his employment and for 24 months after his separation from that employment for any reason; (iii) will not, directly or indirectly, have any ownership interest in, work for, advise, manage, act as an agent or consultant for, or have any business connection or business or employment relationship with any entity or person which competes with Company; (iv) not, directly or indirectly, have any ownership interest in, work for, advise, manage, act as an agent or consultant for, or have any business connection or business or employment relationship with any entity or person which competes with the Company during his employment and (v) will not, directly or indirectly and in a competitive capacity own, manage, finance, operate, control or participate in ownership, management, or operation of, act as an agent, consultant, or be employed with, any business engaged in the design, manufacture, marketing, sale or servicing of any service or product with which Dr. Carrico was involved during his last year of employment with the Company; or which the Company is developing, producing, marketing, selling or servicing (or plans to develop, produce, market, sale or service) and about which Dr. Carrico gained any confidential information in the course of his employment with the Company for a period of 24 months after his separation from the Company.
| 83 |
Non-Employee Director Compensation
The following table presents the compensation awarded to or earned by or paid to all individuals who served as non-employee directors during the year ended December 31, 2023. We do not provide additional compensation to directors who are our employees for also serving as a director.
| Name | Fees Earned ($) | Stock Awards Earned | Total ($) | |||||||||
| Timothy R. Henrichs (1) | 23,836 | 19,863 | 43,699 | |||||||||
| Bradley M. Watkins | 23,836 | 19,863 | 43,699 | |||||||||
| Jane E. Keyser | 23,836 | 19,863 | 43,699 | |||||||||
(1) On January 30, 2024, Timothy R. Henrichs resigned as a member of the board of directors, effective February 2, 2024 and became the Company’s Chief Financial Officer.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth certain information known to us with respect to the beneficial ownership of our common stock as of April 12, 2024 based on 6,594,897 common shares outstanding.
We have determined beneficial ownership in accordance with the rules of the Securities and Exchange Commission, and the information is not necessarily indicative of beneficial ownership for any other purpose. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities as well as any common stock that the person has the right to acquire within 60 days of April 12, 2024 through the exercise of stock options or other rights. These shares are deemed to be outstanding and beneficially owned by the person holding those options for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them.
| Beneficially Ownership Common Stock | ||||||
| Shares | Percentage | |||||
| Name of beneficial owner 5% shareholders: | ||||||
| Masimo Corporation (1) | 821,327 | 11.9 | % | |||
| Brian P. Hannasch (2) | 562,420 | 8.5 | % | |||
| PBF Venture Group LLC (3) | 415,258 | 6.3 | % | |||
| Gary Peterson | 400,120 | 6.1 | % | |||
| 108 Sussex LLC (4) | 372,171 | 5.6 | % | |||
| Named executive officers and directors: | ||||||
| Brian Carrico | 84,118 | 1.3 | % | |||
| Timothy Henrichs | - | * | ||||
| Adrian Miranda | 67,441 | 1.0 | % | |||
| Thomas Carrico | 66,247 | 1.0 | % | |||
| Christopher Robin Brown | 792,837 | 12.0 | % | |||
| Bradley Mitch Watkins | - | * | ||||
| Beth Keyser | - | * | ||||
| Kristin Ferge | - | * | ||||
| All executive officers and directors as a group ( eight (8) persons) | 1,010,643 | 14.9 | % | |||
| * | Less than 1%. |
(1) The business address for Masimo Corporation is 52 Discovery, Irvine, California 92618. Shares of common stock beneficially owned includes 289,779 warrants.
(2) Shares of common stock beneficially owned includes 12,852 warrants.
(3) The business address for PBF Venture Group LLC is 5501 Woodrow Avenue, Austin, Texas 78756.
(4) The business address for 108 Sussex LLC is 304 South Euclid Avenue, Westfield, New Jersey 07090.
| 84 |
Item 13. Certain Relationships and Related Transactions and Director Independence
The following includes a summary of transactions since January 1, 2022, to which we have been a party in which the amount involved exceeded or will exceed the lesser of (i) $120,000 and (ii) one percent (1%) of the average of our total assets at year-end for the prior two fiscal years, and in which any of our directors, executive officers or, to our knowledge, beneficial owners of more than 5% of our capital stock or any member of the immediate family of any of the foregoing persons had or will have a direct or indirect material interest, other than equity and other compensation, termination, change in control and other arrangements, which are described under “Item 11. Executive Compensation.” We also describe below certain other transactions with our directors, executive officers and stockholders.
The Company has two demand notes receivable from its two founding shareholders, Christopher Robin Brown and Gary Peterson, related to the sale of common stock on January 1, 2016. The initial balances of both notes were $506,400, with interest calculated monthly based on applicable federal rates. No payments have been received on the notes. As of December 31, 2023, the balances of both notes were $506,400. The entire $1,012,800 balance has been fully reserved as of December 31, 2023.
The Company has loans payable to Christopher Robin Brown, one of our founders and a member of our board of directors, related to funding needs for operations with original principal amounts of $55,000 and $50,000 each bearing interest at 15% per annum. In August 2023, the Company used a portion of the proceeds from the IPO to repay these loans payable in full, including $37,103 in interest.
Mr. Watkins, Director, provided certain sales, marketing and commercialization consulting services to the Company prior to his appointment to the Board of Directors. For the fiscal years ended December 31, 2023 and 2022, the Company paid Mr. Watkins $11,083 and $3,523, respectively.
John Seale, our former Chief Financial Officer, is also the managing partner of RBSK. Mr. Seale, through RBSK, has provided accounting services since 2017. For the fiscal years ended December 31, 2023 and 2022, the Company paid RBSK $312,803 and $116,542, respectively, for accounting services.
We have adopted a formal policy that our executive officers, directors, holders of more than 5% of any class of our voting securities, and any member of the immediate family of and any entity affiliated with any of the foregoing persons, are not permitted to enter into a related party transaction with us without the prior consent of our Audit Committee, or other independent members of our Board of Directors if it is inappropriate for our audit committee to review such transaction due to a conflict of interest. Any request for us to enter into a transaction with an executive officer, director, principal shareholder, or any of their immediate family members or affiliates, in which the amount involved exceeds the lesser of $120,000 or 1% of the average of our total assets at year-end for the last two completed fiscal years must first be presented to our audit committee for review, consideration and approval. In approving or rejecting any such proposal, our Audit Committee is to consider the relevant facts and circumstances available and deemed relevant to the audit committee, including, but not limited to, whether the transaction is on terms no less favorable than terms generally available to an unaffiliated third party under the same or similar circumstances and the extent of the related party’s interest in the transaction.
Item 14. Principal Accounting Fees and Services
Fees Billed for Audit and Non-Audit Services
The following table presents for each of the last two fiscal years the aggregate fees billed in connection with the audits of our financial statements and other professional services rendered by our independent registered public accounting firm Rosenberg Rich Baker Berman, P.A.
| 2023 | 2022 | |||||||
| Audit Fees (1) | $ | 221,570 | $ | 162,165 |
(1) Audit Fees. These are fees for professional services for the audit of our annual financial statements, and for the review of the financial statements included in our filings on Form 10-K and Form 10-Q, and for services that are normally provided in connection with statutory and regulatory filings or engagements.
| 85 |
PART IV
ITEM 15. EXHIBIT AND FINANCIAL STATEMENT SCHEDULES
| (a) | 1. Financial Statements |
| The financial statements and Report of Independent Registered Public Accounting Firm are listed in the “Index to Financial Statements and Schedules” on page F-1 and included from F-2 onwards. | |
| 2. Financial Statement Schedules | |
| All schedules for which provision is made in the applicable accounting regulations of the Securities and Exchange Commission (the “Commission”) are either not required under the related instructions, are not applicable (and therefore have been omitted), or the required disclosures are contained in the financial statements included herein. | |
| 3. Exhibits (including those incorporated by reference). |
(b) Exhibits
| 86 |
| 87 |
| 21.1 | List of Subsidiaries of Neuraxis, Inc. (incorporated by reference to exhibit 21.1 to Registration Statement on Form S-1, filed on January 10, 2023) | |
| 31.1* | Certification of Chief Executive Officer Pursuant to Section 302 of the Sarbanes-Oxley Act Of 2002 | |
| 31.2* | Certification of Chief Financial Officer Pursuant to Section 302 of the Sarbanes-Oxley Act Of 2002 | |
| 32.1** | Certification of Chief Executive Officer and Chief Financial Officer Pursuant to Section 906 of the Sarbanes-Oxley Act Of 2002 | |
| 97.1* | ||
| 101.INS* | Inline XBRL Instance Document. | |
| 101.SCH* | Inline XBRL Taxonomy Extension Schema Document. | |
| 101.CAL* | Inline XBRL Taxonomy Extension Calculation Linkbase Document. | |
| 101.DEF* | Inline XBRL Taxonomy Extension Definition Linkbase Document. | |
| 101.LAB* | Inline XBRL Taxonomy Extension Label Linkbase Document. | |
| 101.PRE* | Inline XBRL Taxonomy Extension Presentation Linkbase Document. | |
| 104* | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). |
† Executive compensation plan or arrangement.
* Filed herewith.
** Furnished herewith.
ITEM 16. FORM 10-K SUMMARY
Not Applicable.
| 88 |
Index to Financial Statements
FOR THE YEARS ENDED DECEMBER 31, 2023 AND 2022
| 89 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To
the Board of Directors and
Stockholders of Neuraxis, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Neuraxis, Inc. (the Company) as of December 31, 2023 and 2022, and the related statements of operations, stockholders’ equity (deficit), and cash flows for each of the years in the two year period ended December 31, 2023, and the related notes (collectively referred to as the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2023 and 2022, and the results of its operations and its cash flows for each of the years then ended, in conformity with accounting principles generally accepted in the United States of America.
Restatement of Previously Issued Financial Statements
As discussed in Note 19 to the financial statements, the Company restated its 2023 quarterly information for an error in the classification of warrants.
Emphasis of Matter Regarding Going Concern
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company is not profitable, has recorded negative cash flows from operations, and will need substantial capital to support its operations. This raises substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters also are described in Note 2. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
| /s/
|
|
| We have served as the Company’s auditor since 2022. | |
| April 16, 2024 |
| F-1 |
Neuraxis, Inc.
Balance Sheets
| December 31, 2023 | December 31, 2022 | |||||||
| Assets | ||||||||
| Current Assets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Accounts receivable, net | ||||||||
| Inventories | ||||||||
| Prepaids and other current assets | ||||||||
| Total current assets | ||||||||
| Property and Equipment, at cost: | ||||||||
| Less - accumulated depreciation | ( | ) | ( | ) | ||||
| Property and equipment, net | ||||||||
| Other Assets: | ||||||||
| Deferred offering costs | ||||||||
| Operating lease right of use asset, net | ||||||||
| Intangible assets, net | ||||||||
| Total Assets | $ | $ | ||||||
Notes to financial statements are an integral part of these statements.
| F-2 |
Neuraxis, Inc.
Balance Sheets
| December 31, 2023 | December 31, 2022 | |||||||
| Liabilities | ||||||||
| Current Liabilities: | ||||||||
| Accounts payable | $ | $ | ||||||
| Accrued expenses | ||||||||
| Notes payable | ||||||||
| Current portion of operating lease payable | ||||||||
| Notes payable - related party | ||||||||
| Notes payable - convertible notes, net of unamortized discount of $ | ||||||||
| Customer deposits | ||||||||
| Derivative liabilities | ||||||||
| Warrant liabilities | ||||||||
| Total current liabilities | ||||||||
| Non-current Liabilities: | ||||||||
| Operating lease payable, net of current portion | ||||||||
| Total non-current liabilities | ||||||||
| Total liabilities | ||||||||
| Commitments and contingencies (see note 14) | ||||||||
| Stockholders’ Deficit | ||||||||
| Convertible Series A Preferred stock, $ par value; shares authorized; issued and outstanding as of December 31, 2023 and as of December 31, 2022 | ||||||||
| Convertible Series Seed Preferred Stock, $ par value; shares authorized; issued and outstanding as of December 31, 2023 and as of December 31, 2022 | ||||||||
| Common stock, $ par value; shares authorized; issued and outstanding as of December 31, 2023 and as of December 31, 2022 | ||||||||
| Additional paid in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total stockholders’ deficit | ( | ) | ( | ) | ||||
| Total Liabilities and Stockholders’ Deficit | $ | $ | ||||||
Notes to financial statements are an integral part of these statements.
| F-3 |
Neuraxis, Inc.
Statements of Operations
| For the Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Net Sales | $ | $ | ||||||
| Cost of Goods Sold | ||||||||
| Gross Profit | ||||||||
| Selling Expenses | ||||||||
| Research and Development | ||||||||
| General and Administrative | ||||||||
| Operating Loss | ( | ) | ( | ) | ||||
| Other (Expense) Income: | ||||||||
| Financing charges | ( | ) | ( | ) | ||||
| Interest expense | ( | ) | ( | ) | ||||
| Change in fair value of warrant liability | ||||||||
| Change in fair value of derivative liability | ||||||||
| Amortization of debt discount and issuance cost | ( | ) | ( | ) | ||||
| Extinguishment of debt liabilities | ( | ) | ||||||
| Other income | ||||||||
| Other expense | ( | ) | ||||||
| Total other (expense) income, net | ( | ) | ( | ) | ||||
| Net Loss | $ | ( | ) | $ | ( | ) | ||
| Per-share Data | ||||||||
| Basic and diluted loss per share | $ | ) | $ | ) | ||||
| Weighted Average Shares Outstanding | ||||||||
| Basic and diluted | ||||||||
Notes to financial statements are an integral part of these statements.
| F-4 |
Neuraxis, Inc.
Statements of Stockholders’ Deficit
For the Twelve Months Ended December 31, 2023 and 2022
| Convertible Series A Preferred Stock | Convertible Series Seed Preferred Stock | Common Stock | Additional Paid In | Accumulated | Stockholders’ | |||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Capital | Deficit | Deficit | ||||||||||||||||||||||||||||
| Balances as of January 1, 2022 | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||||||||||||||||
| Stock based compensation | — | — | — | |||||||||||||||||||||||||||||||||
| Common stock issued upon signing of notes payable | — | — | ||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | ( | ) | ( | ) | |||||||||||||||||||||||||||||
| Balances as of December 31, 2022 | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||||||||||||||||
| Common stock issued upon initial public offering | — | — | ||||||||||||||||||||||||||||||||||
| Common stock issued upon preferred stock conversion | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||||||||
| Common stock issued upon debt conversion | — | — | ||||||||||||||||||||||||||||||||||
| Common stock issued from agreements | — | — | ||||||||||||||||||||||||||||||||||
| Reclassification of warrant liability to equity | — | — | — | |||||||||||||||||||||||||||||||||
| Warrants exercised | — | — | ||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | ( | ) | ( | ) | |||||||||||||||||||||||||||||
| Balances as of December 31, 2023 | $ | $ | $ | $ | $ | ( | ) | $ | ( | ) | ||||||||||||||||||||||||||
Notes to financial statements are an integral part of these statements.
| F-5 |
Neuraxis, Inc.
Statements of Cash Flows
| For the Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Cash Flows from Operating Activities | ||||||||
| Net Loss | $ | ( | ) | $ | ( | ) | ||
| Adjustments to reconcile net loss to net cash used by operating activities: | ||||||||
| Amortization of debt discount and issuance cost | ||||||||
| Depreciation and amortization | ||||||||
| Provisions for losses on accounts receivable | ||||||||
| Non-cash lease expense | ||||||||
| Non-cash interest expense | ||||||||
| Stock based compensation | ||||||||
| Extinguishment of debt liabilities | ||||||||
| Issuance of common stock for non-cash consideration | ||||||||
| Finance Charges | ||||||||
| Change in fair value of derivative liabilities | ( | ) | ( | ) | ||||
| Change in fair value of warrant liabilities | ( | ) | ( | ) | ||||
| Changes in operating assets and liabilities: | ||||||||
| Accounts receivable | ( | ) | ||||||
| Inventory | ( | ) | ||||||
| Prepaids and other current assets | ( | ) | ||||||
| Accounts payable | ( | ) | ||||||
| Accrued expenses | ( | ) | ||||||
| Customer deposits | ( | ) | ||||||
| Operating lease liability | ( | ) | ( | ) | ||||
| Net cash used by operating activities | ( | ) | ( | ) | ||||
| Cash Flows from Investing Activities | ||||||||
| Additions to property and equipment | ( | ) | ( | ) | ||||
| Additions to intangible assets | ( | ) | ( | ) | ||||
| Net cash used by investing activities | ( | ) | ( | ) | ||||
| Cash Flows from Financing Activities | ||||||||
| Proceeds from issuance of common stock, net of issuance costs | ||||||||
| Deferred offering costs paid | ( | ) | ||||||
| Proceeds from exercised warrants | ||||||||
| Principal payments on notes payable | ( | ) | ( | ) | ||||
| Proceeds from notes payable | ||||||||
| Proceeds from convertible notes | ||||||||
| Financing fees paid | ( | ) | ( | ) | ||||
| Net cash provided by financing activities | ||||||||
| Net Decrease in Cash and Cash Equivalents | ( | ) | ( | ) | ||||
| Cash and Cash Equivalents at Beginning of Period | ||||||||
| Cash and Cash Equivalents at End of Period | $ | $ | ||||||
| Supplemental Disclosure of Non-Cash Operating Activities | ||||||||
| Cash paid for interest | $ | $ | ||||||
| Cash paid for income taxes | ||||||||
| Supplemental Schedule of Non-Cash Investing and Financing Activities | ||||||||
| Fair value of warrant liabilities of warrants from convertible notes | $ | $ | ||||||
| Fair value of derivative liabilities of conversion feature from convertible notes | ||||||||
| Prior year offering costs paid | ||||||||
| Reclassification of warrant liability to equity | ||||||||
| Conversion of preferred stock to common stock | ||||||||
Notes to financial statements are an integral part of these statements.
| F-6 |
Neuraxis, Inc.
Notes to Financial Statements
1. Basis of Presentation, Organization and Other Matters
Neuraxis, Inc. (“we,” “us,” the “Company,” or “Neuraxis”) was established in 2011 and incorporated in the state of Indiana on April 17, 2012, under the name of Innovative Health Solutions, Inc. The name was changed to Neuraxis, Inc. in March of 2022. Additionally, the Company filed a Certificate of Conversion to become a Delaware corporation on June 23, 2022. The authorized shares were increased, and a par value established.
On
January 10, 2023, the Company’s board of directors authorized a
As part of the conversion to a Delaware corporation, the total number of shares of all classes of stock which the Corporation shall have authority to issue is (1) shares of Common Stock, par value $ per share (“Common Stock”) and (ii) shares of Preferred Stock, par value $ per share (“Preferred Stock”), of which is hereby designated as “Series A Preferred Stock” and of which is hereby designated as “Series Seed Preferred Stock” with the rights, preferences, powers, privileges and restrictions, qualifications and limitations set forth in this Article IV of the Delaware Certificate of Incorporation. All share amounts have been retroactively restated to give effect to these changes.
On
August 9, 2023, the Company consummated an initial public offering, conducted on a firm commitment basis, pursuant to which it sold shares of its common stock at a price of $per share, resulting in gross proceeds to the
Company of $
The Company is headquartered in Carmel, Indiana. The Company specializes in the development, production, and sale of medical neuromodulation devices.
The Company has developed three FDA cleared products, the IB-STIM (DEN180057, 2019), the NSS-2 Bridge (DEN170018, 2017), and the original 510(K) clearance (K140530, 2014).
| ● | The IB-STIM is a percutaneous electrical nerve field stimulator (PENFS) device that is indicated in patients 11-18 years of age with functional abdominal pain associated with irritable bowel syndrome. The IB-STIM currently is the only product marketed and sold by the Company. | |
| ● | The
NSS-2 Bridge is a percutaneous nerve field stimulator (PNFS) device indicated for use in the reduction of the symptoms of opioid
withdrawal. The NSS-2 Bridge device was licensed to Masimo Corporation in April 2020, and the Company received a one-time licensing
fee of $ | |
| ● | The original 510(K) device was the EAD, an electroacupuncture device, now called NeuroStim. The EAD is no longer being manufactured, sold or distributed but reserved only for research purposes. |
| F-7 |
Neuraxis, Inc.
Notes to Financial Statements
2. Summary of Significant Accounting Policies
The summary of significant accounting policies of Neuraxis, Inc. is presented to assist in understanding the Company’s financial statements. The financial statements and notes are representations of the Company’s management, who are responsible for their integrity and objectivity. These accounting policies conform to U.S. generally accepted accounting principles and have been consistently applied in the preparation of the financial statements.
Preparing the Company’s financial statements in conformity with accounting principles generally accepted in the United States of America (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period.
Use of Estimates and Critical Accounting Estimates and Assumptions
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the dates of the financial statements and the reported amounts of revenues and expenses during the reporting periods.
These significant accounting estimates or assumptions bear the risk of change due to the fact that there are uncertainties attached to these estimates or assumptions, and certain estimates or assumptions are difficult to measure or value.
Management bases its estimates on historical experience and on various assumptions that are believed to be reasonable in relation to the financial statements taken as a whole under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources.
Management regularly evaluates the key factors and assumptions used to develop the estimates utilizing currently available information, changes in facts and circumstances, historical experience, and reasonable assumptions. After such evaluations, if deemed appropriate, those estimates are adjusted accordingly. The Company uses estimates in accounting for, among other items, revenue recognition, allowance for credit losses, stock-based compensation, income tax provisions, excess and obsolete inventory reserve, and impairment of property and equipment, and intellectual property. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid debt instruments purchased with a maturity of three months or less to be cash equivalents. The Company did not hold any cash equivalents as of December 31, 2023 and 2022.
Trade Accounts Receivable and Allowance for Credit Losses
Trade
accounts receivable are stated at the amount management expects to collect from outstanding balances, net of an allowance for credit
losses. Management evaluates many factors when determining the collectability of specific customer accounts, including, but not limited
to, creditworthiness, past transaction and payment history, current economic industry trends and changes in payment terms. We use assumptions
and judgment based on the best available facts and circumstances to estimate and record an allowance. The allowance for credit losses
was $
| F-8 |
Neuraxis, Inc.
Notes to Financial Statements
Customer Deposits
Customer
deposits consists of billings, payments, and returned devices from clients in advance of revenue recognition. The Company will recognize
the customer deposits over the next year. As of December 31, 2023, and 2022, the Company had customer deposits of $
Inventories
Inventories
are valued at the lower of cost or net realizable value. Cost is determined using the weighted average method. The inventory is comprised
of finished medical devices on hand. Certain components within the devices have an expiration date that are removed from current inventory
and expensed at the date of expiration. For the years ended December 31, 2023 and 2022, $
Deferred Offering Costs
Deferred
offering costs consist of costs incurred in connection with the preparation of an initial public offering. These costs, together with
the underwriting discounts and commissions, were charged to additional paid in capital upon the IPO. As of December 31, 2023 and 2022,
the Company had deferred offering costs of $ and $
Property and Equipment
Property and equipment are recorded at cost less accumulated depreciation. Depreciation is computed using the straight-line method over the estimated useful lives of the assets.
Depreciation is calculated using the following estimated useful lives:
| Classification | Years | |
| Machinery and Equipment | ||
| Furniture and Fixtures | ||
| Computer Hardware | ||
| Leasehold Improvements |
Depreciation
expense was $
Research and Development
Costs for research and development are expensed as incurred. Research and development expenses consist primarily of clinical research studies and new product development.
Intangible Assets
Intangible assets consist of software, patents and a trademark. Intangible assets are stated at their historical cost and amortized on a straight-line basis over their expected useful lives. Capitalized patent costs, net of accumulated amortization, includes legal costs incurred for patent applications. In accordance with ASC 350, once a patent is granted, we amortize the capitalized patent costs over the remaining life of the patent using the straight-line method. If the patent is not granted, we write off any capitalized patent costs at that time.
The
Company entered into an agreement for a trademark related to the Company’s name on July 11, 2022. The agreement called for an initial
payment of $
The
Company entered into an option agreement on April 12, 2023 to enter into a royalty-bearing licensing agreement to bring the optionor’s
invention to commercialization. The agreement required an initial payment of $
| F-9 |
Neuraxis, Inc.
Notes to Financial Statements
We review intangible assets for impairment annually or when events or circumstances indicate that their carrying amount may not be recoverable. During the years ended December 31, 2023, and 2022, the Company recorded no impairment charges for intangible assets.
Amortization
expense was $
Income Taxes
The Company recognizes deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the Company’s financial statements and tax returns. Deferred tax assets and liabilities are determined based upon the differences between the financial statement carrying amounts, and the tax bases of existing assets and liabilities for the loss and credit carryforwards using enacted tax rates expected to be in effect in the year in which the differences are expected to reverse. Deferred tax assets are reduced by a valuation allowance if it is more likely than not that these assets may not be realized. The Company determines whether a tax position will be sustained upon examination. If it is more likely than not that a position will be sustained, none of the benefit attributable to the position is recognized. The tax benefit to be recognized for any tax position that meets the more likely than not recognition threshold is calculated as the largest amount that is more than 50% likely of being realized upon resolution of the contingency. The Company accounts for interest and penalties related to uncertain tax positions as part of its provision for income taxes.
Based on the results of management’s evaluation, adoption of the rules did not have a material effect on the Company’s financial statements. Further, no interest or penalties have been accrued or charged to expense as of December 31, 2023 and 2022 and for the years then ended.
The Company’s income tax returns are subject to examination by the taxing authorities until the expiration of the related statutes of limitations on those tax returns. In general, the federal and state income tax returns have a three-year statute of limitations. As of December 31, 2023, the following tax years are subject to examination:
| Jurisdiction | Open Years for Filed Returns | Return to File in 2024 | ||
| Federal | 2020 – 2022 | 2023 | ||
| Various States | 2020 – 2022 | 2023 |
Advertising Cost
Advertising
costs are expensed as incurred and amounted to $
Derivative Liabilities
The Company accounts for derivative financial instruments as either equity or liabilities in accordance with ASC Topic 815, Derivatives and Hedging, or ASC 815, based on the characteristics and provisions of each instrument. Embedded derivatives are required to be bifurcated from the host instruments and recorded at fair value if the derivatives are not clearly and closely related to the host instruments on the date of issuance. Derivative instrument liabilities are classified in the balance sheets as current or non-current based on whether or not net-cash settlement of the derivative instrument could be required within 12 months of the balance sheet date.
The Company utilizes a Monte Carlo simulation model for the common stock conversion feature of the notes payable that have an option to convert at a variable number of shares to compute the fair value of the derivative and to mark to market the fair value of the derivative at each balance sheet date. The inputs utilized in the application of the Monte Carlo model included a stock price, an expected remaining term of each note as of the valuation date, estimated volatility, drift, and a risk-free rate. The Company records the change in the fair value of the derivative as other income or expense in the Statements of Operations.
| F-10 |
Neuraxis, Inc.
Notes to Financial Statements
Warrant Liabilities
Management evaluates all of the Company’s financial instruments, including issued warrants to purchase its Class A common stock, to determine if such instruments are liabilities or contain features that qualify as embedded derivatives, pursuant to ASC 480 and ASC 815-15. The classification of derivative financial instruments, including whether such instruments should be recorded as liabilities or as equity, is reassessed at the end of each reporting period.
The Company utilizes a Monte Carlo simulation model for warrants that have an option to convert at a variable number of shares to compute the fair value of the derivative and to mark to market the fair value of the derivative at each balance sheet date. The inputs utilized in the application of the Monte Carlo model included a starting stock price, an expected remaining term of each warrant as of the valuation date, estimated volatility, drift, and a risk-free rate. The Company records the change in the fair value of the warrant as other income or expense in the statements of operations.
Fair Value Measurements
The Company accounts for financial instruments in accordance with ASC 820, Fair Value Measurements and Disclosures (“ASC 820”). ASC 820 establishes a fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value. The hierarchy gives the highest priority to unadjusted quoted prices in active markets for identical assets or liabilities (Level 1 measurements) and the lowest priority to unobservable inputs (Level 3 measurements). The three levels of the fair value hierarchy under ASC 820 are described below:
Level 1 – Quoted prices (unadjusted) for identical unrestricted assets or liabilities in active markets that the reporting entity has the ability to access as of the measurement date.
Level 2 – Significant other observable inputs other than Level 1 prices such as quoted prices for similar assets or liabilities: quoted prices in markets that are not active; or financial instruments for which all significant inputs are observable or can be corroborated by observable market date, either directly or indirectly.
Level 3 – Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities. These unobservable inputs reflect that reporting entity’s own assumptions about assumptions that market participants would use in pricing the asset or liability. Level 3 assets and liabilities include financial instruments whose value is determined using pricing models, discounted cash flow methodologies, or similar techniques, as well as instruments for which the determination of fair value require significant management judgment or estimation.
The Company’s Level 1 assets/liabilities include cash, accounts receivable, accounts payable, prepaids, and other current assets. Management believes the estimated fair value of these accounts on December 31, 2023 approximate their carrying value as reflected in the balance sheets due to the short-term nature of these instruments or the use of market interest rates for debt instruments.
The Company’s Level 3 assets/liabilities include derivative and warrant liabilities. Inputs to determine fair value are generally unobservable and typically reflect management’s estimates of assumptions that market participants would use in pricing the asset or liability. The fair values are therefore determined using model-based techniques, including option pricing models and discounted cash flow models. The valuation techniques involve management’s estimates and judgment based on unobservable inputs. The fair value estimates may not be indicative of the amounts that would be realized in a market exchange. Additionally, there may be inherent uncertainties or changes in the underlying assumptions used, which could significantly affect the current or future fair value estimates. Unobservable inputs used in the models are significant to the fair values of the assets and liabilities.
| F-11 |
Neuraxis, Inc.
Notes to Financial Statements
The following tables provides a summary of the relevant assets and liabilities that are measured at fair value on recurring basis:
Fair Value Measurements as of
December 31, 2023
| Total | (Level 1) | (Level 2) | (Level 3) | |||||||||||||
| Liabilities: | ||||||||||||||||
| Warrant liabilities | $ | $ | $ | $ | ||||||||||||
| Derivative liabilities | $ | $ | $ | $ | ||||||||||||
| Total Liabilities | $ | $ | $ | $ | ||||||||||||
Fair Value Measurements as of
December 31, 2022
| Total | (Level 1) | (Level 2) | (Level 3) | |||||||||||||
| Liabilities: | ||||||||||||||||
| Warrant liabilities | $ | $ | $ | $ | ||||||||||||
| Derivative liabilities | $ | $ | $ | $ | ||||||||||||
| Total Liabilities | $ | $ | $ | $ | ||||||||||||
The following table shows the valuation methodology and unobservable inputs for Level 3 assets and liabilities measured at fair value on recurring basis as of December 31, 2023 and December 31, 2022:
| Fair Value As of | Fair Value | Valuation | Unobservable | |||||||||
| December 31, 2023 | December 31, 2022 | Methodology | Inputs | |||||||||
| Warrant liabilities | $ | $ | Monte Carlo model | Project simulated cash flows | ||||||||
| Derivative liabilities | $ | $ | Monte Carlo model | Project simulated cash flows | ||||||||
There were no transfers between any of the levels during the years ended December 31, 2023 and year ended December 31, 2022. In addition to assets and liabilities that are recorded at fair value on a recurring basis, the Company’s assets and liabilities are also subject to nonrecurring fair value measurements. As of December 31, 2023 and 2022, the Company had no assets that were measured on a nonrecurring basis.
Earnings
or loss per share (“EPS”) is computed by dividing net income (loss), net of preferred stock dividends, by the weighted average
number of shares of common stock outstanding during the period. Diluted EPS is computed by dividing net income (loss) by the weighted
average of all potentially dilutive shares of common stock that were outstanding during the periods presented. Preferred stock dividends
(not declared or paid) were $
Basic net loss per common share is computed by dividing net loss attributable to common stockholders by the weighted-average number of common shares outstanding during the period. Diluted net loss per common share is determined using the weighted-average number of common shares outstanding during the period, adjusted for the dilutive effect of common stock equivalents. In periods when losses are reported, which is the case for December 31, 2023 and 2022 presented in these financial statements, the weighted-average number of common shares outstanding excludes common stock equivalents because their inclusion would be anti-dilutive.
| F-12 |
Neuraxis, Inc.
Notes to Financial Statements
| 2023 | 2022 | |||||||
| Convertible Series A Preferred Stock | ||||||||
| Convertible Series Seed Preferred Stock | ||||||||
| Options | ||||||||
| Pre-Funded Warrants for Convertible Series A Preferred Stock | ||||||||
| Warrants | ||||||||
| Convertible Bridge Debt | ||||||||
| Totals | ||||||||
| For the Years Ended December 31, | ||||||||
| 2023 | 2022 | |||||||
| Numerator | ||||||||
| Net loss | $ | ( | ) | ( | ) | |||
| Preferred stock dividends | ( | ) | ||||||
| ( | ) | ( | ) | |||||
| Denominator | ||||||||
| Weighted-average shares of common stock outstanding - basic and diluted | ||||||||
| Basic and diluted net loss per share | $ | ) | $ | ) | ||||
The Company accounts for all stock-based payments and awards at fair value. The Company recognizes its stock-based compensation expense using the straight-line method. Compensation cost is not adjusted for estimated forfeitures, but instead is adjusted upon an actual forfeiture of a stock option.
The Company accounts for the granting of stock options to employees and non-employees using the fair value method whereby all awards are measured at fair value on the date of the grant. The fair value of all employee stock options is expensed over the requisite service period with a corresponding increase to additional paid-in capital. Upon exercise of stock options, the consideration paid by the option holder is recorded in additional paid-in capital, while the par value of the shares received is reclassified from additional paid in capital to common stock.
Stock-based payments to non-employees are measured based on the fair value of the equity instrument issued. Compensation expense for non-employee stock awards is recognized over the requisite service period following the measurement of the fair value on the grant date.
The Company uses the Black-Scholes option-pricing model to calculate the fair value of stock options. The use of the Black-Scholes option-pricing model requires management to make assumptions with respect to the expected term of the option, the expected volatility of the common stock consistent with the expected term of the option, risk-free interest rates, the value of the common stock and expected dividend yield of the common stock. Changes in these assumptions can materially affect the fair value estimate.
Revenue Recognition
Neuraxis, Inc. specializes in the development, production, and sale of medical neuromodulation devices to healthcare providers primarily located in the United States. Patented and trademarked neuromodulation devices is the Company’s major product line. Products are generally transferred at a point in time (rather than over time). Essentially all the Company’s revenue is generated from purchase order contracts.
| F-13 |
Neuraxis, Inc.
Notes to Financial Statements
In accordance with FASB’s ASC 606, Revenue from Contracts with Customers, (“ASC 606”), the Company recognizes revenue when its customer obtains control of promised goods or services, in an amount that reflects the consideration which the Company expects to be entitled in exchange for those goods or services, in an amount that reflects the consideration which the Company expects to be entitled in exchange for those goods or services. To determine revenue recognition for arrangements that the Company determines are within the scope of ASC 606, it performs the following five steps:
| (i) | identify the contract(s) with a customer; | |
| (ii) | identify the performance obligations in the contract; | |
| (iii) | determine the transaction price; | |
| (iv) | allocate the transaction price to the performance obligations in the contract; and | |
| (v) | recognize revenue when (or as) the entity satisfies a performance obligation. |
The Company applies the five-step model to contracts when it determines that it is probable it will collect substantially all the consideration it is entitled to in exchange for the goods or services it transfers to the customer. At contract inception, once the contract is determined to be within the scope of ASC 606, the Company assesses the goods or services promised within each contract and determines those that are performance obligations and assesses whether each promised good or service is distinct. The Company then recognizes as revenue the amount of the transaction price, after consideration of variability and constraints, if any, that is allocated to the respective performance obligation when the performance obligation is satisfied.
The Company estimates credit losses on accounts receivable by estimating expected credit losses over the contractual term of the receivable using a discounted cash flow method. When developing this estimate of expected credit losses, the Company considers all available information (past, current, and future) relevant to assessing the collectability of cash flows.
The Company offers a Patient Assistance Program for patients without insurance coverage for IB-Stim. This program extends potential self-pay discounts for IB-Stim devices, based upon household income and size.
Also, the Company offers providers an opt-in program to address adequate insurance claim payments on IB-Stim devices. This program may extend a rebate or invoice credit where the insurance payment and patient responsibility (i.e., deductible, co-payment, and/or co-insurance amounts required by the Payer) are less than the acquisition cost of the IB-Stim device. The Company recognizes revenue at such a time that collection of the amount due is assured.
The following table disaggregates the Company’s revenue based on the customer’s location by state for the years ended December 31:
| 2023 | 2022 | |||||||||
| California | $ | Wisconsin | $ | |||||||
| Ohio | Ohio | |||||||||
| Wisconsin | California | |||||||||
| Florida | Florida | |||||||||
| Massachusetts | Missouri | |||||||||
| All other states | All other states | |||||||||
| $ | $ | |||||||||
The following economic factors affect the nature, amount, timing, and uncertainty of the Company’s revenue and cash flows as indicated:
Type of customer: Based on dollar amounts of revenue, essentially all of the goods sold by the Company are sold to healthcare customers including hospitals and clinics. Sales to healthcare customers lack seasonality and have a mild correlation with economic cycles.
Geographical location of customers: Sales to customers located within the United States represent essentially all of the Company’s sales.
| F-14 |
Neuraxis, Inc.
Notes to Financial Statements
Type of contract: Sales contracts consist of purchase order contracts that tend to be short-term (i.e., less than or equal to one year in duration).
Company’s Performance Obligations with Customers:
Timing of Satisfaction
The Company typically satisfies its performance obligations as the goods are delivered.
Goods that are shipped to customers are typically shipped FOB shipping point with freight prepaid by the Company. As such, ownership of goods in transit transfer to the customer when shipped and the customer bears the associated risks (e.g., loss, damage, delay).
Shipping and handling costs are recorded as general and administrative expenses in the Statement of Operations.
Significant Payment Terms
Payment for goods sold by the Company is typically due after an invoice is sent to the customer, within 30 days. However, other payment terms are frequently negotiated with customers ranging from due upon receipt to due within 90 days. Some payment terms may call for payment only after the healthcare provider receives their insurance reimbursement. Invoices for goods are typically sent to customers within three calendar days of shipment. The Company does not offer discounts if the customer pays some or all of an invoiced amount prior to the due date.
None of the Company’s contracts have a significant financing component.
Nature
Medical devices that the Company contracts to sell and transfer to customers are manufactured by one specific third-party manufacturer. The manufacturer is located within the state of Indiana. In no case does the Company act as an agent (i.e., the Company does not provide a service of arranging for another party to transfer goods to the customer).
Returns, Refunds, etc.
Orders may not be cancelled after shipment. Customers may return devices within 10 days of delivery if the goods are found to be defective, nonconforming, or otherwise do not meet the stated technical specifications. At the option of the customer, the Company shall either:
| ● | Refund the price paid for any defective or nonconforming products. | |
| ● | Supply and deliver to the customer replacement conforming products. | |
| ● | Reimburse the customer for the cost of repairing any defective or nonconforming products. |
At the time revenue is recognized, the Company estimates expected returns and excludes those amounts from revenue. The Company also maintains appropriate accounts to reflect the effects of expected returns on the Company’s financial position and periodically adjusts those accounts to reflect its actual return experience. Historically, returns have been immaterial, and the Company currently does not provide a provision for this liability.
| F-15 |
Neuraxis, Inc.
Notes to Financial Statements
Warranties
In most cases, goods that customers purchase from the Company are covered by manufacturers’ warranties. The Company does not sell warranties separately.
The manufacturer guarantees the product for the period up to the expiration date printed on the device’s label or twelve months from the date of purchase, whichever comes first. The guarantee applies to flaws of material and workmanship. The Company’s warranties provide customers with assurance that purchased devices comply with published specifications, inspection standards, and workmanship. At the time revenue is recognized, the Company estimates the cost of expected future warranty claims but does not exclude any amounts from revenue. The Company maintains appropriate accounts to reflect the effects of expected future warranty claims on the Company’s financial position and periodically adjusts those accounts to reflect its actual warranty claim experience. Historically, warranty claims have been immaterial, and the Company currently does not provide a provision for this liability.
The Company typically satisfies its performance obligations for goods at a point in time. In most cases, goods are shipped by common carrier to customers under “FOB Shipping Point” terms. As such, customers typically obtain control of the goods upon shipment. The Company’s management exercises judgment in determining when performance obligations for goods have been satisfied. In making such judgments, management typically relies on shipping information obtained from common carriers to evaluate when the customer has obtained control of the goods.
The Company’s contracts with customers typically do not involve variable consideration. The information that the Company uses to determine the transaction price for a contract is similar to the information that the Company’s management uses in establishing the prices of goods to be sold.
Leases
In accordance with Accounting Standards Updated (“ASU”) No. 2016-02, Leases (Topic 842) (“ASU 2016-02” or “ASC 842”), at the inception of an arrangement, the Company determines whether the arrangement is or contains a lease based on the unique facts and circumstances present in the arrangement. Leases with a term greater than one year are recognized on the balance sheet as right-of-use assets and current and non-current lease liabilities, as applicable.
Operating lease liabilities and their corresponding right-of-use assets are initially recorded based on the present value of lease payments over the expected remaining lease term. Certain adjustments to the right-of-use asset may be required for items such as incentives received. The interest rate implicit in lease contracts is typically not readily determinable. As a result, the Company utilizes its incremental borrowing rate to discount lease payments, which reflects the fixed rate at which the Company could borrow on a collateralized basis the amount of the lease payments in the same currency, for a similar term, in a similar economic environment. Prospectively, the Company will adjust the right-of-use assets for straight-line rent expense, or any incentives received and remeasure the lease liability at the net present value using the same incremental borrowing rate that was in effect as of the lease commencement or transition date. The Company has elected not to recognize leases with an original term of one year or less on the balance sheet. The Company typically only includes an initial lease term in its assessment of a lease arrangement. Options to renew a lease are not included in the Company’s assessment unless there is reasonable certainty that the Company will renew.
Assumptions made by the Company at the commencement date are re-evaluated upon occurrence of certain events, including a lease modification. A lease modification results in a separate contract when the modification grants the lessee an additional right of use not included in the original lease and when lease payments increase commensurate with the standalone price for the additional right of use. When a lease modification results in a separate contract, it is accounted for in the same manner as a new lease.
Entities may elect not to separate lease and non-lease components. The Company has elected to account for lease and non-lease components together as a single lease component for all underlying assets and allocate all the contract consideration to the lease component only.
| F-16 |
Neuraxis, Inc.
Notes to Financial Statements
Impairment of Long-Lived Assets
Long-lived assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset group may not be recoverable. If events or changes in circumstances indicate that the carrying amount of an asset group may not be recoverable, we compare the carrying amount of the asset group to future undiscounted net cash flows, excluding interest costs, expected to be generated by the asset group and their ultimate disposition. If the sum of the undiscounted cash flows is less than the carrying value, the impairment to be recognized is measured by the amount by which the carrying amount of the asset group exceeds the fair value of the asset group. Assets to be disposed of are reported at the lower of the carrying amount or fair value, less costs to sell.
Concentrations of Credit Risk
The Company’s business activity consists of the sale of medical neuromodulation devices to doctors, clinics, and hospitals across the country.
Receivables
consist of unsecured amounts due from customers. As of December 31, 2023, accounts receivable from four customers with balances due
in excess of
The
table below sets forth the Company’s customers that accounted for greater than
| Percentage of | Percentage of | |||||||||||
| 2023 | Sales | 2022 | Sales | |||||||||
| Hospital A | $ | $ | ||||||||||
| Hospital B | ||||||||||||
| Hospital C | ||||||||||||
| $ | $ | |||||||||||
From
time to time, the Company’s bank balances may exceed the FDIC limit of $
Going Concern
We
have incurred losses since inception and have funded our operations primarily with a combination of sales, debt, and the sale of capital
stock. As of December 31, 2023, we had a stockholders’ deficit totaling $
On
August 9, 2023, the Company consummated its initial public offering, conducted on a firm commitment basis, pursuant to which it sold
shares of its common stock at a price of $
per share, resulting in gross proceeds to the Company of $
| F-17 |
Neuraxis, Inc.
Notes to Financial Statements
Our future capital requirements will depend upon many factors, including progress with developing, manufacturing, and marketing our technologies, the time and costs involved in preparing, filing, prosecuting, maintaining, and enforcing patent claims and other proprietary rights, our ability to establish collaborative arrangements, marketing activities and competing technological and market developments, including regulatory changes and overall economic conditions in our target markets. Our ability to generate revenue and achieve profitability requires us to successfully market and secure purchase orders for our products from customers currently identified in our sales pipeline and to new customers as well. The primary activity that will drive all customers and revenues is the adoption of insurance coverage by commercial insurance carriers nationally which is a top priority of the Company. These activities, including our planned research and development efforts, will require significant uses of working capital through the rest of 2024 and beyond. Based on our current operating plans, we believe that our existing cash at the time of this filing will only be sufficient to meet our anticipated operating needs through the end of 2024.
Management evaluates whether there are conditions or events that raise substantial doubt about the Company’s ability to continue as a going concern for a period of one year from the date the financial statements are issued.
To date, the Company has experienced operating losses and negative cash flows from operations. Management believes that increased sales and acceptance of their product by insurance providers will allow the Company to achieve profitability in the future.
While the Company believes in the viability of its strategy to further implement its business plan and generate sufficient revenues and in its ability to raise additional funds by way of a public or private offering of its debt or equity securities, there can be no assurance that it will be able to do so on reasonable terms, or at all. The ability of the Company to continue as a going concern is dependent upon its ability to further implement its business plan and generate sufficient revenues and its ability to raise additional funds by way of a public or private offering. Neither future cash generated from operating activities, nor management’s contingency plans to mitigate the risk and extend cash resources through the evaluation period, are considered probable. As a result, substantial doubt is deemed to exist about the Company’s ability to continue as a going concern. As we continue to incur losses, our transition to profitability is dependent upon achieving a level of revenues adequate to support its cost structure. We may never achieve profitability, and unless and until doing so, we intend to fund future operations through additional dilutive or nondilutive financing. There can be no assurances, however, that additional funding will be available on terms acceptable to us, if at all.
The financial statements do not include any adjustments related to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary should the Company be unable to continue as a going concern.
Recently Adopted Accounting Pronouncements
In June 2016, the FASB issued ASU 2016-13, Financial Instruments – Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments, which requires measurement and recognition of expected credit losses for financial assets held and requires enhanced disclosures regarding significant estimates and judgments used in estimating credit losses. In November 2019, the FASB issued ASU 2019-10, Financial Instruments – Credit Losses (Topic 326), Derivatives and Hedging (Topic 815) and Leases (Topic 842): Effective Dates, which amended the effective date of ASU 2016-13. Public business entities meeting the definition of an SEC filer, excluding entities eligible to be a Smaller Reporting Company (“SRC”) as defined by the SEC, are required to adopt the standard for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. All other entities are required to adopt the standard for fiscal years beginning after December 15, 2022, including interim periods within those fiscal years. The Company meets the definition of an SRC and therefore the standard was adopted as of January 1, 2023. The adoption of this guidance did not have a material impact on the Company’s financial statements.
Recently Issued Accounting Pronouncements
In December 2023, the FASB issued ASU 2023-19, Income Taxes (Topic 740): Improvements to Income Tax Disclosures, which requires the enhancement of income tax disclosures to provide better insight into how an entity’s operations and related tax risks, planning and opportunities affect its tax rate and prospects for future cash flows. The enhanced disclosures require (i) specific categories in a tabular rate reconciliation including both amounts and percentages and (ii) additional information for reconciling items and income tax paid that meet a quantitative threshold. Public business entities are required to adopt the standard for annual periods beginning after December 15, 2024. All other entities are required to adopt the standard for annual periods beginning after December 15, 2025. The adoption of the standard is not expected to have a material impact on the Company’s financial statements.
| F-18 |
Neuraxis, Inc.
Notes to Financial Statements
3. Related Party Transactions
The
Company has two demand notes receivable from shareholders related to the sale of common stock on January 1, 2016. Both notes’ initial
balances were $, with interest calculated monthly based on applicable federal rates. No payments have been received on the notes.
Since repayment is not assured, the Company provided an allowance for the entire balance of principal and interest as of December 31,
2019. The current allowance is $
| Loan | Interest | Interest | ||||||||||
| December 31, 2023 | Receivable | Receivable | Income | |||||||||
| Shareholder 1 | $ | $ | $ | |||||||||
| Shareholder 2 | ||||||||||||
| Allowance for Collection Risk | ( | ) | ( | ) | ( | ) | ||||||
| Net Balance | $ | $ | $ | |||||||||
| Loan | Interest | Interest | ||||||||||
| December 31, 2022 | Receivable | Receivable | Income | |||||||||
| Shareholder 1 | $ | $ | $ | |||||||||
| Shareholder 2 | ||||||||||||
| Allowance for Collection Risk | ( | ) | ( | ) | ( | ) | ||||||
| Net Balance | $ | $ | $ | |||||||||
The Company has loans payable to shareholders related to funding needs for operations. The current loan details for all related party loans are as follows:
| Interest & | ||||||||||||||||||||
| Interest | Loan | Service Fee | Interest | |||||||||||||||||
| December 31, 2023 | Due Date | Rate | Balance | Accrued | Paid | |||||||||||||||
| Shareholder 1 | % | $ | $ | $ | ||||||||||||||||
| Shareholder 1 | % | |||||||||||||||||||
| Other Convertibles | % | |||||||||||||||||||
| Total | $ | $ | $ | |||||||||||||||||
| Interest & | ||||||||||||||||||||
| Interest | Loan | Service Fee | Interest | |||||||||||||||||
| December 31, 2022 | Due Date | Rate | Balance | Accrued | Paid | |||||||||||||||
| Shareholder 1 | % | $ | $ | $ | ||||||||||||||||
| Shareholder 1 | % | |||||||||||||||||||
| Other Convertibles | % | |||||||||||||||||||
| Total | $ | $ | $ | |||||||||||||||||
Mr.
Bradley Mitch Watkins, Director, provided certain sales, marketing and commercialization consulting services to the Company prior to his
appointment to the Board of Directors. For the years ended December 31, 2023 and 2022, the Company paid Mr. Watkins $
The
Company’s former Chief Financial Officer is contracted for services through a third-party public accounting firm. He is the firm’s
managing partner and majority shareholder. The firm is engaged by the Company to provide accounting and tax services on a continuous
basis. Fees paid for services were $
| F-19 |
Neuraxis, Inc.
Notes to Financial Statements
4. Property and Equipment
Property and equipment, net consists of the following:
| December 31, 2023 | December 31, 2022 | |||||||
| Machinery and equipment | $ | $ | ||||||
| Furniture and fixtures | ||||||||
| Computer hardware | ||||||||
| Leasehold improvements | ||||||||
| Total property and equipment | ||||||||
| Less: accumulated depreciation | ( | ) | ( | ) | ||||
| Property and equipment, net | $ | $ | ||||||
Depreciation
expense was $
5. Intangible Assets
Intangible assets, net consists of the following:
| December 31, 2023 | December 31, 2022 | |||||||
| Software Implementation | $ | $ | ||||||
| Patents | ||||||||
| Trademark | ||||||||
| Licenses | ||||||||
| Total intangible assets | ||||||||
| Less: accumulated amortization | ( | ) | ( | ) | ||||
| Intangible assets, net | $ | $ | ||||||
Amortization
expense was $
6. Accrued Expenses
Accrued expenses consisted of the following:
| December 31, 2023 | December 31, 2022 | |||||||
| Compensation and benefits | $ | $ | ||||||
| Underwriter fees | ||||||||
| Interest | ||||||||
| Related party interest | ||||||||
| Other | ||||||||
| Total accrued expenses | $ | $ | ||||||
7. Notes Payable
On
February 15, 2023, The Company signed a note with Exchange Listing, LLC in the amount of $
| F-20 |
Neuraxis, Inc.
Notes to Financial Statements
The
Company borrowed $
The lender was granted and assigned a continuing security interest in all the Company’s personal property assets including, but not limited to, business equipment, inventory, accounts, accounts receivable, intellectual property, chattel paper, instruments, deposit accounts, commercial tort claims, contract rights, licenses, claims, and general intangibles.
The
future minimum principal payments to be paid in 2024 is $
Convertible Notes
The following table presents a summary of the Company’s convertible notes:
| December 31, | ||||||||
| 2023 | 2022 | |||||||
| Principal | $ | $ | ||||||
| Less: Original issue discount | ( | ) | ||||||
| Less: Debt discount | ( | ) | ||||||
| Less: Deferred financing fees | ( | ) | ||||||
| $ | $ | |||||||
From March to July of 2023, the Company conducted
multiple closings of a private placement offering to accredited investors for aggregate gross principal amounts of $
As facilitators to the 2023 convertible notes,
Signature Bank received a $
After the closing of the initial public offering
on August 8, 2023, the Company converted the notes payable plus $
In March of 2023, the Company used borrowings from
the 2023 convertible notes to pay the principal balance of some of the 2022 notes totaling $
The remaining investors of the 2022 notes did
not convert their notes and, as such, the Company used proceeds from the initial public offering to pay the remaining promissory
notes back in full, including interest in the amount of $
| F-21 |
Neuraxis, Inc.
Notes to Financial Statements
The convertible notes signed between June 3, 2022
and November 30, 2022 had aggregate gross principal amounts totaling $
The convertible notes signed on December 19, 2022
had aggregate gross principal amounts totaling $
The
investors of the notes signed on December 19, 2022 above did not convert their notes and, as such the Company used proceeds from the
initial public offering to pay the remaining promissory notes back in full, including interest in the amount of $
The Company paid $
The Company has applied ASC 815 due to
the potential for settlement in a variable quantity of shares. Since these convertible notes and warrants have the option to convert or
be exercised at a variable amount, they are subject to derivative liability treatment. The conversion feature has been measured at fair
value using a Monte Carlo model at the date of issuance and is adjusted to fair value at each reporting period. The fair value of the
embedded derivative and the warrant liability at date of issuance was $
The 2023 and 2022 convertible notes transferred
fair value to investors in the form of principal, warrants and common shares totaling $
During the years ended December 31, 2023 and 2022,
the Company accrued interest on these convertible notes totaling $
| F-22 |
Neuraxis, Inc.
Notes to Financial Statements
8. Leases
The Company’s leases are comprised of operating leases for office space. At the inception of the lease, the Company determines whether the lease contract conveys the right to control the use of identified property for a period of time in exchange for consideration. Leases are classified as operating or finance leases at the commencement date of the lease. Operating leases are recorded as operating lease right-of-use assets, other current liabilities, and operating lease liabilities in the Balance Sheets. The Company did not have any finance leases at December 31, 2023 and 2022.
The
Company had three leases primarily consisting of office space in Versailles, Indiana and Carmel, Indiana.
Operating lease right-of-use assets and liabilities are recognized at the commencement date based on the present value of lease payments over the lease term. As the implicit interest rate is generally not readily determinable, the Company uses an incremental borrowing rate based on the information available at the commencement date in determining the present value of lease payments. The incremental borrowing rate reflects the estimated rate of interest that the Company would pay to borrow on a collateralized basis over a similar economic environment. Lease expense for the operating lease is recognized on a straight-line basis over the lease term.
Leases may include renewal options, and the renewal option is included in the lease term if the Company concludes that it is reasonably certain that the option will be exercised. Certain leases may contain rent escalation clauses, either fixed or adjusted periodically for inflation of market rates, that are factored into the calculation of lease payments to the extent they are fixed and determinable at lease inception. The Company also has variable lease payments that do not depend on a rate or index, primarily for items such as common area maintenance and real estate taxes, which are recorded as expenses when incurred.
For
the years ended December 31, 2023 and 2022, the Company recognized $
The following table presents information related to the Company’s operating leases:
| December 31, 2023 | December 31, 2022 | |||||||
| Operating lease right-of-use assets | $ | $ | ||||||
| Other current liabilities | ||||||||
| Operating lease liabilities | ||||||||
| $ | $ | |||||||
| Weighted-average remaining lease term (in years) | ||||||||
| Weighted-average discount rate | % | % | ||||||
As of December 31, 2023, the maturities of the Company’s operating lease liabilities were as follows:
| 2024 | $ | |||
| 2025 | ||||
| 2026 | ||||
| Total lease payments | ||||
| Less: imputed interest | ||||
| Total present value of lease payments | $ |
9. Common Stock and Warrants
The
Company authorized shares
of common stock, of which and shares
were issued and outstanding as of December 31, 2023 and 2022, respectively. In conjunction with common shares issued upon
the completion of the initial public offering on August 8, 2023, the Company also issued (i)
common shares upon conversion of
shares of Convertible Series A Preferred Stock and
shares of Convertible Series Seed Preferred Stock, (ii)
common shares upon conversion of convertible notes, (iii) commons
shares as payment to vendors for services and (iv) common
shares upon the exercise of warrants. On January 10, 2023, the Company’s board of directors authorized a
On
January 10, 2023, the Company’s board of directors authorized a
On August 9, 2023, the Company consummated an initial public offering, conducted on a firm commitment basis, pursuant to which it sold
shares of its common stock at a price of $ per share, resulting in gross proceeds of $
| F-23 |
Neuraxis, Inc.
Notes to Financial Statements
In
connection with a bridge loan, the Company issued a warrant on September 18, 2018 that allows the holder to purchase common
stock from the Company at a share price of $
per share. The number of shares was based on a formula tied to the final amount of loans made by the holder of $
The
Company issued a second warrant on September 6, 2019, under similar terms but is a penny warrant that allows the holder to purchase
The
Company issued a third warrant to Masimo Corporation on April 9, 2020. This warrant was pre-funded in the amount of $
During
2022, the Company issued
From
March to June of 2023, the Company issued
On
August 9, 2023, the Company issued
On
August 14, 2023, the Company issued
The following is a summary of warrant activity for common stock during the years ended December 31, 2023 and 2022:
| Number of Warrants for Common Stock | Weighted-Avg. Exercise Price | Weighted-Avg. Remaining Contractual Life | ||||||||||
| Outstanding as of January 1, 2022 | $ | |||||||||||
| Granted | ||||||||||||
| Cancelled/Expired | — | |||||||||||
| Exercised | — | |||||||||||
| Outstanding as of December 31, 2022 | $ | |||||||||||
| Granted | $ | |||||||||||
| Converted Prefunded Warrants | — | |||||||||||
| Exercised | ( | ) | — | |||||||||
| Outstanding as of December 31, 2023 | $ | |||||||||||
| F-24 |
Neuraxis, Inc.
Notes to Financial Statements
The following is a summary of warrant activity for preferred stock during the years ended December 31, 2023 and 2022:
| Number of | Weighted-Avg. | |||||||
| Warrants for | Exercise | |||||||
| Preferred Stock | Price | |||||||
| Outstanding as of January 1, 2022 | $ | |||||||
| Granted | ||||||||
| Cancelled/Expired | ||||||||
| Exercised | ||||||||
| Outstanding as of December 31, 2022 | $ | |||||||
| Granted | ||||||||
| Cancelled/Expired | ||||||||
| Converted to Common Stock Warrant | ( | ) | ( | ) | ||||
| Outstanding as of December 31, 2023 | $ | |||||||
The following table summarizes the Company’s warrants outstanding and exercisable as of December 31, 2023.
| Number of | ||||||||||
| Warrants | Exercise | Expiration | ||||||||
| Outstanding | Price | Date | ||||||||
| Investor Warrant W-01 | $ | |||||||||
| Investor Warrant PSA-01 | $ | |||||||||
| 2022 Convertible Notes | $ | |||||||||
| 2023 Convertible Notes | $ | |||||||||
| Consulting Agreement Warrants | $ | |||||||||
| Underwriter’s Warrants | $ | |||||||||
10. Preferred Stock
The Company has authorized shares of preferred stock of which has been designated Series A Preferred and has been designated Series Seed Preferred, of which and shares of Series A Preferred and and shares of Series Seed Preferred are issued and outstanding as of December 31, 2023 and 2022, respectively.
The
aggregate purchase price of the Series A Preferred Stock was $
| F-25 |
Neuraxis, Inc.
Notes to Financial Statements
The following is a summary of Preferred Stock terms:
Voting Rights - The Series A Preferred and Series Seed Preferred shall vote together with the Common Stock on an as-converted basis, and not as separate classes.
Conversion - The Series A Preferred and Series Seed initially convert 1:1 to Common Stock at any time at option of holder, subject to adjustments for stock dividends, splits, combinations, and similar events and as described below under “Anti-dilution Provisions.”
Dividends
- The Series A Preferred will carry an annual
Liquidation - In the event of any liquidation, dissolution or winding up of the Company, the proceeds shall be paid in the following priority:
First, to the Series A Preferred in proportion to each holder’s respective pro rata Series A Original Purchase Price, plus any pro rata share of the Accruing Dividend until the entire Series A Original Purchase Price and Accruing Dividend are paid;
Second, to the Series Seed Preferred in proportion to each holder’s respective pro rata Series Seed Original Purchase Price until the entire amount of the Series Seed Original Purchase Price is paid; and
Thereafter, the Series A Preferred and Series Seed Preferred participate with the Common Stock pro rata on an as-converted basis.
A merger or consolidation (other than one in which stockholders of the Company own a majority by voting power of the outstanding shares of the surviving or acquiring corporation) and a sale, lease, transfer, exclusive license or other disposition of all or substantially all of the assets of the Company will be treated as a liquidation event (a “Deemed Liquidation Event”), thereby triggering payment of the liquidation preferences described.
Anti-dilution
Provisions - The Series A Preferred have full-ratchet anti-dilution protection so that the conversion price will be reduced to
In
consideration for shareholders to make an additional investment in the Company, upon the purchase of the Series A Preferred stock by
the shareholder, the Company converted the existing common shares held by shareholders to Series Seed Preferred Stock at a $
As of December 31, 2023, all preferred stock has been converted pursuant to the initial public offering.
The
Company signed a Convertible Promissory Note on November 8, 2023 for a principal amount of $
On October 12, 2017, the Company adopted the 2017 Stock Compensation Plan authorizing the issuance of shares of common stock. On September 13, 2019 the Company entered into a Stock Option Cancellation agreement with all holders of these stock options effective on that date.
This plan was then amended on September 13, 2019, to increase the share amount to . This plan was enacted to enable the Company to retain the services of certain key employees, officers, and directors of the Company. The Plan provides for the grant of stock options, including incentive stock options, or ISOs, and nonqualified stock options, or NSOs and restricted stock, and they were estimated using the Black-Scholes option-pricing model with the following weighted-average assumptions:
| Assumptions as of December 31, 2023 | ||||
| Fully Vested | ||||
| Number of Shares | ||||
| Stock price | $ | |||
| Exercise price | $ | |||
| Expected term | years | |||
| Expected volatility | % | |||
| Risk-free interest rate | % | |||
| Dividend rate | % | |||
| F-26 |
Neuraxis, Inc.
Notes to Financial Statements
| Number of Options | Weighted Avg. Remaining Contractual Life (in years) | Weighted Avg. Exercise Price | Aggregate Intrinsic Value | |||||||||||||
| Outstanding as of January 1, 2022 | $ | $ | ||||||||||||||
| Granted | ||||||||||||||||
| Forfeited | ||||||||||||||||
| Cancelled/Expired | ||||||||||||||||
| Exercised | ||||||||||||||||
| Outstanding as of December 31, 2022 | $ | $ | ||||||||||||||
| Granted | ||||||||||||||||
| Forfeited | ||||||||||||||||
| Cancelled/Expired | ||||||||||||||||
| Exercised | ||||||||||||||||
| Outstanding as of December 31, 2023 | $ | $ | ||||||||||||||
| Vested and Exercisable as of December 31, 2023 | $ | $ | ||||||||||||||
Stock-based compensation expense is classified in the Company’s statements of operations as general and administrative expense. The amounts were $ and $ for the years ended December 31, 2023 and 2022, respectively. As of December 31, 2023, there was unrecognized compensation expense related to unvested options granted under the Company’s share-based compensation plans.
12. Warrant Liabilities
The Company has evaluated financial instruments arising from an adjustable exercise price for warrants that are issued and outstanding as of December 31, 2023 and 2022.
The
Company utilizes a Monte Carlo simulation model for warrants that have an option to convert at a variable number of shares to compute
the fair value of the liability and to mark to market the fair value of the derivative at each balance sheet date. The inputs utilized
in the application of the Monte Carlo model included a starting stock price of $ per share, an expected remaining term of each warrant
as of the valuation date, estimated volatility of
Risk-free interest rate: The Company uses the risk-free interest rate of a U.S. Treasury Note adjusted to be on a continuous return basis to align with the Black-Scholes option-pricing model.
Dividend
yield: The Company uses a
| F-27 |
Neuraxis, Inc.
Notes to Financial Statements
Volatility: The Company calculates the expected volatility based on comparable company’s historical stock prices with a look back period commensurate with the period to maturity.
Expected term: The Company’s remaining term is based on the remaining contractual maturity of the warrants.
The following are the changes in the warrant liabilities during the years ended December 31, 2023 and 2022.
| Level 1 | Level 2 | Level 3 | ||||||||||
| Warrant liabilities as of January 1, 2022 | $ | $ | $ | |||||||||
| Addition | ||||||||||||
| Changes in fair value of warrant liabilities | ( | ) | ||||||||||
| Warrant liabilities as of December 31, 2022 | ||||||||||||
| Addition | ||||||||||||
| Changes in fair value of warrant liabilities | ( | ) | ||||||||||
| Reclassified to equity | ( | ) | ||||||||||
| Warrant liabilities as of December 31, 2023 | $ | $ | $ | |||||||||
13. Derivative Liabilities
The Company has identified derivative instruments arising from the conversion shares discussed in the Convertible Notes section of Note 7 as of December 31, 2023 and 2022.
The
Company utilizes a Monte Carlo simulation model for commitment shares that have an option to convert at a variable number of shares to
compute the fair value of the derivative and to mark to market the fair value of the derivative at each balance sheet date. The inputs
utilized in the application of the Monte Carlo model included a starting stock price of $ per share, an expected remaining term of
each warrant as of the valuation date, estimated volatility of
Risk-free interest rate: The Company uses the risk-free interest rate of a U.S. Treasury Note adjusted to be on a continuous return basis to align with the Black-Scholes option-pricing model.
Dividend
yield: The Company uses a
Volatility: The Company calculates the expected volatility based on comparable company’s historical stock prices with a look back period commensurate with the period to maturity.
Expected term: The Company’s remaining term is based on the remaining contractual maturity of the warrants.
The following are the changes in the warrant liabilities during the years ended December 31, 2023 and 2022.
| Level 1 | Level 2 | Level 3 | ||||||||||
| Derivative liabilities as of January 1, 2022 | $ | $ | $ | |||||||||
| Addition | ||||||||||||
| Changes in fair value of Derivative liabilities | ( | ) | ||||||||||
| Derivative liabilities as of December 31, 2022 | ||||||||||||
| Addition | ||||||||||||
| Changes in fair value of Derivative liabilities | ( | ) | ||||||||||
| Extinguishment of Derivative liabilities | ( | ) | ||||||||||
| Derivative liabilities as of December 31, 2023 | $ | $ | $ | |||||||||
| F-28 |
Neuraxis, Inc.
Notes to Financial Statements
14. Retirement Plan
The
Company sponsors a 401(k)-retirement plan for its employees. Employees are eligible to participate in the elective deferral portion of
the plan after twelve months and 1,000 hours of service. The Company matches the employees’ contribution up to
15. Commitments and Contingencies
Manufacturing Services Agreement
On August 21, 2020, the Company entered into a Manufacturing Services Agreement (MSA) for the manufacture and supply of the Company’s IB-STIM device based upon the Company’s product specifications as set forth in the MSA. This agreement terminated any prior manufacturing agreements.
The Company provides the necessary equipment to the manufacturer and retains ownership. The manufacturer bears the risk of loss of and damage to the equipment and consigned materials. Performance under the MSA is initiated by orders issued by the Company and accepted by the manufacturer.
The term of the MSA is 24 months and shall automatically renew for renewal terms of twelve months unless either party provides a written termination notice to the other party within 180 days prior to the end of the then-current term.
Trademark Agreement
The
Company entered into an agreement for a trademark related to the Company’s name on July 11, 2022. The agreement called for an initial
payment of $
Executive Employment Agreements
The
Company, as authorized by the board of directors, entered into employment agreements with nine key employees to provide incentives to
improve shareholder value and to contribute to the growth and financial success of the Company. The agreements had an employment start
date of October 1, 2022, with initial terms from
The
total base salaries for the nine key employees in the agreements are $
There
are seven key employees that have stock options of the Company totaling shares. These key employees have a provision in their
agreements whereas the Company will pay a special bonus equal to the aggregate of the strike price or exercise price of all their stock
options plus a tax gross-up payment. The special bonus shall be paid in twenty percent (
In April 2023, the Company amended the employee agreements to, among other things, clarify that the special one-time incentive payment and the deferred bonus are contingent upon the effective date of the planned initial public offering. The amendment also sets forth a process for executives to exercise the stock options in accordance with the terms of the stock option agreement in effect as of the date of the employment agreement and to clarify that there is no modification to the stock option agreements.
| F-29 |
Neuraxis, Inc.
Notes to Financial Statements
The Company has recorded the backpay portion of the incentive
bonus notes above. The balance of the incentive bonuses of $
Litigation
From time to time, the Company may be involved in litigation relating to claims arising out of operations in the normal course of business. As of the date of issuance, other than those described below, there were no pending or threatened legal proceedings that could reasonably be expected to have a material effect on the results of the Company’s operations. There are also no proceedings in which any of the Company’s directors, officers or affiliates is an adverse party to the Company or has a material interest adverse to the Company’s interest.
On February 6, 2019, plaintiff Ritu Bhambhani, M.D.,
initiated a lawsuit against Innovative Health Solutions, Inc. and others in the United States District Court for the District of Maryland.
Plaintiffs Bhambhani and Sudhir Rao subsequently amended the complaint, with the Third Amended Complaint (“Complaint”) containing
the most recent set of allegations. The Complaint asserted claims under the RICO Act, as well as of fraudulent misrepresentation, intentional
misrepresentation by concealment, and civil conspiracy and sought compensatory damages in excess of $
On February 11, 2022, the Company filed a motion for summary judgment based upon the plaintiffs not being proper parties to assert claims against the Company. On June 14, 2022, the Court granted the Company’s motion for summary judgment and dismissed the Complaint.
On July 14, 2022, plaintiffs Ritu Bhambhani and Sudhir Rao filed a notice of appeal with the Fourth Circuit Court of Appeals. The Company filed a motion to dismiss. On January 4, 2023, the Court issued an order that stated it was deferring a ruling on the motion to dismiss the appeal and that it would address those arguments at the same time that it addressed the substantive merits of the case. While it is too early to predict the ultimate outcome of this matter, we continue to believe we have meritorious defenses, that the dismissal of the Complaint should be upheld, and intend to continue to defend this matter vigorously.
On July 14, 2022, plaintiffs Ritu Bhambhani, LLC;
Box Hill Surgery Center, LLC; Pain and Spine Specialists of Maryland, LLC; and SimCare ASC, LLC initiated a lawsuit against the Company
and others in the United States District Court for the District of Maryland. The plaintiffs in this lawsuit are business entities owned
or partially owned by the plaintiffs that initiated the litigation described above. The Complaint asserted claims under the RICO Act,
as well as fraudulent misrepresentation, intentional misrepresentation by concealment, and civil conspiracy and seeks compensatory damages
in excess of $
On September 28, 2022, the Company filed a motion to dismiss all claims. On May 25, 2023, the Court issued an Order and a Memorandum Opinion which dismissed the plaintiffs’claims related to the RICO Act. The remaining claims are still pending, and no trial date has been set for the case.
The Court has vacated its Scheduling Order at the parties’ request so that the parties could try to resolve the disputes in both cases through an independent third-party mediator. No mediation date has been set. While it is too early to predict the ultimate outcome of this matter, we believe the Company has meritorious defenses and intends to defend this matter vigorously.
| F-30 |
Neuraxis, Inc.
Notes to Financial Statements
16. Health Benefit Plan
The Company entered into a self-funded program employer agreement in 2018 in conjunction with a group health plan for the benefit of eligible employees. This plan is a level funded plan, and the services and products include:
| ● | A self-funded employer health benefit plan. | |
| ● | Stop loss insurance purchased from a stop loss insurance company. | |
| ● | Third party administrator to provide administrative services with regard to the plan. |
The
Company maintains a stop loss contract that reimburses the Company for claims paid under the plan if they exceed a predetermined level.
The Company makes contributions for health care costs and associated expenses that are expected during the plan year. The amount of contributions
is determined annually based on the Company’s maximum liability for expected claims, administrative expenses, and premiums for
the stop loss policy. The Company paid premiums of $
The
Company is responsible for the monthly premiums, as established, and nothing further. The stop loss policy covers the claims if they
exceed the claims funds. After a certain time, and if there is a surplus in the claims fund, the Company may be entitled to receive a
17. Income Taxes
The Inflation Reduction Act (IRA) was signed into law on August 16, 2022, imposing a 15% corporate alternative minimum tax on adjusted financial statement income and a 1% excise tax on stock repurchases after January 1, 2023. The IRA did not have an impact on the Company’s 2023 or 2022 financial statements.
The Tax Cuts and Jobs Act (TCJA) requires taxpayers
to capitalize and amortize research and developmental (“R&D”) expenditures undersection 174 for tax years beginning after
December 31, 2021 and, therefore, became effective for the Company during the year ended December 31, 2022. The TCJA resulted in the capitalization
of R&D costs of $
Deferred income taxes are provided for temporary differences between the financial reporting basis and the tax basis of the Company’s assets and liabilities. Differences are primarily attributable to net operating loss carryforwards and stock-based compensation. The Company does not reflect any deferred taxes in its financial statements due to a full valuation allowance. The net deferred tax amounts include the following components:
| December 31, 2023 | December 31, 2022 | |||||||
| Net deferred tax assets - Non-current | ||||||||
| Depreciation | $ | ( | ) | $ | ( | ) | ||
| Amortization | ( | ) | ( | ) | ||||
| Accrual to cash | ||||||||
| Stock based compensation | ||||||||
| Expected income tax benefit from NOL carry-forwards | ||||||||
| Less valuation allowance | ( | ) | ( | ) | ||||
| Deferred tax assets, net of valuation allowance | $ | $ | ||||||
| F-31 |
Neuraxis, Inc.
Notes to Financial Statements
A reconciliation of the federal statutory income tax rate and the effective income tax rate as a percentage of income before income taxes is as follows:
| 2023 | 2022 | |||||||
| Federal statutory income tax rate | % | % | ||||||
| State tax rate, net of federal benefit | ||||||||
| Nondeductible expenses | ||||||||
| Nontaxable income | ||||||||
| Change in valuation allowance on net deferred tax assets | ( | ) | ( | ) | ||||
| Effective income tax rate | % | % | ||||||
The
Company had no income tax expense due to the operating loss incurred for the years ended December 31, 2023 and 2022. The
Company’s management has evaluated the positive and negative evidence bearing upon the realizability of its deferred tax
assets and has determined that it is more likely than not that the Company will not recognize the benefits of the net deferred tax
assets. As a result, the Company recorded a full valuation allowance at December 31, 2023 and 2022. The valuation allowance
increased by $
On
December 31, 2023, the Company had federal and state net operating loss (NOL) carryforwards totaling $
Federal and state tax laws impose limitations on the utilization of NOLs and credit carryforwards in the event of an ownership change for tax purposes, as defined in Section 382 of the Internal Revenue Code. Accordingly, the Company’s ability to utilize these carryforwards may be limited as a result of an ownership change. Such an ownership change could result in a limitation in the use of the net operating losses in future years and possibly a reduction of the net operating losses available.
If not used, the state carryforwards will expire as follows:
| 2038 | $ | |||
| 2039 | $ | |||
| 2040 | $ | |||
| 2041 | $ | |||
| 2042 | $ | |||
| 2043 | $ |
18. Subsequent Events
The
Company evaluated events through the date of issuance. Subsequent to December 31, 2023, the Company entered into a series of
Convertible Promissory Notes totaling $
19. Restatement of Interim Financial Statements (Unaudited)
In connection with the preparation of the Company’s financial statements as of and for the year ended December 31, 2023, management identified a material accounting error in the unaudited interim financial statements as of and for the three and nine month periods ended September 30, 2023 relating to the improper classification of the reduction of the fair value of warrant liabilities as a reduction to the extinguishment of debt charge on the Statement of Operations rather than an increase to additional paid in capital on the Balance Sheet.
Upon the consummation of the Company’s IPO,
the exercise price of certain warrants issued prior to the IPO became known. As a result of the exercise price becoming known, our warrant
liability was reduced by $
The Board of Directors of the Company, upon the recommendation of the Audit Committee and after discussion with management, concluded that the Company’s previously issued unaudited financial statements as of and for the three and nine month periods ended September 30, 2023 should no longer be relied upon and should be restated due to identification of the material accounting error.
The following tables present the amounts previously reported, the restatement impact and the restated amount in the respective unaudited financial statements.
| F-32 |
NeurAxis, Inc.
Balance Sheet
| (Unaudited) | ||||||||||||
| As of September 30, | ||||||||||||
| As Reported | Adjustments | As Restated | ||||||||||
| Assets | ||||||||||||
| Current assets: | ||||||||||||
| Cash and cash equivalents | $ | $ | $ | |||||||||
| Accounts receivable, net | ||||||||||||
| Inventories | ||||||||||||
| Prepaid and other current assets | ||||||||||||
| Total current assets | ||||||||||||
| Property and equipment, at cost | ||||||||||||
| Less - accumulated depreciation | ( | ) | ( | ) | ||||||||
| Property and equipment, net | ||||||||||||
| Operating lease right of use asset, net | ||||||||||||
| Intangible assets | ||||||||||||
| Total assets | $ | $ | $ | |||||||||
| Liabilities and Stockholders’ Deficit | ||||||||||||
| Current liabilities: | ||||||||||||
| Accounts payable | $ | $ | $ | |||||||||
| Accrued expenses | ||||||||||||
| Notes payable | ||||||||||||
| Current portion of operating lease payable | ||||||||||||
| Customer deposits | ||||||||||||
| Share liabilities | ||||||||||||
| Warrant liabilities | ||||||||||||
| Total current liabilities | ||||||||||||
| Operating lease payable, net of current portion | ||||||||||||
| Total liabilities | ||||||||||||
| Stockholders’ deficit: | ||||||||||||
| Convertible Series A preferred stock | ||||||||||||
| Convertible Series Seed preferred stock | ||||||||||||
| Common stock | ||||||||||||
| Additional paid in capital | ||||||||||||
| Accumulated deficit | ( | ) | ( | ) | ( | ) | ||||||
| Total stockholders’ deficit | ( | ) | ( | ) | ||||||||
| Total liabilities and stockholders’ deficit | $ | $ | $ | |||||||||
| F-33 |
NeurAxis, Inc.
Statements of Operations
| (Unaudited) | (Unaudited) | |||||||||||||||||||||||
For the Three Months Ended September 30, 2023 | For the Nine Months Ended September 30, 2023 | |||||||||||||||||||||||
| As Reported | Adjustments | As Restated | As Reported | Adjustments | As Restated | |||||||||||||||||||
| Net sales | $ | $ | $ | $ | $ | $ | ||||||||||||||||||
| Cost of goods sold | ||||||||||||||||||||||||
| Gross profit | ||||||||||||||||||||||||
| Selling expenses | ||||||||||||||||||||||||
| Research and development | ||||||||||||||||||||||||
| General and administrative | ||||||||||||||||||||||||
| Operating loss | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||
| Other (expense) income, net: | ||||||||||||||||||||||||
| Financing charges | ( | ) | ( | ) | ||||||||||||||||||||
| Interest expense | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||
| Interest income | ||||||||||||||||||||||||
| Change in fair value of warrant liability | ||||||||||||||||||||||||
| Change in fair value of derivative financial instruments | ||||||||||||||||||||||||
| Amortization of debt discount and issuance cost | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||
| Extinguishment of debt liabilities | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||
| Other income | ||||||||||||||||||||||||
| Other expense | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||||||
| Total other (expense) income, net | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ( | ) | ||||||||||||
| Net loss | $ | ( | ) | $ | ( | ) | $ | ( | ) | $ | ( | ) | $ | ( | ) | $ | ( | ) | ||||||
| F-34 |
NeurAxis, Inc.
Statements of Cash Flows
| (Unaudited) | ||||||||||||
| For the Nine Months Ended September 30, 2023 | ||||||||||||
| As Reported | Adjustments | As Restated | ||||||||||
| Cash flows from operating activities | ||||||||||||
| Net loss | $ | ( | ) | $ | ( | ) | $ | ( | ) | |||
| Adjustments to reconcile net loss to net cash used by operating activities: | ||||||||||||
| Amortization of debt discount and issuance cost | ||||||||||||
| Depreciation and amortization | ||||||||||||
| Provision for losses on accounts receivable | ||||||||||||
| Non-cash lease expense | ||||||||||||
| Non-cash interest expense | ||||||||||||
| Stock based compensation | ||||||||||||
| Extinguishment of derivative liability | ( | ) | ||||||||||
| Issuance of common stock for non-cash consideration | ||||||||||||
| Finance charges | ||||||||||||
| Change in fair value of derivative liabilities | ( | ) | ( | ) | ||||||||
| Change in fair value of warrant liabilities | ( | ) | ( | ) | ||||||||
| Changes in operating assets and liabilities: | ||||||||||||
| Accounts receivable | ||||||||||||
| Inventory | ||||||||||||
| Prepaids and other current assets | ( | ) | ( | ) | ||||||||
| Accounts payable | ( | ) | ( | ) | ||||||||
| Accrued expenses | ( | ) | ( | ) | ||||||||
| Customer deposits | ||||||||||||
| Operating lease liability | ( | ) | ( | ) | ||||||||
| Net cash used by operating activities | ( | ) | ( | ) | ||||||||
| Cash flows from investing activities | ||||||||||||
| Additions to property and equipment | ( | ) | ( | ) | ||||||||
| Additions to intangible assets | ( | ) | ( | ) | ||||||||
| Net cash used by investing activities | ( | ) | ( | ) | ||||||||
| Cash flows from financing activities | ||||||||||||
| Proceeds from issuance of common stock, net of issuance costs | ||||||||||||
| Offering costs in advance of sale of common stock | ( | ) | ( | ) | ||||||||
| Principal payments on notes payable | ( | ) | ( | ) | ||||||||
| Proceeds from notes payable | ||||||||||||
| Proceeds from convertible notes, net of fees | ||||||||||||
| Net cash provided by financing activities | ||||||||||||
| Net decrease in cash and cash equivalents | ||||||||||||
| Cash and cash equivalents at beginning of period | ||||||||||||
| Cash and cash equivalents at end of period | $ | $ | $ | |||||||||
| F-35 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| NEURAXIS, INC. | ||
| Dated: April 16, 2024 | By: | /s/ Brian Carrico |
| Brian Carrico | ||
| Chief Executive Officer | ||
| Dated: April 16, 2024 | By: | /s/ Timothy R. Henrichs |
| Timothy R. Henrichs | ||
| Chief Financial Officer | ||
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ Brian Carrico | Chief Executive Officer and Director | April 16, 2024 | ||
| Brian Carrico | (Principal Executive Officer) | |||
| /s/ Timothy R. Henrichs | Chief Financial Officer (Principal | April 16, 2024 | ||
| Timothy R. Henrichs | Financial Officer and Principal Accounting Officer) | |||
| /s/ Christopher Robin Brown | Director | April 16, 2024 | ||
| Christopher Robin Brown | ||||
| /s/ Bradley Mitch Watkins | Director | April 16, 2024 | ||
| Bradley Mitch Watkins | ||||
| /s/ Beth Keyser | Director | April 16, 2024 | ||
| Beth Keyser | ||||
| /s/ Kristin Ferge | Director | April 16, 2024 | ||
| Kristin Ferge |
| 90 |